Область техники, к которой относится изобретение
Настоящее изобретение относится к соединениям и их применению в терапии.
Уровень техники
Рак является широко распространенным: в Европе диагностировано примерно 3,2 миллионов случаев рака (53% у мужчин, 47% у женщин) и зарегистрировано 1,7 миллионов случаев смерти от рака (56% мужчин, 44% женщин) (Ferlay et al. (2007) Ann. Oncol. 18(3):581-92). В США вероятность развития инвазивного рака в возрасте 70 лет и старше составляет 38% для женщин и 46% для мужчин. В 2006 г. в США ожидается примерно 1,4 миллионов новых случаев рака. Хотя в настоящее время 5-летний коэффициент выживаемости при раке составляет 65%, в середине 1970-х гг. примерно в 50% случаев рак являлся смертельным. По оценкам в 2006 г. в США от рака погибнут 565000 человек (American Cancer Society, Surveillance Research, 2006). Несмотря на значительные успехи в лечении и диагностике рака, рак остается главной проблемой общественного здравоохранения. Поэтому необходимы новые терапевтические средства, активные при раке.
В промышленно развитых странах проявляется другой кризис, связанный со здравоохранением. По мере старения населения этих стран нейродегенеративные заболевания поражают все большее количество людей, что приводит к большой экономической нагрузке на национальные системы здравоохранения. Самым распространенным нейродегенеративным заболеванием является болезнь Альцгеймера; давно проводится поиск лекарственных средств, воздействующих на это заболевание, но до настоящего времени они не обнаружены. Другими нейродегенеративными патологическими состояниями являются болезнь Паркинсона, болезнь Гентингтона, слабоумие с тельцами Леви, которые характеризуются прогрессированием, лишающим пациентов способности к нормальной ежедневной деятельности и в конечном счете приводящим к смерти.
Одной сходной характеристикой многих типов раковых и нейродегенеративных заболеваний является аберрантная экспрессия генов. Показано, что экспрессию генов изменяет целый ряд соединений, включая ингибиторы гистондеацетилазы, который изменяет профиль ацетилирования хроматина гистоном. Показано, что ингибиторы гистондеацетилазы, такие как SAHA (субероиланилид гидроксамовой кислоты), TSA (трихостатин А) и многие другие, меняют экспрессию генов в различных моделях in vitro и in vivo на животных. Другой модификацией, которая участвует в регуляции экспрессии генов, является метилирование гистона. Гистоны могут подвергаться многочисленным модификациям, включая метилирование лизина и аргинина. Недавно показано, что статус метилирования гистонлизинов важен для динамической регуляции экспрессии генов.
В модификациях гистонлизина участвует группа ферментов, известных, как гистонлизинметилтрансферазы и гистонлизиндеметилазы. Недавно было установлено (Shi et al. (2004) Cell 119:941), что одна конкретная гистонлизиндеметилаза человека под названием лизинспецифическая деметилаза-1 (LSD1) участвует в этой критически важной модификации гистона. Инактивация LSD1 у дрозофилы (dLSD1) сильно влияет на общий уровень метилирования моно- и диметил-H3-K4, но не влияет на метил-H3K9, тогда как степень метилирования некоторых других гистонов и количество меток ацетилирования остаются такими же. Инактивация dLSD1 приводит к увеличенной экспрессии субпопуляции генов, включая нейронные гены в ненейронных клетках, аналогичной функциям LSD1 в клетках человека. У дрозофилы dLSD1 не является незаменимым геном, но у мутантных животных выживаемость сильно уменьшается и зависит от пола (Destefano et al. (2007) Curr Biol. 17(9):808-12). Гомозиготные лишенные LSD1 мыши гибнут на стадии эмбрионов.
LSD1 характеризуется значительным структурным сходством и идентичности/гомологии аминокислот с полиаминоксидазами и моноаминоксидазами, каждая из которых (т.е. МАО-А, МАО-В и LSD1) является зависимой от флавина аминоксидазой, которые катализируют окисление связей азот-водород и/или связей азот-углерод. Недавние эксперименты с LSD1 показали, что она участвует в различных процессах, таких как онкогенез (Kahl et al. (2006) Cancer Res. 66:1341-11347) и воспаление сосудов (Reddy et al. (2008) Circ. Res. 103:615). Установлено, что имеющийся в продаже антидепрессант, парнат®, который направленно воздействует на моноаминоксидазау (МАО), при клинически приемлемых концентрациях также ингибирует LSD1 (Lee et al. (2006) Chem. Biol. 13:563-567). Schmidt et al. обнаружили, что ″значения IC50 для 2-РСРА составляют 20,7±2,1 мкМ для LSD1, 2,3±0,2 мкМ для МАО-А и 0,95±0,07 мкМ для МАО-В.″ См. публикацию Schmidt et al. (2007) Biochemistry 46(14)4408-4416. Так, парнат (2-РСРА) является лучшим ингибитором МАО-А и МАО-В, чем LSD1. В публикации Schmidt et al. отмечено, что значения IC50 для необратимых ингибиторов LSD1, таких как парнат, могут сильно зависеть от условий проведения анализа. Кроме того, производные парната также могут ингибировать LSD1 (Gooden et al. (2008) Bioorg. Med. Chem. Let. 18:3047-3051). Другим классом соединений, для которых недавно установлено, что они ингибируют активность LSD1, являются полиамины (Huang et al. (2007) PNAS 104:8023-8028). Эти полиамины умеренно ингибируют LSD1 и показано, что в раковых клетках они приводят к реэкспресии генов, которые в результате аберрации стали молчащими.
LSD1 также участвует в регуляции метилирования лизинов в некоторых белках, которые не являются гистонами, таких Р53 и DNMT1, которые оба играют критически важную роль при раке.
В публикации Lee et al. ((2006) Chem. Biol. 13:563-567) сообщают, что транилципромин ингибирует деметилирование гистона H3K4 и может подавлять экспрессию гена Egr1 в некоторых линиях раковых клеток. Имеется много данных о том, что во многих случаях Egr-1 является геном-супрессором опухолей. В публикации Calogero et al. ((2004) Cancer Cell International 4:1) сообщают, что Egr-1 подвергается понижающей регуляции при раке головного мозга и экзогенная экспрессия Egr-1 приводит к остановке роста и в конечном счете к гибели первичных линий раковых клеток. В публикации Lucerna et al. ((2006) Cancer Research 66, 6708-6713) показано, что длительная экспрессия Egr-1 в некоторых моделях приводит к антиангиогенному эффекту и подавляет рост опухолей. В публикации Ferraro et al. ((2005,) J. Clin. Oncol. Mar 20; 23(9):1921-6) сообщают, что Egr-1 подвергается понижающей регуляции у пациентов, страдающих раком легких с высоким риском рецидива, и может быть более устойчивым к терапии. В публикации Scoumanne et al. ((2007) J Biol Chem. May 25; 282(21): 15471-5) установлено, что LSD1 необходим для пролиферации клеток. Также установлено, что недостаток LSD1 приводит частичной остановке клеточного цикла в G2/M и сенсибилизированных клетках и усилению супрессии, вызванной повреждением ДНК. В публикации Kahl et al. ((2006) Cancer Res. 66(23): 11341-7) установлено, что экспрессия LSD1 коррелирует с агрессивностью рака предстательной железы. В публикации Metzger et al. ((2005) Nature 15; 437(7057):436-9) сообщают, что модулирование LSD1 с помощью сиРНК (малая интерферирующая РНК) и паргилина регулирует андрогенный рецептор (АР) и, вероятно, может использоваться для лечения типов рака, при которых играет роль АР, таких как рак предстательной железы, яичников и головного мозга. Таким образом, имеется много данных о том, что LSD1 участвует в раковых заболеваниях, что указывает на то, что LSD1 является мишенью терапевтического воздействия при раке.
Фенилциклопропиламины были объектом многих исследований, посвященных изучению соотношений структура-активность для ингибирования МАО. В публикациях Kaiser et al. ((1962) J. Med. Chem. 5:1243-1265); Zirkle et al. ((1962) J. Med. Chem. 1265-1284; патенты США №№3365458; 3471522; 3532749) описаны синтез и активность целого ряда соединений, родственных фенилциклопропиламину. В публикации Zirkle et al. ((1962) J. Med. Chem. 1265-1284) сообщают, что по данным исследования по увеличению активности МАО триптамином моно- и дизамещение аминогруппы в транс-2-фенилциклопропиламине метальными группами лишь немного уменьшает активность, тогда как монозамещение более крупными группами, такими как алкильные и арилалкильные группы приводит к значительному уменьшению активности. Также проведены исследования соединений, родственных фенилциклопропиламину, для определения селективности по отношению к МАО-А по сравнению с селективностью по отношению к МАО-В, поскольку ингибиторы МАО-А могут привести к опасным побочным эффектам (см., например, публикации Yoshida et al. (2004) Bioorg. Med Chem. 12(10):2645-2652; Hruschka et al. (2008) Biorg Med Chem. (16):7148-7166; Folks et al. (1983) J. Clin. Psychopharmacol. (3)249; и Youdim et al. (1983) Mod. Probl. Pharmacopsychiatry (19):63). Другие соединения типа фенилциклопропиламина описаны в публикациях Bolesov et al. ((1974) Zhurnal Organicheskoi Khimii 10:8 1661-1669) и патент РФ №230169 (19681030). В публикации Gooden et al. ((2008) Bioorg. Med. Chem. Let. 18:3047-3051) описан синтез производных и аналогов фенилциклопропиламинов, а также их активности по отношению к МАО-А, МАО-В и LSD1. Ни одно из соединений, описанных в публикации Gooden et al., не характеризуется меньшим значением Ki по отношению к LSD1, чем по отношению к МАО-А или МАО-В. Кроме того, большинство описанных в публикации Gooden et al. производных фенилциклопропиламина являются лучшими ингибиторами для МАО-А, чем для МАО-В.
Ввиду отсутствия адекватных средств лечения таких патологических состояний как рак, настоятельно необходимы лекарственные средства, влияющие на заболеваний, и лекарственные средства, которые воздействуют путем ингибирования новых мишеней. Необходима разработка новых селективных ингибиторов LSD1, в особенности таких, которые селективно ингибируют LSD1.
Краткое изложение сущности изобретения
Настоящее изобретение относится к идентификации соединений и к их применению для лечения или предупреждения заболеваний. Настоящее изобретение относится к соединению формулы 1 или его фармацевтически приемлемой соли или сольвату, к фармацевтической композиции, содержащей соединение формулы 1 или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, и к их применению для лечения или предупреждения заболеваний. Одним применением соединения формулы 1 является лечение или предупреждение рака. Другим применением соединения формулы 1 является ингибирование LSD1. Таким образом, настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения рака. Настоящее изобретение также относится к методикам скрининга для идентификации замещенных гетероарил- и арилциклопропиламинов, которые являются селективными ингибиторами LSD1, и к их применению для лечения или предупреждения заболеваний.
Первым объектом настоящего изобретения является соединение формулы 1 или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемая соль или сольват:
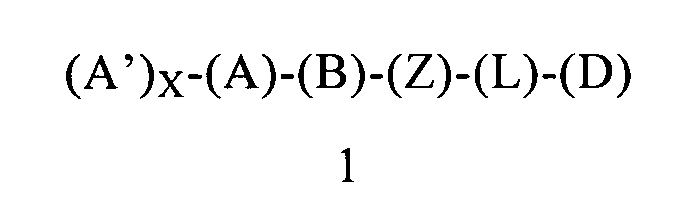
в которой:
(А) обозначает гетероарил или арил, ковалентно связанный с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где каждый (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) или с (D), где (L) обозначает ординарную связь, и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил ковалентно связаны с (L) или с (Z), если (L) обозначает ординарную связь, и где (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), амидогруппу, алкил, галоген, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу;
при условии, что исключены следующие соединения:
N-(2-фенилциклопропил)циклопентанамин;
10,11-дигидро-N-(2-фенилциклопропил)-5Н-дибензо[а,d]циклогептен-5-амин; и
транс-N-(2-фенилциклопропил)циклогексиламин.
Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы 1 или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват, определенное выше, и фармацевтически приемлемый носитель. Предпочтительные варианты осуществления соединения формулы 1, предназначенного для применения в композиции этого первого объекта, определены ниже в этом первом объекте настоящего изобретения и описаны в разделе ″Подробное описание изобретения″.
В одном варианте осуществления этого первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
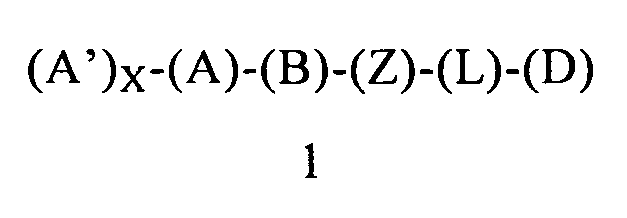
в которой:
(A) обозначает гетероарильную или арильную группу, ковалентно связанную с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) или с (D), где (L) обозначает ординарную связь, и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает циклоалкил, ковалентно связанный с (L) или с (Z), если (L) обозначает ординарную связь, и (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу;
при условии, что исключены следующие соединения:
N-(2-фенилциклопропил)циклопентанамин;
транс-N-(2-фенилциклопропил)циклогексиламин.
В другом варианте осуществления этого первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
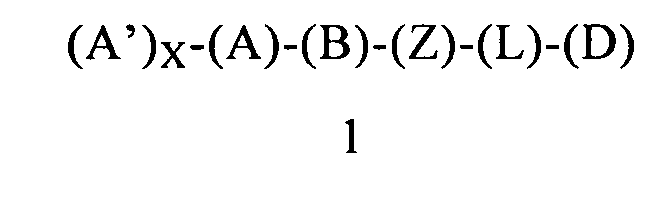
в которой:
(A) обозначает гетероарильную или арильную группу, ковалентно связанную с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (D) и с атомом водорода;
(L) обозначает мостик, который представляет собой ординарную ковалентную связь, связывающую (Z) с (D);
(D) обозначает бензоциклоалкил, ковалентно связанный с (Z), и (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу;
при условии, что исключено следующее соединение:
10,11-дигидро-N-(2-фенилциклопропил)-5Н-дибензо[а,d]циклогептен-5-амин.
В родственном объекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы 1 или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват, определенные выше в первом объекте настоящего изобретения или в его варианте осуществления, и фармацевтически приемлемый носитель.
Вторым объектом настоящего изобретения является способ лечения или предупреждения заболевания или патологического состояния, включающий введение пациенту (предпочтительно человеку), нуждающемуся в лечении или предупреждении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы 1, определенное выше в первом объекте настоящего изобретения или в описанном выше его варианте осуществления, или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель. Этим объектом настоящего изобретения также является соединение формулы 1, предназначенное для применения в качестве лекарственного средства.
Третьим объектом настоящего изобретения является способ ингибирования активности LSD1, включающий введение пациенту (предпочтительно человеку), нуждающемуся в лечении, терапевтически эффективного количества композиции, содержащей соединение формулы 1 или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват:
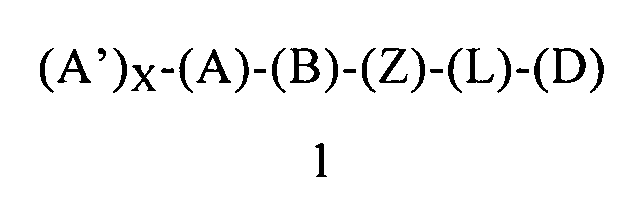
в которой:
(А) обозначает гетероарил или арил, ковалентно связанный с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где каждый (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) или с (D), где (L) обозначает ординарную связь, и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил ковалентно связаны с (L) или с (Z), если (L) обозначает ординарную связь, и где (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу,
и фармацевтически приемлемый носитель, в количестве, достаточном для ингибирования активности LSD1. Этот объект можно переформулировать, как соединение формулы 1, определенное в настоящем изобретении, предназначенное для применения в качестве ингибитора LSD1. Этот объект можно переформулировать, как соединение формулы 1, предназначенное для применения для лечения заболевания, связанного с LSD1. Родственным объектом является способ лечения индивидуума (предпочтительно человека), который включает идентификацию индивидуума, нуждающегося в лечении, и введение указанному индивидууму соединения формулы 1 в терапевтически эффективном количестве. В предпочтительном объекте терапевтически эффективное количество соединения формулы 1 является количеством, достаточным для ингибирования LSD1. Предпочтительные варианты осуществления соединений формулы 1, предназначенных для применения в композиции и способе этого третьего объекта настоящего изобретения, являются такими, как определено выше в первом объекте настоящего изобретения.
Четвертым объектом настоящего изобретения является способ лечения или предупреждения рака, включающий введение пациенту (предпочтительно человеку), нуждающемуся в лечении или предупреждении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы 1, определенное выше в первом объекте настоящего изобретения, или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель. Этим объектом настоящего изобретения также является соединение формулы 1, определенное выше в первом объекте настоящего изобретения, или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемая соль или сольват, предназначенное для применения для лечения или предупреждения рака. Предпочтительно, если рак выбран из группы, включающей рак молочной железы, колоректальный рак, рак легких, рак предстательной железы, тестикулярный рак, рак головного мозга, рак кожи и рак крови. В предпочтительном объекте терапевтически эффективное количество соединения формулы 1 является количеством, достаточным для ингибирования LSD1.
Пятым объектом настоящего изобретения является способ идентификации соединения, которое является селективным ингибитором LSD1, который включает выбор или получение соединения, которое является замещенным гетероарилциклопропиламином или замещенным арилциклопропиламином, и определение способности соединения ингибировать LSD1 и МАО-А и/или МАО-В, где соединение которое ингибирует LSD1 в большей степени, чем МАО-А и/или МАО-В, идентифицируют как селективный ингибитор LSD1. Кроме того, этот способ применим для идентификации двойных ингибиторов LSD1 и МАО-В, которыми являются соединения, которые ингибируют LSD1 и МАО-В в большей степени чем МАО-А.
В контексте этого пятого объекта настоящего изобретения, замещенный гетероарилциклопропиламин или замещенный арилциклопропиламин имеет приведенную ниже формулу 2:
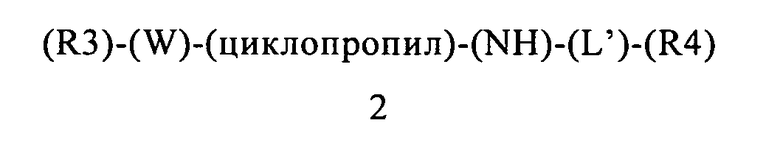
в которой:
(R3) содержится или не содержится, если (R3) содержится, то он выбран из группы, включающей арилалкил, арилалкоксигруппу, гетероциклилалкил и гетероциклилалкоксигруппу, где указанная группа (R3) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, алкил, алкоксигруппу, карбоциклил, циклоалкил, циклоалкоксигруппу, галогеналкил, галогеналкоксигруппу, ациламиногруппу, ацилоксигруппу, алкилтиогруппу, циклоалкилтиогруппу, алкинил, аминогруппу, алкиламиногруппу, арил, арилалкил, арилалкенил, арилалкинил, арилалкоксигруппу, арилоксигруппу, арилтиогруппу, гетероарилтиогруппу, цианогруппу, цианатную группу, галогенарил, гидроксигруппу, гетероциклил, гетероарилоксигруппу, гетероарилалкоксигруппу, изоцианатную группу, изотиоцианатную группу, нитрогруппу, сульфинил, сульфонил, сульфонамидогруппу, тиокарбонил, тиоцианатную группу, тригалогенметансульфонамидогруппу, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил и С-амидогруппу, и указанная группа (R3) ковалентно связана с (W);
(W) обозначает арильную или гетероарильную группу, ковалентно связанную с (R3) и с (циклопропил), где указанный (W) содержит 0, 1 или 2 заместителя, не включая (R3) и (циклопропил), где указанные заместители (W) независимо выбраны из группы, включающей галоген, алкил, алкоксигруппу, карбоциклил, циклоалкил, циклоалкоксигруппу, галогеналкил, галогеналкоксигруппу, ациламиногруппу, ацилоксигруппу, алкилтиогруппу, циклоалкилтиогруппу, алкинил, аминогруппу, алкиламиногруппу, арил, арилалкил, арилалкенил, арилалкинил, арилалкоксигруппу, арилоксигруппу, арилтиогруппу, гетероарилтиогруппу, цианогруппу, цианатную группу, галогенарил, гидроксигруппу, гетероциклил, гетероарилоксигруппу, гетероарилалкоксигруппу, изоцианатную группу, изотиоцианатную группу, нитрогруппу, сульфинил, сульфонил, сульфонамидогруппу, тиокарбонил, тиоцианатную группу, тригалогенметансульфонамидогруппу, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил и С-амидогруппу;
(циклопропил) обозначает циклопропильную группу, ковалентно связанную с (W) и с атомом азота группы (NH);
(NH) обозначает группу -NH- (атом азота, ковалентно связанный с атомом водорода), где атом азота ковалентно связан с (циклопропил) и с (L′), или, если n=0, то (L′) обозначает ординарную связь и атом азота группы -NH-ковалентно связан с (R4);
(L′) обозначает мостик формулы -(CH2)n-, где n равно 0, 1, 2, 3, 4, 5 или 6, где (L′) ковалентно связан с атомом азота группы (NH) и с (R4), или (L′) обозначает ординарную связь, которая ковалентно связывает (NH) и (R4), если n равно 0; и
(R4) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил ковалентно связан с (L′) или с атомом азота группы (NH), если (L′) обозначает ординарную связь, где указанная группа (R4) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, алкил, алкоксигруппу, карбоциклил, циклоалкил, циклоалкоксигруппу, галогеналкил, галогеналкоксигруппу, ациламиногруппу, ацилоксигруппу, алкилтиогруппу, циклоалкилтиогруппу, алкинил, аминогруппу, алкиламиногруппу, арил, арилалкил, арилалкенил, арилалкинил, арилалкоксигруппу, арилоксигруппу, арилтиогруппу, гетероарилтиогруппу, цианогруппу, цианатную группу, галогенарил, гидроксигруппу, гетероциклил, гетероарилоксигруппу, гетероарилалкоксигруппу, изоцианатную группу, изотиоцианатную группу, нитрогруппу, сульфинил, сульфонил, сульфонамидогруппу, тиокарбонил, тиоцианатную группу, тригалогенметансульфонамидогруппу, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил и С-амидогруппу.
Согласно пятому объекту настоящего изобретения, соединение формулы 2 является селективным ингибитором LSD1. Селективный ингибитор LSD1, идентифицированный с помощью способа этого варианта осуществления, можно использовать для приготовления фармацевтической композиции, содержащей указанный селективный ингибитор LSD1 формулы 2 или его фармацевтически приемлемую соль или сольват в терапевтически эффективном количестве и фармацевтически приемлемый носитель. Фармацевтическую композицию можно вводить индивидууму, нуждающемуся в таком лечении. В соответствии с этим настоящее изобретение также относится к способу лечения или предупреждения заболевания или патологического состояния, который включает введение пациенту (предпочтительно человеку), нуждающемуся в лечении или предупреждении, терапевтически эффективного количества фармацевтической композиции, содержащей указанный селективный ингибитор LSD1 формулы 2 и фармацевтически приемлемый носитель. В этом варианте осуществления терапевтически эффективное количество является количеством, достаточным для селективного ингибирования LSD1.
Таким образом, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы 1 или 2, которое является селективным ингибитором LSD1. Селективные ингибиторы LSD1 характеризуются значениями Ki для LSD1, которые не менее, чем в 2 раза меньше, чем значение Ki для МАО-А и/или МАО-В. В одном воплощении этого варианта осуществления значение Ki для LSD1 не менее, чем в 5 раз меньше, чем значение Ki для МАО-А и/или МАО-В. В одном воплощении этого варианта осуществления значение Ki для LSD1 не менее, чем в 10 раз меньше, чем значение Ki для МАО-А и МАО-В. В одном варианте осуществления настоящего изобретения фармацевтическая композиция, содержащая селективный ингибитор LSD1 формулы 1 или 2 или его фармацевтически приемлемую соль или сольват, используют для лечения и/или предупреждения заболевания у индивидуума. В одном объекте терапевтически эффективное количество композиции вводят индивидууму в количестве, достаточном для лечения или предупреждения заболевания. В более предпочтительном объекте заболеванием является рак. В еще более предпочтительном объекте заболеванием является рак, выбранный из группы, включающей рак предстательной железы, тестикулярный рак, рак головного мозга, колоректальный рак, рак легких, рак молочной железы, рак кожи и рак крови. В одном предпочтительном объекте раком является рак предстательной железы. В одном предпочтительном объекте раком является рак легких. В одном предпочтительном объекте раком является рак головного мозга. В одном предпочтительном объекте раком является рак крови (например, лейкоз). В одном предпочтительном объекте раком является рак молочной железы. В одном предпочтительном объекте раком является колоректальный рак.
Согласно пятому объекту настоящего изобретения соединение формулы 2 является двойным ингибитором LSD1 и МАО-В. Двойной ингибитор LSD1/МАО-В, идентифицированный с помощью способа этого варианта осуществления, можно использовать для приготовления фармацевтической композиции, содержащей указанный двойной ингибитор LSD1/МАО-В формулы 2 или его фармацевтически приемлемую соль или сольват в терапевтически эффективном количестве и фармацевтически приемлемый носитель. Фармацевтическую композицию можно вводить индивидууму, нуждающемуся в таком лечении. В этом варианте осуществления терапевтически эффективное количество является количеством, достаточным для ингибирования МАО-В и LSD1.
Таким образом, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы 1 или 2, которое является двойным ингибитором LSD1 и МАО-В. Предпочтительно, если двойные ингибиторы LSD1/МАО-В характеризуются значениями Ki для LSD1 и МАО-В, которые не менее, чем в 2 раза меньше, чем значение Ki для МАО-А. В одном воплощении этого варианта осуществления значения Ki для LSD1 и МАО-В Ki не менее, чем в 5 раз меньше, чем значение Ki для МАО-А. В одном воплощении этого варианта осуществления значения Ki для LSD1 и МАО-В Ki не менее, чем в 10 раз меньше, чем значение Ki для МАО-А.
Предполагается, что соединения формулы 1 или 2, которые обладают ингибирующей активностью по отношению к МАО-В, полезны для лечения заболеваний, при которых ингибирование МАО-В является терапевтически целесообразным, таких как депрессия и нейродегенеративные заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона и болезнь Гентингтона.
Результаты недавних исследований показывают, что LSD1 участвует в вирусной инфекции и реактивации. В частности, показано, что фармакологические ингибиторы LSD1, такие как парнат и сиРНК, подавляющие LSD1, вызывают снижение вирусной нагрузки и снижение реактивации после скрытого периода (Liang et al. (2009) Nat. Med. 15:1312-1317). Поэтому предполагается, что соединения, предлагаемые в настоящем изобретении, включая соединения формулы 1, определенные и описанные в настоящем изобретении, можно использовать для лечения или предупреждения вирусной инфекции. Кроме того, предполагается, что соединения, предлагаемые в настоящем изобретении, можно использовать для лечения или предупреждения реактивации вирусной инфекции после скрытого периода.
Таким образом, в одном варианте осуществления настоящее изобретение относится к способу лечения или предупреждения вирусной инфекции, который включает введение пациенту/индивидууму (предпочтительно человеку), нуждающемуся в лечении или предупреждении, соединения формулы 1, определенного выше в любом из объектов и вариантов осуществления настоящего изобретения, или его фармацевтически приемлемой соли или сольвата в терапевтически эффективном количестве, или фармацевтической композиции, содержащей любое из указанных выше соединений и фармацевтически приемлемый носитель. В соответствии с этим настоящее изобретение также относится к соединению формулы 1, определенному выше в любом из объектов и вариантов осуществления настоящего изобретения, или к его фармацевтически приемлемой соли или сольвату, или к фармацевтической композиции, содержащей любое из указанных выше соединений и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения вирусной инфекции. В предпочтительном варианте осуществления вирусной инфекцией является вирусная инфекция герпеса. В еще более предпочтительном варианте осуществления вирусная инфекция герпеса вызвана вирусом герпеса, выбранным из группы, включающей HSV-1 (вирус простого герпеса типа 1), HSV-2 (вирус простого герпеса типа 2) и вирус Эпштейна-Барра, и/или связана с ним. В одном воплощении этого варианта осуществления вирусная инфекция вызвана ВИЧ (вирус иммунодефицита человека) и/или связана с ним.
В одном варианте осуществления настоящее изобретение относится к способу лечения или предупреждения реактивации вирусной инфекции после скрытого периода, который включает введение пациенту/индивидууму (предпочтительно человеку), нуждающемуся в лечении или предупреждении, соединения формулы 1, определенного выше в любом из объектов и вариантов осуществления настоящего изобретения, или его фармацевтически приемлемой соли или сольвата в терапевтически эффективном количестве, или фармацевтической композиции, содержащей любое из указанных выше соединений и фармацевтически приемлемый носитель. В соответствии с этим настоящее изобретение также относится к соединению формулы 1, определенному выше в любом из объектов и вариантов осуществления настоящего изобретения, или к его фармацевтически приемлемой соли или сольвату, или к фармацевтической композиции, содержащей любое из указанных выше соединений и фармацевтически приемлемый носитель, предназначенной для применения для лечения или предупреждения реактивации вирусной инфекции после скрытого периода. В предпочтительном варианте осуществления вирусом, который реактивируется, является вирус герпеса. В еще более предпочтительном варианте осуществления вирус герпеса, который реактивируется, выбран из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра. В одном воплощении этого варианта осуществления вирусом, который реактивируется, является ВИЧ.
Если не приведено другое определение, то все технические и научные термины, использующиеся в настоящем изобретении, обладают такими же значениями, которые обычно известны специалисту с общей подготовкой в области техники, к которой относится настоящее изобретение. Хотя при практическом осуществлении или тестировании настоящего изобретения можно использовать методики и материалы, сходные с описанными в настоящем изобретении или эквивалентные им, подходящие методики и материалы описаны ниже. В случае противоречий необходимо руководствоваться настоящим описанием, включая определения. Кроме того, материалы, методики и примеры являются лишь иллюстративными и их не следует рассматривать в качестве ограничивающих.
Другие особенности и преимущества настоящего изобретения очевидны из приведенного ниже подробного описания и из формулы изобретения.
Подробное описание изобретения
Настоящее изобретение относится к идентификации соединений и к их применению для лечения или предупреждения заболеваний. Настоящее изобретение относится соединениям формулы 1, фармацевтическим композициям, содержащим соединение формулы 1 или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель, и к их применению для лечения или предупреждения заболеваний. Одним применением соединений формулы 1 является применение для лечения или предупреждения рака. Соединения формулы 1 можно использовать в качестве селективных ингибиторов LSD1, которые ингибируют LSD1 в большей степени, чем МАО-А и МАО-В. Некоторые соединения, предлагаемые в настоящем изобретении, ингибируют LSD1 и МАО-В в большей степени, чем МАО-А. В частности, согласно изобретению было установлено, что производные фенилциклопропиламина формулы 1 дают соединения, которые являются неожиданно активными ингибиторами LSD1. Примеры, описанные в настоящем изобретении, показывают, что все типичные соединения формулы 1 (например соединения примеров 1-16) характеризуются значениями Ki (IC50) для ингибирования LSD1, меньшими, чем 1000 нМ (см. таблицу 1) и многие характеризуются значениями Ki (IC50), меньшими, чем 500 нМ, что делает их примерно в 40-50 раз или более активными для ингибирования LSD1, чем транилципромин. Эти соединения селективны по отношению к LSD1, поскольку они ингибируют LSD1 в большей степени, чем МАО-А и МАО-В.
Первым объектом настоящего изобретения является соединение формулы 1 или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемая соль или сольват:
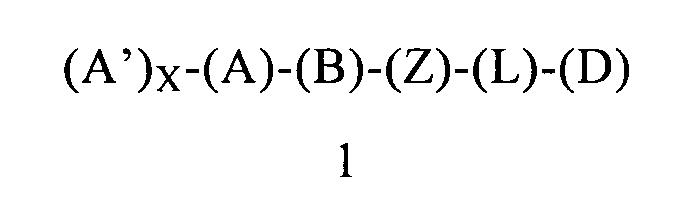
в которой:
(А) обозначает гетероарил или арил, ковалентно связанный с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает атом азота, ковалентно связанный с (В), с (L) и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил ковалентно связаны с (L) или с (Z), если (L) обозначает ординарную связь, и где (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу;
при условии, что исключены следующие соединения:
N-(2-фенилциклопропил)циклопентанамин;
10,11-дигидро-N-(2-фенилциклопропил)-5Н-дибензо[а,d]циклогептен-5-амин; и
транс-N-(2-фенилциклопропил)циклогексиламин.
Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы 1 или его фармацевтически приемлемую соль или сольват, определенное выше, и фармацевтически приемлемый носитель. Предпочтительные варианты осуществления соединения формулы 1, предназначенного для применения в композиции этого первого объекта, определены ниже в этом первом объекте настоящего изобретения.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (А) обозначает арильную группу и другие переменные являются такими, как определено выше в самом широком определении первого объекта настоящего изобретения. В предпочтительном варианте осуществления (А) обозначает фенил. В другом предпочтительном варианте осуществления (А) обозначает нафтил.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (А) обозначает фенильную группу и другие переменные являются такими, как определено выше в самом широком определении первого объекта настоящего изобретения или как определено в других вариантах осуществления первого объекта настоящего изобретения. В предпочтительном варианте осуществления (А) обозначает фенильную группу, содержащую 0, 1, 2 или 3 заместителя (А′), независимо выбранных из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где каждый (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил. В другом предпочтительном варианте осуществления (А) обозначает фенильную группу, содержащую 0, 1, 2 или 3 заместителя (А′), независимо выбранных из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где каждый (А′) содержит 0, 1 или 2 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой X равен 1 или 2, и другие переменные являются такими, как определено выше в самом широком определении первого объекта настоящего изобретения или как определено в других вариантах осуществления первого объекта настоящего изобретения. В предпочтительном варианте осуществления 1 или 2 группы (А′) независимо выбраны из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где каждая группа (А′) содержит 0, 1 или 2 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил. В более предпочтительном варианте осуществления 1 или 2 группы (А′) независимо выбраны из группы, включающей арил и арилалкоксигруппу, где указанная группа (А′) содержит 0, 1 или 2 заместителя, независимо выбранных из группы, включающей независимо галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил. В еще более предпочтительном варианте осуществления 1 или 2 группы (А′) независимо выбраны из группы, включающей фенил, бензилоксигруппу и фенетилоксигруппу, где указанная группа (А′) содержит 0, 1 или 2 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой X обозначает 1; и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила и другие переменные являются такими, как определено выше в самом широком определении первого объекта настоящего изобретения или как определено в других вариантах осуществления первого объекта настоящего изобретения. В одном предпочтительном варианте осуществления указанная группа (А′) содержит 1 заместитель, выбранный из галогена и галогеналкила. В другом предпочтительном варианте осуществления указанная группа (А′) является незамещенной.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (А) обозначает гетероарил и другие переменные являются такими, как определено выше в самом широком определении первого объекта настоящего изобретения или как определено в других вариантах осуществления первого объекта настоящего изобретения. В предпочтительном объекте (А) обозначает гетероарил, выбранный из пиридила, пиримидинила и тиофенила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (А) обозначает гетероарил, выбранный из группы, включающей пиридил, пиримидинил и тиофенил, и другие переменные являются такими, как определено выше в самом широком определении первого объекта настоящего изобретения. В предпочтительном воплощении этого варианта осуществления X равен 0 или 1. В другом предпочтительном объекте X равен О или 1 и (А′) обозначает арильную группу или арилалкоксигруппу, где указанная группа (А′), если она содержится, содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (L) обозначает мостик, выбранный из группы, включающей ординарную связь, -CH2-, -CH2CH2- и -CH2CH2CH2-. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (L) обозначает ординарную связь или -CH2-. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (L) обозначает -CH2CH2-. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (L) обозначает ординарную связь. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли, в которой (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил содержит 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления этого первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (D) обозначает циклоалкильную группу, замещенную амидной группой. Предпочтительно, если циклоалкильной группой является циклопропил, циклобутил, циклопентил или циклогексил. Еще более предпочтительно, если циклоалкилом является циклопропильная группа. В одном предпочтительном варианте осуществления амидная группа присоединена к атому углерода циклоалкильной группы, которая присоединена к (L). В предпочтительном воплощении этого варианта осуществления (А) обозначает арил, ковалентно связанный с (В) и с (А′); каждый (А′) ковалентно связан (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил и арилоксигруппу, где каждый (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, амидогруппу и -CH2C(=O)NH2.
X равен 1 или 2;
(В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-. В предпочтительном воплощении этого варианта осуществления амидная группа, ковалентно связанная с циклоалкильной группой (D), присоединена через тот же атом углерода циклоалкильной группы, что и группа (L).
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В) и где (А) и (Z) находятся в транс-положении по отношению к циклопропильному кольцу (В).
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В), где (А) и (Z) находятся в транс-положении по отношению к циклопропильному кольцу (В) и содержится 1 группа (А′) (X=1) и указанная группа (А′) находится в мета- или пара-положении по отношению к циклопропильному кольцу, где (А) обозначает фенильную группу. Предпочтительно, если одна группа (А′) находится в пара-положении по отношению к циклопропильному кольцу, где указанная группа (А′) выбрана из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа может содержать 0, 1 или 2 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил. Предпочтительно, если 0, 1 или 2 заместителя у (А′) независимо выбраны из галогена и галогеналкила.
Соединения формулы 1, в которой (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил (обозначенный ниже, как ″(карбоцикл)″), обладают общей структурой:
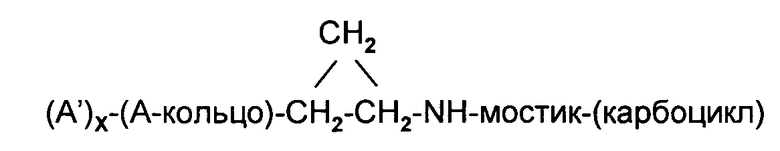
В одном варианте осуществления этого первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
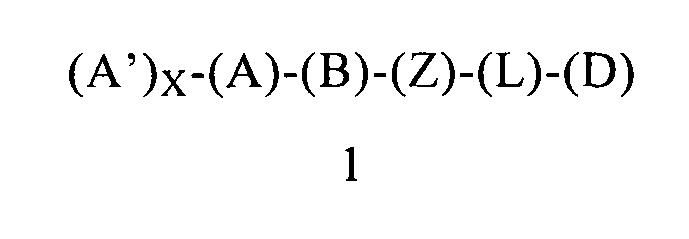
в которой:
(A) обозначает гетероарильную или арильную группу, ковалентно связанную с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где (А′) содержит 0, 1, 2 или 3 заместителя (например, 0, 1 или 2 заместителя), независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает циклоалкил, ковалентно связанный с (L) или с (Z), если (L) обозначает ординарную связь, и (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу;
при условии, что исключены следующие соединения:
N-(2-фенилциклопропил)циклопентанамин; и транс-N-(2-фенилциклопропил)циклогексиламин.
В другом варианте осуществления этого первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
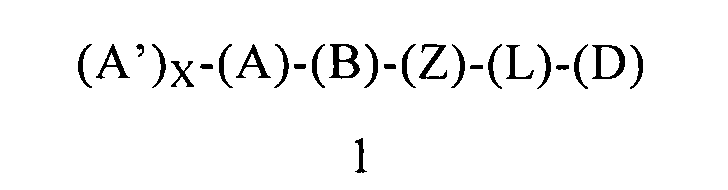
в которой:
(A) обозначает гетероарильную или арильную группу, ковалентно связанную с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где (А′) содержит 0, 1, 2 или 3 заместителя (например, 0, 1 или 2 заместителя), независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает атом азота, ковалентно связанный с (В), с (D) и с атомом азота;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик обозначает ординарную связь;
(D) обозначает бензоциклоалкил, ковалентно связанный с (Z), и (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу;
при условии, что исключено следующее соединение:
10,11-дигидро-N-(2-фенилциклопропил)-5Н-дибензо[а,d]циклогептен-5-амин.
Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и соединение формулы 1 или его фармацевтически приемлемую соль или сольват, определенное выше в первом объекте и родственных вариантах осуществления. Предпочтительные варианты осуществления соединения формулы 1, предназначенного для применения в композиции этого второго объекта, определены ниже в этом втором объекте настоящего изобретения.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (А) обозначает арильную группу. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х-1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (А) обозначает фенильную группу. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой X равен 1 или 2.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой X равен 1 и выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (А) обозначает гетероарил.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (А) обозначает гетероарил выбранный из пиридила, пиримидинила и тиофенила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (L) обозначает мостик, выбранный из группы, включающей -CH2CH2- и -CH2CH2CH2-. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (L) обозначает -CH2CH2-. В предпочтительном варианте осуществления в указанном соединении формулы 1 Х=1 и (А′) выбран из арила и арилалкоксигруппы, где указанная арильная группа или арилалкоксигруппа содержит 0 или 1 заместитель, выбранный из галогена и галогеналкила.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В) и где (А) и (Z) находятся в транс-положении по отношению к циклопропильному кольцу (В).
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, в которой (В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В) и где (А) и (Z) находятся в транс-положении по отношению к циклопропильному кольцу (В), и содержится 1 группа (А′) (X=1) и указанная группа (А′) находится в мета- или пара-положении по отношению к циклопропильному кольцу, где (А) обозначает фенильную группу. Предпочтительно, если одна группа (А′) находится в пара-положении по отношению к циклопропильному кольцу, где указанная группа (А′) выбрана из группы, включающей арил (например, фенил) и арилалкоксигруппу (например, бензилоксигруппа), где указанная арильная группа или арилалкоксигруппа может содержать 0, 1 или 2 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил. Предпочтительно, если 0, 1 или 2 заместителя у (А′) независимо выбраны из группы, включающей галоген и галогеналкил.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1, выбранному из группы, включающей:
N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-5,6-диметокси-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-4,5-диметокси-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин;
6-метокси-N-((транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин;
6-хлор-N-((транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-фенилциклопропил)-6-(трифторметил)-2,3-дигидро-1Н-инден-1-амин;
7-метокси-N-((транс)-2-фенилциклопропил)-1,2,3,4-тетрагидронафталин-1-амин;
N-((транс)-2-(3′-хлорбифенил-4-ил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-(4′-хлорбифенил-4-ил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин;
6-метокси-N-((транс)-2-(3′-метоксибифенил-4-ил)циклопропил)-2,3-дигидро-1Н-инден-1-амин;
N-транс-(2-циклогексилэтил)-2-фенилциклопропанамин;
(транс)-N-(3-циклогексилпропил)-2-фенилциклопропанамин;
(транс)-N-(2-циклогептилэтил)-2-фенилциклопропанамин;
(транс)-2-(4-(3-бромбензилокси)фенил)-N-(2-циклогексилэтил)-циклопропанамин;
N-((транс)-2-(4-(3-бромбензилокси)фенил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин;
(транс)-2-(3′-хлорбифенил-4-ил)-N-(2-циклогексилэтил)циклопропанамин;
(транс)-2-(4′-хлорбифенил-4-ил)-N-(2-циклогексилэтил)циклопропанамин;
(транс)-N-(2-циклогексилэтил)-2-(3′-метоксибифенил-4-ил)циклопропанамин;
N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-7-метокси-1,2,3,4-тетрагидронафталин-1-амин; и
1-((транс)-2-(4-(бензилокси)фенил)циклопропиламино)циклопропан-карбоксамид;
или его фармацевтически приемлемой соли или сольвату.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
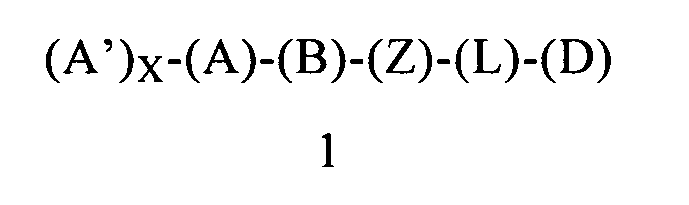
в которой:
(A) обозначает гетероарильную или арильную группу, ковалентно связанную с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу, цианогруппу, где (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает циклоалкильную группу, ковалентно связанную с (L), и (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу. Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы 1, определенное выше, и фармацевтически приемлемый носитель.
В одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
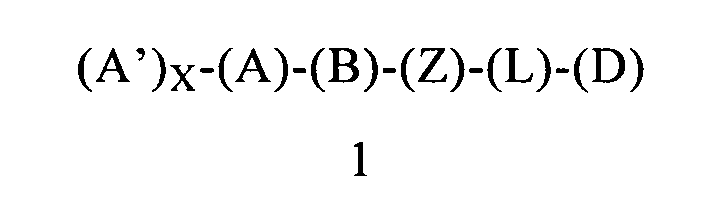
в которой:
(A) обозначает гетероарильную или арильную группу, ковалентно связанную с (В) и с (А′), если они содержатся;
(А′), если он содержится, выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу, цианогруппу, где (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) ковалентно связан (L), где (D) обозначает циклоалкильную группу, выбранную из группы, включающей циклопентил, циклогексил и циклогептил, и
(D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу. Предпочтительно, если заместители у (D) независимо выбраны из группы, включающей -NH2, -NH(C1-C6-алкил) и -N(C1-C6-алкил)(C1-C6-алкил). Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы 1 или его фармацевтически приемлемую соль или сольват, определенное выше, и фармацевтически приемлемый носитель.
В еще одном предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
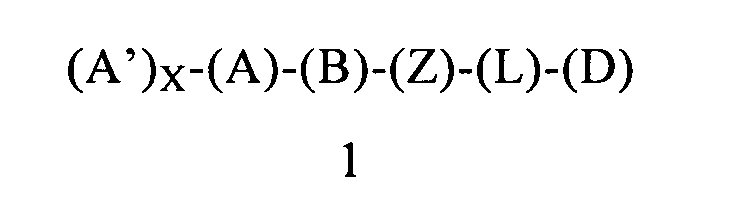
в которой:
(A) обозначает гетероарильную или арильную группу, ковалентно связанную с (В) и с (А′);
(А′) выбран из группы, включающей арильную группу или арилалкоксигруппу, где (А′) содержит 0, 1 или 2 заместителя, независимо выбранных из группы, включающей галоген и галогеналкил;
X равен 1;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) или с (D), где (L) обозначает ординарную связь, и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил ковалентно связаны с (L) или с (Z), если (L) обозначает ординарную связь, и где (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу. Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы 1, определенное выше, и фармацевтически приемлемый носитель.
В другом предпочтительном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
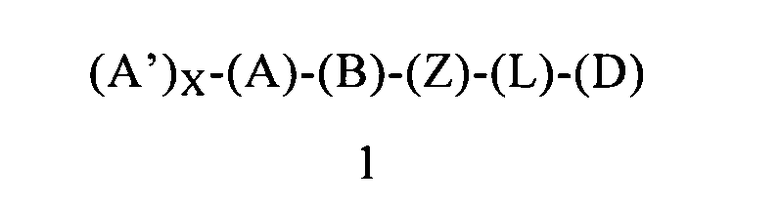
в которой:
(A) обозначает фенильную группу, ковалентно связанную с (В) и с (А′);
(А′) выбран из группы, включающей арильную группу или арилалкоксигруппу, где (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген и галогеналкил;
X равен 1;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В), где группы (А) и (Z) находятся в транс-положении по отношению к циклопропильному кольцу (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (D) и с атомом водорода;
(L) обозначает мостик, который представляет собой ординарную ковалентную связь, связывающую (Z) и (D);
(D) обозначает бензоциклоалкил, ковалентно связанный с (Z), и (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу. Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы 1 или его фармацевтически приемлемую соль или сольват, определенное выше, и фармацевтически приемлемый носитель.
В одном варианте осуществления первого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату:
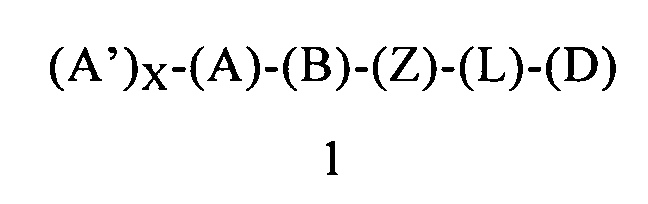
в которой:
(A) обозначает гетероарил, ковалентно связанный с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где каждый (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(B) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) или с (D), где (L) обозначает ординарную связь, и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил ковалентно связаны с (L) или с (Z), если (L) обозначает ординарную связь, и где (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу. Родственным объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы 1 или его фармацевтически приемлемую соль или сольват, определенное выше, и фармацевтически приемлемый носитель.
Вторым объектом настоящего изобретения является способ лечения или предупреждения заболевания или патологического состояния, этот способ включает введение пациенту (предпочтительно человеку), нуждающемуся в лечении или предупреждении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы 1, определенное выше, или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель. Этим объектом настоящего изобретения также является соединение формулы 1, определенное выше, или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемая соль или сольват, предназначенный для применения в качестве лекарственного средства.
Третьим объектом настоящего изобретения является способу ингибирования активности LSD1, включающий введение пациенту (предпочтительно человеку), нуждающемуся в лечении, терапевтически эффективного количества композиции, содержащей соединение формулы 1 или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват:
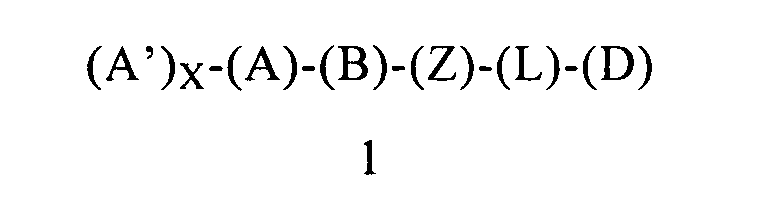
в которой:
(А) обозначает гетероарил или арил, ковалентно связанный с (В) и с (А′), если они содержатся;
каждый (А′), если он содержится, ковалентно связан с (А) и независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где каждый (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил;
X равен 0, 1, 2 или 3;
(В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) или с (D), где (L) обозначает ординарную связь, и с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-;
(D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил ковалентно связаны с (L) или с (Z), если (L) обозначает ординарную связь, и где (D) содержит 0, 1, 2 или 3 заместителя, выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу;
и фармацевтически приемлемый носитель, в количестве, достаточном для ингибирования активности LSD1. Этот объект можно переформулировать, как соединение формулы 1, определенное в настоящем изобретении, предназначенное для применения в качестве ингибитора LSD1. Этот объект можно переформулировать, как соединение формулы 1, предназначенное для применения для лечения заболевания, связанного с LSD1. Родственным объектом является способ лечения индивидуума (предпочтительно человека), который включает идентификацию индивидуума, нуждающегося в лечении, и введение указанному индивидууму соединения формулы 1 в терапевтически эффективном количестве. В предпочтительном объекте терапевтически эффективное количество соединения формулы 1 является количеством, достаточным для ингибирования LSD1. Предпочтительные варианты осуществления соединений формулы 1, предназначенных для применения в композиции и способе этого четвертого объекта настоящего изобретения, определены выше в первом объекте настоящего изобретения.
Четвертым объектом настоящего изобретения является способ лечения или предупреждения рака, который включает введение пациенту (предпочтительно человеку), нуждающемуся в лечении или предупреждении, терапевтически эффективного количества фармацевтической композиции, содержащей соединение формулы 1, определенное выше в первом объекте настоящего изобретения, или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемую соль или сольват и фармацевтически приемлемый носитель. Этот объект можно переформулировать, как соединение формулы 1, определенное выше в первом или втором объекте настоящего изобретения, или его энантиомер, диастереоизомер или их смесь, или его фармацевтически приемлемая соль или сольват, предназначенное для применения для лечения или предупреждения рака. Предпочтительно, если рак выбран из группы, включающей рак молочной железы, колоректальный рак, рак легких, рак предстательной железы, тестикулярный рак, рак головного мозга, рак кожи и рак крови. В предпочтительном объекте терапевтически эффективное количество соединения формулы 1 является количеством, достаточным для ингибирования LSD1.
В одном варианте осуществления четвертого объекта настоящее изобретение относится к соединению формулы 1 или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения рака:
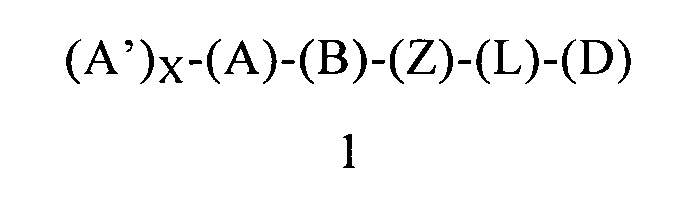
в которой:
(А) обозначает фенильную группу, ковалентно связанную с (В) и с (А′);
(А′) выбран из группы, включающей арил и арилалкоксигруппу, где указанный арил или арилалкоксигруппа содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген и галогеналкил;
X равен 1;
(В) обозначает циклопропильное кольцо, которое ковалентно связано с (А) и с (Z), где (А) и (Z) ковалентно связаны с разными атомами углерода в (В) и где группы (А) и (Z) находятся в транс-положении по отношению к циклопропильному кольцу (В);
(Z) обозначает -NH-; соответственно, (Z) обозначает атом азота, ковалентно связанный с (В), с (L) или с атомом водорода;
(L) обозначает мостик, который связывает (Z) и (D), где указанный мостик выбран из группы, включающей -CH2CH2- и -CH2CH2CH2-;
(D) обозначает циклоалкильную группу, ковалентно связанную с (L), и (D) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу. Родственным объектом настоящего изобретения является фармацевтическая композиция, предназначенная для применения для лечения или предупреждения рака, содержащая соединение формулы 1 или его фармацевтически приемлемую соль или сольват, определенное выше, и фармацевтически приемлемый носитель. В еще более предпочтительном объекте рак, выбран из группы, включающей рак предстательной железы, тестикулярный рак, рак головного мозга, колоректальный рак, рак легких, рак молочной железы, рак кожи и рак крови. В одном предпочтительном объекте раком является рак предстательной железы. В одном предпочтительном объекте раком является рак легких. В одном предпочтительном объекте раком является рак головного мозга. В одном предпочтительном объекте раком является рак крови (например, лейкоз). В одном предпочтительном объекте раком является рак молочной железы. В одном предпочтительном объекте раком является колоректальный рак.
Пятым объектом настоящего изобретения является способ идентификации соединения, которое является селективным ингибитором LSD1, который включает выбор или получение соединения, которое является замещенным гетероарилциклопропиламином или замещенным арилциклопропиламином, и определение способности соединения ингибировать LSD1 и МАО-А и/или МАО-В, где соединение которое ингибирует LSD1 в большей степени, чем МАО-А и/или МАО-В, идентифицируют как селективный ингибитор LSD1.
В контексте этого пятого объекта настоящего изобретения замещенный гетероарилциклопропиламин или замещенный арилциклопропиламин имеет приведенную ниже формулу 2:
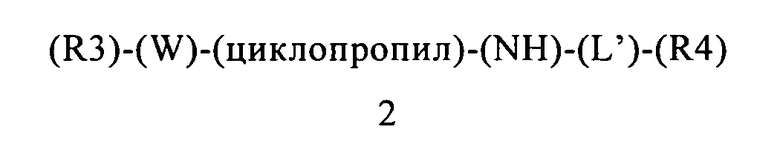
в которой:
(R3) содержится или не содержится, если (R3) содержится, то он выбран из группы, включающей арилалкил, арилалкоксигруппу, арилалкил, гетероциклилалкил и гетероциклилалкоксигруппу, где указанная группа (R3) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, алкил, алкоксигруппу, карбоциклил, циклоалкил, циклоалкоксигруппу, галогеналкил, галогеналкоксигруппу, ациламиногруппу, ацилоксигруппу, алкилтиогруппу, циклоалкилтиогруппу, алкинил, аминогруппу, алкиламиногруппу, арил, арилалкил, арилалкенил, арилалкинил, арилалкоксигруппу, арилоксигруппу, арилтиогруппу, гетероарилтиогруппу, цианогруппу, цианатную группу, галогенарил, гидроксигруппу, гетероциклил, гетероарилоксигруппу, гетероарилалкоксигруппу, изоцианатную группу, изотиоцианатную группу, нитрогруппу, сульфинил, сульфонил, сульфонамидогруппу, тиокарбонил, тиоцианатную группу, тригалогенметансульфонамидогруппу, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил и С-амидогруппу, и указанная группа (R3) ковалентно связана (W);
(W) обозначает арильную или гетероарильную группу, ковалентно связанную с (R3) и с (циклопропил), где указанный (W) содержит 0, 1 или 2 заместителя, не включая (R3) и (циклопропил), и эти заместители независимо выбраны из группы, включающей галоген, алкил, алкоксигруппу, карбоциклил, циклоалкил, циклоалкоксигруппу, галогеналкил, галогеналкоксигруппу, ациламиногруппу, ацилоксигруппу, алкилтиогруппу, циклоалкилтиогруппу, алкинил, аминогруппу, алкиламиногруппу, арил, арилалкил, арилалкенил, арилалкинил, арилалкоксигруппу, арилоксигруппу, арилтиогруппу, гетероарилтиогруппу, цианогруппу, цианатную группу, галогенарил, гидроксигруппу, гетероциклил, гетероарилоксигруппу, гетероарилалкоксигруппу, изоцианатную группу, изотиоцианатную группу, нитрогруппу, сульфинил, сульфонил, сульфонамидогруппу, тиокарбонил, тиоцианатную группу, тригалогенметансульфонамидогруппу, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил и С-амидогруппу;
(циклопропил) обозначает циклопропильную группу, ковалентно связанную с (W) и с атомом азота группы (NH);
(NH) обозначает группу -NH- (атом азота, ковалентно связанный с атомом водорода), где атом азота ковалентно связан с (циклопропил) и с (L′), или с (R4), если (L′) обозначает ординарную связь;
(L′) обозначает мостик формулы -(CH2)n-, где n равно 0, 1, 2, 3, 4, 5 или 6, и, кроме того, где (L′) ковалентно связан с атомом азота группы (NH) и с (R4) (R4); и
(R4) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил ковалентно связан с (L′) или с (Z), если (L′) обозначает ординарную связь, где указанная группа (R4) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, алкил, алкоксигруппу, карбоциклил, циклоалкил, циклоалкоксигруппу, галогеналкил, галогеналкоксигруппу, ациламиногруппу, ацилоксигруппу, алкилтиогруппу, циклоалкилтиогруппу, алкинил, аминогруппу, алкиламиногруппу, арил, арилалкил, арилалкенил, арилалкинил, арилалкоксигруппу, арилоксигруппу, арилтиогруппу, гетероарилтиогруппу, цианогруппу, цианатную группу, галогенарил, гидроксигруппу, гетероциклил, гетероарилоксигруппу, гетероарилалкоксигруппу, изоцианатную группу, изотиоцианатную группу, нитрогруппу, сульфинил, сульфонил, сульфонамидогруппу, тиокарбонил, тиоцианатную группу, тригалогенметансульфонамидогруппу, О-карбамил, N-карбамил, О-тиокарбамил, N-тиокарбамил и С-амидогруппу.
Согласно пятому объекту настоящего изобретения соединение формулы 2 является селективным ингибитором LSD1. Селективный ингибитор LSD1, идентифицированный с помощью способа этого варианта осуществления, используют для приготовления фармацевтической композиции, содержащей указанный селективный ингибитор LSD1 формулы 2 или его фармацевтически приемлемую соль или сольват в терапевтически эффективном количестве и фармацевтически приемлемый носитель. Фармацевтическую композицию можно вводить индивидууму, нуждающемуся в таком лечении. В этом варианте осуществления терапевтически эффективное количество является количеством, достаточным для селективного ингибирования LSD1.
Таким образом, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы 1 или 2, которое является селективным ингибитором LSD1. Селективные ингибиторы LSD1 характеризуются значениями Ki для LSD1, которые не менее, чем в 2 раза меньше, чем значение Ki для МАО-А и/или МАО-В. В одном воплощении этого варианта осуществления значение Ki для LSD1 не менее, чем в 5 раз меньше, чем значение Ki для МАО-А и/или МАО-В. В одном воплощении этого варианта осуществления значение Ki для LSD1 не менее, чем в 10 раз меньше, чем значение Ki для МАО-А и МАО-В. В одном варианте осуществления настоящего изобретения фармацевтическая композиция, содержащая селективный ингибитор LSD1 формулы 1 или 2 или его фармацевтически приемлемую соль или сольват, используют для лечения и/или предупреждения заболевания у индивидуума. В одном объекте терапевтически эффективное количество композиции вводят индивидууму в количестве, достаточном для лечения или предупреждения заболевания. В более предпочтительном объекте заболеванием является рак. В еще более предпочтительном объекте заболеванием является рак, выбранный из группы, включающей рак предстательной железы, тестикулярный рак, рак головного мозга, колоректальный рак, рак легких, рак молочной железы, рак кожи и рак крови. В одном предпочтительном объекте раком является рак предстательной железы. В одном предпочтительном объекте раком является рак легки. В одном предпочтительном объекте раком является рак головного мозга. В одном предпочтительном объекте раком является рак крови (например, лейкоз). В одном предпочтительном объекте раком является рак молочной железы. В одном предпочтительном объекте раком является колоректальный рак.
Согласно пятому объекту настоящего изобретения соединение формулы 2 является двойным ингибитором LSD1 и МАО-В. Двойной ингибитор LSD1/МАО-В, идентифицированный с помощью способа этого варианта осуществления, можно использовать для приготовления фармацевтической композиции, содержащей указанный двойной ингибитор LSD1/МАО-В формулы 2 или его фармацевтически приемлемую соль или сольват в терапевтически эффективном количестве и фармацевтически приемлемый носитель. Фармацевтическую композицию можно вводить индивидууму, нуждающемуся в таком лечении. В этом варианте осуществления терапевтически эффективное количество является количеством, достаточным для ингибирования МАО-В и LSD1.
Таким образом, настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы 1 или 2, которое является двойным ингибитором LSD1 и МАО-В. Предпочтительно, если двойные ингибиторы LSD1/МАО-В характеризуются значениями Ki для LSD1 и МАО-В, которые не менее, чем в 2 раза меньше, чем значение Ki для МАО-А. В одном воплощении этого варианта осуществления значения Ki для LSD1 и МАО-В Ki не менее, чем в 5 раз меньше, чем значение Ki для МАО-А. В одном воплощении этого варианта осуществления значения Ki для LSD1 и МАО-В Ki не менее, чем в 10 раз меньше, чем значение Ki для МАО-А.
Результаты недавних исследований указывают на участие LSD1 в вирусной инфекции и реактивации. В частности, показано, что фармакологические ингибиторы LSD1, такие как парнат и сиРНК, подавляющие LSD1, вызывают снижение вирусной нагрузки и снижение реактивации после скрытого периода (Liang et al. (2009) Nat. Med. 15:1312-1317). Поэтому предполагается, что соединения, предлагаемые в настоящем изобретении, можно использовать для лечения или предупреждения вирусной инфекции. Кроме того, предполагается, что с помощью соединений, предлагаемых в настоящем изобретении, можно лечить или предупреждать реактивацию вирусной инфекции после скрытого периода.
Таким образом, в одном варианте осуществления настоящее изобретение относится к способу лечения или предупреждения вирусной инфекции, который включает введение пациенту/индивидууму (предпочтительно человеку), нуждающемуся в лечении или предупреждении, соединения формулы 1, определенного выше в любом из объектов и вариантов осуществления настоящего изобретения, или его фармацевтически приемлемой соли или сольвата. В соответствии с этим настоящее изобретение также относится к соединению формулы 1, определенному выше в любом из объектов и вариантов осуществления настоящего изобретения, или к его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения вирусной инфекции. В предпочтительном варианте осуществления вирусной инфекцией является вирусная инфекция герпеса. В еще более предпочтительном варианте осуществления вирусная инфекция герпеса вызвана вирусом герпеса, выбранным из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра, и/или связана с ним. В одном воплощении этого варианта осуществления вирусная инфекция вызвана ВИЧ.
В одном варианте осуществления настоящее изобретение относится к способу лечения или предупреждения реактивации вирусной инфекции после скрытого периода, который включает введение пациенту/индивидууму (предпочтительно человеку), нуждающемуся в лечении или предупреждении, соединения формулы 1, определенного выше в любом из объектов и вариантов осуществления настоящего изобретения, или его фармацевтически приемлемой соли или сольвата. В соответствии с этим настоящее изобретение также относится к соединению формулы 1, определенному выше в любом из объектов и вариантов осуществления настоящего изобретения, или к его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения реактивации вирусной инфекции после скрытого периода. В предпочтительном варианте осуществления вирусом, который реактивируется, является вирус герпеса. В еще более предпочтительном варианте осуществления вирус герпеса, который реактивируется, выбран из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра. В одном воплощении этого варианта осуществления вирусом, который реактивируется, является ВИЧ.
Соединения формулы 1 или 2, предлагаемые в настоящем изобретении, не включают ни одно из следующих:
N-(2-фенилциклопропил)циклопентанамин (соответствует регистрационному № CAS 802594-05-4);
10,11-дигидро-N-(2-фенилциклопропил)-5Н-дибензо[а,d]циклогептен-5-амин (соответствует регистрационному № CAS 749796-68-7); и
транс-N-(2-фенилциклопропил)циклогексиламин (соответствует регистрационному № CAS 32751-99-8).
В другом предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы 1:
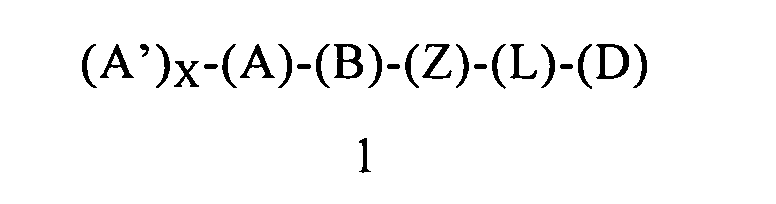
или к его энантиомеру, диастереоизомеру или их смеси, или к его фармацевтически приемлемой соли или сольвату.
Настоящее изобретение также относится к фармацевтической композиции, содержащей любое из указанных выше веществ и фармацевтически приемлемый носитель. Настоящее изобретение также относится к соединению формулы 1 или к его фармацевтически приемлемой соли или сольвату, предназначенному для применения для лечения или предупреждения рака. Кроме того, настоящее изобретение относится к способу лечения или предупреждения рака, который включает введение пациенту (предпочтительно человеку), нуждающемуся в лечении или предупреждении, соединения формулы 1 или его фармацевтически приемлемой соли или сольвата в терапевтически эффективном количестве, или фармацевтической композиции, содержащей любое из указанных выше веществ и фармацевтически приемлемый носитель. Раком может являться, например, рак предстательной железы, тестикулярный рак, рак головного мозга, колоректальный рак, рак легких, рак молочной железы, рак кожи или рак крови.
Для этого предпочтительного варианта осуществления определения и предпочтительные определения групп, содержащихся в формуле 1, приведены ниже.
(А) обозначает гетероарил или арил.
Следует понимать, что термины ″гетероарил″ и ″арил″ необязательно означают одновалентные группы. Соответственно, если X отличается от 0, то фрагмент (А) ковалентно связан с фрагментом (В) и с X фрагментов (А′), как показано в формуле 1.
В предпочтительном воплощении этого предпочтительного варианта осуществления (А) обозначает арил (такой как, например, фенил, нафтил или антраценил) или, более предпочтительно, если (А) обозначает фенил или нафтил, или, еще более предпочтительно, если (А) обозначает фенил.
В другом предпочтительном воплощении этого предпочтительного варианта осуществления (А) обозначает гетероарил (такой как, например, тиофенил (т.е. тиенил), бензо[b]тиенил, нафто[2,3-b]тиенил, тиантренил, фурил, изобензофуранил, хроменил, ксантенил, феноксантиинил, пирролил, имидазолил, пиразолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолизинил, изоиндолил, 3Н-индолил, индолил, индазолил, пуринил, 4Н-хинолизинил, изохинолил, хинолил, фтализинил, нафтиридинил, хинозалинил, циннолинил, птеридинил, карбазолил, бета-карболинил, фенантридинил, акридинил, перимидинил, фенантролинил, феназинил, изотиазолил, фенотиазинил, изоксазолил, фуразанил, феноксазинил, изокумарин, пиридо[1,2-а]пиримидин-4-он, пиразоло[1,5-а]пиримидинил, 1,2-бензоизоксазол-3-ил, бензимидазолил, 2-оксиндолил или 2-оксобензимидазолил) или, более предпочтительно, если (А) выбран из группы, включающей пиридил, пиримидинил и тиофенил.
Каждый (А′), если он содержится, независимо выбран из группы, включающей арил, арилалкоксигруппу, арилалкил, гетероциклил, арилоксигруппу, галоген, алкоксигруппу, галогеналкил, циклоалкил, галогеналкоксигруппу и цианогруппу, где каждая из указанных выше групп (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил. Следует понимать, что, если (А′) содержит 0 заместителей, то соответствующая группа (А′) является незамещенной (т.е. замещена атомом водорода).
Предпочтительно, если каждый (А′), если он содержится, независимо выбран из группы, включающей арил, арилалкил, арилоксигруппу и арилалкоксигруппу, более предпочтительно из группы, включающей арил и арилалкоксигруппу, где указанный арил, указанный арилалкил, указанная арилоксигруппа или указанная арилалкоксигруппа содержит 0, 1, 2 или 3 (предпочтительно, 0, 1 или 2; более предпочтительно, 0 или 1) заместителя. Все указанные заместители независимо выбраны из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил. Предпочтительно, если все указанные заместители независимо выбраны из группы, включающей галоген, галогеналкил, алкил, алкоксигруппу, цианогруппу, сульфонил и сульфинил. Более предпочтительно, если все указанные заместители независимо выбраны из группы, включающей галоген и галогеналкил.
Более предпочтительно, если каждый (А′), если он содержится, независимо выбран из группы, включающей фенил, бензилоксигруппу и фенетилоксигруппу, где указанный фенил, указанная бензилоксигруппа или указанная фенетилоксигруппа содержит 0, 1, 2 или 3 (предпочтительно, 0, 1 или 2; более предпочтительно, 0 или 1) заместителя. Все указанные заместители независимо выбраны из группы, включающей галоген, галогеналкил, галогеналкоксигруппу, арил, арилалкоксигруппу, алкил, алкоксигруппу, амидогруппу, -CH2C(=O)NH2, гетероарил, цианогруппу, сульфонил и сульфинил. Предпочтительно, если все указанные заместители независимо выбраны из группы, включающей галоген, галогеналкил, алкил, алкоксигруппу, цианогруппу, сульфонил и сульфинил. Более предпочтительно, если все указанные заместители независимо выбраны из группы, включающей галоген и галогеналкил.
X равен 0, 1, 2 или 3. Следует понимать, что если X равен 0, то группа (А) не присоединена у какой-либо группе (А′), а вместо этого присоединена к атому водорода.
В предпочтительном воплощении этого предпочтительного варианта осуществления X равен 0, 1, 2 или 3, или более предпочтительно, если X равен 1 или 2, или еще более предпочтительно, если X равен 1.
В другом предпочтительном воплощении этого предпочтительного варианта осуществления X равен 0 или 1, или более предпочтительно, если X равен 0.
Если X равен 1 и (А) обозначает фенил, то (А′) предпочтительно находится в мета-положении или в пара-положении (более предпочтительно в пара-положении) по отношению к циклопропильному кольцу (В).
(В) обозначает циклопропильное кольцо. Фрагменты (А) и (Z) ковалентно связаны с разными атомами углерода циклопропильного кольца (В). Предпочтительно, если (А) и (Z) находятся в транс-положении по отношению к циклопропильному кольцу (В).
(Z) обозначает -NH-, т.е. группу -N(-H)-.
(L) выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2-, -CH2CH2CH2- и -CH2CH2CH2CH2-.
Предпочтительно, если (L) выбран из группы, включающей ординарную связь, -CH2-, -CH2CH2- и -CH2CH2CH2-. Более предпочтительно, если (L) обозначает ординарную связь или -CH2-. Еще более предпочтительно, если (L) обозначает ординарную связь. В другом предпочтительном варианте осуществления (L) обозначает -CH2CH2-.
(D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил; предпочтительно, если (D) обозначает циклоалкил (который предпочтительно выбран из группы, включающей циклопентил, циклогексил, и циклогептил) или бензоциклоалкил (который предпочтительно выбран из группы, включающей индил, 1,2,3,4-тетрагидронафтил и бензоциклогептил, и более предпочтительно выбран из группы, включающей индил и 1,2,3,4-тетрагидронафтил). Указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил (или, соответственно, указанный циклоалкил) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу, предпочтительно выбранных из группы, включающей -NH2, -NH(C1-C6-алкил) и -N(C1-C6-алкил)(C1-C6-алкил).
Следует понимать, что, если указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил (или, соответственно, указанный циклоалкил) содержит 0 заместителей, то соответствующая группа является незамещенной (т.е. замещена атомом водорода).
В предпочтительном воплощении этого предпочтительного варианта осуществления указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил (или, соответственно, указанный циклоалкил) является незамещенным.
В другом предпочтительном воплощении этого предпочтительного варианта осуществления указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил (или, соответственно, указанный циклоалкил) содержит 1, 2 или 3 заместителя, более предпочтительно 1 или 2 заместителя, и еще более предпочтительно 1 заместитель, где указанные заместители независимо выбраны из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу, предпочтительно выбраны из группы, включающей -NH2, -NH(C1-C6-алкил) и -N(C1-C6-алкил)(C1-C6-алкил).
В объем настоящего изобретения также включены все комбинации указанных выше альтернативных предпочтительных объектов. Соответственно, в одном объекте: (А) обозначает арил или, более предпочтительно, если (А) обозначает фенил или нафтил или, еще более предпочтительно, если (А) обозначает фенил; X равен 1, 2 или 3, или более предпочтительно, если X равен 1 или 2, или еще более предпочтительно, если X равен 1; и (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил является незамещенным. В другом объекте: (А) обозначает арил или, более предпочтительно, если (А) обозначает фенил или нафтил или, еще более предпочтительно, если (А) обозначает фенил; X равен 1, 2 или 3, или более предпочтительно, если X равен 1 или 2, или еще более предпочтительно, если X равен 1; и (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил содержит 1, 2 или 3 заместителя, более предпочтительно 1 или 2 заместителя, и еще более предпочтительно 1 заместитель, где указанные заместители независимо выбраны из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу, предпочтительно выбраны из группы, включающей -NH2, -NH(C1-C6-алкил) и -N(C1-C6-алкил)(C1-C6-алкил). В другом объекте: (А) обозначает арил или, более предпочтительно, если (А) обозначает фенил или нафтил или, еще более предпочтительно, если (А) обозначает фенил; X равен О или 1, или более предпочтительно, если X равен 0; и (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил является незамещенным. В другом объекте: (А) обозначает арил или, более предпочтительно, если (А) обозначает фенил или нафтил или, еще более предпочтительно, если (А) обозначает фенил; X равен 0 или 1, или более предпочтительно, если X равен 0; и (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил содержит 1, 2 или 3 заместителя, более предпочтительно 1 или 2 заместителя, и еще более предпочтительно 1 заместитель, где указанные заместители независимо выбраны из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу, предпочтительно выбраны из группы, включающей -NH2, -NH(C1-C6-алкил) и -N(C1-C6-алкил)(C1-C6-алкил). В другом объекте: (А) обозначает гетероарил, или более предпочтительно, если (А) выбран из группы, включающей пиридил, пиримидинил и тиофенил; X равен 1, 2 или 3, или более предпочтительно, если X равен 1 или 2, или еще более предпочтительно, если X равен 1; и (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил является незамещенным. В другом объекте: (А) обозначает гетероарил, или более предпочтительно, если (А) выбран из группы, включающей пиридил, пиримидинил и тиофенил; X равен 1, 2 или 3, или более предпочтительно, если X равен 1 или 2, или еще более предпочтительно, если X равен 1; и (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил содержит 1, 2 или 3 заместителя, более предпочтительно 1 или 2 заместителя, и еще более предпочтительно 1 заместитель, где указанные заместители независимо выбраны из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу, предпочтительно выбраны из группы, включающей -NH2, -NH(C1-C6-алкил) и -N(C1-C6-алкил)(C1-C6-алкил). В другом объекте: (А) обозначает гетероарил, или более предпочтительно, если (А) выбран из группы, включающей пиридил, пиримидинил и тиофенил; X равен 0 или 1, или более предпочтительно, если X равен 0; и (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил является незамещенным. В другом объекте: (А) обозначает гетероарил, или более предпочтительно, если (А) выбран из группы, включающей пиридил, пиримидинил и тиофенил; X равен 0 или 1, или более предпочтительно, если X равен 0; и (D) обозначает алифатическую карбоциклическую группу или бензоциклоалкил, где указанная алифатическая карбоциклическая группа или указанный бензоциклоалкил содержит 1, 2 или 3 заместителя, более предпочтительно 1 или 2 заместителя, и еще более предпочтительно 1 заместитель, где указанные заместители независимо выбраны из группы, включающей -NH2, -NH(C1-C6-алкил), -N(C1-C6-алкил)(C1-C6-алкил), алкил, галоген, амидогруппу, цианогруппу, алкоксигруппу, галогеналкил и галогеналкоксигруппу, предпочтительно выбраны из группы, включающей -NH2, -NH(C1-C6-алкил) и -N(C1-C6-алкил)(C1-C6-алкил).
Следующие соединения исключены из этого предпочтительного варианта осуществления: N-(2-фенилциклопропил)циклопентанамин; 10,11-дигидро-N-(2-фенилциклопропил)-5Н-дибензо[а,d]циклогептен-5-амин; и транс-N-(2-фенилциклопропил)циклогексиламин.
Определения
При использовании в настоящем изобретении термин ″алкил″ означает насыщенный алифатический (т.е. неароматический) ациклический углеводород (т.е. группу, состоящую из атомов углерода и атомов водорода), включая обладающие линейной цепью и/или разветвленной цепью группы, содержащие от 1 до 20 атомов углерода (если это указано в настоящем изобретении, числовой диапазон, такой как ″от 1 до 20″ означает каждое целое число, находящееся в этом диапазоне; например, ″от 1 до 20 атомов углерода″ означает, что алкильная группа может содержать 1 атом углерода, 2 атома углерода, 3 атома углерода и т.п. и включительно до 20 атомов углерода) и не содержащие ни двойные углерод-углеродные связи, ни тройные углерод-углеродные связи. Предпочтительно, если ″алкил″ содержит от 1 до 10 атомов углерода. Более предпочтительно, если ″алкил″ содержит от 1 до 6 атомов углерода и еще более предпочтительно от 1 до 4 атомов углерода.
При использовании в настоящем изобретении термин ″алкенил″ означает ненасыщенный алифатический ациклический углеводород, включая обладающие линейной цепью и/или разветвленной цепью группы, содержащие по меньшей мере одну двойную углерод-углеродную связь и не содержащие тройные углерод-углеродные связи. В более предпочтительном определении алкенильная группа дополнительно определена, как содержащая от 2 до 20 атомов углерода. В другом более предпочтительном определении этой группой является алкенил, содержащий от 2 до 10 атомов углерода. В еще одном более предпочтительном определении этой группой является алкенил, содержащий от 2 до 6 атомов углерода, и в еще одном более предпочтительном определении этой группой является алкенил, содержащий от 2 до 4 атомов углерода.
При использовании в настоящем изобретении термин ″алкинил″ означает ненасыщенный алифатический ациклический углеводород, включая обладающие линейной цепью и/или разветвленной цепью группы, содержащие по меньшей мере одну тройную углерод-углеродную связь и необязательно содержащие одну или большее количество двойных углерод-углеродных связей. В более предпочтительном определении алкинильная группа дополнительно определена, как содержащая от 2 до 20 атомов углерода. В другом более предпочтительном определении этой группой является алкинил, содержащий от 2 до 10 атомов углерода. В еще одном более предпочтительном определении этой группой является алкинил, содержащий от 2 до 6 атомов углерода, и в еще одном более предпочтительном определении этой группой является алкинил, содержащий от 2 до 4 атомов углерода.
При использовании в настоящем изобретении термин ″галоген″ означает хлор, фтор, бром и йод.
При использовании в настоящем изобретении термин ″гидро″ означает атом водорода (группу -Н).
При использовании в настоящем изобретении термин ″алкоксигруппа″ -О-алкильную группу, где ″алкил″ обладает указанным выше значением.
При использовании в настоящем изобретении термин ″галогеналкил″ означает алкильную группу, замещенную с помощью от 1 до 6 галогенидных групп. В предпочтительном варианте осуществления галогеналкил означает группу -СХ3, где X обозначает галогенидную группу. Галогенидные группы могут быть выбраны независимо. В более предпочтительном варианте осуществления галогеналкил означает группу -CF3.
При использовании в настоящем изобретении термин ″галогеналкоксигруппа″ означает алкоксигруппу, замещенную с помощью от 1 до 6 галогенидных групп. В предпочтительном варианте осуществления галогеналкоксигруппа означает группу -ОСХ3, где X обозначает галогенидную группу. Галогенидные группы могут быть выбраны независимо. Предпочтительно, если галогеном является фтор.
При использовании в настоящем изобретении термин ″циклоалкоксигруппа″ означает -О-циклоалкильную группу, где циклоалкильная группа является такой, как определено ниже в настоящем изобретении.
При использовании в настоящем изобретении термин ″арилоксигруппа″ означает -О-арильную группу, где арильная группа является такой, как определено ниже в настоящем изобретении.
При использовании в настоящем изобретении термин ″гетероарилоксигруппа″ означает -О-гетероарильную группу, где гетероарильная группа является такой, как определено ниже в настоящем изобретении.
При использовании в настоящем изобретении термин ″меркаптогруппа″ означает группу -SH.
При использовании в настоящем изобретении термин ″алкилтиогруппа″ означает -S-алкильную группу, где алкильная группа является такой, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″циклоалкилтиогруппа″ означает -S-циклоалкильную группу, где циклоалкильная группа является такой, как определено ниже в настоящем изобретении.
При использовании в настоящем изобретении термин ″арилтиогруппа″ означает -S-арильную группу, где арильная группа является такой, как определено ниже в настоящем изобретении.
При использовании в настоящем изобретении термин ″гетероарилтиогруппа″ означает -S-гетероарильную группу, где гетероарильная группа является такой, как определено ниже в настоящем изобретении.
При использовании в настоящем изобретении термин ″карбонил″ означает группу -C(=O)R″, где R″ является таким, как определено ниже в настоящем изобретении.
Если не указано иное, R″ выбран из группы, включающей гидро, алкил, циклоалкил, арил, гетероарил (присоединенный через кольцевой атом углерода) и гетероциклил (присоединенный через кольцевой атом углерода), где указанные гидро, алкил, циклоалкил, арил, гетероарил и гетероциклил являются такими, как определено в настоящем изобретении.
При использовании в настоящем изобретении термин ″альдегидная группа″ означает карбонильную группу, определенную выше в настоящем изобретении, в которой R″ обозначает гидро.
При использовании в настоящем изобретении термин ″амидная группа″ означает группу -C(=O)NH2.
При использовании в настоящем изобретении термин ″циклокетонная группа″ означает циклоалкильную группу, определенную ниже в настоящем изобретении, в которой один из атомов углерода, образующий кольцо, содержит связанную с ним группу ″=O″, т.е. один из кольцевых атомов углерода атомов углерода циклоалкильной группы входит в группу -С(=O).
При использовании в настоящем изобретении термин ″тиокарбонил″ означает группу -C(=S)R″, в которой R″ является таким, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″О-карбоксигруппа″ означает группу -OC(=O)R″, в которой R″ является таким, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″С-карбоксигруппа″ означает группу -C(=O)OR″, в которой R″ является таким, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″сложный эфир″ означает С-карбоксигруппу, определенную выше в настоящем изобретении, или молекулу, содержащую такую группу, где R″ является таким, как определено выше в настоящем изобретении и R″ не обозначает гидро.
При использовании в настоящем изобретении термин ″соль С-карбоксигруппы″ означает группу -С(=O)O-М+ или молекулу, содержащую такую группу, где М+ выбран из группы, включающей литий, натрий, магний, кальций, калий, барий, железо, цинк и четвертичный аммоний, и, кроме того, где ″+″ в М+ не означает конкретное количество положительных зарядов, которое зависит от соответствующего иона.
При использовании в настоящем изобретении термин ″ацетил″ означает группу -С(=O)СН3.
При использовании в настоящем изобретении термин ″карбоксиалкил″ означает группу -(CH2)rC(=O)OR″, где r равно от 1 до 6 и R″ является таким, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″соль карбоксиалкила″ означает группу -(CH2)rC(=O)O-М+ или молекулу, содержащую такую группу, где r равно от 1 до 6 и М+ выбран из группы, включающей литий, натрий, калий, кальций, магний, барий, железо, цинк и четвертичный аммоний, и, кроме того, где ″+″ в М+ не означает конкретное количество положительных зарядов, которое зависит от соответствующего иона.
При использовании в настоящем изобретении термин ″карбоновая кислоты″ означает С-карбоксигруппу, определенную выше в настоящем изобретении, в которой R″ обозначает гидро.
При использовании в настоящем изобретении термин ″цианогруппа″ означает группу -C≡N.
При использовании в настоящем изобретении термин ″цианатная группа″ означает группу -OCN.
При использовании в настоящем изобретении термин ″изоцианатная группа″ означает группу -NCO.
При использовании в настоящем изобретении термин ″тиоцианатная группа″ означает группу -SCN.
При использовании в настоящем изобретении термин ″изотиоцианатная группа″ означает группу -NCS.
При использовании в настоящем изобретении термин ″сульфинил″ означает группу -S(=O)R″, R″ обозначает C1-C6-алкил.
При использовании в настоящем изобретении термин ″сульфонил″ означает группу -S(=O)2R″, R″ обозначает C1-C6-алкил.
При использовании в настоящем изобретении термин ″сульфонамидная группа″ обозначает -S(=O)2NR17R18, в которой R17 и R18 являются такими, как определено ниже в настоящем изобретении.
Если не указано иное, R17 и R18 независимо выбраны из группы, включающей гидро, алкил, арил, карбоциклил, гетероциклил, -(CH2)арил, -(CH2)карбоциклил и -(CH2)гетероциклил, где гидро, алкил, арил, карбоциклил и гетероциклил являются такими, как определено в настоящем изобретении, и, кроме того, где указанный алкил, арил, карбоциклил, гетероциклил, -(CH2)арил, -(CH2)карбоциклил и -(CH2)гетероциклил необязательно может быть замещенным. В одном воплощении вариантов осуществления, приведенных в настоящем изобретении, указанные группы, из числа которых выбраны R17 и R18, являются незамещенными.
При использовании в настоящем изобретении термин ″тригалогенметансульфонамидная группа″ означает группу -N(R17)S(=O)2CX3, где X обозначает галогенидную группу, определенную выше в настоящем изобретении, и R17 является таким, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″О-карбамил″ означает группу -OC(=O)NR17R18, где R17 и R18 являются такими, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″N-карбамил″ означает группу -N(R17)C(=O)OR18, где R17 и R18 являются такими, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″О-тиокарбамил″ означает группу -OC(=S)NR17R18, где R17 и R18 являются такими, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″N-тиокарбамил″ означает группу -N(R17)C(=S)OR18, где R17 и R18 являются такими, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″аминогруппа″ означает группу -NH2.
При использовании в настоящем изобретении термин ″алкиламиногруппа″ означает группу -NR23R24, где R23 и R24 независимо выбраны из группы, включающей -Н, C1-C6-алкил (т.е. алкил, содержащий от 1 до 8 атомов углерода) и фенил.
При использовании в настоящем изобретении термин ″С-амидная группа″ означает группу -C(=O)NR17R18, где R17 и R18 являются такими, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″N-амидная группа″ означает группу -N(R17)C(=O)R18, где R17 и R18 являются такими, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″нитрогруппа″ означает группу -NO2.
При использовании в настоящем изобретении термин ″четвертичный аммоний″ означает группу -NR20R21R22, где R20, R21 и R22 независимо выбраны из группы, включающей гидро и C1-C6-алкил.
При использовании в настоящем изобретении термин ″метилендиоксигруппа″ означает группу -OCH2O-, где 2 атома кислорода присоединены к соседним кольцевым атомам углерода.
При использовании в настоящем изобретении термин ″этилендиоксигруппа″ означает группу -OCH2CH2O-, где 2 атома кислорода присоединены к соседним кольцевым атомам углерода.
При использовании в настоящем изобретении термин ″алифатическая карбоциклическая группа″ (или также ″алифатический карбоцикл″ или ″алифатический карбоциклил″) означает радикал, образованный из алифатического (т.е. неароматического) углеводородного кольца (т.е. моноциклического углеводородного кольца) или алифатической (т.е. неароматической) углеводородной кольцевой системы, содержащей от 2 до 4 конденсированных колец (т.е. колец, у которых соседняя пара кольцевых атомов углерода является общей), где указанное моноциклическое углеводородное кольцо содержит от 3 до 8 атомов углерода и предпочтительно является насыщенным, и, кроме того, где каждое из колец, образующих указанную углеводородную кольцевую систему независимо содержит от 3 до 8 атомов углерода и предпочтительно является насыщенным. Соответственно, алифатические карбоциклические группы не включают арилы, определенные ниже в настоящем изобретении. Примерами алифатических карбоциклических групп, без наложения ограничений, являются циклоалкилы (такие как циклопропил, циклобутил, циклопентил, циклогексил или циклогептил) и циклоалкенилы (такие как циклогептатриенил, циклопентенил или циклогексадиенил), а также такие группы, как индан.
При использовании в настоящем изобретении термин ″циклоалкил″ означает циклическую насыщенную алифатическую углеводородную группу, которая не содержит двойной углерод-углеродной связи и тройной углерод-углеродной связи. Предпочтительно, если циклоалкил содержит от 3 до 7 атомов углерода. Неограничивающими примерами циклоалкильных групп являются циклопропил, циклобутил, циклопентил, циклогексил или циклогептил. Особенно предпочтительными циклоалкильными группами являются циклопентил или циклогексил.
При использовании в настоящем изобретении термин ″циклоалкенил″ означает циклическую ненасыщенную алифатическую углеводородную группу, которая содержит по меньшей мере одну двойную углерод-углеродную связь и не содержит тройной углерод-углеродной связи. Предпочтительно, если циклоалкенил содержит от 3 до 7 атомов углерода. Неограничивающими примерами циклоалкенильных групп являются циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогексадиенил или циклогептенил.
При использовании в настоящем изобретении термин ″бензоциклоалкил″ означает циклоалкил, определенный выше в настоящем изобретении, и этот циклоалкил сконденсирован с фенильным кольцом (т.е. содержит 2 общих соседних кольцевых атома углерода с фенильным кольцом). Неограничивающими примерами бензоциклоалкилов являются индил (т.е. 2,3-дигидро-1Н-инденил или бензоциклопентил), 1,2,3,4-тетрагидронафтил (т.е. тетралинил или бензоциклогексил) или бензоциклогептил. Предпочтительными примерами бензоциклоалкилов являются индил или 1,2,3,4-тетрагидронафтил. Предпочтительно, если бензоциклоалкил присоединен к остальной части молекулы через циклоалкильный фрагмент (а не через фрагмент фенильного кольца).
При использовании в настоящем изобретении термин ″гетероциклил″ или ″гетероциклический″ означает кольцо или кольцевую систему, содержащую 1 кольцо или от 2 до 4 конденсированных колец (предпочтительно, известное насыщенное или частично насыщенное 3-7-членное моноциклическое кольцо или известную 7-10-членную бициклическую кольцевую систему), которые содержат атомы углерода и от 1 до 4 гетероатомов, независимо выбранных из О, N и S, где гетероатомы азота и серы необязательно могут быть окислены, а атом азота необязательно может быть кватернизован (включая например, любую бициклическую группу, в которой любое из определенных выше гетероциклических колец сконденсировано с бензольным кольцом). Неограничивающие примеры насыщенных или частично насыщенных гетероциклических групп включают тетрагидрофуранильные, пиранильные, пиперидинильные, пиперазинильные, пирролидинильные, имидазолидинильные, имидазолинильные, индолинильные, изоиндолинильные, хинуклидинильные, морфолинильные, изохроманильные, хроманильные, пиразолидинильные, пиразолинильные, тетроноильные и тетрамоильные группы. Примеры ″гетероциклильных″ или ″гетероциклических″ колец также включают, но не ограничиваются только ими, морфолиновую группу, пиперидил, пиперазинил, пирролидинил, тиоморфолиновую группу, гомопиперазинил, имидазолил, имидазолидинил, пиразолидинил, диоксанил и диоксоланил. ″Гетероциклил″ может включать гетероарилы, в которых пи-электронная система гетероциклила является полностью сопряженной.
При использовании в настоящем изобретении термин ″арил″ означает радикал, образованный из углеводородного кольца (т.е. моноциклического углеводородного кольца) или углеводородной кольцевой системы, содержащей от 2 до 4 конденсированных колец, где указанное моноциклическое углеводородное кольцо содержит 5 или 6 атомов углерода и является ароматическим, и, кроме того, где каждое из колец, образующих указанную углеводородную кольцевую систему независимо содержит 5 или 6 атомов углерода. Примерами арильных групп, без наложения ограничений, являются фенил, нафталинил (т.е. нафтил) и антраценил. Особенно предпочтительным арилом является фенил.
При использовании в настоящем изобретении термин ″арилоксигруппа″ означает -О-арил, где ″арил″ является таким, как определено выше.
При использовании в настоящем изобретении термин ″гетероарил″ означает радикал, образованный из кольца (т.е. моноциклического кольца) или кольцевой системы, содержащей от 2 до 4 конденсированных колец, где указанное моноциклическое кольцо содержит 5 или 6 элементов и является ароматическим, и, кроме того, где каждое из колец, образующих указанную кольцевую систему независимо содержит 5 или 6 элементов, и по меньшей мере одно из указанных колец, образующих указанную кольцевую систему является ароматическим, где каждый из указанных элементов (т.е. кольцевых атомов) независимо выбран из С, СН, N, О, S, и по меньшей мере одним из элементов каждого кольца является N, О или S. Неограничивающие примеры гетероарильных групп включают тиенил (тиофенил), бензо[b]тиенил, нафто[2,3-b]тиенил, тиантренил, фурил (фуранил), изобензофуранил, хроменил, ксантенил, феноксантиинил, пирролил, включая без наложения ограничений 2Н-пирролил, имидазолил, пиразолил, пиридил (пиридинил), включая, без наложения ограничений, 2-пиридил, 3-пиридил и 4-пиридил, пиразинил, пиримидинил, пиридазинил, индолизинил, изоиндолил, 3Н-индолил, индолил, индазолил, пуринил, 4Н-хинолизинил, изохинолил, хинолил, фтализинил, нафтиридинил, хинозалинил, циннолинил, птеридинил, карбазолил, бета-карболинил, фенантридинил, акридинил, перимидинил, фенантролинил, феназинил, изотиазолил, фенотиазинил, изоксазолил, фуразанил, феноксазинил, изокумарин, пиридо[1,2-а]пиримидин-4-он, пиразоло[1,5-а]пиримидинил, включая без наложения ограничений пиразоло[1,5-а]пиримидин-3-ил, 1,2-бензоизоксазол-3-ил, бензимидазолил, 2-оксиндолил и 2-оксобензимидазолил. Если гетероарильная группа содержит кольцевой атом азота, такой кольцевой атом азота может находиться в форме N-оксида, например, пиридил-N-оксид, пиразинил-N-оксид и пиримидинил-N-оксид. Предпочтительные гетероарильные группы являются полностью ароматическими и включают, но не ограничиваются только ими, пиридил, тиенил и пиримидинил.
При использовании в настоящем изобретении термин ″арилалкил″ означает любую из C1-С10-алкильных групп, замещенную любой из указанных выше арильных групп, определенных выше. Неограничивающие примеры арилалкильной группы включают бензил, фенетил и нафтилметил.
При использовании в настоящем изобретении термин ″арилалкенил″ означает С2-С10-алкенильную группу, замещенную С6-С14-арильной группой (арильной группой, содержащей от 6 до 14 атомов углерода), определенной выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″арилалкинил″ означает С2-С10-алкинильную группу, замещенную С6-С14-арильной группой (арильной группой, содержащей от 6 до 14 атомов углерода), определенной выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″арилалкоксигруппа″ означает любую из C1-С10-алкоксигрупп, замещенных любой из арильных групп, определенных выше в настоящем изобретении. Примеры арилалкоксигрупп включают бензилоксигруппу и фенетилоксигруппу.
При использовании в настоящем изобретении термин ″арилоксигруппа″ означает кислород, замещенный любой из арильных групп, определенных выше.
При использовании в настоящем изобретении термин ″арилтиогруппа″ означает -S-арильную группу, где арильная группа является такой, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″гетероарилтиогруппа″ означает -S-гетероарильную группу, где гетероарильная группа является такой, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″галогенарил″ означает арильную группу, которая в качестве заместителей содержит от 1 до 6 галогенидных групп, где арильная группа и галогенидные группы являются такими, как определено выше в настоящем изобретении, и, кроме того, где галогенидные группы выбраны независимо.
При использовании в настоящем изобретении термин ″ациламиногруппа″ означает группу -N(R17)C(=O)R18, где R17 и R18 являются такими, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″ацилоксигруппа″ означает группу -O-C(=O)R17, где R17 является таким, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″гетероарилоксигруппа″ означает -О-гетероарильную группу, где гетероарильная группа является такой, как определено выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″гетероарилалкоксигруппа″ означает C1-С10-алкоксигруппу, определенную выше в настоящем изобретении, замещенную гетероарильной группой, определенной выше в настоящем изобретении.
При использовании в настоящем изобретении термин ″предупреждение усиления симптома″ означает недопущение усиления или ухудшения симптома, а также уменьшение скорости усиления симптома. Например, симптом можно охарактеризовать содержанием маркера конкретного заболевания, т.е. белка (например, биомаркера рака). В другом примере симптомом может быть ухудшение познавательной способности. Предупреждение усиления в соответствии с определением, приведенным в настоящем изобретении, означает, что степень проявления симптома (например, содержание белка или степень ухудшения познавательной способности) не усиливается или что уменьшается скорость его усиления.
При использовании в настоящем изобретении термин ″лечение заболевания или нарушения″ означает замедление или обращение прогрессирования заболевания. Лечение заболевания или нарушения включает устранение симптома и/или ослабление симптома заболевания.
″Лечение нарушения или заболевания″ подразумевает, что нарушение или заболевание предположительно имеется или диагностировано у пациента/субъекта. У пациента/субъекта, который предположительно страдает от нарушения или заболевания, обычно проявляются конкретные клинические и/или патологические симптомы, которые специалист в данной области техники легко может приписать конкретному патологическому состоянию (т.е. диагностировать нарушение или заболевание).
″Лечение нарушения или заболевания″ может, например, привести к остановке прогрессирования нарушения или заболевания (например, прекращению ухудшения симптомов) или задержке прогрессирования нарушения или заболевания (в случае, если остановка прогрессирования носит лишь временный характер). ″Лечение нарушения или заболевания″ также может привести к частичной реакции (например, к ослаблению симптомов) или полной реакции (например, к исчезновению симптомов) субъекта/пациента, страдающего от нарушения или заболевания. ″Улучшение протекания″ нарушения или заболевания может, например, привести к остановке прогрессирования нарушения или заболевания или задержке прогрессирования нарушения или заболевания. После такой полной или частичной реакции может последовать рецидив. Следует понимать, что у субъекта/пациента может проявляться широкий диапазон реакций на лечение (например, типичные реакции, описанные выше в настоящем изобретении).
Лечение нарушения или заболевания, в частности, может включать излечивающее лечение (предпочтительно приводящее к полной реакции и, в конечном счете, к излечиванию нарушения или заболевания) и паллиативное лечение (включая ослабление симптомов).
При использовании в настоящем изобретении термин ″предупреждение заболевания или нарушения″ означает замедление заболевания или начала развития заболевания или ослабление симптомов. Предупреждение заболевания или нарушения может включать остановку развития заболевания или его симптомов.
Например, на пациента/субъекта, предположительно предрасположенного к нарушению или заболеванию, определенному в настоящем изобретении, может, в частности, быть оказано благоприятное воздействие путем предупреждения нарушения или заболевания. У пациента/субъекта может быть восприимчивость или предрасположенность к нарушению или заболеванию, включая, но не ограничиваясь только им, наследственную предрасположенность. Наличие такой предрасположенности можно определить с помощью стандартных анализов с использованием, например, генетических маркеров или фенотипических индикаторов. Следует понимать, что нарушение или заболевание, которое необходимо предупредить в контексте настоящего изобретения, не диагностировано или не может быть диагностировано у пациента/субъекта (например, у пациента/субъекта не наблюдаются какие-либо клинические или патологические симптомы). Таким образом, термин ″предупреждение″ включает применение соединений, предлагаемых в настоящем изобретении, до того, как какие-либо клинические и/или патологические симптомы диагностированы или установлены или могут быть диагностированы или установлены лечащим врачом.
При использовании в настоящем изобретении термин ″разовая дозированная форма″ означает отдельные порции, такие как капсулы или таблетки, пригодные для использования в качестве разовых доз для пациентов-людей. Каждая порция содержит заранее заданное количество соединения формулы 1 или 2, которое, как это установлено или предположено, приводит к необходимому фармакокинетическому профилю, который обеспечивает необходимый терапевтический эффект. Дозированная форма содержит соединение формулы 1 вместе по меньшей мере с одним фармацевтически приемлемым носителем, солью, инертным наполнителем или их комбинацией.
При использовании в настоящем изобретении термин ″доза″ или ″дозировка″ означает количество активного ингредиента, который индивидуум принимает или которое вводят за один раз. Например, равная 40 мг доза соединения формулы 1 или 2 в случае режима введения два раза в сутки означает ситуацию, когда индивидуум принимает 40 мг соединения формулы 1 или 2 два раза в сутки, например, 40 мг утром и 40 мг вечером. Равную 40 мг дозу соединения формулы 1 или 2 можно разделить на 2 или большее количество доз, например, 2 дозы по 20 мг соединения формулы 1 или 2 в форме таблетки или 2 дозы по 20 мг соединения формулы 1 или 2 в форме капсулы.
При использовании в настоящем изобретении ″фармацевтически приемлемое пролекарство″ означает соединение, которое при физиологических условиях или путем сольволиза может превратиться в заданное соединение или в фармацевтически приемлемую соль такого соединения.
В объем настоящего изобретения также входят фармацевтически приемлемые пролекарства соединений, описанных и определенных в настоящем изобретении, предпочтительно пролекарства соединений формулы 1 или 2. Пролекарства соединений формулы 1 или 2 являются производными, которые содержат химически или метаболически отщепляющиеся группы и путем сольволиза или при физиологических условиях превращаются в соединения, предлагаемые в настоящем изобретении, которые являются фармацевтически активными in vivo. Пролекарства соединений формулы 1 или 21, предлагаемые в настоящем изобретении, можно получить обычным образом с использованием функциональной группы соединений, таких как аминогруппа, гидроксигруппа или карбоксигруппа. Пролекарственное производное часто обеспечивает преимущества растворимости, совместимости с тканью или замедленного высвобождения в организме млекопитающего (см. публикацию Bundgaard, Н., Design of Prodrugs, pp.7-9, 21-24, Elsevier, Amsterdam 1985). Пролекарства включают производные кислот, хорошо известные специалисту в данной области техники, такие как, например, сложные эфиры, полученные по реакции исходной кислоты с подходящим спиртом, или амиды, полученные по реакции исходной кислоты с подходящим амином. Если соединение, использующееся в настоящем изобретении, содержит карбоксигруппу, то примером пролекарства является сложноэфирное производное, полученное по реакции карбоксигруппы с подходящим спиртом, или амидное производное, полученное по реакции карбоксигруппы с подходящим амином. Особенно предпочтительным сложноэфирным производным в качестве пролекарства является метиловый эфир, этиловый эфир, н-пропиловый эфир, изопропиловый эфир, н-бутиловый эфир, изобутиловый эфир, трет-бутиловый эфир, морфолиноэтиловый эфир, N,N-диэтиленгликольамидоэфир или α-ацетоксиэтиловый эфир. Если соединение, использующееся в настоящем изобретении, содержит гидроксигруппу, то примером пролекарства является ацилоксипроизводное, полученное по реакции гидроксигруппы с подходящим ацилгалогенидом или с подходящим ангидридом кислоты. Особенно предпочтительным ацилоксипроизводным в качестве пролекарства является -ОС(=O)-СН3, -ОС(=O)-С2Н5, -ОС(=O)-(трет-Bu), -OC(=O)-C15H31, -OC(=O)-(m-COONa-Ph), -ОС(=O)-CH2CH2COONa, -O(C=O)-CH(NH2)CH3 или -OC(=O)-CH2-N(CH3)2. Если соединение, использующееся в настоящем изобретении, содержит аминогруппу, то примером пролекарства является амидное производное, полученное по реакции аминогруппы с подходящим галогенангидридом кислоты или с подходящим смешанным ангидридом кислоты. Особенно предпочтительным амидным производным в качестве пролекарства является -NHC(=O)-(CH2)2OCH3 или -NHC(=O)-CH(NH2)CH3.
При использовании в настоящем изобретении ″фармацевтически активный метаболит″ означает фармакологически активный продукт, образовавшийся путем происходящего в организме метаболизма из указанного соединения или его соли. Метаболиты соединения можно идентифицировать по стандартным методикам, известным в данной области техники, и их активность определяют с использованием тестов, таких как описанные в настоящем изобретении.
При использовании в настоящем изобретении ″фармацевтически приемлемая соль″ означает соль, которая сохраняет биологическую эффективность свободных кислот и оснований данного соединения и не является биологически или в ином отношении нежелательной. Соединение, предназначенное для применения в настоящем изобретении, может обладать достаточным количеством кислотных, достаточным количеством основных групп или обоими типами функциональных групп и соответствующим образом взаимодействовать с любым количеством неорганических или органических оснований и неорганических и органических кислот с образованием фармацевтически приемлемой соли. Типичные фармацевтически приемлемые соли включают соли, полученные по реакции соединений, предлагаемых в настоящем изобретении, с неорганической или органической кислотой или с неорганическим основанием, включая такие соли, как сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, йодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себацинаты, фумараты, малеаты, бутин-1,4 диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, гамма-гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин-2-сульфонаты и манделаты.
При использовании в настоящем изобретении ″фармацевтически приемлемый носитель″ означает вещества, не являющиеся АФИ (АФИ означает активный фармацевтический ингредиент), такие как разрыхлители, связующие, наполнители, и смазывающие вещества, использующиеся для приготовления фармацевтических продуктов. Они обычно являются безопасными при введении людям в соответствии с установленными правительственными стандартами, включая введенные в действие Управлением по контролю за качеством пищевых продуктов и медикаментов США и Европейским медицинским агентством.
В объем настоящего изобретения также входят твердые формы соединений формулы 1 или 2 в любой сольватированной форме, включая например, сольваты с водой, например, гидраты, или с органическими растворителями, такими как, например, метанол, этанол или ацетонитрил, т.е. метанолят, этанолят или ацетонитрилат соответственно; или в виде любой полиморфной формы.
Специалист в данной области техники понимает, что некоторые переменные в перечне заместителей повторяются (разные названия для одного и того же заместителя), являются родовыми для других переменных перечня и/или по значению частично перекрываются с другими терминами. Специалист в данной области техники понимает, что в соединениях, предлагаемых в настоящем изобретении, заместители могут быть присоединены к остальной части молекулы в целом ряде положений и предпочтительными являются положения, указанные в примерах.
Кроме того, соединения формулы 1 или 2 могут содержать асимметрические атомы углерода и поэтому могут существовать в рацемических и оптически активных формах. Таким образом, оптические изомеры или энантиомеры, рацематы, таутомеры и диастереоизомеры соединений формулы 1 или 2 также входят в объем настоящего изобретения. Способы, предлагаемые в настоящем изобретении, включают применение всех таких изомеров и их смесей. Методики разделения смесей энантиомеров и диастереоизомеров хорошо известны специалисту в данной области техники. Кроме того, рацемические формы можно разделить с помощью физических методик, таких как, например, фракционная кристаллизация, разделение или кристаллизация диастереоизомеров производных или разделение с помощью хиральной колоночной хроматографии. Отдельные оптические изомеры можно получить из рацематов по обычным методикам, таким как, например, образование соли с оптически активной кислотой с последующей кристаллизацией. В объем настоящего изобретения входят любые выделенные рацемические или оптически активные формы соединений, описанных формулой 1 или 2, или любые их смеси. В одном объекте соединения, предлагаемые в настоящем изобретении, обладают транс-конфигурацией относительно циклопропильного кольца, как в транс-фенилциклопропиламине. В одном объекте соединения, предлагаемые в настоящем изобретении, обладают цис-конфигурацией относительно циклопропильного кольца, как в цис-фенилциклопропиламине. В предпочтительном объекте соединения формулы 1 или 2 обладают трансконфигурацией.
Обычно соединения формулы 1 или 2 могут быть эффективны в количестве, составляющем от примерно 0,01 до примерно 100 мг/(кг сутки) в пересчете на полную массу тела. Активный ингредиент можно вводить одной порцией или можно разделить на несколько меньших доз, вводимых через заранее заданные промежутки времени. Разовая доза, подходящая для каждого введения может составлять, например, от примерно 1 мкг до примерно 2000 мг, предпочтительно от примерно 5 мкг до примерно 1000 мг.
Следует понимать, что указанные выше диапазоны доз являются лишь типичными и не ограничивают объем настоящего изобретения. Терапевтически эффективное количество каждого активного соединения может меняться от таких факторов, включая, но не ограничиваясь только ими, как активность использующегося соединения, стабильность активного соединения в организме пациента, тяжесть патологических состояний, протекание которых облегчают, полная масса подвергающегося лечению пациента, путь введения, легкость всасывания, распределения и выведения активного соединения из организма, возраст и чувствительность пациента, подвергающегося лечению и т.п., что должен понимать специалист в данной области техники. Вводимое количество можно регулировать, поскольку различные факторы меняются во времени.
Для пероральной доставки активные соединения можно ввести в препарат, который включает фармацевтически приемлемые носители, такие как связующие (например, желатин, целлюлоза, трагакантовая камедь), инертные наполнители (например, крахмал, лактоза), смазывающие вещества (например, стеарат магния, диоксид кремния), разрыхлители (например, альгинат, примогель и кукурузный крахмал) и подсластители или ароматизаторы (например, глюкоза, сахароза, сахарин, метилсалицилат и перечная мята). Препарат можно доставлять перорально в виде закрытых капсул из желатина или прессованных таблеток. Капсулы и таблетки можно изготовить по любым обычным методикам. На капсулы и таблетки также можно нанести различные покрытия, известные в данной области техники, для изменения аромата, вкуса, цвета и формы капсул и таблеток. Кроме того, в капсулы можно внести жидкие носители, такие как нелетучее жидкое масло.
Препараты, подходящие для перорального введения также могут находится в форме суспензии, сиропа, жевательной резинки, облатки, эликсира и т.п. При необходимости в них также можно включить обычные агенты, изменяющие аромат, вкус, цвет и форму этих специальных форм препаратов. Кроме того, для удобства введения с помощью зонда для искусственного кормления пациентов, которые не могут глотать, активные соединения можно растворить в приемлемом липофильном растительном масле в качестве разбавителя, таком как оливковое масло, кукурузное масло и сафлоровое масло.
Активные соединения также можно вводить парентерально в виде раствора или суспензии или в лиофилизированной форме, которую перед использованием можно превратить в раствор или суспензию. В таких препаратах можно использовать разбавители или фармацевтически приемлемые носители, такие как стерильная вода и буферный физиологический раствор. Можно включить другие обычные растворители, буферы для регулирования pH, стабилизаторы, антибактериальные средства, поверхностно-активные вещества и антиоксиданты. Например, применяющиеся компоненты включают хлорид натрия, ацетатные, цитратные или фосфатные буферы, глицерин, декстрозу, нелетучие масла, метилпарабены, полиэтиленгликоль, пропиленгликоль, бисульфат натрия, бензиловый спирт, аскорбиновую кислоту и т.п. Препараты для парентерального введения можно хранить в любых обычных контейнерах, таких как флаконы и ампулы.
Пути местного введения включают назальное, трансбуккальное, проводимое через слизистую оболочку, ректальное или вагинальное введение. Для местного введения активные соединения можно приготовить в виде лосьонов, кремов, мазей, гелей, порошков, паст, спреев, суспензий, капель и аэрозолей. В препараты можно включить один или большее количество загущающих агентов, влагоудерживающих веществ и стабилизирующих агентов. Примеры таких агентов включают, но не ограничиваются только ими, полиэтиленгликоль, сорбит, ксантановую камедь, вазелиновое масло, пчелиный воск или минеральное масло, ланолин, сквален и т.п. Специальным типом местной доставки является использование чрескожного пластыря. Методики изготовления чрескожных пластырей описаны, например, в публикации Brown, et al. (1988) Ann. Rev. Med. 39:221-229, которая включена в настоящее изобретение в качестве ссылки.
Подходящим путем введения также может быть подкожная имплантация, предназначенная для пролонгированного высвобождения активных соединений. Она включает хирургические процедуры для имплантации активного соединения в любой подходящей композиции в подкожное пространство, например, под переднюю брюшную стенку. См., например, публикацию Wilson et al. (1984) J. Clin. Psych. 45:242-247. Для пролонгированного высвобождения активных соединений в качестве носителя можно использовать гидрогели. Гидрогели в целом известны в данной области техники. Их обычно получают путем сшивки высокомолекулярных биологически совместимых полимеров в сетку, которая набухает в воде с образованием гелеобразного материала. Предпочтительно, если гидрогели являются биологически разлагающимися или биологически сорбирующимися. Для задач настоящего изобретения можно использовать гидрогели, приготовленные из полиэтиленгликолей, коллагена или сополимера гликоля с L-молочной кислотой. См., например, публикацию Phillips et al. (1984) J. Pharmaceut. Sci., 73:1718-1720.
Соответственно, соединения формулы 1 или 2 или фармацевтические композиции, содержащие соединение формулы 1 или 2 и фармацевтически приемлемый носитель, можно вводить субъекту любым удобным путем системно/периферически или в положение необходимого воздействия, включая, но не ограничиваясь только ими, одним или большим количеством следующих путей: перорально (например, в виде таблетки, капсулы или в виде раствора для проглатывания), местно (например, чрескожно, назально, в глаза, трансбуккально и сублингвально), парентерально (например, по методикам инъекции или по методикам вливания, и включая, например, инъекцию, например, подкожную, внутрикожную, внутримышечную, внутривенную, вунтриартериальную, внутрисердечную, внутриоболочечную, внутрипозвоночную, внутрикапсулярную, подкапсулярную, внутриорбитальную, внутрибрюшинную, внутритрахеальную, подкутикулярную, внутрисуставную, субарахноидальную или надчревную, например, путем имплантации депо, например, подкожно или внутримышечно), в легкие (например, путем лечения посредством ингаляции или вдувания с использованием, например, аэрозоля, вводимого, например, через рот или нос), путем введения в желудочно-кишечный тракт, внутриматочного, внутриглазного, подкожного, глазного (включая введение в стекловидное тело или в камеру), ректального и вагинального введения.
Активные соединения также можно конъюгировать с растворимым в воде неиммуногенным, непептидным обладающим большой молекулярной массой полимером с образованием конъюгата с полимером. Например, активное соединение ковалентно связывают с полиэтиленгликолем с образованием конъюгата. Обычно такой конъюгат обладает улучшенной растворимостью, стабильностью и пониженной токсичностью и иммуногенностью. Таким образом, при введении пациенту активное соединение в конъюгате может обладать более длительным периодом полувыведения из организма и большей эффективностью. См. публикацию Burnham (1994) Am. J. Hosp. Pharm. 15:210-218. Пэгилированные (ПЭГ - полиэтиленгликоль) белки в настоящее время используются в белковозаместительной терапии и для других терапевтических целей. Например, пэгилированные интерферон (ПЭГ-ИНТРОН А®) используют для клинического лечения гепатита В. Пэгилированную аденозиндезаминазу (адаген®) используют для лечения тяжелого комбинированного иммунодефицита (SCIDS). Пэгилированную L-аспарагиназу (онкапспар®) используют для лечения острого лимфобластного лейкоза (ALL). Предпочтительно, если ковалентная связь между полимером и активным соединением и/или в самом полимере гидролитически разрывается при физиологических условиях. Такие конъюгаты, известные в качестве ″пролекарств″, могут легко высвобождать активное соединение внутри организма. Регулируемое высвобождение активного соединения также можно обеспечить путем включения активного ингредиента в микрокапсулы, нанокапсулы или гидрогели, общеизвестные в данной области техники. Другие фармацевтически приемлемые пролекарства соединений, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, сложные эфиры, карбонаты, тиокарбонаты, N-ацилпроизводные, N-ацилоксиалкилпроизводные, четвертичные производные третичных аминов, N-основания Манниха, шиффовы основания, конъюгаты аминокислот, фосфаты, соли металлов и сульфонаты.
В качестве носителей для активных соединений, предлагаемых в настоящем изобретении, также можно использовать липосомы. Липосомы являются мицеллами, образованными из различных липидов, таких как холестерин, фосфолипиды, жирные кислоты и их производные. Также можно использовать различные модифицированные липиды. Липосомы могут уменьшать токсичность активных соединений и повышать их стабильность. Методики приготовления липосомных суспензий, содержащих активные ингредиенты, общеизвестны в данной области техники. См., например, патент США №4522811; Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N. Y. (1976).
Активные соединения также можно вводить в комбинации с другим активным средством, которое синергетически устраняет или предупреждаем такие же симптомы или эффективно для лечения другого заболевания или устранения симптома у подвергающегося лечению пациента, если другое активное средство не мешает активному соединению или неблагоприятно не влияет на воздействие активных соединений, предлагаемых в настоящем изобретении. Такие другие активные средства включают, но не ограничиваются только ими, противовоспалительные средства, противовирусные средства, антибиотики, фунгицидные средства, антитромботические средства, сердечнососудистые средства, средства, снижающие содержание холестерина, противораковые лекарственные средства, гипотензивные лекарственные средства и т.п.
Примеры противоопухолевых средств, которые можно использовать в комбинации с соединениями и способами, предлагаемыми в настоящем изобретении, обычно включают, если это является подходящим, алкилирующие средства, антиметаболиты, эпидофиллотоксины, противоопухолевые ферменты, ингибиторы топоизомеразы, прокарбазины, митоксантроны, координационные комплексы платины, модификаторы биологического ответа и ингибиторы роста, гормональные/антигормональные терапевтические средства и гематопоэтические факторы роста. Типичные классы противоопухолевых средств включают антрациклины, алкалоиды барвинка, митомицины, блеомицины, цитотоксические нуклеозиды, эпотилоны, дискодермолиды, птеридины, диинены и подофиллотоксины. Особенно полезные представители этих классов включают, например, карминомицин, даунорубицин, аминоптерин, метотрексат, метоптерин, дихлорметотрексат, митомицин С, порфиромицин, 5-фторурацил, 6-меркаптопурин, гемцитабин, цитозинарабинозид, подофиллотоксин или производные подофиллотоксина, такие как этопозид, этопозидфосфат или тенипозид, мелфалан, винбластин, винкристин, лейрозидин, виндезин, лейрозин, паклитаксел и т.п. Другие полезные противоопухолевые средства включают эстрамустин, карбоплатин, циклофосфамид, блеомицин, гемцитабин, ифосфамид, мелфалан, гексаметил меламин, тиотепа, цитарабин, идатрексат, триметрексат, дакарбазин, L-аспарагиназа, камптотецин, СРТ-11, топотекан, ara-С, бикалутамид, флутамид, лейпролид, производные пиридобензоиндола, интерфероны и интерлейкины.
Так, в одном варианте осуществления соединение, предлагаемое в настоящем изобретении, предпочтительно соединение формулы 1 или 2, можно использовать в комбинации с другими терапевтическими средствами. Если соединение используют в комбинации со вторым терапевтическим средством, активным по отношению к тому же заболеванию, доза каждого соединения может отличаться от дозы, использующейся, когда соединение применяется по отдельности. Комбинация соединения, предлагаемого в настоящем изобретении, с другим(и) лекарственным средством (средствами) может предполагать введение лекарственного средства (средств) вместе с соединением, предлагаемым в настоящем изобретении. Такое введение может представлять собой одновременное/совместное введение. Однако также возможно последовательное/раздельное введение.
Предпочтительно, если второе терапевтическое средство, которое вводят в комбинации с соединением, предлагаемым в настоящем изобретении, является противораковым лекарственным средством. Противораковым лекарственным средством, вводимым в комбинации с соединением, предлагаемым в настоящем изобретении, может быть: ингибитор ангиогенеза опухоли (например, ингибитор протеазы, ингибитор киназы рецептора эпидермального фактора роста или ингибитор киназы рецептора сосудистого эндотелиального фактора роста); цитотоксическое лекарственное средство (например, антиметаболит, такой как пуриновый и пиримидиновый аналоги антиметаболитов); антимитотическое средство (например, лекарственное средство, стабилизирующее микротрубочки или антимитотический алкалоид); координационный комплекс платины; противоопухолевый антибиотик; алкилирующее средство (например, азотистый иприт или нитрозомочевина); эндокринное средство (например, адренокортикостероид, андроген, антиандроген, эстроген, антиэстроген, ингибитор ароматазы, агонист гонадолиберина или аналог соматостатина); или соединение, которое направленно воздействует на фермент или рецептор, который сверхэкспрессируется и/или другим образом участвует в конкретном метаболическом пути, регуляция которого нарушена в опухолевой клетке (например, ингибиторы АТР (аденозинтрифосфат) и GTP (гуанозинтрифосфат) фосфодиэстеразы, ингибиторы гистондезацетилазы, ингибиторы протеинкиназы (такие как ингибиторы серии-, треонин- и тирозинкиназы (например, протеинтирозинкиназы Абельсона)) и различные факторы роста, их рецепторы и ингибиторы их киназ (такие как ингибиторы киназы рецептора эпидермального фактора роста, ингибиторы киназы рецептора сосудистого эндотелиального фактора роста, ингибиторы фактора роста фибробластов, ингибиторы рецептора инсулиноподобного фактора роста и ингибиторы киназы рецептора тромбоцитарного фактора роста)); метионин; ингибиторы аминопептидазы; ингибиторы протеосомы; ингибиторы циклооксигеназы (например, ингибиторы циклооксигеназы-1 или циклооксигеназы-2); или ингибиторы топоизомеразы (например, ингибиторы топоизомеразы I ингибиторы или топоизомеразы II).
Алкилирующим средством, которое можно использовать в качестве противоракового лекарственного средства в комбинации с соединением, предлагаемым в настоящем изобретении, может быть, например, азотистый иприт (такой как циклофосфамид, мехлорэтамин (хлорметин), урамустин, мелфалан, хлорамбуцил, ифосфамид, бендамустин или трофосфамид), нитрозомочевина (такая как кармустин, стрептозоцин, фотемустин, ломустин, нимустин, преднимустин, ранимустин или семустин), алкилсульфонат (такой как бусульфан, манносульфан или треосульфан), азиридин (такой как гексаметилмеламин (алтретамин), триэтиленмеламин, тиотепа (N,N′N′-триэтилентиофосфорамид), карбоквон или триазиквон), гидразин (такой как прокарбазин), триазен (такой как дакарбазин) или имидазотетразины (такие как темозоломид).
Координационным комплексом платины, который можно использовать в качестве противоракового лекарственного средства в комбинации с соединением, предлагаемым в настоящем изобретении, может быть, например, цисплатин, карбоплатин, недаплатин, оксалиплатин, сатраплатин или триплатинтетранитрат.
Цитотоксическим лекарственным средством, которое можно использовать в качестве противоракового лекарственного средства в комбинации с соединением, предлагаемым в настоящем изобретении, может быть, например, антиметаболит, включая антиметаболиты - аналоги фолиевой кислоты (такие как аминоптерин, метотрексат, пеметрексед или ралтитрексед), антиметаболиты - аналоги пурина (такие как кладрибин, клофарабин, флударабин, 6-меркаптопурин (включая его пролекарственную форму азатиоприн), пентостатин или 6-тиогуанин) и антиметаболиты - аналоги пиримидина (такие как цитарабин, децитабин, 5-фторурацил (включая его пролекарственные формы капецитабин и тегафур), флоксуридин, гемцитабин, эноцитабин или сапацитабин).
Антимитотическим средством, которое можно использовать в качестве противоракового лекарственного средства в комбинации с соединением, предлагаемым в настоящем изобретении, может быть, например, таксан (такой как доцетаксел, ларотаксел, ортатаксел, паклитаксел/таксол или тесетаксел), алкалоид барвинка (такой как винбластин, винкристин, винфлунин, виндезин, или винорелбин), эпотилон (такой как эпотилон А, эпотилон В, эпотилон С, эпотилон D, эпотилон Е или эпотилон F) или аналог эпотилона В (такой как иксабепилон/азаэпотилон В).
Противоопухолевым антибиотиком, который можно использовать в качестве противоракового лекарственного средства в комбинации с соединением, предлагаемым в настоящем изобретении, может быть, например, антрациклин (такой как акларубицин, даунорубицин, доксорубицин, эпирубицин, идарубицин, амрубицин, пирарубицин, валрубицин или зорубицин), антрацендион (такой как митоксантрон или пиксантрон) или противоопухолевый антибиотик, выделенный из Streptomyces (такой как актиномицин (включая актиномицин D), блеомицин, митомицин (включая митомицин С) или пликамицин).
Ингибитором тирозинкиназы, который можно использовать в качестве противоракового лекарственного средства в комбинации с соединением, предлагаемым в настоящем изобретении, может быть, например, акситиниб, босутиниб, цедираниб, дасатиниб, эрлотиниб, гефитиниб, иматиниб, лапатиниб, лестауртиниб, нилотиниб, семаксаниб, сорафениб, сунитиниб или вандетаниб.
Ингибитором топоизомеразы, который можно использовать в качестве противоракового лекарственного средства в комбинации с соединением, предлагаемым в настоящем изобретении, может быть, например, ингибитор топоизомеразы I (такой как иринотекан, топотекан, камптотецин, белотекан, рубитекан или ламелларин D) или ингибитор топоизомеразы II (такой как амсакрин, этопозид, этопозид фосфат, тенипозид или доксорубицин).
В комбинации с соединением, предлагаемым в настоящем изобретении, можно использовать другие противораковые лекарственные средства. Противораковые лекарственные средства могут включать биологические или химические молекулы, такие как родственный TNF индуцирующий апоптоз лиганд (TRAIL), тамоксифен, амсакрин, бексаротен, эстрамустин, ирофульвен, трабектедин, цетуксимаб, панитумумаб, тозитумомаб, алемтузумаб, бевацизумаб, эдреколомаб, гемтузумаб, альвоцидиб, селициклиб, аминолевулиновая кислота, метиламинолевулинат, эфапроксирал, порфимер натрия, талапорфин, темопорфин, вертепорфин, алитретиноин, третиноин, анагрелид, триоксид мышьяка, атрасентан, бортезомиб, кармофур, целекоксиб, демеколцин, элескломол, элсамитруцин, этоглуцид, лонидамин, лукантон, мазопрокол, митобронитол, митогуазон, митотан, облимерсен, омацетаксин, ситимаген, цераденовек, тегафур, тестолактон, тиазофурин, типифарниб и вориностат.
Кроме того, в совместной терапии с соединениями, предлагаемыми в настоящем изобретении, также можно использовать биологические лекарственные средства, такие как антитела, фрагменты антител, конструкции антител (например, одноцепочечные конструкции) и/или модифицированные антитела (такие как привитые к CDR антитела, гуманизированные антитела, ″полностью гуманизированные″ антитела и т.п.), направленные на рак или опухоль маркеры/факторы/цитокины, участвующие в пролиферативных заболеваниях. Примерами таких биологических молекул являются антитела к HER2 (например, трастузумаб, герцептин®), антитела к CD20 (например, ритуксимаб, ритуксан®, мабтера®, редитукс®), конструкции антител к CD19/CD3 (см., например, ЕР-А-1071752) и антитела к TNF (см., например, публикацию Taylor PC. Antibody therapy for rheumatoid arthritis. Curr Opin Pharmacol. 2003. 3(3):323-328). Другие антитела, фрагменты антител, конструкции антител и/или модифицированные антитела для использования в совместной терапии с соединениями, предлагаемыми в настоящем изобретении, указаны в публикации Taylor PC. Curr. Opin. Pharmacol. 2003. 3(3):323-328; Roxana A. Maedica. 2006. 1(1):63-65.
Указанные выше комбинации могут быть предназначены для применения в форме фармацевтического препарата. Отдельные компоненты таких комбинаций можно вводить последовательно или совместно/одновременно в отдельных или объединенных фармацевтических препаратах любым подходящим путем. Если введение является последовательным, то первым можно вводить соединение, предлагаемое в настоящем изобретении, или второе терапевтическое средство. Если введение является одновременным, то комбинацию можно вводить в одной или разным фармацевтических композициях. Следует понимать, что при объединении в одном препарате эти два соединения должны быть стабильными и совместимыми друг с другом и другими компонентами препарата. При раздельном приготовлении они могут быть включены в любой подходящий препарат таким удобным образом, который известен для таких соединений в данной области техники.
В другом варианте осуществления соединения, предлагаемые в настоящем изобретении, предпочтительно соединения формулы 1 или 2, используют в сочетании с физиотерапией, такой как лучевая терапия. Лучевую терапию можно начинать до, после или одновременно с введением соединений. Например, лучевую терапию можно начинать от 1 до 10 мин, от 1 до 10 ч или от 24 до 72 ч после введения соединений. Однако эти временные границы не следует рассматривать, как ограничивающие. На субъекта воздействуют излучением, предпочтительно гамма-излучением, и при этом можно использовать одну дозу или несколько доз излучения, воздействующих через несколько часов, дней и/или недель. Гамма-излучением можно воздействовать в соответствии со стандартными радиотерапевтическими методиками с использованием стандартных доз и режимов. Если не ограничиваться теоретическими соображениями, то можно полагать, что соединения, предлагаемые в настоящем изобретении, можно использовать для того, чтобы сделать клетки, предпочтительно нежелательные пролиферативные/гиперпролиферативные клетки, такие как раковые клетки или опухолевые клетки, более восприимчивыми к такой физиотерапии, например, лучевой терапии.
В соответствии с этим настоящее изобретение относится к соединению формулы 1 или 2 или его фармацевтически приемлемой соли или сольвату, или фармацевтической композиции, содержащей любое из указанных выше веществ в комбинации с фармацевтически приемлемым носителем, предназначенному для применения для лечения или предупреждения рака, и при этом соединение или фармацевтическую композицию следует использовать в сочетании с антипролиферативным лекарственным средством, противораковым лекарственным средством, цитостатическим лекарственным средством, цитотоксическим лекарственным средством и/или лучевой терапией.
В контексте настоящего изобретения, ″субъект″, ″пациент″, или ″индивидуум″, такой как субъект, нуждающийся в лечении или предупреждении, может представлять собой животное, позвоночное животное, млекопитающее, грызунов (например, морскую свинку, хомяка, крысу, мышь), мышиных (например, мышь), собачьих (например, собаку), кошачьих (например, кошку), лошадиных (например, лошадь), приматов, обезьяньих (например, обезьяну или человекообразную обезьяну), мартышек (например, игрунка, павиана), человекообразных обезьян (например, гориллу, шимпанзе, орангутана, гиббона) или человека. Значения терминов ″животное″, ″млекопитающее″ и т.п. хорошо известны в данной области техники и описаны, например, в публикации Wehner und Gehring (1995; Thieme Verlag). В контексте настоящего изобретения следует понимать, что лечению подвергают животных, которые являются важными с экономической, агрономической или научной точки зрения. Важные с научной точки зрения животные включают, но не ограничиваются только ими, мышей, крыс и кроликов. Неограничивающими примерами важных с агрономической точки зрения животных являются овцы, крупный рогатый скот и свиньи, тогда как кошки и собаки можно рассматривать в качестве животных, важных с экономической точки зрения. Предпочтительным субъектом/пациентом/индивидуумом является млекопитающее. Более предпочтительным субъектом/пациентом/индивидуумом является человек.
Описание общего пути синтеза
Соединения, предлагаемые в настоящем изобретении, можно синтезировать общими путями, описанными на схемах 1, 2, 3, 4 и 5. Следует отметить, что соединения формулы (I) и формулы (II), описанные ниже в описаниях синтеза и схемах, отличаются отсоединений формулы 1 и соединений формулы 2, описанных выше в вариантах осуществления/объектах настоящего изобретения (или в формуле изобретения).
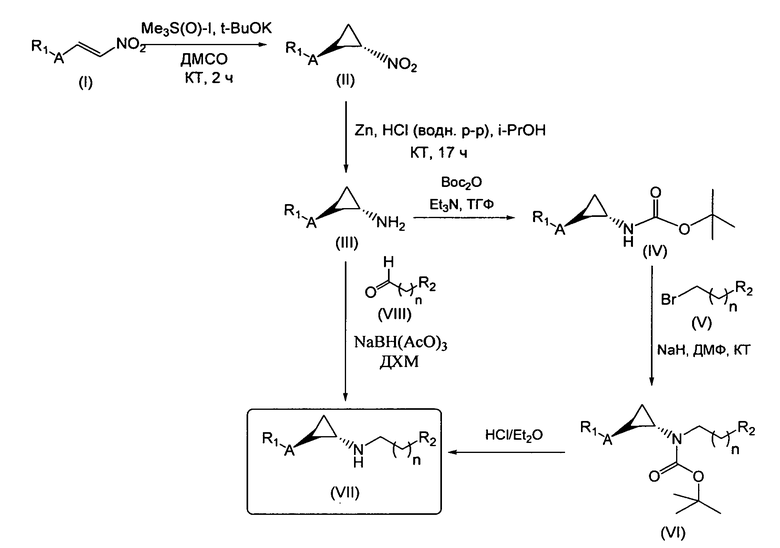
Схема 1: ДМФ (N,N-диметилформамид), ДМСО (диметилсульфоксид), ДХЭ (дихлорэтан), ТГФ (тетрагидрофуран)
Имеющиеся в продаже нитропроизводные формулы (I) вводят в реакцию циклопропанирования с использованием триметилсульфоксониййодида и трет-бутилата калия. Затем нитрогруппу полученных нитроциклопропилпроизводных формулы (II) восстанавливают с использованием цинка в хлористоводородной кислоте и получают производные циклопропиламина формулы (III). Эти соединения формулы (III) можно ввести в реакцию с трет-бутилдикарбонатом при комнатной температуре (КТ) с использованием триэтиламина в качестве основания и тетрагидрофурана в качестве растворителя и получить промежуточный продукт формулы (IV). Алкилирование производных формулы (IV) имеющимися в продаже производными алкилгалогенидов формулы (V) с использованием NaH в качестве основания и ДМФ в качестве растворителя дает промежуточные продукты формулы (VI). Удаление защитной группы Boc с использованием HCl в диэтиловом эфире приводит к образованию производных формулы (VII), которые являются объектом настоящего изобретения.
Альтернативно, реакция производных циклопропиламина формулы (III) с имеющимися в продаже альдегидами формулы (VIII) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента и дихлорэтана в качестве растворителя также приводит к образованию производных формулы (VII), которые являются объектом настоящего изобретения.
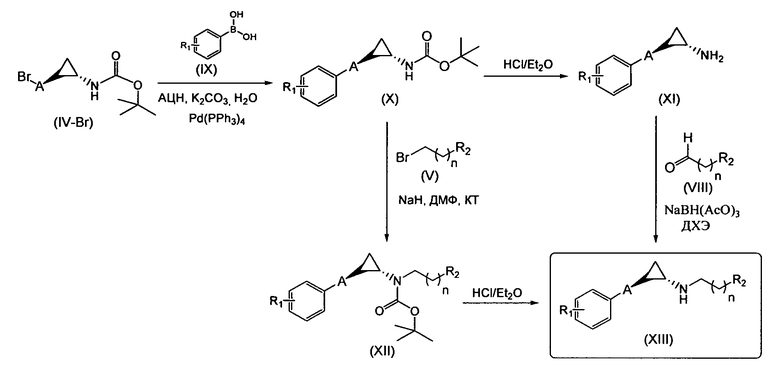
Схема 2: АЦН (ацетонитрил), ДМФ (N,N-диметилформамид), ДХЭ (дихлорэтан)
Реакция промежуточного продукта формулы (IV-Br) с имеющимися в продаже производными бороновых кислот формулы (IX) с использованием ацетонитрила и воды в качестве растворителя, карбоната калия в качестве основания и тетракис(трифенилфосфин)палладия(0) в качестве катализатора приводит к образованию соединений формулы (X). Удаление защитной группы Boc с использованием HCl в Et2O приводит к образованию производных формулы (XI). Эти соединения формулы (XI) можно ввести в реакцию с имеющимися в продаже альдегидами формулы (VIII) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента и дихлорэтана в качестве растворителя, что приводит к образованию соединений формулы (XIII), которые также являются объектами настоящего изобретения.
Альтернативно, алкилирование производных формулы (X) имеющимися в продаже производными алкилгалогенидов формулы (V) с использованием NaH в качестве основания и ДМФ в качестве растворителя дает промежуточные продукты формулы (XII). Последующее удаление защитной группы Boc с использованием HCl в диэтиловом эфире также приводит к образованию производных формулы (XIII), которые также являются объектом настоящего изобретения.
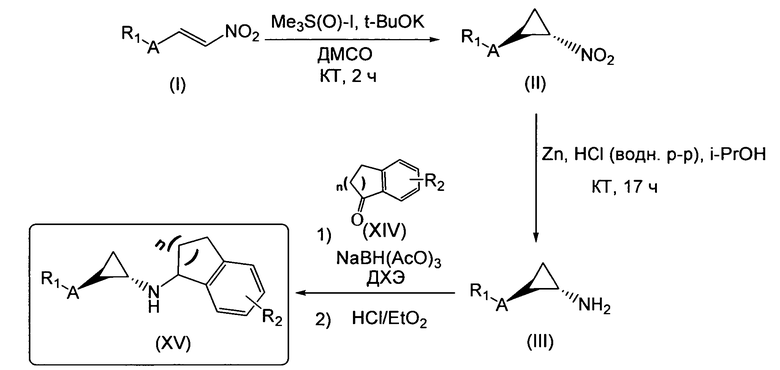
Схема 3: ДМСО (диметилсульфоксид), ДХЭ (дихлорэтан)
Имеющиеся в продаже нитропроизводные формулы (I) вводят в реакцию циклопропанирования с использованием триметилсульфоксониййодида и трет-бутилата калия. Затем нитрогруппу полученных нитроциклопропилпроизводных формулы (II) восстанавливают с использованием цинка в хлористоводородной кислоте и получают производные циклопропиламина формулы (III). Эти соединения формулы (III) можно ввести в реакцию с имеющимися в продаже кетонами формулы (XIV) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента и дихлорэтана в качестве растворителя и получить производные формулы (XV), которые также являются объектом настоящего изобретения.
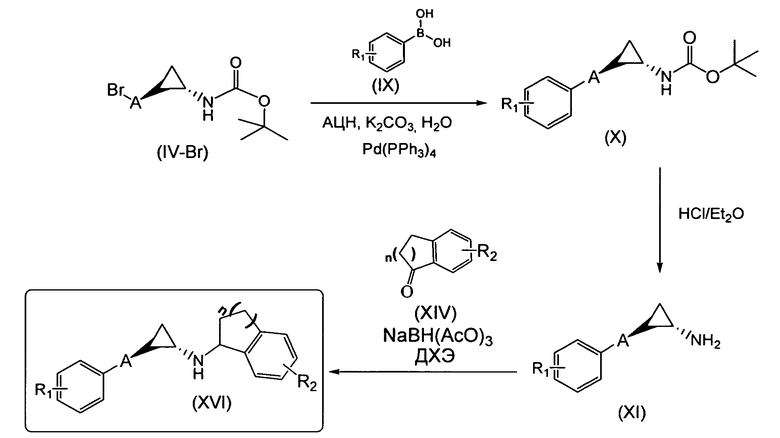
Схема 4: АЦН (ацетонитрил), ДХЭ (дихлорэтан)
Реакция соединений формулы (IV-Br) с имеющимися в продаже производными бороновых кислот формулы (IX) с использованием ацетонитрила и воды в качестве растворителя, карбоната калия в качестве основания и тетракис(трифенилфосфин)палладия(0) в качестве катализатора приводит к образованию соединений формулы (X). Удаление защитной группы Boc с использованием HCl в Et2O приводит к образованию производных формулы (XI). Эти производные (например, формулы (XI)) вводят в реакцию с имеющимися в продаже кетонами формулы (XIV) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента и дихлорэтана в качестве растворителя и получают соединения формулы (XVI), которые также являются объектом настоящего изобретения.
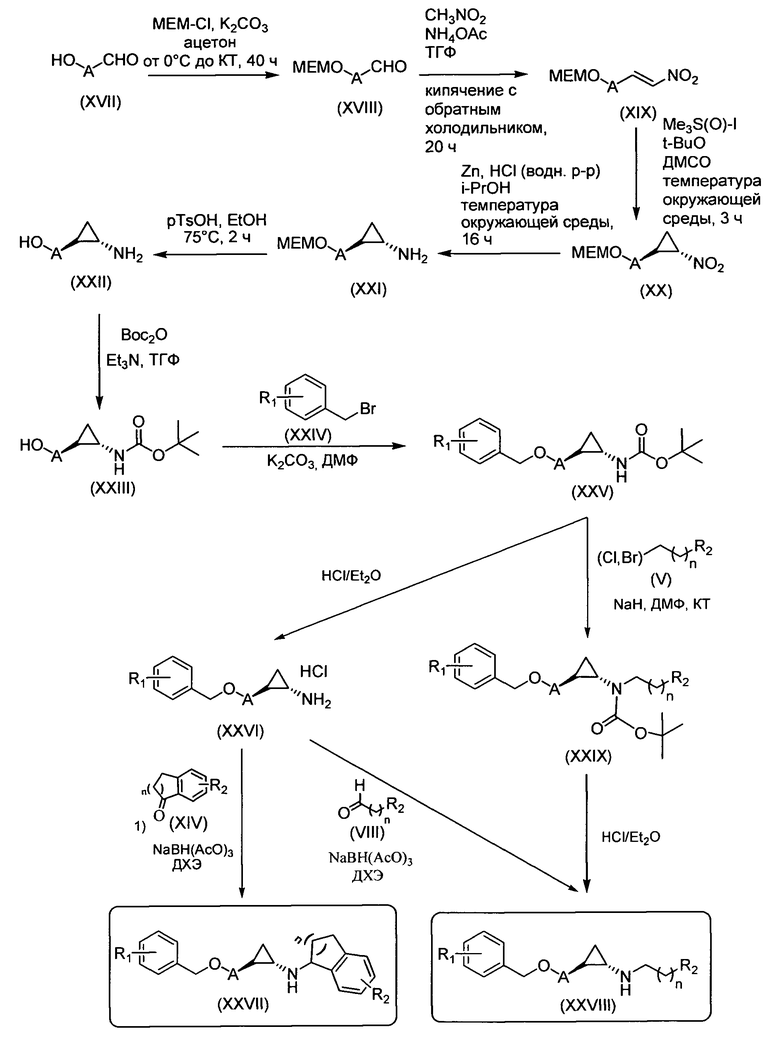
Схема 5: ДХЭ (дихлорэтан), ДМФ (N,N-диметилформамид), ДМСО (диметилсульфоксид), МЕМ-Cl (метоксиэтоксиметилхлорид), p-TsOH (п-толуолсульфоновая кислота), ТГФ (тетрагидрофуран).
Реакция имеющихся в продаже альдегидов формулы (XVII) с метоксиэтоксиметилхлоридом в ацетоне с использованием карбоната калия в качестве основания приводит к образованию производных альдегида формулы (XVIII). Эти соединения вводят в реакцию с нитрометаном и ацетатом аммония в тетрагидрофуране и получают нитровиниловые производные формулы (XIX). Реакция циклопропанирования с использованием триметилсульфоксониййодида и гидрида натрия в диметилсульфоксиде в качестве растворителя приводит к образованию производных (транс)-нитроциклопропана формулы (XX). Затем нитрогруппу восстанавливают с использованием цинка в хлористоводородной кислоте и получают производные (транс)-циклопропиламина формулы (XXI). Удаление защитной группы с использованием п-толуолсульфоновой кислоты в этаноле приводит к образованию производных формулы (XXII). Реакция трет-бутилдикарбоната в тетрагидрофуране с использованием триэтиламина в качестве основания приводит к образованию производных трет-бутил-(транс)-циклопропилкарбамата формулы (XXIII). Алкилирование имеющимися в продаже производными бензилбромида формулы (XXIV) с использованием карбоната калия в качестве основания и N,N-диметилформамида в качестве растворителя приводит к образованию производных формулы (XXV). Удаление у этих производных защитной группы Boc с использованием 2М раствора HCl в диэтиловом эфире и с использованием диэтилового эфира в качестве растворителя приводит к образованию соответствующих производных гидрохлорида (транс)-циклопропиламина формулы (XXVI). Эти производные амина можно алкилировать двумя путями:
1) Имеющимися в продаже кетонами формулы (XIV) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента и дихлорэтана в качестве растворителя с получением соединений формулы (XXVII), которые также являются объектом настоящего изобретения.
2) Имеющимися в продаже альдегидами формулы (VIII) с использованием триацетоксиборогидрида натрия в качестве восстановительного реагента и дихлорэтана в качестве растворителя с получением соединений формулы (XXVIII), которые также являются объектом настоящего изобретения.
Альтернативно, алкилирование производных циклопропилкарбамата формулы (XXV) имеющимися в продаже алкилгалогенидами формулы (V) с использованием NaH в качестве основания и ДМФ в качестве растворителя дает промежуточные продукты формулы (XXIX). Последующее удаление защитной группы Boc с использованием HCl в диэтиловом эфире также приводит к образованию соединений формулы (XXVIII), которые также являются объектом настоящего изобретения.
Специалист в данной области техники знает, как выбрать или синтезировать алкилирующие реагенты, использующиеся вместо реагентов формулы (V), (VIII), (XIV) и (XXIV) для синтеза соединений, предлагаемых в настоящем изобретении.
На приведенных выше схемах группа А может обозначать арильную группу, которая в разделе, посвященном примерам, в качестве примера представлена, как фенильная группа, и группа А также может обозначать, например, гетероарильную группу (например, пиридил или другой гетероарил). Специалист в данной области легко может внести изменения в схемы синтеза, приведенные в настоящем изобретении, и получить соединения, которые содержат гетероарильные группы в положении А.
Примеры
Для образования названий, соответствующих структурам, приведенным ниже в примерах соединений, использовали программу ChemBioDraw Ultra version 11.0.1, выпускающуюся фирмой CambridgeSoft. В случае противоречия между названием и изображенной структурой определяющей является структура. Эта программа образует названия соединений в конфигурации (1S, 2R), что обусловлено конфигурацией вводимой в программу структуры, и вместо термина (1S, 2R), заданного в программе, используют термин ″транс″. Структуры, приведенные ниже в примерах соединений представлены, как обладающие одной определенной стереохимической конфигурацией атомов углерода циклопропильного фрагмента фенилциклопропиламинового ядра, (1S, 2R). Все соединения синтезированные в примерах, являются смесями, содержащими обе конфигурации (1R, 2S) и (1S, 2R), т.е. они обладают ″транс″ - конфигурацией циклопропилового кольца циклопропильной кольцевой системы. Это обусловлено тем, что использующийся исходный фенилциклопропиламин обладает ″транс″-конфигурацией. Предполагается, что в качестве исходного вещества можно использовать соединение в цис-конфигурации или отдельные диастереоизомеры и все они имеются в продаже или их можно синтезировать. Таким образом, настоящее изобретение относится к соединениям, которые обладают указанной стереохимической конфигурацией циклопропильного кольца, например, транс ((1R, 2S) и (1S, 2R)) и цис ((1R, 2R) и (1S, 2S)) или к их отдельным диастереоизомерам. Предпочтительной стереохимической конфигурацией циклопропильного кольца фенилциклопропиламина является транс-конфигурация.
Соединения, указанные в примерах, также можно синтезировать или они поставляются в виде солей. Специалист в данной области техники знает, как получить и может получить солевые формы и/или подвергнуть превращению солевые формы соединений, предлагаемых в настоящем изобретении, включая приведенные в примерах. В некоторых случаях соединения, предлагаемые в настоящем изобретении, включая приведенные в примерах, в форме солей могут быть стабильнее, чем свободные основания.
Приведенные ниже промежуточные продукты (и аналоги промежуточных продуктов или их производные), указанные на схемах синтеза, представленных в настоящем изобретении, можно получить по приведенным ниже методикам. Специалист с общей подготовкой в данной области техники знает, как внести изменения в эти схемы или использовать альтернативные подходы и получить соединения, предлагаемые в настоящем изобретении.
Промежуточный продукт А: (Транс)-2-фенилциклопропанамин
NaOMe (0,80 г, 11,8 ммоля) добавляли к раствору (транс)-2-фенилциклопропанамингидрохлорида (2,00 г, 11,8 ммоля) в МеОН (40 мл) и перемешивали 1 ч. Растворитель удаляли досуха.
Промежуточный продукт В: трет-Бутил-(транс)-2-фенилциклопропил-карбамат
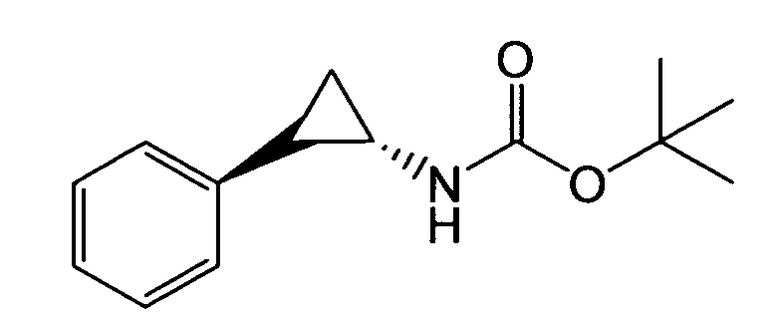
К раствору (транс)-2-фенилциклопропанамина (промежуточный продукт А, 1,14 г, 1 экв.) в 50 мл ТГФ добавляли 1,25 г (1,05 экв.) K2CO3 и перемешивали при комнатной температуре в течение 3 ч. После удаления растворителя добавляли CH2Cl2, затем промывали насыщенным раствором NaHCO3 и рассолом. Органический слой экстрагировали, сушили над MgSO4 и фильтровали. Неочищенное вещество очищали с помощью хроматографии на силикагеле (гексан-МТБЭ (метил-трет-бутиловый эфир 90:10) и получали 1,89 г трет-бутил-(транс)-2-фенилциклопропилкарбамата. Выход: 95%
Промежуточный продукт С: 1-(Бензилокси)-4-[(транс)-2-нитроциклопропил]бензол
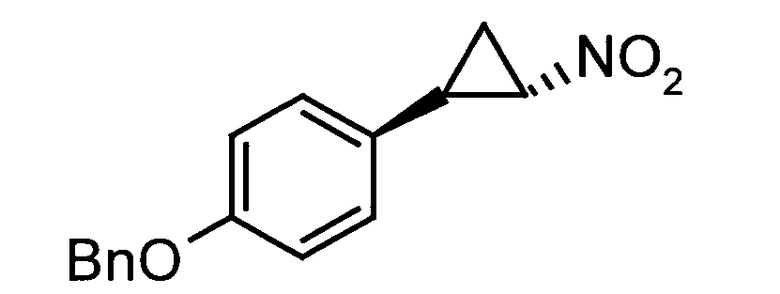
Триметилсульфоксониййодид (0,62 г, 2,82 ммоля) порциями добавляли к раствору t-BuOH (0,32 г, 2,82 ммоля) в сухом ДМСО (5 мл). Через 10 мин через канюлю добавляли раствор 1-(бензилокси)-4-[(Е)-2-нитровинил]бензола (0,60 г, 2,35 ммоля) в ДМСО (5 мл) и смесь перемешивали при комнатной температуре в течение 6 ч. Реакционную смесь выливали в воду (10 мл) и экстрагировали с помощью Et2O (3×10 мл); органические слои промывали рассолом (2×15 мл), сушили над безводным Na2SO4 и фильтровали. После удаления растворителя оставшееся оранжевое масло очищали с помощью колоночной хроматографии на силикагеле (5% EtOAc/гексаны) и получали 0,16 г 1-(бензилокси)-4-[(транс)-2-нитроциклопропил]бензола [Rf=0,5 (20% EtOAc/гексаны), белое твердое вещество, выход 26%].
Промежуточный продукт D: (Транс)-2-[4-(бензилокси)фенил]-циклопропанамин
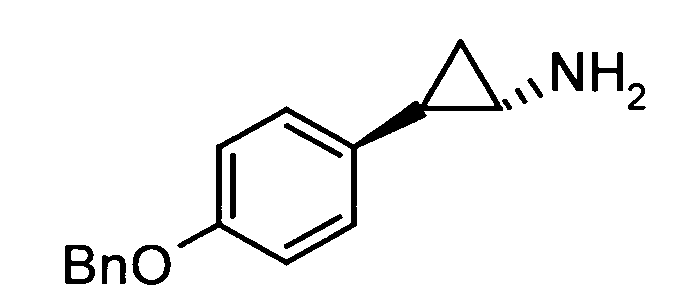
При энергичном перемешивании Zn пыль (1,97 г, 30 молей) в течение 30 мин небольшими порциями добавляли к раствору 1-(бензилокси)-4-[(транс)-2-нитроциклопропил]бензола (промежуточный продукт С, 0,81 г, 3,0 ммоля) в i-PrOH (25 мл) и HCl (11 мл 2,7 н. водного раствора, 30 ммолей). Через 17 ч смесь фильтровали через слой целита, который промывали с помощью 10 мл метанола. Фильтрат концентрировали и добавляли 10 мл воды, промывали с помощью CH2Cl2 (3×15 мл). Органические слои сушили над безводным Na2SO4 и фильтровали. После удаления растворителя неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (10% МеОН/CH2Cl2) и получали 0,50 г (транс)-2-[4-(бензилокси)фенил]циклопропанамин [Rf=0,2 (10% МеОН/CH2Cl2), белое твердое вещество, выход 70%]. 1H ЯМР (ядерный магнитный резонанс) δ част./млн): МеОН 400 МГц: 7,45-7,27 (m, 5Н, ArH); 6,96 (d, J=8,5 Гц, 2Н, ArH); 6,86 (d, J=8,5 Гц, 2Н, ArH); 5,03 (s, 2Н, CH2); 2,41-2,34 (m, 1Н, СН); 1,86-1,76 (m, 1Н, СН); 0,98-0,85 (m, 2Н, CH2).
Промежуточный продукт Е: трет-Бутил-(транс)-2-[4-(бензилокси)фенил]-циклопропилкарбамат
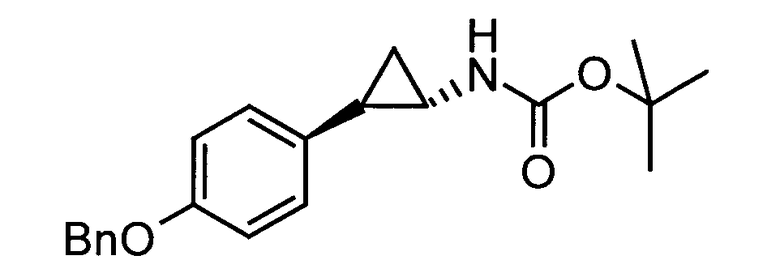
Boc2O (1,65 экв.) добавляли к раствору (транс)-2-[4-(бензилокси)фенил]-циклопропанамина (промежуточный продукт D; 1 экв.) и Et3N (1,65 экв.) в ТГФ и перемешивали в течение 3 ч. После удаления растворителя неочищенный остаток растворяли в EtOAc и последовательно промывали водой и с помощью HCl (10% водный раствор) и рассолом. Органический слой сушили над безводным Na2SO4 и фильтровали; после удаления растворителя остаток очищали с помощью колоночной хроматографии на силикагеле (10-20% EtOAc/гексаны) и получали искомое соединение (выход 78%). 1H ЯМР δ (част./млн): МеОН 400 МГц: 7,45-7,27 (m, 5Н, ArH); 6,93 (d, J=8,5 Гц, 2Н, ArH); 6,86 (d, J=8,5 Гц, 2Н, ArH); 5,03 (s, 2Н, CH2); 2,41-2,34 (m, 1Н, СН); 1,86-1,76 (m, 10Н, СН; tBu); 0,98-0,85 (m, 2Н, CH2).
Промежуточный продукт F: 1-Бром-4-[(транс)-2-нитроциклопропил]бензол
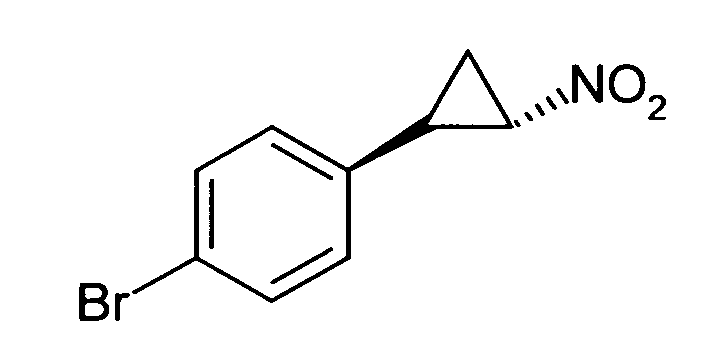
Это соединение синтезировали по такой же методике, как и описанная для промежуточного продукта С, с использованием в качестве исходного вещества имеющегося в продаже (Е)-1-бром-4-(2-нитровинил)бензола. Выход: 27%.
Промежуточный продукт G: (Транс)-2-(4-бромфенил)циклопропанамин
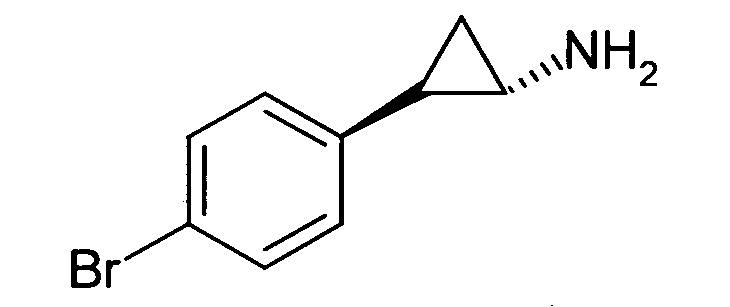
Это соединение синтезировали по такой же методике, как и описанная для промежуточного продукта D, с использованием в качестве исходного вещества 1-бром-4-[(транс)-2-нитроциклопропил]бензола (промежуточный продукт F). Выход: 10%. 1Н ЯМР (CD3OD): 1,45 (m, 2Н), 2,61 (m, 1Н), 2,86 (m, 1H), 6,98 (d, 2H), 7,11 (d, 2H). MC (масс-спектрометрия) (M+H): 211,9.
Промежуточный продукт H: трет-Бутил-(транс)-2-(4-бромфенил)-циклопропилкарбамат
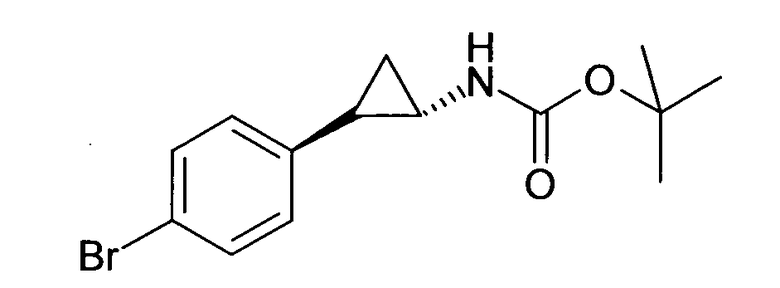
Boc2O (1,65 экв.) добавляли к раствору (транс)-2-(4-бромфенил)циклопропанамина (промежуточный продукт G; 1 экв.) и Et3N (1,65 экв.) в ТГФ и перемешивали в течение 3 ч. После удаления растворителя неочищенный остаток растворяли в EtOAc и последовательно промывали водой и с помощью HCl (10% водный раствор) и рассолом. Органический слой сушили над безводным Na2SO4 и фильтровали; после удаления растворителя остаток очищали с помощью колоночной хроматографии на силикагеле (10-20% EtOAc/гексаны) и получали трет-бутил-(транс)-2-(4-бромфенил)циклопропилкарбамат (выход 85%).
Промежуточный продукт I: 4-((2-Метоксиэтокси)метокси)бензальдегид
2-Метоксиэтоксиметилхлорид (5,10 мл, 45,0 ммоля) медленно добавляли к смеси 4-гидроксибензальдегида (5,00 г, 40,9 ммоля) и K2CO3 (6,20 г, 45,0 ммоля) в ацетоне (70 мл), охлажденной до 0°C. Смеси давали нагреться до комнатной температуры и ее перемешивали в течение 40 ч. После удаления растворителя неочищенный остаток растворяли в EtOAc (50 мл) и последовательно промывали водой (50 мл) и с помощью NaOH (10% водный раствор, 2×20 мл). Органический слой сушили над безводным Na2SO4 и фильтровали. После удаления растворителя получали 6,85 г 4-((2-метоксиэтокси)метокси)бензальдегида [Rf=0,6 (50% AcOEt/гексаны), бесцветное масло, выход 80%], который использовали без дополнительной очистки.
Промежуточный продукт J: (Е)-1-((2-Метоксиэтокси)метокси)-4-(2-нитровинил)бензол
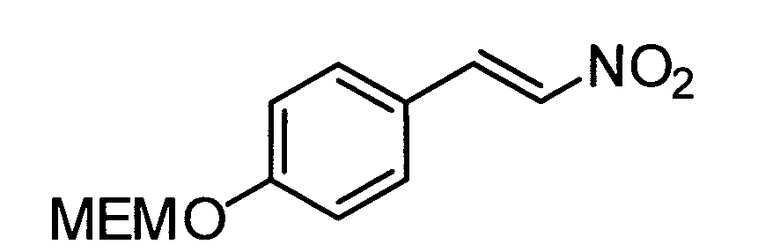
Смесь 4-((2-метоксиэтокси)метокси)бензальдегида (промежуточный продукт I, 1,86 г, 8,85 ммоля) и NH4OAC (0,75 г, 9,73 ммоля) в сухом ТГФ (15 мл) и CH3NO2 (15 мл) кипятили с обратным холодильником в течение 20 ч и давали охладиться до комнатной температуры. Объем реакционной смеси уменьшали примерно до 1/3 в роторном испарителе; полученный раствор выливали в воду (15 мл) и экстрагировали с помощью AcOEt (2×15 мл). Органические слои промывали рассолом (20 мл), сушили над безводным Na2SO4 и фильтровали. После удаления растворителя оставшееся коричневое масло очищали с помощью колоночной хроматографии на силикагеле (15-30% EtOAc/гексаны) и получали 1,77 г (Е)-1-((2-метоксиэтокси)метокси)-4-(2-нитровинил)бензола [Rf=0,7 (50% AcOEt/гексаны), желтое твердое вещество, выход 79%].
Промежуточный продукт K: 1-((2-Метоксиэтокси)метокси)-4-((транс)-2-нитроциклопропил)бензол
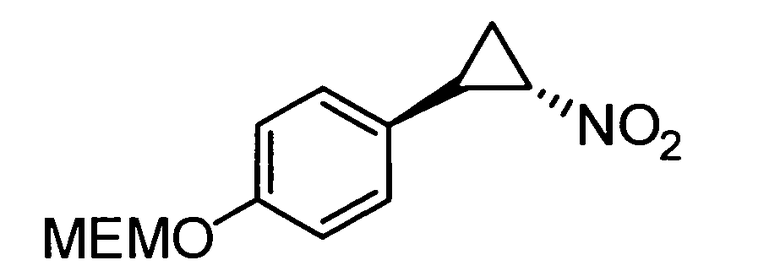
Триметилсульфоксониййодид (0,76 г, 3,44 ммоля) небольшими порциями добавляли к суспензии NaH [0,14 г (60% в минеральном масле), 3,44 ммоля] в сухом ДМСО (5 мл). Смесь перемешивали до прекращения выделения газа и образовывался прозрачный раствор (45 мин). Затем через канюлю добавляли раствор (Е)-1-((2-метоксиэтокси)метокси)-4-(2-нитровинил)бензола (промежуточный продукт J, 0,73 г, 2,86 ммоля) в ДМСО (5 мл) и реакционную смесь перемешивали в течение еще 20 ч. Смесь выливали в воду (20 мл) и экстрагировали с помощью Et2O (3×15 мл). Органические слои промывали рассолом (20 мл), сушили над безводным Na2SO4 и фильтровали; после удаления растворителя оставшееся оранжевое масло очищали с помощью колоночной хроматографии на силикагеле (10-20% EtOAc/гексаны) и получали 0,44 г 1-((2-метоксиэтокси)метокси)-4-((транс)-2-нитроциклопропил)бензола [Rf=0,4 (50% AcOEt/гексаны), бесцветное масло, выход 36%].
Промежуточный продукт L: (Транс)-2-(4-((2-метоксиэтокси)метокси)-фенил)циклопропанамин
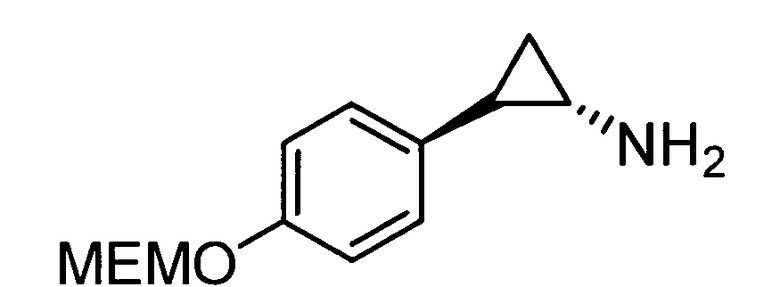
При энергичном перемешивании Zn пыль (0,99 г, 15,1 моля) в течение 20 мин небольшими порциями добавляли к раствору 1-((2-метоксиэтокси)метокси)-4-((транс)-2-нитроциклопропил)бензола (промежуточный продукт K, 0,40 г, 1,51 ммоля) в i-PrOH (15 мл) и HCl (5,6 мл 2,7 н. водного раствора, 15,1 ммоля).
Через 16 ч смесь подщелачивали с помощью NaOH (10% водный раствор, 10 мл) и фильтровали через слой целита, который промывали с помощью 10 мл метанола. Фильтрат концентрировали и добавляли 15 мл воды, экстрагировали с помощью CH2Cl2 (3×15 мл); органические слои промывали рассолом (25 мл), сушили над безводным Na2SO4 и фильтровали. После удаления растворителя неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (2-5% МеОН/CH2Cl2) и получали 0,26 г (транс)-2-(4-((2-метоксиэтокси)метокси)фенил)циклопропанамина [Rf=0,1 (5% МеОН/CH2Cl2), белое твердое вещество, выход 73%].
Промежуточный продукт М: 4-((Транс)-2-аминоциклопропил)фенол
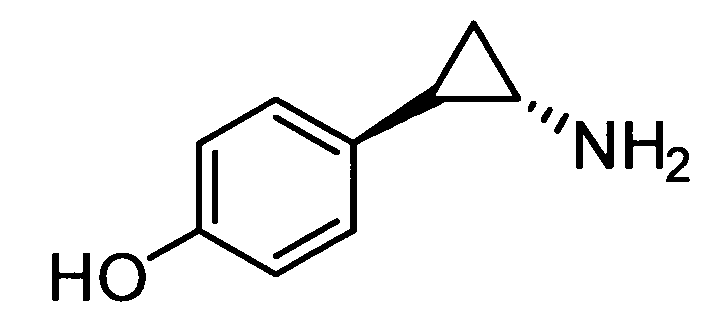
Раствор (транс)-2-(4-((2-метоксиэтокси)метокси)фенил)циклопропанамина (промежуточный продукт L, 62 мг, 0,26 ммоля) м p-TsOH·H2O (60 мг, 0,31 ммоля) в EtOH (5 мл) нагревали при 75°С в течение 2 ч. Значение pH реакционной смеси устанавливали равным 7 с помощью NaOH (10% водный раствор), смесь выливали в воду (10 мл) и экстрагировали с помощью EtOAc (4×10 мл). Органический слой промывали рассолом (10 мл), сушили над безводным Na2SO4 и фильтровали. После удаления растворителя получали коричневатый остаток (44 мг, 4-((транс)-2-аминоциклопропил)фенол, загрязненный с помощью p-TsOH), который использовали на следующей стадии без дополнительной очистки.
Промежуточный продукт N: трет-Бутил-(транс)-2-(4-гидроксифенил)-циклопропилкарбамат
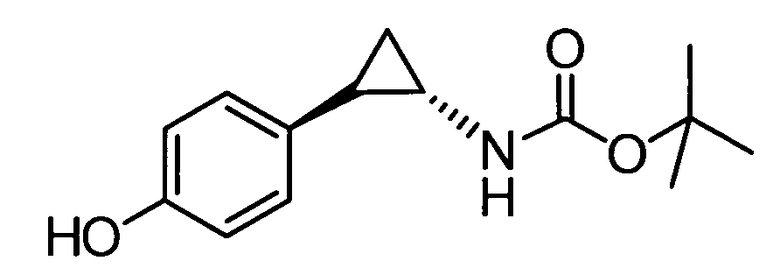
ВоС2О (94 мг, 0,43 ммоля) добавляли к раствору 4-((транс)-2-аминоциклопропил)фенола (промежуточный продукт М, 0,26 ммоля) и Et3N (59 мкл, 0,43 ммоля) в ТГФ (4 мл) и перемешивали в течение 3 ч. После удаления растворителя неочищенный остаток растворяли в EtOAc (10 мл) и последовательно промывали смесью [вода (5 мл) и HCl (10% водный раствор, 1 мл)] и рассолом (5 мл). Органический слой сушили над безводным Na2SO4 и фильтровали; после удаления растворителя остаток очищали с помощью колоночной хроматографии на силикагеле (10-20% EtOAc/гексаны), и получали 26 мг трет-бутил-(транс)-2-(4-гидроксифенил)циклопропилкарбамата [Rf=0,7 (50% AcOEt/гексаны), бесцветное масло, выход 40%].
1H-ЯМР (CDCl3, 250 МГц, δ): 1,10-1,02 (m, 2Н), 1,46 (s, 9Н), 1,99-1,94 (m, 1Н), 2,66 (br, 1Н), 4,90 (br, 1Н), 6,46 (br, 1Н), 6,69 (d, 2Н), 6,93 (d, 2Н).
Пример 1: N-((Транс)-2-(4-(бензилокси)фенил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин дигидрохлорид
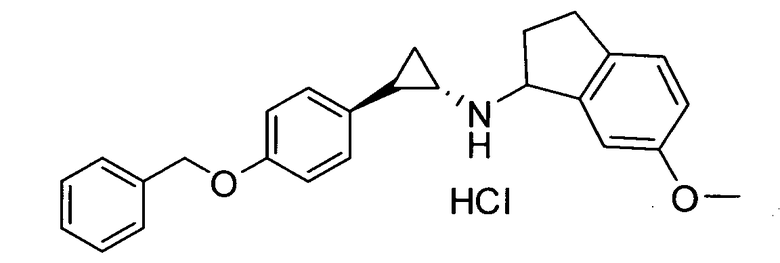
К раствору (транс)-2-(4-(бензилокси)фенил)циклопропанамина (промежуточный продукт D, 150 мг, 0,627 ммоля) и 2,3-дигидро-6-метоксиинден-1-она (132 мг, 0,815 ммоля) в ДХЭ (10 мл) при 0°С медленно добавляли триацетоксиборогидрид натрия (159,5, 0,752 ммоля) и перемешивали при комнатной температуре в течение 5 ч. Реакционную смесь выпаривали при пониженном давлении. Остаток растворяли в метаноле (15 мл) и к реакционной смеси при 0°С медленно добавляли NaBH4 (47,5 мг, 1,25 ммоля) и перемешивали в течение 3 ч. После завершения реакции реакционную смесь выливали в воду (100 мл) и экстрагировали с помощью EtOAc (2×100 мл). Объединенные органические слои промывали рассолом (100 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали при пониженном давлении. Неочищенное вещество очищали с помощью препаративной ВЭЖХ и получали (70 мг, 29,1%) N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амина. Добавляли HCl в диоксане (20 мл) и перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали и получали N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-6-метокси-2,3-дигидро-1Н-индан-1-амин гидрохлорид (72 мг, выход=94%) в виде бледно-желтого твердого вещества. 1H-ЯМР (ДМСО-d6) δ (част./млн): 1,26 (m, 1Н), 1,51-1,59 (m, 1Н), 2,29 (m, 1Н), 2,45 (m, 1Н), 2,80 (m, 2Н), 3,02 (m, 1Н), 3,64 (d, 3Н), 4,85 (br, 1Н), 5,09 (s, 2Н), 6,92 (m, 3Н), 7,09 (t, 2Н), 7,25 (m, 2Н), 7,37 (m, 1Н), 7,42 (m, 5Н), 9,70 (br, 1Н), 9,90 (br, 1Н). МС (М+Н): 386,1.
Приведенные ниже соединения можно синтезировать по методике, описанной в примере 1, с использованием соответствующего производного транс-циклопропиламина и имеющихся в продаже циклических кетонов.
Пример 2: N-((Транс)-2-(4-(бензилокси)фенил)циклопропил)-5,6-диметокси-2,3-дигидро-1Н-инден-1-амин гидрохлорид
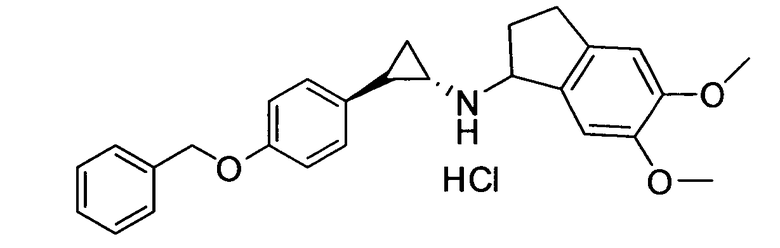
1H-ЯМР (ДМСО-d6) δ (част./млн): 1,25 (m, 1Н), 1,50 (m, 1Н), 2,29 (m, 1Н), 2,42 (m, 1Н), 2,78 (m, 1Н), 2,89 (br, 1Н), 3,03 (m, 1Н), 3,54-3,59 (s, 3Н), 3,74 (d, 3Н), 4,80 (br, 1Н), 5,09 (s, 2Н), 6,92 (m, 3Н), 7,09 (m, 3Н), 7,41 (m, 5Н), 9,49 (br, 1Н), 9,58 (br, 1Н). МС (М+Н): 416,3.
Пример 3: N-((Транс)-2-(4-(бензилокси)фенил)циклопропил)-4,5-диметокси-2,3-дигидро-1Н-инден-1-амин гидрохлорид
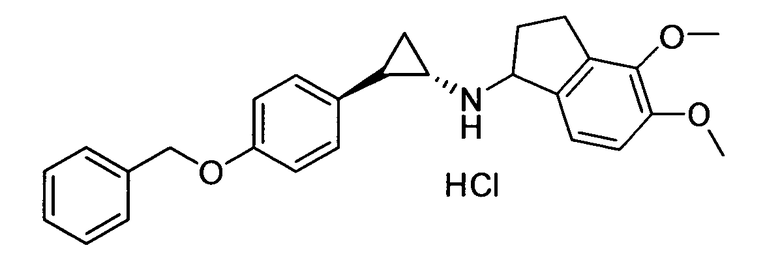
1H-ЯМР (ДМСО-d6) δ (част./млн): 1,24 (m, 1Н), 1,42-1,52 (m, 1Н), 2,33 (m, 1Н), 2,42 (m, 2Н), 2,81 (m, 2Н), 3,04 (m, 1Н), 3,65-3,70 (s, 3Н), 3,79 (s, 3Н), 4,81 (br, 1Н), 5,08 (s, 2Н), 6,95 (m, 3Н), 7,03 (d, 1Н), 7,09 (d, 1Н), 7,27 (d, 1Н), 7,33 (d, 1Н), 7,42 (m, 4Н), 9,48 (br, 1Н), 9,58 (br, 1Н). МС (М+Н): 224,5.
Пример 4: N-((Транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин гидрохлорид
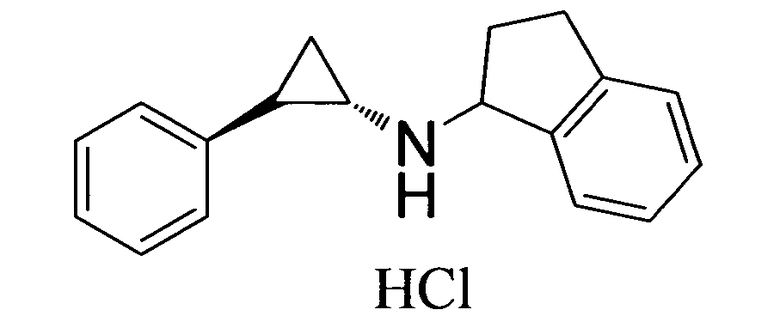
1H-ЯМР (CD3OD) δ (част./млн): 1,43 (qd, 1Н), 1,50-1,59 (m, 1Н), 2,40 (m, 1Н), 2,47 (m, 1Н), 2,59 (m, 1Н), 2,99 (m, 2Н), 3,17 (m, 1H), 4,98 (dd, 1H), 7,12 (q, 2Н), 7,26 (t, 1Н), 7,31 (m, 3Н), 7,37 (m, 2Н), 7,55 (t, 1Н). МС (М+Н): 249,9.
Пример 5: 6-Метокси-N-((транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин
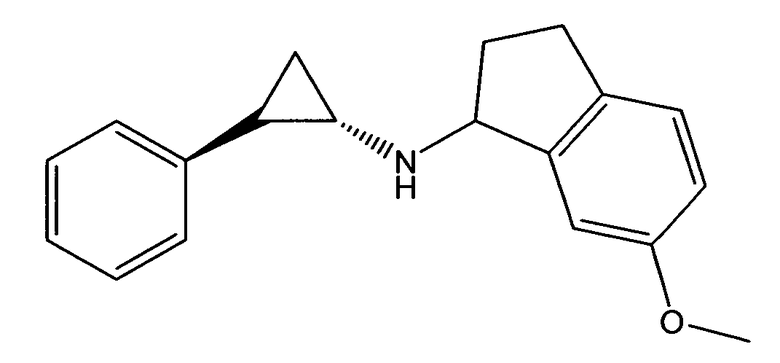
1H-ЯМР (CDCl3) δ (част./млн): 1,03 (m, 1Н), 1,16 (m, 1Н), 1,94-2,00 (m, 2Н), 2,47-2,50 (m, 2Н), 2,74 (m, 1Н), 2,90 (m, 1Н), 3,69-3,76 (s, 3Н), 4,32 (m, 1Н), 6,74 (d, 1Н), 6,81 (s, 1H), 6,89 (s, 1Н), 7,03 (d, 2Н), 7,11 (d, 2Н), 7,16 (d, 1Н), 7,24 (m, 1Н). МС (М+Н): 280,0.
Пример 6: 6-Хлор-N-((транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин гидрохлорид
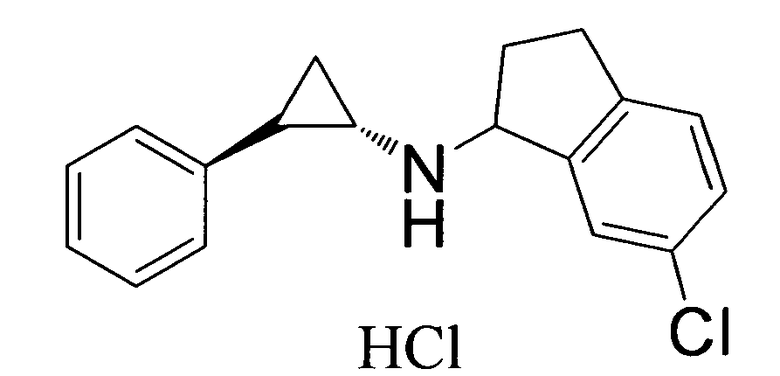
1H-ЯМР (CD3OD) δ (част./млн): 1,43 (qd, 1Н), 1,56 (m, 1Н), 2,45 (m, 2Н), 2,61 (m, 1Н), 2,98 (m, 2Н), 3,15 (m, 1Н), 4,98 (dd, 1Н), 7,12 (q, 2Н), 7,25 (m, 1Н), 7,31 (m, 3Н), 7,36 (m, 1Н), 7,58 (s, 1Н). МС (М+Н): 284,0/286,0.
Пример 7: N-((Транс)-2-фенилциклопропил)-6-(трифторметил)-2,3-дигидро-1Н-инден-1-амин гидрохлорид
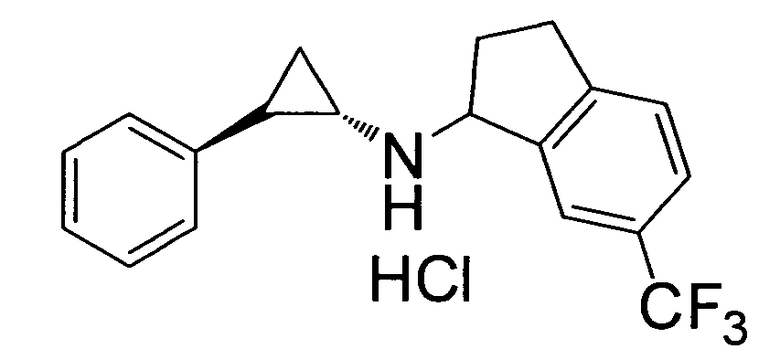
1H-ЯМР (CD3OD) δ (част./млн): 1,47 (m, 1Н), 1,55 (m, 1Н), 2,45 (m, 2Н), 2,66 (m, 1H), 3,07 (m, 2Н), 3,22 (m, 1H), 5,08 (td, 1H), 7,12 (dd, 2Н), 7,26 (d, 1Н), 7,31 (m, 2Н), 7,56 (dd, 1Н), 7,70 (t, 1Н), 7,85 (d, 1Н). МС (М+Н): 318,0.
Пример 8: 7-Метокси-N-((транс)-2-фенилциклопропил)-1,2,3,4-тетрагидронафталин-1-амин
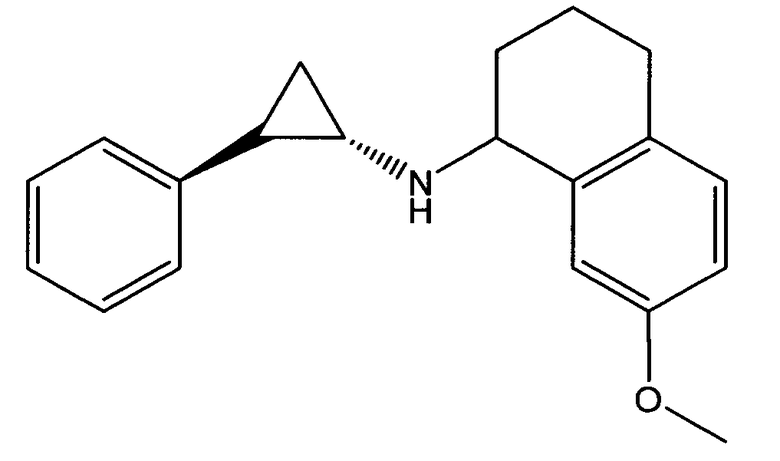
1H-ЯМР (CDCl3) δ (част./млн): 1,04 (m, 1Н), 1,12-1,18 (m, 1H), 1,56 (m, 4Н), 1,72 (m, 1Н), 1,92 (m, 2Н), 1,98 (m, 1Н), 2,42-2,50 (m, 1Н), 2,72 (m, 2Н), 3,66-3,76 (s, 3Н), 3,86 (m, 1Н), 6,72 (m, 1Н), 6,82 (s, 1Н), 6,92 (s, 1Н), 6,98 (d, 1Н), 7,06 (t, 3Н), 7,16 (t, 1Н). МС (М+Н): 294,0.
Пример 9: N-((Транс)-2-(3′-хлорбифенил-4-ил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин гидрохлорид
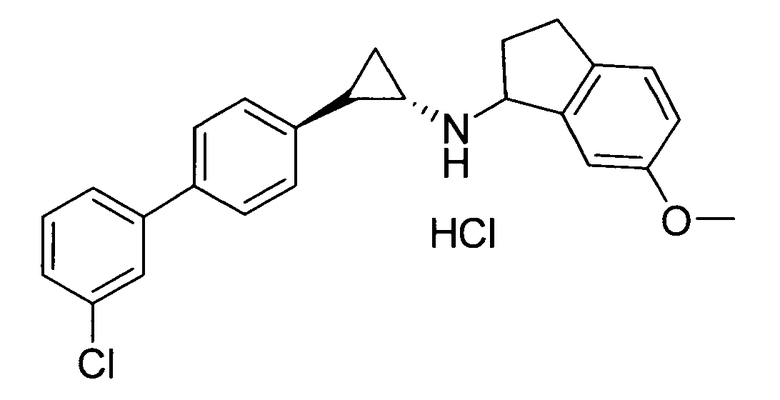
Стадия 1:
Раствор трет-бутил-(транс)-2-(4-бромфенил)циклопропилкарбамата (промежуточный продукт Н, 3 г, 9,6 ммоля), 3-хлорфенилбороновой кислоты (1,8 г, 11,5 ммоля) и K2CO3 (3,9 г, 28,8 ммоля) в смеси ацетонитрил:вода (4:1) дегазировали в течение 20 мин. Добавляли тетракис(трифенилфосфин)палладий (300 мг) и кипятили с обратным холодильником в течение 18 ч. После завершения реакции реакционную смесь выливали в воду со льдом (100 мл) и экстрагировали с помощью EtOAc (2×50 мл). Объединенные органические экстракты промывали водой (50 мл), рассолом (50 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью колоночной хроматографии (SiO2) с использованием смеси EtOAc:петролейный эфир и получали (3 г, 91%) трет-бутил-(транс)-2-(3′-хлорбифенил-4-ил) циклопропилкарбамат в виде белого твердого вещества.
Стадия 2:
К охлажденному раствору трет-бутил-(транс)-2-(3′-хлорбифенил-4-ил) циклопропилкарбамата (3 г) в Et2O (15 мл) при 0°С добавляли HCl в Et2O (15 мл), перемешивали при KT в течение 16 ч. За протеканием реакции следили с помощью ТСХ (тонкослойная хроматография). После завершения реакции растворитель выпаривали, остаток растирали с Et2O и получали (транс)-2-(3′-хлорбифенил-4-ил)циклопропанамин в виде соли с HCl (2,3 г, 95,8%), белое твердое вещество. Соль с HCl превращали в свободное основание с использованием раствора NaHCO3 и использовали в следующей реакции.
Стадия 3:
К раствору (транс)-2-(3′-хлорбифенил-4-ил)циклопропанамина (520 мг, 2,1 ммоля) и 6-метокси-2,3-дигидро-1Н-инден-1-она (381 мг, 2,3 ммоля) в ДХЭ (10 мл) при 0°С медленно добавляли триацетоксиборогидрид натрия (890 мг, 2 экв.) и перемешивали в течение 20 ч. После завершения реакции реакционную смесь выпаривали. Остаток растворяли в метаноле (15 мл) и к реакционной смеси при 0°С медленно добавляли NaBH4 (240 мг, 3 экв.) и перемешивали в течение 3 ч. После завершения реакции реакционную смесь выливали в воду со льдом (20 мл) и экстрагировали с помощью EtOAc (2×20 мл). Объединенные органические слои промывали рассолом (20 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью препаративной ВЭЖХ и получали (110 мг, 13,2%) свободного амина. Добавляли смесь диоксан-HCl (20 мл), перемешивали при KT в течение 15 мин и выпаривали и получали N-((транс)-2-(3′-хлорбифенил-4-ил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин гидрохлорид (100 мг, 83%) в виде бледно-коричневого твердого вещества. 1H-ЯМР (ДМСО-d6) δ (част./млн): 1,41 (m, 1Н), 1,61-1,69 (m, 1Н), 2,33 (m, 1Н), 2,44 (m, 1H), 2,64 (m, 1Н), 2,81 (m, 1Н), 3,03 (m, 2Н), 3,65 (d, 3Н), 4,88 (br, 1Н), 6,92 (m, 1Н), 7,23 (d, 1Н), 7,29 (m, 2Н), 7,42 (d, 1Н), 7,50 (t, 2Н), 7,63 (t, 3Н), 7,71 (s, 1Н), 9,71 (br, 1Н), 9,88 (br, 1Н). МС (М+Н): 390,1.
Приведенные ниже соединения можно синтезировать по методике, описанной в примере 9, с использованием соответствующего производного транс-циклопропиламина и имеющихся в продаже бороновых кислот.
Пример 10: N-((Транс)-2-(4′-хлорбифенил-4-ил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин гидрохлорид
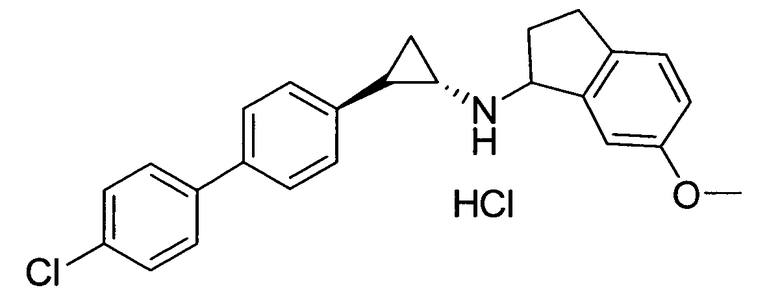
1H-ЯМР (ДМСО-d6) δ (част./млн): 1,41 (m, 1Н), 1,64-1,70 (m, 1H), 2,34 (m, 1Н), 2,45 (m, 1Н), 2,66 (m, 1Н), 2,80 (m, 1Н), 3,05 (m, 2Н), 3,66 (d, 3Н), 4,88 (br, 1H), 6,91 (m, 1Н), 7,25 (m, 4Н), 7,52 (d, 2Н), 7,61 (d, 2Н), 7,68 (d, 2Н), 9,84 (br, 1Н), 10,05 (br, 1Н). МС (М+Н): 390,1.
Пример 11: 6-Метокси-N-((транс)-2-(3′-метоксибифенил-4-ил)циклопропил)-2,3-дигидро-1Н-инден-1-амин гидрохлорид
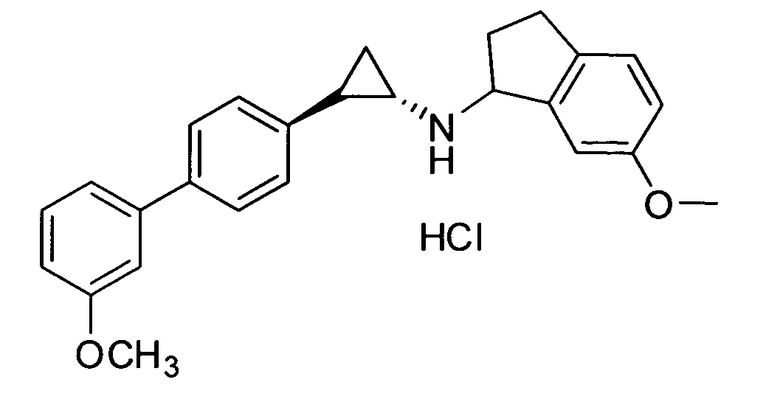
1H-ЯМР (ДМСО-d6) δ (част./млн): 1,40 (m, 1Н), 1,58-1,65 (m, 1H), 2,31 (m, 1H), 2,44 (m, 1H), 2,60 (m, 1H), 2,79 (m, 1H), 3,02 (m, 2H), 3,65 (d, 3H), 3,81 (s, 3H), 4,90 (br, 1H), 6,94 (m, 2H), 7,23 (m, 6H), 7,35 (t, 1H), 7,60 (d, 2H), 9,63 (br, 1H), 9,79 (br, 1H). MC (M+H): 386,1.
Пример 12: N-Транс-(2-циклогексилэтил)-2-фенилциклопропанамин гидрохлорид
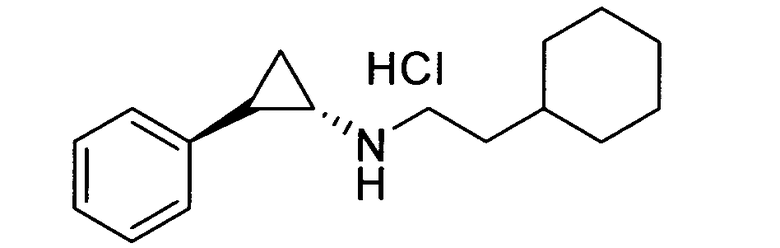
К раствору промежуточного продукта В (1,5 г, 6,42 ммоля) в диметилформамиде (ДМФ, 30 мл) добавляли гидрид натрия (0,38 мг, 9,64 ммоля) и суспензию перемешивали при комнатной температуре в течение 30 мин. Затем добавляли 1-циклогексилэтилбромид (1,2 мл, 7,71 ммоля) и суспензию перемешивали при комнатной температуре в течение 12 ч. Растворители выпаривали и остаток растворяли в дихлорметане (60 мл) и промывали водой, рассолом и водой, сушили и концентрировали. Полученное твердое вещество очищали с помощью колоночной хроматографии и получали продукт, содержащий защитную группу Boc (1,4 г). Это твердое вещество растворяли в дихлорметане и добавляли HCl (15 мл). Осадок отфильтровывали, промывали холодным эфиром и сушили и получали искомый продукт (1,56 г, 88%). 1H-ЯМР (CDCl3) δ (част./млн): 1,05 (m, 13Н), 1,21 (m, 1Н), 1,44 (m, 1Н), 2,41 (m, 1Н), 2,92 (m, 1Н), 3,20 (m, 2Н), 7,16-7,25 (m, 5Н), 8,2 (bs, 2Н). MC (М+Н): 245,0.
Приведенные ниже соединения можно синтезировать по методике, описанной в примере 12, с использованием соответствующих имеющихся в продаже алкилгалогенидов.
Пример 13: (Транс)-N-(3-циклогексилпропил)-2-фенилциклопропанамин гидрохлорид
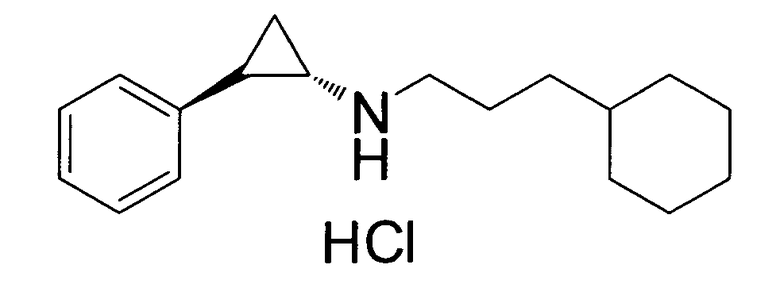
1H-ЯМР (CD3OD) δ (част./млн): 0,94 (m, 1H), 1,30 (m, 6H), 1,35 (q, 1H), 1,47 (m, 1H), 1,72 (m, 8H), 2,43 (m, 1H), 2,93 (квинтет, 1H), 3,14 (t, 2H), 7,17 (d, 2H), 7,22 (t, 1H), 7,30 (t, 2H). МС (M+H): 258,0.
Пример 14: (Транс)-N-(2-циклогептилэтил)-2-фенилциклопропанамин гидрохлорид
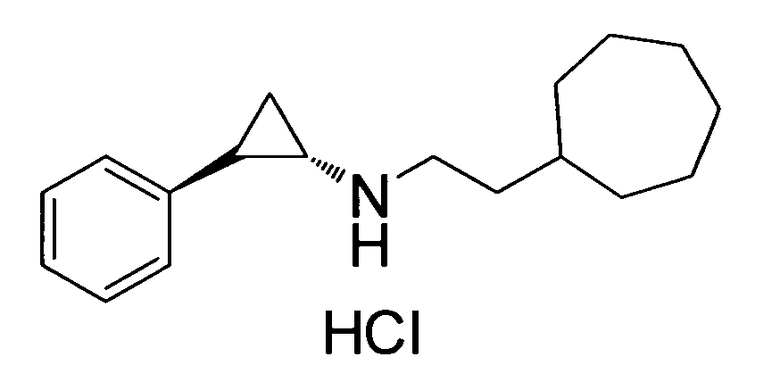
1H-ЯМР (CD3OD) δ (част./млн): 1,10 (m, 2Н), 1,38 (m, 4Н), 1,45 (m, 3Н), 1,53 (m, 2Н), 1,62 (m, 2Н), 1,71 (t, 2Н), 1,79 (m, 3Н), 2,44 (m, 1Н), 2,93 (квинтет, 1Н), 3,16 (t, 2H), 7,16 (d, 2H), 7,23 (t, 1H), 7,31 (t, 2H). MC (M+H): 258,0.
Пример 15: (Транс)-2-(4-(3-бромбензилокси)фенил)-N-(2-циклогексилэтил)циклопропанамин гидрохлорид
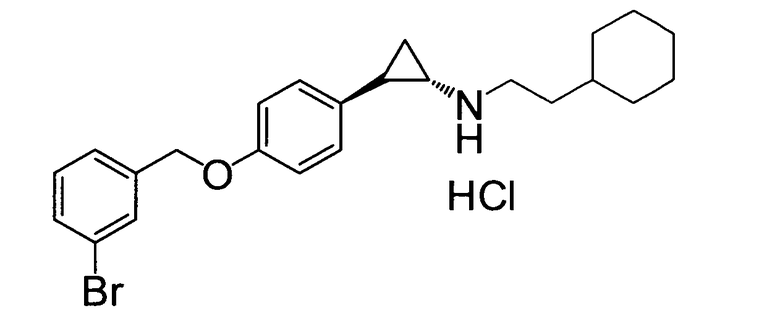
Стадия 1:
К раствору трет-бутил-(транс)-2-(4-гидроксифенил)циклопропилкарбамата (промежуточный продукт N, 5 г, 20,0 ммоля), K2CO3 (6,8 г, 50,0 ммоля) в сухом ДМФ (10 объемов) добавляли 3-бромбензилбромид (5 г, 20,0 ммоля) и перемешивали при КТ в течение 16 ч. После завершения реакции реакционную смесь выливали в воду со льдом (150 мл) и экстрагировали с помощью EtOAc (2×75 мл). Объединенные экстракты промывали водой (100 мл), рассолом (100 мл), сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью флэш-хроматографии и получали (трет-бутил-(транс)-2-(4-(3-бромбензилокси)фенил)циклопропилкарбамат (5,2 г, 62%) в виде белого твердого вещества. Неочищенное вещество использовали на следующей стадии без дополнительной очистки.
Стадия 2:
К охлажденному раствору (трет-бутил-(транс)-2-(4-(3-бромбензилокси)фенил)циклопропилкарбамата (5,2 г) в Et2O (50 мл) при 0°С добавляли HCl в Et2O (50 мл), перемешивали при KT в течение 16 ч. Растворитель выпаривали, остаток растирали с Et2O (2×20 мл) и получали (транс)-2-(4-(3-бромбензилокси)фенил)циклопропанамин гидрохлорид (3,3 г, 73%) в виде белого твердого вещества. Соль с HCl превращали в свободное основание с использованием раствора NaHCO3 и использовали в следующей реакции.
Стадия 3:
К раствору (транс)-2-(4-(3-бромбензилокси)фенил)циклопропанамина (2 г, 6,2 ммоля) и 2-циклогексилацетальдегида (790 мг, 6,2 ммоля) в ДХЭ (20 мл) при 0°С медленно добавляли триацетоксиборогидрид натрия (2,3 г, 2 экв.) и перемешивали в течение 20 ч. После завершения реакции реакционную смесь выпаривали. Остаток растворяли в метаноле (15 мл) и к реакционной смеси при 0°С медленно добавляли NaBH4 (627 мг, 3 экв.) и перемешивали в течение 3 ч. После завершения реакции реакционную смесь выливали в воду со льдом (50 мл) и экстрагировали с помощью EtOAc (2×50 мл). Объединенные органические слои промывали рассолом (50 мл) и сушили над безводным Na2SO4, фильтровали и выпаривали. Неочищенный остаток очищали с помощью колоночной флэш-хроматографии с использованием смеси EtOAc:петролейный эфир и получали (транс)-2-(4-(3-бромбензилокси) фенил)-N-(2-циклогексилэтил) циклопропанамин (500 мг, 21%) в виде бледно-желтой жидкости.
1H-ЯМР (ДМСО-d6) δ (част./млн): 0,90 (q, 2Н), 1,18 (m, 3Н), 1,33 (m, 1Н), 1,47 (m, 1Н), 1,53 (q, 2Н), 1,65 (br, 4Н), 2,41 (m, 1H), 2,86 (m, 1Н), 3,04 (m, 2Н), 3,51 (br, 2Н), 5,10 (s, 2Н), 6,96 (d, 2Н), 7,10 (d, 2Н), 7,35 (t, 1Н), 7,43 (d, 1Н), 7,53 (d, 1Н), 7,63 (s, 1H), 9,16 (br, 2Н). МС (М+Н): 428,2.
Приведенные ниже соединения можно синтезировать по методике, описанной в примере 15, с использованием соответствующего производного транс-циклопропиламина и имеющихся в продаже альдегидов или кетонов.
Пример 16: N-((Транс)-2-(4-(3-бромбензилокси)фенил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин гидрохлорид
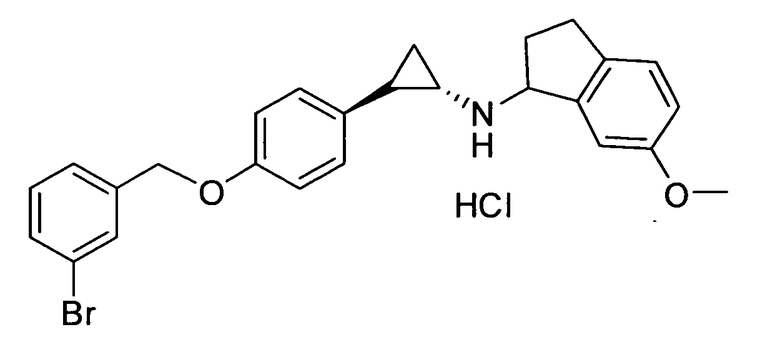
1H-ЯМР (ДМСО-d6) δ (част./млн): 1,28 (m, 1Н), 1,48-1,57 (m, 1Н), 2,29 (m, 1Н), 2,42 (m, 2Н), 2,81 (m, 2Н), 3,01 (m, 1Н), 3,64 (d, 3Н), 4,86 (br, 1H), 5,11 (s, 2Н), 6,91 (d, 1Н), 6,95 (d, 2Н), 7,10 (t, 2Н), 7,22 (m, 2Н), 7,36 (m, 1Н), 7,43 (d, 1Н), 7,52 (d, 1Н), 7,64 (s, 1Н), 9,57 (br, 1Н), 9,74 (br, 1Н). MC (М+Н): 464,1.
Приведенные ниже соединения специалист с общей подготовкой в данной области техники может получить в соответствии с описанными схемами синтеза или их модификациями с использованием соответствующих исходных веществ и реагентов.
Пример 17: (Транс)-2-(3′-хлорбифенил-4-ил)-N-(2-циклогексилэтил)-циклопропанамин
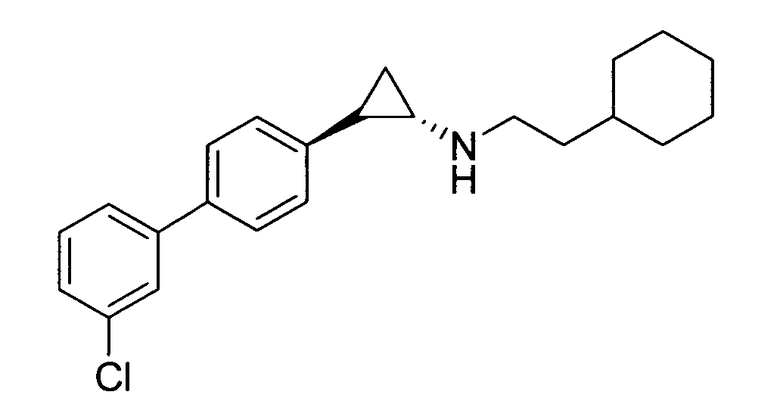
1H-ЯМР (ДМСО-d6) δ (част./млн): 0,91 (q, J=12 Гц, 2Н), 1,13-1,24 (m, 3Н), 1,34 (q, J=7 Гц, 2Н), 1,47-1,60 (m, 3Н), 1,60-1,74 (m, 5Н), 2,54 (brs, 1Н), 3,01 (brs, 1Н), 3,07 (brs, 2Н), 7,28 (d, J=8 Гц, 2Н), 7,41-7,43 (m, 1Н), 7,48 (t, J=8 Гц, 1Н), 7,64 (t, J=8 Гц, 3Н), 7,71 (s, 1Н), 9,21 (brs, 2Н). МС (М+Н): 354,1.
Пример 18: (Транс)-2-(4′-хлорбифенил-4-ил)-N-(2-циклогексилэтил)-циклопропанамин
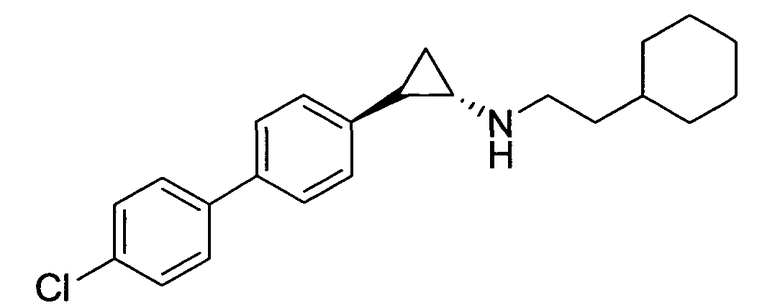
1H-ЯМР (ДМСО-d6) δ (част./млн): 0,92 (q, J=12 Гц, 2Н), 1,10-1,26 (m, 3H), 1,30-1,42 (m, 2Н), 1,48-1,60 (m, 3H), 1,60-1,76 (m, 5H), 2,54 (brs, 1H), 3,02 (brs, 1H), 3,07 (brs, 2Н), 7,28 (d, J=8 Гц, 2Н), 7,51 (d, J=8 Гц, 2Н), 7,62 (d, J=8 Гц, 2Н), 7,68 (d, J=8 Гц, 2Н), 9,18 (brs, 2Н). MC (M+H): 354,1.
Пример 19: (Транс)-N-(2-циклогексилэтил)-2-(3′-метоксибифенил-4-ил)циклопропанамин
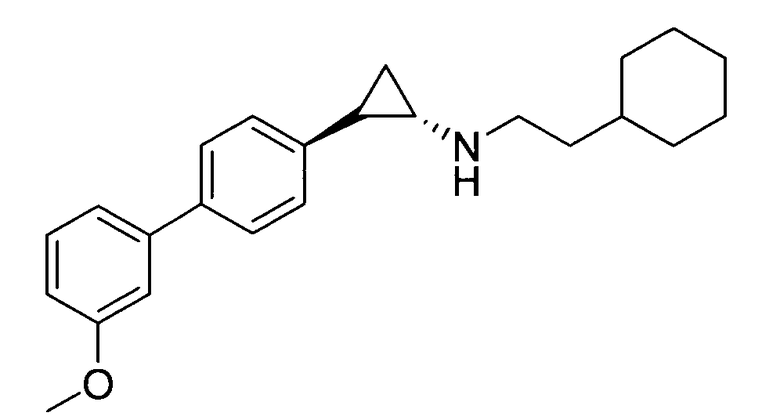
1H-ЯМР (ДМСО-d6) δ (част./млн): 0,91 (q, J=12 Гц, 2Н), 1,10-1,28 (m, 3H), 1,34-1,41 (m, 2Н), 1,47-1,60 (m, 3H), 1,60-1,75 (m, 5H), 2,56 (brs, 1H), 3,00 (brs, 1H), 3,07 (brs, 2Н), 3,82 (s, 3H), 6,88-6,96 (m, 1H), 7,16 (s, 1H), 7,21 (d, J=8 Гц, 1H), 7,26 (d, J=8 Гц, 2Н), 7,37 (t, J=8 Гц, 1H), 7,61 (d, J=8 Гц, 2Н), 9,24 (brs, 2Н). MC (M+H): 350,2.
Пример 20: N-((Транс)-2-(4-(бензилокси)фенил)циклопропил)-7-метокси-1,2,3,4-тетрагидронафталин-1-амин
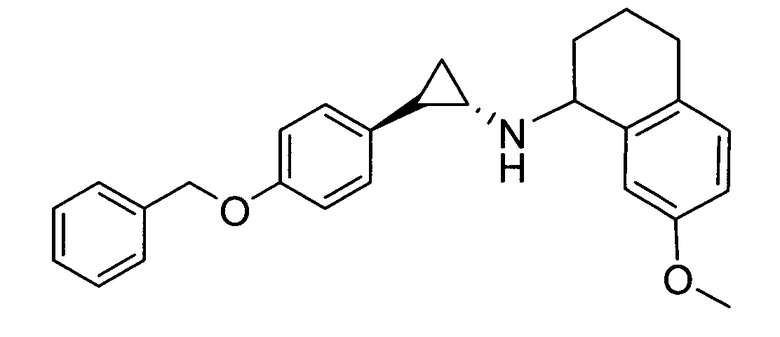
1H-ЯМР (D2O) δ (част./млн): 1,30-1,52 (m, 2Н), 1,80-1,98 (m, 2Н), 2,02-2,17 (m, 2Н), 2,21-2,30 (m, 1Н), 2,43-2,54 (m, 1H), 2,64-2,92 (m, 2H), 3,01-3,08 (m, 1H), 3,38 и 3,46 (2s, 3H), 4,58-4,65 (m, 1H), 5,17 (s, 2H), 6,74-6,82 (m, 1H), 6,88-7,12 (m, 4H), 7,15-7,25 (m, 2H), 7,39-7,56 (m, 5H). MC (M+H): 400,2.
Если не ограничиваться теоретическими соображениями, то можно полагать, что соединения, описанные в приведенных выше примерах, являются активными селективными ингибиторами LSD1 и их можно использовать для лечения или предупреждения заболевания, при котором целесообразно ингибирование LSD1.
Пример 21: 1-((Транс)-2-(4-(бензилокси)фенил)циклопропиламино)-циклопропанкарбоксамид
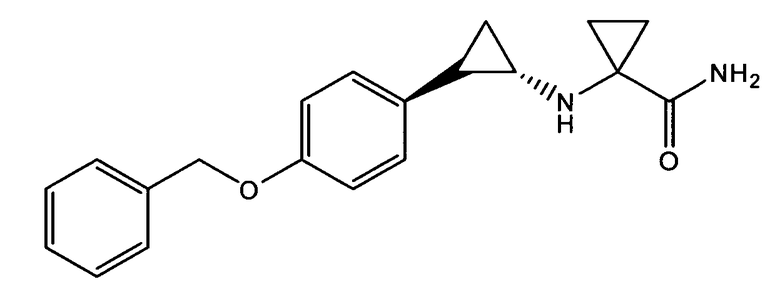
Пример 22: Биологические исследования
Можно исследовать способность соединений, предлагаемых в настоящем изобретении, ингибировать LSD1. Способность соединений, предлагаемых в настоящем изобретении, ингибировать LSD1 можно изучить следующим образом. Рекомбинантный белок LSD1 человека приобретали у фирмы BPS Bioscience Inc. Для изучения ферментативной активности LSD1 и/или степени его ингибирования рассматриваемым ингибитором (ингибиторами), предлагаемым в настоящем изобретении, в качестве субстрата выбран диметилированный пептид Н3-К4 (Millipore). Активность деметилазы оценивали при анаэробных условиях путем определения количества H2O2, образовавшегося при каталитической реакции с использованием набора для исследования Amplex® Red пероксид/пероксидаза (Invitrogen).
Вкратце, методика заключалась в следующем: определенное количество LSD1 инкубировали на льду в течение 15 мин при отсутствии и/или в присутствии ингибитора при разных концентрациях (например, от 0 до 75 мкМ, в зависимости от активности ингибитора). В качестве контроля для ингибирования использовали транилципромин (Biomol International). При проведении эксперимента каждую концентрацию ингибитора исследовали трижды. После введения фермента в реакцию с ингибитором в каждую реакционную смесь добавляли 12,5 мкМ диметилированного пептида Н3-К4 и реакционную смесь выдерживали в темноте при 37°С в течение 1 ч. Реакции ферментов проводили в буфере, содержащем 50 мМ фосфат натрия, pH 7,4. После завершения инкубации к реакционной смеси добавляли реагент Amplex® Red и раствор пероксидазы хрена (ПРХ) в соответствии с инструкциями поставщика (Invitrogen) и инкубацию проводили в течение еще 30 мин при комнатной температуре в темноте. Для проверки эффективности набора использовали 1 мкМ раствор H2O2. За превращением реагента Amplex® Red в резоруфин вследствие наличия H2O2 в реакционной смеси следили по флуоресценции (возбуждение при 540 нм, испускание при 590 нм) с использованием считывающего устройства для микропланшетов (Infinite 200, Tecan). Для определения содержания H2O2, образовавшегося при отсутствии и/или в присутствии ингибитора использовали произвольные единицы.
Максимальная активность деметилазы по отношению к LSD1 наблюдалась при отсутствии ингибитора и вносили поправку на фоновую флуоресценцию при отсутствии LSD1. Значение Ki (IC50) для каждого ингибитора определяли при половине максимальной активности.
В приведенной ниже таблице 1 представлены данные исследования по ингибированию LSD1 целым рядом соединений примеров. Обнаружено, что парнат (2-транс-фенилциклопропиламин) характеризуется значением Ki, равным примерно от 15 до 35 мкМ в зависимости от препарата фермента. Эти исследования показывают, что соединения, предлагаемые в настоящем изобретении, неожиданно активно ингибируют LSD1.
Пример 23: Биологические исследования - использование моноаминоксидазы для определения селективности соединений, предлагаемых в настоящем изобретении, по отношению к LSD1
Рекомбинантные белки моноаминоксидазы человека МАО-А и МАО-В приобретали у фирмы Sigma Aldrich. МАО катализируют окислительное дезаминирование первичных, вторичных и третичных аминов. Для определения ферментативной активности МАО и/или степени их ингибирования рассматриваемым ингибитором (ингибиторами) использовали скрининг ингибиторов на основе флуоресценции. В качестве субстрата выбрано нефлуоресцирующее соединение 3-(2-аминофенил)-3-оксопропанамин (кинураминдигидробромид, Sigma Aldrich). Кинурамин является неспецифическим субстратом для определения активности обоих МАО. При изучении окислительного дезаминирования с помощью МАО кинурамин превращается во флуоресцирующий продукт, 4-гидроксихинолин (4-HQ).
Активность моноаминоксидазы оценивали путем определения степени превращения кинурамина в 4-гидроксихинолин. Исследования проводили в черных 96-луночных планшетах с прозрачным дном (Corning) при конечном объеме, равном 100 мкл. Буфером для исследования являлся 100 мМ HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота), pH 7,5. Каждый эксперимент в каждой серии проводили трижды.
Вкратце, методика заключалась в следующем: определенное количество МАО (0,25 мкг МАО-А и 0,5 мкг МАО-В) инкубировали на льду в течение 15 мин в буфере для проведения реакции при отсутствии и/или в присутствии ингибитора при разных концентрациях (например, от 0 до 50 мкМ, в зависимости от активности ингибитора). В качестве контроля для ингибирования использовали транилципромин (Biomol International).
После введения фермента (ферментов) в реакцию с ингибитором в каждую реакционную смесь добавляли от 60 до 90 мкМ кинурамина для исследования МАО-В и МАО-А соответственно и реакционную смесь выдерживали в темноте при 37°C в течение 1 ч. Окислительное дезаминирование субстрата останавливали путем добавления 50 мкл (об./об.) 2 н. NaOH. За превращением кинурамина в 4-гидроксихинолин следили по флуоресценции (возбуждение при 320 нм, испускание при 360 нм) с использованием считывающего устройства для микропланшетов (Infinite 200, Tecan). Для определения интенсивности флуоресценции при отсутствии и/или в присутствии ингибитора использовали произвольные единицы.
Максимальная активность дезаминирования наблюдалась при определении содержания 4-гидроксихинолина, образовавшегося из кинурамина при отсутствии ингибитора с поправкой на фоновую флуоресценцию при отсутствии ферментов МАО. Значение Ki (IC50) для каждого ингибитора определяли при Vmax/2.
Диапазоны значений Ki, приведенные в Таблице 1, составляют для МАО-А - I = более 40 мкМ и II = от 1 до 40 мкМ; для МАО-В - I = более 40 мкМ, II = от 1 до 40 мкМ, и III = от 0,1 до 1 мкМ; для LSD1 - I = более 40 мкМ, II = от 1 до 40 мкМ, III = от 0,1 до 1 мкМ, и IV от 0,001 до 0,1 мкМ. Соединение примера 7 характеризуется значением Ki для МАО-А, равным примерно 40 мкМ. Если указаны два диапазона, это означает, что значение находится вблизи от границы перекрывания диапазонов, например, II, III означает примерно 1 мкМ.
Для большинства соединений примеров найдены значения Ki (IC50) для МАО-А и МАО-В, равные более 1 мкМ, тогда как значения Ki для LSD1, находились в наномолярном и нижнем наномолярном диапазоне. В этих исследованиях, описанных в настоящем изобретении, для транс-2-фенилциклопропиламина (транилципромина) получены значения Ki для МАО-А, равные примерно 2 мкМ, и значения Ki для МАО-В, равные примерно 0,6 мкМ, и для LSD1 равные примерно 15-35 мкМ.
Поэтому настоящее изобретение относится к ингибиторам, селективным по отношению к LSD1. Селективные ингибиторы LSD1 характеризуются значениями Ki для LSD1, которые не менее, чем в 2 раза меньше, чем значения Ki для МАО-А и/или МАО-В.
Таким образом, соединения, предлагаемые в настоящем изобретении, являются неожиданно активными ингибиторами LSD1 и неожиданно селективными по отношению к LSD1 по сравнению с МАО-А и МАО-В. Имеются соединения, предлагаемые в настоящем изобретении, которые ингибируют и LSD1 и МАО-В в большей степени, чем МАО-А, и поэтому они являются двойными ингибиторами LSD1 и МАО-В.
Некоторые соединения, предлагаемые в настоящем изобретении, обладают активностью по отношению к МАО-В и могут быть применимы для лечения и предупреждения заболеваний, которые можно лечить путем ингибирования МАО-В, таких как депрессия и нейродегенеративные заболевания, такие как болезнь Паркинсон, болезнь Альцгеймера и болезнь Гентингтона.
Пример 24: Исследование линии раковых клеток
Клетки линии рака толстой кишки человека НСТ116 получали от American Type Culture Collection (АТСС; CCL-247). Клетки линии НСТ116 держали в среде МДСИ (модифицированная по методике Дульбекко среда Игла) GlutaMAX (Invitrogen), к которой добавлены 10% фетальной телячьей сыворотки.
Клетки выращивали в инкубаторе во влажной атмосфере при 37°C в 5% CO2.
Исследование с помощью красителя Alamar Blue
Клетки помещали в 96-луночные планшеты при плотности, равной 6000 клеток/лунка, в 100 мкл среды за 24 ч до добавления лекарственных средств. Затем их добавляли при концентрациях, равных от 100 мкМ до 0,45 нМ (каждую концентрацию исследовали трижды). Для этого готовили планшет с разведениями лекарственных средств, равными удвоенным концентрациям, использованным при скрининге. Через 72 ч исследование жизнеспособности с использованием Alamar Blue (Biosource, Invitrogen) проводили по методике изготовителя. Вкратце, методика заключалась в следующем: Alamar Blue разбавленный в среде, добавляли к клеткам с получением 5% раствора. Клетки инкубировали при 37°C в течение 3 ч и при комнатной температуре в течение 30 мин. В качестве контроля использовали клетки, не содержащие лекарственного средства, и клетки, не содержащие лекарственного средства, подвергнутые лизису с помощью triton Х-100. Флуоресценцию регистрировали при возбуждении при длине волны, равной 530 нм, и при испускании при длине волны, равной 590 нм. Количественные результаты получали с использованием считывающего устройства для микропланшетов Infinite F200 (Tecan Group, Ltd.). Значения ЕС50 рассчитывали, как дозу лекарственных средств, необходимых для подавления роста клеток на 50%, с помощью компьютерной программы Origin 7.0.
Значение ЕС50 (мкМ), полученное с использованием соединения №1, составляло примерно 37,7 мкМ, с использованием соединения №2 составляло примерно 50 мкМ, с использованием соединения №3 составляло примерно 37 мкМ, с использованием соединения №9 составляло примерно 35,1 мкМ, с использованием соединения №11 составляло примерно 22,3 мкМ, с использованием соединения №12 составляло примерно 63,8 мкМ и с использованием соединения №15 составляло примерно 13,3 мкМ.
Если не ограничиваться теоретическими соображениями, то авторы настоящего изобретения полагают, что соединения формулы 1, в которой (А′) обозначает ароматическую группу, такую как арилалкил, арил и арилалкоксигруппа превосходно проникают в клетку и обладают высокой активностью. В соединениях формулы 1 группы этого типа могут находиться в мета- или пара-положениях по отношению к циклопропильному кольцу и предпочтительно, если они находятся в пара-положении. Согласно изобретению также было неожиданно установлено, что соединения, такие как указанные в примерах 1, 2, 3, 4 и 5, и другие аналогичные им указанные в примерах, содержащие карбоциклические группы, присоединенные непосредственно к аминогруппе фенилциклопропиламинового ядра, являются активными и селективными ингибиторами LSD1. Кроме того, согласно изобретению также неожиданно было установлено, что производные фенилциклопропиламина, содержащие алкильные группы вместо аминогрупп, где алкильные группы замещены карбоциклами и циклоалкилами, являются неожиданно хорошими ингибиторами LSD1. Кроме того, по данным исследования жизнеспособности клеток с использованием линий раковых клеток соединения, предлагаемые в настоящем изобретении, обладают хорошей активностью.
В предшествующих публикациях об LSD1 установлено, что она участвует в пролиферации и росте клеток. В некоторых исследованиях LSD1 рассматривалась в качестве мишени терапевтического воздействия при раке. В публикации Huang et al. (2007) PNAS 104:8023-8028 установлено, что полиаминовые ингибиторы LSD1 в раковых клетках приводят к умеренной реэкспресии генов, которые в результате аберрации стали молчащими, в особенности при колоректальном раке (Huang et al. Clin Cancer Res. (2009) Dec 1; 15(23):7217-28. Epub 2009 Nov 24. PMID: 19934284). В публикации Scoumanne et al. ((2007) J. Biol. Chem. May 25; 282(21):15471-5) установлено, что недостаток LSD1 приводит частичной остановке клеточного цикла в G2/M и сенсибилизированных клетках и усилению супрессии, вызванной повреждением ДНК. В публикации Kahl et al. ((2006) Cancer Res. 66(23): 11341-7.) установлено, что экспрессия LSD1 коррелирует с агрессивностью рака предстательной железы. В публикации Metzger et al. сообщают, что модулирование LSD1 с помощью сиРНК и паргилина регулирует андрогенный рецептор (АР) и, вероятно, может использоваться для лечения типов рака, при которых играет роль АР, таких как рак предстательной железы, яичников и головного мозга. В публикации Lee et al. ((2006) Chem. Biol. 13:563-567) сообщают, что в некоторых линиях раковых клеток транилципромин дерепрессирует ген Egr-1. Имеется много данных о том, что во многих случаях Egr-1 является геном-супрессором опухолей. Например, в публикации Calogero et al. (2004) Cancer Cell International 4:1) экзогенная экспрессия Egr-1 приводит к остановке роста и в конечном счете к гибели первичных линий раковых клеток; в публикации Lucerna et al. (2006) Cancer Research 66, 6708-6713) показано, что длительная экспрессия Egr-1 в некоторых моделях приводит к антиангиогенному эффекту и подавляет рост опухолей; в публикации Ferraro et al. ((2005) J. Clin. Oncol. Mar 20; 23(9):1921-6) сообщают, что Egr-1 подвергается понижающей регуляции у пациентов, страдающих раком легких с высоким риском рецидива, и может быть более устойчивым к терапии. Таким образом, усиление экспрессии Egr-1 путем ингибирования LSD1 является методикой лечения некоторых типов рака. В недавних исследованиях показано, что LSD1 участвует в раке головного мозга (Schulte et al. (2009) Cancer Res. Mar 1; 69(5):2065-71). В других исследованиях показано, что LSD1 участвует в раке молочной железы (Lims et al. Carcinogenesis. 2009 Dec 30. [Epub ahead of print] PMID: 20042638).
Таким образом, большое количество данных показывает, что LSD1 участвует в целом ряде раковых заболеваний, что указывает на то, что LSD1 является мишенью терапевтического воздействия при раке. Авторы настоящего изобретения обнаружили класс ингибиторов LSD1, которые можно использовать для лечения заболеваний, в которых мишенью для лечения является LSD1, таких как рак. В соответствии с этим фенилциклопропиламины, предлагаемые в настоящем изобретении, можно использовать для лечения таких заболеваний.
Результаты недавних исследований показывают, что LSD1 участвует в вирусной инфекции и реактивации. В частности, показано, что фармакологические ингибиторы LSD1, такие как парнат и сиРНК, подавляющие LSD1, вызывают снижение вирусной нагрузки и снижение реактивации после скрытого периода (Liang et al. (2009) Nat. Med. 15:1312-1317). Поэтому предполагается, что соединения предлагаемые в настоящем изобретении, включая соединения формулы 1, определенные и описанные в настоящем изобретении, можно использовать для лечения или предупреждения вирусной инфекции. Кроме того, предполагается, что соединения, предлагаемые в настоящем изобретении, можно использовать для лечения или предупреждения реактивации вирусной инфекции после скрытого периода
В проведенных ранее исследованиях, описанных в литературе, указано, что замещение аминогруппы фенилциклопропиламинов уменьшает способность соединения ингибировать аминоксидазы, которые характеризуются значительной структурной гомологией с LSD1. Например, в публикации Zirkle et al. ((1962) J. Med. Chem. 1265-1284) установлено, что замещение аминогруппы метильной группой немного уменьшает активность, тогда как замещение более крупными алкильными группами и группами, содержащими кольцевую систему, такими как арилалкильные группы приводит к значительному уменьшению активности МАО. Авторы настоящего изобретения неожиданно установили, что различные заместители аминогруппы фенилциклопропиламина приводят к активным ингибиторам LSD1. Кроме того, соединения формулы 1, содержащие заместители в пара-положении фенильного кольца фенилциклопропиламинового ядра, содержащего ароматическую группу, являются высокоактивными и селективными соединениями. Данные настоящего изобретения показывают, что дополнительные модификации фенилпропиламинового ядра, описанные в настоящем изобретении, могут привести к получению активных ингибиторов LSD1. В число активных селективных ингибиторов LSD1, предлагаемых в настоящем изобретении, также входят производные гетероарилциклопропиламина (соединения формулы 1, в которой (А) обозначает гетероарил, или формулы 2, в которой (W) обозначает гетероарил). В примерах приведены соединения, которые более селективно ингибируют LSD1, чем МАО-А и МАО-В. Таким образом, авторы настоящего изобретения выявили новый класс арил- и гетероарилциклопропиламинов, содержащий ингибиторы LSD1, обладающие неожиданной активностью и селективностью по отношению к LSD1, биологически важной мишенью в онкологии.
Все публикации и заявки на патенты, отмеченные в описании, указывают на уровень специалистов в области техники, к которой относится настоящее изобретение. Все публикации и заявки на патенты включены в настоящее изобретение в качестве ссылки в такой же степени, как, если бы для каждой отдельной публикации или заявки на патент было специально и по отдельности указано, что она включена в настоящее изобретение в качестве ссылки. Простое указание на публикации и заявки на патенты необязательно является признанием того, что они являются предшествующим уровнем для настоящей заявки.
Хотя приведенное выше настоящее изобретение подробно описано в качестве иллюстрации и примера для облегчения понимания, очевидно, что в объеме прилагаемой формулы изобретения в него можно внести некоторые изменения и модификации.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИЗИНСПЕЦИФИЧЕСКИЕ ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ-1 И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2602814C2 |
| ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ LSD1 НА ОСНОВЕ АРИЛЦИКЛОПРОПИЛАМИНА И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2011 |
|
RU2611437C2 |
| (ГЕТЕРО)АРИЛЦИКЛОПРОПИЛАМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ LSD1 | 2012 |
|
RU2668952C2 |
| (ГЕТЕРО)АРИЛЦИКЛОПРОПИЛАМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ LSD1 | 2012 |
|
RU2681211C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ, ВКЛЮЧАЮЩИЕ ПРОИЗВОДНЫЕ ПИРИДО [4,3-d]ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРА HSP90 И ИНГИБИТОРА HER2 | 2009 |
|
RU2532375C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2009 |
|
RU2518462C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2007 |
|
RU2456273C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНОНА/БЕНЗОКСАЗИНОНА И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2333204C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2695664C1 |
| КОНДЕНСИРОВАННЫЕ ТРИАЗОЛАМИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ Р2Х7 | 2010 |
|
RU2533122C2 |
Настоящее изобретение относится к новым соединениям формулы (1) или их фармацевтически приемлемым солям, которые обладают ингибирующей активностью в отношении LSD1 (лизинспецифические ингибиторы деметилазы-1). Указанная активность позволяет их использовать для лечения или предупреждения заболеваний, таких как рак. Указанный рак выбран из группы, включающей рак молочной железы, колоректальный рак, рак легких, рак предстательной железы, тестикулярный рак, рак головного мозга, рак кожи и рак крови. В соединении формулы 1
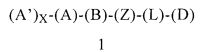
(A) обозначает пиридил или фенил; каждый (А′), если он содержится, независимо выбран из группы, включающей фенил, фенил-C1-C4-алкоксигруппу, фенил-C1-C4-алкил, галоген, C1-C4-алкоксигруппу и галоген-C1-C4-алкил, где каждый (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галоген-C1-C4-алкил, фенил, C1-C4-алкил и C1-C4-алкоксигруппу; X равен 0, 1 или 2; (B) обозначает циклопропильное кольцо, где (А) и (Z) ковалентно связаны с разными атомами углерода в (В); (Z) обозначает -NH-; (L) выбран из группы, включающей одинарную связь, -СН2-, -СН2СН2-, -СН2СН2СН2- и -СН2СН2СН2СН2-; и (D) обозначает C3-C7-циклоалкил или бензо-C5-C7-циклоалкил, где указанный C3-C7-циклоалкил или указанный бензо-C5-C7-циклоалкил, содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил; или его фармацевтически приемлемая соль. При этом соединение, которое замещено гетероарилциклопропиламином или замещенным арилциклопропиламином и соответствует формуле 2 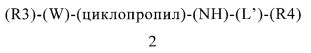
в которой (R3) содержится или не содержится, если (R3) содержится, то он выбран из группы, включающей фенил-C1-C4-алкил и фенил-C1-C4-алкоксигруппу, где указанная группа (R3) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, C1-C4-алкил, C1-C4-алкоксигруппу, галоген-C1-C4-алкил и фенил; (W) обозначает фенил или пиридильную группу, где указанный (W) содержит 0, 1 или 2 заместителя, выбранных из группы, включающей галоген, C1-C4-алкоксигруппу, галоген-C1-C4-алкил и фенил; (L′) обозначает мостик формулы -(CH2)n-, где n равно 0, 1, 2, 3 или 4; и (R4) обозначает C3-C7-циклоалкил или бензо-C5-C7-циклоалкил, где указанный C3-C7-циклоалкил или указанный бензо-C5-C7-циклоалкил содержит 0, 1, 2 или 3 заместителя, выбранных из группы, включающей галоген, C1-C4-алкоксигруппу, галоген-C1-C4-алкил, аминогруппу и С-амидогруппу; могут быть использованы для способа идентификации из группы соединений формулы 2 в качестве селективного ингибитора LSD1, которое ингибирует LSD1 в большей степени, чем МАО-А и/или МАО-В. Указанные соединения могут быть использованы для производства лекарственного средства, обладающего LSD1 ингибирующей активностью для лечения или предупреждения заболевания или патологического состояния. 8 н. и 43 з.п. ф-лы, 1 табл.., 21 пр.
1. Соединение формулы 1
в которой
(A) обозначает пиридил или фенил;
каждый (А′), если он содержится, независимо выбран из группы, включающей фенил, фенил-C1-C4-алкоксигруппу, фенил-C1-C4-алкил, галоген, C1-C4-алкоксигруппу и галоген-C1-C4-алкил, где каждый (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галоген-C1-C4-алкил, фенил, C1-C4-алкил и C1-C4-алкоксигруппу;
X равен 0, 1 или 2;
(B) обозначает циклопропильное кольцо, где (А) и (Z) ковалентно связаны с разными атомами углерода в (В);
(Z) обозначает -NH-;
(L) выбран из группы, включающей одинарную связь, -СН2-, -СН2СН2-, -СН2СН2СН2- и -СН2СН2СН2СН2-; и
(D) обозначает C3-C7-циклоалкил или бензо-C5-C7-циклоалкил, где указанный C3-C7-циклоалкил или указанный бензо-C5-C7-циклоалкил содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил;
или его фармацевтически приемлемая соль;
при условии, что исключены следующие соединения:
N-(2-фенилциклопропил)циклопентанамин; и
транс-N-(2-фенилциклопропил)циклогексиламин.
2. Соединение по п. 1, в котором (D) выбран из группы, включающей циклопентил, циклогексил, циклогептил, индил, 1,2,3,4-тетрагидронафтил и бензоциклогептил, где указанный циклопентил, указанный циклогексил, указанный циклогептил, указанный индил, указанный 1,2,3,4-тетрагидронафтил или указанный бензоциклогептил содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
3. Соединение по п. 1, в котором (D) обозначает C3-C7-циклоалкил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
4. Соединение по п. 3, в котором (D) обозначает циклопентил или циклогексил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
5. Соединение по п. 3, в котором (D) обозначает циклогексил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
6. Соединение по п. 1, в котором (L) обозначает одинарную связь и (D) обозначает бензо-C5-C7-циклоалкил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
7. Соединение по любому из пп. 1-6, в котором (А) обозначает фенильную группу.
8. Соединение по любому из пп. 1-6, в котором X равен 1 и (А′) обозначает фенильную группу или фенил-C1-C4-алкоксигруппу, где указанный (А′) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галоген-C1-C4-алкил, фенил, C1-C4-алкил и C1-C4-алкоксигруппу.
9. Соединение по любому из пп. 1-6, в котором X равен 1 и (А′) обозначает фенильную группу, содержащую 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галоген-C1-C4-алкил, фенил, C1-C4-алкил и C1-C4-алкоксигруппу.
10. Соединение по любому из пп. 1-6, в котором X равен 1 и (А′) обозначает бензилоксигруппу, содержащую 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, галоген-C1-C4-алкил, фенил, C1-C4-алкил и C1-C4-алкоксигруппу.
11. Соединение по любому из пп. 1-6, в котором X равен 0.
12. Соединение по любому из пп. 1-5, в котором (L) обозначает одинарную связь.
13. Соединение по любому из пп. 1-5, в котором (L) выбран из группы, включающей -СН2-, -СН2СН2-, -СН2СН2СН2- и -СН2СН2СН2СН2-.
14. Соединение по любому из пп. 1-6, в котором группа (D) содержит 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
15. Соединение по любому из пп. 1-6, в котором группа (D) содержит 1 заместитель, выбранный из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
16. Соединение по любому из пп. 1-6, в котором группа (D) содержит 1 заместитель, который обозначает -NH2.
17. Соединение по любому из пп. 1-6, в котором (А) обозначает пиридил.
18. Соединение по п. 1, в котором
(А) обозначает фенил и
(D) обозначает C3-C7-циклоалкил или бензо-C5-C7-циклоалкил, где указанный C3-C7-циклоалкил или указанный бензо-C5-C7-циклоалкил содержит 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
19. Соединение по п. 18, в котором (L) обозначает одинарную связь.
20. Соединение по п. 1, в котором
(А) обозначает фенил и
(D) обозначает C3-C7-циклоалкил, где указанный C3-C7-циклоалкил содержит 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
21. Соединение по п. 1, в котором
(А) обозначает фенил;
X равен 0 и
(D) обозначает C3-C7-циклоалкил, где указанный C3-C7-циклоалкил содержит 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
22. Соединение по п. 1, в котором
(А) обозначает фенил;
(L) обозначает ординарную связь и
(D) обозначает C3-C7-циклоалкил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
23. Соединение по п. 1, в котором
(А) обозначает фенил;
X равен 0 и
(D) обозначает циклопентил или циклогексил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
24. Соединение по п. 1, в котором
(А) обозначает фенил;
X равен 0 и
(D) обозначает циклогексил, содержащий 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей -NH2, галоген, амидогруппу, C1-C4-алкоксигруппу и галоген-C1-C4-алкил.
25. Соединение по любому из пп. 1-6 или 18-24, в котором (А) и (Z) находятся в транс-положении по отношению к циклопропильному кольцу (В).
26. Соединение по п. 1, которое выбрано из группы, включающей:
N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-5,6-диметокси-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-4,5-диметокси-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин;
6-метокси-N-((транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин;
6-хлор-N-((транс)-2-фенилциклопропил)-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-фенилциклопропил)-6-(трифторметил)-2,3-дигидро-1H-инден-1-амин;
7-метокси-N-((транс)-2-фенилциклопропил)-1,2,3,4-тетрагидронафталин-1-амин;
N-((транс)-2-(3′-хлорбифенил-4-ил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин;
N-((транс)-2-(4′-хлорбифенил-4-ил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин;
6-метокси-N-((транс)-2-(3′-метоксибифенил-4-ил)циклопропил)-2,3-дигидро-1Н-инден-1-амин;
N-транс-(2-циклогексилэтил)-2-фенилциклопропанамин;
(транс)-N-(3-циклогексилпропил)-2-фенилциклопропанамин;
(транс)-N-(2-циклогептилэтил)-2-фенилциклопропанамин;
(транс)-2-(4-(3-бромбензилокси)фенил)-N-(2-циклогексилэтил)циклопропанамин;
N-((транс)-2-(4-(3-бромбензилокси)фенил)циклопропил)-6-метокси-2,3-дигидро-1Н-инден-1-амин;
(транс)-2-(3′-хлорбифенил-4-ил)-N-(2-циклогексилэтил)циклопропанамин;
(транс)-2-(4′-хлорбифенил-4-ил)-N-(2-циклогексилэтил)циклопропанамин;
(транс)-N-(2-циклогексилэтил)-2-(3′-метоксибифенил-4-ил)-циклопропанамин;
N-((транс)-2-(4-(бензилокси)фенил)циклопропил)-7-метокси-1,2,3,4-тетрагидронафталин-1-амин; и
1-((транс)-2-(4-(бензилокси)фенил)циклопропиламино)циклопропан-карбоксамид;
или его фармацевтически приемлемая соль.
27. Фармацевтическая композиция, обладающая LSD1 ингибирующей активностью, содержащая соединение по любому из пп. 1-26 в терапевтически эффективном количестве и фармацевтически приемлемый носитель.
28. Соединение формулы 1 или его фармацевтически приемлемая соль, как определено в любом из пп. 1-6, 18-24 или 26 для применения в качестве лекарственного средства, обладающего LSD1 ингибирующей активностью.
29. Соединение по любому из пп. 1-6, 18-24 или 26 для применения для лечения или предупреждения рака.
30. Соединение по п. 29, где указанный рак выбран из группы, включающей рак молочной железы, колоректальный рак, рак легких, рак предстательной железы, тестикулярный рак, рак головного мозга, рак кожи и рак крови.
31. Применение соединения по любому из пп. 1-26 для приготовления лекарственного средства, обладающего LSD1 ингибирующей активностью для лечения или предупреждения рака.
32. Применение по п. 31, где указанный рак выбран из группы, включающей рак молочной железы, колоректальный рак, рак легких, рак предстательной железы, тестикулярный рак, рак головного мозга, рак кожи и рак крови.
33. Соединение по любому из пп. 1-6, 18-24 или 26 для применения для лечения или предупреждения вирусной инфекции.
34. Соединение по п. 33, где указанной вирусной инфекцией является вирусная инфекция герпеса.
35. Соединение по п. 34, где указанная вирусная инфекция герпеса вызвана вирусом герпеса, выбранным из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра.
36. Соединение по п. 33, где указанная вирусная инфекция вызвана ВИЧ.
37. Применение соединения по любому из пп. 1-26 для приготовления лекарственного средства, обладающего LSD1 ингибирующей активностью для лечения или предупреждения вирусной инфекции.
38. Применение по п. 37, где указанной вирусной инфекцией является вирусная инфекция герпеса.
39. Применение по п. 38, где указанная вирусная инфекция герпеса вызвана вирусом герпеса, выбранным из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра.
40. Применение по п. 37, где указанная вирусная инфекция вызвана ВИЧ.
41. Соединение по любому из пп. 1-6, 18-24 или 26 для применения для лечения или предупреждения реактивации вирусной инфекции после скрытого периода.
42. Соединение по п. 41, где вирусом, который реактивируется, является вирус герпеса.
43. Соединение по п. 42, где указанный вирус герпеса выбран из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра.
44. Соединение по п. 41, где вирусом, который реактивируется, является вирус ВИЧ.
45. Применение соединения по любому из пп. 1-26 для приготовления лекарственного средства, обладающего LSD1 ингибирующей активностью для лечения или предупреждения реактивации вирусной инфекции после скрытого периода.
46. Применение по п. 45, где вирусом, который реактивируется, является вирус герпеса.
47. Применение по п. 46, где указанный вирус герпеса выбран из группы, включающей HSV-1, HSV-2 и вирус Эпштейна-Барра.
48. Применение по п. 45, где вирусом который реактивируется, является вирус ВИЧ.
49. Применение соединения по любому из пп. 1-26 для приготовления лекарственного средства для ингибирования активности LSD1, где лекарственное средство содержит указанное соединение и фармацевтически приемлемый носитель в количестве, достаточном для ингибирования активности LSD1.
50. Соединение, которое является замещенным гетероарилциклопропиламином или замещенным арилциклопропиламином формулы 2:
в которой
(R3) содержится или не содержится, если (R3) содержится, то он выбран из группы, включающей фенил-C1-C4-алкил и фенил-C1-C4-алкоксигруппу, где указанная группа (R3) содержит 0, 1, 2 или 3 заместителя, независимо выбранных из группы, включающей галоген, C1-C4-алкил, C1-C4-алкоксигруппу, галоген-C1-C4-алкил и фенил;
(W) обозначает фенил или пиридильную группу, где указанный (W) содержит 0, 1 или 2 заместителя, выбранных из группы, включающей галоген, C1-C4-алкоксигруппу, галоген-C1-C4-алкил и фенил;
(L′) обозначает мостик формулы -(CH2)n-, где n равно 0, 1, 2, 3 или 4; и
(R4) обозначает C3-C7-циклоалкил или бензо-C5-C7-циклоалкил, где указанный C3-C7-циклоалкил или указанный бензо-C5-C7-циклоалкил содержит 0, 1, 2 или 3 заместителя, выбранных из группы, включающей галоген, C1-C4-алкоксигруппу, галоген-C1-C4-алкил, аминогруппу и С-амидогруппу;
для способа идентификации из группы соединений формулы 2 селективного ингибитора LSD1, которое ингибирует LSD1 в большей степени, чем МАО-А и/или МАО-В.
51. Применение селективного ингибитора LSD1 формулы 2, определенного в п. 50, для производства лекарственного средства, обладающего LSD1 ингибирующей активностью для лечения или предупреждения заболевания или патологического состояния.
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| BOLESOV, I | |||
| G | |||
| et al | |||
| Cyclopropanes and cyclobutanes | |||
| XVIII | |||
| N-Mono- and N,N-disubstituted 1-phenyl-2-aminocyclopropanes | |||
| Zhurnal Organicheskoi Zhurnal Organicheskoi Khimii, 1974, 10(8), 1661-9 (Russian) | |||
| US 3532749 A, 06.10.1970 | |||
| RU 2008119322 A, 27.11.2009 | |||
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| GOODEN D | |||
| M | |||
| et al.,Facile | |||

