ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ПАТЕНТНЫЕ ЗАЯВКИ
По настоящей патентной заявке испрашивается приоритет предварительной патентной заявки США №60/972877 (поданной 17 сентября 2007) и предварительной патентной заявки США №61/096791 (поданной 13 сентября 2008). Полный текст этих заявок включен в качестве ссылки в эту заявку.
ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится: (a) к соединениям и их солям, которые, среди прочего, применяются в качестве ингибиторов вируса гепатита C (HCV); (b) к промежуточным соединениям, применяемым для получения таких соединений и солей; (c) композициям, содержащим такие соединения и соли; (d) способам получения таких промежуточных соединений, соединений, солей и композиций; (e) способам применения таких соединений, солей и композиций; и (f) наборам, содержащим такие соединения, соли и композиции.
ПРЕДПОСЫЛКИ К СОЗДАНИЮ ИЗОБРЕТЕНИЯ
Гепатит C представляет собой переносимое с кровью инфекционное вирусное заболевание, вызываемое гапатотропным вирусом, называемым HCV. К настоящему времени известно по меньшей мере шесть различных генотипов HCV (с несколькими подтипами в пределах каждого генотипа). В Северной Америке преобладает HCV генотипа 1a, за которым следует HCV генотипов 1b, 2a, 2b и 3a. В Соединенных Штатах, HCV генотипов 1, 2 и 3 являются наиболее распространенными, при этом примерно у 80% пациентов с гепатитом C имеется HCV генотипа 1. В Европе, преобладающим является HCV генотипа 1b, за которым следуют HCV генотипов 2a, 2b, 2c и 3a. HCV генотипов 4 и 5 обнаруживаются почти исключительно в Африке. Как рассмотрено ниже, генотип HCV пациента является клинически важным в определении потенциальной реакции пациента на лечение и требуемой продолжительности такого лечения.
HCV инфекция может вызывать воспаление печени (гепатит), которое зачастую протекает бессимптомно, но в результате хронический гепатит может приводить к циррозу печени (фиброзному рубцеванию печени), раку печени и/или печеночной недостаточности. По оценкам Всемирной Организации Здравоохранения примерно 170 миллионов человек во всем мире хронически инфицированы HCV, примерно от трех примерно до четырех миллионов человек вновь инфицируются каждый год по всему миру. В соответствии с данными Центров по контролю заболеваний и профилактике, примерно четыре миллиона людей в Соединенных Штатах инфицировано HCV. Широко распространено совместное инфицирование вирусом иммунодефицита человека (ВИЧ), и показатели HCV инфекции среди ВИЧ-положительных популяций являются более высокими.
Существует небольшой шанс спонтанного освобождения от вируса, но большинство пациентов с хроническим гепатитом C не избавятся от него без лечения. Показания для лечения обычно включают подтвержденную HCV инфекцию и постоянно не соответствующие норме функциональные пробы печени. Существует две схемы лечения, которые в первую очередь применяются при лечении гепатита C: монотерапия (с использованием интерферонового средства - либо «обычного», либо пэгилированного интерферона длительного действия), и комбинированная терапия (с использованием интерферонового средства и рибавирина). Интерферон, который вводят в кровоток, работает посредством стимуляции иммунного ответа на HCV; и считается, что рибавирин, который применяют перорально, работает путем предотвращения репликации HCV. Принимаемый отдельно, рибавирин эффективно не подавляет уровни HCV, а сочетание интерферон/рибавирин является более эффективным, чем только интерферон. Обычно, гепатит C лечат сочетанием пэгилированного интерферона альфа и рибавирина в течение 24 или 48 недель, в зависимости от генотипа HCV.
Целью лечения является устойчивый вирусологический ответ - означающий, что HCV не определяется в крови после завершения лечения. После лечения сочетанием пэгилированного интерферона альфа и рибавирина, устойчивые показатели эффективности лечения (устойчивый вирусологический ответ) примерно 75% или лучше, встречаются у людей с HCV генотипов 2 и 3 через 24 недели лечения, примерно 50% у людей с HCV генотипа 1 при лечении 48 недель, и примерно 65% у людей с HCV генотипа 4 через 48 недель лечения.
Лечение может быть тяжелым физически, особенно у тех, у кого в анамнезе злоупотребление наркотиками или алкоголем, поскольку и интерферон, и рибавирин имеют многочисленные побочные эффекты. Широко распространенные побочные эффекты, связанные с интерфероном, включают в себя гриппоподобные симптомы, чрезмерную усталость, тошноту, потерю аппетита, проблемы с щитовидной железой, высокий уровень сахара в крови, выпадение волос и кожные реакции в месте введения. Возможные серьезные побочные эффекты, связанные с интерфероном, включают психозы (например, суицидальное поведение), проблемы с сердцем {например, сердечный приступ, низкое кровяное давление), повреждение других внутренних органов, проблемы с кровью (например, опасное снижение формулы крови), и появление или ухудшение аутоиммунного заболевания (например, ревматоидного артрита). Побочные эффекты, связанные с рибавирином, включают анемию, усталость, раздражительность, кожную сыпь, заложенность носа, синусит и кашель. Рибавирин также может вызывать патологии родов, поэтому следует избегать беременности пациентам женского пола и партнерам женского пола пациентов мужчин во время лечения и в течение шести месяцев после.
Некоторые пациенты не заканчивают лечение вследствие серьезных побочных эффектов, рассмотренных выше; другие пациенты (пациенты с отсутствием клинического ответа) несмотря на лечение, продолжают иметь определяемые уровни HCV; а у других пациентов (больных с рецидивом заболевания) во время лечения вирус «выводится», но вирус иногда возвращается после завершения схемы лечения. Таким образом, сохраняется необходимость в альтернативных соединения, композициях и способах лечения (применяемых либо в сочетании, либо вместо интерферонового средства и/или рибавирина) для облегчения симптомов гепатита C, тем самым, обеспечивая частичное или полное купирование симптомов. Настоящее изобретение относится к соединениям (в том числе их солям), композициям и способам лечения, которые главным образом направлены на эту потребность.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, структура которых соответствует формуле I:
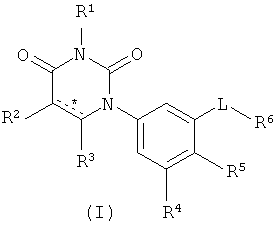
В формуле I:
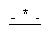 выбрана из группы, состоящей из одинарной углерод-углеродной связи и двойной углерод-углеродной связи;
выбрана из группы, состоящей из одинарной углерод-углеродной связи и двойной углерод-углеродной связи;
R1 выбран из группы, состоящей из водорода, метила и азот-защищающей группы;
R2 выбран из группы, состоящей из водорода, гало, гидрокси, метила, циклопропила и циклобутила;
R3 выбран из группы, состоящей из водорода, гало, оксо, и метила;
R4 выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила, и гетероциклила, где:
(a) амино, аминокарбонил и аминосульфонил необязательно замещены:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила и алкилсульфонила,
или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероциклил, и
(b) алкил, алкенил, алкинил, алкилокси, алкенилокси, алкинилокси и алкилсульфонил, необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из гало, оксо, нитро, циано, азидо, гидрокси, амино, алкилокси, триметилсилила, карбоциклила и гетероциклила, где:
амино необязательно замещен:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилкарбонила, алкилсульфонила, алкилоксикарбонила, карбоциклила, гетероциклила, карбоциклилалкила и гетероциклилалкила, или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл, и
(c) карбоциклил и гетероциклил необязательно замещены заместителями, вплоть до трех, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, гало, оксо, нитро, циано, азидо, гидрокси, амино, алкилокси, триметилсилила, карбоциклила и гетероциклила, где:
амино необязательно замещен:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилкарбонила, алкилсульфонила, алкилоксикарбонила, карбоциклила, гетероциклила, карбоциклилалкил и гетероциклилалкила, или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл;
R5 выбран из группы, состоящей из водорода, гидрокси, алкила, алкенила, алкинила, алкилокси, алкенилокси, алкинилокси, алкилсульфонилокси, карбоциклилсульфонилокси, галоалкилсульфонилокси, и гало;
L выбран из группы, состоящей из связи, C(RA)=C(RB), C≡C, C(O)N(RC), N(RD)C(O), C1-C2-алкилен, C(H)2O, OC(H)2, циклопропил-1,2-ен, C(H)2N(RL), N(RM)C(H)2, C(O)CH2, и CH2C(O);
RA, RB, RL и RM независимо выбраны из группы, состоящей из водорода, C1-C6-алкила, C1-C6-алкилокси, C3-C8-циклоалкила и гало, где:
C1-C6-алкил необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, гидрокси, нитро, оксо, амино, циано, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила и гетероциклила;
RC выбран из группы, состоящей из водорода и алкила;
RD выбран из группы, состоящей из водорода и алкила;
R6 выбран из группы, состоящей из C5-C6-карбоциклила, 5-6-членного гетероциклила, конденсированного 2-кольцевого гетероциклила, и конденсированного 2-кольцевого карбоциклила, где каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK;
каждый RE независимо выбран из группы, состоящей из гало, нитро, гидрокси, оксо, карбокси, циано, амино, имино, азидо и альдегидо, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила;
каждый RF независимо выбран из группы, состоящей из алкила, алкенила и алкинила, где:
каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, имино, нитро, азидо, оксо, аминосульфонила, алкилсульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, имино, аминосульфонил, аминокарбонил, карбоциклил и гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилсульфонила, алкенилсульфонила, алкинилсульфонила, алкилсульфониламино, гидрокси и алкилокси, где:
амино часть алкилсульфониламино необязательно замещена заместителем, выбранным из группы, состоящей из алкила, алкенила и алкинила;
каждый RG независимо выбран из группы, состоящей из карбоциклила и гетероциклила, где:
каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилсульфонила, алкенилсульфонила и алкинилсульфонила;
каждый RH независимо выбран из группы, состоящей из алкилокси, алкенилокси, алкинилокси, алкилсульфонилокси, алкенилсульфонилокси и алкинилсульфонилокси, где:
каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилсульфонила, алкенилсульфонила и алкинилсульфонила;
каждый RI независимо выбран из группы, состоящей из алкилкарбонила, алкенилкарбонила, алкинилкарбонила, аминокарбонила, алкилоксикарбонила, карбоциклилкарбонила и гетероциклилкарбонила, где:
(a) алкилкарбонил, алкенилкарбонил и алкинилкарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, и
(b) аминокарбонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилоксиалкила, карбоциклила, гетероциклила, алкилсульфонила и алкилсульфониламино, где:
карбоциклил и гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из гало, алкила и оксо;
каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилкарбониламино, алкенилкарбониламино, алкинилкарбониламино, алкилоксикарбониламино, алкенилоксикарбониламино, алкинилоксикарбониламино, алкилсульфониламино, алкенилсульфониламино, алкинилсульфониламино, аминокарбониламино, алкилоксикарбониламиноимино, алкилсульфониламиноимино, алкенилсульфониламиноимино и алкинилсульфониламиноимино, где:
(a) амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкенила, алкинила, алкилкарбонила, алкенилкарбонила, алкинилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, карбокси, гидрокси, алкилокси, алкенилокси, алкинилокси, гало, нитро, циано, азидо, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная, алкенильная и алкинильная часть таких заместителей необязательно замещена одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкенилокси и алкинилокси, где:
алкил необязательно замещен одним или несколькими гидрокси;
(c) карбоциклильная и гетероциклильная части таких заместителей необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, карбокси, гидрокси, алкилокси, алкенилокси, алкинилокси, гало, нитро, циано, азидо и амино, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила; и
каждый RK независимо выбран из группы, состоящей из аминосульфонила, алкилсульфонила, алкенилсульфонила и алкинилсульфонила, где:
(a) алкилсульфонил, алкенилсульфонил и алкинилсульфонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила; и
(b) аминосульфонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила.
Настоящее изобретение также относится к солям (в том числе, фармацевтически приемлемым солям) соединений по настоящему изобретению. Настоящее изобретение также относится к композициям (в том числе, фармацевтическим композициям), которые содержат одно или несколько соединений и/или солей по настоящему изобретению, и, необязательно, один или несколько дополнительных терапевтических агентов.
Настоящее изобретение также относится к наборам, которые содержат одно или несколько соединений и/или солей по настоящему изобретению, и, необязательно, один или несколько дополнительных терапевтических агентов.
Настоящее изобретение также относится к способам применения соединений, солей, композиций и/или наборов по настоящему изобретению, например, для ингибирования репликации РНК-вируса (в том числе HCV), лечения заболевания, поддающегося лечению путем ингибирования полимеразы рибонуклеиновой кислоты (РНК) HCV (в том числе, гепатита C). Настоящее изобретение также относится к применению одного или нескольких соединения и/или солей по настоящему изобретению для получения лекарственного средства. Лекарственное средство необязательно может содержать один или несколько дополнительных терапевтических агентов. В некоторых вариантах осуществления, это лекарственное средство применимо для лечения гепатита C. Дополнительные преимущества изобретения Заявителей будут очевидны специалисту в данной области из текста настоящей патентной заявки.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для этанольного сольвата соединения IB-L0-2.3.
Фигура 2 демонстрирует иллюстративный профиль TGA этанольного сольвата соединения IB-L0-2.3.
Фигура 3 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для ацетонитрильного сольвата соединения IB-L0-2.3.
Фигура 4 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для этилацетатного сольвата соединения IB-L0-2.3.
Фигура 5 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для 2-пропанольного сольвата соединения IB-L0-2.3.
Фигура 6 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для метанольного сольвата соединения IB-L0-2.3.
Фигура 7 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для 1-пропанольного сольвата соединения IB-L0-2.3.
Фигура 8 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для кристаллического соединения IB-L0-2.3, свободного от растворителя.
Фигура 9 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для гидрата соединения IB-L0-2.3.
Фигура 10 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца A мононатриевой соли соединения IB-L0-2.3.
Фигура 11 демонстрирует иллюстративный профиль TGA образца A мононатриевой соли соединения IB-L0-2.3.
Фигура 12 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца B мононатриевой соли соединения IB-L0-2.3.
Фигура 13 демонстрирует иллюстративный профиль TGA образца B мононатриевой соли соединения IB-L0-2.3.
Фигура 14 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца C мононатриевой соли соединения IB-L0-2.3.
Фигура 15 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для динатриевой соли соединения IB-L0-2.3.
Фигура 16 демонстрирует иллюстративный профиль TGA динатриевой соли соединения IB-L0-2.3.
Фигура 17 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для монокалиевой соли соединения IB-L0-2.3.
Фигура 18 демонстрирует иллюстративный профиль TGA монокалиевой соли соединения IB-L0-2.3.
Фигура 19 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца A монохолиновой соли соединения IB-L0-2.3.
Фигура 20 демонстрирует иллюстративный профиль TGA образца A монохолиновой соли соединения IB-L0-2.3.
Фигура 21 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца B монохолиновой соли соединения IB-L0-2.3.
Фигура 22 демонстрирует иллюстративный профиль TGA образца B монохолиновой соли соединения IB-L0-2.3.
Фигура 23 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для дихолиновой соли соединения IB-L0-2.3.
Фигура 24 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для девятиводной динатриевой соли соединения IB-L1-1.1.
Фигура 25 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для четырехводной динатриевой соли соединения IB-L1-1.1.
Фигура 26 демонстрирует иллюстративный профиль TGA четырехводной динатриевой соли соединения IB-L1-1.1.
Фигура 27 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для четырехводной дикалиевой соли соединения IB-L1-1.1.
Фигура 28 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для тригидрата монокалиевой соли соединения IB-L1-1.1.
Фигура 29 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для дигидрата монокалиевой соли соединения IB-L1-1.1.
Фигура 30 демонстрирует иллюстративный профиль TGA дигидрата монокалиевой соли соединения IB-L1-1.1.
Фигура 31 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для 1/7 калиевой соли соединения IB-L1-1.1.
Фигура 32 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для четырехводной монодиэтиламинной соли соединения IB-L1-1.1.
Фигура 33 демонстрирует иллюстративный профиль TGA четырехводной монодиэтиламинной соли соединения IB-L1-1.1.
Фигура 34 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца A полиморфного соединения IB-L1-1.1.
Фигура 35 демонстрирует иллюстративный профиль дифференциальной сканирующей калориметрии образца A полиморфного соединения IB-L1-1.1.
Фигура 36 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца B полиморфного соединения IB-L1-1.1.
Фигура 37 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца C полиморфного соединения IB-L1-1.1.
Фигура 38 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца D полиморфного соединения IB-L1-1.1.
Фигура 39 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца A гидрата соединения IB-L1-1.1.
Фигура 40 демонстрирует иллюстративный профиль TGA образца A гидрата соединения IB-L1-1.1.
Фигура 41 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца B гидрата соединения IB-L1-1.1.
Фигура 42 демонстрирует иллюстративный профиль TGA образца B гидрата соединения IB-L1-1.1.
Фигура 43 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца C гидрата соединения IB-L1-1.1.
Фигура 44 демонстрирует иллюстративный профиль TGA образца C гидрата соединения IB-L1-1.1.
Фигура 45 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца D гидрата соединения IB-L1-1.1.
Фигура 46 демонстрирует иллюстративную порошковую дифракционную рентгенограмму для образца E гидрата соединения IB-L1-1.1.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее подробное описание предназначено только для ознакомления специалистов в данной области с изобретением Заявителей, его принципами и его практическим применением, так, чтобы другие специалисты в данной области могли адаптировать и применить настоящее изобретение в его многочисленных формах, которые могут наилучшим образом соответствовать требованиям практического применения. Это описание и его конкретные примеры предназначены только для иллюстрации. Настоящее изобретение, следовательно, не ограничивается вариантами осуществления, описанными в настоящей заявке, и может быть модифицировано различными способами.
А. Определения.
Термин «алкил» (отдельно или в сочетании с другим термином (терминами)) означает насыщенный углеводородный заместитель с прямой или разветвленной цепью, обычно содержащий от 1 до 20 атомов углерода, более характерно от 1 примерно до 8 атомов углерода, и еще более характерно от 1 примерно до 6 атомов углерода. Примеры таких заместителей включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изо-амил и гексил. Как в этом определении, по всему тексту подробного описания Заявители представили наглядные примеры. Предоставление таких наглядных примеров не следует интерпретировать так, что представленные наглядные примеры являются единственными вариантами, доступными специалисту в данной области.
Термин «алкенил» (отдельно или в сочетании с другим термином (терминами)) означает углеводородный заместитель с прямой или разветвленной цепью, содержащий одну или несколько двойных связей, и обычно от 2 примерно до 20 атомов углерода, более характерно примерно от 2 примерно до 8 атомов углерода, и еще более характерно примерно от 2 примерно до 6 атомов углерода. Примеры таких заместителей включают этенил (винил), 2-пропенил, 3-пропенил, 1,4-пентадиенил, 1,4-бутадиенил, 1-бутенил, 2-бутенил и 3-бутенил. Термин «алкинил» (отдельно или в сочетании с другим термином (терминами)) означает углеводородный заместитель с прямой или разветвленной цепью, содержащий одну или несколько тройных связей и обычно от 2 примерно до 20 атомов углерода, более характерно примерно от 2 примерно до 8 атомов углерода, и еще более характерно примерно от 2 примерно до 6 атомов углерода. Примеры таких заместителей включают этинил, 2-пропинил, 3-пропинил, 2-бутинил и 3-бутинил.
Термин «карбоциклил» (отдельно или в сочетании с другим термином (терминами)) означает насыщенный циклический (т.е., «циклоалкильный»), частично насыщенный циклический (т.е., «циклоалкенильный»), или полностью ненасыщенный (т.е., «арильный») углеводородный заместитель, содержащий от 3 до 14 кольцевых атомов углерода («кольцевыми атомами» являются атомы, связанные вместе с образованием кольца или колец циклического заместителя). Карбоциклил может представлять собой одиночное кольцо, которое обычно содержит от 3 до 6 кольцевых атомов. Примеры таких однокольцевых карбоциклилов включают циклопропил (циклопропанил), циклобутил (циклобутанил), циклопентил (циклопентанил), циклопентенил, циклопентадиенил, циклогексил (циклогексанил), циклогексенил, циклогексадиенил и фенил. Альтернативно карбоциклил может представлять собой 2 или 3 кольца, конденсированные вместе, такие как нафталенил, тетрагидронафталенил (тетралинил), инденил, инданил (дигидроинденил), антраценил, фенантренил и декалинил.
Термин «циклоалкил» (отдельно или в сочетании с другим термином (терминами)) означает насыщенный циклический углеводородный заместитель, содержащий от 3 до 14 кольцевых атомов углерода. Циклоалкил может представлять собой одиночное углеродное кольцо, которое обычно содержит от 3 до 6 кольцевых атомов углерода. Примеры однокольцевых циклоалкилов включают циклопропил, циклобутил, циклопентил и циклогексил. Альтернативно циклоалкил может представлять собой 2 или 3 углеродных кольца, конденсированных вместе, например, декалинил.
Термин «арил» (отдельно или в сочетании с другим термином (терминами)) означает ароматический карбоциклил, содержащий от 6 до 14 кольцевых атомов углерода. Примеры арилов включают фенил, нафталенил и инденил.
В некоторых случаях, число атомов углерода в углеводородном заместителе (например, алкиле, алкениле, алкиниле или циклоалкиле) указывается с помощью индекса «Cx-Cy-», где x означает минимальное, а y означает максимальное число атомов углерода в этом заместителе. Таким образом, например, «C1-C6-алкил» относится к алкильному заместителю, содержащему от 1 до 6 атомов углерода. Поясняя дополнительно, C3-C6-циклоалкил означает насыщенное углеводородное кольцо, содержащее от 3 до 6 кольцевых атомов углерода.
Термин «водород» (отдельно или в сочетании с другим термином (терминами)) означает водородный радикал и может обозначаться -H.
Термин «гидрокси» (отдельно или в сочетании с другим термином (терминами)) означает -OH.
Термин «нитро» (отдельно или в сочетании с другим термином (терминами)) означает -NO2.
Термин «циано» (отдельно или в сочетании с другим термином (терминами)) означает -CN, который также может обозначаться как -C=N.
Термин «кето» (отдельно или в сочетании с другим термином (терминами)) означает оксо радикал и может быть обозначен как =O.
Термин «карбокси» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-OH.
Термин «амино» (отдельно или в сочетании с другим термином (терминами)) означает -NH2.
Термин «имино» (отдельно или в сочетании с другим термином (терминами)) означает =NH.
Термин «аминоимино» (отдельно или в сочетании с другим термином (терминами)) означает -NNH2.
Термин «галоген» или «гало» (отдельно или в сочетании с другим термином (терминами)) означает радикал фтора (который может быть обозначен как -F), радикал хлора (который может быть обозначен как -Cl), радикал брома (который может быть обозначен как -Br), или радикал йода (который может быть обозначен как -I).
Заместитель является «замещаемым» если он содержит по меньшей мере один атом углерода или азота, который связан с одним или несколькими атомами водорода. Таким образом, например, водород, галоген и циано не подпадают под это определение. Кроме того, атом серы в гетероциклиле, содержащем такой атом, является замещаемым одним или двумя оксо заместителями.
В тех случаях, если заместитель описан как «замещаемый», не водородный радикал находится на месте водородного радикала на углероде или азоте заместителя. Таким образом, например, замещенный алкильный заместитель представляет собой алкильный заместитель, у которого по меньшей мере один не водородный радикал находится на месте водородного радикала на алкильном заместителе. Для пояснения, монофторалкил представляет собой алкильный заместитель с фтор-радикалом, а дифторалкил представляет собой алкил, замещенный двумя радикалами фтора. Должно быть понятно, что в том случае, когда существует более одной замены на заместителе, такой не водородный радикал может быть идентичным или отличным (если не указано иное).
В том случае, если заместитель описан как «необязательно замещенный », этот заместитель может быть, либо (1) незамещенным, либо (2) замещенным. Если заместитель описан как необязательно замещенный вплоть до конкретного числа не водородных радикалов, этот заместитель может быть, либо (1) незамещенным; либо (2) замещен вплоть до того конкретного числа не водородных радикалов, или вплоть до максимального числа замещаемых положений на заместителе, смотря что меньше. Таким образом, например, если заместитель описывается как гетероарил, необязательно замещенный вплоть до 3 не водородных радикалов, то любой гетероарил менее чем с 3 замещаемыми положениями был бы необязательно замещен только вплоть до такого количества не водородных радикалов, сколько гетероарил имеет замещаемых положений. Для пояснения, тетразолил (который имеет только одно замещаемое положение) может быть необязательно замещен вплоть до одного не водородного радикала. Для дальнейшего пояснения, если аминоазот описывается как необязательно замещенный вплоть до 2 не водородных радикалов, то первичный аминоазот будет необязательно замещен вплоть до 2 не водордных радикалов, тогда как вторичный аминоазот будет необязательно замещен только вплоть до 1 не водородного радикала.
В настоящей патентной заявке используются термины «заместитель» и «радикал» взаимозаменяемо.
Приставка «гало» указывает на то, что заместитель, к которому присоединяется эта приставка, замещен одним или несколькими независимо выбранными галогеновыми радикалами. Например, галоалкил означает алкильный заместитель, в котором по меньшей мере один водородный радикал заменен галогеновым радикалом. Примеры галоалкилов включают хлорметил, 1-бромэтил, фторметил, дифторметил, трифторметил и 1,1,1-трифторэтил. Должно быть понятно, что если заместитель замещен более чем одним галогеновым радикалом, эти галогеновые радикалы могут быть одинаковыми или разными (если не указано иное).
Приставка «пергало» указывает на то, что каждый водородный радикал на заместителе, к которому эта приставка присоединена, заменен независимо выбранными галогеновыми радикалами, т.е., каждый водородный радикал на заместителе заменен галогеновым радикалом. Если все галогеновые радикалы являются одинаковыми, эта приставка обычно будет обозначать галогеновый радикал. Таким образом, например, термин «перфтор» означает, что каждый водородный радикал на заместителе, к которому присоединена эта приставка, замещен радикалом фтора. Для пояснения, термин «перфторалкил» означает алкильный заместитель, в котором радикал фтор находится на месте каждого водородного радикала.
Термин «карбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-.
Термин «аминокарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-NH2.
Термин «окси» (отдельно или в сочетании с другим термином (терминами)) означает эфирный заместитель и может быть обозначен -O-.
Термин «алкокси» (отдельно или в сочетании с другим термином (терминами)) означает алкилэфирный заместитель, т.е., -O-алкил. Примеры такого заместителя включают метокси (-O-CH3), этокси, n-пропокси, изопропокси, n-бутокси, изо-бутокси, втор-бутокси и трет-бутокси.
Термин «алкилкарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-алкил.
Термин «аминоалкилкарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-алкил-NH2.
Термин «алкоксикарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-O-алкил.
Термин «карбоциклилкарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-карбоциклил.
Аналогично, термин «гетероциклилкарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-гетероциклил.
Термин «карбоциклилалкилкарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-алкил-карбоциклил.
Аналогично, термин «гетероциклилалкилкарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-алкил-гетероциклил.
Термин «карбоциклилоксикарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-O-карбоциклил.
Термин «карбоциклилалкоксикарбонил» (отдельно или в сочетании с другим термином (терминами)) означает -C(O)-O-алкил-карбоциклил.
Термин «тио» или «тиа» (отдельно или в сочетании с другим термином (терминами)) означает тиаэфирный заместитель, т.е., эфирный заместитель, где двухвалентный атом серы находится на месте эфирного атома кислорода. Такой заместитель может быть обозначен как -S-. Это, например, «алкил-тио-алкил» означает алкил-S-алкил (алкил-сульфанил-алкил).
Термин «тиол» или «сульфгидрил» (отдельно или в сочетании с другим термином (терминами)) означает сульфгидрильный заместитель, и может быть обозначен как -SH.
Термин «(тиокарбонил)» (отдельно или в сочетании с другим термином (терминами)) означает карбонил, в котором атом кислорода был заменен серой. Такой заместитель может быть обозначен как -C(S)-.
Термин «сульфонил» (отдельно или в сочетании с другим термином (терминами)) означает -S(O)2-.
Термин «аминосульфонил» (отдельно или в сочетании с другим термином (терминами)) означает -S(O)2-NH2.
Термин «сульфинил» или «сульфоксид» (отдельно или в сочетании с другим термином (терминами)) означает -S(O)-.
Термин «гетероциклил» (отдельно или в сочетании с другим термином (терминами)) означает насыщенную (т.е., «гетероциклоалкильную»), частично насыщенную (т.е., «гетероциклоалкенильную»), или полностью ненасыщенную {т.е., «гетероарильную») кольцевую структуру всего из 3-14 кольцевых атомов. По меньшей мере один из атомов кольца представляет собой гетероатом (т.е., кислород, азот или серу), остальные атомы кольца независимо выбраны из группы, состоящей из углерода, кислорода, азота и серы. Гетероциклил может представлять собой одиночное кольцо, которое обычно содержит от 3 до 7 атомов в кольце, более характерно от 3 до 6 атомов в кольце и еще более характерно от 5 до 6 атомов в кольце. Примеры однокольцевых гетероциклилов включают фуранил, дигидрофуранил, тетрагидрофуранил, тиофенил (тиофуранил), дигидротиофенил, тетрагидротиофенил, пирролил, пирролинил, пирролидинил, имидазолил, имидазолинил, имидазолидинил, пиразолил, пиразолинил, пиразолидинил, триазолил, тетразолил, оксазолил, оксазолидинил, изоксазилидинил, изоксазолил, тиазолил, изотиазолил, тиазолинил, изотиазолинил, тиазолидинил, изотиазолидинил, тиодиазолил, оксадиазолил (в том числе,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил (фуразанил), или 1,3,4-оксадиазолил), оксатриазолил (в том числе 1,2,3,4-оксатриазолил или 1,2,3,5-оксатриазолил), диоксазолил (в том числе 1,2,3-диоксазолил, 1,2,4-диоксазолил, 1,3,2-диоксазолил или 1,3,4-диоксазолил), оксатиазолил, оксатиолил, оксатиоланил, пиранил, дигидропиранил, тиопиранил, тетрагидротиопиранил, пиридинил (азинил), пиперидинил, диазинил (в том числе пиридазинил (1,2-диазинил), пиримидинил (1,3-диазинил) или пиразинил (1,4-диазинил)), пиперазинил,триазинил (в том числе 1,3,5-триазинил, 1,2,4-триазинил и 1,2,3-триазинил)), оксазинил (в том числе 1,2-оксазинил, 1,3-оксазинил или 1,4-оксазинил)), оксатиазинил (в том числе 1,2,3-оксатиазинил, 1,2,4-оксатиазинил, 1,2,5-оксатиазинил или 1,2,6-оксатиазинил)), оксадиазинил (в том числе 1,2,3-оксадиазинил, 1,2,4-оксадиазинил, 1,4,2-оксадиазинил или 1,3,5-оксадиазинил)), морфолинил, азепинил, оксепинил, тиепинил и диазепинил.
Гетероциклил альтернативно может представлять собой 2 или 3 кольца, конденсированные вместе, например, такие как индолизинил, пиранопирролил, 4Н-хинолизинил, пуринил, нафтиридинил, пиридопиридинил (в том числе [3,4-b]-пиридинил, пиридо[3,2-b]-пиридинил или пиридо[4,3-b]-пиридинил), и птеридинил. Другие примеры гетероциклилов с конденсированными кольцами, включают бензо-конденсированные гетероциклилы, такие как индолил, изоиндолил (изобензазолил, псевдоизоиндолил), индоленил (псевдоиндолил), изоиндазолил (бензпиразолил), бензазинил (в том числе хинолинил (1-бензазинил) или изохинолинил (2-бензазинил)), фталазинил, хиноксалинил, хиназолинил, бензодиазинил (в том числе циннолинил (1,2-бензодиазинил) или хиназолинил (1,3-бензодиазинил)), бензопиранил (в том числе хроманил или изохроманил), бензоксазинил (в том числе 1,3,2-бензоксазинил, 1,4,2-бензоксазинил, 2,3,1-бензоксазинил или 3,1,4-бензоксазинил), и бензизоксазинил (в том числе 1,2-бензизоксазинил или 1,4-бензизоксазинил).
Термин гетероциклил «с 2-конденсированными кольцами» (отдельно или в сочетании с другим термином (терминами)) означает насыщенный, частично насыщенный или арилгетероциклил, содержащий 2 конденсированных кольца. Примеры гетероциклилов с 2 конденсированными кольцами включают индолизинил, хинолизинил, пуринил, нафтиридинил, птеридинил, индолил, изоиндолил, индоленинил, изоиндазолил, фталазинил, хиноксалинил, хиназолинил, бензодиазинил, бензопиранил, бензотиопиранил, бензоксазолил, антранилил, бензодиоксолил, антранилил, бензодиоксолил, бензодиоксанил, бензоксадиазолил, бензофуранил, изобензофуранил, бензотиазолил, бензотиадиазолил, бензимидазолил, бензотриазолил, бензотриазолил, бензоксазинил и тетрагидроизохинолинил.
Термин «гетероарил» (отдельно или в сочетании с другим термином (терминами)) означает ароматический гетероциклил, содержащий от 5 до 14 атомов в кольце. Гетероарил может представлять собой одиночное кольцо или 2 или 3 конденсированных кольца. Примеры гетероарильных заместителей включают 6-членные кольцевые заместители, такие как пиридил, пиразил, пиримидинил, пиридазинил и 1,3,5-, 1,2,4- или 1,2,3-триазинил; 5-членные кольцевые заместители, такие как имидазил, фуранил, тиофенил, пиразолил, оксазолил, изоксазиолил, тиазолил, 1,2,3-, 1,2,4-, 1,2,5- или 1,3,4-оксадиазолил и изотиазолил; заместители с 6/5-членными конденсированными кольцами, такие как бензотиофуранил, бензизоксазолил, бензоксазолил, пуринил и антранилил; и 6/6-членными конденсированными кольцами, такими как бензопиранил, хинолинил, изохинолинил, циннолинил, хиназолинил и бензоксазинил.
Приставка, присоединенная к многокомпонентному заместителю применяется только к первому компоненту. Для пояснения, термин «алкилциклоалкил» содержит два компонента: алкил и циклоалкил. Таким образом, приставка C1-C6- у C1-C6-алкилциклоалкила означает, что алкильный компонент алкилциклоалкила содержит от 1 до 6 атомов углерода; приставка C1-C6-не описывает циклоалкильный компонент. Для дальнейшего пояснения, приставка «гало» у галоалкоксиалкила указывает, что только алкокси компонент алкоксиалкильного заместителя замещен одним или несколькими галогеновыми радикалами. Если галогеновое замещение может альтернативно или дополнительно происходить на алкильном компоненте, заместитель был бы описан как «галоген-замещенный алкоксиалкил», а не «галоалкоксиалкил». И, наконец, если галогеновое замещение может происходить только на алкильном компоненте, заместитель вместо этого был бы описан как «алкоксигалоалкил». Если заместители описываются как «независимо выбранные» из группы, каждый заместитель выбирается независимо от другого. Каждый заместитель, следовательно, может быть идентичным или отличным от другого заместителя (заместителей). В тех случаях, когда для описания заместителя используют слова, крайний справа описанный компонент заместителя представляет собой компонент, который имеет свободную валентность.
В тех случаях, когда для описания заместителя используют химическую формулу, тире на левой стороне формулы указывает на часть заместителя, которая имеет свободную валентность.
В тех случаях, когда химическую формулу используют для описания связующего элемента между двумя другими элементами изображенной химической структуры, крайнее слева тире у заместителя указывает на часть заместителя, которая связана с левым элементом в изображенной структуре. Тире крайнее справа, с другой стороны, указывает часть заместителя, которая связана с правым элементом в изображенной структуре. Для пояснения, если изображенная химическая структура представляет собой X-L-Y, a L описан как -C(O)-N(H)-, тогда химическое вещество представляло бы собой X-C(O)-N(H)-Y.
При ссылке на использование слов «содержат» или «содержит» или «содержащий» в настоящей патентной заявке (в том числе формуле изобретения). Заявители отмечают, что если по контексту не требуется иное, эти слова используются на основании и четком понимании, что их следует интерпретировать включительно, а не исключительно, и Заявители имеют ввиду, что каждое из этих слов интерпретируется таким образом в толковании настоящей патентной заявки, включая приведенную ниже формулу изобретения.
Программное обеспечение ChemDraw было использования для составления названий соединений в настоящей патентной заявке.
Термин «некристаллическое» применительно к соединению, относится к твердому состоянию, в котором молекулы соединения представлены в неупорядоченной структуре и не образуют различимую кристаллическую решетку или элементарную ячейку. При проведении рентгеновской порошковой дифрактометрии, некристаллическое соединение не дает каких-либо характеристических кристаллических пиков.
Термин «кристаллическая форма» применительно к соединению, относится к твердому состоянию, в котором молекулы соединения расположены с образованием различимой кристаллической решетки (i) содержащей различимые элементарные ячейки, и (ii) дают пики на рентгенограмме, при воздействии рентгеновского излучения.
Термин «чистота», кроме случаев, оговоренных особо, означает химическую чистоту соединения в соответствии с общепринятым ВЭЖХ анализом.
Термин «фазовая чистота» означает чистоту твердого состояния соединения относительно конкретной кристаллической или некристаллической формы соединения, определенную с помощью аналитических способов рентгеновской порошковой дифрактометрии. Термин «фазовочистый» относится к чистоте относительно других форм соединения в твердом состоянии, и не обязательно предполагает высокую степень химической чистоты относительно других соединений.
Термин «PXRD» означает рентгеновскую порошковую дифрактометрию.
Термин «TGA» означает термогравиметрический анализ.
Термин «DSC» означает дифференциальную сканирующую калориметрию.
В. Соединения.
Настоящее изобретение частично относится к соединениям, которые представляют собой производные фенил-урацила, структура которых соответствует формуле I:
В этих соединениях,  выбрана из группы, состоящей из одинарной углерод-углеродной связи и двойной углерод-углеродной связи.
выбрана из группы, состоящей из одинарной углерод-углеродной связи и двойной углерод-углеродной связи.
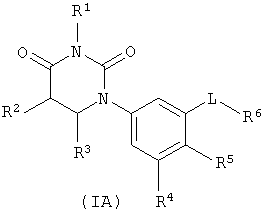
В некоторых вариантах осуществления, представляет собой одинарную углерод-углеродную связь. В этих вариантах осуществления, структура соединений формулы I соответствует следующей формуле (т.е., формуле IA):
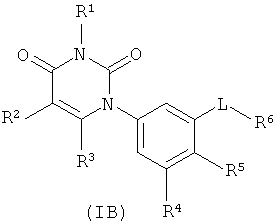
В других вариантах осуществления, представляет собой двойную углерод-углеродную связь. В этих вариантах осуществления, структура соединений формулы I соответствует следующей формуле (т.е., формуле IB):
B1. Заместитель R1.
R1 выбран из группы, состоящей из водорода, метила и азот-защищающей группы.
В некоторых вариантах осуществления, R1 представляет собой водород.
В некоторых вариантах осуществления, R1 представляет собой метил.
В некоторых вариантах осуществления, R1 выбран из группы, состоящей из водорода и метила.
В некоторых вариантах осуществления, R1 представляет собой азот-защищающую группу. В этих вариантах осуществления, эти соединения применимы в качестве промежуточных соединений для получения соединений формулы I. Азот-защищающие группы подходящие для получения соединений формулы I, известны специалистам в данной области.
B2. Заместитель R2.
R2 выбран из группы, состоящей из водорода, гало, гидрокси, метила, циклопропила и циклобутила.
В некоторых вариантах осуществления, R2 представляет собой водород.
В некоторых вариантах осуществления, R2 представляет собой гало. В некоторых таких вариантах осуществления, R2 выбран из группы, состоящей из фтора и хлора. В других таких вариантах осуществления, R2 представляет собой фтор. В других таких вариантах осуществления, R2 представляет собой хлор. В других таких вариантах осуществления, R2 представляет собой бром. В дополнительных таких вариантах осуществления, R2 представляет собой йод.
В некоторых вариантах осуществления, R2 представляет собой гидрокси.
В некоторых вариантах осуществления, R2 представляет собой метил.
В некоторых вариантах осуществления, R2 представляет собой циклопропил.
В некоторых вариантах осуществления, R2 представляет собой циклобутил.
В некоторых вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила, гидрокси и гало. В некоторых таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила, гидрокси, фтора и хлора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила, гидрокси и фтора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила, гидрокси, и хлора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила, гидрокси и брома. В дополнительных таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила, гидрокси и йода. В некоторых вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила и гало. В некоторых таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила, фтора и хлора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила и фтора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила и хлора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила и брома. В дополнительных таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, метила и йода.
В некоторых вариантах осуществления, R2 выбран из группы, состоящей из водорода и галогена. В некоторых таких вариантах осуществления, R2 выбран из группы, состоящей из водорода, фтора и хлора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода и фтора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода и хлора. В других таких вариантах осуществления, R2 выбран из группы, состоящей из водорода и брома. В дополнительных таких вариантах осуществления, R2 выбран из группы, состоящей из водорода йода.
B3. Заместитель R3.
R3 выбран из группы, состоящей из водорода, гало, оксо, и метила. В некоторых таких вариантах осуществления, R3 выбран из группы, состоящей из водорода, фтора, оксо, и метила. В других таких вариантах осуществления, R3 выбран из группы, состоящей из водорода, хлора, оксо, и метила. В других таких вариантах осуществления, R3 выбран из группы, состоящей из водорода, брома, оксо, и метила. В других таких вариантах осуществления, R3 выбран из группы, состоящей из водорода, йода, оксо, и метила. В некоторых вариантах осуществления, R3 выбран из группы, состоящей из водорода, гало, и оксо. В некоторых таких вариантах осуществления, R3 выбран из группы, состоящей из водорода, фтора и оксо. В других таких вариантах осуществления, R3 выбран из группы, состоящей из водорода, хлора и оксо. В других таких вариантах осуществления, R3 выбран из группы, состоящей из водорода, брома и оксо. В других таких вариантах осуществления, R3 выбран из группы, состоящей из водорода, йода и оксо. В некоторых вариантах осуществления, R3 выбран из группы, состоящей из водорода и метила.
В некоторых вариантах осуществления, R3 представляет собой водород.
В некоторых вариантах осуществления, R3 представляет собой метил.
В некоторых вариантах осуществления, R3 представляет собой оксо.
В некоторых вариантах осуществления, R3 представляет собой гало. В некоторых таких вариантах осуществления, R3 представляет собой фтор. В других таких вариантах осуществления, R3 представляет собой хлор. В других таких вариантах осуществления, R3 представляет собой бром. В дополнительных таких вариантах осуществления, R3 представляет собой йод.
B4. Заместитель R4.
R4 выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила, и гетероциклила, где:
(a) амино, аминокарбонил, и аминосульфонил необязательно замещены:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, и алкилсульфонила, или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл,
(b) алкил, алкенил, алкинил, алкилокси, алкенилокси, алкинилокси, и алкилсульфонил, необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из гало, оксо, нитро, циано, азидо, гидрокси, амино, алкилокси, триметилсилила, карбоциклила, и гетероциклила, где:
амино необязательно замещен:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилкарбонила, алкилсульфонила, алкилоксикарбонила, карбоциклила, гетероциклила, карбоциклилалкила, и гетероциклилалкила, или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл, и
(c) карбоциклил и гетероциклил необязательно замещены заместителями, вплоть до трех, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, гало, оксо, нитро, циано, азидо, гидрокси, амино, алкилокси, триметилсилила, карбоциклила, и гетероциклила, где:
амино необязательно замещен:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилкарбонила, алкилсульфонила, алкилоксикарбонила, карбоциклила, гетероциклила, карбоциклилалкила и гетероциклилалкила, или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила и гетероциклила, где:
амино, аминокарбонил, и аминосульфонил необязательно замещены:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила и алкилсульфонила, или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл.
В некоторых вариантах осуществления, R выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила и гетероциклила, где:
алкил, алкенил, алкинил, алкилокси, алкенилокси, алкинилокси и алкилсульфонил, необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из гало, оксо, нитро, циано, азидо, гидрокси, амино, алкилокси, триметилсилила, карбоциклила и гетероциклила, где:
амино необязательно замещен:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилкарбонила, алкилсульфонила, алкилоксикарбонила, карбоциклила, гетероциклила, карбоциклилалкила и гетероциклилалкила, или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила и гетероциклила, где:
карбоциклил и гетероциклил необязательно замещены заместителями, вплоть до трех, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, гало, оксо, нитро, циано, азидо, гидрокси, амино, алкилокси, триметилсилила, карбоциклила и гетероциклила, где:
амино необязательно замещен:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилкарбонила, алкилсульфонила, алкилоксикарбонила, карбоциклила, гетероциклила, карбоциклилалкила и гетероциклилалкила, или
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила и гетероциклила, где:
(a) амино, аминокарбонил, и аминосульфонил необязательно замещены:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила, или,
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл; и
(b) алкил, алкенил, алкинил, алкилокси, алкенилокси, алкинилокси, алкилсульфонил, карбоциклил, и гетероциклил необязательно замещены заместителями, вплоть до трех, независимо выбранными из группы, состоящей из гало, оксо, нитро, циано, азидо, гидрокси, амино, алкилокси, карбоциклила, и гетероциклила, где амино необязательно замещен:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилкарбонила, алкилсульфонила, алкилоксикарбонила, карбоциклила, гетероциклила, карбоциклилалкила, и гетероциклилалкила, или,
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила и гетероциклила, где:
амино, аминокарбонил и аминосульфонил необязательно замещены:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила, или,
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила и гетероциклила, где:
алкил, алкенил, алкинил, алкилокси, алкенилокси, алкинилокси, алкилсульфонил, карбоциклил, и гетероциклил необязательно замещены заместителями, вплоть до трех, независимо выбранными из группы, состоящей из гало, оксо, нитро, циано, азидо, гидрокси, амино, алкилокси, карбоциклила, и гетероциклила, где амино необязательно замещен:
(1) одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилкарбонила, алкилсульфонила, алкилоксикарбонила, карбоциклила, гетероциклила, карбоциклилалкила, и гетероциклилалкила, или,
(2) двумя заместителями, которые вместе с аминоазотом образуют однокольцевой гетероцикл.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, C2-C4-алкенила, C2-C4-алкинила, амино, C1-C4-алкилсульфонила, C3-C6-карбоциклила, и 5-6-членного гетероциклила, где:
(a) амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, и алкилсульфонила,
(b) C1-C4-алкил, C2-C4-алкенил, и C2-C4-алкинил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси, и триметилсилила, и
(c) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены заместителями, вплоть до трех, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, гало, и амино, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, и алкилсульфонила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из C1-C4-алкила, C2-C4-алкенила, C2-C4-алкинила, амино, C1-C4-алкилсульфонила, C3-C6-карбоциклила, и 5-6-членного гетероциклила, где:
(a) амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, и алкилсульфонила,
(b) C1-C4-алкил, C2-C4-алкенил, и C2-C4-алкинил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси, и триметилсилила, и
(c) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены заместителями, вплоть до трех, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, гало, и амино, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, и алкилсульфонила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, C3-C6-карбоциклила, и 5-6-членного гетероциклила, где:
(a) C1-C4-алкил необязательно замещен заместителями, вплоть до трех, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси, и триметилсилила, и
(b) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, гало, и алкилсульфониламино.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, C3-C6-карбоциклила, и 5-6-членного гетероциклила, где:
(a) C1-C4-алкил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси, и триметилсилила, и
(b) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены заместителем, выбранным из группы, состоящей из алкила, гало, и алкилсульфониламино.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из C1-C4-алкила, C3-C6-карбоциклила, и 5-6-членного гетероциклила, где:
(a) C1-C4-алкил необязательно замещен заместителями, вплоть до трех, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси, и триметилсилила, и
(b) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, гало, и алкилсульфониламино.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, трет-бутила, C3-C6-карбоциклила, и 5-6-членного гетероциклила, где:
C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены заместителем, выбранным из группы, состоящей из алкила, гало и алкилсульфониламино.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из трет-бутила, C3-C6-карбоциклила, и 5-6-членного гетероциклила, где:
C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены заместителем, выбранным из группы, состоящей из алкила, гало и алкилсульфониламино.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, алкила, галоалкила, карбоксиалкила, гидроксиалкила, алкилоксиалкила, триметилсилилалкинила, алкилкарбоциклила, карбоциклила, алкилгетероциклила, гетероциклила, галокарбоциклила, алкилсульфониламино, и алкилсульфонила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, алкила, алкенила, алкинила, нитро, циано, азидо, алкилокси, алкенилокси, алкинилокси, амино, аминокарбонила, аминосульфонила, алкилсульфонила, карбоциклила, и гетероциклила. В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, C2-C4-алкенила, C2-C4-алкинила, амино, C1-C4-алкилсульфонила, C3-C6-карбоциклила, и 5-6-членного гетероциклила. В некоторых таких вариантах осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, C2-C4-алкенила, C2-C4-алкинила, амино, C1-C4-алкилсульфонила, C6-карбоциклила, и 5-6-членного гетероциклила. В другом таком варианте осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, C2-C4-алкенила, C2-C4-алкинила, амино, C1-C4-алкилсульфонила, фенила и 5-6-членного гетероарила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из C1-C4-алкила, C2-C4-алкенила, C2-C4-алкинила, амино, C1-C4-алкилсульфонила, C3-C6-карбоциклила, и 5-6-членного гетероциклила. В некоторых таких вариантах осуществления, R4 выбран из группы, состоящей из C1-C4-алкила, C2-C4-алкенила, C2-C4-алкинила, амино, C1-C4-алкилсульфонила, C6-карбоциклила, и 5-6-членного гетероциклила. В другом таком варианте осуществления, R4 выбран из группы, состоящей из C1-C4-алкила, C2-C4-алкенила, C2-C4-алкинила, амино, C1-C4-алкилсульфонила, фенила, и 5-6-членного гетероарила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, C3-C6-карбоциклила и 5-6-членного гетероциклила. В некоторых таких вариантах осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, C6-карбоциклила и 5-6- членного гетероциклила. В других таких вариантах осуществления, R4 выбран из группы, состоящей из гало, C1-C4-алкила, фенила и 5-6- членного гетероарила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из C1-C4-алкила, C3-C6-карбоциклила и 5-6-членного гетероциклила. В некоторых таких вариантах осуществления, R4 выбран из группы, состоящей из C1-C4-алкила, C6-карбоциклила и 5-6-членного гетероциклила. В других таких вариантах осуществления, R4 выбран из группы, состоящей из C1-C4-алкила, фенила и 5-6-членного гетероарила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, трет-бутила, C3-C6-карбоциклила и 5-6-членного гетероциклила. В некоторых таких вариантах осуществления, R4 выбран из группы, состоящей из гало, трет-бутила, C6-карбоциклила и 5-6-членного гетероциклила. В других таких вариантах осуществления, R4 выбран из группы, состоящей из гало, трет-бутила, фенила и 5-6-членного гетероарила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из трет-бутила, C3-C6-карбоциклила и 5-6-членного гетероциклила. В некоторых таких вариантах осуществления, R4 выбран из группы, состоящей из трет-бутила, C6-карбоциклила и 5-6-членного гетероциклила. В других таких вариантах осуществления, R4 выбран из группы, состоящей из трет-бутила, фенила и 5-6-членного гетероарила.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из C3-C6-карбоциклила и 5-6-членного гетероциклила. В некоторых таких вариантах осуществления, R4 выбран из группы, состоящей из C6-карбоциклила и 5-6-членного гетероциклила. В других таких вариантах осуществления, R4 выбран из группы, состоящей из фенила и 5-6-членного гетероарила.
Подходящие карбоциклилы для приведенных выше вариантов осуществления включают, например, циклопропил и фенил.
Подходящие гетероциклилы для приведенных выше вариантов осуществления включают, например, фуранил, тиенил и пиридинил.
В некоторых вариантах осуществления, R4 выбран из группы, состоящей из гало, алкила, и алкилокси.
В некоторых вариантах осуществления, R4 представляет собой алкил. В некоторых вариантах осуществления, R4 представляет собой трет-бутил.
B5. Заместитель R5.
R5 выбран из группы, состоящей из водорода, гидрокси, алкила, алкенила, алкинила, алкилокси, алкенилокси, алкинилокси, алкилсульфонилокси, карбоциклилсульфонилокси, галоалкилсульфонилокси, и галогена.
В некоторых вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и галогена. В некоторых таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и фтора. В других таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и фтора. В других таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и хлора. В других таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и брома. В дополнительных таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и йода.
В некоторых вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, метокси и галогена. В некоторых таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, метокси и фтора. В других таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, метокси и хлора. В других таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, метокси и брома. В дополнительных таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, метокси и йода. В некоторых вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси и алкилокси. В некоторых таких вариантах осуществления, R5 выбран из группы, состоящей из водорода, гидрокси, метокси и этокси.
В некоторых вариантах осуществления, R5 представляет собой водород.
В некоторых вариантах осуществления, R5 представляет собой гидрокси.
В некоторых вариантах осуществления, R5 представляет собой алкилокси.
В некоторых вариантах осуществления, R5 представляет собой метокси.
В некоторых вариантах осуществления, R5 представляет собой этокси.
B6. Заместитель L.
L выбран из группы, состоящей из связи, C(RA)=C(RB), C≡C, C(O)N)(RC), N(RD)C(O), C1-C2-алкилена, C(H)2O, OC(H)2, циклопропил-1,2-ена, C(H)2N(RL), H(RM)C(H)2, C(O)CH2, и CH2C(O), где RA, RB, RC, RD, RL, и RM рассмотрены ниже.
В некоторых вариантах осуществления, L выбран из группы, состоящей из связи, C(RA)=C(RB), C≡C, C(O)N(RC), N(RD)C(O), C1-C2-алкилена, C(H)2O, OC(H)2, циклопропил-1,2-ена, C(H)2N(RL), и N(RM)C(H)2.
В некоторых вариантах осуществления, L выбран из группы, состоящей из C(RA)=C(RB), этилена и циклопропил-1,2-ена.
В некоторых вариантах осуществления, L выбран из группы, состоящей из C(RA)=C(RB), C≡C, C(O)N(RC), N(RD)C(O), C1-C2-алкилена, C(H)2O, OC(H)2, циклопропил-1,2-ена, C(H)2N(RL), N(RM)C(H)2, C(O)CH2 и CH2C(O).
В некоторых вариантах осуществления, L выбран из группы, состоящей из C≡C, C(O)N(RC), N(RD)C(O), C(H)2O, OC(H)2, C(H)2N(RL), и N(RM)C(H)2.
В некоторых вариантах осуществления, L представляет собой связь. В этих вариантах осуществления структура соединений формулы I соответствует формуле I-L0:
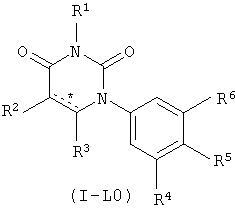 .
.
В некоторых таких вариантах осуществления, структура соединений соответствует следующей формуле (т.е., формуле IA-L0):
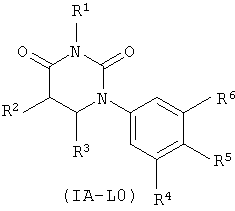 .
.
В других таких вариантах осуществления, структура соединений соответствует следующей формуле (т.е. формуле IB-L0):
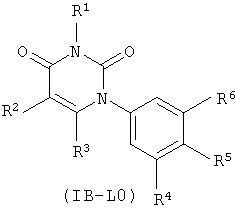 .
.
В некоторых вариантах осуществления, L представляет собой C(RA)=C(RB), где RA и RB рассмотрены ниже. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L1:
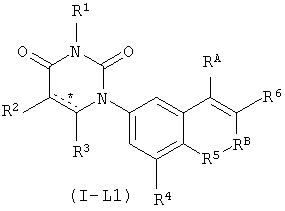 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L1:
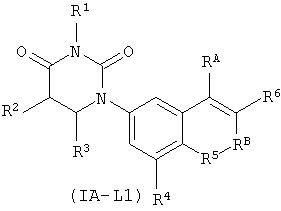 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L1:
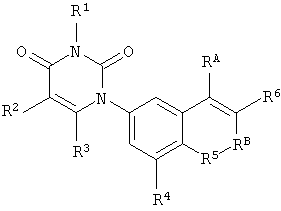 .
.
Обычно, соединения формулы I-L1 являются более эффективными, если R6 и фенил-урацил находятся по разные стороны двойной связи (т.е., в транс-конфигурации относительно двойной связи).
В некоторых вариантах осуществления, L представляет собой C≡C. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L2:
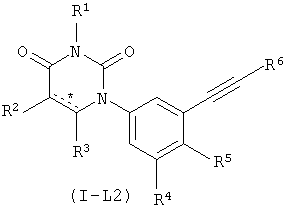 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре IA-L2:
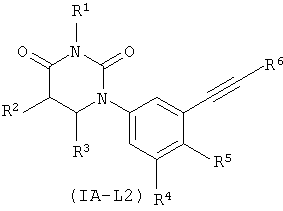 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре форму IB-L2:
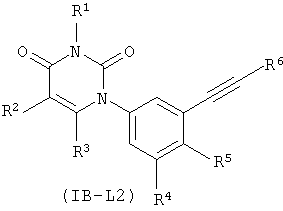 .
.
В некоторых вариантах осуществления, L представляет собой C(O)N(RC), где RC рассмотрен ниже. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L3:
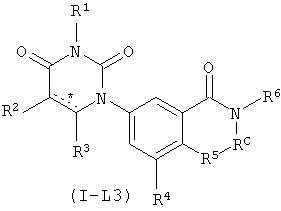 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структур формуле IA-L3:
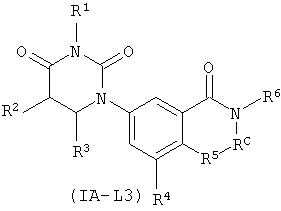 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L3:
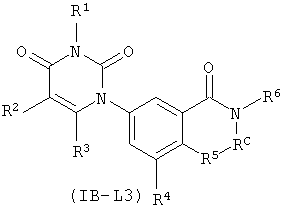 .
.
В некоторых вариантах осуществления, L представляет собой N(RD)C(O), где RD рассмотрен ниже. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L4:
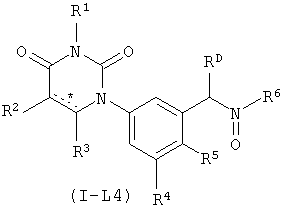 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L4:
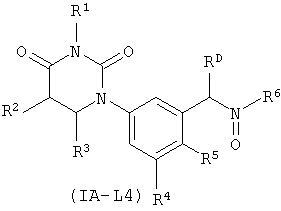 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L4:
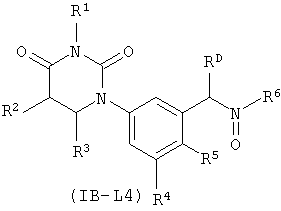 .
.
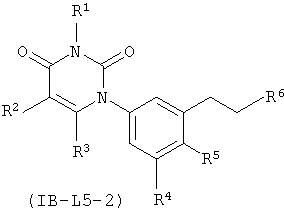
В некоторых вариантах осуществления, L представляет собой C1-C2-алкилен. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L5-1 (если L представляет собой метилен) или I-L5-2 (если L представляет собой этилен):
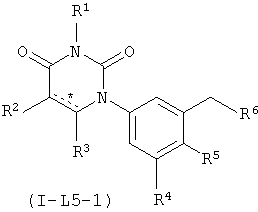
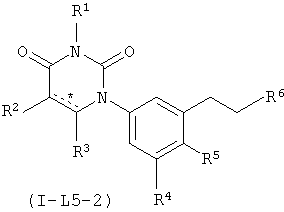
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L5-1 (если L представляет собой метилен) или IA-L5-2 (если L представляет собой):
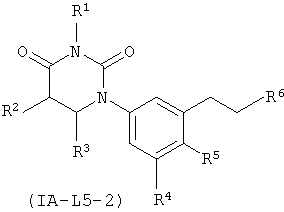
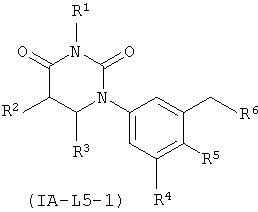 .
. 
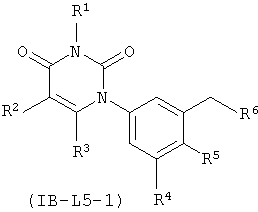
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L5-1 (если L представляет собой метилен) или IB-L5-2 (если L представляет собой этилен):
В некоторых вариантах осуществления, L представляет собой C(H)2O. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L6:
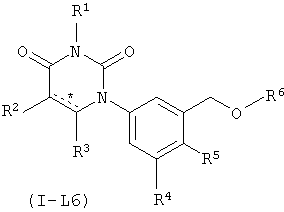 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L6:
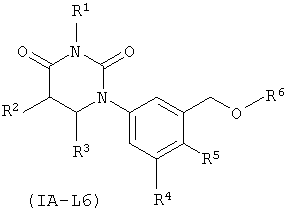 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L6:
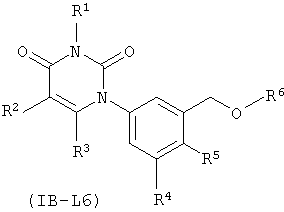 .
.
В некоторых вариантах осуществления, L представляет собой OC(H)2. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L7:
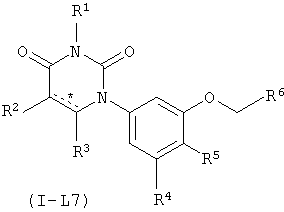 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L7:
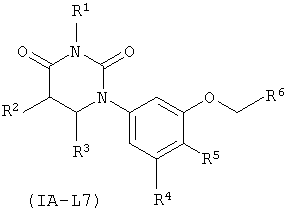 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L7:
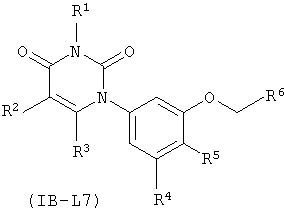 .
.
В некоторых вариантах осуществления, L представляет собой циклопропил-1,2-ен. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L8:
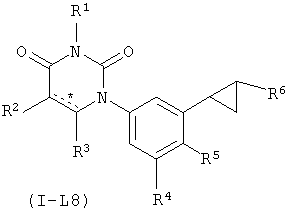 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L8:
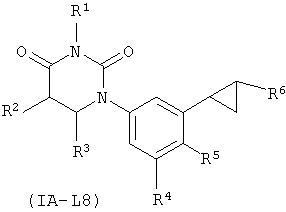 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L8:
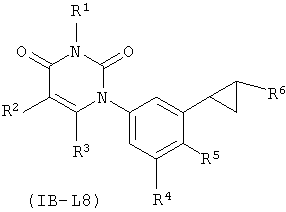 .
.
В некоторых вариантах осуществления, L выбран из группы, состоящей из C=C, этилена, и циклопропил-1,2-ена.
В некоторых вариантах осуществления, L представляет собой C(H)2N(RL). В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L9:
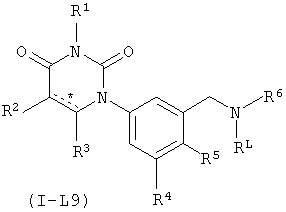 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L9:
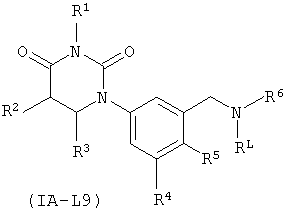 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L9:
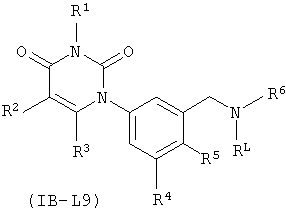 .
.
В некоторых вариантах осуществления, L представляет собой N(RM)C(H)2. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L10:
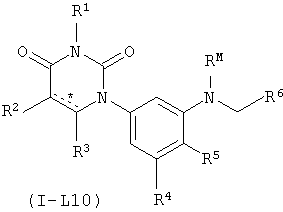 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L10:
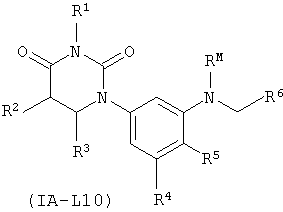 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L10:
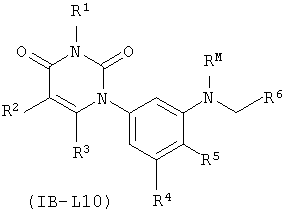 .
.
В некоторых вариантах осуществления, L представляет собой C(O)C(H)2. В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L11:
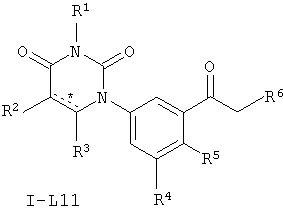 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L11:
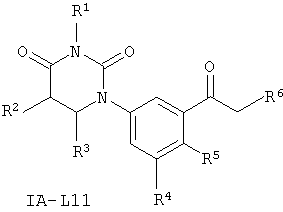 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L11:
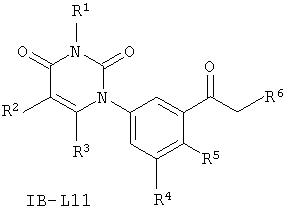 .
.
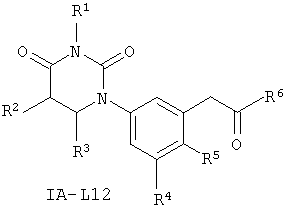
В некоторых вариантах осуществления, L представляет собой C(H)2C(O). В этих вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L12:
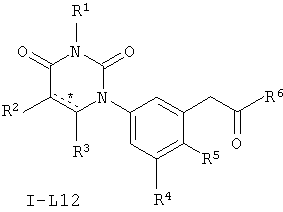 .
.
В некоторых таких вариантах осуществления, соединения соответствуют по структуре формуле IA-L12:
 .
.
В других таких вариантах осуществления, соединения соответствуют по структуре формуле IB-L12:
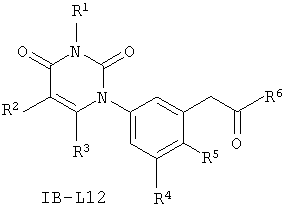 .
.
B7. Заместители RA и RB.
RA и RB независимо выбраны из группы, состоящей из водорода C1-C6-алкила, C1-C6-алкилокси, C3-C8-пиклоалкила, и гало, где:
C1-C6-алкил необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, гидрокси, нитро, оксо, амино, циано, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, и гетероциклила.
В некоторых вариантах осуществления, один из RA и RB представляет собой водород, а другой выбран из группы, состоящей из C1-C6-алкила, C1-C6-алкилокси, C3-C8-циклоалкила и гало, где:
C1-C6-алкил необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, гидрокси, нитро, оксо, амино, циано, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, и гетероциклила.
В некоторых вариантах осуществления, RA и RB независимо выбраны из группы, состоящей из водорода, C1-C6-алкила, C1-C6-алкилокси, C3-C8-циклоалкила, и гало.
В некоторых из представленных выше вариантов осуществления, RA представляет собой водород. В другом из представленных выше вариантов осуществления, RB представляет собой водород.
В некоторых вариантах осуществления, один из RA и RB представляет собой водород, а другой выбран из группы, состоящей из водорода, метила, метокси, и гало.
В некоторых вариантах осуществления, RA представляет собой водород, a RB выбран из группы, состоящей из метила, метокси, и гало. В некоторых таких вариантах осуществления, RB выбран из группы, состоящей из метила, метокси и фтора. В других таких вариантах осуществления, RB выбран из группы, состоящей из метила, метокси, и хлора. В других таких вариантах осуществления, RB выбран из группы, состоящей из метила, метокси, и брома. В дополнительных таких вариантах осуществления, RB выбран из группы, состоящей из метила, метокси, и йода. Еще в таких дополнительных вариантах осуществления, RB выбран из группы, состоящей из метила, метокси, хлора и фтора.
В некоторых вариантах осуществления, RB представляет собой водород, а RA выбран из группы, состоящей из метила, метокси, и гало. В некоторых таких вариантах осуществления, RA выбран из группы, состоящей из метила, метокси и фтора. В других таких вариантах осуществления, RA выбран из группы, состоящей из метила, метокси, и хлора. В других таких вариантах осуществления, RA выбран из группы, состоящей из метила, метокси, и брома. В дополнительных таких вариантах осуществления, RA выбран из группы, состоящей из метила, метокси, и йода. Еще в таких дополнительных вариантах осуществления, RA выбран из группы, состоящей из метила, метокси, хлора и фтора. В некоторых вариантах осуществления, RA представляет собой водород, a RA представляет собой водород.
B8. Заместитель RC.
RC выбран из группы, состоящей из водорода и алкила. В некоторых таких вариантах осуществления, RC выбран из группы, состоящей из водорода и метила.
В некоторых вариантах осуществления, RC представляет собой водород.
В некоторых вариантах осуществления, RC представляет собой алкил. В некоторых таких вариантах осуществления, RC представляет собой метил.
B9. Заместитель RD.
RD выбран из группы, состоящей из водорода и алкила. В некоторых таких вариантах осуществления, RD выбран из группы, состоящей из водорода и метила.
В некоторых вариантах осуществления, RD представляет собой водород.
В некоторых вариантах осуществления, RD представляет собой алкил. В некоторых таких вариантах осуществления, RD представляет собой метил.
B10. Заместитель RL.
RL выбран из группы, состоящей из водорода, C1-C6-алкила, C1-C6-алкилокси, C3-C8-циклоалкила, и гало, где:
C1-C6-алкил необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, гидрокси, нитро, оксо, амино, пиано, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, и гетероциклила.
В некоторых вариантах осуществления, RL выбран из группы, состоящей из водорода, C1-C6-алкил, C1-C6-алкилокси, C3-C8-циклоалкил, и гало.
В некоторых вариантах осуществления, RL выбран из группы, состоящей из водорода, C1-C6-алкил, C1-C6-алкилокси и гало, где:
C1-C6-алкил необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, гидрокси, нитро, оксо, амино, циано, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила и гетероциклила.
В некоторых вариантах осуществления, RL выбран из группы, состоящей из водорода, C1-C6-алкила, C1-C6-алкилокси и гало.
В некоторых из приведенных выше вариантов осуществления, RL представляет собой гало.
В некоторых таких вариантах осуществления, галогеном является фтор. В других таких вариантах осуществления, галогеном является хлор. В других таких вариантах осуществления, галогеном является бром. В дополнительных таких вариантах осуществления, галогеном является йод.
В некоторых из приведенных выше вариантов осуществления, RL представляет собой водород.
В некоторых из приведенных выше вариантов осуществления, RL представляет собой C1-C6-алкил.
В некоторых из приведенных выше вариантов осуществления, RL представляет собой C1-C6-алкилокси.
B11. Заместитель RM.
RM выбран из группы, состоящей из водорода, C1-C6-алкил, C1-C6-алкилокси, C3-C8-циклоалкил и гало, где:
C1-C6-алкил необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, гидрокси, нитро, оксо, амино, циано, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, и гетероциклила.
В некоторых вариантах осуществления, RM выбран из группы, состоящей из водорода, C1-C6-алкила, C1-C6-алкилокси, C3-C8-циклоалкила и галогена.
В некоторых вариантах осуществления, RM выбран из группы, состоящей из водорода, C1-C6-алкила, C1-C6-алкилокси и галогена, где:
C1-C6-алкил необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, гидрокси, нитро, оксо, амино, циано, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила и гетероциклила.
В некоторых вариантах осуществления, RM выбран из группы, состоящей из водорода, C1-C6-алкила, C1-C6-алкилокси и галогена.
В некоторых из приведенных выше вариантов осуществления, RM представляет собой гало.
В некоторых таких вариантах осуществления, галоген представляет собой фтор. В других таких вариантах осуществления, галоген представляет собой хлор. В других таких вариантах осуществления, галоген представляет собой бром. В дополнительных таких вариантах осуществления, галоген представляет собой йод.
В некоторых из приведенных выше вариантов осуществления, RM представляет собой водород.
В некоторых из приведенных выше вариантов осуществления, RM представляет собой C1-C6-алкил.
В некоторых из приведенных выше вариантов осуществления, RM представляет собой C1-C6-алкилокси.
B12. Заместитель R6.
R6 выбран из группы, состоящей из C5-C6-карбоциклила, 5-6-членного гетероциклила, конденсированного 2-кольцевого карбоциклила и конденсированного 2-кольцевого гетероциклила, где каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK где RE, RF, RG, RH, RI, RJ и RK рассмотрены ниже. В некоторых таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил не замещены. В других таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены заместителем, выбранным из группы, состоящей из RE, RF, RG, RH, RI, RJ, и RK. В других таких вариантах осуществления, С5-Сб-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены заместителем, выбранным из группы, состоящей из RE, RF, RI, RJ и RK. В других таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил, и конденсированный 2-кольцевой гетероциклил замещены заместителем, выбранным из группы, состоящей из RE, RF, и RJ. В других таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил, и конденсированный 2-кольцевой гетероциклил замещены заместителем, выбранным из группы, состоящей из RF и RJ. В других таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены RJ. В других таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены двумя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В других таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены двумя заместителями, независимо выбранными из группы, состоящей из RE, RF, RI, RJ и RK. В других таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены двумя заместителями, независимо выбранными из группы, состоящей из RE, RF и RJ. В других таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены двумя заместителями, независимо выбранными из группы, состоящей из RF и RJ. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RI, RJ и RK. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены тремя заместителями, независимо выбранными из группы, состоящей из RE, RF и RJ. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены тремя заместителями, независимо выбранными из группы, состоящей из RF и RJ. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RI, RJ и RK. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF и RJ. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил, 5-6-членный гетероциклил, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RF и RJ.
В некоторых вариантах осуществления, R6 выбран из группы, состоящей из C5-C6-карбоциклила и 5-6-членного гетероциклила, где каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В некоторых таких вариантах осуществления, C5-C6-карбоциклил и 5-6-членныйгетероциклил не замещены. В других таких вариантах осуществления, C5-C6-карбоциклил и 5-6-членный гетероциклил замещены заместителем, выбранным из группы, состоящей из RE, RF, RG, RH, RI, RJ, и RK. В других таких вариантах осуществления, C5-C6-карбоциклил и 5-6-членный гетероциклил замещены двумя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ, и RK. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил и 5-6-членный гетероциклил замещены тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил и 5-6-членный гетероциклил замещены одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK.
В некоторых вариантах осуществления, R6 представляет собой C5-C6-карбоциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В некоторых таких вариантах осуществления, C5-C6-карбоциклил не замещен. В других таких вариантах осуществления, C5-C6-карбоциклил замещен заместителем, выбранным из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В других таких вариантах осуществления, C5-C6-карбоциклил замещен двумя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил замещен тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, C5-C6-карбоциклил замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK.
В некоторых вариантах осуществления, R6 представляет собой 5-6-членный гетероциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В некоторых таких вариантах осуществления, 5-6-членный гетероциклил не замещен. В других таких вариантах осуществления, 5-6-членный гетероциклил замещен заместителем, выбранным из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В других таких вариантах осуществления, 5-6-членный гетероциклил замещен двумя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, 5-6-членный гетероциклил замещен тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, 5-6-членный гетероциклил замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK.
В некоторых вариантах осуществления, R6 выбран из группы, состоящей из конденсированного 2-кольцевого карбоциклила и конденсированного 2-кольцевого гетероциклила, где каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В некоторых таких вариантах осуществления, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил не замещены. В других таких вариантах осуществления, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены заместителем, выбранным из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В других таких вариантах осуществления, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены двумя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. B дополнительных таких вариантах осуществления конденсированный 2-кольцевой карбоциклил и конденсированный 2-кольцевой гетероциклил замещены одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK.
В некоторых вариантах осуществления, R6 представляет собой конденсированный 2-кольцевой карбоциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В некоторых таких вариантах осуществления, конденсированный 2-кольцевой карбоциклил не замещен. В других таких вариантах осуществления, конденсированный 2-кольцевой карбоциклил замещен заместителем, выбранным из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В других таких вариантах осуществления конденсированный 2-кольцевой карбоциклил замещен двумя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, конденсированный 2-кольцевой карбоциклил замещен тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, конденсированный 2-кольцевой карбоциклил замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK.
В некоторых вариантах осуществления, R6 представляет собой конденсированный 2-кольцевой гетероциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В некоторых таких вариантах осуществления, конденсированный 2-кольцевой гетероциклил не замещен. В других таких вариантах осуществления, конденсированный 2-кольцевой гетероциклил замещен заместителем, выбранным из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В других таких вариантах осуществления, конденсированный 2-кольцевой гетероциклил замещен двумя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ и RK. В дополнительных таких вариантах осуществления, конденсированный 2-кольцевой гетероциклил замещен тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ, и RK. В дополнительных таких вариантах осуществления, конденсированный 2-кольцевой гетероциклил замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RG, RH, RI, RJ, и RK.
В некоторых из приведенных выше вариантов осуществления, необязательно замещенный C5-C6-карбоциклил выбран из группы, состоящей из циклопентила, циклопентенила, циклопентадиенила, циклогексила, циклогексенила, циклогексадиенила и фенила. В некоторых таких вариантах осуществления, необязательно замещенный C5-C6-карбоциклил представляет собой фенил.
В некоторых из приведенных выше вариантов осуществления, необязательно замещенный C5-C6-карбоциклил представляет собой C5-карбоциклил. Примеры C5-карбоциклилов включают циклопентил, циклопентенил и циклопентадиенил.
В другом из представленных выше вариантов осуществления, необязательно замещенный C5-C6-карбоциклил представляет собой C6-карбоциклил. Примеры C6-карбоциклилов включают циклогексил, циклогексенил, циклогексадиенил и фенил.
В некоторых из приведенных выше вариантов осуществления, необязательно замещенный 5-6-членный-гетероциклил выбран из группы, состоящей из фуранила, дигидрофуранила, тетрагидрофуранила, тиофенила (тиофуранила), дигидротиофенила, тетрагидротиофенила, пирролила, пирролинила, пирролидинила, оксазолила, дигидрооксазолила, изоксазолила, дигидроизоксазолила, оксазолидинила, изоксазолидинила, тиазолила, изотиазолила, тиазолинила, изотиазолинила, тиазолидинила, изотиазолидинила, имидазолила, имидазолидинила, пиразолила, пиразолинила, пиразолидинила, оксатиолила, оксатиоланила, триазолила, оксадиазолила, фуразанила, тетразолила, оксатриазолила, диоксазолила, оксатиазолила, оксатиазолидинила, дигидрооксадиазолила, диоксазолидинила, пиранила, дигидропиранила, тетрагидропиранила, пиридинила, дигидропиридинила, тетрагидропиридинила, пиперидинила, диазинила, пиразинила, пиридазинила, пиримидинила, дигидропиразинила, тетрагидропиразинила, пиперазинила, триазинила, дигидротриазинила, тетрагидротриазинила, триазинанила, оксазинила, дигидрооксазинила, морфолинила, оксатиазинила, дигидрооксатиазинила, оксатиазинанила, оксадиазинила, дигидрооксадиазинила, оксадиазинанила, тиопиранила, дигидротиопиранила и тетрагидротиопиранила.
В некоторых из приведенных выше вариантов осуществления, необязательно замещенный 5-6-членный -гетероциклил представляет собой 5-членный гетероциклил. Примеры такого 5-членного гетероциклила включают фуранил, дигидрофуранил, тетрагидрофуранил, тиофенил (тиофуранил), дигидротиофенил, тетрагидротиофенил, пирролил, пирролинил, пирролидинил, оксазолил, дигидрооксазолил, изоксазолил, дигидроизоксазолил, оксазолидинил, изоксазолидинил, тиазолил, изотиазолил, тиазолинил, изотиазолинил, тиазолидинил, изотиазолидинил, имидазолил, имидазолидинил, пиразолил, пиразолинил, пиразолидинил, оксатиолил, оксатиоланил, триазолил, оксадиазолил, фуразанил, тетразолил, оксатриазолил, диоксазолил, оксатиазолил, оксатиазолидинил, дигидрооксадиазолил и диоксазолидинил.
В других представленных выше вариантах осуществления, необязательно замещенный 5-6-членный гетероциклил представляет собой 6-членный гетероциклил. Примеры 6-членных гетероциклилов включают пиранил, дигидропиранил, тетрагидропиранил, пиридинил, дигидропиридинил, тетрагидропиридинил, пиперидинил, диазинил, пиразинил, пиридазинил, пиримидинил, дигидропиразинил, тетрагидропиразинил, пиперазинил, триазинил, дигидротриазинил, тетрагидротриазинил, триазинанил, оксазинил, дигидрооксазинил, морфолинил, оксатиазинил, дигидрооксатиазинил, оксатиазинанил, оксадиазинил, дигидрооксадиазинил, оксадиазинанил, тиопиранил, дигидротиопиранил и тетрагидротиопиранил.
В некоторых из приведенных выше вариантов осуществления, необязательно замещенный конденсированный 2-кольцевой карбоциклил выбран из группы, состоящей из нафталенила, дигидронафталенила, тетрагидронафталенила, гексагидронафталенила, октагидронафталенила, декагидронафталенила, инденила, дигидроинденила, гексагидроинденила, октагидроинденила, пенталенила, октагидропенталенила и гексагидропенталенила. В некоторых таких вариантах осуществления, необязательно замещенный конденсированный 2-кольцевой карбоциклил выбран из группы, состоящей из нафталенила и дигидроинденила. В некоторых таких вариантах осуществления, необязательно замещенный конденсированный 2-кольцевой карбоциклил представляет собой нафталенил. В других таких вариантах осуществления, необязательно замещенный конденсированный 2-кольцевой карбоциклил представляет собой дигидроинденил. В дополнительных таких вариантах осуществления, необязательно замещенный конденсированный 2-кольцевой карбоциклил представляет собой инденил. В некоторых из приведенных выше вариантов осуществления, необязательно замещенный конденсированный 2-кольцевой гетероциклил выбран из группы, состоящей из
 ,
,  ,
, 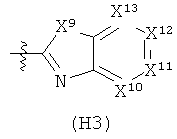 ,
,
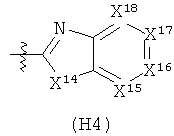 ,
, 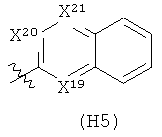 ,
, 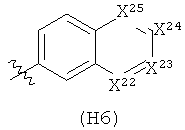 ,
,
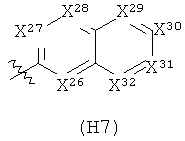 ,
, 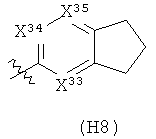 ,
, 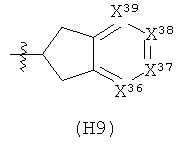 ,
,
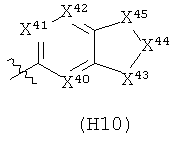 ,
, 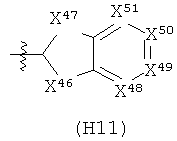 ,
, 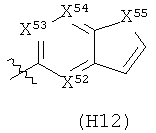 ,
,
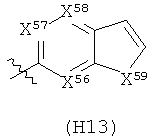 ,
, 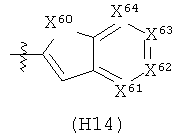 ,
, 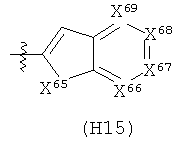 ,
,
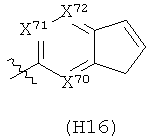 ,
, 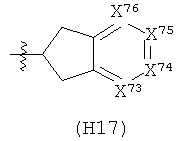 ,
, 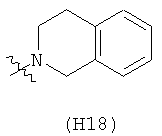 ,
,
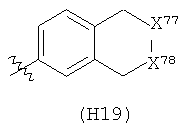 ,
, 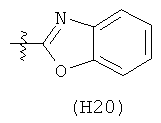 ,
, 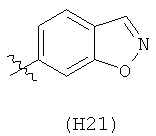 , and
, and
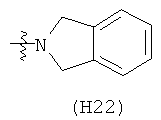 ;
;
X1, X2 и X3 независимо выбраны из группы, состоящей из N и C(H);
X4 выбран из группы, состоящей из N(H), O и S;
X5, X6 и X7 независимо выбраны из группы, состоящей из N и C(H);
X8 выбран из группы, состоящей из N(H), O и S;
X9 выбран из группы, состоящей из N(H), O и S;
X10, Х11, X12 и X13 независимо выбраны из группы, состоящей из N и C(H);
X14 выбран из группы, состоящей из N(H), O и S;
X15, X16, X17 и X18 независимо выбраны из группы, состоящей из N и C(H);
один или несколько из Х19, Х20 и Х21 представляет собой N, а остальной (остальные) представляет/представляют собой C(H);
один или несколько из X22, X23, X24 и X25 представляет собой N, а остальной (остальные) представляет/представляют собой C(H);
один или несколько из X26, X27 и X28 представляет собой N, а остальной (остальные) представляет/представляют собой C(H);
один или несколько из X29, X30, X31 и X32 представляет собой N, а остальной (остальные) представляет/представляют собой C(H);
один или несколько из Х33, Х34 и Х35 представляет собой N, а остальной (остальные) представляет/представляют собой C(H);
один или несколько из X36, X37, X38 и X39 представляет собой N, а остальной (остальные) представляет/представляют собой C(H);
X40, X41 и X42 независимо выбраны из группы, состоящей из N и C(H);
один из X43, X44 и X45 выбран из группы, состоящей из N(H), O и S, а остальные два представляют собой C(H)2;
один из X46 и X47 выбран из группы, состоящей из N(H), O и S, а другой представляет собой C(H)2;
X48, X49, X50 и X51 независимо выбраны из группы, состоящей из N и C(H);
X52, X53 и X54 независимо выбраны из группы, состоящей из N и C(H);
X55 выбран из группы, состоящей из N(H), O и S;
X56, X57 и X58 независимо выбраны из группы, состоящей из N и C(H);
X59 выбран из группы, состоящей из N(H), О и S;
X60 выбран из группы, состоящей из N(H), О и S;
X61, X62, X63 и X64 независимо выбраны из группы, состоящей из N и C(H);
X65 выбран из группы, состоящей из N(H), O и S;
X66, X67, X68 и X69 независимо выбраны из группы, состоящей из N и C(H);
один или несколько из X70, X71 и X72 представляет собой N, а остальной (остальные) представляет/представляют собой C(H);
один или несколько из X73, X74, X75 и X76 представляет собой N, а остальной (остальные) представляет/представляют собой C(H); и
один из X77 и X78 представляет собой N(H), а оставшийся представляет собой C(H)2. В некоторых из приведенных выше вариантов осуществления, необязательно замещенный конденсированный 2-кольцевой гетероциклил выбран из группы, состоящей из
, , ,
, , ,
, , ,
, , ,
, , ,
, and .
В некоторых из приведенных выше вариантов осуществления, необязательно замещенный конденсированный 2-кольцевой гетероциклил выбран из группы, состоящей из:
,
,
,
,
,
,
, , ,
, и .
В некоторых из приведенных выше вариантов осуществления, X1, X2 и X3 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, X5, X6 и X7 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, X10, X11, X12 и X13 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, X15, X16, X17 и X18 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, один из X19, X20 и X21 представляет собой N.
В некоторых из приведенных выше вариантов осуществления, один из X22, X23, X24 и X25 представляет собой N.
В некоторых из приведенных выше вариантов осуществления, один из X26, X27 и X28 представляет собой N и один из X29, X30, X31 и X32 представляет собой N.
В некоторых из приведенных выше вариантов осуществления, X40, X41 и X42 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, X48, X49, X50 и X51 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, X52, X53 и X54 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, X56, X57 и X58 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, X61, X62, X63 и X64 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, X66, X67, X68 и X69 представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, один или несколько из X70, X71 и X72 представляет собой N, а остальной (остальные) представляет/представляют собой C(H).
В некоторых из приведенных выше вариантов осуществления, один или несколько из X73, X74, X75 и X76 представляет собой N, а остальной (остальные) представляет/представляют собой C(H).
B13. Заместитель RE.
Каждый RE независимо выбран из группы, состоящей из гало, нитро, гидрокси, оксо, карбокси, циано, амино, имино, азидо, и альдегидо, где амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила.
В некоторых вариантах осуществления, каждый RE независимо выбран из группы, состоящей из гало, нитро, гидрокси, оксо, карбокси, амино, имино и альдегидо, где амино необязательно замещен одним или двумя независимо выбранными алкилами.
В некоторых вариантах осуществления, каждый RE независимо выбран из группы, состоящей из гало, нитро, гидрокси, оксо, карбокси, амино, имино, альдегидо и алкиламино.
В некоторых вариантах осуществления, каждый RE независимо выбран из группы, состоящей из хлора, фтора, нитро, гидрокси, оксо, карбокси, амино, имино, альдегидо и алкиламино.
В некоторых вариантах осуществления, каждый RE независимо выбран из группы, состоящей из гало, нитро, гидрокси, оксо, карбокси, циано, амино, имино и азидо. В некоторых таких вариантах осуществления, каждый RE представляет собой гало. В других таких вариантах осуществления, каждый RE представляет собой нитро. В других таких вариантах осуществления, каждый RE представляет собой гидрокси. В других таких вариантах осуществления, каждый RE представляет собой оксо. В других таких вариантах осуществления, каждый RE представляет собой карбокси. В других таких вариантах осуществления, каждый RE представляет собой циано. В других таких вариантах осуществления, каждый RE представляет собой амино. В дополнительных таких вариантах осуществления, каждый RE представляет собой имино. Еще в дополнительных вариантах осуществления, каждый RE представляет собой азидо.
В некоторых вариантах осуществления, каждый RE независимо выбран из группы, состоящей из гало, нитро, гидрокси, оксо, карбокси, циано, амино и имино.
B14. Заместитель RF.
Каждый RF независимо выбран из группы, состоящей из алкила, алкенила и алкинила, где:
каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, имино, нитро, азидо, оксо, аминосульфонила, алкилсульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, имино, аминосульфонил, аминокарбонил, карбоциклил и гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилсульфонила, алкенилсульфонила, алкинилсульфонила, алкилсульфониламино, гидрокси и алкилокси, где:
амино часть алкилсульфониламино необязательно замещена заместителем, выбранным из группы, состоящей из алкила, алкенила и алкинила.
В некоторых вариантах осуществления, каждый RF независимо выбран из группы, состоящей из алкила, алкенила и алкинила, где:
каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, имино, нитро, азидо, оксо, аминосульфонила, алкилсульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, имино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилсульфонила, алкенилсульфонила, алкинилсульфонила и алкилсульфониламино, где:
амино часть алкилсульфониламино необязательно замещена заместителем, выбранным из группы, состоящей из алкила, алкенила и алкинила.
В некоторых из приведенных выше вариантов осуществления, каждый RF независимо выбран из группы, состоящей из алкила, алкинила и алкинила, где такие заместители являются незамещенными.
В некоторых вариантах осуществления, каждый RF независимо выбран из группы, состоящей из алкила, алкенила и алкинила, где:
каждый такой заместитель необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, имино, нитро, оксо, аминосульфонила, алкилсульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, имино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкилсульфонила и алкилсульфониламино, где:
амино часть алкилсульфониламино необязательно замещена алкилом.
В некоторых вариантах осуществления, каждый RF представляет собой независимо выбранный алкил, необязательно замещенный заместителем, выбранным из группы, состоящей из карбокси, гидрокси, гало, амино, имино, нитро, оксо, аминосульфонила, алкилсульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, имино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из труппы, состоящей из алкила, алкилсульфонила и алкилсульфониламино, где:
амино часть алкилсульфониламино необязательно замещена алкилом.
В некоторых вариантах осуществления, каждый RF представляет собой независимо выбранный алкил, необязательно замещенный заместителем, выбранным из группы, состоящей из карбокси, гало, амино, имино и аминосульфонила, где:
амино, имино и аминосульфонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкилсульфонила и алкилсульфониламино.
В некоторых вариантах осуществления, каждый RF представляет собой независимо выбранный алкил, необязательно замещенный амино, где амино необязательно замещен алкилсульфонилом.
В некоторых вариантах осуществления, каждый RF представляет собой независимо выбранный алкил, замещенный амино, где амино замещен алкилсульфонилом. В некоторых таких вариантах осуществления, каждый RF представляет собой метилсульфониламинометил.
В некоторых вариантах осуществления, каждый RF независимо выбран из группы, состоящей из алкила, алкенила и алкинила, где:
каждый такой заместитель необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, имино, нитро, азидо, оксо, аминосульфонила, алкилсульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила.
В некоторых вариантах осуществления, каждый RF представляет собой независимо выбранный алкил, замещенный одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, имино, нитро, азидо, оксо, аминосульфонила, алкилсульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила.
B15. Заместитель RG.
Каждый RG независимо выбран из группы, состоящей из карбоциклила и гетероциклила, где:
каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилсульфонила, алкенилсульфонила и алкинилсульфонила.
В некоторых из приведенных выше вариантов осуществления, каждый RG независимо выбран из группы, состоящей из карбоциклила и гетероциклила, где такие заместители являются незамещенными.
В некоторых вариантах осуществления, каждый RG независимо выбран из группы, состоящей из карбоциклила и гетероциклила, где:
каждый такой заместитель необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, гало, амино, нитро, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила и алкилсульфонила.
В некоторых из приведенных выше вариантов осуществления, карбоциклил представляет собой C3-C6-карбоциклил.
В некоторых из приведенных выше вариантов осуществления, гетероциклил представляет собой 5-6-членный гетероциклил.
B16. Заместитель RH.
Каждый RH независимо выбран из группы, состоящей из алкилокси, алкенилокси, алкинилокси, алкилсульфонилокси, алкенилсульфонилокси и алкинилсульфонилокси, где:
каждый такой заместитель необязательно замещен одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилсульфонила, алкенилсульфонила и алкинилсульфонила.
В некоторых из приведенных выше вариантов осуществления, каждый RH независимо выбран из группы, состоящей из алкилокси, алкенилокси, алкинилокси, алкилсульфонилокси, алкенилсульфонилокси и алкинилсульфонилокси, где такие заместители являются незамещенными.
В некоторых вариантах осуществления, каждый RH независимо выбран из группы, состоящей из алкилокси и алкилсульфонилокси, где:
каждый такой заместитель необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила и алкилсульфонила.
В некоторых вариантах осуществления, каждый RH независимо выбран из группы, состоящей из алкилокси и алкилсульфонилокси, где:
каждый такой заместитель необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила и алкилсульфонила.
В некоторых вариантах осуществления, каждый RH независимо выбран из группы, состоящей из алкилокси и алкилсульфонилокси, где:
каждый такой заместитель необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, циано и аминокарбонила.
В некоторых вариантах осуществления, каждый RH представляет собой независимо выбранный алкилокси.
В некоторых вариантах осуществления, каждый RH представляет собой независимо выбранный алкилсульфонилокси.
B17. Заместитель RI.
Каждый RI независимо выбран из группы, состоящей из алкилкарбонила, алкенилкарбонила, алкинилкарбонила, аминокарбонила, алкилоксикарбонила, карбоциклилкарбонила и гетероциклилкарбонила, где:
(a) алкилкарбонил, алкенилкарбонил и алкинилкарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, и
(b) аминокарбонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилоксиалкила, карбоциклила, гетероциклила, алкилсульфонила и алкилсульфониламино, где:
карбоциклил и гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из гало, алкила и оксо.
В некоторых вариантах осуществления, каждый RI независимо выбран из группы, состоящей из алкилкарбонила, алкенилкарбонила, алкинилкарбонила, аминокарбонила, алкилоксикарбонила, карбоциклилкарбонила и гетероциклилкарбонила, где такие заместители являются незамещенными.
В некоторых вариантах осуществления, каждый RI независимо выбран из группы, состоящей из алкилкарбонила, аминокарбонила, алкилоксикарбонила, карбоциклилкарбонила и гетероциклилкарбонила, где:
(a) алкилкарбонил необязательно замещен заместителем, выбранным из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси и аминокарбонила, и
(b) аминокарбонил необязательно замещен заместителем, выбранным из группы, состоящей из алкила, алкилоксиалкила, алкилсульфонила и алкилсульфониламино.
В некоторых вариантах осуществления, каждый RI независимо выбран из группы, состоящей из алкилкарбонила и аминокарбонила, где:
аминокарбонил необязательно замещен заместителем, выбранным из группы, состоящей из алкила, алкилоксиалкила, алкилсульфонила и алкилсульфониламино.
В некоторых вариантах осуществления, каждый RI независимо выбран из группы, состоящей из алкилкарбонила, алкенилкарбонила, алкинилкарбонила и аминокарбонила, где:
(a) алкилкарбонил, алкенилкарбонил и алкинилкарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, и
(b) аминокарбонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила и алкилсульфониламино.
В некоторых из приведенных выше вариантов осуществления, каждый RI независимо выбран из группы, состоящей из алкилкарбонила, алкенилкарбонила, алкинилкарбонила и аминокарбонила, где такие заместители являются незамещенными.
В некоторых вариантах осуществления, каждый RI независимо выбран из группы, состоящей из алкилкарбонила и аминокарбонила, где:
(a) алкилкарбонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, гетероциклила, циано и аминокарбонила, и
(b) аминокарбонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилсульфониламино.
В некоторых вариантах осуществления, каждый RI независимо выбран из группы, состоящей из алкилкарбонила и аминокарбонила, где:
(a) алкилкарбонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, циано и аминокарбонила, и
(b) аминокарбонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилсульфониламино.
В некоторых вариантах осуществления, каждый RI независимо выбран из группы, состоящей из алкилкарбонила и аминокарбонила, где:
алкилкарбонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, гетероциклила, циано и аминокарбонила.
В некоторых вариантах осуществления, каждый RI представляет собой независимо выбранный алкилкарбонил.
В некоторых вариантах осуществления, каждый RI представляет собой независимо выбранный аминокарбонил.
B18. Заместитель RJ.
Каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилкарбониламино, алкенилкарбониламино, алкинилкарбониламино, алкилоксикарбониламино, алкенилоксикарбониламино, алкинилоксикарбониламино, алкилсульфониламино, алкенилсульфониламино, алкинилсульфониламино, аминокарбониламино, алкилоксикарбониламиноимино, алкилсульфониламиноимино, алкенилсульфониламиноимино и алкинилсульфониламиноимино, где:
(a) амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкенила, алкинила, алкилкарбонила, алкенилкарбонила, алкинилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, карбокси, гидрокси, алкилокси, алкенилокси, алкинилокси, гало, нитро, циано, азидо, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная, алкенильная и алкинильная часть таких заместителей необязательно замещена одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкенилокси и алкинилокси, где:
алкил необязательно замещен одним или несколькими гидрокси;
(c) карбоциклильная и гетероциклильная части таких заместителей необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, карбокси, гидрокси, алкилокси, алкенилокси, алкинилокси, гало, нитро, циано, азидо и амино, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилкарбониламино, алкенилкарбониламино, алкинилкарбониламино, алкилоксикарбониламино, алкенилоксикарбониламино, алкинилоксикарбониламино, алкилсульфониламино, алкенилсульфониламино, алкинилсульфониламино, аминокарбониламино, алкилсульфониламиноимино, алкенилсульфониламиноимино и алкинилсульфониламиноимино, где:
(a) амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкенила, алкинила, алкилкарбонила, алкенилкарбонила, алкинилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, карбокси, гидрокси, алкилокси, алкенилокси, алкинилокси, гало, нитро, циано, азидо, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная, алкенильная и алкинильная часть таких заместителей необязательно замещена одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, алкилокси, алкенилокси и алкинилокси, где:
алкил необязательно замещен одним или несколькими гидрокси;
(c) карбоциклильная и гетероциклильная части таких заместителей необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила, алкинила, карбокси, гидрокси, алкилокси, алкенилокси, алкинилокси, гало, нитро, циано, азидо и амино, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила; и
В некоторых из приведенных выше вариантов осуществления, каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилкарбониламино, алкенилкарбониламино, алкинилкарбониламино, алкилоксикарбониламино, алкенилоксикарбониламино, алкинилоксикарбониламино, алкилсульфониламино, алкенилсульфониламино, алкинилсульфониламино, аминокарбониламино, алкилсульфониламиноимино, алкенилсульфониламиноимино и алкинилсульфониламиноимино, где такие заместители являются незамещенными.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилкарбониламино, алкилоксикарбониламино, алкилсульфониламино, аминокарбониламино и алкилсульфониламиноимино, где:
(a) амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная часть таких заместителей необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси;
(c) карбоциклильная и гетероциклильная части таких заместитеей необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано и амино, где:
амино необязательно замещен одним или двумя независимо выбранными алкильными заместителями.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилсульфониламино и алкилсульфониламиноимино, где:
(a) амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная часть таких заместителей необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси;
(c) карбоциклильная и гетероциклильная части таких заместителей необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано и амино, где:
амино необязательно замещен одним или двумя независимо выбранными алкильными заместителями.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилсульфониламино и алкилсульфониламиноимино, где:
амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилсульфониламино и алкилсульфониламиноимино, где:
алкильная часть алкилсульфониламино и алкилсульфониламиноимино необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино, гетероциклилсульфониламино, алкилсульфониламино и алкилсульфониламиноимино, где:
карбоциклильная и гетероциклильная части таких заместителей необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано и амино.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из карбоциклилсульфониламино и гетероциклилсульфониламино, где:
карбоциклильная и гетероциклильная части таких заместителей необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано и амино.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из алкилсульфониламино, алкенилсульфониламино, алкинилсульфониламино и алкилсульфониламиноимино, где:
(a) амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная, алкенильная и алкинильная часть таких заместителей необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламино, где:
(a) амино часть алкилсульфониламино необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная часть алкилсульфониламино необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламино, где:
амино часть алкилсульфониламино необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламино, где:
амино часть алкилсульфониламино необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила.
В некоторых вариантах осуществления, каждый R1 представляет собой независимо выбранный алкилсульфониламино, где:
алкильная часть алкилсульфониламино необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламино, где:
алкильная часть алкилсульфониламино необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламино. В некоторых таких вариантах осуществления, каждый RJ представляет собой метилсульфониламино.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламиноимино, где:
(a) амино часть алкилсульфониламиноимино необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная часть алкилсульфониламиноимино необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламиноимино, где:
амино часть алкилсульфониламиноимино необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламиноимино, где:
амино часть алкилсульфониламиноимино необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламиноимино, где:
алкильная часть алкилсульфониламиноимино необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламиноимино, где:
алкильная часть алкилсульфониламиноимино необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано.
В некоторых вариантах осуществления, каждый RJ представляет собой независимо выбранный алкилсульфониламиноимино. В некоторых таких вариантах осуществления, каждый RJ представляет собой метилсульфониламиноимино.
В некоторых вариантах осуществления, каждый RJ независимо выбран из группы, состоящей из алкилкарбониламино и алкилоксикарбониламино, где:
алкильная часть таких заместителей необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано.
B19. Заместитель RK.
Каждый RK независимо выбран из группы, состоящей из аминосульфонила, алкилсульфонила, алкенилсульфонила и алкинилсульфонила, где:
(a) алкилсульфонил, алкенилсульфонил и алкинилсульфонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, азидо, оксо, аминосульфонила, алкилоксикарбонила, алкенилоксикарбонила, алкинилоксикарбонила, алкилкарбонилокси, алкенилкарбонилокси, алкинилкарбонилокси, алкилокси, алкенилокси, алкинилокси, карбоциклила, гетероциклила, циано и аминокарбонила, где:
амино, аминосульфонил и аминокарбонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила; и
(b) аминосульфонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила.
В некоторых из приведенных выше вариантов осуществления, каждый RK независимо выбран из группы, состоящей из аминосульфонила, алкилсульфонила, алкенилсульфонила и алкинилсульфонила, где такие заместители являются незамещенными.
В некоторых вариантах осуществления, каждый RK независимо выбран из группы, состоящей из аминосульфонила и алкилсульфонила, где:
(a) алкилсульфонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, гетероциклила, циано и аминокарбонила; и
(b) аминосульфонил амино необязательно замещен одним или двумя независимо выбранными алкильными заместителями.
В некоторых вариантах осуществления, каждый RK независимо выбран из группы, состоящей из аминосульфонила и алкилсульфонила.
C. Варианты модификации соединений формулы I.
Различные варианты модификации заместителей R1, R2, R3, R4, R5, L, RA, RB, RC, RD, R6, RE, RF, RG, RH, RI, RJ и RK были рассмотрены выше. Эти варианты модификации заместителей могут быть объединены с получением различных вариантов воплощения соединений формулы I. Все варианты воплощения соединений формулы I, образованные путем объединения вариантов модификации заместителей, рассмотренных выше, входят в объем изобретения Заявителей, и некоторые иллюстративные варианты воплощения соединений формулы I представлены ниже.
В некоторых вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-L0:
;
выбрана из группы, состоящей из одинарной углерод-углеродной связи и двойной углерод-углеродной связи;
R1 выбран из группы, состоящей из водорода и метила;
R2 выбран из группы, состоящей из водорода и галогена
R3 выбран из группы, состоящей из водорода и галогена;
R4 выбран из группы, состоящей из C1-C4-алкила, C3-C6-карбоциклила и 5-6-членного гетероциклила, где:
(a) C1-C4-алкил необязательно замещен заместителями, вплоть до трех, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси и триметилсилила, и
(b) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, гало и алкилсульфониламино;
R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и гало;
R6 выбран из группы, состоящей из C5-C6-карбоциклила, 5-6-членного гетероциклила, конденсированного 2-кольцевого гетероциклила и конденсированного 2-кольцевого карбоциклила, где каждый такой заместитель замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF, RI, RJ и RK;
каждый RE независимо выбран из группы, состоящей из хлора, фтора, нитро, гидрокси, оксо, карбокси, амино, имино, альдегидо и алкиламино;
каждый RF представляет собой независимо выбранный алкил, необязательно замещенный заместителем, выбранным из группы, состоящей из карбокси, гало, амино, имино и аминосульфонила, где:
амино, имино и аминосульфонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкилсульфонила и алкилсульфониламино;
каждый RI независимо выбран из группы, состоящей из алкилкарбонила и аминокарбонила, где:
аминокарбонил необязательно замещен заместителем, выбранным из группы, состоящей из алкила, алкилоксиалкила, алкилсульфонила и алкилсульфониламино;
каждый RJ независимо выбран из группы, состоящей из алкилсульфониламино, алкенилсульфониламино, алкинилсульфониламино и алкилсульфониламиноимино, где:
(a) амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная, алкенильная и алкинильная часть таких заместителей необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где:
алкил необязательно замещен одним или несколькими гидрокси; и
каждый RK независимо выбран из группы, состоящей из аминосульфонила и алкилсульфонила, где:
(a) алкилсульфонил необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гидрокси, гало, амино, нитро, оксо, аминосульфонила, алкилоксикарбонила, алкилкарбонилокси, алкилокси, карбоциклила, гетероциклила, циано и аминокарбонила; и
(b) аминосульфонил необязательно замещен одним или двумя независимо выбранными алкильными заместителями. В некоторых вариантах осуществления, в соединениях формулы I:
выбран из группы, состоящей из одинарной углерод-углеродной связи и двойной углерод-углеродной связи;
R1 выбран из группы, состоящей из водорода и метила;
R2 выбран из группы, состоящей из водорода и галогена;
R3 выбран из группы, состоящей из водорода и галогена;
R4 выбран из группы, состоящей из C1-C4-алкила, C3-C6-карбоциклила и 5-6-членного гетероциклила, где:
(a) C1-C4-алкил необязательно замещен заместителями, вплоть до трех, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси и триметилсилила, и
(b) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, гало и алкилсульфониламино;
R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и гало;
L выбран из группы, состоящей из C(RA)=C(RB), этилена и циклопропил-1,2-ена;
один из RA и RB представляет собой водород, а другой выбран из группы, состоящей из водорода, метила, метокси и гало;
R6 выбран из группы, состоящей из C5-C6-карбоциклила и 5-6-членного гетероциклила, где каждый такой заместитель замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из RE, RF и RJ;
каждый RE независимо выбран из группы, состоящей из хлора, фтора, нитро, гидрокси, оксо, карбокси, амино, имино, альдегидо и алкиламино;
каждый RF представляет собой независимо выбранный алкил, необязательно замещенный заместителем, выбранным из группы, состоящей из карбокси, гало, амино, имино и аминосульфонила, где:
амино, имино и аминосульфонил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, алкилсульфонила и алкилсульфониламино;
каждый RI независимо выбран из группы, состоящей из алкилкарбонила и аминокарбонила, где:
аминокарбонил необязательно замещен заместителем, выбранным из группы, состоящей из алкила, алкилоксиалкила, алкилсульфонила и алкилсульфониламино; и
каждый RJ независимо выбран из группы, состоящей из алкилсульфониламино, алкенилсульфониламино, алкинилсульфониламино и алкилсульфониламиноимино, где:
(a) амино часть таких заместителей необязательно замещена заместителем, независимо выбранным из группы, состоящей из карбоциклилалкила, гетероциклилалкила, алкилкарбонилокси, аминокарбонилалкила, алкила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилоксикарбонила, алкилкарбонилоксиалкила и алкилсульфонила, где:
(1) карбоциклильная часть карбоциклилалкила и гетероциклильная часть гетероциклилалкила необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, карбокси, гидрокси, алкилокси, гало, нитро, циано, оксо и амино, и
(2) амино часть аминокарбонилалкила необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила, алкенила и алкинила,
(b) алкильная, алкенильная и алкинильная часть таких заместителей необязательно замещена одним или двумя заместителями, независимо выбранными из группы, состоящей из карбокси, гало, оксо, амино, алкилоксикарбонила, алкилкарбонилокси, гидрокси, алкилокси, карбоциклила, гетероциклила и циано, где:
амино необязательно замещен одним или двумя заместителями, независимо выбранными из группы, состоящей из алкила и алкилокси, где алкил необязательно замещен одним или несколькими гидрокси.
В некоторых вариантах осуществления, соединения формулы I соответствуют по структуре формуле I-LO:
;
выбрана из группы, состоящей из одинарной углерод-углеродной связи и двойной углерод-углеродной связи;
R1 представляет собой водород;
R2 выбран из группы, состоящей из водорода и галогена
R3 представляет собой водород;
R4 представляет собой трет-бутил;
R5 выбран из группы, состоящей из водорода, гидрокси, метокси и гало;
R6 представляет собой конденсированный 2-кольцевой карбоциклил, выбранный из группы, состоящей из нафталенила, дигидронафталенила, тетрагидронафталенила, гексагидронафталенила, октагидронафталенила, декагидронафталенила, инденила, дигидроинденила, гексагидроинденила, октагидроинденила, пенталенила, октагидропенталенила и гексагидропенталенила, где каждый такой заместитель замещен заместителем, выбранным из группы, состоящей из RF и RJ;
RF представляет собой алкилсульфониламиноалкил; и
RJ представляет собой алкилсульфониламино.
Примеры соединений формулы I (и их солей) приведены в Таблицах 1-38, представленных ниже. Примеры синтеза, приведенные ниже предоставляют пошаговые инструкции по получению некоторых из этих соединений. Остальные соединения получали, используя общее рассмотрение способа получения, конкретные примеры синтеза, приведенные ниже, и/или обсуждение по всему тексту этой заявки.
Таблица 1
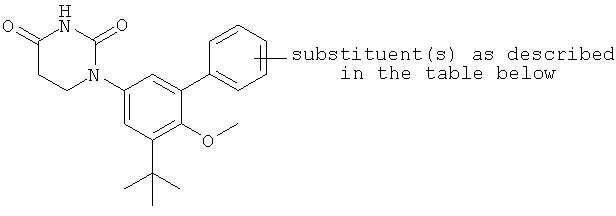
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 2
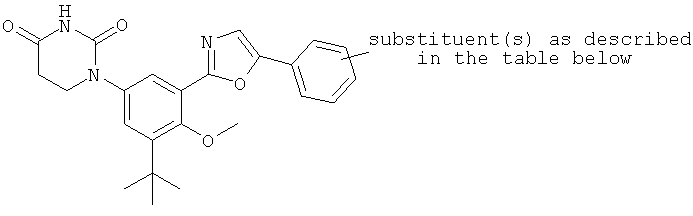
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 3
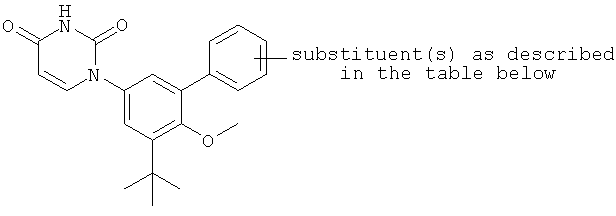
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 4
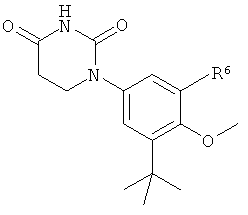
Таблица 5
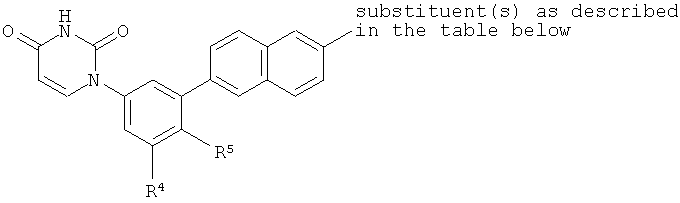
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 6
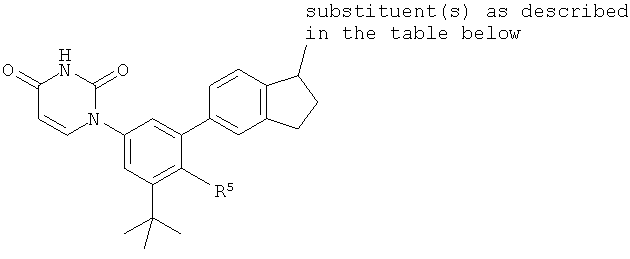
Substituent(s) as described int the table below - Заместители), описанные в приведенной ниже таблице
Таблица 7
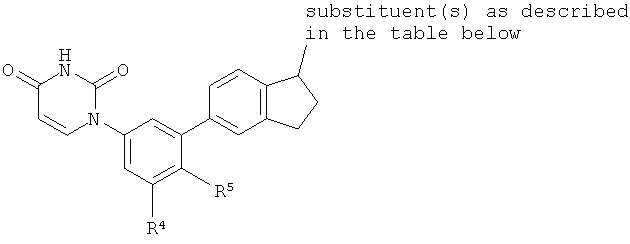
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 8
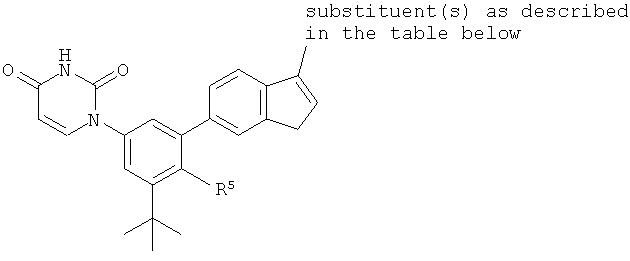
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 9
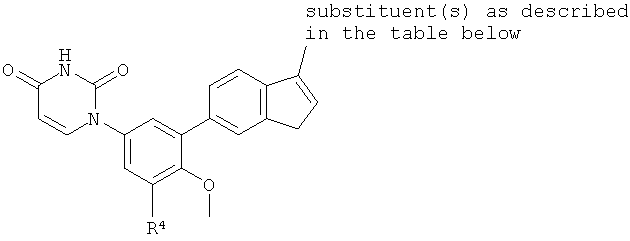
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
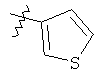
Таблица 10
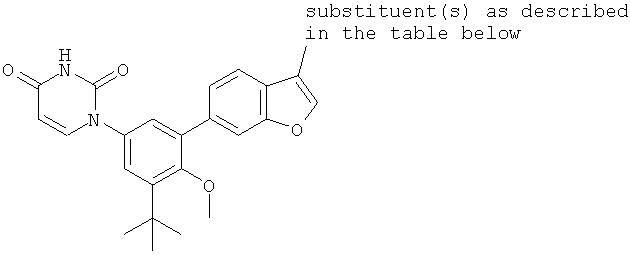
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 11
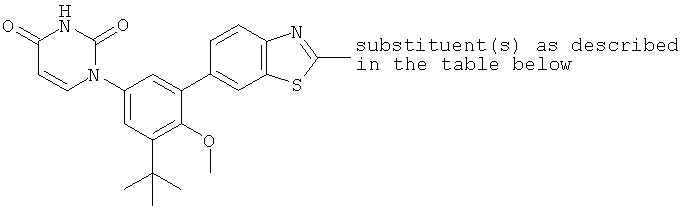
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
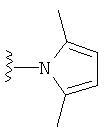
Таблица 12
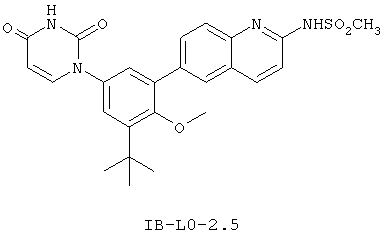
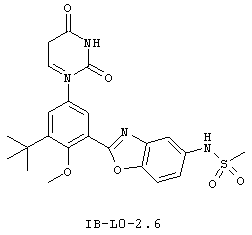
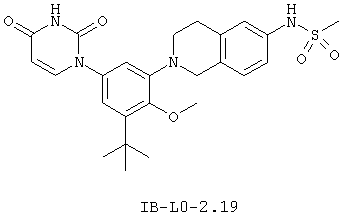
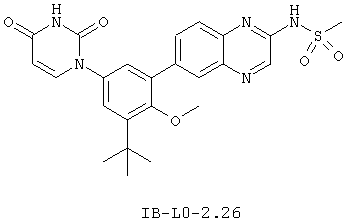
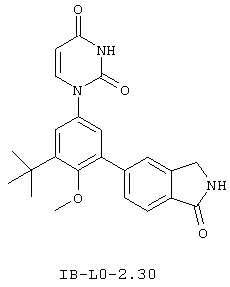

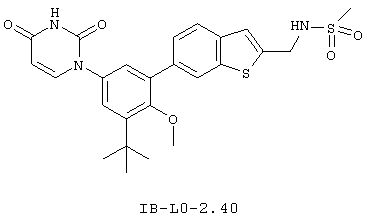
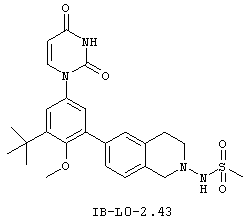
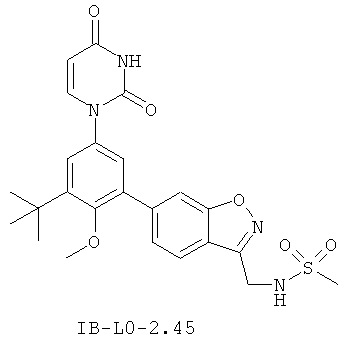
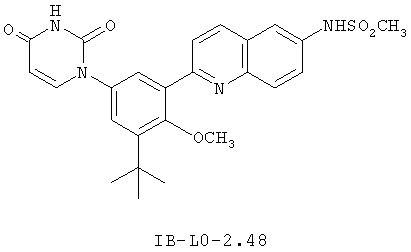
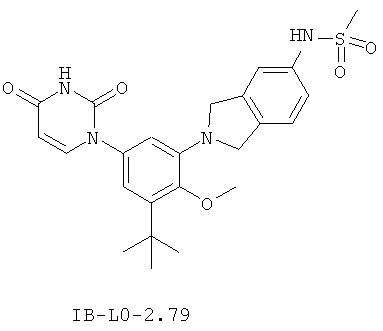
Таблица 13
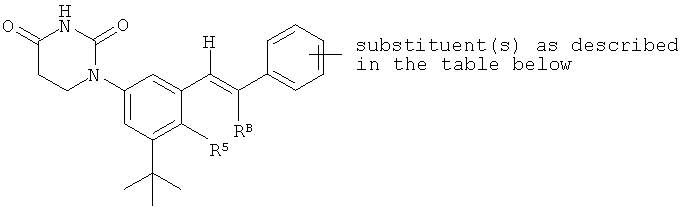
Substituent(s) as described int the table below - Заместителе), описанные в приведенной ниже таблице
Таблица 14
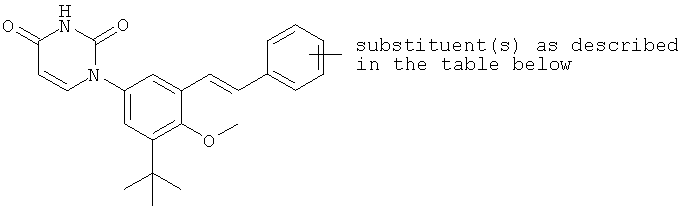
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
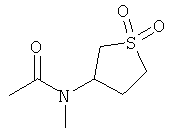 и -4-N(H)S(O)2CH3 [E]
и -4-N(H)S(O)2CH3 [E]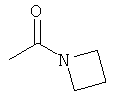 и -4-N(H)S(O)2CH3 [E]
и -4-N(H)S(O)2CH3 [E]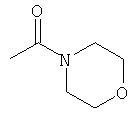 и -4-N(H)S(O)2CH3 [E]
и -4-N(H)S(O)2CH3 [E]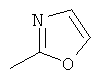 и -4-N(H)S(O)2CH3 [E]
и -4-N(H)S(O)2CH3 [E] и -4-N(H)S(O)2CH3 [E]
и -4-N(H)S(O)2CH3 [E] и -4-N(H)S(O)2CH3 [E]
и -4-N(H)S(O)2CH3 [E] и -4-N(H)S(O)2CH3 [E]
и -4-N(H)S(O)2CH3 [E]
Таблица 15
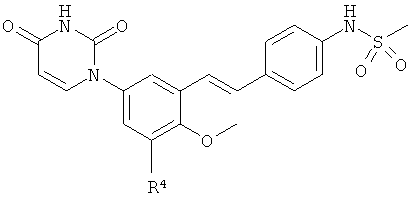
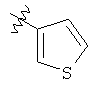 [E]
[E]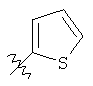 [E]
[E]
Таблица 16
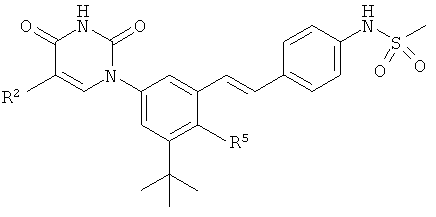
Таблица 17
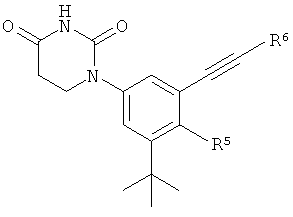
Таблица 18
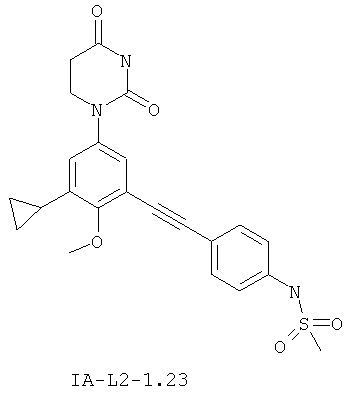
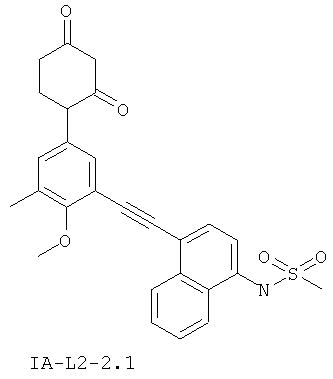
Таблица 19
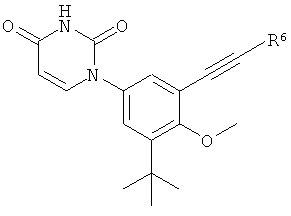
 и -4-N(H)S(O)2CH3
и -4-N(H)S(O)2CH3
Таблица 20
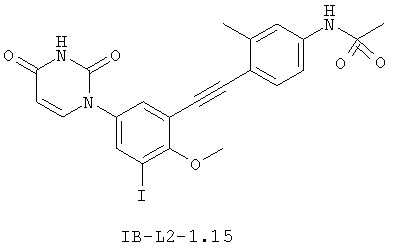
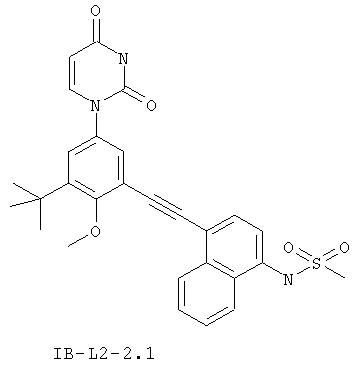
Таблица 21
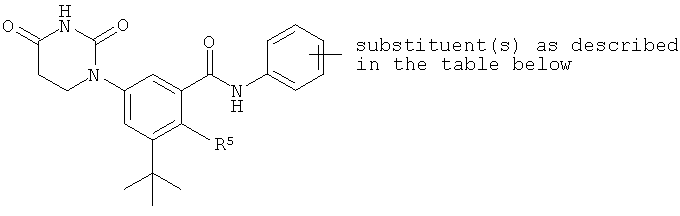
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
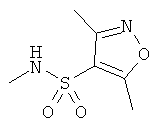
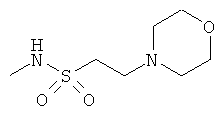
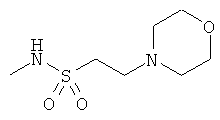
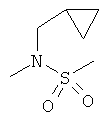
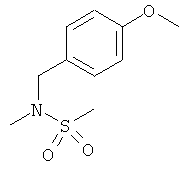
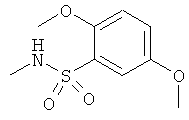
Таблица 22
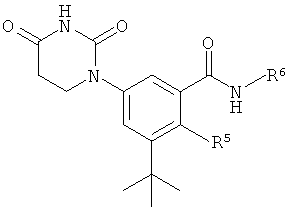
Таблица 23
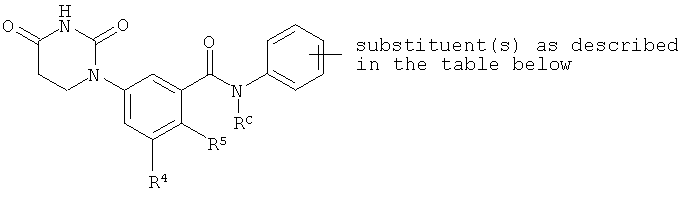
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 24
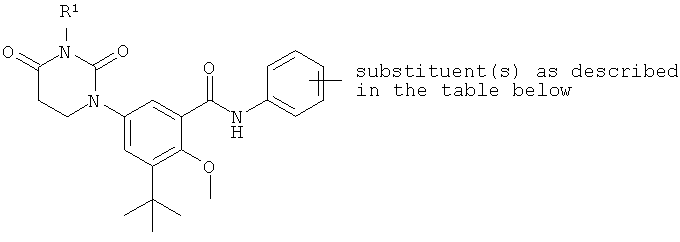
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 25
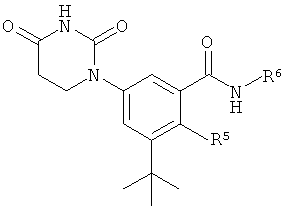
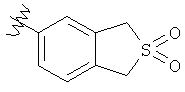
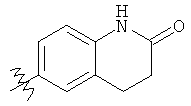
Таблица 26
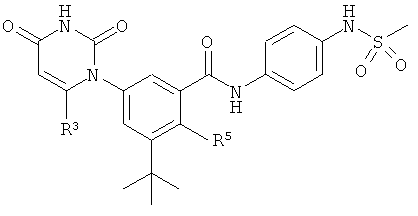
Таблица 27
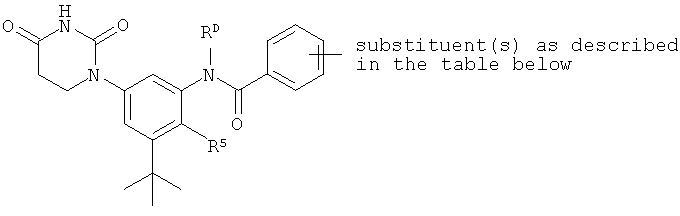
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 28
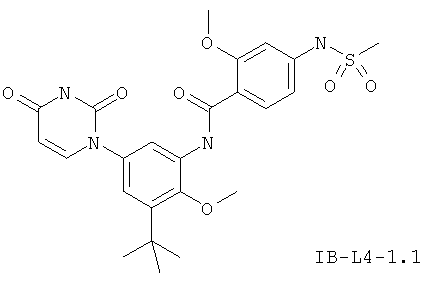
Таблица 29
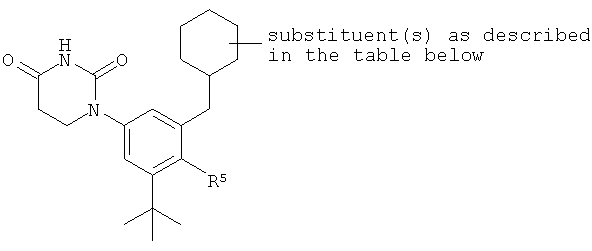
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 30
Таблица 31
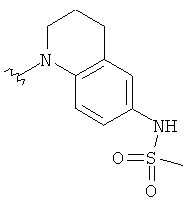
Таблица 32
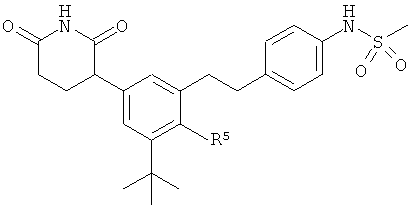
Таблица 33
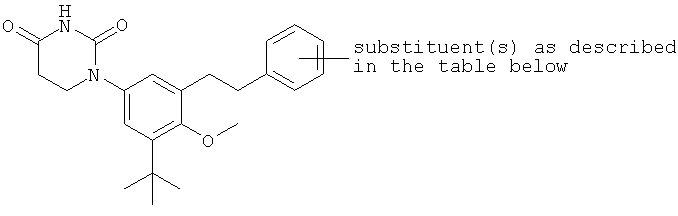
Substituent(s) as described int the table below - Заместител(и), описанные в приведенной ниже таблице
Таблица 34
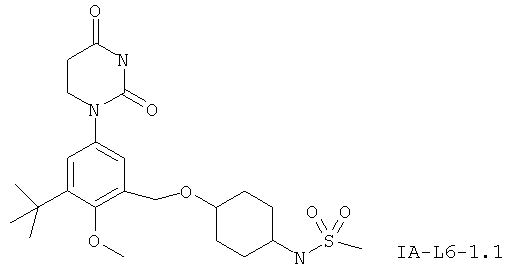
Таблица 35
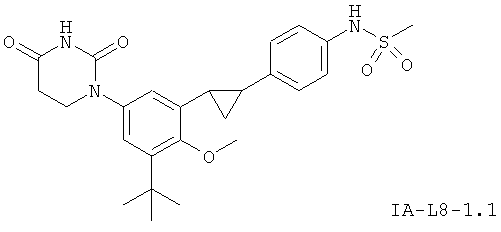
Таблица 36
Таблица 37
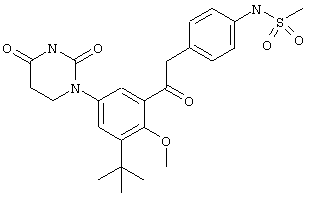
Таблица 38
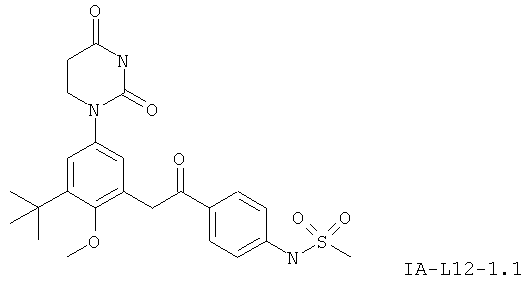
D. Изомеры.
Настоящее изобретение частично относится ко всем изомерам соединений формулы I (и их солям) (т.е., структурным и стереоизомерам). Структурные изомеры включают цепь и положение изомеров. Стереоизомеры включают E/Z изомеры (т.е., изомеры относительно одной или нескольких двойных связей), энантиомеры (т.е., стереоизомеры, которые имеют противоположные конфигурации при всех стереогенных центрах), и диастереоизомеры (т.е., стереоизомеры, которые имеют одинаковую конфигурацию при одном или нескольких стереогенных цетрах, но отличную при других стереогенных центрах).
E. Соли.
Настоящее изобретение также относится, частично, ко всем солям соединений формулы I. Соль соединения может быть предпочтительной благодаря одному или нескольким свойствам соли, например, таким как повышенная фармацевтическая стабильность при различных температурах и влажности, или желаемая растворимость в воде или других растворителях. В тех случаях, когда соль предназначена для введения пациенту (в отличие, например, от применения в контексте in vitro), соль предпочтительно является фармацевтически приемлемой и/или физиологически совместимой. Термин «фармацевтически приемлемая» используется в качестве прилагательного в этой патентной заявке для обозначения того, что определяемое существительное подходит для применения в качестве фармацевтического продукта или в качестве части фармацевтического продукта. Фармацевтически приемлемые соли включают соли, обычно используемые для образования солей щелочных металлов и для образования аддитивных солей свободных кислот или свободных оснований. В основном, эти соли обычно могут быть получены общепринятыми способами, посредством взаимодействия, например, соответствующей кислоты или основания с соединением по изобретению.
Фармацевтически приемлемые кислотно-аддитивные соли соединений формулы I могут быть получены из неорганической или органической кислоты. Примеры во многих случаях подходящих неорганических кислот включают соляную, бромистоводородную, йодистоводородную, азотную, угольную, серную и фосфорную кислоту. Подходящие органические кислоты в основном включают, например, алифатические, циклоалифатические, ароматические, аралифатические, гетероциклические, карбоновые и сульфоновые классы органических кислот.Конкретные примеры во многих случаях подходящих солей органических кислот включают ацетат, трифторацетат, формиат, пропионат, сукцинат, гликолят, глюконат, диглюконат, лактат, малат, винная кислота, цитрат, аскорбат, глюкуронат, малеат, фумарат, пируват, аспартат, глютамат, бензоат, аминобензойную кислоту, мезилат, стеарат, салицилат, p-гидроксибензоат, фенилацетат, соль миндальной кислоты, эмбонат (памоат), этансульфонат, бензолсульфонат, пантотенат, 2-гидроксиэтансульфонат, сульфанилат, циклогексиламиносульфонат, альгиновую кислоту, бета-гидроксимасляную кислоту, галактарат, галактуронат, адипат, альгинат, бисульфат, бутират, камфорат, камфорсульфонат, циклопентанпропионат, додецилсульфат, гликогептаноат, глицерофосфат, гептаноат, гексаноат, никотинат, оксалат, пальмоат, пектинат, 2-нафталсульфонат, 3-фенилпропионат, пикрат, пивалат, тиоцианат, тозилат и ундеканоат.
Фармацевтически приемлемые основные аддитивные соли соединений формулы I включают, например, соли металлов и органические соли. Предпочтительные соли металлов включают соли щелочных металлов (группа Ia), соли щелочноземельных металлов (группа IIa) и другие физиологически приемлемые соли металлов. Такие соли могут быть получены с алюминием, кальцием, литием, магнием, калием, натрием и цинком. Предпочтительные органические соли могут быть получены с аминами, такими как трометамин, диэтиламин, N,N'-дибензилэтилендиамин, хлоропрокаин, холин, диэтаноламин, этилендиамин, меглумин (N-метилглюкамин), и прокаин. Основные азот-содержащие группы могут быть кватернизированы такими агентами, как низшие алкил (C1-C6) галиды (например, метил, этил, пропил и бутил хлориды, бромиды и йодиды), диалкилсульфаты {например, диметил, диэтил, дибутил и диамилсульфаты), галиды с длинной цепью (например, децил, лаурил, миристил и стеарил хлориды, бромиды и йодиды), арилалкил галиды (например, бензил и фенэтил бромиды), и другие.
В некоторых вариантах осуществления, представляет собой натриевую соль N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-пиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой мононатриевую соль N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой динатриевую соль N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-пиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой калиевую соль N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-пиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой монокалиевую соль N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой холиновую соль N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-пиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой монохолиновую соль N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой натриевую соль (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-пиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой динатриевую соль (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой калиевую соль (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, соль представляет собой монокалиевую соль (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
F. Чистота.
Соединения формулы I (и их соли) любого уровня чистоты (в том числе чистые и по существу чистые) входят в объем настоящего изобретения. Термин «по существу чистый» в отношении соединения/соли/изомера, означает, что препарат/композиция, содержащая соединение/соль/изомер содержит примерно более 85% по массе соединения/соли/изомера, предпочтительно примерно более 90% по массе соединения/соли/изомера, предпочтительно примерно более 95% по массе соединения/соли/изомера, предпочтительно примерно более 97% по массе соединения/соли/изомера, и предпочтительно примерно более 99% по массе соединения/соли/изомера.
G. Кристаллические формы некоторых конкретных соединений и солей по настоящему изобретению.
G1. Кристаллические формы N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.3).
Настоящее изобретение также относится, частично, к кристаллическим формам N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.3), а именно к сольватным, гидратным и не содержащим растворитель кристаллическим формам рассмотренным ниже.
G1A. IB-L0-2.3 Сольваты.
Настоящее изобретение также относится, частично, к этанольному сольвату соединения IB-L0-2.3.
В некоторых вариантах осуществления, этанольный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,3±0,2, 9,7±0,2, 10,6±0,2, 13,6±0,2, 17,2±0,2, 19,2±0,2, 22,7±0,2, 26,9±0,2 и 29,4±0,2 градусов два тета (2θ). В некоторых таких вариантах осуществления, этанольный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,3±0,2, 9,7±0,2, 10,6±0,2, 13,6±0,2, 17,2±0,2, 19,2±0,2, 22,7±0,2, 26,9±0,2 и 29,4±0,2 градусов 2θ. В других таких вариантах осуществления, этанольный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,3±0,2, 9,7±0,2, 10,6±0,2, 13,6±0,2, 17,2±0,2, 19,2±0,2, 22,7±0,2, 26,9±0,2 и 29,4±0,2 градусов 2θ.
В некоторых вариантах осуществления, этанольный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,3±0,2, 9,7±0,2, 10,0±0,2, 10,6±0,2, 13,6±0,2, 17,2±0,2, 17,5±0,2, 19,2±0,2, 19,4±0,2, 22,7±0,2, 26,9±0,2 и 29,4±0,2 градусов 2θ. В некоторых таких вариантах осуществления, этанольный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,3±0,2, 9,7±0,2, 10,0±0,2, 10,6±0,2, 13,6±0,2, 17,2±0,2, 17,5±0,2, 19,2±0,2, 19,4±0,2, 22,7±0,2, 26,9±0,2 и 29,4±0,2 градусов 2θ. В других вариантах осуществления, этанольный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,3±0,2, 9,7±0,2, 10,0±0,2, 10,6±0,2, 13,6±0,2, 17,2±0,2, 17,5±0,2, 19,2±0,2, 19,4±0,2, 22,7±0,2, 26,9±0,2 и 29,4±0,2 градусов 2θ. В некоторых вариантах осуществления, этанольный сольват имеет порошковую рентгенограмму, по существу, как показано на Фигуре 1. Величины 2θ для пиков на Фигуре 1 (и их интенсивности) представляют собой следующее: 8,25 (54), 9,67 (74), 9,92 (63), 10,59 (21), 13,64 (49), 17,25 (40), 17,51 (20), 19,19 (66), 19,43 (100), 22,75 (19), 26,92 (25) и 29,39 (18).
Настоящее изобретение также относится, частично, к ацетонитрильному сольвату соединения IB-L0-2.3.
В некоторых вариантах осуществления, ацетонитрильный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,3±0,2, 8,3±0,2, 9,7±0,2, 10,5±0,2, 13,8±0,2, 17,2±0,2, 19,1±0,2 и 19,5±0,2 градусов 2θ. В некоторых таких вариантах осуществления, ацетонитрильный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,3±0,2, 8,3±0,2, 9,7±0,2, 10,5±0,2, 13,8±0,2, 17,2±0,2, 19,1±0,2 и 19,5±0,2 градусов 2θ. В других таких вариантах осуществления, ацетонитрильный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,3±0,2, 8,3±0,2, 9,7±0,2, 10,5±0,2, 13,8±0,2, 17,2±0,2, 19,1±0,2 и 19,5±0,2 градусов 2θ. В некоторых вариантах осуществления, ацетонитрильный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,3±9,2, 8,3±0,2, 9,7±0,2, 10,5±0,2, 13,8±0,2, 17,2±0,2, 17,7±0,2, 19,1±0,2, 19,5±0,2, 22,0±0,2, 22,8±0,2 и 27,2±0,2 градусов 2θ. В некоторых таких вариантах осуществления, ацетонитрильный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,3±0,2, 8,3±0,2, 9,7±0,2, 10,5±0,2, 13,8±0,2, 17,2±0,2, 17,7±0,2, 19,1±0,2, 19,5±0,2, 22,0±0,2, 22,8±0,2 и 27,2±0,2 градусов 2θ. В других таких вариантах осуществления, ацетонитрильный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,3±0,2, 8,3±0,2, 9,7±0,2, 10,5±0,2, 13,8±0,2, 17,2±0,2, 17,7±0,2, 19,1±0,2, 19,5±0,2, 22,0±0,2, 22,8±0,2 и 27,2±0,2 градусов 2θ.
В некоторых вариантах осуществления, ацетонитрильный сольват имеет порошковую рентгенограмму, по существу, как показано на Фигуре 3. Величины 2θ для пиков на Фигуре 3 (и их интенсивности) представляют собой следующее: 5,27 (14), 8,29 (33), 9,72 (100), 10,53 (20), 13,77 (67), 17,25 (38), 17,69 (17), 19,05 (63), 19,47 (58), 22,05 (19), 22,75 (16) и 27,17 (21).
Настоящее изобретение также относится, частично, к этилацетатному сольвату соединения IB-L0-2.3.
В некоторых вариантах осуществления, этилацетатный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 7,9±0,2, 9,3±0,2, 9,7±0,2, 10,6±0,2, 18,7±0,2, 38,5±0,2 и 44,7±0,2 градусов 2θ. В некоторых таких вариантах осуществления, этилацетатный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 7,9±0,2, 9,3±0,2, 9,7±0,2, 10,6±0,2, 18,7±0,2, 38,5±0,2 и 44,7±0,2 градусов 2θ. В других таких вариантах осуществления, этилацетатный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 7,9±0,2, 9,3±0,2, 9,7±0,2, 10,6±0,2, 18,7±0,2, 38,5±0,2 и 44,7±0,2 градусов 2θ.
В некоторых вариантах осуществления, этилацетатный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 7,9±0,2, 9,3±0,2, 9,7±0,2, 10,6±0,2, 13,7±0,2, 17,4±0,2, 18,7±0,2, 21,7±0,2, 22,0±0,2, 28,2±0,2, 38,5±0,2 и 44,7±0,2 градусов 2θ. В некоторых таких вариантах осуществления, этилацетатный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 7,9±0,2, 9,3±0,2, 9,7±0,2, 10,6±0,2, 13,7±0,2, 17,4±0,2, 18,7±0,2, 21,7±0,2, 22,0±0,2, 28,2±0,2, 38,5±0,2 и 44,7±0,2 градусов 2θ. В других таких вариантах осуществления, этилацетатный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 7,9±0,2, 9,3±0,2, 9,7±0,2, 10,6±0,2, 13,7±0,2, 17,4±0,2, 18,7±0,2, 21,7±0,2, 22,0±0,2, 28,2±0,2, 38,5±0,2 и 44,7±0,2 градусов 2θ.
В некоторых вариантах осуществления, сольват этилацетата имеет порошковую рентгенограмму, по существу, как показано на Фигуре 4. Величины 2θ для пиков на Фигуре 4 (и их интенсивности) представляют собой следующее: 7,94 (24), 9,33 (26), 9,72 (13), 10,58 (23), 13,71 (19), 17,40 (28), 18,72 (44), 21,69 (8), 22,04 (10), 28,23 (8), 38,45 (100) и 44,66 (95). Настоящее изобретение также относится, частично, к 2-пропанольному сольвату соединения IB-L0-2.3.
В некоторых вариантах осуществления, 2-пропанольный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 16,3±0,2, 18,1±0,2, 18,6±0,2, 19,4±0,2, 21,6±0,2 и 22,5±0,2 градусов 2θ. В некоторых таких вариантах осуществления, 2-пропанольный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 16,3+0,2, 18,1±0,2, 18,6±0,2, 19,4±0,2, 21,6±0,2 и 22,5±0,2 градусов 2θ. В других таких вариантах осуществления, 2-пропанольный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 16,3±0,2, 18,1±0,2, 18,6±0,2, 19,4±0,2, 21,6±0,2 и 22,5±0,2 градусов 2θ.
В некоторых вариантах осуществления, 2-пропанольный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 16,3±0,2, 18,1±9,2, 18,6±0,2, 19,4±0,2, 21,6±0,2, 22,5±0,2, 23,8±0,2, 26,0±0,2 и 28,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, 2-пропанольный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 16,3±0,2, 18,1±0,2, 18,6±0,2, 19,4±0,2, 21,6±0,2, 22,5±0,2, 23,8±0,2, 26,0±0,2 и 28,0±0,2 градусов 2θ. В других таких вариантах осуществления, 2-пропанольный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 16,3±0,2, 18,1±0,2, 18,6±0,2, 19,4±0,2, 21,6±0,2, 22,5±0,2, 23,8±0,2, 26,0±0,2 и 28,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, 2-пропанольный сольват имеет порошковую рентгенограмму, по существу, как показано на Фигуре 5. Величины 2θ для пиков на Фигуре 5 (и их интенсивности) представляют собой следующее: 8,18 (32), 9,26 (100), 10,12 (81), 16,28 (93), 18,11 (30), 18,59 (63), 19,40 (67), 21,57 (60), 22,51 (31), 23,82 (29), 25,94 (24) и 28,05 (29).
Настоящее изобретение также относится, частично, к метанольному сольвату соединения IB-L0-2.3.
В некоторых вариантах осуществления, метанольный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,4±0,2, 9,7±0,2, 10,1±0,2, 13,8±0,2, 17,4±0,2, 19,3±0,2 и 19,6±0,2 градусов 2θ. В некоторых таких вариантах осуществления, метанольный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,4±0,2, 9,7±0,2, 10,1±0,2, 13,8±0,2, 17,4±0,2, 19,3±0,2 и 19,6±0,2 градусов 2θ. В других таких вариантах осуществления, метанольный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,4±0,2, 9,7±0,2, 10,1±0,2, 13,8±0,2, 17,4±0,2, 19,3±0,2 и 19,6±0,2 градусов 2θ.
В некоторых вариантах осуществления, метанольный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,4±0,2, 9,7±0,2, 10,1±0,2, 13,5±0,2, 13,8±0,2, 17,4±0,2, 19,3±0,2, 19,6±0,2 и 27,1±0,2 градусов 2θ. В некоторых таких вариантах осуществления, метанольный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,4±0,2, 9,7±0,2, 10,1±0,2, 13,5±0,2, 13,8±9,2, 17,4±9,2, 19,3±0,2, 19,6±0,2 и 27,1±0,2 градусов 2θ. В других таких вариантах осуществления, метанольный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,4±0,2, 9,7±0,2, 10,1±0,2, 13,5±0,2, 13,8±0,2, 17,4±0,2, 19,3±0,2, 19,6±9,2 и 27,1±0,2 градусов 2θ.
В некоторых вариантах осуществления, метанольный сольват имеет порошковую рентгенограмму, по существу, как показано на Фигуре 6. Величины 2θ для пиков на Фигуре 6 (и их интенсивности) представляют собой следующее: 8,36 (48), 9,74 (65), 19,05 (74), 13,55 (24), 13,79 (69), 17,40 (32), 19,30 (80), 19,58 (100) и 27,08 (24). Настоящее изобретение также относится, частично, к 1-пропан ольному сольвату соединения IB-L0-2.3.
В некоторых вариантах осуществления, 1-пропанольный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 15,7±0,2, 16,2±0,2, 18,4±0,2, 19,3±0,2, 21,6±9,2 и 22,8±0,2 градусов 2θ. В некоторых таких вариантах осуществления, 1-пропанольный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 15,7±0,2, 16,2±0,2, 18,4±0,2, 19,3±0,2, 21,6±0,2 и 22,8±0,2 градусов 2θ. В других таких вариантах осуществления, 1-пропанольный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,2±9,2, 9,3±9,2, 19,1±0,2, 15,7±9,2, 16,2±0,2, 18,4±9,2, 19,3±0,2, 21,6±0,2 и 22,8±0,2 градусов 2θ.
В некоторых вариантах осуществления, 1-пропанольный сольват имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±0,2, 10,5±0,2, 15,7±0,2, 16,2±0,2, 18,4±0,2, 18,6±0,2, 19,3±0,2, 21,0±0,2, 21,6±0,2 и 22,8±0,2 градусов 2θ. В некоторых таких вариантах осуществления, 1-пропанольный сольват имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,2±0,2, 9,3±0,2, 10,1±9,2, 19,5±9,2, 15,7±9,2, 16,2±9,2, 18,4±0,2, 18,6±0,2, 19,3±0,2, 21,0±0,2, 21,6±0,2 и 22,8±0,2 градусов 2θ. В других таких вариантах осуществления, 1-пропанольный сольват имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,2±9,2, 9,3±9,2, 19,1±9,2, 19,5±9,2, 15,7±9,2, 16,2±9,2, 18,4±0,2, 18,6±0,2, 19,3±0,2, 21,0±0,2, 21,6±0,2 и 22,8±9,2 градусов 2θ.
В некоторых вариантах осуществления, 1-пропанольный сольват имеет порошковую рентгенограмму, по существу, как показано на Фигуре 7. Величины 2θ для пиков на Фигуре 7 (и их интенсивности) представляют собой следующее: 8,15 (27), 9,26 (87), 19,98 (84), 19,47 (62), 15,73 (40), 16,24 (100), 18,37 (41), 18,59 (49), 19,33 (50), 20,97 (28), 21,65 (71) и 22,81 (44).
Настоящее изобретение также относится, частично, к способу получения предствленных выше сольватов путем суспендирования соединения IB-L0-2.3 в соответствующем растворителе.
G1B. IB-L0-2.3, не содержащее растворитель.
Настоящее изобретение также относится, частично, к кристаллической форме соединения IB-L0-2.3, не содержащей растворителя.
В некоторых вариантах осуществления, не содержащее растворитель соединение IB-L0-2.3 имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,2±0,2, 7,9±0,2, 9,9±0,2, 16,2±0,2 и 18,3±0,2 градусов два тета (2θ). В некоторых таких вариантах осуществления, не содержащее растворитель соединение IB-L0-2.3 имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,9±0,2, 9,9±0,2, 16,2±0,2 и 18,3±0,2 градусов 2θ. В других таких вариантах осуществления, не содержащее растворитель соединение IB-L0-2.3 имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,9±0,2, 9,9±0,2, 16,2±0,2 и 18,3±0,2 градусов 2θ.
В некоторых вариантах осуществления, не содержащее растворитель соединение IB-L0-2.3 имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,2±0,2, 7,9±0,2, 9,9±0,2, 10,1±0,2, 14,9±0,2, 16,2±0,2, 18,3±0,2, 19,8±0,2 и 26,5±0,2 градусов 2θ. В некоторых таких вариантах осуществления, не содержащее растворитель соединение IB-L0-2.3 имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,9±0,2, 9,9±0,2, 10,1±0,2, 14,9±0,2, 16,2±0,2, 18,3±0,2, 19,8±0,2 и 26,5±0,2 градусов 2θ. В других таких вариантах осуществления, не содержащее растворитель соединение IB-L0-2.3 имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,9±0,2, 9,9±0,2, 10,1±0,2, 14,9±0,2, 16,2±0,2, 18,3±0,2, 19,8±0,2 и 26,5±0,2 градусов 2θ. В других таких вариантах осуществления, не содержащее растворитель соединение IB-L0-2.3 имеет порошковую рентгенограмму, содержащую восемь или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,9±0,2, 9,9±0,2, 10,1±0,2, 14,9±0,2, 16,2±0,2, 18,3±0,2, 19,8±0,2 и 26,5±0,2 градусов 2θ.
В некоторых вариантах осуществления, не содержащее растворитель соединение IB-L0-2.3 имеет порошковую рентгенограмму, по существу, как показано на Фигуре 8. Величины 2θ для пиков на Фигуре 8 (и их интенсивности) представляют собой следующее: 6,20 (36), 7,85 (66), 9,89 (61), 10,12 (75), 14,87 (27), 16,19 (89), 18,32 (100), 19,82 (77) и 26,53 (34).
Настоящее изобретение также относится, частично, к способу получения кристаллической формы соединения IB-L0-2.3 не содержащей растворителя, путем десольвации одного из сольватов IB-L0-2.3, рассмотренных выше. Сольват может быть десольвирован путем нагревания сольвата в твердом состоянии примерно в течение 10 мин при ~125°C.
G1C. гидрат IB-L0-2.3.
Настоящее изобретение также относится, частично, к гидрату соединения IB-L0-2.3. В некоторых вариантах осуществления, гидрат имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,4±0,2, 12,9±0,2, 17,9±0,2 и 18,9±0,2 градусов 2θ. В некоторых таких вариантах осуществления, гидрат имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,4±0,2, 12,9±0,2, 17,9±0,2 и 18,9±0,2 градусов 2θ.
В некоторых вариантах осуществления, гидрат имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,4±0,2, 12,9±0,2, 17,5±0,2, 17,9±0,2, 18,9±0,2 и 24,4±0,2 градусов 2θ. В некоторых таких вариантах осуществления, гидрат имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,4±0,2, 12,9±0,2, 17,5±0,2, 17,9±0,2, 18,9±0,2 и 24,4±0,2 градусов 2θ. В других таких вариантах осуществления, гидрат имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,4±0,2, 12,9±0,2, 17,5±0,2, 17,9±0,2, 18,9±0,2 и 24,4±0,2 градусов 2θ.
В некоторых вариантах осуществления, гидрат имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,4±0,2, 12,7±0,2, 12,9±0,2, 14,1±0,2, 15,7±0,2, 17,2±0,2, 17,5±0,2, 17,9±0,2, 18,9±0,2, 21,2±0,2, 24,4±0,2 и 25,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, гидрат имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,4±0,2, 12,7±0,2, 12,9±0,2, 14,1±0,2, 15,7±0,2, 17,2±0,2, 17,5±0,2, 17,9±0,2, 18,9±0,2, 21,2±0,2, 24,4±0,2 и 25,0±0,2 градусов 2θ. В других таких вариантах осуществления, гидрат имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,4±0,2, 12,7±0,2, 12,9±0,2, 14,1±0,2, 15,7±0,2, 17,2±0,2, 17,5±0,2, 17,9±0,2, 18,9±0,2, 21,2±0,2, 24,4±0,2 и 25,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, гидрат имеет порошковую рентгенограмму, по существу, как показано на Фигуре 9. Величины 2θ для пиков на Фигуре 9 (и их интенсивности) представляют собой следующее: 6,42 (60), 12,71 (33), 12,89 (58), 14,05 (17), 15,68 (18), 17,22 (44), 17,53 (100), 17,86 (51), 18,87 (77), 21,25 (17), 24,35 (28) и 24,95 (20).
Настоящее изобретение также относится, частично, к способу получения гидрата путем суспендирования в воде описанного выше кристаллического соединения, не содержащего растворителя. Гидрат был получен путем суспендирования 300 мг кристаллического соединения, не содержащего растворителя, в 2 мл воды при 45°C в течение четырех дней.
G2. Кристаллические формы N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида, мононатриевая соль.
Настоящее изобретение также относится, частично, к кристаллическим формам N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида, мононатриевой соли, а именно образцу A, образцу B и образцу C, кристаллических форм, рассмотренных ниже.
Настоящее изобретение относится, частично, к образцу A кристаллической мононатриевой соли.
В некоторых вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 18,6±0,2, 22,8±0,2 и 23,9±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 18,6±0,2, 22,8±0,2 и 23,9±0,2 градусов 2θ. В других таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 18,6±0,2, 22,8±0,2 и 23,9±0,2 градусов 2θ. В некоторых вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 18,6±0,2, 22,8±0,2, 23,3±0,2 и 23,9±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 18,6±0,2, 22,8±0,2, 23,3±0,2 и 23,9±0,2 градусов 2θ. В других таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 18,6±0,2, 22,8±0,2, 23,3±0,2 и 23,9±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 16,0±0,2, 18,6±0,2, 22,8±0,2, 23,3±0,2, 23,9±0,2 и 28,3±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 16,0±0,2, 18,6±0,2, 22,8±0,2, 23,3±0,2, 23,9±0,2 и 28,3±0,2 градусов 2θ. В других таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 16,0±0,2, 18,6±0,2, 22,8±0,2, 23,3±0,2, 23,9±0,2 и 28,3±0,2 градусов 2θ. В других таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую восемь или более пиков, выбранных из группы, состоящей из 4,6±0,2, 10,4±0,2, 12,0±0,2, 15,6±0,2, 16,0±0,2, 18,6±0,2, 22,8±0,2, 23,3±0,2, 23,9±0,2 и 28,3±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, по существу, как показано на Фигуре 10. Величины 2θ для пиков на Фигуре 10 (и их интенсивности) представляют собой следующее: 4,64 (62), 10,41 (38), 12,04 (38), 15,62 (44), 15,99 (44), 18,63 (49), 22,77 (60), 23,29 (40), 23,93 (100) и 28,31 (56).
Настоящее изобретение также относится, частично, к способу получения образца A мононатриевой соли. Образец A мононатриевой соли получали путем добавления 1M водного NaOH (0,548 мл) к соединению IB-L0-2.3 (225,72 мг), кристаллизации полученой в результате суспензии кристаллической динатриевой солью N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида, (полученной, как рассмотрено ниже), и уравновешивания полученной в результате суспензии во внешних условиях. Образец A мононатриевой соли образовался на следующий день способом, опосредованным раствором. Стехиометрия соли предположительно составляет 1:1 на основании процесса кристаллизации. Настоящее изобретение также относится, частично, к образцу B кристаллической мононатриевой соли.
В некоторых вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 28,8±0,2, 29,1±0,2 и 31,8±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 28,8±0,2, 29,1±0,2 и 31,8±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 28,8±0,2, 29,1±0,2 и 31,8±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую восемь или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 28,8±0,2, 29,1±0,2 и 31,8±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 28,8±0,2, 29,1±0,2 и 31,8±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую два или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 29,1±0,2 и 31,8±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую два или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 28,8±0,2 и 31,8±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2 и 31,8±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 19,2±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусов 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,4±0,2, 10,8±0,2, 16,3±0,2, 22,1±0,2 и 23,7±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2 и 16,3±0,2 градусах 2θ. В некоторых таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 16,3±0,2 и 22,1±0,2 градусах 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 16,3±0,2, 22,1±0,2 и 23,7±0,2 градусах 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусах 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусах 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 19,2±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусах 2θ. В других таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0+0,2, 18,8±0,2, 19,2±0,2, 21,6±0,2, 22,1±0,2 и 23,7±0,2 градусах 2θ. В дополнительных таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2 и 31,8±0,2 градусах 2θ. Еще в дополнительных таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 28,8±0,2 и 31,8±0,2 градусах 2θ. Еще в дополнительных таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2±0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 29,1±0,2 и 31,8±0,2 градусах 2θ. Еще в дополнительных таких вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, содержащую пики на 5,4±0,2, 10,8±0,2, 14,4±0,2, 16,3±0,2, 17,0±0,2, 18,8±0,2, 19,2+0,2, 19,6±0,2, 21,6±0,2, 22,1±0,2, 23,7±0,2, 28,8±0,2, 29,1±0,2 и 31,8±0,2 градусах 2θ.
В некоторых вариантах осуществления, образец B мононатриевой соли имеет порошковую рентгенограмму, по существу, как показано на Фигуре 12. Величины 2θ для пиков на Фигуре 12 (и их интенсивности) представляют собой следующее: 5,36 (100), 10,75 (42), 14,43 (20), 16,34 (60), 17,00 (25), 18,83 (18), 19,24 (18), 19,66 (12), 21,64 (29), 22,12 (41), 23,73 (32), 28,83 (9), 29,10 (9) и 31,78 (10).
Настоящее изобретение также относится, частично, к способу получения образца B мононатриевой соли. Образец B мононатриевой соли может быть может быть получен путем суспендирования образца A мононатриевой соли (например, ~30 мг) в различных органических растворителях (например, ~125 мкл ацетонитрила, этанола, 1-пропанола или 2-пропанола) при комнатной температуре. Образец B мононатриевой соли также был получен путем кристаллизации раствора образцом B мононатриевой соли. Соединение IB-LO-2.3 (12,5 г) растворяли в DMSO (37,5 мл) при ~68°C. Добавляли 1,04 г NaOH растворенного в 6,3 мл воды, 6,3 мл 2-пропанола и 12,5 мл смеси 35,2:1 об/об 2-пропанола/воды. Раствор кристаллизировали с использованием 125 мг затравки образца B, суспендированной в 12,5 мл смеси 35,2:1 об/об 2-пропанола/воды, и кристаллизационную суспензию инкубировали при ~68°C в течение ~1,5 ч. 175 мл смеси 35,2:1 об/об 2-пропанола/воды при ~68°C добавляли на протяжении ~7 ч, и кристаллизационную суспензию охлаждали до ~0°C на протяжении не менее 7 ч. Кристаллы выделяли фильтрованием и анализировали с помощью PXRD. Кристаллы затем сушили при ~50°C под вакуумом (приблизительно 3 дюйма ртутного столба). Высушенные кристаллы анализировали с помощью PXRD, у которых не было показано изменений по сравнению с образцом перед высушиванием. Стехиометрию образца B мононатриевой соли подтверждали с помощью ионной хроматографии.
Настоящее изобретение также относится, частично, к образцу C кристаллической мононатриевой соли.
В некоторых вариантах осуществления, образец C мононатриевой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,0±0,2, 12,0±0,2, 17,5±0,2, 18,8±0,2 и 22,7±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец C мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,0±0,2, 12,0±0,2, 17,5±0,2, 18,8±0,2 и 22,7±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец C мононатриевой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,0±0,2, 12,0±0,2, 17,5±0,2, 17,8±0,2, 18,8±0,2 и 22,7±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,0±0,2, 12,0±0,2, 17,5±0,2, 17,8±0,2, 18,8±0,2 и 22,7±0,2 градусов 2θ. В других таких вариантах осуществления, образец A мононатриевой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,0±0,2, 12,0±0,2, 17,5±0,2, 17,8±0,2, 18,8±0,2 и 22.7±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец C мононатриевой соли имеет порошковую рентгенограмму, по существу, как показано на Фигуре 14. Величины 2θ для пиков на Фигуре 14 (и их интенсивности) представляют собой следующее: 4,97 (100), 12,03 (24), 17,55 (32). 17,80 (77), 18,79 (23) и 22,74 (33).
Настоящее изобретение также относится, частично, к способу получения образца C мононатриевой соли. Образец C мононатриевой соли получали следующим образом. Образец B мононатриевой соли (100 мг) растворяли в 400 мкл DMSO и 2 мл смеси 12:1 об/об 2-пропанола/H2O при 70°C. Кристаллы затравки образца B мононатриевой соли добавляли в раствор, и этот раствор затем охлаждали до окружающей температуры на протяжении 20 мин. Фильтрация давала кристаллы образца C мононатриевой соли.
G3. Кристаллическая форма N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида, динатриевая соль.
Настоящее изобретение также относится, частично, к кристаллической форме динатриевой соли N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, динатриевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,8±0,2, 9,6±0,2, 10,5±0,2, 13,0±0,2, 14,6±0,2, 15,4±0,2, 16,8±0,2 и 23,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, динатриевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,8±0,2, 9,6±0,2, 10,5±0,2, 13,0±0,2, 14,6±0,2, 15,4±0,2, 16,8±0,2 и 23,0±0,2 градусов 2θ. В других таких вариантах осуществления, динатриевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,8±0,2, 9,6±0,2, 10,5±0,2, 13,0±0,2, 14,6±0,2, 15,4±0,2, 16,8±0,2 и 23,0±0,2 градусов 2θ. В некоторых вариантах осуществления, динатриевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,8±0,2, 9,6±0,2, 10,5±0,2, 13,0±0,2, 14,6±0,2, 15,4±0,2, 16,8±0,2, 22,7±0,2, 23,0±0,2 и 23,3±0,2 градусов 2θ. В некоторых таких вариантах осуществления, динатриевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,8±0,2, 9,6±0,2, 10,5±0,2, 13,0±0,2, 14,6±0,2, 15,4±0,2, 16,8±0,2, 22,7±0,2, 23,0±0,2 и 23,3±0,2 градусов 2θ. В других таких вариантах осуществления, динатриевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,8±0,2, 9,6±0,2, 10,5±0,2, 13,0±0,2, 14,6±0,2, 15,4±0,2, 16,8±0,2, 22,7±0,2, 23,0±0,2 и 23,3±0,2 градусов 2θ.
В некоторых вариантах осуществления, динатриевая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 15. Величины 2θ для пиков на Фигуре 15 (и их интенсивности) представляют собой следующее: 4,80 (100), 9,59 (10), 10,51 (13), 12,98 (11), 14,56 (8), 15,38 (12), 16,84 (6), 22,68 (10), 23,04 (6) и 23,33 (4).
Настоящее изобретение также относится, частично, к способу получения динатриевой соли. Динатриевая соль была получена путем суспендирования соединения IB-L0-2.3 (52,83 мг) в 1M водном NaOH (1,1 мл) (молярное соотношение соединение:NaOH составляло 1:10). Раствор нагревали до 36°C, и твердое вещество растворялось полностью с образованием прозрачного раствора. Этот раствор естественным образом охлаждали до температуры окружающей среды, и соль кристаллизовалась через 24 ч. Альтернативно, динатриевую соль получали путем суспендирования соединения IB-L0-2.3 (51 мг) в EtOH (1 мл). Добавляли NaOH в 1,2 мл 5:1 об/об EtOH/H2O (2,1 молярный эквивалент). Реакционную смесь концентрировали и 2 мл ацетонитрила добавляли для индукции кристаллизации. Стехиометрию этого твердого вещества определяли с помощью ионной хроматографии.
G4. Кристаллическая форма N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансулъфонамида, монокалиевая соль.
Настоящее изобретение также относится, частично, к кристаллической форме монокалиевой соли N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, монокалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,0±0,2, 9,9±0,2, 11,3±0,2, 13,3±0,2, 16,9±0,2, 18,1±0,2, 19,1±0,2, 20,0±0,2, 21,1±0,2, 23,5±0,2, 24,8±0,2 и 25,7±0,2 градусов 2θ. В некоторых таких вариантах осуществления, монокалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,0±0,2, 9,9±0,2, 11,3±0,2, 13,3±0,2, 16,9±0,2, 18,1±0,2, 19,1±0,2, 20,0±0,2, 21,1±0,2, 23,5±0,2, 24,8±0,2 и 25,7±0,2 градусов 2θ. В других таких вариантах осуществления, монокалиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,0±0,2, 9,9±0,2, 11,3±0,2, 13,3±0,2, 16,9±0,2, 18,1±0,2, 19,1±0,2, 20,0±0,2, 21,1±0,2, 23,5±0,2, 24,8±0,2 и 25,7±0,2 градусов 2θ.
В некоторых вариантах осуществления, монокалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,0±0,2, 9,9±0,2, 11,3±0,2, 13,3±0,2, 16,9±0,2, 18,1±0,2, 19,1±0,2, 20,0±0,2, 21,1±0,2, 21,5±0,2, 23,5±0,2, 24,8±0,2 и 25,7±0,2 градусов 2θ. В некоторых таких вариантах осуществления, монокалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,0±0,2, 9,9±0,2, 11,3±0,2, 13,3±0,2, 16,9±0,2, 18,1±0,2, 19,1±0,2, 20,0±0,2, 21,1±0,2, 21,5±0,2, 23,5±0,2, 24,8±0,2 и 25,7±0,2 градусов 2θ. В других таких вариантах осуществления, монокалиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,0±0,2, 9,9±0,2, 11,3±0,2, 13,3±0,2, 16,9±0,2, 18,1±0,2, 19,1±0,2, 20,0±0,2, 21,1±0,2, 21,5±0,2, 23,5±0,2, 24,8±0,2 и 25,7±0,2 градусов 2θ.
В некоторых вариантах осуществления, монокалиевая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 17. Величины 2θ для пиков на Фигуре 17 (и их интенсивности) представляют собой следующее: 4,97 (100), 9,94 (7), 11,33 (15), 13,28 (7), 16,91 (5), 18,13 (7), 19,14 (4), 20.00 (4), 21,13 (4), 21,45 (4), 23,54 (4), 24,84 (3) и 25,67 (6).
Настоящее изобретение также относится, частично, к способу получения монокалиевой соли. Монокалиевую соль получали в водной среде. 0,366 мл 1M водного КОН добавляли к 150,56 мг соединения IB-L0-2.3 (молярное соотношение 1:1,2). Полученную в результате суспензию уравновешивали в окружающих условиях. Монокалиевая соль образовывалась на следующий день в процессе, опосредованном растворением. Альтернативно, монокалиевую соль получали путем суспендирования соединения IB-L0-2.3 (300 мг) в 3 мл ацетонитрила. Добавляли КОН в 1,3 мл H2O (2,1 молярного эквивалента). Добавляли дополнительный 1 мл H2O для растворения всех твердых веществ. После этого, 12 мл ацетонитрила добавляли для индукции кристаллизации. Стехиометрию соли подтверждали с помощью ионной хроматографии.
G5. Кристаллические формы N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида, монохолиновая соль.
Настоящее изобретение также относится, частично, к кристаллическим формам монохолиновой соли N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида, а именно образцу A и образцу B кристаллических форм, рассмотренных ниже.
Настоящее изобретение относится, частично, к образцу A кристалличесокй монохолиновой соли.
В некоторых вариантах осуществления, образец A монохолиновой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 10,9±0,2, 12,1±0,2, 13,4±0,2, 15,5±0,2, 17,0±0,2, 17,8±0,2, 18,3±0,2, 19,5±0,2 и 21,9±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A монохолиновой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 10,9±0,2, 12,1±0,2, 13,4±0,2, 15,5±0,2, 17,0±0,2, 17,8±0,2, 18,3±0,2, 19,5±0,2 и 21,9±0,2 градусов 2θ. В других таких вариантах осуществления, образец A монохолиновой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 10,9±0,2, 12,1±0,2, 13,4±0,2, 15,5±0,2, 17,0±0,2, 17,8±0,2, 18,3±0,2, 19,5±0,2 и 21,9±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец A монохолиновой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 10,9±0,2, 12,1±0,2, 13,0±0,2, 13,4±0,2, 13,6±0,2, 15,5±0,2, 17,0±0.2, 17,8±0,2, 18,3±0,2, 19,5±0,2, 19,7±0,2 и 21,9±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A монохолиновой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из градусов 2θ. В других таких вариантах осуществления, образец A монохолиновой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из градусов 2θ.
В некоторых вариантах осуществления, образец A монохолиновой соли имеет порошковую рентгенограмму, по существу, как показано на Фигуре 19. Величины 2θ для пиков на Фигуре 19 (и их интенсивности) представляют собой следующее: 10,94 (42), 12,96 (20), 12,96 (26), 13,42 (64), 13,64 (27), 15,51 (18), 16,98 (78), 17,81 (26), 18,32 (100), 19,49 (48), 19,70 (33) и 21,91 (22).
Настоящее изобретение также относится, частично, к способу получения образца A монохолиновой соли. Его получали в смеси растворителей тетрагидрофурана (THF) и метанола. Соединение IB-L0-2.3 (56,79 мг) растворяли в THF при 60°C, добавляли 40,01 мг раствора гидроксида холина (45 масс% в метаноле), получая в результате молярное соотношение 1:1,2. Кристаллы формировались при естестивенном охлаждении до окружающей температуры.
Настоящее изобретение также относится, частично, к образцу B кристаллической монохолиновой соли.
В некоторых вариантах осуществления, образец B монохолиновой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,0±0,2, 9,4±0,2, 11,0±0,2, 13,0±0,2, 13,7±0,2, 15,9±0,2, 17,0±0,2, 18,3±0,2, 18,9±0,2, 19,8±0,2 и 22,1±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец B монохолиновой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,0±0,2, 9,4±0,2, 11,0±0,2, 13,0±0,2, 13,7±0,2, 15,9±0,2, 17,0±0,2, 18,3±0,2, 18,9±0,2, 19,8±0,2 и 22,1±0,2 градусов 2θ. В других таких вариантах осуществления, образец B монохолиновой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,0±0,2, 9,4±0,2, 11,0±0,2, 13,0±0,2, 13,7±0,2, 15,9±0,2, 17,0±0,2, 18,3±0,2, 18,9±0,2, 19,8±0,2 и 22,1±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец B монохолиновой соли имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,0±0,2, 9,4±0,2, 11,0±0,2, 13,0±0,2, 13,3±0,2, 13,7±0,2, 15,9±0,2, 17,0±0,2, 17,4±0,2, 18,3±0,2, 18,9±0,2, 19,8±0,2, 21,8±0,2 и 22,1±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец B монохолиновой соли имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,0±0,2, 9,4±0,2, 11,0±0,2, 13,0±0,2, 13,3±0,2, 13,7±0,2, 15,9±0,2, 17,0±0,2, 17,4±0,2, 18,3±0,2, 18,9±0,2, 19,8±0,2, 21,8±0,2 и 22,1±0,2 градусов 2θ. В других таких вариантах осуществления, образец B монохолиновой соли имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,0±0,2, 9,4±0,2, 11,0±0,2, 13,0±0,2, 13,3±0,2, 13,7±0,2, 15,9±0,2, 17,0±0,2, 17,4±0,2, 18,3±0,2, 18,9±0,2, 19,8±0,2, 21,8±0,2 и 22,1±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец B монохолиновой соли имеет порошковую рентгенограмму, по существу, как показано на Фигуре 21. Величины 2θ для пиков на Фигуре 21 (и их интенсивности) представляют собой следующее: 7,96 (41), 9,38 (34), 10,96 (24), 12,98 (76), 13,34 (33), 13,72 (37), 15,90 (100), 17,03 (60), 17.42 (37), 18,30 (31), 18,85 (93), 19,82 (90), 21,76 (38) и 22,06 (46).
Настоящее изобретение также относится, частично, к способу получения образца B монохолиновой соли. Его получали путем суспендирования некристаллической холиновой соли в этилацетате в течение семи дней.
G6. Кристаллическая форма N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансулъфонамида, дихолиновая соль.
Настоящее изобретение также относится, частично, к кристаллической форме дихолиновой соли N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В некоторых вариантах осуществления, дихолиновая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,6±0,2, 11,0±0,2, 12,9±0,2, 17,0±0,2, 17,5±0,2, 18,9±0,2, 19,8±0,2 и 21,9±0,2 градусов 2θ. В некоторых таких вариантах осуществления, дихолиновая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,6±0,2, 11,0±0,2, 12,9±0,2, 17,0±0,2, 17,5±0,2, 18,9±0,2, 19,8±0,2 и 21,9±0,2 градусов 2θ. В других таких вариантах осуществления, дихолиновая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,6±0,2, 11,0±0,2, 12,9±0,2, 17,0±0,2, 17,5±0,2, 18,9±0,2, 19,8±0,2 и 21,9±0,2 градусов 2θ. В некоторых вариантах осуществления, дихолиновая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 8,6±0,2, 11,0±0,2, 12,9±0,2, 17,0±0,2, 17,5±0,2, 18,9±0,2, 19,8±0,2, 21,9±0,2 и 22,1±0,2 градусов 2θ. В некоторых таких вариантах осуществления, дихолиновая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 8,6±0,2, 11,0±0,2, 12,9±0,2, 17,0±0,2, 17,5±0,2, 18,9±0,2, 19,8±0,2, 21,9±0,2 и 22,1±0,2 градусов 2θ. В других таких вариантах осуществления, дихолиновая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 8,6±0,2, 11,0±0,2, 12,9±0,2, 17,0±0,2, 17,5±0,2, 18,9±0,2, 19,8±0,2, 21,9±0,2 и 22,1±0,2 градусов 2θ.
В некоторых вариантах осуществления, дихолиновая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 23. Величины 2θ для пиков на Фигуре 23 (и их интенсивности) представляют собой следующее: 8,62 (28), 10,98 (29), 12,93 (50), 15,88 (100), 17,03 (42), 17,47 (29), 18,88 (66), 19,82 (57), 21,89 (42), 2,07 (41).
Настоящее изобретение также относится, частично, к способу получения дихолиновой соли. Ее получали путем суспендирования соединения IB-L0-2.3 (200 мг) в 0,75 мл MeOH. Добавляли гидроксид холина в MeOH (210 мл, 45 масс%, 2,10 молярного эквивалента). Реакционную смесь концентрировали, и добавляли 4 мл ацетонитрила и 6 мл изопропилацетатата. Реакционную смесь затем кристаллизовали следовыми количествами кристаллов затравки монокалиевой соли соединения IB-L0-2.3 (рассмотрено выше). Вскоре после этого реакционная смесь начинала кристаллизоваться. Стехиометрию соли определяли с помощью 1H ЯМР в растворе.
G7. Кристаллические формы (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксистирил)фенил)метансульфонамида, динатриевая соль.
Настоящее изобретение также относится, частично, к кристаллическим формам динатриевой соли (E)-N-(4-(3 -трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида, а именно девятиводным и четырехводным кристаллическим формам, рассмотренным ниже.
Настоящее изобретение относится, частично, к девятиводной кристаллической динатриевой соли. Кристаллографические параметры элементарной ячейки девятиводной кристаллической динатриевой соли были определены как следующие: а равно 8,9Ǻ, b равно 9,4Ǻ и c равно 20,7Ǻ (более точно, а равно 8,926(2)Ǻ, b равно 9,415(2)Ǻ, и c равно 20,674(5)Ǻ); углы ячейки составляют: α - 94,8°, β - 93,3°, и γ - 107,0° (более точно, α равен 94,796(4)°, β равен 93,345(4)°, и γ равен 107,013(4)°); и объем ячейки составляет 1649Ǻ3 (более точно, 1649,3(7)Ǻ3). Эта соль кристаллизуется в пространственной группе P-1.
В некоторых вариантах осуществления, девятиводная динатриевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,3±0,2, 10,4±0,2, 10,9±0,2, 11,6±0,2, 12,9±0,2, 14,7±0,2, 16,4±0,2, 17,8±0,2, 19,4±0,2, 19,8±0,2, 20,8±0,2, 21,9±0,2 и 23,5±0,2 градусов 2θ. В некоторых таких вариантах осуществления, девятиводная динатриевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,3±0,2, 10,4±0,2, 10,9±0,2, 11,6±0,2, 12,9±0,2, 14,7±0,2, 16,4±0,2, 17,8±0,2, 19,4±0,2, 19,8±0,2, 20,8±0,2, 21,9±0,2 и 23,5±0,2 градусов 2θ. В других таких вариантах осуществления, девятиводная динатриевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,3±0,2, 10,4±0,2, 10,9±0,2, 11,6±0,2, 12,9±0,2, 14,7±0,2, 16,4±0,2, 17,8±0,2, 19,4±0,2, 19,8±0,2, 20,8±0,2, 21,9±0,2 и 23,5±0,2 градусов 2θ.
В некоторых вариантах осуществления, девятиводная динатриевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,3±0,2, 10,4±0,2, 10,9±0,2, 11,6±0,2, 12,9±9,2, 14,7±0,2, 14,9±0,2, 16,4±0,2, 17,8±0,2, 19,4±0,2, 19,7±0,2, 19,8±0,2, 20,8±0,2, 20,9±0,2, 21,9±0,2, 22,1±0,2 и 23,5±0,2 градусов 2θ. В некоторых таких вариантах осуществления, девятиводная динатриевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,3±0,2, 10,4±0,2, 10,9±0,2, 11,6±0,2, 12,9±0,2, 14,7±0,2, 14,9±0,2, 16,4±0,2, 17,8±0,2, 19,4±0,2, 19,7±0,2, 19,8±0,2, 20,8±0,2, 20,9±0,2, 21,9±0,2, 22,1±0,2 и 23,5±0,2 градусов 2θ. В других таких вариантах осуществления, девятиводная динатриевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,3±0,2, 10,4±0,2, 10,9±0,2, 11,6±0,2, 12,9±0,2, 14,7±0,2, 14,9±0,2, 16,4±0,2, 17,8±0,2, 19,4±0,2, 19,7±0,2, 19,8±0,2, 20,8±0,2, 20,9±0,2, 21,9±0,2, 22,1±0,2 и 23,5±0,2 градусов 2θ.
В некоторых вариантах осуществления, девятиводная динатриевая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 24. Величины 2θ для пиков на Фигуре 24 (и их интенсивности) представляют собой следующее: 4,31 (100), 10,36 (12), 10,91 (23), 11,61 (52), 12,93 (24), 14,73 (65), 14,89 (20), 16,44 (41), 17,80 (38), 19,44 (26), 19,67 (37), 19,83 (59), 20,75 (69), 20,89 (21), 21,92 (43), 22,13 (40) и 22,42 (24).
Настоящее изобретение также относится, частично, к способу получения девятиводной динатриевой соли. Ее получали в водной среде. Водный NaOH (1M, 1,18 мл) добавляли к (Е)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамиду (соединение IB-L1-1.1) (27,82 мг) (молярное соотношение 1:20 кислота:основание). Полученную в результате суспензию уравновешивали в окружающих условиях. Девятиводная динатриевая соль образуется спустя семь дней в процессе, опосредованном растворением. Альтернативно, девятиводную динатриевую соль получали путем суспендирования 278,8 мг соединения IB-L1-1.1 в 1,25 мл THF при нагревании примерно до 50°C. Добавляли водный NaOH (1 н, 1,5 мл, 2,2 молярного эквивалента). Твердое вещество растворялось полностью с образованием прозрачного раствора, который естественным путем охлаждался до окружающих температур. Эта соль кристаллизовалась спонтанно. Молекулярную структуру определяли с помощью монокристальной дифрактометрии.
Настоящее изобретение относится, частично, к четырехводной кристаллической динатриевой соли.
В некоторых вариантах осуществления, четырехводная динатриевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,8±0,2, 12,1±0,2, 14,0±0,2, 17,0±0,2, 17,5±0,2, 20,9±0,2, 21,6±0,2, 25,0±0,2 и 29,5±0,2 градусов 2θ. В некоторых таких вариантах осуществления, четырехводная динатриевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,8±0,2, 12,1±0,2, 14,0±0,2, 17,0±0,2, 17,5±0,2, 20,9±0,2, 21,6±0,2, 25,0±0,2 и 29,5±0,2 градусов 2θ. В других таких вариантах осуществления, четырехводная динатриевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,8±0,2, 12,1±0,2, 14,0±0,2, 17,0±0,2, 17,5±0,2, 20,9+0,2, 21,6±0,2, 25,0+0,2 и 29,5±0,2 градусов 2θ.
В некоторых вариантах осуществления, четырехводная динатриевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,8±0,2, 12,1±0,2, 14,0±0,2, 14,4±0,2, 17,0±0,2, 17,5±0,2, 20,9±0,2, 21,6±0,2, 25,0±0,2, 29,5±0,2 и 34,2±0,2 градусов 2θ. В некоторых таких вариантах осуществления, четырехводная динатриевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,8±0,2, 12,1±0,2, 14,0±0,2, 14,4±0,2, 17,0+0,2, 17,5±0,2, 20,9±0,2, 21,6±0,2, 25,0±0,2, 29,5±0,2 и 34,2±0,2 градусов 2θ. В других таких вариантах осуществления, четырехводная динатриевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,8±0,2, 12,1±0,2, 14,0±0,2, 14,4±0,2, 17,0±0,2, 17,5±0,2, 20,9±0,2, 21,6±0,2, 25,0±0,2, 29,5±0,2 и 34,2±0,2 градусов 2θ.
В некоторых вариантах осуществления, четырехводная динатриевая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 25. Величины 2θ для пиков на Фигуре 25 (и их интенсивности) представляют собой следующее: 4,81 (100), 12,07 (7), 14,01 (27), 14,41 (8), 16,96 (18), 17,53 (11), 20,87 (18), 21,58 (22), 24,99 (11), 29,47 (9) и 34,20 (9).
Настоящее изобретение также относится, частично, к способу получения четырехводной динатриевой соли путем суспендирования девятиводной динатриевой соли в органическом растворителе {например, этаноле, 1-пропаноле или 2-пропаноле).
G8. Кристаллическая форма дикалиевой соли (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансулъфонамида.
Настоящее изобретение также относится, частично, к кристаллической четырехводной дикалиевой соли (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Кристаллографические параметры элементарной ячейки четырехводной дикалиевой соли были определены следующими: a равно 14.5Ǻ, b равно 10,8Ǻ, и c равно 35,8Ǻ (более точно, a равно 14,454(14)Ǻ, b равно 10,763(14)Ǻ, и c равно 35,75(4)Ǻ); угол ячейки составляет: β - 98,8° (более точно, β равено 98,82(3)°); и объем ячейки составляет 5499Ǻ3 (более точно, 5499(11)Ǻ3). Эта соль кристаллизуется в пространственной группе C2/c.
В некоторых вариантах осуществления, четырехводная дикалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,0±0,2, 11,9±0,2, 12,4±0,2, 13,7±0,2, 15,0±0,2, 16,5±0,2, 17,1±0,2, 20,8±0,2, 21,3±0,2, 22,2±0,2, 24,0±0,2, 26,4±0,2 и 29,3±0,2 градусов 2θ. В некоторых таких вариантах осуществления, четырехводная дикалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,0±0,2, 11,9±0,2, 12,4±0,2, 13,7±0,2, 15,0±0,2, 16,5±0,2, 17,1±0,2, 20,8±0,2, 21,3±0,2, 22,2±0,2, 24,0±0,2, 26,4±0,2 и 29,3±0,2 градусов 2θ. В других таких вариантах осуществления, четырехводная дикалиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,0±0,2, 11,9±0,2, 12,4±0,2, 13,7±0,2, 15,0±0,2, 16,5±0,2, 17,1±0,2, 20,8±0,2, 21,3±0,2, 22,2±0,2, 24,0±0,2, 26,4±0,2 и 29,3±0,2 градусов 2θ.
В некоторых вариантах осуществления, четырехводная дикалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,0±0,2, 11,9±0,2, 12,4±0,2, 12,6±0,2, 13,7±0,2, 15,0±0,2, 16,5±0,2, 16,7±0,2, 17,1±0,2, 20,7±0,2, 20,8±0,2, 21,3±0,2, 22,2±0,2, 22,4±0,2, 24,0±0,2, 26,4±0,2 и 29,3±0,2 градусов 2θ. В некоторых таких вариантах осуществления, четырехводная дикалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,0±0,2, 11,9±0,2, 12,4±0,2, 12,6±0,2, 13,7±0,2, 15,0±0,2, 16,5±0,2, 16,7±0,2, 17,1±0,2, 20,7±0,2, 20,8±0,2, 21,3±0,2, 22,2±0,2, 22,4±0,2, 24,0±0,2, 26,4±0,2 и 29,3±0,2 градусов 2θ. В других таких вариантах осуществления, четырехводная дикалиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,0±0,2, 11,9±0,2, 12,4±0,2, 12,6±0,2, 13,7±0,2, 15,0±0,2, 16,5±0,2, 16,7±0,2, 17,1±0,2, 20,7±0,2, 20,8±0,2, 21,3±0,2, 22,2±0,2, 22,4±0,2, 24,0±0,2, 26,4±0,2 и 29,3±0,2 градусов 2θ.
В некоторых вариантах осуществления, четырехводная дикалиевая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 27. Величины 2θ для пиков на Фигуре 27 (и их интенсивности) представляют собой следующее: 5,00 (100), 11,86 (34), 12,39 (32), 12,64 (19), 13,70 (23), 15,03 (21), 16,47 (24), 16,66 (24), 17,12 (28), 20,75 (29), 20,81 (33), 21,34 (22), 22,15 (46), 22,38 (31), 24,02 (24), 26,44 (24) и 29,32 (21).
Настоящее изобретение также относится, частично, к способу получения четырехводной дикалиевой соли путем суспендирования соединения IB-L1-1.1 (261,13 мг) в 1,25 мл THF при нагревании примерно до 50°C. Добавляли КОН (1 н, 1,3 мл, 2,2 молярного эквивалента). Твердое вещество полностью растворялось с образованием прозрачного раствора, который естественным путем охлаждался до окржающих температур. Кристаллизация происходила во время медленного процесса испарения.
G9. Кристаллические формы (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида монокалиевой соли.
Настоящее изобретение также относится, частично, к кристаллическим формам монокалиевой соли (E)-N-(4-(3 -трет-бутил-5 -(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида, а именно трехводным и двуводным кристаллическим формам, рассмотренным ниже.
Настоящее изобретение относится, частично, к трехводной монокалиевой соли. Кристаллографические параметры элементарной ячейки трехводной кристаллической монокалиевой соли были определены следующими: a равно 9,0Ǻ, b равно 8,3Ǻ, и c равно 18,6Ǻ (более точно, a равно 9,0393(16)Ǻ, b равно 8,3332(15)Ǻ, и c равно 18,582(3)Ǻ); углы ячейки составляют: α - 80,5°, β - 85,1°, и γ - 80.5° (более точно, α равен 80,511(2)°, β равен 85,134(3)°, и γ равен 80,531(2)°); и объем ячейки составляет 1359Ǻ3 (более точно, 1359,3(4)Ǻ3). Эта соль кристаллизуется в пространственной группе P-1.
В некоторых вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2 и 23,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2 и 23,0±0,2 градусов 2θ. В других таких вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2 и 23,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 13,6±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2, 22,6±0,2 и 23,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 13,6±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2, 22,6±0,2 и 23,0±0,2 градусов 2θ. В других таких вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 13,6±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2, 22,6±0,2 и 23,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 13,6±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2, 22,6±0,2 и 23,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2 и 23,0±0,2 градусов 2θ. В других таких вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 4,8±0,2, 10,8±0,2, 11,3±0,2, 13,4±0,2, 15,3±0,2, 16,9±0,2, 21,2±0,2, 21,7±0,2, 22,1±0,2, 22,5±0,2 и 23,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, трехводная монокалиевая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 28. Величины 2θ для пиков на Фигуре 28 (и их интенсивности) представляют собой следующее: 4,83 (60), 10,79 (100), 11,31 (22), 13,42 (41), 13,59 (18), 15,32 (21), 16,90 (38), 21,24 (22), 21,68 (20), 21,68 (21), 22,15 (22), 22,55 (29), 22,63 (23) и 23,02 (27).
Настоящее изобретение также относится, частично, к способу получения трехводной монокалиевой соли. Ее получают путем суспендирования соединения IB-L1-1.1 (108,81 мг) в 0,4 мл THF при нагревании примерно до 50°C. Добавляли водный раствор КОН (1 н, 0,278 мл, 1,2 молярного эквивалента). Твердое вещество растворялось полностью с образованием прозрачного раствора. В раствор добавляли дополнительно 1,6 мл THF, который затем естественным путем охлаждали до окружающих температур, и наблюдали кристаллизацию. Альтернативно, трехводную монокалиевую соль получали путем суспендирования соединения IB-L1-1.1 (343,89 мг) в 1,0 мл THF при нагревании до 50°C. Добавляли водный КОН (1 н, 0,878 мл, 1,2 молярного эквивалента). Твердое вещество растворялось полностью с образованием прозрачного раствора. К раствору добавляли по каплям этанол до общего объема 4,0 мл. Этот раствор затем охлаждали естественным путем до окружающей температуры и наблюдали кристаллизацию.
Настоящее изобретение относится, частично, к двуводной монокалиевой соли.
В некоторых вариантах осуществления, двуводная монокалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 7,7±0,2, 8,8±0,2, 16,1±0,2 и 19,7±0,2 градусов 2θ. В некоторых таких вариантах осуществления, двуводная монокалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из градусов 2θ.
В некоторых вариантах осуществления, двуводная монокалиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 7,7±0,2, 8,8±0,2, 12,4±0,2, 14,0±0,2, 16,1±0,2, 17,7±0,2, 19,2±0,2, 19,7±0,2, 23,1±0,2 и 29,2±0,2 градусов 2θ. В некоторых таких вариантах осуществления, двуводная монокалиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 7,7±0,2, 8,8±0,2, 12,4±0,2, 14,0±0,2, 16,1±0,2, 17,7+0,2, 19,2±0,2, 19,7±0,2, 23,1±0,2 и 29,2±0,2 градусов 2θ. В других таких вариантах осуществления, двуводная монокалиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 7,7±0,2, 8,8±0,2, 12,4±0,2, 14,0±0,2, 16,1±0,2, 17,7±0,2, 19,2±0,2, 19,7±0,2, 23,1±0,2 и 29,2±0,2 градусов 2θ.
В некоторых вариантах осуществления, двуводная монокалиевая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 29. Величины 2θ для пиков на Фигуре 29 (и их интенсивности) представляют собой следующее: 7,68 (19), 8,83 (100), 12,40 (7), 13,97 (10), 16,12 (25), 17,75 (9), 19,22 (12), 19,73 (40), 23,05 (9) и 29,21 (7).
Настоящее изобретение также относится, частично, к способу получения двуводной монокалиевой соли. Ее получали путем суспендирования трехводной монокалиевой соли в среде с низкой водной активностью, такой как смесь этанол/H2O (50/1 об/об). Альтернативно, двуводную монокалиевую соль получали путем растворения твердого трехводного калия (1,8 г) в 36 мл IPA и 4 мл воды при 80°C. Полученный в результате раствор охлаждали до 55°C за 1 ч. Этот раствор затем кристаллизовали с использованием 7,5 мг кристаллов дигидрата при 55°C и поддерживали при 55°C в течение 1 ч. Затем добавляли гептан (36 мл) на протяжении 3 ч. Реакционную смесь охлаждали до 0°C, и фильтрование давало вещество, содержащее кристаллы как ди-, так и тригидрата. Твердое вещество затем ресуспендировали в 20 мл 10:1 об/об EtOH/H2O при 50°C в течение 3 ч и охлаждали до 25°C за 5 ч. Суспензию затем перемешивали при 25°C в течение дополнительных 3 дней и охлаждали до 0°C за 3 ч и удерживали при этой температуре в течение 2 ч. Полученные в результате кристаллы фильтровали и сушили на воздухе на делительной воронке в течение 1 ч с получением дигидрата. Двуводную монокалиевую соль также получали путем суспендирования смеси кристаллов дигидрата и тригидрата в 10:1 об/об EtOH/H2O при 80°C в течение 2 дней. Содержание калия подтверждали с помощью ионной хроматографии.
G10. Кристаллическая форма 1/7 калиевой соли (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Настоящее изобретение также относится, частично, к кристаллической форме 1/7 калиевой соли (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, 1/7 калиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 7,7±0,2, 8,3±0,2, 10,1±0,2, 10,6±0,2, 11,4±0,2, 12,0±0,2, 13,4±0,2, 15,6±0,2, 16,3±0,2, 16,7±0,2, 17,2±0,2, 18,3±0,2, 18,8±0,2, 19,4±0,2, 19,9±0,2, 20,2±0,2, 20,5±0,2, 21,2±0,2, 22,1±0,2 и 22,9±0,2 градусов 2θ. В некоторых таких вариантах осуществления, 1/7 калиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 7,7±0,2, 8,3±0,2, 10,1±0,2, 10,6±0,2, 11,4±0,2, 12,0±0,2, 13,4±0,2, 15,6±0,2, 16,3±0,2, 16,7±0,2, 17,2±0,2, 18,3±0,2, 18,8±0,2, 19,4±0,2, 19,9±0,2, 20,2±0,2, 20,5±0,2, 21,2±0,2, 22,1±0,2 и 22,9+0,2 градусов 2θ. В других таких вариантах осуществления, 1/7 калиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 7,7±0,2, 8,3±0,2, 10,1±0,2, 10,6±0,2, 11,4+0,2, 12,0±0,2, 13,4±0,2, 15,6±0,2, 16,3±0,2, 16,7+0,2, 17,2±0,2, 18,3±0,2, 18,8±0,2, 19,4±0,2, 19,9±0,2, 20,2±0,2, 20,5±0,2, 21,2±0,2, 22,1±0,2 и 22,9±0,2 градусов 2θ.
В некоторых вариантах осуществления, 1/7 калиевая соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 7,7±0,2, 8,3±0,2, 10,1±0,2, 10,6±0,2, 11,4±0,2, 12,0±0,2, 13,4±0,2, 15,6±0,2, 16,3±0,2, 16,7±0,2, 17,2±0,2, 18,3±0,2, 18,8±0,2, 19,4±0,2, 19,9±0,2, 20,2±0,2, 20,5±0,2, 20,8±0,2, 21,2±0,2, 22,1±0,2, 22,9±0,2, 24,3±0,2, 24,9±0,2 и 25,1±0,2 градусов 2θ. В некоторых таких вариантах осуществления, 1/7 калиевая соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 7,7±0,2, 8,3±0,2, 10,1±0,2, 10,6±0,2, 11,4±0,2, 12,0±0,2, 13,4±0,2, 15,6±0,2, 16,3±0,2, 16,7±0,2, 17,2±0,2, 18,3±0,2, 18,8±0,2, 19,4±0,2, 19,9±0,2, 20,2±0,2, 20,5±0,2, 20,8±0,2, 21,2±0,2, 22,1±0,2, 22,9±0,2, 24,3±0,2, 24,9±0,2 и 25,1±0,2 градусов 2θ. В других таких вариантах осуществления, 1/7 калиевая соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 7,7±0,2, 8,3±0,2, 10,1±0,2, 10,6±0,2, 11,4±0,2, 12,0±0,2, 13,4±0,2, 15,6±0,2, 16,3±0,2, 16,7±0,2, 17,2±0,2, 18,3±0,2, 18,8±0,2, 19,4±0,2, 19,9±0,2, 20,2±0,2, 20,5±0,2, 20,8±0,2, 21,2±0,2, 22,1±0,2, 22,9±0,2, 24,3±0,2, 24,9±0,2 и 25,1±0,2 градусов 2θ.
В некоторых вариантах осуществления, 1/7 калиевая соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 31. Величины 2θ для пиков на Фигуре 31 (и их интенсивности) представляют собой следующее: 7,71 (19), 8,33 (34), 10,10 (100), 10,66 (29), 11,39 (27), 12,04 (22), 13,39 (39), 15,56 (41), 16,27 (62), 16,69 (70), 17,22 (59), 18,31 (18), 18,78 (47), 19,44 (36), 19,89 (28), 20,19 (33), 20,54 (87), 20,80 (33), 21,15 (47), 22,05 (24), 22,82 (67), 24,32 (22), 24,87 (22) и 25,07 (33).
Настоящее изобретение также относится, частично, к способу получения 1/7 калиевой соли. Ее получали путем суспендирования соединения IB-L1-1.1 (2 г) в 6 мл THF при 50°C. Добавляли один молярный эквивалент КОН, растворенного в 4,3 мл воды, и реакционную смесь нагревали до 65°C до растворения всех твердых частиц. Затем раствор охлаждали до окружающих температур за 2 ч и имела место спонтанная кристаллизация. Затем суспензию охлаждали до 5°C и держали при этой температуре в течение 2 ч. Бледно желтые кристаллы фильтровали и сушили на воздухе 24 ч в окружающих условиях. Содержание калия определяли с помощью ионной хроматографии.
G11. Кристаллическая форма четырехводной монодиэтиламинной соли (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Настоящее изобретение также относится, частично, к кристаллической четырехводной монодиэтиламинной соли (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, четырехводная монодиэтиламинная соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 9,5±0,2, 10,0±0,2, 11,8±0,2, 12,1±0,2, 14,4±0,2, 16,8±0,2, 17,6±0,2, 19,8±0,2, 20,8±0,2, 21,4±0,2, 21,8±0,2 и 29,8±0,2 градусов 2θ. В некоторых таких вариантах осуществления, четырехводная монодиэтиламинная соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 9,5±0,2, 10,0±0,2, 11,8±0,2, 12,1±0,2, 14,4±0,2, 16,8±0,2, 17,6±0,2, 19,8±0,2, 20,8±0,2, 21,4±0,2, 21,8±0,2 и 29,8±0,2 градусов 2θ. В других таких вариантах осуществления, четырехводная монодиэтиламинная соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 9,5±0,2, 10,0±0,2, 11,8±0,2, 12,1±0,2, 14,4±0,2, 16,8±0,2, 17,6±0,2, 19,8±0,2. 20,8±0,2, 21,4±0,2, 21,8±0,2 и 29,8±0,2 градусов 2θ.
В некоторых вариантах осуществления, четырехводная монодиэтиламинная соль имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 9,5±0,2, 10,0±0,2, 11,8±0,2, 12,1±0,2, 14,4±0,2, 16,8±0,2, 17,6±0,2, 19,4±0,2, 19,8±0,2, 20,8±0,2, 21,4±0,2, 21,8±0,2, 21,9±0,2 и 29,8±0,2 градусов 2θ. В некоторых таких вариантах осуществления, четырехводная монодиэтиламинная соль имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 9,5±0,2, 10,0±0,2, 11,8±0,2, 12,1±0,2, 14,4±0,2, 16,8±0,2, 17,6±0,2, 19,4±0,2, 19,8±0,2, 20,8±0,2, 21,4±0,2, 21,8±0,2, 21,9±0,2 и 29,8±0,2 градусов 2θ. В других таких вариантах осуществления, четырехводная монодиэтиламинная соль имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 9,5±0,2, 10,0±0,2, 11,8±0,2, 12,1±0,2, 14,4±0,2, 16,8±0,2, 17,6±0,2, 19,4±0,2, 19,8±0,2, 20,8±0,2, 21,4±0,2, 21,8±0,2, 21,9±0,2 и 29,8±0,2 градусов 2θ.
В некоторых вариантах осуществления, четырехводная монодиэтиламинная соль имеет порошковую рентгенограмму, по существу, как показано на Фигуре 32. Величины 2θ для пиков на Фигуре 32 (и их интенсивности) представляют собой следующее: 9,45 (100), 9,97 (31), 11,85 (67), 12,09 (16), 14,38 (22), 16,80 (9), 17,59 (10), 19,39 (8), 19,83 (21), 20,85 (25), 21,37 (12), 21,75 (34), 21,87 (8) и 29,78 (7).
Настоящее изобретение также относится, частично, к способу получения четырехводной монодиэтиламинной соли. Ее получали в водной среде. Соединение IB-L1-1.1 медленно добавляли к 500 мкл 1 М диэтиламина, до тех пор, пока твердое вещество может растворяться в растворе. Затем раствор медленно упаривали при окружающих температурах, и соль кристаллизовалась спустя 2 дня. Альтернативно, четырехводную монодиэтиламинную соль получали путем суспендирования 64,15 мг соединения IB-L1-1.1 в 400 мкл 1M диэтиленамина при нагревании до 50°C. Добавляли около 5 капель THF (~20 мкл). Твердое вещество полностью растворялось после добавления с образованием прозрачного раствора. Этот раствор затем упаривали при окружающей температуре, и соль кристаллизовалась спустя 4 дня. Стехиометрию этой соли подтверждали с помощью 1H ЯМР в растворе.
G12. Кристаллические формы (Е)-К-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1 (2Н)-ил)-2-метоксистирил)фенил)метансулъфонамида (соединение IB-L1-1.1).
Настоящее изобретение также относится, частично, к кристаллическим формам (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.1), а именно к настоящим полиморфам (образцу A, образцу B, образцу C и образцу D) и гидратным (образец AH, образец ВН, образец СН, и образец DH) кристаллическим формам, рассмотренным ниже.
G12A. Настоящие полиморфы IB-L1-1.1.
Настоящее изобретение относится, частично, образцу A кристаллического (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, образец A полиморфа имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,8±0,2, 9,9±0,2, 11,8±0,2, 12,4±0,2, 14,5±0,2, 18,8±0,2, 22,7±0,2 и 29,2±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A полиморфа имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,8±0,2, 9,9±0,2, 11,8±0,2, 12,4±0,2, 14,5±0,2, 18,8±0,2, 22,7±0,2 и 29,2±0,2 градусов 2θ. В других таких вариантах осуществления, образец A полиморфа имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,8±0,2, 9,9±0,2, 11,8±0,2, 12,4±0,2, 14,5±0,2, 18,8±0,2, 22,7±0,2 и 29,2±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец A полиморфа имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,8±0,2, 9,9±0,2, 11,8±0,2, 12,4±0,2, 14,0±0,2, 14,5±0,2, 15,3±0,2, 18,5±0,2, 18,8±0,2, 22,2±0,2, 22,7±0,2, 23,8±0,2, 26,0±0,2 и 29,2±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A полиморфа имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,8±9,2, 9,9±0,2, 11,8±0,2, 12,4±0,2, 14,0±0,2, 14,5±0,2, 15,3±0,2, 18,5±0,2, 18,8±0,2, 22,2±0,2, 22,7±0,2, 23,8±0,2, 26,0±0,2 и 29,2±0,2 градусов 2θ. В других таких вариантах осуществления, образец A полиморфа имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,8±0,2, 9,9±0,2, 11,8±0,2, 12,4±0,2, 14,0±0,2, 14,5±0,2, 15,3±0,2, 18,5±0,2, 18,8±0,2, 22,2±0,2, 22,7±0,2, 23,8±0,2, 26,0±0,2 и 29,2±0,2 градусов 2θ. В некоторых вариантах осуществления, образец A полиморфа имеет порошковую рентгенограмму, по существу, как показано на Фигуре 34. Величины 2θ для пиков на Фигуре 34 (и их интенсивности) представляют собой следующее: 5,85 (28), 9,88 (51), 11,79 (73), 12,38 (56), 14,03 (38), 14,45 (100), 15,27 (29), 18,52 (39), 18.80 (47), 22,24 (40), 22,72 (77), 23,76 (39), 25,98 (22) и 29,21 (64).
Настоящее изобретение также относится, частично, к способу получения образца A полиморфа. Образец A полиморфа получали как описано ниже в Примере F. Настоящее изобретение относится, частично, образцу В кристаллического (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, образец B полиморфа имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 11,5±0,2, 13,3±0,2, 15,4±0,2, 16,4±0,2, 17,1±0,2, 18,6±0,2, 19,4±0,2, 20,4±0,2, 21,6±0,2, 22,4±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец B полиморфа имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 11,5±0,2, 13,3±0,2, 15,4±0,2, 16,4±0,2, 17,1±0,2, 18,6±0,2, 19,4±0,2, 20,4±0,2, 21,6±0,2, 22,4±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец B полиморфа имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 11,5±0,2, 13,3±0,2, 15,4±0,2, 16,4±0,2, 17,1±0,2, 18,6±0,2, 19,4±0,2, 20,4±0,2, 21,6±0,2, 22,4±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ. В некоторых вариантах осуществления, образец B полиморфа имеет порошковую рентгенограмму, по существу, как показано на Фигуре 36. Величины 2θ для пиков на Фигуре 36 (и их интенсивности) представляют собой следующее: 11,52 (71), 13,30 (87), 15,37 (100), 16,42 (60). 17,13 (69), 18,60 (97), 19,37 (56), 20,40 (62), 21,55 (55), 22,41 (39), 23,99 (33), 26,81 (31) и 28,98 (50).
Настоящее изобретение относится, частично, образцу C кристаллического (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, образец C полиморфа имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 7,7±0,2, 10,1±0,2, 10,6±0,2, 12,0±0,2, 13,4±0,2, 16,2±0,2, 19,4±0,2, 20,5±0,2, 21,4±0,2, 22,0±0,2, 22,6±0,2, 24,3±0,2 и 27,6±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец C полиморфа имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 7,7±0,2, 10,1±0,2, 10,6±0,2, 12,0±0,2, 13,4±0,2, 16,2±0,2, 19,4±0,2, 20,5±0,2, 21,4±0,2, 22,0±0,2, 22,6±0,2, 24,3±0,2 и 27,6±0,2 градусов 2θ. В других таких вариантах осуществления, образец C полиморфа имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 7,7±0,2, 10,1±0,2, 10,6±0,2, 12,0±0,2, 13,4±0,2, 16,2±0,2, 19,4±0,2, 20,5±0,2, 21,4±0,2, 22,0±0,2, 22,6±0,2, 24,3±0,2 и 27,6±0,2 градусов 2θ. В некоторых вариантах осуществления, образец C полиморфа имеет порошковую рентгенограмму, по существу, как показано на Фигуре 37. Величины 2θ для пиков на Фигуре 37 (и их интенсивности) представляют собой следующее: 7,69 (27), 10,13 (27), 10,64 (49), 12,01 (31), 13,39 (33), 16,25 (91), 19,44 (46), 20,49 (100), 21,40 (35), 22,03 (37), 22,60 (30), 24,32 (23) и 27,55 (27).
Настоящее изобретение относится, частично, к образцу D кристаллического (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, образец D полиморфа имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,8±0,2, 10,7±0,2, 11,2±0,2, 15,2±0,2, 16,1±0,2, 16,9±0,2, 19,9±0,2, 22,1±0,2, 24,7±0,2 и 26,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец D полиморфа имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,8±0,2, 10,7±0,2, 11,2±0,2, 15,2±0,2, 16,1±0,2, 16,9±0,2, 19,9±0,2, 22,1±0,2, 24,7±0,2 и 26,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец D полиморфа имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,8±0,2, 10,7±0,2, 11,2±0,2, 15,2±0,2, 16,1±0,2, 16,9±0,2, 19,9±0,2, 22,1±0,2, 24,7±0,2 и 26,0±0,2 градусов 2θ. В некоторых вариантах осуществления, образец D полиморфа имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,8±0,2, 10,7±0,2, 11,2±0,2, 15,2±0,2, 16,1±0,2, 16,9±0,2, 17,1±0,2, 19,9±0,2, 20,1±0,2, 22,1±0,2, 24,7±0,2 и 26,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец D полиморфа имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,8±0,2, 10,7±0,2, 11,2±0,2, 15,2±0,2, 16,1±0,2, 16,9±0,2, 17,1±0,2, 19,9±0,2, 20,1±0,2, 22,1±0,2, 24,7±0,2 и 26,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец D полиморфа имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,8±0,2, 10,7±0,2, 11,2±0,2, 15,2±0,2, 16,1±0,2, 16,9±0,2, 17,1±0,2, 19,9±0,2, 20,1±0,2, 22,1±0,2, 24,7±0,2 и 26,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец D полиморфа имеет порошковую рентгенограмму, по существу, как показано на Фигуре 38. Величины 2θ для пиков на Фигуре 38 (и их интенсивности) представляют собой следующее: 5,81 (24), 10,70 (91), 11,23 (60), 15,17 (28), 16,10 (48), 16,89 (100), 17,10 (42), 19,88 (81), 20,12 (100), 22,12 (59), 24,72 (37) и 25,91 (24).
Настоящее изобретение также относится, частично, к способу получения образца B, C и D полиморфов, путем нагревания образца A полиморфа примерно до 160, примерно до 225 и примерно до 268°C, соответственно, используя DSC.
G12B. Гидраты IB-L1-L1.
Настоящее изобретение также относится, частично, к гидратам соединения IB-L1-1.1, а именно к гидратам A, B, C, D и E, рассмотренным ниже. Настоящее изобретение относится, частично, к образцу A гидрата (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидро- пиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида. В некоторых вариантах осуществления, образец A гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 5,1±0,2, 7,9±0,2, 9,5±0,2, 10,3±0,2, 13,7±0,2, 16,5±0,2, 17,1±0,2, 17,5±0,2, 18,8±0,2, 19,2±0,2, 20,7±0,2, 21,3±0,2, 21,6±0,2, 25,8±0,2, 26,8±0,2 и 28,4±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец A гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 5,1±0,2, 7,9±0,2, 9,5±0,2, 10,3±0,2, 13,7±0,2, 16,5±0,2, 17,1±0,2, 17,5±0,2, 18,8±0,2, 19,2±0,2, 20,7±0,2, 21,3±0,2, 21,6±0,2, 25,8±0,2, 26,8±0,2 и 28,4±0,2 градусов 2θ. В других таких вариантах осуществления, образец A гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 5,1±0,2, 7,9±0,2, 9,5±0,2, 10,3±0,2, 13,7±0,2, 16,5±0,2, 17,1±0,2, 17,5±0,2, 18,8±0,2, 19,2±0,2, 20,7±0,2, 21,3±0,2, 21,6±0,2, 25,8±0,2, 26,8±0,2 и 28,4±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец A гидрата имеет порошковую рентгенограмму, по существу, как показано на Фигуре 39. Величины 2θ для пиков на Фигуре 39 (и их интенсивности) представляют собой следующее: 5,13 (13), 7,87 (80), 9,45 (100), 10,29 (60), 13,7 (28), 16,54 (30), 17,07 (17), 17,51 (40), 18,80 (99), 19,18 (74), 20,69 (21), 21,25 (21), 21,63 (23), 25,85 (32), 26,81 (20) и 28,35 (27).
Настоящее изобретение также относится, частично, к способу получения образца A гидрата путем суспендирования образца A полиморфа (рассмотренного выше) в этилацетате.
Восстановленный образец A гидрата содержит ~1 молекулу воды на молекулу соединения IB-L1-1.1.
Настоящее изобретение также относится, частично, к образцу B гидрата (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-пиримидин-1(2Н)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, образец B гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,3±0,2, 7,7±0,2, 10,4±0,2, 12,7±0,2, 13,3±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 18,9±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец B гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,3±0,2, 7,7±0,2, 10,4±0,2, 12,7±0,2, 13,3±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 18,9±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец B гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,3±0,2, 7,7±0,2, 10,4±0,2, 12,7±0,2, 13,3±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 18,9±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец B гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,3±0,2, 7,7±0,2, 10,4±0,2, 12,7±0,2, 13,3±0,2, 13,5±0,2. 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,5±0,2, 18,6±0,2, 18,9±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец B гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,3±0,2, 7,7±0,2, 10,4±0,2, 12,7±0,2, 13,3±0,2, 13,5±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,5±0,2, 18,6±0,2, 18,9±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец B гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,3±0,2, 7,7±0,2, 10,4±0,2, 12,7±0,2, 13,3±0,2, 13,5±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,5±0,2, 18,6±0,2, 18,9±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 24,0±0,2, 26,8±0,2 и 29,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец B гидрата имеет порошковую рентгенограмму, по существу, как показано на Фигуре 41. Величины 2θ для пиков на Фигуре 41(и их интенсивности) представляют собой следующее: 6,31 (7), 7,72 (14), 10,45 (24), 12,67 (26), 13,30 (88), 13,50 (44), 14,89 (70), 15,40 (100), 16,43 (43), 18,46 (47), 18,63 (86), 18,91 (26), 19,42 (33), 22,52 (47), 23,52 (44), 24,02 (20), 26,82 (40) и 28,97 (49).
Настоящее изобретение также относится, частично, к способу получения образца B гидрата путем суспендирования образца A полиморфа (рассмотренного выше) в ацетонитриле/воде (9/1 об/об). Восстановленный образец B гидрата содержит ~0,7 молекул воды на молекулу соединения IB-L 1-1.1.
Настоящее изобретение также относится, частично, к образцу C гидрата (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксистирил)фенил)метансульфонамида.
В некоторых вариантах осуществления, образец C гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 10,5±0,2, 13,3±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 19,0±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 26,9±0,2 и 29,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец C гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 10,5±0,2, 13,3±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 19,0±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 26,9±0,2 и 29,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец C гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 10,5±0,2, 13,3±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 19,0±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 26,9±0,2 и 29,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец C гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 10,5±0,2, 13,3±0,2, 13,5±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 19,0±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 26,9±0,2 и 29,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец C гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 10,5±0,2, 13,3±0,2, 13,5±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 19,0±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 26,9±0,2 и 29,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец C гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 10,5±0,2, 13,3±0,2, 13,5±0,2, 14,9±0,2, 15,4±0,2, 16,4±0,2, 18,6±0,2, 19,0±0,2, 19,4±0,2, 22,5±0,2, 23,5±0,2, 26,9±0,2 и 29,0±0,2 градусов 2θ. В некоторых вариантах осуществления, образец C гидрата имеет порошковую рентгенограмму, по существу, как показано на Фигуре 43.
Величины 2θ для пиков на Фигуре 43 (и их интенсивности) представляют собой следующее: 10,47 (21), 13,31 (56), 13,49 (31), 14,91 (28), 15,40 (86), 16,43 (48), 18,61 (100), 18,96 (20). 19,44 (19), 22,55 (26), 23,54 (39), 26,84 (29) и 28,99 (54).
Настоящее изобретение также относится, частично, к способу получения образца C гидрата путем суспендирования образца A полиморфа (рассмотренного выше) в воде. Восстановленный образец C гидрата содержит ~1 молекулу воды на молекулу соединения IB-L1-1.1
Настоящее изобретение также относится, частично, к образцу D гидрата (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Кристаллографические параметры элементарной ячейки образца D гидратной соли были определены следующими: a равно 17,8Å, b равно 9,6Å, и c равно 27,0Å (более точно, a равно 17,783(2)Å, b равно 9,5651(12)Å, и c равно 27,014(4)Å); угол ячейки составляет: β - 93,3° (более точно, β равен 93,256(2)); и объем ячейки составляет 4588Å3 (более точно, 4587,5(10)Å3). Эта соль кристаллизуется в пространственной группе C2/c.
В некоторых вариантах осуществления, образец D гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,6±0,2, 10,0±0,2, 10,5±0,2, 11,1±0,2, 11,6±0,2, 12,2±0,2, 14,2±0,2, 16,6±0,2, 17,1±0,2, 17,7±0,2, 18,5±0,2, 18,8±0,2, 19,3±0,2, 21,4±0,2, 22,7±0,2, 23,1±0,2, 23,6±0,2, 24,6±0,2, 25,2±0,2, 27,2±0,2, 29,1±0,2 и 31,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец D гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,6±0,2, 10,0±0,2, 10,5±0,2, 11,1±0,2, 11,6±0,2, 12,2±0,2, 14,2±0,2, 16,6±0,2, 17,1±0,2, 17,7±0,2, 18,5±0,2, 18,8±0,2, 19,3±0,2, 21,4±0,2, 22,7±0,2, 23,1±0,2, 23,6±0,2, 24,6±0,2, 25,2±0,2, 27,2±0,2, 29,1±0,2 и 31,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец D гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,6±0,2, 10,0±0,2, 10,5±0,2, 11,1±0,2, 11,6±0,2, 12,2±0,2, 14,2±0,2, 16,6±0,2, 17,1±0,2, 17,7±0,2, 18,5±0,2, 18,8±0,2, 19,3±0,2, 21,4±0,2, 22,7±0,2, 23,1±0,2, 23,6±0,2, 24,6±0,2, 25,2±0,2, 27,2±0,2, 29,1±0,2 и 31,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец D гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,6±0,2, 10,0±0,2, 10,5±0,2, 11,1±0,2, 11,6±0,2, 12,2±0,2, 12,5±0,2, 14,2±0,2, 16,6±0,2, 17,1±0,2, 17,7±0,2, 18,5±0,2, 18,8±0,2, 19,3±0,2, 21,4±0,2, 22,7±0,2, 22,8±0,2, 23,1±0,2, 23,6±0,2, 24,6±0,2, 24,9±0,2, 25,2±0,2, 27,2±0,2, 29,1±0,2 и 31,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец D гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,6±0,2, 10,0±0,2, 10,5±0,2, 11,1±0,2, 11,6±0,2, 12,2±0,2, 12,5±0,2, 14,2±0,2, 16,6±0,2, 17,1±0,2, 17,7±0,2, 18,5±0,2, 18,8±0,2, 19,3±0,2, 21,4±0,2, 22,7±0,2, 22,8±0,2, 23,1±0,2, 23,6±0,2, 24,6±0,2, 24,9±0,2, 25,2±0,2, 27,2±0,2, 29,1±0,2 и 31,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец D гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,6±0,2, 10,0±0,2, 10,5±0,2, 11,1±0,2, 11,6+0,2, 12,2±0,2, 12,5±0,2, 14,2±0,2, 16,6±0,2, 17,1±0,2, 17,7±0,2, 18,5±0,2, 18,8±0,2, 19,3±0,2, 21,4±0,2, 22,7±0,2, 22,8±0,2, 23,1±9,2, 23,6±0,2, 24,6±0,2, 24,9±0,2, 25,2±0,2, 27,2±0,2, 29,1±0,2 и 31,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец D гидрата имеет порошковую рентгенограмму, по существу, как показано на Фигуре 45. Величины 2θ для пиков на Фигуре 45 (и их интенсивности) представляют собой следующее: 6,55 (10), 9,96 (12), 10,51 (37), 11,09 (31), 11,62 (100), 12,24 (44), 12,54 (40), 14,22 (15), 16,62 (68), 17,07 (22), 17,77 (21), 18,52 (82), 18,84 (47), 19,30 (63), 21,45 (34), 22,67 (30), 22,80 (34), 23,08 (20), 23,57 (58), 24,63 (73), 24,88 (26), 25,24 (21), 27,23 (36), 29,06 (41) и 31,04 (21).
Настоящее изобретение также относится, частично, к способу получения образца D гидрата. Он может быть получен путем суспендирования образца A полиморфа (рассмотренного выше) в этаноле. Альтернативно, он может быть получен путем суспендирования соединения IB-L1-1.1 (103,03 мг) в 400 мкл THF при нагревании примерно до 55°C. Добавляли водный NaOH (1M, 264 мкл, 1,2 молярного эквивалента). Твердое вещество растворялось полностью с образованием прозрачного раствора. К этому раствору добавляли этанол (1,6 мл). Раствр оставляли охлаждаться естественным путем до окружающих температур. Кристаллы формировались во время медленного процесса испарения. Хотя, судя по всему, кристаллическая решетка может разместить вплоть до 0,5 молекул воды на молекулу соединения IB-L1-1.1, восстановленный образец D гидрата содержал ~0,2 молекулы воды на молекулу соединения IB-L1-1.1.
Настоящее изобретение также относится, частично, к образцу E гидрата (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Кристаллографические параметры элементарной ячейки образца E гидрата кристаллической динатриевой соли были определены следующими: a равно 9,5Å, b равно 14,5Å и c равно 17,3Å (более точно, a равно 9,462(2)Å, b равно 14,462(3)Å и c равно 17,281(4)Å); углы ячейки составляют: α - 84,9°, β - 80,8° и γ - 81,8° (более точно, α равен 84,863(4)°, β равен 80,760(4)° и γ равен 81,751(4)°); и объем ячейки составляет 2304Å3 (более точно, 2304,4(9)Å3). Эта соль кристаллизуется в пространственной группе P-1.
В некоторых вариантах осуществления, образец E гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,2±0,2, 7,8±0,2, 10,2±0,2, 10,7±0,2, 12,1±0,2, 16,3±0,2, 19,7±0,2, 20,9±0,2, 21,8±0,2, 24,5±0,2 и 28,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец E гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,8±0,2, 10,2±0,2, 10,7±0,2, 12,1±0,2, 16,3±0,2, 19,7±0,2, 20,9±0,2, 21,8±0,2, 24,5±0,2 и 28,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец E гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,8±0,2, 10,2±0,2, 10,7±0,2, 12,1±0,2, 16,3±0,2, 19,7±0,2, 20,9±0,2, 21,8±0,2, 24,5±0,2 и 28,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец E гидрата имеет порошковую рентгенограмму, содержащую один или несколько пиков, выбранных из группы, состоящей из 6,2±0,2, 7,8±0,2, 10,2±0,2, 10,4±0,2, 10,7±0,2, 12,1±0,2, 16,3±0,2, 19,7±0,2, 20,9±0,2, 21,8±0,2, 24,5±0,2 и 28,0±0,2 градусов 2θ. В некоторых таких вариантах осуществления, образец E гидрата имеет порошковую рентгенограмму, содержащую три или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,8±0,2, 10,2±0,2, 10,4±0,2, 10,7±0,2, 12,1±0,2, 16,3±0,2, 19,7±0,2, 20,9±0,2, 21,8±0,2, 24,5±0,2 и 28,0±0,2 градусов 2θ. В других таких вариантах осуществления, образец E гидрата имеет порошковую рентгенограмму, содержащую пять или более пиков, выбранных из группы, состоящей из 6,2±0,2, 7,8±0,2, 10,2±0,2, 10,4±0,2, 10,7±0,2, 12,1±0,2, 16,3±0,2, 19,7±0,2, 20,9±0,2, 21,8±0,2, 24,5±0,2 и 28,0±0,2 градусов 2θ.
В некоторых вариантах осуществления, образец E гидрата имеет порошковую рентгенограмму, по существу, как показано на Фигуре 46. Величины 2θ для пиков на Фигуре 46 (и их интенсивности) представляют собой следующее: 6,19 (6), 7,81 (18), 10,17 (13), 10,40 (14), 10,68 (39), 12,06 (20), 16,29 (78), 19,72 (32), 20,88 (100), 21,77 (27), 24,52 (25) и 28,01 (27).
Настоящее изобретение также относится, частично, к способу получения образца E гидрата. Его получают путем суспендирования соединения IB-L1-1.1 (56,76 мг) в 200 мкл THF при нагревании. Добавляли водный NaOH (1M, 146 мкл, 1,2 молярного эквивалента), который давал прозрачный раствор. К этому раствору добавляли этанол (800 мкл). Этот раствор оставляли охлаждаться естественным путем до окружающих температур. Кристаллы формировались во время процесса медленного испарения. Хотя, судя по всему, кристаллическая решетка может разместить вплоть до одной молекулы воды на молекулу соединения IB-L1-1.1, восстановленный образец E гидрата содержал -0,25 молекул воды на молекулу соединения IB-L1-1.1.
H. Композиции.
Настоящее изобретение также относится, частично, к композициям, содержащим одно или несколько соединений и/или солей по изобретению (в том числе кристаллических соединений и солей рассмотренных выше в разделе G). В некоторых вариантах осуществления, композиции содержат одну или несколько по существу фазово-чистых кристаллических форм (соединений/солей/сольватов/гидратов) рассмотрены выше в разделе G. Эти композиции могут представлять собой фармацевтические композиции.
В некоторых вариантах осуществления, композиции дополнительно содержат один или несколько дополнительных терапевтических агентов. Такие терапевтические агенты могут, но не обязательно должны быть, дополнительными ингибиторами HCV.
Предпочтительная композиция зависит от способа введения и обычно содержит один или несколько общепринятых фармацевтически приемлемых носителей, адъювантов и/или наполнителей (все вместе называются «эксципиентами»). Технология получения лекарственных средств в основном рассмотрена, например. Hoover, J., Remington's Pharmaceutical Sciences (Mack Publishing Co., 1975) и Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems (Lippincott Williams & Wilkins, 2005).
Твердые лекарственные формы для перорального введения включают, например, капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах соединения или соли обычно объединяют с одним или несколькими эксципиентами. При введении per os, соединения или соли могут быть смешаны, например, с лактозой, сахарозой, порошкообразным крахмалом, сложными эфирами целлюлозы и алкановых кислот, сложными алкилэфирами целлюлозы, тальком, стеариновой кислотой, стеаратом магния, оксидом магния, натриевыми и кальциевыми солями фосфорной и серной кислот, желатином, акациевой камедью, альгинатом натрия, поливинилпирролидоном и/или поливиниловым спиртом, а затем таблетированы или инкапсулированы для удобства применения. Такие капсулы или таблетки могут содержать составы с контролируемым высвобождением, которое может обеспечиваться, например, дисперсией соединения или соли в гидроксипропилметилцеллюлозе. В случае капсул, таблеток и пилюль, лекарственные формы также могут содержать буферные агенты, такие как цитрат натрия или карбонат или бикарбонат магния или кальция. Таблетки и пилюли дополнительно могут быть получены с кишечнорастворимыми покрытиями.
Жидкие лекарственные формы для перорального введения включают, например, фармацевтически приемлемые эмульсии (в том числе, как эмульсии масло-в-воде, так и эмульсии вода-в-масле), растворы (в том числе, как водные растворы, так и неводные растворы), суспензии (в том числе, как водные, так и неводные суспензии), сиропы и эликсиры, содержащие инертные разбавители, обычно применяемые в этой области (например, воду). Такие композиции также могут содержать, например, увлажняющие, эмульгирующие, суспендирующие, вкусо-ароматические (например, подстастители) и/или ароматизирующие добавки.
Парентеральное введение включает подкожные инъекции, внутривенные инъекции, внутримышечные инъекции, внутригрудинные инъекции и инфузию. Инъецируемые препараты (например, стерильные инъецируемые водные или масляные суспензии) могут быть получены в соответствии с известным уровнем техники, используя подходящие диспергирующие, увлажняющие агенты и/или суспендирующие агенты. Подходящие наполнители и растворители включают, например, воду 1,3-бутандиол, раствор Рингера, изотонический раствор хлорида натрия, легкие нелетучие масла (например, синтетические моно- или диглицериды), жирные кислоты (например, олеиновую кислоту), диметилацетамид, поверхностно-активные вещества (например, ионные и неионные детергенты), и/или полиэтиленгликоли.
Составы для парентерального введения, например, могут быть получены из стерильных порошков или гранул, содержащих один или несколько из упомянутых эксципиентов для применения в составах для перорального применения. Соединение или соль по настоящему изобретению можно растворить в воде, полиэтиленгликоле, пропиленгликоле, этаноле, кукурузном масле, хлопковом масле, арахисовом масле, кунжутном масле, бензиловом спирте, хлориде натрия и/или различных буферах. При необходимости, pH можно установить с использованием подходящей кислоты, основания или буфера.
Суппозитории для ректального введения могут быть получены, например, путем перемешивания соединения или соли по настоящему изобретению с подходящим нераздражающим эксципиентом, который является твердым при обычных температурах, но жидким при ректальной температуре, и будет, следовательно, плавиться в прямой кишке с высвобождением лекарственного средства. Подходящие эксципиенты включают, например, масло какао, синтетические моно-, ди-, или триглицериды, жирные кислоты и/или полиэтиленгликоли.
Местное применение включает использование чрескожного введения, такого как чрескожные пластыри или устройства для ионтофореза.
Также могут быть использованы другие эксципиенты и способы введения, известные в фармацевтической области.
Заявителями было установлено, что некоторые соединения I-L1, в которых R6 и фенилурацил находятся в транс-положении отностительно двойной связи, при нахождении в растворе, имеют тенденцию к превращению в соответствующий цис-изомер под воздействием света; следовательно, может быть желательным хранить такие растворы в условиях, при которых уменьшается воздействие света (например, в сосуде из желтого стекла или в темном месте).
Предпочтительная суммарная суточная доза соединения или соли (введенная в однократной или дробных дозах) обычно составляет примерно от 0,001 примерно до 100 мг/кг, более предпочтительно, примерно от 0,001 примерно до 30 мг/кг, и еще более предпочтительно примерно от 0,01 примерно до 10 мг/кг (т.е., мг соединения или соли на кг массы тела). Единичная доза композиций может содержать такие количества или их доли для получения суточной дозы. Во многих случаях, введение соединения или соли будет повторяться большое количество раз. Многократные суточные дозы обычно могут быть использованы для повышения общей суточной дозы, при необходимости.
Факторы, влияющие на предпочтительный режим дозирования, включают в себя тип, возраст, массу тела, пол, рацион питания и состояние пациента; тяжесть патологического состояния; путь введения; фармакологические факторы, такие как активность, эффективность, фармакокинетический и токсикологический профили конкретного применяемого соединения или соли; используется ли система доставки лекарственного средства; и вводится ли соединение или соль как часть комбинации лекарственных средств. Таким образом, фактически используемый режим дозирования может варьировать в широких пределах, и, следовательно, может быть получен из предпочтительного режима дозирования, изложенного выше.
I. Наборы.
Настоящее изобретение также относится, частично, к набору, содержащему одно или несколько соединений и/или солей по изобретению. Набор необязательно может содержать один или несколько дополнительных терапевтических агентов и/или инструкции, например, по применению этого набора.
J. Способы применения.
Настоящее изобретение также относится, частично, к способу ингибирования репликации РНК-вируса. Этот способ включает в себя воздействие на вирус одним или несколькими соединениями и/или солями по настоящему изобретению. В некоторых вариантах осуществления, репликацию РНК-вируса ингибируют in vitro. В других вариантах осуществления, репликацию РНК-вируса ингибируют in vivo. В некоторых вариантах осуществления, РНК-вирус, репликацию которого ингибируют, представляет собой односпиральный положительно полярный РНК-вирус.В некоторых таких вариантах осуществления, РНК-вирус, репликацию которого подавляют, предсталяет собой вирус семейства Flaviviridae. В некоторых таких вариантах осуществления, РНК-вирус, репликацию которого подавляют, представляет собой HCV.
Настоящее изобретение также относится, в части, к способу ингибирования РНК полимеразы HCV. Этот способ включает в себя воздействие на полимеразу одним или несколькими соединениями и/или солями по настоящему изобретению. В некоторых вариантах осуществления, активность РНК полимеразы HCV ингибируют in vitro. В других вариантах осуществления, активность РНК полимеразы HCV ингибируют in vivo. Термин «ингибирование» означает снижение уровня репликации РНК-вируса/активности полимеразы HCV либо in vitro, либо in vivo. Например, если соединение/соль по настоящему изобретению снижает уровень репликации РНК-вируса по меньшей мере примерно на 10% по сравнению с уровнем репликации РНК-вируса до воздействия на вирус соединением/солью, тогда соединение/соль ингибирует репликацию РНК вируса. В некоторых вариантах осуществления, соединение/соль может ингибировать репликацию РНК-вируса по меньшей мере примерно на 20%, по меньшей мере примерно на 30%, по меньшей мере примерно на 40%, по меньшей мере примерно на 50%, по меньшей мере примерно на 60%, по меньшей мере примерно на 70%, по меньшей мере примерно на 80%, по меньшей мере примерно на 90%, или по меньшей мере примерно на 95%.
Настоящее изобретение относится, частично, к способу лечения заболевания, которое можно вылечить путем ингибирования РНК полимеразы HCV. Таким образом, настоящее изобретение также относится, частично, к способу лечения гепатита C у животного, нуждающегося в таком лечении. Эти способы включают в себя введение животногому одного или нескольких соединений и/или солей по настоящему изобретению, и, необязательно, один или несколько дополнительных фармацевтических агентов. В некоторых вариантах осуществления, животному вводят терапевтически эффективное количество соединения(й) и/или соли(солей). «Лечение» означает облегчение, подавление, эрадикацию, профилактику, снижение риска и/или задержку появления заболевания, подвергаемого лечению. Заявители особенно имеют в виду, что термин «лечение» охватывает введение соединений и/или солей по настоящему изобретению, HCV-негативному пациенту, который является кандидатом на трансплантацию органа. Способы лечения в частности подходят для применения у людей, но могут быть использованы у других животных, в частности, млекопитающих. «Терапевтически-эффективным количеством» или «эффективным количеством» является количество, при котором будет достигаться цель лечения целевого состояния.
В некоторых вариантах осуществления, эти способы включают в себя комбинированную терапию, где соединение(я) и/или соль(и) по изобретению вводят совместно со вторым (или даже третьим, четвертым, и т.д.) соединением, например, таким как другое терапевтическое средство, используемое для лечения гепатита C (например, интерферона или комбинации интерферон/рибавирин, или ингибитора HCV, например, такого, как ингибитор HCV полимеразы или ингибитор HCV протеазы). Соединение(я) и/или соль(и) по настоящему изобретению также могут быть введены совместно с терапевтическими средствами, отличными от терапевтических средств, используемых для лечения гепатита C (например, средствами против ВИЧ). В этих вариантах осуществления совместного введения, соединение(я) и/или соль(и) по настоящему изобретению и второе, и т.д., терапевтическое средство(а) можно вводить по существу одновременно (например, в пределах примерно 5 минут друг от друга), последовательно, или и тем, и другим способом. Предполагается, что такие комбинированные способы лечения могут включать введение одного терапевтического средства большое количество раз между введениями другого. Период времени между введением каждого средства может находиться в диапазоне от нескольких секунд (или менее) до нескольких часов или дней, и будет зависеть, например, от свойств каждой композиции и активного ингредиента (например, эффективности, растворимости, биодоступности, периода полужизни и кинетического профиля), а также состояния пациента. Соединение(я) и/или соль(и) по настоящему изобретению и второе, и т.д. терапевтическое средство можно также вводить в одной композиции.
Настоящее изобретение также относится, частично, к применению одного или нескольких соединений и/или солей по настоящему изобретению, и, необязательно, одного или нескольких дополнительных терапевтических средств для получения лекарственного средства. В некоторых вариантах осуществления, лекарственное средство предназначено для совместного введения с одним или несколькими дополнительными терапевтическими агентами.
В некоторых вариантах осуществления, лекарственное средство предназначено для ингибирования репликации РНК-вируса.
В некоторых вариантах осуществления, лекарственное средство предназначено для лечения гепатита C.
Настоящее изобретение также отностится, частично, к одному или нескольким соединениям и/или солям по настоящему изобретению, и, необязательно, одному или нескольким дополнительным терапевтическим средствам, для использования в качестве лекарственного средства. В некоторых вариантах осуществления, лекарственное средство предназначено для ингибирования репликации РНК-вируса. В других вариантах осуществления, лекарственное средство предназначено для лечения гепатита C.
K. Промежуточные соединения.
Настоящее изобретение также относится, частично, к промежуточным соединениям, которые соответствуют по структуре формуле II, которые могут быть использованы для получения соединений формулы I (и их солей), (хотя некоторые промежуточные соединения также могут быть использованы, также как и соединения формулы I, в качестве ингибиторов HCV, и специалист в данной области может определить такую способность соединений формулы II путем использования, например, способов, рассмотренных ниже):
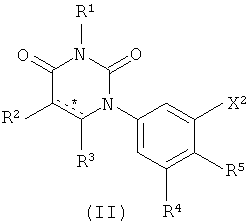 .
.
В формуле II:
, R1, R2, R3, R4 и R5 рассмотрены выше для соединений формулы I; и
X2 представляет собой гало.
Различные варианты воплощения для , R1, R2, R3, R4 и R5 (а также их сочетаний) рассмотренные выше, применимы к соединениям формулы II. Что касается X2, в некоторых вариантах осуществления, X2 выбран из группы, состоящей из хлора, брома и йода. В других вариантах осуществления, X2 выбран из группы, состоящей из хлора и брома. Еще в одних вариантах осуществления, X2 выбран из группы, состоящей из хлора и йода. Еще в одних вариантах осуществления, X2 выбран из группы, состоящей из йода и брома. В дополнительных вариантах осуществления, X2 представляет собой фтор. Еще в дополнительных вариантах осуществления, X2 представляет собой хлор. Еще в дополнительных вариантах осуществления X2 представляет собой бром. И еще в одних вариантах осущесвтвления, X2 представляет собой йод.
Различные варианты воплощения для , R1, R2, R3, R4, R5 и X2 рассмотренные выше, могут быть объединены с получением различных вариантов воплощения соединений формулы II, и всех вариантов воплощения соединений формулы II, полученные таким образом соединения входят в объем предложенного Заявителями изобретения. Некоторые варианты воплощения соединений (и их солей) формулы II, приводимые в качестве примера, рассмотрены ниже.
В некоторых вариантах осуществления, соединения формулы II соответствуют по структуре формуле IIA:
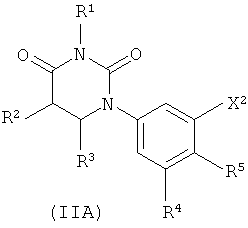 .
.
В других вариантах осуществления, соединения формулы II соответствуют по структуре формуле IIB:
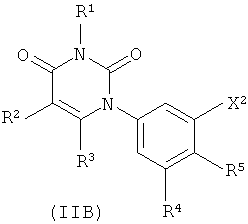 .
.
В некоторых вариантах воплощения соединений формулы II:
R1 выбран из группы, состоящей из водорода, метила и азот-защищающей группы;
R2 выбран из группы, состоящей из водорода и галогена;
R3 выбран из группы, состоящей из водорода и галогена;
R4 выбран из группы, состоящей из C1-C4-алкила, C3-C6-карбоциклила и 5-6-членного гетероциклила, где:
(a) C1-C4-алкил необязательно замещен заместителями, вплоть до трех, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси и триметилсилила, и
(b) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, гало и алкилсульфониламино;
R5 выбран из группы, состоящей из водорода, гидрокси, алкилокси и гало; и
X2 выбран из группы, состоящей из хлора, брома и йода.
В некоторых вариантах воплощения соединений формулы II:
представляет собой двойную углерод-углеродную связь;
R1 представляет собой водород;
R2 выбран из группы, состоящей из водорода и галогена;
R3 представляет собой водород;
R4 представляет собой трет-бутил;
R5 выбран из группы, состоящей из водорода, гидрокси и метокси; и
X2 выбран из группы, состоящей из брома и йода.
В некоторых вариантах воплощения соединений формулы II:
R1 выбран из группы, состоящей из водорода и метила;
R2 выбран из группы, состоящей из водорода и метила;
R3 выбран из группы, состоящей из водорода и метила;
R4 представляет собой трет-бутил;
R5 выбран из группы, состоящей из гидрокси и метокси; и
X2 выбран из группы, состоящей из хлора, брома и йода.
В некоторых вариантах воплощения соединений формулы II:
представляет собой двойную углерод-углеродную связь;
R1 представляет собой водород;
R2 представляет собой водород;
R3 представляет собой водород;
R4 представляет собой трет-бутил;
R5 выбран из группы, состоящей из гидрокси и метокси; и
X2 выбран из группы, состоящей из хлора, брома и йода.
В некоторых вариантах осуществления, соединение формулы II выбрано из группы, состоящей из
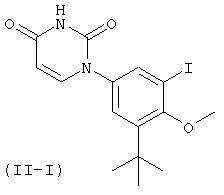 ,
, 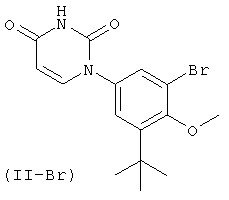 , и
, и 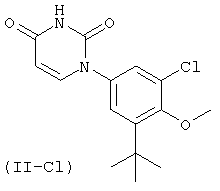 .
.
Приведенное ниже описание предоставляет инструкции по получению промежуточных соединений формулы II (и их солей).
L. Исходные соединения.
Настоящее изобретение также относится, частично, к исходным соединениям, которые соответствуют по структуре формуле III, которые могут быть использованы для получения соединений формул II и I (и их солей):
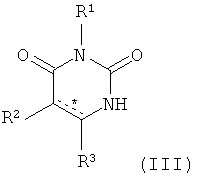 .
.
В формуле III, , R1, R2, и R3 рассмотрены выше для соединений формулы I и II. Различные варианты воплощения для , R1, R2, и R3 (а также их сочетаний) рассмотренные выше, применимы к соединениям формулы III. Различные варианты воплощения для , R1, R2, и R3 рассмотрены выше и могут быть объединены с получением различных вариантов воплощения соединений формулы III, и все варианты воплощения соединений формулы III полученные таким образом, входят в объем представленного Заявителями изобретения. Некоторые варианты воплощения соединений (и их солей) формулы III, приведенные в качестве примера, рассмотрены ниже.
В некоторых вариантах воплощения соединений формулы III:
R1 выбран из группы, состоящей из водорода, метила и азот-защищающей группы;
R2 выбран из группы, состоящей из водорода и галогена; и
R3 выбран из группы, состоящей из водорода и галогена.
В некоторых вариантах воплощения соединений формулы III:
представляет собой двойную углерод-углеродную связь;
R1 выбран из группы, состоящей из водорода;
R2 выбран из группы, состоящей из водорода и галогена; и
R3 выбран из группы, состоящей из водорода.
В некоторых вариантах воплощения соединений формулы III:
R1 выбран из группы, состоящей из водорода и метила;
R2 выбран из группы, состоящей из водорода и метила; и
R3 выбран из группы, состоящей из водорода и метила.
В некоторых вариантах осуществления, соединение формулы III представляет собой урацил.
Настоящее изобретение также относится, частично, к исходным соединениям, которые соответствуют по структуре формуле IV, которые могут быть использованы для получения соединений формул II и I (и их солей):
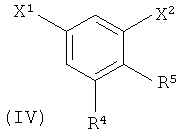 .
.
В формуле IV:
R4, R5, и X2 рассмотрены выше для соединений формулы I и II; и
X1 представляет собой гало.
Различные варианты воплощения для R4, R5, и X2 (а также их сочетаний), рассмотренные выше, применимы для соединений формулы IV. Что касается X1 в некоторых вариантах осуществления, X1 выбран из группы, состоящей из хлора, брома и йода. В других вариантах осуществления, X1 выбран из группы, состоящей из хлора и брома. Еще в одних вариантах осуществления, X1 выбран из группы, состоящей из хлора и йода. В других вариантах осуществления, X1 выбран из группы, состоящей из йода и брома. В дополнительных вариантах осуществления, X1 представляет собой фтор. Еще в дополнительных вариантах осуществления, X1 представляет собой хлор. Еще в дополнительных вариантах осуществления, X1 представляет собой бром. И еще в дополнительных вариантах осуществления, X1 представляет собой йод. Что касается X1 и X2, в некоторых вариантах осуществления, X1 и X2 являются идентичными.
Различные варианты воплощения для R4, R5, X1, и X2 рассмотренные выше, могут быть объединены с получением различных вариантов воплощения соединений формулы IV, и все варианты воплощения соединений формулы III, полученные таким образом, входят в объем представленного Заявителями изобретения. Некоторые варианты воплощения соединений формулы IV (и их солей), приведенные в качестве примера, рассмотрены ниже.
В некоторых вариантах воплощения соединений формулы IV:
R4 выбран из группы, состоящей из C1-C4-алкила, C3-C6-карбоциклила и 5-6-членного гетероциклила, где:
(a) C1-C4-алкил необязательно замещен заместителями, вплоть до трех, независимо выбранными из группы, состоящей из гало, оксо, гидрокси, алкилокси и триметилсилила, и
(b) C3-C6-карбоциклил и 5-6-членный гетероциклил необязательно замещены одним или несколькими заместителями, независимо выбранными из группы, состоящей из алкила, гало и алкилсульфониламино;
R5 выбран из группы, состоящей из водорода, гидрокси и алкилокси;
X1 выбран из группы, состоящей из хлора, брома и йода; и
X2 выбран из группы, состоящей из хлора, брома и йода.
В некоторых вариантах воплощения соединений формулы IV:
R4 выбран из группы, состоящей из трет-бутила;
R5 выбран из группы, состоящей из водорода, гидрокси и метокси;
X1 выбран из группы, состоящей из брома и йода; и
X2 выбран из группы, состоящей из брома и йода.
В некоторых вариантах воплощения соединений формулы IV:
R4 выбран из группы, состоящей из трет-бутила;
R5 выбран из группы, состоящей из гидрокси и метокси;
X1 выбран из группы, состоящей из хлора, брома и йода; и
X2 выбран из группы, состоящей из хлора, брома и йода.
В некоторых вариантах воплощения соединений формулы IV:
R4 представляет собой трет-бутил;
R5 выбран из группы, состоящей из гидрокси и метокси;
X1 выбран из группы, состоящей из хлора, брома и йода; и
X2 выбран из группы, состоящей из хлора, брома и йода.
В некоторых вариантах осуществления, соединение формулы IV выбрано из группы, состоящей из
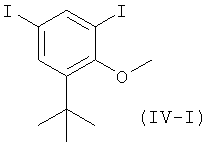 ,
, 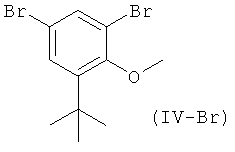 , и
, и 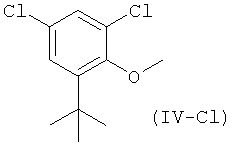 .
.
Приведенное ниже описание предоставляет инструкции по получению исходных соединений формулы IV (и их солей).
L. Способы получения.
Настоящее изобретение также относится, в частности, к способу получения соединений формулы II. Способ включает реакцию соединения формулы III с соединением формулы IV в присутствии (i) в качестве катализатора соли меди (1) и (ii) азотсодержащего гетероарильного лиганда:
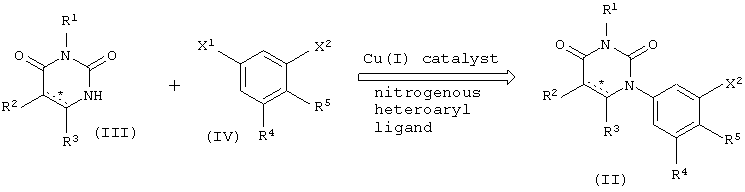
(Cu(I) catalyst - катализатор Cu(I), nitrogenous heteroaryl ligand - азотсодержащий гетероарильный лиганд).
В указанном выше процессе R1, R2, R3, R4, R5, X1, и X2 имеют указанные выше значения.
Заявители установили, что процесс в основном заключается в замещении водорода у атома азота N1 урацилового производного III с получением промежуточного соединения II. Когда X2 промежуточного соединения II представляет собой хлор, бром или иод, тогда соединение II пригодно для последующей реакции (например, реакции сочетания Сузуки с соответствующей бороновой кислотой или боронатным эфиром) с получением соединения формулы I. Другими словами, когда X2 в промежуточном соединении II представляет собой хлор, бром или йод, то указанный выше процесс пригоден для получения соединений формулы I.
В ряде воплощений изобретения соединение III представляет собой урацил и соединение IV соответствует по структуре соединению, выбранному из группы, состоящей из соединения IV-I, IV-Br и IV-Cl, при этом в случае соединений IV-I и IV-Br выход обычно лучше, чем в случае соединения IV-Cl.
Пригодные медные катализаторы Cu(1) включают, например, CuI, CuBr, CuCl, Cu2O и CH3C(O)OCu. В ряде воплощений изобретения катализатор выбирают из группы, состоящей из CuI и CuBr.
В одном из таких воплощений изобретения катализатор представляет собой CuI. В другом из таких воплощений изобретения катализатор представляет собой CuBr. В ряде воплощений изобретения процесс ведут в присутствии основания. В одном из таких воплощений изобретения основание является неорганическим основанием. Пригодные неорганические основания включают, например, соли натрия, калия и цезия (например, K2CO3, K3PO4, Cs2CO3, Na2CO3). В ряде воплощений изобретения основание выбирают из группы, состоящей из соли калия или соли цезия. В одном из таких воплощений изобретения соль выбирают из группы, состоящей из K3PO4 и Cs2CO3. В ряде воплощений изобретения основание включает соль калия. В одном из таких воплощений изобретения соль калия представляет собой K2CO3. В другом из таких воплощений соль калия представляет собой K3PO4. В ряде воплощений изобретения основание включает соль цезия. В одном из таких воплощений соль цезия представляет собой Cs2CO3.
Обычно процесс ведут в присутствии растворителя. Пригодные растворители включают, например, диметилсульфоксид (ДМСО), диметилформамид (ДФА) и ацетонитрил (MeCN). В некоторых воплощениях изобретения растворитель представляет собой ДМСО.
Обычно процесс ведут при температуре приблизительно от 40 приблизительно до 130°C.
В одних воплощениях изобретения азотсодержащий гетероарильный лиганд включает 8-гидроксихинолин. В других воплощениях изобретения лиганд включает 2-(2-пиридил)-бензимидазол. Еще в других воплощениях изобретения лиганд включает пиколинамидное соединение, соответствующее по структуре формуле V:
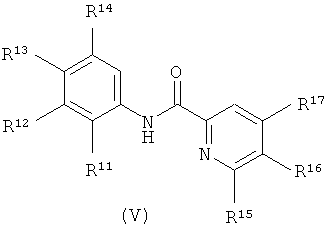 .
.
В формуле V R11, R12, R13, R14, R15, R16 и R17 независимо выбраны из группы, состоящей из водорода, C1-4-перфторалкила, C1-4-алкокси, C1-4-галоалкила, хлора или циано. В некоторых воплощениях изобретения R11, R12, R13, R14, R15, R16 и R17 независимо выбраны из группы, состоящей из водорода, метила, метокси, трифторметила, хлора или циано. В одних воплощениях изобретения лиганд фомулы V включает N-(4-цианофеил)пиколинамид. В других воплощениях изобретения лиганд фомулы V включает N-(2-цианофенил)пиколинамид.
В ряде воплощений изобретения способ включает (a) получение соединения формулы IV; и (b) реакцию соединения формулы III с соединением формулы IV в присутствии (i) в качестве катализатора соли меди (1) и (ii) азотсодержащего гетероарильного лиганда необязательно в присутствии неорганического основания.
Соединение формулы IV-I может быть получено, например, превращением 2-трет-бутилфенола в 2-трет-бутил-4,6-дийодфенол (например, путем его взаимодействия с NaI и NaOCl), и затем превращением 2-трет-бутил-4,6-дииодфенола в 1-трет-бутил-3,5-дийод-2-анизол (например, обработкой его CH3I в присутствии основания, такого как, например, NaOH).
Соединение формулы IV-Br может быть получено, например, превращением 2-трет-бутилфенола в 2,4-дибром-6-трет-бутилфенол (например, путем его взаимодействия с 1,3-дибром-5,5-диметилимидазолидин-2,4-дионом) и затем превращением 2,4-дибром-6-трет-бутилфенола в 1,5-дибром-3-трет-бутил-2-анизол (например, его обработкой СНз1 в присутствии KOtBu).
Дополнительная информация о получении соединений формулы I и II (и их солей) имеется в приведенных ниже общих пояснениях и/или в приведенных ниже конкретных примерах синтеза. В приведенных ниже общих пояснениях R1, R2, R3, R4, R5, L, RA, RB, RC, RD, R6, RE, RF, RG, RH, RI, RJ, RK, X1 и X2 имеют, если не указано иначе, указанные выше значения.
Схема 1
Соединение (1-1), где R7 представляет, например, водород или -CO2Me, и R8 представляет, например, водород или т-бутил, может быть обработано азотной кислотой в растворителе, таком как, например, уксусная кислота или вода, при температуре приблизительно от 0 приблизительно до 35°C в течение примерно от 1 до примерно 5 часов с получением соединения (1-2). Затем соединение (1-2) может быть восстановлено в известных для специалиста условиях с получением соответствующего анилина (1-3). Типичные условия для такого восстановления включают использование водорода при давлении примерно от 1 до примерно 5 атмосфер в присутствии катализатора, такого как, например, палладий или платина на угле, в растворителе, таком как, например, тетрагидрофуран, этилацетат, этанол или гексан при температуре окружающей среды или около нее в течение примерно 1-12 часов. В зависимости от наличия функциональных групп, более оптимальной может быть иная процедура восстановления, например, использование железных опилок в присутствии слабой кислоты, такой как, например, хлорид аммония или разбавленная соляная кислота, при температуре дефлегмации в смеси растворителей, содержащей, например, метанол, воду и/или тетрагидрофуран, в течение, примерно, 1-12 часов. Другие условия восстановления включают использование борогидрида натрия в смеси растворителей, такой как, например, вода и тетрагидрофуран. Еще другие условия восстановления включают использование хлорида олова (II) в присутствии соляной кислоты в таких растворителях, как, например, вода и метанол или их смесь.
Соединение (1-2) перед восстановлением может быть модифицировано. Например, обработка соединения (1-2), где R7 представляет собой водород, хлористым йодом в смеси метанол - вода при температуре окружающей среды в течение примерно 8-24 часов дает соединение (1-4), где X1 представляет собой йод. Альтернативно, соединение (1-2) может быть обработано пербромидом пиридингидробромида в растворителе, таком как, например, уксусная кислота при температуре окружающей среды или вблизи нее в течение примерно 2-16 часов с получением соединения (1-4), где X1 представляет бром. Модификации могут быть осуществлены по фенольному фрагменту соединения (1-4). Например, фенол может быть алкилирован галоидными алкилами (например, метил йодидом), алкилсульфатами (например, метилсульфатом), галоидными алкенилами (например, аллилбромидом), галоидными алкинилами (например, пропаргилбромидом) в присутствии основания, такого как, например, карбонат калия в ацетоне, гидрид натрия в диметилформамиде или т-бутилат калия в тетрагидрофуране, при температуре от примерно 0 до примерно 35°C в течение примерно 1-24 часов с получением соединения (1-5), где R9 представляет, например, алкил, алкенил или алкинил. Альтернативно, алкилирование можно осуществить, используя такой реагент, как (триметилсилил) диазометан в растворителе, таком как, например, метанол или т-бутил метиловый эфир или их смесь, в запаянной трубке при комнатной температуре или около нее в течение примерно 8-24 часов. Соединение (1-5) может быть в дальнейшем восстановлено до соединения (1-6), используя железные опилки или хлорид олова (II) в описанных выше условиях. Альтернативный процесс восстановления использует гидрогенизацию при давлении приблизительно 1 атмосфера в присутствии катализатора, такого как 5% платина на сульфиде углерода, в растворителе, таком как метанол. Защита полученного анилинового соединения (1-6), например, т-бутилкарбаматом может быть осуществлена путем его обработки ди-трет-бутилдикарбонатом в растворителе, таком как, например, тетрагидрофуран или диоксан при температуре приблизительно от 50 до приблизительно 65°C в течение примерно 1 - 8 часов с получением соединения (1-7).
Модификации могут быть осуществлены по фенольному фрагменту соединения (1-2). Специалист в данной области может осуществить алкилирование фенольной части соединения (1-2) используя, например, описанные выше условия с получением соединения (1-8). Соединение (1-8) превращают в соединение (1-9) используя, например, одно или более из описанных выше подходящих условий.
Другая модификация фенольной группы в соединении (1-2) заключается в сульфонилировании до конечного соединения (1-8), где R9 представляет алкилсульфонил, карбоциклилсульфонил или галоид алкилсульфонил. Такое соединение может быть получено обработкой соединения (1-2) сульфохлоридом, таким как, например, метансульфохлорид, циклогексансульфохлорид, бензолсульфохлорид или 3-хлорпропансульфохлорид в присутствии основания, такого как, например, триэтиламин, диизопропилэтиламин или пиридин, в растворителе, таком как, например, дихлорметан при температуре окружающей среды или около нее в течение примерно от 1 до 24 часов. Специалист в данной области может осуществить превращение соединения (1-8) в соединение (1-9), используя подходящие условия восстановления.
Схема 2
Анилин (2-4) может быть получен, используя перегруппировку Курциуса. Для этого соединение (2-1), где R4 не является амино, обрабатывают каталитическим количеством диметилформамида при нагревании с обратным холодильником в тионилхлориде в течение примерно от 1 примерно до 4 часов с получением хлорангидрида (2-2). Соединение (2-2) также может быть получено обработкой при температуре флегмы тионилхлоридом в растворителях, таких как, например, хлороформ или толуол. Соединение (2-2) можно также подвергать взаимодействию с водным раствором азида натрия в среде растворителя, такого как, например, ацетон, в течение примерно 1-8 часов с получением ацилазида (2-3). Затем соединение (2-3) может быть подвергнуто перегруппировке Курциуса в растворителях, таких как диоксан или толуол, при нагревании с обратным холодильником. Промежуточный изоцианат подвергают гидролизу водной кислотой, такой как разбавленная соляная кислота в среде растворителя, такого как диметоксиэтан, с получением соединения (2-4).
Схема 3
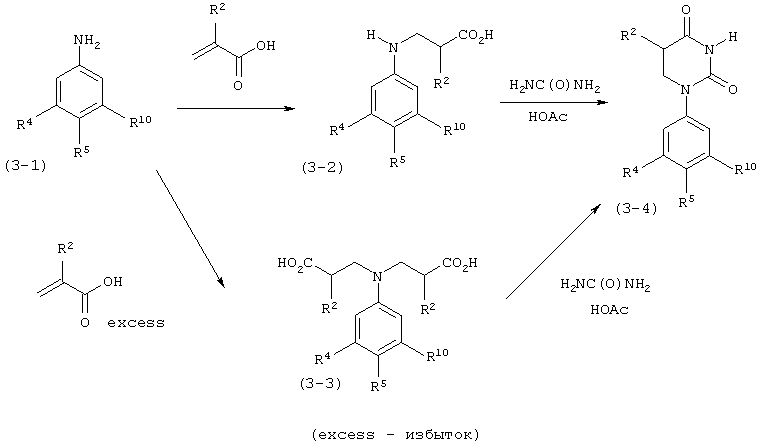
Соединение (3-1), где R10 представляет, например, водород, бром, йод или -CO2Me, может быть обработано акриловой кислотой при температуре окружающей среды или около нее в среде растворителя, такого как, например, толуол с последующим нагреванием с обратным холодильником в течение примерно от 15 примерно до 48 часов с получением соединения (3-2). При использовании избытка акриловой кислоты получают соединение (3-3). Соединение (3-2) или (3-3) можно обработать мочевиной в растворителе, таком как, например, уксусная кислота приблизительно при 100-120°C в течение примерно от 2 примерно до 48 часов с получением соединения (3-4).
Схема 4
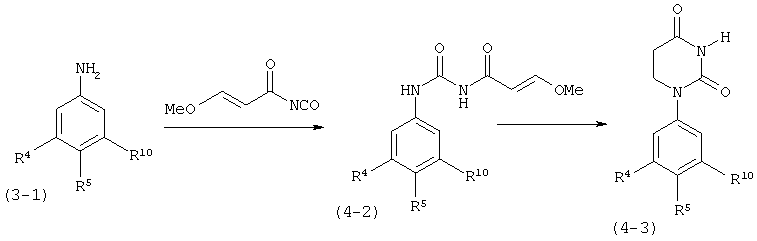
Соединение (4-2) можно получить из соединения (3-1) путем его растворения в растворителях, таких как, например, диметилформамид или диметилацетамид, с последующим добавлением бензольного раствора (Е)-3-метоксиакрилоилизоцианата (полученного как описано в Santana, L.; et al. J. Heterocyclic Chem. 1999, 36, 293-295.) при температуре примерно от -40 примерно до -15°С в инертной атмосфере, и затем нагреванием до температуры окружающей среды в течение примерно от 30 мин примерно до 4 часов. Соединение (4-2) может быть обработано кислотой, такой как, например, серная кислота, в смеси этанол-вода при диапазоне температур примерно от 90 примерно до 110°С в течение примерно 1-8 часов с получением соединения (4-3). Альтернативно, можно осуществить циклизацию соединения (4-2) до урацила (4-3) в основной среде, как описано Ueno, Y.; et al. J. Org. Chem. 70:7925-7935 (2005).
Схема 5
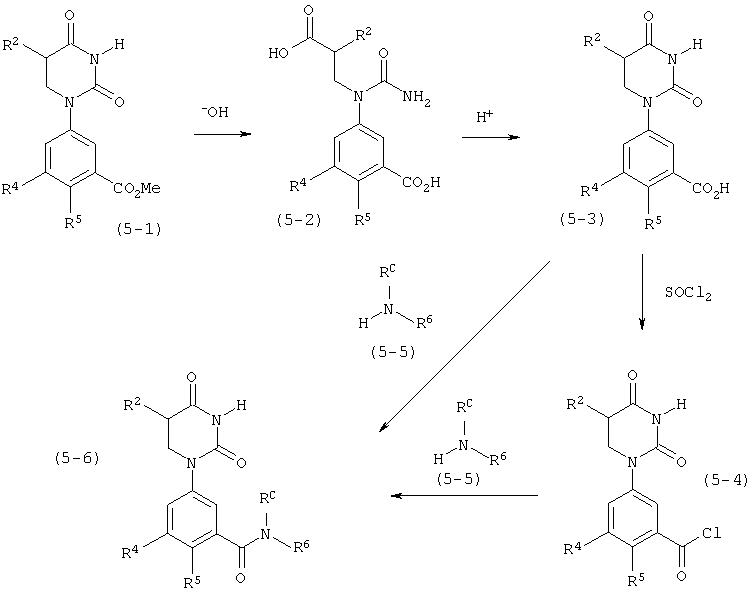
Можно осуществить гиролиз соединения (5-1) основанием, таким как, например, гироксид натрия, гидроксид лития или гидроксид калия, в среде растворителя, такого как, например, метанол, этанол или тетрагидрофуран или их смесь. Полученную реакционную смесь перемешивают в течение примерно 6-48 часов при температуре окружающей среды. При подкислении разбавленной водной кислотой происходит гидролиз эфира и раскрытие тетрагидропиримидинового кольца с получением соединения (5-2).
Циклизацию соединения (5-2) до соединения (5-3) осуществляют обработкой сильной кислотой, такой как, например, концентрированная соляная кислота в диапазоне температур примерно от 90 примерно до 120°С в течение примерно 1-3 часов. Соединение (5-3) можно обработать тионилхлоридом с каталитическим количеством диметилформамида или без него при нагревании с обратным холодильником в течение примерно 1-4 часов с получением хлорангидрида (5-4). Обработка тионилхлоридом в растворителях, таких как, например, хлороформ или толуол, при температуре флегмы также дает соединение (5-4).
Соединение (5-4) может быть обработано амином или соответствующей солью (5-5) в среде растворителя, такого как, например, диоксан, диметилформамид, диметилацетамид или дихлорметан, необязательно в присутствии основания, такого как, например, пиридин, триэтиламин или диизопропилэтиламин, при температуре примерно, от температуры окружающей среды примерно до 100°С в течение примерно 1-24 часов с получением соединения (5-6).
Альтернативно, соединение (5-3) может быть прямо превращено в соединение (5-6) реакцией с эквимолярным количеством амина (5-5) в присутствии связующего агента, такого как, например, бис(2-оксо-3-оксазолидинил)фосфинхлорид (ВОРС1), O-(7-азабензотразол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат (HATU) или O-бензотразол-1-ил-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU), совместно с вспомогательным связующим, таким как, например, 1-гидрокси-7-азабензотриазол (HOAT) или 1-гидроксибензотриазолгидрат (НОВТ) в присутствии или отсутствии основания, такого как, например, N-метилморфолин, диизопропилэтиламин, в растворителях, таких как, например, тетрагидрофуран, N,N-диметилацетамид, N,N-диметилформамид, пиридин и хлороформ. Обычно реакции осуществляют при температуре примерно от 0 до примерно 65°C или в микроволновом реакторе для облегчения связывания.
Схема 6
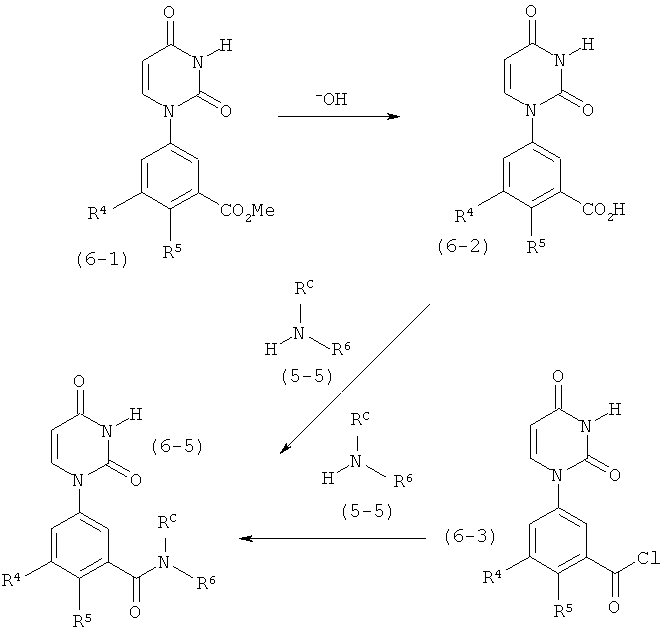
Соединение (6-1) может быть превращено в соединение (6-5), используя преобразования, описанные выше в Схеме 5.
Схема 7
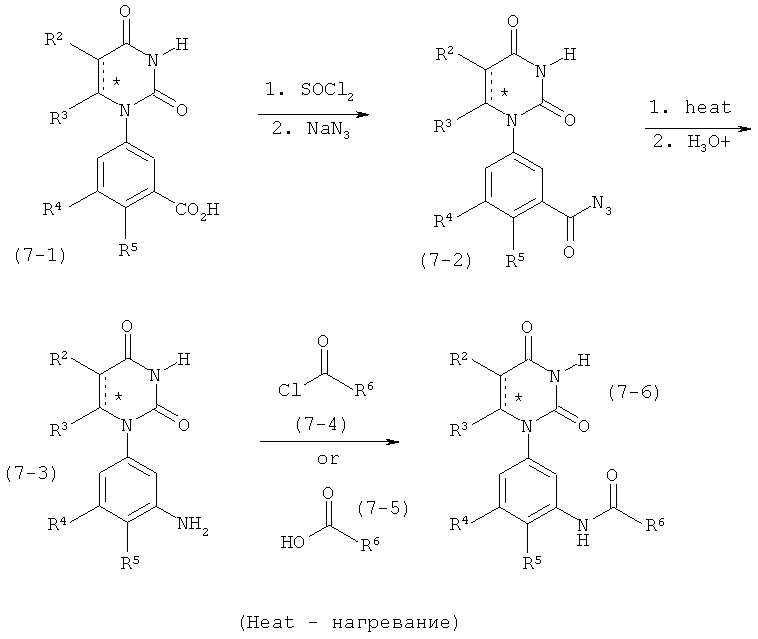
Соединение (7-1) может быть превращено в ацилазид (7-2) с помощью двухстадийного способа. Сначала соединение (7-1) обрабатывают тионилхлоридом при нагревании с обратным холодильником в присутствии каталитического количества диметилформамида или без него, в течение примерно от 1 до 4 часов с получением соответствующего хлорангидрида. Обработка тионилхлоридом при температуре флегмы в растворителях, таких как, например, хлороформ или толуол также приводит к получению желаемого хлорангидрида. Хлорангидрид подвергают взаимодействию с водным раствором азида натрия в растворителе, таком как, например, ацетон, в течение примерно от 1 до 8 часов с получением ацилазида (7-2). Затем соединение (7-2) подвергают перегруппировке Курциуса при нагревании с обратным холодильником в среде растворителей, таких как, например, диоксан или толуол. Промежуточный изоцианат гидролизуют водной кислотой, такой как, например, разбавленная соляная кислота, в растворителе, таком как, например, диметоксиэтан, с получением соединения (7-3). Соединение (7-3) может быть превращено в соединение (7-6), используя или хлорангидрид (7-4), или карбоновую кислоту (7-5) и образование амидной связи в условиях, описанных в Схемах 5 и 6.
Схема 8
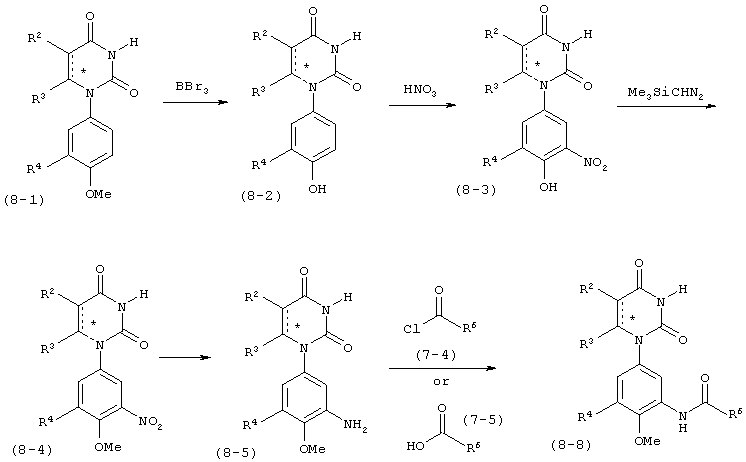
Для осуществления нитрования соединения (8-1) оно может быть активировано удалением метильной группы с помощью добавленного изначально примерно при 0°C BBr3 с последующим нагреванием с обратным холодильником в течение примерно от 10 до примерно 24 часов в растворителе, таком как, например, дихлорметан, с получением соединения (8-2). Фенол (8-2) может быть обработан азотной кислотой в уксусной кислоте в течение примерно от 1 примерно до 10 часов при температуре окружающей среды или около нее, с получением соединения (8-3). Затем Соединение (8-3) может быть превращено в соответствующий метиловый эфир (8-4) обработкой раствором (триметилсилил)диазоэтана в тетрагидрофуране в растворителе, таком как, например, метанол или смесь метанола и тетрагидрофурана, при температуре окружающей среды или около нее в течение примерно 8-24 часов. Соединение (8-4) может быть восстановлено до соединения (8-5), используя описанные в Схеме 1 условия восстановления и пригодные для имеющихся функциональных групп. Соединение (8-5) может быть превращено в соединение (8-8) связыванием с хлорангидридом (7-4) или карбоновой кислотой (7-5), используя условия образования амидной связи, описанные в Схемах 5 и 6.
Схема 9
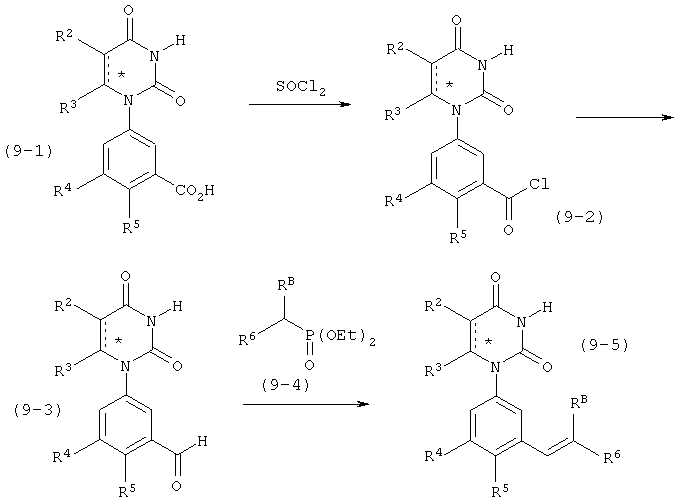
Соединение (9-1) может быть обработано тионилхлоридом при нагревании с обратным холодильником в течение примерно от 1 примерно до 4 часов с получением хлорангидрида (9-2). Обработка тионилхлоридом при температуре флегмы в растворителях, таких как, например, хлороформ или толуол также приводит к образованию соединения (9-2). Соединение (2) превращают в соответствующий альдегид (9-3) восстановлением с помощью литий три-m-бутоксиалюминий гидрида в растворителе, таком как, например, тетрагидрофуран, примерно при -78°C в течение примерно 1-8 часов. Восстановление можно также осуществить, используя обработку хлоридом индия и гидридом трибутилолова в присутствии трифенилфосфина в растворителе, таком как тетрагидрофуран или толуол при температуре примерно от - 40 примерно до 0°C. Соединение (9-3) может быть обработано соединением (9-4) в присутствии основания, такого как m-бутилат калия, в растворителе, таком как дихлорметан, при комнатной температуре или вблизи нее в течение примерно от 1 до примерно 8 часов с получением соединения (9-5).
Схема 10
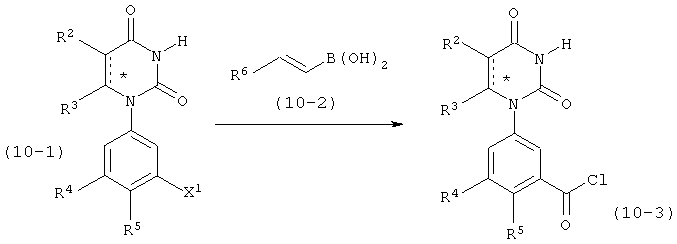
Соединение (10-1), где X1 представляет собой галоген (например, бром, йод) можно подвергнуть реакции сочетания Сузуки с винилбороной кислотой (10-2) с получением соединения (10-3). Обычно реакция требует использования основания и катализатора. Примеры оснований включают, например, карбонат калия, фосфат калия, m-бутилат калия, карбонат натрия, карбонат цезия и фторид цезия. Примеры катализаторов включают, например, трис(дибензилидинацетон)дипалладий (0), ацетат палладия, бис(трифенилфосфин)палладий (II) хлорид, тетракис(трифенилфосфин)палладий, дихлор[1,1'-бис(ди-трет-бутилфосфин)ферроцен] палладий (II) или аддукт дихлор[1,1'-бис(дифенилфосфин)ферроцен]палладий (II)/дихлорметан. Реакцию можно проводить в растворителе, таком как, например, вода, диоксан, диметоксиэтан, диметилформамид, толуол, этанол, тетрагидрофуран и т.д. или их смесь. Реакцию можно проводить при окружающей температуре или выше.
Схема 11
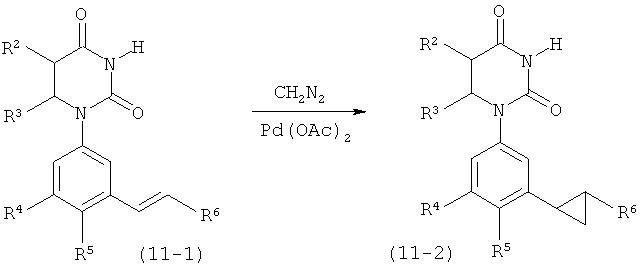
Соединение (11-1) можно превратить в соединение (11-2) обработкой диазометаном в растворителе, таком как, например, тетрагидрофуран, в присутствии ацетата палладия при комнатной температуре или около нее в течение примерно от 30 мин до примерно 4 часов.
Схема 12
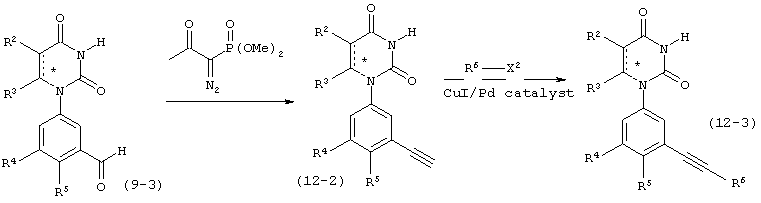
Соединение (9-3) может быть обработано диметил 1-диазо-2-оксопропилфосфонатом (полученным, как описано Ohira, S., Syn. Comm. 19:561-564 (1989)) в присутствии основания, типа карбоната калия, в растворителе, таком как, например, метанол, в течение примерно от 8 до примерно 24 часов при комнатной температуре, или около нее, с получением алкина (12-2). Соединение (12-2) может затем быть обработано R6-X2, где X2 представляет собой йод, бром или -O-трифлат, в присутствии йодида меди (I), палладиевого катализатора, основания и, необязательно, дополнительно трифенилфосфина в инертной атмосфере с получением соединения (12-3). Пригодные палладиевые катализаторы включают, например, трис(дибензилидинацетон)палладий (0), ацетат палладия, бис(трифенилфосфин)палладий (II) хлорид или тетракис(трифенилфосфин)палладий. Основания, которые могут использоваться, включают, например, триэтиламин, диэтиламин, диизопропилэтиламин, карбонат калия, необязательно, в присутствии тетрабутиламмоний бромида и бикарбоната натрия. Используемые растворители включают, например, ацетонитрил, диметилформамид, воду, диоксан и тетрагидрофуран или их смеси. Реакцию можно проводить при температуре от комнатной до температуры флегмы растворителей в течение примерно от 1 примерно до 48 часов. Нагревание примерно от 50 примерно до 120°C в микроволновом реакторе в течение примерно 5-15 минут также приводит к получению соединения (12-3).
Схема 13
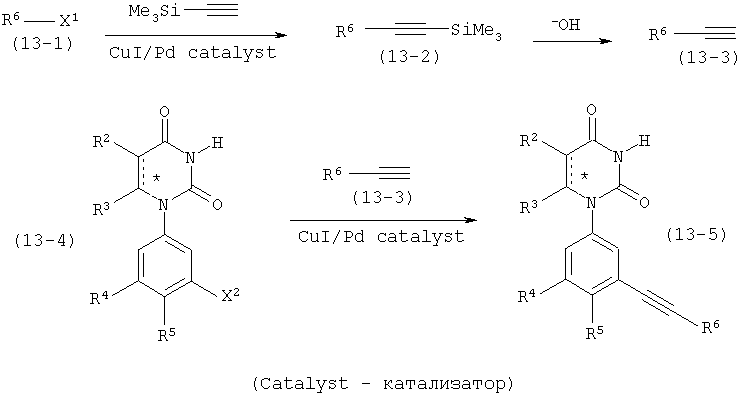
Соединение (13-1), где X1 представляет собой бром или йод, может быть введено во взаимодействие в инертной атмосфере с (триметилсилил)ацетиленом в присутствии катализатора, такого как, например, палладий ацетат/трифенилфосфин или йодид меди /бис(трифенилфосфин)палладий (II) хлорид, и основания, такого как, например, триэтиламин, в растворителе, таком как, например, толуол или ацетонитрил, с получением соединения (13-2). Реакцию можно проводить при нагревании примерно от 70°C примерно до 100°C необязательно в запаянной трубке в течение примерно от 30 мин до примерно 48 часов. Соединение (13-2) может быть превращено в соединение (13-3) обработкой основанием, таким как, например, карбонат калия или гидроксид натрия в растворителе, таком как, например, метанол, при температуре окружающей среды. Соединение (13-3) можно подвергнуть взаимодействию с соединением (13-4), где X2 представляет собой бром или йод, в присутствии йодида меди (I), палладиевого катализатора, основания и, необязательно, дополнительно трифенилфосфина в инертной атмосфере с получением соединения (13-5). Пригодные палладиевые катализаторы включают, например, трис(дибензилидинацетон)дипалладий (0), ацетат палладия, бис(трифенилфосфин)палладий (II) хлорид или тетракис(трифенилфосфин)палладий. Используемые основания включают, например, триэтиламин, диэтиламин, диизопропилэтиламин, калий карбонат, необязательно, в присутствии тетрабутиламмоний бомида, и бикарбонат натрия. Используемые растворители включают, например, ацетонитрил, диметилформамид, воду, диоксан и тетрагидрофуран или их смеси. Реакцию можно проводить при температуре примерно от 40°C до температуры флегмы растворителей в течение примерно от 15 мин примерно до 48 часов. Нагревание под действием микроволнового излучения примерно при 50°C примерно до 120°C примерно от 5 примерно до 15 минут представляет собой альтернативный процесс нагревания с получением соединения (13-5).
Схема 14
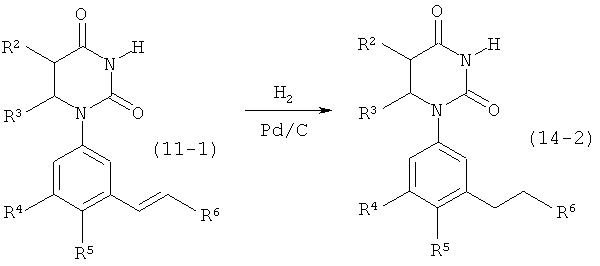
Соединение (11-1) подвергают восстанавлению с получением соединения (14-2). Типичные условия восстановления включают использование водорода при давлении примерно от 1 примерно до 5 атмосфер в присутствии катализатора, такого как, например, палладий или платина на угле, в растворителе, таком как, например, тетрагидрофуран, этилацетат, этанол или гексан при температуре окружающей среды или вблизи нее в течение примерно от 1 примерно до 12 часов.
Схема 15
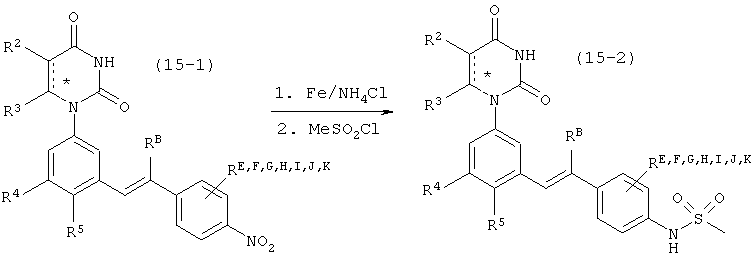
Соединение (15-1) может быть превращено в соединение (15-2) в две стадии. Начальная стадия включает восстановление ароматической нитрогруппы железными опилками в присутствии слабой кислоты, такой как, например, хлорид аммония или разбавленная соляная кислота, при температуре от примерно от 60 до примерно 80°C в смеси растворителей, содержащей, например, метанол, воду и тетрагидрофуран, в течение, примерно от 1 примерно до 12 часов. Вторая стадия представляет собой выдерживание полученного на первой стадии анилина с метансульфонилхлоридом в присутствии основания, такого как пиридин, в растворителе, таком как дихлорметан, при температуре окружающей среды или около нее.
Схема 16

Анилин (16-1) и ароматическое нитросоединение (16-4), где X2 представляет собой, например, бром, йод или трифлат, могут быть превращены в соединение (16-3). Соединение (16-1) может быть трансформировано в соединение (16-2) обработкой метансульфонилхлоридом в присутствии основания, такого как пиридин, в растворителе, таком как дихлорметан. Затем соединение (16-2) может быть превращено в соединение (16-3) обработкой в инертной атмосфере (триметилсилил)ацетиленом в присутствии катализатора, такого как, например, ацетат палладия, бис(трифенилфосфин)палладий (II) хлорид, бис(трифенилфосфин)палладий (II) хлорид, в комбинации с йодидом меди (I) и, когда X2 означает бром, трифенилфосфин, и основанием, таким как, например, триэтиламин, в растворителе, таком как толуол или ацетонитрил при примерно 80°C.
Соединение формулы (16-4) может быть введено в реакцию в инертной атмосфере с (триметилсилил) ацетиленом в присутствии катализатора, такого как, например, бис(трифенилфосфин)палладий (II) хлорид/йодид меди (I), и основания, такого как, например, триэтиламин, в растворителе, таком как, например, ацетонитрил, при приблизительно 80°C с получением соединения (16-5). Соединение (16-5) может быть превращено в соединение (16-3) в две стадии. Начальная стадия включает восстановление ароматической нитро группы железными опилками в присутствии слабой кислоты, такой как, например, хлорид аммония или разбавленная соляная кислота при температуре примерно от 60 примерно до 80°C в смеси растворителей, содержащей, например, метанол, воду и тетрагидрофуран в течение примерно от 1 примерно до 12 часов. Вторая стадия заключается в выдержке полученного на первой стадии анилина с метансульфонилхлоридом в присутствии основания, такого как пиридин, в растворителе, таком как дихлорметан, при температуре окружающей среды или вблизи нее.
Удаление триметилсилильной группы соединения (16-3) осуществляют, как описано для получения соединения (13-3) в указанной выше схеме 13.
Схема 17
Соединение (17-1) может быть обработано метансульфонилхлоридом в присутствии основания, такого как, например, пиридин, в растворителе, таком как, например, дихлорметан, с получением мезилатного соединения (17-2). Выдерживание соединения (17-3) с комплексом боран-диметилсульфид в растворителе, таком как, например, тетрагидрофуран, примерно при 0-10°C приводит к получению соединения (17-4). Соединения (17-2) и (17-4) могут быть связаны с ацетальдегидом в тетрагидрофуране при нагревании с обратным холодильником. Последующая обработка водой при комнатной температуре приводит к получению соединения (17-5).
Схема 18
Карбоновая кислота (18-1) может быть восстановлена боран-тетрагидрофурановым комплексом при нагревании с получением спирта (18-2). Соединение (18-2) может быть превращено в соответствующий бромид (18-3) с помощью N-бромсукцинимида и трифенилфосфина в среде растворителя, такого как, например, дихлорметан, при комнатной температуре в течение нескольких часов. Обработка соединения (18-3) триэтилфосфитом примерно при 120°C в течение примерно от 1 до примерно 3 часов дает соединение (18-4). Соединение (18-4) может быть использовано, например, для получения соединения (9-5) как описано в Схеме 9.
Схема 19
Бензальдегид (19-1) может быть обработан диэтилфосфонатом в присутствии основания, такого как, например, метилат натрия, в растворителе, таком как, например, метанол, при комнатной температуре с получением соединения (19-2). Соединение (19-2) может быть обработано N-хлорсукцинимидом и трифенилфосфином в дихлорметане при комнатной температуре с получением соединения (19-3). Соединение (19-2) можно также ввести в реакцию с (диэтиламино)трифторидом серы (DAST) с получением соединения (19-4).
Соединение (19-1) может также быть обработано п-толуолсульфоновой кислотой и триметилортоформиатом в метаноле примерно при 50°C с получением ацеталя (19-5). Соединение (19-5) может также быть превращено в соединение (19-6) выдерживанием с триэтилфосфитом и диэтилэфиратом трехфтористого бора при температуре примерно от -20°C до примерно температуры окружающей среды.
Соединения (19-3), (19-4) и (19-6) могут быть использованы, например, для получения соединения (9-5), как описано в Схеме 9.
Схема 20
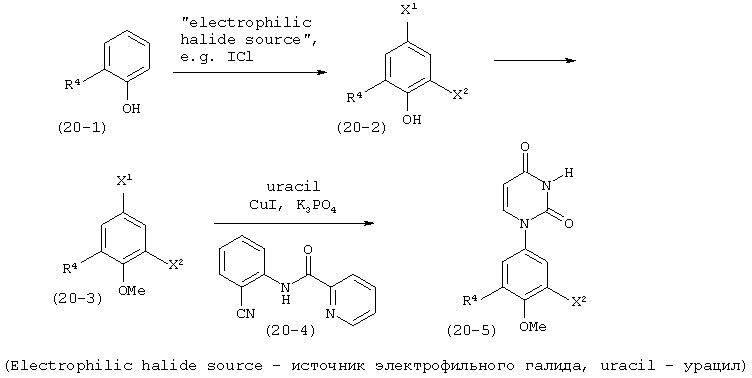
Фенол (20-1), где R4 является отличным от амино, обрабатывают источником электрофильного галида, таким как, например, монохлорид йода, с получением дигалогенированного соединения (20-2), где X1 и X2 независимо представляют бром или йод. Соединение (20-2) превращают в соединение (20-3) взаимодействием алкилирующего агента, такого как, например, метилсульфат, с основанием, таким как, например, карбонат калия, при нагревании с обратным холодильником в ацетоне. Альтернативно, метилйодид в присутствии основания, такого как, например, m-бутилат калия, в растворителе, таком как, например, тетрагидрофуран или диметилформамид, также дает соединение (20-3). Еще в одной альтернативе соединение (20-2) может быть прометилировано (триметилсилил)диазометаном в растворителе, таком как, например, т-бутилметиловый эфир. Соединение (20-3) можно ввести в реакцию с урацилом, лигандом (20-4), йодидом меди (I) и фосфатом калия в диметилсульфоксиде при температуре примерно от 40°C примерно до 100°C с получением соединения (20-5).
Например, когда в соединении (20-3) R представляет собой трет-бутил, X1 представляет собой йод и X2 представляет собой йод или бром, соединение (20-3) можно смешивать с урацилом и соединением (20-4) в присутствии CuI и K2PO4 в ДМСО в течение примерно 15 -24 часов примерно при 60°C с получением соединения (20-5). Вместо лиганда (20-4) для изготовления (20-5) можно использовать 8-гидроксихинолин и 2-(2-пиридил)-бензимидазол.
Схема 21
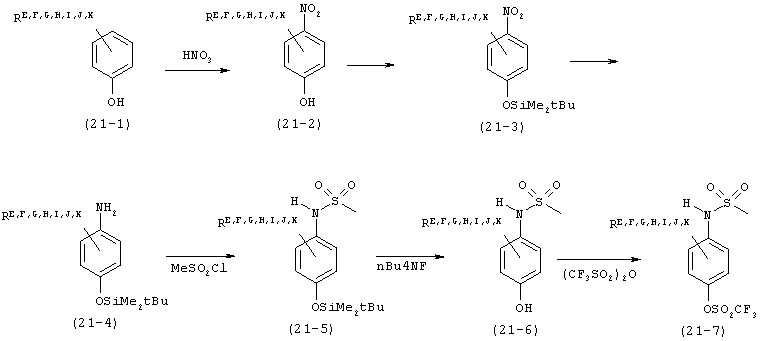
Можно осуществить нитрование соединения (21-1) азотной кислотой в уксусной кислоте при температуре примерно от 10 примерно до 15°C с получением соединения (21-2). Фенольная чать соединения (21-2) может быть защищена силиловым эфиром, например, т-бутилдиметилсилиловым эфиром, путем обработки силилхлоридом, таким как, например, т-бутилдиметилсилилхлорид, и имидазолом в растворителе, таком как, например, диметилформамид при температуре окружающей среды с получением соединения (21-3). Затем соединение (21-3) может быть восстановлено в известных для специалиста условиях с получением соответствующего анилина (21-4). Типичные условия такого восстановления включают использование водорода при давлении примерно 1-5 атмосфер в присутствии катализатора, такого как, например, палладий или платина на угле в растворителе, таком как, например, тетрагидрофуран, этилацетат, этанол, метанол или гексан, при температуре окружающей среды, или вблизи нее, в течение примерно 1-12 часов. В зависимости от наличия функциональных групп, более подходящими могут быть другие условия восстановления, такие как, например, использование железных опилок в присутствии слабой кислоты, такой как, например, хлорид аммония или разбавленная соляная кислота, при нагревании с обратным холодильником в смеси растворителей, содержащей, например, метанол, воду и тетрагидрофуран, в течение примерно 1-12 часов.
Затем можно осуществить сульфонилирование анилина (21-4) метан сульфонилхлоридом в присутствии пиридина в растворителе, таком как, например, дихлорметан. Исходные материалы и реагенты смешивают примерно при 0°C и затем дают постепенно нагреться до температуры окружающей среды с получением соединения (21-5). Защитную силилэфирную группу удаляют в хорошо известных для специалиста условиях. Например, тетрабутиламмонийфторид в тетрагидрофуране при комнатной температуре превращает соединение (21-5) в соединение (21-6). Фенольную группу соединения (21-6) можно сульфонилировать ангидридом трифторметансульфоновой кислоты в присутствии основания, такого как, например, пиридин, в растворителе, таком как, например, дихлорметан, при комнатной температуре с получением соединения (21-7). Соединение (21-7) можно использовать, как описано в Схеме 12, для получения соединения (12-3).
Схема 22
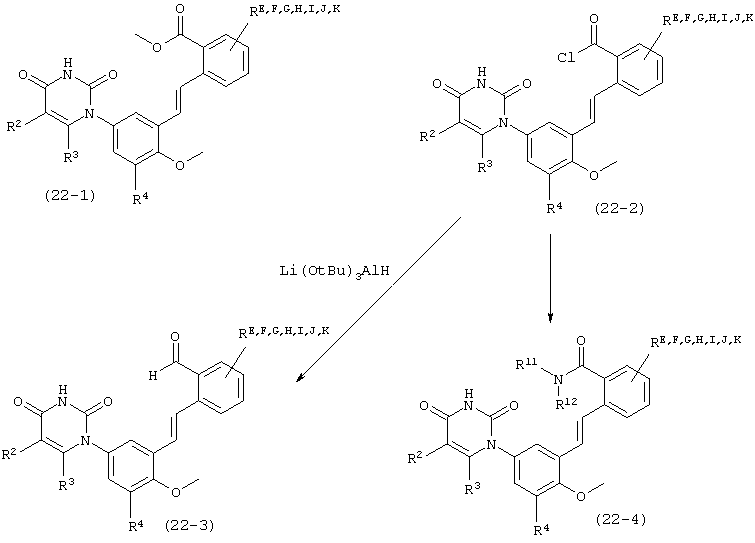
Соединение (22-1) может быть превращено в соединение (22-2) в две стадии. Сначала можно осуществить гидролиз соединения (22-1) с помощью основания, такого как, например, гидроксид натрия, гидроксид лития или гидроксид калия, в растворителе, таком как, например, метанол, этанол или тетрагидрофуран или их смеси. Полученную реакционную смесь перемешивают в течение примерно 6-48 часов при температуре окружающей среды. Затем, полученную карбоновую кислоту нагревают с обратным холодильником в тионилхлориде с каталитическим количеством диметилформамида, или без него, примерно от 1 примерно до 4 часов до получения хлорангидрида (22-2). Обработка тионилхлоридом при нагревании с обратным холодильником в растворителях, таких как, например, хлороформ или толуол, также приводит к получению соединения (22-2). Обработка карбоновой кислоты оксалилхлоридом в дихлорметане с каталитическим количеством диметилформамида также дает соединение (22-2).
Соединение (22-2) может быть обработано амином или соответствующей солью в растворителе, таком как, например, диоксан, диметилформамид, диметилацетамид или дихлорметан, необязательно в присутствии основания, такого как, например, пиридин, триэтиламин или диизопропилэтиламин при температуре от температуры окружающей среды, или близкой к ней, примерно до 100°C в течение примерно 1-24 часов с получением соединения (22-4), где R11 и R12 независимо представляют собой водород или RF, или взятые вместе с азотом, с которым они связаны, образуют 5-6-членный гетероциклил или конденсированный из 2-х колец гетероциклил.
Соединение (22-2) может быть превращено в соответствующий альдегид (22-3) путем восстановления с помощью литий три-m-бутоксиалюминий гидрида в растворителе, таком как, например, тетрагидрофуран, примерно при температур от -60°C примерно до -78°C.
Схема 23
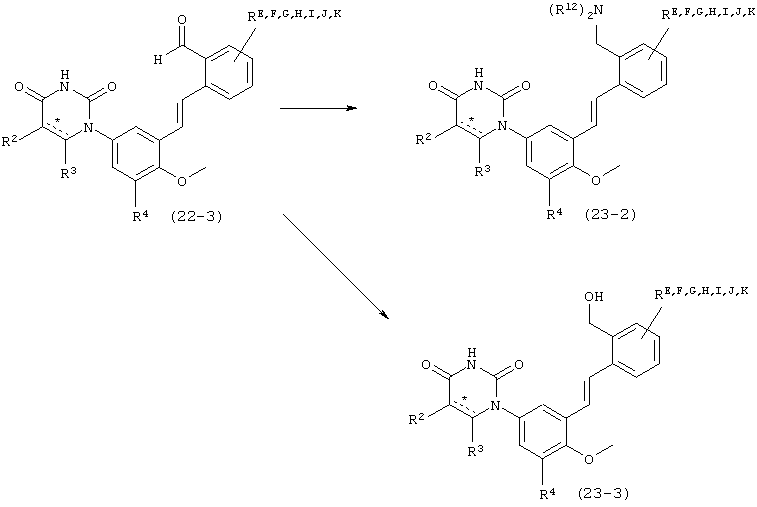
Соединение (22-3) может быть превращено в соединение (23-2), где R11 и R12 независимо предствляют собой водород или RF, или взятые вместе с азотом, с которым они связаны, образуют 5-6-членный гетероциклил или конденсированный из 2-х колец гетероциклил, обработкой амином, N(R11)(R12), в присутствии восстановителя, такого как, например, триацетоксиборогидрид натрия или цианоборогидрид натрия, в растворителе, таком как, например, метанол, этанол, дихлорметан, диметилацетамид или диметилформамид, в течение примерно от 1 примерно до 24 часов. Часто реакция лучше протекает при кислом значении pH, которое может поддерживаться добавлением уксусной кислоты или хлористоводородной кислоты.
Соединение (22-3) может также быть превращено в соединение (23-3) восстановлением с помощью литий три-m-бутоксиалюминий гидрида в растворителе, таком как тетрагидрофуран, при комнатной температуре.
Схема 24
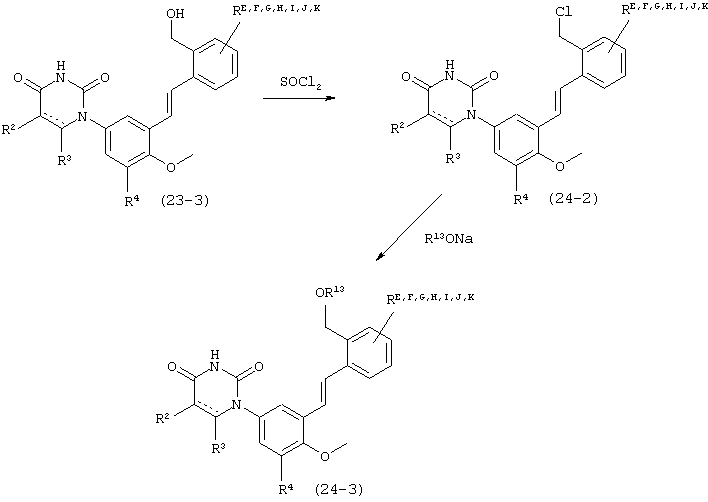
Соединение (23-3) может быть превращено в соединение формулы (24-2) обработкой тионилхлоридом в дихлорметане при комнатной температуре. Соединение (24-2) может быть обработано алкоксидом натрия R13ONa в нагретом растворе соответствующего спирта с получением соединение (24-3), где R13 представляет собой водород или RF.
Схема 25
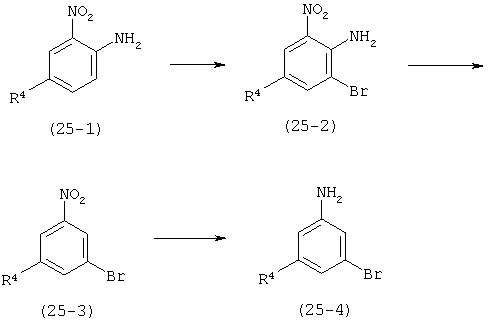
Можно осуществить бромирование соединения (25-1) обработкой, например, пербромидом пиридингидробромида в растворителе, таком как, например, уксусная кислота при температуре окружающей среды, или близкой к ней, в течение примерно 1-8 часов с получением соединения (25-2). Аминогруппа соединения (25-2) может быть удалена выдерживанием с т-бутилнитритом в растворителе, таком как, например, диметилформамид, сначала при температуре окружающей среды с последующим увеличением температуры примерно до 50-65°C с получением соединения (25-3). Можно добавить дополнительные аликвоты т-бутилнитрита при температуре окружающей среды с последующим нагреванием до полного превращения. Соединение (25-3) может быть восстановлено до соединения (25-4), например, обработкой железом и хлоридом амония.
Схема 26
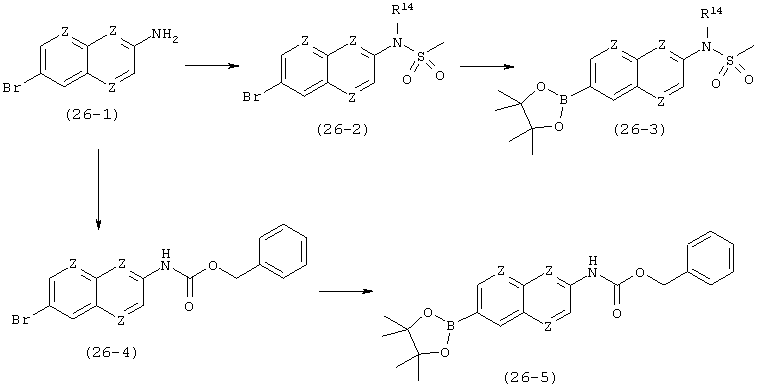
Соединение (26-1), где каждый Z независимо представляет N или СН, может быть превращено в эфир бороновой кислоты для использования в реакциях Сузуки. Например, соединение формулы (26-1) может быть превращено в соединение (26-2), где R14 представляет собой водород или метансульфонил (при использовании избытка метансульфонилхлорида), обработкой метансульфонилхлоридом в пиридине приблизительно при температуре окружающей среды в течение примерно 1-8 часов.
Соединение (26-2) может преобразовано в соединение (26-3) обработкой пинакол-бораном в присутствии катализатора, такого как, например, трис(дибензилидинацетон)дипалладий (0), лиганда, такого как, например, три-т-бутилфомфин, и основания, такого как триэтиламин, в растворителях, таких как, например, тетрагидрофуран, диоксан или толуол, при температуре от температуры окружающей среды до примерно 130°C.
Можно также осуществить взаимодействие соединения (26-2) с бис(пинаколат)дибором в присутствии катализатора, такого как, например, Combiphos® Pd6, аддукт дихлор[1,1'-бис(дифенилфосфин)ферроцен]палладий (II)/дихлорметан или ацетат палладия, в присутствии лиганда, такого как, например, 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (XPhos), и основания, такого как, например, ацетат калия, в растворителях, таких как, например, толуол, диоксан, тетрагидрофуран, диметилформамид или диметилсульфоксид, при температуре примерно от 60 примерно до 130°C с получением соединения (26-3).
Соединение (26-3) может быть превращено в имеющее защиту соединение (26-4) обработкой бензилхлорформиатом примерно при 0°C в присутствии насыщенного водного раствора бикарбоната натрия в смеси ацетона и воды. Смесь может быть нагрета до температуры окружающей среды и быть выдержана при этой температуре в течение примерно от 12 до 24 часов. Затем соединение (26-4) может быть превращено пинаконовый эфир бороновой кислоты (26-5), используя описанные выше условия.
Схема 27
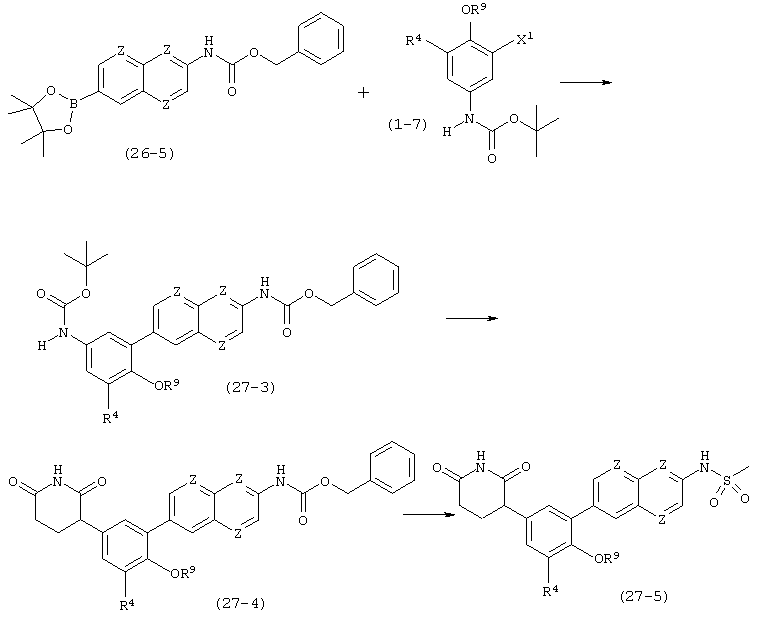
Соединение (26-5), где каждый Z независимо представляет собой N или CH, может быть соединено с соединением (1-7) в условиях реакции сочетания Сузуки с получением соединения (27-3). Такие условия включают, например, использование палладиевого катализатора, такого как, например, трис(дибензилидинацетон)палладий (0), ацетат палладия, бис(трифенилфосфин)палладий (II) хлорид, тетракис(трифенилфосфин)палладий или аддукт дихлор[1,1'-бис(дифенилфосфин)ферроцен]палладий (II)/дихлорметан; основания, такого как, например, карбонат калия, фосфат калия, m-бутилат калия, карбонат натрия, карбонат цезия или фторид цезия; и растворителя, такого как, например, толуол, этанол, вода или тетрагидрофуран или их смеси, нагретые до примерно 40-130°C.
Соединение (27-3) может быть преобразовано в соединение (27-4) в три стадии. Первая стадия включает удаление защитной т-бутоксикарбонильной группы с помощью кислоты, такой как, например, трифторуксусная кислота, в растворителе, таком как, например, дихлорметан или хлористоводородная кислота в диоксане, при комнатной температуре в течение примерно 1-24 часов. Затем может быть введен дигидропиримидиндион, как описано в Схеме 3.
Соединение (27-5) может быть получено из соединения (27-4) в две стадии. Сначала в условиях восстановления с нафтиламина удаляют защитную группу. Как правило, это гидрогенизация (~1 атмосфера) в присутствии катализатора, такого как, например, 10% палладий на угле, в растворителе, таком как, например, эталацетат, при температуре окружающей среды или вблизи нее в течение примерно 8-24 часов. Затем нафтиламин может сразу быть сульфонилирован обработкой метансульфонилхлоридом в присутствии основания, такого как триэтиламин, в растворителе (например, дихлорметан) при комнатной температуре в течение примерно от 20 мин до примерно 4 часов.
Схема 28
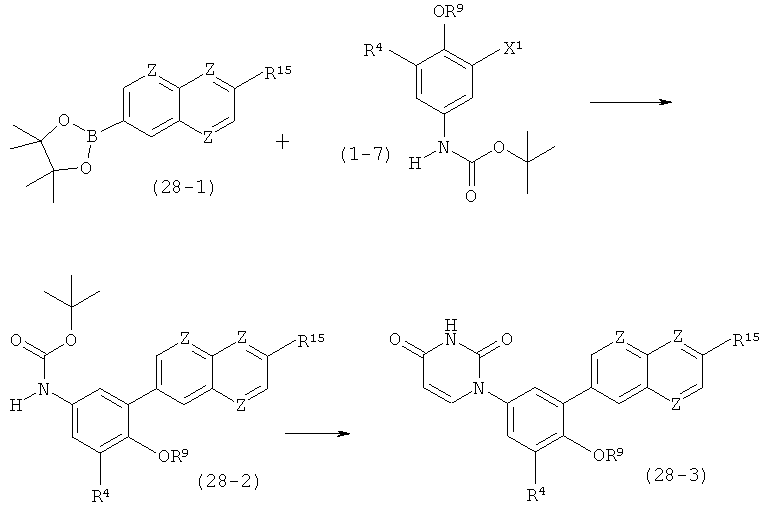
Соединение (28-1), где каждый Z независимо представляет N или CH и R15, например, водород, -NHSO2Me, -N(SO2Me)2 или метокси, может быть соединено с соединением (1-7) в условиях реакции сочетания Сузуки с получением соединения (28-3). Такие условия включают, например, использование палладиевого катализатора, такого как, например, трис(дибензилидинацетон)палладий (0), ацетат палладия, бис(трифенилфосфин)палладий (II) хлорид, тетракис(трифенилфосфин)палладий или аддукт дихлор[1,1'-бис(дифенилфосфин)ферроцен]палладий (II)/дихлорметан; основания, такого как, например, карбонат калия, фосфат калия, m-бутилат калия, карбонат натрия, карбонат цезия или фторид цезия; и растворителя, такого как, например, толуол, этанол, вода или тетрагидрофуран или их смеси и нагревание до примерно 40-130°C. Перед нагреванием, как правило, удаляют кислород с помощью инертного газа, например азота. Нагрев можно осуществлять в обычной стеклянной запаянной трубке или в микроволновом реакторе в течение примерно 1-24 часов.
Соединение (28-2) может быть преобразовано в соединение (28-3) в три стадии. Первая стадия включает удаление защитной т-бутоксикарбонильной группы с помощью кислоты, такой как, например, трифторуксусная кислота, в растворителе, таком как, например, дихлорметан или хлористоводородная кислота в диоксане, при комнатной температуре в течение примерно 1-24 часов. Затем может быть введен урацил, как описано в Схеме 4.
Схема 29
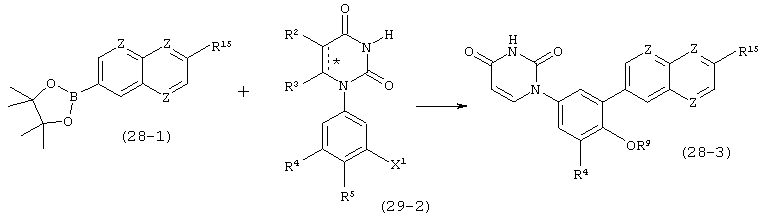
Соединение (28-1), где каждый Z независимо представляет собой N или CH и R15, например, водород, -NHSO2Me, -N(SO2Me)2 или метокси, может быть соединено с соединением (29-2), где X1, например, бром или йод, в условиях реакции сочетания Сузуки с получением соединения формулы (28-3). Такие условия включают, например, использование палладиевого катализатора, такого как, например, трис(дибензилидинацетон)палладий (0), ацетат палладия, бис(трифенилфосфин)палладий (II) хлорид, тетракис(трифенилфосфин)палладий, аддукт дихлор[1,1'-бис(дифенилфосфин)ферроцен]палладий (II)/дихлорметан или бис(дифенилфосфин)ферроцен]палладий (II)/дихлорметан; основания, такого как, например, карбонат калия, фосфат калия, m-бутилат калия, карбонат натрия, карбонат цезия или фторид цезия; и растворителя, такого как, например, толуол, этанол, вода или тетрагидрофуран или их смеси, нагретые до примерно 40-130°C. Перед нагреванием, как правило, удаляют кислород с помощью инертного газа, например азота. Нагрев можно осуществлять в обычной стеклянной запаянной трубке или в микроволновом реакторе в течение примерно 1-24 часов.
Схема 30
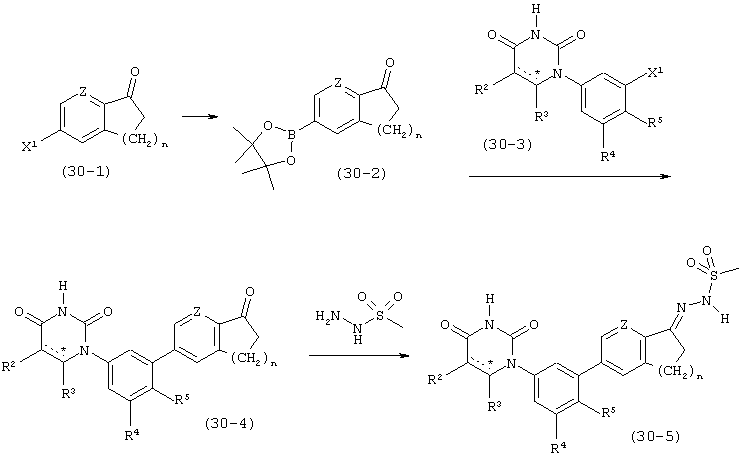
Можно осуществить взаимодействие соединения (30-1), где X1 представляет собой бром или йод, n равно 1 или 2 и Z представляет собой CH или N, с бис(пинаколат)дибором в присутствии катализатора, такого как, например, Combiphos® Pd6, аддукт дихлор[1,1'-бис(дифенилфосфин)ферроцен]палладий (II)/дихлорметан или ацетат палладия, в присутствии лиганда, такого как, например, 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (XPhos), и основания, такого как, например, ацетат калия, в растворителях, таких как, например, толуол, диоксан, тетрагидрофуран, диметилформамид или диметилсульфоксид, при температуре примерно от 60 примерно до 130°C с получением соединения (30-2). Перед нагреванием, как правило, удаляют кислород с помощью инертного газа, например азота. Нагрев можно осуществлять в обычной стеклянной запаянной трубке или в микроволновом реакторе в течение примерно 1-24 часов. Соединение (30-3) можно ввести в реакцию с соединением (30-2) с получением соединения (30-4), используя описанные в Схеме 29 условия.
Обработка соединения (30-4) метансульфонилгидразидом в растворителе, таком как, например, тетрагидрофуран, метанол или этанол или их смесь, при температуре от температуры окружающей среды примерно до 100°C в течение 8-48 часов дает соединение (30-5).
Схема 31
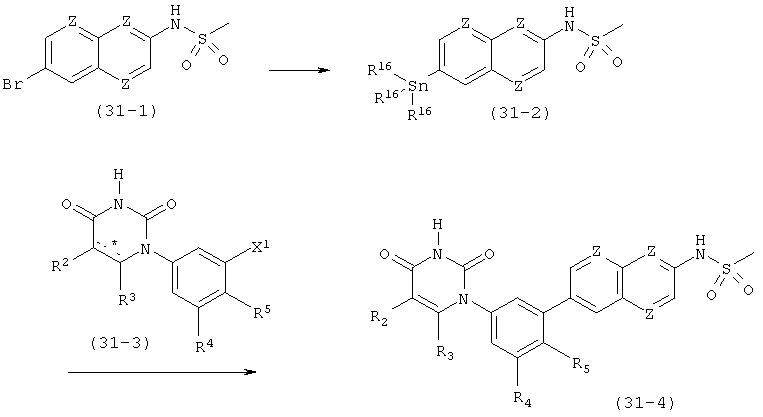
Соединение (31-1) можно обработать гексаметилдиоловом или гексабутилдиоловом в присутствии катализатора, такого как, например, бис(трифенилфосфин)палладий (II) хлорид, в растворителе, таком как, например, толуол или диоксан при примерно от 50 примерно 130°C с получением соединения (31-2). Соединение (31-2) может быть обработано соединением (31-3) в присутствии катализатора, такого как, например, трис(дибензилидинацетон)палладий (0), и лиганда, такого как трис(2-фурил)фосфин в растворителе, таком как, например, толуол, диоксан или тетрагидрофуран, нагретом примерно до 40-130°C с получением соединения (31-4).
Схема 32
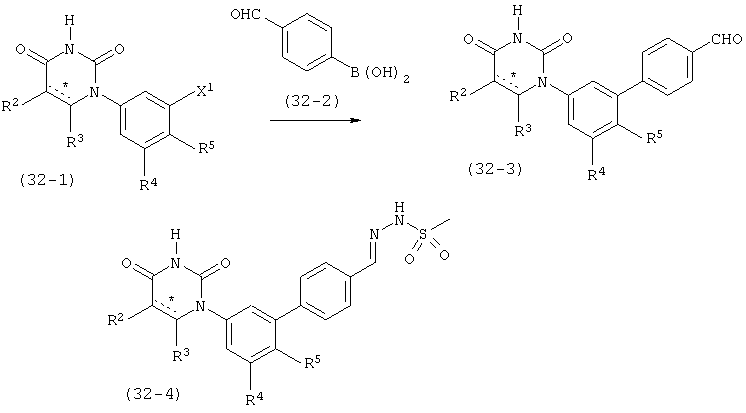
Можно осуществить взаимодействие соединения (32-1) с соединением (32-2) в условиях реакции сочетания Сузуки с получением соединения формулы (32-3). Обработка метансульфонилгидразидом как описано в Схеме 30 дает соединение (32-4).
Схема 33
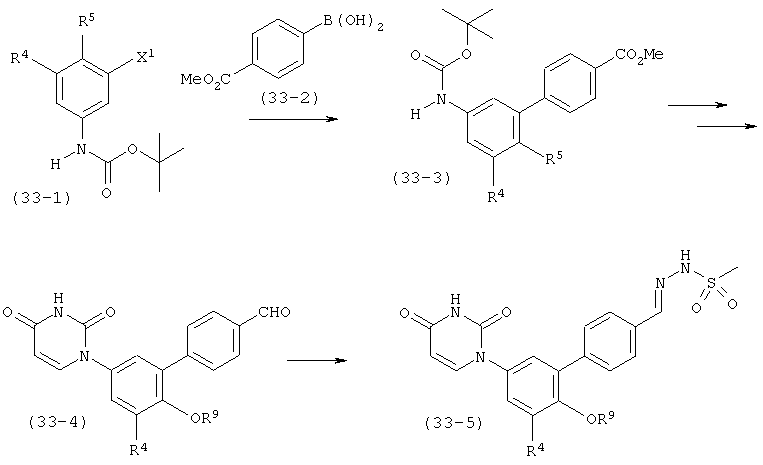
Можно осуществить взаимодействие соединения (33-1) с соединением (33-2) в условиях реакции сочетания Сузуки с получением соединения (33-3). Соединение (33-3) может быть превращено в соединение (33-4) формированием урацилового кольца. Затем метилкарбоксилат может быть превращен в соответствующий диальдегид. Соединение (33-4) может быть обработано метансульфонилгидразидом с получением соединения (33-5).
Схема 34
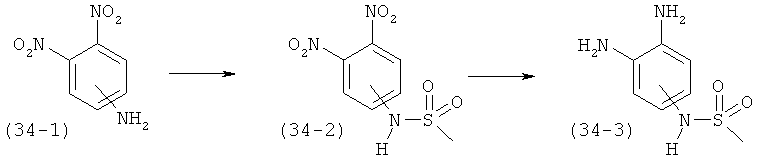
Можно осуществить сульфонилирование динитроанилина (34-1) метансульфонилхлоридом в присутствии основания, например, пиридина, в растворителе, таком как, например, дихлорметан, при комнатной температуре в течение примерно 8-36 часов с получение соединения (34-2). Соединение (34-2) может быть превращено в соединение (34-3), используя железные опилки в присутствии слабой кислоты, такой как, например, хлорид аммония или разбавленная хлористоводородная кислота, при нагревании с обратным холодильником в смеси растворителей, таких как, например, метанол, вода и тетрагидрофуран,в течение примерно 1-12 часов.
Схема 35
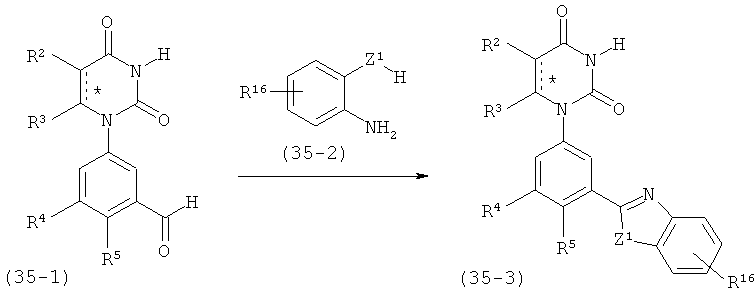
Соединение (35-1) можно ввести во взаимодействие с соединением (35-2), где Z1 представляет собой O, S или NH, и R16 представляет собой водород, -NHSO2Me или NO2, в присутствии находящегося в контакте с воздухом древесного угля в среде растворителя, такого как, например, толуол, при нагревании примерно от 90 до примерно 110°C в течение примерно 24-72 часов с получением соединения (35-3).
Схема 36
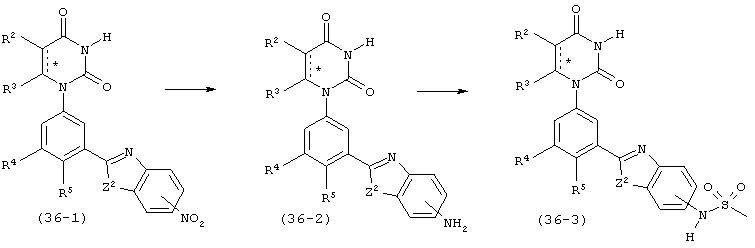
Соединение (36-1), где Z2 представляет собой O или S, может быть восстановлено до соединения (36-2), используя железные опилки в присутствии слабой кислоты, такой как, например, хлорид аммония или разбавленная хлористоводородная кислота при температуре примерно от 60 примерно до 90°C в растворителях, таких как, например, метанол, этанол, вода и тетрагидрофуран или их смеси, в течение примерно от 30 мин примерно до 12 часов. Можно провести сульфонилирование соединения (39-2) метансульфонилхлоридом в присутствии основания, например, пиридина в растворителе, таком как, например, дихлорметан, при комнатной температуре в течение примерно 8-36 часов.
Схема 37
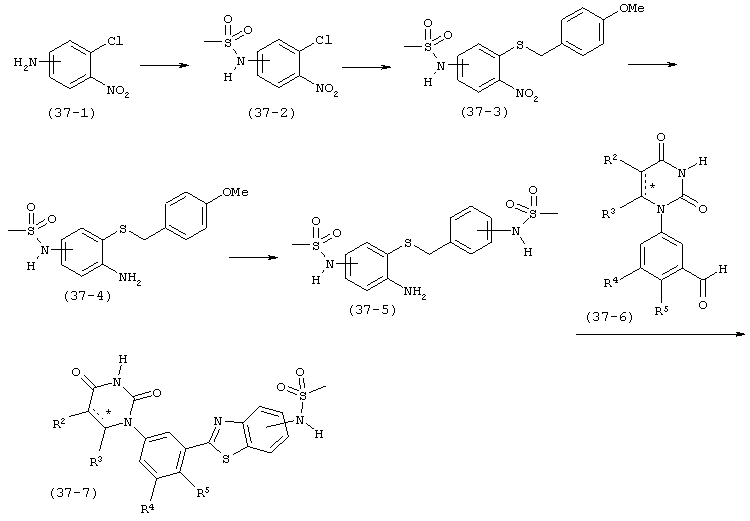
Соединение (37-1) может быть подвергнуто сульфонилированию метансульфонилхлоридом в присутствии основания, например, пиридина, в растворителе, таком как, например, дихлорметан, при комнатной температуре в течение примерно 8-36 часов с получением соединения (37-2). Соединение (37-2) можно подвергнуть взаимодействию с (4-метоксифенил)метантиолом в присутствии основания, такого как, например, карбонат калия, в растворителе, таком как, например, диметилформамид, нагретом до примерно 90-110°C, в течение примерно 8-24 часов с получением соединения (37-3). Соединение (37-3) может быть восстановлено до соединения (37-4), используя железные опилки, в присутствии слабой кислоты, такой как, например, хлорид аммония или разбавленная соляная кислота при температуре примерно от 60 примерно до 90°C, в растворителе, таком как, например, метанол, этанол, вода и тетрагидрофуран или их смеси, в течение примерно от 30 мин примерно до 12 часов. Соединение (37-4) может быть превращено в соединение (37-5) в присутствии ацетата ртути (II), анизола и трифторуксусной кислоты примерно при 0°C в течение примерно 30-90 мин с последующим барботированием сероводорода через эту смесь. Соединение (37-5) может быть обработано соединением (37-6) в присутствии п-толуолсульфокислоты и трифенилфосфина в растворителе, таком как, например, толуол, нагретом до температуры флегмы в течение примерно 2-16 часов с получением соединения (37-7).
Схема 38
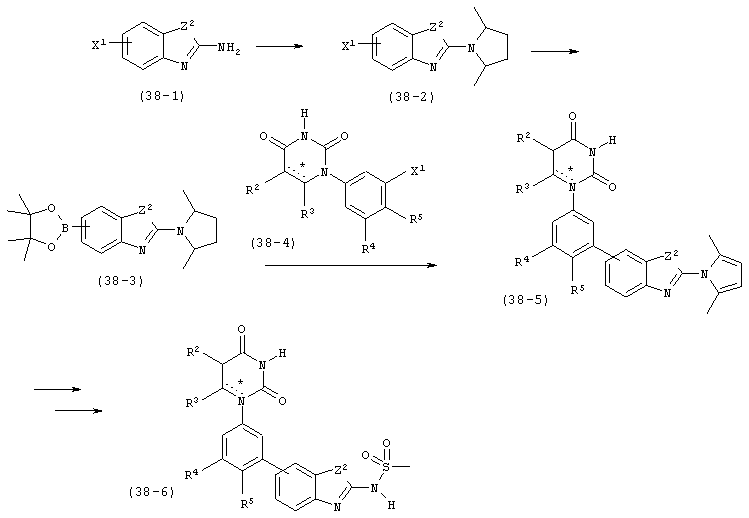
Соединение (38-1), где X1 представляет собой бром или йод и Z2 представляет собой O или S, можно ввести во взаимодействие с 2,5-гександионом в присутствии п-толуолсульфокислоты и пиридина, при нагревании в бензоле с получением соединения формулы (38-2). Соединение (38-2) можно ввести во взаимодействие бис(пинаколат)дибором в присутствии катализатора, такого как, например, Combiphos® Pd6, аддукт дихлор[1,1'-бис(дифенилфосфин)ферроцен]палладий (II)/дихлорметан или ацетат палладия, в присутствии лиганда, такого как, например, 2-дициклогексилфосфин-2',4',6'-триизопропилбифенил (XPhos), и основания, такого как, например, ацетат калия, в растворителе, таком как, например, толуол, диоксан, тетрагидрофуран, диметилформамид или диметилсульфоксид, при температуре примерно от 60 примерно до 130°C с получением соединения (38-3). Соединение (38-3) может быть введено во взаимодействие с соединением (38-4) с получением соединения (38-5) в условиях реакции сочетания Сузуки. Такие условия включают, например, применение палладиевого катализатора, такого как, например, дигидро дихлорбис(ди-т-бутилфосфинито-KP)палладат(2-), трис(дибензилидинацетон)палладий (0), ацетат палладия, бис(трифенилфосфин)палладий (II) хлорид, тетракис (трифенилфосфин)палладий или аддукт дихлор[1,1'-бис(дифенилфосфин)ферроцен] палладий (II)/дихлорметан; основания, такого как, например, ацетат калия, карбонат калия, фосфат калия, m-бутилат калия, карбонат натрия, карбонат цезия или фторид цезия; и растворителя, такого как, например, толуол, этанол, вода или тетрагидрофуран или их смеси, нагретые до примерно 40-130°C.
Соединение (38-5) может быть обработано гидрохлоридом гидроксиламина в нагретом этаноле для удаления защитной пиррольной группы. Затем обработка метансульфонилхлоридом в присутствии основания, такого как, например, пиридин, в растворителе, таком как, например, дихлорметан, при температуре окружающей среды или близкой к ней, дает соединение (38-6).
ПРИМЕРЫ
Следующие примеры являются всего лишь пояснительными, а не ограничивающими это описание каким-либо образом.
Пример A. Получение (E)-N-(3-трет-бутил-5-йод-4-метоксифенилкарбамоил)-3-метокси акриламида.
Часть A. Получение 2-трет-бутил-4-нитрофенола.
К интенсивно перемешанному раствору 2-трет-бутилфенола (10 г, 66,6 ммоль) в гептане (67 мл) добавляли быстрыми каплями раствор 70% азотной кислоты (4,25 мл, 66,6 ммоль) разбавленной водой (4,25 мл). Полученную в результате темнокрасную/коричневую смесь интенсивно перемешивали в течение 2 ч. Суспендированное твердое вещество собирали путем фильтрации, промывали гексаном (300 мл), водой (200 мл) и еще раз гексаном (200 мл) с получением порошка цвета какао, который сушили до постоянной массы (4,65 г, 35,6%).
Часть B. Получение 2-трет-бутил-6-йод-4-нитрофенола.
К продукту из Части A (4,5 г, 23,05 ммоль), растворенному в MeOH (120 мл) и воде (30 мл), добавляли по каплям монохлорид йода (1,155 мл, 23,05 ммоль) за период времени 10 мин. Смесь перемешивали в течение 2 ч и разбавляли в 1 л воды, и оставляли стоять в течение ночи. Твердое вещество собирали фильтрацией и промывали 3×50 мл водой, и сушили под вакуумом в течение ночи с получением твердого вещества желтовато-коричневого цвета (7,14 г, 96%).
Часть C. Получение 1-трет-бутил-3-йод-2-метокси-5-нитробензола.
К охлажденному на ледяной бане раствору продукта из Части B (5,5 г, 17,13 ммоль) в MTBE (15 мл) в 50 мл сосуде высокого давления добавляли 2,0 М TMS диазометан (12,85 мл, 25,7 ммоль) с последующим добавлением по каплям метанола (1,0 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали при комнатной температуре в течение 16 ч, охлаждали и давление сбрасывали. Раствор распределяли между EtOAc и водой. Органический слой промывали 1,0 М HCl, насыщенным раствором карбоната калия и насыщенным NaCl. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали с получением красного масла, которое использовали без очистки (5,4 г, 84%).
Часть D. Получение 3-трет-бутил-5-йод-4-метоксианилина.
Смесь продукта из Части C (5,80 г, 17,31 ммоль), хлорида аммония (1,389 г, 26,0 ммоль), и железа (4,83 г, 87 ммоль) в THF/MeOH/воде (всего 200 мл, 2/2/1) нагревали с обратным холодильником в течение 2 ч, охлаждали и фильтровали через целит. Фильтрат упаривали, и остаток разделяли между водой и EtOAc. Органический слой промывали насыщенным солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением коричневого масла (5,28 г, 100% выход).
Часть E. Получение (E)-N-(3-трет-бутил-5-йод-4-метоксифенилкарбамоил)-3-метокси акриламида.
К раствору продукта из Части D (3,05 г, 10 ммоль) в DMF (50 мл) при -20°C в атмосфере N2 добавляли быстрыми каплями 0,4 M раствор в бензоле (E)-3-метоксиакрилоилизоцианата (50,0 мл, 20,00 ммоль, полученного способом Santana et al., J. Heterocyclic Chem. 36:293 (1999). Раствор перемешивали в течение 15 мин при -20°C, нагревали до комнатной температуры в течение 45 мин и разбавляли в EtOAc. EtOAc слой промывали 4×300 мл водой, 2×100 мл солевым раствором, сушили (Na2SO4) и концентрировали до твердого вещества коричневого цвета. Остаток растирали в порошок в Et2O/гексане с получением мелкого порошка, который собирали фильтрацией и сушили с получением желтовато-коричневого порошка (2,46 г, 57%).
Пример B. Получение 1-(3-трет-бутил-5-йод-4-метоксифенил)дигидропиримидин-2,4(1H,3H)-диона.
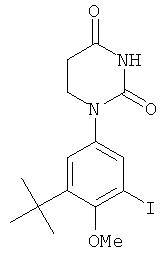
К суспензии продукта из Примера A (2,46 г, 5,69 ммоль) в этаноле (50 мл) добавляли раствор 5,5 мл H2SO4 в 50 мл воды, и смесь нагревали при 110°C в течение 2,5 ч с получением прозрачного раствора. Раствор охлаждали и разбавляли 50 мл воды при перемешивании, с получением беловатого твердого вещества, которое собирали фильтрованием, промывали водой и сушили (2,06 г, 90%).
Пример C. Получение 1-(3-трет-бутил-5-йод-4-метоксифенил)пиримидин-2,4(1Н,3H)-диона.
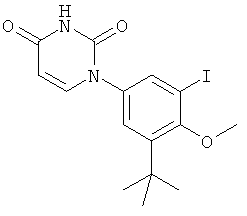
Часть A. Получение 2-трет-бутил-4,6-дийодфенола.
Раствор 2-трет-бутилфенола (20,0 г, 133 ммоль) в метаноле (266 мл) обрабатыали гранулами гидроксида натрия (6,39 г, 160 ммоль). Смесь перемешивали до растворения всего гидроксида натрия и затем охлаждали на бане с ледяной солью до -2°C. Добавляли йодид натрия (15,0 г, 100 ммоль), а затем по каплям добавляли 10% раствор гипохлорита натрия (45 мл, 73,3 ммоль) с такой скоростью, чтобы температура раствора не поднималась выше 1,3°C. Эту последовательность событий повторяли (3×) пока не будет добавлено все 60 г (400 ммоль) йодида натрия, и раствор гипохлорита натрия добавляли до тех пор, пока цвет раствора не изменялся со светлого желто-зеленого цвета до цвета слабого чая со льдом. Для этого требуется почти 16 мл из 180 мл всего отмеренного раствора гипохлорита натрия. Продолжая охлаждение примерно при 2°C, раствор пятиводного тиосульфата натрия (20 г) в воде (100 мл) добавляли по каплям на протяжении 20 мин. После добавления, раствор подкисляли до pH 3 путем добавления по каплям концентрированной соляной кислоты (приблизительно 35 мл требовалось из 40 мл, помещенных в дополнительную воронку). Осадок собирали фильтрованием и промывали с использованием >1 литра воды. Твердое вещество оранжево-желтого цвета отсасывали досуха, и сушили в вакуумной печи при 50°C в течение 18 ч. Эти процедуры давали продукт (49,61 г, 93%) в виде твердого вещества желтовато-коричневого цвета.
Часть B. Получение 1-трет-бутил-3,5-дийод-2-метоксибензола.
Раствор продукта из Части A (20,0 г, 49,7 ммоль) в ацетоне (140 мл) обрабатывали метилйодидом (3,9 мл, 8,83 г, 62,2 ммоль) и 50% (масс/масс) раствором гидроксида натрия (3,02 мл, 4,58 г, 57,2 ммоль) с последующим перемешиванием при окружающей температуре в течение 48 ч. Смесь концентрировали в вакууме до объема приблизительно 50-60 мл, с последующим разбавлением гептаном (80 мл) и водой (50 мл). Слои отделяли, и органический слой экстрагировали насыщенным раствором хлорида натрия. Высушивание (Na2SO4) и концентрирование в вакууме давало продукт (20,59 г, 99%) в виде светло-желтого масла.
Часть С. Получение 1-(3-трет-бутил-5-йод-4-метоксифенил)пиримидин-2,4(1Н,3H)-диона.
Суспензию продукта из Части В (12,04 г, 28,9 ммоль), урацила (3,89 г, 34,7 ммоль), N-(2-цианофенил)пиколинамида (1,29 г, 5,79 ммоль) и трехосновного фосфата калия (12,9 г, 60,8 ммоль) в DMSO (181 мл) дегазировали путем барботирования азота в течение 1 ч. Затем смесь обрабатывали йодидом меди (I) (551 мг, 2,89 ммоль) и дегазирование продолжали еще в течение 10 мин. Затем смесь нагревали при 60°C в течение 18 ч. Затем смесь выливали в воду (600 мл) и подкисляли до pH 3 путем добавления 4 н раствора соляной кислоты. Смесь разбавляли этилацетатом, и органический слой экстрагировали водой (3×), насыщенным раствором хлорида аммония (1×) и насыщенным раствором хлорида натрия. Раствор сушили и обрабатывали (3-меркаптопропил) силикагелем, с последующим перемешиванием в течение 2 ч. Смесь фильтровали и концентрировали в вакууме. Полученное твердое вещество растирали в порошок с эфиром-этилацетатом (>10:1) и собирали фильтрованием и промывали эфиром. После высушивания в вакуумной печи при 50°C в течение 2 ч, эти процедуры давали продукт (2,75 г) в виде твердого вещества белого цвета. Маточные растворы концентрировали в вакууме с получением твердого вещества янтарно-желтого цвета. Это вещество хроматографировали на картридже с силикагелем Flash 65, элюируя 20-100% этилацетатом в гексане. Эти процедуры давали почти белое твердое вещество, которое растирали в порошок с эфиром-гексаном и собирали фильтрованием. После высушивания в вакуумной печи в течение 3 ч, эти процедуры давали еще 4,31 г продукта в виде тведого вещества белого цвета. Суммарный выход: 7,06 г (61%).
Пример D. Получение 1-(3-трет-Бутил-5-йод-4-метоксифенил)пиримидин-2,4(1Н,3H)-диона.
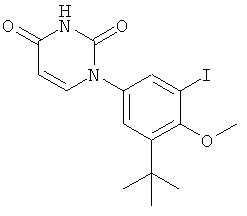
Часть A. Получение 2-/иреот-бутил-4,6-дийодфенола.
2-трет-бутилфенол (99,95 г, 665,36 ммоль) растворяли в 1250 мл метанола и превращали в соответствующий феноксид с использованием 31,96 г (799,0 ммоль, 1,2 экв.) гидроксида натрия, путем перемешивания гранул гидроксида натрия при комнатной температуре, а затем охлаждения реакционной смеси на бане лед/соль. Иодид натрия (299,34 г, 1997,07 ммоль, 3,0 экв.) и 8,3% раствор гипохлорита натрия (1265,83 г, 1411,39 ммоль, 2,1 экв.) добавляли к холодному реакционному раствору четырьмя равными частями, раствор гипохлорита натрия добавляли, поддерживая реакционную смесь при <0°C. 500 мл 20% (масс/масс) раствора тиосульфата натрия добавляли на протяжении периода времени 18 минут, с повышением температуры от -0,6°C до 2,5°C. pH реакционной смеси доводили приблизительно до 3, путем добавления 197,5 мл конц. HCl за период времени 97 мин с температурой реакции, идущей от 1,2°C до 4,1°C. Полученную в результате густую суспензию фильтровали и отфильтрованный осадок промывали ~2 л воды. Отфильтрованный осадок оставляли на воронке Бюхнера под вакуумом в течение ночи (приблизительно 15 ч) с получением на выходе 289,33 г (потенциально возможный скорректированный выход =254,61 г) названного продукта.
Часть B. Получение 1-трет-бутил-3,5-дийод-2-метоксибензола.
Продукт из Части A (93% проба, 21,6 г, 50 ммоль) растворяли в 140 мл ацетона. Добавляли метилйодид (4,2 мл, 67,5 ммоль, 1,35 экв.), с последующим добавлением 50% водного гидроксида натрия (5,0 г, 62,5 ммоль, 1,25 экв.). Реакционную смесь перемешивали в течение ночи, затем концентрировали приблизительно до 50-60 мл. Добавляли 80 мл гептана с последующим добавлением 50 мл воды, и слои встряхивали и отделяли, и водный слой подвергали обратной экстракции 20 мл гептана. Органические слои объединяли и промывали дважды, 50 мл каждый, 10% водным NaCl с получением 91,1 грамм раствора гептана, анализ которого дал 19,1 г названного соединения.
Часть C. Получение 1-(3-трет-бутил-5-йод-4-метоксифенил)пиримидин-2,4(1Н,3H)-диона.
Урацил (33,3 г, 297 ммоль, 1,2 экв.), K3PO4 (106 г, 500 ммоль, 2,1 экв.), CuI (4,6 г, 24,2 ммоль, 0,1 экв.), и N-(2-цианофенил)пиколинамид (6,4 г, 28,7 ммоль, 0,12 экв.) помещали в колбу с инертным аргоном. Разведенный в MeCN 1-трет-бутил-3,5-дийодо-2-метоксибензол растворяют в 1 л DMSO при барботировании аргоном и добавляли к твердому остатку. Реакционную смесь нагревали до 60°C в течение 16 ч. После охлаждения, реакционную смесь разбавляли 2 л EtOAc и промывали 2,6 л воды (обратная экстракция 3×1 л EtOAc). Объединенные органические слои промывали 2×1 л 0,25 М (CuOAc)2 затем 2×830 мл 15% NH4Cl затем 800 мл солевого раствора. Органический слой затем концентрировали и промывали 1 л гептана, затем растирали в порошок при нагревании с обратным холодильником 85:15 (об/об) гептан:iPrOAc в течение 4 ч. После охлаждения, продукт собирали фильтрацией и промывали дополнительными 330 мл 85:15 об/об гептан: EtOAc с выходом после высушивания 66,9 г (70% выход) продукта в виде твердого вещества белого цвета.
Пример E. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
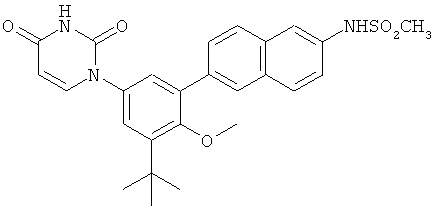
Раствор 100 мл воды и 300 мл THF барботировали азотом, а затем переносили через канюлю и под давлением азота, в колбу, содержащую 19,9965 г (49,96 ммоль) продукта из Примера D, 20,8234 г (59,97 ммоль, 1,20 эквивалентов) N-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)нафтален-2-ил)метансульфонамида и 21,8711 г (103,03 ммоль, 2,06 эквивалентов) фосфата калия, которые были предварительно продуты азотом. Через полученный раствор опять барботируют азот.
THF (100 мл) барботировали азотом, а затем с помощью канюли переносят под давлением азота в колбу, содержащую 462,8 мг (0,51 ммоль, 0,01 эквивалентов) Pd2dba3 и 735,8 мг (2,52 ммоль, 0,05 эквивалентов) 1,3,5,7-тетраметил-6-фенил-2,4,8-триокса-6-фосфаадамантана, которые были предварительно продуты азотом. Через полученный раствор опять барботируют азот.
Исходный раствор THF/вода переносят с помощью канюли под давлением азота в колбу, содержащую катализатор и лиганд в THF. Реакционную смесь нагревали до 50°C и перемешивали в течение ночи под постоянным давлением азота. На следующее утро брали реакционную пробу. Результаты ВЭЖХ образца показали 0,28 РА% исходного йодоурацила, 76,8 РА% продукта и 5,2 РА% бороната.
Реакционную смесь охлаждали до комнатной температуры и промывали, тремя порциями, раствором 5,84 г L-цистеина и 81,4 г хлорида натрия в 550 мл воды, которая была барботирована азотом. Раствор THF фильтровали через слой целита. Слой промывали 100 мл THF, который был объединен с исходным раствором THF. Раствор THF концентрировали на роторном испарителе до 136 г. В белую суспензию добавляли 405 мл этилацетата при хорошем встряхивании. Суспензию фильтровали после перемешивания в течение ночи. Фильтровальный осадок промывали 2×50 мл этилацетата. Твердое вещество, сольват этилацетата, сушили в вакуумной печи при 50°C. Масса составила 25,49 г.
Твердое вещество и 8,7 г 3-меркаптопропил-производного силикагеля перемешивали в 500 мл THF, затем фильтровали через слой целлита. Фильтрат концентрировали на роторном испарителе с получением 13,08 г твердого вещества белого цвета. Твердое вещество, которое было отфильтровано на слое целлита, экстрагировали 500 мл THF при 60°C. Раствор THF концентрировали до 66 г и обрабатывали 206 мл этилацетата. Осажденное твердое вещество фильровали и сушили, с получением 9,13 г продукта. Это твердое вещество объединяли с исходным твердым веществом и суспендировали в 100 мл этанола 200-proof 3A. Фильтровали и сушили в вакуумной печи при 50°C с получением 20,74 г продукта.
Пример F. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
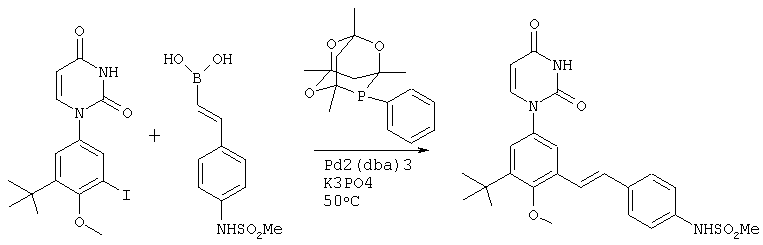
В трехгорлую круглодонную колбу загружали бороновую кислоту (96% концентрации) (3,75 г, 15,6 ммоль, 1,2 экв.), продукт из Примера D (5,0 г, 12,5 ммоль), Cytec лиганд (175 мг, 5 мол%), Pd2(dba)3 (46 мг, 0,4 мол%) и фосфат калия (5,25 г, 25,0 ммоль, 2 экв.). Твердое вещество в течение 10 мин продували азотом и загружали в колбу. Через 75 мл раствора 4:1 THF:вода в течение 10 мин барботировали азот и загружали в колбу. Реакционную смесь перемешивали до растворения твердого вещества с последующим нагреванием смеси при 50°C в темноте в течение ночи. Результаты ВЭЖХ показали, что после перемешивания в течение ночи реакция прошла не полностью (~2% непрореагировавшего йодоурацила). Реакционную смесь разбавляли 375 мл дихлорметана (DCM) и 250 мл 10% лимонной кислоты. Смесь встряхивали в делительной воронке и разделяли слои. Дихлорметановый слой промывали раствором 0,6 г L-цистеина в 250 мл 5% NaHCO3 30 мин, в течение которых дихлорметановый слой менял цвет от оранжевого до желтого. Повторяли обработку дихлорметанового слоя 0,6 г L-цистеина в 250 мл 5% NaHCO3 в течение 30 мин с последующим промыванием 250 мл 5% NaHCO3 и 250 мл 10% NaCl. Дихлорметановый слой обрабатывали 2 г кремнеземтиомочевины в течение 30 мин. Добавляли 1 г угля, перемешивали 5 мин до обесцвечивания и фильтровали через фильтр hy-flo. Отфильтрованный осадок промывали DCM. DCM раствор затем отпаривали с получением 6,74 г светлого желтого твердого остатка. Твердый остаток имел чистоту ~92%. Твердый остаток нагревали в смеси 192 мл DCM и 9 мл MeOH. Он никогда полностью не растворялся. Охлаждали до комнатной температуры при перемешивании. Добавляли 80 мл гептана, и большее количество продукта начинало кристаллизоваться. Суспензию перемешивают с субботы до понедельника. Порциями по 50 мл добавляли суммарно 230 мл гептана. Продукт отфильтровывали. Фильтрат измеряли при 1,21 мг/мл при 210 нм и 1,35 при 220 нм, что равно потере 522-582 мг в жидкости или потере 9-10% по сравнению с теоретической. Отфильтрованный осадок промывали 50 мл смеси 27 мл гептана: 22 мл DCM: 1 мл MeOH. Промывочный раствор содержал 0,5 мг/мл продукта или 25 мг (0,4% по сравнению с теоретическим). Выход продукта составил 5,22 г (88,9%) с чистотой 99,2% PA. При кристаллизации происходит удаление йодоурацила. Образцы были представлены в твердом состоянии для анализа и аналитическом для определения Pd. ЯМР не показал наличия каких-либо остатков растворителя.
Пример 1. Получение (E)-N'-((3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метоксибифенил-4-ил)метилен)метансульфонгидразида (соединение IB-L0-1.1).
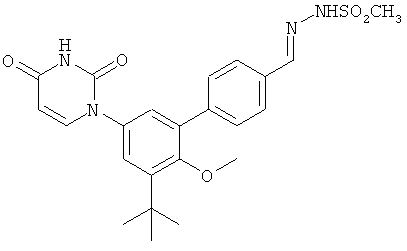
Часть A. Получение 2-трет-бутил-4-нитрофенола.
К интенсивно перемешанному раствору 2-трет-бутилфенола (10 г, 66,6 ммоль) в гептане (67 мл) добавляли быстрыми каплями раствор 70% азотной кислоты (4,25 мл, 66,6 ммоль) разбавленной водой (4,25 мл). Полученную в результате темно-красную/коричневую смесь интенсивно перемешивали в течение 2 ч. Суспендированное твердое вещество собирали фильтрацией, промывали гексаном (300 мл), водой (200 мл) и еще один раз гексаном (200 мл) с получением порошка цвета какао, который сушили до постоянной массы (4,65 г, 35,6%).
Часть B. Получение 2-бром-6-трет-бутил-4-нитрофенола.
Раствор продукта из Части A (1,0 г, 5,12 ммоль) в ледяной уксусной кислоте (10,25 мл) обрабатывали порциями пиридин гидробромид пербромидом (1,80 г, 5,63 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 ч. Дополнительное количество пиридиний гидробромид пербромида (3,80 г) добавляли двумя порциями и после еще 3 ч перемешивания, реакцию завершали. Смесь выливали в ледяную воду, и эту смесь обрабатывали небольшим количеством сульфита натрия. Полученное в результате твердое вещество фильтровали и сушили под вакуумом с получением названного соединения в виде твердого вещества коричневого цвета (1,40 г, 100%).
Часть C. Получение 1-бром-3-трет-бутил-2-метокси-5-нитробензола.
Раствор продукта из Части B (1,40 г, 5,11 ммоль) в 10:1 т-бутилметиловом эфире - метаноле (25,5 мл) обрабатывали 2,0 M триметилсилилдиазометана в эфире (5,1 мл, 10,21 ммоль), с последующим перемешиванием при комнатной температуре в течение 18 ч. Смесь концентрировали под вакуумом с получением желтого масла, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гексаном с получением названного соединения в виде желтого масла (1,36 г, 92%).
Часть D. Получение трет-бутил 3-бром-5-трет-бутил-4-метоксифенилкарбамата.
Раствор продукта из Части C (960 мг, 3,33 ммоль) в метаноле (17 мл) обрабатывали 5% платиной на сульфидированном углероде (100 мг), с последующей гидрогенизацией под давлением из баллонной шины в течение 3 ч, а затем фильтровали через целит и концентрировали под вакуумом с получением 3-бром-5-трет-бутил-4-метоксианилина в виде желтого масла (860 мг, 3,33 ммоль, 100%). Раствор этого вещества в THF (17 мл) обрабатывали ди-трет-бутилдикарбонатом (800 мг, 3,66 ммоль) с последующим нагреванием с обратным холодильником в течение 2 ч. Концентрирование под вакуумом давало твердое вещество бежевого цвета, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гексаном. Твердое вещество растирали в порошок с гексаном, собирали фильтрованием, и сушили под вакуумом с получением названного соединения в виде почти белого твердого вещества (890 мг, 75%).
Часть E. Получение метил 5'-(трет-бутоксикарбониламино)-3'-трет-бутил-2'-метокси бифенил-4-карбоксилата.
Толуол (2 мл) и этанол (2 мл) объединяли с продуктом из Части E (281 мг, 0,78 ммоль), метил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензоатом (411 мг, 1,57 ммоль) и 1M карбонатом натрия (0,78 мл, 0,78 ммоль) и дегазировали в течение 20 мин N2. Добавляли тетракис(трифенил-фосфин)палладий(0) (18 мг, 0,016 ммоль) и дегазирование продолжали в течение 5-10 мин. Нагревали 100°C в герметично закрытой пробирке в течение 18 ч, охлаждали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение (182 мг, 56%).
Часть F. Получение метил 5'-амино-3'-трет-бутил-2'-метоксибифенил-4-карбоксилата. К раствору продукта из Части E (180 мг, 0,43 ммоль) в CH2Cl2 (4 мл) добавляли трифторуксусную кислоту (2 мл). Перемешивали в течение 30 мин и концентрировали под вакуумом. Растворяли в EtOAc и промывали 10% NaHCO3 и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением названного соединения (136 мг, 100%).
Часть G. Получение 3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2'-метокси бифенил-4-карбоксилата.
К раствору продукта из Части F (120 мг, 0,38 ммоль) в DMF (2,5 мл) при -25°C добавляли по каплям (E)-3-метоксиакрилоилизоцианат (1,34 мл, 0,76 ммоль), удерживая температуру ниже -10°C до завершения. Смесь нагревали до комнатной температуры, перемешивали в течение 4 ч и выливали в эфир. Промывали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала (E)-метил-3'-трет-бутил-2'-метокси-5'-(3-(3-метоксиакрилоил)уреид)бифенил-4-карбоксилат (105 мг, 62%). Добавляли этанол (3 мл), H2O (3 мл) и конц. H2SO4 (0,3 мл) и нагревали при 100°C в течение 1 ч. Охлаждали, выливали в H2O и экстрагировали EtOAc. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 2% CH3OH/CHCl3 давала названное соединение (73 мг, 79%).
Часть H. Получение 3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метокси бифенил-4-карбальдегида.
Раствор продукта из Части G (73 мг, 0,18 ммоль) в диоксане (1 мл) обрабатывали 0,5М LiOH (1 мл, 0,36 ммоль) при комнатной температуре в течение 1 ч, выливали в 1 н HCl и экстрагировали EtOAc. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением 3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метоксибифенил-4-карбоновой кислоты (69 мг, 98%). Растворяли в тионилхлориде (2 мл) и нагревали с обратным холодильником в течение 3 ч, охлаждали и концентрировали под вакуумом. Азеотропировали дважды толуолом с получением 3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метоксибифенил-4-карбонилхлорида (72 мг, 100%), который растворяли в THF (1,7 мл) и охлаждали до -78°C. 1M литий три-трет-бутоксиалюминий гидрид (THF) (0,19 мл, 0,19 ммоль) добавляли по каплям и перемешивание продолжали при -78°C в течение 2 ч. Гасили 1 н HCl (1 мл) и нагревали до комнатной температуры. Добавляли воду и экстрагировали EtOAc. Промывали 10% NaHCO3, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 1:1 EtOAc/гексаном давала названное соединение (23 мг, 35%).
Часть I. Получение (Е)-N'-((3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2'-метоксибифенил-4-ил)метилен)метансульфонгидразида.
Раствор продукта из Части Н (23 мг, 0,061 ммоль) в CH3OH (0,8 мл) обрабатывали метансульфонгидразидом (7,7 мг, 0,07 ммоль) при комнатной температуре в течение 1 ч, нагревали до 35°C в течение 2 ч, охлаждали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 5% CH3OH/CHCl3 давала названное соединение (14,8 мг, 52%). 1Н ЯМР (300 MHz CDCl3) ppm 1.44 (s, 9 Н) 3.21 (s, 3 Н) 3.32 (s, 3 Н) 5.82 (d, J=8.09 Hz, 1 Н) 7.14-7.24 (m, 1 Н) 7.35 (d, J=8.09 Hz, 1 H) 7.61 (d, J=8.46 Hz, 2 H) 7.75 (d, J=8.46 Hz, 2 H) 7.79 (s, 1 H) 7.87 (s, 1 H) 8.21 (br s, 1 H). MS (ESI+) m/z 471 (M+H)+.
Пример 2. Получение (E)-N'-((3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2'-метоксибифенил-4-ил)метилен)метансульфонгидразида (соединение IA-L0-1.1).
Часть A. Получение 2-трет-бутил-6-йод-4-нитрофенола.
К продукту из Примера 1, Части A (4,5 г, 23,05 ммоль), растворенного в MeOH (120 мл) и воде (30 мл) добавляли монохлорид йода (1,155 мл, 23,05 ммоль) по каплям за период времени 10 мин. Смесь перемешивали в течение 2 ч и разбавляли в 1 л воды и оставляли стоять в течение ночи. Твердое вещество собирали фильтрацией и промывали 3×50 мл водой и сушили под вакуумом в течение ночи с получением твердого вещества желтовато-коричневого цвета (7,14 г, 96%).
Часть B. Получение 1-трет-бутил-3-йод-2-метокси-5-нитробензола.
К охлажденному на ледяной бане раствору продукта из Части A (5,5 г, 17,13 ммоль) в MTBE (15 мл) в 50 мл сосуде высокого давления добавляли 2,0 M TMS диазометан (12,85 мл, 25,7 ммоль) с последующим добавлением по каплям метанола (1,0 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали при комнатной температуре в течение 16 ч, охлаждали и давление сбрасывали. Раствор разделяли между EtOAc и водой. Органический слой промывали 1,0 M HCl, насыщенным раствором карбоната калия, и насыщенным NaCl. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали с получением красного масла, которое использовали без очистки (5,4 г, 84%).
Часть C. Получение 3-трет-бутил-5-йод-4-метоксианилина.
Смесь продукта из Части B (5,80 г, 17,31 ммоль), хлорида аммония (1,389 г, 26,0 ммоль), и железа (4,83 г, 87 ммоль) в THF/MeOH/воде (всего 200 мл, 2/2/1) нагревали с обратным холодильником в течение 2 ч, охлаждали и фильтровали через целит. Фильтрат упаривали, и остаток разделяли между водой и EtOAc. Органический слой промывали насыщенным солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением коричневого масла (5,28 г, 100% выход).
Часть D. Получение 1-(3-трет-бутил-5-йод-4-метоксифенил)дигидропиримидин-2,4(1Н,3H)-диона.
Продукт из Части C (8,2 г, 26,9 ммоль) обрабатывали акриловой кислотой (5,53 мл, 81 ммоль) и перемешивали в течение ночи с получением чрезвычайно вязкой смеси. Смесь обрабатывали уксусной кислотой (60 мл) и мочевиной (7,3 г, 120 ммоль), нагревали при 120°C в течение 24 ч, охлаждали и концентрировали. Остаток азеотропировали 3×100 мл толуолом с получением твердого вещества коричневого/желтовато-коричневого цвета. Твердое вещество суспендировали в смеси 50 мл EtOAc и 100 мл насыщенного NaHCO3 и перемешивали в течение тридцати мин для нейтрализации какого-либо количества оставшейся уксусной кислоты. Твердое вещество собирали фильтрацией и промывали повторно 50 мл порциями воды и, наконец, 3:1 гексаном/EtOAc (50 мл) с получением беловатого твердого вещества, которое сушили до постоянной массы (7,1 г, 66%).
Часть E. Получение 3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2'-метокси бифенил-4-карбальдегида.
Смесь продукта из Части D (101 мг, 0,25 ммоль), 4-формилфенилбороновой кислоты (56,2 мг, 0,38 ммоль), 1M карбоната натрия (0,25 мл, 0,25 ммоль) и комплекса 1,1'-бис(дифенилфосфино) ферроцен-палладий(II)дихлорид дихлорметан (10,2 мг, 0,013 ммоль) в толуоле/этаноле (2 мл, 1/1) продували барботирующим N2 в течение 5 мин и нагревали в микроволновой печи при 100°C в течение 15 мин. Экстрагировали EtOAc и промывали солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией MeOH/CH2Cl2 (1%-5%) давала названное соединение (92 мг, 97%).
Часть F. Получение (E)-N-((3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2'-метоксибифенил-4-ил)метилен)метансульфонгидразида.
Смесь продукта из Части E (90 мг, 0,24 ммоль) и метансульфонгидразида (29 мг, 0,26 ммоль) в метаноле (4 мл) нагревали при 40°C в течение 2 ч. Упаривали и очищали с помощью колоночной хроматографии на силикагеле, элюируя MeOH/CH2Cl2 (1%-4%) с получением названного соединения (80 мг, 72%). Температура плавления 209-211°C. 1H ЯМР (300 MHz, DMSO-D6) δ 1.39 (s. 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.08 (s, 3 H) 3.24 (s, 3 H) 3.80 (t, J=6.62 Hz, 2 H) 7.17 (d, J=2.57 Hz, 1 H) 7.24 (d, J=2.94 Hz, 1 H) 7.59 (d, J=8.46 Hz, 2 H) 7.77 (d, J=8.46 Hz, 2 H) 8.04 (s, 1 H) 10.33 (s, 1 H) 11.10 (s, 1 H).
Пример 3. Получение N-(6-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IA-L0-2.9).
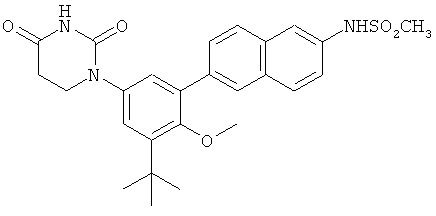
Часть A. Получение 6-бром-2-нафтойной кислоты.
Раствор метил 6-бром-2-нафтоата (7,70 г, 29,0 ммоль) в смеси 2:1 THF:вода (150 мл) обрабатывали гидратом гидроксида лития (2,44 г, 58,1 ммоль) с последующим перемешиванием при комнатной температуре в течение 48 ч. Концентрировали под вакуумом, разбавляли водой и охлаждали до 0°C. Подкисляли до pH 3 с использованием 4 н HCl. Твердые вещества собирали фильтрованием, растворяли в толуоле-EtOAc (приблизительно 2 л) и промывали солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Твердое вещество коричневого цвета растирали в порошок с эфиром, собирали фильтрованием, и сушили под вакуумом с получением названного соединения в виде почти белого твердого вещества (5,07 г, 70%).
Часть B. Получение 6-бромнафтален-2-амина.
Раствор продукта Части A (5,07 г, 20,19 ммоль) и триэтиламина (4,22 мл, 3,07 г, 30,3 ммоль) в сухом DMF (155 мл) обрабатывали дифенилфосфороилазидом (6,55 мл, 8,34 г, 30,3 ммоль) с последующим перемешиванием при комнатной температуре в течение 3 ч. Затем раствор обрабатывали водой (20 мл) с последующим нагреванием при 100°C в течение 1 ч. Раствор охлаждали, и колбу снабжали насадкой для молекулярной перегонки, и DMF удаляли перегонкой под высоким вакуумом. Твердый остаток растворяли в EtOAc и промывали насыщенным раствором бикарбоната натрия. Фильтровали через целит, и фильтрат промывали водой (3×), а затем солевым раствором. Сушили над Na2S04, фильтровали и концентрировали под вакуумом с получением названного соединения в виде твердого вещества бежевого цвета (4,48 г, 100%).
Часть C. Получение бензил 6-бромнафтален-2-илкарбамата.
Смесь продукта из Части B (1,79 г, 8,06 ммоль) и насыщенного раствора бикарбоната натрия (18 мл) в ацетоне (40 мл) при 0°C обрабатывали по каплям бензилхлорформиатом. Смесь перемешивали при 0°C в течение 1 ч, а затем оставляли для постепенного нагревания до комнатной температуры на протяжении 18 ч. Смесь разбавляли EtOAc и водой и слои отделяли. Органический слой экстрагировали водой и промывали солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение в виде твердого вещества розового цвета (1,5 г, 52%).
Часть D. Получение бензил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)нафтален-2-ил карбамата.
Повторно герметизируемый сосуд Шленка, содержащий раствор продукта из Части C (1,42 г, 3,99 ммоль), бис(пинаколато)дибор (1,11 г, 4,39 ммоль), и ацетат калия (1,17 г, 11,96 ммоль) в DMF (28 мл) дегазировали посредством трех циклов замораживания-оттаивания. Раствор обрабатывали комплексом 1,1'-бис(дифенилфосфино)ферроцен палладий (II) хлорид дихлорметан (98 мг, 0,12 ммоль), с последующим дегазированием посредством двух дополнительных циклов замораживания-оттаивания. Сосуд Шленка затем герметично закрывали, и смесь нагревали при 80°C в течение 18 ч. Охлаждали и разбавляли этилацетатом и водой. Смесь обрабатывали Darco G-60, а затем фильтровали через целит. Фильтрат экстрагировали водой (4×) и насыщенным раствором хлорида натрия. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением светло-коричневого масла. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение в виде бесцветного масла (910 мг, 57%).
Часть E. Получение 2-трет-бутил-4-нитрофенола. К интенсивно перемешанному раствору 2-трет-бутилфенола (10 г, 66,6 ммоль) в гептане (67 мл) добавляли быстрыми каплями раствор 70% азотной кислоты (4,25 мл, 66,6 ммоль) разбавленной водой (4,25 мл). Полученную в результате темно-красную/коричневую смесь интенсивно перемешивали в течение 2 ч. Суспендированное твердое вещество собирали фильтрацией, промывали гексаном (300 мл), водой (200 мл) и еще один раз гексаном (200 мл) с получением порошка цвета какао, который сушили до постоянной массы (4,65 г, 35,6%).
Часть F. Получение 2-бром-6-трет-бутил-4-нитрофенола. Раствор продукта из Части E (1,0 г, 5,12 ммоль) в ледяной уксусной кислоте (10,25 мл) обрабатывали порциями пиридин гидробромид пербромидом (1,80 г, 5,63 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 ч. Дополнительное количество пиридиний гидробромид пербромида (3,6 г) добавляли двумя порциями и после еще 3 ч перемешивания, реакцию завершали. Смесь выливали в ледяную воду, и смесь обрабатывали небольшим количеством сульфита натрия. Полученное в результате твердое вещество фильтровали и сушили под вакуумом с получением названного соединения в виде твердого вещества коричневого цвета (1,40 г, 100%).
Часть G. Получение 1-бром-3-трет-бутил-2-метокси-5-нитробензола.
Раствор продукта из Части F (1,40 г, 5,11 ммоль) в смеси 10:1 т-бутилметилэфир-метанол (25,5 мл) обрабатывали 2,0 М триметилсилилдиазометаном в эфире (5,1 мл, 10,21 ммоль), с последующим перемешиванием при комнатной температуре в течение 18 ч. Смесь концентрировали под вакуумом с получением желтого масла, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гексаном с получением названного соединения в виде желтого масла (1,36 г, 92%).
Часть H. Получение трет-бутил 3-бром-5-трет-бутил-4-метоксифенилкарбамата. Раствор продукта из Части G (960 мг, 3,33 ммоль) в метаноле (17 мл) обрабатывали 5% платиной на сульфидированном углероде (100 мг), с последующей гидрогенизацией под давлением из баллонной шины в течение 3 ч, а затем фильтровали через целит и концентрировали под вакуумом с получением 3-бром-5-трет-бутил-4-метоксианилина в виде желтого масла (860 мг, 3,33 ммоль, 100%). Раствор этого вещества в THF (17 мл) обрабатывали ди-трет-бутилдикарбонатом (800 мг, 3,66 ммоль) с последующим нагреванием с обратным холодильником в течение 2 ч. Концентрирование под вакуумом давало твердое вещество бежевого цвета, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гексаном. Твердое вещество растирали в порошок с гексаном, собирали фильтрованием, и сушили под вакуумом с получением названного соединения в виде почти белого твердого вещества (890 м г, 75%).
Часть I. Получение бензил 6-(3-трет-бутил-5-(трет-бутилкарбамоил)-2-метоксифенил) нафтален-2-ил карбамата.
Толуол (928 мкл) и EtOH (928 мкл) объединяли с продуктом из Части H (133 мг, 0,37 ммоль), продуктом из Части D (299 мг, 0,74 ммоль) и 1M карбоната натрия (371 мкл, 0,37 ммоль) и дегазировали в течение 20 мин азотом. Добавляли тетракис(трифенилфосфин)палладий(0) (8,6 мг, 7,4 мкмоль) и дегазирование продолжали 5-10 мин. Нагревали при 85-90°C в течение 18 ч, охлаждали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение (102 мг, 49%).
Часть J. Получение бензил 6-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-илкарбамата.
Раствор продукта из Части I (100 мг, 0,18 ммоль) в CH2Cl2 (1,0 мл) обрабатывали трифторуксусной кислотой (0,5 мл, 6,5 ммоль) при комнатной температуре в течение 1 ч. Концентрировали под вакуумом. Растворяли в этилацетате, промывали 10% NaHCO3, солевым раствором. Сушили над Na2S04, фильтровали и концентрировали под вакуумом. Растворяли в толуоле (1,0 мл) и добавляли Et3N (25 мкл, 0,18 ммоль) и акриловую кислоту (13 мкл, 0,19 ммоль), и смесь нагревали с обратным холодильником в течение 16 ч. Концентрировали под вакуумом. Растворяли в уксусной кислоте (1,0 мл, 17,5 ммоль) и добавляли мочевину (11,9 мг, 0,20 ммоль) и нагревали с обратным холодильником в течение 72 ч. Охлаждали и выливали в ледяную воду, экстрагировали три раза CHCl3, экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле, с элюцией EtOAc/гексаном давала названное соединение (57,5 мг, 58%).
Часть K. Получение N-(6-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
Объединяли продукт из Части J (56 мг, 0,10 ммоль) и EtOAc (1,0 мл) и добавляли 10% палладий на угле (10 мг). Перемешивали под давлением газообразного H2 из баллонной шины в течение 16 ч. Фильтровали через целит и концентрировали под вакуумом. Растворяли в CH2Cl2 (1,0 мл), добавляли Et3N (16 мкл, 0,115 ммоль) и метансульфонилхлорид (8,7 мкл, 0,112 ммоль) и перемешивали при комнатной температуре в течение 30 мин. Концентрировали под вакуумом, и очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение (10 мг, 20%). 1H ЯМР (300 MHz, DMSO-d6) δ 1.34-1.48 (m, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.08 (s, 3 H) 3.21 (s, 3 H) 3.82 (t, J=6.62 Hz, 2 H) 7.26 (s, 2 H) 7.41 (dd, J=8.82, 1.84 Hz, 1 H) 7.59-7.76 (m, 2 H) 7.89-8.04 (m, 3 H) 10.03 (s, 1 H) 10.34 (s, 1 H); MS (ESI+) m/z 496 (M+H)+; (ESI-) m/z 494 (M-H)-.
Пример 4A. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.3).
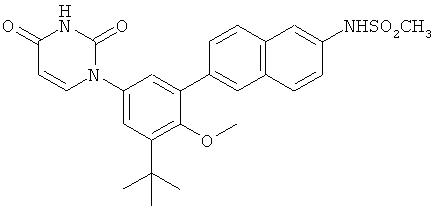
Часть A. Получение N-(6-бромнафтален-2-ил)метансульфонамида.
Раствор продукта из Примера 3, Части B (4,48 г, 20,17 ммоль) в пиридине (100 мл) обрабатывали по каплям метансульфонилхлоридом (1,97 мл, 2,89 г, 25,2 ммоль) с последующим перемешиванием при комнатной температуре в течение 1 ч. Разбавляли толуолом и дважды концентрировали под вакуумом. Остаток экстрагировали EtOAc и промывали водой, 1M лимонной кислотой и солевым раствором. Обрабатывали Darco G-60, сушили над Na2SO4, фильтровали через целит и концентрировали под вакуумом. Твердое вещество растирали в порошок с эфиром-гексаном, собирали фильтрованием и сушили под вакуумом с получением названного соединения в виде твердого вещества бледно-розового цвета (3,32 г, 55%).
Часть B. Получение N-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)нафтален-2-ил) метансульфонамида.
Смесь продукта из Части A (1,00 г, 3,33 ммоль), бис(пинаколато)дибора (1,27 г, 5,00 ммоль), ацетата калия (0,98 г, 9,99 ммоль) и Combiphos Pd6 (84 мг, 0,17 ммоль) в толуоле (22 мл) нагревали с обратным холодильником в течение 3 ч. Охлаждали и разбавляли этилацетатом и водой. Смесь обрабатывали Darco G-60 и фильтровали через целит. Фильтрат промывали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Масло растворяли в эфире и осаждали путем добавления гексана. Продукт собирали фильтрацией и промывали гексаном. Упаривание фильтрата и очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном. Названное соединение в результате кристаллизации и хроматографии получали в виде твердого вещества белого цвета (927 мг, 80%).
Часть C. Получение трет-бутил 3-трет-бутил-4-метокси-5-(6-(метилсульфонамид)нафтален-2-ил)фенилкарбамата.
Объединяли продукт из Примера 3, Части H (87 мг, 0,243 ммоль), продукт из Части B (169 мг, 0,486 ммоль), толуол (1,0 мл), этанол (1,0 мл) и карбонат натрия (0,243 мл, 0,243 ммоль) в герметично закрытой пробирке и дегазировали газообразным N2 в течение 20 мин. Добавляли тетракис(трифенилфосфин)палладий(0) (5,61 мг, 4,86 мкмоль) и дегазирование продолжали еще 5-10 мин. Нагревали при 90-95°C в течение 16 ч. Охлаждали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение (92,2 мг, 76%).
Часть D. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
Раствор продукта из Части C (90 мг, 0,180 ммоль) в CH2Cl2 (2,0 мл) обрабатывали трифторуксусной кислотой (1,0 мл, 12,98 ммоль) при комнатной температуре в течение 1 ч. Концентрировали под вакуумом, остаток растворяли в EtOAc, промывали 10% NaHCO3, и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Растворяли в DMF (1,4 мл) и охлаждали до -25°C и добавляли (E)-3-метокси-акрилоилизоцианат (0,633 мл, 0,361 ммоль) по каплям, поддерживая температуру ниже - 10°C. Нагревали до комнатной температуры и перемешивали в течение 2 ч. Выливали в эфир, промывали водой, и солевым раствором. Сушили над Na2SO4, фильтровали и
концентрировали под вакуумом. Добавляли смесь H2SO4 (0,1 мл, 1,876 ммоль), воды (1,0 мл) и EtOH (1,0 мл), и перемешивали при 100°C 16 ч. Охлаждали и концентрировали под вакуумом. Выливали в воду, экстрагировали EtOAc, экстракты объединяли и промывали солевым раствором. Сушили над NO3SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией MeOH/CHCl3 давала названное соединение (53 мг, 59%). 1H ЯМР (300 MHz DMSO-d6) δ 1.42 (s, 9 H) 3.08 (s, 3 H) 3.25 (s, 3 H) 5.65 (d, J=7.72 Hz, 1 H) 7.34 (dd, J=15.81, 2.57 Hz, 2 H) 7.42 (dd, J=8.82, 1.84 Hz. 1 H) 7.65-7.76 (m, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.96 (t, J=8.27 Hz, 2 H) 8.02 (s, 1 H) 10.04 (s, 1 H) 11.41 (s, 1 H); MS (ESI+) m/z 494 (M+H)+; (ESI-) m/z 492 (M-H)-.
Пример 4B. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.3).
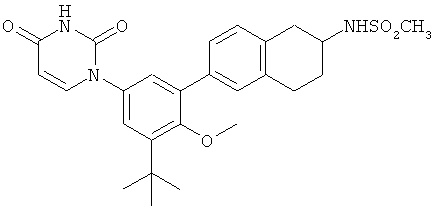
Часть A Получение 2-трет-бутил-6-йод-4-нитрофенола.
К продукту из Примера 3, Части E (4,5 г, 23,05 ммоль) растворенному в MeOH (120 мл) и воде (30 мл) добавляли монохлорид йода (1,155 мл, 23,05 ммоль) по каплям за период времени 10 мин. Смесь перемешивали в течение 2 ч и разбавляли в 1 л воды, и оставляли стоять в течение ночи. Твердое вещество собирали фильтрацией и промывали 3×50 мл водой и сушили под вакуумом в течение ночи с получением твердого вещества желтовато-коричневого цвета (7,14 г, 96%).
Часть B. Получение 1-трет-бутил-3-йод-2-метокси-5-нитробензола.
К охлажденному на ледяной бане раствору продукта из Части A (5,5 г, 17,13 ммоль) в MTBE (15 мл) в 50 мл сосуде высокого давления добавляли 2,0М триметилсилилдиазометан (12,85 мл, 25,7 ммоль) с последующим добавлением по каплям метанола (1,0 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали при комнатной температуре в течение 16 ч, охлаждали и давление сбрасывали. Раствор разделяли между EtOAc и водой. Органический слой промывали 1,0М HCl, насыщенным раствором карбоната калия, и насыщенным NaCl. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали с получением красного масла, которое использовали без очистки (5,4 г, 84%).
Часть C. Получение 3-трет-бутил-5-йод-4-метоксианилина. Смесь продукта из Части B (5,80 г, 17,31 ммоль), хлорида аммония (1,389 г, 26,0 ммоль), и железа (4,83 г, 87 ммоль) в THF/MeOH/воде (всего 200 мл, 2/2/1) нагревали с обратным холодильником в течение 2 ч, охлаждали и фильтровали через целит. Фильтрат упаривали, и остаток разделяли между водой и EtOAc. Органический слой промывали насыщенным солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением коричневого масла (5,28 г, 100% выход).
Часть D. Получение (E)-N-(3-трет-бутил-5-йод-4-метоксифенилкарбамоил)-3-метокси акриламида.
К раствору продукта из Части C (3,05 г, 10 ммоль) в DMF (50 мл) при -20°C в атмосфере N2 добавляли быстрыми каплями 0,4М раствор в бензоле (E)-3-метоксиакрилоилизоцианата (50,0 мл, 20,00 ммоль, полученного по способу Santana et al., J. Heterocyclic. Chem. 36:293 (1999). Раствор перемешивали в течение 15 мин при -20°C, нагревали до комнатной температуры в течение 45 мин и разбавляли EtOAc. Органическую фазу промывали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали до твердого вещества коричневого цвета. Остаток растирали в порошок в Et2O/гексане с получением мелкого порошка, который собирали фильтрованием и сушили под вакуумом с получением названного соединения в виде порошка желтовато-коричневого цвета (2,46 г, 57%).
Часть E. Получение 1-(3-трет-бутил-5-йод-4-метоксифенил)дигидропиримидин-2,4(1Н,3H)-диона.
К суспензии продукта из Части D (2,46 г, 5,69 ммоль) в этаноле (50 мл) добавляли раствор 5,5 мл H2SO4 в 50 мл воды и смесь нагревали при 110°C в течение 2,5 ч с получением прозрачного раствора. Охлаждали и разбавляли 50 мл воды при перемешивании с получением беловатого твердого вещества, которое собирали фильтрованием, промывали водой и сушили под вакуумом с получением названного соединения (2,06 г, 90%).
Часть F. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В микроволновой трубке, продукт из Части E (104 мг, 0,26 ммоль), продукт из Примера 4A, Части B (108 мг, 0,31 ммоль), и 1,0M раствор карбоната натрия (312 мкл, 0,31 ммоль) в смеси 1:1 этанол-толуол (1,7 мл) дегазировали путем барботирования азота в течение 15 мин. Добавляли комплекс 1,1'-бис(дифенилфосфино) ферроцен палладий (II) хлорид дихлорметан (9 мг, 0,011 ммоль), и дегазирование продолжали в течение еще 5 мин. Эту трубку герметично закрывали и нагревали в микроволновой печи при 100°C в течение 1 ч. Разбавляли дихлорметаном и промывали 1М раствором лимонной кислоты и солевым раствором. Органический слой затем перемешивали с (3-меркаптопропил) силикагелем в течение 1 ч. Фильтровали через целит и концентрировали под вакуумом. Растирали в порошок с эфиром, метанолом, а затем снова с эфиром с получением названного соединения в виде почти белого твердого вещества (32 мг, 25%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.41 (d, J=1.84 Hz, 1 Н) 10.04 (s, 1 Н) 8.03 (s, 1 Н) 7.96 (t, J=8.09 Hz, 2 Н) 7.80 (d, J=8.09 Hz, 1 Н) 7.63-7.79 (m, 2 Н) 7.35-7.45 (m, 1 Н) 7.37 (d, J=2.57 Hz. I Н) 7.32 (d, J=2.57 Hz, 1 Н) 5.65 (dd, J=8.09, 2.21 Hz, 1 Н) 3.25 (s, 3 Н) 3.09 (s, 3 Н) 1.43 (s, 9 Н). MS (+ESI) m/z (относительная распространенность): 494 (100, М+Н), 511 (90, M+NH4), 987 (20, 2М+Н), 1009 (8, 2M+Na).
Пример 5. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)хинолин-2-ил)метансульфонамида (соединение IB-L0-2.5).
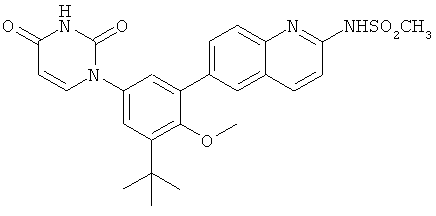
Часть A. Получение (E)-N-(4-бромфенил)-3-метоксиакриламида.
Объединяли 4-броманилин (285 мг, 1,659 ммоль), CH2Cl2 (2,0 мл) и пиридин (0,25 мл, 3,09 ммоль) и медленно добавляли (E)-3-метоксиакрилоилхлорид (200 мг, 1,659 ммоль) и перемешивали при комнатной температуре в течение 2 ч. Полученное в результате твердое вещество желтого цвета отфильтровывали и промывали водой. Твердое вещество сушили под вакуумом с получением названного соединения (406 мг, 96%).
Часть B. Получение 6-бромхинолин-2(1H)-она.
Продукт из Части A (395 мг, 1,542 ммоль) добавляли порциями к H2SO4 (4,5 мл). Перемешивали в течение 3 ч при комнатной температуре, выливали на дробленый лед. Твердое вещество фильтровали, промывали водой и сушили под вакуумом с получением названного соединения (203 мг, 59%).
Часть C. Получение 6-бром-2-хлорхинолина.
К оксихлориду фосфора (2,5 мл, 26,8 ммоль) добавляли, частями, продукт из Части B (200 мг, 0,893 ммоль). Нагревали с обратным холодильником в течение 1 ч, охлаждали до комнатной температуры и выливали на дробленый лед. Экстрагировали CHCl3, экстракты объединяли, сушили над MgSO4, фильтровали и концентрировали под вакуумом с получением названного соединения (173 мг, 80%).
Часть D. Получение 6-бром-2-аминохинолина.
Продукт из Части C (173 мг, 0,713 ммоль), ацетамид (843 мг, 14,27 ммоль) и карбонат калия (493 мг, 3.57 ммоль) объединяли и нагревали при 200°C в течение 2 ч. Охлаждали до комнатной температуры, после чего он застывал. Растворяли в смеси CHCl3 и воды. Водный слой экстрагировали еще два раза CHCl3, экстракты объединяли, промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией MeOH/CHCl3 давала названное соединение (92 мг, 58%).
Часть E. Получение N-(6-бромхинолин-2-ил)-N-(метилсульфонил)метансульфонамида.
Объединяли продукт из Части D (90 мг, 0,403 ммоль) и CH2Cl2 (2,0 мл) и добавляли триэтиламин (0,062 мл, 0,444 ммоль) и метансульфонилхлорид (0,035 мл, 0,444 ммоль). Перемешивали при комнатной температуре 16 ч. Добавляли триэтиламин (0,062 мл, 0,444 ммоль) и метансульфонилхлорид (0,035 мл, 0,444 ммоль) и перемешивали при комнатной температуре в течение 1 ч. Разбавляли EtOAc, промывали 10% лимонной кислотой, 10% NaHCO3 и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Растворяли в EtOAc и выливали в избыток гексана. Твердое вещество собирали фильтрованием с получением названного соединения (94 мг, 61%).
Часть F. Получение N-(метилсульфонил)-N-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хинолин-2-ил)метансульфонамида.
Объединяли продукт из Части E (94 мг, 0,248 ммоль),бис(пинаколато)дибор (94 мг, 0,372 ммоль), ацетат калия (73,0 мг, 0,744 ммоль), Combi-Phos®PD6 (6,22 мг, 0,012 ммоль) и толуол (1,5 мл) и нагревали с обратным холодильником 18 ч. Охлаждали до комнатной температуры, разбавляли EtOAc и водой, фильтровали через целит, отделяли фазы, органическую фазу промывали солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение (67 мг, 63%).
Часть G. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)хинолин-2-ил)метансульфонамида.
Объединяли в микроволновой трубке продукт из Примера 4B, Часть E (27 мг, 0,067 ммоль), продукт из Части F (37,4 мг, 0,088 ммоль), этанол (1,0 мл), толуол (1,0 мл) и 1М карбонат натрия (0,067 мл, 0,067 ммоль) и раствор дегазировали, используя газообразный N2, в течение 20 мин. Добавляли тетракис-(трифенил-фосфин)палладий(0) (1,559 мг, 1,349 мкмоль) и раствор дегазировали дополнительно 5 мин. Пробирку герметично закрывали и нагревали в микроволновой печи при 100°C в течение 45 мин. Охлажденный раствор разбавляли 1:1 EtOAc:водой и фильтровали через целит. Водный слой экстрагировали еще да раза EtOAc, объединяли органические экстракты и промывали солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией MeOH/CHCl3 давала названное соединение (13,7 мг, 41%). 1H ЯМР (300 MHz, CDCl3) δ 1.45 (s, 9 H) 3.18 (s, 3 H) 3.30 (s, 3 H) 5.83 (dd, J=7.91, 2.02 Hz, 1 H) 6.99 (d, J=8.82 Hz, 1 H) 7.21 (d, J=2.57 Hz, 1 H) 7.36 (d, J=7.72 Hz, 1 H) 7.52 (d, J=8.46 Hz, 1 H) 7.82-7.91 (m, 2 H) 7.98 (d, J=9.19 Hz, 1 H) 8.29 (s, 1 H); MS (ESI+) m/z 495 (M+H)+; (ESI-) m/z 493 (M-H)-.
Пример 6. Получение (Е)-N'-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил-иден)метансульфонгидразида (соединение IB-L0-2.4).
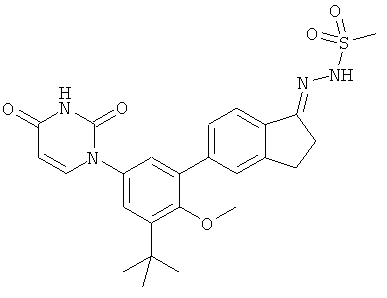
Часть A. Получение 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1H-инден-1-она.
Смесь 5-бром-2,3-дигидро-1H-инден-1-она (2,50 г, 11,85 ммоль), бис(пинаколато) дибора (3,61 г, 14,21 ммоль), ацетата калия (3,49 г, 35,5 ммоль) и Combiphos Pd6 (178 мг, 0,36 ммоль) в толуоле (60 мл) нагревали с обратным холодильником в течение 8 ч. Охлаждали, разбавляли EtOAc и экстрагировали водой (2×) и промывали солевым раствором. Сушили над Na2SO4 и перемешивали в течение 1 ч с (3-меркаптопропил)силикагелем. Фильтровали и концентрировали под вакуумом с получением твердого вещества желтого цвета. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала твердое вещество желтого цвета. Растирали в порошок с холодным гексаном, фильтровали и сушили под вакуумом с получением названного соединения в виде мелкодисперсного твердого вещества почти белого цвета (1,99 г, 65%). Вторую партию кристаллов (140 мг) получали из маточных растворов, получая выход до 70%.
Часть B. Получение 1-(3-трет-бутил-4-метокси-5-(1-оксо-2,3-дигидро-1H-инден-5-ил)фенил)пиримидин-2,4(1Н,3H)-диона.
В микроволновой трубке, суспензию продукта из Примера 4B, Часть Е (130 мг, 0,33 ммоль), продукта из Части A (101 мг, 0,39 ммоль), и 1,0М раствора карбоната натрия (390 мкл, 0,39 ммоль) в смеси 1:1 этанол-толуол (1,20 мл) дегазировали путем барботирования азота в течение 15 мин. Смесь обрабатывали комплексом 1,1'-бис(дифенилфосфино)ферроцен палладий (II) хлори дихлорметан (13 мг, 0,016 ммоль) и дегазирование продолжали в течение еще 5 мин, и нагревали при 100°C в микроволновой печи в течение 1 ч. Охлаждали, разбавляли EtOAc и экстрагировали 1М раствором лимонной кислоты и солевьм раствором. Органический слой затем перемешивали с (3-меркаптопропил)силикагелем в течение 1 ч. Фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение в виде твердого вещества белого цвета (80 мг, 61%).
Часть C. Получение (E)-N'-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1 -ил-иден)метансульфонгидразида.
Суспензию продукта из Части B (77 мг, 0,19 ммоль) и метансульфонилгидразида (22 мг, 0,20 ммоль) в 3:1 THF:MeOH (1,9 мл) нагревали при 60°C в течение 24 ч. Смесь концентрировали под вакуумом и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гексаном с получением названного соединения в виде твердого вещества белого цвета (62 мг, 66%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.40 (d, J=1.84 Hz, 1 H) 9.94 (s, 1 H) 7.76 (dd, J=13.97, 8.09 Hz, 2 H) 7.52-7.59 (m, 1 H) 7.51 (d, J=8.46 Hz, 1 H) 7.11-7.40 (m, 2 H) 3.28 (s, 3 H) 2.96-3.19 (m, 5 H), 2.85 (m, 2 H), 1.40 (s, 9 H). MS (+ESI) m/z (относительная распространенность): 497 (100, М+Н), 1015 (5, 2M+Na).
Пример 7. Получение N-(2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]оксазол-5-ил)метансульфонамида (соединение IB-L0-2.6).
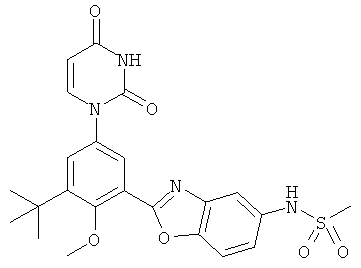
Часть A. Получение метил 3-трет-бутил-2-гидрокси-5-нитробензоата.
Метил 3,5-ди-трет-бутил-2-гидроксибензоат (28,66 г, 108,4 ммоль) растворяли при перемешивании в 430 мл ледяной уксусной кислоты и полученную в результате смесь обрабатывали по каплям дымящей азотной кислотой (90%, 179,26 мл). Когда добавление было завершено, полученную в результате смесь перемешивали в течение 2,5 ч. Реакционную смесь выливали в 2,0 л дробленого льда и оставляли стоять 30 мин. После этого, добавляли 1,0 л воды и смесь льда и воды оставляли расплавляться. Затем смесь фильтровали, промывали водой и сушили с получением названного соединения (24,57 г, 89%).
Часть B. Получение метил 3-трет-бутил-2-метокси-5-нитробензоата.
Добавляли вместе метил 3-трет-бутил-2-гидрокси-5-нитробензоат (11,41 г, 45,0 ммоль), карбонат калия (9,34 г, 67,6 ммоль), ацетон (200 мл), и диметилсульфат (6,46 г, 67,6 ммоль). Полученную в результате смесь затем нагревали с обратным холодильником в течение 16 ч. Смесь затем фильтровали, и твердое вещество промывали этилацетатом. Полученную в результате органическую жидкость затем концентрировали под вакуумом до масла и повторено растворяли в этилацетате (600 мл). Органический раствор затем промывали водой, сушили, фильтровали и концентрировали под вакуумом до масла, которое затем подвергали очистке посредством колоночной хроматографии (градиент 5%-40% EtOAc/гексан) с получением на выходе названного соединения в виде масла (10,42, 87%).
Часть C. Получение метил 5-амино-3-трет-бутил-2-метоксибензоата.
Добавляли вместе метил 3-трет-бутил-2-метокси-5-нитробензоат (10,42 г, 39,0 ммоль), железные опилки (325 меш, 10,89 г, 195 ммоль), хлорид аммония (3,13 г, 58,5 ммоль), воду (30 мл), и метанол (150 мл). Полученную в результате смесь затем нагревали с обратным холодильником в течение 1 ч. Смесь затем охлаждали до комнатной температуры, фильтровали через целит, и целит промывали метанолом. Фильтрат затем концентрировали под вакуумом и растворяли в этилацетате (600 мл). Полученный в результате раствор затем промывали водой и солевым раствором. Органический экстракт затем сушили, фильтровали и концентрировали под вакуумомс получением на выходе названного соединения в виде масла (9,25 г, 100%).
Часть D. Получение (E)-метил 3-трет-бутил-2-метокси-5-(3-(3-метоксиакрилоил)уреид) бензоата.
Продукт, полученный, как описано в Части C (2,0 г, 8,43 ммоль) растворяли в 30 мл N,N-диметилацетамида и охлаждали до -25°C. 0,5 молярный раствор E-3-метоксиакрилоилизоцианата в бензоле (21,9 мл, 10,96 ммоль) добавляли по каплям, и полученный в результате раствор перемешивали при окружающей температуре в течение 4 ч, а затем выливали в воду. Продукт экстрагировали в дихлорметан, промывали солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали под вакуумом с получением 100% выхода.
Часть E. Получение метил 3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксибензоата.
Продукт из Части D (3,1 г, 8,51 ммоль) растворяли в этаноле (60 мл). Серную кислоту (6 мл) добавляли в воду (60 мл), а затем этот раствор добавляли одной порцией в этанол. Гетерогенную смесь нагревали при 100°C в течение 3 ч. Этанол удаляли под вакуумом, а затем водный раствор экстрагировали дихлорметаном и упаривали до сухости. Этот остаток очищали с помощью флэш-хроматографии, элюируя 1% метан олом/дихлорметаном с получением на выходе 1,23 г (44%).
Часть F. Получение 3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксибензойной кислоты.
Продукт из Части E (1,23 г, 3,7 ммоль) помещали в этанол (5 мл) и 1 молярный раствор гидроксида натрия (10 мл) и перемешивали при окружающей температуре в течение 18 ч. Этот раствор разбавляли 1М HCl и полученное в результате твердое вещество фильтровали и сушили с получением 0,945 г (80%).
Часть G. Получение 3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксибензальдегида.
Продукт из Части F (0,945 г, 2,97 ммоль) помещали в тионилхлорид (4.5 мл) и смесь нагревали при 80°C в течение 40 мин. После упаривания до сухости, хлорангидрид растворяли в сухом THF (8 мл) и охлаждали до -78°C. 1 молярный раствор литий три-трет-бутоксиалюминий гидрида в THF (3,0 мл, 3,0 ммоль) добавляли по каплям. Через 45 мин холодную реакционную смесь гасили 1М HCl (5 мл), экстрагировали в этилацетат, и очищали с помощью испарительной колонки, элюируя дихлорметаном, после этого 1% метанолом/дихлорметаном с получением 0,635 г (71%).
Часть H. Получение 1-(3-трет-бутил-4-метокси-5-(5-нитробензо[d]оксазол-2-ил)фенил)пиримидин -2,4(1Н,3H)-диона.
Продукт из Части G (400 мг, 1,323 ммоль), 2-амино-4-нитрофенол (204 мг, 1,323 ммоль), древесный уголь (Darco KB, 191 мг, 15,88 ммоль) и толуол (50 мл) добавляли в колбу и смесь нагревали до 120°C, и перемешивали открыто на воздухе в течение 48 ч. Фильтровали через целит и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией CH2Cl2/MeOH давала названное соединение (300 мг, 52%).
Часть I. Получение N-(2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]оксазол-5-ил)метансульфонамида.
К продукту из Части H (300 мг, 0,687 ммоль), железу (192 мг, 3,44 ммоль), и хлориду аммония (55 мг, 1,031 ммоль) добавляли смесь THF (15 мл), EtOH (15 мл) и воды (4,5 мл). Полученный в результате раствор нагревали до 90°C в течение 45 мин, и охлаждали. Фильтровали через целит, промывали этанолом, и концентрировали под вакуумом. Твердое вещество растворяли в этилацетате, и промывали водой. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией CH2Cl2/MeOH давала анилин. Твердое вещество (75 мг, 0,185 ммоль) растворяли в CH2Cl2 (5 мл), и добавляли пиридин (0,045 мл, 0,554 ммоль) и метансульфонилхлорид (0,025 мл, 0,323 ммоль) и перемешивали при комнатной температуре в течение 16 ч. Добавляли CH2Cl2 с последующим промыванием 1 н HCl. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией CH2Cl2/MeOH давала названное соединение в виде твердого вещества (9,8 мг, 3%, две стадии). 1H ЯМР (300 MHz, DMSO-d6): δ 11.46 (s, 1H), 9.85 (s, 1H), 7.91 (d, J=2.2Hz, 1Н), 7.81 (dd, J=9.9, 8.8 Hz, 2H), 7.68 (d, J=2.2 Hz, 1Н), 7.56 (d, J=2.6 Hz, 1Н), 7.33 (dd, J=8.8, 1.8 Hz, 1Н), 5.68 (d, J=7.7 Hz, 1Н), 3.64 (s, 3H), 3.00 (s, 3H), 1.42 (s, 9Н). MS: m/z 485 (M+H)+.
Пример 8. Получение 1-(3-трет-бутил-4-метокси-5-(6-нитробензо[d]оксазол-2-ил)фенил) дигидропиримидин-2,4(1Н,3H)-диона (соединение IA-L0-2.6).
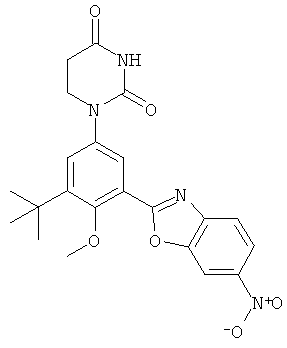
Часть A. Получение 3-(3-трет-бутил-4-метокси-5-(метоксикарбонил)фениламино) пропановой кислоты.
Продукт из Примера 7, Часть C (16,44 г, 69,3 ммоль) растворяли в толуоле (200 мл). Эту смесь нагревали с обратным холодильником и добавляли акриловую кислоту на протяжении периода времени (1 мл акриловой кислоты добавляли каждые 3 ч, всего 5,23 мл, 76,2 ммоль). Затем смесь нагревали с обратным холодильником в течение 24 ч. Затем смесь охлаждали и концентрировали под вакуумом до сухости с получением на выходе масла в качестве неочищенного названного соединения, которое использовали непосредственно в следующей реакции.
Часть B. Получение метил 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензоата.
Добавляли вместе продукт из Части A (21,43 г, 69,3 ммоль), мочевину (10,4 г, 173 ммоль) и уксусную кислоту (ледяную, 200 мл). Смесь затем нагревали до 120°C в течение 18,5 ч с последующим концентрированном под вакуумом до сухости до получения масла. К этому маслу добавляли метанол (13 мл), и этилацетат (350 мл). Полученную в результате смесь оставляли стоять в течение 24-48 ч, за счет чего образовывался осадок. Полученное в результате твердое вещество отфильтровывали и промывали небольшим количеством метанола (10 мл), а затем сушили на воздухе с получением на выходе названного соединения в виде твердого вещества (15,26 г, 66%).
Часть C. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси бензойной кислоты.
Добавляли вместе продукт из Части B (4,52 г, 13,52 ммоль), метанол (70 мл), и тетрагидрофуран (70 мл). Смесь затем интенсивно перемешивали до получения в результате гомогенного раствора. После достижения гомогенности, добавляли раствор водного гидроксида натрия (1,0М, 68 мл). Смесь затем перемешивали в течение 12 ч, смесь затем концентрировали под вакуумом для удаления органического растворителя с последующим добавлением водного раствора соляной кислоты (1,0М, 80 мл), что в результате приводило к образованию твердого вещества. Смесь затем концентрировали под вакуумом. К этому веществу добавляли соляную кислоту (12М, 100 мл) и полученное в результате вещество нагревали до 100°C в течение 1,5 ч. Реакционную смесь затем охлаждали и добавляли воду. Полученное в результате твердое вещество фильтровали, промывали водой, и сушили с получением на выходе названного соединения в виде твердого вещества (3,55 г, 82%).
Часть D. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси бензальдегида.
Продукт, полученный в Части C (4,07 г, 12,71 ммоль) и тионилхлорид (40,82 мл, 559 ммоль) объединяли, и смесь нагревали с обратным холодильником в течение 2 ч, с последующим концентрированном под вакуумом с получением твердого продукта светло-желтого цвета. Твердое вещество растворяли в тетрагидрофуране (125 мл), раствор охлаждали до -78°C и медленно добавляли LiAl(OtBu)3 (1М, 14 мл) на протяжении 10 мин, поддерживая температуру -78°C. Смесь перемешивали при 78°C в течение 2 ч. Реакцию гасили соляной кислотой (водн., 1М, 25 мл) при -78°C. Смесь нагревали до комнатной температуры и добавляли этилацетат. Слои отделяли, и водный слой промывали этилацетатом. Органические экстракты объединяли и промывали полунасыщенным раствором бикарбоната натрия. Органический слой сушили, фильтровали и концентрировали под вакуумом с получением на выходе твердого вещества в качестве названного соединения (3,73 г, 96%).
Часть E. Получение 1-(3-трет-бутил-4-метокси-5-(6-нитробензо [d]оксазол-2-ил)фенил)дигидропиримидин-2,4(1Н,3H)-диона.
Смесь продукта из Части D (75 мг, 0,246 ммоль), 2-амино-5-нитрофенола (38 мг, 0,0246 ммоль) и древесного угля Darco KB (избыток) нагревали с обратным холодильником в толуоле (10 мл) в течение 24 ч под воздействием атмосферного кислорода. Охлаждали, фильтровали и очищали с помощью ВЭЖХ с обращенной фазой, элюируя 40-100% градиентом ацетонитрила в воде (0,1% TFA) с получением названного соединения в виде твердого вещества (96 мг, 64%). 1H ЯМР (300 MHz, DMSO-d6): δ 1.42 (s, 9 H) 2.74 (t, J=6.80 Hz, 2 H) 3.66 (s, 3 H) 3.82-3.88 (m, 2 H) 7.56 (d, J=2.57 Hz, 1 H) 7.91 (d, J=2.57 Hz, 1 H) 8.09 (d, J=8.82 Hz, 1 H) 8.37 (dd, J=8.82, 2.21 Hz, 1 H) 8.84 (d, J=2.21 Hz, 1 H) 10.44 (s, 1 H). MS ESI+ (439) (M+H)+.
Пример 9. Получение N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]оксазол-6-ил)метансульфонамида (соединение IA-L0-2.5).
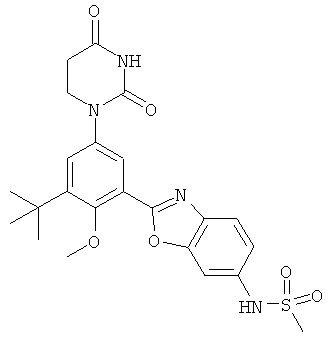
Продукт из Примера 8 (96 мг, 0,219 ммоль) взаимодействовал с железом (0,614 г, 1,10 ммоль), и хлоридом аммония (0,176 г, 0,329 ммоль) в присутствии смеси тетрагидрофурана (5 мл), этанола (5 мл) и воды (3 мл). Суспензию нагревали до 90°C в течение 45 мин, охлаждали до окружающей температуры. Фильтровали через слой целита (10 г), промывали этанолом (20 мл), и фильтрат концентрировали под вакуумом до твердого вещества. Полученное в результате твердое вещество растворяли в этилацетате и промывали водой. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом до твердого вещества желтого цвета, получая соответствующий анилин. Твердое вещество растворяли в дихлорметане (10 мл), добавляли пиридин (0,670 мл, 0,657 ммоль) и метансульфонилхлорид (0,221 мл, 0,329 ммоль) и раствор перемешивали при комнатной температуре 16 ч. Добавляли CH2Cl2 с последующим промыванием 1 н водным раствором HCl. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 98:2 CH2Cl2:MeOH давала названное соединение в виде твердого вещества (25 мг, 21%, две стадии). 1H ЯМР (300 MHz, DMSO-d6): δ 1.41 (s, 9 Н) 2.73 (t, J=6.62 Hz, 2 Н) 3.06 (s, 3 Н) 3.61 (s, 3 Н) 3.83 (t, J=6.62 Hz, 2 Н) 7.28 (dd, J=8.46, 1.84 Hz, 1 Н) 7.48 (d, J=2.57 Hz, 1 Н) 7.65 (d, J=1.84 Hz, 1 Н) 7.80 (d, J=1.47 Hz, 1 Н) 7.82 (d, J=4.04 Hz, 1 Н) 10.03 (s, 1 Н) 10.41 (s, 1 Н). MS ESI+ (487) (M+H)+.
Пример 10. Получение 1-(3-трет-бутил-4-метокси-5-(5-нитробензо[d]оксазол-2-ил)фенил) дигидропиримидин-2,4(1Н,3H)-диона (соединение IA-L0-2.7).
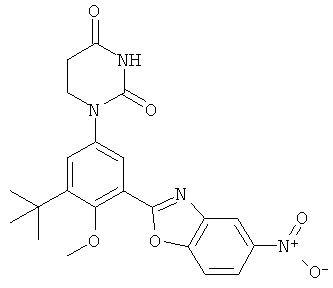
Продукт из Примера 8, Часть D(150 мг, 0,493 ммоль) взаимодействовал с 2-амино-4-нитрофенолом (76 мг, 0,493 ммоль) в соответствии с процедурами из Примера 8, Части E с получением названного соединения в виде твердого вещества (70 мг, 32%). 1H ЯМР (300 MHz, DMSO-d6): δ 1.42 (s, 9 Н) 2.74 (t, J=6.80 Hz, 2 Н) 3.65 (s, 3 Н) 3.85 (t, J=6.62 Hz, 2 Н) 7.55 (d, J=2.57 Hz, 1 Н) 7.89 (d, J=2.94 Hz, 1 Н) 8.12 (d, J=8.82 Hz, 1 Н) 8.40 (dd, J=9.01, 2.39 Hz, 1 Н) 8.76 (d, J=2.21 Hz, 1 Н) 10.43 (s, 1 Н). MS ESI+ (439) (M+H)+.
Пример 11. Получение N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)бензо[d]оксазол-5-ил)метансульфонамида (соединение IA-L0-2.8).
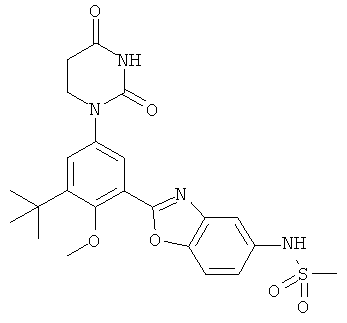
Продукт из Примера 10 (65 мг, 0,148 ммоль) вступал в реакцию в соответствии с процедурами из Примера 9 с образованием названного соединения в виде твердого вещества (42 мг, 44%). 1H ЯМР (300 MHz, DMSO-d6): δ 1.41 (s, 9 Н) 2.73 (t, J=6.43 Hz, 2 Н) 3.01 (s, 3 Н) 3.60 (s, 3 Н) 3.83 (t, J=6.43 Hz, 2 Н) 7.31 (dd, J=8.64, 2.02 Hz, 1 Н) 7.49 (d, J=2.94 Hz, 1 Н) 7.56 (d, J=2.21 Hz, 1 Н) 7.67 (d, J=2.21 Hz, 1 Н) 7.81 (s, 1 Н) 9.82 (s, 1 H) 10.41 (s, 1 Н). MS ESI+ (487) (M+H)+.
Пример 12. Получение 1-(3-(бензо[d]тиазол-2-ил)-5-трет-бутил-4-метоксифенил)дигидро пиримидин-2,4(1Н,3H)-диона (соединение IA-L0-2.3).
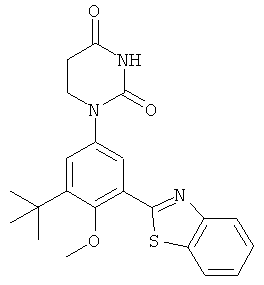
Продукт из Примера 8, Часть D (75 мг, 0,246 ммоль) взаимодействовал с 2-аминобензолтиолом (0,026 мл, 0,246 ммоль) в соответствии с процедурами из Примера 8, Части E с образованием названного соединения в виде твердого вещества (25 мг, 25%). 1H ЯМР (300 MHz, DMSO-d6): δ 1.44 (s, 9 H) 2.73 (t, J=6.43 Hz, 2 H) 3.62 (s, 3 H) 3.84 (t, J=6.62 Hz, 2 H) 7.46 (d, J=2.57 Hz, 1 H) 7.48-7.60 (m, 2 H) 7.86 (d, J=2.57 Hz, 1 H) 8.13 (dd, J=17.28, 7.72 Hz, 2 H) 10.40 (s, 1 H). MS ESI+ (410) (M+H)+.
Пример 13. Получение N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)-1Н-бензо[d]имидазол-5-ил)метансульфонамида (соединение IA-L0-2.1).
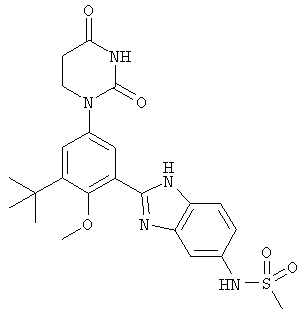
Часть A. Получение N-(3,4-динитрофенил)метансульфонамида.
Смесь 3,4-динитроанилина (5,27 г, 28,8 ммоль), метансульфонилхлорида (3,36 мл, 43,1 ммоль) и пиридина (5,82 мл, 71,9 ммоль) в CH2Cl2 (100 мл) перемешивали в течение 24 ч. Смесь концентрировали под вакуумом с получением сырого полутвердого названного соединения, которое использовали без дальнейшей очистки.
Часть B. Получение N-(3,4-диаминофенил)метансульфонамида.
Продукт из Части A (7,51 г, 28,8 ммоль) взаимодействовал с железом (16 г, 288 ммоль) и NH4Cl (3.84 г, 71.9 ммоль) в нагревающемся с обратным холодильником CH3OH (100 мл) и воде (20 мл) в течение 2 ч. Фильтровали через целит и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией MeOH/CH2Cl2 давала названное соединение в виде темного полутвердого вещества (0,5 г, 8%).
Часть C. Получение N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси фенил)-1H-бензо[d]имидазол-5-ил)метансульфонамида.
Смесь продукта из Примера 8, Части D (200 мг, 0,657 ммоль) взаимодействовала с продуктом из Части B (132 мг, 0,657 ммоль) в соответствии с процедурами из Примера 8, Части E с образованием названного соединения в виде твердого вещества (112 мг, 34%). 1H ЯМР (300 MHz, DMSO-d6): δ 1.43 (s, 9 Н) 2.72 (t, J=6.62 Hz, 2 Н) 2.93 (s, 3 Н) 3.44 (s, 3 Н) 3.82 (t, J=6.43 Hz, 2 Н) 7.07-7.14 (m, 1 H) 7.38 (d, J=2.57 Hz, 1 Н) 7.48-7.64 (m, 2 Н) 7.72 (d, J=2.57 Hz, 1 Н) 9.57 (s, 1 Н) 10.38 (s, 1 Н) 12.55 (s, 1 Н). MS ESI+ (486) (M+H)+.
Пример 14. Получение N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]тиазол-6-ил)метансульфонамида (соединение IA-L0-2.2).
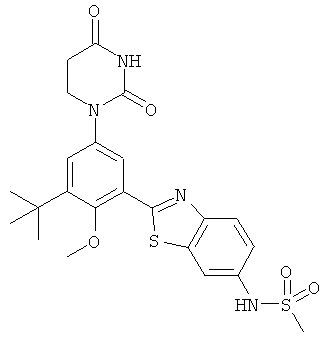
Часть A. Получение N-(3-хлор-4-нитрофенил)метансульфонамида.
Смесь 3-хлор-4-нитроанилина (4,85 г, 28,1 ммоль), метансульфонилхлорида (3,29 мл, 42,2 ммоль) и пиридина (6,82 мл, 84 ммоль) в THF (100 мл) перемешивали в течение 24 ч. Выливали в 1М HCl (500 мл). Полученный в результате осадок фильтровали и сушили на воздухе с получением названного соединения в виде твердого вещества (7,03 г, 100%).
Часть B. Получение N-(3-(4-метоксибензилтио)-4-нитрофенил)метансульфонамида.
Смесь продукта из Части A (7,0 г, 27,9 ммоль), (4-метоксифенил)метантиола (3,89 мл, 27,9 ммоль) и K2CO3 (11,58 г, 84 ммоль) в DMF нагревали при 100°C в течение 12 ч. Охлаждали и выливали в 1М HCl (800 мл). Полученный в результате осадок фильтровали и сушили на воздухе с получением названного соединения в виде твердого вещества желтого цвета (6,98 г, 68%).
Часть C. Получение N-(4-амино-3-(4-метоксибензилтио)фенил)метансульфонамида.
Продукт из Части B (6,98 г, 19,0 ммоль) взаимодействовал в соответствии с процедурами из Примера 13, Части B с получением названного соединения в виде желтого полутвердого вещества (4,44 г, 69%).
Часть D. Получение N,N'-(3,3'-дисульфандиилбис(4-амино-3,1-фенилен))диметан-сульфонамида.
Продукт из Части C (708 мг, 2,09 ммоль) взаимодействовал с ацетатом ртути (II) (667 мг, 2,09 ммоль), анизолом (0,457 мл, 4,18 ммоль) и TFA (10 мл) при 0°C в течение 45 мин. Концентрировали под вакуумом и растворяли в MeOH. Газообразный сероводород барботировали в раствор в течение 1 ч с последующий фильтрацией и концентрированней под вакуумом. Очистка с помощью хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение в виде желтоватого твердого вещества (340 мг, 75%).
Часть E. Получение N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]тиазол-6-ил)метансульфонамида.
Продукт из Части D (100 мг, 0,23 ммоль) взаимодействовал с продуктом из Примера 8, Части D (140 мг, 0,46 ммоль), трифенилфосфином (60,4 мг, 0,23 ммоль) и 4-метилбензолсульфоновой кислотой (0,0054 мл, 0,046 ммоль) в нагревающемся с обратным холодильником толуоле в течение 3 ч. Концентрировали под вакуумом и очищали с помощью ВЭЖХ хроматографии с обращенной фазой, элюируя 40-100% градиентом ацетонитрила в воде (0,1% TFA) с получением названного соединения в виде твердого вещества (99 мг, 43%). 1H ЯМР (300 MHz, DMSO-d6): δ 1.43 (s, 9 H) 2.73 (t, J=6.62 Hz, 2 H) 3.07 (s, 3 H) 3.63 (s, 3 H) 3.83 (t, J=6.62 Hz, 2 H) 7.39 (dd, J=8.82, 2.21 Hz, 1 H) 7.45 (d, J=2.57 Hz, 1 H) 7.83 (d, J=2.57 Hz, 1 H) 7.95 (d, J=2.21 Hz, 1 H) 8.05 (d, J=8.82 Hz, 1 H) 10.03 (s, 1 H) 10.39 (s, 1 H). MS ESI+ (503) (M+H)+.
Пример 15. Получение N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]тиазол-5-ил)метансульфонамида (соединение IA-L0-2.4).
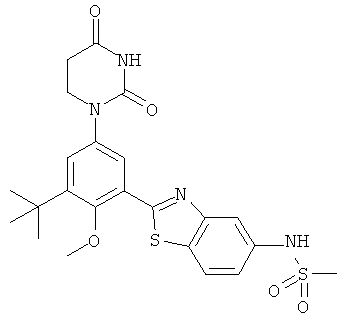
Часть A. Получение N-(4-хлор-3-нитрофенил)метансульфонамида.
Смесь 4-хлор-3-нитроанилина (5,0 г, 29 ммоль), метансульфонилхлорида (2,37 мл, 30,4 ммоль) и пиридина (5,9 мл, 72,4 ммоль) в THF (100 мл) перемешивали в течение 24 ч. Выливали в 1М HCl (500 мл). Полученный в результате осадок фильтровали и сушили на воздухе с получением названного соединения в виде твердого вещества (6,7 г, 92%).
Часть B. Получение N-(4-(4-метоксибензилтио)-3-нитрофенил)метансульфонамида. Смесь продукта из Части A (3,0 г, 12 ммоль), (4-метоксифенил)метантиола (1,67 мл, 12 ммоль) и K2CO3 (4,96 г, 36 ммоль) в DMF нагревали при 100°C в течение 12 ч. Охлаждали и выливали в 1М HCl (800 мл). Полученный в результате осадок фильтровали и сушили на воздухе с получением названного соединения в виде твердого вещества желтого цвета (1,95 г, 44.2%).
Часть C. Получение N-(3-амино-4-(4-метоксибензилтио)фенил)метансульфонамида.
Продукт из Части B (1,43 г, 3,88 ммоль) вступал в реакции в соответствии с процедурами из Примера 13, Части B с образованием названного соединения в виде твердого вещества белого цвета (1,31 г, 100%).
Часть D. Получение N,N'-(4,4'-дисульфандиилбис(3-амино-4,1-фенилен))диметан-сульфонамида.
Продукт из Части C (75 мг, 0,222 ммоль) взаимодействовал с ацетатом ртути (II) (70,6 мг, 0,222 ммоль), анизолом (0,048 мл, 0,443 ммоль) и TFA (10 мл) при 0°C в течение 45 мин. Концентрировали под вакуумом и растворяли в MeOH. Газообразный сероводород барботировали в раствор в течение 1 ч с последующей фильтрацией и концентрированием под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией EtOAc/гексаном давала названное соединение в виде желтоватого твердого вещества (34 мг, 71%).
Часть E. Получение N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси фенил)бензо[d]тиазол-5-ил)метансульфонамида. Продукт из Части D (50 мг, 0,115 ммоль) взаимодействовал с продуктом из Примера 8, Части D (70 мг, 0,230 ммоль), трифенилфосфином (30,2 мг, 0,115 ммоль) и 4-метилбензолсульфоновой кислотой (0,00267 мл, 0,023 ммоль) в нагревающемся с обратным холодильником толуоле в течение 3 ч. Концентрировали под вакуумом и очищали с помощью ВЭЖХ хроматографии с обращенной фазой, элюируя 40-100% градиентом ацетонитрила в воде (0,1% TFA) с получением названного соединения в виде твердого вещества (40 мг, 33%). 1H ЯМР (300 MHz, DMSO-d6): δ 1.43 (s, 9 H) 2.73 (t, J=6.80 Hz, 2 H) 3.05 (s, 3 H) 3.63 (s, 3 H) 3.84 (t, J=6.62 Hz, 2 H) 7.35 (dd, J=8.64, 2.02 Hz, 1 H) 7.46 (d, J=2.94 Hz, 1 H) 7.86 (d, J=2.94 Hz, 1 H) 7.92 (d, J=1.84 Hz, 1 H) 8.10 (d, J=8.46 Hz, 1 H) 9.98 (s, 1 H) 10.40 (s, 1 H). MS ESI+ (503) (M+H)+.
Пример 16. Получение 1-(3-трет-бутил-4-метокси-5-(нафтален-2-ил)фенил) пиримидин-2,4(1Н,3H)-диона (соединение IB-L0-2.1).
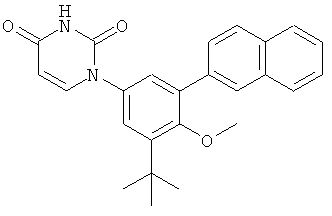
Часть A. Получение трет-бутил 3-трет-бутил-4-метокси-5-(нафтален-2-ил)фенил карбамата. В повторно герметизируемом сосуде Шленка, раствор продукта из Примера 3, Части H (200 мг, 0,56 ммоль), нафтален-2-бороновую кислоту (144 мг, 0,84 ммоль), и 1,0М раствор карбоната натрия (558 мкл, 0,56 ммоль) в толуоле (2.8 мл) дегазировали путем барботирования азота в течение 10 мин. Эту смесь обрабатывали комплексом 1,1'-бис(дифенилфосфино)ферроцен палладий (II) хлорид дихлорметан (14 мг, 0,017 ммоль) и дегазирование продолжали в течение еще 5 мин. Сосуд Шленка герметично закрывали и нагревали при 95°C в течение 18 ч. Охлаждали и разбавляли этилацетатом и водой. Обрабатывали Darco G-60 и фильтровали через целит. Фильтрат экстрагировали водой (2×) и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 10-75% EtOAc в гексане давала названное соединение в виде масла (210 мг, 93%).
Часть B. Получение 3-трет-бутил-4-метокси-5-(нафтален-2-ил)анилина. Продукт из Части A (210 мг, 0,52 ммоль) растворяли в 4 н HCl в диоксане (4,0 мл) и перемешивали при комнатной температуре в течение 1 ч. Концентрирование под вакуумом давало твердое вещество, которое суспендировали в этилацетате и перемешивали насыщенным раствором бикарбоната натрия. Органический слой сушили над Na2S04, фильтровали и концентрировали под вакуумом с получением названного соединения, в виде коричневого масла (111 мг, 70%).
Часть C. Получение (E)-N-(3-трет-бутил-4-метокси-5-(нафтален-2-ил)фенилкарбамоил)-3-метоксиакриламида.
Раствор продукта из Части B (111 мг, 0,36 ммоль) в сухом DMF (2,9 мл) при -20°C обрабатывали раствором (E)-3-метоксиакрилоилизоцианата (0,66 мл, 0,55М в бензоле, 0,36 ммоль) с последующим постепенным нагреванием до комнатной температуры. После перемешивания в течение 30 мин, смесь охлаждали снова до -20°C и добавляли еще раствор (E)-3-метоксиакрилоилизоцианата (1,0 мл, 0,55 ммоль). После повторного нагревания до комнатной температуры в течение 30 мин, реакцию завершали. Разбавляли EtOAc и экстрагировали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 10-100% EtOAc в гексане давала названное соединение в виде светло-желтого масла (144 мг, 92%).
Часть D. Получение 1-(3-трет-бутил-4-метокси-5-(нафтален-2-ил)фенил)пиримидин-2,4(1Н,3H)-диона.
Суспензию продукта из Части C (144 мг, 0,33 ммоль) в смеси 2:2:1 этанол-вода-THF (15 мл) обрабатывали раствром 1 н серной кислоты (3,0 мл) с последующим нагреванием при 100°C в течение 24 ч. Охлаждали и разбавляли EtOAc и экстрагировали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 10-100% EtOAc в гексане давала названное соединение в виде твердого вещества белого цвета (62 мг, 47%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.42 (s, 1 H), 8.08 (s, 1 H), 7.90-8.04 (m, 3 H), 7.81 (d, J=7.72 Hz, 1 H), 7.72 (d, J=8.46 Hz, 1 H), 7.56 (dd, J=6.25, 3.31 Hz, 2 H), 7.39 (d, J=2.57 Hz, 1 H), 7.33 (d, J=2.57 Hz, 1 H), 5.65 (d, J=7.72 Hz, 1 H), 3.24 (s, 3 H), 1.43 (s, 9 H). MS+ESI m/z (относительная распространенность): 401 (100, М+Н), 418 (30, M+NH4).
Пример 17. Получение 1-(3-трет-бутил-4-метокси-5-(6-метоксинафтален-2-ил)фенил) пиримидин-2,4(1Н,3H)-диона (соединение IB-L0-2.2).
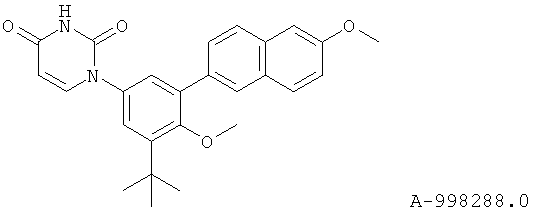
Часть A. Получение трет-бутил 3-трет-бутил-4-метокси-5-(6-метоксинафтален-2-ил)фенил карбамата.
Продукт из Примера 3, Части H (158 мг, 0,44 ммоль) взаимодействовал с 6-метокси-нафтален-2-илбороновой кислотой (107 мг, 0,52 ммоль) в соответствии с процедурами из Примера 16, Части A с образованием названного соединения в виде твердого вещества белого цвета (92 мг, 47%).
Часть B. Получение 3-трет-бутил-4-метокси-5-(6-метоксинафтален-2-ил)анилина. Продукт из Части A (92 мг, 0,21 ммоль) вступал в реакции в соответствии с процедурами из Примера 16, Части B с образованием названного соединения в виде твердого вещества розового цвета (71 мг, 99%).
Часть C. Получение (E)-N-(3-трет-бутил-4-метокси-5-(6-метоксинафтален-2-ил)фенил карбамоил)-3-метоксиакриламида.
Продукт из Части B (71 мг, 0,21 ммоль) вступал в реакции в соответствии с процедурами из Примера 16, Часть C с образованием названного соединения в виде темно-желтого твердого вещества (58 мг, 59%).
Часть D. Получение 1-(3-трет-бутил-4-метокси-5-(6-метоксинафтален-2-ил)фенил) пиримидин-2,4(1Н,3H)-диона.
Раствор продукта из Части C (58 мг, 0,13 ммоль) в смеси 2:1:1 этанол-THF-вода (4,0 мл) обрабатывали 1,0М раствором серной кислоты (3,0 мл) с последующим нагреванием при 95°C в течение 24 ч. Охлаждали и разбавляли EtOAc. Экстрагировали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 10-100% EtOAc в гексане давала продукт в виде бледно-розового твердого вещества (28 мг, 52%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.41 (s, 1 H), 8.00 (s, 1 H), 7.91 (dd, J=8.64, 4.60 Hz, 2 H), 7.80 (d, J=7.72 Hz, 1 H), 7.67 (d, J=8.82 Hz, 1 H), 7.34-7.47 (m, 2 H), 7.21-7.32 (m, 1 H), 7.20 (dd, J=9.01, 2.39 Hz, 1 H), 5.65 (d, J=7.72 Hz, 1 H), 3.90 (s, 3 H), 3.24 (s, 3 H), 1.42 (s, 9 H). MS +ESI m/z (относительная распространенность): 431 (100, М+Н), 448 (45, M+NH4).
Пример 18. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.8).
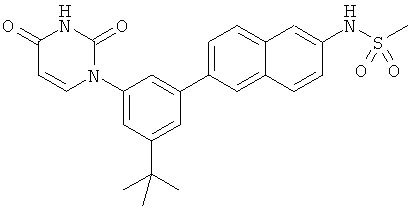
Часть A. Получение 2-бром-4-трет-бутил-6-нитроанилина.
Суспензию 4-трет-бутил-2-нитроанилина (1,033 г, 5,32 ммоль) в ледяной уксусной кислоте (7,8 мл) нагревали с помощью термофена до растворения всех твердых веществ. Раствор затем охлаждали и обрабатывали порциями пиридиний гидробромид пербромидом (1,96 г, 6,12 ммоль). После добавления раствор перемешивали при комнатной температуре в течение 1 ч. Смесь добавляли в воду (50 мл) и обрабатывали небольшим количеством сульфита натрия. После перемешивания в течение 30 мин, осадок собирали фильтрацией. Полученное твердое вещество промывали водой и растворяли в EtOAc. Промывали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением названного соединения в виже желто-оранжевого твердого вещества (1,36 г, 94%).
Часть B. Получение 1-бром-3-трет-бутил-5-нитробензола.
Раствор трет-бутилнитрита (300 мкл 90%, 261 мг, 2,27 ммоль) в сухом DMF (4 мл) нагревали при 50°C и обрабатывали раствором продукта из Части A (414 мг, 1,52 ммоль) в DMF (3,5 мл). После нескольких минут перемешивания, раствор начинал интенсивно выделять пузырьки газа. После нагревания при 50°C в течение 1 ч, добавляли дополнительно (300 мкл) трет-бутилнитрит с последующим нагреванием при 50°C в течение 1 ч. Через 18 ч при комнатной температуре, добавляли трет-бутилнитрит (1,2 мл), и смесь нагревали при 50°C в течение 2 ч. Охлаждали и разбавляли EtOAc. Промывали водой и солевым раствором. Сушили над Na2S04, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 5-40% этилацетатом в гексане давала названное соединение в виде светло-желтого масла (159 мг, 41%).
Часть C. Получение 3-бром-5-трет-бутиланилина.
Раствор продукта из Части B (770 мг, 2,98 ммоль) в смеси 3:3:1 метанол-вода-THF (14.9 мл) обрабатывали хлоридом аммония (239 мг, 4,47 ммоль) и железными опилками (833 мг, 14,92 ммоль) с последующим нагреванием с обратным холодильником в течение 8 ч. Разбавляли EtOAc и водой и фильтровали через целит. Фильтрат экстрагировали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением названного соединения в виде желтого масла.
Часть D. Получение (E)-N-(3-бром-5-трет-бутилфенилкарбамоил)-3-метокси акриламида. Раствор продукта из Части C (681 мг, 2,99 ммоль) в сухом DMF (23 мл) при -30°C обрабатывали по каплям 0,4М раствором (E)-3-метоксиакрилоилизоцианата в бензоле (14,9 мл, 5,96 ммоль). Раствор перемешивали при -30°C в течение 30 мин с последующим постепенным нагреванием до комнатной температуры, и затем перемешивали в течение 18 ч. Разбавляли EtOAc и промывали водой и солевым раствором. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением твердого вещества желтого цвета, которое растирали в порошок с эфиром-гексаном и собирали фильтрованием. Сушили под вакуумом с получением названного соединения в виде светло-коричневого порошка. (951 мг, 90%).
Часть E. Получение 1-(3-бром-5-трет-бутилфенил)пиримидин-2,4(1Н,3H)-диона.
Суспензию продукта из Части D (951 мг, 2,68 ммоль) в этаноле (25 мл) обрабатывали раствором концентрированной серной кислоты (2,60 мл, 4,78 г, 18,22 ммоль) в воде (13,4 мл) с последующим нагреванием при 100°C в течение 1 ч. Охлаждали и концентрировали для удаления этанола. Охлаждали до 0°C и осадок собирали фильтрацией и промывали водой. Сушили под вакуумом с получением названного соединения в виде твердого вещества оранжевого цвета (619 мг, 72%).
Часть F. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)фенил) нафтален-2-ил)метансульфонамида.
В микроволновой трубке, суспензию продукта из Части E (104 мг, 0,32 ммоль), продукта из Примера 4A, Части B (134 мг, 0,39 ммоль), и 1,0М раствора карбоната натрия (386 мкл, 0,39 ммоль) в смеси 1:1 этанол-толуол (2,1 мл) дегазировали путем барботирования азота в течение 10 мин. Раствор обрабатывали 1,1'-бис(ди-трет-бутилфосфино)ферроцен-палладий (II) дихлоридом (20 мг, 0,031 ммоль) и дегазирование продолжали в течение еще 5 мин. Смесь нагревали при 100°C в микроволновой печи в течение 30 мин. Разбавляли EtOAc и промывали водой и солевым раствором. Сушили над Na2SO4 и обрабатывали (3-меркаптопропил) силикагелем в течение 30 мин. Фильтровали и концентрировали под вакуумом с получением твердого вещества янтарного цвета, которое растирали в порошок с эфиром-гексаном. Твердое вещество собирали фильтрованием и сушили под вакуумом с получением названного соединения (81 мг, 54%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.46 (s, 1 H) 10.05 (s, 1 H), 8.25 (s, 1 H) 7.98 (dd, J=11.58, 9.01 Hz, 1 H) 7.86-7.93 (m, 1 H) 7.78-7.85 (m, 2 H) 7.72 (s, 1 H) 7.67 (s, 1 H) 7.31-7.51 (m, 2 H) 5.70 (dd, J=7.72, 2.21 Hz, 1 H) 3.08 (s, 3 H) 1.39 (s, 9 H).
Пример 19. Получение (E)-N'-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)фенил)-2,3-дигидро-1Н-инден-1-илиден)метансульфонгидразида (соединение IB-L0-2.7).
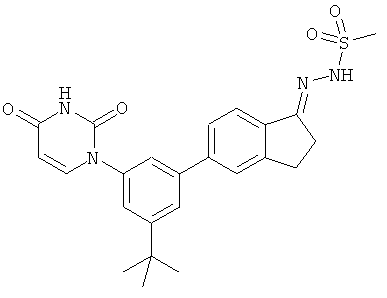
Часть A. Получение 1-(3-трет-бутил-5-(1-оксо-2,3-дигидро-1H-инден-5-ил)фенил) пиримидин-2,4(1Н,3H)-диона.
В микроволновой трубке, суспензию продукта из Примера 18, Части E, продукта из Примера 6, Части A (144 мг, 0,56 ммоль), 1,0М раствора карбоната натрия (557 мкл, 0,56 ммоль) в смеси 1:1 этанол-толуол (3,0 мл) дегазировали путем барботирования азота в течение 15 мин. Добавляли комплекс 1,1'-бис(ди-т-бутилфосфино) ферроцен палладий (II) хлорид (15 мг, 0,023 ммоль) и дегазирование продолжали в течение еще 5 мин. Трубку герметично закрывали, и смесь нагревали при 100°C в микроволновой печи в течение 30 мин. Разбавляли EtOAc и водой. Промывали 1М раствором лимонной кислоты, водой, и солевым раствором. Органическую фазу перемешивали с (3-меркаптопропил)силикагелем в течение 1 ч. Сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Очистка с помощью колоночной хроматографии на силикагеле с элюцией 10-100% EtOAc в гексане давала названное соединение в виде беловатого твердого вещества (86 мг, 50%).
Часть B. Получение (E)-N'-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)фенил)-2,3-дигидро-1H-инден-1-илиден)метансульфонгидразида.
Продукт из Части A (80 мг, 0,21 ммоль) вступал в реакции в соответствии с процедурами из Примера 6, Часть C с образованием названного соединения в виде твердого вещества белого цвета (73 мг, 73%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.44 (s, 1 H) 9.92 (s, 1 H) 7.64 -7.98 (m, 5 H) 7.57 (s, 1 H) 7.45 (s, 1 H) 5.68 (d, J=7.72 Hz, 1 H) 3.00 - 3.20 (m, 5 H) 2.85 (d, J=12.50 Hz, 2 H) 1.36 (s, 9 H). MS +ESI m/z (относительная распространенность): 467 (100, М+Н).
Пример 20. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(4-(метилсульфонамид)фенил)бензамида (соединение IA-L3-1.6).
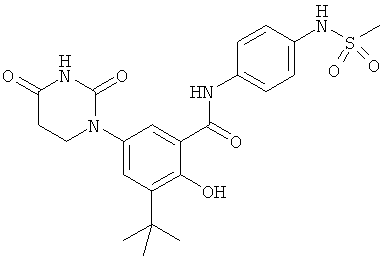
Часть A. Получение метил 3-трет-бутил-2-гидрокси-5-нитробензоата.
Метил 3,5-ди-трет-бутил-2-гидроксибензоат (28,66 г, 108,4 ммоль) растворяли при перемешивании в 430 мл ледяной укусной кислоты и полученную в результате смесь обрабатывали по каплям дымящей азотной кислотой (90%, 179,26 мл). После завершения добавления, полученную в результате смесь перемешивали в течение 2,5 ч. Реакционную смесь выливали в 2,0 л дробленого льда и оставляли стоять 30 мин. После этого, добавляли 1,0 л воды и эту смесь воды и льда оставляли плавиться. Смесь затем фильтровали, промывали водой и сушили с получением названного соединения (24,57 г, 89%).
Часть B. Получение метил 5-амино-3-трет-бутил-2-гидроксибензоата.
Продукт Части A (0,43 г, 1,70 ммоль) обрабатывали каталитическим количеством Pd/C в THF (10 мл) под давлением водорода из баллонной шины в течение 3 ч. Колбу продували азотом, и смесь фильтровали, концентрировали, и очищали с помощью колоночной хроматографии на силикагеле, элюируя 50% гексаном/дихлорметаном, после этого дихлорметаном с получением на выходе 0,37 г (98%).
Часть C. Получение метил 5-(3-амино-3-оксопропиламино)-3-трет-бутил-2-гидрокси бензоата.
Продукт Части B (0,37 г, 1,66 ммоль) и акриловую кислоту (0,12 мкл, 1,74 ммоль) объединяли в толуоле (10 мл) и нагревали с обратным холодильником в течение 20 ч. Раствор концентрировали до сухости.
Часть D. Получение метил 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидроксибензоата.
Продукт Части C растворяли в ледяной уксусной кислоте (5 мл) и обрабатывали мочевиной (0,24 г, 4,0 ммоль) при 120°C в течение 3 ч. Раствор разбавляли холодной водой, экстрагировали в этилацетат, концентрировали, и очищали с помощью колоночной хроматографии на силикагеле, элюируя 1%, затем 2%, затем 4% метанол/дихлор-метаном с получением как продукта (0,25 г, 46%), так и дигидроурацила с открытым циклом (0,112 г, 20%).
Часть E. Получение 3-трет-бутил-5-(1-(2-карбоксиэтил)уреид)-2-гидроксибензойной кислоты.
Продукты из Части D растворяли в метаноле (6 мл) и добавляли 1М раствор гидроксида натрия (15 мл). Через 20 ч, pH этого раствора доводили до рН 2 концентрированной соляной кислотой и экстрагировали в этилацетат, сушили над сульфатом натрия, и концентрировали с получением 0,303 г (89%).
Часть F. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-бензойной кислоты.
Продукт из Части E (0,303 г, 0,93 ммоль) помещали в 7 мл концентрированной соляной кислоты и нагревали в открытой колбе при 120°C в течение 1 ч, в течение этого времени избыток кислоты испарялся и оставался сухой продукт 0,20 г (70%).
Часть G. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(4-(метилсульфонамид)фенил)бензамида.
Продукт из Части F (0,13 г, 0,42 ммоль) нагревали с тионилхлоридом (3 мл) при 90°C в течение 1,5 ч в открытой колбе, с получением сухого хлорангидрида, который помещали в диоксан (4 мл). Добавляли N-(4-амино-фенил)метансульфонамид.HCl (0,070 мг, 0,31 ммоль) и раствор нагревали при 90°C в течение 1 ч. Смесь концентрировали и затем растирали в порошок с дихлорметаном, фильтровали, и сушили с получением 0,071 мг (48%) названного соединения. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.39 (s, 9 H), 2.73 (t, J=6.62 Hz, 2 H), 2.99 (s, 3Н), 3.78 (t, J=6.62 Hz, 2 H), 7.24 (d, J=8.82 Hz, 2 H), 7.40 (d, J=2.21 Hz, 1 H), 7.60 (d, J=9.19 Hz, 2 H), 7.89 (d, J=2.21 Hz, 1 H), 9.74 (s, 1 H), 10.39 (s, 1 H), 10.44 (s, 1 H), 13.30 (s, 1 H).
Пример 21. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(4-(2-метоксиэтилсульфонамид)фенил)бензамида (соединение IA-L3-1.8).
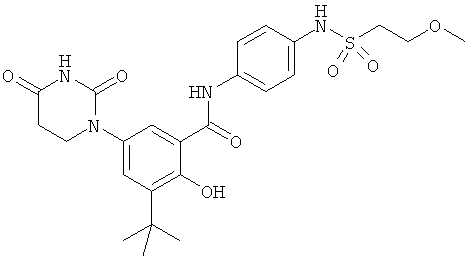
Часть A. Получение трет-бутил 4-(винилсульфонамид)фенилкарбамата.
Раствор трет-бутил 4-аминофенилкарбамата (2,63 г, 12,63 ммоль) и триэтиламина (7,04 мл, 50,51 ммоль) объединяли в дихлорметане (50 мл) и охлаждали на ледяной бане. После добавления по каплям 2-хлорэтансульфонилхлорида (1,45 мл, 13,9 ммоль), раствор перемешивали при окружающей температуре в течение 4 ч, затем разбавляли 0,5М HCl и экстрагировали в дихлорметан. Продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 1% метанолом/дихлорметаном с получением 2,48 г (66%).
Часть B. Получение трет-бутил 4-(2-метоксиэтилсульфонамид)фенилкарбамата.
Продукт из Части A (0,70 г, 2,35 ммоль) нагревали при 60°C в герметично закрытой пробирке с 10 мл метанола и 5 мл 25% по массе метоксида натрия в метаноле, в течение 16 ч. Этот раствор разбавляли водой и доводили до рН 6 с использованием 1М HCl, затем экстрагировали в дихлорметан и концентрировали с получением 0,582 г (75%).
Часть C. Получение N-(4-аминофенил)-2-метоксиэтансульфонамида.
Продукт из Части B (0,582 г, 1,76 ммоль) помещали в 15 мл 4М HCl в диоксане и перемешивали при окружающей температуре в течение 20 ч. Этот раствор разбавляли дихлорметаном и твердый продукт отфильтровывали и сушили с получением 0,395 г (84%).
Часть D. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-(2-метоксиэтилсульфонамид)фенил)бензамида.
Продукт из Примера 20, Часть F (0,05 г, 0,163 ммоль) обрабатывали тионилхлоридом (0,5 мл) и продуктом из Части C (0,038 г, 0,163 ммоль) как в Примере 20, Части G с получением 0,038 г (45%) названного соединения. 1H ЯМР (300 MHz, DMSO-D6): δ ppm 1.39 (s, 9 H), 2.73 (t, J=6.62 Hz, 2 H), 3.20 (s, 3 H), 3.33-3.42 (m, 2 H), 3.67 (t, J=6.25 Hz, 2 H), 3.78 (t, J=6.62 Hz, 2 H), 7.23 (d, J=9.19 Hz, 2 H), 7.40 (d, J=2.21 Hz, 1 H), 7.58 (d, J=9.19 Hz, 2 H), 7.89 (d, J=2.21 Hz, 1 H), 9.80 (s, 1 H), 10.39 (s, 1 H), 10.44 (s, 1 H), 13.30 (s, 1 H).
Пример 22. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-метилсульфонамид)фенил)бензамида (соединение IA-L3-1.51).
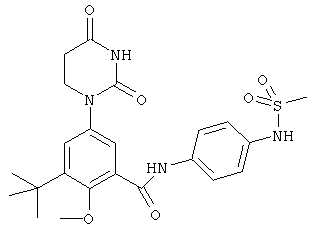
Часть A. Получение метил 3-трет-бутил-2-гидрокси-5-нитробензоата.
Метил 3,5-ди-трет-бутил-2-гидроксибензоат (28,66 г, 108,4 ммоль) растворяли при перемешивании в 430 мл ледяной уксусной кислоты и полученную в результате смесь обрабатывали по каплям дымящей азотной кислотой (90%, 179,26 мл). После завершения добавления полученную в результате смесь перемешивали в течение 2,5 ч. Реакционную смесь выливали в 2,0 л дробленого льда, и оставляли стоять 30 мин. После этого, добавляли 1,0 л воды, и эту смесь воды и льда оставляли плавиться. Смесь затем фильтровали, промывали водой и сушили с получением названного соединения (24,57 г, 89%).
Часть B. Получение метил 3-трет-бутил-2-метокси-5-нитробензоата.
Добавляли вместе продукт из Части A (11,41 г, 45,0 ммоль), карбонат калия (9,34 г, 67,6 ммоль), ацетон (200 мл), и диметилсульфат (6,46 г, 67,6 ммоль). Полученную в результате смесь затем нагревали с обратным холоридильником в течение 16 ч. Смесь затем фильтровали и твердое вещество промывали этилацетатом. Полученную в результате органическую жидкость затем концентрировали под вакуумом до масла и герастворяли в этилацетате (600 мл). Этот органический раствор затем промывали водой, сушили, фильтровали и концентрировали под вакуумом до масла, которое затем подвергали очистке посредством колоночной хроматографии (градиент 5%-40% EtOAc/гексан) с получением на выходе названного соединения в виде масла (10,42, 87%).
Часть C. Получение метил 5-амино-3-трет-бутил-2-метоксибензоата.
Добавляли вместе продукт из Части B (10,42 г, 39,0 ммоль), железные опилки (325 меш, 10,89 г, 195 ммоль), хлорид аммония (3,13 г, 58,5 ммоль), воду (30 мл), и метанол (150 мл). Полученную в результате смесь затем нагревали с обратным холодильником в течение 1 ч. Смесь затем охлаждали до комнатной температуры, фильтровали через целит, и целит промывали метанолом. Фильтрат затем концентрировали под вакуумом и растворяли в этилацетате (600 мл). Полученный в результате раствор затем промывали водой и солевым раствором. Органический экстракт затем сушили, фильтровали и концентрировали под вакуумомс получением на выходе названного соединения в виде масла (9,25 г, 100%).
Часть D. Получение 3-(3-трет-бутил-4-метокси-5-(метоксикарбонил)фениламино) пропановая кислота.
Продукт из Части C (16,44 г, 69,3 ммоль) растворяли в толуоле (200 мл). Эту смесь нагревали с обратным холодильником и добавляли акриловую кислоту на протяжении периода времени (1 мл акриловой кислоты добавляли каждые 3 ч, всего 5,23 мл, 76,2 ммоль). Полученную в результате смесь нагревали с обратным холодильником в течение 24 ч. Смесь охлаждали и концентрировали до сухости под вакуумом с получением на выходе сырого названного соединения в виде масла, которое использовали непосредственного в следующей реакции.
Часть E. Получение метил 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензоата.
Добавляли вместе продукт из Части D (21,43 г, 69,3 ммоль), мочевину (10,4 г, 173 ммоль) и уксусную кислоту (ледяная, 200 мл). Смесь затем нагревали до 120°C в течение 18,5 ч с последующим концентрированием под вакуумом до сухости до получения масла. К этому маслу добавляли метанол (13 мл), и этилацетат (350 мл). Полученную в результате смесь оставляли стоять в течение 24-48 ч, за счет чего образовывался осадок. Полученное в результате твердое вещество отфильтровывали и промывали небольшим количеством метанола (10 мл) и затем сушили на воздухе с получением на выходе названного соединения в виде твердого вещества (15,26 г, 66%).
Часть F. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси бензойной кислоты.
Добавляли вместе продукт из Части E (4,52 г, 13,52 ммоль), метанол (70 мл), и тетрагидрофуран (70 мл). Смесь затем интенсивно перемешивали до получения в результате гомогенного раствора. После достижения гомогенности добавляли раствор водного гидроксида натрия (1,0М, 68 мл). Смесь затем перемешивали в течение 12 ч, затем эту смесь концентрировали под вакуумом для удаления органического растворителя, с последующим добавлением водного раствора соляной кислоты (1,0М, 80 мл), в результате чего образовывалось твердое вещество. Смесь затем концентрировали под вакуумом. К этому веществу добавляли соляную кислоту (12М, 100 мл) и полученное в результате вещество нагревали до 100°C в течение 1,5 ч. Реакционную смесь затем охлаждали и добавляли воду. Полученное в результате твердое вещество фильтровали, промывали водой, и сушили с получением на выходе названного соединения в виде твердого вещества (3,55 г, 82%).
Часть G. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2 метоксибензоилхлорида.
Добавляли вместе продукт из Части F, (2H)-ил)-2-метоксибензойную кислоту (4,07 г, 12,71 ммоль) и тионилхлорид (40,82 мл, 559 ммоль). Смесь затем нагревали с обратным холодильником в течение 2 ч, с последующим концентрированием под вакуумом с получением продукта в виде твердого вещества светло-желтого цвета.
Часть H. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(4-метилсульфонамид)фенил)бензамида.
Продукт, полученный в Части G (0,55 г, 1,71 ммоль) растворяли в CH2Cl2 (35 мл) и добавляли по каплям к суспензии в CH2Cl2 (40 мл), содержащей соль N-(4-аминофенил)метансульфонамид гидрохлорид (0,38 г, 1,71 ммоль) и пиридин (0,41 мл, 5,1 ммоль). Реакционную смесь перемешивали 18 ч при комнатной температуре. Реакционную смесь фильтровали и разбавляли 400 мл CH2Cl2. Органический слой промывали 1 н H3PO4, 10% NaHCO3 и 10% NaCl и сушили над безводным твердым сульфатом натрия. Высушивающее вещество фильтровали и органический слой упаривали в вакууме с получением в остатке названного соединения в виде твердого вещества кремового цвета (474 мг, 57%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.37 (s, 9 Н) 2.71 (t, J=6.62 Hz, 2 Н) 2.95 (s, 3 Н) 3.77 (s, 3 Н) 3.80 (d, 2 Н) 7.20 (d, J=9.19 Hz, 2 Н) 7.28 (d, J=2.57 Hz, 1 Н) 7.33 (d, J=2.94 Hz, 1 Н) 7.69 (d, J=9.19 Hz, 2 Н) 9.59 (s, 1 Н) 10.35 (s, 1 Н) 10.38 (s, 1 Н).
Пример 23. Получение 4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидроксибензамидо)фенилметансульфоната (соединение IA-L3-1.19).
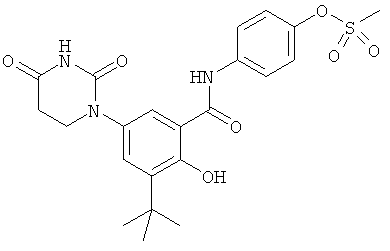
Часть A. Получение 4-(трет-бутоксикарбониламино)фенилметансульфоната.
Трет-бутил-4-гидроксифенилкарбамат (1,0 г, 4,78 ммоль) и триэтиламин (0,80 мл, 5,73 ммоль) объединяли в дихлорметане (50 мл), охлаждали на ледяной бане и обрабатывали метансульфонилхлоридом (0,41 мл, 5,26 ммоль). Раствор перемешивали при окружающей температуре в течение 2 ч, затем промывали 1М HCl, сушили над сульфатом натрия, фильтровали, и концентрировали с получением 1,2 г (87%).
Часть B. Получение 4-аминофенил метансульфонат гидрохлорида. Продукт из Части A (1,2 г, 4,18 ммоль) обрабатывали 4 М HCl в диоксане (10 мл) при окружающей температуре и перемешивали в течение 18 ч. Смесь концентрировали и твердое вещество растирали в порошок с дихлорметаном, фильтровали, и сушили с получением 0,855 г (92%).
Часть C. Получение 4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси бензамидо)фенил метансульфоната.
Продукт из Примера 20, Часть F (0,055 г, 0,18 ммоль) обрабатывали тионилхлоридом (0,4 мл, 5,4 ммоль) при 80°C в течение 35 мин, затем концентрировали до сухости. Этот хлорангидрид растворяли в диоксане (2 мл) и обрабатывали продуктом из Части B (0,060 г, 0,27 ммоль) и пиридином (0,037 мл, 0,45 ммоль). Полученную в результате смесь перемешивали при 80°C в течение 1 ч, разбавляли 1М HCl, экстрагировали в этилацетат, концентрировали и очищали с помощью колоночной хроматографии на силикагеле, элюируя дихлорметаном, а затем 2% метанолом/дихлорметаном с получением 0,055 г (64%) названного соединения. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.39 (s, 9 H), 2.74 (t, J=6.62 Hz, 2 H) 3.40 (s, 3 H), 3.79 (t, J=6.80 Hz, 2 H), 7.36-7.45 (m, 3 H) 7.76 (d, J=9.19 Hz, 2 H), 7.90 (d, J=2.21 Hz, 1 H), 10.40 (s, 1 H), 10.58 (s, 1 H) 13.13 (s, 1 H).
Пример 24. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-метил-N-(метилсульфонамид)фенил)бензамида (соединение IA-L3-1.27).
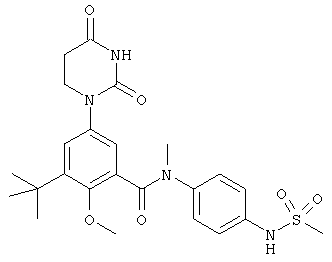
Часть A. Получение трет-бутил 4-аминофенил(метил)карбамата.
Смесь N-метил-4-нитроанилина (1,00 г, 6,57 ммоль), ди-трет-бутил дикарбоната (2,51 г, 11,50 ммоль), и DMAP (40 мг, 0,33 ммоль) в дихлорметане (35 мл) перемешивали при нагревании с обратным холодильником в течение 2 ч. Реакционную смесь промывали водой (20 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток растворяли в смеси THF (12 мл) и метанола (12 мл). К полученному в результате раствору добавляли железные опилки (1,50 г, 27,0 ммоль) и раствор хлорида аммония (0,54 г, 10,11 ммоль) в воде (5 мл). Смесь перемешивали при 70°C в течение 3 ч, охлаждали до комнатной температуры и фильтровали через целит, и концентрировали под вакуумом. Остаток азеотропно сушили, используя толуол (3X) и затем растирали в порошок с эфиром с получением твердого вещества, которое удаляли фильтрованием. Фильтрат концентрировали под вакуумом с получением названного соединения (1,45 г, 99%).
Часть B. Получение N-(4-(метиламино)фенил)метансульфонамид гидрохлорида.
Продукт, полученный в Части A (1,45 г, 6,52 ммоль) растворяли в безводном дихлорметане (25 мл) и обрабатывали пиридином (1,32 мл, 16,31 ммоль) и метансульфонилхлоридом (0,57 мл, 7,18 ммоль). Полученный в результате раствор перемешивали при комнатной температуре в течение 3 ч, и затем выливали в 0,5М водн. HCl (25 мл). Слои отделяли и водную фазу промывали дихлорметаном (2×25 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Колоночная хроматография на силикагеле с использованием 1% метанола в хлороформе в качестве элюента давала трет-бутл метил(4-(метилсульфонамид)фенил) карбамат (1,31 г, 67%), который растворяли в 4 н HCl в 1,4-диоксане (20 мл). Полученный в результате раствор перемешивали при 40°C в течение 1 ч и концентрировали под вакуумом. Остаток растирали в порошок с дихлорметаном с получением названного соединения в виде твердого вещества, которое собирали фильтрацией и сушили под вакуумом (0,99 г, 96%).
Часть C. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-метил-N-(4-(метилсульфонамид)фенил)бензамида.
Продукт, полученный в Примере 22, Части G (40 мг, 0,13 ммоль) и тионилхлорид (0,3 мл, 4 ммоль) нагревали с обратным холодильником в течение 30 мин, с последующим концентрированием под вакуумом. Остаток растворяли в безводном N,N-диметилацетамиде (2 мл), и в полученный в результате раствор добавляли продукт из Части B (30 мг, 0,13 ммоль) и пиридин (0,025 мл, 0,31 ммоль). Смесь перемешивали при 80°C в течение 30 мин, и разделяли между 1 н HCl (5 мл) и этилацетатом (3×5 мл). Органические экстракты объединяли, сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением на выходе сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле, элюируя 19:1 MeOH:CHCl3 с получением названного соединения в виде бесцветного твердого вещества (45 мг, 72%). 1H ЯМР (500 MHz, DMSO-D6) δ ppm 1.07 (s, 9 Н), 2.69 (t, J=6.1 Hz, 2 Н), 2.83 (s, 3 Н) 3.33-3.38 (m, 5 Н) 3.73 (s, 3 Н) 6.92 (d, J=8.5 Hz, 2 Н) 7.01 (d, J=9.2 Hz, 2 Н) 7.06 (d, J=2.4 Hz, 1 Н) 7.12 (d, J=2.4 Hz, 1 Н) 9.52-9.73(m, 1 H) 10.28 (s, 1 Н).
Пример 25. Получение (N-(4-(3-трет-бутил-5-(3-(бутирилоксиметил)-2,4-диоксотетрагидро пиримидин-1(2H)-ил)-2-метоксибензамидо)фенил)метилсульфонамид)метилбутирата (соединение IA-L3-1.88).
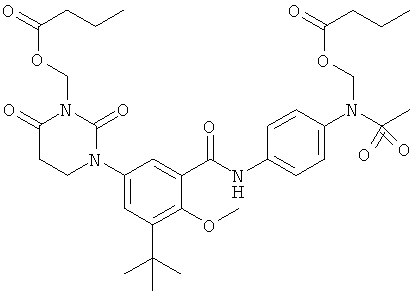
Продукт из Примера 22, Часть G (0,098 г, 0,20 ммоль) растворяли в DMSO (2 мл) и обрабатывали карбонатом калия (0,166 г, 1,20 ммоль) и хлорметилбутиратом (0,411 г, 3,0 ммоль). Смесь перемешивали 20 ч при комнатной температуре. Реакционную смесь разделяли этилацетатом и водой. Органический слой промывали солевым раствором и сушили над безводным твердым сульфатом натрия. Высушивающее вещество фильтровали и растворитель упаривали под вакуумом. Остаток очищали силикагелем, элюируя этилацетатом/гексаном (10%-80%) с получением двух основных фракций. Первую фракцию очищали силикагелем, элюируя метанолом/дихлорметаном (1%-3%) с получением названного соединения в виде пены (0,014 г, 10%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 0.88 (m, 6 Н) 1.38 (s, 9 Н) 1.55 (m, 4 Н) 2.26 (t, J=7.17 Hz, 2 Н) 2.39 (t, J=7.17 Hz, 2 Н) 2.95 (t, J=6.62 Hz, 2 Н) 3.14 (s, 3 Н) 3.77 (s, 3 Н) 3.81 (t, J=6.62 Hz, 2 Н) 5.57 (s, 2 Н) 5.68 (s, 2 Н) 7.35 (d, J=2.57 Hz, 1 H) 7.38 (d, J=2.94 Hz, 1 H) 7.42 (d, J=8.82 Hz, 2 H) 7.79 (d, J=8.82 Hz, 2 H) 10.60 (s, 1 H).
Пример 26. Получение (N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензамидо)фенил)метилсульфонамид)метил бутирата (соединение IA-L3-1.64).
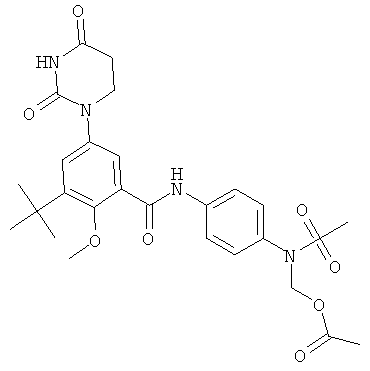
Продукт из Примера 22, Части G (0,098 г, 0,20 ммоль) растворяли в DMSO (1 мл) и обрабатывали карбонатом цезия (0,209 г, 0,64 ммоль) и бромметилацетатом (0,123 г, 0,80 ммоль). Смесь перемешивали 4 ч при комнатной температуре. Реакционную смесь разделяли этилацетатом и водой. Органический слой промывали солевым раствором и сушили над безводным твердым сульфатом натрия. Высушивающее вещество фильтровали и растворитель упаривали под вакуумом. Остаток очищали с помощью HTP группы посредством препаративной ВЭЖХ на Waters Nova-Pak® HR C18 6um 60Ǻ Prep-Pak® картридже-колонке (40 мм × 100 мм). Использовали градиент ацетонитрила (A) и 10 мМ ацетата аммония в воде (B), при скорости потока 70 мл/мин (0-0,5 мин 10% A, 0,5-12,0 мин линейный градиент 10-95% A, 12,0-15,0 мин 95% A, 15,0-17,0 мин линейный градиент 95-10% A) с получением названного соединения в виде твердого вещества белого цвета (0,034 г, 30%). Температура плавления 229-230°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 H) 2.11 (s, 3 H) 2.72 (t, J=6.80 Hz, 2 H) 3.15 (s, 3 H) 3.78 (m, 5 H) 5.55 (s, 2 H) 7.30 (d, J=2.57 Hz, 1 H) 7.35 (d, J=2.57 Hz, 1 H) 7.42 (d, J=9.19 Hz, 2 H) 7.79 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.58 (s, 1 H).
Пример 27. Получение N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-4-(метилсульфонамид)бензамида (соединение IA-L4-1.9).
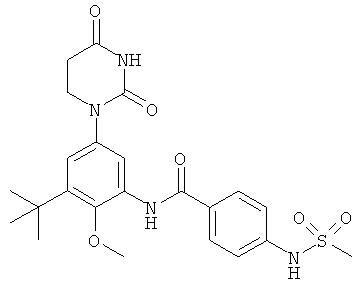
Часть А. Получение 2-трет-бутил-1метокси-4-нитробензола.
Смесь 1:1 AcOH и дымящей HNO3 (0,6 мл) медленно добавляли к раствору 2-трет-бутилфенола (1,0 г, 6,6 ммоль) в циклогексане (3 мл) при 0°C. Полученную в результате темную смесь перемешивали при 0°C в течение 1 ч, с последующим добавлением гексана (5 мл). Полученное в результате твердое вещество собирали фильтрацией и промывали гексаном с получением светлого зеленоватого твердого вещества (0,37 г, 29%). Твердое вещество растворяли в ацетоне (10 мл), и в полученный в результате раствор добавляли K2CO3 (0,3 г, 2.2 ммоль), с последующим добавлением по каплям Me2SO4 (0,27 мл, 2,8 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение ночи, и затем выливали в 1 н HCl (20 мл). Смесь экстрагировали EtOAc (3×20 мл), сушили над Na2SO4, фильтровали и концентрировали с получением названного соединения в виде масла (0,4 г, колич.).
Часть B. Получение 1-(3-трет-бутил-4-метоксифенил)дигидропиримидин-2,4(1H,3H)-диона.
Продукт, описанный в Части A, растворяли (0,4 г, 1,9 ммоль) в EtOAc (10 мл) и обрабатывали 10% Pd на угле (50 мг). Смесь перемешивали при окружающей температуре под давлением 1 атм H2 в течение ночи. Смесь фильтровали через целит и концентрировали под вакуумом с получением сырого продукта, который очищали на силикагеле. Продукт элюировали, используя 1:1 EtOAc:гексан, и выделяли в виде масла (0,23 г, 68%). Акриловую кислоту (0,1 мл, 1,46 ммоль) и толуол (10 мл) добавляли к выделенному маслу и полученную в результате смесь нагревали при 100°C в течение ночи, и затем концентрировали в вакууме с получением темного масла. Масло обрабатывали AcOH (5 мл) и мочевиной (0,2 г, 3,3 ммоль), и смесь нагревали при 120°C в течение 6 ч. Смесь охлаждали до окружающей температуры, выливали в воду (20 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали под вакуумом с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле, используя 1:1 EtOAc:гексан. Названное соединение получали в виде бесцветного твердого вещества (0,144 г, 41%).
Часть C. Получение 1-(3-трет-бутил-4-гидрокси-5-нитрофенил)дигидропиримидин-2,4(1H,3H)-диона.
Продукт, полученный в Части B (1,00 г, 3,62 ммоль) растворяли в CH2Cl2 (25 мл) при 0°C и обрабатыали 1М раствором BBr3 в CH2Cl2 (18 мл, 18 ммоль). Смесь перемешивали при нагревании с обратным холодильником в течение ночи и выливали в воду (50 мл). Смесь экстрагировали 3:1 CH2Cl2:2-PrOH (2×50 мл), и объединенные экстракты сушили над MgSO4, фильтровали и концентрировали под вакуумом. Сырой продукт очищали с помощью колоночной хроматографии на диоксиде кремния, используя 2:1 EtOAc:гексан для элюции этого продукта, получаемого в виде твердого вещества (0,60 г, 63%). Твердое вещество суспендировали в AcOH (20 мл), к которому добавляли дымящую HNO3 (0,105 мл). Полученный в результате раствор перемешивали при комнатной температуре 1 ч и выливали в ледяную воду (100 мл). Смесь экстрагировали 3:1 CH2Cl2:2-PrOH (2×50 мл), и объединенные экстракты сушили над MgSO4, фильтровали и концентрировали под вакуумом. Остаток растирали в порошок с эфиром с получением твердого вещества, которое собирали фильтрацией (0,40 г, 57%).
Часть D. Получение 1-(3-амино-5-трет-бутил-4-метоксифенил)дигидропиримидин-2,4(1H,3H)-диона.
Продукт, полученный в Части C (0,31 г, 1,01 ммоль) растворяли в 1:1 THF:MeOH (50 мл) и обрабатывали 2М раствором триметилсилилдиазометана в THF (1,5 мл, 3,0 ммоль). Полученный в результате раствор перемешивали при окружающей температуре в течение ночи, и концентрировали под вакуумом. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 1:1 EtOAc:гексан, и получали бесцветное твердое вещество (0,235 г, 72%). Это твердое вещество растворяли в 1:1 CH2Cl2:MeOH (50 мл), обрабатывали 10% Pd/C (25 мг), и смесь перемешивали при окружающей температуре под давлением 1 атм H2 в течение 2 ч. Смесь фильтровали через целит и концентрировали под вакуумом с получением названного соединения (0,215 г, колич.).
Часть E. Получение N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-4-нитробензамида.
Продукт, полученный в Части D (0,215 г, 0,74 ммоль) растворяли в безводном CH2Cl2 (50 мл) и обрабатывали 4-нитробензоилхлоридом (0,164 г, 0,88 ммоль) и пиридином (0,07 мл, 0,88 ммоль). Полученную в результате смесь перемешивали при окружающей температуре в течение ночи, промывали водой (50 мл), сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 1:1 EtOAc:гексан с получением названного соединения (0,26 г, 80%).
Часть F. Получение N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-4-(метилсульфонамид)бензамида.
Продукт, полученный в Части E (0,26 г, 0,59 ммоль) растворяли в 2:1 смеси CH2Cl2:MeOH (6 мл), обрабатывали 10% Pd на угле (30 мг) и перемешивали при окружающей температуре под давлением 1 атм H2 в течение 2 ч, фильтровали через целит и концентрировали под вакуумом. Остаток растворяли в безводном CH2Cl2 (10 мл), обрабатывали метансульфонилхлоридом (0,054 мл, 0,70 ммоль) и пиридином (0,056 мл, 0,70 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение ночи, разделяли между водой (20 мл) и 3:1 CH2Cl2:2-PrOH (3×20 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 19:1 CH2Cl2:MeOH с получением названного соединения в виде твердого вещества (0,12 г, 42%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.37 (s, 9 H), 2.70 (t, J=6.62 Hz, 2 H), 3.10 (s, 3 H), 3.71 (s, 3 H), 3.76 (t, J=6.62 Hz, 2 H), 7.11 (d, J=2.57 Hz, 1 H) 7.30 (d, J=8.82 Hz, 2 H) 7.37 (d, J=2.57 Hz, 1 H), 8.02 (d, J=8.82 Hz, 2 H), 9.86 (s, 1 H) 10.20 (s, 1 H) 10.33 (s, 1 H).
Пример 28. Получение N-(3-трет-бутил-5-(2,4-диоксотетрагидро пиримидин-1(2H)-ил)-2-метоксифенил)-4-метилсульфонилметил)бензамида (соединение IA-L4-1.10).
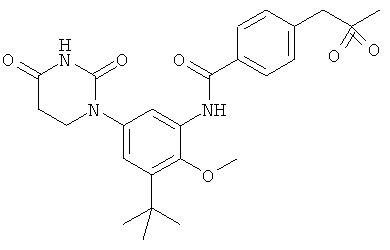
Тионилхлорид (0,31 мл, 4,2 ммоль) и 4-(метилсульфонилметил)бензойную кислоту (0,03 г, 0,14 ммоль) объединяли и нагревали при 85°C в течение 30 мин, затем концентрировали до сухости. Этот хлорангидрид растворяли в N,N- диметилацетамиде (2 мл) с продуктом из Примера 27, Часть D (0,041 г, 0,14 ммоль) и пиридином (0,025 мл, 2,2 ммоль) и нагревали при 100°C в течение 20 мин, затем охлаждали до окружающей температуры и разбавляли 1М HCl. Этот твердый осадок выделяли фильтрацией, растирали в порошок с метанолом и сушили с получением названного соединения) (0,0175 г, 26%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.37 (s, 9 H), 2.71 (t, J=6.62 Hz, 2 H), 2.95 (s, 3 H), 3.72 (s, 3 H), 3.77 (t, J=6.62 Hz, 2 H), 4.60 (s, 2 H), 7.13 (d, J=2.94 Hz, 1 H), 7.38 (d, J=2.57 Hz, 1 H), 7.56 (d, J=8.09 Hz, 2 H) 8.05 (d, J=8.09 Hz, 2 H), 10.03 (s, 1 H), 10.33 (s, 1 H).
Пример 29. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IA-L1-1.9).
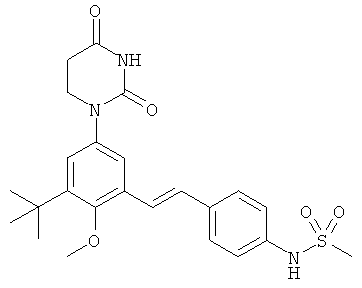
Часть A. Получение метил 3-трет-бутил-2-гидрокси-5-нитробензоата.
Метил 3,5-ди-трет-бутил-2-гидроксибензоат (28,66 г, 108,4 ммоль) растворяли при перемешивании в 430 мл ледяной уксусной кислоты и полученную в результате смесь обрабатывали по каплям дымящей азотной кислотой (90%, 179,26 мл). После завершения добавления полученную в результате смесь перемешивали в течение 2,5 ч. Реакционную смесь выливали в 2,0 л дробленого льда и оставляли стоять 30 мин. После этого, добавляли 1,0 л воды и эту смесь воды и льда оставляли плавиться. Смесь затем фильтровали, промывали водой и сушили с получением названного соединения (24,57 г, 89%).
Часть B. Получение метил 3-трет-бутил-2-метокси-5-нитробензоата.
Добавляли вместе метил 3-трет-бутил-2-гидрокси-5-нитробензоат (11,41 г, 45,0 ммоль), карбонат калия (9,34 г, 67,6 ммоль), ацетон (200 мл), и диметилсульфат (6,46 г, 67,6 ммоль). Полученную в результате смесь затем нагревали с обратным холодильником в течение 16 ч. Смесь затем фильтровали и твердое вещество промывали этилацетатом. Полученную в результате органическую жидкость затем концентрировали под вакуумом до масла и перерастворяли в этилацетате (600 мл). Органический раствор затем промывали водой, сушили, фильтровали и концентрировали под вакуумом до масла, которое затем подвергали очистке посредством колоночной хроматографии (градиент 5% - 40% EtOAc/гексан) с получением на выходе названного соединения в виде масла (10,42, 87%).
Часть C. Получение метил 5-амино-3-трет-бутил-2-метоксибензоата.
Добавляли вместе метил 3-трет-бутил-2-метокси-5-нитробензоат (10,42 г, 39,0 ммоль), железные опилки (325 меш, 10,89 г, 195 ммоль), хлорид аммония (3,13 г, 58,5 ммоль), воду (30 мл), и метанол (150 мл). Полученную в результате смесь затем нагревали с обратным холодильником в течение 1 ч. Смесь затем охлаждали до комнатной температуры, фильтровали через целит, и целит промывали метанолом. Фильтрат затем концентрировали под вакуумом и растворяли в этилацетате (600 мл). Полученный в результате раствор затем промывали водой и солевым раствором. Органический экстракт затем сушили, фильтровали и концентрировали под вакуумом с получением на выходе названного соединения в виде масла (9,25 г, 100%).
Часть D. Получение 3-(3-трет-бутил-4-метокси-5-(метоксикарбонил)фениламино) пропановой кислоты.
Продукт из Части C (16,44 г, 69,3 ммоль) растворяли в толуоле (200 мл). Эту смесь нагревали с обратным холодильником и добавляли акриловую кислоту на протяжении периода времени (1 мл акриловой кислоты добавляли каждые 3 ч, всего 5,23 мл, 76,2 ммоль).
Смесь затем нагревали с обратным холодильником в течение 24 ч. Смесь затем охлаждали и концентрировали под вакуумом до сухости с получением на выходе масла в качестве неочищенного называнного соединения, которое использовали непосредственно в следующей реакции.
Часть E. Получение метил 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензоата.
Добавляли вместе продукт из Части D (21,43 г, 69,3 ммоль), мочевину (10,4 г, 173 ммоль) и уксусную кислоту (ледяную, 200 мл). Смесь затем нагревали до 120°C в течение 18,5 ч с последующим концентрированием под вакуумом с получением масла. К этому маслу добавляли метанол (13 мл), и этилацетат (350 мл). Полученную в результате смесь оставляли стоять в течение 24-48 ч, за счет чего образовывался осадок. Полученное в результате твердое вещество отфильтровывали и промывали небольшим количеством метанола (10 мл) и затем сушили на воздухе с получением на выходе названного соединения в виде твердого вещества (15,26 г, 66%).
Часть F. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси бензойной кислоты.
Добавляли вместе продукт из Части D (4,52 г, 13,52 ммоль), метанол (70 мл), и тетрагидрофуран (70 мл). Смесь затем интенсивно перемешивали до получения в результате гомогенного раствора. После достижения гомогенности добавляли раствор водного гидроксида натрия (1,0М, 68 мл). Смесь затем перемешивали в течение 12 ч, смесь затем концентрировали под вакуумом для удаления органического растворителя, с последующим добавлением водного раствора соляной кислоты (1,ОМ, 80 мл), что в результате приводило к образованию твердого вещества. Смесь затем концентрировали под вакуумом. К этому веществу добавляли соляную кислоту (12М, 100 мл) и полученное в результате вещество нагревали до 100°C в течение 1,5 ч. Реакционную смесь затем охлаждали и добавляли воду. Полученное в результате твердое вещество фильтровали, промывали водой, и сушили с получением на выходе названного соединения в виде твердого вещества (3,55 г, 82%).
Часть G. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-бензальдегида.
Продукт, полученный в Части F (4,07 г, 12,71 ммоль) и тионилхлорид (40,82 мл, 559 ммоль) объединяли, и смесь нагревали с обратным холодильником в течение 2 ч, с последующим концентрированием под вакуумом с получением твердого продукта светло желтого цвета. Твердое вещество растворяли в тетрагидрофуране (125 мл), этот раствор охлаждали до -78°C и медленно добавляли LiAlH(OtBu)3 (1М, 14 мл) на протяжении 10 мин, поддерживая температуру -78°C. Смесь перемешивали при -78°C в течение 2 ч, и реакцию гасили соляной кислотой (водн., 1М, 25 мл) при -78°C. Смесь нагревали до комнатной температуры и добавляли этилацетат. Слои отделяли и водный слой промывали этилацетатом. Органические экстракты объединяли и промывали полу-насыщенным раствором бикарбоната натрия. Органический слой сушили, фильтровали и концентрировали под вакуумом с получением на выходе названного соединения в виде твердого вещества (3,73 г, 96%).
Часть H. Получение 1-(3-трет-бутил-4-метокси-5-(4-нитростирил)фенил)дигидро-пиримидин-2,4(1Н,3H)-диона.
Продукт, полученный в Части G (1,00 г, 3,29 ммоль) и диэтил 4-нитробензил-фосфонат (0,853 г, 3,12 ммоль) растворяли в дихлорметане (50 мл). Твердый трет-бутоксид, калия (0,737 г, 6,57 ммоль) добавляли порциями при комнатной температуре. Полученный в результате темно-красный раствор перемешивали в течение 1,5 ч при комнатной температуре. Добавляли раствор 1 н водной HCl (50 мл) и смесь перемешивали 30 мин, и затем разбавляли дихлорметаном (50 мл). Полученный в результате органический слой отделяли и сушили. Это вещество очищали с помощью колоночной хроматографии на силикагеле, используя 99/1 дихлорметан/метанол в качестве элюента с получением названного соединения в виде твердого вещества (1,12 г, 80%).
Часть I. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидро-пиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Продукт, полученный в Части H (1,1 г, 2.60 ммоль), железо (0,725 г, 12,99 ммоль), и хлорид аммония (0,208 г, 3,90 ммоль) добавляли к смеси тетрагидрофурана (40 мл), этанола (40 мл) и воды (12 мл). Суспензию нагревали до 90°C в течение 45 мин, и затем охлаждали до окружающей температуры. Этот раствор фильтровали через слой целита (10 г), промывали этанолом (20 мл), и фильтрат концентрировали под вакуумом до твердого вещества. Полученное в результате твердое вещество растворяли в этилацетате (100 мл), и этот раствор промывали водой (50 мл) и сушили над Na2SO4. Высушивающее вещество отфильтровывали, и растворитель удаляли под вакуумом с получением анилинового аддукта в виде твердого вещества желтого цвета (830 мг).
Твердое вещество (830 мг, 2,109 ммоль) растворяли в дихдорметане (50 мл), и добавляли пиридин (0,512 мл, 6,33 ммоль) и метансульфонилхлорид (0,181 мл, 2,32 ммоль) и полученный в результате раствор перемешивали при комнатной температуре 16 ч. Добавляли дихлорметан (100 мл) с последующей экстракцией раствором 1 н водн. HCl (2×50 мл). Органический слой сушили, концентрировали под вакуумом и очищали с помощью колоночной хроматографии на силикагеле, используя 98/2 CH2Cl2/MeOH с получением названного соединения в виде твердого вещества (480 мг, 39%, две стадии). Температура плавления =260-261°C (транс-изомер) 1H ЯМР (500 MHz, DMSO-d6): δ ppm 1.37 (s, 9H), 2.71 (t, J=6.7 Hz, 2H), 3.01 (s, 3H), 3.75 (s, 3H), 3.79 (t, J=6.6 Hz, 2H), 7.13 (d, J=16.5 Hz, 1H), 7.15 (d, J=2.4 HZ, 2H), 7.23 (d, J=8.5 Hz, 2H), 7.25 (d, J=16.5 Hz, 1H), 7.51 (d, J=2.4 Hz, 1H), 7.61 (d, J=8.6 Hz, 2H), 9.80 (bs, 1H), 10.30 (s, 1H). (транс-изомер).
Пример 30. Получение (Z)-N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)-1-хлорвинил)фенил)метансульфонамида (соединение IA-L1-1.3).
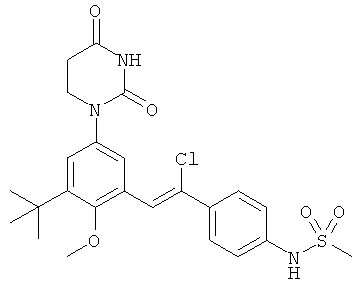
Часть A. Получение диэтилгидрокси(4-нитрофенил)метилфосфоната.
Названное соединение получали, как описано у Taylor, WP, et. Al, Bioorg. Med. Chem. 4:1515-1520 (1996). 4-Нитробензальдегид (3,0 г, 19,85 ммоль) и диэтил фосфонат (2,74 г, 19,85 ммоль) объединяли и обрабатывали 0,5 н раствором метоксида натрия в метаноле (0,993 мл, 0,496 ммоль). Полученный в результате красно-оранжевый раствор перемешивали 12 ч при комнатной температуре. Реакционную смесь экстрагировали дихлорметаном (20 мл), после этого полунасыщенным хлоридом аммония (20 мл). Органический слой отделяли, сушили и концентрировали под вакуумом с получением названного соединения в виде полутвердого вещества (5,1 г, 89%).
Часть B. Получение диэтилхлор(4-нитрофенил)метилфосфоната.
Продукт, полученный в Части A (500 мг, 1,729 ммоль) растворяли в дихлорметане (10 мл) и обрабатывали трифенилфосфином (998 мг, 3,80 ммоль), после этого N-хлорсукцинимидом (462 мг, 3,46 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч. Раствор концентрировали под вакуумом и остаток очищали с помощью колоночной хроматографии, используя силикагель, элюируя смесью 1/1 гексана/этилацетата с получением названного соединения в виде масла (262 мг, 49%).
Часть C. Получение (Z)-N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-1-хлорвинил)фенил)метансульфонамида.
Продукт, полученный в Примере 29, Части G (100 мг, 0,329 ммоль) обрабатывали продуктом, полученным из Части B, используя процедуры, описанные в Примере 29, Части H и Примере 29, Части I с получением 39 мг названного соединения. 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.36 (s, 9H), 2.71 (t, J=6.8 Hz, 2H), 3.06 (s, 3H), 3.71 (s, 3H), 3.78 (t, J=6.8 Hz, 2H), 7.23 (d, J=2.6 Hz, 1H), 7.27 (s, 1H), 7.28 (d, J=8.6 Hz, 2H), 7.48 (d, J=2.6 Hz, 1H), 7.78 d, J=8.8 Hz, 1H), 10.05 (s, 1H), 10.34 (s, 1H).
Пример 31. Получение (E)-1-(3-трет-бутил-5-(4-фторстирил)-4-метоксифенил) дигидропиримидин-2,4(1H,3H)-диона (соединение IA-L1-1.12).
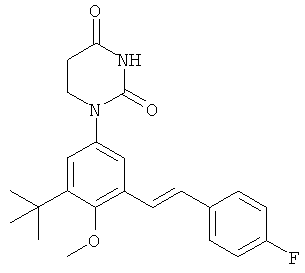
Названное соединение получали в соответствии с процедурами, описанными в Примере 29, Части H и Примере 29, Части I, используя продукт, полученный в Примере 29, Части G (50 мг, 0,164 ммоль) и диэтил 4-фторбензилфосфонат (40,5 мг, 0,164 ммоль). Названное соединение получали в виде твердого вещества (30 мг, 46%). 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.37 (s, 9H), 2.72 (t, J=6.6 Hz, 2H), 3.76 (s, 3H), 3.79 (t, =6.6 Hz, 2H), 7.21 (m, 4H), 7.30 (d, J=16.3 Hz, 1H), 7.53 (d, J=2.6 Hz, 1H), 7.73 (m, 2H), 10.35 (s, 1H).
Пример 32. Получение (Z)-N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-1-фторвинил)фенил)метансульфонамида (соединение IA-L1-1.4).
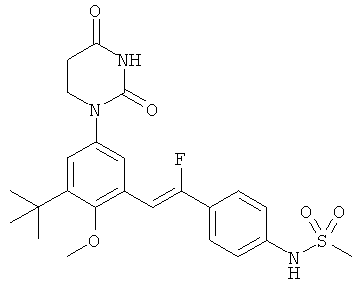
Часть A. Получение диэтилфтор(4-нитрофенил)метилфосфоната. Названное соединение получали, как описано у Taylor, WP, et. Al, Bioorg. Med. Chem. 4:1515-1520 (1996). Продукт из Примера 30, Части А (500 мг, 1,729 ммоль) растворяли в дихлорметане (10 мл) и обрабатывали добавлением по каплям трифторида (диэтиламино)серы (DAST) (2,5 мл, 18,9 ммоль). Смесь перемешивали при комнатной температуре в течение 18 ч. Добавляли раствор полунасыщенного одноосновного фосфата натрия (20 мл), с последующим добавлением дихлорметана (20 мл) и отделением полученной в результате органической фазы. Этот органический раствор сушили и концентрировали под вакуумом, и затем подвергали колоночной хроматографии, используя силикагель, элюируя 1/1 смесью гексана/этилацетата с получением названного соединения в виде масла (215 мг, 43%).
Часть B. Получение (Z)-N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-1-фторвинил)фенил)метансульфонамида.
Продукт, полученный, как описано в Части A (100 мг, 0,329 ммоль) обрабатывали продуктом, полученным в Примере 29, Части G (96 мг, 0,329 ммоль) в соответствии с процедурами, описанными в Примере 29, Части H и Примере 29, Части I с получением 53 мг названного соединения в виде 1/1 смеси цис/транс изомеров. Хроматографическое разделение с помощью ВЭЖХ с обращенной фазой, с использованием 40-100% градиента ацетонитрила в 0,1% водной трифторуксусной кислоте давало названное соединение в виде твердого вещества (20 мг). 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.37 (s, 9H), 2.71 (t, J=6.8 Hz, 2H), 3.06 (s, 3Н), 3.77 (s, 3Н), 3.78 (m, 2H), 6.62 (d, J=40.4 Hz, 1H), 7.18 (d, J=2.6 Hz, 1H), 7.30 (d, J=8.4 Hz, 2H), 7.55 (d, J=2.6 HZ, 1H), 7.75 (d, J=8.8 Hz, 2H), 10.08 (s, 1H), 10.33 (s, 1H).
Пример 33. Получение (E)-N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-1-фторвинил)фенил)метансульфонамида (соединение IA-L1-1.5).
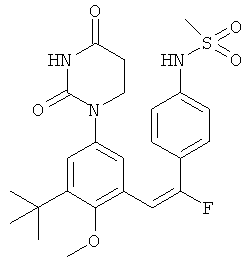
Хроматографическое разделение с помощью ВЭЖХ с обращенной фазой 1/1 смеси цис/транс изомерного вещества (53 мг) из Примера 32, Части A, используя 40-100% градиент ацетонитрила в 0,1% водной трифторуксусной кислоте, давало названное соединение в виде твердого вещества (16,5 мг). 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.33 (s, 9H), 2.60 (t, J=6.6 Hz, 2H), 3.01 (s, 3Н), 3.57 (t, J=6.6 Hz, 2H) 3.79 (s, 3Н), 6.46 (d, J=21.3 Hz, 1H), 6.87 (d, J=2.2 Hz, 1H), 7.14 (m, 3Н), 7.36 (d, J=8.8 Hz, 2H), 10.02 (s, 1H), 10.24 (s, 1H).
Пример 34. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксистирил)-2-фторфенил)метансульфонамида (соединение IA-L1-1.26)
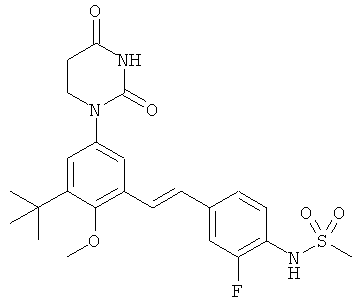
Часть A. Получение 4-(бромметил)-2-фтор-1 -нитробензола.
(3-фтор-4-нитрофенол)метанол (1,24 г, 7,25 ммоль) растворяли в дихлорметане (25 мл) и обрабатывали трифенилфосфином (2,281 г, 8,70 ммоль) после этого N-бромсукцинимидом (1,548 г, 8,70 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли воду (50 мл) и дихлорметан (40 мл), и органический слой отделяли и сушили. Этот раствор концентрировали под вакуумом и очищали с помощью колоночной хроматографии, используя силикагель, элюируя 5/1 смесью гексана/этилацетата с получением названного соединения в виде твердого вещества (1,27 г, 75%).
Часть B. Получение диэтил 3-фтор-4-нитробензилфосфоната.
Продукт, полученный в Части A (1,27 г, 5,43 ммоль) добавляли к триэтилфосфину (8 мл, 54,3 ммоль), и этот раствор нагревали до 120°C в течение 1 ч. После охлаждения, избыток триэтилфосфина удаляли нагреванием под вакуумом и остаток подвергали колоночной хроматографии на силикагеле, используя 99/1 дихлорметан/метанол в качестве элюента с получением неочищенного названного соединения в виде масла (800 мг).
Часть C. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксистирил)-2-фторфенил)метансульфонамида.
Продукт, описанный в Примере 29, Части G (533 мг, 1,751 ммоль) обрабатывали продуктом, описанным в Части B (510 мг, 1,751 ммоль) в соответствии с процедурами, описанными в Примере 29, Части Н и Примере 29, Части I с получением 80 мг названного соединения. 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.37 (s, 9H), 2.71 (t, J=6.5 Hz, 2H), 3.05 (s, 3H), 3.76 (s, 3H), 3.79 (t, J=6.6 HZ, 2H), 7.18 (m, 2H), 7.36 (d, J=16.5 Hz, 1H), 7.39 (m, 1H), 7.44 (m, 1H), 7.52 (d, J=2.6 Hz, 1H), 7.63 (m, 1H), 9.65 (s, 1H), 10.35 (s, 1H).
Пример 35. Получение N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)циклопропил)фенил)метансульфонамида (соединение IA-L8-1.1).
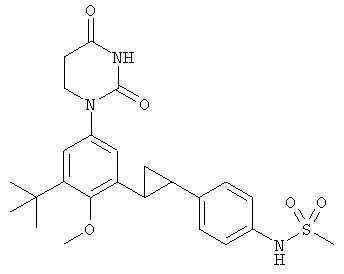
Продукт, полученный, как описано в Примере 29, Части I (30 мг, 0,064 ммоль) растворяли в тетрагидрофуране (2 мл) и обрабатывали 0,95 мл 0,67М эфирного раствора диазометана (0,636 ммоль), после чего ацетатом палладия (0,7 мг, 0,0031 ммоль). Смесь перемешивали в течение 30 мин при комнатной температуре с последующим удалением твердого вещества фильтрованием и концентрированием фильтрата. Фильтрат очищали с помощью колоночной хроматографии на силикагеле, используя 98/2 дихлорметан/метанол в качестве элюента с получением названного соединения в виде твердого вещества (21,6 мг, 70%). Температура плавления 265-266°C. 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.33 (s, 9H) 1.50 (m, 2H), 2.13 (m, 1H). 2.27 (m, 1H), 2.69 (t, J=6.6 Hz, 2H), 2.94 (s, 3H), 3.63 (s, 3H), 3.74 (t, J=6.6 Hz, 2H), 6.84 (d, J=2.6 Hz, 1H), 7.04 (d, J=2.6 Hz, 1H), 7.14 (d, J=8.8 Hz, 2H), 7.20 (d, J=8.8 Hz, 2H), 9.60 (s, 1H), 10.29 (s, 1H).
Пример 36. Получение N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенэтил)фенил)метансульфонамида (соединение IA-L5-2-1.1).
Продукт, полученный, как описано в Примере 29, Части I (415 мг, 0,88 ммоль) растворяли в метаноле (30 мл) и обрабатывали 50 мг 10% палладия на угле. Суспензию перемешивали в течение 48 ч при комнатной температуре под давлением 1 атм водорода. Реакционную смесь фильтровали через целит и концентрировали в вакууме с получением названного соединения в виде твердого вещества (230 мг, 55%). Температура плавления 233-234°C. 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.34 (s, 9H), 2.68 (t, J=6.8 Hz, 2H), 2.86 (s, 4H), 2.93 (s, 3H), 3.70 (m, 2H), 3.74 (s, 3H), 7.11 (m, 4H), 7.23 (m, 2H), 9.59 (s, 1H), 10.29 (s,).
Пример 37. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)стирил)фенил)метансульфонамида (соединение IA-L1-1.16)
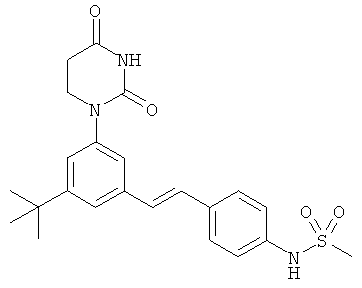
Часть A. Получение метил 3-трет-бутил-5-(хлоркарбонил)бензоата.
Смесь 3-трет-бутил-5-(метоксикарбонил)бензойной кислоты (9,18 г, 38,9 ммоль, полученной по методу Carter et. al., WO 2005021500 A1), тионилхлорида (75 мл) и 1 капли DMF в толуоле (200 мл) нагревали с обратным холодильником в течение 2 ч, охлаждали и концентрировали. Остаток азеотропировали толуолом (3×50 мл) и сушили под высоким вакуумом с получением названного соединения в виде беловатого воскообразного твердого вещества (9,9 г, количественный выход).
Часть B. Получение метил 3-(азидокарбонил)-5-трет-бутилбензоата.
К продукту Части A (9,9 г, 38,9 ммоль) в ацетоне (200 мл) добавляли быстрыми каплями раствор азида натрия (10,12 г, 156 ммоль) растворенного в воде (20 мл). Смесь перемешивали в течение 2 ч и разбавляли EtOAc. Органический слой промывали H2O, насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали с получением названного соединения в виде твердого вещества белого цвета (9,9 г, 97%).
Часть C. Получение метил 3-амино-5-трет-бутилбензоата.
Продукт из Части B (9,9 г, 37,9 ммоль) в толуоле (100 мл) нагревали с обратным холодильником в течение 1 ч и концентрировали с получением промежуточного изоцианата, который растворяли в DME (60 мл), обрабатывали 8% HCl (150 мл) и перемешивали в течение 16 ч. Смесь концентрировали и остаток растворяли в воде, нейтрализовали твердым бикарбонатом натрия и экстрагировали 3×100 мл EtOAc. Органические фазы объединяли, промывали насыщенным NaCl, сушили (Na2SO4), фильтровали и концентрировали. Сырой продукт хроматографировали на силикагеле, элюируя 2:1 гексаном/EtOAc с получением названного соединения в виде масла (2,7 г, 35%).
Часть D. Получение метил 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензоата.
Смесь продукта Части C (2,34 г, 11,29 ммоль) и акриловой кислоты (2,32 мл, 33,9 ммоль) в толуоле (60 мл) нагревали с обратным холодильником в атмосфере азота в течение 24 ч, охлаждали и концентрировали. Полученный в результате остаток затем обрабатывали мочевиной (2,03 г, 33,9 ммоль) в уксусной кислоте (35 мл) и нагревали при 120°C в течение 24 ч, охлаждали и концентрировали. Остаток азеотропировали 3×50 мл толуолом и растворяли в 100 мл EtOAc. Органический слой промывали разбавленным водным NaHCO3, H2O, насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали с получением названного соединения в виде твердого вещества белого цвета (2,1 г, 61%).
Часть E. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)бензойной кислоты.
Смесь продукта из Части D (1,8 г, 5,91 ммоль) и 1М NaOH (29,6 мл, 29,6 ммоль) в МеОН (15 мл) и THF (15 мл) перемешивали в течение 24 ч и концентрировали. Остаток обрабатывали 50 мл 1М HCl и экстрагировали в EtOAc. EtOAc слой промывали НзО, насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали с получением твердого вещества белого цвета. Эту промежуточную мочевину объединяли с 20 мл концентрированной НС1 и нагревали при 100°C в течение 1 ч, охлаждали и разбавляли 75 мл ледяной воды с получением белого порошка, который собирали фильтрацией и сушили до постоянной массы с получением названного соединения (1,6 г, 93%).
Часть F. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил) стирил)фенил)метансульфонамида.
Продукт, описанный в Части E обрабатывали тионилхлоридом и литий три-трет-бутоксиалюминий гидридом в соответствии с процедурами, описанными в Примере 29, Части G с получением 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)бензальдегида. Этот альдегид обрабатывали диэтил 4-нитробензилфосфонатом в соответствии с процедурами, описанными в Примере 29, Части H и Примере 29, Части I с получением названного соединения (85 мг). 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.32 (s, 9 Н) 2.72 (t, J=6.43 Hz, 2 Н) 3.01 (s, 3 Н) 3.82 (t, J=6.62 Hz, 2 Н) 7.18-7.25 (m, 5 Н) 7.39 (s, 1 Н) 7.46 (s, 1 Н) 7.58 (d, J=8.46 Hz, 2 Н) 9.84 (s, 1 Н) 10.37 (s, 1 Н).
Пример 38. Получение (Z)-N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)-1-метоксивинил)фенил)метансульфонамида (соединение IA-L1-1.17).
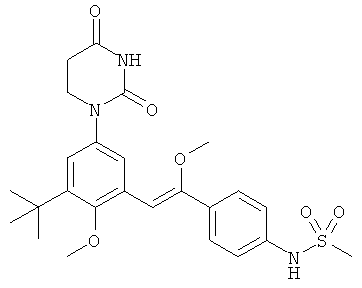
Часть A. Получение 1 -(диметоксиметил)-4-нитробензола.
В колбу, снабженную магнитной мешалкой и колонкой Vigreux помещали 4-нитро-бензальдегид (5,0 г, 33,1 ммоль), пиридиний p-толуолсульфат (1,66 г, 6,62 ммоль), триметоксиметан (3,51 г, 33,1 ммоль) и метанол (100 мл). Смесь нагревали при 50°C в течение 12 ч и концентрировали в вакууме. Остаток перерастворяли в EtOAc и промывали водн. NaOH (1M), H2O и солевым раствором. Смесь сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением на выходе названного соединения в виде прозрачного светло-желтного масляного продукта (6,36 г, 97%).
Часть B. Получение диэтилметокси(4-нитрофенил)метилфосфоната. Продукт из Части A (3,0 г, 15,2 ммоль) и триэтилфосфит (2,53 г, 15,2 ммоль) растворяли в дихлорметане (30 мл) в атмосфере азота, охлаждали до -20°C и обрабатывали по каплям добавлением эфирата трехфтористого бора (2,27 г, 16 ммоль). Смесь оставляли медленно нагреваться до комнатной температуры в течение ночи при перемешивании. Добавляли воду и полученную в результате смесь перемешивали 5 мин, отделяли, и органический слой сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением твердого остатка. Этот остаток очищали на силикагеле (100% EtOAc до 3% CH3OH/EtOAc) с получением на выходе названного соединения в виде светло-желтого масляного продукта (3,78 г, 82%).
Часть C. Получение (Z)-N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-1-метоксивинил)фенил)метансульфонамида.
Продукт, полученный в соответствии с процедурой, описанной в Примере 29, Части G (400 мг, 1,314 ммоль) обрабатывали продуктом, полученным в Части B (399 мг, 1,314 ммоль) в соответствии с процедурами, описанными в Примере 29, Части H и Примере 29, Части I с получением названного соединения (17 мг, 6%). 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.36 (s, 9 Н) 2.71 (t, J=6.62 Hz, 2 Н) 3.05 (s, 3 Н) 3.58 (s, 3 Н) 3.75 (s, 3 Н) 3.76-3.81 (m, 2 Н) 6.25 (s, 1 Н) 7.11 (d, J=2.57 Hz, 1 Н) 7.27 (d, J=8.46 Hz, 2 Н) 7.60 (d, J=8.82 Hz, 2 Н) 7.67 (d, J=2.57 Hz, 1 Н) 9.96 (s, 1 Н) 10.32 (s, 1 Н).
Пример 39. Получение (E)-1-(3-трет-бутил-4-метокси-5-стирилфенил)дигидро-пиримидин-2,4(1H,3H)-диона (соединение IA-L1-1.18).
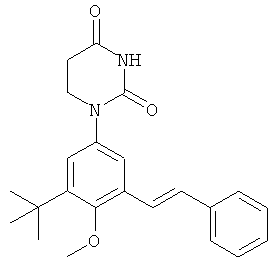
Продукт, полученный в соответствии с процедурой, описанной в Примере 29, Части G (50 мг, 0,164 ммоль) обрабатывали диэтилбензилфосфонатом (0,034 мл, 0,164 ммолье) в соответствии с процедурой, описанной в Примере 29, Части H с получением названного соединения (13 мг, 19%). 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.37 (s, 9 Н) 2.72 (t, J=6.62 Hz, 2 Н) 3.76 (s, 3 Н) 3.80 (t, J=6.80 Hz, 2 Н) 7.16-7.18 (m, 1 Н) 7.21-7.23 (m, 1 Н) 7.29-7.33 (m, 2 Н) 7.36-7.43 (m, 2 Н) 7.54 (d, J=2.57 Hz, 1 Н) 7.64 (d, J=7.35 Hz, 2 Н) 10.35 (s, 1 Н).
Пример 40. Получение (E)-1-(3-трет-бутил-4-метокси-5-(4-метоксистирил)фенил) дигидропиримидин-2,4(1H,3H)-диона (соединение IA-L1-1.14).
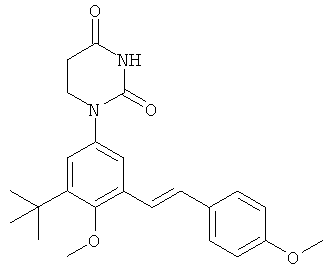
Продукт, полученный в соответствии с процедурой, описанной в Примере 29, Части G (50 мг, 0,164 ммоль) обрабатывали диэтил 4-метоксибензилфосфонатом (0,028 мл, 0,164 ммоль) в соответствии с процедурой, описанной в Примере 29, Части H с получением названного соединения (4 мг, 4%). 1H ЯМР (300 MHz, DMSO-d6): δ ppm 1.37 (s, 9 Н) 2.71 (t, J=6.62 Hz, 2 Н) 3.70-3.81 (m, 8 Н) 6.96 (d, J=8.82 Hz, 2 Н) 7.13 (d, J=2.21 Hz, 1 Н) 7.15 (d, J=2.57 Hz, 2 Н) 7.50 (d, J=2.57 Hz, 1 Н) 7.58 (d, J=8.46 Hz, 2 Н) 10.34 (s, 1 Н).
Пример 41A. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.1).
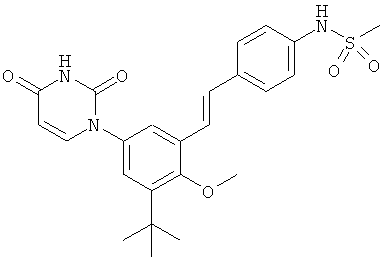
Часть A. Получение (E)-метил 3-трет-бутил-2-метокси-5-(3-(3-метоксиакрилоил)уреид) бензоата.
Продукт, полученный как описано в Примере 29, Части C (2,0 г, 8,43 ммоль) растворяли в 30 мл N,N-диметилацетамида и охлаждали до -25°C. Добавляли по каплям 0,5 молярный раствор E-3-метоксиакрилоилизоцианата в бензоле (21,9 мл, 10,96 ммоль), и полученный в результате раствор перемешивали при окружающей температуре в течение 4 ч, и затем выливали воду. Этот продукт экстрагировали в дихлорметан, промывали солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали под вакуумом с получением названного соединения.
Часть B. Получение метил 3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксибензоата.
Продукт из Части A (3,1 г, 8,51 ммоль) растворяли в этаноле (60 мл). К этому раствору добавляли смесь концентрированной серной кислоты (6 мл) и воды (60 мл). Гетерогенную смесь нагревали при 100°C в течение 3 ч. Этанол удаляли под вакуумом, и затем водный раствор экстрагировали дихлорметаном и упаривали до сухости. Этот остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя 1% метанолом/дихлорметаном с получением на выходе названного соединения (1,23 г, 44%).
Часть C. Получение 3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси бензойной кислоты.
Продукт из Части B (1,23 г, 3,7 ммоль) помещали в этанол (5 мл) и 1М раствор гидроксида натрия (10 мл) и перемешивали при окружающей температуре в течение 18 ч. Этот раствор подкисляли 1М HCl и полученное в результате твердое вещество фильтровали и сушили с получением названного соединения (0,945 г, 80%).
Часть D. Получение 3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси бензальдегида.
Продукт из Части C (0,945 г, 2,97 ммоль) помещали в тионилхлорид (4,5 мл) и смесь нагревали при 80°C в течение 40 мин. После упаривания до сухости, хлорангидрид растворяли в сухом THF (8 мл) и охлаждали до -78°C. Добавляли по каплям 1 М раствор литий три-трет-бутоксиалюминий гидрида в THF (3,0 мл, 3,0 ммоль). Через 45 мин холодную реакционную смесь гасили 1М HCl (5 мл), экстрагировали в этилацетат, и очищали с помощью колоночной хроматографии на силикагеле, элюируя дихлорметаном после чего 1% метанолом/дихлорметаном с получением названного соединения (0,635 г, 71%).
Часть E. Получение (E)-1-(3-трет-бутил-4-метокси-5-(4-нитростирил)фенил)пиримидин-2,4(1H,3H)-диона.
Продукт Части D (0,634 г, 2,1 ммоль) и диэтил 4-нитробензилфосфонат (0,573 г, 2,1 ммоль) объединяли в дихлорметане (25 мл) при окружающей температуре. Добавляли порциями трет-бутоксид калия (0,494 г, 4,4 ммоль) и полученную в результате красную/коричневую гетерогенную смесь перемешивали в течение 1,5 ч. Эту смесь гасили 1М HCl (15 мл), выливали в воду и экстрагировали в этилацетат, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 1% метанолом/дихлорметаном с получением названного соединения (0,735 г, 83%).
Часть F. Получение (E)-1-(3-(4-аминостирил)-5-трет-бутил-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
Продукт из Части E (0,735 г, 1,74 ммоль), хлорид аммония (0,14 г, 2,62 ммоль), и железо (0,487 г, 8,72 ммоль) объединяли в растворе этанола (10 мл), воды (5 мл), и THF (10 мл) и нагревали при 75°C в течение 1 ч. Смесь фильтровали через диатомит, хорошо промывали THF и концентрировали с получением названного соединения.
Часть G. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Продукт из Части F (0,683 г, 1,75 ммоль) и пиридин (0,564 мл, 6,98 ммоль) объединяли в дихлорметане (15 мл) при окружающей температуре. Метансульфонилхлорид (0,163 мл, 2,1 ммоль) добавляли по каплям, и раствор перемешивали в течение 18 ч. Смесь выливали в 1М HCl и экстрагировали в дихлорметан, концентрировали, и очищали с помощью колоночной хроматографии на силикагеле, элюируя 1%, 2% метанолом/дихлорметаном. Растирание с дихлорметаном давало твердое вещество, которое фильтровали и сушили с получением названного соединения в виде бесцветного порошка (0,465 г, 57%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 H), 3.01 (s, 3 H), 3.79 (s, 3 H) 5.65 (d, J=7.72 Hz, 1 H), 7.17-7.28 (m, 5 H), 7.58-7.70 (m, 3 H), 7.75 (d, J=7.72 Hz, 1 H), 9.86 (s, 1 H), 11.42 (s, 1 H).
Пример 41B. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.1).
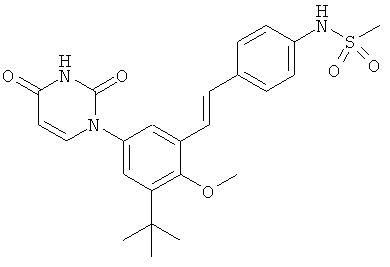
Часть A. Получение N-(4-этинилфенил)метансульфонамида.
В 2 л, 3-горловую круглодонную колбу, снабженную верхнеприводной мешалкой, добавляли 4-этиниланилин (30 г, 256 ммоль) и пиридин (42,5 мл, 525 ммоль) в дихлорметане (512 мл) с получением оранжевого раствора. Смесь охлаждали до 5°C и добавляли по каплям Метансульфонилхлорид (19,96 мл, 256 ммоль) на протяжении 15 мин. Реакционный раствор перемешивали при 5°C в течение 2 ч и промывали 1М водным HCl (3×250 мл). Дихлорметановый слой затем промывали последовательно насыщенным водным NaHCO3, водой, и насыщенным водным NaCl. Дихлорметановый слой сушили над сульфатом натрия и обрабатывали одновременно обесцвечивающим древесным углем в течение 30 мин, этот раствор затем фильтровали через целит и фильтрат концентрировали. Розовое/оранжевое твердое вещество растворяли в минимальном количестве горячего этилацетата (50-75 мл) и медленно разбавляли гексаном (500-600 мл) с получением оранжевых кристаллов, которые собирали фильтрованием и сушили с получением названного соединения (40,0 г, 80%).
Часть B. Получение (E)-4-(метилсульфонамид)стирилбороновой кислоты. (Ссылка: Org. Prep.Proc. Int., 2004, 36, 573-579) В колбу добавляли комплекс боран-метилсульфид (8,03 мл, 85 ммоль), затем тетрагидрофуран (16 мл), и эту смесь затем охлаждали до 0°C. Затем к охлаждаемому льдом раствору добавляли по каплям (1R)-(+)-альфа-пинен (26,2 мл, 169 ммоль) (за 10 мин). Смесь затем перемешивали при 0°C в течение 1 ч с последующим перемешиванием 2 ч при комнатной температуре. Полученную в результате густую белую суспензию охлаждали до -40°C и сушили на бане лед/ацетон, с последующим добавлением по каплям на протяжении 30 мин продукта из Части A (15,0 г, 77 ммоль), растворенного в 60 мл THF. После завершения добавления, смесь перемешивали в течение дополнительного часа при -35°C, затем 1 ч при комнатной температуре. Светло-желтый раствор затем охлаждали до 0°C и добавляли ацетальдегид (61,4 мл, 1088 ммоль), затем смесь нагревали с обратным холодильником при 50°C в течение 18 ч. Затем растворитель удаляли под вакуумом с получением концентрированного раствора оранжевого цвета, к которому добавляли воду (115 мл), и эту гетерогенную смесь перемешивали в течение 3 ч при комнатной температуре. Образовавшееся светло-желтое твердое вещество собирали и промывали водой (250 мл), затем сушили в вакуумной печи в течение ночи. Полученное в результате вещество затем растворяли в кипящем ацетоне (190 мл), что давало гомогенный раствор желтого цвета, с последующим устранением нагревания раствора и добавлением гексана (365 мл) на протяжении периода времени 5 мин. В растворе образовывалось твердое вещество белого цвета, и смесь перемешивали до охлаждения раствора до комнатной температуры, затем белое твердое вещество собирали и сушили в вакуумной печи в течение 1 часа с получением названного соединения (12,1 г, 85%).
Часть C. Получение 2-трет-бутил-4-нитрофенола.
К интенсивно перемешанному раствору 2-трет-бутилфенола (10 г, 66,6 ммоль) в гептане (67 мл) добавляли быстрыми каплями раствор 70% азотной кислоты (4,25 мл, 66,6 ммоль) разбавленной водой (4,25 мл). Полученную в результате темно-красную/коричневую смесь интенсивно перемешивали в течение 2 ч. Суспендированное твердое вещество собирали фильтрацией, промывали гексаном (300 мл), водой (200 мл) и еще один раз гексаном (200 мл) с получением порошка цвета какао, который сушили до постоянной массы (4,65 г, 35,6%).
Часть D. Получение 2-бром-6-трет-бутил-4-нитрофенола.
Раствор продукта из Части C (1,0 г, 5,12 ммоль) в ледяной уксусной кислоте (10,25 мл) обрабатывали порциями пиридин гидробромид пербромидом (1,80 г, 5,63 ммоль) с последующим перемешиванием при комнатной температуре в течение 2 ч. Дополнительное количество пиридиния гидробромида пербромида (3,6 г) добавляли двумя порциями и после еще 3 ч перемешивания реакцию завершали. Смесь выливали в ледяную воду, и эту смесь обрабатывали небольшим количеством сульфита натрия. Полученное в результате твердое вещество фильтровали и сушили под вакуумом с получением названного соединения в виде твердого вещества коричневого цвета (1,40 г, 100%).
Часть E. Получение 1-бром-3-трет-бутил-2-метокси-5-нитробензола.
Раствор продукта из Части D (1,40 г, 5,11 ммоль) в 10:1 т-бутилметиловом эфире-метаноле (25,5 мл) обрабатывали 2,0М триметилсилилдиазометаном в эфире (5,1 мл, 10,21 ммоль), с последующим перемешиванием при комнатной температуре в течение 18 ч. Смесь концентрировали под вакуумом с получением желтого масла, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гексаном с получением названного соединения в виде желтого масла (1,36 г, 92%).
Часть F. Получение трет-бутил 3-бром-5-трет-бутил-4-метоксифенилкарбамата.
Раствор продукта из Части E (960 мг, 3,33 ммоль) в метаноле (17 мл) обрабатывали 5% платиной на сульфидированном углероде (100 мг), с последующий гидрогенизацией под давлением из баллонной шины в течение 3 ч, и затем фильтровали через целит и концентрировали под вакуумом с получением 3-бром-5-трет-бутил-4-метоксианилина в виде желтого масла (860 мг, 3,33 ммоль, 100%). Раствор этого вещества в THF (17 мл) обрабатывали ди-трет-бутил дикарбонатом (800 мг, 3,66 ммоль) с последующим нагреванием с обратным холодильником в течение 2 ч. Концентрирование под вакуумом давало твердое вещество бежевого цвета, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гексаном. Твердое вещество растирали в порошок с гексаном, собирали фильтрованием, и сушили под вакуумом с получением названного соединения в виде почти белого твердого вещества (890 мг, 75%).
Часть G. Получение (E)-N-(3-бром-5-трет-бутил-4-метоксифенилкарбамоил)-3-метоксиакриламида.
Продукт из Части F (2,0 г, 5,58 ммоль) растворяли в дихлорметане (10 мл) и добавляли трифторуксусную кислоту (5 мл). Этот раствор перемешивали при комнатной температуре в течение 1 ч с последующим концентрированном под вакуумом и добавлением 10% водного бикарбоната натрия (50 мл), с последующей экстракцией этилацетатом (3×50 мл). Объединенные органические экстракты сушили и концентрировали с получением остатка, который растворяли в 10 мл N,N-диметилацетамида и охлаждали до -25°C. Добавляли по каплям 0,5 молярный раствор E-3-метоксиакрилоилизоцианата в бензоле (20,3 мл, 11,16 ммоль) и полученный в результате раствор перемешивали при окружающей температуре в течение 4 ч, и затем выливали в воду. Этот продукт экстрагировали в дихлорметан, промывали солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали под вакуумом с получением названного соединения.
Часть H. Получение 1-(3-бром-5-трет-бутил-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
Продукт из Части G (2,15 г, 5,58 ммоль) растворяли в этаноле (10 мл). К этому раствору добавляли смесь концентрированной серной кислоты (1 мл) и воды (10 мл). Эту гетерогенную смесь нагревали при 100°C в течение 2 ч. Этанол удаляли под вакуумом, и затем водный раствор экстрагировали дихлорметаном и упаривали до сухости. Этот остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя 1% метанолом/дихлорметаном с получением на выходе названного соединения (1,35 г, 69%).
Часть I. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Продукт из Части H (8,0 г, 22,65 ммоль), продукт из Части B (5,90 г, 24,46 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий дихлорид (0,738 г, 1,132 ммоль), и фосфат калия (9,62 г, 45,3 ммоль) растворяли в смеси тетрагидрофурана (128 мл) и воды (32 мл). Газообразный азот барботировали через полученную в результате смесь в течение 10 мин с последующим нагреванием этого раствора при 50°C в течение 5 ч в темноте. Реакционную смесь оставляли охлаждаться до комнатной температуры с последующим добавлением насыщенного водного хлорида аммония (50 мл), воды (200 мл), и этот раствор экстрагировали дихлорметаном (600 мл). К органическому экстракту добавляли сульфат магния и 3-меркаптопропил-активированный силикагель (20 г), и полученный в результате раствор перемешивали в темноте в течение 18 ч. Твердые вещества затем удаляли фильтрованием и фильтрат концентрировали под вакуумом и подвергали колоночной хроматографии на силикагеле, используя градиент 99/1-99/2 дихлорметан/метанол с получением названного соединения (7,4 г, 70%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 Н), 3.01 (s, 3 Н), 3.79 (s, 3 Н) 5.65 (d, J=7.72 Hz, 1 Н), 7.17-7.28 (m, 5 Н), 7.58-7.70 (m, 3 Н), 7.75 (d, J=7.72 Hz, 1 Н), 9.86 (s, 1 Н), 11.42 (s, 1 H).
Пример 42. Получение (E)-N-(4-(3-трет-бутил-5-(5-фтор-2,4-диоксо-3,4-дигидро-пиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.2).
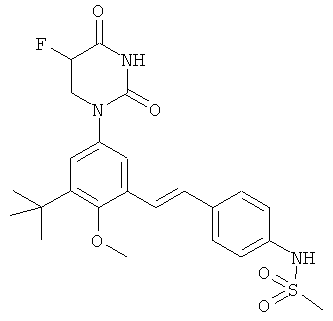
Часть A. Получение метил 3-трет-бутил-5-(5-фтор-6-метокси-2,4-диоксотетрагидро-пиримидин-1(2H)-ил)-2-метоксибензоата.
Процедуру фторирования проводили, как описано у Lal, GS, et al. J. Org Chem., 60:7340-7342 (1995). Продукт из Примера 41A, Части B (0,42 г, 1,26 ммоль) и Selectfluor™ (0,672 г, 1,9 ммоль) объединяли в смеси ацетонитрила (8 мл) и метанола (1 мл) и нагревали при 90°C в атмосфере N2 в течение 5 ч. Раствор разбавляли водой, экстрагировали в этилацетат, промывали раствором бикарбоната натрия, концентрировали и очищали с помощью колоночной хромато графин на силикагеле с получением названного соединения (0,138 г, 29%).
Часть B. Получение метил 3-трет-бутил-5-(5-фтор-2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксибензоата.
Продукт из Части A (0,134 г, 0,35 ммоль) и триэтиламин (1 мл) объединяли в метаноле (4 мл) и перемешивали при окружающей температуре в течение 18 ч. Раствор гасили 1М HCl, экстрагировали в дихлорметан и концентрировали с получением названного соединения (0,113 г, 92%).
Часть C. Получение 3-трет-бутил-5-(5-фтор-2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксибензойной кислоты.
Продукт из Части B (0,113 г, 0,32 ммоль) обрабатывали, как описано в Примере 41A, Части C с получением названного соединения (0,088 г, 81%).
Часть D. Получение 3-трет-бутил-5-(5-фтор-2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксибензальдегида.
Продукт из Части C (0,088 г, 0,26 ммоль) обрабатывали, как описано в Примере 41A, Части D с получением названного соединения (0,075 г, 90%).
Часть E. Получение (E)-1-(3-трет-бутил-4-метокси-5-(4-нитростирил)фенил)-5-фторпиримидин-2,4(1H,3H)-диона.
Продукт Части D (0,075 г, 0,23 ммоль) обрабатывали, как описано в Примере 41A, Части E с получением 0,077 г (75%).
Часть F. Получение (E)-1-(3-(4-аминостирил)-5-трет-бутил-4-метоксифенил)-5-фторпиримидин-2,4(1H,3H)-диона.
Продукт Части E (0,077 г, 0,18 ммоль) обрабатывали, как описано в Примере 41A, Части F с получением названного соединения (0,071 г, 94%).
Часть G. Получение (E)-N-(4-(3-трет-бутил-5-(5-фтор-2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
Продукт Части F (0,071 г, 0,17 ммоль) обрабатывали, как описано в Примере 41A, Части G с получением названного соединения (0,048 г, 57%). 1H ЯМР (300 MHz, DMSO-D6): δ ppm 1.38 (s, 9 H), 3.01 (s, 3 H), 3.79 (s, 3 H) 7.19-7.27 (m, 5 H), 7.62 (d, J=8.82 Hz, 2 H), 7.66 (d, J=2.57 Hz, 1 H), 8.25 (d, J=6.99 Hz, 1H).
Пример 43. Получение N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-3-метилфенил)метансульфонамида (соединение IA-L2-1.9).
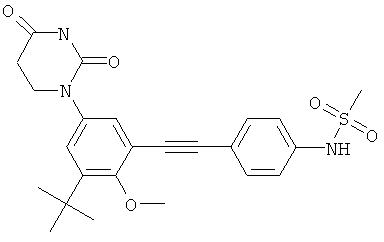
Часть A. Получение 2-трет-бутил-4-нитрофенола.
К интенсивно перемешанному раствору 2-трет-бутилфенола (10 г, 66,6 ммоль) в гептане (67 мл) добавляли быстрыми каплями раствор 70% азотной кислоты (4,25 мл, 66,6 ммоль), разбавленной водой (4,25 мл). Полученную в результате темно-красную/коричневую смесь интенсивно перемешивали в течение 2 ч. Суспендированное твердое вещество собирали фильтрацией, промывали гексаном (300 мл), водой (200 мл) и еще один раз гексаном (200 мл) с получением названного соединения в виде порошка цвета какао, который сушили до постоянной массы (4,65 г, 35,6%).
Часть B. Получение 2-трет-бутил-6-йод-4-нитрофенола.
К продукту из Части A (4,5 г, 23,05 ммоль), растворенному в MeOH (120 мл) и воде (30 мл), добавляли монохлорид йода (1,155 мл, 23,05 ммоль) по каплям за период времени 10 мин. Смесь перемешивали в течение 2 ч и разбавляли в 1 л воды и оставляли стоять в течение ночи. Твердое вещество собирали фильтрацией и промывали 3×50 мл водой и сушили под вакуумом в течение ночи с получением названного соединения в виде твердого вещества желтовато-коричневого цвета (7,14 г, 96%).
Часть C. Получение 1-трет-бутил-3-йод-2-метокси-5-нитробензола.
К охлажденному на ледяной бане раствору продукта из Части B (5,5 г, 17,13 ммоль) в MTBE (15 мл) в 50 мл сосуде высокого давления добавляли 2,0М TMS диазометан (12,85 мл, 25,7 ммоль) с последующим добавлением по каплям метанола (1,0 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали при комнатной температуре в течение 16 ч, охлаждали и давление сбрасывали. Раствор разделяли между EtOAc и водой. Органический слой промывали 1,0М HCl, насыщенным раствором карбоната калия, и насыщенным NaCl. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, с получением названного соединения в виде красного масла, которое использовали без очистки (5,4 г, 84%).
Часть D. Получение 3-трет-бутил-5-йод-4-метоксианилина. Смесь продукта из Части C (5,80 г, 17,31 ммоль), хлорида аммония (1,389 г, 26,0 ммоль), и железа (4,83 г, 87 ммоль) в смеси THF/MeOH/вода (всего 200 мл, 2/2/1) нагревали с обратным холодильником в течение 2 ч, охлаждали и фильтровали через целит. Фильтрат упаривали, и остаток разделяли между водой и EtOAc. Органический слой промывали насыщенным солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением названного соединения в виде коричневого масла (5,28 г, 100% выход).
Часть E. Получение 1-(3-трет-бутил-5-йод-4-метоксифенил)дигидропиримидин-2,4(1Н,3H)-диона.
Продукт из Части D (8,2 г, 26,9 ммоль) обрабатывали акриловой кислотой (5,53 мл, 81 ммоль) и перемешивали в течение ночи с получением чрезвычайно вязкой смеси. Эту смесь обрабатывали уксусной кислотой (60 мл) и мочевиной (7,3 г 120 ммоль), нагревали при 120°C в течение 24 ч, охлаждали и концентрировали. Остаток азеотропировали 3×100 мл толуолом с получением коричневого/желтовато-коричневого твердого вещества. Твердое вещество суспендировали в смеси 50 мл EtOAc и 100 мл насыщенного NaHCO3 и перемешивали в течение 30 мин для нейтрализации какого-либо количества оставшений уксусной кислоты. Твердое вещество собирали фильтрацией и промывали многократно 50 мл порциями воды и, наконец, 3:1 гексаном/EtOAc (50 мл) с получением названного соединения в виде беловатого твердого вещества, которое сушили до постоянной массы (7,1 г, 66%).
Часть F. Получение N-(4-йод-3-метилфенил)метансульфонамида.
Раствор 4-йод-3-метиланилина (4,37 г, 18,75 ммоль) в CH2Cl2 (25 мл) обрабатывали пиридином (6,07 мл, 75 ммоль) с последующим добавлением по каплям метансульфонилхлорида (1,607 мл, 20,63 ммоль) с получением красноватой/оранжевой смеси. Смесь перемешивали в течение 2 ч, концентрировали и разбавляли EtOAc. EtOAc слой промывали 1М HCl, водой, насыщенным NaCl, сушили (Na2SO4) и фильтровали. EtOAc фильтрат обрабатывали активированным углем в течение 30 мин при 50°C и фильтровали через 10 г прокладку из диоксида кремния и концентрировали с получением названного соединения в виде светло-желтого твердого вещества (5,5 г, 94%).
Часть G. Получение N-(3-метил-4-((триметилсилил)этинил)фенил)метансульфонамида.
Смесь продукта из Части F (3,11 г, 10 ммоль), йодида меди (I) (0,067 г, 0,35 ммоль), дихлорбис(трифенилфосфин)палладия (II) (0,351 г, 0,50 ммоль), триэтиламина (6,97 мл, 50,0 ммоль) и триметилсилилацетилена (1,684 мл, 12,0 ммоль) в ацетонитриле (50 мл) продували барботирующим N2 в течение 5 мин и нагревали в атмосфере N2 при 80°C в течение 30 мин. Реакционную смесь выливали в 200 мл EtOAc и разделяли водой, добавляя достаточное количество 1М HCl для приведения pH к 1. Эту смесь интенсивно перемешивали в течение 15 мин и слои отделяли. EtOAc слой промывали последовательно 10% водным NaHCO3, водой, и насыщенным NaCl, сушили над Na2SO4 и фильтровали. Фильтрат обрабатывали 2,0 г силикагеля Silicycle Si-thiol, перемешивали в течение 2 ч и фильтровали через слой силикагеля толчиной 1 дюйм. Фильтрат концентрировали и остаток подвергали флэш-хроматографии на диоксиде кремния, элюируя 9:1 гексаном/EtOAc -->3:1 гексаном/EtOAc с получением названного соединения в виде твердого вещества бежевого цвета (2,7 г, 96%).
Часть H. Получение N-(4-этинил-3-метилфенил)метансульфонамида.
Продукт из Части G (1,13 г, 4,01 ммоль) в MeOH (20,07 мл) обрабатывали 1М NaOH (8,43 мл, 8,43 ммоль), перемешивали в течение 1 ч, распределяли в EtOAc/воду и осторожно подкисляли до pH 3 с использованием 1М HCl. EtOAc слой промывали солевым раствором, сушили (Na2SO4) и концентрировали с получением названного соединения в виде твердого вещества желтовато-коричневого цвета (820 мг, 98%).
Часть I. Получение N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-3-метилфенил)метансульфонамида.
Смесь продуктов из Части E (1,38 г, 3,43 ммоль), Части H (0,79 г, 3,78 ммоль), йодида меди (I) (0,023 г, 0,12 ммоль) дихлорбис(трифенилфосфин)палладия (II) (0,12 г, 0,172 ммоль) и триэтиламина (2,392 мл, 17,16 ммоль) в ацетонитриле (60 мл) продували барботирующим N2 в течение 5 мин и нагревали на масляной бане в атмосфере N2 при 80°C в течение 20 мин. Реакционную смесь выливали в 400 мл теплого EtOAc и разделяли водой, добавляя достаточное количество 1М HCl для приведения pH к 1. Эту смесь интенсивно перемешивали в течение 15 мин и слои отделяли. EtOAc слой промывали последовательно 10% NaHCO3, водой, и насыщенным NaCl. Органический слой сушили (Na2SO4), и фильтровали. Фильтрат обрабатывали 4,0 силикагеля Silicycle Si-thiol, осторожно нагревали с обратным холодильником в течение 2 ч, охлаждали и фильтровали через слой силикагеля толщиной 1 дюйм. Фильтрат концентрировали в твердое вещество желтого цвета, которое перекристаллизовывали, путем растворения в горячем EtOAc/MeOH (270 мл/30 мл), уменьшая объем до 100 мл и оставляли охлаждаться. Полученный в результате осадок собирали фильтрацией и перекристаллизовывали второй раз с получением названного соединения в виде твердого вещества белого цвета (760 мг, 46%). Температура плавления >280°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.35 (s, 9 H) 2.46 (s, 3 H) 2.70 (t, J=6.62 Hz, 2 H) 3.05 (s, 3 H) 3.77 (t, J=6.62 Hz, 2 H) 4.04 (s, 3 H) 7.08 (dd, J=8.46, 1.84 Hz, 1 H) 7.14 (s, 1 H) 7.25 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.57 Hz, 1 H) 7.50 (d, J=8.46 Hz, 1 H) 9.99 (s, 1 H) 10.36 (s, 1 H).
Пример 44. Получение N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)фенил)этинил)-3-хлорфенил)метансульфонамида (соединение IA-L2-1.3).
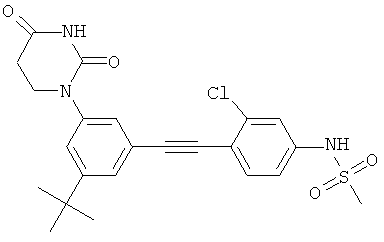
Часть A. Получение метил 3-трет-бутил-5-(хлоркарбонил)бензоата.
Смесь 3-трет-бутил-5-(метоксикарбонил)бензойной кислоты (9,18 г, 38,9 ммоль, полученной по методу Carter et. al., WO 2005021500 A1), тионилхлорида (75 мл) и 1 капли DMF в толуоле (200 мл) нагревали с обратным холодильником в течение 2 ч, охлаждали и концентрировали. Остаток азеотропировали толуолом (3×50 мл) и сушили под высоким вакуумом с получением названного соединения в виде беловатого воскообразного твердого вещества (9,9 г, количественный выход).
Часть B. Получение метил 3-(азидокарбонил)-5-трет-бутилбензоата.
К продукту Части A (9,9 г, 38,9 ммоль) в ацетоне (200 мл) добавляли быстрыми каплями раствор азида натрия (10,12 г, 156 ммоль), растворенного в воде (20 мл). Смесь перемешивали в течение 2 ч и разбавляли EtOAc. Органический слой промывали H2O, насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали с получением названного соединения в виде твердого вещества белого цвета (9,9 г, 97%).
Часть C. Получение метил 3-амино-5-трет-бутилбензоата.
Продукт из Части B (9,9 г, 37,9 ммоль) в толуоле (100 мл) нагревали с обратным холодильником в течение 1 ч и концентрировали с получением промежуточного изоцианата, который растворяли в DME (60 мл), обрабатывали 8% HCl (150 мл) и перемешивали в течение 16 ч. Смесь концентрировали, и остаток растворяли в воде, нейтрализовали твердым бикарбонатом натрия и экстрагировали 3×100 мл EtOAc. Органические экстракты объединяли, промывали насыщенным NaCl, сушили (Na2SO4), фильтровали и концентрировали. Сырой продукт хроматографировали на диоксиде кремния смесью 2:1 гексан/EtOAc с получением названного соединения в виде масла (2,7 г, 35%).
Часть D. Получение метил 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензоата.
Смесь продукта Части C (2,34 г, 11,29 ммоль) и акриловой кислоты (2,32 мл, 33,9 ммоль) в толуоле (60 мл) нагревали с обратным холодильником в атмосфере азота в течение 24 ч, охлаждали и концентрировали. Полученный в результате остаток затем обрабатывали мочевиной (2,03 г, 33,9 ммоль) в уксусной кислоте (35 мл) и нагревали при 120°C в течение 24 ч, охлаждали и концентрировали. Остаток азеотропировали 3×50 мл толуолом и растворяли в 100 мл EtOAc. Органический слой промывали разбавленным бикарбонатом, H2O, насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали с получением названного соединения в виде твердого вещества белого цвета (2,1 г, 61%).
Часть E. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)бензойной кислоты.
Смесь продукта из Части D (1,8 г, 5,91 ммоль) и 1М NaOH (29,6 мл, 29,6 ммоль) в MeOH (15 мл) и THF (15 мл) перемешивали в течение 24 ч и концентрировали. Остаток обрабатывали 50 мл 1М HCl и экстрагировали в EtOAc. EtOAc слой промывали H2O, насыщенным солевым раствором, сушили (Na2SO4), фильтровали и концентрировали с получением твердого вещества белого цвета. Эту промежуточную мочевину объединяли с 20 мл концентрированной HCl и нагревали при 100°C в течение 1 ч, охлаждали и разбавляли 75 мл ледяной воды с получением твердого вещества, которое собирали фильтрацией и сушили до постоянной массы с получением названного соединения в виде бесцветного порошка (1,6 г, 93%).
Часть F. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)бензальдегида.
Раствор продукта Части E (0,8 г, 2,76 ммоль) в сернокислом дихлориде (25 мл) нагревали с обратным холодильником в течение 1,5 ч, охлаждали и концентрировали. Остаток азеотропировали 3×25 мл толуолом с получением белого порошка. Этот хлорангидрид растворяли в безводном THF (25 мл), охлаждали до -78°C в атмосфере азота и обрабатывали по каплям 1М литий три-трет-бутоксиалюминий гидридом (3,03 мл, 3,03 ммоль) в THF. Этот раствор перемешивали при -78°C в течение 3 ч и гасили холодным с использованием 1М HCl, нагревали до окружающей температуры и экстрагировали 3×25 мл EtOAc. Органические экстракты объединяли, промывали водой, 10% бикарбонатом, насыщенным солевым раствором, и сушили сульфатом натрия. EtOAc фильтровали и концентрировали с получением названного соединения в виде твердого вещества белого цвета (0,77 г, количественный выход).
Часть G. Получение 1-(3-трет-бутил-5-этинилфенил)дигидропиримидин-2,4(1Н,3H)-диона.
Смесь продукта из Части G (913 мг, 3 ммоль), диметил 1-диазо-2-оксопропилфосфоната (749 мг, 3,90 ммоль, полученного по методу Ohira, Syn. Comm. 19 (3&4) 561-564 (1989), и карбоната калия (829 мг, 6,00 ммоль) в MeOH (20 мл) перемешивали в течение 16 ч и осторожно подкисляли 1М HCl. Смесь экстрагировали 2×50 мл CH2Cl2. Органические экстракты объединяли, промывали водой, насыщенным NaCl, сушили (Na2SO4), фильтровали и концентрировали. Сырой продукт очищали посредством хроматографии на силикагеле, элюируя смесью 20:1 (CH2Cl2/MeOH) с получением названного соединения в виде твердого вещества белого цвета (415 мг, 46%).
Часть H. Получение N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)фенил)этинил)-3-хлорфенил)метансульфонамида.
Смесь продукта из Части G (40,5 мг, 0,15 ммоль), йодида меди (I) (1,4 мг, 7,5 мкмоль), бис(трифенилфосфин)палладий(II) хлорида (5,26 мг, 7,50 мкмоль), N-(3-хлор-4-йодфенил) метансульфонамида (52,2 мг, 0,158 ммоль, полученного из 3-хлор-4-йоданилина по методу Примера 43, Часть F) и триэтиламина (0,105 мл, 0,750 ммоль) в ацетонитриле (2 мл) объединяли в герметично закрытой 5 мл микроволновой трубке и очищали путем барботирования N2 в течение 5 мин. Смесь нагревали под действием микроволн при 70°C в течение 5 мин, охлаждали и концентрировали. Сырое вещество очищали на 4 г картридже с диоксидом кремния, элюируя 99,5:0,5 CH2Cl2/MeOH --> 97:3 CH2Cl2/MeOH. Желаемые фракции объединяли и концентрировали. Вещество растирали в минимальном количестве EtOAc, и твердое вещество белого цвета собирали фильтрацией и сушили с получением названного соединения (28 мг, 39%). Температура плавления 278-280°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.31 (s. 9 Н) 2.72 (t, 7=6.62 Hz, 2 Н) 3.12 (s, 3 Н) 3.82 (t, 7=6.62 Hz, 2 Н) 7.20 (dd, 7=8.46, 2.21 Hz, 1 Н) 7.35 (s, 2 Н) 7.38-7.46 (m, 2 Н) 7.66 (d, 7=8.46 Hz, 1 Н) 10.31 (s, 1 H) 10.41 (s, 1 H).
Пример 45. Получение N-(6-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)пиридин-3-ил)метансульфонамида (соединение IA-L2-1.25).
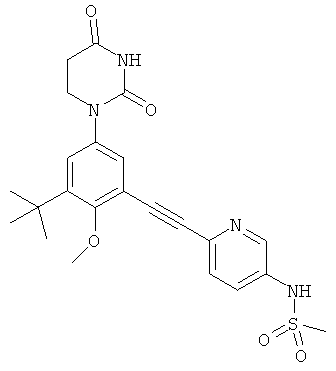
Часть A. Получение N-(6-йодпиридин-3-ил)метансульфонамида.
К раствору 6-йодпиридин-3-амина (1,077 г, 4,90 ммоль) в дихлорметане (40 мл) и пиридина (1,98 мл, 24,49 ммоль) при температуре ледяной бани добавляли метансульфонилхлорид (0,401 мл, 2,444 ммоль). Смесь оставляли нагреваться до комнатной температуры и перемешивали четыре дня. Реакционную смесь обрабатывали 5% уксусной кислотой и оставляли перемешиваться 20 мин при комнатной температуре. Органическую фазу промывали водой (2×50 мл), сушили (MgSO4) и концентрировали в вакууме. Остаток медленно добавляли порциями к быстро перемешиваемой воде (100 мл), и полученное в результате твердое вещество собирали фильтрованием, промывали водой и сушили в вакууме с получением названного соединения (0,7287 г, 49,9%).
Часть B. Получение N-(6-((триметилсилил)этинил)пиридин-3-ил)метансульфонамида.
Продукт из Части A (566 мг, 1,899 ммоль) объединяли с йодидом меди (I) (17 мг, 0,089 ммоль) и бис(трифенилфосфин) палладий (II) хлоридом (73 мг, 0,104 ммоль) в пробирке под давлением. Добавляли безводный ацетонитрил (17 мл), после чего триэтиламин (1,323 мл, 9,49 ммоль). Азот барботировали через полученную в результате желтую суспензию при перемешивании в течение 5 мин, затем добавляли триметилсилилацетилен (0,526 мл, 3,80 ммоль). Сосуд погружали в предварительно нагретую масляную баню при 80°C. Реакционную смесь оставляли перемешиваться с нагреванием в течение 2 ч, затем охлаждали до комнатной температуры и переносили в круглодонную колбу. Летучие компоненты удаляли в вакууме и коричневый остаток фракционировали с помощью (флэш)хроматографии на силикагеле (этилацетат/гексан) с получением названного продукта (0,4539 г, 89%) в виде твердого вещества желтовато-коричневого цвета.
Часть C. Получение N-(6-этинилпиридин-3-ил)метансульфонамида.
Продукт из Части B (0,533 г, 1,984 ммоль) растворяли в метаноле (17 мл) и добавляли по каплям 2 н раствор гидроксида натрия (2 мл, 4,17 ммоль) при комнатной температуре. Смесь оставляли перемешиваться 1 ч. Реакционную смесь концентрировали в вакууме. Остаток распределяли между этилацетатом и водой, и pH доводили до нейтрального ледяной уксусной кислотой. Органическую фазу разбавляли дополнительным количеством этилацетата, затем промывали несколько раз солевым раствором и концентрировали в вакууме с получением названного соединения в виде твердого вещества желтовато-коричневого цвета (0,3266 г, 84%).
Часть D. Получение N-(6-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)пиридин-3-ил)метансульфонамида.
Продукт из Части C (75 мг, 0,382 ммоль) объединяли с продуктом, полученным, как описано в Примере 43, Части E (146 мг, 0,363 ммоль), йодидом меди (I) (7,42 мг, 0,039 ммолье) и бис(трифенилфосфин) палладий (II) хлоридом (18,24 мг, 0,026 ммоль) в толстостенной стеклянной пробирке, и этот сосуд герметично закрывали винтовой крышкой с резиновой прокладкой. В атмосфере азота добавляли безводный ацетонитрил (10 мл), после чего, триэтиламин (0,266 мл, 1,911 ммоль). Азот барботировали через полученную в результате коричневую суспензию в течение 5 мин и затем эту пробирку погружали в предварительно нагретую 80°C масляную баню. За реакцией следили с помощью LC/MS. Дополнительные аликвоты алкина (всего 71 мг, 0,362 ммоль) каждая в THF (1 мл), добавляли через шприц на протяжении 8 ч. Реакционную смесь впоследствии выливали в 150 мл теплого (45°C) этилацетата, разделяли солевым раствором (75 мл) и оставляли перемешиваться 15 мин. Водную фазу экстрагировали этилацетатом (2×25 мл) и объединенную органическую фазу сушили (MgSO4) и фильтровали. Фильтрат обрабатывали граммом Silicycle Sithiol силикагеля и нагревали в атмосфере азота при перемешивании в течение 90 мин. Силикагель удаляли фильтрованием и после концентрирования в вакууме сырой продукт выделяли в виде бледно-оранжевого твердого вещества. Названное соедиенение (0,1315 мг, 73.1%) получали в виде беловатого твердого вещества с помощью (флэш)хроматографии на силикагеле, используя пошаговый градиент этилацетата в гексане, после чего метанола в дихлорметане. Температура плавления 191-192,5°C (d). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.35 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.12 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.07 (s, 3 H) 7.30 (d, J=2.94 Hz, 1 H) 7.40 (d, J=2.57 Hz, 1 H) 7.65 (d, J=1.47 Hz, 2 H) 8.44 (s, 1 H) 10.29-10.34 (m, 1 H) 10.38 (s, 1 H).
Пример 46. Получение N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-3-(трифторметил)фенил)метансульфонамида (соединение IA-L2-1.18).
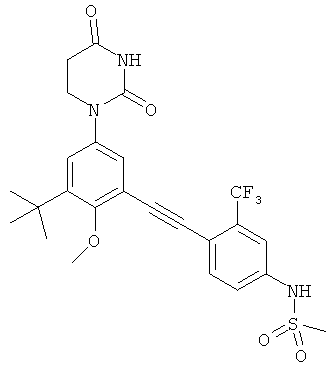
Часть A. Получение N-(4-бром-3-(трифторметил)фенил)метансульфонамида.
Названное соединение получали путем взаимодействия 4-бром-3-(трифторметил)анилина с метансульфонилхлоридом, как описано в Примере 45, Части A.
Часть B. Получение N-(3-(трифторметил)-4-((триметилсилил)этинил)фенил)метансульфонамида.
Продукт из Части A (2,00 г, 6,29 ммоль) объединяли с трифенилфосфином (0,211 г, 0,805 ммоль) и ацетатом палладия (II) (0,099 г, 0,440 ммоль) в 250 круглодонной колбе, снабженной конденсатором, и реакцию проводили, как описано W.B Austin et al, J. Org. Chem., 46 (11):2280 (1981). Добавляли толуол (40 мл), после чего, триэтиламин (80 мл) и триметилсилилацетилен (4,41 мл, 31,4 ммоль). Полученный в результате желтый раствор продували азотом в течение 5 мин при комнатной температуре. Реакционную смесь нагревали в атмосфере азота на масляной бане при в течение 24 ч. Охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали в вакууме и остаток хроматографировали на силикагеле (этилацетат-гексан) с получением названного соединения в виде твердого вещества желтовато-коричневого цвета (1,4554 г, 69%).
Часть C. Получение N-(4-этинил-3-(трифторметил)фенил)метансульфонамида.
Продукт из Части B (0,378 г, 1,126 ммоль) растворяли в метаноле (8 мл) и обрабатывали карбонатом калия (0,322 г, 2,331 ммоль) при комнатной температуре. Через 90 мин реакционную смеь распределяли между этилацетатом и разбавленной HCl. Органическую фазу промывали водой, затем сушили (MgSO4) и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (этилацетат-гексан) с получением названного соединения в виде прозрачного масла, которое медленно кристаллизуется при стоянии (0,2502 г, 84%).
Часть D. Получение N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-3-(трифторметил)фенил)метансульфонамида.
Продукт из Части C (0,2502 г, 0,950 ммоль) обрабатывали продуктом, полученным, как описано в Примере 43, Части E (0,364 г, 0,905 ммоль), как описано в Примере 45, Части D. Названное соединеие (0,3195 г, 65,7%) получали в виде твердого вещества белого цвета, путем растирания сырого продукта с эфиром-дихлорметаном. Температура плавления 257,5-26ГС (d). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.35 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.14 (s, 3 H) 3.78 (t, J=6.43 Hz, 2 H) 4.03 (s, 3 H) 7.29 (d, J=2.57 Hz, 1 H) 7.34 (d, J=2.57 Hz, 1 H) 7.48-7.56 (m, 1 H) 7.60 (s, 1 H) 7.83 (d, J=8.09 Hz, 1 H) 10.37 (s, 1 H) 10.45 (s, 1 H).
Пример 47. Получение N-[4-(ацетил-метансульфонил-амино)-фенил]-3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-бензамида (соединение IA-L3-1.69).
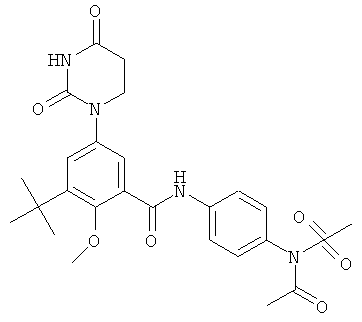
Часть A. Получение 3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-бензоилхлорида.
Добавляли вместе продукт из Примера 29, Части F, (2H)-ил)-2-метоксибензойную кислоту (4,07 г, 12,71 ммоль) и тионилхлорид (40,82 мл, 559 ммоль). Смесь затем нагревали с обратным холодильником в течение 2 ч, с последующим концентрированием под вакуумом с получением продукта в виде светло-желтого твердого вещества.
Часть B. Получение N-[4-(ацетил-метансульфонил-амино)-фенил]-3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-бензамида.
Продукт, полученный из Части A (0,073 г, 0,15 ммоль) растворяли в пиридине (2 мл) и обрабатывали по каплям уксусным ангидридом (0,042 мл, 0,45 ммоль). Смесь перемешивали 3 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме, и остаток растворяли в этилацетате (25 мл). Органический слой промывали водн. HCl, водн. NaHCO3, солевым раствором, и сушили над безводным твердым сульфатом натрия. Высушивающее вещество фильтровали, и растворитель упаривали под вакуумом с получением названного соединения в виде твердого вещества белого цвета (55 мг, 68%). Температура плавления 228-229°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 H) 1.92 (s, 3 H) 2.72 (t, J=6.62 Hz, 2 H) 3.52 (s, 3 H) 3.73-3.82 (m, 5 H) 7.32 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.94 Hz, 1 H) 7.44 (d, J=8.82 Hz, 2 H) 7.83 (d, J=8.82 Hz, 2 H) 10.37 (s, 1 H) 10.64 (s, 1 H).
Пример 48. Получение N-(6-(3-бром-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.69).
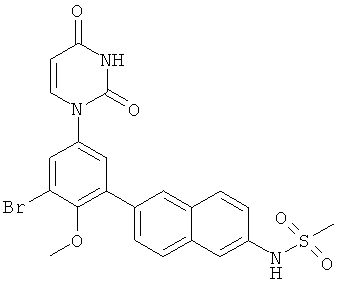
Часть A. Получение 2-бром-4,6-дийодфенола.
В 1 л круглодонную колбу помещали 2-бромфенол (Aldrich, 8,65 г, 50 ммоль) и метанол (100 мл) с получением бесцветного раствора. Добавляли гидроксид натрия (2,40 г, 60,0 ммоль) и перемешивали до растворения всех гранул гидроксида. Этот раствор охлаждали на бане с ледяной водой и добавляли йодид натрия (5,6 г, 37,4 ммоль) с последующим добавлением по каплям гирохлорита натрия (17 мл, 27,5 ммоль) с получением прозрачного коричневого/красного раствора, и постепенного осаждения густого белого твердого вещества. Добавление йодида натрия и раствора гипохлорита натрия повторяли три раза с получением оранжевой смеси, которую перемешивали в течение 2 ч, обрабатывали раствором тиосульфата натрия в воде (20 г в 100 мл), перемешивали в течение 15 мин и обрабатывали по каплям концентрированной HCl до постоянного значения рН 1. Смесь перемешивали в течение 15 мин и фильтровали для сбора белого твердого вещества, которое промывали многократно водой и сушили до постоянной массы (14,7 г, 69%).
Часть B. Получение 1-бром-3,5-дийод-2-метоксибензола.
В 500 мл круглодонную колбу помещали продукт из Части A (14,7 г, 34,6 ммоль), йодметан (2,70 мл, 43,3 ммоль), и гидроксид натрия (2,101 мл, 39,8 ммоль) в ацетоне (96 мл) с получением раствора желтовато-коричневого цвета. Смесь перемешивали в течение 24 ч и концентрировали. Остаток растворяли в этилацетате, промывали водой и насыщенным хлоридом натрия, сушили над сульфатом натрия, фильтровали и концентрировали с получением белого твердого вещества. Это твердое вещество перекристаллизовывали из горячего гексана с получением белого твердого вещества, которое собирали фильтрацией (12,3 г, 81%).
Часть C. Получение 1-(3-бром-5-йод-4-метоксифенил)пиримидин-2,4(1Н,3H)-диона.
В 250 круглодонную колбу помещали продукт из Части B (8,09 г, 18,44 ммоль), пиримидин-2,4(1H,3H)-дион (2,273 г, 20,28 ммоль), N-(2-цианофенил)пиколинамид (0,823 г, 3,69 ммоль), йодид меди (I) (0,351 г, 1,844 ммоль) и фосфат калия (8,22 г, 38,7 ммоль) в DMSO (70 мл). Смесь герметично закрывали, барботировали азотом в течение 15 мин и нагревали при 60°C в течение 16 ч. Смесь разделяли этилацетатом и водой. Органический слой промывали 1М HCl, водой, солевым раствором, сушили сульфатом натрия, и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем (каталог Aldrich # 538086), фильтровали через целит и упаривали с получением беловатого твердого вещества (3,92 г, 50%).
Часть D. Получение N-(6-(3-бром-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части C (212 мг, 0,50 ммоль), продукт из Примера 4A, Части B (17 мг, 0,50 ммоль), фосфат калия (223 мг, 1,05 ммоль), PA-Ph (CAS 97739-46-3, 4,38 мг, 0,015 ммоль) и трис (дибензилиденацетон)дипалладий(О) (4,58 мг, 5,00 мкмоль) в тетрагидрофуране (3,0 мл) и воде (1,0 мл). Трубку герметично закрывали, и смесь барботировали азотом в течение 5 мин, и затем перемешивали в течение 24 ч. Реакционную смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем (каталог Aldrich # 538086), фильтровали через целит и упаривали. Остаток растирали с метанолом/ СН2С12 с получением названного соединения в виде твердого вещества белого цвета (256 мг, 51%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.08 (s, 3 H) 3.43 (s, 3 H) 5.68 (d, J=8.09 Hz, 1 H) 7.43 (dd, J=8.82, 2.21 Hz, 1 H) 7.60 (d, J=2.57 Hz, 1 H) 7.72 (m, 2 H) 7.82 (d, J=3.31 Hz, 1 H) 7.84 (d, J=1.84 Hz, 1 H) 7.96 (m, 2 H) 8.09 (s, 1 H) 10.07 (s, 1 H) 11.49 (s, 1 H). MS (ESI-) m/z 513.9, 515.9 (M-H)+.
Пример 49. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-3-(5-метилфуран-2-ил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.58).
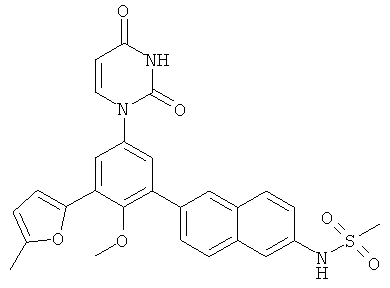
В 5 мл микроволновую трубку добавляли продукт Примера 48 (52 мг, 0,101 ммоль), 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан (0,025 мл, 0,121 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий дихлорид (3,28 мг, 5,04 мкмоль) и фосфат калия (42,8 мг, 0,201 ммоль) в THF (3,0 мл) и воде (1,0 мл). Трубку герметично закрывали и смесь барботировали азотом в течение 5 мин, и затем нагревали при 50°C в течение 3 ч. Охлажденную смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и концентрировали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток очищали с помощью хроматографии с обращенной фазой с получением желаемого продукта в виде твердого вещества белого цвета (23 мг, 44%, Температура плавления 174-178°C.) 1H ЯМР (300 MHz, DMSO-D6) δ ppm 2.38 (s, 3 H) 3.09 (s, 3 H) 3.33 (s, 3 H) 5.69 (dd, J=7.72, 2.21 Hz, 1 H) 6.30 (d, J=3.31 Hz, 1 H) 7.00 (d, J=3.31 Hz, 1 H) 7.43 (m, 2 H) 7.74 (d, J=2.57 Hz, 2 H) 7.78 (dd, J=8.46, 1.84 Hz, 1 H) 7.85 (d, J=8.09 Hz, 1 H) 7.97 (t, J=8.82 Hz, 2 H) 8.12 (s, 1 H) 10.05 (s, 1 H) 11.46 (d, J=2.21 Hz, 1 H). MS (ESI+) m/z 518 (M+H)+.
Пример 50. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(тиофен-3-ил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.53).
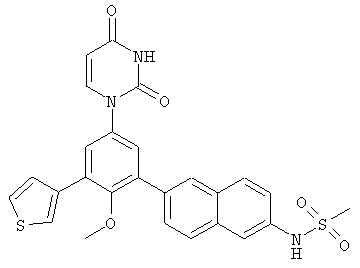
Названное соединение получали в соответствии с процедурой Примера 49, заменяя тиофен-3-илбороновую кислоту на 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан с получением белого твердого вещества (12 мг, 23%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.07 (s, 3 H) 3.22 (s, 3 H) 5.69 (d, J=7.72 Hz, 1 H) 7.41 (dd, J=8.64, 2.02 Hz, 1 H) 7.50 (d, J=2.94 Hz, 1 H) 7.59 (dd, J=5.13, 1.08 Hz, 1 H) 7.69 (m, 3 H) 7.76 (dd, J=8.64, 1.65 Hz, 1 H) 7.89 (d, J-7.72 Hz, 1 H) 7.95 (m, 3 H) 8.09 (s, 1 H) 10.05 (s, 1 H) 11.47 (s, 1 H). MS (ESI+) m/z 520 (M+H)+.
Пример 51. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-3-(тиофен-2-ил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.61).
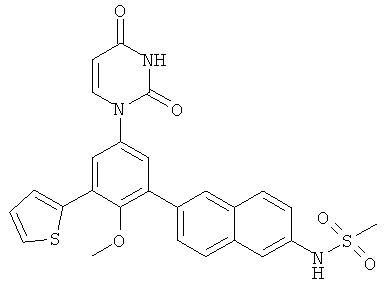
Названное соединение получали в соответствии с процедурой Примера 49, заменяя тиофен-2-илбороновую кислоту на 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан с получением твердого вещества белого цвета (8 мг, 15%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.08 (s, 3 H) 3.30 (s, 3 H) 5.70 (d, J=8.09 Hz, 1 H) 7.19 (dd, J=5.33, 3.86 Hz, 1 H) 7.42 (dd, J=8.82, 2.21 Hz, 1 H) 7.49 (d, J=2.57 Hz, 1 H) 7.69 (dd, J=5.15, 1.20 Hz, 1 H) 7.80 (m, 3 H) 7.88 (d, J=7.72 Hz, 1 H) 7.92 (d, J=2.57 Hz, 1 H) 7.98 (m, 2 H) 8.12 (s, 1 H) 10.06 (s, 1 H) 11.48 (s, 1 H). MS (ESI+) m/z 520 (M+H)+.
Пример 52. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(фуран-2-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.59).
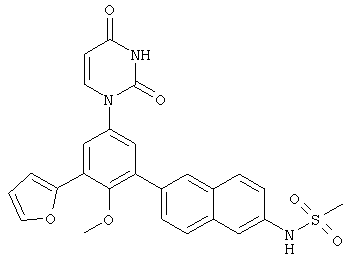
Названное соединение получали в соответствии с процедурой Примера 49, заменяя фуран-2-илбороновую кислоту на 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан с получением твердого вещества белого цвета (16 мг, 32%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.09 (s, 3 H) 3.35 (s, 3 H) 5.69 (d, J=7.72 Hz, 1 H) 6.69 (dd, J=3.31, 1.84 Hz, 1 H) 7.11 (d, J=3.31 Hz, 1 H) 7.43 (dd, J=8.82, 2.21 Hz, 1 H) 7.49 (d, J=2.94 Hz, 1 H) 7.80 (m, 5 H) 7.96 (m, 2 H) 8.13 (s, 1 H) 10.06 (s, 1 H) 11.47 (s, 1 H). MS (ESI-) m/z 502.1 (M-H)+.
Пример 53. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(фуран-3-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.64).
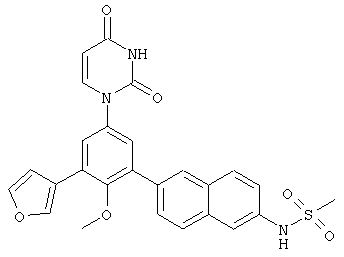
Названное соединение получали в соответствии с процедурой Примера 49, заменяя фуран-3-илбороновую кислоту на 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан с получением твердого вещества белого цвета (6 мг, 12%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.09 (s, 3 H) 3.30 (s, 3 H) 5.69 (dd, J=7.71, 1.83 Hz, 1 H) 7.10 (dd, J=1.74, 0.78 Hz, 1 H) 7.42 (dd, J=8.82, 2.21 Hz, 1 H) 7.46 (d, J=2.57 Hz, 1 H) 7.73 (d, J=2.21 Hz, 1 H) 7.76 (d, J=2.57 Hz, 1 H) 7.78 (d, J=1.84 Hz, 1 H) 7.81 (t, J=1.84 Hz, 1 H) 7.86 (d, J=7.72 Hz, 1 H) 7.96 (t, J=8.82 Hz, 2 H) 8.10 (s, 1 H) 8.28 (s, 1 H) 10.05 (s, 1 H) 11.48 (s, 1 H). MS (ESI-) m/z 502.1 (M-H)+.
Пример 54. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-бифенил-3-ил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.71).
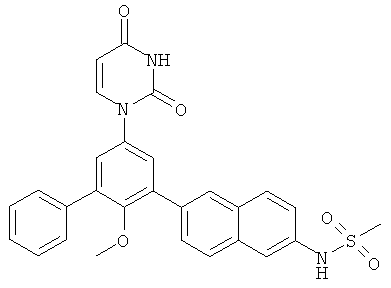
Названное соединение получали в соответствии с процедурой Примера 49, заменяя фенилбороновую кислоту на 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан. Сырой продукт очищали хроматографией на силикагеле, элюируя 3% метанолом/CH2Cl2 с получением твердого вещества белого цвета (10 мг, 8%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.08 (s, 3 Н) 3.12 (s, 3 Н) 5.69 (dd, J=7.81, 1.47 Hz, 1 Н) 7.36 (m, 5 Н) 7.56 (d, J=2.57 Hz, 1 Н) 7.64 (m, 2 Н) 7.74 (d, J=2.21 Hz, 1 Н) 7.78 (dd, J=8.46, 1.84 Hz, 1 Н) 7.94 (m, 3 Н) 8.11 (s, 1 Н) 10.04 (s, 1 Н) 11.47 (s, 1 Н). MS (ESI-) m/z 512 (M-H)+.
Пример 55. Получение N-(6-(3'-хлор-5-(2,4-диоксо-3,4-дигидропиримид,ин-1(2H)-кл)-2-метоксибифенил-3-ил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.74).
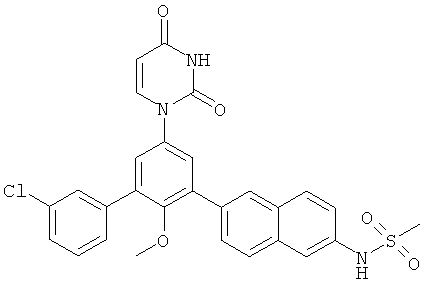
Названное соединение получали в соответствии с процедурой Примера 49, заменяя 3-хлорфенилбороновую кислоту на 4,4,5,5-тетраметил-2-(5-метилфуран-2-ил)-1,3,2-диоксаборолан с получением твердого вещества белого цвета (38 мг, 68%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.09 (s, 3 Н) 3.13 (s, 3 Н) 5.70 (dd, J=8.09, 2.21 Hz, 1 Н) 7.43 (dd, J=8.82, 2.21 Hz, 1 Н) 7.52 (m, 3 Н) 7.62 (m, 2 Н) 7.72 (m, 2 Н) 7.79 (dd, J=8.46, 1.47 Hz, 1 Н) 7.95 (m, 3 Н) 8.12 (s, 1 Н) 10.05 (s, 1 Н) 11.47 (d, J=2.21 Hz, 1 Н). MS (ESI-) m/z 546 (M-H)+.
Пример 56. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-3-(5-метилтиофен-2-ил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.73).
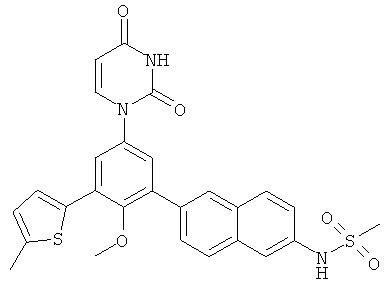
Названное соединение получали в соответствии с процедурой Примера 49, заменяя 4,4,5,5-тетраметил-2-(5-метилтиофен-2-ил)-1,3,2-диоксаборолан на 4,4,5,5-тетраметил-2-(5-метил-фуран-2-ил)-1,3,2-диоксаборолан с получением твердого вещества белого цвета (22 мг, 41%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 2.49 (s, 3 H) 3.09 (s, 3 H) 3.29 (s, 3 H) 5.69 (dd, J=8.09, 2.21 Hz, 1 H) 6.87 (d, J=2.57 Hz, 1 H) 7.43 (m, 2 H) 7.54 (d, J=3.68 Hz, 1 H) 7.76 (m, 2 H) 7.85 (s, 1 H) 7.87 (d, J=5.15 Hz, 1 H) 7.98 (t. J=9.01 Hz, 2 H) 8.11 (s, 1 H) 10.06 (s, 1 H) 11.47 (d, J=2.21 Hz, 1 H). MS (ESI+) m/z 534 (M+H)+.
Пример 57. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(1-гидрокси-2-метилпропан-2-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.54).
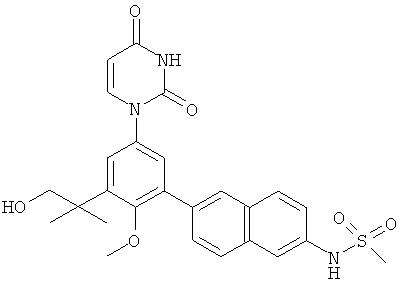
Часть A. Получение 2-(2-гидрокси-3,5-дииодфенил)уксусной кислоты.
В 250 мл круглодонную колбу добавляли 2-(2-гидроксифенил)уксусную кислоту (Aldrich, 3,04 г, 20 ммоль) в ацетонитриле (50 мл) с получением бесцветного раствора. N-йодсукцинимид (9,00 г, 40,0 ммоль) добавляли порциями за 15 мин с получением красного/коричневого прозрачного раствора, который перемешивали в течение 16 ч. Смесь концентрировали, и полученное в результате твердое вещество растирали в 75 мл воды и фильтровали для сбора оранжевого твердого вещества, которое сушили под вакуумом. Сырое твердое вещество перекристализовывали из толуола с получением светлого оранжевого порошка (6,0 г, 74%).
Часть B. Получение метил 2-(3,5-дийод-2-метоксифенил)ацетата.
В 250 мл круглодонную колбу добавляли продукт из Части A (6 г, 14,85 ммоль), карбонат калия (6,16 г, 44,6 ммоль), и диметилсульфат (4,12 г, 32,7 ммоль) в ацетоне (49,5 мл) с получением коричневой суспензии. Нагревали с обратным холодильником в течение 16 ч, охлаждали, концентрировали и остаток разделяли между EtOAc и водой. EtOAc слой промывали солевым раствором, сушили (Na2SO4) и концентрировали до коричневого масла, которое хроматографировали на 40 г картридже с диоксидом кремния, элюируя 3:1 гексаном/EtOAc с получением желтого масла (6,0 г, 94%).
Часть C. Получение метил 2-(3,5-дийод-2-метоксифенил)-2-метилпропаноата.
В 100 мл круглодонную колбу в атмосфере азота добавляли продукт из Части B (1,728 г, 4 ммоль) в безводном THF (20 мл) и HMPA (2 мл) с получением бесцветного раствора. Добавляли метилйодид (1,251 мл, 20,00 ммоль) и раствор охлаждали до -40°C. Добавляли по каплям т-бутоксид калия (12,00 мл, 12,00 ммоль), и смесь перемешивали при температуре от -40 до -20°C в течение 30 мин и гасили 1М HCl до pH 1. Смесь экстрагировали 3×40 мл EtOAc. Экстракты объединяли, промывали солевым раствором, сушили (Na2SO4) и концентрировали. Сырой продукт подвергали флэш-хроматографии на 40 г картридже с диоксидом кремния ISCO, элюируя 9:1 гексаном/EtOAc с получением бис-метилированного продукта в виде желтого масла (1,63 г, 89%).
Часть D. Получение 2-(3,5-дийод-2-метоксифенил)-2-метилпропановой кислоты.
Суспензию продукта из Части C (2,63 г, 5,72 ммоль) в MeOH (40 мл) и THF (40 мл) обрабатывали 4,0М гидроксидом натрия (28 мл, 112 ммоль) и нагревали при 80°C в течение 48 ч. Органический растворитель упаривали и оставшийся водный раствор подкисляли 1М HCl, получая твердое вещество, которое собирали фильтрацией, промывали водой и сушили с получением желаемой карбоновой кислоты (2,46 г, 96%).
Часть E. Получение 2-(3,5-дийод-2-метоксифенил)-2-метилпропан-1-ола.
Раствор продукта из Части D (1,00 г, 2,242 ммоль) в THF (40 мл) обрабатывали по каплям комплексом боран THF 1,ОМ (20 мл, 20 ммоль), и затем нагревали при 50°C в течение 24 ч. Смесь обрабатывали метанолом (20 мл), нагревали с обратным холодильником в течение 30 мин и концентрировали. Полученный в результате остаток промывали водой, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя гексаном/EtOAc (4:1) с получением желаемого продукта (810 мг, 84%).
Часть F. Получение трет-бутил(2-(3,5-дийод-2-метоксифенил)-2-метилпропокси)-диметилсилана.
Раствор продукта из Части E (432 мг, 1,000 ммоль) в DMF (5 мл) обрабатывали трет-бутилдиметилхлорсиланом (301 мг, 2,000 ммоль), и имидазолом (204 мг, 3,00 ммоль) и перемешивали в течение 2 ч. Смесь распределяли между 1М HCl и этилацетатом. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя гексаном/EtOAc (9:1) с получением желаемого продукта (522 мг, 96%).
Часть G. Получение 1-(3-(1-(трет-бутилдиметилсилилокси)-2-метилпропан-2-ил)-5-йод-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
В 50 мл круглодонную колбу добавляли продукт из Части F (520 мг, 0,952 ммоль), пиримидин-2,4(1H,3H)-дион (117 мг, 1,047 ммоль), N-(2-цианофенил)пиколинамид (42,5 мг, 0,190 ммоль), йодид меди (I) (18,13 мг, 0,095 ммоль) и фосфат калия (424 мг, 1,999 ммоль) в DMSO (5 мл). Сосуд герметично закрывали, барботировали азотом и затем нагревали при 60°C в течение 24 ч. Смесь распределяли между 1М HCl и этилацетатом. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя гексаном/EtOAc (3:2) с получением продукта в виде твердого вещества (285 мг, 65%).
Часть H. Получение N-(6-(3-(1-(трет-бутилдиметилсилилокси)-2-метилпропан-2-ил)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части G (50 мг, 0,094 ммоль), продукт из Примера 4A, Части B (32,7 мг, 0,094 ммоль), фосфат калия (42,0 мг, 0,198 ммоль), PA-Ph (CAS 97739-46-3) (0,827 мг, 2,83 мкмоль) и трис(дибензилиденацетон)палладий(0) (0,863 мг, 0,943 мкмоль) в THF (3,0 мл) и воде (1,0 мл). Сосуд герметично закрывали, и смесь барботировали азотом в течение 5 мин и затем нагревали при 50°C в течение 2 ч. Смесь распределяли между 1М HCl и этилацетатом. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя гексаном/EtOAc (3:7) с получением твердого вещества (32 мг, 54%).
Часть I. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(1-гидрокси-2-метилпропан-2-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида.
Продукт из Части H (31 мг, 0,050 ммоль) в THF (2,0 мл) обрабатывали 1М TBAF (0,3 мл, 0,3 ммоль) в THF и перемешивали в течение ночи. Смесь разделяли водой и этилацетатом. Органический слой промывали солевым раствором три раза, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя 2%-8% метанолом в CH2Cl2 с получением твердого вещества (21 мг, 83%). Температура плавления: 256-257°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.35 (s, 6 H) 3.08 (s, 3 H) 3.23 (s, 3 H) 3.67 (d, J=4.78 Hz, 2 H) 4.72 (t, J=4.78 Hz, 1 H) 5.65 (d, J=8.09 Hz, 1 H) 7.36 (m, 3 H) 7.74 (m, 3 H) 7.98 (m, 3 H) 10.04 (s, 1 H) 11.41 (s, 1 H). MS (ESI+) m/z 527 (M+NH4)+.
Пример 58. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(1 -метокси-2-метилпропан-2-ил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.66).
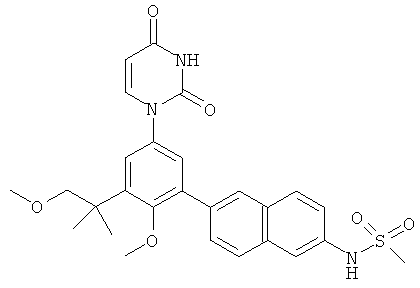
Часть A. Получение 1,5-дийод-2-метокси-3-(1-метокси-2-метилпропан-2-ил)бензола.
В 25 мл круглодонную колбу добавляли продукт из Примера 57, Части E. (259 мг, 0,6 ммоль) и гидрид натрия (28,8 мг, 1,200 ммоль) в THF (5 мл). Смесь перемешивали в течение 30 мин и добавляли йодметан (0,045 1, 0,720 ммоль). Смесь перемешивали в течение 16 ч и распределяли между этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением масла (235 мг, 88%).
Часть B. Получение 1-(3-йод-4-метокси-5-(1-метокси-2-метилпропан-2-ил)фенил)пиримидин-2,4(1H,3H)-диона.
В 25 мл круглодонную колбу добавляли продукт из Части A (230 мг, 0,516 ммоль), пиримидин-2,4(1H,3H)-дион (63,6 мг, 0,567 ммоль), N-(2-цианофенил)пиколинамид (23,02 мг, 0,103 ммоль), йодид меди (I) (9,82 мг, 0,052 ммоль) и фосфат калия (230 мг, 1,083 ммоль) в DMSO (5 мл). Сосуд герметично закрывали, барботировали азотом и нагревали при 60°C в течение 16 ч. Смесь охлаждали и распределяли между этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя 2%-5% метанолом в CH2Cl2 с получением твердого вещества (140 мг, 63%).
Часть C. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-3-(1-метокси-2-метилпропан-2-ил)фенил)нафтален-2-ил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части B (43 мг, 0,100 ммоль), продукт из Примера 4A, Части B (34,7 мг, 0,100 ммоль), фосфат калия (44,6 мг, 0,210 ммоль), PA-Ph (CAS 97739-46-3) (0,876 мг, 3,00 мкмоль) и трис(дибензилиденацетон)палладий(0) (0,915 мг, 0,999 мкмоль) в THF (3,0 мл) и воде (1,0 мл). Сосуд герметично закрывали, барботировали азотом в течение 5 мин и нагревали при 50°C в течение 2 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток растирали с метанолом/CH2Cl2 (1:1) с получением твердого вещества (28 мг, 54%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.39 (s, 6 Н) 3.08 (s, 3 Н) 3.23 (s, 3 Н) 3.25 (s, 3 Н) 3.61 (s, 2 Н) 5.65 (d, J=7.72 Hz, 1 Н) 7.27 (d, J=2.57 Hz, 1 Н) 7.37 (d, J=2.57 Hz, 1 Н) 7.42 (dd, J=8.64, 2.02 Hz, 1 Н) 7.69 (dd, J=8.46, 1.84 Hz, 1 Н) 7.73 (d, J=2.21 Hz, 1 Н) 7.78 (d, J=7.72 Hz, 1 Н) 7.95 (t, J=8.27 Hz, 2 Н) 8.02 (s, 1 Н) 10.04 (s, 1 Н) 11.41 (s, 1 Н). MS (ESI+) m/z 541 (M+NH4)+.
Пример 59. Получение метил 2-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(6-(метилсульфонамид)нафтален-2-ил)фенил)-2-метилпропаноата (соединение IB-L0-2.70).
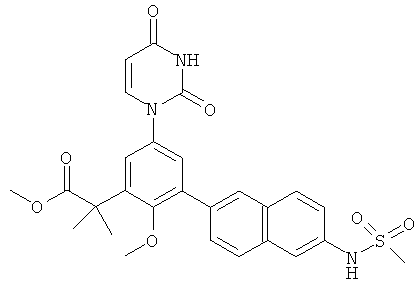
Часть A. Получение метил 2-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-йод-2-метоксифенил)-2-метилпропаноата.
В 100 мл круглодонную колбу в атмосфере N2 добавляли продукт из Примера 57, Части C (410 мг, 0,891 ммоль), 1H-пиримидин-2,4-дион (120 мг, 1,069 ммоль), и трехосновный фосфат калия (397 мг, 1,872 ммоль) в DMSO (5 мл) с получением бесцветной суспензии. Добавляли N-(2-цианофенил) пиколинамид (39,8 мг, 0,178 ммоль), и смесь барботироали N2 в течение 5 мин. Добавляли йодид меди (I) (16,97 мг, 0,089 ммоль), и эту смесь еще раз барботировали в течение 10 мин, помещали в атмосферу N2 и нагревали при 60°C в течение 18 ч. Смесь охлаждали и распределяли между EtOAc и водой, доводили pH до 1 с использованием HCl. Водные слои экстрагировали 2X EtOAc. Органические экстракты объединяли, промывали водой, насыщенным NaHCO3, и насыщенным NaCl. Органический слой сушили (Na2SO4), обрабатывали 3-меркаптопропил-функционализированным диоксидом кремния, фильтровали и концентрировали. Сырой продукт очищали хроматографией на 40 г картридже с диоксидом кремния ISCO, элюируя 3% MeOH в CH2Cl2 с получением белой пены (269 мг, 68%).
Часть B. Получение метил 2-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-3-(6-(метилсульфонамид)нафтален-2-ил)фенил)-2-метилпропаноата.
В 20 мл микроволновую трубку добавляли продукт из Части A (0,444 г, 1,0 ммоль), продукт из Примера 4A, Части B (0,365 г, 1,050 ммоль), и трехосновный фосфат калия (0,446 г, 2,100 ммоль) в смеси 3:1 тетрагидрофуран-вода (12 мл) и дегазировали путем барботирования азота в течение 20 мин. Затем раствор обрабатывали PA-Ph (CAS 97739-46-3) (8,77 мг, 0,030 ммоль) и трис (дибензилиден-ацетон)палладием(0) (9,16 мг, 10,00 мкмоль) с последующим дегазированием в течение еще 5 мин. Эту микроволновую трубку затем герметично закрывали и нагревали при 50°C в течение 18 ч, охлаждали и распределяли между EtOAc и водой, доводя pH до 1 с использованием 1М HCl. EtOAc слой промывали водой, насыщенным NaHCO3, и насыщенным NaCl. Органический слой сушили над сульфатом натрия, перемешивали в течение 1 ч 3-меркаптопропил-функционализированным диоксидом кремния, фильтровали и концентрировали. Сырой продукт очищали хроматографией на 12 г картридже с диоксидом кремния ISCO, элюируя 1-3% MeOH в CH2Cl2 с получением светлых желтовато-коричневых кристаллов (480 мг, 98%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.52 (s, 6 H) 3.08 (s, 3 H) 3.14 (s, 3 H) 3.64 (s, 3 H) 5.67 (dd, J=8.09, 1.84 Hz, 1 H) 7.37-7.48 (m, 3 H) 7.65 (dd, J=8.46, 1.84 Hz, 1 H) 7.73 (d, J=2.21 Hz, 1 H) 7.83 (d, J=8.09 Hz, 1 H) 7.96 (dd, J=8.64, 5.70 Hz, 2 H) 8.01 (s, 1 H) 10.05 (s, 1 H) 11.45 (s, 1 H). MS (ESI-) m/z 536 (M-H)+.
Пример 60. Получение 2-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(6-(метилсульфонамид)нафтален-2-ил)фенил)-2-метилпропановой кислоты (соединение IB-L0-2.77).
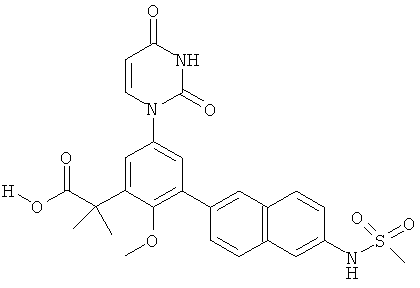
Смесь продукта из Примера 59 (108 мг, 0,2 ммоль) и гидроксида натрия (1 мл, 4,00 ммоль) в метаноле, THF, воду (3:3:1, 10 мл) нагревали при 80°C в течение 18 ч, охлаждали и осторожно подкисляли до pH 1 концентрированной HCl, получая в результате образование белого осадка. Твердое вещество собирали фильтрацией, промывали водой и сушили. Сырой материал растирали в 1 мл 1:1 EtOAc/MeOH, обрабатывали ультразвуком в течение 5 мин, и твердое вещество собирали фильтрацией в виде ярко-белого твердого вещества (58 мг, 54% выход), Температура плавления >300°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.50 (s, 6 H) 3.08 (s, 3 H) 3.18 (s, 3 H) 5.66 (d, J=7.72 Hz, 1 H) 7.34-7.45 (m, 3 H) 7.67 (dd, J=8.64, 1.65 Hz, 1 Н) 7.73 (d, J=1.84 Hz, 1 H) 7.82 (d, J=7.72 Hz, 1 H) 7.96 (dd, J=9.01, 4.60 Hz, 2 H) 8.02 (s, 1 H) 10.04 (s, 1 H) 11.43 (s, 1 H) 12.15 (s, 1 H). MS (ESI-) m/z 522 (M-H)+.
Пример 61. Получение метил 5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(6-(метилсульфонамид)нафтален-2-ил)бензоата (соединение IB-L0-2.72).
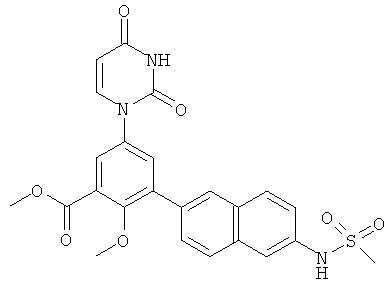
Часть A. Получение метил 3,5-дийод-2-метоксибензоата.
Смесь 2-гидрокси-3,5-дийодбензойной кислты (3,9 г, 10,0 ммоль), карбоната калия (4,15 г, 30,0 ммоль) и диметилсульфата (2,77 г, 22,0 ммоль) в ацетоне (33 мл) нагревали с обратным холодильником в течение 16 ч, охлаждали и концентрировали. Остаток растворяли в EtOAc и промывали водой, солевым раствором, сушили (Na2SO4), фильтровали и концентрировали с получением беловатого твердого вещества (4,2 г, количественный выход).
Часть B. Получение метил 5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-йод-2-метоксибензоата.
В 100 мл круглодонную колбу в атмосфере N2 добавляли продукт из Части A (2,09 г, 5,0 ммоль), 1H-пиримидин-2,4-дион (0,67 г, 6,0 ммоль), и трехосновный фосфат калия (2,2 г, 10,5 ммоль) в DMSO (20 мл) с получением бесцветной суспензии. Добавляли N-(2-цианофенил)пиколинамид (220 мг, 1,0 ммоль) и эту смесь барботировали N2 в течение 5 мин. Добавляли йодид меди (I) (95 мг, 0,5 ммоль), и эту смесь барботировали еще раз в течение 10 мин, помещали в атмосферу N2 и нагревали при 60°C в течение 18 ч. Смесь охлаждали и распределяли между EtOAc и водой, доводя pH до 1, используя HCl. Водный слой экстрагировали 2X EtOAc. Органические экстракты объединяли, промывали водой, насыщенным NaHCO3, и насыщенным NaCl. Органический слой сушили (Na2SO4), обрабатывали 3-меркаптопропил-функционализированным диоксидом кремния, фильтровали и концентрировали. Сырой продукт очищали хроматографией на 40 г картридже с диоксидом кремния ISCO, элюируя 3% MeOH в CH2Cl2 с получением белой пены (1,0 г, 50%).
Часть C. Получение метил 5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(6-(метилсульфонамид)нафтален-2-ил)бензоата.
Смесь продукта из Части B (101 мг, 0,25 ммоль), продукта из Примера 4А, Части B (91 мг, 0,263 ммоль), и трехосновного фосфата калия (111 мг, 0,525 ммоль) в смеси 3:1 тетрагидро-фуран-вода (12 мл) дегазировали путем барботирования азота в течение 20 мин. Затем раствор обрабатывали PA-Ph (CAS 97739-46-3) (2,192 мг, 7,50 мкмоль) и трис(дибензилиденацетон)палладием (0) (2,289 мг, 2,500 мкмоль) с последующим дегазированием в течение еще 5 мин. Микроволновую трубку затем герметично закрывали, нагревали при 50°C в течение 18 ч, охлаждали и распределяли между EtOAc и водой, доводя pH до 1, используя 1М HCl. EtOAc слой промывали водой, насыщенным NaHCO3, и насыщенным NaCl. Органический слой сушили Na2SO4, перемешивали в течение 1 ч с 3-меркаптопропил-функционализированным диоксидом кремния, фильтровали и концентрировали. Сырой продукт очищали с помощью хроматографии на 12 г картридже с диоксидом кремния ISCO, элюируя 3% MeOH в CH2Cl2 с получением беловатой пены (80 мг, 63%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.09 (s, 3 H) 3.45 (s, 3 H) 3.89 (s, 3 H) 5.69 (d, J=7.72 Hz, 1 H) 7.43 (dd, J=8.82, 2.21 Hz, 1 H) 7.68 - 7.79 (m, 4 H) 7.84 (d, J=7.72 Hz, 1 H) 7.89 -8.01 (m, 2 H) 8.09 (s, 1 H) 10.06 (s, 1 H) 11.49 (s, 1 H). MS (ESI-) m/z 494 (M-H)+.
Пример 62. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-йод-2-метоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.57).
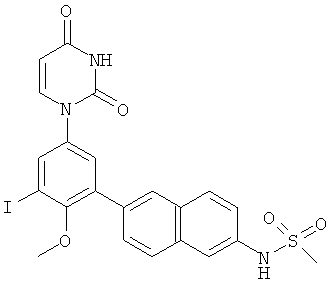
Часть A. Получение 1,3,5-трийод-2-метоксибензола.
В 250 мл сосуд высокого давления добавляли 2,4,6-трийодфенол (5 г, 10,60 ммоль) в МТВЕ (60 мл) с получением желтого раствора. Этот раствор охлаждали на ледяной бане и добавляли быстрыми каплями 2,0М триметилсилилдиазометан (7,95 мл, 15,90 ммоль) с последующим добавлением по каплям метанола (6 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор распределяли между EtOAc и водой и органический слой промывали 1М HCl, насыщенным NaHCO3, и насыщенным NaCl. Слой EtOAc сушили (MgSO4), фильтровали и концентрировали с получением твердого вещества желтовато-коричневого цвета, которое использовали без очистки (4,8 г, 94%).
Часть B. Получение 1-(3,5-дийод-4-метоксифенил)пиримидин-2,4(1Н,3H)-диона. В 100 мл круглодонную колбу в атмосфере N2 добавляли продукт из Части A (3,5 г, 7,2 ммоль), 1H-пиримидин-2,4-дион (0,97 г, 8,64 ммоль), и трехосновный фосфат калия (3,2 г, 15,0 ммоль) в DMSO (50 мл) с получением бесцветной суспензии. Добавляли N-(2-цианофенил)пиколинамид (320 мг, 1,44 ммоль) и смесь барботировали N2 в течение 5 мин. Добавляли йодид меди (I) (137 мг, 0,72 ммоль), и смесь барботировали еще раз в течение 10 мин, помещали в атмосферу N2 и нагревали при 60°C в течение 18 ч. Смесь охлаждали и распределяли между EtOAc и водой, доводя pH до 1, используя HCl. Водный слой экстрагировали 2X, используя EtOAc. Органические экстракты объединяли, промывали водой, насыщенным NaHCOs, и насыщенным NaCl, сушили (Na2SO4), обрабатывали 3-меркаптопропил-функционализированным диоксидом кремния, фильтровали и концентрировали. Полученное в результате твердое вещество растирали в смеси 2:1 гексан/EtOAc с получением беловатого порошка (2,2 г, 62%).
Часть C. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-йод-2-метоксифенил)нафтален-2-ил)метансульфонамида.
Смесь продукта из Части B 1-(3,5-дийод-4-метоксифенил)пиримидин-2,4(1Н,3H)-диона (118 мг, 0,25 ммоль), продукта из Примера 4A, Части B (87 мг, 0,25 ммоль), комплекса 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид CH2Cl2 (10,21 мг, 0,013 ммоль) и карбоната натрия (0,250 мл, 0,25 ммоль) в толуоле (1,0 мл) и этанола (1,0 мл) барботировали азотом в течение 5 мин и нагревали в микроволновой печи при 100°C в течение 30 мин. Смесь охлаждали и разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на диоксиде кремния, элюируя этилацетатом/гексаном (2:3-4:1) с получением названного соединения (16 мг, 11%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.08 (s, 3 Н) 3.35 (s, 3 Н) 5.67 (d, J=8.09 Hz, 1 Н) 7.42 (dd, J=8.82, 2.21 Hz, 1 Н) 7.59 (d, J=2.57 Hz, 1 Н) 7.73 (m, 2 Н) 7.81 (d, J=8.09 Hz, 1 Н) 7.95 (m, 3 Н) 8.09 (s, 1 Н) 10.06 (s, 1 Н) 11.47 (s, 1 Н). MS (ESI-) m/z 562 (M-H)+.
Пример 63. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-((триметилсилил)этинил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.78).
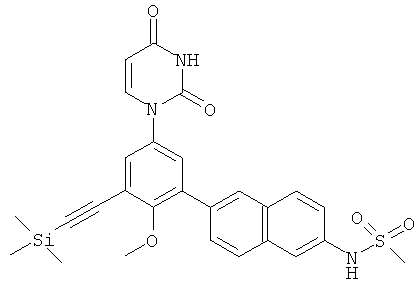
В 5 мл микроволновой трубке объединяли этинилтриметилсилан (0,044 мл, 0,32 ммоль), продукт из Примера 62 (45,1 мг, 0,08 ммоль), йодид меди (I) (0,762 мг, 4,0 мкмоль), бис(трифенил-фосфин)палладий(II) хлорид (2,81 мг, 4,0 мкмоль) и триэтиламин (0,056 мл, 0,40 ммоль) в ацетонитриле (2 мл). Эту смесь барботировали азотом в течение 5 мин, герметично закрывали и нагревали в микроволновой печи при 80°C в течение 20 мин. Реакционную смесь охлаждали и разделяли этилацетатом и водой. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на диоксиде кремния, элюируя 1-4% метанолом в CH2Cl2 с получением твердого вещества, (18 мг, 42%) Температура плавления 175-178°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 0.25 (s, 9 H) 3.07 (s, 3 H) 3.65 (s, 3 H) 5.66 (dd, J=7.91, 2.02 Hz, 1 H) 7.41 (dd, J=8.82, 2.21 Hz, 1 H) 7.58 (m, 2 H) 7.69 (dd, J=8.46, 1.84 Hz, 1 H) 7.72 (d, J=2.21 Hz, 1 H) 7.81 (d, J=7.72 Hz, 1 H) 7.93 (m, 2 H) 8.05 (d, J=1.32 Hz, 1 H) 10.04 (s, 1 H) 11.45 (d, J=2.21 Hz, 1 H). MS (ESI+) m/z 534 (M+H)+.
Пример 64. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(метилсульфонил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.68).
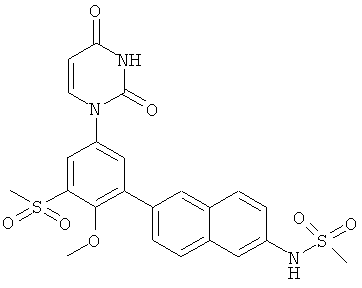
Часть A. Получение 4-нитробензол-2-диазо-1 -оксида.
В 250 мл круглодонную колбу добавляли 2-амино-4-нитрофенол (6,165 г, 40,0 ммоль) в 48% тетрафторборной кислоте (15 мл). Нитрит натрия (2,76 г, 40,0 ммоль) в воде (6 мл) добавляли по каплям при 0°C, и смесь перемешивали при комнатной температуре в течение 30 мин. Твердое вещество собирали фильтрацией, промывали тетрафторборной кислотой и водой. Твердое вещество суспендировали в ацетоне (50 мл), фильтровали и сушили с получением твердого вещества (3,31 г, 50%).
Часть B. Получение 2-(метилтио)-4-нитрофенола.
В 1 л лабораторный стакан добавляли продукт из Части A (2,70 г, 16,35 ммоль) в ледяной воде (250 г) с получением коричневой суспензии. Добавляли медь (0,520 г, 8,18 ммоль) с последующим медленным добавлением тиометоксида натрия (2,292 г, 32,7 ммоль) в воде (50 мл). Смесь перемешивали при комнатной температуре в течение 24 ч. Смесь фильтровали, и фильтрат подкисляли 1М HCl, получая твердое вещество, которое собирали фильтрацией и сушили (2,53 г, 84%).
Часть C. Получение 2-(метилсульфонил)-4-нитрофенола.
В 250 мл круглодонную колбу добавляли продукт из Части B (1,111 г, 6,00 ммоль) в MeOH (20 мл) с получением коричневой суспензии. Медленно добавляли оксон (7,746 г, 12,60 ммоль) в воде (20 мл) при 0°C. Смесь нагревали до комнатной температуры, перемешивали в течение 1 ч и разделяли этилацетатом и 1М HCl. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя 1%-5% метанолом в CH2Cl2 с получением твердого вещества (0,472 г, 36%).
Часть D. Получение 2-йод-6-(метилсульфонил)-4-нитрофенола.
В 50 мл круглодонную колбу добавляли продукт из Части C (470 мг, 2,164 ммоль) в MeOH (10 мл) и воде (2,5 мл). Добавляли по каплям монохлорид йода (0,130 мл, 2,60 ммоль) в CH2Cl2 (2,0 мл), и смесь перемешивали при комнатной температуре, выливали в воду (200 мл) и перемешивали в течение 10 мин. Полученное в результате твердое вещество собирали фильтрацией и сушили (636 мг, 86%).
Часть E. Получение 1-йод-2-метокси-3-(метилсульфонил)-5-нитробензола.
В 50 мл сосуд высокого давления добавляли продукт из Части D (630 мг, 1,836 ммоль) в MTBE (6 мл) с получением желтой суспензии. Смесь охлаждали на ледяной бане, и добавляли 2M триметилсилил-диазометан (1,377 мл, 2,75 ммоль) быстрыми каплями с последующим добавлением по каплям MeOH (0,4 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали при комнатной температуре в течение 1 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением беловатого твердого вещества (655 мг, 100%).
Часть F. Получение 3-йод-4-метокси-5-(метилсульфонил)анилина.
В 250 мл круглодонную колбу добавляли продукт из Части E (0,650 г, 1,820 ммоль), хлорид аммония (0,146 г, 2,73 ммоль), и железо (0,508 г, 9,10 ммоль) в THF/MeOH/воде (50 мл, 2/2/1). Смесь нагревали с обратным холодильником в течение 2 ч, охлаждали и фильтровали. Фильтрат упаривали, и остаток разделяли этилацетатом и водой. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением твердого вещества (590 мг, 99%).
Часть G. Получение (E)-N-(3-йод-4-метокси-5-(метилсульфонил)фенилкарбамоил)-3-метоксиакриламида.
В 100 мл круглодонную колбу добавляли продукт из Части F (500 мг, 1,528 ммоль) в DMF (15,0 мл). Этот раствор охлаждали в атмосфере азота до -20°C, и добавляли по каплям (E)-3-метоксиакрилоилизоцианат (15,28 мл, 6,11 ммоль; полученный, как описано Santana, L.; et al. J. Heterocyclic Chem. 1999, 36, 293-295). Смесь перемешивали при этой температуре в течение 15 мин, затем нагревали до комнатной температуры и перемешивали в течение 45 мин. Смесь разбавляли этилацетатом и промывали водой (3×50 мл), солевым раствором (3×50 мл), сушили сульфатом натрия, фильтровали и упаривали. Остаток растирали с этилацетатом/гексаном с получением твердого вещества (425 мг, 61%).
Часть H. Получение 1-(3-йод-4-метокси-5-(метилсульфонил)фенил)пиримидин-2,4(1H,3H)-диона.
В 100 мл кругло донную колбу добавляли продукт из Части G (420 мг, 0,925 ммоль) в этаноле (10 мл) с получением суспензии. Добавляли концентрированную серную кислоту (1 мл, 18,76 ммоль) в воде (10 мл), и смесь нагревали при 110°C в течение 2 ч. Реакционную смесь охлаждали, разбавляли водой (50 мл) и перемешивали в течение 10 мин. Твердое вещество собирали фильтрацией, промывали водой и сушили с получением твердого вещества белого цвета (325 мг, 83%).
Часть I. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(метилсульфонил)фенил)нафтален-2-ил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части H (63,3 мг, 0,15 ммоль), продукт из Примера 4A, Части B (52,1 мг, 0,150 ммоль), комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорида (6,12 мг, 7,50 мкмоль) и 1M карбонат натрия (0,150 мл, 0,150 ммоль) в растворителях толуоле (1,0 мл) и этаноле (1,0 мл). Сосуд герметично закрывали, и смесь барботировали азотом в течение 5 мин и нагревали в микроволновой печи при 100°C в течение 30 мин. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя 1%-8% метанолом в CH2Cl2 с получением сырого продукта. Конечное растирание в смеси 1:1 метанол/этилацетат давало чистое твердое вещество (26 мг, 34%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.10 (s, 3 Н) 3.44 (s, 3 Н) 3.45 (s, 3 Н) 5.71 (d, J=8.09 Hz, 1 Н) 7.44 (dd, J=8.82, 2.21 Hz, 1 Н) 7.75 (d, J=1.84 Hz, 1 Н) 7.80 (dd, J=8.46, 1.84 Hz, 1 Н) 7.86 (d, J=8.09 Hz, 1 Н) 7.91 (d, J=2.57 Hz, 1 Н) 7.96 (d, J=2.57 Hz, 1 Н) 8.00 (m, 2 Н) 8.16 (d, J=1.47 Hz, 1 Н) 10.10 (s, 1 Н) 11.51 (s, 1 Н). MS (ESI+) m/z 533 (M+NH4)+.
Пример 65. Получение N-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(6-(метилсульфонамид)нафтален-2-ил)фенил)метансульфонамида (соединение IB-L0-2.75).
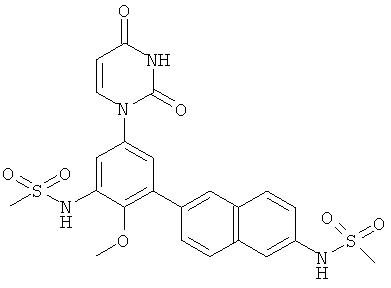
Часть A. Получение 2,4-дийод-6-нитрофенола.
К раствору 2-нитрофенола (2,78 г, 20 ммоль) в MeOH (120 мл) и воде (30 мл) добавляли по каплям раствор монохлорида йода (2,105 мл, 42,0 ммоль) в 10 мл CH2Cl2. Смесь перемешивали в течение 2 ч, выливали в 600 мл воды, перемешивали и обрабатывали ультразвуком в течение 30 мин. Смесь фильтровали для сбора твердого вещества желтого цвета, которое промывали 3× водой (50 мл каждое промывание) и сушили до постоянной массы (7,3 г, 93%).
Часть B. Получение 1,5-дийод-2-метокси-3-нитробензола.
В 50 мл сосуд высокого давления помещали продукт из Части A и МТВЕ (10 мл) с получением раствора желтого цвета. Этот раствор охлаждали на ледяной бане и быстрыми каплями добавляли 2M триметилсилилдиазометан (2,251 мл, 4,50 ммоль), с последующим добавлением по каплям MeOH (0,6 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали, оставляя нагреваться до комнатной температуры на протяжении 4 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением твердого вещества желтого цвета (1,22 г, 100%).
Часть C. Получение 3,5-дийод-2-метоксианилина.
В 250 мл круглодонную колбу добавляли продукт из Части B (0,98 г, 2,420 ммоль), хлорид аммония (0,194 г, 3,63 ммоль), и железо (0,676 г, 12,10 ммоль) в смеси THF/метанол/вода (20 мл/20 мл/10 мл). Смесь нагревали с обратным холодильником в течение 16 ч, охлаждали и фильтровали. Фильтрат упаривали, и остаток разделяли водой и этилацетатом. Органический слой сушили сульфатом натрия, фильтровали и упаривали с получением масла (780 мг, 86%).
Часть D. Получение 1-(3-амино-5-йод-4-метоксифенил)пиримидин-2,4(1Н,3H)-диона.
В 25 мл круглодонную колбу добавляли продукт из Части C (650 мг, 1,734 ммоль), пиримидин-2,4(1Н,3H)-дион (214 мг, 1,907 ммоль), N-(2-цианофенил)пиколинамид (77 мг, 0,347 ммоль), йодид меди (I) (33,0 мг, 0,173 ммоль) и фосфат калия (773 мг, 3,64 ммоль) в DMSO (5 мл). Сосуд герметично закрывали и смесь барботировали азотом в течение 15 мин и нагревали при 60°C в течение 16 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток хроматографировали на диоксиде кремния, элюируя смесью 5:95 метанол/D CH2Cl2CM с получением твердого вещества (125 мг, 20%).
Часть E. Получение N-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-йод-2-метокси-фенил)метансульфонамида.
Раствор продукта из Части D (110 мг, 0,306 ммоль) в пиридине (2 мл) обрабатывали метансульфонилхлоридом (0,048 мл, 0,612 ммоль) и перемешивали в течение 24 ч. Растворитель упаривали и остаток разделяли этилацетатом и 1М HCl. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя 2%-5% метанолом в CH2Cl2 с получением твердого вещества (20 мг, 15%).
Часть F. Получение N-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(6-(метилсульфонамид)нафтален-2-ил)фенил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части E (18 мг, 0,041 ммоль), Примера 4A, Части B (14,30 мг, 0,041 ммоль), фосфат калия (18,35 мг, 0,086 ммоль), PA-Ph (CAS 97739-46-3) (0,361 мг, 1,235 мкмоль) и трис (дибензилиденацетон)дипалладий(0) (0,377 мг, 0,412 мкмоль) в THF (3,0 мл) и воде (1,0 мл). Сосуд герметично закрывали, и смесь барботировали азотом в течение 5 мин и нагревали при 50°C в течение 2 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя 2%-5% метанолом в CH2Cl2 с получением твердого вещества. Конечное растирание в смеси 1:1 метанол/CH2Cl2 давало желаемый продукт (7 мг, 32%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.09 (s, 3 H) 3.17 (s, 3 H) 3.37 (s, 3 H) 5.69 (dd, J=7.91, 2.02 Hz, 1 H) 7.34 (d, J=2.57 Hz, 1 H) 7.43 (dd, J=8.82, 2.21 Hz, 1 H) 7.47 (d, J=2.57 Hz, 1 H) 7.73 (m, 2 H) 7.81 (d, J=8.09 Hz, 1 H) 7.94 (d, J=6.25 Hz. 1 H) 7.97 (d, J=6.62 Hz, 1 H) 8.07 (s, 1 H) 9.45 (s, 1 H) 10.05 (s, 1 H) 11.45 (d, J=1.84 Hz, 1 H). MS (ESI-) m/z 529 (M-H).
Пример 66. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-3-(трифторметил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.56).
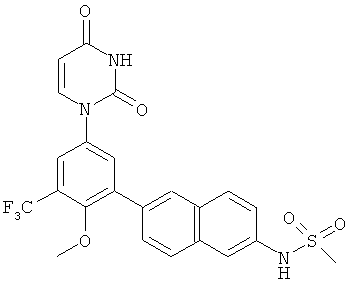
Часть A. Получение 4-йод-2-(трифторметил)фенола.
К раствору 2-(трифторметил) фенола (3,24 г, 20 ммоль) в MeOH (40 мл) добавляли гидроксид натрия (0,960 г, 24,0 ммоль) и перемешивали до растворения гидроксида. Смесь охлаждали до 0°C и добавляли йодид натрия (3,0 г, 20 ммоль) с последующим добавлением по каплям 10% водного гипохлорита натрия (9,0 мл, 14,6 ммоль). Добавление йодида натрия с последующим добавлением гипохлорита натрия повторяли еще дважды. Смесь перемешивали при окружающей температуре в течение 2 ч и обрабатывали по каплям концентрированной HCl до pH 1. Смесь экстрагировали 3X EtOAc. Эти экстракты объединяли, промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя EtOAc/гексаном (1:9) с получением монойодного продукта (5,0 г, 87%).
Часть B. Получение 2-бром-4-йод-6-(трифторметил)фенола.
В 250 мл круглодонную колбу добавляли продукт из Части A (5,00 г, 17,36 ммоль) и 1,3-дибром-5,-диметилгидантоин (2,73 г, 9,55 ммоль) в CHCl3 (80 мл) с получением оранжевого раствора. Смесь перемешивали в течение 2 ч, промывали водой, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Сырой продукт очищали на силикагеле, элюируя этилацетатом/гексаном (5:95) с получением твердого вещества (3,5 г, 54%).
Часть C. Получение 1-бром-5-йод-2-метокси-3-(трифторметил)бензола.
Смесь продукта из Части B (3,2 г, 8,72 ммоль), йодметана (1,36 мл, 21,8 ммоль), и 50% гидроксида натрия (0,507 мл, 9,59 ммоль) в ацетоне (20 мл) перемешивали в течение 24 ч. Растворитель упаривали, и остаток разделяли этилацетатом и водой. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Сырой материал очищали на силикагеле, элюируя этилацетатом/гексаном (5:95) с получением твердого вещества (2,67 г, 80%).
Часть D. Получение 1-(3-бром-4-метокси-5-(трифторметил)фенил)пиримидин-2,4(1Н,3H)-диона.
В 20 мл микроволновую трубку добавляли продукт из Части C (762 мг, 2,0 ммоль), пиримидин-2,4(1Н,3H)-дион (247 мг, 2,2 ммоль), N-(2-цианофенил)пиколинамид (89 мг, 0,4 ммоль), йодид меди (I) (38,1 мг, 0,2 ммоль) и фосфат калия (892 мг, 4,2 ммоль) в DMSO (10 мл). Сосуд герметично закрывали, и смесь барботировали азотом в течение 15 мин и нагревали при 60°C в течение 16 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя этилацетатом/гексаном (2:3) с получением желаемого продукта (63 мг, 9%).
Часть E. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(трифторметил)фенил)нафтален-2-ил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части D (60 мг, 0,164 ммоль), продукт из Примера 4A, Части B (62,8 мг, 0,181 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий дихлорид (5,36 мг, 8,22 мкмоль) и фосфат калия (69,8 мг, 0,329 ммоль) в THF/воде (3 мл/1 мл). Сосуд герметично закрывали, и смесь барботировали азотом в течение 5 мин и нагревали при 60°C в течение 2 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток очищали с помощью хроматографии с обращенной фазой с получением названного соединения в виде твердого вещества (26 мг, 31%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.10 (s, 3 H) 3.37 (s, 3 H) 5.71 (dd, J=7.72, 2.21 Hz, 1 H) 7.44 (dd, J=8.82, 2.21 Hz, 1 H) 7.75 (s, 1 H) 7.78 (d, J=1.84 Hz, 1 H) 7.88 (m, 3 H) 7.98 (d, J=3.31 Hz, 1 H) 8.01 (d, J=3.68 Hz, 1 H) 8.15 (s, 1 H) 10.09 (s, 1 H) 11.51 (d, J=2.21 Hz, 1 H). MS (ESI-) m/z 504.1 (M-H)+.
Пример 67. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(перфторэтил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.60).
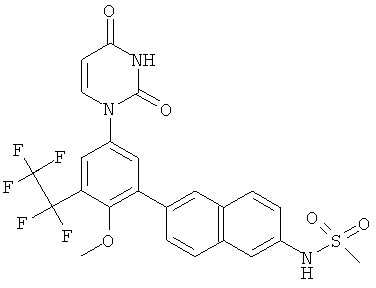
Часть A. Получение 1-метокси-4-нитро-2-(перфторэтил)бензола.
В 250 мл круглодонную колбу добавляли 2-бром-1-метокси-4-нитробензол (3,5 г, 15,08 ммоль), йодид меди (I) (5,75 г, 30,2 ммоль), и 2,2,3,3,3-пентафторпропаноат натрия (5,25 г, 28,2 ммоль) в DMF (75 мл) и толуоле (25 мл) с получением желтовато-коричневой суспензии. Смесь нагревали при 150°C, и толуол удаляли с помощью насадки Дина-Старка. Смесь нагревали при 155°C в течение 6 ч в атмосфере азота, охлаждали и выливали в 100 мл воды и 100 мл эфира, фильтровали через прокладку из целита толщиной 1 дюйм, и эту прокладку промывали эфиром. Слои фильтрата отделяли. Органический слой промывали солевым раствором, сушили (Na2SO4) фильтровали и концентрировали. Темное масло подвергали флэш-хроматографии на картридже с диоксидом кремния Isco 40 г, элюируя 4:1 гексаном/EtOAc с получением желтого масла, которое представляло собой смесь (3:1) желаемого вещества и исходного материала (1,5 г, 37%).
Часть B. Получение 4-нитро-2-(перфторэтил)фенола. В 100 мл круглодонную колбу добавляли продукт из Части A (1,4 г, 5,16 ммоль) и пиридингидрохлорид (4 г, 34,6 ммоль) беспримесный. Смесь нагревали при 210°C в течение 20 мин, охлаждали, и распределяли между EtOAc и водой. Органический слой промывали солевым раствором, сушили (Na2SO4) и концентрировали. Сырой продукт подвергали флэш-хроматографии на картридже с диоксидом кремния Isco 12 г, элюируя 3:2 гексаном/EtOAc с получением желтого масла (1,3 г, 98%).
Часть C. Получение 2-йод-4-нитро-6-(перфторэтил)фенола.
В 100 мл круглодонную колбу добавляли продукт из Части B (1,3 г, 5,06 ммоль) и N-йодсукцинимид (1,251 г, 5,56 ммоль) в ацетонитриле (16,85 мл) с получением желтого раствора. Раствор перемешивали в течение 16 ч, разбавляли 100 мл EtOAc и промывали 2×50 мл 10% тиосульфатом натрия, солевым раствором, сушили (Na2SO4) и концентрировали до полутвердого вещества оранжевого цвета. Это полутвердое вещество подвергали флэш-хроматографии на картридже с диоксидом кремния Isco 40 г, элюируя 3:1 гексаном EtOAc с получением масла густого желтого/оранжевого цвета (1,3 г, 67%).
Часть D. Получение 1-йод-2-метокси-5-нитро-3-(перфторэтил)бензола.
В 100 мл круглодонную колбу добавляли продукт из Части C (1,04 г, 2,72 ммоль) карбонат калия (0,563 г, 4,07 ммоль) и диметилсульфат (0,411 г, 3,26 ммоль) в ацетоне (20 мл) с получением коричневой суспензии. Смесь осторожно нагревали с обратным холодильником в течение 16 ч, охлаждали, разбавляли в EtOAc, промывали водой и солевым раствором. Органический слой сушили Na2SO4, фильтровали и концентрировали до желтого масла, которое очищали с помощью флэш-хроматографии на картридже с диоксидом кремния Isco 40 г, элюируя 9:1 гексаном /EtOAc (600 мг, 56%).
Часть E. Получение 3-йод-4-метокси-5-(перфторэтил)анилина.
В 250 мл круглодонную колбу добавляли продукт из Части D (0,6 г, 1,511 ммоль), железо (0,422 г, 7,56 ммоль), и хлорид аммония (0,121 г, 2,267 ммоль) в смеси растворителей EtOH (9 мл), THF (9 мл) и воды (3 мл) с получением коричневой суспензии, которую нагревали при 95-100°C в течение 2 ч. Реакционную смесь фильтровали через прокладку целита, и целит промывали многократно EtOH. Фильтрат концентрировали, и остаток растворяли в EtOAc, промывали водой, солевым раствором, сушили (Na2SO4), фильтровали и концентрировали с получением масла (560 мг, 99%).
Часть F. Получение 1,5-дийод-2-метокси-3-(перфторэтил)бензола.
В 25 мл круглодонную колбу в атмосфере азота добавляли продукт из Части E (0,565 г, 1,539 ммоль), трет-бутилнитрит (0,293 мл, 2,463 ммоль), йодид меди (I) (0,293 г, 1,539 ммоль), йодид натрия (0,231 г, 1,539 ммоль) и йод (0,195 г, 0,770 ммоль) в DME (15,39 мл) с получением коричневой суспензии. Смесь нагревали при 60°C в течение 3 ч, охлаждали и фильтровали через целит, хорошо промывая слой целлита EtOAc. EtOAc фильтрат обрабатывали 10% тиосульфатом натрия, солевым раствором, сушили (Na2SO4), фильтровали и концентрировали до темного масла. Сырой материал очищали с помощью флэш-хроматографии на картридже с диоксидом кремния Isco 40 г, элюируя 95:5 гексаном/EtOAc с получением желтого масла (360 мг, 49%).
Часть G. Получение 1-(3-йод-4-метокси-5-(перфторэтил)фенил)пиримидин-2,4(1Н,3H)-диона.
В 20 мл микроволновую трубку добавляли продукт из Части F (0,36 г, 0,753 ммоль), 1H-пиримидин-2,4-дион (0,101 г, 0,904 ммоль), трехосновный фосфат калия (0,336 г, 1,582 ммоль) N-(2-цианофенил)пиколинамид (0,034 г, 0,151 ммоль) и йодид меди (I) (0,014 г, 0,075 ммоль в DMSO (7 мл). Сосуд герметично закрывали, и смесь барботировали N2 в течение 30 мин, нагревали при 60°C в течение 24 ч, охлаждали и разбавляли в EtOAc. EtOAc слой промывали 1М HCl, насыщенным NaHCO3, и насыщенным NaCl, сушили (Na2SO4), фильтровали и концентрировали. Остаток подвергали флэш-хроматографии на картридже с диоксидом кремния Isco 40 г, элюируя гексаном --> 1:1 гексаном/EtOAc с получением желтой пены (100 мг, 29%).
Часть H. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(перфторэтил)фенил)нафтален-2-ил)метансульфонамида.
В 5 мл микроволновой трубке объединяли продукт из Части G (0,10 г, 0,216 ммоль), Примера 4A, Части B (0,075 г, 0,216 ммоль), и трехосновный фосфат калия (0,096 г, 0,454 ммоль) в смеси 3:1 тетрагидрофуран-вода (5 мл) и дегазировали путем барботирования азота в течение 10 мин. Смесь затем обрабатывали PA-Ph (CAS 97739-46-3) (1,898 мг, 6,49 мкмоль) и трис(дибензилиденацетон)дипалладием(0) (1,982 мг, 2,164 мкмоль) с последующим дегазированием в течение еще 5 мин. Затем колбу герметично закрывали и перемешивали при 50°C в течение 16 ч, и распределяли между EtOAc и водой. EtOAc слой промывали 0,1М HCl, насыщенным NaHCO3, и насыщенным NaCl. Органическую фазу сушили Na2S04, перемешивали в течение 0,5 ч с 3-меркаптопропил-функционализированным диоксидом кремния для удаления металлов, фильтровали и концентрировали. Сырой продукт очищали с помощью хроматографии на картридже с диоксидом кремния Isco 12 г, элюируя CH2Cl2 --> 3% MeOH в CH2Cl2 с получением светлой желтой пены (84 мг, 99%) Температура плавления 162-165°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 3.10 (s, 3 Н) 3.33 (s, 3 Н) 5.70 (d, J=7.72 Hz, 1 Н) 7.44 (dd, J=8.82, 2.21 Hz, 1 Н) 7.70-7.76 (m, 2 Н) 7.80 (d, J=2.57 Hz, 1 Н) 7.86 (d, J=8.09 Hz, 1 Н) 7.91 (d, J=2.57 Hz, 1 Н) 8.00 (dd, J=8.82, 2.94 Hz, 2 Н) 8.12 (s, 1 Н) 10.10 (s, 1 Н) 11.50 (s, 1 Н). MS (ESI-) m/z 554 (M-H)+.
Пример 68. Получение (E)-N'-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(тиофен-2-ил)фенил)-2,3-Дигидро-1H-инден-1-илиден)метансульфонгидразида (соединение IB-L0-2.51).
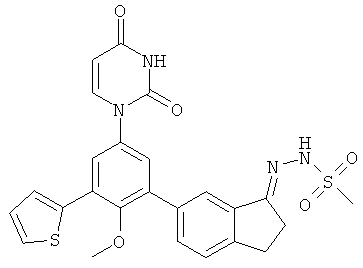
Часть A. Получение 1-(3-бром-4-метокси-5-(1-оксо-2,3-дигидро-1Н-инден-5-ил)фенил)-пиримидин-2,4(1Н,3H)-диона.
В 100 мл круглодонную колбу добавляли продукт из Примера 48, Части C (846 мг, 2,00 ммоль). Примера 6, Части A (516 мг, 2,000 ммоль), фосфат калия (892 мг, 4,20 ммоль), PA-Ph (CAS 97739-46-3) (17,54 мг, 0,060 ммоль) и трис(дибензилиденацетон)-дипалладий(0) (18,31 мг, 0,020 ммоль) в THF (12,0 мл) и воде (4,0 мл). Сосуд герметично закрывали и смесь барботировали азотом в течение 5 мин и перемешивали при окружающей температуре в течение 72 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали через целит и упаривали. Остаток очищали, используя силикагель, элюируя 1-4% метанолом в CH2Cl2 с получением твердого вещества (690 мг, 81%).
Часть B. Получение (E)-N'-(5-(3-бром-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-илиден)метансульфонгидразида.
В 50 мл круглодонную колбу добавляли продукт из Части A (685 мг, 1,603 ммоль) и метансульфонгидразид (194 мг, 1,764 ммоль) в MeOH (20 мл). Смесь нагревали до 40°C и перемешивали в течение 24 ч. Смесь охлаждали, фильтровали и промывали метанолом с получением твердого вещества (569 мг, 68%).
Часть C. Получение (E)-N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-3-(тиофен-2-ил)фенил)-2,3-дигидро-1H-инден-1-илиден)метансульфонгидразида.
В 5 мл микроволновую трубку добавляли продукт из Части B (52 мг, 0,100 ммоль), тиофен-2-илбороновую кислоту (12,81 мг, 0,100 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий дихлорид (3,26 мг, 5,01 мкмоль) и фосфат калия (42,5 мг, 0,200 ммоль) в THF (3,0 мл) и воде (1,0 мл). Смесь барботировали азотом в течение 5 мин и нагревали при 50°C в течение 3 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали через целит и упаривали. Остаток очищали с помощью хроматографии с обращенной фазой AA методом с получением твердого вещества белого цвета (27 мг, 52%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 2.86 (m, 2 H) 3.09 (s, 3 H) 3.14 (m, 2 H) 3.32 (s, 3 H) 5.69 (d, J=7.72 Hz, 1 H) 7.18 (dd, J=5.15, 3.68 Hz, 1 H) 7.41 (d, J=2.57 Hz, 1 H) 7.63 (m, 3 H) 7.75 (m, 2 H) 7.86 (d, J=8.09 Hz, 1 H) 7.91 (d, J=2.94 Hz, 1 H) 9.96 (s, 1 H) 11.48 (s, 1 H). MS (ESI+) m/z 523 (M+H)+.
Пример 69. Получение (E)-N'-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(фуран-2-ил)-2-метоксифенил)-2,3-дигидро-1Н-инден-1-илиден)метансульфонгидразида (соединение IB-L0-2.55).
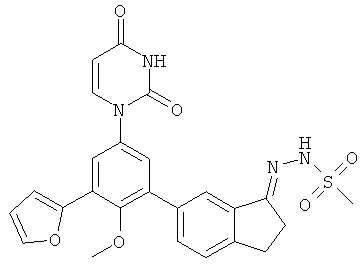
В 5 мл микроволновую трубку добавляли продукт из Примера 68, Части В (52 мг, 0,100 ммоль), фуран-2-илбороновую кислоту (11,20 мг, 0,100 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий дихлорид (3,26 мг, 5,01 мкмоль) и фосфат калия (42,5 мг, 0,200 ммоль) в THF (3,0 мл) и воде (1,0 мл). Смесь барботировали азотом в течение 5 мин и нагревали при 50°C в течение 3 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали через целит и упаривали. Остаток очищали с помощью хроматографии с обращенной фазы АА методом с получением твердого вещества (24 мг, 47%). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 2.86 (m, 2 H) 3.09 (s, 3 H) 3.14 (m, 2 H) 3.36 (s, 3 H) 5.68 (d, J=8.09 Hz, 1 H) 6.69 (dd, J=3.31, 1.84 Hz, 1 H) 7.09 (d, J=3.31 Hz, 1 H) 7.41 (d, J=2.57 Hz, 1 H) 7.62 (m, 2 H) 7.75 (d, J=8.09 Hz, 1 H) 7.80 (d, J=2.57 Hz, 1 H) 7.86 (m, 2 H) 9.97 (s, 1 H) 11.46 (s, 1 H). MS (ESI+) m/z 507 (M+H)+.
Пример 70. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-этоксифенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.23).
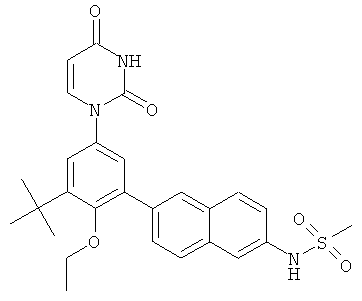
Часть A. Получение 2-трет-бутил-4-йодфенола.
В 250 мл круглодонную колбу добавляли 2-трет-бутилфенол (3,76 г, 25 ммоль) в MeOH (50,0 мл) с получением бесцветного раствора. Добавляли гидроксид натрия (1,200 г, 30,0 ммоль) и смесь перемешивали до полного растворения гидроксида. Раствор охлаждали до 0°C и обрабатывали йодидом натрия (1,75 г, 11,6 ммоль) с последующим добавлением по каплям 10% раствора гипохлорита натрия (7,2 мл, 11,6 ммоль). Добавление йодида натрия с последующим добавлением гипохлорита натрия повторяли дважды, и смесь перемешивали при 0°C в течение 30 мин. Смесь обрабатывали 10% масс/масс раствором тиосульфата натрия, перемешивали в течение 30 мин и обрабатывали по каплям концентрированной HCl до постоянного значения pH 1. Смесь экстрагировали 3X EtOAc. Экстракты объединяли, промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали. Неочищенное масло подвергали флэш-хроматографии на картридже с диоксидом кремния ISCO 80 г, элюируя гексаном -->4:1 гексан/EtOAc с получением желтого масла (5,2 г, 75%).
Часть B. Получение 2-бром-6-трет-бутил-4-йодфенола.
В 250 мл круглодонную колбу добавляли продукт из Части A (4,8 г, 17,38 ммоль) и 1,3-дибром-5,5-диметилгидантион (2,61 г, 9,13 ммоль) в хлороформе (87 мл) с получением оранжевого раствора. Реакционную смесь перемешивали в течение 2 ч, получая в результате черный раствор, который промывали водой, солевым раствором, сушили (Na2SO4) и концентрировали. Черное масло подвергали флэш-хроматографии на картридже с диоксидом кремния 120 г Isco, элюируя гексаном, с получением розоватого твердого вещества (4,84 г, 78%).
Часть C. Получение 1-бром-3-трет-бутил-2-этокси-5-йодбензола.
В 50 мл круглодонную колбу добавляли продукт из Части B (888 мг, 2,5 ммоль), этилйодид (409 мг, 2,63 ммоль), и карбонат калия (415 мг, 3,00 ммоль) в ацетоне (12 мл) с получением зеленой суспензии. Смесь нагревали с обратным холодильником в течение 16 ч, охлаждали и концентрировали. Остаток разделяли между водой и EtOAc. Органический слой промывали дважды солевым раствором, сушили над Na2SO4, фильтровали и концентрировали до красного масла. Масло подвергали флэш-хроматографии на картридже с диоксидом кремния Isco 40 г, элюируя гексаном, с получением прозрачного масла (820 мг, 86%).
Часть D. Получение 1-(3-бром-5-трет-бутил-4-этоксифенил)пиримидин-2,4(1H,3H)-диона.
В 20 мл микроволновую трубку под струей азота добавляли продукт из Части C (0,4 г, 1,044 ммоль), 1H-пиримидин-2,4-дион (0,140 г, 1,253 ммоль), и трехосновный фосфат калия (0,465 г, 2,193 ммоль) в DMSO (5 мл) с получением бесцветной суспензии. Добавляли N-(2-цианофенил)пиколинамид (0,047 г, 0,209 ммоль), и смесь барботировали азотом в течение 10 мин. Добавляли йодид меди (I) (0,020 г, 0,104 ммоль), и смесь барботировали еще раз в течение 10 мин, помещали в атмосферу азота и нагревали при 60°C в течение 18 ч. Смесь охлаждали и распределяли между EtOAc и водой, доводя pH до 1, используя HCl. Водный слой экстрагировали 2X EtOAc. Органические экстракты объединяли, промывали водой, насыщенным NaHCO3, и насыщенным NaCl. Органический слой сушили (Na2SO4), перемешивали с 3-меркаптопропил-функционализированным диоксидом кремния в течение 1 ч, фильтровали и концентрировали. Сырой продукт очищали с помощью хроматографии на картридже с диоксидом кремния ISCO 12 г, элюируя 2% MeOH в CH2Cl2 с получением белого порошка (266 мг, 69%).
Часть E. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-этоксифенил)нафтален-2-ил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части D (55,1 мг, 0,15 ммоль), продукт из Примера 4A, Части B (52,1 мг, 0,150 ммоль), трехосновный фосфат калия (63,7 мг, 0,300 ммоль) и 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий дихлорид (4,89 мг, 7,50 мкмоль) в THF (3 мл) воде (1 мл). Смесь барботировали в течение 10 мин азотом, нагревали герметично закрытой при 50°C в течение 4 ч, охлаждали и разбавляли в EtOAc. EtOAc слой промывали 1М HCl, насыщенным NaHCO3, насыщенным NaCl, сушили (Na2SO4) и одновременно обрабатывали меркаптопропил силикагелем, фильтровали и концентрировали. Остаток подвергали флэш-хроматографии на картридже с диоксидом кремния 12 г Isco, элюируя 2% MeOH в CH2Cl2 с получением твердого вещества, (16 мг, 21%) Температура плавления 196-202°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.00 (t, J=6.99 Hz, 3 H) 1.44 (s, 9 H) 3.09 (s, 3 H) 3.43 (q, J=7.11 Hz, 2 H) 5.64 (dd, J=7.91, 1.29 Hz, 1 H) 7.32 (d, J=2.94 Hz, 1 H) 7.36 (d, J=2.94 Hz, 1 H) 7.41 (dd, J=8.82, 2.21 Hz, 1 H) 7.72 (s, 1 H) 7.74 (d, J=1.47 Hz, 1 H) 7.80 (d, J=7.72 Hz, 1 H) 7.90-8.00 (m, 2 H) 8.05 (s, 1 H) 10.04 (s, 1 H) 11.41 (s, 1 H). MS (ESI-) m/z 506 (M-H)+.
Пример 71. Получение N-(6-(3-трет-бутил-2-хлор-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)фенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.14).
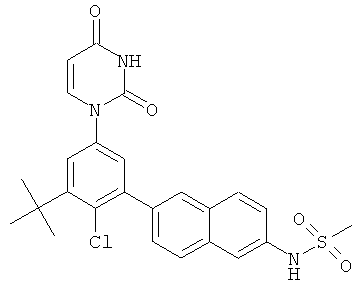
Часть A. Получение 2-бром-6-трет-бутил-4-йоданилина.
В 50 мл круглодонную колбу добавляли 2-бром-6-трет-бутиланилин [полученный по способу Onitsuka, et.al. Organometallics, 25(5), 2006, pp 1270-1278] (1,18 г, 5,17 ммоль) и бикарбонат натрия (0,782 г, 9,31 ммоль) в воде (5 мл). Смесь охлаждали на ледяной бане и добавляли йод (1,444 г, 5,69 ммоль) несколькими порциями. Смесь нагревали до окружающей температуры и перемешивали в течение 16 ч. Смесь обрабатывали водным тиосульфатом натрия, экстрагировали этилацетатом, сушили сульфатом натрия, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя 5% этилацетатом в гексане с получением масла (1,2 г, 65%).
Часть B. Получение 1-бром-3-трет-бутил-2-хлор-5-йодбензола.
К смеси трет-бутилнитрита (0,198 мл, 1,5 ммоль) и хлорида меди (II) (161 мг, 1,2 ммоль) в ацетонитриле (5 мл) добавляли продукт из Части A (354 мг, 1,0 ммоль) в виде раствора в ацетонитриле (5 мл). Смесь нагревали при 60°C в течение 30 мин, охлаждали, разделяли этилацетатом и 1М HCl. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя 5% этилацетатом в гексане с получением этого продукта (300 мг, 81%).
Часть C. Получение 1-(3-бром-5-трет-бутил-4-хлорфенил)пиримидин-2,4(1H,3H)-диона.
В 20 мл микроволновую трубку добавляли продукт из Части B (300 мг, 0,803 ммоль), пиримидин-2,4(1H,3H)-дион (99 мг, 0,884 ммоль), N-(2-цианофенил)пиколинамид (35,9 мг, 0,161 ммоль), йодид меди (I) (15,30 мг, 0,080 ммоль) и фосфат калия (358 мг, 1,687 ммоль) в DMSO (5 мл). Смесь герметично закрывали, продували азотом и нагревали при 60°C в течение 4 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя 10%-40% этилацетатом в гексане с получением твердого вещества (175 мг, 61%).
Часть D. Получение N-(6-(3-трет-бутил-2-хлор-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)фенил)нафтален-2-ил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части C (35,8 мг, 0,10 ммоль), продукт из Примера 4A, Части B (38,2 мг, 0,110 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий дихлорид (3,26 мг, 5,00 мкмоль) и фосфат калия (42,5 мг, 0,200 ммоль) в THF/воде (3 мл:1 мл). Смесь продували азотом в течение 5 мин и нагревали при 60°C в течение 2 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток очищали на силикагеле, элюируя 1:1 этилацетатом/гексаном с получением твердого вещества, которое растирали с 1% метанолом в CH2Cl2 с получением твердого вещества белого цвета (29 мг, 55%), температура плавления: >280°C. 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.53 (s, 9 H) 3.08 (s, 3 H) 5.69 (d, J=7.72 Hz, 1 H) 7.42 (m, 2 H) 7.52 (dd, J=8.46, 1.84 Hz, 1 H) 7.56 (d, J=2.57 Hz, 1 H) 7.74 (d, J=1.84 Hz, 1 H) 7.84 (d, J=7.72 Hz, 1 H) 7.88 (s, 1 H) 7.91 (d, J=8.82 Hz, 1 H) 7.95 (d, J=9.19 Hz, 1 H) 10.04 (s, 1 H) 11.46 (s, 1 H). MS (ESI-) m/z 496 (M-H)+.
Пример 72. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]изоксазол-3-ил)метил)метансульфонамида (соединение IB-L0-2.45).
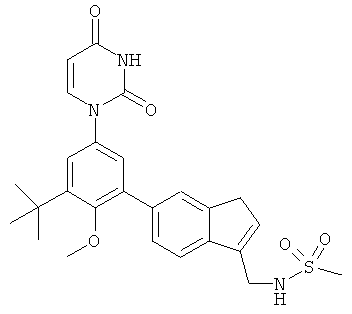
Часть A. Получение N-((6-бромбензо[d]изоксазол-3-ил)метил)-N-(4-метоксибензил)-метансульфонамида.
В кипящий с обратным холодильником раствор 6-бром-3-метилбензо[d]изоксазола (1,0 г, 4,72 ммоль) в CCl4 (25 мл) добавляли 1-бромпирролидин-2,5-дион (0,923 г, 5,19 ммоль) и ангидрид пероксибензойной кислоты (0,114 г, 0,472 ммоль). Смесь нагревали с обратным холодильником в течение 6 ч, и затем охлаждали до комнатной температуры, фильтровали через целит, и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя CH2Cl2 в качестве элюента, с получением этого дибромида в виде твердого вещества (0,84 г, 43%). К раствору этого дибромида (0,20 г, 0,687 ммоль) и N-(4-метоксибензил)метансульфонамида (0,148 г, 0,687 ммоль) в EtOH (3 мл) добавляли 1 н водн. NaOH (0,722 мл, 0,722 ммоль), и полученную в результате смесь перемешивали при 80°C в течение 90 мин. Смесь распределяли между 0,1 н водн. HCl (10 мл) и EtOAc (2×10 мл), и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 2:3 EtOAc:гексан в качестве элюента с получением названного соединения в виде масла (65 мг, 22%).
Часть B. Получение N-(4-метоксибензил)-N-((6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[d]изоксазол-3-ил)метил)метансульфонамида.
Раствор продукта из Части A (56 мг, 0,132 ммоль), бис(пинаколато)дибора (37 мг, 0,145 ммоль), и ацетата калия (39 мг, 0,395 ммоль) в 1,4-диоксане (1,3 мл) дегазировали путем барботирования газообразным N2 в течение 15 мин. Добавляли комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид дихлорметан (3 мг, 0,004 ммоль), и полученную в результате смесь перемешивали при 80°C в течение 16 ч, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 1:2 EtOAc:гексан в качестве элюента с получением названного соединения в виде бесцветного масла (49 мг, 79%).
Часть C. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]изоксазол-3-ил)метил)-N-(4-метоксибензил)метансульфонамида.
Смесь продукта из Примера C (31,8 мг, 0,079 ммоль), продукта из Части B (45 мг, 0,095 ммоль) в EtOH (0,5 мл), толуол (0,5 мл) 1M водн. Na2CO3 (0,095 мл, 0,095 ммоль) дегазировали путем барботирования газообразным N2 в течение 10 мин. Добавляли комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II) дихлорид дихлорметан (2 мг, 2,4 мкмоль), и дегазирование с использованием N2 продолжали в течение 5 мин. Реакционную смесь герметично закрывали и нагревали при 100°C в микроволновом реакторе в течение 1 ч. Смесь концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 1:9 MeOH:CHCl3 в качестве элюента. Названное соединение получали в виде светло-коричневого масла (41 мг, 83%).
Часть D. Получение N-((6-(3-терет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]изоксазол-3-ил)метил)метансульфонамида.
Раствор продукта из Части C (39 мг, 0,063 ммоль) в TFA (0,5 мл) перемешивали при 40°C в течение 6 ч. TFA удаляли в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 4% MeOH в CHCl3 в качестве элюента с получением названного соединения (13 мг, 41%). 1H ЯМР (300 MHz, CDCl3) δ 8.39 (s, 1 Н) 7.74-7.82 (m, 2 Н) 7.57 (dd, J=8.27, 1.65 Hz, 1 H) 7.36 (d, J=7.72 Hz, 1 H) 7.25 (d, J=2.57 Hz, 1 Н) 7.19 (d, J=2.94 Hz, 1 H) 5.82 (dd, J=7.72, 2.21 Hz, 1 H) 5.25-5.33 (m, 1 H) 4.70 (d, J=6.25 Hz, 2 H) 3.29 (s, 3 H) 3.12 (s, 3 H) 1.45 (s, 9 H).
Пример 73. Получение метил 2-(5-(3-теретп-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-177-инден-1-илиден)гидразинкарбоксилата (соединение IB-L0-2.24).
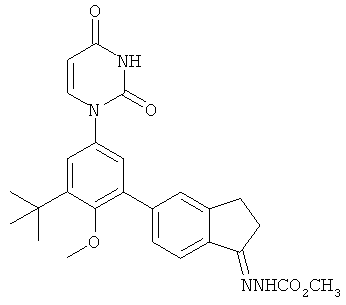
К раствору продукта из Примера 6, Части B (0,05 г, 0,124 ммоль) в MeOH (1 мл) добавляли метилкарбазат (17 мг, 0,185 ммоль). Смесь перемешивали при 60°C в течение 16 ч, и затем концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 5% MeOH в CH2Cl2 в качестве элюента с получением названного соединения (44 мг, 74%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.40 (s, 1 H) 10.05 (s, 1 H) 7.78 (d, J=8.09 Hz, 1 H) 7.69 (d, J=7.72 Hz, 1 H) 7.45-7.57 (m, 2 H) 7.24-7.33 (m, 2 H) 5.64 (d, J=8.09 Hz, 1 H) 3.71 (s, 3 H) 3.28 (s, 3 H) 3.06-3.16 (m, 2 H) 2.78-2.88 (m, 2 H) 1.40 (s, 9H).
Пример 74. Получение 1-(3-трет-бутил-4-метокси-5-(1-оксоизоиндолин-5-ил)фенил)-пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.30).
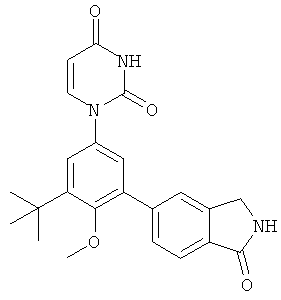
Часть A. Получение 5-бром-2-(2,4-диметоксибензил)изоиндолин-1-она. К раствору метил 4-бром-2-(бромметил)бензоата (1,0 г, 3,25 ммоль) и (2,4-диметоксифенил)метанамина (0,65 г, 3,90 ммоль) в THF (16 мл) добавляли триэтиламин (0,91 мл, 6,5 ммоль), и полученную в результате смесь перемешивали при комнатной температуре в течение 16 ч. Полученное в результате твердое вещество отфильтровывали, и фильтрат концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 1:4 EtOAc:гексан в качестве элюента с получением названного соединения в виде бесцветного твердого вещества (0,52 г, 44%).
Часть B. Получение 2-(2,4-диметоксибензил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изоиндолин-1 -она.
Продукт из Части A (100 мг, 0,276 ммоль) подвергали условиям, описанным для Примера 72, Части B с получением названного соединения в виде масла (107 мг, 95%).
Часть C. Получение 1-(3-трет-бутил-5-(2-(2,4-диметоксибензил)-1-оксоизоиндолин-5-ил)-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
Продукт из Части C (44 мг, 0,111 ммоль) подвергали условиям, описанным для Примера 72, Части C с получением названного соединения (50 мг, 81%).
Часть D. Получение 1-(3-трет-бутил-4-метокси-5-(1-оксоизоиндолин-5-ил)фенил) пиримидин-2,4(1H,3H)-диона.
Раствор продукта из Части C (48 мг, 0,086 ммоль) в CH2Cl2 (0,3 мл) и TFA (0,6 мл, 7,79 ммоль) перемешивали при комнатной температуре в течение 16 ч, и затем концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 5% МеОН в CHCl3 в качестве элюента, с получением названного соединения в виде бесцветного твердого вещества (22 мг, 63%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.41 (d, J=1.84 Hz, 1 H) 8.61 (s, 1 H) 7.72-7.83 (m, 3 H) 7.62-7.69 (m, 1 H) 7.29-7.36 (m, 2 H) 5.65 (dd, J=8.09, 2.21 Hz, 1 H) 4.44 (s, 2 H) 3.25 (s, 3 H) 1.41 (s, 9 H).
Пример 75. Получение N-(2-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1H-инден-3-ил)пропан-2-ил)метансульфонамида (соединение IB-L0-2.41).
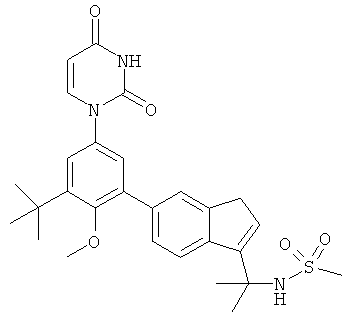
Часть A. Получение 6-бром-1H-инден-3-карбонитрила.
К раствору 5-бром-2,3-дигидро-1H-инден-1-она (1 г, 4,74 ммоль) в безводном THF (15 мл) при -10°C по каплям добавляли 2M диизопропиламид лития в THF (0,242 мл, 0,483 ммоль). Полученную в результате смесь перемешивали при -10°C в течение 15 мин перед добавлением по каплям диэтилцианофосфоната (0,791 мл, 5,21 ммоль). После добавления смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 3 ч. Смесь охлаждали до -78°C и добавляли по каплям диэтилэфират трифторида бора (1,196 мл, 9,52 ммоль). После добавления, смесь перемешивали при -78°C в течение 1 ч и затем оставляли нагреваться до комнатной температуры, и перемешивали при комнатной температуре в течение 16 ч. Смесь концентрировали в вакууме, и остаток распределяли между EtOAc (50 мл) и H2O (2×50 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 9:1 EtOAc:гексан в качестве элюента. Названное соединение получали в виде желтовато-коричневого твердого вещества (0,72 г, 69%).
Часть B. Получение N-(2-(6-бром-1H-инден-3-ил)пропан-2-ил)метансульфонамида. Безводный хлорид церия (III) (0,224 г, 0,909 ммоль) сушили в вакууме пламени и помещали в атмосферу сухого N2. Добавляли безводный THF (1,5 мл), и полученную в результате смесь перемешивали в атмосфере N2 при 45°C в течение 48 ч. Смесь охлаждали до комнатной температуры, и добавляли продукт из Части A (0,1 г, 0,454 ммоль). Полученную в результате смесь охлаждали до -78°C, и по каплям добавляли 1,5M раствор комплекса метил-литий литий бромид (0,757 мл, 1,136 ммоль) в Et2O за период времени 15 мин. После добавления смесь оставляли нагреваться до -20°C и перемешивали в течение 24 ч. Добавляли по каплям концентрированный водный NH4OH (0,3 мл), и смесь оставляли нагреваться до комнатной температуры, перемешивали в течение 30 мин, и затем фильтровали и промывали THF (2×5 мл). Фильтрат концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 5% MeOH в CH2Cl2 в качестве элюента с получением твердого вещества (23 мг, 20%). К раствору этого твердого вещества (23 мг, 0,091 ммоль) в CH2Cl2 (1 мл) добавляли метансульфонилхлорид (0,011 мл, 0,137 ммоль). Смесь охлаждали до 0°C и добавляли по каплям диизопропилэтиламин (0,024 мл, 0,137 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 90 мин, и затем распределяли между 0,1 н водн. HCl (2 мл) и CH2Cl2 (3×2 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле с получением названного соединения (17 мг, 56%).
Часть C. Получение N-(2-(6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-инден-3-ил)пропан-2-ил)метансульфонамида.
Продукт из Части C (50 мг, 0,151 ммоль) подвергали условиям, описанным для Примера 72, Части B с получением названного соединения в виде бесцветного твердого вещества (37 мг, 65%).
Часть D. Получение N-(2-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1H-инден-3-ил)пропан-2-ил)метансульфонамида.
Продукт из Части C (35 мг, 0,093 ммоль) подвергали условиям, описанным для Примера 72, Части C с получением названного соединения в виде бесцветного твердого вещества (41 мг, 84%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.40 (s, 1 H) 7.94 (d, J=8.09 Hz, 1 H) 7.78 (d, J=8.09 Hz, 1 H) 7.65 (d, J=1.50 Hz, 1 H) 7.56 (s, 1 H) 7.48 (dd, J=8.09, 1.47 Hz, 1 H) 7.27 (s, 2 H) 6.48 (s, 1 H) 5.63 (d, J=8.09 Hz, 1 H) 3.43 (s, 2 H) 3.25 (s, 3 H) 2.63 (s, 3 H) 1.68 (s, 6 H) 1.41 (s, 9 H).
Пример 76. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[b]тиофен-3-ил)метил)метансульфонамида (соединение IB-L0-2.11).
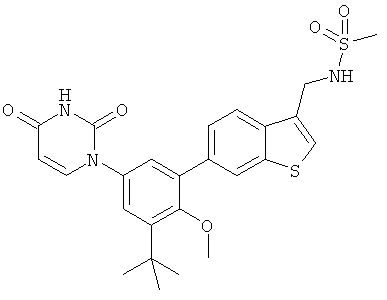
Часть A. Получение этил 6-бромбензо[b]тиофен-2-карбоксилата.
Раствор этилтиогликолята (0,65 г, 5,42 ммоль), 4-бром-2-фторбензальдегида (1,0 г, 4,93 ммоль) и триэтиламина (1,25 мл, 12,3 ммоль) в DMSO (5 мл) нагревали при 75°C в течение 2 ч. Смесь распределяли между H2O (50 мл) и CH2Cl2 (2×50 мл), и объединенные органические слои сушили над Na2SO4. Высушивающее вещество отфильтровывали, и растворитель удаляли в вакууме с получением названного соединения в виде масла (1,29 г, 92%).
Часть B. Получение 6-бромбензо[b]тиофен-2-карбоновой кислоты.
К раствору продукта из Части A (1,21 г, 4,24 ммоль) в THF (10 мл) добавляли а раствор LiOH (0,305 г, 12,73 ммоль) в H2O (4 мл) и полученную в результате смесь перемешивали при 40°C в течение 2 ч. Смесь распределяли между H2O (50 мл) и CH2Cl2 (50 мл). Водный слой доводили до pH=2, используя 1 н HCl, и экстрагировали CH2Cl2 (2×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением названного соединения в виде масла (1,04 г, 95%).
Часть C. Получение 6-бромбензо[b]тиофена.
Продукт из Части B (0,70 г, 2,73 ммоль) и DBU (1,35 мл, 8,94 ммоль) объединяли в DMA (6 мл) в герметично закрытой пробирке и нагревали при 200°C в микроволновом реакторе в течение 70 мин. Полученный в результате темный раствор разбавляли 1 М HCl (20 мл) и экстрагировали CH2Cl2 (2×20 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя CH2Cl2 в качестве элюента с получением названного соединения в виде масла (0,484 г 83%).
Часть D. Получение 6-бром-3-(хлорметил)бензо[b]тиофена.
К раствору продукта из Части C (0,484 г, 2,27 ммоль) в бензоле (0,20 мл) добавляли 37% водн. раствор формальдегида (1 мл) и концентрированную HCl (1 мл). Полученную в результате смесь нагревали при 70°C в течение 1 ч., при этом газообразный HCl барботировали через эту смесь смесь. Смесь распределяли между H2O (20 мл) и CH2Cl2 (2×20 мл), и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя CH2Cl2 с получением названного соединения в виде воскообразного твердого вещества (0,49 г, 82%).
Часть E. Получение N-((6-бромбензо[b]тиофен-3-ил)метил)-N-(2,4-диметоксибензил) метансульфонамида.
К раствору продукта из Части D (275 мг, 1,05 ммоль) и N-(2,4-диметоксибензил)-метансульфонамида (284 мг, 1,15 ммоль) в DMA (6 мл) добавляли K2CO3 (160 мг, 1,15 ммоль), и смесь перемешивали при комнатной температуре в течение 3 ч. Смесь распределяли между H2O (20 мл) и Et2O (2×20 мл), и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 2% EtOAc в CH2Cl2 в качестве элюента с получением названного соединения в виде воскообразного твердого вещества (316 мг, 64%).
Часть F. Получение N-(2,4-диметоксибензил)-N-((6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[b]тиофен-3-ил)метил)метансульфонамида.
Продукт из Части E (300 мг, 0,64 ммоль) подвергали условиям, описанным для Примера 72, Части B с получением названного соединения в виде воскообразного твердого вещества (248 мг, 75%).
Часть G. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[b]тиофен-3-ил)метил)-N-(2,4-диметоксибензил)метансульфонамида.
Продукт из Части F (214 мг, 0,414 ммоль) подвергали условиям, описанным для Примера 72, Части C с получением названного соединения в виде светлого желтого твердого вещества (238 мг, 87%).
Часть H. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[b]тиофен-3-ил)метил)метансульфонамида.
К раствору продукта из Части G (230 мг, 0,34 ммоль) в CH2Cl2 (4 мл) добавляли трифторуксусную кислоту (0,5 мл), и смесь перемешивали при комнатной температуре в течение 30 мин. Раствор разбавляли CH2Cl2 (10 мл) и экстрагировали насыщенным водн. NaHCO3 (2×10 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 3% MeOH в CH2Cl2 с получением названного соединения в виде беловатого твердого вещества (149 мг, 84%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.41 (s, 1 H) 8.16 (d, J=1.10 Hz, 1 H) 8.02 (d, J=8.46 Hz, 1 H) 7.79 (d, J=7.72 Hz, 1 H) 7.71 (s, 1 H) 7.60-7.66 (m, 2 H) 7.29-7.38 (m, 2 H) 5.65 (d, J=7.72 Hz, 1 H) 4.44 (d, J=5.88 Hz, 2 H) 3.24 (s, 3 H) 2.95 (s, 3 H) 1.42 (s, 9 H).
Пример 77. Получение N-(2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1,2,3,4-тетрагидроизохинолин-6-ил)метансульфонамида (соединение IB-L0-2.19).
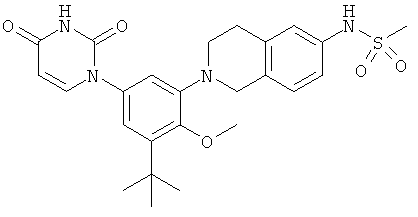
Часть A Получение 1-(3-амино-5-трет-бутил-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
К раствору продукта из Примера 7, Части F (170 мг, 0,534 ммоль) и триэтиламина (223 мкл, 1,6 ммоль) в THF (5 мл) добавляли дифенилфосфорилазид (173 мкл, 0,80 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 1 ч, и затем перемешивали при 45°C в течение 1 ч. Добавляли воду (280 мкл), и полученную в результате смесь перемешивали при 50°C в течение 1 ч, и затем перемешивали при комнатной температуре в течение 16 ч. Раствор разбавляли H2O (10 мл), и полученное в результате твердое вещество отфильтровывали. Твердое вещество суспендировали в 1M водн. HCl и фильтровали с получением аминного продукта в виде HCl соли. Эту соль суспендировали в водн. NaHCO3 (20 мл) и экстрагировали EtOAc (2×20 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением названного соединения в виде бесцветного твердого вещества (55 мг, 36%).
Часть B. Получение 1-(3-трет-бутил-4-метокси-5-(6-нитро-3,4-дигидроизохинолин-2(1H)-ил)фенил)пиримидин-2,4(1H,3H)-диона.
Раствор продукта из Части A (100 мг, 0,28 ммоль) и 2-(2-(метилсульфонилокси)-этил)-4-нитробензилметансульфоната (196 мг, 0,68 ммоль) в безводном DMA (4 мл) перемешивали при 80°C в течение 18 ч. Охлажденную смесь распределяли между H2O (20 мл) и EtOAc (2×20 мл), и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток суспендировали в CH2Cl2 и фильтровали для удаления непрореагировавшего исходного анилинового вещества. Фильтрат концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 1% MeOH в CH2Cl2 с получением названного соединения в виде светлого желтого твердого вещества (39,3 мг, 31%).
Часть C. Получение N-(2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1,2,3,4-тетрагидроизохинолин-6-ил)метансульфонамида.
К раствору продукта из Части B (35 мг, 0,078 ммоль) в THF (0,5 мл), MeOH (0,5 мл) и H2O (0,25 мл) добавляли Fe порошок (17,4 мг, 0,41 ммоль) и NH4Cl (6,2 мг, 0,12 ммоль), и полученную в результате смесь перемешивали при 70°C в течение 1 ч. Горячую смесь фильтровали через целит и промывали с использованием THF и MeOH. Фильтрат концентрировали и сушили в вакууме с получением твердого вещества. К раствору этого твердого вещества (32 мг, 0,076 ммоль) и пиридина (26 мкл, 0,32 ммоль) в CH2Cl2 (1,5 мл) добавляли метансульфонилхлорид (7,7 мкл, 0,099 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 5% MeOH в CH2Cl2 с получением названного соединения в виде светлого желтого твердого вещества (7 мг, 19%). 1H ЯМР (300 MHz, DMSO-d6) δ 7.71 (d, J=8.09 Hz, 1 H) 7.14-7.21 (m, 1 H) 7.05-7.12 (m, 3 H) 6.98 (d, J=2.57 Hz, 1 H) 5.65 (d, J=7.72 Hz, 1 H) 4.18 (s, 2 H) 3.86 (s, 3 H) 3.03 (t, J=4.23 Hz, 2 H) 2.99 (s, 3 H) 1.38 (s, 9 H).
Пример 78. Получение N-(2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)изоиндолин-5-ил)метансульфонамида (соединение IB-L0-2.79).
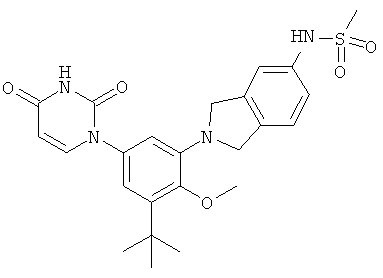
Часть A. Получение (4-нитро-1,2-фенилен)бис(метилен)диметансульфоната.
К раствору 4-нитрофталиевой кислоты (500 мг, 2,37 ммоль) в THF (24 мл) при комнатной температуре добавляли по каплям 1М раствор комплекса BH3·THF (9,95 мл, 9,95 ммоль). Этот раствор перемешивали при 65°C в течение 1 ч, и затем оставляли охлаждаться до комнатной температуры. К смеси добавляли MeOH (1 мл), и смесь перемешивали в течение 30 мин и концентрировали в вакууме. Остаток распределяли между 1М водн. HCl (20 мл) и EtOAc (2×20 мл), и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 3% MeOH в CH2Cl2 с получением масла (253 мг, 58%). К раствору этого масла (250 мг, 2,37 ммоль) и триэтиламина (438 мкл, 3,14 ммоль) в безводном CH2Cl2 (30 мл) при 0°C добавляли по каплям метансульфонилхлорид (234 мкл, 3,0 ммоль). Раствор перемешивали при комнатной температуре в течение 18 ч, и распределяли между 1М водн. HCl (20 мл) и CH2Cl2 (2×20 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2 с получением названного соединения (150 мг, 32%).
Часть B. Получение 1-(3-трет-бутил-4-метокси-5-(5-нитроизоиндолин-2-ил)фенил) пиримидин-2,4(1H,3H)-диона.
К раствору продукта Части A (110 мг, 0,324 ммоль) и продукта Примера 77, Части A (113 мг, 0,389 ммоль) в безводном 1,4-диоксане (4 мл) добавляли бикарбонат натрия (60 мг, 0.71 ммоль) и диизопропилэтиламин (142 мкл, 0,81 ммоль), и полученную в результате смесь перемешивали при 95°C в течение 16 ч. Смесь распределяли между 0,5М водн. HCl (10 мл) и CH2Cl2 (2×10 мл), и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 1% MeOH в CH2Cl2 с получением названного соединения в виде светлого желтого твердого вещества (110 мг, 78%).
Часть C. Получение N-(2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)изоиндолин-5-ил)метансульфонамида.
Продукт из Части B (100 мг, 0,25 ммоль) подвергали условиям, описанным для Примера 77, Части C с получением названного соединения в виде беловатого твердого вещества (53 мг, 45%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.37 (s, 1 H) 9.70 (s, 1 H) 7.71 (d, J=7.72 Hz, 1 H) 7.34 (d, J=8.09 Hz, 1 H) 7.23 (d, J=1.84 Hz, 1 H) 7.13 (dd, J=8.09, 1.84 Hz, 1 H) 6.98 (d, J=2.57 Hz, 1 H) 6.81 (d, J=2.21 Hz, 1 H) 5.62 (d, J=7.72 Hz, 1 H) 4.52 (s, 2 H) 4.50 (s, 2 H) 3.63 (s, 3 H) 2.98 (s, 3 H) 1.38 (s, 9 H).
Пример 79. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1H-инден-3-ил)метил)метансульфонамида (соединение IB-L0-2.13).
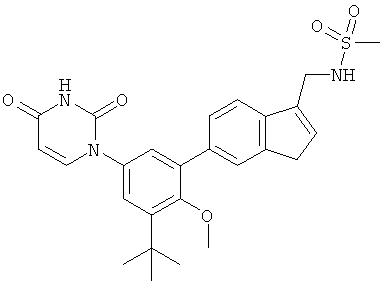
Часть A. Получение 5-бром-1-(триметилсилилокси)-2,3-дигидро-1H-инден-1-карбонитрила.
К раствору 5-бром-2,3-дигидро-1H-инден-1-она (10,0 г, 47,4 ммоль) и N-метил-морфолин N-оксида (1,67 г, 14,21 ммоль) в CH2Cl2 (50 мл) добавляли триметилсилилцианид (7,05 г, 71,1 ммоль), и полученный в результате раствор перемешивали при комнатной температуре в течение 72 ч, и затем концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 5% EtOAc в гексане в качестве элюента, с получением названного соединения в виде бесцветной жидкости (12,65 г, 86%).
Часть B. Получение 1-(аминометил)-5-бром-2,3-дигидро-1H-инден-1-ола.
К раствору продукта из Части A (18,44 г, 59,4 ммоль) в безводном Et2O (250 мл) в атмосфере газообразного N2 при 0°C по каплям добавляли 1M раствор LiAlH4 в Et2O (62,4 мл, 62,4 ммоль) на протяжении 1 ч. После добавления, смесь оставляли нагреваться до комн. темп. и перемешивали при комнатной температуре в течение 2 ч. Смесь охлаждали на ледяной бане при добавлении по каплям H2O (4,3 мл), с последующим добавлением 15% водн. NH4OH (4.3 мл), и затем H2O (13 мл). Смесь перемешивали при комнатной температуре в течение 15 мин, и затем фильтровали через целит и промывали EtOAc. Фильтрат концентрировали в вакууме, и остаток суспендировали в Et2O (40 мл) с получением осадка, который фильтровали и сушили, с получением названного соединения в виде бесцветного твердого вещества (10,0 г, 70%).
Часть C. Получение соли (6-бром-1H-инден-3-ил)метанамингидрохлорида.
К раствору продукта из Части B (10,0 г, 41,3 ммоль) в MeOH (100 мл) добавляли 6 н водн. НС1 (125 мл), и смесь перемешивали при 70°C в течение 3 ч, и затем оставляли охлаждаться до комнатной температуры. MeOH удаляли в вакууме с получением осадка, который собирали фильтрацией, промывали H2O, и сушили в вакууме с получением названного соединения в виде бесцветного твердого вещества (9,89 г, 92%).
Часть D. Получение N-((6-бром-1H-инден-3-ил)метил)метансульфонамида.
К суспензии продукта из Части С (6,46 г, 24,8 ммоль) в безводном CH2Cl2 (260 мл) добавляли метансульфонилхлорид (3,86 мл, 49,6 ммоль) и диизопропилэтиламин (13,0 мл, 74,4 ммоль), и полученную в результате смесь перемешивали при комнатной температуре в течение 10 ч. Раствор промывали 1 н водн. HCl (2×300 мл), и органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток суспендировали в Et2O (100 мл) с получением осадка, который собирали фильтрацией и сушили с получением названного соединения в виде бесцветного твердого вещества (6,25 г, 83%).
Часть E. Получение N-((6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-инден-3-ил)метил)метансульфонамида.
Раствор продукта из Части D (2,0 г, 6,62 ммоль), бис(пинаколато)дибора (1,85 г, 7,28 ммоль), ацетата калия (1,95 г, 19,86 ммоль) и комплекса 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид дихлорметан (0,27 г, 0,331 ммоль) в безводном 1,4-диоксане (80 мл) в атмосфере N2 перемешивали при 95°C в течение 8 ч. Охлажденную смесь фильтровали через целит, промывали EtOAc (2×20 мл) и затем концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 1:2 EtOAc тексан в качестве элюента, с получением названного соединения в виде бесцветного масла (2,02 г, 87%).
Часть F. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1H-инден-3-ил)метил)метансульфонамида.
Смесь продукта из Части E (3,14 г, 8,99 ммоль), продукта из Примера C (3,78 г, 9,44 ммоль), трифосфата калия (3,82, 17,98 ммоль), 1,3,5,7-тетраметил-2,4,8-триокса-6-фосфа-6-фенил-адамантана (Cytec [97739-46-3]) (105 мг, 0,36 ммоль), и трис(дибензилиденацетон)-дипалладия(0) (165 мг, 0,18 ммоль) помещали в атмосферу газообразного N2. К смеси добавляли, через канюлю, смесь THF (45 мл) и H2O (15 мл) которая была дегазирована барботированием газообразного Ar в течение 10 мин. Полученную в результате смесь дополнительно дегазировали барботированием Ar в течение дополнительных 15 мин. Смесь перемешивали при 50°C в течение 1,5 ч при непрерывном барботировании Ar через этот раствор. Добавляли дополнительное количество трис(дибензилиденацетон)дипалладия(0) (55 мг, 0,6 ммоль) в THF (2 мл), и смесь перемешивали при 50°C в течение 1 ч. Смесь оставляли охлаждаться до комн. темп., и распределяли между CH2Cl2 (300 мл) и 1 н водн. HCl (150 мл). К оранжевому органическому слою добавляли 3-меркаптопропил-функционализированный силикагель (10 г, Aldrich) и MgSO4, и смесь перемешивали при комнатной температуре в течение 16 ч, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3:1 EtOAc тексан в качестве элюента с получением названного соединения в виде бесцветного твердого вещества (2,7 г, 61%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.40 (s, 1H), 7.78 (d, J=7.4 Hz, 1H), 7.66 (s, 1H), 7.60 (d, J=7.7 Hz, 1H), 7.50 (m, 2H), 7.25 (m, 2H), 6.56 (m, 1H), 5.64 (dd, J=2.2, 7.7 Hz, 1H), 4.18 (d, J=5.1 Hz, 2H), 3.46 (s, 2H), 3.25 (s, 3H), 2.96 (s, 3H), 1.41 (s, 9H).
Пример 80. Получение N-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)метансульфонгидразида (соединение IB-L0-2.31).
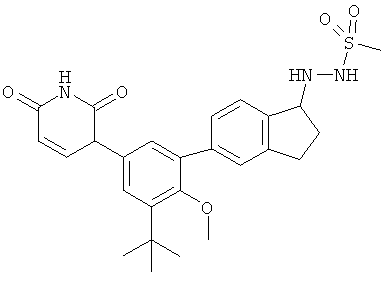
К раствору продукта из Примера 6, Часть C (100 мг, 0,201 ммоль) в THF (2 мл) и MeOH (2 мл) добавляли 2 капли 10% HCl в MeOH, после чего добавляли цианоборгидрид натрия (19 мг, 0,302 ммоль). Смесь доводили до pH 4 добавлением 10% HCl в MeOH, и затем перемешивали при комнатной температуре в течение 1 ч. Полученную в результате смесь распределяли между насыщенным водн. бикарбонатом натрия (5 мл) и CH2Cl2 (20 мл), и органический слой сушили над Na2SO4, фильтровали и концентрировали. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента с получением названного соединения в виде бесцветного твердого вещества (58 мг, 58%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.39 (s, 1H), 8.18 (d, J=3.7 Hz, 1Н), 7.77 (d, J=7.7 Hz, 1H), 7.51 (d, J=8.1 Hz, 1H), 7.38 (m, 2H), 7.27 (d, J=2.6 Hz, 1H), 7.21 (d, J=2.9 Hz, 1H), 5.63 (d, J=7.7 Hz, 1H), 5.25 (m, 1H), 4.39 (m, 1H), 3.27 (s, 3H), 2.98 (m, 1H), 2.83 (s, 3H), 2.78 (m, 1H), 2.22 (m, 1H), 2.07 (m, 1H), 1.40 (s, 9H).
Пример 81. Получение 1-(3-трет-бутил-5-(1-гидрокси-2,3-дигидро-1H-инден-5-ил)-4-метоксифенил)пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.36).
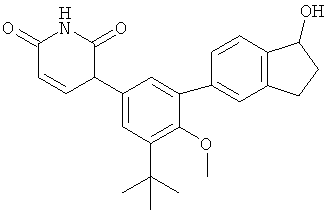
К раствору продукта из Примера 6, Части B (150 мг, 0,371 ммоль) в MeOH (3 мл) и CH2Cl2 (3 мл) добавляли боргидрид натрия (28 мг, 0,742 ммоль), и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь распределяли между 1 н водн. HCl (10 мл) и CH2Cl2 (20 мл), и органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 5% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения в виде бесцветного твердого вещества (90 мг, 60%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.39 (s, 1H), 7.44 (d, J=4.0 Hz, 1H), 7.40 (m, 2H), 7.21 (d, J=2.6 Hz, 1H), 7.26 (d, J=2.6 Hz, 1H), 5.63 (d, J=8.1 Hz, 1H), 5.29 (d, J=5.9 Hz, 1H), 5.09 (m, 1H), 3.26 (s, 3H), 2.97 (m, 1H), 2.79 (m, 1H), 2.38 (m, 1H), 1.83 (m, 1H), 1.40 (s. 9H).
Пример 82. Получение 1-(3-трет-бутил-5-(2-(2,5-диметил-1H-пиррол-1-ил)бензо[d]тиазол-6-ил)-4-метоксифенил)пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.47).
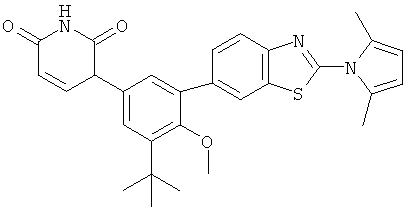
Часть A. Получение 6-бром-2-(2,5-диметил-1H-пиррол-1-ил)бензо[d]тиазола.
Раствор 6-бромбензо[d]тиазол-2-амина (5,75 г, 25,1 ммоль), гексан-2,5-диона (2,95 мл, 25,1 ммоль), и PPTS (0,95 г, 3,76 ммоль) в бензоле (100 мл) нагревали с обратным холодильником в течение 16 ч, в то же время воду удаляли с помощью насадки Дина-Старка. Охлажденную смесь выливали в EtOAc (100 мл) и экстрагировали насыщенным водн. NaHCO3 (2×100 мл) и солевым раствором. Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 9:1 EtOAc:гексан в качестве элюента с получением названного соединения в виде оранжевого масла (6,46 г, 84%).
Часть B. Получение 2-(2,5-диметил-1H-пиррол-1-ил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[d]тиазола.
Смесь продукта из Части A (3,24 г, 10,54 ммоль), бис(пинаколато)дибора (4,01 г, 15,81 ммоль), бис(ди-трет-бутил(гидрокси)фосфино)палладий(II) дихлорида (0,264 г, 0,527 ммоль), и ацетата калия (3,10 г, 31,6 ммоль) в безводном толуоле (25 мл) дегазировали путем барботирования газообразным N2 в течение 15 мин, и затем нагревали с обратным холодильником в атмосфере N2 в течение 72 ч. Охлажденную смесь фильтровали через целит и промывали EtOAc, и фильтрат концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 9:1 EtOAc:гексан в качестве элюента, с получением названного соединения (2,77 г, 74%).
Часть С. Получение 1-(3-трет-бутил-5-(2-(2,5-диметил-1H-пиррол-1-ил)бензо[d]тиазол-6-ил)-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
Продукт из Части B (405 мг, 1,14 ммоль) подвергали условиям, описанным для Примера 72, Части C с получением названного соединения (430 мг, 68%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.43 (d, J=2.21 Hz, 1 Н) 8.32 (d, J=1.47 Hz, 1 Н) 8.12 (d, J=8.46 Hz, 1 Н) 7.80 (d, J=7.72 Hz, 1 Н) 7.76 (dd,.7=8.46, 1.84 Hz, 1 H) 7.35 (q,.7=2.57 Hz, 2 H) 5.97 (s, 2 H) 5.66 (dd,.7=7.72, 2.21 Hz, 1 H) 3.30 (s, 3 H) 2.30 (s, 6 H) 1.43 (s, 9 H).
Пример 83. Получение 1-(3-(2-аминобензо[d]тиазол-6-ил)-5-трет-бутил-4-метокси-фенил)пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.27).
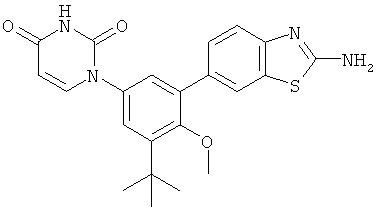
К раствору продукт из Примера 82 (4,0 г, 8,0 ммоль) в трифторуксусной кислоте (50 мл) добавляли несколько капель H2O, и полученную в результате смесь перемешивали при 80°C в течение 2,5 ч, и затем концентрировали в вакууме. Раствор остатка в MeOH нейтрализовали, используя конц. NH4OH, концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 9:1 CH2Cl2:MeOH в качестве элюента, с получением названного соединения (3,3 г, 98%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.40 (s, 1 H) 7.81 (s, 1 H) 7.77 (d, J=8.09 Hz, 1 H) 7.57 (s, 1 H) 7.40 (s, 1 H) 7.33-7.38 (m, 1 H) 7.25 (s, 1 H) 5.60-5.69 (m, 1 H) 3.26 (s, 3 H) 1.40 (s, 9 H).
Пример 84. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]тиазол-2-ил)метансульфонамида (соединение IB-L0-2.28).
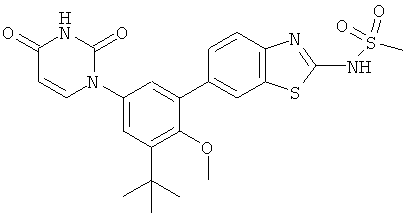
К раствору продукта из Примера 83 (0,35 г, 0,83 ммоль) в безводном CH2Cl2 (50 мл) добавляли метансульфонилхлорид (194 мкл, 2,49 ммоль) и пиридин (1,34 мл, 16,6 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 16 ч и концентрировали в вакууме. Сырой продукт очищали с помощью C-18 ВЭЖХ с обращенной фазой, используя градиент ацетонитрил:H2O (0,1% TFA), с получением названного соединения (19 мг, 4%). 1H ЯМР (300 MHz, DMSO-d6) δ 13.09 (s, 1 H) 11.41 (d, J=1.84 Hz, 1 H) 7.96 (d, J=1.47 Hz, 1 H) 7.77 (d, J=8.09 Hz, 1 H) 7.57 (dd, 1 H) 7.42 (d, J=8.09 Hz, 1 H) 7.25-7.32 (m, 2 H) 5.64 (dd, J=8.09, 2.21 Hz, 1 H) 3.25 (s, 3 H) 3.02 (s, 3 H) 1.40 (s, 9 H).
Пример 85. Получение 1-(3-(бензо[d]тиазол-6-ил)-5-трет-бутил-4-метоксифенил) пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.33).
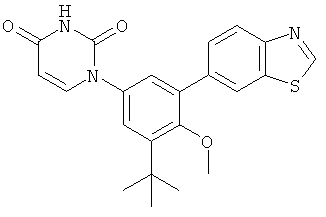
К раствору продукта из Примера 83 (30 мг, 0,071 ммоль) в безводном 1,4-диоксане (3 мл) в атмосфере N2 добавляли изоамилнитрит (19 мкл, 0,142 ммоль). Полученную в результате смесь перемешивали при нагревании с обратным холодильником в течение 1 ч, и концентрировали в вакууме. Сырой продукт очищали с помощью С-18 ВЭЖХ с обращенной фазой, используя градиент ацетонитрил:H2O (0,1% TFA), с получением названного соединения (14 мг, 48%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.42 (d, J=1.84 Hz, 1 H) 9.44 (s, 1 H) 8.34 (d, J=1.47 Hz, 1 H) 8.19 (d, J=8.46 Hz, 1 H) 7.79 (d, J=7.72 Hz, 1 H) 7.73 (dd, J=8.46, 1.84 Hz, 1 H) 7.32-7.37 (m, 2 H) 5.65 (dd, J=7.91, 2.39 Hz, 1 H) 3.24 (s, 3 H) 1.42 (s, 9 H).
Пример 86. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[d]тиазол-2-ил)ацетамида (соединение IB-L0-2.49).
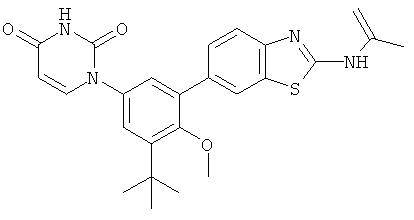
Смесь продукта из Примера 83 (30 мг, 0,071 ммоль) и уксусного ангидрида (3 мл) перемешивали при 100°C в течение 2 ч, и затем оставляли охлаждаться до комнатной температуры. Полученное в результате твердое вещество собирали фильтрацией, промывали Н20, и сушили с получением названного соединения в виде беловатого твердого вещества (29 мг, 88%). 1H ЯМР (300 MHz, DMSO-d6) δ 12.42 (s, 1 H) 11.41 (d, J=2.21 Hz, 1 H) 8.12 (d, J=1.47 Hz, 1 H) 7.82 (d, J=8.46 Hz, 1 H) 7.78 (d, J=8.09 Hz, 1 H) 7.61 (dd, J=8.46, 1.84 Hz, 1 H) 7.31 (q, J=2.70 Hz, 2 H) 5.64 (dd, J=8.09, 2.21 Hz, 1 H) 3.24 (s, 3 H) 2.22 (s, 3 H) 1.41 (s, 9 H).
Пример 87. Получение 1-(3-трет-бутил-4-метокси-5-(2-(пропиламино)бензо[d]тиазол-6-ил)фенил)пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.46).
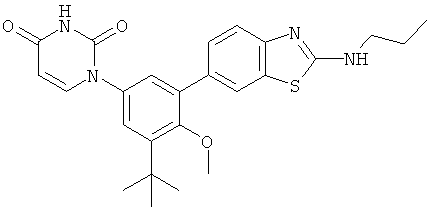
Часть A. Получение 1-(3-трет-бутил-5-(2-хлорбензо[d]тиазол-6-ил)-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
К смеси продукта из Примера 83 (50 мг, 0,118 ммоль) и хлорида меди (II) (24 мг, 0,178 ммоль) в ацетонитриле (3 мл) при 0°C добавляли wpew-бутилнитрит (21 мкл, 0,178 ммоль). Смесь перемешивали при 0°C в течение 1 ч, и затем нагревали до 65°C и перемешивали в течение 2 ч. Смесь концентрировали в вакууме и очищали с помощью колоночной хроматографии на силикагеле, используя 5% MeOH в CH2Cl2 с получением названного соединения в виде беловатого твердого вещества (43 мг, 82%).
Часть B. Получение 1-(3-трет-бутил-4-метокси-5-(2-(пропиламино)бензо[d]тиазол-6-ил)фенил)пиримидин-2,4(1H,3H)-диона.
Смесь продукта из Части A (50 мг, 0,11 ммоль), 1-аминопропана (9 мкл, 0,11 ммоль), и K2CO3 (15,6 мг, 0,11 ммоль) в безводном DMF (5 мл) перемешивали при 100°C в течение 24 ч. Смесь концентрировали в вакууме и очищали с помощью колоночной хроматографии на силикагеле, используя 2% MeOH в EtOAc в качестве элюента, с получением названного соединения в виде беловатого твердого вещества (21 мг, 40%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.39 (d, J=1.84 Hz, 1 H) 8.12 (t, J=5.52 Hz, 1 H) 7.82 (d, J=1.47 Hz, 1 H) 7.77 (d, J=7.72 Hz, 1 H) 7.44 (t, J=9.01 Hz, 1 H) 7.37-7.41 (m, 1 H) 7.25 (s, 2 H) 5.63 (dd, J=7.91, 2.02 Hz, 1 H) 3.33-3.38 (m, 2 H) 3.26 (s, 3 H) 1.56-1.69 (m, 2 H) 1.40 (s, 9 H) 0.94 (t, J=7.35 Hz, 3 H).
Пример 88. Получение 1-(3-трет-бутил-4-метокси-5-(3-метилбензофуран-6-ил)фенил)-пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.42).
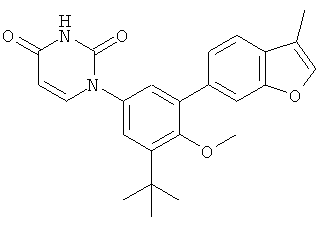
Часть A. Получение метил 2-(2-ацетил-5-бромфенокси)ацетата.
Раствор 1-(4-бром-2-гидроксифенил)этанона (1,35 г, 6,28 ммоль) в безводном DMF (16 мл) обрабатывали несколькими порциями гидридом натрия (377 мг 60% в масле, 226 мг, 9,42 ммоль) с последующим перемешиванием при комнатной температуре в течение 30 мин. Смесь затем обрабатывали метил бромацетатом (871 мкл, 1,45 г, 9,48 ммоль) по каплям (после завершения добавления раствор становился теплым) с последующим перемешиванием при комнатной температуре в течение 18 ч. Смесь разбавляли этилацетатом и экстрагировали водой (4×) и насыщенным раствором хлорида натрия. Сушка (Na2SO4) и концентрирование в вакууме давали почти бесцветное твердое вещество, которое очищали с помощью колоночной хроматограф ии на силикагеле, элюируя 20-100% этилацетатом в гексане. Эти процедуры давали названное соединение в виде бесцветного твердого вещества (1,47 г, 82%).
Часть B. Получение 2-(2-ацетил-5-бромфенокси)уксусной кислоты. Раствор продукта из Части A (1,47 г, 5,12 ммоль) в тетрагидрофуране (26 мл) обрабатывали 1,0 н раствором гидроксида натрия (6,7 мл, 6,7 ммоль) с последующим перемешиванием при комнатной температуре в течение 3 ч, в этот момент реакцию завершали. Смесь концентрировали в вакууме для удаления тетрагидрофурана и затем разбавляли водой и охлаждали до 0°C. Смесь подкисляли до pH 3 путем добавления 1 н раствора соляной кислоты, и затем этот продукт экстрагировали этилацетатом. Органический слой экстрагировали насыщенным раствором хлорида натрия и сушили (Na2SO4). Концентрирование в вакууме давало названное соединение в виде бесцветного твердого вещества (1,36 г, 97%).
Часть C. Получение 6-бром-3-метилбензофурана.
Раствор продукта из Части B (500 мг, 1,83 ммоль) в уксусном ангидриде (9,2 мл) обрабатывали ацетатом натрия (300 мг, 3,66 ммоль) с последующим нагреванием с обратным холодильником в течение 18 ч. Смесь охлаждали до комнатной температуры и разбавляли толуолом и концентрировали в вакууме для азеотропного удаления уксусного ангидрида. Этот процесс повторяли 3×. Смесь затем разбавляли этилацетатом и перемешивали насыщенным раствором бикарбоната натрия в течение 1 ч. Слои отделяли и органический слой экстрагировали насыщенным раствором хлорида натрия. Сушка (Na2SO4) и концентрирование в вакууме давали масло янтарного цвета, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя 8-50% этилацетатом в гексане. Эти процедуры давали названное соединение в виде бесцветной жидкости (316 мг, 82%).
Часть D. Получение 4,4,5,5-тетраметил-2-(3-метилбензофуран-6-ил)-1,3,2-диоксаборолана.
В микроволновой трубке, смесь продукта из Части C (303 мг, 1,44 ммоль), бис(пинаколато)дибора (401 мг, 1,58 ммоль) и ацетата калия (423 мг, 4,31 ммоль) в безводном диоксане (5 мл) дегазировали путем барботирования азота в течение 15 мин. Смесь обрабатывали комплексом 1,1'-бис-(дифенилфосфино)ферроцен палладий (II) хлорид дихлорметан (24 мг, 0,029 ммоль) с последующим дегазированием в течение еще 5 мин. Микроволновую трубку герметично закрывали, и смесь нагревали при 90°C в течение 18 ч. Смесь охлаждали и разбавляли этилацетатом и экстрагировали водой и насыщенным раствором хлорида натрия. Органический слой сушили (Na2SO4) и перемешивали с (3-меркаптопропил) силикагелем в течение 1 ч. Смесь фильтровали и концентрировали в вакууме с получением коричневого полутвердого вещества, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя 8-40% этилацетатом в гексане. Эти процедуры давали названное соединение в виде бесцветного масла, которое медленно становилось твердым при стоянии (307 мг, 83%).
Часть E. Получение 1-(3-трет-бутил-4-метокси-5-(3-метилбензофуран-6-ил)фенил)-пиримидин-2,4(1H,3H)-диона.
В микроволновой трубке, раствор продукта из Части D (307 мг, 1,19 ммоль), продукт из Примера C (414 мг, 1,03 ммоль), 1,3,5,7-тетраметил-2,4,8-триокса-6-фосфа-6-фенил-адамантана (Cytec [97739-46-3]) (15 мг, 0,052 ммоль), и трехосновного фосфата калия (439 мг, 2,07 ммоль) в смеси 3:1 тетрагидрофуран-вода (8 мл) дегазировали путем барботирования азота в течение 20 мин. Смесь обрабатывали трис(дибензилиденацетон)дипалладием (0) (12 мг, 0,012 ммоль) с последующим дегазированием в течение еще 10 мин. За это время раствор превращался из исходного глубокого темно-бордового цвета в зеленовато-коричныевый цвет. Микроволновую трубку герметично закрывали, и раствор нагревали при 50°C в течение 56 ч. Раствор охлаждали и разбавляли этилацетатом и подкисляли 1М раствором лимонной кислоты. Органический слой экстрагировали насыщенным раствором хлорида натрия, сушили (Na2SO4), и затем перемешивали с (3-меркаптопропил) силикагелем в течение 1 ч. После фильтрования и концентрирования в вакууме, полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя 4-20% ацетоном в дихлорметане, после чего второй колоночной хроматографией на силикагеле, элюируя 20-100% этилацетатом в гексане. Эти процедуры давали названное соединение в виде бесцветного твердого вещества (355 мг). 1H ЯМР (300 MHz, DMSO-d6): δ 11.40 (d, J=1.84 Hz, 1 Н) 7.74-7.92 (m, 2 Н) 7.58-7.76 (m, 2 Н) 7.46 (dd, J=8.09, 1.47 Hz, 1 Н) 7.30 (q, J=2.82 Hz, 2 Н) 5.64 (dd, J=8.09, 2.21 Hz, 1 H) 3.22 (s, 3 Н) 2.25 (s, 3H) 1.41 (s, 9H).
Пример 89. Получение N-((6-(3-треот-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензофуран-3-ил)метил)метансульфонамида (соединение IB-L0-2.18).
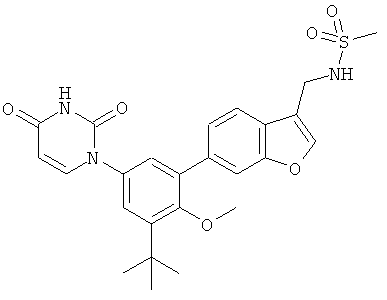
Часть A. Получение 6-бром-3-(бромметил)бензофурана. Раствор продукта из Примера 88, Части C (1,0 г, 4,74 ммоль) и дибензоилпероксида (287 мг, 1,19 ммоль) в хлорбензоле (24 мл) при нагревании с обратным холодильником обрабатывали четырьмя порциями N-бромсукцинимидом (843 мг, 4,74 ммоль) за период времени 30 мин. Смесь затем перемешивали при нагревании с обратным холодильником в течение 2 ч. Смесь охлаждали, фильтровали и концентрировали и очищали с помощью колоночной хроматографии на силикагеле, элюируя 7-30% хлороформом в гексане. Эти процедуры давали названное соединение в виде светло-желтого масла (438 мг, 32%).
Часть B. Получение N-((6-бромбензофуран-3-ил)метил)-N-(4-метоксибензил)метан-сульфонамида.
Раствор продукта из Части A (515 мг, 1,78 ммоль), N-(4-метоксибензил)метан-сульфонамида (421 мг, 1,95 ммоль), и карбоната калия (260 мг, 1,95 ммоль) в безводном DMF (8,9 мл) перемешивали при 70°C в течение 3 ч. Смесь охлаждали и разбавляли этилацетатом и экстрагировали водой (4×). Органический слой затем экстрагировали насыщенным раствором хлорида натрия и сушили (Na2SO4). Концентрирование в вакууме давало твердое вещество бежевого цвета. Это вещество очищали с помощью колоночной хроматографии на силикагеле, элюируя 20-100% этилацетатом в гексане. Эти процедуры давали названное соединение в виде бесцветного твердого вещества (224 мг, 35%).
Часть C. Получение N-(4-метоксибентл)-N-((6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензофуран-3-ил)метил)метансульфонамида.
Продукт из Части B (186 мг, 0,44 ммоль) подвергали условиям, описанным для Примера 88, Части D с получением названного соединения в виде бесцветного твердого вещества (177 мг, 86%).
Часть D. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензофуран-3-ил)метил)-N-(4-метоксибензил)метансульфонамида.
В микроволновой трубке, суспензию продукта из Части C (169 мг, 0,36 ммоль), продукта из Примера C (143 мг, 0,36 ммоль), и 1,0 М раствор карбоната натрия (0,5 мл, 0,50 ммоль) в смеси 1:1 этанол-толуол (3 мл) дегазировали путем барботирования азота в течение 20 мин. Этот раствор обрабатывали комплексом 1,1-бис(дифенилфосфино)ферроцен-палладий(II) хлорид дихлорметан (7 мг, 9 мкмоль) с последующим дегазированием в течение еще 5 мин. Микроволновую трубку герметично закрывали, и смесь нагревали при 100°C в микроволновой печи в течение 1 ч. Смесь разбавляли этилацетатом и водой, и подкисляли 1M раствором лимонной кислоты. Органический слой экстрагировали насыщенным раствором хлорида натрия, сушили (Na2SO4), и оставляли стоять в течение ночи на (3-меркаптопропил)силикагеле. Фильтрование и концентрирование в вакууме давало беловатую пену, которую очищали с помощью колоночной хроматографии на силикагеле, элюируя 5-30% этилацетатом в дихлорметане. Эти процедуры давали названное соединение в виде бесцветного твердого вещества (96 мг, 43%).
Часть E. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензофуран-3-ил)метил)метансульфонамида.
Раствор продукта из Части D (88 мг, 0,14 ммоль) в дихлорметане (1,4 мл) обрабатывали трифторуксусной кислотой (1,4 мл) с последующим перемешиванием при комнатной температуре в течение 18 ч, и затем перемешиванием при 40°C в течение 2 ч. Смесь концентрировали в вакууме с получением темной фиолетово-коричневой пены, которую подвергали колоночной хроматографии на силикагеле, элюируя 5-50% этилацетатом в метиленхлориде с получением содержащего примеси вещества, которое очищали с помощью хроматографии с обращенной фазой на колонке C-18, элюируя 1% водой-TFA/ацетонитрилом. Эти процедуры давали названное соединение в виде твердого вещества (3,9 мг). 1H ЯМР (300 MHz, DMSO-d6): δ 11.31-11.48 (m, 1 Н) 8.01 (s, 1 H) 7.68-7.94 (m, 2 H) 7.40-7.65 (m, 2 H) 7.10-7.38 (m, 2 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 4.33 (d, J=5.88 Hz, 2 H) 3.23 (s, 3 H) 2.95 (s, 3 H) 1.41 (s, 9 H).
Пример 90. Получение N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1-метил-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида (соединение IB-L0-2.25).
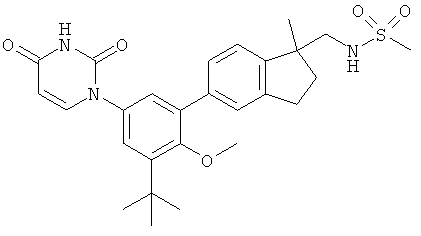
Часть A. Получение 5-бром-1-(1,3-дитиан-2-ил)-2,3-дигидро-1H-инден-1-ола.
Раствор 1,3-дитиана (11,96 г, 99 ммоль) в безводном тетрагидрофуране (100 мл) при -30°C обрабатывали по каплям на протяжении 10 мин н-бутиллитием (2,5M в гексане, 38,4 мл, 96 ммоль) с последующим перемешиванием при -15°C в течение 2 ч. Затем раствор обрабатывали раствором 5-бром-2,3-дигидро-1H-инден-1-она (15 г, 71,1 ммоль) в безводном тетрагидрофуране (250 мл) на протяжении 1 ч, поддерживая температуру между -9°C и 2°C. Смесь затем оставляли в холодильнике при 2-8°C в течение 18 ч. Раствор концентрировали в вакууме с получением темно-бордового масла, которое обрабатывали 1 н раствором соляной кислоты и экстрагировали эфиром. Эфирный слой экстрагировали насыщенным раствором хлорида натрия, сушили (Na2SO4) и концентрировали в вакууме с получением масла янтарного цвета (23,55 г).
Часть B. Получение 2-(5-бром-2,3-дигидро-1H-инден-1-илиден)-1,3-дитиана.
Раствор продукта из Части A (23,55 г, 71,1 ммоль) в бензоле (350 мл) обрабатывали моногидратом п-толуолсульфоновой кислоты (3,0 г) с последующим перемешиванием при нагревании с обратным холодильником в течение 1 ч, при этом удаляя воду с помощью насадки Дина-Старка. Смесь экстрагировали насыщенным раствором бикарбоната натрия и затем насыщенным раствором хлорида натрия. Сушка (Na2SO4) и концентрирование в вакууме давали продукт в виде масла янтарного цвета (22,27 г).
Часть C. Получение 5-бром-2,3-дигидро-1H-инден-1-карбоновой кислоты.
Раствор продукта из Части B (22,27 г, 71,1 ммоль) в ледяной уксусной кислоте (375 мл) обрабатывали концентрированным раствором соляной кислоты (125 мл) с последующим перемешиванием при нагревании с обратным холодильником в течение 3 ч. Смесь охлаждали и концентрировали в вакууме путем азеотропного удаления уксусной кислоты и воды толуолом (3×). Полученное коричневое масло фильтровали через прослойку силикагеля 70-230 меш в 2 л воронке спеченного стекла (объем силикагеля ок. 1800 мл), элюируя дихлорметаном для удаления неполярных примесей (1,3-пропандитиола, среди прочего) и затем этилацетатом для элюции названного соединения, которое получали в виде твердого вещества коричневого цвета (9,85 г, 58%).
Часть D. Получение метил 5-бром-2,3-дигидро-1H-инден-1-карбоксилата.
Суспензию продукта из Части C (9,85 г, 40,9 ммоль) в метаноле (400 мл) обрабатывали 4 н хлоридом азота в 1,4-диоксане (125 мл), и смесь перемешивали при нагревании с обратным холодильником в течение 8 ч. Смесь концентрировали в вакууме с получением коричневого масла, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя 0-30% метил т-бутиловым эфиром в хлороформе. Эти процедуры давали названное соединение в виде масла янтарного цвета (7,99 г, 77%).
Часть Е. Получение метил 5-бром-1-метил-2,3-дигидро-1H-инден-1-карбоксилата.
Раствор продукта из Части D (2,03 г, 7,96 ммоль) в безводном тетрагидрофуране (40 мл) при -78°C в атмосфере N2 обрабатывали по каплям бис(триметилсилил)амидом лития (1,0М в тетрагидрофуране, 9,55 мл, 9,55 ммоль) на протяжении 10 мин. Раствор перемешивали при -78°C в течение 45 мин и затем обрабатывали метилйодидом (1,5 мл, предварительно высушенным путем пропускания через слой щелочного оксида алюминия). Смесь затем оставляли постепенно нагреваться до комн. темп. и перемешивали в течение 18 ч. Смесь гасили путем добавления насыщенного раствора хлорида аммония (2 мл). Смесь концентрировали в вакууме для удаления тетрагидрофурана, и остаток разбавляли этилацетатом. Смесь экстрагировали насыщенным раствором хлорида аммония и насыщенным раствором хлорида натрия. Сушка (Na2SO4) и концентрирование в вакууме давали названное соединение в виде масла янтарного цвета (2,06 г, 96%).
Часть F. Получение 5-бром-1-метил-2,3-дигидро-1H-инден-1-карбоновой кислоты.
Раствор продукта из Части E (2,06 г, 7,65 ммоль) и триметилсиланоата калия (5,5 г 90%, 4,91 г, 38,3 ммоль) в тетрагидрофуране (40 мл) перемешивали при нагревании с обратным холодильником в течение 3 ч. Смесь охлаждали и концентрировали в вакууме для удаления тетрагидрофурана. Темно-бордовый осадок растворяли в воде (ок. 175 мл) и экстрагировали метил т-бутиловым эфиром. Водную фазу охлаждали до 0°C и подкисляли до pH 3 путем добавления концентрированного раствора соляной кислоты. Смесь экстрагировали этилацетатом (2×) и затем насыщенным раствором хлорида натрия. Раствор сушили (Na2SO4) и обрабатывали Darco G-60, с последующим фильтрованием через целит.Фильтрат концентрировали в вакууме с получением названного соединения в виде светлого желтого твердого вещества (1,93 г, 99%).
Часть G. Получение 5-бром-1-метил-2,3-дигидро-1H-инден-1-карбоксамида.
Раствор продукта из Части F (1,56 г, 6,12 ммоль) и DMF (473 мкл, 447 мг, 6,12 ммоль) в гексане (100 мл) обрабатывали оксалилхлоридом (1,61 мл, 2,32 г, 18,4 ммоль) с последующим перемешиванием при комнатной температуре в течение 1 ч. Смесь обрабатывали целитом и затем фильтровали через целит. Фильтрат концентрировали в вакууме и растворяли в ацетоне (75 мл), и охлаждали до 0°C. Этот раствор обрабатывали 28% водным раствором аммиака (75 мл) с последующим перемешиванием при 0°C в течение 30 мин и затем нагреванием до комнатной температуры. Смесь концентрировали в вакууме и экстрагировали этилацетатом. Органический слой экстрагировали насыщенным раствором хлорида натрия и сушили (Na2S04). Концентрирование в вакууме давало названное соединение в виде масла (1,55 г, 100%).
Часть H. Получение (5-бром-1-метил-2,3-дигидро-1H-инден-1-ил)метанамингидрохлорида.
В колбе, снабженной колонной Vigreaux и насадкой для молекулярной перегонки, раствор продукта из Части G (1,21 г, 4,76 ммоль) в безводном тетрагидрофуране (8 мл) нагревали до легкой дефлегмации и обрабатывали по каплям боран-диметилсульфидным комплексом (904 мкл, 723 мг, 9,52 ммоль). Полученную в результате смесь перемешивали при нагревании с обратным холодильником в течение 2 ч. Раствор охлаждали до комн. темп., и осторожно обрабатывали метанолом до прекращения выделения пузырьков газа, с последующей осторожной обработкой 4 н хлороводородом в растворе 1,4-диоксана (4 мл). Смесь затем концентрировали в вакууме. Полученное бесцветное твердое вещество растирали в порошок с эфиром и собирали фильтрованием. После высушивания в вакуумной печи при 50°C в течение 2 ч, названное соединение получали в виде бесцветного твердого вещества (893 мг, 68%).
Часть I. Получение трет-буткл (5-бром-1-метил-2,3-дигидро-1H-инден-1-ил)метил-карбамата.
Суспензию продукта из Части H (893 мг, 3,23 ммоль) в тетрагидрофуране (16 мл) обрабатывали ди-трет-бутил дикарбонатом (846 мг, 3,87 ммоль) и насыщенным раствором бикарбоната натрия (7,2 мл, ок. 6,46 ммоль) с последующим перемешиванием при комнатной температуре в течение 18 ч. Смесь разбавляли этилацетатом и экстрагировали водой и насыщенным раствором хлорида натрия. Раствор сушили (Na2SO4) и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии, элюируя 5-40% этилацетатом в гексане. Эти процедуры давали названное соединение в виде бесцветного твердого вещества (1.03 г, 94%).
Часть J. Получение трет-бутил (1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1H-инден-1-ил)метилкарбамата.
Продукт из Части I (1,03 г, 3,03 ммоль) подвергали условиям, описанным для Примера 88, Части D с получением названного соединения в виде бесцветного твердого вещества (977 мг, 83%).
Часть K. Получение трет-бутил (5-(3-отрет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1-метил-2,3-дигидро-1H-инден-1-ил)метилкарбамата.
Продукт из Части J (965 мг, 2,49 ммоль) подвергали условиям, описанным для Примера 89, Части D с получением названного соединения в виде бесцветного твердого вещества (618 мг, 47%).
Часть L. Получение N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1-метил-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида.
Продукт из Части K (446 мг, 0,84 ммоль) растворяли в 4 н хлороводороде в растворе диоксана (12 мл), с последующим перемешиванием при комнатной температуре в течение 18 ч. Суспензию полученного бесцветного твердого вещества затем концентрировали в вакууме. Это вещество суспендировали в дихлорметане (5 мл) и охлаждали до 0°C, с последующей обработкой триэтиламином (280 мкл, 203 мг, 2,01 ммоль) и метансульфонилхлоридом (81 мкл, 120 мг, 1,05 ммоль). Смесь перемешивали при 0°C в течение 1 ч и затем нагревали до комнатной температуры и разбавляли дихлорметаном. Смесь экстрагировали 1M раствором лимонной кислоты и затем сушили (Na2SO4) и концентрировали в вакууме. Остаток растворяли в 3:1 тетрагидрофуране-воде (8 мл) и обрабатывали карбонатом калия (231 мг, 1,68 ммоль) с последующим перемешиванием при комнатной температуре в течение 1 ч. Смесь концентрировали в вакууме, и остаток разбавляли водой и затем подкисляли до pH ок. 2 путем добавления 1M лимонной кислоты. Этот продукт экстрагировали этилацетатом, и органический слой экстрагировали насыщенным раствором хлорида натрия. Сушка (Na2SO4) и концентрирование в вакууме давали бесцветное твердое вещество, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя 30-100% этилацетатом в гексане. Эти процедуры давали названное соединение в виде бесцветного твердого вещества (184 мг, 43%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.39 (s, 1 H) 7.77 (d, J=7.72 Hz, 1 H) 7.14-7.48 (m, 5 H) 7.06 (t, J=6.62 Hz, 1 H) 5.63 (d, J=7.72 Hz, 1 H) 3.18-3.33 (m, 3 H) 2.96-3.15 (m, 2 H) 2.85-3.00 (m, 2 H) 2.70-2.87 (m, 3 H) 2.10-2.34 (m, 1 H) 1.63-1.90 (m, 1 H) 1.40 (s, 9 H) 1.20-1.34 (m, 3 H).
Пример 91. Получение N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1 -фтор-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида (соединение IB-L0-2.12).
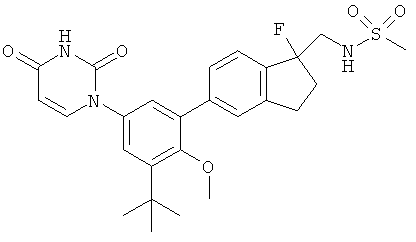
Часть A. Получение 5-(5-бром-2,3-дигидро-1H-инден-1-илиден)-2,2,3,3,7,7,8,8-октаметил-4,6-диокса-3,7-дисиланонана.
К раствору продукта из Примера 90, Части C (1,2 г, 4,98 ммоль) в безводном THF (5 мл) добавляли TBSC1 (1,726 г, 11,45 ммоль), и полученный в результате желтый раствор охлаждали до 0°C на ледяной бане. Добавляли по каплям 1,ОМ раствор LiHMDS в THF (11,95 мл, 11,95 ммоль) на протяжении 5 мин, и полученный в результате темный красный раствор перемешивали при 0°C в течение 90 мин, и затем при комнатной температуре в течение 6 ч. Растворитель удаляли в вакууме и маслянистый полутвердый остаток обрабатывали пентаном (2×35 мл) для осаждения LiCl. Суспензию фильтровали, и растворитель удаляли в вакууме с получением названного соединения в виде коричневого масла (2,3 г).
Часть B. Получение 5-бром-1-фтор-2,3-дигидро-1H-инден-1-карбоновой кислоты.
К смеси 1-хлорметил-4-фтор-1,1-диазониабицикло[2.2.2.]октан бис(тетрафтор-бората)(Selectfluor, 2,26 г, 6,37 ммоль в CH3CN (20 мл) добавляли продукт из Части A (2,3 г, 4,90 ммоль) в CH3CN (6 мл). Полученный в результате желто-оранжевый раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в 50 мл 1 н HCl (водный), экстрагировали EtOAc (2×35 мл). Объединенные органические экстракты промывают 0,5 н NaOH (3×30 мл). Объединенные водные экстракты промывают EtOAc (2×25 мл), затем pH смеси доводят до 1, используя 5 н HCl (10 мл). Полученный в результате мутный коричневый раствор экстрагировали EtOAc (2×50 мл), объединенные органические слои промывали 10% NaCl и затем обрабатывали обесцвечивающим углем, и перемешивали в течение 1 ч. Смесь сушили над безводным Na2SO4(s), фильтровали через целит и растворитель удаляли в вакууме с получением названного соединения в виде остающегося желтого масла (0,84 г).
Часть C. Получение 5-бром-1-фтор-2,3-дигидро-1H-инден-1-карбонилхлорида.
К раствору продукта из Части В (0,95 г, 3,67 ммоль) в CH2Cl2 добавляли оксалилхлорид (0,96 мл, 11,00 ммоль), с последующим добавлением DMF (0,28 мл). Полученный в результате, выделяющий пузырьки газа, раствор перемешивали при комнатной температуре в течение 2 ч, фильтровали через целит, и растворитель удаляли в вакууме с получением названного соединения в виде коричневого масла (0,99 г).
Часть D. Получение 5-бром-1-фтор-2,3-дигидро-1H-инден-1-карбоксамида.
К раствору продукта из Части C (0,99 г, 3,57 ммоль) в ацетоне (20 мл) и при 0°C добавляли водный NH4OH (28%, 0,28 мл, 3,57 ммоль), и полученную в результате темную коричневую смесь перемешивали при 0°C в течение 1 ч. Реакционную смесь концентрировали в вакууме, и остаток разделяли между водой и EtOAc (2×50 мл). Объединенные органические экстракты промывали 1N H3PO4, 10% NaHCO3 (водн), 10% NaCl, и сушили над безводным Na2SO4(s), фильтровали и концентрировали в вакууме. Твердое вещество коричневого цвета очищали с помощью колоночной хроматографии на силикагеле, используя градиент растворителей CH2Cl2/MeOH (99/1-96/4). Названное соединение получали в виде твердого вещества коричневого цвета (0,205 г, 22%).
Часть E. Получение трет-бутил (5-бром-1-фтор-2,3-дигидро-1H-инден-1-ил)метил-карбамата.
К раствору продукта из Части D (0,234 г, 0,907 ммоль) в безводном THF (5 мл) при 80°C добавляли по каплям комплекс боран-DMS (0,172 мл, 1,813 ммоль). Реакционную колбу снабжали конденсером для молекулярной перегонки, и смесь перемешивали при нагревании с обратным холодильником в течение 2 ч, собирая THF и DMS. Смесь затем охлаждали до комнатной температуры и добавляли MeOH (5 мл), с последующим добавлением 4 н HCl в 1,4-диоксане (5 мл). Растворитель удаляли в вакууме с получением бесцветного твердого вещества (0,25 г, 98%). Это твердое вещество растворяли в THF (5 мл), и к этому раствору добавляли триэтиламин (0,137 мл, 0,980 ммоль), с последующим добавлением ди-трет-бутил дикарбоната (0,214 г, 0,980 ммоль). Мутную смесь перемешивали при комнатной температуре в течение 30 мин, и добавляли 10% водн. NaHCO3 (1 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение 18 ч и затем концентрировали в вакууме до получения маслянистого остатка. Остаток растворяли в EtOAc (50 мл), промывали водой, 1 н H3PO4, 10% NaCl, и сушили над безводным Na2SO4(s). Высушивающее вещество отфильтровывали, и растворитель удаляли в вакууме с получением названного соединения в виде масла (0,27 г, 88%).
Часть F. Получение трет-бутил (1-фтор-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1H-инден-1-ил)метилкарбамата.
Продукт из Части E (0,27 г, 0,784 ммоль) подвергали условиям, описанным для Примера 72, Части B с получением названного соединения в виде твердого вещества желтовато-коричневого цвета (0,159 г, 52%).
Часть G. Получение трет-бутил (5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1-фтор-2,3-дигидро-1H-инден-1-ил)метилкарбамата.
К раствору продукта из Части F (0,159 г, 0,405 ммоль), продукта из Примера С (0,162 г, 0,405 моль), 1,3,5,7 тетраметил-2,4,8-триокса-6-фосфа-6-фениладамантана (PA-Ph, CAS 97739-46-3) (3,55 г, 0,012 ммоль) в THF (3 мл) добавляли K3PO4 (0,181 г, 0,851 ммоль) и воду (1 мл), с последующим добавлением катализатора трис(дибензилиденацетон)дипалладий(0) (3,71 мг, 0,00405 ммоль). Полученную в результате смесь дегазировали барботированием N2 в течение 20 мин, и затем перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь разбавляли EtOAc (50 мл), промывали 1 н H3PO4, 10% NaHCO3, 10% NaCl, и сушили над безводным Na2SO4(s). Смесь фильтровали, и растворитель удаляли в вакууме с получением коричневого масла, которое очищали с помощью колоночной хроматографии на силикагеле, элюируя 98/2 CH2Cl2/MeOH. Названное соединение выделяли в виде бесцветного твердого вещества (0,118 г, 54%).
Часть H. Получение N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1-фтор-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида.
Продукт из Части G (0,118 г, 0,219 ммоль) растворяли в 4 н HCl в 1,4-диоксане (2 мл) и перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли в вакууме, и остаток суспендировали в CH2Cl2 и упаривали (2×4 мл) с получением бесцветного твердого вещества (0,10 г, 96%). Это твердое вещество растворяли в CH2Cl2 (1 мл), и полученную в результате суспензию перемешивали на ледяной бане. К этой суспензии добавляли триэтиламин (0,059 мл, 0,422 ммоль), получая в результате прозрачный раствор, и к этому раствору добавляли метансульфонилхлорид (0,02 мл, 0,253 ммоль). Полученную в результате смесь перемешивали на ледяной бане в течение 1 ч. Реакционную смесь разбавляли CH2Cl2 50 мл, промывали 1 н H3PO4, 10% NaHCO3, 10% NaCl, и сушили над безводным Na2SO4(s). Высушивающее вещество отфильтровывали, и растворитель удаляли в вакууме, оставляя сырой продукт, который очищали с помощью колоночной хроматографии на силикагеле, элюируя градиентом 1:1-3:7 гексан:EtOAc. Названное соединение получали в виде бесцветного твердого вещества (64 мг, 62%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.39 (s, 1 Н) 7.77 (d, J=7.72 Hz, 1 Н) 7.30-7.48 (m, 3 Н) 7.12-7.32 (m, 3 Н) 5.63 (d, J=7.72 Hz, 1 H) 3.27 (s, 3 H) 2.94-3.08 (m, 4 H) 2.91 (s, 3 H) 2.17-2.38 (m, 1 H) 1.76-1.97 (m, 1 H) 1.40 (s, 9 H).
Пример 92. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-3,4-дигидроизохинолин-2(1H)-ил)метансульфонамида (соединение IB-L0-2.43).
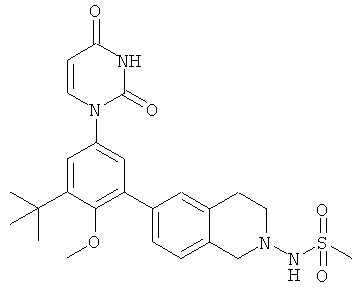
Часть A. Получение N-(3-бромфенэтил)-2,2,2-трифторацетамида.
К раствору 2-(3-бромфенил)этанамина (10 г, 50,0 ммоль) в дихлорметане (200 мл) при 0°C добавляли 2,6-лутидин (6,40 мл, 55,0 ммоль) и затем трифторацетальдегид (7,77 мл, 55,0 ммоль) по каплям, и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду при 0°C, и реакционную смесь промывали 1М HCl, H2O, и нас. NaHCO3. Органическую фазу сушили над MgSO4, фильтровали и концентрировали с получением названного соединения в виде твердого вещества желтовато-коричневого цвета (14,7 г, 99%).
Часть B. Получение 1-(6-бром-3,4-дигидроизохинолин-2(1H)-ил)-2,2,2-трифтор-этанона.
К продукту из Части A (14,70 г, 49,6 ммоль) и параформальдегиду (2,39 г, 80 ммоль) добавляли смесь уксусной кислоты (81 мл) и серной кислоты (53,7 мл) при комнатной температуре. Суспензию перемешивали в течение 60 ч, во время чего она превращалась в раствор. Реакционную смесь выливали в холодную воду. Реакционную смесь разбавляли этилацетатом и промывали водой, нас.NaHCO3, и солевым раствором. Органический слой сушили над MgSO4, фильтровали и концентрировали с получением названного соединения, загрязненного 8-бром изомером, в виде бесцветного масла (10,5 г, 67%).
Часть C. Получение 6-бром-1,2,3,4-тетрагидроизохинолина. К раствору продукта из Части B (9,5 г, 30,8 ммоль) в метаноле (231 мл) и воде (77 мл) при комнатной температуре добавляли карбонат калия (8,52 г, 61,7 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь разбавляли водой и 25% изопропанолом в хлороформе и pH доводили до 9, используя 1 н HCl. Смесь экстрагировали дважды 25% изопропанолом в хлороформе. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали с получением названного соединения, загрязненного 8-бром изомером (6,55 г, количественный).
Часть D. Получение 6-бром-2-нитрозо-1,2,3,4-тетрагидроизохинолина.
К раствору продукта из Части C (6,55 г, 30,9 ммоль) в уксусной кислоте (61,8 мл) и 3 н водн. соляной кислте (10,29 мл, 30,9 ммоль) при 0°C добавляли по каплям 1,9М нитрит натрия (20,64 мл, 39,2 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель упаривали, и реакционную смесь разбавляли 25% изопропанолом в хлороформе и нас. NaHCO3. Водный слой экстрагировали дважды 25% изопропанолом в хлороформе. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали с получением названного соединения, загрязненного 8-бром изомером (6,97 г, 94%).
Часть E. Получение 6-бром-3,4-дигидроизохинолин-2(1H)-амина.
К раствору продукта из Части D (0,5 г, 2,074 ммоль) в метаноле (4,15 мл) добавляли цинк (0,542 г, 8,30 ммоль) и реакционную смесь охлаждали до 0°C, с последующим добавлением по каплям AcOH (4,15 мл). Реакционную смесь нагревали до комн. темп., и реакционную смесь перемешивали в течение 2,5 ч. Реакционную смесь фильтровали и твердое вещество промывали метанолом. Фильтрат упаривали, и остаток разбавляли водой и добавляли 25% изопропанол в хлороформе и насыщенный NaHCO3. Твердое вещество белого цвета удаляли фильтрацией, и водный слой экстрагировали дважды 25% изопропанолом в хлороформе. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали с получением названного соединения, загрязненного 8-бром изомером (0,472 г, количественный).
Часть F. Получение трет-бутил 6-бром-3,4-дигидроизохинолин-2(1H)-илкарбамата.
Раствор продукта из Части E (0,472 г, 2,078 ммоль) в THF (20,78 мл) охлаждали до 0°C с последующим добавлением ди-трет-бутил дикарбоната (0,531 мл, 2,286 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле (выделенный низкий Rf продукт), используя градиент, начинающийся дихлорметаном и заканчивающийся 10% этилацетатом в дихлорметане с получением названного соединения (49 мг, 73%).
Часть G. Получение трет-бутил 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,4-дигидроизохинолин-2(1H)-илкарбамата.
Раствор продукта из Части F (100 мг, 0,306 ммоль), бис(пинаколато)дибора (85 мг, 0,336 ммоль), и ацетата калия (57,3 мкл, 0,917 ммоль) в 1,4-диоксане (3,0 мл) дегазировали путем барботирования газообразного N2 в течение 15 мин. Добавляли комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид дихлорметан (11,18 мг, 0,015 ммоль), и полученную в результате смесь перемешивали при 95°C в течение 16 ч. Охлажденный раствор разбавляли 25% изопропанолом в хлороформе и промывали водой. Органический слой сушили над MgSO4, фильтровали и концентрировали в вакууме. Продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя градиентом, начиная с дихлорметана и заканчивая 25% этилацетатом в дихлорметане с получением названного соединения (70 мг, 61%).
Часть H. Получение трет-бутил 6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-3,4-дигидроизохинолин-2(1H)-илкарбамата.
Смесь продукта из Примера C (74,8 мг, 0,187 ммоль), продукта из Части G (70 мг, 0,187 ммоль) в EtOH (1,0 мл), толуола (1,0 мл) 1М водн. Na2CO3 (281 мкл, 0,281 ммоль) дегазировали путем барботирования газообразного N2 в течение 10 мин. Добавляли комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II) дихлорид дихлорметан (6,84 мг, 9,35 мкмоль), и дегазирование с использованием N2 продолжали в течение 5 мин. Реакционную смесь герметично закрывали и нагревали при 78°C в течение 16 ч. Реакционную смесь охлаждали и разбавляли 25% изопропанолом в хлороформе и промывали водой. Органический слой сушили над MgSO4, фильтровали и концентрировали. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя градиентом, начиная с дихлорметана и заканчивая этилацетатом с получением названного соединения (53 мг, 54%).
Часть I. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-3,4-дигидроизохинолин-2(1H)-ил)метансульфонамида.
К раствору продукта из Части H (25 мг, 0,048 ммоль) в дихлорметане (0,5 мл) при комнатной температуре добавляли TFA (0,5 мл), и реакционную смесь перемешивали в течение 30 мин, и затем концентрировали в вакууме. Остаток разбавляли 25% изопропанолом в хлороформе и промывали нас.NaHCO3. Органический слой сушили над MgSO4, фильтровали и концентрировали с получением твердого вещества (17,8 мг, 88%). К раствору этого твердого вещества в пиридине (0,5 мл) при 0°C добавляли метансульфонилхлорид (12,6 мкл, 0,162 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Добавляли метанол и реакционную смесь перемешивали в течение 10 мин. Остаток разбавляли 25% изопропанолом в хлороформе и промывали нас. K2HCO3. Органический слой сушили над MgSO4, фильтровали и концентрировали, и этот продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя градиентом, начиная с дихлорметана и заканчивая этилацетатом, с получением названного соединения (11 мг, 52%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.39 (s, 1 H) 8.53 (s, 1 H) 7.76 (d, J=7.72 Hz, 1 H) 7.11-7.42 (m, 5 H) 5.63 (d, J=7.72 Hz, 1 H) 4.04 (s, 2 H) 3.28 (s, 3 H) 3.10 (d, J=5.52 Hz, 2 H) 2.98 (s, 3 H) 2.90-3.05 (m, 2 H) 1.40 (s, 9 H).
Пример 93. Получение N-((6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(фуран-2-ил)-2-метоксифенил)-1H-инден-3-ил)метил)метансульфонамида (соединение IB-L0-2.65).
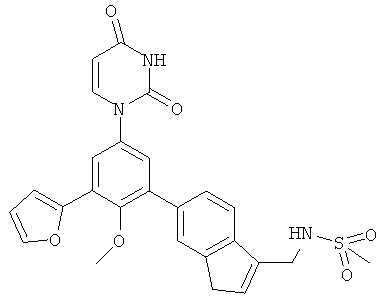
Часть A. Получение N-((6-(3-бром-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1H-инден-3-ил)метил)метансульфонамида.
Продукт из Примера 48, Части C (0,242 г, 0,573 ммоль) и продукт из Примера 79, Части E (0,200 г, 0,57 ммоль) подвергали условиям, описанным для Примера 79, Части F с получением названного соединения в виде беловатого твердого вещества (0,104 г, 35%).
Часть B. Получение N-((6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(фуран-2-ил)-2-метоксифенил)-1H-инден-3-ил)метил)метансульфонамида.
Раствор продукта из Части A (25,2 мг, 0,049 ммоль) в смеси 3:1 об/об THF-вода (1,3 мл) объединяли в микроволновой трубке при комнатной температуре с фуран-2-илбороновой кислотой (6,91 мг, 0,062 ммоль) и фосфатом калия (16,84 мг, 0,097 ммоль). К этому раствору добавляли 1,1'-бис(ди-трет-бутил-фосфино)ферроцен палладий дихлорид (1,65 мг, 2,53 мкмоль). Трубку герметично закрывали, и полученную в результате смесь продували азотом в течение 4 мин, и затем нагревали в течение 16,5 ч на масляной бане при 50°C. Реакционную смесь распределяли между разбавленной HCl и этилацетатом, и органическую фазу сушили (MgSO4) и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (этилацетат-гексан) с получением названного соединения в виде беловатого твердого вещества (11,4 мг, 46%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.45 (s, 1 H) 7.80-7.89 (m, 2 H) 7.73-7.79 (m, 2 H) 7.56-7.63 (m, 2 H) 7.50 (t, J=6.07 Hz, 1 H) 7.38 (d, J=2.94 Hz, 1 H) 7.09 (d, J=3.31 Hz, 1 H) 6.68 (dd, J=3.68, 1.84 Hz, 1 H) 6.58 (s, 1 H) 5.68 (d, J=7.72 Hz, 1 H) 4.19 (d, J=5.15 Hz, 2 H) 3.48 (s, 2 H) 3.34 (s, 3 H) 2.96 (s, 3 H).
Пример 94. Получение N-((6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(тиофен-2-ил)фенил)-1H-инден-3-ил)метил)метансульфонамида (соединение IB-L0-2.63).
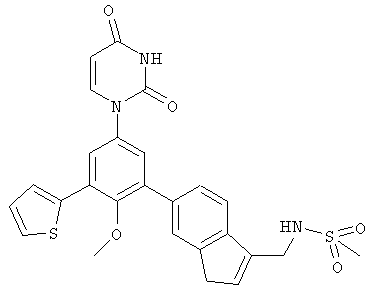
Продукт из Примера 93, Части A (26,5 мг, 0,051 ммоль) взаимодействовал с тиофен-2-ил бороновой кислотой (8,3 мг, 0,065 ммоль), как описано в Примере 93, Части B с получением названного соединения в виде беловатого твердого вещества (8,6 мг, 32%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.47 (s, 1 H) 7.86 (d, J=7.72 Hz, 2 H) 7.55-7.78 (m, 5 H) 7.50 (t, J=6.25 Hz, 1 H) 7.38 (d, J=2.57 Hz, 1 H) 7.16-7.21 (m, 1 H) 6.58 (s, 1 H) 5.69 (d, J=7.72 Hz, 1 H) 4.19 (d, J=4.78 Hz, 2 H) 3.48 (s, 2 H) 3.30 (s, 3 H) 2.96 (s, 3 H).
Пример 95. Получение N-((6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(тиофен-3-ил)фенил)-1H-инден-3-ил)метил)метансульфонамида (соединение IB-L0-2.62).
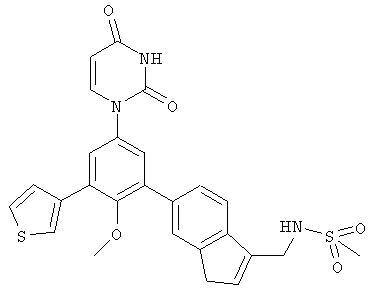
Продукт из Примера 93, Части A (25,9 мг, 0,050 ммоль) взаимодействовал с тиофен-3-ил бороновой кислотой (8,1 мг, 0,063 ммоль), как описано в Примере 93, Части B с образованием названного соединения в виде off-solid (8,6 мг, 33%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.45 (d, J=1.84 Hz, 1 H) 7.93 (d, J=2.94 Hz, 1 H) 7.87 (d, J=7.72 Hz, 1 H) 7.53-7.75 (m, 6 Н) 7.49 (t, J=6.25 Hz, 1 H) 7.39 (d, J=2.57 Hz, 1 H) 6.57 (s, 1 H) 5.68 (dd, J=7.91, 2.02 Hz, 1 H) 4.19 (d, J=5.15 Hz, 2 H) 3.47 (s, 2 H) 3.21 (s, 3 H) 2.96 (s, 3 H).
Пример 96. Получение N-((6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(фуран-3-ил)-2-метоксифенил)-1H-инден-3-ил)метил)метансульфонамида (соединение IB-L0-2.67).
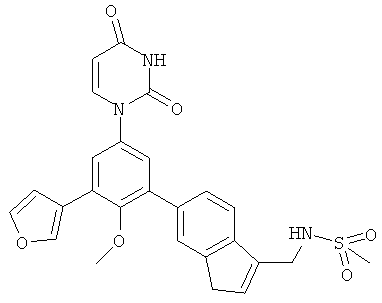
Продукт из Примера 93, Части A (25,9 мг, 0,050 ммоль) взаимодействовал с фуран-3-ил бороновой кислотой (7,2 мг, 0,064 ммоль), как описано в Примере 93, Части B с получением названного соединения в виде беловатого твердого вещества (10,6 мг, 45%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.46 (s, 1 H) 7.84 (d, J=8.09 Hz, 1 H) 7.80 (t, J=1.84 Hz, 1 H) 7.68-7.75 (m, 2 H) 7.54-7.64 (m, 2 H) 7.50 (t, J=6.07 Hz, 1 H) 7.35 (d, J=2.57 Hz, 1 H) 7.08 (d, J=1.47 Hz, 1 H) 6.57 (s, 1 H) 5.68 (d, J=8.09 Hz, 1 H) 3.47 (s, 2 H) 3.30 (s, 3 H) 2.96 (s, 3 H).
Пример 97. Получение 1-(3-трет-бутил-4-метокси-5-(1-(метилсульфонил)индолин-5-ил)фенил)пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.32).
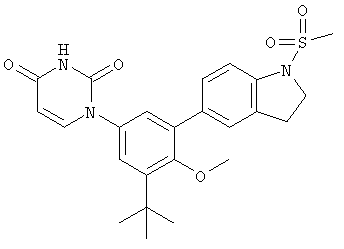
Часть A. Получение 5-бром-1-(метилсульфонил)индолина.
К DMF (5,0 мл) добавляли гидрид натрия (53 мг, 1,3 ммоль) и раствор перемешивали при комнатной температуре в течение 30 мин. Добавляли 5-броминдолин (240 мг, 1,2 ммоль), и этот раствор перемешивали при комнатной температуре в течение 30 мин. Добавляли метансульфонилхлорид (94 мкл, 1,2 ммоль), и этот раствор перемешивали при комнатной температуре в течение ночи, затем концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 2% CH3OH/CHCl3 с получением названного соединения (202 мг, 60%).
Часть B. Получение 1-(метилсульфонил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)индолина.
Продукт из Части A (192 мг, 0,70 ммоль) подвергали условиям, описанным для Примера 72, Части B с получением названного соединения (114 мг, 51%).
Часть C. Получение 1-(3-трет-бутил-4-метокси-5-(1-(метилсульфонил)индолин-5-ил)фенил) пиримидин-2,4(1H,3H)-диона.
Продукт из Примера C (58 мг, 0,145 ммоль) и продукт из Части B (56,2 мг, 0,174 ммоль) подвергали условиям, описанным для Примера 72, Части C с получением названного соединения в виде бесцветного твердого вещества (12 мг, 18%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.40 (d, J=1.84 Hz, 1 H) 7.76 (d, J=7.72 Hz, 1 H) 7.53-7.67 (m, 1 H) 7.45 (s, 1 H) 7.32-7.41 (m, 2 H) 7.23 (dd, J=13.60, 2.57 Hz, 2 H) 5.63 (dd, J=8.09, 2.21 Hz, 1 H) 3.99 (t, J=8.46 Hz, 2 H) 3.29 (s, 3 H) 3.18 (t, J=8.46 Hz, 2 H) 3.04 (s, 3 H).
Пример 98. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)хиноксалин-2-ил)метансульфонамида (соединение IB-L0-2.26).
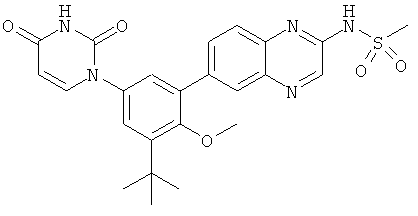
Часть A. Получение N-(4-бром-2-нитрофенил)-3-оксобутанамида.
Раствор дикетена (0,32 мл, 4,15 ммоль) в толуоле (2 мл) добавляли к 80°C раствору 4-бром-2-нитроанилина (900 мг, 4,15 ммоль) в толуоле (7 мл), и этот раствор нагревали с обратным холодильником в течение 5 ч. Добавляли триэтиламин (0,58 мл, 4,15 ммоль) в толуоле (2 мл), и нагревание с обратным холодильником продолжали в течение 30 мин. Охлажденный раствор концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 2:1 гексаном/EtOAc с получением названного соединения в виде твердого вещества желтого цвета (920 мг, 74%).
Часть B. Получение 6-бромхиноксалин-2(1H)-она.
К раствору гидроксида натрия (337 мг, 8,4 ммоль) в H2O (2.1 мл) добавляли продукт из Части A (423 мг, 1,4 ммоль), и перемешивание продолжали при 65°C в течение 1 ч. Охлажденный раствор разбавляли H2O (4 мл) и добавляли боргидрид натрия (31,9 мг, 0,84 ммоль) и перемешивание продолжали при комнатной температуре в течение 1,5 ч. В этот раствор добавляли лед, с последующим добавлением по каплям 6 н HCl до подкисления. Полученное в результате твердое вещество собирали фильтрацией, промывали H2O, и сушили в вакуумной печи с получением названного соединения (273 мг, 86%).
Часть C. Получение 6-бром-2-хлорхиноксалина.
В колбу, содержащую хлорангидрид фосфорной кислоты (3,4 мл, 36,5 ммоль) добавляли продукт из Части B (255 мг, 1,1 ммоль), и этот раствор нагревали при 60°C в течение ночи. Раствор охлаждали до комнатной температуры, выливали на лед и полученное в результате твердое вещество собирали фильтрованием с получением названного соединения (239 мг, 87%).
Часть D. Получение 6-бром-N-(4-метоксибензил)хиноксалин-2-амина.
К раствору продукта из Части C (2,8 г, 11,5 ммоль) в этаноле (58 мл) добавляли (4-метоксифенил)метанамин (7,5 мл, 57,5 ммоль), и этот раствор перемешивали при комнатной температуре в течение 1 ч. Растворитель концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 20% EtOAc/гексаном с получением названного соединения (1,97 г, 50%).
Часть E. Получение N-(4-метоксибензил)-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиноксалин-2-амина.
Продукт из Части D (500 мг, 1,45 ммоль) подвергали условиям, описанным для Примера 72, Части B с получением названного соединения (378 мг, 66%).
Часть F. Получение 1-(3-трет-бутил-4-метокси-5-(2-(4-метоксибензиламино)хиноксалин-6-ил)фенил)пиримидин-2,4(1H,3H)-диона.
Продукт из Части E (133 мг, 0,34 ммоль) подвергали условиям, описанным для Примера 72, Части C с получением названного соединения (125 мг, 82%).
Часть G. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)хиноксалин-2-ил)метансульфонамида.
К раствору продукта из Части F (87 мг, 0,16 ммоль) в CH2Cl2 (1,6 мл) и H2O (0,07 мл) добавляли DDQ (40,4 мг, 0,18 ммоль) и интенсивно перемешивали при комнатной температуре в течение 1 ч. Раствор фильтровали через целит, и темное твердое вещество собранное на целите, растворяли в 5 мл CH3OH. Метанольный раствор фильтровали, растворитель удаляли в вакууме, и сырое промежуточное соединение растворяли в пиридине (0,6 мл). Добавляли метансульфонилхлорид (11 мкл, 0,14 ммоль) и этот раствор нагревали при 60°C в течение ночи. Охлажденный раствор концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 2% CH3OH/CHCl3 с получением названного соединения (7,7 мг, 12%). 1H ЯМР (300 MHz, CDCl3) δ 8.42 (s, 1 H) 8.29 (s, 1 H) 8.13 (s, 1 H) 7.88 (d, 1 H) 7.54 (s, 1 H) 7.19-7.43 (m, 4 H) 5.83 (dd, J=7.91, 2.39 Hz, 1H) 3.32 (s, 3 H) 3.27 (s, 3 H) 1.46 (s, 9 H).
Пример 99. Получение N-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)метансульфонамида (соединение IB-L0-2.44).
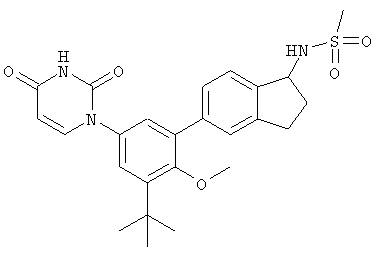
Часть A. Получение 5-бром-2,3-дигидро-1H-инден-1-ола.
Суспензию 5-бром-2,3-дигидро-1H-инден-1-она (2,07 г, 9,81 ммоль) в этаноле (49 мл) обрабатывали боргидридом натрия (186 мг, 4,90 ммоль) одновременно. Через несколько минут, раствор слегка нагревался, и все твердые вещества растворялись. После перемешивания при комнатной температуре в течение 1 ч, смесь концентрировали в вакууме для удаления этанола. Полученное смолистое выделение распределяли между этилацетатом и водой. Органический слой экстрагировали насыщенным раствором бикарбоната натрия (2×) и насыщенным раствором хлорида натрия. Сушка (Na2SO4) и концентрирование в вакууме давали названное соединение (3,05 г, 98%) в виде бесцветного масла, которое кристаллизовалось при прокачке под высоким вакуумом в течение ночи.
Часть B. Получение 1-азидо-5-бром-2,3-дигидро-1H-индена.
Раствор продукта из Части A (1,01 г, 4,73 ммоль) в толуоле (8,1 мл) обрабатывали дифенилфосфороилазидом (1,23 мл, 1,56 г, 5,67 ммоль) с последующим охлаждением до 0°C. Раствор обрабатывали по каплям DBU (855 мкл, 863 мг, 5,67 ммоль) с последующим перемешиванием при 0°C в течение 2 ч, и затем нагреванием до комнатной температуры в течение 48 ч. Смесь разбавляли этилацетатом и экстрагировали водой и 1 M раствором лимонной кислоты, и затем насыщенным раствором хлорида натрия. Сушка (Na2SO4) и концентрирование в вакууме давали коричневое масло, которое очищали с помощью флэш-хроматографии, элюируя 5-50% этилацетатом в гексане. Эти процедуры давали названное соединение (889 мг, 79%) в виде светло-желтого масла.
Часть C. Получение 5-бром-2,3-дигидро-1H-инден-1-амина.
К -15°C раствору 1М литий алюминий гидрида в THF (0,84 мл, 0,84 ммоль) в THF (0,88 мл) добавляли по каплям раствор продукта из Части B (200 мг, 0,84 ммоль), и этот раствор нагревали до комнатной температуры и перемешивали в течение ночи. Раствор охлаждали до -10°C и 4:1 THF:H2O (0,5 мл) добавляли по каплям. Раствор перемешивали при комнатной температуре в течение 4 ч, фильтровали через целит, и фильтрат концентрировали в вакууме с получением названного соединения (151 мг, 85%).
Часть D. Получение N-(5-бром-2,3-дигидро-1H-инден-1-ил)метансульфонамида.
К раствору продукта из Части C (150 мг, 0,71 ммоль) в пиридине (3,5 мл) добавляли метансульфонилхлорид (61 мкл, 0,78 ммоль), и этот раствор перемешивали при комнатной температуре в течение ночи. Раствор концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 20% EtOAc/гексаном с получением названного соединения (111 мг, 54%).
Часть E. Получение N-(5-{3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)метансульфонамида.
Продукт из Части D (109 мг, 0,38 ммоль) подвергали условиям, описанным для Примера 72, Части B и Части C с получением названного соединения (39 мг, 60%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.39 (d, J=1.84 Hz, 1 Н) 7.77 (d, J=7.72 Hz, 1 Н) 7.58 (d, J=8.82 Hz, 1 Н) 7.39-7.48 (m, 3 Н) 7.27 (d, J=2.57 Hz, 1 Н) 7.19-7.23 (m, 1 Н) 5.63 (dd, J=8.09, 2.21 Hz, 1 Н) 4.86 (q, J=7.97 Hz, 1 Н) 3.27 (s, 3 Н) 3.04 (s, 3 Н) 2.90-3.01 (m, 1 Н) 2.71-2.90 (m, 1 H) 2.52-2.62 (m, 1 Н) 1.85-1.98 (m, 1 H) 1.40 (s, 9 H).
Пример 100. Получение N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида (соединение IB-L0-2.17).
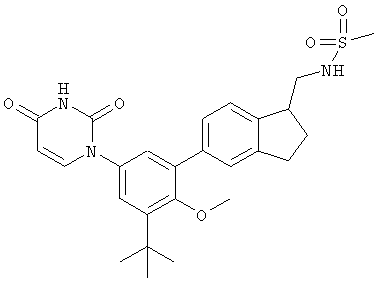
Часть A. Получение (E)-5-бром-1-(метоксиметилен)-2,3-дигидро-1H-индена.
К суспензии хлорида (метоксиметил)трифенилфосфония (39,7 г, 116 ммоль) в THF (210 мл) при -20°C добавляли по каплям 1М т-бутоксид калия (95 мл, 95 ммоль), и этот раствор перемешивали при -20°C в течение 20 мин. К этому раствору добавляли по каплям раствор 5-бром-2,3-дигидро-1H-инден-1-она (10,0 г, 47,4 ммоль) в THF (230 мл) и перемешивание продолжали при -20°C в течение 30 мин, затем нагревали до комнатной температуры и перемешивали в течение 2 ч. Раствор фильтровали через целит, и фильтрат концентрировали в вакууме с получением сырого продукта, который очищали хроматографией на картридже с силикагелем, элюируя CH2Cl2/гексаном, с получением названного соединения (10,56 г, 93%).
Часть B. Получение 5-бром-2,3-дигидро-1H-инден-1-карбальдегида.
К раствору продукта из Части A (1,44 г, 6,0 ммоль) в CH2Cl2 (30 мл) при -78°C добавляли по каплям 1M трехбромистый бор в CH2Cl2 (13,8 мл, 13,8 ммоль) и перемешивание продолжали при -78°C в течение 4 ч. Раствор выливали в смесь льда и насыщенного бикарбоната натрия и интенсивно перемешивали. Слои отделяли, и водный слой экстрагировали CH2Cl2 (2×), органические экстракты объединяли, сушили (Na2SO4), и концентрировали в вакууме с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле, элюируя 10% EtOAc/гексаном с получением названного соединения (604 мг, 45%).
Часть C. Получение 1-(5-бром-2,3-дигидро-1H-инден-1-ил)-N-(4-метоксибензил)-метанамина.
К раствору продукта из Части B (300 мг, 1,3 ммоль) в CH3OH (18,5 мл) добавляли 4-метоксибензиламин (0,17 мл, 1,3 ммоль) и декаборан (49 мг, 0,4 ммоль) и перемешивание продолжали при комнатной температуре в течение 1 ч, растворитель концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 3% CH3OH/CHCl3 с получением названного соединения (264 мг, 57%).
Часть D. Получение N-((5-бром-2,3-дигидро-1H-инден-1-ил)метил)-N-(4-метокси-бензил)метансульфонамида.
К раствору продукта из Части C (88 мг, 0,25 ммоль) в CH2Cl2 (1,0 мл) добавляли триэтиламин (39 мкл, 0,28 ммоль) и метансульфонилхлорид (22 мкл, 0,28 ммоль) и перемешивание продолжали при комнатной температуре в течение 1 ч, растворитель концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя EtOAc/гексаном с получением названного соединения (55 мг, 51%).
Часть E. Получение N-(4-метоксибензил)-N-((5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида.
Продукт из Части D (1,15 г, 2,71 ммоль) подвергали условиям, описанным для Примера 72, Части B с получением названного соединения (840 мг, 66%).
Часть F. Получение N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида.
Продукт из Части E (840 мг, 2,1 ммоль) подвергали условиям, описанным для Примера 72, Части C, и выделенное вещество (1,28 г, 2,07 ммоль) растворяли в CH2Cl2 (10 мл) и медленно добавляли трифторуксусную кислоту (10 мл). После перемешивания при комнатной температуре в течение 1 ч, растворитель концентрировали в вакууме, и сырой продукт суспендировали в 10% NaHCO3, экстрагировали CH2Cl2 (3×), органические экстракты объединяли, сушили (Na2SO4), и растворитель концентрировали в вакууме с получением сырого продукта, который очищали с помощью колоночной хроматографии на силикагеле, элюируя 2% CH3OH/CHCl3 с получением названного соединения (0,84 г, 81%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.39 (s, 1 H) 7.77 (d, J=8.09 Hz, 1 H) 7.29-7.59 (m, 3 H) 7.25 (d, J=2.94 Hz, 1 H) 7.10-7.22 (m, 2 H) 5.63 (dd, J=7.72, 1.84 Hz, 1 H) 3.93 (s, 3 H) 3.26 (s, 2 H) 3.23-3.40 (m, 1 H) 2.89 (s, 3 H) 2.71-3.09 (m, 2 H) 2.14-2.32 (m, 1 H) 1.75-1.95 (m, 1 H) 1.40 (s, 9 H).
Пример 101. Получение 5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-N-(метилсульфонил)-2,3-дигидро-1H-инден-1-карбоксамида (соединение IB-L0-2.34).
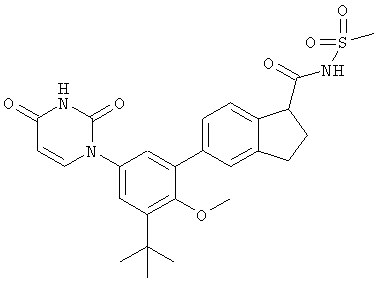
Часть A. Получение 5-бром-2,3-дигидро-1H-инден-1-карбоновой кислоты.
К раствору продукта из Примера 100, Части B (300 мг, 1,3 ммоль) и 2-метил-2-пентена (8 мл) в трет-бутаноле (32 мл) добавляли раствор хлорита натрия (1,36 г, 0,12 ммоль) в H2O (12 мл) содержащего однозамещенный фосфорнокислый натрий (1,07 г, 8,9 ммоль), и смесь интенсивно перемешивали в течение 20 мин при комнатной температуре. Растворители концентрировали в вакууме, и остаток разбавляли H2O, экстрагировали EtOAc (3×), экстракты объединяли, сушили (Na2SO4), и концентрировали в вакууме с получением названного соединения (180 мг, 56%).
Часть B. Получение 5-бром-N-(метилсульфонил)-2,3-дигидро-1H-инден-1-карбоксамида.
К раствору продукта из Части A (100 мг, 0,42 ммоль) в CH2Cl2 (1,7 мл) добавляли карбонилдиимидазол (67,3 мг, 0,42 ммоль) и реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Добавляли метансульфонамид (39,5 мг, 0,42 ммоль) и DBU (62,5 мг, 0,42 ммоль) и перемешивание продолжали при комнатной температуре в течение 2 ч. Раствор разбавляли CH2Cl2, промывали 1 н HCl, солевым раствором, сушили (Na2SO4), концентрировали в вакууме, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя 20% EtOAc/гексаном с получением названного соединения (121 мг, 92%).
Часть C. Получение N-(метилсульфонил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2,3 -дигидро-1H-инден-1-карбоксамида.
Продукт из Части B (159 мг, 0,5 ммоль) подвергали условиям, описанным для Примера 72, Части B с получением названного соединения (144 мг, 79%).
Часть D. Получение 5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-N-(метилсульфонил)-2,3-дигидро-1H-инден-1-карбоксамида.
Продукт из Части C (134 мг, 0,34 ммоль) подвергали условиям, описанным для Примера 72, Части C с получением названного соединения (14 мг, 8%). 1H ЯМР (300 MHz, CDCl3) δ 8.11 (m, 1 H) 7.08-7.57 (m, 7 H) 5.80 (dd, J=7.91, 2.39 Hz, 1 H) 4.07 (dd, J=9.01, 6.07 Hz, 1 H) 3.33 (s, 3 H) 3.08 (s, 3 H) 2.91-3.22 (m, 1 H) 2.35-2.74 (m, 1 H) 1.44 (s, 9H) 1.17-1.34 (m, 1 H) 0.60-1.00 (m, 1 H).
Пример 102. Получение 1-(3-(2-аминобензо[d]тиазол-6-ил)-5-трет-бутил-4-метокси-фенил)пиримидин-2,4(1H,3H)-диона (соединение IB-L0-2.39).
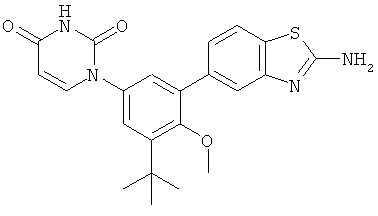
Названное соединение получали, используя процедуры, описанные для получения Примера 83, заменяя 5-бром[d]тиазол-2-амин на 6-бромбензо[d]тиазол-2-амин. 1H ЯМР (300 MHz, DMSO-d6) δ 11.40 (d, J=1.84 Hz, 1 H) 8.40 (s, 2 H) 7.84 (d, J=8.09 Hz, 1 H) 7.78 (d, J=7.72 Hz, 1 H) 7.54 (d, J=1.47 Hz, 1 H) 7.27-7.32 (m, 3 H) 5.64 (dd, J=8.09, 2.21 Hz, 1 H) 3.27 (s, 3 H) 1.41 (s, 9 H).
Пример 103. Получение N-(2-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)пропан-2-ил)метансульфонамида (соединение IB-L0-2.29).
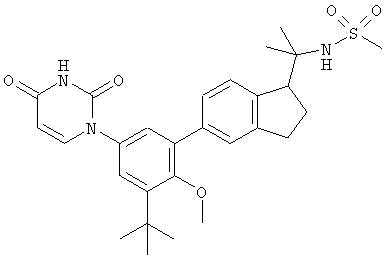
К раствору продукта из Примера 75, Части D (20 мг, 0,038 ммоль) в смеси 1:1 бензол:MeOH (0,6 мл) добавляли оксид платины (IV) (1 мг). Полученную в результате смесь перемешивали под давлением 1 атм H2 при комнатной температуре в течение 1 ч, и затем фильтровали через целит, и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CHCl3 в качестве элюента, с получением названного соединения в виде твердого вещества (14 мг, 70%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.39 (s, 1 H) 7.77 (d, J=7.72 Hz, 1 H) 7.58 (d, J=8.09 Hz, 1 H) 7.28-7.38 (m, 2 H) 7.21-7.26 (m, 2 H) 7.07 (s, 1 H) 5.63 (d, J=7.72 Hz, 1 H) 3.61 (dd, J=8.64, 5.33 Hz, 1 H) 3.25 (s, 3 H) 3.00 (s, 3 H) 2.75-2.98 (m, 2 H) 1.97-2.21 (m, 2 H) 1.40 (s, 9 H) 1.24 (d, J=8.46 Hz, 6 H).
Пример 104. Получение (S)-N-(2-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)пропан-2-ил)метансульфонамида (соединение IB-L0-2.22).
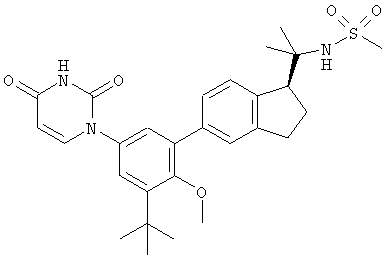
Продукт из Примера 103 (10 мг) подвергали хиральной хроматографии (колонка Chiralpak AD-H; элюируя 1:3 2-PrOH:гексаном (0,1% TFA)). Выделение компонента ранней элюции давало названное соединение (4,4 мг). 1H ЯМР идентичен продукту из Примера 103.
Пример 105. Получение (R)-N-(2-(5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)пропан-2-ил)метансульфонамида (соединение IB-L0-2.37).
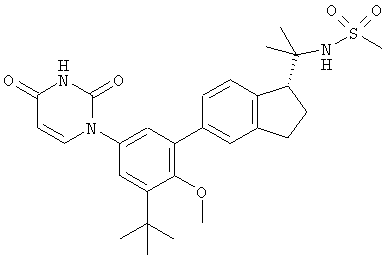
Продукт из Примера 103 (10 мг) подвергали хиральной хроматографии (колонка Chiralpak AD-H; элюируя 1:3 2-PrOH:гексаном (0,1% TFA)). Выделение компонента поздней элюции давало названное соединение (4,2 мг). 1H ЯМР идентичен продукту из Примера 103.
Пример 106. Получение (S)-N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида (соединение IB-L0-2.9).
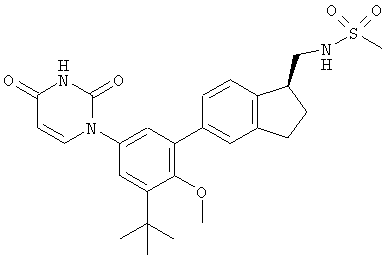
Продукт из Примера 100, Части F (20 мг) подвергали хиральной хроматографии (колонка Chiralpak AD-H; элюируя 1:4 2-PrOH:гексаном (0,1% TFA)). Выделение компонента ранней элюции давало названное соединение (5,3 мг). 1H ЯМР идентичен продукту из Примера A-100, Части F.
Пример 107. Получение (R)-N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида (соединение IB-L0-2.15).
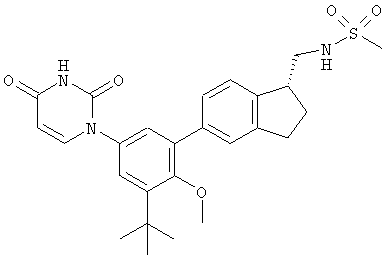
Продукт из Примера 100, Части F (20 мг) подвергали хиральной хроматографии (колонка Chiralpak AD-H; элюируя 1:4 2-PrOH:гексаном (0,1% TFA)). Выделение компонента поздней элюции давала названное соединение (5,7 мг). 1H ЯМР идентичен продукту из Примера 100, Части F.
Пример 108. Получение (S)-N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1-фтор-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида (соединение IB-L0-2.20).
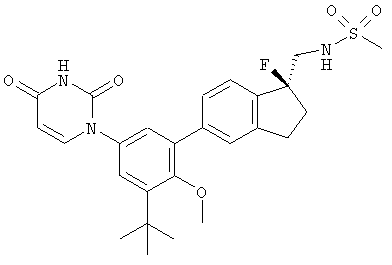
Продукт из Примера 91, Часть Н подвергали условиям, описанным в Примере 104 с получением названного соединения. 1H ЯМР идентичен продукту из Примера 91, Части H.
Пример 109. Получение (R)-N-((5-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1-фтор-2,3-дигидро-1H-инден-1-ил)метил)метансульфонамида (соединение IB-L0-2.10).
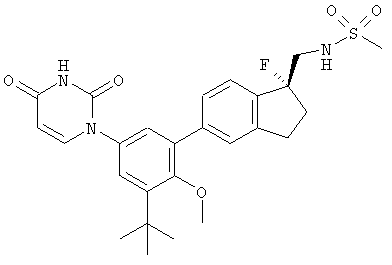
Продукт из Примера 91, Части Н подвергали условиям, описанным в Примере 104 с получением названного соединения. 1H ЯМР идентичен продукту из Примера 91, Часть H.
Пример 110. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-трет-пентилфенил)нафтален-2-ил)метансульфонамида (соединение IB-L0-2.52).
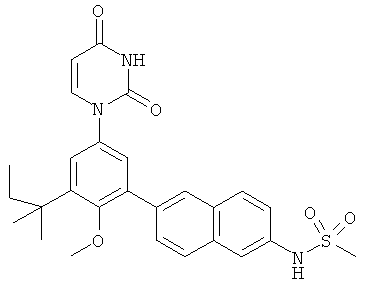
Часть A. Получение 1-(3-трет-бутил-5-йод-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
2-трет-амилфенол (5,0 г, 30 ммоль) вступал в реакцию в соответствии с процедурой из Примера C, Части A, Части B, и Части C с образованием названного продукта в виде бесцветного твердого вещества (6,7 г, 56% суммарный выход для 3 стадий).
Часть B. Получение N-(6-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-трет-пентилфенил)нафтален-2-ил)метансульфонамида.
Продукт из Части A (100 мг, 0,241 ммоль), продукт из Примера 4A, Части B (92 мг, 0,266 ммоль), карбонат натрия (38,4 мг, 0,362 ммоль) и комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(П)дихлорид дихлорметан (9,9 мг, 0,012 ммоль) растворяли в смеси растворителей толуол (4 мл) и этанол (4 мл), которую барботировали азотом в течение 10 мин, затем смесь нагревали до 85°C в течение 18 ч. К этому раствору затем добавляли CH2Cl2 (20 мл) с последующим добавлением 1 н водного HCl (10 мл), органический слой отделяли 3-меркаптопропил силикагелем (100 мг) и добавляли сульфат магния. Раствор концентрировали и очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения в виде бесцветного твердого вещества (71 мг, 58%). 1H ЯМР (300 MHz, DMSO-d6): δ 11.41 (s, 1H), 10.04 (s, 1H), 8.03 (s, 1H), 7.95 (t, J=8.7 Hz, 2H), 7.79 (d, J=7.7 Hz, 1H), 7.73 (d, J=1.8 Hz, 1 H), 7.69 (dd, J=8.8, 1.6 Hz, 1 H), 7.42 (dd, J=8.8, 2.2 Hz, 1H), 7.37 (d, J=2.6 Hz, 1H), 7.25 (d, J=2.6 Hz, 1H), 5.65 (dd, J=8.1, 1.6 Hz, 1H), 3.22 (s, 3H), 3.08 (s, 3H), 1.84 (m, 2H), 1.38 (s, 6H), 0.73 (t, J=7.5 Hz, 3H).
Пример 111. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1H-инден-3-ил)метил)-N-метилметансульфонамида (соединение IB-L0-2.16).
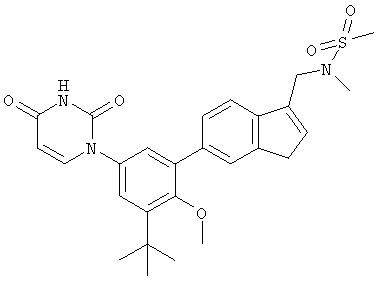
Часть A. Получение N-метил-N-((6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-инден-3-ил)метил)метансульфонамида.
К раствору продукта из Примера 79, Части E (210 мг, 0,60 ммоль) в безводном THF (5 мл) добавляли 1,0M раствор литий бис(триметилсилил)амида в толуоле (0,60 мл, 0,60 ммоль), и полученную в результате смесь перемешивали при комнатной температуре в течение 5 мин. Добавляли йодметан (0,075 мл, 1,20 ммоль), и смесь перемешивали при комнатной температуре в течение 2 ч, и распределяли между этилацетатом и водой. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя градиентом этилацетата/в гексане (10%-25%) с получением названного соединения в виде твердого вещества (125 мг, 57%).
Часть B. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-1H-инден-3-ил)метил)-N-метилметансульфонамида.
Смесь продукта из Примера C (60,0 мг, 0,15 ммоль), продукта Части A (54,5 мг, 0,15 ммоль), фосфата калия (66,9 мг, 0,315 ммоль), PA-Ph (CAS 97739-46-3, 1,32 мг, 4,5 мкмоль) и трис(дибензилиденацетон)дипалладия(0) (1,37 мг, 1,5 мкмоль) в тетрагидрофуране (3,0 мл) и воде (1,0 мл) продували N2 в течение 30 мин. Смесь перемешивали при 50°C в течение 2 ч, и затем распределяли между этилацетатом и 1M HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали через целит и концентрировали в вакууме. Сырой продукт очищали колоночной хроматографией с обращенной фазой на силикагеле C-18, используя градиент растворителей 10-100% ацетонитрила в воде(0,1% TFA) с получением названного соединения в виде твердого вещества (19 мг, 24%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.40 (d, J=1.84 Hz, 1 Н) 7.78 (d, J=7.72 Hz, 1 Н) 7.65 (m, 2 Н) 7.49 (dd, J=7.72, 1.47 Hz, 1 Н) 7.26 (m, 2.57 Hz, 2 Н) 6.63 (s, 1 Н) 5.64 (dd, J=7.72, 2.21 Hz, 1 Н) 4.26 (s, 2 Н) 3.51 (s, 2 Н) 3.26 (s, 3 Н) 3.01 (s, 3 Н) 2.72 (s, 3 Н) 1.41 (s, 9 Н).
Пример 112. Получение N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[b]тиофен-2-ил)метил)метансульфонамида (соединение IB-L0-2.40).
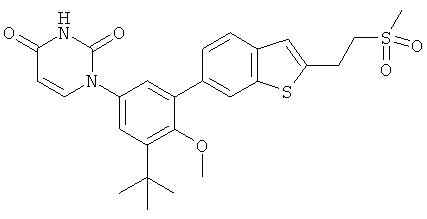
Часть A. Получение этил 6-бромбензо[6]тиофен-2-карбоксилата.
К раствору 4-бром-2-фторбензальдегида (1,02 г, 4,83 ммоль) в DMSO (4 мл), добавляли этил 2-меркаптоацетат (0,58 мл, 5,31 ммоль), с последующим добавлением Et3N (1,35 мл, 9,65 ммоль), и смесь нагревали при 80°C в течение 3 ч. Полученную в результате темную смесь выливали в воду (50 мл) и экстрагировали EtOAc (2×50 мл). Объединенные органические экстракты промывали 10% NaCl, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме с получением названного соединения в виде светлого желтого воскообразного твердого вещества (1,29 г, 94%).
Часть B. Получение (6-бромбензо[b]тиофен-2-ил)метанола.
К раствору продукта из Части A (0,82 г, 2,88 ммоль) в Et2O (20 мл) при 0°C добавляли по каплям 1М раствор литий алюминий гидрида в Et2O (3,16 мл, 3,16 ммоль), и полученную в результате суспензию перемешивали между 5 и 10°C в течение 1 ч. Эту суспензию обрабатывали 0,3 мл H2O, 0,3 мл 15% водн. NaOH, 0,7 мл H2O, перемешивали 30 мин, фильтровали и концентрировали в вакууме с получением названного соединения в виде бесцветного твердого вещества (0,58 г, 83%).
Часть C. Получение 6-бром-2-(бромметил)бензо[b]тиофена.
Смесь продукта из Части B (85 мг, 0,35 ммоль), N-бромсукцинимида (74 мг, 0,413 ммоль) и трифенилфосфина (106 мг, 0,403 ммоль) в CH2Cl2 (2 мл) перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли 50 мл CH2Cl2, промывали водой, 10% NaHCO3 и 10% NaCl, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя 9:1 гексаном:EtOAc с получением на выходе названного соединения в виде твердого вещества белого цвета (96 мг, 89%).
Часть D. Получение N-(4-метоксибензил)метансульфонамида.
К раствору (4-метоксифенил)метанамина (1,317 г, 9,60 ммоль) в CH2Cl2 (10 мл) добавляли по каплям метансульфонилхлорид (0,34 мл, 4,36 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли 50 мл CH2Cl2, промывали 1 н H3PO4, 10% NaCl, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме с получением названного соединения в виде твердого вещества белого цвета (0,84 г, 89%).
Часть E. Получение N-((6-бромбензо[b]тиофен-2-ил)метил)-N-(4-метоксибензил)-метансульфонамида.
Раствор продукта из Части D (0,223 г, 1,037 ммоль) в EtOH (2 мл) и 1,0М NaOH (1,1 мл, 1,1 ммоль) добавляли к суспензии, содержащей продукт из Части С (0,317 г, 1,037 ммоль) в EtOH (4 мл). Полученную в результате суспензию нагревали с обратным холодильником в течение 1 ч, и затем концентрировали в вакууме с получением пастообразного твердого вещества. Остаток распределяли между 40 мл воды и 40 мл EtOAc. Органический слой промывали 1 н H3PO4, 10% NaHCO3, 10% NaCl, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме, с получением желтого масла. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя CH2Cl2 с получением названного соединения в виде бесцветного твердого вещества (0,15 г, 33%).
Часть F. Получение N-(4-метоксибензил)-N-((6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[b]тиофен-2-ил)метил)метансульфонамида.
Продукт из Части E (0,15 г, 0,34 ммоль) подвергали условиям, описанным для получения Примера 72, Части B с получением названного соединения в виде бесцветного твердого вещества (0,121 г, 73%).
Часть G. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[b]тиофен-2-ил)метил)-N-(4-метоксибензил)метансульфонамида.
Продукт из Части F (24 мг, 0,049 ммоль) подвергали условиям, описанным для получения Примера 72, Части C с получением названного соединения в виде бесцветного твердого вещества (20 мг, 65%).
Часть H. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[b]тиофен-2-ил)метил)метансульфонамида.
Раствор продукта из Части G (14 мг, 0,022 ммоль) в CH2Cl2 (0,3 мл) и TFA (0,3 мл) перемешивали при комнатной температуре в течение 4 ч и затем концентрировали в вакууме. Остаток распределяли между 10 мл CH2Cl2 и 2 мл 10% водн. NaHCO3 и органический слой концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью 99:1 CH2Cl2:MeOH с получением названного соединения в виде бесцветного твердого вещества (5 мг, 44%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.40 (s, 1 H) 8.09 (s, 1 H) 7.82-7.97 (m, 3 H) 7.79 (d, J=7.72 Hz, 1 H) 7.47-7.63 (m, 1 H) 7.40 (s, 1 H) 7.26-7.34 (m, 1 H) 5.64 (d, J=7.72 Hz, 1 H) 4.48 (d, J=5.88 Hz, 2 H) 3.23 (s, 3 H) 2.95 (s, 3 H) 1.41 (s, 9 H).
Пример 113. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[b]тиофен-3-ил)метил)-N-метилметансульфонамида (соединение IB-L0-2.21).
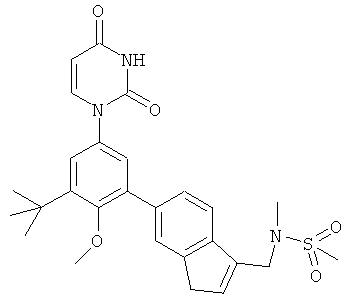
Часть A. Получение N-((6-бромбензо[b]тиофен-3-ил)метил)-N-метилметансульфонамида.
Смесь продукта из Примера 76, Части D (0,100 г, 0,382 ммоль), N-метилметан-сульфонамида (45,9 мг, 0,421 ммоль) и карбоната калия (0,127 г, 0,918 ммоль) в N,N-диметилацетамиде (5 мл) перемешивали при 80°C в течение 11 ч, охлаждали до комнатной температуры и распределяли между диэтиловьм эфиром и водой (3×), сушили над MgSO4, фильтровали и концентрировали в вакууме с получением названного соединения в виде бесцветного воскообразного твердого вещества (0,128 г, колич.).
Часть B. Получение N-метил-N-((6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензо[b]тиофен-3-ил)метил)метансульфонамида.
Продукт из Части A (0,128 г, 0,382 ммоль) подвергали условиям, описанным для получения Примера 72, Части B с получением названного соединения в виде бесцветного кристаллического твердого вещества (0,120 г, 82%).
Часть C. Получение N-((6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)бензо[b]тиофен-3-ил)метил)-N-метилметансульфонамида.
Продукт из Части B (50,6 мг, 0,133 ммоль) подвергали условиям, описанным для получения Примера 79, Части F с получением названного соединения в виде бесцветного твердого вещества (61,5 мг, 88%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.41 (s, 1 H) 8.17 (d, J=1.47 Hz, 1 H) 8.09 (d, J=8.09 Hz, 1 H) 7.74-7.85 (m, 2 H) 7.63 (dd, J=8.46, 1.47 Hz, 1 H) 7.29-7.36 (m, 2 H) 5.65 (d, J=7.72 Hz, 1 H) 4.52 (s, 2 H) 3.24 (s, 3 H) 3.03 (s, 3 H) 2.70 (s, 3 H) 1.42 (s, 9 H).
Пример 114. Получение (E)-N-(4-(3-бром-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.52).
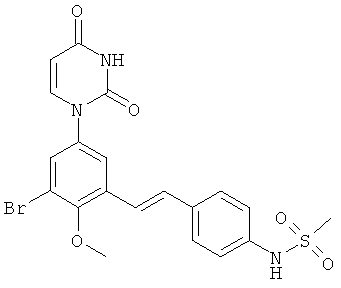
Часть A. Получение 2-бром-4,6-дийодфенола.
В 1 л круглодонную колбу помещали 2-бромфенол (8,65 г, 50 ммоль) и метанол (100 мл) с получением бесцветного раствора. Добавляли гидроксид натрия (2,40 г, 60,0 ммоль) и перемешивали до полного растворения гранул гидроксида. Раствор охлаждали на ледяной бане и добавляли йодид натрия (5,6 г, 37,4 ммоль) с последующим добавлением по каплям гипохлорита натрия (17 мл, 27,5 ммоль) с получением прозрачного коричневого/красного раствора, и постепенного осаждения густого белого твердого веещства. Добавление йодида натрия и раствора гипохлорита натрия повторяли 3 раза с получением оранжевой смеси, которую перемешивали в течение 2 ч, обрабатывали раствором тиосульфата натрия в воде (20 г в 100 мл), перемешивали в течение 15 мин и обрабатывали по каплям концентрированной HCl до постоянного pH 1. Смесь перемешивали в течение 15 мин и фильтровали для сбора твердого вещества белого цвета, которое промывали многократно водой и сушили до постоянной массы (14,7 г, 69%).
Часть B. Получение 1-бром-3,5-дийод-2-метоксибензола. В 500 мл круглодонную колбу помещали продукт из Части A (14,7 г, 34,6 ммоль), йодметан (2,70 мл, 43,3 ммоль), и гидроксид натрия (2,101 мл, 39,8 ммоль) в ацетоне (96 мл) с получением желтовато-коричневого раствора. Смесь перемешивали в течение 24 ч и концентрировали. Остаток растворяли в этилацетате, промывали водой и насыщенным хлоридом натрия, сушили над сульфатом натрия, фильтровали и концентрировали с получением твердого вещества белого цвета. Твердое вещество перекристаллизовывали из горячего гексана с получением твердого вещества белого цвета, которое собирали фильтрованием (12,3 г, 81%).
Часть C. Получение 1-(3-бром-5-йод-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
В 250 мл круглодонную колбу помещали продукт из Части B (8,09 г, 18,44 ммоль), пиримидин-2,4(1H,3H)-дион (2,273 г, 20,28 ммоль), N-(2-цианофенил)пиколинамид (0,823 г, 3,69 ммоль), йодид меди (I) (0,351 г, 1,844 ммоль) и фосфат калия (8,22 г, 38,7 ммоль) в DMSO (70 мл). Смесь герметично закрывали, барботировали азотом в течение 15 мин и нагревали при 60°C в течение 16 ч. Смесь разделяли этилацетатом и водой. Органический слой промывали 1M HCl, водой, солевым раствором, сушили сульфатом натрия, и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем (Aldrich catalog #538086), фильтровали через целит и упаривали с получением беловатого твердого вещества (3,92 г, 50%).
Часть D. Получение (E)-N-(4-(3-бром-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида.
В 100 мл круглодонную колбу добавляли продукт из Части C (846 мг, 2,0 ммоль), продукт из Примера 41B, Части B (482 мг, 2,000 ммоль), фосфат калия (892 мг, 4,20 ммоль), 1,3,5,7-тетраметил-6-фенил-2,4,8-триокса-6-фосфаадамант (PA-Ph) (CAS 97739-46-3) (17,54 мг, 0,060 ммоль) и трис(дибензилиденацетон)дипалладий(0) (18,31 мг, 0,020 ммоль) в THF (12,0 мл) и воде (4,0 мл). Колбу герметично закрывали, и смесь барботировали азотом в течение 5 мин и перемешивали при окружающей температуре в течение 72 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток растирали с минимальным количеством метанола/CH2Cl2 с получением названного соединения в виде твердого вещества белого цвета (595 мг, 60%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.03 (s, 3 H) 3.82 (s, 3 H) 5.69 (dd, J=7.72, 1.50 Hz, 1 H) 7.24 (d, J=8.46 Hz, 2 H) 7.35 (m, 2 H) 7.61 (d, J=8.46 Hz, 2 H) 7.69 (d, J=2.21 Hz, 1 H) 7.78 (d. J=8.09 Hz, 1 H) 7.87 (d, J=2.21 Hz, 1 H) 9.90 (s, 1 H) 11.50 (s, 1 H). MS (ESI-) m/z 490,492 (M-H)+.
Пример 115. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(тиофен-2-ил)стирил)фенил)метансульфонамида (соединение IB-L1-1.48).
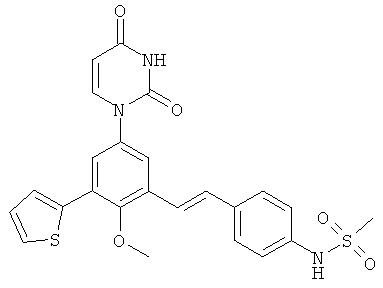
В 5 мл микроволновую трубку добавляли продукт из Примера 114, Части D (40 мг, 0,081 ммоль), тиофен-2-илбороновую кислоту (10,40 мг, 0,081 ммоль), 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладий дихлорид (2,65 мг, 4,.06 мкмоль) и фосфат калия (34,5 мг, 0,162 ммоль) в THF (3,0 мл) и воде (1,0 мл). Сосуд герметично закрывали, и смесь барботировали азотом в течение 5 мин и нагревали при 50°C в течение 3 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали через целит и упаривали. Остаток очищали с помощью хроматографии с обращенной фазой с получением названного соединения в виде твердого вещества белого цвета (20 мг, 50%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.03 (s, 3 H) 3.70 (s, 3 H) 5.70 (dd, J=7.72, 2.21 Hz, 1 H) 7.18 (dd, J=5.43, 4.05 Hz, 1 H) 7.25 (d, J=8.82 Hz, 2 H) 7.35 (s, 2 H) 7.63 (d, J=8.82 Hz, 2 H) 7.68 (m, 2 H) 7.77 (m, 2 H) 7.83 (d, J=7.72 Hz, 1 H) 9.89 (s, 1 H) 11.49 (d, J=2.21 Hz, 1 H). MS (ESI+) m/z 496 (M+H)+.
Пример 116. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(фуран-2-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.46).
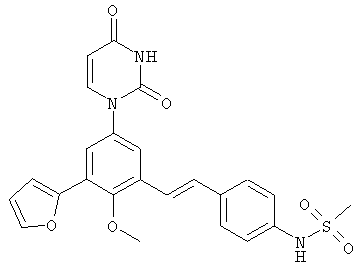
Названное соединение получали в соответствии с процедурой Примера 115 заменяя фуран-2-илбороновую кислоту на тиофен-2-илбороновую кислоту с получением твердого вещества белого цвета (22 мг, 56%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.03 (s, 3 H) 3.76 (s, 3 H) 5.69 (d, J=7.72 Hz, 1 H) 6.69 (dd, J=3.31, 1.84 Hz, 1 H) 7.08 (d, J=2.57 Hz, 1 H) 7.25 (d, J=8.46 Hz, 2 H) 7.36 (m, 2 H) 7.63 (d, J=8.82 Hz, 2 H) 7.67 (d, J=2.57 Hz, 1 H) 7.77 (d, J=2.57 Hz, 1 H) 7.82 (m, J=7.72 Hz, 2 H) 9.88 (s, 1 H) 11.48 (s, 1 H). MS (ESI+) m/z 497 (M+NH4)+.
Пример 117. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(пиридин-4-ил)стирил)фенил)метансульфонамида (соединение IB-L1-1.55).
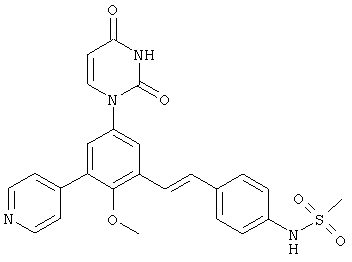
Названное соединение получали в соответствии с процедурой Примера 115, заменяя 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин на тиофен-2-илбороновую кислоту с получением твердого вещества белого цвета (15 мг, 38%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.03 (s, 3 Н) 3.49 (s, 3 Н) 5.72 (dd, J=7.72, 2.21 Hz, 1 Н) 7.25 (d, J=8.46 Hz, 2 Н) 7.38 (d, J=4.41 Hz, 2 Н) 7.51 (d, J=2.57 Hz, 1 Н) 7.63 (d, J=8.82 Hz, 2 Н) 7.80 (d, J=5.88 Hz, 2 Н) 7.85 (d, J=7.72 Hz, 1 Н) 7.97 (d, J=2.57 Hz, 1 Н) 8.77 (d, J=6.25 Hz, 2 Н) 9.90 (s, 1 Н) 11.51 (d, J=2.21 Hz, 1 Н). MS (ESI+) m/z 491 (M+H)+.
Пример 118. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(пиридин-3-ил)стирил)фенил)метансульфонамида (соединение IB-L1-1.53).
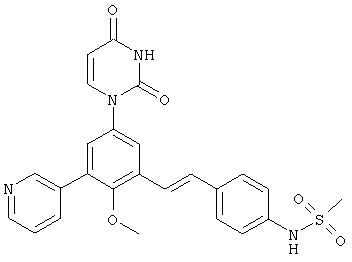
Названное соединение получали в соответствии с процедурой Примера 115 заменяя 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин на тиофен-2-илбороновую кислоту с получением твердого вещества белого цвета (19 мг, 48%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.02 (s, 3 Н) 3.45 (s, 3 Н) 5.71 (dd, J=8.09, 2.21 Hz, 1 Н) 7.24 (d, J=8.46 Hz, 2 Н) 7.37 (d, J=2.94 Hz, 2 Н) 7.47 (d, J=2.57 Hz, 1 Н) 7.63 (m, 3 Н) 7.85 (d, J=7.72 Hz, 1 Н) 7.93 (d, J=2.57 Hz, 1 H) 8.15 (m, 1 Н) 8.68 (dd, J=4.80 Hz, 1.47 Hz, 1 Н) 8.86 (d, J=1.84 Hz, 1 Н) 9.89 (s, 1 Н) 11.50 (d, J=2.21 Hz, 1 H). MS (ESI+) m/z 491 (M+H)+.
Пример 119. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(тиофен-3-ил)стирил)фенил)метансульфонамида (соединение IB-L1-1.47).
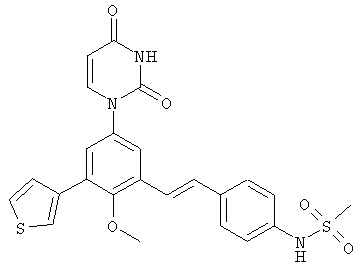
Названное соединение получали в соответствии с процедурой Примера 115, заменяя тиофен-3-илбороновую кислоту на тиофен-2-илбороновую кислоту с получением твердого вещества белого цвета (19 мг, 38%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.02 (s, 3 H) 3.55 (s, 3 H) 5.69 (d, J=8.09 Hz, 1 H) 7.24 (d, J=8.46 Hz, 2 H) 7.36 (s, 2 H) 7.55 (m, 2 H) 7.61 (d, J=8.46 Hz, 2 H) 7.67 (dd, J=5.15, 2.94 Hz, 1 H) 7.78 (d, J-2.57 Hz, 1 H) 7.83 (d, J=7.72 Hz, 1 H) 7.93 (dd, J=2.57, 0.96 Hz, 1 H) 9.88 (s, 1 H) 11.48 (s, 1 H). MS (ESI-) m/z 494 (M-H)+.
Пример 120. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-3-(фуран-3-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.50).
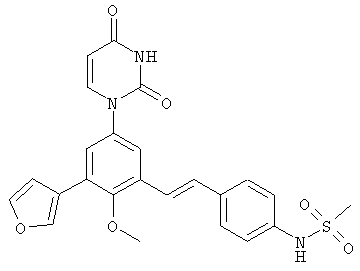
Названное соединение получали в соответствии с процедурой Примера 115, заменяя фуран-3-илбороновую кислоту на тиофен-2-илбороновую кислоту с получением твердого вещества белого цвета (14 мг, 29%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.02 (s, 3 H) 3.69 (s, 3 H) 5.69 (d, J=8.09 Hz, 1 H) 7.05 (dd, J=2.57, 0.90 Hz, 1 H) 7.24 (d, J=8.82 Hz, 2 H) 7.34 (s, 2 H) 7.61 (m, 3 H) 7.74 (d, J=2.57 Hz, 1 H) 7.80 (m, 2 H) 8.25 (s, 1 H) 9.88 (s, 1 H) 11.49 (s, 1 H). MS (ESI-) m/z 478 (M-H)+.
Пример 121. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(1-гидрокси-2-метилпропан-2-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.45).
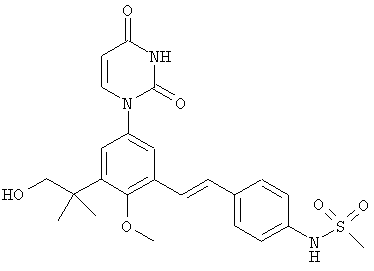
Часть A. Получение 2-(2-гидрокси-3,5-дийодфенил)уксусной кислоты.
В 250 мл круглодонную колбу добавляли 2-(2-гидроксифенил)уксусную кислоту (Aldrich, 3,04 г, 20 ммоль) в ацетонитрил (50 мл) с получением бесцветного раствора. N-йодсукцинимид (9,00 г, 40,0 ммоль) добавляли порциями за период времени 15 мин с получением красного/коричневого прозрачного раствора, который перемешивали в течение 16 ч. Смесь концентрировали, и полученное в результате твердое вещество растирали в 75 мл воды и фильтровали для сбора оранжевого твердого вещества, которое сушили под вакуумом. Это неочищенное твердое вещество перекристаллизовывали из толуола с получением светлого оранжевого порошка (6,0 г, 74%).
Часть B. Получение метил 2-(3,5-дийод-2-метоксифенил)ацетата.
В 250 мл круглодонную колбу добавляли продукт из Части A (6 г, 14,85 ммоль), карбонат калия (6,16 г, 44,6 ммоль), и диметилсульфат (4,12 г, 32,7 ммоль) в ацетоне (49,5 мл) с получением коричневой суспензии. Эту суспензию нагревали с обратным холодильником в течение 16 ч, охлаждали, концентрировали, и остаток распределяли между EtOAc и водой. EtOAc слой промывали солевым раствором, сушили (Na2SO4) и концентрировали до коричневого масла, которое хроматографировали на картридже с диоксидом кремния 40 г, элюируя 3:1 гексаном/EtOAc с получением желтого масла (6,0 г, 94%).
Часть C. Получение метил 2-(3,5-дийод-2-метоксифенил)-2-метилпропаноата.
В 100 мл круглодонную колбу в атмосфере азота добавляли продукт из Части B (1,728 г, 4 ммоль) в безводном THF (20 мл) и HMPA (2 мл) с получением бесцветного раствора. Добавляли метилйодид (1,251 мл, 20,00 ммоль), и этот раствор охлаждали до -40°C. Добавляли по каплям т-бутоксид калия (12,00 мл, 12,00 ммоль), и смесь перемешивали при температуре от -40 до -20°C в течение 30 мин и гасили 1М HCl до pH 1. Смесь экстрагировали 3×40 мл EtOAc. Экстракты объединяли, промывали солевым раствором, сушили (Na2SO4) и концентрировали. Сырой продукт подвергали флэш-хроматографии на картридже с диоксидом кремния 40 г ISCO, элюируя 9:1 гексаном/EtOAc с получением бис-метилированного продукта в виде желтого масла (1,63 г, 89%).
Часть D. Получение 2-(3,5-дийод-2-метоксифенил)-2-метилпропановой кислоты.
Суспензию продукта из Части C (2,63 г, 5,72 ммоль) в MeOH (40 мл) и THF (40 мл) обрабатывали 4,0 М гидроксидом натрия (28 мл, 112 ммоль) и нагревали при 80°C в течение 48 ч. Органический растворитель упаривали и оставшийся водный раствор подкисляли 1M HCl, получая твердое вещество, которое собирали фильтрацией, промывали водой и сушили с получением желаемой карбоновой кислоты (2,46 г, 96%).
Часть E. Получение 2-(3,5-дийод-2-метоксифенил)-2-метилпропан-1-ола.
Раствор продукта из Части D (1,00 г, 2,242 ммоль) в THF (40 мл) обрабатывали по каплям комплексом боран THF 1,0M (20 мл, 20 ммоль) и затем нагревали при 50°C в течение 24 ч. Смесь обрабатывали метанолом (20 мл), нагревали с обратным холодильником в течение 30 мин и концентрировали. Полученный в результате остаток промывали водой, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя гексаном/EtOAc (4:1) с получением желаемого продукта (810 мг, 84%).
Часть F. Получение трет-бутил(2-(3,5-дийод-2-метоксифенил)-2-метилпропокси)-диметилсилана.
Раствор продукта из Части E (432 мг, 1,000 ммоль) в DMF (5 мл) обрабатывали трет-бутилдиметилхлорсиланом (301 мг, 2,000 ммоль), и имидазолом (204 мг, 3,00 ммоль) и перемешивали в течение 2 ч. Смесь распределяли между 1М HCl и этилацетатом. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя гексаном/EtOAc (9:1) с получением желаемого продукта (522 мг, 96%).
Часть G. Получение 1-(3-(1-(трет-бутилдиметилсилилокси)-2-метилпропан-2-ил)-5-йод-4-метоксифенил)пиримидин-2,4(1H,3H)-диона.
В 50 мл круглодонную колбу добавляли продукт из Части F (520 мг, 0,952 ммоль), пиримидин-2,4(1H,3H)-дион (117 мг, 1,047 ммоль), N-(2-цианофенил)пиколинамид (42,5 мг, 0,190 ммоль), йодид меди (I) (18,13 мг, 0,095 ммоль) и фосфат калия (424 мг, 1,999 ммоль) в DMSO (5 мл). Сосуд герметично закрывали, барботировали азотом и затем нагревали при 60°C в течение 24 ч. Смесь распределяли между 1М HCl и этилацетатом. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя гексаном/EtOAc (3:2) с получением продукта в виде твердого вещества (285 мг, 65%).
Часть H. Получение (E)-N-(4-(3-(1-(трет-бутилдиметилсилилокси)-2-метилпропан-2-ил)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида. В 5 мл микроволновую трубку добавляли продукт из Части G (53 мг, 0,1 ммоль), продукт из Примера 41B, Части B (24 мг, 0,1 ммоль), фосфат калия (44,0 мг, 0,2 ммоль), PA-Ph (CAS 97739-46-3) (0,87 мг, 3,0 мкмоль) и трис(дибензилиденацетон)палладий(0) (0,9 мг, 1 мкмоль) в THF (3,0 мл) и воде (1,0 мл). Сосуд герметично закрывали, и смесь барботировали азотом в течение 5 мин и затем нагревали при 50°C в течение 2 ч. Смесь распределяли между 1М HCl и этилацетатом. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя гексаном/EtOAc (1:1) с получением твердого вещества (50 мг, 83%).
Часть I. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-(1-гидрокси-2-метилпропан-2-ил)-2-метоксистирил)фенил)метансульфонамида.
Раствор продукта из Части H (120 мг, 0,20 ммоль) в THF (5,0 мл) обрабатывали 1М TBAF (0,800 мл, 0,800 ммоль) в THF и перемешивали в течение 16 ч. Смесь разделяли водой и этилацетатом. Органический слой промывали (3 X солевым раствором), сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя 4% метанолом в CH2Cl2 с получением твердого вещества (85 мг, 88%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.30 (s, 6 H) 3.01 (s, 3 H) 3.62 (d, J=5.52 Hz, 2 H) 3.77 (s, 3 H) 4.67 (t, J=5.33 Hz, 1 H) 5.66 (d, J=8.09 Hz, 1 H) 7.21 (m, 5 H) 7.62 (m, 3 H) 7.72 (d, J=8.09 Hz, 1 H) 9.85 (s, 1 H) 11.42 (s, 1 H). MS (ESI+) m/z 503 (M+NH4)+.
Пример 122. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-3-йод-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.51).
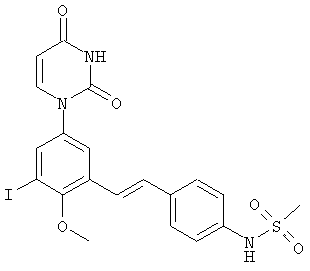
Часть A. Получение 1,3,5-трийод-2-метоксибензола. В 250 мл сосуд высокого давления добавляли 2,4,6-трийодфенол (5 г, 10,60 ммоль) в МТВЕ (60 мл) с получением желтого раствора. Раствор охлаждали на ледяной бане и добавляли быстрыми каплями 2,ОМ триметилсилилдиазометан (7,95 мл, 15,90 ммоль) с последующим добавлением по каплям метанола (6 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали при комнатной температуре в течение 4 ч. Реакционный раствор распределяли между EtOAc и водой и органический слой промывали 1М HCl, насыщенным NaHCO3, и насыщенным NaCl. Слой EtOAc сушили (MgSO4), фильтровали и концентрировали с получением твердого вещества желтовато-коричневого цвета, которое использовали без очистки (4,8 г, 94%).
Часть B. Получение 1-(3,5-дийод-4-метоксифенил)пиримидин-2,4(1H,3H)-диона. В 100 мл круглодонную колбу в атмосфере N2 добавляли продукт из Части A (3,5 г, 7,2 ммоль), 1H-пиримидин-2,4-дион (0,97 г, 8,64 ммоль), и трехосновный фосфат калия (3,2 г, 15,0 ммоль) в DMSO (50 мл) с получением бесцветной суспензии. Добавляли N-(2-цианофенил)пиколинамид (320 мг, 1,44 ммоль) и смесь барботировали N2 в течение 5 мин. Добавляли йодид меди (I) (137 мг, 0,72 ммоль), и эту смесь барботировали еще раз в течение 10 мин, помещали в атмосферу N2 и нагревали при 60°C в течение 18 ч. Смесь охлаждали и распределяли между EtOAc и водой, доводя pH до 1, используя HCl. Водный слой экстрагировали 2X EtOAc. Органические экстракты объединяли, промывали водой, насыщенным K2HCO3, и насыщенным NaCl, сушили (Na2SO4), обрабатывали 3-меркаптопропил-функционализированным диоксидом кремния, фильтровали и концентрировали. Полученное в результате твердое вещество растирали в 2:1 гексане/EtOAc с получением беловатого порошка (2,2 г, 62%).
Часть C. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-йод-2-метоксистирил)фенил)метансульфонамида.
В 5 мл микроволновой трубке перемешивали продукт из Части B (141 мг, 0,30 ммоль), продукт из Примера 41B, Части B (72,3 мг, 0,300 ммоль), комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорид CH2Cl2 (12,25 мг, 0,015 ммоль) и фосфат калия (70,0 мг, 0,330 ммоль) в THF (3,0 мл) и воде (1.0 мл). Смесь барботировали азотом в течение 5 мин и нагревали при 50°C в течение 2 ч. Смесь разделяли этилацетатом и 1M HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток хроматографировали на диоксиде кремния, элюируя 5% метанолом в CH2Cl2 с получением твердого вещества (47 мг, 29%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.02 (s, 3 H) 3.77 (s, 3 H) 5.67 (d, J=7.72 Hz, 1 H) 7.28 (m, 4 H) 7.60 (d, J=8.82 Hz, 2 H) 7.76 (d, J=8.09 Hz, 1 H) 7.81 (d, J=2.57 Hz, 1 H) 7.86 (d, J=2.21 Hz, 1 H) 9.90 (s, 1 H) 11.48 (s, 1 H). MS (ESI-) m/z 538 (M-H)+.
Пример 123. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(метилсульфонил)стирил)фенил)метансульфонамида (соединение IB-L1-1.49).
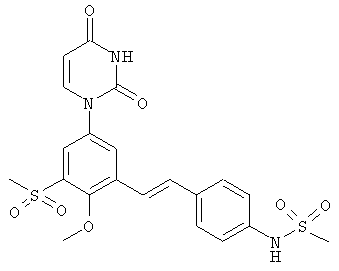
Часть A. Получение 4-нитробензол-2-диазо-1-оксида.
В 250 мл круглодонную колбу добавляли 2-амино-4-нитрофенол (6,165 г, 40,0 ммоль) в 48% тетрафторборной кислоте (15 мл). Добавляли по каплям нитрит натрия (2,76 г, 40,0 ммоль) в воду (6 мл) при 0°C, и смесь перемешивали при комнатной температуре в течение 30 мин. Твердое вещество собирали фильтрацией, промывали тетрафторборной кислотой и водой. Твердое вещество суспендировали в ацетоне (50 мл), фильтровали и сушили с получением твердого вещества (3,31 г, 50%).
Часть B. Получение 2-(метилтио)-4-нитрофенола.
В 1 л лабораторный стакан добавляли продукт из Части A (2,70 г, 16,35 ммоль) в ледяной воде (250 г) с получением коричневой суспензии. Добавляли медь (0,520 г, 8,18 ммоль), с последующим медленным добавлением тиометоксида натрия (2,292 г, 32,7 ммоль) в воде (50 мл). Смесь перемешивали при комнатной температуре в течение 24 ч. Смесь фильтровали, и фильтрат подкисляли 1M HCl, получая твердое вещество, которое собирали фильтрованием, и сушили (2,53 г, 84%).
Часть C. Получение 2-(метилсульфонил)-4-нитрофенола.
В 250 мл круглодонную колбу добавляли продукт из Части B (1,111 г, 6,00 ммоль) в MeOH (20 мл) с получением коричневой суспензии. Медленно добавляли оксон (7,746 г, 12,60 ммоль) в воде (20 мл) при 0°C. Смесь нагревали до комнатной температуры, перемешивали в течение 1 ч и разделяли этилацетатом и 1М HCl. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали. Остаток хроматографировали на силикагеле, элюируя 1%-5% метанолом в CH2Cl2 с получением твердого вещества (0,472 г, 36%).
Часть D. Получение 2-йод-6-(метилсульфонил)-4-нитрофенола.
В 50 мл круглодонную колбу добавляли продукт из Части С (470 мг, 2,164 ммоль) в MeOH (10 мл) и воду (2,5 мл). Добавляли по каплям монохлорид йода (0,130 мл, 2,60 ммоль) в CH2Cl2 (2.0 мл), и смесь перемешивали при комнатной температуре, выливали в воду (200 мл) и перемешивали в течение 10 мин. Полученное в результате твердое вещество собирали фильтрованием и сушили (636 мг, 86%).
Часть E. Получение 1-йод-2-метокси-3-(метилсульфонил)-5-нитробензола.
В 50 мл сосуд высокого давления добавляли продукт из Части D (630 мг, 1,836 ммоль) в MTBE (6 мл) с получением желтого раствора. Смесь охлаждали на ледяной бане и добавляли быстрыми каплями 2M триметилсилил-диазометан (1,377 мл, 2,75 ммоль) с последующим добавлением по каплям MeOH (0,4 мл), получая в результате спокойное выделение пузырьков газа. Сосуд герметично закрывали и перемешивали при комнатной температуре в течение 1 ч. Смесь разделяли этилацетатом и 1М HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением беловатого твердого вещества (655 мг, 100%).
Часть F. Получение 3-йод-4-метокси-5-(метилсульфонил)анилина.
В 250 мл круглодонную колбу добавляли продукт из Части E (0,650 г, 1,820 ммоль), хлорид аммония (0,146 г, 2,73 ммоль), и железо (0,508 г, 9,10 ммоль) в THF/MeOH/воде (50 мл, 2/2/1). Смесь нагревали с обратным холодильником в течение 2 ч, охлаждали и фильтровали. Фильтрат упаривали, и остаток разделяли этилацетатом и водой. Органический слой промывали солевым раствором, сушили сульфатом натрия, фильтровали и упаривали с получением твердого вещества (590 мг, 99%).
Часть G. Получение (E)-N-(3-йод-4-метокси-5-(метилсульфонил)фенилкарбамоил)-3-метоксиакриламида.
В 100 мл круглодонную колбу добавляли продукт из Части F (500 мг, 1,528 ммоль) в DMF (15,0 мл). Раствор охлаждали в атмосфере азота до -20°C и добавляли по каплям (E)-3-метоксиакрилоилизоцианат (15,28 мл, 6,11 ммоль; полученный, как описано Santana, L.; et al. J. Heterocyclic Chem. 1999, 36, 293-295). Смесь перемешивали при этой температуре в течение 15 мин, затем нагревали до комнатной температуры и перемешивали в течение 45 мин. Смесь разбавляли этилацетатом и промывали водой (3×50 мл), солевым раствором (3×50 мл), сушили сульфатом натрия, фильтровали и упаривали. Остаток растирали с этилацетатом/гексаном с получением твердого вещества (425 мг, 61%).
Часть H. Получение 1-(3-йод-4-метокси-5-(метилсульфонил)фенил)пиримидин-2,4(1H,3H)-диона.
В 100 мл круглодонную колбу добавляли продукт из Части G (420 мг, 0,925 ммоль) в этанол (10 мл) с получением суспензии. Добавляли концентрированную серную кислоту (1 мл, 18,76 ммоль) в воду (10 мл), и смесь нагревали при 110°C в течение 2 ч. Реакционную смесь охлаждали, разбавляли водой (50 мл) и перемешивали в течение 10 мин. Твердое вещество собирали фильтрацией, промывали водой и сушили с получением твердого вещества белого цвета (325 мг, 83%).
Часть I. Получение (E)-N-(4-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-3-(метилсульфонил)стирил)фенил)метансульфонамида.
В 5 мл микроволновую трубку добавляли продукт из Части H (63,3 мг, 0,15 ммоль), продукт из Примера 41B, Части B (36,2 мг, 0,150 ммоль), фосфат калия (66,9 мг, 0,315 ммоль), PA-Ph (CAS 97739-46-3) (1,315 мг, 4,50 мкмоль) и трис(дибензилиденацетон)дипалладий(0) (1,374 мг, 1,500 мкмоль) в THF (3,0 мл) и воде (1,0 мл). Сосуд герметично закрывали, и смесь барботировали азотом в течение 5 мин и нагревали при 50°C в течение 2 ч. Смесь разделяли этилацетатом и 1M HCl. Органический слой промывали насыщенным бикарбонатом натрия, солевым раствором, сушили сульфатом натрия и фильтровали. Фильтрат обрабатывали 3-меркаптопропил-функционализированным силикагелем, фильтровали и упаривали. Остаток растирали с метанолом/CH2Cl2 с получением твердого вещества (62 мг, 84%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.03 (s, 3 Н) 3.37 (s, 3 Н) 3.94 (s, 3 Н) 5.72 (d, J=7.72 Hz, 1 Н) 7.26 (m, 3 Н) 7.45 (m, 1 Н) 7.65 (d, J=8.46 Hz, 2 Н) 7.77 (d, J=2.57 Hz, 1 Н) 7.81 (d, J=8.09 Hz, 1 Н) 8.21 (d, J=2.57 Hz, 1 Н) 9.93 (s, 1 Н) 11.52 (s, 1 H). MS (ESI+) m/z 509 (M+NH4)+.
Пример 124. Получение (E)-метил 2-(3-отреот-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-5-(метилсульфонамид)бензоата (соединение IB-L1-1.7).
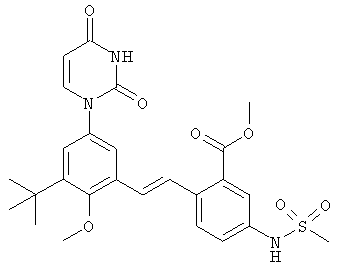
Часть A. Получение метил 2-((диэтоксифосфорил)метил)-5-нитробензоата.
К раствору метил 2-метил-5-нитробензоата (0,40 г, 2,05 ммоль) в CCl4 (20 мл) добавляли N-бромсукцинимид (365 мг, 2,05 ммоль) и 2,2'-азобисизобутиронитрил (34 мг, 0,21 ммоль). Полученную в результате смесь перемешивали при нагревании с обратным холодильником в течение 18 ч, охлаждали до комнатной температуры и распределяли между EtOAc (50 мл) и H2O (50 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 1:3 EtOAc тексан в качестве элюента с получением этого бромида в виде масла (345 мг, 61%). Это масло помещали в триэтилфосфит (5 мл) и нагревали при перемешивании при 120°C в течение 3 ч. Смесь оставляли охлаждаться до комнатной температуры, и сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 5% MeOH в CH2Cl2 в качестве элюента. Названное соединение получали в виде масла (313 мг, 75%).
Часть B. Получение (E)-метил 2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-5-нитробензоата.
К раствору продукта из Части A (360 мг, 1,09 ммоль) и продукта из Примера 41A, Части D (329 мг, 1,09 ммоль) в безводном CH2Cl2 (10 мл) добавляли трет-бутоксид калия (305 мг, 2,72 ммоль). Полученный в результате темный красный раствор перемешивали при комнатной температуре в течение 1 ч, и затем выливали в 1н водн. HCl (10 мл). Полученную в результате смесь экстрагировали CH2Cl2 (10 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением твердого вещества. Раствор этого твердого вещества в тионилхлориде (2,3 мл) нагревали при 85°C в течение 30 мин, и тионилхлорид удаляли в вакууме. Остаток перемешивали в смеси 2:1 CH2Cl2 и MeOH (3 мл) в течение 30 мин, и упаривали в вакууме до сухости. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения (350 мг, 69%).
Часть C. Получение (E)-метил 2-(3-треот-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-5-(метилсульфонамид)бензоата.
К раствору продукта из Части B (465 мг, 0,97 ммоль) в смеси 2:2:1 THF:MeOH:H2O (10 мл) добавляли железные опилки (271 мг, 4,85 ммоль), и хлорид аммония (78 мг, 1,46 ммоль). Смесь нагревали при 80°C в течение 45 мин, фильтровали через целит, и концентрировали до сухости в вакууме. Остаток объединяли с метансульфонилхлоридом (0,16 мл, 2,0 ммоль) и триэтиламином (0,392 мл, 4,85 ммоль) в безводном CH2Cl2 (10 мл) и полученную в результате смесь перемешивали при комнатной температуре в течение 3 ч. Смесь распределяли между 1 н HCl (20 мл) и CH2Cl2 (20 мл), и органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента с получением названного соединения (270 мг, 53%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.42 (s, 1 Н) 10.07 (s, 1 Н) 7.90 (d, J=8.82 Hz, 1 Н) 7.66-7.79 (m, 3 Н) 7.52 (d, J=2.57 Hz, 1 H) 7.44 (dd, J=8.64, 2.39 Hz, 1 H) 7.14-7.26 (m, 2 H) 5.65 (dd, J=7.72, 1.84 Hz, 1 H) 3.86 (s, 3 H) 3.79 (s, 3 H) 3.04 (s, 3 H) 1.38 (s, 9 H).
Пример 125. Получение (E)-2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-5-(метилсульфонамид)бензойной кислоты (соединение IB-L1-1.4).
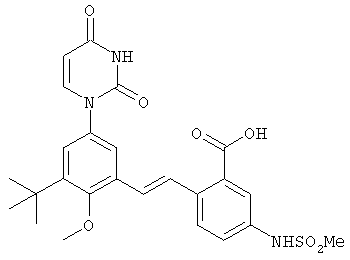
Раствор продукта из Примера 124 (55 мг, 0,104 ммоль) в THF (1 мл) и 1 н водн. NaOH (1 мл) перемешивали в темноте при комнатной температуре в течение 1,5 ч. Добавляли 1 н водный HCl до pH 3, и полученную в результате смесь экстрагировали EtOAc (2×2 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением названного соединения (53 мг, 99%). 1H ЯМР (300 MHz, DMSO-d6) δ 13.22 (br s, 1 Н) 11.40 (d, J=2.21 Hz, 1 H) 10.02 (s, 1 H) 7.72-7.91 (m, 3 H) 7.68 (d, J=2.57 Hz, 1 H) 7.49 (d, J=2.57 Hz, 1 H) 7.42 (dd, J=8.64, 2.39 Hz, 1 H) 7.21 (d, J=2.57 Hz, 1 H) 7.16 (d, J=16.18 Hz, 1 H) 5.64 (dd, J=7.72, 2.21 Hz, 1 H) 3.79 (s, 3 H) 3.04 (s, 3 H) 1.38 (s, 9 H).
Пример 126. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-(морфолин-4-карбонил)фенил)метансульфонамида (соединение IB-L1-1.23).
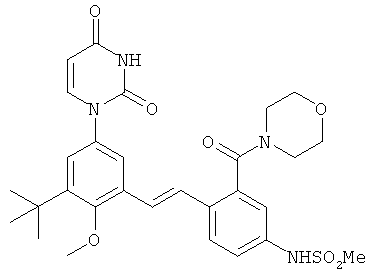
Часть A. Получение (E)-2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-5-(метилсульфонамид)бензоилхлорида.
Раствор продукта из Примера 125 (257 мг, 0,50 ммоль) в тионилхлориде (1,5 мл) нагревали при 85°C в течение 40 мин и затем концентрировали и сушили в вакууме с получением названного соединения в виде твердого вещества (0,27 г).
Часть B. Пол учение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-(морфолин-4-карбонил)фенил)метансульфонамида.
К раствору продукта из Части A (24 мг, 0,045 ммоль) в безводном CH2Cl2 (1 мл) добавляли морфолин (0,02 мл, 0,226 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч, и затем распределяли между 1 н водн. HCl (5 мл) и EtOAc (2×5 мл).
Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 4% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения (19 мг, 71%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 11.41 (d, J=1.84 Hz, 1 Н) 10.04 (s, 1 Н) 7.85 (d, J=8.46 Hz, 1 Н) 7.75 (d, J=8.09 Hz, 1 Н) 7.52 (d, J=2.57 Hz, 1 Н) 6.99-7.34 (m, 5 Н) 5.65 (dd, J=7.72, 1.84 Hz, 1 Н) 3.76 (s, 3 Н) 3.56-3.71 (m, 4 Н) 3.40-3.51 (m, 2 Н) 3.11-3.22 (m, 2 Н) 3.06 (s, 3 H) 1.38 (s, 9 H).
Пример 127. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-(гидроксиметил)фенил)метансульфонамида (соединение IB-L1-1.10).
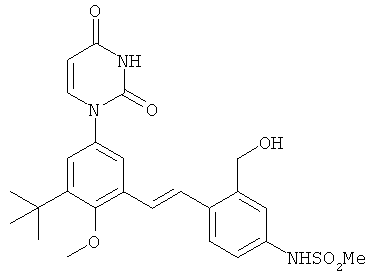
К раствору продукта из Примера 126, Части A (375 мг, 0,705 ммоль) в безводном THF (5 мл) при 0°C в атмосфере газообразного N2 добавляли по каплям 1,0 М раствор литий трет-бутоксиалюминий гидрида (1,8 мл, 1,8 ммоль). Полученную в результате смесь перемешивали при 0°C в течение 30 мин, и затем оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Смесь распределяли между 1 н водн. HCl (10 мл) и EtOAc (2×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения (220 мг, 63%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 11.41 (s, 1 Н) 9.82 (s, 1 Н) 7.73 (t,.J=8.27 Hz, 2 Н) 7.66 (d, J=2.57 Hz, 1 Н) 7.31-7.39 (m, 2 Н) 7.20 (d, J=2.57 Hz, 1 Н) 7.12-7.19 (m, 2 Н) 5.65 (d, J=8.09 Hz, 1 Н) 5.28 (t, J=5.52 Hz, 1 H) 4.65 (d, J=5.52 Hz, 2 H) 3.79 (s, 3 H) 3.00 (s, 3 H) 1.38 (s, 9 H).
Пример 128. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-(метоксиметил)фенил)метансульфонамида (соединение IB-L1-1.13).
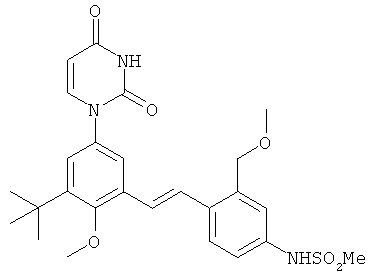
К раствору продукта из Примера 127 (32 мг, 0,064 ммоль) в безводном СН2С1з (1 мл) добавляли тионилхлорид (23 мкл, 0,32 ммоль), и полученную в результате смесь перемешивали при комнатной температуре в течение 30 мин. Смесь распределяли между насыщенным водн. NaHCO3 (5 мл) и CH2Cl2 (5 мл), и органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток растворяли в MeOH (1 мл), и добавляли раствор 25% NaOMe в MeOH (58 мкл, 0,254 ммоль). Полученную в результате смесь перемешивали при 50°C в течение 2 ч. Смесь распределяли между 1 н водн. HCl (10 мл) и EtOAc (2×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения (15 мг, 46%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.43 (s, 1 Н) 9.86 (s, 1 Н) 7.62-7.87 (m, 3 Н) 7.12-7.39 (m, 5 Н) 5.66 (d, J=7.72 Hz, 1 Н) 4.58 (s, 2 Н) 3.78 (s, 3 Н) 3.35 (s, 3 Н) 3.00 (s, 3 Н) 1.38 (s, 9H).
Пример 129. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-((изопентиламино)метил)фенил)метансульфонамида (соединение IB-L1-1.31).
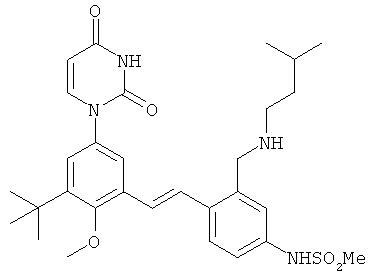
Часть А. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидцн-1(2H)-ил)-2-метоксистирил)-3-формилфенил)метансульфонамида.
К раствору продукта из Примера 127 (0,60 г, 1,20 ммоль) в безводном DMA (15 мл) добавляли 2-йодоксибензойную кислоту (336 мг, 1,20 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч, и затем распределяли между EtOAc (20 мл) и H2O (2×20 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 2% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения в виде бесцветного твердого вещества (395 мг, 66%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 11.43 (d, J=2.21 Hz, 1 Н) 10.45 (s, 1 Н) 10.15 (s, 1 Н) 8.06 (d, J=16.18 Hz, 1 Н) 7.97 (d, J=8.82 Hz, 1 Н) 7.73-7.78 (m, 2 Н) 7.69 (d, J=2.57 Hz, 1 Н) 7.51 (dd, J=8.64, 2.39 Hz, 1 H) 7.30 (d, J=16.18 Hz, 1 H) 7.26 (d, J=2.57 Hz, 1 H) 5.66 (dd, J=7.72, 2.21 Hz, 1 H) 3.81 (s, 3 H) 3.07 (s, 3 H) 1.39 (s, 9 H).
Часть B. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-((изопентиламино)метил)фенил)метансульфонамида.
К раствору продукта из Части A (50 мг, 0,10 ммоль) и 3-метилбутан-1-амина (12 мкл, 0,10 ммоль) в безводном THF (3 мл) добавляли триацетоксиборгидрид натрия (32 мг, 0,15 ммоль) и AcOH (9 мкл, 0,15 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 4 ч, и затем распределяли между H2O (10 мл) и EtOAc (2×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента с получением названного соединения (37 мг, 65%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.45 (d, J=1.84 Hz, 1 H) 10.04 (s, 1 H) 8.80-8.87 (m, 1 H) 7.88 (d, J=8.46 Hz, 1 H) 7.71-7.77 (m, 2 H) 7.41-7.48 (m, 1 H) 7.37 (d, J=2.21 Hz, 1 H) 7.21-7.29 (m, 3 H) 5.67 (dd, J=7.91, 2.02 Hz, 1 H) 4.30-4.38 (m, 2 H) 3.80 (s, 3 H) 3.10 (s, 3 H) 2.95-3.04 (m, 2 H) 1.49-1.67 (m, 3 H) 1.38 (s, 9 H) 0.86 (d, J=6.25 Hz, 6 H).
Пример 130. Получение N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-((E)-(метоксиимино)метил)фенил)метансульфонамида (соединение IB-L1-1.19).
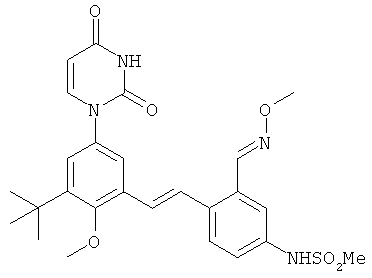
К раствору продукта из Примера 129, Часть A (35 мг, 0,070 ммоль) в EtOH (2 мл) добавляли (9-метоксиламин гидрохлорид (29 мг, 0,35 ммоль) и бикарбонат натрия (30 мг, 0,35 ммоль). Полученную в результате смесь перемешивали при 70°C в течение 2 ч. К смеси добавляли 1 н водн. HCl (1 мл) с получением бесцветного осадка, который фильтровали и сушили с получением названного соединения в виде бесцветного твердого вещества (24 мг, 64%). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 11.43 (d, J=2.21 Hz, 1 H) 9.94 (s, 1 H) 8.74 (s, 1 H) 7.79-7.85 (m, 2 H) 7.76 (d, J=7.72 Hz, 1 H) 7.57-7.65 (m, 2 H) 7.32 (dd, J=8.64, 2.39 Hz, 1 H) 7.23 (d, J=2.57 Hz, 1 H) 7.18 (d, J=16.18 Hz, 1 H) 5.66 (dd, J=7.72, 2.21 Hz, 1 H) 3.93 (s, 3 H) 3.79 (s, 3 H) 3.03 (s, 3H) 1.38 (s, 9H).
Пример 131. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-(оксазол-2-ил)фенил)метансульфонамида (соединение IB-L1-1.26).
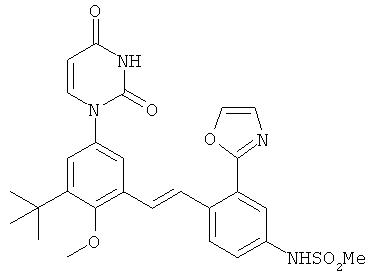
К раствору продукта из Примера 126, Части A (80 мг, 0,15 ммоль) в тетраметиленсульфоне (1,5 мл) добавляли 1H-1,2,3-триазол (10 мкл, 0,17 ммоль) и карбонат калия (73 мг, 0,53 ммоль). Смесь нагревали в течение 35 мин при 130°C в микроволновом реакторе. После охлаждения до комнатной температуры, смесь распределяли между 1 н водным HCl (10 мл) и EtOAc (2×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента с получением названного соединения (37 мг, 46%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.41 (d, J=1.84 Hz, 1 Н) 10.10 (s, 1 Н) 8.29 (d, J=1.10 Hz, 1 Н) 8.05 (d, J=16.18 Hz, 1 H) 7.95 (d, J=8.82 Hz, 1 H) 7.82 (d, J=2.21 Hz, 1 H) 7.74 (d, J=8.09 Hz, 1 H) 7.51 (d, J=2.57 Hz, 1 H) 7.46 (d, J=0.74 Hz, 1 H) 7.39 (dd, J=8.64, 2.39 Hz, 1 H) 7.20-7.30 (m, 2 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 3.80 (s, 3 H) 3.07 (s, 3 H) 1.38 (s, 9 H).
Пример 132. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-3-(1H-имидазол-2-ил)фенил)метансульфонамида (соединение IB-L1-1.16).
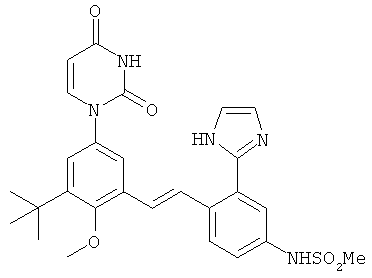
К раствору продукта из Примера 129, Части A (50 мг, 0,10 ммоль) в EtOH (2 мл) добавляли глиоксаль (57 мкл, 0,50 ммоль) и концентрированный водный NH4OH (70 мкл, 0,50 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 16 ч. В смесь добавляли 1 н водн. HCl до pH=7, и смесь распределяли между H2O (10 мл) и EtOAc (2×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 5% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения (27 мг, 50%). 1H ЯМР (300 MHz, DMSO-d6) δ 12.39 (s, 11 H) 11.40 (d, J=1.84 Hz, 1 Н) 9.98 (s, 1 Н) 7.89 (d, J=8.82 Hz, 1 Н) 7.66-7.76 (m, 2 Н) 7.38 (t, J=2.21 Hz, 2 H) 7.23-7.31 (m, 2 H) 7.06-7.21 (m, 3 H) 5.63 (dd, J=8.09, 1.84 Hz, 1 H) 3.78 (s, 3 H) 3.07 (s, 3H) 1.37 (s, 9H).
Пример 133. Получение (E)-трет-бутил 2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-5-(метилсульфонамид)фенилкарбамата (соединение IB-L1-1.32).
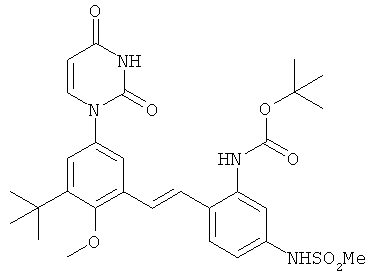
К раствору продукта из Примера 125 (75 мг, 0,146 ммоль) в трет-бутаноле (4 мл) добавляли дифенилфосфорилазид (47 мкл 0,219 ммоль) и триэтиламин (31 мкл, 0,219 ммоль). Полученную в результате смесь перемешивали при 80°C в течение 18 ч. Охлажденную смесь распределяли между H2O (10 мл) и EtOAc (2×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 3% MeOH в CH2Cl2 в качестве элюента, с получением названного соединения (16 мг, 19%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.45 (d, J=1.84 Hz, 1 H) 9.86 (s, 1 H) 9.03 (s, 1 H) 7.75 (d, J=7.72 Hz, 2 H) 7.55 (d, J=2.57 Hz, 1 H) 7.10-7.33 (m, 4 H) 7.04 (dd, J=8.64, 2.39 Hz, 1 H) 5.66 (dd, J=7.91, 2.02 Hz, 1 H) 3.78 (s, 3 H) 3.02 (s, 3 H) 1.45 (s, 9 H) 1.38 (s, 9 H).
Пример 134. Получение (E)-N-(3-амино-4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)фенил)метансульфонамида (соединение IB-L1-1.28).
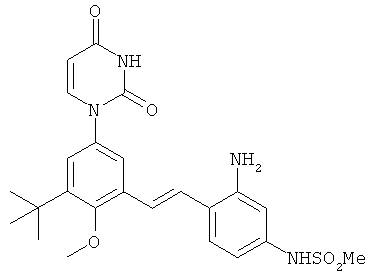
Процедура, описанная для получения Примера 133 давала названное соединение, которое очищали с помощью колоночной хроматографии на силикагеле, используя 5% метанол в CH2Cl2 в качестве элюента (6 мг, 9%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.44 (d, J=2.21 Hz, 1 H) 9.55 (s, 1 H) 7.77 (d, J=2.57 Hz, 1 H) 7.75 (d, J=8.09 Hz, 1 H) 7.45 (d, J=8.46 Hz, 1 H) 7.33 (d, J=15.81 Hz, 1 H) 7.15 (d, J=2.57 Hz, 1 H) 7.00 (d, J=16.18 Hz, 1 H) 6.56 (d, J=2.21 Hz, 1 H) 6.44 (dd, J=8.46, 2.21 Hz, 1 H) 5.66 (dd, J=7.91, 2.02 Hz, 1 H) 5.56 (s, 2 H) 3.78 (s, 3 H) 2.97 (s, 3 H) 1.37 (s, 9H).
Пример 135. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-2-фторфенил)метансульфонамида (соединение IB-L1-1.5).
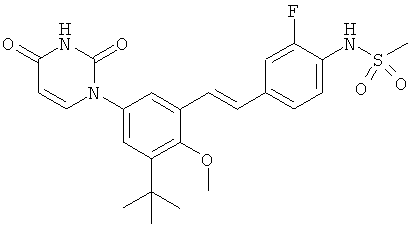
Часть A. Получение (3-фтор-4-нитрофенил)метанола.
К раствору 3-фтор-4-нитробензойной кислоты (2,0 г, 10,8 ммоль) в THF (50 мл) при 0°C добавляли по каплям комплекс ВН3·Me2S (2,215 мл, 22,15 ммоль). Смесь перемешивали при 0°C в течение 3 ч, и затем перемешивали при 65°C в течение 18 ч. К охлажденной смеси добавляли лед (50 г), с последующим добавлением 1 н водн. HCl (100 мл), и полученную в результате смесь экстрагировали EtOAc (200 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением названного соединения в виде твердого вещества белого цвета (1,79 г, 97%).
Часть B. Получение 4-(бромметил)-2-фтор-1-нитробензола.
Раствор продукта из Части A (1,79 г, 10,46 ммоль), N-бромсукцинимида (2,234 г, 12,55 ммоль) и трифенилфосфина (3,29 г, 12,55 ммоль) в CH2Cl2 (100 мл) и THF (50 мл) перемешивали при комнатной температуре в течение 3 ч. Смесь распределяли между H2O (200 мл) и EtOAc (400 мл), и органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 1:1 EtOAc:гексан в качестве элюента с получением названного соединения (1,14 г, 47%).
Часть C. Получение диэтил 3-фтор-4-нитробензилфосфоната.
Продукт из Части B (1,25 г, 5,34 ммоль) подвергали условиям, описанным для Примера 34, Части B с получением названного продукта (0,75 г, 48%).
Часть D. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-2-фторфенил)метансульфонамида.
Продукт из Части C (0,193 г, 0,662 ммоль) подвергали условиям, описанным для Примера 41A, Части E, Части F, и Части G с получением названного продукта в виде бесцветного твердого вещества (15 мг, 5%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.43 (s, 1H), 9.67 (s, 1H), 7.76 (d, J=8.1 Hz, 1H), 7.62 (m, 2H), 7.41 (m, 2H), 7.38 (m, 1H), 7.23 (m, 2H), 5.66 (dd, J=8.0, 2.0 Hz, 1H), 3.80 (s, 3H), 3.05 (s, 3H), 1.38 (s, 9H).
Пример 136. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-2-фтор-5-метилфенил)метансульфонамида (соединение IB-L1-1.15).
Часть A. Получение N-(4-бром-2-фтор-5-метилфенил)метансульфонамида.
К раствору 4-бром-2-фтор-5-метиланилина (2,04 г, 10,0 ммоль) в безводном CH2Cl2 (20 мл) и пиридине (3,23 мл, 40,0 ммоль) добавляли метансульфонилхлорид (0,86 мл, 11,0 ммоль), и полученную в результате смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли в вакууме, и остаток распределяли между EtOAc и 1М водн. HCl. Органический слой промывали насыщенным водным NaHCO3, солевым раствором и затем сушили над Na2SO4. Высушивающее вещество отфильтровывали, и фильтрат концентрировали с получением названного соединения в виде твердого вещества (2,80 г, 99%).
Часть B. Получение N-(4-этинил-2-фтор-5-метилфенил)метансульфонамида.
Смесь продукта из Части A (3,0 г, 10,63 ммоль), трифенилфосфина (0,279 г, 1,06 ммоль), триметилсилилацетата (6,0 мл, 42,5 ммоль) и ацетата палладия(II) (0,12 г, 0,53 ммоль) в триэтиламине (30 мл) и толуоле (15 мл) в атмосфере N; нагревали при 80°C в течение 5 ч. Смесь оставляли охлаждаться до комнатной температуры, и распределяли между EtOAc и 1М водн. HCl. Органический слой промывали насыщенным NaHCO3 и солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя градиент растворителей 10%-35% EtOAc в гексане с получением масла (3,0 г, 94%). К раствору этого масла (3,0 г, 10,0 ммоль) в MeOH (50 мл) добавляли 1М водн. NaOH (21 мл, 21,0 ммоль), и полученную в результате смесь перемешивали при комнатной температуре в течение 45 мин. Смесь распределяли между EtOAc и 1М водн. HCl, и органический слой промывали солевым раствором и сушили над Na2SO4. Высушивающее вещество отфильтровывали, и фильтрат концентрировали в вакууме с получением названного соединения в виде твердого вещества (2,3 г, колич.).
Часть C. Получение (E)-5-фтор-2-метил-4-(метилсульфонамид)стирилбороновой кислоты.
Продукт из Части B (0,20 г, 0,88 ммоль) подвергали условиям, описанным для получения Примера 41B, Части B с получением названного соединения (42 мг, 17%).
Часть D. Получение (E)-N-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксистирил)-2-фтор-5-метилфенил)метансульфонамида.
Продукт из Части C (40 мг, 0,15 ммоль) подвергали условиям, описанным для получения Примера 41B, Части I с получением названного соединения (51 мг, 83%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.42 (d, J=2.21 Hz, 1 H) 9.59 (s, 1 H) 7.70-7.78 (m, 2 H) 7.66 (d, J=11.77 Hz, 1 H) 7.20-7.32 (m, 3 H) 5.65 (dd, J=7.72, 2.21 Hz, 1 H) 3.79 (s, 3 H) 3.05 (s, 3 H) 2.38 (s, 3 H) 1.38 (s, 9H).
Пример 137. Получение метил 2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенэтил)-5-(метилсульфонамид)бензоата (соединение IB-L5-2-1.1).
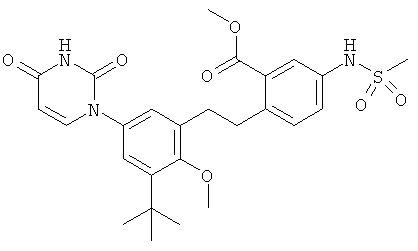
К раствору продукта из Примера 124 (40 мг, 0,076 ммоль) в MeOH (2 мл) и THF (2 мл) добавляли 10% Pd/C (20 мг), и полученную в результате смесь перемешивали при комнатной температуре под давлением 1 атм H2 в течение 16 ч. Смесь фильтровали через целит и концентрировали в вакууме с получением твердого вещества (27,5 мг, 68%). 1H ЯМР (300 MHz, DMSO-d6) δ 11.39 (s, 1 H) 9.88 (s, 1 H) 7.61-7.71 (m, 2 H) 7.28-7.36 (m, 2 H) 7.20 (d, J=2.57 Hz, 1 H) 7.13 (d, J=2.94 Hz, 1 H) 5.64 (d, J=7.72 Hz, 1 H) 3.83 (s, 3 H) 3.75 (s, 3 H) 3.14 (dd, J=10.30, 5.88 Hz, 2 H) 2.96 (s, 3 H) 2.83-2.92 (m, 2 H) 1.34 (s, 9 H).
Пример 138. Получение N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенэтил)фенил)метансульфонамида (соединение IB-L5-2-1.2).
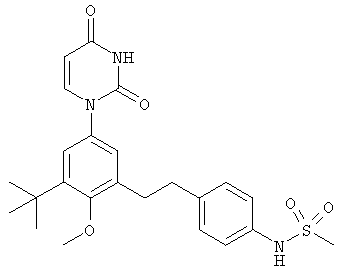
Продукт из Примера 41B, Части M (200 мг, 0,426 ммоль) растворяли в MeOH (10 мл) с последующим добавлением 10% палладия на активированном угле (50 мг). Полученную в результате смесь вакуумировали и присоединяли водородную шину, затем перемешивали при комнатной температуре в течение 48 ч. Смесь затем фильтровали через целит и фильтрат концентрировали под вакуумом до масла, которое растворяли в этаноле (4 мл), затем добавляли 1 н раствор водного гидроксида натрия (3,8 мл, 3,8 ммоль), и этот раствор перемешивали при комнатной температуре в течение 18 ч. Этанол затем удаляли под вакуумом и добавляли 1 н раствор водной соляной кислоты (4 мл) для подкисления смеси с последующей экстракцией с использованием EtOAc (2×10 мл). Органические экстракты объединяли, сушили и очищали с помощью колоночной хроматографии на силикагеле, используя 5% MeOH в CH2Cl2 в качестве элюента с получением названного соединения в виде бесцветного твердого вещества (82 мг, 41%). 1H ЯМР (300MHz, DMSO-d6) δ 11.39 (s, 1H), 9.60 (s, 1H), 7.65 (d, J=8.1Hz, 1H), 7.23 (m, 3H), 7.17 (m, 3H), 5.64 (d, J=7.7Hz, 1H), 3.77 (s, 3H), 2.93 (s, 3H), 2.88 (br s, 4H), 1.35 (s, 9H).
Пример 139. Получение (E)-N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-этоксистирил)фенил)метансульфонамида (соединение IB-L1-1.30).
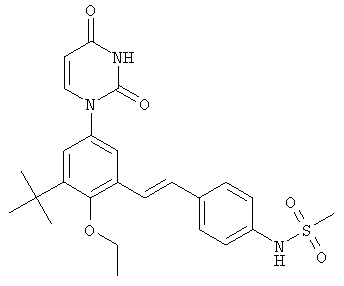
Часть A. Получение 2-трет-бутил-4-йодфенола.
В 250 мл круглодонную колбу добавляли 2-трет-бутилфенол (3,76 г, 25 ммоль) в MeOH (50,0 мл) с получением бесцветного раствора. Добавляли гидроксид натрия (1,200 г, 30,0 ммоль), и смесь перемешивали до полного растворения гидроксида. Раствор охлаждали до 0°C и обрабатывали йодидом натрия (1,75 г, 11,6 ммоль) с последующим добавлением по каплям 10% раствора гипохлорита натрия (7,2 мл, 11,6 ммоль). Добавление йодида натрия с последующим добавлением гипохлорита натрия повторяли дважды, и смесь перемешивали при 0°C в течение 30 мин. Смесь обрабатывали 10% масс/масс раствором тиосульфата натрия, перемешивали в течение 30 мин и обрабатывали по каплям концентрированной HCl до постоянного рН 1. Смесь экстрагировали 3× EtOAc. Экстракты объединяли, промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали. Неочищенное масло подвергали флэш-хроматографии на картридже с диоксидом кремния Isco 80 г, элюируя гексаном до >4:1 гексан/EtOAc с получением желтого масла (5,2 г, 75%).
Часть B. Получение 2-бром-6-трет-бутил-4-йодфенола.
В 250 мл круглодонную колбу добавляли продукт из Части A (4,8 г, 17,38 ммоль) и 1,3-дибром-5,5-диметилгидантоин (2,61 г, 9,13 ммоль) в хлороформе (87 мл) с получением оранжевого раствора. Реакционную смесь перемешивали в течение 2 ч, получая в результате черный раствор, который промывали водой, солевым раствором, сушили (Na2SO4) и концентрировали. Черное масло подвергали флэш-хроматографии на картридже с диоксидом кремния 120 г Isco, с получением розоватого твердого вещества (4,84 г, 78%).
Часть C. Получение 1-бром-3-трет-бутил-2-этокси-5-йодбензола.
В 50 мл круглодонную колбу добавляли продукт из Части B (888 мг, 2,5 ммоль), этилйодид (409 мг, 2,63 ммоль), и карбонат калия (415 мг, 3,00 ммоль) в ацетоне (12 мл) с получением зеленой суспензии. Смесь нагревали с обратным холодильником в течение 16 ч, охлаждали и концентрировали. Остаток разделяли между водой и EtOAc. Органический слой промывали дважды солевым раствором, сушили над Na2SO4, фильтровали и концентрировали до красного масла. Это масло подвергали флэш-хроматографии на картридже с диоксидом кремния Isco 40 г, элюируя гексаном с получением прозрачного масла (820 мг, 86%).
Часть D. Получение 1-(3-бром-5-трет-бутил-4-этоксифенил)пиримидин-2,4(1H,3H)-диона. В 20 мл микроволновую трубку под струей азота добавляли продукт из Части C (0,4 г, 1,044 ммоль), 1H-пиримидин-2,4-дион (0,140 г, 1,253 ммоль), и трехосновный фосфат калия (0,465 г, 2,193 ммоль) в DMSO (5 мл) с получением бесцветной суспензии. Добавляли N-(2-цианофенил)пиколинамид (0,047 г, 0,209 ммоль), и смесь барботировали азотом в течение 10 мин. Добавляли йодид меди (I) (0,020 г, 0,104 ммоль), и эту смесь барботировали еще раз в течение 10 мин, помещали в атмосферу азота и нагревали при 60°C в течение 18 ч. Смесь охлаждали и распределяли между EtOAc и водой, доводя pH до 1, используя HCl. Водный слой экстрагировали 2× EtOAc. Органические экстракты объединяли, промывали водой, насыщенным NaHCO3, и насыщенным NaCl. Органический слой сушили (Na2SO4), перемешивали с 3-меркаптопропил-функционализированным диоксидом кремия в течение 1 ч, фильтровали и концентрировали. Сырой продукт очищали с помощью хроматографии картридже с диоксидом кремния Isco 12 г, элюируя 2% MeOH в CH2Cl2 с получением белого порошка (266 мг, 69%).
Часть E. Получение (E)-N-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-этоксистирил)фенил)метансульфонамида.
Смесь продукта из Части D (55,1 мг, 0,15 ммоль), продукта из Примера 41 В, Части В (36,2 мг, 0,150 ммоль), трехосновного фосфата калия (63,7 мг, 0,300 ммоль) и 1,1'-бис(ди-трет-бутилфосфино)ферроцен палладия дихлорида (4,89 мг, 7,50 мкмоль) в THF (3 мл) и воде (1 мл) барботировали в течение 10 мин азотом, и затем герметично закрывали и нагревали при 50°C в течение 4 ч. Смесь охлаждали до комнатной температуры и разбавляли в EtOAc. EtOAc слой промывали 1М HCl, насыщенным NaHCO3, насыщенным NaCl, сушили (Na2SO4) и одновременно обрабатывали меркаптопропилсиликагелем, фильтровали и концентрировали. Сырой продукт очищали с помощью колоночной хроматографии на силикагеле, используя 2% MeOH в CH2Cl2 в качестве элюента с получением названного соединения в виде твердого вещества (40 мг, 55%) Температура плавления 265-266°C. 1H ЯМР (300 MHz, DMSO-d6) δ 11.42 (s, 1 H) 9.87 (s, 1 H) 7.76 (d, J=8.09 Hz, 1 H) 7.55-7.66 (m, 3 H) 7.17-7.27 (m, 5 H) 5.65 (dd, J=7.72, 1.47 Hz, 1 H) 3.89 (q, J=6.74 Hz, 2 H) 3.02 (s, 3 H) 1.45 (t, J=6.99 Hz, 3 H) 1.39 (s, 9 H).
Пример 140. Получение N-(4-((3-циклопропил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)фенил)метансульфонамида (соединение IA-L2-1.23).
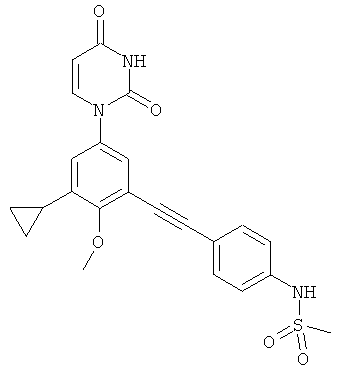
Часть A. Получение 3-бром-2-метокси-5-нитробензальдегида.
В сосуд для реакций под давлением соотверствующего размера, помещали коммерчески доступный 3-бром-2-гидрокси-5-нитробензальдегид (2,46 г, 10,00 ммоль), йодметан (6,23 мл, 100 ммоль), оксид серебра (2,317 г, 10,00 ммоль) и ацетонитрил (50 мл). Этот сосуд герметично закрывали и нагревали с перемешиванием при 50°C в течение 2 ч. Реакционную смесь фильтровали через целит, и фильтрат концентрировали с получением твердого остатка, который растирали с гексаном, собирали фильтрованием и сушили. Полученное в результате твердое вещество очищали с помощью хроматографии на силикагеле (гексан-этилацетат) с получением названного соединения (1,2 г, 46%).
Часть B. Получение 3-циклопропил-2-метокси-5-нитробензальдегида.
Продукт из Части A (0,65 г, 2,50 ммоль) объединяли с циклопропилбороновой кислотой (0,279 г, 3,25 ммоль), фосфатом калия (1,85 г, 8,75 ммоль), трициклогексилфосфин тетрафторборатом (0,092 г, 0,250 ммоль), ацетатом палладия (II) (0,028 г, 0,125 ммоль) и смесью толуол-вода, 20:1 об/об (12 мл) в микроволновой трубке. Реакционную смесь продували азотом в течение 5 мин, затем нагревали при 100°C в микроволновом реакторе в течение 20 мин. Смесь в дальнейшем разделяли этилацетатом и водой. Органическую фазу промывали солевым раствором и концентрировали в вакууме с получением остатка, который очищали с помощью хроматографии на силикагеле (гексан-этилацетат) с получением названного соединения в виде твердого вещества белого цвета (0,47 г, 85%).
Часть C. Получение 1-циклопропил-3-этинил-2-метокси-5-нитробензола.
Продукт из Части B (0,20 г, 0,904 ммоль) взаимодействовал с 1-диазо-2-оксопропилфосфонатом (0,182 г, 0,949 ммоль), полученным по методу Ohira, Syn. Comm. 19 (3&4) 561-564 (1989), как описано в Примере 44 Части G. Сырой продукт очищали с помощью хроматографии на силикагеле (этилацетат-гексан) с получением названного соединения в виде бледно-желтого масла, которое кристаллизуется при стоянии (0,155 г, 79%).
Часть D. Получение N-(4-((3-циклопропил-2-метокси-5-нитрофенил)этинил)фенил)метансульфонамида.
Продукт из Части C (0,155 г, 0,712 ммоль) объединяли в круглодонной колбе с N-(4-йодфенил)метансульфонамидом (0,212 г, 0,712 ммоль), полученным, как в Примере 43, Части F, заменяя 4-йоданилин на 4-йод-3-метиланилин. В эту смесь добавляли йодид меди (I) (10,31 мг, 0,054 ммоль), бис(трифенилфосфин)палладий(П)хлорид (0,034 г, 0,048 ммоль), триэтиламин (0,496 мл, 3,56 ммоль) и безводный ацетонитрил (14 мл). Присоединяли обратный холодильник, и смесь продували азотом в течение 5 мин, и затем нагревали в атмосфере азота на масляной бане при 80°C в течение 1 ч. Реакционную смесь концентрировали в вакууме, и сырой продукт очищали с помощью хроматографии на колонке с силикагелем, элюируя этилацетатом-гексаном с получением названного соединения в виде твердого вещества желтого цвета (0,1887 г, 68,6%).
Часть E. Получение N-(4-((5-амино-3-циклопропил-2-метоксифенил)этинил)фенил)метансульфонамида.
Продукт из Части D (0,184 г, 0,475 ммоль) объединяли с железом (0,133 г, 2,38 ммоль) и хлоридом аммония (0,050 г, 0,932 ммоль) в круглодонной колбе. В эту колбу добавляли этанол (1 мл), тетрагидрофуран (THF) (2 мл) и воду (2 мл), и полученную в результате смесь нагревали при интенсивном перемешивании в атмосфере азота до 80°C на масляной бане всего 4,5 ч. При завершении нагревания реакционную смесь фильтровали через слой песка и целита. Фильтровальный слой промывали THF и объединенные фильтраты концентрировали в вакууме. Остаток разделяли между водой и дихлорметаном. Органические слои промывали водой и затем сушили (MgSO4), и концентрировали. Остаток очищали с помощью хроматографии на силикагеле, элюируя этилацетатом-гексаном с получением названного соединения в виде масла янтарного цвета (0,1098 г, 65%).
Часть F. Получение N-(4-((3-циклопропил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)фенил)метансульфонамида.
Продукт из Части E (0,1098 г, 0,308 ммоль) взаимодействовал с акриловой кислотой (1,27 мл, 18,48 ммоль), как описано в Примере 43 Части E. Сырой продукт в акриловой кислоте обрабатывали мочевиной (0,094 г, 1,558 ммоль) и ледяной уксусной кислотой (1,3 мл), затем нагревали в атмосфере азота до 120°C на масляной бане в течение одиннадцати часов. Реакционную смесь концентрировали в вакууме и разбавляли водой. Полученное в результате твердое вещество собирали вакуумной фильтрацией, промывали водой и сушили в вакууме с получением названного соединения в виде твердого вещества желтого цвета (0,0877 г, 63%). Очистка с помощью хроматографии на силикагеле с использованием этилацетата-гексана, затем дихлорметана-метанола давала аналитический материал. 1H ЯМР (300 MHz, DMSO-d6) δ 10.36 (s, 1 H) 10.08 (s, 1 H) 7.53 (d, J=8.82 Hz, 2 H) 7.26 (dd, J=5.52, 2.94 Hz, 3 H) 6.84 (d, J=2.57 Hz, 1 H) 3.94 (s, 3 H) 3.73 (t, J=6.62 Hz, 2 H) 3.06 (s, 3 H) 2.68 (t, J=6.62 Hz, 2 H) 2.16 (s, 1 H) 0.94-1.03 (m, 2 H) 0.63-0.74 (m, 2 H).
С использованием представленного выше описания были получены следующие соединения:
(E)-N'-(1-(3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2'-метоксибифенил-4-ил) этилиден)метансульфонгидразид (соединение IA-L0-1.2). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.26 (s, 3 H) 2.70 (t, J=6.62 Hz, 2 H) 3.10 (s, 3 H) 3.24 (s, 3 H) 3.80 (t, J=6.62 Hz, 2 H) 7.17 (d, J=2.57 Hz, 1 H) 7.24 (d, J=2.57 Hz, 1 H) 7.57 (d, J=8.46 Hz, 2 H) 7.87 (d, J=8.46 Hz, 2 H) 10.12 (s, 1 H) 10.33 (s, 1 H).
(E)-N-((3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-3,5-дифтор-2'-метокси бифенил-4-ил)метилен)метансульфонгидразид (соединение IA-L0-1.3). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.08 (s, 3 H) 3.30 (s, 3 H) 3.80 (t, J=6.43 Hz, 2 H) 7.26 (d, J=2.57 Hz, 1 H) 7.34 (m, 3 H) 8.11 (s, 1 H) 10.36 (s, 1 H) 11.39 (s, 1 H).
(E)-N'-((3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-3-фтор-2'-метокси бифенил-4-ил)метилен)метансульфонгидразид (соединение IA-L0-1.4). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.11 (s, 3 H) 3.28 (s, 3 H) 3.80 (t, J=6.80 Hz, 2 H) 7.22 (d, J=2.57 Hz, 1 H) 7.28 (d, J=2.57 Hz, 1 H) 7.45 (m, 2 H) 7.94 (t, J=8.09 Hz, 1 H) 8.20 (s, 1 H) 10.35 (s, 1 H) 11.32 (s, 1 H).
(E)-N'-((3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2',4-диметокси-бифенил-3-ил)метилен)метансульфонгидразид (соединение IA-L0-1.5). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.03 (s, 3 H) 3.24 (s, 3 H) 3.78 (t, J=6.80 Hz, 2 H) 3.90 (s, 3 H) 7.12 (d, J=2.57 Hz, 1 H) 7.22 (m, 2 H) 7.54 (dd, J=8.46, 2.21 Hz, 1 H) 7.86 (d, J=2.21 Hz, 1 H) 8.34 (s, 1 H) 10.32 (s, 1 H) 11.05 (s, 1 H).
(E)-N'-((3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2,3-дифтор-2'-метокси бифенил-4-ил)метилен)метансульфонгидразид (соединение IA-L0-1.6). 1H ЯМР (500 MHz, DMSO-d6) δ ppm 1.38 (s, 9 H) 2.70 (t, J=6.41 Hz, 2 H) 3.11 (s, 3 H) 3.28 (s, 3 H) 3.79 (t, J=6.71 Hz, 2 H) 7.18 (d, J=2.44 Hz, 1 H) 7.35 (m, 2 H) 7.74 (t, J=7.32 Hz, 1 H) 8.19 (s, 1 H) 10.32 (s, 1 H) 11.44(s, 1 H).
(E)-N'-((3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2'-метоксибифенил-3-ил) метилен)метансульфонгидразид (соединение IA-L0-1.7). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.39 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.07 (s, 3 H) 3.23 (s, 3 H) 3.80 (t, J=6.43 Hz, 2 H) 7.17 (d, J=2.57 Hz, 1 H) 7.25 (d, J=2.57 Hz, 1 H) 7.54 (m, 2 H) 7.73 (m, 2 H) 8.06 (s, 1 H) 10.33 (s, 1 H) 11.11 (s, 1H).
(Z)-N'-(1-(3'-трет-бутил-5'-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2'-метоксибифенил-3-ил) этилиден)метансульфонгидразид (соединение IA-L0-1.8). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.26 (s, 3 H) 2.70 (t, J=6.80 Hz, 2 H) 3.07 (s, 3 H) 3.22 (s, 3 H) 3.80 (t, J=6.62 Hz, 2 H) 7.18 (d, J=2.57 Hz, 1 H) 7.24 (d, J=2.94 Hz, 1 H) 7.46-7.59 (m, 2 H) 7.79 (d, J=7.35 Hz, 1 H) 7.84 (s, 1 H) 10.11 (s, 1 H) 10.33 (s, 1 H).
N-(3-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил) оксазол-5-ил)фенил)метансульфонамид (соединение IA-L0-1.9)
1-(3-трет-бутил-4-метокси-5-(5-(3-нитрофенил)оксазол-2-ил)фенил)дигидропиримидин-2,4(1Н,3H)-дион (соединение IA-L0-1.10)
1-(3-трет-бутил-4-метокси-5-(5-(4-нитрофенил)оксазол-2-ил)фенил)дигидропиримидин-2,4(1Н,3H)-дион (соединение IA-L0-1.11)
N-(3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метоксибифенил-4-ил) метансульфонамид (соединение IB-L0-1.2). 1H ЯМР (300 MHz, CDCl3) δ ppm 1.43 (s, 9H) 3.07 (s, 3H) 3.32 (s, 3H) 5.82 (dd, J=8.09, 2.21 Hz, 1H) 6.79 (s, 1H) 7.14 (d, J=2.94 Hz, 1H) 7.21 (d, J=2.57 Hz, 1H) 7.28 (d, 2H) 7.35 (d, J=8.09 Hz, 1H) 7.54 (d, J=8.46 Hz, 2H) 8.31 (s, 1H).
(E)-N'-((3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-фтор-2'-метокси бифенил-4-ил)метилен)метансульфонгидразид (соединение IB-L0-1.3). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 11.42 (s, 1 H) 11.33 (s, 1 H) 8.21 (s, 1 H) 7.95 (s, 1 H) 7.78 (d, J=7.72 Hz, 1 H) 7.39-7.59 (m, 2 H) 7.35 (s, 2 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 3.20-3.44 (m, 3 H) 3.11 (s, 3 H) 1.40 (s, 9H).
N-(2-(3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2''-метоксибифенил-4-ил)этил)метансульфонамид (соединение IB-L0-1.4). 1H ЯМР (300 MHz, CDCl3) δ ppm 1.43 (s, 9H) 2.86-2.99 (m, 2H) 2.91 (s, 3H) 3.31 (s, 3H) 3.38-3.51 (m, 2H) 5.80 (dd, J=8.09, 2.21 Hz, 1H) 7.12 (d, J=2.57 Hz, 1H) 7.21 (d, J=2.57 Hz, 1H) 7.26-7.32 (m, 1H) 7.35 (d,.7-8.09 Hz, 1H) 7.52 (d,.7=8.09 Hz, 2H) 7.78 (d, J=7.72 Hz, 1H) 8.15 (s. 1H).
(E)-N'-(1-(3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метокси бифенил-4-ил)этилиден)метансульфонгидразид (соединение IB-L0-1.5). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 11.40 (s, 1 H) 10.13 (s, 1 H) 7.88 (d, J=8.09 Hz, 2 H) 7.78 (d, J=7.72 Hz, 1 H) 7.60 (d, J=8.09 Hz, 2 H) 7.29 (d, J=4.04 Hz, 2 H) 5.64 (dd, J=7.72, 1.84 Hz, 1 H) 3.27 (s, 3 H) 3.10 (s, 3 H) 2.27 (s, 3H) 1.40 (s, 9H).
(E)-N'-((3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2',4-диметокси бифенил-3-ил)метилен)метансульфонгидразид (соединение IB-L0-1.6). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 11.38 (s, 1 H) 11.05 (s, 1 H) 8.34 (s, 1 H) 7.88 (s, 1 H) 7.75 (d, J=8.09 Hz, 2 H) 7.41-7.66 (m, 1 H) 7.00-7.37 (m, 3 H) 5.63 (d, J=7.72 Hz, 1 H) 3.91 (s, 3 H) 3.27 (s. 3 H) 3.03 (s, 3H) 1.40 (s, 9H).
N-(3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метоксибифенил-4-илкарбамоил)метансульфонамид (соединение IB-L0-1.7). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9H) 2.50 (s, 3H) 3.26 (s, 3H) 5.63 (dd, J=7.91, 2.02 Hz, 1H) 7.23 (d, 2H) 7.38-7.63 (m, 5H) 7.76 (d, J=8.09 Hz, 1H) 8.95 (s, 1H) 10.46 (s, 1H) 11.39 (d, J=2.21 Hz, 1H).
N-(3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метоксибифенил карбонил) метансульфонгидразид (соединение IB-L0-1.8). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.41 (s, 9H) 3.03 (s, 3H) 3.26 (s, 3H) 5.65 (dd,.7=7.72, 1.84 Hz, 1H) 7.27-7.36 (m, 2H) 7.73 (dd, J=28.31,8.09 Hz, 4H) 8.01 (d, J=8.09 Hz, 2H) 9.65 (s, 1H) 10.82 (s, 1H) 11.41 (s, 1H).
(E)-N'-(1-(3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метокси бифенил-3-ил)этилиден)метансульфонгидразид (соединение IB-L0-1.9). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 11.40 (s, 1 H) 10.12 (s, 1 H) 7.82-7.99 (m, 1Н) 7.62-7.82 (m, 2 H) 7.35-7.71 (m, 2 H) 7.30 (s, 2 H) 5.64 (dd, J=7.91, 2.02 Hz, 1 H) 3.26 (s, 3 H) 3.07 (s, 3 H) 2.27 (s, 3 H) 1.40 (s, 9 H).
(E)-N'-((3'-трет-бутил-5'-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2'-метокси бифенил-3-ил)метилен)метансульфонгидразид (соединение IB-L0-1.10). 1H ЯМР (300 MHz, CDCl3) δ ppm 1.44 (s, 9H) 3.19 (s, 3H) 3.31 (s, 3H) 5.44 (s, 1H) 5.82 (d, J=7.72 Hz, 1H) 7.17 (d, J=2.57 Hz, 1H) 7.24 (d, J=2.57 Hz, 1H) 7.36 (d, J=7.72 Hz, 1H) 7.48 (t, J=7.72 Hz, 1H) 7.63 (d, J=7.72 Hz, 1H) 7.70 (d, J=7.72 Hz, 1H) 7.80 (s, 1H) 7.88 (s, 1H) 8.32 (s, 1H).
N-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-1Н-бензо[d]имидазол-5-ил)-N-(метилсульфонил)метансульфонамид (соединение IA-L0-2.10) 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.45 (s, 9 Н) 2.73 (t, J=6.62 Hz, 2 Н) 3.48 (s, 3 Н) 3.56 (s, 6 Н) 3.83 (t, J=6.80 Hz, 2 Н) 4.05 (s, 1 Н) 7.38 (dd, J=8.46, 1.84 Hz, 1 Н) 7.46 (d, J=2.57 Hz, 1 Н) 7.71 (d, J=8.46 Hz, 1 Н) 7.76 (d, J=2.57 Hz, 1 Н) 7.82 (d, J=1.84 Hz, 1 Н) 10.41 (s, 1 Н)
N-((6-(3-трет-бутил-2-хлор-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)фенил)бензо[b]тиофен-3-ил)метил)метансульфонамид (соединение IB-L0-2.35). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.52 (s, 9 Н) 2.95 (s, 3 Н) 4.44 (d, J=5.88 Hz, 2 Н) 5.68 (d, J=8.09 Hz, 1 Н) 7.40 (d, J=2.57 Hz, 1 Н) 7.46 (dd, J=8.09, 1.47 Hz, 1 Н) 7.56 (d, J=2.57 Hz, 1 Н) 7.62 (t, J=6.07 Hz, 1 Н) 7.72 (s, 1 Н) 7.83 (d, J=8.09 Hz, 1 Н) 8.01 (m, 2 Н) 11.46 (s, 1 H).
1-(3-трет-бутил-5-(2-хлорбензо[d]тиазол-6-ил)-4-метоксифенил)пиримидин-2,4(1H,3H)-дион (соединение IB-L0-2.38). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.41 (s, 9 Н) 3.24 (s, 3 Н) 5.65 (dd, J=8.09, 2.21 Hz, 1 Н) 7.34 (s, 2 Н) 7.73 (dd, J=8.64, 1.65 Hz, 1 Н) 7.79 (d, J=8.09 Hz, 1 H) 8.07 (d, J=8.46 Hz, 1 H) 8.30 (d, J=1.84 Hz, 1 H) 11.42 (d, J=1.84 Hz, 1 H)
N-(2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)хинолин-6-ил)метансульфонамид (соединение IB-L0-2.48).
1-(3-трет-бутил-4-метокси-5-(1-оксо-2,3-дигидро-1H-инден-5-ил)фенил)пиримидин-2,4(1H,3H)-дион (соединение IB-L0-2.50).
N,N'-(6,6'-(5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-1,3-фенилен) бис(нафтален-6,2-диил))диметансульфонамид (соединение IB-L0-2.76). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 3.08 (s, 6 H) 3.13 (s, 3 H) 5.72 (d, J=8.18 Hz, 1 H) 7.43 (dd, J=8.46, 1.84 Hz, 2 H) 7.59 (s, 2 H) 7.79 (m, 4 H) 7.96 (m, 5 H) 8.14 (s, 2 H) 10.05 (s, 2 H) 11.48 (s, 1 H).
(E)-N-(4-(1-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил) проп-1-ен-2-ил)фенил)метансульфонамид (соединение IA-L1-1.6). 1H ЯМР (300 MHz, DMSO-d6) δ 2.14 (s, 3 H) 2.70 (t, J=6.62 Hz, 2 H) 3.01 (s, 3 H) 3.68 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 6.82 (s, 1 H) 7.10-7.17 (m, 2 H) 7.23 (d, J=8.46 Hz, 2 H) 7.59 (d, J=8.46 Hz, 2 H) 9.78 (s, 1 H) 10.32 (s, 1 H). (Z)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксистирил)фенил) метансульфонамид (соединение IA-L1-1.10). 1H ЯМР (300 MHz, DMSO-d6) δ 10.23 (s, 1 H) 9.74 (s, 1 H) 7.23 (d, J=8.46 Hz, 2 H) 7.13 (d, J=2.57 Hz, 1 H) 7.06 (d, J=8.82 Hz, 2 H) 6.92 (d, J=2.57 Hz, 1 H) 6.54-6.67 (m, 2 H) 3.78 (s, 3 H) 3.57 (t, J=6.62 Hz, 2 H) 2.96 (s, 3 H) 2.60 (t, J=6.80 Hz, 2 H) 1.34 (s, 9 H).
(Е)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксистирил) фенил)-N-(метилсульфонил)ацетамид (соединение IA-L1-1.11). 1H ЯМР (300 MHz, DMSO-d6) δ 10.36 (s, 1 H) 7.77 (d, J=8.46 Hz, 2 H) 7.56 (d, J=2.21 Hz, 1 H) 7.39-7.50 (m, 3 H) 7.25 (d, J=16.55 Hz, 1 H) 7.19 (d, J=2.57 Hz, 1 H) 3.74-3.85 (m, 5 H) 3.54 (s, 3 H) 2.72 (t, J=6.62 Hz, 2 H) 1.94 (s, 3 H) 1.38 (s, 9 H).
(Е)-1-(3-(4-аминостирил)-5-трет-бутил-4-метоксифенил)дигидропиримидин-2,4(1H,3H)-дион (соединение IA-L1-1.13). 1H ЯМР (300 MHz, DMSO-d6) δ 1.36 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.74 (s, 3 H) 3.77 (t, J=6.62 Hz, 2 H) 5.34 (s, 1 H) 6.57 (d, J=8.46 Hz, 2 H) 6.98 (s, 1 H) 7.07 (d, J=2.21 Hz, 1 H) 7.17 (s, 2 H) 7.30 (d, J=8.09 Hz, 2 H) 7.45 (d, J=2.21 Hz, 1 H) 10.32 (s, 1 H).
(Z)-N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксистирил)фенил) метансульфонамид (соединение IA-L1-1.20). 1H ЯМР (500 MHz, DMSO-d6): δ ppm 1.37 (s, 9H), 2.71 (t, J=6.7Hz, 2H), 3.01 (s, 3H), 3.75 (s, 3H), 3.79 (t, J=6.6Hz, 2H), 7.13 (d, J=16.5Hz, 1H), 7.15 (d, J=2.4Hz, 2H), 7.23 (d, J=8.5Hz, 2H), 7.25 (d, J=16.5 Hz, 1H), 7.51 (d, J=2.4Hz, 1H), 7.61 (d, J=8.6 Hz, 2H), 9.80 (bs, 1H), 10.30 (s, 1H).
N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-1-фторвинил)фенил)метансульфонамид (соединение IA-L1-1.21). (рацемическая смесь (1:1) соединений IA-L1-1.4 и IA-L1-1.5).
(E)-1-(3-трет-бутил-4-метокси-5-(4-нитростирил)фенил)дигидропиримидин-2,4(1H,3H)-дион (соединение IA-L1-1.22).
1-{3-трет-бутил-5-[(Z)-2-хлор-2-(4-нитро-фенил)-винил]-4-метокси-фенил}-дигидро-пиримидин-2,4-дион (соединение IA-L1-1.23).
1-{3-трет-бутил-4-метокси-5-[(Z)-2-(4-нитро-фенил)-пропенил]-фенил}-дигидро-пиримидин-2,4-дион (соединение IA-L1-1.24).
1-{3-трет-бутил-5-[(E)-2-(4-нитро-фенил)-винил]-фенил}-дигидро-пиримидин-2,4-дион (соединение IA-L1-1.25). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.33 (s, 9 H) 2.70-2.77 (m, 2 H) 3.84 (t, J=6.80 Hz, 2 H) 7.33 (s, 1 H) 7.49 (d, J=4.04 Hz, 2 H) 7.56 (d, J=5.88 Hz, 2 H) 7.89 (d, J=8.82 Hz, 2 H) 8.25 (d, J=8.82 Hz, 2 H) 10.40 (s, 1 H)
N-(4-{(E)-2-[3-трет-бутил-5-(диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-фенил]-винил}-3-метокси-фенил)-метансульфонамид (соединение IA-L1-1.27). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 10.33 (s, 1 H) 9.86 (s, 1 H) 7.64 (d, J=8.46 Hz, 1 H) 7.45 (d, J=2.21 Hz, 1 H) 7.26 (s, 2 H) 7.12 (d, J=2.21 Hz, 1 H) 6.89 (s, 1 H) 6.85 (dd, J=8.46, 1.84 Hz, 1 H) 3.84 (s, 3 H) 3.78 (t, J=6.80 Hz, 2 H) 3.74 (s, 3 H) 3.04 (s, 3 H) 2.71 (t, J=6.62 Hz, 2 H) 1.37 (s, 9 H)
N-(4-{(E)-2-[3-трет-Бутил-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-2-метокси-фенил]-винил}-3-формил-фенил)-метансульфонамид (соединение IB-L1-1.6). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.39 (s, 9 H) 3.07 (s, 3 H) 3.81 (s, 3 H) 5.66 (dd, J=7.72, 2.21 Hz, 1 H) 7.26 (d, J=2.57 Hz, 1 H) 7.30 (d, J=16.18 Hz, 1 H) 7.51 (dd, J=8.64, 2.39 Hz, 1 H) 7.69 (d, J=2.57 Hz, 1 H) 7.73-7.78 (m, 2 H) 7.97 (d, J=8.82 Hz, 1 H) 8.06 (d, J=16.18 Hz, 1 H) 10.15 (s, 1 H) 10.45 (s, 1 H)11.43(d,J=2.21 Hz, 1 H)
N-[4-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-2-метокси-фенил]-винил}-3-(гидроксиимино-метил)-фенил]-метансульфонамид (соединение IB-L1-1.8). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 3.03 (s, 3 H) 3.79 (s, 3 H) 5.66 (dd, J=7.91, 2.02 Hz, 1 H) 7.16 (d, J=15.81 Hz, 1 H) 7.22 (d, J=2.57 Hz, 1 H) 7.26 (dd, J=8.64, 2.39 Hz, 1 H) 7.59 (d, J=16.18 Hz, 1 H) 7.63 (d, J=2.21 Hz, 1 H) 7.73-7.83 (m, 3 H) 8.64 (s, 1 H) 9.96 (s, 1 H) 11.42 (d, J=2.21 Hz, 1 H) 11.50 (s, 1 H).
2-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил}-5-метансульфониламино-N-(2-метокси-этил)-бензамид (соединение IB-L1-1.9). 1H ЯМР (300 MHz, DMSO-D6) δ 1.38 (s, 9 H) 3.05 (s, 3 H) 3.20 (s, 3 H) 3.37-3.49 (m, 4 H) 3.78 (s, 3 H) 5.64 (d, J=7.72 Hz, 1 H) 7.15 (d, J=2.57 Hz, 1 H) 7.20 (d, J=2.57 Hz, 1 H) 7.24 (s, 2 H) 7.28 (dd, J=8.46, 2.21 Hz, 1 H) 7.42 (d, J=2.57 Hz, 1 H) 7.73 (d, J=7.72 Hz, 1 H) 7.87 (d, J=8.82 Hz, 1 H) 8.49 (t, J=5.15 Hz, 1 H) 9.99 (s, 1 H) 11.42 (s, 1 H).
Этиловый эфир 2-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил}-5-метансульфониламино-бензойной кислоты (соединение IB-L1-1.11). 1H ЯМР (300 MHz, DMSO-d6) δ 1.31 (t, J=7.17 Hz, 3 H) 1.38 (s, 9 H) 3.05 (s, 3 H) 3.79 (s, 3 H) 4.33 (q, J=7.23 Hz, 2 H) 5.65 (dd, J=7.72, 2.21 Hz, 1 H) 7.15-7.25 (m, 2 H) 7.46 (dd, J=8.64, 2.39 Hz. 1 H) 7.52 (d, J=2.57 Hz, 1 H) 7.68 (d, J=2.57 Hz, 1 H) 7.71-7.81 (m, 2 H) 7.90 (d, J=8.46 Hz, 1 H) 10.06 (s, 1 H) 11.42 (d, J=1.84 Hz, 1 H).
N-(4-{(E)-2-[3-трет-бутил-2-хлор-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-фенил]-винил}-фенил)-метансульфонамид (соединение IB-L1-1.12). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.49 (s, 9 H) 3.02 (s, 3 H) 5.69 (d, J=7.72 Hz, 1 H) 7.22 (m, 3 H) 7.41 (d, J=2.21 Hz, 1 H) 7.51 (d, J=16.18 Hz, 1 H) 7.59 (d, J=8.82 Hz, 2 H) 7.78 (d, J=2.21 Hz, 1 H) 7.80 (d, J=8.09 Hz, 1 H) 9.90 (s, 1 H) 11.47 (s, 1 H).
2-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил}-5-метансульфониламино-N,N-диметил-бензамид (соединение IB-L1-1.14). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.76 (s, 3 H) 3.03 (s, 3 H) 3.05 (s, 3 H) 3.76 (s, 3 H) 5.64 (dd, J=7.91, 1.65 Hz. 1 H) 6.95 (d, J=16.55 Hz, 1 H) 7.02 (d, J=2.21 Hz, 1 H) 7.17-7.25 (m, 2 H) 7.27 (dd, J=8.64, 2.39 Hz, 1 H) 7.48 (d, J=2.57 Hz, 1 H) 7.74 (d, J=8.09 Hz, 1 H) 7.82 (d, J=8.82 Hz, 1 H) 10.03 (s, 1 H) 11.39-11.43 (m, 1 H).
2-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил}-5-метансульфониламино-N-метил-бензамид (соединение IB-L1-1.17). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 2.77 (d, J=4.41 Hz, 3 H) 3.06 (s, 3 H) 3.77 (s, 3 H) 5.64 (dd, J=7.72, 1.84 Hz, 1 H) 7.16-7.33 (m, 5 H) 7.43 (d, J=2.21 Hz, 1 H) 7.73 (d, J=7.72 Hz, 1 H) 7.84 (d, J=8.46 Hz, 1 H) 8.37 (q, J=4.41 Hz, 1 H) 10.00 (s, 1 H) 11.40 (d, J=1.84 Hz, 1 H).
2-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил}-N-(1,1-диоксо-тетрагидро-1 лямбда*6*-тиофен-3-ил)-5-метансульфониламино-N-метил-бензамид (соединение IB-L1-1.18). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.17-2.47 (m, 2 H) 2.70 (s, 3 H) 3.06 (s, 3 H) 3.15-3.31 (m, 2 H) 3.36-3.51 (m, 2 H) 3.77 (s, 3 H) 5.37 (dt, J=17.74, 8.96 Hz, 1 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 6.93 (d, J=16.18 Hz, 1 H) 7.05 (d, J=2.21 Hz, 1 H) 7.19-7.35 (m, 3 H) 7.50 (d, J=2.57 Hz, 1 H) 7.76 (d, J-8.09 Hz, 1 H) 7.87 (d, J=8.82 Hz, 1 H) 10.04 (s, 1 H) 11.38 (d, J=2.21 Hz, 1 H).
N-(4-{(E)-2-[3-трет-бутил-5-(5-хлор-2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-2-метокси-фенил]-винил}-фенил)-метансульфонамид (соединение IB-L1-1.20). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 11.31 (s, 1 H) 9.77 (s, 1 H) 7.53 (d, J=8.09 Hz, 1 H) 7.23 (d, J=8.46 Hz, 2 H) 7.17 (d, J=2.57 Hz, 1 H) 7.06 (d, J=8.82 Hz, 2 H) 7.01 (d, J=2.57 Hz, 1 H) 6.53-6.71 (m, 2 H) 5.56 (d, J=7.72 Hz, 1 H) 3.81 (s, 3 H) 2.96 (s, 3 H) 1.35 (s, 9 H)
2-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-2-метокси-фенил]-винил}-5-метансульфониламино-бензамид (соединение IB-L1-1.21). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 3.07 (s, 3 H) 3.78 (s, 3 H) 5.64 (d, J=7.72 Hz, 1 H) 7.18-7.34 (m, 5 H) 7.43 (d, J=2.21 Hz, 1 H) 7.54 (s, 1 H) 7.73 (d, J=7.72 Hz, 1 H) 7.84 (d, J=8.46 Hz, 1 H) 7.93 (s, 1 H).
N-(3-(азетидин-1-карбонил)-4-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-2-метокси-фенил]-винил}-фенил)-метансульфонамид (соединение (соединение IB-L1-1.22). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 3.07 (s, 3 H) 3.78 (s, 3 H) 5.64 (d, J=7.72 Hz, 1 H) 7.18-7.34 (m, 5 H) 7.43 (d, J=2.21 Hz, 1 H) 7.54 (s, 1 H) 7.73 (d, J=7.72 Hz, 1 H) 7.84 (d, J=8.46 Hz, 1 H) 7.93 (s, 1 H).
2-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-2-метокси-фенил]-винил}-5-метансульфониламино-N-(2-метокси-этил)-N-метил-бензамид (соединение IB-L1-1.24). 1H ЯМР (300 MHz, DMSO-d6) δ 1.40 (s, 9 H) 2.81 (s, 3 H) 3.07 (s, 3 H) 3.23 (s, 3 H) 3.29 (t, J=5.33 Hz, 1 H) 3.39 (t, J=4.96 Hz, 1 H) 3.62 (t, J=4.78 Hz, 2 H) 3.82 (s, 3 H) 5.68 (d, J=8.09 Hz, 1 H) 6.96-7.07 (m, 1 H) 7.09-7.17 (m, 1 H) 7.23-7.38 (m, 3 H) 7.49 (dd, J=16.55, 2.57 Hz, 1 H) 7.71-7.76 (m, 1 H) 7.83-7.94 (m, 1 H).
N-(4-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил }-3-изопропоксиметил-фенил)-метансульфонамид (соединение IB-L1-1.25). 1H ЯМР (300 MHz, DMSO-d6) δ 1.16 (d, J=5.88 Hz, 6 H) 1.38 (s, 9 H) 3.01 (s, 3 H) 3.69 (dt, J=12.13, 6.07 Hz, 1 H) 3.79 (s, 3 H) 4.59 (s, 2 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 7.13-7.29 (m, 4 H) 7.32-7.40 (m, 1 H) 7.59 (d, J=2.57 Hz, 1 H) 7.75 (d, J=8.09 Hz, 2 H) 9.86 (s, 1 H) 11.43 (d, J=1.84 Hz, 1 H).
N-[4-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил}-3-(пирролидин-1-карбонил)-фенил]-метансульфонамид (соединение IB-L1-1.27). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 1.73-1.89 (m, 4 H) 3.03-3.12 (m, 5 H) 3.51 (t, J=6.80 Hz, 2 H) 3.76 (s, 3 H) 5.64 (dd, J=7.91, 2.02 Hz, 1 H) 6.99-7.06 (m, 1 H) 7.08 (d, J=2.21 Hz, 1 H) 7.19-7.31 (m, 3 H) 7.46 (d, J=2.57 Hz, 1 H) 7.75 (d, J=8.09 Hz, 1 H) 7.82 (d, J=8.82 Hz, 1 H) 10.01 (s, 1 H) 11.41 (d, J=2.21 Hz, 1 H).
N-[4-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1 -ил)-2-метокси-фенил]-винил}-3-(3-гидрокси-азетидин-1-илметил)-фенил]-метансульфонамид (соединение IB-L1-1.29). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 2.78-2.85 (m, 2 H) 2.99 (s, 3 H) 3.50-3.58 (m, 2 H) 3.71 (s, 2 H) 3.79 (s, 3 H) 4.19 (td, J=12.41, 6.07 Hz, 1 H) 5.29 (d, J=6.25 Hz, 1 H) 5.66 (d, J=8.09 Hz, 1 H) 7.10-7.18 (m, 2 H) 7.20 (t, J=2.21 Hz, 2 H) 7.35 - 7.42 (m, I H) 7.63 (d, J=2.57 Hz, 1 H) 7.69 (d, J=8.46 Hz, 1 H) 7.76 (d, J=7.72 Hz, 1 H) 9.78 (s, 1 H) 11.42 (s, 1 H).
N-(4-{(E)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил}-3-пирролидин-1-илметил-фенил)-метансульфонамид (соединение IB-L1-1.33). 1H ЯМР (500 MHz, DMSO-d6) δ 1.40 (s, 9 H) 1.72-1.95 (m, 4 H) 2.84 (s, 2 H) 2.88-2.98 (m, 2 H) 3.01 (s, 3 H) 3.81 (s, 3 H) 3.86-4.23 (m, 2 H) 5.63 (d, J=7.81 Hz, I H) 7.17 (d, J-15.63 Hz, I H) 7.21-7.28 (m, 2 H) 7.32-7.38 (m, 1 H) 7.47 (d, J=16.11 Hz, 1 H) 7.53-7.59 (m, 1 H) 7.61 (d, J=7.81 Hz, 1 H) 7.70 (d, J=6.35 Hz, 1 H) 9.42 (s, 1 H) 10.88 (s, 1 H).
N-(4-{(Z)-2-[3-трет-бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-2-метокси-фенил]-винил}-фенил)-метансульфонамид (соединение IB-L1-1.34 1H ЯМР (300 MHz, DMSO-D6) δ ppm 11.31 (s, 1 H) 9.77 (s, 1 H) 7.53 (d, J=8.09 Hz, 1 H) 7.23 (d, J=8.46 Hz, 2 H) 7.17 (d, J=2.57 Hz, 1 H) 7.06 (d, J=8.82 Hz, 2 H) 7.01 (d, J=2.57 Hz, 1 H) 6.53-6.71 (m, 2 H) 5.56 (d, J=7.72 Hz, 1 H) 3.81 (s, 3 H) 2.96 (s, 3 H) 1.35 (s, 9 H)
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)фенил)этинил)фенил)-метансульфонамид (соединение IA-L2-1.1). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.30 (s, 9 H) 2.72 (t, J=6.43 Hz, 2 H) 3.06 (s, 3 H) 3.82 (t, J=6.62 Hz, 2 H) 7.24 (d, J=8.82 Hz, 2 H) 7.33 (s, 1 H) 7.39 (d, J=1.47 Hz, 2 H) 7.54 (d, J=8.82 Hz. 2 H) 10.08 (s, 1 H) 10.40 (s, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)фенил)этинил)-3-метил-фенил)метансульфонамид (соединение IA-L2-1.2). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.30 (s, 9 H) 2.44 (s, 3 H) 2.72 (t, J=6.62 Hz, 2 H) 3.05 (s, 3 H) 3.82 (t, J=6.62 Hz, 2 H) 7.07 (dd, J=8.46, 1.47 Hz, 1 H) 7.13 (s, 1 H) 7.33 (s, 1 H) 7.39 (d, J=1.47 Hz, 2 H) 7.48 (d, J=8.09 Hz, 1 H) 9.98 (s, 1 H) 10.40 (s, 1 H)
1-(3-трет-бутил-4-метокси-5-(фенилэтинил)фенил)дигидропиримидин-2,4(1H,3H)-дион (соединение IA-L2-1.4). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.78 (t, J=6.43 Hz, 2 H) 4.07 (s, 3 H) 7.27 (d, J=2.57 Hz, 1 H) 7.38 (d, J=2.57 Hz, 1 H) 7.41-7.49 (m, 3 H) 7.55-7.63 (m, 2 H) 10.37 (s, 1 H).
N-(3-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил) этинил)фенил)метансульфонамид (соединение IA-L2-1.7). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.04 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.06 (s, 3 H) 7.23-7.35 (m, 3 H) 7.35-7.46 (m, 3 H) 9.94 (s, 1 H) 10.37 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил) этинил)-фенил)метансульфонамид (соединение IA-L2-1.8). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.35 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.06 (s, 3 H) 3.77 (t, J=6.62 Hz, 2 H) 4.05 (s, 3 H) 7.25 (dd, J=5.52, 2.94 Hz, 3 H) 7.35 (d, J=2.57 Hz, 1 H) 7.55 (d, J=8.46 Hz, 2 H) 10.09 (s, 1 H) 10.37 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил) этинил)-2-метилфенил)метансульфонамид (соединение IA-L2-1.10). 1H ЯМР (300 MHz, DMSO-d6) δ 1.35 (s, 9 H) 2.32 (s, 3 H) 2.70 (t, J=6.62 Hz, 2 H) 3.03 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.06 (s, 3 H) 7.26 (d, J=2.57 Hz, 1 H) 7.32-7.44 (m, 3 H) 7.48 (s, 1 H) 9.23 (s, 1 H) 10.37 (s, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил) этинил)-3-этилфенил)метансульфонамид (соединение IA-L2-1.11). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.15-1.28 (m, 3 H) 1.36 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 2.81 (q, J=7.72 Hz, 2 H) 3.05 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.04 (s, 3 H) 7.10 (dd, J=8.27, 2.02 Hz, 1 H) 7.14 (s, 1 H) 7.25 (d, J=2.57 Hz, 1 H) 7.35 (d, J=2.21 Hz, 1 H) 7.50 (d, J=8.09 Hz, 1 H) 10.01 (s, 1 H) 10.36 (s, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил) этинил)-3-фторфенил)метансульфонамид (соединение IA-L2-1.12). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.35 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.13 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.05 (s, 3 H) 6.98-7.20 (m, 2 H) 7.28 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.57 Hz, 1 H) 7.61 (t, J=8.27 Hz, 1 H) 10.37 (s, 2 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил) этинил)-2-фторфенил)метансульфонамид (соединение IA-L2-1.13). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.35 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.10 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.05 (s, 3 H) 7.28 (d, J=2.57 Hz, 1 H) 7.38 (d, J=2.57 Hz, 1 H) 7.42-7.61 (m, 3 H) 9.89 (s, 1 H) 10.38 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил) этинил)-3-хлорфенил)метансульфонамид (соединение IA-L2-1.14). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.35 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.12 (s, 3 H) 3.78 (t, J=6.80 Hz, 2 H) 4.07 (s, 3 H) 7.21 (dd, J=8.46, 2.21 Hz, 1 H) 7.28 (d, J=2.57 Hz, 1 H) 7.36 (dd, J=4.60, 2.39 Hz, 2 H) 7.67 (d, J=8.46 Hz, 1 H) 10.32 (s, 1 H) 10.37 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил) этинил)-2-хлорфенил)метансульфонамид (соединение IA-L2-1.15). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.35 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.10 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.06 (s, 3 H) 7.29 (d, J=2.94 Hz, 1 H) 7.39 (d, J=2.57 Hz, 1 H) 7.48-7.59 (m, 2 H) 7.75 (s, 1 H) 9.65 (s, 1 H) 10.38 (s, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил) этинил)-3-метоксифенил)метансульфонамид (соединение IA-L2-1.16). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.35 (s, 9 Н) 2.63-2.76 (m, 2 Н) 3.08 (s, 3 Н) 3.77 (t, J=6.80 Hz, 2 Н) 3.82 (s, 3 Н) 4.08 (s, 3 Н) 6.79-6.89 (m, 1 Н) 6.91 (d, J=1.84 Hz, 1 Н) 7.22 (d, J=2.57 Hz, 1 Н) 7.30 (d, J=2.94 Hz, 1 Н) 7.45 (d, J=8.46 Hz, 1 Н) 10.06 (s, 1 Н) 10.35 (s, 1 Н).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)этинил)-2-(трифторметокси)фенил)метансульфонамид (соединение IA-L2-1.17). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.35 (s, 9 Н) 2.70 (t, J=6.62 Hz, 2 Н) 3.13 (s, 3 Н) 3.78 (t, J=6.62 Hz, 2 Н) 4.05 (s, 3 Н) 7.29 (d, J=2.57 Hz, 1 Н) 7.40 (d, J=2.57 Hz, 1 Н) 7.52-7.69 (m, 3 Н) 10.06 (s, I H) 10.38 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-2-(трифторметил)фенил)метансульфонамид (соединение IA-L2-1.19). 1H ЯМР (300 MHz, DMSO-d6) 8 ppm 1.35 (s, 9 H) 2.72 (d, J=6.62 Hz, 2 H) 3.09 (s, 3 H) 3.78 (s, 2 H) 4.06 (s, 3 H) 7.29 (s, 1 H) 7.41 (s, 1 H) 7.57-7.73 (m, 1 H) 7.80-7.94 (m, 2 H) 9.62 (s, 1 H) 10.38 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-2-фтор-5-метилфенил)метансульфонамид (соединение IA-L2-1.20). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.36 (s, 9 H) 2.45 (s, 3 H) 2.71 (t, J=6.43 Hz, 2 H) 3.09 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.05 (s, 3 H) 7.28 (d, J=2.57 Hz, 1 H) 7.36 (d, J=8.09 Hz, 1 H) 7.39 (d, J=2.57 Hz, 1 H) 7.47 (d, J=11.03 Hz, 1 H) 9.80 (s, 1 H) 10.37 (s, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-3-хлор-2-фторфенил)метансульфонамид (соединение IA-L2-1.21). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 2.71 (t, J=6.43 Hz, 2 H) 3.14 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.08 (s, 3 H) 7.31 (d, J=2.94 Hz, 1 H) 7.39-7.46 (m, 1 H) 7.48 (d, J=7.72 Hz, 1 H) 7.51-7.61 (m, 1 H) 10.15 (s, 1 H) 10.38 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)этинил)-2-фтор-5-(трифторметил)фенил)метансульфонамид (соединение IA-L2-1.22). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.35 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.17 (s, 3 H) 3.78 (t, J=6.43 Hz, 2 H) 4.03 (s, 3 H) 7.32 (d, J=2.57 Hz, 1 H) 7.37 (d, J=2.21 Hz, 1 H) 7.83 (d, J=7.72 Hz, 1 H) 7.88 (d, J=10.66 Hz, 1 H) 10.27 (s, 1 H) 10.38 (s, 1 H)
N-(6-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)этинил)-5-метилпиридин-3-ил)метансульфонамид (соединение IA-L2-1.24). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.13 (s, 3 H) 3.79 (t, J=6.80 Hz, 2 H) 4.07 (s, 3 H) 7.30 (d, J=2.57 Hz, 1 H) 7.41 (d, J=2.57 Hz, 1 H) 7.55 (s, 1 H) 8.29 (s, 1 H) 10.24 (s, 1 H) 10.38 (s, 1 H).
N-(5-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-пиридин-2-ил)метансульфонамид 2,2,2-трифторацетат (соединение IA-L2-1.26). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.35 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.78 (t, J=6.80 Hz, 2 H) 4.05 (s, 3 H) 7.01 (d, J=8.82 Hz, 1 H) 7.27 (d, J=2.57 Hz, 1 H) 7.37 (d, J=2.57 Hz, 1 H) 7.93 (dd, J=8.82, 2.21 Hz, 1 H) 8.50 (s, 1 H) 10.37 (s, 1 H) 10.93 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-нафтален-1-ил)метансульфонамид (соединение IA-L2-2.1). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.73 (t, J=6.62 Hz, 2 H) 3.08 (s, 3 H) 3.82 (t, J=6.62 Hz, 2 H) 4.13 (s, 3 H) 7.32 (d, J=2.57 Hz, 1 H) 7.55 (d, J=2.57 Hz, 1 H) 7.58 (d, J=7.72 Hz, 1 H) 7.72 (m, 2 H) 7.87 (d, J=7.72 Hz, 1 H) 8.35 (d, J=8.82 Hz, 1 H) 8.41 (d, J=8.09 Hz, 1 H) 9.99 (s, 1 H) 10.39 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)этинил)-3-метилфенил)метансульфонамид (соединение IB-L2-1.1). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.36 (s, 9 H) 2.46 (s, 3 H) 3.05 (s, 3 H) 4.09 (s, 3 H) 5.64 (d, J=7.72 Hz, 1 H) 7.09 (dd, J=8.27, 2.02 Hz, 1 H) 7.14 (s, 1 H) 7.31 (d, J=2.57 Hz, 1 H) 7.51 (m, 2 H) 7.73 (d, J=7.72 Hz, 1 H) 10.00 (s, 1 H) 11.42 (s, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-3-хлорфенил)метансульфонамид (соединение IB-L2-1.2) 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.36 (s, 9 H) 3.12 (s, 3 H) 4.11 (s, 3 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 7.22 (dd, J=8.46, 2.21 Hz, 1 H) 7.34 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.21 Hz, 1 H) 7.50 (d, J=2.57 Hz, 1 H) 7.68 (d, J=8.82 Hz, 1 H) 7.74 (d, J=8.09 Hz, 1 H) 10.33 (s, 1 H) 11.42 (d, J=1.84 Hz, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-2-фтор-5-метилфенил)метансульфонамид (соединение IB-L2-1.3). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.36 (s, 9 H) 2.45 (s, 3 H) 3.09 (s, 3 H) 4.09 (s, 3 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 7.36 (m, 2 H) 7.48 (d, J=10.66 Hz, 1 H) 7.53 (d, J=2.57 Hz, 1 H) 7.74 (d, J=7.72 Hz, 1 H) 9.81 (s, 1 H) 11.43 (d, J=1.84 Hz, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-2,6-дифторфенил)метансульфонамид (соединение IB-L2-1.4). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.36 (s, 9 H) 3.10 (s, 3 H) 4.10 (s, 3 H) 5.66 (dd, J=7.72, 2.21 Hz, 1 H) 7.37 (d, J=2.57 Hz, 1 H) 7.46-7.58 (m, 3 H) 7.74 (d, J=8.09 Hz, 1 H) 9.74 (s, 1 H) 11.44 (d, J=1.84 Hz, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-2-фтор-5-(трифторметил)фенил)метансульфонамид (соединение IB-L2-1.5). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.36 (s, 9 H) 3.17 (s, 3 H) 4.07 (s, 3 H) 5.66 (dd, J=7.91, 2.02 Hz, 1 H) 7.38 (d. J=2.57 Hz, 1 H) 7.50 (d, J=2.57 Hz, 1 H) 7.74 (d, J=8.09 Hz, 1 H) 7.83 (d, J=7.72 Hz, 1 H) 7.89 (d, J=10.66 Hz, 1H) 10.28 (s, 1 H) 11.43 (d, J=1.84 Hz, 1 H)
N-(4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-3-гидроксифенил)метансульфонамид (соединение IB-L2-1.6). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 3.04 (s, 3 H) 4.12 (s, 3 H) 5.64 (dd, J=7.72, 2.21 Hz, 1 H) 6.68 (dd, J=8.46, 2.21 Hz, 1 H) 6.88 (d, J=1.84 Hz, 1 H) 7.26 (d, J=2.57 Hz, 1 H) 7.35 (d, J=8,46 Hz, 1 H) 7.41 (d, J=2.57 Hz, 1 H) 7.73 (d, J=7.72 Hz, 1 H) 9.97 (s, 1 H) 10.30 (s, 1 H) 11.41 (d, J=2.21 Hz, 1H).
метил 2-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)-этинил)-5-(метилсульфонамид)бензоат (соединение IB-L2-1.7). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 3.09 (s, 3 H) 3.88 (s, 3 H) 4.11 (s, 3 H) 5.65 (dd, J=7.72, 2.21 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 7.46 (m, 2 H) 7.74 (m, 3 H) 10.30 (s, 1 H) 11.42 (d, J=1.84 Hz, 1 H).
N-(6-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)этинил)-пиридин-3-ил)метансульфонамид (соединение IB-L2-1.8). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 3.13 (s, 3 H) 4.11 (s, 3 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 7.35 (d, J=2.57 Hz, 1 H) 7.53 (d, J=2.57 Hz, 1 H) 7.66 (d, J=1.47 Hz, 2 H) 7.75 (d, J=7.72 Hz, 1 H) 8.45 (s, 1 H) 10.33 (s, 1 H) 11.43 (d, J=1.84 Hz, 1 H).
N-(5-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-пиразин-2-ил)метансульфонамид (соединение IB-L2-1.9). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 3.38 (s, 3 H) 4.12 (s, 3 H) 5.65 (dd, J=8.09, 2.21 Hz, 1 H) 7.38 (d, J=2.94 Hz, 1 H) 7.55 (d, J=2.57 Hz, 1 H) 7.75 (d, J=7.72 Hz, 1 H) 8.32 (s, 1 H) 8.62 (d, J=1.47 Hz, 1 H) 11.43 (s, 2H).
N-(3-трет-бутил-4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метокси-фенил)этинил)фенил)метансульфонамид (соединение IB-L2-1.10). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 1.49 (s, 9 H) 3.06 (s, 3 H) 4.08 (s, 3 H) 5.64 (d, J=7.72 Hz, 1 H) 7.13 (dd, J=8.27, 2.02 Hz, 1 H) 7.30 (dd, J=6.80, 2.39 Hz, 2 H) 7.43 (d, J=2.57 Hz, 1 H) 7.56 (d, J=8.09 Hz, 1 H) 7.74 (d, J=8.09 Hz, 1 H) 10.01 (s, 1 H) 11.41 (s, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)этинил)-3-(морфолин-4-карбонил)фенил)метансульфонамид (соединение IB-L2-1.11). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 3.11 (s, 3 H) 3.19 (t, J=4.92 Hz, 2 H) 3.50 (t, J=4.95 Hz, 2 H) 3.65 (m, 4 H) 4.01 (s, 3 H) 5.65 (dd, J=7.91, 2.02 Hz, 1 H) 7.13 (d, J=1.84 Hz, 1 H) 7.29 (m, 2 H) 7.42 (d, J=2.57 Hz, 1 H) 7.62 (d, J=8.82 Hz, 1 H) 7.75 (d, J=8.09 Hz, 1 H) 10.28 (s, 1H) 11.43 (d, J=1.56 Hz, 1H).
N-(2-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)этинил)-5-(метилсульфонамид)фенил)ацетамид (соединение IB-L2-1.12) 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 2.10 (s, 3 H) 3.07 (s, 3 H) 4.08 (s, 3 H) 5.66 (dd, J=7.72, 2.21 Hz, 1 H) 7.04 (dd, J=8.46, 2.21 Hz, 1 H) 7.31 (d, J=2.94 Hz, 1 H) 7.53 (m, 2 H) 7.67 (s, 1 H) 7.73 (d, J=8.09 Hz, 1 H) 9.50 (s, 1 H) 10.13 (s, 1 H) 11.44 (d, J=2.21 Hz, 1 H).
N-(4-((5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-йод-2-метоксифенил)этинил)-3-метилфенил)метансульфонамид (соединение IB-L2-1.15). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 2.46 (s, 3 H) 3.06 (s, 3 H) 3.95 (s, 3 H) 5.65 (dd, J=8.09, 1.47 Hz, 1 H) 7.09 (dd, J=8.46, 2.19 Hz, 1 H)7.15 (d, J=1.84 Hz, 1 H) 7.49 (d, J=8.46 Hz, 1 H) 7.67 (d, J=2.57 Hz, 1 H) 7.73 (d, J=7.72 Hz, 1 H) 7.94 (d, J=2.57 Hz, 1 H) 10.04 (s, 1 H) 11.47 (d, J=1.26 Hz, 1 H).
N-(4-((3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)этинил)-нафтален-1-ил)метансульфонамид (соединение IB-L2-2.1). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.40 (s, 9 H) 3.08 (s, 3 H) 4.17 (s, 3 H) 5.67 (dd, J=7.91, 2.02 Hz, 1 H) 7.37 (d, J=2.57 Hz, 1 H) 7.58 (d, J=7.72 Hz, 1 H) 7.71 (m, 3 H) 7.78 (d, J=8.09 Hz, 1 H) 7.88 (d, J=7.72 Hz, 1 H) 8.35 (d, J=8.46 Hz, 1 H) 8.42 (d, J=8.09 Hz, 1 H) 10.00 (s, 1 H) 11.45 (d, J=1.84 Hz, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-метил-N-фенил-бензамид (соединение IA-L3-1.1) 1H ЯМР (300 MHz, DMSO-d6) δ 1.27 (s, 9 H) 2.53-2.61 (m, 5 H) 3.28 (t, J=6.80 Hz, 2 H) 6.69 (d, J=2.57 Hz, 1 H) 7.06 (d,.1=2.57 Hz, 1 H) 7.16-7.25 (m, 3 H) 7.26-7.34 (m, 2 H) 10.22 (s, 1 H) 10.32 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-метил-N-(4-(метил-сульфонамид)фенил)бензамид (соединение IA-L3-1.2). 1H ЯМР (300 MHz, DMSO-d6) δ 1.28 (s, 9 H) 2.57 (t, J=6.62 Hz, 2 H) 2.95 (s, 3 H) 3.33-3.45 (m, 5 H) 6.68 (d, J=1.10 Hz, 1 H) 7.08 (d, J=8.82 Hz, 3 H) 7.14-7.19 (m, 2 H) 9.76 (s, 1 H) 10.21 (s, 1 H) 10.43 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-фенилбензамид (соединение IA-L3-1.3). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.74 (t, J=6.80 Hz, 2 H) 3.79 (t, J=6.62 Hz, 2 H) 7.20 (t, J=7.35 Hz, 1 H) 7.35-7.47 (m, 3 H) 7.64 (d, J=7.35 Hz, 2 H) 7.91 (d, J=2.21 Hz, 1 H) 10.39 (s, 1 H) 10.45 (s, 1 H) 13.27 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(4-(N-метил метилсульфонамид)фенил)бензамид (соединение IA-L3-1.4). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.74 (t, J=6.62 Hz, 2 H) 2.96 (s, 3 H) 3.24 (s, 3 H) 3.79 (t,.7=6.62 Hz, 2 H) 7.38-7.49 (m, 3 H) 7.69 (d, J=8.82 Hz, 2 H) 7.90 (d, J=2.57 Hz, 1 H) 10.39 (s, 1 H) 10.51 (s, 1 H) 13.21 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(3-(метилсульфонамидо) фенил)бензамид (соединение IA-L3-1.5). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.74 (t, J=6.62 Hz, 2 H) 3.02 (s, 3 H) 3.79 (t, J=6.62 Hz, 2 H) 7.03 (d, J=7.72 Hz, 1 H) 7.30-7.47 (m, 3 H) 7.54-7.62 (m, 1 H) 7.90 (d, J=2.57 Hz, 1 H) 9.86 (s, 1 H) 10.39 (s, 1 H) 10.49 (s, 1 H) 13.20 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(4-(2-метил пропилсульфонамид)фенил)бензамид (соединение IA-L3-1.7). 1H ЯМР (300 MHz, DMSO-d6) δ 1.00 (d, J=6.62 Hz, 6 H) 1.39 (s, 9 H) 2.14 (ddd, J=19.76, 13.14, 6.80 Hz, 1 H) 2.73 (t, J=6.62 Hz, 2 H) 2.98 (d, J=6.25 Hz, 2 H) 3.78 (t, J=6.62 Hz, 2 H) 7.22 (d, J=8.82 Hz, 2 H) 7.40 (d, J=2.21 Hz, 1 Н) 7.59 (d, J=8.82 Hz, 2 H) 7.89 (d, J=2.21 Hz, 1 H) 9.80 (s, 1 H) 10.39 (s, 1 H) 10.43 (s, 1 H) 13.30 (s, 1H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-(2,2,2-трифторэтилсульфонамид)фенил)бензамид (соединение IA-L3-1.9). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.73 (t, J=6.62 Hz, 2 H) 3.78 (t, J=6.43 Hz, 2 H) 4.52 (q, J=9.44 Hz, 2 H) 7.25 (d, J=8.82 Hz, 2 H) 7.40 (d, J=1.84 Hz, 1 H) 7.62 (d, J=8.82 Hz, 2 H) 7.89 (d, J=2.21 Hz, 1 H) 10.39 (s, 1 H) 10.45 (d, J=2.57 Hz, 2 H) 13.28 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-(фенилсульфон-амидо)фенил)бензамид (соединение IA-L3-1.10). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 3.76 (t, J=6.62 Hz, 2 H) 7.11 (d, J=8.82 Hz, 2 H) 7.38 (d, J=2.21 Hz, 1 H) 7.48 (d, J=8.82 Hz, 2 H) 7.52-7.64 (m, 3 H) 7.76 (d, J-6.62 Hz, 2 H) 7.83 (d, J=2.21 Hz, 1 H) 10.29 (s, 1 H) 10.37 (d, J=1.84 Hz, 2 H) 13.21 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(3-(метилсульфон-амидометил)фенил)бензамид (соединение IA-L3-1.11). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.74 (t, J=6.62 Hz, 2 H) 2.89 (s, 3 H) 3.79 (t, J=6.62 Hz, 2 H) 4.18 (d, J=6.62 Hz, 2 H) 7.18 (d, J=7.72 Hz, 1 H) 7.35-7.43 (m, 2 H) 7.57-7.66 (m, 3 H) 7.93 (d, J=2.21 Hz, 1 H) 10.39 (s, 1 H) 10.49 (s, 1 H) 13.27 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(4-(фенилметил-сульфонамид)фенил)бензамид (соединение IA-L3-1.12). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.74 (t, J=6.80 Hz, 2 H) 3.79 (t, J=6.62 Hz, 2 H) 4.47 (s, 2 H) 7.22 (d, J=8.82 Hz, 2 H) 7.26-7.33 (m, 2 H) 7.34-7.44 (m, 4 H) 7.60 (d, J=8.82 Hz, 2 H) 7.90 (d, J=1.10 Hz, 1 H) 9.87 (s, 1 H) 10.39 (s, 1 H) 10.44 (s, 1 H) 13.32 (s, 1 H).
3-трет-бутил-N-(4-(3,5-диметилизоксазол-4-сульфонамид)фенил)-5-(2,4-диоксотетрагидро-пиримидин-1(2Н)-ил)-2-гидроксибензамид (соединение IA-L3-1.13). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 2.22 (s, 3 H) 2.44 (s, 3 H) 2.73 (t, J=6.62 Hz, 2 H) 3.77 (t, J=6.43 Hz, 2 H) 7.13 (d, J=8.82 Hz, 2 H) 7.40 (d, J=2.21 Hz, 1 H) 7.59 (d, J=8.82 Hz, 2 H) 7.87 (d, J=1.84 Hz, 1 H) 10.38 (s, 1 H) 10.40 (s, 1 H) 10.45 (s, 1 H) 13.19 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-(2-морфолино этилсульфонамид)фенил)бензамид (соединение IA-L3-1.14). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.25-2.37 (m, 4 H) 2.63-2.81 (m, 4 H) 3.20-3.29 (m, 2 H) 3.44-3.53 (m, 4 H) 3.78 (t, J=6.80 Hz, 2 H) 7.25 (d, J-8.82 Hz, 2 H) 7.40 (d, J=2.21 Hz, 1 H) 7.59 (d, J=9.19 Hz, 2 H) 7.89 (d, J=2.57 Hz, 1 H) 9.83 (s, 1 H) 10.39 (s, 1 H) 10.44 (s, 1 H) 13.30 (s, 1 H).
2-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидроксибензамидо)фенил)-уксусная кислота (соединение IA-L3-1.15). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.73 (t, J=6.62 Hz, 2 H) 3.57 (s, 2 H) 3.78 (t, J=6.80 Hz, 2 H) 7.28 (d, J=8.46 Hz, 2 H) 7.39 (d, J=0.74 Hz, 1 H) 7.58 (d, J=8.46 Hz, 2 H) 7.90 (s, 1 H) 10.38 (s, 1 H) 10.44 (s, 1 H) 12.34 (s, 1 H) 13.31 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(4-(N-метил сульфамоилметил)фенил)бензамид (соединение IA-L3-1.16). 1H ЯМР (300 MHz, DMSO-d6) δ 2.50 (s, 9 H) 2.58 (d, J=5.15 Hz, 3 H) 2.74 (t, J=6.80 Hz, 2 H) 3.79 (t, J=6.62 Hz, 2 H) 4.33 (s, 2 H) 6.95 (q, J=4.78 Hz, 1 H) 7.36-7.44 (m, 3 H) 7.66 (d, J=8.82 Hz, 2 H) 7.91 (d, J=2.21 Hz, 1 H) 10.39 (s, 1 H) 10.49 (s, 1 H) 13.25 (s, 1 H).
3-трет-бутил-N-(4-(цианометокси)фенил)-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидроксибензамид (соединение IA-L3-1.17). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.39 (s, 9 H) 2.73 (t, J-6.62 Hz, 2 H) 3.79 (t, J=6.80 Hz, 2 H) 5.18 (s, 2 H) 7.08 (m, 2 H) 7.40 (d, J=2.57 Hz, 1 H) 7.60 (m, 2 H) 7.89 (d, J=2.21 Hz, 1 H) 10.39 (s, 1 H) 10.44 (s, 1 H) 13.33 (s, 1 H)
N-(4-(2-амино-2-оксоэтокси)фенил)-3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидроксибензамид (соединение IA-L3-1.18). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.39 (s, 9 H) 2.73 (t, J=6.62 Hz, 2 H) 3.78 (t, J=6.80 Hz, 2 H) 4.43 (s, 2 H) 6.97 (m, 2 H) 7.40 (m, 2 H) 7.53 (m, 3 H) 7.89 (d, J=2.21 Hz, 1 H) 10.40 (m, 2 H) 13.41 (s, 1 H)
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(2-(метил сульфонил)фенил)бензамид (соединение IA-L3-1.20) 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.74 (t, J=6.62 Hz, 2 H) 3.27 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 7.45 (d, J=1.47 Hz, 1 H) 7.57 -7.66 (m, 1 H) 7.77 (d, J=1.84 Hz, 1 H) 7.81-7.89 (m, 2 H) 8.02 (d, J=7.72 Hz, 1 H) 10.40 (s, 1 H) 10.64 (s, 1 H) 12.99 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-(метил сульфонил)фенил)бензамид (соединение IA-L3-1.21). 1H ЯМР (300 MHz, DMSO-d6) δ 1.40 (s, 9 H) 2.74 (t, J=6.62 Hz, 2 H) 3.22 (s, 3 H) 3.79 (t, J=6.80 Hz, 2 H) 7.44 (d, J=2.21 Hz, 1 H) 7.92 (d, J=2.21 Hz, 1 H) 7.96 (s, 4 H) 10.41 (s, 1 H) 10.75 (s, 1 H) 12.89 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(2-сульфамоилфенил)бензамид (соединение IA-L3-1.22). 1H ЯМР (300 MHz, DMSO-d6) δ 1.40 (s, 9 H) 2.74 (t, J=6.62 Hz, 2 H) 3.78 (t, J=6.62 Hz, 2 H) 7.39-7.49 (m, 2 H) 7.61-7.74 (m, 4 H) 7.93 (d, J=8.09 Hz, 1 H) 8.01 (d, J=8.09 Hz, 1 H) 10.39 (s, 1 H) 10.42 (s, 1 H) 12.93 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-фенилбензамид (соединение IA-L3-1.24). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.80 Hz, 2 H) 3.70-3.86 (m, 5 H) 7.10 (t, J=7.35 Hz, 1 H) 7.24-7.44 (m, 4 H) 7.73 (d, J=7.35 Hz, 2 H) 10.36 (s, 1 H) 10.39(s, 1 H).
3-трет-бутил-N-[4-(метансульфонил-метил-амино)-фенил]-2-метокси-5-(3-метил-2,4-диоксо-тетрагидро-пиримидин-1-ил)-бензамид (соединение IA-L3-1.25). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.37 (s, 9 H) 2.84 (t, J=6.80 Hz, 2 H) 2.94 (s, 3 H) 3.04 (s, 3 H) 3.22 (s, 3 H) 3.71-3.81 (m, 5 H) 7.30 (d, J=2.57 Hz, 1 H) 7.35 (d, J=2.57 Hz, 1 H) 7.39 (d, J=8.82 Hz, 2 H) 7.75 (d, J=8.82 Hz, 2 H) 10.51 (s, 1 H)
3-трет-бутил-2-метокси-5-(3-метил-2,4-диоксотетрагидропиримидин-1(2Н)-ил)-N-(4-(N-метилметилсульфонамид)фенил)бензамид (соединение IA-L3-1.26). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.84 (t, J=6.80 Hz, 2 H) 2.94 (s, 3 H) 3.04 (s, 3 H) 3.22 (s, 3 H) 3.71-3.81 (m, 5 H) 7.30 (d, J=2.57 Hz, 1 H) 7.35 (d, J=2.57 Hz, 1 H) 7.39 (d, J=8.82 Hz, 2 H) 7.75 (d, J=8.82 Hz, 2 H) 10.51 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-N-(2-этилфенил)-2-метокси бензамид (соединение IA-L3-1.28).
2-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензамидо) фенил)- уксусная кислота (соединение IA-L3-1.30). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.53 (s, 2 H) 3.73-3.82 (m, 5 H) 7.23 (d, J=8.46 Hz, 2 H) 7.28 (d, J=2.57 Hz, 1 H) 7.33 (d, 1 H) 7.66 (d, J=8.46 Hz, 2 H) 10.35 (s, 1 H) 10.37 (s, 1 H) 12.29 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(N-метил сульфамоилметил)фенил)бензамид (соединение IA-L3-1.31). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.58 (d, J=4.78 Hz, 3 H) 2.71 (t, J=6.62 Hz, 2 H) 3.72-3.84 (m, 5 H) 4.29 (s, 2 H) 6.89 (q, J=4.78 Hz, 1 H) 7.29 (d, J=2.57 Hz, 1 H) 7.31-7.40 (m, 3 H) 7.72 (d, J=8.46 Hz, 2 H) 10.36 (s, 1 H) 10.46 (s, 1H).
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-N-(4-трифторметил-фенил)-бензамид (соединение IA-L3-1.32).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-N-(4-гидроксифенил)-2-метоксибензамид (соединение IA-L3-1.33). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.76 (s, 3 H) 3.76-3.82 (m, 2 H) 6.73 (d, J=8.82 Hz, 2 H) 7.25 (d, J=2.57 Hz, 1 H) 7.31 (d, J=2.57 Hz, 1 H) 7.50 (d, J=9.19 Hz, 2 H) 9.25 (s, 1 H) 10.11 (s, 1 H) 10.35 (s, 1 H).
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-N-(2-метокси-фенил)-бензамид (соединение IA-L3-1.34).
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-N-(3-метокси-фенил)-бензамид (соединение IA-L3-1.35).
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-N-(4-метокси-фенил)-бензамид (соединение IA-L3-1.36).
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-N-(2-этокси-фенил)-2-метокси-бензамид (соединение IA-L3-1.37).
3-трет-бутил-N-(4-(цианометокси)фенил)-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензамид (соединение IA-L3-1.38). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.76 (m, 5 H) 5.15 (s, 2 H) 7.07 (m, 2 H) 7.29 (d, J=2.94 Hz, 1 H) 7.33 (d, J=2.57 Hz, 1 H) 7.69 (m, 2 H) 10.35 (m, 2 H)
N-(4-(2-амино-2-оксоэтокси)фенил)-3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензамид (соединение IA-L3-1.39). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.80 Hz, 2 H) 3.78 (m, 5 H) 4.40 (s, 2 H) 6.95 (m, 2 H) 7.27 (d, J=2.57 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 7.39 (s, 1 H) 7.51 (s, 1 H) 7.64 (m, 2 H) 10.26 (s, 1 H) 10.35 (s, 1 H)
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-N-(4-трифторметокси-фенил)-бензамид (соединение IA-L3-1.40).
4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензамидо)фенил метансульфонат (соединение IA-L3-1.41). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 3.43 (s, 3 H) 3.73-3.83 (m, 5 H) 7.30 (d, J=2.94 Hz, 1 H) 7.33-7.39 (m, 3 H) 7.82 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.58 (s, 1 H).
N-(4-ацетилфенил)-3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-бензамид (соединение IA-L3-1.42). 1H ЯМР (300 MHz, DMSO d6) δ ppm 1.38 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.75 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 6.02 (s, 1 H) 6.55 (d, J=8.82 Hz, 1 H) 7.34 (dd, J=11.77, 2.57 Hz, 2 H) 7.66 (d, J=8.82 Hz, 1 H) 7.83-7.92 (m, 2 H) 7.92-8.04 (m, 2 H) 10.37 (s, 1 H) 10.75 (s, 1H).
Этил 3-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензамидо)фенил)-3-оксопропаноат (соединение IA-L3-1.43). 1H ЯМР (300 MHz, DMSO d6) δ ppm 1.19 (t, J=7.17 Hz, 3 H) 1.38 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.61-3.83 (m, 5 H) 4.06-4.20 (m, 4 H) 7.35 (dd, J=12.13, 2.57 Hz, 2 H) 7.84-7.91 (m, 2 H) 7.96-8.06 (m, 2 H) 10.37 (s, 1 H) 10.80 (s, 1 H).
3-трет-бутил-N-(3-карбамоил-фенил)-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-бензамид (соединение IA-L3-1.44).
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-N-(4-фтор-фенил)-2-метокси-бензамид (соединение IA-L3-1.45).
3-трет-бутил-N-(4-хлор-фенил)-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-бензамид (соединение IA-L3-1.46).
N-(4-ацетамидофенил)-3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-бензамид (соединение IA-L3-1.47). 1H ЯМР (300 MHz, DMSO d6) δ 10.35 (s, 1 H), 10.31 (s, 1 H), 9.91 (s, 1 H), 7.59-7.72 (m, 2 H), 7.41-7.59 (m, 2 H), 7.33 (d, J=2.57 Hz, 1 H), 7.28 (d, J=2.94 Hz, 1 H), 3.58-3.93 (m, 5 H), 2.71 (t, J=6.62 Hz, 2 H), 2.03 (s, 3 H), 1.37 (s, 9 H).
2-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензамидо)фенил-амино)-2-оксоэтилацетат (соединение IA-L3-1.48). 1H ЯМР (300 MHz, DMSO d6) δ 10.35 (s, 2 H), 10.06 (s, 1 H), 7.64-7.76 (m, 2 H), 7.54 (d, J=9.19 Hz, 1 H), 7.33 (d, J=2.57 Hz, 1 Н), 7.28 (d, J=2.94 Hz, 1 H), 4.63 (s, 2 H), 3.60-3.90 (m, 5 H), 2.71 (t, J=6.62 Hz, 2 H), 2.12 (s, 3 H), 1.39 (s, 9 H).
Метил 4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензамидо) фенилкарбамат (соединение IA-L3-1.49). 1H ЯМР (300 MHz, DMSO d6) δ 10.35 (s, 1 H), 10.28 (s, 1 H), 9.59 (s, 1 H), 7.62 (d, J=9.19 Hz, 2 H), 7.41 (d, J=8.82 Hz, 2 H), 7.32 (d, J=2.94 Hz, 1 H), 7.27 (d, J=2.57 Hz, 1 H), 3.69-3.88 (m, 5 H), 3.66 (s, 3 H), 2.71 (t, J=6.80 Hz, 2 H), 1.37 (s, 9 H).
Трет-бутил 4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензамидо)-фенилкарбамат (соединение IA-L3-1.50). 1H ЯМР (300 MHz, DMSO d6) δ 1.37 (s, 9 H) 1.47 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.71-3.84 (m, 5 H) 7.27 (d, J=2.57 Hz, 1 H) 7.32 (d, J=2.94 Hz, 1 H) 7.40 (d, J=8.82 Hz, 2 H) 7.59 (d, J=8.82 Hz, 2 H) 9.29 (s, 1 H) 10.25 (s, 1 H) 10.35 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(2-метилпропил-сульфонамид)фенил)бензамид (соединение IA-L3-1.52). 1H ЯМР (300 MHz, DMSO d6) δ 0.99 (d, J=6.62 Hz, 6 H) 1.37 (s, 9 H) 2.03-2.25 (m, 1 H) 2.71 (t, J=6.80 Hz, 2 H) 2.94 (d, J=6.25 Hz, 2 H) 3.69-3.84 (m, 5 H) 7.18 (d, J=8.82 Hz, 2 H) 7.28 (d, J=2.57 Hz, 1 H) 7.33 (d, J=2.57 Hz, 1 H) 7.67 (d, J=8.82 Hz, 2 H) 9.66 (s, 1 H) 10.36 (s, 1 H) 10.37 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-N-(4-(2-гидроксиэтилсульфонамид)фенил)-2-метоксибензамид (соединение IA-L3-1.53). 1H ЯМР (300 MHz, DMSO-D6) δ 1.37 (s, 9 H) 2.71 (t, J=6.80 Hz, 2 H) 3.19 (t, J=6.80 Hz, 2 H) 3.69-3.86 (m, 7 H) 4.93 (t, J=5.70 Hz, 1 H) 7.20 (d, J=8.82 Hz, 2 H) 7.28 (d, J=2.94 Hz, 1 H) 7.33 (d, J=2.57 Hz, 1 H) 7.68 (d, J=8.82 Hz, 2 H) 9.60 (s, 1 H) 10.36 (s, 1 H) 10.38 (s, 1 H)
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(2-метоксиэтил сульфонамид)фенил)бензамид (соединение IA-L3-L54). 1H ЯМР (300 MHz, DMSO d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.21 (s, 3 H) 3.25-3.31 (m, 2 H) 3.66 (t, J=6.07 Hz, 2 H) 3.71-3.83 (m, 5 H) 7.19 (d, J=8.82 Hz, 2 H) 7.28 (d, J=2.57 Hz, 1 H) 7.33 (d, J=2.57 Hz, 1 H) 7.67 (d, J=8.82 Hz, 2 H) 9.67 (s, 1 H) 10.35 (s, 1 H) 10.37 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(2,2,2-трифтор-этилсульфонамид)фенил)бензамид (соединение IA-L3-1.55). 1H ЯМР (300 MHz, DMSO d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.71-3.85 (m, 5 H) 4.46 (q, J=9.93 Hz, 2 H) 7.20 (d, J=8.82 Hz, 2 H) 7.28 (d, J=2.57 Hz, 1 H) 7.33 (d, J=2.57 Hz, 1 H) 7.70 (d, J=8.82 Hz, 2 H) 10.32 (s, 1 H) 10.36 (s, 1 H) 10.41 (s, 1 H).
N-(4-(2-(бис(2-гидроксиэтил)амино)этилсульфонамид)фенил)-3-трет-бутил-5-(2,4-диоксо-тетрагидропиримидин-1(2Н)-ил)-2-метоксибензамид (соединение IA-L3-1.56). 1H ЯМР (300 MHz, DMSO d6) δ 1.37 (s, 9 H) 2.44-2.49 (m, 2 H) 2.71 (t, J=6.62 Hz, 2 H) 2.90-2.96 (m. 2 H) 3.18 (dd, J=9.01, 5.70 Hz, 2 H) 3.35-3.41 (m, 4 H) 3.73-3.81 (m, 5 H) 4.41 (s, 2 H) 7.20 (d, J=8.82 Hz, 2 H) 7.28 (d, J=2.57 Hz, 1 H) 7.33 (d, J=2.57 Hz, 1 H) 7.68 (d, J=9.19 Hz, 2 H) 9.65 (s, 1 H) 10.35 (s, 1 H) 10.38 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(фенилметил-сульфонамид)фенил)бензамид (соединение IA-L3-1.57). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 3.72-3.84 (m, 5 H) 4.43 (s, 2 H) 7.18 (d, J=8.82 Hz, 2 H) 7.25-7.41 (m, 7 H) 7.68 (d, J=8.82 Hz, 2 H) 9.75 (s, 1 H) 10.36 (s, 1 H) 10.38 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(2-морфолиноэтилсульфонамид)фенил)бензамид (соединение IA-L3-1.58). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.28-2.35 (m, 4 H) 2.63-2.75 (m, 4 H) 3.20-3.28 (m, 2 H) 3.46-3.54 (m, 4 H) 3.72-3.82 (m, 5 H) 7.20 (d, J=8.82 Hz, 2 H) 7.28 (d, J=2.57 Hz, 1 H) 7.33 (d, J=2.57 Hz, 1 H) 7.68 (d, J=8.82 Hz, 2 H) 9.70 (s, 1 H) 10.35 (s, 1 H) 10.37 (s, 1 H).
3-трет-бутил-N-[4-(3,5-диметил-изоксазол-4-сульфониламино)-фенил]-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-бензамид (соединение IA-L3-1.59). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 H) 2.22 (s, 3 H) 2.44 (s, 3 H) 2.73 (t, J=6.62 Hz, 2 H) 3.77 (t, J=6.43 Hz, 2 H) 7.13 (d, J=8.82 Hz, 2 H) 7.40 (d, J=2.21 Hz, 1 H) 7.59 (d, J=8.82 Hz, 2 H) 7.87 (d, J=1.84 Hz, 1 H) 10.38 (s, 1 H) 10.40 (s, 1 H) 10.45 (s, 1 H) 13.19 (s, 1 H)
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(4-(фенилсульфонамид) фенил)бензамид (соединение IA-L3-1.60). 1H ЯМР (300 MHz, DMSO-d6) δ 1.35 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 3.71 (s, 3 H) 3.75 (t, J=6.62 Hz, 2 H) 7.05 (d, J=8.82 Hz, 2 H) 7.24 (d, J=2.57 Hz, 1 H) 7.31 (d, J=2.57 Hz, 1 H) 7.51-7.64 (m, 5 H) 7.70-7.80 (m, 2 H) 10.17 (s, 1 H) 10.31 (s, 1 H) 10.34 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(N-метилметил сульфонамид)фенил)бензамид (соединение IA-L3-1.62). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 2.94 (s, 3 H) 3.22 (s, 3 H) 3.73-3.84 (m, 5 H) 7.29 (d, J=2.57 Hz, 1 H) 7.34 (d, J=2.57 Hz, 1 H) 7.40 (d, J=8.82 Hz, 2 H) 7.75 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.50 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-N-(4-(N-этилметилсульфонамид)фенил)-2-метоксибензамид (соединение IA-L3-1.63). 1H ЯМР (300 MHz, DMSO d6) δ ppm 1.01 (t, J=6.99 Hz, 3 H) 1.38 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 2.97 (s, 3 H) 3.64 (d, J=7.35 Hz, 2 H) 3.74-3.83 (m, 5 H) 7.22-7.48 (m, 4 H) 7.77 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.53 (s, 1 H).
(N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензамидо)фенил) метилсульфонамид)метил пивалат (соединение IA-L3-1.65). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.19 (s, 9 H) 1.38 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.17 (s, 3 H) 3.74-3.82 (m, 5 H) 5.56 (s, 2 H) 7.30 (d, J=2.57 Hz, 1 H) 7.35 (d, J=2.57 Hz, 1 H) 7.41 (d, J=8.82 Hz, 2 H) 7.80 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.58 (s, 1 H)
3-трет-бутил-N-(4-(N-(циклопропилметил)метилсульфонамид)фенил)-5-(2,4-диоксотетрагидро пиримидин-1(2Н)-ил)-2-метоксибензамид(соединение IA-L3-1.66). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 0.09 (d, J=4.78 Hz, 2 H) 0.40 (d, J=7.72 Hz, 2 H) 0.84 (d, 1 H) 1.38 (s, 9 H) 2.72 (t, J=6.43 Hz, 2 H) 2.97 (s, 3 H) 3.47 (d, J=6.62 Hz, 2 H) 3.68-3.88 (m, 5 H) 7.19-7.54 (m, 4 H) 7.77 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.53 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(4-(N-(4-метокси-бензил)метилсульфонамид)фенил)бензамид (соединение IA-L3-1.67). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 2.64-2.79 (m, 2 H) 3.06 (s, 3 H) 3.69 (s, 3 H) 3.76 (s, 3 H) 3.75-3.83 (m, 2 H) 4.75 (s, 2 H) 6.83 (d, J=8.46 Hz, 2 H) 7.16 (d, J=8.46 Hz, 2 H) 7.24-7.39 (m, 4 H) 7.65 (d, J=8.82 Hz, 2 H) 10.35 (s, 1 H) 10.45 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(4-N-(метилсульфонил)пропионамидо)фенил)бензамид (соединение IA-L3-1.70). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 0.93 (t, J=7.35 Hz, 3 H) 1.38 (s, 9 H) 2.12 (q, J=7.35 Hz, 2 H) 2.72 (t, J=6.62 Hz, 2 H) 3.52 (s, 3 H) 3.78 (m, 5 H) 7.31 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.57 Hz, 1 H) 7.44 (d, J=8.46 Hz, 2 H) 7.83 (d, J=8.82 Hz, 2 H) 10.37 (s, 1 H) 10.64 (s, 1 H)
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(N-(метилсульфонил)бутирамидо)фенил)бензамид (соединение IA-L3-1.71). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 0.79 (t, J=7.35 Hz, 3 H) 1.38 (s, 9 H) 1.48 (m, 2 H) 2.09 (t, J=7.17 Hz, 2 H) 2.72 (t, J=6.62 Hz, 2 H) 3.52 (s, 3 H) 3.78 (m, 5 H) 7.32 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.57 Hz, 1 H) 7.43 (d, J=8.82 Hz, 2 H) 7.83 (d, J=8.82 Hz, 2 H) 10.37 (s, 1 H) 10.65 (s, 1 H)
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-N-(метилсульфонил)изобутирамидо)фенил)бензамид (соединение IA-L3-1.72). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 0.99 (d, J=6.62 Hz, 6 H) 1.38 (s, 9 H) 2.29-2.41 (m, 1 H) 2.72 (t, J=6.62 Hz, 2 H) 3.51 (s, 3 H) 3.75-3.83 (m, 5 H) 7.32 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.57 Hz, 1 H) 7.48 (d, J=8.82 Hz, 2 H) 7.84 (d, J=8.82 Hz, 2 H) 10.37 (s, 1 H) 10.66 (s, 1 H)
Метил 4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензамидо) фенил(метилсульфонил)карбамат (соединение IA-L3-1.73). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 3.56 (s, 3 H) 3.72 (s, 3 H) 3.74-3.84 (m, 5 H) 7.31 (d, J=2.57 Hz, 1 H) 7.35 (dd, J=5.70, 2.76 Hz, 3 H) 7.77 (d, J=8.82 Hz, 2 H) 10.37 (s, 1 H) 10.58 (s, 1 H)
Этил 4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензамидо)-фенил(метилсульфонил)карбамат (соединение IA-L3-1.74). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.16 (t, J=7.17 Hz, 3 H) 1.38 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.56 (s, 3 H) 3.73-3.84 (m, 5 H) 4.20 (q, J=6.99 Hz, 2 H) 7.26-7.41 (m, 4 H) 7.77 (d, J=8.82 Hz, 2 H) 10.37 (s, 1 H) 10.58 (s, 1 H)
Изобутил 4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензамидо)фенил(метилсульфонил)карбамат (соединение IA-L3-1.76). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 0.79 (d, J=6.99 Hz, 6 H) 1.38 (s, 9 H) 1.74-1.92 (m, 1 H) 2.71 (t, J=6.62 Hz, 2 H) 3.56 (s, 3 H) 3.73-3.83 (m, 5 H) 3.94 (d, J=6.62 Hz, 2 H) 7.29-7.41 (m, 4 H) 7.78 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.58 (s, 1 H)
2-метоксиэтил 4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси бензамидо)фенил(метилсульфонил)карбамат (соединение IA-L3-1.77). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.19 (s, 3 H) 3.46-3.52 (m, 2 H) 3.57 (s, 3 H) 3.72-3.84 (m, 5 H) 4.28 (dd, J=5.52, 3.68 Hz, 2 H) 7.27-7.42 (m, 4 H) 7.78 (d, J=8.82 Hz, 2 H) 10.36(s, 1 H) 10.58 (s, 1 H)
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(метилсульфонил) фенил)бензамид (соединение IA-L3-1.78). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 3.19 (s, 3 H) 3.74 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 7.35 (dd, 2 H) 7.95 (dd, 4 H) 10.37 (s, 1 H) 10.86 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(2-(метилсульфонил) фенил)бензамид (соединение IA-L3-1.79). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.72 (t, J=6.43 Hz, 2 H) 3.29 (s, 3 H) 3.77-3.85 (m, 5 H) 7.43 (d, J=2.57 Hz, 1 H) 7.45-7.49 (m, 1 H) 7.50 (d, J=2.57 Hz, 1 H) 7.77-7.85 (m, 1 H) 7.95 (d, J=8.46 Hz, 1 H) 8.45 (d, J=8.46 Hz, 1 H) 10.39 (s, 2 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(2-сульфамоилфенил)бензамид (соединение IA-L3-1.80). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 3.73-3.89 (m, 5 H) 7.34 (t, J=7.72 Hz, 1 H) 7.42 (d, J=2.94 Hz, 1 H) 7.49 (d, J=2.57 Hz, 1 H) 7.66 (t, J=7.17 Hz, 1 H) 7.72 (s, 2 H) 7.91 (d, J=8.09 Hz, 1 H) 8.48 (d, J=8.09 Hz, 1 H) 10.31 (s, 1 H) 10.39 (s, 1 H).
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-N-(3-гидрокси-2-метил-фенил)-2-метокси-бензамид (соединение IA-L3-1.81).
3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-N-(4-фтор-2-метил-фенил)-2-метокси-бензамид (соединение IA-L3-1.82).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(2-метил-4-(метил сульфонамид)фенил)бензамид (соединение IA-L3-1.83). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 H) 2.26 (s, 3 H) 2.72 (t, J=6.62 Hz, 2 H) 2.98 (s, 3 H) 3.79 (t, J=6.62 Hz, 2 H) 3.83 (s, 3 H) 7.04 (m, 2 H) 7.33 (m, 2 H) 7.47 (d, J=8.09 Hz, 1 H) 9.65 (s, 1 H) 9.78 (s, 1 H) 10.36 (s, 1 H)
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)фенил)-4-(метилсульфонамид) бензамид (соединение IA-L3-1.84). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 1.38 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 3.09 (s, 3 H) 3.79 (t, J=6.62 Hz, 2 H) 3.84 (s, 3 H) 7.33 (d, J=2.21 Hz, 1 H) 7.37 (d, J=2.57 Hz, 1 H) 7.55 (m, 2 H) 7.67 (d, J=8.82 Hz, 1 H) 10.03 (s, 1 H) 10.18 (s, 1 H) 10.38 (s, 1 H)
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(2-метокси-4-(метил сульфонамид)фенил)бензамид (соединение IA-L3-1.85). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.68-2.76 (m, 2 H) 3.00 (s, 3 H) 3.75-3.87 (m, 8 H) 6.84 (dd, J=8.46, 2.21 Hz, 1 H) 6.94 (d, J=2.21 Hz, 1 H) 7.39 (d, J=2.94 Hz, 1 H) 7.47 (d, J=2.57 Hz, 1 H) 7.93-7.99 (m, 1 H) 9.59 (s, 1 H) 9.68 (s, 1 H) 10.37 (s, 1 H).
N-(5-ацетиламино-2-метокси-фенил)-3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-бензамид (соединение IA-L3-1.86).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(метилсульфон-амидо)-3-нитрофенил)бензамид (соединение IA-L3-1.87). 1H ЯМР (300 MHz, DMSO-d6) δ 10.84 (s, 1 H) 10.37 (s, 1 H) 9.71 (s, 1 H) 8.52 (d, J=2.57 Hz, 1 H) 7.98 (dd, J=9.01, 2.39 Hz, 1 H) 7.62 (d, J=8.82 Hz, 1 H) 7.35 (dd, J=12.50, 2.57 Hz, 2 H) 3.71-3.83 (m, 5 H) 3.11 (s, 3 H) 2.71 (t, J=6.62 Hz, 2 H) 1.38 (s, 9 H).
Масляной кислоты ({4-[3-трет-бутил-5-(3-бутирилоксиметил-2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-метокси-бензоиламино]-фенил}-метансульфонил-амино)-метиловый эфир (соединение IA-L3-1.88). 1H ЯМР (300 MHz, DMSO-D6) δ ppm 0.88 (m, 6 H) 1.38 (s, 9 H) 1.55 (m, 4 H) 2.26 (t, J=7.17 Hz, 2 H) 2.39 (t, J=7.17 Hz, 2 H) 2.95 (t, J=6.62 Hz, 2 H) 3.14 (s, 3 H) 3.77 (s, 3 H) 3.81 (t, J=6.62 Hz, 2 H) 5.57 (s, 2 H) 5.68 (s, 2 H) 7.35 (d, J=2.57 Hz, 1 H) 7.38 (d, J=2.94 Hz, 1 H) 7.42 (d, J=8.82 Hz, 2 H) 7.79 (d, J=8.82 Hz, 2 H) 10.60 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(4-(метилсульфонамид метил)-3-сульфамоилтиофен-2-ил)бензамид (соединение IA-L3-1.89). 1H ЯМР (300 MHz, DMSO-d6) δ 1.40 (s, 9 H) 2.70-2.78 (m, 2 H) 2.97 (s, 3 H) 3.78 (t, J=6.43 Hz, 2 H) 4.36 (d, J=6.99 Hz, 2 H) 7.14 (s, 1 H) 7.42 (d, J=2.21 Hz, 1 H) 7.47 (d, J=2.21 Hz, 1 H) 7.60 (t, J=6.43 Hz, 1 H) 7.82 (s, 2 H) 10.42 (s, 1 H) 11.43 (s, 1 H) 11.93 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(метилсульфонамид метил)-3-сульфамоилтиофен-2-ил)бензамид (соединение IA-L3-1.90). 1H ЯМР (300 MHz, DMSO-d6) δ 1.40 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 2.97 (s, 3 H) 3.73 (s, 3 H) 3.80 (t, J=6.80 Hz, 2 H) 4.36 (d, J=5.52 Hz, 2 H) 7.06 (s, 1 H) 7.47 (d, J=2.57 Hz, 1 H) 7.57 (t, J=6.43 Hz, 1 H 7.61 (d, J=2.57 Hz, 1 H) 7.70 (s, 2 H) 10.40 (s, 1 H) 11.56 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-(метилсульфонамид метил)тиофен-2-ил)бензамид (соединение IA-L3-1.91). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.74 (t, J=6.80 Hz, 2 H) 2.86 (s, 3 H) 3.79 (t, J=6.62 Hz, 2 H) 4.10 (d, J=6.25 Hz, 2 H) 6.98 (s, 1 H) 7.04 (d, J=1.47 Hz, 1 H) 7.42 (d, J=2.57 Hz, 1 H) 7.55 (t, J=6.25 Hz, 1 H) 7.89 (d, J=2.21 Hz, 1 H) 10.41 (s, 1 H) 11.68 (s, 1 H) 12.95 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(4-(метилсульфонамид метил)тиофен-2-ил)бензамид (соединение IA-L3-1.94). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.80 Hz, 2 H) 2.85 (s, 3 H) 3.67 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.07 (d, J=5.88 Hz, 2 H) 6.82 (d, J=1.84 Hz, 1 H) 6.87 (s, 1 H) 7.28-7.32 (m, 1 H) 7.36 (d, J=2.57 Hz, 1 H) 7.49 (t, J=6.25 Hz, 1 H) 10.37 (s, 1 H) 11.59 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-((N-метилметил сульфонамид)метил)тиофен-2-ил)бензамид (соединение IA-L3-1.95). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 2.70 (s, 3 H) 2.74 (t, J=6.80 Hz, 2 H) 2.92 (s, 3 H) 3.79 (t, J=6.80 Hz, 2 H) 4.18 (s, 2 H) 7.01 (m, 2 H) 7.42 (d, J=2.21 Hz, 1 H) 7.89 (d, J=2.57 Hz, 1 H) 10.41 (s, 1 H) 11.68 (s, 1 H) 12.92 (s, 1 H).
трет-бутил (5-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1 (2Н)-ил)-2-метоксибензамидо) тиофен-3-ил)метил(метилсульфонил)карбамат (соединение IA-L3-1.96). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 1.48 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.30 (s, 3 H) 3.67 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.66 (s, 2 H) 6.77-6.87 (m, 2 H) 7.30 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.57 Hz, 1 H) 10.37 (s, 1 H) 11.63 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(4-((N-метилметил сульфонамид)метил)тиофен-2-ил)бензамид (соединение IA-L3-1.97). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.70 (m, 5 H) 2.91 (s, 3 H) 3.68 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.15 (s, 2 H) 6.80 (d, J=1.47 Hz, 1 H) 6.94 (d, J=1.47 Hz, 1 H) 7.31 (d, J=2.57 Hz, 1 H) 7.36 (d, J=2.94 Hz, 1 H) 10.37 (s, 1 H) 11.59 (s, 1 H).
трет-бутил (5-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1 (2Н)-ил)-2-гидроксибензамидо) тиофен-3-ил)метил(метилсульфонил)карбамат (соединение IA-L3-1.98). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 H) 1.47 (s, 9 H) 2.74 (t, J=6.80 Hz, 2 H) 3.35 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 4.68 (s, 2 H) 6.96 (s, 1 H) 7.04 (d, J=1.47 Hz, 1 H) 7.41 (d, J=2.21 Hz, 1 H) 7.88 (d, J=2.21 Hz, 1 H) 10.40 (s, 1 H) 11.70 (s, 1 H) 12.92 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(6-(метилсульфонамид)пиридин-3-ил)бензамид (соединение IA-L3-1.99). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.28 (s, 3 H) 3.77 (m, 5 H) 7.01 (d, J=8.82 Hz, 1 H) 7.31 (d, J=2.57 Hz, 1 H) 7.35 (d, J=2.57 Hz, 1 H) 8.07 (dd, J=8.82, 2.57 Hz, 1 H) 8.61 (d, J=2.57 Hz, 1 H) 10.36 (s, 1 H) 10.49 (s, br, 1 H) 10.50 (s, 1 H).
N-(6-аминопиридин-3-ил)-3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси бензамид (соединение IA-L3-1.100). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.74 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 7.06 (d, J=9.56 Hz, 1 H) 7.31 (d, J=2.57 Hz, 1 H) 7.37 (d, J=2.57 Hz, 1 H) 7.99 (s, 2 H) 8.04 (dd, J=9.56, 2.21 Hz, 1 H) 8.53 (d, J=1.84 Hz, 1 H) 10.38 (s, 1 H) 10.67 (s, 1 H) 13.64 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(5-(N-(метилсульфонил)метилсульфонамид)пиридин-2-ил)бензамид (соединение IA-L3-1.101). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.38 (s, 9 H) 2.72 (t, J=6.62 Hz, 2 H) 3.58 (s, 6 H) 3.75 (s, 3 H) 3.80 (t, J=6.62 Hz, 2 H) 7.36 (m, 2 H) 8.06 (dd, J=8.82, 2.57 Hz, 1 H) 8.30 (d, J=8.82 Hz, 1 H) 8.51 (d, J=2.21 Hz, 1 H) 10.37 (s, 1 H) 11.10 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(5-(метилсульфонамид)пиридин-2-ил)бензамид (соединение IA-L3-1.102). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.02 (s, 3 H) 3.74 (s, 3 H) 3.79 (t, J=6.62 Hz, 2 H) 7.35 (m, 2 H) 7.71 (dd, J=8.82, 2.57 Hz, 1 H) 8.18 (m, 2 H) 9.81 (s, 1 H) 10.36 (s, 1 H) 10.74 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-((1r,4r)-4-(метилсульфонамидо)циклогексил)бензамид (соединение IA-L3-1.103). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.32 (d, 11 H) 1.91 (s, 2 H) 2.65-2.78 (m, 4 H) 2.91 (s, 3 H) 3.07 (s, 2 H) 3.39 (s, 2 H) 3.66-3.80 (m, 5 H) 7.03 (d, J=7.35 Hz, 1 H) 7.12 (d, J=2.57 Hz, 1 H) 7.25 (d, J=2.57 Hz, 1 H) 8.19 (d, J=8.09 Hz, 1 H) 10.32 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(тиазол-2-ил)бензамид (соединение IA-L3-1.104). 1H ЯМР (300 MHz, DMSO-d6) δ 1.56-1.68 (m, 9 H) 2.92 (t, J=6.62 Hz, 2 H) 3.90-4.07 (m, 2 H) 7.27-7.70 (m, 2 H) 7.80 (d, J=4.04 Hz, 1 H) 8.03 (s, 1 H) 10.51 (s, 1 H) 14.06 (d, J=116.92 Hz, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-N-(4-(метилсульфонамид)фенил)бензамид (соединение IA-L3-1.105). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.34 (s, 9 H) 2.74 (t, J=6.62 Hz, 2 H) 2.96 (s, 3 H) 3.86 (t, J=6.62 Hz, 2 H) 7.09-7.90 (m, 7 H) 9.62 (s, 1 H) 10.24 (s, 1 H) 10.43 (s, 1 H).
N-(4-аминофенил)-3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси бензамид (соединение IA-L3-1.107). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.71-3.83 (m, 5 H) 7.26-7.39 (m, 4 H) 7.82 (d, J=8.82 Hz, 2 H) 9.95 (s, 1 H) 10.36 (s, 1H) 10.57 (s, 1 H).
N-(4-(N-аллилметилсульфонамид)фенил)-3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензамид (соединение IA-L3-1.108). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.01 (s, 3 H) 3.70-3.83 (m, 5 H) 4.25 (d, J=5.88 Hz, 2 H) 5.00-5.24 (m, 2 H) 5.68-5.84 (m, 1 H) 7.29 (d, J=2.57 Hz, 1 H) 7.30-7.41 (m, 3 H) 7.74 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.51 (s, 1 H).
5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-3-изопропил-2-метокси-N-(4-(метилсульфонамид) фенил)бензамид (соединение IA-L3-1.111). 1H ЯМР (500 MHz, DMSO-d6) δ ppm 1.22 (d, J=6.71 Hz, 6 Н) 2.72 (t, J=6.71 Hz, 2 H) 2.95 (s, 3 H) 3.23-3.39 (m, 1 H) 3.75 (s, 3 H) 3.79 (t, J=6.71 Hz, 2 H) 7.19 (d, J=9.16 Hz, 2 H) 7.31 (d, J=2.44 Hz, 1 H) 7.38 (d, J=2.44 Hz, 1 H) 7.69 (d, J=9.16 Hz, 2 H) 9.55 (s, 1 H) 10.29 (s, 1 H) 10.34 (s, 1H).
5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-3-этил-2-метокси-N-(4-(метилсульфонамид)фенил)бензамид (соединение IA-L3-1.112). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.20 (t, J=7.35 Hz, 3 H) 2.60-2.78 (m, 4 H) 2.95 (s, 3 H) 3.69-3.84 (m, 5 H) 7.19 (d, J=9.19 Hz, 2 H) 7.27-7.41 (m, 2 H) 7.69 (d, J=8.82 Hz, 2 H) 9.59 (s, 1 H) 10.31 (s, 1 H) 10.38 (s, 1 H).
5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-3-этил-2-гидрокси-N-(4-(метилсульфонамид) фенил)бензамид (соединение IA-L3-1.113). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.17 (t, J=7.54 Hz, 3 H) 2.54-2.66 (m, 2 H) 2.73 (t, J=6.62 Hz, 2 H) 2.99 (s, 3 H) 3.78 (t, J=6.62 Hz, 2 H) 7.24 (d, J=8.82 Hz, 2 H) 7.37 (d, J=1.84 Hz, 1 H) 7.62 (d, J=8.82 Hz, 2 H) 7.87 (d, J=2.21 Hz, 1 H) 9.72 (s, 1 H) 10.40 (s, 1 H) 10.43 (s, 1 H) 12.70 (s, 1 H).
5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-(метилсульфонамид)фенил)бензамид (соединение IA-L3-1.114). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 2.72 (t, J=6.80 Hz, 2 H) 2.97 (s, 3 H) 3.76 (t, J=6.80 Hz, 2 H) 6.99 (d, J=8.82 Hz, 1 H) 7.22 (d, J=9.19 Hz, 2 H) 7.40 (dd, J=8.82, 2.57 Hz, 1 H) 7.65 (d, J=8.82 Hz, 2 H) 7.87 (d, J=2.57 Hz, 1 H) 9.66 (s, 1 H) 10.38 (d, J=1.84 Hz, 2 H) 11.83 (s, 1 H).
3-трет-бутил-5-(3-(2-(этиламино)-2-оксоэтил)-2,4-диоксотетрагидропиримидин-1(2Н)-ил)-N-(4-(N-(2-(этиламино)-2-оксоэтил)метилсульфонамид)фенил)-2-метоксибензамид (соединение IA-L3-1.115). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 0.92-1.04 (m, 6 H) 1.37 (s, 9 H) 2.88 (t, J=6.62 Hz, 2 H) 3.01-3.08 (m, 4 H) 3.09 (s, 3 H) 3.75 (s, 3 H) 3.81 (t, J=6.62 Hz, 2 H) 4.22 (d, J=5.52 Hz, 4 H) 7.30 (d, J=2.57 Hz, 1 H) 7.31-7.37 (m, 1 H) 7.47 (d, J=8.82 Hz, 2 H) 7.74 (d, J=8.82 Hz, 2 H) 7.88-8.04 (m, 2 H) 10.53 (s, 1 H).
(3-(3-трет-бутил-4-метокси-5-(4-(N-(пивалоилоксиметил)метилсульфонамид)фенилкарбамоил)фенил)-2,6-диоксотетрагидропиримидин-1(2Н)-ил)метил пивалат (соединение IA-L3-1.116). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.11 (s, 9 H) 1.19 (s, 9 H) 1.38 (s, 9 H) 2.95 (t, J=6.62 Hz, 2 H) 3.17 (s, 3 H) 3.77 (s, 3 H) 3.81 (t, J=6.62 Hz, 2 H) 5.56 (s, 2 H) 5.67 (s, 2 H) 7.35 (d, J=2.57 Hz, 1 H) 7.38 (d, J=2.57 Hz, 1 H) 7.41 (d, J=9.19 Hz, 2 H) 7.80 (d, J=8.82 Hz, 2 H) 10.61 (s, 1 H).
5-(3-((1,3-диоксолан-2-ил)метил)-2,4-диоксотетрагидропиримидин-1(2H)-ил)-3-трет-бутил-2-метокси-N-(4-(метилсульфонамид)фенил)бензамид (соединение IA-L3-1.117). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.87 (t, J=6.62 Hz, 2 H) 2.95 (s, 3 H) 3.78 (s, 3 H) 3.76-3.85 (m, 4 H) 3.87-3.96 (m, 4 H) 5.07 (t, J=4.96 Hz, 1 H) 7.20 (d, J=8.82 Hz, 2 H) 7.32 (dd, J=13.97, 2.57 Hz, 2 H) 7.69 (d, J=8.82 Hz, 2 H) 9.60 (s, 1 H) 10.40 (s, 1 H).
5-(3-аллил-2,4-диоксотетрагидропиримидин-1(2Н)-ил)-N-(4-(N-аллилметилсульфонамид)фенил)-3-трет-бутил-2-метоксибензамид (соединение IA-L3-1.118). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 2.78-2.93 (m, 2 H) 3.01 (s, 3 H) 3.78 (s, 3 H) 3.76-3.85 (m, 2 H) 4.25 (d, J=5.88 Hz, 4 H) 4.94-5.29 (m, 4 H) 5.67-5.96 (m, 1 H) 7.00-7.21 (m, 1 H) 7.31 (d, J=2.57 Hz, 1 H) 7.35-7.42 (m, 3 H) 7.74 (d, J=8.82 Hz, 2 H) 10.52 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидрокси-N-(2-метокси-4-(метил сульфонамид)фенил)бензамид (соединение IA-L3-1.119). 1H ЯМР (300 MHz, DMSO-d6) δ 1.38 (s, 9 H) 2.73 (t, J=6.62 Hz, 2 H) 3.05 (s, 3 H) 3.74-3.81 (m, 5 H) 6.84 (dd, J=8.46, 2.21 Hz, 1 H) 6.95 (d, J=2.21 Hz, 1 H) 7.30 (d, J=8.46 Hz, 1 H) 7.39 (d, J=2.21 Hz, 1 H) 7.89 (d, J=2.57 Hz, 1 H) 9.82 (s, 1 H) 10.06 (s, 1 H) 10.37 (s, 1 H) 13.50 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(3-(метилсульфонамид метил)фенил)бензамид (соединение IA-L3-1.120). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 2.89 (s, 3 H) 3.69-3.88 (m, 5 H) 4.15 (d, J=6.25 Hz, 2 H) 7.00-7.16 (m, 1 H) 7.26-7.40 (m, 3 H) 7.59 (t, J=6.43 Hz, 1 H) 7.65 (d, J=8.82 Hz, 1 H) 7.74 (s, 1 H) 10.36 (s, 1 H) 10.44 (s, 1 H).
3-трет-бутил-N-(4-(2,5-диметоксифенилсульфонамид)фенил)-5-(2,4-диоксотетрагидро-пиримидин-1(2Н)-ил)-2-метоксибензамид (соединение IA-L3-1.121). 1H ЯМР (300 MHz, DMSO d6) δ 1.35 (s, 9 H) 2.69 (t, J=6.80 Hz, 2 H) 3.71 (s, 6 H) 3.75 (t, J=6.62 Hz, 2 H) 3.85 (s, 3 H) 7.06 (d, J=8.82 Hz, 2 H) 7.13 (s, 2 H) 7.22 (dd, J=3.68, 2.21 Hz, 2 H) 7.31 (d, J=2.57 Hz, 1 H) 7.53 (d, J=8.82 Hz, 2 H) 9.87 (s, 1 H) 10.28 (s, 1 H) 10.34 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-N-(4-(2-гидроксиэтилсульфонамид)фенил)-2-метоксибензамид (соединение IA-L3-1.122). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.80 Hz, 2 H) 3.19 (t, J=6.80 Hz, 2 H) 3.69-3.86 (m, 7 H) 4.93 (t, J=5.70 Hz, 1 H) 7.20 (d, J=8.82 Hz, 2 H) 7.28 (d, J=2.94 Hz, 1 H) 7.33 (d, J=2.57 Hz, 1 H) 7.68 (d, J=8.82 Hz, 2 H) 9.60 (s, 1 H) 10.36 (s, 1 H) 10.38 (s, 1 H).
(N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензамидо)фенил)метилсульфонамид)метил бутират (соединение IA-L3-1.123). 1H ЯМР (300 MHz, DMSO-d6) δ 0.90 (t, J=7.54 Hz, 3 H) 1.38 (s, 9 H) 1.57 (m, 2 H) 2.39 (t, J=7.35 Hz, 2 H) 2.71 (t, J=6.62 Hz, 2 H) 3.14 (s, 3 H) 3.77 (m, 5 H) 5.57 (s, 2 H) 7.30 (d, J=2.57 Hz, 1 H) 7.35 (d, J=2.57 Hz, 1 H) 7.41 (d, J=8.82 Hz, 2 H) 7.79 (d, J=8.82 Hz, 2 H) 10.36 (s, 1 H) 10.57 (s, 1 H)
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидрокси-N-(хинолин-6-ил)бензамид (соединение IA-L3-2.1). 1H ЯМР (300 MHz, DMSO-d6) δ 1.41 (s, 9 H) 2.75 (t, J=6.62 Hz, 2 H) 3.77-3.86 (m, 2 H) 7.45 (d, J=2.21 Hz, 1 H) 7.68 (dd, J=8.46, 4.41 Hz, 1 H) 7.98 (d, J=2.57 Hz, 1 H) 8.11 (s, 2 H) 8.44 (s, 1 H) 8.58 (d, J=8.46 Hz, 1 H) 8.96 (dd, J=4.41, 1.47 Hz, 1 H) 10.41 (s, 1 H) 10.81 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метокси-N-(2-оксоиндолин-5-ил) бензамид (соединение IA-L3-2.2). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.62 Hz, 2 H) 3.50 (s, 2 H) 3.75 (s, 3 H) 3.77 (t, J=6.62 Hz, 2 H) 6.78 (d, J=8.46 Hz, 1 H) 7.26 (d, J=2.21 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 7.48 (dd, J=8.46, 1.84 Hz, 1 H) 7.64 (s, 1 H) 10.24 (s, 1 H) 10.34 (s, 1 H) 10.35 (s, 1 H).
3-трет-бутил-N-(2,2-диоксо-1,3-дигидробензо[c]тиофен-5-ил)-5-(2,4-диоксотетрагидро пиримидин-1(2H)-ил)-2-метоксибензамид (соединение IA-L3-2.3). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.71 (t, J=6.43 Hz, 2 H) 3.70-3.85 (m, 5 H) 4.45 (s, 2 H) 4.53 (s, 2 H) 7.29 (d, J=2.57 Hz, 1 H) 7.32-7.40 (m, 2 H) 7.62 (dd, J=8.27, 1.65 Hz, 1 H) 7.86 (s, 1 H) 10.36 (s, 1 H) 10.54 (s, 1 H).
3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метокси-N-(2-оксо-1,2,3,4-тетрагидрохинолин-6-ил)бензамид (соединение IA-L3-2.4). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.44 (t, 2 H) 2.71 (t, J=6.62 Hz, 2 H) 2.86 (t, J=7.35 Hz, 2 H) 3.77 (t, 2 H) 3.75 (s, 3 H) 6.82 (d, J=8.46 Hz, 1 H) 7.26 (d, J=2.57 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 7.45 (dd, J=8.46, 2.21 Hz, 1 H) 7.57 (d, 1 H) 10.05 (s, 1 H) 10.24 (s, 1 H) 10.35 (s, 1 H).
3-трет-бутил-N-(2,2-диоксо-1,3-дигидробензо[c]тиофен-5-ил)-5-(2,4-диоксотетрагидро пиримидин-1(2H)-ил)-2-гидроксибензамид (соединение IA-L3-2.5). 1H ЯМР (300 MHz, DMSO-d6) δ 1.39 (s, 9 H) 2.74 (t, J=6.80 Hz, 2 H) 3.79 (t, J=6.62 Hz, 2 H) 4.49 (s, 2 H) 4.55 (s, 2 H) 7.42 (dd, J=4.96, 2.76 Hz, 2 H) 7.59 (dd, J=8.27,1.65 Hz, 1 H) 7.74 (s, 1 H) 7.90 (d, J=1.84 Hz, 1 H) 10.40 (s, 1 H) 10.54 (s, 1 H) 13.15 (s, 1 H).
3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метокси-N-(4-(метилсульфонамидо)фенил)бензамид (соединение IB-L3-1.1). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 2.50 (s, 9 H) 2.94 (s, 3 H) 3.79 (s, 3 H) 5.66 (d, J=8.09 Hz, 1 H) 7.14-7.25 (m, 2 H) 7.39 (s, 2 H) 7.62-7.75 (m, 3 H) 9.60 (s, 1 H) 10.44 (s, 1 H) 11.42 (s, 1 H).
3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-гидрокси-N-(4-(метилсульфон-амид)фенил)бензамид (соединение IB-L3-1.2). 1H ЯМР (300 MHz, DMSO-d6) δ 1.40 (s, 9 H) 2.99 (s, 3 H) 5.70 (dd, J=7.72, 2.21 Hz, 1 H) 7.24 (d, J=8.82 Hz, 2 H) 7.46 (d, J=2.21 Hz, 1 H) 7.61 (d, J=8.82 Hz, 2 H) 7.76 (d, J=7.72 Hz, 1 H) 8.03 (d, J=2.21 Hz, 1 H) 9.75 (s, 1 H) 10.45 (s, 1 H) 11.48 (d, J=2.21 Hz, 1 H) 13.52 (s, 1 H)
3-трет-бутил-2-метокси-5-(6-метил-2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-N-(4-(метил сульфонамид)фенил)бензамид (соединение IB-L3-1.3). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.37 (s, 9 H) 1.82 (s, 3 H) 2.96 (s, 3 H) 3.80 (s, 3 H) 5.63 (s, 1 H) 7.20 (d. J=8.82 Hz, 2 H) 7.34 (s, 2 H) 7.69 (d, J=8.82 Hz, 2 H) 9.60 (s, 1 H) 10.41 (s, 1 H) 11.27 (s, 1 H).
3-трет-бутил-2-гидрокси-5-(6-метил-2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-N-(4-(метил сульфонамид)фенил)бензамид (соединение IB-L3-1.4). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.39 (s, 9 Н) 1.86 (s, 3 Н) 2.99 (s, 3 Н) 5.68 (s, 1 Н) 7.24 (d, J=8.82 Hz, 2 Н) 7.40 (s, 1 Н) 7.61 (d, J=8.82 Hz, 2 Н) 7.95 (s, 1 Н) 9.74 (s, 1 Н) 10.39 (s, 1 Н) 11.35 (s, 1 Н) 13.57 (s, 1 Н).
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)фенил)-4-(метилсульфонамид) бензамид (соединение IA-L4-1.1). 1H ЯМР (300 MHz, DMSO-d6) δ 1.29 (s, 9 Н) 2.72 (t, J=6.62 Hz, 2 Н) 3.09 (s, 3 Н) 3.79 (t, J=6.62 Hz, 2 Н) 7.08 (t, J=1.84 Hz, 1 Н) 7.30 (d, J=8.82 Hz, 2 Н) 7.67 (dd, J=6.99, 1.84 Hz, 2 Н) 7.95 (d, J=8.82 Hz, 2 Н) 10.16 (s, 1 Н) 10.19 (s, 1 Н) 10.35 (s, 1 Н)
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидроксифенил)бензамид (соединение IA-L4-1.2). 1H ЯМР (300 MHz, DMSO-d6) δ 10.28 (s, 1 Н) 10.17 (s, 1 H) 8.99 (s, 1 Н) 8.04 (d, J=6.99 Hz, 2 H) 7.49-7.67 (m, 3 H) 7.22 (d, J=2.57 Hz, 1 H) 7.06 (d, J=2.57 Hz, 1 H) 3.73 (t, J=6.80 Hz, 2 H) 2.70 (t, J=6.80 Hz, 2 H) 1.40 (s, 9 H).
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидроксифенил)-4-(метилсульфонамидо)бензамид (соединение IA-L4-1.3). 1H ЯМР (300 MHz, DMSO-d6) δ 10.29 (s, 1 Н) 10.25 (br s, 1 Н) 10.11 (s, 1 Н) 9.02 (s, 1 Н) 8.02 (d, J=8.82 Hz, 2 H) 7.30 (d, J=8.82 Hz, 2 H) 7.19 (d, J=2.21 Hz, 1 H) 7.06 (d, J=2.57 Hz, 1 H) 3.73 (t, J=6.80 Hz, 2 H) 3.10 (s, 3 H) 2.71 (t, J=6.62 Hz, 2 H) 1.39 (s, 9 H).
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидроксифенил)-4-(метил сульфонилметил)бензамид (соединение IA-L4-1.4). 1H ЯМР (300 MHz, DMSO-d6) δ 1.40 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 2.95 (s, 3 H) 3.73 (t, J=6.80 Hz, 2 H) 4.61 (s, 2 H) 7.07 (d, J=2.57 Hz, 1 H) 7.21 (d, J=2.57 Hz, 1 H) 7.57 (d, J=8.46 Hz, 2 H) 8.05 (d, J=8.09 Hz, 2 H) 8.95 (s, 1 H) 10.16 (s, 1 H) 10.29 (s, 1 H).
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидроксифенил)-4-нитро бензамид (соединение IA-L4-1.5). 1H ЯМР (300 MHz, DMSO-d6) δ 10.28 (s, 1 H) 10.26 (s, 1 H) 8.91 (s, 1 H) 8.38 (d, J=8.82 Hz, 2 H) 8.26 (d, J=9.20 Hz, 2 H) 7.19 (d, J=2.57 Hz, 1 H) 7.09 (d, J=2.57 Hz, 1 H) 3.73 (t, J=6.62 Hz, 2 H) 2.70 (t, J=6.80 Hz, 2 H) 1.40 (s, 9 H).
4-амино-N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-гидроксифенил) бензамид (соединение IA-L4-1.6). 1H ЯМР (300 MHz, DMSO-d6) δ 10.29 (s, 1 H) 9.95 (s, 1 H) 9.46 (s, 1 H) 7.79 (d, J=8.82 Hz, 2 H) 7.16 (d, J=2.57 Hz, 1 H) 7.03 (d, J=2.21 Hz, 1 H) 6.61 (d, J=8.46 Hz, 2 H) 5.90 (s, 2 H) 3.72 (t, J=6.80 Hz, 2 H) 2.70 (t, J=6.62 Hz, 2 H) 1.39 (s, 9 H).
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)-N-метил-4-(метилсульфонамид)бензамид (соединение IA-L4-1.7). 1H ЯМР (300 MHz, DMSO-d6) δ 10.36 (s, 1 H) 9.81 (s, 1 H) 7.36 (d, J=2.21 Hz, 1 H) 7.15 (d, J=8.46 Hz, 2 H) 7.05 (d, J=2.21 Hz, 1 H) 6.92 (d, J=8.46 Hz, 2 H) 3.65-3.90 (m, 2 H) 3.41 (s, 3 H) 3.17 (d, J=5.52 Hz, 3 H) 2.88 (s, 3 H) 2.66-2.76 (m, 2 H) 1.03 (s, 9 H).
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)бензамид (соединение IA-L4-1.8). 1H ЯМР (300 MHz, DMSO-d6) δ 10.33 (s, 1 H) 9.98 (s, 1 H) 8.00-8.07 (m, 2 H) 7.49-7.64 (m, 3 H) 7.38 (d, J=2.57 Hz, 1 H) 7.13 (d, J=2.57 Hz, 1 H) 3.77 (t, J=6.62 Hz, 2 H) 3.72 (s, 3 H) 2.71 (t, J=6.62 Hz, 2 H) 1.37 (s, 9 H).
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)-2-метокси-4-(метилсульфонамид)бензамид (соединение IA-L4-1.11). 1H ЯМР (300 MHz, DMSO-d6) δ 10.33 (s, 1 H) 10.31 (s, 1 H) 10.20 (s, 1 H) 8.21 (d, J=2.57 Hz, 1 H) 8.02 (d, J=8.82 Hz, 1 H) 7.01-7.07 (m, 2 H) 6.96 (dd, J=8.46, 1.84 Hz, 1 H) 4.03-4.07 (m, 3 H) 3.79-3.82 (m, 3 H) 3.76 (t, J=6.80 Hz, 2 H) 3.14 (s, 3 H) 2.71 (t, J=6.62 Hz, 2 H) 1.39 (s, 9 H).
N-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)-2-хлор-4-(метилсульфонамид)бензамид (соединение IA-L4-1.12). 1H ЯМР (300 MHz, DMSO-d6) δ 10.33 (s, 1 H) 10.23 (s, 1 H) 10.02 (s, 1 H) 7.52-7.65 (m, 2 H) 7.20-7.33 (m, 2 H) 7.09 (d, J=2.57 Hz, 1 H) 3.71-3.82 (m, 5 H) 3.11 (s, 3 H) 2.71 (t, J=6.62 Hz, 2 H) 1.35 (s, 9 H).
N-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)-2-метокси-4-(метилсульфонамид)бензамид (соединение IB-L4-1.1). 1H ЯМР (300 MHz, DMSO-d6) δ 1.40 (s, 9 H) 3.14 (s, 3 H) 3.84 (s, 3 H) 4.06 (s, 3 H) 5.65 (dd, J=7.72, 2.21 Hz, 1 H) 6.96 (dd, J=8.46, 1.84 Hz, 1 H) 7.04 (d, J=1.84 Hz, 1 H) 7.09 (d, J=2.94 Hz, 1 H) 7.71 (d, J=7.72 Hz, 1 H) 8.01 (d, J=8.82 Hz, 1 H) 8.28 (d, J=2.57 Hz, 1 H) 10.27 (s, 1 H) 10.32 (s, 1 H) 11.41 (d, J=2.21 Hz, 1 H).
{1-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пиперидин-4-ил}-карбаминовой кислоты трет-бутиловый эфир (соединение IA-L5-1-1.1). 1H ЯМР (300 MHz, CDCl3) δ ppm 7.36 (s, 1 H) 7.03 (d, J=2.94 Hz, 1 H) 6.77 (d, J=2.57 Hz, 1 H) 4.43 (s, 1 H) 3.79 (t, J=6.62 Hz, 2 H) 3.66 (s, 2 H) 3.44-3.61 (m, 1 H) 2.88-3.01 (m, 1 H) 2.81 (t, J=6.62 Hz, 2 H) 2.22 (s, 2 H) 1.98 (s, 2 H) 1.44 (s, 9 H) 1.39 (s, 9 H) 1.28-1.71 (m, 2 H)
N-{1-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пиперидин-3-илметил}-метансульфонамид (соединение IA-L5-1-1.2). 1H ЯМР (300 MHz, CDCl3) δ ppm 7.45 (s, 1 H) 7.03 (d, J=2.57 Hz, 1 H) 6.78 (d, J=2.21 Hz, 1 H) 4.37 (s, 1 H) 3.81 (t, 7=6.80 Hz, 2 H) 3.58-3.73 (m, 2 H) 3.07 (s, 2 H) 2.92 (s, 3 H) 2.81 (t, J=6.62 Hz, 2 H) 1.72-1.95 (m, 4 H) 1.49-1.72 (m, 4 H) 1.39 (s, 9 H)
Этиловый эфир 1-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пиперидин-3-карбоновой кислоты (соединение IA-L5-1-1.3). 1H ЯМР (300 MHz, CDCl3) δ ppm 7.39 (s, 1 H) 7.03 (d, J=2.57 Hz, 1 H) 6.78 (d, J=2.57 Hz, 1 H) 4.09-4.22 (m, 2 H) 3.79 (t, J=6.62 Hz, 2 H) 3.67 (s, 2 H) 3.05 (s, 1 H) 2.81 (t, J=6.80 Hz, 2 H) 2.51-2.69 (m, 1 H) 2.38 (s, 1 H) 2.15 (s, 1 H) 1.88-2.07 (m, 1 H) 1.70-1.85 (m, 1 H) 1.46-1.69 (m, 3 H) 1.39 (s, 9 H) 1.21-1.30 (m, 3 H)
1-[3-трет-бутил-4-гидрокси-5-(3-метил-пиперидин-1-илметил)-фенил]-дигидро-пиримидин-2,4-дион; соединение с трифторуксусной кислотой (соединение IA-L5-1-1.4).
метиловый эфир 1-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пиперидин-4-карбоновой кислоты (соединение IA-L5-1-1.5). 1H ЯМР (300 MHz, CDCl3) δ ppm 7.38 (s, 1 H) 7.03 (d, J=2.57 Hz, 1 H) 6.77 (d, J=2.57 Hz, 1 H) 3.79 (t, J=6.80 Hz, 2 H) 3.69 (s, 3 H) 3.66 (s, 2 H) 2.97 (s, 2 H) 2.80 (t, J=6.80 Hz, 2 H) 2.30-2.46 (m, 1 H) 2.17 (s, 2 H) 1.91-2.03 (m, 2 H) 1.83 (s, 2 H) 1.39 (s, 9 H)
1-[3-трет-бутил-4-гидрокси-5-((R)-3-гидрокси-пиперидин-1-илметил)-фенил] -дигидро-пиримидин-2,4-дион; соединение с трифторуксусной кислотой (соединение IA-L5-1-1.6).
1-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пиперидин-3-карбоновой кислоты диэтиламид, соль трифторуксусной кислоты (соединение IA-L5-1-1.7).
Амид 1-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пиперидин-3-карбоновой кислоты, соль трифторуксусной кислоты (соединение IA-L5-1-1.8).
трет-бутиловый эфир 4-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пиперазин-1-карбоновой кислоты (соединение IA-L5-1-1.10.) 1H ЯМР (300 MHz, CDCl3) δ ppm 7.37 (s, 1 H) 7.05 (d, J=2.57 Hz, 1 H) 6.80 (d, J=2.57 Hz, 1 H) 3.79 (t, J=6.62 Hz, 2 H) 3.69 (s, 2 H) 3.34-3.61 (m, 2 H) 2.81 (t, J=6.62 Hz, 2 H) 2.52 (s, 2 H) 1.56 (s, 4 H) 1.46 (s, 9 H) 1.39 (s, 9 H)
N-{1-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пирролидин-3-ил}-метансульфонамид (соединение IA-L5-1-1.11). 1H ЯМР (300 MHz, CDCl3) δ ppm 7.48 (s, 1 H) 7.04 (d, J=2.57 Hz, 1 H) 6.81 (d, J=2.21 Hz, 1 H) 4.66 (s, 1 H) 4.04-4.17 (m, 2 H) 3.80 (t, J=6.62 Hz, 2 H) 2.96 (s, 3 H) 2.85-2.93 (m, 1 H) 2.82 (t, J=6.62 Hz, 2 H) 2.65-2.76 (m, 1 H) 2.50-2.64 (m, 1 H) 2.34-2.49 (m. 1 H) 1.73-1.89 (m, 1 H) 1.39 (s, 9 H)
трет-бутиловый эфир {1-[3-трет-бутил-5-(2,4-диоксо-тетрагидро-пиримидин-1-ил)-2-гидрокси-бензил]-пирролидин-3-ил}-карбаминовой кислоты (соединение IA-L5-1-1.12). 1H ЯМР (300 MHz, CDCl3) δ ppm 7.38 (s, 1 H) 7.01-7.04 (m, 1 H) 6.77-6.80 (m, 1 H) 4.66-4.75 (m, 1 H) 4.16-4.27 (m, 1 H) 3.80 (t, 2 H) 3.68-3.87 (m, 2 H) 2.81 (t, 2 H) 2.26-2.96 (m, 5 H) 1.49-1.74 (m, 2 H) 1.43 (s, 9 H) 1.40 (s, 9 H)
1-(3-трет-бутил-5-((2,6-диметилморфолино)метил)-4-гидроксифенил)дигидро пиримидин-2,4(1Н,3H)-дион 2,2,2-трифторацетат (соединение IA-L5-1-1.13).
1-(3-трет-бутил-4-гидрокси-5-(морфолинометил)фенил)дигидропиримидин-2,4(1Н,3H)-дион 2,2,2-трифторацетат (соединение IA-L5-1-1.14).
1-(3-трет-бутил-4-метокси-5-((1-метил-1Н-индол-3-ил)метил)фенил)дигидропиримидин 2,4(1Н,3H)-дион (соединение IA-L5-1-2.1). 1H ЯМР (300MHz, DMSO-d6): δ 10.23 (s, 1H), 7.48 (d, J=8.1 Hz, 1H), 7.38 (d, J=8.1Hz, 1H), 7.17 (m, 1H), 7.08 (d, J=2.6Hz, 1H), 7.04 (s, 1H), 6.98 (m, 2Н), 4.05 (s, 2H), 3.76 (s, 3H), 3.72 (s, 3H), 3.66 (t, J=6.6 HZ, 1H), 2.62 (t, J=6.6 HZ, 1H), 1.37 (s, 9H)
N-(1-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидроксибензил)-1,2,3,4-тетрагидрохинолин-6-ил)метансульфонамид (соединение IA-L5-1-2.2). 1H ЯМР (300 MHz, CDCl3) δ 9.38 (s, 1 H) 7.46 (s, 1 H) 7.09 (d, J=2.21 Hz, 1 H) 6.94-7.06 (m, 2 H) 6.91 (d, J=2.57 Hz, 1 H) 6.23-6.31 (m, 1 H) 5.37 (d, J=6.99 Hz, 1 H) 3.77-3.89 (m, 3 H) 3.04-3.12 (m, 2 H) 2.97 (s, 3 H) 2.78-2.96 (m, 3 H) 1.94-2.04 (m, 2 H) 1.39 (s, 9 H).
N-(4-(3 -трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)фенэтил)фенил)метан сульфонамид (соединение IA-L5-2-1.2). 1H ЯМР (300 MHz, DMSO-d6) δ 1.25 (s, 9 H) 2.69 (t, J=6.62 Hz, 2 H) 2.83 (s, 4 H) 2.91 (s, 3 H) 3.75 (t, J=6.62 Hz, 2 H) 6.99-7.21 (m, 7 H) 9.60 (s, 1 H) 10.31 (s, 1 H).
метил 2-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенэтил)-5-(метилсульфонамид)бензоат (соединение IB-L5-2-1.1). 1H ЯМР (300 MHz, DMSO-d6) δ 1.34 (s, 9 H) 2.83-2.92 (m, 2 H) 2.96 (s, 3 H) 3.14 (dd, J=10.30, 5.88 Hz, 2 H) 3.75 (s, 3 H) 3.83 (s, 3 H) 5.64 (d, J=7.72 Hz, 1 H) 7.13 (d, J=2.94 Hz, 1 H) 7.20 (d, J=2.57 Hz, 1 H) 7.28-7.36 (m, 2 H) 7.61-7.71 (m, 2 H) 9.88 (s, 1 H) 11.39 (s, 1 H)
N-(4-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенэтил)фенил) метансульфонамид (соединение IB-L5-2-1.2). 1H ЯМР (300 MHz, DMSO-d6): δ 11.39 (s, 1H), 9.60 (s, 1H), 7.65 (d, J=8.1Hz, 1H), 7.23 (m, 3H), 7.17 (m, 3H), 5.64 (d, J=7.7 Hz, 1H), 3.77 (s, 3H), 2.93 (s, 3H), 2.88 (bs, 4H), 1.35 (s, 9H)
N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксибензилокси)фенил) метансульфонамид (соединение IA-L6-1.1). 1H ЯМР (300 MHz, DMSO-d6) δ ppm 1.36 (s, 9 H) 2.69 (t, J=6.62 Hz, 2 H) 2.89 (s, 3 H) 3.71-3.76 (m, 2 H) 3.78 (s, 3 H) 5.05 (s, 2 H) 6.96-7.12 (m, 2 H) 7.10-7.21 (m, 2 H) 7.23 (d, J=2.94 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 9.39 (s, 1 H) 10.32 (s, 1 H).
1-(3-трет-бутил-5-((циклогексил(этил)амино)метил)-4-гидроксифенил)дигидропиримидин-2,4(1H,3H)-дион 2,2,2-трифторацетат (соединение IA-L9-1.1).
1-(3-трет-бутил-5-((циклогексил(метил)амино)метил)-4-гидроксифенил)дигидро пиримидин-2,4(1H,3H)-дион 2,2,2-трифторацетат (соединение IA-L9-1.2).
N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-гидроксибензиламино)фенил)метансульфонамид (соединение IA-L9-1.3). 1H ЯМР (300 MHz, DMSO-d6) δ 1.37 (s, 9 H) 2.66 (t, J=6.62 Hz, 2 H) 2.82 (s, 3 H) 3.65 (t, J=6.62 Hz, 2 H) 4.24 (d, J=5.15 Hz, 2 H) 6.10 (t, J=5.52 Hz, 1 H) 6.64 (d, J=8.82 Hz, 2 H) 6.98 (d, J=8.82 Hz, 2 H) 7.03 (s, 2 H) 8.79 (s, 1 H) 9.04 (s, 1 H) 10.22 (s, 1 H).
N-(4-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксибензиламино)фенил) метансульфонамид (соединение IA-L9-1.4). 1H ЯМР (500 MHz, DMSO-d6) δ 1.36 (s, 9H), 2.65 (t, J=6.7 Hz, 2H), 2.80 (s, 3H), 3.68 (t, 6.7 Hz, 2H), 3.79 (s, 3H), 4.25 (d, J 5.5 Hz, 2H), 6.10 (m, 1H), 6.55 (d, J=8.5 Hz, 2H), 6.94 (d, J=8.5 Hz, 2H), 7.13 (d, J=2.5 Hz, 1H), 7.19 (d, J=2.4 Hz, 1H), 8.92 (s, 1H), 10.23 (s, 1H).
N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2Н)-ил)-2-метоксифенил)-2-оксоэтил)фенил)метансульфонамид (соединение IA-L11-1.1). 1H ЯМР (300 MHz, DMSO-d6) δ 1.36 (s, 9 H) 2.70 (t, J=6.62 Hz, 2 H) 2.96 (s, 3 H) 3.64 (s, 3 H) 3.76 (t, J=6.62 Hz, 2 H) 4.27 (s, 2 H) 7.10-7.26 (m, 4 H) 7.32-7.41 (m, 2 H) 9.67 (s, 1 H) 10.37 (s, 1 H).
N-(4-(2-(3-трет-бутил-5-(2,4-диоксотетрагидропиримидин-1(2H)-ил)-2-метоксифенил)ацетил)фенил)метансульфонамид (соединение IA-L12-1.1). 1H ЯМР (300 MHz, DMSO-d6) δ 1.33 (s, 9 H) 2.68 (t, J=6.62 Hz, 2 H) 3.12 (s, 3 H) 3.61 (s, 3 H) 3.72 (t, J=6.62 Hz, 2 H) 4.36 (s, 2 H) 7.01 (d, J=2.94 Hz, 1 H) 7.15 (d, J=2.57 Hz, 1 H) 7.29 (d, J=8.82 Hz, 2 H) 8.04 (d, J=8.82 Hz, 2 H) 10.29 (s, 1 H) 10.35 (s, 1 H).
Следующие соединеня могут быть получены с использованием представленного выще описания:
Таблица А
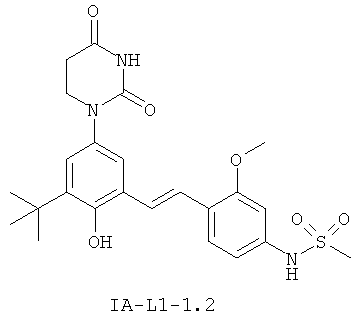
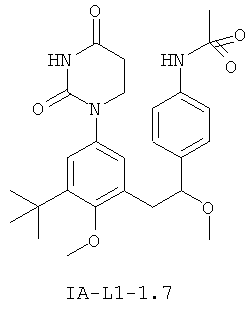
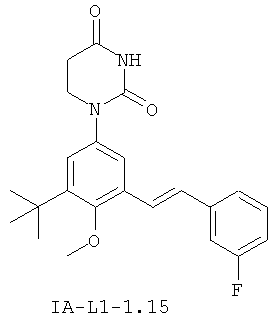
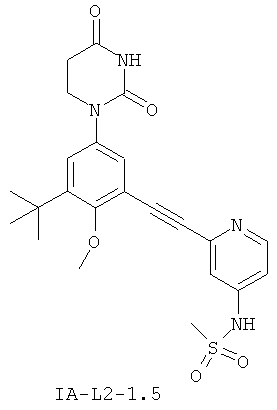
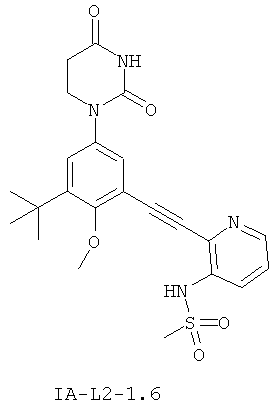
Таблица В
Substituent(s) as described in the table below - заместитель(и), как описано в приведенной ниже таблице
Исследование ингибирования HCV полимеразы
Либо двухкратные серийные разведения (исследование парциального ингибирования), либоболее узкий интервал разведении, перекрывающий IC50 ингибитора (исследование прочности связывания) ингибиторов инкубировали с 20 мМ Трис-Cl рН 7,4, 2 мМ MnCl2, 1 мМ дитиотреитолом, 1 мМ этилендиаминтетрауксусной кислотой (EDTA), 60-125 мкМ GTP и 20-50 нМ ∆21 NS5B (HCV штамм 1B (ВК, Genbank регистрационный номер М58335, или H77, Genbank регистрационный номер AF011751)) в течение 15 мин при комнатной температуре. Реакцию инициировали добавлением 20 мкМ СТР, 20 мкМ АТР, 1 мкМ 3H-UTP (10 мКю/мкмоль), 5 нМ матричной РНК и 0,1 Ед/мкл ингибитора РНазы (RNasin, Promega), и оставляли для протекания реакции в течение 2 - 4 ч при комнатной температуре. Объем реакции составил 50 мкл. Реакцию заканчивали путем добавления 1 объема 4 мМ спермина в 10 мМ Трис-Cl pH 8,0, 1 мМ EDTA. После инкубирования по меньшей мере в течение 15 мин при комнатной температуре, осажденную РНК собирали фильтрованием через GF/B фильтр (Millipore) в 96 луночном формате. Фильтровальную пластинку промывали три раза 200 мкл каждого из 2 мМ спермина, 10 мМ Трис-Cl pH 8,0, 1 мМ EDTA, и 2 раза этанолом. После сушки на воздухе в каждую лунку добавляли, 30 мкл сцинтилляционной смеси Microscint 20 (Packard), и удержанное число импульсов в минуту определяли с помощью сцинтилляционного счета. Значения IC50 рассчитывали с помощью уравнения нелинейной регрессии с двумя независимыми переменными, используя неингибированный контрольный образец и полностью ингибированный контрольный образец для определения минимума и максимума для этой кривой. Исследования прочности связывания проводили на тех соединениях, которые демонстрируют значения IC50 менее 0,005 мкМ в исследовании парциального ингибирования для более точного измерения значений IC50. Удержанное число импульсов в минуту наносили на график в зависимости от концентрации ингибитора и аппроксимировали к уравнению 1, используя нелинейную регрессию (ссылка 1) для получения значений IC50:
Удержанное число импульсов в минуту=A[sqrt{(IC50+It-Et)^2+4*IC50*Et}-(IC50+It-Et)] (уравнение 1)
где A=Vmax[S]/2(Km+[S]); It=суммарная концентрация ингибитора и Et=суммарная активная концентрация фермента.
Ссылка: Morrison, J. F. и S. R. Stone. 1985. Approaches to the study and analysis of the inhibition of enzymes by slow- and tight-binding inhibitors. Comments Mol. Cell. Biophys. 2: 347-368. Последовательность используемой матричной РНК представляла собой: 5'-GGGCGAAUUG GGCCCUCUAG AUGCAUGCUC GAGCGGCCGC CAGUGUGAUG GAUAUCUGCA GAAUUCGCCC UUGGUGGCUC CAUCUUAGCC CUAGUCACGG CUAGCUGUGA AAGGUCCGUG AGCCGCUUGA CUGCAGAGAG UGCUGAUACU GGCCUCUCUG CAGAUCAAGUC-3'
При тестировании указанным выше способом, соединения по настоящему изобретению ингибировали HCV полимеразу 1А и/или 1B. Обозначения в приведенной ниже Таблице представляют собой следующее: А -- IC50≤0,01 мкМ; В - 0,1 мкМ≥IC50≥0,01 мкМ; С - 1 мкМ≥IC50≥0,1 мкМ; и D -- IC50>1 мкМ; ND - не определено.
Исследование репликона HCV полимеразы
Для характеристики соединения в клеточной культуре использовали две стабильные клеточне линии, несущие субгеномные репликоны: одие получен из генотипа 1a-Н77 и один получен из генотипа 1b-Con1 (получен от Apath, LLC, St. Louis, МО). Все конструкты репликона представляли собой бицистронные субгеномные репликоны, аналогичне тем, которые описаны Bartenschlager и соавторами (Lohmann et al., Replication of Subgenomic Hepatitis C Virus RNAs in a Hepatoma Cell Line, science 285:110-3(1999)). Конструкт репликона генотипа 1а содержит NS3-NS5B кодирующую область, происходящую из Н77 штамма HCV (1a-H77) (Blight et al., Efficient Replication of Hepatitis C Virus Genotype la RNAs in Cell Culture, J. virol. 77:3181-90 (2003)). Этот репликон также имеет репотерную люциферазу светлячка и селектируемый маркер неомицин фосфотрансферазу (Neo). Эти две кодирующие области, разделенные FMDV 2a протеазой, содержат первый цистрон бицистронного конструкта репликона со вторым цистроном, содержащим NS3-NS5B кодирующую область с дополнительными адаптивными мутациями Е1202 G, K1691R, K2040R и S2204I. 1b-Con1 конструкт репликона идентичен 1a-H77 репликону, за исключением того, что NS3-NS5B кодирующая область была получена из штамма 1b-Con1, и адаптивными мутациями являются Е1202 G, T1280I и S2204I. Репликон-содержащие клеточные линии подращивали в модифицированной по Дульбекко среде Игла (DMEM), содержащей 10% (об/об) эмбриональной сыворотки теленка (FBS), 100 МЕ/мл пенициллина, 100 мг/мл стрептомицина (Invitrogen), и 200 мг/мл G418 (Invitrogen). Ингибирующие действия соединений на репликацию HCV определяли путем измерения активности репортерного гена люциферазы. Вкратце, репликон-содержащие клетки высевали в 96-луночные планшеты с плотностью 5000 клеток на лунку в 100 мкл DMEM, содержащей 5% FBS. 16-24 ч позже, соединения разбавляли в диметилсульфоксиде (DMSO) для получения 200х маточного раствора в сериях восьми полулогарифмических разведении. Серии разведении затем дополнительно разбавляли в 100 раз в среде, содержащей 5% FBS. Добавляли среду с ингибитором на ночь в планшеты для клеточных культур, уже содержащие 100 мкл DMEM с 5% FBS. В исследованиях, в которых измеряют ингибирующую активность в присутствии плазмы человека, ночную среду из планшетов для клеточных культур заменяли на DMEM, содержащую 40% плазму человека и 5% FBS. Клетки инкубировали в течение трех дней в термостатах для клеточных культур, а затем подвергали лизису для экстракции РНК. Для люциферазного исследования в каждую лунку добавляли 30 мкл пассивного буфера для лизиса (Passive Lysis buffer) (Promega), и затем планшеты инкубировали в течение 15 мин с раскачиванием для лизиса клеток. Раствор люциферина (50 - 100 мкл, Promega) добавляли в каждую лунку и активность люциферазы измеряли с помощью люминометра Victor II (Perkin-Elmer). Процент ингибирования HCV РНК репликации рассчитывали для каждой концентрации соединения и значение EC.so рассчитывали, используя кривую нелинейной регрессии, аппроксимированную к 4-параметрическому логарифмическому уравнению и программное обеспечение GraphPad Prism 4.
При тестировании указанным выше способом, соединения по настоящему изобретению ингибируют HCV полимеразу 1A и/или 1B. Обозначения в приведенной ниже Таблице, представляют собой следующее: А -- EC50≤0,01 мкМ; В - 0,1 мкМ≥EC50≥0,01 мкМ; С - 1 мкМ≥EC50≥0,1 мкМ; и D -- EC50≥1 мкМ; ND - не определено.
Все ссылки (пантентные и непатентные), процитированные выше, включены в качестве ссылки в настоящую патентную заявку. Описание этих ссылок предназначено только лишь для обобщения притязаний, сделанных их авторами. Не признается тот факт, что какая-либо ссылка (или часть какой-либо ссылки) является релевантной предшествующему уровню техники (или уровню техники вообще). Заявители сохраняют за собой право оспаривать правильность и релевантность процитированных ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ УРАЦИЛА ИЛИ ТИМИНА ДЛЯ ЛЕЧЕНИЯ ГЕПАТИТА С | 2008 |
|
RU2543620C2 |
| N-ФЕНИЛ-ДИОКСО-ГИДРОПИРИМИДИНЫ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРА ВИРУСА ГЕПАТИТА С (HCV) | 2008 |
|
RU2542099C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2571662C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2552533C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2010 |
|
RU2541571C2 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2647592C2 |
| ДИАРИЛГИДАНТОИНЫ | 2011 |
|
RU2638833C2 |
| ИНГИБИТОРЫ БРОМДОМЕНА | 2012 |
|
RU2671571C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2800292C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2597364C2 |
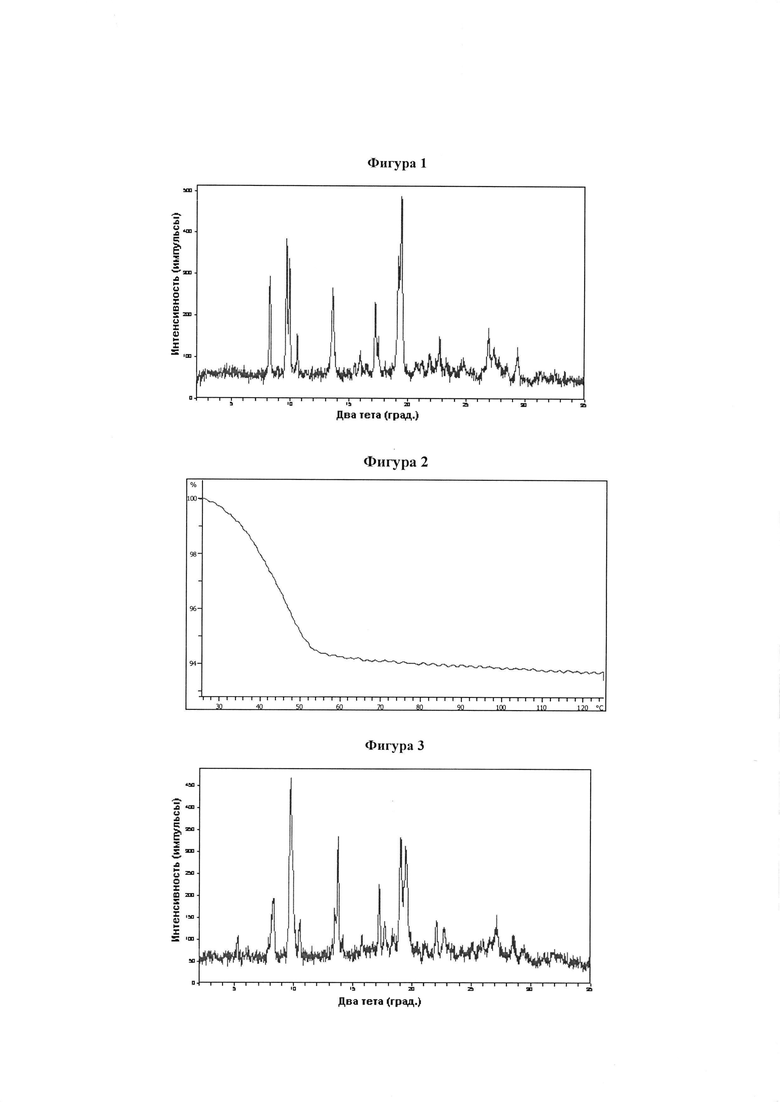
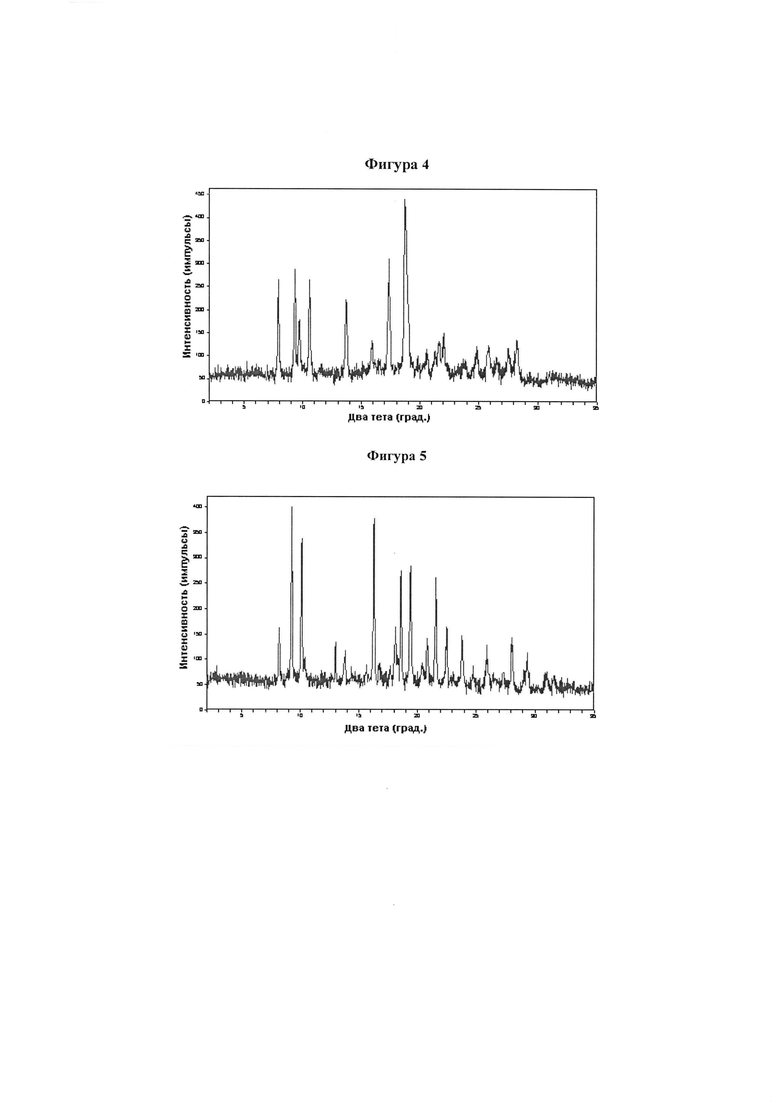
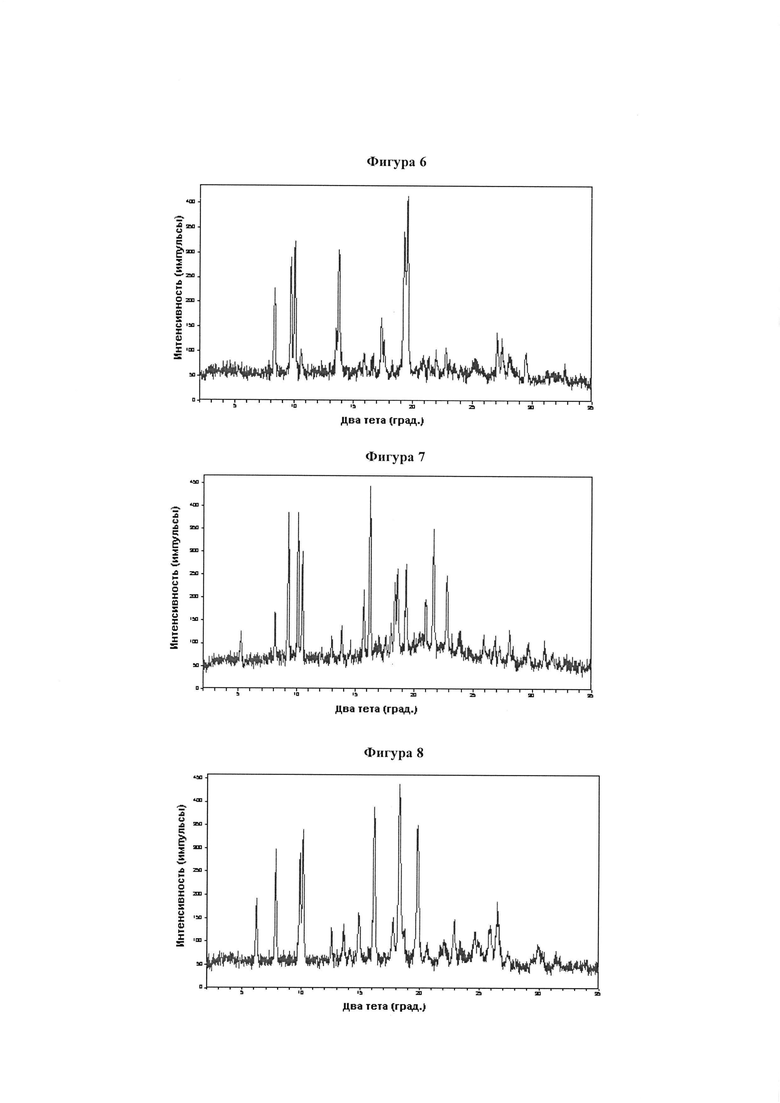
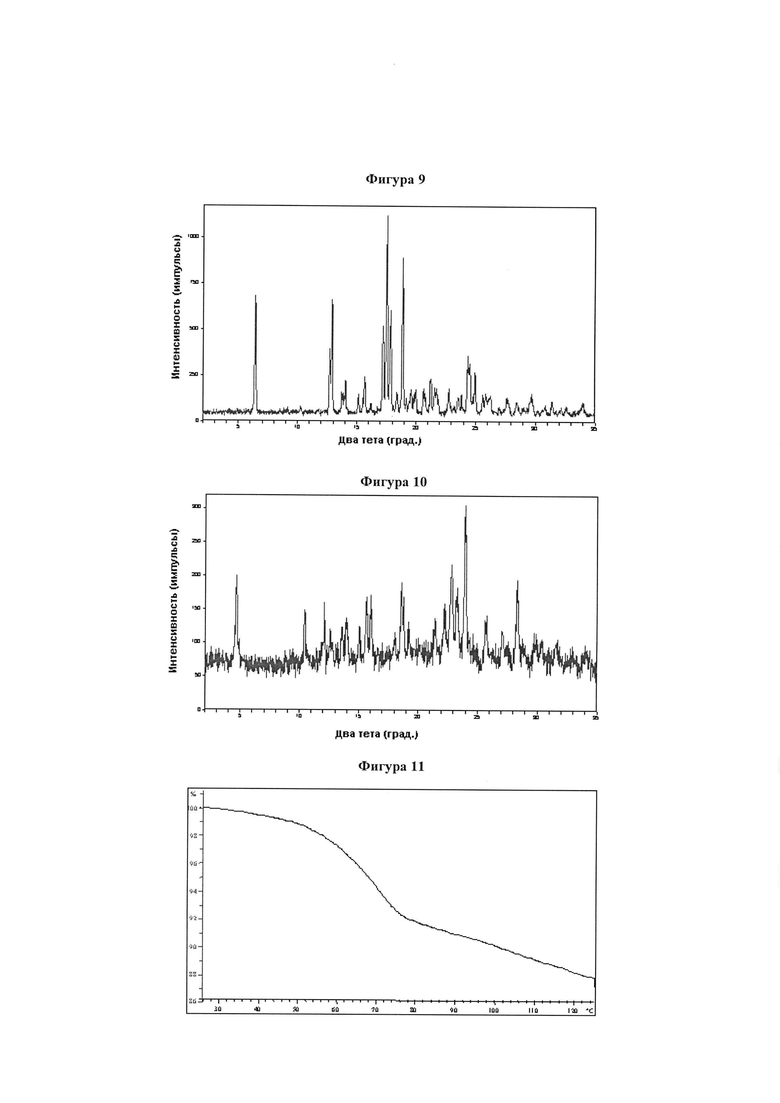
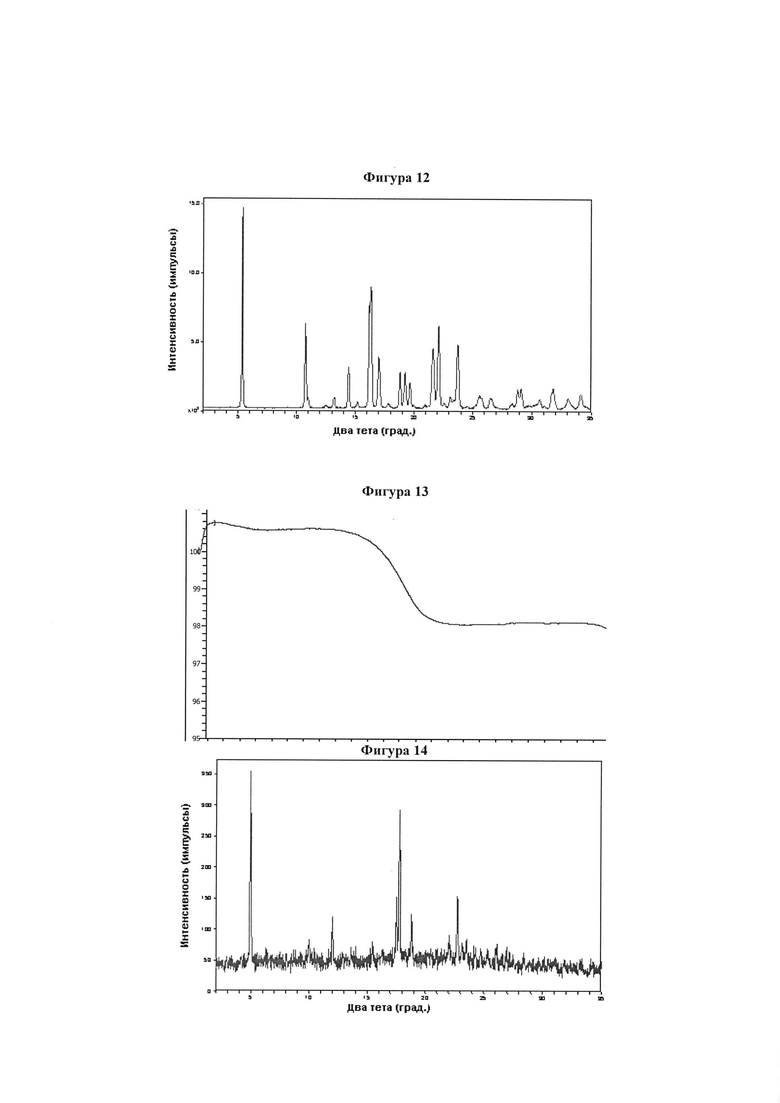
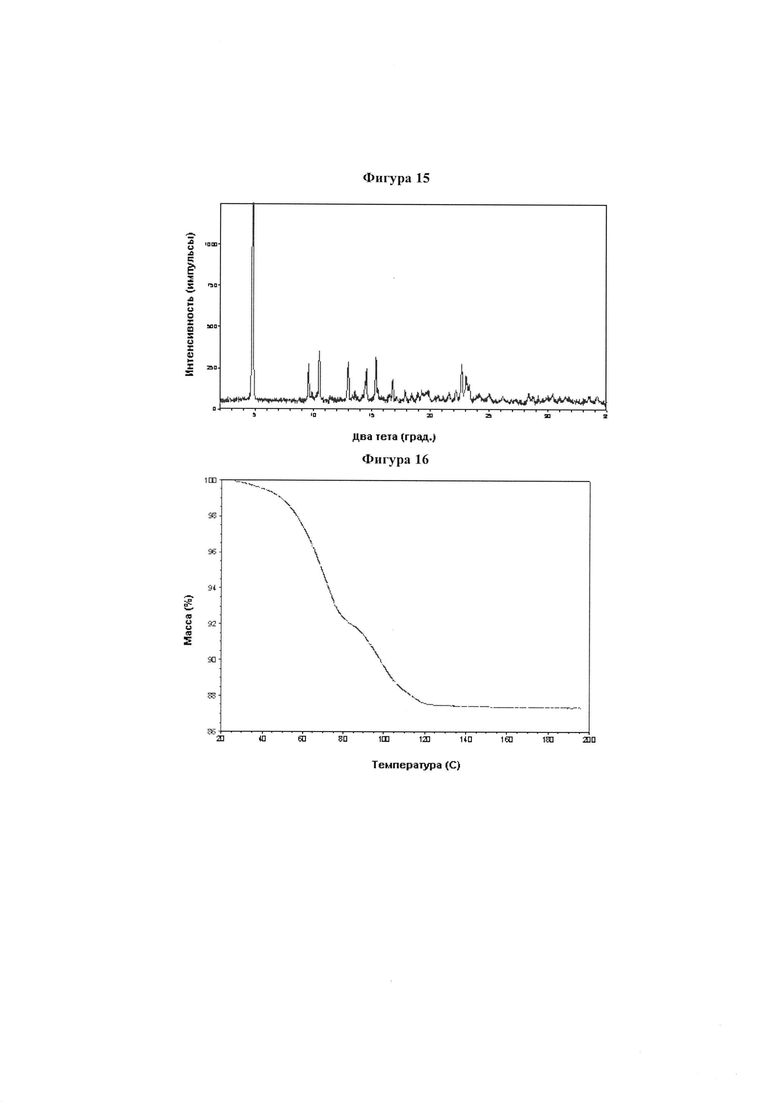
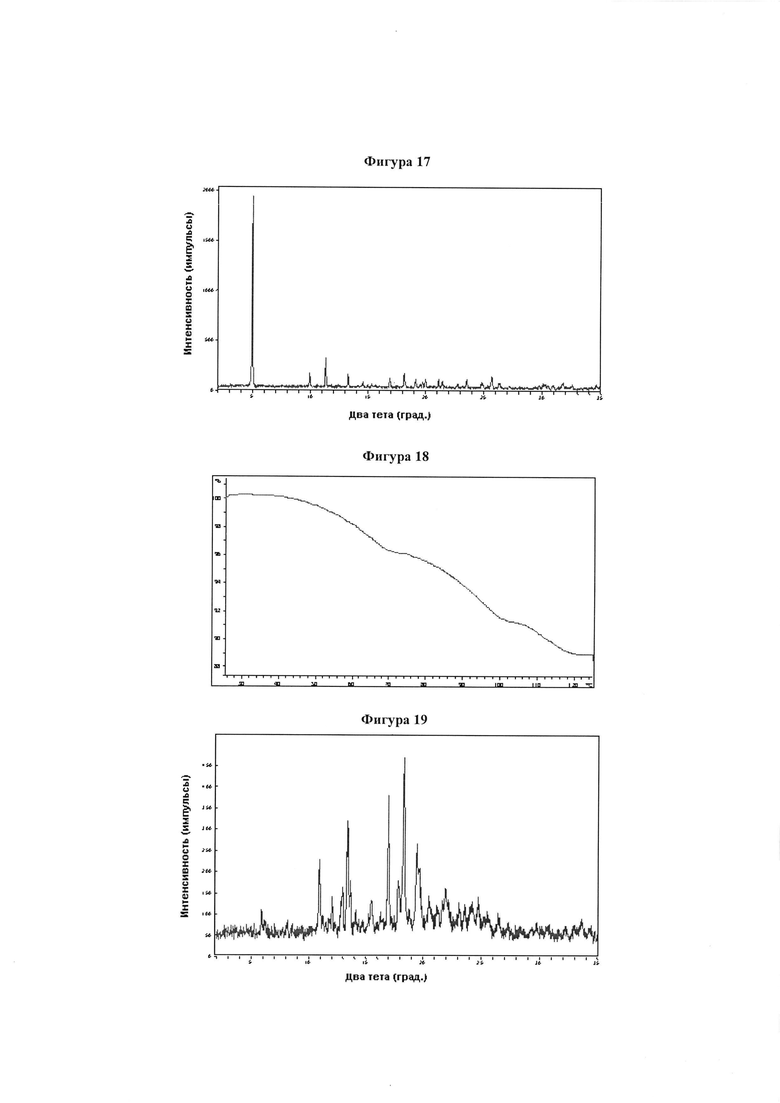
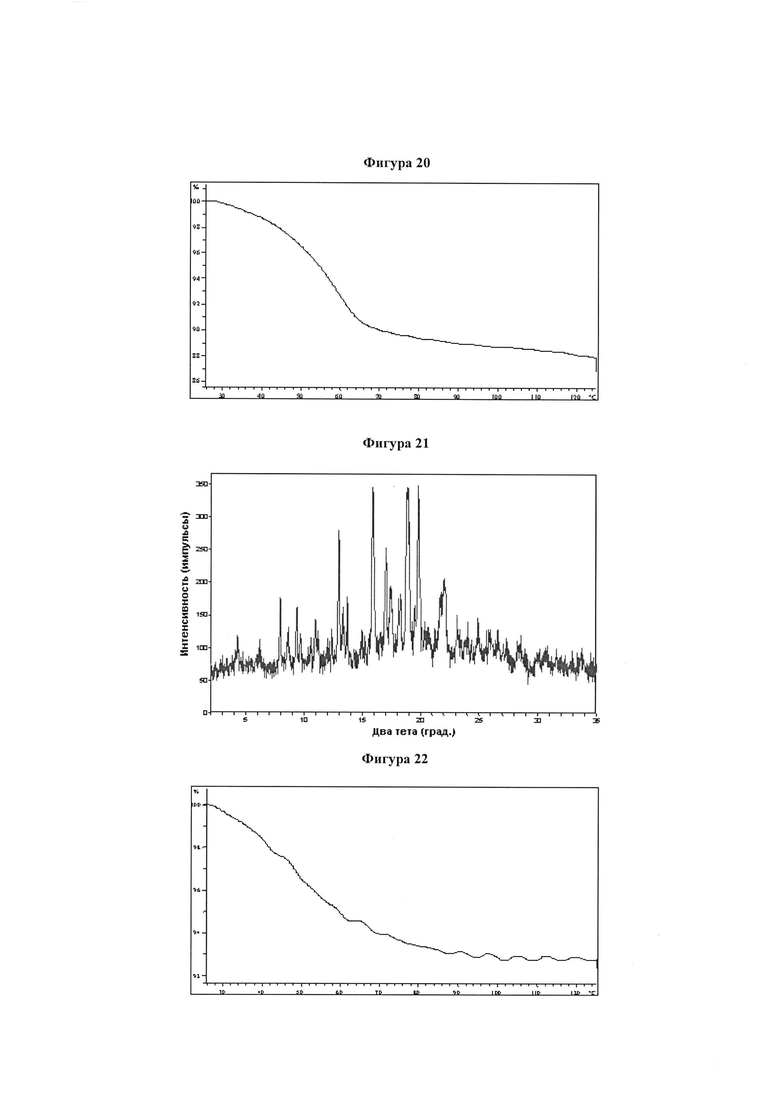
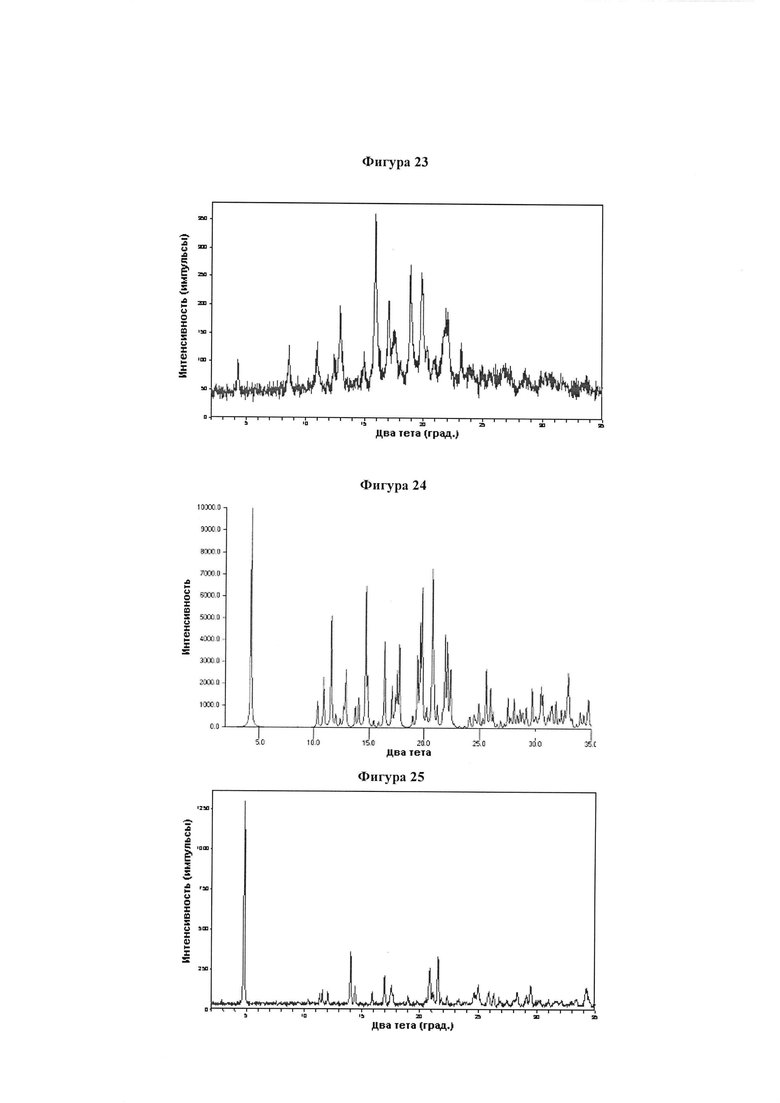
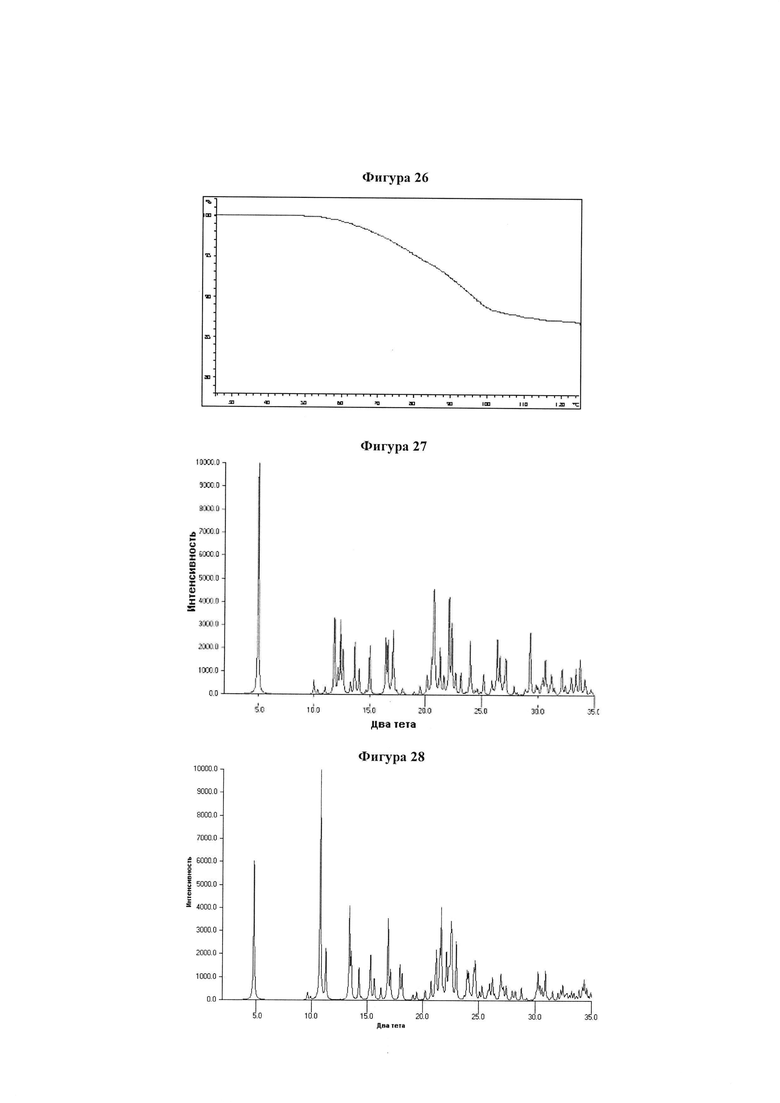
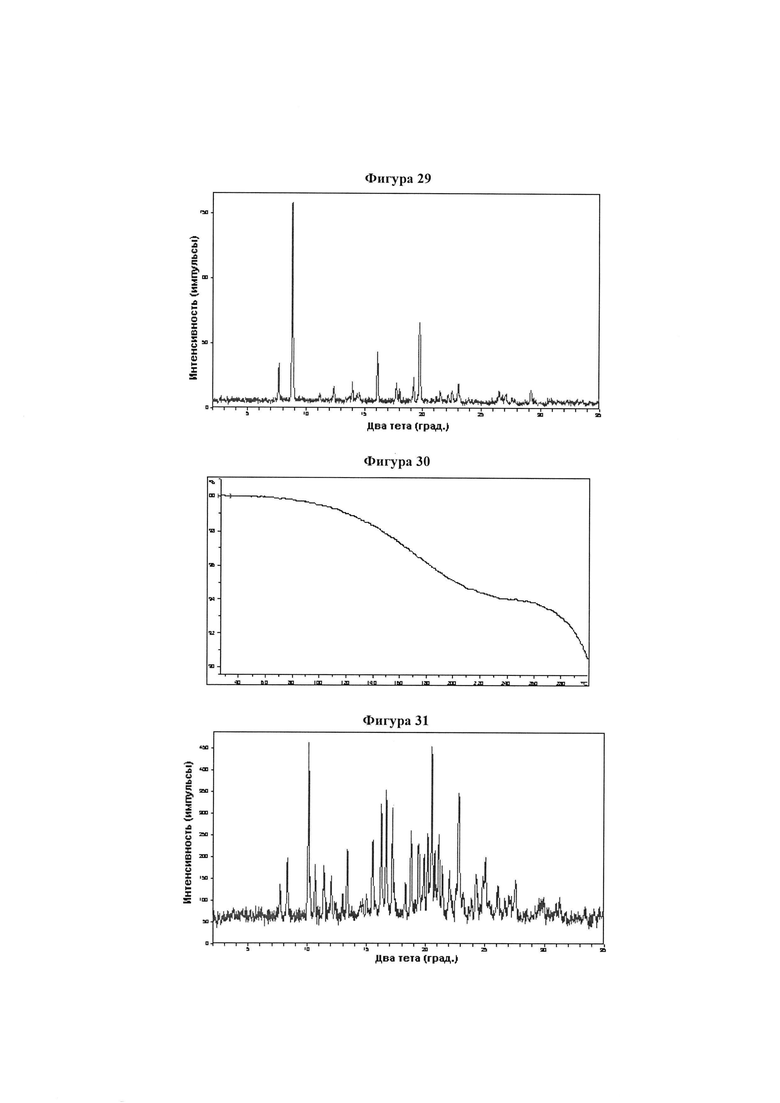
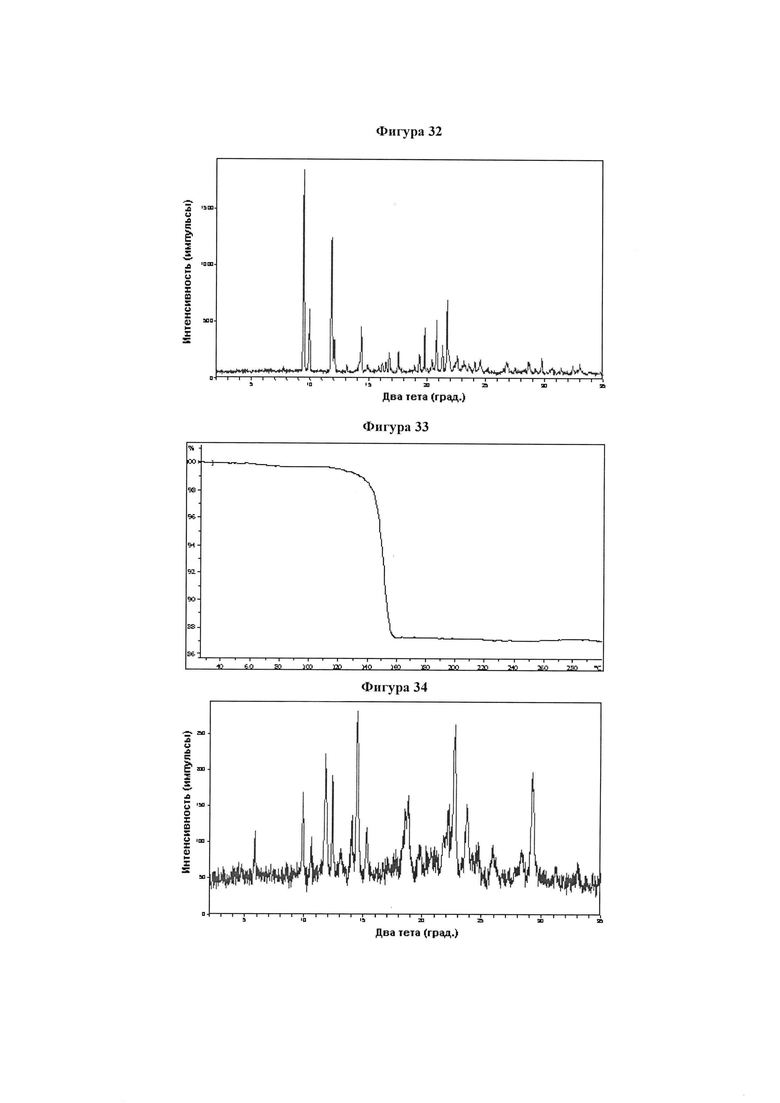
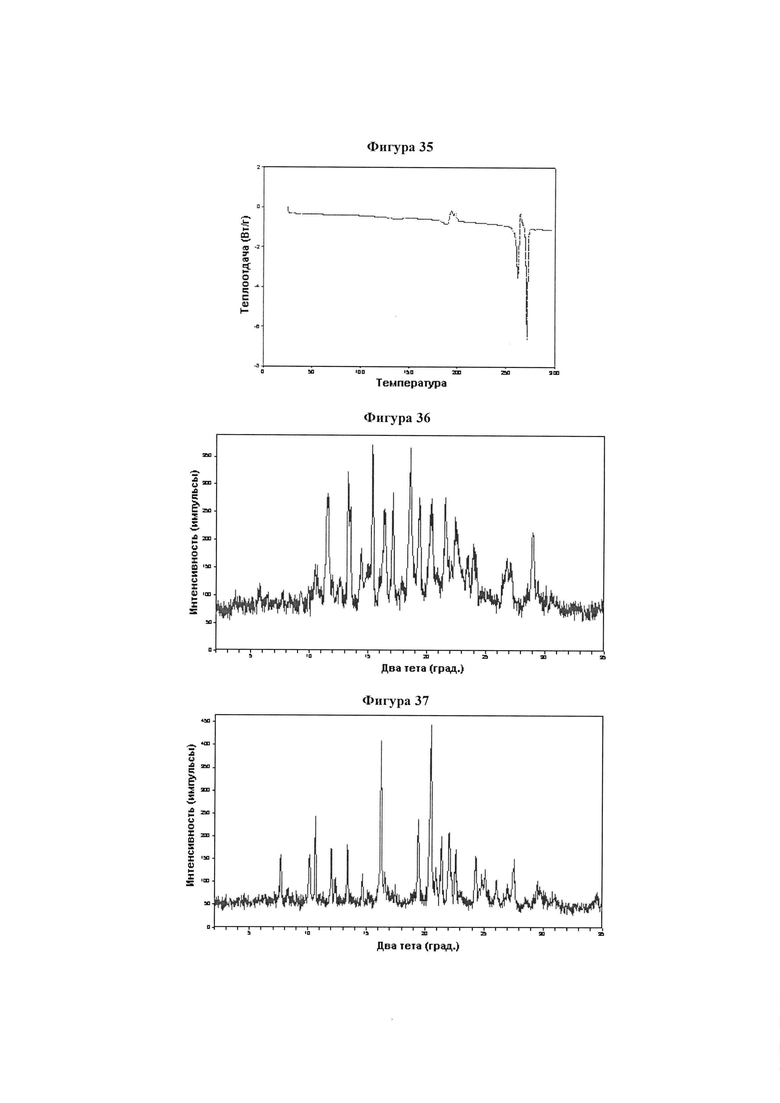
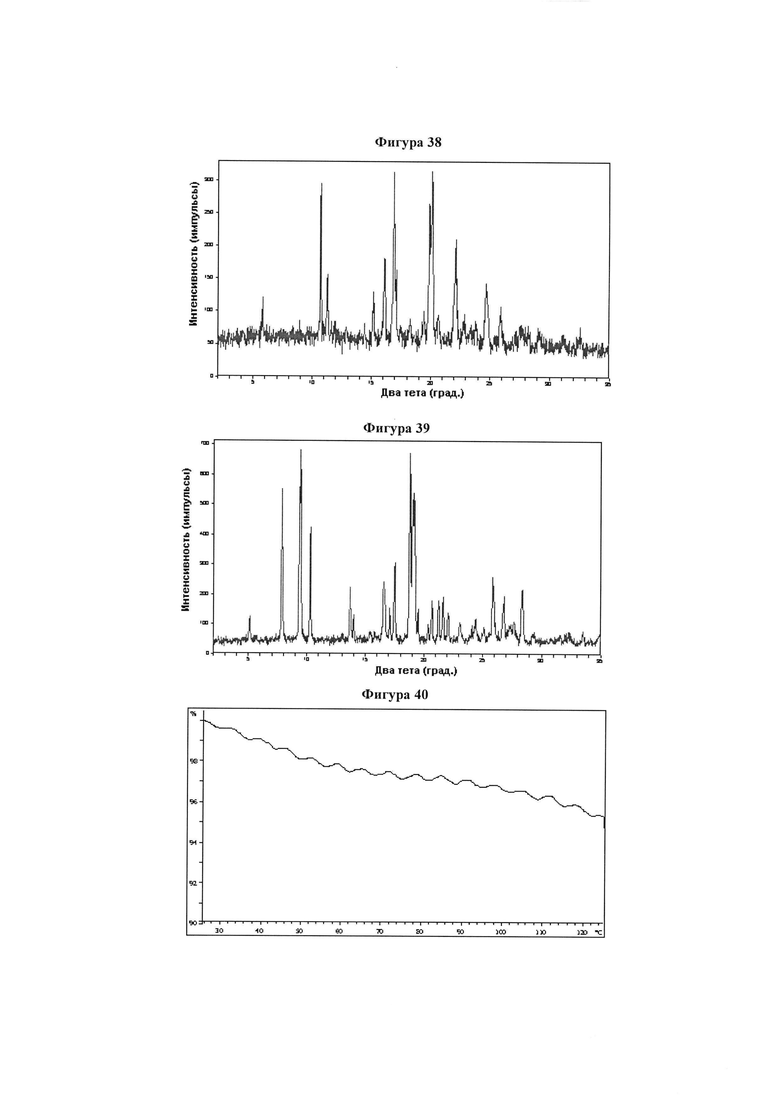
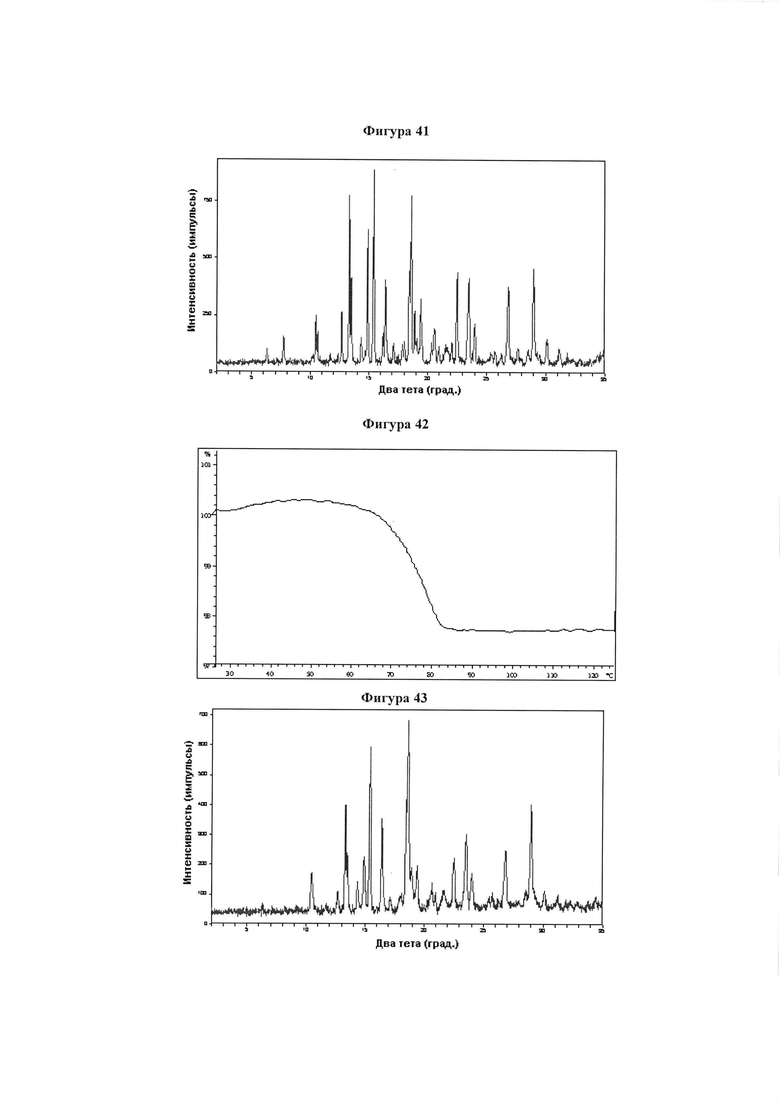
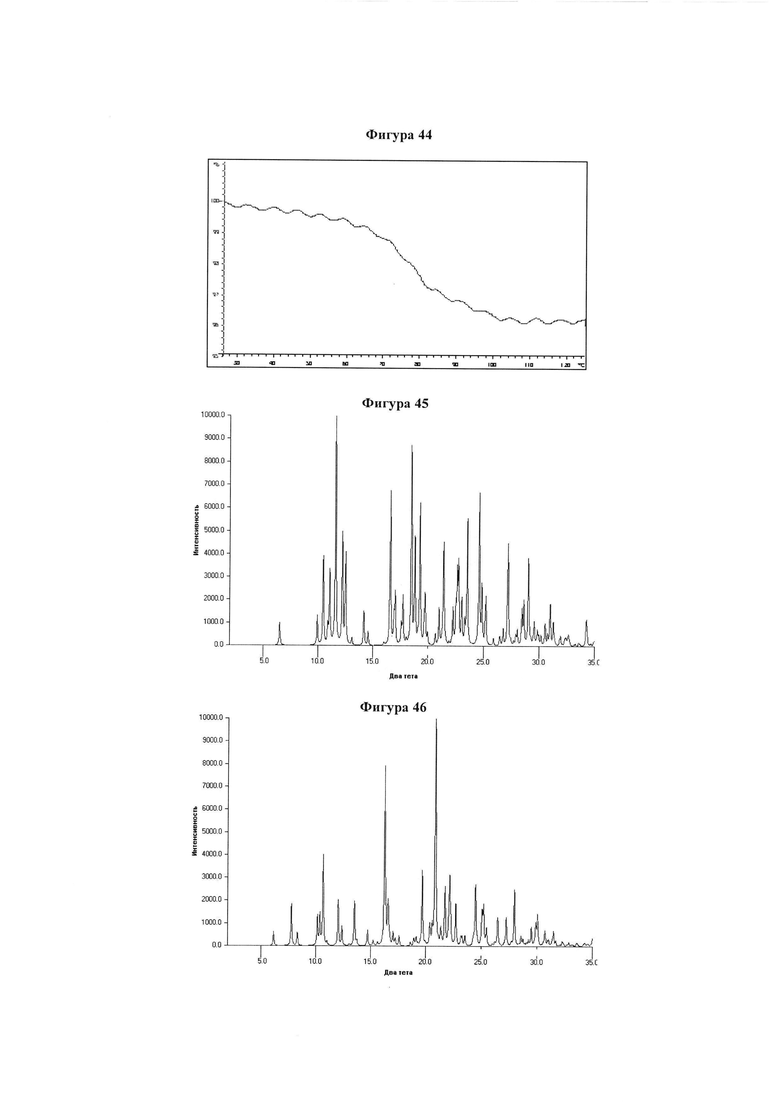
Изобретение относиться к соединению N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида или его фармацевтически приемлемой соли для применения в качестве лекарственного средства для ингибирования репликации вируса гепатита С (HCV), а также к лекарственному средству, содержащему терапевтически эффективное количество указанного соединения или его фармацевтически приемлемой соли. Технический результат - устойчивые показатели эффективности лечения, облегчение симптомов гепатита С, обеспечение частичного или полного купирования симптомов. 2 н. и 3 з.п. ф-лы, 46 ил., 40 табл., 140 пр.
1. Лекарственное средство, обладающее ингибиторной активностью в отношении полимеразы вируса гепатита С (HCV), содержащее терапевтически эффективное количество N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамида или его фармацевтически приемлемой соли.
2. Лекарственное средство по п. 1, в котором фармацевтически приемлемая соль представляет собой мононатриевую соль.
3. Лекарственное средство по п. 1, которое дополнительно содержит один или несколько дополнительных терапевтических агентов, выбранных из группы, состоящей из интерферонового агента, рибавирина, ингибитора HCV, и ингибитора HTV.
4. Лекарственное средство по п. 1, которое дополнительно содержит один или несколько дополнительных терапевтических агентов, которые представляют собой ингибитор HCV.
5. N-(6-(3-трет-бутил-5-(2,4-диоксо-3,4-дигидропиримидин-1(2Н)-ил)-2-метоксифенил)нафтален-2-ил)метансульфонамид или его фармацевтически приемлемая соль для применения в качестве лекарственного средства для ингибирования репликации вируса гепатита С (HCV).
| US 4239888 A, 16.12.1980 | |||
| M.W.MILLER ET AL., "Anticoccidial activity of 1-phenyluracils", Journal of Medicinal Chemistry, 26(7), 1983, р.1075-1076 | |||
| R.S.BALTRUSIS ET AL., "Bromo derivatives of 1-(4-hydroxyphenyl)dihydrouracil and 1-(4-hydroxyphenyl)-5- or -6-methyldihydrouracils", Chemistry of Heterocyclic Compounds, 18(9), 1982, p.1251-1254 | |||
| RU |

