Область техники
Настоящее изобретение относится к области биотехнологии и фармацевтической промышленности, точнее к конструированию и получению циклических пептидов, предназначенных для диагностики и лечения онкологических заболеваний или любых других патологий, включающих нежелательную клеточную пролиферацию.
Предпосылки создания изобретения
Злокачественное новообразование является заболеванием, характеризуемым неконтролируемым клеточным делением и ростом. Злокачественные новообразования приобретают способность инвазировать орган, из которого они произошли, распространяться через кровоток и лимфу в дистальные органы и фиксироваться и расти на них. Это является высоко гетерогенным процессом, но общим для более чем 200 типов злокачественных новообразований с совершенно различной эволюцией. Для развития заболевания необходимо одновременное изменение нескольких генов. Все эти свойства усложняют изучение и раскрытие механизмов возникновения злокачественных новообразований, и, таким образом, изучение злокачественных новообразований является широкой и мультидисциплинарной областью и включает несколько направлений исследований. Важно отметить, что это заболевание занимает второе место в структуре смертности во всем мире и, как ожидается, займет первое место к 2020 году, опережая смертность от сердечно-сосудистых заболеваний (Forteza F (2004) Avances médicos de Cuba. 40:33).
Действительно, злокачественные новообразования уже занимают первое место в структуре смертности в развитых странах и второе место - в развивающихся странах (World Health Organization. The Global Burden of Disease: 2004 Update. Geneva: World Health Organization; 2008). Заболеваемость растет в последних из упомянутых странах в связи с ростом стареющего населения, и еще чаще из-за предрасположенного к возникновению злокачественных новообразований образа жизни - гиподинамии, курения и «западных» диет.
По данным GLOBOCAN 2008 зафиксировано 12,7 миллионов пациентов, имеющих злокачественные новообразования, и 7,6 миллионов смертельных исходов в 2008 году; из них 56% пациентов и 64% смертельных исходов наблюдается в развивающихся странах (Ferlay J, Shin HR, Bray F, Forman D, Mathers CD, Parkin D. GLOBOCAN 2008, Cancer Incidence and Mortality Worldwide: IARC Cancer Base No. 10. Lyon, France: International Agency for Research on Cancer; Available from: http://globocan.iarc.fr.2010. Accessed 8/17/2010).
Выживаемость при злокачественных новообразованиях, как правило, ниже в развивающихся странах, вероятно из-за сочетания поздней диагностики и ограниченного доступа к своевременному и подходящему лечению, невзирая на цитотоксические средства, уже имеющиеся в продаже и оптимизированные для лечения злокачественных новообразований. Новые биологические молекулы необходимы для создания лекарственных средств против злокачественных новообразований нового поколения, более эффективных и безопасных в ближайшем будущем и способных в значительной степени проникать на рынок терапевтических средств против злокачественных новообразований.
На сегодняшний день принято считать, что, для того чтобы быть эффективным, лечение злокачественных новообразований должно сочетать различные принципы действия, такие как прямое воздействие на опухолевые клетки и влияние на окружающие опухоль условия. Это может быть достигнуто сочетанием молекул, в отдельности обладающих каждым из этих свойств или одновременно демонстрирующих оба из них. Безусловно, этот последний тип молекул является преимущественным с фармакологической и экономической точки зрения. Доклинические исследования ингибиторов ангиогенеза, предназначенных для прекращения доставки кислорода и питательных веществ в опухоль, продемонстрировали многообещающие результаты, зачастую достигая полной или частичной регрессии опухоли в отсутствие резистентности к ингибитору. До настоящего времени главным достижением клинических испытаний была продолжительная компенсация заболевания в течение заданного периода времени. С этой целью, антиангиогенные средства используются в качестве вспомогательного лечения в сочетании с другими противоопухолевыми средствами.
Результаты клинических испытаний показали, что однонаправленного действия регуляторов ангиогенеза недостаточно для продолжительного ингибиторного ответа. Возрастает потребность в более эффективных антиангиогенных средствах, способных заблокировать и также реверсировать опухолевый рост, для достижения значительного увеличения продолжительности жизни и качества жизни пациента в сравнении с общепринятыми способами лечения.
Имеющиеся в настоящее время пептиды представляют небольшую фракцию среди бесчисленного множества средств, используемых в терапевтических целях. Фактически, потенциал пептидов совершенствуется с помощью новых технологий для модификации их структуры, фармакокинетики, биораспределения, стабильности и доклинического применения. В частности, они приобрели актуальность в лечении злокачественных новообразований благодаря новым методологиям, имеющимся для их модификации и повышения их противораковой эффективности (Li, Zhi J.; Cho, Chi H. Current Pharmaceutical Design, 16 (10), April 2010, pp. 1180-1189).
Несколько исследований продемонстрировали возможность использования пептидов в диагностике и лечении злокачественных новообразований. Некоторые из них находятся на этапе близкой к завершению фазы клинических испытаний, и другие средства нового поколения уже появляются в последние годы с весьма перспективными результатами доклинических исследований.
Цитотоксическая активность литического пептида, сконструированного для связывания рецептора эпидермального ростового фактора, была продемонстрирована на некоторых раковых клеточных линиях человека. Было доказано, что конформационные изменения, возникающие при связывании литического пептида, увеличивали селективность его ассоциации с мембраной раковых клеток, и результатом этого приобретенного синергетического действия была селективная деструкция опухолевых клеток. Обработка литическим пептидом, связывающим рецептор эпидермального ростового фактора, обладала цитотоксической активностью in vitro против раковых клеток, устойчивых к ингибиторам тирозинкиназы, с мутациями K-ras (Kohno, Masayuki. European Journal of Cancer 47(5), p.773, Mar 2011).
Пенетрирующие клетку пептиды, как правило, соединены с олигонуклеотидами для повышения их эффективности в лечении злокачественных новообразований. С этой целью, пенетрирующие клетку пептиды конструируют с тем, чтобы глутаматный пептид был соединен с N-концом Oct6 NLS, который демонстрирует совместную локализацию в ядре клетки и также захват клеточными линиями рака поджелудочной железы и простаты (Lewis, H Dan. BMC Biotechnology, 10(1), p.79, Oct 2010).
Пептидный фрагмент из ингибитора сигнального пути тканевого фактора (TFPI), который является естественным антикоагулирующим белком, был способен блокировать опухолевый рост и ангиогенез в моделях in vivo. Более того, он ингибировал метастазирование опухоли и рост новых кровеносных сосудов без явного воздействия на нормальные сосуды (HEMBROUGH Todd A.; RUIZ Jose F.; SWERDLOW Bonnie M.; SWARTZ Glenn M.; HAMMERS Hans J.; ZHANG Li; PLUM Stacy M.; WILLIAMS Mark S.; STRICKLAND Dudley K.; PRIBLUDA Victor S. Blood A. 2004, vol. 103, n° 9, pp. 3374-3380).
Разработка более селективных веществ для визуализации и лечения различных опухолей является современной тенденцией в лечении и диагностике злокачественных новообразований. В этом смысле пептиды являются небольшими аминокислотными последовательностями, которые могут быть получены или сконструированы для связывания с заданной молекулой-мишенью, и они потенциально способны препятствовать ее функции. Эти специфические пептиды могут ингибировать компоненты специфических сигналов, необходимых для развития и прогрессирования злокачественных новообразований.
Серрализин является основным внеклеточным белком бактерии Serratia marcescens CMIB4202 и связан с патогенностью данного микроорганизма у людей с приписываемыми противоопухолевыми свойствами, зависящими от его каталитической активности (Wu Jun, Akaike T, Hayashida K, et al., (2001) Japanese J. Cancer Res. 92:439-451). В этом штамме (S. marcescens CMIB4202) наиболее высоко представленным белком является белок р50, который принадлежит семейству Serralysins (SERMA). Известно, что полипептид, содержащий С-концевой некаталитический домен серрализина (обозначаемый р25), является мощным ингибитором эндотелиальной пролиферации и роста первичных опухолей и метастазирования in vivo (Abrahantes-Pérez MC et al., «Pharmaceutical composition containing polypeptide fragments of serralysins». Международная патентная заявка № WO 2006/005268). Этот полипептид был назван CIGB370r в экспериментах по экспрессии в виде рекомбинантного белка в Escherichia coli.
Существует огромная потребность в идентификации и получении более эффективных противоопухолевых веществ в связи с увеличением числа вновь регистрируемых случаев этого заболевания с целью замены или дополнения современного лечения злокачественных новообразований для нуждающихся в нем пациентов, несмотря на наличие огромного числа лекарственных средств, предназначенных для этой цели.
Подробное описание изобретения
Данное изобретение вносит вклад в решение вышеупомянутых проблем путем предоставления циклических пептидов с противоопухолевым и антиангиогенным свойством. В настоящем документе рассмотрено конструирование и получение этих пептидных соединений, при этом демонстрируя их эффективность на некоторых животных моделях рака.
Поразительно, что противоопухолевая активность полипептида р25 S. Marcescens воспроизводилась структурно ограниченным пептидным фрагментом, который был едва доступен внутри области взаимодействия N- и C-концевых доменов Serralisine. Это дало основание предположить, что структурная конформация в этой области полипептида являлась минимальной функционально активной структурной единицей полипептида р25, погруженной вовнутрь Serralysin и, вероятно, становясь доступной во время белкового аутокатализа или оказавшись в протеолитическом окружении опухоли. Данные, представленные в настоящем документе, демонстрируют, что структурно ограниченные пептиды по настоящему изобретению обладают прямой цитотоксической активностью в отношении опухолевых клеток и антиангиогенной активностью, и предполагают возможный механизм и новую парадигму об опухолевой регрессии, опосредованной инфекцией.
Пептиды являются очень пластичными молекулами и, соответственно, могут принимать различные структуры. Одна или несколько таких возможных структур могут иметь специфическую биологическую значимость. Для определения возможных значимых конформаций необходимо заключить пептиды в один-единственный участок конформационного пространства для дальнейшего определения значимой формы. В конечном итоге, путем скрининга некоторых из этих конформаций возможно найти биологически значимую конформацию.
Существуют новые методологии создания более точных синтетических структур. Необходимо принимать во внимание определенную гибкость в подходе. То есть, если конструируемая структура является слишком жесткой, другой структуре нельзя присвоить свойства, заданные для ее биологической активности in vivo, в виду того, что слегка пластичная структура способна к такому виду корректировки. Такое важное понимание требований, подлежащих применению в отношении рецепторов пептидов, активных сайтов ферментов и большого разнообразия других биологических процессов, обеспечивается использованием адекватных методик и методологий конструирования синтетических пептидов.
Свойства, проявляемые пептидом в биологических системах, зависят от структуры пептида. Следовательно, способность использовать рациональную конструкцию для получения пригодных пептидов зависит от соответствующих профессиональных знаний по установлению взаимосвязи между молекулярной структурой и ее биологической активностью. Профессиональные знания по признанию таких взаимоотношений основаны на некоторых неопределенностях, обусловленных не только системами биологического анализа, но также интерпретацией данных. Более сложным фактором, вовлеченным в данную проблему, является трудность определения трехмерной структуры самого пептида. Многие пептиды от природы являются пластичными и принимают широкий спектр конформаций в растворе. Проблема заключается в определении того, какая среди всех возможных конформаций ответственна за наблюдаемую активность пептида, рассматривая многие пептиды, активные в более чем одной конформации. Использование конформационных ограничений показало пригодность в выявлении таких взаимоотношений структура-функция. Если пептид ограничивается именно определенной конформацией или таковой, полностью похожей на семейство активных конформаций, тогда измеряемая активность непосредственно отражает эффект данной структуры. Даже когда невозможно получить молекулу с абсолютно жесткой структурой, можно начать с приписывания определенных биологических активностей вызывающим их структурам путем конструирования аналогов с заданными структурными мотивами.
В настоящем изобретении физическое картирование функциональных сайтов внутри последовательности белка р50/р25 путем использования синтетических пептидов длиной 20 а/к, перекрывающихся на участке длиной 10, и дальнейшего поиска цитотоксической активности in vitro на опухолевых клетках (см. пример 2) указывало на то, что пептид Gly255-Ser274 (N06P87) является активным. Однако активность этого пептида in vivo была ниже таковой, проявляемой белком р25. Кроме того, замена сегмента Gly266-Asp268 трипептидом Ala-Ala-Ala как в р25-подобном рекомбинантном полипептиде CIGB370r, так и в синтетическом пептиде N06P87 устраняла биологическую активность обеих молекул, указывая на то, что этот сегмент необходим для противораковой активности. Более того, исходя из данного результата, полагают, что требуется одна или несколько боковых цепей на остатках Arg267 и Asp268 для взаимодействия с рецептором(ами), еще не идентифицированных, несмотря на вероятный негативный эффект тройной мутации на соответствующий пептид и биологически активные белковые конформации. В этом смысле, если допускать, что локальная, биологически значимая структура трипептида подобна таковой в кристаллографической структуре р50, тогда замена Gly266 на Ala весьма невыгодна, поскольку основная цепь на этом остатке образует положительные торсионные углы, недопустимые для аланиновой аминокислоты (фиг.14). Кроме того, результаты, показанные в примере 2, доказывают то, что наличие самого по себе фрагмента Gly266-Asp268 недостаточно для достижения биологического эффекта и необходимы также другие остатки. Как показано в таблице 2, синтетический пептид F07P16, содержащий последовательность Thr265-Trp284 и перекрывающийся по 10 остаткам с линейным пептидом N06P87 (Gly255-Ser274), является неактивным, несмотря на имеющуюся в своем составе последовательность Gly266-Asp268.
В настоящем изобретении вызывает удивление идентификация Gly255-Ser274 как часть функционального сайта, ответственного за противораковую активность р50/р25, из-за криптической природы этого сегмента внутри 3D-структуры белка р50. Большинство аминокислот сегмента Gly255-Ser274 полностью или частично маскировано внутри 3D-структуры белка р50, включая остатки Thr257, Tyr258, Gly259, Phe260, Thr265, Arg267, Phe269, Leu270 и Thr272. Изгиб Arg267-Leu270 является частью поверхности стыка N- и C-концевых доменов белка. Остатки Arg267 и Asp268 образуют соляные мостики (каждый двойные водородные мостики) с остатками Asp98 и Arg171 N-концевого домена, соответственно. Поверхность стыка также содержит гидрофобные междоменные взаимодействия с участием остатка Phe269 в С-концевом домене и остатков Ala232 и Ala233 в N-концевом домене. Кроме того, криптическая природа данного сайта соответствует высокой эффективностью белка р25 по сравнению с таковой р50, поскольку у р25 отсутствует N-концевой домен и сегмент Gly255-Ser274 более доступен (см. пример 3, фиг.5 и таблицу 3).
В настоящем изобретении было доказано, что конформация пептида N06P87 существенно важна для его биологической активности. Значимость конформации пептида N06P87 для его биологической активности подтверждается результатами примера 4, демонстрирующего, что его активность зависит от фланкирующих областей, которые обеспечивают правильную укладку молекулы. С другой стороны, полипептид р25 не проявлял никакой активности при экспрессии в виде рекомбинантной молекулы и после ренатурации в отсутствие кальция. Следовательно, неструктурированные препараты, соответствующие полипептидам в отсутствие атомов кальция, не обладают активностью (связывание кальция необходимо для правильной укладки белка и стабилизации). Дополнительно в примере 4 показано, что введение дисульфидного мостика в пептид N06P87 - путем добавления цистеинового остатка на N-конце и другого на С-конце - обусловливает потерю биологической активности пептида (пептид N06P87 в таблице 4, фиг.7). Циклизация, обеспечиваемая этими способами, уменьшает конформационное пространство, доступное для пептида в растворе; тем не менее, циклизация несовместима с конформацией, принимаемой сегментом Gly255-Ser274 в уложенной структуре р50. Расстояние между аминоконцом и карбоксильным концом сегмента Gly255-Ser274 в кристаллографической структуре белка составляет 24,4 Å (фиг.10Е), что несовместимо со стереохимией дисульфидного мостика (расстояние между альфа углеродами соединяемых цистеинов никогда не бывает более 7 Å). Эта модификация, таким образом, предполагает значительное изменение структурных свойств пептидной цепи в N06P87. Эти результаты и их соответствующий анализ дают основание предполагать, что активная конформация N06P87 могла бы быть подобной таковой сегмента Gly255-Ser274 в нативном полипептиде р25.
Как показано в примерах 2, 4 и 6, практически возможно сконструировать пептидные аналоги со сходной биологической активностью полипептида р25. В данном изобретении представлено создание семейства потенциально активных пептидов короткого и среднего размера (длиной от 9 до 25 остатков), в основе которых лежит структура пептида N06P87 и модифицированные посредством введения/замены определенных химических групп и/или структурных ограничений (таблица 5), что позволяет этим пептидам проявлять значения эффективности и потенциальной активности, подобные или даже лучшие таковых полипептида р25.
Помимо эффективности и потенциальной активности, пептиды короткого и среднего размера по настоящему изобретению имеют ряд преимуществ в качестве противораковых средств по сравнению с нативными полноразмерными белками. Как правило, размер молекулы влияет на фармакокинетические свойства противораковых веществ (такие как биораспределение). Документально доказанными примерами являются рекомбинантные одноцепочечные антитела (r-sc-Fv) при сравнении с их соответствующими антителами, где первые из упомянутых демонстрируют лучшую доступность к тканям и опухолям, которые являются едва доступными для полноразмерных антител (Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De Baetselier P, Muyldermans S, Revets H; (2004). Cancer Res. 64(8):2853-7).
Терапия антителами оказала особенно ограниченное влияние на лечение солидных опухолей (Stern M, Herrmann R; (2005). Crit Rev Oncol Hematol. 54(1):11-29). В целом, экспериментальные данные указывают на то, что фармакокинетические свойства лиганда улучшаются при уменьшении его размера (Reilly R. M., Sandhu J., Alvarez-Diez T. M., et al. (1995). Clin. Pharmacokinet. 28: 126142). Пептиды короткого и среднего размера (обычно от 1 до 3 кДа) могут преодолеть, по крайней мере, частично эти трудности, с которыми сталкиваются при проведении лечения злокачественных новообразований с использованием антител (Ladner R. C., Sato A. K., Gorzelany J., de Souza M. (2004). Drug Discov. Today 9: 525529). В частности, пептиды могут обладать лучшей способностью проникать внутрь опухоли, более низким неспецифическим захватом и вызывать более низкий иммунный ответ.
Таким образом, пептиды по настоящему изобретению конструируют для оптимизации взаимодействия с их рецептором и, что особенно важно, для обеспечения эффективного биораспределения.
Как правило, пептиды короткого и среднего размера длиной до 20-25 остатков являются слабо иммуногенными, что не относится к гетерологичным белкам и особенно антигенам микроорганизмов, как р25. Использование таких белков в качестве терапевтических средств может вызвать иммунный ответ у пациентов с последующей индукцией синтеза антител, которые могут нейтрализовать терапевтический эффект белка. Этот эффект особенно значим при лечении хронических заболеваний, требующих повторного использования терапевтических средств. С другой стороны, если микроорганизм является патогеном для людей, то вполне вероятно, что фракция популяции индуцирует образование нейтрализующих антител, наличие которых до начала лечения потребует увеличения терапевтических доз лекарственного средства. В этой связи, поскольку значительная часть молекулярной поверхности сегмента Gly255-Ser274 нарушена внутри области взаимодействия N- и С-концевых доменов белка р50, и поэтому маскирована в нативной структуре белка. Следовательно, получаемый в результате пептид N06P87 потенциально является слабоиммуногенным, иначе говоря, антитела против белка р50 неэффективны в узнавании (нейтрализации) пептида N06P87. Исходя из вышеизложенного и рассматривая антигенный/иммуногенный потенциал терапевтической молекулы, более предпочтительным является использование пептидов вместо полноразмерных белков, особенно, когда пептиды способны вызывать биологический эффект подобный таковому природного белка.
Существенно важный аспект конструирования эффективных противораковых средств в настоящем изобретении включает конструирование циклических пептидов, то есть, они имеют аминокислоты, связанные ковалентными связями, включая химические группы, расположенные в боковых цепях, и/или группы на N- и С-концах. Таким образом, пептиды, сконструированные в настоящем документе, структурно ограничены посредством циклизации, что значительно снижает структурную пластичность этих молекул. В большинстве случаев использование пептидов в качестве терапевтических средств имеет несколько недостатков. Именно так обстоит дело для внутренней пластичности пептидов, особенно пептидов короткого и среднего размера, которые являются гораздо более гибкими, чем уложенные белки, и, следовательно, их процесс связывания с белками или другими рецепторными макромолекулами включает значительную потерю конформационной энтропии. Этот факт обусловливает у этих молекул, как правило, более низкую аффинность связывания, нежели таковую белок-белкового взаимодействия. Более низкая аффинность, демонстрируемая пептидами (и, следовательно, более низкая эффективность), может быть также связана с тем фактом, что белок-рецепторная контактная поверхность меньше по сравнению с белок-рецепторной областью взаимодействия, в частности, когда пептиды содержат фрагмент нативного белка. По этим причинам, реконструирование и химическая модификация пептидов необходимы для увеличения их аффинности связывания рецептора (и, следовательно, эффективности). Ранее был идентифицирован полипептид, полученный из опосредованной инфекцией модели регрессии опухоли, который был назван р25, оказывающий антиангиогенное и непосредственное воздействие на опухолевые клетки (международная патентная заявка № WO 2006/005268). В настоящем изобретении была разработана платформа, основанная на пептидах, имитирующих активный мотив полипептида р25, и демонстрирующая несколько усовершенствований по сравнению с исходной молекулой. Нативный полипептид бактериального происхождения может быть применен только ограниченное число раз для лечения злокачественных новообразований из-за потенциальной индукции иммунных ответов, которые могут нейтрализовать его активность, препятствуя пролонгированному лечению, требуемому при хронических заболеваниях, таких как злокачественные новообразования. Это является причиной, почему в настоящем изобретении область исследования была сфокусирована на создании молекул, полученных из инфекционно-опосредованной опухолевой регрессии и пригодных для лечения злокачественных новообразований, путем идентификации минимальной функционально активной единицы в полипептиде р25, которые мало чем отличаются от его противораковой активности, но не обладают способностью индуцировать негативный иммунный ответ во время пролонгированного применения лечения онкологических патологий или нежелательной клеточной пролиферации.
Значительный вклад настоящего изобретения заключается в возможности на практике создать пептидные молекулы длиной до 25 аминокислот, которые в структурном плане имитируют минимальную функционально активную единицу противоопухолевых белков, полученных из инфекционно-опосредованной опухолевой регрессии. Поражает то, что эти небольшие молекулы не проявляют своей активности в отношении опухоли иммуно-опосредованными механизмами, как это ранее рассматривалось в качестве парадигмы для инфекционно-опосредованной опухолевой регрессии (Paglia P, y Guzman CA. Cancer Immunol. Immunother. 1998. 46:88-92). Более того, другой новый аспект настоящего изобретения заключается в том, что эти активные области не расположены на экспонируемой поверхности бактериальных белков, но становятся поверхностными, как только происходит ферментативное расщепление белка. Этот процесс может происходить в богатой металлопротеиназами окружающей опухоль области, инициирующей интенсивное цитотоксическое воздействие на опухолевые клетки и ассоциированный с опухолью ангиогенез. Это обстоятельство также может вносить вклад в потенциальную противоопухолевую активность, ранее приписываемую инфекционно-опосредованной опухолевой регрессии, области исследования, ожидающей пригодность молекул для лечения злокачественных новообразований и способность стать новыми биотехнологическими продуктами в лечении онкологических заболеваний на протяжении более века. В настоящем документе продемонстрирована обоснованность этой гипотезы, поскольку обнаружено противоопухолевое действие пептидов по настоящему изобретению in vitro и in vivo при пролонгированном лечении без признаков наличия нейтрализующих антител, которые могут ограничивать их продолжительное применение. Масштаб используемой технологии их получения различен. Преимуществами этих пептидов являются:
• Широкий спектр действия на опухолевые клетки различного гистологического происхождения.
• Непосредственное воздействие на опухолевый ангиогенез и непосредственное воздействие на опухолевые клетки.
• Р53-независимый механизм действия.
• Цитотоксическое воздействие на клетки, выделенные из метастазов опухоли человека, и последующее антиметастатическое воздействие.
• Индуцируют апоптоз на опухолевых клетках и специфичны для клеток, у которых активирована пролиферация.
• Противоопухолевый эффект либо при системном, либо внутриопухолевом применении. Снижают скорость роста ксенографтных опухолей и пролонгируют выживаемость имеющих опухоль животных.
• Полная регрессия опухоли в целом ряде опухолей.
• Отсутствие токсичности при повторных инъекциях у животных в течение длительного периода времени.
• Имеет величину распределения выше таковой молекулы происхождения.
• Профиль биораспределения обеспечивает лечение злокачественных опухолей различных патологий.
• Экономически доступные технологии производства.
• Более практически осуществимая и более быстрая фармацевтическая разработка, нежели таковая молекул, полученных рекомбинантными технологиями.
Субъектами по настоящему изобретению являются циклические пептиды с антинеопластической и антиангиогенной активностью, где указанные циклические полипептиды характеризуются аминокислотной последовательностью, содержащей:
а) сегмент А с аминокислотной последовательностью:
X1-Asn-Thr-X2-Arg-Asp-Phe-X3-X4,
где
X1 является аминокислотой, выбранной из группы, содержащей Ser, Cys, Lys, Asp, Glu и искусственную аминокислоту, боковая цепь которой содержит функциональную сульфгидрильную группу, аминогруппу или карбоксильную группу; или последовательностью, выбранной из группы, содержащей тетрапептид, пентапептид и гексапептид,
X2 является аминокислотой Gly или D-Ala,
X3 является аминокислотой, выбранной из группы, содержащей Leu, Cys, Lys, Asp, Glu и искусственную аминокислоту, боковая цепь которой содержит функциональную сульфгидрильную группу, аминогруппу или карбоксильную группу
X4 является дополнительной аминокислотой, которая может быть выбрана из группы, содержащей Ser, Cys, Lys, Asp, Glu и искусственную аминокислоту, боковая цепь которой содержит функциональную сульфгидрильную группу, аминогруппу или карбоксильную группу;
b) N-концевой сегмент, дополнительный и расположенный до сегмента, описанного в а), с аминокислотной последовательностью:
X-5-Asp-Thr-Val-X-4-X-3-X-2-X-1, где
X-1 является аминокислотой, выбранной из группы, содержащей Asn, D-Asp, D-Glu, D-Gln и D-Ala, и связанной пептидной связью с остатком X1, описанным в а), и указанная пептидная связь содержит карбонильную группу на основной цепи остатка X1 и аминогруппу на основной цепи остатка X1 сегмента, описанного в а);
X-2 является аминокислотой, выбранной из группы, содержащей Phe, Cys, Lys, Asp, Glu и искусственную аминокислоту, боковая цепь которой содержит функциональную сульфгидрильную группу, аминогруппу или карбоксильную группу,
X-3 является аминокислотой, выбранной из группы, содержащей Gly и D-Ala,
X-4 является аминокислотой, выбранной из группы, содержащей Tyr, Cys, Lys, Asp, Glu и искусственную аминокислоту, боковая цепь которой содержит функциональную сульфгидрильную группу, аминогруппу или карбоксильную группу,
X-5 является аминокислотой, выбранной из группы, содержащей Gly и D-Ala,
c) C-концевой сегмент, дополнительный и расположенный после сегмента, описанного в а), аминокислотная последовательность которого выбрана из группы, содержащей Thr-X+1X+2, Thr-X+1-X+2-X+3 и Thr-X+1-X+2-X+3-X+4,
где
N-концевой остаток Thr в указанном С-концевом сегменте соединен с сегментом, описанном в а), пептидной связью, которая содержит аминогруппу основной цепи указанного N-концевого остатка Thr и карбонильную группу основной цепи остатка X4 сегмента, описанного в а),
X+1 является аминокислотой, выбранной из группы, содержащей Thr, Gly и Ala,
X+2 является аминокислотой, выбранной из группы, содержащей Ser, Asn, Cys, Lys, Asp, Glu и искусственную аминокислоту, содержащую в боковой цепи функциональную сульфгидрильную группу, аминогруппу или карбоксильную группу,
X+3 является аминокислотой, выбранной из группы, содержащей Cys, Gln, Arg, Asn, Lys, Asp, Glu и искусственную аминокислоту, содержащую в боковой цепи функциональную сульфгидрильную группу, аминогруппу или карбоксильную группу,
X+4 является аминокислотой, выбранной из группы, содержащей Gln, Arg, Asn и Lys,
d) по меньшей мере ковалентную связь, выбранную из группы, содержащей пептидную связь, образованную аминогруппой и карбонильной группой N- и С-конца пептида, присутствующего, если последовательность X1 сегмента, описанного в а), является последовательностью тетрапептида, пентапептида или гексапептида; ковалентный дисульфидный мостик, содержащий сульфгидрильные группы в боковой цепи остатков X1 и X4, или X-4 и X3, или X-2 и X+2, или X-2 и X+3, если указанные X1 и X4, или X-4 и X3, или X-2 и X+2, или X-2 и X+3 являются цистеинами или искусственной аминокислотой, боковая цепь которой содержит сульфгидрильную группу; амидную связь, содержащую карбонильную группу и аминогруппу в боковых цепях остатков X1 и X4, или X-4 и X3, или X-2 и X+2, или X-2 и X+3, если указанные X1 и X4, или X-4 и X3, или X-2 и X+2, или X-2 и X+3 являются Lys (или искусственная аминокислота, боковая цепь которой содержит аминогруппу) и Glu (или Asp или искусственная аминокислота, боковая цепь которой содержит карбонильную группу), или указанные остатки являются соответственно Glu (или Asp или искусственной аминокислотой, боковая цепь которой содержит карбонильную группу) и Lys (или искусственная аминокислота, боковая цепь которой содержит аминогруппу); и амидную связь, содержащую карбонильную концевую группу пептида и аминогруппу боковой цепи остатка X-2, и указанная амидная связь присутствует, если X+2 является остатком на карбоксильном конце пептида, Asn аминокислотой и X-2 является аминокислотой Lys или искусственной аминокислотой, боковая цепь которой содержит аминогруппу.
В предпочтительном варианте осуществления настоящего изобретения циклические пептиды содержат пептидную связь между аминогруппой и карбонильной группой N- и С-конца пептида, и последовательность X1 указанных пептидов является аминокислотной последовательностью тетрапептида, предпочтительно, последовательностью, выбранной из группы, содержащей (D-Ser)-Pro-Thr-Pro, (D-Ala)-Pro-Thr-Pro и Gly-Pro-Thr-Pro.
В другом варианте осуществления изобретения указанные циклические пептиды содержат пептидную связь между аминогруппой и карбонильной группой на N- и С-концевых остатках пептида, и последовательность X1 указанных пептидов является пентапептидной аминокислотной последовательностью, предпочтительно последовательностью, выбранной из группы, содержащей Arg-Arg-Pro-Asn-Ser, Arg-Arg-Pro-(D-Ala)-Ser, Lys-Lys-Pro-Asn-Ser и Lys-Lys-Pro-(D-Ala)-Ser. В другом варианте осуществления изобретения указанные циклические пептиды содержат пептидную связь между аминогруппой и карбонильной группой на N- и С-концах пептида, и последовательность X1 указанных пептидов имеет гексапептидную аминокислотную последовательность, предпочтительно, последовательность, выбранную из группы, содержащей Thr-Pro-(D-Ala)-Gln-Asn-Ser, Arg-Pro-(D-Ala)-Gln-Asn-Ser, Thr-Pro-(D-Ala)-(BmGln)-(NmAsn)-Ser и Arg-Pro-(D-Ala)-(BmGln)-(NmAsn)-Ser, где BmGln представляет собой аминокислоту L-b-метилглутамин и NmAsn представляет собой аминокислоту L-N-метиласпарагин.
В данном изобретении циклические пептиды могут иметь N-конец, ковалентно связанный с ацетильной группой, пироглутаминовой аминокислотой, с липидом или полимером, предпочтительно, полиэтиленгликолем, и связь может быть образована напрямую или посредством спейсерной группы, предпочтительно, аминокислоты Gly. Более того, циклические пептиды по изобретению могут иметь С-конец в амидной форме или быть ковалентно связанными с липидом или полимером, предпочтительно, полиэтиленгликолем, и связь образуется напрямую или посредством спейсера, предпочтительно, аминокислоты Gly.
В другом варианте осуществления изобретения циклические пептиды могут содержать ковалентную связь между пептидом и липидом или любым полимером, предпочтительно, полиэтиленгликолем, и указанная связь может содержать сульфгидрильную группу, аминогруппу или карбоксильную группу на боковой цепи остатка X1, X3, X4, X-2, X-4, X+2 или X+3, и указанный остаток X1, X3, X4, X-2, X-4, X+2 или X+3 представляет собой аминокислоту Cys, Lys, Asp, Glu или искусственную аминокислоту, боковая цепь которой содержит функциональную сульфгидрильную группу, аминогруппу или карбоксильную группу.
В другом варианте осуществления изобретения циклическим пептидам могут быть свойственны остатки X1, X3, X4, X-2, X-4, X+2 или X+3, выбранные из группы, содержащей аминокислоту цистеин, (2R)-2-амино-3-сульфанилбутановую кислоту, (2R)-2-амино-3-метил-3-сульфанилбутановую кислоту, (2S)-2-амино-4-сульфанилбутановую кислоту, 2-амино-5-сульфанилпентановую кислоту, 2-амино-3-сульфанилпентановую кислоту, 2-амино-4-метил-3-сульфанилпентановую кислоту, 2-амино-3-метил-4-сульфанилпентановую кислоту, 2-амино-3,4-диметил-3-сульфанилпентановую кислоту, 2-амино-3-этил-3-сульфанилпентановую кислоту, (2R)-2-амино-3-метил-3-сульфанилпентановую кислоту, (4S)-4-амино-2-метил-5-сульфанилпентановую кислоту, (4R)-4-амино-2-метил-5-сульфанилпентановую кислоту, (4R)-4-амино-5-сульфанилпентановую кислоту, и (4S)-4-амино-5-сульфанилпентановую кислоту. В другом варианте осуществления изобретения циклическим пептидам могут быть свойственны остатки X1, X3, X4, X-2, X-4, X+2 или X+3, выбранные из группы, содержащей аминокислоту Lys, 2-[бис(3-аминопропил)амино]уксусную кислоту, (2S)-2,5-диаминопентановую кислоту, 2,2-диаминоуксусную кислоту, (3S)-3,4-диаминобутановую кислоту, (2R)-2,4-диаминобутановую кислоту, (2S)-2,4-диаминобутановую кислоту, (2S)-2,3-диаминопропановую кислоту, (2R)-2,3-диаминопропановую кислоту, 2-[(2-аминоэтил)амино]уксусную кислоту, 2-[(3-аминопропил)амино]уксусную кислоту, 2-[(4-аминобутил)амино]уксусную кислоту, (4S)-4,8-диаминооктановую кислоту, (2S)-2-амино-3-(4-аминофенил)пропановую кислоту, (2S)-2-амино-3-[4-(2-аминоэтокси)фенил]пропановую кислоту, 2-(пиперидин-4-иламино)уксусную кислоту, (2S)-2-амино-4-[(5R)-2,2-диметил-1,3-оксазолидин-5-ил]бутановую кислоту, (2S)-2-амино-6-(метиламино)гексановую кислоту, (2R,4R)-4-аминопирролидин-2-карбоновую кислоту или (2R,4S)-4-аминопирролидин-2-карбоновую кислоту, 2-(4-аминопиперидин-4-ил)уксусную кислоту, 4-аминопиперидин-4-карбоновую кислоту, (2S,4R)-4-аминопирролидин-2-карбоновую кислоту и имидазолидин-2-карбоновую кислоту.
В другом предпочтительном варианте осуществления изобретения циклическим пептидам могут быть свойственны остатки X1, X3, X4, X-2, X-4, X+2 или X+3, выбранные из группы, содержащей аминокислоту Glu, Asp, 3-[(карбоксиметил)амино]пропановую кислоту, 2-[(карбоксиметил)амино]уксусную кислоту, 3-[(2-карбоксиэтил)амино]пропановую кислоту, (3R)-3-аминогександиовую кислоту, 4-аминогептандиовую кислоту, 4-аминопиперидин-1,4-дикарбоновую кислоту, (2S,4S)-4-аминопирролидин-2-карбоновую кислоту, 2-[(карбоксиметил)амино]уксусную кислоту, (2S)-2-амино-6-[(карбоксиметил)амино]гексановую кислоту, 3-[(2-карбоксиэтил)амино]пропановую кислоту, (2S)-2-аминогептандиовую кислоту, (2S)-2-аминооктандиовую кислоту, (2R)-2-амино-3-[(2-карбоксиэтил)сульфанил]пропановую кислоту, (2R)-2-амино-3-[(карбоксиметил)сульфанил]пропановую кислоту, 4-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}бутановую кислоту и (2S)-2-амино-3-[4-(карбоксиметокси)фенил]пропановую кислоту.
В частном варианте осуществления изобретения циклические пептиды с противоопухолевым и антиангиогенным эффектом имеют аминокислотную последовательность, выбранную из последовательностей SEQ ID 1-76.
Субъектом настоящего изобретения является использование указанных циклических пептидов для приготовления лекарственного препарата с целью лечения злокачественных новообразований или для лечения патологий, связанных с нежелательной клеточной пролиферацией, или для приготовления антиангиогенного лекарственного препарата.
Другой вариант осуществления изобретения включает способ лечения злокачественных новообразований, патологий, связанных с нежелательной клеточной пролиферацией, и нежелательный ангиогенез, где указанный способ включает назначение фармацевтической композиции, содержащей эффективное количество по меньшей мере одного из циклических пептидов по изобретению, нуждающемуся в нем индивидууму. Фармацевтические композиции, содержащие по меньшей мере один из пептидов по настоящему изобретению и вспомогательные вещества или фармацевтически пригодные наполнители лекарственной формы также являются субъектом настоящего изобретения.
Изобретение также представляет соединения для диагностики рака, содержащие по меньшей мере один из пептидов по изобретению и вещество для визуализации, где указанное вещество для визуализации выбирают из группы, содержащей флуоресцентную группу, нефлуоресцентную группу, полупроводниковую флуоресцентную частицу, парамагнитное или суперпарамагнитное вещество и радиоизотоп.
Другой аспект настоящего изобретения включает фармацевтическую комбинацию, содержащую по меньшей мере один из пептидов по изобретению в сочетании по меньшей мере с одним средством для лечения, таким как противораковые лекарственные вещества и гормоны. В варианте осуществления изобретения в указанной комбинации пептид конъюгирован напрямую с лечебным средством ковалентными связями. В других случаях пептид конъюгирован с лечебным средством соединительным элементом.
Изобретение также включает фармацевтические комбинации, содержащие по меньшей мере один из пептидов по изобретению в сочетании с продигиозинами и их производными. Образующие такие комбинации составляющие могут быть назначены нуждающимся в них индивидуумам в рамках курса медикаментозного лечения либо последовательно, либо одновременно.
Большое количество лекарственных средств должно быть назначено парентеральным способом, например: внутривенной инъекцией, внутримышечным или подкожным способом, для достижения предполагаемого терапевтического эффекта. Для некоторых терапевтических средств использование контролируемого высвобождения наполнителя лекарственной формы может повысить эффективность лекарственного средства и степень удовлетворенности пациента. Молекулярная самосборка недавно была изучена с целью конструирования материалов для инкапсулирования и контролируемого высвобождения терапевтических средств. Существует огромный прогресс в конструировании платформ материалов самосборки, основанных на пептидах и полимерах (Monica C. Branco a,b, Joel P. Schneider. Acta Biomaterialia 5 (2009) 817-831). Некоторые из пептидов по настоящему изобретению, такие как J08P48, обладают амфипатическими свойствами, обеспечивая их самосборку, и, таким образом, позволяя быть частью систем контролируемого высвобождения для терапевтических молекул или в области нанотехнологии. Таким образом, субъектом настоящего изобретения являются новые инкапсулированные композиции в форме липосом или микросфер для контролируемого высвобождения этих пептидов в качестве лекарственных средств для комбинированного лечения злокачественных новообразований. И также комплексы наночастиц с контролируемой системой определения мишени для диагностических/терапевтических целевых сайтов, или сами проявляя эти виды активности специфическим способом.
Краткое описание чертежей
Фигура 1. Цитотоксическая активность пептида N06P87 в отношении опухолевых клеток различного гистологического происхождения. А: клетки Colo 205; В: клетки HEp-2. Полипептид CIGB370r и цисплатин были использованы в качестве положительных контролей противоопухолевых средств.
Фигура 2. Цитотоксическая активность N06P87 в отношении клеточной линии А549 немелкоклеточного рака легких (А) и клеточной линии рака толстой кишки (В). Другие пептиды библиотеки не проявляли активности при оценке их воздействия на пролиферацию клеточной линии А549.
Фигура 3. Методы оценки активности ряда пептидов из библиотеки, предмета исследования, в отношении человеческих опухолевых клеточных линий М14 (А) и Ls174T (B). Полипептид CIGB370r был использован в качестве положительного контроля в обоих методах.
Фигура 4. Цитотоксическая активность пептида E07P04 в отношении клеточной линии меланомы М14. Полипептид CIGB370r был использован в качестве положительного контроля в данном методе.
Фигура 5. Вычисления доступности поверхностей N-концевого (1srpN) и С-концевого (1srpC) доменов основной протеазы Serratia в сравнении с нативным белком (1srp).
Фигура 6. Последовательности, фланкирующие 20-аминокислотный лидерный пептид (ак.) N06P87.
Фигура 7. Цитотоксическая активность в отношении клеток меланомы. Лидерный пептид N06P87 был единственным, способным ингибировать клеточную пролиферацию в концентрации 50 мкМ.
Фигура 8. Оценка противоопухолевой способности линейного пептида N06P87, полученного из полипептида р25, в сравнении с исходной молекулой. Схема назначений препарата (А); Противоопухолевое действие, оцененное измерением объема опухоли (В); И противоопухолевое действие, оцененное по выживаемости инокулированных мышей (С). Используемая модель: клеточная линия Ls174T рака толстой кишки человека, имплантированная в бестимусных мышей NIH.
Фигура 9. Определение функциональных областей сегмента Gly255-Ser274 (линейный пептид N06P87). Первичный сегмент пептида является центральной областью, фланкируемой вторичными N- и C-концевыми сегментами.
Фигура 10. Моделирование циклических пептидов J08P48s-s, A08P28s-s, A08P25s-s, J08P46s-s и линейного пептида N06P87 по настоящему изобретению, сконструированных из сегмента Gly255-Ser274 белка р50. Модели были получены с помощью компьютерной программы MODELLER путем использования кристаллографической структуры белка р50 в качестве шаблона. Дисульфидные мостики были внесены в позиции, ранее идентифицированные методом MODIP. A: моделирование N06P87 (линейного пептида) в соответствии с трехмерной структурой (3D) сегмента Gly255-Ser274 белка р50; В: моделирование пептида J08P48s-s; С: моделирование пептида A08P28s-s; D: моделирование пептида A08P25s-s; Е: моделирование пептида J08P46s-s, указывая расстояние между N- и C-концом пептида.
Фигура 11. Структурное моделирование циклического пептида 33 в таблице 5. Лизин 7 образует амидную связь с карбонильной концевой группой.
Фигура 12. Пространственное изображение структурного моделирования циклического пептида 69 в таблице 5. Исходный сегмент Asn264-Ser271 циклизуют гексапептидным линкером Arg-Pro-(dAla)-Gln-Asn-Ser. Остаток dAla является стереоизомером D-Ala.
Фигура 13. Пространственное изображение структурного моделирования циклического пептида 76 в таблице 5. Исходный сегмент Asn264-Ser271 циклизуют пентапептидным линкером Arg-Arg-Pro-Asn-Ser (подчеркнутые остатки).
Фигура 14. Диаграмма Рамачандрана, соответствующая 3D-структуре сегмента Gly255-Ser274 белка р50.
Фигура 15. Основная диаграмма химической структуры сконструированных циклических пептидов по настоящему изобретению. Линии указывают на наличие ковалентной связи. В случае соединения двух остатков пептида линии означают, что эти остатки циклизованы, т.е. ковалентно связаны. Линии, расположенные сверху последовательности пептида, соответствуют циклизации основной цепи, и таковые ниже отражают связи для циклизации, вовлекая по меньшей мере одну боковую цепь. Если линия указывает на остаток Xi, это означает, что ковалентная связь образована боковой цепью этого остатка Xi. Если линия начинается от пунктира между двумя остатками, это означает, что ковалентная связь включает в себя аминогруппу или карбонильную группу остатка до или после пунктира, соответственно. Линия, указывающая на пунктир до X1, отображает амидную ковалентную связь, содержащую аминогруппу остатка X1; в этом случае пептид не имеет вторичного N-конца. Линии позади X4 и X+2 означают ковалентную связь, содержащую карбонильную группу X4 и X+2, дополнительно отображая отсутствие вторичного С-конца позади X4, и что остаток является карбоксильным концом пептида позади X+2. В фигурных скобках указаны группы, вовлеченные в ковалентную связь и тип связи: амидные связи или дисульфидные мостики, также демонстрируя предпочтительные последовательности для пептидных соединительных частей. Затененные области пептидной последовательности означают ранг от N- к С-концу функциональных сегментов, определенный в данном изобретении: (а) вторичный N-концевой сегмент, (b) первичный сегмент и (с) вторичный С-концевой сегмент. Треугольник Р означает сайты, способные быть модифированными путем ковалентного соединения с полимерами или другими химическими группами, такие модификации возможны, если остаток или группа на задействованной основной цепи не участвует в связи циклизации. Пептид может нести одну или несколько связей циклизации и одну или несколько модификаций и/или химических групп. Аминокислотными аналогами, приведенными на фигуре в качестве возможно занимающих позиции замены X-4, X-2, X1, X3, X4, X+2 и X+3, являются аминокислоты, боковые цепи которых включают аминогруппу или карбоксильную группу или сульфгидрильную группу.
Фигура 16. Оптимизированные циклические пептиды, полученные из полипептида р25, увеличивают выживаемость леченых животных. Животным, имеющим подкожную опухоль ТС-1, вводили инъекцию в виде единственной дозы величиной 80 мкМ внутрь опухоли (i.t.). Каждая группа получала различный пептид или полипептид CIGB370r или вспомогательное вещество. Выживаемость показана для всех групп.
Фигура 17. Оптимизированные циклические пептиды, полученные из полипептида р25, увеличивают выживаемость леченых животных независимо от способа введения препарата. Животным, имеющим подкожную опухоль ТС-1, вводили инъекцию в виде единственной дозы величиной 80 мкМ внутрь опухоли (i.t.) или интраперитонеально (i.p.). Каждая группа получала различный пептид или полипептид CIGB370r или вспомогательное вещество. Выживаемость показана для всех групп.
Фигура 18. Биологическая активность J08P48s-s in vitro на человеческих опухолевых клеточных линиях различного гистологического происхождения.
Фигура 19. Оптимизированные циклические пептиды, полученные из полипептида р25, увеличивают противоопухолевую активность и выживаемость у животных, леченных линейным пептидом, и их активности аналогичны таковой полипептида CIGB370r. (A) Схема назначений препарата; (В) Противоопухолевое действие, оцененное измерением объема опухоли (В); и Противоопухолевое действие, оцененное по выживаемости инокулированных мышей (С). Модель: клеточная линия Ls174T рака толстой кишки человека, имплантированная в бестимусных мышей NIH.
Фигура 20. Действие двух циклических пептидов по настоящему изобретению на клеточную дифференцировку эндотелиальных клеток в матригеле. Клетки HMEC культивировали в условиях активации (10 нг/мл EGF, 1 мкг/мл гидрокортизона) в присутствии двух циклических пептидов по настоящему изобретению или вспомогательного вещества. Пептиды обладали способностью ингибировать образование тубулярных структур эндотелиальными клетками, активированными для пролиферации, свидетельствуя об антиангиогенной активности этих пептидов, в то время как вспомогательное вещество допускало образование таких тубулярных структур.
Фигура 21. Микрофотография циклических пептидов по настоящему изобретению в условиях, обеспечивающих их самосборку.
Подробное описание вариантов осуществления изобретения/примеры
Примеры и данные демонстрируют несколько аспектов и свойств, связанных с получением циклических пептидов, начиная с библиотеки линейных синтетических пептидов и третичной структуры полипептида, соответствующего С-концу серрализина PRZN_SERMA (Braunagel SC, and Benedik MJ (1990). Mol. Gen. Genet. 222:446-451), обозначаемого р25, с доказанными фармацевтическими потенциальными возможностями (Abrahantes-Pérez MC et al., «Pharmaceutical composition containing polypeptide fragments of serralysins». Международная патентная заявка № WO 2006/005268).
Примеры, представленные ниже, касаются соединений и/или способов по настоящему изобретению, включая использование молекул, полученных из этих пептидов, оптимизированных и/или производных. При сравнении с предыдущим уровнем техники, соединения и способы, показанные в настоящем документе, обеспечивают удивительные и перспективные ожидаемые результаты. Пригодность изобретения проиллюстрирована использованием этих соединений в фармацевтической области. Указанные соединения имеют преимущества по сравнению с другими соединениями, известными в данной области техники.
Пример 1. Конструирование и синтез библиотеки линейных пептидов, полученных из полипептида р25.
Были сконструированы линейные пептиды длиной 20 аминокислот (ак.), перекрывающиеся по 10 аминокислотам, нацеленные на идентификацию минимальной функционально активной единицы полипептида р25 (Abrahantes-Pérez MC y col., «Pharmaceutical composition containing polypeptide fragments of serralysins». Международная патентная заявка № WO 2006/005268) и получение новых молекул из этой области с улучшенными фармакологическими и фармакодинамическими свойствами для лечения злокачественных новообразований. Это означает идентификацию активных участков, входящих в состав первичной последовательности длиной от 10 до 20 ак. Таблица 1 демонстрирует первичную последовательность каждого пептида, их код синтеза и молекулярную массу после синтеза и очистки. Молекулярная масса препаратов полученных в итоге пептидов была проверена масс-спектрометрией.
Конструирование и синтез библиотеки линейных пептидов, начиная с первичной последовательности полипептида р25
Пептиды были синтезированы в твердой фазе на смоле Fmoc-AM-MBHA путем использования методики Fmoc/tBu (Barany, G. and Merrifield, R. B. J Am Chem Soc. 99 (1977) 7363-7365). Аминокислоты были соединены методом DIC/HOBt-опосредованной активации, и завершенность реакции связывания была проверена нингидриновой реакцией (Kaiser, E., Colescott, R. L., Bossinger, C. D., Cook, P. I. Anal Biochem. 34 (1970) 595-598). Пептиды были отсоединены от смолы раствором TFA/EDT/H2O/TIS (94%/2,5%/2,5%/1%) с последующей либо преципитацией, либо лиофилизацией в течение 72 ч. Циклизацию осуществляли путем образования дисульфидного мостика посредством окисления диметилсульфоксида (DMSO) (Andreu, D., Albericio, F., Solé, N. A., Munson, M. C., Ferrer, M. and Barany, G., Pennington, M. W. and Dunn, B. M. (Eds), Peptide Synthesis Protocols, Methods in Molecular Biology, Totowa, NJ, 1994, pp. 91-169), и пептиды были дополнительно очищены с помощью ОФ-ВЭЖХ. Собранные фракции анализировали независимо с помощью аналитической ОФ-ВЭЖХ, и конечный препарат для каждого пептида был получен пулированием всех соответствующих фракций, демонстрирующих чистоту свыше 99%.
Пример 2. Отбор первичных последовательностей из библиотеки синтетических пептидов, полученных из полипептида р25, исходя из их цитотоксической активности in vitro в отношении опухолевых клеток.
Цитотоксическая активность библиотеки синтетических пептидов, полученной из полипептида р25, была определена на опухолевых клетках методом с использованием сульфородамина В (SRB) (Skehan P, Storeng R, Scudiero D, et al., (1990) J. Natl. Cancer Inst. 82: 1107-1112; Monks A, Scudiero D, Skehan P, et al., (1991). J Natl Cancer Inst. 83:757-66; Tesei A, Ulivi P, Fabbri F, et al., (2005). J Transl Med. 3:7). Клетки отрицательного контроля культивировали с использованием объема наполнителя лекарственной формы, равного таковому экспериментальных образцов. Кривая «x-y» (доза-ответ) была построена по проценту оставшихся в живых клеток по сравнению с клетками отрицательного контроля, и оценивали следующие параметры: 50% ингибирование роста (GI50); суммарное ингибирование роста (TGI); и летальная концентрация 50 (LC50), которая является концентрацией, вызывающей 50% клеточную гибель (Boyd MR, Paull KD, and Rubinstein LR (1992) “Data display and analysis strategies for the NCI Disease Oriented In-Vitro Antitumor Drug Screen, in Cytotoxic Anticancer Drugs: Models and Concept for Drug Discovery and Development” (Baleriote FA, Corbett TH and Baker LH eds) pp 11-34, Kluwer Academia Publishers, Boston).
Цитотоксическими пептидами рассматривались все те пептиды, которые обладали способностью ингибировать на 50% клеточную пролиферацию в зависимости от дозы, и демонстрировали значения GI50 ниже 100 мкМ по меньшей мере в одной из исследуемых клеточных линий. Использовали следующие опухолевые клеточные линии человека: HEp-2 (рак гортани), А549 (эпителиальная аденокарцинома легких), М14 (меланома), Colo 205 (аденокарцинома толстой кишки), Ls174T (аденокарцинома толстой кишки), LnCAP (карцинома простаты), PC-3 (карцинома простаты) и Н 125 (немелкоклеточная аденокарцинома легких). Результаты этого пептидного скрининга показаны в таблице 2. Представлены первичные последовательности для конструируемых пептидов, используемый для их получения код и их соответствующая цитотоксическая способность в отношении опухолевых клеток человека.
Скрининг пептидов, основанный на цитотоксической активности, демонстрируемой на опухолевых клетках человека in vitro методом SRB
Пептид GDTVYGFNSNTGRDFLSTTS-амид (Код N06P87/J07P73) был положительным (+) для нескольких опухолевых клеточных линий, в то время как пептид NASSNVTDLSVNIGGHQAPD-амид (Код E02P04) только проявлял активность на клеточной линии меланомы человека М14. Остальные пептиды рассматривались негативными (-). Фигуры 1 и 2 отражают потенциальную возможность N06P87 ингибировать пролиферацию опухолевых клеток человека различного гистологического происхождения, из клеточных линий Colo 205, HEp-2, A549 и Ls174T. Значение GI50 было ниже 100 мкМ для всех случаев. Две партии цисплатина были использованы в качестве положительного контроля в случае карциномы гортани HEp-2. Это является продуктом первой линии, имеющимся в продаже для лечения рака гортани. В этом анализе была продемонстрирована его пригодность в этом типе патологии и также достоверность анализа.
Результаты оценки воздействия некоторых пептидов из библиотеки в таблице 1 на опухолевые клеточные линии человека М14 и Ls174T представлены на фиг.3. Эта панель пептидов была неспособна ингибировать клеточную пролиферацию анализируемых клеточных линий.
Помимо перекрывающейся области пептида N06P87, 20 ак. область, кодируемая пептидом E07P04 (таблица 1), демонстрировала цитотоксическую активность, но только на клеточной линии М14 и для GI50 выше 100 мкМ, как показано на фиг.4.
Таблица 2 суммирует все результаты пептидного скрининга по цитотоксической активности на опухолевых клетках, доказывая, что пептид GDTVYGFNSNTGRDFLSTTS-амид, включающий участок Gly255-Ser274 на С-концевом домене серрализина PRZN_SERMA и расположенный на N-концевом участке полипептида р25, активен на опухолевых клеточных линиях различного гистологического происхождения, воспроизводя широкий спектр цитотоксической активности, которой обладает исходная молекула р25. Все эти данные предполагают, что пептид, идентифицируемый как цитотоксический в настоящем документе, может представлять собой минимальную структурно-активную последовательность на полипептиде р25, подлежащую дальнейшей оптимизации для фармакологических применений против рака.
Пример 3. Вклад удаления N-концевого остатка на экспонирование в растворе остатков белка р50.
Хорошо известно, что С-концевая область серрализинов (например, серрализин PRZN_SERMA) отвечает за большую часть цитотоксической активности на опухолевых клетках сразу после того, как отщепляется N-конец от белка посредством аутокатализа, химического расщепления бромистым цианогеном, или путем экспрессии С-конца в Escherichia coli (международная патентная заявка № WO 2006/005268). В связи с этим предполагают, что минимальная функционально активная единица в С-концевой области церрализинов обеспечивала их доступность к опухолевым клеткам сразу после отсоединения от N-конца белка. Таким образом, было решено идентифицировать аминокислотные остатки белка, опосредующие его доступность поверхности в растворе после расщепления N- и С-концов нативного белка, также подтвердить, имеет ли пептид Gly255-Ser274 (N06P87) такие остатки. Чтобы проверить эту гипотезу, были проведены вычисления доступности для поверхностей N- и С-концов основной протеазы Serratia (Hamada K, Hata Y, Katsuya Y, Hiramatsu H, Fujiwara T, Katsube Y. (1996) J. Biochem. 119:844-851).
Были идентифицированы аминокислотные области внутренних частей N- и C-концевых доменов, которые становятся доступными сразу после того, как образуются обе молекулы после протеолитического расщепления протеазы Serratia. Вычисления доступности поверхности для остатков были проведены с использованием компьютерной программы DSSP (Kabsch W, Sander C. 1983. Biopolymers 22:2577-2637). Значения доступности для остатков были выражены в ангстремах (Å2). Как показано на фиг.5, основные различия в значениях доступности соответствовали аминокислотам, расположенным в С-концевой области в N-концевом протеолитическом домене (Gln210, Phe211, Asn226, His229, Leu236, Ile239, Ser251) и в N-концевой области в С-концевом непротеолитическом домене (Thr252, Phe269, Ile280, Trp284, Arg302 и Phe310) основной протеазы Serratia. Остатки, демонстрирующие наиболее значительные изменения значений доступности, представлены в таблице 3.
Аминокислотные (ак.) остатки, демонстрирующие наиболее значительные изменения значений доступности
Вычисления доступности поверхностей N-конца (1srpN) и C-конца (1srpC) основной протеазы Serratia по сравнению с нативным белком (1rp). Остатки упорядочены в соответствии со значением различия в экспонировании (колонка Порядок). Представлены 64 остатка, демонстрирующих наивысшие значения повышенного экспонирования.
Пары остатков Asp98 и Arg267, и Asp225 и Lys317 участвуют в образовании двух дисульфидных мостиков между N- и С-концевыми доменами. Более того, эти остатки демонстрируют среднее значение различия величины доступности в 78,2Å2. Средние значения различия величин доступности для протеолитического N-конца и непротеолитического С-конца основной протеазы Serratia составили 5,1±15,4 Å2 и 7,1±20,1 Å2, соответственно. Другими рассматриваемыми позициями были: Ile22, Asn32, Gln94 и Arg171. На данный момент никакой биологической активности не приписывают этим остаткам.
Напротив, ни остатки активного сайта (His176, Glu177 и His180), расположенные в α-спирали Е в N-концевом протеолитическом домене, ни остатки Gly183 и His186, включенные в цинк-связывающий мотив HEXXHXXGXXH, не демонстрировали каких-либо изменений в их соответствующих значениях доступности. Эти данные согласуются с экспериментальными результатами; эффективно демонстрируя, что цитотоксическая активность белка р50 (который принадлежит семейству серрализинов) не зависит от его протеолитической активности и связана с его непротеолитической С-концевой областью, которая увеличивает его доступность в растворителе сразу после отщепления от протеолитического домена. С другой стороны, сегмент Gly255-Ser274 (пептид N06P87) имеет три остатка (Arg267, Asp268 Phe269) из тех, что демонстрируют наибольшее увеличение экспонирования (таблица 3), в частности, среди наибольших 14 значений, дополнительно подтверждая очевидную роль этого сегмента как структурно функциональную единицу внутри белка р25, ответственную за противоопухолевую активность.
Пример 4. Модификации сегмента Gly255-Ser274 пептида N06P87
Пептидная библиотека, представленная в примере 1, включает сегменты длиной 20 ак., перекрывающиеся по 10 ак., начиная от N-конца полипептида р25. Эта конструкция была создана для идентификации линейного участка из 10 ак., связанного с цитотоксической активностью, предмета скрининга. Однако перекрывающиеся 10 ак., входящие в состав N06P87, не демонстрировали никакой цитотоксической активности (таблица 2) в другом контексте (фиг.6, пептиды E07P06 и F07P16). Это свидетельствует о том, что 20 ак., включающие область Gly255-Ser274 (пептид N06P87), необходимы для формирования правильного экспонирования мотива для взаимодействия с опухолевыми клетками или правильных позиций для вторичных взаимодействий, фланкирующих активный сайт, увеличивая таким способом их аффинность и/или специфичность связывания потенциального рецептора. Рассматривая этот критерий, пептиды были сконструированы и синтезированы с введенными модификациями для оценки их возможного влияния на цитотоксическую активность в отношении опухолевых клеток и для частичной характеристики пептида N06P87 для дальнейших оптимизаций. Такие модификации включали замену амидной группы на карбоксильную группу на С-конце пептида N06P87 и циклизацию пептида N06P87 путем вставки одного цистеина в оба конца пептида. Полученные пептиды и их молекулярные массы представлены в таблице 4.
Модификации в 20 ак. лидерном пептиде N06P87
Неспособность пептида N06P87 (циклизованного двумя цистеинами, расположенными на обоих N- и С-концах) ингибировать клеточную пролиферацию в опухолевых клетках меланомы человека М14, показана на фиг.7. Это предполагает необходимость дальнейших ограничений, налагаемых на другие позиции в последовательности пептида N06P87, поддерживая мотивацию имитации третичной структуры, которую имеет эта область у предполагаемой структуры полипептида р25. На той же фиг.7 также продемонстрирована необходимость блокирования С-конца (например, амидной группой или другой модификацией) для достижения цитотоксической способности, ожидаемой у представляющего интерес участка пептида.
Пример 5. Композиции лидерного пептида не увеличивают его цитотоксическую активность
Исследование было проведено с целью оценки влияния буферных условий, рН и вспомогательных веществ нескольких парентеральных композиций, содержащих лидерный пептид, на его цитотоксическую активность в опухолевых клетках. Среди протестированных буферов были: глициновый, фосфатный, цитратный и другие, включающие широкий диапазон значений рН, от кислых до высоко основных. Также были протестированы несколько вспомогательных веществ в рамках использования для парентеральных композиций, такие как: глицин, сахароза, декстран, глутамат натрия, сорбит, циклодекстрин, полиэтиленгликоль, ЭДТК, неионные детергенты и другие. Пептид N06P87, составляющий смесь с этими вспомогательными веществами, не демонстрировал повышенной цитотоксической активности в опухолевых клетках различного гистологического происхождения.
Пример 5. Активность in vivo линейного пептида N06P87
Опухолевая модель рака толстой кишки Ls174T в бестимусных мышах была использована для оценки потенциального эффекта линейного сегмента Gly255-Ser274 (пептид N06P87) при сравнении с полипептидом CIGB370r на моделях опухолей человека. Опухолевые клетки человека вводили подкожно, и исследуемые молекулы или наполнитель вводили внутрь опухоли (100 мкл). Спустя 13 дней, когда опухоли были имплантированы и пальпируемы, начинали применять исследуемые молекулы по назначенной схеме (фиг.8А). Пептид N06P87 применяли с понижением дозы: два назначения по 600 мкМ каждые 48 ч с последующими 4 назначениями по 330 мкМ каждые 48 ч и последние 4 назначения по 90 мкМ каждые 72 ч. Пептид CIGB370r применяли еженедельно в течение 4 недель.
Спустя тридцать пять дней после начала лечения были выявлены значимые различия в объеме опухоли (р<0,001) среди групп, леченных N06P87 и наполнителем (фиг.8В). Более того, обнаружены высоко значимые различия (р<0,0001) между группами, леченными CIGB370r и получавшей наполнитель, согласно одностороннему ANOVA с апостериорным критерием Бонферрони. Также были выявлены высоко значимые различия между группами, получавшими обе молекулы (р<0,0001).
На фиг.8С представлена выживаемость среди животных, леченных этими молекулами или получавших наполнитель. Обе молекулы обладали способностью значимо повышать выживаемость (логранговый критерий: р<0,05) леченых животных при сравнении с группой, получавшей наполнитель. При этом, в случае пептида N06P87, отношение T/C (где Т является средним значением выживаемости леченых животных и С является таковым животных, получавших только наполнитель) составило 117%. Эти данные указывают на то, что данный линейный пептид не квалифицируют как потенциально пригодную молекулу для лечения злокачественных новообразований у людей, где отношение Т/C должно составлять по меньшей мере 120%. В случае полипептида CIGB370r отношение Т/C составило 142%, что отражает ожидаемые свойства молекулы, потенциально пригодной для лечения злокачественных новообразований человека.
Пептид, даже не отвечающий критериям при вычислении отношения Т/С, соотносили с объемом опухоли на 35 день (последний день, когда все животные были еще живы), демонстрируя отношение T/C, равное 72%, с тем же параметром, будучи 35% для полипептида CIGB370r. В этом случае отношение T/C характеризует в значительной степени активное соединение при диапазоне ниже 40-50% (Marie Suggitt and Michael C. Bibby. 50 Years of Preclinical Anticancer Drug Screening: Empirical to Target-Driven Approaches Clinical Cancer Research. 2005. Vol. 11, 971-981). Это означало то, что идентифицируемой пептидной последовательности требовалась дополнительная оптимизация для пролонгируемой выживаемости, подобной той, которая достигается в случае применения полипептида CIGB370r, и для обеспечения его дальнейшего терапевтического использования.
Пример 7. Определение функциональных остатков/участков в пептиде N06P87
Экспериментальные результаты, представленные в примерах 2 и 4, наряду с трехмерным структурным анализом белка р50 (пример 3 и раздел Подробное описание изобретения) позволяют определить три сегмента или отличающиеся по химической структуре участки пептида N06P87 в соответствии с их влиянием на структурно-функциональные отношения пептида: а) первичный участок связывания (центральная петля); b) вторичный участок связывания на С-конце; и с) вторичная N-концевая область для связывания и/или поддержания структуры. Это схематически отображено на фиг.9.
Центральная петля содержит сегмент Gly266-Asp268, существенно важный для его биологической активности, а также остатки Phe269 и Leu270, которые увеличивают его экспонирование как в р25, так и р50. С-концевая область представляет собой петлю, принимающую растянутую конформацию в белке р50. Остатки в этом сегменте, по-видимому, участвуют во взаимодействии с рецептором, как показано в примере 4, где С-концевая амидная группа пептида N06P87 заменена карбоксильной группой, приводя к потере биологической активности. Такая модификация подразумевает внесение нового локального положительного заряда, и также потерю группы доноров водородного мостика в пептиде. Также возможным является то, что этот участок может играть структурную роль: а) Leu270 устанавливает гидрофобные взаимодействия с Phe269, Tyr259 и алифатической боковой цепью Arg267 в центральной петле; b) Thr272 устанавливает гидрофобные контакты с Phe261 в N-концевой области; и с) Ser271 образует три водородных мостика с основаниями центральной петли, аминогруппа Ser271 является донором водородных мостиков для карбонильной группы Arg267, и группа OG на боковой цепи Ser271 также является донором для карбонила Arg267 и акцептором группы ОН для боковой цепи Thr265. N-концевой сегмент играет структурную роль: водородные мостики основной цепи Asp256 (карбонильной группы) и Val258 (аминогруппы) с атомами ND2 и OD1 боковых цепей остатка Asn264, карбонильной группы (акцептора) остатка Val258 в качестве донора амина (основной цепи) для Asn264 и атома OG Ser263.
Пример 8. Введение или структурные ограничения, циклизация, связи боковых цепей и/или искусственные аминокислоты
Ключевой аспект создания сильнодействующих противораковых средств по настоящему изобретению включает пептиды, конструируемые в циклической форме, то есть, они содержат аминокислоты, соединенные посредством ковалентных связей, включая химические группы боковых цепей и/или аминоконец и карбоксильный конец. Следовательно, пептиды, сконструированные в настоящем документе, структурно ограничены посредством циклизации, которая в значительной степени снижает структурную пластичность этих молекул. Как правило, использование пептидов в качестве терапевтических средств имеет ряд недостатков. Это характерно для свойственной пептидам пластичности; особенно короткого и среднего размера, которые являются более пластичными нежели уложенные белки. Поэтому процесс их связывания с белками или какими-либо другими рецепторными макромолекулами зачастую ограничен более высокой потерей конформационной энтропии. Это обстоятельство обусловливает у этих молекул проявление, как правило, аффинности связывания ниже таковой белок-белкового взаимодействия. Проявляемая этими пептидами более низкая аффинность (и следовательно более низкая эффективность) может быть также связана с тем фактом, что контактирующая поверхность пептид-рецептор меньше таковой, требуемой для контактирующей поверхности белок-рецептор, в частности, когда пептиды включают сегмент нативного белка. По этим причинам, как правило, требуется повторно конструировать и химически модифицировать пептиды для увеличения их аффинности для связывания рецептора (и следовательно, эффективности).
В настоящем изобретении циклизация пептида предпочтительно осуществляется посредством: а) амидных связей между боковыми цепями Lys и Asp/Glu (пептиды 5-16, 19-22, 40-43 в таблице 5) или между боковой цепью Lys и карбоксильной концевой группой (пептид 32); и b) вставкой дисульфидных мостиков (пептиды 1-4, 17-18, 23-31, 33-39, 44-50). В таблице 5 представлены последовательности репрезентативных пептидов.
Создание циклических пептидов, аналогичных линейному пептиду N06P87
Уникальные аминокислоты, подходящие для использования аналогично остаткам Lys и Asp/Glu для циклизации боковых цепей и/или химической модификации пептида посредством ковалентной связи с полимером или боковой цепью
Определенные аминокислоты, подходящие для использования аналогично цистеину для циклизации пептида посредством дисульфидных мостиков и/или для химической модификации пептида посредством связи с полимером боковой цепью
Во всех случаях структурные ограничения, вводимые в структуру конструируемого пептида, должны быть совместимы с биологически активной конформацией молекулы. Таким образом, дизайн циклизаций по настоящему изобретению включает как отбор потенциальных позиций на последовательности для замены/вставки аминокислот, подлежащих связыванию с боковыми цепями (позиции замены), так и тип(ы) аминокислот(ы), подлежащих внесению в последовательность (остатки присоединения). Расстояния между остатками, соответствующими позициям замены в пептиде, должны быть совместимы со стереохимической природой выбранных остатков присоединения. В частном случае введения дисульфидных мостиков, например, в качестве потенциальных позиций замены не рассматриваются те остатки, у которых расстояние между альфа-углеродными атомами выше 7 Å или ниже 3,8 Å в активной конформации (Vardhan S. Dani, C. Ramakrishnan and Raghavan Varadarajan. Protein Engineering vol. 16 no.3 pp. 187-193, 2003). Подобным образом, позиции с расстоянием между бета-углеродными атомами в диапазоне от 3,6 Å до 4,7 Å рассматривают в качестве предпочтительных. Стереохимические характеристики позиций замены, таких как альфа- и бета-углеродные атомы, должны поддерживать торсионные углы на боковых цепях остатков присоединения достаточным образом, чтобы принимать подходящие значения, как только образуется ковалентная связь между остатками, что означает существование подходящих нековалентных взаимодействий (Ван-дер-Ваальс).
Были рассмотрены следующие этапы конструирования пептидов, циклизованных посредством дисульфидных мостиков (цистеинами) по настоящему изобретению: а) выбор потенциальных пар позиций замены пептида N06P87, пригодных для замены цистеинами, связанными дисульфидными мостиками, b) трехмерное структурное моделирование модифицированных пептидов/минимизированной энергии моделей и с) оценка энергии и/или стереохимических качественных параметров моделей. Позиции замены на цистеины в белке N06P87 были выбраны с использованием MODIP (Vardhan S. Dani, C. Ramakrishnan and Raghavan Varadarajan. Protein Engineering vol. 16 no.3 pp. 187-193, 2003), программного обеспечения, разработанного для конструирования дисульфидных мостиков белка. Метод присваивает балл потенциальным дисульфидным мостикам путем использования эмпирической энергии в зависимости от межатомного расстояния для альфа и бета атомов углерода, а также значения для торсионных углов χ1, χ2, χSS, χ1' и χ2'(R. Sowdhamini, N. Srinivasan, B. Shoichet, D.V. Santi, C. Ramakrishnan and P. Balaram (1989). Prot. Engng., 3, 95-103). В зависимости от значений энергии, вычисленных для потенциальных дисульфидных мостиков, MODIP присваивает показатель качества А (идеальная стереохимия), В (надлежащая геометрия, но имеется стереохимическое кручение) или С (достаточно близко, чтобы позволить образование дисульфидных мостиков), где А представляет наивысшее качество и С наименьшее. Трехмерные (3D) структурные модели конструируемых пептидов были получены с использованием программного обеспечения молекулярного моделирования и (предпочтительно) могут быть получены ядерным магнитным резонансом. В настоящем изобретении было использовано программное обеспечение MODELLER для моделирования пептидов (Sali A, Blundell TL, 1993, J Mol Biol 234:779-815) y WHATIF (Vriend G, 1990, J Mol Graph. 8(1):52-6, 29).
Кристаллографическая структура p50 была использована в качестве отправной точки для этого анализа - файл PDB 1SRP. Как обсуждалось ранее в разделе «Подробное описание изобретения», экспериментальные данные свидетельствуют о том, что биологически активная конформация пептида N06P87 аналогична таковой, принимаемой фрагментом Gly255-Ser274 в белке p50/p25. В таблице 8 представлены результаты для прогнозирования потенциальных позиций замещения в этом сегменте.
Прогнозирование позиций замещения на цистеины, связанные дисульфидными мостиками в пептиде N06P87 с помощью MODIP
*, Прогноз был сделан для модели пептида N06P87, модифицированного заменой Thr19→Gly; §, Степень стереохимического качества для прогнозируемого дисульфидного мостика, А, наивысшая, С - более низкого качества.
Пары 2-4 предсказаны, исходя из нативной структуры, и демонстрируют показатели качества А и В, в то время как пара 1 требует небольшого конформационного изменения на остатке T19 и, таким образом, получаемый в итоге дисульфидный мостик имеет более низкое качество. Таким образом, настоящее изобретение включает пептиды, содержащие замены T19→G или T19→А в пользу принятия благоприятной конформации для установления дисульфидного мостика. Замена Gly позволяет увеличить локальную пластичность в пользу конформационных изменений на карбоксильном конце, способствуя образованию мостика. Замена Ala является адекватной, учитывая благоприятное свойство этого остатка принимать геликоидальные конформации, как прогнозируется для остатка 19 в соответствии с моделями, полученными для структуры пептида 4 в таблице 5 (торсионные углы фи-пси модели -69, -38). Введение Ala, по сравнению с заменой T19→G, характеризуется более низкой потерей энтропии конфигурации во время укладки и/или связывания рецептора. На фиг.10 представлены модели для группы циклических пептидов, сконструированных по прогнозам MODIP, показанных в таблице 8.
Те же позиции замены, используемые для конструирования дисульфидных мостиков, также подходят для конструирования циклических пептидов с остатками присоединения, образующими амидные связи, как остатки Lys и Asp/Glu. Поскольку число атомов, связывающих альфа-углеродные атомы остатков присоединения, выше в данном случае, чем таковые, существующие в дисульфидных мостиках, стереохимические ограничения, определяющие их введение в конструкцию пептидов, являются менее строгими, и, следовательно, позиции замены, прогнозируемые MODIP, также являются адекватными в качестве основного подхода. Сразу после выбора пары или остатков для
данной связи, получаемый пептид моделируют и оценивают с энергетической точки зрения (и/или оценивают качество в соответствии со стереохимическими параметрами). Пептид 33 в таблице 5 был сконструирован по аналогичному протоколу, этот пептид содержит амидную связь между Lys7 и концевой карбонильной группой (фиг.11).
Другой предпочтительный в настоящем изобретении способ циклизации включает введение ковалентной связи между амино- и карбонильной группами N- и С-концов пептидов. Стереохимические ограничения пептидной связи вызывают необходимость введения ак. линкеров, что облегчает принятие структуры, совместимой с биологической активностью пептидов. Например, чем больше расстояние между альфа-углеродными атомами «якорных» остатков в трехмерной структуре присоединяемого белка p25/p50 (dCA-CA) по сравнению с расстоянием 3,9 Å, существующим между CA пептидной единицы, тем выше количество требуемых остатков линкера (Ncon). Если адекватные остатки линкера не введены между двумя якорными остатками, чтобы подходить для соответствующих ограничений расстояния, весьма вероятно, что пептид не сможет принять наблюдаемую в экспериментальных условиях конформацию, это, весьма вероятно, оказывает негативное влияние на биологическую активность. Остатки линкера были сконструированы в соответствии со следующим протоколом:
I. Выбор присоединяемых остатков пептида N06P87: соседние остатки в трехмерной структуре, предпочтительно те, что не включены в сегмент первичного связывания или первичный сегмент;
II. Определение требуемого минимального числа остатков линкера: Ncon>dCA-CA/3,9;
III. Выбор якорных остатков: присоединяемые остатки пептида N06P87 и соседние остатки, с 2-3 остатками, выбранными на обоих концах;
IV. Поиск сегментов из N остатков (Ncon+4>N>Ncon+6) в недублируемой базе данных трехмерной структуры белка, таким образом, чтобы структура основной цепи из 2-3 остатков на концах фрагмента была аналогична структуре якорных остатков.
V. Выбор последовательности и структуры сегмента линкера: выбирают структуру наиболее распространенных сегментов линкера, наблюдаемую на IV этапе, и вычисляют параметры выбора для каждой аминокислоты в каждой позиции, то есть соотношение между числом появлений аминокислоты и ожидаемым значением
в соответствии с относительной распространенностью аминокислоты в базе данных. Для каждой позиции выбирают аминокислоты (одна или несколько), демонстрирующие самые высокие показатели предпочтения. Можно модифицировать остатки, граничащие с сегментом линкера, следуя критерию наибольшего значения параметра предпочтения. В случаях, включающих позиции с положительными торсионными углами, возможно ввести D- стереоизомеры. Кроме того, можно ввести другие искусственные аминокислоты (такие как бета- и N-метилированные аминокислоты) для увеличения структурной предрасположенности остатка принимать адекватную конформацию на основной цепи для требуемой структуры.
Поскольку эти пептиды циклизуют амидными связями между N- и С-концами, циклические пермутации последовательности содержат идентичные пептиды. В одном из примеров настоящего изобретения пептиды конструируют на основе первичного сегмента связывания, определенного для пептида N06P87, и указанный сегмент соединен тетрапептидом (два дипептида) общей последовательности Z1Z2X3X4 (или X3X4 у Z1Z2, соответственно, пептид 64 в таблице 5). Эти пептиды содержат до 11 аминокислот. Последовательности линкера по настоящему изобретению могут также включать D-ак. в тех позициях, в которых остатки принимают положительные торсионные углы (пептиды 65 и 66 в таблице 5). Последовательность тетрапептида линкера предпочтительна, но не только, dSer-Pro-Thr-Pro (пептид 65 в таблице 5, dS является D-Ser стереоизомером), хотя он также может быть Gly-Pro-Thr-Pro (пептид 71 в таблице 5) или dAla-Pro-Thr-Pro (пептид 70 в таблице 5, dAla является D-Ala стереоизомером). В другом иллюстративном примере пептиды по настоящему изобретению содержат последовательность, соответствующую сегменту Asn262-Ser271 полипептида p50, соединенных тетрапептидом. Получаемые в результате пептиды имеют 14 остатков. Предпочтительная последовательность тетрапептидного линкера может быть, но не только, Thr-Pro-Gly-Gln (пептид 72 в таблице 5), альтернативно, но не только, Arg-Pro-(dAla)-Gln (пептид 69, см. фиг.12) или Arg-Pro-Gly-Gln (пептид 73 в таблице 5) или Thr-Pro-(dAla)-Gln (пептид 68 в таблице 5). Таким образом, последовательность этих пептидов может быть описана как сегмент Asn264-Ser271, циклизованный посредством линкерного гексапептида, последовательность которого предпочтительно представляет собой: (Thr или Arg)-Pro-(Gly или dAla)-Gln-Asn-Ser. Альтернативно, остатки 4 и 5 в гексапептидном линкере могут быть, соответственно, аминокислотами BmGln и NmAsn, где BmGln и NmAsn являются аминокислотами L-b-метилглутамином (L-глутамин, метилированный по его бета-углеродному атому) и L-n-метиласпарагин (N-метил-L-аспарагин), соответственно. Бета-метилированные аминокислоты более склонны к принятию растянутых конформаций (бета-подобных структур) и N-метилированные склонны к принятию полипролин-подобных структур, которые являются структурами, принимаемыми остатками 4 и 5 в модельных конструкциях пептида, получаемых по описанному протоколу (этапы I-IV) для конструирования сегментов линкера.
В другом иллюстративном примере настоящего изобретения представлены пептиды, последовательность которых соответствует сегменту Asn262-Ser271 p50, соединенные трипептидом (или по аналогии сегменту Asn264-Ser271, соединенные пентапептидом). Трипептидный линкер предпочтительно имеет, но не исключая прочее, последовательность (Arg или Lys)-(Arg или Lys)-Pro (пептиды 76-77 в таблице 5, фиг.13), соответствующий пентапептидный линкер, имеющий последовательность (Arg или Lys)- (Arg или Lys)-Pro-Asn-Ser.
Пример 9. Стереоизомеры
С целью повышения структурной стабильности, и, следовательно, аффинности/эффективности пептидов, модификация, рассмотренная в настоящем изобретении, включает замену природных L-аминокислот в исходной последовательности N06P87 их соответствующими стереоизомерами (D-аминокислотами, D-ак.). Поскольку D-ак. принимают подходящие положительные торсионные углы, подлежащими замене остатками-кандидатами N06P87 являются те, что принимают такие торсионные углы в структуре нативного белка (и/или структурных моделях пептидов). На фиг.14 представлена диаграмма Рамачандрана, соответствующая трехмерной структуре сегмента Gly255-Ser274 р50. С помощью значений определения областей диаграммы Рамачандрана, полученных Rooman и Wodak (Rooman MJ, Kocher JP, Wodak SJ. 1991. J Mol Biol. 221(3):961-79), остатки N8 и G12 находятся в области L (геликоидальная область с левым вращением) и G6 в области Е растянутой конформации (значения фи и пси G6 являются также недопустимыми для остальных L-аминокислот). Таким образом, замена N8, G12 и G6 на D-Ala выгодна, учитывая предрасположенность этого остатка принимать такие конформации основной цепи и/или уменьшать связанную с укладкой/связыванием потерю конфигурационной энтропии. Остатки L-Glu, L-Gln и L-Asp весьма склонны принимать геликоидальные конформации (правое вращение), когда эти остатки находятся в петлях белка (Swindells МВ, MacArthur MW, Thornton JM. 1995. Nat Struct Biol. 2(7):596-603), по аналогии, D-Glu, D-Gln и D-Asp склонны принимать геликоидальные конформации с левым вращением (как в случае с остатком N8). Пептиды 17, 23, 25, 27-31, 45-51 (таблица 5) имеют от одной до трех D-ак., сконструированных в соответствии с ранее упомянутыми критериями. Введение D-ак. в структуру пептидов по настоящему изобретению также оказывает благоприятное влияние на конформацию и, следовательно, на аффинность, положительным образом влияя на фармакокинетические свойства пептидов путем увеличения их устойчивости к протеолизу in vivo по двум основным причинам: а) прямое воздействие, поскольку эндопротеазы сыворотки расщепляют стерео-специфичные субстраты; и б) меньшая пластичность пептидов, что обусловливает их меньшую восприимчивость к протеолитическому расщеплению.
Пример 10. Блокирование концов пептида и/или конъюгация с полимерами
Пептиды характеризуются более низким значением периода полураспада in vivo (меньший размер, почечное выведение), наибольшей пластичностью, также обусловливая повышенную восприимчивость к протеолизу и, следовательно, более низкую биодоступность. Следовательно, предлагается введение химических модификаций в пептиды для улучшения их биодоступности. Настоящее изобретение включает конструирование ковалентно модифицированных пептидов с полиэтиленгликолевой(ыми) цепью(ями) (PEG), предпочтительно путем модификации концов пептида, но не без исключения других модификаций, как, например, боковых цепей. Примеры пегилированных пептидов по настоящему изобретению представлены в таблице 5 (пептиды 62 и 63). Пегилированные пептиды по настоящему изобретению, таким образом, демонстрируют лучший профиль устойчивости к протеолизу, сниженную почечную фильтрацию, более низкую вероятность взаимодействия с антителами и последующей нейтрализации их активности и сниженную антигенность и иммуногенность.
Пегилирование увеличивает время циркуляции небольших молекул, было показано, что небольшие пептиды, которым свойственна быстрая экскреция, зачастую обладают почечной токсичностью, когда они радиоактивно меченные (Blumenthal R.D., Sharkey R.M., Goldenberg D.M. Goldenberg D.M. eds. Cancer Therapy with Radiolabeled Antibodies, 295-314, CRC Press Boca Raton, FL 1995). Пегилирование было использовано для модификации синтетических лекарственных препаратов, таких как интерфероны и антитела. Настоящее изобретение включает презентацию пептидов посредством конструирования мультимерных структур, используя линейные или разветвленные PEG, которые обеспечивают более высокую авидность связывания пептида с рецептором.
Пегилирование увеличивает наблюдаемый молекулярный размер пептидов, снижая скорость почечной фильтрации (Knauf M.J., Bell D. P., Hirtzer P., Luo Z. P., Young J. D., Katre N. V. J. Biol. Chem., 263: 15064-15070, 1988; Behr T. M., Goldenberg D. M., Becker W. Eur. J. Nucl. Med., 25: 201-212, 1988). Пептиды по настоящему изобретению модифицируют предпочтительно PEG, увеличивая их молекулярный размер до значений, равных или выше 50 кДа, чтобы резко снизить клубочковую фильтрацию данной молекулы.
Пегилирование, в дополнение к увеличению времени циркуляции, уменьшает антигенность и восприимчивость к протеолизу терапевтических молекул (Delgado С., Francis G. E., Fischer D. Crit. Rev. Ther. Drug Carrier Syst., 9: 249-304, 1992.) и приводит к увеличению их растворимости и сосудистой проницаемости (Francis G.E., Delgado С., Fischer D., Malik F., Agrawal A. K. J. Drug Target., 3: 321-340, 1996), высоко требуемое свойство для антинеопластических препаратов.
Существует ряд экзопептидаз в крови, почках и печени (Werle M, Bernkop-Schnrch A. Amino Acids. 2006 Jun; 30(4):351-67) и, следовательно, модификация N- и С-концов пептида может значительно увеличить его протеолитическую стабильность. N- ацетилирование является модификацией, рекомендуемой в настоящем изобретении, а также введение пироглутамата на N-конец, или D- стереоизомера аминокислоты и т.д. Пептиды по настоящему изобретению в основном С-амидированы (амид на С-конце), что обеспечивает им устойчивость к карбоксипептидазе.
Модификация N- и/или С-концов может заключаться в пегилировании, что увеличивает устойчивость к протеазам и, в частности, к экзопептидазам путем модификации концов, помимо имеющих к этому отношение и ранее упомянутых свойств, связанных с увеличенным размером и сниженной антигенностью/иммуногенностью. Кроме того, добавление PEG вызывает увеличение устойчивости к протеолизу в целом (эндо и экзо) путем создания стерического препятствия для протеолитических ферментов. Для этой цели PEG используют либо линейными, либо разветвленными. Циклизация также защищает пептиды от протеолиза экзопептидазами, образуя ковалентную связь между N- и С-концами, как описано ранее (пептиды 64-66, 68-69 в таблице 5), или путем ковалентного связывания одного конца с боковой цепью (пептиды 32-33 в таблице 5). Устойчивость к экзопептидазам также достигается путем замены концевых аминокислот D-ак. (пептид 67 в таблице 5).
В качестве альтернативы модификации с PEG, также возможна конъюгация с N-ацетилнейраминовой кислотой (сиаловой кислотой), являющейся встречающимся в природе полимером и высоко гидрофильной, биоразрушаемой, без рецепторов в организме человека, и которая может увеличить устойчивость к протеазам (стабилизации плазмы) и период полураспада (Gregoriadis G, Fernandes A, Mital M, McCormack В (2000) Cell Mol Life Sci 57: 1964-1969; Fernandes Al, Gregoriadis G (1997) Biochim Biophys Acta 1341: 26-34). Другая приемлемая модификация пептидов по настоящему изобретению заключается в N-концевой замене жирными кислотами или липидизацией (пептиды 52-353 в таблице 5). Эта стратегия увеличивает устойчивость пептидов к протеолизу экзопептидазами и облегчает взаимодействие пептидов с мембранами. В зависимости от природы липидов это может способствовать взаимодействию с определенными мембранными доменами, облегчая накопление пептидов в доменах, изобилующих на рецепторе - мишени фармакологического действия пептидов. Липидизация также способствует образованию супрамолекулярных структур, обладающих лучшими фармакокинетическими и фармакодинамическими свойствами (мицеллы, агрегаты, частицы, пузырьки).
Пример 11. Описание химической структуры пептидов по настоящему изобретению
На фиг.15 показано общее представление о химической структуре пептидов по настоящему изобретению. Конструкция основана, во-первых, на первичном сегменте, определенном для осуществления на практике в примере 7. В общем, пептидная последовательность Asn264-Leu270 представлена в различных структурных контекстах, включающих различные степени ограничения, посредством циклизации, в которую вовлечены основная цепь и/или боковые цепи пептидов. Циклизации осуществляют посредством связей различной химической природы: амидной или дисульфидной связей, но позиции замены, занимаемые остатками циклических боковых цепей и самими боковыми цепями, тщательно подбирают для воспроизведения структуры и топографии функционального сайта белка p50/p25 белка, идентифицированного в настоящем изобретении. Частное решение заключается в циклизации основной цепи, осуществляемой выбором якорных сайтов и последовательностей линкера, которые обладают высокой структурной предрасположенностью принимать конформацию, совместимую со структурой первичного сегмента. Вторичные сегменты также используются дополнительно, в основном для поддержания позиций замены циклическими боковыми цепями. Кроме того, вторичные сегменты могут обеспечивать дополнительную контактную поверхность для взаимодействия с потенциальными рецепторами, в соответствии с топографией функционального сайта р50/25. Пример включает введение остатков, несущих положительный заряд, и/или доноров водородных мостиков в положения Х+3 и Х+4 С-конца вторичного сегмента. Пептиды могут включать один или несколько циклов, показанных на фиг.15, хотя только моноциклы были включены в качестве примеров, представленные в таблице 5. Пептиды по настоящему изобретению могут включать искусственные или специальные ак., боковые цепи которых включают амино, карбоксильную или сульфгидрильную группы, направленные на замещение позиций замены для циклизации или любой другой химической модификации. Это также может включать стереоизомеры D-ак. в позициях полипептидной цепи, способных принимать положительные торсионные углы, либо для первичного, либо для вторичного сегмента. Кроме того, пептиды по настоящему изобретению могут быть модифицированы ковалентной связью с полимерами, такими как PEG и/или другими химическими группами. Такие модификации могут быть сделаны на концах и/или боковых цепях позиций замены, если они не заняты циклизованными боковыми цепями.
Пептиды 1, 2, 3 и 4 в таблице 5 (кодируемые в процессе их синтеза и очистки, как: A08P25s-s, A08P28s-s, J08P46s-s и J08P48s-s, соответственно) являются осуществлением на практике примеров, касающихся существенных аспектов пептидов по изобретению, включая: различные размеры, наличие или отсутствие вторичных сегментов и основные позиции замены. Эти пептиды были выбраны для демонстрации их эффектов в моделях опухоли in vivo: структурная имитация криптического фрагмента пептида, обладающего цитотоксической активностью в полипептиде p25, генерирует молекулы пептида длиной до 20 ак., которые эффективно воспроизводят противоопухолевую активность p25, преимущественно, для использования в лечении злокачественных новообразований.
Пример 12. Оценка противоопухолевой активности оптимизированных циклических пептидов в сингенных и ксенотрансплантатных моделях опухолей
С целью оценки противоопухолевой активности нового семейства циклических пептидов по настоящему изобретению, эти пептиды, имитирующие структуру активной области полипептида p25, были выбраны 4 пептидных модели, включающие различные варианты циклизации и вставки этого семейства. Они представляли собой пептиды 1, 2, 3 и 4 в таблице 5 (кодируемые для анализа активности как A08P25s-s, A08P28s-s, J08P46s-s и J08P48s-s, соответственно). В качестве группы положительного контроля использовали группу, обработанную полипептидом CIGB370r.
6-8-недельные самки мышей C57BL/6 весом 22 г были предоставлены Национальным центром по выращиванию животных для лабораторных целей (CENPALAB, Havana, Cuba) и содержались в стерильных условиях в виварии Центра Генной Инженерии и Биотехнологии (CIGB). Эксперименты были выполнены в соответствии с правилами надлежащего обращения с лабораторными животными и ухода за ними. Объем опухоли был измерен в каждом случае (вычислен по формуле: объем = a2x(b/2), где a представляет собой ширину и b представляет собой длину опухоли), и была оценена выживаемость животных.
Выживаемость сравнивали между группами с помощью логрангового теста. Статистические параметры были получены с помощью программного обеспечения GraphPad Prism 4.0 (GraphPad Software, San Diego, CA, USA).
Мышам C57BL/6 инъецировали в правый бок подкожно (п.к.) 50000 клеток ТС-1. Когда опухоли становились детектируемыми, мыши были рандомизированы по лечебным группам для оценки лечения исследуемыми пептидами, чтобы дать им оценку при различных способах введения. Имеющие опухоль животные получали 6 назначений в разные дни, применяя 80 мкМ исследуемого пептида, в качестве контроля применяли наполнитель. Пять мышей использовали для каждой группы. Животных содержали в стерильных условиях, и процедуры были проведены в соответствии с рекомендуемыми нормами содержания и ухода за животными в лабораторных условиях.
Выживание было статистически значимым во всех леченых группах по сравнению с группой, получавшей наполнитель. На фиг.16 представлены три блока данных: 1) блок данных с самыми высокими значениями выживаемости, включая группы, леченные пептидом J08P48s-s и CIGB370r, соответственно, без статистически значимых различий между ними; 2) блок данных с промежуточными значениями выживаемости, включая группы, леченные пептидами A08P28s-s, A08P25s-s и J08P46s-s, соответственно, без статистически значимых различий между ними, и отличный от 1 блока; и 3) группа отрицательного контроля. Эти результаты демонстрируют, что новое семейство пептидов, описанных в настоящем документе, представленные пептидами, которым была дана оценка в этой модели, способны достигать активность, описанную для исходного полипептида.
Помимо внутриопухолевого способа введения, в данной модели ТС-1 были изучены другие способы применения пептидов, такой как внутрибрюшинный способ введения. Фиг.17 демонстрирует, что циклические пептиды (например, пептид J08P48s-s), воспроизводят эффекты, наблюдаемые в этой модели для полипептида CIGB370r, когда их применяли обоими способами. Это свидетельствует об эффективности циклических пептидов по настоящему изобретению для лечения дистальных солидных опухолей.
Эффекты линейного сегмента Gly255-Ser274 (пептид N06P87) и два других смоделированных циклических пептида по настоящему изобретению сравнивали с эффектом полипептида CIGB370r в модели рака толстой кишки человека Ls174T у бестимусных мышей NIH, для которых на фиг.19 представлены дозы и схемы применения. Через 13 дней, после того как опухоли были имплантированы и стали пальпируемыми, начинали вводить биомолекулы (фиг.19А). Пептиды вводили в уменьшенной дозе: 2 применения по 600 мкМ каждые 48 ч с последующими 4 применениями по 330 мкМ каждые 48 ч, и 4 завершающих применения по 90 мкМ каждые 72 ч. CIGB370r вводили еженедельно в течение 4 недель.
В этом случае, и в отличие от линейного пептида, циклические пептиды, показанные на фиг.19, обладали способностью значительно увеличивать выживаемость леченых животных (логранговый критерий: р<0,05) по сравнению с группой, получавшей носитель, и линейным пептидом N06P87. Значения T/C, относящиеся к среднему значению объема опухоли (фиг.19В), составили: 35%, 35%, 37% и 72% для пептидов A08P25s-s, J08P48s-s, CIGB370r и N06P87, соответственно. Эти результаты демонстрируют, что циклические пептиды по настоящему изобретению, имитирующие трехмерную структуру противоопухолевого активного сайта p25, пригодны для разработки антинеопластической терапии. Пептиды по настоящему изобретению демонстрируют ряд фармакокинетических и фармакодинамических преимуществ по сравнению с их нативными белками.
Пример 13. Широкий спектр действия циклических пептидов по настоящему изобретению на клеточные линии человеческих опухолей различного гистологического происхождения
Пептиды по настоящему изобретению были исследованы на различных клеточных линиях опухолей человека различного гистологического происхождения, и используя метод сульфородамина В (Skehan P, Storeng R, Scudiero D, et al., (1990) "New colorimetric cytotoxicity assay for anticancer-drug screening". J. Natl. Cancer Inst. 82: 1107-1112).
Фиг.18 демонстрирует, в качестве примера, пептид широкого спектра действия на исследуемые клеточные линии, показывая дозозависимый эффект. Это свидетельствует о том, что эти новые молекулы пригодны для лечения злокачественных опухолей различного гистологического происхождения, таких как: карцинома гортани (НЕр-2), мелкоклеточный рак легкого (H82), немелкоклеточный рак легкого (H125), рак шейки матки (HeLa, Caski) и глиомы (U87), среди прочих.
Пример 14. Ингибирование образования тубулярной структуры в матригеле
В настоящее время международное научное сообщество предоставило обоснованные доказательства того, что комбинирование является ключевым фактором в лечении злокачественных новообразований, особенно путем комбинирования прямого воздействия на опухолевые клетки (ингибирование их пролиферации или инициирование их гибели) вместе с действием на окружение опухоли для ингибирования ангиогенеза. По этой причине антиангиогенную активность пептидов по настоящему изобретению оценивали методом оценки образования эндотелиальных клеточных тяжей человеческими эндотелиальными клетками сосудистого происхождения (HMEC) на матригеле (Crum R, Szabo S, Folkman J. (1985). Science. 230:1375-8). Фиг.20 демонстрирует способность циклических пептидов по настоящему изобретению ингибировать образование тубулярных структур в зависимости от используемой концентрации, и показывая их антиангиогенную активность.
Пример 15. Пептидные структуры со свойствами самосборки, основанные на циклических пептидах, полученных из Serratia marcescens
Пептиды по настоящему изобретению обладают амфипатическими свойствами, обусловленными сегрегацией гидрофобных и гидрофильных участков на поверхности пептидов. Гидрофобный участок формируется в основном боковыми цепями остатков Tyr259, Phe269 и Leu270, которые напоминают их структуры на трехмерной структуре белка p50. Остальная часть поверхности образована, главным образом, полярными или заряженными остатками. Это амфипатическое свойство позволяет пептидам образовывать нанометровые и/или микрометровые супрамолекулярные конгломераты, как показано на микрофотографии фиг.21. Агрегация пептидов может быть дополнительно опосредована образованием межмолекулярных водородных мостиков, включая сегменты основной цепи и боковых цепей. Растянутые структуры N- и С-концов вторичных сегментов пептидов могут содействовать типу агрегации при определенных условиях формирования для новых приложений в лечении злокачественных новообразований, таких как: комбинированная терапия и в области нанобиотехнологии, и систем контролируемого высвобождения (Monica С Branco, Joel P. Schneider. Acta Biomaterialia 5 (2009) 817-831).
Пример 16. Противоопухолевые и антиангиогенные циклические пептиды в сочетании с продигиозином, демонстрирующие синергический цитотоксический эффект на опухолевые клетки
Продигиозины, выделенные из Serratia marcescens CMIB4202 (Abrahantes-Pérez MC, Reyes-González J, Véliz Ríos G, et al., (2006) Cytotoxic proteins combined with prodigiosin obtained from Serratia marcescens have both broad and selective cytotoxic activity on tumor cells. J Chemother. 18: 172-81), комбинировали с циклическими пептидами по настоящему изобретению приемлемыми композициями, либо посредством ковалентных связей, либо путем прямого химического синтеза, либо в результате химических реакций между отдельными молекулами. Циклические пептиды, такие как A08P25s-s, A08P28s-s, J08P46s-s и J08P48s-s, усиливали цитотоксическую активность продигиозина на различных опухолевых клеточных линиях человека (как A549, A375, PC-3, U87 и т.д.), оцениваемую методом SRB (Skehan P, Storeng R, Scudiero D, et al., (1990) "New colorimetric cytotoxicity assay for anticancer-drug screening". J. Natl. Cancer Inst. 82: 1107-1112), показывая значения GI50 в нанометровом диапазоне, и снижая цитотоксическую активность в результате действия продигиозина на первичные фибробласты, по меньшей мере на один порядок. Это дает основание предположить, что механизмы действия обеих молекул отличаются друг от друга и, следовательно, они могут быть комбинированы, обеспечивая эффективные результаты в лечении злокачественных новообразований.
Пример 17. Получение фармацевтической композиции противоопухолевых и антиангиогенных циклических пептидов, инкапсулированных с полимерами семейства сополимеров полимолочной и гликолевой кислоты (PLGA)
Для того чтобы изменить фармакокинетику и биораспределение пептидов по настоящему изобретению в соответствии с онкологическими терапевтическими критериями, требуемыми для каждого конкретного случая, была исследована инкапсуляция этих циклических пептидов вместе с полимерами в микросферах PLGA. С этой целью был приготовлен раствор, содержащий сополимер молочной кислоты и гликолевой кислоты 50:50 в концентрации 10% (вес./об.) путем растворения 1 г полимера в дихлорметане. Один миллилитр полимерного раствора смешивали с 200 мкл водного раствора, содержащего по меньшей мере один из пептидов по настоящему изобретению, таких как A08P25s-s, A08P28s-s, J08P46s-s и J08P48s-s пептиды, в диапазоне концентраций 20-40 мг/мл. Эту смесь обрабатывали ультразвуком в течение 30 секунд с использованием ультразвукового наконечника. Полученную эмульсию выливали в 40 мл 1% поливинилового спирта, и вторая эмульсия была получена (в/м/в) перемешиванием двух фаз при 14000 в гомогенизаторе Ultraturrax T8. Двойную эмульсию выливали в 140 мл 0,1% поливинилового спирта 30000-70000 и перемешивали в гомогенизаторе со скоростью 300 об/мин в течение 1 часа для испарения дихлорметана. Наконец, микросферы были собраны фильтрованием, промыты пять раз 50 мл дистиллированной воды и заморожены/высушены в лиофилизаторе. Сухие микросферы хранили при 4°С до начала использования.
Микросферы, содержащие циклические пептиды, такие как A08P25s-s, A08P28s-s, J08P46s-s, J08P48s-s, со вспомогательными веществами, были получены, как описано, но с добавлением Pluronic F-127 (10 мг) и NaCl (0,5 мг) в центральную часть водной фазы. Оба типа микросфер вводили подкожно вблизи опухоли у бестимусных мышей, имеющих имплантированные опухоли меланомы человека A375. Разовую дозу вводили, когда опухоли достигали объема выше 200 мм3, достигая результатов, аналогичных тем, что были получены после многократных введений (трижды в неделю в течение 4 недель) для отношений T/C ниже 10%
для объема опухоли и выше 170% для выживания.
Пример 18. Получение обычных липосом, содержащих противоопухолевые и антиангиогенные циклические пептиды
Фосфатидилхолин в концентрации 10 мг/мл был растворен в абсолютном этаноле в круглодонной колбе, объемом 50 мл. Липид был высушен с помощью роторного испарителя при комнатной температуре до образования сухого слоя на стенках накопителя. С целью инкапсуляции в липосомы по меньшей мере одного из циклических пептидов, описанных в настоящем изобретении, таких как A08P25s-s, A08P28s-s, J08P46s-s и J08P48s-s, сухой липидный слой был гидратирован гомогенизацией буферным раствором, содержащим по меньшей мере один из ранее упомянутых пептидов. Чтобы уменьшить размер липосом, препарат, содержащий липосомы, заполненные указанными пептиды, подвергали последовательным этапам экструзии через поликарбонатную мембрану с размером пор в среднем 100 нм в диаметре до тех пор, пока липосомы не были размером приблизительно 100 нм.
Свободные пептиды были отделены от пептид-содержащих липосом центрифугированием суспензии при 100000×g в течение 40 мин при 4°C. Супернатант был собран в чистую пробирку, и осадок ресуспендировали фосфатным буферным солевым раствором, рН 7,2. После второго этапа центрифугирования в тех же условиях полученный супернатант собирали в чистую пробирку и смешивали с супернатантом первого центрифугирования. Осадок (липосомы, содержащие циклические пептиды по настоящему изобретению) ресуспендировали в фосфатном буферном солевом растворе, рН 7,2. Этот конечный препарат хранили при 4°С до начала использования. Липосомы, содержащие по меньшей мере один из пептидов по настоящему изобретению, вводили в виде однократной дозы мышам, имеющим опухоль ТС-1, воспроизводя результаты, полученные после многократных введений неинкапсулированных пептидов (см. пример 12).
Пример 19. Противоопухолевые и антиангиогенные циклические пептиды со способностью связывать ионы металлов и направлять лекарственный препарат в специфический орган
Аминокислотный состав пептидов по настоящему изобретению обеспечивает способность связывать ионы металлов, как радиометаллы и парамагнитные металлы, для их использования в качестве фармацевтических препаратов, не влияя на их биологические свойства.
Комплекс пептид-иона металла может быть получен различными физико-химическими методами в участках, указанных в предыдущих примерах. Среди радиометаллов были найдены изотопы Tc, Re, Au, Ag, Pd, As, Cu, Hg и Ru. Продукт реакции между ионом металла и пептидом представляет собой комплекс между двумя молекулами, которые продемонстрировали направленное специфическое действие на опухоли и метастазы различного гистологического происхождения на животных моделях (мышах C57BL/6), имеющих опухоль ТС-1. Эти комплексы могут быть использованы для диагностики и лечения злокачественных новообразований очень специфическим образом, сводя к минимуму их захват физиологически нормальными тканями и органами.
В случае комплексов пептидов по настоящему изобретению с радиоизотопами для диагностики и лечения злокачественных новообразований, радиоактивными изотопами могут быть, например: 99Tc и 131I. Металл-связанные пептиды, как описано в данном изобретении, могут быть использованы для непосредственного введения, либо конъюгированы с другими молекулами-носителями.
Чтобы направить пептиды по настоящему изобретению непосредственно в опухоль для достижения их накопления внутри нее, по сравнению с нормальными тканями и органами, был синтезирован комплекс, образованный пептидами, такими как A08P25s-s, A08P28s-s, J08P46s-s и J08P48s-s, ковалентно связанные с полиэтиленгликолем в соответствии с примером 8. Они дополнительно образовывали смесь с наночастицами оксида железа, тем самым создавая магнитный векторный комплекс наночастиц, основными компонентами которого были: противоопухолевые циклические пептиды по настоящему изобретению, полимерная молекула и наночастица оксида железа. Комплекс вводили внутривенно (в хвостовую вену мыши) мышам C57BL/6, имеющим подкожную опухоль ТС-1 (расположенную в правом боку животных), когда объем опухоли был выше 200 мм3. Оказывали местное воздействие внешним магнитным полем в области опухоли, и комплекс транспортировался током крови и концентрировался в опухоли (Lübbe AS, Bergemann C, Alexiou С. Targeting tumors with magnetic drugs. In: Pagé M, editor. Cancer drug discovery and development: tumor targeting in cancer therapy. Totowa NJ: Humana Press Inc; 2003. pp. 379-88). Схема назначений и доза, аналогичные таковым, представленным в примере 12, были использованы как контрольная группа, и группа отрицательного контроля получала только наполнитель, лишенный противоопухолевых пептидов. Было показано, что противоопухолевый эффект в группе, леченной магнитным вектором наночастиц, несущим противоопухолевые пептиды, проявлялся в уменьшении объема опухоли раньше положительных контролей (р<0,05), и выживаемость была значительно выше, чем в остальных группах (р<0,05). Эти результаты продемонстрировали потенциал данной технологии для достижения терапевтического эффекта, описанного для циклических пептидов по настоящему изобретению, основанной на способности концентрировать более высокие количества терапевтических молекул на опухоли.
Пример 20. Синергетический эффект объединения сочетания противоопухолевых и антиангиогенных циклических пептидов со стандартными цитостатиками
Была проведена оценка синергизма противоопухолевого действия пептидов по настоящему изобретению (таких как A08P25s-s, A08P28s-s, J08P46s-s, J08P48s-s), когда по отдельности комбинировали с группой стандартных цитостатиков, при следующих экспериментальных условиях. Клетки А549 (клетки немелкоклеточного рака легких) культивировали в 96-луночных планшетах в присутствии одного из пептидов, упомянутых ранее в диапазоне концентраций 200-12,5 мкМ. Одновременно по меньшей мере один из цитостатиков, выбранных для этого исследования, был добавлен (Цисплатин, Паклитаксел, 5-фторурацил, Винкристин, Доксорубицин, Циклофосфамид, Митомицин С, Иматиниб, Велкейд, Иресса) в концентрациях в диапазоне 1-2000 нМ, и инкубация длилась в течение 72. В это время жизнеспособность клеток была оценена методом МТТ. Наконец, оптическую плотность измеряли при 570 нм для всех случаев, и были построены соответствующие кривые зависимости доза-эффект. Значения доз, снижающие на 50% клеточную пролиферацию (IC50) для каждого цитостатика, были снижены на один или два порядка величины, когда их одновременно комбинировали по меньшей мере с одним из пептидов по изобретению. Результаты этих анализов показывают усиление антинеопластического эффекта фармацевтической комбинации, содержащей циклические пептиды по настоящему изобретению вместе с цитостатическими соединениями, упомянутыми в данном изобретении.
Пример 21. Усиление противоопухолевого эффекта фармацевтической комбинации в животной модели рака
Для этой цели 5×106 клеток А549 инокулировали подкожно в области спины 6-8-недельным бестимусным мышам (мышам Balb/C). Через 10 дней, когда опухоли были определены (приблизительно 30 мм3), применяли фармацевтическую комбинацию по изобретению. Внутрибрюшинным способом применяли компоненты комбинации, включающей по меньшей мере один из циклических пептидов по настоящему изобретению (такие как: A08P25s-s, A08P28s-s, J08P46s-s и J08P48s-s), составленной в виде смеси с соответствующим наполнителем и применяемой по тем же схемам и в той же дозе, как показано в примере 12. Цитостатики, как Цисплатин, Циклофосфамид и Митомицин С, были одновременно введены внутрибрюшинным способом ежедневно в количестве 1 мг/кг массы тела с той же частотой лечения. Цитостатики были растворены в том же наполнителе, что и пептиды. Наконец, объемы опухолевых масс были измерены и нанесены на график в виде зависимости от времени для оценки антинеопластического эффекта in vivo. Результаты показали, что фармацевтическая комбинация по настоящему изобретению вызывает усиление противоопухолевого эффекта, способствуя полной регрессии опухолевой массы в случае, когда оба ингредиента применяют одновременно. Было выявлено значительное ингибирование роста опухоли, а также наблюдалось значительное увеличение выживаемости животных, по сравнению с группой плацебо. Все эти данные демонстрируют, что синергическое действие между компонентами фармацевтической комбинации по изобретению также эффективны in vivo согласно результатам, полученным в соответствующей и прогнозируемой доклинической модели злокачественных новообразований.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИТЕЛА ПРОТИВ РЕЦЕПТОРА CCR7 ДЛЯ ЛЕЧЕНИЯ РАКА | 2006 |
|
RU2404808C2 |
| СРЕДСТВО, ИНДУЦИРУЮЩЕЕ ИММУНИТЕТ | 2012 |
|
RU2614386C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ | 2011 |
|
RU2623038C2 |
| АГЕНТ, ИНДУЦИРУЮЩИЙ ИММУНИТЕТ | 2012 |
|
RU2634862C2 |
| ГУМАНИЗИРОВАННЫЕ АНТИТЕЛА ПРОТИВ CD79b И ИММУНОКОНЪЮГАТЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2008 |
|
RU2557319C2 |
| ЦИКЛИЧЕСКИЙ С-КОНЦЕВОЙ ПЕПТИД АЦЕТИЛХОЛИНЭСТЕРАЗЫ В ЛЕЧЕНИИ ИЛИ ПРЕДУПРЕЖДЕНИИ РАКА ИЛИ МЕТАСТАЗИРОВАНИЯ | 2015 |
|
RU2711161C2 |
| ЛЕЧЕНИЕ РАКА С ПОМОЩЬЮ АНТИТЕЛА К СЕМАФОРИНУ 4D В КОМБИНАЦИИ С ЭПИГЕНЕТИЧЕСКИМ МОДУЛИРУЮЩИМ АГЕНТОМ | 2018 |
|
RU2783535C2 |
| АНТИТЕЛА ПРОТИВ TENB2, СКОНСТРУИРОВАННЫЕ С ЦИСТЕИНОМ, И КОНЪЮГАТЫ АНТИТЕЛО - ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2008 |
|
RU2505544C2 |
| ПОЛИПЕПТИДЫ | 2011 |
|
RU2581800C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ МОДУЛЯЦИИ АКТИВАЦИИ ФАКТОРА РОСТА ГЕПАТОЦИТОВ | 2005 |
|
RU2405041C2 |





















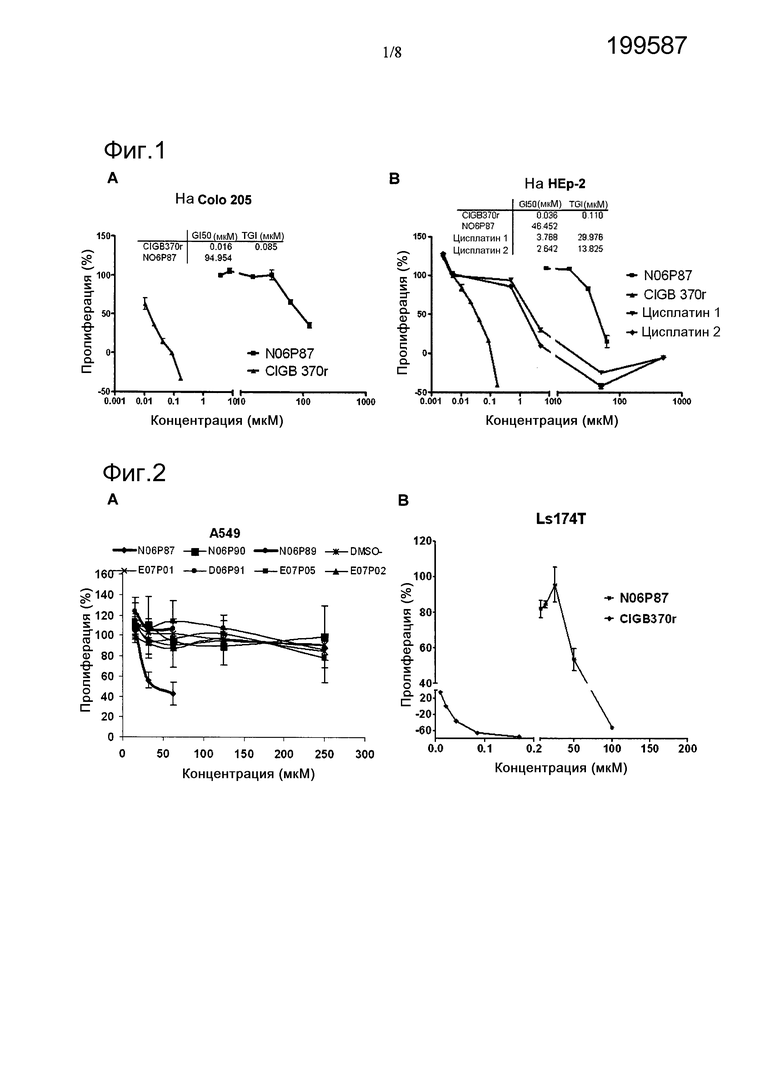
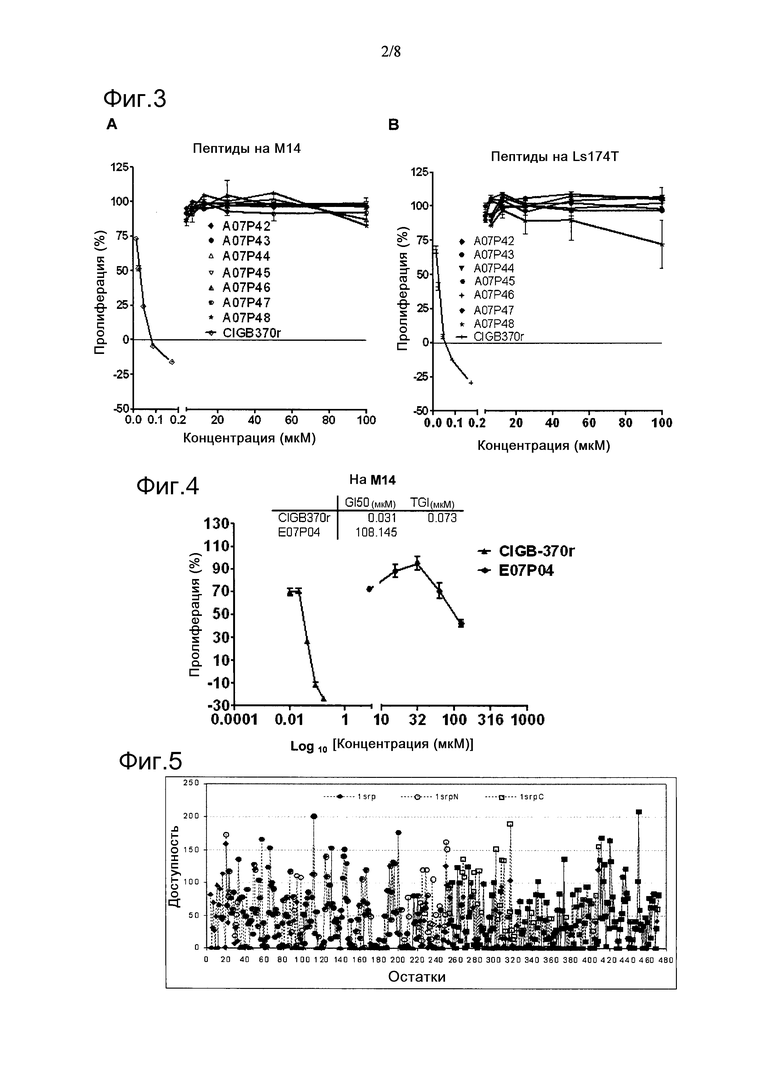
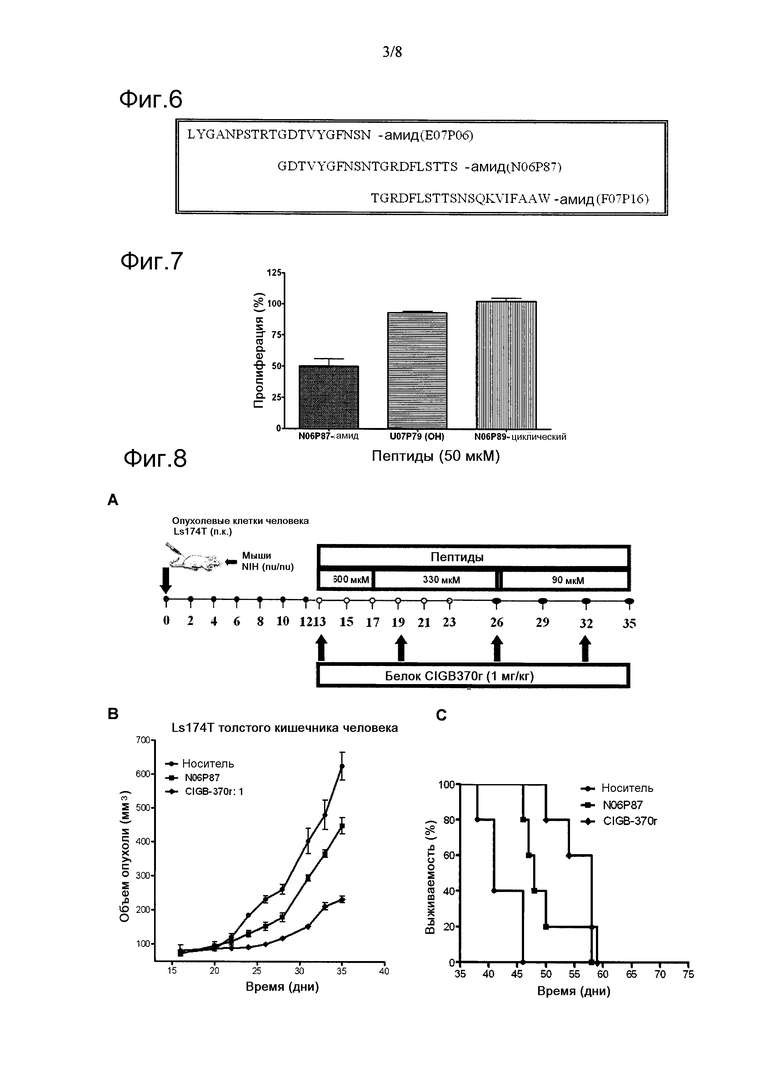
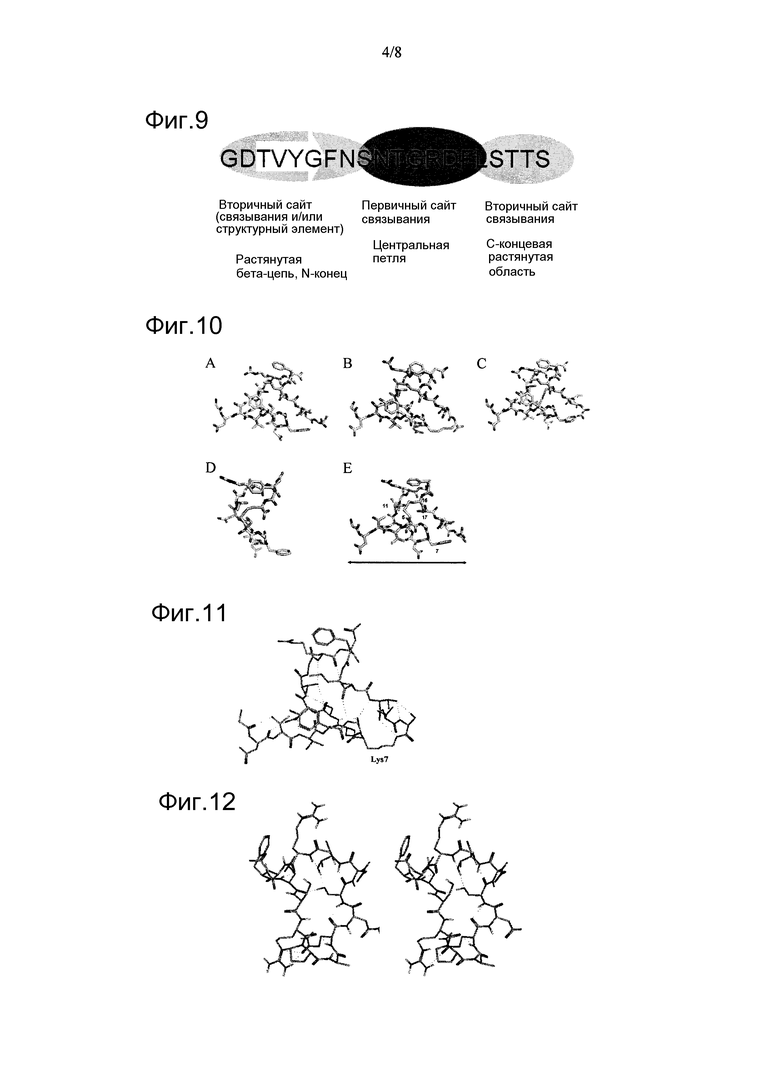
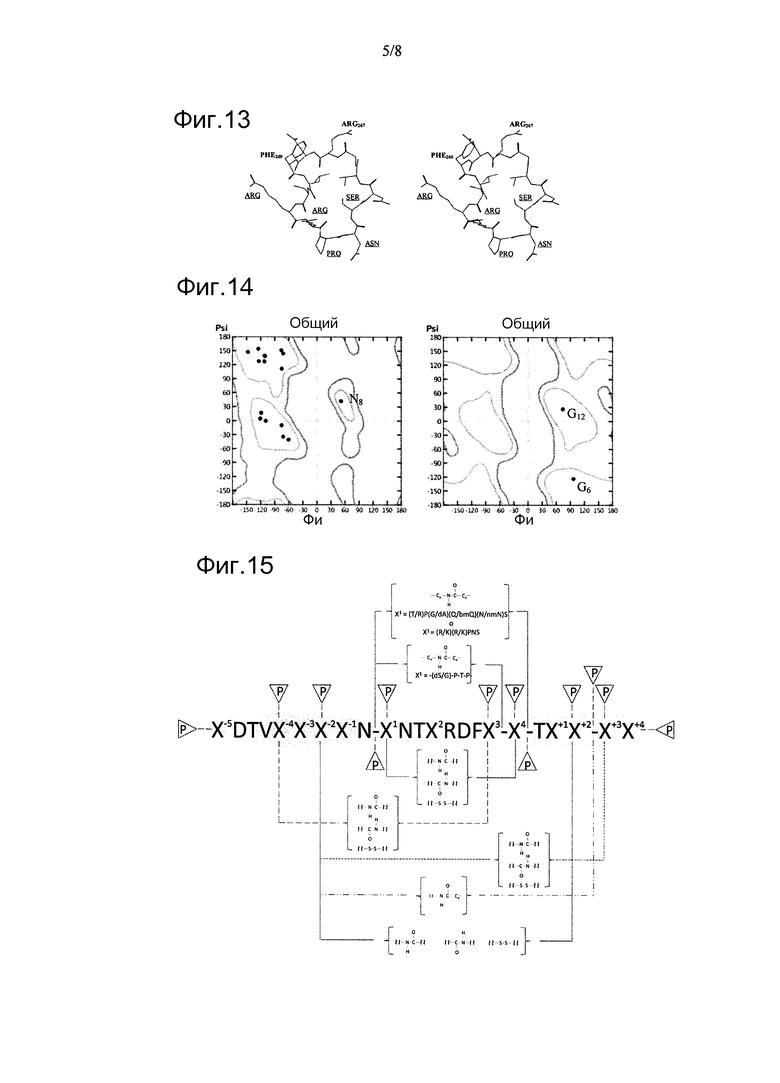
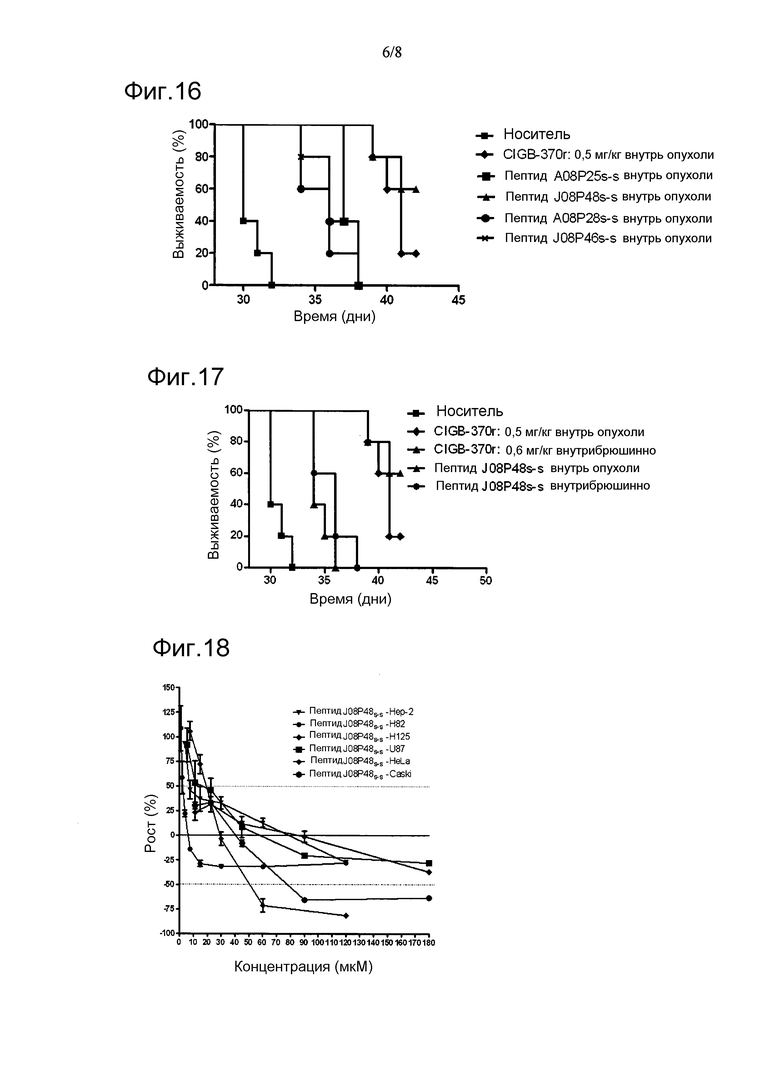
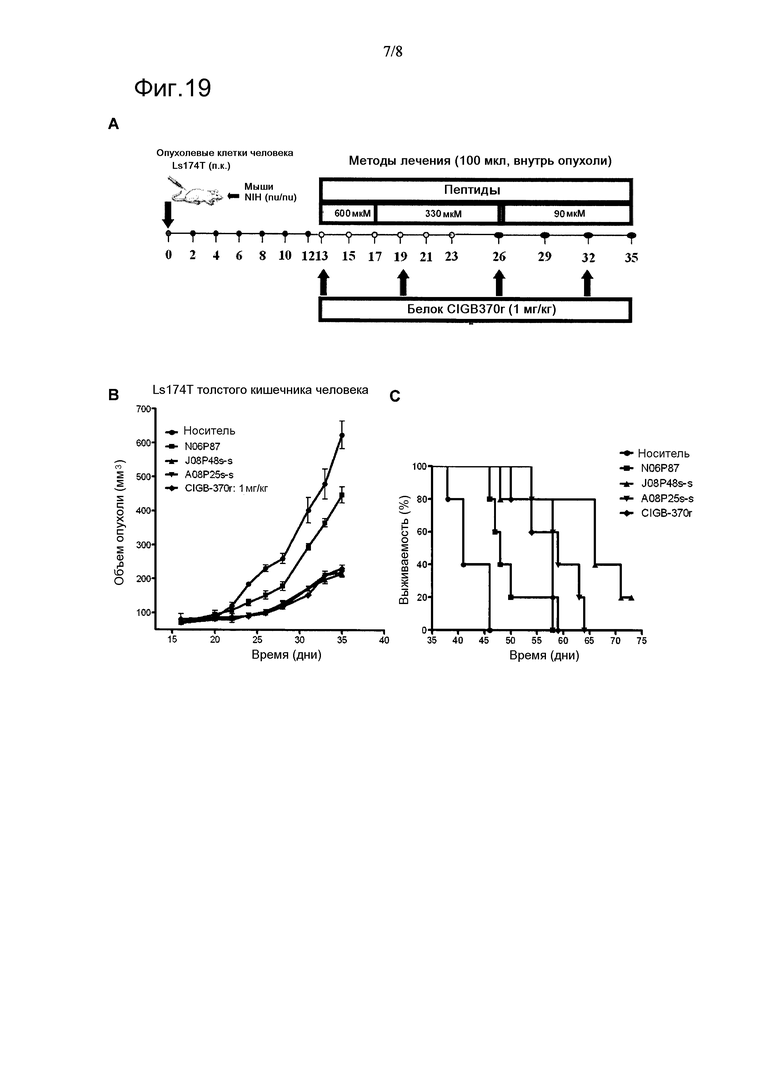
Настоящее изобретение относится к биохимии, в частности к циклическому пептиду, обладающему антинеопластической и антиангиогенной активностями, имеющему одну из SEQ ID NO:1-76. Настоящее изобретение раскрывает фармацевтическую композицию, фармацевтическую комбинацию и применение указанного циклического пептида для лечения злокачественных новообразований или нарушений, связанных с нежелательной клеточной пролиферацией или нежелательным ангиогенезом. Фармацевтическая комбинация указанного циклического пептида в концентрации 12,5-200 мкМ с противораковым лекарственным средством в концентрации 1-2000 нМ обладает повышенной антинеопластической активностью. Настоящее изобретение раскрывает средство для диагностики злокачественных новообразований, содержащее указанный циклический пептид и радиофармацевтическое средство, например флуоресцентную частицу. Настоящее изобретение позволяет расширить арсенал средств для борьбы со злокачественными новобразованиями. 6 н. и 8 з.п. ф-лы, 21 ил., 8 табл., 21 пр.
1. Циклический пептид, обладающий антинеопластической и антиангиогенной активностями, характеризующийся аминокислотной последовательностью, выбранной из группы, состоящей из последовательностей, обозначенных в списке последовательностей как последовательности SEQ ID 1-76.
2. Применение циклического пептида по п. 1 для приготовления лекарственного препарата для лечения злокачественных новообразований или для лечения нарушений, связанных с нежелательной клеточной пролиферацией или нежелательным ангиогенезом.
3. Применение по п. 2, где по меньшей мере один из указанных пептидов вводят в или подготавливают для введения в эффективном количестве нуждающемуся в этом индивидууму.
4. Применение по п. 3, где указанное введение или подготовка дополнительно содержит вещество, выбранное из группы, содержащей нефлуоресцентную группу, флуоресцентную полупроводниковую частицу, парамагнитное или суперпарамагнитное вещество и радиоизотоп.
5. Фармацевтическая композиция для лечения злокачественного новообразования, содержащая эффективное количество пептида по п. 1 и вспомогательные вещества или фармацевтически приемлемые носители.
6. Фармацевтическая композиция по п. 5, отличающаяся тем, что пептид является частью систем контролируемого высвобождения и систем, относящихся к области нанобиотехнологии.
7. Фармацевтическая композиция по п. 6, где указанные системы контролируемого высвобождения могут быть образующимися липосомами, микросферами или структурами самосборки в сочетании с пептидом по п. 1.
8. Фармацевтическая композиция по п. 5, отличающаяся тем, что дополнительно включает вещества, выбранные из группы, состоящей из полупроводниковой частицы, парамагнитного или суперпарамагнитного вещества и радиоизотопа.
9. Соединение для диагностики злокачественных новообразований, отличающееся содержанием пептида по п. 1 и радиофармацевтического средства, где указанное радиофармацевтическое средство выбрано из группы, состоящей из флуоресцентной группы, нефлуоресцентной группы, полупроводниковой флуоресцентной частицы, парамагнитного или суперпарамагнитного вещества и радиоизотопа.
10. Фармацевтическая комбинация, обладающая повышенной антинеопластической активностью, содержащая 12,5-200 мкМ пептида по п. 1 в сочетании с по меньшей мере одним лекарственным веществом, выбранным из группы, состоящей из противораковых лекарственных средств, выбранных из следующей группы: Цисплатин, Паклитаксел, 5-фторурацил, Винкристин, Доксорубицин, Циклофосфамид, Митомицин С, Иматиниб, Велкейд, Иресса в концентрации 1-2000 нМ.
11. Комбинация по п. 10, отличающаяся тем, что указанный пептид напрямую конъюгирован с указанным лекарственным веществом ковалентными связями.
12. Комбинация по п. 10, отличающаяся тем, что указанный пептид конъюгирован с указанным лекарственным веществом соединительным элементом.
13. Фармацевтическая комбинация по п. 12, отличающаяся тем, что соединительное вещество выбрано из группы, содержащей аминокислотный остаток и гидрокарбил и замещенный гидрокарбил.
14. Фармацевтическая комбинация для лечения злокачественных новообразований, содержащая эффективное количество пептида по п. 1 и продигиозинов или их производных.
| WO 2006005268, 19.01.2006 | |||
| US 20100015058 A1, 21.01.2010 | |||
| ABRAHANTES PÉREZ M.C., et al., B05 CIGB370, a novel polypeptide derived from Serratia marcescens with potent antitumor and antimetastasic activity in lung cancer, Annals of Oncology, 2009, 20 (Supplement 3), iii30-iii31; | |||
| ТЮРИН Ю.А., и др., Природная устойчивость бактерий к |

