Настоящее изобретение относится к пиперазиниловым соединениям, особенно полезным при лечении рака, к содержащим их композициям и способу их получения.
С увеличением продолжительности жизни одной из основных причин смертности становятся онкологические заболевания, они влияют на все большее число людей, и их по-прежнему трудно лечить.
Серьезную проблему, значительно затрудняющую лечение различных типов рака, представляет собой развитие устойчивости к противораковым агентам. Снижение толерантности к агенту часто сопровождается перекрестной устойчивостью к разнообразных другим лекарствам. Такая множественная устойчивость к противораковым агентам называется множественной лекарственной устойчивостью (Multidrug Resistance, MDR) и возникает по различным механизмам, только небольшая часть которых была охарактеризована. Эти механизмы включают усиление выведения лекарства, повышение способности клеток к детоксикации, изменение структуры молекулярным мишеней, на которые действуют эти противораковые вещества, модификация системы репарации ДНК и путей апоптоза (Baguley, Mol. Biotechnol., 2010, 46, 308-316; Gatti et al., Methods Mol. Med. 2005, 111, 127-148; Longley et al., J. Pathol. 2005, 205, 275-292; Kohno et al., Eur. J. Cancer 2005, 41, 2577-2586).
Разработка противораковых веществ, способных избежать резистентности, развивающейся по этим механизмам, серьезно затруднена, и, по данным проводимых клинических исследований, до настоящего времени в этой области получено мало результатов.
Противораковые агенты, предназначенные, в частности, для лечения рака, устойчивого к химиотерапии, описаны в WO 2009/150248. Эти вещества соответствуют следующей общей формуле
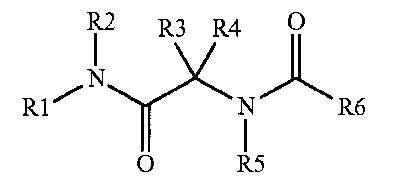
где R1 и R2, совместно с атомом азота, к которому они прикреплены, могут образовывать гетероцикл, такой как возможно замещенная пиперазинильная группа, причем в примерах представлено единственное соединение, возможно замещенное по атому азота пиперазина.
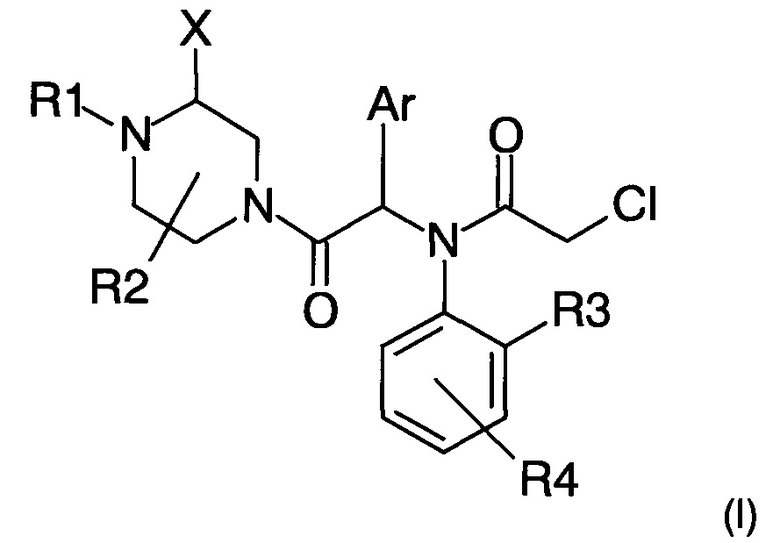
Авторы настоящего изобретения с удивлением обнаружили, что включение заместителя X в альфа-положение относительно второго атома азота пиперазина (см. формулу (I) ниже) позволяет улучшить физико-химические свойства соединений, в частности их растворимость, фармакокинетические свойства и биологическую активность.
Предмет настоящей патентной заявки, таким образом, более конкретно представляет собой замещенное пиперазинильное соединение следующей общей формулы (I)
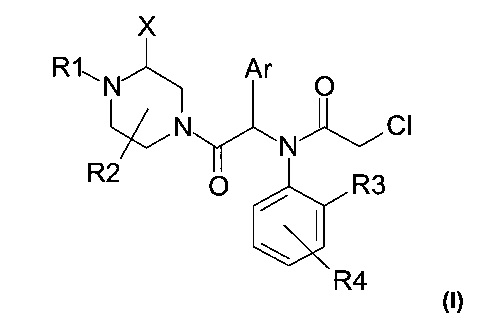
и его фармацевтически приемлемые соли, стереоизомеры или смеси стереоизомеров в любом соотношении, в частности энантиомерная смесь и в особенности рацемическая смесь, где:
- X представляет собой группу (C1-C6) алкил, фенил, бензил, C(O)OR5 или C(O)NHR5;
- R1 представляет собой атом водорода или группу С(O)Н, C(O)R6 или C(O)OR6;
- R2 представляет собой атом водорода или группу (С1-С6)алкил;
или R2 совместно с R1 или X формируют насыщенную углеводородную цепь, которая образует 5- или 6-членный цикл, в частности 5-членный цикл;
- R3 представляет собой атом водорода или галогена или группу (С1-С6)алкил или (С1-С6)акокси;
- R4 представляет собой атом водорода или галогена, CN, NO2 или группу (С1-С6)алкил, (С1-С6)алкокси, арилокси, бензилокси или гетероарилокси, причем указанная группа возможно замещена одним или более атомами галогена;
- Ar представляет собой тиофенильную или фенильную группу возможно замещенную одним или более атомами галогена; и
- R5 и R6 независимо друг от друга представляют собой группу (C1-С6)алкил, арил-(С1-С6)алкил или арил, причем указанная группа возможно замещена одним или более атомами галогена.
Термин "галоген" в контексте настоящего изобретения означает атом фтора, брома, хлора или иода. Преимущественно, это атом фтора, брома или хлора.
Термин "алкильная" группа в контексте настоящего изобретения означает любую насыщенную линейную или разветвленную углеводородную группу, преимущественно содержащую от 1 до 6, предпочтительно от 1 до 4 атомов углерода. К числу таких групп, в частности, относятся группы метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, н-пентил, неопентил или н-гексил. Преимущественно это группы метил, этил, изопропил, трет-бутил или изобутил.
В некоторых случаях алкильная группа может быть замещена одним или несколькими атомами галогена, в частности брома, хлора или фтора, преимущественно фтора. В частности такой группой является группа -CF3.
Термин "алкокси" группа в контексте настоящего изобретения означает алкильную группу, такую как описана выше, связанную с остальной частью молекулы через атом кислорода. Примерами алкокси-группы являются метокси, этокси, изопропокси или трет-бутокси группы. Преимущественно, это метокси или трет-бутокси группы, еще более преимущественно, это метокси группа.
Иногда, алкокси-группа может быть замещена одним или больше атомами фтора. В таком случае, преимущественно, это группа -OCHF2 или -OCF3, в частности -OCF3.
Термин "арильная" группа в контексте настоящего изобретения означает ароматическую группу, предпочтительно содержащую от 5 до 10 атомов углерода, а также один или более сопряженных циклов. Предпочтительно, это фенильная группа.
Термин "гетероарильная" группа в контексте настоящего изобретения означает любую арильную группу, такую как описана выше, в которой один или больше атомов углерода замещены одним или несколькими гетероатомами, предпочтительно от 1 до 4, более предпочтительно от 1 до 2, таких как, например, атомы серы, азота или кислорода. Преимущественно, это группы фурил, теофенил, пиридинил, пиримидил, хинолинил, 1,2,3-тиадиазолил, бензоимидазолил, индазолил или 1,2,3-бензотриазолил.
Термин "арилокси" в контексте настоящего изобретения означает арильную группу, такую как описана выше, связанную с остальной частью молекулы через атом кислорода. Преимущественно, это фенилокси группа.
Термин "гетероарилокси" группа в контексте настоящего изобретения означает гетероарильную группу, такую как описана выше, связанную с остальной частью молекулы через атом кислорода. Преимущественно, это пиридинилокси группа.
Термин "арил-(С1-С6)алкильная" группа в контексте настоящего изобретения означает арильную группу, такую как описана выше, связанную с остальной частью молекулы через алкильную группу, такую как описана выше, содержащую от 1 до 6 атомов углерода. Преимущественно, это группа бензил или 1-фенэтил, более предпочтительно бензил.
Термин "фармацевтически приемлемый" в контексте настоящего изобретения означает полезный для получения фармацевтической композиции, которая в целом безопасна, нетоксична, не является нежелательной в биологическом или ином смысле и приемлема для использования в ветеринарии и для лечения людей.
Термин "фармацевтически приемлемые соли" соединения в контексте настоящего изобретения означает соли, фармацевтически приемлемые в соответствии с данным здесь определением и проявляющие желательную фармакологическую активность родительского соединения. Такие соли включают:
(1) Гидраты и сольваты;
(2) Соли присоединения кислоты, образованные с такими неорганическими кислотами, как соляная, бромоводородная, серная, азотная, фосфорная кислота и подобные им; или образованные с такими органическими кислотами, как уксусная, бензолсульфоновая, бензойная, камфоросульфоновая, лимонная, этан-сульфоновая, фумаровая, глюкогептоновая, глюконовая, глутаминовая, гликолевая, гидроксинафтоевая, 2-гидроксиэтансульфоновая, молочная, малеиновая, яблочная, манделовая, метансульфоновая, муконовая, 2-нафталинсульфоновая, пропионовая, салициловая, янтарная, дибензоил-L-тартаровая, тартаровая, п-толуолсульфоновая, триметилуксусная, трифторуксусная кислота и подобные им, предпочтительно соляная кислота; а также
(3) Соли, получаемые таким образом, что кислый протон родительского соединения либо замещается ионом металла, например ионом щелочного металла (например Na+, K+ или Li+), ионом щелочно-земельного металла (таким как Са2+ или Mg2+) или ионом алюминия; либо координируется с органическим или неорганическим основанием. Приемлемые органические основания включают диэтаноламин, этаноламин, N-метилглюкамин, триэтаноламин, трометамин и подобные им. Приемлемые неорганические основания включают гидроксид алюминия, кальция, калия, натрия и карбонат натрия.
Термин "стереоизомеры" в контексте настоящего изобретения означает диастереоизомеры или энантиомеры. Таким образом, это оптические изомеры. Стереоизомеры, которые не являются зеркальными отображениями друг друга, называются диастереоизомерами, а стереоизомеры, которые представляют собой несовмещаемые между собой зеркальные отображения, называются энантиомерами.
Атом углерода, связанный с четырьмя различными заместителями, называется хиральным центром.
Эквимолярная смесь двух энантиомеров называется рацемической смесью.
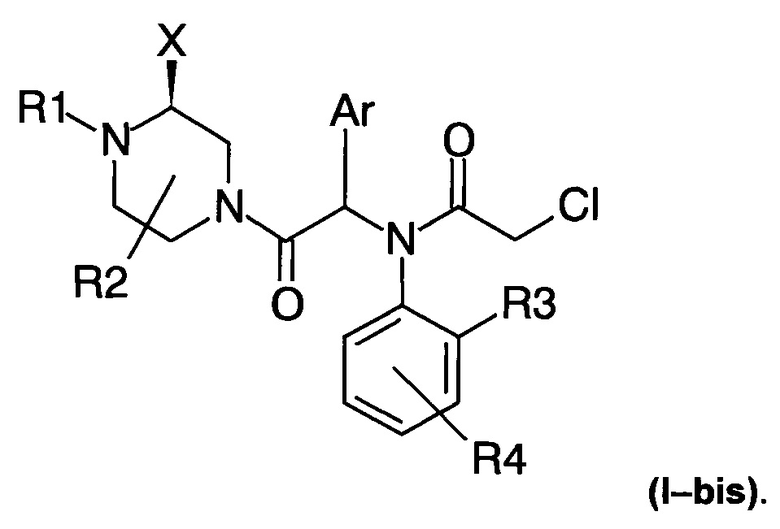
Соединения по настоящему изобретению могут, в частности, соответствовать следующей формуле (I-bis)
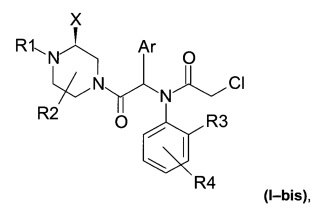
где атом азота, к которому прикреплена группа X, находится в (S) конфигурации.
Предпочтительно X представляет собой (С1-С6)алкил, в частности (C1-С4)алкил, фенил или бензил.
Предпочтительно R1 представляет собой атом водорода или группу C(O)R6 или C(O)OR6, в частности атом водорода.
Предпочтительно R2 представляет собой атом водорода или группу (C1-С6)алкил, например метил.
Предпочтительно R3 представляет собой атом водорода или группу (C1-С6)алкил, например метил.
Предпочтительно R4 представляет собой атом водорода или галогена, или группу (С1-С6)алкил, (С1-С6)акокси или арилокси, причем указанная группа возможно замещена одним или более атомами галогена, в особенности, фтора.
Предпочтительно Ar представляет собой тиофенильную или фенильную группу, замещенную одним или более атомами фтора, такую как 4-фторфенил.
В соответствии с одним конкретным воплощением изобретения, X представляет собой группу (С1-С6)алкил, фенил, бензил, C(O)OR5, C(O)NHR5; R1 представляет собой атом водорода; R2 представляет собой атом водорода или группу (С1-С6)алкил, предпочтительно (С1-С4)алкил или совместно с R1 или X образует насыщенную углеводородную цепь, которая образует 5-членный цикл; R3 представляет собой атом водорода или галогена, или группу (С1-С6)алкил, в частности (С1-С3)алкил или (С1-С6)алкокси, например метокси; R4 представляет собой атом галогена, группу CN, NO2 или (С1-С6)алкил, (С1-С6)алкокси, арилокси, бензилокси или гетероарилокси, причем указанная группа возможно замещена одним или более атомами галогена; Ar представляет собой тиофенильную или фенильную группу, возможно замещенную галогеном; a R5 и R6 независимо друг от друга представляют собой группу (С1-С6)алкил, арил-(C1-С6)алкил или арил, причем указанная группа возможно замещена одним или более атомами галогена.
Более предпочтительно X представляет собой группу (С1-С6)алкил, фенил, бензил, C(O)OR5, C(O)NHR5; R1 представляет собой атом водорода; R2 представляет собой атом водорода или группу (С1-С6)алкил, предпочтительно (C1-С4)алкил; R3 представляет собой атом водорода или галогена, или группу (C1-С6)алкил, в частности (С1-С3)алкил, или (С1-С6)алкокси, например метокси; R4 представляет собой атом галогена или группу (С1-С6)алкил, (C1-C6)алкокси, арилокси, бензилокси или гетероарилокси, причем указанная группа возможно замещена одним или более атомами галогена; Ar представляет собой тиофенильную или фенильную группу, возможно замещенную галогеном; a R5 и R6 независимо друг от друга представляют собой группу (С1-С6)алкил, арил-(C1-С6)алкил или арил, причем указанная группа возможно замещена одним или более атомами галогена.
Еще более предпочтительно X представляет собой группу (С1-С6)алкил, фенил или бензил; R1 и R2 представляют собой атом водорода; R3 представляет собой атом водорода или галогена или группу (С1-С6)алкил, в частности (C1-С3)алкил; R4 представляет собой атом галогена или группу (С1-С6)алкил, (C1-С6)алкокси, арилокси или бензилокси, причем указанная группа возможно замещена одним или более атомами галогена; Ar представляет собой тиофенильную или фенильную группу, возможно замещенную галогеном; a R5 и R6 независимо друг от друга представляют собой группу (С1-С6)алкил, арил-(С1-С6)алкил или арил, причем указанная группа возможно замещена одним или более атомами галогена.
Предпочтительно X представляет собой группу (С1-С6)алкил, фенил или бензил; R1 и R2 представляют собой атом водорода; R3 представляет собой атом водорода или группу (С1-С6)алкил, в частности (С1-С3)алкил; R4 представляет собой атом галогена или группу (С1-С6)алкил, (С1-С6)алкокси, арилокси или бензилокси, причем указанная группа возможно замещена одним или более атомами галогена; Ar представляет собой тиофенильную или фенильную группу, возможно замещенную атомом фтора, такую как 4-фторфенил; a R5 и R6 независимо друг от друга представляют собой группу (С1-С6)алкил, арил-(C1-С6)алкил или арил, причем указанная группа возможно замещена одним или более атомами фтора.
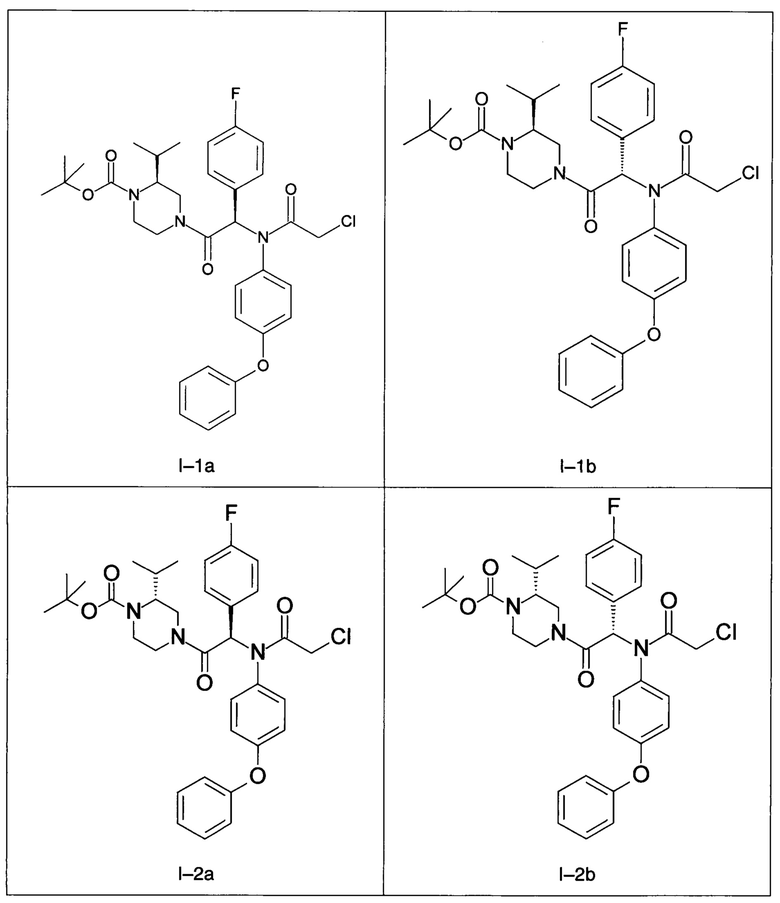
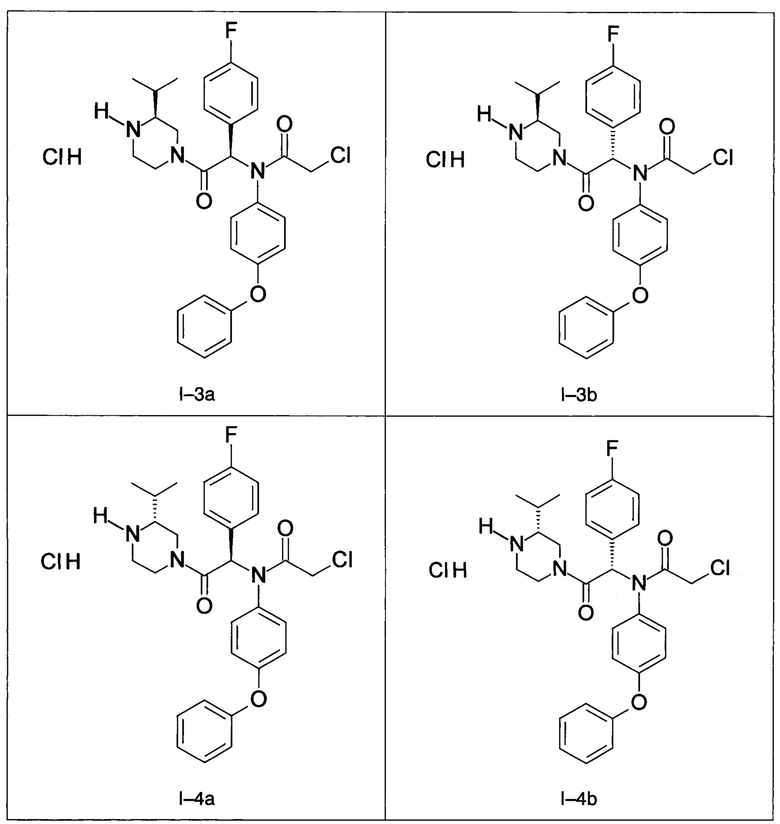
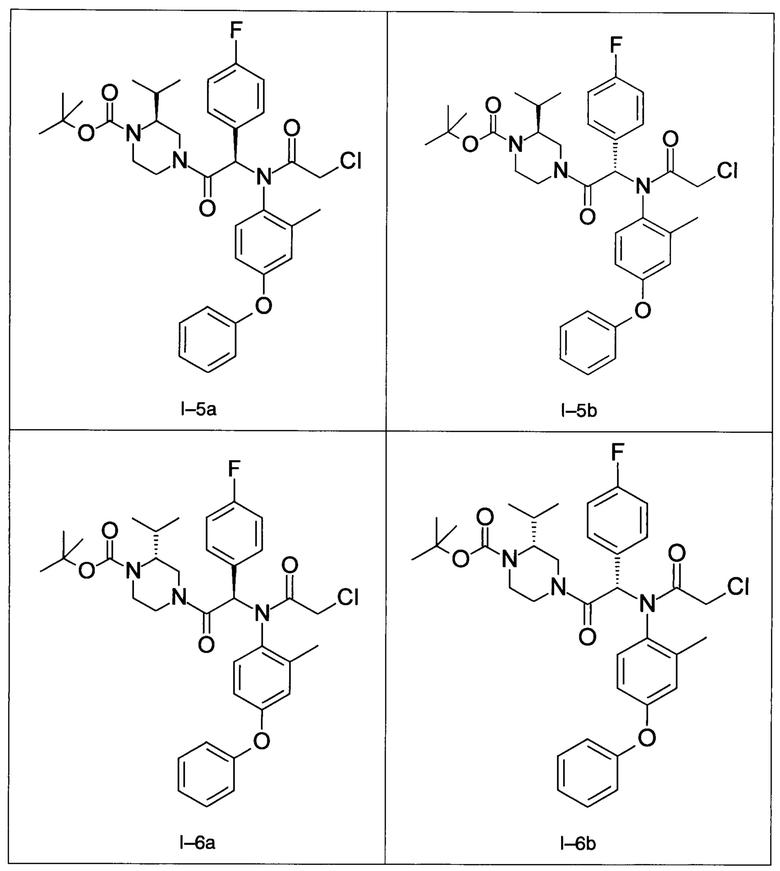
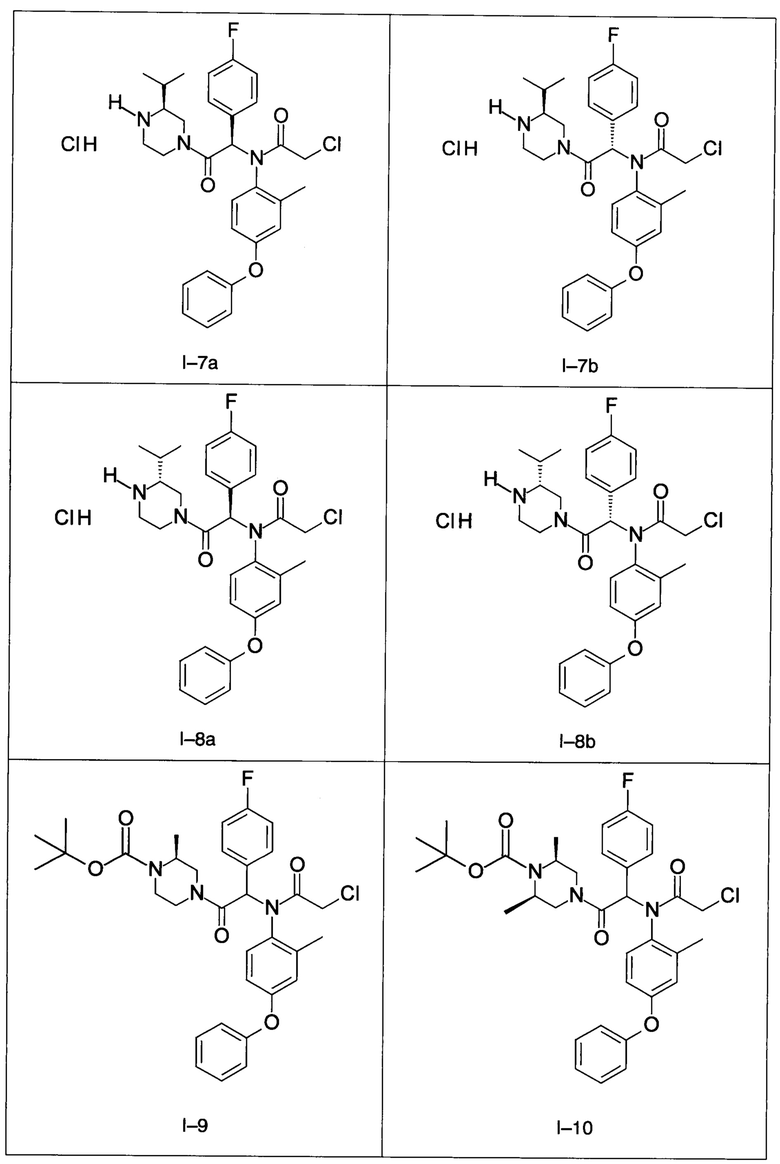
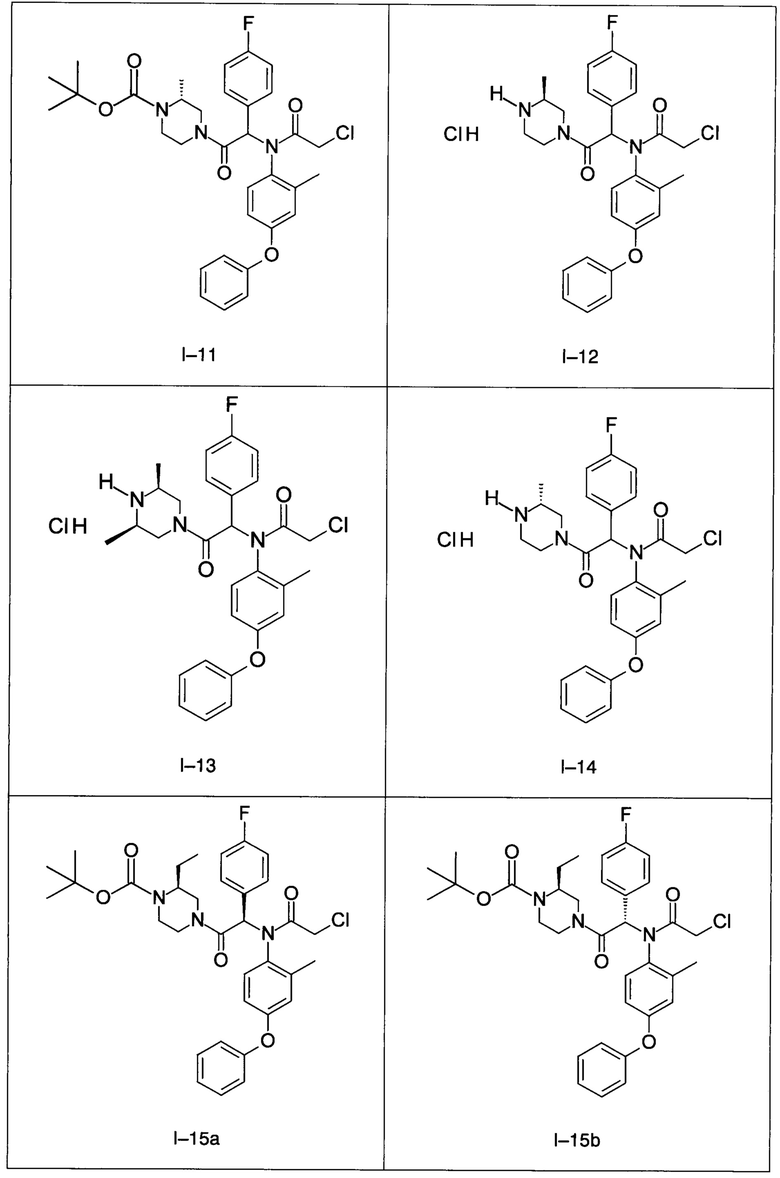
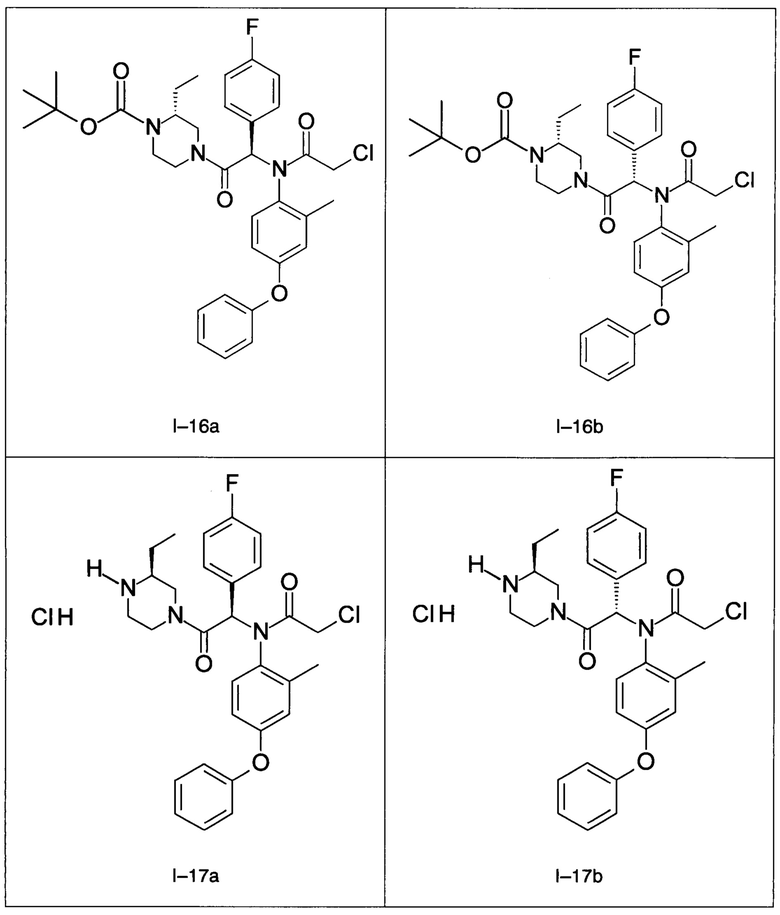
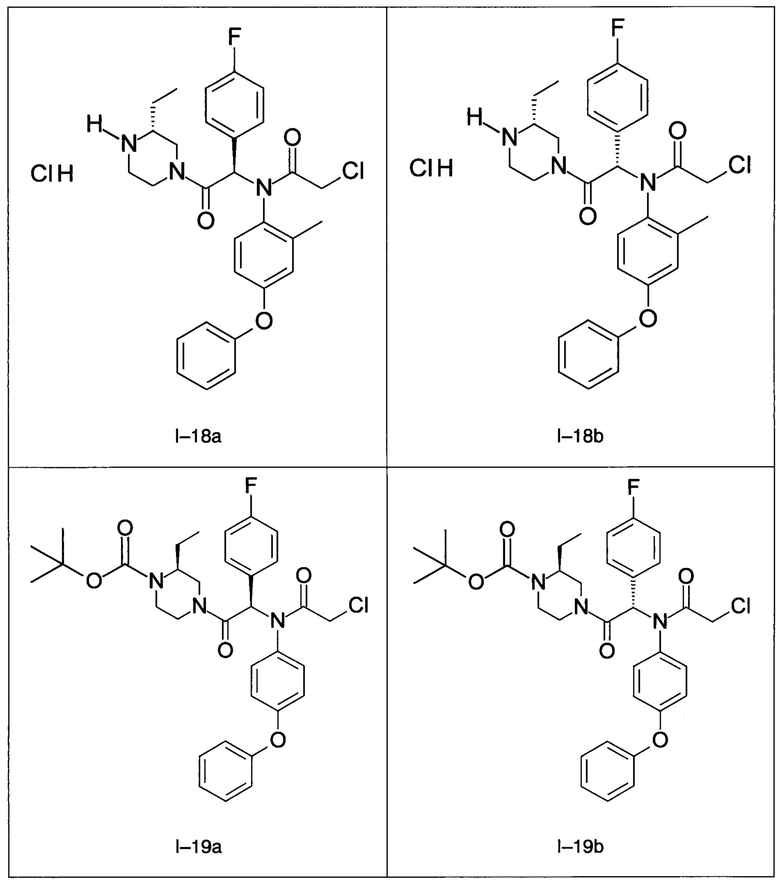
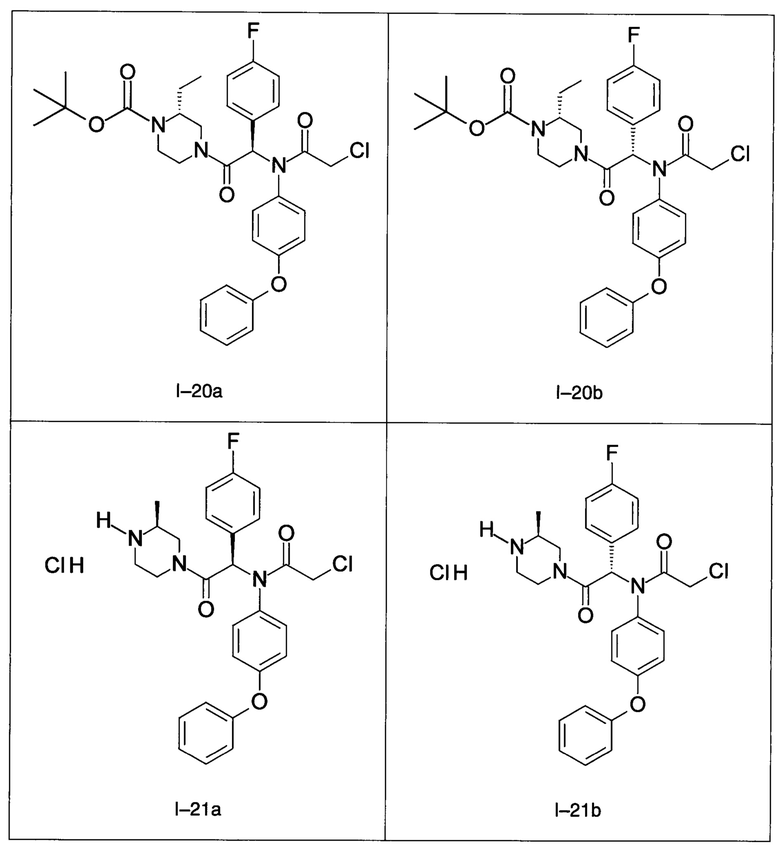
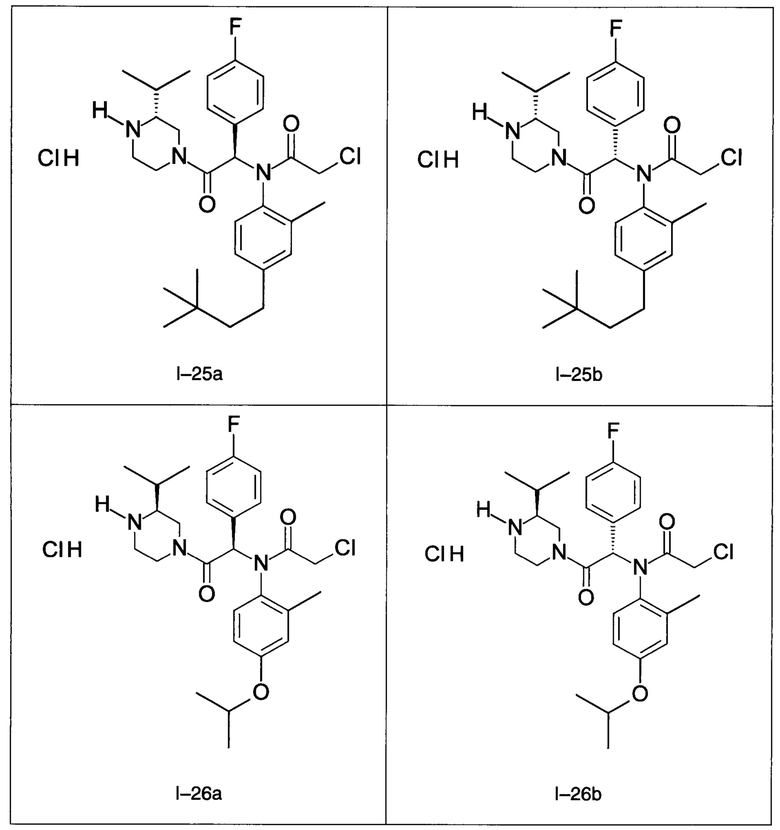
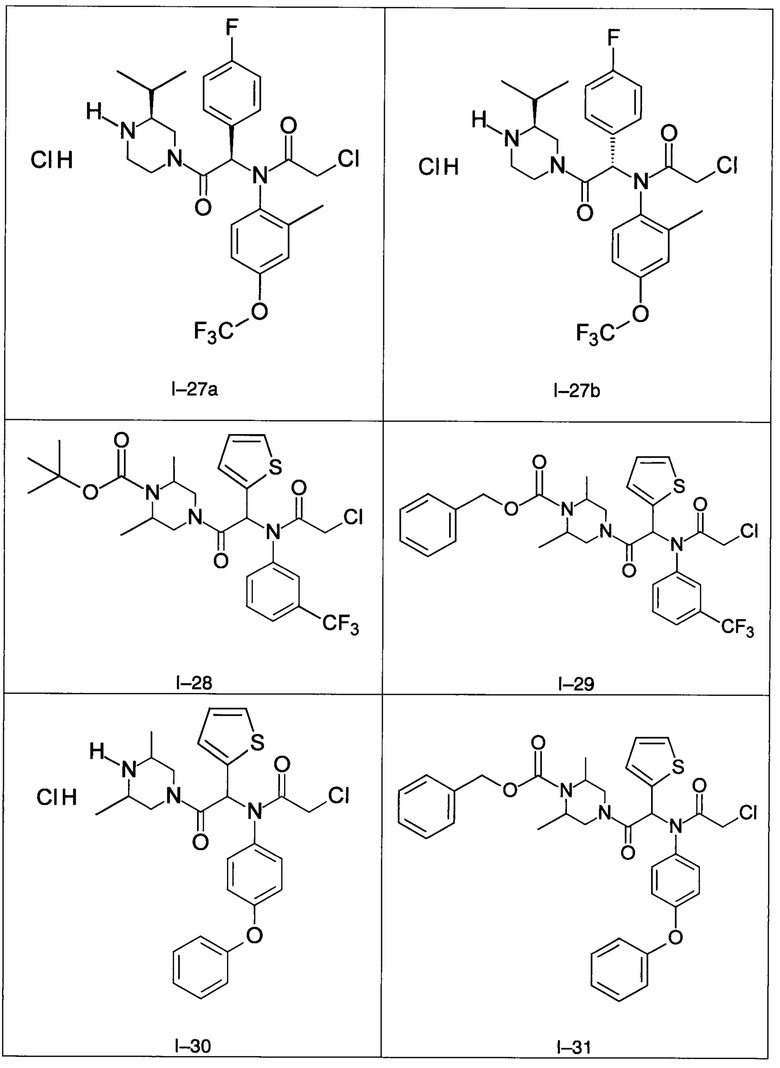
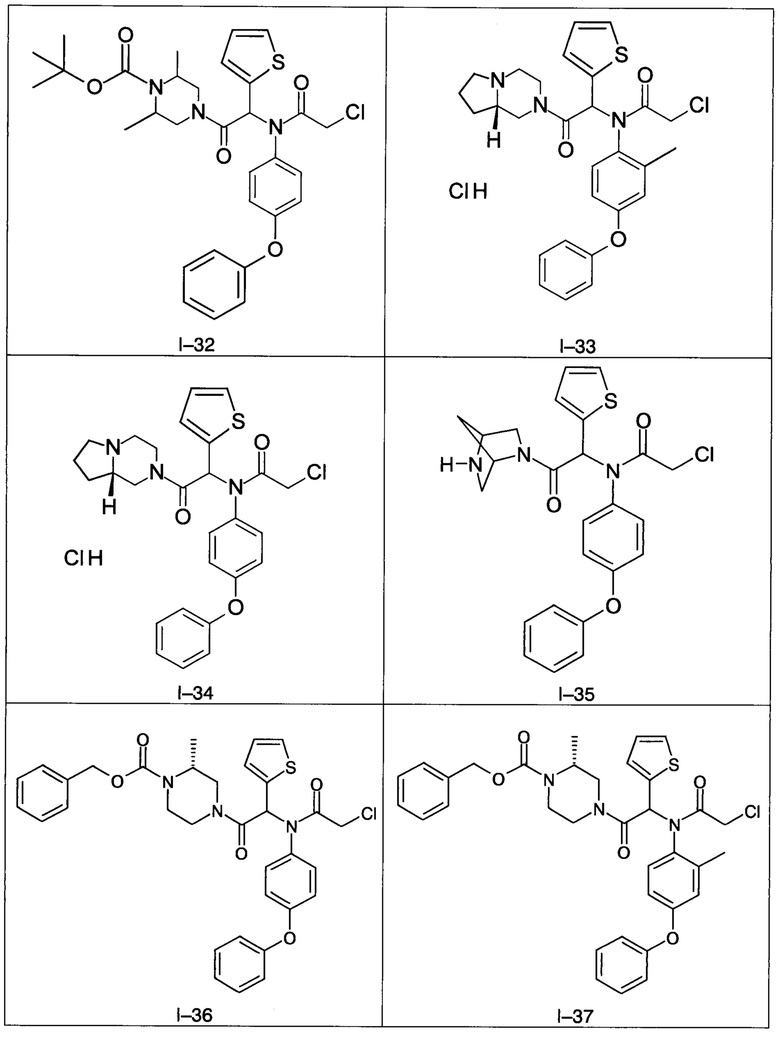
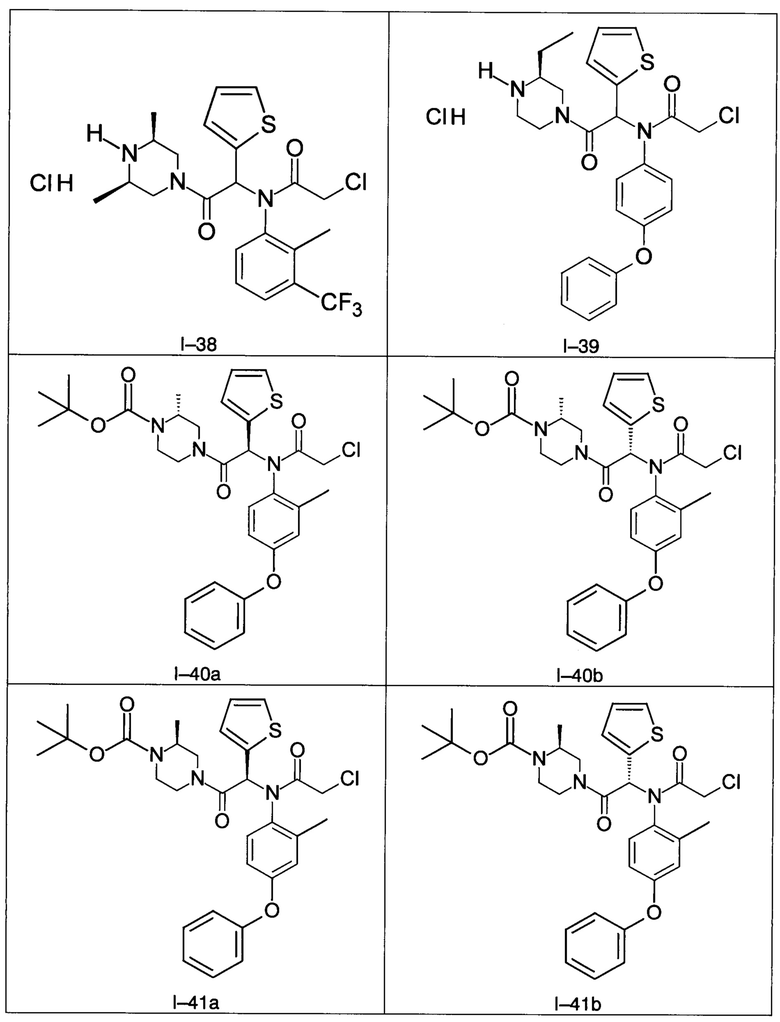
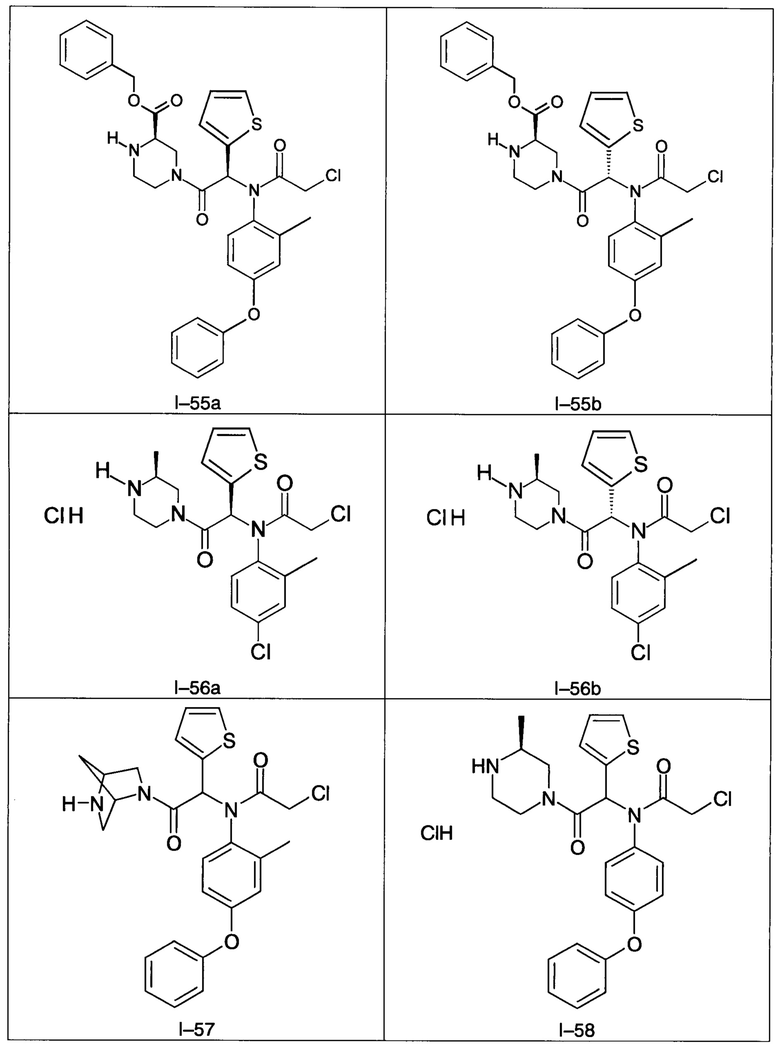
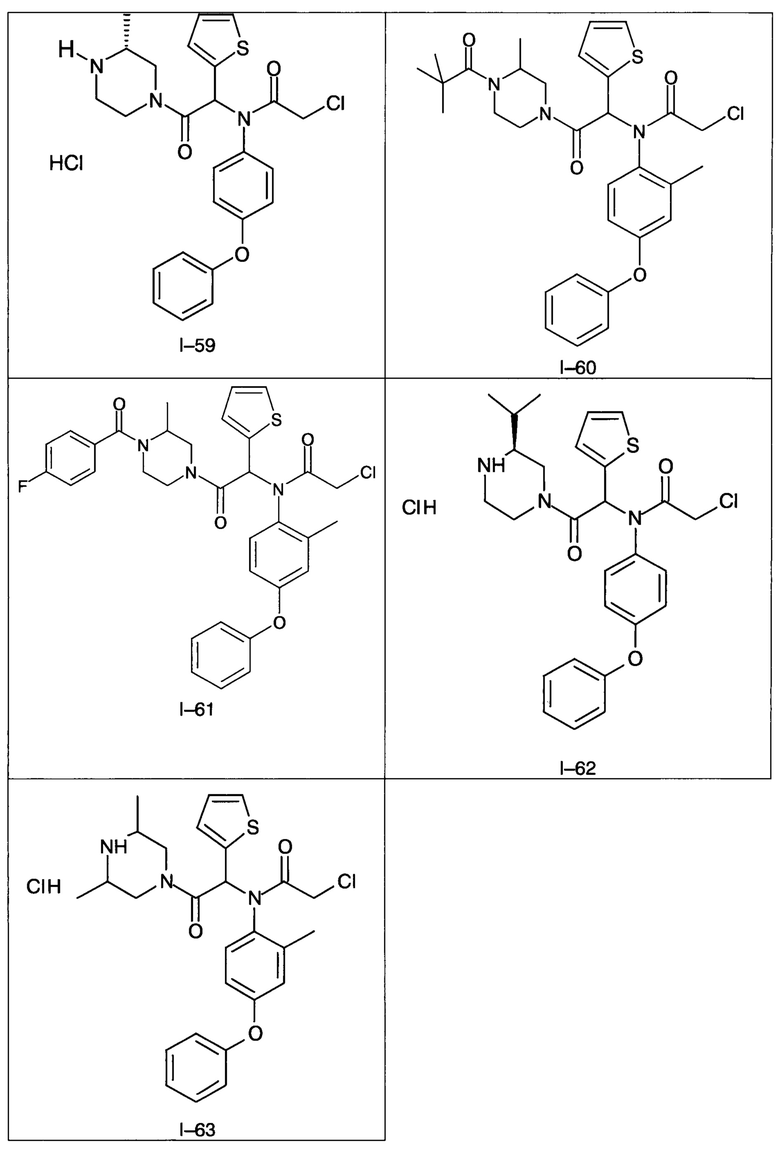
В частности, изобретение относится к одному из соединений из примеров от I-1а до I-63, описанных в экспериментальной части, или одному из их фармацевтически приемлемых солей, одному из их стереоизомеров или смесей стереоизомеров в любой пропорции, в частности смеси энантиомеров и в особенности рацемической смеси.
Настоящее изобретение относится также к соединению формулы (I), такому как описано выше, для применения в качестве лекарства, предназначенного, в частности, для лечения или профилактики рака, и в особенности для лечения рака, устойчивого к химиотерапии.
Настоящее изобретение относится также к соединению формулы (I), такому как описано выше, для изготовления лекарства, в частности, предназначенного для лечения или профилактики рака, в особенности для лечения рака, устойчивого к химиотерапии.
Настоящее изобретение относится также к способу лечения или профилактики рака, в особенности рака, устойчивого к химиотерапии, включающему введение достаточного количества соединения формулы (I), такому как описано выше, нуждающемуся в этом пациенту.
Еще один объект изобретения представляет собой фармацевтическую композицию, содержащую по меньшей мере одно соединение формулы (I), такое как описано выше, совместно с одним или более фармацевтически приемлемым эксципиентом.
В соответствии с одним конкретным воплощением, эта композиция может содержать по меньшей мере один дополнительный активный ингредиент.
В частности этот или эти активные ингредиенты могут представлять собой противораковые агенты, обычно используемые для лечения рака. Эти противораковые агенты могут быть выбраны, в частности, из цисплатина и его производных, таких как карбоплатин и оксалиплатин; таксанов, таких как таксол, таксотер, паклитаксел и доцетаксел; алкалоидов барвинка, таких как винбластин, винкристин и винорелбин; аналогов пурина, таких как меркаптопурин, тиогуанин, пентостатин и 2-хлородезоксиаденозин; ингибиторов топоизомеразы I, таких как соединения камптотецина, например иринотекан и топотекан; ингибиторов топоизомеразы II, таких как эпидофиллотоксин, подофиллотоксин и их производные, например этопозид и тенипозид; противораковых производных нуклеозидов, таких как 3-фтороурацил, лейковорин, гемцитабин или капецитабин; алкилирующих агентов, таких как азотистые иприты, например циклофосфамид, мехлорэтамин, хлороамбуцил и мелфалан, нитрозомочевин, таких как кармустин, ломустин и стрептозоцин, алкилсульфонатов, таких как бусульфан, этиленимины и метилмеламины, такие как тиотепа и гексаметилмеламин, и тетразины, такие как дакарбазин; производных противораковых антрациклинов, таких как даунорубицин, адриамицин, доксил, идарубицин и митоксантрон; молекул, действующих на рецептор IGF-I, таких как пикроподофиллин; производных тетракарцина, таких как тетрокарцин А; кортикостероидов, таких как преднизон; антител, таких как трастузумаб (анти-HER2 антитело), ритуксимаб (анти-CD20 антитело), гемтузумаб, цетуксимаб, пертузумаб и бевацизумаб; антагонистов или селективных модуляторов эстрогеновых рецепторов, таких как тамоксифен, фулвестрант, торемифен, дролоксифен, фаслодекс и ралоксифен; ингибиторов ароматазы, таких как экземестан, анастрозол, летрозол и ворозол; дифференцирующих агентов, таких как ретиноиды, например, ретиноевая кислота или витамин D, и агенты, блокирующие метаболизм ретиноевой кислоты, такие как аккутан; ингибиторов ДНК метил-трансферазы, таких как азацитидин и децитабин; антифолатов, таких как перметрексед динатрия; антибиотиков, таких как антиномицин D, блеомицин, митомицин С, актиномицин D, карминомицин, дауномицин и пликамицин; антиметаболитов, таких как хлофарабин, аминоптерин, цитозина арабинозид, флоксуридин и метотрексат; агентов, индуцирующих апоптоз и анти-ангиогенных ингибиторов Bcl-2, таких как YC 137, ВН 312, АВТ 737, госсипол, НА 14-1, TW 37 и деканоевая кислота; агентов, связывающихся с тубулином, таких как комбрестатин, производные колхицина и нокодазол; ингибиторов киназ, таких как флавопиридол, иматиниба мезилат, эрлотиниб и гефитиниб; ингибиторов фарнезил трансферазы, таких как типифарниб; ингибиторов гистондеацетилаз, таких как бутират натрия, субероиланилид гидроксамовой кислоты, депсипептид, NVP-LAQ824, R306465, JNJ-26481585 и трихостатин А; ингибиторов убиквитин-протеасомной системы, таких как MLN.41, бортезомиб и йонделис; а также ингибиторов теломеразы, таких как теломестатин.
Путь введения соединений по изобретению может быть оральным, сублингвальным, парентеральным, подкожным, внутримышечным, внутривенным, трансдермальным, местным или ректальным.
В фармацевтической композиции по настоящему изобретению, предназначенной для введения по оральному, сублингвальному, парентеральному, подкожному, внутримышечному, внутривенному, трансдермальному, местному или ректальному путям, активный ингредиент может вводиться животным или людям в форме единичной дозированной формы в смеси с обычными фармацевтическими носителями. Подходящие единичные дозированные формы включают такие формы для перорального введения, как таблетки, капсулы, порошки, гранулы и пероральные растворы или суспензии, а также формы для сублингвального или буккального введения, формы для парентерального, подкожного, внутримышечного, внутривенного, интраназального или внутриглазного введения, а также формы для ректального введения.
Если твердую композицию готовят в виде таблеток, главный активный ингредиент смешивают с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналоги. Таблетки можно покрыть сахарозой или другим подходящим материалом, или их можно обработать так, чтобы придать им свойство задержанного или длительного высвобождения, чтобы из них непрерывно выделялось определенное количество активного ингредиента.
Капсулу получают, смешивая активный ингредиент с разбавителем и наполняя полученной смесью мягкие или твердые капсулы.
Сироп или эликсир могут содержать активный ингредиент совместно с подсластителем, антисептиком и усилителем вкуса, и подходящим красителем.
Диспергируемые в воде порошки или гранулы могут содержать активный ингредиент в виде смеси с диспергирующими агентами, увлажняющими агентами или суспендирующими агентами, а также с усилителями вкуса или подсластителями.
Для ректального введения готовят суппозитории, используя для этого связующие вещества, которые плавятся при температуре в прямой кишке, например кокосовое масло или полиэтилен гликоли.
Для парентерального, интраназального или внутриглазного введения применяют водные суспензии, изотонические физиологические растворы или стерильные растворы для инъекций, содержащие фармакологически совместимые диспергирующие агенты и/или увлажнители.
Активный ингредиент можно также включать в микрокапсулы, возможно с одним или несколькими дополнительными носителями.
Соединения по изобретению можно применять в дозах от 0,01 мг до 1000 мг в сутки, в виде однократной суточной дозы или в виде нескольких доз на протяжении суток, например равные количества два раза в день. Суточная доза предпочтительно составляет от 5 до 500 мг, более предпочтительно от 10 до 200 мг. Может возникнуть необходимость в дозировках за пределами этих диапазонов, и специалист, знакомый с уровнем техники, сможет их определить.
Другим объектом изобретения является фармацевтическая композиция, содержащая:
(i) по меньшей мере одно соединение формулы (I), такое как определено выше;
и
(ii) по меньшей мере один другой активный ингредиент
в виде комбинированного продукта, предназначенного для одновременного, раздельного или поочередного применения.
Часто для лечения рака эффективно применяют двухкомпонентную или трехкомпонентную терапию. В частности, может быть полезно применять молекулы по изобретению совместно с одним или несколькими противораковыми соединениями прежде всего для лечения рака и, во-вторых, для профилактики появления устойчивых раковых клеток.
В частности, этот или эти активные ингредиенты могут представлять собой противораковые агенты, обычно используемые для лечения рака. Эти противораковые агенты могут быть выбраны, в частности, из цисплатина и его производных, таких как карбоплатин и оксалиплатин; таксанов, таких как таксол, таксотер, паклитаксел и доцетаксел; алкалоидов барвинка, таких как винбластин, винкристин и винорелбин; аналогов пурина, таких как меркаптопурин, тиогуанин, пентостатин и 2-хлородезоксиаденозин; ингибиторов топоизомеразы I, таких как соединения камптотецина, например, иринотекан и топотекан; ингибиторов топоизомеразы II, таких как эпидофиллотоксин, подофиллотоксин и их производные, например, этопозид и тенипозид; противораковых производных нуклеозидов, таких как 3-фтороурацил, лейковорин, гемцитабин или капецитабин; алкилирующих агентов, таких как азотистые иприты, например, циклофосфамид, мехлорэтамин, хлороамбуцил и мелфалан, нитрозомочевин, таких как кармустин, ломустин и стрептозоцин, алкилсульфонатов, таких как бусульфан, этиленимины и метилмеламины, такие как тиотепа и гексаметилмеламин, и тетразины, такие как дакарбазин; производных противораковых антрациклинов, таких как даунорубицин, адриамицин, доксил, идарубицин и митоксантрон; молекул, действующих на рецептор IGF-I, таких как пикроподофиллин; производных тетракарцина, таких как тетрокарцин А; кортикостероидов, таких как преднизон; антител, таких как трастузумаб (анти-HER2 антитело), ритуксимаб (анти-CD20 антитело), гемтузумаб, цетуксимаб, пертузумаб и бевацизумаб; антагонистов или селективных модуляторов эстрогеновых рецепторов, таких как тамоксифен, фулвестрант, торемифен, дролоксифен, фаслодекс и ралоксифен; ингибиторов ароматазы, таких как экземестан, анастрозол, летрозол и ворозол; дифференцирующих агентов, таких как ретиноиды, например, ретиноевая кислота или витамин D и агенты, блокирующие метаболизм ретиноевой кислоты, такие как аккутан; ингибиторов ДНК метил-трансферазы, таких как азацитидин и децитабин; антифолатов, таких как перметрексед динатрия; антибиотиков, таких как антиномицин D, блеомицин, митомицин С, актиномицин D, карминомицин, дауномицин и пликамицин; антиметаболитов, таких как хлофарабин, аминоптерин, цитозина арабинозид, флоксуридин и метотрексат; агентов, индуцирующих апоптоз и анти-ангиогенных ингибиторов Bcl-2, таких как YC 137, ВН 312, АВТ 737, госсипол, НА 14-1, TW 37 и деканоевая кислота; агентов, связывающихся с тубулином, таких как комбрестатин, производные колхицина и нокодазол; ингибиторов киназ, таких как флавопиридол, иматиниба мезилат, эрлотиниб и гефитиниб; ингибиторов фарнезил трансферазы, таких как типифарниб; ингибиторов гистондеацетилаз, таких как бутират натрия, субероиланилид гидроксамовой кислоты, депсипептид, NVP-LAQ824, R306465, JNJ-26481585 и трихостатин А; ингибиторов убиквитин-протеасомной системы, таких как MLN.41, бортезомиб и йонделис; а также ингибиторов теломеразы, таких как теломестатин.
Еще одним объектом изобретения является фармацевтическая композиция, такая как описана выше, для применения в качестве лекарства для лечения или профилактики рака, в частности рака, устойчивого к химиотерапии.
Настоящее изобретение также относится к способу получения соединения формулы (I), такого как описано выше, включающему следующие последовательные этапы:
а) взаимодействие амина следующей формулы (II)
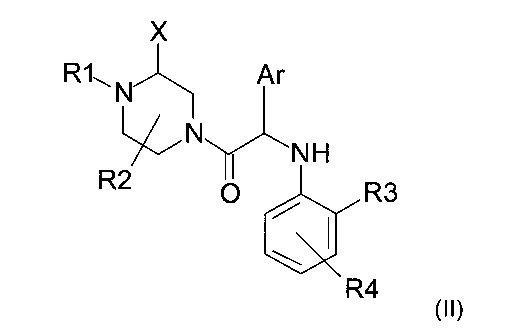
где X, R1, R2, R3, R4 и Ar представляют собой такие, как определено ранее, и R1 не является атомом водорода;
с хлорацетилхлоридом в присутствии основания с получением соединения формулы (I), где R1 ≠ Н; и
b) возможное снятие защиты с атома азота, несущего группу R1 ≠ Н, с получением соединения формулы (I), где R1=Н.
Этап а):
Основание, используемое на этом этапе предпочтительно является слабым основанием, таким как NaHCO3.
Амин формулы (II) можно получить в результате взаимодействия пиперазина следующей формулы (III)
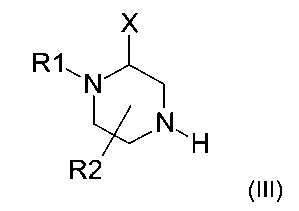
где X, R1 и R2 представляют собой такие, как определено ранее, и R1 не является атомом водорода,
с кислотой следующей формулы (IV)
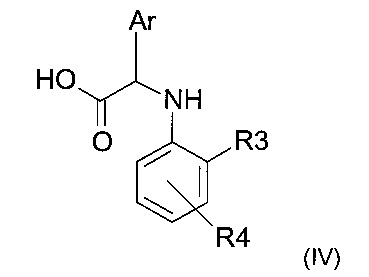
где R3, R4 и Ar представляют собой такие, как определено ранее.
Эту реакцию можно проводить в условиях пептидного сочетания, хорошо известных специалисту.
Таким образом, сочетание предпочтительно проводят в присутствии агента сочетания, такого как диизопропилкарбодиимид (DIC), дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDC), карбонилдиимидазол (CDI), 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HBTU), 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония тетрафторборат (TBTU) или O-(7-азобензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HATU), возможно ассоциированного с вспомогательным агентом сочетания, таким как N-гидроксисукцинимид (NHS), N-гидроксибензотриазол (HOBt), 3,4-дигидро-3-гидрокси-4-оксо-1,2,3-бензотриазол (HOOBt), 1-гидрокси-7-азабензотриазол (HAt) или N-гидроксисульфоскцинимид (сульфо NHS). Предпочтительно, это HBTU.
Может также присутствовать такое основание, как диизопропилэтиламин (DIPEA).
Пиперазин формулы (III) либо закупают, либо получают способами, хорошо известными специалистам в уровне техники.
Кислоту формулы (IV) можно получить, выполняя следующие последовательные этапы:
i) взаимодействие кетоэфира следующей формулы (V)
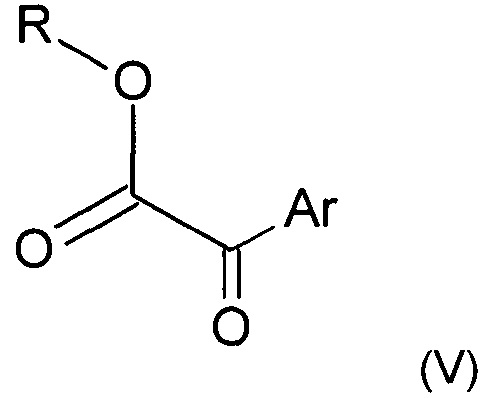
где Ar представляет собой такой, как определено ранее, a R представляет собой группу (С1-С6)алкил, например этил,
с анилином формулы (VI)
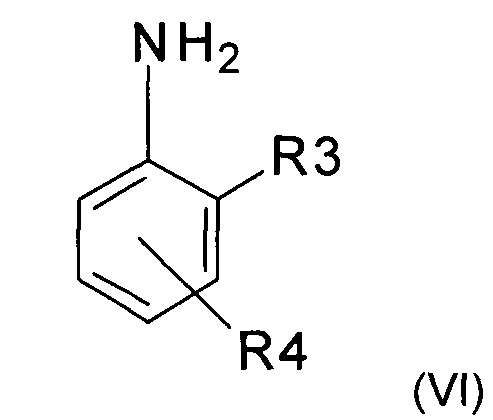
где R3 и R4 представляют собой такие, как определено ранее,с получением имина следующей формулы (VII)
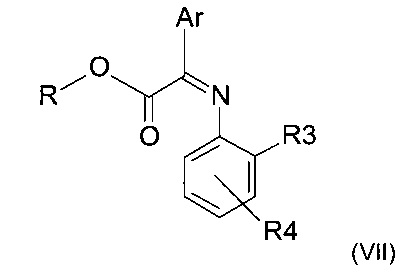
где R, R3, R4 и Ar представляют собой такие, как определено ранее;
ii) восстановление имина формулы (VII), полученного на предыдущем этапе, с получением амина следующей формулы (VIII)
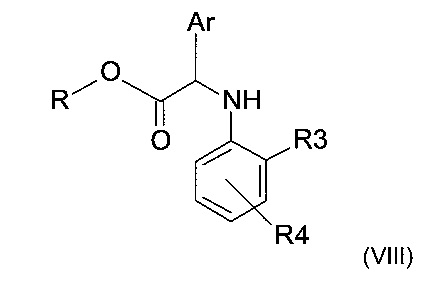
где R, R3, R4 и Ar представляют собой такие, как определено ранее; и
iii) сапонификация эфирной функциональной группы соединения формулы (VIII), полученного на предыдущем этапе, с получением кислоты формулы (IV).
Этап i) можно проводить в присутствии кислоты, такой как паратолуолсульфоновая кислота (PTSA). Реакцию можно проводить в полярном растворителе, таком как толуол. Предпочтительно кипятить реакционную среду с обратным холодильником в аппарате Дина-Старка, чтобы удалить воду по мере ее образования в ходе реакции.
Используемый в реакции кетоэфир (V) либо покупают, либо получают посредством реакции Фиделя-Крафтса с использованием этилоксалилхлорида и соответствующего ароматического соединения в присутствии кислоты Льюиса, такой как хлорид алюминия AlCl3.
Используемый в реакции анилин (VI) либо покупают, либо получают способами, хорошо известными специалистам в уровне техники.
Этап ii) восстановления можно проводить в присутствии восстановителя, хорошо известного специалистам в уровне техники, такого как цианоборгидрид натрия.
Этап iii) сапонификации можно проводить в условиях, хорошо известных опытным специалистам, в частности в присутствии основания, такого как NaOH, КОН или LiOH.
Этап b):
Этот этап предпочтительно проводят с использованием соединения формулы (I), в котором R1=CO2R6, например CO2tBu, обрабатывая его кислотой, такой как HCl.
Полученные таким способом соединения можно выделить из реакционной смеси с помощью способов, хорошо известным специалистам, например экстракцией, упариванием растворителя или осаждением и фильтрованием.
При необходимости соединение можно также очистить, используя способы, хорошо известные специалистам, например перекристаллизацию, если соединение кристаллическое, перегонку, хроматографию на силикагеле или высокоэффективную жидкостную хроматографию (ВЭЖХ).
Способ получения соединений по настоящему изобретению, где R1 ≠ Н, показан на следующей схеме реакции:
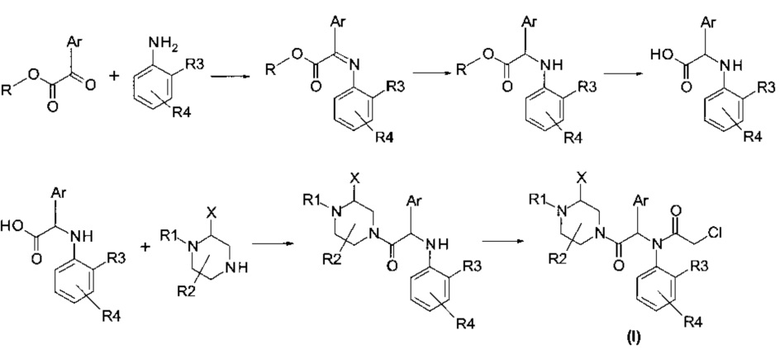
Приводимые далее примеры иллюстрируют изобретение, но не ограничивают его.
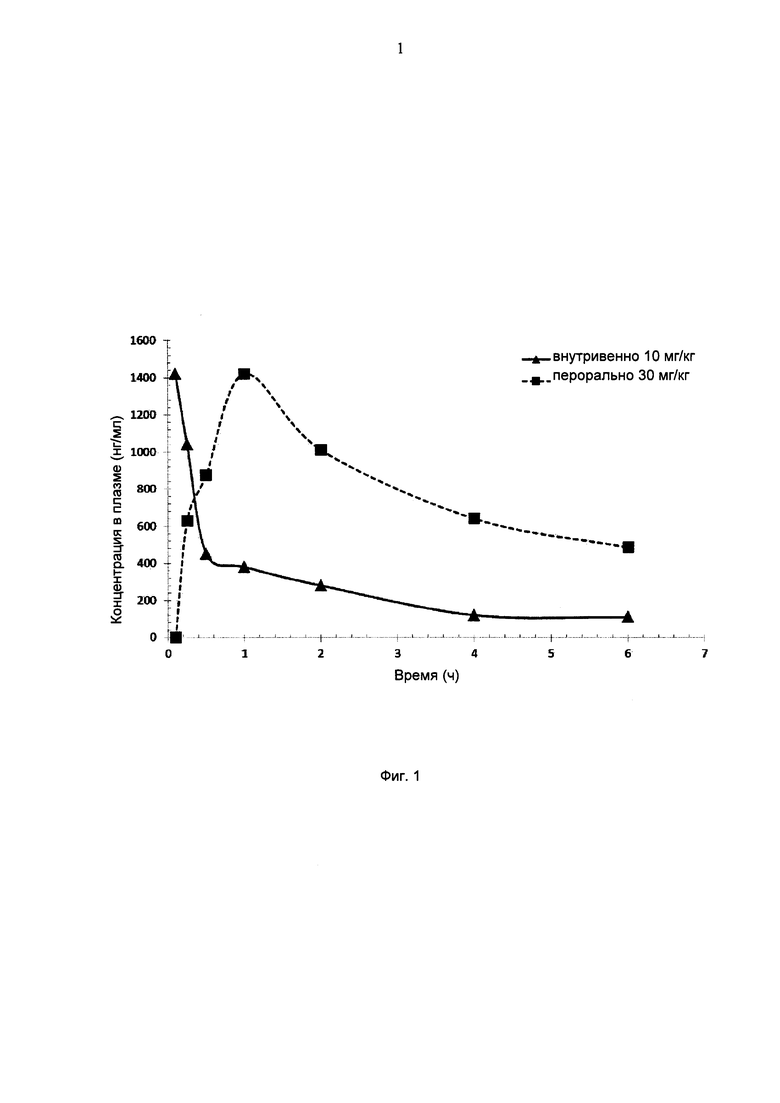
Фигура:
На фигуре 1 представлены кривые зависимости концентрации вещества в плазме от времени для мышей, получавших соединение I-43 диа2 внутривенно (IV) в дозе 10 мг/кг или перорально (РО) в дозе 30 мг/кг.
Примеры:
I - Синтез соединений по изобретению
В следующем разделе при описании диастереоизомеров соединения по изобретению использовали две различные номенклатуры.
- а/b обозначают конкретную структуру отдельного диастереоизомера;
- диа1/диа2 соответственно обозначает менее полярный и более полярный диастереоизомер в использовавшейся хроматографической системе. Конкретная стереохимия каждого диастереоизомера определена не была. Таким образом, связать конкретную структуру а или b с каждым выделенным диастереоизомером диа1 или диа2 не представляется возможным. Именно поэтому использовалась двойная номенклатура.
В настоящем разделе использовались следующие сокращения:
HBTU 2-(1Н-Бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат
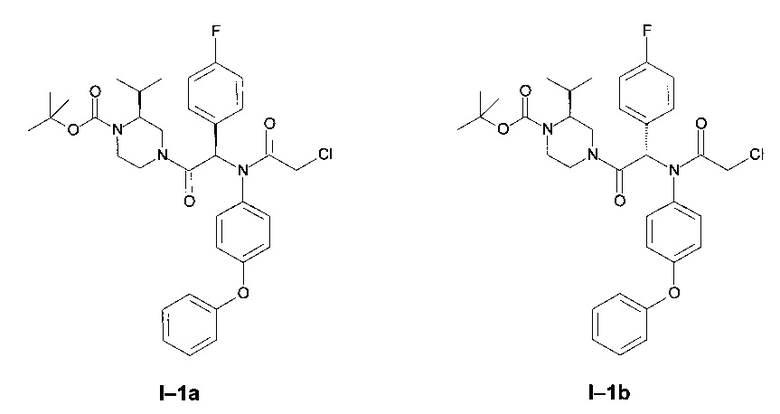
Примеры I-1а и I-1b: диастереоизомеры трет-бутилового эфира 4-[2-[(2-хлорацетил)-(4-фенокси-фенил)-амино]-2-(4-фторфенил)-ацетил]-(S)-2-изопропил-пиперазин-1-карбоновой кислоты.
Стадия 1: Этиловый эфир (4-фторфенил)-оксо-уксусной кислоты (1):
К раствору хлорида алюминия (21,13 г; 160 ммоль) в DCM (200 мл) при 0°С в атмосфере аргона по каплям в течение 10 минут добавили этилоксалилхлорид (17,9 мл; 160 ммоль). Систему перемешивали еще 10 минут. По каплям при 0°С добавили фторбензол (14,7 мл; 160 ммоль), растворенный в 30 мл DCM. Смесь перемешивали при комнатной температуре в течение 12 часов. Затем ее промыли водой, и высушили органическую фазу над MgSO4. После упаривания образовавшееся масло очистили флэш-хроматографией на силикагеле, элюирование проводили смесью циклогексан-этилацетат в соотношении 90:10. Получено желтое масло (17,08 г; 54%).
1Н ЯМР (300 МГц, CDCl3): δ 8,04-8,14 (m; 1,8 Н); 7,15-7,24 (m; 1,9 Н); 4,46 (q; J=7,2 Hz; 2,0 Н); 1,44 (t; J=7,2 Hz; 3,0 H).
Стадия 2: Этиловый эфир (4-фторфенил)-[(Z)-4-фенокси-фенилимино]-уксусной кислоты (2):
К раствору 1 (3,92 г; 20 ммоль) в толуоле (25 мл) в присутствии молекулярного сита последовательно добавили пара-толуолсульфоновую кислоту (200 мг; 1 ммоль) и 4-феноксифенил-анилин (3,70 г; 20 ммоль). Смесь поместили в аппарат Дина-Старка и 20 часов кипятили с обратным холодильником. Затем ее промыли водой, и высушили органическую фазу над MgSO4. После упаривания образовавшееся масло очистили флэш-хроматографией на силикагеле, элюирование проводили смесью циклогексан-этил ацетат в соотношении 90:10.
Получено желтое масло (6,27 г; 86%).
LCMS [М+Н]=364 (C22H18FNO3)
Сталия 3: Этиловый эфир (4-фторфенил)-(4-фенокси-фениламино)-уксусной кислоты (3):
К раствору 2 (6,27 г; 17,26 ммоль) в метаноле (75 мл) и уксусной кислоте (7,5 мл) добавили цианоборгидрид натрия (1,63 г; 26 ммоль). Смесь 1 час перемешивали при комнатной температуре. Метанол частично упарили, нейтрализовали раствор Na2CO3 с добавлением воды при необходимости. Среду экстрагировали DCM, и органическую фазу высушили над MgSO4. После упаривания образовавшееся масло очистили флэш-хроматографией на силикагеле, элюирование проводили смесью циклогексан-этил ацетат в соотношении 95:5.
Получено желтое масло (5,91 г; 93%).
LCMS [М+Н]=366 (C22H20FNO3)
1Н ЯМР (300 МГц, CDCl3): δ 7,45-7,54 (m; 1,9 Н); 7,23-7,31 (m; 1,9 Н); 6,97-7,11 (m; 2,9 Н); 6,81-6,94 (m; 3,9 Н); 6,54 (d; J=9,0 Hz; 2,0 Н); 5,01 (br; 1,0 Н); 4,90 (br; 0,9 Н); 4,10-4,32 (m; 2,0 Н); 1,23 (t; J=7,0 Hz; 3,0 Н).
Стадия 4: (4-фторфенил)-(4-фенокси-фениламино)-уксусной кислоты (4):
К раствору 3 (8,04 г; 22 ммоль) в 130 мл ацетонитрила добавили 66 мл 1 М раствора LiOH (3 эквив.). Реакционную смесь перемешивали от 2 до 3 часов, завершение реакции контролировали по ТСХ (циклогексан-этил ацетат 60:40). Ацетонитрил частично упарили, среду закислили 1М раствором HCl в 200 мл воды. Смесь профильтровали, полученное твердое вещество три раза промыли водой и высушили в вакуумном сушильном шкафу в присутствии P2O5.
Получен белый порошок (7,17 г; 97%).
LCMS [М+Н]=338 (C20H16FNO3)
1Н ЯМР (300 МГц, ДМСО): δ 7,55 (dd; J=8,5 Hz; J=5,6 Hz; 2,1 H); 7,28 (t; J=7,9 Hz; 2,1 H); 7,20 (t; J=8,5 Hz; 2,1 H); 6,99 (t; J=7,0 Hz; 1,1 H); 6,74-6,90 (m; 4,0 H; 6,62-6,70 (m; 2,0 H); 5,10 (s 1,0 H).
Стадия 5: Трет-бутиловый эфир 4-[2-(4-фторфенил)-2-(4-фенокси-фениламино)-ацетил]-(S)-2-изопропил-пиперазин-1-карбоновой кислоты (5):
К раствору 4 (7,17 г; 21,2 ммоль) в DCM (150 мл) в присутствии одного эквивалента DIEA (3,7 мл) добавили раствор Вос-альфа-(S)-изопропил-пиперазин гидрохлорида (5,63 г; 21,26 ммоль) в присутствии 1 эквивалента DIEA (3,7 мл) в 50 мл DCM, а затем HBTU (8,06 г; 21,2 ммоль). Систему перемешивали 12 часов. Затем ее промыли водой, и высушили органическую фазу над MgSO4. После упаривания образовавшееся масло очистили флэш-хроматографией на силикагеле, элюирование проводили смесью циклогексан-этилацетат в соотношении 80:20. Получена белая пена (11,90 г; 100%).
LCMS [М+Н]=548 (C32H38FN3O4).
Стадия 6: Трет-бутиловый эфир of 4-[2-[(2-хлорацетил)-(4-фенокси-фенил)-амино]-2-(4-фторфенил)-ацетил]-(S)-2-изопропил-пиперазин-1-карбоновой кислоты
К раствору 5 (11,86 г; 22,66 ммоль) в 250 мл DCM в присутствии NaHCO3 (7,30 г; 87,0 ммоль) добавили хлорацетилхлорид (3,45 мл; 43,3 ммоль). Систему перемешивали 12 часов. Затем ее промыли водой, и высушили органическую фазу над MgSO4. После упаривания образовавшееся масло очистили флэш-хроматографией на силикагеле, элюирование проводили градиентом циклогексан-этилацетат в соотношении от 95:5 до 50:50 с получением двух диастереоизомеров по отдельности в форме бесцветной пены:
Менее полярный диастереоизомер (I-1 диа1)
(3,80 г; 28%)
LCMS [М+Н]=625 (C34H39CIFN3O5).
1Н ЯМР (300 МГц, CDCl3): δ 7,92-8,01 (m; 1,0 Н); 7,30-7,40 (m; 2,0 Н); 7,10-7,18 (m; 1,1 Н); 7,01-7,09 (m; 1,1 Н); 6,84-7,00 (m; 6,1 Н); 6,55-6,65 (m; 1,1 Н); 6,32-6,48 (m; 2,1 Н); 4,72 (d; J=13,5 Hz; 0,5 H) 4,63 (d; J=13,5 Hz; 0,4 H); 3,52-3,96 (m; 4,0 H); 3,10-3,27 (m; 0,5 H); 2,85-3,07 (m; 0,4 H); 2,23-2,85 (m; 0,5 H + 0,7 H + 0,4 H); 1,87-2,14 (m; 0,6 H); 1,42 (s; 8,7 H); 1,17 (d; J=6,6 Hz; 1,0 H); 1,03 (d; J=6,6 Hz; 1,3 H); 0,88 (d; J=6,6 Hz; 1,1 H); 0,69 (d; J=6,6 Hz; 1,3 H).
Более полярный диастереоизомер (I-1 диа2)
(3,29 г; 24%)
LCMS [М+Н]=625 (C34H39CIFN3O5)
1Н ЯМР (300 МГц, CD2Cl2): δ 7,85-8,00 (m; 1,0 Н); 7,36 (t; J=7,6 Hz; 2,1 H); 6,99-7,21 (m; 3,2 H); 6,81-6,98 (m; 5,2 H); 6,63 (br; 1,1 H); 6,35-65,5 (m; 2,1 H); 4,65 (d; J=13,1 Hz; 0,6 H)4,42 (d; J=13,1 Hz; 0,3 H); 3,50-4,16 (m; 4,9 H); 3,00-3,43 (m; 0,9 H); 2,57-2,90 (m; 1,9 H); 1,98-2,18 (m; 0,7 H); 1,36-1,49 (m; 10,0 H); 1,73 (d; J=6,5 Hz; 2,1 H); 0,90 (d; J=6,5 Hz; 2,1 H); 0,63 (d; J=6,5 Hz; 1,0 H); 0,20 (d; J=6,5 Hz; 0,9 H).
Примеры I-2a and I-2b: Диастереоизомеры трет-бутилового эфира 4-[-2-[(2-хлорацетил)-(4-фенокси-фенил)-амино]-2-(4-фторфенил)-ацетил]-(R)-2-изопропил-пиперазин-1-карбоновой кислоты.
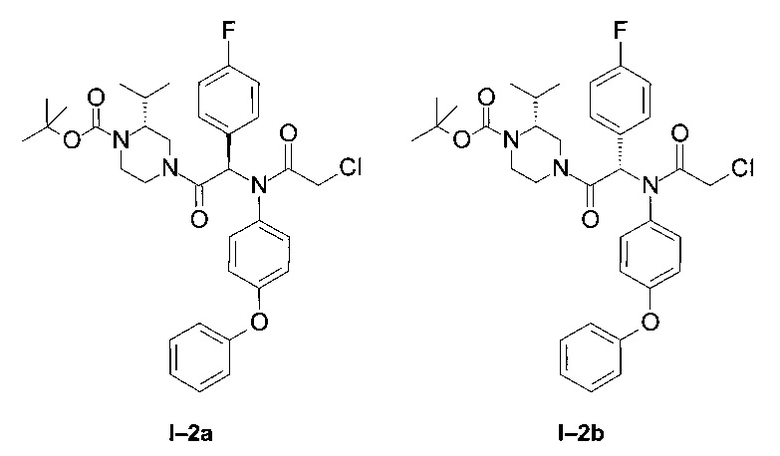
Стадия 1: Трет-бутиловый эфир 4-[2-(4-фторфенил)-2-(4-фенокси-фениламино)-ацетил]-(R)-2-изопропил-пиперазин-1-карбоновой кислоты (6):
К раствору (4-фторфенил)-(4-фенокси-фениламино)-уксусной кислоты 4 (253 мг; 0,75 ммоль) в DCM (10 мл) в присутствии одного эквивалента DIEA (131 мкл) добавили раствор Вос-альфа-(R)-изопропил-пиперазина (171 мг; 0,75 ммоль) в 5 мл DCM также в присутствии одного эквивалента DIEA (131 мкл), а затем ввели HBTU (285 мг; 0,75 ммоль). Систему перемешивали 12 часов. Затем ее промыли водой, и высушили органическую фазу над MgSO4. После упаривания образовавшееся масло очистили флэш-хроматографией на силикагеле, элюирование проводили системой циклогексан-этилацетат в соотношении 80:20. Получена белая пена (369 мг; 90%).
LCMS [М+Н]=548 (C32H38FN3O4)
Стадия 2: Трет-бутиловый эфир 4-[-2-[(2-хлорацетил)-(4-фенокси-фенил)-амино1-2-(4-фторфенил)-ацетил]-(R)-2-изопропил-пиперазин-1-карбоновой кислоты:
Оба диастереоизомера получили из 6 тем же способом, который использовался в Примере 1 (стадия 6).
Оба диастереоизомера выделены по отдельности в виде бесцветной пены.
Менее полярный диастереоизомер (I-2 диа1) (195 мг; 42%)
LCMS [М+Н]=625 (C34H39CIFN3O5)
1Н ЯМР (300 МГц, CD2Cl2): δ 7,85-8,00 (m; 1,0 Н); 7,36 (t; J=7,6 Hz; 2,0 Н); 6,99-7,21 (m; 3,1 Н); 6,81-6,98 (m; 4,9 Н); 6,63 (br; 1,0 Н); 6,35-6,55 (m; 2,1 Н); 4,65 (d; J=13,0 Hz; 0,7 Н) 4,42 (d; J=13,0 Hz; 0,2 H); 3,50-4,16 (m; 4,9 H); 3,00-3,43 (m,; 0,8 H); 2,57-3,90 (m; 2,0 H); 1,98-2,18 (m; 0,8 H); 1,36-1,49 (m; 10,5 H); 1,73 (d; J=6,5 Hz; 2,0 H); 0,90 (d; J=6,5 Hz; 2,0 H); 0,63 (d; J=6,5 Hz; 0,8 H);0,20 (d; J=6,5 Hz; 0,8 H).
Более полярный диастереоизомер (I-2 диа2) (122 мг; 26%)
LCMS [М+Н]=625 (C34H39CIFN3O5)
1Н ЯМР (300 МГц, CDCI3): δ 7,92-8,01 (m; 1,0 Н); 7,30-7,40 (m; 2,0 Н); 7,10-7,18 (m; 1,0 Н); 7,01-7,09 (m; 1,1 Н); 6,84-7,00 (m; 6,0 Н); 6,55-6,65 (m; 1,1 Н); 6,32-6,48 (m; 2.1 Н); 4,72 (d; J=13,5 Hz; 0,4 Н) 4,63 (d; J=13,5 Hz; 0,3 H); 3,52-3,96 (m; 4,7 H); 3,10-3,27 (m; 0,7 H); 2,85-3,07 (m; 0,5 H); 2,23-2,85 (m; 0,5 H + 0,6 H + 0,8 H); 1,87-2,14 (m; 0,9 H); 1,42 (s; 8,6 H); 1,17 (d; J=6,6 Hz; 1,4 H); 1,03 (d; J=6,6 Hz; 2,1 H); 0,88 (d; J=6,6 Hz; 2,1 H); 0,69 (d; J=6,6 Hz; 1,6 H).
Примены I-3a и I-3b: Гидрохлориды диастереоизомеров 2-хлор-N-[1-(4-фторфенил)-2-((S)-3-изопропил-пиперазин-1-ил)-2-оксо-этил]-N-(4-фенокси-фенил)-ацетамида.
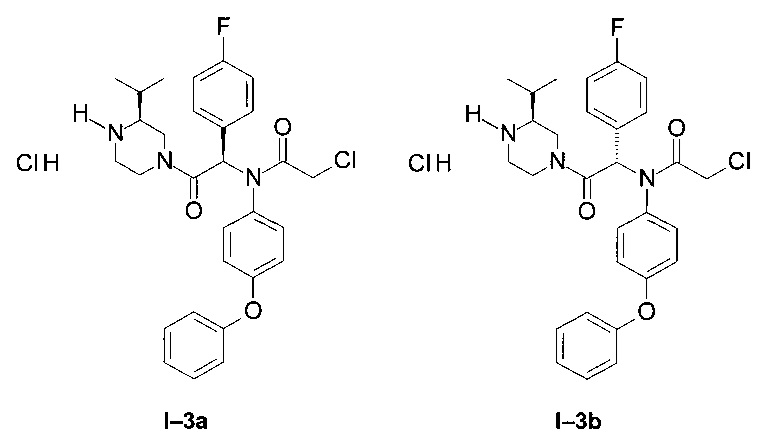
Через раствор диастереоизомера I-1диа2 (3,24 г; 5,2 ммоль) в 50 мл DCM пропустили газообразный HCl посредством пробулькивания. Реакционную смесь 12 часов перемешивали при КТ. DCM упарили, и полученное масло осадили в эфире. После фильтрования продукт I-3 диа2 выделили в форме белого порошка: (2,53 г; 87%).
LCMS [М+Н]=524 (C29H32Cl2FN3O3).
1Н ЯМР (300 МГц, ДМСО): δ 8,60-9,35 (m; 1,6 Н); 7,77 (br; 0,8 Н); 7,30-7,40 (m; 2,0 Н); 7,00-7,23 (m; 5,1 Н); 6,80-7,00 (m; 3,1 Н); 6,54-6,76 (m; 3,0 Н); 4,56 (d; J=13,3 Hz; 1,0 Н); 3,88-4,16 (m; 3,0 Н); 3,00-3,30 (m; 3,1 Н); 2,65-2,96 (m; 1,7 Н); 1,52-2,00 (m; 1,6 Н); 1,00 (t; J=7,4 Hz; 2,4 Н); 0,59 (dd; J=15,6 Hz; J=6,7 Hz; 3,5 H) В результате применения описанного способа к веществу 1-1диа1, после фильтрования выделили продукт I-3 диа1 в виде белого порошка (63 мг).
LCMS [М+Н]=524 (C29H32Cl2FN3O3).
1Н ЯМР (300 МГц, ДМСО): δ 8,65-9,6 (br; 1,2 Н); 7,77 (br,; 0,8 Н); 7,30-7,40 (m; 2,0 Н); 6.36- 7,25 (m; 11,7 Н); 4,40-4,60 (m; 0,8 Н); 4,00-4,12 (m; 2,0 Н); 3,76-3,98 (m; 0,9 Н); 3.37-3,63 (m; 0,9 Н); 2,65-3,30 (m; 4,0 Н); 1,77-2,06 (m; 1,6 Н); 0,89-1,06 (m; 6,1 Н).
Примеры I-4а и I-4b: Гидрохлориды диастереоизомеров 2-хлор-N-[1-(4-фторфенил)-2-((R)-3-изопропил-пиперазин-1-ил)-2-оксо-этил]-N-(4-фенокси-фенил)-ацетамид.
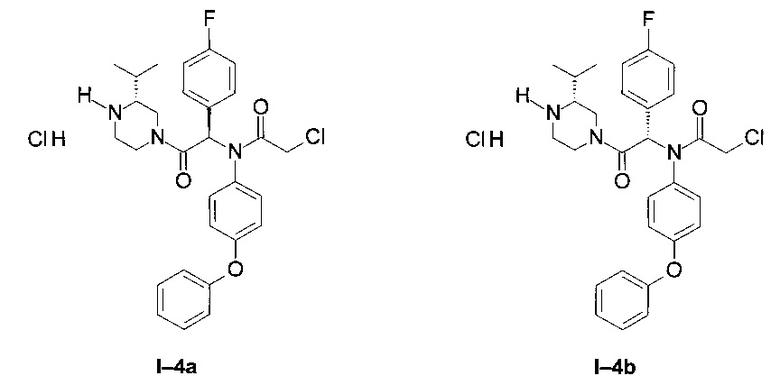
Использовался протокол, описанный для примеров I-3а и I-3b, но начиная от диастереоизомеров I-2а и I-2b
Начиная от первого диастереоизомера Примера I-2 (I-4 диа1):
после фильтрования выделили белый порошок: (95 мг)
LCMS [М+Н]=524 (C29H32Cl2FN3O3)
1Н ЯМР (300 МГц, ДМСО): δ 8,65-9,6 (br; 1,3 Н),; 7,77 (br; 0,4 Н); 7,30-7,40 (m; 2,0 Н); 6.36- 7,25 (m; 11,8 Н); 4,40-4,60 (m; 0,9 Н); 4,00-4,12 (m; 2,0 Н); 3,76-3,98 (m; 1,0 Н); 3.37- 3,63 (m; 1,0 Н); 2,65-3,30 (m; 3,8 Н); 1,77-2,06 (m; 1,8 Н); 0,89-1,06 (m; 6,1 Н).
Начиная от второго диастереоизомера примера I-2 (I-4 диа2):
после фильтрования выделили белый порошок: (95 мг)
LCMS [М+Н]=524 (C29H32Cl2FN3O3)
1Н ЯМР (300 МГц, ДМСО): δ 8,60-9,35 (m; 1,7 Н); 7,77 (br; 0,9 Н); 7,30-7,40 (m; 2,0 Н); 7,00-7,23 (m; 5,0 Н); 6,80-7,00 (m; 3,0 Н); 6,54-6,76 (m; 2,9 Н); 4,56 (d; J=13,3 Hz; 1,0 Н); 3,88-4,16 (m; 3,0 Н); 3,00-3,30 (m; 3,1 Н); 2,65-2,96 (m; 1,7 Н); 1,52-2,06 (m; 1,9 Н); 1,00 (t; J=7,2 Hz; 2,4 Н); 0,59 (dd; J=15,4 Hz; J=6,7 Hz; 3,3 H).
Примеры I-5а и I-5b: Диастереоизомеры трет-бутилового эфира 4-[-2-[(2-хлорацетил)-(2-метил-4-фенокси-фенил)-амино]-2-(4-фторфенил)-ацетил]-(S)-2-изопропил-пиперазин-1-карбоновой кислоты.
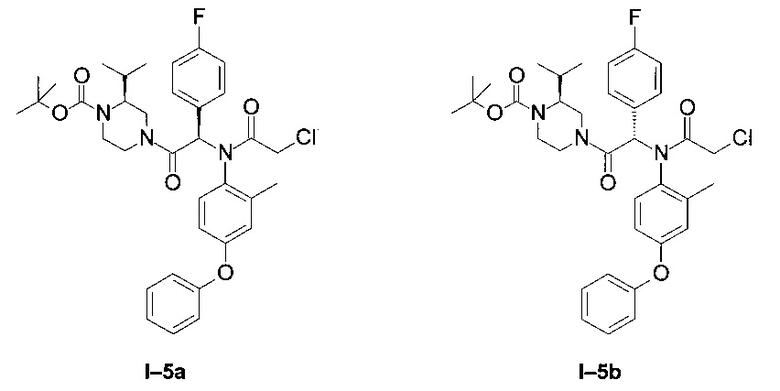
Стадия 1: Трет-бутиловый эфир 4-[2-(4-фторфенил)-2-(2-метил-4-фенокси-фениламино-ацетил]-(S)-2-изопропил-пиперазин-1-карбоновой кислоты (8):
К раствору (4-фторфенил)-{2-метил-4-фенокси-фениламино)-уксусной кислоты (9,29 г; 26,4 ммоль) в DCM (150 мл) в присутствии одного эквивалента DIEA (4,6 мл) добавили раствор Вос-альфа-(S)-изопропил-пиперазин гидрохлорида (7,00 г; 26,4 ммоль) в 50 мл DCM также в присутствии одного эквивалента DIEA (4,6 мл), а затем ввели HBTU (10,00 г; 26,4 ммоль). Систему перемешивали 12 часов. Затем ее промыли водой, и высушили органическую фазу над MgSO4. После упаривания образовавшееся масло очистили флэш-хроматографией на силикагеле, элюирование проводили системой циклогексан-этилацетат в соотношении 80:20. Получена белая пена (14,13 г; 95%).
LCMS [М+Н]=562 (C33H40FN3O4)
Стадия 2: Трет-бутиловый эфир 4-[-2-[(2-хлорацетил)-(2-метил-4-фенокси-фенил)-амино]-2-(4-фторфенил)-ацетил]-(S)-2-изопропил-пиперазин-1-карбоновой кислоты
К раствору 8 (14,13 г; 25,1 ммоль) в 250 мл DCM в присутствии NaHCO3 (8,40 г; 100,0 ммоль) добавили хлорацетил хлорид (4,00 мл; 50,0 ммоль). Систему перемешивали 12 часов. Затем ее промыли водой, и высушили органическую фазу над MgSO4. После упаривания образовавшееся масло очистили флэш-хроматографией на силикагеле, элюирование проводили в градиенте циклогексан-этилацетат от 90:10 до 50:50.
Оба диастереоизомера выделены в форме бесцветной пены:
Менее полярный диастереоизомер (I-5 диа1) (3,83 г; 24%)
LCMS [М+Н]=639 (C35H41ClFN3O5)
1Н ЯМР (300 МГц, CD2Cl2): δ 7,94-8,57 (m; 1,0 Н); 7,35 (t; J=7,9 Hz; 2,0 H); 7,07-7,27 (m; 3,0 H); 6,74-6,95 (m; 5,0 H); 6,58 (br d; J=2,6 Hz; 1,1 H); 6,51 (br s; 0,2 H); 6,41 (s; 0,8 H); 6,31 (br s; 0,3 H); 4,62 (d; J=13,5 Hz; 0,7 H) 4,39 (d; J=13,5 Hz; 0,3 H); 3,53-4,05 (m; 4,8 H); 3,04-3,46 (m; 0,8 H); 2,41-2,96 (m; 2,1 H); 2,04-2,23 (m; 0,8 H); 1,82-1,95 (m; 2,2 H); 1,43 (brs; 10,1 H); 1,07 (d; J=6,5 Hz; 2,1 H); 0,90 (d; J=6,5 Hz; 2,3 H); 0,63 (d; J=6,5 Hz; 1,0 H); 0,29 (d; J=6,5 Hz; 0,8 H).
Более полярный диастереоизомер (I-5 диа2) (4,40 г; 27%)
LCMS [М+Н]=639 (C35H41ClFN3O5)
1Н ЯМР (300 МГц, CDCl3): δ 8,00-8,10 (m; 1,0 Н); 7,30-7,40 (m; 2,1 Н); 6,98-7,18 (m; 3,2 Н); 6,73-6,90 (m; 5,3 Н); 6,52-6,58 (m; 1,0 Н); 6,34-6,39 (m; 1,0 Н); 4,71 (d; J=13,5 Hz; 0,7 Н); 4,49 (d; J=13,5 Hz; 0,4 H); 3,50-4,00 (m; 4,7 H); 3,10-3,30 (m; 0,7 H); 2,86-3,07 (m; 0,4 H); 2,54-2,85 (m; 1,5 H); 2,30-2,47 (m; 0,4 H); 1,80-1,87 (m; 2,8 H); 1,54-1,60 (m; 2,5 H); 1,42 (br s; 8,8 H); 1,19 (d; J=6,6 Hz; 1,1 H); 1,00 (d; J=6,6 Hz; 1,5 H); 0,88 (d; J=6,6 Hz; 1,2 H); 0,64 (d; J=6,6 Hz; 1,5 H).
Примеры I-6a и I-6b: диастереоизомеры трет-бутилового эфира 4-[-2-[(2-хлорацетил)-(4-фенокси-фенил)-амино]-2-(4-фторфенил)-ацетил]-(R)-2-изопропил-пиперазин-1-карбоновой кислоты
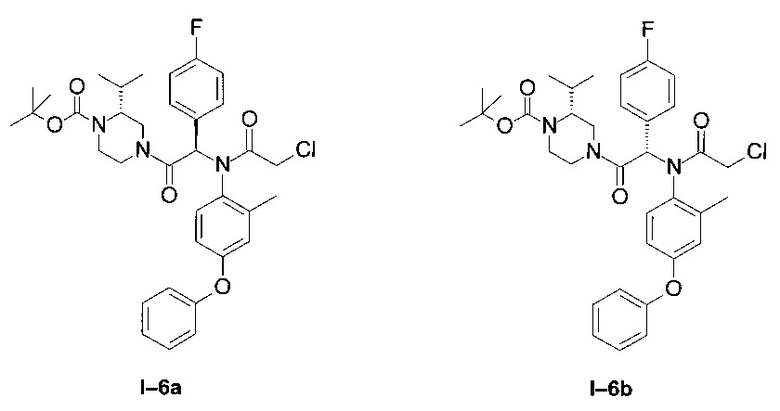
Оба этих диастереоизомера были получены в виде бесцветной пены тем же способом, что и в предыдущем примере:
Менее полярный диастереоизомер (I-6 диа1) (97 мг; 30%)
LCMS [М+Н]=639 (C35H41ClFN3O5)
1Н ЯМР (300 МГц, CD2Cl2): δ 7,94-8,57 (m; 0,9 Н); 7,35 (t; J=7,9 Hz; 1,9 H); 7,05-7,25 (m; 3,1 H); 6,72-6,93 (m; 5,0 H); 6,58 (br d; J=2,6 Hz; 1,1 H); 6,41 (s; 0,8 H); 6,31 (br s; 0,3 Н); 4,63 (d; J=13,5 Hz; 0,8 H) 4,40 (d; J=13,5 Hz; 0,3 H); 3,51-4,06 (m; 4,8 H); 3,03-3,45 (m; 1,0 H); 2,41-2,96 (m; 1,6 H); 2,02-2,21 (m; 0,8 H); 1,82-1,95 (m; 2,1 H); 1,43 (br s; 10,1 H); 1,07 (d; J=6,5 Hz; 2,1 H); 0,90 (d; J=6,5 Hz; 2,3 H); 0,63 (d; J=6,5 Hz; 1,0 H); 0,29 (d; J=6,5 Hz; 0,8 H).
Более полярный диастереоизомер (I-6 диа2) (90 мг; 28%)
LCMS [М+Н]=639 (C35H41ClFN3O5)
1Н ЯМР (300 МГц, CDCl3): δ 8,00-8,10 (m; 1,0 Н); 7,30-7,40 (m; 2,1 Н); 6,98-7,18 (m; 3,1 Н); 6,73-6,90 (m; 5,1 Н); 6,52-6,58 (m; 1,0 Н); 6,34-6,39 (m; 1,0 Н); 4,70 (d; J=13,5 Hz; 0,7 Н); 4,49 (d; J=13,5 Hz; 0,4 H); 3,50-4,00 (m; 4,8 H); 3,10-3,30 (m; 0,7 H); 2,86-3,07 (m; 0,4 H); 2,54-2,85 (m; 1,5 H); 2,30-2,47 (m; 0,4 H); 1,80-1,87 (m; 2,8 H); 1,54-1,60 (m; 2,6 H); 1,42 (br s; 8,6 H); 1,20 (d; J=6,6 Hz; 1,1 H); 1,01 (d; J=6,6 Hz; 1,5 H); 0,89 (d; J=6,6 Hz; 1,2 H); 0,64 (d; J=6,6 Hz; 1,5 H).
Примеры I-7a и I-7b: Гидрохлориды диастереоизомеров 2-xлop-N-[-1-(4-фторфенил)-2-((S)-3-изопропил-пиперазин-1-ил)-2-оксо-этил]-N-(2-метил-4-фенокси-фенил)-ацетамида
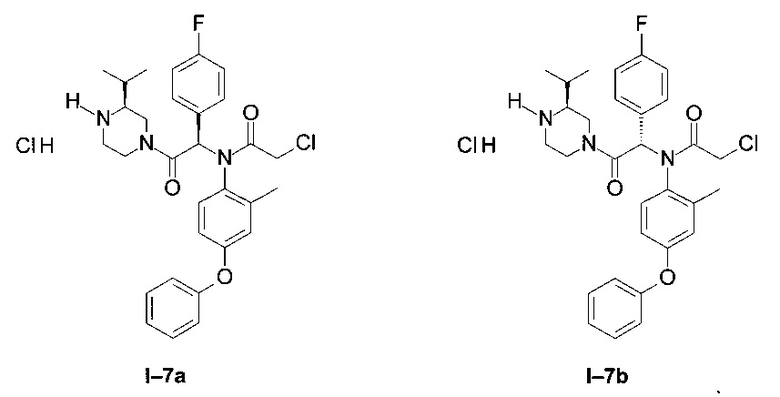
Через раствор одного из диастереоизомеров I-5а и I-5b в 50 мл DCM пропустили газообразный хлороводород методом пробулькивания. Реакционную смесь 12 часов перемешивали при КТ. DCM упарили, и полученное масло осадили в этиловом эфире.
Начиная от первого диастереоизомера примера I-5 (I-7 диа1):
После фильтрования выделили белый порошок: (26 мг)
LCMS [М+Н]=538 (C30H34Cl2FN3O3)
1Н ЯМР (300 МГц, ДМСО): δ 8,79-9,33 (m; 1,3 Н); 7,83 (t; J=9,0 Hz; 1,0 Н); 7,24-7,40 (m; 4,0 Н); 6,97-7,15 (m; 3,1 Н); 6,73-6,89 (т; 3,2 Н); 6,64 (d; J=2,7 Hz; 0,9 Н); 6,51-6,59 (m; 1,0 Н); 4,40-4,55 (br m; 1,1 Н); 3,86-4,09 (m; 3,6 Н); 3,45-3,60 (m; 0,7 Н); 2,78-3,05 (m; 2,8 H); 1,79-2,00 (m; 4,5 H); 1,61-1,77 (m; 0,7 H); 0,97 (d; J=6,7 Hz; 6,0 H).
Начиная от второго диастереоизомера примера I-5 (1-7 диа2):
После фильтрования выделили белый порошок: (2,62 г)
LCMS [М+Н]=538 (C30H34Cl2FN3O3)
1Н ЯМР (300 МГц, ДМСО): δ 8,78-9,51 (m; 1,9 Н); 7,82 (t; J=8,9 Hz; 0,9 Н); 7,20-7,41 (m; 4,0 Н); 6,97-7,16 (m; 3,1 Н); 6,71-6,90 (m; 3,1 Н); 6,61-6,70 (m; 1,9 Н); 4,46-4,60 (br m; 1,0 Н); 3,85-4,15 (m; 3,1 Н); 3,00-3,30 (m; 3,0 Н); 2,57-2,96 (m; 1,8 Н); 1,43-1,98 (m; 4,3 Н); 1,00 (dd; J=8,8 Hz; J=7,0 Hz; 2,7 H); 0,71 (d; J=6,8 Hz; 1,6 H); 0,65 (d; J=6,8 Hz; 1,5 H).
Пример I-8: Гидрохлорид 2-хлор-N-1-(4-фторфенил)-2-((R)-3-изопропил-пиперазин-1-ил)-2-оксо-этил]-N-(2-метил-4- фенокси-фенил)-ацетамида
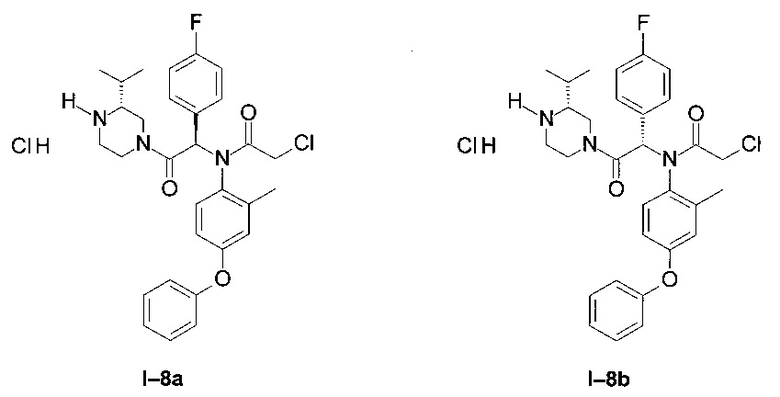
Использовался протокол, описанный в предыдущем примере, но начиная от обоих диастереоизомеров примера I-6.
Начиная от первого диастереоизомера примера I-6 (I-8 диа1):
После фильтрования выделили белый порошок: (34 мг; 56%)
LCMS [М+Н]=538 (C30H34Cl2FN3O3)
1Н ЯМР (300 МГц, ДМСО): δ 8,79-9,33 (m; 1,3 Н); 7,83 (t; J=9,0 Hz; 0,9 Н); 7,24-7,40 (m; 4,0 Н); 6,97-7,15 (m; 3,1 Н); 6,73-6,89 (m; 3,1 Н); 6,64 (d; J=2,7 Hz; 1,0 Н); 6,51-6,59 (m; 1,0 Н); 4,41-4,56 (br m; 1,1 Н); 3,86-4,09 (m; 3,4 Н); 3,45-3,60 (m; 0,7 Н); 2,78-3,05 (m; 2,8 Н); 1,79-2,00 (m; 4,4 Н); 1,61-1,77 (m; 0,8 Н); 0,97 (d; J=6,7 Hz; 6,0 Н).
Начиная от второго диастереоизомера примера I-6 (I-8 диа2):
После фильтрования выделили белый порошок: (30 мг; 54%)
LCMS [М+Н]=538 (C30H34Cl2FN3O3)
1Н ЯМР (300 МГц, ДМСО): δ 8,78-9,51 (m; 1,5 Н); 7,82 (t; J=8,9 Hz; 1,0 Н); 7,20-7,41 (m; 4,0 Н); 6,97-7,16 (m; 3,1 Н); 6,71-6,90 (m; 3,2 Н); 6,61-6,70 (m; 1,9 Н); 4,46-4,60 (br m; 1,0 H); 3,85-4,15 (m; 3,1 H); 3,00-3,30 (m; 2,9 H); 2,57-2,96 (m; 1,8 H); 1,43-1,98 (m; 4,3 H); 1,00 (dd; J=8,8 Hz; J=7,0 Hz; 2,7 H); 0,71 (d; J=6,8 Hz; 1,6 H); 0,65 (d; J=6,8 Hz; 1,5 H).
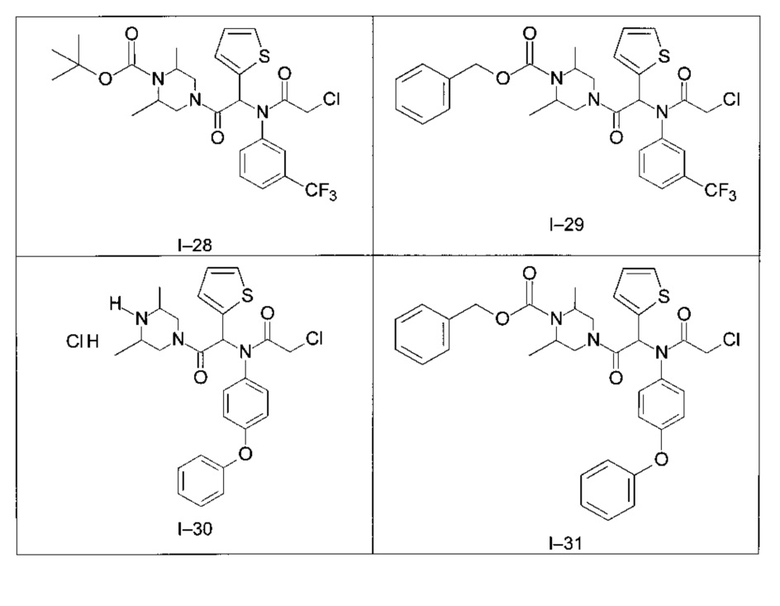
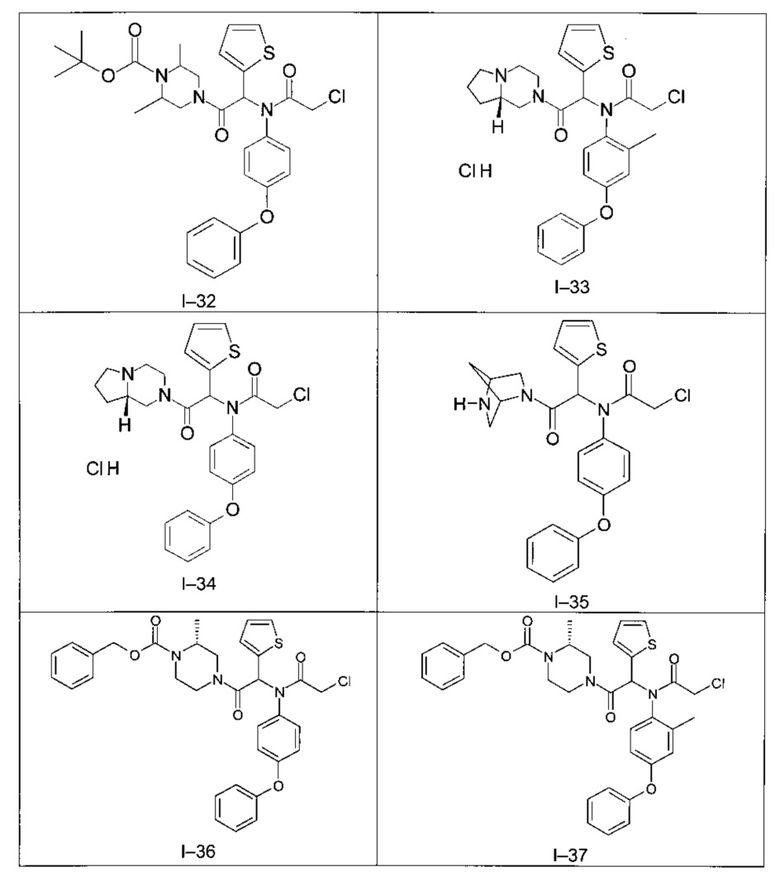
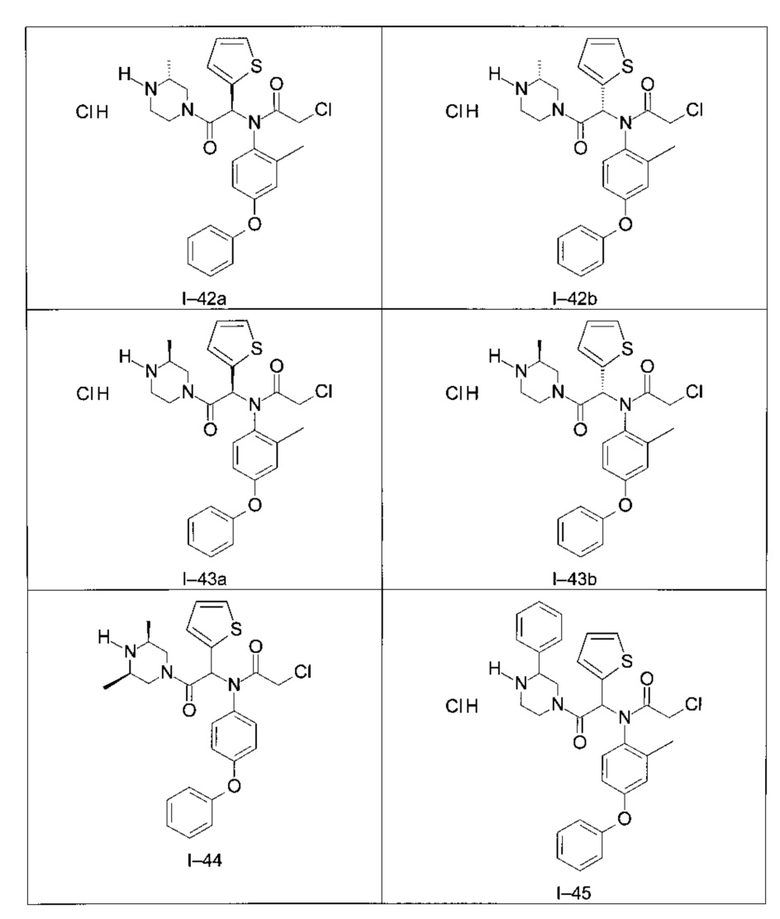
Используя такие же способы синтеза и разделения (хроматография на силикагеле), как описаны выше, из разнообразных замещенных анилинов и пиперазинов были получены следующие примеры. Они были выделены либо в форме смеси двух или четырех диастереоизомеров (один номер примера для одинаковых химических структур), либо в виде отдельных диастереоизомеров. В последнем случае для обозначения каждого диастереоизомера использовали номенклатуру а/b.
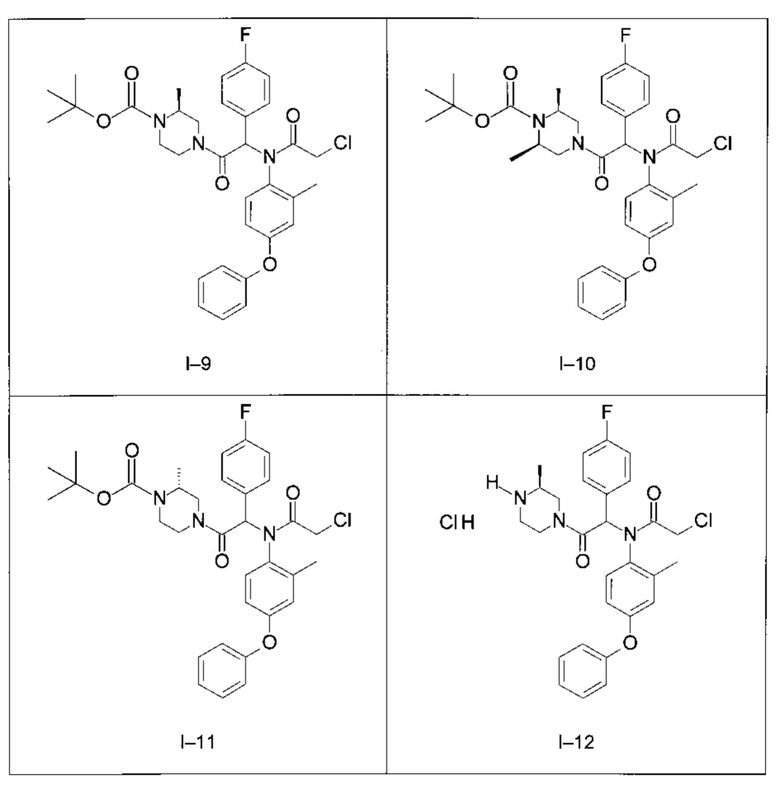
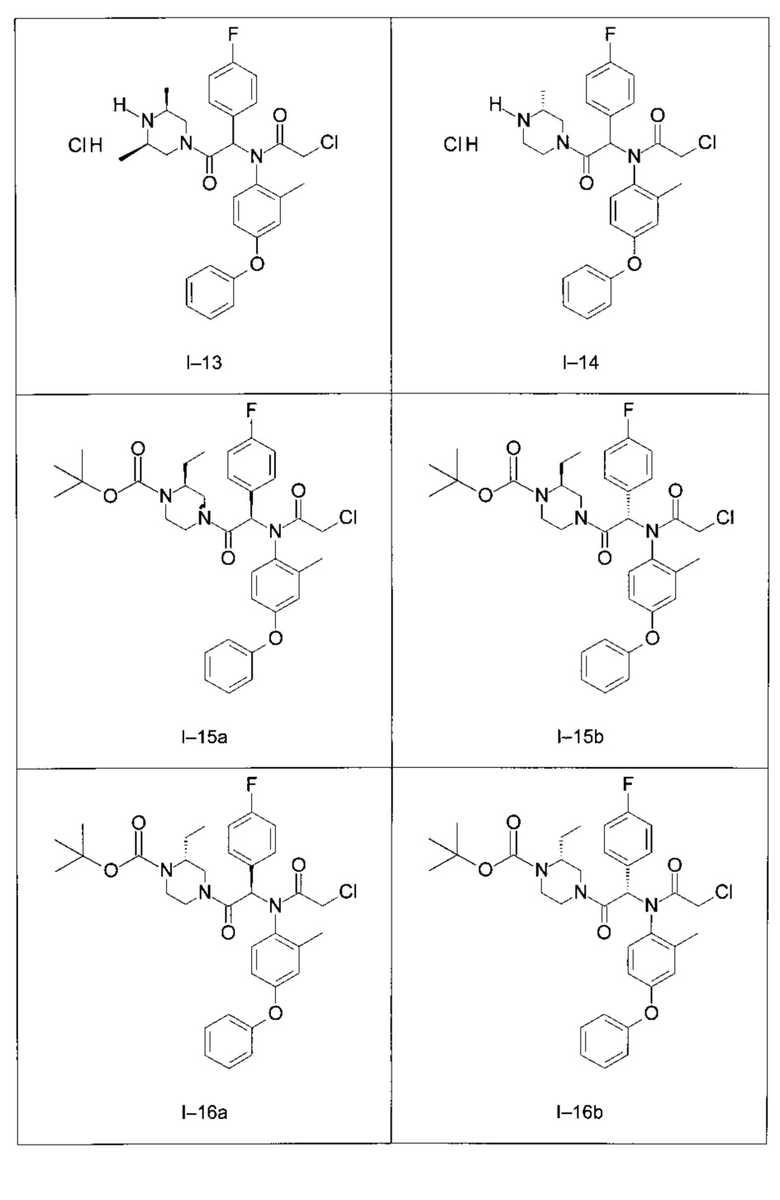
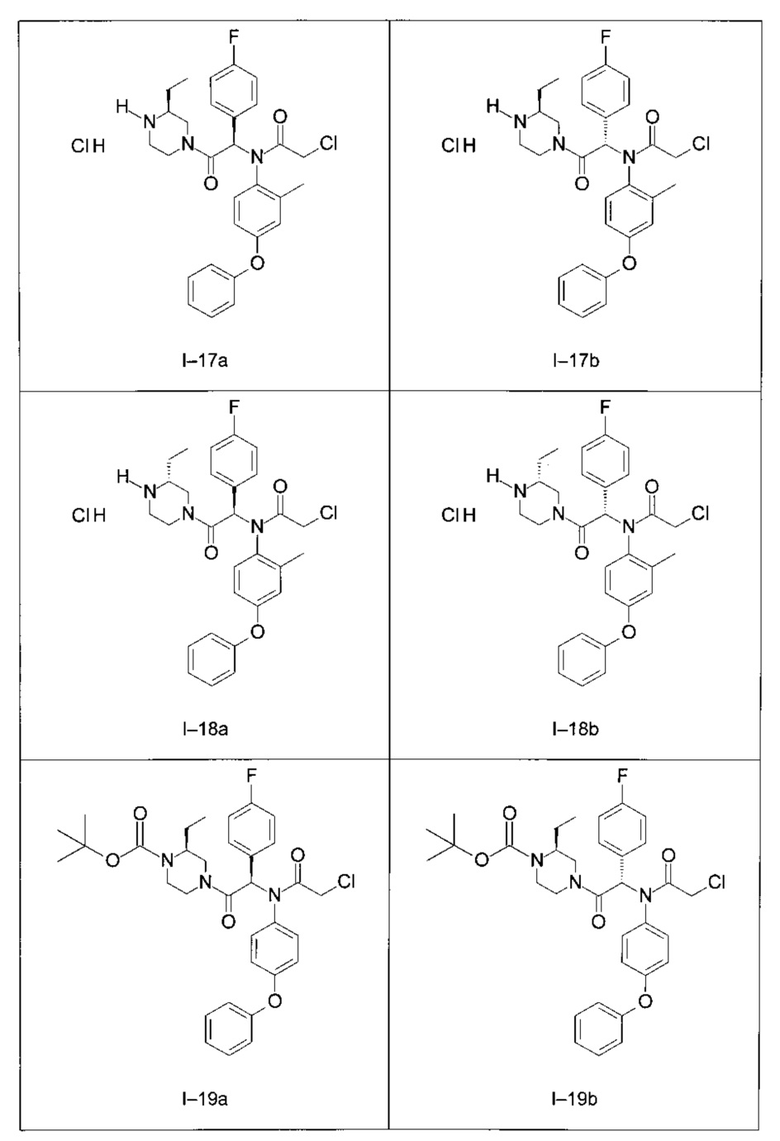
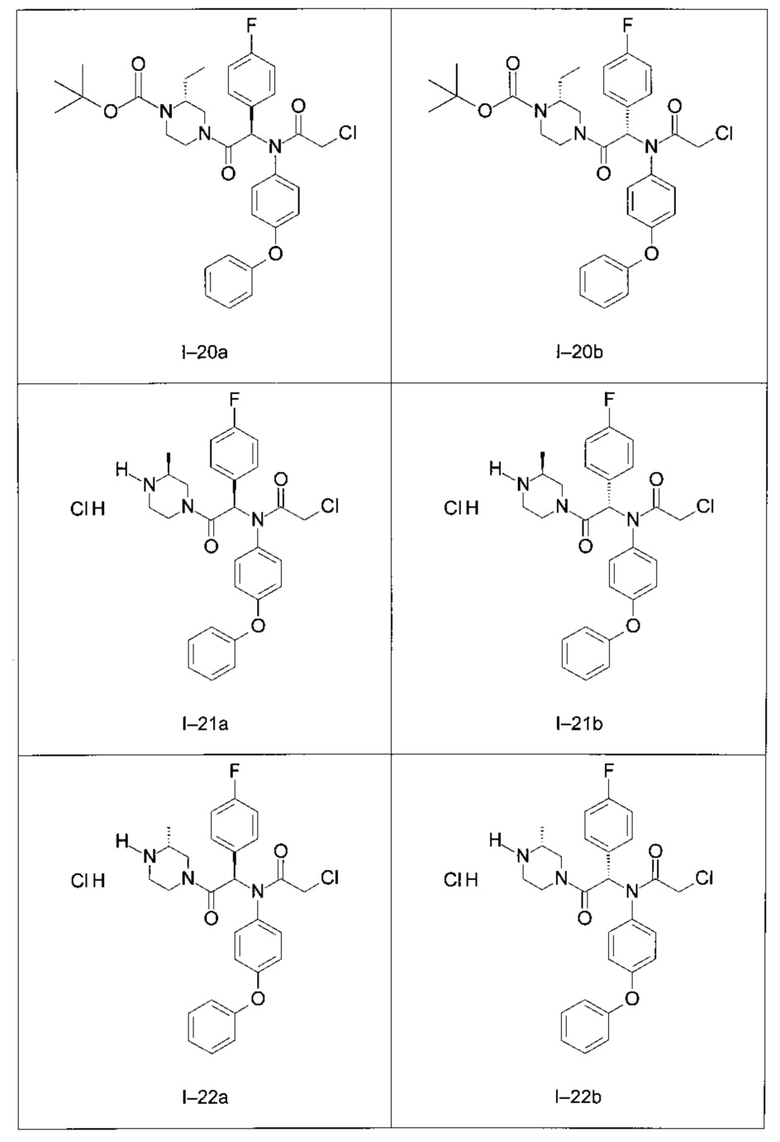
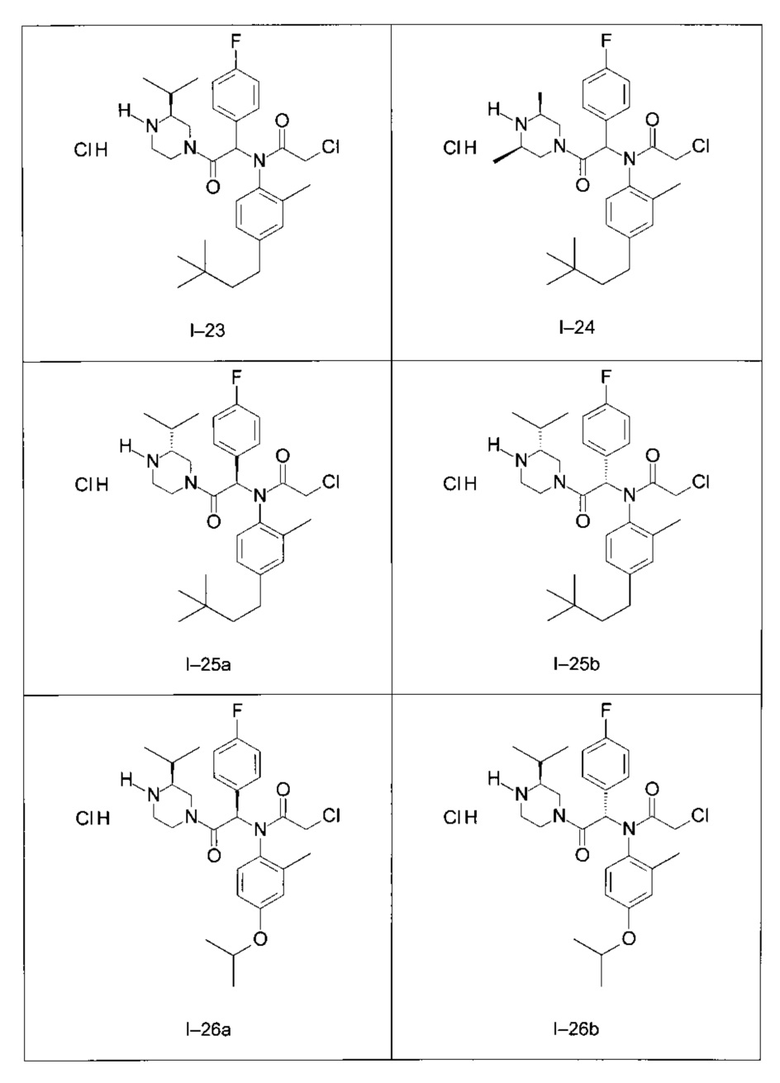
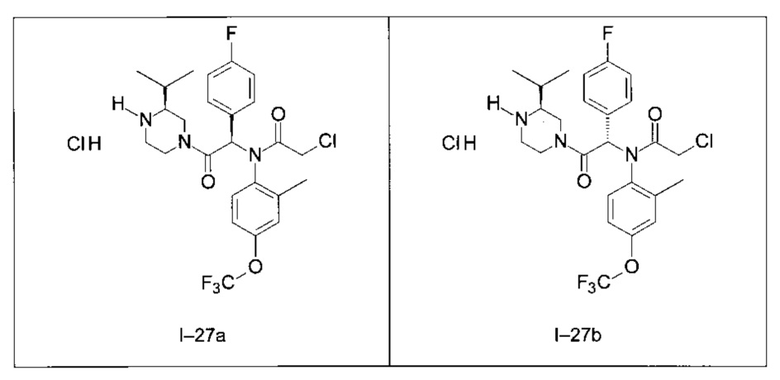
Следующие примеры были получены при замещении этилового эфира (4-фторфенил)-оксо-уксусной кислоты этилтиофен-2-глиоксатом, используя описанные выше способы работы.

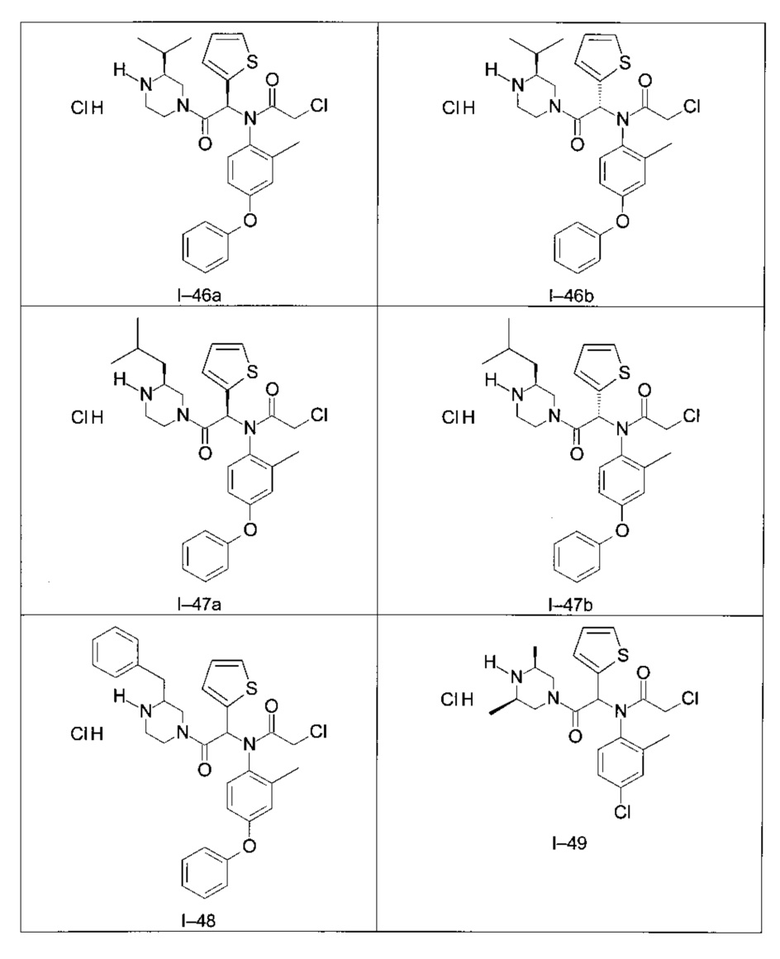
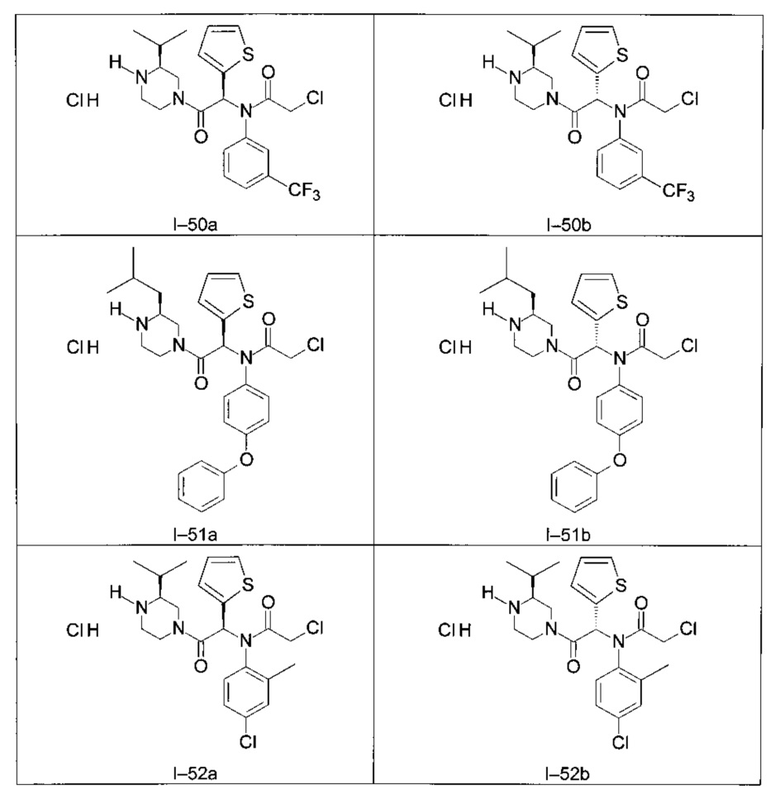
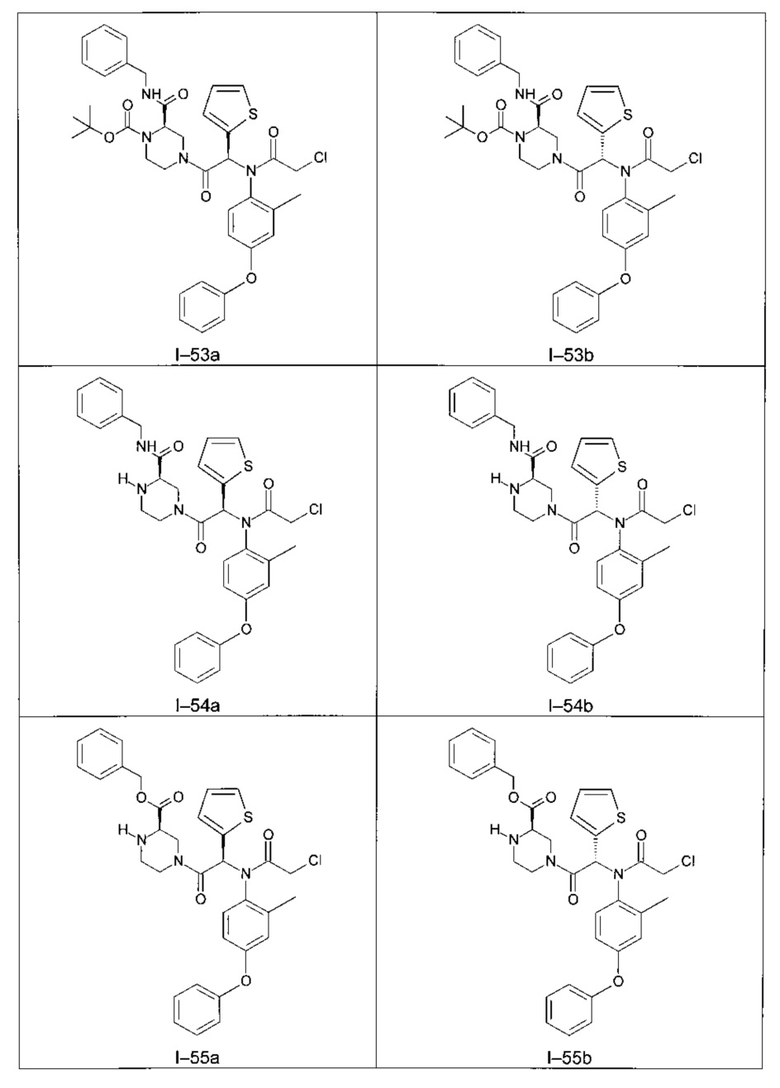
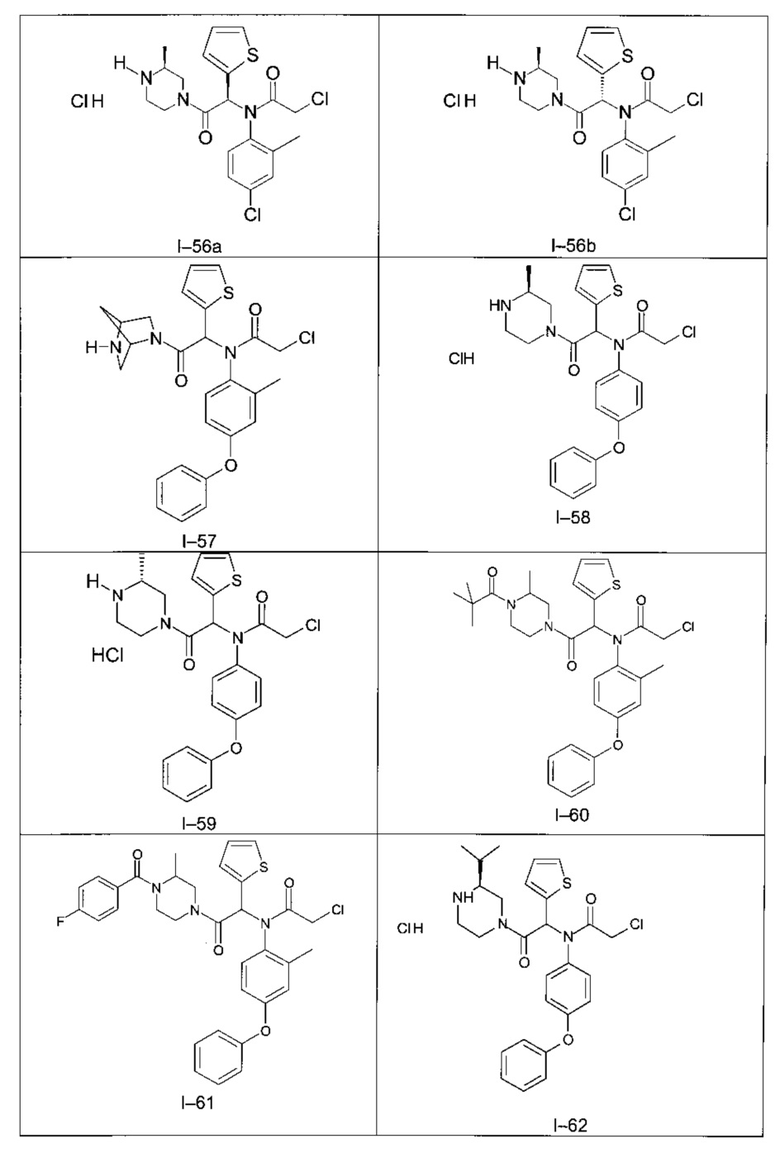
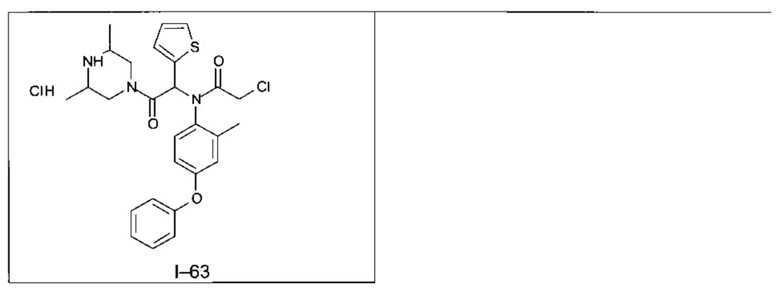
II - Фармакологическое исследование соединений по настоящему изобретению
1) Исследования цитотоксичности
Влияние соединений по изобретению на пролиферацию раковых клеток было изучено на различных линиях раковых клеток из различных тканей (MCF-7: рак молочной железы, MCF-7/adr- адриамицин-устойчивый рак молочной железы, HL-60: острая промиелоцитарная лейкемия, HL-60/R10: доксорубицин-устойчивая острая промиелоцитарная лейкемия, НТ29: аденокарцинома толстой кишки, Mia Раса2: опухоль поджелудочной железы, Panc-1: экзокринная опухоль поджелудочной железы, SK-OV-3: циспластин- и адриамицин-устойчивый рак яичников). Использовавшиеся в настоящем исследовании раковые клетки инкубировали при 37°С в присутствии одного из соединений по изобретению, которое добавляли к культуральной среде в различных концентрациях.
Раковые клеточные линии были получены в АТСС (Американская коллекция типовых культур, линия MCF-7), в Госпитале Питье Сальпетриер (Hôpital de la Pitié Salpetrière, линия MCF-7/adr), и в компании "Онкодизайн" (Oncodesign, Дижон, Франция, линии HL-60, HL-60/R10, НТ29, MiaPaCa2, Panc-1 и SK-OV-3). Клетки выращивали в среде RPMI 1640, содержащей 2 мМ L-глутамина с 10% сывороткой эмбриона теленка ("Лонза", Вервье, Бельгия). Все клеточные линии растили при 37°С во влажной атмосфере, содержащей 5% CO2. Пролиферацию клеток анализировали с использованием набора ViaLight® Plus Assay Kit ("Лонза"; Вервье, Бельгия) в соответствии с инструкциями производителя. Клетки засеяли в 96-луночные культуральные планшеты, на которых возможно определять люминесценцию (белые планшеты с прозрачным дном) в пропорции от 5000 до 10000 клеток на лунку в 100 мкл культуральной среды. После преинкубации в течение 24 часов при 37°С, соединения по изобретению растворили в диметилсульфоксиде (ДМСО) и по отдельности добавили в каждую лунку в пропорции 0,5 мкл на лунку. Через 72 часа инкубации при 37°С во влажной атмосфере с 5% CO2 в каждую лунку добавили по 50 мкл лизирующего буфера и через 15 минут еще по 100 мкл агента для определения АТФ. Чтобы определить выживаемость клеток, люминесценция планшетов была измерена с использованием люминометра. Полученные данные обработали на компьютере с получением значений ЕС50, то есть концентрации каждого соединения, индуцирующую 50% выживаемость клеток, по сравнению с контрольным значением (0,5% одного ДМСО).
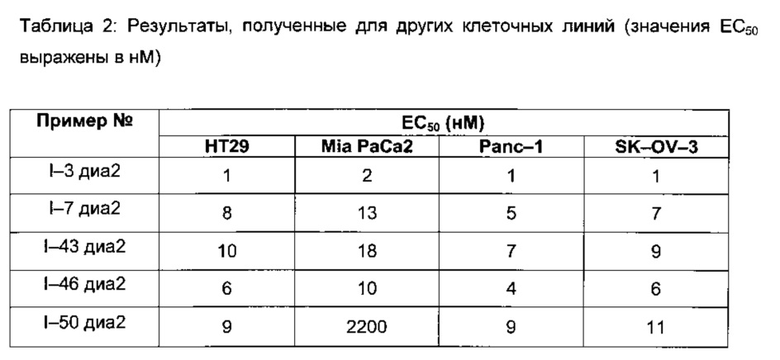
Полученные результаты приведены в следующих таблицах 1 и 2.
Таблица 1: Результаты, полученные для клеточных линий HL-60, HL60/R10, MCF-7 and MCF-7/adr (значения ЕС50 выражены в нМ)
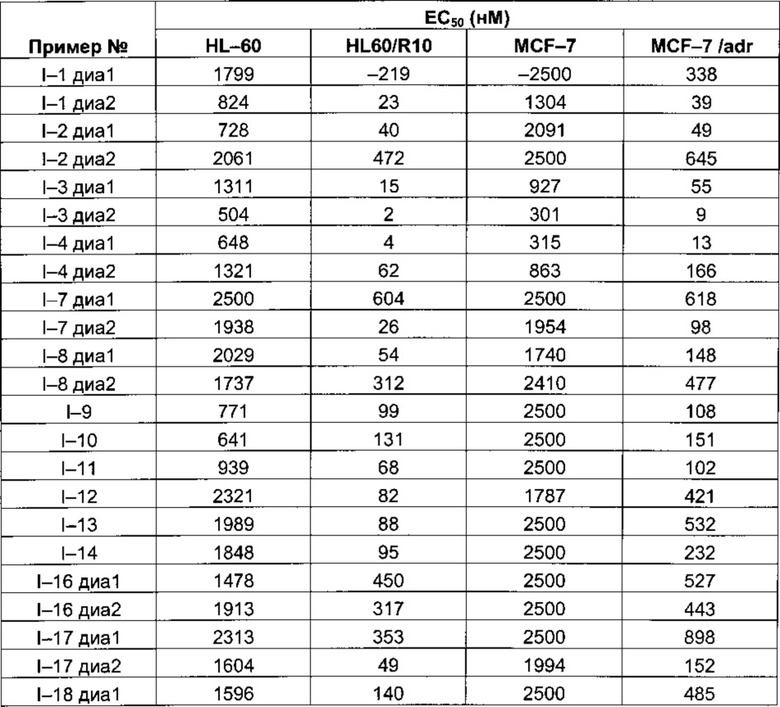
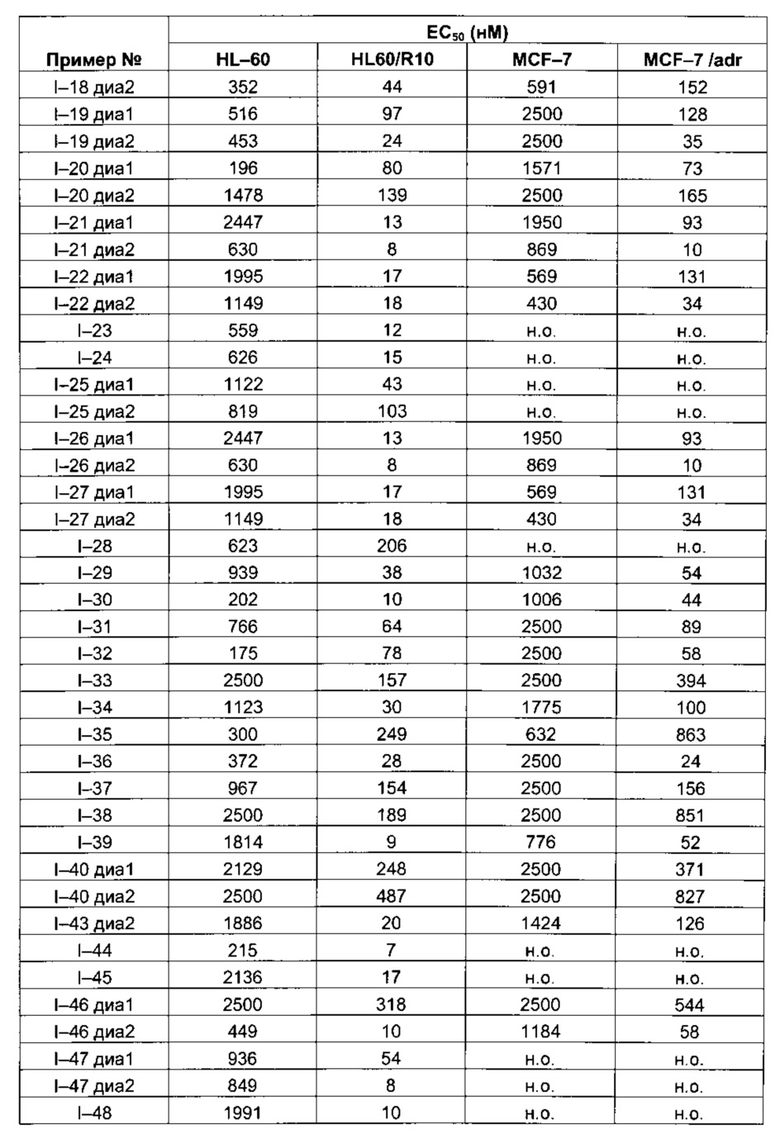
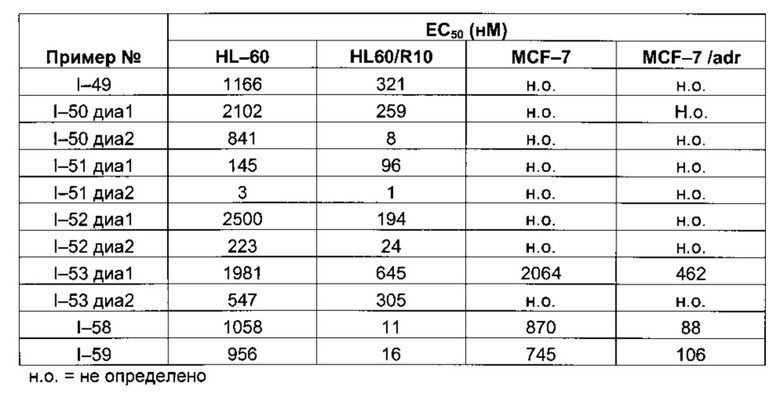
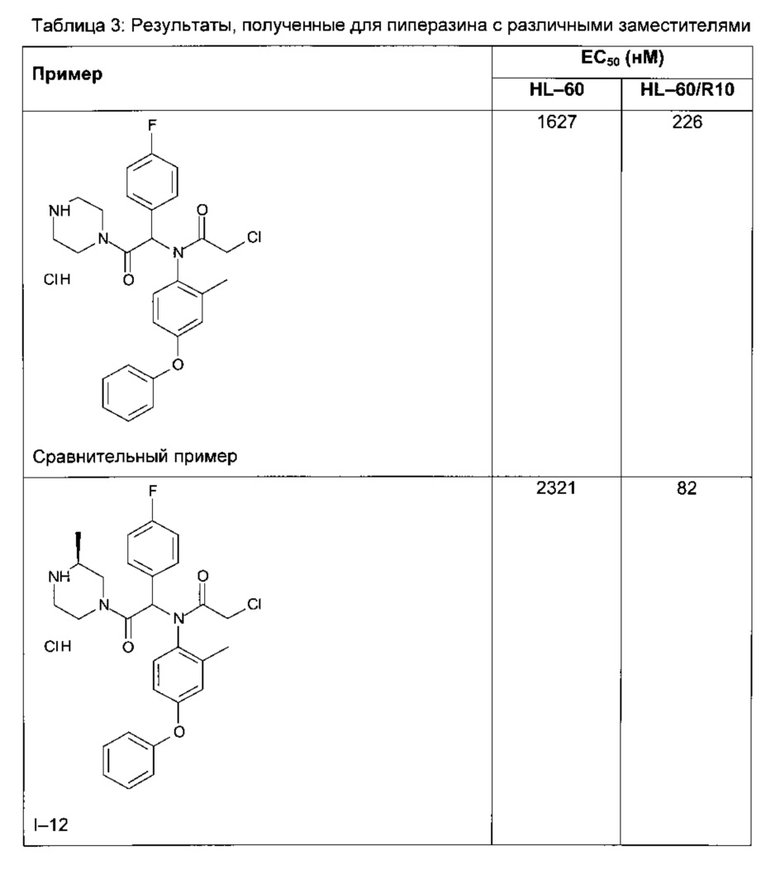
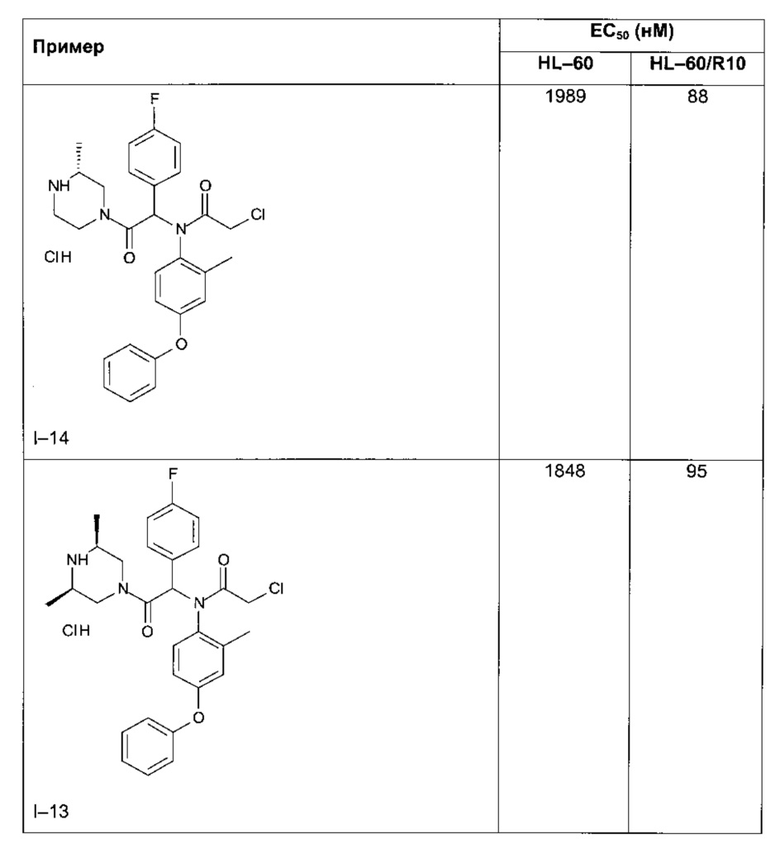
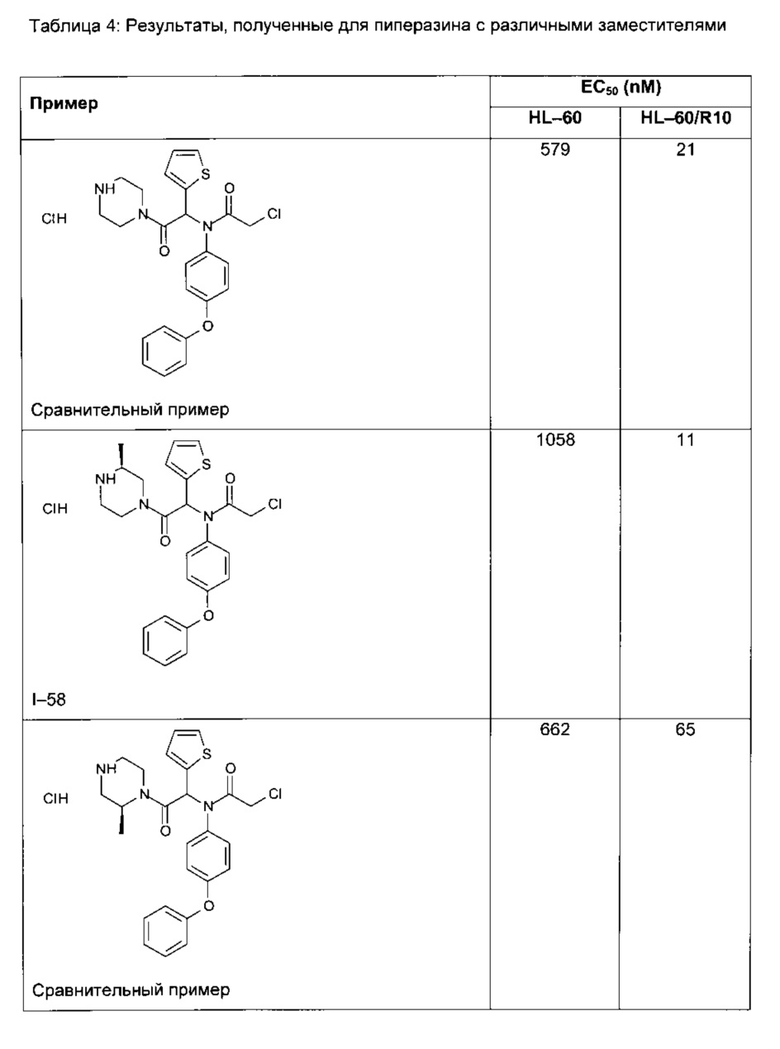
Следующие далее таблицы 3 и 4 иллюстрируют усиление цитотоксической активности по отношению к устойчивой линии HL60/R10, причем активность измерялась для соединений, содержащих пиперазин, замещенный в альфа-положении азота 4 пиперазинового цикла, по сравнению с пиперазином, незамещенным и/или замещенным в других положениях. Наилучшая цитотоксическая активность получена для абсолютной конфигурации (S) такого замещения.
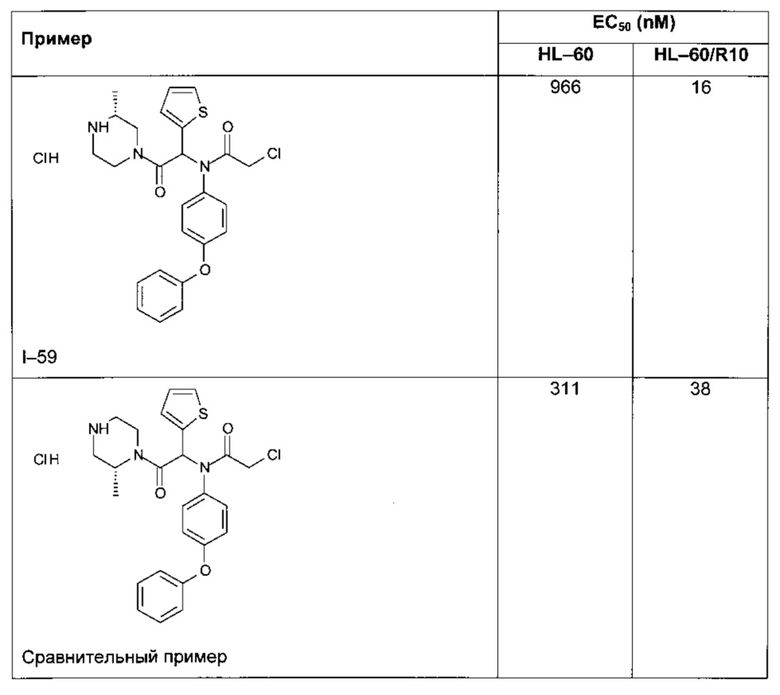
2) Определение растворимости в воде
Растворимость в воде является основным физико-химическим параметром, над которым работают при улучшении свойств ADME (Поглощение, Распределение, Метаболизм и Выведение) молекулы (Drug-like properties: concepts, structure design and methods, Edward Harvel Kerns, Li Di; Academic Press, 2008).
Водную растворимость всех соединений измеряли при рН 7,4. Ее определяли, проводя ВЭЖХ супернатанта, который был получен после центрифугирования насыщенного раствора, а его, в свою очередь, готовили 24 часа перемешивая избыток вещества в среде при температуре 20°С. Приготовление и обработка образцов были роботизированы.
В таблице 5 демонстрируется более высокая водная растворимость соединения I-58 по изобретению, по сравнению с пиперазинами, незамещенными или замещенными по другим положениям.
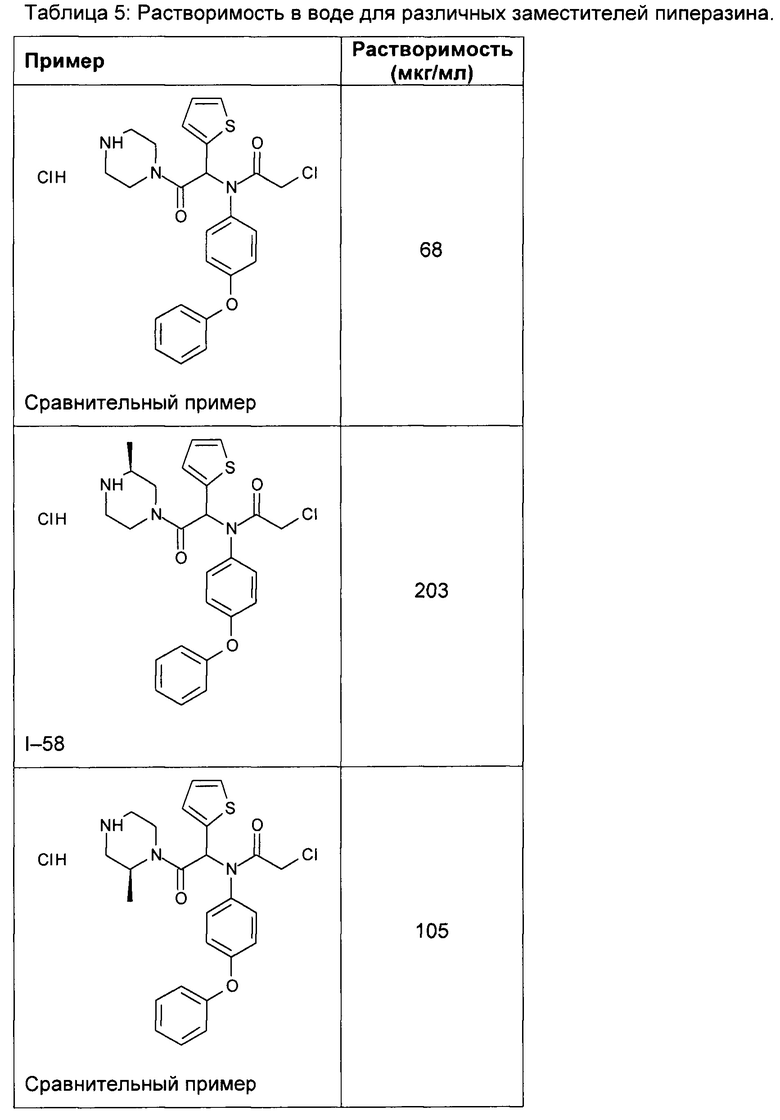
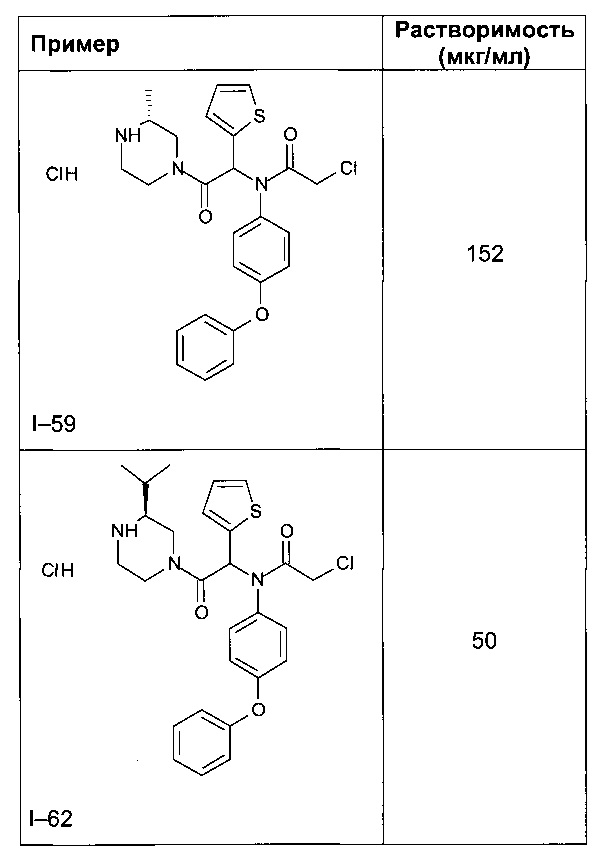
3) Фармакокинетические характеристики соединений для мышей
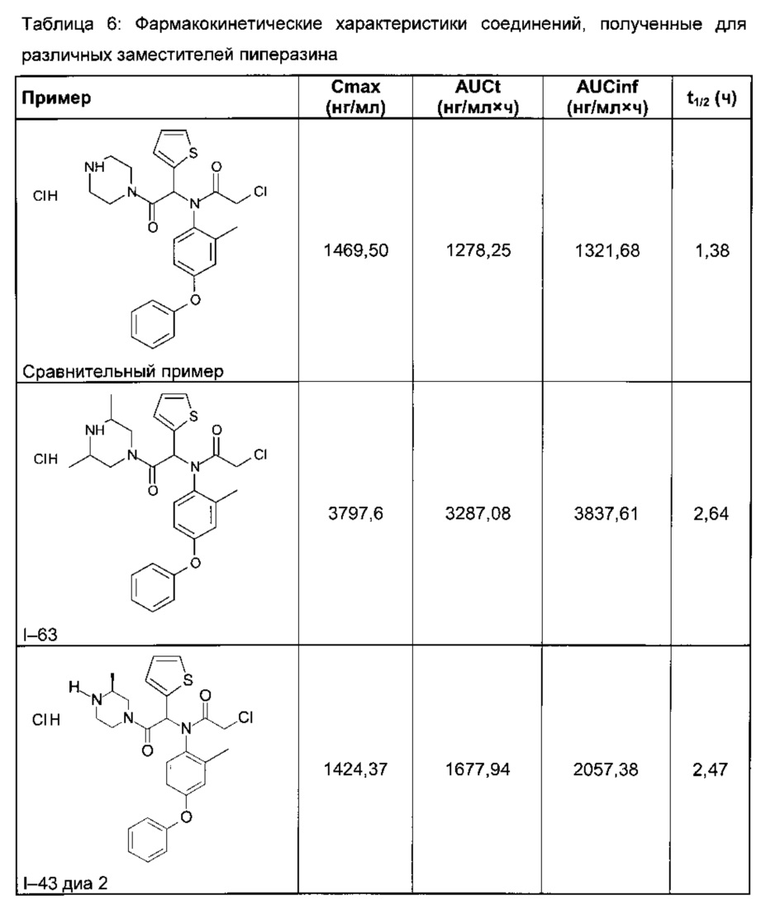
Знание фармакокинетики соединений необходимо для их разумного использования в экспериментах in vivo. Растворы соединений в ДМСО вводили внутривенно (ВВ) или перорально (ПО) мышам линии balb/c. В различные промежутки времени от 5 минут до 6 часов после введения у животных брали кровь, отделяли плазму, и определяли в ней концентрацию соединений методом ЖХ/МС/МС. Полученные данные позволили построить график зависимости концентрации от времени и определить фундаментальные характеристики, такие как период полужизни соединения в плазме (Т½), площадь под кривой на данный момент времени (AUCt) и максимальную достигнутую концентрацию (Сmах). В таблице 6 показано, насколько заместители пиперазина улучшают фармакокинетические характеристики соединений, вводимых внутривенно в дозе 10 мг/кг.
На Фигуре 1 представлены кривые зависимости концентрации в плазме от времени после введения соединения I-43 диа2 по ВВ и ПО маршрутам. Соединение I-43 диа2 демонстрирует на мышах хорошую биодоступность, особенно при пероральном введении.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРИДИНА ДЛЯ ЛЕЧЕНИЯ МЕТАБОЛИЧЕСКИХ НАРУШЕНИЙ, СВЯЗАННЫХ С УСТОЙЧИВОСТЬЮ К ДЕЙСТВИЮ ИНСУЛИНА ИЛИ ГИПЕРГЛИКЕМИЕЙ | 2007 |
|
RU2448093C2 |
| ПОДГОТОВКА И ИСПОЛЬЗОВАНИЕ ИНГИБИТОРА КИНАЗЫ | 2016 |
|
RU2691401C2 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2690853C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2018 |
|
RU2797808C2 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2760554C1 |
| ЛИЗИНСПЕЦИФИЧЕСКИЕ ИНГИБИТОРЫ ДЕМЕТИЛАЗЫ-1 И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2602814C2 |
| НОВЫЕ ГИДРОКСИСЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2734418C2 |
| НОВЫЕ ГИДРОКСИКИСЛОТНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2745430C1 |
| ПИРИМИДИЛЦИКЛОПЕНТАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ AKT-ПРОТЕИНКИНАЗЫ | 2008 |
|
RU2486178C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИСПИРОИНДОЛИНОНОВ | 2018 |
|
RU2682678C1 |
Изобретение относится к соединению общей формулы (I)
в которой X представляет собой группу (С1-С6)алкил, фенил, бензил, C(O)OR5 или C(O)NHR5; R1 представляет собой атом водорода или группу C(O)R6 или C(O)OR6; R2 представляет собой атом водорода или группу (С1-С6)алкил; или R2 совместно с R1 или X образуют насыщенную углеводородную цепь, которая образует 5- или 6-членный цикл; R3 представляет собой атом водорода или группу (С1-С6)алкил; R4 представляет собой атом галогена или группу (С1-С6)алкил, (C1-С6)алкокси или фенилокси, причем указанная группа возможно замещена одним или более атомами галогена; Ar представляет собой тиофенильную или фенильную группу, возможно замещенную одним атомом галогена; и R5 и R6 независимо друг от друга представляют собой группу (С1-С6)алкил, фенил-(С1-С6)алкил или фенил, возможно замещенный одним атомом галогена. Изобретение также относится к фармацевтической композиции и к способу получения соединений общей формулы (I). Технический результат: получены новые пиперазиниловые соединения общей формулы (I), которые могут применяться при лечении или профилактике рака молочной железы, лейкемии, рака толстой кишки, рака поджелудочной железы и рака яичников. 3 н. и 9 з.п. ф-лы, 1 ил., 6 табл., 8 пр.
1. Соединение следующей общей формулы (I)
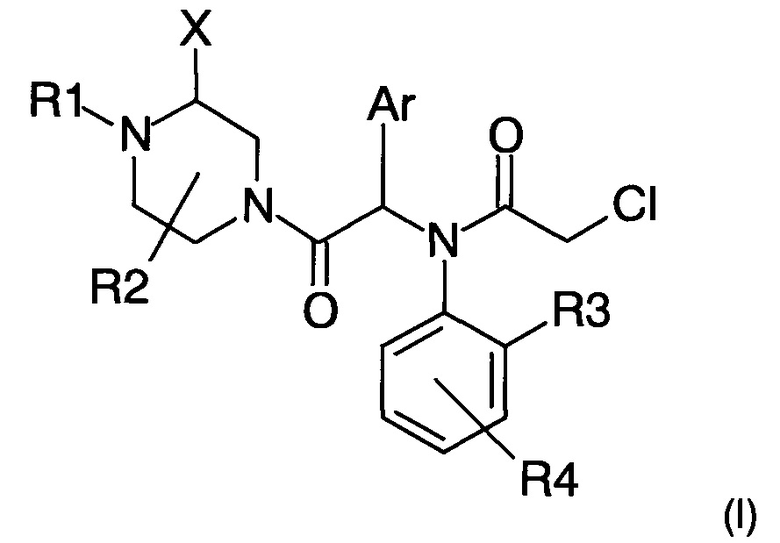
и его фармацевтически приемлемые соли, стереоизомеры или смеси стереоизомеров в любом соотношении, в частности смесь энантиомеров и в особенности рацемическая смесь, где
- X представляет собой группу (С1-С6)алкил, фенил, бензил, C(O)OR5 или C(O)NHR5;
- R1 представляет собой атом водорода или группу C(O)R6 или C(O)OR6;
- R2 представляет собой атом водорода или группу (С1-С6)алкил; или R2 совместно с R1 или X образуют насыщенную углеводородную цепь, которая образует 5- или 6-членный цикл, в частности 5-членный цикл;
- R3 представляет собой атом водорода или группу (С1-С6)алкил;
- R4 представляет собой атом галогена или группу (С1-С6)алкил, (C1-С6)алкокси или фенилокси, причем указанная группа возможно замещена одним или более атомами галогена;
- Ar представляет собой тиофенильную или фенильную группу, возможно замещенную одним атомом галогена; и
- R5 и R6 независимо друг от друга представляют собой группу (С1-С6)алкил, фенил-(С1-С6)алкил или фенил, возможно замещенный одним атомом галогена.
2. Соединение по п. 1, соответствующее следующей формуле (I-bis)
3. Соединение по п. 1, где Ar представляет собой тиофенильную или фенильную группу, замещенную одним атомом фтора, такую как 4-фторфенил.
4. Соединение по п. 1, где R4 представляет собой атом галогена или группу (С1-С6)алкил, (С1-С6)алкокси или фенилокси, причем указанная группа возможно замещена одним или более атомами фтора.
5. Соединение по п. 1, где R3 представляет собой атом водорода или метил.
6. Соединение по п. 1, где X представляет собой группу (С1-С6)алкил, фенил или бензил; R1 и R2 представляют собой атом водорода; R3 представляет собой атом водорода или группу (С1-С6)алкил, в частности (С1-С3)алкил; R4 представляет собой атом галогена или группу (С1-С6)алкил, (С1-С6)алкокси или фенилокси, причем указанная группа возможно замещена одним или более атомами галогена; Ar представляет собой тиофенильную или фенильную группу, возможно замещенную одним атомом фтора, такую как 4-фторфенил; и R5 и R6 независимо друг от друга представляют собой группу (С1-С6)алкил или фенил-(С1-С6)алкил.
7. Соединение по п. 1, выбранное из следующих соединений:

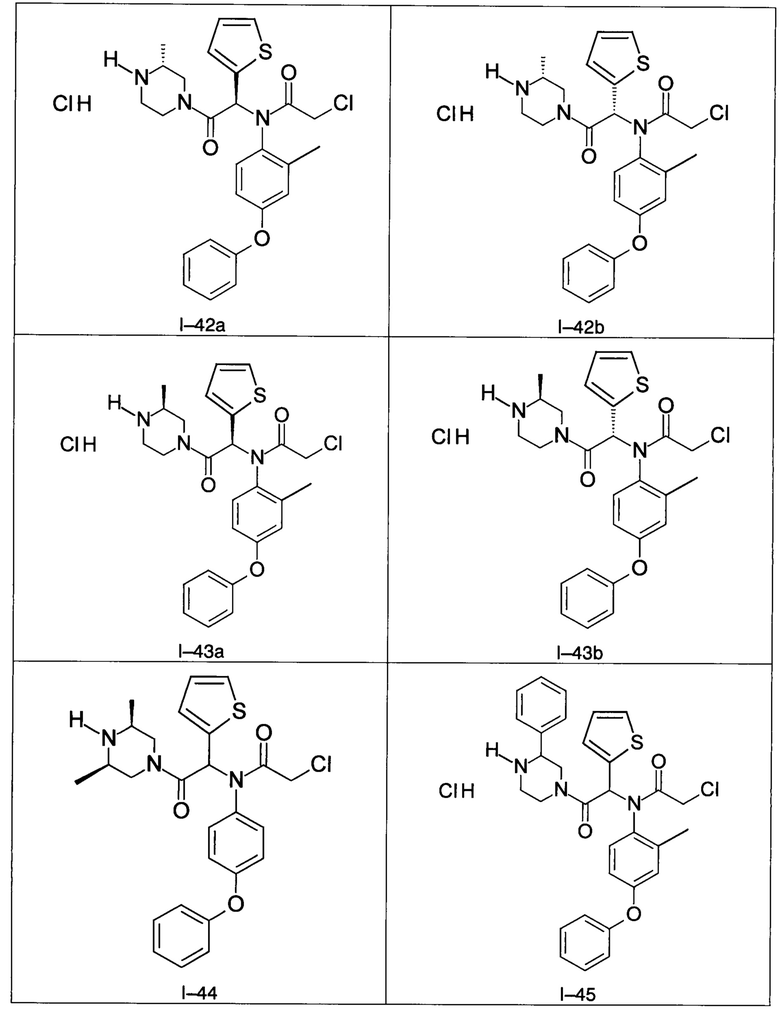
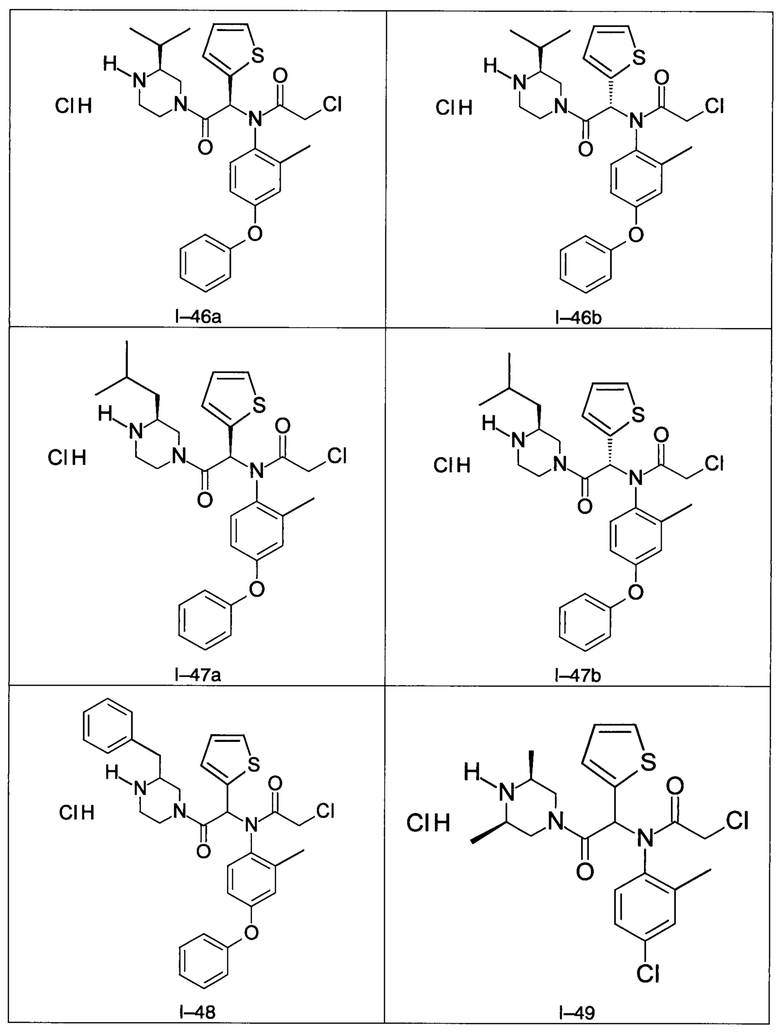
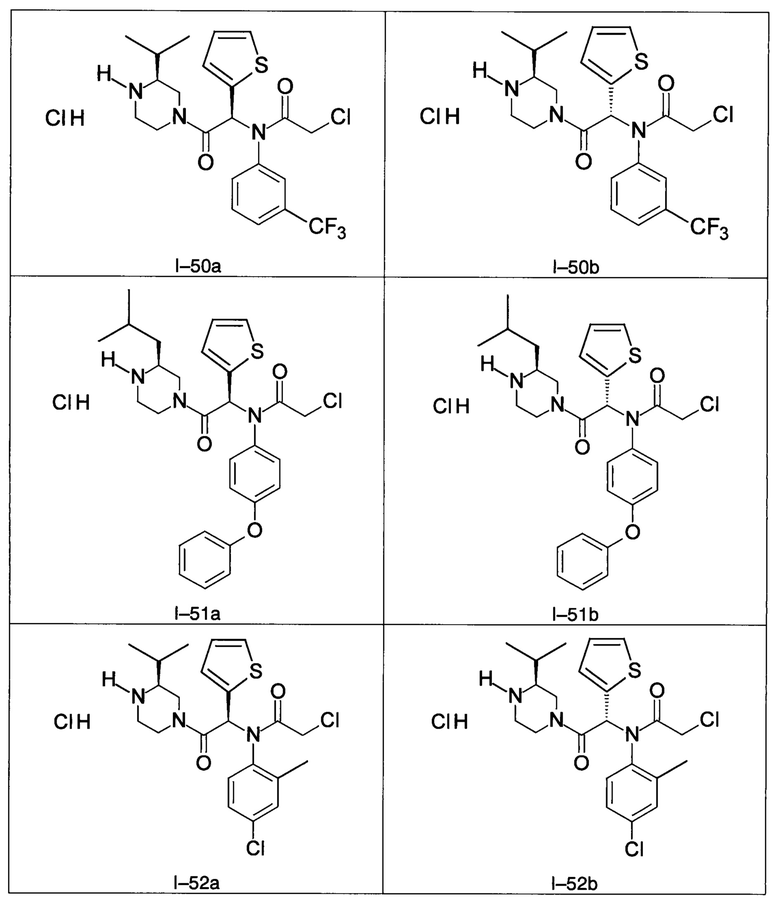
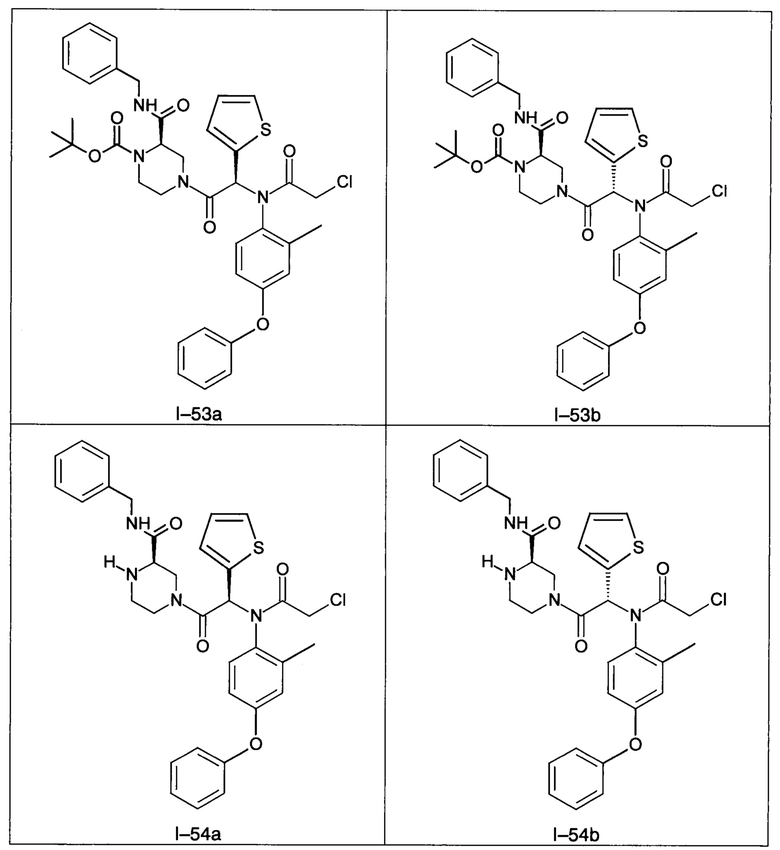
8. Соединение общей формулы (I) по любому из пп. 1-7 для применения в лечении или профилактике рака, а также рака, устойчивого к химиотерапии, где рак выбран из группы, состоящей из рака молочной железы, лейкемии, рака толстой кишки, рака поджелудочной железы и рака яичников.
9. Фармацевтическая композиция для лечения или профилактики рака, выбранного из группы, состоящей из рака молочной железы, лейкемии, рака толстой кишки, рака поджелудочной железы и рака яичников, содержащая по меньшей мере одно соединение общей формулы (I) по п. 1 совместно с одним или более фармацевтически приемлемыми эксципиентами.
10. Фармацевтическая композиция по п. 9 для применения в качестве лекарства для лечения или профилактики рака, в особенности для лечения рака, устойчивого к химиотерапии, где рак выбран из группы, состоящей из рака молочной железы, лейкемии, рака толстой кишки, рака поджелудочной железы и рака яичников.
11. Способ получения соединения формулы (I) по любому из пп. 1-7, включающий следующие последовательные этапы:
a) взаимодействие амина следующей формулы (II)
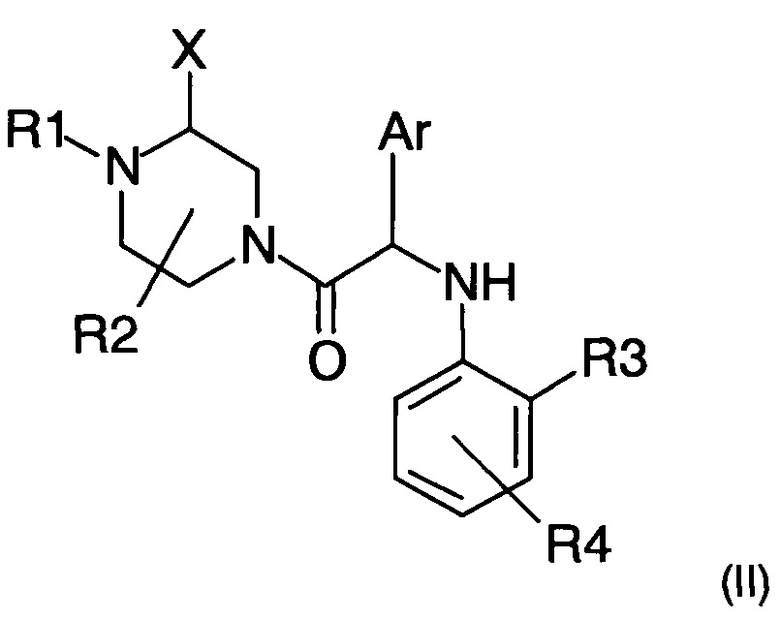
где X, R1, R2, R3, R4 и Ar представляют собой такие, как определено в п. 1, R1 не является атомом водорода;
с хлорацетилхлоридом в присутствии основания с получением соединения формулы (I), где R1≠H; и
b) возможное снятие защиты с атома азота, несущего группу R1≠Н, с получением соединения формулы (I), где R1=Н.
12. Способ по п. 11, включающий следующие последовательные этапы:
i) взаимодействие кетоэфира следующей формулы (V)
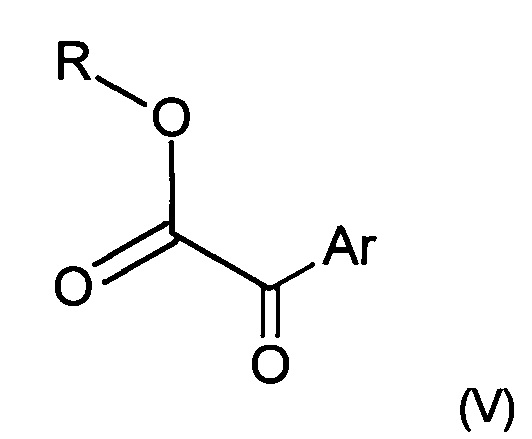
где Ar представляет собой такой, как определен в п. 1, a R представляет собой группу (С1-С6)алкил, такую как этил,
с анилином следующей формулы (VI)
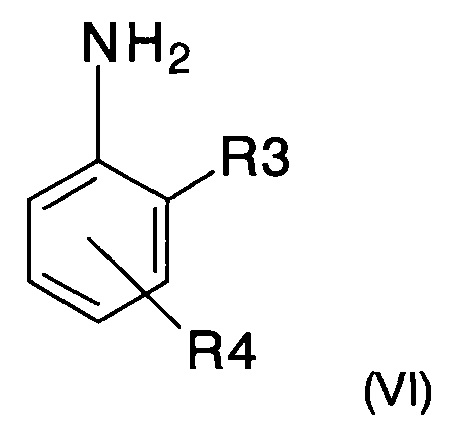
где R3 и R4 представляют собой такие, как определено в п. 1,
с получением имина следующей формулы (VII):
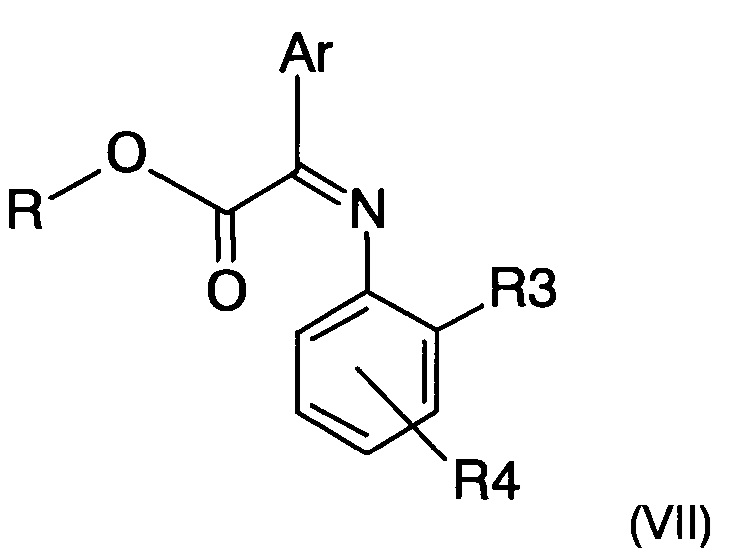
где R, R3, R4 и Ar представляют собой такие, как определено в п. 1;
ii) восстановление имина формулы (VII), полученного на предыдущем этапе, с получением амина следующей формулы (VIII)
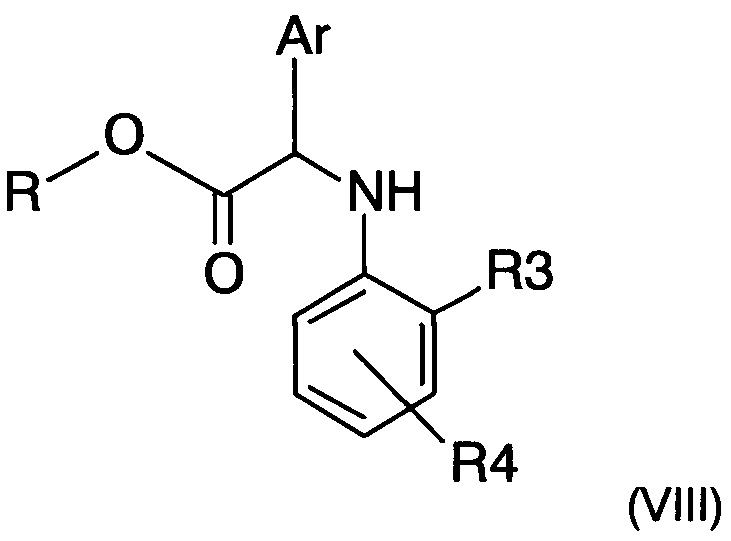
где R, R3, R4 и Ar представляют собой такие, как определено в п. 1; и
iii) сапонификация эфирной функциональной группы соединения формулы (VIII), полученного на предыдущем этапе, с получением кислоты следующей формулы (IV)
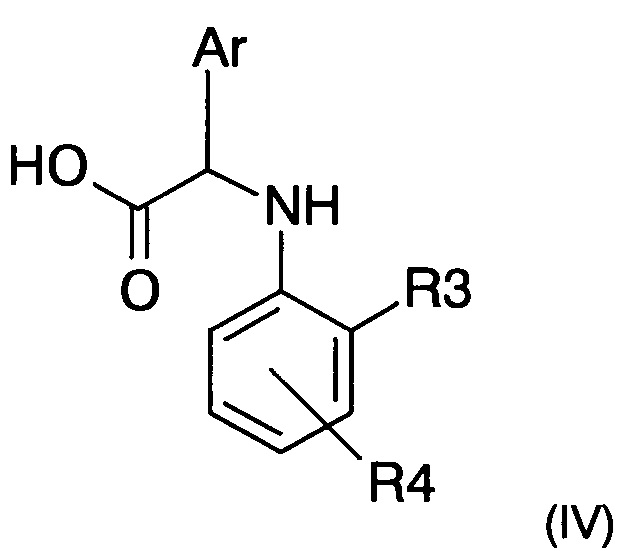
где R3, R4 и Ar представляют собой такие, как определено в п. 1;
iv) взаимодействие кислоты формулы (IV), полученной на предыдущем этапе, с пиперазином следующей формулы (III)
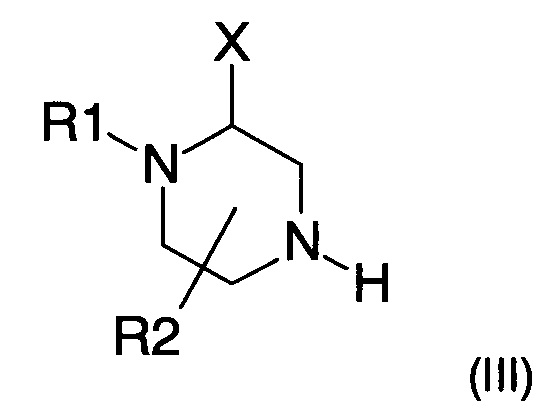
где X, R1 и R2 представляют собой такие, как определено в п. 1, R1 не является атомом водорода,
с получением амина формулы (II) в соответствии с п. 11;
v) взаимодействие амина формулы (II), полученного на предыдущем этапе, с хлорацетилхлоридом в присутствии основания с получением соединения формулы (I), где R1≠Н; и
vi) возможное снятие защиты с атома азота, несущего группу R1≠Н, с получением соединения формулы (I), где R1=Н.
| WO 2009150248 A1, 17.12.2009 | |||
| WO 2007087585 A1, 02.08.2007 | |||
| RU 2006125380 А, 27.01.2008. |

