ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицинской химии. В частности, настоящее изобретение относится к приготовлению и использованию нового ингибитора киназы.
УРОВЕНЬ ТЕХНИКИ
Нарушение клеточного цикла является признаком рака. Циклин-зависимая киназа (CDK) представляет собой класс серин/треонинкиназ, играющих центральную роль в клеточном цикле, участвуя в активации, развитии и окончании клеточного цикла. Семейство CDK включает в себя киназы CDK1-13.
Во многих случаях рака киназа CDK4/6 проявляет себя сверхактивно, что приводит к неконтролируемости пролиферации клеток. Исследования показывают, что рак пищевода может сопровождаться сверхэкспрессией CyclinD1 и CDK4, и повышение экспрессии обоих зависит от степени дифференциации рака пищевода. Обычно экспрессия киназы CDK4 наблюдается как при доброкачественных, так и при злокачественных эндокринных опухолях поджелудочной железы. Уровень экспрессии CDK4 в легочных раковых тканях также значительно выше, чем в нормальных тканях легких. Степень высокой положительной экспрессии положительно коррелирует с гистопатологической классификацией рака легких, лимфатическим метастазированием и злокачественным развитием опухоли на клинической стадии, что является потенциально неблагоприятным прогностическим фактором. Сверхэкспрессия киназы CDK6 наблюдается во множестве опухолевых клеток, например, обнаружено, что CDK6 сверхэкспрессирует в мужских гормон-чувствительных клеточных линиях рака предстательной железы. Экзогенная сверхэкспрессия CDK6 приводит к ускоренному росту опухолевой клетки, в то время как скорость роста опухолевых клеток, конфликтующих с CDK6, значительно меньше.
CDK4 и CDK6 имеют 71%-ную гомологию с составом аминокислоты, что говорит об их функциональном сходстве. Недавно проведенные исследования также показывают, что вещество CDK4/6-CyclinD может фосфорилировать транскрипционный фактор FOXM1, повышая его стабильность и активность в меланоме. Таким образом, ингибирование киназы CDK4/6 может приводить к ингибированию пролиферации клеток, начиная с конца сигнального пути. Сочетание применения ингибиторов CDK4/6 и эндокринной терапии может достигать двойного ингибирующего эффекта, и доклинические исследования также подтверждает, что данное сочетание имеет значительный синергетический эффект.
За последние 20 лет ингибиторы семейства CDK получили широкое распространение в качестве потенциальной мишени при лечении опухолей. Однако первое поколение ингибиторов CDK не обладает способностью к избирательной активации и является пан-ингибитором. Примером является флавопиридол, который может ингибировать киназы CDK1, CDK2, CDK4, CDK6, CDK7 и CDK9. Несмотря на то, что флавопиридол способен вызывать остановку клеточного цикла и проявлять цитотоксичность, его клиническая эффективность неудовлетворительна. Второе поколение ингибиторов CDK выполнено с возможностью повышения избирательной способности, в частности, значительное внимание было обращено на селективные ингибиторы, адресно воздействующие на CDK4/6, которые обладают большей клинической эффективностью и меньшими побочными эффектами токсичного воздействия. Двойной ингибитор киназ CDK4/6 Palbociclib (торговое название Ibrance) компании Pfizer стал первым зарегистрированным ингибитором киназ CDK4/6 и был одобрен FDA (Управление по контролю за продуктами и лекарствами) как препарат первой линии для лечения ER-положительного НЕR2-негативного рака молочной железы.
Препарат Novartis LEE011 - это двойной ингибитор киназ CDK4/CDK6, проявляющий наибольшую чувствительность в случаях злокачественной рабдоидной опухоли и нейробластомы. LEE011 в основном используется в сочетании с ингибиторами ароматазы и ингибиторами PI3K и может проявлять большую противоопухолевую активность во время клинических испытаний. Для лечения метастатического HR-положительного/HER2-негативного рака молочной железы на стадии III LEE011 используется в сочетании с летрозолом. Для лечения метастатического HR-положительного рака молочной железы на стадии Ib/II LEE011 используется в сочетании с BYL719 и летрозолом.
Кроме того, клиническое исследование стадии III проходит препарат LY-2835219 компании Lilly. Если данные клинические испытания достигнут желаемых результатов, то двойной ингибитор киназ CDK4/CDK6 обеспечит благоприятные показатели выживаемости большому числу пациентов с распространенным раком молочной железы.
Как было упомянуто выше, разработка селективного двойного ингибитора киназ CDK4/CDK6 является приоритетным направлением в исследованиях противоопухолевых лекарств. Поэтому существует острая потребность в разработке новых ингибиторов киназ CDK.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является разработка принципиально нового ингибитора киназ CDK, метода его приготовления и применения.
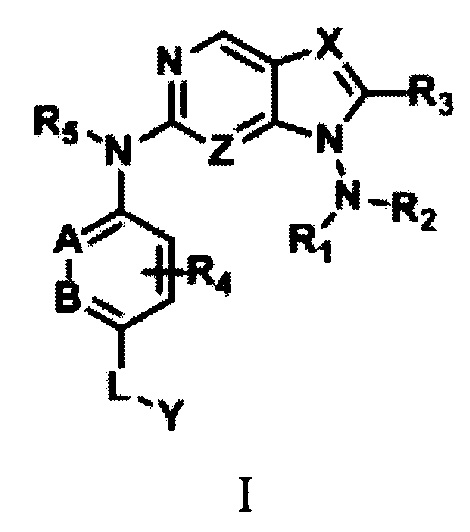
В первом аспекте настоящего изобретения предложено соединение формулы I или его фармацевтически приемлемая соль:
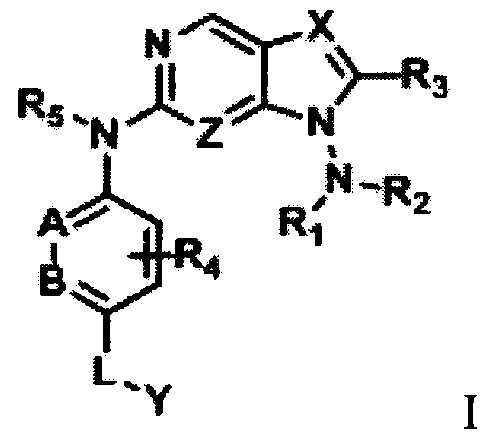
где
R1 и R2 независимо выбраны из Н, замещенного или незамещенного С1-С8-алкила, C(O)OR8, CONR9R10, C(O)R11, замещенного или незамещенного 5-8-членного арила, замещенного или незамещенного 5-8-членного гетероарила, замещенного или незамещенного 3-12-членного насыщенного или ненасыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S; а упомянутый гетероцикл содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S;
кроме того, R1 и R2 могут быть связаны с соседним атомом N, образуя кольцевую структуру, которая включает в себя замещенный или незамещенный 3-12-членный насыщенный или ненасыщенный гетероцикл или кольцо с внутренним мостиком или спиро-кольцо; причем упомянутый гетероцикл относится к кольцевой структуре, содержащей 0-3 гетероатома, выбранных из группы, состоящей из N, О или S, в дополнение к атому азота, присоединенному к исходному ядру;
R3 выбран из Н, замещенного или незамещенного С1-С8-алкила, CN, C(O)OR12, CONR13R14, C(O)R15, замещенного или незамещенного 5-8-членного арила, замещенного или незамещенного 5-8-членного гетероарила, замещенного или незамещенного 3-12-членного насыщенного или ненасыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S; а упомянутый гетероцикл содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S;
R4 выбран из Н, замещенного или незамещенного С1-С4-алкила, замещенного или незамещенного С1-С8-алкокси, галогена, ОН, CN, C(O)OR12, CONR13R14, C(O)R15, замещенного или незамещенного 5-8-членного арила, замещенного или незамещенного 5-8-членного гетероарила, замещенного или незамещенного 3-12-членного насыщенного или ненасыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S; а упомянутый гетероцикл содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S;
R5 выбран из Н или С1-С4-алкила;
X - это CR16 или N;
А, В и Z независимо выбраны из N или CR16;
R16 - это Н, С1-С4-алкил или С1-С4-галогеналкил;
L выбран из группы, состоящей из ничего, С1-С6-алкилена, С(О), CONR17 или S(O)2;
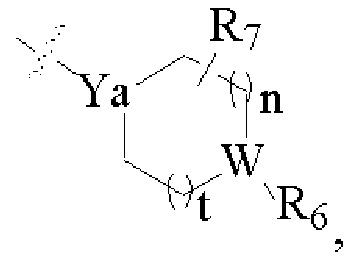
Y - это Н, R18, NR19R20, ОН, или Y выбран из части группы, состоящей из:

где
R6 - ничто, Н, замещенный или незамещенный С1-С8-алкил, замещенный или незамещенный С1-С8-алкокси, замещенный или незамещенный С2-С6-ацил, замещенный или незамещенный С2-С6-сульфонил, замещенный или незамещенный С1-С6-гидроксиалкилен, CONR22R23 или C(O)R24;
R7 может быть заместителями 0-3, a R7 - это замещенный или незамещенный С1-С8-алкил, кислород или галоген; или два или более R7 образуют циклоалкил с внутренним мостиком; W - это CR21, N или О (когда W - О, R6 отсутствует);
Ya - это CR21 или N; R21 - это Н или галоген;
R8, R9, R10, R11, R12, R13, R14, R15, R17, R18, R19, R20, R22, R23 и R24 независимо выбраны из Н, замещенного или незамещенного С1-С8-алкила, замещенного или незамещенного С1-С8-алкокси, замещенного или незамещенного С1-С6-алкиленомино, замещенного или незамещенного С1-С6-алкиленгидрокси, замещенного или незамещенного 5-8-членного арила, замещенного или незамещенного 5-8-членного гетероарила, замещенного или незамещенного 3-12-членного насыщенного или ненасыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из N, О или S; а упомянутый гетероцикл содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из N, О или S;
n и t - 0, 1 или 2 соответственно;
во всех упомянутых веществах определение «замещенный» означает, что один или несколько атомов водорода в группе замещены заместителем (заместителями), выбранными из группы, состоящей из галогена, ОН, NH2, CN, незамещенного или галогенированного С1-С8-алкила, С1-С8-алкокси, незамещенного или галогенированного С2-С6-алкенила, незамещенного или галогенированного С2-С6-алкинила, незамещенного или галогенированного С2-С6-ацила, незамещенного или галогенированного 5-8-членного арила, незамещенного или галогенированного 5-8-членного гетероарила, незамещенного или галогенированного 3-12-членного насыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S, а упомянутый гетероцикл содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S.
В другом предпочтительном варианте осуществления изобретения  представляет собой замещенную или незамещенную группу, выбранную из группы, состоящую из
представляет собой замещенную или незамещенную группу, выбранную из группы, состоящую из

где m равно 0, 1, 2, 3, 4, 5, 6, 7, 8 или 9, а используемый термин «замещенный» определяется, как описано выше.
В другом предпочтительном варианте осуществления изобретения R1 и R2 вместе с соседним атомом азота образуют 4-12-членную кольцевую структуру.
В другом предпочтительном варианте осуществления изобретения R1 и R2 вместе с соседним атомом азота образуют 5-7-членную кольцевую структуру.
В другом предпочтительном варианте осуществления изобретения R1 и R2 вместе с соседним атомом азота образуют 6-членную кольцевую структуру.
В другом предпочтительном варианте осуществления изобретения R13 и R14 вместе с соседним атомом азота образуют 4-6-членную кольцевую структуру.
В другом предпочтительном варианте осуществления изобретения, при котором L отсутствует, Y представляет собой 6-членный гетероцикл, содержащий атом азота.
В другом предпочтительном варианте осуществления изобретения А, В, L, X, Y, Z, R1, R2, R3, R4 или R5 представляет собой соответствующую группу в конкретных соединениях, описанных в приведенных примерах.
В другом предпочтительном варианте осуществления изобретения соединение формулы I выглядит следующим образом:
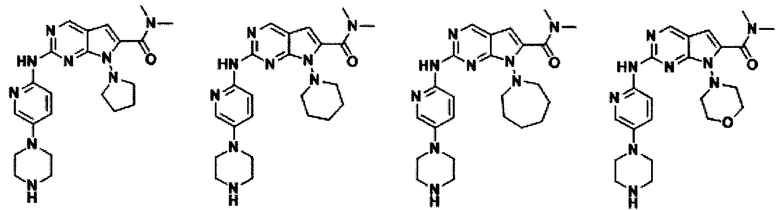



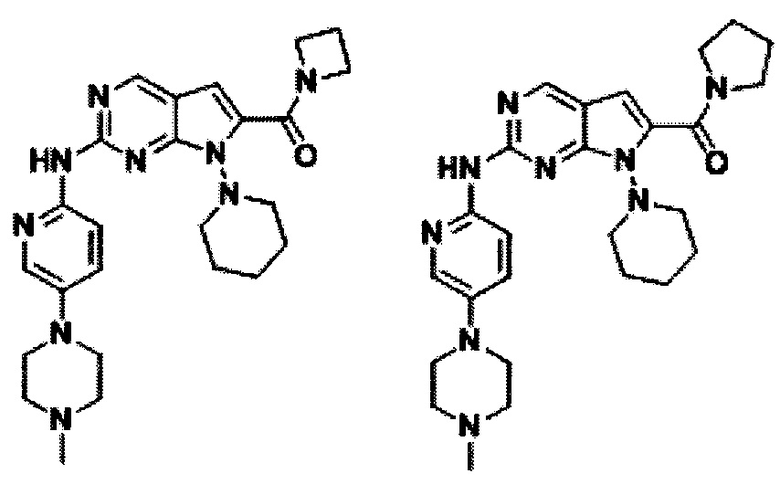


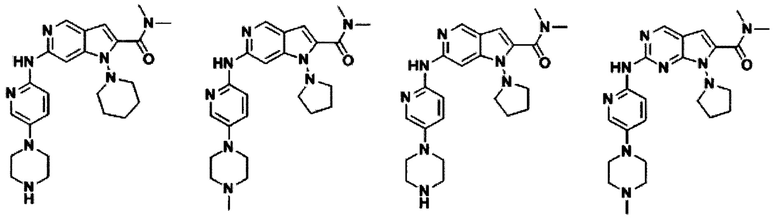


Во втором аспекте настоящего изобретения предлагается способ получения соединения формулы I, описанного в первом аспекте. Данный способ включает в себя следующие этапы:

а) соединение формулы I-6 вступает в реакцию с соединением формулы I-7 в инертном растворителе с образованием соединения формулы I, при этом каждая группа определяется в соответствии с вышеприведенным описанием.
В другом предпочтительном варианте осуществления изобретения инертный растворитель выбирается из группы, включающей толуол, ксилол, диметиловый эфир гликоля, диоксан, тетрагидрофуран (ТГФ), диметилформамид (ДМФА), диметилсульфоксид (ДМСО), N-метилпирролидон или их сочетание.
В другом предпочтительном варианте осуществления изобретения реакцию проводят в присутствии палладиевого катализатора.
В другом предпочтительном варианте осуществления изобретения палладиевый катализатор выбирают из группы, состоящей из Pd(PPh3)4, Pd2(dba)3, Pd(dba)2, Pd(OAc)2, Pd(PPh3)2Cl2, Pd(dppe)Cl2, Pd(dppf)Cl2, Pd(dppf)Cl2⋅CH2Cl2 или их сочетание.
В другом предпочтительном варианте осуществления изобретения реакцию проводят в присутствии лиганда.
В другом предпочтительном варианте осуществления изобретения лиганд представляет собой монодентатный фосфиновый лиганд или бидентатный фосфиновый лиганд; предпочтительно, лиганд выбирают из группы, включающей трифенилфосфин, триметилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, X-Phos, S-Phos, бинафтилдифенилфосфин, 1,1'-бис(дифенилфосфино)ферроцен, 1,2-бис(дифенилфосфино)этан, Xant-Phos или их сочетание.
В другом предпочтительном варианте осуществления изобретения реакцию проводят в присутствии базы.
В другом предпочтительном варианте осуществления изобретения основание выбирают из группы, состоящей из Na2CO3, K2CO3, Cs2CO3, LiHMDS, NaHMDS, KHMDS, трет-бутоксида натрия, трет-бутоксида калия, триэтиламина, диизопропиламина, диизопропилэтиламина или их сочетания.
Предпочтительный способ получения включает следующие этапы:

(1) Соединение A3 может быть получено в результате реакции соединения А1 с соответствующим гидразином А2 в присутствии основания (включая, без ограничений, диизопропилэтиламин, триметиламин) в инертном растворителе (этаноле, ТГФ и т.д.).
(2) Соединение А5 может быть получено в результате реакции сочетания Соногаширы (время реакции составляет 2-8 ч) соединения A3 и соответствующего терминального алкина А4 в инертном растворителе (таком как ТГФ, ДМФ, ДМСО, диоксан и т.д.) в присутствие катализатора (например, тетракис(трифенилфосфин)палладия, трис(дибензилиденацетон)дипалладия (Pd2(dba)3), бис(дибензилиденацетон)палладия, дихлорбис(трифенилфосфин)палладия, бис(три-о-толилфосфин)палладий дихлорида, 1,2-бис(дифенилфосфино)этандихлорпалладия, дихлорид[1,1'-бис(дифенилфосфино)ферроцен]палладия, [1,1'-бис(дифенилфосфино)ферроцен]дихлорметана, комплекса дихлорметана и т.д.), катализатора b (например, йодида меди, хлорида цинка, оксида серебра, карбоната серебра и т.д.) и щелочи (например, карбоната калия, фторида калия, карбоната цезия, фторида цезия, фторида натрия, фосфата калия, гидратированного фосфата калия, карбоната натрия, бикарбоната натрия, 1,8-диазабицикло[5.4.0]ундец-7-ен, триэтиламина, диизопропиламина, диизопропилэтиламина, пиридина или их сочетания и т.д.);
(3) Соединение А6 может быть получено в результате реакции соединения А5 в инертном растворителе (дихлорметане, ТГФ, ацетонитриле и т.д.) при нагревании с добавлением фторида тетрабутиламмония (ТБАФ).
(4) Соединение А8 может быть получено в результате реакции сочетания Бухвальда-Хартвига (время реакции составляет 2-8 часов) соединения А6 и соответствующего ароматического амина А7 в инертном растворителе (таком как толуол, ТГФ, ДМФ, ДМСО, диоксан и т.д.) в присутствии катализатора (такого как тетракис(трифенилфосфин)палладий, трис(дибензилиденацетон)дипалладий (Pd2(dba)3), бис(дибензилиденацетон)палладий, дихлорбис(трифенилфосфин)палладий, бис(три-о-толилфосфин)палладий дихлорид, 1,2-бис(дифенилфосфино)этан дихлорпалладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий дихлорид, [1,1'-бис(дифенилфосфино)ферроцен]дихлорметан и т.д.), лиганда (такой как триметилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, 2,2'-бис(дифенилфосфино)-1,1'-бинафтил и т.д.) и основания (такого как карбонат калия, фторид калия, карбонат цезия, фторид цезия, фторид натрия, фосфат калия, гидратированный фосфат калия, карбонат натрия, бикарбонат калия, трет-бутоксид натрия, трет-бутоксид калия, 1,8-диазабицикло[5.4.0]ундец-7-ен, триэтиламин, диизопропиламин, диизопропилэтиламин, пиридин, их счетание и т.д.).
В третьем аспекте настоящего изобретения предлагается способ использования соединения формулы I, описанного в первом аспекте настоящего изобретения. Соединение формулы I используется для:
(a) приготовления лекарственного средства для лечения заболевания, связанного с активностью киназы CDK или величиной экспрессии;
(b) приготовления ингибитора, адресно воздействующего на киназы CDK;
(c) нетерапевтического ингибирования активности киназ CDK in vitro;
(d) нетерапевтического ингибирования пролиферации опухолевых клеток in vitro; и/или
(e) лечения заболевания, связанного с активностью киназ CDK или величиной экспрессии;
В другом предпочтительном варианте осуществления изобретения киназа CDK выбирается из группы, состоящей из CDK4, CDK6 или их сочетания; и/или
опухолевая клетка является лейкемической клеточной линией, предпочтительно клеточной линией миелоидного лейкоза и более предпочтительно клеточной линией миелоидного лейкоза KG1.
В четвертом аспекте настоящего изобретения представлена фармацевтическая композиция, включающая в себя: (i) эффективное количество соединения формулы I или его фармацевтически приемлемой соли; и (ii) фармацевтически приемлемый носитель.
В пятом аспекте настоящего изобретения предлагается способ ингибирования активности киназ CDK, в рамках которого производится введение пациенту ингибирующего эффективного количества соединения формулы I, описанного в первом аспекте настоящего изобретения, или его фармацевтически приемлемой соли, или введение пациенту ингибирующего эффективного количества фармацевтической композиции, описанной в четвертом аспекте настоящего изобретения.
В шестом аспекте настоящего изобретения предлагается способ ингибирования раковых клеток in vitro, в рамках которого производится введение пациенту ингибирующего эффективного количества соединения формулы I, описанного в первом аспекте настоящего изобретения, или его фармацевтически приемлемой соли, или введение пациенту ингибирующего эффективного количества фармацевтической композиции, описанной в четвертом аспекте настоящего изобретения.
Следует понимать, что каждый из вышеперечисленных технических признаков изобретения и каждый технический признак, более подробно описанный ниже (например, в примерах), могут быть объединены друг с другом в рамках настоящего изобретения, чтобы создать новое или предпочтительное техническое решение, отдельное описание каждого из которых не требуется из-за ограничений по размеру документа.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения провели долгосрочное и интенсивное исследование для получения класса соединений, имеющих структуру, представленную формулой I, и обнаружили, что они обладают ингибирующей активностью против киназ CDK. Соединения оказывают ингибирующее действие на ряд киназ CDK при очень низкой концентрации (до ≤100 нМ), и т.к. они обладают очень хорошей ингибирующей активностью, то могут быть использованы для лечения заболеваний, связанных с активностью киназ CDK или величиной экспрессии, например опухолей. Авторы подготовили настоящее изобретение на основе вышеприведенных результатов.
Термины
Используемый в данном документе термин «С1-С6-алкил» обозначает линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, например метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил и т.п.
Используемый в данном документе термин «С2-С6-ацил» обозначает линейный или разветвленный алкилкарбонил, содержащий от 1 до 6 атомов углерода, например, ацетил, пропионил, бутирил и т.п.
Термин «С1-С6-алкилен» обозначает группу, образованную после потери описанным выше С1-С6-алкилом одного атома водорода, например -СН2-, -СН2-СН2- и т.п.
Термин «С6-С10-арилен» обозначает группу, образованную после потери арилом, имеющим от 6 до 10 атомов углерода, одного атома водорода, включая моноциклический или бициклический арилен, например фенилен, нафтилен и т.п.
Термин «6-членный арил» обозначает фенил.
Термин «5-8-членный арил» обозначает заместителя карбоновой ненасыщенной системы, имеющего 5-8-членное кольцо, например фенил или т.п.
Термин «5-8-членный гетероарил» обозначает заместителя ненасыщенной кольцевой системы, имеющего 5-8-членную кольцевую систему с одним или несколькими гетероатомами, выбранными из О, S, N или Р, например пиридил, тиенил и т.п.
Термин «насыщенный 3-12-членный карбоцикл» обозначает заместителя насыщенной карбоциклической кольцевой системы, имеющего от 3 до 12 атомов углерода, например фенил или т.п.
Термин «3-12-членный гетероцикл» обозначает заместителя насыщенной кольцевой системы, имеющего 3-12-членную кольцевую систему с одним или несколькими гетероатомами, выбранными из О, S, N или Р, например пиперидинил, пирролил и т.п.
Термин «галоген» обозначает F, Cl, Br и I.
В настоящем изобретении термины «содержит(-ат)», «включает(-ют) в себя» или «содержит(-ат)» обозначают, что различные ингредиенты могут использоваться вместе в смесях или композициях настоящего изобретения. Таким образом, термины «в основном состоит из», «состоит из» охватывается термином «содержит(-ат)».
В настоящем изобретении термин «фармацевтически приемлемый» ингредиент обозначает, что препарат подходит для использования в отношении людей и/или животных без проявления чрезмерных побочных эффектов (например, токсичность, раздражение и аллергическая реакция), то есть вещество обладает разумным соотношением эффективности и рисков.
В настоящем изобретении термин «эффективное количество» обозначает количество терапевтического средства, которое может использоваться для лечения, облегчения или предотвращения целевого заболевания или состояния, или количество, при использовании которого может достигаться обнаруживаемый терапевтический или профилактический эффект. Точное эффективное количество для конкретного пациента определяется в зависимости от типа тела и состояния здоровья пациента, природы и стадии заболевания, а также выбранного терапевтического средства и/или сочетания терапевтических средств. Поэтому бесполезно заранее выбирать точное эффективное количество. Однако при данном условии эффективное количество может быть определено врачом в результате проведения стандартных экспериментов.
В настоящем изобретении, если не указано иное, термин «замещенный» означает, что один или несколько атомов водорода в группе замещены заместителем(-ями), выбранными из группы, состоящей из галогена, незамещенного или галогенированного С1-С6-алкила, незамещенного или галогенированного С2-С6-ацила и незамещенного или галогенированного С1-С6-алкилгидрокси.
Если не указано иное, то все соединения, раскрытые в настоящем изобретении, предназначены для включения всех возможных оптических изомеров, таких как однохиральное соединение или смеси различных хиральных соединений (т.е. рацемата). Во всех соединениях настоящего изобретения каждый хиральный атом углерода в качестве варианта может быть R-конфигурацией или S-конфигурацией или смесью R-конфигурации и S-конфигурации.
Используемый в данном документе термин «соединение изобретения» обозначает соединение формулы I. Этот термин также относится к различным кристаллическим формам, фармацевтически приемлемой соли, гидрату или сольвату соединения формулы I.
Используемый в данном документе термин «фармацевтически приемлемая соль» обозначает соль, которая подходит для использования в медицине и образована соединением изобретения и кислотой или основанием. К фармацевтически приемлемым солям относятся неорганические соли и органические соли. Предпочтительная соль образуется соединением изобретения и кислотой. К кислотам, подходящим для образования солей, относятся, среди прочих, неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, фтористоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.д.; органические кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, пикриновая кислота, метансульфоновая кислота, бензолметансульфоновая кислота, бензолсульфоновая кислота и т.д.; и кислые аминокислоты, такие как аспарагиновая кислота, глутаминовая кислота и т.п.
Фармацевтическая композиция и ее введение
Соединения изобретения обладают превосходной ингибирующей активностью против киназ CDK, таких как CDK4, CDK6, поэтому соединения изобретения и различные кристаллические формы, фармацевтически приемлемые неорганические или органические соли, гидрат или сольват и фармацевтические композиции, содержащие соединения изобретения в качестве основного активного ингредиента, могут быть использованы для лечения, профилактики и облегчения заболеваний, связанных с активностью CDK или величиной экспрессии. Согласно данному уровню техники, соединения изобретения могут быть использованы для лечения следующих заболеваний: рак молочной железы, рак эндометрия, рак желудка, рак мочевого пузыря, лимфома, рак головы и шеи и т.д.; в частности, соединения также могут быть объединены с PI3K, B-RAF, FGFR и другими ингибиторами киназы с целью преодоления устойчивости к ингибиторам киназы и могут быть использованы для лечения устойчивых к лечению препаратами направленного действия меланомы, рака молочной железы, немелкоклеточного рака легких, рака печени, глиомы, рака толстой кишки и других опухолей.
Фармацевтическая композиция изобретения содержит соединение изобретения или его фармацевтически приемлемую соль в безопасном и эффективном диапазоне доз и фармацевтически приемлемый эксципиент или носитель. При этом «безопасная и эффективная доза» обозначает количество соединения, достаточное для улучшения состояния пациента и не вызывающее серьезного побочного эффекта. Как правило, фармацевтическая композиция содержит 1-2000 мг соединения изобретения/дозы, более предпочтительно от 5 до 200 мг соединения изобретения/дозы. Предпочтительно, чтобы упомянутая «одна доза» представляла собой одну капсулу или таблетку.
«Фармацевтически приемлемый носитель» означает один или несколько совместимых твердых или жидких наполнителей или желеобразных материалов, подходящих для применения для людей, а также обладающих достаточной чистотой и достаточно низкой токсичностью. «Совместимость» здесь означает, что каждый компонент композиции можно смешивать с соединением изобретения или друг с другом без значительного снижения эффективности соединения. Некоторые примеры фармацевтических приемлемых носителей включают целлюлозу и ее производные (например, карбоксиметилцеллюлоза натрия, натрий-этил целлюлоза, ацетат целлюлозы и т.п.), желатин, тальк, твердые смазочные вещества (например, стеариновая кислота, стеарат магния), сульфат кальция, растительные жиры (например, соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.п.), полиолы (например, пропилен-гликоль, глицерин, маннитол, сорбитол и т.п.), эмульгаторы (например, Tween®), смачивающий реагент (например, додецилсульфат натрия), красящие добавки, ароматизаторы, стабилизаторы, антиоксиданты, консерванты, апирогенная вода и т.п.
Способ введения соединений или фармацевтических композиций изобретения специально не ограничен, а репрезентативный способ введения включает в себя (без ограничений) следующие способы: пероральный, внутриутробный, ректальный, парентеральный (внутривенный, внутримышечный или подкожный) и местное введение.
Твердая лекарственная форма для перорального введения включает в себя капсулы, таблетки, пилюли, порошки и гранулы. В данных твердых лекарственных формах активные соединения смешиваются как минимум с одним традиционным инертным эксципиентом (или носителем), например, цитратом натрия или дикальцийфосфатом, или со следующими компонентами: (а) наполнители или совместители, например крахмал, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (b) связующие вещества, например гидроксиметилцеллюлоза, альгинат, желатин, поливинилпирролидон, сахароза и гуммиарабик; (с) увлажнители, такие как глицерин; (d) дезинтегрирующие агенты, такие как агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые сложные силикаты и карбонат натрия; (е) агенты, замедляющие растворение, такие как парафин; (f) ускорители абсорбции, например соединения четвертичного аммония; (g) смачивающие агенты, такие как цетиловый спирт и одинарный глицерилстеарат; (h) адсорбенты, например каолин; и (i) смазывающие вещества, такие как тальк, стеариновый кальций, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия или их смеси. В капсулах, таблетках и пилюлях лекарственные формы могут также содержать буферные вещества.
Твердые лекарственные формы, такие как таблетки, сахарные пилюли, капсулы, пилюли и гранулы, могут иметь покрытие и оболочку, например из кишечнорастворимого материала или других материалов, используемых для данной цели. Они могут содержать опаковый агент, задерживающий высвобождение активных соединений или соединений в композициях в пищеварительном тракте. В качестве заливочных компонентов могут быть использованы полимеры и воски. При необходимости активные соединения и один или несколько вышеуказанных вспомогательных веществ могут образовывать микрокапсулы.
Жидкие лекарственные формы для перорального введения включают в себя фармацевтически приемлемые эмульсии, растворы, взвеси, сиропы или вытяжки. Кроме активных соединений, жидкие лекарственные формы могут содержать современные традиционные инертные разбавители, например, воду или иные растворители, сжижающие реагенты и эмульгаторы, например, этанол, изопропанол, угольноэтиловый эфир, этилацетат, пропилен-гликоль, 1,3-бутандиол, диметилформамид, а также масло, в частности, хлопковое масло, арахисовое масло, масло из кукурузных зерен, оливковое масло, касторовое масло и кунжутное масло или их сочетания.
Кроме данных инертных разбавителей, композиция также может содержать присадки, например, смачивающие реагенты, эмульгаторы и суспендирующее вещество, подсластители, ароматизаторы и отдушки.
Кроме активных соединений, взвесь может содержать суспендирующее вещество, например, этоксилированный изооктадеканол, полиоксиэтиленовый сорбитол и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метанол, алюминий и агар или их сочетания.
Композиции для парэнтеральных инъекций могут содержать физиологические приемлемые стерильные водные или безводные растворы, дисперсии, взвеси или эмульсии, а также стерильные порошки, которые можно повторно растворить в стерильных инъекционных растворах или дисперсиях. Подходящие водные и безводные носители, разбавители, растворители или эксципиенты включают в себя воду, этанол, полиол и их подходящие смеси.
Лекарственные формы соединений изобретения для местного введения включают в себя мази, порошки, пластыри, аэрозоли и ингалянты. Активные ингредиенты смешивают с физиологически приемлемыми носителями и любыми консервантами, буферами или, при необходимости, пропеллентами в стерильных условиях.
Соединения изобретения можно вводить отдельно или в сочетании с другими фармацевтически приемлемыми соединениями.
При использовании фармацевтических композиций применяется безопасное и эффективное количество соединений настоящего изобретения в отношении нуждающихся в этом млекопитающих (таких как человек), при этом применяемая доза является фармацевтически эффективной. Для человека весом 60 кг суточная доза обычно составляет 1-2000 мг, рекомендуется 5-500 мг. Конечно, конкретная доза также зависит от других факторов, таких, как, например, путь введения, состояние здоровья пациента. Данные факторы определяются опытным терапевтом.
Соединение формулы I
В настоящем изобретении предложено соединение формулы I или его фармацевтически приемлемая соль:

где
R1 и R2 независимо выбраны из Н, замещенного или незамещенного С1-С8-алкила, C(O)OR8, CONR9R10, C(O)R11, замещенного или незамещенного 5-8-членного арила, замещенного или незамещенного 5-8-членного гетероарила, замещенного или незамещенного 3-12-членного насыщенного или ненасыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S; а упомянутый гетероцикл содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S;
кроме того, R1 и R2 могут быть связаны с соседним атомом N, образуя кольцевую структуру, которая включает в себя замещенный или незамещенный 3-12-членный насыщенный или ненасыщенный гетероцикл или кольцо с внутренним мостиком или спиро-кольцо; причем упомянутый гетероцикл относится к кольцевой структуре, содержащей 0-3 гетероатома, выбранных из группы, состоящей из N, О или S, в дополнение к атому азота, присоединенному к исходному ядру;
R3 выбран из Н, замещенного или незамещенного С1-С8-алкила, CN, C(O)OR12, CONR13R14, C(O)R15, замещенного или незамещенного 5-8-членного арила, замещенного или незамещенного 5-8-членного гетероарила, замещенного или незамещенного 3-12-членного насыщенного или ненасыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S; а упомянутый гетероцикл содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S;
R4 выбран из Н, замещенного или незамещенного С1-С4-алкила, замещенного или незамещенного С1-С8-алкокси, галогена, ОН, CN, C(O)OR12, CONR13R14, C(O)R15, замещенного или незамещенного 5-8-членного арила, замещенного или незамещенного 5-8-членного гетероарила, замещенного или незамещенного 3-12-членного насыщенного или ненасыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S; а упомянутый гетероцикл содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S;
R5 выбран из Н или С1-С4-алкила;
X - это CR16 или N;
А, В и Z независимо выбраны из N или CR16;
R16 - это Н, С1-С4-алкил или С1-С4-галогеналкил;
L выбран из группы, состоящей из ничего, С1-С6-алкилена, С(О), CONR17 или S(O)2;
Y - это Н, R18, NR19R20, ОН, или Y выбран из части группы, состоящей из:

где
R6 - ничто, Н, замещенный или незамещенный С1-С8-алкил, замещенный или незамещенный С1-С8-алкокси, замещенный или незамещенный С2-С6-ацил, замещенный или незамещенный С2-С6-сульфонил, замещенный или незамещенный С1-С6-гидроксиалкилен, CONR22R23 или C(O)R24;
R7 может быть заместителями 0-3, a R7 - это замещенный или незамещенный С1-С8-алкил, кислород или галоген; или два или более R7 образуют циклоалкил с внутренним мостиком; W - это CR21, N или О (когда W - О, R6 отсутствует);
Ya - это CR21 или N; R21 - это Н или галоген;
R8, R9, R10, R11, R12, R13, R14, R15, R17, R18, R19, R20, R22, R23 и R24 независимо выбраны из Н, замещенного или незамещенного С1-С8-алкила, замещенного или незамещенного С1-С8-алкокси, замещенного или незамещенного С1-С6-алкиленомино, замещенного или незамещенного С1-С6-алкиленгидрокси, замещенного или незамещенного 5-8-членного арила, замещенного или незамещенного 5-8-членного гетероарила, замещенного или незамещенного 3-12-членного насыщенного или ненасыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из N, О или S; а упомянутый гетероцикл содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из N, О или S;
n и t - 0, 1 или 2 соответственно;
во всех упомянутых веществах определение «замещенный» означает, что один или несколько атомов водорода в группе замещены заместителем (заместителями), выбранными из группы, состоящей из галогена, ОН, NH2, CN, незамещенного или галогенированного С1-С8-алкила, С1-С8-алкокси, незамещенного или галогенированного С2-С6-алкенила, незамещенного или галогенированного С2-С6-алкинила, незамещенного или галогенированного С2-С6-ацила, незамещенного или галогенированного 5-8-членного арила, незамещенного или галогенированного 5-8-членного гетероарила, незамещенного или галогенированного 3-12-членного насыщенного гетероцикла или карбоцикла; причем упомянутый гетероарил содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S, а упомянутый гетероцикл содержит 1-3 гетероатома, выбранных из группы, состоящей из N, О или S.
В другом предпочтительном варианте осуществления изобретения  представляет собой замещенную или незамещенную группу, выбранную из группы, состоящую из
представляет собой замещенную или незамещенную группу, выбранную из группы, состоящую из
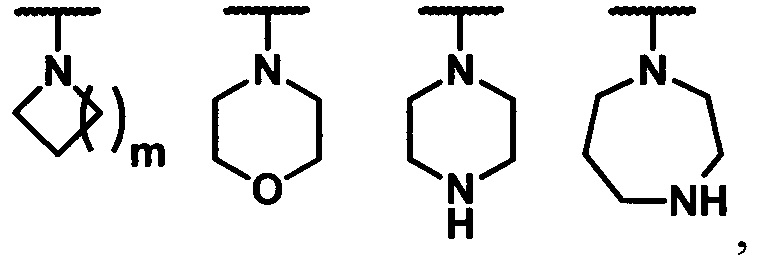
где m равно 0, 1, 2, 3, 4, 5, 6, 7, 8 или 9, а используемый термин «замещенный» определяется, как описано выше.
В другом предпочтительном варианте осуществления изобретения R1 и R2 вместе с соседним атомом азота образуют 4-12-членную кольцевую структуру.
В другом предпочтительном варианте осуществления изобретения R1 и R2 вместе с соседним атомом азота образуют 5-7-членную кольцевую структуру.
В другом предпочтительном варианте осуществления изобретения R1 и R2 вместе с соседним атомом азота образуют 6-членную кольцевую структуру.
В другом предпочтительном варианте осуществления изобретения R13 и R14 вместе с соседним атомом азота образуют 4-6-членную кольцевую структуру.
В другом предпочтительном варианте осуществления изобретения, при котором L отсутствует, Y представляет собой 6-членный гетероцикл, содержащий атом азота.
В другом предпочтительном варианте осуществления изобретения А, В, L, X, Y, Z, R1, R2, R3, R4 или R5 представляет собой соответствующую группу в конкретных соединениях, описанных в приведенных примерах.
В другом предпочтительном варианте осуществления изобретения соединение формулы I выглядит следующим образом:










Способ получения соединения формулы I
В настоящем изобретении предлагается способ приготовления соединения формулы I, включающий следующие стадии:

а) соединение формулы I-6 вступает в реакцию с соединением формулы I-7 в инертном растворителе с образованием соединения формулы I, при этом каждая группа определяется в соответствии с вышеприведенным описанием.
В другом предпочтительном варианте осуществления изобретения инертный растворитель выбирается из группы, включающей толуол, ксилол, диметиловый эфир гликоля, диоксан, тетрагидрофуран (ТГФ), диметилформамид (ДМФА), диметилсульфоксид (ДМСО), N-метилпирролидон или их сочетание.
В другом предпочтительном варианте осуществления изобретения реакцию проводят в присутствии палладиевого катализатора.
В другом предпочтительном варианте осуществления изобретения палладиевый катализатор выбирают из группы, состоящей из Pd(PPh3)4, Pd2(dba)3, Pd(dba)2, Pd(OAc)2, Pd(PPh3)2Cl2, Pd(dppe)Cl2, Pd(dppf)Cl2, Pd(dppf)Cl2⋅CH2Cl2 или их сочетание.
В другом предпочтительном варианте осуществления изобретения реакцию проводят в присутствии лиганда.
В другом предпочтительном варианте осуществления изобретения лиганд представляет собой монодентатный фосфиновый лиганд или бидентатный фосфиновый лиганд; предпочтительно, лиганд выбирают из группы, включающей трифенилфосфин, триметилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, X-Phos, S-Phos, бинафтилдифенилфосфин, 1,1'-бис(дифенилфосфино)ферроцен, 1,2-бис(дифенилфосфино)этан, Xant-Phos или их сочетание.
В другом предпочтительном варианте осуществления изобретения реакцию проводят в присутствии базы.
В другом предпочтительном варианте осуществления изобретения основание выбирают из группы, состоящей из Na2CO3, K2CO3, Cs2CO3, LiHMDS, NaHMDS, KHMDS, трет-бутоксида натрия, трет-бутоксида калия, триэтиламина, диизопропиламина, диизопропилэтиламина или их сочетания.
Предпочтительный способ получения включает следующие этапы:

(1) Соединение A3 может быть получено в результате реакции соединения А1 с соответствующим гидразином А2 в присутствии основания (включая, кроме прочего, диизопропилэтиламин, триметиламин) в инертном растворителе (этаноле, ТГФ и т.д.).
(2) Соединение А5 может быть получено в результате реакции сочетания Соногаширы (время реакции составляет 2-8 ч) соединения A3 и соответствующего терминального алкина А4 в инертном растворителе (таком как ТГФ, ДМФ, ДМСО, диоксан и т.д.) в присутствие катализатора (например, тетракис(трифенилфосфин)палладия, трис(дибензилиденацетон)дипалладия (Pd2(dba)3), бис(дибензилиденацетон)палладия, дихлорбис(трифенилфосфин)палладия, бис(три-о-толилфосфин)палладий дихлорида, 1,2-бис(дифенилфосфино)этандихлорпалладия, дихлорид[1,1'-бис(дифенилфосфино)ферроцен]палладия, [1,1'-бис(дифенилфосфино)ферроцен]дихлорметана, комплекса дихлорметана и т.д.), катализатора b (например, йодида меди, хлорида цинка, оксида серебра, карбоната серебра и т.д.) и щелочи (например, карбоната калия, фторида калия, карбоната цезия, фторида цезия, фторида натрия, фосфата калия, гидратированного фосфата калия, карбоната натрия, бикарбоната натрия, 1,8-диазабицикло[5.4.0]ундец-7-ен, триэтиламина, диизопропиламина, диизопропилэтиламина, пиридина или их сочетания и т.д.);
(3) Соединение А6 может быть получено в результате реакции соединения А5 в инертном растворителе (дихлорметане, ТГФ, ацетонитриле и т.д.) при нагревании с добавлением фторида тетрабутиламмония (ТБАФ).
(4) Соединение А8 может быть получено в результате реакции сочетания Бухвальда-Хартвига (время реакции составляет 2-8 часов) соединения А6 и соответствующего ароматического амина А7 в инертном растворителе (таком как толуол, ТГФ, ДМФ, ДМСО, диоксан и т.д.) в присутствии катализатора (такого как тетракис(трифенилфосфин)палладий, трис(дибензилиденацетон)дипалладий (Pd2(dba)3), бис(дибензилиденацетон)палладий, дихлорбис(трифенилфосфин)палладий, бис(три-о-толилфосфин)палладий дихлорид, 1,2-бис(дифенилфосфино)этан дихлорпалладий, [1,1'-бис(дифенилфосфино)ферроцен]палладий дихлорид, [1,1'-бис(дифенилфосфино)ферроцен]дихлорметан и т.д.), лиганда (такой как триметилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, 2,2'-бис(дифенилфосфино)-1,1'-бинафтил и т.д.) и основания (такого как карбонат калия, фторид калия, карбонат цезия, фторид цезия, фторид натрия, фосфат калия, гидратированный фосфат калия, карбонат натрия, бикарбонат калия, трет-бутоксид натрия, трет-бутоксид калия, 1,8-диазабицикло[5.4.0]ундец-7-ен, триэтиламин, диизопропиламин, диизопропилэтиламин, пиридин, их счетание и т.д.).
Применение соединения формулы I
В настоящем изобретении предлагается следующее применение соединения формулы I:
(a) приготовление лекарственного средства для лечения заболевания, связанного с активностью киназы CDK или величиной экспрессии;
(b) приготовление ингибитора, адресно воздействующего на киназы CDK;
(c) нетерапевтическое ингибирование активности киназ CDK in vitro;
(d) нетерапевтическое ингибирование пролиферации опухолевых клеток in vitro; и/или
(e) лечение заболевания, связанного с активностью киназ CDK или величиной экспрессии;
В другом предпочтительном варианте осуществления изобретения киназа CDK выбирается из группы, состоящей из CDK4, CDK6 или их сочетания; и/или
опухолевая клетка является лейкемической клеточной линией, предпочтительно клеточной линией миелоидного лейкоза и более предпочтительно клеточной линией миелоидного лейкоза KG1.
Основными преимуществами настоящего изобретения являются:
1. Разработка соединения формулы I.
2. Разработка нового ингибитора киназ CDK, способного ингибировать активность различных типов киназ CDK при очень низкой концентрации, способов его приготовления и применения.
3. Разработка фармацевтической композиции для лечения заболеваний, связанных с активностью киназ CDK.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что данные примеры предназначены только для иллюстрации изобретения, а не для ограничения объема изобретения. Способы проведения экспериментов без особых условий, описанные в следующих примерах, обычно подразумевают проведение в обычных условиях или в соответствии с инструкциями изготовителя. Если не указано иное, части и процентные доли рассчитываются по весу.
В каждом примере:
Прибор для LCMS (жидкостная хроматомасс-спектрометрия): Детектор PumpAgilent1100UV: Agilent1100DAD;
Масс-спектрометр: API3000;
хроматографическая колонка: WaterssunfireC18, 4,6×50 мм, 5 мкм;
подвижная фаза: А - ацетонитрил; В - H2O (0,1% FA).
Пример 1

В сухую трехгорлую колбу на 250 мл добавили соединение 1 (5,00 г, 21,94 ммоль) и этанол (100,0 мл), а затем при -20°С медленно добавили по каплям 1-аминопиперидин 2 (3,30 г, 32,91 ммоль) и N,N-диизопропилэтиламин (4,25 г, 32,91 ммоль). Реакционную систему перемешивали при -20°С в течение 3 часов, а затем реакционный раствор выпарили и очистили с помощью колонки из силикагеля с получением соединения 3 (2,9 г, 45,24%) в виде белого твердого вещества. LCMS: 293(М+Н)+, RT=0,50 мин.
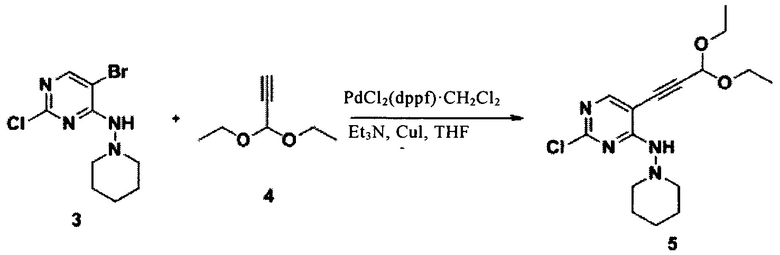
Растворили соединение 3 (2,5 г, 8,56 ммоль) и комплекс [1,1'-бис(дифенилфосфино)ферроцен]-палладий(II) дихлорид-дихлорметана (0,35 г, 0,4281 ммоль) в тетрагидрофуране (12 мл), добавили триметиламин (1,3 г, 12,84 ммоль) и 3,3-диэтокси-1-пропин 4 (1,64 г, 12,84 ммоль) при комнатной температуре. Воздух реакционной системы заменяли азотом в течение 1 минуты, а затем в течение 10 минут перемешивали реакционную систему при комнатной температуре. Затем добавили иодид меди (65,1 мг, 0,3425 ммоль) и воздух трижды заменили азотом. Реакционную систему подвергали СВЧ-излучению при 100°С в течение 6 часов. Смесь смешали с силикагелем и очистили с помощью колонки с получением соединения 5 (1,512 г, 52,25%) в виде желтого масла. LCMS:339(M+H)+, RT=1,72 мин.

Соединение 5 (1,512 г, 4,47 ммоль) растворили в тетрагидрофуране (80 мл). При комнатной температуре добавили фторид тетрабутиламмония (7,13 г, 27,29 ммоль). Реакционную систему перемешивали при 65°С в течение 2 часов, затем реакционный раствор выпарили и очистили с помощью колонки из силикагеля с получением соединения 6 (1,208 г, 79,89%) в виде белого желтого масла. LCMS:339(M+H)+, RT=1,72 мин.
1Протонный магнитный резонанс (CD3Cl3, 400 МГц) δ (чнм) 8,723 (s, 1Н), 6,512 (s, 1H), 5,728 (s, 1Н), 3,967 (t, 2Н, J=11 Гц), 3,646-3,729 (m, 4Н), 3,104 (d, 2Н, J=10 Гц), 1,689-1,838 (m, 6Н), 1,261 (t, 6Н, J=7 Гц).
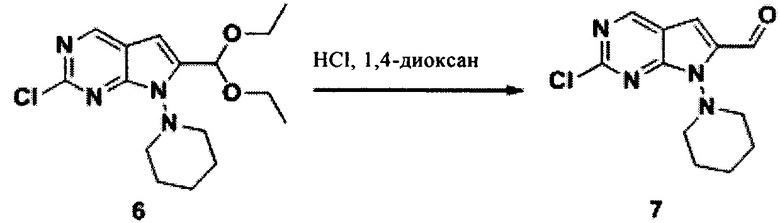
Соединение 6 (0,98 г, 2,899 ммоль) растворили в 1,4-диоксане (15 мл) и добавили концентрированную соляную кислоту (8 мл) при комнатной температуре. Реакционную систему перемешивали в течение 10 минут, разбавили водой (60 мл) и затем дважды экстрагировали этилацетатом (80 мл). Органическую фазу соединили, высушили над безводным Na2SO4 и выпарили с получением соединения 7 (0,765 г, 100%) в виде коричневого твердого вещества, которое использовали для следующей стадии без очистки. LCMS:265(M+H)+, RT=1,34 мин.

Соединение 7 (0,765 г, 2,899 ммоль) растворили в N, N-диметилформамиде (5 мл) и добавили пероксимоносульфат калия (1,96 г, 3,1875 ммоль) при комнатной температуре. Реакционную систему перемешивали при комнатной температуре в течение ночи. К реакционному раствору добавили воду, выделили из твердого вещества осадок и отфильтровали, получив соединение 8 (0,812 г, 100%) в виде желтого твердого вещества. LCMS:281(M+H)+, RT=0,92 мин.

Растворили соединение 8 (0,54 г, 1,929 ммоль) и гидрохлорид диметиламина 9 (0,189 г, 2,3143 ммоль) в N,N-диметилформамиде (6 мл), затем добавили 2-(7-азобензотриазол)-N,N,N', N'-тетраметилуроний гексафторфосфат (0,733 г, 1,929 ммоль) и N,N-диизопропилэтиламин (0,784 г, 5,786 ммоль) при комнатной температуре. Реакционную систему перемешивали в течение 1 часа. Реакционную систему выпарили с помощью масляного насоса и очистили с помощью колонки с получением соединения 10 (0,118 г, 19,93%) в виде желтого твердого вещества. LCMS:308(M+H)+, RT=1,42 мин.
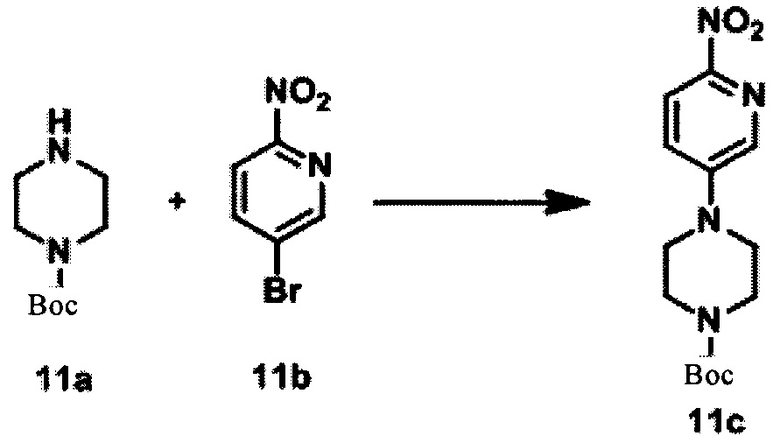
В ДМСО (100 мл) добавили соединение 11а (13,7 г, 73,9 ммоль), соединение 11b (10 г, 49,3 ммоль), йодид калия (81,8 мг, 0,493 ммоль) и карбонат калия (13,6 г, 98,6 ммоль). Реакционный раствор перемешивали при 120°С в течение ночи, затем охладили до комнатной температуры, довели до рН 7 соляной кислотой (1 моль) и экстрагировали дихлорметаном. Водную фазу подщелачили насыщенным раствором карбоната натрия и снова экстрагировали дихлорметаном. Органическую фазу объединили, высушили над безводным Na2SO4, концентрировали и затем суспендировали водой, получив соединение 11с (9,2 г, 60%). LCMS:309(M+H)+, RT=1,710 мин.
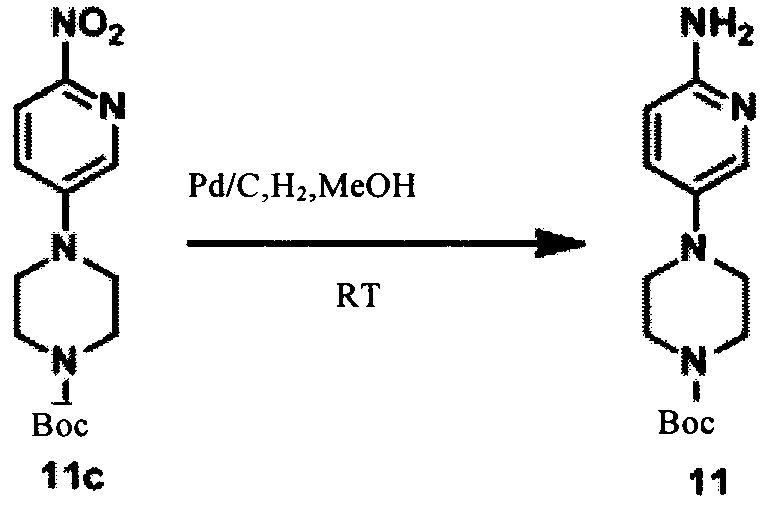
В метанол (100 мл) добавили соединение 11с (9,2 г, 29,9 ммоль) и влажный палладиевый углерод (2 г), воздух в реакционном растворе заменяли водородом четыре-пять раз, а затем реакционную систему перемешивали в водородной атмосфере при комнатной температуре в течение ночи. Реакционный раствор отфильтровали, осадок на фильтре промыли небольшим количеством метанола, фильтрат сконцентрировали, получив соединение 11 (7,1 г, 85%). LCMS:279(M+H)+, RT=1,120 мин.
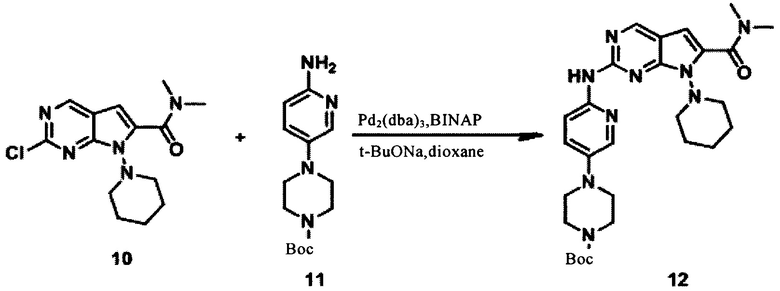
Соединение 10 (90 мг, 0,2932 ммоль), 4-(6-аминопиридин-3-ил)пиперазин-1-карбоновой кислоты трет-бутиловый эфир 11 (122,2 мг, 0,4397 ммоль) и Трис(дибензилиденацетон)дипалладий (26,8 мг, 0,02932 ммоль) были растворены в 1,4-диоксане (1 мл), а затем были добавлены трет-бутоксид натрия (50,7 мг, 0,5277 ммоль) и бис(дифенилфосфино)-1,1'-бинафталин (36,5 мг, 0,05863 ммоль), трижды подвергнутые замещению азотом. Реакционную систему подвергли СВЧ-излучению при 110°С в течение 1,5 часов. Смесь выпарили и очистили, получив соединение 12 (68 мг, 42,3%) в виде коричневого твердого вещества. LCMS:550(M+H)+, RT=1,41 мин.
1Протонный магнитный резонанс (CDCl3, 400 МГц) δ (чнм) 8,931 (s, 1Н), 8,280 (s, 1H), 8,118 (s, 1Н), 7,916 (s, 1H), 6,455 (s, 1Н), 3,719 (s, 4H), 3,647 (s, 2H), 3,300 (s, 5H), 3,147 (s, 3H), 3,006 (s, 3H), 3,243 (t, 1H, J=7,6 Гц), 1,990-2,018 (m, 4H), 1,481 (s, 9Н), 1,427 (s, 2Н).

Соединение 12 (68 мг, 0,1239 ммоль) растворили в дихлорметане (10 мл), добавили трифторуксусную кислоту (2 мл), и затем реакционную систему перемешивали при комнатной температуре в течение ночи. Реакционный раствор выпарили, получив сырой продукт, который очистили с помощью предварительной ТСХ, чтобы получить соединение 13 в виде желтого твердого вещества, выход вещества: 59,34%. LCMS:450(M+H)+, RT=1,12 мин.
1Протонный магнитный резонанс (MeOD, 400 МГц) δ (чнм) 8,680 (s, 1Н), 8,308 (d, 1Н, J=9,2 Гц), 7,972(d, 1H, J=2,8 Гц), 7,498-7,528 (m, 1Н), 6,413 (s, 1Н), 3,990 (s, 2Н), 3,344 (s, 1H), 3,138 (t, 6Н, J=4,8 Гц), 3,056 (s, 3Н), 2,987-3,011 (m, 4Н), 2,121-2,205 (m, 1Н), 1,986-2,088 (m, 1Н), 1,581-1,737 (m, 6Н).
С использованием аналогичных методов могут быть получены следующие соединения:
N,N-диметил-2-((5-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид

LCMS:464(M+H)+, RT=1,13 мин.
1Протонный магнитный резонанс (ДМСО, 400 МГц) δ (чнм) 9,361 (s, 1Н), 8,732 (s, 1Н), 8,240 (d, 1H, J=9,2 Гц), 8,006 (d, 1Н, J=2,4 Гц), 7,465-7,493 (m, 1Н), 6,379 (s, 1H), 3,857 (s, 2H), 3,132 (s, 5H), 3,020 (s, 3H), 2,952 (m, 3H), 2,479-2,490 (m, 4H), 2,234 (s, 3H), l,196(s, 1H), 1,547-1,703 (m, 6H).
7-(гексаметиленимин-1-ил)N,N-диметил-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид

LCMS:464(M+H)+, RT=1,18 мин.
1Протонный магнитный резонанс (MeOD, 400 МГц) δ (чнм) 8,686 (s, 1Н), 8,373 (d, 1H, J=8,8 Гц), 8,006 (d, 1H, J=2,8 Гц), 7,565-7,595 (m, 1Н), 6,397 (s, 1Н), 5,338 (t, 1Н, J=4,8 Гц), 3,958 (s, 2Н), 3,221-3,245 (m, 4Н), 3,158 (s, 3Н), 3,065 (s, 3Н), 2,156-2,209 (m, 1H), 2,002-2,048 (m, 2Н), l,781 (s, 6Н), 1,581-1,674 (m, 4Н).
Азетидин-1-ил-(2-((5-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7H-пирроло[2,3-d]пиримидин-6-ил)метанон
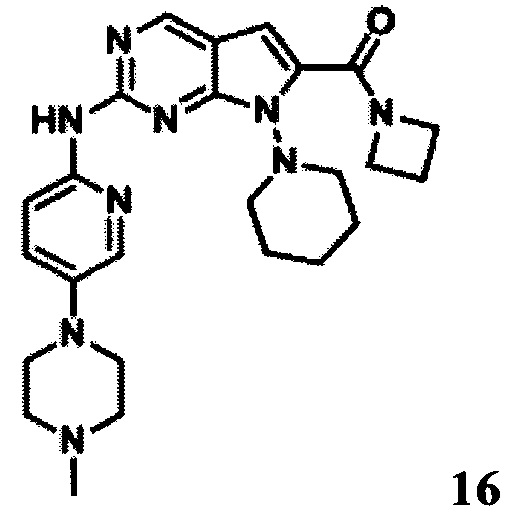
LCMS:476(M+H)+, RT=1,16 мин.
1Протонный магнитный резонанс (MeOD, 400 МГц) δ (чнм) 8,696 (s, 1Н), 8,316 (d, 1H, J=8,8 Гц), 7,985 (d, 1H, J=2,8 Гц), 7,514-7,544 (m, 1Н), 6,593 (s, 1H), 4,288 (t, 2Н, J=7,6 Гц), 4,208 (t, 2Н, J=7,6 Гц), 3,953 (s, 2Н), 3,232 (t, 4Н, J=4,8 Гц), 2,710 (s, 3Н), 2,367-2,424 (m, 4Н), 2,152-2,187 (m, 1H), 2,019-2,033 (m, 1H), 1,787 (s, 5Н), 1,598 (m, 1H).
Азетидин-1-ил-(2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-ил)метанон

LCMS:462(M+H)+, RT=1,19 мин.
1Протонный магнитный резонанс (MeOD, 400 МГц) δ (чнм) 8,706 (s, 1Н), 8,351 (d, 1H, J=8,4 Гц), 8,008 (d, 1H, J=2,8 Гц), 7,543-7,573 (m, 1Н), 6,602 (s, 1Н), 4,290 (t, 2Н, J=7,6 Гц), 4,210 (t, 2Н, J=8 Гц), 3,952 (s, 2Н), 3,215-3,229 (m, 6Н), 2,349-2,427 (m, 2Н), 2,152-2,205 (m, 2Н), 2,008-2,033 (m, 1H), 1,788 (m, 1H), l,787(s, 5Н), 1,598 (t, 1Н, J=6,4 Гц).
N-метил-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид
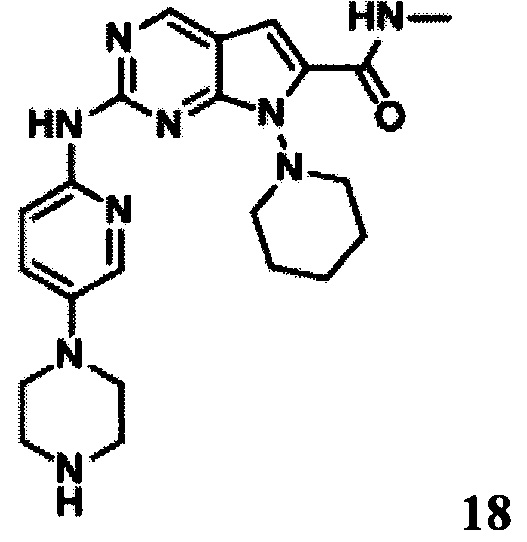
LCMS:436(M+H)+, RT=1,15 мин.
1Протонный магнитный резонанс (MeOD, 400 МГц) δ (чнм) 8,777(s, 1Н), 8,7251 (d, 1H, J=9,2 Гц), 7,994 (d, 1Н, J=2,8 Гц), 7,511-7,542 (m, 1Н), 7,130 (s, 1Н), 4,201 (t, 2Н, J=10,8 Гц), 3,132-3,166 (m, 5Н), 3,004 (s, 5Н), 2,187 (t, 2Н, J=7,6 Гц), 2,019-2,003 (m, 1Н), 1,944 (s, 2Н), l,790 (d, 2Н, J=13,6 Гц), 1,580-1,616 (m, 2Н).
Трет-бутил 4-(6-((6-(диметилкарбамоил)-7-(пирролидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилат

LCMS:536(M+H)+, RT=1,36 мин
1Протонный магнитный резонанс (CDCl3, 400 МГц) δ (чнм) 8,723 (s, 1Н), 8,395 (d, 1Н, J=9,2 Гц), 7,713 (d, 1H, J=9,2 Гц), 7,593 (s, 1H), 6,374 (s, 1Н), 3,604 (d, 7H, J=13,6 Гц), 3,494 (s, 1H), 3,163 (s, 3H), 3,121 (s, 4H), 3,031 (s, 3H), 2,075 (s, 4H), 1,497 (s, 9H).
2-((5-(3,3-диметилпиперазин-1-ил)пиридин-2-ил)амино)-N,N-диметил-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид

LCMS:478(M+H)+, RT=1,20 мин
1Протонный магнитный резонанс (MeOD, 400 МГц) δ (чнм) 8,720 (s, 1Н), 8,277 (s, 1H), 8,018 (s, 1H), 7,624 (s, 1H), 6,434 (s, 1H), 4,003 (m, 2Н), 3,477 (m, 1H), 3,141 (m, 4H), 3,049 (s, 3Н), 2,177 (d, 2H, J=8 Гц), 2,026 (d, 4Н, J=5,6 Гц), 1,735-1,757 (m, 3Н), 1,580-1,616 (m, 3Н), 1,471 (s, 4H), 1,401 (s, 2H).
(2-((5-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-ил)-пирролидин-1-илметанон

1Протонный магнитный резонанс (400 МГц, ДМСО-d6) δ 9,37 (d, 1Н, J=3,2 Гц), 8,72 (s, 1H), 8,24 (d, 1Н, J=5,6 Гц), 8,00 (d, 1H, J=0,8 Гц), 7,48 (m, 1H), 6,45 (s, 1H), 3,45 (m, 4Н), 3,15 (m, 6Н), 2,28 (m, 4Н), 1,96 (s, 3Н), 1,84 (m, 6Н), 1,64 (m, 8Н).
N,N-диметил-2-((5-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-7-морфолино-7H-пирроло[2,3-d]пиримидин-6-карбоксамид
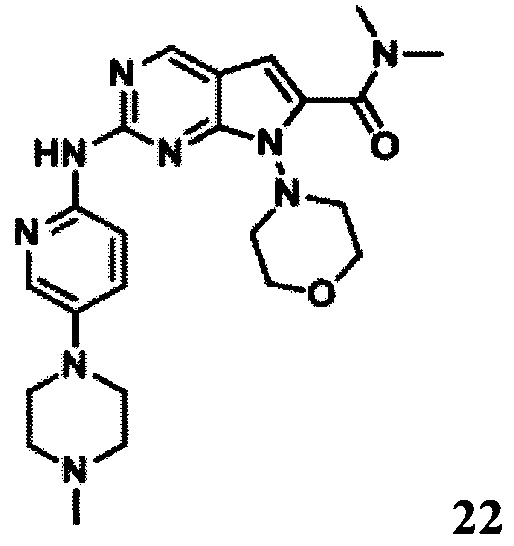
1Протонный магнитный резонанс (400 МГц, ДМСО-d6) δ 9,36 (s, 1Н), 8,74 (s, 1Н), 8,16 (d, 1Н, J=9,2 Гц), 8,00 (d, 1H, J=2,4 Гц), 7,39 (dd, 1Н, J=2,8 Гц), 6,42 (s, 1Н), 3,71 (m, 4Н), 3,12 (t, 4Н, J=4,6 Гц), 3,03 (s, 3Н), 2,97 (s, 3Н), 2,47 (m, 8Н), 2,23 (s, 3Н).
(2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-ил)-пирролидин-1-илметанон

1Протонный магнитный резонанс (400 МГц, ДМСО-d6) δ 9,38 (s, 1Н), 8,73 (s, 1H), 8,23 (d, 1H, J=8,4 Гц), 8,00 (d, 1Н, J=1,2 Гц), 7,46 (dd, 1Н, J=6 Гц), 6,45 (s, 1Н), 3,48 (m, 4Н), 3,10 (m, 8Н), 2,93 (m, 6Н), 1,65 (m, 8Н).
N,N-диметил-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7-(пирролидин-1-ил)-7H-пирроло[2,3-d]пиримидин-6-карбоксамид

1Протонный магнитный резонанс (400 МГц, MeOD-d4) δ 8,69 (s, 1Н), 8,20 (d, 1Н, J=9,2 Гц), 7,97 (d, 1Н, J=2,4 Гц), 7,52 (dd, 1Н, J=2,8 Гц), 6,43 (s, 1H), 3,63 (m, 4H), 3,14 (m, 4H), 3,13 (s, 3H), 3,08 (s, 3H), 3,00 (m, 4H), 2,06 (m, 4H).
N,N-диметил-2-((5-морфолинопиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид
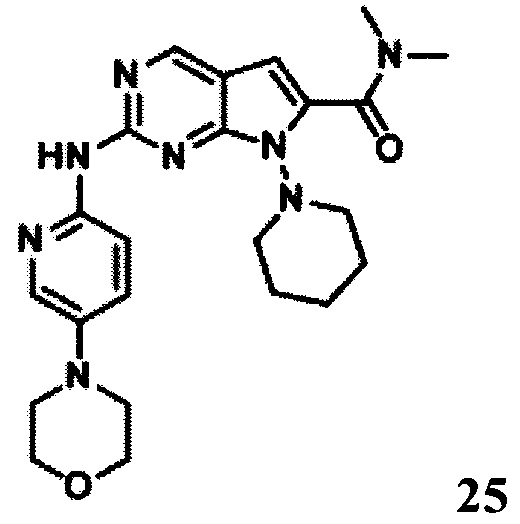
LCMS:450(M+H)+, RT=6,41 мин.
1Протонный магнитный резонанс (CDCl3, 400 МГц) δ (чнм) 8,661 (s, 1Н), 8,414 (d, 1H, J=8,8 Гц), 8,014 (d, 1H, J=2,4 Гц), 7,895 (s, 1H), 7,349-7,320 (m, 1H), 6,288 (s, 1H), 3,967-3,882 (m, 6H), 3,240-3,133 (m, 9H), 3,016 (s, 3H), 1,776-1,630 (m, 6H).
N,N-диметил-2-((5-(4-этилпиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид

LCMS:477(M+H)+, RT=5,56 мин
1Протонный магнитный резонанс (ДМСО-d6, 400 МГц) δ (чнм) 9,385 (s, 1Н), 8,730 (s, 1Н), 8,248 (d, J=8,8 Гц, 1Н), 8,009 (s, 1Н), 7,493 (d, 1H, J=8,4 Гц), 6,377 (s, 1Н), 3,146 (s, 6Н), 3,014-2,949 (m, 7Н), 2,567 (m, 5Н), 1,706-1,552 (m, 4Н), 1,235 (s, 4Н), 1,054 (s, 3Н).
2-((5-((3S,5R)-3,5-диметилпиперазин-1-ил)пиридин-2-ил)амино)-N,N-диметил-7-(пиперидин-1-ил)-7H-пирроло[2,3-d]пиримидин-6-карбоксамид

LCMS:478(M+H)+, RT=1,20 мин.
1Протонный магнитный резонанс (d6-ДМСО, 400 МГц) δ (чнм) 9,30 (s, 1Н), 8,72 (s, 1Н), 8,22 (d, 1H, J=8,0 Гц), 7,97 (s, 1Н), 7,46 (d, 1Н, J=8,0 Гц), 6,37 (s, 1H), 3,83 (s, 1Н), 3,49 (d, 2Н, J=8,0 Гц), 3,01 (s, 3Н), 2,94 (s, 3Н), 2,89 (m, 2Н), 2,33 (s, 1Н), 2,14 (m, 2H), 2,01 (m, 2H), 1,23 (s, 6H), 1,04 (d, 6H, J=8,0 Гц), 0,85 (m, 1H).
N,N-диметил-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7-морфолино-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид

1Протонный магнитный резонанс (400 МГц, ДМСО-d6) δ 9,47 (s, 1Н), 8,77 (s, 1H), 8,17 (d, 1H, J=4 Гц), 8,01 (s, 1H), 7,40 (m, 1H), 6,43 (s, 1H), 3,71 (m, 8H), 3,03 (s, 6H), 3,00 (m, 4H), 2,87 (m, 4H).
N,N-диэтил-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид
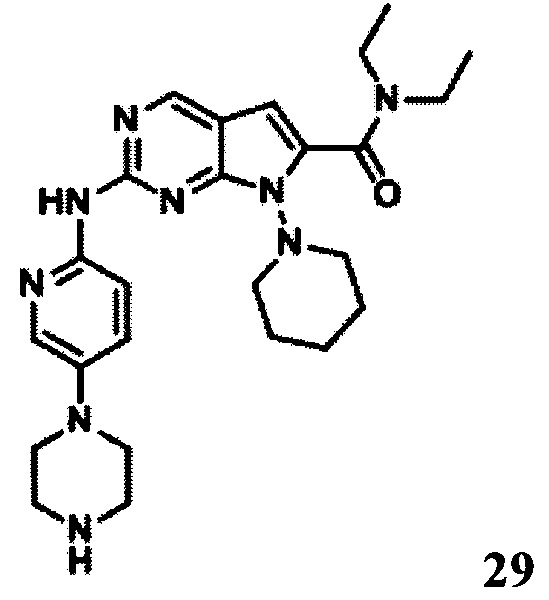
1Протонный магнитный резонанс (400 МГц, MeOD-d4) δ 8,72 (s, 1Н), 8,35 (d, 1Н, J=4 Гц), 8,02 (d, 1H, J=4 Гц), 7,55 (t, 1Н, J=3 Гц), 6,42 (s, 1Н), 4,01 (m, 2H), 3,61 (q, 2H, J=8 Гц), 3,39 (m, 4H), 3,17 (m, 4H), 3,03 (m, 4H), 1,83 (m, 4H), 1,64 (m, 4H), 1,34 (m, 8H).
N,N-диметил-2-((5-(4-дейтерометилпиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид

1Протонный магнитный резонанс (ДМСО-d6, 400 МГц) δ (чнм) 9,34 (s, 1Н), 8,72 (s, 1Н), 8,23 (d, J=9,2 Гц, 1H), 8,00 (s, 1Н), 7,47 (dd, J=9,2 Гц, 1H), 6,37 (s, 1Н), 3,51 (m, 2Н), 3,21 (s, 6Н), 3,01 (s, 3Н), 2,95 (s, 3Н), 1,68 (m, 10Н).
N,N-диметил-2-((5-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-7-(гомопиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид
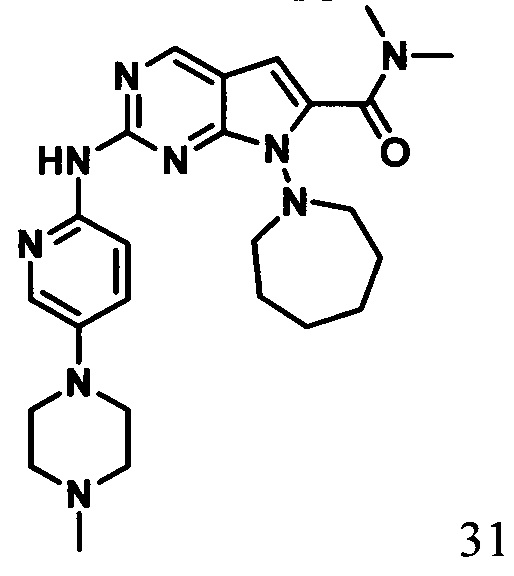
1Протонный магнитный резонанс (ДМСО-d6, 400 МГц) δ (чнм) 9,34 (s, 1Н), 8,71 (s, 1Н), 8,29 (d, J=9,2 Гц, 1H), 8,01 (s, 1Н), 7,50 (m, 1H), 6,34 (s, 1Н), 3,83 (m, 2Н), 3,20 (m, 8Н), 3,03 (s, 3Н), 2,95 (s, 3Н), 1,67 (m, 10Н).
N,N-диметил-2-((5-(4-метил-1,4-гомопиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид
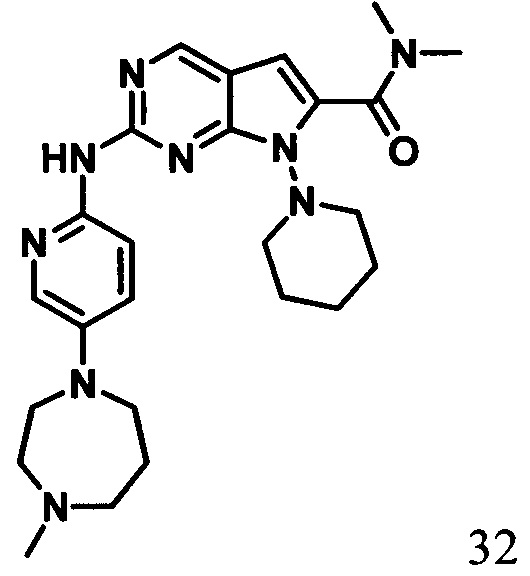
1Протонный магнитный резонанс (ДМСО-d6, 400 МГц) δ (чнм) 9,16 (s, 1Н), 8,70 (s, 1H), 8,15 (d, J=8,0 Гц, 1H), 7,87 (d, J=2,8 Гц, 1Н), 7,27 (m, 1H), 6,36 (s, 1H), 3,66 (s, 3Н), 3,48 (m, 3Н), 3,01 (m, 10Н), 2,60 (s, 3Н), 2,08 (s, 3Н), 1,64 (m, 6Н).
N,N-диметил-2-((5-(4-метилпиперазин-1-ил)метил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7H-пирроло[2,3-d]пиримидин-6-карбоксамид

1Протонный магнитный резонанс (метанол-d4, 400 МГц) δ (чнм) 8,98 (s, 1Н), 8,31 (s, 1Н), 8,20 (d, J=8,4 Гц, 1Н), 7,53 (d, J=8,8 Гц, 1H), 6,64 (s, 1H), 3,98 (s, 2Н), 3,71 (s, 2Н), 3,57-3,43 (m, 2Н), 3,34 (m, 2Н), 3,16 (s, 6Н), 3,06 (s, 4Н), 2,91 (s, 3Н), 2,52 (s, 2Н), 1,76-1,60 (s, 6Н).
N,N-диметил-2-((4-(4-метилпиперазин-1-ил)фенил)амино)-7-(пиперидин-1-ил)-7H-пирроло[2,3-d]пиримидин-6-карбоксамид

1Протонный магнитный резонанс (ДМСО-d6, 400 МГц) δ (чнм) 9,37 (s, 1Н), 8,69 (s, 1H), 7,75 (d, J=8,0 Гц, 2Н), 6,95 (d, J=8,4 Hz, 2Н), 6,33 (s, 1H), 3,86 (m, 2H), 3,26-3,13 (m, 6H), 3,01 (s, 4H), 2,95 (s, 6H), 2,57 (s, 3H), 1,73-1,55 (m, 6H).
N,N-диметил-2-((5-(4-циклопропилпиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид

1Протонный магнитный резонанс (ДМСО-d6, 400 МГц) δ (чнм) 9,31 (s, 1Н), 8,72 (s, 1H), 8,23 (d, J=8,8 Гц, 1Н), 7,99 (d, J=2,8 Гц, 1H), 7.48 (m, 1Н), 6,37 (s, 1H), 3,18 (d, J=5,6 Гц, 4Н), 3,01 (s, 3Н), 2,95 (s, 3Н), 2,70 (t, J=8,8 Гц, 4Н), 1,67 (m, 8Н), 0,45 (d, J=4,4 Гц, 2Н), 0,35 (d, J=2,8 Гц, 2Н).
N,N-диметил-2-((6-(4-метилпиперазин-1-ил)пиридазин-3-ил)амино)-7-(пиперидин-1-ил)-7H-пирроло[2,3-d]пиримидин-6-карбоксамид

1Протонный магнитный резонанс (ДМСО-d6, 400 МГц) δ (чнм) 9,85 (s, 1Н), 8,73 (s, 1Н), 8,29 (d, 1Н, J=10 Гц), 7,45 (d, 1Н, J=9,6 Гц), 6,39 (s, 1Н), 3,80-3,71 (m, 2Н), 4,45 (s, 4Н), 3,02 (s, 3Н), 2,95 (s, 3Н), 2,45 (s, 4Н), 2,24 (s, 3Н), 1,66-1,58 (m, 6Н), 1,24 (s, 2Н),
2-((5-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-ил)метанол
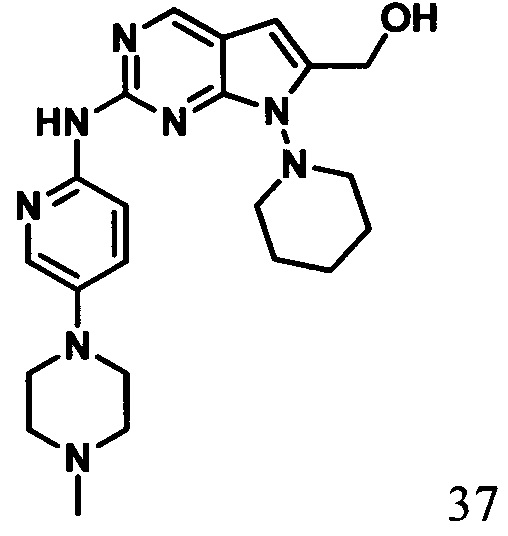
1Протонный магнитный резонанс (ДМСО-d6, 400 МГц) δ (чнм) 11,02 (s, 1H), 10,23 (s, 1Н), 8,83 (s, 1H), 8,01 (s, 1H), 7,89 (d, 1H, J=8,4 Гц), 7,71 (d, 1H, J=8,8 Гц), 6,44 (s, 1H), 4,61 (s, 2H), 4,06-3,79 (m, 6H), 3,27-2,89 (m, 6H), 2,89 (s, 3H), 1,77-1,65 (m, 6H).
N,N-диметил-2-((5-(4-метилпиперазин-1-ил)пиридин-2-ил)амино)-5-метил-7-(пиперидин-1-ил)-7Н-пирроло[2,3-d]пиримидин-6-карбоксамид

1Протонный магнитный резонанс (CDCl3, 400 МГц) δ (чнм) 8,58 (s, 1Н), 7,93 (s, 2Н), 7,37 (s, 1Н), 3,98-3,93 (t, 3Н, J=8,8 Гц, 10,8 Гц), 3,49 (s, 9Н), 3,18-2,71 (m, 12Н), 2,44 (s, 3Н), 2,04-1,62 (m, 6Н).
Аналогичным образом могут быть получены следующие соединения:



Пример 2 Определение активности соединений настоящего изобретения против киназы CDK
1. Экспериментальный материал
Киназа CDK, используемая в данном эксперименте: CDK4/CyclinD1 (Invitrogen, Спец. №: PV4400); CDK6/CyclinD1 (Invitrogen, Спец. №: PV4401); CDK1/CyclinB (Invitrogen, Спец. №: PV3292).
Использованные реагенты: Субстрат - ULight-4E-BP1 (PerkinElmer, Спец. №: TRF0128); антитело - Eu-labeledanti-phospho-eIF4E-bindingprotein1 (Thr37/46) (Perkin-Elmer, Спец. №: TRF0216).
2. Экспериментальный способ
Тестируемое соединение растворили в диметилсульфоксиде (концентрация диметилсульфоксида - 4%), затем раствор разбавили до каждого градиента концентрации буфером (50 мМ HEPES, 10 мМ MgCl2, 1 мМ EGTA, 2 мМ DTT и 0,01% Tween20) в соответствии с требованиями испытания. Затем АТФ и субстрат ULight-4Е-ВР1 разбавили буфером для получения смеси 800 мкМ АТФ и 200 нм субстрата для дальнейшего использования. В лунки добавили 2,5 мкл смеси субстрата и АТФ или 2,5 мкл субстрата, затем добавили 2,5 мкл соединения или 4%-ный буфер диметилсульфоксида, а также 5 мкл фермента (конечная концентрация составляла 0,66 мкг/мл), инкубировали, избегая света, при комнатной температуре в течение 60 минут. В каждую лунку добавили 5 мкл стоп-буфера EDTA (конечная концентрация 6 нМ), разбавленного 1х детекторным буфером (LANCEDetectionBuffer, 10х, PerkinElmer, CR97-100), затем добавили 5 мкл антитела (конечная концентрация 2 нМ), разбавленного 1х детекторным буфером, и инкубировали, избегая света, при комнатной температуре в течение 60 минут. Для измерения пластин был использован прибор Perkin Elmer EnVision® TRFRETmode (Длина волны возбуждения: 320 нм, длина волны излучения: 615 нм и 665 нм). Скорость ингибирования образца определили по следующей формуле:

Значения IC50 были рассчитаны с использованием программного обеспечения GraphPadPrism.
3. Результаты
Результаты приведены в Таблице 1. Символ «+» обозначает IC50 менее 100 нМ, символ «++» обозначает IC50 от 100 нм до 500 нм, символ «+++» обозначает IC50 больше 500 нм, а «н/д» обозначает отсутствие данных.

Результаты показывают, что соединения настоящего изобретения могут эффективно ингибировать активность киназы CDK4 при очень низкой концентрации (≤100 нМ) и обладают слабой ингибирующей активностью против киназы CDK1.
Пример 3 Определение ингибирующей пролиферацию активности против клеточной линии рака толстой кишки человека Colo205 для соединений настоящего изобретения
1. Экспериментальный способ
Анализ клеток in vitro, описанный ниже, позволяет определять ингибирующую активность пролиферации тестируемых соединений к клеточной линии рака толстой кишки человека, и их активность может быть представлена через значение IC50.
Клетки Colo205 (Фонд клеток Комитета по хранению типичных культур Китайской академии наук) инокулировали в 96-луночный планшет с подходящей концентрацией - 2000 клеток на отверстие, 140 мкл среды на лунку, затем инкубировали с диоксидом углерода в инкубаторе при 37°С в течение ночи. Добавляли 10 мкл различных концентраций тестируемых соединений и подвергали взаимодействию в течение 96 часов, а затем устанавливали контрольную группу растворителей (группу отрицательного контроля). Активность ингибирования пролиферации тестируемых соединений против опухолевых клеток была проверена с помощью CCK8 (CellCounting Kit-8, Спец. №: CK04, приобретен у компании Tongren Chemical) через 96 часов. Для считывания использовался полноволновый микропланшет-ридер SpectraMax190, длина волны измерения составляла 450 нм.
Скорость ингибирования образца определили по следующей формуле:

Значение IC50 было рассчитано с помощью четырехпараметрической регрессии с использованием микропланшет-ридера со случайным программным обеспечением.
2. Результаты
Результаты приведены в Таблице 2. Символ «+» обозначает IC50 менее 0,5 мкМ, символ «++» обозначает IC50 от 0,5 мкМ до 2 мкМ, символ «+++» обозначает IC50 больше 2 мкМ, а «н/д» обозначает отсутствие данных.


Результаты показывают, что соединения настоящего изобретения могут эффективно ингибировать пролиферацию опухолевых клеток при низкой концентрации (≤2 мкМ).
Все публикации, упомянутые в данном документе, включены в качестве ссылки. Следует также понимать, что после прочтения вышеприведенных положений настоящего изобретения специалистами в данной области техники могут быть внесены различные изменения или модификации, эквиваленты которых входят в объем формулы изобретения, прилагаемой далее.
| название | год | авторы | номер документа |
|---|---|---|---|
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ИНГИБИТОРЫ CDK | 2011 |
|
RU2621674C2 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ, ЕГО ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2732576C2 |
| СОЕДИНЕНИЕ 2-АМИНОПИРИМИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ ДАННОГО СОЕДИНЕНИЯ | 2015 |
|
RU2704129C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, ИСПОЛЬЗУЕМОЕ КАК ИНГИБИТОР FGFR | 2017 |
|
RU2742485C2 |
| ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2719428C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| Соединение в качестве ингибитора циклинзависимой киназы 9 и его применение | 2020 |
|
RU2819762C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОНАФТАЛИНА И ТЕТРАГИДРОИЗОХИНОЛИНА В КАЧЕСТВЕ РАЗРУШИТЕЛЕЙ ЭСТРОГЕНОВОГО РЕЦЕПТОРА | 2017 |
|
RU2797244C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
Изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям, которые обладают ингибирующей активностью в отношении киназы CDK4. В формуле I R1 и R2 связаны с соседним атомом N, образуя замещенный С1-С8-алкилом или незамещенный 3-12-членный насыщенный гетероцикл, причем упомянутый гетероцикл относится к кольцевой структуре, содержащей 0-3 гетероатома, выбранных из группы, состоящей из N или О, в дополнение к атому азота, присоединенному к исходному ядру; R3 выбран из С1-С8-алкила, CN, CONR13R14, C(O)R15; R13 и R14 независимо выбраны из Н или незамещенного С1-С8-алкила; R15 выбран из незамещенного 3-12-членного насыщенного гетероцикла, причем упомянутый гетероцикл содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из N и О; R4 представляет собой Н; R5 представляет собой Н; X представляет собой CR16; А, В и Z независимо выбраны из N или CR16; R16 представляет собой Н или С1-С4-алкил; L отсутствует или представляет собой С1-С6-алкилен; Y представляет собой  где когда W представляет собой О, то R6 отсутствует, когда W представляет собой N, то R6 представляет собой Н, незамещенный С1-С8-алкил или C(O)R24; R24 представляет собой незамещенный С1-С8-алкокси; R7 может быть 0-3 заместителями, представляющими собой незамещенный С1-С8-алкил; Ya представляет собой N; n и t независимо представляют собой 0, 1 или 2. Изобретение относится также к способу получения указанных соединений, их применению для приготовления лекарственного средства для лечения заболевания, связанного с активностью киназы CDK4 или величиной экспрессии; для приготовления ингибитора, адресно воздействующего на киназу CDK4; и/или для лечения заболевания, связанного с активностью киназы CDK4 или величиной экспрессии, фармацевтической композиции и способам ингибирования активности киназы CDK4 и опухолевых клеток in vitro с использованием указанных соединений. 6 н. и 4 з.п. ф-лы, 2 табл., 3 пр.
где когда W представляет собой О, то R6 отсутствует, когда W представляет собой N, то R6 представляет собой Н, незамещенный С1-С8-алкил или C(O)R24; R24 представляет собой незамещенный С1-С8-алкокси; R7 может быть 0-3 заместителями, представляющими собой незамещенный С1-С8-алкил; Ya представляет собой N; n и t независимо представляют собой 0, 1 или 2. Изобретение относится также к способу получения указанных соединений, их применению для приготовления лекарственного средства для лечения заболевания, связанного с активностью киназы CDK4 или величиной экспрессии; для приготовления ингибитора, адресно воздействующего на киназу CDK4; и/или для лечения заболевания, связанного с активностью киназы CDK4 или величиной экспрессии, фармацевтической композиции и способам ингибирования активности киназы CDK4 и опухолевых клеток in vitro с использованием указанных соединений. 6 н. и 4 з.п. ф-лы, 2 табл., 3 пр.
1. Соединение, представляющее собой соединение формулы I
 ,
,
где
R1 и R2 связаны с соседним атомом N, образуя замещенный С1-С8-алкилом или незамещенный 3-12-членный насыщенный гетероцикл, причем упомянутый гетероцикл относится к кольцевой структуре, содержащей 0-3 гетероатома, выбранных из группы, состоящей из N или О, в дополнение к атому азота, присоединенному к исходному ядру;
R3 выбран из С1-С8-алкила, CN, CONR13R14, C(O)R15; R13 и R14 независимо выбраны из Н или незамещенного С1-С8-алкила; R15 выбран из незамещенного 3-12-членного насыщенного гетероцикла, причем упомянутый гетероцикл содержит по меньшей мере один гетероатом, выбранный из группы, состоящей из N и О;
R4 представляет собой Н;
R5 представляет собой Н;
X представляет собой CR16;
А, В и Z независимо выбраны из N или CR16;
R16 представляет собой Н или С1-С4-алкил;
L отсутствует или представляет собой С1-С6-алкилен;
Y представляет собой
 где
где
когда W представляет собой О, то R6 отсутствует,
когда W представляет собой N, то R6 представляет собой Н, незамещенный С1-С8-алкил или C(O)R24; R24 представляет собой незамещенный С1-С8-алкокси;
R7 может быть 0-3 заместителями, представляющими собой незамещенный С1-С8-алкил;
Ya представляет собой N;
n и t независимо представляют собой 0, 1 или 2;
или соединение  , или соединение
, или соединение  ,
,
или фармацевтически приемлемую соль любого из вышеуказанных соединений.
2. Соединение по п.1, представляющее собой
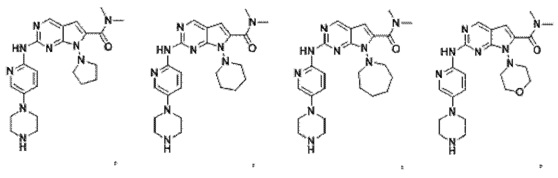

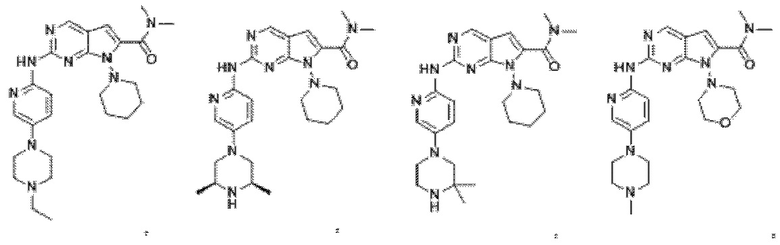
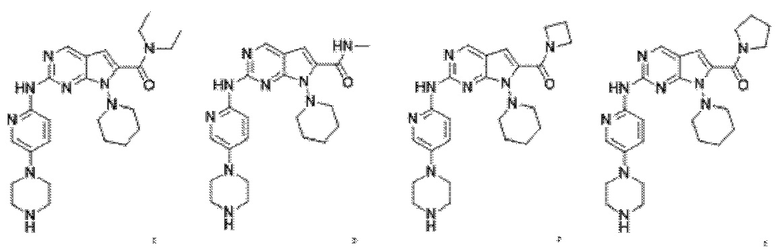






3. Способ получения соединения по п.1, в котором способ включает в себя следующий этап:

а) соединение формулы I-6 вступает в реакцию с соединением формулы I-7 в инертном растворителе с образованием соединения формулы I, при этом каждая группа определяется в соответствии с вышеприведенным п.1.
4. Способ по п.3, в котором инертный растворитель выбирается из группы, включающей толуол, ксилол, диметиловый эфир гликоля, диоксан, ТГФ, ДМФА, ДМСО, N-метилпирролидон или их сочетания.
5. Способ по п.3, в котором способ обладает одной или несколькими следующими характеристиками:
реакция проводится в присутствии палладиевого катализатора, который выбирается из группы, состоящей из Pd(PPh3)4, Pd2(dba)3, Pd(dba)2, Pd(OAc)2, Pd(PPh3)2Cl2, Pd(dppe)Cl2, Pd(dppf)Cl2, Pd(dppf)Cl2⋅CH2Cl2 или их сочетания;
реакция проводится в присутствии лиганда, представляющего собой монодентатный фосфиновый лиганд или бидентатный фосфиновый лиганд; лиганд выбирают из группы, включающей трифенилфосфин, триметилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, X-Phos, S-Phos, бинафтилдифенилфосфин, 1,1'-бис(дифенилфосфино)ферроцен, 1,2-бис(дифенилфосфино)этан, Xant-Phos или их сочетание; и/или
реакция проводится в присутствии основания, которое выбирают из группы, состоящей из Na2CO3, K2CO3, Cs2CO3, LiHMDS, NaHMDS, KHMDS, трет-бутоксида натрия, трет-бутоксида калия, триэтиламина, диизопропиламина, диизопропилэтиламина или их сочетания.
6. Применение соединения по п.1, в котором соединение используется для:
(a) приготовления лекарственного средства для лечения заболевания, связанного с активностью киназы CDK4 или величиной экспрессии;
(b) приготовления ингибитора, адресно воздействующего на киназу CDK4; и/или
(c) лечения заболевания, связанного с активностью киназы CDK4 или величиной экспрессии.
7. Применение по п.6, в котором заболевание, связанное с активностью киназы CDK4 или величиной экспрессии, представляет собой рак толстой кишки.
8. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении киназы CDK4, включающая в себя: (i) эффективное количество соединения по п.1 и (ii) фармацевтически приемлемый носитель.
9. Способ ингибирования активности киназы CDK4, в котором способ включает следующие этапы: введение пациенту ингибирующего эффективного количества соединения по п.1 или введение пациенту ингибирующего эффективного количества фармацевтической композиции по п.8.
10. Способ ингибирования опухолевых клеток in vitro, в котором способ включает в себя введение пациенту ингибирующего эффективного количества соединения по п.1 или фармацевтической композиции по п.8.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| CN 103864792 A, 18.06.2014 | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДИНА, КАК ИНГИБИТОРЫ ЦИКЛИН-ЗАВИСИМОЙ КИНАЗЫ | 2005 |
|
RU2414472C9 |

