Объектом настоящего изобретения является новый способ получения травопроста.
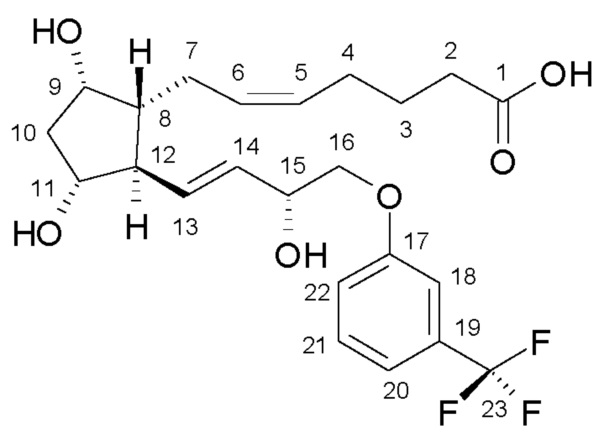
Травопрост формулы (I):
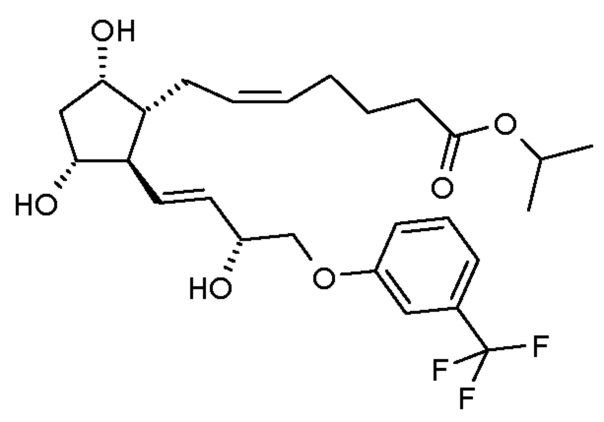
является известным производным простагландина, применяющимся для лечения глаукомы и высокого внутриглазного давления (US 5510383).
Способы получения травопроста раскрыты, например, в EP 2143712, WO 2011/046569, WO 2011/055377.
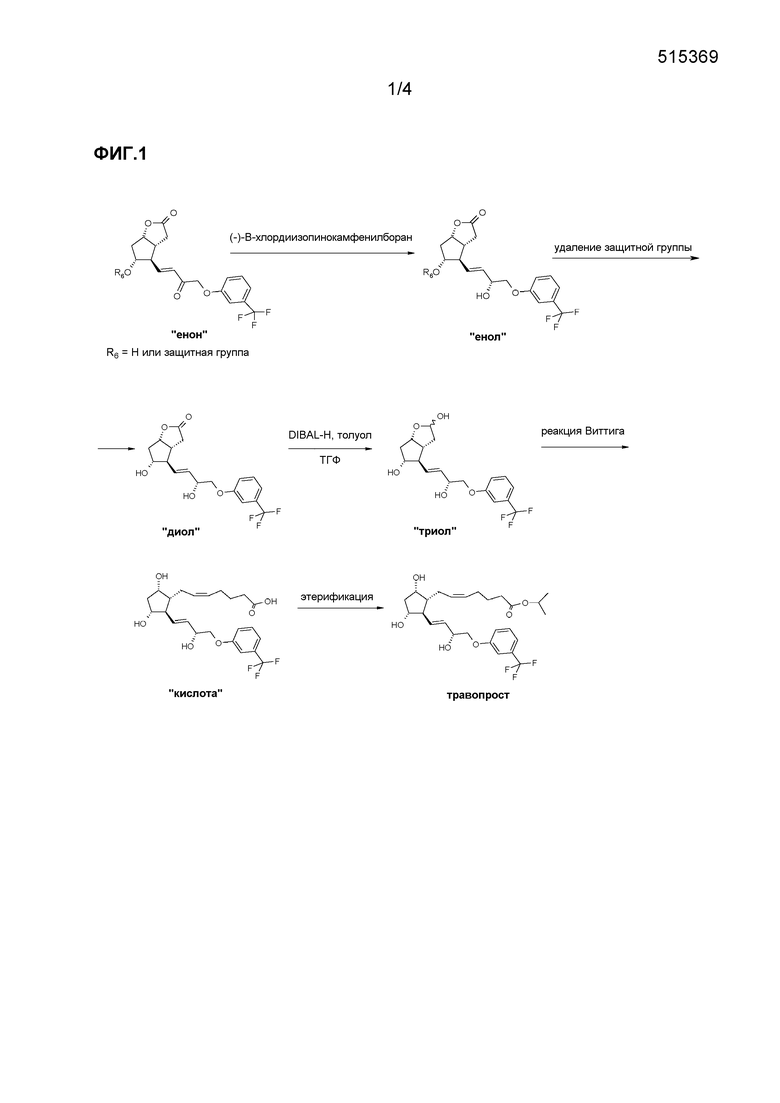
Способ, соответствующий EP 2143712, представлен на фиг. 1.
Стереоселективность восстановления енон→енол составляет 88,7% (пример 10).
В соответствии со способом, раскрытым в WO 2011/046569, примесь 15-эпи удаляют путем защиты групп OH диола трет-бутилдиметилсилильной группой (TBDMS) и кристаллизации полученного таким образом защищенного диола.
В способе, описанном в WO 2011/055377, преобразование енон→енол проводят с помощью комплекса N,N-диэтиланилин/боран в качестве восстановительного реагента в присутствии катализатора Corey (CBS-оксазаборолидин). Продукт очищают с помощью препаративной ВЭЖХ.
Полный выход равен 7%.
Задачей настоящего изобретения является разработка способа, имеющего более высокую стереоселективность и лучший выход.
Объектом настоящего изобретения является получение травопроста формулы (I):
с помощью
стереоселективного восстановления соединения формулы (II):
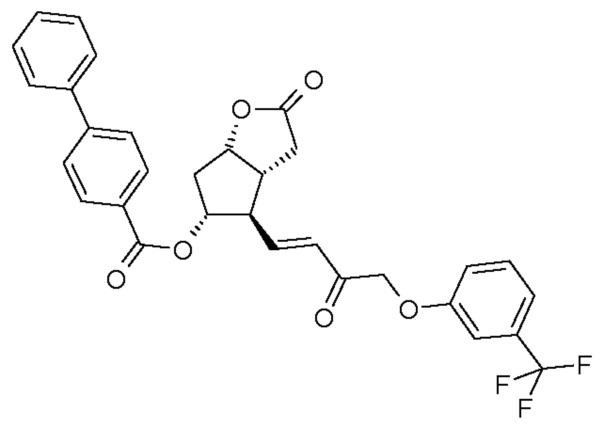
восстановления лактонной группы полученного соединения формулы (III):
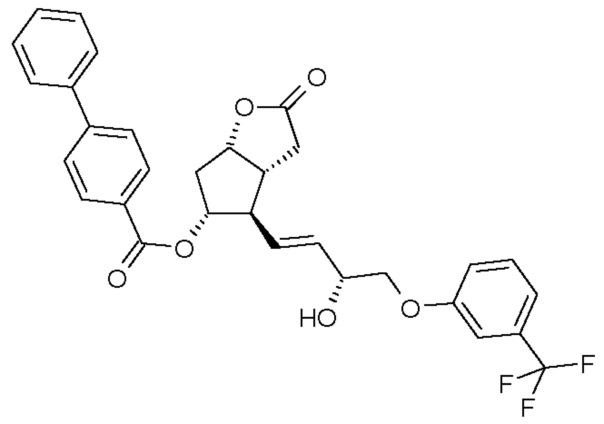
удаления п-фенилбензоильной защитной группы из полученного таким образом соединения формулы (IV):
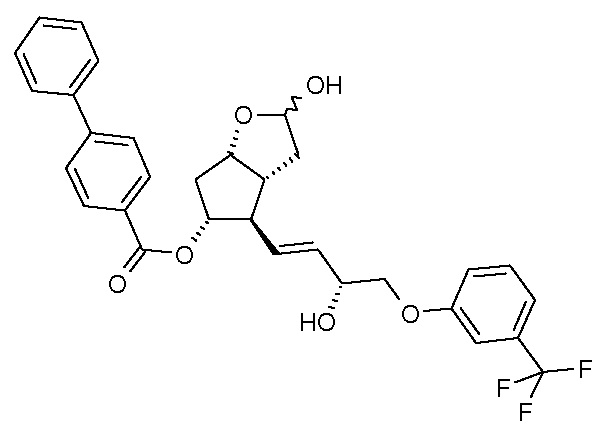
преобразования полученного триола формулы (V) по реакции Виттига:
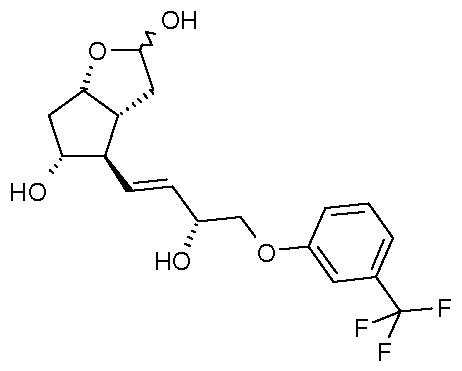
в кислоту формулы (VI):
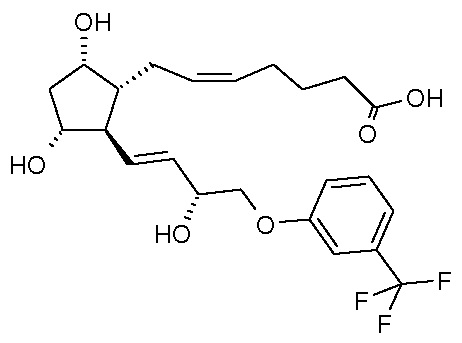
которую затем этерифицируют.
Исходное соединение формулы (II) можно получить, например, путем окисления PPB-Corey-лактона формулы (XII):
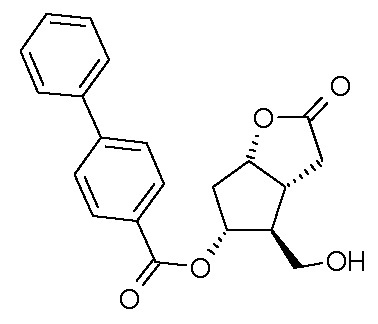
в альдегид, который затем превращают в фосфонат формулы (XIII):
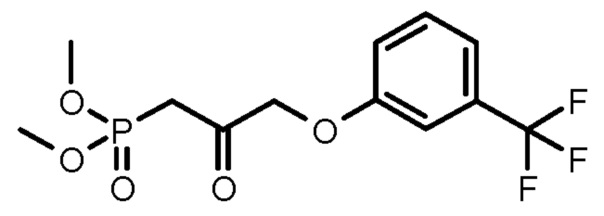
по реакции HWE в безводной среде в присутствии твердого гидроксида калия и затем в соединение формулы (II).
В одном варианте осуществления способа, основанного на настоящем изобретении, PPB-Corey-лактон окисляют по реакции Pfitzner-Moffatt в альдегид (Pfitzner, K.E., Moffatt J.G.; J.Am.Chem.Soc. 1963, 85, 3027), затем меньшую цепь формируют по реакции Horner-Wadsworth-Emmons (HWE) (Wadsworth, W.; Org. React., 1977, 25, 73) - с использованием подходящего фосфоната - в безводной среде в присутствии твердого гидроксида калия. Для депротонирования фосфоната - вместо использования подробно описанных гидрида натрия, трет-бутилата калия, карбоната лития, DBU, галогенидов лития или магния, триэтиламина, гексаметилдисилазида калия (KHMDS) или краун-эфиров - оснований заявители настоящего изобретения использовали твердый гидроксид калия, который является экономичным и может безопасно использоваться в промышленном масштабе.
Реакцию HWE проводят в апротонном органическом растворителе при температуре в диапазоне от 40 до -50°C, предпочтительно при -10°C, с использованием в качестве растворителя ароматического углеводорода, такого как толуол, или простого эфира, такого как тетрагидрофуран, метилтетрагидрофуран, циклопентилметиловый эфир, диметоксиэтан, трет-бутилметиловый эфир, диизопропиловый эфир, диэтиловый эфир или их смеси.
В другом варианте осуществления настоящего изобретения селективное восстановление соединения формулы (II) проводят восстановительным реагентом типа борана.
В качестве восстановительного реагента типа борана можно использовать боран-диметилсульфид, (-)-В-хлордиизопинокамфенилборан (DIP-C1), катехолборан, предпочтительно катехолборан. В другом варианте осуществления способа восстановление соединения формулы (II) проводят в присутствии хирального катализатора. В качестве хирального катализатора можно использовать CBS-оксазаборолидин. Реакцию проводят в присутствии органического растворителя, при температуре от -10°C до -90°C, от 10°C до -80°C, предпочтительно от -10 до -20°C. В качестве растворителя можно использовать
толуол, гексан, гептан, пентан, тетрагидрофуран, метилтетрагидрофуран, циклопентилметиловый эфир, диметоксиэтан, трет-бутилметиловый эфир, диизопропиловый эфир, диэтиловый эфир или их смеси, в частности, используют смеси толуол/тетрагидрофуран.
Полученное соединение формулы (III) очищают с помощью кристаллизации, при которой количество нежелательного изомера в значительной степени уменьшается. Кристаллическая форма соединения формулы (III) ранее не была известна, она является новой формой. Кристаллизацию проводят в полярных или аполярных растворителях или в их смеси.
В одном варианте осуществления способа, предлагаемого в настоящем изобретении, кристаллизацию проводят при температуре от -20 до 70°C таким образом, что вещество растворяют в спирте при кипячении с обратным холодильником и кристаллизуют путем постепенного охлаждения. Затем кристаллы отфильтровывают, промывают и сушат.
Восстановление соединения формулы (III) можно провести диизобутилалюминийгидридом (DIBAL-H). В качестве растворителя можно использовать инертные апротонные растворители, такие как ТГФ, толуол, гексан и гептан. Реакцию проводят при температуре от -80 до -50°C, предпочтительно от -80 до -70°C.
Продукт восстановления с помощью DIBAL-H, промежуточное соединение формулы (IV), является новым соединением.
Защитную группу РРВ можно удалить по известным методикам путем метанолиза в щелочной среде, предпочтительно в присутствии карбоната калия.
В другом варианте осуществления способа полученное промежуточное соединение формулы (V) очищают с помощью кристаллизации, при которой количество нежелательного изомера уменьшается до необходимого ограниченного значения. Кристаллическая форма соединения формулы (V) ранее не была описана, она является новой формой. Кристаллизацию проводят в смеси полярных и аполярных растворителей. В качестве смеси полярных и аполярных растворителей можно использовать смесь этилацетат/гексан.
Превращение соединения формулы (V) в соединение формулы (VI) проводят по реакции Виттига, а этерификацию соединения формулы (VI) проводят изопропилйодидом.
В реакции этерификации в качестве растворителей используют циклические третичные амиды, такие как N-метилпирролидон и/или 1,3-диметилимидазолидинон. Этерификацию проводят при температуре от 20 до 90°C, предпочтительно в диапазоне от 40 до 50°C.
Другим объектом настоящего изобретения является новое соединение формулы (IV):
и его применение для получения травопроста.
Кроме того, объектом настоящего изобретения является кристаллическое соединение формулы (III):
имеющее температуру плавления 129,5-134,5°C, и его применение для получения травопроста.
Кроме того, объектом настоящего изобретения является кристаллическое соединение формулы (V):
имеющее температуру плавления 85,4-86,6°C, и его применение для получения травопроста.
Один вариант осуществления полного синтеза травопроста, предлагаемого в настоящем изобретении, представлен ниже на схеме 1:
Схема 1
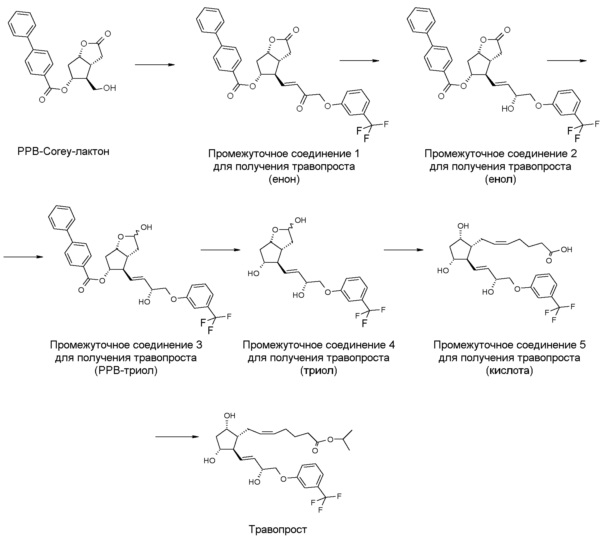
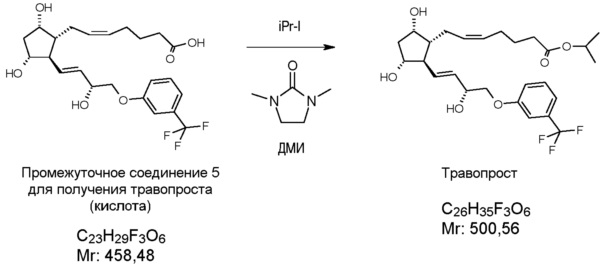
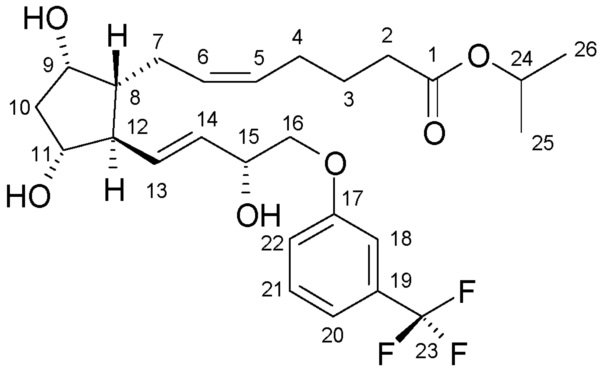
В одном варианте осуществления настоящего изобретения, который начинается с PPB-Corey-лактона, меньшую цепь формируют с помощью подходящего фосфоната по реакции Horner-Wadsworth-Emmons. Для депротонирования фосфоната используют недорогой и безопасно использующийся в промышленном масштабе твердый гидроксид калия. Восстановление полученного промежуточного соединения 1 для получения травопроста (енон - соединение формулы (II)) проводят в присутствии 2-метил-CBS-оксазаборолидинового катализатора восстановительным реагентом типа борана, таким как катехолборан, и обеспечивают стереоселективность, равную 90%. Полученное таким образом промежуточное соединение 2 для получения травопроста (енол - соединение формулы (III)) очищают с помощью кристаллизации и восстанавливают диизобутилалюминийгидридом (DIBAL-H). Из полученного промежуточного соединения 3 для получения травопроста (PPB-триол - соединение формулы (IV)) удаляют защитную группу PPB, и полученное таким образом промежуточное соединение 4 для получения травопроста (триол - соединение формулы (V)) очищают с помощью кристаллизации. Промежуточное соединение 5 для получения травопроста (кислота - соединение формулы (VI)) получают по реакции Виттига. В заключение проводят этерификацию изопропилйодидом в ДМИ (1,3-диметилимидазолидин-2-он) и получают эфир (травопрост - соединение формулы (I)).
Преимущества способа, предлагаемого в настоящем изобретении:
- В реакции HWE, для получения исходного соединения формулы (II) депротонирование фосфоната проводят недорогим и безопасно использующимся в промышленном масштабе твердым гидроксидом калия - вместо дорогого и огнеопасного гидрида натрия, который обычно и широко используется в современной практике.
- Применение CBS-оксазаборолидина и катехолборана для восстановления 15-оксогруппы при синтезе травопроста является новым подходом, не использовавшимся ранее, с помощью которого можно обеспечить диастереоизомерный избыток (ДИ), даже превышающий 90-92%. В способе, описанном в EP 2143712, при использовании DIP-Cl селективность составляет ДИ(S)=88,7%. В способе, раскрытом в WO 2011/055377 A1, кроме катализатора CBS, используют комплекс N,N-диэтиланилинборана, но степень стереоселективности не указана.
- Методика очистка является совершенно новой, поскольку удаление примеси 15-эпи проводят путем кристаллизации, без хроматографии, с высоким выходом, в отличие от методик MPLC (методика очистки с помощью хроматографии среднего давления) (WO 2011/046569 A1) или препаративной ВЭЖХ (WO 2011/055377 A1), известных в литературе.
- Кристаллическая форма соединения формулы (III) и форма соединения формулы (V) ранее не описаны в литературе. В способе, предлагаемом в настоящем изобретении, кристаллическая форма также используется для очистки промежуточных соединений и удаления нежелательного изомера.
- На стадии этерификации в качестве нового растворителя используют 1,3-диметилимидазолидинон (ДМИ), который не сильно токсичен в отличие от обычно использующегося диметилформамида (EP 2143712 A1, WO 2011/046569 A1). ДМИ является растворителем, использующимся в косметической промышленности. Дополнительным преимуществом является то, что при использовании ДМИ не образуются содержащие формил примеси, которые образуются из широко использующегося растворителя диметилформамида. Реакцию этерификации можно провести с очень высокой степенью превращения без образования новых примесей (приблизительно 100%).
- Полный выход нового способа является очень высоким, 16%, что более чем вдвое больше выхода, описанного в WO 2011/055377 A1 (7%).
- В приведенных ниже примерах приведено дополнительное подробное описание настоящего изобретения, но оно не ограничивается приведенными ниже примерами.
Примеры
1. Формирование меньшей цепи (окисление и реакция HWE)
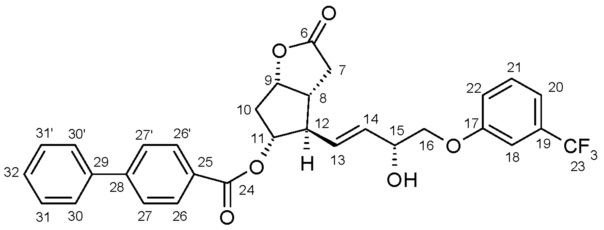
Получение (3aR,4R,5R,6aS)-гексагидро-2-оксо-4-[(1E)-3-оксо-4-[3-(трифторметил)фенокси]-1-бутен-1-ил]-2H-циклопента[b]фуран-5-илового эфира [1,1'-бифенил]-4-карбоновой кислоты (соединения формулы (II))
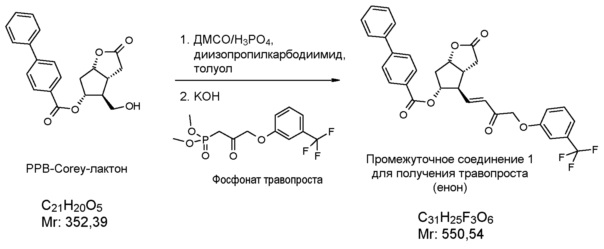
1069 г PPB-Corey-лактона суспендируют в инертной атмосфере в 11,1 л безводного толуола. К этой суспензии добавляют 1,4 л диизопропилкарбодиимида и затем 0,855 л диметилсульфоксида в фосфорной кислоте. Реакционную смесь нагревают до 50°C и порциями добавляют еще 0,34 л диметилсульфоксида в фосфорной кислоте. После завершения реакции окисления смесь охлаждают до -10°C и, поддерживая это значение температуры, добавляют 316 г гидроксида калия и затем 1,45 кг раствора фосфоната травопроста в толуоле. После завершения реакции HWE, реакционную смесь выливают в 1 M раствор хлористоводородной кислоты, и смесь перемешивают. Осадившиеся кристаллы отфильтровывают и промывают. Фазы фильтрата разделяют, органическую фазу промывают 1 M раствором гидрокарбоната натрия и затем разбавляют раствором хлористоводородной кислоты. Органическую фазу выпаривают и очищают с помощью хроматографии на колонке с силикагелем (элюент: смесь толуол/этилацетат). Главную фракцию выпаривают и кристаллизуют из смеси этилацетат/гексан.
Выход: 915 г, 55%.
Температура плавления: 112,5-114,5°C.
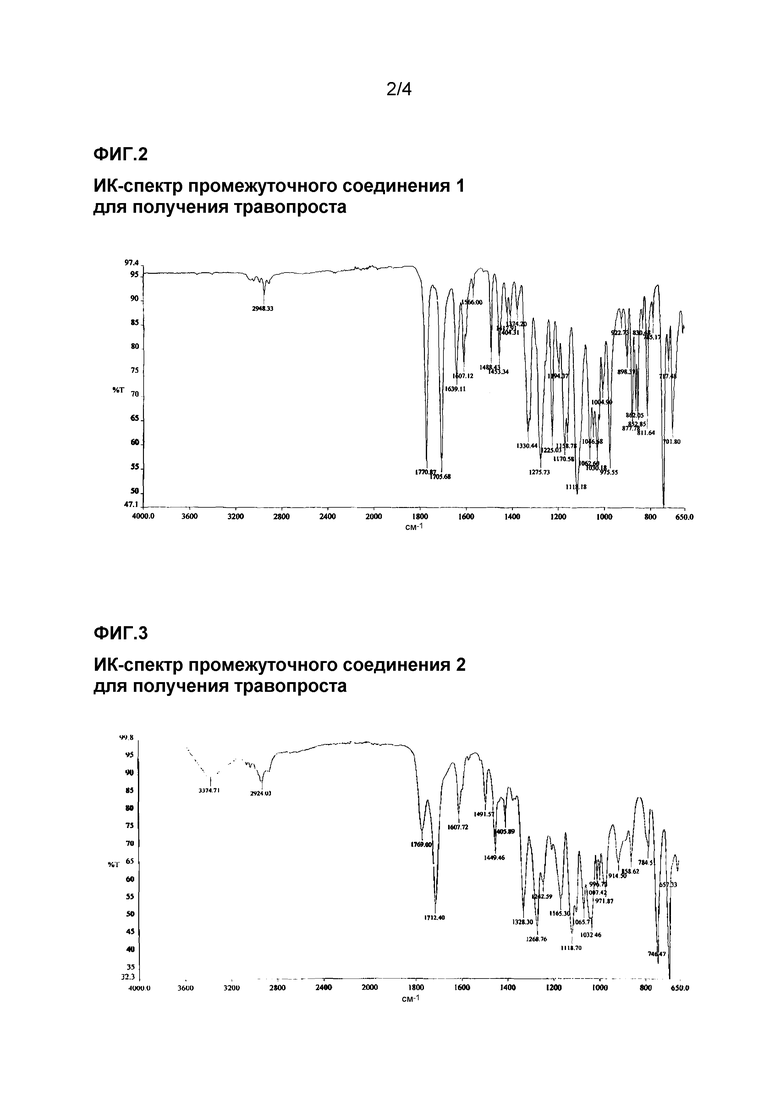
ИК-спектр промежуточного соединения 1 для получения травопроста приведен на фиг. 2.
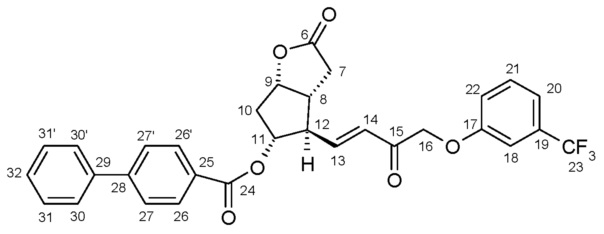
Промежуточное соединение 1 для получения травопроста (енон формулы (II)):
(м.д.)
(м.д.)
(Гц)
(±0,2 Гц)
α: 2,55
1
д
α: 2,14
1
дд
2. Восстановление 15-оксогруппы (стереоселективное восстановление)
Получение (3aR,4R,5R,6aS)-гексагидро-4-[(1E,3R)-3-гидрокси-4-[3-(трифторметил)фенокси]-1-бутен-1-ил]-2-оксо-2H-циклопента[b]фуран-5-илового эфира [1,1'-бифенил]-4-карбоновой кислоты (соединения формулы (III))
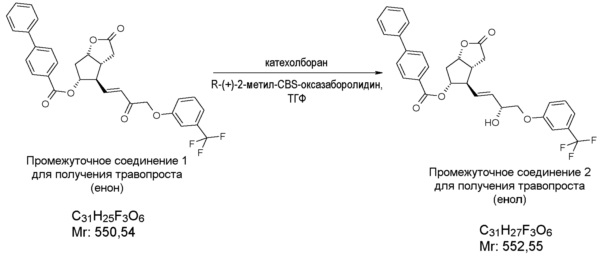
279 мл катехолборана растворяют в 4,6 л тетрагидрофурана (ТГФ) и к нему добавляют 549 мл 1 M раствора R-(+)-2-метил-CBS-оксазаборолидина в толуоле. Смесь охлаждают до -10°C и, поддерживая это значение температуры, добавляют 915 г промежуточного соединения 1 для получения травопроста (енон - соединение формулы (II)) в 6,9 л ТГФ. После завершения реакции смесь разлагают путем перемешивания с 13 л 1 M раствора NaHSO4. Затем добавляют этилацетат, и фазы разделяют. Органическую фазу промывают раствором NaOH и затем раствором хлористоводородной кислоты. Органическую фазу сушат над сульфатом натрия, фильтруют, выпаривают и для удаления нежелательного изомера кристаллизуют сначала из смеси гексан/ацетон, затем из метанола ДИ(S)92% - >ДИ(S)98%.
(ДИ означает: диастереоизомерный избыток)
Выход: 701 г, 55% ДИ(S): 98%.
Температура плавления: 129,5-134,5°C.
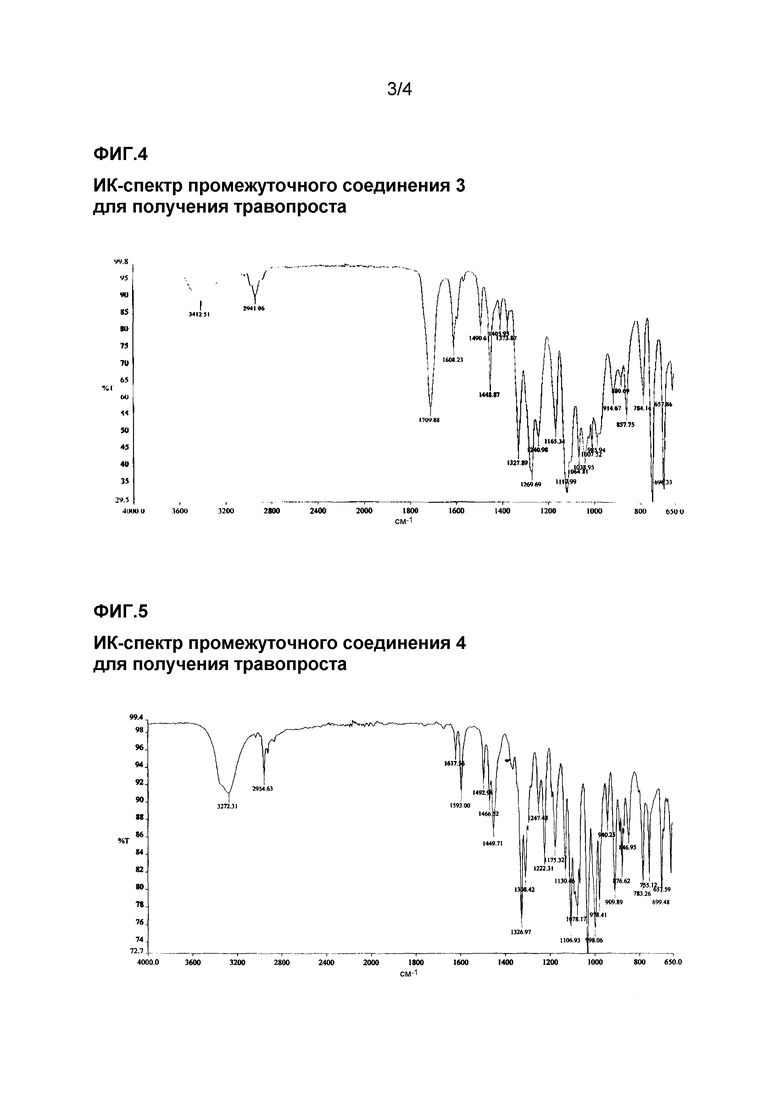
ИК-спектр промежуточного соединения 2 для получения травопроста приведен на фиг. 3.
(м.д.)
(м.д.)
(Гц)
(±0,2 Гц)
α: 2,46
1
дд
J7α,8=0,9
α: 2,05
1
м (дд)
J10α,11=4,6
b: 3,90
1
дд
J15,16b=6,7
3. Восстановление лактона (получение лактола)
Получение (3aR,4R,5R,6aS)-гексагидро-4-[(1E,3R)-3-гидрокси-4-[3-(трифторметил)фенокси]-1-бутен-1-ил]-2-гидроксициклопента[b]фуран-5-илового эфира [1,1'-бифенил]-4-карбоновой кислоты (соединения формулы (IV))
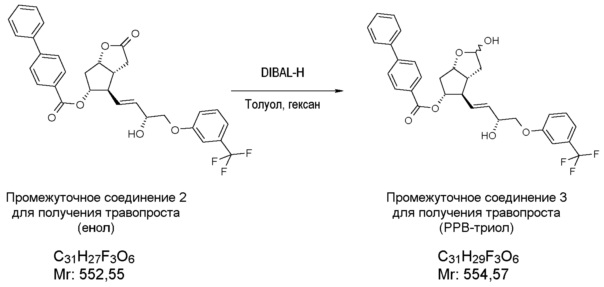
В многогорлую колбу в атмосфере азота помещают 701 г енола, который затем при комнатной температуре растворяют в 6,8 л ТГФ. Прозрачный раствор охлаждают до -75°C, и к нему в течение примерно 30 мин добавляют предварительно охлажденный (-75°C) 1 M раствор 2921 мл диизобутилалюминийгидрида (DIBAL-H) в гексане. Реакционную смесь перемешивают при -75°C до завершения реакции. После достижения подходящей степени превращения реакционную смесь выливают в смесь раствора NaHSO4 и этилацетата. Фазы разделяют, водную фазу экстрагируют этилацетатом, объединенную органическую фазу промывают раствором NaHCO3 и разбавленным раствором хлористоводородной кислоты и затем выпаривают, добавив к ней триэтиламин (ТЭА). Получают 639,5 г масла.
Выход: 639,5 г, 91%.
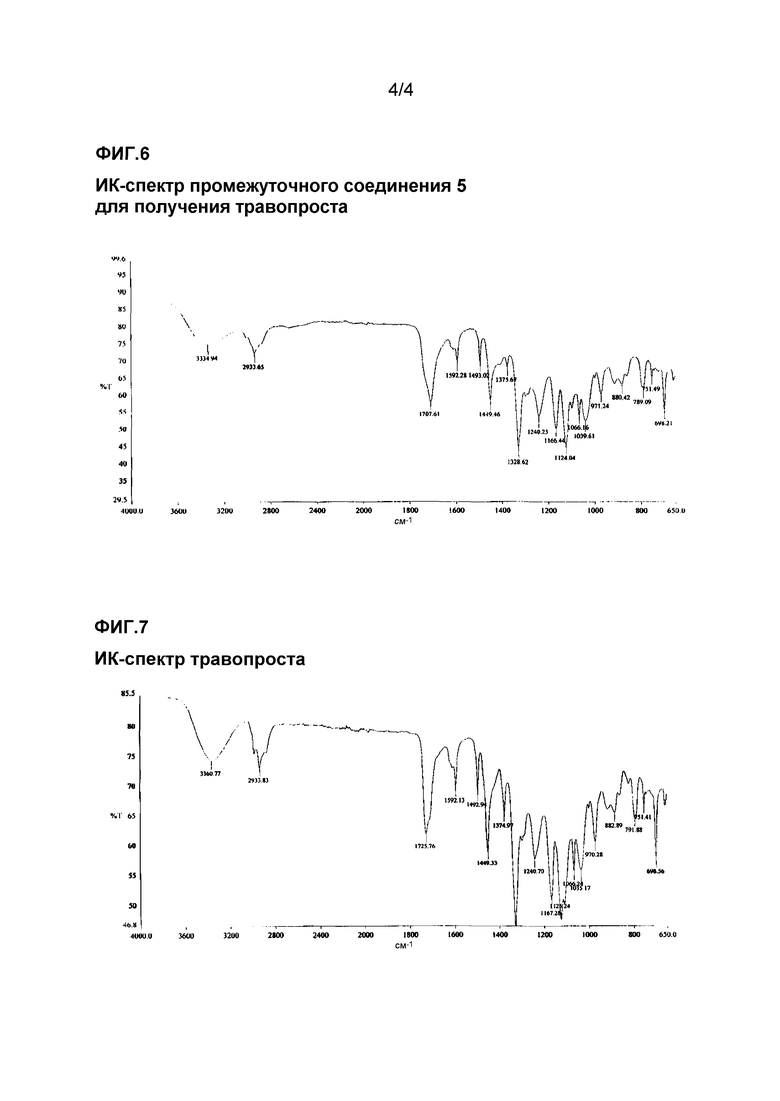
ИК-спектр промежуточного соединения 3 для получения травопроста приведен на фиг. 4.
А)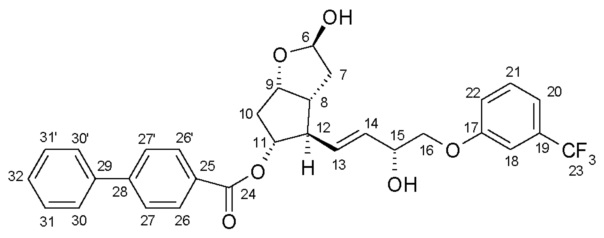
В)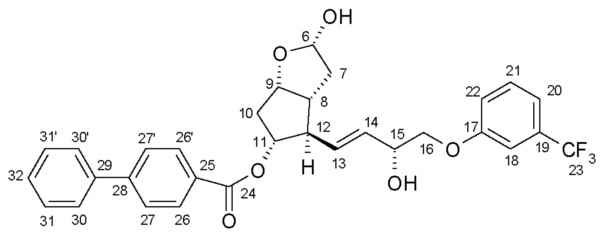
(м.д.)
(м.д.)
(±0,2 Гц)
b: 1,89*
1
м
α: 1,74***
1
м (ддд)
J10α,11=6,9
b: 3,87#
1
м (дд)
J15,16b=6,7
(с, 3)
$$: перекрывающиеся сигналы 13C-ЯМР.
(м.д.)
(м.д.)
(Гц)
(±0,2 Гц)
α: 1,73***
1
м
Jgem~11,8; J6,7α=1,9
α: 1,90*
1
м
b: 3,84#
1
м (дд)
J15,16b=6,6
(с, 3%)
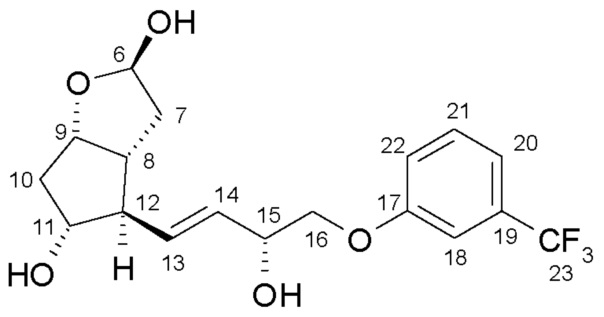
4. Удаление защитной группы (получение триола)
4a
Получение (3aR,4R,5R,6aS)-гексагидро-4-[(1E,3R)-3-гидрокси-4-[3-(трифторметил)фенокси]-1-бутен-1-ил]-2H-циклопента[b]фуран-2,5-диола (соединения формулы (V))
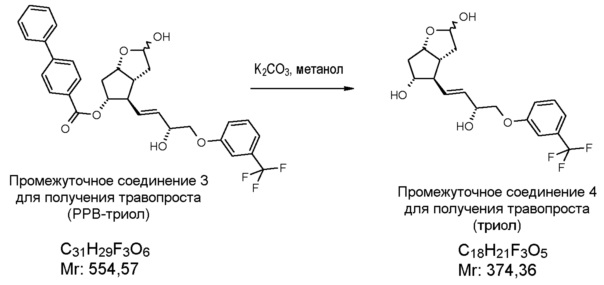
639,5 г PPB-триола растворяют в 6,4 л метанола, и раствор нагревают при 40°C. Добавляют 95 г K2CO3, и смесь перемешивают при 40°C до завершения реакции. После достижения подходящей степени превращения реакционную смесь охлаждают до 2°C, и порциями добавляют раствор фосфорной кислоты. Осадившиеся кристаллы PPB-метилового эфира отфильтровывают и промывают. Фильтрат концентрируют, добавляют воду и этилацетат, и фазы разделяют. Водную фазу экстрагируют этилацетатом, сушат над Na2SO4, и раствор выпаривают. Неочищенное масло кристаллизуют из смеси этилацетат/гексан. Осадившиеся кристаллы отфильтровывают, промывают смесью гексан/этилацетат и сушат.
Выход: 367 г, 85%.
Температура плавления: 85,4-86,6°C.
4b
Перекристаллизация (3aR,4R,5R,6aS)-гексагидро-4-[(1E,3R)-3-гидрокси-4-[3-(трифторметил)фенокси]-1-бутен-1-ил]-2H-циклопента[b]фуран-2,5-диола (соединения формулы (V) - триола)
Осадившиеся кристаллы растворяют в 10-кратном количестве этилацетата, затем добавляют 10-кратное количество н-гексана, и раствор перемешивают при комнатной температуре. К полученной суспензии кристаллов добавляют 20-кратное количество н-гексана и перемешивают при комнатной температуре. Осадившиеся кристаллы фильтруют, промывают смесью гексан/этилацетат и сушат. В случае проводимого в любое время повторения указанной выше процедуры количество нежелательного изомера может быть уменьшено до любого значения, меньшего пренебрежимо малого предельного значения (<0,05%).
Выход: 52-85% (в зависимости от количества операций перекристаллизации).
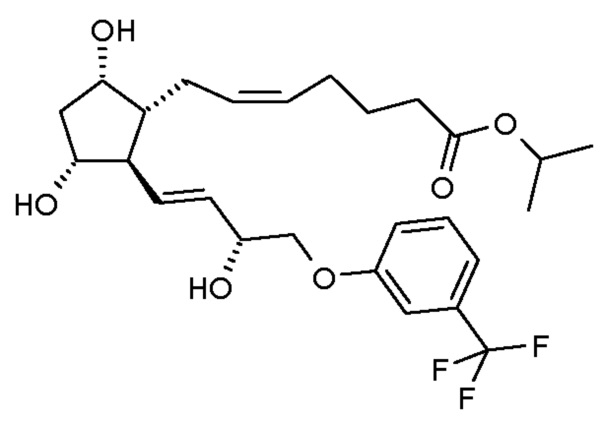
ИК-спектр промежуточного соединения 4 для получения травопроста приведен на фиг. 5.
(м.д.)
(м.д.)
(Гц)
(±0,2 Гц)
α: 1,44
1
м (ддд)
b: 3,92#
1
м (дд)
J15,16b=7,0
(с, 3)
$$: перекрывающийся сигнал 13C-ЯМР.
(м.д.)
(м.д.)
(Гц)
(±0,2 Гц)
α: 1,61
1
м
Jgem=12,9; J6,7α~1,5
α: 1,72*
1
м (ддд)
b: 3,92#
1
м (дд)
J15,16b=6,9
5. Формирование большой цепи (получение травопроста в форме кислоты)
Получение (5Z)-7-[(1R,2R,3R,5S)-3,5-дигидрокси-2-[(1E,3R)-3-гидрокси-4-[3-(трифторметил)фенокси]-1-бутен-1-ил]циклопентил]-5-гептановой кислоты (соединения формулы (VI))
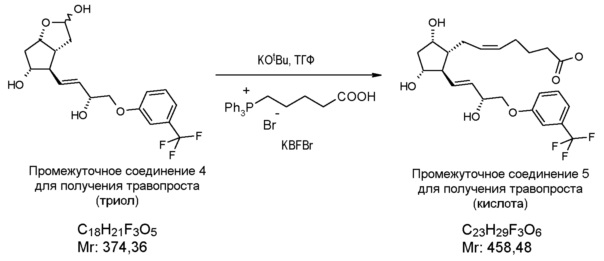
В атмосфере азота 1509 г 4-карбоксибутилфосфонийбромида (KBFBr) растворяют в 12,8 л ТГФ, раствор охлаждают до 0°C и, поддерживая это значение температуры, к нему порциями добавляют 1,12 кг трет-бутилата калия. После перемешивания в течение 15 мин, реакционную смесь охлаждают до -10°C, затем добавляют 367 г триола, растворенного в 2,24 л ТГФ, и смесь перемешивают при -10°C. После завершения реакции реакционную смесь разлагают водой и добавляют толуол. Водную фазу экстрагируют дихлорметаном (ДХМ) и подкисляют раствором NaHSO4. Затем добавляют этилацетат, фазы разделяют, и водную фазу экстрагируют этилацетатом. Объединенную органическую фазу промывают разбавленным раствором хлорида натрия, сушат над Na2SO4, осушающий материал отфильтровывают, фильтрат промывают, и раствор фильтрата выпаривают. Остаток кристаллизуют из смеси ацетон/диизопропиловый эфир. Кристаллы отфильтровывают, промывают смесью диизопропиловый эфир/ацетон. Маточный раствор выпаривают.
Выход: 463 г, 103%.
ИК-спектр промежуточного соединения 5 для получения травопроста приведен на фиг. 6.
(м.д.)
(м.д.)
(Гц)
(±0,2 Гц)
a: 1,96***
1
м
a: 3,93+
1
дд
J15,16a=6,6
(с, 3)
6. Получение травопроста (соединения формулы (I))
463 г травопроста в форме кислоты растворяют в 2,3 л 1,3-диметилимидазолидинон (ДМИ), и добавляют 420 г K2CO3 и 300 мл изопропилйодида. Реакционную смесь перемешивают при 45°C. После завершения реакции добавляют раствор NaHSO4, воду, гексан и этилацетат. Смесь встряхивают, затем фазы разделяют, и нижнюю водную фазу экстрагируют смесью гексан/этилацетат. Объединенную органическую фазу промывают водой, сушат над Na2SO4, осушающий материал отфильтровывают, и раствор выпаривают. Продукт очищают с помощью хроматографии на силикагеле, с использованием смеси диизопропиловый эфир, ацетон, дихлорметан, изопропанол в качестве элюента.
Выход: 338,7 г, 67%.
ИК-спектр травопроста приведен на фиг. 7.
(м.д.)
(м.д.)
(Гц)
(±0,2 Гц)
a: 1,96***
1
м
a: 1,44**
1
ддд
J9,10b=5,9; J10a,11=5,7; J9,10a=2,3;
b: 3,95
1
м
(с, 3)
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения дибензо[f,h]фуразано[3,4-b]хиноксалина и его замещенных производных, обладающих зарядотранспортными полупроводниковыми свойствами | 2019 |
|
RU2723014C1 |
| ЗАМЕЩЕННЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ АУТОРЕПАРАЦИИ И РЕГЕНЕРАЦИИ ТКАНИ | 2015 |
|
RU2728782C2 |
| 5-Арилзамещенный 4-(5-нитрофуран-2-ил)пиримидин, обладающий широким спектром антибактериальной активности, способ его получения и промежуточное соединение, обладающее широким спектром антибактериальной активности | 2016 |
|
RU2626647C1 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2013 |
|
RU2651369C1 |
| ПРОИЗВОДНЫЕ ИНДОЛА, ИХ ТАУТОМЕРЫ, СМЕСИ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С АНТИОПУХОЛЕВОЙ ИЛИ ИНГИБИРУЮЩЕЙ ПРОТЕИН-ТИРОЗИНКИНАЗУ АКТИВНОСТЬЮ И СПОСОБ ТОРМОЖЕНИЯ ЗАВИСЯЩЕГО ОТ ПРОТЕИН-ТИРОЗИНКИНАЗЫ ЗАБОЛЕВАНИЯ ИЛИ БОРЬБЫ С АБЕРРАНТНЫМ РОСТОМ КЛЕТОК МЛЕКОПИТАЮЩЕГО ИЛИ ЧЕЛОВЕКА. | 1993 |
|
RU2155187C2 |
| ПРОТИВОГРИБКОВЫЕ СОЕДИНЕНИЯ НА ОСНОВЕ ПРОИЗВОДНЫХ 3,5,8-ТРИОКСАБИЦИКЛО[5.1.0]ОКТАНА | 2013 |
|
RU2538962C1 |
| ЗАМЕЩЕННЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДОТВРАЩЕНИЯ И ЛЕЧЕНИЯ ОСТЕОПОРОЗА | 2015 |
|
RU2709205C2 |
| НОВЫЕ ГЕМ-ДИФТОРИРОВАННЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2369612C2 |
| МОЛЕКУЛЫ С ОПРЕДЕЛЕННОЙ ПЕСТИЦИДНОЙ АКТИВНОСТЬЮ И ОТНОСЯЩИЕСЯ К НИМ ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, КОМПОЗИЦИИ И СПОСОБЫ | 2014 |
|
RU2650498C2 |
| ПРИМЕНЕНИЕ СОЕДИНЕНИЙ ПИКОЛИНАМИДА В КАЧЕСТВЕ ФУНГИЦИДОВ | 2015 |
|
RU2702088C2 |
Изобретение относится к способу получения травопроста формулы (I), отличающемуся тем, что соединение формулы (II) стереоселективно восстанавливают восстановительным реагентом типа борана в присутствии хирального катализатора, полученное соединение формулы (III) при желании кристаллизуют, лактонную группу соединения формулы (III) восстанавливают диизобутилалюминийгидридом, п-фенилбензоильную защитную группу полученного таким образом соединения формулы (IV) удаляют путем метанолиза, полученный триол формулы (V) при желании после кристаллизации превращают по реакции Виттига в
кислоту формулы (VI), которую затем этерифицируют в растворителях типа циклического третичного амида изопропилйодидом. Способ имеет более высокую селективность и лучший выход.
 (I)
(I) 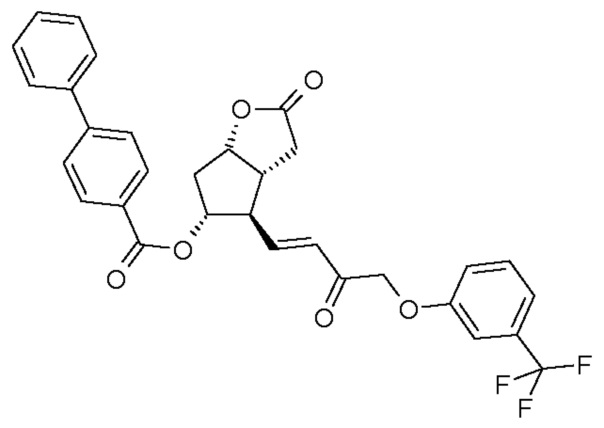 (II)
(II)
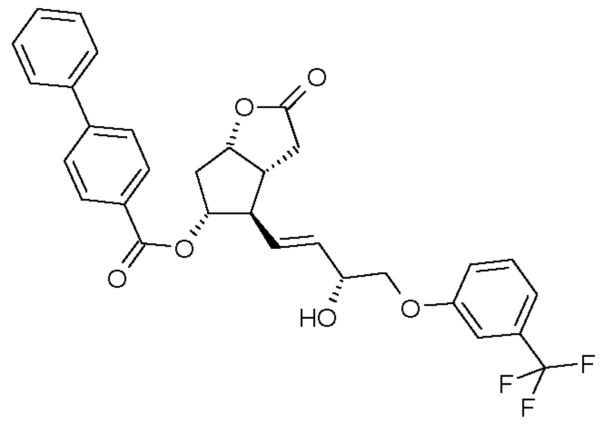 (III)
(III) 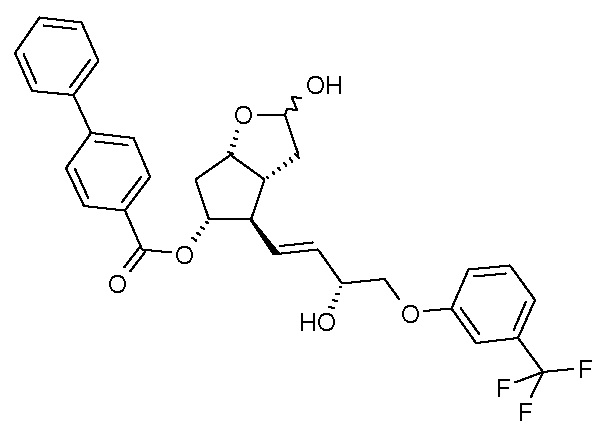 (IV)
(IV)
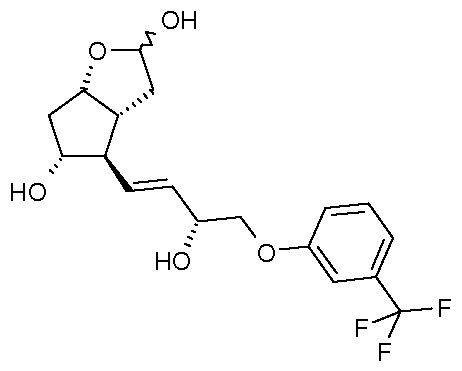 (V)
(V) 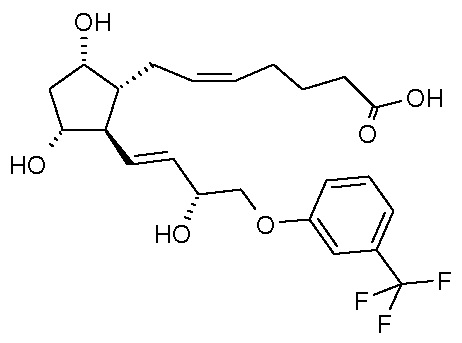 (VI)
(VI)
4 н. и 24 з.п. ф-лы, 7 ил., 6 пр.
1. Способ получения травопроста формулы (I):
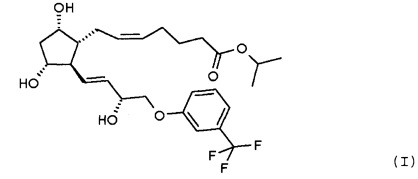
отличающийся тем, что соединение формулы (II):
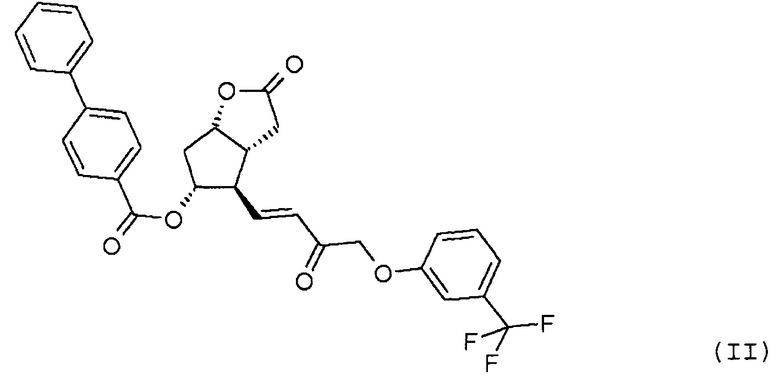
стереоселективно восстанавливают восстановительным реагентом типа борана в присутствии хирального катализатора, полученное соединение формулы (III):
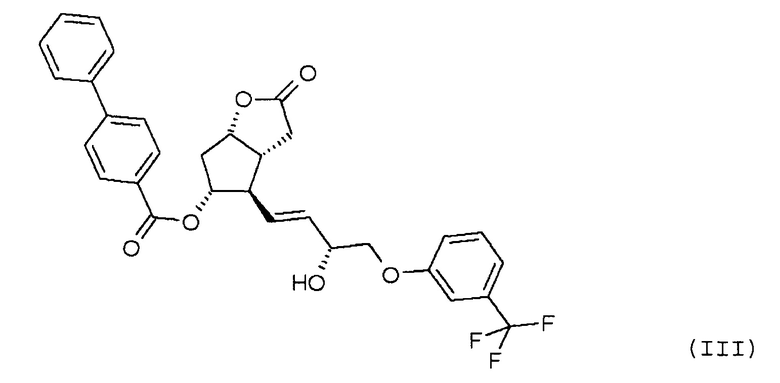
при желании кристаллизуют, лактонную группу соединения формулы (III) восстанавливают диизобутилалюминийгидридом, п-фенилбензоильную защитную группу полученного таким образом соединения формулы (IV):
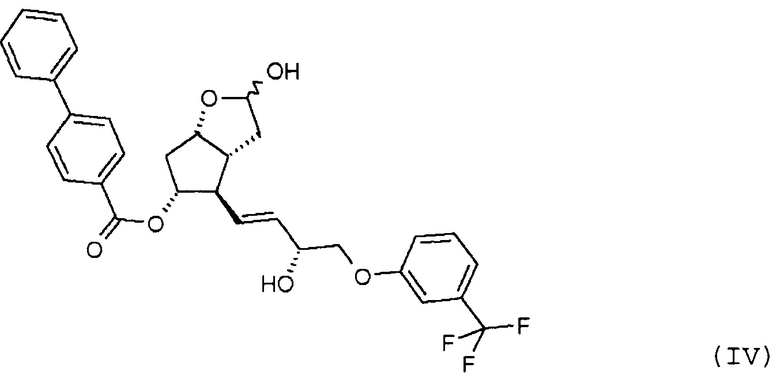
удаляют путем метанолиза, полученный триол формулы (V):
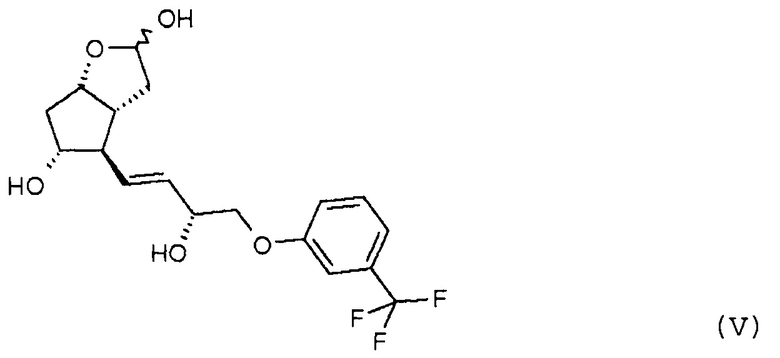
при желании после кристаллизации превращают по реакции Виттига в
кислоту формулы (VI):
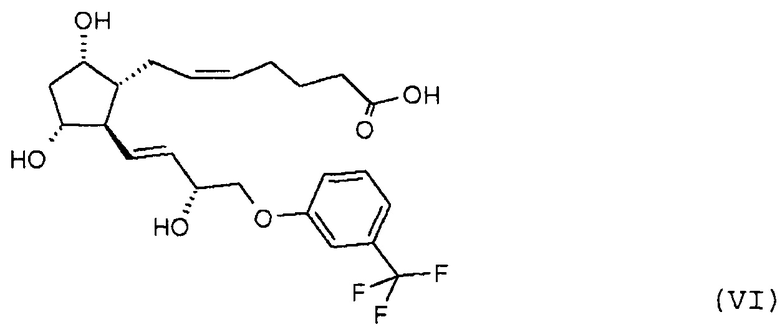
которую затем этерифицируют в растворителях типа циклического третичного амида изопропилйодидом.
2. Способ по п. 1, отличающийся тем, что в качестве восстановительного реагента типа борана используют катехолборан.
3. Способ по п. 1, отличающийся тем, что в качестве хирального катализатора используют CBS-оксазаборолидин.
4. Способ по пп. 1-3, отличающийся тем, что восстановление проводят в растворителях типа углеводорода или простого эфира.
5. Способ по п. 4, отличающийся тем, что восстановление проводят в толуоле, гексане, гептане, пентане, тетрагидрофуране, метилтетрагидрофуране, циклопентилметиловом эфире, диметоксиэтане, трет-бутилметиловом эфире, диизопропиловом эфире, диэтиловом эфире или в их смеси.
6. Способ по п. 5, отличающийся тем, что восстановление проводят в смеси толуол/тетрагидрофуран.
7. Способ по пп. 1-3, 5 или 6, отличающийся тем, что восстановление проводят при температуре от -10 до -90°C.
8. Способ по п. 7, отличающийся тем, что восстановление проводят при температуре от -10 до -20°C.
9. Способ по пп. 1-3, 5 или 6, отличающийся тем, что восстановление проводят при температуре от 10 до -80°C.
10. Способ по пп. 1-3, 5, 6 или 8, отличающийся тем, что полученное соединение формулы (III) очищают с помощью кристаллизации.
11. Способ по п. 10, отличающийся тем, что кристаллизацию проводят в растворителях типа углеводорода, хлорированного углеводорода, простого эфира, сложного эфира, кетона или спирта или в их смеси.
12. Способ по п. 11, отличающийся тем, что кристаллизацию проводят многократно в различных растворителях или в их смеси.
13. Способ по п. 12, отличающийся тем, что кристаллизацию проводят в смеси гексан/ацетон и/или в метаноле.
14. Способ по пп. 11-13, отличающийся тем, что кристаллизацию проводят при температуре от -20 до 70°C таким образом, что вещество растворяют в спирте при кипячении с обратным холодильником, кристаллизуют путем постепенного охлаждения и затем отфильтровывают, промывают и сушат.
15. Способ по п. 1, отличающийся тем, что п-фенилбензоильную защитную группу удаляют в присутствии карбоната калия.
16. Способ по п. 1, отличающийся тем, что промежуточное соединение формулы (V) очищают с помощью кристаллизации.
17. Способ по п. 16, отличающийся тем, что кристаллизацию проводят в смеси полярных и аполярных растворителей.
18. Способ по п. 17, отличающийся тем, что кристаллизацию проводят в смеси этилацетат/гексан.
19. Способ по п. 17 или 18, отличающийся тем, что при надлежащем количестве повторений процедуры кристаллизации количество нежелательного изомера уменьшается до значения,
меньшего пренебрежимо малого предельного значения (0,05%).
20. Способ по п. 1, отличающийся тем, что в качестве растворителя типа циклического третичного амида используют N-метилпирролидон или 1,3-диметилимидазолидинон.
21. Способ по п. 1 или 20, отличающийся тем, что этерификацию проводят при температуре в диапазоне от 20 до 90°C.
22. Способ по п. 21, отличающийся тем, что этерификацию проводят при температуре в диапазоне от 40 до 50°C.
23. Способ по п. 1, отличающийся тем, что продукт формулы (I) очищают с помощью хроматографии.
24. Способ по п. 23, отличающийся тем, что продукт очищают с помощью гравиметрической хроматографии на силикагеле.
25. Способ по п. 24, отличающийся тем, что хроматографическую очистку проводят с использованием смеси диизопропилового эфира, ацетона, дихлорметана, изопропанола в качестве элюентов.
26. Соединение формулы (IV):
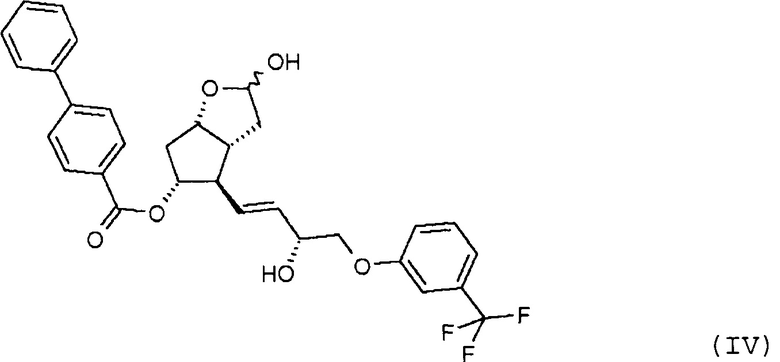
27. Применение соединения формулы (IV) по п. 26 для получения травопроста формулы (I).
28. Способ получения соединения формулы (IV), отличающийся тем, что соединение формулы (II):
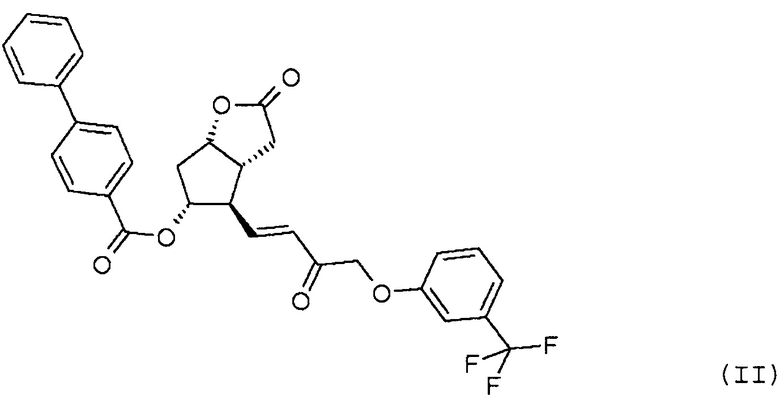
стереоселективно восстанавливают восстановительным реагентом типа борана в присутствии хирального катализатора и лактонную группу полученного соединения формулы (III):
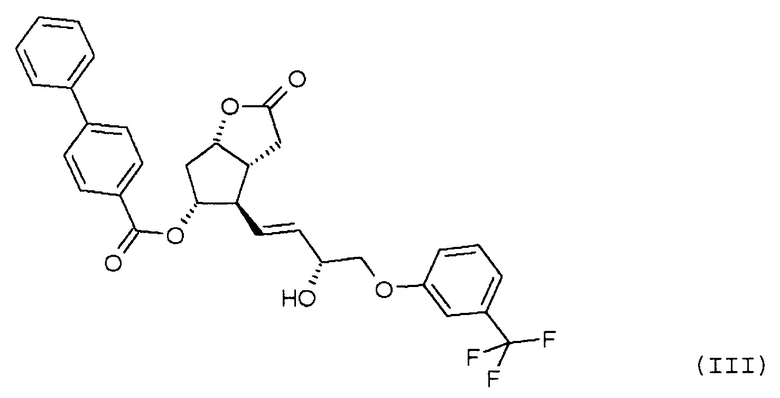
восстанавливают диизобутилалюминийгидридом.
| Aswathanarayanappa et al | |||
| "Diastereoselective reduction of the enone intermediate of travoprost" Organic Process Research & Development, 15(5), p | |||
| АППАРАТ, СЛУЖАЩИЙ ДЛЯ ОДНОВРЕМЕННОГО ВОСПРОИЗВЕДЕНИЯ С ОДНОЙ И ТОЙ ЖЕ ЛЕНТЫ ЗВУКА, ЦВЕТА И СТЕРЕОСКОПИЧНОСТИ ДВИЖУЩЕГОСЯ ПРЕДМЕТА | 1921 |
|
SU1085A1 |
| КАМЕРА ОБЛУЧЕНИЯ И/ИЛИ ПОДСЧЕТА ДЛЯ НЕЙТРОННОГО АНАЛИЗА | 1995 |
|
RU2143712C1 |
| УРОВНЕМЕР ДЛЯ ИНДИКАЦИИ РАЗДЕЛА ФАЗ ЖИДКОСТЕЙ | 0 |
|
SU239696A1 |
| Легкоплавкая пробка для паровых котлов | 1928 |
|
SU10595A1 |

