ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицины. Конкретные аспекты изобретения относятся к соединениям, фармацевтическим композициям и их применению для ауторепарации ткани и/или регенерации ткани пораженного органа, для генерации образования роста ткани и/или для модуляции экспрессии маркеров ауторепарации ткани и/или маркеров регенерации ткани, таких как металлопротеиназы и факторы роста.
УРОВЕНЬ ТЕХНИКИ
Регенерация ткани содержит известные маркеры, такие как металлопротеиназы и факторы роста, в том числе, без ограничения этим, ФРГ (фактор роста гепатоцитов), ЛОК (лизилоксидаза), MMP1, MMP2, MMP9, MMP13, PLAT (tPA), PLAU (uPA), Serpin A1 (AAT), Serpin E1 (PAI-1), TIMP3, ИСК (интегрин-связанную киназу).
Влияние ФРГ на репарацию и регенерацию ткани хорошо описано в научном обзоре: The discovery of Hepatocyte Growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine от Nakamura and Mizuno, Proc.Jpn. Acad. Ser B86 (2010). В этой обзорной статье описана роль ФРГ в регенерации ткани в печени, почках, сердце и легких. Кроме этого, ФРГ необходим для ауторепарации после поражений кожи, желудка, кишечника, мышц и хрящей, а также участвует в развитии органа (органогенезе, в том числе митогенезе, мотогенезе и морфогенезе). ФРГ также участвует в регенерации пораженной ткани путем его модуляции фермента регенерации (металлопротеиназы), а также путем ингибирования апоптоза. Кроме того, недавние сообщения подтверждают, что ФРГ обладает противовоспалительным действием и ослабляет клеточное старение. Таким образом, генотерапия ФРГ или соединение, повышающее экспрессию и секрецию ФРГ, может представлять собой омолаживающую терапию при сердечно-сосудистых заболеваниях (Nakagami, Morishita, 2009). ФРГ также применяют для ускорения заживления ран (Li et al., BioMed Research International, Volume 2013 (2013), Article ID 470418.
Ферменты регенерации (в том числе металлопротеиназы) также очень важны в репарации и регенерации пораженных органов.
В недавней публикации (реферат, представленный на Plastic surgery meeting 2014 Radtke et al., озаглавленный Single treatment With Alpha-1 antitrypsin Enhances Nerve Regeneration After Peripheral Nerve Injury) было продемонстрировано, что AAT улучшает регенерацию периферических нервов. Применение ААТ в острой модели аксотомии привело к значительно улучшенной регенерации аксонов и повторной миелинизации, по сравнению с животными контрольной группы. Более того, после прямой инъекции AAT после острого поражения периферического нерва наблюдалось не только гистологическое, но и функциональное улучшение. Их результаты показывают, что AAT, доставленный в пораженный периферический нерв, участвует в репарации нейронов.
Старение кожи является сложным явлением, ответственным за прогрессивные изменения кожи. Старение кожи происходит в результате двух процессов: (1) внутренний процесс, соответствующий хронологическому старению, и (2) внешний процесс, возникающий главным образом из-за повреждающего эффекта стрессов от влияния факторов среды. Генетические факторы, ультрафиолетовое воздействие, климатические факторы (жесткость/ветер/холод/тепло), загрязнение (химические вещества, свободные радикалы, загрязняющие вещества, окись азота, металлы), потребление алкоголя или курение являются факторами, связанными с кожным старением.
Воздействие раздражителей компрометирует барьерную функцию рогового слоя и снижает его способность защищать кожу от стрессов от влияния факторов среды (например, ультрафиолетового облучения, инфекционных средств и т. д.). Повторная и длительная экспозиция по отношению к раздражителям окружающей среды приводит к денатурированным белкам кожи, дезорганизации слоев липидных пластинок, удалению защитных межклеточных липидов, потере естественных увлажняющих факторов и уменьшению когезии между клетками. Эти повреждения также ответственны за потерю функции ферментов, ответственных за десквамацию корнеоцитов. Существует акцентирование на этих проблемах, связанных с воздействием загрязнения, холодом, солнцем, ветром, низкой влажностью или химическими средствами. Раздражающим является любое средство, способное вызывать повреждение клеток при воздействии в течение достаточного времени и в достаточных концентрациях. Тяжесть повреждения зависит от типа и интенсивности воздействия этих раздражающих факторов. Существуют также эндогенные факторы, которые с помощью внешних факторов делают человека восприимчивым к поврежденной коже. Эти факторы содержат активное заболевание кожи, такое как экзема, состояния врожденной сухой кожи, предыдущую историю болезни кожи, чувствительную кожу и/или более старший возраст.
Для стимулирования ауторепарации ткани и регенерации ткани в поврежденном органе необходимы новые соединения и лекарственные препараты.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Общие аспекты изобретения относятся к фармацевтическому применению соединений, в соответствии с Формулой I, как определено в настоящем документе, и их фармацевтически приемлемых солей.
Конкретные аспекты изобретения относятся к применению соединений и композиций для ауторепарации ткани и/или регенерации ткани пораженного органа и/или для модуляции экспрессии маркеров ауторепарации ткани и/или маркеров регенерации ткани, таких как металлопротеиназы и факторы роста, в том числе, без ограничения этим, ФРГ, ЛОК (лизилоксидаза), MMP1, MMP2, MMP9, MMP13, PLAT (tPA), PLAU (uPA), Serpin A1 (AAT), Serpin E1 (PAI-1), TIMP3 и ИСК (интегрин-связанная киназа).
Способ ауторепарации ткани или регенерации ткани органа у нуждающегося в этом субъекта, включающий этап введения нуждающемуся в этом субъекту соединения, представленного Формулой I, или его фармацевтически приемлемой соли.
В соответствии с другим аспектом, изобретение относится к способу ауторепарации ткани или регенерации ткани органа у нуждающегося в этом субъекта, включающему введение соединения, представленного Формулой I, или его фармацевтически приемлемой соли, как определено в настоящем документе, указанному субъекту. В варианте реализации изобретения, изобретение относится к способу ауторепарации ткани органа у нуждающегося в этом субъекта, включающему введение соединения, представленного Формулой I, или его фармацевтически приемлемой соли, как определено в настоящем документе, указанному субъекту. В варианте реализации изобретения, изобретение относится к способу ремоделирования ткани органа у нуждающегося в этом субъекта, включающему введение соединения, представленного Формулой I, или его фармацевтически приемлемой соли, как определено в настоящем документе, указанному субъекту. В варианте реализации изобретения, изобретение относится к способу регенерации ткани органа у нуждающегося в этом субъекта, включающему введение соединения, представленного Формулой I, или его фармацевтически приемлемой соли, как определено в настоящем документе, указанному субъекту.
В соответствии с другим аспектом, изобретение относится к способу стимуляции генерации роста ткани с помощью соединения, представленного Формулой I, или его фармацевтически приемлемой соли, как определено в настоящем документе.
В соответствии с другим аспектом, изобретение относится к способу стимуляции экспрессии маркеров ауторепарации ткани и/или маркеры регенерации ткани, с помощью соединения, представленного Формулой I, или его фармацевтически приемлемой соли, как определено в настоящем документе. Более конкретно, указанные маркеры содержат, без ограничения этим, металлопротеиназы, факторы роста, фактор роста гепатоцитов (ФРГ), ЛОК (лизилоксидаза), MMP1, MMP2, MMP9, MMP13, PLAT (tPA), PLAU (uPA), Serpin A1 (AAT), Serpin E1 (PAI-1), TIMP3 и ИСК (интегрин-связанную киназу).
В соответствии с другим аспектом, изобретение относится к способу повышения уровня ФРГ в органе, включающем этап введения в указанный орган соединения, представленного Формулой I, или его фармацевтически приемлемой соли, как определено в настоящем документе. Орган содержат, без ограничения этим, почки, сердце, печень, легкие, кожу, желудок, кишечник, мышцы и хрящи.
В соответствии с другим аспектом, изобретение относится к способу повышения уровня AAT в органе, включающем этап введения в указанный орган соединения, представленного Формулой I, или его фармацевтически приемлемой соли, как определено в настоящем документе.
Дополнительные аспекты изобретения будут очевидны специалисту в данной области техники из нижеследующего описания, формулы изобретения и обобщений.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 представлена иллюстрация влияния Соединения I на увеличение экспрессии мРНК фактора роста гепатоцитов (ФРГ), причем фактор роста вовлечен в ауторепарацию и регенерацию ткани.
На Фиг. 2 представлена иллюстрация влияния Соединения I на модуляцию маркеров генерации, экспрессируемых в поврежденном фибробласте (НФКЧ), вовлеченном в ауторепарацию и регенерацию ткани.
На Фиг. 3 представлена иллюстрация влияния Соединения I на модуляцию маркеров генерации, экспрессируемых в поврежденных эпителиальных клетках (HK-2), вовлеченных в ауторепарацию и регенерацию ткани.
На Фиг. 4 проиллюстрировано, что Соединение I может увеличивать экспрессию мРНК Serpin A1 (AAT), вовлеченного в генерацию нервов.
На Фиг. 5 представлено отображение повышения функции органа (СКФ), наблюдаемое с Соединением I, и указывает на регенерацию ткани поврежденной почки.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящем изобретении описаны соединения Формулы I, их фармацевтически приемлемые соли, композиции, содержащие их, и применения их. Различные варианты реализации настоящего изобретения включают:
Соединения по изобретению
В соответствии с одним аспектом, изобретение относится к фармацевтическим применениям соединений, представленных Формулой I, или их фармацевтически приемлемых солей:
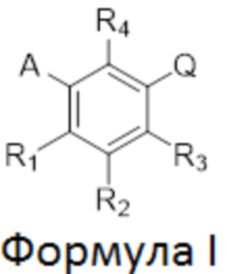
где
А представляет собой C5 алкил, C6 алкил, C5 алкенил, C6 алкенил, C(O)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3 где n равен 3 или 4; или представляет собой C5 алкил, C5 алкенил, C(O)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где n равен 3; или представляет собой C6 алкил, C6 алкенил, C(O)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где равен 4;
R1 представляет собой Н, F или OH; или представляет собой Н или OH;
R2 представляет собой Н, F, OH, C5 алкил, C6 алкил, C5 алкенил, C6 алкенил, C(O)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где n равен 3 или 4; или представляет собой C5 алкил, C5 алкенил, C(O)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где n равен 3; или представляет собой C6 алкил, C6 алкенил, C(O)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где n равен 4
R3 представляет собой Н, F, OH или CH2Ph; или представляет собой Н, F или OH; или представляет собой Н или OH;
R4 представляет собой Н, F или OH; или представляет собой Н или OH;
Q представляет собой
1) (CH2)mC(O)OH, где m равен 1 или 2,
2) CH(CH3)C(O)OH,
3) C(CH3)2C(O)OH,
4) CH(F)-C(O)OH,
5) CF2-C(O)OH или
6) C(O)-C(O)OH.
В соответствии с конкретным вариантом реализации изобретения, А представляет собой C5 алкил или C6 алкил. Предпочтительно, C5 алкил представляет собой прямую цепь C5 алкил.
В соответствии с конкретным вариантом реализации изобретения, R1 представляет собой Н или OH.
В соответствии с конкретным вариантом реализации изобретения, R2 представляет собой Н, F, OH, C5 алкил или C6 алкил.
В соответствии с конкретным вариантом реализации изобретения, R3 представляет собой Н или OH.
В соответствии с конкретным вариантом реализации изобретения, R4 представляет собой Н или OH.
В соответствии с конкретным вариантом реализации изобретения, Q представляет собой:
1) (CH2)mC(O)OH, где m равен 1 или 2,
2) CH(F)-C(O)OH,
3) CF2-C(O)OH или
4) C(O)-C(O)OH.
В соответствии с конкретным вариантом реализации изобретения, Q представляет собой (CH2)mC(O)OH, где m равен 1 или 2.
В соответствии с другим вариантом реализации изобретения, соединение представляет собой соединение Формулы I, где А представляет собой C5 алкил или C6 алкил; R1 представляет собой Н, F или OH; R2 представляет собой Н, F, OH, C5 алкил или C6 алкил; R3 представляет собой Н, OH или CH2Ph; R4 представляет собой Н, F или OH; и Q представляет собой (CH2)mC(O)OH, где m равен 1 или 2.
В соответствии с другим вариантом реализации изобретения, соединение представляет собой соединение Формулы I; где А представляет собой C5 алкил; R1 представляет собой Н; R2 представляет собой Н или C5 алкил; R3 представляет собой Н; R4 представляет собой Н; и Q представляет собой (CH2)mC(O)OH, где m равен 1.
Используемый в настоящем документе, термин “алкил” предназначен для того, чтобы включать насыщенную алифатическую углеводородную группу с прямой цепью, имеющую определенное число атомов углерода в линейном расположении, и насыщенную алифатическую углеводородную группу с разветвленной цепью, имеющую определенное число атомов углерода в нелинейной расположении, или насыщенную алифатическую углеводородную группу с циклической цепью, имеющую определенное число атомов углерода в циклическом положении.
Используемый в настоящем документе, термин “алкенил” предназначен для того, чтобы означать ненасыщенные углеводородные группы с прямой цепью, имеющие определенное число атомов углерода в них, и в которых по меньшей мере два атома углерода связаны друг с другом двойной связью и имеют либо региохимию E или Z, либо их комбинации.
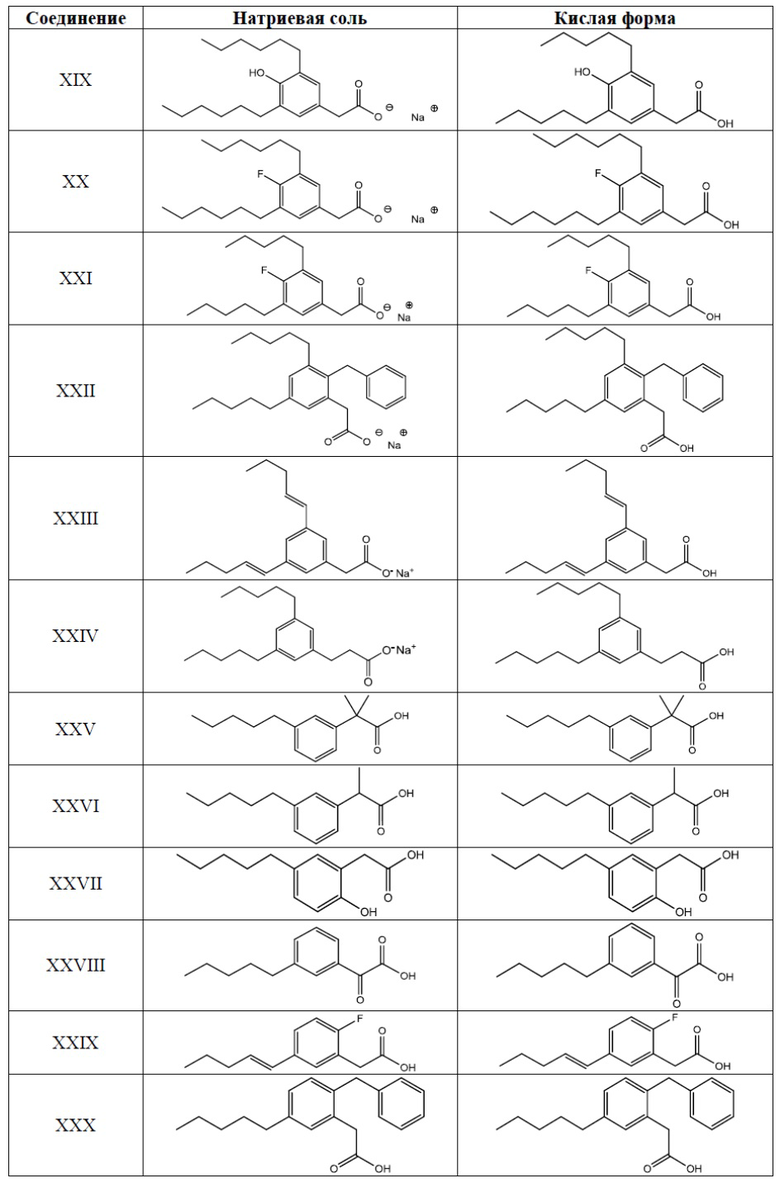
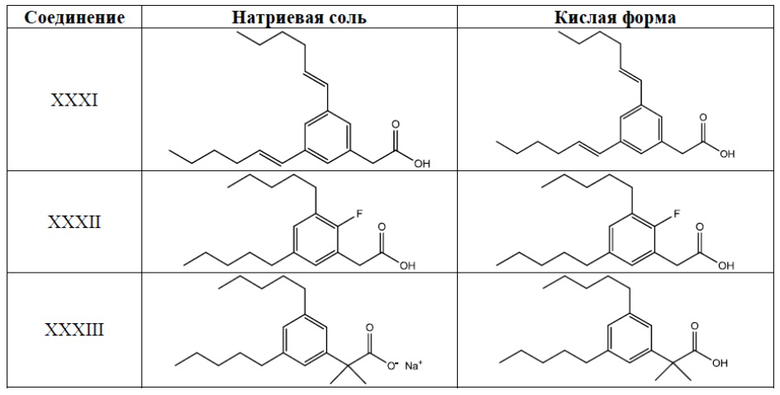
Примеры соединений Формулы I включают, но не ограничиваясь этим, Соединения I-XXXIII и их кислую форму, приведенные в Таблице 1 ниже.
Таблица 1: Типичные соединения Формулы I их кислая форма


Соли
Используемый в настоящем документе, термин “фармацевтически приемлемая соль” предназначен для того, чтобы означать соли присоединения основания. Пример фармацевтически приемлемых солей также описан, например, у Berge et al., “Pharmaceutical Salts”, J. Pharm. Sci. 66, 1-19 (1977). Фармацевтически приемлемые соли могут быть синтезированы из исходного средства, которое содержит кислую функциональную группу, с помощью обычных химических способов. Как правило, такие соли получают путем реакции свободных кислых форм этих средств со стехиометрическим количеством приемлемого основания в воде или в органическом растворителе, или в смеси двух. Соли могут быть получены in situ, во время окончательного выделения или очистки средства или путем отдельного взаимодействия очищенного соединения по изобретению в его свободной кислой форме с желаемым соответствующим основанием и выделения образовавшейся таким образом соли.
Фармацевтически приемлемая соль соединений Формулы I может быть выбрана из группы, состоящей из солей присоединения основания натрия, калия, кальция, магния, лития, аммония, марганца, цинка, железа или меди. В предпочтительных вариантах реализации изобретения, фармацевтически приемлемая соль соединений в соответствии с изобретением может быть натриевой, калиевой, кальциевой, магниевой или литиевой солью. Более предпочтительно, фармацевтически приемлемая соль является натриевой.
Соединения Формулы I, описанные в данном документе, могут быть в любой форме, в том числе в любой кислой, солевой или других ионных и неионных формах. Например, если соединение показано в данном документе как кислота, также содержатся солевые формы соединения. Аналогично, если соединение показано в виде соли, также содержатся кислые формы.
Пролекарства
В некоторых вариантах реализации изобретения, соединения Формулы I, описанные в данном документе, где указанные соединения присутствуют в форме свободной карбоновой кислоты, могут также включать все фармацевтически приемлемые соли, изостерические эквиваленты, такие как тетразол и их формы пролекарств. Примеры последних содержат фармацевтически приемлемые сложные эфиры или амиды, полученные при взаимодействии спиртов или аминов, в том числе аминокислот, со свободными кислотами Формулы I.
Хиральность
Соединения Формулы I, описанные в данном документе, их фармацевтически приемлемые соли или их пролекарства могут содержать один или несколько асимметричных центров, хиральных осей и хиральных плоскостей и, таким образом, могут способствовать образованию энантиомеров, диастереомеров и других стереоизомерных форм и могут быть определены в терминах абсолютной стереохимии, например, (R)- или (S)-. Настоящее изобретение подразумевает содержание всех таких возможных изомеров, а также их рацемических и оптически чистых форм. Оптически активные (+) и (-), (R)- и (S)- изомеры могут быть получены с применением хиральных синтонов или хиральных реагентов или разделены с применением традиционных технологий, таких как обращенно-фазовая ВЭЖХ. Рацемические смеси могут быть получены, а затем разделены на отдельные оптические изомеры, или эти оптические изомеры могут быть получены путем хирального синтеза. Энантиомеры могут быть разделены с помощью способов, известных специалистам в данной области, например, путем образования диастереоизомерных солей, которые затем могут быть разделены с помощью кристаллизации, газожидкостной или жидкостной хроматографией, селективной реакцией одного энантиомера с энантиомерным специфическим реагентом. Специалисту в данной области техники также будет понятно, что если желаемый энантиомер превращается в другой химический объект с помощью технологии разделения, то требуется дополнительный этап для получения желаемой энантиомерной формы. Кроме того, отдельные энантиомеры могут быть синтезированы с помощью асимметричного синтеза с применением оптически активных реагентов, субстратов, катализаторов или растворителей или с помощью преобразования одного энантиомера в другой с помощью асимметрического преобразования.
Некоторые соединения Формулы I или их фармацевтически приемлемые соли, описанные в данном документе, могут существовать в цвиттерионной форме, а настоящее изобретение включает применение цвиттерионных форм этих соединений и их смесей.
Гидраты
К тому же, соединения Формулы I или их фармацевтически приемлемые соли, описанные в данном документе, также могут существовать в гидратированных и безводных формах. Настоящее изобретение включает применение гидратов любых соединений Формулы I или их фармацевтически приемлемых солей, описанных в настоящем документе, которые могут существовать в виде моногидрата или в форме полигидрата.
Способы получения
В общем, все соединения Формулы I или их фармацевтически приемлемые соли, описанные в данном документе, могут быть получены любыми общепринятыми способами с применением легкодоступных и/или традиционно получаемых исходных материалов, реагентов и обычных методик синтеза. Особый интерес представляет работа Hundertmark, T.; Littke, A.F.; Buchwald, S.L.; Fu, G.C. Org. Lett. 12, 1729-1731 (2000).
В разделе иллюстраций в данном документе предлагаются общие схемы и конкретные, но не ограничивающие, примеры синтеза Соединений I-XXXIII.
Фармацевтические применения
Соединения Формулы I или их фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе, являются применимыми: при ауторепарации ткани и/или регенерации ткани пораженного органа, ткани или клетки, при стимуляции генерации новых клеток в in vitro клеточной культуре и/или при модуляции экспрессии маркеров ауторепарации ткани и/или маркеров регенерации ткани, таких как металлопротеиназы и факторы роста. В соответствии с вариантом реализации изобретения, Соединения Формулы I или их фармацевтически приемлемые соли, описанные в данном документе, являются применимыми для лечебно-профилактического ухода, предотвращающего старение кожи. В варианте реализации изобретения, лечение, предпочтительно, включает введение Соединения Формулы I или его фармацевтически приемлемых солей, описанных в данном документе, или их комбинации или фармацевтической композиции, содержащей терапевтически эффективное количество одного или более соединений Формулы I или их фармацевтически приемлемых солей, описанных в данном документе. Выражения “ауторепарация ткани” и “регенерация ткани”, применяемые в данном документе, могут также относиться к процессам, участвующим в лечебно-профилактического уходе, предотвращающем старение кожи. Типичные Соединения в соответствии с Формулой I, описанные в данном документе, как было установлено, стимулируют экспрессию известных маркеров, связанных с омолаживанием, регенерацией ткани и ауторепарацией ткани, а также стимулируют генерацию новых клеток.
В варианте реализации изобретения, пораженный орган, ткань или клетка не являются органом, тканью или клеткой, пораженными воспалительным заболеванием. В варианте реализации изобретения, пораженный орган, ткань или клетка не являются органом, тканью или клеткой, пораженными раком.
В варианте реализации изобретения, повреждение органа, ткани или клеток происходит в результате физического повреждения (т.е. после острого воздействия внешнего фактора или стресса, что приводит к некоторой форме повреждения/поражения органа, ткани или клетки), например, орган, ткань или клетка, пораженные путем физической травмы/удара (например, пореза, прикуса, удара, разрывания, прокола, перфорации, ожога (теплового или химического), замерзания, излучений, поражения электрическим током, физического перенапряжение) или хирургического вмешательства. Физическое поражение, как применяют в данном документе, исключает повреждения органов, тканей или клеток, являющиеся результатом (то есть, в случае которых является основной причиной повреждения органов, тканей или клеток) основного заболевания, например, воспалительных или аутоиммунных заболеваний, таких как воспалительные заболевания кишечника, гломерулонефрит, васкулит, псориатический артрит, системная красная волчанка (СКВ), идиопатическая тромбоцитопеническая пурпура (ИТП), псориаз, болезнь Крона, воспалительное заболевание кишечника, анкилозирующий спондилоартрит, синдром Шегрена, болезнь Стилла (синдром активации макрофагов), увеит, склеродермия, миозит, синдром Рейтера и синдром Вегенера. Однако, Соединения Формулы I или их фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе, могут применяться для стимулирования ауторепарации ткани и/или регенерации ткани с целью лечения вторичных повреждений/поражений ткани, являющихся результатом первоначального физического повреждения, например, вторичных повреждений/поражений ткани, вызванных воспалением, которое может происходить после первоначального физического поражения.
Таким образом, в другом аспекте, настоящее изобретение предлагает способ лечения физического поражения в органе, ткани или клетке (например, для стимулирования ауторепарации и/или регенерации ткани пораженных органа, ткани или клетки), причем способ включает приведение в контакт органа, ткани или клетки с эффективным количеством соединения Формулы I или его фармацевтически приемлемой соли (или содержащей тоже самое композиции), описанные в данном документе.
В другом аспекте, настоящее изобретение предлагает применение соединения Формулы I или его фармацевтически приемлемой соли (или содержащей тоже самое композиции), описанных в данном документе, для лечения физического поражения в органе, ткани или клетке (например, для стимулирования ауторепарации и/или регенерации ткани пораженных органа, ткани или клетки). В другом аспекте, настоящее изобретение предлагает соединение Формулы I или его фармацевтически приемлемую соль (или содержащую тоже самое композицию), описанные в данном документе, для применения при лечении физического поражения в органе, ткани или клетке (например, для стимулирования ауторепарации и/или регенерации ткани пораженных органа, ткани или клетки).
В варианте реализации изобретения, (физически) пораженный орган, ткань или клетка не относятся к почкам или почечной ткани. В другом варианте реализации изобретения, (физически) пораженный орган, ткань или клетка не относятся к кости или костной ткани. В варианте реализации изобретения, (физически) пораженный орган, ткань или клетка относятся к коже, мышце, сухожилиям, связкам, печени, сердцу, поджелудочной железе, органу/ткани пищеварительного/ желудочно-кишечного тракта (например, рту, пищеводу, желудку, кишечнику), желчному пузырю, печени, органу дыхательных путей (например, легким), спинному мозгу, селезенке, молочной железе, ткани глаза, кровеносного сосуда, ткани периодонта, слизистой оболочке (например, слизистой оболочке полости рта, слизистой оболочке носа) и/или хрящей.
В варианте реализации изобретения, соединения Формулы I или их фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе, применяют/вводят безотлагательно, то есть вскоре после травмы. В варианте реализации изобретения, соединения Формулы I или их фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе, применяют/вводят для стимулирования ауторепарации ткани и/или регенерации ткани до развития фиброза в поврежденном органе, ткани или клетке, например, до развития фиброзного заболевания.
В варианте реализации изобретения, соединения Формулы I или их фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе, являются применимыми для стимулирования лечения раны.
В другом варианте реализации изобретения, пораженный орган, ткань или клетка представляет собой орган, ткань или клетку нервной системы (например, ткань нейронов), например, орган, ткань или клетку центральной нервной системы или периферической нервной системы. В варианте реализации изобретения, соединения Формулы I или их фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе, являются применимыми для ауторепарации ткани и/или регенерации ткани после поражения нейронов, например, поражения спинного мозга, поражения периферического нерва или поражения нейронов, связанных с рассеянным склерозом.
В варианте реализации изобретения, соединения Формулы I или их фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе, являются применимыми для ауторепарации ткани и/или регенерации ткани кожи, например, после обрезания кожи, прокола, синяка или ожога.
В варианте реализации изобретения, пораженные орган, ткань или клетка представляет собой орган, ткань или клетку дыхательной системы, например, легких.
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к печени или печеночной ткани.
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к мочевому пузырю или ткани мочевого пузыря.
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к яичникам или ткани яичников.
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к предстательной железе или ткани предстательной железы.
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к селезенке или ткани селезенки.
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к молочной железе или ткани молочной железы.
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к мышце, например, мышце, пораженной мышечным напряжением, мышечным разрывом и/или любым другим типом физической мышечной травмы.
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к кровеносному сосуду (например, артерии).
В варианте реализации изобретения, пораженные орган, ткань или клетка относятся к органу/ткани пищеварительного/желудочно-кишечного тракта например, рта, пищевода, желудка, кишечника).
В конкретных вариантах реализации изобретения, описанные в данном документе способы и применения не предназначены для ремоделирования и/или регенерации островков Лангерганса. В конкретном варианте реализации изобретения, ткань является отличной от костной. В варианте реализации изобретения, ткань является отличной от ткани поджелудочной железы.
Авторы настоящего изобретения показали, что типичные соединения формулы I или их фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе, повышают маркеры, которые стимулируют ауторепарацию ткани и регенерацию ткани пораженного органа у субъекта. В варианте реализации изобретения, соединения формулы I, описанные в данном документе, оказывают тканевосстановительную активность.
В другом аспекте, настоящее изобретение относится к косметической композиции, содержащей соединение формулы I или его фармацевтически приемлемые соли (или содержащая тоже самое композиция), описанные в данном документе. В другом аспекте, настоящее изобретение относится к композиции для ухода за кожей, содержащей соединение формулы I или его фармацевтически приемлемые соли (или содержащей тоже самое композиции), описанные в данном документе. В другом аспекте, настоящее изобретение относится к омолаживающей композиции для ухода за кожей, содержащей соединение формулы I или его фармацевтически приемлемые соли (или содержащей тоже самое композиции), описанные в данном документе.
В другом аспекте, настоящее изобретение относится к упоминаемому выше соединению формулы I или его фармацевтически приемлемым солям (или содержащей тоже самое композиции) для применения в омолаживающем средстве для ухода за кожей. В другом варианте реализации изобретения, упоминаемое выше соединение формулы I или содержащая тоже самое композиция предназначены для применения при стимуляции репарации и/или регенерации кожи после связанного со старением повреждения кожи. В другом варианте реализации изобретения, упоминаемое выше соединение или композиция предназначены для применения при стимуляции репарации и/или регенерации кожи после повреждения или поражения кожи. В варианте реализации изобретения, повреждение или поражение кожи происходит в результате воздействия УФ-излучения, например, воздействия солнца (например, солнечных ожогов).
В варианте реализации изобретения, способы и применения, описанные в данном документе, дополнительно включают идентификацию субъекта, имеющего поврежденный орган, ткань или клетку, и который нуждается в лечении с помощью упоминаемого выше соединения формулы I или его фармацевтически приемлемых солей (или содержащей тоже самое композиции) для стимулирования ауторепарации и/или регенерации ткани в пораженном органе, ткани или клетке. Способ может включать идентификацию в полученном от субъекта образце, таком как образец органа, ткани или клетки, пониженного уровня одного или более маркеров ауторепарации ткани и/или регенерации ткани, таких как металлопротеиназы и факторы роста, в том числе, без ограничения этим, ФРГ, ЛОК (лизилоксидаза), MMP1, MMP2, MMP9, MMP13, PLAT (tPA), PLAU (uPA), Serpin A1 (AAT), Serpin E1 (PAI-1), TIMP3 и ИСК (интегрин-связанная киназа), и приведение в контакт органа, ткани или клетки с эффективным количеством соединения формулы I или его фармацевтически приемлемых солей (или содержащей тоже самое композиции), описанных в данном документе.
Термин “субъект” включает нуждающиеся в лечении живые организмы, как описано в данном документе, например, те, у которых поражен орган. Термин “субъект” включает таких животных, как млекопитающие или птицы. Предпочтительно, субъект представляет собой млекопитающее, в том числе, но без ограничения этим, человека, лошадь, собаку и кошку. В некоторых вариантах реализации изобретения млекопитающее не является мышью. Более предпочтительно, млекопитающее является человеком.
Фармацевтические композиции и составы
В варианте реализации изобретения, соединения Формулы I или их фармацевтически приемлемые соли, описанные в данном документе, содержатся в фармацевтических композициях, содержащий терапевтически эффективное количество соединений или их фармацевтически приемлемых солей. Как указано в данном документе, фармацевтические композиции могут быть применимыми: при ауторепарации ткани и/или регенерации ткани пораженного органа, при стимуляции генерации новых клеток в in vitro клеточной культуре и/или при модуляции экспрессии маркеров ауторепарации ткани и/или маркеров регенерации ткани, таких как металлопротеиназы и факторы роста.
Используемый в настоящем документе, термин “терапевтически эффективное количество” обозначает количество соединения, которое, при введении субъекту для лечения или профилактики конкретного расстройства, заболевания или состояния или для оказания биологического эффекта (например, для стимуляции ауторепарации ткани и/или генерации новых клеток в in vitro клеточной культуре и/или для модуляции (повышения) экспрессии маркеров ауторепарации ткани и/или маркеров регенерации ткани), является достаточным для осуществления такого лечения или профилактики этого расстройства, заболевания или состояния или для оказания биологического эффекта. Дозировки и терапевтически эффективные количества могут изменяться, например, в зависимости от ряда факторов, в том числе активности конкретного применяемого средства, возраста, массы тела, общего состояния здоровья, пола и рациона субъекта, времени введения, пути введения, скорости выделения и любой комбинации лекарственных средств, если это применимо, эффекта, который специалист желает оказать на субъекта, свойств соединений (например, биодоступности, стабильности, активности, токсичности, и т. д.) и конкретного(ых) расстройства(расстройств), от которого(ых) субъект страдает. К тому же, терапевтически эффективное количество может зависеть от параметров крови пациента (например, уровня кальция, профиля липидов, уровней инсулина, гликемии), тяжести состояния заболевания, функции органа или основного заболевания или осложнений. Такие подходящие дозы могут быть определены с применением любых доступных анализов, в том числе анализов, описанных в данном документе. Когда одно или более соединений Формулы I или их фармацевтически приемлемые соли, описанные в данном документе, должны быть введены людям, причем врач может, например, сначала назначить относительно низкую дозу, затем увеличивать дозу, пока не будет получен приемлемый ответ. Доза, подлежащая введению, в конечном счете, будет на усмотрение медицинского персонала. В общем, однако, предполагается, что доза для соединений Формулы I или их фармацевтически приемлемых солей, описанных в настоящем документе, может находиться в диапазоне от 1 до 50 мг/кг в сутки у человека. В выбранном варианте реализации изобретения, диапазон может составлять от 1 до 30 мг/кг в день у человека. В выбранном варианте реализации изобретения, диапазон может составлять от 1 до 20 мг/кг в день у человека. В выбранном варианте реализации изобретения, диапазон может составлять от 5 до 18 мг/кг в день у человека. В выбранном варианте реализации изобретения, диапазон может составлять от 1 до 18 мг/кг в день у человека.
Используемый в настоящем документе, термин “фармацевтическая композиция” соответствует присутствию по меньшей мере одного соединения в соответствии с Формулой I или его фармацевтически приемлемых солей, как определено в настоящем документе, и по меньшей мере одного фармацевтически приемлемого носителя, разбавителя, наполнителя или вспомогательного вещества. Используемый в настоящем документе, термин “фармацевтически приемлемый носитель”, “фармацевтически приемлемый разбавитель” или “фармацевтически приемлемое вспомогательное вещество” предназначен для того, чтобы означать, без ограничения, любой адъювант, носитель, вспомогательное вещество, смазывающее средство, подсластитель, разбавитель, консервант, краситель/красящее средство, усилитель вкуса, поверхностно-активное вещество, смачивающее средство, диспергирующее средство, суспендирующее средство, стабилизатор, изотоническое средство, растворитель, эмульгатор или инкапсулирующее средство, такое как липосома, циклодекстрины, инкапсулирующие полимерные системы доставки или полиэтиленгликолевая матрица, которая является приемлемой для применения на субъектах, предпочтительно, людях. Это предпочтительно относится к соединению или композиции, которые одобрены или одобряемы регулирующим органом федерального правительства или правительства штата или приведено в фармакопее США или другой общепризнанной фармакопее для применения на животных и, более конкретно, на людях. Фармацевтически приемлемый наполнитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла. Дополнительные примеры фармацевтически приемлемых наполнителей включают, но не ограничиваясь этим: Воду USP для инъекций; водные наполнители, такие как, но без ограничения этим, инъекция хлорида натрия, инъекция Рингера, инъекция декстрозы, инъекция декстрозы и натрия хлорида и лактатный раствор Рингера для инъекций; смешивающиеся с водой наполнители, такие как, но без ограничения этим, этиловый спирт, полиэтиленгликоль и полипропиленгликоль; и неводные наполнители, такие как, но без ограничения этим, кукурузное масло, хлопковое масло, арахисовое масло, кунжутное масло, этилолеат, изопропилмиристат и бензилбензоат. Предотвращение действия микроорганизмов может быть достигнуто с помощью добавления антибактериальных и противогрибковых средств, например, парабенов, хлорбутанола, фенола, аскорбиновой кислоты, тимеросала и тому подобных. Во многих случаях в композиции содержатся изотонические средства, например, сахара, хлорид натрия или полиспирты, такие как маннит и сорбит. Пролонгированное всасывание инъекцируемых композиций может быть вызвано путем применения в композициях средства, задерживающего абсорбцию, например, моностеарата алюминия или желатина.
Композиция по настоящему изобретению может содержать одно или более соединений Формулы I, как определено в настоящем документе, или фармацевтически приемлемые производные, соли, пролекарства, аналоги, изомеры или их энантиомеры. Составы на основе активного соединения могут быть получены таким образом, чтобы обеспечить фармацевтическую композицию в форме, подходящей для энтерального, мукозального (в том числе орального, сублингвального, глазного, назального, легочного и ректального), парентерального (в том числе внутримышечного, внутрикожного, подкожного и внутривенное) или местного (в том числе мази, кремы, лосьоны или капли) введения. Этот состав может быть, при необходимости, удобно представлен в дискретных дозированных единицах и может быть получен любым из способов, хорошо известных в области фармацевтического состава. Все способы включают этап объединения вместе активного фармацевтического ингредиента с жидкими носителями или тонко измельченными твердыми носителями или ими обоими, как того требуют обстоятельства. При необходимости, описанные выше составы могут быть адаптированы таким образом, чтобы обеспечить пролонгированное высвобождение активного фармацевтического ингредиента. Композиции замедленного высвобождения, хорошо известные в данной области техники, содержат применение болюсной инъекции, непрерывной инфузии, биосовместимых полимеров или липосом.
Вышеупомянутое соединение или композиция могут быть составлены в применимой местно косметической композиции (например, композиции для местного применения). Неограничивающие примеры таких применимых местно композиций содержат крем для ухода за кожей, очищающий крем, мазь, лосьон для ухода за кожей, гель для ухода за кожей, пену для ухода за кожей, солнцезащитный состав, солнцезащитный крем, крем для снятия макияжа, лосьон для снятия макияжа, крем-основу, жидкий тональный крем, средство для ванны и душа, дезодорирующий состав, состав против пота, композицию средства для бритья, гель или лосьон после бритья, композицию косметического средства, крем для удаления волос, композицию мыла, композицию очистителя для рук, очищающее мыло, средство по уходу за ребенком, средство по уходу за волосами, шампунь, лосьон для укладки волос, лосьон, оказывающий лечебно-профилактическое действие, крем для волос, гель для волос, окрашивающую композицию, композицию, восстанавливающую структуру, композицию для перманента или любую другую композицию, которая адаптирована для применения в актуальном косметическом режиме. Такие композиции могут дополнительно содержать один или более космецевтически приемлемых наполнителей.
Кремы, как хорошо известно в области фармацевтической и космецевтической композиции, являются вязкими жидкостями или полутвердыми эмульсиями, либо типа "масло в воде", либо типа "вода в масле". Основы кремов являются смываемыми водой и содержат масляную фазу, эмульгатор и водную фазу. Масляная фаза, также называемая "внутренней" фазой, обычно состоит из вазелина и жирного спирта, такого как цетиловый или стеариловый спирт. Водная фаза обычно, хотя и не обязательно, превышает масляную фазу по объему и, как правило, содержит увлажнитель. Эмульгатор в составе крема обычно представляет собой неионное, анионное, катионное или амфотерное поверхностно-активное вещество.
Лосьоны представляют собой препараты, которые наносят на поверхность кожи без трения и обычно представляют собой жидкие или полужидкие препараты, в которых твердые частицы, в том числе активное средство, присутствуют в водной или спиртовой основе. Лосьоны обычно представляют собой суспензии твердых веществ и, предпочтительно, для этой цели содержат жидкую масляную эмульсию типа "масло в воде". Лосьоны представляют собой предпочтительные составы для лечения больших областей тела, из-за легкости применения более жидкой композиции. Обычно необходимо, чтобы нерастворимое вещество в лосьоне было тонко измельчено. Лосьоны обычно содержат суспендирующие средства для получения лучших дисперсий, а также соединения, применимые для локализации и удерживания активного средства в контакте с кожей, например, с метилцеллюлозой, натрийкарбоксиметилцеллюлозой или тому подобным.
Растворы представляют собой гомогенные смеси, полученные растворением одного или более химических веществ (растворенных веществ) в жидкости таким образом, что молекулы растворенного вещества диспергированы среди растворителей. Раствор может содержать другие космецевтически приемлемые химические вещества для буферизации, стабилизации или сохранения растворенного вещества. Типичными примерами растворителей, применяемых при получении растворов, являются этанол, вода, пропиленгликоль или любые другие космецевтически приемлемые наполнители.
Гели представляют собой полутвердые суспензионные системы. Однофазные гели содержат органические макромолекулы, распределенные, по существу, равномерно по всей жидкости носителя, которая обычно является водной, но также, предпочтительно, содержат спирт и, необязательно, масло. "Органические макромолекулы", то есть гелеобразующие средства, представляют собой сшитые полимеры акриловой кислоты, такие как семейство "карбомеров" полимеров, например, карбоксиполилиалкилены, которые могут быть коммерчески получены под маркой CarbopolTM. Другими примерами являются гидрофильные полимеры, такие как полиэтиленоксиды, сополимеры полиоксиэтилена и полиоксипропилена и поливиниловый спирт; целлюлозные полимеры, такие как гидроксипропилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилметилцеллюлоза, фталат гидроксипропилметилцеллюлозы и метилцеллюлоза; смолы, такие как трагакант и ксантановая смола; альгинат натрия; и желатин. Для получения однородного геля могут быть добавлены диспергирующие средства, такие как спирт или глицерин, либо путем растирания, механического смешивания или перемешивания или их комбинаций может быть диспергировано гелеобразующее средство.
Мази представляют собой полутвердые препараты, которые обычно основаны на вазелине или других производных продуктов переработки углеводородного сырья. Специальная мазевая основа, которая будет применяться, как будет понятно специалистам в данной области техники, является такой, которая обеспечит ряд желательных характеристик, например, смягчающее действие или тому подобное. Как и другие носители или наполнители, основа мази должна быть инертной, стабильной, не раздражающей и не сенсибилизирующей. Как объяснено в Remington: The Science and Practice of Pharmacy, 19th Ed. (Easton, Pa.: Mack Publishing Co., 1995), на страницах 1399-1404, и мазевые основы могут быть сгруппированы в четыре класса: масляные основы; эмульгирующие основы; эмульсионные основы; и водорастворимые основы. Маслянистые мазевые основы содержат, например, растительные масла, жиры, полученные от животных, и полутвердые углеводороды, полученные из продуктов переработки углеводородного сырья. Эмульгирующие основы мазей, также известные как абсорбирующие мазевые основы, содержат мало или совсем не содержат воду и содержат, например, сульфат гидроксистеарина, безводный ланолин и гидрофильный вазелин. Эмульсионные мазевые основы представляют собой эмульсии типа "вода в масле" (В/М) или эмульсии типа "масло в воде" (М/В) и содержат, например, цетиловый спирт, моностеарат глицерина, ланолин и стеариновую кислоту. Предпочтительные водорастворимые основы мази получают из полиэтиленгликолей с различной молекулярной массой; снова, для дополнительной информации смотри Remington: The Science and Practice of Pharmacy.
Пасты представляют собой полутвердые лекарственные формы, в которых активное средство суспендируют в подходящем основании. В зависимости от природы основы, пасты разделяют между пастами на основе жира или такими, которые сделаны из однофазных водных гелей. Основа в пасте на основе жира обычно представляет собой вазелин или гидрофильный вазелин или тому подобное. Пасты, сделанные из однофазных водных гелей, обычно содержат в качестве основы карбоксиметилцеллюлозу или тому подобное.
Составы также могут быть получены с липосомами, мицеллами и микросферами. Липосомы представляют собой микроскопические везикулы с липидной стенкой, содержащей липидный бислой, и в данном контексте инкапсулируют один или более компонентов омолаживающих составов. Липосомальные препараты в этом документе содержат катионные (положительно заряженные), анионные (отрицательно заряженные) и нейтральные препараты. Катионные липосомы легко доступны. Например, липосомы N[1-2,3-диолеилокси)пропил]-N,N,N-триэтиламмоний (DOTMA) доступны под торговой маркой Lipofectin™ (GIBCO BRL, Гранд-Айленд, Н.Й.). Аналогичным образом легко доступны анионные и нейтральные липосомы, например, от Avanti Polar Lipids (Birmingham, Ala.), или могут быть легко получены с применением легкодоступных материалов. Такие материалы содержат, среди прочих, фосфатидилхолин, холестерин, фосфатидилэтаноламин, диолеоилфосфатидилхолин (DOPC), диолеоилфосфатидилглицерин (DOPG) и диолеоилфолфатидилэтаноламин (DOPE). Эти материалы также можно смешивать с DOTMA в соответствующих соотношениях. Способы получения липосом с применением этих материалов хорошо известны в данной области техники.
Мицеллы известны в данной области техники как состоящие из молекул поверхностно-активных веществ, расположенных таким образом, что их полярные головные группы образуют внешнюю сферическую оболочку, тогда как гидрофобные углеводородные цепи ориентированы по направлению к центру сферы, образуя ядро. Мицеллы образуют в водном растворе, содержащем поверхностно-активное вещество при достаточно высокой концентрации, таким образом, что, естественно, получают мицеллы. Поверхностно-активные вещества, применимые для образования мицелл, содержат, но без ограничения этим, лаурат калия, октанолсульфонат натрия, декансульфонат натрия, додекансульфонат натрия, лаурилсульфат натрия, докузат натрия, бромид децилтриметиламмония, бромид додецилтриметиламмония, бромид тетрадецилтриметиламмония, хлорид тетрадецилтриметиламмония, хлорид додециламмония, полиоксил-8-додециловый эфир, полиоксил-12-додециловый эфир, ноноксинол 10 и ноноксинол 30.
Подобным образом, в настоящих составах смогут содержаться микросферы. Подобно липосомам и мицеллам, микросферы по существу инкапсулируют один или более компонентов настоящих составов. Они, как правило, хотя и не обязательно, образованы из липидов, предпочтительно заряженных липидов, таких как фосфолипиды. Получение липидных микросфер хорошо известно в данной области техники и описано в соответствующих текстах и литературе.
Наборы
Соединение(я) Формулы I или его(их) фармацевтически приемлемые соли, описанные в данном документе, могут быть упакованы как часть набора, необязательно, в том числе емкости (например, упаковки, коробки, флакона и т. д.). Набор может быть коммерчески применен в соответствии со способами, описанными в настоящем документе, и может содержать инструкции для применения в способе, описанном в данном документе. Дополнительные компоненты набора могут содержать кислоты, основания, буферные средства, неорганические соли, растворители, антиоксиданты, консерванты или металлохелаторы. Дополнительные компоненты набора присутствуют в виде чистых композиций, либо в виде водных, либо в виде органических растворов, которые содержат один или более дополнительных компонентов набора. Любой или все компоненты набора необязательно дополнительно содержат буферы.
Соединение(я) Формулы I или его(их) фармацевтически приемлемые соли, описанные в данном документе, могут быть или не могут быть введены пациенту одновременно или одним и тем же путем введения. Поэтому способы изобретения включают наборы, которые при применении врачом могут упростить введение пациенту соответствующих количеств двух или более активных ингредиентов.
Типичный набор по изобретению содержит стандартную лекарственную форму по меньшей мере одного соединения Формулы I, как указано в настоящем документе, или его фармацевтически приемлемую соль и единичную дозированную форму по меньшей мере одного дополнительного активного ингредиента. Примеры дополнительных активных ингредиентов, которые могут применяться в сочетании с соединениями по изобретению, включают, но не ограничиваясь этим, любые лекарственные средства, указанные в данном документе, которые могут быть применены в сочетании с соединением(ями) Формулы I или его(их) фармацевтически приемлемыми солями, как определено в настоящем документе.
Наборы по изобретению могут дополнительно содержать фармацевтически приемлемые наполнители, которые могут применяться для введения одного или более активных ингредиентов. Например, если активный ингредиент представлен в твердой форме, которая должна быть восстановлена для парентерального введения, набор может содержать герметичную емкость или подходящий наполнитель, в котором активный ингредиент может быть растворен с образованием стерильного раствора, не содержащего макрочастиц, который подходит для парентерального введения. Примеры фармацевтически приемлемых наполнителей предложены в данном документе ранее.
ПРИМЕРЫ
Следующие примеры дополнительно иллюстрируют практику этого изобретения, но не предназначены для его ограничения.
Пример 1: Экспериментальные процедуры для получения некоторых типичных соединений
Все хроматограммы ВЭЖХ и масс-спектры записывали на инструменте ЖХ-МС HP 1100 Agilent™ с применением аналитической колонки C18 (250 × 4,6 мм, 5 микрон) с градиентом в течение 5 мин. от 15 до 99% CH3CN-H2O с 0,01% TFA в качестве элюента и потоком 2 мл/мин.
Соединение I: Синтез натриевой соли (3-пентилфенил)уксусной кислоты с применением модифицированной процедуры Соногашира

Этап 1
К раствору/суспензии 3-бромфенилуксусной кислоты (5,02 г, 23,33 ммоль) в этаноле (100 мл) при комнатной температуре добавляли концентрированную серную кислоту (1 мл). Бесцветное твердое вещество затем перемешивали в течение ночи при 80°C. Раствор концентрировали при пониженном давлении. Остаток разбавляли с помощью этилацетата (25 мл), воды (25 мл) и разделяли два слоя. Водный слой экстрагировали с помощью этилацетата (2 × 25 мл) и солевого раствора (20 мл). Объединенные органические слои промывали насыщенным раствором NaHCO3 (2 × 25 мл), солевым раствором (25 мл) и высушивали над сульфатом натрия. После фильтрации раствор выпаривали досуха. Это давало светло-желтое масло (5,4 г, 95%). 1H-ЯМР (400 МГц, CDCl3): δ 1,26 (т, J = 4,7 Гц, 3H), 3,57 (с, 2H), 4,15 (КВ, J = 7,0 и 14,3 Гц, 2H), 7,17-7,26 (м, 2H), 7,38-7,44 (м, 1H), 7,44 (д, J = 1,56 Гц, 1H).
Этап 2
Смесь этил(3-бромфенил)ацетата (0,3 г, 1,24 ммоль) и гидрата фторида тетрабутиламмония (0,97 г, 3,72 ммоль) обрабатывали с помощью PdCl2(PPh3)2 (26 мг, 0,037 ммоль; 3% моль) и 1-пентина (367 μл, 3,72 ммоль) в герметично закрытой пробирке. Пробирку нагревали при 80°C в течение 2 ч. Смесь обрабатывали водой и экстрагировали с помощью диэтилового эфира. Органический экстракт высушивали над сульфатом натрия, отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке 25 M Biotage™ (силикагель), элюируя с помощью этилацетата/гексана от 0:1 до 2:98, давало этил(3-(пентин-1-ил)фенил)ацетата в виде бледно-желтого масла (0,23 г, 79%).
Этап 3
К этил[3-[пентин-1-ил]фенил]-ацетату (0,23 г, 0,98 ммоль) в этаноле (5 мл) в атмосфере азота добавляли Pd на углероде (10%, 25 мг, 10% мас./мас.). Смесь энергично перемешивали в атмосфере водорода при комнатной температуре в течение ночи. Раствор отфильтровывали и промывали палладиевый катализатор на углеродном носителе этанолом (20 мл). Фильтрат концентрировали с помощью силикагеля. Неочищенный продукт очищали с помощью флеш-хроматографии с применением смеси 10% гексан/этилацетат. Получали прозрачное масло (0,21 г, 90%).
Этап 4
К раствору сложного эфира (0,2 г, 0,9 ммоль) в тетрагидрофуране (5 мл), метаноле (1,5 мл) и воде (1,5 мл) добавляли гидроксид лития (0,09 г, 3,6 ммоль) при 0°C. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Нерастворимые вещества отфильтровывали, а фильтрат концентрировали при пониженном давлении. Остаток затем обрабатывали с помощью 2 M HCl и экстрагировали с помощью этилацетата. Органическую фазу высушивали над сульфатом натрия и выпаривали при пониженном давлении. Неочищенный материал очищали на колонке 40 L Biotage (силикагель) с применением этилацетата/гексана (от 0:10 до 4:6) в качестве элюента. Это давало чистую (3-пентилфенил)уксусную кислоту (0,19 г, 99%) в виде белого клейкого твердого вещества. 1H ЯМР (400 МГц, CD3OD): δ 0,90 (т, J = 7,0 Гц, 3H), 1,28-1,38 (м, 4H), 1,61 (кв, J = 7,6 Гц, 15,0 Гц, 2H), 2,58 (т, J = 7,6 Гц, 2H), 3,56 (с, 2H), 7,07 (м, 3H), 7,20 (м, 1H); МСНР (ESI): m/z 207 (MH+); ВЭЖХ: 4 мин.
Этап 5
К перемешиваемому раствору кислоты (0,19 г, 0,82 ммоль) в этаноле (4 мл) и воде (1 мл) добавляли бикарбонат натрия (0,07 г, 0,82 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали, а белое клейкое твердое вещество растворяли в воде и лиофилизировали раствор. Это давало чистую натриевую соль (3-пентилфенил)уксусной кислоты (0,17 г, 92%) в виде белого твердого вещества. т. пл. 110-112°C; 1H ЯМР (400 МГц, CD3OD): δ 0,89 (т, J = 6,8 Гц, 3H), 1,28-1,37 (м, 4H), 1,60 (кв, J = 7,4 Гц, 15,0 Гц, 2H), 2,56 (т, J = 7,6 Гц, 2H), 3,43 (с, 2H), 6,96 (м, 1H), 7,12 (м, 3H); МСНР (ESI): m/z 207 ((MH+); ВЭЖХ: 4 мин.
Соединение II: Натриевая соль 3-(3-пентилфенил)пропионовой кислоты
Указанное выше соединение получали таким же образом, что и в случае Соединения I, начиная со сложного этилового эфира 3-оксо-3-бромфенилпропионовой кислоты. Кетоновую группу и двойную связь восстанавливали одновременно с применением палладиевого катализатора на углеродном носителе в этаноле под давлением водорода. Белое твердое вещество; 1H ЯМР (400 МГц, CDCl3): δ 7,14-7,10 (м, 1H), 7,04-7,00 (м, 2H), 6,95-6,93 (м, 1H), 2,88-2,84 (м, 2H), 2,55 (т, J = 7,4 Гц, 2H), 2,44-2,40 (м, 2H), 1,63-1,55 (м, 2H), 1,35-1,28 (м, 4H), 0,90 (м, 3H); 13C ЯМР (101 МГц, CD3OD): δ 179,3, 141,2, 140,8, 126,7, 126,4, 124,0, 123,8, 38,6, 34,2, 31,2, 29,9, 29,8, 20,9, 11,7; МСНР (ESI): m/z 203 (MH+-CO-NaOH); ВЭЖХ: 4,5 мин.
Соединение III: Натриевая соль 3-(3-бутилфенил)пропионовой кислоты
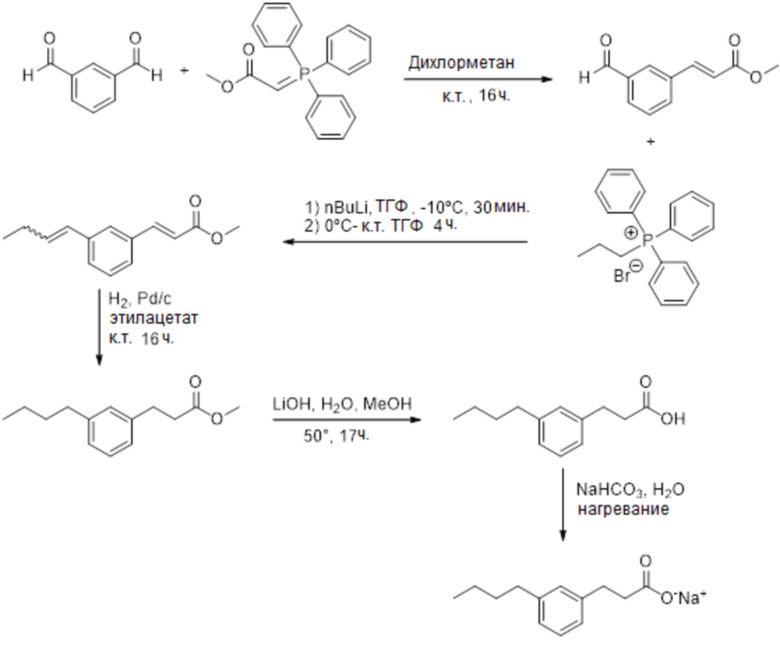
Этап 1
В круглодонную колбу (250 мл) взвешивали изофталевый альдегид (1,0 г, 7,5 ммоль), а после чего дихлорметан (100 мл). Через делительную воронку с равновесием давления добавляли метил(трифенил-фосфоранилиден)ацетат (2,7 г, 8,2 ммоль) в дихлорметане (25 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь отфильтровывали на подушке силикагеля и промывали дихлорметаном (150 мл). Растворитель затем выпаривали при пониженном давлении, а неочищенный продукт применяли на следующем этапе без дополнительной очистки.
Этап 2
Бромид пропилтрифенилфосфония (3,2 г, 8,2 ммоль) помещали в круглодонную колбу, под азотом, и добавляли сухой ТГФ (5 мл). Колбу охлаждали на бане со льдом/ацетоном (-10ºC) и медленно добавляли н-бутиллитий (2,5 M в гексане, 3,28 мл, 8,2 ммоль). Смесь темнела при перемешивании в течение 30 минут. На баню со льдом/ацетоном (-10ºC) помещали неочищенную реакционную смесь с предыдущего этапа в сухом ТГФ (5 мл) под азотом. К раствору альдегида при -10ºC медленно добавляли раствор фосфония, а реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 4 ч. Добавляли насыщенный раствор хлорида аммония (10 мл) и экстрагировали органический слой с помощью этилацетата (3x). Органический слой высушивали над безводным сульфатом натрия, отфильтровывали и добавляли силикагель для получения сухой смеси. Соединение очищали с помощью SP1 (этилацетат/гексан). Это давало ожидаемый продукт (8,8 г, 54%). 1H ЯМР (400 МГц, CDCl3): δ 7,70-7,65 (м, 1H), 7,45-7,24 (м, 4,5H), 6,45-6,28 (м, 2,5H), 5,70-5,67 (м, 0,5H), 3,78 (м, 3H), 2,34-2,20 (м, 2H), 1,10-1,03 (м, 3H).
Этап 3
В круглодонную колбу (25 мл) помещали ненасыщенный сложный эфир (140 мг, 0,65 ммоль), растворенный в этилацетате (10 мл). К этому раствору добавляли 10% палладиевый катализатор на активированном угле Pd/C (10 мг). Колбу накрывали мембранной перегородкой, а сверху помещали емкость с атмосферой водорода. Колбу продували трижды водородом, а реакционную смесь перемешивали при комнатной температуре в течение ночи. Твердое вещество затем отфильтровывали через Celite™. Добавляют силикагель и получали сухую смесь. Очистка с помощью флеш-хроматографии с применением 0-20% этилацетата/гексана давала желаемый продукт (124 мг, 87%). МСНР (ESI): m/z 221 (MH+); ВЭЖХ: 5,0 мин.
Этап 4
В круглодонную колбу помещали сложный эфир (124 мг, 0,56 ммоль), а после чего метаноле(4 мл) и гидроксид лития (118 мг, 2,8 ммоль). Добавляли воду (1 мл), а реакционную смесь нагревали при 50ºC при перемешивании в течение 17 ч. Реакционную смесь переносили в делительную воронку, подкисляли до pH ниже 4 с помощью HCl (1M) и экстрагировали с помощью этилацетата (3x). Органический слой высушивали над безводным сульфатом натрия, отфильтровывали и выпаривали. Неочищенный материал очищали с помощью ВЭЖХ/Waters. Это давало белое твердое вещество (80 мг, 70%). 1H ЯМР (400 МГц, CD3OD): δ 7,16-7,12 (м, 1H), 7,01-6,96 (м, 3H), 2,88-2,84 (м, 2H), 2,57-2,53 (м, 4H), 1,60-1,52 (м, 2H), 1,37-1,28 (м, 2H), 0,91(т, 3H, J = 7,3 Гц); МСНР (ESI): m/z 205 (M-H); ВЭЖХ: 4,2 мин.
Этап 5
В колбу (20 мл) помещали кислоту (80 мг, 0,39 ммоль), а после чего NaHCO3 (33 мг, 0,39 ммоль) и воду (8 мл). К смесям добавляли ацетонитрил (3 мл), а реакционную смесь обрабатывали ультразвуком, нагревали и встряхивали, пока почти все твердые вещества не оказывались в растворе. Раствор отфильтровывали через нейлоновый фильтр. Вода затвердевала путем погружения пробирки в ванну с сухим льдом/ацетоном и лиофилизировали в течение ночи. Это давало желаемый продукт в виде белого твердого вещества. 1H ЯМР (400 МГц, CD3OD): δ 7,14-7,10 (м, 1H), 7,04-6,93 (м, 3H), 2,88-2,84 (м, 2H), 2,57-2,54 (м, 2H), 2,44-2,40 (м, 4H), 1,61-1,53 (м, 2H), 1,39-1,30 (м, 2H), 0,93(т, 3H, J = 7,3 Гц);13C ЯМР (101 МГЦ, CD3OD): δ 142,7, 142,4, 128,2, 128,0, 125,6, 125,4, 125,3, 40,1, 35,5, 33,9, 32,7, 22,2, 13,1; МСНР (ESI): m/z 251,0 (м, MNa+), 229,0 (w, MH+), 189,2 (100%, ион ацила [M – Na+ + 2H+ -H2O]); ВЭЖХ: 4,1мин.
Соединение IV: Натриевая соль E-(3-пент-1-енил-фенил)уксусной кислоты.
Указанное выше соединение получали таким же образом, что и в случае Соединения I, начиная с метилового сложного эфира E-(3-пент-1-енил-фенил)уксусной кислоты. Последний был получен путем взаимодействия метилового сложного эфира 3-бромфенилуксусной кислоты со сложным пинаколовым эфиром транс-1-пентенилбороновой кислоты в условиях Сузуки. Белое твердое вещество 1H ЯМР (400 МГц, CD3OD): δ = 7,32 (с, 1H), 7,11-7,18 (м, 3H), 6,35 (д, J = 15,7 Гц, 1H), 6,20-6,27 (м, 1H), 3,44 (с, 2H), 2,19 (м, 2H), 1,45-1,54 (м, 2H), 0,96 (т, J = 7,4, 3H); 13C ЯМР (101 МГц, CD3OD): δ = 179,26, 138,25, 137,92, 130,32, 130,04, 128,06, 127,59, 126,60, 123,52, 45,21, 35,06, 22,52, 12,89; МСНР (ESI): m/z 205 (MH+); ВЭЖХ: 4,1 мин.
Соединение V: Натриевая соль 2-(3-(гекс-1-енил]фенил)уксусной кислоты.
Указанное выше соединение получали с помощью сочетания Сузуки метил-2-(3-бромфенил)ацетата и сложного пинаколового эфира (E)-гекс-1-енилбороновой кислоты таким же образом, что и в случае Соединения VII; с последующим гидролизом сложного эфира и образованием натриевой соли в качестве Соединения I. Белое твердое вещество: 1H ЯМР (400 МГц, CD3OD): δ 7,33 (с, 1H), 7,12-7,19 (м, 3H), 6,35 (д, J = 15,8 Гц, 1H), 6,20 (дт, J = 15,8, 6,8 Гц, 1H), 3,46 (с, 2H), 2,17-2,22 (м, 2H), 1,33-1,49 (м, 4H), 0,93 (т, J = 7,2 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 179,35, 138,27, 137,95, 130,27, 130,16, 128,10, 127,61, 126,64, 123,56, 45,24, 32,66, 31,67, 22,16, 13,22; МСНР (ESI): m/z 263,1 (100%, M + Na+); ВЭЖХ : 4,4 мин.
Соединение VI: Натриевая соль 2-(3-гексилфенил)уксусной кислоты
Указанное выше соединение получали с помощью сочетания Сузуки метил-2-(3-бромфенил)ацетата и сложного пинаколового эфира (E)-гекс-1-енилбороновой кислоты таким же образом, что и в случае Соединения VII; с последующим гидрированием, гидролизом сложного эфира и образованием натриевой соли в качестве Соединения I. Белое твердое вещество; 1H ЯМР (400 МГц, D2O): δ 7,14 (дд, J = 7,8, 7,6 Гц, 1H), 7,01 (с, 1H), 7,00 (д, J = 7,8 Гц, 1H), 6,96 (д, J = 7,6 Гц, 1H), 3,34 (с, 2H), 2,46 (д, J = 7,5 Гц, 2H), 1,41-1,48 (м, 2H), 1,10-1,18 (м, 6H), 0,70 (т, J = 6,8 Гц, 3H); 13C ЯМР (101 МГц, D2O): δ 181,23, 143,98, 137,46, 129,47, 128,73, 126,63, 126,48, 44,58, 35,14, 31,12, 30,94, 28,23, 22,13, 13,53; МСНР (ESI): m/z 265 (100%, M + Na+); ВЭЖХ: 4,6 мин.
Соединение VII: Натриевая соль 3-гидрокси-5-пентилфенилуксусной кислоты

Этап 1
Раствор метил[3,5-дигидроксифенил]ацетата (2,1 г, 11,5 ммоль) в ацетоне (100 мл) обрабатывали с помощью карбоната калия (2,4 г, 17,4 ммоль), йодида калия (383 мг, 2,31 ммоль) и бензилбромид (1,5 мл, 12,7 ммоль), а смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой и экстрагировали дихлорметаном (x3). Объединенные органические экстракты высушивали над сульфатом натрия и выпаривали in vacuo. Неочищенный материал очищали на колонке 40M Biotage™ (силикагель), элюируя с помощью 40% этилацетата/гексана, для получения метил[3-бензилокси-5-гидроксифенил]ацетата (1,0 г, 33%). 1H ЯМР (400 МГц, CDCl3): δ 7,32-7,42 (м, 5H), 6,48 (д, J = 1,4 Гц, 1H), 6,38-6,39 (м, 2H), 4,99 (с, 2H), 3,69 (с, 3H), 3,53 (с, 2H).
Этап 2
Раствор бензилового эфира (1,04 г, 3,8 ммоль) в дихлорметане (15 мл) при 0°C обрабатывали с помощью N-фенил-бис(трифторсульфонил)имида (1,40 г, 3,9 ммоль), а затем медленно добавляли триэтиламин (0,6 мл, 4,1 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 ч, и затем при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли водой, а затем экстрагировали диэтилэфиром (x 2). Объединенные органические экстракты промывали с помощью 1M водного гидроксида натрия, воды (x 2) и насыщенного водного хлорида натрия, затем высушивали над сульфатом натрия, отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке 40M Biotage™ (силикагель), элюируя с помощью 25% этилацетата/гексана, давало метил[3-бензилокси-5-трифторметансульфонилоксифенил]ацетат (1,2 г, 79%). 1H ЯМР (400 МГц, CDCl3): δ 7,36-7,46 (м, 5H), 6,98 (с, 1H), 6,97 (с, 1H), 6,84 (с, 1H), 5,06 (с, 2H), 3,72 (с, 3H), 3,63 (с, 2H).
Этап 3
Раствор сложного пинаколового эфира E-1-пентен-1-илбороновой кислоты (0,8 г, 3,9 ммоль) в диметоксиэтане (5 мл) обрабатывали с помощью раствора трифлата (1,2 г, 3,0 ммоль) в диметоксиэтане (5 мл). Раствор обрабатывали с помощью палладия(0) (0,7 г, 0,6 ммоль) и 2M водного карбоната натрия (1,3 мл, 2,6 ммоль). Смесь затем нагревали при 90°C в течение 3 дней. Реакционную смесь охлаждали до комнатной температуры и отфильтровывали сквозь Celite™. Фильтрат выпаривали in vacuo, а неочищенный материал очищали на колонке 25M Biotage™ (силикагель), элюируя с помощью 5% этилацетата/гексана, для получения метил[3-бензилокси-5-[пент-1-енил]фенил]ацетата (0,4 г, 40%). 1H ЯМР (400 МГц, CDCl3): δ 7,36-7,47 (м, 5H), 6,90-6,92 (м, 2H), 6,79 (дд, J = 2,0, 2,0 Гц, 1H), 6,35 (д, J = 15,9 Гц, 1H), 6,24 (дт, J = 15,9, 6,8 Гц, 1H), 5,07 (с, 2H), 3,70 (с, 3H), 3,59 (с, 2H), 2,20 (тд, J = 7,4, 6,8 Гц, 2H), 1,51 (дт, J = 7,4 Гц, 2H), 0,98 (т, J = 7,4 Гц, 3H).
Этап 4
Раствор алкена (0,4 г, 1,2 ммоль) в этаноле (13 мл) обрабатывали с помощью 1% палладиевого катализатора на углеродном носителе (40 мг). Смесь перемешивали под 1 атм. водорода при комнатной температуре в течение ночи. Реакционную смесь отфильтровывали, выпаривали in vacuo и очищали на колонке 25S Biotage™ (силикагель), элюируя с помощью 15% этилацетата/гексана, для получения метил[3-гидрокси-5-пентилфенил]ацетата (0,3 г, 93%). 1H ЯМР (400 МГц, CDCl3): δ 6,64 (с, 1H), 6,58-6,60 (м, 2H), 3,70 (с, 3H), 3,55 (с, 2H), 2,51 (т, J = 7,7 Гц, 2H), 1,55-1,59 (м, 2H), 1,28-1,34 (м, 4H), 0,88 (т, J = 7,0 Гц, 3H).
Этап 5
Раствор сложного эфира (0,3 г, 1,3 ммоль) в этаноле (12 мл) обрабатывали с помощью воды (3 мл) и гидроксида лития (155 мг, 6,4 ммоль), а смесь энергично перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли водой (100 мл); промывали дихлорметаном; затем подкисляли до pH 1 с помощью 1М водной хлористоводородной кислоты и экстрагировали дихлорметаном (x 3). Объединенные органические экстракты высушивали над сульфатом натрия (0,3 г, 95%). Этот материал применяли без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3): δ 6,66 (с, 1H), 6,58-6,59 (м, 2H), 3,55 (с, 2H), 2,52 (т, J = 7,7 Гц, 2H), 1,55-1,59 (м, 2H).
Этап 6
Раствор кислоты (0,27 г, 1,23 ммоль) в этаноле (6 мл) и воде (6 мл) обрабатывали с помощью бикарбоната натрия (0,1 г, 1,2 ммоль), а реакционную смесь перемешивали при комнатной температуре в течение нескольких часов. Растворитель концентрировали in vacuo, а раствор разбавляли водой, отфильтровывали (0,2 мкм) и лиофилизировали для получения [3-гидрокси-5-пентилфенил]ацетата натрия в виде белого твердого вещества (0,3 г, 95%). т. пл. 63-66°C; 1H ЯМР (400 МГц, CD3OD): δ 6,63 (с, 1H), 6,58 (с, 1H), 6,42 (с, 1H), 3,36 (с, 2H), 2,48 (т, J = 7,6 Гц, 2H), 1,55-1,62 (м, 2H), 1,26-1,38 (м, 4H), 0,89 (т, J = 6,8 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 177,79, 155,31, 142,36, 137,62, 119,08, 111,66, 111,18, 43,70, 34,17, 29,95, 29,56, 20,87, 11,64; МСНР (ESI): m/z 445,2 (2M - 2Na+ + 3H+), m/z 223 (M - Na+ + 2H+); ВЭЖХ: 3,5 мин.
Соединение VIII: Натриевая соль 2-(4-гидрокси-3-пентилфенил)уксусной кислоты
Указанное выше соединение получали с помощью сочетания Сузуки бензил-2-(4-(бензилокси)-3-бромфенил)ацетата и сложных пинаколовых эфиров (E)-пент-1-енилбороновой кислоты, например, VII; с последующим гидрированием. Белое твердое вещество; температура плавления 192-195°C; 1H ЯМР (400 МГц, CD3OD): δ 7,01 (д, J = 2,3 Гц, 1H), 6,93 (дд, J = 8,2, 2,3 Гц, 1H), 6,64 (д, J = 8,2 Гц, 1H), 3,35 (с, 2H), 2,53 (т, J = 7,7 Гц, 2H), 1,54-1,61 (м, 2H), 1,30-1,37 (м, 4H), 0,90 (т, J = 7,2 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 180,25, 153,20, 130,54, 128,80, 128,76, 127,10, 114,49, 44,45, 31,84, 30,10, 29,73, 22,52, 13,31; МСНР (ESI): m/z 245,2 (55%, MH+), 177,4 (100%, M – CO2Na); ВЭЖХ: 1,9 мин.
Соединение IX: Натриевая соль 2-(2-гидрокси-3-пентилфенил)уксусной кислоты
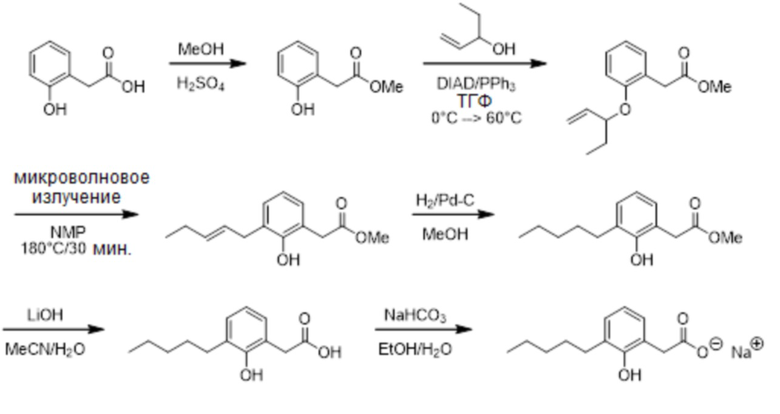
Этап 1
Раствор 2-(2-гидроксифенил)уксусной кислоты (3,00 г, 19,7 ммоль) в метаноле (40 мл) обрабатывали с помощью серной кислоты (0,95 мл, 17,8 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 18 часов. Реакционную смесь разбавляли с помощью этилацетата (250 мл), а раствор промывали водой (2 x 150 мл) и насыщенным водным хлоридом натрия (150 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Перекристаллизация из горячего гексана давало метил-2-(2-гидроксифенил)ацетат (2,83 г, 87%). 1H ЯМР (400 МГц, CDCl3): δ 7,20 (ддд, J = 7,7, 7,4, 1,8 Гц, 1H), 7,09-7,11 (м, 1H), 6,94 (дд, J = 8,0, 1,2 Гц, 1H), 6,88 (ддд, J = 7,4, 7,4, 1,2 Гц, 1H), 3,75 (с, 3H), 3,69 (с, 2H).
Этап 2
Раствор метил-2-(2-гидроксифенил)ацетата (1,00 г, 6,0 ммоль), трифенилфосфина (2,37 г, 9,0 ммоль) и пент-1-ен-3-ола (0,78 г, 9,0 ммоль) в тетрагидрофуране (30 мл) охлаждали до 0°C под азотом и добавляли по каплям диизопропилазодикарбоксилат (1,86 мл; 9,0 мл) за 10 минут. Реакционную смесь затем нагревали до 60°C в течение 21,5 часов. Растворитель выпаривали in vacuo и экстрагировали остаток с помощью 5% этилацетата в гексане. Экстракт отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на системе SP1 Biotage™ (кассета с 120 г силикагеля), элюируя с помощью 0-3% этилацетата в гексане, давало метил-2-(2-(пент-1-ен-3-илокси)фенил)ацетат (0,39 г, 28%). 1H ЯМР (400 МГц, CDCl3): δ 7,21-7,26 (м, 1H), 7,20 (д, J = 7,6 Гц, 1H), 6,91 (ддд, J = 7,4, 7,4, 1,0 Гц, 1H), 6,87 (д, J = 8,0 Гц, 1H), 5,84 (ддд, J = 17,4, 10,7, 6,0 Гц, 1H), 5,26 (д, J = 17,4 Гц, 1H), 5,22 (д, J = 10,7 Гц, 1H), 4,63 (дт, J = 6,0, 6,0 Гц, 2H), 3,70 (с, 3H), 3,68 (с, 2H), 1,71-1,87 (м, 2H), 1,02 (т, J = 7,5 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 172,58,. 156,28, 137,75, 131,19, 128,50, 123,87, 120,52, 116,66, 113,18, 79,76, 52,00, 36,61, 28,71, 9,62.
Этап 3
Раствор метил-2-(2-(пент-1-ен-3-илокси)фенил)ацетата (0,24 г, 1,0 ммоль) в N-метил-2-пирролидине (1,0 мл) облучали микроволновым излучением в Biotage Initiator при 180°C в течение 30 минут, затем в течение 15 минут. Раствор разбавляли с помощью этилацетата (25 мл), затем промывали водой (4 x 25 мл) и насыщенным водным хлоридом натрия (25 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на системе SP1 Biotage™ (кассета с 40 г силикагеля), элюируя с помощью 0-7% этилацетата в гексане, давало метил-(E)-2-(2-гидрокси-3-(пент-2-енил)фенил)ацетат (0,89 г, 37%). 1H ЯМР (400 МГц, CDCl3): δ 7,09 (с, 1H), 7,08 (дд, J = 7,4, 1,6 Гц, 1H), 7,01 (дд, J = 7,6, 1,6 Гц, 1H), 6,85 (дд, J = 7,6, 7,4 Гц, 1H), 5,59-5,70 (м, 2H), 3,75 (с, 3H), 3,69 (с, 2H), 3,41 (д, J = 4,7 Гц, 2H), 2,04-2,11 (м, 2H), 1,01 (т, J = 7,4 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 174,31, 153,53, 134,44, 129,86, 129,32, 128,62, 127,13, 121,08, 120,82, 52,79, 37,59, 34,17, 25,77, 13,97.
Этап 4
Mетил-(E)-2-(2-гидрокси-3-(пент-2-енил)фенил)ацетата (0,14 г, 0,6 ммоль) гидрировали таким же образом, что и в случае Соединения I, этап 3, но с применением метанола в качестве растворителя, для получения метил-2-(2-гидрокси-3-пентилфенил)ацетата (0,11 г, 76%). 1H ЯМР (400 МГц, CDCl3): δ 7,57 (с, 1H), 7,11 (дд, J = 7,4, 1,6 Гц, 1H), 6,96 (дд, J = 7,4, 1,6 Гц, 1H), 6,84 (дд, J = 7,4, 7,4 Гц, 1H), 3,76 (с, 3H), 3,70 (с, 2H), 2,68 (т, J = 7,8 Гц, 2H), 1,61-1,67 (м, 2H), 1,36-1,43 (м, 4H), 0,93 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 175,01, 153,48, 131,75, 129,98, 128,75, 120,74, 120,60, 53,01, 38,30, 32,10, 30,50, 29,91, 22,87, 14,34.
Этап 5
Метил-2-(2-гидрокси-3-пентилфенил)ацетат (0,11 г, 0,5 ммоль) гидрировали таким же образом, что и в случае Соединения I, этап 4, с применением ацетонитрила/воды (4:1) в качестве растворителей, для получения 2-(2-гидрокси-3-пентилфенил)уксусной кислоты (0,57 г, 57%). 1H ЯМР (400 МГц, CDCl3): δ 8,70 (br с, 1H), 7,09 (дд, J = 7,6, 1,6 Гц, 1H), 6,98 (дд, J = 7,4, 1,6 Гц, 1H), 6,84 (дд, J = 7,6, 7,4 Гц, 1H), 3,68 (с, 2H), 2,62 (т, J = 7,8 Гц, 2H), 1,57-1,65 (м, 2H), 1,31-1,40 (м, 4H), 0,91 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 179,89, 152,79, 130,92, 130,04, 128,98, 121,08, 120,24, 37,74, 32,02, 30,34, 29,78, 22,80, 14,30.
Этап 6
2-(2-Гидрокси-3-пентилфенил)уксусную кислоту (22 мг, 0,098 ммоль) преобразовывали в натриевую соль таким же образом, что и в случае Соединения I, этап 5, для получения 2-(2-гидрокси-3-пентилфенил)ацетата натрия (24 мг, 98%). 1H ЯМР (400 МГц, CD3OD): δ 6,91 (дд, J = 7,5, 1,6 Гц, 1H), 6,87 (дд, J = 7,5, 1,6 Гц, 1H), 6,66 (дд, J = 7,5, 7,5 Гц, 1H), 3,49 (с, 2H), 2,59 (т, J = 7,7 Гц, 2H), 1,55-1,62 (м, 2H), 1,28-1,38 (м, 4H), 0,90 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 180,26, 154,27, 130,75, 128,21, 127,90, 124,24, 119,23, 42,91, 31,83, 30,21, 29,82, 22,51, 13,29; МСНР (Анионная ESI): m/z 220,8 (100%, M –Na+); СВЭЖХ (Система A): 5,0 мин. СВЭЖХ Система A: Подвижная фаза A = 10 mM водный раствор формиата аммония; Подвижная фаза B = ацетонитрил; неподвижная фаза = HSS T3 колонка; градиент = 5-100% B в A в течение 10 минут.
Соединение X: Натриевая соль 2-(3-фтор-5-пентилфенил)уксусной кислоты
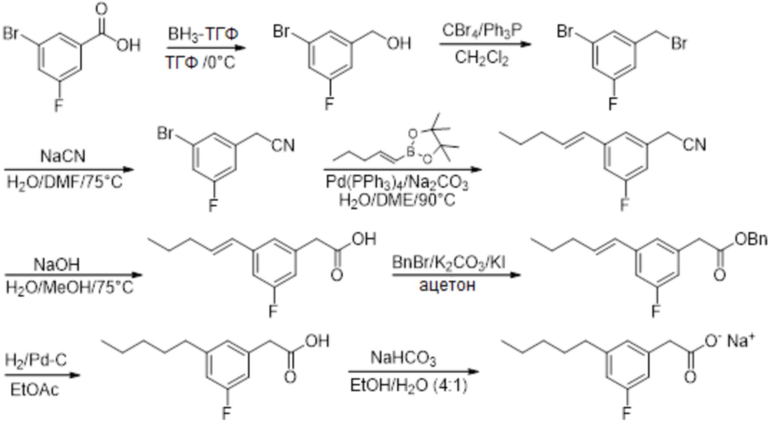
Этап 1
Раствор 3-бром-5-фторбензойной кислоты (2,74 г, 12,5 ммоль) в тетрагидрофуране (6 мл), при 0°C под азотом, обрабатывали с помощью комплекса боран-тетрагидрофуран (1M, 15 мл, 15 ммоль) небольшими порциями в течение 12 минут, а реакционную смесь затем перемешивали при 0°C в течение 70 минут и при комнатной температуре в течение 22 ч. Реакционную смесь гасили путем добавления метанола (10 мл), а метанольную смесь перемешивали при комнатной температуре в течение 3 ч. и затем выпаривали in vacuo с совместным выпариванием из метанола, затем из этилацетата, для получения неочищенного продукта. Материал растворяли в этилацетате (200 мл), и раствор промывали с помощью 0,5M водного гидроксида натрия (200 мл) и насыщенного водного хлорида натрия (100 мл); затем высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения 3-бром-5-фторбензилового спирта (1,79 г, 67%). 1H ЯМР (400 МГц, CDCl3): δ 7,29 (с, 1H), 7,15 (ддд, JHF = 8,2 Гц, JHH = 2,2, 1,8 Гц, 1H), 7,00-7,02 и 7,02-7,04 (dм, JHF = 9,2 Гц, JHH = не определено, 1H), 4,66 (с, 2H), 2,04 (br с, 1H); 19F ЯМР (377 МГц, CDCl3): δ -111,05 (дд, JHF = 9,3, 8,0 Гц, 1F); 13C ЯМР (101 МГц, CDCl3): δ 162,87 (д, JCF = 250,6 Гц), 145,42 (д, JCF = 6,9 Гц), 125,45 (д, JCF = 3,1 Гц), 122,69 (д, JCF = 9,2 Гц), 118,01 (д, JCF = 24,6 Гц), 112,51 (д, JCF = 21,5 Гц), 63,60 (д, JCF = 2,3 Гц).
Этап 2
Раствор 3-бром-5-фторбензилового спирта (1,79 г, 8,39 ммоль) и трифенилфосфина (3,65 г, 10,10 ммоль) в дихлорметане (45 мл) обрабатывали с помощью тетрабромида углерода (3,34 г, 10,10 ммоль) небольшими порциями в течение 10 мин., а реакционную смесь затем перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали in vacuo, а остаток обрабатывали с помощью диэтилового эфира (50 мл). Полученную белую суспензию перемешивали при комнатной температуре, а затем отфильтровывали сквозь Celite™. Остаток промывали диэтилэфиром (2 x 50 мл), а объединенные фильтрат и промывочные растворы выпаривали in vacuo для получения неочищенного продукта. Очищение на подушке силикагеля, элюируя с помощью 2% этилацетата/гексана, давало 3-бром-5-фторбензилбромид (2,21 г, 98%). 1H ЯМР (400 МГц, CDCl3): δ 7,33 (с, 1H), 7,18 (ддд, JHF = 8,2 Гц, JHH = 2,0, 2,0 Гц, 1H), 7,05 (ддд, JHF = 9,0 Гц, JHH = 1,8, 1,6 Гц, 1H), 4,38 (с, 2H); 19F ЯМР (377 МГц, CDCl3): δ -110,19 to -110,14 (м, 1F); 13C ЯМР (101 МГц, CDCl3): δ 162,67 (д, JCF = 252,1 Гц), 141,61(д, JCF = 8,5 Гц), 128,17 (д, JCF = 3,1 Гц), 122,94 (д, JCF = 10,0 Гц), 119,39 (д, JCF = 24,6 Гц), 115,34 (д, JCF = 22,3 Гц), 31,31 (д, JCF = 2,3 Гц).
Этап 3
Суспензию цианида натрия (0,38 г, 7,73 ммоль) в воде (0,35 мл) обрабатывали с помощью раствора 3-бром-5-фторбензилбромида (1,38 г, 5,15 ммоль) в диметилформамиде (2,6 мл), а реакционную смесь нагревали при 75°C в герметично закрытой пробирке в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и разделяли между этилацетатом (50 мл) и 2,5% мас./об. водным бикарбонатом натрия (100 мл). Водную фазу экстрагировали с помощью дополнительной части этилацетата (50 мл); а объединенные экстракты промывали водой (2 x 50 мл) и насыщенным водным хлоридом натрия (50 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке 40iM Biotage™ (силикагель), элюируя с помощью 10% этилацетата/гексана, давало 2-[3-бром-5-фторфенил]ацетонитрил (0,64 г, 58%). 1H ЯМР (400 МГц, CDCl3): δ 7,26-7,28 (м, 1H), 7,17-7,19 & 7,19-7,21 (дм, JHF = 8,0 Гц, JHH = не определено, 1H), 6,98-7,00 & 7,00-7,02 (дм, JHF = 8,8 Гц, JHH = не определено, 1H), 3,73 (с, 2H); 19F ЯМР (377 МГц, CDCl3): δ -109,46 (дд, JHF = 8,0, 8,0 Гц, 1F); 13C ЯМР (101 МГц, CDCl3): δ 162,90 (д, JCF = 252,1 Гц), 133,95 (д, JCF = 8,5 Гц), 127,24 (д, JCF = 3,8 Гц), 123,53 (д, JCF = 10,0 Гц), 119,22 (д, JCF = 23,8 Гц), 117,00, 114,50 (д, JCF = 23,1 Гц), 23,30 (д, JCF = 1,5 Гц).
Этап 4
Раствор арилбромида (0,55 г, 2,58 ммоль) и сложного пинаколового эфира (E)-1-пентен-1-илбороновой кислоты (0,61 г, 3,13 ммоль) в диметоксиэтане (13 мл) обрабатывали с помощью раствора карбоната натрия (0,55 г, 5,17 ммоль) в воде (3 мл). Раствор дезоксигенировали с помощью азота и обрабатывали с помощью тетракис(трифенилфосфин)палладия (0,15 г, 0,13 ммоль; 5 % моль). Смесь затем нагревали при 90°C в герметично закрытой пробирке в течение 17 ч. Реакционную смесь охлаждали до комнатной температуры и разделяли между этилацетатом (50 мл) и 1M водной хлористоводородной кислоты (50 мл). Органическую фазу промывали насыщенным водным хлоридом натрия (30 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке 40iM Biotage™ (силикагель), элюируя с помощью (3%) этилацетата/гексана, давало (E)-2-[3-фтор-5-[пент-1-енил]фенил]ацетонитрил (0,43 г, 82%). 1H ЯМР (400 МГц, CDCl3): δ 7,04 (с, 1H), 6,97 (ддд, JHF = 9,8 Гц, JHH = 2,0, 1,5 Гц, 1H), 6,82-6,85 (м, 1H), 6,31 (д, J = 15,8 Гц, 1H), 6,25 (ддд, J = 15,8, 5,9, 0 Гц, 1H), 3,68 (с, 2H), 2,18 (тд, J = 7,2, 5,4 Гц, 2H), 1,49 (кв, J = 7,4, 7,4 Гц, 2H), 0,95 (т, J = 7,4 Гц, 3H); 19F ЯМР (377 МГц, CDCl3): δ -112,93 (дд, JHF = 10,6, 9,3 Гц, 1F); 13C ЯМР (101 МГц, CDCl3): δ 163,43 (д, JCF = 246,0 Гц), 141,44 (д, JCF = 8,5 Гц), 133,99, 132,37 (д, JCF = 8,5 Гц), 128,42 (д, JCF = 2,3 Гц), 121,60 (д, JCF = 3,1 Гц), 117,66, 113,40 (д, JCF = 23,1 Гц), 112,21 (д, JCF = 22,3 Гц), 35,22, 23,49 (д, JCF = 2,3 Гц), 22,51, 13,94.
Этап 5
Раствор производного фенилацетонитрила (0,43 г, 2,10 ммоль) в метаноле (42 мл) обрабатывали с помощью водного гидроксида натрия (5M; 21 мл, 105 ммоль), а смесь нагревали при 75°C в герметично закрытой пробирке в течение 4,5 ч. Реакционную смесь охлаждали до комнатной температуры и гасили с помощью 6M водной хлористоводородной кислоты (21 мл); перемешивали при комнатной температуре в течение 10 мин.; затем экстрагировали с помощью этилацетата (2 x 75 мл). Органический экстракт промывали насыщенным водным хлоридом натрия (75 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке 40iM Biotage™ (силикагель), элюируя с помощью 70% этилацетата/гексана, давало сложный метиловый эфир желаемого продукта (0,09 г, 18%) и ~95% чистой (E)-2-[3-фтор-5-[пент-1-енил]фенил]уксусной кислоты (0,22 г, 48%). 1H ЯМР (400 МГц, CDCl3): δ 11,17 (br с, 1H), 7,02 (с, 1H), 6,98 (ддд, JHF = 9,8 Гц, JHH = 2,0, 1,8 Гц, 1H), 6,85 (ддд, JHF = 9,0 Гц, JHH = 1,8, 1,6 Гц, 1H), 6,33 (д, J = 15,8 Гц, 1H), 6,25 (дт, J = 15,8, 6,4 Гц, 1H), 3,62 (с, 2H), 2,17-2,22 (м, 2H), 1,51 (кв, J = 7,4, 7,4 Гц, 2H), 0,96 (т, J = 7,4 Гц, 3H); 19F ЯМР (377 МГц, CDCl3): δ -114,10 (дд, JHF = 9,3, 9,3 Гц, 1F).
Этап 6
Раствор частично очищенной кислоты (0,28 г, 1,26 ммоль) в ацетоне (5 мл) обрабатывали с помощью карбоната калия (0,26 г, 1,90 ммоль), йодида калия (0,04 г, 0,25 ммоль) и бензилбромида (0,18 мл, 1,5 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 18 ч. Реакционную смесь разделяли между этилацетатом (25 мл) и 1M водной хлористоводородной кислотой (25 мл). Органическую фазу затем промывали насыщенным водным хлоридом натрия (25 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке 40iM Biotage™ (силикагель), элюируя с помощью 5% этилацетата/гексана давало бензил-(E)-2-[3-фтор-5-[пент-1-енил]фенил]ацетат (0,3 г, 75%). 1H ЯМР (400 МГц, CDCl3): δ 7,32-7,40 (м, 5H), 7,03 (с, 1H), 6,97 (ддд, JHF = 10,0 Гц, JHH = 2,3, 1,5 Гц, 1H), 6,86 (ддд, JHF = 9,0 Гц, JHH = 2,0, 1,7 Гц, 1H), 6,33 (д, J = 15,8 Гц, 1H), 6,23 (дт, J = 15,8, 6,5 Гц, 1H), 5,16 (с, 2H), 3,64 (с, 2H), 2,17-2,23 (м, 2H), 1,52 (кв, J = 7,4, 7,4 Гц, 2H), 0,97 (т, J = 7,4 Гц, 3H); 19F ЯМР (377 МГц, CDCl3): δ -114,34 (дд, JHF = 9,3, 9,3 Гц, 1F); 13C ЯМР (101 МГц, CDCl3): δ 171,08, 163,32 (д, JCF = 244,4 Гц), 140,65 (д, JCF = 7,7 Гц), 136,17 (д, JCF = 8,5 Гц), 135,93, 133,05, 128,95 (д, JCF = 3,1 Гц), 128,84, 128,52 (д, JCF = 9,2 Гц), 128,48, 123,09 (д, JCF = 2,3 Гц), 114,78 (д, JCF = 22,3 Гц), 111,46 (д, JCF = 22,3 Гц), 67,04, 41,26 (д, JCF = 1,5 Гц), 35,27, 22,63, 14,00.
Этап 7
Раствор сложного бензилового эфира (0,16 г, 0,50 ммоль) в этилацетате (2 мл) обрабатывали с помощью палладиевого катализатора на углеродном носителе (1% мас./мас. Pd; 15 мг). Смесь дегазировали водородом и перемешивали под 1 атмосферой водорода при комнатной температуре в течение ночи. Реакционную смесь отфильтровывали и выпаривали in vacuo для получения 2-[3-фтор-5-пентилфенил]-уксусной кислоты (0,11 г, 97%). 1H ЯМР (400 МГц, CDCl3): δ 11,47 (br с, 1H), 6,89 (с, 1H), 6,81-6,86 (м, 2H), 3,62 (с, 2H), 2,60 (т, J = 7,8 Гц, 2H), 1,58-1,66 (м, 2H), 1,28-1,41 (м, 4H), 0,92 (т, J = 6,8 Гц, 3H); 19F ЯМР (377 МГц, CDCl3): δ -114,34 (дд, JHF = 9,3, 9,3 Гц, 1F); 13C ЯМР (101 МГц, CDCl3): δ 178,15, 163,08 (д, JCF = 246,0 Гц), 145,02 (д, JCF = 7,7 Гц), 135,04 (д, JCF = 8,5 Гц), 125,49 (д, JCF = 2,3 Гц), 114,49 (д, JCF = 20,8 Гц), 113,83 (д, JCF = 22,3 Гц), 41,01 (д, JCF = 1,5 Гц), 35,87 (д, JCF = 1,5 Гц), 31,67, 31,03, 22,74, 14,24.
Этап 8
Раствор кислоты (0,11 г, 0,49 ммоль) в этаноле (3 мл) обрабатывали с помощью раствора бикарбоната натрия (0,041 г, 0,49 ммоль) в воде (0,75 мл) и перемешивали реакционную смесь при комнатной температуре в течение 17 ч. Этанол выпаривали in vacuo, а остаточный водный концентрированный раствор разбавляли водой (10 мл), отфильтровывали (0,2 μи) и лиофилизировали для получения 2-[3-фтор-5-пентилфенил]ацетата натрия в виде белого твердого вещества (0,12 г, 99%). т. пл. 120-123°C; 1H ЯМР (400 МГц, CD3OD): δ 6,94 (с, 1H), 6,87 (ддд, JHF = 9,8 Гц, JHH = 2,0, 2,0 Гц, 1H), 6,70 (ддд, JHF = 10,0 Гц, JHH = 2,0, 2,0 Гц, 1H), 3,45 (с, 2H), 2,56 (т, J = 7,7 Гц, 2H), 1,58-1,63 (м, 2H), 1,26-1,39 (м, 4H), 0,90 (т, J = 7,0 Гц, 3H); 19F ЯМР (377 МГц, CD3OD): δ -117,54 (дд, JHF = 10,0, 10,0 Гц, 1F); 13C ЯМР (101 МГц, CD3OD): δ 178,66, 163,04 (д, JCF = 242,9 Гц), 145,07 (д, JCF = 7,7 Гц), 140,42 (д, JCF = 8,5 Гц), 125,03 (д, JCF = 2,3 Гц), 112,99 (д, JCF = 22,3 Гц), 112,30 (д, JCF = 20,8 Гц), 44,96, 35,53 (д, JCF = 1,5 Гц), 31,46, 31,00, 22,45, 13,30; ВЭЖХ: 1,2 мин.
Соединение XI: Натриевая соль 2-(2-фтор-3-пентилфенил)уксусной кислоты
Указанное выше соединение получали таким же образом, что и в случае Соединения X, начиная с 3-бром-2-фторбензойной кислоты. Белое твердое вещество; 1H ЯМР (400 МГц, CD3OD): δ 7,13 (ддд, JHF = 7,0 Гц, JHH = 7,4, 1,9 Гц, 2H), 7,03 (ддд, JHF = 7,0 Гц, JHH = 7,4, 1,9 Гц, 1H), 6,97 (дд, JHH = 7,4, 7,4 Гц, 1H), 3,51 (д, JHF = 1,4 Гц, 2H), 2,61 (т, J = 7,6 Гц, 2H), 1,56-1,63 (м, 2H), 1,28-1,40 (м, 4H), 0,90 (т, J = 6,9 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 178,21, 159,70 (д, JCF = 242,9 Гц), 129,07 (д, JCF = 4,6 Гц), 128,88, 128,43 (д, JCF = 5,4 Гц), 125,02 (д, JCF = 17,7 Гц), 123,31 (д, JCF = 4,6 Гц), 37,89 (д, JCF = 3,8 Гц), 31,55, 29,98, 28,91 (д, JCF = 3,1 Гц), 22,41, 13,26; 19F ЯМР (377 МГц, CD3OD): δ -126,09 to -126,05 (м, 1F); МСНР (ESI): m/z 220,0 (M – CO2Na + ацетонитрил), 179,4 (M – CO2Na); ВЭЖХ: 1,2 мин.
Соединение XII: Натриевая соль 2-(4-фтор-3-пентилфенил)уксусной кислоты
Указанное выше соединение получали из метил-2-(3-бром-4-фторфенил)ацетата с помощью сочетания Сузуки таким же образом, что и в случае Соединения Соединения VII; с последующим гидрированием, гидролизом сложного эфира и образованием соли в качестве Соединения I. Исходный сложный эфир получали с помощью реакции 2-(3-бром-4-фторфенил)уксусной кислоты с метанолом в присутствии серной кислоты. Белое твердое вещество; 1H ЯМР (400 МГц, CD3OD): δ 7,16 (дд, JHF = 7,4 Гц, JHH = 2,3 Гц, 2H), 7,08 (ддд, JHF = 5,0 Гц, JHH = 8,3, 2,3 Гц, 1H), 6,88 (дд, JHF = 10,1 Гц, JHH = 8,3 Гц, 1H), 3,40 (с, 2H), 2,59 (т, J = 7,7 Гц, 2H), 1,55-1,63 (м, 2H), 1,28-1,40 (м, 4H), 0,90 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 179,12, 159,88 (д, JCF = 240,6 Гц), 133,88 (д, JCF = 3,8 Гц), 131,26 (д, JCF = 4,6 Гц), 128,78 (д, JCF = 16,1 Гц), 127,96 (д, JCF = 8,5 Гц), 114,26 (д, JCF = 23,1 Гц), 44,38, 31,51, 30,00, 28,76 (д, JCF = 1,5 Гц), 22,36, 13,18; 19F ЯМР (377 МГц, CD3OD): δ -126,45 to -126,40 (м, 1F); МСНР (ESI): m/z 225,2 (M – Na+ + 2H+); ВЭЖХ: 1,9 мин.
Соединение XIII: Натриевая соль (RS)-2-фтор-2-(3-пентилфенил)уксусной кислоты
Указанное выше соединение получали из этил-2-фтор-2-(3-пентилфенил)ацетата таким же образом, что и в случае Соединения I. Сложный эфир получали с помощью реакции этил-2-(3-пентилфенил)ацетата с диизопропиламидом лития и N-фторбензолсульфонимидом при -78°C в тетрагидрофуране. Белое твердое вещество; 1H ЯМР (400 МГц, CD3OD): δ 7,34 (с, 1H), 7,30 (дд, J = 7,6, 1,4 Гц, 1H), 7,24 (дд, J = 7,6, 7,6 Гц, 1H), 7,13 (дд, J = 7,4, 1,0 Гц, 1H), 5,53 (д, JHF = 51,3 Гц, 1H), 2,60 (т, J = 7,7 Гц, 2H), 1,59-1,65 (м, 2H), 1,27-1,39 (м, 4H), 0,76 (т, J = 6,9 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 173,73 (д, JCF = 23,9 Гц), 141,34, 136,37 (д, JCF = 20,0 Гц), 126,79 (д, JCF = 2,3 Гц), 126,40, 125,41 (д, JCF = 5,4 Гц), 122,84 (д, JCF = 5,4 Гц), 90,34 (д, JCF = 183,4 Гц), 34,13, 29,91, 29,65, 20,85, 11,64; 19F ЯМР (377 МГц, CD3OD): δ -168,83 (д, JHF = 51,7 Гц, 1F); МСНР (Анионная ESI): m/z 223,0 (100%, M – Na+); ВЭЖХ: 4,1 мин.
Соединение XIV: 2-[3,5-Дипентилфенил]ацетат натрия
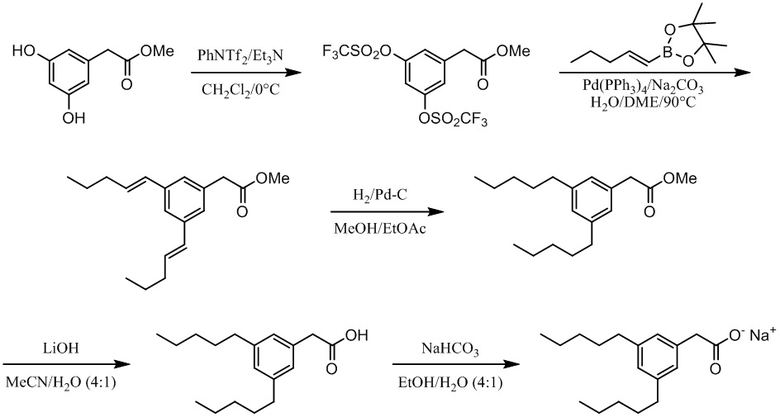
Этап 1
Суспензию метил-2-[3,5-дигидроксифенил]ацетата (1,00 г, 5,49 ммоль) и N-фенил-бис(трифторметилсульфонил)имида (4,31 г, 12,1 ммоль) в дихлорметане (20 мл), при 0°C под азотом, обрабатывали с помощью триэтиламина (1,68 мл, 12,1 ммоль). Образовался прозрачный раствор. Реакционную смесь затем перемешивали под азотом при 0°C в течение 2 ч. и при комнатной температуре в течение 21 ч. Реакционную смесь разбавляли с помощью этилацетата (100 мл), а раствор промывали с помощью 0,5M водного гидроксида натрия (2 x 100 мл) и насыщенным водным хлоридом натрия (75 мл); затем высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке 40iM Biotage™ (силикагель), элюируя с помощью этилацетата/гексана от 0:1 до 1:9, давало метил-2-[3,5-бис(трифторметилсульфонилокси)фенил]ацетат (2,23 г, 91%) в виде белесого масла. 1H ЯМР (400 МГц, CDCl3): δ 7,32 (д, J = 2,2 Гц, 2H), 7,18 (дд, J = 2,2, 2,2 Гц, 1H), 3,72 (с, 5H); 19F ЯМР (377 МГц, CDCl3): δ -73,20 (с, 3F); 13C ЯМР (101 МГц, CDCl3): δ 170,05, 149,48, 139,01, 122,95, 118,87 (кв, JCF = 320,5 Гц), 114,42, 52,62, 40,29.
Этап 2
Раствор арилбис(трифлата) (2,23 г, 4,99 ммоль) и сложного пинаколового эфира (E)-1-пентен-1-илбороновой кислоты (2,45 г, 12,5 ммоль) в 1,2-диметоксиэтанe (25 мл) обрабатывали с помощью раствора карбоната натрия (1,59 г, 15,0 ммоль) в воде (8 мл). Раствор дезоксигенировали с помощью азота, а затем обрабатывали с помощью тетракис(трифенилфосфин)палладия (0,58 г, 0,50 ммоль). Смесь нагревали при 90°C в герметично закрытой пробирке в течение 17 ч. Реакционную смесь охлаждали до комнатной температуры и разделяли между этилацетатом (200 мл) и 1M водной хлористоводородной кислоты (150 мл). Органическую фазу промывали с помощью 5% водного бикарбоната натрия (150 мл) и насыщенным водным хлоридом натрия (150 мл); затем высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке 40iL Biotage™ (силикагель), элюируя с помощью этилацетата/гексана от 0:1 до 3:97, давало метил-2-[3,5-ди[(E)-1-пент-1-енил]фенил]ацетат в виде неразделимой смеси 10:4 с избытком сложного пинаколового эфира (E)-1-пентен-1-илбороновой кислоты (1,12 г, 61%). 1H ЯМР (400 МГц, CDCl3): δ 7,21 (с, 1H), 7,10 (д, J = 1,3 Гц, 2H), 6,34 (д, J = 15,8 Гц, 1H), 6,22 (дд, J = 15,8, 6,7 Гц, 1H), 3,65 (с, 3H), 3,55 (с, 2H), 2,18 (tdd, J = 6,8, 6,8, 1,0 Гц, 2H), 1,49 (кв, J = 7,4, 7,2 Гц, 2H), 0,96 (т, J = 7,4 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 172,04, 138,59, 134,47, 131,34, 129,97, 125,57, 122,75, 52,07, 41,32, 35,39, 22,77, 13,97.
Этап 3
Раствор ненасыщенного соединения (1,12 г, 78,5% мас./мас., 3,07 ммоль) в этилацетате (1 мл) и метаноле (1 мл) обрабатывали с помощью палладиевого катализатора на углеродном носителе (10% мас./мас. Pd; 0,12 г). Смесь дегазировали водородом и перемешивали под 1 атм. водорода при комнатной температуре в течение 22 ч. Реакционную смесь отфильтровывали и выпаривали in vacuo для получения метил-2-[3,5-дипентилфенил]ацетата в виде неразделимой смеси 10:4 со сложным пинаколовым эфиром пентилбороновой кислоты (0,86 г, 76%). 1H ЯМР (400 МГц, CDCl3): δ 6,93 (с, 3H), 3,70 (с, 3H), 3,59 (с, 2H), 2,58 (т, J = 7,9 Гц, 2H), 1,58-1,66 (м, 2H), 1,32-1,38 (м, 4H), 0,91 (т, J = 6,8 Гц, 3H).
Этап 4
Раствор сложного метилового эфира (0,86 г, 79% мас./мас., 2,34 ммоль) в ацетонитриле (24 мл) обрабатывали с помощью раствора гидроксида лития (0,28 г, 11,7 ммоль) в воде (6 мл) и перемешивали реакционную смесь при комнатной температуре в течение 22 ч. Реакционную смесь гасили с помощью 1М водной хлористоводородной кислоты (55 мл), а затем экстрагировали с помощью этилацетата (100 мл). Органический экстракт промывали насыщенным водным хлоридом натрия (50 мл); затем высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на колонке из оксида кремния SiliaSep, элюируя с помощью этилацетата/гексана от 0:1 до 1:4, давало 2-[3,5-дипентил]фенил]уксусную кислоту в виде бесцветного масла (0,55 г, 84%). 1H ЯМР (400 МГц, CDCl3): δ 6,99 (с, 3H), 3,65 (с, 2H), 2,63 (т, J = 7,8 Гц, 2H), 1,64-71 (м, 2H), 1,36-1,44 (м, 4H), 0,97 (т, J = 6,9 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 178,96, 143,55, 133,21, 127,93, 127,06, 41,47, 36,13, 31,94, 31,47, 22,86, 14,34.
Этап 5
Раствор кислоты (0,48 г, 1,75 ммоль) в этаноле (12 мл) обрабатывали с помощью раствора бикарбоната натрия (0,15 г, 1,75 ммоль) в воде (3 мл) и перемешивали реакционную смесь при комнатной температуре в течение 3 д. Этанол выпаривали in vacuo, а остаточный водный концентрированный раствор разбавляли водой (50 мл), отфильтровывали (PES, 0,2 мкм) и лиофилизировали для получения 2-[3,5-дипентилфенил]ацетата натрия в виде белого твердого вещества (0,52 г, количественно). т. пл. 225-230°C; 1H ЯМР (400 МГц, CD3OD + D2O): δ 6,92 (с, 2H), 6,76 (с, 1H), 3,41 (с, 2H), 2,50 (т, J = 7,5 Гц, 2H), 1,52-1,59 (м, 2H), 1,23-1,33 (м, 4H), 0,85 (т, J = 6,9 Гц, 3H); 13C ЯМР (101 МГц, CD3OD + D2O): δ 179,99, 142,66, 137,63, 126,66, 126,16, 45,11, 35,61, 31,36, 31,19, 22,41, 13,47; МСНР (ESI): m/z 277,5 (w, [M – Na+ + 2H+]), 231,1 (100%, ион тропилия из-за потери карбоксильной группы); ВЭЖХ: 3,0 мин.
Соединение XV: Натриевая соль 2-(3,5-дигексилфенил)уксусной кислоты
Указанное выше соединение получали из сложного пинаколового эфира (E)-гекс-1-енилбороновой кислоты таким же образом, что и в случае Соединения XIV. Белое твердое вещество; 1H ЯМР (400 МГц, CD3OD): δ 6,96 (с, 2H), 6,79 (с, 1H), 3,43 (с, 2H), 2,54 (д, J = 7,7 Гц, 4H), 1,55-1,63 (м, 4H), 1,28-1,36 (м, 12H), 0,89 (т, J = 6,8 Гц, 6H); 13C ЯМР (101 МГц, CD3OD): δ 179,68, 142,38, 137,82, 126,55, 126,07, 45,30, 35,87, 31,83, 31,67, 29,02, 22,61, 13,42; МСНР (ESI): m/z 322,0 (100%, M - Na+ + H+ + NH4+) и 259,0 (35%, M – CO2Na); СВЭЖХ (Система A): 8,9 мин. СВЭЖХ Система A: Подвижная фаза A = 10 mM водный раствор гидрокарбоната аммония; Подвижная фаза B = ацетонитрил; неподвижная фаза = HSS T3 колонка; градиент = 5-100% B в A в течение 10 минут.
Соединение XVI: Натриевая соль 2-(2-гидрокси-3,5-дипентилфенил)уксусной кислоты
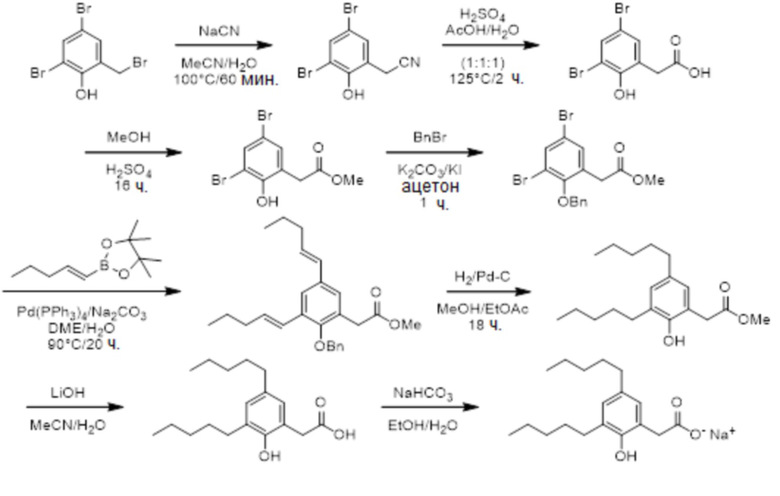
Этап 1
Раствор 2,4-дибром-6-(бромметил)фенола (3,5 г, 10,0 ммоль) в ацетонитриле (17 мл) обрабатывали с помощью раствора цианида натрия (2,5 г, 50,0 ммоль) и нагревали реакционную смесь при 100°C с обратным холодильником в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в воду (100 мл). pH доводили от 10 до 8 с помощью 1М водной хлористоводородной кислоты, а смесь экстрагировали с помощью этилацетата (3 x 250 мл). Объединенные экстракты промывали с помощью 1М водной хлористоводородной кислоты (250 мл) и насыщенным водным хлоридом натрия (250 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Экстрагирование с помощью ацетона; фильтрация; и выпаривание in vacuo давало 2-(3,5-дибром-2-гидроксифенил)ацетонитрил (2,6 г, 90%). 1H ЯМР (400 МГц, d6-ацетон): δ 8,75 (br с, 1H), 7,69 (д, J = 2,3 Гц, 1H), 7,54 (д, J = 2,3 Гц, 1H), 3,92 (с, 2H); 13C ЯМР (101 МГц, d6-ацетон): δ 151,31, 134,51, 131,92, 122,80, 117,43, 111,89, 111,53, 18,70.
Этап 2
2-(3,5-Дибром-2-гидроксифенил)ацетонитрил (2,6 г, 9,0 ммоль) обрабатывали с помощью смеси серной кислоты (2,5 мл), уксусной кислоты (2,5 мл) и воды (2,5 мл), а реакционную смесь нагревали при 125°C с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в смесь льда (50 мл) и воды (50 мл), а затем перемешивали до тех пор, пока лед не растает. Смесь экстрагировали с помощью этилацетата (250 мл); а экстракт затем промывали водой (100 мл) и насыщенным водным хлоридом натрия (100 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенной 2-(3,5-дибром-2-гидроксифенил)уксусной кислоты (3,1 г). Этот материал применяли непосредственно на следующем этапе без дополнительных очистки и исследования.
Этап 3
Раствор неочищенной 2-(3,5-дибром-2-гидроксифенил)уксусной кислоты (3,1 г, 9,0 ммоль) в метаноле (17 мл) обрабатывали с помощью серной кислоты (0,43 мл, 8,1 ммоль) и перемешивали реакционную смесь при температуре окружающей среды в течение 16 ч. Метанол выпаривали in vacuo, а остаток растворяли в этилацетате (270 мл). Раствор промывали водой (2 x 200 мл) и насыщенным водным хлоридом натрия (130 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на системе SP1 Biotage™ (кассета с 120 г силикагеля), элюируя с помощью 0-20% этилацетата в гексане, давало метил-2-(3,5-дибром-2-гидроксифенил)ацетат (1,4 г, 49%). 1H ЯМР (400 МГц, CDCl3): δ 7,52 (д, J = 2,2 Гц, 1H), 7,23 (д, J = 2,2 Гц, 1H), 6,42 (br с, 1H), 3,72 (с, 3H), 3,65 (с, 2H); 13C ЯМР (101 МГц, CDCl3): δ 172,06, 150,60, 133,74, 133,50, 123,94, 112,62, 111,77, 52,78, 36,61.
Этап 4
Раствор метил-2-(3,5-дибром-2-гидроксифенил)ацетата (0,5 г, 1,54 ммоль) в ацетоне (5 мл) обрабатывали с помощью карбоната калия (0,26 г, 1,86 ммоль), йодида калия (0,05 г, 0,32 ммоль) и бензилбромида (0,20 мл, 1,7 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 1 ч. Ацетон выпаривали in vacuo, а остаток разделяли между этилацетатом (50 мл) и 1M водной хлористоводородной кислотой (50 мл). Органическую фазу промывали насыщенным водным хлоридом натрия (50 мл); высушивали над сульфатом натрия; отфильтровывали и выпаривали in vacuo для получения неочищенного продукта. Очищение на системе SP1 Biotage™ (кассета с 40 г силикагеля), элюируя с помощью 0-10% этилацетата в гексане, давало метил-2-(2-(бензилокси)-3,5-дибромфенил)ацетат (0,6 г, 95%). 1H ЯМР (400 МГц, CDCl3): δ 7,67 (д, J = 2,4 Гц, 1H), 7,48-7,51 (м, 2H), 7,37 (д, J = 2,4 Гц, 1H), 7,34-7,43 (м, 3H), 4,99 (с, 2H), 3,66 (с, 3H), 3,60 (с, 2H); 13C ЯМР (101 МГц, CDCl3): δ 171,26, 153,79, 136,56, 135,38, 133,57, 132,04, 128,82, 128,64, 128,52, 118,69, 117,56, 75,53, 52,50, 35,86.
Этап 5
Метил-2-(2-(бензилокси)-3,5-дибромфенил)ацетат (0,3 г, 0,73 ммоль) и сложный пинаколовый эфир (E)-пент-1-енилбороновой кислоты (0,4 г, 1,79 ммоль) соединяли таким же образом, что и в случае Соединения I, этап 2, для получения метил-2-(2-(бензилокси)-3,5-ди((E)-пент-1-енил)фенил)ацетата (0,21 мг, 72%). 1H ЯМР (400 МГц, CDCl3): δ 7,50 (д, J = 7,2 Гц, 2H), 7,44 (дд, J = 7,2, 7,2 Гц, 2H), 7,43 (д, J = 2,1 Гц, 1H), 7,38 (дд, J = 7,2, 7,2 Гц, 1H), 7,18 (д, J = 2,1 Гц, 1H), 6,72 (д, J = 15,8 Гц, 1H), 6,39 (д, J = 15,8 Гц, 1H), 6,32 (дт, J = 15,8, 7,0 Гц, 1H), 6,22 (дт, J = 15,8, 6,8 Гц, 1H), 4,87 (с, 2H), 3,69 (с, 3H), 3,67 (с, 2H), 2,20-2,29 (м, 4H), 1,50-1,60 (м, 4H), 1,01 (т, J = 7,3 Гц, 3H), 1,00 (т, J = 7,4 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 172,49, 153,59, 137,58, 134,35, 132,91, 131,91, 130,84, 129,53, 128,78, 128,32, 128,30, 128,24, 127,26, 125,21, 123,89, 75,89, 52,21, 35,94, 35,74, 35,42, 22,87, 22,77, 14,07, 14,06.
Этап 6
Метил-2-(2-(бензилокси)-3,5-ди((E)-пент-1-енил)фенил)ацетат (0,2 г, 0,53 ммоль) гидрировали таким же образом, что и в случае Соединения I, этап 3, для получения метил-2-(2-гидрокси-3,5-дипентилфенил)ацетата (0,12 г, 73%). 1H ЯМР (400 МГц, CDCl3): δ 7,37 (с, 1H), 6,92 (д, J = 2,1 Гц, 2H), 6,77 (д, J = 2,1 Гц, 1H), 3,76 (с, 3H), 3,67 (с, 2H), 2,65 (т, J = 7,8 Гц, 2H), 2,51 (т, J = 7,8 Гц, 2H), 1,58-1. 66 (м, 4H), 1,31-1,41 (м, 8H), 0,93 (т, J = 7,0 Гц, 3H), 0,92 (т, J = 6,9 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 175,01, 151,27, 135,14, 131,48, 129,92, 128,52, 120,30, 52,95, 38,35, 35,34, 32,15, 31,86, 31,74, 30,61, 30,03, 22,87, 22,83, 14,34, 14,31.
Этап 7
Метил-2-(2-гидрокси-3,5-дипентилфенил)ацетат (0,2 г, 0,53 ммоль) гидрировали таким же образом, что и в случае Соединения I, этап 4, для получения неочищенного продукта, смешанного с лактонизированным материалом. Небольшую порцию очищали на системе SP1 Biotage™ (кассета с 120 г силикагеля), элюируя с помощью 0-100% этилацетата в гексане, для получения 2-(2-гидрокси-3,5-дипентилфенил)уксусной кислоты (13,5 мг). 1H ЯМР (400 МГц, CDCl3): δ 10,5 (br с, 1H), 6,89 (д, J = 2,2 Гц, 1H), 6,78 (д, J = 2,2 Гц, 1H), 6,32 (br с, 1H), 3,66 (с, 2H), 2,58 (т, J = 7,9 Гц, 2H), 2,48 (т, J = 7,8 Гц, 2H), 1,52-1. 63 (м, 4H), 1,26-1,37 (м, 8H), 0,90 (т, J = 7,0 Гц, 3H), 0,88 (т, J = 6,8 Гц, 3H).
Этап 8
2-(2-Гидрокси-3,5-дипентилфенил)уксусную кислоту (13,5 мг, 0,046 ммоль) преобразовывали в натриевую соль таким же образом, что и в случае Соединения I, этап 5, для получения 2-(2-гидрокси-3,5-дипентилфенил)ацетата натрия (11 мг, 77%). 1H ЯМР (400 МГц, CD3OD): δ 6,72 (д, J = 2,0 Гц, 1H), 6,69 (д, J = 2,0 Гц, 1H), 3,46 (с, 2H), 2,56 (т, J = 7,6 Гц, 2H), 2,44 (т, J = 7,6 Гц, 2H), 1,50-1. 61 (м, 4H), 1,25-1,37 (м, 8H), 0,90 (т, J = 6,8 Гц, 3H), 0,88 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 180,33, 151,94, 133,47, 130,37, 128,21, 127,81, 123,99, 42,90, 34,97, 31,81, 31,60, 31,40, 30,25, 29,88, 22,51, 22,45, 13,29, 13,24; МСНР (Анионная ESI): m/z 291,2 (100%, M –Na+); СВЭЖХ (Система B): 7,7 мин. СВЭЖХ Система B: Подвижная фаза A = 0,1% водный раствор муравьиной кислоты; Подвижная фаза B = 0,1% formic acid in ацетонитрил; неподвижная фаза = HSS T3 колонка; градиент = 5-100% B в A в течение 10 минут.
Соединение XVII: Натриевая соль 2-(3,5-дигексил-2-гидроксифенил)уксусной кислоты
Указанное выше соединение получали таким же образом, что и в случае Соединения XVI, с применением сложного пинаколового эфира (E)-гекс-1-енилбороновой кислоты. 1H ЯМР (400 МГц, CD3OD): δ 6,72 (д, J = 2,0 Гц, 1H), 6,69 (д, J = 2,0 Гц, 1H), 3,46 (с, 2H), 2,56 (т, J = 7,6 Гц, 2H), 2,44 (т, J = 7,5 Гц, 2H), 1,50-1. 60 (м, 4H), 1,27-1,37 (м, 12H), 0,89 (т, J = 6,6 Гц, 3H), 0,88 (т, J = 6,80 Гц, 3H); МСНР (Анионная ESI): m/z 319 (100%, M – Na+); СВЭЖХ (Система B): 8,7 мин. ULC Система B: Подвижная фаза A = 0,1% водный раствор муравьиной кислоты; Подвижная фаза B = 0,1% formic acid in ацетонитрил; неподвижная фаза = HSS T3 колонка; градиент = 5-100% B в A в течение 10 минут.
Соединение XVIII: Натриевая соль 2-(4-гидрокси-3,5-дипентилфенил)уксусной кислоты
Указанное выше соединение получали таким же образом, что и в случае Соединения XVI, из 2-(3,5-дибром-4-гидроксифенил)уксусной кислоты. 1H ЯМР (400 МГц, CD3OD): δ 6,87 (с, 2H), 3,33 (с, 2H), 2,55 (т, J = 7,7 Гц, 4H), 1,53-1. 61 (м, 4H), 1,31-1,37 (м, 8H), 0,90 (т, J = 7,0 Гц, 6H); МСНР (Анионная ESI): m/z 291,1 (100%, M – Na+); СВЭЖХ (Система B): 6,8 мин. СВЭЖХ Система B: Подвижная фаза A = 0,1% водный раствор муравьиной кислоты; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3 колонка; градиент = 5-100% B в A в течение 10 минут.
Соединение XIX: Натриевая соль 2-(3,5-дигексил-4-гидроксифенил)уксусной кислоты
Указанное выше соединение получали таким же образом, что и в случае Соединения XVI, из 2-(3,5-дибром-4-гидроксифенил)уксусной кислоты и сложного пинаколового эфира (E)-гекс-1-енилбороновой кислоты. 1H ЯМР (400 МГц, CD3OD): δ 6,72 (д, J = 2,0 Гц, 1H), 6,69 (д, J = 2,0 Гц, 1H), 3,46 (с, 2H), 2,56 (т, J = 7,6 Гц, 2H), 2,44 (т, J = 7,5 Гц, 2H), 1,50-1,60 (м, 4H), 1,27-1,37 (м, 12H), 0,89 (т, J = 6,6 Гц, 3H), 0,88 (т, J = 6,8 Гц, 3H); МСНР (Анионная ESI): m/z 319,1 (100%, M – Na+); СВЭЖХ (Система B): 7,6 мин. СВЭЖХ Система B: Подвижная фаза A = 0,1% водный раствор муравьиной кислоты; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3 колонка; градиент = 5-100% B в A в течение 10 минут.
Соединение XX: Натриевая соль 2-(4-фтор-3,5-дигексилфенил)уксусной кислоты
Указанное выше соединение получали таким же образом, что и в случае Соединения XVI, исходя из 3,5-дибром-4-фторбензилбромида и сложного пинаколового эфира (E)-гекс-1-енилбороновой кислоты. 3,5-Дибром-4-фторбензилбромида получали с помощью бромирования 3,5-дибром-4-фтортолуола с N-бромсукцинимидом и азобисизобутиронитрилом в ацетонитриле при 80°C. 1H ЯМР (400 МГц, CD3OD): δ 6,98 (д, JHF = 7,0 Гц, 2H), 3,38 (с, 2H), 2,57 (т, J = 7,7 Гц, 4H), 1,54-1,61 (м, 4H), 1,28-1,37 (м, 12H), 0,89 (т, J = 6,7 Гц, 6H); 19F ЯМР (377 МГц, CD3OD): δ -132,17 (д, JHF = 6,6 Гц, 1F); 13C ЯМР (101 МГц, CD3OD): δ 179,44, 158,11 (д, JCF = 239,8 Гц), 133,26 (д, JCF = 3,8 Гц), 128,73 (д, JCF = 5,4 Гц), 128,56 (д, JCF = 16,9 Гц), 44,52, 31,69, 30,35 (д, JCF = 1,5 Гц), 28,98, 28,97 (д, JCF = 3,1 Гц), 22,51, 13,29; МСНР (Анионная ESI): m/z 321,0 (100%, M – Na+); СВЭЖХ (Система B): 9,2 мин. СВЭЖХ Система B: Подвижная фаза A = 0,1% водный раствор муравьиной кислоты; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3 колонка; градиент = 5-100% B в A в течение 10 минут.
Соединение XXI: Натриевая соль 2-(4-фтор-3,5-дипентилфенил)уксусной кислоты
Указанное выше соединение получали таким же образом, что и в случае Соединения XVI, исходя из 3,5-дибром-4-фторбензилбромида. 1H ЯМР (400 МГц, CD3OD): δ 6,98 (д, JHF = 6,8 Гц, 2H), 3,37 (с, 2H), 2,57 (т, J = 7,6 Гц, 4H), 1,54-1,62 (м, 4H), 1,28-1,37 (м, 8H), 0,90 (т, J = 7,0 Гц, 6H); 19F ЯМР (377 МГц, CD3OD): δ -132,34 (д, JHF = 6,6 Гц, 1F); 13C ЯМР (101 МГц, CD3OD): δ 179,41, 158,10 (д, JCF = 239,8 Гц), 133,26 (д, JCF = 3,8 Гц), 128,72 (д, JCF = 4,6 Гц), 128,56 (д, JCF = 16,9 Гц), 44,51, 31,54, 30,07, 28,92 (д, JCF = 3,1 Гц), 22,38, 13,22; МСНР (Анионная ESI): m/z 293,0 (100%, M – Na+); СВЭЖХ (Система B): 8,4 мин. СВЭЖХ Система B: Подвижная фаза A = 0,1% водный раствор муравьиной кислоты; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3 колонка; градиент = 5-100% B в A в течение 10 минут.
Соединение XXII: Натриевая соль 2-(2-бензил-3,5-дипентилфенил)уксусной кислоты
Указанное в названии соединение получали таким же образом, что и в случае Соединения XIV, из метил-2-(2-бензил-3,5-ди((E)-пент-1-енил)фенил)ацетата. Последний был выделен в виде побочного продукта (1,1% выхода) при масштабировании Соединения XIV. 1H ЯМР (400 МГц, CD3OD): δ 7,17 (дд, J = 7,3, 7,3 Гц, 2H), 7,09 (дд, J = 7,3, 7,3 Гц, 1H), 6,97-6,99 (м, 3H), 6,86 (д, J = 1,8 Гц, 1H), 4,13 (с, 2H), 3,40 (с, 2H), 2,55 (т, J = 7,7 Гц, 2H), 2,49 (т, J = 7,8 Гц, 2H), 1,59-1,67 (м, 2H), 1,31-1,45 (м, 6H), 1,21-1,26 (м, 4H), 0,91 (т, J = 7,0 Гц, 3H), 0,82 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 179,48, 141,46, 141,24, 140,47, 137,46, 133,70, 128,36, 128,05, 127,86, 127,75, 125,42, 43,25, 35,54, 33,90, 33,61, 31,86, 31,65, 31,25, 30,96, 22,49, 22,40, 13,31, 13,23; МСНР (Анионная ESI): m/z 365,0 (20%, M – Na+), 321,1 (100%, M – CO2Na); СВЭЖХ (Система B): 9 мин. (СВЭЖХ Система B: Подвижная фаза A = 0,1% водный раствор муравьиной кислоты; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3; градиент = 5-100% B в A в течение 10 мин.)
Соединение XXIII: 2-[3,5-Ди[(E)-пент-1-енил]фенил]ацетат натрия
Указанное в названии соединение получали с применением той же самой процедуры, что и в случае Соединения XIV, но с пропуском этапа гидрирования. т. пл. 226-30°C; 1H ЯМР (400 МГц, CD3OD): δ 7,18 (д, J = 1,2 Гц, 2H), 7,11 (д, J = 1,2 Гц, 1H), 6,34 (д, J = 15,9 Гц, 2H), 2,23 (дт, J = 15,9, 6,7 Гц, 2H), 3,44 (с, 2H), 2,14-2,19 (м, 4H), 1,49 (tкв, J = 7,4, 7,4 Гц, 4H), 0,95 (т, J = 7,3 Гц, 6H); 13C ЯМР (101 МГц, CD3OD): δ 179,41, 138,34, 138,06, 130,30, 130,16, 125,26, 121,60, 45,24, 35,10, 22,55 & 12,98; МСНР (анионная ионизация): m/z 271 (w, [M – Na+]), 227,2 (100%, [M – Na+– CO2]); СВЭЖХ: 8 мин. (СВЭЖХ; Условия растворитель A = 0,1% муравьиная кислота в воде; Растворитель B = 0,1% муравьиная кислота в ацетонитриле; Градиент: 5-100% B в A течение 10 мин. при 0,7 мл/мин.)
Соединение XXIV: 3-[3,5-Дипентилфенил]пропаноат натрия
Указанное в названии соединение получали с применением той же самой процедуры, что и в случае Соединения XIV, исходя из 3-[3,5-дибромфенил]пропановой кислоты. т. пл. 211-217°C; 1H ЯМР (400 МГц, CDCl3): δ 6,73 (с, 1H), 6,68 (с, 2H), 2,73-2,77 (м, 2H), 2,42-2,46 (м, 2H), 2,38 (т, J = 7,8 Гц, 4H), 1,43-1,51 (м, 4H), 1,19-1,28 (м, 8H), 0,83 (т, J = 6,9 Гц, 6H); 13C ЯМР (101 МГц, CDCl3): δ 182,55, 142,93, 141,85, 125,96, 125,77, 39,80, 36,13, 32,77, 31,99, 31,47, 22,79 & 14,27; МСНР (анионная ионизация): m/z 289,4 (100%, [M – Na+]); СВЭЖХ: 9 мин. (СВЭЖХ: Условия растворитель A = 0,1% муравьиная кислота в воде, растворитель B = 0,1% муравьиная кислота в ацетонитриле, Градиент: 5-100% B в A в течение 10 мин.при 0,7 мл/мин.
Соединение XXV: Натриевая соль 2-метил-2-(3-пентилфенил)пропановой кислоты
Указанное в названии соединение получали из метил-2-[3-бромфенил]ацетата, что и в случае соединения XIV, с дополнительным этапом алкилирования интермедиата метил-2-[3-пентилфенил]ацетата с гидридом натрия и метилйодидом; и с температурой этапа гидролиза сложного эфира, поднимаемой до 50°C. Беловатое твердое вещество: 1H ЯМР (400 МГц, D2O): δ 7,11 (дд, J = 7,7, 7,7 Гц, 1H), 7,07 (с, 1H), 7,02 (д, J = 7,6 Гц, 1H), 6,95 (д, J = 7,4 Гц, 1H), 2,44 (т, J = 7,7 Гц, 2H), 1,43 (tт, J = 7,4, 7,4 Гц, 2H), 1,28 (с, 6H), 1,09-1,17 (м, 4H), 0,68 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, D2O): δ 186,51, 148,17, 143,67, 128,48, 126,27, 126,24, 123,26, 48,67, 35,33, 30,90, 30,77, 27,20, 22,01, 13,46; МСНР (ESI +ve): m/z 189,1 (100%, MH+ - CO2Na); ВЭЖХ: 5 мин (15-99% ацетонитрил в воде в течение 5 мин (трифторуксусная кислота в обоих растворителях).
Соединение XXVI: Натриевая соль (RS)-2-(3-пентилфенил)пропановой кислоты

Этап 1
Смесь йодида меди(I) (17 мг, 0,09 ммоль), 2-пиколиновой кислоты (22 мг, 0,18 ммоль) и карбоната цезия (1,7 г, 5,30 ммоль), под аргоном, обрабатывали с помощью безводного 1,4-диоксана (3 мл), диэтилмалоната (0,54 мл, 3,5 ммоль) и 1-бром-3-йодбензола (0,23 мл, 1,77 ммоль). Реакционную смесь затем нагревали при 70°C, под аргоном, в течение 15 ч. Неочищенную реакционную смесь выпаривали на силикагеле и очищали на колонке SiO2 SiliaSep, элюируя с помощью этилацетата в гексане (0-12%) для получения диэтил-2-[3-бромфенил]малоната (0,34 г, 64%). 1H ЯМР (400 МГц, CDCl3): δ 7,30-7,47 (м, 3H), 7,20-7,26 (м, 1H), 4,16-4,24 (м, 4H), 3,36 (с, 1H), 1,23-1,29 (м, 6H).
Этап 2
Суспензию гидрида натрия (60% мас./мас.; 0,53 г, 13,3 ммоль) в безводном ТГФ (16 мл) охлаждали до 0°C под аргоном и обрабатывали с помощью раствора диэтил-2-[3-бромфенил]малоната (3,0 г, 9,52 ммоль) в безводном ТГФ (20 мл). Реакционную смесь перемешивали при 0°C в течение 30 мин., а затем по каплям обрабатывали с помощью метилйодида (0,8 мл, 13,3 ммоль). Реакционную смесь затем нагревали до комнатной температуры и перемешивали при комнатной температуре, под аргоном, в течение ночи. Реакционную смесь гасили с помощью насыщенного водного раствора хлорида аммония (100 мл), а смесь экстрагировали с помощью этилацетата (3 x 100 мл). Объединенные органические экстракты высушивали (сульфат магния) и выпаривали in vacuo для получения неочищенного соединения. Очищение на колонке SiO2 SiliaSep, элюируя с помощью этилацетата в гексане (0-5%), давало диэтил-2-[3-бромфенил]-2-метилмалонат (2,6 г, 82%). 1H ЯМР (400 МГц, CDCl3): δ 7,52 (ддд, J = 1,9, 1,9, 0,4 Гц, 1H), 7,43 (ддд, J = 7,9, 1,9, 1,0 Гц, 1H), 7,31 (ддд, J = 8,0, 1,9, 1,0 Гц, 1H), 7,20 (ддд, J = 7,9, 7,9, 0,4 Гц, 1H), 4,21-4,26 (м, 4H), 1,84 (с, 3H), 1,26 (т, J = 7,2 Гц, 6H).
Этап 3
Диэтил-2-[3-бромфенил]-2-метилмалонат (2,6 г, 7,8 ммоль) соединяли со сложным пинаколовым эфиром (E)-1-пентен-1-илбороновой кислоты (2,1 г, 10,9 ммоль) с применением способа, описанного в случае соединения X, Этап 4, для получения диэтил(E)-2-метил-2-[3-[пент-1-енил]фенил]малоната (1,7 г, 68%). 1H ЯМР (400 МГц, CDCl3): δ 7,24-7,32 (м, 3H), 7,21 (ддд, J = 7,1, 1,9, 1,9 Гц, 1H), 6,37 (д, J = 15,9 Гц, 1H), 6,20 (дт, J = 15,9, 6,9 Гц, 1H), 4,21-4,26 (м, 4H), 2,15-2,21 (м, 2H), 1,87 (с, 3H), 1,49 (tт, J = 7,3, 7,3 Гц, 2H), 1,26 (т, J = 7,2 Гц, 6H), 0,95 (т, J = 7,4 Гц, 3H).
Этап 4
Диэтил-(E)-2-метил-2-[3-[пент-1-енил]фенил]малонат (1,4 г, 4,27 ммоль) гидрировали с применением способа, описанного в случае соединения I, Этап 3, для получения диэтил-2-метил-2-[3-пентилфенил]малоната (1,2 г, 91%). 1H ЯМР (400 МГц, CDCl3): δ 7,24 (дд, J = 7,3, 7,3 Гц, 1H), 7,16 (д, J = 7,3 Гц, 1H), 7,15 (с, 1H), 7,10 (д, J = 7,6 Гц, 1H), 4,20-4,25 (м, 4H), 2,59 (т, J = 7,9 Гц, 2H), 1,85 (с, 3H), 1,49 (tт, J = 7,6, 7,6 Гц, 2H), 1,28-1,34 (м, 4H), 1,25 (т, J = 7,0 Гц, 6H), 0,88 (т, J = 7,0 Гц, 3H).
Этап 5
Раствор диэтил-2-метил-2-[3-пентилфенил]малоната (1,1 г, 3,5 ммоль) в ацетонитриле (9 мл), метаноле (3 мл) и воде (3 мл) обрабатывали с помощью гидроксида лития (1,3 г, 52,8 ммоль), а смесь нагревали при 50°C в течение 48 ч. Реакционную смесь концентрировали in vacuo, разбавляли водой (10 мл), а затем промывали дихлорметаном (15 мл). pH водной фазы затем доводили до pH 4 с помощью 1М водной хлористоводородной кислоты, а смесь экстрагировали дихлорметаном (3 x 25 мл). Объединенные органические экстракты высушивали (сульфат магния) и выпаривали in vacuo для получения неочищенного соединения. Очищение на колонке SiO2 SiliaSep, элюируя с помощью этилацетата в гексане (0-20%), давало (RS)-2-[3-пентилфенил]пропановую кислоту (0,4 г, 52%). 1H ЯМР (400 МГц, CD3OD): δ 7,20 (дд, J = 7,6, 7,6 Гц, 1H), 7,03-7,12 (м, 3H), 3,66 (кв, J = 7,1 Гц, 1H), 2,58 (т, J = 7,8 Гц, 2H), 1,60 (tт, J = 7,6, 7,6 Гц, 2H), 1,42 (д, J = 7,1 Гц, 3H), 1,27-1,38 (м, 4H), 0,90 (т, J = 7,1 Гц, 3H).
Этап 6
(RS)-2-[3-Пентилфенил]пропановую кислоту (0,4 г, 1,8 ммоль) преобразовывали в натриевую соль с применением способа, описанного в случае соединения I, Этап 5, для получения (RS)-2-[3-пентилфенил]пропаноата натрия (0,44 г, количественно). 1H ЯМР (400 МГц, CD3OD): δ 7,19 (с, 1H), 7,14-7,17 (м, 1H), 7,13 (дд, J = 7,5, 7,5 Гц, 1H), 6,95 (д, J = 6,9 Гц, 1H), 3,54 (кв, J = 7,1 Гц, 1H), 2,56 (т, J = 7,8 Гц, 2H), 1,60 (tт, J = 7,5, 7,5 Гц, 2H), 1,39 (д, J = 7,2 Гц, 3H), 1,29-1,35 (м, 4H), 0,90 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 182,18, 144,23, 142,49, 127,76, 127,55, 125,82, 124,73, 49,17, 35,85, 31,54, 31,33, 22,43, 18,95, 13,22; ВЭЖХ: 5 мин (15-99% ацетонитрил в воде в течение 5 мин (трифторуксусная кислота в обоих растворителях).
Соединение XXVII: Натриевая соль 2-(2-гидрокси-5-пентилфенил)уксусной кислоты
Указанное выше соединение получали таким же образом, как и соединение VII, Этапы 3-6, с применением метил-2-[2-(бензилокси)-5-бромфенил]ацетата (полученного на 2 этапах из 2-[5-бром-2-гидроксифенил]уксусной кислоты. Белое твердое вещество: 1H ЯМР (400 МГц, CD3OD): δ 6,82-6,88 (м, 2H), 6,69 (д, J = 8,6 Гц, 1H), 3,47 (с, 2H), 2,47 (т, J = 7,7 Гц, 2H), 1,51-1,59 (м, 2H), 1,24-1,36 (м, 4H), 0,89 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 180,04, 154,04, 134,05, 130,25, 127,36, 124,15, 116,57, 42,50, 34,90, 31,59, 31,42, 22,44, 13,23; МСНР (ESI -ve): m/z 221,1 (100%, M - Na+), 177,1 (м, M - Na+ - CO2); ВЭЖХ: 2 мин (Градиент 70-99% ацетонитрил в воде в течение 5 мин и трифторуксусная кислота в обоих растворителях).
Соединение XXVIII: Натриевая соль 2-оксо-2-[3-пентилфенил]уксусной кислоты
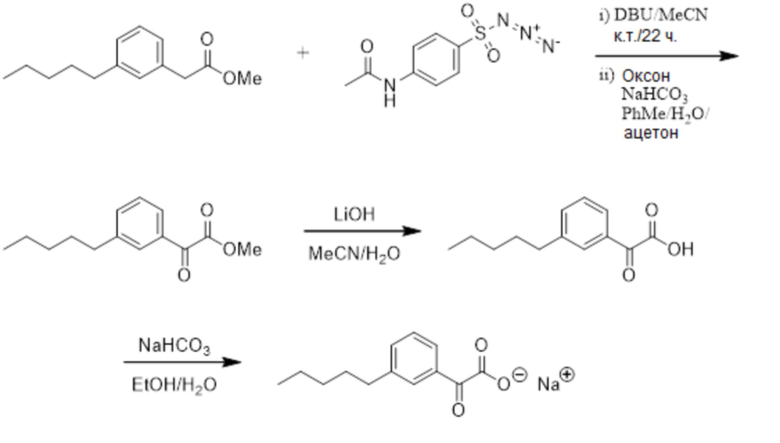
Этап 1:
i) Раствор метил-2-[3-пентилфенил]ацетата (0,5 г, 2,0 ммоль) в ацетонитриле (15 мл), под азотом, обрабатывали с помощью 1,8-диазабицикло[5,4,0]ундец-7-ена (0,22 мл, 1,5 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 15 мин. Реакционную смесь охлаждали до 0°C и медленно добавляли 4-ацетамидобензолсульфонилазид (0,6 г, 2,4 ммоль). Реакционную смесь затем нагревали до комнатной температуры и перемешивали, под азотом, в течение 22,5 ч.
ii) Раствор интермедиата метил-2-диазо-2-[3-пентилфенил]ацетата разбавляли толуолом (15 мл), ацетоном (11 мл) и водой (15 мл), а затем обрабатывали с помощью бикарбоната натрия (6,4 г, 75,7 ммоль). Медленно добавляли оксон (12,1 г, 19,7 ммоль), а реакционную смесь затем энергично перемешивали при комнатной температуре в течение 25 мин. Реакционную смесь разбавляли водой (30 мл), а затем экстрагировали с помощью этилацетата (3 x 30 мл). Объединенные экстракты промывали насыщенным водным хлоридом натрия (30 мл), высушивали над сульфатом натрия и выпаривали in vacuo для получения неочищенного продукта. Экстракция с помощью дихлорметана и очистка на колонке SiO2 SiliaSep, элюируя с помощью этилацетата в гексане (0-2%), давали метил-2-оксо-2-[3-пентилфенил]ацетат (0,13 г, 30%). 1H ЯМР (400 МГц, CDCl3): δ 7,79-7,82 (м, 2H), 7,47 (д, J = 7,6 Гц, 1H), 7,66 (дд, J = 7,6, 7,6 Гц, 1H), 3,97 (с, 3H), 2,66 (д, J = 7,8 Гц, 2H), 1,58-1,64 (м, 2H), 1,27-1,35 (м, 4H), 0,88 (т, J = 6,9 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 186,61, 164,48, 144,17, 135,53, 132,61, 129,88, 129,01, 127,97, 52,96, 35,87, 31,58, 31,18, 22,70, 14,22.
Этап 2
Метил-2-оксо-2-[3-пентилфенил]ацетат (64 мг, 0,8 ммоль) гидрировали таким образом, как это описано в случае Соединения IX, Этап 5, для получения 2-оксо-2-[3-пентилфенил]уксусной кислоты (60 мг, колич.). 1H ЯМР (400 МГц, CDCl3): δ 10,32 (br с, 1H), 7,98 (д, J = 7,4 Гц, 1H), 7,96 (с, 1H), 7,43 (д, J = 7,5 Гц, 1H), 7,36 (дд, J = 7,4, 7,4 Гц, 1H), ), 2,60 (д, J = 7,7 Гц, 2H), 1,52-1,59 (м, 2H), 1,20-1,29 (м, 4H), 0,81 (т, J = 6,8 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 185,51, 164,18, 144,28, 136,10, 132,04, 130,81, 129,12, 128,85, 35,90, 31,59, 31,19, 22,71, 14,23.
Этап 3
2-Оксо-2-[3-пентилфенил]уксусную кислоту (57 мг, 0,3 ммоль) преобразовывали в натриевую соль с применением способа, описанного в случае соединения I, Этап 5, для получения 2-оксо-2-[3-пентилфенил]ацетата натрия (51 мг, 95%). 1H ЯМР (400M Гц, CD3OD): δ 7,79-7,81 (м, 2H), 7,45 (ддд, J = 7,6, 1,5, 1,5 Гц, 1H), 7,41 (ддд, J = 7,8, 7,8, 1,0 Гц, 1H), 2,67 (т, J = 7,6 Гц, 2H), 1,64 (tт, J = 7,5, 7,5 Гц, 2H), 1,28-1,39 (м, 4H), 0,90 (т, J = 7,1 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 196,19, 172,77, 143,54, 133,89, 133,76, 129,34, 128,47, 127,03, 35,45, 31,32, 31,06, 22,38, 13,20; МСНР (ESI -ve): m/z 219,1 (100%, M – Na+); ВЭЖХ: 3,3 мин (Градиент 15-99% ацетонитрил в воде в течение 5 мин и трифторуксусная кислота в обоих растворителях).
Соединение XXIX: Натриевая соль (E)-2-[2-фтор-5-[пент-1-енил]фенил]уксусной кислоты
Указанное выше соединение получали из метил-2-[2-фтор-5-бромфенил]ацетата, таким же образом, что и в случае соединение XIV, с пропуском этапа гидрирования. Белое твердое вещество; 1H ЯМР (400 МГц, CD3OD): δ 7,32 (дд, JHF = 7,4 Гц, JHH = 2,1 Гц, 1H), 7,15-7,18 (м, 1H), 6,92 (дд, JHF = 9,4 Гц, JHH = 8,8 Гц, 1H), 6,33 (д, J = 15,8 Гц, 1H), 6,16 (дд, J = 15,8, 7,0 Гц, 1H), 2,16 (тд, J = 7,1, 7,1 Гц, 2H), 1,48 (tт, J = 7,3, 7,3 Гц, 2H), 0,95 (т, J = 7,3 Гц, 3H); 19F ЯМР (377 МГц, CD3OD): δ -122,74 to -122,26 (м, 1F), 13C ЯМР (101 МГц, CD3OD): δ 177,91, 160,51 (д, JCF = 243,6 Гц), 134,08 (д, JCF = 3,8 Гц), 129,87 (д, JCF = 1,5 Гц), 129,23, 128,94 (д, JCF = 4,6 Гц), 125,09-125,26 (м, 2C), 114,63 (д, JCF = 22,3 Гц), 37,75 (д, JCF = 1,5 Гц), 35,00, 22,50, 12,87; МСНР (ESI -ve): m/z 176,9 (100%, M – Na+ – CO2); ВЭЖХ: 6 мин (СВЭЖХ Градиент: Подвижная фаза A = 0,1% муравьиная кислота в воде; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3; градиент = 5-100% B в A в течение 10 мин).
Соединение XXX: Натриевая соль 2-[2-бензил-5-пентилфенил]уксусной кислоты
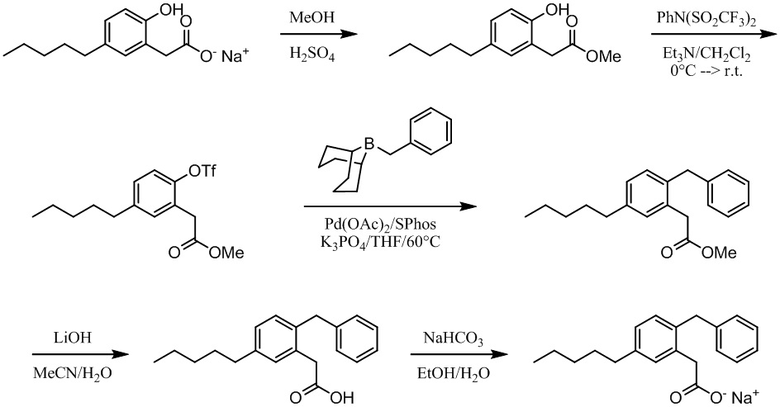
Этап 1
Соединение XXVII (2,4 г, 10,0 ммоль) этерифицировали таким же образом, что и соединение IX, Этап 1, для получения метил-2-[2-гидрокси-5-пентилфенил]ацетата (2,3 г, 96%). 1H ЯМР (400 МГц, CDCl3): δ 7,24 (br с, 1H), 6,98 (дд, J = 8,2, 2,3 Гц, 1H), 6,90 (д, J = 2,3 Гц, 1H), 6,83 (д, J = 8,2 Гц, 1H), 3,73 (с, 3H), 3,65 (с, 2H), 2,50 (т, J = 7,9 Гц, 2H), 1,52-1,60 (м, 2H), 1,25-1,36 (м, 4H), 0,86-0,90 (м, 3H).
Этап 2
Метил-2-[2-гидрокси-5-пентилфенил]ацетат (2,3 г, 9,6 ммоль) преобразовывали в производное трифторметансульфоната таким же образом, что и в случае Соединения VII, Этап 2, для получения метил-2-[5-пентил-2-(трифторметилсульфонилокси)фенил]ацетата (3,4 г, 97%). 1H ЯМР (400 МГц, CDCl3): δ 7,20 (д, J = 8,6 Гц, 1H), 7,18 (д, J = 2,4 Гц, 1H), 7,16 (дд, J = 8,6, 2,4 Гц, 1H), 3,72 (с, 3H), 3,71 (с, 2H), 2,60 (т, J = 7,8 Гц, 2H), 1,56-1,64 (м, 2H), 1,27-1,37 (м, 4H), 0,89 (т, J = 6,9 Гц, 3H); 19F ЯМР (377 МГц, CDCl3): δ -73,92 (с, 3F); 13C ЯМР (101 МГц, CDCl3): δ 170,59, 146,25, 143,76, 132,42, 129,30, 126,95, 121,31, 118,76 (кв, JCF = 319,8 Гц), 52,38, 35,70, 35,40, 31,62, 31,08, 22,66, 14,10.
Этап 3
В заполненный азотом под давлением сосуд загружали последовательно трехосновный фосфат калия (5,4 г, 25,3 ммоль), ацетат палладия(II) (74 мг, 0,33 ммоль), 2-дициклогексилфосфино-2’,6’-диметокси-1,1’-бифенил (0,14 г, 0,33 ммоль), раствор метил-2-[5-пентил-2-(трифторметилсульфонилокси)фенил]ацетата (3,1 г, 8,3 ммоль) в безводном тетрагидрофуране (20 мл) и 0,5M раствор 9-бензил-9-борабицикло[3,3,1]нонана в тетрагидрофуране (34 мл, 17 ммоль). Сосуд затем герметизировали, а реакционную смесь нагревали при 60°C. Спустя 17 ч. реакционную смесь охлаждали до комнатной температуры и разделяли между этилацетатом (300 мл) и 0,5M водным гидроксидом натрия (250 мл). Органическую фазу промывали насыщенным водным хлоридом натрия (200 мл), высушивали над сульфатом натрия, отфильтровывали и выпаривали in vacuo для получения неочищенного соединения. Очищение на колонке SiO2 SiliaSep, элюируя с помощью этилацетата в гексане (0-2%), давало метил-2-[2-бензил-5-пентилфенил]ацетат (2,5 г, 96%). 1H ЯМР (400 МГц, CDCl3): δ 7,29 (дд, J = 7,4, 7,0 Гц, 2H), 7,21 (дд, J = 7,4, 7,0 Гц, 1H), 7,13-7,15 (м, 2H), 7,08-7,09 (м, 3H), 4,04 (с, 2H), 3,63 (с, 3H), 3,60 (с, 2H), 2,61 (т, J = 7,8 Гц, 2H), 1,61-1,68 (м, 2H), 1,34-1,39 (м, 4H), 0,93 (т, J = 7,1 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 172,29, 141,66, 140,65, 136,61, 132,84, 131,21, 130,87, 129,03, 128,67, 127,83, 126,28, 52,19, 39,01, 39,00, 35,73, 31,89, 31,40, 22,84, 14,35.
Этап 4
Метил-2-[2-бензил-5-пентилфенил]ацетат (2,9 г, 9,3 ммоль) гидрировали таким образом, как это описано в случае Соединения IX, Этап 5, для получения 2-[2-бензил-5-пентилфенил]уксусной кислоты (2,48 г, 90%). 1H ЯМР (400 МГц, CDCl3): δ 7,26 (дд, J = 7,3, 7,3 Гц, 2H), 7,16 (дд, J = 7,5, 7,5 Гц, 1H), 7,10-7,13 (м, 2H), 7,05-7,07 (м, 3H), 4,01 (с, 2H), 3,58 (с, 2H), 2,58 (т, J = 7,8 Гц, 2H), 1,57-1,65 (м, 2H), 1,30-1,37 (м, 4H), 0,90 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CDCl3): δ 178,67, 141,80, 140,51, 136,84, 132,20, 131,37, 130,95, 129,08, 128,75, 128,13, 136,39, 39,07, 38,98, 35,74, 31,93, 31,41, 22,87, 14,38.
Этап 5
2-[2-Бензил-5-пентилфенил]уксусную кислоту (2,5 г, 8,4 ммоль) преобразовывали в натриевую соль с применением способа, описанного в случае соединения I, Этап 5, для получения 2-[2-бензил-5-пентилфенил]ацетата натрия (2,5 г, 93%). 1H ЯМР (400 МГц, CD3OD): δ 7,22 (дд, J = 8,4, 7,4 Гц, 2H), 7,09-7,15 (м, 3H), 6,92-6,93 (м, 3H), 4,03 (с, 2H), 3,47 (с, 2H), 2,55 (т, J = 7,8 Гц, 2H), 1,57-1,65 (м, 2H), 1,28-1,38 (м, 4H), 0,90 (т, J = 7,0 Гц, 3H); 13C ЯМР (101 МГц, CD3OD): δ 179,25, 141,25, 140,60, 136,90, 136,48, 130,45, 129,78, 128,83, 128,13, 126,13, 125,64, 42,70, 38,49, 35,49, 31,64, 31,32, 22,51, 13,35; МСНР (ESI -ve): m/z 295,2 (40%, M – Na+), 251,2 (100%, M – Na+ – CO2); ВЭЖХ: 5,0 мин (Градиент 70-99% MeCN в воде в течение 5 мин и трифторуксусная кислота в обоих растворителях).
Соединение XXXI: Натриевая соль 2-(3,5-ди((E)-гекс-1-енил)фенил)уксусной кислоты
Указанное в названии соединение получали тем же самым способом, что и в случае соединения II, но с пропуском этапа гидрирования. Off-Беловатое твердое вещество: 1H ЯМР (400 МГц, CD3OD): δ 7,17 (д, J = 1,1 Гц, 2H), 7,10 (с, 1H), 6,33 (д, J = 15,8 Гц, 2H), 6,22 (дт, J = 15,8, 6,7 Гц, 2H), 3,44 (с, 2H), 2,16-2,21 (м, 4H), 1,34-1,46 (м, 8H), 0,93 (т, J = 7,3 Гц, 6H); 13C ЯМР (101 МГц, CD3OD): δ 179,44, 138,34, 138,07, 130,37, 130,13, 125,27, 121,60, 45,26, 32,70, 31,67, 22,19, 13,27; МСНР (анионная ESI): m/z 299,2 (м, M - Na+) и 255,2 (100%, M – Na+ – CO2); СВЭЖХ: 8,7 мин. (СВЭЖХ Условия растворитель A = 0,1% муравьиная кислота в воде; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3; градиент = 5-100% B в A в течение 10 мин)
Соединение XXXII: Натриевая соль 2-(2-фтор-3,5-дипентилфенил)уксусной кислоты

Этап 1:
Метил-2-амино-3,5-дибромбензоат (10,0 г, 32,4 ммоль) соединяли со сложным пинаколовым эфиром (E)-1-пентен-1-илбороновой кислоты (15,2 г, 77,7) с применением способа, описанного в случае соединения I, для получения метил-2-амино-3,5-ди[(E)-пент-1-енил]бензоата (6,00 г, 64%). 1H ЯМР (400 МГц, CDCl3): δ 7,76 (д, J = 2,2 Гц, 1H), 7,37 (д, J = 2,2 Гц, 1H), 6,35 (д, J = 15,4 Гц, 1H), 6,26 (д, J = 15,8 Гц, 1H), 6,08 (дт, J = 15,6, 7,0 Гц, 1H), 6,06 (дт, J = 15,8, 7,0 Гц, 1H), 5,5-6,5 (br с, 2H), 3,87 (s. 3H), 2,19-2,25 (м, 2H), 2,13-2,18 (м, 2H), 1,43-1,56 (м, 8H), 0,97 (т, J = 7,3 Гц, 3H), 0,94 (т, J = 7,3 Гц, 3H).
Этап 2:
Метил-2-амино-3,5-ди[(E)-пент-1-енил]бензоат (5,7 г, 19,9 ммоль) гидрировали таким образом, как описано в случае соединения I, для получения метил-2-амино-3,5-дипентилбензоата (5,50 г, 95%). 1H ЯМР (400 МГц, CDCl3): δ 7,50 (д, J = 2,2 Гц, 1H), 6,95 (д, J = 2,2 Гц, 1H), 5,5-6,1 (br с, 2H), 3,79 (s. 3H), 2,40 (т, J = 7,2 Гц, 4H), 1,45-1,58 (м, 4H), 1,20-1,32 (м, 8H), 0,84 (т, J = 7,2 Гц, 3H), 0,82 (т, J = 7,1 Гц, 3H).
Этап 3:
Метил-2-амино-3,5-дипентилбензоат (4,5 г, 15,4 ммоль) обрабатывали с помощью водной тетрафторборной кислоты (5,5M, 3,7 мл, 20 ммоль) и водной хлористоводородной кислоты (8,5M, 3,3 мл, 28 ммоль). Смесь охлаждали до 0°C, а затем обрабатывали по каплям водным раствором нитрита натрия (2,1M, 8,8 мл, 18,5 ммоль) за 2 минуты. Спустя 60 минут при 0°C, реакционную смесь экстрагировали с помощью ксилолов (30 мл). Экстракт ксилолов высушивали над сульфатом натрия, а затем нагревали от 60°C до 120°C за 55 минут. Фильтрование и выпаривание ксилолов in vacuo давало неочищенное соединение, которое очищали на колонке SiO2 SiliaSep, элюируя с помощью этилацетата в гексане (0-5%), для получения метил-2-фтор-3,5-дипентилбензоата (3,1 г, 69%). 1H ЯМР (400 МГц, CDCl3): δ 7,50 (дд, JHF = 6,5 Гц, JHH = 2,4 Гц, 1H), 7,15 (дд, JHF = 6,5 Гц, JHH = 2,4 Гц, 1H), 3,91 (s. 3H), 2,62 (тд, JHH = 7,7 Гц, JHF = 1,2 Гц, 2H), 2,56 (т, J = 7,7 Гц, 2H), 1,55-1,63 (м, 4H), 1,26-1,37 (м, 8H), 0,89 (т, J = 7,0 Гц, 6H); 19F ЯМР (377 МГц, CDCl3): δ -121,31 (дд, JHF = 6,6, 6,6 Гц, 1F).
Этап 4:
Раствор метил-2-фтор-3,5-дипентилбензоата (3,1 г, 10,6 ммоль) в безводном тетрагидрофуране (60 мл) охлаждали до -78°C и медленно обрабатывали литийалюминийгидридом (0,5 г, 13,8 ммоль). Реакционную смесь перемешивали при -78°C в течение 25 минут, затем при 0°C в течение 30 минут. Реакционную смесь гасили путем добавления этилацетата. Смесь промывали водным тартратом калия-натрия (1M, 100 мл) и насыщенным водным хлоридом натрия (100 мл); а затем высушивали над сульфатом натрия, отфильтровывали и выпаривали in vacuo для получения неочищенной соединение. Очищение на колонке SiO2 SiliaSep, элюируя с помощью этилацетата в гексане (3-20%), давало 2-фтор-3,5-дипентилбензиловый спирт (1,8 г, 65%). 1H ЯМР (400 МГц, CDCl3): δ 7,02 (дд, JHF = 6,8 Гц, JHH = 2,3 Гц, 1H), 6,92 (дд, JHF = 7,1 Гц, JHH = 2,4 Гц, 1H), 4,71 (s. 2H), 2,59 (тд, JHH = 7,6 Гц, JHF = 1,2 Гц, 2H), 2,54 (т, J = 7,8 Гц, 2H), 1,73 (с, 1H), 1,54-1,62 (м, 4H), 1,25-1,36 (м, 8H), 0,894 (т, J = 7,0 Гц, 3H), 0,890 (т, J = 7,1 Гц, 3H); 19F ЯМР (377 МГц, CDCl3): δ -131,25 (дд, JHF = 6,7, 6,6 Гц, 1F); 13C ЯМР (101 МГц, CDCl3): δ 157,41 (д, JCF = 242,9 Гц), 138,48 (д, JCF = 4,3 Гц), 130,07 (д, JCF = 5,4 Гц), 129,33 (д, JCF = 16,2 Гц), 127,33 (д, JCF = 15,6 Гц), 126,67 (д, JCF = 4,6 Гц), 59,84 (д, JCF = 5,4 Гц), 35,50, 31,86, 31,77, 31,62, 30,21, 29,21 (д, JCF = 2,4 Гц), 22,80, 22,74, 14,28 (2C).
Этап 5:
Раствор 2-фтор-3,5-дипентилбензилового спирта (1,4 г, 5,3 ммоль) в безводном дихлорметане (35 мл) охлаждали до 0°C и обрабатывали по каплям метансульфонилхлоридом (0,5 мл, 5,8 ммоль) за 10 минут. Реакционную смесь перемешивали при 0°C в течение 20 минут, а затем гасили путем добавления ледяной воды (35 мл). Органическую фазу промывали с помощью водной хлористоводородной кислоты (1M, 35 мл), насыщенного водного бикарбоната натрия (35 мл) и насыщенного водного хлорида натрия (35 мл); а затем высушивали над сульфатом натрия, отфильтровывали и выпаривали in vacuo для получения неочищенного 2-фтор-3,5-дипентилбензилметансульфоната (1,7 г, 93%). Этот материал применяли на следующем этапе без очистки. 1H ЯМР (400 МГц, CDCl3): δ 7,02-7,05 (м, 2H), 5,26 (д, JHF = 1,0 Гц, 2H), 2,98 (s. 3H), 2,52-2,63 (м, 2H), 2,54 (т, J = 7,8 Гц, 2H), 1,54-1,62 (м, 4H), 1,27-1,37 (м, 8H), 0,892 (т, J = 7,0 Гц, 3H), 0,888 (т, J = 7,0 Гц, 3H).
Этап 6:
pH раствора цианида натрия (0,4 г, 7,4 ммоль) в воде (5 мл) доводили до pH 10 с помощью 6M водной хлористоводородной кислоты. Затем добавляли раствор 2-фтор-3,5-дипентилбензилметансульфоната (1,7 г, 4,9 ммоль) в ацетонитриле (25 мл), а реакционную смесь нагревали при 60°C в течение 2 ч. Реакционную смесь концентрировали до 15 мл in vacuo и экстрагировали с помощью этилацетата (100 мл). Органический экстракт промывали водой (100 мл) и насыщенным водным хлоридом натрия (100 мл); а затем высушивали над сульфатом натрия, отфильтровывали и выпаривали in vacuo для получения неочищенной соединение. Очищение на колонке SiO2 SiliaSep, элюируя с помощью этилацетата в гексане (1-10%), давало 2-[2-фтор-3,5-дипентилфенил]ацетонитрил (0,7 г, 55%). 1H ЯМР (400 МГц, CDCl3): δ 7,04 (дд, JHF = 6,9 Гц, JHH = 2,2 Гц, 1H), 6,96 (дд, JHF = 7,1 Гц, JHH = 2,2 Гц, 1H), 3,72 (s. 2H), 2,59 (тд, JHH = 7,7 Гц, JHF = 0,9 Гц, 2H), 2,55 (т, J = 7,8 Гц, 2H), 1,54-1,62 (м, 4H), 1,27-1,37 (м, 8H), 0,90 (т, J = 7,0 Гц, 6H); 19F ЯМР (377 МГц, CDCl3): δ -131,25 (ддд, JHF = 7,0, 7,0, 0,8 Гц, 1F); 13C ЯМР (101 МГц, CDCl3): δ 157,02 (д, JCF = 244,5 Гц), 139,16 (д, JCF = 4,7 Гц), 130,84 (д, JCF = 4,6 Гц), 129,93 (д, JCF = 16,1 Гц), 126,97 (д, JCF = 3,1 Гц), 117,52, 116,79 (д, JCF = 16,2 Гц), 35,38, 31,74, 31,66, 31,54, 30,06, 29,16 (д, JCF = 2,4 Гц), 22,74, 22,68, 17,90 (д, JCF = 6,1 Гц), 14,26, 14,23.
Этап 7:
Смесь 2-[2-фтор-3,5-дипентилфенил]ацетонитрила (0,7 г, 2,7 ммоль), уксусной кислоты (4 мл) и воды (4 мл) обрабатывали по каплям с помощью концентрированной серной кислоты (4 мл); а смесь затем нагревали при 125°C в течение 3,5 ч. Реакционную смесь охлаждали до комнатной температуры и затем гасили путем добавления льда (40 мл). Смесь экстрагировали с помощью этилацетата (40 мл), а органический экстракт затем промывали насыщенным водным хлоридом натрия (40 мл); высушивали над сульфатом натрия, отфильтровывали и выпаривали in vacuo для получения 2-[2-фтор-3,5-дипентилфенил]уксусной кислоты (537 мг, 67%). 1H ЯМР (400 МГц, CDCl3): δ 6,84 (дд, JHF = 7,0 Гц, JHH = 2,3 Гц, 1H), 6,80 (дд, JHF = 6,8 Гц, JHH = 2,2 Гц, 1H), 3,59 (д, JHF = 1,2 Гц, 2H), 2,52 (т, J = 7,5 Гц, 2H), 2,45 (т, J = 7,8 Гц, 2H), 1,46-1,55 (м, 4H), 1,20-1,30 (м, 8H), 0,80-0,84 (м, 6H).
Этап 8:
2-[2-Фтор-3,5-дипентилфенил]уксусной кислоты (537 мг, 1,8 ммоль) преобразовывали в натриевую соль таким образом, как описано в случае соединения I, для получения 2-[2-фтор-3,5-дипентилфенил]ацетата натрия (465 мг, 81%) в виде бледно-коричневого, липкого твердого вещества: 1H ЯМР (400 МГц, CD3OD): δ 6,94 (дд, JHF = 6,9 Гц, JHH = 2,2 Гц, 1H), 6,83 (дд, JHF = 7,0 Гц, JHH = 2,3 Гц, 1H), 3,48 (д, JHF = 1,1 Гц, 2H), 2,58 (т, J = 7,6 Гц, 2H), 2,51 (т, J = 7,6 Гц, 2H), 1,54-1,62 (м, 4H), 1,28-1,38 (м, 8H), 0,90 (т, J = 7,0 Гц, 3H), 0,89 (т, J = 7,0 Гц, 3H); 19F ЯМР (377 МГц, CD3OD): δ -130,71 (дд, JHF = 6,6, 6,6 Гц, 1F); 13C ЯМР (101 МГц, CD3OD): δ 178,31, 157,95 (д, JCF = 240,6 Гц), 137,64 (д, JCF = 3,8 Гц), 128,72 (д, JCF = 4,6 Гц), 128,42 (д, JCF = 17,7 Гц), 128,21 (д, JCF = 5,4 Гц), 124. 50 (д, JCF = 17,7 Гц), 37,94 (д, JCF = 3,1 Гц), 35,05, 31,52, 31,45, 31,37, 30,00, 28,96 (д, JCF = 2,3 Гц), 22,43, 22,38, 13,23, 13,21; МСНР (анионная ESI): m/z 293 (w, M - Na+) и 249,1 (100%, M – Na+ – CO2); СВЭЖХ: 8,4 мин (СВЭЖХ Условия Подвижная фаза A = 0,1% муравьиная кислота в воде; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3; градиент = 5-100% B в A в течение 10 мин.
Соединение XXXIII: Натриевая соль 2-(3,5-дипентилфенил)-2-метилпропановой кислоты
[001] Указанное выше соединение получали таким же образом, как и соединение I, с дополнительным этапом алкилирования интермедиата метил-2-[3,5-дипентилфенил]ацетата с помощью гидрида натрия и метилйодида; и с температурой этапа гидролиза сложного эфира, поднимаемой до 100°C. Off-Беловатое твердое вещество: 1H ЯМР (400 МГц, CD3OD): δ 7,04 (д, J = 1,3 Гц, 2H), 6,76 (с, 1H), 2,54 (т, J = 7,7 Гц, 4H), 1,55-1,63 (м, 4H), 1,46 (с, 6H), 1,27-1,38 (м, 8H), 0,90 (т, J = 7,0 Гц, 6H); 13C ЯМР (101 МГц, CD3OD): δ 184,58, 148,51, 141,98, 125,57, 123,46, 36,02, 48,26, 31,59, 31,42, 27,57, 22,47, 13,29; МСНР (анионная ESI): m/z 303,1 (100%, M - Na+); СВЭЖХ: 8,9 мин (СВЭЖХ Условия Подвижная фаза A = 0,1% муравьиная кислота в воде; Подвижная фаза B = 0,1% муравьиная кислота в ацетонитриле; неподвижная фаза = HSS T3; градиент = 5-100% B в A в течение 10 мин).
Пример 2: Влияние типичных соединений формулы I на экспрессию фактора роста гепатоцитов (ФРГ) для ауторепарации, регенерации и омолаживания ткани.
Эксперименты проводили для определения влияния соединений на экспрессию фактора роста гепатоцитов in vitro нормальных фибробластов кожи человека (НФКЧ) от взрослого донора (Clonetics #CC-2511). НФКЧ выдерживали на голодном рационе в течение ночи в DMEM/F12 + 0,5% FBS и обрабатывали с или без rhTGF-β1 (10 нг/мл) и соединения I (500 мкM) в течение 24 ч. РНК выделяли с помощью набора miRNeasy® (QIAGEN®), в том числе этапа расщепления с помощью ДНКаз на колонке. Синтез кДНК осуществляли (0,5 мкг РНК/реакцию) с применением набора First Strand RT2 (QIAGEN® #330401). ПЦР в реальном времени выполняли таким образом, как описано в руководстве RT2 Profiler PCR Array для ДНК-амплификатора в реальном времени AB-7900HT. Данные ПЦР в реальном времени анализировали с применением способа ΔΔCt на веб-портале RT2 Profiler PCR Array Data Analysis. Все Ct > 35 или не амплифицированные величины изменяли на граничное значение 35. Конститутивными генами, применяемыми для нормализации, являются GAPDH и RPLP0. Контрольная группа состоит из обработанных с помощью TGF-β1 клеток.
Как проиллюстрировано на Фиг. 1, Соединение I повышает экспрессию ФРГ, фактор роста, связанный с репарацией, регенерацией иомоложением ткани. Следующая ниже Таблица 2 демонстрирует, что экспрессию ФРГ в клетках НФКЧ (необработанных) снижали с помощью TGF-β1, что корректировали или увеличивали с помощью типичных Соединений формулы I, описанных в данном документе (Соединение №).
Таблица 2

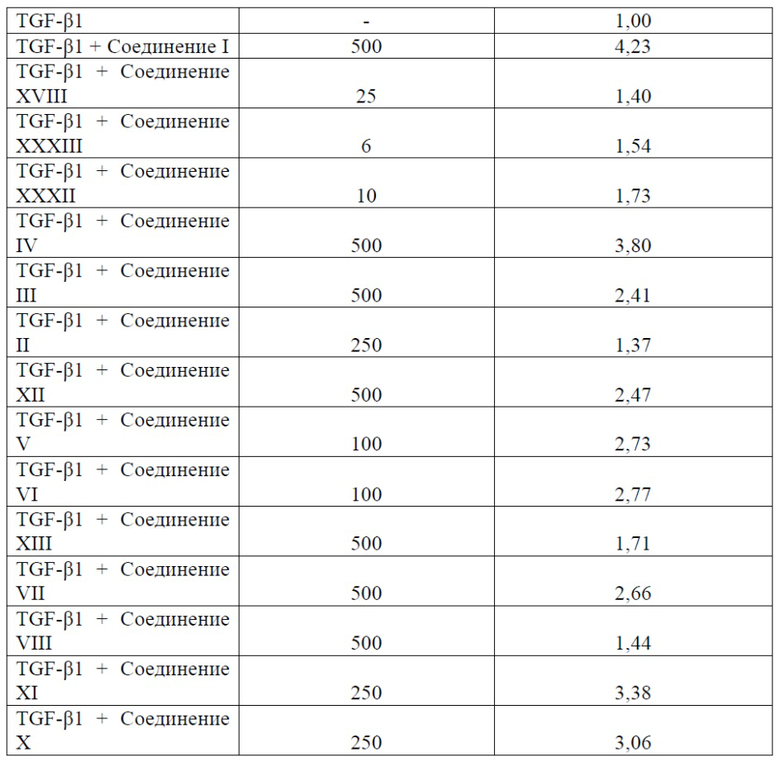
Проводили эксперимент по определению влияния соединений на экспрессию маркеров регенерации. Этот эксперимент проводили с НФКЧ (нормальными фибробластами кожи человека) и эпителиальными клетками человека (почечно-трубчатые эпителиальные клетки, HK-2), участвующими в регенерации ткани после однократного, множественного или постоянного поражения. Поражение моделировали путем инкубации клеток с TGF-β1. НФКЧ применяли таким образом, как описано выше, а клетки HK-2 проксимальных канальцев эпителия человека (ATCC #CRL-2190) выдерживали на голодном рационе в течение ночи в DMEM/F12 + 0,2% FBS и обрабатывали с или без rhTGF-β1 (10 нг/мл) и соединения I (500 мкM) в течение 24 ч. Результаты показали, что соединение I приводит уровень экспрессии маркеров регенерации при нормальном контрольном уровне, указывая на механизм ауторепарации пораженных клеток. В НФКЧ (Фиг. 2), все из ЛОК, MMP13, PLAU (uPA), serpin E1, TIMP3 и ИСК экспрессируются при нормальном уровне, дополнительно в клетках HK-2 (Фиг. 3), помимо этого, все из ЛОК, MMP1, MMP2, MMP9, MMP13, TIMP3 и PLAT (tPA) экспрессируются при уровне, близком к нормальному уровню, наблюдаемому в здоровых клетках.
Пример 3: Влияние соединения I на эндогенное получение ААТ и регенерацию нервной ткани.
Как упоминалось выше, AAT может вызывать нервную регенерацию. Через кПЦР-панель на НФКЧ (способ, описанный в Примере 2) Соединение I продемонстрировало способность увеличивать экспрессию мРНК AAT (Фиг. 4) в пораженных клетках, что указывает на то, что Соединение I может увеличивать регенерацию мозга или других пораженных тканей. Соединение I является типичным для соединений формулы I, описанных в данном документе. Поэтому соединения формулы I, описанные в настоящем документе, могут увеличить регенерация нервов посредством получения эндогенного ААТ в месте повреждения.
* * *
Заголовки включены в данный документ для справки и для облегчения поиска определенных разделов Эти заголовки не предназначены для ограничения объема концепций, описанных в настоящем документе, и эти понятия могут быть применимы в других разделах на протяжении всей спецификации. Таким образом, настоящее изобретение не предназначено для ограничения вариантов реализации изобретения, показанных в данном документе, но должно соответствовать самому широкому объему, совместимому с принципами и новыми признаками, раскрытыми в настоящем документе.
Формы единственного числа включают ссылки на соответствующее множественное число, если из контекста явно не следует иное.
Если не указано иное, все числа, выражающие количества ингредиентов, условия реакции, концентрации, свойства и так далее, применяемые в описании и формуле изобретения, следует понимать как модифицированные во всех случаях термином “около”. Как минимум, каждый числовой параметр должен быть истолкован, по крайней мере, в свете количества приведенных значащих цифр и с применением обычных способов округления. Соответственно, если не указано иное, численные параметры, указанные в настоящем описании и прилагаемой формуле изобретения, являются приближениями, которые могут варьироваться в зависимости от свойств, которые должны быть получены. Несмотря на то, что численные диапазоны и параметры, представляющие широкий объем вариантов реализации изобретения, являются приблизительными, числовые значения, изложенные в конкретных примерах, сообщаются как можно точнее. Однако любое числовое значение по своей сути содержит определенные ошибки, возникающие в результате вариаций в экспериментах, измерений в ходе исследований, статистического анализа и т. д.
Понятно, что примеры и варианты реализации изобретения, описанные в настоящем документе, предназначены только для иллюстративных целей и что специалистам в данной области техники будут предложены различные модификации или изменения в свете этого и они должны быть включены в настоящее изобретение и объем прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДОТВРАЩЕНИЯ И ЛЕЧЕНИЯ ОСТЕОПОРОЗА | 2015 |
|
RU2709205C2 |
| ЗАМЕЩЕННЫЕ АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДОТВРАЩЕНИЯ И ЛЕЧЕНИЯ ДИАБЕТА | 2015 |
|
RU2723486C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ | 2021 |
|
RU2817736C1 |
| СОЕДИНЕНИЯ ФЕНИЛКЕТОНКАРБОКСИЛАТА И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПРЕДОТВРАЩЕНИЯ И ЛЕЧЕНИЯ ОСТЕОПОРОЗА | 2015 |
|
RU2712027C2 |
| ПЕРОРАЛЬНО БИОДОСТУПНЫЕ ПРОЛЕКАРСТВА (+)-3- ГИДРОКСИМОРФИНАНА ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2008 |
|
RU2430089C2 |
| СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ ОКСАДИАЗОЛ, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2789456C2 |
| Оксазолидиноны на основе производных пиридоксина, обладающие антибактериальной активностью | 2024 |
|
RU2836570C1 |
| ПРОИЗВОДНЫЕ ДИГИДРОИМИДАЗО[5,1-А]- БЕТА-КАРБОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2298010C2 |
| НОВОЕ ИНДАНСУЛЬФАМИДНОЕ ПРОИЗВОДНОЕ | 2013 |
|
RU2619937C2 |
| ПРОТИВООПУХОЛЕВЫЕ АНАЛОГИ ЛАМЕЛЛАРИНА | 2003 |
|
RU2328500C2 |
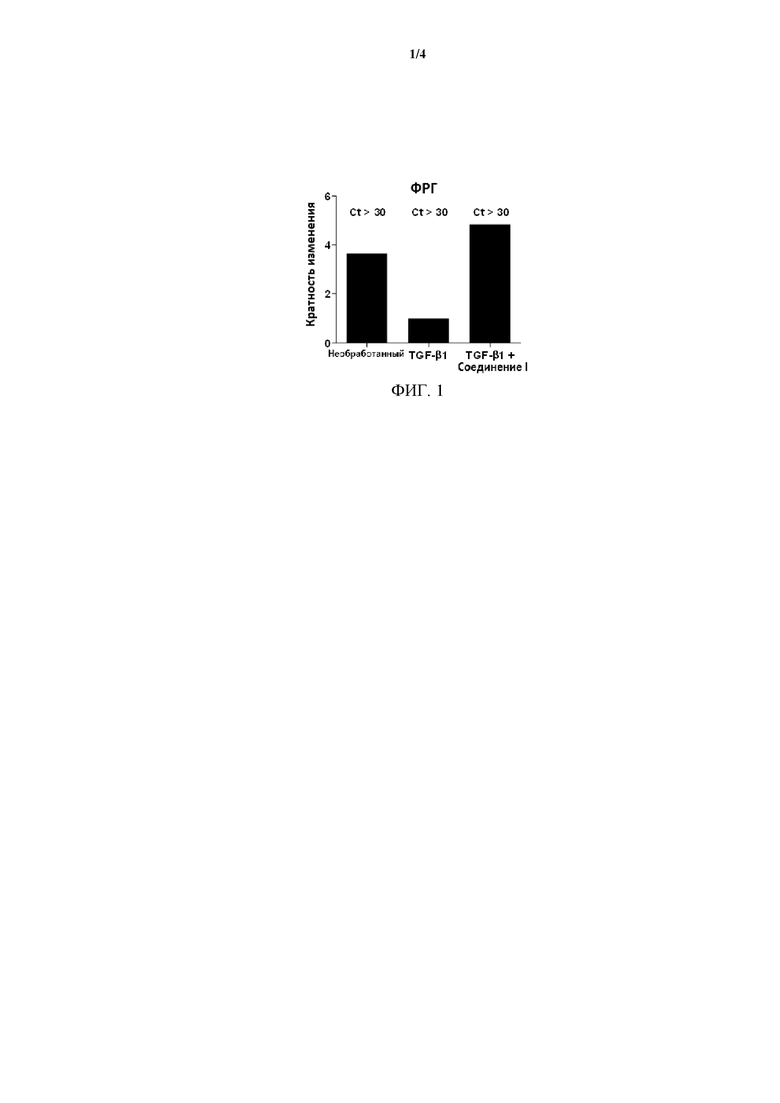

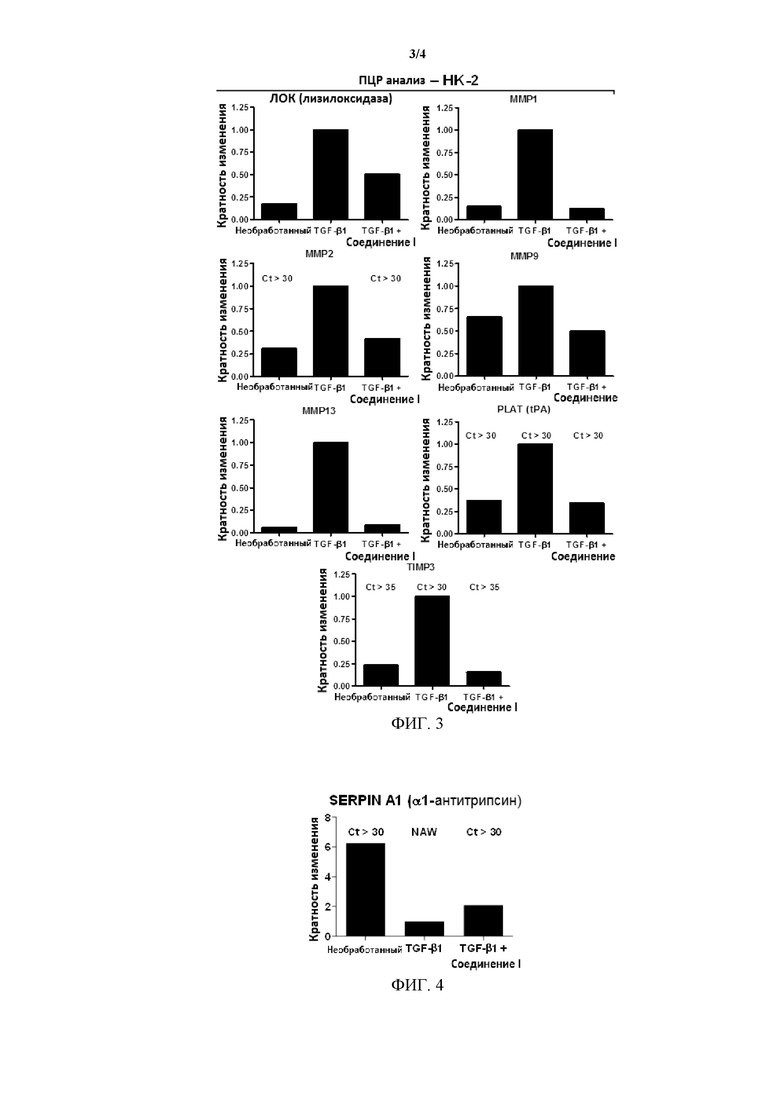
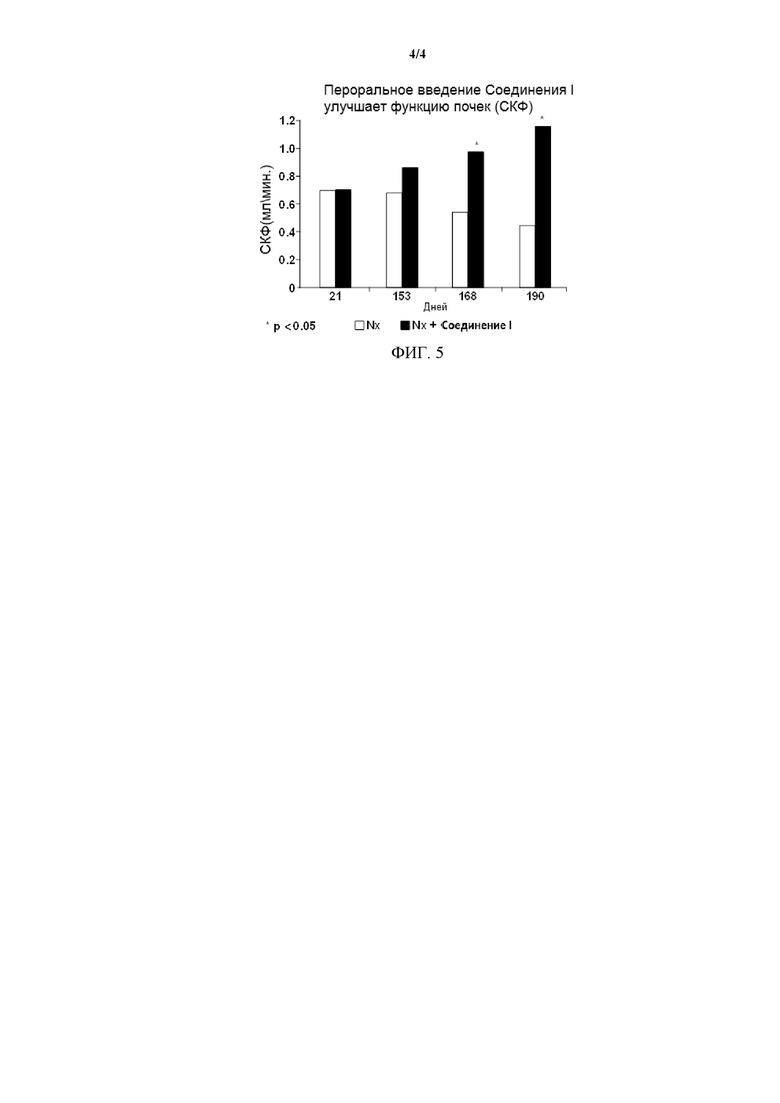
Изобретение относится к способу стимуляции регенерации органа или ткани в поврежденном органе или ткани у нуждающегося в этом субъекта, где поврежденный орган или ткань представляет собой сердце, печень, легкое, кожу, желудок, кишечник, мышцы или хрящи, включающему введение нуждающемуся в этом субъекту соединения формулы I, где А представляет собой C5алкил, C6алкил, C5алкенил, C6алкенил, C(О)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где n равен 3 или 4; R1 представляет собой Н, F или OH; R2 представляет собой Н, F, OH, C5алкил, C6алкил, C5алкенил, C6алкенил, C(О)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где n равен 3 или 4; R3 представляет собой Н, F, OH, или CH2Ph; R4 представляет собой H, F или OH; Q представляет собой (CH2)mC(О)OH, где m равен 1 или 2, CH(CH3)C(О)OH, C(CH3)2C(О)OH, CH(F)-C(О)OH, CF2-C(О)OH или C(О)-C(О)OH, его фармацевтически приемлемой соли или их комбинации. Также изобретение относится к применению соединения формулы I для изготовления лекарственного средства. Технический результат – применение соединений формулы I, их солей или их комбинаций для стимуляции регенерации органа или ткани в поврежденном органе или ткани, где поврежденный орган или ткань представляет собой сердце, печень, легкое, кожу, желудок, кишечник, мышцы или хрящи. 2 н. и 27 з.п. ф-лы, 2 табл., 3 пр., 5 ил.
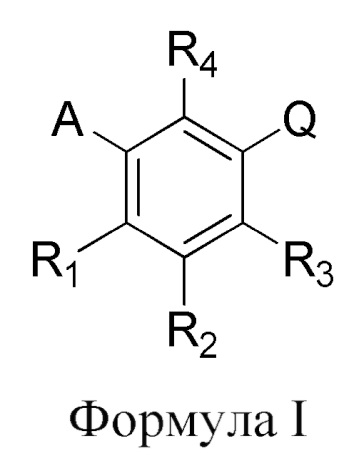
1. Способ стимуляции регенерации органа или ткани в поврежденном органе или ткани у нуждающегося в этом субъекта, где поврежденный орган или ткань представляет собой сердце, печень, легкое, кожу, желудок, кишечник, мышцы или хрящи, включающий введение нуждающемуся в этом субъекту соединения, представленного Формулой I, его фармацевтически приемлемой соли или их комбинации:

где А представляет собой C5 алкил, C6 алкил, C5 алкенил, C6 алкенил, C(O)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где n равен 3 или 4;
R1 представляет собой Н, F или OH;
R2 представляет собой Н, F, OH, C5 алкил, C6 алкил, C5 алкенил, C6 алкенил, C(O)-(CH2)n-CH3 или CH(OH)-(CH2)n-CH3, где n равен 3 или 4;
R3 представляет собой Н, F, OH или CH2Ph;
R4 представляет собой Н, F или OH;
Q представляет собой
1) (CH2)mC(O)OH, где m равен 1 или 2,
2) CH(CH3)C(O)OH,
3) C(CH3)2C(O)OH,
4) CH(F)-C(O)OH,
5) CF2-C(O)OH или
6) C(O)-C(O)OH.
2. Способ по п. 1, где А представляет собой C5 алкил или C6 алкил.
3. Способ по п. 1, где R2 представляет собой Н, F, OH, C5 алкил или C6 алкил.
4. Способ по п. 1, где R3 представляет собой Н, OH или CH2Ph.
5. Способ по п. 1, где Q представляет собой (CH2)mC(O)OH, где m равен 1 или 2.
6. Способ по п. 1, где А представляет собой C5 алкил или C6 алкил; R1 представляет собой Н, F или OH; R2 представляет собой Н, F, OH, C5 алкил или C6 алкил; R3 представляет собой Н, OH или CH2Ph; R4 представляет собой Н, F или OH и Q представляет собой (CH2)mC(O)OH, где m равен 1 или 2.
7. Способ по п. 1, где А представляет собой C5 алкил; R1 представляет собой Н; R2 представляет собой Н или C5 алкил; R3 представляет собой Н; R4 представляет собой Н; и Q представляет собой (CH2)mC(O)OH, где m равен 1.
8. Способ по п. 1, отличающийся тем, что указанное соединение выбрано из группы, состоящей из соединений, представленных следующими структурами:
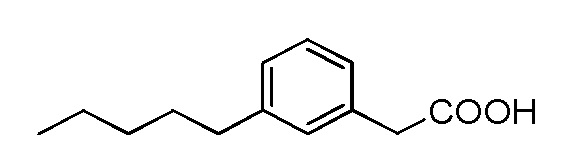 ;
; ;
; ;
;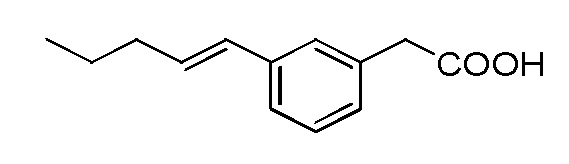 ;
; ;
; ;
;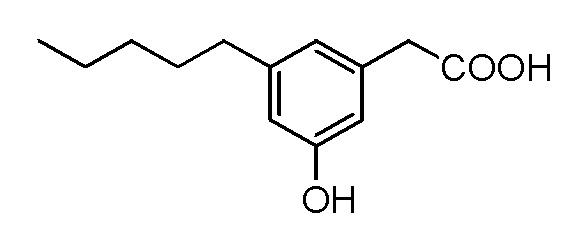 ;
; ;
;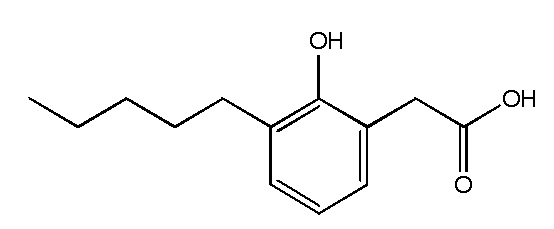 ;
;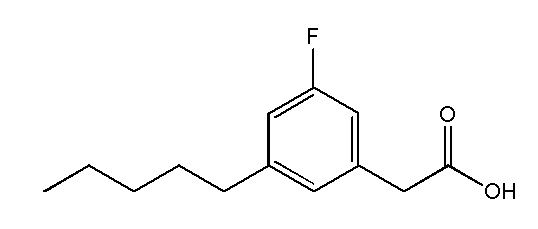 ;
;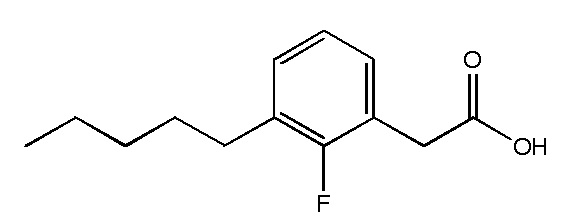 ;
;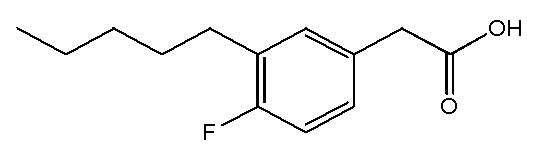 ;
; ;
; ;
; ;
; ;
;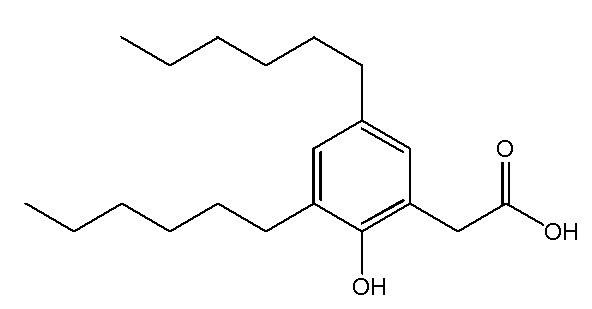 ;
; ;
; ;
; ;
; ;
; ;
; ;
;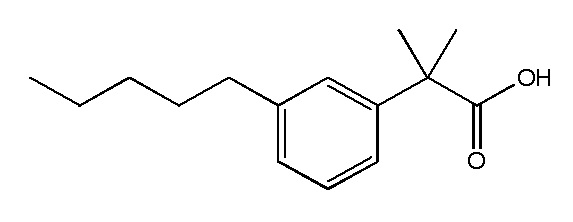 ;
; ;
;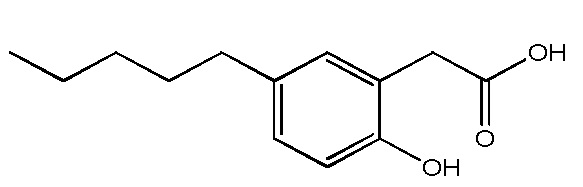 ;
; ;
; ;
; ;
;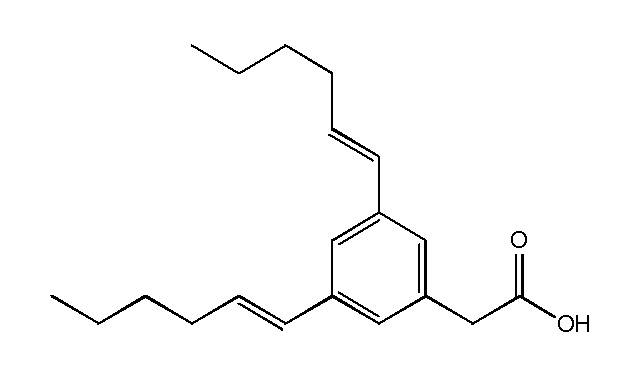 ;
;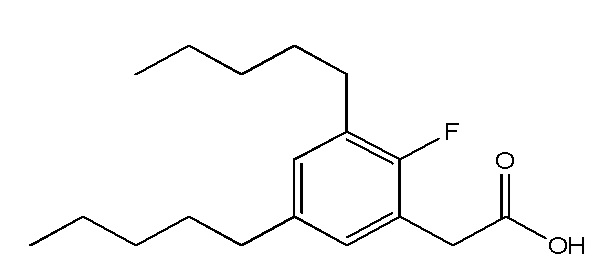 ;
; ,
,
и их фармацевтически приемлемых солей.
9. Способ по п. 1, отличающийся тем, что указанное соединение представлено следующей структурой:

или ее фармацевтически приемлемой солью.
10. Способ по п. 1, отличающийся тем, что указанное соединение представлено следующей структурой:

или ее фармацевтически приемлемой солью.
11. Способ по п. 1, отличающийся тем, что фармацевтически приемлемая соль представляет собой соль присоединения основания, содержащую противоион металла, выбранный из группы, состоящей из натрия, калия, кальция, магния, лития, аммония, марганца, цинка, железа или меди.
12. Способ по п. 1, отличающийся тем, что фармацевтически приемлемая соль представляет собой соль натрия.
13. Способ по любому из пп. 1-12, отличающийся тем, что указанный поврежденный орган или ткань представляет собой сердце.
14. Способ по любому из пп. 1-12, отличающийся тем, что указанный поврежденный орган или ткань представляет собой печень.
15. Способ по любому из пп. 1-12, отличающийся тем, что указанный поврежденный орган или ткань представляет собой легкое.
16. Способ по любому из пп. 1-12, отличающийся тем, что указанный поврежденный орган или ткань представляет собой кожу.
17. Способ по любому из пп. 1-12, отличающийся тем, что указанный поврежденный орган или ткань представляет собой желудок.
18. Способ по любому из пп. 1-12, отличающийся тем, что указанный поврежденный орган или ткань представляет собой кишечник.
19. Способ по любому из пп. 1-12, отличающийся тем, что указанный поврежденный орган или ткань представляет собой мышцы.
20. Способ по любому из пп. 1-12, отличающийся тем, что указанный поврежденный орган или ткань представляет собой хрящи.
21. Применение соединения или его фармацевтически приемлемой соли, определенных по любому из пп. 1-12, или их комбинации для изготовления лекарственного препарата для регенерации поврежденного органа или ткани у нуждающегося в этом субъекта, где поврежденный орган или ткань представляет собой сердце, печень, легкое, кожу, желудок, кишечник, мышцы или хрящи.
22. Применение по п. 21, отличающееся тем, что указанный поврежденный орган или ткань представляет собой сердце.
23. Применение по п. 21, отличающееся тем, что указанный поврежденный орган или ткань представляет собой печень.
24. Применение по п. 21, отличающееся тем, что указанный поврежденный орган или ткань представляет собой легкое.
25. Применение по п. 21, отличающееся тем, что указанный поврежденный орган или ткань представляет собой кожу.
26. Применение по п. 21, отличающееся тем, что указанный поврежденный орган или ткань представляет собой желудок.
27. Применение по п. 21, отличающееся тем, что указанный поврежденный орган или ткань представляет собой кишечник.
28. Применение по п. 21, отличающееся тем, что указанный поврежденный орган или ткань представляет собой мышцы.
29. Применение по п. 21, отличающееся тем, что указанный поврежденный орган или ткань представляет собой хрящи.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| GHALY A | |||
| et al.: "Ischemia-reperfusion modulates inflammation and fibrosis of skeletal muscle after contusion injury" Int.J | |||
| Exp.Pathol., 2010, vol.91, pp.244-255 | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |

