Изобретение относится к области медицины и генной терапии и может быть использовано для лечения инфекции, вызванной вирусом гепатита С (ВГС). Патентуемая композиция представляет собой комплексы молекул малых интерферирующих РНК (миРНК), подавляющих репликацию вирусной РНК, и носителя - липопептидов, способствующих проникновению синтетических молекул миРНК внутрь клеток-мишеней.
Предпосылками изобретения явилось следующее.
Вирусный гепатит С - антропонозная вирусная инфекция из условной группы трансфузионных гепатитов, характеризующаяся поражением печени, безжелтушным, легким и среднетяжелым течением в острой фазе и частой склонностью к хронизации, развитию циррозов печени и первичных гепатокарцином (Chevaliez et al., 2012).
Возбудитель - РНК-содержащий вирус, включенный в состав рода Hepacivirus семейства Flaviviridae (Gower et al., 2014). Выделяют 7 генотипов и более чем 67 субтипов вируса (Simmonds 2004; Smith, Bukh et al. 2014; Echeverria, Moratorio et al. 2015). Наиболее типичным для РФ является генотип 1 (1а и 1b), на долю которого приходится до 60% в структуре хронических гепатитов С (ХГС), на долю генотипов 2, 3 остается 40% (Pimenov, Vdovin et al. 2013).
Актуальность проблемы гепатита С определяется высокой эпидемиологической и социально-экономической значимостью этого заболевания, широким и повсеместным распространением, активным вовлечением в эпидемический процесс лиц репродуктивного, наиболее трудоспособного возраста, значительными расходами государства на лечение лиц, инфицированных ВГС. По данным ВОЗ, вирусом гепатита С инфицировано около 180 млн людей во всем мире, это около 3% населения, 130 млн являются хроническими носителями вируса, 3-4 млн заражаются ВГС ежегодно [(http://www.who.int)].
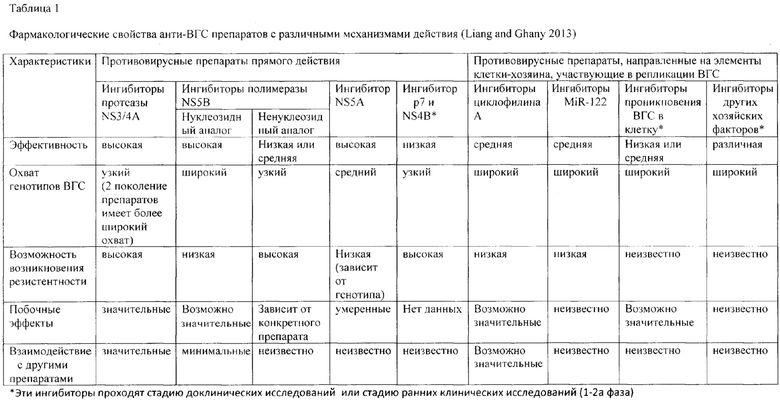
Устойчивый вирусный ответ (УВО) определяется как состояние, при котором РНК ВГС не детектируется в сыворотке крови в течение 24 недель после окончания терапии (Cheng et al., 2014). УВО является стандартным маркером успешной противовирусной терапии при проведении клинических испытаний. В начале 2000-х годов комбинация пегилированного интерферона и рибавирина (ПР) стала стандартом анти-ВГС терапии (Garg and Kar 2009; Pawlotsky 2013). Однако интерфероновая терапия связана со множеством побочных эффектов (гриппоподобные симптомы, усталость и т.д.). Уровень УВО при такой терапии составляет 70- 80% среди пациентов с ВГС-инфекцией, вызванной генотипами 2, 3 и 70% среди пациентов с другими генотипами ВГС (Ghany, Strader et al. 2009). Поэтому есть необходимость в разработке новых противовирусных препаратов. Каждый этап жизненного цикла вируса можно рассматривать как мишень для новых противовирусных препаратов. Современные достижения в понимании жизненного цикла ВГС привели к разработке высокоэффективных и хорошо переносимых противовирусных средств различных механизмов действия (Табл. 1).
Интерференция РНК - это естественный механизм регуляции экспрессии генов в клетке с участием фермента Dicer и молекул малых интреферирующих РНК (миРНК). Интерференция РНК (РНКи) инициируется ферментом Dicer, который разрезает двухцепочечную (ДЦ) молекулу РНК на миРНК размером 21-26 пн, обеспечивающие специфическое постранскрипционное подавление экспрессии генов-мишеней посредством комплементарного связывания и деградации соответствующих мРНК или посредством ингибирования трансляции мРНК (Хаитов М.Р., 2012).
Едва ли не главной проблемой при использовании лекарственных средств, работающих на основе РНК, и является внутриклеточная доставка молекул миРНК в клетку. Большинство методов и средств доставки миРНК в клетку упоминаются в патентах (патент US 20110305772 А1, патент US 20110189180 А1, патент US 20120202871 А1). Данные патенты касаются частных аспектов широко распространенных в молекулярной биологии методов или новых способов их применения.
Препараты на основе миРНК разрабатываются для лечения рака, инфекционных заболеваний и других патологий, которые ассоциированы с нарушениями в функциях специфических генов. На сегодняшний день запатентован целый ряд препаратов на основе миРНК для профилактики и лечения различных вирусных патологий. Так, например, датская компания Santaris Pharma A/S, разработала препарат Miravirsen для лечения ВГС-инфекции (Lna oligomers for improvement in hepatic function, patent WO 2013068348 A1). В основе препарата - модифицированный 15 нуклеотидный олигодезоксинуклеотид - ингибитор клеточной микроРНК miR-122, экспрессирующейся в печени и защищающей геном вируса гепатита С от деградации экзонуклеазой Xrn1 (Li, Masaki et al. 2013). Препарат представляет собой антисмысловой олигонуклеотид в виде замкнутой нуклеиновой кислоты (Locked Nucleic Acid), модифицированной фосфоротиоатной группой и направленной к miR-122. Препарат недавно успешно прошел стадию 2а клинических испытаний.
В охранном документе US 20120308642 А1, который является прототипом заявленной композиции, описываются комбинации миРНК к неперекрывающимся фрагментам 5' нетранслируемого региона генома ВГС, обеспечивающие продолжительный противовирусный эффект. Внутривенное назначение препарата осуществляется в комплексе с липидными наночастицами, модифицированными холестеролом. Заявленное преимущество данного способа доставки по сравнению с описанными ранее вариантами наночастиц заключается в разработке протокола подготовки вспомогательных компонентов лекарственного средства, обеспечивающего уменьшение их размера и повышение доли частиц размером менее 150 нм. В соответствии с документом данная модификация протокола приводит к повышению эффективности доставки миРНК в клетки печени и позволяет снизить концентрацию препарата.
Отличительной особенностью заявленной композиции является структура молекулы миРНК, а также структура ее носителя. В основе вспомогательного компонента патентуемой композиции используются липодипептиды, которые самопроизвольно формируют в водной среде бислойные агрегаты, размер которых не превышает 100 нм. При этом наночастицы не нуждаются в дополнительной модификации.
Задачей предлагаемого изобретения является создание малотоксичного и эффективного средства для лечения ВГС-инфекции на основе молекул миРНК.
Заявленная композиция для лечения ВГС-инфекции, содержащая комплексы липопептидов OrnGlu(С16Н33)2, выступающих в качестве носителя, и молекул миРНК, представленных последовательностью SEQ ID NO 1, подавляющих репликацию ВГС посредством механизма интерференции РНК. Размеры комплексов не превышают 100 нм.
Техническими результатами предлагаемого способа является создание малотоксичной композиции и эффективное воздействие вводимых молекул миРНК, представленных последовательностью SEQ ID NO 1, в комплексах с липопептидами Y14 состава OrnGlu(C16H33)2, на подавление репликации ВГС. Преимуществом заявленной композиции является высокая биодоступность (до 70%) при подкожном способе введения.
Краткое описание чертежей
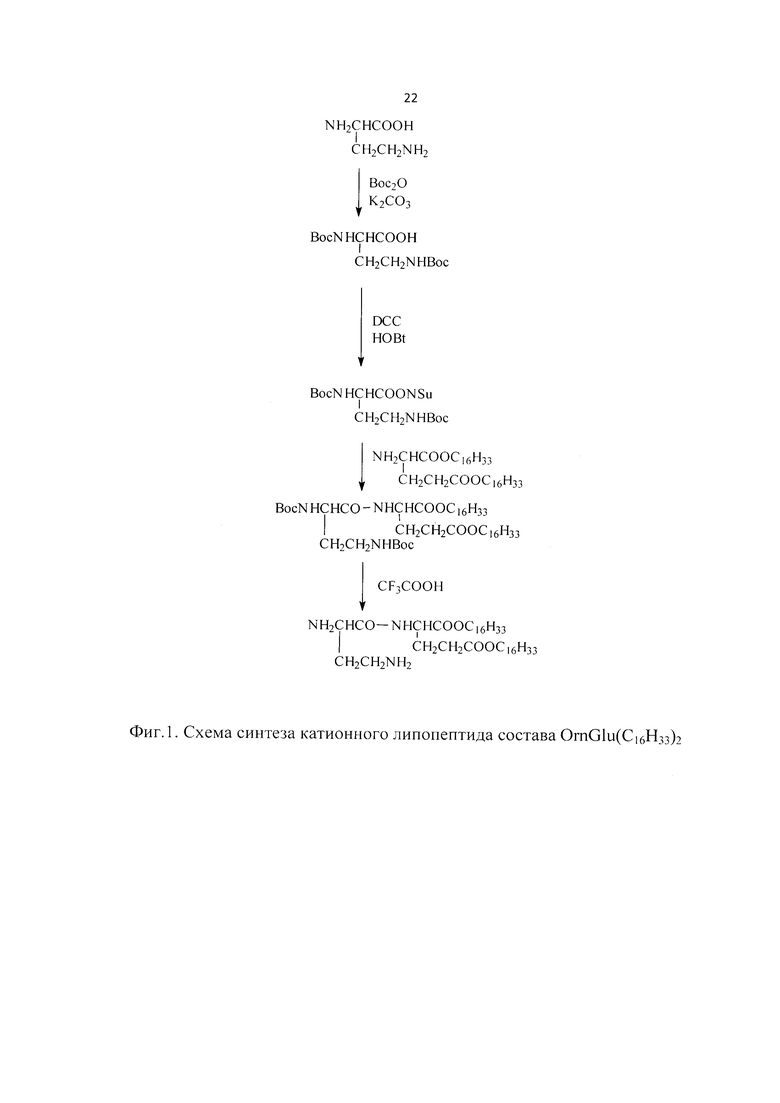
Фиг. 1. Схема синтеза липопептидов состава OrnGlu(C16H33)2.
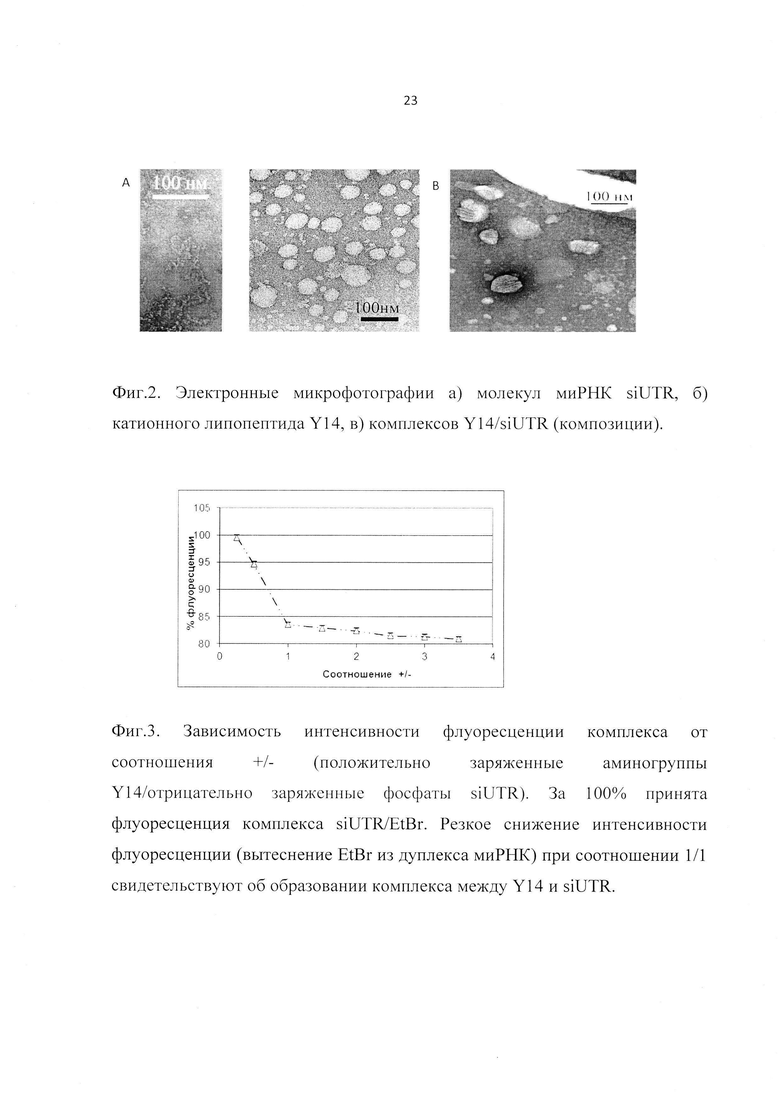
Фиг. 2. Электронные микрофотографии а) молекул миРНК siUTR, б) липопептидов Y14, в) комплексов Y14/siUTR (композиции).
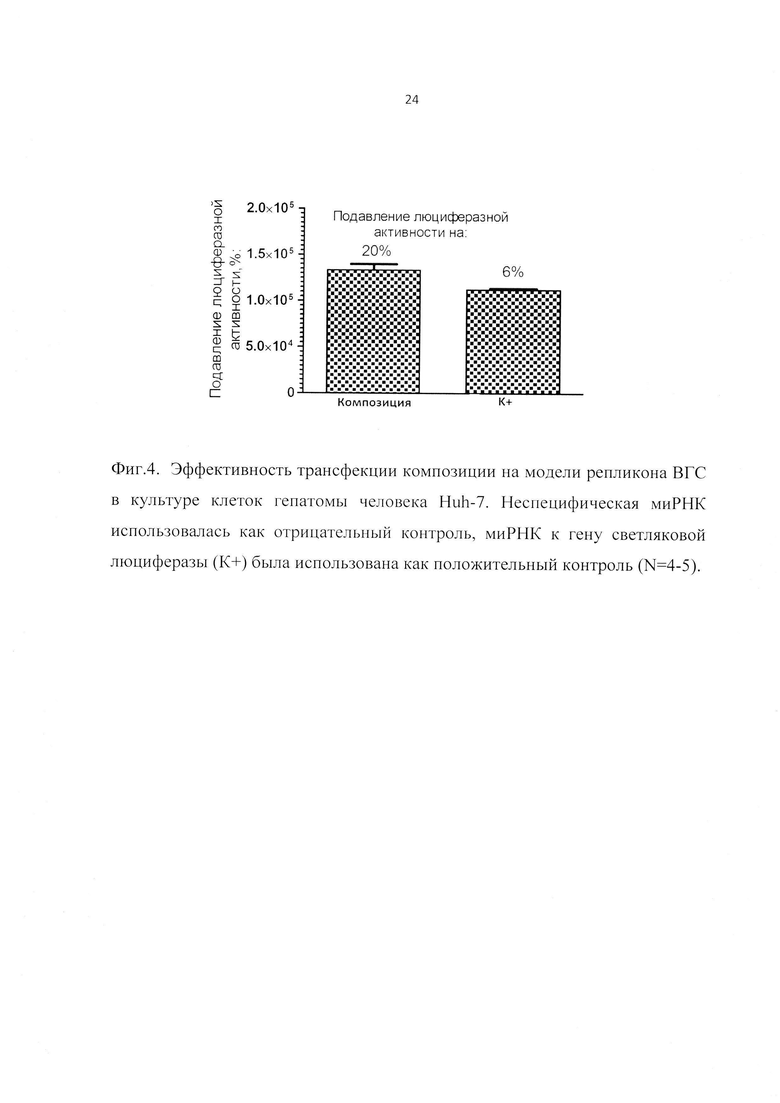
Фиг. 3. Зависимость интенсивности флуоресценции комплекса от соотношения +/- (положительно заряженные аминогруппы YH/отрицательно заряженные фосфаты siUTR).
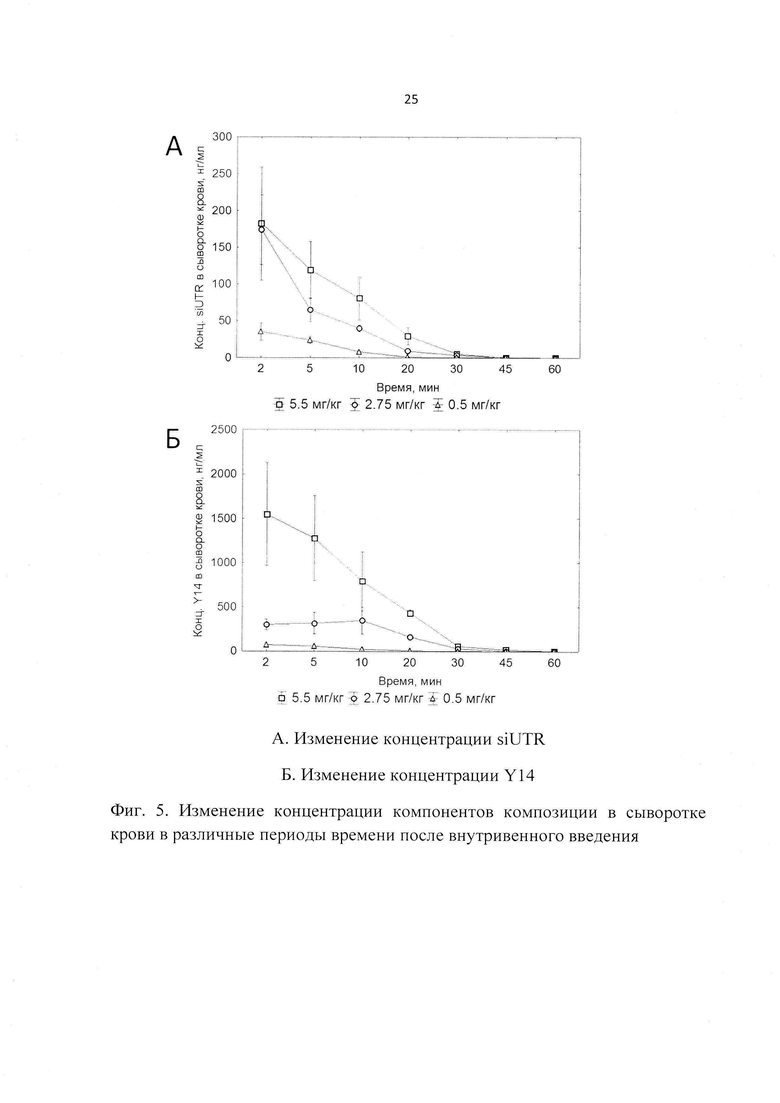
Фиг. 4. Эффективность трансфекции композиции на модели репликона ВГС в культуре клеток гепатомы человека Huh-7.
Фиг. 5. Изменение концентрации компонентов композиции в сыворотке крови в различные периоды времени после внутривенного введения.
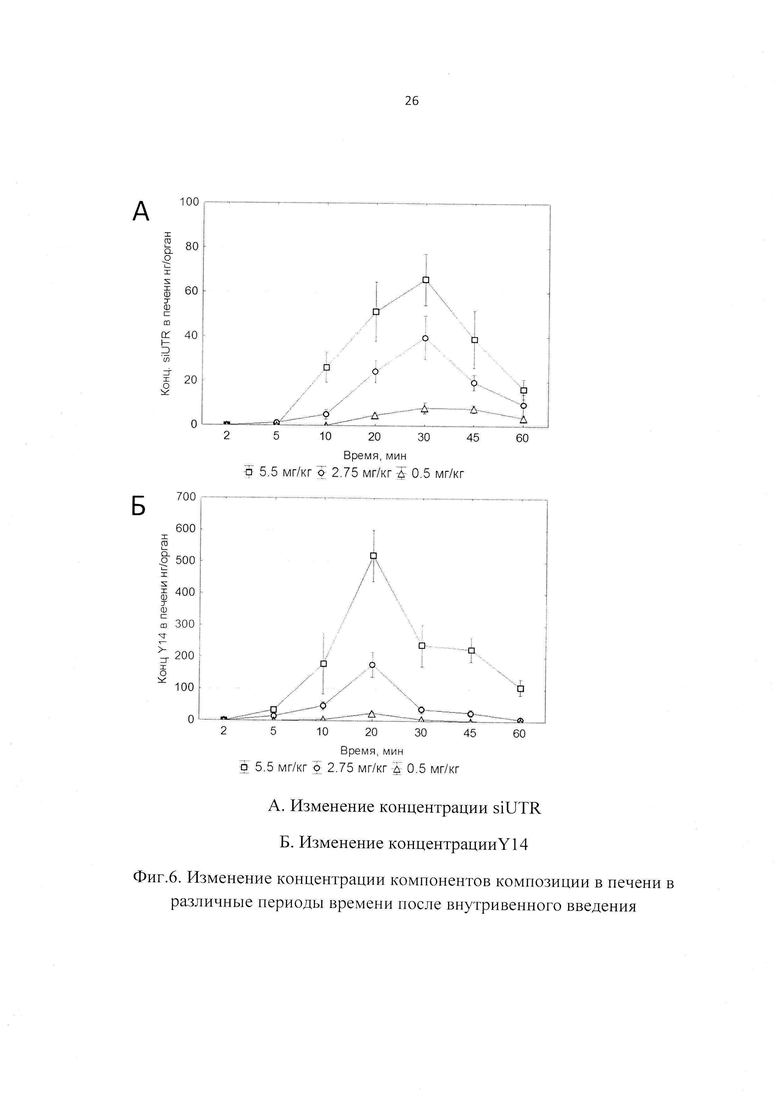
Фиг. 6. Изменение концентрации компонентов композиции в печени в различные периоды времени после внутривенного введения.
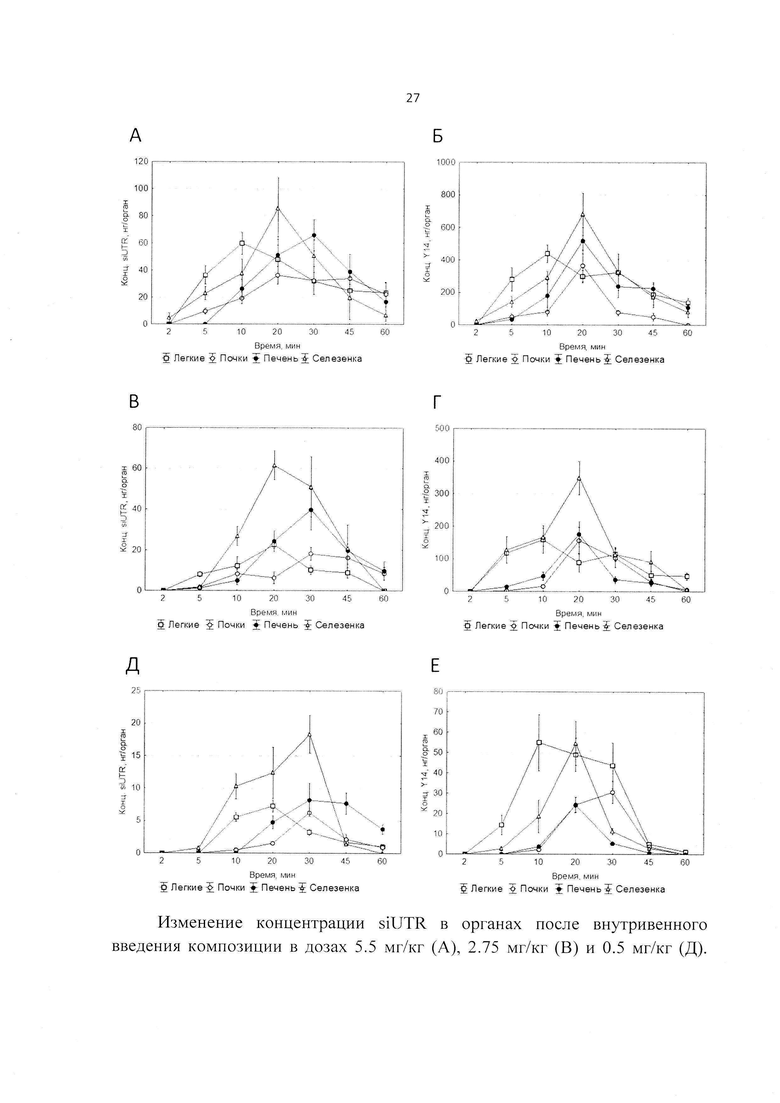
Фиг. 7. Изменение концентрации компонентов композиции в органах (легкие, печень, почки, селезенка) в различные периоды времени после внутривенного введения.
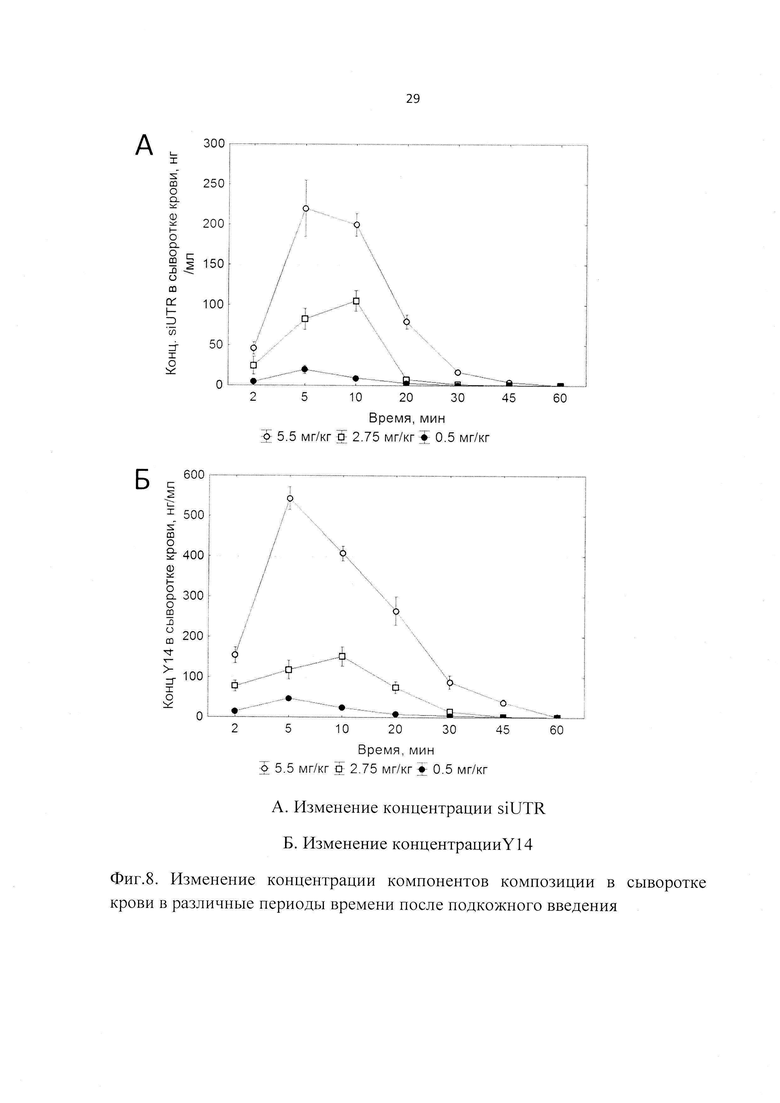
Фиг. 8. Изменение концентрации компонентов композиции в сыворотке крови в различные периоды времени после подкожного введения
Подробное описание изобретения.
Пример 1. Синтез молекул миРНК
Производство миРНК (siUTR) основано на твердофазном химическом синтезе (на твердом носителе), который включает повторяющиеся операции для наращивания олигонуклеозидной цепи, поэтому эти реакции можно проводить с использованием коммерческих автоматических синтезаторов, а также использовать для производства ручные (manual) реакторы, не в автоматическом режиме. Все сырьевые компоненты (реагенты и растворители) взвешивали, отмеряли и загружали в реактор согласно технологическим стадиям, полупродукты появляются после последней стадии синтеза при выгрузке их из реактора. При синтезе получали две комплементарных цепи миРНК, из которых готовили дуплекс путем смешивания эквимолярных количеств смысловой и антисмысловой цепей миРНК siUTR, которые прогревали при 65°C в течение 5 минут, а затем резко охлаждали при 4°C. Готовые дуплексы (siUTR) использовали для приготовления композиции.
Пример 2. Синтез липопептидов
Синтез липопептидов Y14 состава OrnGlu(С16Н33)2 осуществляли по схеме, представленной на Фиг. 1. Boc-замещенное производное аминокислоты получали действием ди-трет-бутилоксикарбоната. Побочными продуктами реакции присоединения Вос-группы являются трет-бутанол и углекислый газ, которые легко отделяются от целевого продукта. Структура синтезированного соединения была подтверждена данными 1Н-ЯМР-, ИК-спектроскопии.
В ИК-спектре наблюдались характерные полосы поглощения амидных связей (1660 см-1, С=O «I амидная полоса», 1658 см-1, N-H «II амидная полоса»), карбоксильной группы (1750 см-1 С=O, 1420 см-1 ОН). Сигнал протонов трет-бутильных групп в 1Н-ЯМР-спектре соединения (25) представлен синглетом с химическим сдвигом 1,4 м. д.
Реакцию получения N-гидроксисукцинимидного эфира (26) проводили в стандартных условиях в присутствии дициклогексилкарбодиимида в качестве конденсирующего агента. Выпавший осадок дициклогексилмочевины отфильтровывали. Полученный продукт давал положительную реакцию в специальном проявителе на N-гидроксисукцинимидную группу. Продукт без дополнительной очистки использовали на следующей стадии.
Реакцию образования амидной связи между активированным эфиром (26) и дигексадециловым эфиром L-глутаминовой кислоты проводили при небольшом избытке последнего (соотношение реагентов 1:1,1 соответственно) в тетрагидрофуране в присутствии водного раствора KHCO3, необходимого для создания слабой щелочной среды. За ходом реакции наблюдали по данным ТСХ. Продукты реакции выделяли с помощью колоночной хроматографии в системе растворителей толуол-хлороформ-метилэтилкетон-изопропанол 10:6:3:1. Структура полученного соединения подтверждалась данными ИК- и 1Н-ЯМР-спектроскопии. В 1Н-ЯМР-спектре присутствовали сигналы протонов алифатических цепей (δ, м. д.: 1,25 (с, 52Н, СН2), 0,84 (т, 6Н, СН3)), протонов трет-бутильных групп (δ, м. д.: 1,45 (с, 18Н, СН3)), амидной связи (δ, м. д.: 5,7 (д, 1Н, NH)), углеводородных цепей орнитина и глутамина (δ, м. д.: 2,5 (м, 2Н, СН2), 2,28 (м, 4Н, СН2), 3,4-4,2 (м, 2Н, СН), 4,36 (м, 2Н, СН2)).
Удаление трет-бутилоксикарбонильной защиты проводили действием безводной трифторуксусной кислоты. В 1Н-ЯМР-спектре наблюдалось исчезновение сигналов протонов, соответствовавших защитным группам при сохранении содержательной части спектров оставшейся структуры. В ИК-спектре наблюдались характерные полосы поглощения амидных связей (1654 см-1, С=O «I амидная полоса», 1681 см-1, N-H «II амидная полоса»), карбоксильной группы (1735 см-1 С=O), свободной аминогруппы (3330 см-1 NH2), алифатических цепей и углеводородных групп орнитина и глутамина (2912, 2847, 1462, 1390 см-1). В масс-спектре MALDI присутствовал сигнал молекулярного иона целевого соединении (М+) 710,85. Для получения липосом 50 мг OrnGlu(C16)2 растворяли в 20 мл хлороформа, который затем медленно отгоняли на ротационном испарителе для формирования тонкой липидной пленки, которую дополнительно высушивали 4 часа на вакуумном насосе. К полученной липидной пленке добавляли 5 мл дистиллированной воды и выдерживали в течение 30 минут. Обрабатывали получаемую дисперсию ультразвуком (3×25 мин). Использовали миниэкструдер с поликарбонатными фильтрами пористостью 400 нм для гомогенизации частиц и стерилизации. Готовые липопептиды использовали для получения композиции.
Пример 3. Состав композиции
Заявленная композиция представляет собой комплексы, в состав которого входят липопептиды Y14 состава OrnGlu(С16Н33)2 (молекулярная масса 710 г/моль, концентрация 0.8 мг/мл, объем 1.75 мл) и дуплексы миРНК siUTR (SEQ ID NO 1, концентрация 0.08 мг/мл, объем 1.75 мл). Массовое соотношение Y14/siUTR=10/1. Растворы обоих компонентов приготовлены в 5% растворе глюкозы.
Пример 4. Физико-химические характеристики композиции
Перед использованием композиции Y14/siUTR в условиях in vitro и in vivo были исследованы ее физико-химические характеристики. Для комплекса Y14/siUTR определены соотношения компонентов, при которых наблюдается максимальная степень связывания. Эксперименты проводились с использованием флуоресцентной спектроскопии.
Смеси состава 1:2-2:1 моль/моль (+/-, положительно заряженные аминогруппы YH/отрицательно заряженные фосфаты siUTR) инкубировались в течение 15 мин, достаточных для формирования используемых в экспериментах по трансфекции комплексов.
В результате экспериментов по флуоресцентной спектроскопии было показано, что для липопептида Y14 минимальное соотношения, при которых наблюдается комплексообразование составляют 1:1 (+/-) (Фиг. 3).
Размер и форма комплексов определялись методом трансмиссионной электронной микроскопии с контрастированием уранилацетатом. Было показано, что комплексы Y14/siUTR, входящие в состав заявленной композиции, представляют собой сэндвичевую структуру (Фиг. 2), размер которой не превышает 100 нм.
Пример 5. Оценка эффективности трансфекции in vitro
Исследования жизненного цикла ВГС затрудняются отсутствием адекватных моделей. Единственной распространенной клеточной системой является искусственный вирусный репликон, представляющий собой (+) цепь вирусной РНК, у которой структурная область заменена на ген устойчивости к неомицину и ген светляковой люциферазы, и, кроме того, область внутренней посадки рибосомы (IRES) заменена на таковую вируса энцефаломиокардита. Изучение специфической биологической активности композиции в экспериментах in vitro проводили с использованием перевиваемой культуры клеток гепатомы человека Huh-7, которая экспрессирует субгеномный репликон ВГС, соответствующий генотипу 1б, изоляту Con1 и содержащий мутации E1202G, T1280I и K1846T, которые усиливают репликацию РНК. Наличие в используемом репликоне репортерного гена светляковой люциферазы, экспрессия которого коррелирует с уровнем экспрессии РНК ВГС, позволяет использовать измерение уровня люминесценции в клетках как быстрый и удобный тест для измерения эффективности трансфекции. При этом в качестве положительного контроля во всех экспериментах была использована миРНК, направленная к гену люциферазы (siLuc), а в качестве отрицательного контроля - неспецифическая миРНК, направленная к гену NP вируса гриппа A H5N1.
Результаты данной серии экспериментов представлены на Фиг. 4. Для оценки эффективности трансфекции использовали 12.5 мкл композиции (что соответствует 0.5 мкг миРНК на лунку), а также эквивалентное количество контрольных миРНК K+ и K-, смешанных в соотношении 1:10 с препаратом липопептида Y14 (по аналогии с составом композиции). Результаты, полученные в серии экспериментов, представляли в виде интенсивности люминисценции люциферазы по сравнению со значением, полученным обработкой комплексами на основе неспецифической миРНК (K-), направленной к NP-гену вируса гриппа A H5N1. Результаты выражали в виде процента остаточной люциферазной активности в клетках, обработанных специфическими миРНК (композиция и K+) по сравнению с неспецифической (Фиг. 4). Наибольшую эффективность трансфекции продемонстрировала заявленная композиция. Ее применение вызывает снижение люминисценции до 20%.
Таким образом, в данной серии экспериментов была показана специфическая противовирусная активность заявленной композиции, а также продемонстрирована потенциальная возможность создания терапевтических препаратов на основе миРНК, которые могут в дальнейшем использоваться как самостоятельно, так и в комбинации с интерферонами для лечения хронической инфекции ВГС.
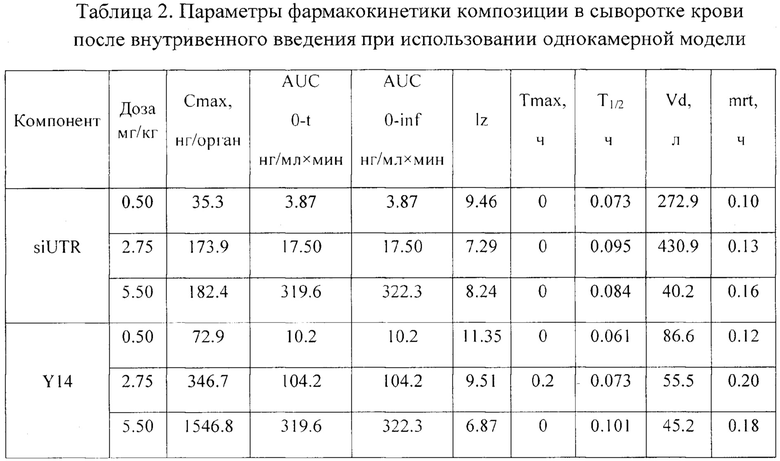
Пример 6. Фармакокинетика композиции при внутривенном и подкожном введении in vivo.
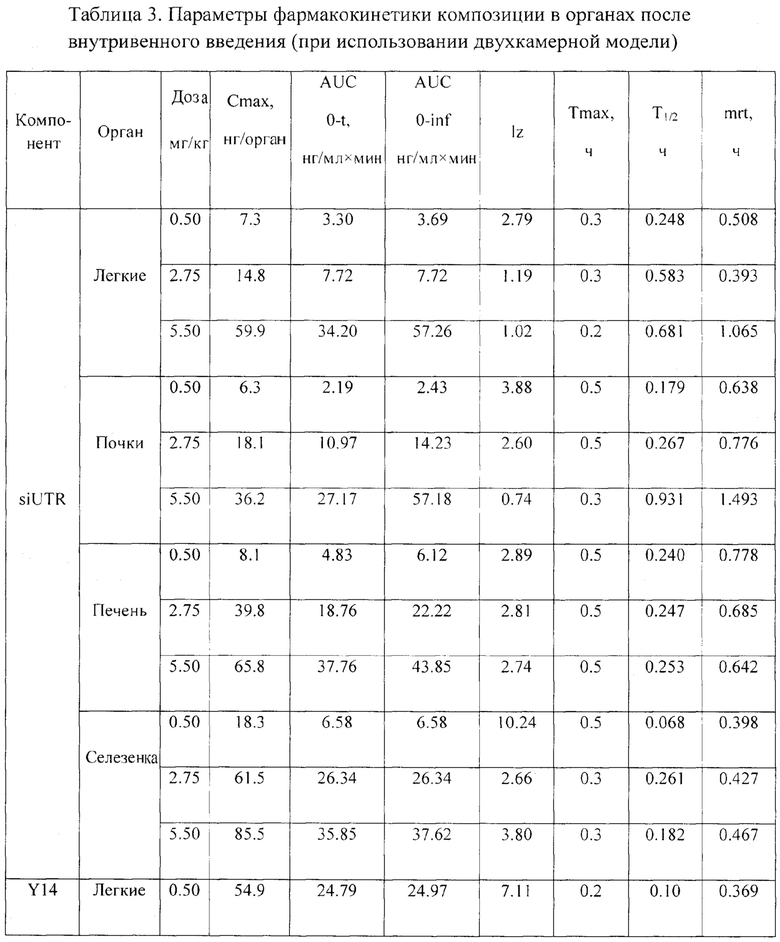
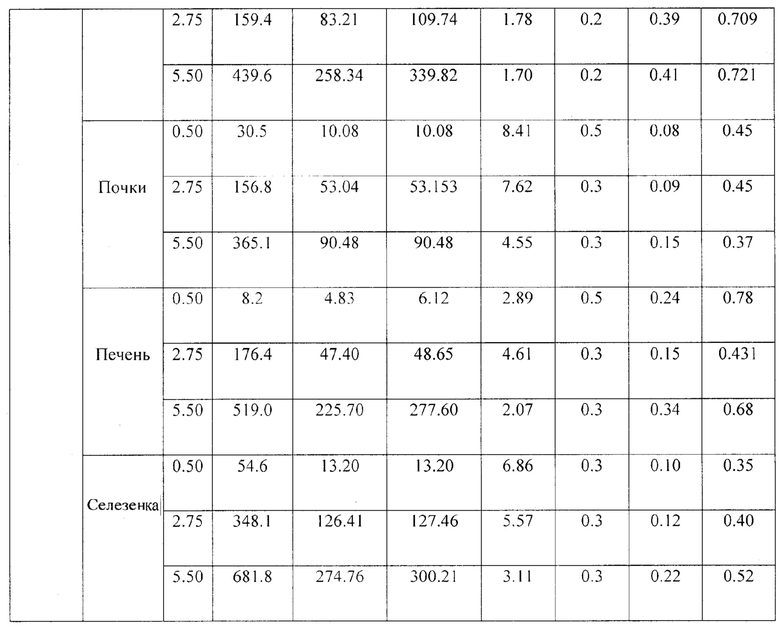
Были проведены исследования фармакокинетики заявленной композиции при внутривенном пути введения. Мышам вводили композицию в дозах 5.5 мг/кг, 2.75 мг/кг и 0.5 мг/кг, что соответствует 11, 5.5 и 1 терапевтической дозе (ТД) в объеме 0.25 мл на мышь внутривенно, и через 2, 5, 10, 20, 30, 45 и 60 мин у них отбирали кровь (не менее 300 мкл) для последующего получения сыворотки (не менее 50 мкл). Животных умерщвляли через указанные интервалы времени после введения композиции и помимо крови у них изымали внутренние органы (легкие, почки, печень, селезенку, тимус, мышца бедра и головной мозг). Органы гомогенизировали в 1.7 мл физ. р-ра, полученные гомогенаты хранили при - 20°C до анализа методом хроматографии с флуоресцентной детекцией. С применением хроматографа с флуоресцентным детектором была оценена концентрация компонентов препарата (siUTR и Y14) в выбранных биологических образцах в различные периоды времени.
В результате были получены следующие данные. Максимальная концентрация компонентов препарата в крови выявлена сразу после введения препарата (через 2 мин), а через 60 мин после введения компоненты препарата не детектировались в крови (Фиг. 5). Период полувыведения для обоих компонентов испытуемого препарата был сходен и составлял от 0.078 до 0.095 часа для siUTR и от 0.061 до 0.101 часа для Y14, что свидетельствует об устойчивости комплексов Y14/siUTR в крови (Табл. 2).
При изучении распределения компонентов заявленной композиции в органе-мишени (печени) было установлено, что Тмах для siUTR приходится на 30 мин после внутривенного введения, а Тмах для Y14 достигается несколько раньше - 20 мин. Период полувыведения siUTR из печени составляет примерно 0.25 ч и не зависит от вводимой дозы препарата, а для Y14 период полувыведения колеблется от 0.15 до 0.34 в зависимости от дозы (Фиг. 6, 7).
При изучении распределения препарата по различным органам были рассмотрены иммунные органы: тимус и селезенка, и не иммунные органы: легкие, головной мозг и мышца бедра, а также органы выведения: почки и печень. При изучении распределения компонентов препарата в органах было установлено, что оба компонента максимально накапливаются в селезенке с Тмах в среднем 20 мин. Однако выводятся siUTR и Y14 из селезенки быстрее, чем из печени: 0.17 против 0.25 ч для siUTR и 0.15 против 0.24 для Y14 (Табл. 3). Предположительно именно селезенка является тропным органом для исследуемой композиции. В другом органе иммунной системы - тимусе - не дектировались ни Y14, ни siUTR.
Следующими органами, где наблюдались высокие концентрации компонентов композиции, были печень, почки и легкие (Фиг. 7, Табл. 3). Вероятнее всего, выведение метаболитов (или пустой флуоресцентной метки) производится почками, т.к. обе метки детектируются в моче.
В мышце бедра и головном мозге компоненты препарата не детектировались. Это свидетельствует о том, что препарат не проходит гематоэнцефалический барьер и не накапливается в мышечной ткани, следовательно, не он не оказывает токсического эффекта на центральную нервную систему.
Изучалась также фармакокинетика заявленной композиции при подкожном пути введения как рекомендованном для клинического использования. Подкожно вводились три дозы препарата 5.5, 2.75 и 0.5 мг/кг, что соответствовало 11, 5,5 и 1 ТД. В результате были получены следующие данные. Максимальная концентрация компонентов препарата в крови выявлялась через 5 (для доз 5.5 и 0.5 мг/кг) и 10 мин (для дозы 2.75 мг/кг) после введения препарата, а через 60 мин после введения компоненты не детектировались в крови (Фиг. 8). Период полувыведения для обоих компонентов был сходен и составлял в среднем порядка 0.1 ч (6 мин), что свидетельствует об устойчивости комплексов Y14/siUTR в крови (Табл. 4).
Полученные данные свидетельствуют о том, что изучаемый препарат достигает органа мишени - печени. Следует отметить, что кинетика препарата характерна для большинства РНК-содержащих препаратов, что заключается в небольшом периоде выведения.
При исследовании распределения препарата по органам было установлено, что препарат более интенсивно накапливается в селезенке. В органе-мишени – печени - компоненты препарата также детектируются в значительных количествах, более того, период полувыведения компонентов исследуемого препарата из печени один из самых высоких, в сравнении с другими органами.
Также данные о накоплении компонентов препарата в почках и детекции обоих флуоресцентных меток в моче позволяет заключить, что именно почки являются основным органом выведения.
При подкожном введении наблюдается относительная высокая биодоступность (около 70%), что свидетельствует о правильно выбранном терапевтическом пути введения. Высокий кажущийся объем распределения говорит о хорошем распределении по тканям внутренних органов, что говорит о правильном выборе средства доставки. При этом относительно низкий период полувыведения говорит о том, что препарат не кумулирует в организме и, как следствие, низка вероятность передозировки.

Перечень последовательностей
<110> ФГБУ "ГНЦ Институт иммунологии" ФМБА
<120> Композиция для лечения вирусного гепатита С
<210> SEQ ID NO 1
<211> 21
<212> RNA
<213> Viral
<400> SEQUENCE 1
AAAUCUCCAGGCAUUGAGCtt
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРОГНОЗИРОВАНИЯ ФОРМИРОВАНИЯ ВИРУСОЛОГИЧЕСКОГО ОТВЕТА У БОЛЬНЫХ ХРОНИЧЕСКИМ ГЕПАТИТОМ С | 2011 |
|
RU2461834C1 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ВИРУСНОГО ГЕПАТИТА С | 2014 |
|
RU2568872C1 |
| ПРИМЕНЕНИЕ ЦИТОКИНА ИЗ СЕМЕЙСТВА ИНТЕРЛЕЙКИНА-6 ДЛЯ ПОЛУЧЕНИЯ КОМПОЗИЦИИ ДЛЯ КОМБИНИРОВАННОГО ВВЕДЕНИЯ С ИНТЕРФЕРОНОМ-АЛЬФА | 2006 |
|
RU2413529C2 |
| Способ диагностики цирроза печени (стадии F4) в исходе хронического вирусного гепатита С | 2019 |
|
RU2724595C1 |
| Способ прогноза отсутствия регресса фиброза печени у больных хроническим гепатитом С | 2019 |
|
RU2723387C1 |
| СПОСОБЫ УЛУЧШЕНИЯ ФАРМАКОКИНЕТИКИ | 2010 |
|
RU2591830C2 |
| СПОСОБ ЛЕЧЕНИЯ ВИРУСНОГО ГЕПАТИТА С | 2012 |
|
RU2496512C1 |
| ПРОТИВОВИРУСНОЕ СОЕДИНЕНИЕ МНОЖЕСТВЕННОГО ДЕЙСТВИЯ, ЕГО СОСТАВ И СПОСОБ ЛЕЧЕНИЯ ВИРУСНЫХ ЗАБОЛЕВАНИЙ | 2012 |
|
RU2597150C2 |
| ПЕГИЛИРОВАННЫЙ ИНТЕРФЕРОН ЛЯМБДА, ОБЛАДАЮЩИЙ ВЫСОКОЙ БИОДОСТУПНОСТЬЮ ПРИ ПЕРОРАЛЬНОМ ПРИМЕНЕНИИ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2017 |
|
RU2678332C1 |
| Иммунобиологическое средство для повышения клеточного ответа против вируса гепатита С | 2023 |
|
RU2811136C1 |

Изобретение относится к области медицины и генной терапии, а именно к иммунологии, и может быть использовано для лечения инфекции, вызванной вирусом гепатита С (ВГС). Композиция для лечения ВГС-инфекции посредством механизма РНКи состоит из комплексов липопептидов состава OrnGlu(С16Н33)2, выступающих в качестве носителя, и молекул миРНК, представленных 5'-AAAUCUCCAGGCAUUGAGCtt-3' (SEQ ID NO 1). Применение композиции на перевиваемой культуре клеток гепатомы человека Huh-7, экспрессирующей субгеномный репликон ВГС, вызывает достоверное снижение на 20% экспрессии вирусного репликона, обеспечивая высокую биодоступность при подкожном введении композиции. 1 з.п. ф-лы, 8 ил., 4 табл., 5 пр.,
1. Композиция для лечения ВГС-инфекции, содержащая комплексы липопептидов OrnGlu(С16Н33)2, выступающих в качестве носителя, и молекул миРНК, представленных последовательностью SEQ ID NO 1, подавляющие репликацию ВГС посредством механизма интерференции РНК.
2. Композиция по п. 1, отличающаяся тем, что размеры комплексов липопептидов с молекулами миРНК не превышают 100 нм.
| WO 2013068348 A1, 16.05.2013 | |||
| US 20120308642 А1, 06.12.2012 | |||
| СПОСОБЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ИНФЕКЦИИ ГЕПАТИТА С | 2006 |
|
RU2440822C2 |
| КОЛОСКОВА О.О | |||
| и др | |||
| Дизайн адресной липосомальной композиции // Молодой учёный, 24 (104), декабрь-2, 2015 г., найдено: https://moluch.ru/archive/104/24556/. | |||

