Настоящее изобретение касается способа получения сложных диэфиров терефталевой кислоты посредством взаимодействия терефталевой кислоты по меньшей мере с одним спиртом.
Сложные эфиры терефталевой кислоты находят применение в качестве пластификатора и характеризуются благоприятными токсикологическими свойствами.
Известно, что сложные эфиры карбоновых кислот получают в результате взаимодействия карбоновых кислот со спиртами. Эта реакция может проводиться автокаталитически или каталитически, например, при помощи кислот Бренстеда или Льюиса. Независимо от типа катализа всегда существует зависящее от температуры равновесие между исходными веществами (карбоновой кислотой и спиртом) и продуктами (сложным эфиром и водой).
Чтобы сдвинуть равновесие в сторону сложного эфира (или соответственно полного сложного эфира в случае многоосновных кислот), как правило, используют средство для азеотропной отгонки, с помощью которого из загруженной массы удаляют воду. Если одно из исходных веществ (спирт или карбоновая кислота) кипит ниже, чем образующийся сложный эфир, и образует с водой область несмешиваемости, то исходное вещество может быть использовано в качестве средства для азеотропной отгонки и после отделения воды снова подано обратно в загруженную массу. При этерификации более высокомолекулярных алифатических карбоновых кислот, ароматических карбоновых кислот или двух- или многоосновных карбоновых кислот, как правило, средством для азеотропной отгонки является используемый спирт.
Если в качестве средства для азеотропной отгонки служит используемый спирт, то обычно речь идет о том, что выпар из реактора по меньшей мере частично конденсируют, конденсат разделяют на водную фазу и органическую фазу, в основном состоящую из применяемого для этерификации спирта, и органическую фазу возвращают по меньшей мере частично в реактор.
В европейском патенте ЕР-А 1186593 описан способ получения сложных эфиров карбоновых кислот посредством реакции ди- или поликарбоновых кислот или их ангидридов со спиртами, причем реакционную воду удаляют посредством азеотропной дистилляции со спиртом. Выведенное из реакции посредством азеотропной дистилляции количество жидкости восполняют полностью или частично спиртом.
В международной заявке WO 2010/076192 А1 предлагается удалять легкокипящую фракцию из возвращаемой органической фазы, чтобы избежать ее накопления в системе реакторов.
В патенте US 7,276,621 В2 описывается способ катализируемой титанатом этерификации терефталевой кислоты 2-этилгексанолом. Через реакционную смесь пропускают инертный газ для содействия удалению воды.
В японском патенте JP 4956945 В2 также описывается способ этерификации терефталевой кислоты 2-этилгексанолом. Терефталевую кислоту при этом непрерывно или периодически подают в виде взвеси в реакционную систему. При этом добавление осуществляют с той же скоростью, с которой терефталевая кислота превращается в продукт.
В патенте US 7,799,942 В2 описывается способ получения сложных диэфиров терефталевой кислоты в реакторе при атмосферном давлении с использованием дистилляционной колонны, установленной на реактор. Дополнительно через реакционную смесь продувают инертный газ.
В международной заявке WO 2010/076193 А1 описывается способ очистки сырых сложноэфирных продуктов реакции этерификации, в котором используют металлсодержащий катализатор этерификации.
Растворимость терефталевой кислоты в высших спиртах является низкой. Например, в 2-этилгексаноле при 180°С растворяется менее 0,65% масс. терефталевой кислоты. Взаимодействие терефталевой кислоты с высшими спиртами протекает только с той частью терефталевой кислоты, которая находится в спирте в растворенном состоянии. Для достижения высоких конверсий является обязательным обеспечение постоянного перемешивания гетерогенной смеси терефталевой кислоты и спирта и эффективного подведения тепла в реакционную систему. Кроме того, важным является поддержание низкого содержания воды в реакционной смеси, чтобы имелась возможность смещать равновесие реакции в сторону продуктов, и в случае использования чувствительных к гидролизу катализаторов этерификации, предотвратить гидролиз катализатора. Дозирование твердой терефталевой кислоты в реактор с кипящим спиртом, например через подающий шнек, при котором порошок на свободном конце шнека падает в реактор в свободном падении, возможно только с трудностями из-за опасности комкования терефталевой кислоты. В случае высоких реакторов большого объема установка накопительного резервуара для терефталевой кислоты выше реактора часто связана с конструктивными сложностями.
Поэтому в основу настоящего изобретения была положена задача, заключающаяся в предоставлении способа получения сложных диэфиров терефталевой кислоты, который допускает простое внесение терефталевой кислоты в реактор, обеспечивает эффективное перемешивание реакционной смеси и эффективное подведение тепла в реакционную систему, а также позволяет достигать полной конверсии терефталевой кислоты. Другой задачей настоящего изобретения является предоставление способа, который может быть проведен в имеющихся в распоряжении реакторах для реакций этерификаций посредством незначительных доработок.
Задача решается посредством способа получения сложных диэфиров терефталевой кислоты в результате взаимодействия терефталевой кислоты по меньшей мере с одним спиртом, причем
a) терефталевую кислоту суспендируют в спирте в диспергирующем сосуде, причем получают предварительную суспензию,
b) предварительную суспензию из диспергирующего сосуда направляют в реактор и подвергают взаимодействию в присутствии катализатора этерификации, причем получают реакционную суспензию,
c) поток реакционной суспензии выводят из реактора, предпочтительно из самого глубокого места реактора, направляют через расположенный вне реактора теплообменник, предпочтительно в противоположном силе тяжести направлении, и нагревают, а нагретую реакционную суспензию возвращают в реактор, и
d) реакционную воду в виде водно-спиртового азеотропа отводят дистилляцией вместе с выпаром, выпар по меньшей мере частично конденсируют, конденсат разделяют на водную фазу и органическую фазу и органическую фазу по меньшей мере частично возвращают в реактор.
Способ согласно изобретению может быть проведен в периодическом или непрерывном режиме, однако предпочтительно проводят в периодическом режиме.
Данный способ избегает проблем, связанных с дозированием в реактор твердой терефталевой кислоты, таких как комкование терефталевой кислоты и закупоривание подающего шнека или других подающих механизмов. Данный способ предполагает получение предварительной суспензии в диспергирующем сосуде. Терефталевую кислоту дозируют в реактор не в виде твердого вещества, а в форме суспензии.
Для получения предварительной суспензии порошкообразную терефталевую кислоту суспендируют в диспергирующем сосуде в частичном количестве спирта. Для этого используют подходящее перемешивающее устройство. Так часть терефталевой кислоты может быть смешана со спиртом при помощи мешалки, в качестве альтернативы может быть использован диспергирующий насос. При этом, например, все количество терефталевой кислоты может быть суспендировано на одной стадии или терефталевую кислоту суспендируют порционно в течение способа. Для порционного суспендирования терефталевая кислота может быть добавлена, например, при помощи подающего шнека в диспергирующий сосуд.
Смешивание может быть осуществлено однако также и в закрытой камере в результате взаимодействия вращающегося ротора и статора, причем непрерывно смешивают между собой в каждом случае только инкрементное количество компонентов, и после этого из камеры выходит суспензия.
В качестве спирта для приготовления предварительной суспензии может быть использован свежий спирт и/или возвращенный спирт, т.е. органическая фаза, которая получена после конденсации выпара и разделения конденсата на фазы.
Диспергирующий сосуд в большинстве случаев состоит из металлических материалов, причем предпочтительной является высококачественная сталь. Диспергирующий сосуд может быть соединен с реактором с возможностью прохождения газа.
Предварительную суспензию направляют в реактор с использованием насоса или под действием силы тяжести. В качестве насосов в принципе могут быть использованы все известные специалисту в данной области подающие насосы, которые в данном случае могут быть рассмотрены в качестве пригодных при принятии во внимание свойств подаваемой суспензии. Предпочтительно в качестве подающих насосов могут быть использованы лопастные, поршневые, шнековые, пластинчатые или рукавные насосы. Дозирование предварительной суспензии в реактор может быть осуществлено порционно или непрерывно. Предпочтительно дозирование осуществляют непрерывно. Предварительная суспензия в принципе может быть подана в любое место реактора, предпочтительно однако предварительную суспензию подают в верхнюю часть реактора, в частности выше уровня жидкости в реакторе. Таким образом может быть максимально предотвращен возвратный поток против направления дозирования.
В случае реактора речь может идти о любом реакторе, который подходит для проведения химических взаимодействий в жидкой фазе. В качестве реакторов подходят реакторы без обратного перемешивания, такие как трубчатые реакторы или приемные резервуары, снабженные встроенными элементами, однако предпочтительны реакторы с обратным перемешиванием, такие как аппараты с мешалкой, реакторы с внутренним контуром циркуляции, струйные реакторы с внутренним контуром циркуляции или реакторы со струйным смешиванием. При необходимости несколько реакторов также могут быть объединены в одну многоступенчатую установку. Такими реакторами являются, например, реакторы с внутренним контуром циркуляции со встроенным сетчатым дном, соединенные в каскад резервуары, трубчатые реакторы с промежуточной подачей или колонны с перемешивающим устройством.
Предпочтительно применяют реакционный аппарат с мешалкой. Реакционные аппараты с мешалкой в большинстве случаев состоят из металлических материалов, причем предпочтительной является высококачественная сталь.
В частности, предпочтительным является применение существующих реакционных систем, которые, например, применяют для этерификации фталевого ангидрида, и в результате незначительного переоборудования могут быть использованы для этерификации терефталевой кислоты. Необходимое переоборудование касается в основном установки диспергирующего сосуда.
В реакторе предварительную суспензию и катализатор этерификации приводят в контакт, причем получают реакционную суспензию. В одном из вариантов осуществления способа при этом i) в незаполненный реактор направляют предварительную суспензию, ii) нагревают предварительную суспензию до кипения и iii) добавляют катализатор этерификации. При необходимости последовательность стадий ii) и iii) может быть обратной.
В одном предпочтительном варианте осуществления способа катализатор этерификации находится в реакторе в частичном количестве спирта, например 15-50% общего количества спирта, предпочтительно 25-40%. Смесь катализатора со спиртом сначала нагревают до кипения, а затем начинают дозирование предварительной суспензии. В качестве альтернативы предварительную суспензию добавляют к смеси катализатора со спиртом и затем нагревают. При необходимости нагрев смеси катализатора со спиртом и дозирование предварительной суспензии можно проводить параллельно.
В течение взаимодействия реакционная суспензия в реакторе характеризуется температурой, близкой к температуре кипения реакционной смеси, например температурой от 150°С до 250°С, предпочтительно от 185°С до 220°С. Температура кипения реакционной суспензии зависит от соотношения сложного диэфира терефталевой кислоты к спирту и повышается в ходе реакции.
Подведение тепла к реакционной системе согласно изобретению осуществляют посредством направления реакционной суспензии через установленный вне реактора теплообменник
При этом реакционную суспензию с применением насоса выводят из реактора и направляют через теплообменник. В качестве альтернативы подачу реакционной суспензии через теплообменник осуществляют посредством естественной циркуляции. Предпочтительно реакционную суспензию направляют в противоположном силе тяжести направлении через теплообменник. Насос соединен с теплообменником с возможностью сообщения текучей средой. Реакционную суспензию предпочтительно выводят в самом глубоком месте реактора. При этом реактор выполнен таким образом, что реакционную суспензию выводят в геодезически самом глубоком месте и в реакторе отсутствуют застойные зоны, обусловленные локально самыми глубокими местами. Насос для выведения реакционной суспензии может быть расположен в принципе в различных местах вне реактора. Например, насос располагают в геодезически самом глубоком месте циркуляционного контура, состоящего из реактора, насоса и соединительных линий.
В качестве насоса пригодными являются в принципе все известные специалисту в данной области подающие насосы, которые могут быть использованы для проведения способа согласно изобретению и считаются подходящими при принятии во внимание свойств подаваемой реакционной суспензии. Предпочтительно в качестве подающих насосов могут быть использованы лопастные, поршневые, шнековые, пластинчатые или рукавные насосы. Особо предпочтительными являются аксиальные и радиальные лопастные насосы. Теплообменник для возвращения нагретой реакционной суспензии в реактор соединен с реактором с возможностью сообщения текучей средой.
В результате выведения реакционной суспензии в самом глубоком месте реактора избегают осаждения терефталевой кислоты. Терефталевую кислоту в виде суспензии в спирте выводят из нижней области реактора и направляют через теплообменник. После повторного дозирования в реактор терефталевая кислота находится также в суспендированном в спирте состоянии и готова к взаимодействию.
Возврат реакционной суспензии из теплообменника в реактор в принципе может быть осуществлен в любое место реактора, целесообразно однако осуществлять возврат в верхней области реактора, например на высоте уровня жидкости реакционной суспензии или в области от высоты уровня жидкости реакционной суспензии до 30% ниже. Объемную скорость потока, который пропускают через нагревательное устройство, выбирают таким образом, что перекачивание содержимого реактора полностью осуществляют в период времени от 1 до 60 минут, предпочтительно от 1 до 10 минут. В результате постоянного перекачивания содержимого реактора обеспечивается эффективное перемешивание реакционной суспензии.
Предпочтительно поток реакционной суспензии пропускают через расположенный вне реактора теплообменник в противоположном силе тяжести направлении, т.е. снизу вверх. В результате указанного направления потока, противоположного силе тяжести, предотвращают осаждение терефталевой кислоты в теплообменнике.
Реакционную суспензию посредством направления через теплообменник нагревают до температуры, при которой на поверхности реакционной смеси образуется достаточно большой поток выпара, чтобы выводить реакционную воду, например до температуры от 150 до 250°С, предпочтительно от 180 до 220°С.
При необходимости перемешивание реакционной суспензии может быть поддержано посредством введения инертного газа в реактор, в частности в самом нижнем месте реактора, и/или в поток реакционной суспензии. Введение инертного газа, в частности, при перебоях в работе насоса для выведения реакционной суспензии, например при выходе насоса из строя, способствует предотвращению осаждения терефталевой кислоты на дне реактора и/или в трубопроводах. Предпочтительно введение инертного газа осуществляют на стороне всасывания насоса. В качестве альтернативы введение может быть осуществлено на стороне нагнетания насоса. Это способствует образованию циркуляции через теплообменник. Инертными газами являются все газы, которые в реакционных условиях не обладают реакционной способностью по отношению к компонентам реакционной суспензии, в частности азот или аргон. Предпочтительно инертный газ вводят в количестве от 0,01 до 5 объемных единиц инертного газа на объемную единицу реакционной суспензии в час.
Во время реакции водно-спиртовой азеотроп вместе с выпаром отделяют дистиляцией, выпар по меньшей мере частично конденсируют, конденсат разделяют на водную фазу и органическую фазу, и органическую фазу по меньшей мере частично возвращают в реактор.
Для конденсации или соответственно частичной конденсации выпара могут быть использованы все подходящие конденсаторы. Эти конденсаторы могут охлаждаться с помощью любых охлаждающих сред. Конденсаторы с воздушным охлаждением и/или водяным охлаждением являются предпочтительными, причем воздушное охлаждение является особенно предпочтительным.
Полученный конденсат подвергают разделению на водную фазу и органическую фазу. Обычно для этого конденсат подают в фазовый сепаратор (декантер), где он в результате механического оседания распадается на две фазы, которые могут быть выведены по отдельности. Водная фаза отделяется и может, при необходимости после обработки, выбрасываться или применяться в качестве воды для отгонки легких фракций при последующей обработке сложного эфира.
Органическую фазу направляют предпочтительно через колонну (так называемую колонну возвратного спирта) обратно в реактор, в которой органическую фазу направляют противотоком к по меньшей мере части выпара. В случае колонны возвратного спирта речь может идти, например, о тарельчатой колонне, насадочной колонне или колонне с насадочными телами. Небольшого числа ступеней разделения, как правило, достаточно.
Подходящей является, например, колонна, имеющая от 2 до 10 теоретических ступеней разделения. Предпочтительно колонну устанавливают на головной части реактора, то есть в непосредственном соединении с реактором. В целесообразном варианте органическую фазу вводят в колонну возвратного спирта в головной части или в верхней области. Стекающий конденсат из колонны возвратного спирта поступает обратно в реактор. Подача органической фазы обратно через колонну возвратного спирта обладает тем преимуществом, что возвращаемая органическая фаза предварительно подогревается и освобождается от следов воды, которые остаются в органической фазе после разделения фаз или соответственно растворены в органической фазе в соответствии со своей термодинамической растворимостью. Содержание воды в возвращаемой органической фазе составляет значение, меньшее, чем максимальная растворимость воды в спирте, предпочтительно менее 3% масс., в частности менее 0,5% масс.
В способе согласно изобретению используют линейные, разветвленные или циклические алифатические спирты с 4-18 атомами углерода, в частности 8-14 атомами углерода, или ароматические спирты. Спирты представляют собой моноолы и/или полиолы и могут быть третичными, вторичными и первичными.
Используемые спирты могут быть получены от различных источников. Подходящими исходными веществами являются спирты жирного ряда, спирты из процесса ALFOL или спирты или спиртовые смеси, которые получены в результате гидрирования насыщенных или ненасыщенных альдегидов, в частности таких, синтез которых включает стадию гидроформилирования.
Алифатическими спиртами, которые используют в способе согласно изобретению, являются, например, н-бутанол, изобутанол, пентанолы, гексанолы, гептанолы, октанолы, такие как н-октанол, 2-этилгексанол, нонанолы, дециловые спирты или тридеканолы, полученные в результате гидроформилирования или альдольной конденсации и последующего гидрирования. Спирты могут быть использованы в виде чистых веществ, в виде смесей изомерных соединений или в виде смесей соединений с различным числом атомов углерода. Примером спиртовой смеси такого рода является смесь спиртов C9/С11.
Ароматическими спиртами, которые могут быть использованы в способе согласно изобретению, являются, например, фенол, бензиловый спирт, 1-нафтол, 2-нафтол, 1,2-дигидроксибензол, 1,3-дигидроксибензол, 1,4-дигидроксибензол, 1,4-нафтогидрохинон, 2,4,6-тринитрофенол, первичный фенилэтиловый спирт, вторичный фенилэтиловый спирт, фенилпропиловый спирт, о-толиловый спирт, п-толиловый спирт, куминовый спирт, п-нитрофенол, м-, о- или п-алкилфенол, например, м-, о- или п-метилфенол или м-, о- или п-этилфенол, м-, о- или п-галогенфенол, например м-, о- или п-хлорфенол или м-, о- или п-бромфенол. Кроме того, могут быть использованы п-нитробензиловый спирт, м-, о- или п-алкилбензиловый спирт, например м-, о- или п-метилбензиловый спирт или м-, о- или п-этилбензиловый спирт, м-, о- или п-галогенбензиловый спирт, например м-, о- или п-хлорбензиловый спирт или м-, о- или п-бромбензиловый спирт, 2-этоксифенол, 3-этоксифенол, 4-этоксифенол, 2-пропоксифенол, 3-пропоксифенол, 4-пропоксифенол, 2-этоксибензиловый спирт, 3-этоксибензиловый спирт, 4-этоксибензиловый спирт, 2-пропоксибензиловый спирт, 3-пропоксибензиловый спирт или 4-пропоксибензиловый спирт.
Полиолами, которые могут быть использованы в способе согласно изобретению, являются, например, 1,2-пропандиол, 1,3-пропандиол, 1,2-бутандиол, 1,3-бутандиол, 1,4-бутандиол, неопентилгликоль, 1,5-пентандиол, 1,6-гександиол, 1,10-декандиол, диэтиленгликоль, 2,2,4-триметилпентан-1,5-диол, 2,2-диметилпропан-1,3-диол, 1,4-диметилолциклогексан, 1,6-диметилолциклогексан, глицерин, триметилолпропан, эритрит, пентаэритрит и сорбит.
Особо предпочтительными спиртами являются 2-этилгексанол, 2-пропилгептанол, изомерная смесь изононанолов, изомерная смесь деканолов и смесь спиртов С9/С11.
Подвергаемый взаимодействию спирт, который служит средством для азеотропной отгонки, может быть использован в стехиометрическом избытке. Предпочтительно количество используемого спирта выбирают таким образом, чтобы в неочищенном продукте взаимодействия оставалось от 10 до 35% масс. спирта в пересчете на теоретическую полную конверсию терефталевой кислоты.
Этерификацию согласно изобретению проводят в присутствии катализатора этерификации.
В предпочтительном варианте осуществления способа согласно изобретению катализатор этерификации растворим в спирте.
В подходящем варианте катализатор этерификации выбирают из кислот Льюиса, таких как алкоголяты, карбоксилаты и хелатные соединения титана, циркония, гафния, олова, алюминия и цинка, бортрифторид, эфиратов бортрифторида, минеральных кислот, таких как серная кислота, фосфорная кислота, сульфоновых кислот, таких как метансульфоновая кислота и толуолсульфоновая кислота, и ионных текучих сред.
В подходящем варианте катализатор этерификации выбирают из алкоголятов, карбоксилатов и хелатных соединений титана, циркония, гафния, олова, алюминия и цинка. Пригодными являются тетраалкилтитанаты, такие как тетраметилтитанат, тетраэтилтитанат, тетра-н-пропилтитанат, тетра-изопропилтитанат, тетра-н-бутилтитанат, тетра-изобутилтитанат, тетра-втор-бутилтитанат, тетраоктилтитанат, тетра-(2-этилгексил)титанат, диалкилтитанаты ((RO)2TiO, в которых R означает изо-пропил, н-бутил, изо-бутил, такие как изопропил-н-бутил, хелаты титанацетилацетоната, такие как диизопропокси-бис(ацетилацетонат)титанат, диизопропокси-бис(этилацетилацетонат)титанат, ди-н-бутил-бис(ацетилацетонат)титанат, ди-н-бутил-бис(этилацетоацетат)титанат, три-изопропоксид-бис(ацетилацетонат)титанат, цирконий тетраалкилаты, такие как цирконий тетраэтилат, цирконий тетрабутилат, цирконий тетрабутират, цирконий тетрапропилат, цирконий карбоксилаты, такие как цирконий диацетат, хелаты цирконий ацетилацетоната, такие как цирконий тетра(ацетилацетонат), трибутоксицирконий ацетилацетонат, дибутоксицирконий (бис-ацетилацетонат), алюминий трисалкилаты, такие как алюминий триизопропилат, алюминий трисбутилат, хелаты алюминий ацетилацетоната, такие как алюминий трис(ацетилацетонат) и алюминий трис(этил-ацетилацетонат). В частности, используют изопропил-н-бутил титанат, тетра(изопропил)орто-титанат или тетра(бутил)орто-титанат или их смеси.
Пригодными ионными текучими средами (ионными жидкостями) являются, например, трифлат метилимидазолийбутансульфоновой кислоты и гидросульфат 1-этил-3-метилимидазолия.
Концентрация катализатора зависит от типа катализатора. В случае предпочтительно используемых соединений титана она составляет от 0,001 до 1,0% мол. в пересчете на количество терефталевой кислоты, в частности от 0,01 до 0,2% мол.
Температура реакции находится в диапазоне от 150°С до 250°С. Оптимальные температуры зависят от исходных веществ, протекания реакции и концентрации катализатора. Для каждого отдельного случая они могут быть легко определены при помощи эксперимента. Более высокие температуры повышают скорость реакции и способствуют протеканию побочных реакций, как, например, образование олефинов или образование окрашенных побочных продуктов. Для удаления реакционной воды необходимо, чтобы спирт можно было выводить из реакционной смеси дистилляцией.
Желаемая температура или желаемый температурный диапазон может быть установлен при помощи давления в реакторе. Поэтому в случае низкокипящих спиртов взаимодействие может быть проведено при избыточном давлении, а в случае более высоко кипящих спиртов при пониженном давлении. Например, в случае взаимодействия терефталевой кислоты с 2-этилгексанолом процесс ведут при температуре в диапазоне от 180°С до 220°С и давлении в диапазоне от 300 мбар до 2 бар.
В целесообразном варианте реактор и диспергирующий сосуд эксплуатируют при практически одинаковом давлении, в частности примерно атмосферном давлении. При необходимости реактор и диспергирующий сосуд можно эксплуатировать также при разных давлениях.
Предпочтительно способ согласно изобретению проводят до практически полной конверсии терефталевой кислоты. Определение конверсии можно осуществлять посредством измерения кислотного числа реакционной суспензии. Кислотное число измеряют посредством нейтрализации образца реакционной суспензии гидроксидом тетрабутиламмония. Через израсходованную при нейтрализации массу гидроксида тетрабутиламмония можно определить количество вещества израсходованного гидроксида тетрабутиламмония и через стехиометрическое соотношение количество вещества свободных кислотных групп не вступившей в реакцию терефталевой кислоты. Исходя из известного количества вещества использованной терефталевой кислоты, можно таким образом рассчитать конверсию. Дополнительной возможностью для определения конверсии является ВЭЖХ-анализ и измерение помутнения реакционной суспензии в результате встроенного измерения помутнения. В способе согласно изобретению предпочтительно достигают конверсии более 99%.
После окончания реакции реакционная смесь, которая в основном состоит из желаемого сложного эфира и избыточного спирта, помимо катализатора и/или продуктов его превращения, содержит незначительные количества карбоновой кислоты со сложноэфирными группами и/или не вступившую в реакцию карбоновую кислоту.
Для обработки этой неочищенной сложноэфирной смеси неочищенный ди(С4-С18-алкил)терефталат смешивают с водным основанием, из полученной смеси испаряют воду, полученную жидкую фазу смешивают с водой с образованием эмульсии вода-в-масле, из этой эмульсии отделяют дистилляцией воду и фильтруют ди(С4-С18-алкил)терефталат.
Сначала катализатор этерификации в результате добавления водного основания дезактивируется и выпадает в осадок. Одновременно не вступившая в реакцию этерификации кислота или неполные сложные эфиры кислоты превращаются в соли.
Добавление водного основания может быть осуществлено любым подходящим образом. Его осуществляют предпочтительно ниже поверхности жидкости неочищенного эфира. Для этого подходят, например, перфорированные трубки или форсунки, которые установлены на дне устройства или стенке устройства. Смесь затем интенсивно перемешивают, например, при помощи мешалки или циркуляционного насоса.
Добавляемое количество водного основания определяют таким образом, чтобы обеспечивалась полная нейтрализация кислотных компонентов неочищенного сложного эфира. На практике используют более или менее значительный избыток основания. Общее количество кислотных компонентов неочищенного сложного эфира целесообразно определять при помощи кислотного числа (в мг КОН/г). Предпочтительно при помощи водного основания вносить от 100 до 300%, в частности от 130 до 220% эквивалентов нейтрализации, в пересчете на кислотное число неочищенного сложного эфира. При этом под эквивалентом нейтрализации понимают количество основания, которое может образовывать такое же количество протонов, как и 1 мг КОН. Другими словами, используют избыток основания до 200%, предпочтительно от 30 до 120%.
В качестве водного основания пригодными являются растворы гидроксидов, карбонатов, гидрокарбонатов щелочных и щелочно-земельных металлов. Водные растворы гидроксидов щелочных металлов являются в основном предпочтительными. Водный раствор гидроксида натрия по причине своей легкой доступности является особо предпочтительным.
Концентрация водного основания не является критичной, однако при использовании концентрированных щелочных растворов в месте ввода основания это может привести к гидролизу сложного эфира. С другой стороны, концентрация водного основания не должна быть слишком низкой, так как внесенную с водным основанием воду на последующих стадиях необходимо опять удалять. Поэтому предпочтительными являются водные основания с концентрацией от умеренной до низкой, например таковые с концентрацией от 0,5 до 25% масс., в частности от 1 до 10% масс. Водный раствор гидроксида натрия с концентрацией от 1 до 5% масс. является особо предпочтительным.
Часто выпадающее в осадок твердое вещество, которое состоит из продуктов разрушения катализатора и солей не вступившей в реакцию кислоты или соответственно неполных сложных эфиров, находится в тонкодисперсной, тяжело фильтруемой форме. В целесообразном варианте мелкие частицы агломерируют в более большие, легко отделимые частицы.
Для этого жидкую фазу смешивают с водой с образованием эмульсии вода-в-масле. Вода распределяется в качестве дисперсной фазы в форме мелких капелек в жидкой органической фазе. Мелкие частицы твердого вещества перемещаются на границу разделения фаз между водными капельками и объединенной органической фазой. При последующем испарении воды мелкие частицы агломерируются и образуют крупные, хорошо отделимые частицы.
Чтобы образовывалась отдельная водная фаза, количество добавляемой воды должно быть больше, чем соответствующая растворимость воды в органической фазе. Растворимость воды в органической фазе зависит помимо прочего от содержания не вступившего в реакцию спирта, так как спирт действует в качестве способствующего растворению средства. Чем выше содержание спирта, тем больше воды необходимо добавить для образования эмульсии. В случае обычного содержания остаточного спирта от 20 до 30% масс. подходящими в основном количествами являются от 10 до 80 г воды, предпочтительно от 30 до 60 г, в пересчете на 1 кг неочищенного сложного эфира.
Водную фазу распределяют на мелкие капельки при помощи мешалки или гомогенизатора или в результате перекачивания эмульсии с использованием циркуляционного насоса. Полученные капельки воды предпочтительно имеют средний размер капель менее 1000 мкм. В качестве мешалки с высокой относительной эффективностью перемешивания пригодны, например, дисковые мешалки. В качестве альтернативы особенно при непрерывной методике проведения способа можно применять смесительную форсунку, в случае которой через диспергирующий клапан воду подают непосредственно в поток неочищенного сложного эфира.
Образование эмульсии осуществляют предпочтительно при примерно нормальном давлении.
Из полученной таким образом эмульсии на следующей стадии воду опять отделяют дистилляцией.
После указанной обработки твердое вещество находится в хорошо фильтруемой форме, и при фильтрации мелкая фракция не просачивается. Для фильтрования сложного эфира подходят все пригодные фильтры, такие как камерные фильтр-прессы, ленточный фильтр, патронный фильтр или тарельчатый фильтр. Для непрерывного проведения способа особо подходят тарельчатые фильтры с центробежным выведением осадка. Отделенное твердое вещество выбрасывают.
После фильтрации сложный эфир может быть подвергнут различной дополнительной обработке, как, например, отгонке с водяным паром или тому подобному.
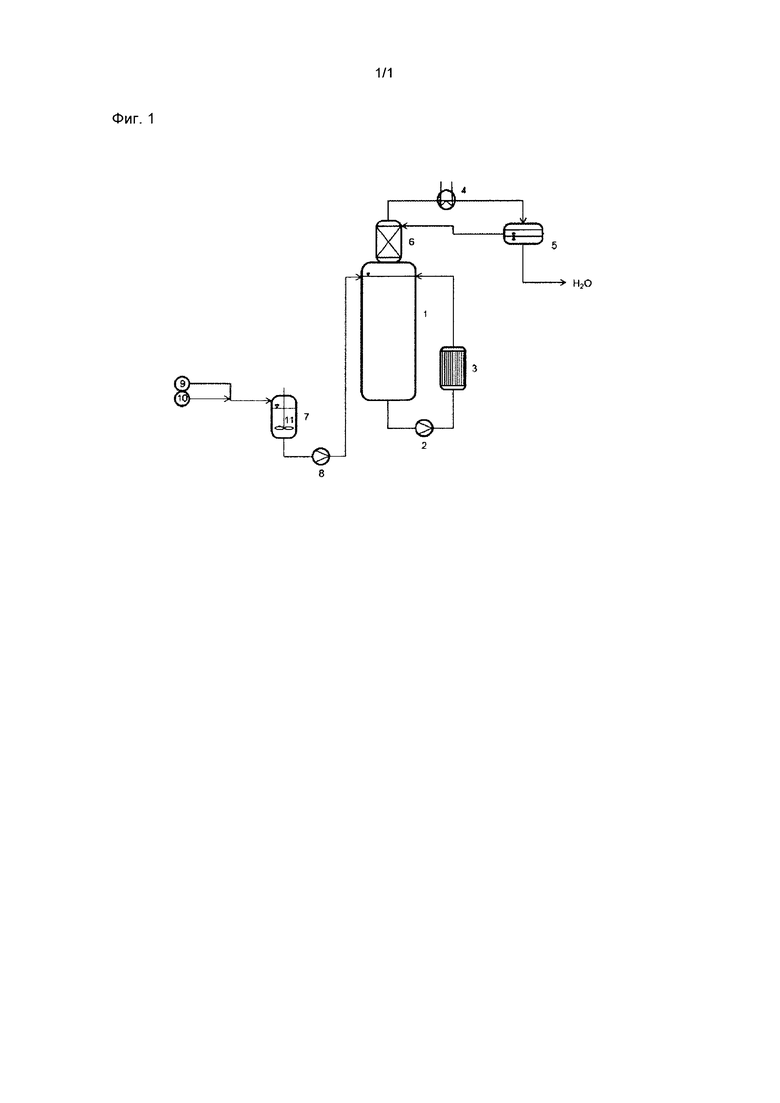
Настоящее изобретение более подробно разъясняется посредством прилагаемой фигуры.
Фигура 1 демонстрирует пригодную для проведения способа согласно изобретению установку.
Согласно фигуре 1 в диспергирующий сосуд 7 подают спирт из резервуара 9 и терефталевую кислоту из резервуара 10 и с применением мешалки 11 смешивают в предварительную суспензию. Предварительную суспензию при помощи насоса 8 направляют в верхнюю область реактора 1. В реакторе 1 находится другая часть спирта и катализатор этерификации. Предпочтительно в самом глубоком месте реактора 1 реакционную суспензию с применением насоса 2, установленного вне реактора, выводят из реактора 1 и пропускают через расположенный вне реактора теплообменник. Нагретую в теплообменнике 3 реакционную суспензию опять возвращают в реактор в его верхней области. Выпар проходит через колонну 6 и по меньшей мере частично конденсируется в конденсаторе 4. В фазовом сепараторе 5 конденсат разделяют на водную и органическую фазу. Водную фазу выбрасывают, а органическую фазу возвращают через колонну 6 в реактор.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ДИЭФИРОВ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ С ПЕРЕКАЧИВАНИЕМ РЕАКЦИОННОЙ СМЕСИ | 2015 |
|
RU2668964C1 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ДИЭФИРОВ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ С ОБОГАЩЕНИЕМ ВОЗВРАТНЫМ СПИРТОМ | 2015 |
|
RU2665579C1 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ДИЭФИРОВ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ С ОБЕЗВОЖИВАНИЕМ ВОЗВРАТНОГО СПИРТА | 2015 |
|
RU2668223C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЦЕТОНЦИАНГИДРИНА И ЕГО ПРОИЗВОДНЫХ ПРОДУКТОВ ПУТЕМ ЦЕЛЕНАПРАВЛЕННОГО ОХЛАЖДЕНИЯ | 2007 |
|
RU2491272C2 |
| КАТАЛИЗАТОР И СПОСОБ | 2003 |
|
RU2316396C2 |
| ДИСТИЛЛЯЦИОННАЯ ОБРАБОТКА АЦЕТОНЦИАНГИДРИНА И СПОСОБ ПОЛУЧЕНИЯ АЛКИЛОВЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ И ПРОИЗВОДНЫХ ПРОДУКТОВ | 2007 |
|
RU2495868C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ КАРБОНОВЫХ КИСЛОТ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПЛАСТИФИКАТОРОВ | 2014 |
|
RU2636586C1 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ СЛОЖНЫХ АЛКИЛОВЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ | 2007 |
|
RU2486173C2 |
| СПОСОБ СИНТЕЗА БИОРЕСУРСНЫХ СЛОЖНЫХ ЭФИРОВ АКРИЛОВОЙ КИСЛОТЫ | 2009 |
|
RU2514422C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЦЕТОНЦИАНГИДРИНА | 2007 |
|
RU2497805C2 |
Настоящее изобретение касается способа получения сложного диэфира терефталевой кислоты посредством взаимодействия терефталевой кислоты по меньшей мере с одним спиртом, причем a) терефталевую кислоту суспендируют в спирте в диспергирующем сосуде с получением предварительной суспензии, b) предварительную суспензию из диспергирующего сосуда направляют в реактор и подвергают взаимодействию в присутствии катализатора этерификации с получением реакционной суспензии, c) поток реакционной суспензии выводят из реактора, направляют через расположенный вне реактора теплообменник и нагревают, а нагретую реакционную суспензию возвращают в реактор, и d) реакционную воду в виде водно-спиртового азеотропа отводят дистилляцией вместе с выпаром, выпар по меньшей мере частично конденсируют, конденсат разделяют на водную фазу и органическую фазу и органическую фазу по меньшей мере частично возвращают в реактор. Способ допускает простое внесение терефталевой кислоты в реактор, обеспечивает эффективные перемешивание реакционной смеси и подведение тепла. 14 з.п. ф-лы, 1 ил.
1. Способ получения сложного диэфира терефталевой кислоты посредством взаимодействия терефталевой кислоты по меньшей мере с одним спиртом, причем
a) терефталевую кислоту суспендируют в спирте в диспергирующем сосуде с получением предварительной суспензии,
b) предварительную суспензию из диспергирующего сосуда направляют в реактор и подвергают взаимодействию в присутствии катализатора этерификации с получением реакционной суспензии,
c) поток реакционной суспензии выводят из реактора, направляют через расположенный вне реактора теплообменник и нагревают, а нагретую реакционную суспензию возвращают в реактор, и
d) реакционную воду в виде водно-спиртового азеотропа отводят дистилляцией вместе с выпаром, выпар по меньшей мере частично конденсируют, конденсат разделяют на водную фазу и органическую фазу и органическую фазу по меньшей мере частично возвращают в реактор.
2. Способ по п. 1, причем поток реакционной суспензии выводят из самого глубокого места реактора.
3. Способ по п. 1, причем органическую фазу перед возвратом в реактор направляют через колонну, в которой органическую фазу направляют противотоком к по меньшей мере части выпара.
4. Способ по п. 1, причем возвращенная в реактор органическая фаза имеет содержание воды, которое меньше, чем растворимость воды в спирте.
5. Способ по п. 1, причем возвращенная в реактор органическая фаза имеет содержание воды менее 3% масс.
6. Способ по п. 1, причем катализатор этерификации выбирают из кислот Льюиса, минеральных кислот, сульфоновых кислот и ионных текучих сред.
7. Способ по п. 6, причем катализатор этерификации выбирают из алкоголятов, карбоксилатов и хелатных соединений титана, циркония, гафния, олова, алюминия и цинка, бортрифторида, эфиратов бортрифторида, серной кислоты, фосфорной кислоты, метансульфоновой кислоты и толуолсульфоновой кислоты.
8. Способ по любому из пп. 1-5, причем катализатор этерификации выбирают из кислых ионообменных материалов, цеолитов, оксидов и/или гидроксидов магния, алюминия, цинка, титана, кремния, олова, свинца, сурьмы, висмута, молибдена и марганца.
9. Способ по п. 1, причем катализатор этерификации растворим в спирте.
10. Способ по п. 1, причем спирт выбирают из линейных, разветвленных и циклических алифатических спиртов с 4-18 атомами углерода и ароматических спиртов.
11. Способ по п. 1, причем спирт используют в таком стехиометрическом избытке, что неочищенный продукт этерификации содержит от 15 до 35% масс. спирта.
12. Способ по п. 1, причем для флюидизации в реактор и/или поток реакционной суспензии дозируют инертный газ.
13. Способ по п. 12, причем инертный газ дозируют в количестве от 0,1 до 5 объемных единиц инертного газа на объемную единицу реакционной суспензии в час.
14. Способ по п. 1, причем неочищенный сложный диэфир терефталевой кислоты обрабатывают посредством смешивания с водным основанием, испарения из полученной смеси воды, смешивания полученной жидкой фазы с водой с образованием эмульсии вода-в-масле, отделения дистилляцией воды из указанной эмульсии и фильтрации сложного диэфира терефталевой кислоты.
15. Способ по п. 1, причем реакционную суспензию пропускают через теплообменник при объемной скорости потока, выбранной таким образом, что перекачивание содержимого реактора полностью осуществляют в период времени от 1 до 10 минут.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕСИ КАРБОНОВАЯ КИСЛОТА/ДИОЛ, ПОДХОДЯЩЕЙ ДЛЯ ПРИМЕНЕНИЯ ПРИ ПРОИЗВОДСТВЕ СЛОЖНЫХ ПОЛИЭФИРОВ | 2003 |
|
RU2330014C2 |

