Область техники, к которой относится изобретение
Фармацевтическая комбинация, включающая (a) ингибитор фосфатидилинозитол-3-киназы (PI3K) соединение формулы (I), определение которого приведено в данном описании, или его фармацевтически приемлемую соль и (b) паклитаксел или его фармацевтически приемлемую соль, для одновременного, раздельного или последовательного применения, с целью лечения рака головы и шеи; фармацевтическая композиция, содержащая указанную комбинацию; применение указанной комбинации для приготовления лекарственного средства, предназначенного для лечения рака головы и шеи; способ лечения или предотвращения рака головы и шеи, включающий введение совместно терапевтически эффективного количества подобной комбинации нуждающемуся в этом субъекту; и промышленная упаковка для нее.
Уровень техники
Рак головы и шеи включает все виды рака, связанные с верхними отделами желудочно-кишечного тракта и дыхательных путей. Плоскоклеточные карциномы, возникающие на слизистых поверхностях, составляют более 90% случаев заболеваний. Частота возникновения заболеваний, вызванных плоскоклеточным раком головы и шеи (HNSCC), постоянно растает в течение последних трех десятилетий. Это пятая основная причина возникновения онкологических заболеваний и шестая основная причина смертности от рака в мире. Методы терапии HNSCC включают хирургию, лучевую терапию и химиотерапию. В случае HNSCC в прогрессирующей стадии лишь от 35% до 55% пациентов выживают и не имеют признаков заболевания в течение трех лет, несмотря на агрессивное лечение. Локальный рецидив опухоли развивается у 30%-40% пациентов, а отдаленные метастазы развиваются у 12%-22% пациентов. Паллиативное лечение рецидивирующего/метастатического HNSCC остается в значительной степени неэффективным, и достигнут незначительный прогресс.
Хотя HNSCC можно считать химиочувствительным заболеванием, о чем свидетельствует степень реагирования на агрессивные индукционные методы лечения (например, сочетание 5-FU, цисплатина и доцетаксела), результаты при рецидиве заболевания плохие. Несмотря на прогресс в первичном лечении путем комбинирования химиотерапии, хирургии, лучевой терапии и поддерживающего лечения, частота рецидивов составляет от 35-50%. У пациентов рецидив, как правило, возникает локально, и развиваются такие симптомы, как трудности при глотании, в процессе приема пищи и при разговоре. Медиана выживаемости пациентов с рецидивирующим заболеванием составляет шесть месяцев и может достигать 10 месяцев у пациентов с хорошим общим статусом. Таким образом, улучшение клинического результата для пациентов, страдающих от рака головы и шеи, важно для улучшения качества жизни пациента.
Сейчас необходимы более эффективные средства и средства направленного действия для лечения рака головы и шеи, в особенности HNSCC.
Авторы настоящего изобретения полагают, что сочетание соединения формулы (I) и паклитаксела обеспечит улучшенное и эффективное лечение, по сравнению с каждой отдельной монотерапией, для пациентов, страдающих от рака головы и шеи, в частности, пациентов, которые страдают от рака головы и шеи или плоскоклеточного рака головы и шеи, устойчивого к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе препаратов платины или их комбинации.
Сущность изобретения
Настоящее изобретение относится к фармацевтической композиции, включающей (a) ингибитор фосфатидилинозитол-3-киназы (PI3K) соединение формулы (I), определенное в данном описании, или его фармацевтически приемлемую соль и (b) паклитаксел или его фармацевтически приемлемую соль, для одновременного, раздельного или последовательного применения при лечении или предотвращении рака головы и шеи.
В одном варианте его осуществления настоящее изобретение относится также к применению комбинации по настоящему изобретению для приготовления фармацевтической композиции или лекарственного средства, предназначенного для лечения или предотвращения рака головы и шеи.
В одном варианте осуществления настоящего изобретения предлагается фармацевтическая комбинация, включающая (a) ингибитор фосфатидилинозитол-3-киназы (PI3K) соединение формулы (I) и (b) паклитаксел или его фармацевтически приемлемую соль, для применения при лечении или предотвращении рака головы и шеи.
В одном варианте осуществления настоящего изобретения предлагается комбинация по настоящему изобретению для применения при лечении или предотвращении рака головы и шеи.
Один вариант осуществления настоящего изобретения относится к способу лечения или предотвращения рака головы и шеи, который включает введение нуждающемуся в этом субъекту совместно терапевтически эффективного количества комбинации по настоящему изобретению.
Один вариант осуществления настоящего изобретения касается фармацевтической композиции, содержащей количество комбинации по настоящему изобретению, которое совместно терапевтически эффективно против рака головы и шеи.
Один вариант осуществления настоящего изобретения относится также к продажной упаковке, содержащей в качестве терапевтических агентов комбинацию по настоящему изобретению, вместе с инструкциями для ее одновременного, раздельного или последовательного введения при использовании для лечения или предупреждения рака головы и шеи.
Краткое описание чертежей
На фигуре 1 приведены значения IC50 для Соединения А для нескольких клеточных линий рака.
На фигуре 2 показаны результаты введения соединения А в клетки плоскоклеточного рака.
Подробное описание изобретения
Настоящее изобретение относится к фармацевтической композиции, включающей (a) ингибитор фосфатидилинозитол-3-киназы (PI3K) соединение формулы (I), которое определено в данном описании, или его фармацевтически приемлемую соль и (b) паклитаксел или его фармацевтически приемлемую соль для одновременного, раздельного или последовательного применения при лечении или предотвращении рака головы и шеи.
Общие термины, используемые в данном описании, определены со следующими значениями, если явно не указано иное:
Термины "содержащий" и "включающий" используются в данном описании в их широком и не ограничивающем смысле, если не указано иное.
Термины, относящиеся к неопределенному количеству и другие подобные ссылки в контексте описания изобретения (в частности, в контексте приведенной ниже формулы изобретения) следует истолковывать как охватывающие как единственное, так и множественное число, если не указано иное или если нет явного противоречия с контекстом. В тех случаях, когда форма множественного числа используется для соединений, солей и т.п., следует понимать, что она также означает одно соединение, одну соль или т.п.
Термин "комбинация" или "фармацевтическая комбинация", используемый в данном описании, определяет либо фиксированную комбинацию в виде одной готовой лекарственной формы, либо комплект агентов для комбинированного введения, при этом соединение формулы (I), в частности, Соединение А и паклитаксел можно вводить независимо друг от друга в одно и то же время или по отдельности за интервалы времени, в течение которые терапевтические агенты (т.е., соединение формулы (I), в частности, Соединение А и паклитаксел и их фармацевтически приемлемые соли) способны продемонстрировать совместный, например, синергический эффект.
Термин "фармацевтическая композиция" означает в данном описании смесь или раствор, содержащий, по меньшей мере, один терапевтический агент, предназначенный для введения субъекту, например, млекопитающему или человеку, с целью предотвращения или лечения конкретного заболевания или состояния, оказывающего влияние на млекопитающего.
Термин "фармацевтически приемлемый" в данном описании используется для обозначения тех соединений, веществ, композиций и/или лекарственных форм, которые по результатам тщательной медицинской оценки подходят для контактирования с тканями субъекта, например, млекопитающего или человека, не оказывая чрезмерного токсического действия, не вызывая аллергической реакции и других проблемных осложнений, соразмерных с допустимым соотношением благоприятное действие/риск.
Термин "комбинированный препарат" в данном описании относится, в частности, к "набору агентов" в том смысле, что терапевтические агенты (a) и (b), определение которых приведено выше, могут дозироваться независимо или за счет применения различных фиксированных комбинаций с различными количествами терапевтических агентов (a) и (b), в частности, одновременно или в различные моменты времени. Части набора агентов могут, например, вводиться одновременно или в определенном хронологическом порядке, т.е. в различные моменты времени и с одинаковыми или различными интервалами времени для любой части набора агентов. Отношение общего количества терапевтического агента (a) к терапевтическому агенту (b), которое должно быть установлено в комбинированном препарате, может меняться, например, для того, чтобы соответствовать потребностям субпопуляции пациентов, лечение которых проводят, или потребностям конкретного пациента.
Термин "совместное введение", используемый в данном описании, определяется таким образом, что он охватывает назначение выбранных терапевтических агентов одному пациенту и, как следует понимать, включает режимы лечения, при которых агенты необязательно вводят одним и тем же способом или в одно и то же время.
Термин "лечение" или "терапия" в данном описании включает лечение, которое ослабляет, снижает или облегчает, по меньшей мере, один симптом у субъекта или задерживает прогрессирование рака головы и шеи. Например, лечение может заключаться в уменьшении одного или нескольких симптомов рака головы и шеи или в полной ликвидации рака головы и шеи. В соответствии с настоящим изобретением, термин "лечить" также обозначает прекращение, задержку начала (т.е. периода, предшествующего клиническим проявлениям рака головы и шеи) и/или снижение риска развития или ухудшения рака головы и шеи. Термин "предотвращение", используемый в данном описании, обозначает предотвращение, задержку или лечение при развитии длительного онкологического заболевания или при обострении рака головы и шеи у субъекта.
Термин "совместное терапевтическое действие" или "совместно терапевтически эффективный" означает, что терапевтические агенты можно вводить по отдельности (в хронологически установленном порядке, в частности, в определенной последовательности) в такие промежутки времени, которые удобны для лечения теплокровного животного, в частности, человека, при этом агенты все еще демонстрируют (предпочтительно, оказывают синергическое действие) взаимодействие (совместное терапевтическое действие). Происходит ли это на самом деле, можно, в частности, определить затем по уровням в крови, которые показывают, что оба или все терапевтические агенты присутствуют в крови человека, лечение которого проводят, по крайней мере, в течение определенных промежутков времени.
Термин "эффективное количество" комбинации терапевтических агентов (например, соединения формулы (I) и паклитаксела или их фармацевтически приемлемых солей) обозначает количество, достаточное для обеспечения наблюдаемого улучшения, по сравнению с исходными клинически наблюдаемыми симптомами и признаками рака головы и шеи, при лечении с использованием указанной комбинации.
Термин "субъект" или "пациент", используемый в данном описании, включает животных, которые могут страдать от рака головы и шеи или подвержены раку головы и шеи или любому расстройству, прямо или косвенно включающему рак головы и шеи. Примеры субъектов включают млекопитающих, например, людей, собак, коров, лошадей, свиней, овец, коз, кошек, мышей, кроликов, крыс и трансгенных животных, отличных от человека. В предпочтительном варианте осуществления настоящего изобретения субъектом является человек, например, человек, страдающий от рака головы и шеи, который подвержен риску рака головы и шеи или потенциально способен пострадать от рака головы и шеи.
Термин "приблизительно" или "примерно" обычно означает в пределах 20%, более предпочтительно, в пределах 10% и, наиболее предпочтительно, в пределах 5% от заданного значения или диапазона. В качестве альтернативы, особенно в биологических системах, термин "приблизительно" означает в пределах логарифма (т.е. в пределах порядка величины), предпочтительно, отличается в два раза от заданной величины.
Настоящее изобретение относится к фармацевтической комбинации, включающей (a) ингибитор фосфатидилинозитол-3-киназы (PI3K) соединение формулы (I), как определено в данном описании, или его фармацевтически приемлемую соль и (b) паклитаксел или его фармацевтически приемлемую соль, для одновременного, раздельного или последовательного применения при лечении или предотвращении рака головы и шеи.
WO 07/084786 описывает конкретные пиримидиновые производные, которые, как было установлено, ингибируют активность PI3K. Соединение 5-(2,6-диморфолин-4-ил-пиримидин-4-ил)-4-трифторметилпиридин-2-иламин (далее обозначают как "Соединение А") имеет химическую структурную формулу (I)
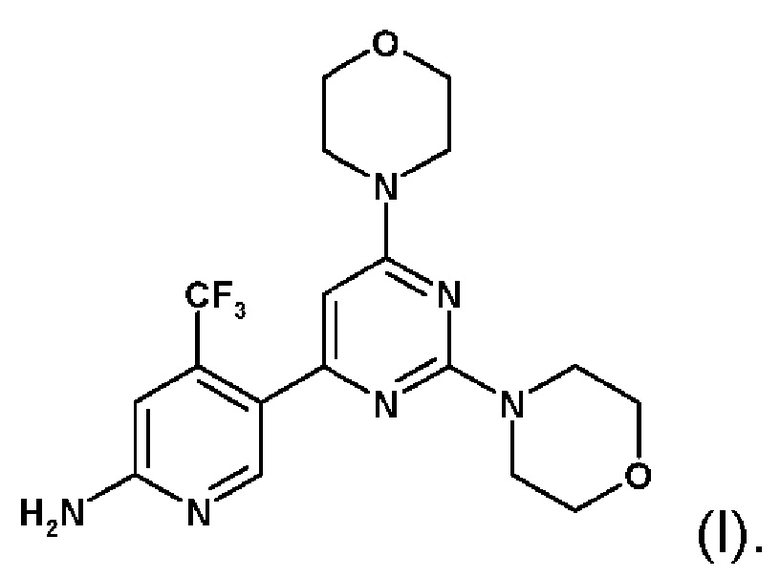
Указанное соединение, его соли, его применение в качестве ингибитора PI3K и синтез соединения 5-(2,6-диморфолин-4-ил-пиримидин-4-ил)-4-трифторметилпиридин-2-иламина описаны в документе WO 2007/084786, который во всей своей полноте, например, как пример 10, включен в настоящее описание посредством ссылки.
Ингибитор фосфатидилинозитол-3-киназы соединение формулы (I) может присутствовать в фармацевтической комбинации в форме свободного основания или его фармацевтически приемлемой соли. Подобные соли могут быть получены in situ в процессе конечного выделения и очистки соединений или путем отдельного взаимодействия основной или кислотной функциональной группы с подходящей органической или неорганической кислотой или основанием, соответственно. Подходящие соли соединения формулы (I) включают, однако этим не ограничиваясь, следующие соли: ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат и ундеканоат. Кроме того, основные азотсодержащие группы могут быть кватернизованы такими агентами, как алкилгалогениды, такие как метил-, этил-, пропил-, бутилхлорид, бромиды и иодиды; диалкилсульфаты, такие как диметил-, диэтил-, дибутил- и диамилсульфаты, длинноцепочечные галогензамещенные соединения, такие как децил-, лаурил-, миристил- и стеарилхлориды, бромиды и иодиды, аралкилгалогениды, такие как бензил- и фенетилбромиды и др.
Примеры кислот, которые могут быть использованы для образования фармацевтически приемлемых кислотно-аддитивных солей, включают такие неорганические кислоты, как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, серная кислота и фосфорная кислота, и такие органические кислоты, как муравьиная кислота, уксусная кислота, трифторуксусная кислота, фумаровая кислота, винная кислота, щавелевая кислота, малеиновая кислота, метансульфоновая кислота, янтарная кислота, яблочная кислота, метансульфоновая кислота, бензолсульфоновая кислота, и п-толуолсульфоновая кислота, лимонная кислота и кислые аминокислоты, такие как аспарагиновая кислота и глутаминовая кислота.
Фармацевтически приемлемые соли включают, однако этим не ограничиваясь, соли, включающие катионы щелочных и щелочноземельных металлов, таких как натрий, литий, калий, кальций, магний, соли алюминия и т.п., а также нетоксичные соли аммония, четвертичного аммония и аминовые катионы, в том числе, однако этим не ограничиваясь, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, этиламин и т.п. Другие типичные органические амины, пригодные для образования основно-аддитивных солей, включают диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин, пиридин, пиколин, триэтаноламин и т.п. и основные аминокислоты, такие как аргинин, лизин и орнитин.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (I) существует в виде его гидрохлоридной соли.
Паклитаксел (TAXOL®) представляет собой натуральный продукт, обладающий противоопухолевой активностью. Паклитаксел получают посредством полусинтетического способа из Taxus baccata. Химическое название паклитаксела 5β,20-эпокси-1,2α,4,7β,10β,13α-гексагидрокситакс-11-ен-9-он-4,10-диацетат-2-бензоат 13-эфир с (2R,3S)-N-бензоил-3-фенилизосерином. Также включены общие формы паклитаксела и различные лекарственные формы паклитаксела. Различные лекарственные формы паклитаксела включают, однако этим не ограничиваясь, альбуминовые наночастицы паклитаксела, которые продаются в виде препаратов ABRAXANE® и ONXOL®.
Далее в данном описании фармацевтическую комбинацию соединения формулы (I) или его фармацевтически приемлемой соли и паклитаксела называют комбинацией по настоящему изобретению.
Если не указано иное, или явно следует из текста, или не может быть применено, ссылка на терапевтические агенты, пригодные для комбинации по настоящему изобретению, включают как свободное основание соединений и все фармацевтически приемлемые соли соединений.
Настоящее изобретение, в частности, относится к комбинации по настоящему изобретению, пригодной для раздельного, одновременного или последовательного введения нуждающемуся в этом субъекту при лечении или предотвращении рака головы и шеи.
Настоящее изобретение дополнительно относится к применению комбинации по настоящему изобретению для получения фармацевтической композиции или лекарственного средства, предназначенного для лечения или предотвращения рака головы и шеи.
Настоящее изобретение относится к способу лечения или предотвращения рака головы и шеи, включающему введение нуждающемуся в этом субъекту совместно терапевтически эффективного количества комбинации по настоящему изобретению.
В настоящем изобретение предлагается также промышленная упаковка, включающая в качестве терапевтических агентов комбинацию по настоящему изобретению вместе с инструкциями для ее одновременного, раздельного или последовательного введения при использовании для лечения или предупреждения рака головы и шеи.
Настоящее изобретение, в частности, относится к комбинации по настоящему изобретению, пригодной для лечения или предотвращения рака головы и шеи у нуждающегося в этом субъекта. В этом варианте осуществления настоящего изобретения комбинацию по настоящему изобретению используют для лечения или предотвращения рака головы и шеи, и он включает назначение указанному субъекту комбинированной терапии с использованием эффективного количества комбинации соединения формулы (I) или его фармацевтически приемлемой соли и паклитаксела. Предпочтительно, указанные терапевтические агенты вводят в терапевтически эффективных дозах, которые при их объединении обеспечивают благоприятное воздействие. Введение может быть раздельным, одновременным или последовательным.
В одном варианте осуществления настоящего изобретения комбинация по настоящему изобретению пригодна для лечения или предотвращения рака головы и шеи. Термин "рак головы и шеи" используется в данном описании для обозначения широкого спектра опухолей, возникающих в верхних отделах желудочно-кишечного тракта и дыхательных путей. Примеры подобных опухолей включают, однако этим не ограничиваясь, рак или опухоль полости рта, губ, глотки (в том числе носоглотки, ротоглотки, гортанной части глотки), гортани, околоносовых пазух, полости носа, горла и слюнных желез. Кроме того, в зависимости от типа опухоли и конкретной используемой комбинации может быть достигнуто уменьшение объема опухоли. Комбинация по настоящему изобретению, раскрытая в данном описании, также пригодна для предотвращения метастазирования опухолей и роста или развития микрометастазов. В предпочтительном варианте осуществления настоящего изобретения раскрытая в данном описании комбинация по настоящему изобретению используется для лечения или предотвращения рака головы и шеи.
В другом варианте осуществления настоящего изобретения рак головы и шеи представляет собой плоскоклеточный рак. Термин плоскоклеточный рак головы и шеи используется в данном описании для обозначения рака головы и шеи, который возникает в плоскоклеточных клетках. Примеры плоскоклеточного рака головы и шеи включают, однако этим не ограничиваясь, рак плоскоклеточного клеток или опухоли полости рта, губ, глотки (в том числе носоглотки, ротоглотки и гортанной части глотки), гортани, околоносовых пазух, носовой полости, горла и слюнных желез.
В другом варианте осуществления настоящего изобретения рак головы и шеи резистентен к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU) и/или терапии на основе платины. Фраза "резистентен к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU) и/или терапии на основе платины" используется для обозначения прогрессирования рака или опухоли у пациента, страдающего от указанного рака или опухоли, в тот момент времени, когда пациент получал лечения с использованием паклитаксела, фторурацила (5-FU) или терапии на основе платины. Примеры известных ранее методов лечения на основе платины включают, однако этим не ограничиваясь, предшествующий курс лечения с использованием цисплатина, карбоплатина, оксалиплатина или их комбинации. Онкологическое заболевание, резистентное к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU) и/или терапии на основе платины, определяется как рост или прогрессирование онкологического заболевания, на которое воздействуют паклитакселом, фторурацилом (5-FU) и/или терапией на основе платины. В предпочтительном варианте осуществления настоящего изобретения рак головы и шеи представляет собой плоскоклеточный рак головы и шеи, резистентный к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
В предпочтительном варианте осуществления настоящего изобретения, комбинация по настоящему изобретению, раскрытая в данном описании, применяется для лечения или предотвращения плоскоклеточного рака головы и шеи.
Комбинация по настоящему изобретению, раскрытая в данном описании, подходит для лечения или предотвращения состояния у пациентов с неблагоприятным прогнозом, в частности больных с неблагоприятным прогнозом, страдающих от рака головы и шеи. Таким образом, в еще одном варианте осуществления настоящего изобретения рак головы и шеи представляет собой плоскоклеточный рак головы и шеи. В предпочтительном варианте осуществления настоящего изобретения рак головы и шеи представляет собой плоскоклеточный рак головы и шеи, который резистентен к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
Комбинация по настоящему изобретению особенно пригодна для лечения или предотвращения рака головы и шеи, имеющего генетические изменения в метаболических путях фосфатидилинозитол-3-киназы, такие как, например, амплификация PI3K альфа, соматическая мутация PIK3CA, генеративные мутации или соматические мутации PTEN или мутации и транслокация p85-альфа, которые служат для активирования комплекса p85-p110.
Таким образом, в одном варианте осуществления настоящего изобретения рак головы и шеи отличается амплификацией PI3K альфа, соматической мутацией PIK3CA, генеративными мутациями или соматическими мутациями PTEN или мутациями и транслокацией p85-альфа, которые служат для активирования комплекса p85-p110.
В другом варианте осуществления настоящего изобретения рак головы и шеи представляет собой плоскоклеточный рак головы и шеи, который резистентен к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации и отличается амплификацией PI3K альфа, соматической мутацией PIK3CA, генеративными мутациями или соматическими мутациями PTEN или мутациями и транслокацией p85-альфа, которые служат для активации комплекса р85-p110.
Один вариант осуществления настоящего изобретения относится к способу лечения или предотвращения рака головы и шеи, который включает введение совместно терапевтически эффективного количества комбинации по настоящему изобретению нуждающемуся в этом субъекту. В каждом варианте осуществления настоящего изобретения следует понимать, что субъект, нуждающийся в конкретном лечении, включает субъектов, страдающих от рака головы и шеи, или субъектов с установленным диагнозом рака головы и шеи в таком варианте осуществления настоящего изобретения.
Еще один вариант осуществления настоящего изобретения относится к способу лечения или предотвращения рака головы и шеи, отличающегося амплификацией PI3K альфа, соматической мутацией PIK3CA, генеративными мутациями или соматическими мутациями PTEN, или мутациями и транслокацией p85-альфа, который включает введение совместно терапевтически эффективного количества комбинации по настоящему изобретению нуждающемуся в этом субъекту.
Другой вариант осуществления настоящего изобретения относится к способу лечения или предотвращения рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации, который включает введение совместно терапевтически эффективного количества комбинации по настоящему изобретению нуждающемуся в этом субъекту. В каждом варианте осуществления настоящего изобретения следует понимать, что субъект, нуждающийся в конкретном лечении, включает субъектов, страдающих от рака головы и шеи, или субъектов с установленным диагнозом рака головы и шеи в таком варианте осуществления настоящего изобретения.
Один вариант осуществления настоящего изобретения относится к способу лечения или предотвращения плоскоклеточного рака головы и шеи, который включает введение совместно терапевтически эффективного количества комбинации (a) соединения формулы (I) или его фармацевтически приемлемой соли и (b) паклитаксела нуждающемуся в этом субъекту.
Другой вариант осуществления настоящего изобретения относится к способу лечения или предотвращения плоскоклеточного рака головы и шеи, отличающегося амплификацией PI3K альфа, соматической мутацией PIK3CA, генеративными мутациями или соматическими мутациями PTEN или мутациями и транслокацией P85-альфа, который включает введение совместно терапевтически эффективного количества комбинации по настоящему изобретению нуждающемуся в этом субъекту.
Еще один вариант осуществления настоящего изобретения относится к способу лечения или предотвращения плоскоклеточного рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации, который включает введение совместно терапевтически эффективного количества комбинации (a) соединения формулы (I) или его фармацевтически приемлемой соли и (b) паклитаксела нуждающемуся в этом субъекту.
Природа рака является многофакторной. При определенных обстоятельствах могут быть объединены лекарственные средства с различными механизмами действия. Тем не менее, один лишь учет любой комбинации терапевтических агентов, оказывающих различный тип воздействия, не обязательно приводит к комбинациям, оказывающим благоприятное действие.
Введение фармацевтической комбинации по настоящему изобретению, может привести не только к благоприятному эффекту, в частности, синергическому терапевтическому эффекту, например, касающемуся облегчения, задержки в прогрессировании или ингибирования симптомов, но также и к другим неожиданным благоприятным эффектам, например, к меньшим побочным эффектам, более длительной ответной реакции, улучшению качества жизни или снижению заболеваемости, по сравнению с монотерапией с применением лишь одного из фармацевтических терапевтических агентов, используемых в комбинации по настоящему изобретению.
Предпочтительно, возникает, по меньшей мере, один благоприятный эффект, например, взаимное усиление воздействия терапевтического агента (a) и (b), в частности, синергизм (в частности, более чем аддитивный эффект), дополнительные благоприятные эффекты, меньшие побочные эффекты, комбинированный терапевтический эффект при использовании неэффективной дозы одного или обоих терапевтических агентов (a) и (b), и наиболее предпочтительно, выраженный синергизм терапевтических агентов (a) и (b).
Термин "синергический эффект" или "синергизм", используемый в данном описании, относится к действию двух терапевтических агентов, таких как, например, соединение формулы (I), в частности, Соединение А, и паклитаксел, которое производит эффект, например, эффект замедления симптоматического прогрессирования пролиферативного заболевания или его симптомов, больший, чем простое добавление эффектов каждого вводимого лекарственного средства самого по себе. Синергический эффект может быть рассчитан, например, с использованием подходящих методов, таких как сигмоидное уравнение для вычисления максимальной эффективности (Holford, N.H.G. and Scheiner, L.B., Clin. Pharmacokinet. 6: 429-453 (1981)), уравнение аддитивности Loewe (Loewe, S. and Muischnek, H., Arch. Exp. Pathol. Pharmacol. 114: 313-326 (1926)) и уравнение среднего эффекта (Chou, T.C. and Talalay, P., Adv. Enzyme Regul. 22: 27-55 (1984)). Каждое вышеуказанное уравнение может быть применено к экспериментальным данным для построения соответствующего графика, помогающего оценить действие комбинации лекарственных средств. Соответствующие графики, связанные с вышеуказанными уравнениями, представляют собой кривую зависимости эффекта от концентрации, кривую изоболограммы и кривую показателя аддитивности, соответственно.
С помощью известных тестов на моделях может быть показано, что комбинация по настоящему изобретению приводит благоприятным эффектам, указанным ранее в данном описании. Специалист в данной области техники вполне способен выбрать соответствующую модель для проведения исследования, с целью показать подобные благоприятные эффекты. Фармакологическая активность комбинации по настоящему изобретению может быть, например, продемонстрирована в клиническом исследовании или с использованием животной модели, как подробно рассматривается ниже в данном описании.
Определение синергического взаимодействия между одним или несколькими компонентами, оптимального диапазона проявления эффекта и абсолютного диапазона доз каждого компонента для проявления эффекта может быть точно осуществлено путем введения компонентов в разных диапазонах масс./масс. соотношения доз для нуждающихся в лечении пациентов. Для людей сложность и стоимость проведения клинических исследований на пациентах может сделать непрактичным использование данной формы тестирования как основной модели для определения синергизма. Тем не менее, наблюдение синергизма у одного вида может быть использовано для прогнозирования данного эффекта у других видов и в существующих животных моделях, как указано в данном описании, с целью измерения синергического эффекта, а результаты подобных исследований также могут быть использованы для прогнозирования эффективных диапазонов соотношении доз и абсолютных доз и концентраций в плазме, необходимых для других видов, путем применения фармакокинетических/фармакодинамических методов. Установленные корреляции между моделями опухолей и наблюдаемыми эффектами у человека заставляют предположить, что синергизм у животных может быть, например, продемонстрирован с помощью моделей ксенотрансплантатов или в соответствующих клеточных линиях.
Подходящие клинические исследования, например, представляют собой нерандомизованные исследования без контроля плацебо с увеличением дозы или многоцентровые, рандомизованные, двойные слепые, плацебо-контролируемые исследования больных с раком головы и шеи. Подобные исследования могут доказать аддитивность или синергизм активных ингредиентов комбинации по настоящему изобретению. Благотворное воздействие на пролиферативные заболевания можно определить непосредственно по результатам подобных исследований или по изменениям в планировании эксперимента, которые известны специалистам в данной области техники. Подобные исследования, в частности, пригодны для сравнения воздействий монотерапии с использованием активных ингредиентов и комбинации по настоящему изобретению. Предпочтительно, терапевтический агент (a) вводят в виде фиксированной дозы, а дозу терапевтического агента (b) увеличивают до тех пор, пока не будет достигнута максимально переносимая доза.
Соединение формулы (I), предпочтительно, вводят ежедневно с дозой в диапазоне от 1,0 до 30 мг/кг массы тела. В одном предпочтительном варианте осуществления настоящего изобретения дозировка соединения формулы (I) находится в диапазоне от приблизительно 60 мг/сутки до приблизительно 120 мг/сутки, в особенности, если теплокровным животным является взрослый человек. Предпочтительно, дозировка соединения формулы (I) находится в диапазоне от приблизительно 80 мг/сутки до приблизительно 100 мг/сутки для взрослого человека. Соединение формулы (I) может назначаться перорально взрослому человеку один раз в день непрерывно (ежедневно) или с перерывами (например, 5 из 7 дней) с подходящей дозой.
В случае паклитаксела диапазон доз для взрослого человека соответствует диапазону доз от приблизительно 15 до 200 мг/м2, в частности, от приблизительно 50 до 175 мг/м2, от приблизительно 60 до 100 мг/м2 или от приблизительно 70 до 100 мг/м2 в неделю. Предпочтительно, доза составляет 80 мг/м2 в неделю.
Один вариант осуществления настоящего изобретения касается фармацевтической композиции или комбинированного препарата, содержащего совместно терапевтически эффективное количество комбинации по настоящему изобретению и необязательно, по меньшей мере, один фармацевтически приемлемый носитель для применения при лечении или предотвращении рака головы и шеи. В указанной композиции или комбинированном препарате терапевтические агенты, соединение формулы (I), в частности, Соединение А или его фармацевтически приемлемую соль и паклитаксел, можно вводить вместе в одной композиции или в одной дозированной лекарственной форме, вводить одновременно, но по отдельности, или вводить последовательно любым подходящим способом.
Терапевтически эффективное количество терапевтических агентов в комбинации по настоящему изобретению может вводиться одновременно или последовательно и в любом порядке, и компоненты могут быть введены по отдельности или в виде фиксированной комбинации. Например, способ лечения или предотвращения рака головы и шеи, согласно настоящему изобретению, может включать (i) введение первого терапевтического агента в свободной форме или форме фармацевтически приемлемой соли и (ii) введение второго терапевтического агента в свободной форме или в форме фармацевтически приемлемой соли, одновременно или последовательно в любом порядке, в совместно терапевтически эффективных количествах, предпочтительно, в синергически эффективных количествах. Индивидуальные терапевтические агенты комбинации по настоящему изобретению могут быть введены раздельно в разное время в течение курса терапии или одновременно в разделенных или единых комбинированных формах. Таким образом, следует понимать, что настоящее изобретение охватывает все такие режимы одновременного или переменного лечения, и термин "введение" должен интерпретироваться соответственно. Предпочтительно, соединение формулы (I) и паклитаксел вводят раздельно.
Фармацевтические композиции по настоящему изобретению могут быть получены известным способом и представляют собой композиции, пригодные для энтерального, такого как пероральное или ректальное, и парентерального введения млекопитающим (теплокровным животным), в том числе человеку. В качестве альтернативы, когда агенты вводят раздельно, один агент может представлять собой энтеральный препарат, а другой агент может быть введен парентерально.
Новая фармацевтическая композиция содержит, например, от приблизительно 10% до приблизительно 100%, предпочтительно, от приблизительно 20% до приблизительно 60% активных ингредиентов. Фармацевтические препараты для комбинированной терапии, предназначенные для энтерального или парентерального введения, представляют собой, например, препараты в стандартных лекарственных формах, таких как таблетки с сахарным покрытием, таблетки, капсулы или суппозитории, а также ампулы. Если не указано иное, их получают известным способом, например, с помощью обычных методов смешивания, гранулирования, нанесения сахарного покрытия, растворения или лиофилизации. Следует иметь в виду, что единичное содержание одного из терапевтических агентов, которые находятся в индивидуальной дозе каждой лекарственной формы, необязательно само по себе составляет эффективное количество, поскольку необходимое эффективное количество может быть достигнуто путем введения множества единиц дозирования.
При приготовлении композиций для пероральной лекарственной формы могут быть использованы любые из обычных фармацевтически приемлемых носителей, такие как, например, вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители; или такие носители, как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, лубриканты, связующие вещества, разрыхлители и т.п. в случае пероральных твердых препаратов, таких как, например, порошки, капсулы и таблетки, при этом твердые пероральные препараты более предпочтительны, чем жидкие препараты. Благодаря легкости их введения, таблетки и капсулы представляют собой наиболее удобную пероральную лекарственную форму, и в этом случае, очевидно, используют твердые фармацевтические носители.
Специалист в данной области техники может выбрать один или несколько из вышеуказанных носителей в соответствии с конкретными требуемыми свойствами лекарственной формы путем обычных экспериментов и без каких-либо чрезмерных затрат. Количество каждого из используемых носителей может меняться в диапазонах, которые обычны в данной области техники. Следующие источники, которые включены в данное описание посредством ссылки, раскрывают методы и наполнители, используемые при разработке пероральных лекарственных форм. См. The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); и Remington: the Science and Practice of Pharmacy, 20th edition, Gennaro, Ed., Lippincott Williams & Wilkins (2003).
Примеры фармацевтически приемлемых разрыхлителей включают, однако этим не ограничиваясь, крахмалы; глины; целлюлозы; альгинаты; природные смолы; поперечно-сшитые полимеры, в частности, поперечно-сшитый поливинилпирролидон или кросповидон, например, POLYPLASDONE XL от компании International Specialty Products (Wayne, NJ);); поперечно-сшитое натриевое производное карбоксиметилцеллюлозы или натриевое производное кроскармеллозы, например, AC-DI-SOL от компании FMC; и поперечно-сшитое кальциевое производное карбоксиметилцеллюлозы; соевые полисахариды; и гуаровую камедь. Разрыхлитель может присутствовать в количестве от приблизительно 0% до приблизительно 10% от массы композиции. В одном варианте осуществления настоящего изобретения разрыхлитель присутствует в количестве от приблизительно 0,1% до приблизительно 5% от массы композиции.
Примеры фармацевтически приемлемых связующих включают, однако этим не ограничиваясь, крахмалы; целлюлозы и их производные, например, микрокристаллическую целлюлозу, в частности, AVICEL PH от компании FMC (Philadelphia, PA), гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу и гидроксипропилметилцеллюлозу METHOCEL от компании Dow Chemical Corp. (Midland, Ml); сахарозу; декстрозу; кукурузный сироп; полисахариды; и желатин. Связующее может присутствовать в количестве от приблизительно 0% до приблизительно 50%, например, в диапазоне 2-20% от массы композиции.
Примеры фармацевтически приемлемых лубрикантов и фармацевтически приемлемых глидантов включают, однако этим не ограничиваясь, коллоидный диоксид кремния, трисиликат магния, крахмалы, тальк, трехосновный фосфат кальция, стеарат магния, стеарат алюминия, стеарат кальция, карбонат магния, оксид магния, полиэтиленгликоль, порошкообразную целлюлоза и микрокристаллическую целлюлозу. Лубрикант может присутствовать в количестве от приблизительно 0% до приблизительно 10% от массы композиции. В одном варианте осуществления настоящего изобретения лубрикант может присутствовать в количестве от приблизительно 0,1% до приблизительно 1,5% от массы композиции. Глидант может присутствовать в количестве от приблизительно 0,1% до приблизительно 10% масс.
Примеры фармацевтически приемлемых наполнителей и фармацевтически приемлемых разбавителей включают, однако этим не ограничиваясь, сахарную глазурь, прессованный сахар, декстраты, декстрин, декстрозу, лактозу, маннит, микрокристаллическую целлюлозу, порошкообразную целлюлозу, сорбит, сахарозу и тальк. Наполнитель и/или разбавитель, в частности, может присутствовать в количестве от приблизительно 0% до приблизительно 80% от массы композиции.
Эффективная доза каждого из используемых терапевтических агентов в комбинации по настоящему изобретению может меняться в зависимости от конкретного соединения или используемой фармацевтической композиции, способа введения, состояния и тяжести подлежащего лечению состояния. Таким образом, схему приема лекарственной комбинации по настоящему изобретению выбирают в соответствии с множеством факторов, включающих путь введения и функции почек и печени пациента. Практикующий врач или терапевт обычной квалификации может легко определить и прописать эффективное количество отдельных терапевтических агентов, необходимых, чтобы облегчить симптомы, противодействовать прогрессированию состояния или остановить прогрессирование состояния.
Оптимальные соотношения, индивидуальные и комбинированные дозы и концентрации терапевтических агентов (a) и (b) и необязательно (c) в комбинации по настоящему изобретению, которые обеспечивают эффективность без токсичности, основываются на кинетике доступности терапевтических агентов для участков-мишеней, и их определяют с использованием способов, известных специалистам из данной области техники.
Эффективная доза каждого из терапевтических агентов может потребовать более частое введение одного соединений, по сравнению с другими соединениями в комбинации. Таким образом, чтобы обеспечить соответствующую дозировку, упакованные фармацевтические препараты могут содержать одну или несколько лекарственных форм, которые содержат комбинацию соединений, и одну или несколько лекарственных форм, которые содержат одну из комбинаций соединений, но не другое(ие) соединение(ия) комбинации.
Когда терапевтические агенты, которые используются в комбинации по настоящему изобретению, применяются в форме, поступающей в продажу в виде отдельных препаратов, их дозировка и способ введения могут соответствовать информации, приведенной в листе-вкладыше соответствующего продаваемого препарата, если не указано иное.
Оптимальная доза каждого из терапевтических агентов для лечения или предотвращения пролиферативного заболевания может быть определена эмпирически для каждого индивида с помощью известных методов и зависит от множества факторов, в том числе, однако этим не ограничиваясь, от степени развития заболевания; возраста, массы тела, общего состояния здоровья, пола и рациона индивида; времени и пути введения; и от других лекарств, которые индивид принимает. Оптимальные дозы могут быть установлены с помощью обычных испытаний и методик, которые хорошо известны из данной области техники.
Количество каждого терапевтического агента в комбинации по настоящему изобретению, которое может быть объединено с веществами-носителями для получения стандартной лекарственной формы, будет изменяться в зависимости от подлежащего лечению индивида и конкретного способа введения. В некоторых вариантах осуществления настоящего изобретения стандартные лекарственные формы, содержащие комбинацию агентов, рассмотренную в данном описании, будут содержать количество каждого терапевтического агента из комбинации, которые обычно вводят, когда терапевтические агенты назначают индивидуально.
Частота дозирования может меняться в зависимости от используемого терапевтического агента и конкретного состояния, подлежащего лечению или предотвращению. За пациентами обычно наблюдают для установления терапевтической эффективности, используя анализы, подходящие для состояния, подлежащего лечению или предотвращению, которые знакомы специалистам в данной области техники.
Один вариант осуществления настоящего изобретения относится к применению комбинации по настоящему изобретению для лечения или предотвращения рака головы и шеи.
Другой вариант осуществления настоящего изобретения относится к применению комбинации по настоящему изобретению для лечения или предотвращения рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их сочетания.
Другой вариант осуществления настоящего изобретения относится к применению комбинации по настоящему изобретению для лечения или предотвращения плоскоклеточного рака головы и шеи.
Предпочтительный вариант осуществления настоящего изобретения относится к применению фармацевтической комбинации, включающей Соединение А или его фармацевтически приемлемую соль и паклитаксел или его фармацевтически приемлемую соль, для лечения или предотвращения плоскоклеточного рака головы и шеи. Предпочтительно, плоскоклеточный рак головы и шеи резистентен к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
Один вариант осуществления настоящего изобретения относится к применению комбинации по настоящему изобретению для приготовления лекарственного средства, предназначенного для лечения или предотвращения рака головы и шеи.
Другой вариант осуществления настоящего изобретения относится к применению комбинации по настоящему изобретению для приготовления лекарственного средства, предназначенного для лечения или предотвращения рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
Еще один вариант осуществления настоящего изобретения относится к применению комбинации по настоящему изобретению для приготовления лекарственного средства, предназначенного для лечения или предотвращения плоскоклеточного рака головы и шеи.
Другой вариант осуществления настоящее изобретение относится к применению комбинации по настоящему изобретению для приготовления лекарственного средства, предназначенного для лечения или предотвращения плоскоклеточного рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
Предпочтительный вариант осуществления настоящего изобретения относится к применению фармацевтической комбинации, содержащей Соединение А или его фармацевтически приемлемую соль и паклитаксел или его фармацевтически приемлемую соль, для получения лекарственного средства, предназначенного для лечения или предотвращения плоскоклеточного рака головы и шеи. Предпочтительно, плоскоклеточный рак головы и шеи резистентен к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
Кроме того, в настоящем изобретении предлагается промышленная упаковка, включающая в качестве активных ингредиентов комбинацию по настоящему изобретению вместе с инструкциями для ее одновременного, раздельного или последовательного введения при использовании для лечения или предупреждения рака головы и шеи.
Другой вариант осуществления настоящего изобретения относится к продажной упаковке, включающей в качестве активных ингредиентов комбинацию по настоящему изобретению вместе с инструкциями для ее одновременного, раздельного или последовательного введения при использования для лечения или предупреждения рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
В соответствии с другими аспектами, в настоящем изобретении предлагается:
- фармацевтическая комбинация, включающая (a) комбинацию по настоящему изобретению, в которой активные ингредиенты присутствуют в каждом случае в свободной форме или в форме фармацевтически приемлемой соли, и необязательно, по меньшей мере, один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного применения, с целью лечения или предотвращения рака головы и шеи;
- фармацевтическая комбинация, включающая (a) комбинацию по настоящему изобретению, в которой активные ингредиенты присутствуют в каждом случае в свободной форме или в форме фармацевтически приемлемой соли, и необязательно, по меньшей мере, один фармацевтически приемлемый носитель; для одновременного, раздельного или последовательного применения, с целью лечения или предотвращения рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации;
- фармацевтическая композиция, содержащая количество комбинации по настоящему изобретению, которое совместно терапевтически эффективно против рака головы и шеи, и, по меньшей мере, один фармацевтически приемлемый носитель;
- фармацевтическая композиция, содержащая количество комбинации по настоящему изобретению, которое совместно терапевтически эффективно против рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации, и, по меньшей мере, один фармацевтически приемлемый носитель;
- комбинированный препарат, содержащий (a) одну или несколько стандартных лекарственных форм терапевтического соединения формулы (I) или его фармацевтически приемлемой соли и (b) паклитаксела или его фармацевтически приемлемой соли, для применения при лечении или предотвращении рака головы и шеи;
- комбинированный препарат, содержащий (a) одну или несколько стандартных лекарственных форм терапевтического соединения формулы (I) или его фармацевтически приемлемой соли и (b) паклитаксела или его фармацевтически приемлемой соли, для применения при лечении или предотвращении рака головы и шеи, резистентного к предшествующему курсу лечения с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
Следующие примеры иллюстрируют настоящее изобретение, описанное выше; однако они не ограничивают каким-либо образом объем настоящего изобретения. Благотворное воздействие комбинации по настоящему изобретению может также быть определено с использованием других моделей испытаний, известных специалисту из данной области техники.
Пример 1: Чувствительность клеточных линий рака головы и шеи к Соединению A и комбинации паклитаксела и Соединения А.
Как показано на фигуре 1, два набора анализов независимо проверяют на чувствительность к Соединению А. Большинство клеточных линий дает значения IC50 менее 1 мкМ для клинически значимых концентраций (концентрация, доставленная пациентам, которых лечат с использованием ежедневной дозы 100 мг, как ожидается, составит около 1 мкМ). (Красный = KRAS мутант = UMSCC-74A. Желтый = PI3KCA мутант = CAL33).
Как показано на фигуре 2, лечение ксенотрансплантата FaDu (плоскоклеточный рак гортаноглотки) с использованием соединения A в количестве 30 мг в день (эквивалентно 100 мг в день у пациентов) показывает ингибирование pAKT в опухолевой ткани, подтверждающее сокращение метаболического пути PI3K при лечении. Лечение с использованием паклитаксела и Соединения А в клеточных линиях рака головы и шеи демонстрирует эффект комбинации с потенциальным синергизмом в некоторых случаях. Клетки высевают в 24-луночные планшеты с плотностью от 5×104 до 1×105 клеток на лунку и выращивают в DMEM с 10% FBS и 1% PSF. На следующий день после посева (1 день), Соединение А - 1 мкмол/л серийно разводят в 10 раз, получая 6 концентраций, и препарат добавляют отдельно или в комбинации с одной концентрацией паклитаксела 1 нг/мл. Данные сравнивают с не подвергнутыми обработке контролями. Клетки подсчитывают в день, когда добавляют препарат, и по прошествии 5 дней, и указанные 2 подсчета сравнивают. Клетки собирают, обработав трипсином, и сразу подсчитывают с помощью счетчика частиц Coulter Z2 (Beckman Coulter Inc., Fullerton, CA, USA).
Процент ингибирования роста, определяемый по уравнению 100×[1-(поколения в обработанных ячейках/поколения в необработанных контрольных образцах)] определяют, как описано ранее (Finn, et al., 2009). Эксперименты проводят дважды. В таблице ниже приведены подробные результаты.
Ингибирование роста раковых клеток
(1 нг/мл)
Пример 2: Клиническое исследование
Клиническое исследование с использованием (a) ингибитора фосфатидилинозитол-3-киназы Соединения А или его гидрохлоридной соли в сочетании с (b) паклитакселом для лечения больных с рецидивирующим или метастатическим раком HNSCC, который развивался после предшествующей схемы лечения на основе соединений платины.
Фазу II многоцентрового, рандомизованного, двойного слепого, плацебо-контролируемого испытания комбинации, включающей (a) Соединение А или его гидрохлоридную соль и (b) паклитаксел, проводят для пациентов с рецидивирующим или метастатическим раком HNSCC, который развивался после предшествующей схемы лечения с использованием соединений на основе платины. Пациентов с гистологически/цитологически подтвержденным HNSCC, рецидивирующим или метастатическим заболеванием, прогрессирующим после предшествующей терапии первой линии на основе соединений платины, рандомизуют в соотношении 1:1 в 2 различные клинические группы, которые должны принимать слепым способом один из двух типов лечений: (a) Соединение А или его гидрохлоридная соль в сочетании с паклитакселом или (b) плацебо в комбинации с паклитакселом. Для участия в исследовании зачисляют приблизительно 150 человек, однако эффективность комбинированного лечения может быть оценена и по результатам с меньшим общим числом пациентов. Пациенты могут быть стратифицированы в зависимости от количества предварительных линий обработки (1 против 2) и места расположения исследователей. Пациенты продолжают получать лечение по программе исследований в соответствии с рандомизацией до тех пор, пока не будет установлено прогрессирование заболевания (оценивается по RECIST версии 1.1), неприемлемая токсичность, смерть или прекращение лечения в программе исследования по любой другой причине (например, отзыв согласия, начало новой противораковой терапии или по усмотрению исследователя). Мониторинг эффективности и безопасности продолжают согласно графику обследования у врача. Оценки опухоли проводят через 4 недели после начала лечения в рамках исследования, а затем каждые 6 недель до подтвержденного рентгеновскими методами прогрессирования заболевания.
Для клинического исследования выражение "развивался после предшествующей схемы лечения с использованием соединений на основе платины" определяется как прогрессирование во время проведения химиотерапии с использованием соединений на основе платины, которую осуществляют в рецидивирующей/метастатической обстановке.
Следующие критерии включения и исключения определяют, какие пациенты подходят для исследования:
Критерии включения: Пациенты, пригодные для включения в данное исследование, отвечают всем из следующих критериев:
1. Возраст пациента ≥18 лет.
2. Письменное информированное согласие получено до проведения каких-либо судебных мероприятий и в соответствии с местными нормативами.
3. Пациент имеет гистологически/цитологически подтвержденный HNSCC.
4. Пациент располагает взятыми из архива или свежими опухолевыми тканями для проведения анализов связанных с PI3K биомаркеров.
5. Пациенты с рецидивирующим или метастатическим заболеванием, резистентным к химиотерапии на основе платины (определяется как прогрессирование во время проведения химиотерапии с использованием соединений на основе платины, которую осуществляют в рецидивирующей/метастатической обстановке). Допускается предшествующий курс лечения с использованием цетуксимаба (как часть химиолучевой терапии, терапии первой линии или поддержки или как монотерапия второй линии).
6. Измеряемые проявления болезни, которые определяют в соответствии с критерием RECIST версии 1.1. Если единственным местом измеримого проявления болезни является ранее облученное поражение, требуется документально подтвержденное прогрессирование заболевания и 4-недельный период после завершение стадии лучевой терапии.
7. Адекватная функция костного мозга и функция органов, как показано:
- Абсолютное количество нейтрофилов (ANC)≥1,5×109/л
- Гемоглобин ≥9 г/дл (может быть достигнуто при переливании)
- Тромбоциты ≥100×109 /л (что может быть достигнуто путем переливания)
- INR≤1,5
- Калий, кальций (с поправкой на сывороточный альбумин) и магний в пределах норм (WNL) для назначения терапии
- Аланинаминотрансфераза (AST) и аспартатаминотрансфераза (ALT) ниже или равно верхнему пределу диапазона нормальных значений (или <3,0×ULN, если в печени присутствуют метастазы)
- Общий сывороточный билирубин ниже или равно верхнему пределу диапазона нормальных значений (или ≤1,5×ULN, если присутствуют метастазы в печени); или общий билирубин ≤3,0×ULN с прямым билирубином ниже или в пределах нормального диапазона у больных с надежно подтвержденным синдромом Жильбера, который определяется как наличие случаев неконъюгированной гипербилирубинемии с нормальными результатами подсчета CBC (включая нормальное количество ретикулоцитов и нормальный клинический анализ крови), нормальные результаты тестирования функции печени и отсутствие других способствующих патологических процессов на момент постановки диагноза (см. Приложение в итоговом протоколе)
- Сывороточный креатинин ≤1,5×ULN или рассчитанный или непосредственно измеренный CrCl≥50% LLN (нижний предел диапазона нормальных значений)
- Глюкоза в плазме натощак (FPG)≤120 мг/дл, или ≤6,7 ммоль/л
- HbA1с≤8%
8. Статус показателя ECOG≤1.
9. Пациент в состоянии проглотить и удержать пероральные лекарственные средства (в том числе пациенты, способные проглотить пероральные лекарственные средства, но в основном пациенты, самокормящиеся посредством желудочной или тощекишечной питательной трубки).
Критерии исключения : Пациенты, пригодные для участия в исследованиях не отвечают ни одному из следующих критериев:
1. Пациент получал предыдущее лечение с любым AKT, ингибиторами мишени рапамицина в клетках млекопитающих (mTOR) или ингибиторами метаболических путей фосфатидилинозитол-3-киназы (PI3K).
2. Пациент получал лечение с таксаном как часть предшествующего лечения метастатического заболевания.
3. Пациента получал ранее лечение по более чем одной схеме химиотерапевтического лечения рецидивирующего/метастатического заболевания (т.е. химиотерапии, химиотерапии в сочетании с биологическим/целевым агентом). Тем не менее, пациенты, получавшие лечение по схеме адъювантной/неоадъювантной химиотерапии и/или сопутствующей химиолучевой терапии, которая могла включать биологические/целевых агенты, пригодны для участия в исследованиях, и допускается монотерапия с использованием цетуксимаба в метастатической обстановке.
4. Пациент имеет симптоматический метастазы в ЦНС. Пациенты с бессимптомными метастазами ЦНС могут принимать участие в данном исследовании. Пациент должен завершить любое предшествующее местное лечение метастазов ЦНС≥28 дней до начала лечения в рамках исследования (в том числе лучевую терапию и/или хирургию) и должен иметь стабильные низкие дозы кортикостероидной терапии.
5. Пациент, который получал широкоугольную лучевую терапию ≤4 недель или ограниченоугольную лучевую терапию для временного облегчения ≤2 недели до начала исследования лекарственных средств или который не восстановился до 1 степени или лучше от связанных побочных эффектов подобной терапии (за исключением алопеции).
6. Пациент не восстановился до 1 степени или лучше (за исключением алопеции) от побочных эффектов любой предшествующей противоопухолевой терапии.
7. Пациенту была проведена полостная операция в течение 14 дней до начала исследования лекарственного средства или он не оправился от серьезных побочных эффектов.
8. Пациент в данное время получает усиленное или постоянное лечение (>5 дней) с использованием кортикостероидов или других иммунодепрессантов, поскольку длительное применение кортикостероидов (>5 дней) может индуцировать CYP3A4.
- Разрешены следующие применения кортикостероидов: однократные дозы; стандартная медикаментозная подготовка для паклитаксела; местное применение (например, сыпь), ингаляционные аэрозоли (например, обструктивные заболевания дыхательных путей), глазные капли или местные инъекции (например, внутрисуставные инъекции)
9. Пациент принимает терапию в начале лечения в рамках исследования с использованием любого из следующих лекарственных средств:
- Препараты, известные как умеренные и сильные ингибиторы или стимуляторы изофермента CYP3A4, включая травяные лекарства (список запрещенных ингибиторов и стимуляторов CYP3A4 будет предоставлен в итоговом протоколе)
- Лекарственные препараты с известным риском вызвать двунаправленную веретенообразную желудочковую тахикардию.
Примечание: Пациент должен прекратить принимать сильные стимуляторы, по крайней мере, за одну неделю до начала лечения, и должен прекратить принимать сильные ингибиторы прежде, чем начнется лечение в рамках исследования. Допускается переключение на другие лекарства до начала лечения в рамках исследования.
10. Пациент в данное время получает варфарин или другой полученный из кумарина антикоагулянт, для лечения, профилактики или и т.п. Допускается терапия с применением гепарина, низкомолекулярного гепарина (LMWN) или фондапаринукса.
11. Пациент обладает известной гиперчувствительностью и/или имеет противопоказания против использования паклитаксела, против стандартной подготовки к лечению паклитакселом или других продуктов, содержащих Cremophor.
12. Пациенты, которые имеют другие сопутствующие тяжелые и/или неконтролируемые медицинские состояния, которые, по мнению исследователя, противопоказаны для участия пациента в клиническом исследовании (например, активная или неконтролируемая острая инфекция, хронический активный гепатит, ослабленная иммунная система, острый или хронический панкреатит, неконтролируемое высокое кровяное давление, интерстициальное заболевание легкого и т.д.).
13. Пациент имеет известную историю ВИЧ-инфекции (тестирование не является обязательным).
14. Пациент имеет любую из следующих сердечно-сосудистых патологий:
- симптоматическая хроническая сердечная недостаточность,
- история документально подтвержденной хронической сердечной недостаточности (функциональная классификация III-IV Нью-Йоркской кардиологической ассоциации), документально подтвержденная кардиомиопатия,
- фракция выброса левого желудочка (LVEF) <50%, как определено сканированием радиоизотопной вентрикулографией (MUGA) или эхокардиографией (ECHO)
- инфаркт миокарда ≤6 месяцев до регистрации,
- нестабильная стенокардия,
- серьезные неконтролируемые нарушения сердечного ритма,
- симптоматический перикардит,
- QTcF>480 мс при скрининге ЭКГ (с использованием формулы QTcF),
- в данное время получает лечение с помощью лекарств, которые обладают известным риском удлинения интервала QT или индуцирования двунаправленной веретенообразной желудочковой тахикардии, и лечение не может быть прекращено или переключено на другие лекарственные средства до начала исследования лекарства. Список запрещенных препаратов будет предоставлен в итоговом протоколе.
15. Пациент имеет нарушения функции желудочно-кишечного тракта (GI) или болезнь желудочно-кишечного тракта, которые могут существенно изменить всасывание исследуемого препарата (например, язвенные заболевания, неконтролируемая тошнота, рвота, диарея, синдром мальабсорбции или резекция тонкой кишки).
16. Пациент набирает ≥12 баллов при ответе на вопросник PHQ-9.
17. Пациент выбирает ответ из "1, 2 или 3" на вопрос № 9 в анкете PHQ-9, который отражает потенциал для суицидальных мыслей или замыслов (независимо от общего балла по PHQ-9).
18. Пациент имеет величину ≥15 по шкале GAD-7.
19. Пациент имеет медицинскую документированную историю или активный депрессивный эпизод тяжелой степени, биполярное расстройство (I или II), обсессивно-компульсивное расстройство, шизофрению, историю попытки или замысла самоубийства или гомицидальных идей (в частности, риск причинения вреда себе или другим); пациенты с активными тяжелыми расстройствами личности (определяют в соответствии с DSM-IV) не могут быть включены в исследование.
Примечание: для пациентов, принимающих лечение с использованием психотропных препаратов на базовом уровне, доза и схема приема препаратов не должны изменяться в течение предыдущих 6 недель до начала лечения в рамках исследования.
20. Пациент имеет степень тревоги ≥3 по CTCAE.
21. Пациент имеет предшествующее или одновременное злокачественное заболевание (за исключением следующих случаев: прошедшее адекватное лечение базальных клеток или плоскоклеточного рака кожи или другое адекватное лечение рака in situ или любого другого онкологического заболевания, от которого пациент освободился ≥3 года).
22. Пациент имеет историю несоблюдения медицинского режима или не может предоставить согласие.
23. Пациент одновременно использует другой из одобренных или исследуемых противоопухолевых агентов.
24. Беременные или кормящие (вскармливающие грудью женщины), где беременность определяется как состояние женщины после оплодотворения яйцеклетки и до окончания беременности, подтвержденное лабораторным положительным тестом hCG (>5 миллимеждународных единиц/мл). Пациенты с повышенной величиной hCG, по сравнению с базовым значением, что, как считается, связано с опухолью, могут быть включены в исследование, если уровни hCG не показывают ожидаемого удвоения, когда проводят повторное исследование по прошествии 5-7 дней, или если беременность была исключена при исследовании методом вагинальной ультразвуковой биолокации.
25. Пациент, который не применяет высоко эффективную контрацепцию во время исследования и позднее, как указано ниже, после последней дозы лечения в рамках исследования:
- Мужчины должны использовать эффективный метод контрацепции и не зачать ребенка в процессе исследования и вплоть до шести месяцев после лечения, им также рекомендуется обратиться за консультацией по вопросам сохранения спермы до начала лечения паклитакселом, в соответствии с инструкцией по медицинскому применению лекарственного препарата.
- Женщины репродуктивного потенциала, которые определяются как все женщины, физиологически способных забеременеть, должны использовать высоко эффективную контрацепцию во время исследования и, по меньшей мере, через 4 недели после последней дозы лечения в рамках исследования или как указано в инструкции о порядке назначения лекарственного средства для паклитаксела (например, в течение 6 месяцев после назначения последней дозы паклитаксела в соответствии с Pl/SmPC из Франции и Великобритании).
- Высокая эффективность контрацепции определяется как любое из:
1. Полное воздержание: Если это находится в соответствии с предпочтительным и обычным образом жизни субъекта. [Периодическое воздержание (например, по графику, овуляция, симптотермальный метод, послеовуляционные методы) и прерываемое совокупление не являются приемлемыми методами контрацепции].
2. Женская стерилизация: хирургическая билатеральная овариэктомия (вместе с гистерэктомией или без гистерэктомии) или лигирование маточных труб, по крайней мере, за шесть недель до участия в лечении в рамках исследования. В случае лишь овариэктомии, только тогда, когда репродуктивный статус женщины был подтвержден последующей оценкой уровня гормона.
3. Стерилизации партнера-мужчины (с соответствующим после вазэктомии документальным подтверждением отсутствия сперматозоидов в эякуляте). [Для принимающих участие в исследованиях женщин партнер-мужчина, подвергнутый вазэктомии, должен быть единственным партнером для данного пациента].
4. Использование сочетания следующих (одновременно a+b):
a. Размещение внутриматочного контрацептива (IUD) или внутриматочной системы (IUS).
b. Барьерные методы контрацепции: презерватив или окклюзионный противозачаточный колпачок (мембранные или шейные/влагалищные колпачки) вместе со спермицидной пеной/гелем/пленкой/кремом/вагинальным суппозиторием.
Примечание: Гормональные методы контрацепции (например, пероральные, инъекционные, имплантационные) не допускаются, поскольку Соединение А снижает эффективность гормональных контрацептивов.
Женщины считаются находящимися в постклимактерическом состоянии и не обладающими репродуктивным потенциалом, если у них естественным образом (спонтанно) отсутствует менструация в течение 12 месяце вместе с соответствующим клиническим профилем (например, соответствующим возрастом, историей вазомоторных симптомов) или же они подвергались хирургическому двустороннему удалению яичников (вместе с гистерэктомией или без гистерэктомии), по крайней мере, шесть недель назад. В случае лишь удаления яичников, женщина считается не обладающей репродуктивным потенциалом, лишь тогда, когда репродуктивный статус женщины был подтвержден последующей оценкой уровня гормона.
Отбор проводится в течение 1-35 дней до начала лечения (за исключением рентгеновских оценок опухолей, которые проводятся в течение 1-28 дней до начала лечения). Повторное обследование разрешается только один раз для пациента, если пациент не был зарегистрирован как начавший фазу лечения. Повторные лабораторные исследования в пределах времени отбора разрешены для результатов, выходящих за пределы определенного диапазона.
Основной целью является оценка эффективности лечения комбинации, включающей назначение один раз в день Соединения А или его гидрохлорида и паклитаксела, на выживаемость без прогрессирования заболевания (PFS) (на основании местного рентгеновского исследования) у вышеуказанных пациентов. Основной переменной эффективности является выживаемость без прогрессирования заболевания, которую оценивают по результатам местного рентгеновского исследования в соответствии с RECIST версии 1.1. PFS определяется как время от даты рандомизации до объективного прогрессирования опухоли или смерти от любой причины. Дата прогрессирования заболевания представляет собой самое ранее время, когда наблюдается любое определяемое с помощью RECIST прогрессирование осложнения (т.е. подтвержденного рентгеновскими методами прогрессирование или смерть) не более чем с одной ранее пропущенной оценкой.
Оценки опухоли проводят на основе критериев RECIST. При оценке опухоли, возможность измерения определяют как наличие, по крайней мере, одного измеримого узлового или не узлового поражения и, если оно ограничивается изолированным поражением, то его онкологическая природа должна быть надежно подтверждена цитологически/гистологически.
Вторичные цели включают оценку общей выживаемости (время от рандомизации до дня смерти по любой причине), общий уровень положительного клинического ответа (часть пациентов с наилучшим общим ответом, связанным с полной ремиссией (CR) или частичной ремиссией (PR) по оценке исследователя); степень контролирования заболевания (доля пациентов с наилучшим общим ответом CR, PR или стабилизацией заболевания (SD) по оценке исследователя и по продолжительности ответной реакции (Определяется только для подгруппы пациентов с лечебным эффектом, т.е. для пациентов с подтвержденным CR или PR по оценке исследователя. Оно представляет собой фактическую длительность от даты первого документированного ответа до следующий даты события, определяемого как первое документированное прогрессирование или смерть вследствие первопричинного онкологического заболевания).
После отбора пациентов случайным образом распределяют в одну из двух лечебных групп, и подобная рандомизация остается конфиденциальной. Пациентов лечат с помощью либо: (a) Соединения А или его гидрохлоридной соли и паклитаксела, либо (b) плацебо, соответствующего Соединению А, и паклитаксела до прогрессирования заболевания, неприемлемой токсичности, смерти или прекращения участия в исследовании по любой другой причине. Визиты к врачу и соответствующие оценки, которые отличаются ±3 дня от плановой даты (за исключением дня 1) не считаются отклонениями от протокола. Сканирования MRI/CT проводят в цикле 2 дня 1 (±3 дня) и каждые 6 недель (±4 дня) до прогрессирования заболевания, до прекращения участия, до неспособности продолжить участие в исследовании, до начала другого противоопухолевого лечения или смерти, в зависимости от того, какое событие наступит раньше. Лабораторные исследования, проведенные как часть оценки при отборе и в течение 7 дней назначения первой дозы исследуемого препарата не требуется повторять в первый день дозирования.
Соединение A или его гидрохлоридную соль вводят перорально один раз в день по схеме непрерывного дозирования, начиная со дня 1, в сочетании с один раз в неделю вводимым паклитакселом с дозой 80 мг/м2 (дни 1, 8, 15 и 22) из 28-дневного цикла. Соединение А или его гидрохлоридную соль назначают с дозой 100 мг свободного основания Соединения А.
Паклитаксел вводят путем внутривенной инфузии. Паклитаксел поставляется в виде многодозовых флаконов для инъекций. Паклитаксел разводят с помощью 0,9%-ного раствора хлорида натрия для инъекций, USP; 5%-ного раствора декстрозы для инъекций, USP; 5%-ного раствора декстрозы и 0,9%-ного раствора хлорида натрия для инъекций, USP или 5%-ного декстрона в растворе Рингера для инъекций до конечной концентрации от 0,3 до 1,2 мг/мл. Паклитаксел вводят каждую неделю в виде 1-часовой (±15 мин) внутривенного вливания после стандартной медикаментозной подготовки в день 1 каждого цикла. Перед введением паклитаксела пациентам проводят медикаментозную подготовку в соответствии со стандартной практикой, принятой в лечебном заведении, или в соответствии с инструкцией по применению лекарственного препарата с тем, чтобы предотвратить серьезные аллергические реакции. Антиаллергическую терапию можно назначать перед проведением ЭКГ в каждом цикле. Опции включают: 20 мг дексаметазона, который вводят перорально за 12 и 6 час до начала введения паклитаксела, или дифенгидрамин (или эквивалент): 50 мг вводят внутривенно приблизительно за 30-60 мин до начала введения паклитаксела, или ранитидин: 50 мг вводят внутривенно приблизительно за 30-60 мин до начала введения паклитаксела, или циметидин: 300 мг внутривенно вводят приблизительно за 30-60 мин до начала введения паклитаксела. Циметидин следует вводить в виде одной дозы и только тогда, когда не может быть найдена другая альтернатива. Если при введении паклитаксела возникает гиперчувствительность, то необходимо воспользоваться следующими рекомендациями по проведению терапии:
- Для мягких симптомов (в частности, легкое покраснение, сыпь, зуд) можно завершить вливание под строгим наблюдением врача.
- Для умеренных симптомов (в частности, умеренная сыпь, покраснение, легкая одышка, умеренный дискомфорт в области грудной клетки, легкая гипотензия): (1) остановить вливание паклитаксела и назначить 25-50 мг дифенгидрамина внутривенно и 125 мг метилпреднизолона внутривенно, (2) после того, как симптомы исчезают, возобновить вливания паклитаксела со скоростью введения, составляющей 10% от исходной скорости, в течение 15 мин, затем 25% от первоначальной скорости в течение 15 минут. Если дальнейшие симптомы не развиваются, продолжить с исходной скоростью до полного завершения вливания.
- Для тяжелых симптомов (в частности, один или несколько из следующих симптомов: расстройство дыхания, требующее лечения, аллергическая сыпь, ангионевротический отек, артериальная гипотензия, требующая лечения): (1) остановить вливание паклитаксела и назначить дифенгидрамин и метилпреднизон, как описано выше. Использовать эпинефрин или бронхолитические средства, если имеется показание, или (2) не назначать повторно паклитаксел пациенту.
Cоответствующее Соединению A плацебо может быть введено перорально один раз в день по режиму непрерывного дозирования, начиная с дня 1.
Лечение продолжают до прогрессирования заболевания (подтвержденного рентгеновскими методами в соответствии с RECIST версии 1.1) или до прекращения исследования по любой другой причине. Полный цикл лечения определяется как 28 календарных дней, в течение которых соединение А, или его гидрохлоридную соль, или его плацебо назначают один раз в день, а паклитаксел назначают один раз в неделю. Последним днем полного цикла лечения является день 29. День 1 следующий цикл начинается день 29. Регулярно проводится мониторинг эффективности и безопасности. Оценки размера опухоли осуществляют через 4 недели после начала лечения в рамках исследования и затем каждые 6 недель до подтвержденного рентгеновскими методами прогрессирования заболевания.
Эффективность и ответную реакцию опухоли определяют в соответствии с конкретными рекомендациями Критериев оценки объективного ответа при солидных опухолях (RECIST), основанными на RECIST версии 1.1.
Безопасность контролируют путем врачебного осмотра, оценки основных показателей состояния организма, массы, показателей общего состояния, ЭКГ, визуализации сердца, с помощью лабораторных оценок, включая мониторинг глюкозы и исследования пациента по рейтингу шкалы настроения, а также побочных явлений (серьезных и несерьезных).
Участие в исследовании препарата пациентов, которым требуется задержать назначение дозы Соединения A, или его гидрохлоридной соли, или плацебо >28 дней, окончательно прекращается. Побочные явлений 4 степени приводят к полному прекращению участия независимо от времени восстановления. Кроме того, разрешаются следующие максимум 3 сокращения дозы Соединения А или его гидрохлоридной соли, и каждая доза отражает количество дозы Соединения A в виде свободного основания:
Уровень дозы - 1
Уровень дозы - 2
Уровень дозы - 3
80 мг/день непрерывно
100 мг/день 5 дней из 7
80 мг/день 5 дней из 7**
** Снижение дозы ниже 80 мг/день 5 дней из 7 не допускается. Если требуется снижение дозы ниже уровня дозы 3, то назначение пациенту Соединения A/плацебо должно быть окончательно прекращено.
Переходу от непрерывной схемы к прерывистой (5 дней из 7) предшествуют 2 дня лечения без Соединения A. Модификации дозы и прерывание дозы разрешается в соответствии со следующими указаниями:
(Степень CTCAE 4.03)
2 степень (ANK<1,5-1,0×109/л)
4 степень (ANK<0,5×109/л)
- Если восстановится в течение ≤7 дней, то затем поддерживать уровень дозы
- Если восстановится в течение >7 дней, то ↓ до уровня дозы 1
(ANK <1,0×109/л, при однократной температуре ≥38,3°С или при устойчивой температуре ≥38°C в течение более чем одного часа)
2 степень (PLT<75-50×109/л)
- Если восстановится в течение ≤7 дней, то затем поддерживать уровень дозы
- Если восстановится в течение >7 дней, то ↓ до уровня дозы 1
- Если восстановится в течение ≤7 дней, то затем поддерживать уровень дозы
- Если восстановится в течение >7 дней, то ↓ до уровня дозы 1
(*для пациентов с синдромом Жильбера указанные изменения дозы применимы к изменениям только прямой билирубин)
будет разделен на фракции, если повышен
- Если восстановится в течение ≤7 дней, то затем поддерживать уровень дозы
- Если восстановится в течение >7 дней, то ↓ до уровня дозы 1
- Если восстановится в течение ≤7 дней, то ↓ до уровня дозы 1
- Если восстановится в течение >7 дней, то прекратить назначать пациенту соединение А/плацебо
- Если восстановится в течение ≤7 дней, то затем поддерживать уровень дозы
- Если восстановится в течение >7 дней, то ↓ до уровня дозы 1
- Если восстановится в течение ≤7 дней, то затем поддерживать уровень дозы
- Если восстановится в течение >7 дней, то ↓ до уровня дозы 1
Контроль гепатической токсичности (* для пациентов с синдром Жильбера: должны контролироваться общий и прямой билирубин, усиленный мониторинг относится к изменениям только в прямом билирубине; мониторинг включает в себя следующие LFTs: альбумин, ALT, AST, общий билирубин (фракционированный, если общий билирубин>2,0×ULN), щелочную фосфатазу (фракционированную, если щелочная фосфатаза имеет сорт 2 или выше) и GGT):
- Цикл 1 и 2: каждую неделю (если программа посещения врача позволяет более частый контроль, то следует его рассмотреть) или чаще, если клинически показано, в особенности для пациентов с пограничными приемлемыми значениями AST/ALT/билирубин*
- Цикл 3 и далее: ежемесячно или чаще, если клинически показано
В случае увеличения ≥2 степени ALT/AST/билирубин* исследование функции печени необходимо проводить еженедельно или чаще, если клинически показано, пока не восстановится до ≤1 степени.
В случае увеличения ≥3 степени ALT/AST/билирубина* исследование функции печени необходимо контролировать еженедельно или чаще, если клинически показано, пока не восстановлено до ≤1 степени; далее мониторинг должен быть продолжен раз в неделю или чаще, если клинически показано, до конца лечения исследуемым препаратом.
Пациентов, которые прекратили участие в лечении в рамках исследования, необходимо контролировать еженедельно, в том числе по параметрам LFTs*, или более часто, если клинически показано, пока они не восстановятся до ≤1 степени или до стабилизации (без изменения степени по CTCAE в течение 4 недель).
- инициировать или усилить прием лекарств вместе с соответствующим антидиабетическим лечением по усмотрению исследователя
- проинструктировать пациента о соблюдении диеты в соответствии с местными стандартами и/или стандартами учреждения для лечения сахарного диабета (такими, как рекомендации Американской Диабетической Ассоциации) во время проведения исследований
- рассмотреть возможность использования пероральной антигипергликемической терапии, например, с использованием метформина (или интенсифицировать прием уже используемых лекарств)
- проверять FPG, по крайней мере, еженедельно в течение 8 недель, затем продолжить проверку, по крайней мере, каждые 2 недели
- поддерживать уровень дозы и осуществлять мониторинг FPG, по крайней мере, еженедельно до тех пор, пока FPG не восстановится до ≤1 степени
- инициировать или усилить прием лекарств вместе с соответствующим антидиабетическим лечением, таким как метформин; рассмотреть вопрос о добавлении второго перорального агента, если нет улучшения после нескольких дней
- проинструктировать пациента о соблюдении диеты в соответствии с местными стандартами и/или стандартами учреждения для лечения сахарного диабета (такими, как рекомендации Американской Диабетической Ассоциации) во время проведения данных исследований
- если FPG не восстанавливается до ≤1 степени в течение 14 дней после назначения соответствующего антидиабетического лечения, снизить Соединение А/плацебо на 1 уровень дозы
- продолжить антидиабетическое лечение и проверять FPG, по крайней мере, еженедельно в течение 8 недель, а затем продолжить проверку, по крайней мере, каждые 2 недели
- назначить внутривенное восполнение потери жидкости и соответствующие клинические процедуры для электролита/кетоацидоза/гиперосмолярных возмущений
- продолжить перерыв в приеме Соединения А/плацебо
- осуществлять мониторинг FPG, по крайней мере, два раза в неделю до тех пор, пока FPG не восстановится до ≤1 степени
- если FPG восстанавливается до ≤1 степени в течение 7 дней или меньше, затем вновь начать назначать Соединение А/плацебо и ↓ 1 уровень дозы
- Если FPG остается на уровне больше, чем 1 степень тяжести более 7 дней, то прекратить назначать пациенту Соединение А/плацебо
- начать или продолжить антидиабетическое лечение соответствующими способами
- проинструктировать пациента о соблюдении диеты в соответствии с местными стандартами и/или стандартами учреждения для лечения сахарного диабета (такими, как рекомендации Американской Диабетической Ассоциации) во время проведения данных исследований
- рассмотреть возможность применения пероральной антигипергликемической терапии, такой как использование метформина
- проверять FPG, по крайней мере, еженедельно в течение 8 недель, а затем продолжить проверки, по крайней мере, каждые 2 недели
Для глюкозы в плазме не натощак >250-500 мг/дл (>13,9-27,8 ммоль/л), сопровождаемой признаками/симптомами гипергликемии (например, изменения психического состояния, чрезмерная жажда, учащенное мочеиспускание) или присутствием крови или кетоновых тел в моче, прекратить назначать Соединение А/плацебо и следовать рекомендациям лечения 3 степени глюкозы в плазме натощак (FPG)
-назначить внутривенное восполнение потери жидкости и соответствующие клинические процедуры для электролита/кетоацидоза/гиперосмолярных возмущений
-прекратить назначать пациенту Соединение А/плацебо
-проинструктировать пациента о соблюдении диеты в соответствии с местными стандартами и/или стандартами учреждения для лечения сахарного диабета (такими, как рекомендации Американской Диабетической Ассоциации) во время проведения данных исследований
-рассмотреть возможность применения пероральной антигипергликемической терапии, такой как использование метформина
-проверять FPG, по крайней мере, еженедельно в течение 8 недель, а затем продолжить проверки, по крайней мере, каждые 2 недели, если клинически показано
Для глюкозы в плазме не натощак >500 мг/дл (>27,8 ммоль/л), сопровождаемой признаками/симптомами гипергликемии (например, изменения психического состояния, чрезмерная жажда, учащенное мочеиспускание) или присутствием крови или кетоновых тел в моче, прекратить назначать Соединение А/плацебо и следовать рекомендациям лечения 4 степени глюкозы в плазме натощак (FPG)
Повторить LVEF в течение 4 недель или клинически подходящим образом
- Измерение LVEF следует повторить, если не восстановится* в течение 3 недель, прекратить лечение пациента с использованием Соединения A
* осложнение считается восстановленным, когда пациент не имеет симптомов, имеет фракцию выброса в состоянии покоя ≥40% и ≤20% снижение, по сравнению с базовым уровнем
или>60 мс изменение по сравнению с базовым уровнем, по меньшей мере, в двух отдельных ЭКГ
- Прервать назначение Соединения А/плацебо
- Провести анализ калия и магния в сыворотке, и если меньше нижней границы нормы, скорректировать дополнениями до нормальных пределов. Необходимо рассмотреть сопутствующую медикаментозную терапию.
- Провести повторное ЭКГ в течение одного часа при первом QTcF >500 мс или>60 мс, по сравнению с базовым уровнем
- Если QTcF остается >500 мс или >60 мс, по сравнению с базовым уровнем, повторить ЭКГ, как показано клинически, но, по крайней мере, один раз в день до тех пор, пока QTcF не вернется к <480 мс. Получить информацию от кардиолога.
- После того, как QTcF удлинение восстановится, можно возобновить назначать Соединение А/плацебо с одним из меньших уровней дозы
Второе появление:
- Прекратить назначать пациенту Соединение A/плацебо
- Предусмотреть консультацию у психиатра по усмотрению исследователя и назначить наилучший медицинский уход
- Предусмотреть консультацию у психиатра по усмотрению исследователя и назначить наилучший медицинский уход
- Первое событие: если восстановится до ≤1 степени или исходного состояния, продолжить совместное медикаментозное лечение, а затем сохранить уровень дозы
- Второе и последующие события: если состояние восстановится до ≤1 степени или до исходного состояния, продолжать совместное медикаментозное лечение, а затем ↓ до уровня дозы 1
- Требуется консультация психиатра и наилучший медицинский уход
- Если состояние улучшается до ≤1 степени или до исходного состояния, продолжать совместное медикаментозное лечение, а затем ↓ до уровня дозы 1
- Требуется консультация психиатра и наилучший медицинский уход
- Если восстановится в течение ≤2 недель, поддерживать уровень дозы.
- Если восстановится более чем через 2 недели, ↓ до уровня дозы 1.
Второе появление: ↓ до уровня дозы 1
Начать/активизировать подходящую терапию кожной токсичности (например, использовать антигистаминные препараты, кортикостероиды местно)
Второе появление: окончательно прекратить назначать пациенту Соединение A/плацебо
По усмотрению исследователей, могут быть взяты парные биопсии кожи (как с обработанных, так и незатронутых областей кожи для местной патоморфологической оценки), если клинически уместно.
По усмотрению исследователей, могут быть взяты парные биопсии кожи (как с обработанных, так и незатронутых областей кожи для местной патоморфологической оценки), если клинически уместно.
- Если восстановится в течение ≤7 дней, поддерживать уровень дозы
- Если восстановится >7 дней, ↓ до уровня дозы 1
2 степень: 2: Сократить введение Соединения А/плацебо до уровня дозы 1 до восстановления <1 степени. Лечение в рамках исследования может быть прервано. Пациенты прекращают прием препаратов, если они не восстановятся до <1 степени в течение 3 недель.
3 степень: Остановить лечение с использованием Соединения A/плацебо до восстановления до <1 степени. Можно начать повторно лечение в рамках исследования в течение 3 недель при пониженном уровне дозы (на один уровень), если имеются доказательства клинической эффективности.
4 степень: Прекратить лечение с использованием Соединения А/плацебо.
Примечание: Пропустить дозы при ≥3 степени рвоты или ≥3 степени тошноты, только если с рвотой или тошнотой нельзя справиться с помощью оптимального противорвотного средства
Модификации дозы паклитаксела разрешаются в случае возникновения побочных эффектов, если, как предполагается, они вызваны паклитакселом. Следует придерживаться следующих принципов:
- Паклитаксел следует назначать, лишь если ANK>1500/мм3 (1,5×109/л) и уровень тромбоцитов >100000/мм3 (100×109/л).
- В случае опасных для жизни осложнений, рассмотреть вопрос о прекращении назначения паклитаксела.
- В случае 3 степени или 4 степени AEs несмотря на терапию:
- Остановить назначение паклитаксела до тех пор, пока осложнение не восстановится до 1 степени или лучше, затем вновь продолжить с уменьшенной дозой.
- В случае второго такого же осложнения с 3 или 4 степенью, рассмотреть возможность прекращения назначения паклитаксела.
- В случаях негематологических осложнений 2 степени (кроме алопеции), которые стойко проявляются, несмотря на терапию, рассмотреть вопрос о приостановке назначения паклитаксел до восстановления до 1 степени или лучше, а затем возобновить с уменьшенной дозой.
- Минимальная доза паклитаксела, допускаемая в процессе исследования и первый уровень снижения дозы составляют 65 мг/м2 (т.е. разрешается только одно снижение дозы паклитаксела до 65 мг/м2).
Кроме того, следует регулировать дозу паклитаксела, по мере необходимости, в соответствии с инструкцией по медицинскому применению данного препарата и местной практикой.
Допускается сопутствующая терапия, необходимая для ухода за пациентом, за исключением:
- Системное лечение кортикостероидами не допускается, за исключением местного применения, применения ингаляционных спреев, глазных капель или местных инъекций; системные кортикостероиды ≤ противовоспалительной действенностью 4 мг дексаметазона; или в случае медикаментозной подготовки для паклитаксела.
- Необходимо контролировать лекарственные препараты, метаболизируемые ферментами CYP450.
- Некоторые неферментные стимулирующие анти-эпилептические препараты.
- Другая противоопухолевая терапия или другие экспериментальные методы лечения не допускаются.
- Профилактическое применение гемопоэтических факторов роста не допускается. Разрешается применение в случае чрезвычайной ситуации.
- Терапевтические дозы варфарина натрия или каких-либо других антикоагулянтов на основе производных кумарина не допускаются.
- Ферментные стимулирующие анти-эпилептические препараты не допускаются.
- Лекарственные препараты с известным риском вызвать двунаправленную веретенообразную желудочковую тахикардию не допускаются.
- Умеренные и сильные ингибиторы CYP3A не допускаются.
- Растительные препараты/лекарственные средства не допускаются.
Пациенты должны быть выведены из исследования, если возникает любой из следующих случаев: негативное событие, утрата возможности связаться с пациентом, несоблюдение режима лечения в рамках исследования, решение врача, беременность, прогрессирующая болезнь, отклонение от протокола, прекращение исследования спонсором, технические проблемы, решение субъекта/опекуна, смерть. Пациенты должны быть выведены из исследования, если возникает любой из следующих случаев: регулирование лечения в рамках исследования, которое приводит к исключению пациента из исследования, использование запрещенных препаратов, прерывание лечения в рамках исследования на период >28 дней от предполагаемого дня введения следующей запланированной дозы.
Для всех пациентов, которые прекращают участие в лечении в рамках исследования вследствие прогрессирования заболевания, прогрессирование их заболевания должно быть точно задокументировано в соответствии с критериями, указанными в RECIST версии 1.1. Если пациент прекращает участие в лечении в рамках исследования не вследствие прогрессирования заболевания, смерти, начала новых противоопухолевых методов лечения, утраты возможности связаться с пациентом или отзыва согласия вследствие невозможности эффективного продолжения, то необходимо продолжать оценки опухоли каждые 6 недель до начала новой противораковой терапии, прогрессирования болезни, смерти, утраты возможности связаться с пациентом или отзыва согласия вследствие невозможности эффективного продолжения.
Кроме того, все новые противораковые терапии, назначаемые после последней дозы во время лечения в рамках исследования, вплоть до прогрессирования заболевания, смерти, утраты возможности связаться с пациентом или отзыва согласия вследствие невозможности эффективного продолжения, будут записаны в виде электронных индивидуальных регистрационных карт клинических исследований (eCRFs).
После прекращения лечения всех пациентов приглашают для оценки безопасности (т.е. оценки побочных эффектов и/или серьезных побочных эффектов, сопутствующих медицинских препаратов) в течение 30 дней после назначения последней дозы исследуемого препарата. За пациентами, лечение которых прервано или окончательно прекращено из-за побочных эффектов, наблюдают, по крайней мере, один раз в неделю в течение 4 недель, а затем с 4-недельными интервалами до нормализация состояния или стабилизации осложнения в зависимости от того, что наступит раньше.
Для пациентов, которые не прекращают принимать участие в лечении в рамках исследования вследствие прогрессирования заболевания, смерти, начала новой противоопухолевой терапии, утраты возможности связаться с пациентом или отзыва согласия вследствие невозможности эффективного продолжения, оценки опухоли проводят каждые 6 недель до начала новой антираковой терапии, прогрессирования болезни, смерти, утраты возможности связаться с пациентом или отзыва согласия вследствие невозможности эффективного продолжения.
Всех пациентов наблюдают на предмет статуса выживаемости каждые 3 месяца, независимо от причины прекращения лечения вплоть до наступления смерти, утраты возможности связаться с пациентом или отзыва согласия на последующие наблюдения и сбор данных по продолжительности выживаемости. Перед сбором информации о выживаемости, окончательная информация после лечения (в частности, информация о побочном эффекте, утрате возможности связаться с пациентом, решении врача, беременности, отклонении от протокола, технических проблемах, решении субъекта/опекуна, смерти, новой терапии для получения показаний, прогрессировании заболевания, прекращении исследования спонсором) собирается по завершении фазы исследования.
| название | год | авторы | номер документа |
|---|---|---|---|
| Применение липосомы митоксантрона гидрохлорида | 2021 |
|
RU2821030C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ, СОДЕРЖАЩАЯ TNO155 И РИБОЦИКЛИБ | 2020 |
|
RU2813111C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2001 |
|
RU2284818C2 |
| КОМБИНАЦИЯ АНТАГОНИСТА PD-1 И ИНГИБИТОРА IDO1 ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2744880C1 |
| КОМБИНАЦИЯ ИНОТУЗУМАБА ОЗОГАМИЦИНА И ТОРИЗЕЛА ДЛЯ ЛЕЧЕНИЯ РАКА | 2012 |
|
RU2607594C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2006 |
|
RU2429838C2 |
| КОМБИНАЦИЯ, ВКЛЮЧАЮЩАЯ N-{5-[4-(4-МЕТИЛПИПЕРАЗИНОМЕТИЛ)БЕНЗОИЛАМИДО]-2-МЕТИЛФЕНИЛ}-4-(3-ПИРИДИЛ)-2-ПИРИМИДИНАМИН И ХИМИОТЕРАПЕВТИЧЕСКИЙ АГЕНТ | 2002 |
|
RU2318517C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2010 |
|
RU2587013C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2801665C2 |
| ДУОКАРМИЦИНОВЫЕ ADC, ДЕМОНСТРИРУЮЩИЕ УЛУЧШЕННУЮ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ IN VIVO | 2015 |
|
RU2689779C2 |

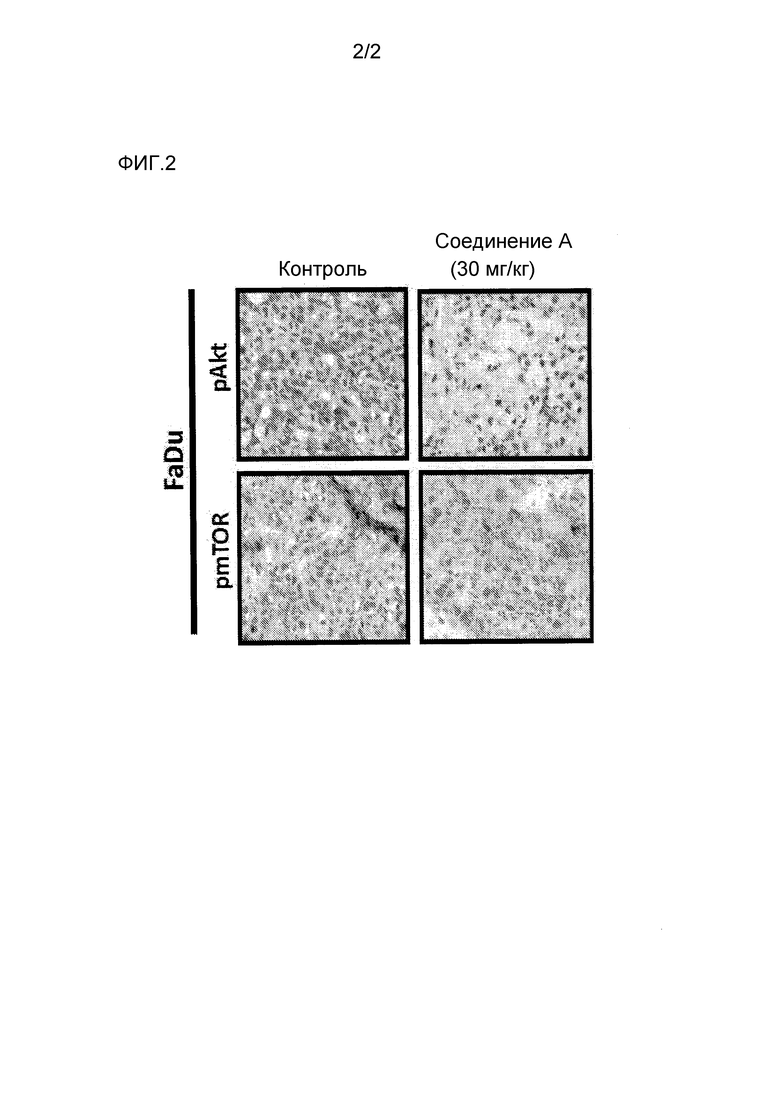
Изобретение относится к фармацевтической комбинации, включающей ингибитор фосфатидилинозитол-3-киназы (PI3K) соединение формулы (I), как определено в данном описании, или его фармацевтически приемлемую соль и паклитаксел или его фармацевтически приемлемую соль, для одновременного, раздельного или последовательного применения при лечении рака головы и шеи; фармацевтической композиции, включающей указанную комбинацию; использованию указанной комбинации для получения лекарственного средства, предназначенного для лечения рака головы и шеи; способу лечения или предотвращения рака головы и шеи, включающему введение совместно терапевтически эффективного количества комбинации нуждающемуся в этом субъекту. 7 н. и 8 з.п. ф-лы, 2 пр., 1 табл., 2 ил.
1. Фармацевтическая комбинация, включающая (a) соединение формулы (I)
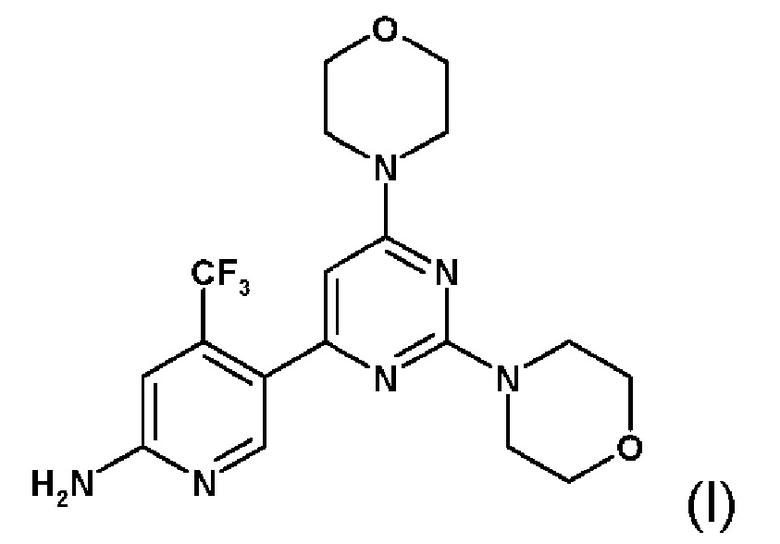
или его фармацевтически приемлемую соль и (b) паклитаксел или его фармацевтически приемлемую соль, для одновременного, раздельного или последовательного применения при лечении или предотвращении рака головы и шеи.
2. Фармацевтическая комбинация по п. 1, где соединение формулы (I) или его фармацевтически приемлемую соль вводят взрослому человеку с дозой в диапазоне от приблизительно 60 мг/день до приблизительно 120 мг/день.
3. Фармацевтическая комбинация по п. 1, где паклитаксел вводят взрослому человеку с дозой в диапазоне от приблизительно 15 до 200 мг/м2 в неделю.
4. Фармацевтическая комбинация по любому из пп. 1-3, где рак головы и шеи выбран из группы, включающей рак или опухоль полости рта, губ, глотки (в том числе носоглотки, ротоглотки, гортанной части глотки), гортани, околоносовых пазух, полости носа, горла и слюнных желез.
5. Фармацевтическая комбинация по любому из пп. 1-3, где рак головы и шеи представляет собой плоскоклеточный рак головы и шеи.
6. Фармацевтическая комбинация по любому из пп. 1-3, где рак головы и шеи резистентен к предшествующему лечению с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
7. Фармацевтическая композиция, содержащая совместно терапевтически эффективное количество фармацевтической комбинации по п. 1 и по меньшей мере один фармацевтически приемлемый носитель, предназначенная для применения при лечении или предотвращении рака головы и шеи.
8. Комбинированный препарат, который включает (a) одну или несколько лекарственных форм соединения формулы (I) по п. 1 или его фармацевтически приемлемой соли и (b) одну или несколько лекарственных форм паклитаксела или его фармацевтически приемлемой соли, предназначенный для применения при лечении или предотвращении рака головы и шеи.
9. Применение фармацевтической комбинации по п. 1 для приготовления лекарственного средства, предназначенного для применения при лечении или предотвращении рака головы и шеи.
10. Применение фармацевтической комбинации по п. 1 для лечения или предотвращения рака головы и шеи.
11. Применение по п. 9 или 10, где рак головы и шеи выбран из группы, которая включает рак или опухоль полости рта, губ, глотки (в том числе носоглотки, ротоглотки, гортанной части глотки), гортани, околоносовых пазух, полости носа, горла и слюнных желез.
12. Применение по п. 9 или 10, где рак головы и шеи представляет собой плоскоклеточный рак головы и шеи.
13. Применение по п. 9 или 10, где рак головы и шеи резистентен к предшествующему лечению с использованием паклитаксела, фторурацила (5-FU), терапии на основе платины или их комбинации.
14. Способ лечения или предотвращения рака головы и шеи, включающий введение нуждающемуся в этом субъекту совместно терапевтически эффективного количества фармацевтической комбинации по п. 1.
15. Набор, содержащий фармацевтическую комбинацию по п. 1 вместе с инструкциями для ее одновременного, раздельного или последовательного введения при использовании для лечения или предупреждения рака головы и шеи.
| СПОСОБ ПОЛУЧЕНИЯ ЭНЕРГИИ, ВОДОРОДА И АЛМАЗОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2003 |
|
RU2261223C2 |
| WO 2009155659 A1, 09.10.2013 | |||
| Sauveur-Michel Maira et al | |||
| Кровля из глиняных обожженных плит с арматурой из проволочной сетки | 1921 |
|
SU120A1 |

