ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к частицам фенилэфрина, пригодным для твердых, полутвердых или жидких лекарственных форм. Частицы фенилэфрина, которые могут иметь покрытие, высвобождают фенилэфрин со скоростью, которая обеспечивает фармацевтически приемлемые концентрации в плазме в течение длительного периода времени. Настоящее изобретение также относится к способу изготовления лекарственных форм, содержащих частицы фенилэфрина, и к способам облегчения заложенности носа и затруднений дыхания у пациента-человека с помощью перорального введения лекарственных форм. Лекарственные формы дополнительно могут содержать один или более дополнительных терапевтически активных агентов, выбранных из одной или более групп, состоящих из антигистаминных средств, противоотечных средств, анальгетиков, противовоспалительных средств, жаропонижающих, противокашлевых и отхаркивающих средств.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Фенилэфрин является мощным сосудосуживающим, обладающим прямыми и косвенными симпатомиметическими эффектами [Hoffman 2001]. Основным и прямым эффектом является агонизм α1-адренорецепторов. Стимуляция α1-адренорецепторов, расположенных на емкостных кровеносных сосудах слизистой оболочки носа (посткапиллярных венул), приводит к вазоконстрикции, снижению объема циркулирующей крови и снижению объема слизистой оболочки носа (заложенности носа) [Johnson 1993]. Суженные кровеносные сосуды позволяют меньшему количеству жидкости попасть в нос, горло и выстилку полостей, что приводит к снижению воспаления слизистой оболочки носа, а также снижение выработки слизи [Johnson 1993]. Таким образом, вызывая сужение кровеносных сосудов, главным образом, тех, которые расположены в носовых проходах, фенилэфрин вызывает уменьшение заложенности носа [Hoffman 2001, Empey 1 981].
Фенилэфрин принадлежит к категории I (как правило, рассматривается в качестве безопасного и эффективного (GRASE)) безрецептурного (OTC) средства против заложенности носа для перорального применения. В целом, фенилэфрин был доступен начиная с 1960-х годов, а с 1996 года фенилэфрин широко используется в Соединенных Штатах. Фенилэфрина гидрохлорид, который широко используется как безрецептурное средство для взрослых и детей от кашля и простуды, как известно, используется взрослыми и детьми для временного облегчения заложенности носа из-за простуды, сенной лихорадки или других аллергий с проявлением в верхних дыхательных путях (аллергический ринит). Препарат коммерчески доступен в виде 10-мг таблеток для перорального применения у взрослых. Режим дозирования - одна доза 10 мг фенилэфрина каждые четыре часа, но не превышая 60 мг (шесть доз) в течение 24 часов. Полная информация доступна в фармакопейной статье безрецептурного лекарственного средства для утвержденных препаратов.
Фенилэфрин, химическое название (R)-1-(3-гидроксифенил)-2-метиламиноэтанол, является коммерчески доступным в виде гидрохлоридной соли. Эмпирическая формула C9H13NO2⋅HCl и молекулярный вес 203,67. Соединение, которое представляет собой кристаллический порошок от белого до грязно-белого цвета, имеет следующую химическую структуру:
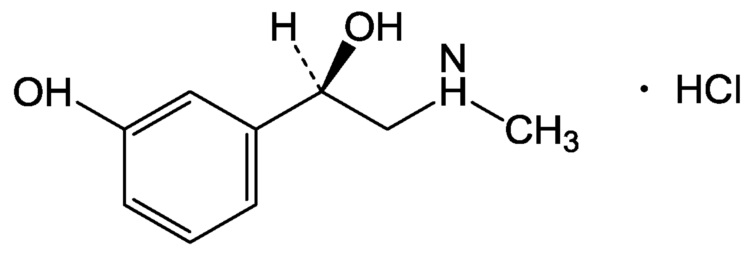
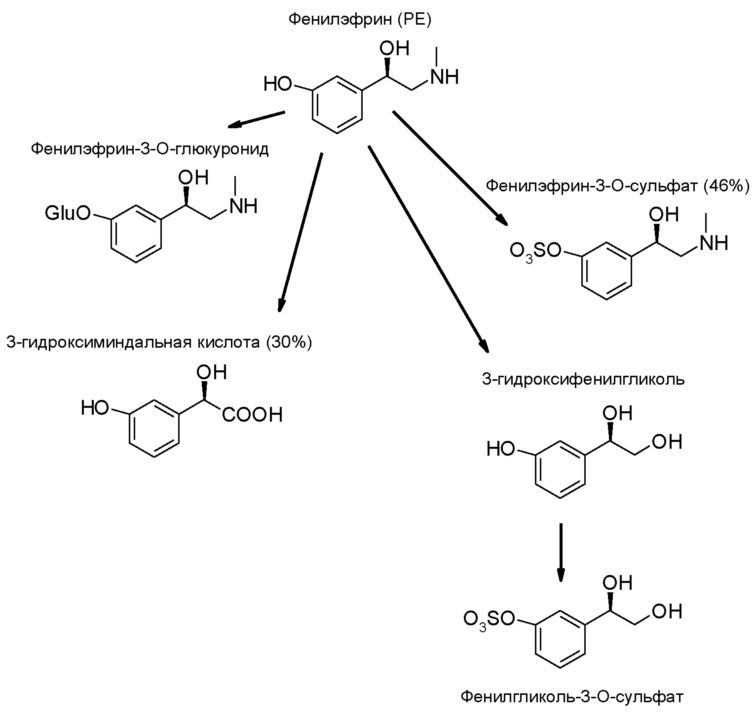
Основными путями метаболизма фенилэфрина являются сульфатное сопряжение (в основном в стенке кишечника) и окислительное дезаминирование формами А и В моноаминоксидазы [Suzuki 1979]. Глюкуронизация также происходит, но в меньшей степени. В одном исследовании следующие дозы в 30 мг вводились перорально в течение восьми часов [Ibrahim 1983], фенилэфрин метаболизировался до фенилэфрин-сульфата, м-гидроксиминдальной кислоты, фенилэфрин-глюкуронида и м-гидрокси-фенилгликоль-сульфата в 47%, 30%, 12% и 6% от дозы соответственно. Удаление аминогруппы является преобладающим метаболическим путем после внутривенного введения фенилэфрина [Hengstmann 1982], в то время как сульфатное сопряжения является преобладающим путем после введения перорального. Метаболиты фенилэфрина фаз I и II в организме человека приведены ниже. Процентные значения в схеме соответствуют процентам при пероральном введении, согласно исследованиям Ibrahim.
Данные об эффективности клинических исследований немедленного освобождения фенилэфрина при применении у взрослых показывают, что фенилэфрин является эффективным средством от заложенности носа.
Ацетаминофен является пара-аминофенольной производной, имеющей анальгезирующее и жаропонижающее действие. Он используется для временного облегчения незначительных болей и болей, связанных с простудой, болей в спине, головной боли, зубной боли, менструальных спазмов и мышечных болей; и для временного облегчения слабой боли при артрите и уменьшения лихорадки. Взрослая доза ацетаминофена в США составляет 1000 мг каждые четыре-шесть часов с максимумом 4000 мг в течение 24 часов. Взрослая доза ацетаминофена замедленного высвобождения составляет 1300 мг каждые восемь часов с максимумом 3900 мг в течение 24 часов.
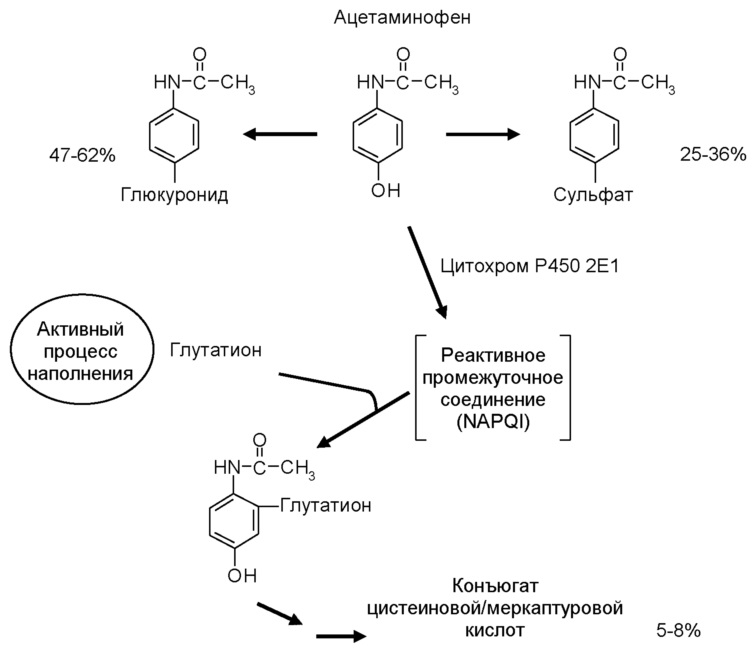
Ацетаминофен прежде всего метаболизируется в печени с помощью трех основных параллельных путей: глюкуронидации, сульфатации и окисления [Miners 1983; Slattery 1989; Lee 1992; Miners 1992]. Глюкуроновый и окислительные пути придерживаются процесса скорости первого порядка, что означает, что концентрация метаболизированного ацетаминофена повышается с повышением концентрации в печени. Сульфатный путь придерживается кинетики Михаэлиса-Ментена, что означает, что концентрация метаболизированного ацетаминофена остается постоянной, как только концентрация в печени увеличивается выше уровня насыщения.
Схема метаболизма ацетаминофена показана ниже. Менее 9% терапевтической дозы выводится в неизмененном виде с мочой [Miners 1992]. Основным метаболическим путем является глюкуронирование, при котором от 47% до 62% дозы ацетаминофена конъюгирует с глюкуронидом. Эти глюкуронидные конъюгаты неактивны и нетоксичны [Koch-Weser 1976], выделяются с желчью и выводятся с мочой. Глюкуронидная конъюгация катализируется главным образом одной изоформой глюкуронтрансферазы (UGT1A6) [Court 2001] с уридин-5'-дифосфоглюкуроновой кислотой в качестве основного кофактора.
Вторым основным путем метаболизма ацетаминофена является сульфирование, где от 25% до 36% дозы конъюгируют с сульфатом. Эти сульфатные конъюгаты также неактивны и нетоксичны [Koch-Weser 1976] и легко выводятся из организма с мочой. Сульфатация опосредуется сульфотрансферазами, которые являются гетерогенными цитозольными ферментами, кофактором является 3'-фосфоаденозин 5'-фосфат. Скорость-регулирующим фактором ацетаминофен-сульфатации является скорее активность сульфотрансфераз, а не снижение количества сульфата [Blackledge 1991].
Третий путь - окисление, при котором от 5% до 8% дозы ацетаминофена метаболизируется с помощью ферментной системы цитохрома Р-450. Изофермент цитохрома Р-450, в первую очередь ответственный за метаболизм ацетаминофена - это CYP2E1 [Manyike 2000]. При метаболизировании ацетаминофен CYP2E1, он образует высокореактивное промежуточное соединение, N-ацетил-p-бензохинонеимин (NAPQI). NAPQI обладает высокой реакционной способностью, он не может быть измерен вне печени и не может накапливаться. Это промежуточное соединение быстро инактивируется гепатоцеллюлярными запасами глутатиона с образованием цистеина и конъюгатов меркаптурата, которые неактивны и нетоксичны [Koch-Weser 1976]. Эти конъюгаты выводятся с мочой [Mitchell 1 974].
Существует необходимость сократить частоту доставки фенилэфрина. Менее частые введения приводят к улучшению соблюдения пациентом режима лечения. Кроме того, постоянные терапевтические концентрации активных компонентов в плазме могут быть более эффективными и даже чрезвычайно эффективными по сравнению с колебаниями, которые заметны при применении нескольких стандартных доз немедленного высвобождения. Устойчивые эффективные уровни могут уменьшить тяжесть и частоту заметных побочных эффектов с высокими пиками уровня вещества в плазме. Таким образом, необходимы лекарственные препараты, изготовленные на основе фенилэфрина, которые можно вводить менее часто, например, один раз в 6, 8, 12, 16, 20 или 24 часа.
Существует также потребность обеспечить соответствие продолжительности действия фенилэфрина с действиями других активных препаратов, время высвобождения которых больше, чем у фенилэфрина немедленного высвобождения.
Опубликованная заявка на патент США № 20070281020, корпорация Schering-Plough, раскрывает введение пациенту-человеку таблетированной формы лекарственного препарата с замедленным высвобождением, содержащей 30 мг фенилэфрина, гидроксипропилметилцеллюлозу, натрий карбоксиметилцеллюлозу, Kollidon CL-M, коллоидный диоксид кремния и стеарат магния, и сравнение действия таблетки с замедленным высвобождением с действием трех доз 10 мг фенилэфрина немедленного освобождения.
В патенте США № 8282957, McNeil-PPC, Inc, раскрываются частицы фенилэфрина с покрытием, содержащие фенилэфрин HCl, модифицированный крахмал и Eudragit NE30D™, покрытые первым слоем покрытия, содержащим Eudragit RS PO, ацетилтрибутилцитрат и стеарат магния, и вторым слоем, содержащим Eudragit NE30D™, Eudragit FS30D™, стеарат магния, лаурилсульфат натрия и симетикон, и использование их в фармацевтических лекарственных формах, в том числе лекарственных формах, содержащих ацетаминофен.
В патенте США № 6001392, Компания Warner Lambert, раскрывается комплекс действующее вещество/смола, который содержит смесь мелованной и немелованной смолы Amberlite™ IR69, сшитой с дивинилбензолом.
В опубликованной заявке на патент США № 20100068280, корпорация Schering-Plough, описаны фармацевтические лекарственные формы, содержащие фенилэфрин в форме с замедленным высвобождением. В соответствии с вариантом одна доза фенилэфрина в таблетке, содержащей 30 мг фенилэфрина, моногидрат лактозы, Methocel K100M CR, Klucel EXF и стеарат магния, сравнивалась с двумя таблетками 10 мг фенилэфрина с немедленным высвобождением, введенными с разницей в 4 часа, в исследовании биоэквивалентности.
В опубликованных заявках на патент США № 20050266032 и № 20060057205, Sovereign Pharmaceuticals, описаны фармацевтические лекарственные формы, содержащие фенилэфрин. В соответствии с вариантом осуществления фенилэфрин включен в комплекс ионообменной смолы с использованием, например, полистирол сульфоната натрия, и покрыт полимером, обеспечивающим замедленное высвобождение, например, Eudragit® L 100, Kollidon® МАЕ и Aquacoat® cPD. Формула в этом варианте содержит 45 мг фенилэфрина с замедленным высвобождением и 15 мг фенилэфрина немедленного освобождения.
Патент США № 8062667, Tris Pharma, Inc, раскрывает комплексы препарат-ионообменная смола, имеющие покрытие. В соответствии с вариантом фенилэфрин включен в комплекс ионообменной смолы с использованием полистирол сульфоната натрия и покрыт оболочкой из Kollicoat™ SR-30D, триацетина и воды.
Патент США № 8394415, McNeil-PPC, Inc, раскрывает жидкий состав, содержащий ибупрофен немедленного высвобождения и комплекс фенилэфрина и конкретной ионообменной смолы замедленного высвобождения, покрытый первым и вторым слоями покрытия, включающей указанные ингредиенты.
Заявка на патент США № 11/761698, McNeil-PPC, Inc, описывает твердую композицию, содержащую ибупрофен (НВ) и фенилэфрин, покрытую первым слоем покрытия, содержащим этилцеллюлозу, и вторым слоем, содержащим защитное покрытие.
Заявка на патент США № 20100068280, Schering-Plough Healthcare Products, Inc., раскрывает исследование биодоступности, в котором сравнивается 10 мг фенилэфрина HCl, доставленного в капсулах Enterion™, 10 мг Sudafed PE™ и 30 мг фенилэфрина HCl, доставленного в капсулах Enterion™.
Заявка на патент США № 2007014239, Coating Place, Inc., раскрывает способ и композицию для помещения одного или нескольких препаратов на одну или более частиц ионообменной смолы для формирования частиц, содержащих препарат.
По-прежнему есть необходимость в продуктах, содержащих фенилэфрин, имеющих атрибуты, описанные выше.
ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к частицам фенилэфрина, которые обеспечивают доставку фенилэфрина или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом, для обеспечения максимальной концентрации фенилэфрина в плазме от примерно 0,1 до примерно 16 часов, предпочтительно от около 0,5 до около 5 часов, более предпочтительно от примерно 1 до примерно 4,5 часов после приема, отличающихся тем, что фенилэфрин поддерживается на уровне выше чем приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл, по меньшей мере около 6, приблизительно 8, приблизительно 12, приблизительно 16, приблизительно 20 и/или около 24 часов после приема.
В соответствии с предпочтительным вариантом осуществления настоящее изобретение направлено на разработку частиц фенилэфрина резината с покрытием, которые обеспечивают доставку фенилэфрина или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом, для обеспечения максимальной концентрации фенилэфрина в плазме от примерно 0,1 до примерно 16 часов, предпочтительно от около 0,5 до около 5 часов, более предпочтительно от примерно 1 до примерно 4,5 часа после приема, отличающихся тем, что фенилэфрин поддерживается на уровне выше чем приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл, по меньшей мере около 6, приблизительно 8, приблизительно 12, приблизительно 16, приблизительно 20 и/или около 24 часов после приема.
Настоящее изобретение также относится к фармацевтическим лекарственным формам, содержащим частицы фенилэфрина, которые обеспечивают доставку фенилэфрина или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом, для обеспечения максимальной концентрации фенилэфрина в плазме от примерно 0,1 до примерно 16 часов, предпочтительно от около 0,5 до около 5 часов, более предпочтительно от примерно 1 до примерно 4,5 часа после приема, отличающихся тем, что фенилэфрин поддерживается на уровне выше чем приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл, по меньшей мере около 6, приблизительно 8, приблизительно 12, приблизительно 16, приблизительно 20 и/или около 24 часов после приема.
В другом варианте осуществления частицы фенилэфрина, которые обеспечивают замедленное высвобождение фенилэфрина, совмещены с частицами фенилэфрина в форме, обеспечивающей немедленное высвобождение.
В другом варианте осуществления частицы фенилэфрина совмещены с одним или более дополнительными терапевтическими средствами для немедленного или замедленного высвобождения. Такой агент или агенты могут быть приготовлены для немедленного высвобождения после приема, для замедленного высвобождения, для высвобождения в толстой кишке одновременно с по меньшей мере частью фенилэфрина или для высвобождения в любой комбинации. В одном варианте осуществления дополнительный терапевтический агент не имеет покрытия. В другом варианте осуществления дополнительный терапевтический агент имеет покрытие.
Дополнительное терапевтическое средство может быть одним из следующих: антигистамин, противозастойное, анальгетик, противовоспалительное, жаропонижающее, противокашлевое, отхаркивающее, или любым другим терапевтическим агентом или комбинацией таких агентов, полезны для облегчения симптомов простуды, сезонных и других аллергий, сенной лихорадки или проблем в пазухах, любой из которых может привести к увеличению выделения из носа. Предпочтительно, одним или более из дополнительных терапевтических агентов является ацетаминофен.
Примеры антигистаминных средств и деконгестантов включают, помимо прочего, бромофенирамин, хлорциклизин, дексбромфенирамин, бромгексан, фениндамин, фенирамин, пириламин, тонзиламин, приполидин, эфедрин, псевдоэфедрин, фенилпропаноламин, хлорфенирамин, декстрометорфан, дифенгидрамин, доксиламин, астемизол, терфенадин, фексофенадин, нафазолин, оксиметазолин, монтелукаст, пропилгексадрин, трипролидин, клемастин, акривастин, прометазин, оксомемазин, меквитазин, буклизин, бромгексин, кетотифен, эбастин, оксатамид, ксиломеазолин, лоратидин, дезлоратидин и цетиризин, их изомеры, фармацевтически приемлемые соли и эфиры.
Примеры соответствующих обезболивающих, противовоспалительных и жаропонижающих средств включают, помимо прочего, нестероидные противовоспалительные препараты (НПВП), такие как производные пропионовой кислоты (например, ибупрофен, напроксен, кетопрофен, флурбипрофен, фенбуфен, фенопрофен, индопрофен, флупрофен, пирпрофен, карпрофен, оксапрозин, пранопрофен и супрофен) и ингибиторы циклооксигеназы, такие как целекоксиб, ацетаминофен, ацетилсалициловая кислота, производные уксусной кислоты, такие как индометацин, диклофенак, сулиндак и толметин, производные фенамовой кислоты, такие как мефенамовая кислота, меклофенамовая кислота и флуфенамовая кислота, производные бифенилкарбодиловой кислоты, такие как дифлунизал и флуфенизал; оксикамы, такие как пироксикам, судоксикам, изоксикам и мелоксикам, их изомеры, а также фармацевтически приемлемые соли и пролекарственные формы.
Примеры средств от кашля и отхаркивающих средств включают, помимо прочего, дифенгидрамин, декстрометорфан, носкапин, клофедианол, ментол, бензонатат, этилморфон, кодеин, ацетилцистеин, карбоцистеин, амброксол, алкалоиды красавки обыкновенной, собренол, гваякол и гвайфенезин, их изомеры, фармацевтически приемлемые соли и пролекарственные формы.
Другой аспект настоящего изобретения представляет собой способ лечения симптомов гриппа, простуды, аллергии или не-аллергического ринита у пациента, нуждающегося в этом, включающий введение частиц фенилэфрина по настоящему изобретению. В некоторых вариантах осуществления частицы фенилэфрина вводят примерно каждые 6, 8, 12, 16, 20 или 24 часа. В одном предпочтительном варианте осуществления частицы фенилэфрина вводят примерно каждые 12 часов. В другом предпочтительном варианте осуществления частицы фенилэфрина резината вводят примерно каждые 8 часов.
Некоторые варианты осуществления настоящего изобретения относятся к способам поддержания устойчивой биодоступности фенилэфрина у пациента, включающим пероральное введение пациенту частиц фенилэфрина, отличающимся тем, что по меньшей мере часть фенилэфрина поглощается из толстой кишки, и отличающимся тем, что концентрация фенилэфрина в плазме пациента составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл примерно через 6 часов после введения композиции. В конкретных вариантах осуществления концентрация фенилэфрина в плазме крови пациента составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл примерно через 8 часов после введения композиции. В конкретных вариантах осуществления концентрация фенилэфрина в плазме крови пациента составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл примерно через 12 часов после введения композиции. В конкретных вариантах осуществления концентрация фенилэфрина в плазме крови пациента составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл примерно через 20 часов после введения композиции. В конкретных вариантах осуществления концентрация фенилэфрина в плазме крови пациента составляет по меньшей мере приблизительно 20, приблизительно 40, приблизительно 60, приблизительно 80, приблизительно 100, приблизительно 120, приблизительно 140, приблизительно 160, приблизительно 180 или приблизительно 200 пг/мл примерно через 24 часа после введения композиции. Некоторые другие варианты осуществления настоящего изобретения представляют способы введения фенилэфрина пациенту, включающие пероральное введение частиц фенилэфрина, где указанная композиция доставляет по меньшей мере часть фенилэфрина в толстую кишку, где фенилэфрин высвобождается в толстой кишке и всасывается из толстой кишки.
Настоящее изобретение может быть лучше понято посредством ссылки на фигуры, подробное описание и примеры, которые следуют.
КРАТКОЕ ОПИСАНИЕ ФИГУР
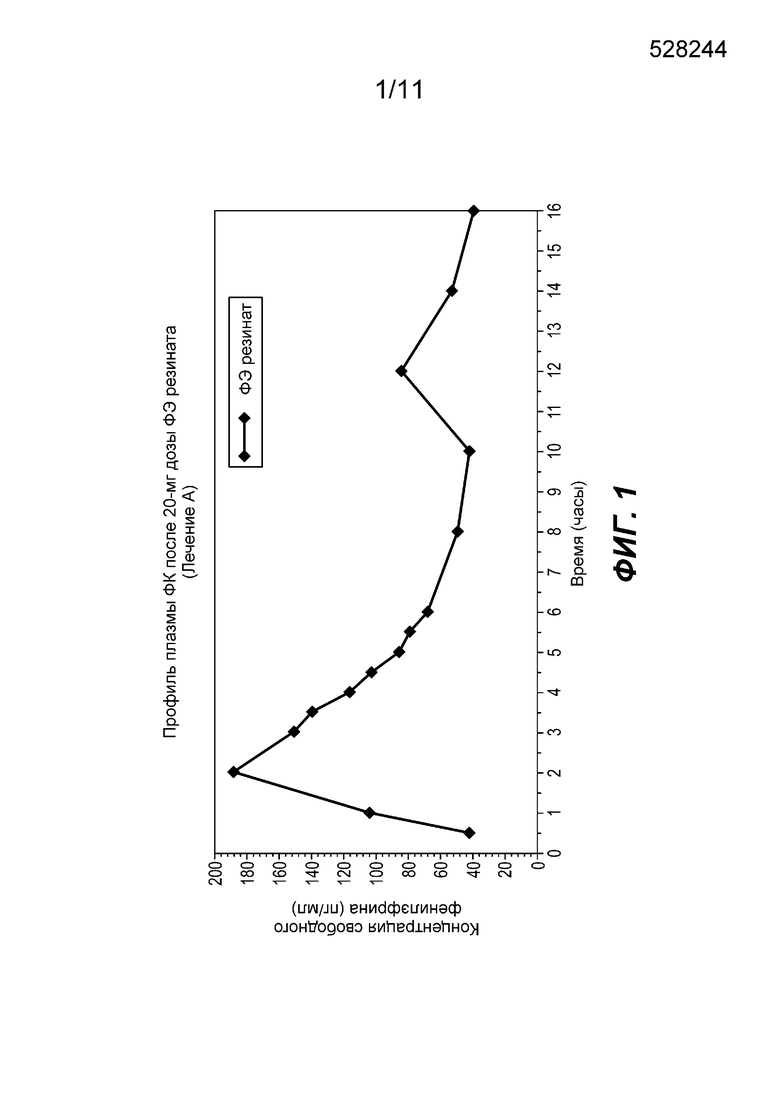
Фиг. 1 показывает средний профиль концентрации фенилэфрина в плазме при введении частиц фенилэфрина резината замедленного высвобождения (ЗВ) с покрытием, содержащих 20 мг фенилэфрина. Как показано на фиг. 1, ось y представляет собой концентрацию свободного фенилэфрина в плазме в пикограммах (пг) на миллилитр (мл). По оси x показано время в часах. На фиг. 1 показано, что средняя концентрация фенилэфрина достигла пика (Cmax) через приблизительно 2 часа. Фиг. 1 также показывает вторичный пик через примерно 12 часов.
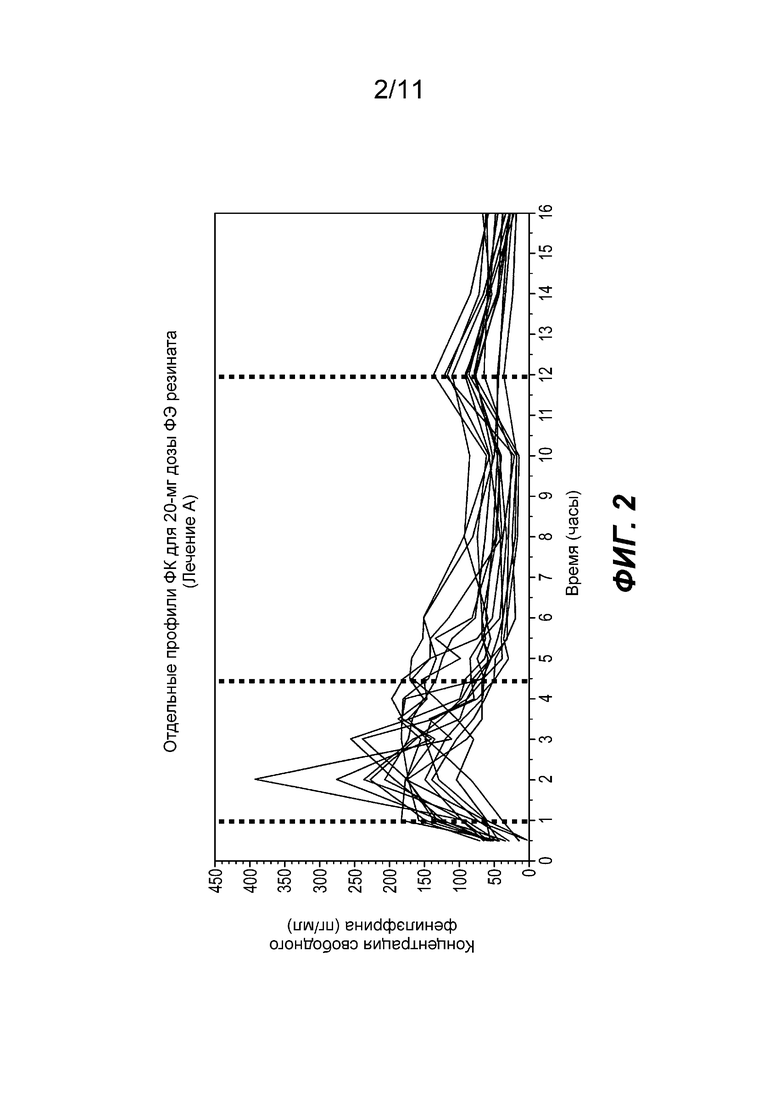
На фиг. 2 показаны отдельные профили концентрации фенилэфрина в плазме при введении частиц фенилэфрина резината ЗВ с покрытием, содержащих 20 мг фенилэфрина. Ссылаясь на фиг. 2, межличностная вариабельность признана хорошей для модифицировано высвобождаемого фенилэфрина. Диапазон Cmax проявился на интервале от примерно 1 часа до примерно 4,5 часа. Вторичный пик через около 12 часов наблюдался у всех пациентов.
Фиг. 3 показывает средний профиль концентрации фенилэфрина в плазме при введении фенилэфрина HCl частиц ЗВ с покрытием, содержащих 20 мг фенилэфрина. Ссылаясь на фиг. 3, пунктирная линия показывает профиль с фиг. 1 для сравнительных целей. Наблюдалось несколько более высокая Cmax с частицами фенилэфрина резината. Вторичный пик через около 12 часов наблюдается в обоих профилях. Это может быть результатом меньшего количества фенилэфрина, метаболизированного персистемно стенкой кишечника в результате быстрого движения частиц вниз по желудочно-кишечному тракту. Высвобождение фенилэфрина в толстой кишке может привести к более высоким уровням поглощения в более позднее время.
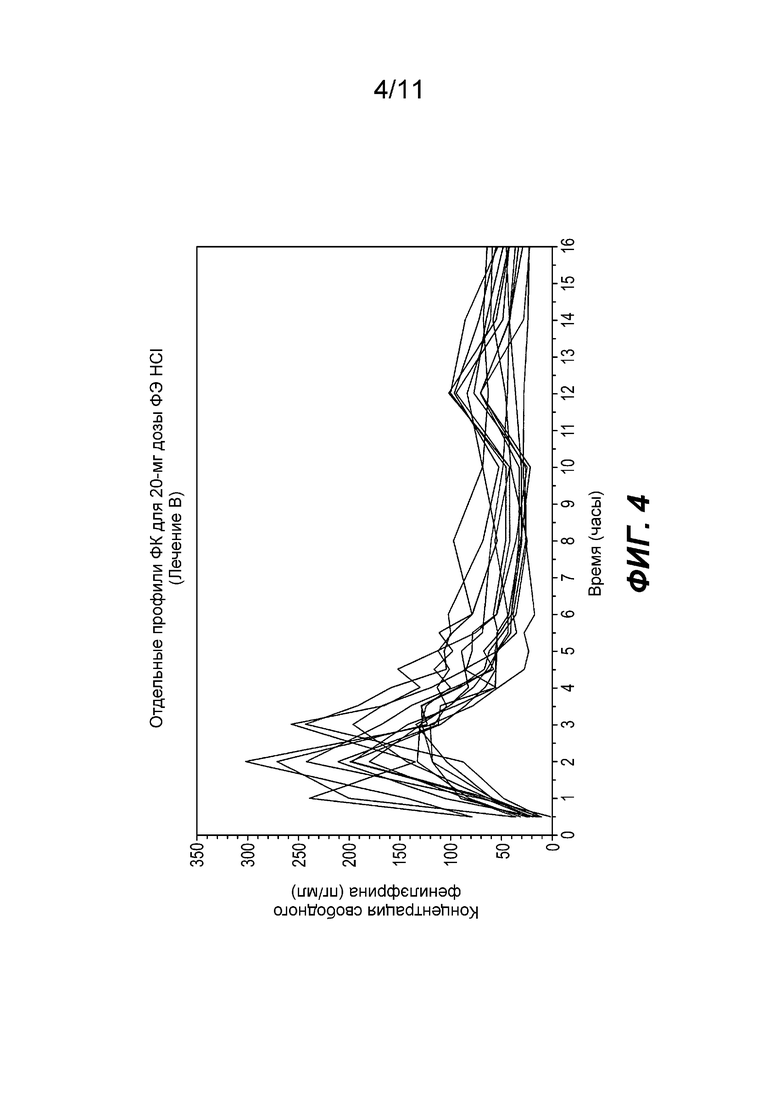
Фиг. 4 показывает отдельные профили концентрации фенилэфрина в плазме при введении фенилэфрина HCl частиц ЗВ с покрытием, содержащих 20 мг фенилэфрина.
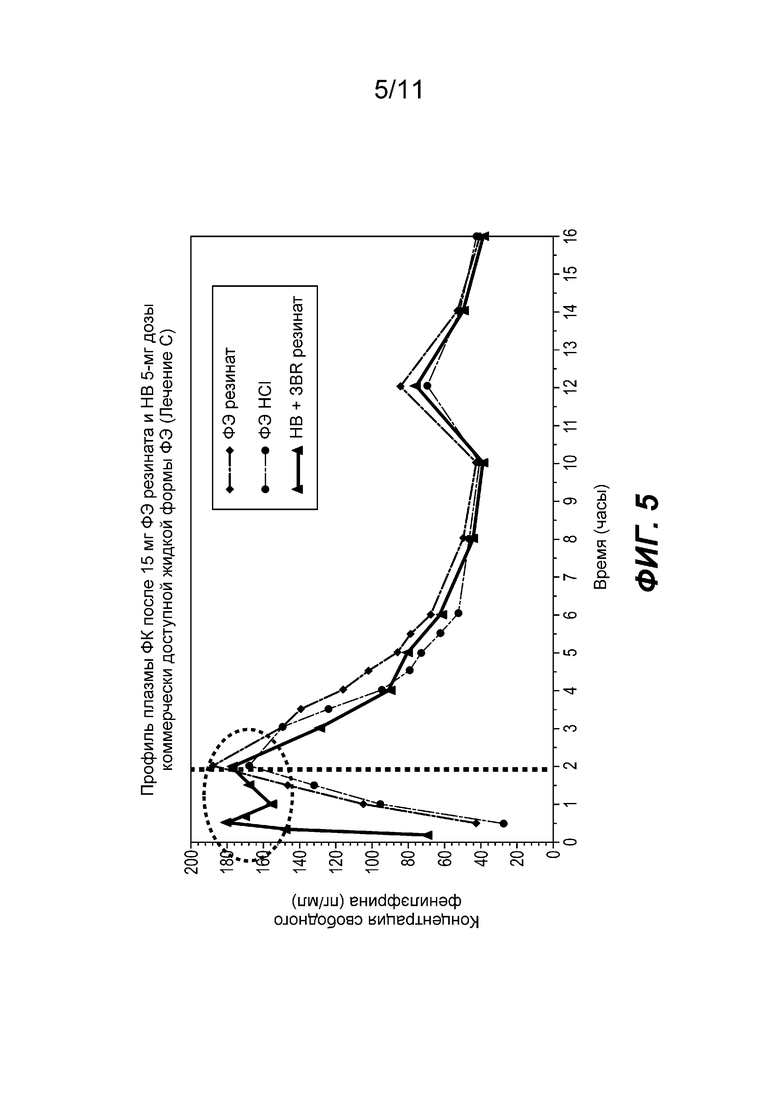
На фиг. 5 показан средний профиль концентрации фенилэфрина в плазме при введении частиц фенилэфрина резината ЗВ с покрытием, содержащих 15 мг фенилэфрина вместе с жидкой формой фенилэфрина HCl НВ, содержащей 5 мг фенилэфрина (далее «ЗВ-НВ смесь»). Ссылаясь на фиг. 5, непрерывная линия представляет ЗВ-НВ смесь. Опять же, кривая для этого лечения соответствует тому, что было видно с резинатом и лекарственным составом, приготовленным с HCl. Для ЗВ-НВ смеси существуют два пика в течение первых 2 часов; один главный от дозы НВ и другой от накопления доз НВ и ЗВ. Cmax достигалась быстрее и сохранялась в течение более длительного периода времени. Таким образом ЗВ-НВ смесь представляется полезной с точки зрения наступления эффективности.
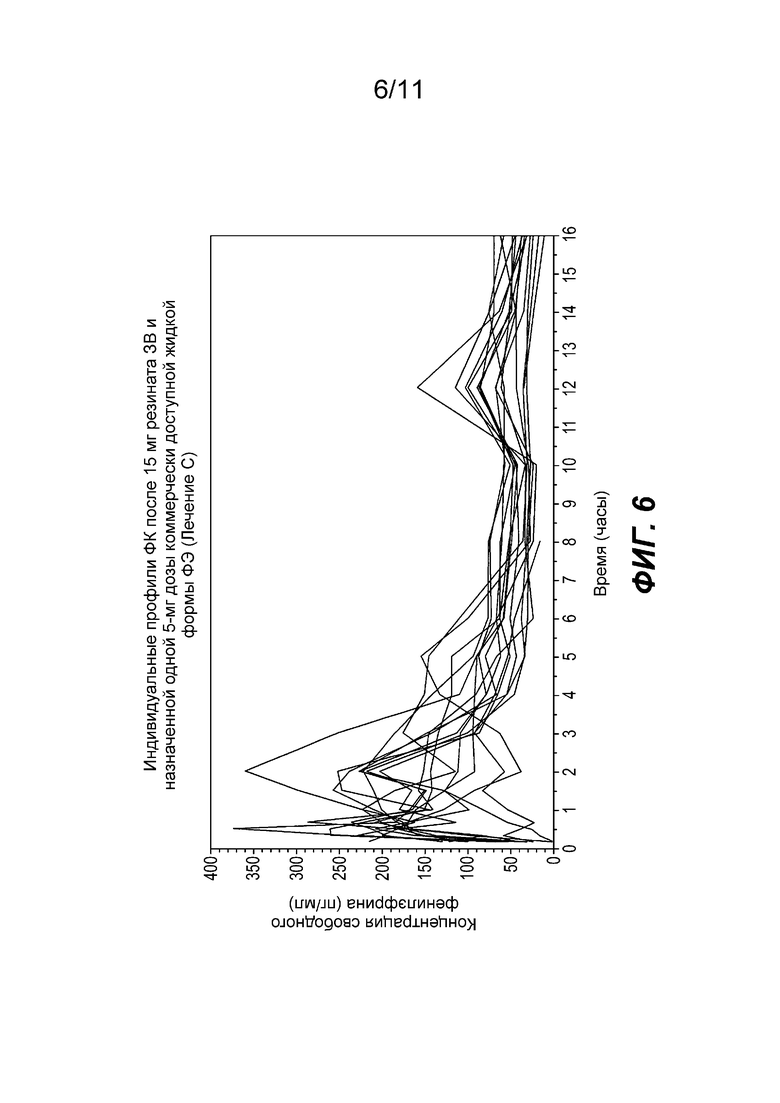
На фиг. 6 показаны отдельные профили концентрации фенилэфрина в плазме при введении частицы фенилэфрина резината ЗВ с покрытием, содержащих 15 мг фенилэфрина вместе с жидкой формой фенилэфрина HCl НВ, содержащей 5 мг фенилэфрина.
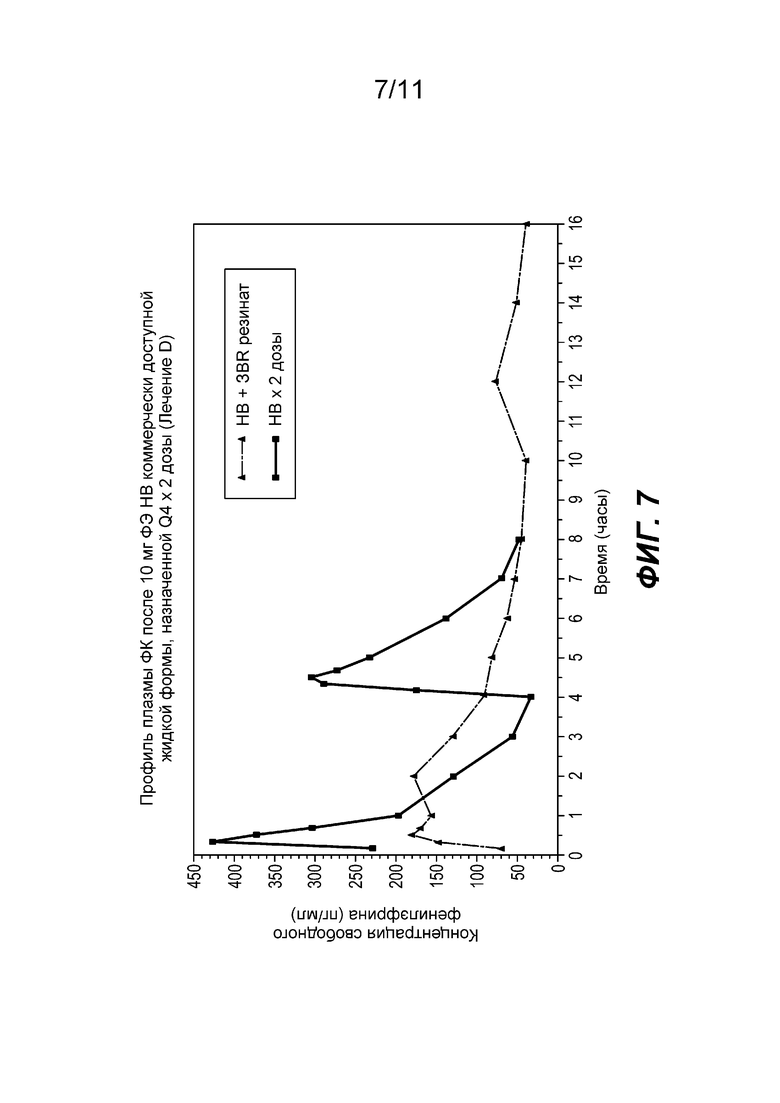
На фиг. 7 показан средний профиль концентрации фенилэфрина в плазме при введении жидкой формы фенилэфрина HCl НВ, содержащей 20 мг фенилэфрина. Ссылаясь на фиг. 7, непрерывная линия представляет собой профиль представленных в настоящее время на рынке жидких лекарственных продуктов НВ и пунктирная линия представляет собой профиль с фиг. 5, приведенный для сравнения. Cmax в ЗВ-НВ смеси ниже Cmax в форме НВ.
На фиг. 8 показаны отдельные профили концентрации фенилэфрина в плазме при введении жидкой формы фенилэфрина HCl НВ, содержащей 20 мг фенилэфрина.
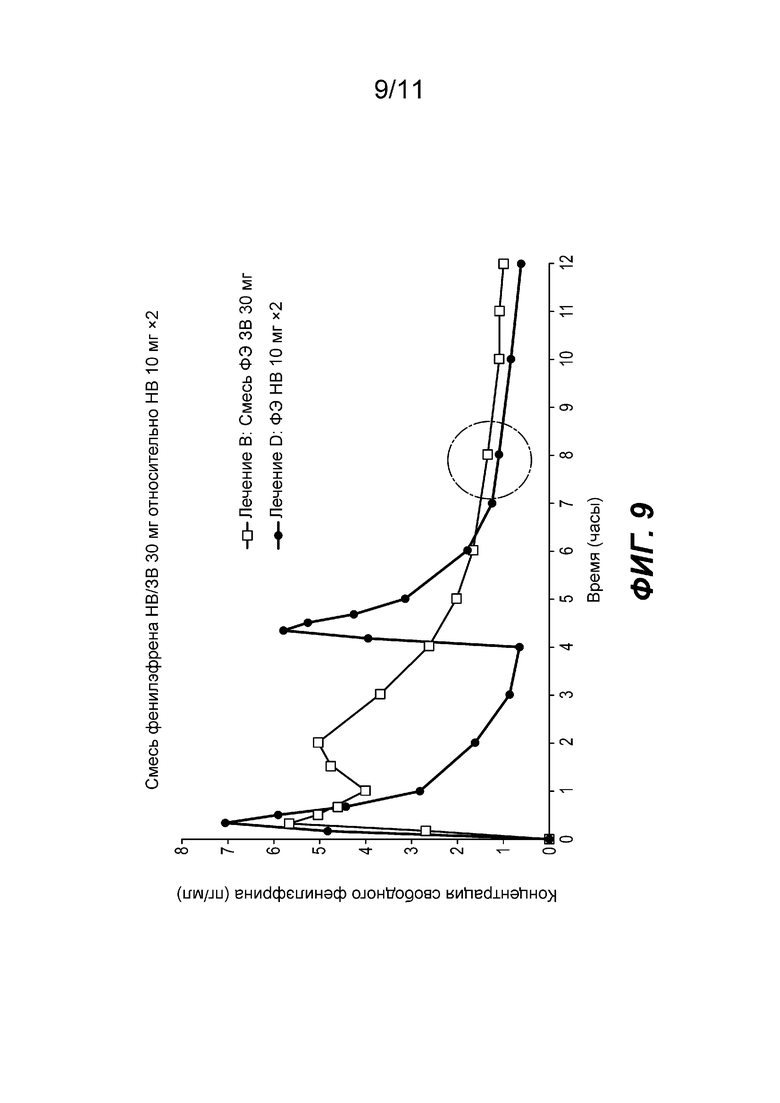
На фиг. 9 показан средний профиль концентрации фенилэфрина в плазме при введении частиц фенилэфрина резината ЗВ с покрытием, содержащих 22,5 мг фенилэфрина вместе с жидкой формой фенилэфрина HCl НВ, содержащей 7,5 мг фенилэфрина (далее «ЗВ-НВ смесь»), по сравнению с жидкой формой фенилэфрина HCl НВ, содержащей 20 мг фенилэфрина.
Фиг. 10А и 10В сравнивают средний профиль концентрации фенилэфрина в плазме (1) при введении частиц фенилэфрина резината ЗВ с покрытием, содержащих 15 мг фенилэфрина и жидкой формы фенилэфрина HCl НВ, содержащей 5 мг фенилэфрина (10А) и (2) при введении частиц фенилэфрина резината ЗВ с покрытием, содержащих 22,5 мг фенилэфрина вместе с жидкой формой фенилэфрина HCl НВ, содержащей 7,5 мг фенилэфрина (фиг. 10b) с (3) жидкой формой фенилэфрина HCl НВ, содержащей 20 мг фенилэфрина.
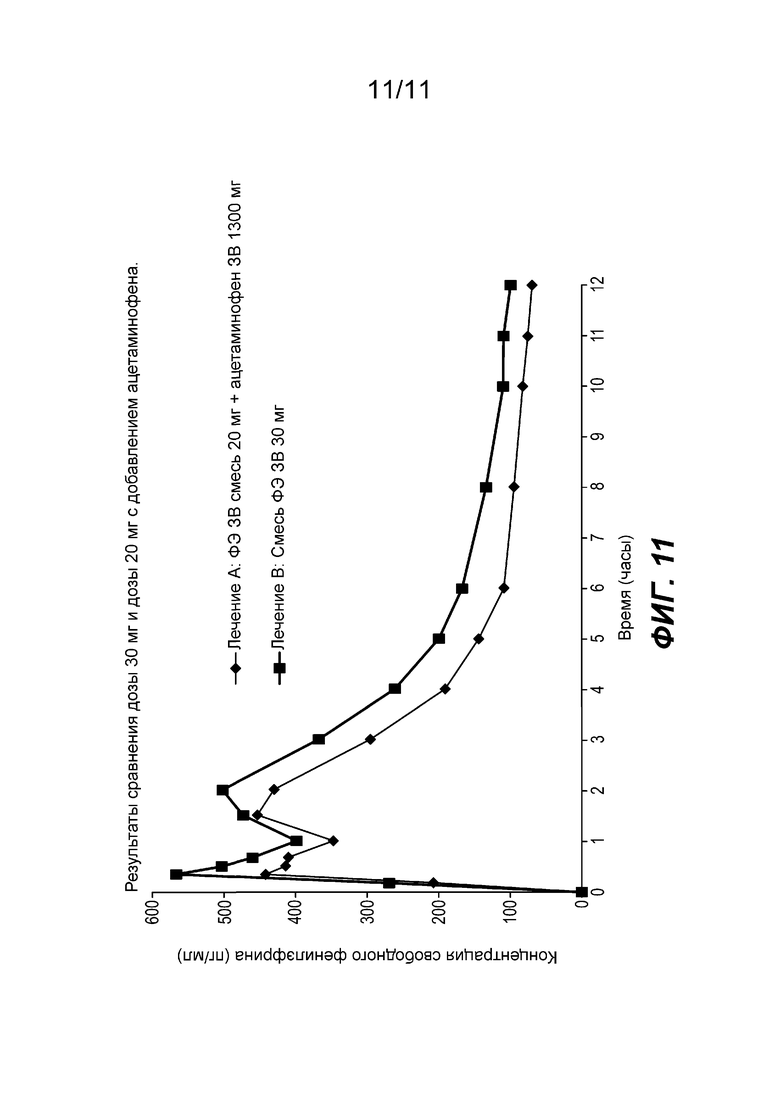
Фиг. 11 сравнивает средний профиль концентрации фенилэфрина в плазме после введения (1) частиц фенилэфрина резината ЗВ с покрытием, содержащих 15 мг фенилэфрина и жидкой формы фенилэфрина HCl НВ, содержащей 5 мг фенилэфрина с (2) комбинацией (а) частиц фенилэфрина резината ЗВ с покрытием, содержащих 15 мг фенилэфрина, (b) жидкой формы фенилэфрина HCl НВ, содержащей 5 мг фенилэфрина и (с) 1300 мг ацетаминофена ЗВ.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Считается, что специалист в данной области, основываясь на представленном в настоящем документе описании, сможет использовать настоящее изобретение в самом полном объеме. Представленные ниже конкретные варианты осуществления следует рассматривать лишь в качестве примеров, которые ни в коей мере не ограничивают остальную часть описания.
Если не определено иное, все технические и научные термины, применяемые в настоящем документе, имеют общепринятое значение, понятное среднему специалисту в области, к которой относится настоящее изобретение. Кроме того, все публикации, заявки на патенты, патенты и другие материалы, упомянутые в настоящем документе, включены в настоящий документ путем ссылки. В настоящем документе все процентные значения, если не указано иное, даны по весу. Кроме того, все указанные в настоящем документе диапазоны предполагают включение любых комбинаций значений между двумя конечными значениями включительно.
Определения
Используемый здесь термин «фармацевтически приемлемая соль фенилэфрина» включает, но не ограничивается, следующими: гидрохлорид фенилэфрина, битартат фенилэфрина, таннат фенилэфрина и т.д. В одном предпочтительном варианте осуществления фармацевтически приемлемая соль фенилэфрина представляет собой гидрохлорид фенилэфрина.
Термин «AUC», используемый здесь, для любого данного лекарственного средства означает «площадь под кривой концентрация-время» дозировки или активации препарата на момент времени, вычисляется по правилу трапеций. AUC является параметром, показывающим кумулятивную концентрацию лекарственного средства в плазме в течение долгого времени, и является показателем общего количества и доступности препарата в плазме.
Используемый здесь термин «Cmax» означает максимальную (пиковую) концентрацию, при которой препарат достигает в тестируемой области после введения и до введения второй дозы.
Используемый здесь термин «кристаллическая форма» означает не-аморфную форму активного ингредиента, показывающую кристаллические свойства, включая, но не ограничиваясь, следующими: способность дифрагировать видимый свет. Термин «кристаллический» также может быть использован для описания активного ингредиента в чистом виде, то есть, например, без добавления других эксципиентов.
Под термином «замедленное высвобождение» подразумевается то, что после приема препарата существует по меньшей мере один период времени, когда активный ингредиент не высвобождается из лекарственной формы, т.е. высвобождение активного ингредиента (ингредиентов) происходит не сразу после перорального приема.
Используемый здесь термин «среда растворения» означает любую подходящую жидкую среду, в которой суспензионная лекарственная форма по настоящему изобретению может быть растворена, такую как, например, среда растворения in vitro, используемая для тестирования продукта, или желудочно-кишечные жидкости. Подходящая среда растворения in vitro, используемая для тестирования растворения активного ингредиента или ингредиентов суспензионной лекарственной формы по настоящему изобретению, включает описанные в Фармакопее США.
Термины «дозировка», «лекарственная форма» или «доза», используемый здесь, означает количество лекарственного препарата, содержащего терапевтически активный агент(ы), вводимый одновременно. Термины «дозировка», «лекарственная форма» или «доза» включают введение одной или нескольких единиц лекарственного препарата, вводимых одновременно. В одном варианте осуществления лекарственная форма представляет собой таблетку. В одном варианте осуществления лекарственная форма представляет собой многослойную таблетку. В варианте, содержащем многослойную таблетку, один слой может содержать порцию немедленного высвобождения, а другой слой может содержать порцию замедленного высвобождения. В варианте, содержащем многослойную таблетку, один слой может содержать частицы фенилэфрина резината, а другой слой может содержать форму немедленного высвобождения фенилэфрина и/или второго активного ингредиента. В одном варианте осуществления лекарственная форма, содержащая частицы фенилэфрина резината, является заполненным жидкостью мягким гелем.
Используемый здесь термин «комплекс действующее вещество-смола» означает связанную форму активного ингредиента, в том числе, но не ограничиваясь, фармацевтически активных ингредиентов и ионообменной смолы. Комплекс действующее вещество-смола также упоминается в данной области техники как «резинат». Ионообменной смолой, которую можно использовать в соответствии с изобретением, является смола Amberlite™ IRP 69, The Dow Chemical Company, нерастворимая сильнокислая натриевая форма катионообменной смолы, которую получают из сульфированного сополимера стирола и дивинилбензола. Мобильным или обменным катионом является натрий, который может быть обменен или заменен на многие базовые виды катионов, в том числе, например, медь, цинк, железо, кальций, стронций, магний и литий. Адсорбция препарата на частицах ионообменной смолы с образованием комплекса действующее вещество-смола является хорошо известной техникой, как показано в патентах США № 2990332 и № 4221778. В целом препарат смешивают с водной суспензией смолы, и комплекс затем промывают и сушат. Адсорбция препарата на смоле может быть обнаружена путем измерения изменения рН реакционной среды или путем измерения изменения концентрации натрия или препарата. Сформированный комплекс действующее вещество-смола может быть собран и промыт этанолом и/или водой для удаления любого несвязанного лекарства. Комплексы, как правило, сушат на воздухе в лотках при комнатной или повышенной температуре. Комплекс действующее вещество-смола содержит соотношение фенилэфрина к смоле от около 0,25:1 до примерно 0,65:1, предпочтительно от примерно 0,30:1 до примерно 0,55:1, предпочтительно от примерно 0,35:1 до примерно 0,45:1.
«Энтеросолюбильный» означает возможность растворения при рН больше, чем примерно 5,0, или больше, чем примерно 5,5, или больше, чем примерно 6,0, или при уровне рН кишечника.
Под термином «замедленное высвобождение» имеется в виду то, что после введения активный ингредиент высвобождается из лекарственной формы по существу непрерывно и регулируемо, и требуется больше времени до полного высвобождения, т.е. истощения, активного ингредиента из лекарственной формы, чем в случае лекарственной формы с тем же действующим веществом с немедленным высвобождением. Виды замедленного высвобождения включают в себя контролируемое высвобождение, поддерживаемое, продленное, высвобождение нулевого порядка, первого порядка, пульсирующее и тому подобное.
Используемый здесь термин «немедленное высвобождение» означает, что характеристики растворения по меньшей мере одного активного ингредиента соответствуют спецификациям ФСША для таблеток с немедленным высвобождением, содержащим активный ингредиент. Активный ингредиент, имеющий непосредственное свойство высвобождения, может быть растворен в желудочно-кишечном содержимом без намерения задержки или продления растворения активного ингредиента.
«Жидкие лекарственные формы» может включать в себя неисключительно суспензии или эликсиры, отличающиеся тем, что один или более из активных ингредиентов находится в растворенном, частично растворенном или в нерастворенном или взвешенном состоянии.
Используемый здесь термин «модифицированное высвобождение» применяется к изменению высвобождения или растворения активного ингредиента в среде для растворения, например, желудочно-кишечных жидкостях. Типы модифицированного высвобождения включают в себя: 1) замедленное высвобождение; или 2) отложенное высвобождение. В целом, дозированные лекарственные формы с модифицированным высвобождением создаются с тем, чтобы сделать активные ингредиенты доступными в течение продолжительного периода времени после их приема, что таким образом позволяет снижать частоту приема в сравнении с приемом того же самого активного ингредиента(ов) в традиционной дозированной форме. Лекарственные формы с модифицированным высвобождением также позволяют использовать комбинации активных ингредиентов, отличающиеся тем, что продолжительность одного активного ингредиента может отличаться от продолжительности другого активного ингредиента.
Используемый здесь термин «фармакодинамика» или «ФД» является изучением взаимосвязи между концентрацией лекарственного средства на участке действия и полученным в результате эффектом.
Используемый здесь термин «фармакокинетика» или «ФК» является изучением временного хода всасывания препарата, распределения, метаболизма и экскреции.
Используемый здесь термин «фенилэфрин» означает бензолметанол, 3-гидрокси-α-[(метиламино)метил], и включает в себя, но не ограничивается ими, фармацевтически приемлемые соли, сложные эфиры, изомеры или их производные.
Используемый здесь термин «скорость высвобождения» препарата относится к количеству высвобожденного лекарственного вещества из лекарственной формы за единицу времени, например, миллиграмм высвобожденного лекарственного вещества в час (мг/ч). Скорости высвобождения лекарственного средства рассчитываются по растворению лекарственной формы in vitro в условиях тестирования, известных в данной области техники. Скорость высвобождения препарата, полученная в заданное время «после введения», относится к скорости высвобождения лекарства in vitro, полученной за заданное время после начала соответствующего теста на растворение, например, одном из тестов согласно ФСША 24 (Фармакопея США 24, United States Pharmacopeia Convention, Inc., Rockville, MD).
Используемый здесь термин «полупроницаемая» означает, что сквозь нее могут проходить молекулы воды и другие молекулы, в том числе молекулы солей и активных ингредиентов, описанных здесь, которые имеют возможность медленно диффундировать через такую мембрану, когда мембрана находится в контакте с соответствующей средой для растворения, например, желудочно-кишечными жидкостями или в среде растворения in vitro.
Термин «полутвердые лекарственные формы» означает лекарственные формы с высокой вязкостью, которые обладают некоторыми из свойств жидкостей, в том числе, но не ограничиваясь (1) возможностью сопротивляться приложенному давлению с изменением формы; и (2) значительно меньшей текучестью в сравнении с жидкостями. Полутвердые лекарственные формы также имеют некоторые свойства твердых тел, в том числе, но не ограничиваясь, имеющих более высокую плотность и определенную форму. Полутвердые вещества могут неисключительно включать гели, жевательные лекарственные формы, жевательные формы на основе пектина, кондитерские жевательные формы, поддающиеся формированию формы желатинового типа.
«Твердые лекарственные формы» означает лекарственные формы, являющиеся по существу твердым при комнатной температуре и имеющие плотность по меньшей мере около 0,5 г/см. Твердые лекарственные формы могут неисключительно включать сформированные таблетки, лекарственные средства в виде капсул, капсулы, наполненные порошком или гранулами, саше, наполненные порошком или гранулами, прессованные таблетки, таблетки с покрытием, жевательные лекарственные формы и быстрорастворимые лекарственные формы.
Используемый здесь термин «по существу покрытая» по отношению к частицам означает, что менее приблизительно 20%, например, менее чем примерно 15% или менее чем примерно 1,0% от площади поверхности частицы открыто, например, не покрыто желаемым покрытием. Используемый здесь термин «по существу покрывает» или «по существу непрерывно» при использовании для описания покрытия означает, что покрытие, как правило, непрерывно и в целом охватывает всю поверхность ядра или подстилающего слоя, так что активный ингредиент или подстилающий слой практически не открыт воздействию. Покрытия, которые применяются к частицам, могут быть слоистыми, где каждый слой приготавливается в смеси водных (на основе воды) или органических растворителей и последовательно добавляется до тех пор, пока толщина покрытия не достигнет требуемого уровня.
Используемый здесь термин «терапевтический эффект» означает любой эффект или действие активного ингредиента, предназначенное для диагностики, лечения, смягчения или предотвращения заболевания, или влияет на структуру или любую функцию тела.
Конкретные варианты осуществления настоящего изобретения проиллюстрированы представленными ниже примерами. Настоящее изобретение не ограничено конкретными ограничениями, установленными в данных примерах.
ПРИМЕРЫ
Частицы фенилэфрина с замедленным высвобождением были разработаны для приготовления жидких и твердых лекарственных форм. Частицы фенилэфрина с замедленным высвобождением можно использовать для соответствия длительности действия таковой у других активных компонентов (в частности, обезболивающих), которые могут действовать более длительный срок, чем фенилэфрин. Такие активные вещества включают, но не ограничиваются следующими: ацетаминофен, ибупрофен, напроксен и их соли и производные.
Пример 1: Подготовка лекарственного препарата, содержащего частицы фенилэфрина замедленного высвобождения с покрытием
Был подготовлен лекарственный препарат, который содержит частицы фенилэфрина, покрытые полимерным покрытием. Способ изготовления лекарственного препарата, который обеспечивает высвобождение фенилэфрина в течение продолжительного периода времени, показал стабильность при 25°C/60% ОВ в течение 24 месяцев и при 40°C/75% ОВ в течение 3 месяцев. Многие гранулированные препараты фенилэфрина не являются стабильными в течение долгого времени и подвергаются значительному распаду.
Партию 3,203 кг частиц фенилэфрина с покрытием получали в соответствии с формулой в таблице 1. Количественная формула и формула партии, соответственно, представлены в таблице 1.
Частицы фенилэфрина замедленного высвобождения с покрытием1
1: Состав на одну дозу частиц, содержащих 20 мг фенилэфрина HCl, составляет приблизительно 377,4 мг. Фактический вес зависит от анализируемого количества фенилэфрина HCl в частицах.
2: Сухой остаток веса.
3: Содержит этилцеллюлозу, цетиловый спирт и лаурилсульфат натрия.
4: Очищенная вода, ацетон и изопропиловый спирт удаляют в процессе обработки.
Наслоение частиц:
1. Очищенную воду ФСША добавляли в подходящего размера контейнер из нержавеющей стали.
2. Этилакрилат НФ и сополимерную дисперсию метилметакрилата НФ (Eudragit® NE 30D) добавляли при осторожном перемешивании.
3. Фенилэфрин HCl ФСША добавляли при сильном перемешивании и перемешивали.
4. Смесь из шага 3 использовали для покрытия (слоя) прежелатинизированного модифицированного крахмала НФ.
Сушка и просеивание:
5. Прослоенный фенилэфрин HCl/прежелатинизированный модифицированный крахмал, полученный на шаге 4, сушили и просеивали через сито № 20.
Покрытие прослоенных частиц покрывающим раствором этилцеллюлозы:
6. Следующие вещества добавили в том порядке, в котором они представлены, в контейнер соответствующего размера при легком помешивании: изопропиловый спирт ФСША, далее - ацетон НФ, далее - ацетилтрибутилцитрат НФ.
7. Этилцеллюлоза НФ (Ethocel® Standard Premium 10) была добавлена при перемешивании, и перемешивание проводилось до получения прозрачного раствора.
8. Стеарат магния добавляли к раствору при перемешивании.
9. Просеянные прослоенные частицы фенилэфрина/прежелатинизированного модифицированного крахмала из шага 5 были покрыты раствором из шага 8 с помощью подходящего элемента с перегородками Вустера для инкапсуляции в псевдоожиженном слое.
Отверждение:
10. Частицы из шага 9 были высушены в духовке.
Покрытие частиц, покрытых Ethocel®, веществами Eudragit® NE30D и Aquacoat ECD®:
11. Далее в подходящего размера контейнер был добавлен Eudragit® NE30D, после чего - очищенная вода ФСША и водная дисперсия этилцеллюлозы НФ (Aquacoat ECD®), все было смешано при осторожном перемешивании.
12. Частицы, покрытые Ethocel® из шага 10, были покрыты раствором для покрытия с помощью подходящего элемента с перегородками Вустера для инкапсуляции в псевдоожиженном слое.
13. Покрытые оболочкой частицы из шага 12 были смешаны с коллоидной двуокисью кремния НФ.
Отверждение:
14. Частицы из шага 13 были высушены в духовке.
Анализ растворения
Покрытые оболочкой частицы фенилэфрина из шага 14 анализировали на растворение от 0 до 14 часов с использованием аппарата, описанного в Фармакопее США <Общие положения Глава <711> Растворение>, Apparatus II с вращающимися лопастями, используя УФ-детектирование при 274 нм. Среда растворения представляла собой 750 мл 0,1 н. HCl в течение первого часа, а затем со второго по четырнадцатый час - 1000 мл 0,05 М натрийфосфатного буфера, рН 6,8. Температура была 37°С, скорость вращения составляла 50 оборотов в минуту. Растворение показало, что высвобожденный процент фенилэфрина в сравнении со стандартом, изготовленным из 100% фенилэфрина в композиции, был меньше или равен 50% в течение 1 часа, больше или равна 30% в течение 3 часов и больше или равен 50% в течение 8 часов. Используемый способ описан ниже, и результаты показаны в таблице 2 ниже.
Способ растворения ФСША Apparatus (2 лопасти, 50 оборотов в минуту)
1. Убедитесь, что температура среды растворения достигла целевого значения (37°C).
2. Взвесьте образцы, эквивалентные 45 мг фенилэфрина HCl. Добавьте образцы (на поверхности среды раствора) в каждый сосуд, содержащий 750 мл 0,1 н. соляной кислоты, и начните испытание на растворимость при скорости лопастей 50 оборотов в минуту. После 1 часа воздействия в 0,1 н. соляной кислоты, завершить измерение в точке времени 1 час. Немедленно перейти к стадии буфера, добавив 250 мл 0,20 М трехосновного фосфата натрия. pH буферной среды составляет 6,8±0,05.
3. Измерьте УФ поглощения фенилэфрина HCl, высвободившегося в среде, с помощью волоконно-оптической системы LEAP с линией зондов для измерения УФ при 274 нм.
4. Количество растворенного фенилэфрина HCl может быть определено с помощью УФ-поглощения раствора образца при испытании, по сравнению с таковым у стандартного раствора при длине волны 274 нм. Количество растворенного фенилэфрина HCl также может быть определено с использованием способа определения количественного состава, указанного ниже.
Способ определения количественного состава
Приготовление образца
1. Точно взвесить примерно 1600 мг частиц фенилэфрина HCl и поместить в мерную колбу 200 мл. (Рекомендуется добавить 1 мл 1%-ного раствора уксусной кислоты/воды для смачивания частиц, чтобы избежать образования твердых комков).
2. Добавить 70 мл 1%-ного раствора уксусной кислоты/ацетонитрила; трясти колбу на качалке с платформой на низкой скорости в течение 1 часа. Примечание: периодически вращать колбу, чтобы удалить частицы, собравшиеся над уровнем растворителя.
3. Добавить около 50 мл 1%-ного раствора уксусной кислоты/воды в колбу, и непрерывно трясти колбу на низкой скорости в течение 1 часа.
4. Разбавить до нужного объема раствором 1% уксусной кислоты/воды и тщательно перемешать.
5. Отфильтровать аликвоту, используя 0,45 мкм фильтр Millipore MILLEX PVDF. Утилизировать первые 1-2 мл фильтрата перед сбором фильтрата для дальнейшего разбавления.
6. Пипеткой перенести 6 мл фильтрата в мерную колбу на 50 мл, разбавить до объема 1% уксусной кислоты/воды и хорошо перемешать.
Анализ фенилэфрина
Стандарт (0,05 мг/мл фенилэфрина HCl в 1% уксусной кислоты/воды) и образцы вводят в подходящую систему ВЭЖХ при условиях, аналогичных предлагаемым ниже. Параметры могут быть изменены для оптимизации хроматографии. Провести анализ фенилэфрина HCl, используя пиковые области тестируемых образцов раствора и сравнить их с пиковыми областями стандартного раствора.
Условия хроматографии ВЭЖХ
(рН 4,6): ацетонитрил (65:35, об/об)
Способ продуктов распада
Приготовление образца
1. Точно взвесить примерно 1600 мг частиц фенилэфрина HCl и поместить в мерную колбу 200 мл. (Рекомендуется добавить 1 мл 1%-ного раствора уксусной кислоты/воды для смачивания частиц, чтобы избежать образования твердых комков).
2. Добавить 70 мл 1%-ного раствора уксусной кислоты/ацетонитрила; трясти колбу на качалке с платформой на низкой скорости в течение 1 часа. Примечание: периодически вращать колбу, чтобы удалить частицы, собравшиеся над уровнем растворителя.
3. Добавить около 50 мл 1%-ного раствора уксусной кислоты/воды в колбу, и непрерывно трясти колбу на низкой скорости в течение 1 часа.
4. Разбавить до нужного объема раствором 1% уксусной кислоты/воды и тщательно перемешать.
5. Отфильтровать аликвоту, используя 0,45-мкм фильтр Millipore MILLEX PVDF. Утилизировать первые 1-2 мл фильтрата перед сбором фильтрата для дальнейшего разбавления.
6. Пипеткой перенести 6 мл фильтрата в мерную колбу на 50 мл, разбавить до объема 1% уксусной кислоты/воды и хорошо перемешать.
Анализ фенилэфрина
Стандарт (0,00025 мг/мл фенилэфрина HCl в 1% уксусной кислоты/воды) и образцы вводят в подходящую систему ВЭЖХ при условиях, аналогичных предлагаемым ниже. Параметры могут быть изменены, чтобы оптимизировать хроматографии. Провести анализ фенилэфрина HCl, используя пиковые области тестируемых образцов раствора и сравнить их с пиковыми областях стандартного раствора.
Условия хроматографии ВЭЖХ
B: [буферный раствор 100 мМ формиат аммония рН 2,9:ацетонитрил (50:50)]
Пример 2: Подготовка частиц фенилэфрина резината замедленного высвобождения с покрытием
Частицы, которые содержат фенилэфрин и катионообменную смолу, были подготовлены и дополнительно покрыты полупроницаемой мембраной. Соотношение количеств покрытия ингредиентов, которое может быть до определенной степени изменено, может быть, например, ацетат целлюлозы:гидроксипропилцеллюлоза 2:1, 3:1, 4:1 или 5:1. Уровень покрытия, который может быть до определенной степени изменен, может быть, например, 50%, 45%, 40%, 35%, 30%, 25% или 20% от массы частицы с покрытием. Большинство частиц в начальной катионообменной смоле имело размер частиц между приблизительно 74 мкм и примерно 177 мкм (микрон).
Частицы фенилэфрина резината, которые обеспечивают высвобождение фенилэфрина в течение продолжительного периода времени, показали стабильность при 25°C/60% ОВ в течение 24 месяцев и при 40°C/75% ОВ в течение 3 месяцев. Многие гранулированные препараты фенилэфрина не являются стабильными в течение долгого времени и подвергаются значительному распаду.
Партию 3,846 кг частиц фенилэфрина резината с покрытием получали в соответствии с формулой в таблице 3. Количественная формула и формула партии представлены в таблице 3 и таблице 4 соответственно.
Количественный состав фенилэфрин резината с покрытием
мг/ед
мг/ед
1: Стандартные дозы частиц, содержащих 20 мг (А) и 15 мг (B) фенилэфрин HCl, составляют примерно 84,2 мг и 63,2 мг соответственно. Фактический вес зависит от анализируемого количества фенилэфрина HCl в частицах.
2: Количество представляет собой свободное основание (1 мг фенилэфрина HCl эквивалентно 0,821 мг свободного основания фенилэфрина).
3: Ацетон и очищенную воду удаляют в процессе обработки.
Формула партии фенилэфрин резината с покрытием
1: Один грамм гидрохлорида фенилэфрина эквивалентен 0,821 г свободного основания фенилэфрина.
2: Ацетон и очищенную воду удаляют в процессе обработки.
Покрытые частицы фенилэфрина резината были произведены, используя следующие шаги обработки:
Просеивание:
1. Полистирол сульфонат натрия ФСША, имеющий желательные размеры частиц, пропускали через сито 170 меш и собрали фракцию, оставшуюся на сите.
Промывка:
2. Полистирол сульфонат натрия ФСША из шага 1 диспергировали в очищенной воде и перемешивали.
3. При перемешивании часть суспензии из шага 2 отфильтровывали и промывали очищенной водой ФСША. Фильтрование продолжали до тех пор, пока большая часть воды не была удалена.
4. Смолу переместили в контейнер.
5. Шаги 3 и 4 повторяли до тех пор, пока вся суспензия не была удалена.
Помещение лекарственного вещества:
6. Очищенную воду ФСША добавляли в подходящего размера контейнер из нержавеющей стали.
7. При перемешивании фенилэфрин HCl был добавлен в контейнер и перемешан до растворения.
8. Промытую смолы из шага 5 добавляли при непрерывном перемешивании и перемешивали в суспензию.
9. При перемешивании часть суспензии из шага 8 была удалена и промыта очищенной водой ФСША. Фильтрование продолжали до тех пор, пока большая часть воды не была удалена.
10. Промытый отфильтрованный фенилэфрина резинат из шага 9 переносили в контейнер.
11. Шаги 9 и 10 были повторены, пока вся суспензии не была отфильтрована.
Сушка:
12. Фенилэфрин резинат был высушен.
Приготовление покрывающего раствора:
13. Очищенная вода ФСША и ацетон НФ были добавлены в соответствующего размера контейнер из нержавеющей стали.
14. Гидроксипропилцеллюлозу НФ медленно добавляли в контейнер и смешивали до полного растворения. Ацетат целлюлозы НФ медленно добавляли и перемешивали до растворения.
15. Ацетон НФ был добавлен до желаемой массы раствора.
Покрытие:
16. Фенилэфрин резинат из шага 12 был покрыт раствором для покрытия из шага 15 с помощью подходящего оборудования с перегородками Вустера для инкапсуляции в псевдоожиженном слое.
17. Фенилэфрин резинат с покрытием выгружали в контейнер.
Сушка:
18. Фенилэфрин резинат с покрытием высушили.
Просеивание:
19. Высушенный покрытый фенилэфрин резинат просеивали через стандартное сито США № 40 меш, и прошедшую через сито фракцию собирали.
Анализ растворения
Покрытые оболочкой частицы фенилэфрина резината из шага 19 анализировали на растворение от 0 до 14 часов с использованием аппарата, описанного в Фармакопее США <Общие положения Глава <711> Растворение>, Apparatus II с вращающимися лопастями, используя УФ-детектирование при 274 нм. Среда растворения представляла собой 750 мл 0,1 н. HCl в течение первого часа, а затем со второго по четырнадцатый час - 1000 мл 0,05 М натрийфосфатного буфера, рН 6,8. Температура была 37°С, скорость вращения составляла 50 оборотов в минуту. Растворение показало, что высвобожденный процент фенилэфрина в сравнении со стандартом, изготовленным из 100% фенилэфрина в композиции, был меньше или равен 50% в течение 1 часа, больше или равен 30% в течение 3 часов и больше или равен 50% в течение 8 часов. Используемый способ описан ниже, и результаты показаны в таблице 5 ниже.
Способ растворения ФСША Apparatus 2 (лопасти), 50 об/мин
1. Убедитесь, что температура среды растворения достигла целевого значения.
2. Добавьте образец (на поверхности среды раствора) в каждый сосуд, содержащий 750 мл 0,1 н. соляной кислоты, и начните испытание на растворимость при скорости лопастей 50 оборотов в минуту. После 1 часа воздействия в 0,1 н. соляной кислоты, извлеките подвергавшийся воздействию в течение часа образец и сразу же перейдите к стадии буфера добавлением 250 мл 0,20 М трехосновного фосфата натрия. pH среды должен составлять 6,8±0,05.
3. Извлеките 10 мл раствора образца растворения из каждой емкости через 1 час, 3 часа, 6 часов (опционально) и 8 часов. Профильтруйте образцы раствора через полнопроточный фильтр Varian (10 мкм).
4. Количество растворенного фенилэфрина может быть определено с помощью УФ-поглощения по сравнению с таковым у стандартного раствора при длине волны 274 нм. Количество растворенного фенилэфрина может быть также определено с использованием способа определения количественного состава фенилэфрина.
5. Можно корректировать растворенное количество на отметке 3, 6 и 8 часов добавлением количества, извлеченного в более ранние моменты времени. Используйте программу DISSL (или эквивалент) или проведите корректировку вручную для промежуточного отбора проб.
Пример 3: Анализ гранулометрического состава
Несколько партий смолы и частиц на основе смолы анализировали на распределение частиц по размерам. Образцы включали (1) смолу Amberlite™ IRP69, коммерчески доступную у The Dow Chemical Company, (2) чистую смолу, имеющую выбранные размеры частиц (подготовленную согласно процессу A или процессу B соответственно), и (3) частицы смолы, содержащие препарат (т.е. содержащие фенилэфрин). Распределение частиц по размерам анализировали с использованием образцов массой примерно 75 грамм в анализаторе FMC Syntron Sieve (FMC Technologies, Houston, TX), с настройками на 90 вольт в течение 11 минут. Сита обрабатывают легким напылением стеарата магния с целью недопущения прилипания во время работы. Полученные результаты показаны в таблицах 6 и 7.
Распределение частиц по размерам может быть проанализировано в меньшем масштабе с использованием, например, ATM L3P Sonic Sifter (Advantech Manufacturing, New Berlin, WI), которая работает с помощью звуковых импульсов в сочетании с механическим перемешиванием для обеспечения эффективного разделения частиц.
Гранулометрический анализ чистой смолы без изменений и процессы отбора A и B
Партия 11
Партия 21
Процесс A2
Процесс A2
Процесс B3
Процесс B3
1. Коммерчески доступная смола Amberlite™ IRP69 без изменений
2. Смола Amberlite™ IRP69 после выбора размера частиц «процесс А»
3. Смола Amberlite™ IRP69 после выбора размера частиц «процесс B»
4. D10, D50 и D90 определяли с помощью GRADISTAT, Blott, S.J. и Pye, K. (2001) GRADISTAT: a grain size distribution and statistics package for the analysis of unconsolidated sediments. Earth Surface Processes and Landforms 26, 1237-1248.
Гранулометрический анализ смолы, содержащей препарат, по процессам отбора А и B
Процесс A1
Процесс A1
Процесс B2
Процесс B2
Процесс B2
Процесс B2
Процесс B2
1. Смола Amberlite™ IRP69 после выбора размера частиц «процесс А»
2. Смола Amberlite™ IRP69 после выбора размера частиц «процесс B»
3. D10, D50 и D90 определяли с помощью GRADISTAT, Blott, S.J. и Pye, K. (2001) GRADISTAT: a grain size distribution and statistics package for the analysis of unconsolidated sediments. Earth Surface Processes and Landforms 26, 1237-1248.
Наблюдалось воздействие соотношения действующее вещество-смола на эффективность помещения препарата на смолу. Результаты показаны в таблицах 8 и 9 ниже.
Влияние соотношения действующее вещество-смола на процесс помещения препарата на смолу для представленных партий помещения препарата на смолу
Выводы о влиянии соотношения действующее вещество-смола на содержание лекарственного вещества в процессе изготовления
2. Основываясь на свободном основании фенилэфрина.
Способ определения количественного состава фенилэфрина - Измерения для таблиц 8 и 9
Приготовление образца
1. Точно взвесить соответствующее количество образца фенилэфрина резината с покрытием (содержащего эквивалент 25 мг фенилэфрина HCl), и перенести взвешенный образец в мерную колбу на 500 мл.
2. Добавить 400 мл разбавителя (1 н. HCl); трясти колбу на качалке с платформой при низкой скорости в течение не менее 2 часов.
3. Чтобы убедиться, что частицы не собираются выше уровня растворителя, периодически смывать частицы в раствор струей растворителя.
4. Довести до нужного объема растворителем и хорошо перемешать.
5. Отфильтровать аликвоту, используя 0,45-мкм шприцевой фильтр Millipore Millex PVDF или его эквивалент. Удалить первые приблизительно 5 мл фильтрата перед сбором остатка в емкость для анализа ВЭЖХ.
Анализ фенилэфрина
Стандарт (0,05 мг/мл фенилэфрина HCl в 1 н. HCl) и образцы вводят в подходящую систему ВЭЖХ при условиях, аналогичных предлагаемым ниже. Параметры могут быть изменены для оптимизации хроматографии. Аналитические результаты действительны при соблюдении характеристик пригодности системы.
Условия хроматографии ВЭЖХ
(рН 4,6): ацетонитрил (65:35, об/об)
Пример 4 - Анализ растворения материала ФК исследования
Покрытые оболочкой частицы фенилэфрина резината, используемые в первом исследовании ФК, втором исследовании ФК и исследовании ФД из примера 5 были проанализированы на растворение в течение от нуля до 8 часов с использованием метода, описанного в примере 2. Результаты показаны в таблице 10А ниже.
Анализ растворения (50 об/мин)
Покрытые оболочкой частицы фенилэфрина резината, используемые в первом исследовании ФК, втором исследовании ФК и исследовании ФД из примера 5 были также проанализированы на растворение в течение от нуля до 8 часов с использованием метода, описанного ниже. Результаты показаны в таблице 10B ниже.
Способ растворения ФСША Apparatus 2 (лопасти), 75 мин
1. Убедитесь, что температура среды растворения достигла целевого значения.
2. Добавьте образец (непосредственно в раствор среды с использованием подходящей трубки) в каждый сосуд, содержащий 750 мл 0,1 н. соляной кислоты, и начните испытание на растворимость при скорости лопастей 75 оборотов в минуту. После 1 часа воздействия в 0,1 н. соляной кислоты, извлеките подвергавшийся воздействию в течение часа образец и сразу же перейдите к стадии буфера добавлением 250 мл 0,20 М трехосновного фосфата натрия. pH среды должен составлять 6,8±0,05.
3. Извлеките 10 мл раствора образца растворения из каждой емкости через 1 час, 3 часа, 6 часов (опционально) и 8 часов. Профильтруйте образцы раствора через полнопроточный фильтр Varian (10 мкм).
4. Определите количество растворенного фенилэфрина с помощью УФ-поглощения по сравнению с таковым у стандартного раствора при длине волны 274 нм.
Количество растворенного фенилэфрина может быть также определено с использованием способа определения количественного состава фенилэфрина.
5. Можно корректировать растворенное количество на отметке 3, 6 и 8 часов добавлением количества, извлеченного в более ранние моменты времени. Используйте программу DISSL (или эквивалент) или проведите корректировку вручную для промежуточного отбора проб.
Анализ растворения (75 об/мин)
Анализ устойчивости
Покрытые оболочкой частицы фенилэфрина резината, используемые в первом исследовании ФК и втором исследовании ФК примера 5, были проанализированы на стабильность при хранении в течение 1 месяца при 25°С и относительной влажности 60% и в течение 1 месяца при 40°С и относительной влажности 75%. Для всех образцов уровни 3-гидроксибензальдегида составляли меньше или равны 0,5%; уровни фенилэфрин 4,6-изомера (N-метил-4,6-дигидрокси-1,2,3,4-тетрагидроксиизохинолон HCl) и фенилэфрин 4,8-изомеров (N-метил-4,8-дигидрокси-1,2,3,4-тетрагидроксиизохинолон HCl) составляли меньше или равно 2,0%. Общее количество продуктов распада по сравнению с фенилэфрином составило меньше или равно 2,0% через 1 месяц в каждой среде.
Способ продуктов распада
Подготовка образца для способа продуктов распада
1. Точно взвесить соответствующее количество образца фенилэфрина резината с покрытием (содержащего эквивалент 25 мг фенилэфрина HCl), и перенести взвешенный образец в мерную колбу на 500 мл.
2. Добавить 400 мл разбавителя (1 н. HCl); трясти колбу на качалке с платформой при низкой скорости в течение не менее 2 часов.
3. Чтобы убедиться, что частицы не собираются выше уровня растворителя, периодически смывать частицы в раствор струей растворителя.
4. Довести до нужного объема растворителем и хорошо перемешать.
5. Отфильтровать аликвоту, используя 0,45-мкм шприцевой фильтр Millipore Millex PVDF или его эквивалент. Удалить первые приблизительно 5 мл фильтрата перед сбором остатка в емкость для анализа ВЭЖХ.
Анализ фенилэфрина для способа продуктов распада
Стандарт (0,00025 мг/мл фенилэфрина HCl в 1 н. HCl) и образцы вводят в подходящую систему ВЭЖХ при условиях, аналогичных предлагаемым ниже. Параметры могут быть изменены для оптимизации хроматографии. Аналитические результаты действительны при соблюдении характеристик пригодности системы.
Условия хроматографии ВЭЖХ
размер пор 100 ангстрем
B: [буферный раствор 100 мМ формиат аммония рН 2,9:ацетонитрил (50:50)]
Пример 5. Клинические исследования
Было проведено два фармакокинетических (ФК) исследования и фармакодинамическое (ФД) исследование.
A. Первое исследование ФК
Пилотное исследование было проведено на шестнадцати пациентах для определения фармакокинетического профиля, биодоступности и метаболизма частиц фенилэфрина замедленного высвобождения с покрытием по примеру 1 и частиц фенилэфрина резината замедленного высвобождения с покрытием по примеру 2. Пациентам были назначены четыре процедуры лечения после ночного голодания. Между четырьмя периодами были семидневные периоды вымывания. В обоих случаях частицы с покрытием, эквивалентные дозе 20 мг фенилэфрина HCl, были введены пациентам в яблочном пюре. Кроме того, была оценена комбинация частиц фенилэфрина резината замедленного высвобождения с покрытием по примеру 2 и коммерческой жидкости с немедленным высвобождением. При комбинированной терапии, частицы фенилэфрина резината с покрытием, эквивалентные 15 мг фенилэфрина HCl, были введены в яблочном пюре, и 10 мл жидкости, эквивалентной 5 мг фенилэфрина HCl, были введены с помощью шприца для перорального введения.
Частицы фенилэфрина замедленного высвобождения с покрытием по примеру 1 и частицы фенилэфрина резината замедленного высвобождения с покрытием по примеру 2 сравнили с жидким средством от заложенности носа McNeil-PPC, Inc.'s Non-Drowsy Children's Sudafed PE® (фенилэфрин HCl 2,5 мг/5 мл). Таблица 11 суммирует процедуры в первом исследовании ФК.
1. Стандартная доза примерно 84,2 мг частиц фенилэфрина резината ЗВ с покрытием эквивалентна в дозе 20 мг фенилэфрина HCl. Стандартная доза примерно 63,2 мг частиц фенилэфрина резината ЗВ с покрытием эквивалентна в дозе 15 мг фенилэфрина HCl, и эту последнюю единичную дозу вводили с 10 мл жидкой формы фенилэфрина 2,5 м/5 мл, в общей дозе, эквивалентной 20 мг фенилэфрина HCl.
Частицы фенилэфрина резината ЗВ с покрытием и частицы фенилэфрина HCl ЗВ вводили перорально после помещения отмеренного количества в чашку с яблочным пюре объемом 118 мл (4 унции) непосредственно перед дозированием. Эти разовые дозы были проглочены не разжевывая, и пациенты запили их 240 мл воды. Жидкая форма фенилэфрина HCl вводилась перорально с помощью шприцов для перорального введения. Для стандартизации условий дозирования эталонного лечение, после первой из двух перорально вводимых 10 мг доз жидкости, пациентам выдали чашки с яблочным пюре объемом 118 мл (4 унции) и 240 мл воды.
Серийные образцы крови собирали в пробирки K3-EDTA в определенные моменты времени в течение 8 или 16 часов после дозы.
B. Второе исследование ФК
Было проведено второе пилотное исследование: (i) для определения того, может ли 30 мг фенилэфрина достичь схожих максимальных концентраций лекарственного препарата по сравнению с двумя дозами 10 мг фенилэфрина немедленного освобождения, полученными через 4 часа друг от друга; и (II) для оценки профиля ЗВ ФК и биодоступности 20 мг фенилэфрина и 1300 мг ацетаминофена.
Второе пилотное исследование было проведено на двадцати пациентах для определения фармакокинетических профилей, биодоступности и метаболизма (1) комбинации (а) частиц фенилэфрина резината замедленного высвобождения с покрытием по примеру 2, эквивалентных 15 мг фенилэфрина HCl, (b) 10 мл фенилэфрина в жидкой форме, эквивалентных 5 мг фенилэфрина HCl, и (с) 1300 мг ацетаминофена замедленного высвобождения; (2) сочетание (а) частиц фенилэфрина резината замедленного высвобождения с покрытием по примеру 2, эквивалентных 22,55 мг фенилэфрина HCl, и (b) фенилэфрина в жидкой форме, эквивалентного 7,5 мг фенилэфрина HCl, (3) сочетание (а) фенилэфрина в жидкой форме, эквивалентного 20 мг фенилэфрина HCl, и (b) 1300 мг ацетаминофена замедленного высвобождения; и (4) фенилэфрина в жидкой форме, эквивалентного 20 мг фенилэфрина HCl. Таблица 12 суммирует процедуры во втором исследовании ФК.
2 Таблетки ацетаминофена с замедленным высвобождением представляли собой ту же гранулированную композицию, которая является коммерчески доступной под маркой Tylenol® Arthritis.
Серийные образцы крови собирали в пробирки K3-EDTA в определенные моменты времени в течение 12 или 20 часов.
Результаты
Результаты исследований ФК показаны на фиг. 1-11 и в таблице 13 ниже.
Сравнение средних параметров первого исследования ФК
(20 мг ФЭ резината)
(20 мг ФЭ HCl)
(5 IR + 15 мг ЗВ резината)
(10 мг IR Q4H × 2 дозы)
(пг-ч/мл)
(пг/мл)
(час)
Примечание: Лечение А, В и С = AUC в течение 16 ч. Лечение D = AUC в течение 8 часов
Цифры округлены
В целом, полученные результаты показывают, что:
ЗВ-НВ смесь, содержащая 20 мг фенилэфрина, показала Сmax, составляющую 50% от таковой у дозы НВ 10 мг, и AUCinf был на 15% больше, чем у двух 10-мг доз НВ (20 мг).
ЗВ-НВ смесь, содержащая 30 мг фенилэфрина, показала Cmax, составляющую 85% от таковой у дозы НВ 10 мг, и AUCinf был на 61% больше, чем у двух 10-мг доз НВ (20 мг).
ЗВ-НВ смесь, содержащая 20 мг фенилэфрина и 1300 мг ацетаминофена, показала Сmax, составляющую 80% от таковой у дозы 10 мг фенилэфрина НВ, и AUCinf, который был на 22% больше, чем у двух 10-мг доз НВ (20 мг).
Полученные результаты показывают, что композиция по настоящему изобретению обеспечивает эффективность в течение длительного периода времени.
Эти результаты также показывают, что композиция по настоящему изобретению может соответствовать продолжительности длительного высвобождения ацетаминофена.
Результаты также показывают, что воздействие фенилэфрина увеличивается и ФК профиль фенилэфрина улучшается по сравнению с 10-мг дозой немедленного высвобождения фенилэфрина при введении фенилэфрина в сочетании с ацетаминофеном. Это может происходить из-за конкуренции за метаболизм в стенки кишечника, что приводит к увеличению поглощения фенилэфрина и не оказывает влияния на ацетаминофен; и длительное высвобождение обеспечивает увеличение поглощения фенилэфрина в нижней части желудочно-кишечного тракта из-за избежания метаболизма в стенки кишечника.
Фармакодинамическое исследование
Проведено рандомизированное, двойное слепое плацебо-контролируемое исследование с целью определения эффективности фенилэфрина и комбинации фенилэфрин-ацетаминофен с замедленным высвобождением у пациентов с заложенным носом и болевыми симптомами, обусловленными инфекциями верхних дыхательных путей. Доза 30 мг ЗВ, доза 45 мг ЗВ и доза 30 мг ЗВ при совместном введении с 1300 мг ацетаминофена были оценены и сравнены с плацебо. В каждом примере использовались частицы фенилэфрина резината ЗВ с покрытием по настоящему изобретению. Доза ЗВ 30 мг, доза 45 мг ЗВ и доза 30 мг ЗВ, введенные совместно с 1300 мг ацетаминофена, показали хорошие результаты в сравнении с плацебо при оценке тяжести следующих симптомов: (1) забитость/заложенность носа; (2) давление в пазухах/чувствительность пазух; и (3) «тяжесть» в голове, обусловленная приливом крови, от 0 до 12 часов на 1 день.
Указанные выше примеры не должны ограничивать область настоящего изобретения, которая может определяться пунктами формулы изобретения. В частности, специалисту в данной области известны различные эквиваленты и заменители, применимые в вышеизложенном описании, и они будут попадать в рамки сферы действия изобретения.
Литература
Blackledge HM, O'Farrell J, Minton NA с соавторами The effect of therapeutic doses of paracetamol on sulphur metabolism in man. Hum Exp Toxicol 1991 май; 10(3): 159-65.
Court MH, Duan SX, Von Moltke LL с соавторами Interindividual variability in acetaminophen glucuronidation by human liver microsomes: Identification of relevant acetaminophen UDP-glucuronosyltransferase isoforms. J Pharmacol Exp Ther 2001; 299(3):998-1006.
Empey DW и Medder KT. Nasal Decongestants. Drugs 1981, 21:438-443.
Hengstmann JH, Goronzy J. Pharmacokinetics of 3H-phenylephrine in man. Eur J Clin Pharmacol. 1982, 21, 335-341.
Hoffman BB. Раздел 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists. В: Goodman & Gilman's The Pharmacologic Basis of Therapeutics - 10 издание. Hardman JG and Limbird LE, изд. McGraw-Hill, Medical Publishing Division, США, 2001.
Ibrahim KE, Midgley JM, Crowley IR и Willaims CM. The mammalian metabolism of R-(-)-m-synephrine. J Pharm Pharmacol. 1983; 35:144-147.
Johnson DA, Hricik JG. The pharmacology of a-adrenergic decongestants. Pharmacother 1993; 13: стр.110-115.
Koch-Weser J. Medical Intelligence: Drug Therapy. N Engl J Med 2 декабря 1976; 295(23):1297-1300.
Manyike PT, Kharasch ED, Kalhorn TF с соавторами Contribution of CYP2E1 and CYP3A to acetaminophen reactive metabolite formation. Clin Pharmacol Ther март 2000; 67(3):275-282.
Miners JO, Atwood J, Birkett DJ. Influence of sex and oral contraceptive steroids on paracetamol metabolism. Br J Clin Pharmacol 1983; 16:503-509.
Miners JO, Osborne NJ, Tonkin AL с соавторами Perturbation of paracetamol urinary metabolic ratios by urine flow rate. Br J Clin Pharmacol 1992; 34:359-362.
Mitchell JR, Thorgeirsson SS, Potter WZ с соавторами Acetaminophen-induced injury: Protective role of glutathione in man and rationale for therapy. Clin Pharmacol Ther 1974; 16:676-684.
Slattery JT, McRorie TI, Reynolds R с соавторами Lack of effect of cimetidine on acetaminophen disposition in humans. Clin Pharmacol Ther ноябрь 1989; 46(5):591-597.
Suzuki O. Matsumoto T. Oya M, Katsumata Y. Oxidation of synephrine by type A and type B monoamine oxidase. Experientia 1979; 35:1283-1284.



Группа изобретений относится к области фармацевтической промышленности, а именно к комплексу действующее вещество-смола с замедленным высвобождением действующего вещества, который покрыт покрытием, а также к способу его получения и к фармацевтическому составу, содержащему указанный комплекс совместно с фармацевтически приемлемым носителем. В предлагаемом комплексе с нанесенным покрытием действующее вещество представляет собой фенилэфрин, смолой является катионнообменная смола, полученная из сульфированного сополимера стирола и дивинилбензола, по меньшей мере 90% которой до комбинирования с фенилэфрином находится в виде частиц размером 74-177 мкм, а покрытие включает ацетат целлюлозы и гидроксипропилцеллюлозу при их массовом соотношении соответственно 2:1, 3:1, 4:1 или 5:1, причем количество покрытия составляет 20, 25, 30, 35, 40, 45 или 50 мас.% от массы покрытого комплекса. Указанный комплекс обеспечивает биодоступность действующего вещества в течение 8 часов и более после введения субъекту и проявляет вторичный пик Сmax через 12 часов после введения субъекту. 3 н. и 3 з.п. ф-лы, 11 ил., 16 табл., 5 пр.
1. Комплекс действующее вещество-смола с замедленным высвобождением действующего вещества, который покрыт покрытием, характеризующийся тем, что указанным действующим веществом является фенилэфрин, указанной смолой является катионнообменная смола, полученная из сульфированного сополимера стирола и дивинилбензола, при этом по меньшей мере 90% указанной катионнообменной смолы до комбинирования с фенилэфрином находится в виде частиц размером от 74 мкм до 177 мкм, указанное покрытие включает ацетат целлюлозы и гидроксипропилцеллюлозу при их массовом соотношении в покрытии соответственно 2:1, 3:1, 4:1 или 5:1, при этом количество указанного покрытия составляет 20, 25, 30, 35, 40, 45 или 50 мас.% от массы покрытого комплекса, причем указанный комплекс с замедленным высвобождением действующего вещества проявляет биодоступность в течение по меньшей мере 8 часов после введения субъекту и проявляет вторичный пик Сmax через 12 часов после введения субъекту.
2. Комплекс по п.1, характеризующийся тем, что катион выбран из группы, состоящей из натрия, меди, цинка, железа, кальция, стронция, магния и лития.
3. Комплекс по п.2, характеризующийся тем, что катион является натрием.
4. Фармацевтический состав, содержащий комплекс с замедленным высвобождением действующего вещества по п.1 и фармацевтически приемлемый носитель.
5. Фармацевтический состав по п.4, характеризующийся тем, что дополнительно включает форму фенилэфрина с немедленным высвобождением.
6. Способ изготовления комплекса с замедленным высвобождением действующего вещества по п.1, включающий нанесение покрытия на комплекс действующее вещество-смола.
| WO 2008064192 A2, 29.05.2008 | |||
| WO 2010115015 A1, 07.10.2010 | |||
| US 6001392 A, 14.12.1999 | |||
| КОМПОЗИЦИИ С МОДИФИЦИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ, СОДЕРЖАЩИЕ КОМПЛЕКСЫ ЛЕКАРСТВЕННОЕ ВЕЩЕСТВО - ИОНООБМЕННАЯ СМОЛА | 2007 |
|
RU2435569C2 |
| US 20060057205 A1, 16.03.2006. | |||

