Настоящее изобретение относится к композициям для перорального введения. Предпочтительно изобретение относится по меньшей мере к одной не вызывающей зависимости композиции для доставки лекарственного средства, имеющего потенциал к вызыванию зависимости, к связанным с ними способам получения этих дозированных форм и к способам лечения пациента, нуждающегося в этом, включающим введение пациенту композиций по изобретению. Более предпочтительно, эти композиции включают по меньшей мере один неопиоидный аналгетик и по меньшей мере один ограниченный опиоидный аналгетик.
УРОВЕНЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Зависимость от отпускаемых по рецепту лекарственных средств стала проблемой общественного здравоохранения во многих сообществах. Опиоиды являются одним из распространенных классов лекарственных средств, которые вызывают зависимость. Опиоиды являются основным классом аналгетиков, используемым для устранения от умеренной до тяжелой боли в США вследствие их эффективности, простоты титрования и благоприятного соотношения риска и пользы.
Одним из эффектов введения опиоидов является способность таких лекарственных средств у некоторых индивидов изменять настроение и ощущения таким образом, чтобы обеспечивалось желаемое ощущение "благополучия", отличающееся от терапевтических улучшающих эффектов. Повторяющееся незаконное применение средств, вызывающих зависимость, приводит у некоторых потребителей к пристрастию к опиоидам. Аналогично опиоидам, многие другие классы лекарственных средств также вызывают зависимость, хотя характер и эффекты зависимости варьируют.
Таким образом, в данной области описаны различные способы и составы для уменьшения или устранения зависимости различного характера, такой как зависимость, связанная со случайным или намеренным быстрым высвобождением в спирте, измельчением и вдыханием и т.д.
В патентной заявке США 11/625705 и заявке PCT PCT/US07/60864, поданной 22 января 2007 года, которые включены в настоящий документ в качестве ссылок в полном объеме для всех целей, описаны различные способы и композиции не вызывающих зависимости составов, имеющих вызывающие зависимость лекарственные средства. В этих патентных заявках использовали обширный скрининг составов для идентификации пригодных экструдируемых составов, проявляющих двухфазное растворение лекарственного средства in vitro (>30% через 1 ч, >80% через 8 ч) для наркотического лекарственного средства гидрокодона битартрата 2,5-гидрата. Однако было выявлено, что растворение второго лекарственного средства ацетаминофена, также известного как парацетамол или APAP, не удовлетворяло критерию двухфазного растворения лекарственного средства (с >30% через 1 ч, >80% через 8 ч). Хотя оба лекарственных средства, гидрокодон битартрат 2,5-гидрат и ацетаминофен, экструдировали и каландрировали из гомогенно перемешанной смеси твердых веществ, во всех исследованиях полученных дозированных форм было показано, что два активных ингредиента высвобождались с различными скоростями. Эти данные in vitro также были подтверждены в исследованиях на экспериментальных животных (карликовые свиньи) и в клинических испытаниях, проведенных с этими дозированными формами. Клиническое испытание также показало, что хотя требуемая кинетика достигалась для гидрокодона битартрата 2,5-гидрата, этого не происходило в случае ацетаминофена. Таким образом, необходимо было найти новые концепции изготовления составов для достижения требуемого двухфазного профиля растворения лекарственного средства также для ацетаминофена. Кроме того, также было выявлено, что в большинстве случаев каландрируемые экструдируемые таблетки, изготовленные в соответствии с патентными заявками US 11/625705 и PCT/US07/60864, имеют шероховатые поверхности и, таким образом, исходя из их внешнего вида, они не во всех случаях удовлетворяли критериям сбываемых на рынке таблеток. Таким образом, также понятна необходимость в усовершенствовании в этом отношении.
Хотя существует множество композиций, составов и способов, обращенных на зависимость от лекарственных средств, все композиции, составы и способы имеют ограничения в большей или меньшей степени. Таким образом, существует необходимость в предоставлении новых и/или усовершенствованных составов, композиций и способов для предотвращения зависимости от лекарственных средств, имеющих потенциал в отношении зависимости. Более конкретно, существует необходимость в разработке пероральных составов, которые удовлетворяли бы двухфазному профилю растворения лекарственного средства и также имели признаки, которые включают сдерживание от применения лекарственного средства и желательный внешний вид для удовлетворения критериям для сбываемых на рынке таблеток. Эта информация об уровне техники предоставлена для того, чтобы сделать известной некоторую информацию, которая, как полагают заявители, возможно имеет отношение к настоящему изобретению. Не подразумевается допущения, а также не следует истолковывать, что любая представленная информация составляет предшествующий уровень техники для настоящего изобретения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Определенные предпочтительные варианты осуществления настоящего изобретения относятся к дозированным формам и способам для доставки лекарственных средств, в частности вызывающих зависимость лекарственных средств, характеризующимся устойчивостью к экстракции растворителем, сдавлению, дроблению или растиранию и обеспечивающим исходную волну высвобождения лекарственного средства с последующим длительным периодом контролируемого высвобождения лекарственного средства. Предпочтительно, дозированная форма включает по меньшей мере один неопиоидный аналгетик и по меньшей мере один ограниченный опиоидный аналгетик.
В одном предпочтительном варианте осуществления, настоящее изобретение относится к фармацевтической композиции, имеющей центральную часть и нецентральный слой, включающей: (a) гидрокодон, его фармацевтически приемлемую соль или гидрат и (b) ацетаминофен или ибупрофен. В этом варианте осуществления, по меньшей мере 75% от всего гидрокодона, его фармацевтически приемлемой соли или гидрата находится в центральной части, и ацетаминофен или ибупрофен находятся в нецентральном слое. Кроме того, эта композиция адаптирована так, чтобы она была пригодна для перорального введения человеку 3, 2 или 1 раз в сутки. Предпочтительно, более чем 90% гидрокодона, его фармацевтически приемлемой соли или гидрата находится в центральной части. Более предпочтительно, по существу весь гидрокодон, его фармацевтически приемлемая соль или гидрат находятся в центральной части. В другом варианте осуществления, центральная часть дополнительно содержит ацетаминофен или ибупрофен. Более предпочтительно, центральная часть дополнительно содержит ацетаминофен.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда единичная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,4 нг/мл/мг и Cmax для ацетаминофена приблизительно от 2,8 нг/мл/мг до 7,9 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,4 нг/мл/мг до приблизительно 1,9 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,0 нг/мл/мг до приблизительно 10,4 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,0 нг/мл/мг и Cmax для ацетаминофена от приблизительно 3,0 нг/мл/мг до приблизительно 5,2 нг/мл/мг, после однократной дозы. Другие варианты осуществления дозированной формы включают приблизительно 5-20 мг гидрокодона битартрата пентагемигидрата и приблизительно 400-600 мг ацетаминофена. Другой вариант осуществления дозированной формы включает 10-15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500-600 мг ацетаминофена.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. При введении пациенту-человеку дозированная форма обеспечивает AUC для гидрокодона от приблизительно 9,1 нг*ч/мл/мг до приблизительно 19,9 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,6 нг*ч/мл/мг до приблизительно 59,1 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 7,0 нг*ч/мл/мг до приблизительно 26,2 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 18,4 нг*ч/мл/мг до приблизительно 79,9 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 11,3 нг*ч/мл/мг до приблизительно 18,7 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,7 нг*ч/мл/мг до приблизительно 53,5 нг*ч/мл/мг. Предпочтительно в этом варианте осуществления, скорость высвобождения фармацевтической композиции in vitro имеет двухфазный профиль высвобождения, и где для каждой фазы скорость высвобождения in vitro имеет нулевой порядок или первый порядок для ацетаминофена и нулевой порядок или первый порядок для гидрокодона битартрата пентагемигидрата.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Дозированная форма обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,18 нг/мл/мг до приблизительно 1,51 нг/мл/мг и концентрацию в плазме C1 через 1 час для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 7,24 нг/мл/мг. В предпочтительных вариантах осуществления, таких как состав 15, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,32 нг/мл/мг до приблизительно 1,51 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 5,50 нг/мл/мг.
В некоторых других вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Дозированная форма обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,30 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 5,57 нг/мл/мг. В предпочтительных вариантах осуществления, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,45 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 4,43 нг/мл/мг.
В других вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 3,63 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена натощак. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 2,76 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,79 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,23 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена 1,80±0,42 мкг/мл с 95% доверительным интервалом для среднего значения от приблизительно 1,61 мкг/мл до приблизительно 2,00 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. 95% доверительный интервал для комбинированной C1 для гидрокодона и ацетаминофена в случае предпочтительных вариантов осуществления и контроля перекрывается. 95% доверительный интервал для среднего значения комбинированной C1 для гидрокодона и ацетаминофена для контроля находится в диапазоне приблизительно от 1,46 до 1,96 мкг/мл, после введения в качестве однократной дозы 15 мг гидрокодона и 500 мг ацетаминофена пациенту-человеку. Контроль обеспечивает достаточные уровни в плазме опиоидного и неопиоидного аналгетика для обеспечения снижения интенсивности боли в пределах приблизительно 1 часа после введения. При введении популяции здоровых северных американцев или западных европейцев, в частности, когда состав адаптирован, чтобы он был пригоден или предназначен для введения человеку каждые 12 часов при необходимости, приблизительно 20-45% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 20-45% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. В другом варианте осуществления, приблизительно 25-35% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 25-35% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. Кроме того, в другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 12 часов, и по меньшей мере от 60% до приблизительно 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 6 часов до приблизительно 8,5 часов. В другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 11 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 8 часов до приблизительно 11 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтических композиций в течение от приблизительно 9 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 9 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 10 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 10 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 11 часов до приблизительно 12 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 11 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 13 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 13 часов.
Однако, когда вариант с замедленным высвобождением состава адаптирован, чтобы он был пригоден или предназначен для введения человеку два раза в сутки, при необходимости, тогда по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 18 часов до приблизительно 23 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 18 часов до приблизительно 23 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 20 часов до приблизительно 25 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 20 часов до приблизительно 25 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 21 часа до приблизительно 22 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 21 часа до приблизительно 22 часов. В другом варианте осуществления этого варианта осуществления с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 22 часов до приблизительно 26 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 22 часов до приблизительно 26 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 27 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 27 часов.
В предпочтительном варианте осуществления, настоящее изобретение относится к композиции, где центральный слой содержит эксципиент или смесь эксципиентов, способных контролировать высвобождение лекарственного средства, и нецентральный слой содержит эксципиент, способный немедленно высвобождать лекарственное средство. Кроме того, в предпочтительном варианте осуществления, центральный слой изготовлен путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, и нецентральный слой нанесен распылением на центральный слой. Наиболее предпочтительно, композиция содержит приблизительно 500 мг ацетаминофена и приблизительно 15 мг гидрокодона битартрата пентагемигидрата.
В другом иллюстративном варианте осуществления, настоящее изобретение относится к фармацевтической композиции, имеющей центральный и нецентральный слой, содержащей: (a) вызывающее зависимость лекарственное средство, его фармацевтически приемлемую соль или гидрат и не вызывающее зависимость лекарственное средство или его фармацевтически приемлемую соль в центральном слое, и (b) не вызывающее зависимость лекарственное средство, его фармацевтически приемлемую соль или гидрат в нецентральном слое. Предпочтительно, эта композиция характеризуется по меньшей мере одним из следующих признаков:
i) количество вызывающего зависимость лекарственного средства, которое экстрагируется из композиции 40% водным раствором этанола в течение одного часа при 37°C in vitro, является меньшим или равно количеству вызывающего зависимость лекарственного средства, которое экстрагируется 0,01 н. хлористо-водородной кислотой in vitro в течение одного часа при 37°C, умноженному на 1,5,
ii) композиция не разрушается при воздействии силы 150 Н, предпочтительно 300 Н, более предпочтительно 450 Н, более предпочтительно 500 Н при измерении с помощью устройства для испытания на твердость "Pharma Test PTB 501",
iii) композиция высвобождает по меньшей мере 20% вызывающего зависимость лекарственного средства и не более чем 45% вызывающего зависимость лекарственного средства в течение первого часа тестирования растворения in vitro и предпочтительно также в течение первого часа тестирования in vivo,
iv) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства в течение от 1 до 2 часов после однократной дозы,
v) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства и/или вызывающего зависимость лекарственного средства через 1 час и через 12 часов после однократной дозы,
vi) в композиции высвобождение вызывающего зависимость лекарственного средства при измельчении повышается менее чем в 2-3 раза, по сравнению с целой таблеткой, когда композицию измельчают в течение 1 минуты кофемолкой при 20000-50000 об/мин, в 40% водном растворе этанола в течение 1 часа при 37°C,
vii) измельченная композиция содержит частицы размером от приблизительно 2 см до приблизительно 355 микрометров для приблизительно 20% фракции, более чем приблизительно 63 микрометра и менее чем приблизительно 355 микрометров для приблизительно 66% фракции и менее чем приблизительно 63 микрометра для приблизительно 14% фракции, при измерении с помощью теста просеиванием, или
viii) композиция является по существу однородной, где среднее значение центральной линии (CLA) составляет от приблизительно 0,1 до приблизительно 0,6, предпочтительно от приблизительно 0,1 до приблизительно 0,4 и наиболее предпочтительно от приблизительно 0,1 до приблизительно 0,2.
В этой композиции, количество вызывающего зависимость лекарственного средства, которое экстрагируется из состава 40% водным раствором этанола в течение одного часа при 37°C, составляет от приблизительно 70% до приблизительно 130% от количества лекарственного средства, которое экстрагируется 0,01 н. хлористо-водородной кислотой в течение одного часа при 37°C. В другом варианте осуществления, количество вызывающего зависимость лекарственного средства, которое экстрагируется из состава 40% водным раствором этанола в течение одного часа при 37°C, составляет от приблизительно 70% до приблизительно 90% от количества лекарственного средства, которое экстрагируется 0,01 н. хлористо-водородной кислотой в течение одного часа при 37°C. В другом варианте осуществления, вызывающее зависимость лекарственное средство, которое экстрагируется из состава 40% водным раствором этанола в течение одного часа при 37°C, составляет от приблизительно 75% до приблизительно 90% от количества лекарственного средства, которое экстрагируется 0,01 н. хлористо-водородной кислотой в течение одного часа при 37°C.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, имеющей центральный слой и нецентральный слой. В этой композиции центральный слой содержит смесь (a) по меньшей мере одного опиоида и (b) по меньшей мере одного изменяющего скорость фармацевтически приемлемого полимера, сополимера или их сочетаний. Нецентральный слой содержит по меньшей мере один неопиоидный аналгетик. Кроме того, эти композиции адаптированы для того, чтобы они были пригодными для перорального введения человеку 3, 2 или 1 раз в сутки. Предпочтительно, центральный слой дополнительно содержит по меньшей мере один неопиоидный аналгетик. В предпочтительном варианте осуществления, композиция характеризуется по меньшей мере одним из следующих признаков:
i) количество вызывающего зависимость лекарственного средства, которое экстрагируется из композиции 40% водным раствором этанола в течение одного часа при 37°C in vitro, является меньшим или равно количеству вызывающего зависимость лекарственного средства, которое экстрагируется 0,01 н. хлористо-водородной кислотой in vitro в течение одного часа при 37°C, умноженному на 1,5,
ii) композиция не разрушается при воздействии силы 150 Н, предпочтительно 300 Н, более предпочтительно 450 Н, более предпочтительно 500 Н при измерении с помощью устройства для испытания на твердость "Pharma Test PTB 501",
iii) композиция высвобождает по меньшей мере 20% вызывающего зависимость лекарственного средства и не более чем 45% вызывающего зависимость лекарственного средства в течение первого часа тестирования растворения in vitro и предпочтительно также в течение первого часа тестирования in vivo,
iv) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства в течение от 1 до 2 часов после однократной дозы,
v) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства и/или вызывающего зависимость лекарственного средства через 1 час и через 12 часов после однократной дозы,
vi) в композиции высвобождение вызывающего зависимость лекарственного средства при измельчении повышается менее чем в 2-3 раза, по сравнению с целой таблеткой, когда композицию измельчают в течение 1 минуты кофемолкой при 20000-50000 об/мин, в 40% водном растворе этанола в течение 1 часа при 37°C,
vii) измельченная композиция содержит частицы размером от приблизительно 2 см до приблизительно 355 микрометров для приблизительно 20% фракции, более чем приблизительно 63 микрометра и менее чем приблизительно 355 микрометров для приблизительно 66% фракции и менее чем приблизительно 63 микрометра для приблизительно 14% фракции, при измерении с помощью теста просеиванием, или
viii) композиция является по существу однородной, где среднее значение центральной линии (CLA) составляет от приблизительно 0,1 до приблизительно 0,6, предпочтительно от приблизительно 0,1 до приблизительно 0,4 и наиболее предпочтительно от приблизительно 0,1 до приблизительно 0,2.
В одном варианте осуществления, опиоид выбран из группы, состоящей из альфентанила, аллилпродина, альфапродина, анилеридина, бензилморфина, безитрамида, бупренорфина, буторфанола, клонитазена, кодеина, циклазоцина, дезоморфина, декстроморамида, дезоцина, диампромида, дигидрокодеина, дигидроморфина, дименоксадола, димефептанола, диметилтиамбутена, диоксафетила бутирата, дипипанона, эптазоцина, этогептазина, этилметилтиамбутена, этилморфина, этонитазена, фентанила, героина, гидрокодона, гидроморфона, гидроксипетидина, изометадона, кетобемидона, леваллорфана, левофенацилморфана, леворфанола, лофентанила, меперидина, мептазинола, метазоцина, метадона, метопона, морфина, мирофина, налбуфина, нарцеина, никоморфина, норпипанона, опиума, оксикодона, оксиморфона, папвретума, пентазоцина, фенадоксона, феназоцина, феноморфана, феноперидина, pиминодина, пропирама, пропоксифена, суфентанила, тилидина, трамадола, их солей, гидратов и смесей.
Кроме того, неопиоидный аналгетик выбран из группы, состоящей из ацетаминофена, аспирина, фентанила, ибупрофена, индометацина, каторолака, напроксена, фенацетина, пироксикама, суфентанила, сунлиндака, интерферона альфа, их солей, гидратов и смесей. Предпочтительно, опиоид представляет собой гидрокодон, и неопиоидный аналгетик представляет собой ацетаминофен или ибупрофен. Более предпочтительно, опиоид представляет собой гидрокодон, и неопиоидный аналгетик представляет собой ацетаминофен.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,4 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,8 нг/мл/мг и 7,9 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,4 нг/мл/мг до приблизительно 1,9 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,0 нг/мл/мг до приблизительно 10,4 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,0 нг/мл/мг и Cmax для ацетаминофена от приблизительно 3,0 нг/мл/мг до приблизительно 5,2 нг/мл/мг, после однократной дозы. В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. При введении пациенту-человеку дозированная форма обеспечивает AUC для гидрокодона от приблизительно 9,1 нг*ч/мл/мг до приблизительно 19,9 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,6 нг*ч/мл/мг до приблизительно 59,1 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 7,0 нг*ч/мл/мг до приблизительно 26,2 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 18,4 нг*ч/мл/мг до приблизительно 79,9 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 11,3 нг*ч/мл/мг до приблизительно 18,7 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,7 нг*ч/мл/мг до приблизительно 53,5 нг*ч/мл/мг. Предпочтительно в этом варианте осуществления, скорость высвобождения фармацевтической композиции in vitro имеет двухфазный профиль высвобождения, и где для каждой фазы скорость высвобождения in vitro имеет нулевой порядок или первый порядок для ацетаминофена и нулевой порядок или первый порядок для гидрокодона битартрата пентагемигидрата.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,18 нг/мл/мг до приблизительно 1,51 нг/мл/мг и концентрацию в плазме через 1 час C1 для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 7,24 нг/мл/мг. В предпочтительных вариантах осуществления, таких как состав 15, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,32 нг/мл/мг до приблизительно 1,51 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 5,50 нг/мл/мг.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,30 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 5,57 нг/мл/мг. В предпочтительных вариантах осуществления, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,45 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 4,43 нг/мл/мг.
В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 3,63 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 2,76 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,79 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,23 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена 1,80±0,42 мкг/мл с 95% доверительным интервалом для среднего значения от приблизительно 1,61 мкг/мл до приблизительно 2,00 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. 95% доверительный интервал для комбинированной C1 для гидрокодона и ацетаминофена в случае предпочтительных вариантов осуществления и контроля перекрывается. 95% доверительный интервал для среднего значения комбинированной C1 для гидрокодона и ацетаминофена для контроля находится в диапазоне приблизительно от 1,46 до 1,96 мкг/мл, после введения в качестве однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена пациенту-человеку. Контроль обеспечивает достаточные уровни в плазме опиоидного и неопиоидного аналгетика для обеспечения снижения интенсивности боли в пределах приблизительно 1 часа после введения.
При введении популяции здоровых северных американцев или западных европейцев, в частности, когда состав адаптирован, чтобы он был пригоден или предназначен для введения человеку каждые 12 часов при необходимости, приблизительно 20-45% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 20-45% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. В другом варианте осуществления, приблизительно 25-35% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 25-35% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. Кроме того, в другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 12 часов, и по меньшей мере от 60% до приблизительно 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 6 часов до приблизительно 8,5 часов. В другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 11 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 8 часов до приблизительно 11 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 9 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 9 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 10 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 10 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 11 часов до приблизительно 12 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 11 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 13 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 13 часов.
Однако, когда вариант с замедленным высвобождением состава адаптирован, чтобы он был пригоден или предназначен для введения человеку два раза в сутки, при необходимости, тогда по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 18 часов до приблизительно 23 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 18 часов до приблизительно 23 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 20 часов до приблизительно 25 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 20 часов до приблизительно 25 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 21 часа до приблизительно 22 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 21 часа до приблизительно 22 часов. В другом варианте осуществления этого варианта осуществления с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 22 часов до приблизительно 26 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 22 часов до приблизительно 26 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 27 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 27 часов.
В предпочтительном варианте осуществления, настоящее изобретение относится к композиции, где центральный слой содержит эксципиент, способный контролировать высвобождение лекарственного средства, и нецентральный слой содержит эксципиент, способный немедленно высвобождать лекарственное средство. Кроме того, в предпочтительном варианте осуществления, центральный слой изготовлен путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, и нецентральный слой нанесен распылением на центральный слой. Наиболее предпочтительно, композиция содержит приблизительно 500 мг ацетаминофена и приблизительно 15 мг гидрокодона битартрата пентагемигидрата.
В другом варианте осуществления, настоящее изобретение относится к фармацевтической композиции, имеющей центральный слой и нецентральный слой. В этой композиции, центральный слой содержит смесь (a) по меньшей мере одного опиоида и по меньшей мере одного первого неопиоидного аналгетика; (b) по меньшей мере одного изменяющего скорость фармацевтически приемлемого полимера, сополимера или их сочетаний. Нецентральный слой содержит по меньшей мере один второй неопиоидный аналгетик. Кроме того, композиция адаптирована таким образом, чтобы она был пригодна для перорального введения человеку 3, 2 или 1 раз в сутки. В этом варианте осуществления, предпочтительно, опиоид включает гидрокодон, и первый и второй неопиоидный аналгетик включает ацетаминофен или ибупрофен. Более предпочтительно, опиоид включает гидрокодон, и первый и второй неопиоидный аналгетик включает ацетаминофен. Кроме того, в этом варианте осуществления, нецентральный слой содержит (a) ацетаминофен и (b) по меньшей мере один изменяющий скорость фармацевтически приемлемый полимер, сополимер или их сочетания. Предпочтительно, полимер или сополимер выбран из группы, состоящей из гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, гидроксиэтилцеллюлозы, полиметакрилата, поливинилового спирта, окиси полиэтилена и их сочетаний. Более предпочтительно, полимер или сополимер выбран из группы, состоящей из гидроксипропилметилцеллюлозы и поливинилового спирта или их сочетаний. Более предпочтительно, полимер или сополимер выбран из группы, состоящей из привитых сополимеров поливинилового спирта и окиси полиэтилена. Кроме того, в этом варианте осуществления, соотношение ацетаминофена и контролирующего скорость полимера или сополимера или их сочетания составляет от приблизительно 1:1 до приблизительно 10:1. Более предпочтительно, соотношение ацетаминофена и контролирующего скорость полимера или сополимера или их сочетания составляет от приблизительно 3:1 до приблизительно 5:1. Как подразумевают для настоящего изобретения, в одном предпочтительном варианте осуществления, нецентральный слой имеет по меньшей мере один из следующих признаков:
(a) по существу не растрескивается после 3 месяцев при 40°C, 75% относительной влажности в индукционно закрытых бутылках HDPE;
(b) по существу сухой (не клейкий);
обеспечивает быстрое растворение в 0,01 н. HCl при 37°C, открывая центральный слой,
высвобождает по меньшей мере 80% ацетаминофена в нецентральном слое в пределах 20 минут после введения пациенту-человеку или
(с) обеспечивает белую пигментацию состава без дополнительных пигментов.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,4 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,8 нг/мл/мг и 7,9 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,4 нг/мл/мг до приблизительно 1,9 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,0 нг/мл/мг до приблизительно 10,4 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,0 нг/мл/мг и Cmax для ацетаминофена от приблизительно 3,0 нг/мл/мг до приблизительно 5,2 нг/мл/мг, после однократной дозы.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. При введении пациенту-человеку дозированная форма обеспечивает AUC для гидрокодона от приблизительно 9,1 нг*ч/мл/мг до приблизительно 19,9 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,6 нг*ч/мл/мг до приблизительно 59,1 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 7,0 нг*ч/мл/мг до приблизительно 26,2 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 18,4 нг*ч/мл/мг до приблизительно 79,9 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 11,3 нг*ч/мл/мг до приблизительно 18,7 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,7 нг*ч/мл/мг до приблизительно 53,5 нг*ч/мл/мг. Предпочтительно в этом варианте осуществления, скорость высвобождения фармацевтической композиции in vitro имеет двухфазный профиль высвобождения, и где для каждой фазы скорость высвобождения in vitro имеет нулевой порядок или первый порядок для ацетаминофена и нулевой порядок или первый порядок для гидрокодона битартрата пентагемигидрата.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,18 нг/мл/мг до приблизительно 1,51 нг/мл/мг и концентрацию в плазме C1 через 1 час для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 7,24 нг/мл/мг.
В предпочтительных вариантах осуществления, таких как состав 15, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,32 нг/мл/мг до приблизительно 1,51 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 5,50 нг/мл/мг.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,30 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 5,57 нг/мл/мг. В предпочтительных вариантах осуществления, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,45 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 4,43 нг/мл/мг. В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 3,63 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 2,76 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,79 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,23 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена 1,80±0,42 мкг/мл с 95% доверительным интервалом для среднего значения от приблизительно 1,61 мкг/мл до приблизительно 2,00 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. 95% доверительный интервал для комбинированной C1 для гидрокодона и ацетаминофена в случае предпочтительных вариантов осуществления и контроля перекрывается. 95% доверительный интервал для среднего значения комбинированной C1 для гидрокодона и ацетаминофена для контроля находится в диапазоне приблизительно от 1,46 до 1,96 мкг/мл, после введения в качестве однократной дозы 15 мг гидрокодона и 500 мг ацетаминофена пациенту-человеку. Контроль обеспечивает достаточные уровни в плазме опиоидного и неопиоидного аналгетика для обеспечения снижения интенсивности боли в пределах приблизительно 1 часа после введения
При введении популяции здоровых северных американцев или западных европейцев, в частности, когда состав адаптирован, чтобы он был пригоден или предназначен для введения человеку каждые 12 часов при необходимости, приблизительно 20-45% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 20-45% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. В другом варианте осуществления, приблизительно 25-35% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 25-35% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. Кроме того, в другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 12 часов, и по меньшей мере от 60% до приблизительно 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 6 часов до приблизительно 8,5 часов. В другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 11 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 8 часов до приблизительно 11 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 9 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 9 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 10 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 10 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 11 часов до приблизительно 12 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 11 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 13 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 13 часов.
Однако когда вариант с замедленным высвобождением состава адаптирован, чтобы он был пригоден или предназначен для введения человеку два раза в сутки, при необходимости, тогда по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 18 часов до приблизительно 23 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 18 часов до приблизительно 23 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 20 часов до приблизительно 25 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 20 часов до приблизительно 25 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 21 часа до приблизительно 22 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 21 часа до приблизительно 22 часов. В другом варианте осуществления этого варианта осуществления с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 22 часов до приблизительно 26 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 22 часов до приблизительно 26 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 27 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 27 часов.
В предпочтительном варианте осуществления, настоящее изобретение относится к композиции, где центральный слой содержит эксципиент, способный контролировать высвобождение лекарственного средства, и нецентральный слой содержит эксципиент, способный немедленно высвобождать лекарственное средство. Кроме того, в предпочтительном варианте осуществления, центральный слой изготовлен путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, и нецентральный слой нанесен распылением на центральный слой. Наиболее предпочтительно, композиция содержит приблизительно 500 мг ацетаминофена и приблизительно 15 мг гидрокодона битартрата пентагемигидрата.
Эти и другие задачи, преимущества и признаки изобретения будут очевидны специалисту в данной области после прочтения подробного описания способов и композиций по изобретению, используемых в нем, как более подробно описано ниже.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На фиг.1 показано, что нанесение покрытия на экструдированные таблетки приводило к значительному сглаживанию поверхности таблетки.
На фиг.2 представлена схема для вычисления шероховатости поверхности с использованием подхода среднего значения центральной линии (CLA).
На фиг.3 представлено среднее значение центральной линии (CLA) для состава без покрытия. Для состава без покрытия CLA=36,1, когда (N=69).
На фиг.4 представлено среднее значение центральной линии (CLA) для состава без покрытия. Для состава с покрытием CLA=10,4, когда (N=69).
На фиг.5 представлены предварительные профили средняя концентрация гидрокодона-время для составов 15 и 16 и контроля 1 в течение (a) 48 часов и (b) 12 часов.
На фиг.6 представлены предварительные профили концентрация ацетаминофена-время для составов 15 и 16 и контроля 1 в течение (a) 48 часов и (b) 12 часов.
На фиг.7 представлены профили высвобождения лекарственного средства in vitro для гидрокодона и ацетаминофена для составов 17 и 18, контроля 2 и состава без покрытия VM-1 в течение 480 минут.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение не ограничивается конкретными описанными способами, протоколами, испытаниями на животных и реагентами, которые могут варьировать. Также следует понимать, что терминология, используемая в настоящем документе, предназначена только для целей описания конкретных вариантов осуществления и не предназначена для ограничения объема настоящего изобретения, который ограничивается только прилагаемой формулой изобретения. Необходимо отметить, что, как используют в настоящем документе и в прилагаемой формуле изобретения, формы единственного числа включают формы множественного числа, если контекст ясно не указывает на иное. Таким образом, например, указание на "соединение" включает множество таких соединений и их эквивалентов, известных специалистам в данной области, и т.д. Также, термины "один или несколько" и "по меньшей мере один" могут быть использованы в настоящем документе взаимозаменяемо. Также следует отметить, что термины "содержащий", "включающий" и "имеющий" могут быть использованы взаимозаменяемо.
Если нет иных указаний, все технические и научные термины, используемые в настоящем документе, имеют те же значения, которые обычно понятны специалисту в области, к которой относится изобретение. Хотя любые способы и материалы, сходные или эквивалентные способам и материалам, описанным в настоящем документе, можно использовать для осуществления на практике или тестирования настоящего изобретения, предпочтительные способы и материалы описаны в настоящем документе. Все публикации, упомянутые в настоящем документе, включены в настоящий документ в качестве ссылок для целей описания и раскрытия химических веществ, животных, инструментов, статистического анализа и способов, которые описаны в публикациях, которые можно использовать применительно к изобретению. Ничто в настоящем документе не следует истолковывать как допущение того, что настоящее изобретение не дает право датировать такую публикацию более ранним числом на основании предшествующего изобретения.
Торговые названия используют в этом описании в качестве удобного сокращения для хорошо известных материалов. Как поймет специалист в данной области, следующие названия торговых марок указывают на указанные вещества:
EUDRAGIT®: Полимеры, образованные из сложных эфиров акриловой и метакриловой кислоты
METHOCEL®: Метил- или метоксилцеллюлоза
KOLLICOAT®: Привитые сополимеры поливиниловый спирт-полиэтиленгликоль
PLASDONE®: Полимер или сополимер поливинилпирролидона
LAUROGLYCOL®: Сложный эфир лаурата и пропиленгликоля
SPAN®: Сложные эфиры сорбитана и жирных кислот
CREMOPHOR®: Полиэтоксилированное касторовое масло
POLOXAMER®: Блок-сополимеры полиоксиэтилена и полиоксипропилена или полиоксиэтилена и полипропиленгликоля
TWEEN®: Сложные эфиры полиэтоксилированного сорбитана
KLUCEL®: Гидроксипропилцеллюлоза
KOLLIDON®: Гомо- и сополимеры поливинилпирролидона
XYLITOL®: (2,3,4,5)тетрагидроксипентанол
ISOMALT®: Эквимолярная композиция 6-0-α-D-глюкопиранозидо-D-сорбит (1,6-GPS) и 1-0-α-D-глюкопиранозидо-D-маннит-дигидрат (1,1-GPM-дигидрат)
POLYOX®: Растворимые в воде смолы на основе полиэтиленоксида
XYLIT®: (2,3,4,5)тетрагидроксипентанол
PLUROL OLETQUE®: Олеиновые сложные эфиры полиглицерина
LUTROL®: Блок-сополимеры полиоксиэтилена и полиоксипропилена или полиоксиэтилена и полипропиленгликоля
ETHOCEL®: Этилцеллюлоза
PRIMOJEL®: Натрия крахмала гликолят
Настоящее изобретение относится к улучшенному твердому веществу или твердому раствору, пероральному дозированному составу, который обеспечивает замедленное высвобождение in vivo фармацевтически активных соединений ("лекарственных средств"), которые обладают свойствами, обеспечивающими им вероятность вызывать зависимость, или для которых было показано, что они часто вызывают зависимость, а также их солей, сложных эфиров, пролекарств и других их фармацевтически приемлемых эквивалентов.
Термин "AUC" относится к площади по кривой зависимости концентрации от времени, вычисленной с использованием правила трапеции и Clast/k, где Clast представляет собой последнюю наблюдаемую концентрацию, и k представляет собой вычисленную константу скорости выведения.
Термин "AUCt" относится к площади под кривой зависимости концентрации от времени для последней наблюдаемой концентрации, вычисленной с использованием правила трапеции.
Термин "Cmax" относится к концентрации в плазме указанного вызывающего зависимость лекарственного средства в Tmax, выраженной в нг/мл и мкг/мл, соответственно, к которой приводит пероральный прием композиции по этому изобретению. Если конкретно не указано иное, Cmax относится к общей максимальной наблюдаемой концентрации.
Термин "Cmin" относится к минимальной наблюдаемой концентрации в определенном интервале дозирования, например в интервале дозирования двенадцать часов для состава, указанного в качестве пригодного для дозирования каждые 12 часов или при необходимости, дозированной формы по этому изобретению, вводимой в течение непрерывных интервалов дозирования из 5 доз.
Термин "нг*ч/мл/мг" относится к количеству вещества, измеряемому в количестве нанограмм, умноженному на количество часов на миллилитр крови, деленному на количество миллиграмм вызывающего зависимость лекарственного средства, вводимого животному или человеку.
Как используют в настоящем документе, выражение "возрастающая скорость высвобождения" относится к скорости растворения, которая, главным образом, повышается с течением времени, так что лекарственное средство растворяется в жидкости в применяемой среде со скоростью, которая, главным образом, возрастает со временем, а не остается постоянной или не снижается, до истощения приблизительно 80% лекарственного средства дозированной формы.
При применении в указанных выше и других способах лечения, можно использовать терапевтически эффективную дозу одного из соединений по настоящему изобретению в чистой форме или, когда такая форма существует, в форме фармацевтически приемлемой соли, сложного или пролекарства. Выражение "терапевтически эффективная доза" соединения по изобретению означает достаточное количество соединения для лечения нарушений, при приемлемом соотношении польза/риск, применимом для любого медицинского способа лечения. Однако будет понятно, что решение об общем суточном применении соединений и композиций по настоящему изобретению принимает лечащий врач в соответствии с его медицинским мнением. Конкретный уровень терапевтически эффективной дозы для любого конкретного пациента будет зависеть от множества факторов, включая нарушение, подвергаемое лечению, и тяжесть нарушения; активность конкретного используемого соединения; конкретная используемая композиция; возраст, масса тела, общее состояние здоровья, пол и режим питания пациента; время введения, путь введения и скорость выведения конкретного используемого соединения; длительность лечения; лекарственные средства, используемые в сочетании или одновременно с конкретным используемым соединением; и сходные факторы, хорошо известные в области медицины.
В одном предпочтительном варианте осуществления, это изобретение относится к дозированным формам, которые ингибируют экстракцию из состава лекарственного средства обычными растворителями, например, но не ограничиваясь этим, дистиллированным водным этанолом. Состав предотвращает зависимость вследствие ограничения способности лиц экстрагировать опиоид из состава (либо преднамеренно, либо непреднамеренно), так что опиоид нельзя легко концентрировать для парентерального введения. Также эти обеспечивающие устойчивость к зависимости составы нельзя легко раздробить на более мелкие частицы или форму порошка, которые легко вызывают зависимость при вдыхании через нос. Такой обеспечивающий устойчивость к зависимости состав не требует включения опиоидного антагониста (хотя опиоидный антагонист можно добавлять к препарату для дополнительного предотвращения зависимости). Без связи с какой-либо конкретной теорией, полагают, что включение алкилцеллюлоз, таких как (но не ограничиваясь ими) гидроксиметилцеллюлозы и предпочтительно гидроксипропилметилцеллюлозы, приводит к устойчивости состава к экстракции в спирте, в частности в 20% или 40% водном растворе этанола. Алкилцеллюлоза предпочтительно обладает по меньшей мере 12% замещением алкильным заместителем, более предпочтительно по меньшей мере 16% замещением алкильным заместителем и наиболее предпочтительно по меньшей мере 19% замещением алкильным заместителем. Алкильное замещение целлюлозы, составляющее менее приблизительно 40% и более предпочтительно менее приблизительно 30%, является предпочтительным в контексте этого изобретения. Кроме того, предпочтительно алкильный заместитель представляет собой C1-C6, более предпочтительно C1, C2 или C4 и наиболее предпочтительно C3, и он может быть прямым или разветвленным, когда алкильный заместитель содержит 3 или более атомов углерода.
В другом предпочтительном варианте осуществления, дозированные формы необязательно являются устойчивыми к разрезанию, измельчению, пульверизации и т.п. Удобным показателем этого аспекта данного изобретения является "прочность", как определяют посредством устройства для тестирования твердости "Pharma Test PTB 501". Состав по этому изобретению предпочтительно обладает прочностью по меньшей мере 150 Ньютон (150 Н). Более предпочтительно, состав по этому изобретению обладает прочностью по меньшей мере 300 Н, более предпочтительно по меньшей мере 450 Н и более предпочтительно по меньшей мере 500 Н.
Прочность в соответствии с настоящим изобретением можно определять с помощью таблетки диаметром 10 мм и толщиной 5 мм в соответствии со способом определения прочности таблеток, опубликованным в European Pharmacopoeia 1997, страница 143, 144, способ № 2.9.8. Предпочтительное устройство, используемое для измерения прочности, представляет собой устройство для тестирования материалов "Zwick 22.5", Fmax=2,5 кН, максимальное перемещение 1150 мм с установкой, содержащей колонку и измерительный наконечник, клиренсом менее 100 мм и скоростью тестирования 0,1800 мм/мин. Измерение можно проводить с использованием нагнетательного поршня с вворачиваемой вставкой и цилиндра (диаметром 10 мм), силового измерительного преобразователя (Fmax 1 кН, диаметр=8 мм, класс 0,5 от 10 Н, класс 1 от 2 н. для ISO 7500-1, суммарная сила Zwick Fmax=1,45 кН). Устройство необязательно может быть получено из Zwick GmbH & Co. KG, Ulm, Germany.
Для получения композиции по изобретению можно использовать любой пригодный способ. В предпочтительном варианте осуществления, состав предпочтительно подвергают плавлению и более предпочтительно экструдированию плавлением, а затем в любом случае сразу придают форму без дробления или измельчения состава. Несмотря на указанное выше, подразумевают, что таблетки состава, которым сразу придали форму, можно необязательно покрывать добавкой для облегчения проглатывания, такой как, но не ограничиваясь этим, желатиновое покрытие. Без связи с какой-либо конкретной теорией, полагают, что прямое придание формы для предотвращения образования нежелательных сильно выступающих элементов на составе без промежуточной стадии измельчения приводит к наиболее высокой прочности состава. Кроме того, варианты осуществления состава по изобретению необязательно обеспечивают дополнительную прочность с помощью по меньшей мере двух подвергнутых плавлению полимеров. Без связи с конкретной теорией, полагают, что второй подвергнутый плавлению полимер предпочтительно взаимодействует с первым подвергнутым плавлению полимером так, чтобы преимущественно изменять температуру перехода в стеклообразное состояние композиции в целом в ходе образования таблетки.
В одном варианте осуществления, в составе можно использовать полимер, или сополимер, или их сочетание для создания подвергнутого плавлению и более предпочтительно экструдированного плавлением прямо сформованного состава. Также можно использовать полимеры, которые являются фармакологически неактивными и обеспечивают растворимое в кишечнике покрытие или профиль замедленного освобождения состава. В одном варианте осуществления, пригодные полимеры/сополимеры включают поли(мет)акрилат, например, такой как Eudragit L- или S-типа, которые являются фармакологически неактивными.
EUDRAGIT® представляет собой торговое название некоторых предпочтительных полимеров, которые пригодны для применения в данном изобретении и которые образованы из сложных эфиров акриловой и метакриловой кислоты. Свойства полимеров EUDRAGIT, главным образом, определяются функциональными группами, включенными в мономеры полимеров EUDRAGIT. Отдельные классы EUDRAGIT® отличаются по их соотношению нейтральных, щелочных и кислых групп и, таким образом, по их физико-химическим свойствам. Можно использовать аммоний-алкильные метакрилатные сополимеры или метакрилатные сополимеры, имеющие следующую формулу:
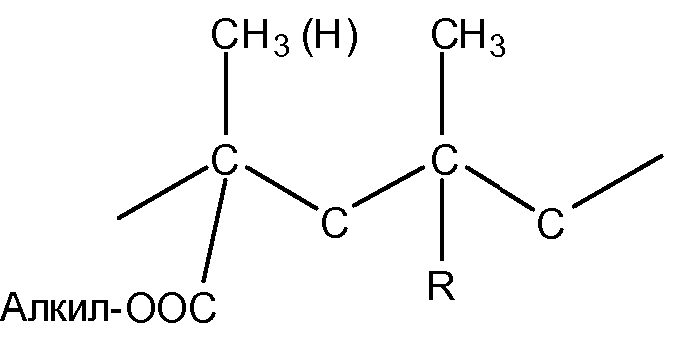
Полимеры Eudragit удовлетворяют описаниям/требованиям, указанным в USP. В соответствии с 2007 US Pharmacopoeia Eudragit определяют согласно с USP 30/NF 25
Сополимер метакриловой кислоты, тип A NF = Eudragit L-100
Сополимер метакриловой кислоты, тип B NF = Eudragit S-100
Сополимер метакриловой кислоты, тип C NF = Eudragit L-100-55 (содержит небольшое количество детергента)
Аммоний-метакрилатный сополимер, тип A NF = Eudragit RL-100 (гранулы)
Аммоний-метакрилатный сополимер, тип A NF = Eudragit RL-PO (порошок)
Аммоний-метакрилатный сополимер, тип B NF = Eudragit RS-100 (гранулы)
Аммоний-метакрилатный сополимер, тип B NF = Eudragit RS-PO (порошок)
Полиакрилатная дисперсия, 30% Ph. Eur. = Eudragit NE30D (=30% водная дисперсия)
Основной бутилированный метакрилатный сополимер Ph. Eur. = Eudragit E-100,
где функциональная группа обладает группой четвертичного аммония (триметиламмонийэтилметакрилат) или R=COOCH2CH2N+(CH3)3Cl- [коммерчески доступный в качестве EUDRAGIT® (RL или RS)] или функциональная группа представляет собой карбоновую кислоту, или R = COOH [коммерчески доступный как EUDRAGIT® (L)]. Когда функциональная группа представляет собой группу карбоновой кислоты, полимер EUDRAGIT® (L) является устойчивым в желудке и растворим в кишечнике. Таким образом, составы с использованием EUDRAGIT® (L) будут устойчивыми к желудочному соку и будут высвобождать активное вещество в кишечнике. Когда функциональная группа представляет собой триметиламмонийэтилметакрилатную группу, полимеры EUDRAGIT® (RL или RS) являются нерастворимыми, проницаемыми, диспергируемыми и pH независимыми. Таким образом, эти полимеры EUDRAGIT® (RL или RS) можно использовать для замедленного высвобождения лекарственного средства в составах с замедленным высвобождением. EUDRAGIT® продается в различных формах, таких как твердая форма (EUDRAGIT® L100/S100/L-100-55, EUDRAGIT® E PO, EUDRAGIT® RL PO, Eudragit RS PO), гранулы (EUDRAGIT® E 100, EUDRAGIT® RL 100/RS 100), дисперсии (L 30 D-55/FS 30D 30%, EUDRAGIT® NE 30 D/40 D 30%/40% содержание полимера, EUDRAGIT® RL 30 D RS 30 D 30%) и органические растворы (EUDRAGIT® L12.5, EUDRAGIT® E12.5, EUDRAGIT® RL 12.5/RS 12.5 - 12,5% органический раствор).
Когда используют по меньшей мере два подвергнутых плавлению полимера, один из них предпочтительно представляет собой производное целлюлозы, более предпочтительно производное гидроксиалкилцеллюлозы и необязательно гидроксипропилметилцеллюлозы, и, независимо, другой полимер предпочтительно представляет собой (мет)акрилатный полимер (такой как любой пригодный полимер Eudragit). Среди (мет)акрилатных полимеров, полимеры, предпочтительные в контексте этого изобретения, представляют собой Eudragit L и Eudragit RS. Одним более предпочтительным полимером в контексте этого изобретения является Eudragit RL. Полимеры Eudragit можно использовать в сочетаниях, при этом смеси Eudragit RS и RL являются предпочтительными.
У лиц, которые (хотя и ненамеренно) выпивают существенные количества алкогольных напитков при приеме назначенных врачом лекарственных средств, может существенно измениться состав желудочных соков, находящихся в желудке, и в крайних случаях желудочные соки могут содержать вплоть 40% спирта. Преимущественно, варианты осуществления, предотвращающего зависимость состава по этому изобретению, необязательно содержат подвергнутую плавлению смесь по меньшей мере одного вызывающего зависимость лекарственного средства, по меньшей мере одного простого эфира целлюлозы или сложного эфира целлюлозы, и по меньшей мере одного (мет)акрилового полимера, где количество лекарственного средства, которое экстрагируется из состава 20% водным раствором этанола, или 40% водным раствором этанола, или и тем и другим, в течение одного часа при 37°C, является меньшим или равным двойному количеству лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой в течение одного часа при 37°С или при 25°C или при обеих этих температурах. Устойчивость к экстракции 40% этанолом является преимущественной в тех случаях, когда индивид целенаправленно пытается экстрагировать вызывающее зависимость лекарственное средство из препарата, содержащего вызывающее зависимость лекарственное средство.
Протоколы экстракции 20% или 40% водным раствором этанола или 0,01н. хлористоводородной кислотой соответственно приведены в экспериментальном разделе, представленном ниже. В более предпочтительных вариантах осуществления, количество лекарственного средства, которое экстрагируется из состава 20% или 40% водным раствором этанола, является меньшим или равным 1,5-кратному количеству лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой в течение одного часа. В других более предпочтительных вариантах осуществления, количество лекарственного средства, которое экстрагируется из состава 20% или 40% водным раствором этанола, является меньшим или равным количеству лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой в течение одного часа. В других более предпочтительных вариантах осуществления, количество лекарственного средства, которое экстрагируется из состава 20% или 40% водным раствором этанола, является меньшим или равным 0,9-кратному количеству лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой в течение одного часа.
Также настоящее изобретение относится к составу с замедленным высвобождением по меньшей мере одного вызывающего зависимость лекарственного средства, который препятствует экстракции лекарственного средства из состава, когда экстракцию проводят посредством экстракции растворителем с помощью широко доступных бытовых растворителей для экстракции, таких как изопропиловый спирт, дистиллированные спирты, примерами которых является водка, белый уксус, вода и водный раствор этанола (например, 20% этанол). В то время как состав является в значительной степени устойчивым к экстракции растворителем, он, тем не менее, обеспечивает надлежащее высвобождение лекарственного средства в водных растворах, таких как желудочные жидкости. Этот состав при дроблении или измельчении также обеспечивает надлежащее высвобождение лекарственного средства в водных растворах, таких как желудочные жидкости.
Хорошо, что в некоторых предпочтительных вариантах осуществления изобретения, количество вызывающего зависимость лекарственного средства, высвобождаемого с момента помещения в 3 унции одного, или двух или трех, или более трех бытовых растворителей, приведенных выше (т.е. в момент 0 часов) до 1 часа не более чем на 15% превышает количество, высвобождаемое за то же время при проглатывании обычным человекам, или от более 1 часа до приблизительно 4 часов оно не более чем на 15% превышает количество, высвобожденное в то же время при проглатывании обычным человеком, или оба этих варианта.
Иллюстративные предпочтительные композиции по этому изобретению содержат простые эфиры целлюлозы и сложные эфиры целлюлозы, которые можно использовать отдельно или в сочетании в этом изобретении и которые обладают предпочтительной молекулярной массой в диапазоне от 50000 до 1250000 Дальтон. Простые эфиры целлюлозы предпочтительно выбирают из алкилцеллюлоз, гидроксиалкилцеллюлоз, гидроксиалкилалкилцеллюлоз или их смесей, таких как этилцеллюлоза, метилцеллюлоза, гидроксипропилцеллюлоза (NF), гидроксиэтилцеллюлоза (NF) и гидроксипропилметилцеллюлоза (USP), или их сочетания. Пригодные простые эфиры целлюлозы представляют собой, но не ограничиваются ими, ацетат целлюлозы (NF), целлюлозы ацетата бутират, целлюлозы ацетата пропионат, фталат гидроксипропилметилцеллюлозы, гидроксипропилметилцеллюлозы ацетата фталат, и их смеси. Наиболее предпочтительно, можно использовать неионные полимеры, такие как гидроксипропилметилцеллюлоза.
Количество групп заместителей на элементах ангидроглюкозы в целлюлозе можно обозначать по среднему количеству групп заместителей, присоединенных к кольцу, эта концепция известна специалистам в области химии целлюлозы как "степень замещения" (D.S.). Если замещены все три доступных положения на каждом элементе, D.S. обозначают как 3, если в среднем два из каждых колец вступают в реакцию, D.S. обозначают как 2 и т.д.
В предпочтительных вариантах осуществления, простой эфир целлюлозы обладает степенью замещения алкилом от 1,3 до 2,0 и молярным замещением гидроксиалкилом вплоть до 0,85.
В предпочтительных вариантах осуществления, замещающий алкил представляет собой метил. Кроме того, предпочтительный замещающий гидроксиалкил представляет собой гидроксипропил. Эти типы полимеров с различными степенями замещения для замещений метокси и гидроксипропокси обобщенно представлены в фармакопеях, например USP под названием "гипромеллоза".
Метилцеллюлоза доступна под торговым названием METHOCEL A. METHOCEL A обладает D.S. метила (или метоксила) от 1,64 до 1,92. Эти типы полимеров приведены в фармакопеях, например USP, под названием "метилцеллюлоза".
Особенно предпочтительный простой эфир целлюлозы представляет собой гидроксипропилметилцеллюлозу. Гидроксипропилметилцеллюлоза доступна под торговым названием METHOCEL E (D.S. для метила составляет приблизительно 1,9, молярное замещение гидроксипропилом составляет приблизительно 0,23), METHOCEL F (D.S. для метила составляет приблизительно 1,8, молярное замещение гидроксипропилом составляет приблизительно 0,13), и METHOCEL K (D.S. для метила составляет приблизительно 1,4, молярное замещение гидроксипропилом составляет приблизительно 0,21). METHOCEL F и METHOCEL K представляют собой предпочтительные гидроксипропилметилцеллюлозы для применения в настоящем изобретении.
Пригодный акриловый полимер включает гомополимеры и сополимеры (этот термин включает полимеры, имеющие более двух различных повторяющихся элементов), содержащие мономеры акриловой кислоты и/или алкакриловой кислоты и/или алкил(алк)акрилата. Как используют в настоящем документе, термин "алкил(алк)акрилат" относится либо к соответствующему акрилатному или алкакрилатному сложному эфиру, который обычно образуются из соответствующих акриловых или алкакриловых кислот соответственно. Иными словами, термин "алкил(алк)акрилат" относится либо к алкилалкакрилату, либо к алкилакрилату. Предпочтительно, алкил(алк)акрилат представляет собой (C1-C22)алкил((C1-C10)алк)акрилат. Примеры C1-C22алкильных групп алкил(алк)акрилатов включают метил, этил, н-пропил, н-бутил, изобутил, трет-бутил, изопропил, пентил, гексил, циклогексил, 2-этилгексил, гептил, октил, нонил, децил, изодецил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил, гептадецил, октадецил, нонадецил, эйкозил, бегенил и их изомеры. Алкильная группа может быть прямой или разветвленной. Предпочтительно, (C1-C22)алкильная группа представляет собой (C1-C6)алкильную группу, как определено выше, более предпочтительно (C1-C4)алкильную группу, как определено выше. Примеры C1-10алкгрупп алкил(алк)акрилатов включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, пентил, гексил, циклогексил, 2-этилгексил, гептил, октил, нонил, децил и их изомеры. Алкгруппы могут быть прямыми или разветвленными. Предпочтительно, (C1-C10)алкгруппа представляет собой (C1-C6)алкгруппу, как определено выше, более предпочтительно (C1-C4)алкгруппу, как определено выше.
Предпочтительно, алкил(алк)акрилат представляет собой (C1-C4)алкил((C1-C4)алк)акрилат, наиболее предпочтительно (C1-C4)алкил(мет)акрилат. Будет понятно, что термин (С1-C4)алкил(мет)акрилат относится либо к (C1-C4)алкилакрилату, либо к (C1-C4)алкилметакрилату. Примеры (C1-C4)алкил(мет)акрилата включают метилметакрилат (MMA), этилметакрилат (EMA), н-пропил метакрилат (PMA), изопропилметакрилат (IPMA), н-бутилметакрилат (BMA), изобутилметакрилат (IBMA), трет-бутилметакрилат (TBMA), метилакрилат (MA), этилакрилат (EA), н-пропилакрилат (PA), н-бутилакрилат (BA), изопропилакрилат (IPA), изобутилакрилат (IBA) и их сочетания.
Предпочтительно, мономер алкакриловой кислоты представляет собой (C1-C10)алкакриловую кислоту. Примеры (C1-C10)алкакриловых кислот включают метакриловую кислоту, этакриловую кислоту, н-пропакриловую кислоту, изопропакриловую кислоту, н-бутакриловую кислоту, изобутакриловую кислоту, трет-бутакриловую кислоту, пентакриловую кислоту, гексакриловую кислоту, гептакриловую кислоту и их изомеры. Предпочтительно, (C1-C10)алкакриловая кислота представляет собой (C1-C4)алкакриловую кислоту, наиболее предпочтительно метакриловую кислоту.
В определенных вариантах осуществления, алкильные группы могут быть замещены арильными группами. Как используют в настоящем документе, "алкильная" группа относится к неразветвленным, разветвленным или циклическим, насыщенным или ненасыщенным алифатическим углеводородам. Алкильная группа имеет 1-16 атомов углерода, и она может быть незамещенной или замещенной одной или несколькими группами, выбранными из галогена, гидрокси, алкокси, карбонила, амидо, алкиламидо, диалкиламидо, нитро, амино, алкиламино, диалкиламино, карбоксила, тио и тиоалкила. Группа "гидрокси" относится к группе OH. Группа "алкокси" относится к -O-алкильной группе, где алкил является таким, как определено выше. Группа "тио" относится к группе -SH. Группа "тиоалкила" относится к группе -SR, где R представляет собой алкил, как определено выше. Группа "амино" относится к группе -NH2. Группа "алкиламино" относится к группе -NHR, где R представляет собой алкил, как определено выше. Группа "диалкиламино" относится к группе -NRR', где все R и R' являются такими, как определено выше. Группа "амидо" относится к -CONH2. Группа "алкиламидо" относится к группе -CONHR, где R представляет собой алкил, как определено выше. Группа "диалкиламидо" относится к группе -CONRR', где R и R' представляют собой алкил, как определено выше. Группа "нитро" относится к группе NO2. Группа "карбоксила" относится к группе COOH.
В определенных вариантах осуществления, алкильные группы могут быть замещены арильными группам. Как используют в настоящем документе, "арил" включает как карбоциклические, так и гетероциклические ароматические кольца, как моноциклические, так и конденсированные полициклические, где ароматические кольца могут представлять собой 5- или 6-членные кольца. Репрезентативные моноциклические арильные группы включают, но не ограничиваются ими, фенил, фуранил, пирролил, тиенил, пиридинил, пиримидинил, оксазолил, изоксазолил, пиразолил, имидазолил, тиазолил, изотиазолил и т.п. Конденсированные полициклические арильные группы представляют собой ароматические группы, которые включают 5- или 6-членное ароматическое или гетероароматическое кольцо, в качестве одного или нескольких колец в системе конденсированных колец. Репрезентативные кондесированные полициклические арильные группы включают нафталин, антрацен, индолизин, индол, изоиндол, бензофуран, бензотиофен, индазол, бензимидазол, бензтиазол, пурин, хинолин, изохинолин, циннолин, фталазин, хиназолин, хиноксазолин, 1,8-нафтиридин, птеридин, карбазол, акридин, феназин, фенотиазин, феноксазин и азулен. Так же, как используют в настоящем документе, арильная группа также включает арилалкильную группу. Кроме того, как используют в настоящем документе "арилалкил" относится к группам, таким как бензил, где ароматическая группа связана с алкильной группой.
Предпочтительно, акриловый полимер представляет собой акриловый сополимер. Предпочтительно, акриловый сополимер содержит мономеры, образованные из алкил(алк)акрилата, и/или акриловой кислоты и/или алкакриловой кислоты, как определено в настоящем документе выше. Наиболее предпочтительно, акриловый сополимер содержит мономеры, образованные из алкил(алк)акрилата, т.е. сополимеризуемые алкилакрилатные и алкилалкакрилатные мономеры, как определено в настоящем документе, выше. Особенно предпочтительные акриловые сополимеры включают (C1-C4)алкилакрилатный мономер и сополимеризуемый (C1-C4)алкил(C1-C4)алкакрилатный сомономер, в частности сополимеры, образованные из метилметакрилата и сополимеризуемого сомономера метилакрилата и/или этилакрилата и/или н-бутилакрилата.
Предпочтительно, (мет)акриловый полимер представляет собой ионный (мет)акриловый полимер, в частности катионный метакриловый полимер. Ионный (мет)акриловый полимер получают сополимеризацией (мет)акриловых мономеров, обладающих ионными группами, с (мет)акриловыми мономерами. Ионные группы предпочтительно представляют собой группы четвертичного аммония.
(Мет)акриловые полимеры, главным образом, являются нерастворимыми в воде, но поддаются набуханию и являются проницаемыми в водных растворах и пищеварительных жидкостях. Молярное отношение катионных групп к нейтральным (мет)акриловым сложным эфирам позволяет контроль проницаемости воды в составе. В предпочтительных вариантах осуществления (мет)акриловый полимер представляет собой сополимер или смесь сополимеров, где молярное соотношение катионных групп и нейтральных (мет)акриловых сложных эфиров находится в диапазоне в среднем приблизительно от 1:20 до 1:35. Соотношение можно корректировать выбором соответствующего коммерчески доступного катионного (мет)акрилового полимера или смешиванием катионного (мет)акрилового полимера с пригодным количеством нейтрального (мет)акрилового полимера.
Пригодные (мет)акриловые полимеры являются коммерчески доступными от Rohm Pharma под торговым названием Eudragit, предпочтительно Eudragit RL и Eudragit RS. Eudragit RL и Eudragit RS представляют собой сополимеры акрилового и метакрилового сложных эфиров с низким содержанием групп четвертичного аммония, при этом молярное соотношение групп аммония и остальных нейтральных (мет)акриловых сложных эфиров составляет 1:20 в Eudragit RL и 1:40 в Eudragit RS. Средняя молекулярная масса составляет приблизительно 150000.
Помимо (мет)акриловых полимеров, дополнительные фармацевтически приемлемые полимеры можно включать составы по этому изобретению в целях коррекции свойств состава и/или упрощения его изготовления. Эти полимеры могут быть выбраны из группы, содержащей гомополимеры N-виниллактамов, особенно поливинилпирролидон (PVP), сополимеры N-виниллактама и одного или нескольких сомономеров, сополимеризуемых с ними, при этом сомономеры выбраны из азотсодержащих мономеров и кислородсодержащих мономеров; особенно сополимер N-винилпирролидона и винилкарбоксилата, при этом предпочтительные примеры представляют собой сополимер N-винилпирролидона и винилацетата или сополимер N-винилпирролидона и винилпропионата; привитые сополимеры, такие как поливиниловый спирт-полиэтиленгликоль (доступные, например, как Kollicoat® IR от BASF AG, Ludwigshafen, Germany); высокомолекулярные окиси полиалкилена, такие как окись полиэтилена и окись полипропилена и сополимеры окиси этилена и окиси пропилена; полиакриламиды; полимеры винилацетата, такие как сополимеры винилацетата и кротоновой кислоты, частично гидролизуемый поливинилацетат (также называемый омыленным "поливиниловым спиртом"); поливиниловый спирт; поли(гидроксикислоты), такие как поли(молочная кислота), поли(гликолевая кислота), поли(3-гидроксибутират) и поли(3-гидроксибутират-со-3-гидроксивалерат); или смеси одной или нескольких из них. PVP образует N-оксид гидрокодона в ходе экструдирования, таким образом применение полимеров и сополимеров PVP не всегда является предпочтительным. Однако когда используют небольшое количество (0,2-0,6% мас./мас. от всего состава) антиоксиданта, тогда предпочтительно можно использовать PVP.
Подразумевают, что "вызывающее зависимость лекарственное средство" означает любой биологически эффективный ингредиент, распространение которого подвергается регулятивным ограничениям. Вызывающие зависимость лекарственные средства, которые обычно можно получать в контексте этого изобретения, включают, но не ограничиваются ими, псевдоэфедрин, антидепрессанты, сильные стимуляторы, диетологические лекарственные средства, стероиды и нестероидные противовоспалительные средства. В категории сильных стимуляторов, метамфетамин представляет собой одно из лекарственных средств, которое недавно получило внимание общества в качестве вызывающего зависимость лекарственного средства. Также в настоящее время существуют некоторые опасения относительно потенциала в отношении зависимости атропина, хиосциамина, фенобарбитала, скополамина и т.п. Другим большим классом вызывающих зависимость лекарственных средств является класс аналгетиков, особенно опиодиов.
Под термином "опиоид" понимают вещество как агонист, так и антагонист, или смешанный агонист-антагонист, которое реагирует с одним или несколькими участками рецептора, связываемого эндогенными опиоидными пептидами, такими как энкефалины, эндорфины и динорфины. Опиоиды включают, но не ограничиваются ими, альфентанил, аллилпродин, альфапродин, анилеридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, циклазоцин, дезоморфин, декстроморамид, дезоцин, диампромид, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, диоксафетила бутират, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, фентанил, героин, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леваллорфан, левофенацилморфан, леворфанол, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, нальбуфин, нарцеин, никоморфин, норпипанон, опиум, оксикодон, оксиморфон, папвретум, пентазоцин, фенадоксон, феназоцин, феноморфан, феноперидин, пиминодин, пропирам, пропоксифен, суфентанил, тилидин и трамадол и их соли и смеси.
В некоторых предпочтительных вариантах осуществления, состав по этому изобретению включает по меньшей мере одно дополнительное лекарственное средство. В более предпочтительных вариантах осуществления, дополнительное лекарственное средство может быть выбрано, но не ограничено ими, из группы, состоящей из нестероидных неопиоидных аналгетиков, и далее необязательно выбрано из группы, состоящей из ацетаминофена, аспирина, фентанила, ибупрофена, индометацина, каторолака, напроксена, фенацетина, пироксикама, суфентанила, сулиндака и интерферона альфа. Особенно предпочтительными являются сочетания лекарственных средств, продаваемых в обществе в настоящее время в качестве сочетаний с фиксированной дозой под контролем пригодной национальной или региональной организации, такой как (в качестве примера пример) US Food and Drug Administration. Такие лекарственные средства включают, но не ограничиваются ими, сочетание (с фиксированной дозой) гидрокодона и ацетаминофена, или сочетание (с фиксированной дозой) гидрокодона и ибупрофена.
Вызывающее зависимость лекарственное средство(а) предпочтительно диспергировано в матриксе, который предпочтительно образован простым эфиром целлюлозы или сложным эфиром целлюлозы, и одним акриловым или метакриловым полимером, а также другими необязательными ингредиентами состава. Также подразумевается, что это описание включает системы, имеющие небольшие частицы, как правило, с диаметром менее 1 мкм, лекарственного средства в матриксной фазе. Эти системы предпочтительно не содержат значительных количеств активных опиоидных ингредиентов в их кристаллическом или микрокристаллическом состоянии, как показано термическим анализом (DSC) или рентгенодиффракционным анализом (WAXS). По меньшей мере 98% (по массе) от общего количества лекарственного средства предпочтительно представлено в аморфном состоянии. Если в составе в соответствии с настоящим изобретением дополнительно содержится дополнительное не вызывающее зависимости активное лекарственное средство, например, такое как ацетаминофен, это дополнительное активное лекарственное средство(а) может находиться в кристаллическом состоянии, погруженное в состав.
Когда дисперсия компонентов является такой, что система является химически и физически однородной или по существу полностью гомогенной или состоит из одной термодинамической фазы, такая дисперсия называется "твердым раствором". Твердые растворы вызывающих зависимость активных веществ являются предпочтительными.
Также состав может содержать одну или несколько добавок, выбранных из спиртов сахаров или их производных, мальтодекстринов, фармацевтически приемлемых поверхностно-активных веществ, регуляторов текучести, дезинтегрирующих веществ, наполнителей и смазывающих веществ. Примерами пригодных спиртов сахаров являются маннит, сорбит, ксилит; пригодные производные спирта сахара включают, но не ограничиваются ими изомальтозу, гидрогенизированную, конденсированную палатинозу и другие производные, которые либо сходны, либо не сходны с ними.
Фармацевтически приемлемые поверхностно-активные вещества предпочтительно представляют собой фармацевтически приемлемое неионное поверхностно-активное вещество. Включение поверхностно-активных веществ является особенно предпочтительным в случае матриксов, содержащих малорастворимые в воде активные ингредиенты и/или для придания составу смачиваемости. Поверхностно-активное вещество может осуществлять мгновенное эмульгирование активного ингредиента, высвобождаемого из дозированной формы, и предотвращать осаждение активного ингредиента в водных жидкостях желудочно-кишечного тракта.
Некоторые предпочтительные добавки включают алкильные простые эфиры полиоксиэтилена, например лауриловый простой эфир полиоксиэтилена (3), цетиловый простой эфир полиоксиэтилена (5), стеариловый простой эфир полиоксиэтилена (2), стеариловый простой эфир полиоксиэтилена (5); алкиларильные простые эфиры полиоксиэтилена, например нонилфениловый простой эфир полиоксиэтилена (2), нонилфениловый простой эфир полиоксиэтилена (3), нонилфениловый простой эфир полиоксиэтилена (4) или октилфениловый простой эфир полиоксиэтилена (3); сложные эфиры жирных кислот и полиэтиленгликоля, например монолаурат PEG-200, дилаурат PEG-200, дилаурат PEG-300, дилаурат PEG-400, дистеарат PEG-300 или диолеат PEG-300; сложные моноэфиры жирных кислот и алкиленгликоля, например моно- и дилаурат пропиленгликоля (Lauroglycol®); сложные эфиры жирных кислот и сахарозы, например монoстеарат сахарозы, дистеарат сахарозы, монолаурат сахарозы или дилаурат сахароза; сложные моно и ди-эфиры жирных килсот и сорбитана, такие как монолаурата сорбитана (Span® 20), моноолеат сорбитана, монопальмитат сорбитана (Span® 40), или стеарат сорбитана, полиоксиэтиленовые производные касторового масла, например полиоксиэтиленглицерина тририцинолеат или полиоксил-35-касторовое масло (Cremophor® EL; BASF Corp.) или полиоксиэтиленглицерола оксистеарат, такой как полиэтиленгликоль-40-гидрогенизированное касторовое масло (Cremophor® RH 40) или полиэтиленгликоль-60-гидрогенизированное касторовое масло (Cremophor® RH 60); или блок-сополимеры оксида этилена и оксида пропилена, так же известные, как блок-сополимеры полиоксиэтилена и полиоксипропилена или полиоксиэтилена и полипропиленгликоля, такие как Pluronic® F68, Pluronic® F127, Poloxamer® 124, Poloxamer® 188, Poloxamer® 237, Poloxamer® 388, или Poloxamer® 407 (BASF Wyandotte Corp.); или сложные моноэфиры жирных кислот полиоксиэтилена (20) сорбитана, например полиоксиэтилена (20) сорбитана моноолеат (Tween® 80), полиоксиэтилена (20) сорбитана моностеарат (Tween® 60), полиоксиэтилена (20) сорбитана монопальмитат (Tween® 40), полиоксиэтилена (20) сорбитана монолаурат (Tween® 20) и т.п., а также смеси двух, трех, четырех, пяти или более из них.
В расплав можно включать различные другие добавки, например регуляторы текучести, такие как коллоидный диоксид кремния; смазывающие вещества, наполнители, дезинтегрирующие вещества, пластификаторы, стабилизаторы, такие как антиоксиданты, светостабилизаторы, акцепторы радикалов или стабилизаторы против воздействия микробов. Кроме того, поскольку содержащий ацетаминофен наружный слой обладает горьким вкусом вследствие самого ацетаминофена, можно использовать подсластители и/или вкусовые добавки и т.д. в качестве добавок для уменьшения этого горького вкуса. Одним предпочтительным способом уменьшения горького вкуса является дополнительное тонкое, не содержащее ацетаминофена, покрытие.
Составы по изобретению можно получать любым пригодным процессом плавления, таким как применение нагретого пресса, и их предпочтительно получают экструдированием плавлением. В целях достижения гомогенного распределения и достаточной степени диспергирования лекарственного средства, содержащий лекарственное средство расплав можно держать в нагретой бочке устройства для экструдирования плавлением в течение достаточного времени нахождения. Плавление происходит при переходе в жидкое или каучукоподобное состояние, при котором возможно гомогенное погружение одного компонента в другой. Плавление, как правило, вовлекает нагревание выше точки размягчения простого эфира/сложного эфира целлюлозы или (мет)акрилового полимера. Расплав можно получать различными способами.
Как правило, температура плавления находится в диапазоне от 70 до 250°C, предпочтительно от 80 до 180°C, наиболее предпочтительно от 100 до 140°C.
Когда процесс плавления включает экструдирование плавлением, плавление и/или смешивание может происходить в устройстве, обычно используемом для этой цели. В частности, пригодными являются экструдеры или месильные машины. Пригодные экструдеры включают одношнековые экструдеры, экструдеры со сцепленными шнеками и многошнековые экструдеры, предпочтительно двухшнековые экструдеры, которые могут вращаться в одном направлении или вращаться в противоположных направлениях и необязательно оборудованы дисками для перемешивания. Будет понятно, что рабочие температуры также будут определяться типом экструдера или типом используемой конфигурации экструдера. Часть энергии, требуемой для плавления, смешивания и растворения компонентов в экструдере может быть предоставлена нагреванием элементов. Однако трение и напряжение сдвига материала в экструдере также может привести к смеси со значительным количеством энергии и способствовать образованию гомогенного расплава компонентов.
В другом варианте осуществления, это изобретение относится к пероральной дозированной форме с замедленным высвобождением, отличающейся тем, что она обладает по меньшей мере двумя из следующих признаков: (a) лекарственное средство, которое экстрагируется из состава этаноловым растворителем, например 40% или 20% водным раствором этанола или обоими из них в течение одного часа при 37°С, при встряхивании или без него, является меньшим или равным двойному количеству лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой в течение одного часа при 37°C; (b) дозированная форма является устойчивой к сдавлению и не разрушается под действием силы 300 Ньютон, предпочтительно 600 Ньютон, более предпочтительно 1200 Ньютон, как измеряют тестером твердости "Pharma Test PTB 501"; и (c) дозированная форма высвобождает по меньшей мере 15%, более предпочтительно 18%, и необязательно 24% лекарственного средства, однако не более 45%, более предпочтительно 38% и необязательно 34% лекарственного средства в течение 30 минут, первого часа, или первых двух часов при тестировании растворения in vitro и необязательно также in vivo (т.е. в пищеварительном тракте животного или человека). Без связи с какой-либо конкретной теорией полагают, что высокой первоначальной скорости высвобождения лекарственного средства из состава достигают, обеспечивая высокую нагрузку лекарственным средством в составе. Нагрузка лекарственным средством для одного активного ингредиента, такого как ацетаминофен, в некоторых вариантах осуществления состава по данному изобретению может составлять более чем приблизительно 60%, 70%, 75%, 80%, 85% по массе. Нагрузка лекарственным средством для ацетаминофена может быть ограничена 80%.
Предпочтительный вариант осуществления этой дозированный формы представляет собой монолитную форму или твердый раствор. Термин "монолитный" образован от корней, означающих "единый" и "камень". Монолитная форма или твердое вещество предпочтительно обладают по меньшей мере одним размером, который составляет более 5 мм. В монолитных вариантах осуществления этого изобретения, вызывающее зависимость лекарственное средство предпочтительно находится в элементе единого твердого вещества, или единого твердого раствора. Монолитное твердое вещество или твердый раствор необязательно могут быть покрыты другими материалами или комбинированы с ними. Эти другие материалы предпочтительно не содержат значительного количества вызывающего зависимость лекарственного средства и эти материалы предпочтительно по существу не влияют на скорость растворения или диспергирования вызывающего зависимость лекарственного средства in vivo или in vitro. Скорости высвобождения in vitro и/или in vivo, вызывающего зависимость лекарственного средства или вызывающих зависимость лекарственных средств, после приблизительно первого часа предпочтительно являются по существу постоянными в течение по меньшей мере приблизительно 6, 8, 10, 12 или 16 часов. Таким образом, варианты осуществления этого изобретения обеспечивают однофазный состав лекарственного средства, который можно адаптировать для обеспечения быстрого выброса вызывающего зависимость лекарственного средства(средств) в целях возможности быстрого достижения терапевтических уровней лекарственного средства в крови пациента или животного, и для поддержания в целях обеспечения терапевтических количеств в течение по меньшей мере приблизительно 8, 12 или 24 часов. Кроме того, состав лекарственного средства предпочтительно является пригодным для повторяющегося введения человеку или животному один раз, два раза или три раза в сутки.
Преимущественно, предпочтительные варианты осуществления дозированной формы по этому изобретению высвобождают по существу полное количество вызывающего зависимость лекарственного средства, включенного в дозированную форму. Например, дозированная форма по этому изобретению может быть адаптирована для доставки более чем 90%, и предпочтительно 95%, лекарственного средства при тестировании растворения in vitro в течение приблизительно 16, и необязательно 12 или 9 часов. Совокупную концентрацию в крови, или AUC, нельзя прямо узнать исходя из времени, в течение которого из состава высвобождается 90% лекарственного средства, однако, как правило, более высокие AUC на 1 мг, вызывающего зависимость лекарственного средства, могут быть достигнуты, когда состав лекарственного средства высвобождает по существу все, или все, вызывающее зависимость лекарственное средство в отделах пищеварительного тракта, способных всасывать лекарственное средство в кровеносную систему пациента (или животных).
В другом предпочтительном варианте осуществления, это изобретение относится к процессу изготовления устойчивого к зависимости дозированного состава лекарственного средства, включающему экструдирование плавлением состава, содержащего по меньшей мере одно терапевтическое средство, далее включающему прямое придание формы экструдату дозированной формы без (промежуточной) стадии измельчения. Продукт экструдирования плавлением предпочтительно содержит производное целлюлозы и предпочтительно также содержит полимер Eudragit. Предпочтительные полимеры Eudragit включают Eudragit L или Eudragit RS, или оба из них, и особенно предпочтительным является Eudragit RL или сочетание Eudragit RL и Eudragit RS.
Расплав может варьировать от пастообразного до тягучего. Перед тем, как позволить расплаву затвердеть, расплаву необязательно можно придавать практически любую требуемую форму. Удобно, что придание формы экструдату необязательно можно проводить посредством каландра, предпочтительно посредством двух противоположно вращающихся валиков с взаимно совпадающими углублениями на их поверхности. Широкий диапазон таблетированных форм можно получать с использованием валиков с различными формами углублений. Альтернативно экструдат можно нарезать на фрагменты, либо до ("горячее нарезание"), либо после затвердения ("холодное нарезание") или использовать в процессе штамповки. Процессы плавления, вовлекающие нагретые прессы, также необязательно могут быть каландрированными.
Образующийся расплав необязательно можно покрывать материалами, которые не содержат значительного количества лекарственного средства с потенциалом в отношении зависимости. Например, монолитную дозированную форму, содержащую вызывающее зависимость лекарственное средство, можно покрывать цветным покрытием, способствующим проглатыванию добавкой или другим слоем фармацевтически приемлемых материалов. Слой материалов, нанесенных на монолитную форму, предпочтительно существенно не изменяет скорость высвобождения активного ингредиента из дозированной формы.
В целях упрощения введения такой дозированной формы млекопитающим, предпочтительным является придание дозированной форме соответствующей формы. Крупные таблетки, которые могут быть удобно проглочены, таким образом, предпочтительно являются удлиненными, а не округлыми по форме.
Пленочное покрытие на дозированной форме далее приводит упрощению проглатывания. Также пленочное покрытие улучшает вкус и обеспечивает приятный внешний вид. Если желательно, пленочное покрытие может представлять собой растворимое в кишечнике покрытие. Пленочное покрытие обычно включает полимерный пленкообразующий материал, такой как гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза и акрилатные или метакрилатные сополимеры. Помимо пленкообразующего полимера, пленочное покрытие далее может содержать пластификатор, например полиэтиленгликоль, поверхностно-активное вещество, например типа Tween®, и необязательно пигмент, например диоксид титана или оксиды железа. Пленочное покрытие также может содержать тальк в качестве антиадгезивного средства. Обычно пленочное покрытие составляет менее приблизительно 5% по массе дозированной формы.
ИЛЛЮСТРАТИВНЫЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Определенные иллюстративные варианты осуществления настоящего изобретения относятся к монолитным дозированным составам, имеющим двухфазный профиль высвобождения для хорошо растворимых в воде лекарственных средств, имеющих содержащую полимер таблетку, получаемую экструдированием и каландрированием. Предпочтительно составы имеют сочетание составов с немедленным высвобождением и контролируемым высвобождением композиций гидрокодона и ацетаминофена. Этот монолитный дозированный состав, особенно имеющий наркотические лекарственные средства, может иметь профили, предотвращающие зависимость, так что растворение лекарственного средства дозированных форм имеет сниженное/минимальное быстрое высвобождение дозы в 40% водном растворе этанола. Более предпочтительно, эти составы могут обеспечить воспроизводимые процессы изготовления, позволяющие быстрое переключение на производственный масштаб.
Требуемого двухфазного растворения лекарственного средства ацетаминофена, при сохранении монолитной дозированной формы, можно достигать путем включения активного ингредиента (ацетаминофена) в два состава с различными скоростями высвобождения, а затем комбинирования их с получением двухслойной или многослойной таблетки. Процессы, пригодные для этой цели, включают способы совместного экструдирования для получения многослойных таблеток, как описано в EP 0857062, конкретно для экструдируемых дозированных форм. Одним из недостатков этого способа является то, что два устройства для экструдирования должны работать одновременно и их и весовые и объемные потоки должны быть скоординированы с большой точностью. В особенности при придании формы таблетке в каландрах, два расплава должны быть объединены друг с другом в соотношении, сохраняющемся очень точно для обеспечения соответствия анализу и требованиям однообразия содержимого таблеток, как указано в фармакопеях (например, USP, Ph. Eur.).
Для этого требуются усилия на высоком уровне.
Также можно изготавливать часть с быстрым высвобождением ацетаминофена в отдельной таблетке, которую затем включают во все еще пластичный расплав части лекарственного средства с замедленным высвобождением в ходе каландрирования. После охлаждения получают каландрированную экструдированную таблетку, которая содержит отдельно включенный компонент с быстрым высвобождением. Дозированные формы этого типа описаны в US 6001391, конкретно для экструдированных дозированных форм. Одним из недостатков этого подхода является то, что таблетка ацетаминофена с быстрым высвобождением должна быть включена очень точно в отдельные полости каландра для предотвращения ее полного окружения расплавом. Только если этот компонент ацетаминофена с быстрым высвобождением расположен непосредственно на поверхности таблетки, растворение лекарственного средства из этой отдельной части таблетки может начаться достаточно быстро при контакте с водной средой.
Также можно получить компонент ацетаминофена с быстрым высвобождением в таблетке нанесением пленочного покрытия, содержащего ацетаминофен. Изготовление покрытых пленкой экструдированных дозированных форм описано в различных патентных заявках США. Однако в этих патентных заявках не описано содержащее лекарственное средство пленочное покрытие, предназначенное конкретно для достижения двухфазного растворения лекарственного средства. Результаты клинического испытания экструдированной дозированной формы, полученной в соответствии с патентными заявками 11/625705 и PCT/US07/60864, показали, что приблизительно 20% ацетаминофена, содержащегося в таблетке, необходимо было превратить в состав с быстрым высвобождением для достижения требуемого двухфазного растворения лекарственного средства (например, > приблизительно 30% через 1 ч, > приблизительно 80% через 8 ч). При общем содержании ацетаминофена от приблизительно 500 мг на таблетку, это означает, что приблизительно 100 мг ацетаминофена должно высвобождаться быстро. Применение приблизительно 100 мг активного ингредиента в форме быстрого высвобождения в таблетках является трудным и возможным только при удовлетворении нескольким следующим требованиям.
Содержание лекарственного средства в составе пленочного покрытия должно быть очень высоким, так чтобы слои не стали слишком толстыми.
Содержащий лекарственное средство раствор или дисперсия, используемые для пленочного покрытия, должны обладать высокой концентрацией для избежания большой продолжительности процесса, которая в ином случае делает процесс неэкономичным.
Слой пленочного покрытия должен обеспечивать достаточную механическую стабильность, даже при большой толщине слоя, он не должен быть клейким и т.д. и он должен быть достаточно пластичным, чтобы не происходило растрескивания даже при толстых слоях. Также необходимо обеспечить хорошую адгезию к поверхности экструдированной центральной части.
Растворение лекарственного средства из слоя пленочного покрытия также должно быть быстрым при использовании толстых слоев (приблизительно максимум 1 ч в предпочтительном варианте осуществления).
Органолептические свойства слоя пленочного покрытия также, главным образом, должны не изменяться при большой толщине слоя и в ходе хранения в течение длительных периодов времени при повышенной температуре, высокой или очень низкой относительной влажности или их сочетании (т.е. без растрескивания, адгезии, расслоения покрытия и т.д.).
Неожиданно, в данной работе было выявлено, что указанные выше требования могут быть удовлетворены, если для слоев пленочного покрытия использовать тонкоизмельченный ацетаминофен, вместе с относительно небольшими количествами пригодного растворимого в воде или набухающего в воде полимера. Было выявлено, что можно получать составы этого типа с высоким содержанием активного ингредиента, и что вязкость растворов для распыления была заметно низкой даже при очень высоких содержаниях твердых веществ, составляющих более 30% по массе, и что даже толстые слои пленочного покрытия (200 микрометров и более) можно наносить в относительно короткое время, тем самым делая процесс экономичным. Растворение лекарственного средства также было достаточно быстрым в слоях, содержащих указанные выше 100 мг ацетаминофена.
Таким образом, был возможным высокоточный контроль количества напыленного ацетаминофена и таким образом также профиля растворения лекарственного средства (т.е. высвобождения в течение первого часа) посредством толщины слоя пленочного покрытия. Другим неожиданным открытием было то, что составы для пленочного покрытия в соответствии с изобретением были способны с высокой эффективностью сглаживать шероховатые поверхности экструдированных таблеток, т.е. пленочное покрытие закрывало углубления на поверхности таблеток с высокой эффективностью. Это было удивительным, учитывая то, что практически все коммерчески доступные пленочные покрытия и полимеры, используемые для их получения, в действительности не обладают и не предназначены для обладания этим свойством. Известные полимеры и составы пленочных покрытий предназначены для детального воспроизведения рельефных элементов (логотипов, и т.д.) и линий разлома. Иными словами, "заполнение" углублений, присутствующих, в частности, в изготавливаемых общепринятыми способами таблетках, является нежелательным и оно должно полностью избегаться (см. WO 2006/002808; конкретная ссылка приведена на этот факт во всех примерах, см. пример 4, стр. 18: "Рельеф хорошо воспроизводился без эффектов смазывания и образования перемычек"). Пригодные полимеры для изготовления составов пленочных покрытий представляют собой растворимые в воде и набухающие в воде фармацевтически приемлемые полимеры, которые уже используются в настоящее время для изготовления пленочных покрытий. Основное требование состоит в том, чтобы образовывались распыляемые, предпочтительно чисто водные растворы или суспензии, которые имеют общее содержание твердых веществ (= сумму всех растворенных или суспендированных составляющих компонентов, включая активный ингредиент) по меньшей мере 20% по массе (предпочтительно 25%, в частности предпочтительно 30% или более). Содержание активного ингредиента в общем содержании твердых веществ раствора или дисперсии также должно составлять по меньшей мере 50% (предпочтительно 60%, в частности предпочтительно 70% или более). Также являются возможными неводные растворы или суспензии, если используют нетоксичный, фармацевтически приемлемый растворитель, такой как этанол. Также являются возможными смеси этих органических растворителей с водой. Однако, как правило, предпочтительными являются чисто водные растворы или суспензии.
Особенно предпочтительными являются полимеры, которые образуют растворы со сравнительно низкой вязкостью в водном растворе, даже при высоких концентрациях, для поддержания вязкости распыляемого раствора в диапазоне, в котором все еще обеспечиваются приемлемые свойства спрея раствора или суспензии, даже при использовании высокого общего содержания твердых веществ, упомянутых выше. Пригодные полимеры включают: неионные полимеры целлюлозы, такие как гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза; катионные полиметакрилаты, такие как Eudragit® E, Eudragit® NE30D, Eudragit® RL, Eudragit® RS; поливиниловый спирт; окись полиэтилена (высокомолекулярные полиэтиленгликоли с молекулярной массой (MW) >100,000); привитые сополимеры поливиниловый спирт/окись полиэтилена (Kollicoat® IR). Предпочтительно, пригодные полимеры выбирают из гидроксипропилметилцеллюлозы, Eudragit® NE30D и поливинилового спирта, или их сочетаний. Более предпочтительно, пригодными полимерами являются привитые сополимеры поливиниловый спирт/окись полиэтилена (например, Kollicoat® IR, BASF).
Активный ингредиент (предпочтительно: ацетаминофен) должен быть растворимым в указанных выше высоких концентрациях в водных или водных/органических или чисто органических растворителях. Если (как в случае ацетаминофена) растворимость в воде не является достаточной, предпочтительно можно использовать суспензии или дисперсии лекарственного средства. Однако в этом случае, решающим фактором является то, что распределение размера частиц активного ингредиента должно быть достаточно небольшим, поскольку иное является нежелательным, т.е. происходит слишком быстрое осаждение суспендированного активного ингредиента в распыляемом растворе и/или насадки для распыления в устройстве для нанесения пленочного покрытия блокируются. Предпочтительными размерами частиц являются: не более чем 10% частиц более 0,25 мм (особенно предпочтительно: не более чем 5%), не более чем 20% (особенно предпочтительно не более чем 10%) частиц более 0,1 мм и не более чем 35% (особенно предпочтительно не более чем 20%) частиц более 0,063 мм. Для достижения этого размера частиц лекарственные средства можно измельчать в ходе процессов измельчения (пригодным является сухое и влажное измельчение).
Неожиданно было выявлено, что слои пленочного покрытия в соответствии с изобретением не только очень хорошо сцепляются с таблетками, но также не становятся хрупкими или клейкими и не демонстрируют растрескивания даже при хранении при повышенных температурах, составляющих вплоть до 60°C. Также не происходило отслоения слоя покрытия от центральной части таблетки.
Различные иллюстративные варианты осуществления представлены ниже. Эти примеры предоставлены только для иллюстративных целей и их не следует считать сужающими объем изобретения.
Пример 1: Получение таблеток для пленочного покрытия
Гомогенную порошковую смесь, состоящую из 61,8% по массе ацетаминофена, 12,6% по массе Eudragit® RL, 12,6% по массе ксилита, 6% по массе гидроксипропилметилцеллюлозы (Methocel® K100), 6% по массе гидроксипропил метилцеллюлоза (Methocel® K100M) и 1,0% по массе Aerosil® 200 отмеряли со скоростью 20 кг/ч в двухшнековый экструдер с вращением в одном направлении (ZSK-40) и экструдировали при температуре приблизительно 140°C с получением гомогенной белой расплавленной ленты. Пока она еще находилась в пластичном состоянии, эту расплавленную ленту подавали в щель ролика формующего роликового каландр с противоположным вращением, ролики которого имели углубления на их поверхности, из которых могли образовываться таблетки непосредственно из расплавленной ленты. Полученные таблетки имели среднюю массу 720 мг после охлаждения и галтования. Поверхность таблеток была шероховатой и местами неравномерной.
Пример 2
Ацетаминофен с размером частиц, на 13% превышающим 0,25 мм и на 68% превышающим 0,063 мм, суспендировали в воде перемешиванием. Активный ингредиент оседал очень быстро после выключения мешалки. Суспензию измельчали и гомогенизировали, пропуская ее через коллоидную мельницу. После измельчения, к этой суспензии добавляли твердый порошковый полимер (Kollicoat® IR, BASF) (отношение масс ацетаминофен/Kollicoat® IR = 75:25) с получением общей концентрации твердых веществ 30% по массе. Даже после добавления полимера, ацетаминофен все еще демонстрировал выраженную тенденцию к осаждению. При непрерывном перемешивании эту суспензию распыляли на таблетки, описанные в примере 1 (6 кг) в устройстве для нанесения пленочного покрытия (Driam). Образцы таблеток отбирали после нанесения 30, 50, 70 и 90 мг ацетаминофена на пленочное покрытие. Во всех случаях наблюдали очень хорошее сцепление покрытия с таблетками, хотя поверхность покрытых чисто белой пленкой таблеток была все еще немного шероховатой вследствие все еще относительно больших частиц ацетаминофена. Уменьшение при высушивании таблеток составляло 1% по массе до и после нанесения пленки для всех форм. Параметры процесса нанесения пленочного покрытия:
6 кг центральных частей таблеток
Скорость барабана: 12 об/мин
Поступающий воздух: 1200 м3/ч
Температура поступающего воздуха: 65°C
Скорость распыления: 40-45 г/мин
Давление при распылении: 4,5 бар
Пример 3
Ацетаминофен с размером частиц 1% - более чем 0,25 мм, 5% - более чем 0,1 мм и 16% - более чем 0,063 мм, суспендировали в воде перемешиванием. Активный ингредиент показал сниженную тенденцию к осаждению после выключения мешалки по сравнению с материалом, который использовали в примере 2. Затем к этой суспензии добавляли твердый порошковый полимер (Kollicoat® IR, BASF) (отношение масс ацетаминофен/Kollicoat IR® = 75:25) с получением общей концентрации твердых веществ 30% по массе. После добавления полимера ацетаминофен почти не показал какой-либо тенденции к осаждению. Затем эту суспензию распыляли на таблетки (6 кг), которые получали, как описано в примере 1, однако с немного модифицированной геометрией таблеток, в устройстве для нанесения пленочного покрытия (Driam) (параметры процесса как в примере 2). Образцы таблеток отбирали после нанесения 30, 50, 70, 90 и 120 мг ацетаминофена на пленочное покрытие. Во всех случаях наблюдали очень высокую адгезию покрытия на таблетках. Поверхность покрытых чисто белой пленкой таблеток была гладкой и однородной.
Пример 4: Растворение лекарственного средства таблеток
Растворение лекарственного средства таблеток в соответствии с примером 1 определяли в устройстве согласно фармакопее США (USP Dissolution Apparatus II (лопастное), USP XXV; 37°C, 0,01M HCl, 50 об./мин). Количество активного ингредиента, высвобождаемого из таблеток в среду водного раствора HCl, определяли посредством ВЭЖХ с различными интервалами.
Таблетки без нанесения пленочного покрытия
Растворение лекарственного средства, измеренное через 30 минут: 7%
Растворение лекарственного средства, измеренное через 60 минут: 11%
Растворение лекарственного средства, измеренное через 120 минут: 17%
Растворение лекарственного средства, измеренное через 240 минут: 27%
Пример 5: Растворение лекарственного средства таблеток с пленочным покрытием
Растворение лекарственного средства таблеток в соответствии с примером 2 определяли в устройстве согласно фармакопее США (USP Dissolution Apparatus II (лопастное), USP XXV; 37°C, 0,01M HCl, 50 об./мин). Количество активного ингредиента, высвобождаемого из таблеток в среду водного раствора HCl, определяли посредством ВЭЖХ с различными интервалами.
Покрытая пленкой таблетка с 90 мг ацетаминофена в пленочном покрытии:
Растворение лекарственного средства, измеренное через 30 минут: 16%
Растворение лекарственного средства, измеренное через 60 минут: 20%
Растворение лекарственного средства, измеренное через 120 минут: 27%
Растворение лекарственного средства, измеренное через 240 минут: 36%
Скорости растворения лекарственного средства возрастали приблизительно на 10% при каждом тестируемом интервале вследствие исходно быстрого высвобождения активного ингредиента, присутствующего в пленочном покрытии.
Пример 6: Растворение лекарственного средства покрытых пленкой таблеток
Растворение лекарственного средства таблеток в соответствии с примером 3 определяли в устройстве согласно фармакопее США (лопастной способ, USP XXV; 37°C, 0,01M HCl, 50 об/мин). Количество активного ингредиента, высвобождаемого из таблеток в среду водного раствора HCl, определяли посредством ВЭЖХ с различными интервалами.
Таблетка без нанесения пленочного покрытия:
Растворение лекарственного средства, измеренное через 30 минут: 7%
Растворение лекарственного средства, измеренное через 60 минут: 12%
Растворение лекарственного средства, измеренное через 120 минут: 19%
Растворение лекарственного средства, измеренное через 240 минут: 29%
Растворение лекарственного средства, измеренное через 360 минут: 37%
Растворение лекарственного средства, измеренное через 480 минут: 43%
Покрытая пленкой таблетка со 120 мг ацетаминофена в пленочном покрытии:
Растворение лекарственного средства, измеренное через 30 минут: 28%
Растворение лекарственного средства, измеренное через 60 минут: 35%
Растворение лекарственного средства, измеренное через 120 минут: 43%
Растворение лекарственного средства, измеренное через 240 минут: 53%
Растворение лекарственного средства, измеренное через 360 минут: 62%
Растворение лекарственного средства, измеренное через 480 минут: 69%
Скорости растворения лекарственного средства возрастали приблизительно на 25% при каждом тестируемом интервале вследствие быстрого исходного высвобождения активного ингредиента, присутствующего в пленочном покрытии.
Пример 7
Тестирование проводили, как и в примере 3, однако вместо Kollicoat® IR использовали твердый порошок на основе гидроксипропилметилцеллюлозы, который содержал небольшую часть цветных пигментов на основе оксида железа. Вследствие значительно более высокой вязкости водной суспензии, общую концентрацию твердых веществ можно было доводить только до 20% по массе, в результате чего время распыления возрастало, а другие параметры процесса оставались неизмененными. Наблюдали очень высокое сцепление покрытия с таблетками. Поверхность красноватых/коричневатых покрытых пленкой таблеток была гладкой и однородной.
Пример 8
Тест проводили, как в примере 3, однако вместо Kollicoat® IR использовали твердый порошок на основе поливинилового спирта, содержащий небольшую часть пигментов диоксида титана. Вследствие немного более высокой вязкости водной суспензии, общую концентрацию твердых вещество можно было доводить только до 25% по массе, в результате чего время распыления возрастало, в то время как другие параметры процесса оставались неизмененными. Наблюдали очень высокое сцепление покрытия с таблетками. Поверхность покрытых чисто белой пленкой таблеток была гладкой и однородной.
Пример 9
Покрытые пленкой таблетки, полученные в соответствии с примерами 3, 7 и 8, хранили в закрытых стеклянных бутылках при температурах 40°C и 60°C. Через 1 месяц не было заметно трещин на таблетках и не наблюдали клейкости. Растворение лекарственного средства, измеренное способом, описанным в примере 4, не показало изменений по сравнению со значениями, зарегистрированными в начале хранения.
Пример 10
Получали образцы покрытых пленкой таблеток, изготовленных в соответствии с примером 3 (90 мг ацетаминофена в слое пленочного покрытия) и делали тонкий срез таблетки в поперечном направлении с помощью микротома и исследовали под микроскопом. На изображениях слой пленочного покрытия был легко отличим от центральной части таблетки. С помощью изображений определили, что слой пленочного покрытия составлял 300 микрометров. Сглаживающий эффект суспензии для покрытия на поверхности шероховатой таблетки был особенно заметен, что также видно на фиг.1, 3 и 4.
Пример 11: Растворение в HCl и водном растворе этанола
Ниже представлено описание иллюстративных способов исследования скорости растворения определенных композиций в HCl и 20% водном растворе этанола. Для исследования скорости растворения в 40% водном растворе этанола можно использовать сходные способы.
Для растворения в 0,01н. хлористоводородной кислоте и 20/40% водном растворе этанола использовали следующее устройство и способы:
(I) Растворение в 0,01н. HCl
Устройство: USP Dissolution Apparatus II (лопастное)
Скорость вращения: 50 об/мин
Среда: 0,01н. HCl
Объем среды: 900 мл
Температура: 37°C
Время забора образцов для тестирования высвобождения в течение 30 ч: 30/60/120/180/240/360/420/480/600/720/840/1080/1320/1560/1800 минут
Объем образца: 10 мл (без замещения объема)
Приготовление образца: используется как есть
Окончание анализа: УФ-детекция, длина волны 280 нм
(II) Растворение в 20 или 40% водном растворе этанола
Устройство: USP Dissolution Apparatus II (лопастное)
Скорость вращения: 50 об/мин
Среда: 20 или 40% водный раствор этанола
Объем среды: 500 мл
Температура: 37°C
Время забора образцов для тестирования высвобождения в течение 30 ч: 30/60/120/180/240/360/420/480/600/720/840/1080/1320/1560/1800 минут
Объем образца: 10 мл (без замещения объема)
Приготовление образца: используется как есть
Окончание анализа: УФ-детекция, длина волны 280 нм
III. Тестирование растворения целых таблеток в 0,01 н. HCl при 37°C
a) Состав с быстрым высвобождением (в отношении ацетаминофена) в 0,01 н. HCl при 37°C представлен на в таблице X. В таблице IX представлен состав центральной части и покрытия состава 5.
В таблице X представлены данные по растворению для гидрокодона (X(a)) и ацетаминофена (X(b)).
b) Состав с замедленным высвобождением (в отношении ацетаминофена) в 0,01 н. HCl при 37°C представлен в таблице XII. В таблице XI представлен состав центральной части и покрытия состава 6.
Данные растворения для гидрокодона (XII(a) и ацетаминофена XII(b))
IV. Тестирование растворения целых таблеток в 40% водном растворе этанола при 37°C
a) Состав с быстрым высвобождением (в отношении ацетаминофена) в 40% водном растворе этанола при 37°C представлен в таблице XIV. В таблице XIII представлен состав центральной части и покрытия состава 5.
В таблице XIV представлены данные растворения для гидрокодона (XIV (a)) и ацетаминофена (XIV (b)).
b) Состав с замедленным высвобождением (в отношении ацетаминофена) в 40% водном растворе этанола при 37°C представлен в таблице XVI. В таблице XV представлен состав центральной части и покрытия состава 8.
В таблице XVI представлены данные растворения для гидрокодона (XVI(a)) и ацетаминофена (XVI(b)).
V. Тестирование растворения измельченных таблеток (кофемолка, 60 с) в 40% водном растворе этанола при 37°C
В бытовой кофемолке 3 экструдированных таблетки измельчали в течение 60 с при ~20000-50000 об/мин. Порошок собирали и количество, эквивалентное одной таблетке, переносили в емкость для растворения для тестирования высвобождения.
Для анализа определения размера частиц образца порошок собирали и просеивали через сито с размером ячеек 355 мкм. Материал, который проходил через сито, просеивали через сито с размером ячеек 63 мкм. Получали следующие фракции:
Фракция 1: размер частиц >355 мкм (~20% от общего количества порошка)
Фракция 2: размер частиц >63 мкм и <355 мкм (~66% от общего количества порошка)
Фракция 3: размер частиц <63 мкм (~14% от общего количества порошка)
a) Состав с быстрым высвобождением (в отношении ацетаминофена) в 40% водном растворе этанола при 37°C представлен в таблице XVII. Данные по растворению для гидрокодона (XVII(a)) и ацетаминофена (XVII(b)) представлены ниже.
b) Состав с замедленным высвобождением (в отношении ацетаминофена) в 40% водном растворе этанола при 37°C представлен в таблице XVIII. Данные по растворению для гидрокодона (XVIII(a)) и ацетаминофена (XVIII(b)) представлены ниже.
VI. Тестирование растворения целых таблеток в 0,01н. HCl при 4°C
a) Состав с быстрым высвобождением (в отношении ацетаминофена) в 0,01 н. HCl при 4°C представлен в таблице XIX. Данные по растворению для гидрокодона (XIX(a)) и ацетаминофена (XIX(b)) представлены ниже.
b) Состав с замедленным высвобождением (в отношении ацетаминофена) в 0,01 н. HCl при 4°C представлен в таблице XX. Данные по растворению для гидрокодона (XX(a)) и ацетаминофена (XX(b)) представлены ниже.
VIII. Шероховатость поверхности
Покрытие экструдированных таблеток приводило к значительному сглаживанию поверхности таблетки, как можно видеть на фиг.1.
Для определения изменения шероховатости поверхности таблетки с покрытием и без него разрезали пополам вдоль малой оси. Поверхность этого поперечного среза измельчали с получением плоской и гладкой поверхности. Оптические микроснимки поперечного среза использовали для определения средней шероховатости поверхности. Для анализа использовали подход среднего значения центральной линии (CLA), как представлено на фиг.2, в котором определяли среднюю высоту на единицу длины от центральной линии. Центральную линию помещали в микрофотографии так, чтобы площадь над и под линией была приблизительно равной.
CLA вычисляли с использованием образцов в положениях с равными расстояниями, в соответствии со следующим уравнением:
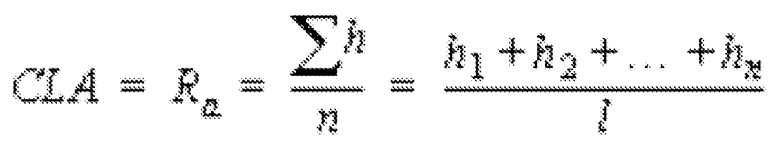
Общая длина l составляла 4,69 мм, расстояние между шагами составляло 68 мкм. Для состава без покрытия CLA=0,56, когда (N=69), как показано на фиг.3. Между тем для состава с покрытием CLA=0,15, когда (N=69), как показано на фиг.4.
IX. Тестирование растворения целых таблеток в 0,01н. HCl при 37°C для различной толщины покрытия
a) Состав с замедленным высвобождением (в отношении ацетаминофена) в 0,01н. Cl при 37°C представлен для различных составов 9-12 в таблицах XXII и XXIII. Композиции составов представлены в таблице XXI.
Eur./NF
Ph. Eur./NF
Ph. Eur./NF
X. Тестирование растворения целых таблеток без покрытия в 0,01н. HCl при 37°C
a) Состав с быстрым высвобождением (в отношении ацетаминофена) в 0,01н. HCl при 37°C представлен в таблице XXV. В таблице XXIV представлен состав центральной части состава 13.
Данные по растворению для гидрокодона (XXV(a)) и ацетаминофена (XXV(b)) представлены ниже:
b) Состав с замедленным высвобождением (в отношении ацетаминофена) в 0,01н. HCl при 37°C представлен в таблице XXVII. В таблице XXVI представлен состав центральной части состава 13.
Данные по растворению для гидрокодона (XXVII(a)) и ацетаминофена (XXVII(b)) представлены ниже.
Пример 12: Сравнение биодоступности тестируемых составов с контролем
Задачей исследования было сравнение биодоступности двух тестируемых составов 15 и 16 с биодоступностью контроля для сравнения, представленного в таблице. Схема испытания включала открытое, состоящее из трех периодов, перекрестное испытание однократной дозы натощак у 21 субъекта. Схема A включала одну таблетку состава 15; схема B включала одну таблетку состава 16; схема C включала одну таблетку контроля 1. Образцы крови собирали через 0, 0,25, 0,5, 0,75, 1, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36 и 48 часов после дозы на 1 сутки испытания. В представленной ниже таблице XXVIII проиллюстрированы составы тестируемых составов 15, 16 и контроля 1. См. также фиг.5 и 6 для средних концентраций гидрокодона и ацетаминофена для составов 15, 16 и контроля 1. Составы 5, 7 и 15 являются по существу идентичными друг другу, однако они пронумерованы по-разному исходя из различной нумерации тестов и экспериментов. Аналогично, составы и 6, 8 и 16 являются по существу идентичными друг другу, однако они были пронумерованы по-разному исходя из различной нумерации тестов и экспериментов. Также аналогично контроли 1 и 2 по существу идентичны друг другу, однако они были пронумерованы по-разному исходя из различной нумерации тестов и экспериментов. В одном варианте осуществления изобретения, предпочтительной дозированной формой является состав 15, поскольку состав 15 обеспечивает лучшие свойства смешивания, чем состав 16, как для смешивания гидрокодона битартрата пентагемигидрата и HPMC, так и для смешивания всех компонентов. Кроме того, смесь состава 15 обеспечивает свойства лучшей текучести, чем состав 16 в экструдере. Также состав 15 обеспечивает свойство лучшего прямого образования формы, чем состав 16, поскольку состав 15 является менее клейким, чем состав 16. Более того, ожидается, что состав 15 имеет лучшие удерживающие от зависимости свойства, чем состав 16.
Предварительные фармакокинетические параметры для составов 15, 16 и контроля 1 представлены ниже в таблице XXIX.
(ч)
(нг/мл)
(нг*ч/мл)
(ч)
(33%)
(17%)
(19%)
(l8%)
(18%)
(19%)
(32%)
(19%)
(20%)
(20%)
(22%)
(18%)
(63%)
(20%)
(18%)
(18%)
(19%)
(16%)
(ч)
(мкг/мл)
(мкг*ч/мл)
(ч)
Предварительные данные по относительной биодоступности составов 15 и 16 против контроля 1 представлены ниже в таблице XXX.
Тест против эталона
Состав 16 против контроля 1
Сmax
13,240
12,626
1,049
0,985-1,116
Состав 16 против контроля 1
AUCt
203,905
206,338
0,988
0,937-1,042
Состав 16 против контроля 1
AUC∞
208,867
210,187
0,994
0,944-1,046
Состав 16 против контроля 1
Сmax
2,395
2,193
1,092
1,018-1,172
Состав 16 против контроля 1
AUCt
22,363
21,732
1,029
0,975-1,086
Состав 16 против контроля 1
AUC∞
22,402
21,987
1,023
0,956-1,095
+ - Антилогарифм разницы (тест минус эталон) среднеквадратичных средних значений для логарифмов
Исходя из предварительных данных два тестируемых состава 15 и 16 являются биологически эквивалентными контролю 1 в отношении как Cmax, так и AUC∞. Первоначальная скорость всасывания гидрокодона является немного более низкой для тестируемых составов 15 и 16 по сравнению с контролем 1.
Пример 13: Профили высвобождения лекарственного средства in vitro
Следующие составы 17 и 18, как представлено ниже в таблице XXXI, исследовали для профилей высвобождения лекарственного средства in vitro и этот профиль сравнивали с центральной частью без покрытия VM-I и контролем 2, как показано на фиг.7(a) и (b).
(650 мг)
(500 мг)
(2,2%)
(2,2%)
(55,9%)
(65,4%}
(13,5%)
(9,3%)
(11,0%)
(9,3%)
(3,0%)
(13,4%)
(3,0%)
(1,0%)
(1,0%)
Пример 14: Получение таблеток экструдированием из расплава, галтованием и нанесением пленочного покрытия
Для каждого из примеров в соответствии с таблицей XXXII получали гомогенную порошковую смесь, содержащую все ингредиенты. В случае примеров с 14A по 16A проводили двухстадийное смешивание для обеспечения равномерного распределения низкой дозы компонента API (гидрокодона битартрата 2,5 гидрата) в конечной смеси. Процесс смешивания описан в таблице XXXIII. В случае примеров 14A-16A общее количество 5 образцов порошка из каждой конечной порошковой смеси перед эксрудированием анализировали в отношении однородности содержимого гидрокодона битартрата 2,5 гидрата.
В таблице XXXII представлен состав порошковых смесей до экструдирования и конечной экструдированной таблетки (после экструдирования из расплава и прямого придания формы). Все ингредиенты тестировали, и они высвобождались как указано согласно фармакопее США (USP, NF) и/или европейской фармакопее (Ph. Eur.).
(Тип: Methocel®K100)
(Тип: Methocel® K100M)
(Тип: Eudragit RL PO)
(Тип: Klucel® EF)
(Тип Xylisorb® 90)
(Тип: Lutrol® F68)
(Тип: Aerosil® 200)
Конечную смесь из примеров 14B-17B дозировали в двухшнековый экструдер с вращением в одном направлении с постоянной скоростью подачи. Гомогенному содержащему белое лекарственное средство расплаву, выходящему из насадки экструдера, прямо придавали форму удлиненных таблеток каландрованием между двумя вращающимися в противоположном направлении роликами, имеющими углубления на их поверхности в соответствии с размерами, приведенными в таблице XXXIV. Установки параметров процесса для экструдирования из расплава и каландрования приведены в таблице XXXIV. В таблице XXXIV представлен процесс экструдирования из расплава и прямого придания формы (каландрования).
(диаметр шнека)
(длина/ширина/высота)
Таблетки согласно примерам 14C, 15C и 17C переносили в устройство для нанесения пленочного покрытия Driam 600. На первой стадии таблетки вращали в устройстве для нанесения покрытия при максимальной скорости вращения для полирования таблеток и для удаления неровностей вокруг таблеток, которые образуются при процессе придания скорости при каландровании. Этот материал, который удаляли из таблеток, удаляли из барабана для нанесения покрытия вместе с выходящим воздухом. После этой стадии "галтования" сразу начинали стадию нанесения пленочного покрытия на таблетки в том же устройстве для нанесения покрытия. В случае примера 16C таблетки помещали в закрытый контейнер из нержавеющей стали и вращали в течение 10 минут после завершения удаления краев и неровностей. Затем таблетки просеивали через сито и переносили в то же устройство для нанесения пленочного покрытия Driam, как и в случае других примеров. Состав слоя пленочного покрытия и установка параметров процесса стадии галтования и последующего нанесения пленочного покрытия приведены в таблице XXXV.
В таблице XXXV представлено галтование таблеток после каландрования.
Изготовление суспензии пленочного покрытия для примеров 14E - 16E главным образом проводили посредством следующих стадий. Сначала, ацетаминофен диспергировали в воде при комнатной температуре при перемешивании. К этой суспензии добавляли полимер (Kollicoat® IR) и перемешивание продолжали до образования гомогенной суспензии. Эту суспензию использовали непосредственно для пленочного покрытия. Перемешивание продолжали в ходе всего процесса нанесения пленочного покрытия. Для примеров 14E-17E использовали готовый для применения порошок ацетаминофена (Rhodia, ацетаминофен "тонкодисперсный порошок"). Не проводили никакого дополнительного просеивания или тонкого измельчения. Состав суспензий для нанесения пленочного покрытия обобщенно представлен в таблице XXXVI.
В таблице XXXVI представлен состав суспензии пленочного покрытия
5%>0,1 мм
16%>0,063 мм
Нанесение пленочного покрытия таблеток после галтования проводили в устройстве для нанесения пленочного покрытия Driam 600. Условия процесса, установка параметров и данные для конечных покрытых пленкой таблеток представлены в таблице XXXVII. В случае всех примеров 14F-17F образцы получали в различные моменты времени в ходе фазы нанесения пленочного покрытия. Это делали для исследования влияния различной величины толщины слоя покрытия на высвобождение лекарственного средства как ацетаминофена, так и гидрокодона битартрата, из покрытых пленкой таблеток. Скорость распыления в ходе основной фазы нанесения пленочного покрытия представляла собой максимальную скорость перистальтического насоса, дозирующего суспензию ацетаминофена/Kollicoat® IR. Должны быть возможны более высокие скорости распыления.
В таблице XXXVII представлен процесс нанесения пленочного покрытия.
7,82 мм
7,07 мм
7,32 мм
6,41 мм
10,66 мм
7,71 мм
7,62 мм
7,23 мм
Главным образом, определенные предпочтительные варианты осуществления настоящего изобретения относятся к дозированным формам и способам доставки лекарственных средств, в частности вызывающих зависимость лекарственных средств, характеризующихся устойчивостью к экстракции растворителем: сдавлению, дроблению или растиранию, и обеспечивающих первоначальную волну высвобождения лекарственного средства с последующим длительным периодом контролируемого высвобождения лекарственного средства.
Кроме того, как показано ниже в таблице XXXVIII, в одном предпочтительном варианте осуществления, настоящее изобретение относится к фармацевтической композиции, имеющей центральный и нецентральный слой, содержащей: (a) гидрокодон, его фармацевтически приемлемую соль или гидрат, и (b) ацетаминофен или ибупрофен. В этом варианте осуществления, по меньшей мере 75% всего гидрокодона, его фармацевтически приемлемой соли или гидрата находится в центральной части, а ацетаминофен или ибупрофен находится в нецентральном слое. Кроме того, эта композиция адаптирована так, чтобы она была пригодна для перорального введения человеку 3, 2 или 1 раз в сутки. Предпочтительно, более чем 90% гидрокодона, его фармацевтически приемлемой соли или гидрата находятся в центральной части. Более предпочтительно, по существу весь гидрокодон, его фармацевтически приемлемая соль или гидрат находится в центральной части. В другом варианте осуществления, центральная часть дополнительно содержит ацетаминофен или ибупрофен. Более предпочтительно, центральная часть дополнительно содержит ацетаминофен.
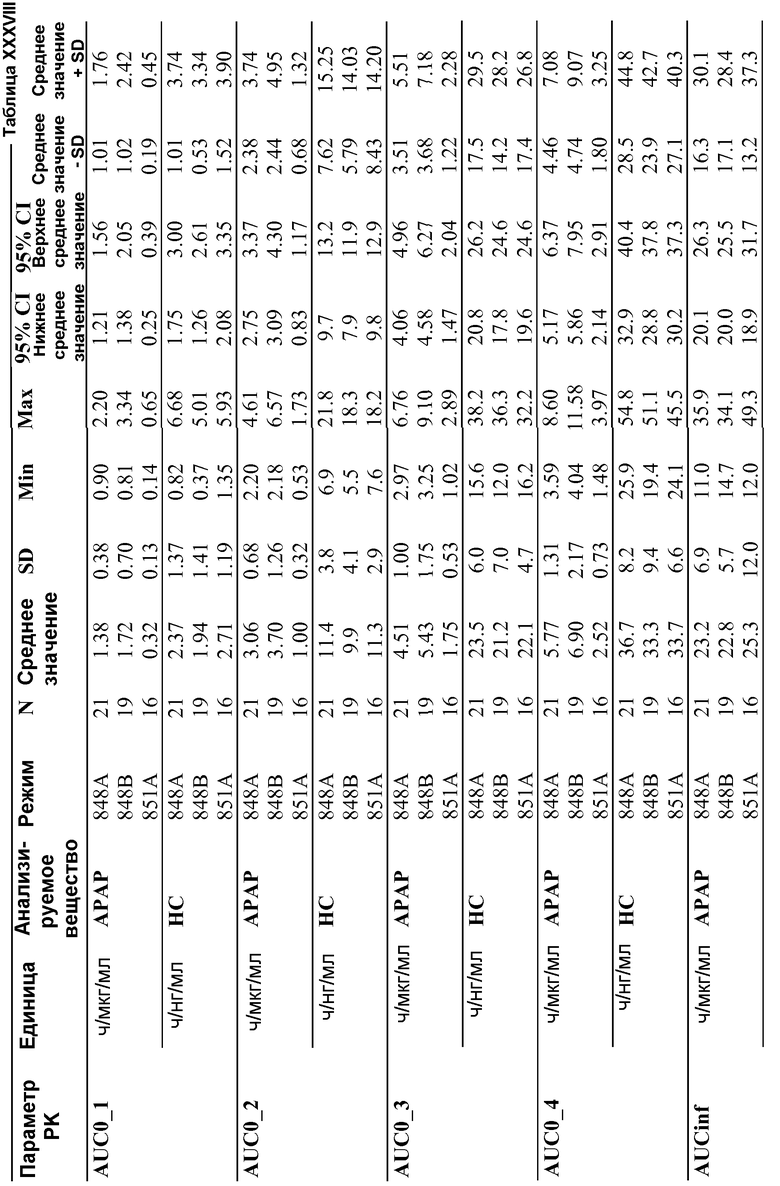
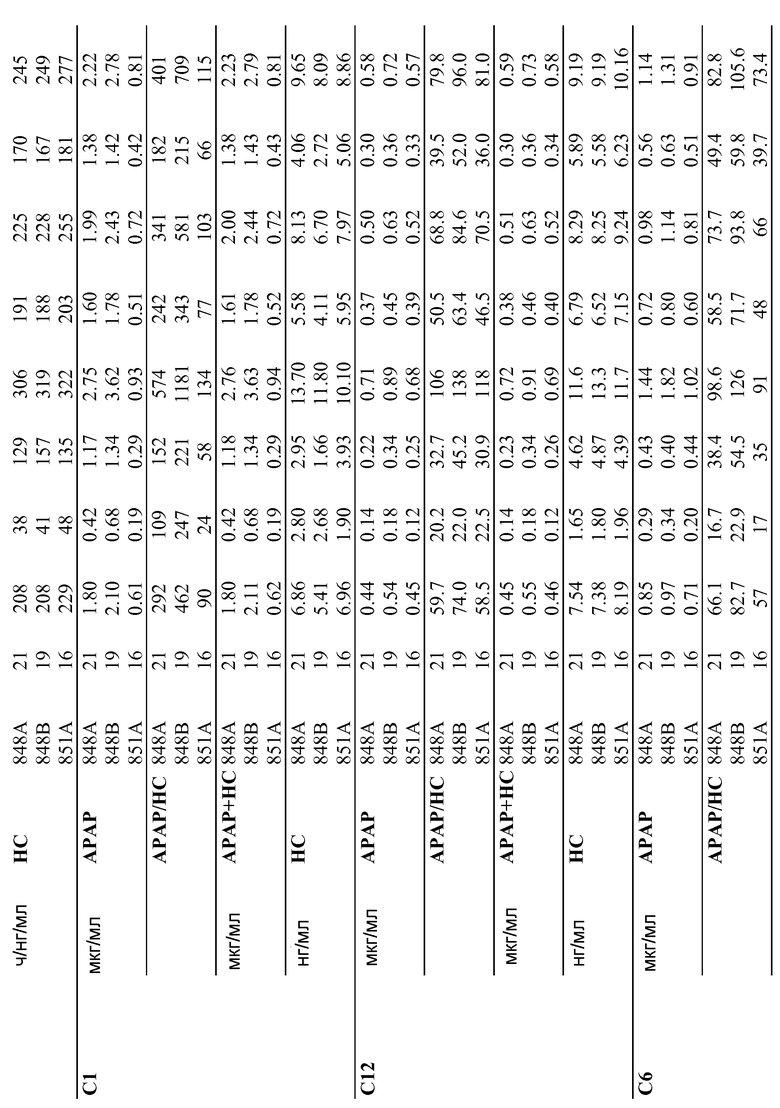
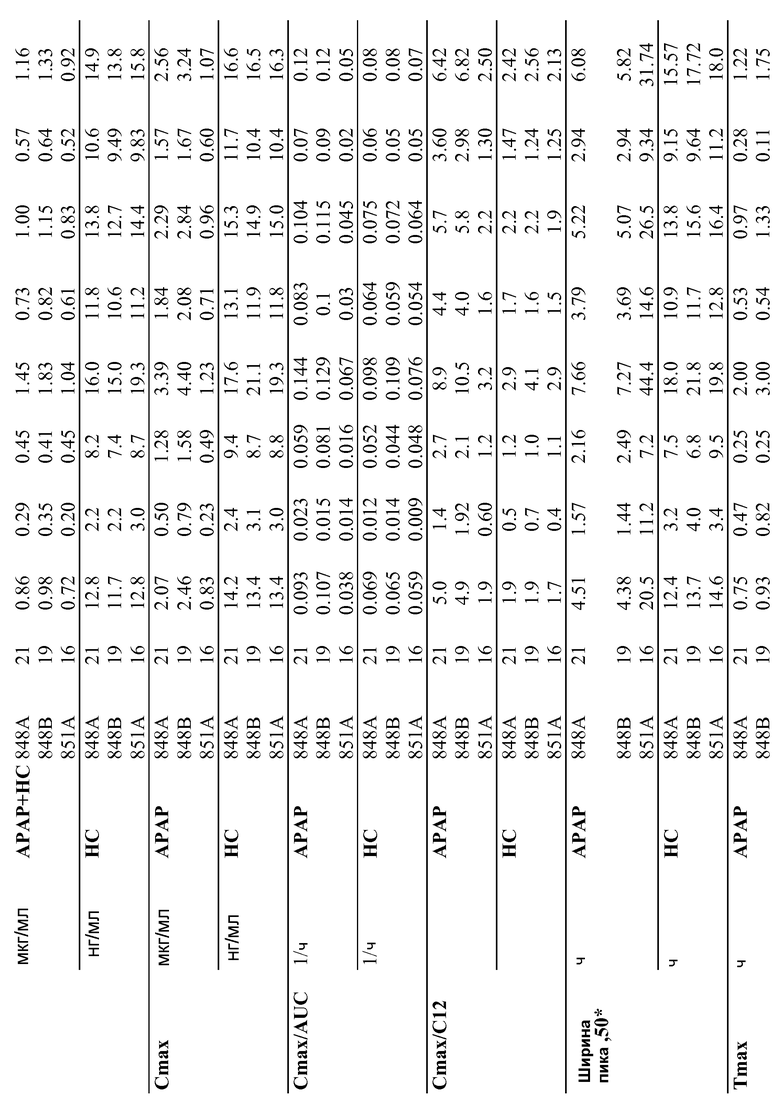

В определенных вариантах осуществления предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,4 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,8 нг/мл/мг и 7,9 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,4 нг/мл/мг до приблизительно 1,9 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,0 нг/мл/мг до приблизительно 10,4 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,0 нг/мл/мг и Cmax для ацетаминофена от приблизительно 3,0 нг/мл/мг до приблизительно 5,2 нг/мл/мг, после однократной дозы. Другие варианты осуществления дозированной формы включают приблизительно 5-20 мг гидрокодона битартрата пентагемигидрата и приблизительно 400-600 мг ацетаминофена. Другой вариант осуществления дозированной формы включает 10-15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500-600 мг ацетаминофена.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. При введении пациенту-человеку, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 9,1 нг*ч/мл/мг до приблизительно 19,9 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,6 нг*ч/мл/мг до приблизительно 59,1 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 7,0 нг*ч/мл/мг до приблизительно 26,2 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 18,4 нг*ч/мл/мг до приблизительно 79,9 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 11,3 нг*ч/мл/мг до приблизительно 18,7 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,7 нг*ч/мл/мг до приблизительно 53,5 нг*ч/мл/мг. Предпочтительно в этом варианте осуществления, скорость высвобождения фармацевтической композиции in vitro имеет двухфазный профиль высвобождения, и где для каждой фазы скорость высвобождения in vitro имеет нулевой порядок или первый порядок для ацетаминофена и нулевой порядок или первый порядок для гидрокодона битартрата пентагемигидрата.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,18 нг/мл/мг до приблизительно 1,51 нг/мл/мг, и концентрацию в плазме C1 через 1 час для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 7,24 нг/мл/мг. В предпочтительных вариантах осуществления, таких как состав 15, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,32 нг/мл/мг до приблизительно 1,51 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 5,50 нг/мл/мг. В некоторых других вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,30 нг/мл/мг до приблизительно 1,06 нг/мл/мг, и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 5,57 нг/мл/мг. В предпочтительных вариантах осуществления, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,45 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 4,43 нг/мл/мг. В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 3,63 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 2,76 мкг/мл после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,79 мкг/мл после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,23 мкг/мл после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена 1,80±0,42 мкг/мл с 95% доверительным интервалом для среднего значения от приблизительно 1,61 мкг/мл до приблизительно 2,00 мкг/мл после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. 95% доверительный интервал для комбинированной C1 для гидрокодона и ацетаминофена в случае предпочтительных вариантов осуществления и контроля перекрывается. 95% доверительный интервал для среднего значения комбинированной C1 для гидрокодона и ацетаминофена для контроля находится в диапазоне приблизительно от 1,46 до 1,96 мкг/мл, после введения в качестве однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена пациенту-человеку. Контроль обеспечивает достаточные уровни в плазме опиоидного и неопиоидного аналгетика для обеспечения снижения интенсивности боли в пределах приблизительно 1 часа после введения.
При введении популяции здоровых северных американцев или западных европейцев, в частности, когда состав адаптирован, чтобы он был пригоден или предназначен для введения человеку каждые 12 часов при необходимости, приблизительно 20-45% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 20-45% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. В другом варианте осуществления, приблизительно 25-35% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 25-35% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. Кроме того, в другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 12 часов, и по меньшей мере от 60% до приблизительно 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 6 часов до приблизительно 8,5 часов. В другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 11 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 8 часов до приблизительно 11 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 9 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 9 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 10 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 10 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 11 часов до приблизительно 12 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 11 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 13 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 13 часов.
Однако когда вариант с замедленным высвобождением состава адаптирован, чтобы он был пригоден или предназначен для введения человеку два раза в сутки, при необходимости, тогда по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 18 часов до приблизительно 23 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 18 часов до приблизительно 23 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 20 часов до приблизительно 25 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 20 часов до приблизительно 25 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 21 часов до приблизительно 22 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 21 часов до приблизительно 22 часов. В другом варианте осуществления этого варианта осуществления с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 22 часов до приблизительно 26 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 22 часов до приблизительно 26 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 27 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 27 часов.
В предпочтительном варианте осуществления, настоящее изобретение относится к композиции, где центральный слой содержит эксципиент или смесь эксципиентов, способных контролировать высвобождение лекарственного средства, и нецентральный слой содержит эксципиент, способный немедленно высвобождать лекарственное средство. Кроме того, в предпочтительном варианте осуществления, центральный слой получен путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, и нецентральный слой нанесен распылением на центральный слой. Наиболее предпочтительно, композиция содержит приблизительно 500 мг ацетаминофена и приблизительно 15 мг гидрокодона битартрата пентагемигидрата. В другом варианте осуществления, нецентральный слой, или покрытие таблетки, можно получать другим способом. В этом способе, слой пленочного покрытия изготавливают отдельно экструдированием и экструдату придают форму пленки. Эту пленку подают в каландр при изготовлении центральных частей. Этот способ особенно пригоден для толстых слоев (что устраняет длительное время нанесения покрытия распылением) и он представляет собой процесс без растворителя. Эта технология также известна как технология Xellex.
В другом иллюстративном варианте осуществления, настоящее изобретение относится к фармацевтической композиции, имеющей центральный и нецентральный слой, содержащей: (a) вызывающее зависимость лекарственное средство, его фармацевтически приемлемую соль или гидрат и не вызывающее зависимость лекарственное средство или его фармацевтически приемлемую соль в центральном слое, и (b) не вызывающее зависимость лекарственное средство, его фармацевтически приемлемую соль или гидрат в нецентральном слое. Предпочтительно, эта композиция характеризуется по меньшей мере одним из следующих признаков:
i) количество вызывающего зависимость лекарственного средства, которое экстрагируется из композиции 40% водным раствором этанола в течение одного часа при 37°C in vitro является меньшим или равно количеству вызывающего зависимость лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой in vitro в течение одного часа при 37°C, умноженному на 1,5,
ii) композиция не разрушается при воздействии силы 150 Н, предпочтительно 300 Н, более предпочтительно 450 Н, более предпочтительно 500 Н при измерении с помощью устройства для испытания на твердость "Pharma Test PTB 501",
iii) композиция высвобождает по меньшей мере 20% вызывающего зависимость лекарственного средства и не более чем 45% вызывающего зависимость лекарственного средства в течение первого часа тестирования растворения in vitro и предпочтительно также в течение первого часа тестирования in vivo,
iv) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства в течение от 1 до 2 часов после однократной дозы,
v) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства и/или вызывающего зависимость лекарственного средства через 1 час и через 12 часов после однократной дозы,
vi) в композиции высвобождение вызывающего зависимость лекарственного средства при измельчении повышается менее чем в 2-3 раза, по сравнению с целой таблеткой, когда композицию измельчают в течение 1 минуты кофемолкой при 20000-50000 об/мин, в 40% водном растворе этанола в течение 1 часа при 37°C,
vii) измельченная композиция содержит частицы размером от приблизительно 2 см до приблизительно 355 микрометров для приблизительно 20% фракции, более чем приблизительно 63 микрометров и менее чем приблизительно 355 микрометров для приблизительно 66% фракции и менее чем приблизительно 63 микрометров для приблизительно 14% фракции, при измерении с помощью теста просеиванием, или
viii) композиция является по существу однородной, где среднее значение центральной линии (CLA) составляет от приблизительно 0,1 до приблизительно 0,6, предпочтительно от приблизительно 0,1 до приблизительно 0,4, и наиболее предпочтительно от приблизительно 0,1 до приблизительно 0,2.
В этой композиции, количество вызывающего зависимость лекарственного средства, которое экстрагируется из состава 40% водным раствором этанола в течение одного часа при 37°C, составляет от приблизительно 70% до приблизительно 130% от количества лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой в течение одного часа при 37°C. В другом варианте осуществления, количество вызывающего зависимость лекарственного средства, которое экстрагируется из состава 40% водным раствором этанола в течение одного часа при 37°C, составляет от приблизительно 70% до приблизительно 90% от количества лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой в течение одного часа при 37°C. В другом варианте осуществления, вызывающее зависимость лекарственное средство, которое экстрагируется из состава 40% водным раствором этанола в течение одного часа при 37°C, составляет от приблизительно 75% до приблизительно 90% от количества лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой в течение одного часа при 37°C.
Другой вариант осуществления настоящего изобретения относится к фармацевтической композиции, имеющей центральный слой и нецентральный слой. В этой композиции центральный слой содержит смесь: (a) по меньшей мере одного опиоида; и (b) по меньшей мере одного изменяющего скорость фармацевтически приемлемого полимера, сополимера или их сочетаний. Нецентральный слой содержит по меньшей мере один неопиоидный аналгетик. Кроме того, эти композиции адаптированы для того, чтобы они были пригодными для перорального введения человеку 3, 2 или 1 раз в сутки. Предпочтительно, центральный слой дополнительно содержит по меньшей мере один неопиоидный аналгетик. В предпочтительном варианте осуществления, композиция характеризуется по меньшей мере одним из следующих признаков:
i) количество вызывающего зависимость лекарственного средства, которое экстрагируется из композиции 40% водным раствором этанола в течение одного часа при 37°C in vitro, является меньшим или равно количеству вызывающего зависимость лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой in vitro в течение одного часа при 37°C, умноженному на 1,5,
ii) композиция не разрушается при воздействии силы 150 Н, предпочтительно 300 Н, более предпочтительно 450 Н, более предпочтительно 500 Н при измерении с помощью устройства для испытания на твердость "Pharma Test PTB 501",
iii) композиция высвобождает по меньшей мере 20% вызывающего зависимость лекарственного средства и не более чем 45% вызывающего зависимость лекарственного средства в течение первого часа тестирования растворения in vitro и предпочтительно также в течение первого часа тестирования in vivo,
iv) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства в течение от 1 до 2 часов после однократной дозы,
v) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства и/или вызывающего зависимость лекарственного средства через 1 час и через 12 часов после однократной дозы,
vi) в композиции высвобождение вызывающего зависимость лекарственного средства при измельчении повышается менее чем в 2-3 раза, по сравнению с целой таблеткой, когда композицию измельчают в течение 1 минуты кофемолкой при 20000-50000 об/мин, в 40% водном растворе этанола в течение 1 часа при 37°C,
vii) измельченная композиция содержит частицы размером от приблизительно 2 см до приблизительно 355 микрометров для приблизительно 20% фракции, более чем приблизительно 63 микрометров и менее чем приблизительно 355 микрометров для приблизительно 66% фракции и менее чем приблизительно 63 микрометров для приблизительно 14% фракции, при измерении с помощью теста просеиванием, или
viii) композиция является по существу однородной, где среднее значение центральной линии (CLA) составляет от приблизительно 0,1 до приблизительно 0,6, предпочтительно от приблизительно 0,1 до приблизительно 0,4, и наиболее предпочтительно от приблизительно 0,1 до приблизительно 0,2.
В одном варианте осуществления, опиоид выбран из группы, состоящей из альфентанила, аллилпродина, альфапродина, анилеридина, бензилморфина, безитрамида, бупренорфина, буторфанола, клонитазена, кодеина, циклазоцина, дезоморфина, декстроморамида, дезоцина, диампромида, дигидрокодеина, дигидроморфина, дименоксадола, димефептанола, диметилтиамбутена, диоксафетила бутирата, дипипанона, эптазоцина, этогептазина, этилметилтиамбутена, этилморфина, этонитазена, фентанила, героина, гидрокодона, гидроморфона, гидроксипетидина, изометадона, кетобемидона, леваллорфана, левофенацилморфана, леворфанола, лофентанила, меперидина, мептазинола, метазоцина, метадона, метопона, морфина, мирофина, налбуфина, нарцеина, никоморфина, норпипанона, опиума, оксикодона, оксиморфона, папвретума, пентазоцина, фенадоксона, феназоцина, феноморфана, феноперидина, pиминодина, пропирама, пропоксифена, суфентанила, тилидина и трамадола, и их солей, гидратов и смесей.
Кроме того, неопиоидный аналгетик выбран из группы, состоящей из ацетаминофена, аспирина, фентанила, ибупрофена, индометацина, каторолака, напроксена, фенацетина, пироксикама, суфентанила, сунлиндака, интерферона альфа, и их солей, гидратов и смесей. Предпочтительно, опиоид представляет собой гидрокодон, и неопиоидный аналгетик представляет собой ацетаминофен или ибупрофен. Более предпочтительно, опиоид представляет собой гидрокодон, и неопиоидный аналгетик представляет собой ацетаминофен.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,18 нг/мл/мг до приблизительно 1,51 нг/мл/мг, и концентрацию в плазме C1 через 1 час для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 7,24 нг/мл/мг. В предпочтительных вариантах осуществления, таких как состав 15, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,32 нг/мл/мг до приблизительно 1,51 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 5,50 нг/мл/мг.
В некоторых других вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,30 нг/мл/мг до приблизительно 1,06 нг/мл/мг, и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 5,57 нг/мл/мг. В предпочтительных вариантах осуществления, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,45 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 4,43 нг/мл/мг.
В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 3,63 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 2,76 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,79 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,23 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена 1,80±0,42 мкг/мл с 95% доверительным интервалом для среднего значения от приблизительно 1,61 мкг/мл до приблизительно 2,00 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. 95% доверительный интервал для комбинированной C1 для гидрокодона и ацетаминофена в случае предпочтительных вариантов осуществления и контроля перекрываются. 95% доверительный интервал для среднего значения комбинированной C1 для гидрокодона и ацетаминофена для контроля находится в диапазоне приблизительно от 1,46 до 1,96 мкг/мл, после введения в качестве однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена пациенту-человеку. Контроль обеспечивает достаточные уровни в плазме опиоидного и неопиоидного аналгетика для обеспечения снижения интенсивности боли в пределах приблизительно 1 часа после введения.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,4 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,8 нг/мл/мг и 7,9 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,4 нг/мл/мг до приблизительно 1,9 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,0 нг/мл/мг до приблизительно 10,4 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,0 нг/мл/мг и Cmax для ацетаминофена от приблизительно 3,0 нг/мл/мг до приблизительно 5,2 нг/мл/мг, после однократной дозы. В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. При введении пациенту-человеку, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 9,1 нг*ч/мл/мг до приблизительно 19,9 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,6 нг*ч/мл/мг до приблизительно 59,1 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 7,0 нг*ч/мл/мг до приблизительно 26,2 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 18,4 нг*ч/мл/мг до приблизительно 79,9 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 11,3 нг*ч/мл/мг до приблизительно 18,7 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,7 нг*ч/мл/мг до приблизительно 53,5 нг*ч/мл/мг. Предпочтительно в этом варианте осуществления, скорость высвобождения фармацевтической композиции in vitro имеет двухфазный профиль высвобождения, и где для каждой фазы скорость высвобождения in vitro имеет нулевой порядок или первый порядок для ацетаминофена и нулевой порядок или первый порядок для гидрокодона.
При введении популяции здоровых северных американцев или западных европейцев, в частности, когда состав адаптирован, чтобы он был пригоден или предназначен для введения человеку каждые 12 часов при необходимости, приблизительно 20-45% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 20-45% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. В другом варианте осуществления, приблизительно 25-35% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 25-35% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. Кроме того, в другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 12 часов, и по меньшей мере от 60% до приблизительно 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 6 часов до приблизительно 8,5 часов. В другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 11 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 8 часов до приблизительно 11 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 9 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 9 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 10 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 10 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 11 часов до приблизительно 12 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 11 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 13 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 13 часов.
Однако когда вариант с замедленным высвобождением состава адаптирован, чтобы он был пригоден или предназначен для введения человеку два раза в сутки, при необходимости, тогда по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 18 часов до приблизительно 23 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 18 часов до приблизительно 23 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 20 часов до приблизительно 25 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 20 часов до приблизительно 25 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 21 часов до приблизительно 22 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 21 часов до приблизительно 22 часов. В другом варианте осуществления этого варианта осуществления с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 22 часов до приблизительно 26 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 22 часов до приблизительно 26 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 27 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 27 часов.
В предпочтительном варианте осуществления, настоящее изобретение относится к композиции, где центральный слой содержит эксципиент, способный контролировать высвобождение лекарственного средства и нецентральный слой содержит эксципиент, способный немедленно высвобождать лекарственное средство. Кроме того, в предпочтительном варианте осуществления, центральный слой изготовлен путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, и нецентральный слой нанесен распылением на центральный слой. Наиболее предпочтительно, композиция содержит приблизительно 500 мг ацетаминофена и приблизительно 15 мг гидрокодона битартрата пентагемигидрата.
В другом варианте осуществления, настоящее изобретение относится к фармацевтической композиции, имеющей центральный слой и нецентральный слой. В этой композиции, центральный слой содержит смесь (a) по меньшей мере одного опиоида и по меньшей мере одного первого неопиоидного аналгетика; (b) по меньшей мере одного изменяющего скорость фармацевтически приемлемого полимера, сополимера или их сочетаний. Нецентральный слой содержит по меньшей мере один второй неопиоидный аналгетик. Кроме того, композиция адаптирована таким образом, чтобы она была пригодна для перорального введения человеку 3, 2 или 1 раз в сутки. В этом варианте осуществления, предпочтительно, опиоид включает гидрокодон и первый и второй неопиоидный аналгетик включает ацетаминофен или ибупрофен. Более предпочтительно, опиоид включает гидрокодон и первый и второй неопиоидный аналгетик включает ацетаминофен. Кроме того, в этом варианте осуществления, нецентральный слой содержит: (a) ацетаминофен; и (b) по меньшей мере одного изменяющего скорость фармацевтически приемлемого полимера, сополимера или их сочетаний. Предпочтительно, полимер или сополимер выбран из группы, состоящей из гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, гидроксиэтилцеллюлозы; полиметакрилата, поливинилового спирта, окиси полиэтилена и их сочетаний. Более предпочтительно, полимер или сополимер выбран из группы, состоящей из гидроксипропилметилцеллюлозы и поливинилового спирта, или их сочетаний. Более предпочтительно, полимер или сополимер выбран из группы, состоящей из привитых сополимеров поливинилового спирта и окиси полиэтилена. Кроме того, в этом варианте осуществления, соотношение ацетаминофена и контролирующего скорость полимера или сополимера или их сочетания составляет приблизительно 1:1 до приблизительно 10:1. Более предпочтительно, соотношение ацетаминофена и контролирующего скорость полимера или сополимера или их сочетания составляет приблизительно 3:1 до приблизительно 5:1. Как обеспечивается настоящим изобретением, в одном предпочтительном варианте осуществления, нецентральный слой имеет по меньшей мере один из следующих признаков:
(a) по существу не растрескивается после 3 месяцев при 40°C, 75% относительной влажности в индукционно закрытых бутылках HDPE;
(b) по существу сухой (не клейкий);
обеспечивает быстрое растворение в 0,01 н. HCl при 37°C, открывая центральный слой высвобождает по меньшей мере 80% ацетаминофена в нецентральном слое в пределах 20 минут после введения пациенту-человеку; или
(с) обеспечивает белую пигментацию состава без дополнительных пигментов.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,18 нг/мл/мг до приблизительно 1,51 нг/мл/мг, и концентрацию в плазме C1 через 1 час для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 7,24 нг/мл/мг.
В предпочтительных вариантах осуществления, таких как состав 15, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,32 нг/мл/мг до приблизительно 1,51 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,34 нг/мл/мг до приблизительно 5,50 нг/мл/мг.
В некоторых других вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает концентрацию в плазме через 1 час (C1) для гидрокодона от приблизительно 0,30 нг/мл/мг до приблизительно 1,06 нг/мл/мг, и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 5,57 нг/мл/мг. В предпочтительных вариантах осуществления, дозированная форма обеспечивает C1 для гидрокодона от приблизительно 0,45 нг/мл/мг до приблизительно 1,06 нг/мл/мг и C1 для ацетаминофена от приблизительно 2,75 нг/мл/мг до приблизительно 4,43 нг/мл/мг.
В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 3,63 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,18 мкг/мл до приблизительно 2,76 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В определенных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,79 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена от приблизительно 1,38 мкг/мл до приблизительно 2,23 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена.
В предпочтительных вариантах осуществления, дозированная форма обеспечивает комбинированную C1 для гидрокодона и ацетаминофена 1,80±0,42 мкг/мл с 95% доверительным интервалом для среднего значения от приблизительно 1,61 мкг/мл до приблизительно 2,00 мкг/мл, после однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена. 95% доверительный интервал для комбинированной C1 для гидрокодона и ацетаминофена в случае предпочтительных вариантов осуществления и контроля перекрываются. 95% доверительный интервал для среднего значения комбинированной C1 для гидрокодона и ацетаминофена для контроля находится в диапазоне приблизительно от 1,46 до 1,96 мкг/мл, после введения в качестве однократной дозы 15 мг гидрокодона битартрата пентагемигидрата и 500 мг ацетаминофена пациенту-человеку. Контроль обеспечивает достаточные уровни в плазме опиоидного и неопиоидного аналгетика для обеспечения снижения интенсивности боли в пределах приблизительно 1 часа после введения.
В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. Предпочтительно при введении пациенту-человеку фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,4 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,8 нг/мл/мг и 7,9 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,4 нг/мл/мг до приблизительно 1,9 нг/мл/мг и Cmax для ацетаминофена от приблизительно 2,0 нг/мл/мг до приблизительно 10,4 нг/мл/мг, после однократной дозы. В другом варианте осуществления, фармацевтическая композиция обеспечивает профиль в плазме, характеризующийся Cmax для гидрокодона от приблизительно 0,6 нг/мл/мг до приблизительно 1,0 нг/мл/мг и Cmax для ацетаминофена от приблизительно 3,0 нг/мл/мг до приблизительно 5,2 нг/мл/мг, после однократной дозы. В определенных вариантах осуществления, предпочтительно проявляется представленный ниже фармакокинетический профиль, когда однократная доза содержит приблизительно 15 мг гидрокодона битартрата пентагемигидрата и приблизительно 500 мг ацетаминофена, при введении пациенту натощак. При введении пациенту-человеку, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 9,1 нг*ч/мл/мг до приблизительно 19,9 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,6 нг*ч/мл/мг до приблизительно 59,1 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 7,0 нг*ч/мл/мг до приблизительно 26,2 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 18,4 нг*ч/мл/мг до приблизительно 79,9 нг*ч/мл/мг. В другом варианте осуществления, дозированная форма обеспечивает AUC для гидрокодона от приблизительно 11,3 нг*ч/мл/мг до приблизительно 18,7 нг*ч/мл/мг и AUC для ацетаминофена от приблизительно 28,7 нг*ч/мл/мг до приблизительно 53,5 нг*ч/мл/мг. Предпочтительно в этом варианте осуществления, скорость высвобождения фармацевтической композиции in vitro имеет двухфазный профиль высвобождения, и где для каждой фазы скорость высвобождения in vitro имеет нулевой порядок или первый порядок для ацетаминофена и нулевой порядок или первый порядок для гидрокодона. При введении популяции здоровых северных американцев или западных европейцев, в частности, когда состав адаптирован, чтобы он был пригоден или предназначен для введения человеку каждые 12 часов при необходимости, приблизительно 20-45% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 20-45% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. В другом варианте осуществления, приблизительно 25-35% гидрокодона высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа и приблизительно 25-35% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение приблизительно 1 часа в 0,01 н. HCl при 50 об/мин при 37°C. Кроме того, в другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 12 часов, и по меньшей мере от 60% до приблизительно 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 6 часов до приблизительно 8,5 часов. В другом варианте осуществления, по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 8 часов до приблизительно 11 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 8 часов до приблизительно 11 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 9 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 9 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 10 часов до приблизительно 12 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 10 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 11 часов до приблизительно 12 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 11 часов до приблизительно 12 часов. В другом варианте осуществления, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 13 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 13 часов.
Однако когда вариант с замедленным высвобождением состава адаптирован, чтобы он был пригоден или предназначен для введения человеку два раза в сутки, при необходимости, тогда по меньшей мере 90% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 18 часов до приблизительно 23 часов, и по меньшей мере 90% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 18 часов до приблизительно 23 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 20 часов до приблизительно 25 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 20 часов до приблизительно 25 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 95% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 21 часов до приблизительно 22 часов, и по меньшей мере 95% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 21 часов до приблизительно 22 часов. В другом варианте осуществления этого варианта осуществления с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение от приблизительно 22 часов до приблизительно 26 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение от приблизительно 22 часов до приблизительно 26 часов. В другом варианте осуществления состава с замедленным высвобождением, по меньшей мере 99% гидрокодона высвобождается из фармацевтической композиции в течение менее чем приблизительно 27 часов, и по меньшей мере 99% ацетаминофена высвобождается in vitro из фармацевтических композиций в течение менее чем приблизительно 27 часов.
В предпочтительном варианте осуществления, настоящее изобретение относится к композиции, где центральный слой содержит эксципиент, способный контролировать высвобождение лекарственного средства и нецентральный слой содержит эксципиент, способный немедленно высвобождать лекарственное средство. Кроме того, в предпочтительном варианте осуществления, центральный слой изготовлен путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, и нецентральный слой нанесен распылением на центральный слой. Наиболее предпочтительно, композиция содержит приблизительно 500 мг ацетаминофена и приблизительно 15 мг гидрокодона битартрата пентагемигидрата. В предпочтительном варианте осуществления, композиция характеризуется по меньшей мере одним из следующих признаков:
i) количество вызывающего зависимость лекарственного средства, которое экстрагируется из композиции 40% водным раствором этанола в течение одного часа при 37°C in vitro, является меньшим или равно количеству гидрокодона, которое экстрагируется 0,01 н. хлористоводородной кислотой in vitro в течение одного часа при 37°C, умноженному на 1,5,
ii) композиция не разрушается при воздействии силы 150 Н, предпочтительно 300 Н, более предпочтительно 450 Н, более предпочтительно 500 Н при измерении с помощью устройства для испытания на твердость "Pharma Test PTB 501",
iii) композиция высвобождает по меньшей мере 20% гидрокодона и не более чем 45% гидрокодона в течение первого часа тестирования растворения in vitro и предпочтительно также в течение первого часа тестирования in vivo,
iv) композиция высвобождает терапевтически эффективную дозу ацетаминофена в течение от 1 до 2 часов после однократной дозы,
v) композиция высвобождает терапевтически эффективную дозу ацетаминофена и/или вызывающего зависимость лекарственного средства через 1 час и через 12 часов после однократной дозы,
vi) в композиции высвобождение гидрокодона при измельчении возрастает более чем в 2-3 раза по сравнению с целой таблеткой, когда композицию измельчают в течение 1 минуты кофемолкой при 20000-50000 об/мин, в 40% водном растворе этанола в течение 1 часа при 37°C,
vii) измельченная композиция содержит частицы размером от приблизительно 2 см до приблизительно 355 микрометров для приблизительно 20% фракции, более чем приблизительно 63 микрометров и менее чем приблизительно 355 микрометров для приблизительно 66% фракции и менее чем приблизительно 63 микрометров для приблизительно 14% фракции, при измерении с помощью теста просеиванием, или
viii) композиция является по существу однородной, где среднее значение центральной линии (CLA) составляет от приблизительно 0,1 до приблизительно 0,6, предпочтительно от приблизительно 0,1 до приблизительно 0,4, и наиболее предпочтительно от приблизительно 0,1 до приблизительно 0,2.
Указанное выше подробное описание и прилагаемые примеры являются только иллюстративными и не предназначены для ограничений объема изобретения, который определяется только прилагаемой формулой изобретения и ее эквивалентами. Различные изменения и модификации описанных вариантов осуществления будут очевидны специалистам в данной области и являются частью настоящего изобретения. Такие изменения и модификации, включая, но не ограничиваясь ими, изменения и модификации, связанные с химической структурой, заместителями, производными, промежуточными соединениями, синтезом, составами и/или способами применения изобретения, можно проводить без отклонения от его сущности и объема.
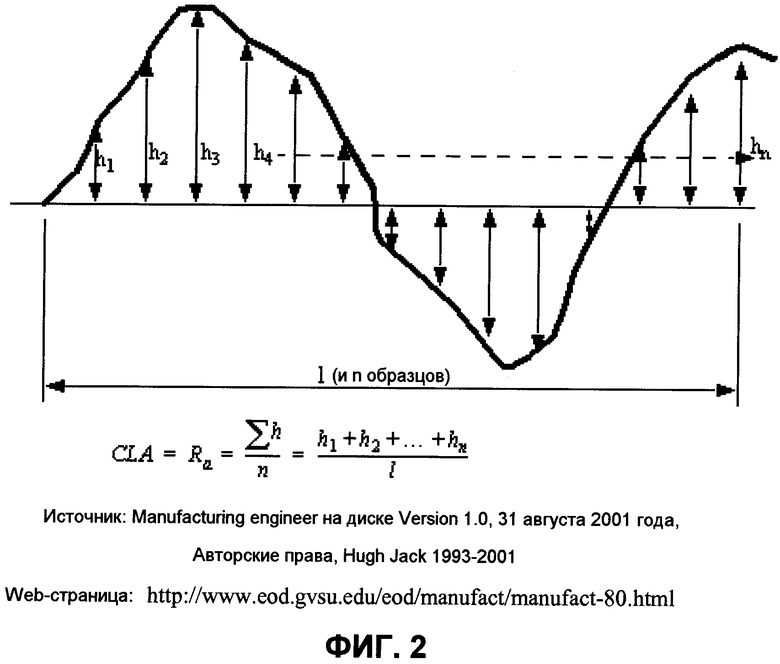
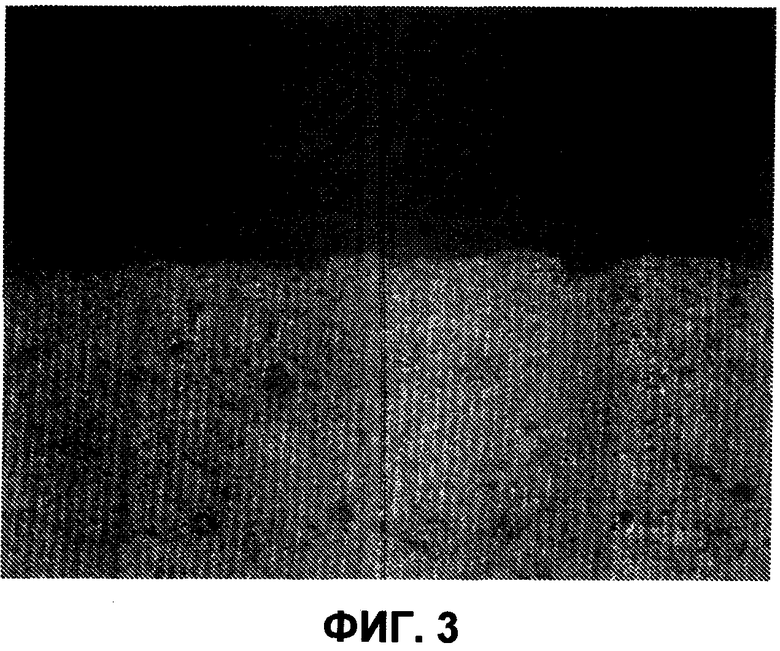
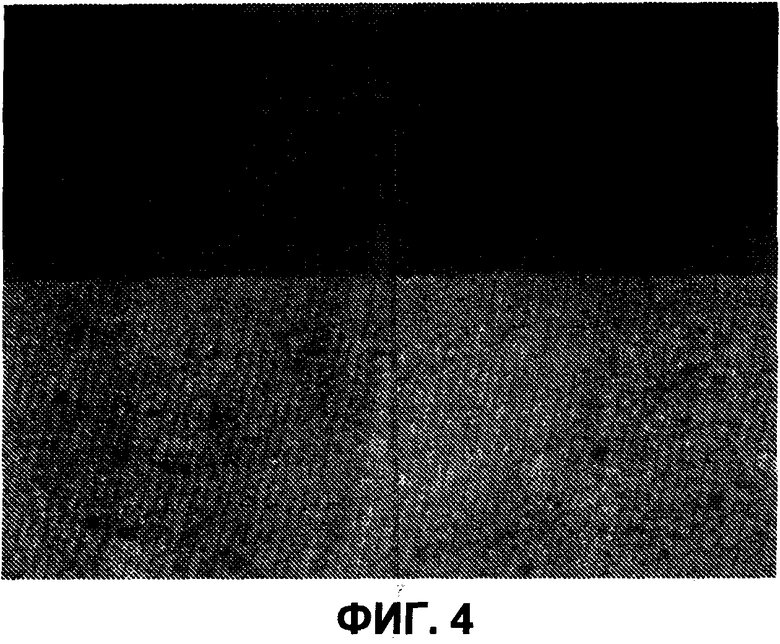
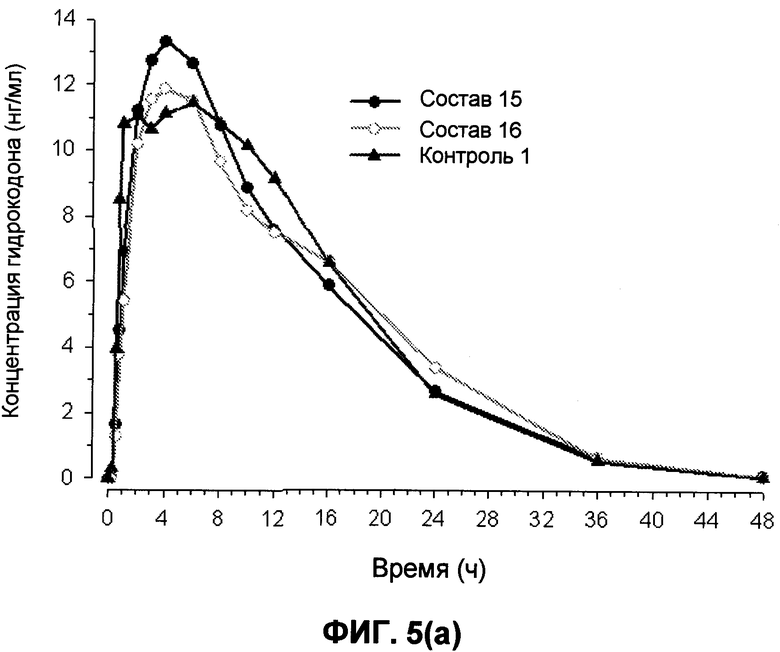
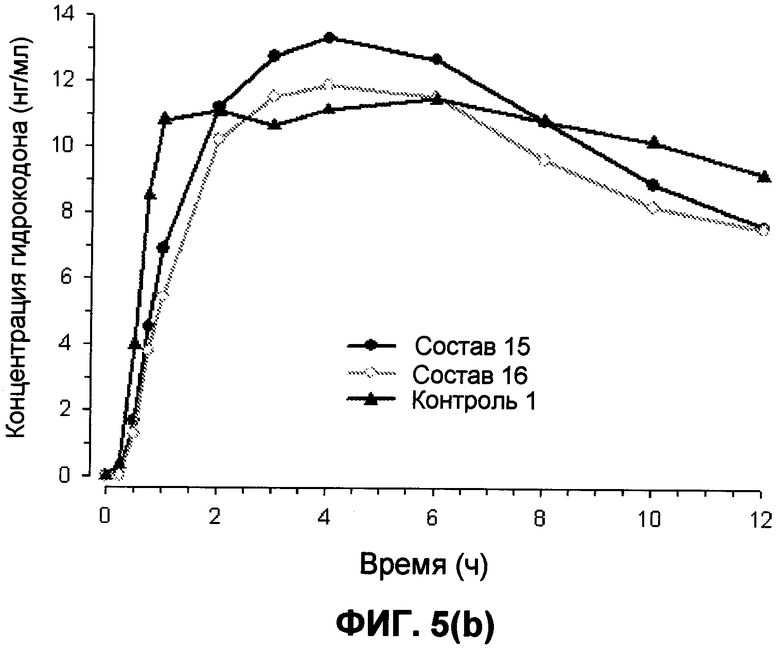
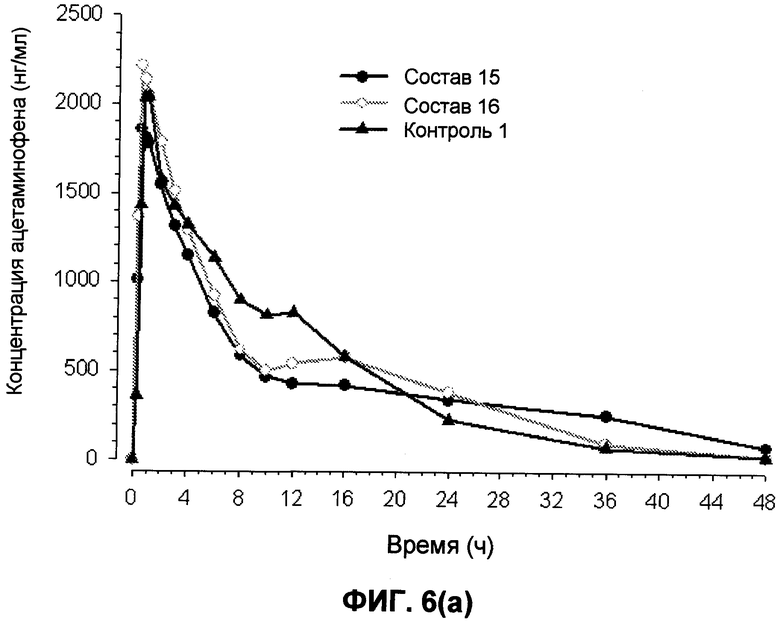
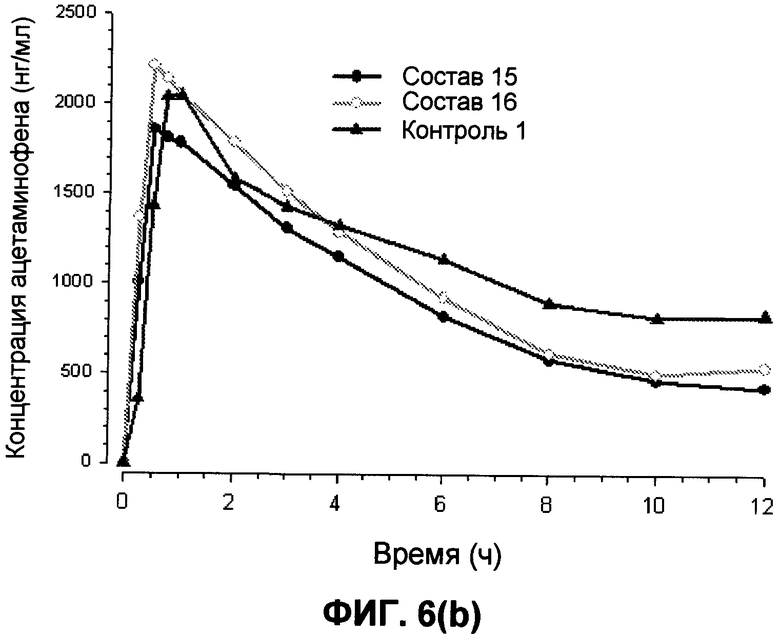
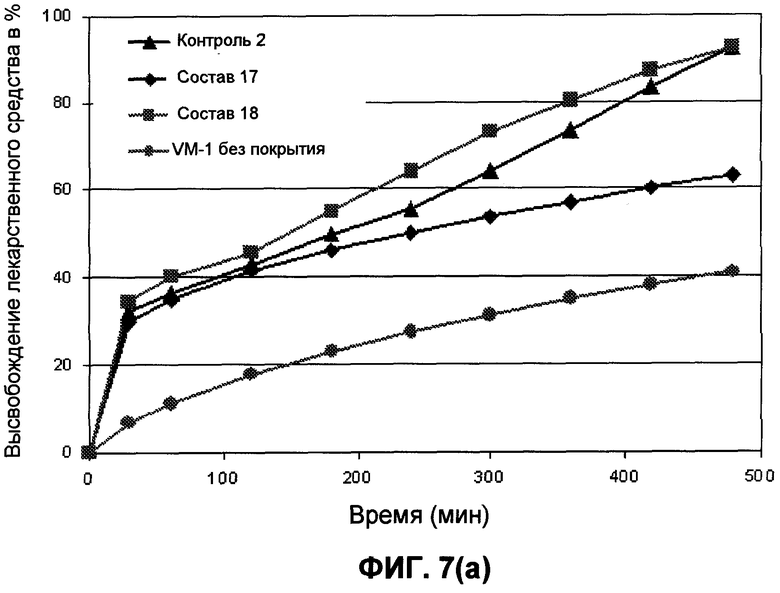
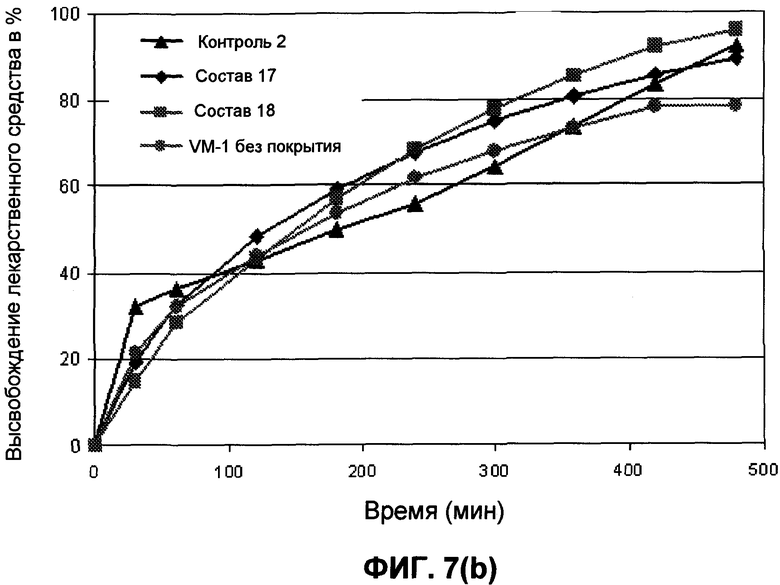
Настоящее изобретение относится к медицине и описывает фармацевтические композиции, имеющие центральный и нецентральный слой, для доставки лекарственных средств, в частности вызывающих зависимость лекарственных средств, имеющих вызывающее зависимость лекарственное средство, по существу заключенное в центральную часть, и не вызывающее зависимость лекарственное средство в нецентральной части. Данные композиции имеют сниженный потенциал к зависимости. В композиции предпочтительно вызывающее зависимость лекарственное средство представляет собой опиоид, а не вызывающее зависимость лекарственное средство представляет собой ацетаминофен или ибупрофен. Более предпочтительно, опиоид представляет собой гидрокодон, и не вызывающий зависимость аналгетик представляет собой ацетаминофен, в определенных предпочтительных вариантах осуществления дозированные формы характеризуются устойчивостью к экстракции растворителем, сдавлению, дроблению или растиранию. Определенные варианты осуществления изобретения обеспечивают первоначальную волну высвобождения лекарственного средства с последующим длительным периодом контролируемого высвобождения лекарственного средства. 4 н. и 23 з.п. ф-лы, 38 табл., 14 пр., 7 ил.
1. Фармацевтическая композиция, имеющая центральный и нецентральный слой, содержащая:
(a) гидрокодон, его фармацевтически приемлемую соль или гидрат, и
(b) ацетаминофен или ибупрофен,
где по меньшей мере 75% гидрокодона, его фармацевтически приемлемой соли или гидрата находится в центральной части,
где ацетаминофен или ибупрофен находится в нецентральном слое, и
где композиция адаптирована так, чтобы она была пригодна для перорального введения человеку 3, 2 или 1 раз в сутки, где центральный слой является монолитным центральным слоем, изготовленным путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, без промежуточной стадии дробления или измельчения,
причем монолитный центральный слой содержит по меньшей мере один изменяющий скорость (мет)акриловый полимер и по меньшей мере одно производное целлюлозы и активные ингредиенты в центральном слое представлены в виде твердого раствора образованного полимерами в центральном слое, где центральный слой представляет собой элемент единого твердого раствора и
где нецентральный слой представляет собой гладкий внешний нецентральный слой, содержащий по меньшей мере один неопиоидный анальгетик и привитой сополимер поливинилового спирта с полиэтиленгликолем, где нецентральный слой не содержит опиоидный анальгетик и где нецентральный слой наносится на центральный слой в виде водного раствора или суспензии с общей концентрацией твердых веществ по меньшей мере 20% по массе и содержание активного ингредиента составляет по меньшей мере 50%.
2. Композиция по п.1, где более чем 90% гидрокодона, его фармацевтически приемлемой соли или гидрата находится в центральной части.
3. Композиция по п.1, где, по существу, весь гидрокодон, его фармацевтически приемлемая соль или гидрат находится в центральной части.
4. Композиция по п.1, где, кроме того, центральная часть дополнительно содержит ацетаминофен.
5. Композиция по п.1, где, кроме того, центральная часть дополнительно содержит ацетаминофен или ибупрофен.
6. Композиция по любому из пп.1 или 5, где центральный слой содержит эксципиент, способный контролировать высвобождение лекарственного средства, и нецентральный слой содержит эксципиент, способный немедленно высвобождать лекарственное средство.
7. Композиция по любому из пп.1-5, где центральный слой изготовлен путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, и нецентральный слой нанесен распылением на центральный слой.
8. Композиция по любому из пп.1-5, где композиция содержит приблизительно 500 мг ацетаминофена и приблизительно 15 мг гидрокодона битартрата пентагемигидрата.
9. Фармацевтическая композиция, имеющая центральный и нецентральный слой, содержащая:
(а) вызывающее зависимость лекарственное средство, его фармацевтически приемлемую соль или гидрат и не вызывающее зависимость лекарственное средство или его фармацевтически приемлемую соль в центральном слое, и
(b) не вызывающее зависимость лекарственное средство, его фармацевтически приемлемую соль или гидрат в нецентральном слое,
где композиция адаптирована так, чтобы она была пригодна для перорального введения человеку 3, 2 или 1 раз в сутки; и
где композиция характеризуется по меньшей мере одним из следующих признаков:
i) количество вызывающего зависимость лекарственного средства, которое экстрагируется из композиции 40%-ным водным раствором этанола в течение одного часа при 37°C in vitro, является меньшим или равно количеству вызывающего зависимость лекарственного средства, которое экстрагируется 0,01 н. хлористоводородной кислотой in vitro в течение одного часа при 37°C, умноженному на 1,5,
ii) композиция не разрушается при воздействии силы 150 Н, предпочтительно 300 Н, более предпочтительно 450 Н, более предпочтительно 500 Н при измерении с помощью устройства для испытания на твердость "Pharma Test PTB 501",
iii) композиция высвобождает по меньшей мере 20% вызывающего зависимость лекарственного средства и не более чем 45% вызывающего зависимость лекарственного средства в течение первого часа тестирования растворения in vitro и предпочтительно также в течение первого часа тестирования in vivo,
iv) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства в течение от 1 до 2 ч после однократной дозы,
v) композиция высвобождает терапевтически эффективную дозу не вызывающего зависимость лекарственного средства и/или вызывающего зависимость лекарственного средства через 1 ч и через 12 ч после однократной дозы,
vi) в композиции высвобождение вызывающего зависимость лекарственного средства при измельчении повышается менее чем в 2-3 раза, по сравнению с целой таблеткой, когда композицию измельчают в течение 1 мин кофемолкой при 20000-50000 об/мин, в 40%-ном водном растворе этанола в течение 1 часа при 37°C,
vii) измельченная композиция содержит частицы размером от приблизительно 2 см до приблизительно 355 мкм для приблизительно 20% фракции, более чем приблизительно 63 мкм и менее чем приблизительно 355 мкм для приблизительно 66% фракции и менее чем приблизительно 63 мкм для приблизительно 14% фракции, при измерении с помощью теста просеиванием, или
viii) композиция является, по существу, однородной, где среднее значение центральной линии (CLA) составляет от приблизительно 0,1 до приблизительно 0,6, предпочтительно от приблизительно 0,1 до приблизительно 0,4, и наиболее предпочтительно от приблизительно 0,1 до приблизительно 0,2,
где центральный слой является монолитным центральным слоем, изготовленным путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву без промежуточной стадии дробления или измельчения,
причем монолитный центральный слой содержит по меньшей мере один изменяющий скорость (мет)акриловый полимер и по меньшей мере одно производное целлюлозы, и активные ингредиенты в центральном слое представлены в виде твердого раствора, образованного полимерами в центральном слое,
где центральный слой представляет собой элемент единого твердого раствора и где нецентральный слой представляет собой гладкий внешний нецентральный слой, содержащий по меньшей мере один неопиоидный анальгетик и привитой сополимер поливинилового спирта с полиэтиленгликолем, где нецентральный слой не содержит опиоидный анальгетик и где нецентральный слой наносится на центральный слой в виде водного раствора или суспензии с общей концентрацией твердых веществ по меньшей мере 20% по массе и содержание активного ингредиента составляет по меньшей мере 50%,
причем вызывающее зависимость лекарственное средство выбрано из группы, состоящей из следующих веществ: альфентанил, аллилпродин, альфапродин, анилеридин, бензилморфин, безитрамид, бупренорфин, буторфанол, клонитазен, кодеин, циклазоцин, дезоморфин, декстроморамид, дезоцин, диампромид, дигидрокодеин, дигидроморфин, дименоксадол, димефептанол, диметилтиамбутен, диоксафетила бутират, дипипанон, эптазоцин, этогептазин, этилметилтиамбутен, этилморфин, этонитазен, фентанил, героин, гидрокодон, гидроморфон, гидроксипетидин, изометадон, кетобемидон, леваллорфан, левофенацилморфан, леворфанол, лофентанил, меперидин, мептазинол, метазоцин, метадон, метопон, морфин, мирофин, налбуфин, нарцеин, никоморфин, норпипанон, опиум, оксикодон, оксиморфон, папвретум, пентазоцин, фенадоксон, феназоцин, феноморфан, феноперидин, пиминодин, пропирам, пропоксифен, суфентанил, тилидин и трамадол, их соли, гидраты и их смеси, а
не вызывающее зависимость лекарственное средство выбрано из группы, состоящей из ацетаминофена, аспирина, фентанила, ибупрофена, индометацина, каторолака, напроксена, фенацетина, пироксикама, суфентанила, сулиндака, интерферона альфа, и их солей, гидратов и их смесей.
10. Фармацевтическая композиция, имеющая центральный слой и нецентральный слой,
(А) где центральный слой содержит смесь:
(a) по меньшей мере одного опиоида;
(b) по меньшей мере одного изменяющего скорость фармацевтически приемлемого полимера, сополимера или их сочетаний;
(B) где нецентральный слой содержит по меньшей мере один неопиоидный апалгетик; и
(C) где композиция адаптирована так, чтобы она была пригодна для пероралыюго введения человеку 3, 2 или 1 раз в сутки,
где центральный слой является монолитным центральным слоем, изготовленным путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву без промежуточной стадии дробления или измельчения, причем монолитный центральный слой содержит по меньшей мере один изменяющий скорость (мет)акриловый полимер и по меньшей мере одно производное целлюлозы, и активные ингредиенты в центральном слое представлены в виде твердого раствора, образованного полимерами в центральном слое, где центральный слой представляет собой элемент единого твердого раствора, и
где нецентральный слой представляет собой гладкий внешний нецентральный слой, содержащий по меньшей мере один неопиоидный анальгетик и привитой сополимер поливинилового спирта с полиэтиленгликолем, где нецентральный слой не содержит опиоидный анальгетик и где нецентральный слой наносится на центральный слой в виде водного раствора или суспензии с общей концентрацией твердых веществ по меньшей мере 20% по массе и содержание активного ингредиента составляет по меньшей мере 50%.
11. Композиция по п.10, где центральный слой дополнительно содержит по меньшей мере один неопиоидный аналгетик.
12. Композиция по п.10, где композиция характеризуется по меньшей мере одним из следующих признаков:
i) количество опиоида, которое экстрагируется из композиции 40%-ным водным раствором этанола в течение одного часа при 37°C in vitro, является меньшим или равно количеству опиоида, которое экстрагируется 0,01 н. хлористоводородной кислотой in vitro в течение одного часа при 37°C, умноженному на 1,5,
ii) композиция не разрушается при воздействии силы 150 Н, предпочтительно 300 Н, более предпочтительно 450 Н, более предпочтительно 500 Н при измерении с помощью устройства для испытания на твердость "Pharma Test PTB 501",
iii) композиция высвобождает по меньшей мере 20% опиоида и не более чем 45% опиоида в течение первого часа тестирования растворения in vitro и предпочтительно также в течение первого часа тестирования in vivo,
iv) композиция высвобождает терапевтически эффективную дозу неопиоидного аналгетика в течение от 1 до 2 ч после однократной дозы,
v) композиция высвобождает терапевтически эффективную дозу неопиоидного аналгетика и/или опиоида через 1 ч и через 12 ч после однократной дозы,
vi) в композиции высвобождение опиоида при измельчении повышается менее чем в 2-3 раза, по сравнению с целой таблеткой, когда композицию измельчают в течение 1 мин кофемолкой при 20000-50000 об/мин, в 40%-ном водном растворе этанола в течение 1 ч при 37°C,
vii) измельченная композиция содержит частицы размером от приблизительно 2 см до приблизительно 355 мкм для приблизительно 20% фракции, более чем приблизительно 63 мкм и менее чем приблизительно 355 мкм для приблизительно 66% фракции и менее чем приблизительно 63 мкм для приблизительно 14% фракции, при измерении с помощью теста просеиванием, или
viii) композиция является, по существу, однородной, где среднее значение центральной линии (CLA) составляет от приблизительно 0,1 до приблизительно 0,6, предпочтительно от приблизительно 0,1 до приблизительно 0,4, и наиболее предпочтительно от приблизительно 0,1 до приблизительно 0,2.
13. Композиция по п.10, где опиоид выбран из группы, состоящей из альфентанила, аллилпродина, альфапродина, анилеридина, бензилморфина, безитрамида, бупренорфина, буторфанола, клонитазена, кодеина, циклазоцина, дезоморфина, декстроморамида, дезоцина, диампромида, дигидрокодеина, дигидроморфина, дименоксадола, димефептанола, диметилтиамбутена, диоксафетила бутирата, дипипапона, эптазоцина, этогептазина, этилметилтиамбутена, этилморфина, этонитазена, фентанила, героина, гидрокодона, гидроморфона, гидроксипетидина, изометадона, кетобемидона, леваллорфана, левофенацилморфана, леворфанола, лофентанила, меперидина, мептазинола, метазоцина, метадона, метопона, морфина, мирофина, налбуфина, нарцеина, никоморфина, норпипанона, опиума, оксикодона, оксиморфона, папвретума, пентазоцина, фенадоксона, феназоцина, феноморфана, феноперидина, риминодина, пропирама, пропоксифена, суфентанил, тилидин, и трамадол, и их солей, гидратов и смесей, и неопиоидный аналгетик выбран из группы, состоящей из ацетаминофена, аспирина, фентанила, ибупрофена, индометацина, каторолака, напроксена, фенацетина, пироксикама, суфентанила, сулиндака, интерферона альфа, и их солей, гидратов и смесей.
14. Композиция по п.10, где опиоид представляет собой гидрокодон, и неопиоидный аналгетик представляет собой ацетаминофен или ибупрофен.
15. Композиция по п.10, где опиоид представляет собой гидрокодон, и неопиоидный аналгетик представляет собой ацетаминофен.
16. Фармацевтическая композиция, имеющая центральный слой и нецентральный слой, (А) где центральный слой содержит смесь
(a) по меньшей мере одного опиоида и по меньшей мере одного первого неопиоидного аналгетика;
(b) по меньшей мере одного изменяющего скорость фармацевтически приемлемого полимера, сополимера или их сочетаний;
(B) где нецентральный слой содержит по меньшей мере один второй неопиоидный аналгетик; и
(C) где композиция адаптирована так, чтобы она была пригодной для перорального введения человеку 3, 2 или 1 раз в сутки, где центральный слой является монолитным центральным слоем, изготовленным путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву без промежуточной стадии дробления или измельчения, причем монолитный центральный слой содержит по меньшей мере один изменяющий скорость (мет)акриловый полимер и по меньшей мере одно производное целлюлозы, и активные ингредиенты в центральном слое представлены в виде твердого раствора, образованного полимерами в центральном слое, где центральный слой представляет собой элемент единого твердого раствора и
где нецентральный слой представляет собой гладкий внешний нецентральный слой, содержащий по меньшей мере один неопиоидный анальгетик и привитой сополимер поливинилового спирта с полиэтиленгликолем, где нецентральный слой не содержит опиоидный анальгетик и где нецентральный слой наносится на центральный слой в виде водного раствора или суспензии с общей концентрацией твердых веществ по меньшей мере 20% по массе и содержание активного ингредиента составляет по меньшей мере 50%.
17. Композиция по п.16, где опиоид включает гидрокодон и первый и второй неопиоидный аналгетик включает ацетаминофен или ибупрофен.
18. Композиция по п.16, где опиоид включает гидрокодон и первый и второй неопиоидный аналгетик включает ацетаминофен.
19. Композиция по п.16, где нецентральный слой содержит:
ацетаминофен; и
по меньшей мере один изменяющий скорость фармацевтически приемлемый полимер, сополимер или их сочетания.
20. Композиция по п.19, где полимер или сополимер выбран из группы, состоящей из: гидроксипропилцеллюлозы, гидроксипропилметилцеллюлозы, гидроксиэтилцеллюлозы; полиметакрилата, поливинилового спирта, окиси полиэтилена и их сочетаний.
21. Композиция по п.19, где полимер или сополимер выбран из группы, состоящей из: гидроксипропилметилцеллюлозы и поливинилового спирта, или их сочетаний.
22. Композиция по п.19, где полимер или сополимер выбран из группы, состоящей из: привитых сополимеров поливинилового спирта и окиси полиэтилена.
23. Композиция по п.19, где соотношение ацетаминофена и контролирующего скорость полимера или сополимера или их сочетания составляет от приблизительно 1:1 до приблизительно 10:1.
24. Композиция по п.19, где соотношение ацетаминофена и контролирующего скорость полимера или сополимера или их сочетания составляет от приблизительно 3:1 до приблизительно 5:1.
25. Композиция по любому из пп.16-24, где нецентральный слой имеет по меньшей мере один из следующих признаков:
(a) по существу, не растрескивается после 3 месяцев при 40°C, 75% относительной влажности в индукционно закрытых бутылках HDPE;
(b) по существу, сухой (не клейкий);
обеспечивает быстрое растворение в 0,01 н. HCl при 37°C, открывая центральный слой,
высвобождает по меньшей мере 80% ацетаминофена в нецентральном слое в пределах 20 мин после введения пациенту-человеку; или
(c) обеспечивает белую пигментацию состава без дополнительных пигментов.
26. Композиция по любому из пп.16-24, где композиция характеризуется по меньшей мере одним из следующих признаков:
i) количество гидрокодона, которое экстрагируется из композиции 40%-ным водным раствором этанола в течение одного часа при 37°C in vitro, является меньшим или равно количеству гидрокодона, которое экстрагируется 0,01н. хлористоводородной кислотой in vitro в течение одного часа при 37°С, умноженному на 1,5,
ii) композиция не разрушается при воздействии силы 150 Н, предпочтительно 300 Н, более предпочтительно 450 Н, более предпочтительно 500 Н при измерении с помощью устройства для испытания на твердость "Pharma Test PTB 501",
iii) композиция высвобождает по меньшей мере 20% гидрокодона и не более чем 45% гидрокодона в течение первого часа тестирования растворения in vitro и предпочтительно также в течение первого часа тестирования in vivo,
iv) композиция высвобождает терапевтически эффективную дозу ацетаминофена в течение от 1 до 2 ч после однократной дозы,
v) композиция высвобождает терапевтически эффективную дозу ацетаминофена и/или опиоида через 1 ч и через 12 ч после однократной дозы,
vi) в композиции высвобождение гидрокодона при измельчении возрастает более чем в 2-3 раза по сравнению с целой таблеткой, когда композицию измельчают в течение 1 мин кофемолкой при 20000-50000 об/мин, в 40%-ном водном растворе этанола в течение 1 ч при 37°C,
vii) измельченная композиция содержит частицы размером от приблизительно 2 см до приблизительно 355 мкм для приблизительно 20% фракции, более чем приблизительно 63 мкм и менее чем приблизительно 355 мкм для приблизительно 66% фракции и менее чем приблизительно 63 мкм для приблизительно 14% фракции, при измерении с помощью теста просеиванием, или
viii) композиция является, по существу, однородной, где среднее значение центральной линии (CLA) составляет от приблизительно 0,1 до приблизительно 0,6, предпочтительно от приблизительно 0,1 до приблизительно 0,4, и наиболее предпочтительно от приблизительно 0,1 до приблизительно 0,2.
27. Композиция по любому из пп.16-24, где центральный слой изготовлен путем экструдирования из расплава с последующим прямым приданием формы содержащему лекарственное средство расплаву, и нецентральный слой нанесен распылением на центральный слой.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ПРЕПАРАТИВНЫЕ ФОРМЫ ГИДРОКОДОНА С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ | 2000 |
|
RU2230556C2 |
| МАШКОВСКИЙ М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 2002, ч.1, с.7-11 | |||
| Planas E | |||
| et al | |||
| Non-steroidal anti-inflammatory drugs antagonize the constipating effects of tramadol | |||
| Eur J Pharmacol | |||
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| Abstract | |||
| Найдено из PubMed. | |||

