РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка испрашивает приоритет предварительной заявки на патент США USSN 61/776440, зарегистрированной 11 марта 2013, содержание которой включается в контекст в качестве ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение относится к ряду новых гетероциклических аминов, которые являются полезными при лечении гиперпролиферативных заболеваний, таких как рак, у млекопитающих. Настоящее изобретение включает в себя также применение таких соединений при лечении гиперпролиферативных заболеваний у млекопитающих, особенно у людей, и фармацевтические композиции, содержащие такие соединения.
УРОВЕНЬ ТЕХНИКИ
Протеинкиназы образовывают большое семейство структурно родственных ферментов, которые являются ответственными за регуляцию большого числа процессов трансдукции сигналов в клетке (Hardie, G. and Hanks, S. (1995) The Protein Kinase Facts Book. I and II, Academic Press, San Diego, CA). Киназы можно классифицировать на семейства посредством субстратов, которые они фосфорилируют (например, протеин-тирозин, протеин-серин/треонин, липиды и т.д.). Были идентифицированы фрагменты последовательностей, которые обычно соответствуют каждому из этих семейств киназ (см., например, Hanks, S.K., Hunter, T., FASEB J., 9:576-596 (1995); Knighton, et al., Science, 253:407-414 (1991); Hiles, et al., Cell, 70:419-429 (1992); Kunz, et al., Cell, 73:585-596 (1993); Garcia-Bustos, et al., EMBO J., 13:2352-2361 (1994)). Протеинкиназы можно характеризовать посредством их механизмов регуляции. Эти механизмы включают в себя, например, аутофосфорилирование, трансфосфорилирование другими киназами, взаимодействия белка с белком, взаимодействия белка с липидом и взаимодействия белка с полинуклеотидом. Индивидуальная протеинкиназа может регулироваться более чем одним механизмом.
Киназы регулируют многие различные процессы клеток, включающие в себя, но не ограничивающиеся указанными, пролиферацию, дифференциацию, апоптоз, подвижность, транскрипцию, трансляцию и другие процессы передачи сигналов, добавлением фосфатных групп к целевым белкам. Эти события фосфорилирования действуют как молекулярные включения и/или выключения, которые могут модулировать или регулировать биологическую функцию целевого белка. Фосфорилирование целевых белков имеет место в ответ на различные внеклеточные сигналы (гормоны, нейротрансмиттеры, факторы роста и дифференциации и т.д.), события клеточного цикла, внешние стрессовые воздействия или стрессы, вызванные нарушением питания, и т.д. Подходящая протеинкиназа функционирует в путях передачи сигналов для активации или инактивации (либо непосредственно, либо опосредованно), например, метаболического фермента, регуляторного белка, рецептора, белка цитоскелета, ионного канала или насоса или фактора транскрипции. Нерегулируемая передача сигнала вследствие нарушенного регулирования фосфорилирования белка имела место в ряде заболеваний, включающих в себя, например, воспаление, рак, аллергию/астму, заболевания и состояния иммунной системы, заболевания и состояния центральной нервной системы и развитие кровеносных сосудов.
Протеинкиназа 70S6K, рибосомальная протеинкиназа p70S6K 70 кДа (известная также как S6K, киназа p70/p85 S6, рибосомальная киназа S6 р70/р85 и p70S6K) является членом подсемейства AGC протеинкиназ. p70S6K является серин-треонинкиназой, которая является компонентом пути фосфатидилинозитол-3-киназы (Pl3K)AKT. p70S6K находится вниз по ходу трансляции Pl3K, и активация происходит посредством фосфорилирования у ряда сайтов в ответ на многочисленные митогены, гормоны и факторы роста. Активность p70S6K находится также под контролем содержащего mTOR комплекса (TORC1), поскольку рапамицин действует, ингибируя активность p70S6K. p70S6K регулируется Pl3K, расположенным вверх по ходу трансляции мишеней AKT и PKCζ. Akt непосредственно фосфорилирует и инактивирует TSC2, тем самым активируя mTOR. Кроме того, исследования с мутантными аллелями p70S6K, которые ингибируются вортманнином, но не рапамицином, предполагают, что путь Pl3K может оказывать действия на p70S6K, независимо от регуляции активности mTOR.
Фермент p70S6K модулирует синтез белков фосфорилированием рибосомального белка S6. Фосфорилирование S6 коррелирует с повышенной трансляцией мРНК, кодирующих компоненты устройства трансляции, включающие в себя рибосомальные белки и факторы трансляционной элонгации, повышенная экспрессия которых является важной для роста и пролиферации клеток. Эти мРНК содержат олигопиримидиновый путь у их 5'-транскрипционного начала (названного 5'TOP), который, как было показано, является важным для их регуляции при трансляционном уровне.
Помимо его участия в трансляции, активация p70S6K участвовала также в регуляции клеточного цикла, дифференциации нейрональных клеток, регуляции подвижности клеток и клеточной реакции, которая является важной в опухолевых метастазах, иммунной реакции и заживлении ткани. Антитела к p70S6K устраняют митогенную реакцию, запускаемую вхождением фибробластов крысы в S-фазу, это является показанием того, что функция p70S6K является важной для развития от фазы G1 до фазы S в клеточном цикле. Кроме того, ингибирование пролиферации клеточного цикла от фазы G1 до фазы S клеточного цикла рапамицином было идентифицировано как последствие ингибирования продуцирования гиперфосфорилированной, активированной формы p70S6K.
Роль p70S6K в пролиферации опухолевых клеток и защите клеток от апоптоза подтверждается на основе его участия в трансдукции, сверхэкспрессии и активации сигнала рецептора фактора роста в опухолевых тканях. Например, анализы Нортерна и Вестерна показали, что амплификация гена pS6K сопровождалась соответствующими увеличениями экспрессии мРНК и белка, соответственно (Cancer Res. (1999) 59: 1408-11-Localization of pS6K to Chromosomal Region 17q23 and Determination of Its Amplification in Breast Cancer).
Хромосома 17q23 амплифицируется до количества в 20% в первичных опухолях молочной железы, в количестве 87% в опухолях молочной железы, содержащих мутации BRCA2, и в количестве до 50% в опухолях, содержащих мутации BRCA1, а также других типах рака, таких как опухоли поджелудочной железы, мочевого пузыря и нейробластома (см. M. Barlund, O. Monni, J. Kononen, R. Cornelison, J. Torhorst, G. Sauter, O.-P. Kallioniemi and Kallioniemi A., Cancer Res., 2000, 60:5340-5346). Показано, что амплификации 17q23 в раковой опухоли молочной железы включает в себя участие генов PAT1, RAD51C, PS6K и SIGMA1B (Cancer Res. (2000): 60, pp. 5371-5375).
Ген p70S6K был идентифицирован как мишень амплификации и сверхэкспрессии в этом участке, и наблюдали статистически значимую связь между амплификацией и плохим прогнозом. Клиническое ингибирование активации p70S6K наблюдали у пациентов с почечной карциномой, подвергнутых лечению CCl-779 (сложным эфиром рапамицина), ингибитором вверх по ходу трансляции mTOR киназы. Описывается существенная линейная связь между развитием заболевания и ингибированием активности p70S6K. В ответ на воздействие энергии супрессор опухоли LKB1 активирует AMPK, который фосфорилирует комплекс TSC1/2, и позволяет ему инактивировать путь mTOR/p70S6K. Мутации в LKB1 обуславливают синдром Peutz-Jeghers (PJS), у пациентов с PJS развитие рака является в 15 раз более вероятным, чем у общей популяции. Кроме того, 1/3 аденокарцином легких являются местом мутаций инактивирующих LKB1. P70S6K принимал участие в метаболических заболеваниях и нарушениях. Было описано, что отсутствие p70S6K защищает от вызванного старостью и питанием ожирения при повышении чувствительности к инсулину. Роль p70S6K при метаболических заболеваниях и нарушениях, таких как ожирение, диабет, метаболический синдром, инсуинорезистентность, гипергликемия, гипераминоацидемия и гиперлипидемия, подтверждается на основе полученных данных.
Соединения, описанные как подходящие для ингибирования p70S6K, указываются в WO 03/064397, WO 04/092154, WO 05/054237, WO 05/056014, WO 05/033086, WO 05/117909, WO05/039506, WO 06/120573, WO 06/136821, WO 06/071819, WO 06/131835, WO 08/140947, WO 10/056563, WO 10/093419, WO 12/013282, WO 12/016001 и WO 12/069146.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В одном аспекте изобретение относится к соединению формулы (I)
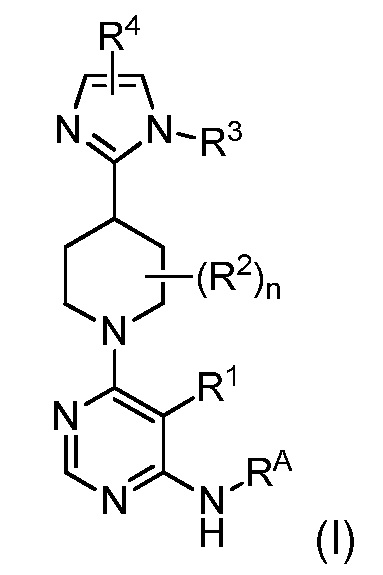
и его фармацевтически приемлемым солям, сольватам, сольватам солей или пролекарствам, у которых каждый из R1, R2, R3, R4, RA и n имеет значения, указанные в контексте.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Задачей настоящего изобретения является предоставление новых соединений, которые модулируют активность киназ. Эта модуляция протеинкиназ включает в себя, но не ограничивается указанным, ингибирование p70S6K и ингибирование Akt соединениями, которые применимы при лечении гиперпролиферативных заболеваний, особенно заболеваний, связанных с гиперактивностью указанных выше протеинкиназ, таких как рак у млекопитающих, и обладают лучшими фармакологическими свойствами, как в отношении их активностей, так и их характеристик растворимости, метаболического клиренса и биологической доступности.
В результате этого, данное изобретение предоставляет новые гетероциклические аминосоединения и их фармацевтически приемлемые соли, сольваты или пролекарства, которые являются ингибиторами киназ и применимы при лечении указанных выше заболеваний.
В одном аспекте изобретение относится к соединению формулы (I)

и его фармацевтически приемлемым солям, сольватам, сольватам солей или пролекарствам, у которых
R1 представляет собой Hal, LA, OH, O(LA), NH2 и/или NH(LA), N(LA)2, NO2, CN, OCN, COOH, COO(LA), CONH2, CONH(LA), CON(LA)2, NHCO(LA), NHCONH(LA), NHCONH2, NHSO2(LA), CHO, CO(LA) или моно- или бициклический, алифатический или ароматический гомо- или гетероцикл, имеющий 0, 1, 2, 3 или 4 атома N и/или O и 4, 5 или 6, 7, 8, 9 или 10 атомов скелета, которые могут быть незамещенными или независимо друг от друга моно-, ди- или тризамещенными Hal, LA, OH, O(LA), NH2 и/или NH(LA), N(LA)2, NO2, CN, OCN, COOH, COO(LA), CONH2, CONH(LA), CON(LA)2, NHCO(LA), NHCONH(LA), NHCONH2, CHO и/или CO(LA), или неразветвленный или разветвленный линейный или циклический алкил, имеющий 1, 2, 3, 4, 5, 6, 7 или 8 C-атомов, у которого одна или две CH2-группы могут быть заменены атомом O и/или группой -NH-, NH(LA), -CO-, -NHCO- или -CH=CH-, и/или у которого CH-группа может быть заменена на -N-;
RA представляет собой H; или RA и R1 вместе с атомами, к которым они присоединены, образуют 3-7-членное гетероциклическое кольцо, имеющее 1-4 гетероатома, независимо выбранные из азота, кислорода или серы, или 5-6-членное моноциклическое гетероарильное кольцо, имеющее 1-4 гетероатома, независимо выбранные из азота, кислорода или серы; которое является необязательно замещенным;
каждый R2 представляет собой независимо Hal, OH или A;
A представляет собой неразветвленную или разветвленную алкильную группу, имеющую 1, 2, 3, 4, 5 или 6 C-атомов, в которой 1-4 атома H могут быть заменены независимо друг от друга на Hal;
R3 представляет собой H или неразветвленную или разветвленную линейную или моно- или бициклическую алкильную группу, имеющую 1, 2, 3 ,4, 5, 6, 7, 8 или 9 C-атомов, у которой одна или две CH2-группы могут быть заменены атомом -O- или группой -NH-, и/или у которой одна или две CH-группы могут быть заменены атомом -N-, и/или у которой 1, 2 или 3 атома H могут быть заменены на Hal или OH; или R3 представляет собой неразветвленную или разветвленную алкильную группу, имеющую 1, 2, 3, 4, 5 или 6 атомов C, которая замещена 3-6 членным гетероциклическим кольцом, которое может быть дополнительно необязательно замещенным;
R4 представляет собой C5-10арильное, 3-8-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, 3-7-членное гетероциклическое кольцо, имеющее 1-4 гетероатома, независимо выбранные из атома азота, кислорода или серы, или 5-6-членное моноциклическое гетероарильное кольцо, имеющее 1-4 гетероатома, независимо выбранные из атомов азота, кислорода или серы; каждый из которых является моно-, ди- или тризамещенным Hal, LA, OH, O(LA), NH2 и/или NH(LA), N(LA)2, NO2, CN, OCN, COOH, COO(LA), CONH2, CONH(LA), CON(LA)2, NHCO(LA), NHCONH(LA), NHCONH2, NHSO2(LA), CHO или CO(LA);
LA представляет собой неразветвленную или разветвленную, насыщенную или частично ненасыщенную линейную углеводородную цепь, имеющую 1, 2, 3 или 4 атома C, где 1, 2 или 3 атома H могут быть заменены на Hal;
Hal представляет собой F, Cl, Br или I и
n равно 1 или 2.
В некоторых вариантах осуществления R1 представляет собой Hal, LA, OH, O(LA), NH2 и/или NH(LA), N(LA)2, NO2, CN, OCN, COOH, COO(LA), CONH2, CONH(LA), CON(LA)2, NHCO(LA), NHCONH(LA), NHCONH2, NHSO2(LA), CHO или CO(LA). В некоторых вариантах осуществления R1 представляет собой моно- или бициклический, алифатический или ароматический гомо- или гетероцикл, имеющий 0, 1, 2, 3 или 4 атома N и/или O и 4, 5 или 6, 7, 8, 9 или 10 атомов скелета, которые могут быть незамещенными или независимо друг от друга моно-, ди- или тризамещенными Hal, LA, OH, O(LA), NH2 и/или NH(LA), N(LA)2, NO2, CN, OCN, COOH, COO(LA), CONH2, CONH(LA), CON(LA)2, NHCO(LA), NHCONH(LA), NHCONH2, CHO и/или CO(LA). В некоторых вариантах осуществления R1 представляет собой неразветвленный или разветвленный линейный или циклический алкил, имеющий 1, 2, 3, 4, 5, 6, 7 или 8 атомов C, в котором одна или две CH2-группы могут быть заменены атомом O и/или группой -NH-, NH(LA), -CO-, -NHCO- или -CH=CH-, и/или в котором CH-группа может быть заменена на -N-;
В некоторых вариантах осуществления R1 представляет собой CN, CONH2, Hal, O(LA) или неразветвленный или разветвленный линейный или циклический алкил, имеющий 1, 2, 3, 4, 5, 6, 7 или 8 атомов C, у которого одна или две CH2-группы могут быть заменены атомом O и/или группой -NH-, NH(LA), -CO-, -NHCO- или -CH=CH-, и/или у которого CH-группа может быть заменена на -N-.
В некоторых вариантах осуществления R1 представляет собой метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил с неразветвленной или разветвленной цепью или гексил с неразветвленной или разветвленной цепью.
В некоторых вариантах осуществления R1 выбирают из таблицы 1.
Таблица 1: заместители для R1 в формуле (I)
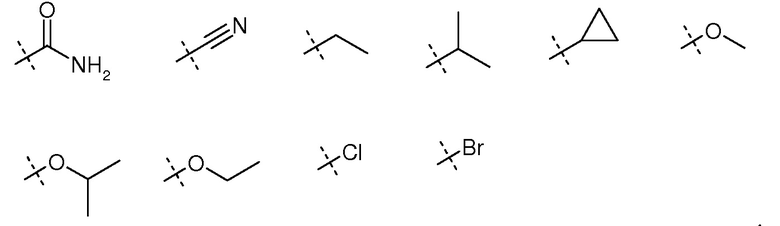
В некоторых вариантах осуществления каждый R2 независимо представляет собой Hal. В некоторых вариантах осуществления каждый R2 независимо представляет собой OH. В некоторых вариантах осуществления каждый R2 независимо представляет собой неразветвленную или разветвленную алкильную группу, имеющую 1, 2, 3, 4, 5 или 6 атомов C, у которой 1-4 атома Н могут быть заменены независимо друг от друга на Hal.
В некоторых вариантах осуществления каждый R2 независимо представляет собой Hal, OH, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил с неразветвленной или разветвленной цепью или гексил с неразветвленной или разветвленной цепью.
В некоторых вариантах осуществления каждый R2 независимо представляет собой F, OH или метил.
В некоторых вариантах осуществления R3 представляет собой Н.
В некоторых вариантах осуществления R3 представляет собой неразветвленную или разветвленную линейную или моно- или бициклическую алкильную группу, имеющую 1, 2, 3,4, 5, 6, 7, 8 или 9 атомов C, в которой одна или две CH2-группы могут быть заменены группой -O- или -NH-, и/или в которой одна или две CH-группы могут быть заменены на -N-, и/или в которой 1, 2 или 3 атома H могут быть заменены на Hal или OH.
В некоторых вариантах осуществления R3 представляет собой неразветвленную или разветвленную алкильную группу, имеющую 1, 2, 3, 4, 5 или 6 атомов C, которая замещена 3-6 членным гетероциклическим кольцом, которое может быть необязательно дополнительно замещено. В некоторых вариантах осуществления замещенное гетероциклическое кольцо является азиридином, азетидином, пирролидином, имидазолидином, пиразолидином, оксазолидином, изоксазолидином, тиазолидином, изотиазолидином, пиперидином, морфолином, пиперазином, гексагидропиримидином или гексагидропиридазином.
в некоторых вариантах осуществления R3 выбирают из таблицы 2.
Таблица 2: заместители для R3 в формуле (I)
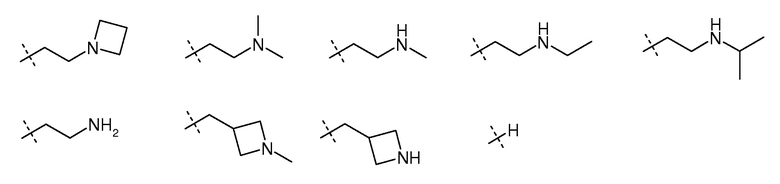
В некоторых вариантах осуществления R4 представляет собой C5-10арильное, 3-8-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, 3-7-членное гетероциклическое кольцо, имеющее 1-4 гетероатома, независимо выбранных из атомов азота, кислорода или серы, или 5-6-членное моноциклическое гетероарильное кольцо, имеющее 1-4 гетероатома, независимо выбранные из атомов азота, кислорода или серы, каждое из которых является моно-, ди- или тризамещенным Hal, LA, OH, O(LA), NH2 и/или NH(LA), N(LA)2, NO2, CN, OCN, COOH, COO(LA), CONH2, CONH(LA), CON(LA)2, NHCO(LA), NHCONH(LA), NHCONH2, NHSO2(LA), CHO или CO(LA).
В некоторых вариантах осуществления R4 представляет собой C5-10арильное или 5-6-членное моноциклическое гетероарильное кольцо, имеющее 1-4 гетероатома, независимо выбранные из атома азота, кислорода или серы, каждое из которых является моно-, ди- или тризамещенным Hal, LA, OH, O(LA), NH2 и/или NH(LA), N(LA)2, NO2, CN, OCN, COOH, COO(LA), CONH2, CONH(LA), CON(LA)2, NHCO(LA), NHCONH(LA), NHCONH2, NHSO2(LA), CHO или CO(LA).
В некоторых вариантах осуществления R4 представляет собой фенил или пиридил, каждый из которых является моно-, ди- или тризамещенным Hal, LA, OH, O(LA), NH2 и/или NH(LA), N(LA)2, NO2, CN, OCN, COOH, COO(LA), CONH2, CONH(LA), CON(LA)2, NHCO(LA), NHCONH(LA), NHCONH2, NHSO2(LA), CHO или CO(LA).
В некоторых вариантах осуществления R4 выбирают из таблицы 3.
Таблица 3: заместители для R4 в формуле (I)
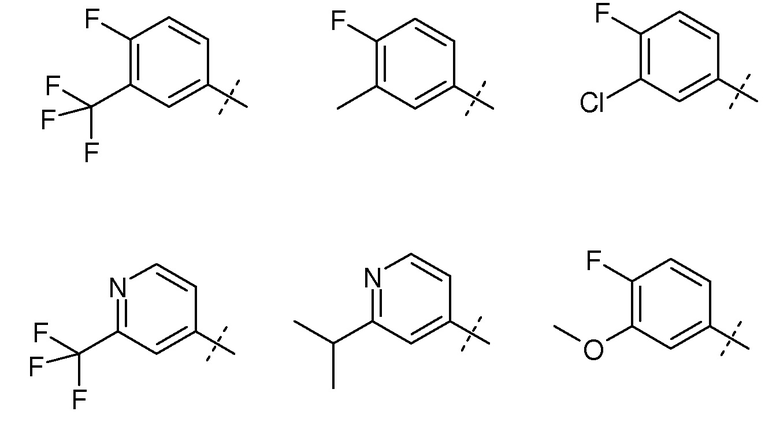
В некоторых вариантах осуществления RA представляет собой H.
В некоторых вариантах осуществления RA и R1 вместе с атомами, к которым они присоединены, образуют 3-7-членное гетероциклическое кольцо, имеющее 1-4 гетероатома, независимо выбранные из атомов азота, кислорода или серы. В некоторых вариант осуществления RA и R1 вместе с атомами, к которым они присоединены, образуют 5-6-членное моноциклическое гетероарильное кольцо, имеющее 1-4 гетероатома, независимо выбранные из атомов азота, кислорода или серы.
В некоторых вариантах осуществления RA и R1 вместе с атомами, к которым они присоединены, образуют 5-членное гетероарильное кольцо, которое является необязательно замещенным.
В некоторых вариантах осуществления RA и R1 вместе образуют 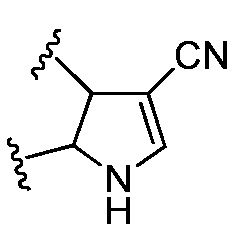 .
.
Обычно все заместители, которые присутствуют в количестве более одного, могут быть одинаковыми или разными, т.е. являются независимыми друг от друга.
В некоторых вариантах осуществления изобретение относится к соединению формулы (II)
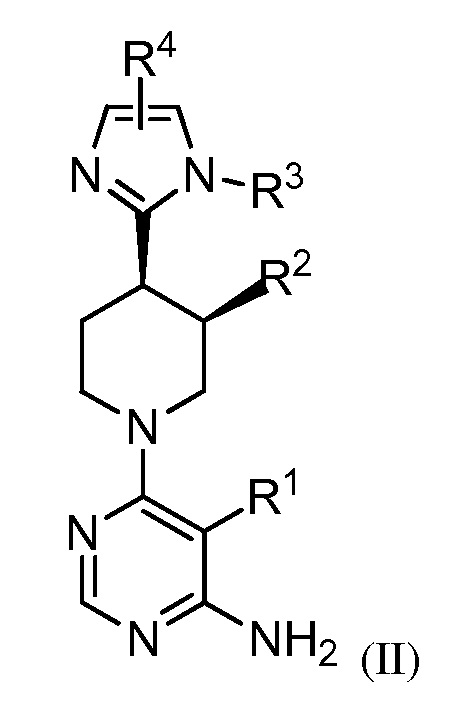
или его фармацевтически приемлемой соли, где каждый из R1, R2, R3 и R4 имеет значения, указанные выше, и описан в указанных выше вариантах осуществления, классах или подклассах и контексте, по отдельности или в комбинации.
В некоторых вариантах осуществления соединение формулы (II) является рацемическим. В некоторых вариантах осуществления соединение формулы (II) является хиральным. В некоторых вариантах осуществления R2 представляет собой F. В некоторых вариантах осуществления R2 представляет собой метил.
В некоторых вариантах осуществления изобретение относится к соединению формулы (III)
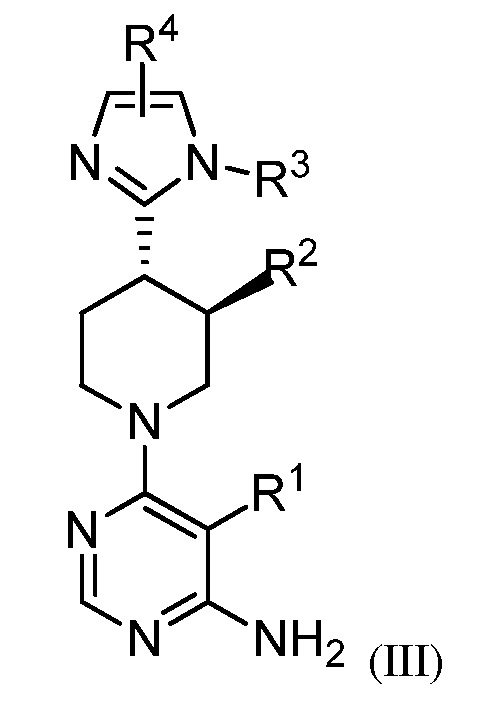
или его фармацевтически приемлемой соли, где каждый из R1, R2, R3 и R4 имеет значения, указанные выше, и описан в указанных выше вариантах осуществления, классах или подклассах и контексте, по отдельности или в комбинации.
В некоторых вариантах осуществления соединение формулы (III) является рацемическим. В некоторых вариантах осуществления соединение формулы (III) является хиральным. В некоторых вариантах осуществления R2 представляет собой F. В некоторых вариантах осуществления R2 представляет собой метил.
В некоторых вариантах осуществления изобретение относится к соединению формулы (IV)
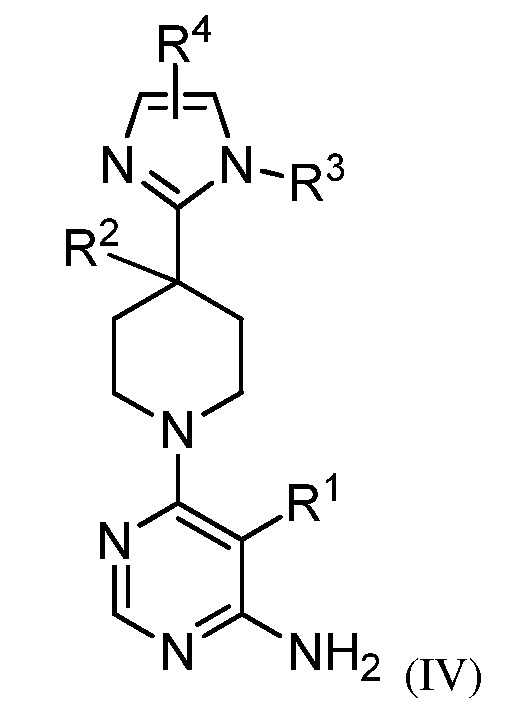
или его фармацевтически приемлемой соли, где каждый из R1, R2, R3 и R4 имеет значения указанные выше и описан в указанных выше вариантах осуществления, классах или подклассах и контексте, по отдельности или в комбинации.
В некоторых вариантах осуществления R2 представляет собой OH.
В некоторых вариант осуществления изобретение относится к соединению формулы (V)
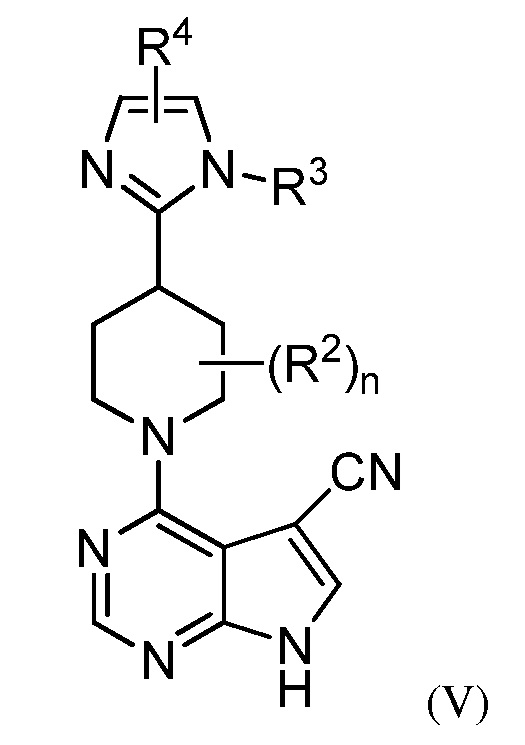
или его фармацевтически приемлемой соли, где каждый из R2, R3, R4 и n имеет значения указанные выше и описан в указанных выше вариантах осуществления, классах и подклассах и контексте, по отдельности или в комбинации.
В некоторых вариантах осуществления R2 представляет собой F.
В другом варианте осуществления изобретение относится к соединениям согласно формуле (I), которые выбирают из группы, состоящей из
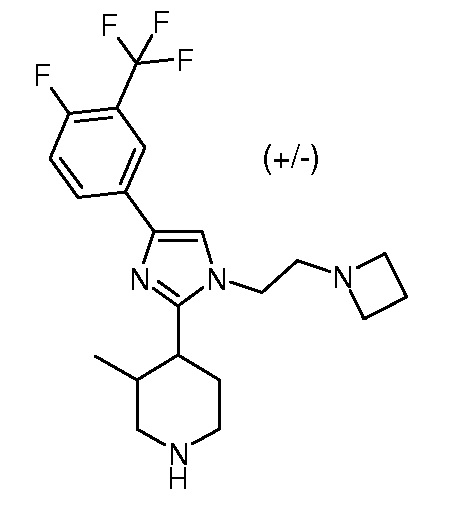
4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“1”);
4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоксамида (рацемического) (“2”);
4-амино-6-[(3R,4S)-4-[1-[2-(азетидин-1-ил)этил]-4-[4-фтор-3-(трифторметил)фенил]имидазол-2-ил]-3-фтор-1-пиперидил]пиримидин-5-карбоксамида (хирального) (“3”);
4-амино-6-[(3S,4R)-4-[1-[2-(азетидин-1-ил)этил]-4-[4-фтор-3-(трифторметил)фенил]имидазол-2-ил]-3-фтор-1-пиперидил]пиримидин-5-карбоксамида (хирального) (“4”);
4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“5”);
4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-ил}пиримидин-5-карбоксамида (рацемического) (“6”);
4-амино-6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“7”);
4-амино-6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоксамида (рацемического) (“8”);
4-амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-метилфенил)имидазол-2-ил]-3-фтор-1-пиперидил]}пиримидин-5-карбонитрила (рацемического) (“9”);
6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламина (рацемического) (“10”);
6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламина (рацемического) (“11”);
5-хлор-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина (рацемического) (“12”);
6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этилпиримидин-4-иламина (рацемического) (“13”);
4-амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“14”);
4-амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“15”);
6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламина (рацемического) (“16”);
6-{цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“17”);
4-амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“18”);
амида 4-амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (рацемического) (“19”);
амида 4-амино-6-{(3S,4R)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (хирального) (“20”);
амида 4-амино-6-{(3R,4S)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (хирального) (“21”);
6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламина (хирального) (“22”);
6-{(3S,4R)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламина (хирального) (“23”);
6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“24”);
5-хлор-6-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина (рацемического) (“25”);
6-{цис-3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“26”);
амида 4-амино-6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-пиримидин-5-карбоновой кислоты (хирального) (“27”);
амида 4-амино-6-{(3S,4R)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-пиримидин-5-карбоновой кислоты (хирального) (“28”);
5-хлор-6-{(3R,4S)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина (хирального) (“29”);
5-хлор-6-{(3S,4R)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина (хирального) (“30”);
6-{(3S,4R)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (хирального) (“31”);
6-{(3R,4S)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (хирального) (“32”);
6-{цис-4-[1-(2-аминоэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“33”);
6-{(3S,4R)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (хирального) (“34”);
6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (хирального) (“35”);
6-{(цис-4-[1-(2-аминоэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламина (рацемического) (“36”);
6-{цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-изопропиламиноэтил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“37”);
6-{цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-метиламиноэтил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“38”);
6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропоксипиримидин-4-иламина (рацемического) (“39”);
4-амино-6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (хирального) (“40”);
6-{цис-4-[1-азетидин-3-илметил-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“41”);
цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-7H-пирроло[2,3-d]пиримидин-5-карбонитрила (рацемического) (“42”);
цис-4-[1-(2-азетидин-1-илэтил)-4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-7H-пирроло[2,3-d]пиримидин-5-карбонитрила (рацемического) (“43”);
4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-гидроксипиперидин-1-ил}-пиримидин-5-карбонитрила (“44”);
1-(6-амино-5-изопропилпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-4-ола (“45”);
1-(6-амино-5-этилпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-4-ола (“46”);
1-(6-амино-5-хлорпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-4-ола (“47”);
4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}пиримидин-5-карбонитрила (“48”);
амида 4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}-пиримидин-5-карбоновой кислоты (“49”) и
6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (“50”);
и их фармацевтически приемлемых солей, сольватов или пролекарств.
Соединения настоящего изобретения могут быть в форме пролекарственного соединения. “Пролекарственное соединение” означает производное, которое превращается в биологически активное соединение согласно настоящему изобретению в физиологических условиях в живом организме, например, окислением, восстановлением, гидролизом или т.д., причем каждая из реакций проводится ферментативно или без участия ферментов. Примерами пролекарств являются соединения, у которых аминогруппа в соединении настоящего изобретения является ацилированной, алкилированной или фосфорилированный, например, является эйкозаноиламино-, аланиламино-, пивалоилоксиметиламиногруппой, или у которых гидроксильная группа является ацилированной, алкилированной, фосфорилированной или превращенной в борат, например, является ацетилокси-, пальмитоилокси-, пивалоилокси-, сукцинилокси-, фумарилокси-, аланилоксигруппой, или у которых карбоксильная группа является этерифицированной или амидированной, или у которых сульфгидрильная группа образует дисульфидный мостик с молекулой носителя, например пептида, который доставляет лекарственное средство селективно к мишени и/или к цитозолу клетки. Эти соединения можно получать из соединений настоящего изобретения согласно хорошо известным методам. Другими примерами пролекарств являются соединения, у которых карбоксилатная группа в соединении настоящего изобретения превращается, например, в группу алкилового, арилового, холинового эфира, амида, ацилоксиметилового эфира, линоленоилового эфира.
Метаболиты соединений настоящего изобретения также находятся в пределах объема настоящего изобретения.
Когда может иметь место таутомеризм, например, кето-енольный таутомеризм, соединений настоящего изобретения или их пролекарств, индивидуальные формы, например, кетоформа или енольная форма, заявляются по отдельности и вместе в виде смесей в любом отношении. То же самое применяют для стереоизомеров, например, энантиомеров, цис/транс-изомеров, конформеров и тому подобных изомеров. При желании, изомеры можно разделить способами, хорошо известными в данной области, например, жидкостной хроматографией. То же самое применяют для энантиомеров, например, с использованием хиральных неподвижных фаз. Кроме того, энантиомеры можно выделить превращением их в диастереомеры, например, сочетанием с энантиомерно чистым вспомогательным соединением, затем разделением образовавшихся диастереомеров и отщеплением вспомогательного остатка. Альтернативно, любой энантиомер соединения настоящего изобретения можно получить стереоселективным синтезом с применением оптически чистых исходных веществ.
Соединения настоящего изобретения могут быть в форме фармацевтически приемлемой соли или сольвата. Термин “фармацевтически приемлемые соли” относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включающих в себя неорганические основания или кислоты и органические основания или кислоты. В случаях, когда соединения настоящего изобретения содержат одну или несколько кислотных или основных групп, изобретение содержит также их соответствующие фармацевтически или токсикологически приемлемые соли, в частности их фармацевтически приемлемые соли. Таким образом, соединения настоящего изобретения, которые содержат кислотные группы, могут присутствовать в форме соли и их можно применять согласно изобретению, например, в виде солей щелочных металлов, солей щелочноземельных металлов или в виде аммониевых солей. Более определенные примеры таких солей включают в себя натриевые соли, калиевые соли, кальциевые соли, магниевые соли или соли с аммиаком или органическими аминами, такими как, например, этиламин, этаноламин, триэтаноламин, или аминокислотами. Соединения настоящего изобретения, которые содержат одну или несколько основных групп, т.е. группы, которые можно протонировать, могут присутствовать в форме соли и их можно применять согласно изобретению в форме их аддитивных солей с неорганическими или органическими кислотами. Примеры подходящих кислот включают в себя хлорид водорода, бромид водорода, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, нафталинсульфоновую кислоту, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, пивалиновую кислоту, диэтилуксусную кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты, известные специалисту в данной области. Если соединения настоящего изобретения одновременно содержат кислотные и основные группы в молекуле, изобретение включает в себя также помимо указанных форм солей, внутренние соли или бетаины (цвиттерионы). Соответствующие соли можно получить обычными способами, которые известны специалисту в данной области, например, контактированием их с органической или неорганической кислотой или основанием в растворителе или дисперсии, или анионообменом или катинообменом с другими солями. Настоящее изобретение включает в себя также все соли соединений настоящего изобретения, которые вследствие их низкой физиологической совместимости не являются непосредственно пригодными для применения в фармацевтических средствах, но которые можно применять, например, в качестве промежуточных продуктов для химических реакций или для получения фармацевтически приемлемых солей.
Термин ”замещенный” предпочтительно относится к замещению вышеуказанными заместителями, где возможным является множество различных степеней замещения, если не указано иначе.
Все физиологически приемлемые соли, производные, сольваты, сольваты солей и стереоизомеры этих соединений, в то числе смеси их во всех отношениях, также включены в изобретение.
Соединения формулы (I) могут иметь один или несколько центров хиральности. Они могут соответственно присутствовать в различных энантиомерных формах и могут быть в рацемической или оптически активной форме. Изобретение, следовательно, относится также к оптически активным формам (стереоизомерам), энантиомерам, рацематам, диастереомерам и гидратам и сольватам этих соединений.
Поскольку фармацевтическая активность рацематов или стереоизомеров соединений согласно изобретению могут отличаться, может быть желательным применение энантиомеров. В этих случаях конечный продукт или даже промежуточные продукты можно разделить на энантиомерные соединения химическими или физическими способами, известными специалисту в данной области, или даже применять как таковые в синтезе.
В случае рацемических аминов, диастереомеры образуются из смеси реакцией с оптически активным агентом разделения. Примерами подходящих агентов разделения являются оптически активные кислоты, такие как R- и S-формы винной кислоты, диацетилвинной кислоты, дибензоилвинной кислоты, миндальной кислоты, яблочной кислоты, молочной кислоты, подходящие N-защищенные аминокислоты (например, N-бензоилпролин или N-бензолсульфонилпролин) или различные оптически активные камфорасульфоновые кислоты. Подходящим является также хроматографическое энантиомерное разделение с помощью оптически активного агента разделения (например, динитробензоилфенилглицина, триацетата целлюлозы или других производных углеводов или хирально дериватизированных метакрилатных полимеров, иммобилизованных на силикагеле). Подходящими элюентами для этой цели являются смеси водных или спиртовых растворителей, таких как, например, гексан/изопропанол/ацетонитрил, например, в отношении 82:15:3. В способе разделения рацематов, содержащих сложноэфирные группы (например, ацетиловых эфиров), применяют ферменты, в частности, эстеразы.
Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим соединение настоящего изобретения или его пролекарство, или фармацевтически приемлемую соль или его сольват в качестве активного ингредиента вместе с фармацевтически приемлемым носителем.
“Фармацевтическая композиция” означает один или несколько активных ингредиентов и один или несколько инертных ингредиентов, которые составляют носитель, а также любой продукт, который является результатом непосредственного или опосредованного комбинирования, комплексообразования или агрегации любых двух или более ингредиентов, или диссоциации одного или нескольких ингредиентов или других типов реакций или взаимодействий одного или нескольких ингредиентов. Соответственно этому, фармацевтические композиции настоящего изобретения включает в себя любую композицию, полученную смешиванием соединения настоящего изобретения и фармацевтически приемлемого носителя.
Фармацевтическая композиция настоящего изобретения может дополнительно содержать одно или несколько других соединений в качестве активных ингредиентов, таких как один или несколько дополнительных соединений настоящего изобретения или соединение-пролекарство или другие ингибиторы p70S6K. Фармацевтические композиции включают в себя композиции, подходящие для перорального, ректального, местного, парантерального (включая подкожное, внутримышечное и внутривенное введение), глазного (офтальмического), легочного (назальной или трансбуккальной ингаляцией) или назального введения, хотя наиболее подходящий путь в любом данном случае будет зависеть от природы и тяжести состояний, которые подвергают лечению, и от природы активного ингредиента. Они могут быть преимущественно представлены в виде дозированной лекарственной формы и их получают любым из способов, хорошо известных в области фармации.
В одном варианте осуществления указанные соединения и фармацевтическую композицию применяют для лечения рака, такого как рак головного мозга, легких, ободочной кишки, эпидермоида, клеток простого сквамозного эпителия, мочевого пузыря, желудка, поджелудочной железы, молочной железы, головы, шеи, почки, печени, яичников, простаты, колоректальной кишки, матки, ректальной кишки, пищевода, яичка, женских половых органов, щитовидной железы, меланомы, гематологических злокачественностей, таких как острый миелоидный лейкоз, множественная миелома, хронический миелоидный лейкоз, лейкоз миелоидных клеток, глиома, саркома Капоши или любой другой тип солидных или жидких опухолей. Рак, который лечат, предпочтительно выбирают из рака молочной железы, колоректального рака, рака легких, простаты, поджелудочной железы или глиобластомы.
Изобретение относится также к применению соединений согласно изобретению для получения лекарственного средства для лечения гиперпролиферативных заболеваний, связанных с гиперактивностью p70S6K, а также заболеваний, модулируемых каскадом p70S6K, у млекопитающих, или нарушений, опосредуемых абберантной пролиферацией, таких как рак или воспаление.
Изобретение относится также к соединению или фармацевтической композиции для лечения заболевания, связанного с образованием и развитием сосудов или развитием кровеносных сосудов у млекопитающего, которая содержит терапевтически эффективное количество соединения настоящего изобретения или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтический приемлемый носитель.
В одном варианте осуществления указанное соединение или фармацевтическую композицию применяют для лечения заболевания, выбранного из группы, состоящей из развития кровеносных сосудов опухоли, хронического воспалительного заболевания, такого как ревматоидный артрит, воспалительное заболевание кишечника, атеросклероз, кожных болезней, таких как псориаз, экзема и склеродерма, диабета, диабетической ретинопатии или ретинопатии недоношенности и связанной со старостью дегенерации желтого пятна.
Изобретение относится также к соединению или фармацевтической композиции для ингибирования аномального роста клеток у млекопитающего, которая содержит количество соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата или пролекарства в комбинации с количеством другого противоракового терапевтического средства, где количества соединения, соли, сольвата или пролекарства и химиотерапевтического средства вместе являются эффективными при ингибировании аномального роста клеток. Многие противораковые терапевтические средства являются в настоящее время известными в данной области. В одном варианте осуществления противораковым терапевтическим средством является химиотерапевтическое средство, выбранное из группы, состоящей из митотических ингибиторов, алкилирующих агентов, антиметаболитов, интеркалярных антибиотиков, ингибиторов фактора роста, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомеразы, модификаторов биологических ответных реакций, антигормонов, ингибиторов развития кровеносных сосудов и антиандрогенов. В другом варианте осуществления противораковым терапевтическим средством является антитело, выбранное из группы, состоящей из бевацизумаба, CD40-специфичных антител, chTNT-1/B, деносумаба, занолимумаба, IGF1R-специфичных антител, линтузумаба, эдерколомаба, WX G250, ритуксимаба, тицилимумаба, трастузумаба и цетуксимаба. Еще в одном варианте осуществления противораковым терапевтически средством является ингибитор другой протеинкиназы, такой как Akt, Ax1, Aurora A, Aurora B, dyrk2, epha2, fgfr3, igf1r, IKK2, JNK3, Vegfr1, Vegfr2, Vegfr3 (известный также как Flt-4), KDR, MEK, MET, Plk1, RSK1, Src, TrkA, Zap70, cKit, bRaf, EGFR, Jak2, Pl3K, NPM-Alk, c-Abl, BTK, FAK, PDGFR, TAK1, LimK, Flt-3, PDK1 и Erk.
Данное изобретение далее относится к способу ингибирования аномального роста клеток у млекопитающего или лечения гиперпролиферативного нарушения, который содержит введение млекопитающему количества соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата, или пролекарства в комбинации с радиационной терапией, где количество соединения, соли, сольвата или пролекарства в комбинации с радиационной терапией является эффективным при ингибировании аномального роста клеток или лечения гиперпролиферативного нарушения у млекопитающего. Способы применения радиационной терапии являются известными в данной области и эти способы можно применять в описанной в контексте комбинированной терапии. Введение соединения изобретения в данной комбинированной терапии можно определить, как описано в контексте. Считается, что соединения настоящего изобретения могут сделать аномальные клетки более чувствительными к лечению радиацией с целью уничтожения и/или ингибирования роста таких клеток.
Соответственно этому, данное изобретение далее относится к способу сенсибилизации аномальных клеток у млекопитающего к лечению радиацией, который содержит введение млекопитающему количества соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата, или пролекарства, причем такое количество является эффективным для сенсибилизации аномальных клеток к лечению радиацией. Количество соединения, соли или сольвата в данном способе можно определить согласно способу для установления эффективных количеств описанных в контексте таких соединений. Изобретение относится также к способу ингибирования роста аномальных клеток у млекопитающего, который содержит применение количества соединения настоящего изобретения или его фармацевтически приемлемой соли или сольвата, его пролекарства или его меченого изотопом производного и количества одного или нескольких веществ, выбранных из агентов против развития кровеносных сосудов, ингибиторов трансдукции сигнала и антипролиферативных агентов.
При практическом применении соединения настоящего изобретения можно применять в качестве активного ингредиента в тесной смеси с фармацевтическим носителем согласно общепринятым способам приготовления фармацевтических смесей. Носитель может иметь различные формы в зависимости от формы препарата, описанного для введения, например, для перорального или парентерального (в том числе внутривенного) введения. При получении композиций для пероральной лекарственной формы можно применять любую обычную фармацевтическую среду, такую как, например, вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и тому подобное. В случае пероральных жидких препаратов можно применять любую из обычных фармацевтических сред, такую как, например, суспензия, эликсир и раствор; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие вещества, связывающие вещества, дезинтегрирующие агенты и тому подобное. В случае пероральных твердых препаратов композиция может иметь такую форму, как, например, порошки, твердые и мягкие капсулы и таблетки, причем твердые пероральные препараты являются предпочтительными по сравнению с жидкими препаратами.
Вследствие легкости их введения, таблетки и капсулы представляют собой наиболее подходящую пероральную дозированную лекарственную форму, в которой, как очевидно, применяют твердые фармацевтические носители. При желании, таблетки можно покрывать оболочкой стандартными водными или неводными способами. Такие композиции и препараты должны содержать по меньшей мере 0,1 процента активного соединения. Процент активного соединения в этих композициях можно, конечно, изменять и он может быть преимущественно между приблизительно 2 процентами и приблизительно 60 процентами массы унифицированной дозы. Количество активного соединения в таких терапевтически применимых композициях является таким, чтобы можно было получить эффективную дозу. Активные соединения можно также вводить в нос, например, в виде жидких капель или спрея.
Таблетки, пилюли, капсулы и т.д. могут содержать также связывающее вещество, такое как трагакантовую камедь, аравийскую камедь, кукурузный крахмал или желатин; эксципиенты, такие как дикальцийфосфат; дезинтегрирующие агенты, такие как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, такое как стеарат магния, и подслащивающий агент, такой как сахароза, лактоза или сахарин. Когда дозированной лекарственной формой является капсула, она может содержать помимо веществ указанного выше типа, жидкий носитель, такой как жидкое масло.
Могут присутствовать различные другие вещества в качестве оболочек (покрытий) или для модификации физической формы унифицированной дозы. Например, таблетки можно покрывать шеллаком, сахаром или тем и другим. Сироп или эликсир может содержать помимо активного ингредиента сахарозу в качестве подслащивающего агента, метил- и пропилпарабены в качестве консервантов, краситель и корригент, такой как вишневый или апельсиновый корригент.
Соединения настоящего изобретения можно также вводить парентерально. Растворы или суспензии этих активных соединений можно получить в воде, подходящим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии можно также получать в глицерине, жидких полиэтиленгликолях и их смесях с маслами. При обычных условиях хранения и применения указанные препараты содержат консервант для предотвращения роста микроорганизмов.
Фармацевтические формы, подходящие для инъекционного применения, включают в себя стерильные водные растворы или дисперсии и стерильные порошки для импровизированного получения стерильных инъецируемых растворов или дисперсий. Во всех случаях форма должна быть стерильной и должна быть жидкой до такой степени, чтобы она легко обладала способностью находиться в шприце. Она должна быть стабильной в условиях изготовления и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носителем может быть растворитель или дисперсионная среда, содержащая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), подходящие их смеси и растительные масла. Для обеспечения млекопитающего, особенно человека, эффективной дозой соединения настоящего изобретения можно применять любой подходящий путь введения. Например, можно применять пероральный, ректальный, местный, парентеральный, глазной, легочный, назальный и подобный путь введения. Лекарственные формы включают в себя таблетки, пастилки, дисперсии, суспензии, растворы, капсулы, кремы, мази, аэрозоли и тому подобное. Соединения настоящего изобретения предпочтительно вводят перорально.
Эффективная доза применяемого активного ингредиента может изменяться в зависимости от конкретного применяемого соединения, способа введения, подвергаемого лечению состояния и тяжести подвергаемого лечению состояния. Такую дозу может легко установить специалист в данной области.
При лечении или профилактике рака, воспаления или других пролиферативных заболеваний, для которых показаны соединения настоящего изобретения, удовлетворительные результаты обычно получают, когда соединения настоящего изобретения вводят при суточной дозе от приблизительно 0,01 миллиграмма до приблизительно 100 миллиграммов на килограмм массы тела животного, предпочтительно введенной в виде разовой суточной дозы. Для большинства крупных млекопитающих общая суточная доза составляет от приблизительно 0,2 миллиграмма до приблизительно 2000 миллиграммов, предпочтительно от приблизительно 0,5 миллиграмма до приблизительно 1000 миллиграммов. В случае взрослого человека массой 70 кг общая суточная доза обычно может быть от приблизительно 0,5 миллиграмма до приблизительно 1000 миллиграммов. Эти вышеуказанные схемы приема лекарственного средства можно регулировать для обеспечения оптимального терапевтического результата.
Изобретение относится также к комплекту (набору), состоящему из отдельных упаковок
(а) эффективного количества соединения согласно изобретению или его физиологически приемлемой соли, сольвата или пролекарства и
(b) эффективного количества дополнительного активного ингредиента лекарственного средства. Комплект содержит подходящие контейнеры, такие как коробки, отдельные пузырьки, пакетики или ампулы. Комплект может, например, содержать отдельные ампулы, причем каждая содержит эффективное количество соединения согласно изобретению и/или его фармацевтически пригодные производные, сольваты и стереоизомеры, включая их смеси во всех отношениях, и эффективное количество дополнительного активного ингредиента лекарственного средства в растворенной или лиофилизованной форме.
В другом варианте осуществления настоящего изобретения описывается фармацевтическая композиция или лекарственный препарат, содержащий соединение согласно формуле (I) или его фармацевтически приемлемую соль, сольват или пролекарство в качестве активного ингредиента вместе с фармацевтически приемлемым носителем.
В другом варианте осуществления настоящего изобретения описывается применение соединения, фармацевтической композиции или лекарственного средства, содержащего соединение согласно формуле (I), для применения при лечении рака.
В другом варианте осуществления настоящего изобретения описывается применение соединения, фармацевтической композиции или лекарственного средства, содержащего соединение согласно формуле (I), для получения лекарственного препарата для лечения рака.
В другом варианте осуществления настоящего изобретения описывается способ лечения рака, содержащий введение субъекту соединения, фармацевтической композиции или лекарственного средства, содержащего соединение согласно формуле (I). В некоторых вариантах осуществления указанный рак выбирают из группы, состоящей из рака головного мозга, легких, ободочной кишки, эпидермоида, клеток простого сквамозного эпителия, мочевого пузыря, желудка, поджелудочной железы, молочной железы, головы, шеи, почки, печени, яичников, простаты, колоректальной кишки, матки, ректальной кишки, пищевода, яичка, женских половых органов, щитовидной железы, меланомы, гематологических злокачественностей, таких как острый миелоидный лейкоз, множественная миелома, хронический миелоидный лейкоз, лейкоз миелоидных клеток, глиома и саркома Капоши.
В другом варианте осуществления настоящего изобретения описывается комплект (набор), содержащий отдельные упаковки (а) эффективного количества соединения согласно формуле (I) или его фармацевтически приемлемой соли, сольвата или пролекарства и (b) эффективного количества дополнительного активного ингредиента лекарственного средства.
Экспериментальный раздел
Некоторые аббревиатуры, которые могут встречаться в заявке, являются следующими.
Аббревиатуры
Соединения настоящего изобретения можно получать согласно процедурам нижеследующих схем и примеров с применением подходящих веществ, примеры получения соединений приводятся в нижеследующих конкретных примерах.
Кроме того, дополнительные соединения настоящего изобретения, указанные в контексте, можно легко получить описанными в контексте процедурами в сочетании со средним уровнем компетентности в данной области. Соединения, иллюстрированные в примерах, однако, не следует рассматривать как образующие только род соединений, который рассматривают в качестве изобретения. Примеры далее иллюстрируют подробности получения соединений настоящего изобретения. Специалистам в данной области будет легко понятно, что для получения этих соединений можно применять известные варианты условий и способов нижеследующих процедур получения.
Полученные таким образом соединения обычно выделяют в форме их фармацевтически приемлемых солей, таких как соли, описанные выше. Основания в виде свободных аминов, соответствующие выделенным солям, обычно получают нейтрализацией солей подходящим основанием, таким как водный гидрокарбонат натрия, карбонат натрия, гидроксид натрия и гидроксид калия, и экстракцией выделенного основания в виде свободного амина в органический растворитель с последующим выпариванием последнего. Основание в виде свободного амина, выделенное таким способом, можно далее превратить в другую фармацевтически приемлемую соль растворением в органическом растворителе с последующим добавлением подходящей кислоты и затем выпариванием, осаждением или кристаллизацией.
Изобретение будет иллюстрировано, но не ограничено, обращением к конкретным вариантам осуществления, описанным в нижеследующих схемах и примерах. Если не указано иное, в схемах символы имеют такие же значения, какие описаны выше. Если не оговорено особо, все исходные вещества получают от коммерческих поставщиков и применяют без дополнительной очистки. Если не указано особо, все температуры указаны в °C и все реакции проводят при комнатной температуре. Соединения очищали либо хроматографией на диоксиде кремния, либо препаративной ВЭЖХ.
Настоящее изобретение относится также к способам получения соединений формулы (I) согласно описанным ниже схемам и рабочим примерам.
Синтетические схемы, описывающие соединения, являющиеся промежуточными и конечными продуктами
Пиперидиновые промежуточные продукты получают согласно синтезу, описанному на схеме 1 и схеме 2.
Схема 1
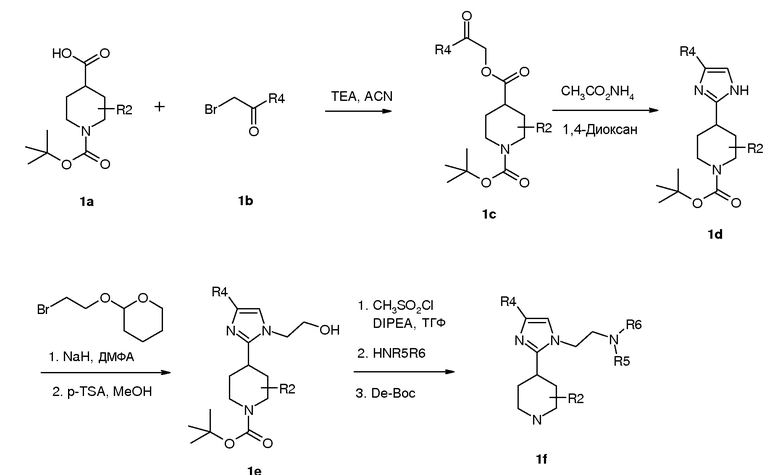
1-трет-Бутиловый эфир замещенной пиперидин-1,4-дикарбоновой кислоты 1a подвергали реакции с 1-замещенным 2-бромэтаноном 1b в присутствии основания, получая при этом эфир 1c, который затем подвергали реакции с ацетатом аммония, получая при этом производное имидазола 1d. Алкилирование соединения 1d 2-(2-бромэтокси)тетрагидропираном в присутствии гидрида натрия в ДМФА с последующим удалением защитной группы п-толуолсульфоновой кислотой в метаноле давало спирт 1e. Спирт 1e превращали в метансульфонатный промежуточный продукт и затем в Boc-защищенный амин, у которого удаляли защитную группу, получая при этом производное пиперидина 1f.
Схема 2

2-Бром-1-(4-фтор-3-метилфенил)этанон 2a подвергали реакции с 1-трет-бутиловым эфиром 4-гидроксипиперидин-1,4-дикарбоновой кислоты в присутствии основания, получая при этом эфир 2b, который затем подвергали затем реакции с ацетатом аммония, получая при этом производное имидазола 2c. Защита гидроксильной группы реакцией с TBDMSCI давала соединение 2d, которое алкилировали 2-(2-бромэтокси)тетрагидропираном в присутствии гидрида натрия в ДМФА с последующим удалением защитной группы п-толуолсульфоновой кислотой в метаноле, получая при этом спирт 2f. Спирт 2f превращали в метансульфонатный промежуточный продукт и затем в Boc-защищенный амин, у которого удаляли защитную группу, получая при этом производное пиперидина 2g.
Конечные соединения формулы (I) получают согласно синтетического пути, показанного на схеме 3.
Схема 3
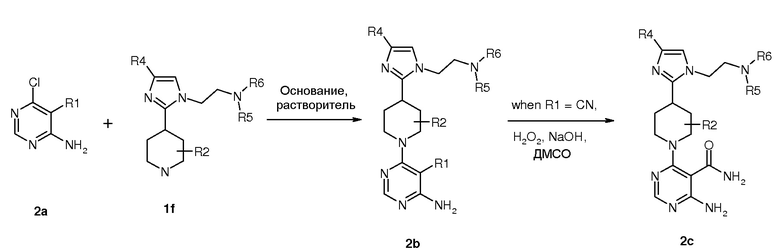
5-Замещенный 4-амино-6-хлорпиримидин 3a подвергали реакции с производным пиперидина в основных условиях, получая при этом требуемое производное пиримидина 3b. 5-нитрилопиримидин 3b (если R1 = нитрил) далее превращали в 5-карбоксамидопиримидин 3c реакцией с пероксидом водорода и гидроксидом натрия в ДМСО.
Аналитическая методология
Аналитический анализ ЖХ/МС проводили с применением следующих трех способов:
Способ A: применяли колонку Discovery C18, 5 мкм, 3×30 мм при скорости потока 400 мкл/мин, ввод 5 мкл образца в петлю, подвижная фаза: (A) вода с 0,1% муравьиной кислоты, подвижная фаза (B): метанол с 0,1% муравьиной кислоты; времена удерживания указываются в минутах. Подробности способа: (I) опыты проводили с 4-ступенчатым насосом G1311A (Agilent), с диодным матричным УФ/VIS-детектором G1315B (Agilent) и детектором Финнигана LCQ Duo MS в ESI + метод с UV-детектированием при 254 и 280 нм с градиентом 15-95% (B) в линейном градиенте 3,2 мин, (II) поддерживание в течение 1,4 мин при 95% (B), (III) снижение от 95 до 15% (B) при линейном градиенте 0,1 мин, (IV) поддерживание в течение 2,3 мин при 15% (B).
Способ В: применяли колонку Waters Symmetry C18, 3,5 мкм, 4,6×75 мм при скорости потока 1 мл/мин, ввод образца в петлю 10 мкл, подвижной фазой (A) является вода с 0,05% TFA, подвижной фазой (B) является ACN с 0,05% TFA; времена удерживания указываются в минутах. Подробности способа: (I) опыты проводили с 2-ступенчатым насосом G1312A (Agilent), с диодным матричным УФ/VIS-детектором G1315B (Agilent) и МС-детектором Agilent G1956B (SL) в ESI + метод с UV-детектированием при 254 и 280 нм с градиентом 20-85% (B) в линейном градиенте 10 мин, (II) поддерживание в течение 1 мин при 85% (B), (III) снижение от 20 до 85% (B) при линейном градиенте 0,2 мин, (IV) поддерживание в течение 3,8 мин при 20% (B).
Способ C: градиент: 4,2 мин/скорость потока: 2 мл/мин 99:01 - 0:100 воды + 0,1% (об.) TFA; ацетонитрила + 0,1% (об.) TFA; от 0,0 до 0,2 мин: 99:01; от 0,2 до 3,8 мин: 99:01×0:100; от 3,8 до 4,2 мин: 0:100; колонка: Chromolith Performance RP18e; длина 100 мм, диаметр 3 мм; длина волны: 220 нм.
Аналитическая хиральная ВЭЖХ
Аналитическую хиральную ВЭЖХ проводили с применением колонки ChiralPak AD-H (250×4,6 мм) от Daicel Chemical Industries, Ltd. на системе an Agilent 1100 Series. В способе применяли ввод объемом 5,0 мкл, скорость потока 1 мл/мин 100% метанола в течение 15 мин при 25°C и УФ-детектор при 254 и 280 нм.
Препаративная ВЭЖХ
Препаративную ВЭЖХ проводили с применением либо колонки Waters Atlantis dC18 OBD™, 10 мкМ (30×250 мм), либо колонки Waters Sunfire Prep C18 OBD 10 мкМ (30×250 мм). Колонки применяли при скорости потока 60 мл/мин на системе Waters Prep LC 4000, снабженной петлей для ввода пробы (10 мл) и УФ-VIS-детектором ISCO UA-6. Подвижную фазу подавали из двух резервуаров для растворителей, содержащих (а) воду и (В) ацетонитрил сорт ВЭЖХ. В типичном препаративном опыте применяли линейный градиент (например, 0-60% растворителя B в течение 60 мин).
ПРИМЕРЫ
Рабочие примеры, представленные ниже, предназначены для иллюстрации конкретных вариантов осуществления изобретения и во всяком случае не предназначены для ограничения объема описания изобретения или формулы изобретения.
Пример 1. Получение промежуточных продуктов
цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин (рацемический)
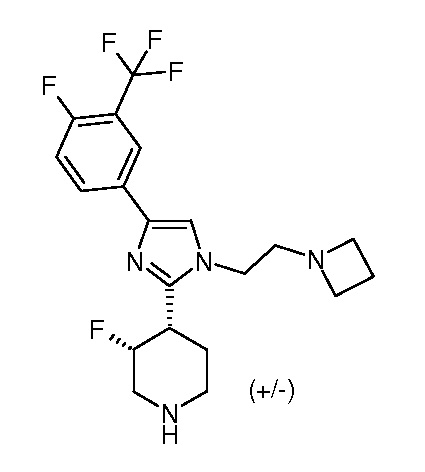
Стадия 1: 1-трет-бутиловый эфир-4-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтиловый] эфир 3-фторпиперидин-1,4-дикарбоновой кислоты
2-Бром-1-(4-фтор-3-трифторметилфенил)этанон (2,23 г; 8,07 ммоль; 1.0 экв.) добавляли к раствору 1-трет-бутилового эфира 3-фторпиперидин-1,4-дикарбоновой кислоты (2,0 г; 8,07 ммоль; 1,0 экв.) и триэтиламина (1,36 мл; 9,68 ммоль; 1,20 экв.) в ацетонитриле (20 мл) при КТ в виде одной порции. Образовавшуюся смесь перемешивали при КТ в течение 1,5 час. Реакционную смесь разбавляли 60 мл этилацетата, промывали один раз насыщенным раствором карбоната натрия и дважды насыщенным раствором соли. Органический слой сушили над MgSO4 и затем концентрировали, получая при этом указанное в заголовке соединение в виде светло-коричневого твердого вещества (3,75 г, выход 103%).
Стадия 2: трет-бутиловый эфир 3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты
Раствор 1-трет-бутилового эфира-4-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтилового] эфира 3-фторпиперидин-1,4-дикарбоновой кислоты (3,60 г; 7,98 ммоль; 1,0 экв.) и ацетата аммония (6,15 г; 79,75 ммоль; 10,0 экв.) в 1,4-диоксане(20 мл) перемешивали при 110°C в течение 3 час. После охлаждения до КТ реакционную смесь разбавляли 100 мл этилацетата, промывали насыщенным раствором бикарбоната натрия один раз и насыщенным раствором соли два раза, сушили над MgSO4 и концентрировали. Остаток сушили в сушильном шкафу на протяжении ночи, получая при этом светло-коричневое твердое вещество в качестве указанного в заголовке соединения (3,5 г, выход 102%), которое применяли для следующей стадии без очистки.
Стадия 3: трет-бутиловый эфир транс-3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты и трет-бутиловый эфир цис-3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты
Гидрид натрия (60% дисперсия в минеральном масле, 417,21 мг; 10,43 ммоль; 3,0 экв.) добавляли к раствору трет-бутилового эфира 3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (1,50 г; 3,48 ммоль; 1,0 экв.) в ДМФА (8,0 мл) при КТ. После перемешивания в течение 30 мин по каплям добавляли 2-(2-бромэтокси)тетрагидропиран (1,58 мл; 10,43 ммоль; 3,00 экв.). Образовавшуюся смесь перемешивали при КТ на протяжении ночи. Реакционную смесь разбавляли 70 мл этилацетата, промывали один раз водой и два раза насыщенным раствором соли. Отделенный органический слой сушили над MgSO4 и концентрировали, получая при этом трет-бутил-3-фтор-4-(4-(4-фтор-3-(трифторметил)фенил)-1-(2-((тетрагидро-2H-пиран-2-ил)окси)этил)-1H-имидазол-2-ил)пиперидин-1-карбоксилат.
Раствор указанного выше трет-бутил-3-фтор-4-(4-(4-фтор-3-(трифторметил)фенил)-1-(2-((тетрагидро-2H-пиран-2-ил)окси)этил)-1H-имидазол-2-ил)пиперидин-1-карбоксилата и гидрата толуол-4-сульфоновой кислоты (0,99 г, 5,22 ммоль, 1,5 экв.) в метаноле (10 мл) перемешивали при к.т. в течение 1,5 час. После удаления растворителя остаток растворяли в этилацетате (100 мл) и насыщенном растворе хлорида аммония (50 мл), органический слой промывали дважды насыщенным раствором соли, сушили над MgSO4 и концентрировали. Сырой продукт очищали препаративной ВЭЖХ (Waters, основное условие), получая при этом трет-бутиловый эфир цис-3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (440 мг, выход 31%) в качестве первой фракции и трет-бутиловый эфир транс-3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (490 мг, выход 34%) в качестве второй фракции.
Стадия 4: цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин
Метансульфонилхлорид (0,07 мл; 0,95 ммоль; 1,5 экв.) добавляли к раствору трет-бутилового эфира цис-3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (300,0 мг; 0,63 ммоль; 1,0 экв.) и диизопропилэтиламина (0.23 мл; 1,26 ммоль; 2,0 экв.) в ТГФ (3 мл) при КТ в течение 3 час. Азетидин (360 мг; 6,31 ммоль; 10,0 экв.) добавляли к реакционной смеси и смесь перемешивали при КТ на протяжении ночи. Реакционную смесь концентрировали для применения в следующей стадии без очистки.
Указанное выше неочищенное соединение растворяли в смеси 1:1 (об./об.) трифторуксусной кислоты и дихлорметана (2 мл) и перемешивали при КТ в течение 2 час. Реакционную смесь концентрировали и очищали препаративной ВЭЖХ (Waters, основное условие), получая при этом указанное в заголовке соединение в виде не совсем белого твердого вещества (196 мг, выход 75%).
цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин (рацемический)
Стадия 1: 1-трет-бутиловый эфир-4-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтиловый] эфир 3-метилпиперидин-1,4-дикарбоновой кислоты
К раствору 2-бром-1-(4-фтор-3-трифторметилфенил)этанона (1000,00 мг; 3,51 ммоль; 1.00 экв.) и 1-трет-бутилового эфира 3-метилпиперидин-1,4-дикарбоновой кислоты (981,62 мг; 4,03 ммоль; 1,15 экв.) в 10 мл ацетона добавляли карбонат цезия (1714,6 мг; 5,26 ммоль; 1,50 экв.). Образовавшуюся смесь перемешивали при КТ в течение 30 мин. Анализ ЖХ-МС показал, что реакция была проведена с образованием желаемого продукта. Реакционный раствор выливали в 50 мл этилацетата и промывали 5% водным раствором NaHCO3, затем насыщенным раствором соли. Органическую фазу сушили и концентрировали, получая при этом указанное в заголовке соединение, которое непосредственно применяли для реакции следующей стадии. ЖХ-МС (M+H = 448, найдено = 448).
Стадия 2: трет-бутиловый эфир 4-[4-(4-фтор-3-трифторметилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]-3-метилпиперидин-1-карбоновой кислоты
Смесь 1-трет-бутилового эфира-4-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтилового] эфира 3-метилпиперидин-1,4-дикарбоновой кислоты (1800,00 мг; 4,02 ммоль; 1,00 экв.), ацетата аммония (3101,09 мг; 40,23 ммоль; 10,00 экв.) и 1-бутанола (3 мл) в микроволновой пробирке помещали в микроволновое устройство и выдерживали при 125°C в течение 20 мин. Реакционную смесь очищали ВЭЖХ, получая при этом указанное в заголовке соединение (300 мг). ЖХ-МС (M+H = 472, найдено = 472).
Стадия 3: трет-бутиловый эфир 4-{4-(4-фтор-3-трифторметилфенил)-1-[2-(тетрагидропиран-2-илокси)этил]-1H-имидазол-2-ил}-3-метилпиперидин-1-карбоновой кислоты
К раствору трет-бутилового эфира 4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-карбоновой кислоты (1000,00 мг; 2,34 ммоль; 1,00 экв.) в ДМФА (8 мл) добавляли NaH (280,72 мг; 7,02 ммоль; 3,00 экв.). После перемешивания при КТ в течение 30 мин добавляли 2-(2-бромэтокси)тетрагидропиран (1467,45 мг; 7,02 ммоль; 3,00 экв.). Реакционную смесь перемешивали при 85°C в течение 1 час, анализ ЖХ-МС показал, что реакция была завершена. Реакционную смесь охлаждали, выливали в воду и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, сушили и концентрировали, получая при этом указанное в заголовке соединение, которое непосредственно применяли для реакции следующей стадии без дополнительной очистки. ЖХ-МС (M+H = 556, найдено = 556).
Стадия 4: трет-бутиловый эфир 4-[4-(4-фтор-3-трифторметилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]-3-метилпиперидин-1-карбоновой кислоты
К раствору трет-бутилового эфира 4-(4-(4-фтор-3-трифторметилфенил)-1-[2-(тетрагидропиран-2-илокси)этил]-1H-имидазол-2-ил}-3-метилпиперидин-1-карбоновой кислоты (1290,00 мг; 2,32 ммоль; 1,00 экв.) в метаноле (20 мл) добавляли толуол-4-сульфоновую кислоту (39,98 мг; 0,23 ммоль; 0,10 экв.). Реакционную смесь перемешивали при КТ на протяжении ночи. Так как анализ ЖХ-МС показал, что исходное соединение было основным компонентом, добавляли еще 2 экв. толуол-4-сульфоновой кислоты (800 мг) и смесь перемешивали при КТ в течение 30 мин при мониторинге реакции ЖХ-МС. После удаления растворителя добавляли 50 мл этилацетата и смесь промывали 10% водным раствором карбоната натрия, затем насыщенным раствором соли. Органическую фазу сушили и концентрировали, получая при этом сырой остаток, который очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение (200 мг, выход 18,3%). ЖХ-МС (M+H = 472, найдено = 472).
Стадия 5: трет-бутиловый эфир 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-карбоновой кислоты
К трет-бутиловому эфиру 4-[4-(4-фтор-3-трифторметилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (200,00 мг; 0,44 ммоль; 1,00 экв.) в ТГФ (2 мл) добавляли этилдиизопропиламин (0,16 мл; 0,87 ммоль; 2,00 экв.) с последующим добавлением метансульфонилхлорида (0,05 мл; 0,66 ммоль; 1,50 экв.). Реакционную смесь перемешивали при КТ на протяжении ночи. Анализ ЖХ-МС показал, что образование мезилата было завершено. Добавляли азетидин (249,61 мг; 4,37 ммоль; 10,00 экв.) и смесь перемешивали при КТ в течение 8 час. Реакционную смесь очищали ВЭЖХ, получая при этом указанное в заголовке соединение (90 мг, выход 38%). ЖХ-МС (M+H = 511, найдено = 511).
Стадия 6: тригидрохлорид 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидина
К раствору трет-бутилового эфира 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-карбоновой кислоты (70,00 мг; 0,14 ммоль; 1,00 экв.) в метаноле (0,5 мл) добавляли 0,5 мл 4,0 М HCl в диоксане и реакционную смесь перемешивали при КТ в течение 3 час. Реакционную смесь концентрировали, получая при этом указанное в заголовке соединение в виде белого твердого вещества (71,3 мг). ЖХ-МС (M+H = 411, найдено = 411).
цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин (рацемический)

Стадия 1: 1-трет-бутиловый эфир-4-[2-(4-фтор-3-метилфенил)-2-оксоэтиловый) эфир 3-фторпиперидин-1,4-дикарбоновой кислоты
2-Бром-1-(4-фтор-3-метилфенил)этанон (9344,79 мг; 40,44 ммоль; 1,00 экв.) добавляли к раствору 1-трет-бутилового эфира 3-фторпиперидин-1,4-дикарбоновой кислоты (10,00 г; 40,44 ммоль; 1,00 экв.) и триэтиламина (6,82 мл; 48,53 ммоль; 1,20 экв.) в MeCN (100,00 мл; 1914,60 ммоль; 47,34 экв.) при КТ в виде одной порции. Образовавшуюся смесь перемешивали при КТ в течение 2,5 час. Мониторинг реакции проводили способом ЖХ-МС. Реакционную смесь разбавляли 300 мл этилацетата и промывали насыщенным водным раствором NaHCO3 один раз и затем насыщенным раствором соли два раза. Органический слой сушили над MgSO4 и концентрировали. Светло-коричневое твердое вещество в виде смеси транс- и цис-изомеров применяли непосредственно для реакции следующей стадии (14,80 г; 37,24 ммоль)). ЖХ-МС (M+H = 398, найдено = 398).
Стадия 2: трет-бутиловый эфир цис-3-Фтор-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты, рацемический
Раствор 1-трет-бутилового эфира-4-[2-(4-фтор-3-метилфенил)-2-оксоэтилового] эфира 3-фторпиперидин-1,4-дикарбоновой кислоты (14,80 г; 37,24 ммоль; 1,00 экв.) и ацетата аммония (28706,37 мг; 372,41 ммоль; 10,00 экв.) в диоксане (100,00 мл; 1173,60 ммоль; 31,51 экв.) перемешивали при 100°C. Реакция завершалась спустя 2 час по данным мониторинга способом ЖХ-МС. После охлаждения до КТ реакционную смесь разбавляли 300 мл этилацетата, промывали два раза насыщенным водным раствором бикарбоната натрия и два раза насыщенным раствором соли. Органическую фазу сушили над MgSO4 и концентрировали. Остаток (содержащий как транс-, так и цис-продукты и побочный продукт) сушили в сушильном шкафу на протяжении ночи, получая при этом светло-коричневое твердое вещество, добавляли DCM (20 мл) и смесь перемешивали в течение 10 мин и добавляли этилацетат (40 мл) и перемешивали еще в течение 1 час. Осадок собирали фильтрованием и промывали небольшим количеством этилацетата, получая при этом бледно-желтое твердое вещество в виде цис-рацематного продукта. (M+H = 378, найдено = 378).
Стадия 3: трет-бутиловый эфир цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]-пиперидин-1-карбоновой кислоты, рацемический
К раствору рацемического трет-бутилового эфира цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (5310,00 мг; 14,07 ммоль; 1,00 экв.) в ДМФА (30 мл) добавляли NaH (1406,77 мг; 35,17 ммоль; 2,50 экв.). После перемешивания при КТ в течение 30 мин по каплям добавляли 2-(2-бромэтокси)тетрагидропиран (4,26 мл; 28,14 ммоль; 2,00 экв.). Реакционную смесь перемешивали при КТ на протяжении ночи. Реакционную смесь выливали в воду (200 мл) и смесь экстрагировали этилацетатом (200 мл × 2). Объединенный органический слой промывали насыщенным раствором соли, сушили и концентрировали, получая при этом рацемический трет-бутиловый эфир цис-3-фтор-4-{4-(4-фтор-3-метилфенил)-1-[2-(тетрагидропиран-2-илокси)этил]-1H-имидазол-2-ил}пиперидин-1-карбоновой кислоты. ЖХ-МС (M+H = 506, найдено = 506). К указанному выше соединению в метаноле (100 мл) добавляли гидрат толуол-4-сульфоновой кислоты (4014,24 мг; 21,10 ммоль; 1,50 экв.). Образовавшуюся смесь перемешивали при КТ в течение 2 час. Анализ ЖХ-МС показал, что реакция была завершена. После удаления растворителя остаток разбавляли этилацетатом (200 мл), промывали 5% водным раствором Na2CO3 и затем насыщенным раствором соли. Органическую фазу сушили и концентрировали, концентрат затем обрабатывали 10 мл простого эфира, получая при этом не совсем белое твердое вещество в качестве указанного в заголовке соединения (4,4 г). ЖХ-МС (M+H = 422, найдено = 422).
Стадия 4: трет-бутиловый эфир цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-карбоновой кислоты, рацемический
К раствору трет-бутилового эфира цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (4400,00 мг; 10,44 ммоль; 1,00 экв.) и этилдиизопропиламина (3,75 мл; 20,88 ммоль; 2,00 экв.) в ТГФ (50 мл) добавляли при 0°C метансульфонилхлорид (1,21 мл; 15,66 ммоль; 1,50 экв.). Образовавшуюся смесь перемешивали при КТ в течение 3 час. Анализ ЖХ-МС показал, что трет-бутиловый эфир цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-метансульфонилоксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты был образован в виде чистого продукта. Реакционную смесь охлаждали на ледяной бане и по каплям добавляли азетидин (5960,26 мг; 104,39 ммоль; 10,00 экв.) и затем перемешивали при КТ на протяжении ночи. Реакционную смесь разбавляли этилацетатом (150 мл), промывали 5% водным раствором NaHCO3 и затем насыщенным раствором соли. Органическую фазу сушили и концентрировали, получая при этом указанное в заголовке соединение, которое применяли непосредственно для реакции следующей фазы. ЖХ-МС (M+H = 461, найдено = 461).
Стадия 5: цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин, рацемический
К раствору трет-бутилового эфира цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-карбоновой кислоты (4700,00 мг; 10,20 ммоль; 1,00 экв.) в метаноле (15 мл) добавляли 4,0 М HCl в диоксане (17,86 мл; 71,43 ммоль; 7,00 экв.) при 0°C. Реакционную смесь затем перемешивали при КТ в течение 2 час. После удаления растворителей остаток обрабатывали простым эфиром, получая при этом бледно-желтое твердое вещество в качестве тетрагидрохлорида цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидина. ЖХ-МС (M+H = 361, найдено = 361).
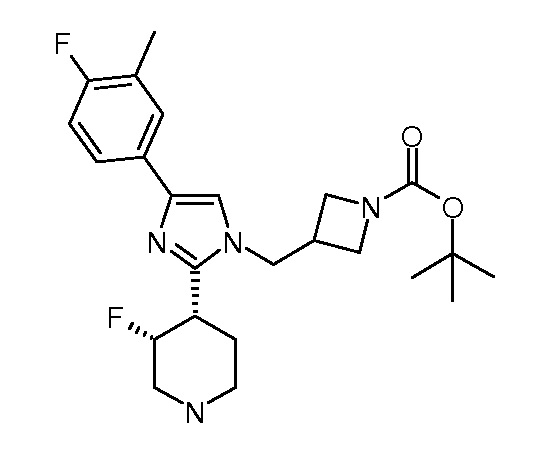
трет-Бутиловый эфир цис-3-[4-(4-фтор-3-метилфенил)-2-(цис-3-фторпиперидин-4-ил)имидазол-1-илметил]азетидин-1-карбоновой кислоты (рацемический)
Стадия 1: бензиловый эфир цис-3-Фтор-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты, рацемический
К раствору рацемического трет-бутилового эфира цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (1230,00 мг; 3,26 ммоль; 1,00 экв.) в DCM (5 мл) добавляли трифторуксусную кислоту (3715,90 мг; 32,59 ммоль; 10,00 экв.). Смесь перемешивали при КТ в течение 1 час, и анализ ЖХ-МС показал, что реакция была завершена. Реакционную смесь концентрировали, получая при этом цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин в виде соли TFA.
К раствору указанной выше соли TFA (2300,00 мг; 3,14 ммоль; 1,00 экв.) в DCM (25 мл) добавляли бензил-(2,5-диоксопирролидин-1-ил)карбонат (1172,2 мг, 4,7 ммоль; 1,5 экв.) и этилдиизопропиламин (3,94 мл; 21,95 ммоль; 7,00 экв.). Реакционную смесь перемешивали при КТ на протяжении ночи, разбавляли DCM, промывали 5% NaHCO3, затем насыщенным раствором соли. Органическую фазу сушили и концентрировали, получая при этом указанное в заголовке соединение, которое непосредственно применяли в реакции следующей стадии. ЖХ-МС (M+H = 546, найдено = 546).
Стадия 2: бензиловый эфир цис-4-[1-(1-трет-бутоксикарбонилазетидин-3-илметил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-карбоновой кислоты, рацемический
К раствору рацемического бензилового эфира цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (1200,00 мг; 2,92 ммоль; 1,00 экв.) в ДМФА (12 мл) добавляли NaH (349,96 мг; 8,75 ммоль; 3,00 экв.). После перемешивания при КТ в течение 30 мин по каплям добавляли трет-бутиловый эфир 3-иодметилазетидин-1-карбоновой кислоты (1,13 мл; 5,25 ммоль; 1,80 экв.). Реакционную смесь перемешивали при КТ в течение 2 час. Реакционную смесь гасили насыщенным водным раствором NH4Cl и добавляли этилацетат (60 мл), промывали водой, 5% NaHCO3, затем насыщенным раствором соли. Органическую фазу сушили и концентрировали. Остаток очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 581, найдено = 581).
Стадия 3: трет-бутиловый эфир цис-3-[4-(4-фтор-3-метилфенил)-2-(цис-3-фторпиперидин-4-ил)имидазол-1-илметил]азетидин-1-карбоновой кислоты, рацемический
К раствору рацемического бензилового эфира цис-4-[1-(1-трет-бутоксикарбонилазетидин-3-илметил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-карбоновой кислоты (120,00 мг; 0,21 ммоль; 1,00 экв.) в метаноле,(5 мл) добавляли трифторуксусную кислоту (47,13 мг; 0,41 ммоль; 2,00 экв.). После перемешивания0 в течение 5 мин добавляли влажный 10% Pd/C (120 мг) с последующим добавлением формиата аммония (130,31 мг; 2,07 ммоль; 10,00 экв.). Реакционную смесь перемешивали при 25°C в течение 1 час. После удаления катализатора и концентрирования остаток очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 447, найдено = 447).
цис-4-[1-(2-Азетидин-1-илэтил)-2-(цис-3-фторпиперидин-4-ил)-1H-имидазол-4-ил]-2-трифторметилпиридин (рацемический)

Стадия 1: цис-1-трет-Бутил-4-{2-оксо-2-[2-(трифторметил)пиридин-4-ил]этил}пиперидин-1,4-дикарбоксилат
К раствору 1-трет-бутилового эфира цис-3-фторпиперидин-1,4-дикарбоновой кислоты (3690,07 мг; 14,92 ммоль; 1,00 экв.) и 2-бром-1-(2-трифторметилпиридин-4-ил)этанона (4000,00 мг; 14,92 ммоль; 1,00 экв.) в ТГФ (50 мл) добавляли по каплям этилдиизопропиламин (5,21 мл; 29,85 ммоль; 2,00 экв.) при КТ. Реакционную смесь перемешивали при КТ в течение еще 1 час, когда анализ ЖХ-МС показал, что реакция была завершена. Реакцию гасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом (200 мл). Отделенный органический слой промывали насыщенным водным раствором NH4Cl (100 мл × 4), сушили и концентрировали, получая при этом указанное в заголовке соединение в виде желтого масла (5400 мг, выход 100%). ЖХ-МС (M+H = 435, найдено = 435).
Стадия 2: трет-бутиловый эфир цис-3-фтор-4-[4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты
К раствору 1-трет-бутилового эфира-4-[2-оксо-2-(2-трифторметилпиридин-4-ил)этилового] эфира цис-3-фторпиперидин-1,4-дикарбоновой кислоты (5000,00 мг; 11,51 ммоль; 1,00 экв.) в диоксане (100 мл) добавляли ацетат аммония (8872,72 мг; 115,11 ммоль; 10,00 экв.). Реакционную смесь перемешивали при 110°C в течение 1,5 час при проведении мониторинга ее способом ЖХ-МС для определения завершения реакции. После охлаждения до КТ реакционную смесь разбавляли 150 мл этилацетата, промывали насыщенным раствором соли и затем насыщенным раствором бикарбоната натрия и насыщенным раствором соли. Органическую фазу сушили над MgSO4 и концентрировали, получая при этом указанное в заголовке соединение (3830 мг, выход 80,7%). ЖХ-МС (M+H = 415, найдено = 415).
Стадия 3: трет-бутиловый эфир цис-3-фтор-4-[1-(2-гидроксиэтил)-4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты
К раствору трет-бутилового эфира цис-3-фтор-4-[4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (3830,00 мг; 9,24 ммоль; 1,00 экв.) в ДМФА (18 мл) добавляли NaH (831,74 мг; 20,80 ммоль; 2,25 экв.). После перемешивания при 0°C в течение 30 мин по каплям добавляли 2-(2-бромэтокси)тетрагидропиран (2,10 мл; 13,86 ммоль; 1,50 экв.). Реакционную смесь перемешивали при КТ на протяжении ночи. Реакцию гасили насыщенным водным раствором NH4Cl (50 мл) и экстрагировали этилацетатом (100 мл × 2). Объединенный органический слой промывали насыщенным раствором соли, сушили и концентрировали. Остаток очищали Biotage (колонка SNAP, элюировали DCM, содержащим 2% метанола и 0,2% TEA), получая при этом трет-бутиловый эфир цис-3-фтор-4-[1-[2-(тетрагидропиран-2-илокси)этил]-4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (1,7 г).
К указанному выше продукту, растворенному в метаноле
(20 мл), добавляли гидрат толуол-4-сульфоновой кислоты (879,03 мг; 4,62 ммоль; 0,50 экв.). Реакционную смесь перемешивали при КТ в течение 1 час. После удаления растворителя к остатку добавляли этилацетат (60 мл) и 10% водный раствор Na2CO3 (20 мл) и смесь перемешивали в течение 20 мин. Отделенный органический слой промывали насыщенным раствором соли, сушили и концентрировали, получая при этом указанное в заголовке соединение в виде желтого твердого вещества (1300 мг, выход 30,7%). ЖХ-МС (M+H = 459, найдено = 459).
Стадия 4: трет-бутиловый эфир цис-4-[1-(2-азетидин-1-илэтил)-4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]-3-фторпиперидин-1-карбоновой кислоты
К раствору трет-бутилового эфира цис-3-хлор-4-[1-(2-гидроксиэтил)-4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (1300,00 мг; 2,84 ммоль; 1,00 экв.) и этилдиизопропиламина (0,76 мл; 4,25 ммоль; 1,50 экв.) в ТГФ (10 мл) добавляли метансульфонилхлорид (0,30 мл; 3,83 ммоль; 1,35 экв.) при 0°C. Реакционную смесь перемешивали при такой же температуре в течение 30 мин и затем при КТ в течение 2 час, когда анализ способом ЖХ-МС показал, что реакция мезилирования была завершена. К указанной выше реакционной смеси добавляли азетидин (2,02 мл; 28,36 ммоль; 10,00 экв.) и перемешивали при КТ в течение 72 час. После удаления растворителя к остатку добавляли 50 мл этилацетата и промывали насыщенным раствором соли, 10% раствором лимонной кислоты, 10% раствором NaHCO3 и насыщенным раствором соли. Органическую фазу сушили и концентрировали, остаток очищали колонкой SNAP (DCM, содержащий 1% метанола и 0,2% TEA), получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 498, найдено = 498).
Стадия 5: 4-[1-(2-азетидин-1-илэтил)-2-(цис-3-фторпиперидин-4-ил)-1H-имидазол-4-ил]-2-трифторметилпиридин
К раствору трет-бутиливого эфира цис-4-[1-(2-азетидин-1-илэтил)-4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]-3-фторпиперидин-1-карбоновой кислоты (650,00 мг; 1,31 ммоль; 1,00 экв.) в метаноле (3 мл) по каплям добавляли 4 М HCl в диоксане (2,29 мл; 9,15 ммоль; 7,00 экв.) при 0°C. Реакционную смесь затем перемешивали при КТ в течение 2 час, когда анализ ЖХ-МС показал, что реакция была завершена. После удаления растворителей остаток растворяли в воде (10 мл) и промывали DCM.
К водной фазе добавляли карбонат натрия (692,34 мг; 6,53 ммоль; 5,00 экв.) и DCM (20 мл) и смесь перемешивали при КТ в течение 1 час. Отделенный органический слой промывали насыщенным раствором соли, сушили и концентрировали, получая при этом указанное в заголовке соединение в виде желтого твердого вещества (430 мг, выход 82,8%). ЖХ-МС (M+H = 398, найдено = 398).
4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин
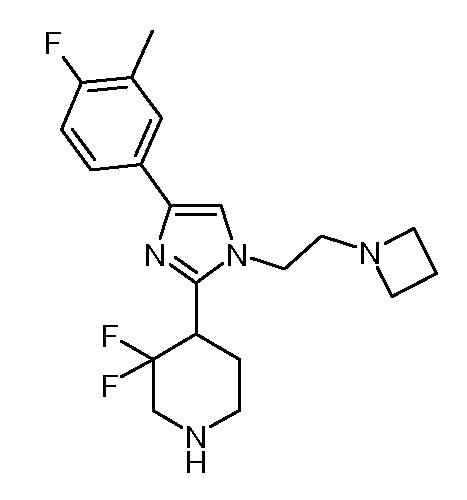
Стадия 1: 1-трет-бутиловый эфир-4-[2-(4-фтор-3-метилфенил)-2-оксоэтиловый] эфир 3,3-дифторпиперидин-1,4-дикарбоновой кислоты
К раствору 1-трет-бутилового эфира 3,3-дифторпиперидин-1,4-дикарбоновой кислоты (3000,00 мг; 11,31 ммоль; 1,00 экв.) и триэтиламина (1,91 мл; 13,57 ммоль; 1,20 экв.) в ACN (30 мл) добавляли 2-бром-1-(4-фтор-3-метилфенил)этанон (2561,03 мг; 11,08 ммоль; 0,98 экв.). Смесь перемешивали при КТ в течение 30 мин. Реакционный раствор выливали в этилацетат (100 мл) и промывали 5% водным раствором NaHCO3, затем насыщенным раствором соли. Органическую фазу сушили над Na2SO4 и концентрировали, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 416, найдено = 416).
Стадия 2: трет-бутиловый эфир 3,3-дифтор-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-пиперидин-1-карбоновой кислоты
К раствору 1-трет-бутилового эфира-4-[2-(4-фтор-3-метилфенил)-2-оксоэтилового] эфира 3,3-дифторпиперидин-1,4-дикарбоновой кислоты (5000,00 мг; 12,04 ммоль; 1,00 экв.) в диоксане (50 мл) добавляли ацетат аммония (9278,08 мг; 120,36 ммоль; 10,00 экв.). Реакционную смесь перемешивали при 110°C в течение 2 час, добавляли еще 2 г ацетата аммония и перемешивали при 120°C в течение еще 1 час. После охлаждения до КТ реакционную смесь выливали в этилацетат (100 мл) и воду (50 мл). Отделенный органический слой промывали 5% водным раствором NaHCO3, затем насыщенным раствором соли, сушили и концентрировали, получая при этом указанное в заголовке соединение в виде желтого твердого вещества (4796 мг, выход 92%), которое непосредственно применяли для реакции следующей стадии. ЖХ-МС (M+H = 396, найдено = 396).
Стадия 3: трет-бутиловый эфир 3,3-дифтор-4-[4-(4-фтор-3-метилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты
К раствору трет-бутилового эфира 3,3-дифтор-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-пиперидин-1-карбоновой кислоты (4330,00 мг; 10,95 ммоль; 1,00 экв.) в ДМФА (18 мл), добавляли NaH (1094,95 мг; 27,38 ммоль; 2,50 экв.). После перемешивания при КТ в течение 30 мин по каплям добавляли 2-(2-бромэтокси)тетрагидропиран (3,32 мл; 21,90 ммоль; 2,00 экв.). Реакционную смесь перемешивали при КТ на протяжении ночи. Анализ ЖХ-МС показал, что реакция была завершена. Реакционную смесь разбавляли 100 мл этилацетата, промывали насыщенным водным хлоридом аммония и насыщенным раствором соли, сушили над MgSO4 и концентрировали, получая при этом остаток в качестве трет-бутилового эфира 3,3-дифтор-4-{4-(4-фтор-3-метилфенил)-1-[2-(тетрагидропиран-2-илокси)этил]-1H-имидазол-2-ил}пиперидин-1-карбоновой кислоты.
Реакционную смесь указанного выше продукта и гидрата толуол-4-сульфоновой кислоты (3124,45 мг; 16,43 ммоль; 1,50 экв.) в метаноле (30 мл) перемешивали при КТ в течение 2 час. После удаления растворителя остаток растворяли в этилацетате, промывали 10% водным Na2CO3. Органическую фазу сушили и концентрировали. Остаток очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение (1190 мг, выход 24%). ЖХ-МС (M+H = 440, найдено = 440).
Стадия 4: 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин
К раствору трет-бутилового эфира 3,3-дифтор-4-[4-(4-фтор-3-метилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (1200,00 мг; 2,73 ммоль; 1,00 экв.) и этилдиизопропиламина (0,98 мл; 5,46 ммоль; 2,00 экв.) в ТГФ добавляли метансульфонилхлорид (0,32 мл; 4,10 ммоль; 1,50 экв.). Реакционную смесь перемешивали при КТ в течение 3 час и мониторинг завершения мезилирования проводили ЖХ-МС. Добавляли азетидин (1,95 мл; 27,31 ммоль; 10,00 экв.) и затем смесь перемешивали при КТ на протяжении ночи. После удаления растворителя к остатку добавляли DCM (5,0 мл) и затем трифторуксусную кислоту (3113,46 мг; 27,31 ммоль; 10,00 экв.). Смесь перемешивали при КТ в течение 2 час. После концентрирования остаток очищали препаративной ВЭЖХ, получая при этом при этом указанное в заголовке соединение. ЖХ-МС (M+H = 379, найдено = 379).
4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-(трет-бутилдиметилсиланилокси)пиперидин

Стадия 1: 1-трет-бутиловый эфир-4-[2-(4-фтор-3-метилфенил)-2-оксоэтиловый] эфир 4-гидроксипиперидин-1,4-дикарбоновой кислоты
2-Бром-1-(4-фтор-3-метилфенил)этанон (2633,06 мг; 11,40 ммоль; 1,00 экв.) добавляли к раствору 1-трет-бутилового эфира 4-гидроксипиперидин-1,4-дикарбоновой кислоты (2796,0 мг; 11,40 ммоль; 1,00 экв.) и триэтиламина (1,92 мл; 13,67 ммоль; 1,20 экв.) в ацетонитриле (30 мл) при КТ в виде одной порции. Образовавшуюся смесь перемешивали при КТ в течение 100 мин. Реакционную смесь разбавляли 100 мл этилацетата и промывали насыщенным раствором NaHCO3 один раз и насыщенным раствором соли два раза. Органический слой сушили над MgSO4 и затем концентрировали, получая при этом указанное в заголовке соединение в виде светло-коричневого твердого вещества, которое применяли непосредственно для реакции следующей стадии. ЖХ-МС (M+H = 396, найдено = 396)
Стадия 2: трет-бутиловый эфир 4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-гидроксипиперидин-1-карбоновой кислоты
К раствору 1-трет-бутилового эфира-4-[2-(4-фтор-3-метилфенил)-2-оксоэтилового] эфира 4-гидроксипиперидин-1,4-дикарбоновой кислоты (4900,00 мг; 12,39 ммоль; 1,00 экв.) в диоксане (30 мл) добавляли ацетат аммония (9551,99 мг; 123,92 ммоль; 10,00 экв.). Смесь перемешивали при 120°C в течение 2 час. После охлаждения до КТ реакционную смесь разбавляли этилацетатом (100 мл) и промывали насыщенным раствором соли, 5% NaHCO3, затем насыщенным раствором соли. Органическую фазу сушили и концентрировали, получая при этом сырой продукт, который обрабатывали простым эфиром и перемешивали в течение 1 час. Осадок собирали, получая при этом указанное в заголовке соединение (4652 мг, выход 43%). ЖХ-МС (M+H = 376, найдено = 376).
Стадия 3: трет-бутиловый эфир 4-(трет-бутилдиметилсиланилокси)-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты
Смесь трет-бутилового эфира 4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-гидроксипиперидин-1-карбоновой кислоты (1600,00 мг; 4,26 ммоль; 1,00 экв.), 1H-имидазола (870,37 мг; 12,79 ммоль; 3,00 экв.) и трет-бутилхлордиметилсилана (1927,00 мг; 12,79 ммоль; 3,00 экв.) в ДМФА (5 мл) перемешивали при 60°C в течение 10 дней и мониторинг реакции проводили способом ЖХ-МС. Реакционную смесь выливали в воду и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором соли, сушили и концентрировали, получая остаток, который обрабатывали гексаном, получая при этом указанное в заголовке соединение в виде белого твердого вещества (2087 мг, выход 91%). ЖХ-МС (M+H = 490, найдено = 490).
Стадия 4: трет-бутиловый эфир 4-(трет-бутилдиметилсиланилокси)-4-{4-(4-фтор-3-метилфенил)-1-[2-(тетрагидропиран-2-илокси)этил]-1H-имидазол-2-ил}пиперидин-1-карбоновой кислоты
К перемешиваемому раствору трет-бутилового эфира 4-(трет-бутилдиметилсиланилокси)-4-[4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (1,50 г; 3,06 ммоль; 1,00 экв.) в ДМФА (8 мл) добавляли порциями NaH (0,28 г; 7,05 ммоль; 2,30 экв.) при 0°C. Реакционную смесь перемешивали при такой же температуре в течение 30 мин. Затем добавляли 2-(2-бромэтокси)тетрагидропиран (0,70 мл; 4,59 ммоль; 1,50 экв.). Реакционную смесь перемешивали при КТ на протяжении ночи. Реакционный раствор выливали в воду и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором соли, сушили и концентрировали, получая при этом указанное в заголовке соединение (1,9 г), которое непосредственно применяли в реакции следующей стадии. ЖХ-МС (M+H = 618, найдено = 618).
Стадия 5: трет-бутиловый эфир 4-(трет-бутилдиметилсиланилокси)-4-[4-(4-фтор-3-метилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты
К раствору трет-бутилового эфира 4-(трет-бутилдиметилсиланилокси)-4-{4-(4-фтор-3-метилфенил)-1-[2-(тетрагидропиран-2-илокси)этил]-1H-имидазол-2-ил}пиперидин-1-карбоновой кислоты (1,9 г, 3,06 ммоль) в метаноле (10 мл) добавляли гидрат толуол-4-сульфоновой кислоты (0,87 г; 4,59 ммоль; 1,50 экв.). Смесь перемешивали при КТ в течение 1 час. После удаления растворителя к остатку добавляли этилацетат (100 мл), промывали 5% раствором NaHCO3, затем насыщенным раствором соли, сушили и концентрировали. Сырой продукт обрабатывали простым эфиром и осадок собирали, получая при этом указанное в заголовке соединение в виде светло-желтого твердого вещества (1,64 г). ЖХ-МС (M+H = 534, найдено = 534).
Стадия 6: 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-(трет-бутилдиметилсиланилокси)пиперидин
К раствору трет-бутилового эфира 4-(трет-бутилдиметилсиланилокси)-4-[4-(4-фтор-3-метилфенил)-1-(2-гидроксиэтил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (1600,00 мг; 3,00 ммоль; 1,00 экв.) и этилдиизопропиламина (0,81 мл; 4,50 ммоль; 1,50 экв.) в ТГФ (50 мл) добавляли метансульфонилхлорид (0,31 мл; 4,05 ммоль; 1,35 экв.) при 0°C. Полученную смесь перемешивали при такой же температуре в течение 30 мин и при КТ в течение 2 час. Анализ ЖХ-МС показал, что мезилирование было завершено. Добавляли азетидин (2,14 мл; 29,98 ммоль; 10,00 экв.) и реакционную смесь перемешивали при КТ в течение 7 дней при мониторинге реакции способом ЖХ-МС. После удаления растворителя остаток разбавляли 50 мл этилацетата и промывали насыщенным раствором соли, сушили и концентрировали, получая при этом трет-бутиловый эфир 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-(трет-бутилдиметилсиланилокси)пиперидин-1-карбоновой кислоты (1717 мг).
К указанному выше продукту, растворенному в метаноле (30 мл) и охлажденному на ледяной бане, добавляли 4,0 М HCl в диоксане (5,25 мл; 20,98 ммоль; 7,00 экв.) в течение 10 мин. Полученную смесь перемешивали при КТ в течение 90 мин. После удаления растворителей остаток нейтрализовали 10% раствором Na2CO3 до pH=9 и экстрагировали этилацетатом (50 мл × 3). Объединенный органический слой промывали насыщенным раствором соли, сушили и концентрировали, получая при этом указанное в заголовке соединение в виде желтого масла (1417 мг). ЖХ-МС (M+H = 473, найден. = 473).
Пример 2. Соединения
4-Амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрил (рацемический)(“1”)

К раствору цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидина (130,0 мг; 0,31 ммоль; 1,0 экв.) в ацетонитриле (2 мл) добавляли диизопропилэтиламин (0,08 мл; 0,47 ммоль; 1,5 экв.) с последующим добавлением 4-амино-6-хлорпиримидин-5-карбонитрила (48,5 мг; 0,31 ммоль; 1,0 экв.). Образовавшуюся смесь перемешивали при КТ в течение 3 час при проведении мониторинга способом ЖХ-МС. Осадок собирали фильтрованием, промывали ацетонитрилом, получая при этом чистое указанное в заголовке соединение (белое твердое вещество, 150 мг, выход 90%). ЖХ-МС (M+H = 533, найдено = 533). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,13-8,00 (м, 10H), 7,79 (с, 4H), 7,48 (дд, J=10,7, 8,8 Гц, 4H), 7,25 (с, 7H), 4,34 (дд, J=13,2, 6,3 Гц, 4H), 3,92 (тт, J=14,6, 7,3 Гц, 7H), 3,79 (дд, J=13,1, 3,4 Гц, 3H), 3,71 (д, J=9,9 Гц, 2H), 3,35 (дд, J=8,4, 4,2 Гц, 3H), 3,13 (д, J=7,0 Гц, 10H), 2,98 (д, J=6,5 Гц, 1H), 2,74-2,63 (м, 4H), 2,43 (д, J=7,1 Гц, 1H), 2,20 (с, 5H), 1,95 (д, J=7,0 Гц, 4H), 1,85 (д, J=10,0 Гц, 5H), 1,00-0,89 (м, 15H), 0,72 (д, J=6,9 Гц, 10H).
4-Амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоксамид (рацемический) (“2”)
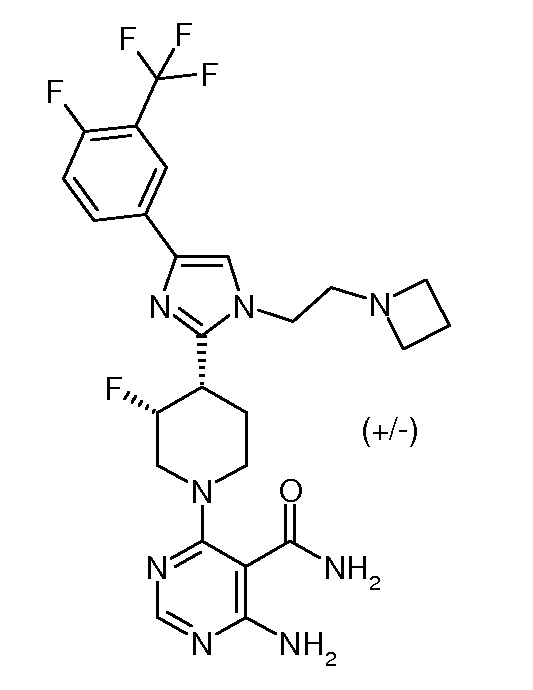
К раствору 4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (50,0 мг; 0,09 ммоль; 1,0 экв.) в ДМСО (5 мл) добавляли пероксид водорода (0,09 мл; 0,94 ммоль; 10,0 экв.) с последующим добавлением 2,0 M гидроксида натрия (0,47 мл; 0,94 ммоль; 10,0 экв.). Реакционную смесь перемешивали при КТ в течение 2 час. Сырой продукт очищали препаративной ВЭЖХ (Waters, основное условие), получая при этом указанное в заголовке соединение в виде белого твердого вещества (41 мг, выход 79%). ЖХ-МС (M+H = 551, найдено = 551).1H ЯМР (400 МГц, ДМСО-d6) δ 8,04 (д, J=1,9 Гц, 3H), 7,79 (д, J=1,7 Гц, 1H), 7,69 (с, 1H), 7,60 (с, 1H), 7,49 (т, J=9,7 Гц, 1H), 6,89 (с, 2H), 5,03-4,78 (м, 1H), 4,38-4,19 (м, 1H), 3,98 (с, 2H), 3,86-3,72 (м, 1H), 3,10 (т, J=10,8 Гц, 2H), 2,93-2,56 (м, 2H), 1,95 (дд, J=27,3, 13,6 Гц, 4H).
4-Амино-6-[(3R,4S)-4-[1-[2-(азетидин-1-ил)этил]-4-[4-фтор-3-(трифторметил)фенил]имидазол-2-ил]-3-фтор-1-пиперидил]пиримидин-5-карбоксамид (хиральный, абсолютная конфигурация неопределенная) (“3”)
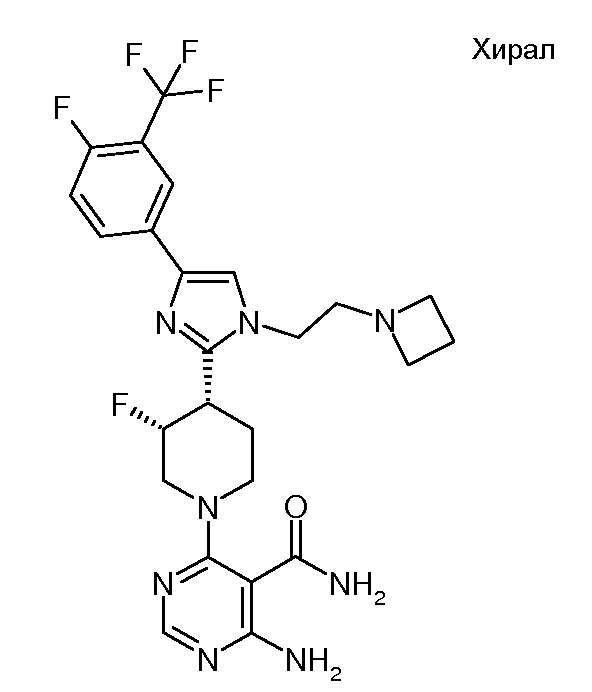
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоксамида. ЖХ-МС (M+H = 551, найдено = 551).
4-Амино-6-[(3S,4R)-4-[1-[2-(азетидин-1-ил)этил]-4-[4-фтор-3-(трифторметил)фенил]имидазол-2-ил]-3-фтор-1-пиперидил]пиримидин-5-карбоксамид (хиральный, абсолютная конфигурация неопределенная) (“4”)

Указанное в заголовке соединение получали хиральным разделением SFC рацемического 4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоксамида. ЖХ-МС (M+H = 551, найдено = 551).
4-Амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-ил}пиримидин-5-карбонитрил (рацемический) (“5”)
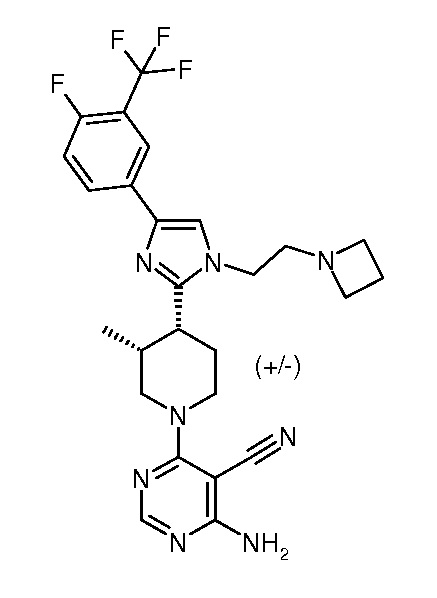
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “1”, с применением рацемического цис-4-(1-(2-(азетидин-1-ил)этил)-4-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-2-ил)-3-метилпиперидина вместо 3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидина. ЖХ-МС (M+H = 529, найдено = 529). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,16-7,98 (м, 6H), 7,79 (с, 2H), 7,48 (дд, J=10,7, 8,8 Гц, 2H), 7,25 (с, 4H), 4,34 (дд, J=13,2, 6,3 Гц, 2H), 3,92 (дт, J=13,3, 7,1 Гц, 4H), 3,79 (дд, J=13,1, 3,4 Гц, 2H), 3,71 (д, J=9,9 Гц, 1H), 3,12 (т, J=7,0 Гц, 8H), 2,72 (дд, J=11,9, 5,9 Гц, 3H), 2,20 (с, 3H), 1,96 (p, J=6,9 Гц, 4H), 1,89-1,73 (м, 3H), 1,01-0,87 (м, 9H), 0,72 (д, J=6,9 Гц, 6H).
4-Амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-ил}пиримидин-5-карбоксамид (рацемический)(“6”)
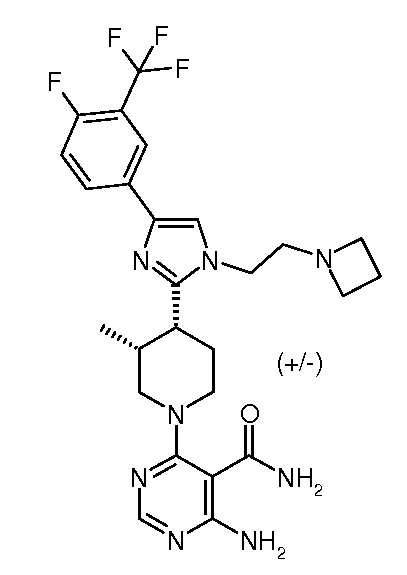
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения 2 с применением 4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-ил}пиримидин-5-карбонитрила. ЖХ-МС (M+H = 547, найдено = 547). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,12-8,01 (м, 4H), 7,98 (с, 2H), 7,77 (с, 2H), 7,48 (дд, J=10,3, 8,0 Гц, 6H), 6,73 (с, 3H), 4,02-3,76 (м, 5H), 3,44 (дд, J=12,8, 3,4 Гц, 2H), 3,26-3,19 (м, 1H), 3,11 (т, J=7,0 Гц, 6H), 2,70 (h, J=6,2 Гц, 3H), 2,24-2,02 (м, 3H), 1,95 (p, J=6,9 Гц, 3H), 1,78 (с, 2H), 0,69 (д, J=6,9 Гц, 4H).
4-Амино-6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрил (рацемический)(“7”)

Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “1” с применением транс-4-[1-(2-азетидин-1-ил-этил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидина. ЖХ-МС (M+H = 533, найден. = 533).
4-Амино-6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоксамид (рацемический) (“8”)
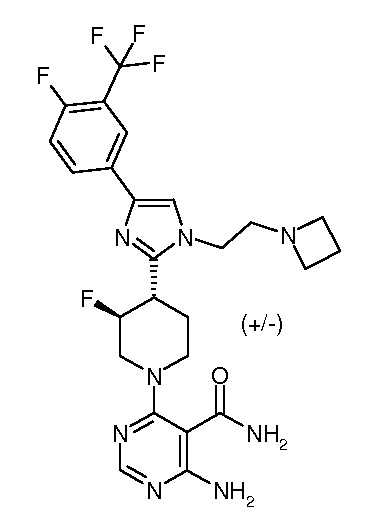
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “2” с применением 4-амино-6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила. ЖХ-МС (M+H = 551, найдено = 551). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,04 (д, J=1,9 Гц, 3H), 7,79 (д, J=1,7 Гц, 1H), 7,69 (с, 1H), 7,60 (с, 1H), 7,49 (т, J=9,7 Гц, 1H), 6,89 (с, 2H), 5,06-4,94 (м, 0,5H), 4,88 (кв., J=7,3, 5,0 Гц, 0,5H), 4,38-4,27 (м, 1H), 3,98 (с, 2H), 3,88-3,71 (м, 2H), 3,10 (т, J=10,8 Гц, 2H), 2,96-2,60 (м, 3H), 1,95 (дд, J=27,3, 13,6 Гц, 4H).
4-Амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-метилфенил)имидазол-2-ил]-3-фтор-1-пиперидил]}пиримидин-5-карбонитрил (рацемический) (“9”)
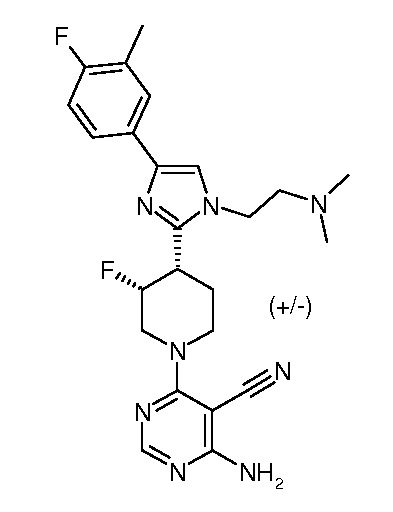
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “1”, с применением рацемического цис-2-(4-(4-фтор-3-метилфенил)-2-(3-фторпиперидин-4-ил)-1H-имидазол-1-ил)-N,N-диметилэтанамина. ЖХ-МС (M+H = 467, найдено = 467).
6-{3,4-цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламин (рацемический) (“10”)
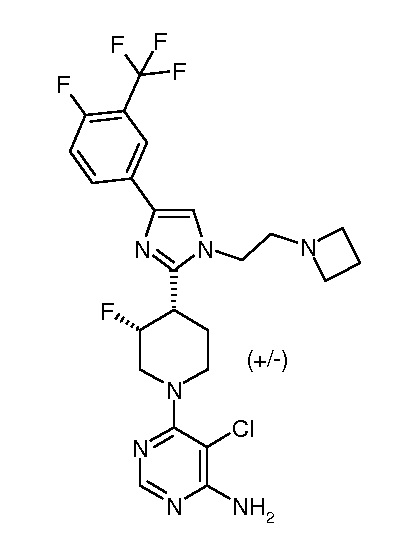
Реакционную смесь 5,6-дихлорпиримидин-4-иламина (28,00 мг; 0,17 ммоль; 1,00 экв.), рацемата цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидина (84,91 мг; 0,20 ммоль; 1,20 экв.) и этилдиизопропиламина (0,06 мл; 0,34 ммоль; 2,00 экв.) в ACN (1,5 мл) перемешивали при 70°C в течение 2 дней. Реакционную смесь концентрировали и очищали ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 542, найдено = 542). H ЯМР (400 МГц, ДМСО-d6) δ 8,13-7,96 (м, 3H), 7,81 (д, J=1,5 Гц, 1H), 7,48 (т, J=9,7 Гц, 1H), 6,82 (с, 2H), 5,13-4,85 (м, 1H), 4,38-4,17 (м, 2H), 3,94 (т, J=6,3 Гц, 2H), 3,53-3,47 (м, 1H), 3,43 (д, J=13,7 Гц, 1H), 3,11 (т, J=6,9 Гц, 5H), 2,74 (д, J=6,6 Гц, 2H), 2,66-2,51 (м, 1H), 1,95 (p, J=7,0 Гц, 2H), 1,80 (д, J=13,9 Гц, 1H).
6-{3,4-транс-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламин (рацемический) (“11”)
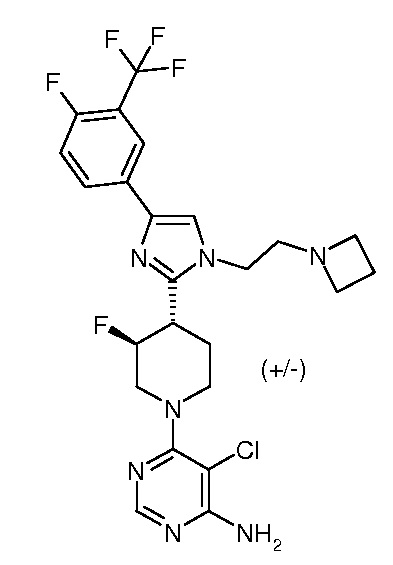
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “10” с применением рацемического транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидина. ЖХ-МС (M+H = 542, найдено = 542). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,05 (кв., J=6,4 Гц, 3H), 7,78 (с, 1H), 7,48 (т, J=9,7 Гц, 1H), 6,92 (с, 2H), 4,95 (дтд, J=48,0, 9,8, 4,6 Гц, 1H), 4,33 (дт, J=11,7, 5,2 Гц, 1H), 4,04 (д, J=13,4 Гц, 1H), 3,93 (т, J=6,4 Гц, 2H), 3,21-2,96 (м, 6H), 2,70 (д, J=7,0 Гц, 2H), 2,02 (т, J=13,5 Гц, 1H), 1,99-1,78 (м, 3H).
5-Хлор-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламин (рацемический) (“12”)
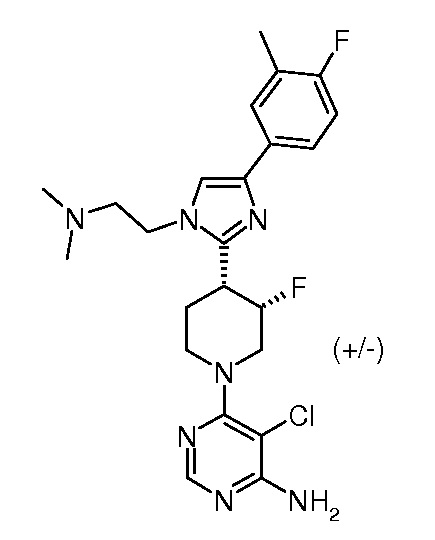
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “10”, с применением рацемического цис-2-(4-(4-фтор-3-метилфенил)-2-(3-фторпиперидин-4-ил)-1H-имидазол-1-ил)-N,N-диметилэтанамина. ЖХ-МС (M+H = 476, найдено = 476).
6-{3,4-цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этилпиримидин-4-иламин (рацемический) (“13”)
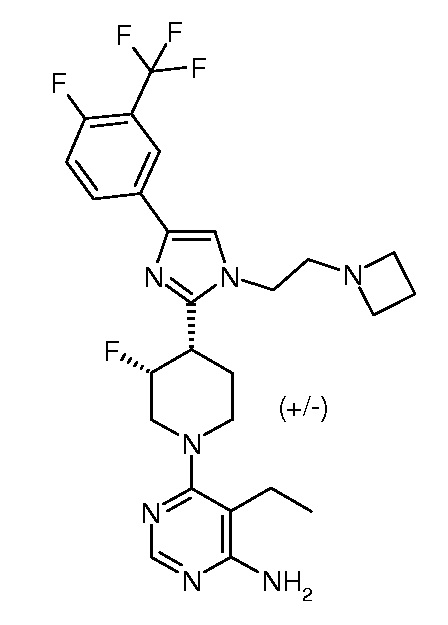
Реакционную смесь 6-хлор-5-этилпиримидин-4-иламина (30,00 мг; 0,19 ммоль; 1,00 экв.), рацемата цис 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидина (78,89 мг; 0,19 ммоль; 1,00 экв.), 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепина (0,05 мл; 0,38 ммоль; 2,00 экв.) и NMP (0,4 мл) в микроволновой пробирке на 5 мл помещали в микроволновое устройство и выдерживали при 135°C в течение 8 час. Реакционную смесь очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 536, найдено = 536). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,12-7,93 (м, 3H), 7,81 (с, 1H), 7,52-7,42 (м, 1H), 6,27 (с, 2H), 5,16-4,90 (м, 1H), 4,04-3,87 (м, 5H), 3,65 (д, J=12,8 Гц, 2H), 3,09 (дт, J=16,3, 7,0 Гц, 4H), 2,80-2,57 (м, 4H), 2,01-1,88 (м, 1H), 1,82 (д, J=13,4 Гц, 1H), 1,12 (т, J=7,3 Гц, 3H).
4-Амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрил (рацемический) (“14”)

Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “1”, с применением 4-амино-6-хлорпиримидин-5-карбонитрила и рацемического цис-2-(4-(4-фтор-3-трифторметилфенил)-2-(3-фторпиперидин-4-ил)-1H-имидазол-1-ил)-N,N-диметилэтанамина. ЖХ-МС (M+H = 521, найдено = 521). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,13-7,96 (м, 5H), 7,84 (с, 1H), 7,47 (т, J=9,7 Гц, 2H), 7,33 (с, 3H), 5,12-4,92 (м, 2H), 4,87 (дд, J=14,4, 9,3 Гц, 2H), 4,71 (д, J=13,6 Гц, 2H), 4,09 (т, J=6,5 Гц, 3H), 3,68 (д, J=14,3 Гц, 1H), 3,59 (д, J=13,8 Гц, 2H), 3,51 (д, J=10,4 Гц, 1H), 3,36 (д, J=12,8 Гц, 1H), 2,63 (гепт., J=5,7 Гц, 3H), 2,47-2,34 (м, 1H), 2,22 (с, 8H), 1,86 (д, J=12,4 Гц, 2H).
4-Амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрил (рацемический) (“15”)
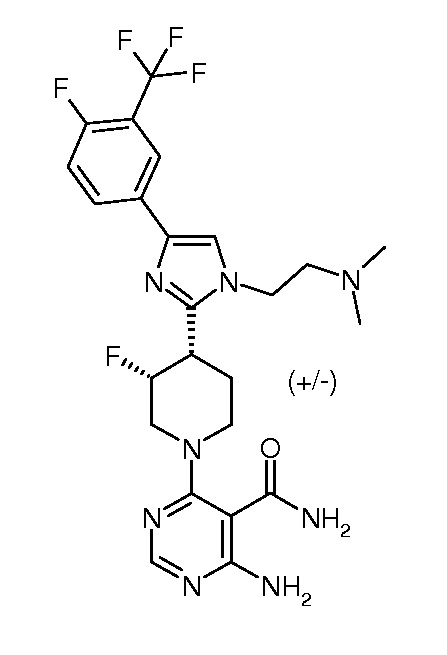
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “2”, с применением 4-амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила. ЖХ-МС (M+H = 539, найдено = 539). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,10-7,97 (м, 3H), 7,82 (с, 1H), 6,89 (с, 2H), 4,96 (д, J=47,6 Гц, 1H), 4,18 (д, J=13,2 Гц, 1H), 4,08 (д, J=6,5 Гц, 2H), 4,03-3,88 (м, 1H), 3,59-3,38 (м, 2H), 3,19 (т, J=11,5 Гц, 1H), 2,60 (гепт., J=6,4 Гц, 2H), 2,21 (с, 6H), 1,83-1,70 (м, 1H), 6,97-6,83 (м, 2H).
6-{3,4-цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламин (рацемический) (“16”)
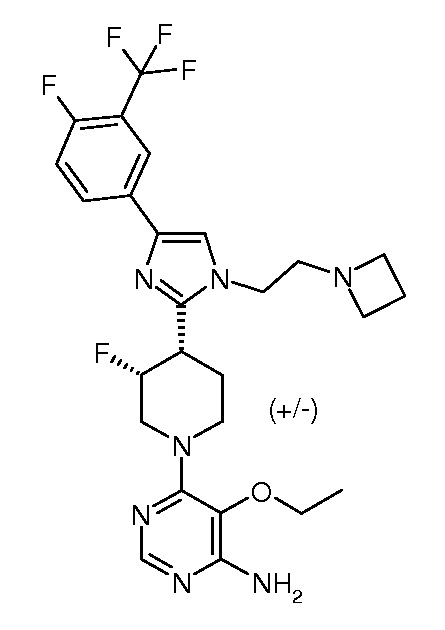
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “13”, с применением 6-хлор-5-этоксипиримидин-4-амина вместо 6-хлор-5-этилпиримидин-4-амина. ЖХ-МС (M+H = 552, найдено = 552). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,11-7,96 (м, 2H), 7,81 (д, J=2,0 Гц, 2H), 7,48 (дд, J=10,7, 8,5 Гц, 1H), 6,19 (с, 2H), 5,01 (д, J=47,9 Гц, 1H), 4,61 (т, J=11,3 Гц, 1H), 4,46 (д, J=13,2 Гц, 1H), 3,95 (кв., J=7,7, 6,4 Гц, 2H), 3,78 (кв., J=7,0 Гц, 2H), 3,59-3,35 (м, 1H), 3,11 (т, J=7,0 Гц, 4H), 2,79-2,68 (м, 2H), 1,95 (p, J=6,9 Гц, 2H), 1,79 (д, J=12,9 Гц, 1H), 1,31 (т, J=7,0 Гц, 3H).
6-{цис-4-[1-(2-Диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин (рацемический) (“17”)
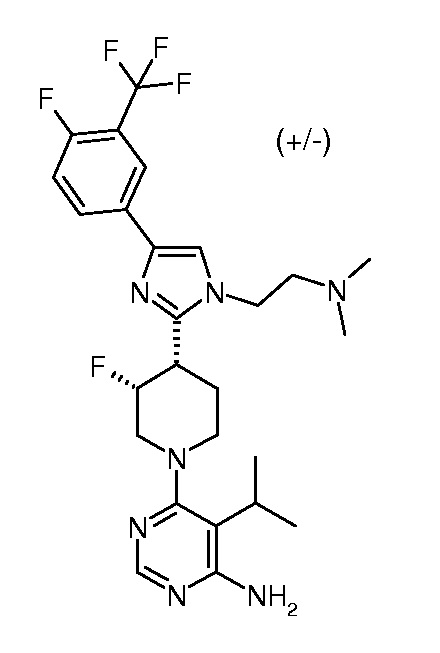
К раствору рацемического цис-3-фторпиперидин-4-ил)-4-(4-фтор-3-трифторметилфенил)имидазол-1-ил]этил}диметиламина (82,06 мг; 0,20 ммоль; 1,00 экв.) в NMP (0,3 мл ) в 5 мл микроволновой ампуле добавляли 6-хлор-5-изопропилпиримидин-4-иламин (35,00 мг; 0,20 ммоль; 1,00 экв.) и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (0,06 мл; 0,41 ммоль; 2,00 экв.). Реакционную смесь помещали в микроволновое устройство и выдерживали при 130°C в течение 12 час. Сырой продукт очищали ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 552, найдено = 552). 1H ЯМР (400 МГц, метанол-d4) δ 8,11-7,94 (м, 10H), 7,61 (д, J=1,1 Гц, 2H), 7,33 (кв., J=9,3 Гц, 2H), 6,22 (д, J=3,1 Гц, 1H), 5,05 (с, 2H), 4,93 (с, 2H), 4,21 (кв., J=7,8, 7,0 Гц, 3H), 3,94 (д, J=3,4 Гц, 1H), 3,65-3,51 (м, 3H), 3,44 (кв., J=9,0, 7,3 Гц, 2H), 3,38 (д, J=6,5 Гц, 1H), 3,24 (д, J=13,6 Гц, 1H), 2,85-2,60 (м, 6H), 2,32-2,26 (м, 3H), 1,95-1,87 (м, 2H), 1,45-1,34 (м, 11H), 1,31 (с, 1H).
4-Амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрил (рацемический) (“18”)
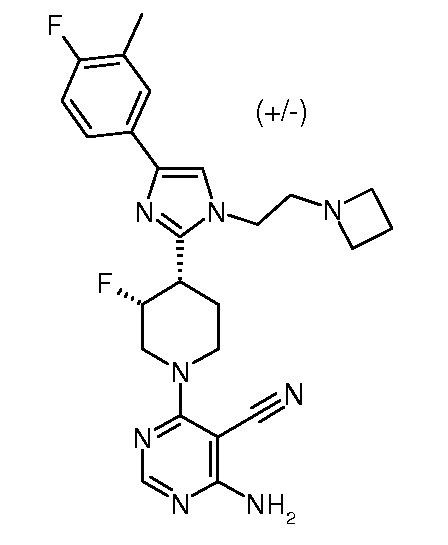
К раствору цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидина (80,00 мг; 0,22 ммоль; 1,00 экв.) в ацетонитриле добавляли этилдиизопропиламин (0,06 мл; 0,33 ммоль; 1,50 экв.) и 4-амино-6-хлорпиримидин-5-карбонитрил (34,30 мг; 0,22 ммоль; 1,00 экв.). Смесь перемешивали при КТ в течение 1 час. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 477, найдено = 477). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,09 (с, 2H), 7,63 (д, J=7,5 Гц, 2H), 7,55 (д, J=11,4 Гц, 4H), 7,33 (с, 3H), 7,07 (т, J=9,2 Гц, 2H), 5,05 (д, J=47,4 Гц, 2H), 4,86 (дд, J=14,3, 9,5 Гц, 2H), 4,71 (д, J=13,7 Гц, 2H), 4,11-3,99 (м, 2H), 3,99-3,85 (м, 3H), 3,71-3,40 (м, 4H), 3,36 (с, 1H), 3,29 (с, 2H), 3,18 (д, J=5,5 Гц, 2H), 3,10 (т, J=7,0 Гц, 7H), 2,71 (кв., J=7,9, 7,1 Гц, 4H), 2,44 (д, J=13,1 Гц, 1H), 2,25 (с, 5H), 1,95 (p, J=7,0 Гц, 4H), 1,90-1,78 (м, 2H), 1,19 (т, J=7,2 Гц, 2H).
Амид 4-амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (рацемический) (“19”)

К раствору 4-амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (70,00 мг; 0,15 ммоль; 1,00 экв.) в ДМСО (8 мл) добавляли H2O2 (0,14 мл; 1,46 ммоль; 10,00 экв.) и 2,0 М водный NaOH (0,73 мл; 1,46 ммоль; 10,00 экв.) при КТ. Образовавшуюся смесь перемешивали при КТ в течение 2 час. Очистка сырого продукта ВЭЖХ (основн.) давала указанное в заголовке соединение. ЖХ-МС (M+H = 497, найдено = 497). 1H ЯМР (400 МГц, метанол-d4) δ 8,06 (д, J=1,2 Гц, 2H), 7,61 (д, J=7,5 Гц, 2H), 7,54 (кв., J=8,1, 6,6 Гц, 1H), 7,40 (с, 1H), 7,00 (т, J=9,0 Гц, 2H), 5,06 (с, 1H), 4,94 (с, 1H), 4,29-4,13 (м, 3H), 4,03 (т, J=6,6 Гц, 3H), 3,59-3,37 (м, 4H), 3,26 (т, J=7,2 Гц, 8H), 2,91-2,79 (м, 3H), 2,77-2,59 (м, 2H), 2,30 (с, 4H), 2,12 (p, J=7,3 Гц, 3H), 1,93-1,84 (м, 2H).
Амид 4-амино-6-{(3S,4R)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (хиральный, абсолютная конфигурация неопределенная) (“20”)
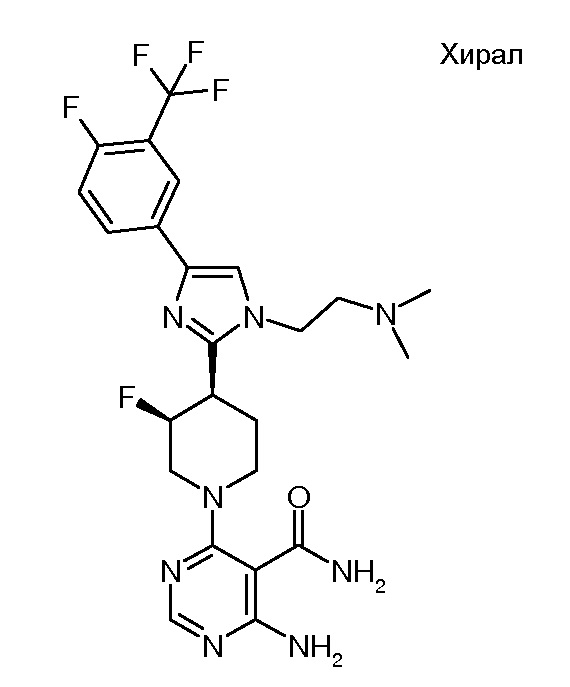
Указанное в заголовке соединение получали хиральным разделением SFC рацемического амида 4-амино-6-{цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты. ЖХ-МС (M+H = 539, найдено = 539).
Амид 4-амино-6-{(3R,4S)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-пиримидин-5-карбоновой кислоты (хиральный, абсолютная конфигурация неопределенная) (“21”)
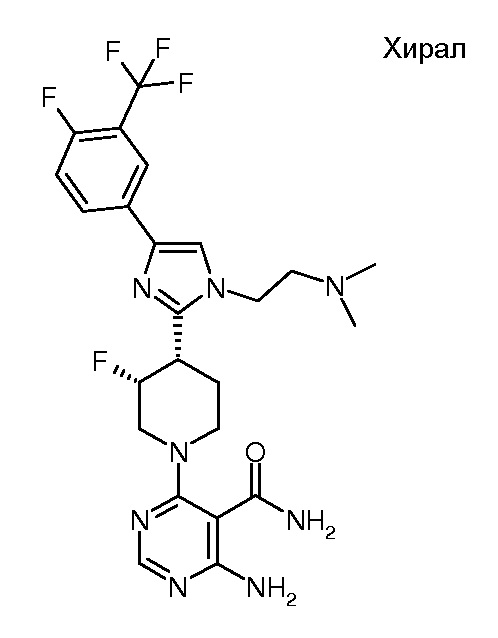
Указанное в заголовке соединение получали хиральным разделением SFC рацемического амида 4-амино-6-{цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты. ЖХ-МС (M+H = 539, найдено = 539).
6-{(3R,4S)-4-[1-(2-Азетидин-1-ил-этил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламин (хиральный, абсолютная конфигурация неопределенная) (“22”)
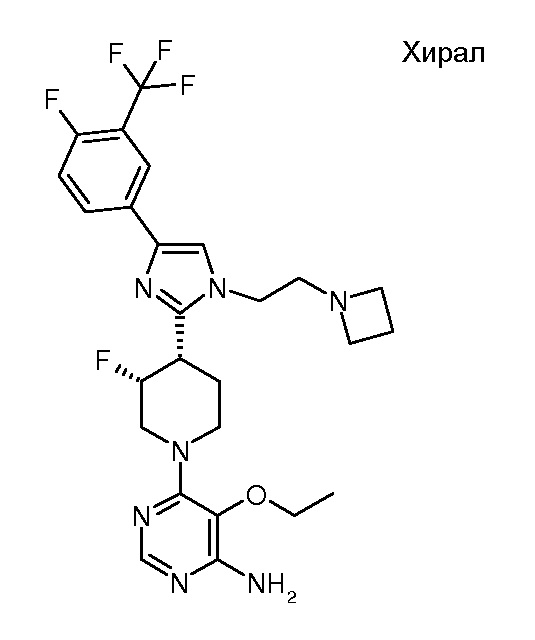
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламина. ЖХ-МС (M+H = 552, найдено = 552).
6-{(3S,4R)-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламин(хиральный, неопределенная абсолютная конфигурация) (“23”)

Указанное в заголовке соединение получали хиральным разделением SFC рацемического 6-{цис-4-[1-(2-азетидин-1-ил-этил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламина. ЖХ-МС (M+H = 552, найдено = 552).
6-{цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемический) (“24”)
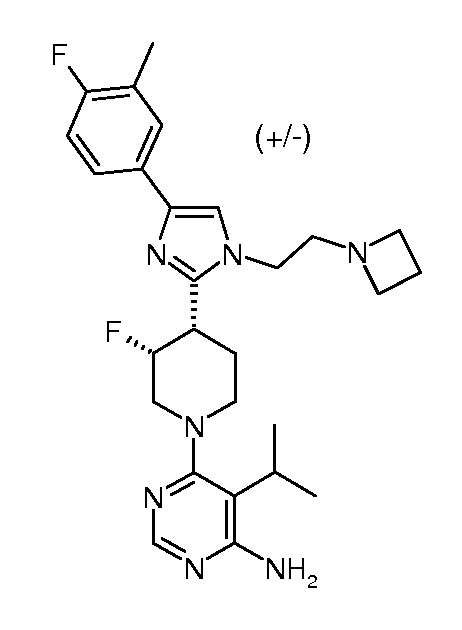
К раствору цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидина (100,81 мг; 0,28 ммоль; 1,00 экв.) в NMP (0,3 мл) добавляли 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (0,08 мл; 0,56 ммоль; 2,00 экв.) и 6-хлор-5-изопропилпиримидин-4-иламин (48,00 мг; 0,28 ммоль; 1,00 экв.). Реакционную смесь помещали в микроволновую печь и выдерживали при 130°C в течение 7 час. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 496, найдено = 496).
5-Хлор-6-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламин(рацемический) (“25”)
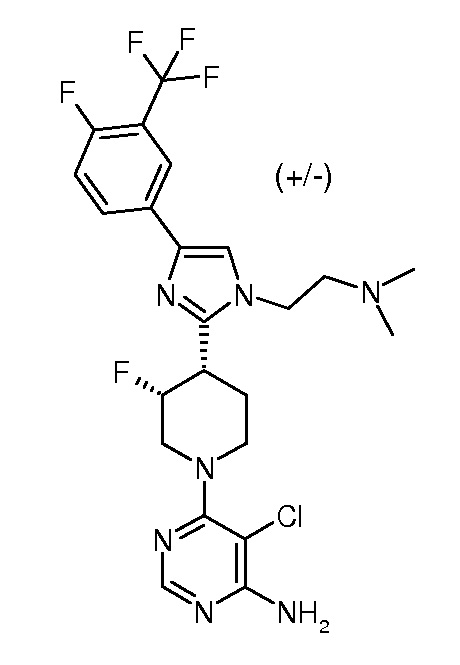
К раствору {2-[2-((цис-3-фторпиперидин-4-ил)-4-(4-фтор-3-трифторметилфенил)-имидазол-1-ил]этил}диметиламина (рацемическому) (82,00 мг; 0,20 ммоль; 1,00 экв.) в ацетонитриле (1 мл) добавляли этилдиизопропиламин (0,05 мл; 0,30 ммоль; 1,50 экв.) и 5,6-дихлорпиримидин-4-иламин (32,60 мг; 0,20 ммоль; 1,00 экв.). Смесь перемешивали при 100°C на протяжении ночи. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 530, найдено = 530). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,11-7,97 (м, 3H), 7,85-7,78 (м, 1H), 7,48 (т, J=9,6 Гц, 1H), 6,83 (с, 2H), 5,07-4,84 (м, 1H), 4,37-4,19 (м, 2H), 4,10 (кв., J=7,7, 6,5 Гц, 2H), 3,51 (д, J=14,0 Гц, 1H), 3,43 (т, J=10,4 Гц, 1H), 3,12 (т, J=12,0 Гц, 1H), 2,62 (тд, J=13,5, 7,0 Гц, 3H), 2,31-2,17 (м, 5H), 2,19-2,11 (м, 1H), 1,80 (д, J=13,1 Гц, 1H).
6-{цис-3-Фтор-4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламин (рацемический) (“26”)
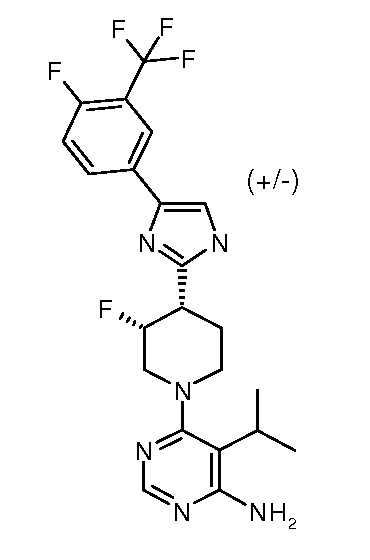
Реакционную смесь 6-хлор-5-изопропилпиримидин-4-иламина (110,00 мг; 0,64 ммоль; 1,00 экв.), цис-3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидина (рацемического) (212,33 мг; 0,64 ммоль; 1,00 экв.) и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепина (0,18 мл; 1,28 ммоль; 2,00 экв.) в NMP (0,4 мл) помещали в микроволновое устройство и выдерживали при 135°C в течение 10 час. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 467, найдено = 467). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,09 (дд, J=7,4, 5,1 Гц, 2H), 8,03 (с, 1H), 7,74 (с, 1H), 7,49 (т, J=10,0 Гц, 1H), 6,19 (с, 2H), 5,09 (д, J=48,0 Гц, 1H), 3,53 (дд, J=14,4, 9,4 Гц, 1H), 3,39 (дд, J=20,8, 10,3 Гц, 2H), 3,24-3,02 (м, 1H), 2,91 (т, J=11,8 Гц, 1H), 2,41-2,28 (м, 1H), 2,00-1,90 (м, 1H), 1,36-1,20 (м, 6H).
Амид 4-амино-6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (хиральный, неопределенная абсолютная конфигурация) (“27”)
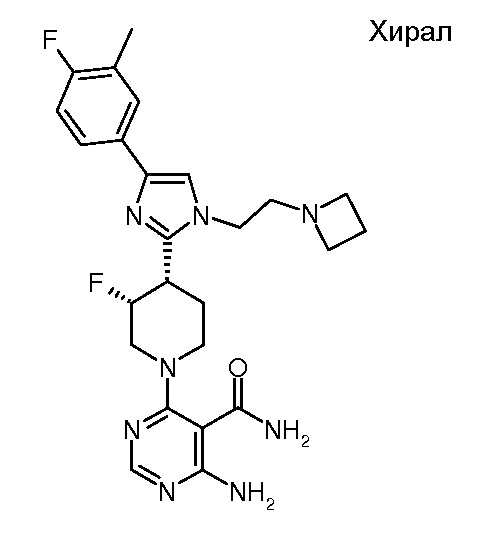
Указанное в заголовке соединение получали хиральным разделением SFC рацемического амида 4-амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты. ЖХ-МС (M+H = 497, найдено = 497).
Амид 4-амино-6-{(3S,4R)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (хиральный, неопределенная абсолютная конфигурация) (“28”).

Указанное в заголовке соединение получали хиральным разделением SFC рацемического амида 4-амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты. ЖХ-МС (M+H = 497, найдено = 497).
5-Хлор-6-{(3R,4S)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламин (хиральный, неопределенная абсолютная конфигурация) (“29”)
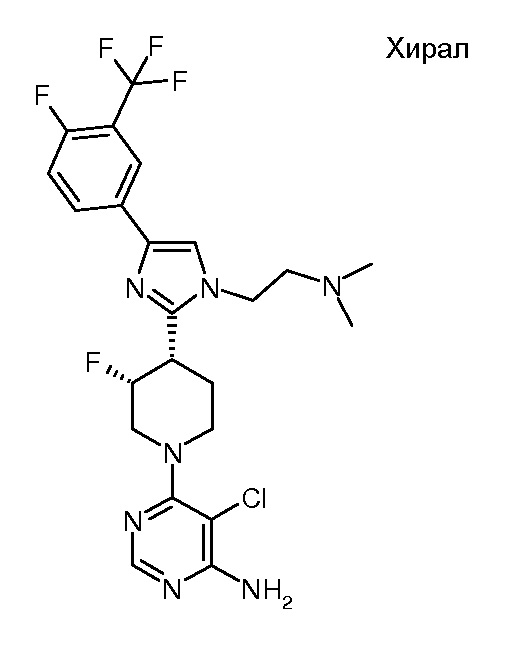
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 5-хлор-6-{цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина. ЖХ-МС (M+H = 530, найдено = 530).
5-Хлор-6-{(3S,4R)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламин (хиральный, неопределенная абсолютная конфигурация) (“30”).
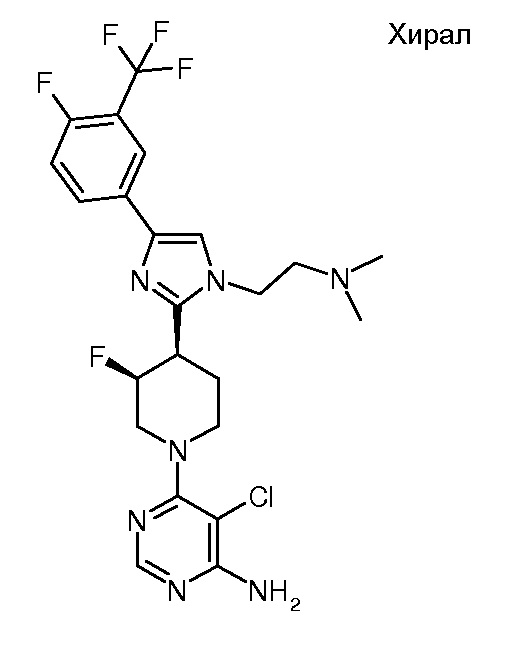
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 5-хлор-6-{цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина. ЖХ-МС (M+H = 530, найдено = 530).
6-{(3S,4R)-4-[1-(2-Диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин (хиральный, неопределенная абсолютная конфигурация) (“31”)
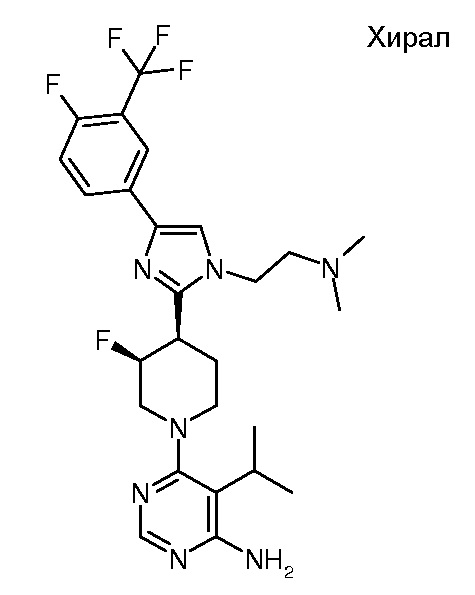
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина. ЖХ-МС (M+H = 538, найдено = 538).
6-{(3R,4S)-4-[1-(2-Диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин (хиральный, неопределенная абсолютная конфигурация) (“32”)
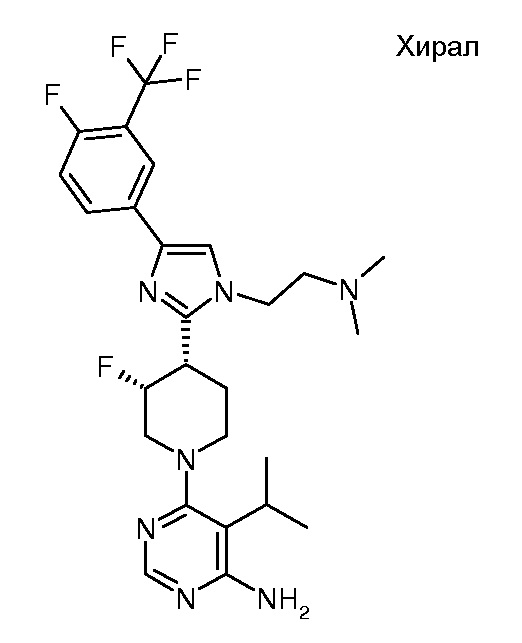
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина. ЖХ-МС (M+H = 538, найдено = 538).
6-{цис-4-[1-(2-Аминоэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин (рацемический) (“33”)

Стадия 1:2-[цис-3-фторпиперидин-4-ил)-4-(4-фтор-3-трифторметилфенил)имидазол-1-ил]этиламин
К раствору трет-бутилового эфира 3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (3000,00 мг; 6,95 ммоль; 1,00 экв.) в ДМФА (10,00 мл; 129,70 ммоль; 18,65 экв.) добавляли NaH (639,72 мг; 15,99 ммоль; 2,30 экв.). После перемешивания при КТ в течение 30 мин добавляли трет-бутиловый эфир (2-бромэтил)карбаминовой кислоты (2805,09 мг; 12,52 ммоль; 1,80 экв.) и смесь перемешивали при КТ в течение 10 дней. Реакционную смесь разбавляли этилацетатом (100 мл) и промывали насыщенным раствором соли. Органическую фазу сушили и концентрировали, получая при этом трет-бутиловый эфир (4-[1-(2-трет-бутоксикарбонилминоэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-карбоновой кислоты.
К указанному выше продукту в DCM (10 мл) добавляли трифторуксусную кислоту (1189,40 мг; 10,43 ммоль; 1,50 экв.). Реакционную смесь перемешивали при КТ в течение 1 час. После удаления растворителя остаток очищали препаративной ВЭЖХ (основн.), получая при этом указанное в заголовке соединение. ЖХ-МС (М+Н = 375, найдено = 375).
Стадия 2: 6-{цис-4-[1-(2-аминоэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин (рацемический)
Смесь 2-[2-цис-3-фторпиперидин-4-ил)-4-(4-фтор-3-трифторметилфенил)имидазол-1-ил]этиламина (80,00 мг; 0,21 ммоль; 1,00 экв.), 6-хлор-5-изопропилпиримидин-4-иламина (34,84 мг; 0,20 ммоль; 0,95 экв.) и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепина (0,06 мл; 0,43 ммоль; 2,00 экв.) в NMP (0,2 мл) помещали в микроволновое устройство и выдерживали в нем при 130°C в течение 11 час. Сырой продукт очищали ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 510, найдено = 510).
6-{(3S,4R)-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин (хиральный, неопределенная абсолютная конфигурация) (“34”)
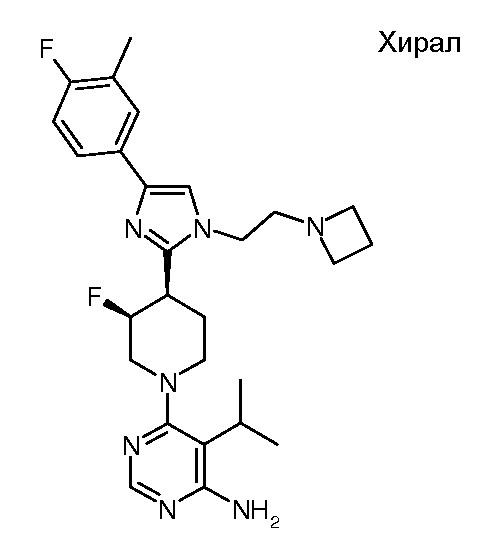
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина. ЖХ-МС (M+H = 496, найдено = 496).
6-{(3R,4S)-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин(хиральный, неопределенная абсолютная конфигурация) (“35”)
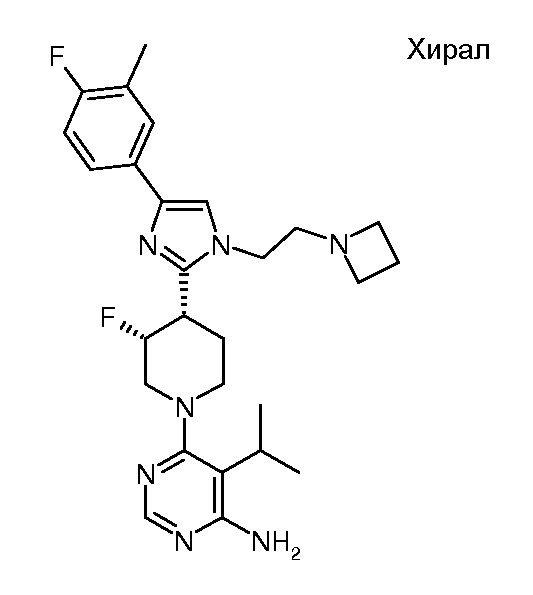
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина. ЖХ-МС (M+H = 496, найдено = 496).
6-{(цис-4-[1-(2-Аминоэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламин (рацемический) (“36”)
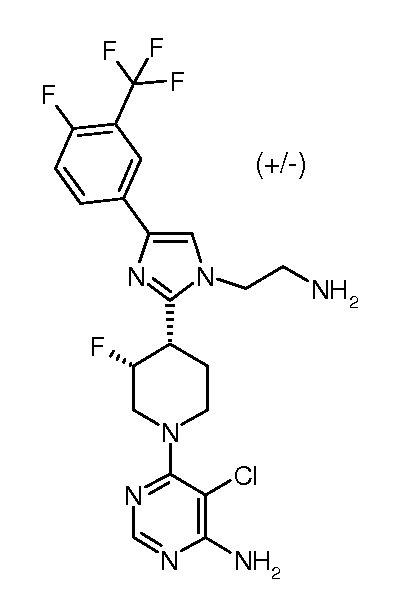
Реакционную смесь 5,6-дихлорпиримидин-4-иламина (35,00 мг; 0,21 ммоль; 1,00 экв.), рацемического 2-[2-(цис-3-фторпиперидин-4-ил)-4-(4-фтор-3-трифторметилфенил)имидазол-1-ил]этиламина (87,89 мг; 0,23 ммоль; 1,10 экв.) и этилдиизопропиламина (0,08 мл; 0,43 ммоль; 2,00 экв.) в ACN (1,5 мл) перемешивали при 70°C в течение 48 час. После удаления растворителя сырой продукт очищали ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 502, найдено = 502).
6-{цис-3-Фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-изопропиламиноэтил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламин (рацемический) (“37”)
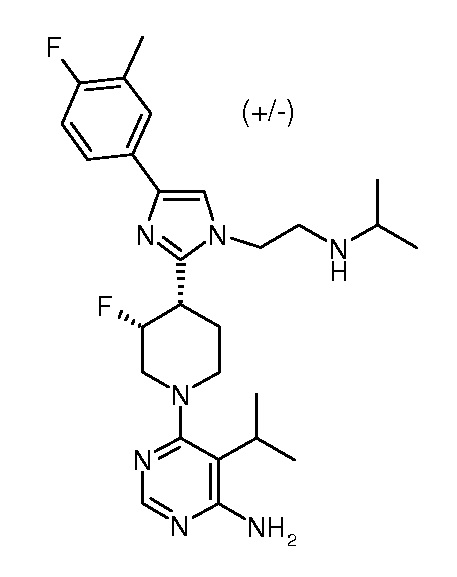
Стадия 1: 2-[4-(4-фтор-3-метилфенил)-2-(цис-3-фторпиперидин-4-ил)имидазол-1-ил]этанол
К раствору рацемического трет-бутилового эфира цис-3-фтор-4-{4-(4-фтор-3-метилфенил)-1-[2-(тетрагидропиран-2-илокси)этил]-1H-имидазол-2-ил}пиперидин-1-карбоновой кислоты (4226,79 мг; 8,36 ммоль; 1,00 экв.) в DCM (15 мл) добавляли трифторуксусную кислоту (11438,82 мг; 100,32 ммоль; 12,00 экв.). Реакционную смесь перемешивали при КТ в течение 1 час. После удаления растворителя остаток очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение (1100 мг, выход 40,9%). ЖХ-МС (M+H = 322, найдено = 322).
Стадия 2: 2-[2-[(цис-1-(6-амино-5-изопропилпиримидин-4-ил)-3-фторпиперидин-4-ил]-4-(4-фтор-3-метилфенил)имидазол-1-ил]этанол
Смесь 2-[4-(4-фтор-3-метилфенил)-2-(цис-3-фторпиперидин-4-ил)-имидазол-1-ил]этанола (337,04 мг; 1,05 ммоль; 1,00 экв.) и 6-хлор-5-изопропилпиримидин-4-иламина (180,00 мг; 1,05 ммоль; 1,00 экв.) в NMP (0,5 мл) помещали в микроволновое устройство и выдерживали при 140°C в течение 10 час. Остаток очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 457, найдено = 457).
Стадия 3: 6-{цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-изопропиламиноэтил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламин (рацемический)
К раствору 2-[2-[цис-1-(6-амино-5-изопропилпиримидин-4-ил)-3-фторпиперидин-4-ил]-4-(4-фтор-3-метилфенил)имидазол-1-ил]этанола (210,00 мг; 0,46 ммоль; 1,00 экв.) и этилдиизопропиламина (0,17 мл; 0,92 ммоль; 2,00 экв.) в ТГФ (1 мл) добавляли метансульфонилхлорид (0,05 мл; 0,69 ммоль; 1,50 экв.). Реакционную смесь перемешивали при КТ в течение 3 час. Анализ ЖХ-МС показал, что реакция мезилирования была завершена. К реакционной смеси добавляли изопропиламин (0,34 мл; 4,60 ммоль; 10,00 экв.) и смесь затем перемешивали при КТ на протяжении ночи и при 40°C в течение еще 24 час. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 478, найдено = 478). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,03 (с, 1H), 7,71-7,61 (м, 1H), 7,57 (д, J=14,3 Гц, 2H), 7,08 (т, J=9,2 Гц, 1H), 6,16 (с, 2H), 5,01 (д, J=47,8 Гц, 1H), 4,00 (т, J=6,6 Гц, 2H), 3,56-3,40 (м, 2H), 3,25-3,08 (м, 1H), 2,93-2,81 (м, 3H), 2,72 (кв., J=6,2 Гц, 1H), 1,90-1,64 (м, 3H), 1,29 (дд, J=19,8, 7,2 Гц, 6H), 0,97 (дд, J=6,3, 2,5 Гц, 6H).
6-{цис-3-Фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-метиламиноэтил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламин (рацемический) (“38”)
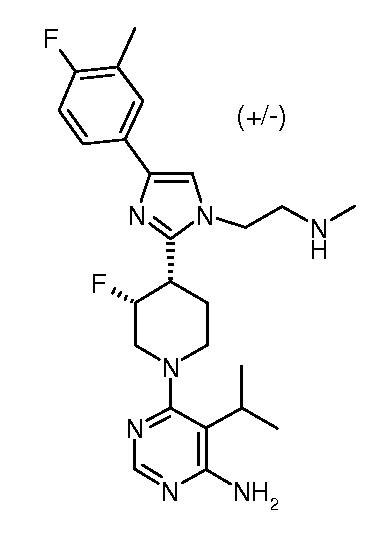
К раствору 2-[2-[цис-1-(6-амино-5-изопропилпиримидин-4-ил)-3-фторпиперидин-4-ил]-4-(4-фтор-3-метилфенил)имидазол-1-ил]этанола (102,00 мг; 0,22 ммоль; 1,00 экв.) и этилдиизопропиламина (0,08 мл; 0,45 ммоль; 2,00 экв.) в ТГФ (1 мл) добавляли метансульфонилхлорид (0,025 мл; 0,336 ммоль; 1,50 экв.). Реакционную смесь перемешивали при КТ в течение 3 час. Анализ ЖХ-МС показал, что реакция мезилирования была завершена. Добавляли метиламин (2,0 M в ТГФ, 1,12 мл; 2,24 ммоль; 10,00 экв.). Реакционную смесь перемешивали при КТ в течение 24 час и затем при 40°C в течение еще 24 час. После удаления растворителя сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 470, найдено = 470).
6-{цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропоксипиримидин-4-иламин (рацемический) (“39”)
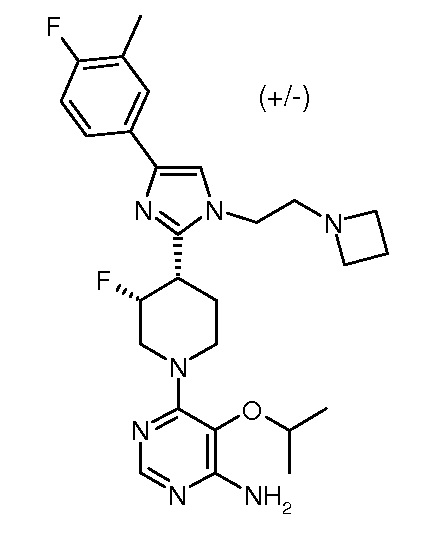
Раствор цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидина (86,45 мг; 0,24 ммоль; 1,00 экв.), 6-хлор-5-изопропоксипиримидин-4-иламина (45,00 мг; 0,24 ммоль; 1,00 экв.) и 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепина (73 мг; 0,48 ммоль; 2,00 экв.) в NMP (0,5 мл) помещали в микроволновое устройство и выдерживали в нем при 140°C в течение 10 час. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 512, найдено = 512). 1H NMR (400 МГц, ДМСО-d6) δ 7,82 (с, 2H), 7,62 (д, J=9,0 Гц, 3H), 7,54 (д, J=9,6 Гц, 5H), 7,07 (т, J=8,9 Гц, 3H), 6,14 (д, J=11,5 Гц, 4H), 5,05 (с, 1H), 4,93 (с, 1H), 4,48 (с, 2H), 4,32 (тд, J=13,8, 12,1, 7,8 Гц, 3H), 3,91 (т, J=6,2 Гц, 3H), 3,09 (кв., J=7,2 Гц, 7H), 3,03 (т, J=7,0 Гц, 2H), 2,67 (дт, J=28,6, 7,7 Гц, 5H), 2,25 (с, 8H), 1,93 (др, J=12,7, 6,7 Гц, 5H), 1,86-1,68 (м, 3H), 1,29-1,10 (м, 13H).
4-Амино-6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрил (хиральный, неопределенная абсолютная конфигурация) (“40”)
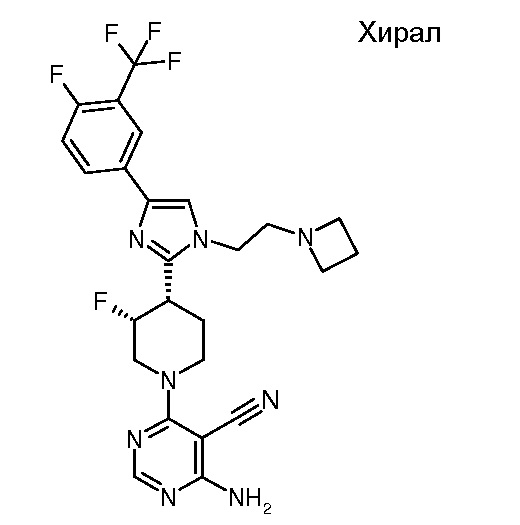
Указанное в заголовке соединение получали хиральным разделением SFC рацемического 4-амино-6-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила. ЖХ-МС (M+H = 533, найдено = 533).
6-{цис-4-[1-Азетидин-3-илметил-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин (рацемический) (“41”)
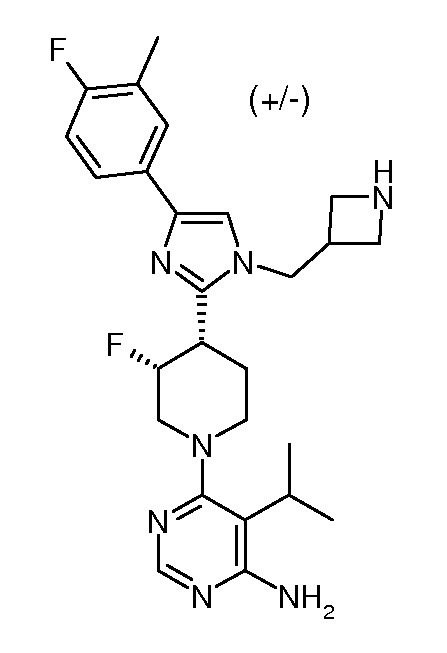
К раствору рацемического трет-бутилового эфира цис-3-[4-(4-фтор-3-метилфенил)-2-(3-фторпиперидин-4-ил)имидазол-1-илметил]азетидин-1-карбоновой кислоты (62,44 мг; 0,14 ммоль; 1,00 экв.) в NMP (0,3 мл), добавляли 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепин (0,04 мл; 0,28 ммоль; 2,00 экв.) и 6-хлор-5-изопропилпиримидин-4-иламин (24,00 мг; 0,14 ммоль; 1,00 экв.). Реакционную смесь помещали в микроволновое устройство и выдерживали в нем при 135°C в течение 19 час. Сырой продукт очищали препаративной ВЭЖХ, получая при этом трет-бутиловый эфир цис-3-[2-1-(6-амино-5-изопропилпиримидин-4-ил)-3-фторпиперидин-4-ил]-4-(4-фтор-3-метилфенил)имидазол-1-илметил]азетидин-1-карбоновой кислоты. ЖХ-МС (M+H = 582, найдено = 582).
К раствору указанного выше соединения в DCM (2 мл) добавляли трифторуксусную кислоту (1 мл). Смесь перемешивали при КТ в течение 2 час. После удаления растворителей остаток очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 482, найдено = 482). 1H ЯМР (400 МГц, метанол-d4) δ 8,06 (д, J=4,4 Гц, 1H), 7,63 (д, J=7,9 Гц, 1H), 7,55 (т, J=6,6 Гц, 1H), 7,36 (с, 1H), 7,01 (т, J=9,0 Гц, 1H), 5,01 (с, 1H), 4,41-4,23 (м, 2H), 3,76 (дкв., J=30,7, 11,4, 9,6 Гц, 2H), 3,66-3,55 (м, 2H), 3,48 (ддт, J=27,4, 11,3, 5,8 Гц, 3H), 3,27-3,15 (м, 2H), 3,09 (т, J=12,3 Гц, 1H), 2,83 (кв., J=13,5, 12,7 Гц, 1H), 1,95-1,86 (м, 1H), 1,38 (дд, J=13,1, 7,2 Гц, 6H).
цис-4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-7H-пирроло[2,3-d]пиримидин-5-карбонитрил (рацемический) (“42”)
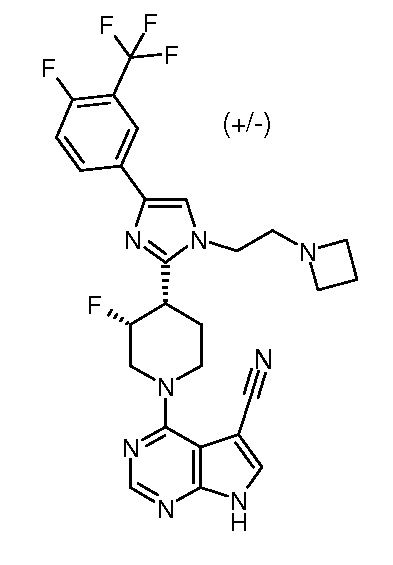
Реакционную смесь 4-хлор-7H-пирроло[2,3-d]пиримидин-5-карбонитрила (24,61 мг; 0,14 ммоль; 1,02 экв.), цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидина (56,00 мг; 0,14 ммоль; 1,00 экв.) и этилдиизопропиламина (22,70 мг; 0,18 ммоль; 1,30 экв.) в ACN (1 мл) перемешивали при 50°C в течение 36 час. После охлаждения до КТ осадок собирали фильтрованием, получая при этом указанное в заголовке соединение в виде желтого твердого вещества. ЖХ-МС (M+H = 557, найдено = 557).
цис-4-[1-(2-Азетидин-1-илэтил)-4-(2-трифторметилпиридин-4-ил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-7H-пирроло[2,3-d]пиримидин-5-карбонитрил (рацемический) (“43”)

Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “42”, с применением цис-4-[1-(2-азетидин-1-илэтил)-2-(3,4)-цис-3-фторпиперидин-4-ил)-1H-имидазол-4-ил]-2-трифторметилпиридина. ЖХ-МС (M+H = 540, найдено = 540).
4-Амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-гидроксипиперидин-1-ил}пиримидин-5-карбонитрил (“44”)

К раствору 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-(трет-бутилдиметилсиланилокси)пиперидина (100,00 мг; 0,21 ммоль; 1,00 экв.) в ацетонитриле (2 мл) добавляли этилдиизопропиламин (0,06 мл; 0,32 ммоль; 1,50 экв.) и затем 4-амино-6-хлорпиримидин-5-карбонитрил (33 мг, 0.21 ммоль, 1,0 экв.). Реакционную смесь перемешивали при КТ на протяжении ночи. После удаления растворителя остаток непосредственно применяли для реакции следующей стадии.
К указанному выше продукту добавляли 2 мл TBAF (1,0 M раствор а ТГФ). Образовавшуюся смесь перемешивали при КТ на протяжении ночи. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 515, найдено = 515). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,06 (с, 1H), 7,67-7,58 (м, 1H), 7,55 (д, J=9,5 Гц, 2H), 7,28 (с, 2H), 7,08 (дд, J=11,7, 6,5 Гц, 1H), 4,32 (дт, J=13,2, 4,0 Гц, 2H), 4,18 (кв., J=6,8 Гц, 2H), 3,72-3,53 (м, 2H), 3,23-3,03 (м, 5H), 2,77 (т, J=6,6 Гц, 2H), 2,28-2,23 (м, 3H), 2,23-2,11 (м, 2H), 1,97 (дд, J=10,9, 4,0 Гц, 4H), 1,62-1,47 (м, 1H), 1,32 (h, J=7,3 Гц, 1H), 0,95 (т, J=7,3 Гц, 2H).
1-(6-Амино-5-изопропилпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-4-ол (“45”)

К раствору 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-(трет-бутилдиметилсиланилокси)пиперидина (100,00 мг; 0,21 ммоль; 1,00 экв.) в ДМСО (1 мл) добавляли 6-хлор-5-изопропилпиримидин-4-иламин (36,31 мг; 0,21 ммоль; 1,00 экв.) и карбонат цезия (103,39 мг; 0,32 ммоль; 1,50 экв.). Смесь перемешивали при 120°C в течение 48 час. Сырой продукт очищали ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 494, найдено = 494). 1H NMR (400 МГц, ДМСО-d6) δ 7,99 (с, 1H), 7,63 (дд, J=7,3, 1,9 Гц, 1H), 7,55 (д, J=4,4 Гц, 2H), 7,08 (дд, J=9,7, 8,5 Гц, 1H), 6,05 (с, 2H), 5,60 (с, 1H), 4,18 (т, J=6,7 Гц, 2H), 3,16 (кв., J=7,9, 7,0 Гц, 6H), 2,78-2,69 (м, 2H), 2,26 (д, J=1,8 Гц, 4H), 2,05-1,85 (м, 4H), 1,28 (д, J=7,2 Гц, 6H).
1-(6-Амино-5-этилпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-4-ол (“46”)
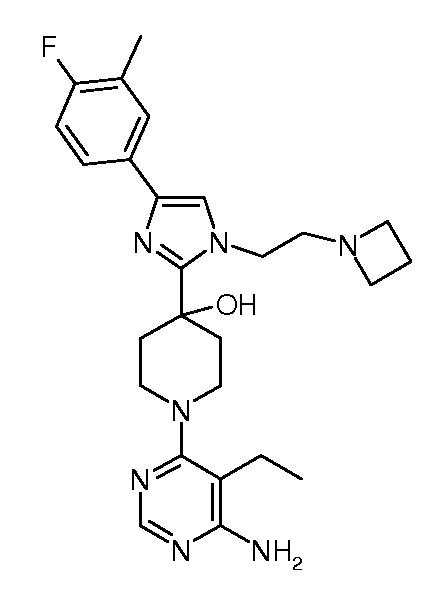
Указанное в заголовке соединение получали согласно процедуре, описанной для получения соединения “45”, с применением 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-(трет-бутилдиметилсиланилокси)пиперидина, который подвергали реакции с 6-хлор-5-этилпиримидин-4-иламином. ЖХ-МС (M+H = 480, найдено = 480). 1H NMR (400 МГц, ДМСО-d6) δ 7,98 (с, 1H), 7,68-7,60 (м, 1H), 7,60-7,47 (м, 2H), 7,08 (дд, J=9,7, 8,5 Гц, 1H), 6,19 (с, 2H), 5,61 (с, 1H), 4,18 (т, J=6,7 Гц, 2H), 3,32-3,19 (м, 4H), 3,15 (т, J=7,0 Гц, 4H), 2,76 (т, J=6,7 Гц, 2H), 2,46 (кв., J=7,1 Гц, 2H), 2,31-2,20 (м, 5H), 2,05-1,85 (м, 4H), 1,11 (т, J=7,3 Гц, 3H).
1-(6-Амино-5-хлорпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-пиперидин-4-ол (“47”)
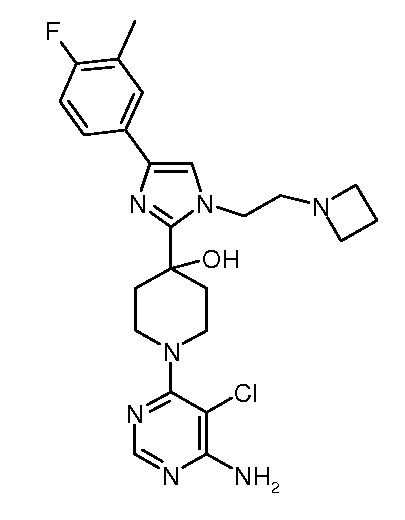
К раствору 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-(трет-бутилдиметилсиланилокси)пиперидина (100,00 мг; 0,21 ммоль; 1,00 экв.) в ацетонитриле (2 мл) добавляли этилдиизопропиламин (0,06 мл; 0,32 ммоль; 1,50 экв.), затем 5,6-дихлорпиримидин-4-иламин (34,69 мг; 0,21 ммоль; 1,00 экв.). Реакционную смесь перемешивали при 100°C в течение 24 час. Сырой продукт очищали ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 486, найдено 486). 1H ЯМР (400 МГц, ДМСО-d6 δ 7,98 (с, 1H), 7,62 (дд, J=7,7, 2,2 Гц, 1H), 7,59-7,48 (м, 2H), 7,08 (т, J=9,1 Гц, 1H), 6,77 (с, 2H), 4,18 (т, J=6,6 Гц, 2H), 3,84 (дт, J=13,2, 4,1 Гц, 2H), 3,25-3,02 (м, 8H), 2,76 (т, J=6,6 Гц, 2H), 2,30-2,15 (м, 5H), 2,06-1,83 (м, 4H), 1,57 (тд, J=10,7, 9,7, 5,9 Гц, 4H), 1,32 (h, J=7,3 Гц, 4H), 0,95 (т, J=7,3 Гц, 5H).
4-Амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}пиримидин-5-карбонитрил (“48”)
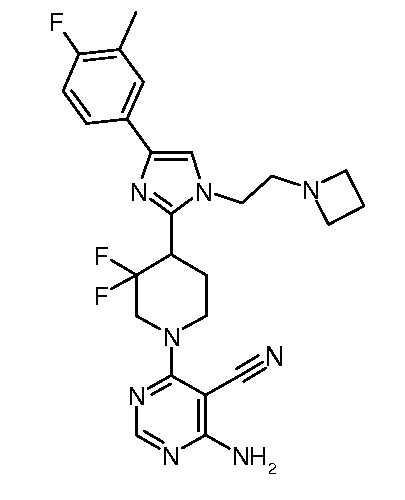
К раствору 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидина (140,00 мг; 0,37 ммоль; 1,00 экв.) в ацетонитриле (3 мл) добавляли этилдиизопропиламин (0,10 мл; 0,55 ммоль; 1,50 экв.) и 4-амино-6-хлорпиримидин-5-карбонитрил (57,18 мг; 0,37 ммоль; 1,00 экв.). Смесь перемешивали при КТ на протяжении ночи. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 497, найдено = 497).1H ЯМР (400 МГц, ДМСО-d6) δ 8,12 (д, J=1,3 Гц, 1H), 7,64 (д, J=9,2 Гц, 2H), 7,61-7,51 (м, 1H), 7,45 (д, J=23,3 Гц, 1H), 7,09 (т, J=9,2 Гц, 1H), 4,90-4,71 (м, 1H), 4,58 (д, J=13,8 Гц, 1H), 4,02-3,81 (м, 4H), 3,58 (т, J=12,1 Гц, 1H), 3,12 (дкв., J=19,6, 6,8 Гц, 4H), 2,69 (дтд, J=18,1, 12,2, 6,4 Гц, 2H), 2,37 (д, J=11,4 Гц, 1H), 2,25 (с, 3H), 2,06 (д, J=14,1 Гц, 1H), 1,95 (кв., J=7,0 Гц, 2H).
Амид 4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}-пиримидин-5-карбоновой кислоты (“49”)

К реакционной смеси 4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}пиримидин-5-карбонитрила (150,00 мг; 0,30 ммоль; 1,00 экв.) в ДМСО (40 мл), перемешиваемой при КТ, добавляли H2O2 (0,29 мл; 3,02 ммоль; 10,00 экв.) и 2,0 M водный раствор NaOH (1,51 мл; 3,02 ммоль; 10,00 экв.). Реакционную смесь перемешивали при КТ в течение 2 час. Сырой продукт очищали препаративной ВЭЖХ (основн.), получая при этом указанное в заголовке соединение (60 мг). ЖХ-МС (M+H = 515, найдено = 515). 1H ЯМР (400 МГц, ДМСО-d6). δ 8,04 (д, J=1,3 Гц, 2H), 7,63 (т, J=6,9 Гц, 9H), 7,55 (т, J=6,7 Гц, 3H), 7,08 (т, J=9,1 Гц, 2H), 6,87 (с, 4H), 4,22-3,99 (м, 5H), 3,99-3,87 (м, 4H), 3,81 (ддт, J=21,1, 10,5, 5,7 Гц, 1H), 3,64 (дд, J=25,7, 13,4 Гц, 2H), 3,11 (дкв., J=19,1, 6,8 Гц, 9H), 2,80-2,58 (м, 4H), 2,35 (д, J=11,4 Гц, 2H), 2,31 (с, 0H), 2,25 (с, 5H), 1,95 (p, J=7,0 Гц, 6H).
6-{4-[1-(2-Азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}-5-изопропилпиримидин-4-иламин (“50”)
Реакционную смесь 6-хлор-5-изопропилпиримидин-4-иламина (55,00 мг; 0,32 ммоль; 1,00 экв.), 4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидина (121,27 мг; 0,32 ммоль; 1,00 экв.), 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепина (0,09 мл; 0,64 ммоль; 2,00 экв.) и NMP (0,4 мл) в 5 мл микроволновой пробирке помещали в микроволновое устройство и выдерживали в нем при 135°C в течение 10 час. Сырой продукт очищали препаративной ВЭЖХ, получая при этом указанное в заголовке соединение. ЖХ-МС (M+H = 514, найдено = 514). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,12-8,02 (м, 1H), 7,71-7,49 (м, 3H), 7,10 (т, J=9,3 Гц, 1H), 6,30 (с, 2H), 3,92 (д, J=7,3 Гц, 2H), 3,71 (д, J=22,8 Гц, 1H), 3,51 (д, J=10,8 Гц, 1H), 3,16-3,03 (м, 4H), 2,96 (т, J=11,5 Гц, 1H), 2,75-2,57 (м, 3H), 2,27 (с, 3H), 2,04-1,84 (м, 3H), 1,29 (дд, J=23,7, 7,2 Гц, 6H).
Биологическая активность
Анализ фермента P70S6K
Соединения-ингибиторы P70S6K разводили и помещали в 96-луночные планшеты. Затем к соединению в планшете для инициирования ферментативной реакции добавляли реакционную смесь, включающую в себя следующие компоненты: P70S6K (3 нМ, мутант T412E, Millipore), смешанный с 24 мкМ АТФ в буфере для анализа, содержащем 100 мМ Hepes (pH 7,5), 5 мМ MgCl2, 1 мМ DTT, 0,015% Brij и 1 мкмМ субстратного пептида FITC-AHA-AKRRRLSSLRA-OH (получен из последовательности рибосомального белка S6, FITC = флуоресцеинизотиоцианат, AHA = 6-аминогексановая кислота). Реакционную смесь инкубировали в течение 90 мин при 25oC перед добавлением 10 мМ EDTA для останавливания реакции. Пропорцию субстрата и продукта (фосфорилированного) пептида анализировали на чипе Caliper Life Sciences Lab Chip 3000, применяя давление 9652,7 Па (1,4 фунт/кв. дюйм), и верхнее и нижнее напряжения - 3000 и - 700 соответственно. Пики продукта разрешали до разрешения пиков субстрата на полученных хроматограммах.
Анализ фермента AKT
Инструмент для обращения с жидкостью TTP Mosquito применяли для помещения 125 нл подходящей концентрации ингибитора в 100% ДМСО (для вычисления кривой доза-ответная реакция) в каждую лунку 384-луночного планшета. К этой реакционной смеси добавляли следующие компоненты до конечного объема 12,5 мкл:
0,1 нг/мкл His-AKT (полноразмерного), (Invitrogen, Part # P2999, Lot # 641228C).
160 мкМ АТФ (Fluka, 02055).
1 мМ DTT (Sigma, D0632).
1 мМ MgCl2 (Sigma, M1028).
1 мкМ субстратный пептид (последовательность FITC-AHA-GRPRTSSFAEG-NH2), синтезированный устройством Tufts Peptide Synthesis.
100 мМ HEPES, pH 7,5 (Calbiochem, 391338).
0,015% Brij-35 (Sigma, B4184).
Реакционную смесь инкубировали в течение 90 мин при 25°C и затем реакцию останавливали добавлением 70 мкл буфера для останавливания реакции (100 мМ HEPES, pH 7,5, 0,015% Brij-35, 10 мМ EDTA (Sigma, E7889)).
Прочтение планшета проводили на Caliper LC 3000 в формате анализа Off-Chip mobility shift, применяя следующие параметры для 12-sipper чип: давление скрининга 15857,9 Па (-2,3 фунт/кв. дюйм), нижнее напряжение -500 и верхнее напряжение -3000. Указанные условия заставляют нефосфорилированный пептидный субстрат и фосфорилированный пептидный продукт разрешаться в виде отдельных пиков, что позволяет проводить непосредственное измерение процента превращения субстрата в продукт. Процентное превращение наносят на диаграмму в зависимости от концентрации ингибитора для получения сигмоидальной кривой доза-ответная реакция, из которой вычисляют IC50.
Величины анализа ингибирования ферментов p70S6K и AKT для соединений, определенные в экспериментальном разделе, представлены в таблице 4.
Ингибирование ферментов p70S6K и AKT соединением, описанным формулой (I)
p70S6K (нМ)
AKT (нМ)
Все патенты, заявки на патенты, публикации патентов, непатентные публикации и любая другая ссылка, цитированная в контексте, таким образом включены в качестве ссылок.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПИРИМИДИНИМИДАЗОЛАМИНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ КИНАЗЫ | 2015 |
|
RU2690679C2 |
| N-(ПИРИД-4-ИЛ)АМИДЫ И N-(ПИРИМИДИН-4-ИЛ)АМИДЫ И ИХ ФАРМАЦЕВТИЧЕСКОЕ И КОСМЕТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2012 |
|
RU2625789C2 |
| СОЕДИНЕНИЯ | 2016 |
|
RU2725616C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK II ТИПА И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2810338C2 |
| КОНДЕНСИРОВАННОЕ 4-ОКСОПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ | 2005 |
|
RU2358969C2 |
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2006 |
|
RU2383545C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-[2-(БЕНЗИМИДАЗОЛ-1-ИЛ)ХИНОЛИН-8-ИЛ]ПИПЕРИДИН-4-ИЛАМИНА | 2004 |
|
RU2323214C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
Изобретение относится к области органической химии, а именно к гетероциклическому соединению формулы (I) или к его фармацевтически приемлемой соли, в которой R1 представляет собой Hal, LA, O(LA), CN, CONH2; RА представляет собой H; каждый R2 представляет собой независимо Hal, OH или A; A представляет собой алкильную группу, имеющую 1 C-атом; R3 представляет собой 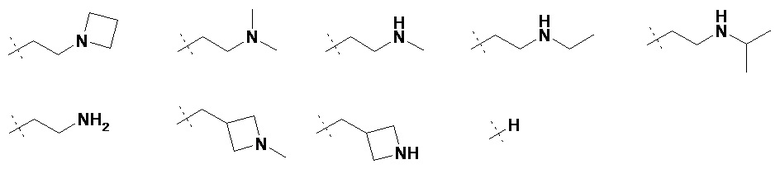 ; R4 представляет собой C6арильное или 6-членное моноциклическое гетероарильное кольцо, имеющее 1 гетероатом, независимо выбранный из атомов азота, каждый из которых является моно- или дизамещенным Hal, LA; LA представляет собой неразветвленную или разветвленную насыщенную линейную углеводородную цепь, имеющую 1, 2, 3 атома C, где 3 атома H могут быть заменены на Hal; Hal представляет собой F, Cl; и n равно 1 или 2. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I), его применению и способу лечения рака. Технический результат: получено новое гетероциклическое соединение, обладающее полезными биологическими свойствами. 4 н. и 14 з.п. ф-лы, 4 табл., 2 пр.
; R4 представляет собой C6арильное или 6-членное моноциклическое гетероарильное кольцо, имеющее 1 гетероатом, независимо выбранный из атомов азота, каждый из которых является моно- или дизамещенным Hal, LA; LA представляет собой неразветвленную или разветвленную насыщенную линейную углеводородную цепь, имеющую 1, 2, 3 атома C, где 3 атома H могут быть заменены на Hal; Hal представляет собой F, Cl; и n равно 1 или 2. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I), его применению и способу лечения рака. Технический результат: получено новое гетероциклическое соединение, обладающее полезными биологическими свойствами. 4 н. и 14 з.п. ф-лы, 4 табл., 2 пр.
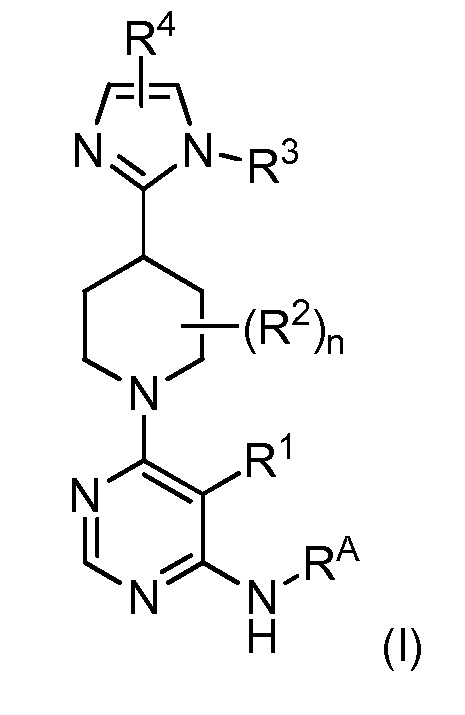
1. Соединение формулы (I)
 ,
,
в которой
R1 представляет собой Hal, LA, O(LA), CN, CONH2;
RА представляет собой H;
каждый R2 представляет собой независимо Hal, OH или A;
A представляет собой алкильную группу, имеющую 1 C-атом;
R3 представляет собой
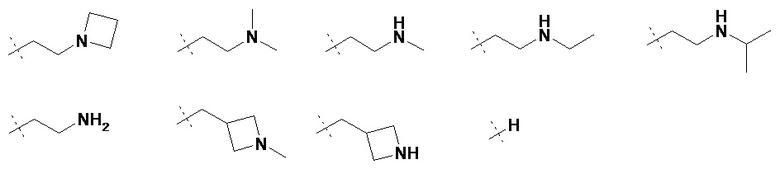 ;
;
R4 представляет собой C6арильное, или 6-членное моноциклическое гетероарильное кольцо, имеющее 1 гетероатом, независимо выбранный из атомов азота, каждый из которых является моно- или дизамещенным Hal, LA;
LA представляет собой неразветвленную или разветвленную насыщенную линейную углеводородную цепь, имеющую 1, 2, 3 атома C, где 3 атома H могут быть заменены на Hal;
Hal представляет собой F, Cl; и
n равно 1 или 2;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где R1 представляет собой CONH2, Hal, O(LA).
3. Соединение по п.2, где R1 выбирают из таблицы 1
Таблица 1: заместители для R1 в формуле (I):
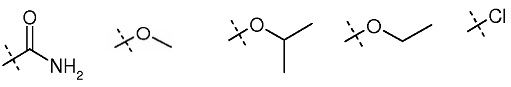
4. Соединение по п.1, где каждый R2 представляет собой независимо Hal, OH, метил.
5. Соединение по п.4, где каждый R2 представляет собой независимо F, OH или метил.
6. Соединение по п.1, где R3 представляет собой H.
7. Соединение по п.1, где R4 представляет собой C6арильное или 6-членное моноциклическое гетероарильное кольцо, имеющее 1 гетероатом, независимо выбранный из атома азота, каждое из которых является моно- или дизамещенным Hal, LA.
8. Соединение по п.7, где R4 выбирают из таблицы 3
Таблица 3: заместители для R4 в формуле (I):
9. Соединение по п.1, формулы (II)
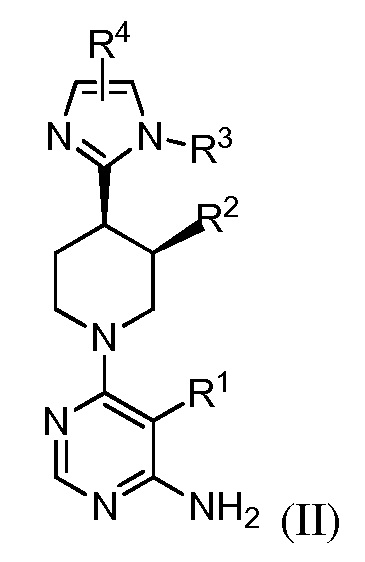
или его фармацевтически приемлемая соль.
10. Соединение по п.1, формулы (III)
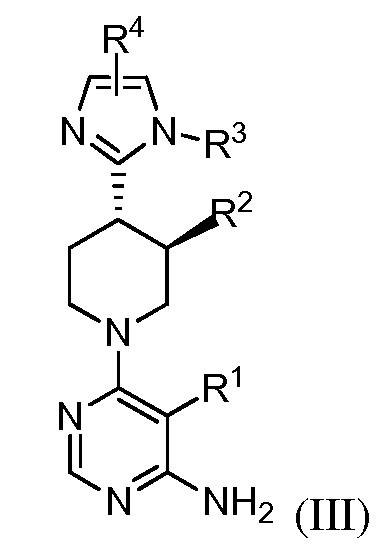
или его фармацевтически приемлемая соль.
11. Соединение по п.1, формулы (IV)
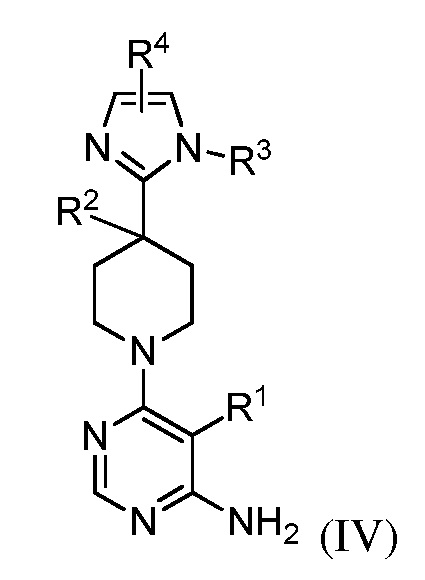
или его фармацевтически приемлемая соль.
12. Соединение по п.1, выбранное из группы, состоящей из
4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“1”);
4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоксамида (рацемического) (“2”);
4-амино-6-[(3R,4S)-4-[1-[2-(азетидин-1-ил)этил]-4-[4-фтор-3-(трифторметил)фенил]имидазол-2-ил]-3-фтор-1-пиперидил]пиримидин-5-карбоксамида (хирального) (“3”);
4-амино-6-[(3S,4R)-4-[1-[2-(азетидин-1-ил)этил]-4-[4-фтор-3-(трифторметил)фенил]имидазол-2-ил]-3-фтор-1-пиперидил]пиримидин-5-карбоксамида (хирального) (“4”);
4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“5”);
4-амино-6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-метилпиперидин-1-ил}пиримидин-5-карбоксамида (рацемического) (“6”);
4-амино-6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“7”);
4-амино-6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоксамида (рацемического) (“8”);
4-амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-метилфенил)имидазол-2-ил]-3-фтор-1-пиперидил]}пиримидин-5-карбонитрила (рацемического) (“9”);
6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламина (рацемического) (“10”);
6-{3,4-транс-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламина (рацемического) (“11”);
5-хлор-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина (рацемического) (“12”);
6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этилпиримидин-4-иламина (рацемического) (“13”);
4-амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“14”);
4-амино-6-{3,4-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“15”);
6-{3,4-цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламина (рацемического) (“16”);
6-{цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“17”);
4-амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (рацемического) (“18”);
амида 4-амино-6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (рацемического) (“19”);
амида 4-амино-6-{(3S,4R)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (хирального) (“20”);
амида 4-амино-6-{(3R,4S)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбоновой кислоты (хирального) (“21”);
6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламина (хирального) (“22”);
6-{(3S,4R)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-этоксипиримидин-4-иламина (хирального) (“23”);
6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“24”);
5-хлор-6-цис-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина (рацемического) (“25”);
6-{цис-3-фтор-4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“26”);
амида 4-амино-6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-пиримидин-5-карбоновой кислоты (хирального) (“27”);
амида 4-амино-6-{(3S,4R)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-пиримидин-5-карбоновой кислоты (хирального) (“28”);
5-хлор-6-{(3R,4S)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина (хирального) (“29”);
5-хлор-6-{(3S,4R)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-4-иламина (хирального) (“30”);
6-{(3S,4R)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (хирального) (“31”);
6-{(3R,4S)-4-[1-(2-диметиламиноэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (хирального) (“32”);
6-{цис-4-[1-(2-аминоэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“33”);
6-{(3S,4R)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (хирального) (“34”);
6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (хирального) (“35”);
6-{(цис-4-[1-(2-аминоэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-хлорпиримидин-4-иламина (рацемического) (“36”);
6-{цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-изопропиламиноэтил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“37”);
6-{цис-3-фтор-4-[4-(4-фтор-3-метилфенил)-1-(2-метиламиноэтил)-1H-имидазол-2-ил]пиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“38”);
6-{цис-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропоксипиримидин-4-иламина (рацемического) (“39”);
4-амино-6-{(3R,4S)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}пиримидин-5-карбонитрила (хирального) (“40”);
6-{цис-4-[1-азетидин-3-илметил-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3-фторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (рацемического) (“41”);
4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-4-гидроксипиперидин-1-ил}-пиримидин-5-карбонитрила (“44”);
1-(6-амино-5-изопропилпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-4-ола (“45”);
1-(6-амино-5-этилпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-4-ола (“46”);
1-(6-амино-5-хлорпиримидин-4-ил)-4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]пиперидин-4-ола (“47”);
4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}пиримидин-5-карбонитрила (“48”);
амида 4-амино-6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}-пиримидин-5-карбоновой кислоты (“49”) и
6-{4-[1-(2-азетидин-1-илэтил)-4-(4-фтор-3-метилфенил)-1H-имидазол-2-ил]-3,3-дифторпиперидин-1-ил}-5-изопропилпиримидин-4-иламина (“50”);
или его фармацевтически приемлемая соль.
13. Фармацевтическая композиция, модулирующая киназную активность, содержащая эффективное количество соединения по любому из пп.1-12 или его фармацевтически приемлемой соли, в качестве активного ингредиента, вместе с фармацевтически приемлемым носителем.
14. Соединение по любому из пп.1-12 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства, модулирующего киназную активность.
15. Соединение по любому из пп.1-12 или его фармацевтически приемлемая соль для применения при лечении рака.
16. Применение соединения по любому из пп.1-12 или его фармацевтически приемлемая соль для получения лекарственного препарата для лечения рака.
17. Способ лечения рака, включающий введение субъекту соединения по любому из пп.1-12 или его фармацевтически приемлемой соли.
18. Способ по п.17, в котором рак выбирают из группы, состоящей из рака головного мозга, легких, ободочной кишки, эпидермоида, клеток простого сквамозного эпителия, мочевого пузыря, желудка, поджелудочной железы, молочной железы, головы, шеи, почки, печени, яичников, простаты, колоректальной кишки, матки, ректальной кишки, пищевода, яичка, женских половых органов, щитовидной железы, меланомы, гематологических злокачественностей, таких как острый миелоидный лейкоз, множественная миелома, хронический миелоидный лейкоз, лейкоз миелоидных клеток, глиома и саркома Капоши.
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| WO 2008075109 A1, 26.06.2008 | |||
| RU 2006133264 A, 10.04.2008. | |||

