ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ДАННОЕ ИЗОБРЕТЕНИЕ
Данное изобретение относится к области лечения поражений сетчатки грудных детей.
УРОВЕНЬ ТЕХНИКИ
Неоваскуляризация и отслоение сетчатки глаза являются ключевыми симптомами ретролентальной фиброплазии (РЛФ). Данным заболеванием с рождения страдают 50-65% недоношенных детей, весящих менее 1250 г. РЛФ является одной из основных причин возникновения детской слепоты по всему миру.
В зависимости от степени тяжести РЛФ, стандартные подходы к лечению данного заболевания включают как наблюдение пациента, так и хирургическое вмешательство. Основной задачей лечения является восстановление функции сетчатки и сохранение зрения. Обычно грудным детям со слабо- или средневыраженным аномальным ростом сосудов не назначается лечения, поскольку у подавляющего большинства таких пациентов заболевание не прогрессирует, и с течением времени самопроизвольно купируется.
Более поздние стадии развития РЛФ, ассоциированные с высокой степенью аномальной неоваскуляризации сетчатки, часто сопровождаются частичным или полным отслоением сетчатки и требуют лечебного воздействия - обычно применяются криотерапия либо лазерная фотокоагуляционная терапия (ЛФТ). Оба варианта терапии приводят к разрушению как минимум части периферической зоны сетчатки.
Недавно было опубликовано клиническое исследование БУАУ-РЛФ (BEAT-ROP), сравнивавшее эффект от интравитреальной монотерапии бевацизумабом с эффектом традиционной ЛФТ на грудных детях, больных РЛФ 3+ стадии (Mintz-Hittner et al. (2011) N Engl J Med. 364(7):603-15). У грудных детей, получавших бевацизумаб интравитреально, было показано значительное улучшение в зоне 1, но не в зоне 2. Периферические сосуды сетчатки продолжили свой рост и после терапии интравитреальным бевацизумабом, а традиционная лазерная фотокоагуляционная терапия вызвала необратимое разрушение периферической зоны сетчатки. В исследовании БУАУ-РЛФ (и подавляющем большинстве более поздних работ) недоношенным новорожденным интравитреально вводилась половина взрослой дозировки бевацизумаба.
Нелицензированный бевацизумаб широко применяется при лечении возрастной дегенерации желтого пятна и других хореоретинальных патологических состояний сетчатки взрослых, несмотря на то, что изначально данный препарат был создан с целью системного использования при лечении колоректального рака. Часто достаточно трудной задачей является предсказание эффектов от применения применяемого для лечения взрослых лекарственного препарата в детской популяции, особенно - среди детей младшего возраста (0-12 лет). Поскольку обычно бевацизумаб при лечении заболеваний глаз вводится интравитреально, высказывались опасения того, что небольшое количество антител антагониста VEGF может попасть в головной мозг, и оказать влияние на нормальное развитие головного мозга ребенка (Sivaprasad et al. (2008) Br J Ophthalmol. 92:451-54). Также высказывались сомнения в отношении системного воздействия антител антагониста VEGF на организм ребенка в случае его применения при лечении детских заболеваний (Lyall et al. (2010) Eye 24: 1730-31).
Таким образом, задачей данного изобретения стало представление улучшенных последующих методик лечения поражений сетчатки грудных детей, решающих хотя бы некоторые из вышеописанных затруднений.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к использованию антагониста VEGF при лечении патологической неоваскуляризации сетчатки, диагностированной у грудных детей. В частности, данное изобретение представляет методику лечения недоношенных новорожденных, страдающих РЛФ, где данная методика заключается во введении в глаз грудного ребенка антагониста VEGF, который либо не проникает в систему кровообращения пациента, либо быстро выводится из нее. антагонист VEGF может вводиться интравитреально, например - посредством инъекций, либо наружно, например - посредством глазных капель.
Пациент
Данное изобретение относится к лечению грудных детей, у которых было диагностировано неоваскулярное поражение сетчатки. Термин “грудной ребенок” здесь и далее используется для обозначения детей раннего возраста с момента рождения и вплоть до возраста в 12 месяцев.
В частности, данное изобретение относится к лечению РЛФ недоношенных детей, либо новорожденных, родившихся раньше срока. Термины “недоношенный ребенок” и “новорожденный, родившийся раньше срока” обычно обозначают грудных детей, родившихся ранее чем на 37 неделе внутриутробного возраста плода.
В ряде случаев определение лечения будет зависеть от гестационного возраста грудного ребенка, проходящего лечение РЛФ. Гестационным возрастом называется количество времени, прошедшее с первого дня последнего менструального цикла и по момент родов (внутриутробный возраст плода) плюс количество времени, прошедшее с момента родов (хронологический возраст). Гестационный возраст обычно записывается в неделях. Например, для родившегося раньше срока новорожденного, родившегося при внутриутробном возрасте плода в 33 недели, возраст которого на данный момент равен 10 неделям (хронологический возраст), постменструальный возраст составит 43 недели.
Антагонист VEGF
VEGF - это детально описанный в литературе сигнальный белок, стимулирующий ангиогенез. К использованию на человеке допущено два антитела - антагониста VEGF, а именно - ранибизумаб (Люцентис®) и бевацизумаб (Авастин®).
Несмотря на то, что ранибизумаб и бевацизумаб имеют сходный иммунный клиренс из глаз в кровоток, ранибизумаб с высокой скоростью экскретируется из системного кровотока; в отличие от него, бевацизумаб сохраняет свой уровень и может вызывать понижение системного уровня VEGF в течение нескольких недель. Если рассмотреть более подробно, время системного полураспада ранибизумаба невелико, и составляет около 2 часов, в то время как время системного полураспада бевацизумаба составляет приблизительно 20 дней. В развивающемся организме, таком как организм грудного ребенка, продолжительное системное подавление VEGF может иметь нежелательное побочное действие на нормальный процесс развития.
Таким образом, в одном из своих аспектов изобретение относится к использованию антагониста VEGF при лечении неоваскулярных поражений сетчатки, где антагонист VEGF либо не попадает в системный кровоток грудного ребенка, либо с большой скоростью выводится из него. В соответствии с изобретением, выведение антагониста VEGF считается достаточно быстрым в том случае, когда время системного полураспада антагониста VEGF составляет от 7 дней до приблизительно 1 часа. При том предпочтительным является время системного полураспада антагониста VEGF менее 7 дней, более предпочтительным является время системного полураспада менее 1 дня, наиболее предпочтительным является время системного полураспада менее 3 часов. Предпочтительным антителом-антагонистом VEGF является ранибизумаб.
Также, в качестве антагониста VEGF может выступать антагонист VEGF, не являющийся антителом. К антагонистам, не являющимся антителами, относятся, например, иммуноадгезины. Одним из таких иммуноадгезинов, обладающих антагонистическим к VEGF действием, является афлиберцепт (Эйлеа®), разрешенный для применения на человеке, и также известный как VEGF-ловушка (Holash et al. (2002) PNAS USA 99:11393-98; Riely & Miller (2007) Clin Cancer Res 13:4623-7s). Афлиберцепт имеет время системного полураспада, равное приблизительно 5-6 дням, и является предпочтительным антагонистом VEGF, не являющимся антителом, для использования с изобретением. Афлиберцепт представляет собой рекомбинантный растворимый гибридный белок-рецептор VEGF человека, состоящий из частей внеклеточных доменов рецепторов VEGF человека 1 и 2, сшитых с Fc-фрагментом IgG1 человека. Это димерный гликопротеин, молекуляная масса белковой части которого равна 97 килодальтон (кДа), и гликозилирующая часть составляет дополнительные 15% от общей молекулярной массы - таким образом, суммарная молекулярная масса равна 115 кДа. Он беспрепятственно производится в форме гликопротеина, посредством экспрессии в рекомбинантных клетках CHO K1. Каждый мономер может иметь следующую аминокислотную последовательность (ИД № ПОСЛ: 1):
SDTGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEKDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPG
и дисульфидные мостики могут образоваться между остатками 30-79, 124-185, 246-306 и 352-410 внутри каждого мономера, и между остатками 211-211 и 214-214 - между мономерами.
Также на этапе доклинической разработки в настоящее время находится другой не являющийся антителом антагонист VEGF - иммуноадгезин, являющийся рекомбинантным растворимым гибридным белком-рецептором VEGF человека, сходный с VEGF-ловушкой, содержащий внеклеточные домены связывания лигандов 3 и 4 из белков VEGFR2/KDR соответственно, и домен 2 из VEGFR1/Flt-1; данные домены сшиты с Fc-фрагментом IgG человека (Li et al. (2011) Molecular Vision 17:797-803). Данный антагонист связывается с изоформами VEGF-A, VEGF-B и VEGF-C. Молекула может быть получена посредством двух раздельных производственных процессов, различающихся характером гликозилирования конечных белков. Данные две гликоформы имеют названия KH902 (конберцепт) и KH906. Гибридный белок может иметь следующую аминокислотную последовательность (ИД № ПОСЛ: 2):
MVSYWDTGVLLCALLSCLLLTGSSSGGRPFVEMYSEIPEIIHMTEGRELVIPCRVTSPNITVTLKKFPLDTLIPDGKRIIWDSRKGFIISNATYKEIGLLTCEATVNGHLYKTNYLTHRQTNTIIDVVLSPSHGIELSVGEKLVLNCTARTELNVGIDFNWEYPSSKHQHKKLVNRDLKTQSGSEMKKFLSTLTIDGVTRSDQGLYTCAASSGLMTKKNSTFVRVHEKPFVAFGSGMESLVEATVGERVRLPAKYLGYPPPEIKWYKNGIPLESNHTIKAGHVLTIMEVSERDTGNYTVILTNPISKEKQSHVVSLVVYVPPGPGDKTHTCPLCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKATPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
И, как и VEGF-ловушка, может быть представлен в виде димера. Данный гибридный белок и относящиеся к нему молекулы более подробно описаны в EP1767546.
Также к не являющимся антителами антагонистам VEGF относятся миметики антител (например, молекулы Аффибоди®, аффилины, аффитины, антикалины, авимеры, пептиды, имеющие в своем составе домен Куница, и монотела), обладающие сходным с антагонистами VEGF характером действия. В силу их малых размеров, миметики антител обычно выводятся из системного кровотока с достаточно большой скоростью (от нескольких минут до нескольких часов). Пегилирование - один из способов повышения продолжительности локального и системного времени полураспада.
Таким образом, термин “не являющиеся антителами антагонисты VEGF” описывает рекомбинантные белки связывания, в состав которых входят домены анкириновых повторов, связывающиеся с VEGF-A и препятствующие его связыванию с VEGFR-2. Примером такой молекулы является DARPin® MP0112. Анкириновый домен связывания может иметь следующую аминокислотную последовательность (ИД № ПОСЛ: 3):
GSDLGKKLLEAARAGQDDEVRILMANGADVNTADSTGWTPLHLAVPWGHLEIVEVLLKYGADVNAKDFQGWTPLHLAAAIGHQEIVEVLLKNGADVNAQDKFGKTAFDISIDNGNEDLAEILQKAA
Рекомбинантные белки связывания, в состав которых входит домен с анкириновым повтором, связывающиеся с VEGF-A и препятствующие его связыванию с VEGFR-2 более подробно описаны в WO2010/060748 и WO2011/135067. Пегилирование повышает продолжительность системного полураспада DARPin® до 1-3 дней.
Также специфичными миметиками антител, обладающими сходным характером действия с антагонистами VEGF, являются пегилированный Антикалин® PRS-050 с молекулярной массой 40 кДа (Mross et al. (2011) Molecular Cancer Therapeutics 10: Supplement 1, Abstract A212) и монотело пегдинетаниб (также известный как Ангиоцепт, или CT-322, см. Dineen et al. (2008) BMC Cancer 8:352).
Вышеописанные, не являющиеся антителами антагонисты VEGF, могут быть модифицированы с целью улучшения их фармакокинетических свойств. Так, например, не являющийся антителом антагонист VEGF может быть химически модифицирован, смешан с биоразлагаемым полимером либо заключен в микрочастицы в целях более длительного интравитреального удержания данного вещества и снижения системного воздействия не являющегося антителом антагониста VEGF.
Различные версии указанных выше антагонистов VEGF, обладающие улучшенными в отношении предпочтительной методики применения свойствами могут быть получены посредством добавления либо удаления аминокислот. Обыкновенно, получаемые варианты аминокислотных последовательностей будут иметь степень аминокислотной идентичности не менее 60% - при сопоставлении с аминокислотными последовательностями ИД № ПОСЛ: 1, ИД № ПОСЛ: 2 либо с ИД № ПОСЛ: 3, предпочтительной при том является степень аминокислотной идентичности не менее 80%, более предпочтительной - не менее 85%, еще более предпочтительной - не менее 90%, и наиболее предпочтительной будет являться степень аминокислотной идентичности не менее 95%; включая в данный перечень, например, степень аминокислотной идентичности, равную 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, и 100%. Идентичность, или гомология, в отношении данной последовательности здесь и далее определяется как процент аминокислотных остатков в кандидатной последовательности, таких же, как в ИД № ПОСЛ: 1, ИД № ПОСЛ: 2 либо с ИД № ПОСЛ: 3 - после выравнивания последовательностей и, по необходимости, включения пропусков с тем, чтобы достичь максимального процента идентичности последовательности, и без учета консервативных замен как составляющей идентичности последовательности.
Идентичность последовательности может быть определена посредством стандартных методов, обыкновенно использующихся для сравнения сходства по позициям аминокислот двух полипептидов. Посредством компьютерной программы, например - BLAST, или FASTA, два полипептида выравниваются таким образом, чтобы достичь оптимального расположения друг относительно друга их соответствующих друг другу аминокислотных остатков (друг против друга сопоставляются одна или обе полные последовательности, либо предопределенные фрагменты одной или обеих последовательностей). Данные программы имеют заданные по умолчанию значения штрафов за открытие пропуска и за каждую дополнительную ячейку пропуска; в сочетании с данными программами возможно использовать стандартные матрицы оценок сопоставления замен, такие как PAM 250 (см. Dayhoff et al. (1978) Atlas of Protein Sequence and Structure, ч. 5, дополн. 3). Так, например, процент идентичности может быть посчитан как: общее количество идентичных совпадений, умноженное на 100, и затем поделенное на сумму длины более длинной из последовательностей внутри сопоставленного фрагмента и количества пропусков, вставленных в более короткие последовательности с тем, чтобы обеспечить выравнивание данных двух последовательностей.
В том случае, когда при реализации данного изобретения используется не являющийся антителом антагонист VEGF, то не являющийся антителом антагонист VEGF связывается с VEGF посредством одного или большего числа белковых доменов, происходящих не от антиген-связывающих доменов антитела. Не являющийся антителом антагонист VEGF предпочтительно является белковоподобным, однако может содержать модификации, не являющиеся белковоподобными (например - пегилирование, гликозилирование). В некоторых вариантах осуществления данного изобретения желательно, чтобы описанный в данном изобретении антагонист VEGF не имел в своем составе Fc-фрагмент антитела, поскольку наличие Fc-фрагмента в ряде случаев повышает время полураспада антагониста VEGF и увеличивает время, в течение которого антагонист VEGF находится в кровотоке.
Пегилирование
В силу меньших размеров, миметики антител обычно выводятся из кровотока с достаточно большой скоростью (от нескольких минут до нескольких часов). Таким образом, в некоторых вариантах осуществления данного изобретения, в частности - в которых антагонист VEGF является миметиком антитела, одна или большее количество молекул полиэтиленгликоля могут быть присоединены к различным позициям молекулы антагониста VEGF.
Данное присоединение может быть обеспечено посредством реакции с аминами, тиолами либо иными подходящими группами реактивов. Тиольная группа может быть представлена в остатке цистеина, и аминогруппа может быть, например, являться первичным амином, находящимся на N-конце полипептида, либо аминогруппа может находится на боковой цепи аминокислоты, такой как, например, лизин либо аргинин.
Присоединение молекул полиэтиленгликоля (ПЭГ) (пегилирование) может быть позиционно-направленным. К примеру, подходящая реакционная группа может быть включена в антагонист VEGF с целью создания позиции, в которой предпочтительно может происходить пегилирование. Так, например, такой миметик антитела, являющийся антагонистом VEGF (например, DARPin® MP0112) может быть модифицирован включением в его состав в нужной позиции остатка цистеина, что позволит провести позиционно-специфичное пегилирование цистеина, например - посредством реакции с дериватом ПЭГ, выполняющим функцию имида малеиновой кислоты. Также возможно, что подходящая реакционная группа может уже изначально присутствовать в антагонисте VEGF.
Частица ПЭГ может иметь широко варьирующуюся молекулярную массу (а именно - от приблизительно 1 кДа до приблизительно 100 кДа) и может быть ветвящейся либо линейной. Предпочтительно, чтобы частица ПЭГ имела молекулярную массу от приблизительно 1 до приблизительно 50 кДа, более предпочтительно - от 10 до приблизительно 40 кДа, еще более предпочтительно - от приблизительно 15 до приблизительно 30 кДа, и наиболее предпочтительной является молекулярная масса, приблизительно равная 20 кДа. Так, например, было показано, что присоединение частицы ПЭГ в 20 кДа дает повышение времени системного полураспада DARPin® вплоть до 20 часов, в то время как частицы ПЭГ больших размеров, 40-60 кДа, дают повышение времени системного полураспада вплоть до 50 часов.
Дозировка
Ранибизумаб обычно вводится взрослым интравитреально, в дозировке 0,5 мг объемом 50мкл. Афлиберцепт также вводится посредством интравитреальной инъекции, и обычно доза для взрослого равна 2 мг. (разведенным в 0,05 мл. буферного раствора, состоящего из 40 мг/мкл на 10 мМ фосфата натрия, 40 мМ хлорида натрия, 0,03% полисорбата 20, и 5% сукрозы, pH 6.2).
Однако, обычная дозировка и/или ее объем могут быть уменьшены при лечении посредством антагониста VEGF детей младшего возраста, и особенно - при лечении посредством антагониста VEGF грудных детей, в силу меньшего интравитреального объема их глаз и повышенного риска, ассоциированного с системным действием антагониста VEGF. Обычно при лечении детей применяется препарат для взрослых, и дозировка просто меняется посредством уменьшения объема, вводимого ребенку. Так, например, в исследовании БУАУ-РЛФ (и в подавляющем большинстве последующих работ на недоношенных новорожденных, больных РЛФ) недоношенным новорожденным вводилась половина взрослой дозировки бевацизумаба.
Однако не всегда удачным решением является изменение дозировки вводимого антагониста VEGF посредством простого уменьшения вводимого количества препарата. Исходя из вышеизложенного, в одном из вариантов осуществления данного изобретения снижается только дозировка антагониста VEGF (например, чтобы уменьшить системное воздействие антагониста VEGF), в то время как вводимый объем остается неизменным. Понижение дозировки может быть обеспечено посредством разведения препарата антагониста VEGF для взрослых посредством смешивания со стерильным буферным раствором (оптимально использование буфера, идентичного тому, в котором антагонист VEGF поставляется в препарате для взрослых). В других вариантах осуществления данного изобретения может вводиться такая же дозировка антагониста VEGF, однако объем вводимого препарата следует уменьшать (поправка на меньший размер глаза грудного ребенка). Предпочтительно, чтобы производилось понижение как дозировки антагониста VEGF, так и объема, в котором она вводится. Например, дозировка и объем препарата могут быть уменьшены пропорционально различиям интравитреального объема глаза, соответственно возрасту ребенка, который будет проходить лечение - в целях обеспечения той же интравитреальной концентрации препарата в глазе, для которой было показано наличие требуемого эффекта у взрослых. Например, 6мг/мкл препарат ранибизумаба является особенно подходящим для формирования дозировок и объемов, адаптированных для различных возрастных групп и особенностей пациентов (например, 0,06 мг, 0,12 мг, 0,18 мг и 0,24 мг в, соответственно, 10 мкл, 20 мкл, 30 мкл и 40 мкл).
Меньшим объемом препарата иногда сложнее манипулировать, и это может приводить к различиям в количестве действительно введенного пациенту антагониста VEGF. Исходя из этого, в некоторых вариантах осуществления данного изобретения, дозировка понижена без сопутствующего уменьшения объема, используемого для введения антагониста VEGF.
Предпочтительным является то, чтобы дозировка, использующаяся при лечении грудного ребенка антагонистом VEGF в соответствии с изобретением составляла менее 50% дозировки, обычно вводимой взрослому (например, менее 40%, предпочтительно - чтобы она составляла менее 30%, и более предпочтительным является, чтобы дозировка составляла менее 20%). Понижение дозировки пропорционально меньшему интравитреальному объему глаза грудного ребенка обычно не является достаточным для предотвращения степени системного воздействия антагониста VEGF, превосходящей определенный на взрослой популяции безопасный максимум. Системное воздействие коррелирует с массой тела пациента. Таким образом, при определении конкретных дозировок, предназначенных для введения грудным детям, возможность недостаточного воздействия, сравнимая с референсным уровнем витреального воздействия на взрослых (пониженный эффект воздействия) должна балансироваться относительно повышенного воздействия на сыворотку крови (повышенного риска). Таким образом, в соответствии с изобретением, дозировка, вводимая грудному ребенку, понижается в большей степени, нежели можно было бы предположить, основываясь на пропорциональном уменьшении в соответствии с пониженным интравитреальным объемом глаза грудного ребенка - с целью обеспечения безопасного уровня системного воздействия антагониста VEGF. Обычно дозировка антагониста VEGF, вводимая грудному ребенку, составляет от 10% до 25% от обычной взрослой дозировки. Например, данная дозировка может быть уменьшена, и достигать от одной четверти до одной восьмой обычной взрослой дозировки (например, одна пятая, одна шестая, либо одна седьмая от обычной взрослой дозировки).
В конкретном варианте осуществления данного изобретения, меньшие дозировки ранибизумаба могут обеспечивать сходные результаты в отношении контроля РЛФ, не вызывая системного подавления уровня VEGF в той же степени, что и прежде применявшиеся способы лечения. Для лечения грудных детей, у которых диагностирована РЛФ, посредством интравитреальных инъекций ранибизумаба предпочтительными являются дозировки менее 0,25 мг, и на каждую дозировку вводится 0,1-0,2 мг ранибизумаба. Так, например, дозировка ранибизумаба может быть снижена до 0,20 мг, предпочтительно снижение дозировки до 0,12 мг, более предпочтительно понижение дозировки до 0,06 мг - посредством введения 30 мкл, 20 мкл либо 10 мкл стандартного раствора ранибизумаба 6 мг/мл. В некоторых случаях для достижения эффективного воздействия могут потребоваться дозировки больших размеров (например, 0,25 мг ранибизумаба в 25 мкл, либо вплоть до 0,24 мг ранибизумаба в 40 мкл). В качестве иного варианта лечения грудные дети, больные РЛФ, могут получать 0,15 мг, предпочтительно - 0,1 мг, более предпочтительно - 0,075г. ранибизумаба. Для достижения данных дозировок, вводится 15 мкл, 10 мкл и 7,5 мкл стандартного раствора ранибизумаба 10 мг/мл соответственно.
Способы введения
Антагонист VEGF, описанный в данном изобретении, обычно будет вводиться посредством интравитреальных инъекций. Обыкновенным является введение в жидкой форме, с обычным объемом введения 5-50 мкл., например – 7,5 мкл, 10 мкл, 15 мкл, 20 мкл, 25 мкл, либо 30 мкл. Инъекции могут проводиться посредством иглы 30-мерок на 125 мм иглы (0,3 мм × 13 мм).
В одной из форм данного изобретения, антагонист VEGF поставляется в виде предварительно наполненного шприца, готового для инъекции. Предпочтительным является, чтобы данный шприц содержал малое количество кремния. Более предпочтительным является, чтобы в составе шприца отсутствовал кремний. Данный шприц может быть изготовлен из стекла. Использование для введения заранее наполненного шприца имеет преимущество в отношении того, что это позволяет избежать любой контаминации стерильного раствора антагониста VEGF до введения. Также предварительно наполненные шприцы удобнее в обращении для вводящего лекарственное средство офтальмолога.
В соответствии с данным изобретением, предварительно наполненный шприц будет содержать подходящую дозировку и объем антагониста VEGF, описанного в данном изобретении. Обычно как дозировка, так и объем лекарственного средства в предварительно наполненном шприце составляют менее 50% от обычной дозировки и объема антагониста VEGF, вводимого взрослому. Обычно объем антагониста VEGF в предварительно наполненном шприце составляет 5-50 мкл, например – 7,5 мкл, 10 мкл, 15 мкл, 20 мкл, 25 мкл, либо 30 мкл. Например, предварительно наполненный шприц может содержать 6 мг/мл препарат ранибизумаба (например, содержащий 0,06 мг, 0,12 мг, 0,18 мг и 0,24 мг в, соответственно, 10 мкл, 20 мкл, 30 мкл, и в 40 мкл). Либо, предварительно наполненный шприц может содержать 10 мг/мл препарат ранибизумаба (например, содержащий 0,2 мг, 0,15 мг, 0,1 мг либо 0,075 мг в, соответственно, 20 мкл, 15 мкл, 10 мкл и 7,5 мкл).
В предпочтительном варианте осуществления данного изобретения, предварительно наполненный лекарственным средством в низкой дозировке шприц имеет номинальный максимальный объем заполнения, равный 0,2 мл, и специально доработан для точного введения объемов менее 50 мкл.
Медленно высвобождающиеся формы
Антагонист VEGF может поставляться в виде медленно высвобождающегося препарата. Медленно высвобождающиеся формы лекарственного средства обычно получаются посредством смешивания терапевтического агента с биоразлагаемым полимером, либо посредством заключения в микрочастицы. Посредством изменения условий производства основанных на полимерах препаратов возможно получать кинетические свойства высвобождения препарата определенного характера. Добавление полимерного носителя также понижает вероятность того, что любое количество антагониста VEGF попадет в кровоток, либо достигнет развивающегося головного мозга ребенка.
Медленно высвобождающаяся лекарственная форма, в соответствии с данным изобретением, обычно состоит из антагониста VEGF, полимерного носителя, и модификатора высвобождения, позволяющего варьировать скорость высвобождения антагониста VEGF из полимерного носителя. Полимерный носитель обычно состоит из одного или большего количества биоразлагаемых полимеров, сополимеров, либо их сочетаний. Так, например, в качестве полимерного носителя можно использовать полилактидную кислоту (ПЛК), полигликолиевую кислоту (ПГК), полилактидкогликолид (ПЛГК), полиэстр, поли(ортоэфир), поли(фосфазин), поли(фосфатный эфир), поликапролактон, либо их комбинацию. Предпочтительным полимерным носителем является ПЛГК. Модификатор высвобождения обычно является длинноцепочечным жирным спиртом, предпочтительно содержащий от 10 до 40 атомов углерода. К использующимся в практике модификаторам высвобождения относятся каприловый спирт, пеларгоновый спирт, каприновый спирт, лауриловый спирт, миристиловый спирт, цетиловый спирт, пальмитолеиновый спирт, стеариловый спирт, изостеариловый спирт, эладиловый спирт, олеиловый спирт, линолеиловый спирт, полиненасыщенный элаилиноиловый спирт, полиненасыщенный линолениловый спирт, элаидолинолениловый спирт, полиненасыщенный рицинолеиловый спирт, арахидиловый спирт, бегениловый спирт, эрициловый спирт, лигноцериловый спирт, цериловый спирт, монтаниловый спирт, клуйтуловый спирт, мелиссиловый спирт, мелиссиловый спирт, и геддиловый спирт.
В одном варианте осуществления данного изобретения антагонист VEGF находится в препарате задержанного высвобождения, основанном на микросферах. Микросферы предпочтительно изготовлены из ПЛГК. Количество антагониста VEGF, присоединенного к микросферам, и скорость высвобождения VEGF возможно менять посредством изменения условий, при которых создаются микросферы. Процессы производства подобного рода препаратов медленного высвобождения подробно описаны в US 2005/0281861 и US 2008/0107694.
Необходимые изменения в дозировке и скорости высвобождения для препарата медленного высвобождения, подходящего для введения грудным детям, могут быть оценены посредством описанных в данном документе моделей воздействия на глаз и системного воздействия.
Режим приема
В соответствии с данным изобретением, антагонист VEGF вводится один или большее количество раз в начале, и впоследствии дополнительно вводится “по необходимости”, в зависимости от эффективности первичного курса лечения. В предпочтительном варианте осуществления данного изобретения, исходный курс ограничивается единичной интравитреальной инъекцией антагониста VEGF. Предпочтительным является, чтобы антагонист VEGF вводился в ходе монотерапии (т.е., без параллельного применения иных методов лечения, таких как лазерная фотокоагуляционная терапия).
Применение в курсе лечения дополнительных инъекций “по необходимости” снижает общее количество инъекций, что позволяет понизить риск потенциального возникновения побочных явлений, которые могут быть вызваны, например, применением общего наркоза, который может потребоваться для безопасного введения антагониста грудным детям.
В некоторых случаях, единичная инъекция антагониста VEGF в соответствии с данным изобретением может быть достаточной для улучшения состояния пациента, либо для предотвращения прогрессирования заболевания. В других случаях, пациенту производится одна инъекция, и необходимость в одной или большем количестве дополнительных инъекций оценивается через 4-16 недель после первой инъекции. Обыкновенно следует избегать повторного лечения ранее, чем через 4 недели после первичной инъекции, чтобы предотвратить повышенный уровень системного воздействия, вызванный накоплением интравитреального антагониста VEGF. В том случае, когда требуется несколько дополнительных инъекций, данные дополнительные инъекции также следует производить с промежутком не менее 4 недель. Лечение может быть прекращено в том случае, когда полностью исчезают все признаки неоваскуляризации сетчатки. Например, лечение может быть прекращено в том случае, когда не менее чем в течение 12-24 недель, например, в течение 16 недель отсутствуют признаки повторной неоваскуляризации сетчатки. В частности, лечение прекращается в том случае, когда на 54 неделе постменструального возраста отсутствуют признаки повторной РЛФ.
Индивидуальный режим введения “по необходимости” определяется по суждению лечащего врача на основании наличествующих повреждений/характере активности заболевания, что оценивается на основании регрессии неоваскуляризации сетчатки с течением времени от первичного момента (т.е., после того, как была введена исходная доза антагониста VEGF), например, начиная с 4 недель, и до 12 месяцев включительно. Так, например, антагонист VEGF вводится грудному ребенку в первый раз после того, как первично поставлен диагноз “патологическая неоваскуляризация сетчатки”. Диагноз нарушения неоваскуляризации сетчатки, такой как, например, РЛФ, может быть поставлен в ходе осмотра глаза при офтальмоскопии.
Оптимально, наличие повреждений/прогрессирование заболевания проверяется еженедельно, не менее одного раза после исходного введения лекарственного средства и вплоть до 16 недель после него, после чего производятся ежемесячные осмотры, вплоть до 12 месяцев включительно. Первичные признаки наличия ответа на лечение посредством антагониста VEGF можно наблюдать уже через 7 дней после первой инъекции; таким образом, ранний осмотр на 7й день после введения исходной дозировки даст возможность раннего повторного лечения, в том случае, Если не наблюдается уменьшение проявлений повреждений/заболевания. Так, например, если первичное интравитреальное введение 0,06 либо 0,075 мг ранибизумаба не приведет к какому-либо улучшению, дополнительная инъекция такой же либо более высокой дозировки (например, вдвое или втрое большей, чем исходная дозировка) может быть произведена уже через 7 дней после первой инъекции.
Более ожидаемым является, что течение болезни будет проверяться каждые 4-6 недель после первичного введения антагониста VEGF. Второе, третье либо последующие введения антагониста VEGF производятся только в том случае, когда осмотр глаза показывает наличие незатухающего либо повторного нарушения неоваскуляризации сетчатки, в особенности - РЛФ. Характеристики течения заболевания (например, характеристики активного ангиогенеза, экссудации либо пропотевания жидкости через сосуды) оцениваются по характеру изменений в сравнении с первичным моментом и по анатомичным конечным критериям оценки с течением времени, начиная с первичного момента (т.е., после того, как была введена первая доза антагониста VEGF), например, начиная с 4й недели, и до 12 месяцев.
Последующие введения антагониста VEGF не считаются необходимыми в тех случаях, когда наблюдается регрессия неоваскуляризации сетчатки, например - при повторном осмотре обнаружено снижение количества новообразованных кровеносных сосудов по сравнению с количеством новообразованных кровеносных сосудов, наблюдавшимся в первичный момент. В случае повторной неоваскуляризации сетчатки, не наблюдается снижения активности образования новых кровеносных сосудов, либо регрессия сочтена недостаточной для предотвращения дальнейшего ущерба сетчатке, производятся вторая, третья либо последующие введения антагониста VEGF.
Комбинированная терапия
Соединения, описанные в данном изобретении, могут вводиться параллельно с одним или большим количеством методик лечения, в особенности - если не наблюдается удовлетворительной реакции на монотерапию пациента посредством антагониста VEGF.
Применение дополнительного лечения (например, ЛФТ либо криотерапия) и введение антагониста VEGF не должны происходить одновременно, т.е., одно из них будет происходить ранее другого. Начало дополнительного лечения и введения антагониста VEGF может происходить в течение 2 и 24 недель, например - в течение 4, 8 либо 16 недель друг относительно друга. Обычно лечение посредством антагониста VEGF применяется до дополнительного лечения.
В некоторых случаях, дополнительное лечение применяется по необходимости. Например, дополнительное лечение может проводиться только в том случае, когда осмотр глаза показывает наличие признаков неснижающейся либо повторной неоваскуляризации после одной или двух (например, двух, трех либо четырех) введений антагониста VEGF. Параметры течения заболевания (например, характеристики активного ангиогенеза, экссудации либо пропотевания жидкости через сосуды) оцениваются по характеру изменений в сравнении с первичным моментом и по анатомичным конечным критериям оценки с течением времени, начиная с первичного момента (т.е., после того, как была введена первая доза антагониста VEGF), например, начиная с 4й недели, и до 12 месяцев, как описано выше.
Например, дополнительное лечение может быть применено в том случае, если течение заболевания сохраняется на прежнем уровне, либо ухудшается на 4 неделе, 6 неделе, 12 неделе либо 16 неделе после исходного введения антагониста VEGF. Ухудшение течения заболевания наблюдается в том случае, когда количество новообразованных сосудов в момент осмотра возросло по сравнению с первичным моментом (т.е., после того, как была введена первая доза антагониста VEGF).
В некоторых случаях, дополнительное лечение производится до введения антагониста VEGF. Например, дополнительное лечение, такое как криоретинопексия, склеральное вдавливание либо витрэктомия могут быть произведены в первую очередь после обнаружения отслоения сетчатки, с тем, чтобы предотвратить потерю зрения. Лечение посредством антагониста VEGF в таком случае производится после дополнительного лечения, с целью предотвращения повторных событий неоваскуляризации сетчатки либо отслоения сетчатки.
В одном из вариантов данного изобретения, лечение посредством описанного в данном изобретении антагониста VEGF может быть использовано в сочетании с ЛФТ в качестве дополнительного лечения.
При ЛФТ лазерное излучение используется для контролируемых повреждений сетчатки с тем, чтобы получить положительный терапевтический эффект. Короткие вспышки лазерного излучения могут закупоривать пропотевающие кровеносные сосуды, уничтожать аномальные кровеносные сосуды, закрывать разрывы роговицы либо уничтожать аномальные ткани на дне глаза. Методики и установки для ЛФТ в настоящее время доступны всем офтальмологам (Lock et al. (2010) Med J Malaysia 65:88-94).
Панретинальная ЛФТ обычно используется для остановки неоваскуляризации у грудных детей, больных РЛФ, посредством нанесения серии ожогов по периферической сетчатке. Обычно используются размеры лазерных пятен 50-500 мкм, со временем свечения 50-200 мс., используя длины волн от желтого к зеленому, например, используя аргоновый газовый лазер (514.5 нм), криптоновый желтый лазер (568.2 нм) либо вариабельный цветной лазер (изменяемая длина волны). В ряде случаев может применяться красный лазер - если зеленый либо желтый лазер исключен (например, в случае наличия кровотечений внутри сетчатки).
Во втором варианте осуществления данного изобретения, лечение посредством описанного в данном изобретении антагониста VEGF может применяться в сочетании с криотерапией в качестве дополнительного лечения.
Криотерапия используется для замораживания и рубцевания периферической сетчатки, что приводит к остановке аномального роста кровеносных сосудов. В ходе криотерапии металлический щуп, который был погружен в криоген (обычно применяется жидкий азот), помещается на склеру.
Общее
Термин “включающий в себя” сосредотачивает в себе как “включает”, так и “состоит из”, например, словосочетание “включающий в себя” X может обозначат то, что в состав входит только X, либо может содержать что-то дополнительное, например, X+Y.
Термин “приблизительно” в отношении к численному значению x является необязательным, и означает, например, x±10%.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
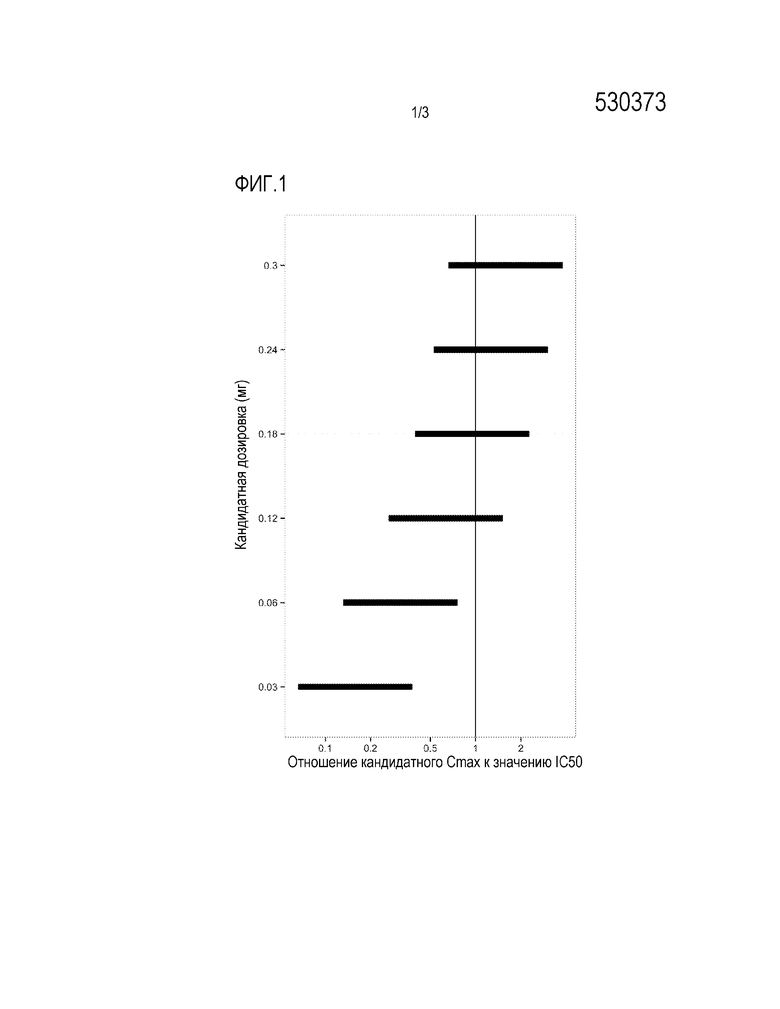
Фигура 1: предсказанные уровни воздействия на максимальную концентрацию ранибизумаба в сыворотке крови (Cmax) для грудных детей, получающих единичные интравитреальные двухсторонние инъекции ранибизумаба в дозировках 0,03-0,3 мг, соответственно референсному in vitro уровню IC50=11 нг/мл. Предсказанные диапазоны воздействия показывают неразрешенную часть предположений модели.
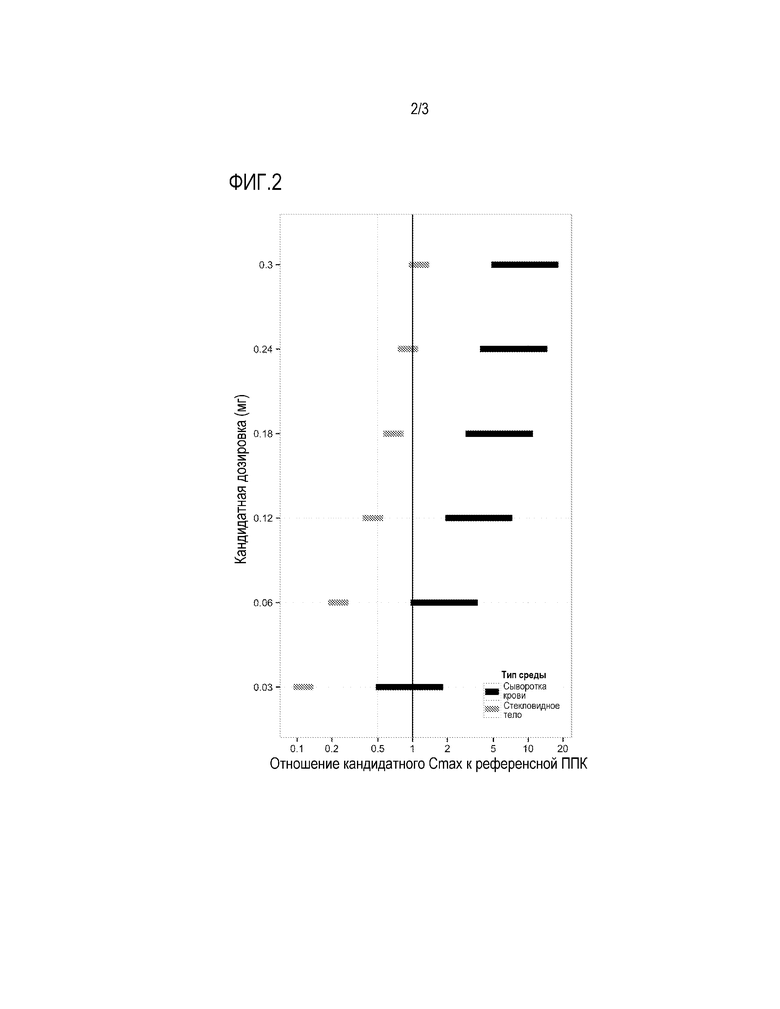
Фигура 2: предсказанные уровни воздействия для площади под кривой (ППК) ранибизумаба в сыворотке крови (черное) и интравитреально (серое) грудных детей, получающих единичные интравитреальные двухсторонние инъекции ранибизумаба в дозировках 0,03-0,3 мг, соответственно референсной ППК для ранибизумаба в сыворотке крови взрослых, получающих единичные интравитреальные инъекции ранибизумаба в один глаз в дозировке 0,5 мг. Предсказанные диапазоны воздействия показывают неразрешенную часть предположений модели.
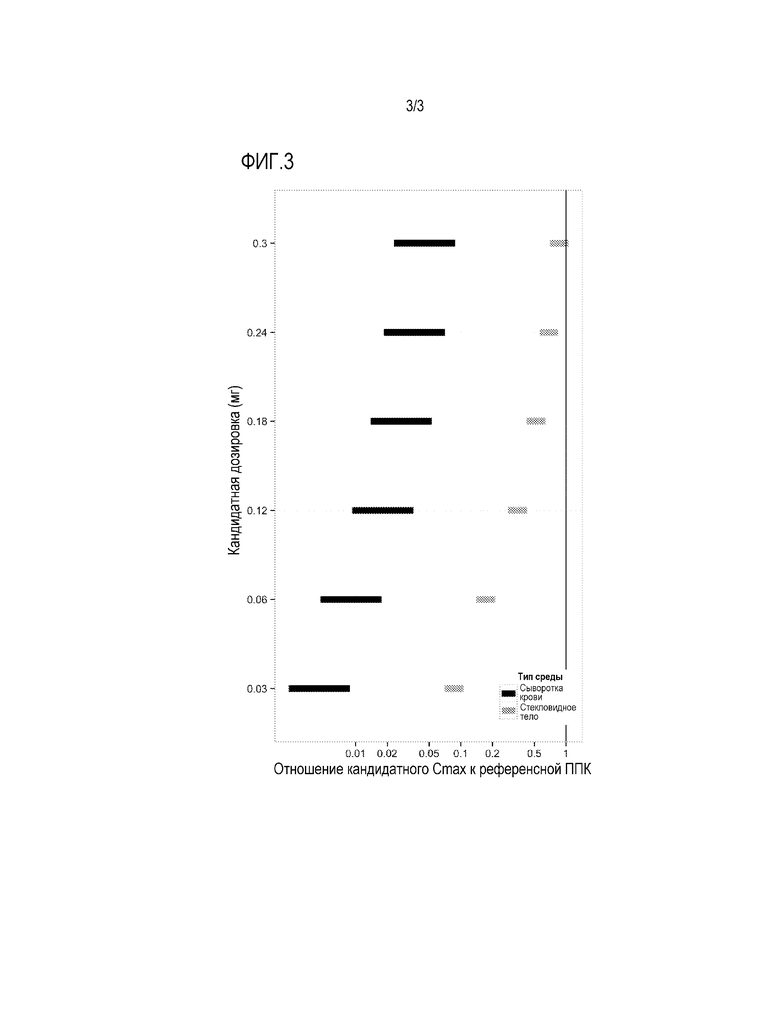
Фигура 3: предсказанные уровни воздействия для (ППК) ранибизумаба в сыворотке крови (черное) и интравитреально (серое) грудных детей, получающих единичные интравитреальные двухсторонние инъекции ранибизумаба в дозировках 0,03-0,3 мг, соответственно референсной ППК для ранибизумаба в сыворотке крови грудных детей, получающих единичные интравитреальные инъекции ранибизумаба в оба глаза в дозировке 0,625 мг. Предсказанные диапазоны воздействия показывают неразрешенную часть предположений модели.
ВАРИАНТЫ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Пример 1.
Фармакокинетическая модель для предсказания глазного и системного воздействия интравитреально введенного грудным детям ранибизумаба.
Чтобы иметь возможность смоделировать глазное и системное воздействие ранибизумаба и бевацизумаба на организм грудных детей, на основании опубликованных данных было утверждено две основных взаимосвязи:
1 - взаимосвязь между возрастом ребенка и глубиной камеры сетчатки и плотностью стекловидного тела, с тем, чтобы предсказать скорость выведения лекарственного средства из глаза и его концентрацию в стекловидном теле.
2 - взаимосвязь между возрастом и весом тела ребенка, и фармакокинетическими параметрами системного характера (аллометрическое шкалирование) с тем, чтобы предсказывать системные концентрации лекарственного средства.
Витреальная концентрация ранибизумаба и бевацизумаба вычислялась, используя объем стекловидного тела. Объем стекловидного тела был посчитан как объем частичной сферы, высота которой равняется глубине стекловидной камеры глазного яблока (ГСК), а диаметр которой равен аксиальной длине (АД) глаза. ГСК и АД недоношенных детей на 10й неделе после рождения скоррелирована с весом тела ребенка при рождении посредством модели линейной регрессии и опубликованных данных (Fledelius (1992) Acta Ophthalmol Suppl 204:10-15). ГСК и АД взрослых скоррелирована с возрастом посредством модели линейной регрессии и опубликованных данных по взрослым (Neelam et al. (2006) Vision Res 46(13):2149-2156). АД глаза вычислялась как соотношение сторон, равное соотношению средних значений АД и ГСК, полученных из вышецитированных работ.
Скорость выведения ранибизумаба и бевацизумаба из глаза человека была посчитана посредством одномерной модели диффузии и конвекции в пористой среде Zhao & Nehorai (2006) IEEE Trans Signal Process 54(6):2213-2225; Dechadilok & Deen (2006) Ind Eng Chem Res 45(21):6953-6959). Согласно данной модели, глаз представляется в виде цилиндра, ось симметрии которого совпадает с передне-задней осью глаза. Передняя грань цилиндра является гиалоидной мебраной глаза, рядом с передней камерой, и задней гранью цилиндра является сетчатка. Длина цилиндра равна ГСК. В дополнение к ГСК, скорость выведения из глаза в данной модели определяется плотностью стекловидного тела. Взаимосвязь между плотностью стекловидного тела и скоростью выведения лекарственного средства из глаза была определена посредством опубликованных данных (Tan et al. (2011) Invest Ophthalmol Vis Sci, 52(2):1111-1118). Взаимосвязь между возрастом и плотностью стекловидного тела была определена на основании опубликованных данных (Oyster (1999) The Human Eye, Sinauer Associates Incorporated, pp. 530-544). Данная модель была дополнительно откалибрована с тем, чтобы привести ее в соответствие с кинетикой в глазе интравитреально вводимого взрослым ранибизумаба и бевацизумаба (популяционная модель фармакокинетики Novartis для ранибизумаба и Zhu et al. (2008) Ophthalmology 115(10):1750-1755).
Системное распространение ранибизумаба и бевацизумаба было описано посредством популяционных моделей фармакокинетики, определенных для каждого из соответствующих антагонистов VEGF, являющихся антителами (популяционная модель фармакокинетики Novartis для ранибизумаба и Lu et al. (2008) Cancer Chemother Pharmacol 62(5):779-786). Системная биологическая доступность бевацизумаба оценивалась посредством опубликованных данных (U.S. Federal Drug Administration (2004) Review and Evaluation of Toxicology Data: Bevacizumab (Avastin), BLA STN#125085; Bakri et al (2007) Ophthalmology 114(5):855-859). Взаимосвязь между массой тела и системной скоростью выведения лекарственного средства моделировалась посредством стандартных методов аллометрического шкалирования (Anderson & Holford (2008) Annu Rev Pharmacol Toxicol 48(1):303-332). Взаимосвязь между возрастом и весом недоношенных новорожденных на момент интравитреальной инъекции была посчитана посредством данных по респределению массы тела ребенка в момент рождения (U.S. Center for Disease Control (2010) http://www.cdc.gov/nchs/VitalStats.htm) и уравнения постнатальной кривой роста (Riddle et al (2006) J Perinatol 26(6):354-358). Масса тела взрослых считалась посредством установленной взаимосвязи между возрастом и параметрами распределения массы тела (Portier et al (2007) Risk Anal 27(1):11-26).
Симуляции модели проводились для типичных пациентов, и позволили нам определить ожидаемое среднее воздействия. Типичным недоношенным новорожденным считался ребенок, рожденный в возрасте 24.2 недель (гестационный возраст), и с массой тела 929 г. Далее, согласно модели, грудному ребенку производилась интравитреальная инъекция ранибизумаба либо бевацизумаба в возрасте 34.5 недель (гестационный возраст) в целях лечения РЛФ, и , на основании типичной кривой роста, ребенок на момент инъекции имел вес 2092 г. Типичный взрослый считался имеющим возраст 70 лет.
Воздействие симулировалось для спектра подобных параметров модели, которые, как ожидалось, оказывают наибольшее влияние на предсказанную степень воздействия. Экспоненты аллометрических шкал взаимосвязей между системной скоростью выведения лекарственного средства, объемом вводимого лекарства и массой тела варьировали между 0,37-0,75 (выведение) и 0,41-1 (объем). Потенциально более высокая проницаемость незрелых мембран глаз детей младшего возраста отражалась в модели посредством повышения скорости выведения лекарственного средства из глаза на 50% по сравнению с данным значением для взрослого. Системная биологическая доступность интравитреально введенного бевацизумаба варьировала от 0,65 до 0,92 (среднее значение 0,77).
Пример 2.
Определение дозировки ранибизумаба для лечения грудных детей, у которых была диагностирована РЛФ.
Посредством описанной в Примере 1 фармакокинетической модели, предсказанная степень воздействия на глаз и степень системного воздействия на грудных детей, получающих интравитреальные инъекции ранибизумаба, сравнивалась с воздействием на взрослых, которым производились интравитреальные инъекции 0,5 мг ранибизумаба, поскольку для взрослых определены степень эффективности воздействия и безопасные уровни концентраций на данном уровне дозировок и при данном режиме введения лекарственного средства.
Степени воздействия ранибизумаба считались для трех различных параметров: (i) максимальная концентрация в сыворотке крови (Cmax), позволяющая оценивать степень острой токсичности, (ii) площадь под кривой (ППК) для сыворотки крови, что позволяет измерять потенциальную долговременную токсичность, ассоциированную с длительным системным ингибированием VEGF, и (iii) ППК в стекловидном теле, что позволяет измерять степень эффективности воздействия, ассоциированного с длительным ингибированием VEGF в глазе.
Отношение степени предсказанного воздействия для грудных детей к степени предсказанного воздействия для взрослых является мерой вероятности глазной и системной токсичности, и может применяться для определения сравнительного преимущества/риска для детских дозировок. Дозировки со степенью системного воздействия, равные либо меньшие 1, считаются имеющими необходимый уровень безопасности. Концентрация лекарственного средства в сыворотке крови также должна быть ниже, чем in vitro IC50 для ранибизумаба, которая находится в диапазоне 11-27 нг/мл. Дозировки со степенью воздействия на стекловидное тело, близкие к 1, считаются имеющими позволительный уровень эффективности воздействия.
Предсказанная максимальная концентрация ранибизумаба в сыворотке крови грудных детей (Cmax) была сходной с in vitro IC50, при дозировках менее 0,3 мг. Однако, основываясь на отношении частоты воздействия Cmax к IC50 в сыворотке крови, предпочтительной является дозировка, равная менее чем 0,24 мг. Дозировка 0,06 мг является еще более предпочтительной, поскольку только в этом случае отношение кандидатного Cmax и IC50 <1 (см. Фиг. 1).
Степени воздействия для ППК в сыворотке крови выше 1 для всех моделировавшихся детских дозировок, в то время как степени воздействия для ППК в стекловидном теле были менее 1 (Фиг. 2). При определении конкретных дозировок для введения грудным детям, вероятность недостаточного воздействия в отношении к референсному воздействию на стекловидное тело для взрослых (пониженный уровень эффективности) следует балансировать относительно повышенной ППК для сыворотки крови (повышенный риск). Однако, поскольку дозировки, учитывавшиеся моделью, варьируют около in vitro IC50 для ранибизумаба, дозировки вплоть до 0,3 мг считаются имеющими в целом приемлемый уровень безопасности, при том, что было показано, что достигаемые при их применении уровни воздействия на стекловидное тело для взрослых являются эффективными в отношении терапевтического воздействия. Это позволяет считать, что все упомянутые дозировки имеют приемлемое отношение положительного воздействия к риску. Таким образом, переход к дозировкам, превышающим 0,06 мг (т.е., вплоть до 0,3 мг) может происходить в контексте клинического исследования, в случаях недостаточной терапевтической эффективности при использовании более низких дозировок и в отсутствие настораживающих явлений.
Существенно, что ППК для сыворотки крови для всех дозировок ранибизумаба для грудных детей заметно ниже, нежели ППК для сыворотки крови при использовании интравитреальных инъекций 0,625 мг. Бевацизумаба, использовавшихся при лечении РЛФ в работе БУАУ-РЛФ (Фиг. 3). Это свидетельствует о меньшей степени системного подавления VEGF, нежели в примерах ранее применявшихся методик лечения, при сохранении сходного уровня терапевтической эффективности.
Поправки для дозировок других антагонистов VEGF, нежели ранибизумаб, применяющихся для лечения грудных детей, могут быть определены посредством описанных в данном тексте предсказанных данных по воздействию ранибизумаба на глаз и системному воздействию.
Пример 3
Клиническое исследование с целью определения эффектов двух различных дозировок ранибизумаба, применяющихся для интравитреальных инъекций новорожденным, больным РЛФ.
Предлагаемое исследование позволит изучить в 16-недельный период исследования с момента инъекции, (i) вызывает ли ранибизумаб эффекты, сходные с показанными в работе БУАУ-РЛФ для бевацизумаба терапевтическими эффектами при лечении РЛФ; (ii) могут ли более низкие дозировки ранибизумаба дать сходные результаты в отношении контроля РЛФ, и (iii) будут ли две дозировки ранибизумаба отличаться в отношении эффекта системного подавления VEGF. За данным первым периодом следует время невмешательства, равное 5 годам, в течение которых прошедшие лечение новорожденные будут осмотрены дважды (в возрасте 2 года и в возрасте 5 лет) на предмет наличия продолжительного офтальмологического и педиатрического развития, в том числе - непосредственное обследование VEGF-зависимых органов, таких как сердце, легкие, кровеносная система и головной мозг.
В данное исследование будут включены 40 больных РЛФ грудных детей, которым, согласно текущим руководствам Немецкого Офтальмологического общества, требуется лечение РЛФ в зоне I (стадии 1+, 2+, 3+/- либо АП-РЛФ) либо в центральной зоне II (стадия 3+). Способы определения лечения будут оцениваться независимо двумя офтальмологами, обладающими опытом диагностики и лечения РЛФ. В исследование будут включены только грудные дети, для которых оба офтальмолога независимо друг от друга согласны с диагнозом стадии РЛФ и назначают одинаковое лечение. Грудные дети делятся случайным образом на две группы лечения, в отношении 1:1.
Грудные дети, отнесенные к группе лечения 1, получают единичную интравитреальную инъекцию 0,06 мг ранибизумаба на день 0, Грудные дети, отнесенные к группе лечения 2, получают единичную интравитреальную инъекцию 0,18 мг ранибизумаба на день 0, Для приготовления обеих упомянутых инъекций будет использоваться стандартный раствор 6 мг/мл.
Интравитреальные инъекции будут производиться офтальмологами, которым неизвестно содержимое шприцев, посредством которых будет вводиться ранибизумаб. Лечащие офтальмологи будут получать два стерильных шприца (0,1-0,2 мл в каждом шприце, без каких-либо пометок) - один из шприцев предназначен для инъекции в левый глаз, другой для инъекции в правый глаз.
Грудные дети в обеих группах лечения пройдут осмотр переднего отрезка глаза и офтальмоскопию с целью обнаружения признаков связанных с лечением имеющихся местных осложнений (эндофтальмиты, повреждение хрусталика, отслоение сетчатки, кровотечения, помутнение сред, недостаточный ответ на лечение) и неонатальный осмотр на предмет связанных с лечением имеющихся системных осложнений в дни 1 и 3.
На 7й день и далее еженедельно, в недели 2-16, грудные дети в обеих группах будут проходить осмотр переднего отрезка глаза, офтальмоскопию и диагностирование стадии РЛФ, а также неонатальные осмотры.
Ключевой точкой определения эффективности воздействия является наличие повторности РЛФ в одном либо в обоих глазах, требующей повторного лечения, до постменструального возраста, равного 54 неделям. Повторное лечение будет заключаться либо в инъекциях бевацизумаба, либо в проведении лазерной фотокоагуляционной терапии, в соответствии с настоящими рекомендациями Немецкого Общества Сетчатки (RG), Федеральной Ассоциации Немецких Офтальмологов (BVA) и Немецкого Офтальмологического Общества (DOG).
В качестве вторичной ключевой точки исследования будет использоваться изменение уровней VEGF в периферической крови в течение первых 16 недель после интравитреальной инъекции. VEGF будет измеряться посредством основанного на методе твердофазного иммуноферментного анализа теста на VEGF-A человека - один раз до инъекции, и затем через 1 неделю, через 2 недели, через 4 недели, через 6 недель, через 8 недель, через 12 недель и через 16 недель после интравитреальной инъекции.
Неинтервенционная стадия исследования, с момента наступления 16й недели после инъекции и до 2 лет и до 5 лет включает в себя оценку офтальмологического развития (острота зрения, оценка состояния мышц глаза, циклоплегическая ретиноскопия, осмотр оптических сред глаза с помощью щелевой лампы, измерение внутриглазного давления, офтальмоскопия, оптическая когерентная томография, фотографирование глазного дна и мультифокальная электроретинография), и оценку педиатрического развития (основные этапы физического и умственного развития, вес, рост, развитие когнитивных, моторных и сенсорных функций).
Пример 4
Сравнение альтернативных дозировок ранибизумаба по степени безопасности и эффективности воздействия ретролентальной фиброплазию (САРЭ-РЛФ)
Данное исследование построено как исследовательская работа в целях оценки степени безопасности и эффективности воздействия двух различных дозировок противо-VEGF-агента ранибизумаба (0,12 мг против 0,20 мг) при лечении грудных детей, у которых была диагностирована ретролентальная фиброплазия.
В данное исследование включены 40 грудных детей, у которых диагностирована ретролентальная фиброплазия (РЛФ) обоих глаз в зоне I (стадии 1+, 2+, 3+/- либо АП-РЛФ) либо в центральной (=постериорной) зоне II (стадия 3+, АП-РЛФ). Зона I определяется как двойное темпорально измеренное расстояние от диска зрительного нерва до центральной ямки, постериорная зона II определяется как тройное темпорально измеренное расстояние от диска зрительного нерва до центральнлй ямки.
Грудные дети не проходили лечение в тех случаях, когда (i) имелись педиатрические затруднения, не позволяющие применять к данному грудному ребенку лечение агентами анти-VEGF либо множественные взятия крови на анализ, что определяется специалистом по неонатальной реаниматологии и офтальмологом исследования; (ii) имеются поражения головного мозга, значительно нарушающие функцию зрительного нерва; (iii) имеется тяжелая гидроцефалия, сопровождающаяся значительно повышенным внутричерепным давлением; (iv) они находились на поздних стадиях развития РЛФ, сопровождающихся частичным либо полным отслоением сетчатки (РЛФ на стадии 4 или 5); (v) наблюдается РЛФ, поразившая только периферическую сетчатку (например, периферическая зона II либо зона III); (vi) наблюдается гиперэргическая реакция к изучаемому лекарственному средству либо к лекарственным средствам со сходной химической структурой; (vii) имеются противопоказания к интравитреальным инъекциям, согласно краткой характеристике лекарственного средства ранибизумаба; (viii) присутствует системное использование анти-VEGF терапевтических средств; (ix) к ним применялись другие исследуемые лекарственные средства - за исключением витаминов и минералов - на момент включения в исследование, либо в течение 30 дней либо 5 периодов полураспада до момента включения в исследование - в течение того из указанных временных периодов, который продолжительнее.
Грудные дети затем случайным образом были отнесены к одной из двух лечебных групп:
(1) Грудные дети в лечебной группе 1 получают интравитреальную инъекцию 0,12 мг (20 мкл при 6 мг/мл) ранибизумаба на день 0. После проявления первичного ответа дозировка, идентичная первой, может быть дополнительно введена не менее чем через четыре недели после инъекции. Максимально может быть произведено 3 дополнительных регулярных инъекции.
(2) Грудные дети в лечебной группе 2 получают интравитреальную инъекцию 0,20 мг (20 мкл при 10 мг/мл) ранибизумаба на день 0. После проявления первичного ответа дозировка, идентичная первой, может быть дополнительно введена не менее чем через четыре недели после инъекции. Максимально может быть произведено 3 дополнительных регулярных инъекции.
Основным результатом исследования является определение степени эффективности лечения. Степень эффективности лечения определяется по количеству грудных детей, которым не требуется срочное лечение вплоть до 24й недели после первой инъекции. Дополнительные инъекции исследуемых дозировок не считаются срочным лечением в том случае, когда они производятся не менее чем через 4 недели после предыдущей инъекции. Вторичными результатами исследования являются (i) регрессия плюс-болезни; (ii) регрессия преретинального васкуляризованного бугорка; (iii) прогрессия периферической внутрисетчаточной васкуляризации за пределами бугорка; (iv) количество и типы побочных эффектов и существенных побочных эффектов; (v) изменения уровней фактора роста эндотелия сосудов (VEGF) в системном кровотоке; (vi) количество повторных инъекций исследуемой дозировки; (vii) количество пациентов, прогрессировавших до 4 или 5 стадии РЛФ; (viii) количество пациентов с полной васкуляризацией периферической сетчатки вплоть до диаметра в один диск зубчатой линии. Вторичные результаты оцениваются в течение временного периода до 24 недель после первой инъекции. Также, дополнительными критериями эффективности являются количество поздних повторностей РЛФ в течение периода последующего наблюдения, количество пациентов, прогрессировавших до 4 или 5 стадии РЛФ после окончания основной части исследования, количество пациентов с полной васкуляризацией периферической сетчатки вплоть до диаметра в один диск зубчатой линии после окончания основной части исследования, оценку долгосрочного офтальмологического развития (острота зрения (если возможно), оценка состояния мышц глаза, циклоплегическая ретиноскопия, рефракция, измерение внутриглазного давления, офтальмоскопия, включающая фотографирование глазного дна (в возрасте в один год и в 5 лет посещается офтальмолог); оценка долгосрочного педиатрического развития: тест Бейли, вес, рост, развитие когнитивных, моторных и сенсорных функций; количество и типы побочных эффектов и сильных побочных эффектов в каждой группе в период между окончанием основной части наблюдательного исследования и окончанием периода последующего наблюдения. Дополнительные результаты оцениваются в течение вплоть до 5 лет после первой инъекции.
Пример 5
Это открытое, рандомизированное исследование по доказательству более высокой эффективности на параллельных группах, оценивающее степень эффективности воздействия и безопасность ранибизумаба в дозировке 0,1 мг, ранибизумаба в дозировке 0,2 мг и лазерной терапии при лечении ретролентальной фиброплазии
Целью данного исследования является демонстрация более высокой эффективности воздействия ранибизумаба в сравнении со стандартным методом лечения посредством лазерной терапии, что оценивается по пропорции пациентов в каждой из лечебных групп, не проявляющих признаков активной РЛФ и нежелательных структурных исходов через 24 недели после первого лечения.
Первичной целью данного исследования является проверка более высокой эффективности лечения посредством ранибизумаба в дозировке 0,2 мг по сравнению с лечением посредством лазерной терапии. Ключевыми вторичными целями данного исследования является проверка более высокой эффективности лечения посредством ранибизумаба в дозировке 0,1 мг по сравнению с лечением посредством лазероной терапии, и проверка более высокой эффективности лечения посредством ранибизумаба в дозировке 0,2 мг по сравнению с лечением посредством ранибизумаба в дозировке 0,1 мг. Также будут оценены глазная и системная безопасность данных методов лечения.
В данном исследовании применено три лечебных группы, в каждую из которых было включено по 80 пациентов. Пациенты являются недоношенными новорожденными мужского и женского пола с диагностированной РЛФ, требующей лечения.
Группа 1: единичная интравитреальная инъекция 0,1 мг ранибизумаба (10 мг/мл) в каждый глаз в первичный момент времени.
Группа 2: единичная интравитреальная инъекция 0,2 мг ранибизумаба (10 мг/мл) в каждый глаз в первичный момент времени.
Группа 3: Контроль. Лазерная фотокоагуляционная терапия каждого глаза в первичный момент времени.
Вторичными конечными критериями оценки являются доли пациентов на 24 неделе после начала исследуемого лечения, которые: потребовали срочного лечения, демонстрируют отсутствие активной РЛФ, демонстрируют отсутствие нежелательных структурных исходов, либо требуют 1, 2 или 3 дополнительных лечений посредством ранибизумаба.
Частота возникновения побочных эффектов в глазах и системных побочных эффектов оценивается на 24 неделе. Частота повторного возникновения заболевания оценивается на 24 неделе. Время до первого повторного проявления РЛФ в каждой лечебной группе считается до 24 недели.
Лазерная коагуляционная терапия может применяться в качестве срочного лечения в том случае, когда у пациентов не наблюдается эффекта лечения ранибизумабом.
Пример 6
Целью данного исследования является оценка работы с детьми младше одного года, проходящими лечение анти-VEGF в Jules Stein Eye Institute/Калифорнийский Университет в Лос-Анджелесе (КУЛА) (Wong, Ryan K.; Tsui, Irena, ARVO 2014).
Метод
Был проведен ретроспективный анализ медицинских карт последующих детей младше одного года, проходившим осмотр на предмет диагностирования ретролентальной фиброплазии в отделении педиатрической реаниматологии в Медицинском Центре КУЛА Рональда Рейгана с января 2012 по декабрь 2013. Были найдены младенцы, у которых было диагностировано предпороговое заболевание 1 типа или более существенная форма, и кльлоые проходили лечение посредством терапии анти-VEGF. В исследование включались все дети младше одного года с периодом последующего наблюдения в 6 месяцев и дольше на момент публикации.
Результаты
В данное исследование было включено шесть глаз (4 ребенка было рождено вне родильного дома). Среднее арифметическое массы тела при рождении составило 605 грамм (в диапазоне: 500-690 грамм), среднее арифметическое гестационного возраста было равно 23,4 недель (в диапазоне: 23,0-24,3 недель), и среднее арифметическое возраста на момент инъекции анти-VEGF лекарственного средства (в 4 глаза вводился ранибизумаб (0,25 мг) и в 2 глаза - бевацизумаб (0,625 мг)) было равно 34,2 недель (в диапазоне: 31,6-36,3 недель). Для всех глаз была диагностирована ретролентальная фиброплазия (РЛФ) на стадии 2 или 3, в постериорной зоне 2, с плюс-болезнью. Все глаза продемонстрировали первичное купирование плюс-болезни и регрессию РЛФ после лечения. Все 6 глаз потребовали дополнительного лечения посредством рлазерной терапии, в среднем возрасте, равном 44,4 недель (в диапазоне: 42,9-50,4 недель). Симптомом к дополнительной лазерной терапии являлось повторное проявление РЛФ в 3 глазах (50%), в среднем через 6,1 недель после лечения посредством анти-VEGF лекарственного средства, и сохранение РЛФ стадии 1, в зоне 3 в 3 глазах (50%), в среднем через 12,9 недель после лечения посредством анти-VEGF лекарственного средства.
Выводы
Грудным детям после анти-VEGF терапии РЛФ часто может потребоваться дополнительная лазерная фотокоагуляционная терапия - в случае повторного проявления либо сохранения активности заболевания в течение 3 месяцев.
Понимается, что данное изобретение было описано только посредством примеров, и в него могут быть внесены изменения в рамках объема и сущности данного изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРИМЕНЕНИЕ АНТАГОНИСТА VEGF ПРИ ЛЕЧЕНИИ ХОРИОРЕТИНАЛЬНЫХ НЕОВАСКУЛЯРНЫХ НАРУШЕНИЙ И НАРУШЕНИЙ ПРОНИЦАЕМОСТИ У ПЕДИАТРИЧЕСКИХ ПАЦИЕНТОВ | 2014 |
|
RU2676274C2 |
| ЛИПОСОМАЛЬНЫЕ ПРЕПАРАТЫ | 2013 |
|
RU2817350C2 |
| ЛИПОСОМАЛЬНЫЕ ПРЕПАРАТЫ | 2013 |
|
RU2680096C2 |
| ПРЕДВАРИТЕЛЬНО ЗАПОЛНЕННЫЙ ПЛАСТИКОВЫЙ ШПРИЦ, СОДЕРЖАЩИЙ АНТАГОНИСТ VEGF | 2015 |
|
RU2702748C2 |
| ПРИМЕНЕНИЕ СИРОЛИМУСА ДЛЯ ЛЕЧЕНИЯ ЭКССУДАТИВНОЙ ВОЗРАСТНОЙ МАКУЛЯРНОЙ ДЕГЕНЕРАЦИИ С ПЕРСИСТИРУЮЩИМ ОТЕКОМ | 2017 |
|
RU2765728C2 |
| СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНЕЙ ГЛАЗ | 2015 |
|
RU2771900C2 |
| СПОСОБ ЛЕЧЕНИЯ БОЛЕЗНЕЙ ГЛАЗ | 2015 |
|
RU2722643C2 |
| НУТРИЦЕВТИЧЕСКАЯ ОФТАЛЬМОЛОГИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЙ СЕТЧАТКИ С НЕОВАСКУЛЯРНЫМ КОМПОНЕНТОМ | 2019 |
|
RU2806096C2 |
| АНТАГОНИСТЫ ИНТЕГРИНОВЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2721907C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ГЛАЗНЫХ ЗАБОЛЕВАНИЙ | 2019 |
|
RU2776850C2 |
Изобретение относится к использованию ранибизумаба при лечении патологической неоваскуляризации сетчатки грудных детей. Способ лечения грудных детей с ретролентальной фиброплазией (РЛФ) включает введение в глаз ребенка ранибизумаба в дозе, составляющей менее 0,25 мг. Изобретение обеспечивает лечение грудных детей с ретролентальной фиброплазией за счет оптимально подобранных доз ранибизумаба. 7 з.п. ф-лы, 3 ил., 6 пр.
1. Способ лечения грудных детей с ретролентальной фиброплазией (РЛФ),
включающий введение в глаз ребенка ранибизумаба в дозе, составляющей менее 0,25 мг.
2. Способ по п.1, где доза составляет 0,05-0,25 мг.
3. Способ по п.1, где доза составляет 0,20 мг.
4. Способ по п.1, где доза составляет 0,1 мг.
5. Способ по п.1, где доза ранибизумаба, вводимая грудному ребенку, составляет 0,06 мг в 10 мкл, 0,075 мг в 7,5 мкл, 0,1 мг в 10 мкл, 0,12 мг в 20 мкл, 0,15 мг в 15 мкл, 0,18 мг в 30 мкл, 0,20 мг в 20 мкл, 0,25 мг в 25 мкл или 0,24 мг в 40 мкл.
6. Способ по п.1, дополнительно включающий проведение лазерной фотокоагуляционной терапии (ЛФТ) или криотерапии.
7. Способ по п.6, где начало ЛФТ или криотерапии и начало введения ранибизумаба происходит в пределах 2-24 недель между ними.
8. Способ по п.6, где ЛФТ или криотерапию проводят только, если при осмотре подвергающегося лечению глаза выявлены признаки сохранения или рецидива неоваскуляризации сетчатки.
| CHUN-JU LIN et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Регулятор для ветряного двигателя в ветроэлектрических установках | 1921 |
|
SU136A1 |
| RADHA P | |||
| KOHLY et al | |||
| MANAGEMENT OF PEDIATRIC CHOROIDAL NEOVASCULAR MEMBRANES WITH INTRAVITREAL ANTI-VEGF AGENTS: A RETROSPECTIVE CONSECUTIVE CASE SERIES/ Canadian Journal of Ophthalmology, 2011, V.46 | |||
| RUDOLF AUTRATA et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| WO 2010060748 A1, 03.06.10 | |||
| WO 2010045506 A2, 22.04.10 | |||
| ЛЕЧЕБНОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ АККОМОДАЦИЙ "STIAK" | 2010 |
|
RU2469734C2 |
| WO 2007038453 A2, 05.04.07. | |||

