Настоящее изобретение относится к способу получения многослойной красочной системы посредством получения пленки базового покрытия или двух или большего количества нанесенных непосредственно друг на друга пленок базового покрытия, непосредственно на металлической основе, покрытой затвердевшей системой электроосажденного покрытия, к получению пленки покровного лака непосредственно на одной или на самой верхней из двух или большего количества пленок базового покрытия, и затем к совместному отверждению одной или двух или большего количества пленок базового покрытия и пленки покровного лака. Настоящее изобретение дополнительно относится к многослойной красочной системе, полученной посредством способа в соответствии с изобретением.
Предшествующий уровень техники,
Известны многослойные красочные системы на металлических основах, примерами которых при этом являются многослойные красочные системы в секторе автомобильной промышленности. Вообще говоря, многослойные красочные системы указанных видов, если их рассматривать от металлической основы наружу, включают электроосажденное покрытие, покрытие, которое нанесено непосредственно на электроосажденное покрытие, и которое обычно упоминается как грунтовочный слой, по меньшей мере одно покрытие, которое содержит цветные пигменты и/или эффектные пигменты, и которое, как правило, упоминается как базовое покрытие, а также покровный лак.
Основные композиции и функции указанных покрытий, и покрывающих материалов, необходимых для формирования указанных покрытий - то есть, материалов, предназначенных для электроосаждения, шпатлевок, покрывающих материалов, содержащих цветные и/или эффектные пигменты, и известных при этом в качестве материалов базового покрытия, а также материалов покровного лака - являются известными. Так, например, основной целью покрытия, нанесенного методом электроосаждения, является защита основы от коррозии. Основной функцией грунтовочного слоя является обеспечение защиты от механических повреждений, например, таких как удары мелких камней, а также заполнение неровностей в основе. Следующее покрытие, которое называют базовым покрытием, в основном отвечает за получение эстетических качеств, таких как цвет и/или оптические эффекты, такие как флоп-эффект, в то время, как покровный лак, который следует далее, в частности, служит для обеспечения многослойной красочной системы стойкостью к царапанию, а также для придания блеска.
Получение указанных многослойных красочных систем, как правило, вначале включает осаждение или электрофоретическое нанесение материала, предназначенного для электроосаждения, в частности, катодного материала для электроосаждения, на металлическую основу, например, такую как автомобильный кузов. Перед осаждением материала, предназначенного для электроосаждения, металлическая основа может подвергаться различным предварительным обработкам - например, могут наноситься известные покрытия, химически взаимодействующие с основой, такие как фосфатные покрытия, в частности, цинкфосфатные покрытия. Операция осаждения материала для электроосаждения, как правило, происходит в соответствующих резервуарах для электроосаждения. После нанесения, покрытую основу удаляют из резервуара и необязательно споласкивают и подвергают самоиспарению и/или промежуточной сушке, и наконец, электроосажденный материал затвердевает. Целью в этом случае является толщина пленок, составляющая примерно 15-25 микрометров. Затем непосредственно на затвердевшее электроосажденное покрытие наносят материал шпатлевки, и необязательно подвергают его самоиспарению и/или промежуточной сушке, и после этого он затвердевает. Для того чтобы позволить затвердевшему грунтовочному слою выполнять указанные выше задачи, толщина пленок должна составлять, к примеру, 25 - 45 микрометров. В последующем, на затвердевший грунтовочный слой непосредственно наносится материал базового покрытия, содержащий цветные и/или эффектные пигменты, который необязательно подвергают самоиспарению и/или промежуточной сушке, при этом материал покровного лака наносится непосредственно на полученную таким образом пленку базового покрытия, без отдельного отверждения. В последующем, пленка базового покрытия и любая пленка покровного лака, которая также была подвергнута самоиспарению и/или промежуточной сушке до этого, затвердевают вместе (метод нанесения покрытия по влажному нижнему покрытию). Причем затвердевшее базовое покрытие в принципе имеет сравнительно небольшую толщину пленок, составляющую 10-20 микрометров, то, например, для затвердевшего покровного лака задается толщина пленок, которая составляет 30 - 60 микрометров, например, для того чтобы достичь описанных технологических потребительских свойств. Нанесение шпатлевки, базового покрытия, и материалов покровного лака может осуществляться, например, посредством способов пневматического распыления и/или электростатического напыления, которые являются известными специалисту в данной области. В настоящее время, шпатлевка и материалы базового покрытия уже все чаще применяют в виде водных материалов покрытия, по экологическим соображениям.
Многослойные красочные системы указанных видов и способы их получения описаны, например, в DE 199 48 004 А1, строка 37 страницы 17 - строка 22 страницы 19, или также в DE 100 43 405 С1, параграф [0018] колонки 3, и параграф [0052] колонки 8 - параграф [0057] колонки 9, совместно с параграфом [0039] колонки 6 - параграфом [0050] колонки 8.
Хотя при этом многослойные красочные системы, полученные таким образом, как правило, способны соответствовать требованиям, предъявляемым автомобильной промышленностью относительно технологических потребительских свойств и эстетических характеристик, в настоящее время все большее значение приобретает охрана окружающей среды и экономические факторы, в связи с чем в центр внимания автопроизводителей входит упрощение описанного сравнительно сложного способа получения.
Таким образом, существуют подходы, где предпринимаются попытки обойтись без отдельной стадии отверждения покрывающего материала, нанесенного непосредственно на затвердевшее электроосажденное покрытие (в контексте стандартного способа, описанного выше, покрывающий материал упоминается как шпатлевка), а также, необязательно, уменьшить толщину покрывающей пленки, полученной из этого покрывающего материала. В пределах уровня техники, эта покрывающая пленка, которая не затвердевает отдельно, часто упоминается как пленка базового покрытия (а больше не как пленка шпатлевки) или упоминается как первая пленка базового покрытия, для того чтобы отличить ее от второй пленки базового покрытия, которую наносят на нее. В некоторых случаях, фактически, предпринимаются попытки вполне обойтись без этой покрывающей пленки (в случае чего, непосредственно на электроосажденном покрытии получают только одну так называемую пленку базового покрытия, и затем она покрывается сверху материалом покровного лака без применения отдельной стадии отверждения, что означает то, что, по сути, также отказываются от отдельной стадии отверждения). Таким образом, предполагается, что вместо отдельной стадии отверждения и вместо дополнительной окончательной стадии отверждения, после нанесения всех покрывающих пленок, нанесенных на электроосажденное покрытие, должна быть только одна окончательная стадия отверждения.
Отказ от отдельной стадии отверждения покрывающего материала, нанесенного непосредственно на электроосажденное покрытие, является очень преимущественным из экологических и экономических причин. Дело в том, что это приводит к сохранению электроэнергии, и конечно, вся технологическая операция, как правило, может происходят с большей точностью.
Тогда, является преимущественным, когда вместо указанной отдельной стадии отверждения покрывающая пленка, полученная непосредственно на электроосажденном покрытии, просто подвергается самоиспарению при комнатной температуре и/или промежуточной сушке при повышенных температурах, без осуществления процесса отверждения, который, как известно, как правило, включает повышенные температуры отверждения и/или длительное время отверждения.
Проблема, однако, состоит в том, что при такой форме изготовления, в настоящее время часто невозможно достичь требуемых технологических свойств и эстетических характеристик.
Например, отказ от отдельного отверждения покрывающей пленки, нанесенной непосредственно на электроосажденное покрытие, такого как, например, отверждение первой пленки базового покрытия перед нанесением дополнительных материалов покрытия, таких как второй материал базового покрытия и материал покровного лака, например, может приводить к возникновению нежелательных включений воздуха, растворителя и/или влаги, и при этом указанные включения могут становиться заметными в виде пузырьков под поверхностью всей красочной системы и они могут лопаться в процессе окончательного отверждения. Полученные в результате отверстия в красочной системе, которые также называют порами и пузырями, приводят к непривлекательному внешнему виду. В результате, количество органического растворителя и/или воды, а также количество воздуха, которое было введено в результате процедуры нанесения всей системы, охватывающей первое базовое покрытие, второе базовое покрытие, и покровный лак, является слишком большим, с тем, чтобы все это количество было способно выделиться из многослойной красочной системы в ходе стадии окончательного отверждения, без образования дефектов. В случае традиционной технологической операции, описанной выше, где пленка шпатлевки сохнет отдельно, до получения обычно сравнительно тонкой пленки базового покрытия (которая, таким образом, содержит только сравнительно небольшое количество воздуха, органических растворителей и/или воды), решение этой проблемы, конечно, является гораздо менее сложным.
Однако, даже при получении многослойных красочных систем, где применение покрывающего материала, упоминаемого в стандартном способе в качестве шпатлевки, полностью исключено, другими словами, систем, где непосредственно на затвердевшее электроосажденное покрытие наносят только материал базового покрытия, описанные проблемы в отношении пор и пузырей также часто встречаются. Причина состоит в том, что в зависимости от нанесения и эксплуатации полученной многослойной красочной системы, в случае полного исключения покрытия, упоминаемого как грунтовочный слой в стандартном способе, требуемая толщина пленки базового покрытия, как правило, является больше, по сравнению со стандартными системами, с тем, чтобы получить желательные свойства. Таким образом, в этом случае, полная толщина покрывающей пленки, которая должна затвердевать на стадии окончательного отверждения, как правило, также более высокая, чем при стандартной операции.
При этом другие соответствующие характеристики также не всегда достигаются в достаточной степени, при формировании многослойных красочных систем с применением описанного способа. Соответственно, задача состоит, например, в достижении качественного общего внешнего вида, на который, в частности, влияет хорошая текучесть применяемых покрывающих материалов. В этом случае реологические свойства покрывающих материалов должны подбираться соответственно описанному рабочему режиму. Подобное применяется в отношение механических характеристик, таких, как адгезия. В связи с этим, аналогично, большой задачей является достижение подходящего качества.
Кроме того, экологические свойства таких многослойных красочных систем все еще является предметом улучшения. Замена в водных материалах покрытия значительной фракции органических растворителей водой уже привносит соответствующий вклад. Но существенное улучшение свойств может быть достигнуто в результате повышения в таких покрывающих материалах содержания твердых частиц. Тем не менее, особенностью водных материалов базового покрытия, содержащих цветные и/или эффектные пигменты, является то, что повышение содержания твердых частиц в то же время сохранение соответствующих реологических свойств и, следовательно, хорошего внешнего вида, является довольно таки сложным.
Соответственно, будет преимущественным иметь в своем распоряжении способ получения многослойных красочных систем, который позволил бы отказаться от отдельной стадии отверждения покрывающего материала, нанесенного непосредственно на электроосажденное покрытие, как описано выше, и чтобы полученная при этом многослойная красочная система демонстрировала отличные технологические потребительские свойства и эстетические характеристики.
Задача
Задачей настоящего изобретения, соответственно, было обеспечение способа получения многослойной красочной системы на металлических основах, где покрывающий материал, нанесенный непосредственно на систему электроосажденного покрытия, не затвердевает отдельно, а вместо этого указанный покрывающий материал затвердевает на совместной стадии отверждения вместе с дополнительными покрывающими пленками, нанесенными после этого. Несмотря на такое технологическое упрощение, полученные многослойные красочные системы должны демонстрировать отличную устойчивость в отношении пор. Кроме того, в зависимости от требований и отдельной области применения, в результате, должно быть возможным обеспечивать многослойные красочные системы, где одна покрывающая пленка или две, или большее количество покрывающих пленок, расположенные между электроосажденным покрытием и покровным лаком, могут иметь различную толщину пленок, и в которых при этом, в частности, не существует проблем с порами, которые возникают даже при относительно высокой толщине пленок. Другие характеристики многослойных красочных систем, в частности общий внешний вид и адгезия, также должны быть на высоком уровне, по меньшей мере они должны быть на уровне, который может быть достигнут посредством стандартного способа, описанного выше. Техническое решение
Было выявлено, что указанные задачи могут достигаться посредством нового способа получения многослойной красочной системы (М) на металлической основе (S), содержащего
(1) получение затвердевшего электроосажденного покрытия (Е.1) на металлической основе (S) посредством электрофоретического нанесения материала для электроосаждения (е.1) на основу (S), и последующего отверждения материала для электроосаждения (е.1),
(2) получение (2.1) пленки базового покрытия (В.2.1) или (2.2) двух или большего количества нанесенных непосредственно друг на друга пленок базового покрытия (В.2.2.x) непосредственно на затвердевшем электроосажденном покрытии (Е.1), посредством (2.1) нанесения водного материала базового покрытия (b.2.1) непосредственно на электроосажденное покрытие (Е.1) или посредством (2.2) непосредственного нанесения на электроосажденное покрытие (Е.1) двух или большего количества материалов базового покрытия (b.2.2.х) друг на друга,
(3) получение пленки покровного лака (K) непосредственно на (3.1) пленке базового покрытия (В.2.1), или на (3.2) самой верхней пленке базового покрытия (В.2.2.x), посредством нанесения материала покровного лака (k) непосредственно на (3.1) пленку базового покрытия (В.2.1) или на (3.2) самую верхнюю пленку базового покрытия (В.2.2.x),
(4) совместное отверждение (4.1) пленки базового покрытия (В.2.1) и пленки покровного лака (К), или (4.2) пленок базового покрытия (В.2.2.x) и покровного лака (K),
где
материал базового покрытия (b.2.1) или по меньшей мере один из материалов базового покрытия (b.2.2.х) содержит по меньшей мере одну водную полиуретан-полимочевинную дисперсию (ПД), которая включает частицы полиуретана и полимочевины, где частицы полиуретана и полимочевины, которые присутствуют в дисперсии (ПД), содержат анионные группы и/или группы, которые могут превращаться в анионные группы, и имеют средний размер частиц, который составляет 40 - 2000 им, а также гель-фракцию, которая составляет по меньшей мере 50%.
Указанных выше способ также упоминается ниже как способ в соответствии с изобретением, и соответственно, является объектом настоящего изобретения. Предпочтительные варианты осуществления способа в соответствии с изобретением можно найти в описании, представленном далее ниже, а также в зависимых пунктах формулы изобретения.
Дополнительным объектом настоящего изобретения является многослойная красочная система, полученная с применением способа в соответствии с изобретением.
Способ в соответствии с изобретением позволяет получать многослойные красочные системы без отдельной стадии отверждения покрывающей пленки, полученной непосредственно на электроосажденном покрытии. Для большей простоты понимания, указанная покрывающая пленка в контексте настоящего изобретения указана как пленка базового покрытия. Вместо отдельного отверждения, указанная пленка базового покрытия затвердевает вместе с любым количеством последующих пленок базового покрытия, расположенных под пленкой покровного лака, и с пленкой покровного лака. При этом, в результате применения способа в соответствии с изобретением, полученные многослойные красочные системы демонстрируют отличные устойчивость в отношении пор. Точно также, общий внешний вид и адгезия указанных многослойных красочных систем являются отличными, и находятся по меньшей мере на уровне многослойных красочных систем, полученных посредством описанного выше стандартного способа.
Подробное описание
Прежде всего будет объяснен ряд терминов, используемых в контексте настоящего изобретения.
Нанесение покрывающего материала на основу, и получение на основе покрывающих пленок, понимают следующим образом. Покрывающий материал, о котором идет речь, наносят таким образом, что покрывающая пленка, полученная из него, располагается на основе, но при это необязательно должна находиться в прямом контакте с основой. Например, между покрывающей пленкой и основой, могут располагаться другие покрытия. Например, на стадии (1), на металлической основе (S) получают затвердевшее электроосажденное покрытие (Е.1), но между основой и электроосажденным покрытием также может находиться покрытие, химически взаимодействующее с основой, как описано далее ниже, такое как цинкфосфатное покрытие.
Тот же принцип применяется к нанесению покрывающего материала (b) на покрывающую пленку (А), полученную с помощью другого покрывающего материала (а), и к получению покрывающей пленки (В) на другой покрывающей пленке (А). Покрывающая пленка (В) необязательно должна находиться в контакте с покрывающей пленкой (А), при этом просто необходимо, чтобы она располагалась выше нее, другими словами, на стороне покрывающей пленки (А), то есть, удаленной от основы.
В отличие от этого, нанесение покрывающего материала непосредственно на основу, или получение покрывающей пленки непосредственно на основе, понимают следующим образом. Покрывающий материал, о котором идет речь, наносят таким образом, что покрывающая пленка, полученная из него, располагается на основе и находится в прямом контакте с основой. Таким образом, в частности, другое покрытие, расположенное между покрывающей пленкой и основой, отсутствует.
Конечно, то же применяется к нанесению покрывающего материала (b) непосредственно на покрывающую пленку (А), полученную посредством нанесения другого покрывающего материала (а), и к получению покрывающей пленки (В) непосредственно на другой покрывающей пленке (А). В этом случае две покрывающие пленки находятся в прямом контакте, располагаясь таким образом непосредственно друг на друге. В частности, между покрывающими пленками (А) и (В) отсутствует дополнительное покрытие. Конечно, тот же принцип применяется к непосредственному нанесению друг на друга покрывающих материалов и к получению нанесенных непосредственно друг на друга покрывающих пленок.
Самоиспарение, промежуточная сушка, и отверждение в контексте настоящего изобретения понимаются как имеющие то же семантическое содержание, которое известно специалисту в данной области в связи со способами получения многослойных красочных систем.
Соответственно, термин "самоиспарение" понимается, в принципе, как обозначающий пассивное или активное испарение органических растворителей и/или воды из покрывающего материала, нанесенного в качестве части получения красочной системы, которое обычно происходит при температуре окружающей среды (то есть, при комнатной температуре), например, при 15-35°С, на протяжении, например, 0,5 - 30 минут. Таким образом, самоиспарение сопровождается испарением органических растворителей и/или воды, которые присутствуют в нанесенном покрывающем материале. Поскольку покрывающий материал все еще является жидким, по меньшей мере непосредственно после нанесения и на начало процесса самоиспарения, то в ходе самоиспарения он может растекаться. Причина состоит в том, что по меньшей мере один покрывающий материал, нанесенной посредством распыления, как правило, наносят в виде капель, и при этом он не обладает однородной толщины. При этом, по причине содержания органических растворителей и/или воды, материал является жидким и, таким образом, может подвергаться растеканию, с получением однородной, гладкой покрывающей пленки. В то же время, происходит последующее испарение органических растворителей и/или воды, возникающее после фазы самоиспарения в сравнительно гладкой покрывающей пленке, которая уже содержит меньше воды и/или растворителя, по сравнению с нанесенным покрывающим материалом. Однако, после самоиспарения, покрывающая пленка все же не находится в готовом к эксплуатации состоянии. В то время, как она больше не является текучей, она все еще является, например, мягкой и/или липкой, и возможно только частично высохшей. В частности, покрывающая пленка еще не отверждена, как описано далее ниже.
Таким образом, промежуточная сушка также понимается как такая, которая относится к пассивному или активному испарению органических растворителей и/или воды из покрывающего материала, нанесенного в качестве части получения красочной системы, обычно при температуре, повышенной относительно температуры окружающей среды и достигающей, например, 40 - 90°С, на протяжении времени, составляющего, например, 1-60 минут. В результате, точно также, в ходе промежуточной сушки нанесенный покрывающий материал будет терять фракцию органических растворителей и/или воды. В зависимости от конкретного покрывающего материала, в общем, если сравнивать промежуточную сушку с самоиспарением, она происходит, например, при более высоких температурах и/или на протяжении более продолжительного периода времени, в результате, по сравнению с самоиспарением, из нанесенной покрывающей пленки выделяется более высокая фракция органических растворителей и/или воды. При этом, даже промежуточная сушка не приводит к получению покрывающей пленка в готовом к эксплуатации состоянии, другими словами, покрывающая пленка не является затвердевшей, как описано далее ниже. Обычная последовательность самоиспарения и промежуточной сушки будет, например, такой как, самоиспарение нанесенной покрывающей пленки при температуре окружающей среды на протяжении 5 минут, а затем ее промежуточная сушка при температуре 80°С на протяжении 10 минут. Однако, окончательное разграничение двух понятий друг от друга, не является ни необходимым, ни желательным. Для четкости понимания, указанные термины используют для того, чтобы было ясно, что различное и последовательное кондиционирование покрывающей пленки может осуществляться перед отверждением, которое описано ниже. В этом случае, в зависимости от покрывающего материала, температуры испарения и времени испарения, могут испаряться большие или меньшие фракции органических растворителей и/или воды, которые присутствуют в покрывающем материале. В этом случае даже возможно, необязательно, что фракция полимеров, которые присутствуют в качестве связующих веществ в покрывающем материале, будет подвергаться сшиванию или переплетаться друг с другом, как описано ниже. При этом, как во время самоиспарения, так и во время промежуточной сушки не получают готовой для эксплуатации покрывающей пленки, как это происходит в случае отверждения, описанного ниже. Соответственно, отверждение однозначно разграничивается от самоиспарения и промежуточной сушки.
Отверждение покрывающей пленки, соответственно, понимают как превращение такой пленки в готовое для эксплуатации состояние, другими словами, в состояние, в котором основа, обладающая покрывающей пленкой, о которой идет речь, может перемещаться, храниться, и применяться для предполагаемой цели. В этом случае, в частности, затвердевшая покрывающая пленка, больше не является мягкой или липкой, а вместо этого имеет нужную физическую кондицию в качестве твердой покрывающей пленки, которая, даже после дополнительного подвержения условиям отверждения, как описано далее ниже, больше не демонстрирует каких-либо существенных изменений своих свойств, таких как твердость или адгезия к основе.
Как известно, покрывающие материалы могут, в принципе, затвердевать физически и/или химически, в зависимости от присутствующих компонентов, таких как связующие вещества и сшивающие агенты. В случае химического отверждения, имеется в виду термохимическое отверждение и актинично-химическое отверждение. В случае, если, например, покрывающий материал является химически термоотверждаемым, то он может быть самосшивающимся и/или внешне сшивающимся. Указание на то, что покрывающий материал является самосшивающимся и/или внешне сшивающимся, в контексте настоящего изобретения, означает, что указанный покрывающий материал содержит в качестве связующих веществ полимеры, и необязательно сшивающие агенты, которые способны сшиваться друг с другом, соответственно. Базовые механизмы, а также связующие вещества и сшивающие агенты, которые при этом могут применяться, описаны далее ниже.
В контексте настоящего изобретения, термин "отверждаемый физически" или "физическое отверждение" означает образование затвердевшей покрывающей пленки в результате выделения растворителя из полимерных растворов или полимерных дисперсий, где при этом отверждение достигается в результате переплетения цепей полимера. Как правило, покрывающие материалы указанных видов составляются в качестве однокомпонентных покрывающих материалов.
В контексте настоящего изобретения, термин "химически термоотверждаемый" или "термохимическое отверждение" означает перекрестное сшивание покрывающей пленки (образование затвердевшей покрывающей пленки), вызываемое посредством химической реакции реакционноспособных функциональных групп, где энергетическая активация указанной химической реакции возможна посредством тепловой энергии. Различные функциональные группы, которые являются дополняющими друг для друга, могут в этом случае вступать в реакцию друг с другом (дополняющие функциональные группы), и/или образование затвердевшего покрытия происходит на основе реакции автореакционноспособных групп, другими словами, функциональных групп, которые вступают в реакцию между собой с группами своего собственного вида. Примеры подходящих дополняющих реакционноспособных функциональных групп и автореакционноспособных функциональных групп известны из заявки на получение патента Германии DE 199 30 665 А1, например, строка 28 страницы 7 - строка 24 страницы 9.
Такое сшивание может быть самосшивающимся и/или внешне сшивающимся. В случае, если, например, дополняющих реакционноспособные функциональные группы уже присутствуют в органическом полимере, который применяют в качестве связующего вещества, например, в сложном полиэфире, полиуретане, или поли(мет)акрилате, то получают самосшивание. Внешнее сшивание получают, например, когда (первый) органический полимер, содержащий определенные функциональные группы, например, гидроксильные группы, вступает в реакцию с сшивающим агентом, известным как таковой, например, с полиизоцианатом и/или меламиновой смолой. Тогда, сшивающий агент содержит реакционноспособные функциональные группы, которые являются дополняющими к реакционноспособным функциональным группам, которые присутствуют в (первом) органическим полимере, который применяют в качестве связующего вещества.
В случае внешнего сшивания, в частности, предполагаются однокомпонентные и многокомпонентные системы, в частности, двухкомпонентные системы, которые являются известными как таковые.
В химически термоотверждаемых однокомпонентных системах, компоненты для сшивания, такие, как например, органические полимеры в качестве связующих веществ и сшивающие агенты, присутствуют рядом друг с другом, другими словами, в виде одного компонента. Необходимость состоит в том, чтобы компоненты, подлежащие сшиванию, вступали в реакцию друг с другом -, то есть, вступали в реакции отверждения - только при относительно высоких температурах, составляющих, например, более 100°С. В ином случае будет необходимо хранить компоненты для сшивания отдельно друг от друга и смешивать их друг с другом только непосредственно перед нанесением на основу, для того чтобы предотвратить преждевременное, по меньшей мере частичное, термохимическое отверждение (сравни с двухкомпонентными системами). В качестве примерной комбинации, могут быть упомянуты сложные полиэфиры с гидроксильными функциональными группами и/или полиуретаны с меламиновой смолы и/или блокированные полиизоцианаты в качестве сшивающих агентов.
В химически термоотверждаемых двухкомпонентных системах, компоненты, которые подлежат сшиванию, такие, как например, органические полимеры в качестве связующих веществ, и сшивающие агенты, присутствуют отдельно друг от друга по меньшей мере в виде двух компонентов, которые не объединяют до непосредственного нанесения. Указанную форму выбирают тогда, когда компоненты для сшивания подвергаются реакции друг с другом даже при температурах окружающей среды или немного повышенных температурах, составляющих, например, 40 - 90°С. В качестве примерной комбинации, могут упоминаться сложные полиэфиры с гидроксильными функциональными группами и/или полиуретаны и/или поли(мет)акрилаты со свободными полиизоцианатами в качестве сшивающего агента.
Также возможно, когда органический полимер в качестве связующего вещества имеет как самосшивающиеся, так и внешне сшивающиеся функциональные группы, и затем объединяется со сшивающими агентами.
В контексте настоящего изобретения, термин "отверждаемый актинично-химически", или "актинично-химическое отверждение", относится к тому факту, что отверждение возможно с применением актиничного излучения, причем оно представляет собой электромагнитное излучение, такое как ближняя инфракрасная область спектра (БИК) и УФ-излучение, в частности, УФ-излучение, а также корпускулярное излучение, такое как пучки электронов для отверждения. Отверждение УФ-излучением обычно вызывается с помощью радикальных или катионных фотоинициаторов. Типичными актинично затвердевающими функциональными группами являются углерод-углеродные двойные связи, причем в этом случае обычно применяют радикальные фотоинициаторы. Актиничное отверждение также основано на химическом сшивании.
Конечно, во время отверждения покрывающего материала, указанного как отверждаемый химически, также всегда будет происходить физическое отверждение, другими словами, переплетение цепей полимера. В этом случае, тем не менее, покрывающий материал указанного вида указывается как отверждаемый химически.
Из приведенного выше следует, что в соответствии с природой покрывающего материала и компонентов, которые он содержит, отверждение, происходит посредством разных механизмов, которые, конечно, также вызывают необходимость в разных условиях на стадии отверждения, в частности, разных температурах отверждения и времени отверждения.
В случае исключительно физически отверждающегося покрывающего материала, отверждение происходит предпочтительно в диапазоне между 15 и 90°С на протяжении периода времени, который составляет 2-48 часов. В этом случае, отверждение отличается от самоиспарения и/или промежуточной сушки, если это целесообразно, исключительно продолжительностью кондиционирования покрывающей пленки. Кроме того, различие между самоиспарением и промежуточной сушкой, является не ощутимым. Например, может быть возможным, если покрывающая пленка, полученная в результате нанесения отверждаемого физически покрывающего материала, например, вначале подвергается самоиспарению или промежуточной сушке при температуре 15-35°С на протяжении времени, составляющего 0,5 - 30 минут, а затем затвердевает при температуре 50°С на протяжении времени, составляющего 5 часов.
Тем не менее, является предпочтительным, если по меньшей мере некоторые покрывающие материалы, предназначенные для применения в контексте способа в соответствии с изобретением, другими словами, материалы для электроосаждения, водные материалы базового покрытия, и материалы покровного лака, являются химически термоотверждаемыми, и особенно предпочтительно, являются химически термоотверждаемыми и внешне сшивающимися.
В принципе, и в контексте настоящего изобретения, отверждение химически термоотверждаемых однокомпонентных систем предпочтительно осуществляется при температурах, которые составляют 100 - 250°С, предпочтительно 100 - 180°С, на протяжении времени, составляющего 5-60 минут, предпочтительно 10 - 45 минут, так как указанные условия, как правило, необходимым для реакций химического сшивания, для того чтобы превращать покрывающую пленку в затвердевшую покрывающую пленку. Соответственно, указанное происходит, когда при этом фаза самоиспарения и/или промежуточной сушки, которая осуществляется перед отверждением, происходит при более низких температурах и/или на протяжении более короткого периода времени. В данном случае, например, самоиспарение может происходить при температуре 15 - 35°С на протяжении времени, составляющего, например, 0,5 - 30 мину, и/или промежуточная сушка может происходить при температуре, составляющей, например, 40 - 90°С, на протяжении времени, составляющего, например, 1 -60 минут.
В принципе, и в контексте настоящего изобретения, отверждение химически термоотверждаемых двухкомпонентных систем осуществляется при температурах, которые составляют, например, 15-90°С, предпочтительно 40 - 90°С, на протяжении времени, составляющего 5-80 минут, предпочтительно 10 - 50 минут. Соответственно, указанное имеет место, когда фаза самоиспарения и/или промежуточной сушки, которая происходит перед отверждением, осуществляется при более низких температурах и/или на протяжении более короткого периода времени. В таком случае, например, уже больше нет смысла делать какие-либо различия между понятиями самоиспарения и промежуточной сушки. Фаза самоиспарения или промежуточной сушки, которая предшествует отверждению, может происходить, например, при температуре 15-35°С на протяжении периода времени, составляющего, например, 0,5 - 30 минут, но в любом случае, при более низких температурах и/или на протяжении более короткого периода времени, чем отверждение, которое затем следует.
Это, конечно, не исключает возможности того, что химически термоотверждаемая двухкомпонентная система может затвердевать при более высоких температурах. Например, на стадии (4) способа в соответствии с изобретением, как описано более подробно далее ниже, пленка базового покрытия или две, или большее количество пленок базового покрытия затвердевают вместе с пленкой покровного лака. В случае, когда в пленках присутствуют как химически термоотверждаемые однокомпонентные системы, так и двухкомпонентные системы, например, однокомпонентный материал базового покрытия и двухкомпонентный материал покровного лака, то, например, совместное отверждение, конечно, будет зависеть от условий отверждения, необходимых для однокомпонентной системы.
Все температуры, которые указаны в контексте настоящего изобретения, необходимо понимать, как температуру помещения, в котором находится покрываемая основа. Таким образом, указанное не означает, что необходимо, чтобы сама основа имела температуру, о которой идет речь.
Если в контексте настоящего изобретения делается ссылка на официальный стандарт, без указания официального срока действия, то ссылка, конечно, относится к версии стандарта, действующего на дату подачи заявки или, если отсутствует действующая версия на указанную дату, к наиболее поздней действующее версии.
Способ в соответствии с изобретением
В способе в соответствии с изобретением, многослойная красочная система создается на металлической основе (S).
Рассматриваемые металлические основы (S), по сути, включают основы, содержащие или, состоящие из, например, железа, алюминия, меди, цинка, магния, и их сплавов, а также стали, в любом из самых разнообразных форм и составов. Предпочтительные основы представляют собой основы, изготовленные из железа и стали, примерами при этом являются обычные железные и стальные основы, которые применяются в секторе автомобильной промышленности. Основы сами по себе могут быть любой формы - то есть, они, например, могут быть простыми металлическими панелями или сложными элементами, такими как, в частности, автомобильные кузова и их части.
До стадии (1) способа в соответствии с изобретением, металлические основы (S) могут быть предварительно обработаны традиционным способом - то есть, например, очищены и/или обеспечены известными покрытиями, химически взаимодействующими с основой. Очищение может осуществляться механически, например, посредством зачистки, шлифовки и/или полировки, и/или химически посредством методов травления, посредством начального протравливания в кислотных или щелочных ваннах, например, с помощью использования соляной или серной кислоты. Очищение органическими растворителями или водными растворами для очистки поверхности, конечно, также возможно. Предварительная обработка также может происходить посредством нанесения покрытий, химически взаимодействующих с основой, в частности, посредством фосфатирования и/или хроматирования, предпочтительно фосфатирования. В любом случае, металлические основы предпочтительно обеспечивают покрытиями, химически взаимодействующими с основой, в частности, фосфатируют, предпочтительно обеспечивают цинкфосфатным покрытием.
На стадии (1) способа в соответствии с изобретением, электрофоретическое нанесение материала для электроосаждения (е.1) на основу (S) и последующее отверждение материала для электроосаждения (е.1) применяют для того, чтобы получить на металлической основе (S) затвердевшее электроосажденное покрытие (Е.1).
Материал для электроосаждения (е.1), который применяют на стадии (1) способа в соответствии с изобретением, может быть катодным или анодным материалом для электроосаждения. Предпочтительным является катодный материал для электроосаждения. Материалы для электроосаждения были давно известны специалистам в данной области. Они представляют собой водные материалы покрытия, содержащие анионные или катионные полимеры в качестве связующих веществ. Указанные полимеры включают функциональные группы, которые потенциально являются анионными, что означает то, что они могут превращаться в анионные группы, например, группы карбоновой кислоты, или включают функциональные группы, которые потенциально являются катионными, что означает то, что они могут превращаться в катионные группы, например, аминогруппы. Превращение в заряженные группы, как правило, достигается в результате применения соответствующих нейтрализующих агентов (органических аминов (анионных), органических карбоновых кислот, таких как муравьиная кислота (катионные)), где при этом в результате получают анионные или катионных полимеры. Материалы для электроосаждения, как правило, и следовательно предпочтительно, дополнительно содержат обычные противокоррозионные пигменты. Катодные материалы для электроосаждения, которые являются предпочтительными в соответствии с изобретением, предпочтительно содержат катионные полимеры в качестве связующих веществ, в частности, простые полиэфирамины с гидроксильными функциональными группами, которые предпочтительно имеют ароматические структурные звенья. Такие полимеры, как правило, получают посредством реакции соответствующих эпоксидных смол на основе бисфенола с аминами, такими как, например, моно- и диалкиламины, алканоламины и/или диалкиламиноалкиламины. Указанные полимеры, в частности, применяются в комбинации с традиционными блокированными полиизоцианатами. В качестве примера можно сослаться на материалы для электроосаждения, описанные в публикациях WO 9833835 А1, WO 9316139 Al, WO 0102498 А1, и WO 2004018580 А1.
Таким образом, материал для электроосаждения (е.1) предпочтительно представляет собой по меньшей мере химически термоотверждаемый покрывающий материал, и в частности, является внешне сшивающимся. Предпочтительно, материал для электроосаждения (е.1) представляет собой химически термоотверждаемый однокомпонентный покрывающий материал.
Материал для электроосаждения (е.1), в качестве связующего вещества, предпочтительно содержит эпоксидную смолу с гидроксильными функциональными группами, и полностью блокированный полиизоцианат в качестве сшивающего агента. Предпочтительно, эпоксидная смола является катодной, в частности, содержит аминогруппы.
Также известным является электрофоретическое нанесение материала для электроосаждения (е.1) указанного вида, которое происходит на стадии (1) способа в соответствии с изобретением. Нанесение осуществляется электрофоретически. Это означает, что вначале металлическую деталь, предназначенную для покрытия, погружают в ванну для погружения, которая содержит покрывающий материал, и между металлической деталью и противоэлектродом применяется электрическое поле постоянного тока. Таким образом, деталь служит в качестве электрода; вследствие заряда на полимерах, применяемых в качестве связующих веществ, в результате действия электрического поля нелетучие составляющие компоненты материала для электроосаждения перемещаются к основе, и осаждаются на основе, в результате чего получают электроосажденную пленку. В случае катодного материала для электроосаждения, основа, например, соответственно, подсоединяется в качестве катода, а ионы гидроксида, которые образуются в результате электролиза воды, осуществляют нейтрализацию катионного связующего вещества, способствуя его осаждению на основе, и при этом формируется электроосажденная пленка. Таким образом, способ представляет один из способов нанесения посредством электрофоретического осаждения.
После нанесения материала для электроосаждения (е.1), покрытую основу (S) удаляют из резервуара, необязательно споласкивают, например, ополаскивающими растворами на основе воды, и затем необязательно подвергают самоиспарению и/или промежуточной сушке, и наконец, нанесенный материал для электроосаждения затвердевает.
Нанесенный материал для электроосаждения (е.1) (или нанесенную, но все еще не затвердевшую электроосажденную пленку) подвергают самоиспарению, например, при температуре 15 - 35°С, на протяжении периода времени, составляющего, например, 0,5 - 30 минут, и/или промежуточной сушке при температуре, составляющей предпочтительно 40 - 90°С, на протяжении периода времени, составляющего, например, 1-60 минут.
Материал для электроосаждения (е.1), нанесенный на основу (или нанесенная, но все еще не затвердевшая электроосажденная пленка), предпочтительно затвердевает при температурах, которые составляют 100 - 250°С, предпочтительно 140 - 220°С, на протяжении периода времени, составляющего 5-60 минут, предпочтительно 10-45 минут, в результате чего получают затвердевшее электроосажденное покрытие (Е.1).
Указанные условия самоиспарения, промежуточной сушка, и отверждения, в частности, применяются в предпочтительном случае, когда материал для электроосаждения (е.1) содержит химически термоотверждаемый однокомпонентный покрывающий материал, как описано выше. Это, однако, не исключает применения материала для электроосаждения, который представляет собой иным образом затвердевающий покрывающий материал, и/или применения отличающихся условий самоиспарения, промежуточной сушки, и отверждения.
Толщина пленки затвердевшего электроосажденного покрытия составляет, например, 10-40 микрометров, предпочтительно 15-25 микрометров. Любую толщину пленок, указанную в контексте настоящего изобретения, необходимо понимать, как толщину сухих пленок. Такой является толщина затвердевшей пленки в каждом случае. Следовательно, если указано, что покрывающий материал наносят с определенной толщиной пленки, то указанное означает, что покрывающий материал наносят таким образом, чтобы получать указанную толщину пленки после отверждения.
На стадии (2) способа в соответствии с изобретением, получают (2.1) пленку базового покрытия (В.2.1), или получают (2.2) две или большее количество нанесенных непосредственно друг на друга пленок базового покрытия (В.2.2.x). Пленки получают посредством нанесения (2.1) водного материала базового покрытия (b.2.1) непосредственно на затвердевшее электроосажденное покрытие (Е.1), или посредством (2.2) нанесения на затвердевшее электроосажденное покрытие (Е.1) двух или большего количества материалов базового покрытия (b.2.2.х) непосредственно друг на друга.
Таким образом, непосредственное нанесение друг на друга двух или большего количества материалов базового покрытия (b.2.2.x) на затвердевшее электроосажденное покрытие (Е.1) означает, что вначале непосредственно на электроосажденное покрытие наносят первый материал базового покрытия, и после этого непосредственно на пленку первого материала базового покрытия наносят второй материал базового покрытия. Затем непосредственно на пленку второго материала базового покрытия наносят необязательный третий материал базового покрытия. Таким же образом указанный способ может затем повторяться для последующих материалов базового покрытия (т.е. четвертого, пятого, и т.д., материала базового покрытия).
После этого, пленка базового покрытия (В.2.1) или первая пленка базового покрытия (В.2.2.x) располагается непосредственно на затвердевшем электроосажденном покрытии (Е.1).
Термины материал базового покрытия и пленка базового покрытия, в отношении покрывающих материалов, нанесенных и покрывающих пленок, полученных на стадии (2) способа в соответствии с изобретением, используют для большей простоты понимания. Пленки базового покрытия (В.2.1) и (В.2.2.x) не затвердевают отдельно, а вместо этого затвердевают вместе с материалом покровного лака. Отверждение, таким образом, происходит по аналогии с отверждением материалов базового покрытия, которые применяются в стандартном способе, описанном во введении. В частности, покрывающие материалы, которые применяются на стадии (2) способа в соответствии с изобретением, не затвердевают отдельно, подобно покрывающим материалам, указанным в качестве шпатлевок в стандартном способе.
Водный материал базового покрытия (b.2.1), который применяют на стадии (2.1), описан подробно далее ниже. При этом, в первом предпочтительном варианте осуществления, он является по меньшей мере химически термоотверждаемым, и более особо предпочтительно, он является внешне сшивающимся. В этом случае, материал базового покрытия (b.2.1) предпочтительно представляет собой однокомпонентный покрывающий материал. Материал базового покрытия (b.2.1) в этом случае предпочтительно содержит комбинацию по меньшей мере одного полимера с гидроксильными функциональными группами в качестве связующего вещества, выбранного из группы, состоящей из полиуретанов, сложных полиэфиров, полиакрилатов, и сополимеров указанных полимеров, примерам при этом являются полиуретан-полиакрилаты, а также по меньшей мере одна меламиновая смола в качестве сшивающего агента. Указанный вариант осуществления изобретения является особенно подходящим, когда, например, многослойная красочная система в соответствии с изобретением должна иметь чрезвычайно хорошей адгезией для склеивания стекла. Применение отверждаемых химически материалов базового покрытия означает, что вся конструкция, содержащая многослойную красочную систему и адгезионный слой, на который она нанесена, является в значительной степени более устойчивой, и в частности, не разрушается под действием механической растягивающей нагрузке, возникающей в пределах красочной системы, например, в пределах базового покрытия.
В зависимости от области применения, в равной степени возможным и, следовательно, вторым предпочтительным вариантом осуществления, является применение материалов базового покрытия (b.2.1), которые содержат только небольшое количество, составляющее менее 5 мас. %, предпочтительно менее 2,5 мас. %, из расчета общей массы материала базового покрытия, сшивающих агентов, такие как, в частности, меламиновые смолы. Дополнительно предпочтительным в этом варианте осуществления является то, что сшивающие агенты не присутствуют вообще. Несмотря на это, во всей конструкции достигается отличное качество. Дополнительное преимущество случая, когда не применяют сшивающие агенты и, следовательно, имеют менее сложный в составе покрывающий материал, заключается в увеличении свободы составления материала базового покрытия. Также может быть улучшен срок годности, вследствие предотвращения возможных реакций на части реакционноспособных компонентов.
Материал базового покрытия (b.2.1) может наноситься посредством способов, известных специалисту в данной области, предназначенных для нанесения жидких покрывающих материалов, например, посредством погружения, нанесения ножевым устройством, распыления, нанесения с помощью роликов, или подобного. Предпочтение отдают применению нанесения посредством методов распыления, таких как, например, распыление с использованием сжатого воздуха (пневматическое нанесение), распыление без использования воздуха, нанесение с использованием высокоскоростного вращения, нанесение с помощью электростатического напыления (ESTA), необязательно совместно с распылением с подогревом, например, с применением горячего воздуха (горячее распыление), например. Особенно предпочтительно, материал базового покрытия (b.2.1) наносят посредством пневматического нанесения посредством распыления или посредством нанесения с помощью электростатического напыления. Нанесение материала базового покрытия (b.2.1) соответственно обеспечивает пленку базового покрытия (В.2.1), другими словами, пленку материала базового покрытия (b.2.1), который наносят непосредственно на электроосажденное покрытие (Е.1).
После нанесения, нанесенный материал базового покрытия (b.2.1) или соответствующую пленку базового покрытия (В.2.1) подвергают самоиспарению, например, при температуре 15-35°С, на протяжении периода времени, составляющего, например, 0,5 - 30 минут, и/или промежуточной сушке при температуре, составляющей предпочтительно 40 - 90°С, на протяжении периода времени, составляющего, например, 1-60 минут.Предпочтение отдают самоиспарению, изначально при температуре 15 - 35°С на протяжении периода времени, составляющего 0,5 - 30 минут, за чем следует промежуточная сушка при температуре 40 - 90°С на протяжении периода времени, составляющего, например, 1-60 минут. Описанные условия самоиспарения и промежуточной сушки, в частности, применимы к предпочтительному случаю, где материал базового покрытия (b.2.1) является химически термоотверждаемым однокомпонентным покрывающим материалом. Указанное, однако, не исключает возможности, когда материал базового покрытия (b.2.1) является иным образом затвердевающим покрывающим материалом, и/или возможности применения отличающихся условий самоиспарения и/или промежуточной сушки.
Во время стадии (2) способа в соответствии с изобретением, пленка базового покрытия (В.2.1) не затвердевает, т.е. предпочтительно не подвергается действию температур, составляющих более 100°С, на протяжении периода времени, являющегося более длительным, чем 1 минута, и более предпочтительно, вообще не подвергается действию температур, составляющих более 100°С. Указанное является прямым и понятным следствием стадии (4) способа в соответствии с изобретением, которая описана далее ниже. Поскольку пленка базового покрытия затвердевает только на стадии (4), то она не может уже затвердевать на стадии (2), поскольку в этом случае отверждение на стадии (4) будет больше невозможным.
Водные материалы базового покрытия (b.2.2.х), которые применяются на стадии (2.2) способа в соответствии с изобретением, также описаны подробно далее ниже. В первом предпочтительном варианте осуществления, по меньшей мере один из материалов базового покрытия, который применяют на стадии (2.2), является по меньшей мере химически термоотверждаемым, и более предпочтительно является внешне сшивающимся. Более предпочтительно, указанное справедливо для всех материалов базового покрытия (b.2.2.х). Предпочтение в этом случае отдают по меньшей мере одному материалу базового покрытия (b.2.2.х), который при этом является однокомпонентным покрывающим материалом, и даже более предпочтительно, указанное справедливо для всех материалов базового покрытия (b.2.2.х). Предпочтительно, в этом случае по меньшей мере один из материалов базового покрытия (b.2.2.х), в качестве связующего вещества, содержит комбинацию по меньшей мере одного полимера с гидроксильными функциональными группами, выбранного из группы, состоящей из полиуретанов, сложных полиэфиров, полиакрилатов, и сополимеров указанных полимеров, таких, как например, полиуретан-полиакрилаты, а также по меньшей мере одну меламиновую смолу в качестве сшивающего агента. Более предпочтительно, указанное применяется для всех материалов базового покрытия (b.2.2.х). В свою очередь, указанный вариант осуществления изобретения является подходящим, когда цель состоит в достижении исключительно хорошей адгезии для склеивания стекла.
Тем не менее, также возможным и, следовательно, также предпочтительным вариантом осуществления, в зависимости от области применения, является применение по меньшей мере одного материала базового покрытия (b.2.2.х), который содержит только небольшое количество, составляющее менее 5 мас. %, предпочтительно менее 2,5 мас. %, сшивающих агентов, таких в частности, как меламиновые смолы, из расчета общей массы материала базового покрытия. Даже более предпочтительным в указанном варианте осуществления является, когда сшивающие агенты не содержатся вообще. Указанное выше применяется предпочтительно для всех материалов базового покрытия (b.2.2.х), которые применяют. Несмотря на это, во всей системе достигается отличное качество. Другие преимуществами являются отсутствие ограничений при составлении и стабильность при хранении.
Материалы базового покрытия (b.2.2.х) могут наноситься посредством способов, известных специалисту в данной области, предназначенных для нанесения жидких покрывающих материалов, например, посредством погружения, нанесения ножевым устройством, распыления, нанесения с помощью роликов или подобного. Предпочтение отдают применению нанесения посредством методов распыления, таких как, например, распыление с использованием сжатого воздуха (пневматическое нанесение), распыление без использования воздуха, нанесение с использованием высокоскоростного вращения, нанесение с помощью электростатического напыления (ESTA), необязательно совместно с распылением с подогревом, например, таким как использование горячего воздуха (горячее распыления). Особенно предпочтительно, материалы базового покрытия (b.2.2.х) наносят посредством пневматического распыления и/или нанесения с помощью электростатического напыления.
На стадии (2.2) способа в соответствии с изобретением, приняты следующие обозначения. Материалы базового покрытия и пленки базового покрытия, как правило, обозначаются как (b.2.2.х) и (В.2.2.x), причем х может заменяться другими буквами, которые соответственно подходят при обозначении определенных отдельных материалов базового покрытия и пленок базового покрытия.
Первый материал базового покрытия и первая пленка базового покрытия может обозначаться как а; самый верхний материал базового покрытия и самая верхняя пленка базового покрытия может обозначаться как z. Эти два материала базового покрытия и пленки базового покрытия в любом случае присутствуют на стадии (2.2). Любым пленкам между ними может предоставляться последовательное обозначение, такое как b, с, d и так далее.
В результате нанесения первого материала базового покрытия (b.2.2.а), соответственно, непосредственно на затвердевшем электроосажденном покрытии (Е.1) получают пленку базового покрытия (В.2.2,а). Затем, непосредственно на пленке базового покрытия (В.2.2.а) получают по меньшей мере одну дополнительную пленку базового покрытия (В.2.2.x). В случае, когда получают две или большее количество дополнительных пленок базового покрытия (В.2.2.x), то их получают в прямой последовательности. Например, можно получать только одну дополнительную пленку базового покрытия (В.2.2.x), в случае чего, указанная пленка располагается в многослойной красочной системе непосредственно под пленкой покровного лака (К), по сути, которая получена и, таким образом, может называться пленкой базового покрытия (B.2.2.z) (смотри также Фигуру 2). Также возможным, например, является получение двух дополнительных пленок базового покрытия (В.2.2.x), в случае чего, пленка, полученная непосредственно на базовом покрытии (В.2.2.а), может обозначаться как (B.2.2.b), и при этом пленка, расположенная непосредственно под пленкой покровного лака (K), в свою очередь, может обозначаться как (B.2.2.Z) (смотри также Фигуру 3).
Материалы базового покрытия (b.2.2.х) могут быть одинаковыми или разными. Также возможно при этом получать две или большее количество пленок базового покрытия (В.2.2.x) с применением одинакового материала базового покрытия, и одну или большее количество дополнительных пленок базового покрытия (В.2.2.x) получать с применением одного или большего количества разных материалов базового покрытия.
Нанесенные материалы базового покрытия (b.2.2.х), как правило, отдельно и/или совместно друг с другом, подвергают самоиспарению и/или промежуточной сушке. На стадии (2.2), предпочтительно, самоиспарение осуществляют при температуре 15-35°С на протяжении периода времени, составляющего 0,5 - 30 мин, и промежуточную сушку осуществляют при температуре 40 - 90°С на протяжении периода времени, составляющего, например, 1-60 мин. Последовательность самоиспарения и/или промежуточной сушки отдельных, или совместно двух или большего количества пленок базового покрытия (В.2.2.x) может быть подобрана в соответствии с требованиями конкретного случая. Описанные выше предпочтительные условия самоиспарения и промежуточной сушки применяются, в частности, к предпочтительному случаю, где по меньшей мере один материал базового покрытия (b.2.2.х), предпочтительно все материалы базового покрытия (b.2.2.х), содержат химически термоотверждаемые однокомпонентные покрывающие материалы. Указанное не исключает, однако, что материалы базового покрытия (b.2.2.х) при этом представляют собой покрывающие материалы, которые могут затвердевать разными способами, и/или не исключает применение разных условий самоиспарения и/или промежуточной сушки.
Если первую пленку базового покрытия получают посредством нанесения первого материала базового покрытия, и при этом дополнительную пленку базового покрытия получают посредством нанесения того же материала базового покрытия, то очевидно, что обе пленки основываются на одинаковом материале базового покрытия. Но при этом очевидно, что нанесение происходит в две стадии, что означает то, что материал базового покрытия, о котором идет речь, в смысле способа в соответствии с изобретением, соответствует первому материалу базового покрытия (b.2.2.а) и дополнительному материалу базового покрытия (b.2.2.z). Описанная система также часто упоминается как однослойная система пленки базового покрытия, полученная в два нанесения. Однако, поскольку во время фактического лакокрасочного покрытия на поточной линии (ППО), технические условия в случае автоматической линии для окраски всегда предписывают наличие определенного промежутка времени между первым нанесением и вторым нанесением, во время которого основу, например, автомобильной кузов доводят до нужной физической кондиции, например, при температуре 15-35°С, то есть при этом происходит самоиспарение, то формально будет более понятным охарактеризовать указанную систему как систему двухслойного базового покрытия. Таким образом, описанный рабочий режим должен быть отнесен ко второму варианту осуществления способа в соответствии с изобретением.
Ряд предпочтительных вариантов последовательностей пленок базового покрытия для материалов базового покрытия (b.2.2.х) может быть описан следующим образом.
Является возможным получить первую пленку базового покрытия, например, посредством нанесения с помощью электростатического напыления (ESTA) первого материала базового покрытия непосредственно на затвердевшее электроосажденное покрытие, осуществить самоиспарение и/или промежуточную сушку, как описано выше, и затем получить вторую пленку базового покрытия посредством непосредственного нанесения второго материала базового покрытия, который отличается от первого материала базового покрытия. Второй материал базового покрытия также может наноситься посредством электростатического напыления, тем самым получая вторую пленку базового покрытия непосредственно на первой пленке базового покрытия. Между и/или после нанесений является, конечно, возможным осуществлять самоиспарение и/или промежуточную сушку снова. Предпочтительно, указанный вариант осуществления стадии (2.2) выбирают, когда прежде всего подготовительная для нанесения цвета пленка базового покрытия, как описано более подробно далее ниже, должна быть получена непосредственно на электроосажденном покрытии, а затем непосредственно на первой пленке базового покрытия должна быть получена придающая цвет- и/или эффект пленка базового покрытия, как описано более подробно далее ниже. В этом случае первая пленка базового покрытия получена на основе подготовительного для нанесения цвета материала базового покрытия, вторая пленка базового покрытия получена на основе придающего цвет- и/или эффект материала базового покрытия. Является также возможным, например, применять указанный второй материал базового покрытия в две стадии, как описано выше, тем самым получая две дополнительные, нанесенные непосредственно друг на друга пленки базового покрытия, непосредственно на первой пленке базового покрытия.
Является также возможным получать три пленки базового покрытия, в прямой последовательности, непосредственно на затвердевшем электроосажденном покрытии, причем пленки базового покрытия представляют собой пленки на основе трех разных материалов базового покрытия. Например, могут быть получены подготовительная для нанесения цвета пленка базового покрытия, дополнительная пленка на основе придающего цвет- и/или эффект материала базового покрытия, и дополнительная пленка на основе второго придающего цвет- и/или эффект материала базового покрытия. Между и/или после отдельных нанесений и/или после всех трех нанесений, в свою очередь, возможно осуществлять самоиспарение и/или промежуточную сушку. Таким образом, предпочтительные в контексте настоящего изобретения варианты осуществления содержат получение на стадии (2.2) способа в соответствии с изобретением двух или трех пленок базового покрытия. В этом случае является предпочтительным, когда пленка базового покрытия, полученная непосредственно на затвердевшем электроосажденном покрытии, основана на подготовительном для нанесения цвета материале базового покрытия. Вторая и любая третья пленка основываются либо на одном и том же придающем цвети/или эффект материале базового покрытия, или на первом придающем цвети/или эффект материале базового покрытия и на отличающемся втором придающем цвет- и/или эффект материале базового покрытия. В одном предпочтительном варианте осуществления, материалы базового покрытия, которые наносят на пленку на основе подготовительного для нанесения цвета материала базового покрытия, в любом случае содержат, но не обязательно исключительно, эффектные пигменты и/или хроматические пигменты. Хроматические пигменты представляют собой часть группы цветных пигментов, последние при этом также включают ахроматические цветные пигменты, такие как черные или белые пигменты.
Во время стадии (2) способа в соответствии с изобретением, пленки базового покрытия (В.2.2.x) не затвердевают - то есть, предпочтительно они не подвергаются действию температур, составляющих более 100°С, на протяжении периода времени, являющегося более продолжительным, чем 1 минута, и предпочтительно не подвергаются действию температур, составляющих более 100°С, вообще. Это ясно и непосредственно видно из стадии (4) способа в соответствии с изобретением, описанной далее ниже. По той причине, что пленки базового покрытия затвердевают только на стадии (4), они не могут быть уже затвердевшими на стадии (2), поскольку в этом случае отверждение на стадии (4) будет больше невозможным.
Материалы базового покрытия (b.2.1) и (b.2.2.х) наносят таким образом, что пленка базового покрытия (В.2.1), а также отдельные пленки базового покрытия (В.2.2.x), после отверждения, которое происходило на стадии (4), имеют толщину пленки, которая составляет, например, 5-50 микрометров, предпочтительно 6 -40 микрометров, особенно предпочтительно 7-35 микрометров. На стадии (2.1), предпочтение отдают получению пленок более высокой толщины, составляющей 15-50 микрометров, предпочтительно 20 - 45 микрометров. На стадии (2.2), отдельные пленки базового покрытия имеют тенденцию к меньшей толщине пленок, в то время как вся система при этом снова в результате имеет толщину пленок, которые находятся в пределах величины одной пленки базового покрытия (В.2.1). В случае двух пленок базового покрытия, например, первая пленка базового покрытия (В.2.2.а) предпочтительно имеет толщину, составляющую 5 - 35, в частности, 10-30, микрометров, и вторая пленка базового покрытия (B.2.2.Z) предпочтительно имеет толщину, составляющую 5 -35 микрометров, в частности, 10-30 микрометров, при этом вся толщина пленок не превышает 50 микрометров.
На стадии (3) способа в соответствии с изобретением, пленку покровного лака (K) получают непосредственно (3.1) на пленке базового покрытия (В.2.1) или (3.2) на самой верхней пленке базового покрытия (B.2.2.z). Указанное достигается посредством соответствующего нанесения материала покровного лака (k).
Материал покровного лака (k) может быть любым желательным прозрачным покрывающим материалом, известным в указанном смысле специалисту в данной области. "Прозрачный" означает, что пленка, образованная таким покрывающим материалом, не является непрозрачно окрашенной, а вместо этого имеет такую структуру, что цветная располагающаяся под ней система базового покрытия является видимой. Однако, как известно, указанное не исключает возможности включения в материал покровного лака, в незначительных количествах, пигментов, при этом указанные пигменты, например, могут поддерживать интенсивность цвета всей система.
Покрывающие материалы, о которых идет речь, являются водными или содержащими растворитель прозрачными покрывающими материалами, которые могут составляться не только в качестве однокомпонентных, а также в качестве двухкомпонентных или многокомпонентных покрывающих материалов. Также, кроме того, подходящими являются материалы покровного лака в виде суспензии порошка. При этом основанные на растворителях материалы покровного лака являются предпочтительными.
Материалы покровного лака (k), которые применяют, в частности, могут быть химически термоотверждаемыми и/или актинично-химически отверждаемыми. В частности, они являются химически термоотверждаемыми и внешне сшивающимися. Предпочтение отдают химически термоотверждаемым двухкомпонентным материалам покровного лака.
Таким образом, обычно и предпочтительно, материалы покровного лака содержат по меньшей мере один (первый) полимер в качестве связующего вещества, имеющий функциональные группы, и по меньшей мере один сшивающий агент, имеющий функциональную группу, дополняющую функциональные группы связующего вещества. Предпочтительно, в качестве связующего вещества применяют по меньшей мере один поли(мет)акрилатный полимер с гидроксильными функциональными группами, и свободный полиизоцианат в качестве сшивающего агента.
Подходящие материалы покровного лака описаны, например, в публикациях WO 2006042585 Al, WO 2009077182 А1, или также WO 2008074490 А1.
Материал покровного лака (k) наносят посредством способов, известных специалисту в данной области, предназначенных для нанесения жидких покрывающих материалов, например, посредством погружения, нанесения ножевым устройством, распыления, нанесение с помощью роликов, или подобного. Предпочтение отдают применению нанесение посредством методов распыления, таких как, например, распыление с использованием сжатого воздуха (пневматическое нанесение), и нанесение с помощью электростатического напыления (ESTA).
После нанесения, материал покровного лака (k) или соответствующую пленку покровного лака (K) подвергают самоиспарению и/или промежуточной сушки, предпочтительно при температуре 15 - 35°С на протяжении периода времени, составляющего 0,5 - 30 минут. Указанные условия самоиспарения и промежуточной сушки в частности, применяются в том предпочтительном случае, когда материал покровного лака (к) содержит химически термоотверждаемый двухкомпонентный покрывающий материал. Но при этом указанное не исключает применение материала покровного лака (k), который представляет собой покрывающий материал, затвердевающий иным образом, и/или применение других условий самоиспарения и/или промежуточной сушки.
Материал покровного лака (k) наносят таким образом, что пленка покровного лака после отверждения, которое происходило на стадии (4), имеет толщину пленки, которая составляет, например, 15-80 микрометров, предпочтительно 20 - 65 микрометров, особенно предпочтительно 25 -60 микрометров.
В способе в соответствии с изобретением, конечно, не исключены дополнительные материалы покрытия, такие, как например, дополнительные материалы покровного лака, которые наносятся после нанесения материала покровного лака (k), а также дополнительных покрывающих пленок, таких, как например, дополнительные пленки покровного лака, которые получают указанным способом. Такие дополнительные покрывающие пленки также затем затвердевают на стадии (4), описанной ниже. Тем не менее, является предпочтительным, когда наносят только один материал покровного лака (k), и затем он затвердевает, как описано на стадии (4).
На стадии (4) способа в соответствии с изобретением происходит совместное отверждение (4.1) пленки базового покрытия (В.2.1) и пленки покровного лака (K) или (4.2) пленок базового покрытия (В.2.2.x) и пленки покровного лака (K).
Совместное отверждение происходит предпочтительно при температурах, которые составляют 100 - 250°С, предпочтительно 100 - 180°С, на протяжении периода времени, составляющего 5-60 минут, предпочтительно 10-45 минут. Указанные условия отверждения применяют, в частности, в том предпочтительном случае, когда пленка базового покрытия (В.2.1) или по меньшей мере одна из пленок базового покрытия (В.2.2.x), предпочтительно все пленки базового покрытия (В.2.2.x), основаны на химически термоотверждаемом однокомпонентном покрывающем материале. Причина состоит в том, что, как описано выше, такие условия, как правило, необходимы для достижения отверждения однокомпонентного покрывающего материала указанного вида, как описано выше. В случае, когда материал покровного лака (k), например, также является химически термоотверждаемым однокомпонентным покрывающим материалом, то конечно, соответствующая пленка покровного лака (K) также затвердевает в указанных условиях. То же самое верно и для предпочтительного случая, где покровный лак (k) является химически термоотверждаемым двухкомпонентным покрывающим материалом.
Однако, сделанные выше указания не исключают применения материалов базового покрытия (b.2.1) и (b.2.2.х), а также материалов покровного лака (k), которые представляют собой иным образом затвердевающие покрывающие материалы, и/или применения других условий отверждения.
Результатом после завершения стадии (4) способа в соответствии с изобретением является многослойная красочная система в соответствии с изобретением (смотри также Фигуры 1-3).
Материалы базового покрытия, предназначенные для применения в соответствии с изобретением
Материал базового покрытия (b.2.1), предназначенной для применения в соответствии с изобретением, содержит по меньшей мере одну, предпочтительно ровно одну, определенную водную полиуретан-полимочевинную дисперсию (ПД).
Таким образом, полимерные частицы, которые присутствуют в дисперсии, основаны на полиуретане и полимочевине. В принципе, такие полимеры можно получить, например, посредством традиционного полиприсоединения полиизоцианатов с полиолами, а также полиаминами. При этом, что касается дисперсии (ПД), предназначенной для применения в соответствии с изобретением, а также полимерных частиц, присутствующих в ней, существуют определенные условия, которые должны быть соблюдены, и которые будут объяснены ниже.
Частицы полиуретана и полимочевины, которые присутствуют в водной полиуретан-полимочевинной дисперсии (ПД), содержат гель-фракцию, которая составляет по меньшей мере 50% (относительно метода определения смотри раздел Примеров). Более того, частицы полиуретана и полимочевины, которые присутствуют в дисперсии (ПД), имеют средний размер частиц, который составляет 40 - 2000 нанометров (нм) (относительно метода определения смотри раздел Примеров).
Таким образом, дисперсии (ПД) в соответствии с изобретением являются микрогелевыми дисперсиями. Микрогелевая дисперсия, на самом деле, как известно, представляет собой полимерную дисперсию, в которой, во-первых, полимер присутствует в виде сравнительно небольших частиц, имеющих размеры, составляющие, например, 0,02 - 10 микрометров ("микро"-гель). Во-вторых, полимерные частицы являются, по меньшей мере частично, сшитыми внутримолекулярно. Значение последней фразы состоит в том, что полимерные структуры, которые присутствуют в пределах частицы, приравниваются к обычной макроскопической сетка с трехмерной сетчатой структурой. Однако, с макроскопической точки зрения, микрогелевая дисперсия указанного вида при этом является дисперсией полимерных частиц в дисперсионной среде, например, воде. В то время, как частицы также могут частично иметь сшивающие друг с другом мостики (это вряд ли можно исключаться, не в последнюю очередь вследствие способа получения), система, при этом, по меньшей мере представляет собой дисперсию с присутствующими в ней дискретными частицами, которые имеют поддающийся измерению средний размер частиц. Однако, вследствие своей молекулярной природы, указанные частицы растворяются в подходящих органических растворителях; макроскопические сетки, в отличие от этого, будут только набухать.
Поскольку микрогели представляют собой структуры, которые находятся между разветвленными и макроскопически сшитыми системами, и, следовательно, объединяют свойства макромолекул с сетчатой структурой, которые растворяются в подходящих органических растворителях, и свойства нерастворимых макроскопических сеток, то фракция сшитых полимеров может, например, определяться только после выделения твердого полимера, посредством удаления воды и любых органических растворителей, а также последующей экстракции. Используемый в этом случае механизм состоит в том, что микрогелевые частицы, изначально растворимые в подходящих органических растворителях, сохраняют свою внутреннюю сетчатую структуру после выделения, и ведут себя в твердом виде подобно макроскопической сетке. Сшивание может подтверждаться посредством получаемой экспериментально гель-фракции. Гель-фракция, по сути, является той частью полимера дисперсии, который, в виде выделенного твердого вещества, не может молекулярно дисперсионно растворяться в растворителе. В указанном контексте необходимо исключить возможность дополнительного увеличения гель-фракции в результате последующих реакций сшивания во время выделения полимерного твердого вещества. В свою очередь, указанная нерастворимая фракция соответствует фракции полимера, которая присутствует в дисперсии в виде внутримолекулярно сшитых частиц или фракций частиц.
В контексте настоящего изобретения выяснилось, что только микрогелевые дисперсии, включающие при этом полимерные частицы с размерами, находящимися в диапазоне, имеющем значение для изобретения, имеют все необходимые эксплуатационные свойства. Таким образом, важное значение, в частности, имеет комбинация частиц относительно небольших размеров с существенной при этом сшитой фракцией или гель-фракцией. Только таким образом возможно достичь преимущественных характеристик, в частности, комбинации хороших оптических и механических свойств многослойных красочных систем, с одной стороны, и высокого содержания твердых частиц, а также хорошей стабильности при хранении водных материалов базового покрытия, с другой стороны. Таким образом, например, дисперсии, которые при этом имеют сравнительно более крупные частицы, составляющие примерно более 2 микрометров (средний размер частиц), демонстрируют, к примеру, повышенное протекание процесса осаждения и, следовательно, более низкую стабильность при хранении.
Частицы полиуретана и полимочевины, которые присутствуют в водной полиуретан-полимочевинной дисперсии (ПД), предпочтительно содержат гель-фракцию, которая составляет по меньшей мере 60%, более предпочтительно по меньшей мере 70%, особенно предпочтительно по меньшей мере 80%. Гель-фракция может, таким образом, составлять до 100% или примерно 100%, например, 99% или 98%. Тогда, в таком случае, весь, или практически весь, полиуретан-полимочевинный полимер представлен в виде сшитых частицы.
Частицы полиуретана и полимочевины, которые присутствуют в дисперсии (ПД), предпочтительно имеют средний размер частиц, который составляет 40 -1500 нм, более предпочтительно 100 - 1000 нм, предпочтительно включая при этом 110 - 500 нм, и более предпочтительно 120 - 300 нм. Особенно предпочтительный диапазон составляет от 130 до 250 нм.
Полученная полиуретан-полимочевинная дисперсия (ПД) является водной. Выражение "водная" является известной специалисту в данной области в указанном контексте. Оно, по сути, относится к системе, которая в качестве своей дисперсионной среды исключительно не содержит, или в основном не содержит, органических растворителей (которые также называют растворителями), а которая, вместо этого, в качестве дисперсионной среды включает значительную фракцию воды. Предпочтительные варианты осуществления водной дисперсии, которые определяются посредством максимального содержания органических растворителей и/или посредством содержания воды, описаны далее ниже.
Частицы полиуретана и полимочевины содержат анионные группы и/или группы, которые могут превращаться в анионные группы (то есть, группы, которые, в результате применения известных нейтрализующих агентов, указанных далее ниже, таких как основания, могут превращаться в анионные группы).
Как известно специалисту в данной области, группами, о которых идет речь, в данном случае представляют собой, например, группы карбоновой, сульфоновой и/или фосфоновой кислоты, особенно предпочтительными являются группы карбоновой кислоты (функциональные группы, которые могут превращаться в анионные группы посредством нейтрализующих агентов), а также анионные группы, полученные из упомянутых выше функциональных групп, такие как, в частности, карбоксилатные, сульфонатные и/или фосфонатные группы, предпочтительно карбоксилатные группы. Известным эффектом от введения таких групп является повышение дисперсности в воде. В зависимости от выбранных условий, указанные группы могут присутствовать частично, или почти полностью, в одном виде (например, карбоновой кислоты) или в другом виде (карбоксилатные). Определяющим влияющим фактором является, например, применение упомянутых выше нейтрализующих агентов, дополнительные подробности в отношении которых приведены в описании ниже. Независимо от того, какой вид имеют указанные группы, часто, в контексте настоящего изобретения, для большей простоты понимания, выбирается единая номенклатура. Например, в случае, когда для полимера указано конкретное кислотное число, или, когда полимер указан как такой, который имеет карбоксильные функциональные группы, то в этом случае всегда ссылаются как на группы карбоновой кислоты, так и на карбоксилатные группы. Если в этом отношении нужна какая-либо дифференциация, то это осуществляют, применяя, например, степень нейтрализации.
Указанные группы могут вводиться в полимеры, такие как частицы полиуретана и полимочевины, например, посредством известного применения соответствующих исходных соединений для получения полимеров. В таком случае, исходные соединения содержат группы, о которых идет речь, например, группы карбоновой кислоты, и сополимеризуются в полимер посредством дополнительных функциональных групп, например, гидроксильных групп. Подробности при этом описаны далее ниже.
Предпочтительные анионные группы и/или группы, которые могут превращаться в анионные группы, представляют собой карбоксилатные группы и группы карбоновой кислоты, соответственно. На основе содержания твердых частиц, полиуретан-полимочевинный полимер, который присутствует в дисперсии в виде частиц, предпочтительно имеет кислотное число, которое составляет 10 - 35 мг КОН/г, в частности, которое составляет 15 - 23 мг КОН/г (относительно метода определения смотри раздел Примеров).
Частицы полиуретана и полимочевины, которые присутствуют в дисперсии (ПД), предпочтительно содержат, в каждом случае в прореагировавшем виде, (Z.1.1) по меньшей мере один преполимер полиуретана, включающий изоцианатные группы и имеющий анионные группы и/или группы, которые могут превращаться в анионные группы, а также (Z.1.2) по меньшей мере один полиамин, содержащий две первичные аминогруппы и одну или две вторичные аминогруппы.
Если в контексте настоящего изобретения указано, что полимеры, такие как частицы полиуретана и полимочевины дисперсии (ПД), например, содержат определенные компоненты в прореагировавшем виде, то это означает, что указанные определенные компоненты применяют в качестве исходных соединений при получении полимеров, о которых идет речь. В зависимости от природы исходных соединений, определенные реакции, в результате которых получают заданный полимер, происходят в соответствии с разными механизмами. Несомненно, затем, при получении частиц полиуретана и полимочевины или полиуретан-полимочевинных полимеров, компоненты (Z.1.1) и (Z.1.2) вступают в реакцию друг с другом в результате реакции изоцианатных групп компонента (Z.11) с аминогруппами компонента (Z.12), с получением мочевинных связей. В этом случае, полимер, конечно, содержит аминогруппы и изоцианатные группы, которые присутствуют до этого, в виде групп мочевины -то есть, в их соответственно прореагировавшем виде. По сути, тем не менее, полимер содержит два компонента (Z.1.1) и (Z.1.2), поскольку, кроме прореагировавших изоцианатных групп и аминогрупп, компоненты остаются неизменными. Для простоты понимания, соответственно, говорят, что полимер, о котором идет речь, содержит компоненты, в каждом случае в прореагировавшем виде. Значение выражения "полимер содержит компонент (X) в прореагировавшем виде" может, таким образом, соответствовать значению выражения "при получении полимера, применяли компонент (X)".
Из приведенного выше следует, что анионные группы и/или группы, которые могут превращаться в анионные группы, вводятся в частицы полиуретана и полимочевины, предпочтительно посредством упомянутого выше преполимера полиуретана, содержащего изоцианатные группы.
Частицы полиуретана и полимочевины предпочтительно состоят из двух компонентов (Z.1.1) и (Z.1.2) - то есть, их получают на основе указанных двух компонентов.
Предпочтительно, водную дисперсию (ПД) могут получать посредством определенного трехстадийного способа. В качестве части описания указанного способа, точно также указаны предпочтительные варианты осуществления компонентов (Z.1.1) и (Z.1.2).
Способ при этом содержит i
(I)
изготовление состава (Z), который включает, в каждом случае, из расчета общего количества состава (Z),
(Z.1) 15 - 65 мас. % по меньшей мере одного промежуточного соединения, содержащего изоцианатные группы и имеющего при этом блокированные первичные аминогруппы, полученные в результате реакции (Z.1.1) по меньшей мере одного преполимера полиуретана, содержащего изоцианатные группы и содержащего анионные группы и/или группы, которые могут превращаться в анионные группы, с (Z.1.2а) по меньшей мере одним полиамином, содержащим две блокированные первичные аминогруппы и одну или две свободные вторичные аминогруппы
посредством реакции присоединения изоцианатных групп компонента (Z.1.1) со свободными вторичными аминогруппами из компонента (Z.1.2), (Z.2) 35 - 85 мас. % по меньшей мере одного органического растворителя, который имеет растворимость в воде, составляющую не более 38 мас. %, при температуре 20°С
(II)
диспергирование состава (Z) в водной фазе, и
(III)
по меньшей мере частичное удаление из дисперсии, полученной на стадии (II), по меньшей мере одного органического растворителя (Z.2).
Таким образом, на первой стадии (I) указанного способа получают определенный состав (Z).
Состав (Z) содержит по меньшей мере одно, предпочтительно ровно одно, определенное промежуточное соединение (Z.1), которое включает изоцианатные группы и имеет блокированные первичные аминогруппы.
Получение промежуточного соединения (Z.1) включает реакцию по меньшей мере одного преполимера полиуретана (Z.1.1), содержащего изоцианатные группы и содержащего анионные группы и/или группы, которые могут превращаться в анионные группы, по меньшей мере с одним полиамином (Z.1.2а), который получают из полиамина (Z.1.2), и который содержит по меньшей мере две блокированные первичные аминогруппы и по меньшей мере одну свободную вторичную аминогруппу.
Полиуретановые полимеры, содержащие изоцианатные группы и содержащие анионные группы и/или группы, которые могут превращаться в анионные группы, являются, в принципе, известными. В контексте настоящего изобретения, для большей простоты понимания, компонент (Z.1.1) упоминается как преполимер. Этот компонент фактически является полимером, который может быть упомянут как предшественник, поскольку его применяют в качестве исходного компонента для получения другого компонента, в частности, промежуточного соединения (Z.1).
Для получения преполимеров полиуретана (Z.1.1), содержащих изоцианатные группы и содержащих анионные группы и/или группы, которые могут превращаться в анионные группы, является возможным применять алифатические, циклоалифатические, алифатические-циклоалифатические, ароматические, алифатические-ароматические и/или циклоалифатические-ароматические полиизоцианаты, которые известны специалисту в данной области. Предпочтение отдают применению диизоцианатов. Следующие диизоцианаты могут быть указаны в качестве примера: 1,3- или 1,4-фенилен диизоцианат, 2,4- или 2,6-толилендиизоцианат, 4,4'- или 2,4'-дифенилметандиизоцианат, 1,4- или 1,5-нафтилендиизоцианат, простой диизоцианатодифениловый эфир, триметилендиизоцианат, тетраметилендиизоцианат, этилэтилендиизоцианат, 2,3-диметилэтилендиизоцианат, 1 -метилтриметилендиизоцианат, пентаметилендиизоцианат, 1,3-циклопентилендиизоцианат, гексаметилендиизоцианат, циклогексилендиизоцианат, 1,2-циклогексилендиизоцианат, октаметилендиизоцианат, триметилгександиизоцианат, тетраметилгександиизоцианат, декаметилендиизоцианат, додекаметилендиизоцианат,
тетрадекаметилендиизоцианат, изофорондиизоцианат (ИФДИ), 2-изоцианато-пропилциклогексилизоцианат, дициклогексилметан 2,4'-диизоцианат, дициклогексилметан 4,4'-диизоцианат, 1,4- или 1,3-бис(изоцианатометил)циклогексан, 1,4- или 1,3- или 1,2-диизоцианатоциклогексан, 2,4- или 2,6-диизоцианато-1-метилциклогексан, 1-изоцианатометил-5-изоцианато-1,3,3-триметилциклогексан, 2,3-бис(8-изоцианатооктил)-4-октил-5-гексилциклогексен, тетраметилксилилендиизоцианаты (ТМКДИ), такие как м-тетраметилксилилендиизоцианат, или смеси указанных полиизоцианатов. Конечно, также возможно применение разных димеров и тримеров указанных диизоцианатов, таких как уретдионы и изоцианураты. Полиизоцианаты с большим количеством изоцианатных функциональных групп также могут применяться. Их примерами являются трис(4-изоцианатофенил)метан, 1,3,4-триизоцианатобензол, 2,4,6-триизоцианатотолуол, 1,3,5-трис(6-изоцианатогексилбиурет), бис-(2,5-диизоцианато-4-метилфенил)метан. Необязательно, количество функциональных групп может понижаться посредством реакции с моноспиртами и/или вторичными аминами. Предпочтение, однако, отдают применению диизоцианатов, более предпочтительно применению алифатических диизоцианатов, таких как гексаметилендиизоцианат, изофорондиизоцианат (ИФДИ), дициклогексилметан 4,4-диизоцианат, 2,4- или 2,6-диизоцианато-1-метилциклогексан, и м-тетраметилксилилендиизоцианат (м-ТМКДИ). Изоцианат называют алифатическим, если изоцианатные группы присоединены к алифатическим группам; другими словами, когда отсутствует ароматический углерод, который обычно присутствует в альфа-положении по отношению к изоцианатной группе.
При получении преполимеров (Z.1.1), полиизоцианаты вступают в реакцию с полиолами, в частности, с диолами, как правило, с образованием уретанов.
Примеры подходящих полиолов представляют собой насыщенные или олефинненасыщенные сложные полиэфиры полиолов и/или простые полиэфиры полиолов. В частности, применяемыми в качестве полиолов являются сложные полиэфиры полиолов, особенно те, которые имеют среднечисленную молекулярную массу, которая составляет 400 - 5000 г/моль (относительно метода определения смотри раздел Примеров). Такие сложные полиэфиры полиолов, предпочтительно сложные полиэфиры диолов, могут быть получены известным способом в результате реакции соответствующих поликарбоновых кислот, предпочтительно дикарбоновых кислот, и/или их ангидридов, с соответствующими полиолами, предпочтительно диолами, посредством эстерификации. Необязательно, конечно, возможно дополнительно, даже частично, применять для получения монокарбоновые кислоты и/или моноспирты. Предпочтительно, сложные полиэфиры диолов являются насыщенными, в частности, насыщенными и неразветвленными.
Примерами подходящих ароматических поликарбоновых кислот для получения таких сложных полиэфиров полиолов, предпочтительно сложных полиэфиров диолов, являются фталевая кислота, изофталевая кислота, а также терефталевая кислота, из которых изофталевая кислота является преимущественной и, таким образом, применяется предпочтительно. Примерами подходящих алифатических поликарбоновых кислот являются щавелевая кислота, малоновая кислота, янтарная кислота, глутаровая кислота, адипиновая кислота, пимелиновая кислота, субериновая кислота, азелаиновая кислота, себациновая кислота, ундекандикарбоновая кислота, и додекандикарбоновая кислота, или также гексагидрофталевая кислота, 1,3-циклогександикарбоновая кислота, 1,4-циклогександикарбоновая кислота, 4-метилгексагидрофталевая кислота, трициклодекандикарбоновая кислота, и тетрагидрофталевая кислота. В качестве дикарбоновых кислот является также возможным применять димерные жирные кислоты, или димеризованные жирные кислоты, которые, как это является известным, представляют собой смеси, полученные в результате димеризации ненасыщенных жирных кислот, и являются доступными под торговым наименованиями Radiacid (от компании Oleon) или Pripol (от компании Croda), например. В контексте настоящего изобретения, применение таких димерных жирны кислот для получения сложных полиэфиров диолов является предпочтительным. Полиолы, которые предпочтительно применяют для получения преполимеров (Z.1.1), таким образом, представляют собой сложные полиэфиры диолов, которые получали, применяя димерные жирные кислоты. Особенно предпочтительными являются те сложные полиэфиры диолов, при получении которых применяют по меньшей мере 50 мас. %, предпочтительно 55 to 75 мас. %, дикарбоновых кислот, которые представляют собой димерные жирные кислоты.
Примерами соответствующих полиолов для получения сложных полиэфиров полиолов, предпочтительно сложных полиэфиров диолов, являются этиленгликоль, 1,2-, или 1,3-пропандиол, 1,2-, 1,3-, или 1,4-бутандиол, 1,2-, 1,3-, 1,4-, или 1,5-пентандиол, 1,2-, 1,3-, 1,4-, 1,5-, или 1,6-гександиол, неопентилгидроксипивалат, неопентилгликоль, диэтиленгликоль, 1,2-, 1,3-, или 1,4-циклогександиол, 1,2-, 1,3-, или 1,4-циклогександиметанол, и триметилпентандиол. Предпочтение, таким образом, отдают применению диолов. Такие полиолы и/или диолы могут, конечно, также применяться непосредственно для получения преполимера (Z.1.1), другими словами, они вступают в реакцию непосредственно с полиизоцианатами.
Дополнительно возможными для применения при получении преполимеров (Z.1.1) являются полиамины, такие как диамины и/или аминоспирты. Примеры диаминов включают гидразин, алкил- или циклоалкилдиамины, такие как пропилендиамин и 1-амино-3-аминометил-3,5,5-триметилциклогексан, и примеры аминоспиртов включают этаноламин или диэтаноламин.
Преполимеры (Z.1.1) содержат анионные группы и/или группы, которые могут превращаться в анионные группы. Для того чтобы ввести указанные группы, является возможным, во время получения преполимеров (Z. 1.1), применять исходные соединения, которые также, как и группы, применяемые для реакции при получении уретановых связей, предпочтительно гидроксильные группы, дополнительно содержат упомянутые выше группы, например, группы карбоновой кислоты. Таким образом группы, о которых идет речь, вводятся в преполимер.
Соответствующие соединения, предполагаемые для введения предпочтительных групп карбоновой кислоты, представляют собой простые полиэфиры полиолов и/или сложные полиэфиры полиолов, при условии, что они содержат карбоксильные группы. Однако, соединения, который применяют предпочтительно, представляют собой по меньшей мере низкомолекулярные соединения, которые имеют по меньшей мере одну группу карбоновой кислоты и по меньшей мере одну функциональную группу, реакционноспособную в отношении изоцианатных групп, предпочтительно гидроксильные группы. В контексте настоящего изобретения, выражение "низкомолекулярное соединение", которое противопоставляется высокомолекулярным соединениям, в частности полимерам, необходимо понимать, как такое, которое означает соединение, которое может иметь дискретную молекулярную массу, предпочтительно, мономерные соединения. Таким образом, в частности, низкомолекулярное соединение не является полимером, поскольку последнее всегда является смесью молекул и его необходимо описывать, применяя среднюю молекулярную массу. Предпочтительно, термин "низкомолекулярное соединение" понимают, как такое, которая означает, что соответствующие соединения имеют молекулярную массу, которая составляет менее 300 г/моль. Диапазон от 100 до 200 г/моль является предпочтительным.
Соединения, предпочтительные в указанном смысле, например, представляют собой монокарбоновые кислоты, содержащий две гидроксильные группы, такие как дигидроксипропионовая кислота, дигидроксиянтарная кислота, и дигидроксибензойная кислота, например. В частности, примерами являются альфа,альфа-диметилолалкановые кислоты, такие как 2,2-диметилолуксусная кислота, 2,2-диметилолпропионовая кислота, 2,2-диметилолмасляная кислота, и 2,2-диметилолпентановая кислота, в частности 2,2-диметилолпропионовая кислота.
Таким образом, преполимеры (Z.1.1) предпочтительно имеют карбоксильные функциональные группы. Предпочтительно они имеют кислотное число, на основе содержания твердых частиц, которое предпочтительно составляет 10 - 35 мг КОН/г, в частности, 15 - 23 мг КОН/г.
Среднечисленная молекулярная масса преполимеров может варьироваться в широком диапазоне, и, например, находится в диапазоне от 2000 до 20000 г/моль, предпочтительно от 3500 до 6000 г/моль (относительно метода определения смотри раздел Примеров).
Преполимер (Z. 1.1) содержит изоцианатные группы. Предпочтительно, он имеет содержание изоцианатов, на основе содержания твердых частиц, которое составляет 0,5 - 6,0 мас. %, предпочтительно 1,0 - 5,0 мас. %, особенно предпочтительно 1,5 - 4,0 мас. % (относительно метода определения смотри Пример раздел).
Поскольку преполимер (Z.1.1) содержит изоцианатные группы, гидроксильное число преполимера will очевидно быть очень низкие в качестве general rule. Гидроксильное число преполимера, на основе содержания твердых частиц, предпочтительно составляет менее 15 мг КОН/г, в частности, менее 10 мг КОН/г, и дополнительно предпочтительно менее 5 мг КОН/г (относительно метода определения смотри раздел Примеров).
Преполимеры (Z.1.1) могут быть получены посредством известных и общепризнанных способов, в виде твердого вещества или в виде раствора, особенно предпочтительно посредством реакции исходных соединений в органических растворителях, таких как метилэтилкетон, предпочтительно, при температурах, которые составляют, например, 60 - 120°С, и необязательно с применением катализаторов, обычных при получении полиуретана. Такие катализаторы являются известными специалисту в данной области; примером является лаурат дибутилолова. В этом случае, конечно, необходимо выбрать пропорцию исходных компонентов, таким образом, чтобы продукт, другими словами, преполимер (Z.1.1), содержал изоцианатные группы. Также конечно очевидно, что растворители необходимо выбирать таким образом, чтобы они не вступали в какие-либо нежелательные реакции с функциональными группами исходных соединений, другими словами, были инертными в отношении указанных групп, чтобы они не препятствовали реакции указанных функциональных групп. Получение предпочтительно фактически осуществляют в органическом растворителе (Z.2), как описано далее ниже, так как этот растворитель должен в любом случае присутствовать в составе (Z) для получения преполимера на стадии (I) способа.
Как уже указано выше, группы в преполимере (Z.1.1), которые могут превращаться в анионные группы, также могут частично присутствовать как соответствующие анионные группы, например, как результат применения нейтрализующего агента. Таким образом, возможно регулировать диспергируемость в воде преполимеров (Z. 1.1) и также, следовательно, промежуточного соединения (Z.1).
Предполагаемые нейтрализующих агенты, в частности, включают известные основные нейтрализующие агенты, такие как, например, карбонаты, гидрокарбонаты, или гидроксиды щелочных металлов и щелочноземельных металлов, такие как LiOH, NaOH, КОН, или Са(ОН)2, например. Также подходящими для нейтрализации и применяемыми предпочтительно в контексте настоящего изобретения являются органические основания, содержащие азот, такие как амины, такие как аммиак, триметиламин, триэтиламин, трибутиламины, диметиланилин, трифениламин, диметилэтаноламин, метилдиэтаноламин, или триэтаноламин, а также их смеси.
Нейтрализация преполимера (Z.1.1) нейтрализующими агентами, в частности, органическими основаниями, содержащими азот, может происходить после получения преполимера в органической фазе, другими словами, в растворе с органическим растворителем, в частности, с растворителем (Z.2), как описано ниже. Нейтрализующий агент, конечно, также может добавляться во время или до начала фактической полимеризации, в случае чего, например, нейтрализуются исходные соединения, содержащие группы карбоновой кислот.
Если желательна нейтрализация групп, которые могут превращаться в анионные группы, в частности, групп карбоновой кислоты, то нейтрализующий агент может добавляться, например, в таком количестве, что нейтрализуется часть групп, составляющая 35% - 65% (степень нейтрализации). Предпочтительным является диапазон от 40% до 60% (в отношении метода вычисления смотри раздел Примеров).
Преполимер (Z.1.1) предпочтительно нейтрализуют, как описано, после его изготовления и до его применения для получения промежуточного соединения (Z.1).
Получение промежуточного соединения (Z.1), описанное в этом документе, включает реакцию описанного выше преполимера (Z. 1.1) по меньшей мере с одним, предпочтительно ровно одним, полиамином (Z.1.2а), полученным из полиамина (Z.1.2).
Полиамин (Z.l.2а) содержит две блокированные первичные аминогруппы и одну или две свободные вторичные аминогруппы.
Как известно, блокированные аминогруппы представляют собой группы, в которых водородные остатки на азоте, которые, как правило, присутствуют в свободных аминогруппах, были замещены в результате обратной реакции с блокирующим агентом. Вследствие блокирования, аминогруппы не могут вступать в реакцию подобно свободным аминогруппам, в результате реакций конденсации или реакций присоединения, и таким образом в указанном отношении являются нереакционноспособными, что отличает их от свободных аминогрупп. Несомненно, затем, известные как таковые реакции аминогрупп возможны только после того как, обратимо присоединяемый блокирующий агент снова удаляют, тем самым, в свою очередь, получая свободные аминогруппы. Сам принцип, таким образом, напоминает принцип закрытых или блокированных изоцианатов, которые также известны в сфере химии полимеров.
Первичные аминогруппы полиамина (Z.1.2а) могут блокироваться с применением традиционных блокирующих агентов, таких как, например, кетоны и/или альдегиды. В этом случае, такое блокирование с выделением воды, дает кетимины и/или альдимины, которые больше не содержат каких-либо азот-водородных связей, что означает то, что обычные реакции конденсации или реакции присоединения аминогруппы с дополнительной функциональной группой, такой как изоцианатная группа, не могут происходить.
Условия реакции для получения блокированного первичного амина указанного вида, например, кетимина, являются известными. Таким образом, например, указанное блокирование может осуществляться посредством введения тепла в смесь первичного амина с избытком кетона, который, вместе с тем, действует в качестве растворителя амина. Воду, которая образуется в результате реакции, предпочтительно удаляют во время реакции, для того чтобы предотвратить возможность, в ином случае, обратной реакции (деблокирование) обратимого блокирования.
Условия реакции для деблокирования блокированных первичных аминогрупп также известны, как таковые. Например, обычное перенесение блокированного амина в водную фазу является достаточным, для того чтобы сместить равновесие обратно в сторону деблокирования, как результат концентрированного давления, которое затем возникает в результате воздействия воды, и тем самым образовать свободные первичные аминогруппы, а также свободный кетон, по мере потребления воды.
Из приведенного выше следует, что в контексте настоящего изобретения, проводится четкое различие между блокированными и свободными аминогруппами. Тем не менее, в случае, когда аминогруппа не указана ни как блокированная, ни как свободная, то считается, что ссылаются на свободную аминогруппу.
Предпочтительными блокирующими агентами для блокирования первичных аминогрупп полиамина (Z.1.2а) являются кетоны. В частности, предпочтительными среди кетонов являются те, которые составляют органический растворитель (Z.2), как описано далее ниже. Причина состоит в том, что указанные растворители (Z.2) должны присутствовать в составе (Z) для изготовления на стадии (I) способа в любом случае. Уже было указано выше, что получение соответствующих первичных аминов, блокированных кетоном, в частности, приводит к хорошему эффекту в избытке кетона. Таким образом, в результате применения кетонов (Z.2) для блокирования, является возможным применять соответствующий предпочтительный способ получения блокированных аминов, без какой-либо необходимости в дорогостоящем и неудобном удалении блокирующего агента, что может быть нежелательным. Вместо этого, раствор блокированного амина может применяться непосредственно, для того чтобы получить промежуточное соединение (Z.1). Предпочтительные блокирующие агенты представляют собой ацетон, метилэтилкетон, метилизобутилкетон, диизопропилкетон, циклопентанон, или циклогексанон, в частности, предпочтительными блокирующими агентами являются кетоны (Z.2) метилэтилкетон и метилизобутилкетон.
Предпочтительное блокирование с применением кетонов и/или альдегидов, в частности, кетонов, и сопутствующее получение кетиминов и/или альдиминов, имеет преимущество, более того, указанные первичные аминогруппы блокируются избирательно. Присутствующие вторичные аминогруппы, несомненно, не могут блокироваться, и поэтому остаются свободными. Следовательно, полиамин (Z.1.2а), который, как и по меньшей мере две блокированные первичные аминогруппы, также содержит одну или две свободные вторичные аминогруппы, легко может быть получен в результате указанных предпочтительных реакций блокирования из полиамина (Z.1.2), который содержит свободные вторичные и первичные аминогруппы.
Полиамины (Z.1.2а) могут быть получены в результате блокирования первичных аминогрупп полиаминов (Z.1.2), содержащих две первичные аминогруппы и одну или две вторичные аминогруппы. По сути, подходящими являются все традиционные алифатические, ароматические, или аралифатические (смешанные алифатические-ароматические) полиамины (Z.1.2), имеющие две первичные аминогруппы и одну или две вторичные аминогруппы. Это означает, что, как и указанные аминогруппы, как таковые, могут присутствовать любые алифатические, ароматические, или аралифатические группы. Возможны, например, одновалентные группы, расположенные в качестве концевых групп на вторичной аминогруппе, или двухвалентные группы, расположенные между двух аминогрупп.
В контексте настоящего изобретения, алифатический представляет собой термин, относящийся ко всем органическим группам, которые не являются ароматическими. Например, присутствующие группы, как и указанные аминогруппы, могут быть группами алифатических углеводородов, другими словами, группами, которые состоят исключительно из углерода и водорода, и которые не являются ароматическими. Указанные группы алифатических углеводородов могут быть неразветвленными, разветвленными или циклическими, и могут быть насыщенными или ненасыщенными. Указанные группы, конечно, также могут содержать циклические и неразветвленные или разветвленные фрагменты. Является также возможным, что алифатические группы содержат гетероатомы, в частности, в виде мостиковых групп, таких как группы простого эфира, сложного эфира, амида и/или уретана. Возможные ароматические группы также являются известными, и не требуют дополнительного разъяснения.
Полиамины (Z.1.2а) предпочтительно содержат две блокированные первичные аминогруппы и одну или две свободные вторичные аминогруппы, и предпочтительно они содержат, в качестве первичных аминогрупп, исключительно блокированные первичные аминогруппы и, в качестве вторичных аминогрупп, исключительно свободные вторичные аминогруппы.
В общем, полиамины (Z.1.2а) предпочтительно содержат три или четыре аминогруппы, указанные группы при этом выбирают из группы блокированных первичных аминогрупп и свободных вторичных аминогрупп.
Особенно предпочтительными полиаминами (Z.1.2а) являются те, которые состоят из двух блокированных первичных аминогрупп, одной или двух свободных вторичных аминогрупп, а также алифатически насыщенных углеводородных групп.
Таким же образом, предпочтительные варианты осуществления применяются к полиаминам (Z.1.2), где они содержат свободные первичные аминогруппы вместо блокированных первичных аминогрупп.
Примерами предпочтительных полиаминов (Z.1.2), из которых в результате блокирования первичных аминогрупп могут быть получены полиамины (Z.1.2а), являются диэтилентриамин, 3-(2-аминоэтил)аминопропиламин, дипропилентриамин, а также N1-(2-(4-(2-аминоэтил)пиперазин-1-ил)этил)этан-1,2-диамин (одна вторичная аминогруппа, две первичные аминогруппы для блокирования) и триэтилентетрамин, а также N,N'-бис(3-аминопропил)-этилендиамин (две вторичные аминогруппы, две первичные аминогруппы для блокирования).
Специалисту в данной области понятно, что не в последнюю очередь по причинам, связанным с чистым техническим синтезом, нельзя всегда получать теоретически идеальное количественное превращение первичных аминогрупп при блокировании. Например, если определенное количество полиамина блокируется, то часть первичных аминогрупп, которые блокируются во время процесса блокирования, может составлять, например, 95 мол. % или большее количество (которое можно определить посредством ИК-спектроскопии; смотри раздел Примеров). В случае, когда полиамин в неблокированном состоянии, например, имеет две свободные первичные аминогруппы, и где первичные аминогруппы определенного количества указанного амина затем блокируются, то в контексте настоящего изобретения говорят, что указанный амин имеет две блокированные первичные аминогруппы, если при этом блокируются фракция, составляющая более 95 мол. % первичных аминогрупп, которые присутствуют в применяемом количестве. С одной стороны, это происходит благодаря тому уже указанному факту, что с точки зрения технического синтеза, количественное превращение не всегда может осуществляться. С другой стороны, тот факт, что блокируется более 95 мол. % первичных аминогрупп, означает то, что основная фракция общего количества аминов, которые применяют для блокирования, фактически содержит исключительно блокированные первичные аминогруппы, а именно две блокированные первичные аминогруппы.
Получение промежуточного соединения (Z.1) включает реакцию преполимера (Z.1.1) с полиамином (Z.1.2а) посредством реакции присоединения изоцианатных групп из компонента (Z.1.1) со свободными вторичными аминогруппами из компонента (Z.1.2а). Эта реакция, которая известна как таковая, затем приводит к прикреплению полиамина (Z.1.2а) на преполимер (Z.1.1), с образованием мочевинных связей, по сути образованием промежуточного компонента (Z.1). При этом будет совершенно очевидным, что при получении промежуточного соединения (Z.1), предпочтение в результате отдают тому, чтобы не применять какие-либо другие амины, имеющие свободные или блокированные вторичные или свободные или блокированные первичные аминогруппы.
Промежуточное соединение (Z.1) может быть получено посредством известных и распространенных способов в виде твердого вещества или раствора, особенно предпочтительно посредством реакции компонента (Z.11) с (Z.1.2а) в органических растворителях. Сразу очевидно, что растворители должны выбираться таким образом, чтобы они не вступали в какие-либо нежелательные реакции с функциональными группами исходных соединений и, таким образом, были инертными или в основном инертными по своему поведению в отношении указанных групп. В качестве растворителя при получении, предпочтение отдают применению, по меньшей мере частично, органического растворителя (Z.2), как описано далее ниже, в частности, метилэтилкетона, даже на этой стадии, поскольку указанный растворитель должен в любом случае присутствовать в составе (Z), который должен быть получен на стадии (I) способа в соответствии с изобретением. Предпочтительно раствор преполимера (Z.1.1) в растворителе (Z.2) смешивают с раствором полиамина (Z.1.2а) в растворителе (Z.2), и при этом описанная реакция может происходить.
Конечно же, полученное таким образом промежуточное соединение (Z.1)-может нейтрализоваться во время или после получения, применяя нейтрализующие агенты, которые уже описаны выше, в соответствии со способом, который также описан выше для преполимера (Z.1.1). Тем не менее, является предпочтительным, когда преполимер (Z.1.1) нейтрализуется перед его применением, с целью получения промежуточного соединения (Z.1), в соответствии со способом, описанным выше, так, что нейтрализация во время или после получения (Z.1) больше не нужна. Таким образом, в указанном случае степень нейтрализации преполимера (Z.1.1) может соответствовать степени нейтрализации промежуточного соединения (Z.1). В случае, когда нет дополнительного добавления нейтрализующих агентов вообще, в контексте способа в соответствии с изобретением, то степень нейтрализации полимеров, которые присутствуют в полученной дисперсии (ПД) в соответствии с изобретением, также может соответствовать степени нейтрализации преполимера (Z.1.1).
Промежуточное соединение (Z.1) имеет блокированные первичные аминогруппы. Несомненно, указанное может достигаться тем, что свободные вторичные аминогруппы реагируют во время реакции преполимера (Z.1.1) и полиамина (Z.1.2а), но при этом блокированные первичные аминогруппы не вступают в реакцию. На самом деле, как уже описано выше, эффект блокирования состоит в том, что обычные реакции конденсации или реакции присоединения с другими функциональными группами, такими как изоцианатные группы, не могут происходить. Это, конечно, означает, что условия реакции должны выбираться таким образом, чтобы блокированные аминогруппы также оставались блокированными, для того чтобы тем самым обеспечивать промежуточное соединение (Z.1). Специалист в данной области знает, каким образом установить такие условия, которые обусловлены, например, реакцией в органических растворителях, что является предпочтительным в любом случае.
Промежуточное соединение (Z.1) содержит изоцианатные группы. Соответственно, для реакции компонента (Z.11) и (Z.1.2а), соотношение указанных компонентов должно, конечно, выбираться таким образом, чтобы продукт - то есть, промежуточное соединение (Z.1) - содержало изоцианатные группы.
Поскольку, как описано выше, в реакции компонента (Z.11) с компонентом (Z.1.2а), свободные вторичные аминогруппы вступают в реакцию с изоцианатными группами, но при этом первичные аминогруппы не вступают в реакцию вследствие блокирования, то прежде всего сразу понятно, что в указанной реакции молярное соотношение изоцианатных групп из компонента (Z.1.1) к свободным вторичным аминогруппам из компонента (Z.1.2а) должно быть больше 1. Эта особенность возникает косвенным образом, тем не менее, явно и непосредственно, исходя из признака, существенного для изобретения, а именно того, что промежуточное соединение (Z.1) содержит изоцианатные группы.
Тем не менее, предпочтительно, чтобы во время реакции присутствовал избыток изоцианатных групп, как определено ниже. Молярные количества (n) изоцианатных групп, свободных вторичных аминогрупп, а также блокированных первичных аминогрупп, в указанном предпочтительном варианте осуществления, удовлетворяют следующему условию: [n (изоцианатные группы из компонента (Z.1.1)) - n (свободные вторичные аминогруппы из компонента (Z.1.2а))] / n (блокированные первичные аминогруппы из компонента (Z.1.2а))=1,2/1 - 4/1, предпочтительно 1,5/1 - 3/1, очень предпочтительно 1,8/1 - 2,2/1, даже более предпочтительно 2/1.
В указанном предпочтительном варианте осуществления, промежуточное соединение (Z.1), образованное посредством реакции изоцианатных групп из компонента (Z.1.1) со свободными вторичными аминогруппами из компонента (Z.1.2а), имеет избыток изоцианатных групп относительно блокированных первичных аминогрупп. Указанный избыток, по сути, достигается в результате выбора молярного соотношения изоцианатных групп из компонента (Z.1.1) к общему количеству свободных вторичных аминогрупп и блокированных первичных аминогрупп из компонента (Z.1.2а), таким образом, чтобы оно было достаточно большим, чтобы даже после получения промежуточного соединения (Z.1) и соответствующего использования изоцианатных групп в результате реакции со свободными вторичными аминогруппами, оставался соответствующий избыток изоцианатных групп.
В случае, когда, например, полиамин (Z.1.2a) имеет одну свободную вторичную аминогруппу и две блокированные первичные аминогруппы, молярное соотношение между изоцианатными группами из компонента (Z.1.1) к полиамину (Z.1.2a) в особенно предпочтительном варианте осуществления устанавливают как 5/1. Тогда использование одной изоцианатной группы в реакции со свободной вторичной аминогруппой будет означать, что для указанного выше условия применяли 4/2 (или 2/1).
Фракция промежуточного соединения (Z.1) составляет от 15 до 65 мас. %, предпочтительно от 25 до 60 мас. %, более предпочтительно от 30 до 55 мас. %, особенно предпочтительно от 35 до 52,5 мас. %, и, в одном конкретном варианте осуществления, от 40 до 50 мас. %, в каждом случае, из расчета общего количества состава (Z).
Определение фракции промежуточного соединения (Z.1) может осуществляться следующим образом: Определяют содержание твердых частиц смеси, которая кроме промежуточного соединения (Z.1), содержит только органические растворители (относительно метода определения для определения твердых частиц (что также называют содержанием твердых частиц) смотри раздел Примеров). В таком случае содержание твердых частиц соответствует количеству промежуточного соединения (Z.1). Таким образом, принимая во внимание содержание твердых частиц в смеси, возможно определить или установить фракцию промежуточного соединения (Z.1) в составе (Z). Принимая во внимание, что в любом случае, промежуточное соединение (Z.1) предпочтительно получают в органическом растворителе, и поэтому, после получения, оно в любом случае присутствует в смеси, содержащей и органические растворители, это и есть выбранный метод.
Состав (Z) дополнительно содержит по меньшей мере один конкретный органический растворитель (Z.2).
Растворители (Z.2) имеют растворимость в воде, составляющую не более 38 мас. %, при температуре 20°С (относительно метода определения смотри раздел Примеров). Растворимость в воде при температуре, составляющей 20°С, предпочтительно составляет менее 30 мас. %. Предпочтительный диапазон составляет от 1 до 30 мас. %.
Растворитель (Z.2), соответственно, имеет довольно умеренную растворимость в воде, будучи при этом, в частности, не полностью смешивается с водой или не имеет при этом бесконечной растворимости в воде. Растворитель полностью смешивается с водой, когда он может смешиваться с водой в любой пропорции, без появления разделения, другими словами, без образования двух фаз.
Примерами растворителей (Z.2) являются метилэтилкетон, метилизобутилкетон, диизобутилкетон, простой диэтиловый эфир, простой дибутиловый эфир, простой диметиловый эфир дипропиленгликоля, простой диэтиловый эфир этиленгликоля, толуол, метилацетат, этилацетат, бутилацетат, пропиленкарбонат, циклогексанон, или смеси указанных растворителей. Предпочтение отдают метилэтилкетону, который имеет растворимость в воде, составляющую 24 мас. %, при температуре 20°С.
Таким образом, среди растворителей (Z.2) отсутствуют такие растворители, как ацетон, N-метил-2-пирролидон, N-этил-2-пирролидон, тетрагидрофуран, диоксан, N-формилморфолин, диметилформамид, или диметилсульфоксид.
Особый эффект выбора определенных растворителей (Z.2) только с ограниченной растворимость в воде состоит в том, что, когда состав (Z) диспергируется в водной фазе, на стадии (II) способа, гомогенный раствор не может образовываться непосредственно. Предполагают, что присутствующая вместо этого дисперсия обеспечивает возможность прохождения в ограниченном объеме реакций сшивания, которые возникают как часть стадии (II) (реакции присоединения свободных первичных аминогрупп и изоцианатных групп с получением мочевинных связей), тем самым, по сути позволяя образование микрочастиц, как определено выше.
Точно также, описанные предпочтительные растворители (Z.2), имеющие растворимость в воде, имеют точку начала кипения, которая составляет не более 120°С, более предпочтительно, составляющую не более 90°С (в условиях атмосферного давления, другими словами, 1,013 бар). Это имеет преимущества в контексте стадии (III) способа, при этом указанная стадия описана далее ниже, другими словами, по меньшей мере частичное удаление по меньшей мере одного органического растворителя (Z.2) из дисперсии, полученной на стадии (II) способа. Очевидно, что во время применения растворителей (Z.2), которые являются предпочтительными в указанном контексте, указанные растворители могут удаляться посредством дистилляции, например, без одновременного удаления значительных количеств воды, введенной на стадии (II) способа.
Поэтому нет необходимости, например, в трудоемком повторном добавлении воды, с тем, чтобы сохранить водную природу дисперсии (ПД).
Фракция по меньшей мере одного органического растворителя (Z.2) составляет от 35 до 85 мас. %, предпочтительно от 40 to до 75 мас. %, более предпочтительно от 45 до 70 мас. %, особенно предпочтительно от 47,5 до 65 мас. %, и, в одном конкретном варианте осуществления, от 50 до 60 мас. %, в каждом случае, из расчета общего количества состава (Z).
В контексте настоящего изобретения выяснилось, что в результате определенной комбинации фракции, как определено выше для промежуточного соединения (Z.1) в составе (Z), и в результате выбора определенных растворителей (Z.2), после описанных ниже стадий (II) и (III), возможно обеспечивать полиуретан-полимочевинные дисперсии, которые содержат частицы полиуретана и полимочевины, имеющие требуемый размер частиц, которые дополнительно имеют требуемую гель-фракцию.
Описанные компоненты (Z.1) и (Z.2) предпочтительно составляют в общем по меньшей мере 90 мас. % состава (Z). Предпочтительно два компонента составляют по меньшей мере 95 мас. %, в частности, по меньшей мере 97,5 мас. %, состава (Z). Особенно предпочтительно, состав (Z) состоит из указанных двух компонентов. В указанном контексте необходимо отметить, что, в случае, когда применяют нейтрализующие агенты, как описано выше, то указанные нейтрализующие агенты приписывают промежуточному соединению, при расчете количества промежуточного соединения (Z.1). Причина состоит в том, что в этом случае, промежуточное соединение (Z.1) по меньшей мере имеет анионные группы, которые получают в результате применения нейтрализующего агента. Соответственно, катион, который присутствует после того, как образовались указанные анионные группы, также приписывается к промежуточному соединению.
В случае, если состав (Z), дополнительно к компонентам (Z.1) и (Z.2), включает другие компоненты, то эти другие компоненты предпочтительно просто представляют собой органические растворители. Таким образом, содержание твердых частиц состава (Z) предпочтительно соответствует фракции промежуточного соединения (Z.1) в составе (Z). Состав (Z), таким образом, предпочтительно имеет содержание твердых частиц, которое составляет 15-65 мас. %, предпочтительно, которое составляет 25 - 60 мас. %, более предпочтительно, которое составляют 30 - 55 мас. %, особенно предпочтительно, которое составляет 35 - 52,5 мас. %, и, в одном особенно предпочтительном варианте осуществления, 40 - 50 мас. %.
Особенно предпочтительный состав (Z), таким образом, в общем содержит по меньшей мере 90 мас. % компонентов (Z.1) и (Z.2) и, кроме промежуточного соединения (Z.1), включает исключительно органические растворители.
Преимущество состава (Z) состоит в том, что он может быть получен без применения неблагоприятных для окружающей среды и вредных для здоровья органических растворителей, таких как N-метил-2-пирролидон, диметилформамид, диоксан, тетрагидрофуран, и Ы-этил-2-пирролидон. Соответственно, предпочтительно состав (Z) содержит менее 10 мас. %, предпочтительно менее 5 мас. %, более предпочтительно менее 2,5 мас. % органических растворителей, выбранных из группы, состоящей из N-метил-2-пирролидона, диметилформамида, диоксана, тетрагидрофурана, и N-этил-2-пирролидона. Предпочтительно, состав (Z) является полностью свободным от указанных органических растворителей.
На второй стадии (II) способа, описанной в этом случае, состав (Z) диспергируется в водной фазе.
Таким образом, известно, а также следует из уже указанного выше, что на стадии (II), происходит деблокирование блокированных первичных аминогрупп промежуточного соединения (Z.1). На самом деле, в результате перенесения блокированного амина в водную фазу, по мере потребления воды, выделяется обратимо присоединенный блокирующий агент, и образуются свободные первичные аминогруппы.
Таким образом, также понятно, что полученные свободные первичные аминогруппы затем вступают в реакцию с изоцианатными группами, которые также присутствуют в промежуточном соединении (Z.1), или в деблокированном промежуточном соединении, образованном из промежуточного соединение (Z.1), посредством реакции присоединения, с образованием мочевинных связей.
Также известно, что перенесение в водную фазу означает, что изоцианатные группы в промежуточном соединении (Z.1), или в деблокированном промежуточном соединении, образованном из промежуточного соединения (Z.1), в принципе, имеют возможность вступать в реакцию с водой, с выделением диоксида углерода, с получением свободных первичных аминогрупп, которые, в свою очередь, могут затем вступать в реакцию с изоцианатными группами, которые все еще присутствуют.
Конечно, реакции и превращения, которые упомянуты выше, происходят параллельно друг с другом. В конечном счете, в результате, например, межмолекулярной и внутримолекулярной реакции или сшивания, образуется дисперсия, которая содержит частицы полиуретана и полимочевины с определенным средним размером частиц и с определенной степенью сшивания, или гель-фракция.
Затем, на стадии (II) способа, описанного в этом случае, состав (Z) диспергируется в воде, при этом происходит деблокирование блокированных первичных аминогрупп промежуточного соединения (Z.1) и реакция полученных свободных первичных аминогрупп с изоцианатными группами промежуточного соединения (Z.1), а также с изоцианатными группами деблокированного промежуточного соединения, образованное из промежуточного соединения (Z.1), посредством реакции присоединения.
Стадия (II) способа, описанного в этом случае, другими словами, диспергирование в водной фазе, может происходить любым желательным способом. Это означает, что по сути имеет значение только то, что состав (Z) смешивают с водой или с водной фазой. Предпочтительно, состав (Z), который после получения может, например, находиться при комнатной температуре, другими словами, при температуре 20 - 25°С, или при температуре, повышенной относительно комнатной температуры, составляющей 30 - 60°С, например, может быть подмешан к воде, с получением дисперсии. Уже введенная вода, например, имеет комнатную температуру. Диспергирование может происходить в чистой воде (деионизированной воде), что означает то, что водная фаза состоит исключительно из воды, причем это является предпочтительным. Конечно, кроме воды, водная фаза частично также может включать традиционные вспомогательные вещества, такие как обычные эмульгаторы и защитные коллоиды. Обобщение подходящих эмульгаторов и защитных коллоидов можно найти, например, в Houben Weyl, Methoden der organischen Chemie [Методы органической химии], том XIV/1 Makromolekulare Stoffe [Макромолекулярные соединения], Georg Thieme Verlag, Штуттгарт 1961, стр. 411 и последующие страницы.
Является преимуществом, если на стадии (II) способа, другими словами, при диспергировании состава (Z) в водной фазе, соотношение массы органических растворителей и воды выбирают таким образом, что полученная дисперсия имеет соотношение массы воды к массе органических растворителей, которое составляет больше 1, предпочтительно, которое составляет 1,05 - 2/1, особенно предпочтительно, которое составляет 1,1 - 1,5/1.
На стадии (III) способа, описанного в этом случае, из дисперсии, полученной на стадии (II), по меньшей мере частично, удаляют по меньшей мере один органический растворитель (Z.2). Конечно, стадия (III) способа также может включать удаление других растворителей, которые точно также возможно присутствуют, например, в составе (Z).
Удаление по меньшей мере одного органического растворителя (Z.2) и любых дополнительных органических растворителей может осуществляться любым известным способом, таким, как например, вакуумная дистилляции при температурах, немного повышенных относительно комнатной температуры, которые составляют, например, 30 - 60°С.
Полученная полиуретан-полимочевинная дисперсия (ПД) является водной (в отношении основного определение "водной" смотри выше).
Особое преимущество дисперсии (ПД), предназначенной для применения в соответствии с изобретением, состоит в том, что она может составляться только с только с очень небольшими фракциями органических растворителей, что все же обеспечивает преимущества в соответствии с изобретением, описанные в начале. Дисперсия (ПД), предназначенная для применения в соответствии с изобретением, предпочтительно содержит не более 15,0 мас. %, особенно предпочтительно не более 10 мас. %, очень предпочтительно не более 5 мас. %, и более предпочтительно не более 2.5 мас. % органических растворителей (относительно метода определения смотри раздел Примеров).
Фракция полиуретан-полимочевинного полимера в дисперсии (ПД) предпочтительно составляет 25 - 55 мас. %, предпочтительно 30 - 50 мас. %, более предпочтительно 35 - 45 мас. %, в каждом случае, из расчета общего количества дисперсии (установленная, как и в случае определения, описанного выше для промежуточного соединения (Z.1), посредством определения содержания твердых частиц).
Фракция воды в дисперсии (ПД) предпочтительно составляет 45 - 75 мас. %, предпочтительно 50 - 70 мас. %, более предпочтительно 55 - 65 мас. %, в каждом случае, из расчета общего количества дисперсии.
Особое преимущество дисперсии (ПД), предназначенной для применения в соответствии с изобретением, состоит в том, что она может составляться таким образом, что она состоит по меньшей мере из 85 мас. %, предпочтительно по меньшей мере из 90,0 мас. %, очень предпочтительно по меньшей мере из 95 мас. %, и даже более предпочтительно по меньшей мере из 97,5 мас. % частиц полиуретана и полимочевины и воды (соответствующее значение получают в результате суммирования количества частиц (то есть полимера, определенного посредством содержания твердых частиц) и количества воды). Выяснилось, что несмотря на такую небольшую фракцию дополнительных компонентов, такие как, в частности, органические растворители, дисперсии, в любом случае, являются очень стабильными, в частности, стабильными при хранении. Таким образом, объединены два соответствующих преимущества. Прежде всего, обеспечивают дисперсии, которые могут применяться в водных материалах базового покрытия, что приводит к эксплуатационным преимуществам, описанным в начале, а также в примерах, приведенных ниже. Во-вторых, однако, соответствующее отсутствие ограничений при составлении достигается при изготовлении водных материалов базового покрытия. Это означает, что в материалах базового покрытия могут применяться дополнительные фракции органических растворителей, которые при этом являются необходимыми, например, для того чтобы обеспечивать подходящее составление различных компонентов. Это, однако, не затрагивает по сути водную природу материала базового покрытия. Напротив, материалы базового покрытия могут при этом составляться с применением сравнительно небольших фракций органических растворителей и, таким образом, имеют, в частности, хорошие экологические свойства.
Даже более предпочтительно, если дисперсия, другая, чем дисперсия полимера, включает только воду и любые органические растворители, например, в виде остаточных фракций, не полностью удаленных на стадии (III) способа. Содержание твердых частиц дисперсии (ПД), таким образом, предпочтительно составляет 25% - 55%, предпочтительно 30% - 50%, более предпочтительно 35% - 45%, и более предпочтительно все еще находится в соответствии с фракцией полимера в дисперсии.
Преимущество дисперсии (ПД) состоит в том, что она может быть получена без применения неблагоприятных для окружающей среды и вредных для здоровья органических растворителей, таких как N-метил-2-пирролидон, диметилформамид, диоксан, тетрагидрофуран, и N-этил-2-пирролидон. Соответственно, дисперсия (ПД) содержит предпочтительно менее 7,5 мас. %, предпочтительно менее 5 мас. %, более предпочтительно менее 2,5 мас. % органических растворителей, выбранных из группы, состоящей из N-метил-2-пирролидона, диметилформамида, диоксана, тетрагидрофурана, и N-этил-2-пирролидона. Предпочтительно, дисперсия (ПД) является полностью свободной от указанных органических растворителей.
Полиуретан-полимочевинный полимер, который присутствует в дисперсии, предпочтительно содержит очень незначительное количество каких-либо гидроксильных групп, или не содержит их вовсе. Гидроксильное число полимера, на основе содержания твердых частиц, предпочтительно составляет менее 15 мг КОН/г, в частности, менее 10 мг КОН/г, даже более предпочтительно менее 5 мг КОН/г (относительно метода определения смотри раздел Примеров).
Фракция одной или большего количества дисперсий (ПД), из расчета общей массы водного материала базового покрытия (b.2.1), предпочтительно составляет 5-60 мас. %, более предпочтительно 15-50 мас. %, и очень предпочтительно 20 - 45 мас. %.
Фракция полиуретан-полимочевинных полимеров, полученных из дисперсии (ПД), из расчета общей массы водного материала базового покрытия (b.2.1), предпочтительно составляет от 2,0 до 24,0 мас. %, более предпочтительно от 6,0 до 20,0 мас. %, и очень предпочтительно 8,0 - 18,0 мас. %.
Определение или установление в материале базового покрытия фракции полиуретан-полимочевинных полимеров, полученных из дисперсии в соответствии с изобретением, может осуществляться посредством определения содержания твердых частиц дисперсии (ПД) в соответствии с изобретением, подлежащей применению в материале базового покрытия.
В случае возможной конкретизации в отношении материалов базового покрытия, содержащего предпочтительные дисперсии (ПД) в определенном диапазоне пропорций, применяется следующее. Дисперсии (ПД), которые не попадают в предпочтительную группу, конечно, все же могут присутствовать в материале базового покрытия. В этом случае, определенный диапазон пропорций применяется только к предпочтительной группе дисперсий (ПД). Тем не менее, является предпочтительным, чтобы общая доля дисперсий (ПД), состоящая из дисперсий из предпочтительной группы и дисперсий, которые не являются частью предпочтительной группы, также была объектом определенного диапазона пропорций.
Соответственно, в случае ограничения диапазона пропорций, составляющего 15-50 мас. %, и предпочтительной группы дисперсий (ПД), этот диапазон пропорций несомненно изначально применяется только к предпочтительной группе дисперсий (ПД). В этом случае, однако, будет предпочтительным также диапазон от 15 до 50 мас. % в общем количестве всех присутствующих изначально содержащихся дисперсий, состоящих из дисперсий из предпочтительной группы и дисперсий, которые не являются частью предпочтительной группы. Таким образом, в случае, когда применяют 35 мас. % дисперсий (ПД) предпочтительной группы, может применяться не более 15 мас. % дисперсий группы, которая не является предпочтительной.
Для целей настоящего изобретения, указанный принцип действует для всех указанных компонентов материала базового покрытия, а также для их диапазонов пропорций - например, для пигментов, которые определены далее ниже, или также для сшивающих агентов, которые определены далее ниже, таких как меламиновые смолы.
Материал базового покрытия (b.2.1), предназначенный для применения в соответствии с изобретением, предпочтительно содержит по меньшей мере один пигмент. В данном случае ссылаются на традиционные пигменты, которые обеспечивают цвет и/или оптический эффект.
Такие цветные пигменты и эффектные пигменты известны специалистам в данной области, и описаны, например, в Rompp-Lexikon Lacke und Druckfarben, Georg Thieme Verlag, Штуттгарт, Нью Йорк, 1998, страницы 176 и 451. Термины "красящий пигмент" и "цветной пигмент" являются взаимозаменямыми, точно также, как и термины "оптически эффектный пигмент" и "эффектный пигмент".
Предпочтительные эффектные пигменты, например, представляют собой пластинчатые металлизированные эффектные пигменты, такие как пластинчатые алюминиевые пигменты, золотистые бронзы, оксидированные бронзы и/или пигменты на основе оксида железа и алюминия, перламутровые пигменты, такие как жемчужная эссенция, основный карбонат свинца, оксихлорид висмута и/или пигменты на основе оксида металла и слюды и/или другие эффектные пигменты, такие как пластинчатый графит, пластинчатый оксид железа, многослойные эффектные пигменты, состоящие из ПВД пленок и/или жидкокристаллические полимерные пигменты. В частности, предпочтительными являются пластинчатые металлизированные эффектные пигменты, в частности, пластинчатые алюминиевые пигменты.
Обычные цветные пигменты, в частности, включают неорганические красящие пигменты, такие как белые пигменты, такие как диоксид титана, цинковые белила, сульфид цинка или литопон; черные пигменты, такие как углеродная сажа, железомагний черный, или черная шпинель; хроматические пигменты, такие как оксид хрома, оксигидрат хрома зеленый, кобальт зеленый или ультрамарин зеленый, кобальт синий, ультрамарин синий или магний синий, ультрамарин фиолетовый или кобальт фиолетовый и магний фиолетовый, оксид железа красный, сульфоселенид кадмия, молибден красный или ультрамарин красный; коричневый оксид железа, смешанный коричневый, фазы шпинеля и фазы корунда или хром оранжевый; или желтый оксид железа, титаноникель желтый, титанохром желтый, сульфид кадмия, сульфид цинка и кадмия, хром желтый или ванадат висмута.
Фракция пигментов предпочтительно находится в диапазоне от 1,0 до 40,0 мас. %, предпочтительно 2,0 - 35,0 мас. %, более предпочтительно 5,0 -30,0 мас. %, из расчета общей массы водного материала базового покрытия (b.2.1) в каждом случае.
Водный материал базового покрытия (b.2.1) предпочтительно дополнительно содержит по меньшей мере один полимер в качестве связующего вещества, то есть, отличающийся от полиуретан-полимочевинных полимеров, которые присутствуют в дисперсии (ПД), в частности, по меньшей мере один полимер, выбранный из группы, состоящей из полиуретанов, сложных полиэфиров, полиакрилатов и/или сополимеров указанных полимеров, в частности, сложные полиэфиры и/или полиуретанполиакрилаты. Предпочтительные сложные полиэфиры описаны, например, в DE 4009858 А1 на строке 53 колонки 6 - строке 61 колонки 7, и строке 24 колонки 10 - строке 3 колонки 13, или в WO 2014/033135 А2, строка 24 страницы 2 - строка 10 страницы 7, и строка 13 страницы 28 - строка 13 страницы 29. Предпочтительные полиуретан-полиакрилатные сополимеры (акрилированные полиуретаны) и их получение описаны, например, в WO 91/15528 А1, строка 21 страницы 3 - строка 33 страницы 20, и в DE 4437535 А1, строка 27 страницы 2 - строка 22 страницы 6. Описанные в качестве связующих веществ полимеры предпочтительно обладают гидроксильными функциональными группами, и особенно предпочтительно имеют гидроксильное число в диапазоне от 15 до 200 мг КОН/г, более предпочтительно от 20 до 150 мг КОН/г.Более предпочтительно, материалы базового покрытия содержат по меньшей мере один полиуретан-полиакрилатный сополимер с гидроксильными функциональными группами, даже более предпочтительно по меньшей мере один полиуретан-полиакрилатный сополимер с гидроксильными функциональными группами, а также по меньшей мере один сложный полиэфир с гидроксильными функциональными группами.
Пропорция дополнительных полимеров в качестве связующих веществ может варьироваться в широком диапазоне, и предпочтительно находится в диапазоне от 1,0 до 25,0 мас. %, более предпочтительно от 3,0 до 20,0 мас. %, очень предпочтительно от 5,0 до 15,0 мас. %, в каждом случае, из расчета общей массы материала базового покрытия (Ь.2.1)
Материал базового покрытия (b.2.1) может дополнительно содержать по меньшей мере один обычный сшивающий агент, известный как таковой. Если он содержит сшивающий агент, то указанный агент предпочтительно содержит по меньшей мере одну аминопластовую смолу и/или по меньшей мере один блокированный полиизоцианат, предпочтительно аминопластовую смолу. Среди аминопластовых смол, меламиновые смолы, в частности, являются предпочтительными.
Если материал базового покрытия (b.2.1) содержит сшивающие агенты, то пропорция указанных сшивающих агентов, в частности, аминопластовых смол и/или блокированных полиизоцианатов, очень предпочтительно аминопластовых смол и, предпочтительно, меламиновых смол, предпочтительно находится в диапазоне от 0,5 до 20,0 мас. %, более предпочтительно от 1,0 до 15,0 мас. %, очень предпочтительно от 1,5 до 10,0 мас. %, в каждом случае, из расчета общей массы материала базового покрытия (b.2.1).
Материал базового покрытия (b.2.1) может дополнительно содержать по меньшей мере один загуститель. Подходящие загустители представляют собой неорганические загустители из группы листовых силикатов, таких, как литиево-алюминиево-магниевые силикаты. Тем не менее, известно, что покрывающие материалы, чей профиль реологических свойств определяется посредством предварительного или преимущественного применения таких неорганических загустителей, нуждаются в улучшении с точки зрения содержания твердых частиц, другими словами, они могут составляться только с явно низким содержанием твердых частиц, которое составляет, например, менее 20%, не затрагивая при этом важные эксплуатационные свойства. Особое преимущество материала базового покрытия (b.2.1) состоит в том, что он может составляться без, или без большой фракции таких неорганических листовых силикатов, которые применяются в качестве загустителей. Соответственно, фракция неорганических листовых силикатов, которые применяются в качестве загустителей, предпочтительно составляет менее 0,7 мас. %, особенно предпочтительно менее 0,3 мас. %, и даже более предпочтительно менее 0,1 мас. %, из расчета общей массы материала базового покрытия. Особенно предпочтительно, материал базового покрытия полностью свободен от таких неорганических листовых силикатов, применяемых в качестве загустителей.
Вместо этого, материал базового покрытия может содержать по меньшей мере один органический загуститель, такой, как например, загуститель на основе сополимера (мет)акриловой кислоты и (мет)акрилата или загуститель на основе полиуретана. Применяемыми предпочтительно являются ассоциированные загустители, такие как, например, ассоциированные загустители на основе полиуретана, известные как таковые. Ассоциированные загустители, как известно, представляют собой растворимые в воде полимеры, которые имеют сильно гидрофобные группы на концах цепи или в боковых цепях, и/или чьи гидрофильные цепи содержат гидрофобные блоки или их концентрации внутри. Как результат, указанные полимеры имеют поверхностно-активную природу, и способны образовывать мицеллы в водной фазе. Подобно поверхностно-активным веществам, гидрофильные участки остаются в водной фазе, в то время, как гидрофобные участки поступают в частицы полимера дисперсии, адсорбируют на поверхности других твердых частиц, таких как пигменты и/или наполнители, и/или образуют мицеллы в водной фазе. По сути, загущающее действие достигается без какого-либо повышения осаждения. Загустители указанного вида доступны коммерчески, такие, как например, под торговым наименованием Adekanol (от компании Adeka Corporation).
Пропорция органических загустителей предпочтительно находится в диапазоне от 0 до 5,0 мас. %, более предпочтительно от 0 до 3,0 мас. %, очень предпочтительно от 0 до 2,0 мас. %, в каждом случае, из расчета общей массы материала базового покрытия.
Очень особое преимущество материалов базового покрытия (Ь.2.1), которые применяют в соответствии с изобретением, состоит в том, что они могут составляться без применения какого-либо загустителя, и при этом имеют отличные характеристики в отношении своих реологических свойств. В результате достигают уменьшения сложности получения покрывающего материала и/или повышения степени отсутствия ограничений при составлении материала базового покрытия.
Кроме того, материал базового покрытия (b.2.1) может дополнительно содержать по меньшей мере одну дополнительную активирующую добавку. Примерами таких активирующих добавок являются соли, которые термически разлагаются без остатка или, в основном без остатка, полимеры в качестве связующих веществ, которые могут затвердевать физически, термически и/или с применением актиничного излучения, и которые отличаются от полимеров, уже указанных в качестве связующих веществ, дополнительные сшивающие агенты, органические растворители, реакционноспособные разбавители, прозрачные пигменты, наполнители, растворимые в молекулярной дисперсии красящие вещества, наночастицы, стабилизаторы света, замедляющие окисление вещества, деаэрирующие агенты, эмульгаторы, добавки скольжения, ингибиторы полимеризации, инициаторы радикальной полимеризации, усилители адгезии, регулирующие расход агенты, пленкообразующие вспомогательные вещества, контролирующие образование натеков вещества (SCA), замедлители горения, ингибиторы коррозии, воски, сиккативы, биоциды, и матирующие агенты. Такие активирующие добавки применяют в традиционных и известных количествах.
Содержание твердых частиц материала базового покрытия (b.2.1) может варьироваться в соответствии с требованиями конкретного случая.. Содержание твердых частиц зависит в основном от необходимой для нанесения вязкости, в частности, нанесения посредством распыления. Особое преимущество состоит в том, что материал базового покрытия в соответствии с изобретением, при сравнительно высоком содержании твердых частиц способен, тем не менее, иметь вязкость, позволяющую осуществлять подходящее нанесение.
Содержание твердых частиц материала базового покрытия, если он содержит по меньшей мере один сшивающий агент, предпочтительно составляет по меньшей мере 25%, более предпочтительно по меньшей мере 27,5%, особенно предпочтительно по меньшей мере 30%.
Если материал базового покрытия не содержит какого-либо сшивающего агента, то содержание твердых частиц предпочтительно составляет по меньшей мере 15%, более предпочтительно по меньшей мере 18%, даже более предпочтительно по меньшей мере 21%.
В указанных условиях, другими словами, с указанным содержанием твердых частиц, предпочтительные материалы базового покрытия (b.2.1) имеют вязкость, которая составляет 40 - 150 мПа⋅с, в частности, 70 - 110 мПа⋅с, при 23°С в условиях усилий сдвига, которые составляют 1000 1/с (для дополнительных подробностей в отношении метода определения смотри раздел Примеров). Для целей настоящего изобретения, вязкость в пределах указанного диапазона в указанных условиях усилий сдвига упоминается как вязкость распыления (рабочая вязкость). Как известно, покрывающие материалы наносят при вязкости распыления, что означает то, что в указанных имеющихся условиях (большие усилия сдвига), они имеют вязкость, которая, в частности, не слишком высокая, с тем, чтобы получить эффективное нанесение. Это означает, что установление вязкости распыления является важным для того, чтобы позволить краске, в принципе, наноситься посредством методов распыления, и обеспечивать возможность получения на основе, которая подлежит покрытию, цельной, однородной покрывающей пленки. Особое преимущество состоит в том, что даже материал базового покрытия (b.2.1), доведенный до вязкости распыления, имеет высокое содержание твердых частиц. Таким образом, предпочтительные диапазоны содержания твердых частиц, в частности, нижние интервалы значений, предполагают, что в применимом состоянии, предпочтительно, материал базового покрытия (b.2.1) имеет сравнительно высокое содержание твердых частиц.
Материал базового покрытия в соответствии с изобретением является водным (в отношении определения "водного" смотри выше).
Фракция воды в материале базового покрытия (b.2.1) предпочтительно составляет от 35 до 70 мас. %, и более предпочтительно 42 - 63 мас. %, в каждом случае, из расчета общей массы материала базового покрытия.
Даже более предпочтительным является, когда сумма процентов содержания твердых частиц материала базового покрытия и фракции воды в материале базового покрытия составляет по меньшей мере 70 мас. %, предпочтительно по меньшей мере 75 мас. %. Среди указанных количественных параметров, предпочтение отдают диапазонам, составляющим 75 - 95 мас. %, в частности, 80 -90 мас. %. В этом представлении, содержание твердых частиц, которое традиционно имеет только единицу измерения "%", указано в "мас. %". Поскольку содержание твердых частиц, по сути, также представляет собой количественный параметр, выраженный в процентах от массы, то эта форма представления оправдана. В случае, если материал базового покрытия, например, имеет содержание твердых частиц, которое составляет 35%, и содержание воды, которое составляет 50 мас. %, то указанная выше сумма процентов в отношении содержания твердых частиц материала базового покрытия и фракции воды в материале базового покрытия составляет 85 мас. %.
В частности, это означает, что предпочтительные материалы базового покрытия, в принципе, включают только небольшие пропорции вредных в экологическом отношении компонентов, таких как, в частности, органические растворители, относительно содержания твердых веществ материала базового покрытия. Соотношение летучей органической фракции материала базового покрытия (в мас. %) к содержанию твердых веществ материала базового покрытия (по аналогии с указанным выше, здесь в мас. %) предпочтительно составляет от 0,05 до 0,7, более предпочтительно от 0,15 до 0,6. Летучую органическую фракцию для целей настоящего изобретения рассматривают как фракцию материала базового покрытия, которая не считается ни частью воды, ни частью содержания твердых частиц.
Другое преимущество материала базового покрытия (b.2.1) состоит в том, что он может быть получен без применения неблагоприятных для окружающей среды и вредных для здоровья органических растворителей, таких как N-метил-2-пирролидон, диметилформамид, диоксан, тетрагидрофуран, и Т-этил-2-пирролидон. Соответственно, материал базового покрытия предпочтительно содержит менее 10 мас. %, более предпочтительно менее 5 мас. %, даже более предпочтительно менее 2,5 мас. % органических растворителей, выбранных из группы, состоящей из N-метил-2-пирролидона, диметилформамида, диоксана, тетрагидрофурана, и N-этил-2-пирролидона. Предпочтительно, материал базового покрытия является полностью свободным от указанных органических растворителей.
Материалы базового покрытия могут быть получены с применением смесительных аппаратов и способов смешивания, которые являются обычными и известными для получения материалов базового покрытия.
По меньшей мере один из материалов базового покрытия (b.2.2.х), который применяют в способе в соответствии с изобретением, имеет признаки, имеющие значение для изобретения, как описано в случае материала базового покрытия (b.2.1). В частности, это означает то, что по меньшей мере один из материалов базового покрытия (b.2.2.х) содержит по меньшей мере одну водную полиуретан-полимочевинную дисперсию (ПД). Предпочтительные признаки и варианты осуществления, описанные при описании материала базового покрытия (b.2.1), также предпочтительно применяются по меньшей мере для одного из материалов базового покрытия (b.2.2.х).
В предпочтительных вариантах осуществления стадии (2.2) способа в соответствии с изобретением, описанной выше, прежде всего наносят первый материал базового покрытия (b.2.2.а), и при этом он также может называться подготовительным для нанесения цвета материалом базового покрытия. Таким образом, он служит в качестве основы для цветной и/или эффектной пленки базового покрытия, которая затем следует, причем в результате она представляет собой пленку, которая способна оптимально исполнять свою функцию в отношении придания цвета и/или визуального эффекта.
В одном конкретном варианте осуществления, подготовительный для нанесения цвета материал базового покрытия в основном не содержит хроматических пигментов и эффектных пигментов. В частности, предпочтительно, материал базового покрытия указанного вида содержит менее 2 мас. %, предпочтительно менее 1 мас. %, хроматических пигментов и эффектных пигментов, в каждом случае, из расчета общей массы водного материала базового покрытия. В указанном варианте осуществления, подготовительный для нанесения цвета материал базового покрытия предпочтительно содержит черные и/или белые пигменты, особенно предпочтительно оба вида указанных пигментов. Предпочтительно он содержит 5 - 30 мас. %, предпочтительно 10-25 мас. %, белых пигментов, и 0,01 -1,00 мас. %, предпочтительно 0,1 - 0,5 мас. %, черных пигментов, в каждом случае, из расчета общей массы материала базового покрытия. Полученный белый, черный, и в частности, серый цвет, который может регулироваться на разных этапах яркости в результате соотношения белых пигментов и черных пигментов, представляет собой отдельно регулируемую основу для системы пленки базового покрытия, которая следует далее, позволяя оптимально выражать цвет и/или эффект, обеспеченный посредством последующей системы базового покрытия. При этом пигменты известны специалисту в данной области, и также были описаны выше. Предпочтительным белым пигментом в этом случае является диоксид титана, предпочтительным черным пигментом является углеродная сажа. Однако, как уже описано, указанный материал базового покрытия, конечно, также может содержать хроматические и/или эффектные пигменты. Указанный вариант осуществления является особенно подходящим, когда полученная многослойная красочная система должна иметь высокохроматический цвет, такой, как например, интенсивно глубокий красный или желтый цвет. В случае, когда пигменты с соответственно хроматическим цветом также добавляют к подготовительному для нанесения цвета материалу базового покрытия, то при этом можно достичь дополнительно улучшенного окрашивания.
Цветной и/или эффектный материал базового покрытия (материалы базового покрытия) для второй пленки базового покрытия или для второй и третьей пленки базового покрытия в указанном варианте осуществления подбирают в соответствии с желательным окрашиванием всей системы. В случае, если при этом желательным является белый, черный, или серый цвет, то по меньшей мере один дополнительный материал базового покрытия содержит соответствующие пигменты, и относительно состав пигментов, по сути, он мало чем отличается от подготовительного для нанесения цвета материала базового покрытия. В случае, когда желательна хроматическая и/или эффектная красочная система, такая, как например, хроматическая красочная система ровного цвета или красочная система с металлическим эффектом, то соответствующие хроматические и/или эффектные пигменты применяют в количествах, составляющих, например, 1 -15 мас. %, предпочтительно 3-10 мас. %, в каждом случае, из расчета общей массы материала базового покрытия. Материалы базового покрытия указанного вида, конечно, также могут включать черные и/или белые пигменты, которые также подходят для регулирования яркости.
Способ в соответствии с изобретением позволяет получать на металлических основах многослойные красочные системы, без отдельной стадии отверждения. При этом, применение способа в соответствии с изобретением приводит к получению многослойных красочных систем, которые демонстрируют отличную устойчивость в отношении образования пор, что означает то, что можно получать даже относительно большую толщину соответствующих пленок базового покрытия, не затрагивая при этом эстетические качества. Такие характеристики, как адгезия или общий внешний вид, также являются отличными.
Настоящее изобретение также относится к водному составу лака для смешивания для получения водных материалов базового покрытия. Состав лака для смешивания, в каждом случае содержит, из расчета общей массы водного состава лака для смешивания,
10-25 мас. % по меньшей мере одного полиуретан-полимочевинного полимера, который получают по меньшей мере из одной дисперсии (ПД),
0-15 мас. % сшивающего агента, выбранного из группы аминопластовых смол и блокированных полиизоцианатов,
3-15 мас. % по меньшей мере одного сложного полиэфира, имеющего гидроксильное число в диапазоне от 15 до 200 мг КОН/г,
2-10 мас. % по меньшей мере одного полиуретан-полиакрилатного сополимера, имеющего гидроксильное число в диапазоне от 15 до 200 мг КОН/г,
45 - 55 мас. % воды, и
5-15 мас. % по меньшей мере одного органического растворителя, при этом в общем описанные компоненты составляют по меньшей мере
90 мас. %, предпочтительно по меньшей мере 95 мас. %, состава лака для смешивания.
Состав лака для смешивания предпочтительно в основном не содержит пигментов, то есть, при этом содержит менее 1 мас. % пигментов. Особенно предпочтительно, совсем не содержит пигментов.
Выяснилось, что состав лака для смешивания отлично подходит для применения при получении водных материалов базового покрытия, благодаря индивидуально подобранному дополнению, в частности, пигментов и необязательно различных добавок. Таким образом, можно применять один состав лака для смешивания для получения разных водных материалов базового покрытия, посредством последующего, отдельного дополнения. Как результат, естественно, происходит значительное уменьшение трудоемкости и, следовательно, повышение рентабельности при составлении материалов базового покрытия, в частности, в промышленном масштабе. Состав лака для смешивания может изготавливаться и храниться отдельно, а затем дополняться, например, соответствующими пигментными пастами по мере необходимости.
Настоящее изобретение, соответственно, также относится к способу изготовления водных материалов базового покрытия, содержащему добавление пигментов, в частности, в виде пигментной пасты, к составу лака для смешивания, как описано выше.
Примеры
Методы определения
1. Содержание твердых частиц
Если не указано иное, содержание твердых частиц, которое также далее упоминается как твердая фракция, определяли в соответствии со стандартом DIN EN ISO 3251 при температуре 130°С; 60 мин, начальная масса 1,0 г. Если в контексте настоящего изобретения ссылаются на официальный стандарт, то это, конечно, означает версию стандарта, которая была действующей на дату подачи заявки, или же, если на указанную дату нет действующей версии, то последнюю действующую версию.
2. Содержание изоцианатов
Содержание изоцианатов, которое также упоминается ниже как NCO-содержание, определяли посредством добавления избытка 2%-й концентрации раствора N,N-дибутиламина в ксилоле к однородному раствору образцов в ацетоне/N-этилпирролидоне (1:1 об. %), посредством потенциометрического обратного титрования избытка амина 0,1 N соляной кислотой, применяя метод на основе стандарта DIN EN ISO 3251, DIN EN ISO 11909, и DIN EN ISO 14896. NCO-содержание полимера, на основе твердых веществ, можно вычислить обратно с помощью фракции полимера (содержание твердых частиц) в растворе.
3. Гидроксильное число
Гидроксильное число определяли в соответствии с R.-P.  , R. Gnauck and R. Algeier, Plaste und Kautschuk, 20, 274 (1982), с помощью уксусного ангидрида в присутствии 4-диметиламинопиридина в качестве катализатора в растворе тетрагидрофурана (ТГФ)/диметилформамида (ДМФ) при комнатной температуре, посредством полного гидролиза избытка уксусного ангидрида, который остается после ацетилирования, и проведения потенциометрического обратного титрования уксусной кислоты спиртовым раствором гидроксида калия. Время ацетилирования, которое составляло 60 минут, было достаточным во всех случаях для обеспечения полного превращения.
, R. Gnauck and R. Algeier, Plaste und Kautschuk, 20, 274 (1982), с помощью уксусного ангидрида в присутствии 4-диметиламинопиридина в качестве катализатора в растворе тетрагидрофурана (ТГФ)/диметилформамида (ДМФ) при комнатной температуре, посредством полного гидролиза избытка уксусного ангидрида, который остается после ацетилирования, и проведения потенциометрического обратного титрования уксусной кислоты спиртовым раствором гидроксида калия. Время ацетилирования, которое составляло 60 минут, было достаточным во всех случаях для обеспечения полного превращения.
4. Кислотное число
Кислотное число определяли на основе стандарта DIN EN ISO 2114 в однородном растворе тетрагидрофурана (ТГФ)/воды (9 объемных частей ТГФ и 1 объемная часть дистиллированной воды) с этанольным раствором гидроксида калия.
5. Степень нейтрализации
Степень нейтрализации компонента х вычисляли, исходя из количества вещества групп карбоновой кислоты, которые присутствуют в компоненте (установленное посредством кислотного числа), и количества вещества нейтрализующего агента, который применяют.
6. Эквивалентная масса амина
Эквивалентная масса амина (раствор) служит для определения содержания амина в растворе, и ее определяли следующим образом. Образец для анализа растворяли при комнатной температуре в ледяной уксусной кислоте и титровали в 0,1N перхлорной кислоты в ледяной уксусной кислоте в присутствии кристаллического фиолетового. Начальная масса образца и расход перхлорной кислоты давало эквивалентную массу амина (раствор), массу раствора основного амина, которая необходима для того, чтобы нейтрализовать один моль перхлорной кислоты.
7. Степень блокирования первичных аминогрупп
Степень блокирования первичных аминогрупп определяли посредством ИК-спектроскопии, применяя ИК-спектрометр Nexus FT (от компании Nicolet) с помощью детектора ИК излучения (d=25 м, окно KВr) с максимальным поглощением при 3310 см-1 на основе применения серии растворов аминов, и стандартизации на максимальное поглощение при 1166 см-1 (внутренний стандарт) при температуре 25°С.
8. Содержание растворителей
Количество органического растворителя в смеси, например, в водных дисперсиях, определяли посредством газовой хроматографии (Agilent 7890А, 50 м кварцевая капиллярная колонка с полиэтиленгликолефой фазой или 50 м кварцевая капиллярная колонка с полидиметилсилоксановой фазой, газ-носитель гелий, инжектор для ввода проб с делением потока 250°С, температурный режим термостата 40 - 220°С, пламенно-ионизационный детектор, температура детектора 275°С, н-пропилгликоль в качестве внутреннего стандарта).
9. Среднечисленная молярная масса
Если не указано иное, среднечисленную молярную массу (Мn) определяли посредством парофазного осмометра 10.00 (от компании Knauer) на серии растворов в толуоле при температуре 50°С, с бензофеноном в качестве калибровочного вещество для определения экспериментальной калибровочной постоянной применяемого инструмента в соответствии с методом Е.  , G.
, G.  , K.-F. Arndt, "Leitfaden der Polymercharakterisierung" [Принципы определения характеристик полимера], Akademie-Verlag, Берлин, стр. 47 - 54, 1982.
, K.-F. Arndt, "Leitfaden der Polymercharakterisierung" [Принципы определения характеристик полимера], Akademie-Verlag, Берлин, стр. 47 - 54, 1982.
10. Средний размер частиц
Средний размер частиц (средний по объему) полиуретана и полимочевины, которые присутствуют в дисперсии (ПД) в соответствии с изобретением, определяли в контексте настоящего изобретения посредством фотонной корреляционной спектроскопии (ФКС).
Для измерения, в частности, применяли Malvern Nano S90 (от компании Malvern Instruments) при температуре 25 ± 1°С. Оборудование охватывает диапазон размеров от 3 - 3000 нм, и было оснащено 4 мВт He-Ne лазером с длиной волны 633 нм. Дисперсии (ПД) разбавляли не содержащей частиц, деионизированной водой в качестве дисперсионной среды, до того, как подвергнуть их измерению в 1 мл полистирольной кювете с подходящей интенсивностью рассеянного излучения. Оценку осуществляли, применяя цифровой коррелятор, с помощью программы анализа серии анализаторов Zetasizer, версия 6.32 (от компании Malvern Instruments). Измерение выполняли пять раз, и потом измерения повторяли на втором, свеже изготовленном образце.
Стандартное отклонение 5-кратного измерения составляло  . Максимальное отклонение среднего арифметического среднего по объему (V-среднее значение) пяти отдельных измерений составляло ±15%. Указанный средний размер частиц (средний по объему) представляет собой среднее арифметическое среднего размера частиц (среднего по объему) отдельных составов. Проверку осуществляли, применяя полистирольные стандарты, имеющие подтвержденные размеры частиц в диапазоне между 50 - 3000 нм.
. Максимальное отклонение среднего арифметического среднего по объему (V-среднее значение) пяти отдельных измерений составляло ±15%. Указанный средний размер частиц (средний по объему) представляет собой среднее арифметическое среднего размера частиц (среднего по объему) отдельных составов. Проверку осуществляли, применяя полистирольные стандарты, имеющие подтвержденные размеры частиц в диапазоне между 50 - 3000 нм.
11. Гель-фракция
Гель-фракцию частиц полиуретана и полимочевины (микрогелевые частицы), которые присутствуют в дисперсии (ПД) в соответствии с изобретением, определяют гравиметрически, в контексте настоящего изобретения. В этом случае, прежде всего, присутствующий полимер отделяли от образца водных дисперсий (ПД) (начальная масса 1,0 г) посредством сушки вымораживанием. Следующее определение температуры замерзания -температуры, после которой электрическое сопротивление образца не демонстрирует какого-либо дополнительного изменения при последующем понижении температуры - полностью замороженный образец подвергался своей основной сушке, обычно в диапазоне давлений вакуумной сушки, находящемся между 5 мбар и 0,05 мбар, при температуре сушки, ниже на 10°С, чем температура замерзания. Посредством постепенного повышения температуры нагреваемых поверхностей под полимером до 25°С, достигали быстрой сушки полимеров вымораживанием; после истечения времени сушки, которое составляло обычно 12 часов, количество выделенного полимер (твердая фракция, определенная посредством сушки вымораживанием) было постоянным, и больше не подвергалось какому-либо изменению, даже после продолжительной сушки вымораживанием. Последующая сушка при температуре поверхности под полимером, которая составляет 30°С с уменьшением окружающего давления до максимального значения (обычно в диапазоне между 0,05 и 0,03 мбар) обеспечивала оптимальную сушку полимера.
Выделенный полимер в последующем спекали в камере с принудительной подачей воздуха при температуре 130°С на протяжении одной минуты, и после этого экстрагировали на протяжении 24 часов при температуре 25°С, в избытке тетрагидрофурана (соотношение тетрагидрофурана и твердой фракции =300:1). Нерастворимую фракцию выделенного полимер (гель-фракцию) затем отделяли на подходящей фритте, сушили в камере с принудительной подачей воздуха при температуре 50°С на протяжении 4 часов, и в последующем повторно взвешивали.
Кроме того, было установлено, что при температуре спекания, составляющей 130°С, изменяя время спекания в пределах от одной минуты до двадцати минут, гель-фракция, которая была определена в отношении микрогелевых частиц, не зависит от времени спекания. Поэтому можно исключить, что реакции сшивания, следующие после выделения полимерного твердого вещества, дополнительно повышают объем гель-фракции.
Гель-фракцию, определенную таким образом в соответствии с изобретением, также называют гель-фракцией (высушенной вымораживанием).
Параллельно, гель-фракцию, которую далее также называют гель-фракция (130°С), определяли гравиметрически, посредством выделения полимерного образца из водной дисперсии (начальная масса 1,0 г), при температуре 130°С на протяжении 60 минут (содержание твердых частиц). Массу полимера определяли, после чего полимер экстрагировали в избытке тетрагидрофурана при температуре 25°С, по аналогии со способом, описанным выше, на протяжении 24 часов, после чего нерастворимую фракцию (гель-фракцию) отделяли, сушили, и повторно взвешивали.
12. Растворимость в воде
Растворимость органического растворителя в воде определяли при температуре 20°С следующим образом. Соответствующий органический растворитель и воду объединяли в подходящем стеклянном сосуде, смешивали, и смесь затем уравновешивали. Количества воды и растворителя выбирали таким образом, что после уравновешивание получали две фазы, отдельные друг от друга. После уравновешивания, образец извлекали из водной фазы (то есть, фазы, содержащей больше воды, чем органического растворителя), применяя шприц, и указанный образец разбавляют тетрагидрофураном с соотношением 1/10, при этом фракцию растворителя определяют посредством газовой хроматографии (в отношении условий смотри раздел 8. Содержание растворителей).
Если две фазы не образуются, независимо от количества воды и растворителя, то растворитель смешивают с водой с любым соотношением по массе. Указанный растворитель, который, в результате, является бесконечно растворимым в воде (например, ацетон), по меньшей мере, не является растворителем (Z.2).
13. Объемное содержание твердых частиц (вычисленное):
Объемное содержание твердых частиц вычисляли в соответствии с VdL-RL 08 [German Paint Industrial Association Guideline], "Determining the solids volume of anticorrosion coating materials as basis for productivity calculations", Verband der Lackindustrie e.V. версия дек. 1999 г. Объем твердых частиц VSC (объем твердой фазы) вычисляли в соответствии со следующей формулой, включающей физические характеристики соответствующих применяемых материалов (плотность растворителей, плотность твердых веществ):
VSC = (плотность (влажного покрытия) х твердая фракция (влажного покрытия))/плотность (термообработанного покрытия) VSC - объемное содержание твердых частиц в % плотность (влажного покрытия) - вычисленная плотность влажного покрывающего материала на основании плотности отдельных компонентов (плотность растворителей и плотность твердых веществ) в г/см
твердая фракция (влажного покрытия) - содержание твердых частиц (в %) влажного покрытия, определенное в соответствии со стандартом DIN EN ISO 3251 при температуре 130°С, 60 минут, начальная масса 1,0 г. плотность (термообработанного покрытия): плотность термообработанного покрывающего материала на металлической панели в г/см3. Получение дисперсии (ПД)
Дисперсию (ПД) изготавливали следующим образом:
а) Получение частично нейтрализованного раствора преполимера
В реакционном сосуде, оснащенном мешалкой, внутренним термометром, обратным холодильником, и электрическим нагревом, растворяли 559,7 части по массе неразветвленного сложного полиэфира полиола и 27,2 части по массе диметилолпропионовой кислоты (от компании GEO Speciality Chemicals) в 344,5 частей по массе метилэтилкетона, в атмосфере азота. Неразветвленный сложный полиэфир диола получали до этого из димеризованной жирной кислоты (Pripol® 1012, от компании Croda), изофталевой кислоты (от компании BP Chemicals), и гексан-1,6-диола (от компании BASF SE) (соотношение массы исходных материалов: доменной жирной кислоты к изофталевой кислоте и к гексан-1,6-диолу =54,00:30,02:15,98), и при этом он имел гидроксильное число, составляющее 73 мг КОН/г на основе твердой фракции, кислотное число, составляющее 3,5 мг КОН/г на основе твердой фракции, рассчитанную среднечисленную молярную массу, составляющую 1379 г/моль, и среднечисленную молярную массу, которая была определено посредством парофазной, составляющую 1350 г/моль.
К полученному раствору при 30°С последовательно добавляли 213,2 части по массе дициклогексилметан 4,4'-диизоцианата (Desmodur® W, компания Bayer MaterialScience) с содержанием изоцианатов, составляющим 32,0 мас. %, и 3,8 части по массе дилаурата дибутилолова (от компании Merck). Смесь затем нагревали до 80°С при перемешивании. Перемешивание продолжали при этой температуре до тех пор, пока содержание изоцианатов в растворе не становилось постоянным и составляло 1,49 мас. %. После этого к преполимеру добавляли 626,2 части по массе метилэтилкетона, и реакционную смесь охлаждали до 40°С. Когда достигали 40°С, на протяжении двух минут добавляли по каплям 11,8 частей по массе триэтиламина (от компании BASF SE) и смесь перемешивали на протяжении дополнительных 5 минут.
б) Реакция преполимера с диэтилентриаминдикетимином Затем 30,2 части по массе 71,9 мас. % разбавленного раствора диэтилентриаминдикетимина в метилизобутилкетоне смешивали на протяжении одной минуты (соотношение изоцианатных групп преполимера к диэтилентриаминдикетимину (имеющему вторичную аминогруппу): 5:1 моль/моль, что соответствует двум NCO-группам на блокированную первичную аминогруппу), и температура реакции повышалась на 1°С сразу же после добавления к раствору преполимера. Разбавленный раствор диэтилентриаминдикетимина в метилизобутилкетоне изготавливали до этого посредством азеотропного удаления воды из реакции во время реакции диэтилентриамина (от компании BASF SE) с метилизобутилкетоном в метилизобутилкетоне при температуре 110 - 140°С. Доведение до эквивалентной массы амина (раствор), составляющей 124,0 г/экв, осуществляли посредством разбавления метилизобутилкетоном. Блокирование первичных аминогрупп, составляющее 98,5%, определяли посредством ИК-спектроскопии, на основе остаточного поглощения при 3310 см-1. Было выявлено, что содержание твердых частиц раствора полимера, содержащего изоцианатные группы, составляло 45,3%.
в) Диспергирование и вакуумная дистилляция
После 30 минут перемешивания при температуре 40°С, содержимое химического реактора диспергировали в 1206 частей по массе деионизированной воды (23°С) на протяжении 7 минут. Метилэтилкетон отгоняли из полученной дисперсии при пониженном давлении при температуре 45°С, и любые потери растворителя и воды компенсировали деионизированной водой, с тем, чтобы получить содержание твердых частиц, составляющее 40 мас. %.
Была получена белая, стабильная, насыщенная твердыми частицами, маловязкая дисперсия, содержащая сшитые частицы, которая при этом ни в малейшей степени не демонстрировала образования осадка, даже по истечении 3 месяцев.
Свойства полученной микрогелевой дисперсии были следующими:
Содержание твердых частиц (130°С, 60 минут, 1 г): 40,2 мас. %
Содержание метилэтилкетона (GC): 0,2 мас. %
Содержание метилизобутилкетона (GC): 0,1 мас. %
Вязкость (23°С, ротационный вискозиметр, скорость сдвига =1000/с): 15 мПа⋅с
Кислотное число 17,1 мг КОН/г на
основе содержания твердых частиц
Степень нейтрализации (вычисленная) 49%
рН (23°С) 7,4
Размер частиц (фотокорреляционная спектроскопия, средний по объему) 167 нм
Гель-фракция (высушенная вымораживанием) 85,1 мас. %
Гель-фракция (130°С) 87,3 мас. %
Получение водных материалов базового покрытия Компоненты, перечисленные в Таблице 1, перемешивали вместе в указанном порядке, с тем, чтобы получить водные составы лаков для смешивания. В то время, как состав лака для смешивания 1 включает в качестве сшивающего агента меламиновую смолу, состав лака для смешивания 2 является полностью свободным от сшивающих агентов. Оба состава лаков для смешивания содержат дисперсию (ПД), описанную выше, и полностью свободны от загустителей, таких как, например, неорганические загустители.

Применяя составы лаков для смешивания, описанные в Таблице 1, изготавливали различные водные материалы базового покрытия ровного цвета, а также цветные и эффектные водные материалы базового покрытия. Для этой цели, составы лаков для смешивания составляли с желательными пастами для подцветки, и необязательно с применением дополнительных добавок и растворителей. Соответственно, в зависимости от требований, например, возможно применять добавки, защищающие от УФ-излучения и/или добавки для изменения текучести или для уменьшения поверхностного натяжения.
Таблицы 2-5 показывают составы водных материалов базового покрытия, изготовленные, с применением указанных компонентов, которые смешивали в указанном порядке. Составляющие компоненты составов лаков для смешивания также здесь перечислены отдельно, поскольку хотя и применение составов лаков для смешивания является преимущественным, тем не менее, оно не является обязательным. В результате соответствующего объединения отдельных компонентов в указанном порядке получают одинаковые материалы базового покрытия.
Все водные материалы базового покрытия (БП) имели рН, составляющую 7,8 - 8,6, и вязкость распыления, составляющую 70 - 110 мПа⋅с, в условиях усилий сдвига, которые составляют 1000 с-1, установленную посредством применения ротационного вискозиметра (прибор Rheomat RM 180 от компании Mettler-Toledo), при температуре 23°С 

Материалы базового покрытия 1 и 2 являются стабильными после хранения при температуре 40°С по меньшей мере на протяжении 4 недель, что означает то, что в это время они не демонстрируют какой-либо склонности к образованию осадка и не испытывают существенных изменений (менее 15%) вязкости в условиях низкой скорости сдвига (усилие сдвига составляет 1 с-1, что было установлено с применением ротационного вискозиметра). Материал базового покрытия 1 имеет содержание твердых частиц, составляющее 42%, и вычисленный объем твердых частиц, который составляет 35%. Материал базового покрытия 2 имеет содержание твердых частиц, составляющее 47%, и вычисленный объем твердых частиц, который составляет 35%. 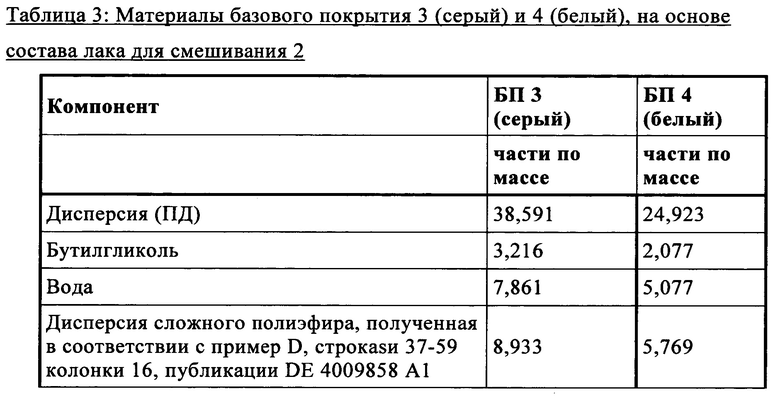
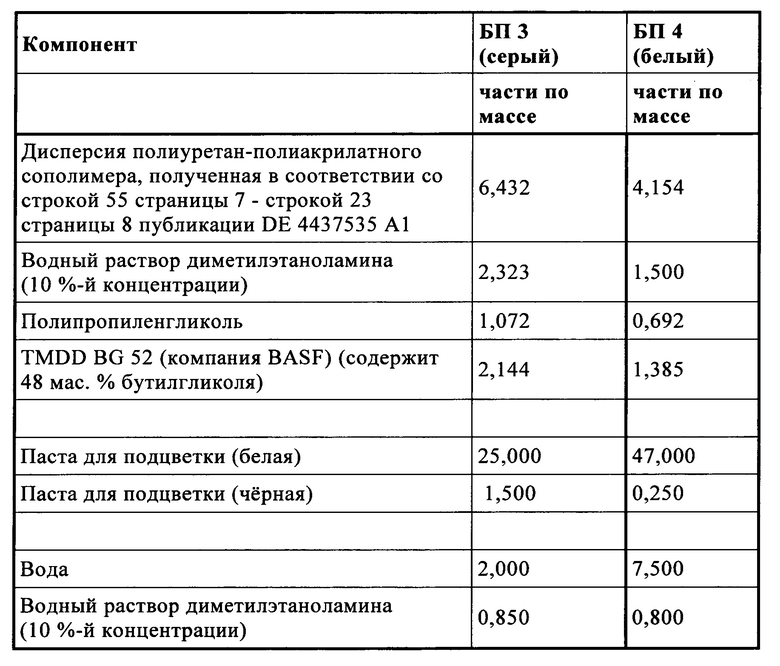
Материалы базового покрытия 3 и 4 являются стабильными после хранения при температуре 40°С на протяжении по меньшей мере 4 недель, что означает то, что в это время они не демонстрируют какой-либо склонности к образованию осадка и не претерпевают существенных изменений (менее 15%) вязкости в условиях низкой скорости сдвига (усилие сдвига, составляющее 1 с-1, что было установлено посредством применения ротационного вискозиметра). Материал базового покрытия 3 имеет содержание твердых частиц, составляющее 38%, и вычисленный объем твердых частиц, составляющий 32%. Материал базового покрытия 4 имеет содержание твердых частиц, составляющее 42%, и вычисленный объем твердых частиц, составляющий 31%.

Материалы базового покрытия 5 и 6 являются стабильными после хранения при температуре 40°С на протяжении по меньшей мере 4 недель, что означает то, что в это время они не демонстрируют какой-либо склонности к образованию осадка и не претерпевают существенных изменений (менее 15%) вязкости в условиях низкой скорости сдвига (усилие сдвига, составляющее 1 с-1, что было установлено посредством применения ротационного вискозиметра). Материал базового покрытия 5 имеет содержание твердых частиц, составляющее 31%, и вычисленный объем твердых частиц, составляющий 27%. Материал базового покрытия 6 имеет содержание твердых частиц, составляющее 38%, и вычисленный объем твердых частиц, составляющий 34%. 


Материалы базового покрытия 7 и 8 являются стабильными после хранения при температуре 40°С на протяжении по меньшей мере 4 недель, что означает то, что в это время они не демонстрируют какой-либо склонности к образованию осадка и не испытывают существенных изменений (менее 15%) вязкости в условиях низкой скорости сдвига (усилие сдвига, составляющее 1 с-1, что было установлено посредством применения ротационного вискозиметра). Материал базового покрытия 7 имеет содержание твердых частиц, составляющее 19%, и вычисленный объем твердых частиц, составляющий 22%. Материал базового покрытия 8 имеет содержание твердых частиц, составляющее 24%, и вычисленный объем твердых частиц, составляющий 21%.
Изготовление упомянутых выше паст для подцветки:
Пасту для подцветки (черную) изготавливали из 25 частей по массе акрилированной полиуретановой дисперсии, полученной в соответствии с международной заявкой на получение патента WO 91/15528, дисперсии связующего вещества А, 10 частей по массе черной углеродной сажи, 0,1 части по массе метилизобутилкетона, 1,36 частей по массе диметилэтаноламина (10% в деминерализованной воде), 2 частей по массе доступного на рынке простого полиэфира (Pluriol® Р900 от компании BASF SE), и 61,45 частей по массе деионизированной воды.
Пасту для подцветки (белую) изготавливали из 43 частей по массе акрилированной полиуретановой дисперсии, полученной в соответствии с международной заявкой на получение патента WO 91/15528, дисперсии связующего вещества А, 50 частей по массе титана рутильной формы 2310, 3 частей по массе 1-пропокси-2-пропанола, и 4 частей по массе деионизированной воды.
Пасту для подцветки (красную) изготавливали из 38,4 частей по массе акрилированной полиуретановой дисперсии, полученной в соответствии с международной заявкой на получение патента WO 91/15528, дисперсии связующего вещества А, 47,1 части по массе Bayferrox® 13 ВМ/Р, 0,6 частей по массе диметилэтаноламина (10%-й концентрации в ДИ воде), 4,7 частей по массе доступного на рынке простого полиэфира (Pluriol® Р900 от компании BASF SE), 2 частей по массе бутилгликоля, и 7,2 частей по массе деионизированной воды. Получение многослойных красочных систем с применением материалов базового покрытия 1 - 8, и исследование эффективности указанных красочных систем (а) Получение посредством способа в соответствии с изобретением двух пленок базового покрытия
Основы, которые применяли для получения красочной системы, представляли собой стальные панели, на которых получали затвердевшее электроосажденное покрытие, применяя доступный на рынке катодный материал для электроосаждения.
Прежде всего, в качестве подготовительного для нанесения цвета материала базового покрытия, наносили серый материал базового покрытия (БП 1 или БП 3), посредством электростатического напыления, где толщина пленки составляла 20 микрометров, и затем она самоиспарялась при комнатной температуре на протяжении 3 минут. Сверху указанной первой пленки базового покрытия наносили цветной и/или эффектный материал базового покрытия (БП 2, БП 4 - БП 8), в каждом случае посредством электростатического напыления, где толщина пленок составляла 20 микрометров, при этом каждая пленка самоиспарялась при комнатной температуре на протяжении 4 минут, и подвергалась промежуточной сушке при 60°С на протяжении 5 минут. Сверху указанной подвергшейся промежуточной сушке пленки базового покрытия с помощью электростатического напыления наносили доступный на рынке двухкомпонентный материал покровного лака, где толщина пленки составляла 35 - 45 микрометров, и вся система затем снова подвергалась самоиспарению при комнатной температуре на протяжении 10 минут, и в последующем затвердевала при температуре 140°С на протяжении 20 минут.
Более того, для определения предела образования пор, получали многослойные красочные системы, в которых, в отличие от описанных выше красочных систем, второй материал базового покрытия наносили в виде клина (толщина пленки составляла до 40 микрометров).
Многослойные красочные системы исследовали относительно текучести и внешнего вида, применяя измерительный инструмент WaveScan (от компании Byk-Gardner) (коротковолновой, длинноволновой), где при этом низкие значения соответствовали лучшей текучести. Дополнительно, исследовали предел образования пор. Склонность к образованию пор увеличивалась с повышением толщины покрывающих пленок (в этом случае, вторая пленка базового покрытия), поскольку, соответственно, при этом из пленки выделяется более высокое количество воздуха, органических растворителей и/или воды. Толщина указанной пленки, выше которой наблюдаются поры, упоминается как предел образования пор. Очевидно, чем выше предел образования пор, тем лучше качество относительно устойчивости к образованию пор.
Также исследовали адгезионные свойства. Проводимые исследования представляли собой испытание на поперечный надрез в соответствии со стандартом DIN EN ISO 2409, испытание PV3.14.7 на растрескивание в соответствии со стандартом DIN EN ISO 20567-1, испытание PV1503 струей пара, адаптированное к стандарту DIN 55662, необязательно в комбинации с исследованием PV3.16.1 конденсации воды (CWT) в соответствии со стандартом DIN EN ISO 6270-2. Низкие значения в этом случае соответствуют хорошей адгезии. 

Результаты показывают, что текучесть многослойных красочных систем является отличной. Предел образования пор с толщиной пленки второго материала базового покрытия, которая составляла 40 микрометров, также не был достигнут, и это является очень хорошим показателем. То же применимо к адгезионным свойствам многослойных красочных систем.
(б) Получение посредством способа в соответствии с изобретением одной пленки базового покрытия
Основы, которые применяли для получения красочной системы, представляли собой стальные панели, на которых получали затвердевшее электроосажденное покрытие, применяя доступный на рынке катодный материал для электроосаждения.
В каждом случае, прежде всего наносили цветной и/или эффектный материал базового покрытия (БП 2, БП 5) посредством применения электростатического напыления, при этом толщина пленок составляла 35 микрометров, затем их подвергали самоиспарению при комнатной температуре на протяжении 4 минут, и в последующем подвергали промежуточной сушке при температуре 60°С на протяжении 5 минут. Сверху указанной промежуточно высушенной пленки базового покрытия, посредством электростатического напыления, наносили доступный на рынке двухкомпонентный материал покровного лака, где толщина пленки составляла 35 - 45 микрометров, и затем всю систему снова подвергали самоиспарению при комнатной температуре на протяжении 10 минут, и в последующем подвергали затвердеванию при температуре 140°С на протяжении 20 минут.
Адгезионные свойства исследовали, как указано в (а). Таблица В показывает следующие результаты.

Очевидно, что полученные таким образом многослойные красочные системы демонстрируют очень хорошую адгезию.
(В) Получение в соответствии со стандартным способом предшествующего уровня техники
Основы, которые применяли для получения красочной системы, представляли собой стальные панели, на которых получали затвердевшее электроосажденное покрытие, применяя доступный на рынке катодный материал для электроосаждения.
Прежде всего, посредством электростатического напыления наносили доступную на рынке серую шпатлевку, с толщиной пленки, которая составляла 30 микрометров, за чем следовало самоиспарение при комнатной температуре на протяжении 10 минут, а затем следовало отверждение при температуре 155°С на протяжении 20 минут. Сверху этого затвердевшего грунтовочного слоя наносили цветной и/или эффектный материал базового покрытия, в каждом случае посредством электростатического напыления, при этом толщина пленок составляла 20 микрометров (БП 2 и БП 3) или 15 микрометров (БП 5 и БП 7), каждую пленку при этом подвергали самоиспарению при комнатной температуре на протяжении 3 минут, а затем подвергали промежуточной сушке при температуре 80°С на протяжении 5 минут. Сверху этой промежуточно-высушенной пленки базового покрытия, посредством электростатического напыления, наносили доступный на рынке двухкомпонентный материал покровного лака, с толщиной пленки, которая составляла 35 - 45 микрометров, и затем всю систему снова подвергали самоиспарению при комнатной температуре на протяжении 10 минут, и после этого подвергали затвердеванию при температуре 150°С на протяжении 20 минут.
Адгезионные свойства и образование пор исследовали как в (а).

Результаты показывают, что характеристики являются хорошими даже в случае, когда применяют стандартный способ, хотя при этом указанный способ отличается от способа в соответствии с изобретением необходимостью в дополнительной стадии отверждения. Рассматривая результаты в целом, является очевидным, что многослойные красочные системы в соответствии с изобретением, полученные посредством способа в соответствии с изобретением, по меньшей мере сопоставимы по своим характеристикам с характеристиками систем, полученных посредством стандартного способа, но при этом могут быть получены более экономичным способом. Соответственно, в результате настоящего изобретения достигается успех в обеспечении способа, который объединяет экономичный технологический прием с отличными свойствами полученных красочных систем.
Краткое описание графических материалов
Фигура 1:
Схематическое формирование многослойной красочной системы (М) в соответствии с изобретением, расположенной на металлической основе (S), при этом система (М) содержит затвердевшее электроосажденное покрытие (Е.1), а также пленку базового покрытия (В.2.1) и пленку покровного лака (K), которые были совместно отверждены.
Фигура 2:
Схематическое формирование многослойной красочной системы (М) в соответствии с изобретением, расположенной на металлической основе (S), при этом система (М) содержит затвердевшее электроосажденное покрытие (Е.1), две пленки базового покрытия (В.2.2.x), а именно первую пленку базового покрытия (b.2.2.а) и самую верхнюю пленку базового покрытия (b.2.2.z), расположенную на ней, и пленку покровного лака (K), которые были совместно отверждены.
Фигура 3:
Схематическое формирование многослойной красочной системы (М) в соответствии с изобретением, расположенной на металлической основе (S), при этом система (М) содержит затвердевшее электроосажденное покрытие (Е.1), три пленки базового покрытия (В.2.2.x), а именно первую пленку базового покрытия (b.2.2.а), пленку базового покрытия (b.2.2.b), расположенную на ней, и самую верхнюю пленку базового покрытия (b.2.2.z), а также пленку покровного лака (K), которые были совместно отверждены.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2016 |
|
RU2700867C1 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ НА ПЛАСТМАССОВЫХ ОСНОВАХ | 2016 |
|
RU2686904C1 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2016 |
|
RU2708852C1 |
| ВОДНОЕ БАЗОВОЕ ПОКРЫТИЕ И ПОЛУЧЕНИЕ МНОГОСЛОЙНЫХ КРАСОЧНЫХ СИСТЕМ ПОСРЕДСТВОМ ПРИМЕНЕНИЯ БАЗОВОГО ПОКРЫТИЯ | 2017 |
|
RU2746776C2 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2014 |
|
RU2665510C1 |
| ВОДНАЯ ДИСПЕРСИЯ ПОЛИУРЕТАН-ПОЛИМОЧЕВИНА И КРАСКА НА ВОДНОЙ ОСНОВЕ, СОДЕРЖАЩАЯ УКАЗАННУЮ ДИСПЕРСИЮ | 2015 |
|
RU2678207C2 |
| КРАСКИ НА ВОДНОЙ ОСНОВЕ, ВКЛЮЧАЮЩИЕ СШИВАЮЩИЕ ПОЛИУРЕТАНОВЫЕ СВЯЗУЮЩИЕ ВЕЩЕСТВА И ОПРЕДЕЛЕННУЮ КОМПОЗИЦИЮ РАСТВОРИТЕЛЕЙ | 2016 |
|
RU2690855C1 |
| ВОДНАЯ ДИСПЕРСИЯ ПОЛИУРЕТАН-ПОЛИМОЧЕВИНЫ И КРАСКА НА ВОДНОЙ ОСНОВЕ, СОДЕРЖАЩАЯ УКАЗАННУЮ ДИСПЕРСИЮ | 2015 |
|
RU2678012C2 |
| СПОСОБ ПОЛУЧЕНИЯ МНОГОСЛОЙНОЙ КРАСОЧНОЙ СИСТЕМЫ | 2014 |
|
RU2675915C1 |
| ВОДНЫЕ БАЗОВЫЕ ПОКРЫТИЯ С ПОВЫШЕННОЙ УСТОЙЧИВОСТЬЮ К ВОЗДЕЙСТВИЮ ЦИРКУЛЯЦИОННОГО ТРУБОПРОВОДА | 2017 |
|
RU2758381C2 |
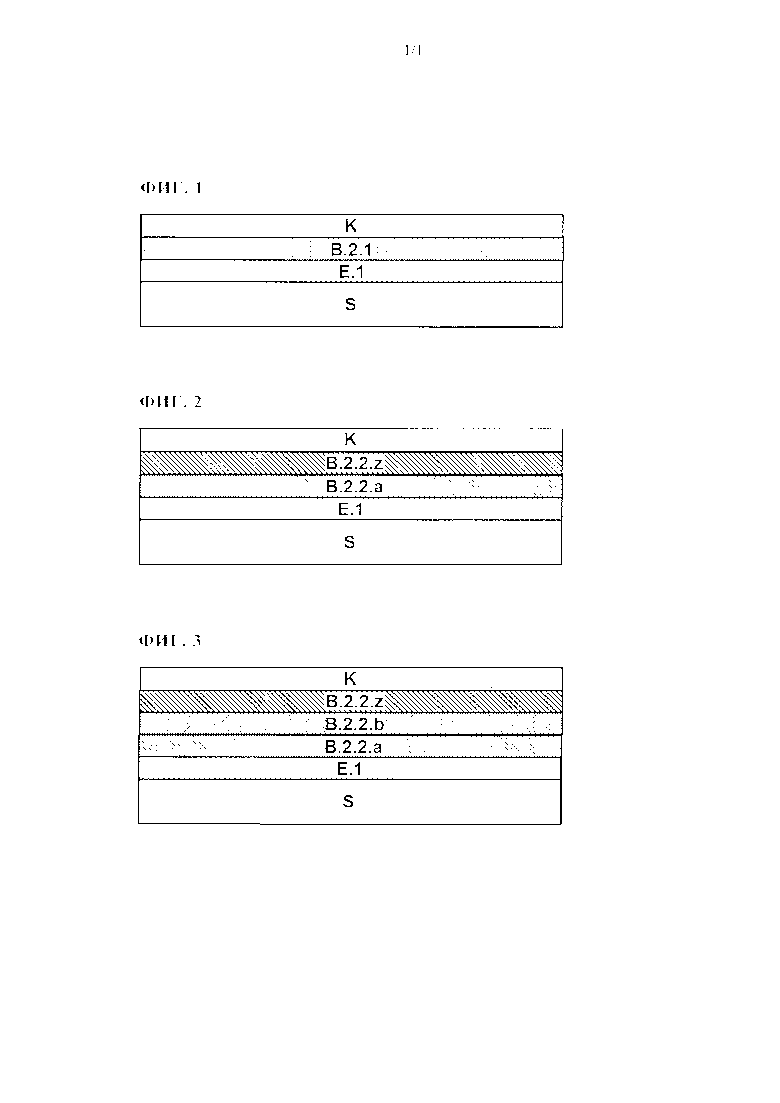
Настоящее изобретение относится к способу получения многослойной красочной системы на металлической основе, а также к многослойной красочной системе. Способ включает (1) получение затвердевшего электроосажденного покрытия на металлической основе, (2) получение пленки базового покрытия или двух или большего количества нанесенных непосредственно друг на друга пленок базового покрытия непосредственно на затвердевшем электроосажденном покрытии, (3) получение пленки покровного лака непосредственно на пленке базового покрытия или на самой верхней пленке базового покрытия и (4) совместное отверждение пленки базового покрытия и пленки покровного лака или пленок базового покрытия и пленки покровного лака. Материал базового покрытия или по меньшей мере один из материалов базового покрытия содержит по меньшей мере одну водную полиуретан-полимочевинную дисперсию, включающую частицы полиуретана и полимочевины. Указанные частицы полиуретана и полимочевины в прореагировавшем виде содержат преполимер полиуретана, включающий изоцианатные группы и содержащий анионные группы и/или группы, которые могут превращаться в анионные группы, а также по меньшей мере один полиамин, содержащий две первичные и одну или две вторичные аминогруппы. Средний размер частиц составляет 40-2000 нм и содержание гель-фракции составляет по меньшей мере 50%. Указанная дисперсия состоит из по меньшей мере 90 мас.% частиц полиуретана и полимочевины, а также воды. Заявленный способ позволяет упростить технологию получения многослойных красочных систем, а полученные многослойные красочные системы демонстрируют отличную устойчивость в отношении пор, а также хорошую адгезию и внешний вид. 2 н. 12 з.п. ф-лы, 3 ил., 9 табл., 8 пр.
1. Способ получения многослойной красочной системы (М) на металлической основе (S), содержащий
(1) получение затвердевшего электроосажденного покрытия (Е.1) на металлической основе (S) посредством электрофоретического нанесения материала для электроосаждения (е.1) на основу (S) и последующего отверждения материала для электроосаждения (е.1),
(2) получение (2.1) пленки базового покрытия (В.2.1) или (2.2) двух или большего количества нанесенных непосредственно друг на друга пленок базового покрытия (В.2.2.x) непосредственно на затвердевшем электроосажденном покрытии (Е.1) посредством (2.1) нанесения водного материала базового покрытия (b.2.1) непосредственно на электроосажденное покрытие (Е.1) или посредством (2.2) нанесения на электроосажденное покрытие (Е.1) двух или большего количества материалов базового покрытия (b.2.2.x) непосредственного друг на друга,
(3) получение пленки покровного лака (K) непосредственно на (3.1) пленке базового покрытия (В.2.1) или на (3.2) самой верхней пленке базового покрытия (В.2.2.x) посредством нанесения материала покровного лака (к) непосредственно на (3.1) пленку базового покрытия (В.2.1) или на (3.2) самую верхнюю пленку базового покрытия (В.2.2.x),
(4) совместное отверждение (4.1) пленки базового покрытия (В.2.1) и пленки покровного лака (K) или (4.2) пленок базового покрытия (В.2.2.x) и пленки покровного лака (K),
где
материал базового покрытия (b.2.1) или по меньшей мере один из материалов базового покрытия (b.2.2.х) содержит по меньшей мере одну водную полиуретан-полимочевинную дисперсию (ПД), которая включает частицы полиуретана и полимочевины, где частицы полиуретана и полимочевины, которые присутствуют в дисперсии (ПД), в каждом случае в прореагировавшем виде, содержат
(Z.1.1) по меньшей мере один преполимер полиуретана, включающий изоцианатные группы, содержит при этом анионные группы и/или группы, которые могут превращаться в анионные группы, а также
(Z.1.2) по меньшей мере один полиамин, содержащий две первичные аминогруппы и одну или две вторичные аминогруппы, и имеют средний размер частиц, который составляет 40-2000 нм, а также гель-фракцию, которая составляет по меньшей мере 50%,
и при этом дисперсия (ПД) состоит из по меньшей мере 90 мас. % частиц полиуретана и полимочевины, а также воды.
2. Способ по п. 1, где анионные группы и/или группы, которые могут превращаться в анионные группы, представляют собой карбоксилатные группы и/или группы карбоновой кислоты.
3. Способ по п. 1, где полиамин (Z.1.2) состоит из одной или двух вторичных аминогрупп, двух первичных аминогрупп, а также алифатически насыщенных углеводородных групп.
4. Способ по любому из пп. 1-3, где частицы полиуретана и полимочевины, которые присутствуют в дисперсии, имеют средний размер частиц, который составляет 110-500 нм, и гель-фракцию, которая составляет по меньшей мере 80%.
5. Способ по любому из пп. 1-4, где материал базового покрытия (b.2.1) или по меньшей мере один из материалов базового покрытия (b.2.2.х), предпочтительно все материалы базового покрытия (b.2.2.х), дополнительно содержат, в качестве связующего вещества, по меньшей мере один полимер с гидроксильными функциональными группами, выбранный из группы, состоящей из полиуретанов, сложных полиэфиров, полиакрилатов и сополимеров указанных полимеров.
6. Способ по любому из пп. 1-5, где материал базового покрытия (b.2.1) или по меньшей мере один из материалов базового покрытия (b.2.2.х), предпочтительно все материалы базового покрытия (b.2.2.х), являются однокомпонентными покрывающими материалами.
7. Способ по любому из пп. 1-6, где совместное отверждение (4) осуществляется при температурах, которые составляют 100-250°С, на протяжении периода времени, составляющего 5-60 мин.
8. Способ по любому из пп. 1-7, где (2.2) получают по меньшей мере две пленки базового покрытия, первую пленку базового покрытия (В.2.2.а), которая при этом содержит белые пигменты и черные пигменты, непосредственно на электроосажденном покрытии (Е.1), и по меньшей мере одну дополнительную пленку базового покрытия (В.2.2.x), которая при этом содержит эффектные пигменты.
9. Способ по любому из пп. 1-8, где материал базового покрытия (b.2.1) и материалы базового покрытия (b.2.2.х) в случае, когда они содержат по меньшей мере один сшивающий агент, имеют содержание твердых частиц, которое составляет по меньшей мере 25%, и в случае, когда материал базового покрытия (b.2.1) и материалы базового покрытия (b.2.2.х) не содержат сшивающего агента, имеют содержание твердых частиц, которое составляет по меньшей мере 15%.
10. Способ по п. 9, где материалы базового покрытия при температуре 23°С в условиях усилия сдвига, которое составляют 1000 1/с, имеют вязкость, составляющую 40-150 мПа⋅с.
11. Способ по любому из пп. 1-10, где материал базового покрытия (b.2.1) или по меньшей мере один из материалов базового покрытия (b.2.2.х), предпочтительно все материалы базового покрытия (b.2.2.х), содержат по меньшей мере один сшивающий агент, выбранный из группы блокированных полиизоцианатов и аминопластовых смол.
12. Способ по любому из пп. 1-11, где преполимер (Z.1.1) содержит по меньшей мере один сложный полиэфир диола, полученный с применением диолов и дикарбоновых кислот, при этом по меньшей мере 50 мас. %, предпочтительно 55-75 мас. %, дикарбоновых кислот, которые применяют при получении сложных полиэфиров диолов, являются димерными жирными кислотами.
13. Способ по любому из пп. 1-12, где материал базового покрытия (b.2.1) или материалы базового покрытия (b.2.2.х) наносят посредством электростатического напыления или пневматическое распыления.
14. Многослойная красочная система (М), полученная посредством способа по любому из пп. 1-13.
| WO 2004035646 A1, 29.04.2004 | |||
| EP 1736246 A1, 27.12.2006 | |||
| ВОДНАЯ ПОЛИУРЕТАНОВАЯ ДИСПЕРСИЯ, НЕ СОДЕРЖАЩАЯ N-МЕТИЛПИРРОЛИДОНА И РАСТВОРИТЕЛЕЙ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2006 |
|
RU2412213C2 |

