Область техники
Данное изобретение относится к производным хиназолина, фармацевтическим композициям, способам их получения и фармацевтическим применениям.
Уровень техники
Киназы являются важными сигнальными молекулами клеток. Мутации киназ могут привести к заболеваниям или состояниям, включая иммунодефициты, рак, сердечнососудистые заболевания и нарушения эндокринной системы, такие как болезнь Паркинсона, нарушения обмена веществ, онкогенез, болезнь Альцгеймера, порок сердца, диабет, дистрофию (вырождение), воспаление, болезнь почек, атеросклероз и заболевание дыхательных путей.
Рак возникает в результате нарушения регулировки сигнальных путей, медиирующих рост клеток и запрограммированную гибель клеток (апоптоз). Протеинкиназы представляют собой большое семейство белков, играющих важную роль в сигнальных путях, регулирующих ряд различных клеточных функций, таких как рост клеток, дифференциация и смерть.
Гиперактивность протеинкиназ влечен за собой ряд различных видов рака человека. Например, гиперэкспрессия Akt2 киназы была установлена при раке яичника (J.Q. Cheung и др., Proc. Natl. Acad. Sci. U.S.A. 89: 9267-9271 (1992)) и раке поджелудочной железы (J.Q. Cheung и др., Proc. Natl. Acad. Sci. U.S.A. 93: 3636-3641 (1996)), а гиперэкспрессия Akt3 киназы была показана в клеточных линиях рака молочной железы и рака простаты (Nakatani и др., J. Biol. Chem. 274: 21528-21532 (1999)).
Было показано, что различные ингибиторы протеинкиназ эффективно лечат определенные виды рака. Например, Gleevec™ (имантиниб, Novartis) можно использовать для лечения хронического миелобластного лейкоза (CML) (Kumar и др.), а ингибитор Raf киназы (BAY-43-9006), как было установлено, можно использовать для лечения солидных опухолей и миелоидного лейкоза (WO 2004/022562).
Тирозинкиназа представляет собой группу ферментов для катализа фосфорилирования тирозиновых остатков белков, она играет критическую роль во внутриклеточной передаче сигналов (сигнальной трансдукции). Тирозинкиназа участвует в регуляции нормальных клеток, передаче сигналов и росте, а также связана с пролиферацией, дифференциацией, миграцией и апоптозом раковых клеток. Дисфункции тирозинкиназ могут вызвать нарушение пролиферации клеток, ведущее, в конечном счете, к образованию опухоли.
Многие рецепторные тирозинкиназы вовлечены в образование опухолей по причинам, включающим генные мутации, транслокацию (перемещение) хромосом или гиперэкспрессию киназы.
Ингибиторы тирозинкиназ разработаны в соответствии с белковыми структурами тирозинкиназ, и большинство из них относятся к конкурентным ингибиторам АТФ (аденозинтрифосфата). Эти ингибиторы воздействуют на внутриклеточную область белка тирозинпротеинкиназы, так что они по существу блокируют дальнейшую передачу сигнала, медиируемую тирозинкиназой, и таким образом ингибируют рост, ангиогенез и метастаз опухоли.
Вандетаниб (Vandetanib) представляет собой синтетическое анилин-хиназолиновое соединение, которое может одновременно воздействовать на тирозинкиназные рецепторы опухолевых клеток: VEGFR (рецептор фактора роста сосудистого эндотелия, рецептор ФРЭС), EGFR (рецептор эпидермального фактора роста) и RET (рецептор, вовлеченный в рак щитовидной железы), а также может селективно ингибировать другие тирозинкиназы и серин/треонинкиназы.
Таким образом, препараты направленного действия, которые могут ингибировать протеинкиназы, представляют собой новое поколение химиотерапевтических средств для специфических молекулярных объектов. Потенциально они могут обеспечить большую эффективность лечения различных видов рака с меньшими побочными эффектами, чем обычные химиотерапевтические средства.
Сущность изобретения
Согласно одному из аспектов, данное изобретение предусматривает соединение формулы (I) или его фармацевтически приемлемую соль:

где: R1 представляет собой -O(СН2)nR3, где n равно 0, 1, 2, 3, 4 или 5, и
R3 представляет собой:
(1) арил, такой как фенил, необязательно замещенный Ra и/или Rb, где Ra и Rb каждый независимо выбран из группы, включающей галоалкил такой как трифторметил, циано и насыщенный гетероциклоалкил, такой как насыщенный гетероциклоалкил, содержащий один или более гетероатомов, выбранных из О, N и S, например, морфолино или Ra и Rb соединены вместе с образованием -О-СН2-О-;
(2) гетероарил, необязательно замещенный Rc и/или Rd, где Rc и Rd каждый независимо выбран из группы, включающей алкил и насыщенный гетероциклоалкил-карбонил, такой как насыщенный гетероциклоалкил-карбонил, содержащий один или более гетероатомов, выбранных из О, N и S, например морфолино-карбонил;
(3) -NReRf, где Re и Rf каждый независимо выбран из группы, включающей водород и алкил, при условии, что они оба не являются водородом, или Re и Rf соединены вместе с образованием -(СН2)4-;
(4) -CONRgRh, где Re и Rh каждый независимо выбран из группы, включающей водород и алкил; или
(5) насыщенный гетероциклоалкил, такой как тетрагидропиран; и
R2 представляет собой 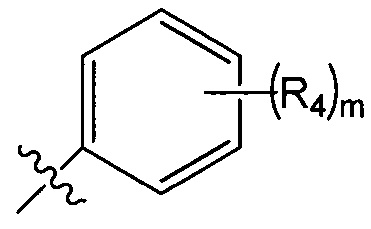 , где R4 независимо выбран из F, Cl и Br; и m=2.
, где R4 независимо выбран из F, Cl и Br; и m=2.
Согласно другому аспекту данное изобретение предусматривает фармацевтическую композицию, содержащую соединение Формулы (I) или его фармацевтически приемлемую соль.
Согласно еще одному аспекту, данное изобретение предусматривает использование соединения Формулы (I) или его фармацевтически приемлемой соли в производстве лекарственного средства для лечения рака, такого как рак щитовидной железы, немелкоклеточный рак, эпидермоидная карцинома, меланома, рак толстой кишки, карцинома желудка, рак пищевода, рак поджелудочной железы, гипернефрома, рак печени, рак легких или рак яичников.
Согласно еще одному аспекту, данное изобретение предусматривает способ получения соединения Формулы (I) или его фармацевтически приемлемой соли, где способ включает проведение реакции соединения формулы (III) или его соли с соединением формулы (IV) или его солью в растворителе и, необязательно, в присутствии одного или более катализаторов, оснований и поверхностно-активных веществ для получения соединения Формулы (I):

где:
R1, R2, R3 и n определены (заданы) в пункте 1;
L представляет собой галоген, гидроксил, мезилокси или водород; и
р=0 или 1, при условии, что когда р равно 1, L является водородом.
Детальное описание изобретения
Термины и определения
Термины, использованные далее в описании и в Формуле изобретения, имеют следующее значение, если не определено особо:
Термин «алкил» относится к радикалу линейного или разветвленного насыщенного углеводорода, содержащего 1-10 атомов углерода. Термин «С1-6 алкил» означает линейный или разветвленный насыщенный углеводород, содержащий 1-6 атомов углерода. Типичные примеры алкилов включают, но не ограничены только этими: метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, 3-метилгексил, 2,2диметилпентил, диметилпентил, 2,3-диметил пентил, н-гептил, н-октил, н-нонил и н-децил.
Термин «насыщенный гетероциклоалкил» означает моноциклический или бициклический насыщенный гетероциклоалкил без двойной и/или тройной связи. Число атомов в кольце насыщенного гетероциклоалкила может составлять 3, 4, 5, 6, 7 или 8. Помимо атомов углерода, кольцо может содержать 1, 2 или 3 гетероатома, независимо выбранных из N, О и S. Примеры насыщенных гетероциклоалкилов включают, но не ограничены только этими: азетидинил, 1,3-диоксанил, 1,3-дитиациклогексанил, 1,3-дитиациклопентил, 1,3-диоксоланил, 1,2-тиазинил, 1,3-тиазинил, азиридинил, пирролидил, пиразолидинил, азепанил, оксазолидинил оксадиазолидинил, диазепанил, имидазолидинил, пиперидил, пиперазинил, тиазолидинил, тиадиазолидинил, тетрагидропиранил, тетрагидрофуранил, тетрагидротиофенил, изоксазолидинил, изотиазолидинил и тритианил. Насыщенный гетероциклоалкил согласно данному изобретению присоединяют к центральной структуре через любой атом углерода или азота в кольце, и он может быть замещенным или незамещенным.
Использованный здесь термин «арил» означает фенил или нафтил.
Использованный здесь термин «гетероарил» означает 5- или 6-членный моноциклический и 9- или 10-членный бициклический гетероарил, который может содержать 1, 2 или 3 гетероатома, независимо выбранных из N, О и S. Типичные примеры моноциклических гетероарилов включают, но не ограничены только этими, фурил, имидазолил, изоксазолил, изотиазолил, оксадиазолил, 1,3-оксазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, пирролил, пиразолил, тетразолил, тиадиазолил, 1,3-тиазолил, тиофенил, триазолил и триазинил. Типичные примеры бициклических гетероарилов включают, но не ограничены только этими: бензофуранил, бензотиофенил, бензоксазолил, бензимидазолил, бензоксадизолил, 6,7-дигидро-1,3-бензотиазолил, имидазо[1,2-а]пиридинил, индазолил, индолил, изоиндолил, изохинолил, нафтиридинил, пиридимидазолил, хинолил, тиазоло[5,4-b]пиридин-2-ил, тиазоло[5,4-b]пиримидин-2-ил, и 5,6,7,8-тетрагидрохинолин-5-ил. Моноциклический и бициклический гетероарил согласно данному изобретению может быть замещенным или незамещенным, и присоединен к центральной структуре через любой атом углерода или атом азота в кольце.
Использованный здесь термин «карбонил» означает -СО-.
Термин «циано» означает -CN.
Термин «галоген» или «гало» означает Cl, Br, I или F.
Использованный здесь термин «галоалкил» означает алкил, как определено выше, в котором 1, 2, 3, 4, 5 или 6 атомов водорода замещены галогеном. Типичные примеры галоалкилов включают, но не ограничены только этими: хлорметил, 2-фторэтил, 2,2,2-трифторэтил, трифторметил, дифторметил, пентафторэтил, 2-хлор-3-фторпентил и трифторпропил, такой как 3,3,3-трифторпропил.
Термин «гетероатом» включает N, О и S.
Термин «соль» включает гидрохлорид, гидробрмид, сульфат, сульфит, фосфат, мезилат, п-тозилат, малеат, тартрат, малат, фумарат, цитрат, трифторацетат и аналогичные.
Соединения по данному изобретению
У соединений могут быть геометрические изомеры. Соединения по данному изобретению могут содержать двойные связи углерод-углерод или углерод-азот в Е или Z конфигурации, где термин "Е" означает, что заместители более высокого порядка расположены по разным сторонам двойной связи углерод-углерод или углерод-азот, а термин "Z" означает, что заместители более высокого порядка расположены по одну сторону двойной связи углерод-углерод или углерод-азот в соответствии с номенклатурой Кана-Ингольда-Прелога. Соединения по данному изобретению могут также существовать в виде смеси "Е" и "Z" изомеров. Заместители вокруг циклоалкила или гетероциклоалкила могут находиться в цис или транс конфигурации.
Соединения по данному изобретению могут содержать асимметрически замещенный атом углерода в R или S конфигурации, где термины "R" и "S" определены в соответствии с рекомендациями ИЮПАК (IUPAC) 1974 года, Раздел Е, Фундаментальная Стереохимия, Pure Appl. Chem. (1976) 45, 13-10. Соединения, имеющие асимметрически замещенный атом углерода с одинаковым количеством R и S конфигураций, представляют собой рацематы по этим атомам углерода. Если атомы в какой-либо конфигурации превалируют над атомами в другой конфигурации, за конфигурацию принимают конфигурацию, присутствующую в большем количестве, которая предпочтительно составляет примерно 85%-90%, более предпочтительно примерно 95%-99%, и еще более предпочтительно, превышает примерно 99%. Соответственно, данное изобретение предусматривает рацемические смеси, относительные и абсолютные стереоизомеры и смеси относительных и абсолютных стереоизомеров.
Соединения по данному изобретению, содержащие NH, С(O)ОН, ОН или SH группы, можно присоединить к фрагментам, образующим пролекарство. In vivo образующие пролекарство фрагменты удаляются в результате метаболических процессов, а высвобождающиеся соединения имеют свободный гидроксил, амино или карбоновую кислоту. Пролекарства полезны для регулирования таких фармакокинетических свойств соединений, как растворимость и/или гидрофобность, абсорбция в желудочно-кишечном тракте, биодоступность, проникновение в ткани и коэффициент очищения.
Соединения по данному изобретению могут быть в форме меченных или обогащенных изотопом, где присутствует один или более атомов с атомной массой или массовым числом, отличающимся от атомной массы или массового числа, наиболее часто встречающихся в природе. Изотопы могут быть радиоактивными или нерадиоактивными изотопами. Изотопы атомов, таких как водород, углерод, фосфор, сера, фтор, хлор и иод, включают, но не ограничены только этими: 2Н, 3Н, 13С, 14С, 15N, 18O, 32Р, 35S, 18F, 36Cl, и 125I. Изобретение также предусматривает соединения, содержащие другие изотопы этих и/или других атомов.
Согласно другому варианту выполнения изобретения меченные изотопом соединения содержат дейтерий (2Н), тритий (3Н) или 14С изотопы. Меченные изотопом соединения по данному изобретению можно получить способами, широко известными специалистам. Можно, например, упомянуть следующие работы: Lizondo, J и др., Drugs Fut, 21 (11), 1116 (1996); Brickner, SJ и др., J Med Chem, 39 (3), 673 (1996); Mallesham, В и др., Org Lett, 5 (7), 963 (2003).
Меченные изотопом соединения согласно изобретению можно использовать в качестве стандартов для определения эффективности Bcl-2 ингибиторов в анализах связывания. Соединения, содержащие изотоп, использовали в фармацевтическом исследовании для определения in vivo метаболического пути соединений для выяснения механизма действия и метаболического пути немеченных изотопом исходных соединений (Blake и др. J. Pharm. Sci. 64, 3, 367-391 (1975)). Такие исследования метаболизма важны при разработке безопасных и эффективных терапевтических лекарств, так как это позволяет проверить, являются ли введенные пациентам соединения, активные in vivo, или их метаболиты, получившиеся из исходных соединений, токсичными или канцерогенными. (Kushner и др., Can. J. Physiol. Pharmacol, 77, 79-88 (1999); Foster и др., Advances in Drug Research Vol.14, pp.2-36, Academic press, London, 1985; Kato и др., J. Labelled Comp. Radiopharmaceut., 36 (10): 927-932 (1995).
Кроме того, лекарства, содержащие нерадиоактивный изотоп, такие как дейтерированные лекарства, называемые «тяжелыми лекарствами», можно использовать для лечения заболеваний и состояний, связанных с Bcl-2 активностью. Увеличение количества изотопа, присутствующего в описанных выше соедиениях, выше природного содержания, называют обогащением. Примеры обогащения включают от примерно 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 16, 21, 25, 29, 33, 37, 42, 46, 50, 54, 58, 63, 67, 71, 75, 79, 84, 88, 92, 96 до примерно 100 мол. %.
Мечение лекарства стабильным изотопом может изменить его физико-химические свойства, такие как pKa и растворимость в липидах. Эти эффекты и изменения могут влиять на фармакодинамический ответ молекулы лекарства, если изотопное замещение влияет на область, вовлеченную во взаимодействие лиганд-рецептор. Несмотря на то, что некоторые физические свойства молекул, меченных стабильным изотопом, отличаются от свойств немеченых молекул, их химические и биологические свойства одинаковы за одним важным исключением: из-за повышенной массы тяжелого изотопа любая связь с участим тяжелого изотопа и другого атома будет прочнее, чем та же связь между легким изотопом и этим атомом. Соответственно, введение изотопа в сайт метаболизма или трансформации фермента замедлит соответствующие реакции и потенциально может изменить фармакокинетический профиль или эффективность по сравнению с соединением, не меченым изотопом.
Амиды, эфиры и пролекарства
Пролекарства представляют собой производные активных лекарств, которые разработаны для улучшения некоторых выявленных нежелательных физических или биологических свойств. Физическими свойствами являются обычно растворимость (слишком большая или недостаточная растворимость в липидах или в воде), или они связаны со стабильностью, в то время как проблемные биологические свойства включают слишком быстрый метаболизм или низкую биодоступность, которые сами по себе могут быть связаны с физикохимическим свойством.
Пролекарства обычно получают следующим образом: а) получают сложный эфир, полуэфиры, сложные эфиры угольной кислоты, нитроэфиры, амиды, гидроксамовые кислоты, карбаматы, имины, основания Манниха, фосфаты, эфиры фосфорной кислоты и енамины активного лекарства, b) проводят функционализацию лекарства азо, гликозидной, пептидной и эфирной функциональными группами, с) используют лекарство в форме аминалов, гемиаминалов, полимеров, солей, комплексов, фосфорамидов, ацеталей, гемиацеталей и кеталей. Например, см. публикацию Andrejus Korolkovas "Essentials of Medicinal Chemistry", John Wiley-Interscience Publications, John Wiley и Sons, New York (1988), pp.97-118, во всей своей полноте включенную в данное изобретение в виде ссылки.
Сложные эфиры можно получить из субстратов, содержащих либо гидроксильную группу, либо карбоксильную группу, известными специалистам способами. Типичными реакциями этих соединений являются реакции замещения одного из гетероатомов другим атомом. Аналогичным образом можно получить амиды из субстратов, содержащих либо амино-группу, либо карбоксильную группу. Сложные эфиры также могут взаимодействовать с аминами или аммиаком с образованием амидов. Другой способ получения амидов состоит в совместном нагревании карбоновых кислот и аминов.
Данное изобретение предусматривает соединение формулы (I) или его фармацевтически приемлемую соль:
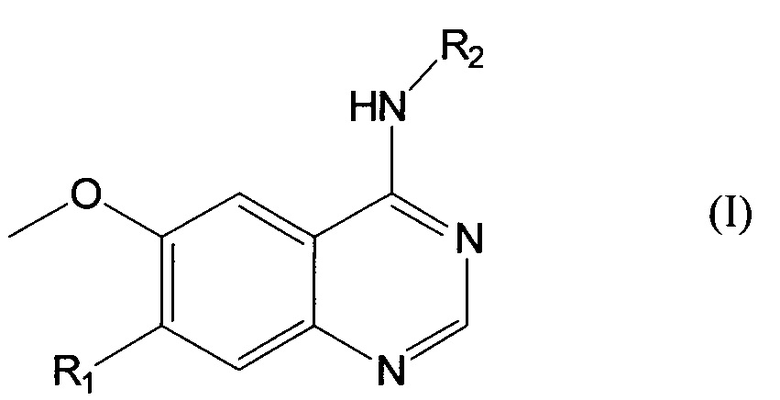
где R1 представляет собой -O(CH2)nR3, где n равно 0, 1, 2, 3, 4 или 5, и
R3 представляет собой:
(1) арил, такой как фенил, необязательно замещенный Ra и/или Rb, где Ra и Rb каждый независимо выбран из группы, включающей галоалкил такой как трифторметил, циано и насыщенный гетероциклоалкил, такой как насыщенный гетероциклоалкил содержащий один или более гетероатомов, выбранных из О, N и S, например морфолино, или Ra и Rb соединены вместе с образованием -О-СН2-О-;
(2) гетероарил, необязательно замещенный Rc и/или Rd, где Rc и Rd каждый независимо выбран из группы, включающей алкил, и насыщенный гетероциклоалкил-карбонил, такой как насыщенный гетероциклоалкил-карбонил, содержащий один или более гетероатомов, выбранных из О, N и S, например морфолино-карбонил;
(3) -NReRf, где Re и Rf каждый независимо выбран из группы, включающей водород и алкил, при условии, что они оба не являются водородом, или Re и Rf соединены вместе с образованием -(СН2)4-;
(4) -CONRgRh, где Rg и Rh каждый независимо выбран из группы, включающей водород и алкил; или
(5) насыщенный гетероциклоалкил, такой как тетрагидропиран; и
R2 представляет собой 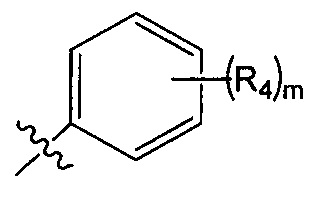 , где R4 независимо выбран из F, Cl и Br; и m=2.
, где R4 независимо выбран из F, Cl и Br; и m=2.
Согласно одному из вариантов выполнения изобретения, указанный R2 представляет собой 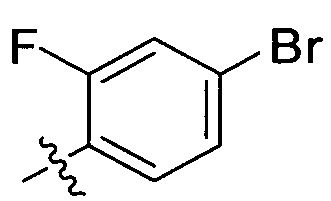 или
или  .
.
Согласно одному из вариантов выполнения изобретения, указанный арил представляет собой фенил или нафтил.
Согласно одному из вариантов выполнения изобретения, указанный насыщенный гетероциклоалкил представляет собой 5-7-ми членный насыщенный гетероциклоалкил, содержащий 1, 2 или 3 гетероатома, независимо выбранных из О, N и S.
Согласно одному из вариантов выполнения изобретения, указанный гетероарил представляет собой моноциклический или бициклический гетероарил, предпочтительно 5- или 6-членный моноциклический или 9- или 10-членный бициклический гетероарил, содержащий 1, 2 или 3 гетероатома, независимо выбранных из О, N и S, более предпочтительно он является пиридинилом, имидазолилом, тиазолилом или бензимидазолилом.
Согласно одному из вариантов выполнения изобретения, указанный алкил представляет собой С1-6 алкил.
Согласно одному из вариантов выполнения изобретения, указанный R1 выбран из:

 и
и

В соединениях формулы (I), R1, R2, R3, R4, Ra, Rb, Rc, Rd, Re, Rf, Rг, Rh, m и n могут быть независимо выбраны подходящим образом. Описанные здесь варианты выполнения изобретения могут быть скомбинированы, и любая комбинация вариантов выполнения изобретения находится в рамках данного изобретения. Например, вариант выполнения изобретения, в котором задан любой из R1, R2, R3, R4, Ra, Rb, Rc, Rd, Re, Rf, Rг, Rh, m и n, и другой вариант выполнения изобретения, в котором задан любой из R1, R2, R3, R4, Ra, Rb, Rc, Rd, Re, Rf, Rг, Rh, m и n, могут быть скомбинированы, чтобы получить новый вариант выполнения изобретения. Если новый вариант выполнения изобретения не является неосуществимым, его следует рассматривать как раскрытый в данном изобретении и составляющий часть изобретения.
Согласно одному из вариантов выполнения изобретения, предусмотрено соединение формулы (I), выбранное из следующих соединений или их фармацевтически приемлемых солей:
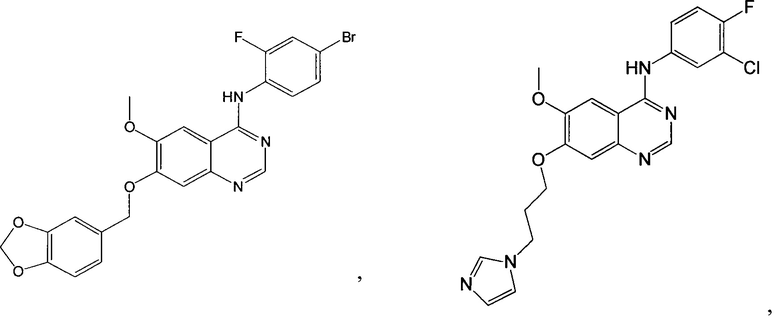




 и
и 
Фармацевтические композиции
Данное изобретение также предусматривает фармацевтические композиции, содержащие соединение Формулы (I) и его фармацевтически приемлемую соль.
Фармацевтическую композицию можно вводить перорально, например, в форме гранул, таблеток или капсул, или парентеральной инъекцией, например, внутривенной инъекцией, подкожной инъекцией, внутримышечной инъекцией или интратекальной инъекцией, или трансфузией, например, в форме стерильных растворов, суспензий или эмульсий, или использовать местное введение, например, в форме мази или крема, или вводить ректально, например, в форме свечей. Как правило, упомянутые выше композиции можно получить традиционными способами с обычными наполнителями.
Фармацевтическую композицию по данному изобретению можно использовать для лечения опухолей.
Использование в медицине
Соединение (включая его фармацевтически приемлемую соль) и фармацевтическую композицию по данному изобретению можно использовать для лечения опухолей, в частности, рака щитовидной железы, немелкоклеточного рака, эпидермоидной карциномй, меланомы, рака толстой кишки, рака печени, рака легких или рака яичников.
Данное изобретение предусматривает способ лечения рака или опухолей, таких как рак щитовидной железы, немелкоклеточный рак, эпидермоидная карцинома, меланома, рак толстой кишки, карцинома желудка, рак пищевода, рак поджелудочной железы, гипернефрома, рак печени, рак легких или рак яичников, включающий терапевтически эффективного количества соединения (включая его фармацевтически приемлемую соль) или фармацевтической композиции по данному изобретению субъекту, нуждающемуся в лечении.
Данное изобретение также предусматривает применение соединения (включая его фармацевтически приемлемую соль) или фармацевтической композиции по данному изобретению в производстве лекарства для лечения рака, такого как рак щитовидной железы, немелкоклеточный рак, эпидермоидная карцинома, меланома, рак толстой кишки, карцинома желудка, рак пищевода, рак поджелудочной железы, гипернефрома, рак печени, рак легких или рак яичников.
Способ получения
Данное изобретение также предусматривает способ получения соединения или его фармацевтически приемлемой соли, включающий взаимодействие (реакцию) соединения формулы (III) или его соли с соединением формулы (IV) или его солью в растворителе и, необязательно, в присутствии одного или более катализаторов, оснований и поверхностно-активных веществ для получения соединения Формулы (I),
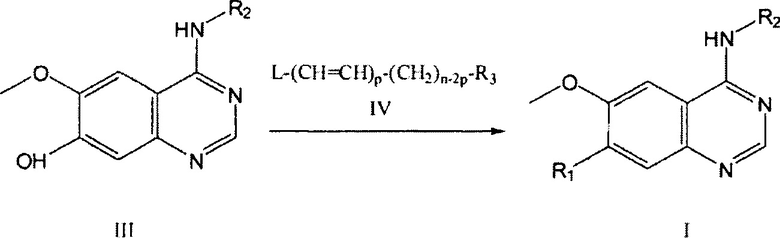
где R1, R2, R3 и n определены (заданы) в пункте 1;
L представляет собой галоген, гидроксил, мезилокси и водород; и
р=0 или 1, при условии, что когда р=1, L является водородом.
В предпочтительном варианте выполнения данного изобретения соединение формулы (III) имеет следующую формулу(III-1) или (III-2):

Согласно одному из вариантов выполнения изобретения соединения формулы (I) получены конденсацией соединения формулы (III) и соединения формулы (IV) в растворителе, где растворитель выбран из:
воды или
органического растворителя, например:
-
тспирта (например, метанола, этанола, изопропилового спирта и аналогичных),
- просого эфира (например, диэтилового эфира, метил-трет-бутилового эфира, тетрагидрофурана, 1,4-диоксана и аналогичных),
- сложного эфира (например, этилацетата и аналогичных),
- ароматического углеводорода (например, толуола, ксилола и аналогичных),
- галогенированного алкана (например, хлористого метилена, хлороформа, четыреххлористого углерода и аналогичных),
- апротонного растворителя (например, ацетона, бутанона, ацетонитрила, N,N-диметилформамида (ДМФА), диметилсульфоксида, N-метилпирролидона и аналогичных),
Предпочтительно растворителем является вода, этанол, тетрагидрофуран, толуол, хлористый метилен, ацетонитрил, N,N-диметилформамид (ДМФА), диметилсульфоксид или N-метилпирролидон.
Более предпочтительно растворителем является тетрагидрофуран, хлористый метилен, ацетонитрил, N,N-диметилформамид (ДМФА) или N-метилпирролидон.
Температура реакции составляет от 0 до 200°C, предпочтительно от 10°C до 150°C, более предпочтительно 20°C до 120°C.
Время реакции составляет 1-72 часа, предпочтительно 2-48 часов.
В упомянутом выше варианте выполнения изобретения в одном случае основание добавляют в реакцию, причем основание выбрано из органического основания или неорганического основания.
Органическое основание выбрано из триэтиламина, N,N-диизопропилэтиламина (DIPEA), пиридина, 4-диметиламинопиридина, морфолина, N-метилморфолина, 1,8-диазабицикло(5.4.0)-ундек-7-ена (DBU), 1,5-диазабицикло[4.3.0]нон-5-ена (DBN) и аналогичных.
Неорганическое основание выбрано из карбоната натрия, карбоната калия, карбоната цезия, гидроксида калия, гидроксида натрия, гидроксида цезия, гидроксида лития, гидрида натрия и аналогичных.
Предпочтительно, основание выбрано из N,N-диизопропилэтиламина (DIPEA), 1,8-диазабицикло(5.4.0)-ундек-7-ена (DBU), карбоната натрия, карбоната калия, карбоната цезия, гидроксида калия и гидроксида натрия.
Более предпочтительно основание выбрано из 1,8-диазабицикло(5.4.0)-ундек-7-ена (DBU), карбоната калия и карбоната цезия.
В упомянутом выше варианте выполнения изобретения в другом случае в реацию добавляют конденсирующий агент, причем указанный конденсирующий агент выбран из N,N'-дициклогексилкарбодиимида (DCC), N,N'-диизопропилкарбодиимида (DIC) или гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDCI или EDCI⋅HCl). Предпочтительно, конденсирующий агент представляет собой N,N'-дициклогексилкарбодиимид (DCC).
В упомянутом выше варианте выполнения изобретения в ином случае в реакцию добавляют катализатор, например, трифенилфосфин (PPh3) или трибутилфосфин, вместе с производным азодикарбонила (таким как диизопропилазодикарбоксилат (DIAD), диэтилазодикарбоксилат (DEAD), ди-трет-бутилазодикарбоксилат (DBAD), азодикарбиксидиморфолид (ADDM) и дипипиридид азодикарбоновой кислоты (ADDP), или другой катализатор (такой как йодистая медь, бромистая медь, иодид калия и иодид натрия). Предпочтительно, катализатором является трифенилфосфин вместе со сложным эфиром азодикарбоновой кислоты, иодидом меди, иодидом калия или иодидом натрия. Более предпочтительно, катализатором является трифенилфосфин вместе с диизопропилазодикарбоксилатом (DIAD) или иодидом меди.
В упомянутом выше варианте выполнения изобретения, в ином случае в реакцию добавляют поверхностно-активное вещество (сурфактант), например, тетрабутиламмоний хлорид, тетрабутиламмоний бромид, тетрабутиламмоний иодид, кислый сульфат тетрабутиламмония или тетрабутиламмоний гидроксид. Предпочтительно, поверхностно-активное вещество представляет собой тетрабутиламмоний бромид или тетрабутиламмоний иодид. Более предпочтительно, поверхностно-активным веществом является тетрабутиламмоний бромид.
Конкретнее, в предпочтительном варианте выполнения изобретения способ получения соединения Формулы (I) или его фармацевтически приемлемой соли включает взаимодействие соединения Формулы (III) или его соли с соединением Формулы (IV) или его солью в присутствии растворителя и катализатора.
Например, катализатором может быть трифенилфосфин и диизопропилазодикарбоксилат (DIAD):
Например, соединение Формулы (III) (центральная структура), соединение Формулы (IV) (боковая цепь) и трифенилфосфин растворяют в растворителе (например, безводном тетрагидрофуране) при комнатной температуре (15-30°C, далее аналогично). Диизопропилазодикарбоксилат добавляют по каплям к раствору под азотом. Затем полученную смесь перемешивают при температуре от комнатной температуры до 40°C для проведения реакции в течение, например, 18 часов. После завершения реакции последующими стадиями могут быть следующие: реакционную смесь охлаждают до комнатной температуры и фильтруют под вакуумом (с отсасыванием) для получения продукта; если в процессе охлаждения твердые продукты не выделяются, реакционный раствор концентрируют при пониженном давлении до сухого остатка (досуха) и разделяют с помощью колоночной хроматографии на силикагеле для получения продукта.
Согласно другому предпочтительному варианту выполнения изобретения, способ получения соединения Формулы (I) или его фармацевтически приемлемой соли включает взаимодействие (реакцию) соединения Формулы (III) с соединением Формулы (IV) в присутствии растворителя, основания, необязательно катализатора, и необязательно поверхностно-активного вещества.
Например, основанием может быть 1,8-диазабицикло(5.4.0)-ундек-7-ен (DBU):
Например, соединение Формулы (III) (центральная структура), соединение Формулы (IV) (боковая цепь) и 1,8-диазабицикло(5.4.0)-ундек-7-ен (DBU) растворяют в растворителе, таком как этанол или N-метилпирролидон (необходимо поверхностно-активное вещество, такое как тетрабутиламмоний бромид). Реакционную смесь нагревают до температуры, такой как 85°C, и перемешивают в течение от 24 до 48 часов для осуществления реакции. Последующими стадиями могут быть следующие. В том случае, если растворителем является этанол, реакционную смесь можно сразу концентрировать для получения неочищенного продукта. В том случае, когда растворителем является N-метилпирролидон, реакционную смесь охлаждают до комнатной температуры, выливают в воду и экстрагируют (например, этилацетатом). Полученные органические слои объединяют, промывают (например, насыщенным раствором NaCl), сушат (например, над безводным сульфатом натрия) и фильтруют. Фильтрат концентрируют под пониженным давлением досуха для получения неочищенного продукта. Неочищенный продукт выделяют с помощью колоночной хроматографии, а затем очищают препаративной тонкослойной хроматографией для получения конечного продукта.
Основанием может быть также карбонат цезия:
Например, соединение Формулы (III) (центральная структура), соединение Формулы (IV) (боковая цепь), карбонат цезия и катализатор, такой как иодид меди, растворяют в растворителе, таком как N,N-диметилформамид (ДМФА). Полученную смесь затем нагревают до температуры, такой как 120°C, и перемешивают в течение, например, 18 часов для осуществления реакции. Последующими стадиями могут быть следующие. Реакционную смесь охлаждают до комнатной температуры, выливают в воду и экстрагируют (например, этилацетатом). Полученные органические слои объединяют, сушат (например, над безводным сульфатом натрия) и фильтруют под вакуумом. Фильтрат концентрируют под пониженным давлением досуха для получения неочищенного продукта. Неочищенный продукт очищают препаративной тонкослойной хроматографией для получения конечного продукта.
Основанием также может быть K2CO3:
Например, соединение Формулы (III) (центральная структура), соединение Формулы (IV) (боковая цепь) и K2CO3 растворяют в растворителе, таком как N,N-диметилформамид (ДМФА) или ацетонитрил. Полученную смесь нагревают до температуры от 60°C до 120°C и перемешивают в течение от 2 до 18 часов. Последующими стадиями могут быть следующие: реакционную смесь охлаждают до комнатной температуры, выливают в воду и экстрагируют (например, этилацетатом). Органические слои объединяют, сушат (например, над безводным сульфатом натрия) и фильтруют под вакуумом. Фильтрат концентрируют под пониженным давлением досуха для получения неочищенного продукта. Неочищенный продукт очищают с помощью колоночной хроматографии на силикагеле для получения конечного продукта.
В предпочтительном варианте выполнения изобретения данные соединения можно получить из соединения Формулы (II), являющегося интермедиатом, который можно получить из соединения Формулы (III). Соединение Формулы (II), соединение боковой цепи, такое как соединение первичного или вторичного амина, HNReRf, где Re и Rf заданы, как указано выше, предпочтительно соединение вторичного амина (например, диметиламин гидрохлорид или тетрагидропиррол) и K2CO3 растворяют в растворителе, таком как ацетонитрил. Полученную смесь нагревают до температуры от 60°C до 120°C и перемешивают в течение от 2 до 18 часов. Последующими стадиями могут быть следующие стадии. Реакционную смесь концентрируют, и добавляют воду. Полученный раствор экстрагируют (например, дихлорметаном). Объединенные органические фазы сушат (например, над безводным сульфатом натрия), и концентрируют для получения неочищенного продукта. Неочищенный продукт очищают колоночной хроматографией на силикагеле для получения конечного продукта.

Соединение формулы (II) получают следующим образом:
Стадия 1:
Соединение Формулы (III) растворяют в растворителе, таком как N,N-диметилформамид (ДМФА). Добавляют K2CO3 и 4-бромбутилацетат при комнатной температуре. Полученную смесь оставляют для реакции при температуре, такой как 50°C, на время, составляющее, например, 2 часа. Реакционную смесь охлаждают до комнатной температуры, выливают в воду и экстрагируют (например, дихлорметаном). Органические слои объединяют, сушат (например, над безводным сульфатом натрия), концентрируют под пониженным давлением досуха для получения неочищенного продукта (в виде желтого масла). Неочищенный продукт используют напрямую на следующей стадии.
Стадия 2:
Продукт, полученный на стадии 1, диспергируют в метаноле, и добавляют воду и гидроксид лития. Полученную смесь оставляют для реакции при комнатной температуре на ночь. Реакционную смесь концентрируют досуха под пониженным давлением. Добавляют воду и этилацетат. При перемешивании полученной смеси выделяется твердое вещество, которое отфильтровывают под вакуумом. Остаток на фильтре сушат для получения продукта, который напрямую используют на следующей стадии.
Стадия 3:
Продукт, полученный на стадии 2, диспергируют в дихлорметане и добавляют триэтиламин. Затем по каплям на ледяной бане добавляют метилсульфонил хлорид. После добавления по каплям полученную смесь оставляют для реакции на 3 часа при комнатной температуре. Добавляют воду, и полученный раствор экстрагируют дихлорметаном. Объединенные органические слои сушат (например, над безводным сульфатом натрия), и концентрируют под пониженным давлением досуха для получения продукта.
ПРИМЕРЫ
Сокращения, используемые в Примерах, имеют следующее значение:
ТГФ Тетрагидрофуран
ДМФА N,N-диметилформамид
ДБУ (DBU) 1,8-диазабицикло(5.4.0)-ундек-7-ен
DIPEA N,N-диизопропилэтиламин
DBN 1,5-диазабицикло[4.3.0]нон-5-ен
DCC N,N'-дициклогексилкарбодиимид
EDC 1-(3-диметиламинопропил)-3-этил-карбодиимид
DIC N,N'-диизопропилкарбодиимид
DIAD диизопропилазодикарбоксилат
DCM хлористый метилен
EDCI или 1-(3-диметиламинопропил)-3-этилкарбодиимид
EDCI⋅HCl гидрохлорид
НОВТ 1 -гидроксибензотризол
ПРИМЕРЫ ПОЛУЧЕНИЯ
В приведенных далее примерах (4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина и его трифторацетат (соль трифторуксусной кислоты) (соединение Формулы(III)-1, центральная структура 1, и его трифторацетат) можно получить аналогично получению центральной структуры 2 и ее трифторфцетата, их также можно приобрести у NANJING CHICO. Другие исходные материалы коммерчески доступны или получены в лаборатории. Способы получения описаны далее.
4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолин или его соль трифторуксусной кислоты (соединение Формулы(III)-2, центральная структура 2, и его соль, трифторфцетат) можно получить в лаборатории следующим способом:
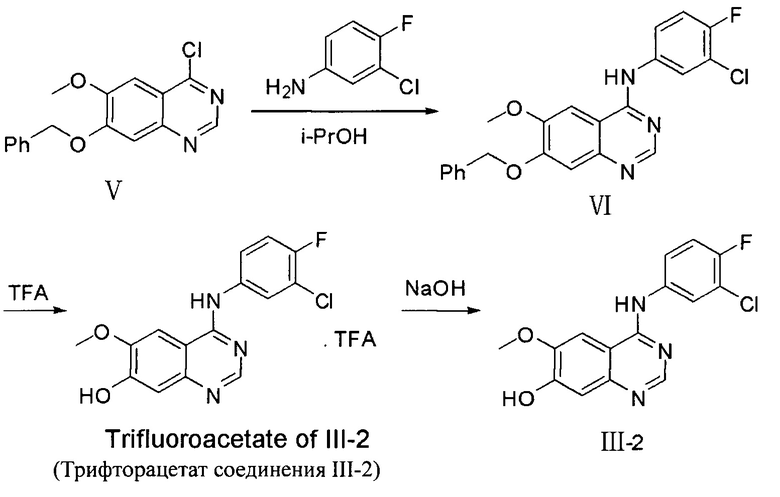
Стадия 1
4-хлор-6-метокси-7-бензилоксихиназолин (формулы V, 30.0 г, 99.8 ммоль) и 3-хлор-4-фторанилин (17.4 г, 119.7 ммоль) диспергировали в изопропиловом спирте (i-PrOH) (600 мл) при комнатной температуре. Полученную дисперсию нагревали с обратным холодильником в течение 18 часов для осуществления реакции. Реакционную смесь охладили до комнатной температуры, отфильтровали под пониженным давлением и сушили для получения грязнобелого твердого вещества (36.0 г), т.е., 4-(3-хлор-4-фторанилино)-6-метокси-7-бензилоксихиназолина (формула VI).
Стадия 2
Соединение Формулы VI (36.0 г), полученное на стадии 1, растворили в трифторуксусной кислоте (300 мл), смесь нагрели до 70°C и перемешивали в течение 18 часов. Полученную смесь концентрировали досуха для получения неочищенного продукта, трифторацетата соединения формулы III-2. Неочищенный продукт вводили в метил-трет-бутиловый эфир для образования суспензии. Затем суспензию фильтровали и сушили для получения гразнобелого твердого вещества (29.7 г), представляющего собой трифторацетат соединения III-2 (общий выход на двух стадиях: 69%).
Стадия 3
Очищенную воду (200 мл) добавляют к твердому (23.0 г) трифторацетату соединения III-2, полученному на предыдущей стадии, для образования суспензии. К суспензии при перемешивании по каплям добавляют водный раствор 1N NaOH для доведения рН до 8. Полученную смесь экстрагируют этилацетатом. Органический слой сушат над безводным Na2SO4 и концентрируют для получения 15.7 г грязнобелого твердого вещества, т.е., центральной структуры III-2, выход: 93%.
Синтез 4-хлор-N,N-диметилбутан-1-амин гидрохлорида.
4-гидрокси-N,N-диметилбутиламин (1.0 г, 8.53 ммоль) по каплям добавляли к тионилхлориду (5 мл) на водяной бане со льдом, температуру окружающей среды в процессе добавления по каплям поддерживали ниже 10°C. После завершения добавления по каплям смеси давали нагреться до комнатной температуры естественным образом, а затем перемешивали в течение 12 часов при комнатной температуре. Реакционную смесь по каплям вводили в этанол (100 мл), предварительно охлажденный до 0-5°C. Полученную смесь концентрировали для получения целевого соединения в виде белого твердого вещества (1.3 г, выход: 89%).
Синтез N-гидроксибутилпирролидина
4-хлор-н-бутанол (5.0 г, 46.1 ммоль), тетрагидропиррол (6.6 г, 92.6 ммоль) и карбонат калия (12.7 г, 92.6 ммоль) добавили к ацетонитрилу (150 мл) при комнатной температуре, и перемешивали смесь в течение 18 часов при 80°C. Затем реакционную смесь охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали для получения коричневого масла (6.8 г), которое напрямую использовали на следующей стадии.
Синтез N-метил-6-бром-гексанамида
6-бром-н-капроновую (гексановую) кислоту (9.5 г, 50 ммоль) и N,N-диметилформамид (ДМФА, 5 мл) растворяли в хлористом метилене (DCM, 150 мл) при комнатной температуре. К полученной смеси медленно и по каплям добавили оксалилхлорид (12.6 г, 100 ммоль). После завершения добавления по каплям смесь перемешивали в течение 1 часа при комнатной температуре для осуществления реакции. Реакционную смесь концентрировали, и остаток растворяли в хлористом метилене (DCM, 100 мл). Полученную смесь добавляли в реакционный сосуд, содержащий раствор метиламина в этаноле (50 мл). Смесь перемешивали в течение 2 часов при комнатной температуре для осуществления реакции, затем добавляли хлористый метилен (DCM, 500 мл). Затем полученную смесь последовательно промывали 1N раствором соляной кислоты (500 мл), насыщенным водным раствором NaHCO3 (500 мл) и насыщенным раствором соли (500 мл), сушили над безводным Na2SO4 и концентрировали для получения белого твердого вещества (4.0 г), которое напрямую использовали на следующей стадии.
Синтез N,N-диметил-6-бром-гексанамида
6-бромкапроновую кислоту (1.94 г, 10 ммоль), метиламин гидрохлорид (1.21 г, 15 ммоль), триэтиламин (2 г, 20 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDCI, 2.11 г, 11 ммоль) и 1-гидроксибензотриазол (НОВТ, 0.5 г) растворяли в хлористом метилене (DCM, 150 мл). Смесь перемешивали в течение 3 час при 15°C для осуществления реакции, и к реакционной смеси добавляли хлористый метилен (DCM, 150 мл). Полученную смесь последовательно промывали 1N разбавленной соляной кислотой (150 мл), насыщенным водным раствором NaHCO3 (150 мл) и насыщенным раствором соли (150 мл), сушили над безводным сульфатом натрия и концентрировали для получения бесцветного масла (2.0 г), которое напрямую использовали на следующей стадии.
Синтез 5-бромпентанамида
5-бромвалериановую кислоту (5 г, 27.6 ммоль) растворили в хлористом метилене (50 мл). К полученному раствору добавили N,N-диметилформамид (ДМФА). Полученную смесь охладили до 0-5°C на водяной бане со льдом. К охлажденной смеси медленно, по каплям добавили оксалилхлорид (10 г, 82.8 ммоль). После завершения добавления по каплям смесь перемешивали в течение 2 часов при 40°C для осуществления реакции. Реакционную смесь концентрировали под пониженным давлением для получения желтого твердого вещества. Полученное в результате твердое вещество растворили в тетрагидрофуране (ТГФ, 50 мл). Полученный раствор медленно, по каплям добавили к водному раствору аммиака (10 мл). Полученную смесь перемешивали в течение 2 часов при комнатной температуре и экстрагировали хлористым метиленом. Органическую фазу сушили над безводным сульфатом натрия и концентрировали для получения белого твердого вещества (4.85 г, выход: 98%).
Синтез 4-хлорбутанамида
Стадия 1:
При комнатной температуре 4-хлорбутановую (хлормасляную) кислоту (20 г, 164 ммоль) растворили в хлористом метилене (200 мл). Полученный раствор охладили до 0-5°C. К охлажденному раствору по каплям добавили оксалилхлорид (41 г, 328 ммоль). После завершения добавления по каплям полученную смесь перемешивали в течение 2 часов при комнатной температуре и концентрировали для получения неочищенного продукта, 4-хлорбутаноил хлорида (22 г), который напрямую использовали на следующей стадии.
Стадия 2:
К водному раствору аммиака (20 мл) в тетрагидрофуране (ТГФ, 100 мл), охлажденному до 0-5°C на бане со льдом, по каплям добавили раствор 4-хлорбутаноил хлорида (22 г) в тетрагидрофуране (ТГФ, 100 мл). После завершения добавления по каплям смесь оставили для реакции на 1 час при температуре. Реакционную смесь вылили в воду (1 литр). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения белого твердого вещества (7 г, выход: 35%).
Синтез 3-(1Н-имидазол-1-ил)пропан-1-ола:
При комнатной температуре 3-бромпропан-1-ол (10 г, 72.4 ммоль), имидазол (4.92 г, 72.4 ммоль) и K2CO3 (25 г, 181 ммоль) диспергировали в ацетонитриле (150 мл). Полученную смесь нагревали с обратным холодильником при перемешивании 18 час, а затем охладили до комнатной температуры. Охлажденную смесь вылили в воду (1 литр). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения бесцветного масла (4 г).
Синтез 2-метил-5-бромметилтиазола:
Стадия 1: Синтез 2-метил-5-гидроксиметилтиазола:
При комнатной температуре алюмогидрид лития (8.88 г, 234 ммоль) диспергировали в безводном тетрагидрофуране (ТГФ, 100 мл). Полученную смесь охлаждали до 0-5°C на водяной бане со льдом. К охлажденной смеси по каплям добавили раствор 2-метил-5-этоксиформилтиазола (20 г, 117 ммоль) в безводном тетрагидрофуране (ТГФ, 100 мл). После завершения добавления по каплям реакционную смесь естественным образом нагрели до комнатной температуры и перемешивали в течение 18 часов при комнатной температуре. К реакционной смеси по каплям добавили воду (10 мл) при 0-5°C. После завершения добавления по каплям полученную смесь отфильтровали. Фильтрат концентрировали для получения желтого маслянистого вещества (12 г), которое напрямую использовали на следующей стадии.
Стадия 2: Синтез 2-метил-5-бромметилтиазола
При комнатной температуре 2-метил-5-гидроксиметилтиазол (12 г) растворили в хлористом метилене (500 мл), и полученный раствор охладили до 0-5°C на водяной бане со льдом. К охлажденной смеси последовательно порцияли добавили трифенилфосфин (52 г, 200 ммоль) и четырехбромистый углерод (66 г, 200 ммоль). После завершения добавления реакционную смесь естественным образом нагрели до комнатной температуры, перемешивали в течение 2 часов при комнатной температуре, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 2:1) для получения твердого желтого вещества (3.3 г, общий выход по двум стадиям: 15%).
Синтез 2-метил-4-хлорметилтиазола:
Тиоацетамид (2 г, 26.6 ммоль) и дихлорацетон (4.05 г, 31.9 ммоль) растворили в этаноле (60 мл) при комнатной температуре. Полученный раствор нагрели до 80°C и оставили для реакции на 4 часа. После завершения реакции реакционную смесь концентрировали. К остатку добавили 100 мл очищенной воды. Полученную смесь довели до рН 8 бикарбонатом натрия. Смесь экстрагировали метил-трет-бутиловым эфиром, и органический слой сушили над безводным сульфатом натрия и концентрировали для получения продукта (1.2 г).
Синтез 1-(2-хлорэтил)-1Н-имидазола
Гидроксид калия (11.2 г), карбонат калия (8.84 г) и тетрабутиламмоний бромид (0.21 г) помещали в трехгорлую колбу при комнатной температуре. К смеси при перемешивании добавили 1,2-дихлорэтан (80 мл). Полученную смесь нагрели 50°C. Затем добавили имидазол (2.04 г). Смесь оставили для реакции на 2 часа при 50°C. После завершения реакции реакционную смесь охладили до комнатной температуры и отфильтровали. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения маслянистого вещества (1.45 г).
Синтез 2-метил-4-метилсульфонилоксиэтилтиазола
Стадия 1: Синтез этил 2-(2-метилтиазол-4-ил)-ацетата.
Этилхлорацетоацетат (5.0 г, 30.5 ммоль) и тиоацетамид (2.3 г, 30.5 ммоль) растворили в абсолютном (безводном) этаноле (50 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 24 часов и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 1:1) для получения белого воскообразного твердого вещества (3.0 г, выход: 53%).
Стадия 2: Синтез 2-метил-4-гидроксиэтилтиазола
При комнатной температуре алюмогидрид лития (1.2 г, 32.4 ммоль) диспергировали в безводном тетрагидрофуране (ТГФ, 20 мл). Смесь охладили до 0-5°C на водяной бане со льдом. К охлажденной смеси по каплям добавили раствор этил 2-(2-метилтиазол-4-ил)ацетата (3.0 г, 16.2 ммоль) в безводном тетрагидрофуране (ТГФ, 10 мл). После завершения добавления по каплям реакционную смесь естественным образом нагрели до комнатной температуры, перемешивали в течение 18 часов при комнатной температуре, а затем охладили до 0-5°C. К охлажденной смеси по каплям добавили воду (1.5 мл) для остановки реакции. После завершения добавления по каплям полученную смесь фильтровали под вакуумом. Фильтрат концентрировали для получения желтой маслянистой жидкости (1.6 г), которую напрямую использовали на следующей стадии.
Стадия 3: Синтез 2-метил-4-метилсульфонилоксиэтилтиазола
2-метил-4-гидроксиэтилтиазол (1.6 г, 11.2 ммоль) и триэтиламин (2.3 г, 22.4 ммоль) растворили в хлористом метилене (20 мл). Смесь охладили до 0-5°C. К охлажденной смеси добавили метансульфонил хлорид (1.9 г, 16.8 ммоль). После завершения добавления по каплям полученную смесь естественным образом охладили до комнатной температуры, перемешивали в течение 1 часа при комнатной температуре и вылили в воду (100 мл). Затем полученную смесь экстрагировали хлористым метиленом, органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения желтого воскообразного твердого вещества (2.0 г).
ПРИМЕР 1
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-((1,3-бензодиоксол-5-ил)-метокси)хиназолина (Соединение 1)

Соль 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина и трифторуксусной кислоты (400 мг, 0.84 ммоль), 5-бромметилбензо[d][1,3]диоксол (181 мг, 0.84 ммоль) и карбонат калия (289 мг, 2.09 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 5 мл). Смесь перемешивали в течение 18 часов при 60°C, поддерживая постоянную температуру, а затем охладили до комнатной температуры. Охлажденную смесь вылили в воду (100 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 1:1) для получения грязно-белого твердого вещества (115 мг, выход: 27%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.54 (s, 1H), 8.35 (s, 1H), 7.81 (s, 1H), 7.66 (dd, 1H, J=9.8, 1.8 Hz), 7.53 (t, 1H, J=8.2 Hz), 7.49-7.43 (m, 1H), 7.29 (s, 1H), 7.06 (s, 1H), 6.99 (d, 1H, J=8.0 Hz), 6.95 (d, 1H, J=7.6 Hz), 6.04 (s, 2H), 5.17 (s, 2H), 3.94 (s, 3H).
MS: m/z 498, 500 (M+1).
ПРИМЕР 2
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-((2-пирролидин-1-ил)этокси)хиназолина (Соединение 2)

Соль 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина и трифторуксусной кислоты (200 мг, 0.42 ммоль), N-хлорэтилпирролидин гидрохлорид (71 мг, 0.42 ммоль) и карбонат калия (232 мг, 1.68 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл). Смесь перемешивали в течение 5 часов, поддерживая температуру 60°C, а затем охладили до комнатной температуры. Охлажденную смесь вылили в воду (75 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения неочищенного продукта (300 мг). Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 10:1) для получения светложелтого твердого вещества (120 мг, выход: 62%).
1H-NMR (400 MHz, CDCl3) δ: 8.66 (s, 1H), 8.41 (t, 1H, J=8.4 Hz), 7.59-7.47 (br s, 1H), 7.38-7.32 (m, 2H), 7.25 (s, 1H), 7.11 (s, 1H), 4.57-4.50 (m, 2H), 4.01 (s, 3H), 3.41-3.33 (m, 2H), 3.27-3.07 (m, 4H), 2.09-1.96 (m, 4H).
MS 231, 232 (1/2M+1).
ПРИМЕР 3
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(3-цианобензилокси)хиназолина (Соединение 3)

Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (300 мг, 0.63 ммоль), 3-(бромметил)бензонитрил (147 мг, 0.75 ммоль) и карбонат калия (261 мг, 1.89 ммоль) диспергировали в ацетонитриле (10 мл) при комнатной температуре. Смесь нагревали с обратным холодильником при перемешивании в течение 2 часов. Реакционную смесь охладили до комнатной температуры и вылили в воду (100 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения неочищенного продукта. Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 10:1) для получения твердого белого вещества (120 мг, выход: 40%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.61 (s, 1H), 8.37 (s, 1H), 7.97 (s, 1H), 7.91-7.82 (m, 3H), 7.72-7.62 (m, 2H), 7.58-7.44 (m, 2H), 7.32 (s, 1H), 5.37 (s, 2H), 3.96 (s, 3H).
MS 479, 481 (M+1).
ПРИМЕР 4
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(4-трифторметил-бензилокси)хиназолина (Соединение 5)
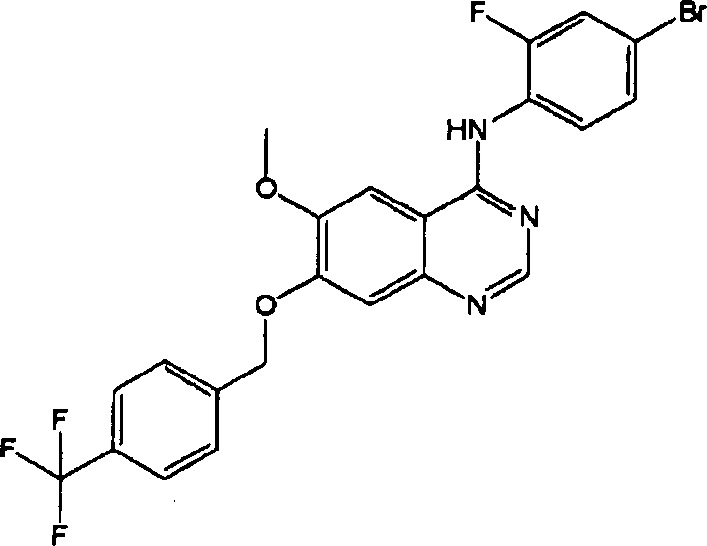
4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (500 мг, 1.37 ммоль), 1-(трифторметил)-4-бромметилбензол (393 мг, 1.64 ммоль) и карбонат калия (568 мг, 4.11 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 15 мл). Полученную смесь нагревали с обратным холодильником в течение 4 часов для осуществления реакции. Реакционную смесь охладили до комнатной температуры и вылили в воду (80 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта (800 мг). Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 10:1) для получения твердого белого вещества (287 мг, выход: 40%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.57 (s, 1H), 8.36 (s, 1H), 7.85-7.80 (m, 3H), 7.74-7.72 (m, 2H), 7.66 (dd, 1H, J=10.0, 2.0 Hz), 7.53-7.45 (m, 2H), 7.30 (s, 1H), 5.43 (s, 2H), 3.97 (s, 3H).
MS 522, 524 (M+1).
ПРИМЕР 5
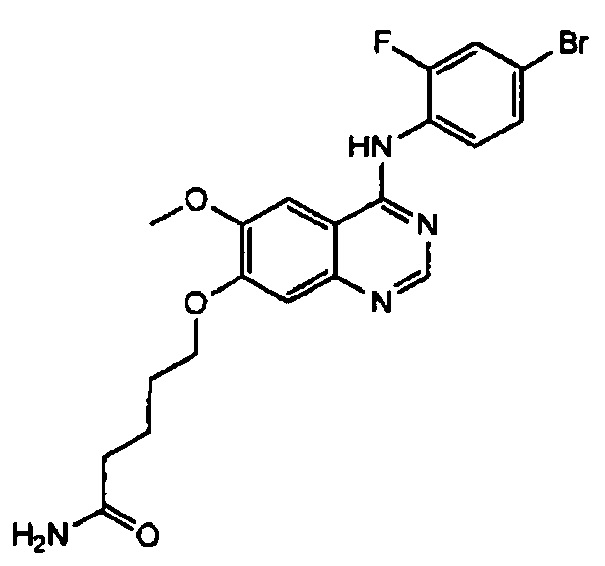
Синтез 6-(4-(4-бром-2-фторанилино)-6-метоксихиназолин-7-ил-окси)гексанамида (Соединение 6)

Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (400 мг, 0.84 ммоль), 6-бромгексанамид (326 мг, 1.68 ммоль) и карбонат калия (232 мг, 1.68 ммоль) диспергировали в ацетонитриле (10 мл). Смесь перемешивали в течение 18 часов при 80°C, поддерживая постоянную температуру. Реакционную смесь охлаждали до комнатной температуры и выливали в воду (75 мл). Твердую часть отделяли. Полученную смесь фильтровали и сушили для получения грязно-белого твердого вещества (170 мг, выход: 42%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.53 (s, 1H), 8.35 (s, 1H), 7.79 (s, 1H), 7.66 (dd, 1H, J=10.0, 2.0 Hz), 7.53 (t, 1H, J=8.2 Hz), 7.46 (dd, 1H, J=8.4, 1.6 Hz) 7.25 (brs, 1H), 7.18 (s, 1H), 6.70 (brs, 1H), 4.13 (t, 2H, J=6.6 Hz), 3.94 (s, 3H), 2.80 (t, 2H, J=7.2 Hz), 1.85-1.75 (m, 2H), 1.63-1.52 (m, 2H), 1.49-1.39 (m, 2H).
MS 477, 479 (M+1).
ПРИМЕР 6
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-((6-метилпиридин-2-ил)метокси)- хиназолина (Соединение 7)
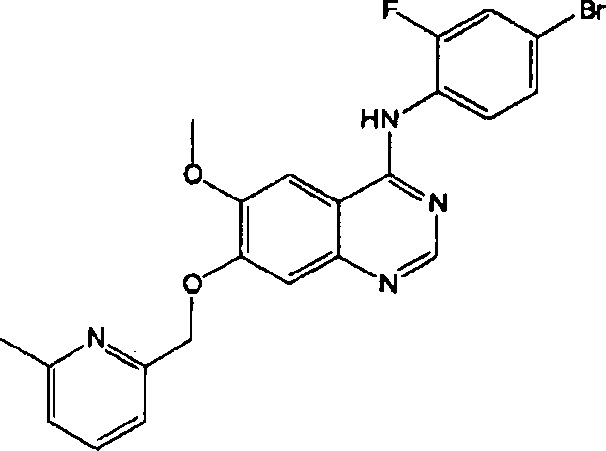
Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (400 мг, 0.84 ммоль), 6-метил-2-гидроксиметилпиридин (103 мг, 0.84 ммоль) и карбонат калия (289 мг, 2.09 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 5 мл) при комнатной температуре. Смесь перемешивали в течение 18 часов при 60°C, поддерживая постоянную температуру. Реакционную смесь охлаждали до комнатной температуры и выливали в воду (100 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 50:1) для получения светло-коричневого твердого вещества (215 мг, выход: 55%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.55 (s, 1H), 8.35 (s,1H), 7.84 (s, 1H), 7.74 (t, 1H, J=8.0 Hz), 7.70-7.62 (m, 1H), 7.58-7.42 (m, 2H), 7.35 (d, 1H, J=7.6 Hz), 7.30-7.20 (m, 2H), 5.30 (s, 2H), 3.97 (s, 3H), 2.50 (s, 3H).
MS 469, 471 (M+1).
ПРИМЕР 7
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(2-(2-метил-1Н-бензо[d]имидазол-1-ил)-этокси)хиназолина (Соединение 8)
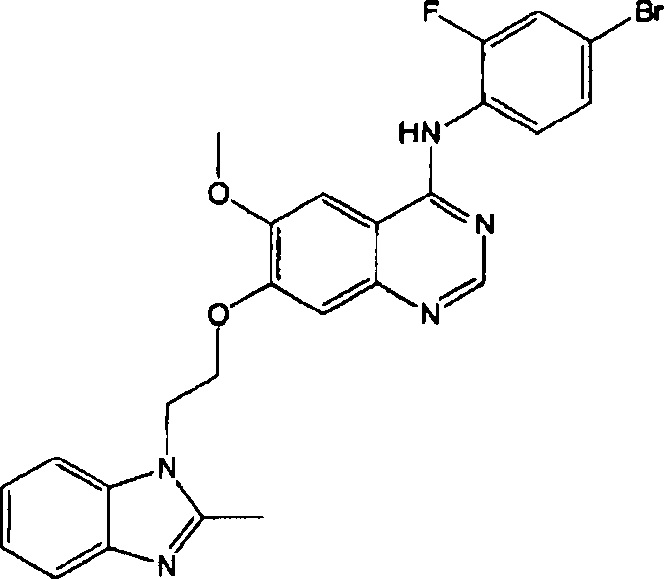
4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (200 мг, 0.55 ммоль), 2-(2-метил-1Н-бензо[d]имидазол-1-ил)этанол (97 мг, 0.55 ммоль), трифенилфосфин (172 мг, 0.65 ммоль) и диизопропилазодикарбоксилат (DIAD, 131 мг, 0.65 ммоль) растворяли в безводном тетрагидрофуране (ТГФ, 5 мг). Полученную смесь перемешивали в течение 18 часов при 40°C под азотом для осуществления реакции. Реакционную смесь охлаждали до комнатной температуры. Твердое вещество отделяли. Смесь фильтровали и сушили для получения грязно-белого твердого вещества (249 мг, выход: 87%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.52 (s, 1H), 8.34 (s, 1H), 7.77 (s, 1H), 7.66-7.62 (m, 2H), 7.54-7.44 (m, 3H), 7.22-7.12 (m, 3H), 4.69 (t, 2H, J=4.6 Hz), 4.47 (t, 2H, J=4.8 Hz), 3.91 (s, 3H), 2.70 (s, 3H).
MS 522, 524 (M+1).
ПРИМЕР 8
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(4-морфолинобензилокси)хиназолина (Соединение 12)

4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (500 мг, 1.37 ммоль), (4-морфолинофенил)метанол (396 мг, 2.05 ммоль), трифенилфосфин (538 мг, 2.05 ммоль) и диизопропилазодикарбоксилат (DIAD, 415 мг, 2.05 ммоль) растворяли в безводном тетрагидрофуране (ТГФ, 10 мл) при комнатной температуре, и раствор перемешивали в течение 18 часов при 40°C под азотом для осуществления реакции. Затем полученную смесь концентрировали и очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения желтого твердого вещества (340 мг, выход: 46%).
1H-NMR (400 MHz, DMSO-d6) δ: 8.50 (s, 1H), 7.86 (s, 1H), 7.51 (d, 1H, J=9.6 Hz), 7.35 (d, 1H, J=8.0 Hz), 7.20 (d, 3H, J=8.0 Hz), 6.91 (d, 3H, J=8.8 Hz) 5.26 (s, 2H), 3.87 (s, 3H), 3.70 (t, 4H, J=4.6 Hz), 3.07 (t, 4H, J=4.4 Hz).
MS 539, 541 (М+1).
ПРИМЕР 9
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(3-(1Н-имидазол-1-ил)пропокси) хиназолина (Соединение 14)
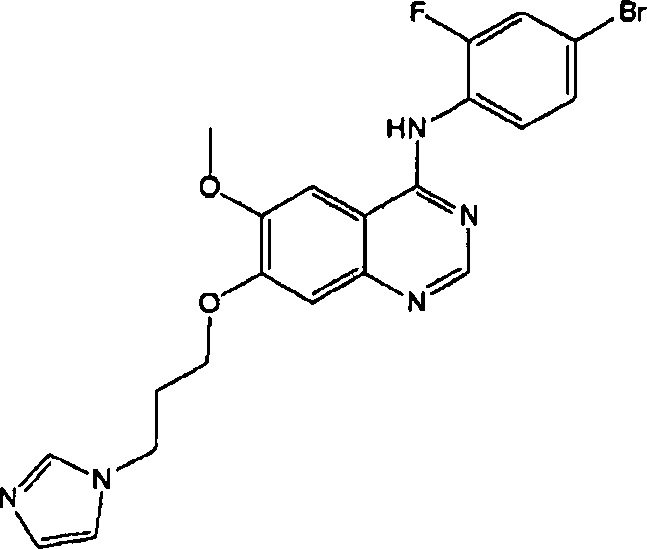
Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (400 мг, 0.84 ммоль), 3-(1Н-имидазол-1-ил)пропан-1-ол (400 мг, 3.17 ммоль), трифенилфосфин (330 мг, 1.26 ммоль) и диизопропилазодикарбоксилат (DIAD, 255 мг, 1.26 ммоль) растворили в безводном тетрагидрофуране (ТГФ, 10 мл) при комнатной температуре. Полученную смесь перемешивали в течение 18 часов при комнатной температуре под азотом и сушили выпариванием (испарением) для получения неочищенного продукта. Неочищенный продукт перекристаллизовывали из смеси метанол/этилацетат (v/v=1:1 по объему) для получения светло-желтого твердого вещества (220 мг, выход: 55%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.56 (s, 1H), 8.35 (s, 1H), 7.82 (s, 1H), 7.70-7.62 (m, 2H), 7.56-7.44 (m, 2H), 7.25-7.15 (m, 2H), 6.90 (s, 1H), 4.17 (t, 2H, J=7.0 Hz), 4.09 (t, 2H, J=6.6 Hz), 3.96 (s, 3H), 2.31-2.21 (m, 2H).
MS 472, 474 (M+1).
ПРИМЕР 10
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-((тетрагидропиран-4-ил)метокси)-хиназолина (Соединение 15)

Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (400 мг, 0.84 ммоль), 4-иодометилтетрагидропиран (190 мг, 0.84 ммоль) и карбонат калия (289 мг, 2.09 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 5 мл). Смесь перемешивали в течение 15 часов при 60°C для осуществления реакции. Полученную реакционную смесь охладили до комнатной температуры и вылили в воду (100 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 50:1) для получения твердого белого вещества (170 мг, выход: 44%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.53 (s, 1H), 8.35 (s, 1H), 7.80 (s, 1H), 7.66 (dd, 1H, J=9.8, 2.0 Hz), 7.53 (t, 1H, J=8.4 Hz), 7.47 (dd, 1H, J=8.4, 1.6 Hz), 7.20 (s, 1H), 4.02 (d, 2H, J=6.4 Hz), 3.95 (s, 3H), 3.89 (dd, 2H, J=11.4, 3.0 Hz), 3.41-3.33 (m, 2H), 2.16-2.03 (m, 1H), 1.76-1.66 (m, 2H), 1.45-1.32 (m, 2H).
MS 462, 464 (M+1).
ПРИМЕР 11
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-((2-метилтиазол-4-ил)метокси)-хиназолина (Соединение 17)

Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.05 ммоль), 2-метил-4-хлорметилтиазол (185 мг, 1.26 ммоль) и карбонат калия (363 мг, 2.63 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл). Смесь нагрели до 60°C и перемешивали в течение 18 часов. Полученную реакционную смесь охладили до комнатной температуры и вылили в воду (75 мл). Твердую часть отделили. Полученную смесь фильтровали и сушили для получения твердого желтого вещества (165 мг, выход: 33%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.56 (s, 1H), 8.37 (s, 1H), 7.82 (s, 1H), 7.70-7.63 (m, 2H), 7.54 (t, 1H, J=8.2 Hz), 7.48 (dd, 1H, J=8.4, 2.0 Hz), 7.38 (s, 1H), 5.28 (s, 2H), 3.94 (s, 3H), 2.69 (s, 3H).
MS 475, 477 (M+1).
ПРИМЕР 12
Синтез (6-(4-(4-бром-2-фторанилино)-6-метоксихиназолин-7-илокси)пиридин-3-ил)-(морфолино)метанона (Соединение 18)
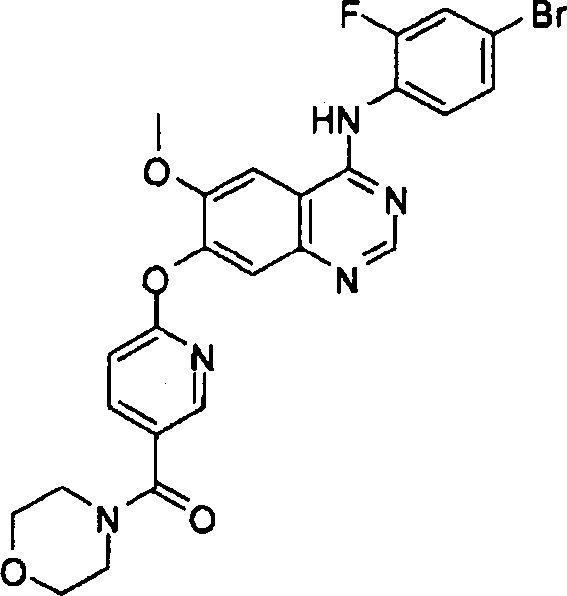
4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (500 мг, 1.37 ммоль), (2-хлорпиридин-5-ил)-(морфолино)метанон (373 мг, 1.64 ммоль), карбонат цезия (1.12 г, 3.43 ммоль) и иодид меди (19 мг, 0.1 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл). Смесь перемешивали в течение 18 часов при 120°C для осуществления реакции. Реакционную смесь охладили до комнатной температуры и вылили в воду (75 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 10:1) для получения твердого белого вещества (174 мг, выход: 23%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.77 (s, 1H), 8.43 (s, 1H), 8.18 (s, 1H), 8.03 (s, 1H), 7.96 (d, 1H, J=8.0 Hz), 7.70 (d, 1H, J=10.4 Hz), 7.57-7.52 (m, 3H), 7.21 (d, 1H, J=8.0 Hz), 3.88 (s, 3H), 3.62-3.32 (m, 8H).
MS 554, 556 (M+1).
ПРИМЕР 13
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(3-(пирролидин-1-ил)-пропокси)-хиназолина (Соединение 19)

Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.05 ммоль), N-хлорпропилпирролидин гидрохлорид (193 мг, 1.05 ммоль) и карбонат калия (363 мг, 2.63 ммоль) добавили в N,N-диметилформамид (ДМФА, 5 мл). Смесь перемешивали в течение 18 часов при 80°C для осуществления реакции. Реакционную смесь охладили до комнатной температуры и вылили в воду (50 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 10:1) для получения твердого белого вещества (160 мг, выход: 32%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.54 (s, 1H), 8.35 (s, 1H), 7.79 (s, 1H), 7.66 (dd, 1H, J=10.0, 2.0 Hz), 7.55-7.45 (m, 2H), 7.18 (s, 1H), 4.18 (t, 2H, J=6.4 Hz), 3.94 (s, 3H), 2.56 (t, 2H, J=7.0 Hz), 2.48-2.45 (m, 4H), 1.98-1.90 (m, 2H), 1.78-1.62 (m, 4H).
MS 238, 239 (1/2M+1).
ПРИМЕР 14
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(2-(пирролидин-1-ил)этокси)-хиназолина (Соединение 20)

Трифторацетат 4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.15 ммоль), N-хлорэтилпирролидин гидрохлорид (294 мг, 1.73 ммоль) и карбонат калия (637 мг, 4.61 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 5 мл). Смесь перемешивали в течение 18 часов при 80°C, поддерживая постоянную температуру. Реакционную смесь охладили до комнатной температуры и вылили в воду (50 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения неочищенного продукта. Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 10:1) для получения твердого белого вещества (160 мг, выход: 33%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.61 (s, 1H), 8.50 (s, 1H), 8.14 (dd, 1H, J=7.0, 2.6 Hz), 7.88-7.78 (m, 2H), 7.45 (t, 1H, J=9.0 Hz), 7.24 (s, 1H), 4.33-4.21 (m, 2H), 3.97 (s, 3H), 3.05-2.85 (m, 2H), 2.78-2.55 (m, 4H), 1.73 (m, 4H).
MS 209, 210 (1/2M+1).
ПРИМЕР 15
Синтез 3-(4-(4-бром-2-фторанилино)-6-метоксихиназолин-7-илокси)-пропанамида (Соединение 22)
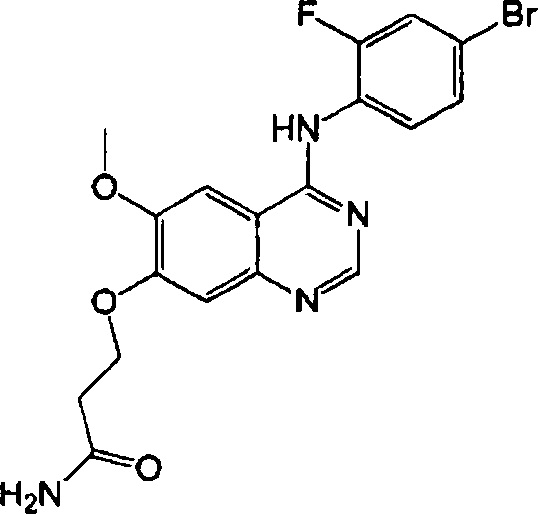
4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (1.0 г, 2.75 ммоль), акриламид (1.95 г, 27.5 ммоль) и 1,8-диазабицикло(5.4.0)-ундек-7-ен (DBU, 837 мг, 5,50 ммоль) растворили в этаноле (10 мл) при комнатной температуре. Смесь перемешивали в течение 48 часов при 85°C для осуществления реакции. Полученную реакционную смесь концентрировали и очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения твердого желтого вещества (151 мг, выход: 13%).
1H-NMR (400 MHz, CD3OD) δ: 8.40 (s, 1Н), 7.60-7.40 (m, 4H), 6.63 (s, 1H), 4.58 (t, 2H, J=6.2 Hz), 3.97 (s, 3H), 2.87 (t, 2H, J=6.2 Hz).
MS 435, 437 (M+1).
ПРИМЕР 16
Синтез 4-(4-(4-бром-2-фторанилино)-6-метоксихиназолин-7-ил-окси)бутанамида (Соединение 23)
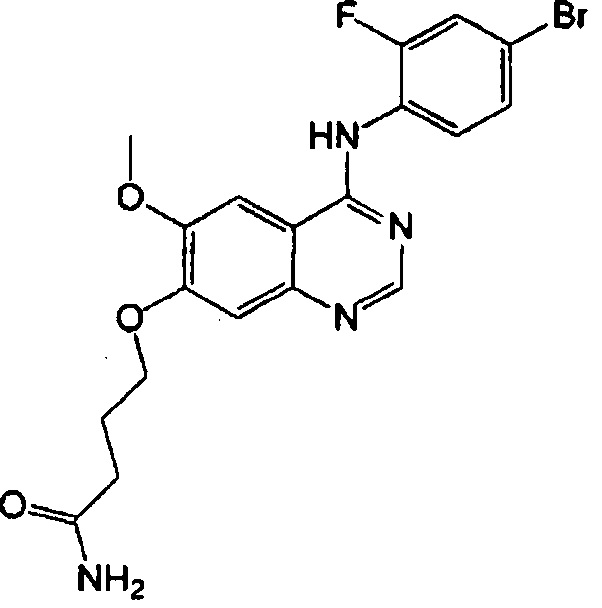
Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (1.5 г, 3.14 ммоль), 4-хлорбутанамид (687 мг, 5.65 ммоль), тетрабутиламмоний иодид (232 мг, 0.63 ммоль) и 1,8-диазабицикло(5.4.0)-ундек-7-ен (DBU, 955 мг, 6.28 ммоль) растворяли в N-метилпирролидоне (20 мл). Смесь перемешивали в течение 24 часов при 85°C, поддерживая постоянную температуру. Реакционную смесь охладили до комнатной температуры и вылили в воду (150 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 50:1) для получения светло-коричневого твердого вещества (110 мг, выход: 8%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.58 (s, 1H), 8.35 (s, 1H), 7.81 (s, 1H), 7.67 (dd, 1H, J=10.0, 1.6 Hz), 7.60-7.40 (m, 2H), 7.37 (br s, 1H), 7.18 (s, 1H), 6.82 (br s, 1H), 4.14 (t, 2H, J=6.4 Hz), 3.95 (s, 3H), 2.27 (t, 2H, J=7.4Hz), 2.10-1.92 (m, 2H).
MS 449, 451 (M+1).
ПРИМЕР 17
Синтез 6-(4-(3-хлор-4-фторанилино)-6-метоксихиназолин-7-илокси)гексанамида (Соединение 24)

Трифторацетат 4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.15 ммоль), 6-хлоргексанамид (336 мг, 2.25 ммоль) и карбонат калия (398 мг, 2.88 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 5 мл) при комнатной температуре. Смесь перемешивали в течение 18 часов при 80°C, поддерживая постоянную температуру. Реакционную смесь охладили до комнатной температуры и вылили в воду (50 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 50:1) для получения твердого белого вещества (220 мг, выход: 44%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.55 (s, 1H), 8.50 (s, 1H), 8.13 (dd, 1H, J=6.8, 2.4 Hz), 7.88-7.75 (m, 2H), 7.45 (t, 1H, J=9.0 Hz), 7.25 (br s, 1H), 7.19 (s, 1H), 6.70 (br s, 1H), 4.13 (t, 2H, J=6.6 Hz), 3.97 (s, 3H), 2.08 (t, 2H, J=7.4Hz), 1.88-1.72 (m, 2H), 1.65-1.50 (m, 2H), 1.50-1.35 (m, 2H).
MS 433, 435 (M+1).
ПРИМЕР 18
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(3-(1Н-имидазол-1-ил)пропокси)-хиназолина (Соединение 25)
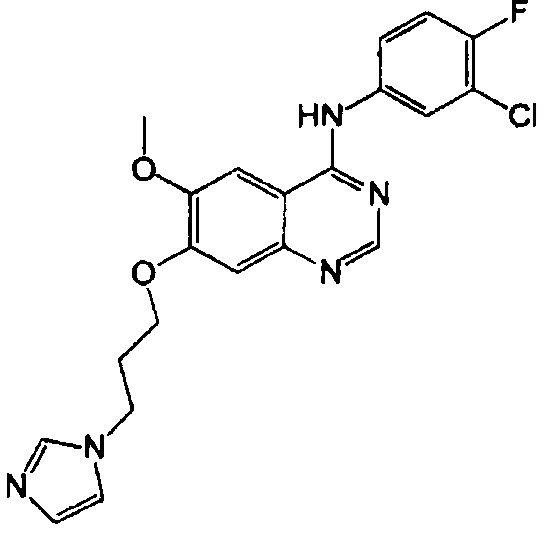
Трифторацетат 4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.15 ммоль), 3-(1Н-имидазол-1-ил)-пропан-1-ол (400 мг) и трифенилфосфин (454 мг, 1.73 ммоль) растворяли в безводном тетрагидрофуране (ТГФ, 10 мл) при комнатной температуре. К полученному раствору по каплям добавили диизопропилазодикарбоксилат (DIAD, 350 мг, 1.73 ммоль) при комнатной температуре под азотом. После завершения добавления по каплям полученную смесь перемешивали в течение 18 часов при 30°C для осуществления реакции. Реакционную смесь концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 50:1) для получения твердого белого вещества (107 мг, выход: 22%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.60 (s, 1H), 8.50 (s, 1H), 8.13 (dd, 1H, J=6.8, 2.8 Hz), 7.90-7.75 (m, 2H), 7.64 (s, 1H), 7.45 (t, 1H, J=9.2 Hz), 7.21 (d, 2H, J=11.6 Hz), 6.90 (s, 1H), 4.17 (t, 2H, J=7.0 Hz), 4.10 (t, 2H, J=6.2 Hz), 3.99 (s, 3H), 2.27 (t, 2H, J=6.4 Hz).
MS 428, 430 (M+1).
ПРИМЕР 19
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(2-(2-метилтиазол-4-ил)этокси)-хиназолина (Соединение 26)
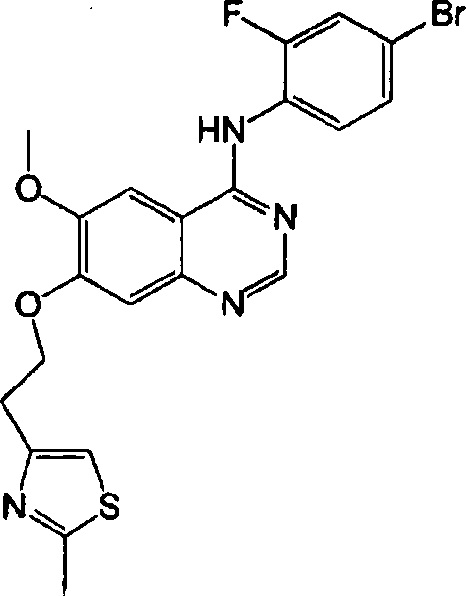
Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (800 мг, 1.67 ммоль), 2-метил-4-метилсульфонилоксиэтилтиазол (555 мг, 2.51 ммоль) и карбонат калия (578 мг, 4.18 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл) при комнатной температуре. Смесь перемешивали в течение 18 часов при 80°C, поддерживая постоянную температуру. Реакционную смесь охладили до комнатной температуры и вылили в воду (50 мл). Твердую часть отделили. Полученную смесь фильтровали и сушили для получения неочищенного продукта. Неочищенный продукт смешали с этилацетатом для образования суспензии. Суспензию отфильтровали и сушили для получения коричневого твердого вещества (320 мг, выход: 39%).
1H-NMR (400 MHz,CDCl3) δ: 8.69 (s, 1H), 8.52 (t, 1H, J=8.6 Hz), 7.40-7.30 (m, 3H), 7.27-7.24 (m, 1H), 7.02-6.92 (m, 2H), 4.51 (t, 2H, J=7.0 Hz), 4.02 (s, 3H), 3.36 (t, 2H, 7=6.8 Hz), 2.71 (s, 3H).
MS 489, 491 (M+1).
ПРИМЕР 20
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-((2-метилтиазол-5-ил)метокси)-хиназолина (Соединение 27)
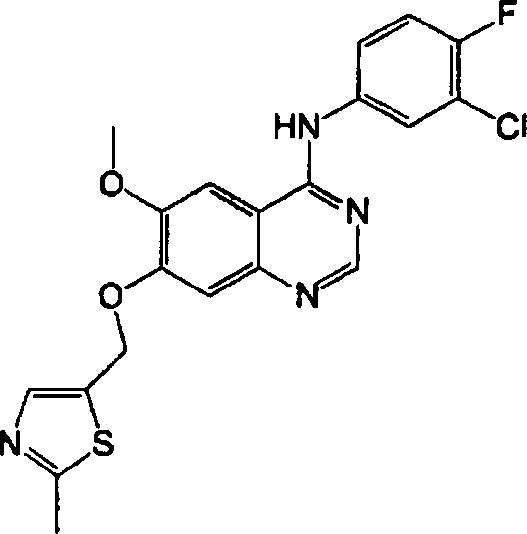
Трифторацетат 4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.15 ммоль), 2-метил-5-бромметилтиазол (332 мг, 1.73 ммоль) и карбонат калия (397 мг, 2.88 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 5 мл). Смесь перемешивали в течение 18 часов при 60°C, поддерживая постоянную температуру. Реакционную смесь охладили до комнатной температуры и выливали в воду (50 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения неочищенного продукта. Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 20:1) для получения твердого белого вещества (120 мг, выход: 24%).
1H-NMR (400 MHz, CDCl3) δ: 8.60 (s, 1H), 7.92-7.88 (m, 1H), 7.70 (s, 1H), 7.65-7.50 (m, 2H), 7.36 (s, 1H), 7.20-7.10 (m, 2H), 5.37 (s, 2H), 4.04 (s, 3H), 2.71 (s, 3H).
MS 431, 433 (M+1).
ПРИМЕР 21
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-((2-метилтиазол-4-ил)метокси)-хиназолина (Соединение 27')
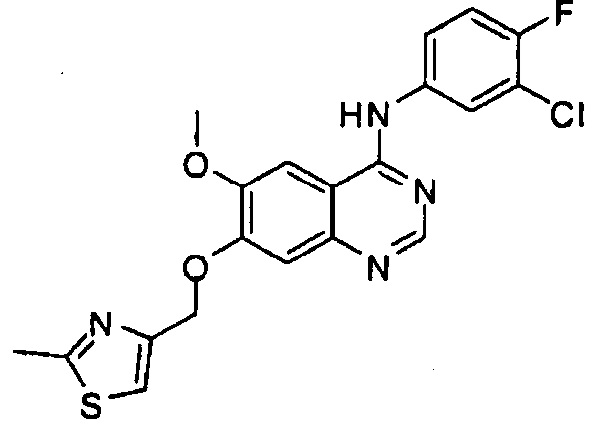
4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолин (60 мг, 0.188 ммоль), 2-метил-4-хлорметилтиазол (100 мг, 0.68 ммоль) и карбонат калия (70 мг) диспергировали в N,N-диметилформамиде (ДМФА, 5 мл). Смесь нагрели до 90°C и перемешивали в течение 2 часов для осуществления реакции. После завершения реакции реакционную смесь охладили до комнатной температуры. Добавили очищенную воду (20 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, концентрировали и очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения целевого соединение (22 мг).
1H-NMR (600 MHz, DMSO) δ: 9.83 (s, 1H), 8.50 (m, 1H), 8.20 (s, 1H), 7.99-7.88 (m, 2H), 7.64 (s, 1H), 7.45-7.38 (m, 2H), 5.27 (s, 2H), 3.98 (s, 3H), 3.04 (s, 3H).
MS: 431, 433 (M+1).
ПРИМЕР 22
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(4-(диметиламино)бутокси)-хиназолина (Соединение 28)
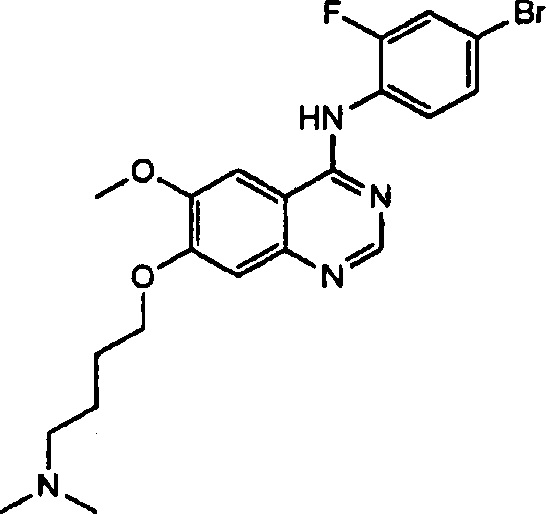
Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.05 ммоль), 4-хлор-N,N-диметилбутан-1-амин гидрохлорид (268 мг, 1.57 ммоль) и карбонат калия (362 мг, 2.62 ммоль) добавили в N,N-диметилформамид (ДМФА, 5 мл) при комнатной температуре. Смесь перемешивали в течение 18 часов при 120°C для осуществления реакции. Реакционную смесь охладили до комнатной температуры и вылили в воду (50 мл). Полученную смесь экстрагировали этилацетатом и концентрировали для получения неочищенного продукта. Неочищенный продукт диспергировали в водном насыщенном расторе бикарбоната натрия для нейтрализации. Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения светло-коричневого твердого вещества (102 мг, выход: 21%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.59 (s, 1H), 8.35 (s, 1H), 7.82 (s, 1H), 7.66 (d, 1H, J=9.2 Hz), 7.59-7.41 (m, 2H), 7.19 (s, 1H), 4.16 (t, 2H, J=6.6 Hz), 3.95 (s, 3H), 2.39-2.29 (m, 2H), 2.18 (s, 6H), 1.87-1.74 (m, 2H), 1.66-1.53 (m, 2H).
MS 463, 465 (M+1).
ПРИМЕР 23
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(3-(диметиламино)-пропокси)хиназолина (Соединение 29)

Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.05 ммоль), 3-хлор-N,N-диметилпропан-1-амин гидрохлорид (246 мг, 1.57 ммоль) и карбонат калия (362 мг, 2.62 ммоль) ввели в N,N-диметилформамид (ДМФА, 5 мл) при комнатной температуре. Смесь перемешивали в течение 5 часов при 80°C для осуществления реакции. Реакционную смесь охладили до комнатной температуры и выливали в воду (80 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения твердого белого вещества (180 мг, выход: 38%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.75 (s, 1H), 8.35 (s, 1H), 7.90 (s, 1H), 7.65 (d, 1H, J=9.6 Hz), 7.59-7.41 (m, 2H), 7.18 (s, 1H), 4.18 (t, 2H, J=6.2 Hz), 3.96 (s, 3H), 2.61-2.51 (m, 2H), 2.29 (s, 6H), 2.07-1.94 (m, 2H).
MS 225, 226 (1/2M+1).
ПРИМЕР24
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(4-(пирролидин-1-ил)бутокси)-хиназолина (Соединение 30)

Трифторацетат 4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.05 ммоль), N-гидроксибутилпирролидин (600 мг) и трифенилфосфин (413 мг, 1.58 ммоль) растворяли в тетрагидрофуране (ТГФ, 10 мл) при комнатной температуре. Полученную смесь охладили до 0-5°C. К смеси по каплям добавили диизопропилазодикарбоксилат (DIAD, 319 мг, 1.58 ммоль). Полученную смесь перемешивали в течение 18 часов при 40°C для осуществления реакции. Реакционную смесь концентрировали и очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения неочищенного продукта. Неочищенный продукт диспергировали в водном насыщенном растворе бикарбоната натрия для нейтрализации, а затем смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения светло-коричневого твердого вещества (120 мг, выход: 23%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.54 (s, 1H), 8.35 (s, 1H), 7.80 (s, 1H), 7.66 (dd, 1H, J=10.0, 2.0 Hz), 7.57-7.43 (m, 2H), 7.19 (s, 1H), 4.16 (t, 2H, J=6.4 Hz), 3.94 (s, 3H), 2.49-2.35 (m, 6H), 1.89-1.76 (m, 2H), 1.73-1.57 (m, 6H).
MS 245, 246 (1/2M+1).
ПРИМЕР 25
Синтез N-метил-6-(4-(4-бром-2-фторанилино)-6-метоксихиназолин-7-илокси)-гексанамида (Соединение 31)
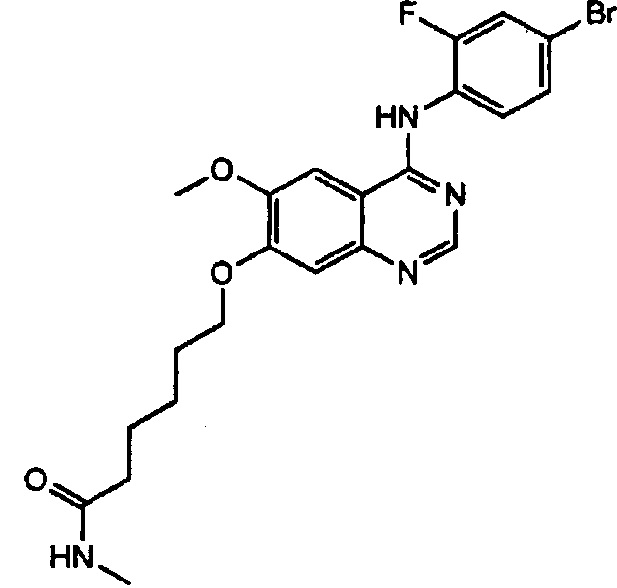
4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (728 мг, 2 ммоль), N-метил-6-бромгексанамид (416 мг, 2 ммоль) и карбонат калия (690 мг, 5 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл). Полученную смесь нагревали с обратным холодильником при перемешивании в течение 18 часов для осуществления реакции. Реакционную смесь охладили до комнатной температуры и выливали в воду (100 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта (500 мг). Неочищенный продукт перекристаллизовали из метанола и отфильтровали для получения твердого белого вещества (230 мг, выход: 23%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.55 (s, 1H), 8.35 (s, 1H), 7.83-7.63 (m, 3H), 7.57-7.44 (m, 2H), 7.18 (s, 1H), 4.12 (t, 1H, J=6.4 Hz), 3.94 (s, 3H, J=11.6 Hz), 2.56 (d, 3Н, J=4.8 Hz), 2.09 (t, 3H, J=6.4 Hz), 1.79 (m, 2H), 1.58 (m, 2H), 1.42 (m, 2H).
MS 491, 493 (M+1).
ПРИМЕР 26
Синтез N,N-диметил-6-(4-(4-бром-2-фторанилино)-6-метоксихиназолин-7-илокси)-гексанамида (Соединение 32)

4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (364 мг, 1.0 ммоль), N,N-диметил-6-бромгексанамид (222 мг, 1.0 ммоль) и карбонат калия (276 мг, 2.0 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл) при комнатной температуре. Смесь нагрели до 80°C и перемешивали в течение 3 часов, поддерживая постоянную температуру. К реакционной смеси добавили воду (50 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали под пониженным давлением для получения неочищенного продукта. Неочищенный продукт перекристаллизовали из метанола для получения твердого белого вещества (160 мг, выход: 32%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.54 (br s, 1H), 8.35 (s, 1H), 7.79 (s, 1H), 7.67 (dd, 1H, J=10.0, 2.0 Hz,), 7.7.58-7.41 (m, 2H), 4.13 (t, 2H, J=6.4 Hz), 3.94 (s, 3H), 2.96 (s, 3H), 2.81 (s, 3H), 2.32 (t, 2H, J=7.2 Hz), 1.88-1.75 (m, 2H), 1.64-1.40 (m, 4H).
MS 505, 507 (M+1).
Пример 27
Синтез 5-(4-(4-бром-2-фторанилино)-6-метоксихиназолин-7-илокси)пентанамида (Соединение 33)
4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (1.8 г, 4.95 ммоль), 5-бромпентанамид (1.78 г, 9.89 ммоль) и карбонат калия (1.8 г, 13.02 ммоль) добавили в N,N-диметилформамид (ДМФА, 15 мл) при комнатной температуре. Смесь нагрели до 50°C и перемешивали в течение 2 часов для осуществления реакции. К реакционной смеси добавили воду (30 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт перекристаллизовали из метанола для получения целевого соединения (1.4 г, выход: 61%).
1H-NMR (600 MHz, DMSO-d6) δ: 9.53 (s, 1H), 8.35 (s, 1H), 7.79 (s, 1H), 7.66 (dd, 1H, J=10.2 2.4 Hz), 7.54 (t, 1H, J=2.4 Hz), 7.46 (dd, 1H, 7=8.4, 1.2 Hz) 7.28 (br s, 1H), 7.19 (s, 1H), 6.74 (br s, 1H), 4.14 (t, 2Н, J=6.6 Hz), 3.95 (s, 3H), 2.15 (t, 2H, J=7.2 Hz), 1.81-1.78 (m, 2H), 1.69-1.67 (m, 2H).
MS: m/z 463.4, 465.3 (M+1)
ПРИМЕР 28
Синтез 4-(4-бром-2-фторанилино)-6-метокси-7-(2-(1Н-имидазол-1-ил)этокси)-хиназолина (Соединение 34)

4-(4-бром-2-фторанилино)-6-метокси-7-гидроксихиназолин (1.00 г, 2.75 ммоль), 1-(2-хлорэтил)-1Н-имидазол (0.72 г, 5.5 ммоль) и карбонат калия (0.95 г, 6.88 ммоль) добавили в N,N-диметилформамид (ДМФА, 15 мл) при комнатной температуре. Смесь нагрели до 60°C и перемешивали в течение 3 часов для осуществления реакции. После завершения реакции в реакционную смесь добавили воду (30 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт перекристаллизовали из метанола для получения целевого соединения (0.91 г, выход: 72%).
1H-NMR (600 MHz, DMSO-d6) δ: 9.56 (s, 1H), 8.36 (s, 1H), 7.82 (s, 1H), 7.72-7.65 (m, 2H), 7.53-7.46 (m, 2H), 7.29-7.23 (m, 2H), 6.91 (s, 1H), 4.45 (m, 4H), 3.95 (s, 3H).
MS: m/z 458.4, 460.4 (M+1)
ПРИМЕР 29
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(2-(1H-имидазол-1-ил)этокси)-хиназолина (Соединение 36)
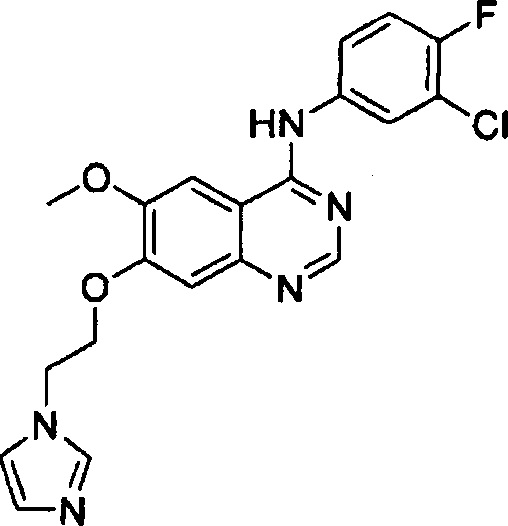
Трифторацетат 4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолина (500 мг, 1.15 ммоль), 1-(2-хлорэтил)-1Н-имидазол гидрохлорид (289 мг, 1.73 ммоль) и карбонат калия (636 г, 4.60 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 5 мл) при комнатной температуре. Смесь нагрели до 80°C и перемешивали в течение 18 часов для осуществления реакции. Реакционную смесь охладили до комнатной температуры и выливали в воду (100 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения светло-желтого твердого вещества (120 мг, выход: 25%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.58 (br s, 1H), 8.50 (s, 1H), 8.13 (dd, 1H, J=7.0, 2.6 Hz), 7.85-7.76 (m, 2H), 7.72 (s, 1H), 7.45 (t, 1H, J=9.2 Hz), 7.29 (s, 1H), 7.24 (s, 1H), 6.90 (s, 1H), 4.45 (s, 4H), 3.97 (s, 3H).
MS 207.5, 208.5 (1/2M+1).
ПРИМЕР 30
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(4-(диметиламино)бутокси)хиназолина (Соединение 37)

Стадия 1: Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-((4-ацетилокси)-бутокси)хиназолина.
4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолин (10 г, 31.3 ммоль) растворили в N,N-диметилформамиде (ДМФА, 100 мл) при комнатной температуре. В полученную смесь добавили карбонат калия (13 г, 93.9 ммоль) и 4-бромбутилацетат (7.3 г, 37.6 ммоль). Смесь оставили для реакции на 2 часа при 50°C. Реакционную смесь охладили до комнатной температуры и вылили в воду (200 мл). Полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения желтого масла (12 г), которое напрямую использовали на следующей стадии.
Стадия 2: Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(4-гидрокси-бутокси)хиназолина
Продукт, полученный на стадии 1 (12 г), растворили в метаноле (50 мл). В смесь добавили воду (10 мл) и моногидрат гидроксида лития (1.4 г, 33.2 ммоль). Полученную смесь оставили для реакции при комнатной температуре на ночь. Реакционную смесь концентрировали. В полученную смесь при перемешивании добавили воду (200 мл) и этилацетат (40 мл). При перемешивании смеси выделилось твердое вещество. Смесь отфильтровали, и полученный на фильтре остаток сушили для получения твердого желтого вещества (6.2 г, выход: 51%).
Стадия 3: Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-((4-метил-сульфонилокси)бутокси)хиназолина
Продукт, полученный на стадии 2 (6.2 г, 15.8 ммоль), растворили в дихлорметане (50 мл). В смесь добавили триэтиламин (3.2 г, 31.6 ммоль), а затем по каплям на ледяной бане добавили метилсульфонил хлорид (2.7 г, 23.7 ммоль). После завершения добавления по каплям полученную смесь оставили для реакции на 3 часа при комнатной температуре. В реакционную смесь добавили воду (150 мл). Смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения твердого желтого вещества (7.2 г, выход: 96%).
Стадия 4: Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(4-(диметиламино)-бутокси)хиназолина
Продукт, полученный на стадии 3 (2.5 г, 5.3 ммоль), диметиламин гидрохлорид (644 мг, 7.95 ммоль) и карбонат калия (2.9 г, 21.2 ммоль) диспергировали в ацетонитриле (15 мл) при комнатной температуре. Смесь перемешивали при 80°C в течение ночи. Реакционную смесь концентрировали. В полученную смесь добавили воду (100 мл). Смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения светло-желтого твердого вещества (700 мг, выход: 32%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.56 (br s, 1H), 8.50 (s, 1H), 8.13 (dd, 1H, J=6.8, 2.8 Hz), 7.85-7.77 (m, 2H), 7.45 (t, 1H, J=9.2 Hz), 7.20 (s, 1H), 4.16 (t, 2H, J=56.4 Hz), 3.97 (s, 3H), 2.27 (t, 2H, J=7.2 Hz), 2.13 (s, 6H), 1.83-1.78 (m, 2H), 1.60-1.60 (m, 2H).
MS 210, 211 (1/2M+1).
ПРИМЕР 31
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(3-(диметиламино)-пропокси)хиназолина (Соединение 38)
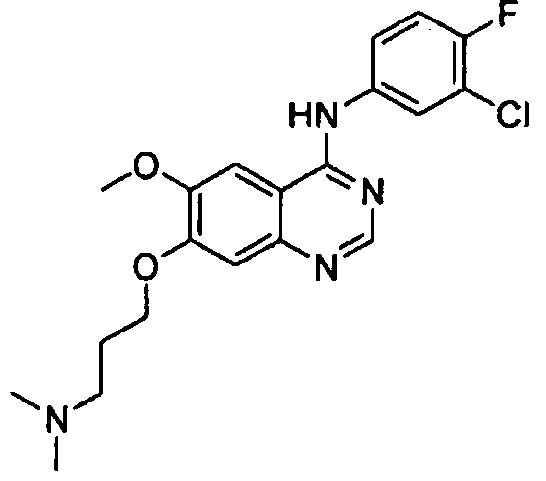
4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолин (2 г, 6.26 ммоль), 3-хлор-N,N-диметилпропан-1-амин гидрохлорид (1.2 г, 7.51 ммоль) и карбонат калия (3.5 г, 25.04 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 20 мл). Смесь перемешивали при 80°C в течение ночи. Реакционную смесь охладили до комнатной температуры и выливали в воду (80 мл). Полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения твердого желтого вещества (325 мг, выход: 13%).
1H-NMR (400 MHz, DMSO-d6) δ: 10.08 (br s, 1H), 8.49 (s, 1H), 8.29 (dd, 1H, J=6.8, 2.4 Hz), 8.14 (s, 1H), 8.01-7.97 (m, 1H), 7.42 (t, 1H, J=9.2 Hz), 7.17 (s, 1H), 4.17 (t, 2H, J=6.4 Hz), 4.01 (s, 3H), 2.42 (t, 2H, J=7.0 Hz), 2.19 (s, 6H), 1.98-1.91 (m, 2H).
MS 203, 204 (1/2M+1).
ПРИМЕР 32
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(3-(пирролидин-1-ил)пропокси) хиназолина (Соединение 39)
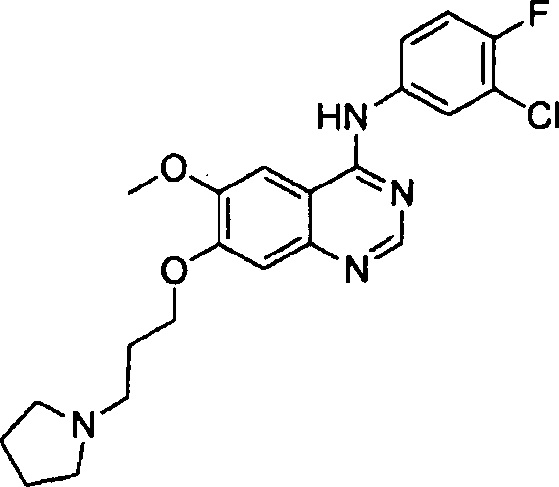
4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолин (2 г, 6.26 ммоль), N-хлорпропилпирролидин гидрохлорид (1.72 г, 9.39 ммоль) и карбонат калия (2.59 г, 18.78 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл). Смесь перемешивали при 80°C в течение ночи. Реакционную смесь охладили до комнатной температуры и выливали в воду (50 мл). Полученную смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения твердого желтого вещества (400 мг, выход: 15%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.59 (s, 1H), 8.51 (s, 1H), 8.14 (dd, 1H, J=7.0, 2.4 Hz), 7.87-7.76 (m, 2H), 7.45 (t, 1H, J=9.0 Hz), 7.20 (s, 1H), 4.20 (t, 2H, J=6.2 Hz), 3.98 (s, 3H), 2.62-2.53 (m, 4H), 2.02-1.99 (m, 2H), 1.73 (m, 4H).
MS 216, 217 (1/2M+1).
ПРИМЕР 33
Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(4-(пирролидин-1-ил)бутокси)-хиназолина (Соединение 40)
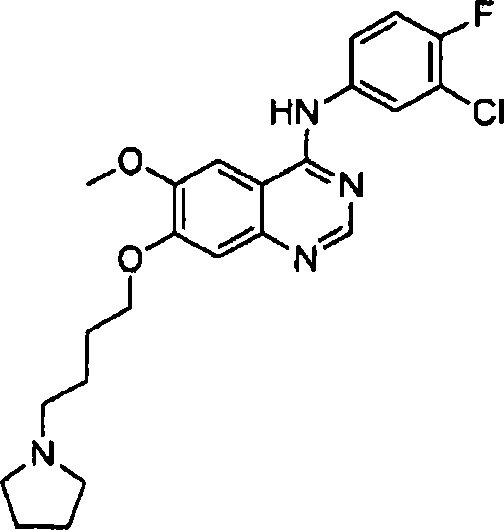
Стадии 1, 2, и 3 идентичны соответствующим стадиям получения соединения 37.
Стадия 4: Синтез 4-(3-хлор-4-фторанилино)-6-метокси-7-(4-(пирролидин-1-ил)-бутокси)хиназолина.
Продукт, полученный на стадии 3 (3.0 г, 6.4 ммоль), тетрагидропиррол (681.6 мг, 9.6 ммоль) и карбонат калия (2.6 г, 19.2 ммоль) диспергировали в ацетонитриле (20 мл) при комнатной температуре. Смесь перемешивали при 80°C в течение ночи. Реакционную смесь концентрировали. В полученную смесь добавили воду (100 мл). Смесь экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (хлористый метилен : метанол = 10:1) для получения светло-желтого твердого вещества (500 мг, выход: 18%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.62 (br s, 1H), 8.49 (s, 1H), 8.13 (dd, 1H, J=6.8, 2.4 Hz), 7.85-7.77 (m, 2H), 7.44 (t, 1H, J=9.0 Hz), 7.19 (s, 1H), 4.16 (t, 2H, J=6.6 Hz), 3.96 (s, 3H), 2.50-2.40 (m, 6H), 1.85-1.81 (m, 2H), 1.68-1.60 (m, 6H).
MS 223, 224 (1/2М+1).
ПРИМЕР 34
Синтез 5-(4-(3-хлор-4-фторанилино)-6-метоксихиназолин-7-илокси)пентанамида (Соединение 41)
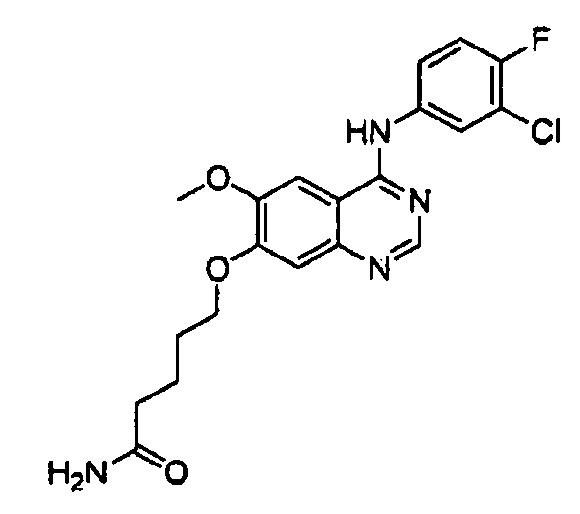
В одногорлую колбу объемом 25 мл последовательно при комнатной температуре добавили 5-бромпентанамид (193 мг, 1.07 ммоль), трифторацетат 4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолина (420 мг, 0.97 ммоль), карбонат калия (401 мг, 2.91 ммоль), иодид калия (32 мг, 0.19 ммоль) и N,N-диметилформамид (ДМФА, 5 мл). Смесь нагрели до 80°C и оставили для реакции на ночь. Реакционную смесь вылили в ледяную воду (20 мл). Полученную смесь отфильтровали. Остаток на фильтре сушили для получения твердого желтого вещества (120 мг, выход: 30%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.56 (br s, 1H), 8.50 (s, 1H), 8.12 (dd, 1H, J=6.8, 2.4 Hz), 7.84-7.77 (m, 2H), 7.45 (t, 1H, J=9.0 Hz), 7.28 (br s, 1H), 7.20 (s, 1H), 6.74 (br s, 1H), 4.15 (t, 2H, J=6.2 Hz), 3.97 (s, 3H), 2.14 (t, 2H, J=7.4 Hz), 1.86-1.62 (m, 4H).
MS: 419 (M+1).
ПРИМЕР 35
Синтез N-метил-6-(4-(3-хлор-4-фторанилино)-6-метоксихиназолин-7-илокси)-гексанамида (Соединение 42)

Трифторацетат 4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолина (867 мг, 2 ммоль), N-метил-6-бромгексанамид (416 мг, 2 ммоль) и карбонат калия (690 мг, 5 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл). Полученную смесь нагревали с обратным холодильником при перемешивании в течение 18 часов для осуществления реакции. Реакционную смесь охладили до комнатной температуры и вылили в воду (100 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта (500 мг). Неочищенный продукт перекристаллизовали из метанола и фильтровали для получения твердого белого вещества (130 мг, выход: 15%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.57 (s, 1H), 8.50 (s, 1H), 8.13 (dd, 1H, J=6.8, 2.4 Hz), 7.83-7.72 (m, 3H), 7.45 (t, 1H, J=9.0 Hz), 7.19 (s, 1H), 4.13 (t, 2H, J=6.4 Hz), 3.97 (s, 3H, J=4.8 Hz), 2.56 (d, 3H, J=4.4 Hz), 2.09 (t, 2H, J=7.4 Hz), 1.79 (m, 2H), 1.58 (m, 2H), 1.42 (m, 2H).
MS 447 (M+1).
ПРИМЕР 36
Синтез N,N-диметил-6-(4-(3-хлор-4-фторанилино)-6-метоксихиназолин-7-илокси)-гексанамида (Соединение 43)

4-(3-хлор-4-фторанилино)-6-метокси-7-гидроксихиназолин (320 мг, 1.0 ммоль), N,N-диметил-6-бромгексанамид (222 мг, 1.0 ммоль) и карбонат калия (276 мг, 2 ммоль) диспергировали в N,N-диметилформамиде (ДМФА, 10 мл). Смесь нагрели до 80°C и перемешивали в течение 3 часов для осуществления реакции. В реакционную смесь добавили воду (100 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт перекристаллизовали из метанола и отфильтровали для получения твердого белого вещества (120 мг, выход: 26%).
1H-NMR (400 MHz, DMSO-d6) δ: 9.56 (s, 1H), 8.50 (s, 1H), 8.14-8.12 (dd, 1H, J=6.8, 2.8 Hz), 7.84-7.78 (m, 2H), 7.45 (t, 1H, J=9.2 Hz,), 7.20 (s, 1H), 4.14 (t, 2H, J=6.4 Hz), 3.97 (s, 3H), 2.96 (s, 3H), 2.81 (s, 3H), 2.32 (t, 2H, J=7.2 Hz), 1.88-1.75 (m, 2H), 1.64-1.40 (m, 4H).
MS 461 (M+1).
Сравнительный Пример 1
Синтез 4-(2-фтор-4-хлоранилино)-6-метокси-7-(3-(пирролидин-1-ил)-пропокси)хиназолина (Соединение 44)

Стадия 1: Синтез 4-(2-фтор-4-хлоранилино)-6-метокси-7-бензилоксихиназолина 6-метокси-7-бензилоксихиназолин-4-он (6.5 г, 23.0 ммоль) добавили в толуол (60 мл). В полученную смесь добавили трибутиламин (5.2 г, 27.6 ммоль). Смесь нагрели до 60°C. В нагретую смесь по каплям добавили хлорокись фосфора (3.5 г, 23.0 ммоль). После завершения добавления по каплям полученную смесь нагрели до 120°C и перемешивали в течение 1 часа для осуществления реакции. Реакционную смесь охладили до 57°C. К охлажденной смеси добавили 4-хлор-2-фторанилин (5.0 г, 34.4 ммоль). Полученную смесь затем нагрели до 95°C и оставили для реакции на 0.5 часа. Реакционную смесь охладили до комнатной температуры и фильтровали для получения светло-желтого продукта (8.0 г, выход 84.8%).
Стадия 2: Синтез 4-(2-фтор-4-хлоранилино)-6-метокси-7-гидроксихиназолина В трехгорлую колбу добавили 4-(2-фтор-4-хлоранилино)-6-метокси-7-бензилоксихиназолин (6.0 г, 14.6 ммоль) и затем трифторуксусную кислоту (30 мл). Смесь перемешивали до растворения, нагрели до 75°C и оставили для реакции на 1 час. После завершения реакции, реакционную смесь охладили до комнатной температуры и затем концентрировали под пониженным давлением для получения маслянистой субстанции. При добавлении к маслянистой субстанции метил-трет-бутилового эфира (150 мл) выделяется твердое вещество. Смесь отфильтровали и сушили для получения светло-желтого твердого вещества (4.0 г, выход: 85.8%).
Стадия 3: Синтез 4-(2-фтор-4-хлоранилино)-6-метокси-7-(3-(пирролидин-1-ил)-пропокси)хиназолина
При комнатной температуре в трехгорлую колбу добавоместили 4-(2-фтор-4-хлоранилино)-6-метокси-7-гидроксихиназолин (1.4 г, 4.4 ммоль), гидрохлорид N-хлор-пропилпирролидина (809 мг, 4.04 ммоль) и карбонат калия (1.2 г, 8.8 ммоль), а затем добавили N,N-диметилформамид (ДМФА, 20 мл). Смесь нагрели до 80°C и перемешивали в течение 18 часов для осуществления реакции. После завершения реакции реакционную смесь охладили до комнатной температуры. В реакционную смесь добавили очищенную воду (50 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 10:1) для получения твердого белого вещества (1.0 г, выход: 53%).
1H-NMR (600 MHz, DMSO-d6) δ: 9.55 (s, 1H), 8.36 (s, 1H), 7.80 (s, 1H), 7.55-7.61 (m, 2H), 7.35-7.36 (m, 1H), 7.19 (s, 1H), 4.18-4.20 (m, 2H), 3.95 (s, 3H), 2.56-2.58 (m, 2H), 2.46-2.51 (m, 4H), 1.95-1.99 (m, 2H), 1.69 (s, 4H).
MS 431.2 (M+1).
СРАВНИТЕЛЬНЫЙ ПРИМЕР 2
Синтез 4-(2-фтор-4-броманилино)-6-метокси-7-(3-(пиперидин-1-ил)-пропокси)хиназолина (Соединение 45)

Стадия 1: Синтез метил 3-(пиперидин-1-ил)пропаноата
В трехгорлую колбу добавили пиперидин (5 г, 58.7 ммоль), а затем ввели метилакрилат (15 мл). Смесь нагрели до 80°C и оставили для реакции на 2 часа. После завершения реакции реакционную смесь охладили до комнатной температуры и концентрировали под пониженным давлением для получения желтого масла (10 г), которое напрямую использовали на следующей стадии.
Стадия 2: Синтез 3-(пиперидин-1-ил)пропанола
В трехгорлую колбу налили безводный тетрагидрофуран (80 мл), после чего окружающую атмосферу охладили до 0°C. В колбу порциями добавили алюмогидрид лития (3.5 г, 93.4 ммоль) и по каплям раствор метил 3-(пиперидин-1-ил)пропаноата (8.0 г, 46.7 ммоль) в тетрагидрофуране. После завершения добавления по каплям смесь нагрели до комнатной температуры и оставили для реакции на 2 часа. После завершения реакции, реакционную смесь охладили до 0°C. К охлажденной смеси последовательно добавляли очищенную воду (10 мл), водный раствор NaOH (15%, 10 мл) и очищенную воду (30 мл). Твердое белое вещество отделили. Полученную смесь отфильтровали. Фильтрат концентрировали для получения желтого масла (4.0 г), которое напрямую использовали на следующей стадии.
Стадия 3: Синтез 3-(пиперидин-1-ил)пропил 4-метил-бензенсульфоната
3-(Пиперидин-1-ил)пропанол (3.0 г, 20.9 ммоль) добавили в дихлорметан (30 мл). Полученную смесь охладили до 0°C. К охлажденной смеси последовательно добавляли водный раствор триэтиламина (4.2 г, 41.8 ммоль) и тозилхлорид (4.0 г, 20.9 ммоль). Смесь оставили при 0°C на 4 часа для реакции. После завершения реакции реакционную смесь нагрели до комнатной температуры. В реакционную смесь добавили очищенную воду (50 мл). Полученная смесь разделилась на два слоя, и водный слой экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения желтого масла (3.0 г), которое без дальнейшей обработки напрямую использовали на следующей стадии.
Стадия 4: Синтез 4-(2-фтор-4-броманилино)-6-метокси-7-(3-(пиперидин-1-ил)-пропокси)хиназолина
4-(2-фтор-4-броманилино)-6-метокси-7-гидроксихиназолин (2.0 г, 5.6 ммоль), 3-(пиперидин-1-ил)пропил-4-метилбензосульфонат (2.0 г, 6.7 ммоль) и карбонат калия (1.2 г, 8.4 ммоль) добавили в ДМФА (15 мл). Смесь нагрели до 80°C и перемешивали в течение 2 часов для осуществления реакции. После завершения реакции в реакционную смесь добавили очищенную воду (50 мл). Полученную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали для получения неочищенного продукта. Неочищенный продукт очищали препаративной тонкослойной хроматографией (хлористый метилен : метанол = 10:1) для получения твердого белого вещества (1.0 г, выход: 36.5%).
1H-NMR (600 MHz, DMSO-d6) δ: 9.57 (s, 1H), 8.35 (s, 1H), 7.81 (s, 1H), 7.56-7.58 (m, 2H), 7.34-7.35 (m, 1H), 7.11 (s, 1H), 4.15-4.19 (m, 2H), 3.94 (s, 3H), 2.42-2.49 (m, 4H), 2.00 (s, 2H), 1.42-1.66 (m, 8H).
MS 490.3 (M+1).
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Исследование 1: Эксперимент на клетках in vitro
Клеточные линии:

Методы (МТТ-анализ)
Клетки в логарифмической фазе роста высевали с определенной плотностью в 96-луночный планшет (200 мкл/лунка). Клеткам давали возможность расти в течение 24 часов, а затем обрабатывали различными концентрациями лекарств (включая холостую (сравнительную) группу и контрольную группу) и инкубировали в течение 72 часов при 37°C. Каждую концентрацию в данном эксперименте тестировали трижды. Через 72 часа экспозиции в среду добавляли 20 мкл реагента МТТ для окрашивания (конечная концентрации МТТ составляла 0.5 мг/мл), и инкубировали клетки дополнительно 4 часа при 37°C. После этого отбирали 180 мкл среды и добавляли в каждую лунку 130 мкл ДМСО (DMSO) или среду удаляли полностью и в каждую лунку добавляли 150 мкл ДМСО. Затем планшет встряхивали и перемешивали на микроосцилляторе. Оптическую плотность (OD) каждого образца измеряли с помощью микропланшетного ридера при 550 нм. Степень ингибирования клеток рассчитывали по следующей формуле:

На основании степеней ингибирования рассчитали IC50 с помощью LOGIT метода. Эксперименты повторяли дважды, и данные представляли в ±SD (стандартное отклонение).
Тестировали влияние соединений на ингибирование клеток перечисленных выше линий, результаты приведены в Таблице 2.

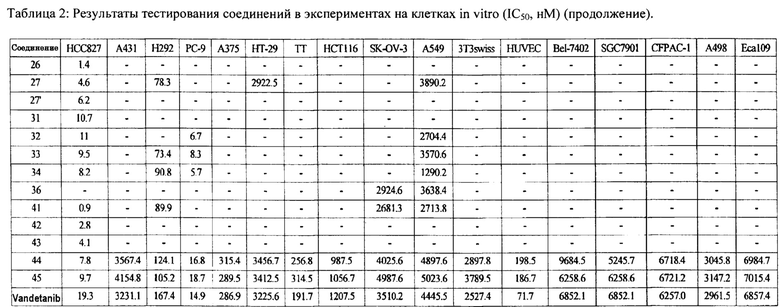
-: No data
Испытание 2: Ферментативные исследования in vitro
Термины:
RET: Рецептор, вовлеченный в рак щитовидной железы
KDR: Рецептор фактора роста сосудистого эндотелия человека 2
FLT-1: Рецептор фактора роста сосудистого эндотелия человека 1
FGFR-1: Рецептор фактора роста фибробластов 1
EGFR: Человеческий рецептор эпидермального фактора роста 1
PDGFRβ: Рецептор фактора роста тромбоцитов β
ВТК: Тирозинкиназа Брутона
AXL: Киназа анапластической лимфомы
Методы:
ELISA (иммуно-ферментный анализ): Планшет для ферментативной реакции покрывали субстратом фермента (поли(Glu, Tyr)4:1) с плотностью 20 мкг/мл. затем фермент, образец и 5 мкМ АТФ смешивали в лунке для реакции. После завершения реакции фосфорилированный субстрат определяли с помощью моноклонального антитела к фосфотирозину (4G10). На следующей стадии в лунку последовательно добавляли козьи антитела к IgG мыши, конъюгированные с пероксидазой хрена (HRP), и тетраметилбензидин (ТМВ) для определения степени фосфорилирования субстрата по интенсивности окрашивания. В ходе эксперимента были созданы две группы: группа без тирозинкиназы и контрольная группа с соответствующей концентрацией DMSO. Наконец, в каждую лунку для остановки реакции добавили 0.18 М H2SO4 (50 мкл/на лунку) Оптическую плотность каждого образца определяли с помощью микропланшетного ридера при 450 нм.

Определили относительную степень ингибирования тирозинкиназ тестируемым соединением.
На основании степеней ингибирования, определенных при разных концентрациях, рассчитали IC50, используя метод LOGIT. Эксперименты повторяли три раза, в качестве окончательного коэффициента ингибирования брали среднее из трех значений IC50, свидетельствующее о способности ингибировать. Результаты приведены в Таблице 3.
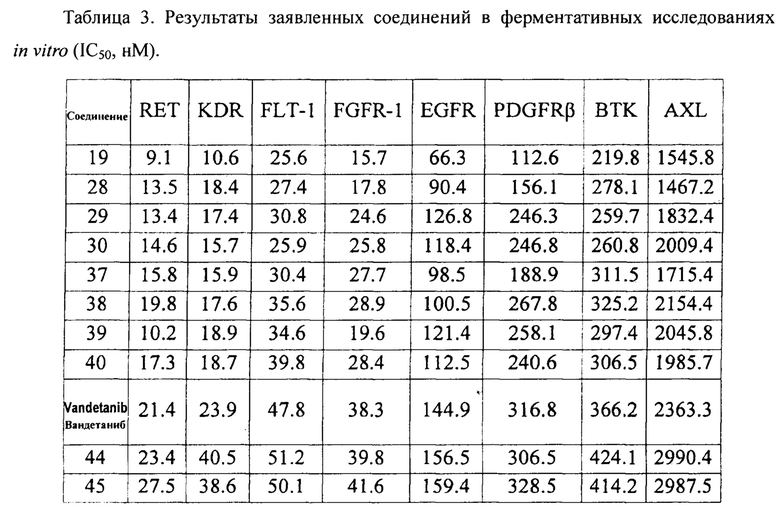
Исследование 3: Исследования максимально переносимой дозы (MTD) у мышей.
В MTD экспериментах мышам вводили заявленные соединения.
Выбрали здоровых мышей, мало различающихся по весу. Выбранных мышей поместили в клетки на разные стеллажи в соответствии с массой их тела. На каждом стеллаже мышей разделили на группы случайным образом. В каждой группе было три мыши.
Вводили следующие различающиеся в два раза дозы: 1000, 500, 250, 125, 62.5 и 31.25 мг/кг. Каждую дозу вводили трем мышам. Тестируемые соединения вводили мышам в течение 7 дней, а затем наблюдали мышей в течение последующих 7 дней, не вводя соединения.
Маркировка животных и клеток.
Маркировка клеток: на каждую клетку маркером наносили название соединения, вводимую дозу и группу мышей.
Метки на животных: Мышей в каждой группе пометили пикриновой кислотой. No. 1: слева спереди, No. 2: слева сзади, и No. 3: справа спереди.
Взвешивание животных: Мышей каждой группы взвешивали, результаты записывали.
Введение соединений: Соединения вводили мышам перорально в соответствии с дозировкой и массой тела.
Результат: Максимально переносимые дозы соединений приведены в Таблице 4.
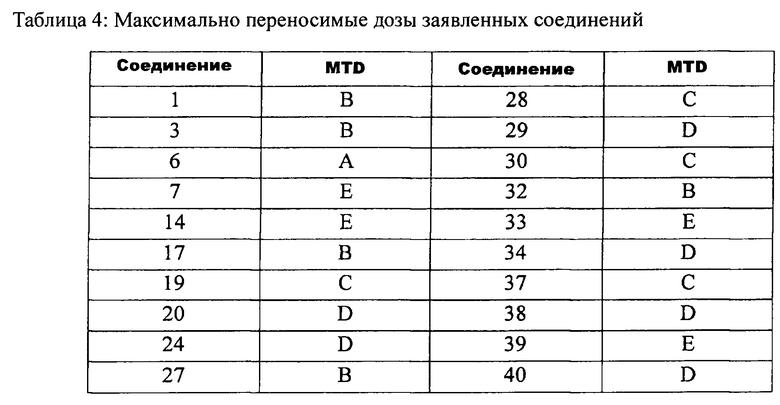
А: >1000 мг/кг;
В: >500 мг/кг и ≤1000 мг/кг;
С: >250 мг/кг и ≤500 мг/кг;
D: >125 мг/кг и ≤250 мг/кг;
Е: >62.5 мг/кг и ≤125 мг/кг;
F: >31.25 мг/кг и ≤62.5 мг/кг;
G: ≤31.25 мг/кг.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2690853C2 |
| БЕНЗОТИА(ДИ)АЗЕПИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ЖЕЛЧНЫХ КИСЛОТ | 2019 |
|
RU2785867C2 |
| БЕНЗОПРОИЗВОДНЫЕ С ШЕСТИЧЛЕННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА DPP-4 И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2702644C2 |
| ПИРАЗОЛ-4-ИЛ-ГЕТЕРОЦИКЛИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2012 |
|
RU2638552C2 |
| ПРОИЗВОДНОЕ N-(АЗААРИЛ)ЦИКЛОЛАКТАМ-1-КАРБОКСАМИДА, МЕТОД ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2765785C2 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2597364C2 |
| Соединение-антагонист PD-L1 | 2020 |
|
RU2823231C1 |
| БИ-АРИЛ-МЕТА-ПИРИМИДИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2006 |
|
RU2589878C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ПОДАВЛЕНИЯ СЛИПАНИЯ ТРОМБОЦИТОВ И КОМПОЗИЦИЯ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1996 |
|
RU2169146C2 |
| ПРОТИВОИНФЕКЦИОННЫЕ СОЕДИНЕНИЯ | 2014 |
|
RU2734760C2 |
Изобретение относится к новому соединению формулы (I) и его фармацевтически приемлемой соли. Соединения обладают свойствами ингибитора киназной активности и могут быть использованы в производстве лекарства для лечения рака щитовидной железы, немелкоклеточной карциномы, эпидермоидной карциномы, меланомы, рака толстой кишки, карциномы желудка, рака пищевода, рака поджелудочной железы, рака почки, рака печени, рака легких или рака яичников. В общей формуле (I)

R1 представляет собой -O(СН2)nR3, где n равно 3, 4 или 5; R3 представляет собой NReRf, где Re и Rf каждый независимо выбран из группы, включающей C1-6 алкил, или Re и Rf соединены вместе с образованием -(СН2)4-; R2 представляет собой  или
или 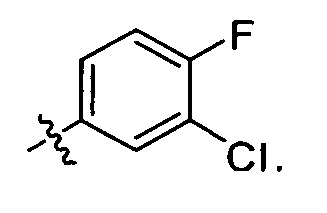 . Соединения формулы (I) получают взаимодействием соединения формулы (III) или его соли с соединением формулы (IV) или его солью в растворителе в присутствии одного или более из катализатора, основания и поверхностно-активного вещества согласно следующей схеме:
. Соединения формулы (I) получают взаимодействием соединения формулы (III) или его соли с соединением формулы (IV) или его солью в растворителе в присутствии одного или более из катализатора, основания и поверхностно-активного вещества согласно следующей схеме:
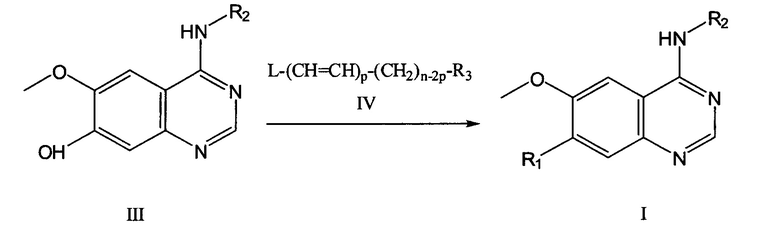
В указанных структурных формулах R1, R2, R3 и n определены выше, L является галогеном, гидроксилом, мезилокси или водородом и р=0 или 1 при условии, что, когда р=1, L является водородом. Соединения формулы (I) проявляют более высокую активность по сравнению с близкими по структуре соединениями. 6 н. и 2 з.п. ф-лы, 4 табл., 37 пр.
1. Соединение формулы (I):

или его фармацевтически приемлемая соль, где:
R1 представляет собой -O(СН2)nR3, где n равно 3, 4 или 5,
R3 представляет собой NReRf, где Re и Rf каждый независимо выбран из группы, включающей C1-6 алкил, или Re и Rf соединены вместе с образованием -(СН2)4-; и
R2 представляет собой 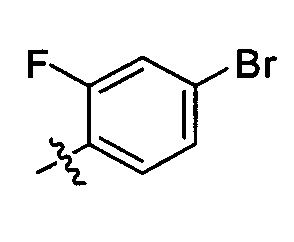 или
или 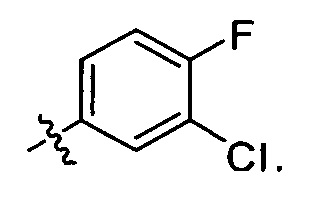
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R3 представляет собой -NReRf, где Re и Rf представляют собой C1-6 алкил.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где
R1 выбран из:
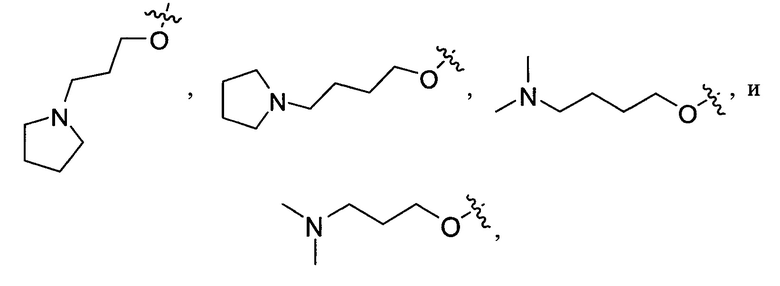
4. Соединение или его фармацевтически приемлемая соль, где соединение выбрано из группы, включающей:

5. Фармацевтическая композиция для лечения рака, содержащая соединение по любому из пп. 1-4 или его фармацевтически приемлемую соль и наполнитель.
6. Применение соединения по любому из пп. 1-4 или его фармацевтически приемлемой соли в производстве лекарства для лечения рака.
7. Применение соединения по любому из пп. 1-4 или его фармацевтически приемлемой соли в производстве лекарства для лечения рака щитовидной железы, немелкоклеточной карциномы, эпидермоидной карциномы, меланомы, рака толстой кишки, карциномы желудка, рака пищевода, рака поджелудочной железы, рака почки, рака печени, рака легких или рака яичников.
8. Способ получения соединения по п. 1 или его фармацевтически приемлемой соли, включающий взаимодействие соединения формулы (III) или его соли с соединением формулы (IV) или его солью в растворителе в присутствии одного или более из катализатора, основания и поверхностно-активного вещества для получения соединения Формулы (I),

где:
R1, R2, R3 и n определены в пункте 1,
L является галогеном, гидроксилом, мезилокси или водородом и
р=0 или 1 при условии что, когда р=1, L является водородом.
| Garofalo, Antonio; et al.,Synthesis and Structure-Activity Relationships of (Aryloxy)quinazoline Ureas as Novel, Potent, and Selective Vascular Endothelial Growth Factor Receptor-2 Inhibitors | |||
| Journal of Medicinal Chemistry, 2012, 55(3),pp, 1189-1204 | |||
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СПОСОБ ДОСТИЖЕНИЯ АНТИАНГИОГЕННОГО И/ИЛИ ЭФФЕКТА СНИЖЕНИЯ ПРОНИЦАЕМОСТИ КРОВЕНОСНЫХ СОСУДОВ У ТЕПЛОКРОВНОГО ЖИВОТНОГО | 1997 |
|
RU2198879C2 |
| DATABASE REGISTRY (online): RN 1356905-14-0,Entered STN: 16 Feb 2012, Retrieved from STN | |||
| DATABASE REGISTRY (online): RN 1356905-13-9 Entered STN: 16 Feb 2012, Retrieved from STN | |||
| CN 102532107 A, 04.07.2012 | |||
| CN 101838245 A, 22.09.2010 | |||
| WO 2004006846 A2, 22.01.2004 | |||
| CN 1205694 A, 20.01.1999 & RU 2194701 C2, 20.12.2002 & EP0873319 B1, 25.07.2001 | |||
| CN 1882577 A, 20.12.2006 | |||
| CN 1863534 A, 15.11.2006 & WO 2005013998 A1,17.02.2005 & US2007027145 A1, 01.02.2007 | |||
| WO 2009094210 A1, 30.07.2009 | |||
| Сырьевая смесь для изготовления древесноминеральных плит | 1983 |
|
SU1211239A1 |
| Перекатываемый затвор для водоемов | 1922 |
|
SU2001A1 |
| HENNEQUIN L F; ET AL Design and Structure-Activity Relationship of a New Class of Potent VEGF Receptor Tyrosine Kinase Inhibitors, JOURNAL OF MEDICINAL CHEMISTRY, 1999, Vol: 42, Page(s): 5369 - 5389 | |||
| Mingzhang Gao; et al., Radiosynthesis of [C]Vandetanib and [C]chloro-Vandetanib as new potential PET agents for imaging of VEGFR in cancer Bioorganic & Medicinal Chemistry Letters, 2011, Vol: 21, Nr:11, Page(s): 3222 - 3226 | |||
| Gang Chen et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ получения деталей с рельефными полостями | 1983 |
|
SU1212684A1 |

