Изобретение относится к биотехнологии. Конкретно изобретение относится к способу получения концентрата вакцины от гриппа, содержащему ВПЧ, где гены гемагглютинина, нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/Michigan/45/2015, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/Novosibirsk/01/2014, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/HongKong/4801/2014, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма B/Phuket/3073/13, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009 или ген гемагглютинина получен из штамма B/Brisbane/60/2008, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009. Изобретение также относится к концентрату вакцины, содержащему такие ВПЧ. Изобретение позволяет получить ВПЧ вируса гриппа имеющего большую гемагглютинирующую активность. Также способ позволяет повысить выход антигена. Кроме того, при осуществлении способа сохраняется жизнеспособность и высокая продуктивность целевых белков без использования регулируемой экспрессии у клеток-продуцентов ВПЧ гриппа.
Грипп человека - высококонтагиозная болезнь. Передача вируса происходит воздушно-капельным путем, а также путем контакта с предметами, контаминированными патогеном (игрушки, дверные ручки и др.), с последующим попаданием в верхние дыхательные пути или конъюнктивальную слизистую. Рецепторы клеток, к которым вирусы гриппа человека имеют предпочтение, экспрессируются на эпителиальных клетках на всем протяжении дыхательных путей: в слизистой оболочке носа, околоносовых пазухах, глотке, трахее, бронхах, бронхиолах и альвеолах, но их количество различно на разных участках [1].
Вирус гриппа имеет негативный геном, состоящий из 8 фрагментов одноцепочечной вирионной РНК (вРНК), кодирующих по крайней мере 11 белков. Если все фрагменты расположить в порядке убывания молекулярного веса, то первые три фрагмента кодируют белки полимеразного комплекса (РВ2, РВ1 и РА), четвертый - гемагглютинин (НА), пятый - нуклеопротеин (NP), шестой - нейраминидазу (NA), седьмой - матриксный белок (Ml) и неструктурный белок (М2), восьмой кодирует два белка (NS1 и NS2). Второй фрагмент РВ1 кодирует еще один белок PB1-F2, образующийся в результате альтернативного сплайсинга мРНК, этот белок вызывает апоптоз через нарушение структуры митохондрий [2]. В составе вириона все фрагменты вРНК находятся в виде рибонуклеопротеида (вРНП) - вРНК, ассоциированного с белком NP и белками полимеразного комплекса [3]. В состав полимеразного комплекса, проявляющего активность РНК-зависимой РНК-полимеразы, входят белки РВ1, РВ2 и РА. Проникновение вируса гриппа (ВГ) в клетку происходит путем эндоцитоза вирионов с последующим переносом всех 8 фрагментов в рибонуклеопротеид (РНП), сначала в цитоплазму, а затем в ядро инфицированной клетки [4, 5].
В процессе упаковки вируса важную роль играет взаимодействие вРНП, M1, НА и NA. Сегменты вирусных РНК содержат уникальные сигналы упаковки, которые частично перекрываются рамками считывания в определенных сегментах РНК, что позволяет координировать упаковку вирусного генома. НА и NA являются важными структурами для определения формы вириона, однако механизм селективного отбора фрагментов вРНП при упаковке их в вирионы до сих пор не известен [6, 7]. Наиболее важными антигенами ВГ типов А и В, изменчивость которых способствует появлению новых эпидемий, являются НА и NA. Первый ответствен за взаимодействие вируса с поверхностью клетки и в процессе размножения в организме вызывает индукцию нейтрализующих его антител. Фермент NA в антигенном отношении отличается от НА. Антитела к NA не нейтрализуют инфекционности вируса (кроме очень высоких концентраций), но сильно замедляют освобождение вируса из инфицированных клеток, и эти антитела могут играть важную роль в снижении репликации вируса in vivo и в предотвращении распространения инфекции [8].
Отличительной чертой ВГ является высокая изменчивость генома, что позволяет ему уклоняться от специфического иммунитета и вследствие этого вызывать ежегодные эпидемии, реже - пандемии при появлении в популяции вируса с новым генотипом, к которому большинство населения не имеют специфического иммунитета [9]. Одна из причин высокой изменчивости вируса связана с РНК-зависимой РНК-полимеразой. Этот фермент совершает одну ошибку на каждые 10 000 нуклеотидов и в отличие от ДНК- зависимой ДНК-полимеразы не может исправлять их. Если единичная клетка, инфицированная ВГ, производит около 10 000 новых вирусных частиц, то в теории по этому коэффициенту ошибок будет произведено 10 000 новых вирусных мутантов. Последствием этого является антигенный дрейф ВГ, благодаря которому за 3-4 года поверхностные белки вируса изменяются настолько сильно, что специфический иммунитет к прежним вариантам не может защитить человека от инфицирования новыми мутантными штаммами [10]. В этом случае говорят об антигенном дрейфе ВГ. Наличие сегментированного генома является причиной другого типа антигенной изменчивости ВГ типа А - антигенного сдвига - появления вирусов с новым набором генов. Это становится возможным в результате обмена сегментами РНК между генетически различными ВГ при инфицировании ими одной клетки [11]. В настоящее время большинство исследователей разделяют точку зрения, что пандемические ВГ появляются в результате обмена генами между ВГ типа А человека и животных. Убедительное подтверждение эта гипотеза получила в 2009 г., когда молекулярно-генетический анализ показал, что новый пандемический штамм ВГ типа A(H1N1)pdm 09 - это тройной реассортант, несущий гены ВГ птиц, человека и свиньи [12]. Однако остается неясным, почему из 16 подтипов НА и 9 подтипов NA, распространенных в ВГ птиц, ВГ человека содержат только три подтипа НА и два подтипа NA. Так, пандемия «испанки» 1918 г. была вызвана ВГ типа A(H1N1), пандемия «азиатского» гриппа 1957 г. - вирусом A(H2N2), пандемия «гонконгского» гриппа 1968 г. - вирусом A(H3N2). Причиной следующих двух пандемий - «русского» гриппа 1978 г. и «свиного» гриппа 2009 г. - вновь был вирус A(H1N1), хотя по молекулярно-биологическим и антигенным характеристикам эти вирусы и вирус «испанки» 1918 г. значительно отличаются друг от друга.
НА - основной поверхностный гликопротеин ВГ. Он инициирует инфекцию, связываясь с клеточным рецептором, вызывает слияние вирусной и эндосомальной мембран, необходимое для выхода РНП в цитоплазму зараженной клетки. Рецепторная специфичность НА - важный фактор в определении круга хозяев. Роль рецептора ВГ на клетках хозяина выполняют сиаловые кислоты. Сиаловая кислота - это N-ацетил-производное нейраминовой кислоты, или аминосахар, прикрепленный ко множеству различных белков на поверхности клетки. Сиаловая кислота всегда является последним остатком в цепочке сахаров, который прикрепляется к белку; предпоследним является галактоза. ВГ человека преимущественно связываются с сиаловой кислотой, которая связана с галактозой связью а2-6 [13]. Эти рецепторы представлены в большом количестве на эпителиальных клетках в трахее человека.
Напротив, ВГ птиц преимущественно распознают сиаловые кислоты, связанные с галактозой связью а2-3, которые распространены на эпителиальных клетках в кишечнике водоплавающей дичи (главном участке репликации ВГ птиц). По-видимому, недостаток рецепторов для ВГ птиц в верхних дыхательных путях человека может быть одним из факторов, предотвращающих продуктивную передачу ВГ птиц от человека к человеку.
Протеолитическая активация НА - определяющий фактор патогенности ВГ А [14]. НА низкопатогенных вирусов имеют сайт расщепления, обычно являющийся агригином, но иногда - лизином [15]. Разрезание молекулы происходит при помощи трипсиноподобных протеаз, представленных лишь в эпителиальных тканях дыхательных путей и кишечного тракта. Высокопатогенные ВГ птиц имеют сайт расщепления, состоящий из 4 аминокислотных остатков, разрезание молекулы НА происходит при помощи фурина, представителя семейства серин-эндопротеаз [16]. Повсеместность этого фермента в клетках объясняет способность ВГ птиц реплицироваться в различных тканях.
Ежегодно в мире гриппом болеют от 3 до 5 млн человек. Эпидемии гриппа возникают с периодичностью 1 - 3 года, но эпидемические вспышки отмечаются ежегодно и наносят большой ущерб здоровью населения, приводят к огромным финансовым затратам на лечение и реабилитацию больных, особенно среди групп риска.
Пандемия гриппа A(H1N1) в 1918-1920-х гг. («испанка»). Пандемия, названная «испанской болезнью», началась в конце Первой мировой войны - в 1918 г. Место ее появления точно установить пока не удалось, но, во всяком случае, первичным эпидемическим очагом была не Испания. Название «испанка» появилось случайно. Так как военная цензура обеих борющихся сторон не допускала сообщений о начавшейся в армии и среди населения эпидемии, то первые известия о ней появились в печати в мае - июне 1918 г. в нейтральной Испании.
Первая волна пандемии была относительно мягкой, в последующие две тяжесть возросла, и уровень смертности достиг 3% (во время обычных вспышек он бывает менее 0,1%). Основной причиной смерти была пневмония, часто осложненная бактериальной инфекцией [17].
Пандемия обошла земной шар трижды. Первая волна началась в конце апреля 1918 г. в Париже и почти сразу вспыхнула по всей Европе. В мае «испанка» появилась в северной Африке, а в июне - в Индии. Этим временно закончилось дальнейшее развитие эпидемии. Число случаев болезни, достигавшее довольно крупных цифр при очень малой смертности, стало убывать. В августе количество заболевших резко снизилось, что было принято за конец пандемии.
После нескольких месяцев затишья подошла вторая волна пандемии, во время которой заболеваемость была ниже, но смертность выше. Сначала болезнь появилась на западном побережье Африки в Сьерра-Леоне в конце августа 1918 г., откуда болезнь распространилась на все западное побережье Африки. В Северной Америке вторая волна началась в октябре 1918 г. в Бостоне, куда, как тогда считали, ее занесли возвратившиеся из Европы солдаты. К концу второй волны из населенных мест остались нетронутыми лишь Мадагаскар, Австралия и Новая Каледония, по-видимому, благодаря рациональным и строго проводимым мерам профилактики. Окончание второй волны относят на конец декабря 1918 г.
Третья волна пандемии началась в феврале - марте 1919 г., захватив на этот раз и уцелевшие до тех пор островные территории. Эта вспышка также отличалась высокой смертностью. Окончилась она в разных местностях не одновременно, и в отдельных районах фиксировалась до июня и даже до августа 1919 г. Пандемия полностью прекратилась только в 1920 г. Невозможно было определить точное количество людей, перенесших грипп. По-видимому, болело не меньше 550 млн человек, а погибло, по разным подсчетам, от 20 до 50 млн - более 2,5 % всей численности населения планеты того времени, составлявшей 1850 млн человек [17].
Пандемия «гонконгского гриппа» A(H3N2) (1968-1970 гг.). Эта пандемия развивалась тремя волнами (1968, 1969 и 1970 гг.) и была вызвана вирусом нового серотипа A(H3N2). Заболевание появилось в Сингапуре в начале августа 1968 г. и к сентябрю уже распространилось на другие сопредельные страны, в частности на Индию и северные территории Австралии, но в большинстве этих районов оно носило клинически легкую форму. Осенью 1968 г. новый вирус достиг Европы, но и там до начала зимы не было зарегистрировано ни одной большой вспышки. Наиболее опасной была вторая волна пандемии. Третья волна пандемии дала о себе знать в конце 1970 г. Затем эпидемические волны резко пошли на убыль. Что касается смертности, пандемия «гонконгского гриппа», как и предыдущая, была относительно мягкой [17].
Пандемия Русского гриппа A(H1N1) (1977 - 1978 гг.). В мае 1977 г. вирус A(H1N1) вновь вызвал вспышку в районе российско-китайской границы, которая достигла Северной Америки в 1978 г. Вирус по антигенным и молекулярно-биологическим характеристикам был близок к изолятам 1950-х гг. [18, 19]. Эта пандемия была необычной. В начале 1978 г. вирусы серотипа (H3N2) вызвали эпидемии в Западной Европе, Африке и Азии, а на Американском континенте (США и Канада) обосновался вирус гриппа серотипа A(H1N1), но уже к марту во многих странах, в том числе и в СССР, изолировали одновременно штаммы вирусов гриппа A(H1N1), A(H3N2) и гриппа В.
Подавляющее число заболевших в эпидемию 1977-1978 гг. приходилось на людей в возрасте до 20 лет, т.е. на ту часть населения, которая не имела контакта с вирусами гриппа серотипа (H1N1), исчезнувшими из циркуляции более 20 лет тому назад. Напротив, лица старше 30 лет составили только 20 % больных, хотя их доля в общей численности населения превышала 50 %, т.е., учитывая низкую заболеваемость в эту эпидемию вообще, люди зрелого и пожилого возраста, имевшие в прошлом контакт с вирусами гриппа H1N1, практически не болели.
Первая пандемия в XXI в. началась, как и прежде, неожиданно, хотя к ней готовились все последнее десятилетие. В двух образцах, независимо собранных в Южной Калифорнии в середине апреля, в Центре контроля заболеваемости (CDC), Атланта, США, идентифицировали вирус гриппа свиного происхождения, первоначально названный S-OIV (Swine-Origin Influenza Virus, который в последствии по предложению ВОЗ был назван A(H1N1)pdm09). Вирус распространился по планете с такой быстротой, что 11 июня 2009 г. ВОЗ подняла уровень готовности к пандемии гриппа до фазы 6, официально объявив первую пандемию гриппа в XXI в. [20]. К тому времени насчитывалось 30000 подтвержденных случаев заболевания в 74 странах. Менее чем через месяц ВОЗ подтвердила уже более 77000 случаев заболевания.
Антигенно пандемический вирус A(H1N1) pdm09 подобен классическому свиному вирусу и тройному реассортантному вирусу свиней A(H1N1), имеющему гены вирусов птиц и человека и циркулирующему в США более 10 лет. В то же время практически не было серологического перекреста пандемического вируса с вирусом сезонного гриппа A(H1N1). Характерной чертой пандемии 2009 г. было диспропорциональное поражение детей и молодых людей по сравнению со старшими возрастными группами [78; 79]. Вероятно, это возрастное распределение связано с наличием иммунитета к этому вирусу в популяции пожилых людей [82]. Так, показано, что в США 33 % людей старше 60 лет имеют антитела, перекрестно реагирующие с A(H1N1) pdm09 в РТГА и реакции нейтрализации [21].
Важнейшей мерой защиты от гриппозной инфекции и ограничения ее распространения является вакцинопрофилактика. Современные гриппозные вакцины индуцируют, как правило, образование антител к поверхностным антигенам вируса гриппа - гемагглютинину и нейраминидазе.
Существующие гриппозные вакцины можно разделить на два вида: аттенуированные (живые) и инактивированные, включая субъединичные. Все они достаточно широко применяются при вакцинации населения и хорошо зарекомендовали себя. Аттенуированные вакцины представляют собой вирусы гриппа с ослабленной вирулентностью. При приготовлении инактивированных субъединичных вакцин также используются эпидемически актуальные штаммы вируса, хотя применение высокопатогенных штаммов ограничено высокими требованиями к биологической безопасности производства. Традиционные способы получения вакцинных штаммов вируса гриппа А имеют ряд недостатков.
Использование как аттенуации, основанной на адаптации вирусов к организму гетерологичного хозяина, так и реассортации при коинфекции эпидемическими штаммами и донорами аттенуации не всегда позволяет сохранить баланс между уровнем вирулентности исходного вируса и его иммуногенностью. Чрезмерная аттенуация может привести к получению штаммов, утративших способность репродуцироваться в клетках дыхательных путей человека.
Альтернативным способом получения вакцинных штаммов является использование методов обратной генетики. Обратная генетика позволяет воссоздать биологически активную вирусную частицу путем коинфекции пермиссивных линий клеток плазмидами, содержащими гены, кодирующие вирусные белки. Внося изменения в эти гены, можно менять вирулентность и антигенные свойства вируса гриппа. С помощью методов обратной генетики можно получить реассортанты вирусов гриппа. Например, плазмидами, кодирующими сегменты генома пандемического или циркулирующего сезонного штамма и аттенуированного вакцинного штамма вируса гриппа А, трансфицируют пермиссивные эукариотические клетки. В результате происходит сборка полноценных вирионов вируса, несущих набор белков как вакцинного, так и патогенного штаммов. С помощью данного метода удалось получить и исследовать вирус гриппа A (H1N1), вызвавший в 1918 году пандемию («испанский грипп»).
Методы обратной генетики позволяют снижать вирулентность вируса путем введения мутаций в различные вирусные гены. Например, мутации в двух генах, кодирующих полимеразные белки РВ1 и РВ2 вируса гриппа птиц A/guinea fowl/Hong Kong/WF 10199 (H9N2), привели к утрате патогенности вируса для кур. Делеция неструктурного белка NS1 обеспечила аттенуацию вируса гриппа А. Полученная таким способом вакцина успешно прошла первую фазу клинических испытаний. Внесения мутаций в белок М2, необходимый для формирования ионного канала, также приводят к аттенуации вируса. Изменение последовательности аминокислот в сайте нарезания НА высокопатогенного вируса гриппа А Н5 при помощи направленного мутагенеза привело к приобретению им характеристик низкопатогенных вирусов.
Методы обратной генетики хорошо зарекомендовали себя при получении аттенуированных штаммов вирусов гриппа. Однако использование реассортации в случае вакцинных штаммов поднимает вопрос биобезопасности в связи с возможностью мутаций, восстанавливающих или повышающих вирулентность вируса. Кроме того, широкое применение живых аттенуированных гриппозных вакцин вызывает опасение ввиду возможной реассортации живой вакцины с циркулирующими штаммами вирусов гриппа человека. В препаративных количествах вакцинные штаммы вируса гриппа чаще всего получают в куриных эмбрионах, что делает невозможным вакцинацию лиц с аллергией на куриный белок. Зависимость технологического процесса от продуктивности куриного стада также относится к недостаткам вакцин, получаемых с помощью куриных эмбрионов.
Решить проблемы, связанные с применением куриных эмбрионов и необходимостью аттенуации патогенных штаммов вируса гриппа, можно с использованием рекомбинантных субъединичных вакцин. Одним из новых подходов к получению субъединичных вакцин против вирусов гриппа является применение различных систем экспрессии для быстрой продукции отдельных вирусных белков в препаративных количествах [22].
В одной из популярных систем экспрессии гриппозные антигены продуцируют в клетках насекомых при помощи бакуловирусных векторов, несущих гены целевых антигенов. Наиболее широко используют вирус множественного ядерного полиэдроза калифорнийской совки (AcMNPV). Для работы с AcMNPV обычно используют линии клеток Sf9, полученные из яичника гусениц Spodoptera frugiperda. В этой системе можно продуцировать различные антигены вируса гриппа А. Иммунизация мышей рекомбинантным НА вируса гриппа H5N1, полученным в бакуловирусной системе экспрессии, приводила к индукции высокого уровня вируснейтрализующих антител. Однако для достижения значимых уровней антител требовалось либо применение адъюванта, либо прайм-буст-иммунизации с помощью инактивированного вируса гриппа H5N1 или рекомбинантного аденовируса, несущего ген НА вируса гриппа.
Недостатком рекомбинантных субъединичных вакцин, как и традиционных субъединичных вакцин, являются низкая иммуногенность и, как следствие, необходимость многократной вакцинации и применения адъювантов.
Вирусоподобные частицы (ВПЧ) представляют собой антигенные детерминанты вирионов без фрагментов геномной РНК. Благодаря отсутствию генетического материала ВПЧ не способны инфицировать клетки человека и животных, что обеспечивает их безопасность. Поверхностные белки гриппозных ВПЧ могут представлять конформационные эпитопы клеткам иммунной системы как нативные вирионы. В ряде исследований показано, что в формировании гриппозных ВПЧ ключевую роль играет участие внутреннего белка вируса гриппа - M1. Этот белок связывается с липидным участком апикального домена плазматической мембраны, взаимодействует с поверхностными гликопротеинами вируса гриппа и инициирует сборку и почкование ВПЧ, содержащих липидную мембрану клетки-хозяина с инкорпорированными в нее тремя трансмембранными белками вируса гриппа.
Гриппозные ВПЧ получены в различных системах экспрессии. Для эффективного освобождения из клеток млекопитающих гриппозных ВПЧ, содержащих НА, необходима либо одновременная экспрессия NA, либо добавление экзогенной NA. Это связано со способностью активной NA расщеплять сиаловые кислоты на поверхности клеточной мембраны. В клетках насекомых гриппозные ВПЧ, содержащие НА, можно получить даже в отсутствие экспрессии NA, так как в этих клетках сиаловые кислоты не связываются с N-гликанами в процессе посттрансляционной модификации.
Один из подходов к получению гриппозных ВПЧ в клетках насекомых предполагает использование рекомбинантных бакуловирусов. На животных моделях показано, что поверхностные гриппозные антигены в составе ВПЧ, полученные при помощи рекомбинантных бакуловирусов, индуцировали выработку как антигемагглютинирующих и вируснейтрализующих антител, так и эффекторов клеточного иммунного ответа. Кроме того, вакцина на основе гриппозных ВПЧ индуцировала протективный иммунитет против гомологичных и гетерологичных штаммов вируса гриппа А. Вакцина на основе ВПЧ, несущих антигены пандемичного вируса гриппа A H1N 1(2009), прошла вторую фазу клинических испытаний на 4563 здоровых взрослых добровольцах и показала безопасность и иммуногенность [23].
Использование рекомбинантных бакуловирусов для экспрессии белков вируса гриппа в клетках насекомых приводит к накоплению в культуральной жидкости не только ВПЧ, но и бакуловирусов. Поскольку эти структуры близки между собой по размерам, возникают проблемы с очисткой ВПЧ от бакуловирусных частиц. Гриппозные ВПЧ могут быть получены в клетках млекопитающих с помощью других ДНК- и РНК-вирусных векторов. Например, разработана система продукции гриппозных ВПЧ в клетках Vero при помощи ДНК-векторов, несущих гены НА, NA, белков M1 и М2 вируса гриппа. Описано использование модифицированного вируса осповакцины Анкара для получения ВПЧ, содержащих белки вируса гриппа H5N1 (НА, NA, Ml) в клетках млекопитающих. Такие ВПЧ способны формировать протективный иммунный ответ у мышей.
Таким образом, получение ВПЧ представляет перспективное направление при разработке гриппозных вакцин нового типа.
Вирусные векторы представляют собой рекомбинантные вирусы, в геном которых встроен целевой ген с набором регуляторных элементов. Среди существующих систем доставки антигенов вирусные векторы занимают особое место, поскольку обладают следующими свойствами: имеют природный механизм взаимодействия с клеткой и проникновения в нее; переносят чужеродный генетический материал в ядро клетки; способны обеспечивать длительную экспрессию антигена; вирусная оболочка защищает генетический материал, кодирующий антиген.
Вакцины на основе вирусных векторов эффективно активируют цитотоксические Т- лимфоциты, что особенно важно при вакцинации против внутриклеточных патогенов. Такие вакцины могут реализовать широкий спектр действия за счет индукции Т-клеточного ответа на консервативные эпитопы, потенциально способные обеспечить защиту от различных штаммов патогенов (в том числе, вируса гриппа).
Вирусные векторы обладают способностью активировать врожденный иммунитет путем связывания генетического материала или белков их оболочки с паттерн-распознающими рецепторами (TLR, RIG-1 и др.). Вирусные векторы распознаются такими TLR, как TLR2, TLR3, TLR4, TLR7, TLR8, TLR9.
При взаимодействии этих рецепторов с лигандами активируются различные факторы транскрипции, что приводит к формированию очага воспаления и быстрой активации защитных реакций организма.
При выборе вирусного вектора для генетической иммунизации необходимо руководствоваться следующими критериями: такая вакцина не должна вызывать симптомов заболевания, она должна быть безопасной для людей с ослабленным иммунитетом, пожилых и детей; собственные белки рекомбинантного вируса не должны вызывать сильного иммунного ответа; вирусный вектор должен быть простым для генетических манипуляций и позволять включать большие фрагменты чужеродной ДНК; полученные препараты должны иметь высокий титр и обеспечивать высокий уровень экспрессии целевых антигенов; при иммунизации ДНК вирусного вектора не должна интегрировать в геном клетки-хозяина, и сам вектор должен полностью выводиться из организма после индукции иммунного ответа. Кроме того, нежелательно наличие предсуществующего иммунного ответа к белкам вирусного вектора у иммунизируемых индивидов, так как он может существенно снизить уровень иммунного ответа на целевой антиген.
Не все вирусы обладают свойствами, необходимыми для создания эффективных векторов. Для создания гриппозных вакцин на основе вирусных векторов в настоящее время наиболее широко используются поксвирусы, вирус болезни Ньюкасла и аденовирусы.
Поксвирусы (Poxviridae) - это ДНК-содержащие вирусы с большим геномом. Среди поксвирусов, используемых в качестве вирусного вектора, наиболее популярен вирус осповакцины, к преимуществам которого относятся простота и дешевизна получения, а также высокая пакующая емкость (до 25 т.п.н.). Для получения вакцин используют аттенуированные вирусы осповакцины, такие, как модифицированный вирус осповакцины Анкара (MVA) и аттенуированный штамм NYVAC, полученный на основе штамма Копенгаген. Аттенуирование MVA было достигнуто путем многократного пассирования в фибробластах куриных эмбрионов, что привело к потере ряда генов, не существенных для репликации в клетках птиц, и снижению репродукции в клетках человека. Аттенуация штамма NYVAC была достигнута путем делеции 18 генов, в результате чего вирус стал репликативнодефектным для клеток человека.
Серьезный недостаток векторов на основе вируса осповакцины - предсуществующий иммунитет к этому вирусу, который сформировался в человеческой популяции в результате иммунизации против оспы. Поэтому целесообразно использовать векторы на основе таких поксвирусов, как вирус оспы канарейки (Сапагурох) и вирус оспы домашней птицы (Flowpox), к которым в человеческой популяции отсутствуют предсуществующие антитела. Иммунизация кур и уток рекомбинантным Flowpox-BHpycoM, в геном которого введен ген НА вируса гриппа А птиц, защищала птиц от заражения летальными дозами гомологичных вирусов гриппа. Высокая пакующая емкость поксвирусов позволяет вводить в геном сразу несколько трансгенов, например, гены НА и NP вируса гриппа А. Однако Сапагурох и Flowpox индуцируют более слабый иммунный ответ на целевые антигены, чем вирус осповакцины, и требуют многократного введения или использования адъювантов.
Вирус болезни Ньюкасла (NDV) относится к семейству Paramyxoviridae. Это вирус с несегментированным одноцепочечным РНК-геном, который содержит шесть генов, кодирующих семь белков: NP, Р- и V-белки, М-белок, белок слияния, или F-белок, HA-NA и большой полимеразный белок L. Так как уровень экспрессии каждого вирусного белка снижается в направлении от 3'- до 5-конца генома, при использовании NDV в качестве вектора уровень экспрессии чужеродного гена можно контролировать по его положению в вирусном геноме. Уровень вирулентности и тропизм NDV зависят от сайта нарезания протеазами F-белка, необходимого для слияния вирусной оболочки и клеточной мембраны.
NDV в природе инфицирует только птиц, поэтому антитела к этому вирусу у человека отсутствуют. Следовательно, проблема предсуществующего иммунного ответа для этого вирусного вектора не стоит. Однако существенным недостатком вакцинного вектора NDV является то, что последствия введения рекомбинантных NDV недостаточно изучены, и не ясно, безопасны ли гриппозные вакцины на основе NDV для человека. Кроме того, NDV характеризуется низкой пакующей емкостью и сложностью получения векторов, несущих несколько целевых антигенов. Препаративные количества NDV получают в куриных эмбрионах, что, как отмечено выше, имеет ряд недостатков.
Рекомбинантные аденовирусы (Adenoviridae) - наиболее хорошо изученные и наиболее часто используемые рекомбинантные вирусные векторы. Вирионы аденовирусов состоят из молекулы двухцепочечной ДНК, окруженной белковым капсидом. Многие типы аденовирусов подробно охарактеризованы, в том числе и на генетическом уровне. Нуклеотидная последовательность геномов большинства из них полностью расшифрована. Подробные данные о структуре, физико-химических и биологических свойствах аденовирусов позволяют использовать их для создания рекомбинантных вакцин и генно-терапевтических препаратов. Среди генетических вакцин, проходящих клинические испытания, на долю вакцин на основе рекомбинантных аденовирусов приходится около 24%.
Аденовирусы обладают такими важными для вакцинных векторов свойствами, как способность обеспечивать высокий уровень экспрессии целевого трансгена в клетке-мишени и трансдуцировать как делящиеся, так и постмитотические клетки. При этом ДНК аденовируса остается во внехромосомной форме. Аденовирусы способны накапливаться в культуре клеток в высоких титрах. Процесс получения нового рекомбинантного аденовируса занимает несколько недель, что может позволить реагировать на меняющуюся эпидемиологическую обстановку в максимально сжатые сроки.
В настоящее время в различных странах ведутся разработки вакцин на основе рекомбинантных аденовирусов против различных серотипов вируса гриппа А. Одна из таких вакцин успешно прошла первую стадию клинических испытаний в США, оказалась безопасной для человека и высокоиммуногенной по отношению к вирусу гриппа A H5N2.
Проблема создания гриппозных вакцин, формирующих гетеросубтипический иммунитет, способный защитить от различных вариантов вируса гриппа, актуальна, и в последние годы появились новые подходы для ее разрешения. Выявлены консервативные для различных субтипов вируса гриппа А конформационные эпитопы НА, на которые могут вырабатываться антитела широкого спектра действия как после перенесенной инфекции, так и при иммунизации живыми вакцинами. Иммунизация рекомбинантными аденовирусными векторами имитирует заражение клеток слизистой оболочки верхних дыхательных путей, обеспечивая экспрессию антигенов с нативной третичной структурой, что позволяет индуцировать образование таких перекрестных антител. Рекомбинантные аденовирусные вакцины также способны формировать сильный Т-клеточный иммунный ответ, который характеризуется более широким, чем гуморальный, спектром действия.
Лентивирусы индуцируют широкое разнообразие патологий у различных видов животных. Общей чертой этих вирусов является способность поражать неделящиеся и дифференцированные клетки - чрезвычайно полезное свойство для целей генной терапии. Название лентивирусов (лат. lenti = медленный) связано с длительным промежутком времени между инфицированием и завершением болезни, который может длиться нескольких месяцев или даже лет. Вирусы, принадлежащие к роду Lentivirus, представлены у приматов, копытных и кошачьих.
В целом, мишенями лентивирусов являются разнообразные типы клеток, что предполагает потенциально широкое использование этих вирусов: от противораковых стратегий до вакцинации и противовирусного иммунитета. Поскольку лентивирусы воздействуют на неделящиеся клетки, они должны обладать механизмом преодоления интактной ядерной мембраны - для доступа к клеточному геному. Среди ретровирусов лентивирусы приобрели наиболее эффективный механизм для достижения этой цели, и это свойство используется в генной терапии [24].
Жизненный цикл вируса разделяют на две основные фазы: первая - перенос вирусного генома в клетку хозяина, а вторая - его экспрессия и размножение вируса. Главный интерес для целей генной терапии представляет первая фаза вместе с ранними этапами вирусного жизненного цикла. Такой процесс определяют терминами генная трансдукция (в генной терапии) или однораундовая инфекция (в вирусологии). В случае ретровирусов инфекция начинается после связывания специфического клеточного рецептора с белками вирусной оболочки, а завершается интеграцией синтезированной ДНК в геном клетки хозяина. Связывание клеточного рецептора и вирусного Env-белка запускает слияние мембраны клетки и оболочки вирусной частицы, в результате в цитоплазму клетки-мишени попадает вирусный нуклеопротеиновый комплекс (вирусный геном в комплексе с вирусными и клеточными белками). Проблемой инфицирования неделящихся клеток является преодоление ядерной мембраны для доступа вирусного генома к ДНК хозяина. Этот этап, ключевой для целей генной терапии, назван ядерным импортом. При вирусной инфекции ядерная мембрана является дополнительным препятствием, которое вирусы преодолевают либо путем перемещения во время деления клетки, либо путем преодоления ядерных пор. Последний вариант является единственным в случае неделящихся клеток, впрочем, механизм до сих пор не ясен [24].
Несмотря на то, что большинство ретровирусов способны инфицировать неделящиеся клетки, именно лентивирусы наиболее эффективны для осуществления ядерного импорта [24].
В основе развития лентивирусных векторов лежит тенденция к оптимизации эффективности генного переноса и, в то же время, уменьшению потенциальной опасности, обусловленной использованием ретровирусных векторов. В ходе оптимизации были разработаны третье и четвертое поколения векторов, характеризующихся минимальным риском образования способных к репликации рекомбинантов. А введение SIN-мутаций не только уменьшает вероятность образования таких рекомбинантов, но и снижает неспецифическую активность энхансеров вирусного промотора [24].
Дальнейшие усилия оптимизации конструкции лентивирусных векторов направлены на улучшение интеграции целевого гена в геном клетки-мишени. Ретровирусы имеют необычную способность размещать гетерологичные оболочечные белки на своей оболочке, которая называется псевдотипированием, в результате чего вирусные частицы могут приобретать новый клеточный тропизм и внутриклеточное поведение. Псевдотипирование характерно для всех ретровирусов, но наибольший интерес представляет псевдотипирование лентивирусов, способных к транедукции неделящихся и дифференцированных клеток [25]. Лентивирусные псевдотипированные векторы нарабатываются на культуре клеток млекопитающих путем ко-трансфекции упаковочными и собственно векторной плазмидами.
Другое свойство псевдотипированных лентивирусных векторов - это способность воздействовать на внутриклеточное поведение вируса. Для достижения эффективного воздействия на клетки в отсутствие главных сигналов активации был разработан ряд стратегий, основанных на искусственно сконструированных оболочках, отличных от VSVg. Общей чертой этих стратегий являются использование особых молекул, расположенных на поверхности вирусной частицы и запускающих сигнал активации при связывании с клеточным рецептором. Сигнал временно активирует клетку и позволяет инфицировать ее. Эта стратегия была успешно применена для трансдукции покоящихся В- и Т-клеток с помощью оболочечных белков вируса кори и лентивирусов с цитокинами на поверхности [26, 27].
Описан пример успешного использования лентивирусных векторов для ретроградного транспорта VEGF на модельных мышах с боковым амиотрофическим склерозом (ALS, болезнь Шарко) [28]. Болезнь Шарко - это заболевание, характеризующееся прогрессирующей дегенерацией мотонейронов в головном и спинном мозге. При лечении этого заболевания одним из важнейших затруднений является доставка терапевтического агента к мотонейронам, для чего была разработана лентивирусная система доставки гена непосредственно к нейтронам за счет тропизма оболочечных белков вируса бешенства. В рассматриваемом исследовании был создан вектор на основе вируса инфекционной анемии лошадей (EIAV), псевдотипированный белком G вируса бешенства. Модельным мышам с нейродегенерацией проводили инъекции препарата лентивирусного вектора, обеспечивающего экспрессию VEGF, в различные мышечные ткани до начала и при прогрессирующей дегенерации нейронов. По результатам испытаний экспрессия репортерного гена была зафиксирована в спинном и головном мозге, и заметное продление развития заболевания установлено для обеих экспериментальных групп. Таким образом, было показано, что лентивирусные векторы на основе EIAV, псевдотипированные оболочечными гликопротеинами G вируса бешенства, представляют собой эффективную систему доставки генов к мотонейронам при внутримышечной инъекции. Исследование также показало терапевтическое действие VEGF на модельных мышах: даже на стадии гибели половины мотонейронов, соответствующей обычной постановке диагноза, применение вирусного вектора для переноса VEGF обеспечивало не только продление жизни экспериментальных животных, но частичное восстановление функции. А микроскопические и гистологические исследования не выявили каких-либо патологий спинного и головного мозга после инъекции препарата, в том числе, аномальной васкуляризации тканей. В дополнение, иммунный ответ на систему доставки наблюдался незначительный даже через пять недель после инъекции.
Таким образом, открытия и исследования в области вирусологии позволяют расширять применение технологию лентивирусных псевдотипированных векторов для получения широкого разнообразия клеток-продуцентов терапевтических препаратов и, в частности, для целей генной терапии.
В последнее время использование неинфекционных вирусоподобных частиц (ВПЧ), которые спонтанно само собираются благодаря взаимодействию вирусных структурных белков, создало хорошую перспективу для усовершенствования вакцин против широкого спектра вирусов, вызывающих заболевания у человека [29]. ВПЧ гриппа, экспрессируемая рекомбинантными бакуловирусами, которые представляют многокомпонентные антигены, включающими гемагглютинин и матриксный белок M1, совместно или без нейраминидазы, способны индуцировать иммунную реакцию против гомологичных или гетерологичных штаммов вируса гриппа, что подробно описано [30, 31, 32, 33, 34, 35, 36]. Кроме того, метод обратной генетики, лицензированный в Европе для создания вакцин против сезонного гриппа, использует систему культивирования на основе клеток млекопитающих, а не в РКЭ [37]. Использование культур клеток млекопитающих, таких как Vero или MDCK, адаптивных хозяев для вакцинных вирусов, имеет ряд преимуществ, не только в связи с увеличением гибкости и согласованности процесса производства, но и в связи с возможностью гликозилирования вирусных антигенов в естественной форме, чего не может быть достигнуто в РКЭ или бакуловирусных системах. В эукариотических клетках гликозилирование белков участвует в правильной сборке и ориентации вновь транслируемых белков, что играет важную роль в функции белка. Генерация ВПЧ гриппа в клетках млекопитающих ранее была получена в результате транзиентной ко-экспрессии гемагглютинина и нейраминидазы в клетках 293Т человека [38]. С целью улучшения производственных характеристик в состав ВПЧ гриппа были включены не только белки гемагглютинина и нейраминидазы, но и белки матрикса M1 и М2 поскольку они играют важную роль в сборке вирусных частиц и их почковании [39, 40, 41, 42, 43, 44]. Включение белков M1 и М2 в ВПЧ гриппа, производимых в клетках млекопитающих, не только увеличивает выход продукции ВПЧ, но и дополняет антигенный портрет ВПЧ антигенами, с консервативными Т- и В-клеточными эпитопами, что позволяет индуцировать иммунитет не только против гомологичных, но и гетерологичных вирусов [45, 46].
Из RU 2335153 от 10.12.2014 известен концентрат вакцины против гриппа и способ его получения. Способ включает заражение куриных эмбрионов и сбор вируссодержащей аллантоисной жидкости, ее очистку микрофильтрацией, концентрирование и очистку, полученных концентратов.
Заявленное изобретение отличается от известных из уровня техники тем, что способ получения концентрата вакцины включает получение ВПЧ на основе штаммов А/Califomia/04/2009(H1N1), А/Novosibirsk/01/2014(H3N2) и В/Phuket/3073/2013. Таким образом, заявленное изобретение относится к способу получения концентрата вакцины от гриппа, содержащему вирусоподобные частицы (ВПЧ) вируса гриппа, построенные из поверхностных белков вируса гриппа: где гены гемагглютинина, нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/Michigan/45/2015, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/Novosibirsk/01/2014, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/HongKong/4801/2014, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма B/Phuket/3073/13, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009 или ген гемагглютинина получен из штамма B/Brisbane/60/2008, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009 и к концентрату поливалентной вакцины, содержащему такие ВПЧ.
Техническим результатом заявленного изобретения является получение ВПЧ вируса гриппа, имеющего большую гемагглютинирующую активность. Также способ позволяет повысить выход антигена.
Кроме того, техническим результатом является сохранение жизнеспособности и высокой продуктивности целевых белков без использования регулируемой экспрессии у клеток-продуцентов ВПЧ гриппа. Что является неожиданным. Специалист в уровне технике, напротив, мог предполагать, что из-за гиперэкспрессии белков вируса гриппа клетки не смогут проходить большое количество пассажей, необходимое для получения рабочего банка. Кроме того, поскольку из уровня техники известно, что при использовании аденовирусных векторов, несущих ген НА A/PR/8/34 (H1N1), при этом инфицирование клеток MDCK вообще регистрировалось только при использовании вектора, несущего ген из штамма гриппа A/PR/8/34 (H1N1); жизнеспособность и продуктивность клеток MDCK ниже других типов клеток млекопитающих (см. RU 2565546), сохранение жизнеспособности на протяжении многих пассажей является неочевидным для специалиста при использовании MDCK клеток.
Изобретение иллюстрируется следующими фигурами.
На Фиг. 1 отображена лентивирусная векторная плазмида, несущая кассету генов гемагглютинина штамма B, нейраминидазы штамма A (H1N1) и M1 белка штамма A (H1N1).
На Фиг. 2 представлена лентивирусная векторная плазмида, несущая кассету генов гемагглютинина штамма A (H3N2), нейраминидазы штамма A (H1N1) и M1 белка штамма A (H1N1).
Фиг. 3 иллюстрирует лентивирусный вектор экспрессии кассеты генов pLenti6.3/TO/HA(H1N1)-M1-NA, несущий кассету генов гемагглютинина штамма A (H1N1), нейраминидазы штамма A (H1N1) и M1 белка штамма A (H1N1).
Фиг. 4 отображает результат электрофоретического разделения в 1,5% геле агарозы амплифицированных фрагментов генов НА и М1.
1 - ПЦР-продукт для полноразмерного матриксного белка (777 п.н.);
2 - ПЦР-продукт для полноразмерного гемагглютинина (1719 п.н.);
М - ДНК маркер, длины в п.н. показаны на левой стороне.
Фиг. 5 иллюстрирует результат электрофоретического разделения в 1,5% геле агарозы амплифицированных фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pDrive_M1 SalI и EcoRI.
1, 12 - ПЦР-продукт М1 (777 п.н.);
2, 3, 4, 5, 6, 7, 8, 9, 10, 11 - независимые клоны плазмиды pDrive_M1 (3830 п.н. + 771 п.н.);
13 - базовая плазмида PDrive (3830 п.н. + 20 п.н.);
М - ДНК маркер, длины в п.н. показаны на левой стороне.
На Фиг. 6 представлен результат электрофоретического разделения в 1,5% геле агарозы амплифицированных фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pDrive_НА SalI и EcoRI.
1, 2, 3, 4, 5, 6 - независимые клоны плазмиды pDrive_HA (3830 п.н. + 1713 п.н.);
М - ДНК маркер, длины в п.н. показаны на левой стороне.
Фиг. 7 иллюстрирует результат электрофоретического разделения в 1,5% геле агарозы независимых клонов рекомбинантной плазмиды pEntry, несущей ген NA.
1 - базовая плазмида pEntry;
2, 3, 4, 5, 6 - независимые клоны плазмиды pEntry, несущей ген NA;
М - ДНК маркер, длины в п.н. показаны на левой стороне.
Фиг. 8 показывает результат электрофоретического разделения в 1,5% геле агарозы амплифицированных фрагментов ДНК НА (H3N2) и НА (В).
1, 2, 3 - три ПЦР-продукта для гемагглютинина H3N2 (607, 599 и 647 п.н.);
4, 5, 6 - три ПЦР-продукта для гемагглютинина В (646, 629 и 638 п.н.);
М - ДНК маркер, длины в п.н. показаны на левой стороне.
Фиг. 9 демонстрирует результат электрофоретического разделения в 1,5% геле агарозы амплифицированных фрагментов ДНК НА (H3N2).
1 - ПЦР-продукт для полноразмерного гемагглютинина H3N2 (1719 п.н.);
М - ДНК маркер, длины в п.н. показаны на левой стороне.
На Фиг. 10 отображен результат электрофоретического разделения в 1,5% геле агарозы фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pEntry, несущей кассету генов НА (N3N2), М1 и NA, эндонуклеазой рестрикции EcoRI (в скобках указаны длины образующихся фрагментов):
1 - базовая плазмида pEntry, несущая кассету генов НА (N1N1), М1 и НА (9875 п.н.);
2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 - независимые клоны плазмиды pEntry, несущей кассету генов НА (N3N2), М1 и NA (22 п.н. + 1060 п.н. + 3033 п.н. + 5760 п.н.);
М - ДНК маркер, длины в п.н. показаны на левой стороне.
На Фиг. 11 показан результат электрофоретического разделения в 1,5% геле агарозы независимых клонов рекомбинантной плазмиды pLenti, содержащей кассету генов HA, M1 и NA:
1, 2, 3, 4, 6, 7, 8, 10,11 - базовая плазмида pLenti6.3/TO/V5-DEST;
5, 9 - независимые клоны плазмиды pLenti, содержащей кассету генов HA, M1 и NA;
М - ДНК маркер, длины в п.н. показаны на левой стороне.
На Фиг. 12 можно увидеть результат электрофоретического разделения в 1,5% геле агарозы фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pLenti, содержащей кассету генов HA (H1N1), M1 и NA, энедонуклеазами рестрикции Xhol, EcoRI, PstI и HindIII (длины образующихся фрагментов указаны в Таблице 2):
1, 2 - независимые клоны плазмиды pLenti, содержащей кассету генов HA (H1N1), M1 и NA, гидролизированной XloI;
3, 4 - независимые клоны плазмиды pLenti, содержащей кассету генов HA (H1N1), M1 и NA, гидролизированной PstI;
5, 6 - независимые клоны плазмиды pLenti, содержащей кассету генов HA (H1N1), M1 и NA, гидролизированной EcoRI;
7, 8 - независимые клоны плазмиды pLenti, содержащей кассету генов HA (H1N1), M1 и NA, гидролизированной HindIII;
М - ДНК маркер, длины в п.н. показаны на левой стороне.
На Фиг. 13 можно увидеть как в ядре клеток MDCK (верхний рисунок) обнаруживаются электронно-плотные включения (стрелка), придающие ему «пятнистый» вид, характерный для пораженных вирусом гриппа клеток. Продукция ВПЧ в клетках НЕК 293FT значительно менее выражена по сравнению с клетками MDCK (нижний рисунок).
На Фиг. 14 представлены основные морфологические формы вирусоподобных частиц: сферические (белая стрелка) и нитевидные (черная стрелка) в клетках MDCK.
Фиг. 15 иллюстрирует разнообразные формы вирусоподобных частиц (стрелки) в клетках MDCK.
Фиг. 16 иллюстрирует апоптоз клетки MDCK. Многочисленные ВПЧ вокруг и на поверхности гибнущей клетки.
На Фиг. 17 А и 17 Б можно наблюдать экспрессию гемагглютинина НА клонами-кандидатами (иммуноблотинг). На Фиг. 17 А изображены клоны клеток MDCK 1F1, 3C5, 5E4 и 5G7. Отрицательный контроль - лизат клеток MDCK. Фиг. 17 Б демонстрирует клоны клеток MDCK 6D6, 6D12, 7F4, 9B3, 10C12, 10E7. Отрицательный контроль - лизат клеток MDCK.
Фиг. 18А и 18Б иллюстрируют экспрессию NA клонами-кандидатами (иммуноблотинг). На Фиг. 18Б изображены клоны клеток MDCK 6D6, 6D12, 7F4, 9B3, 10C12, 10E7. Отрицательный контроль - лизат клеток MDCK.
Фиг. 19А и 19Б показывает экспрессию М1 клонами-кандидатами (иммуноблотинг). На Фиг. 19А изображены клоны клеток MDCK 1F1, 3C5, 5E4, 5G7 и 6D6. Отрицательный контроль - лизат клеток MDCK. Фиг. 19Б демонстрирует клоны клеток MDCK 6D12, 7F4, 9B3, 10C12, 10E7. Отрицательный контроль - лизат клеток MDCK.
На Фиг. 20 можно наблюдать схематическое изображение плазмидной ДНК pLenti6.3/TO/V5-DEST. (https://tools.thermofisher.com/content/sfs/vectors/plenti6_3_to_v5_dest_map.pdf)
На Фиг. 21 представлен профиль элюции ВПЧ в колонке Mustang Q.
Образцы фильтрата брались через каждые 400 мл фильтрации для определения показателей потока. Целью определения показателей потока был контроль перегрузки Mustang Q, что приводит к потере материала во время промывки или фильтрации.
На Фиг. 22 изображен профиль элюции материала из Mustang Q с помощью ЭХ.
На Фиг. 23 - профиль Cellufine S обработки пикового материала, полученного с помощью ЭХ.
На Фиг. 24 отображен профиль элюции с помощью Cellufine S материала с максимальным значением Vo. А = загрузка 0,5 мл. В = загрузка 1 мл. С = загрузка 2 мл.
Фиг. 25 иллюстрирует электрофорез отдельных стадий. Верхнее изображение относится к ВПЧ H1N1M1, демонстрируя исходный вид, после ультрафильтрации, концентрирование на Mustang Q, проскок после Mustang Q, гельфитрацию, аффинную хроматографию, контроль БСА (70 kDa) + лизоцим (15 kDa).
Среднее изображение относится к ВПЧ H3N1M1, демонстрируя исходный вид, концентрат после ионообменной хроматографии (Mustang Q), проскок после ионообменной хроматографии, гельфильтрацию, аффинную хроматографию, контроль БСА (70 kDa) + лизоцим (15 kDa).
Нижнее изображение относится к ВПЧ HA(В)N1M1, демонстрируя вид после фильтрации, концентрат после ионообменной хроматографии (Mustang Q), концентрат на концентраторах Millipore, аффинную хроматографию, контроль БСА (70 kDa) + лизоцим (15 kDa).
На Фиг. 26 отображены результаты очистки трех типов ВПЧ в одном электрофорезе:
- ВПЧ H1N1M1 после всех стадий очистки;
- ВПЧ H3N1M1 после всех стадий очистки;
- ВПЧ HA(В)N1M1 после всех стадий очистки;
- Человечий сывороточный альбумин;
- ЧСА + альфа 2-интерферон.
Фиг. 27 отображает результаты анализа концентрата очищенного ВПЧ H1N1M1 методом электронной микроскопии.
Фиг. 28 отображает результаты анализа концентрата очищенного ВПЧ H3N1M1 методом электронной микроскопии.
Фиг. 29 отображает результаты анализа концентрата очищенного ВПЧ HA(B)N1M1 методом электронной микроскопии.
Фиг. 30 демонстрирует результат электрофоретического разделения в 1.2 % геле агарозы продуктов LR рекомбинации между плазмидными ДНК pEntry_HA(H1N1-Mich)-M1-NA и pLenti6.3/TO/V5-DEST.
1, 2, 3, 5, 6, 7, 8, 11, 12, 13 - нецелевые продукты рекомбинации;
4, 9 - целевой продукт рекомбинации pLenti6.3/TO/HA(H1N1-Mich)-M1-NA;
М - 1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия)
На Фиг. 31 представлена карта рекомбинантной плазмидной ДНК pLenti6.3/TO/HA(A/HongKong)-M1-NA.
Основные генетические элементы, содержащиеся в структуре плазмидной ДНК:
IRES - участок внутренней посадки рибосомы;
WPRE - Woodchuck Posttranscriptional Regulatory Element, регуляторный элемент для увеличения экспорта мРНК из ядра и усиления трансгенной экспрессии;
CMV/TO - гибридный промотор, состоящий из цитомегаловирусного промотора и двух тандемных тетрациклиновых оператора, для регулируемой высокоэффективной экспрессии целевых генов;
НА (A-HongKong) - ген гемагглютинина штамма A/Hong Kong/4801/2014 (H3N2);
NA - ген нейраминидазы штамма A/California/04/2009(H1N1);
M1 - ген M1 штамма A/California/04/2009(H1N1).
На Фиг. 32 - карта рекомбинантной плазмидной ДНК pLenti6.3/TO/HA(B-Brisbane)-M1-NA.
Основные генетические элементы, содержащиеся в структуре плазмидной ДНК:
IRES - участок внутренней посадки рибосомы;
WPRE - Woodchuck Posttranscriptional Regulatory Element, регуляторный элемент для увеличения экспорта мРНК из ядра и усиления трансгенной экспрессии;
CMV/TO - гибридный промотор, состоящий из цитомегаловирусного промотора и двух тандемных тетрациклиновых оператора, для регулируемой высокоэффективной экспрессии целевых генов;
НА (B- Brisbane) - ген гемагглютинина штамма B/Brisbane/60/2008;
NA - ген нейраминидазы штамма A/California/04/2009(H1N1);
M1 - ген M1 штамма A/California/04/2009(H1N1).
На Фиг. 33 отображена карта рекомбинантной плазмидной ДНК pLenti6.3/TO/HA(H1N1-Mich)-M1-NA.
Основные генетические элементы, содержащиеся в структуре плазмидной ДНК:
IRES - участок внутренней посадки рибосомы;
WPRE - Woodchuck Posttranscriptional Regulatory Element, регуляторный элемент для увеличения экспорта мРНК из ядра и усиления трансгенной экспрессии;
CMV/TO - гибридный промотор, состоящий из цитомегаловирусного промотора и двух тандемных тетрациклиновых оператора, для регулируемой высокоэффективной экспрессии целевых генов;
НА (H1N1- Mich) - ген гемагглютинина штамма A/Michigan/45/2015;
NA - ген нейраминидазы штамма A/California/04/2009(H1N1);
M1 - ген M1 штамма A/California/04/2009(H1N1).
На Фиг. 34 по оси ординат отображаются показатели показателя (мг глюкозы/г диска)/ч; по оси абцисс - даты забора образцов.
Даты измерения параметров:
• 30 марта = помещение клеток в биореактор
• 01 апреля = замена на бессывороточную питательную среду OptiMEM
• 18 апреля = температура уменьшена с 37°С до 34°С
 Изменения привели к снижению потребления глюкозы (как и ожидалось), но с течением времени оно снова возросло
Изменения привели к снижению потребления глюкозы (как и ожидалось), но с течением времени оно снова возросло
• 18 апреля = снижение СО2 с 5% до 2,5%
Потребление глюкозы не изменилось. Концентрация СО2 изменялась с целью лучшего контроля рН
• 24 апреля = потребление глюкозы незначительно изменилось
• В этих условиях активность НА была обнаружена через 3 недели после посева.
На Фиг. 35 отображен результат электрофоретического разделения в 1.5 % геле агарозы фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, эндонуклеазами рестрикции XhoI, EcoRI, PstI и HindIII (в скобках указаны длины образующихся фрагментов).
1 - плазмида pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, гидролизованная XhoI (13496 п.н.);
2 - плазмида pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, гидролизованная EcoRI (9091 п.н. + 4405 п.н.);
3 - плазмида pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, гидролизованная HindIII (5580 п.н. + 3344 п.н. + 2639 п.н. + 584 п.н. + 556 п.н. + 481 п.н. + 312 п.н.);
4 - плазмида pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, гидролизованная PstI (11686 п.н. + 1810 п.н.).
Фиг. 36 иллюстрирует результат электрофоретического разделения в 1.2 % геле агарозы независимых клонов рекомбинантной плазмиды pEntry_HA(В-Brisbane)-M1-NA, содержащей кассету генов HA (B/Brisbane/60/2008), M1 (California/04/2009) и NA (California/04/2009).
1, 2, 3, 4, 5 - независимые клоны плазмиды pEntry_HA(В-Brisbane)-M1-NA, несущие кассету генов HA (B/Brisbane/60/2008), M1(California/04/2009) и NA (California/04/2009);
М - 1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия)
На Фиг. 37 показан результат электрофоретического разделения в 1.2 % геле фрагментов ДНК, полученных после гидролиза независимых клонов рекомбинантной плазмиды pEntry_HA(В-Brisbane)-M1-NA, содержащей кассету генов HA (B/Brisbane/60/2008), M1 (California/04/2009) и NA (California/04/2009), эндонуклеазами рестрикции SalI и PstI (в скобках указаны длины образующихся фрагментов).
1, 2, 3, 4, 5 - независимые клоны плазмиды pEntry_HA(В-Brisbane)-M1-NA, несущие кассету генов HA (B/Brisbane/60/2008), M1(California/04/2009) и NA (California/04/2009) (8164 п.н. + 1768 п.н.);
М - 1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия).
На Фиг. 38 отображен результат электрофоретического разделения в 1.2 % геле агарозы продуктов LRрекомбинации между плазмидными ДНК pEntry_HA(В-Brisbane)-M1-NA и pLenti6.3/TO/V5-DEST.
1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 13 - нецелевые продукты рекомбинации;
11 - целевой продукт рекомбинации pLenti6.3/TO/HA(В-Brisbane)-M1-NA;
М -1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия).
На Фиг. 39 отображен результат электрофоретического разделения в 1,5 % геле агарозы фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pLenti6.3/TO/HA(B-Brisbane)-M1-NA, эндонуклеазами рестрикции XhoI, EcoRI, PstI и HindIII(в скобках указаны длины образующихся фрагментов).
1 - плазмида pLenti6.3/TO/HA(B-Brisbane)-M1-NA, гидролизованная HindIII (5580 п.н. + 3344 п.н. + 2639 п.н. + 584 п.н. + 556 п.н. + 481 п.н. + 312 п.н.);
2 - плазмида pLenti6.3/TO/HA(B-Brisbane)-M1-NA, гидролизованная EcoRI (9091 п.н. + 4405 п.н.);
3 - плазмида pLenti6.3/TO/HA(B-Brisbane)-M1-NA, гидролизованная XhoI (13496 п.н.);
4 - плазмида pLenti6.3/TO/HA(B-Brisbane)-M1-NA, гидролизованная PstI (11686 п.н. + 1810 п.н.);
М -1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия).
На Фиг. 40 отображен результат электрофоретического разделения в 1.2 % геле фрагментов ДНК, полученных после гидролиза независимых клонов рекомбинантной плазмиды pEntry_HA(HongKong/4801)-M1-NA, содержащей кассету генов HA (HongKong/4801), M1 (California/04/2009) и NA (California/04/2009), эндонуклеазами рестрикции SalI и PstI (в скобках указаны длины образующихся фрагментов).
1, 2, 3, 4, 5 - независимые клоны плазмиды pEntry_HA(HongKong/4801)-M1-NA, несущие кассету генов HA (HongKong/4801), M1(California/04/2009) и NA (California/04/2009) (8164 п.н. + 1711 п.н.);
М - 1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия).
Фиг. 41 демонстрирует результат электрофоретического разделения в 1.2 % геле агарозы продуктов LR рекомбинации между плазмидными ДНК pEntry_HA(HongKong/4801)-M1-NA и pLenti6.3/TO/V5-DEST.
1, 2, 3 - нецелевые продукты рекомбинации;
4, 5, 6, 7 - целевой продукт рекомбинации pEntry_HA(HongKong/4801)-M1-NA;
М.1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия)
Фиг. 42 иллюстрирует результат электрофоретического разделения в 1.5 % геле агарозы фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pLenti6.3/TO/HA(A/HongKong)-M1-NA, эндонуклеазами рестрикции XhoI, EcoRI, PstI и HindIII (в скобках указаны длины образующихся фрагментов).
1 - плазмида pLenti6.3/TO/HA(A/HongKong)-M1-NA, гидролизованная PstI (11682 п.н. + 1761 п.н.);
2 - плазмида pLenti6.3/TO/HA(A/HongKong)-M1-NA, гидролизованная HindIII (5576 п.н. + 3344 п.н. + 2590 п.н. + 584 п.н. + 556 п.н. + 481 п.н. + 312 п.н.);
3 - плазмида pLenti6.3/TO/HA(A/HongKong)-M1-NA, гидролизованная EcoRI (9087 п.н. + 4356 п.н.);
4 - плазмида pLenti6.3/TO/HA(A/HongKong)-M1-NA, гидролизованная XhoI (8063 п.н. + 5380 п.н.);
М - 1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия).
На Фиг. 43 отображен результат электрофоретического разделения в 1.2 % геле агарозы независимых клонов рекомбинантной плазмиды pEntry_HA(H1N1-Mich)-M1-NA, содержащей кассету генов HA (A/Michigan/45/2015(H1N1)pdm09), M1 (California/04/2009) и NA (California/04/2009).
1, 2, 3, 4, 5, 6 - независимые клоны плазмиды pEntry_HA(H1N1-Mich)-M1-NA, несущие кассету генов HA(A/Michigan/45/2015(H1N1)pdm09), M1(California/04/2009) и NA (California/04/2009);
М - 1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия).
На Фиг. 44 представлен результат электрофоретического разделения в 1.2 % геле фрагментов ДНК, полученных после гидролиза независимых клонов рекомбинантной плазмиды pEntry_HA(H1N1-Mich)-M1-NA, содержащей кассету генов HA (A/Michigan/45/2015(H1N1)pdm09), M1 (California/04/2009) и NA (California/04/2009), эндонуклеазами рестрикции SalI и PstI (в скобках указаны длины образующихся фрагментов).
1, 2, 3, 4, 5 - независимые клоны плазмиды pEntry_HA(H1N1-Mich)-M1-NA, несущие кассету генов HA (A/Michigan/45/2015(H1N1)pdm09), M1(California/04/2009) и NA (California/04/2009) (8164 п.н. + 1711 п.н.);
М - 1 KbДНК маркер, длины в п.н. показаны на левой стороне («СибЭнзим», Россия).
Также изобретение иллюстрируется следующим списком последовательностей:
SEQ ID NO: 1 - Последовательность гемагглютинина H1 ДНК
SEQ ID NO: 2 - Последовательность нейраминидазы N1 ДНК
SEQ ID NO: 3 - Последовательность белка M1 ДНК
SEQ ID NO: 4 - Последовательность нуклеиновых кислот гемагглютинина НА (H3N2) из штамма A/Novosibirsk/01/2014 (H3N2)
SEQ ID NO: 5 - Последовательность нуклеиновых гемагглютинина вируса гриппа В из штамма B/Phuket/3073/2013
SEQ ID NO: 6 - Нуклеотидная последовательность искусственного гена гемагглютинина штамм A/HongKong/4801/2014(H3N2)
SEQ ID NO: 7 - Нуклеотидная последовательность искусственного гена гемагглютинина штамм B/Brisbane/60/2008
SEQ ID NO: 8 - Нуклеотидная последовательность искусственного гена гемагглютинина штамма A/Michigan/45/2015(H1N1)pdm09
ПРИМЕРЫ
Для разработки клеток-продуцентов был выбран вектор с тетрациклин регулируемой экспрессией целевых генов. Технология тетрациклин регулируемой экспрессии целевого гена в клетках млекопитающих основана на связывании тетрациклина с Tet репрессором, что приводит к дерепрессии промотора CMV/TO, контролирующего экспрессию целевого гена. Гибридный промотор CMV/TO содержит CMV промотор и два тетрациклиновых оператора. Если в клетке идет экспрессия тетрациклинового репрессора, то последний блокирует тетрациклиновый оператор. После введения тетрациклина блокируется сам тетрациклиновый репрессор, и тетрациклиновый оператор деблокируется, обеспечивая экспрессию целевого гена.
Однако при исследовании свойств клеток-продуцентов ВПЧ гриппа неожиданно оказалось, что они сохраняли жизнеспособность и высокую продуктивность целевых белков без использования регулируемой экспрессии. Поэтому в разработанных нами клетках-продуцентах белков вируса гриппа тетрациклиновый репрессор не вводился и тетрациклиновый оператор, хотя и присутствует в конструкции вектора, но деблокирован и не участвует в регуляции экспрессии. Наличие подобного элемента в конструкции вектора позволит при необходимости модифицировать клетку-продуцент и обеспечить тетрациклин регулируемую экспрессию целевых генов, если возникнет такая необходимость.
ПРИМЕР 1. Метод получения лентивирусных векторов
1.1. Вирусы, бактериальные штаммы
В работе использовали бактериальные штаммы Escherichia coli XL2-Blue {recAl endAl gyrA96 thi-1 hsdR17 supE44 relAl lac[F' proAB lacqZΔM15 Tn10(Tet') Amy Cam']c,d} и Stb13 {F mcrB mrrhsdS20(rB'-, mB') recA13 supE44 ara-14 galK2 lacY1 proA2 rpsL20 (StrR)xyl- 5 Kleumtl-1}; вирус гриппа штаммов А и Виз коллекции ФБУН ГНЦ ВБ "Вектор".
1. 2. Выделение вирусной РНК
Вирусную РНК выделяли с помощью набора QIAamp MinElute Virus Spin Kit (QLAGEN, США) в соответствии с рекомендациями производителя. К 200 мкл вируссодержащей жидкости добавляли 25 мкл Протеазы и 200 мкл раствора AL, перемешивали и помещали в термостат при 56°С на 15 минут. Затем добавляли 250 мкл этанола (96 %) и перемешивали. Раствор наносили на сорбирующий слой колонки и центрифугировали при 6000 g в течение 1 минуты в центрифуге типа "Эппендорф". Элюат удаляли и сорбирующий слой колонки промывали 500 мкл раствора AW1 центрифугированием при 6000 g в течение 1 минуты в центрифуге типа "Эппендорф". Элюат удаляли и сорбирующий слой колонки промывали 500 мкл раствора AW2 центрифугированием при 6000 g в течение 1 минуты в центрифуге типа "Эппендорф". Элюат удаляли и сорбирующий слой колонки промывали 500 мкл этанола (96%) центрифугированием при 6000 g в течение 1 минуты в центрифуге типа "Эппендорф" с последующим дополнительным центрифугированием при 13000 g в течение 3 минут. РНК- материал элюировали с колонки 100 мкл раствора АЕ центрифугированием при 13000 g в течение 1 минуты в центрифуге типа "Эппендорф".
1.3. ПЦР и ОТ-ПЦР
Для проведения ПЦР реакционная смесь, объемом 50 мкл, содержала буфер для проведения ПЦР (СибЭнзим, Россия) (60 мМ Tris-HCl рН 8.5, 25 мМ КС1, 1.5 мМ MgCb, ЮмМ 2-меркаптоэтанол, 0.1% Тритон Х-100), 0.2 мМ каждого dNTP, олигонуклеотидные праймеры в количестве 20 пмоль каждого, 5 ед.а. Taq-полимеразы, 1 мкл раствора ДНК. Реакцию проводили в программируемом термостате GeneAmp PCR-system 6700 с использованием следующей программы:
ОТ-ПЦР проводили с помощью набора Qiagen OneStep RT-PCR Kit (Qiagen, США) в соответствии с рекомендациями производителя. Реакционная смесь, объемом 50 мкл, содержала lxQIAGEN OneStep RT-PCR Buffer, 0.4 мМ каждого dNTP, олигонуклеотидные праймеры в количестве 30 пмоль каждого, 2.0 мкл QIAGEN OneStep RT-PCR Enzyme Mix, 5 мкл вирусной РНК. Реакцию проводили в программируемом термостате GeneAmp PCR- system 6700 с использованием следующей программы:
1.4. Электрофоретический анализ нуклеиновых кислот в агарозном геле
Электрофоретический анализ фрагментов ДНК проводили в горизонтальных пластинах при напряжении 6-10 V/см в течение 1 часа, используя 1.0-2.0% агарозный гель в буфере ТАЕ. Гель окрашивали бромистым этидием (0.2 мкг/мл) и фотографировали при помощи системы визуализации изображений Image Station 440CF (Kodak, США).
1.5. Элюция фрагментов ДНК из агарозного геля
Элюцию фрагментов ДНК из агарозного геля проводили при помощи набора Gel Extraction Kit (Qiagen, США) в соответствии с рекомендациями производителя. К вырезанной полосе агарозного геля, содержащей нужный фрагмент ДНК, добавляли трехкратный объем раствора QG для расплавления агарозы, перемешивали и помещали в термостат при 50°С на 10 минут до полного растворения геля. Затем добавляли один объем изопропанола и перемешивали. Раствор наносили на сорбирующий слой колонки и центрифугировали при 13000 g в течение 1 минуты в центрифуге типа "Эппендорф". Элюат удаляли и сорбирующий слой колонки промывали 750 мкл раствора РЕ центрифугированием при 13000 g в течение 1 минуты в центрифуге типа "Эппендорф". ДНК-материал элюировали с колонки 10-50 мкл раствора ТЕ центрифугированием при 13000 g в течение 1 минуты в центрифуге типа "Эппендорф".
1. 6. Лигирование
Реакционная смесь, объемом 20 мкл, содержала 0.1 мкг векторной плазмидной ДНК и 0.5 мкг фрагмента ДНК, гидролизованных соответствующими рестриктазами (вектор и фрагмент, после рестрикции были очищены с помощью Gel Extraction Kit (раздел 1.6.)), 50 ед.а. ДНК-лигазы фага Т4 (СибЭнзим, Россия), 2 мкл лигазного буфера 10-кратной концентрации (СибЭнзим, Россия). Реакционную смесь инкубировали в течение 6 часов при 16°С.
1.7. LR рекомбинация
Реакционная смесь, объемом 10 мкл, содержала 0.15 мкг векторной плазмидной ДНК pLenti6.3/TO/V5-DEST и 0.15 мкг векторной плазмидной ДНК pEntry, содержащей целевой фрагмент ДНК, 2 мкл Gateway LR Clonase II Plus Enzyme Mix. Реакционную смесь инкубировали в течение 2 часов при 25°С. Далее к реакционной смеси добавляли 1 мкл Протеиназы К с последующей инкубацией в течение 10 минут при 37°С.
1.8. Приготовление компетентных клеток E.coli
Индивидуальную колонию, выращенную на чашке Петри на агаризованной среде, засевали в ночь в 5 мл JLB-бульона. Затем 500 мкл полученной ночной культуры добавляли в 50 мл свежего LB-бульона и инкубировали при 37°С до оптической плотности Ds65 = 0.8 - 0.85 при интенсивной аэрации. Полученную суспензию клеток охлаждали во льду в течение 30 минут, затем осаждали центрифугированием при 3000 об/мин при 4°С в течение 10 минут в роторе JA-20 в центрифуге J2-21 (Beckman, США). Супернатант тщательно удаляли, осадок ресуспендировали в 15 мл буфера RF-1 и инкубировали во льду в течение 15 минут. Клетки осаждали центрифугированием при 3000 об/мин при 4°С в течение 10 минут в роторе JA-20 в центрифуге J2-21, после чего супернатант тщательно удаляли. Клетки ресуспендировали в 3 мл буфера RF-2 и инкубировали во льду 10 минут. Затем клетки расфасовывали в 1.5 мл пробирки по 200 мкл и хранили в кельвинаторе при минус 70°С.
1.9. Трансформация компетентных клеток E.coli
Компетентные клетки объемом 200 мкл размораживали во льду, добавляли 20 мкл лигазной смеси или 10 мкл продукта LR рекомбинации, или 5 мкл раствора, содержащего плазмидную ДНК в концентрации 0.1 мкг/мл, аккуратно перемешивали и инкубировали 30 минут во льду. Далее клетки подвергали тепловому шоку. Для этого микропробирки помещали в микротермостат с температурой 42°С на 2 минуты, затем охлаждали до 0°С на льду в течение 5 минут. Затем клетки рассевали на чашках Петри с соответствующим антибиотиком.
1.10. Выделение плазмидной ДНК в аналитическом варианте
Плазмидную ДНК очищали с помощью набора QIAprep Spin Miniprep Kit (Qiagen, США) в соответствии с рекомендациями производителя. 3 мл ночной культуры осаждали центрифугированием при 6800 g в течение 3 минут. Супернатант удаляли, осадок ресуспендировали в 250 мкл раствора Р1. Затем к лизату добавляли 250 мкл раствора Р2, аккуратно перемешивали и инкубировали 5 минут при комнатной температуре. После этого к суспензии добавляли 350 мкл раствора N3, аккуратно перемешивали и осаждали при 13000 g в течение 10 минут. Супернатант наносили на сорбирующий слой колонки и центрифугировали при 13000 g в течение 1 минуты в центрифуге типа "Эппендорф". Элюат удаляли и сорбирующий слой колонки промывали 500 мкл раствора РВ центрифугированием при 13000 g в течение 1 минуты в центрифуге типа "Эппендорф". Элюат удаляли и сорбирующий слой колонки промывали 750 мкл раствора РЕ центрифугированием при 13000 g в течение 1 минуты в центрифуге типа "Эппендорф". ДНК-материал элюировали с колонки 50 мкл раствора ЕВ центрифугированием при 13000 g в течение 1 минуты в центрифуге типа "Эппендорф".
1.11. Гидролиз ДНК эндонуклеазами рестрикции
Реакционная смесь содержала 0.1-5.0 мкг ДНК в объеме из расчета не менее 10 мкл на 1 мкг ДНК, эндонуклеазу рестрикции из расчета 1-5 е.а. на 1 мкг ДНК, 1/10 от общего объема реакционной смеси соответствующего буфера 10-кратной концентрации (СибЭнзим, Россия). Гидролиз проводили при 37°С в течение 2-4 часов.
1.12. Секвенирование ДНК
Реакцию секвенирования проводили с использованием набора BigDye 3.1 (Applied Biosystems, США) в соответствии с указаниями фирмы производителя. Каждая реакционная смесь объемом 5 мкл содержала следующие компоненты: 2 мкл раствора из набора для проведения секвенирующей реакции, 5 пкмоль олигонуклеотидного праймера и 0.5 мкг ДНК. Реакцию проводили в программируемом термостате GeneAmp PCR-system 6700 с использованием следующей программы:
После амплификации реакционную смесь очищали от не включившихся флуоресцентно меченных нуклеотидов очисткой на сефадексе G-50 superfine. Секвенирование проводили по обеим цепям ДНК. Расшифровку первичных данных секвенирования (хроматограмм) проводили с помощью программы Sequencher 4.0.5. (Gene Codes, США).
1.13. Очистка плазмидной ДНК от эндотоксинов
Плазмидную ДНК очищали с помощью набора EndoFree Plasmid Maxi Kit (Qiagen, США) в соответствии с рекомендациями производителя. 100 мл ночной культуры осаждали центрифугированием при 6000 g при 4°С в течение 15 минут. Супернатант удаляли, осадок ресуспендировали в 10 мл раствора Р1. Затем к лизату добавляли 10 мл раствора Р2, аккуратно перемешивали и инкубировали 5 минут при комнатной температуре. После этого к суспензии добавляли 10 мл раствора РЗ для выделения плазмидной ДНК, аккуратно перемешивали, переносили лизат в "QIAfilter Cartidge" и инкубировали 10 минут при комнатной температуре с последующим фильтрованием лизата. К осветленному лизату добавляли 2.5 мл раствора ER для удаления эндотоксинов, перемешивали и инкубировали во льду в течение 30 минут. Далее лизат переносили на предварительно уравновешенную 10 мл раствора QBT колонку "QLAGEN-tip 500". После освобождения колонки самотеком, ее промывали 60 мл раствора QC. Очищенную ДНК элюировали с колонки 15 мл раствора QN в чистую пробирку, добавляли 10.5 мл изопропанола, перемешивали и осаждали при 15000 g при 4°С в течение 30 минут. Осадок промывали 70% этанолом и осаждали при 15000 g при 4°С в течение 10 минут. Далее осадок высушивали при комнатной температуре и растворяли в подходящем объеме раствора ТЕ. Концентрация плазмидной ДНК была измерена спектрофотометрически на приборе Ultrospec 3000 pro (GE Healthcare Life Sciences, США).
ПРИМЕР 2. Протоколы получения векторов
2.1. Дизайн и синтез кассеты генов НА, Ml и NA
Дизайн кассеты генов для последующего получения соответствующего лентивирусного вектора. Схематично кассета генов состоит из следующих элементов:

attL1 и attL2 - необходимые сайты в векторной плазмиде pEntry для переноса генетического материала в векторную плазмиду pLenti6.3/TO/V5-DEST по сайтам attRl и attR2 с помощью LR рекомбинации;
IRES - участок внутренней посадки рибосомы;
WPRE - Woodchuck Posttranscriptional Regulatory Element, регуляторный элемент для увеличения экспорта мРНК из ядра и усиления трансгенной экспрессии;
CMV/TO - гибридный промотор, состоящий из цитомегаловирусного промотора и двух тандемных тетрациклиновых операторов, для регулируемой высокоэффективной экспрессии целевых генов;
SalI/PstI - сайты узнавания соответствующих эндонуклеаз рестрикции для замены НА в кассете генов при необходимости;
EcoRI/MluI - сайты узнавания соответствующих эндонуклеаз рестрикции для замены NA в кассете генов при необходимости;
XmaI/XmaI - сайты узнавания соответствующих эндонуклеаз рестрикции для удаления NA из кассеты генов при необходимости;
AgeI/XmaI - сайты узнавания соответствующих эндонуклеаз рестрикции для удаления M1 и NA из кассеты генов при необходимости;
НА - гемагглютинин вируса гриппа с последовательностью Козак и стоп-кодонами;
M1 - матриксный белок вируса гриппа с последовательностью Козак и стоп-кодонами;
NA - нейраминидаза вируса гриппа с последовательностью Козак и стоп-кодонами.
Таким образом, в разработанных конструкциях векторов после рекомбинации кассеты генов вируса гриппа в состав pLenti6.3/TO/V5-DEST, гены гемагглютинина и матриксного белка вируса гриппа экспрессируются с гибридного промотора CMV/TO, находящегося в составе векторной плазмиды, а ген нейраминидазы вируса гриппа экспрессируется с гибридного промотора CMV/TO, находящегося в составе кассеты генов. При этом мРНК с генами НА и M1 кэпирована регуляторным элементом WPRE, находящимся в составе кассеты генов, а мРНК с геном NA кэпирована регуляторным элементом WPRE, находящимся в составе векторной плазмиды. Трансляция матриксного белка осуществляется с участка внутренней посадки рибосомы. Тетрациклин зависимая регуляция экспрессии целевых генов отсутствует.
Известно, что оптимизация кодонового состава генов может увеличить скорость синтеза белка на соответствующей мРНК и, как следствие, увеличить уровень синтеза целевого белка в клетках млекопитающих. Поэтому, гены НА, M1 и NA, входящие в состав кассеты генов, были рассчитаны с модификацией кодонового состава, оптимизированной для экспрессии в клетках млекопитающих.
Кассета генов была синтезирована в ЗАО «Евроген Ру» в составе плазмиды pEntry, подходящей для LR рекомбинации с плазмидной ДНК pLenti6.3/TO/V5-DEST.
2.2. Получение и анализ рекомбинантных плазмид pEntry, содержащих индивидуальные гены HA (H1N1), M1 и NA
Для создания рекомбинантных молекул ДНК, содержащих гены НА, M1 и NA вируса гриппа, на первом этапе работы был произведен расчет олигонуклеотидных праймеров. Дизайн праймеров производился посредством программы «Oligo» (версия 6.31) фирмы «Molecular Biology Insights», США. На настоящий момент установлено, что последовательность около инициаторного кодона влияет на уровень трансляции эукариотической мРНК. Остаток аденина в -3 положении имеет доминантный эффект для достижения высокого уровня трансляции, поэтому в нуклеотидную последовательность рассчитанных праймеров была вставлена последовательность Козак, оптимизированная для экспрессии генов в эукариотических клетках.
Также в структуру олигонуклеотидных праймеров были дополнительно введены сайты узнавания эндонуклеаз рестрикции, позволяющие встраивать фрагменты ДНК в векторные плазмиды по «липким» концам при использовании эндонуклеаз рестрикции SalI и EcoRI. Выбор данных ферментов обусловлен отсутствием сайтов узнавания в нуклеотидной последовательности клонируемых генов (анализ нуклеотидной последовательности генов на наличие сайтов гидролиза эндонуклеаз рестрикции проводили с использованием компьютерной программы Vector NTI (версия 9.0.0) фирмы «InforMax», США).
Первичные последовательности олигонуклеотидных праймеров, использованных в работе, приведены в Таблице 1.
GTCGAC - сайт эндонуклеазы рестрикции SalI
GAATTC - сайт эндонуклеазы рестрикции EcoRI CTGCAG - сайт эндонуклеазы рестрикции
PstI ATCCAT - сайт эндонуклеазы рестрикции Zsp2I
Индивидуальные гены НА, M1 и NA были получены в результате проведения ПЦР или ОТ-ПЦР (Фиг. 4). Для получения НА использовалась пара праймеров HA_SalI_upper и HA_EcoRI_lower и плазмида, содержащая кДНК НА вируса гриппа (A/California/04/2009 (H1N1), в качестве матрицы; для M1 использовалась пара праймеров Ml_SalI_upper и Ml_EcoRI_lower и вирусная РНК (H1N1) в качестве матрицы; для NA использовалась пара праймеров NA_SalI_upper и NA_EcoRI_lower и кассета генов в качестве матрицы.
Далее полученные фрагменты вирусного генома встраивали в плазмиду pDrive (Qiagen, США) с использованием фирменного набора, согласно протоколу фирмы производителя. Отбор клонов осуществляли рестрикционным анализом с помощью эндонуклеаз рестрикции SalI и EcoRI (Фиг. 5 и 6). Правильность нуклеотидных последовательностей всех рекомбинантных плазмид были подтверждены секвенированием. Определение нуклеотидной последовательности проводили на автоматическом секвенаторе 310 Genetic analyzer (Applied Biosystems, США). Для дальнейшей работы использовали клоны рекомбинантных плазмидных ДНК pDrive с подтвержденной нуклеотидной последовательностью встроенных генов НА, M1 и NA.
Далее выделенные из рекомбинантных плазмид фрагменты НА, M1 и NA по сайтам SalI и EcoRI клонировали в плазмиду pEntry предварительно гидролизованную данными рестриктазами (Фиг. 7). Структуру полученных плазмид подтверждали рестрикционным анализом (эндонуклеазами рестрикции SalI и EcoRI) и секвенированием.
Получение и анализ рекомбинантной плазмиды pEntry, содержащей кассету генов НА (H3N2), M1 и NA.
Индивидуальные гены НА (H3N2) получали с помощью ОТ-ПЦР в два раунда. В первом раунде провели независимые три реакции ОТ-ПЦР для получения трех коротких перекрывающихся фрагментов ДНК для каждого типа НА с использованием следующих пар праймеров для H3N2: HA_H3N2_SalI_upper и HA_H3N2_frl_lower, HA_H3N2_fr2_upper и HA_H3N2_fr2_lower, HA_H3N2_fr3_upper и HA_H3N2_PstI_lower; для В: HA_B_SalI_upper и HA_B_frl_lower, HA_B_fr2_upper и HA_B_fr2_lower, HA_B_fr3_upper и HA_B_Zsp2I_lower (Фиг. 8).
Во втором раунде провели общую ПЦР для трех перекрывающихся фрагментов ДНК для H3N2 с добавлением внешних праймеров (HA_H3N2_SalI_upper и HA_H3N2_PstI_lower) (Фиг. 9). Далее полученный амплификационный продукт НА встраивали в плазмиду pDrive с последующим отбором клона без аминокислотных замен относительно референс-штамма с помощью секвенирования.
На следующем этапе фрагмент НА (H3N2) вырезали по сайтам SalI и PstI из выбранного клона и встраивали в рекомбинантную плазмиду pEntry, несущую кассету генов, предварительно гидролизованную данными рестриктазами (взамен НА (H1N1)). Структуру полученной рекомбинантной плазмиды подтверждали рестрикционным анализом с помощью EcoRI (последовательность гемагглютинина H3N2 имеет три дополнительных сайта узнавания EcoRl в сравнении с последовательностью гемагглютинина H1N1, Фиг. 10) и секвенированием.
Получение и анализ рекомбинантной плазмиды pEntry, содержащей кассету генов НА (В), M1 и NA
Ген гемагглютинина вируса гриппа В был синтезирована в ЗАО «Евроген Ру». Синтезированный ген вырезали по сайтам Sad и Pstl из выбранного клона и встраивали в рекомбинантную плазмиду pEntry, несущую кассету генов, предварительно гидролизованную данными рестриктазами (взамен НА (H1N1)). Структуру полученной рекомбинантной плазмиды подтверждали рестрикционным анализом и секвенированием.
ПРИМЕР 3.
3.1.1. Проектирование и синтез гена гемагглютинина штамм A/HongKong/4801/2014(H3N2)
Проектирование нуклеотидной последовательность гена гемагглютинина штамм A/HongKong/4801/2014(H3N2) осуществляли на основе имеющихся нуклеотидных последовательностей в базе данных GISAID EpiFlu (Фиг. 17).
Далее была проведена оптимизация первичной нуклеотидной последовательности гена, кодирующего гемагглютинин, с учетом частоты встречаемости различных кодонов для повышения уровня экспрессии гена в эукариотической системе экспрессии. Нуклеотидная последовательность была синтезирована ЗАО «Евроген Ру» (Российская Федерация). Нуклеотидная последовательность гена представлена SEQ ID NO: 6.
3.1.2. Проектирование и синтез гена гемагглютинина штамма B/Brisbane/60/2008
Проектирование нуклеотидной последовательность гена гемагглютинина штамм B/Brisbane/60/2008 осуществляли на основе имеющихся нуклеотидных последовательностей в базе данных GISAID EpiFlu (Фиг. 18).
Далее была проведена оптимизация первичной нуклеотидной последовательности гена, кодирующего гемагглютинин, с учетом частоты встречаемости различных кодонов для повышения уровня экспрессии гена в эукариотической системе экспрессии. Нуклеотидная последовательность была синтезирована ЗАО «Евроген Ру» (Российская Федерация). Нуклеотидная последовательность гена представлена SEQ ID NO: 7.
3.1.3. Проектирование и синтез гена гемагглютинина штамма A/Michigan/45/2015(H1N1)pdm09
Проектирование нуклеотидной последовательность гена гемагглютинина штамм A/Michigan/45/2015(H1N1)pdm09 осуществляли на основе имеющихся нуклеотидных последовательностей в базе данных GISAID EpiFlu (Фиг. 19).
Далее была проведена оптимизация первичной нуклеотидной последовательности гена, кодирующего гемагглютинин, с учетом частоты встречаемости различных кодонов для повышения уровня экспрессии гена в эукариотической системе экспрессии. Нуклеотидная последовательность была синтезирована ЗАО «Евроген Ру» (Российская Федерация). Нуклеотидная последовательность гена представлена SEQ ID NO: 8.
3.2.1. Клонирование гена гемагглютинина штамма A/HongKong/4801/2014 (H3N2) в вектор pLenti6.3/TO/HA-M1-NA (California/04/2009).
Искусственный ген, кодирующий гемагглютинин штамма A/HongKong/4801/2014 (H3N2) был рассчитан и синтезирован в ЗАО «Евроген Ру» в составе векторной плазмиды.
Далее полученную фирменную векторную плазмиду, содержащую ген, гемагглютинина штамма A/HongKong/4801/2014 (H3N2) гидролизовали эндонуклеазами рестрикции SalI и PstI и встраивали в рекомбинантную плазмиду pEntry_HA-M1-NA (California/04/2009), несущую кассету генов, предварительно гидролизованную данными эндонуклеазами рестрикции (взамен HA(California/04/2009)). Структуру полученной рекомбинантной плазмиды подтверждали рестрикционным анализом и секвенированием.
Для получения рекомбинантной плазмидной ДНК. pLenti6.3/TO/HA(HongKong/4801)-M1-NA, проводили LR рекомбинацию фирменной плазмидной ДНК pLenti6.3/TO/V5-DEST (Фиг. 10) с плазмидной ДНК pEntry_ HA(HongKong/4801)-M1-NA. Проводили анализ продуктов рекомбинации двух плазмид при помощи электрофоретического анализа, рестрикционного анализа и секвенирования. В результате был отобран один клоновый вариант плазмиды pLenti6.3/TO/HA(A/HongKong)-M1-NA (схематическое изображение плазмиды отображено в паспорте), проведена наработка плазмиды и очистка от эндотоксинов при помощи «EndoFree Plasmid Maxi Kit» («Qiagen», Германия). Далее проводили проверку подлинности выделенной плазмидной ДНК при помощи рестрикционного анализа и секвенирования.
3.2.2. Клонирование гена гемагглютинина штамма B/Brisbane/60/2008 в вектор pLenti6.3/TO/HA-M1-NA (California/04/2009).
Искусственный ген, кодирующий гемагглютинин штамма B/Brisbane/60/2008 (H3N2) был рассчитан и синтезирован в ЗАО «Евроген Ру» в составе векторной плазмиды.
Далее полученную фирменную векторную плазмиду, содержащую ген, гемагглютинина штамма B/Brisbane/60/2008 гидролизовали эндонуклеазами рестрикции SalI и PstI и встраивали в рекомбинантную плазмиду pEntry_HA-M1-NA (California/04/2009), несущую кассету генов, предварительно гидролизованную данными эндонуклеазами рестрикции (взамен HA(California/04/2009)). Структуру полученной рекомбинантной плазмиды подтверждали рестрикционным анализом и секвенированием.
Для получения рекомбинантной плазмидной ДНК pLenti6.3/TO/HA(В-Brisbane)-M1-NA, проводили LR рекомбинацию фирменной плазмидной ДНК pLenti6.3/TO/V5-DEST (Фис. 10) с плазмидной ДНК pEntry_ HA(В-Brisbane)-M1-NA. Проводили анализ продуктов рекомбинации двух плазмид при помощи электрофоретического анализа, рестрикционного анализа и секвенирования. В результате был отобран один клоновый вариант плазмиды pLenti6.3/TO/HA(В-Brisbane)-M1-NA (схематическое изображение плазмиды отображено в паспорте), проведена наработка плазмиды и очистка от эндотоксинов при помощи «EndoFree Plasmid Maxi Kit» («Qiagen», Германия). Далее проводили проверку подлинности выделенной плазмидной ДНК при помощи рестрикционного анализа и секвенирования.
3.2.3. Клонирование гена гемагглютинина штамма A/Michigan/45/2015 (H1N1)pdm09 в вектор pLenti6.3/TO/HA-M1-NA (California/04/2009).
Искусственный ген, кодирующий гемагглютинин штамма A/Michigan/45/2015 (H1N1)pdm09 был рассчитан и синтезирован в ЗАО «Евроген Ру» в составе векторной плазмиды.
Далее полученную фирменную векторную плазмиду, содержащую ген, гемагглютинина штамма A/Michigan/45/2015 (H1N1)pdm09 гидролизовали эндонуклеазами рестрикции SalI и PstI и встраивали в рекомбинантную плазмиду pEntry_HA-M1-NA (California/04/2009), несущую кассету генов, предварительно гидролизованную данными эндонуклеазами рестрикции (взамен HA(California/04/2009)). Структуру полученной рекомбинантной плазмиды подтверждали рестрикционным анализом и секвенированием.
Для получения рекомбинантной плазмидной ДНК pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, проводили LR рекомбинацию фирменной плазмидной ДНК pLenti6.3/TO/V5-DEST (Фис. 10) с плазмидной ДНК pEntry_ HA(H1N1-Mich)-M1-NA. Проводили анализ продуктов рекомбинации двух плазмид при помощи электрофоретического анализа, рестрикционного анализа и секвенирования. В результате был отобран один клоновый вариант плазмиды pLenti6.3/TO/HA(H1N1-Mich)-M1-NA (схематическое изображение плазмиды отображено в паспорте), проведена наработка плазмиды и очистка от эндотоксинов при помощи «EndoFree Plasmid Maxi Kit» («Qiagen», Германия). Далее проводили проверку подлинности выделенной плазмидной ДНК при помощи рестрикционного анализа и секвенирования.
3.3.1. Получение и анализ рекомбинантной плазмиды pEntry, содержащей кассету генов HA (B/Brisbane/60/2008), M1(California/04/2009) и NA(California/04/2009)
В качестве векторной плазмиды в работе использовалась рекомбинантная плазмидная ДНК pEntry_HA-M1-NA (California/04/2009). Данная плазмида содержит кассету генов, состоящую из следующих структурных элементов:
• attL1 и attL2-нуклеотидные последовательности, необходимые для LR-рекомбинации между плазмидами pEntry и pLenti6.3/TO/V5-DEST;
• IRES -участок внутренней посадки рибосом;
• CMV - цитомегаловирусный промотор;
• HA - ген, кодирующий гемагглютинин вируса гриппа (California/04/2009);
• SalI/PstI - сайты узнавания соответствующих эндонуклеаз рестрикции для замены HA(California/04/2009) в кассете генов при необходимости;
• M1 - ген, кодирующий матриксный белок вируса гриппа (California/04/2009);
• NA - ген, кодирующий нейраминидазу вируса гриппа (California/04/2009).
Ген, кодирущий геммагглютинин штамма B/Brisbane/60/2008 (H3N2), был синтезирован в ЗАО «ЕврогенРу» в составе векторной плазмиды, при этом ген фланкирован сайтами узнавания эндонуклеаз рестрикции SalI/PstI для дальнейшего переклонирования в необходимый вектор. Кроме того была произведена оптимизация нуклеотидной последовательности гена для увеличения его экспрессии в эукариотических клетках. При оптимизации использовалась нуклеотидная последовательность гена гемагглютинина штамма B/Brisbane/60/2008 (H3N2) из базы данных GISAID EpiFlu , оптимизация нуклеотидного состава гена не приводит к аминокислотным заменам в соответствующем белке.
Для получения рекомбинантной плазмидной ДНК pEntry_HA(В-Brisbane)-M1-NA, векторную плазмиду pEntry_HA-M1-NA (California/04/2009)гидролизовалиэндонуклеазами рестрикции SalI и PstI. С использованием указанных эндонуклеаз рестрикции проводили гидролиз плазмидной ДНК, полученной от ЗАО «ЕврогенРу», содержащей ген, кодирующий гемагглютинин штамма B/Brisbane/60/2008.Далее проводили очистку гидролизованной векторной плазмидной ДНК и гидролизованного фрагмента ДНК, кодирующего гемагглютинин, путем выделения соответствующих фрагментов из агарозного геля при помощи набора GelExtractionKit («QIAGEN», Германия) в соответствии с рекомендациями производителя. Лигировали полученные фрагменты ДНК, трансформировали компетентные клетки E.coli XL-10Gold. Далее проводили выделение плазмидной ДНК из индивидуальных колоний E.coli. Отбор клонов осуществляли рестрикционным анализом с помощью эндонуклеаз рестрикции SalI и PstI (Фиг. 3). Правильность нуклеотидных последовательностей всех рекомбинантных плазмид были подтверждены секвенированием. Определение нуклеотидной последовательности проводили на автоматическом секвенаторе 310 Geneticanalyzer («AppliedBiosystems», США). Для дальнейшей работы использовали клоны рекомбинантных плазмидных ДНК pEntry_HA(В-Brisbane)-M1-NA с подтвержденной нуклеотидной последовательностью встроенного гена, кодирующего гемагглютинин.
3.3.2. Получение и анализ рекомбинантной плазмиды pLenti6.3, содержащей кассету генов HA (B/Brisbane/60/2008), M1(California/04/2009) и NA(California/04/2009)
Для получения рекомбинантной плазмидной ДНК pLenti6.3, содержащей кассету генов HA (B/Brisbane/60/2008), M1(California/04/2009) и NA (California/04/2009), провели LR рекомбинацию между фирменной плазмидной ДНК pLenti6.3/TO/V5-DEST и плазмидной ДНК pEntry_HA(В-Brisbane)-M1-NA, содержащей кассету генов. Далее проводили трансформацию компетентных клеток E.coli штамм Stbl3 смесью плазмидных ДНК, полученных после рекомбинации. Затем осуществляли выделение плазмидной ДНК из индивидуальных колоний E.coli. Потенциально один клоновый вариант являлся целевым. Для подтверждения структуры полученной плазмиды проводили расширенный рестрикционный анализа с использованием эндонуклеаз рестрикции XhoI, EcoRI, PstI и HindIII (длины полученных ДНК-фрагментов после гидролиза соответствуют теоретическим длинам,. Кроме того правильность нуклеотидной последовательности подтверждена секвенированием. Для дальнейшей работы данная плазмида была наработана и очищена от эндотоксинов при помощи набора «EndoFreePlasmidMaxiKit» («QIAGEN», Германия).
Результат электрофоретического разделения в 1.5 % геле агарозы фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pLenti6.3/TO/HA(B-Brisbane)-M1-NA, эндонуклеазами рестрикции XhoI, EcoRI, PstI и HindIII. В таблице приведены теоретические длины фрагментов ДНК, образующихся после гидролиза соответствующими эндонуклеазами рестрикции.
1: плазмида pLenti6.3/TO/HA(B-Brisbane)-M1-NA, гидролизованная HindIII;
2: плазмида pLenti6.3/TO/HA(B-Brisbane)-M1-NA, гидролизованная EcoRI;
3: плазмида pLenti6.3/TO/HA(B-Brisbane)-M1-NA, гидролизованная XhoI;
4: плазмида pLenti6.3/TO/HA(B-Brisbane)-M1-NA, гидролизованная PstI;
М. ДНК маркер 1 Kb («СибЭнзим», Россия).
3.3.3. Получение и анализ рекомбинантной плазмиды pEntry, содержащей кассету генов HA (A/HongKong/4801/2014 (H3N2)), M1(California/04/2009) и NA(California/04/2009)
Схема получения рекомбинантной плазмидной ДНК pEntry_HA(HongKong/4801)-M1-NA аналогична схеме получения pEntry_HA(В-Brisbane)-M1-NA.
В качестве векторной плазмиды также использовалась плазмидная ДНК pEntry_HA-M1-NA (California/04/2009), в которой ген, кодирующий гемагглютинин штамма California/04/2009заменяли на ген, кодирующий гемагглютинин штамма A/HongKong/4801/2014(H3N2).
Ген, кодирующий гемагглютинин штамма A/HongKong/4801/2014(H3N2), был синтезирован в ЗАО «ЕврогенРу», с оптимизацией кодонового состава, для увеличения уровня экспрессии гена в эукариотических клетках. Информация о природной нуклеотидной последовательности гена, кодирующего гемагглютинин, получена из базы данных GISAID EpiFlu.
Плазмидную ДНК pEntry_HA-M1-NA (California/04/2009) и плазмиду синтезированную ЗАО «ЕврогенРу», содержащую ген гемагглютинина необходимого штамма, гидролизовали эндонуклеазами рестрикции SalI и PstI. После чего проводили очистку векторной плазмиды и гена, кодирующего гемагглютинин, из агарозного геля. Лигировали два полученных фрагмента ДНК, затем трансформировали компетентные клетки E.coli штамма XL-10Gold лигазной смесью. Далее из индивидуальных колоний E.coli проводили выделение плазмидных ДНК. Подлинность полученной плазмидной ДНК подтверждали рестрикционным анализом (Фиг. 8) и секвенированием. Для дальнейшей работы был отобран клон без нуклеотидных замен.
3.3.4. Получение и анализ рекомбинантной плазмиды pLenti6.3, содержащей кассету генов HA (A/HongKong/4801/2014(H3N2)), M1(California/04/2009) и NA(California/04/2009)
Для получения плазмидной ДНКpLenti6.3/TO/HA(A/HongKong)-M1-NA была проведена LR-рекомбинация между плазмидной ДНК pLenti6.3/TO/V5-DEST и плазмидной ДНКpEntry_HA(HongKong/4801)-M1-NA. Трансформировали компетентные клетки E.coli штамм Stbl3 продуктами реакции рекомбинации. Далее проводили выделение плазмидной ДНК из индивидуальных клонов E.coli. В результате электрофоретического анализы выделенных плазмидных ДНК, установлено, что потенциально 4 клона, являются целевыми продуктами рекомбинации. Далее один из клонов секвенировали и проводили рестрикционный анализа с использованием эндонуклеаз рестрикции XhoI, EcoRI, PstI и HindIII. Выбранные клоновый вариант pLenti6.3/TO/HA(A/HongKong)-M1-NA не содержит нуклеотидных замен, относительно теоретической последовательности, кроме того длины ДНК-фрагментов, полученных после гидролиза указанными эндонуклеазами рестрикции, соответствуют теоретическим длинам. Для дальнейшей работы данная плазмида была наработана и очищена от эндотоксинов при помощи набора «EndoFreePlasmidMaxiKit» («QIAGEN», Германия).
Результат электрофоретического разделения в 1.5 % геле агарозы фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pLenti6.3/TO/HA(A/HongKong)-M1-NA, эндонуклеазами рестрикции XhoI, EcoRI, PstI и HindIII. В таблице приведены теоретические длины фрагментов ДНК, образующихся после гидролиза соответствующими эндонуклеазами рестрикции.
1: плазмида pLenti6.3/TO/HA(A/HongKong)-M1-NA, гидролизованная PstI;
2: плазмида pLenti6.3/TO/HA(A/HongKong)-M1-NA, гидролизованная HindIII;
3: плазмида pLenti6.3/TO/HA(A/HongKong)-M1-NA, гидролизованная EcoRI;
4: плазмида pLenti6.3/TO/HA(A/HongKong)-M1-NA, гидролизованная XhoI;
М. ДНК маркер 1 Kb («СибЭнзим», Россия)
3.3.5. Получение и анализ рекомбинантной плазмиды pEntry, содержащей кассету генов HA (A/Michigan/45/2015 (H1N1)pdm09), M1(California/04/2009) и NA(California/04/2009)
Для получения рекомбинантной плазмидной ДНК pLenti6.3/TO/HA(H1N1-Mich)-M1-NA на первом этапе работы был произведен синтез гена, кодирующего гемагглютинин штамма A/Michigan/45/2015(H1N1)pdm09. Информация о нуклеотидной последовательности гена гемагглютинина была получена из базы данных GISAID EpiFlu (Фиг. 11). Далее была проведена работа по оптимизации кодонового состава, указанного гена, с целью повышения уровня его экспрессии в эукариотических клетках. Ген был рассчитан и синтезирован в составе плазмидной ДНК в ЗАО «ЕврогенРу». Затем проводили гидролиз полученной плазмидной ДНК по сайтам эндонуклеаз рестрикции SalI и PstI. Кроме того гидролизовали плазмидную ДНК pEntry_HA-M1-NA (California/04/2009) указанными эндонуклеазами рестрикции. После гидролиза векторную ДНК и фрагмент, кодирующий гемагглютинин штамма A/Michigan/45/2015(H1N1)pdm09, выделяли из агарозного геля при помощи набора GelExtractionKit («QIAGEN», Германия), лигировали выделенные фрагменты и лигазной смесью трансформировали компетентные клетки E.coli штамма XL-10Gold. Проводили выделение клоновых вариантов плазмидной ДНК из индивидуальных колоний E.coli. Подлинность выделенной плазмидной ДНК подтверждали рестрикционным анализом (гидролиз эндонуклеазами рестрикции SalI и PstI, и секвенированим. Клон без нуклеотидных замен использовался для дальнейшей работы.
3.3.6. Получение и анализ рекомбинантной плазмиды pLenti6.3, содержащей кассету генов HA (A/Michigan/45/2015 (H1N1)pdm09), M1(California/04/2009) и NA(California/04/2009).
Рекомбинантная плазмидная ДНК pLenti6.3/TO/HA(H1N1-Mich)-M1-NA была получена по схеме, описанной выше. На первом этапе работы проводили LR рекомбинацию между полученной плазмидной ДНК pEntry_HA-M1-NA (California/04/2009) и плазмидной ДНК pLenti6.3/TO/V5-DEST, затем смесью плазмид, полученной после реакции рекомбинации проводили трансформацию компетентных клеток E.coli штамм Stbl3. Проводили выделение клоновых вариантов плазмидной ДНК из индивидуальных колоний E.coli. В результате электрофоретического анализа установлено, что потенциально два клоновых варианта плазмидной ДНК являются целевыми плазмидами pLenti6.3/TO/HA(H1N1-Mich)-M1-NA (Фиг. 14). Один из клонов плазмидной ДНК гидролизовали эндонуклеазами рестрикции XhoI, EcoRI, PstI и HindIII (рис. 15). В результате показано, что длина фрагментов ДНК, полученных после гидролиза, соответствует теоретическим длинам продуктов гидролиза. Правильность нуклеотидной последовательности рекомбинантной плазмиды были подтверждены секвенированием. Для дальнейшей работы данная плазмида была наработана и очищена от эндотоксинов при помощи набора «EndoFreePlasmidMaxiKit» («QIAGEN», Германия).
Результат электрофоретического разделения в 1.5 % геле агарозы фрагментов ДНК, полученных после гидролиза рекомбинантной плазмиды pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, эндонуклеазами рестрикции XhoI, EcoRI, PstI и HindIII. В таблице приведены теоретические длины фрагментов ДНК, образующихся после гидролиза соответствующими эндонуклеазами рестрикции.
1: плазмида pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, гидролизованная PstI;
2: плазмида pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, гидролизованная HindIII;
3: плазмида pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, гидролизованная EcoRI;
4: плазмида pLenti6.3/TO/HA(H1N1-Mich)-M1-NA, гидролизованная XhoI;
М. ДНК маркер 1 Kb («СибЭнзим», Россия).
ПРИМЕР 4. Протоколы исследования физико-химических и молекулярно-биологических свойств лентивирусных векторов
Для получения рекомбинантной плазмидной ДНК pLenti6.3/TO/HA(H1N1)-M1-NA, содержащей кассету генов НА, M1 и NA, провели LR рекомбинацию фирменной плазмидной ДНК pLenti6.3/TO/V5-DEST и синтезированной ранее плазмидной ДНК pEntry, содержащей кассету генов (Фиг. 11). При отборе клонов с помощью ПЦР-анализа (праймеры CMV_upper и HA_lower - ПЦР-продукт длиной 1869 п.н.; праймеры NA_upper и V5 (C-term)_lower - ПЦР-продукт длиной 1538 п.н.) и расширенного рестрикционного анализа с использованием эндонуклеаз рестрикции XhoI, EcoRI, PstI и HindIII (Таблица 2) выявили 2 целевых клона из 40 проанализированных (Фиг. 12). Правильность нуклеотидной последовательности подтверждена секвенированием.
Таким образом, был создан лентивирусный плазмидный вектор, несущий кассету генов гемагглютинина, нейраминидазы и белка M1 вируса гриппа A (H1N1pdm09), и экспрессирующий полипротеин, состоящий из белков НА (H1), NA (N1) и М1.
Плазмидная конструкция H1N1M1 включает в себя три участка белков вируса гриппа, соединенных вместе с помощью двух линкеров 2А. В результате достигается экспрессия одного длинного белка, который затем разделяется в процессе переработки на три функциональных белка. Эталонными последовательностями белков вируса гриппа являются:
• Белок НА = H1: № банка генов ACS45035 (H1N1)
• Белок NA = N1:№ банка генов AGU92835 (H1N1)
• Белок Ml = Ml: № банка генов ААТ70589 (H5N1)
Полученные три лентивирусных вектора использовались в дальнейшем для создания клеток-продуцентов вирусоподобных частиц (ВПЧ) вируса гриппа.
Сайты рестрикции кассетных векторов экспрессии
Сайты рестрикции кассетных векторов экспрессии
Сайты рестрикции кассетных векторов экспрессии
В плазмидах используют следующие последовательности нуклеиновых/аминокислот:
SEQ ID NO: 1 - Последовательность гемагглютинина H1 ДНК;
SEQ ID NO: 2 - Последовательность нейраминидазы N1 ДНК;
SEQ ID NO: 3 - Последовательность белка M ДНК;
SEQ ID NO: 4 - Последовательность нуклеиновых кислот гемагглютинина НА (H3N2) из штамма (H3N2);
SEQ ID NO: 5 - Последовательность нуклеиновых гемагглютинина вируса гриппа В из штамма.
ПРИМЕР 5. Селекция линий клеток-продуцентов ВПЧ гриппа на основе клеточных линий высших млекопитающих (HEK293FT, Vero, MDCK). Создание промышленного штамма-продуцента для синтеза ВПЧ.
Было использовано три упаковочных плазмиды, содержащих гены белков ВИЧ. Лентивирусный вектор - плазмида pLenti6.3 - доставляется в клетку мишень путем трансфекции комплексов с липофектамином, она содержит только неструктурные элементы генома ВИЧ. Отказ от использования упаковочных плазмид полностью исключает возможность формирования лентивирусной частицы.
В качестве клеток-мишеней для трансдукции генами белков вируса гриппа были использованы клетки млекопитающих НЕК 293FT, Vero и MDCK.
Культура клеток MDCK была выделена из клеток почки взрослой здоровой самки коккерспаниеля в 1958 году.
Культура клеток НЕК293 получена в начале 70-х годов в Нидерландах в результате трансформации нормальных клеток почки эмбриона человека генетическим материалом аденовируса 5-го типа, размер вставки вирусного генома в геном клетки составляет примерно 4500 п.н. Ген Е1А аденовируса экспрессируется в этих клетках и участвует в трансактивации некоторых вирусных промоторов, в связи с чем данные клетки продуцируют большое количество белка. Широкое использование клеток НЕК293 в научных целях связано с их высочайшей способностью к трансфекции, которая достигает эффективности 100%. Вариант клеток НЕК293FT содержит большой Т-антиген вируса SV40, который обеспечивает эписомальную репликацию плазмид, содержащих SV40ori сайт репликации. Это свойство обеспечивает высокий уровень амплификации трансфецированных плазмид внутри клеток 293FT и последующую временную экспрессию продукта целевого гена. Культура клеток НЕК293 широко не применяется при производстве вирусных вакцин.
5.1. Методы трансфекции, трансдукции и протоколы описания процедуры
5.1.1. Метод ФОЕ определения количества трансдуцирующих единиц иммунохимическим окрашиванием трансдуцированных клеток
Монослой культуры клеток-мишеней, предназначенных для трансдукции, выращивается в 96-ти луночных планшетах в поддерживающей среде (ДМЕМД00 ед/мл пенецилина,100 мкг/мл стрептомицина, 300 мкг/мл L-глютамин , 10 мкг/мл ДЕАЕ- декстрана (Sigma), 5 мкг/мл трипсина ТРСК). Отдельно в стерильных микропробирках готовятся 10-кратные серийные разведения суспензии псевдовируса путем добавления 50 мкл суспензии псевдовируса к 450 мкл разводящей поддерживающей среды. В лунки планшета с монослоем клеток MDCK вносятся 10-ти кратные разведения псевдовирусной суспензии. Каждое разведение псевдовируса вносится в 4 лунки по 100 мкл. Через 18 час инкубации в СО2 атмосфере при температуре 37°С удаляется среда из лунок и планшет 2 раза промывается ФБР. Добавляется по 100 мкл охлажденного ацетона (80%) на 10-15 мин, после чего ацетон удаляется, планшет ополаскивается дважды ФБР. Готовится разведение 1:5000 моноклональных антител к белку НА1 вируса гриппа А в ФБР, содержащем 1% бычьего сывороточного альбумина и 0.1% Твин 20, и по 100 мкл добавляется на 60 мин в каждую лунку, после чего лунки промываются 4 раза и покрываются аффинно очищенными конъюгированными пероксидазой хрена антителами козы против IgG мыши. Через 60 мин инкубации лунки промываются 4 раза, вносится раствор субстрата 3-amino-9-ethylcarbazole (АЕС). Через 30 мин инкубации субстратный раствор удаляется, планшет промывается 1 раз ФБР, и инфицированные клетки, окрашенные в розово-коричневый цвет, подсчитываются в инвертированном микроскопе. Количество трансдуцирующих единиц рассчитывается после усреднения количества фокусобразующих единиц (окрашенных инфекционных центров) по 4-м лункам по формуле Т=ФОЕСр*10N+1 ФОЕ/мл, где N- номер 10-кратного разведения псевдовируса, в котором проводился учет ФОЕ.
5.1.2. Протокол описания процедуры получения клеток-продуцентов ВПЧ гриппа путем трансдукции клеток млекопитающих (HEK293FT, Vero, MDCK)
Клетки млекопитающих (HEK293FT, Vero, MDCK), которые предполагается трансдуцировать, выращивают в полной питательной среде. В день трансдукции вынимают из никотемператрурного холодильника пробирку с псевдовирусом, оттаивают и, если необходимо, разбавляют до необходимой множественности заражения. Суспензию псевдовируса не взбалтывать. Удаляют культуральную среду с клеток, осторожно добавляют суспензию псевдовируса к монослою клеток. Для увеличения трансдуцирующей активности можно добавить Polybrene® до конечной концентрации до 10 мкг/мл. Аккуратно покачивают планшеклетками и инкубируют при 37°С в увлажненной 5% С02-атмосфере в течение ночи. На следующий день удаляют вируссодержащую суспензию и заменяют свежей полной питательной средой, продолжают инкубацию при 37°С в течение ночи. На следующий день удаляют питательную среду и заменяют ее на среду, содержащую бластицидин. Замену среды проводят каждые 3-4 дня до тех пор, пока устойчивые к антибиотикам колонии могут быть идентифицированы (обычно через 10-12 дней). Выбирают по крайней мере 5 устойчивых к антибиотикам колоний и каждую пересевают в отдельный культуральный флакон. После формирования монослоя проводят оценку продукции ВПЧ в супернатанте для каждого клона в реакции гемагглютинации (РГА) и в электронном микроскопе.
5.2. Методы оценки клеточных культур и имеющихся в них экспрессирующих конструкций (анализ экспрессии репортерного гена, интеграция генетической конструкции в геном клеток-мишеней) в производстве иммунобиологических препаратов
5.2.1. Метод определения концентрации ВПЧ гриппа в реакции гемагглютинации
Для постановки РГА используют 1%-ную суспензию отмытых эритроцитов петуха. Для приготовления суспензии отмытых эритроцитов используют 1 мл свежей крови петуха, к которой добавляется 9 мл ФБР, суспензия центрифугируется при 1200 об/мин, супернатант и осадок клеток «белой» крови удаляется, процедура повторяется еще 2 раза. Для получения 1%-ной суспензии эритроцитов из 1 мл крови необходимо отмытые эритроциты развести в 60 мл ФБР, что соответствует концентрации 60 ООО клеток/мл.
В 96-луночные круглодонные планшеты добавляется по 50 мкл фосфатного буферного раствора (ФБР) в каждую лунку. Готовятся серийные двукратные разведения исследуемых образцов супернатанта культуры клеток-продуцентов ВПЧ гриппа, путем переноса 50 мкл пробы, из лунок последнего ряда удаляется 50 мкл. Во все лунки планшета добавляется по 50 мкл 1%-ной суспензии эритроцитов петуха, планшет слегка покачивается для перемешивания проб в лунках, закрывается и инкубируется 60 мин при комнатной температуре, после чего проводится учет результатов агглютинации. Титром РГА считается наибольшее разведение исследуемой сыворотки, при котором наблюдается агглютинация эритроцитов («зонтик»).
5.2.2. Метод ФОЕ определения количества клеток, экспрессирующих репортерный (целевой) ген, иммунохимическим окрашиванием трансдуцированных клеток
Монослой культуры клеток-продуцентов ВПЧ гриппа выращивается в 96-ти луночных планшетах в поддерживающей среде (ДМЕМД00 ед/мл пенициллина, 100 мкг/мл стрептомицина, 300 мкг/мл L-глютамин , 10 мкг/мл ДЕАЕ-декстрана (Sigma), 5 мкг/мл трипсина ТРСК) путем инкубации в СО2 атмосфере при температуре 37°С. Удаляется среда из лунок и планшет 2 раза промывается ФБР. Добавляется по 100 мкл охлажденного ацетона (80%) на 10-15 мин, после чего ацетон удаляется, планшет ополаскивается дважды ФБР. Готовится разведение 1:5000 моноклональных антител к белку вируса гриппа в ФБР, содержащем 1% бычьего сывороточного альбумина и 0.1% Твин 20, и по 100 мкл добавляется на 60 мин в каждую лунку, после чего лунки промываются 4 раза и покрываются аффинно очищенными конъюгированными пероксидазой хрена антителами козы против IgG мыши. Через 60 мин инкубации лунки промываются 4 раза, вносится раствор субстрата 3-amino-9-ethylcarbazole (АБС). Через 30 мин инкубации субстратный раствор удаляется, планшет промывается 1 раз ФБР, и трансдуцированные клетки, окрашенные в розово-коричневый цвет, подсчитываются в инвертированном микроскопе.
5.2.3. Метод электронной микроскопии клеток-продуцентов ВПЧ гриппа
Образцы клеток-продуцентов ВПЧ гриппа фиксировали в 4%-ном растворе параформальдегида при +4°С в течение 48 ч. Затем, дополнительно фиксировали 1%-ным раствором осмиевой кислоты, обезвоживали по стандартной методике в растворах этилового спирта возрастающей концентрации и ацетоне, и заливали в смесь эпон-аралдит. Ультратонкие срезы готовили на микротоме Райхерт-Янг (Австрия). Полученные срезы и контрастировали уранилацетатом и цитратом свинца. Ультратонкие срезы исследовали в электронном микроскопе JEM 1400 (Jeol, Япония), фотосъемку и анализ изображения проводили с помощью встроенной цифровой камеры Veleta (SIS, Германия) и программного пакета iTEM (SIS, Германия).
5.2.4. Метод ПЦР для определения целевых генов HA, NA, интегрированных в геном клеток-продуцентов
Для выделения клеточных ядер клетки соскабливаются с монослоя, примерно 106-107 клеток, промываются в холодном ФБР дважды, используя центрифугирование при 1500 об/мин. Осадок клеток ресуспендируют в 500 мкл гипотонического буферного раствора, пипетируя несколько раз, и инкубируют на льду в течение 15 минут (20 mM Tris-HCl, рН 7.4, 10 mМ NaCl, 3 mМ MgCl2). Добавляется 25 мкл детергента (10% NP4O) и встряхивается на вортексе 10 секунд. После чего гомогенат центрифугируют в течение 10 минут при 10000 об/мин при 4°С, супернатант удаляют, а осадок, содержащий ядерную фракцию, используют для экстракции геномной ДНК клеток-продуцентов.
Экстракция геномной ДНК из ядерной фракции клеток-продуцентов ВПЧ гриппа проводится с использованием набора реагентов «АмплиСенс РИБО-сорб». Добавяют в пробирки по 450 мкл лизирующего раствора. Вносят в пробирки с лизирующим раствором по 100 мкл исследуемых образцов клеток-продуцентов, используя для каждого образца отдельный наконечник с фильтром. Перемешивают пипетированием. Инкубируют при комнатной температуре 3 мин. Плотно закрыв крышки, тщательно перемешивают на вортексе. Осаждают капли на вортексе. Помещают пробирки в термостат с температурой 60°С на 10 мин. Перемешивают, затем осаждают капли на вортексе. Ресуспендируют сорбент, интенсивно перемешивая на вортексе. Добавляют в каждую пробирку отдельным наконечником по 25 мкл ресуспендированного сорбента, плотно закрывают крышки.
Перемешивают на вортексе, ставят в штатив на 1 мин, еще раз перемешивают и оставляют в штативе на 5 мин. Центрифугируют пробирки на микроцентрифуге в течение 1 мин при 7 000 g. Не захватывая сорбент, удаляют надосадочную жидкость из каждой пробирки отдельным наконечником без фильтра на 200 мкл, используя вакуумный отсасыватель. Добавляют в пробирки по 400 мкл раствора для отмывки, плотно закрывают крышки, перемешивают на вортексе до полного ресуспендирования сорбента. Центрифугируют пробирки на микроцентрифуге в течение 30 с при 7000 g. Не захватывая сорбент, удаляют надосадочную жидкость из каждой пробирки отдельным наконечником без фильтра на 200 мкл, используя вакуумный отсасыватель. Добавляют в пробирки по 400 мкл раствора для отмывки, плотно закрывают крышки, перемешивают на вортексе до полного ресуспендирования сорбента. Центрифугируют пробирки на микроцентрифуге в течение 30 с при 7 000 g. Не захватывая сорбент, удаляют надосадочную жидкость из каждой пробирки отдельным наконечником без фильтра на 200 мкл, используя вакуумный отсасыватель. Повторяют процедуру отмывки. Помещают пробирки с открытыми крышками в термостат с температурой 60°С на 15 мин для подсушивания сорбента. Добавляют в пробирки по 50 мкл буфера для элюции А. Перемешивают на вортексе до полного ресуспендирования сорбента. Помещают в термостат с температурой 60°С на 5 мин. Допускается увеличение объема элюции до 120 мкл. Центрифугируют пробирки на микроцентрифуге в течение 2 мин при 12 тыс g. Надосадочная жидкость содержит очищенные ДНК. Пробы готовы к постановке реакции ПЦР.
Проведение ПЦР-амплификации с детекцией в режиме «реального времени» осуществляется на наборах реагентов производства ФБУН ЦНИИ Эпидемиологии Роспотребнадзора. Общий объем реакционной смеси - 30 мкл, включая объем пробы клеточной ДНК - 10 мкл. Отбирают необходимое количество пробирок с ПЦР-смесью-FL для амплификации ДНК исследуемых и контрольных проб. На поверхность воска вносят по 10 мкл ПЦР-буфера-А, при этом он не должна проваливаться под воск и смешиваться с ПЦР-смесью-FL. В подготовленные пробирки вносят по 10 мкл проб ДНК. Ставят контрольные реакции. Подготовка пробирок для проведения амплификации при помощи комплектов реагентов «ПЦР-комплект» вариант FRT-50 F. Общий объем реакционной смеси - 25 мкл, включая объем пробы ДНК - 10 мкл. Размораживают необходимое количество пробирок с ПЦР-смесью-FL. Перемешивают содержимое пробирок с ПЦР-смесью-FL, ПЦР-буфером-В и полимеразой (TaqF) и осаждают капли кратковременным центрифугированием (1-2 с) с помощью центрифуги/вортекса. Отобрать необходимое количество пробирок или стрипов для амплификации кДНК исследуемых и контрольных проб. Для проведения N реакций смешивают в отдельной пробирке 10*(N+1) мкл ПЦР-смеси-FL, 5*(N+1) мкл ПЦР-буфера-В, и 0,5*(N+1) mkh полимеразы (TaqF). Перемешивают подготовленную смесь на вортексе и осаждают капли кратковременным центрифугированием с помощью центрифуги/вортекса. Вносят в каждую пробирку по 15 мкл подготовленной смеси. В подготовленные пробирки вносят по 10 мкл кДНК, полученных в реакции обратной транскрипции РНК. Ставят контрольные реакции. Проведение амплификации с детекцией осуществляется в режиме «реального времени». Следует запрограммировать прибор (амплификатор с системой детекции в режиме «реального времени») для выполнения амплификации и детекции флуоресцентного сигнала в соответствии с инструкцией к набору реагентов, учитывая рекомендованные каналы для флуорофоров. Устанавливают пробирки в ячейки реакционного модуля прибора. Перед установкой в амплификатор планшетного типа необходимо осадить капли со стенок пробирок кратковременным (1-3 с) центрифугированием на центрифуге/вортексе. Запускают выполнение программы амплификации с детекцией флуоресцентного сигнала. По окончании выполнения программы приступают к анализу и интерпретации результатов.
Результаты. Протоколы исследования полученных клеточных культур-продуцентов
5.3.2.1. Анализ продукции ВПЧ гриппа клонами H1N1M1 методом электронной микроскопии
Дальнейшее подтверждение максимальной продуктивности ВПЧ в клетках MDCK было получено при проведении исследований культур клеток MDCK и HEK-293FT на наличие вирусоподобных частиц (ВПЧ) методом электронной микроскопии. На ультратонких срезах клетки имеют крупные ядра и полный набор цитоплазматических органоидов, отличительной особенностью морфологии клеток MDCK является наличие большого количества эндосомальных структур, формирование многочисленных выростов цитоплазмы разнообразной формы и микроворсинок. Клетки сохраняют эпителиоидный тип строения. В ядрах некоторых клеток выявляются агрегаты электронно-плотных частиц, придающие ядру «пятнистый» вид (Фиг. 13).
На поверхности и в окружающем пространстве примерно 40% клеток MDCK и 5% клеток HEK293FT обнаруживаются частицы, морфологически напоминающие вирус гриппа (Фиг. 13). Различаются два типа вирусоподобных частиц: сферические и нитевидные (Фиг. 14). Нитевидная форма частиц преобладает. Кроме того, в некоторых участках обнаруживаются группы частиц, имеющих плеоморфную форму (Фиг. 15).
Размер сферических частиц типичен для вируса гриппа и составляет 80 - 120 нм, нитевидные ВПЧ диаметром 90-100 нм и длиной (на срезах) до микрометра. Вирусоподобные частицы имеют хорошо выраженную мембранную оболочку, на которой видны шипики (spikes) длиной приблизительно 15 нм и диаметром около 5 нм. Характерной особенностью являлось значительное, по сравнению с типичным для вируса гриппа, снижение электронной плотности внутренней части «вириона», соответствующей расположению вирусного генома.
Около 10% клеток в культуре повреждены, в них накапливались многочисленные аутофагосомы, прослеживалось уплотнение хроматина, фрагментация ядер и формирование апоптозных телец. Гибель клеток путем апоптоза была основной (Фиг. 16).
Результаты проведенного исследования методом электронной микроскопии показывают, что морфология вирусоподобных частиц в культуре клеток MDCK и НЕК- 293FT принципиально не отличается от морфологии вируса гриппа, культивированного на тех же культурах клеток. Особенностью строения ВПЧ является снижение электронной плотности их внутренней части.
ПРИМЕР 6. Протоколы определения в ПЦР целевых генов HI, N1, интегрированных в геном клетки-продуцента
В Таблицах 3, 4, приведены результаты ПЦР детекции наличия целевых генов HI, N1, соответственно, в ядерной фракции клеток-продуцентов ВПЧ. Измеренное пороговое значение Ct для пяти изученных клонов существенно меньше значения, выявляемого для положительного контрольного образца, что указывает на наличие встроенных генов вируса гриппа в геном клетки-продуцента. Основываясь на данных о максимальной продуктивности ВПЧ гриппа на культуре MDCK, определение в ПЦР целевых генов было выполнено только для этой культуры.
Таким образом, приведенные результаты ПЦР диагностики клеточной ДНК свидетельствуют об интеграции генов вируса гриппа в геном изученных клонов клеток-продуцентов ВПЧ.
Далее в качестве клеток-мишеней для трансдукции генов вируса гриппа использовали только клетки MDCK, которые продемонстрировали максимальную продуктивность среди трех изученных линий клеток млекопитающих - НЕК 293FT, Vero и MDCK. В качестве лентивирусного вектора использовалась только плазмида pLenti6.3, три упаковочные плазмиды не использовались. На этом этапе для получения клеток-продуцентов ВПЧ вируса гриппа субтипов A(H1N1) и A(H3N2) и типа В использовали три векторных плазмиды pLenti6.3/TO/HA(HlNl)-Ml-NA, pLenti6.3/TO/HA(H3N2)-M1 -NA, pLenti6.3/TO/HA(B-Phuket)-M1-NA, структура которых описана выше.
Протокол описания процедуры получения клеток-продуцентов.
За день до проведения трансфекции проводится посев 1-3* 105 клеток MDCK на культуральный флакон Т25. Согласно протоколу для трансфецирующего реагента Липофектамин 2000, готовится смесь плазмиды и трансфектанта в 200 мкл поддерживающей среды и инкубируется при комнатной температуре 10-15 минут. После этого однократно промывается монослой клеток фосфатным буферным раствором и наносится приготовленный комплекс липофектамина и плазмиды на монослой клеток. Инкубация осуществляется в термостате (37°С) в течение 3 часов, после чего в культуральный флакон добавляется 5 мл поддерживающей среды. На 3- сутки после трансфекции монослой клеток пересевается в несколько 24-луночных планшетов. Через 3, 5, 7 и 9 суток инкубации планшетов атмосфере СО2 при 37°С из каждой лунки забирается аликвота питательной среды для оценки продукции ВПЧ в РГА. Лунка, в которой наблюдается наибольший уровень продукции рассевается предельными разведениями в несколько 96-луночных планшетов. Через 1-2 недели выбирается 10-15 клонов с максимальной продукцией по РГА. Каждый из выбранных клонов рассевается в несколько лунок 96-луночного планшета, проводится подтверждение наличия продуктов экспрессии всех трех генов методом иммунохимического окрашивания монослоя. Выбранные несколько клонов с максимальной продукцией ВПЧ рассеваются на флаконы, проводится оценка продукции ВПЧ методом электронной микроскопии. Выбирается наиболее продуктивный по данным РГА, иммунохимического окрашивания и электронной микроскопии клон, который рассевается на флаконы 75 см2, и суспензия клеток передается для формирования банка клеток продуцентов ВПЧ вируса гриппа.
Клоны, выделенные селекцией предельными разведениями, сохраняли свою способность продуцировать ВПЧ гриппа на том же уровне, что и в случае трансдукции лентивирусными псевдовирусами. В Таблице 6 приведены результаты оценки продуктивности ВПЧ пятнадцати клонов MDCK.
Пять клонов, трансфецированных плазмидой pLenti6.3/TO/HA(HlNl)-Ml-NA, продуцируют ВПЧ с титром в диапазоне от 1:16 до 1:64. ВПЧ вируса гриппа субтипа A(H3N2) нарабатываются с большей гемагглютинирующей активностью, титр в РГА варьирует от 1:32 до 1:128. Наименьшую продуктивность показали пять клонов, трансфецированных плазмидой pLenti6.3/TO/HA(B- Phuket)-M1-NA. Таким образом, клоны клеток-продуцентов ВПЧ, полученные из клеток MDCK путем трансфекции одной векторной плазмидой, сохраняют продуктивность по ВПЧ близкую к таковой на клонах, полученных в результате инфицирования лентивирусными псевдовирусами.
Проведенное исследование показало возможность получения высокопродуктивных по ВПЧ гриппа клонов клеток MDCK без использования лентивирусных псевдовирусных частиц в качестве системы доставки целевого гена в геном клетки-мишени.
На первом этапе исследования по результатам иммунохимического окрашивания было установлено, что 99,5-99,9 % клеток, подвергнутых трехкратной ко-инфекции псевдовирусами, содержали продукты экспрессии генов гемагглютинина, нейраминидазы и белка M1. Исследованные клоны культур клеток MDCK, Vero и НЕК 293FT, трансдуцированных с помощью лентивирусных псевдовирусов генами гемагглютинина, нейраминидазы и белка M1 вируса гриппа A(H1N1) демонстрировали наличие продукции ВПЧ гриппа, определяемой в РГА супернатанта этих клеток. Только в составе ВПЧ молекулы гемагглютинина способны обеспечить поливалентность конструкции по отношению к сиалозидам на мембране эритроцита и вызвать агглютинацию последних.
Также исследования показали, что клетки MDCK являются наилучшими продуцентами ВПЧ вируса гриппа, обеспечивая достижение титра ВПЧ в РГА в диапазоне от 1:64 до 1:256, тогда как клоны, полученные из клеток HEK293FT и Vero, характеризуются низкой продуктивностью. Результаты проведенного исследования методом электронной микроскопии показывают, что морфология вирусоподобных частиц, продуцируемых в культуре клеток MDCK и HEK-293FT, принципиально не отличается от морфологии вируса гриппа. На поверхности и в окружающем пространстве примерно 40% клеток MDCK и 5% клеток HEK293FT обнаруживаются частицы, морфологически напоминающие вирус гриппа. Различаются два типа вирусоподобных частиц: сферические и нитевидные. Особенностью строения ВПЧ является снижение электронной плотности их внутренней части. Результаты исследования клеточной ДНК клеток MDCK методом ПЦР свидетельствуют об интеграции генов вируса гриппа в геном изученных клонов клеток- продуцентов ВПЧ.
5.4. Методы контроля качества культуры клеток-продуцентов и ВПЧ гриппа
Для контроля качества культур клеток-продуцентов были выбраны двенадцать клонов по интенсивности продукции НА и характеристикам роста. Эти клоны были обозначены как: 5Е4, 7F4, 10C12, 10E7, 3C5, 6D6, 9B3, 5G75G7, 6D12, 1F1. Часть популяции каждого клона подвергалась криоконсервации, а другая часть культивировалась для дальнейшего анализа ВПЧ гриппа.
Белки ВПЧ гриппа разделялись на 4-12% геле Bis-Tris с буфером из морфолинпропансульфоновой кислоты (далее IX MOPS) (реагенты Invitrogen). Гель переносился на мембраны из ПВДФ и блокировался. В Таблице 7 перечислены реагенты и концентрации, которые использовались для обнаружения белков вируса гриппа с помощью вестерн-блоттинга.
На Фиг. 17А и Фиг. 17Б показаны результаты анализа экспрессии НА клонами- кандидатами методом вестерн-блоттиига. Отрицательный контроль (лизат клеток MDCK, не подвергнутых трансдукции) использовался для выявления неспецифического связывания антител. Ожидаемая молекулярная масса белка НА после лизирования составляет примерно 80 кДа (показано стрелкой). Ожидаемая масса нелизированного полипротеина составляет примерно 140 кДа. Отрицательный контроль - лизат клеток MDCK демонстрирует неспецифическое связывание. Отрицательный контроль использовался для выявления неспецифического связывания антител. Отрицательный контроль - лизат клеток MDCK демонстрирует неспецифическое связывание, и полоса положительного контроля NA свидетельствует об активном связывании антител. Белки, синтезированные в клетках MDCK, гликозилированы, поэтому они будут подниматься выше, чем негликозилированные белки. На Фиг. 18А показана активная экспрессия НА клонами-кандидатами 1F1, ЗС5, 5G7, а на Фиг. 18Б показана активная экспрессия NA клоном-кандидатом 6D12. На Фиг. 19А и Фиг. 19Б показаны результаты анализа экспрессии M1 клонами-кандидатами методом вестерн-блоттинга. Отрицательный контроль (лизат клеток MDCK, не подвергнутых трансдукции) использовался для выявления неспецифического связывания антител. Ожидаемая молекулярная масса обработанного белка M1 составляет примерно 25 кДа (показано стрелкой). На Фиг. 19А показана активная экспрессия M1 клонами-кандидатами ЗС5, 6D6 и слабая экспрессия M1 клонами-кандидатами 1F1 и 5G7. Отрицательный контроль клон 7 MDCK демонстрирует неспецифическое связывание. Вместе с тем, видны массивные полосы нелизированного белка. Это может быть связано с тем, что обработка белков трансфецированной базовой клеточной линии может быть неэффективной. На Фиг. 19Б показана активная экспрессия M1 клоном-кандидатом 6D12. Отрицательный контроль лизат клеток MDCK демонстрирует отсутствие специфического связывания.
В Таблице 8 обобщаются данные по экспрессии гемагглютинина, нейраминидазы и белка Ml клонами-кандидатами. Знаки плюс обозначают уровень экспрессии. Дополнительные плюсы обозначают более активную экспрессию.
5.5. Протокол исследования биологических свойств клеток-продуцентов ВПЧ гриппа
5.5.1. Оценка параметров роста клеток.
Целью данного анализа является определение параметров роста клеток. В частности, определялось время удвоения популяции клеток (время удвоения). Каждый клон-кандидат культивировался в 6 лунках 12-луночного планшета. В каждую лунку помещались 2*105 клеток. Каждые 24 часа производили сбор и подсчет клеток в двух лунках. После высева клетки из каждой лунки собирались через 24 часа, 48 часов и 72 часа. В каждой точке времени определялись средние показатели двух лунок, и полученное значение отражало общее количество клеток, образовавшихся в течение 24 часов. Сбор клеток через 24 часа считался точкой времени 0 для стандартизации дня посева клеток в предыдущий день. Показатели времени удвоения рассчитывались на веб-сайте doubling- time, com, используя все временные точки.
Удвоения колеблется в широком диапазоне - от примерно 18 часов до 41 часа. Так как эти клетки будут выращиваться в биореакторе, производство будет более эффективным в случае быстрого размножения клеток и, наоборот. Наилучшие показатели были у клонов 5G7, 1F1, 6D12. 4.3.2.
Рейтинг отдельных клеточных клонов.
Данные по экспрессии белка и характеристикам роста обобщаются в Таблице 9. В таблице показаны уровни экспрессии белков HA/NA/M1 и скорость роста клонов в форме времени удвоения.
Рейтинг клонов-кандидатов определяется по двум факторам. Первым фактором является наличие всех трех белков ВГТЧ (НА, NA, M1) по данным вестерн-блоттинга. ВПЧ не будут формироваться без этих трех белков. Вторым фактором является время удвоения. Это связано с культивированием клеточной линии в биореакторе. Медленный рост клеточной линии будет ограничивать использование реактора. По этим двум показателям клон-кандидат 5G7 был наилучшим, так как он демонстрировал активную экспрессию всех трех белков и наилучшее время удвоения. Клоны 6D12 и 1F1 имели рейтинг 2 и 3. Клон-кандидат 3С5 был интересен в связи с высоким уровнем экспрессии белка. Вместе с тем, время удвоения клона 3С5 было самым худшим.
5.6. Оценка клонов-продуцентов ВПЧ H1N1M1.
Каждый из клонов H1N1M1 с наилучшим рейтингом оценивался на стерильность, жизнеспособность после размораживания и контаминацию микоплазмой. Было установлено, что каждый из клонов не был контаминирован бактериями, грибками и микоплазмой, а также их показатель жизнеспособности после размораживания был больше, чем 85%.
5.6.1. Посев клеток, продуцирующих ВПЧ гриппа, в биореактор.
Использовался биореактор FibraStage, который является недорогой, нетрудоемкой системой культивирования клеток одноразового использования, предназначенной для получения белков, вирусов или клеточной массы из субстрат-зависимых и суспензионных клеточных культур. В системе используются одноразовые бутылки объемом 500 мл, с предварительно помещенными дисками FibraCel, которые являются уникальной твердой матрицей для роста клеток. Клетки остаются фиксированными в слое дисков на протяжении всего процесса, что упрощает замену среды и извлечение конечного продукта. Система исключает автоклавирование, очищение, длительное обучение, дорогостоящий запуск, перекрестную контаминацию между партиями и требует только применения стандартного СО2- инкубатора.
Стабильная клеточная линия из клона 6D12, экспрессирующая ВПЧ гриппа, была использована для посева в двух системах Fibra-Stage. Клетки культивировались в среде DMEM с высоким содержанием глюкозы (Hyclone) с добавлением 10% ФБС/4 мМ глутамина/1 мМ пирувата натрия. В каждую систему Fibra-Stage помещались 21108 клеток. Общий объем среды составлял 450 мл. В одной емкости системы Fibra-Stage содержится 10 г Fibra-дисков. Плотность посева клеток на Fibra-диски составляет 2*107. На пульте управления были установлены следующие параметры: 1,5 мм/с с положением вверху 10 секунд и положением внизу 2 минуты. СО2-инкубатор был настроен на температуру 37°С и концентрацию СО2 5%. После посева клеток в Fibra-Stage была добавлена следующая среда: DMEM с высоким содержанием глюкозы (Hyclone) с добавлением 5% ФБС, 4 мМ глутамина/1 мМ пирувата натрия/1 мМ HEPES.
5.6.2. Рост и поддержание клеток, продуцирующих ВПЧ гриппа.
Для контроля условий и жизнеспособности клеток, ежедневно брались образцы среды и оценивались с помощью анализатора Bio Profile компании «NOVA». Тщательно контролировались уровни глюкозы/глютамина/лактата/рН.
Данные параметры поддерживались в следующих пределах:
По мере роста клеток концентрация глюкозы станет ниже 1,5 г/л до смещения других параметров за указанные пределы нормы. Когда уровень глюкозы становится ниже 1,5 г/л, среда полностью заменяется. В дальнейшем по мере роста клеток вся глюкоза будет потребляться в течение 24 часов. Это произошло примерно через 3 недели после помещения клеток в Fibra-Stage. В этот момент температура понижается с 37°С до 34°С. Настройка инкубатора изменяется с 5% СО2 до 2,5%. Целью изменений является усиление продукции ВПЧ и торможение роста клеток. Данные изменения условий приведут к замедлению роста клеток на несколько дней. Тем не менее, потребление глюкозы снова начнет расти. В этот момент концентрация ФБС в среде также изменяется с 5% до 2,5%. В течение 24 часов рН с 7,2 понизится до 6,8. Полная замена среды приведет к восстановлению рН 7.2.
В каждом реакторе Fibra-stage содержится 10 граммов fibra-дисков. Начальным уровнем глюкозы является количество глюкозы в свежей среде. Клетки растут в течение приблизительно 24 часов без замены среды. В течение этого времени измеряется и регистрируется концентрация глюкозы. На Фиг. 34 показано количество миллиграммов глюкозы, потреблявшейся в расчете на один грамм дисков в час в емкости № 1 FibraStage.
5.6.3. Сбор клеток и количественный анализ образцов из Fibra-Stage на содержание НА.
Пробы были взяты после двух недель культивирования клеток в Fibra-Stage. Через две недели, содержание НА в образцах было низким, но через три недели РГА титр был 1:128, что является приемлемой концентрацией НА для проведения очистки. К концу рабочего цикла Fibra-Stage титр в РГА был примерно 1:256.
Для очищения были подготовлены два отдельных клеточных пула, полученных из каждого устройства - № 1 и № 2.
ПРИМЕР 6. Доработка технологии очистки ВПЧ гриппа для получения моновалентных балков из супернатанта клеток-продуцентов ВПЧ гриппа
6.1. Протоколы выбора и обоснования параметров процесса очистки ВПЧ гриппа для получения моновалентных балков из супернатанта MDCK клеток-продуцентов ВПЧ гриппа
6.1.1. Протокол 1
Принципиальная схема выделения ВПЧ из клеточного пула после культивирования:
1. Материал клеточного пула 3,5 л
2. Осветление Фильтр Sartopore 2
3. Обработка бензоназой 10 Ед/мл на протяжении ночи при +4°С
4. Прямая загрузка в капсулу 5 мл ионообменного хроматографа Mustang Q
5. ЭХ, S-500
6. Аффинная хроматография Cellufine S
Осветление. Материал клеточного пула осветляли с помощью фильтра Sartopore 2 0,8/0,45 мкм и обрабатывался бензоназой (10 Ед/мл) в течение ночи при температуре 4°С. После обработки бензоназой концентрация NaCl в осветленном материале доводилась до 250мМ.
Mustang Q. 3580 мл кондиционированного и осветленного материала загружали в капсулу 5 мл Mustang Q XT 5MSTGQPM6 («Pall Corp.»).Скорость загрузки была 20 мл/мин, а скорость элюирования 10 мл/мин. Захваченные ВПЧ элюировали ступенчатым градиентом В 40% (В = 3 М NaCl в 50 мМ Трис-HCl, рН 7,4).
Профиль элюции ВПЧ в колонке Mustang Q представлен на Фиг. 21.
фильтрации в мл
(титр РГА)
Эксклюзионная хроматография (ЭХ). Элюированный материал из колонки Mustang Q подвергался ЭХ для обмена буфера и дальнейшей очистки. 15 мл материала из колонки Мустанг Q с максимальной концентрацией вещества было загружено в колонку К16 с 120 мл Sephacryl S-500. Скорость загрузки и разрешение составляли 2 мл/мин. Трис- HCI 50 мМ + 100 мМ буфера NaCI использовали в качестве подвижной фазы. На Фиг. 22 показан профиль ЭХ пикового материала капсулы Mustang Q XT. Был получен образец с максимальным показателем Vo.
Аффинная хроматография. В колонку Cellufine S с объемом 1 мл были загружены 2 мл образца ЭХ с максимальным показателем Vo. После того, как был получен несвязанный материал, был применен ступенчатый градиент 40% для вытеснения связанного материала. Пиковый образец после обработки ступенчатым градиентом 40% диализировался с помощью 1 л буфера А (50 мМ Трис НС1 + 100 мМ NaCl, рН 7,4). На Фиг. 23 показан профиль Cellufine S обработки пикового материала, полученного с помощью ЭХ. Был получен пиковый материал после обработки ступенчатым градиентом 40%. Пиковый образец после обработки ступенчатым градиентом 40% диализировали с помощью 1 л буфера А (50 мМ Трис НС1 + 100 мМ NaCl, рН 7,4).
На Фиг. 24 показан профиль 2 мл материала ЭХ с пиковым значением Vo, загруженного в колонку Cellufine 1 мл. Для того, чтобы выявить перегрузку колонки, было проведено несколько прогонов через одну и ту же колонку (с промывками между прогонами) различных объемов материала ЭХ с пиковым показателем Vo.
В прогоне с загрузкой 0,5 мл второй (ВПЧ) максимальный показатель оптической плотности (ОД) составил 10 милли-единиц в условиях элюции 2 мл. В прогоне с загрузкой 1 мл, второй (ВПЧ) максимальный показатель ОД составлял 20 милли-единиц в условиях элюции 2,3 мл, и в прогоне с загрузкой 2 мл второй (ВПЧ) максимальный показатель ОД составлял 40 мили-единиц в условиях элюции 3,5 мл. Существует прямая зависимость между загруженным объемом и значением, что означает отсутствие перегрузки колонки насыщения ВПЧ. На каждом этапе брались образцы и анализировались на активность НА. Полученные результаты отражены в Таблице 12.
Количество НА может быть рассчитано с помощью положительного контроля НА, который использовался на каждой пластине. Выход (%) рассчитывался по общему количеству антигена после каждого этапа относительно предыдущего. Также было учтено, что на последнем этапе было загружено 2 мл в колонку Cellufine S из 37 мл материала после колонки ЭХ. Нормализация показывает, что в процессе очищения активность НА уменьшилась после колонки Mustang Q до 78%, после ЭХ - до 42%, потери практически отсутствовали после колонки Cellufine S (выход - 97%). Таким образом, общие потери антигена при очистке и концентрировании по протоколу 1 составили 67%.
6.1.2. Протокол 2.
С целью увеличения выхода антигена при хроматографической очистке и концентрировании были введены некоторые модификации в процесс.
Ультрафильтрация супернатанта клеток-продуцентов.
Ультрафильтрацию супернатанта клеток-продуцентов, содержащего ВПЧ гриппа H1N1 Ml проводят в два этапа путем прокачки материала через напорную емкость объемом 1 л насосом под давлением 2 bar. На первом этапе на выходе из напорной емкости устанавливается фильтровальная капсула Sartopure GF+ 1.2 pm компании Sartorius stedim biotech, через которую прокачивается материал. На втором этапе материал прокачивается через фильтровальную капсулу Sartopure GF+ 0.65 pm компании Sartorius stedim biotech. Процедура ультрафильтрования происходит при комнатной температуре. Отбирают пробу объемом 1 мл для последующего анализа.
Концентрирование ВПЧ гриппа.
Концентрирование ВПЧ гриппа H1N1M1 проводят методом ионообменной хроматографии. Ионообменную хроматографию осуществляют с помощью хроматографической системы GE Akta Purifier, состоящей из градиентных насосов Р-900, детектора UV-900, блока рН-метрии и кондуктометрии рН/С-900 и системы вводы пробы с вводными и выводными клапанами Вох-900. Управление системой ведется путем программного обеспечения Unicorn software. В качестве сорбционной хроматографической колонки используется ионно-обменная капсула Mustang Q ХТ5 объемом 5 ml компании Pall Corporation. Детектирование ведут при двух длинах волн - 210 нм и 280 нм.
Условия проведения ионно-обменной хроматографии (ИОХ).
Общее давление в хроматографической системе не превышает 0.8 МПа. Максимальная скорость потока элюента 10 мл/мин. Раствор Al: 1 М NaOH, Раствор А2: 50 шМ Tris-HCl, рН 7.5, Раствор В1: ультрафильтрованный супернатант с клеток, содержащий ВПЧ гриппа, Раствор В2: 50 mM Tris-HCl, рН 7.5 + 1М NaCl.
Перед началом хроматографии капсулу уравновешивают раствором А1 (не менее 6 объемов колонки) до постоянных значений оптической плотности, рН и кондуктивности, после чего поток элюата останавливают на 30 минут. После выдерживания капсулы в растворе А1 по истечении 30 минут капсулу промывают раствором А2 (не менее 10 объемов колонки) до наступления постоянных значений оптической плотности, рН и кондуктивности. После промывки через капсулу продавливают раствор В1, содержащий ВПЧ гриппа, в объеме, не превышающим 100 объемов капсулы. Сорбированный на капсулу биологический материал элюируется раствором В2. Формируемый хроматографический пик собирают в отдельную емкость в объем, не превышающий 15 мл (не более 3-х объемов колонки). Из собранного концентрата отбирают пробу объемом 500 мкл для последующего анализа.
Промежуточный анализ концентрата ВПЧ и проскока после ИОХ.
Концентрат и проскок от ИОХ анализируют в реакции гемагглютинации (РГА) с эритроцитами петуха. В конических лунках планшета смешивают 50 мкл анализируемой пробы и 50 мкл раствора эритроцитов в 0.01 фосфатно-солевом буфере с добавлением 0.135 М NaCl, рН 6.5-7. В результате серийных двукратных разведений анализируемой пробы в эритроцитах определяется РГА-титр ВПЧ. Заполненный планшет выдерживается 30 минут при температурном режиме +4°С. РГА-титр определяют в трех пробах: ультрафильтрофанный супернатант, концентрат после ИОХ, проскок после ИОХ.
Первичная хроматографическая очистка ВПЧ гриппа.
Первичную очистку ВПЧ гриппа из концентрата проводят методом Гель-проникающей хроматографии (Гель-фильтрация) с помощью хроматографической системы GE Akta Purifier, состоящей из градиентных насосов Р-900, детектора UV-900, блока рН-метрии и кондуктометрии рН/С-900 и системы вводы пробы с вводными и выводными клапанами Вох-900. Управление системой ведется путем программного обеспечения Unicom software. В качестве препаративной колонки используется Pharmacia Fine Chemicals 250 х 25 мм, которая заполняется сорбентом типа Sepharose CL-4B (компания Pharmacia Fine Chemicals). Детектирование ведут при двух длинах волн - 210 и 280 нм.
Условия проведения хроматографии.
Скорость потока элюента 5 мл/мин. Раствор А1: 50 mM Tris-HCl, рН 7.5. Раствор А2: концентрат ВПЧ гриппа. Перед началом очистки хроматографическую колонку уравновешивают посредством пропускания раствора А1 до постоянных значений оптической плотности, рН и кондуктивности.
Сбор целевой фракции с ВПЧ гриппа.
В колонку вводят не более 5 мл (0.04 объема сорбента) раствора А2 и начинают хроматографический процесс, используя в качестве элюента раствор А1 и наблюдая за оптической плотностью. Фракции собирают в отдельные емкости - всего три фракции, первая из которых содержит ВПЧ гриппа. По 500 мкл каждой фракции отбирается на анализ в РГА.
Вторичная хроматографическая очистка ВПЧ гриппа.
Вторичную очистку ВПЧ гриппа из первично выделенной фракции ВПЧ проводят методом Псевдо-аффинной хроматографии с помощью хроматографической системы GE Akta Purifier, состоящей из градиентных насосов Р-900, детектора UV-900, блока рН-метрии и кондуктометрии рН/С- 900 и системы вводы пробы с вводными и выводными клапанами Вох-900. Управление системой ведется путем программного обеспечения Unicorn software. В качестве препаративной колонки используется Thermo Scientific объемом 10 мл, которая заполняется сорбентом Cellufine Sulfate (компания JNC Corporation). Детектирование ведут при двух длинах волн - 210 и 280 нм.
Условия проведения хроматографии.
Раствор А1: 2 М NaCl, Раствор А2: 50 mM Tris-HCl, рН 7.5, Раствор В1: Фракционированная на этапе гель-фильтрации проба с ВПЧ гриппа. Раствор В2: 50 шМ Tris-HCl, рН 7.5 + 0.6 М NaCl. Перед началом хроматографии колонку промывают раствором А1 (не менее 2 объемов колонки) до постоянных значений оптической плотности и кондуктивности. Далее колонку промывают раствором А2 (не менее 2-х объемов колонки) до наступления постоянных значений оптической плотности, рН и кондуктивности. Скорость потока меняют с 5 мл/мин на 1.5 мл/мин. После промывки через колонку пропускают раствор В1, содержащий ВПЧ гриппа, в объеме, не превышающим 2.5 объема колонки. Сорбированный на колонку биологический материал элюируется раствором В2. Формируемый хроматографический пик собирают в отдельную емкость в объем, не превышающий 10 мл (не более 1-го объема колонки). Из собранного концентрата отбирают пробу объемом 500 мкл для последующего анализа. Анализ очищенного препарата.
Готовая проба анализируется на степень концентрирования, гемагглютинирующую и нейраминидазную активности, а также на степень очистки в аналитической гель-фильтрации и ступенчатом электрофорезе.
6.2. Итоговые результаты очистки ВПЧ гриппа
В таблице 13 представлены результаты оценки концентрирования и активности ВПЧ трех типов. Во всех трех случаях концентрирование происходило в 200 раз, при этом максимальная активность в РГА составляла 1:2048 для ВПЧ-H1N1M1 и ВПЧ-НА(В)N1M1, при концентрации гемагглютинина 450 и 540 мкг/мл, соответственно. ВПЧ-H3N1M1 при концентрации по гемагглютинину 450 мкг/мл, проявляли меньшую агглютинирующую активность - 1:1024.
Результаты электрофореза по Леммли.
Процесс очистки и концентрирования ВПЧ контролировали в электрофорезе как для отдельных стадий процесса, так и только очищенных проб (Фиг. 25). Пробы денатурировались в SDS и бета-меркаптоэтаноле с кипячением 10 минут. Верхний гель - 5%, Нижний гель - 15%. На представленных рисунках электрофореграммы видно, что исходный продукт достаточно чистый, после концентрирования видно проявление примесных белков, которые исчезают после гель-фильтрации. На Фиг. 26 представлены результаты очистки трех типов ВПЧ в одном электрофорезе. Следы примесных белков не регистрируются, а максимальная оптическая плотность полос гемагглютинина соответствует ВПЧ-НА(В)N1M1, затем меньшая оптическая плотность соответствует ВПЧ-H1N1M1, а наименьшая из трех ВПЧ регистрируется для ВПЧ-H3N1M1.
Определение количества остаточной клеточной ДНК в очищенном концентрате трех типов ВПЧ гриппа. Измерение концентрации ДНК проводили двумя методами.
Метод 1 (флюориметрический) с использованием Qubit© dsDNA HS (High Sensitivity) Assay Kit, ThermoFisher Scientific, USA и флюориметр Qubit (1.0). ДНК из суспензии ВПЧ была выделена с использованием PureLink Genomic DNA Mini kit (Invitrogen).
Метод 2 (RT-PCR) с использованием ПЦР-набора resDNASEQ Quantitative MDCK DNA Kit, AB-4464335 для количественного определения ДНК собаки, 100 реакций
ДНК из суспензии ВПЧ была выделена с использованием PureLink Genomic DNA Mini kit (Invitrogen). В Таблице 15 представлены результаты измерения остаточной клеточной ДНК в ВПЧ после всех стадий концентрирования и очистки. Измерения по методу 1 дают достаточно высокие значения примесной ДНК для ВПЧ- H1N1M1 и ВПЧ- H3N1M1 равные 87 и 98 нг/мл соответственно, в концентрате ВПЧ HA(B)N1M1 уровень примесной ДНК находится ниже предела обнаружения для этого метода (<10). ПЦР метод является более чувствительным и специфичным при обнаружении клеточной ДНК, в наших измерениях мы использовали специальный набор для обнаружения ДНК клеток MDCK. Результаты указывают на хорошее соответствие измерений, полученных методом 1 и методом 2, для Bn4-H3N1M1 и ВПЧ- HA(B)N1M1, и существенно более низкие значения по примесной клеточной ДНК для концентрата ВПЧ- H1N1M1. Такое различие возможно связано с наличием двуспиральной клеточной РНК, которая в методе 1 также флюоресцирует. Пересчет на одну дозу вакцины показывает, что суммарная масса ДНК трех типов ВПЧ в одной дозе составляет 3.39 нг, что не превышает порогового значения 10 нг. Следует обратить внимание на тот факт, что в готовой ЛФ клеточная ДНК вообще не обнаруживается, что возможно связано с тем, что ДНК сорбируется на алюминия гидроксиде.
Электронно-микроскопическая характеризация очищенного концентрата трех типов ВПЧ гриппа.
На Фиг. 27-29 представлены результаты анализа концентрата очищенных ВПЧ методом электронной микроскопии. На ВПЧ H1N1M1 (Фиг. 27) присутствуют в изобилии шипики гемагглютинина. ВПЧ имеют неправильную форму гантелей, с шипиками на головках гантелей, диаметр головок - 100 нм, длина гантелей - 200-300 нм. Концентрация ВПЧ H1N1M1-1010 штук/мл. На ВПЧ H3N1M1 (Фиг. 28) присутствуют в изобилии шипики гемагглютинина. ВПЧ имеют неправильную форму гантелей, с шипиками на головках гантелей, диаметр головок - 100 нм, длина гантелей - 200-300 нм. Концентрация ВПЧ - 109 штук/мл. На ВПЧ H3N1M1 (Фиг. 29) присутствуют в изобилии шипики гемагглютинина. ВПЧ имеют неправильную форму гантелей, с шипиками на головках гантелей, диаметр головок - 100 нм, длина гантелей - 200-300 нм. Концентрация ВПЧ - 109 штук/мл.
После завершения разработки технологии получения и культивирования клеток- продуцентов ВПЧ важнейшим этапом создания готовой лекарственной формы становится технология очистки ВПЧ гриппа для получения моновалентных балков из супернатанта клеток-продуцентов ВПЧ гриппа. Мы исследовали два протокола концентрирования и очистки ВПЧ гриппа из супернатанта клеток-продуцентов с целью обеспечения максимального выхода ВПЧ и минимального содержания примесных клеточных белков и ДНК. Оба протокола содержат основные этапы очистки - осветление фильтрованием, концентрирование в ионообменной хроматографии, очистка концентрата гель-фильтрацией и псевдоаффинной хроматографией. Полученный полуфабрикат ВПЧ проходит концентрирование по объему в 200-400 раз. Очищенный по протоколу №1 препарат ВПЧ активен в РГА с титром 1:512, тогда как концентрирование и очистка по протоколу №2 обеспечивает больший титр в РГА от 1:1024 до 1:2048, концентрацию по гемагглютинину около 500 мкг/мл, при этом примесные клеточные белки не регистрируются в электрофорезе моновалентных балков ВПЧ. Уровень примесной клеточной ДНК в препаратах, очищенных по протоколу №2 в пересчете на одну дозу ГЛФ составляет 3.4 нг. Электронно-микроскопическое исследование ВПЧ показало, что после концентрирования и очистки по протоколу №2 достигается концентрация ВПЧ 109-1010 штук/мл, сами ВПЧ имеют неправильную форму преимущественно в виде булав и гантелей, редко встречаются ВПЧ сферической или икосаэдрической формы.
Таким образом, разработанные и примененные методы концентрирования и очистки ВПЧ из супернатанта клеток-продуцентов позволяют создать продукт - моновалентные концентраты вакцины от гриппа - фармакопейного качества.
ПРИМЕР 7. Получение поливалентной вакцины от гриппа
Полученные как указано выше в примерах моновалентные концентраты вакцины от гриппа, содержащие ВПЧ: очищенные ВПЧ содержащие гемагглютинин H1N1; очищенные ВПЧ содержащие гемагглютинин H3N2 или очищенные ВПЧ содержащие гемагглютинин B, разбавляют в ФСБР (рН 7,4) до необходимой концентрации ВПЧ. Смешивание разбавленных моновалентных концентратов очищенных ВПЧ. Проведение стерилизующей фильтрации полученной смеси трех видов ВПЧ. Подготовка суспензии гидроксида алюминия в ФСБР. Проведение сведения суспензии ВПЧ и гидроксида алюминия, сорбция. Выпуск готового препарата.
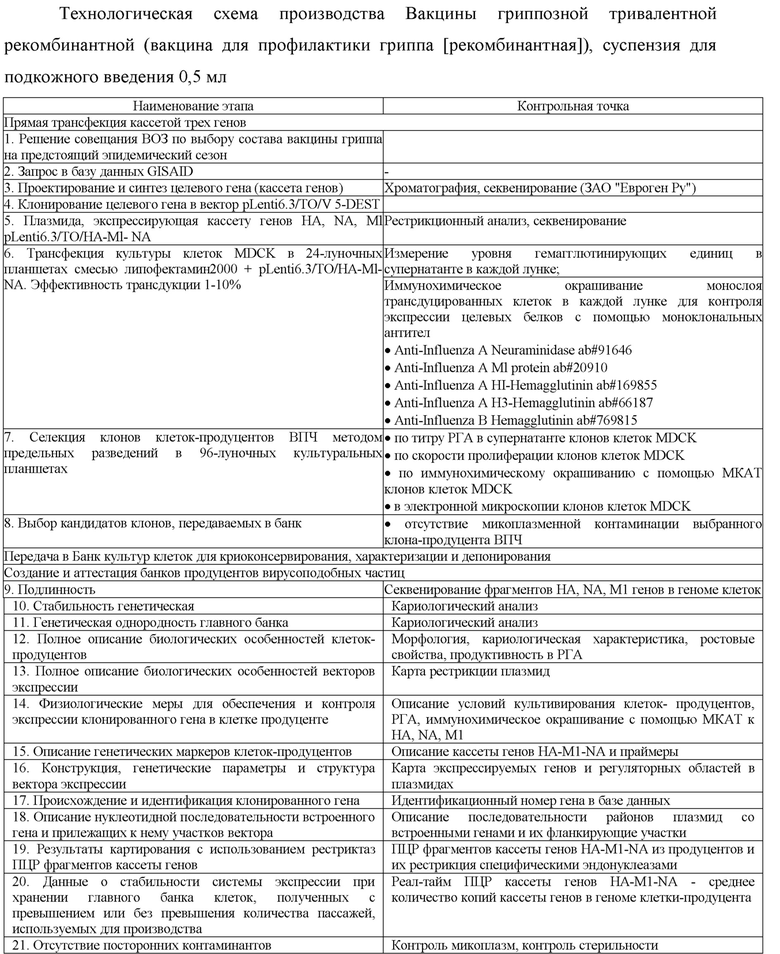
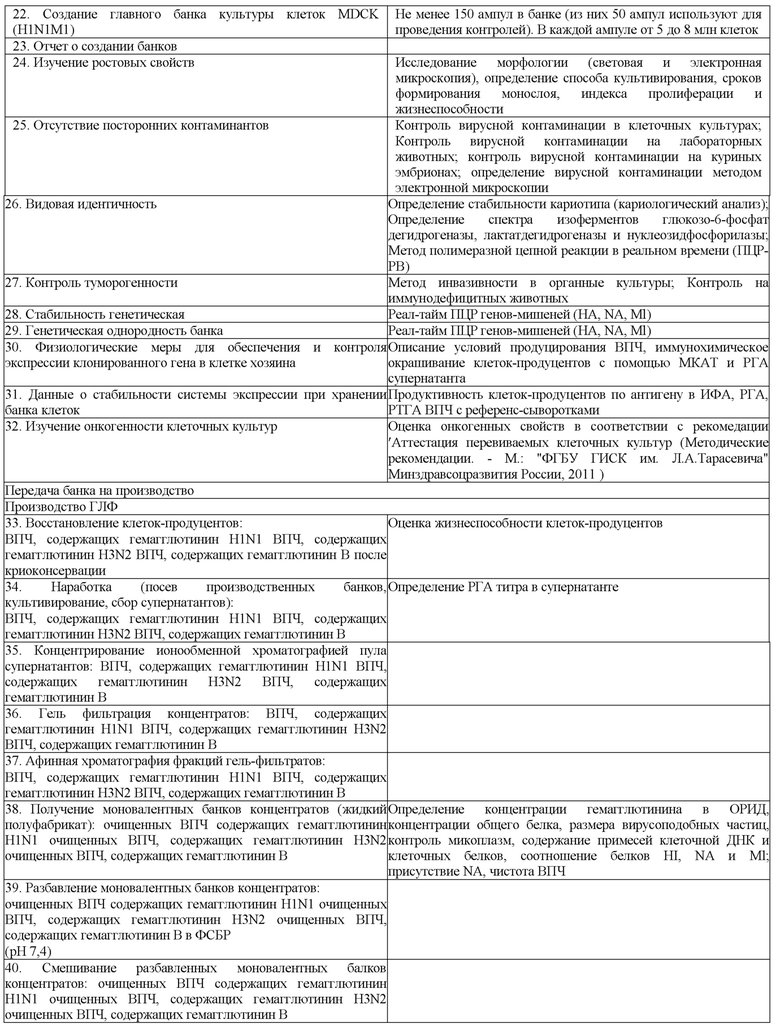
СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ
1. Shinya К, Ebina М, Yamada S, Ono М, Kasai N, Kawaoka Y. Influenza virus receptors in the human airway. Nature. 2006; 440:435-6.
2. Henklein P, Bruns K, Nimtz M, Wray V, Tessmer U, Schubert U. Influenza A vims protein PB1-F2: synthesis and characterization of the biologically active full length protein and related peptides. J Pept Sci. 2005; 11(8):481-90.
3. Noda T, Sagara H, Yen A, Takada A, Kida H, Cheng RH, et al. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature. 2006; 439:490-2.
4. Deng T, Sharps J, Fodor E, Brownlee GG. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A vims polymerase subunits into a functional trimeric complex. J Virol. 2005; 79:8669-74.
5. Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses. Microbes Infect. 2004; 6:929-36.
6. Zhang J, Leser GP, Pekosz A, Lamb RA. The cytoplasmic tails of the influenza virus spike glycoproteins are required for normal genome packaging. Virology. 2000; 269:325-34.
7. Bancroft CT, Parslow TG. Evidence for segment-nonspecific packaging of the influenza a virus genome. J Virol. 2002; 76(14):7133-9.
8. Sandbulte MR, Jimenez GS, Boon AC, Smith LR, Treanor JJ, Webby RJ. Crossreactive neuraminidase antibodies afford partial protection against H5N1 in mice and are present in unexposed humans. PLoS Med. 2007; 4:e59.
9. Hay AJ, Gregory V, Douglas AR, Lin YP. The evolution of human influenza viruses. Philos Trans R Soc Lond В Biol Sci. 2001; 356:1861-70.
10. Boni MF. Vaccination and antigenic drift in influenza. Vaccine. 2008;26 (Suppl 3):C8-14. Reid AH, Taubenberger JK. The origin of the 1918 pandemic influenza virus: a continuing enigma. Journal of General Virology. 2003 ;84:2285-92.
11. Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009; 459:1122-5.
12. Rogers GN, Paulson JC. Receptor determinants of human and animal influenza virus isolates: Differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology. 1983; 127:361-73.
13. Guenther I, Glatthaar B, Doller G, Garten W. A HI hemagglutinin of a human influenza A virus with a carbohydrate-modulated receptor binding site and an unusual cleavage site. Virus Res. 1993; 27:147-60.
14. Stieneke-Groeber A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, et al. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992; 11:2407-14.
15. Klenk H.D. et al., 2011. Molecular mechanisms of interspecies trans-mission and pathogenicity of influenza viruses: Lessons from the 2009 pandemic // Bioessays. - V. 33 Issue: 3. - P. 180-188.
16. Nakajima K., et al., 1978. Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950 // Nature. - V. 274. - P. 334-339.
17. Scholtissek C., et al., 1978. Genetic relatedness between the new 1977 epidemic strains (H1N1) of influenza and human influenza strains isolated between 1947 and 1957 (H1N1) // Virology. - V. 89. - P. 613-617.
18. Kilbourne E.D., 1977. Influenza pandemics in perspective // J. Am. Med. Assoc.. - V. 237. P. 1225-1228.
19. Pyle G.F., 1986. The Diffusion of Influenza: Patterns and Paradigms. New Jersey: Rowan & Littlefield.
20. WHO, 2009. World now at the start of 2009 influenza pandemic. [Электронный ресурс]. - Режим доступа: http://www.who.int/mediacentre/news/statements/2009/hlnl_pandemic_phase6_20090611/ en/index.html (Дата обращения: 19.10.2012).
21. Киселев О.И., 2011. Геном пандемического вируса гриппа A/H1N1 v-2009. - М.: Изд-во «Димитрейд График Групп». - 164 с.
22. Kwan-Gett T.S., et al., 2009. Spring 2009 H1N1 influenza outbreak in King country Washington // Disaster Med Pub Health Preparedness. - V. 3. - P. SI09 - 116.
23. Garten R.J., et al., 2009. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. - V. 325. - P. 197-201.
24. Седова Е.С., Щербинин Д.Н., Мигунов А.И., Смирнов Ю.А., Логунов Д.Ю., Шмаров М.М., Цыбалова Л.М., Народицкий Б.С., Киселев О.И., Гинцбург А.Л. Гриппозные рекомбинантные вакцины. // Acta naturae. - 2012. - Т. 4 - № 4 (15) - С. 17-27.
25. Lopez-Macias C. Virus-like particle (VLP)-based vaccines for pandemic influenza: performance of a VLP vaccine during the 2009 influenza pandemic. // Hum. Vaccin. Immunother. 2012. V. 8. № 3. P. 411-414.
26. Durand, S., Cimarelli, A. The Inside Out of Lentiviral Vectors // Viruses. - 2011. - N 3. - P. 132-159. Sandrin, V., Russell, S., Cosset, F. Targeting retroviral and lentiviral vectors // Curr. Top. Microbiol. Immunol. - 2003. N281. - P. 137-178.
27. Frecha, C., Costa, C., Levy, C., Negre, D., Russell, S., Maisner, A., Salles, G., Peng, K., Cosset, F., Verhoeyen, E. Efficient and stable transduction of resting В lymphocytes and primary chronic lymphocyte leukemia cells using measles virus gp displaying lentiviral vectors // Blood. - 2009. - N 114. - P. 3173-3180.
28. Verhoeyen, E., Dardalhon, V., Ducrey-Rundquist, O., Trono, D., Taylor, N., Cosset, F. IL-7 surface-engineered lentiviral vectors promote survival and efficient gene transfer in resting primary T lymphocytes // Blood. - 2003. - N 101. - P. 2167-2174.
29. Azzouz, M., Ralph, G.S., Storkebaum, E., Walmsley, L., Mitrophanous, K., Kingsman, S., Carmeliet, P., Mazarakis, N. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model // Nature. - 2004. - N 429. - P. 413-17.
30. Kang SM, Yoo DG, Lipatov AS, Song JM, Davis CT, et al. (2009) Induction of long-term protective immune responses by influenza H5N1 vims-like particles. PLoS One 4: e4667.
31. Bright RA, Carter DM, Daniluk S, Toapanta FR, Ahmad A, et al. (2007) Influenza vims-like particles elicit broader immune responses than whole virion inactivated influenza virus or recombinant hemagglutinin. Vaccine 25: 3871-3878.
32. Bright RA, Carter DM, Crevar CJ, Toapanta FR, Steckbeck JD, et al. (2008) Cross-clade protective immune responses to influenza vimses with H5N1 HA and NA elicited by an influenza virus-like particle. PLoS One 3: el501.
33. Galarza JM, Latham T, Сиро A (2005) Virus-like particle vaccine conferred complete protection against a lethal influenza vims challenge. Viral Immunol 18: 365-372.
34. Mahmood K, Bright RA, Mytle N, Carter DM, Crevar CJ, et al. (2008) H5N1 VLP vaccine induced protection in ferrets against lethal challenge with highly pathogenic H5N1 influenza vimses. Vaccine 26: 5393-5399.
35. Tao P, Luo M, Zhu D, Qu S, Yang Z, et al. (2009) Virus-like particle vaccine comprised of the HA, NA, and Ml proteins of an avian isolated H5N1 influenza virus induces protective immunity against homologous and heterologous strains in mice. Viral Immunol 22: 273-281.
36. Quan FS, Huang C, Compans RW, Kang SM (2007) Vims-like particle vaccine induces protective immunity against homologous and heterologous strains of influenza virus. J Virol 81:3514-3524.
37. Sambhara S, Stephenson I (2009) Moving influenza vaccines forward. Expert Rev Vaccines 8: 375-377.
38. Chen BJ, Leser GP, Morita E, Lamb RA (2007) Influenza vims hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid- derived virus-like particles. J Virol 81: 7111-7123.
39. Ali A, Avalos RT, Ponimaskin E, Nayak DP (2000) Influenza vims assembly: effect of influenza vims glycoproteins on the membrane association of Ml protein. J Virol 74: 8709-8719.
40. Enami M, Enami К (1996) Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J Virol 70: 6653-6657.
41. Gregoriades A, Frangione В (1981) Insertion of influenza M protein into the viral lipid bilayer and localization of site of insertion. J Virol 40: 323-328.
42. Gomez-Puertas P, Albo C, Perez-Pastrana E, Vivo A, Portela A (2000) Influenzavirus matrix protein is the major driving force in vims budding. J Virol 74:11538-11547.
43. McCown MF, Pekosz A (2006) Distinct domains of the influenza a virus M2protein cytoplasmic tail mediate binding to the Ml protein and facilitateinfectious virus production. J Virol 80: 8178-8189.
44. Ruigrok RW, Barge A, Durrer P, Bmnner J, Ma K, et al. (2000) Membraneinteraction of influenza virus Ml protein. Virology 267: 289-298.
45. Johansson BE, Bucher DJ, Рокоту В A, Mikhail A, Kilboume ED (1989) Identification of PR8 Ml protein in influenza virus high-yield reassortants byMl-specific monoclonal antibodies. Virology 171: 634-636.
46. McMurry JA, Johansson BE, De Groot AS (2008) A call to cellular &humoral arms: enlisting cognate T cell help to develop broad-spectmm vaccines againstinfluenza A. Hum Vaccin 4: 148-157.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИВАЛЕНТНОЙ ВАКЦИНЫ ОТ ГРИППА | 2018 |
|
RU2701953C1 |
| ПОЛИВАЛЕНТНАЯ ВАКЦИНА ПРОТИВ ГРИППА | 2018 |
|
RU2706191C1 |
| Лентивирусная плазмида (варианты), способ ее получения (варианты), набор праймеров для получения лентивирусного плазмидного вектора (варианты) | 2018 |
|
RU2680537C1 |
| СПОСОБ КОНТРОЛЯ ПОЛУЧЕНИЯ ПРОДУКТА ВАКЦИНА ОТ ГРИППА | 2018 |
|
RU2701959C1 |
| Вирусоподобная частица вируса гриппа и способ ее получения | 2018 |
|
RU2681439C1 |
| MDCK клетка-продуцент белков вируса гриппа (варианты) | 2018 |
|
RU2681482C1 |
| Кассета, предназначенная для получения плазмидных векторов, используемых для создания клеток-продуцентов вирусоподобных частиц (ВПЧ) вируса гриппа | 2018 |
|
RU2680703C1 |
| ПОЛУЧЕНИЕ ВИРУСОПОДОБНЫХ ЧАСТИЦ ВИРУСА ГРИППА В РАСТЕНИЯХ | 2014 |
|
RU2742607C1 |
| ПОЛУЧЕНИЕ ВИРУСОПОДОБНЫХ ЧАСТИЦ ВИРУСА ГРИППА В РАСТЕНИЯХ | 2014 |
|
RU2705555C2 |
| Штамм бактерий Escherichia coli - продуцент рекомбинантного белка NS1 | 2021 |
|
RU2789735C1 |












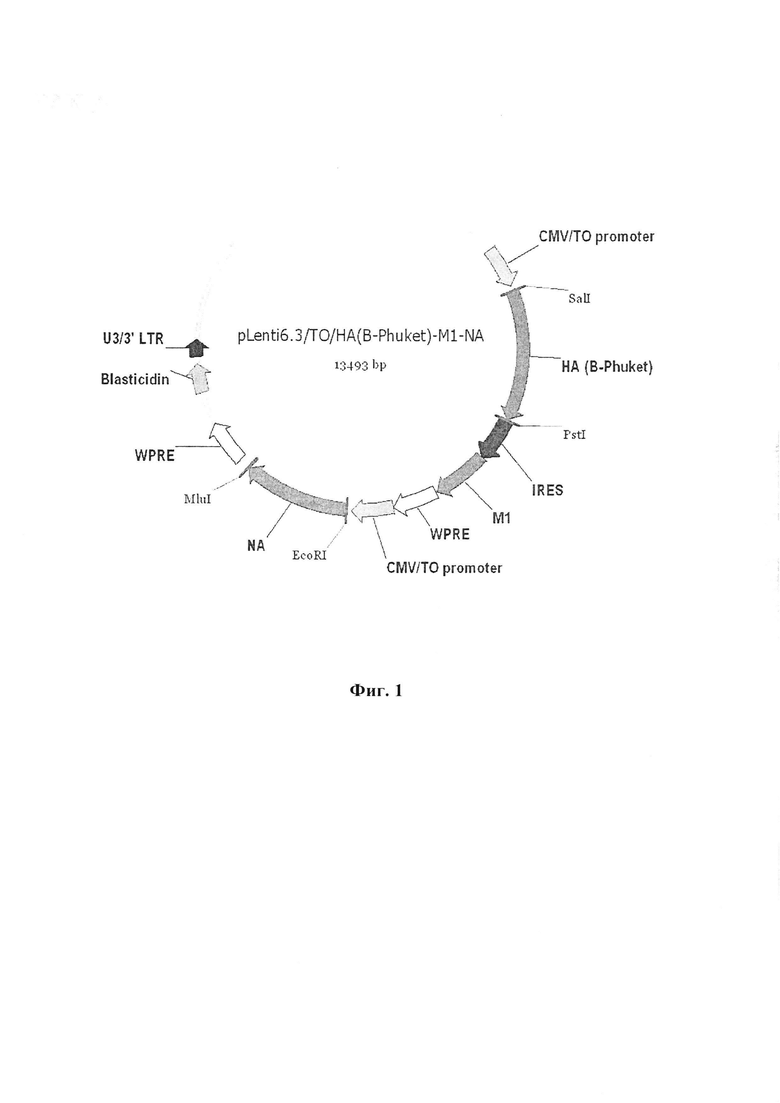
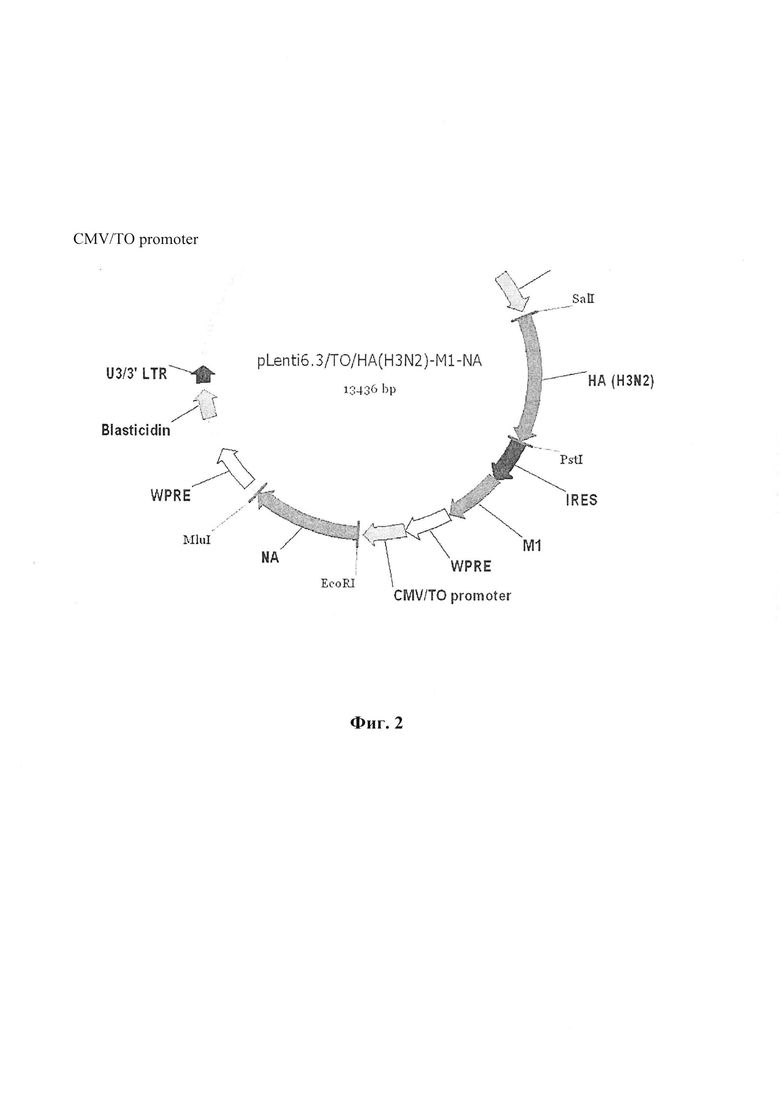
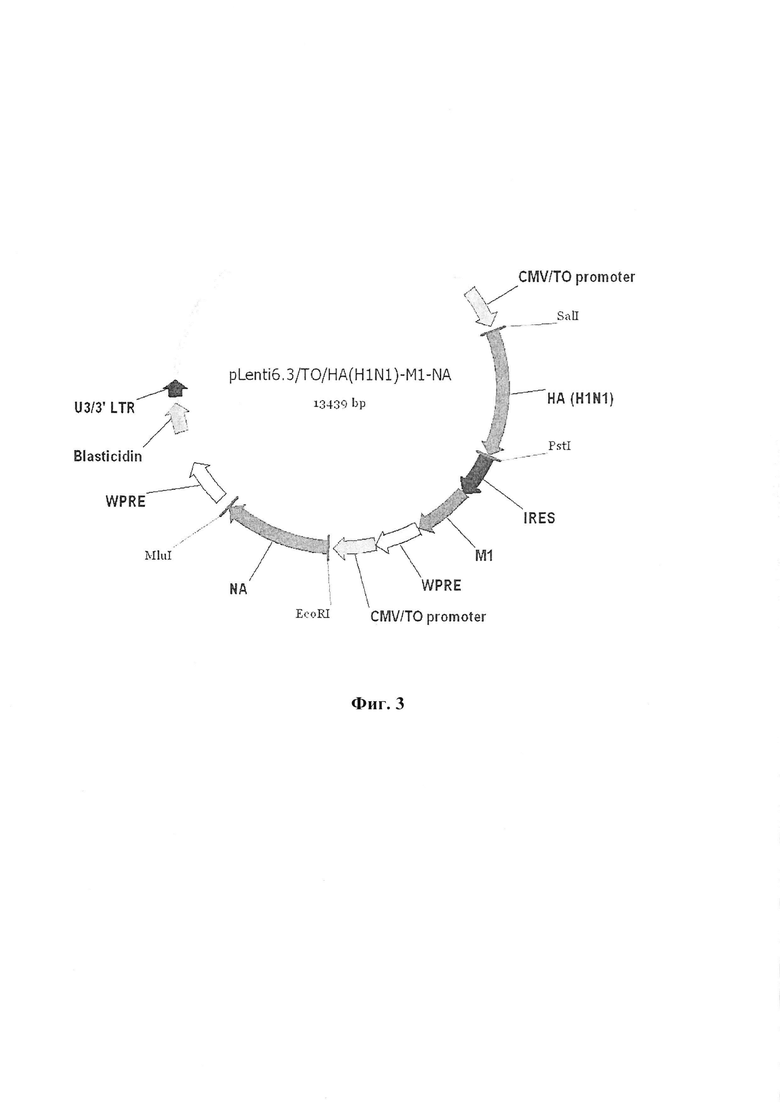
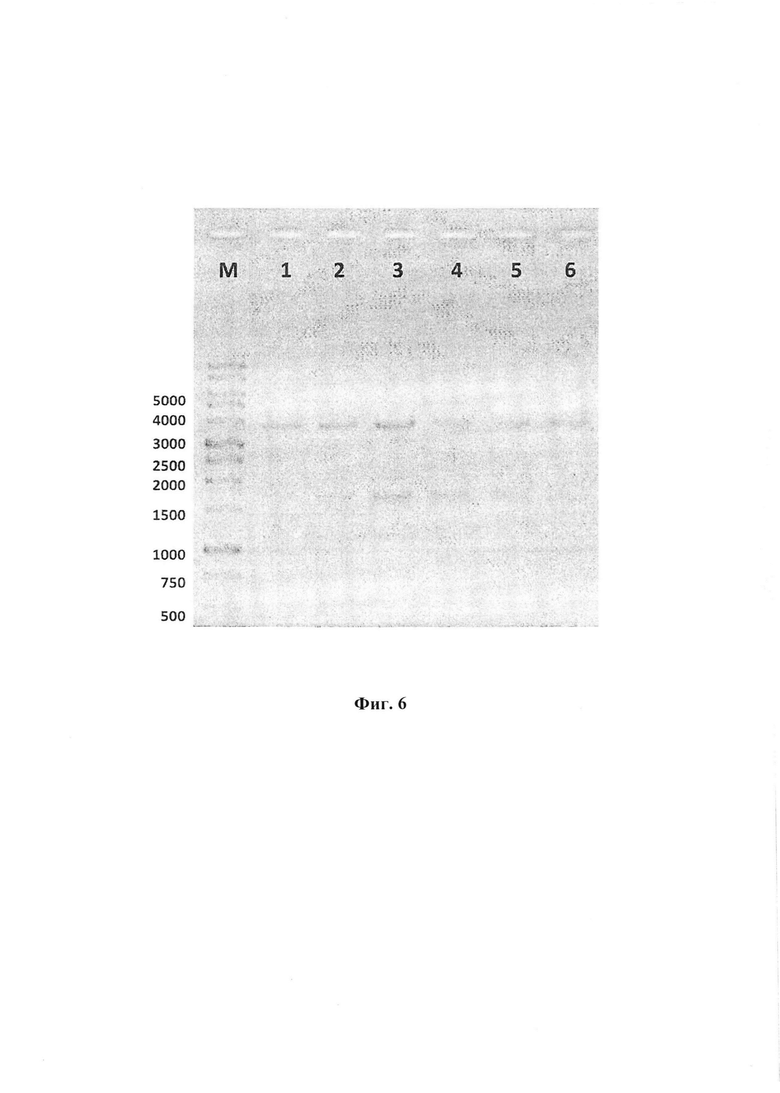




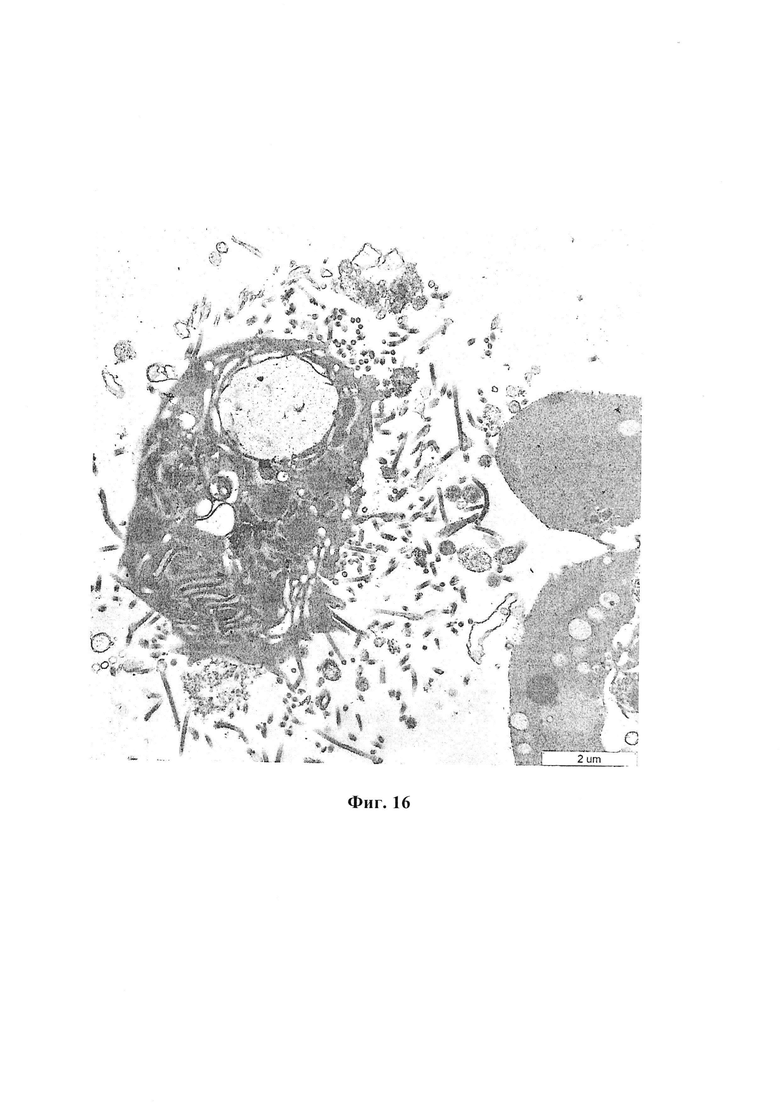
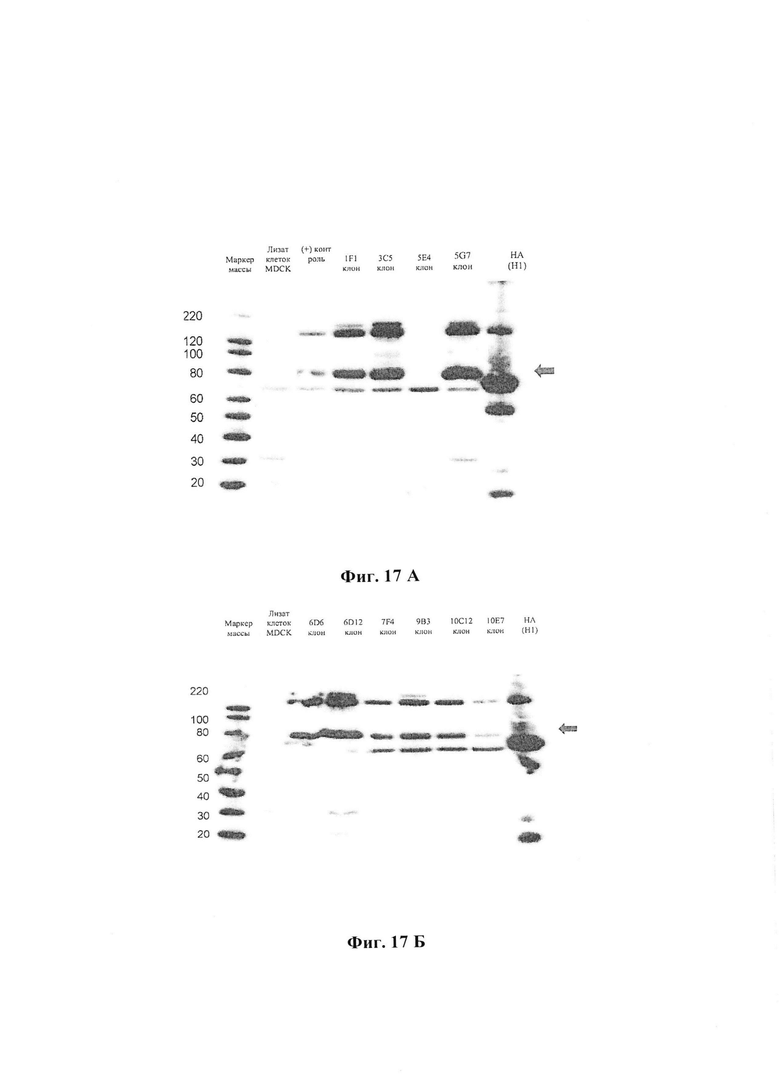
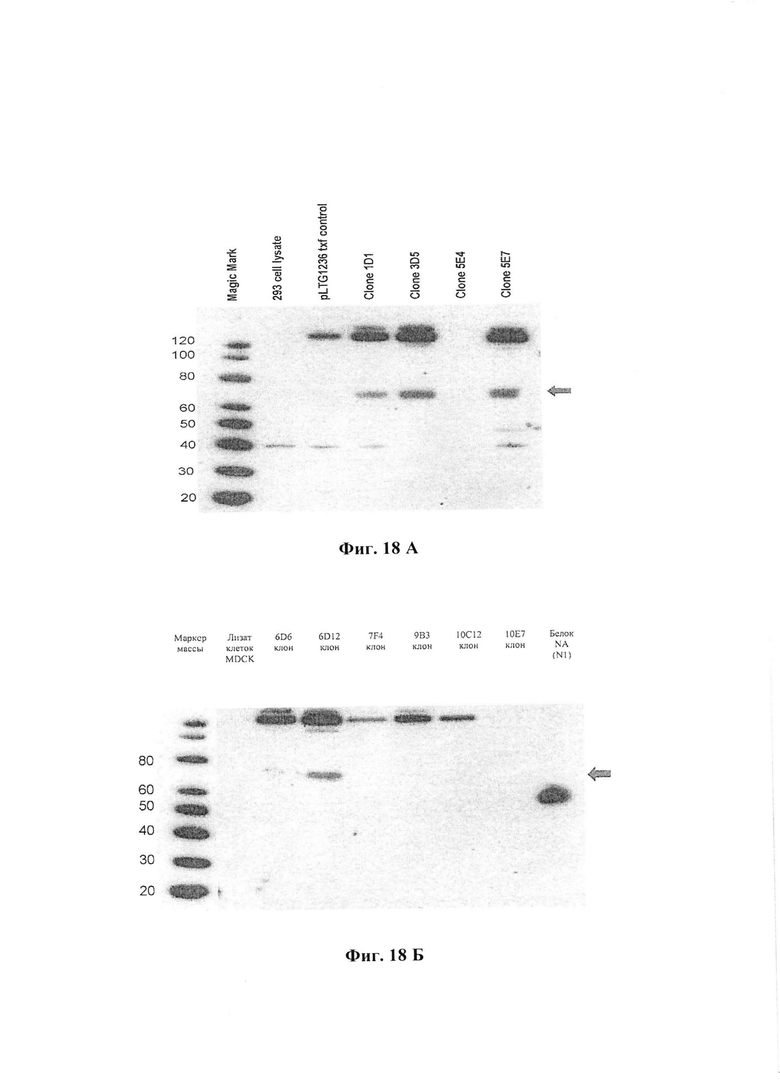
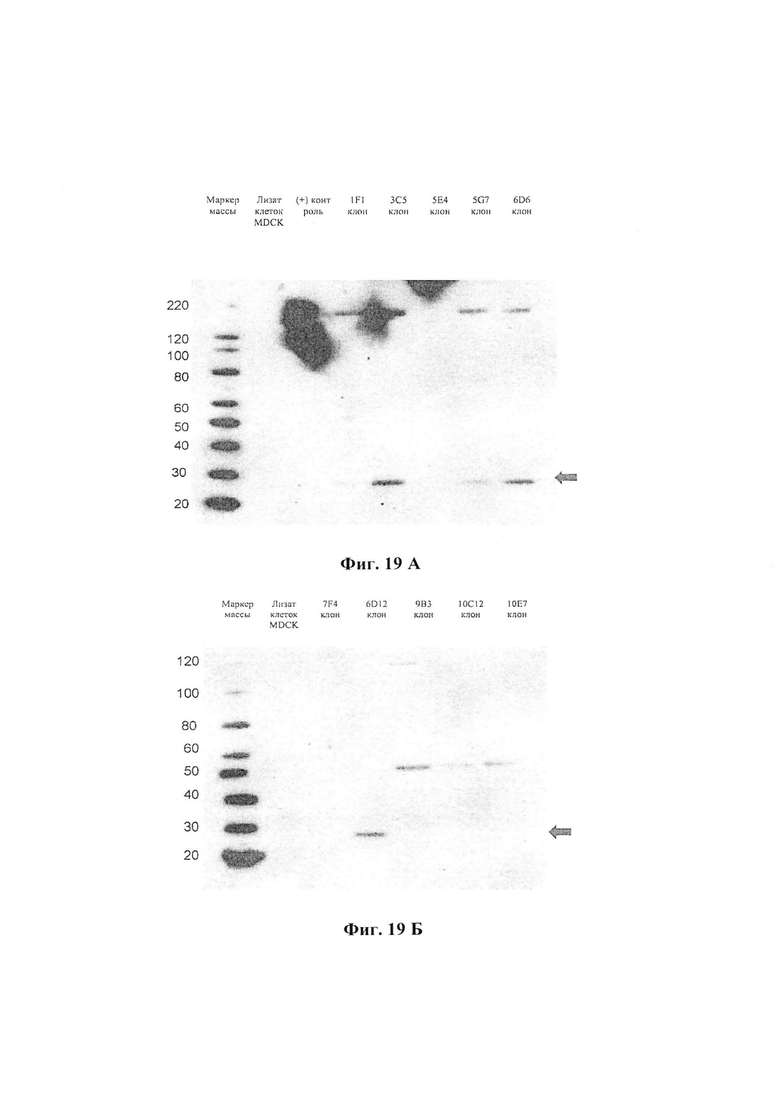




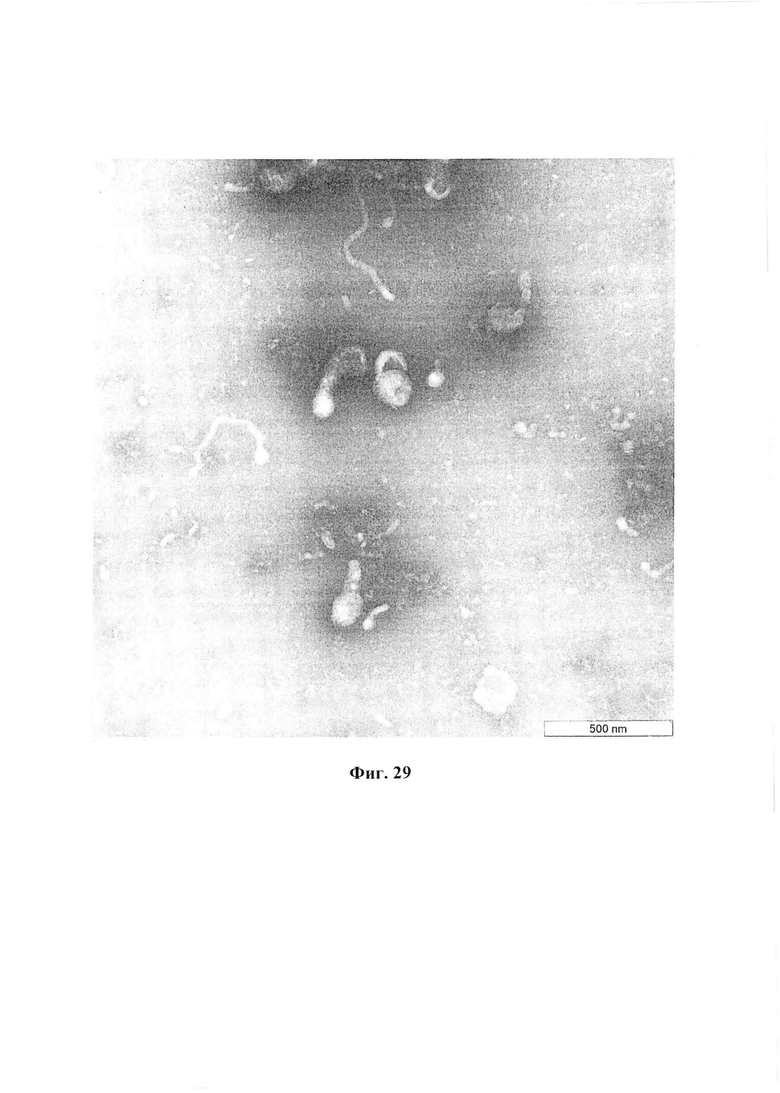
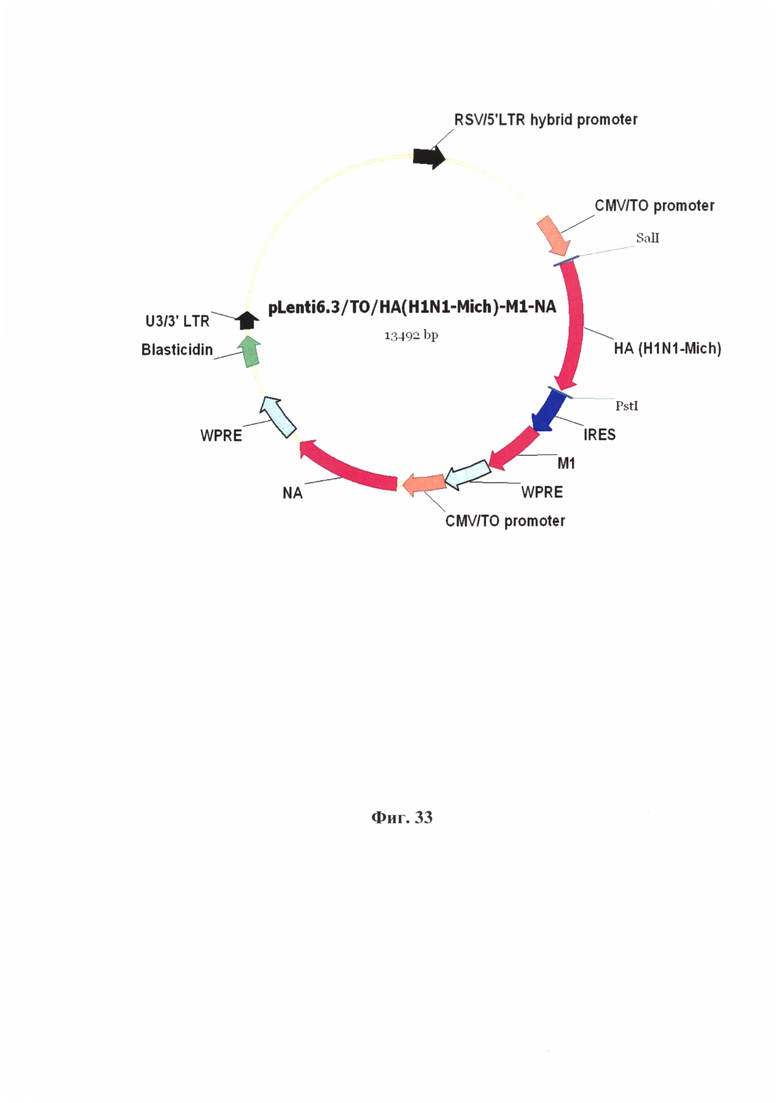

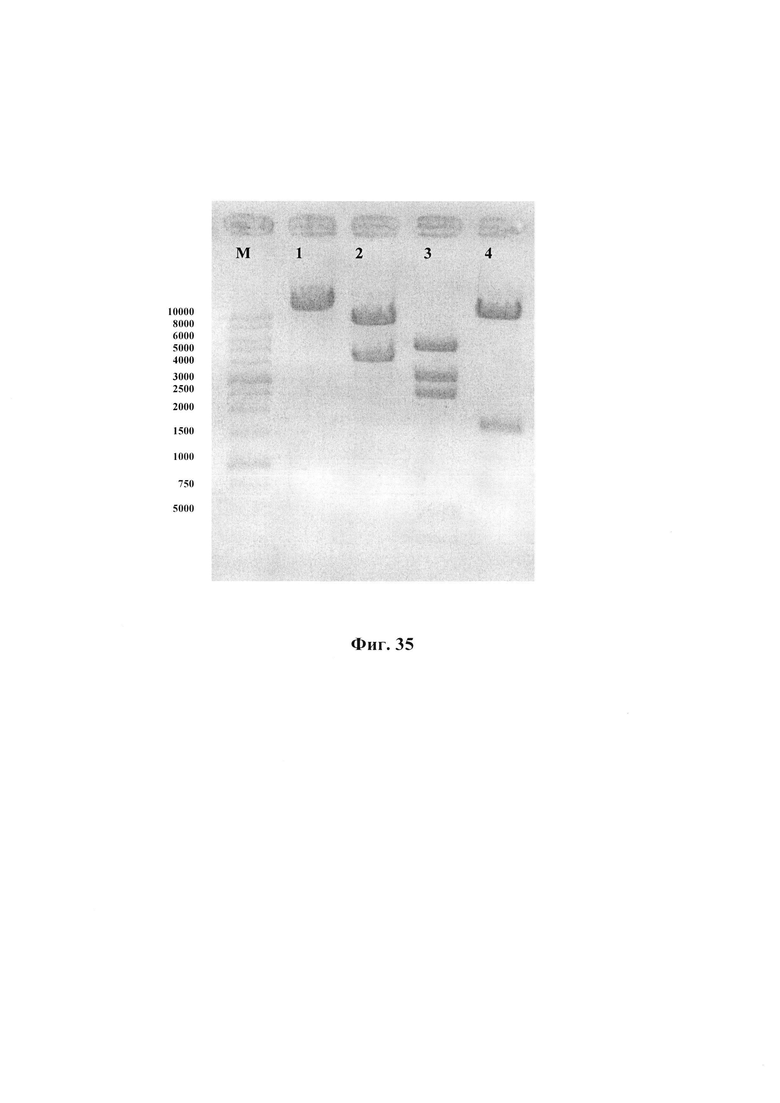



Изобретение относится к биотехнологии. Описан способ получения концентрата вакцины от гриппа, содержащей ВПЧ, где гены гемагглютинина, нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/Michigan/45/2015, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/Novosibirsk/01/2014, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма A/HongKong/4801/2014, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009; ген гемагглютинина получен из штамма B/Phuket/3073/13, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009 или ген гемагглютинина получен из штамма B/Brisbane/60/2008, а гены нейраминидазы и M1 белка получены из штамма A/California/04/2009. Представлен концентрат вакцины против гриппа, содержащий один из указанных типов ВПЧ. Изобретение позволяет получить ВПЧ вируса гриппа, имеющие большую гемагглютинирующую активность. Также способ позволяет повысить выход антигена. Кроме того, при осуществлении способа сохраняется жизнеспособность и высокая продуктивность целевых белков без использования регулируемой экспрессии у клеток-продуцентов ВПЧ гриппа. 2 н. и 2 з.п. ф-лы, 44 ил., 15 табл., 7 пр.
1. Способ получения концентрата вакцины от гриппа, включающий получение MDCK клеток-продуцентов поверхностных белков вируса гриппа, первой MDCK клетки-продуцента, содержащей лентивирусный плазмидный вектор pLenti6.3/TO/HA(H1N1)-M1-NA, содержащий гены гемагглютинина, нейраминидазы и M1 белка вируса гриппа (H1N1), представленный на Фиг. 3 или 33; второй MDCK клетки-продуцента, содержащей лентивирусный плазмидный вектор pLenti6.3/TO/HA(B)-M1-NA, содержащий ген гемагглютинина вируса гриппа В, а также гены нейраминидазы и M1 белка вируса гриппа (H1N1), представленный на фиг. 1 или 32; третьей MDCK клетки-продуцента, содержащей лентивирусный плазмидный вектор pLenti6.3/TO/HA(H3N2)-M1-NA, содержащий клонированный ген гемагглютинина из штамма субтипа H3N2, а также гены нейраминидазы и M1 белка штамма субтипа (H1N1), представленный на Фиг. 2 или 31; наработку и очистку ВПЧ, где ВПЧ нарабатывают путем раздельного культивирования указанных трех клеточных культур MDCK; при этом очистка включает осветление клеточного пула, обработку бензоназой, элюирование ВПЧ ступенчатым градиентом, эксклюзионную хроматографию, аффинную хроматографию, концентрирование путем ионообменной хроматографии и дополнительную очистку ВПЧ в два этапа гель-проникающей хроматографией и псевдоаффинной хроматографией после очистки.
2. Способ по п. 1, где инкубацию культуры MDCK клеток-продуцентов ВПЧ вируса гриппа проводят с посевной концентрацией 3×103 клеток/мл в биореакторах при температуре 37°С в течение 2-3 суток в ростовой питательной среде DMEM, содержащей 10% эмбриональной телячьей сыворотки, до появления монослоя, затем полученные клетки-продуценты промывают фосфатным буферным раствором, вносят поддерживающую питательную среду OptiMEM с добавлением 5 мкг/мл трипсина ТРСК, наработку ВПЧ проводят при температуре 37±1°С в течение 72 ч в режиме непрерывного перемешивания при скорости вращения 10-11 об/мин, до достижения активности РГА 1:64 и выше, при этом процедуру наработки ВПЧ гриппа могут повторять многократно до тех пор, пока урожай ВПЧ в супернатанте по результатам РГА не становится менее 1:64.
3. Способ по п. 1, где ВПЧ содержит: гены гемагглютинина, нейраминидазы и M1 белка, полученные из штамма A/California/04/2009; ген гемагглютинина, полученный из штамма A/Michigan/45/2015, а гены нейраминидазы и M1 белка, полученные из штамма A/California/04/2009; ген гемагглютинина, полученный из штамма A/Novosibirsk/01/2014, а гены нейраминидазы и M1 белка, полученные из штамма A/California/04/2009; ген гемагглютинина, полученный из штамма A/HongKong/4801/2014, а гены нейраминидазы и M1 белка, полученные из штамма A/California/04/2009; ген гемагглютинина, полученный из штамма B/Phuket/3073/13, а гены нейраминидазы и M1 белка, полученные из штамма A/California/04/2009, или ген гемагглютинина, полученный из штамма B/Brisbane/60/2008, а гены нейраминидазы и M1 белка, полученные из штамма A/California/04/2009.
4. Концентрат вакцины от гриппа, полученный способом по пп. 1-3, включающий ВПЧ одного из трех типов, где первый тип ВПЧ имеет белки HA(H1N1), M1 и NA; второй тип ВПЧ имеет белки HA(B), M1 и NA; третий тип ВПЧ имеет белки HA(H3N2), M1 и NA, при этом НА получена из штамма A/California/04/2009; штамма A/Michigan/45/2015; штамма A/Novosibirsk/01/2014; A/HongKong/4801/2014; штамма B/Phuket/3073/13 и из штамма B/Brisbane/60/2008, М1 и NA получены из штамма A/California/04/2009.
| РЕКОМБИНАНТНЫЕ ВИРУСЫ ГРИППА А | 2001 |
|
RU2280690C2 |
| WO 2004110484 A1, 23.12.2004. | |||

