Область техники, к которой относится изобретение
Настоящее раскрытие относится к фармацевтической области, и более конкретно, к наночастицам, содержащим человеческий сывороточный альбумин и гидрофобное лекарственное средство, и способу его получения, и даже более конкретно, к наночастицам альбумина, имеющим специфичные физические свойства, и способу их получения.
Предшествующий уровень техники
Лекарственное средство против злокачественных новообразований, паклитаксел, действует в качестве ингибитора микротрубочек при митозе, и играет важную роль в полимеризации и стабилизации внутриклеточных микротрубочек. На стадии митоза паклитаксел делает невозможным разделение микротрубочек, таким образом, клетки блокируются между фазами G2 и M. В результате, быстро делящиеся опухолевые клетки задерживаются на фазе митоза паклитакселом, что приводит к смерти клеток вследствие затрудненной репликации. Паклитаксел обладает важной клинической активностью по отношению к различным злокачественным новообразованиям (например, раку молочной железы, раку яичника, раку легких и раку мочевого пузыря и т.д.).
Однако, вследствие слабой растворимости в воде, применение паклитаксела в человеческом организме ограничено. Чтобы сделать паклитаксел пригодным для внутривенной инъекции, компания Бристоль-Майерс Сквибб (BMS) разработала ТАКСОЛ®, в котором поверхностно-активное вещество, полиоксиэтиленовое касторовое масло (КРЕМОФОР® EL), и безводный спирт добавляют вместе в качестве сорастворителя для увеличения растворимости паклитаксела. Таксол обладает значительной активностью по отношению к раку яичника, раку молочной железы, раку легких, раку пищевода и раку головы и шеи. Однако было продемонстрировано, что Таксол может индуцировать относящуюся к терапии токсичность, и значительную острую и кумулятивную токсичность, такую как миелосупрессию, фебрильную нейтропению и гиперчувствительность и т.д. Эти побочные эффекты относятся к применяемому поверхностно-активному полиоксиэтиленовому касторовому маслу (Anantbhushan et al., Asia Pac J Clin Oncol. 2013;9:176-181). На основании отчетов по клиническим исследованиям и послепродажных данных по безопасности, суммарная частота случаев гиперчувствительности, индуцированной Таксолом, составляет приблизительно 39%. В настоящее время, когда применяют Таксол, пациентам заблаговременно вводят антигистаминные средства и стероиды, чтобы уменьшить побочные эффекты, обусловленные поверхностно-активным веществом.
Чтобы улучшить растворимость паклитаксела в воде, исследователи также проводят структурные модификации с использованием функциональных групп, которые могут обеспечить более высокую растворимость в воде, получая, например, сульфонатные производные (Kingston et al., Патент США 5059699 (1991)), и сложноэфирные производные аминокислот (Mathew et al., J. Med. Chem. 35:145-151 (1992)). Они проявляют очевидную биологическую активность после модификации. Однако эти модификации могут индуцировать нежелательные побочные эффекты или уменьшать фармацевтическую эффективность помимо увеличения стоимости фармацевтических составов.
Чтобы избежать неблагоприятных воздействий КРЕМОФОР® EL в составах формах паклитаксела, была разработана еще одна другая система доставки лекарственных средств, которая не содержит какого-либо эмульгирующего средства или поверхностно-активного вещества. Такая система находится в форме микрочастиц или наночастиц и содержит альбумин, часть которого образует комплексы с паклитакселом. Однако эта система все еще имеет много недостатков. Например, для состава требуется большое количество человеческого сывороточного альбумина (ЧСА), который может вызывать аллергию. ЧСА все еще получают из человеческой крови, и он обладает потенциальными рисками для безопасности вследствие возможных загрязнений во время сбора и хранения крови. Кроме того, ЧСА является относительно дорогостоящим и в некоторых регионах может быть дефицитным.
Краткое содержание сущности изобретения
Настоящее раскрытие предоставляет очищенные терапевтические наночастицы, композиции, содержащие такие наночастицы, способы получения таких наночастиц или композиций, и способы применения таких наночастиц или композиций.
В одном аспекте настоящее раскрытие предоставляет очищенные терапевтические наночастицы, которые содержат активный ингредиент и человеческий сывороточный альбумин, где массовое отношение человеческого сывороточного альбумина (ЧСА) к активному ингредиенту в терапевтических наночастицах выбирают из 0,01:1, 0,02:1, 0,04:1, 0,05:1, 0,06:1, 0,07:1, 0,08:1, 0,09:1, 0,10:1, 0,11:1, 0,12:1, 0,13:1, 0,14:1, 0,15:1, 0,16:1, 0,17:1, 0,18:1, 0,19:1, 0,2:1, 0,21:1, 0,22:1, 0,23:1, 0,24:1, 0,25:1, 0,26:1, 0,27:1, 0,28:1, 0,29:1, 0,3:1, 0,31:1, 0,32:1, 0,33:1, 0,34:1, 0,35:1, 0,36:1, 0,37:1, 0,38:1, 0,39:1, 0,4:1, 0,41:1, 0,42:1, 0,43:1, 0,44:1, 0,45:1, 0,46:1, 0,47:1, 0,48:1, 0,49:1, 0,5:1, 0,51:1, 0,52:1, 0,53:1, 0,54:1, 0,55:1, 0,56:1, 0,57:1, 0,58:1, 0,59:1, 0,6:1, 0,65:1, 0,70:1, 0,75:1, 0,8:1, 0,85:1, 0,9:1, 0,95:1, 1:1, 1,5:1, 2:1, 2,5:1, 3:1, 3,5:1, 4:1, 4,5:1, 5:1, 5,5:1, 6:1, 6,5:1, 7:1, 7,5:1, 8:1, 8,5:1 или интервала между двумя отношениями, указанными выше, и, где терапевтические наночастицы не содержат по существу свободного ЧСА, который не включен в наночастицы.
В некоторых вариантах осуществления массовое отношение человеческого сывороточного альбумина к активному ингредиенту выбирают из 0,03:1, 0,04:1, 0,05:1, 0,06:1, 0,07:1, 0,08:1, 0,09:1, 0,10:1, 0,11:1, 0,12:1, 0,13:1, 0,14:1, 0,15:1, 0,16:1, 0,17:1, 0,18:1, 0,19:1, 0,2:1, 0,21:1, 0,22:1, 0,23:1, 0,24:1, 0,25:1, 0,26:1, 0,27:1, 0,28:1, 0,29:1, 0,3:1, 0,31:1, 0,32:1, 0,33:1, 0,34:1, 0,35:1, 0,36:1, 0,37:1, 0,38:1, 0,39:1, 0,4:1, 0,41:1, 0,42:1, 0,43:1, 0,44:1, 0,45:1, 0,46:1, 0,47:1, 0,48:1, 0,49:1, 0,5:1,, 0,6:1, 0,7:1, 0,8:1, 0,9:1 или интервала между двумя отношениями, указанными выше.
В некоторых вариантах осуществления наночастицы содержат по большей мере 5% свободного ЧСА (масс.).
В некоторых вариантах осуществления активный ингредиент имеет свойства быть нерастворимым или малорастворимым в воде, но растворимым или легкорастворимым в органическом растворителе.
В некоторых вариантах осуществления активный ингредиент выбирают из таксанов, макролидов, камптотецинов, антрациклиновых антибиотиков, колхицина, димера тиоколхицина, амиодарона, лиотиронина, циклоспорина, эксеместана, флутамида, фулвестранта, ромидепсина, семустина и ибупрофена.
В некоторых вариантах осуществления активный ингредиент представляет собой таксан, такой как паклитаксел или доцетаксел.
В некоторых вариантах осуществления органический растворитель представляет собой чистый растворитель, имеющий низкую растворимость в воде и низкую температуру кипения или его смесь с низкомолекулярными спиртами.
В некоторых вариантах осуществления активный ингредиент инкапсулируют внутри человеческого сывороточного альбумина.
В некоторых вариантах осуществления средний размер частицы терапевтических наночастиц выбирают из: 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 165, 170, 175, 180, 185, 190, 195, 200 нм или интервала между двумя числовыми значениями, приведенными выше.
В еще одном аспекте настоящее раскрытие предоставляет очищенные терапевтические наночастицы, которые содержат активный ингредиент и человеческий сывороточный альбумин, где активный ингредиент инкапсулируют внутри человеческого сывороточного альбумина; массовое отношение человеческого сывороточного альбумина к активному ингредиенту составляет от 0,03:1 до 0,7:1; средний размер частицы терапевтических наночастиц составляет от 50 нм до 190 нм; и наночастицы не содержат по существу свободного ЧСА, который не включен в наночастицы.
В еще одном аспекте настоящее раскрытие предоставляет фармацевтическую композицию, содержащую очищенные терапевтические наночастицы, предоставленные в настоящем описании, где композиция по существу не содержит свободного ЧСА, который не включен в наночастицы.
В некоторых вариантах осуществления фармацевтическая композиция находится в форме жидкости или лиофилизированного порошка.
В некоторых вариантах осуществления где фармацевтическая композиция находится в форме лиофилизированного порошка, она содержит один или несколько эксципиентов лиофилизации, таких как маннит, сахароза, лактоза, мальтоза, трегалоза, декстран или их смесь.
В некоторых вариантах осуществления фармацевтическая композиция содержит по большей мере 5% свободного ЧСА (масс.).
В еще одном аспекте, настоящее раскрытие предоставляет способ получения очищенных терапевтических наночастиц, предоставленных в настоящем описании. Способ включает в себя:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы, и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы, указанных выше;
3) удаление органического растворителя в эмульсии с получением суспензии, содержащей терапевтические наночастицы; и
4) удаление свободного ЧСА, который не включен в наночастицы, из суспензии с получением очищенных терапевтических наночастиц.
В некоторых вариантах осуществления органический растворитель выбирают из одного или нескольких из хлороформа и этанола.
В некоторых вариантах осуществления способ дополнительно включает в себя: между стадиями 3) и 4), стадию диализа суспензии стадии 3) с водным раствором для удаления оставшегося органического растворителя из суспензии.
В некоторых вариантах осуществления водным раствором является вода.
В некоторых вариантах осуществления указанное отделение на стадии 4) проводят, используя метод, выбранный из: центрифугирования, диализа и эксклюзионной хроматографии.
В связанном аспекте настоящее раскрытие предоставляет способ получения фармацевтической композиции, предоставленной в настоящем описании, включающий в себя:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы, и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы, указанных выше;
3) удаление органического растворителя в эмульсии с получением суспензии, содержащей терапевтические наночастицы;
4) удаление свободного ЧСА, который не включен в наночастицы с получением очищенных терапевтических наночастиц;
5) повторное суспендирование очищенных терапевтических наночастиц в растворе, содержащем эксципиент; и
6) необязательно лиофилизацию повторной суспензии очищенных терапевтических наночастиц с получением фармацевтической композиции.
В некоторых вариантах осуществления способ дополнительно включает в себя: между стадиями 3) и 4), стадию диализа суспензии стадии 3) с водным раствором для удаления оставшегося органического растворителя из суспензии.
В некоторых вариантах осуществления водным раствором является вода.
В еще одном связанном аспекте настоящее раскрытие предоставляет способ получения фармацевтической композиции, предоставленной в настоящем описании, включающий в себя:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы, и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы, указанных выше;
3) удаление органического растворителя в эмульсии с получением суспензии, содержащей терапевтические наночастицы;
4) диализ суспензии, полученной после удаления органического растворителя посредством раствора, содержащего эксципиент, для удаления свободного ЧСА, который не включен в наночастицы; и
5) необязательно лиофилизацию диализированной суспензии с получением фармацевтической композиции.
В некоторых вариантах осуществления способ дополнительно включает в себя: между стадиями 3) и 4), стадию диализа суспензии стадии 3) с водным раствором для удаления оставшегося органического растворителя из суспензии.
В некоторых вариантах осуществления водным раствором является вода.
В еще одном аспекте настоящее раскрытие предоставляет способ лечения злокачественного новообразования, включающий в себя: введение терапевтически эффективного количества фармацевтической композиции, предоставленной в настоящем описании, субъекту, нуждающемуся в таком лечении.
В еще одном аспекте настоящее раскрытие предоставляет фармацевтическую композицию, содержащую терапевтические наночастицы, предоставленные в настоящем описании, где композиция дополнительно содержит ЧСА в качестве эксципиента лиофилизации.
Краткое описание чертежей
ФИГ. 1. Изображение сканирующей электронной микроскопии образца Примера 5.
ФИГ. 2. Изображение сканирующей электронной микроскопии наночастиц, полученных с использованием образца Примера 5.
ФИГ. 3. Дифракционная рентгенограмма паклитаксела.
ФИГ. 4. Дифракционная рентгенограмма лиофилизированного порошка альбумина.
ФИГ. 5. Дифракционная рентгенограмма наночастиц паклитаксела-альбумина, соответствующих Примеру 2.
ФИГ. 6. Дифракционная рентгенограмма наночастиц паклитаксела-альбумина, соответствующих Примеру 4.
ФИГ. 7. Дифракционная рентгенограмма наночастиц паклитаксела-альбумина, соответствующих Примеру 6.
ФИГ. 8. Дифракционная рентгенограмма наночастиц паклитаксела-альбумина, соответствующих Примеру 8.
ФИГ. 9. Профили высвобождения in vitro очищенных наночастиц различных лекарственных форм.
ФИГ. 10. Профили высвобождения in vitro очищенных наночастиц и традиционных лекарственных форм.
ФИГ. 11. Фармакокинетические профили очищенных наночастиц и традиционных лекарственных форм у собак.
ФИГ. 12. Концентрация альбумина влияет на ингибирующий эффект лекарственных средств на клетки MCF-7.
ФИГ. 13. Концентрация альбумина влияет на ингибирующий эффект лекарственных средств на клетки SPC-A-1.
ФИГ. 14. Концентрация альбумина влияет на поглощение лекарственных средств человеческими васкулярными эндотелиальными клетками EA.hy 926.
Подробное описание изобретения
Настоящее раскрытие предоставляет очищенные содержащие ЧСА терапевтические наночастицы, композиции, содержащие такие наночастицы, и способы получения или применения таких наночастиц и композиций.
Очищенные терапевтические наночастицы или композиции, которые содержат такие наночастицы, имеют одно или несколько из следующих превосходящих свойств:
(1) В сравнении с ранее известными композициями, которые содержат содержащие ЧСА наночастицы, очищенные терапевтические наночастицы или их композиции уменьшают аллергические реакции у субъектов, которым вводят наночастицы или композиции (см., например, Примеры 25, 27 и 61). Не желая быть связанными соответствием какой-либо гипотезе, авторы полагают, что уменьшение аллергических реакций может быть результатом сниженного количества полимеров ЧСА в очищенных наночастицах или их композициях. Авторы настоящего изобретения установили, что во время получения наночастиц часть мономеров ЧСА образовывали полимеры ЧСА, вызывая более тяжелые аллергические реакции (см., например, Примеры 32 и 60). Очистка наночастиц из первоначального препарата устраняет наибольшую часть свободного ЧСА, который не включен в наночастицы, включая полимеры свободного ЧСА.
(2) В сравнении с ранее известными композициями, которые содержат содержащие ЧСА наночастицы, некоторые композиции, содержащие очищенные терапевтические наночастицы, предоставленные в настоящем описании, являются более стабильными (см., например, Примеры 62 и 63). Данный факт является неожиданным, принимая во внимание значительное снижение отношения ЧСА к активным ингредиентам в очищенных наночастицах и композициях настоящего раскрытия, и учитывая мнение в данной области, что большое количество ЧСА является важным или требуется для стабилизации композиций, которые содержат наночастицы.
(3) При поддержании профилей высвобождения in vitro, максимальных переносимых доз, фармакокинетических свойств и эффективности при испытаниях на животных (см., например, Примеры 22 и 26-31), очищенные терапевтические наночастицы или их композиции, предоставленные в настоящем описании, являются более простыми для доставки или захвата человеческими клетками-мишенями (например, человеческими опухолевыми клетками и человеческими васкулярными эндотелиальными клетками) и позволяют достичь лучших желательных эффектов в этих клетках (см., например, Примеры 58 и 59).
Если не определено иным образом, все научные термины, используемые в настоящем описании, имеют такое же значение, какое является общепринятым для рядовых специалистов в области, к которой принадлежит данное раскрытие.
Несмотря на то, что числовые интервалы и приблизительные значения параметров приведены в широком интервале настоящего раскрытия, все числа в конкретных примерах описаны настолько точно, насколько возможно. Однако по существу в любых числовых значениях присутствует некоторая ошибка, которая может являться результатом стандартного отклонения при измерении для каждого из них. Кроме того, следует понимать, что все интервалы, раскрытые в данном описании, охватывают любые и все возможные поддиапазоны, содержащиеся в них. Например, следует понимать, что интервал "от 1 до 10", описанный в данном документе, охватывает любые и все возможные поддиапазоны между минимально 1 и максимально 10 (включая граничные значения); т.е., все поддиапазоны, начинающиеся от минимально 1 или более, например, от 1 до 6,1, и все поддиапазоны, оканчивающиеся при максимально 10 или менее, например, от 5,5 до 10. Кроме того, следует понимать, что любая ссылка, именуемая как "включенная в данный документ" является включенной в него полностью.
Кроме того, следует отметить, что если иным образом явно и недвусмысленно не утверждается, форма единственного числа включает аналог множественного числа, используемый в настоящем раскрытии. Термин "или" и термин "и/или" используют взаимозаменяемо, если контекст явным образом не указывает на иное.
Термин "наночастица", используемый в настоящем описании, относится к частице с, по меньшей мере, одним размером (например, 1, 2 или 3 размерами) в наномасштабе, например, на уровне приблизительно 1 нм, приблизительно 10 нм или приблизительно 100 нм.
Термин ʺприблизительноʺ означает более или менее чем 10% от конкретного значения. Например, ʺприблизительно 50 нмʺ относится к интервалу от 45 нм до 55 нм.
Термин "терапевтическая наночастица", используемый в настоящем описании, относится к наночастицам, которые могут применяться для лечения или предотвращения заболеваний, где заболевания, например, злокачественные новообразования, предпочтительно выбирают из рака печени, рака простаты и рака легких.
Термин "мономер человеческого сывороточного альбумина" или ʺмономер ЧСАʺ, используемый в настоящем описании, относится к водорастворимому глобулину, состоящему из 585 аминокислот и имеющему приблизительную молекулярную массу около 66000 Дальтонов. Он является наиболее распространенным белком в плазме крови человека. Время удерживания мономера человеческого сывороточного альбумина является самым продолжительным при эксклюзионной по размерам хроматографии, и он является преобладающим среди нормальных продуктов человеческого альбумина. ЧСА имеет множество гидрофобных участков связывания, которые могут связывать разнообразный набор лекарственных средств, особенно, нейтральные и отрицательно заряженные гидрофобные соединения.
Термин "полимер человеческого сывороточного альбумина" или ʺполимер ЧСАʺ, используемый в настоящем описании, относится к сумме полимеров, полимеризуемых с участием мономера человеческого сывороточного альбумина, включающих димер, тример и полимер. Время удерживания полимера человеческого сывороточного альбумина при эксклюзионной по размерам хроматографии является более коротким, чем для мономеров ЧСА. Полимеры ЧСА присутствуют лишь в малом количестве (обычно < 5%) среди продуктов человеческого альбумина.
ЧСА накапливается в различных растущих опухолях и используется как источник энергии и для захвата аминокислот опухолевыми клетками. gp60 (албондин) сверхэкспрессируется в эндотелии кровеносных сосудов и связывает ЧСА, чтобы перенести его в основную ткань посредством трансцитоза. gp60 не экспрессируется в клетках ткани и не вовлечен в транспорт ЧСА в клетках ткани. SPARC (секретируемый кислый белок, обогащенный цистеином), имеет гомологичные аминокислотные последовательности с gp60 и может связываться с ЧСА и антителами против gp60. SPARC сверхэкспрессируется в множестве типов опухолей и многие исследования ясно показывают, что SPARC коррелирует с тканевым клеточным захватом ЧСА. Некоторые исследователи предполагают, что лекарственные средства, связанные с ЧСА, также пересекают эндотелиальный барьер в опухолевой ткани и интернализуются в опухолевые клетки через путь трансмембранного транспорта Gp60-SPARC.
Термин ʺпо существу чистые наночастицыʺ или ʺочищенные наночастицыʺ, используемый в настоящем описании, относится к наночастицам, состоящим из человеческого сывороточного альбумина и активного ингредиента, где менее чем 10% ЧСА (например, менее чем 9%, менее чем 8%, менее чем 7%, менее чем 6%, менее чем 5%, менее чем 4%, менее чем 3%, менее чем 2%, менее чем 1%, менее чем 0,5%, или менее чем 0,1%) представляют собой свободный ЧСА.
Аналогично, термин ʺне содержат по существу свободного человеческого сывороточного альбуминаʺ или ʺне содержащие по существу свободного ЧСАʺ, используемый в настоящем описании, относится к имеющим менее чем 10% свободного ЧСА (например, менее чем 9%, менее чем 8%, менее чем 7%, менее чем 6%, менее чем 5%, менее чем 4%, менее чем 3%, менее чем 2%, менее чем 1%, менее чем 0,5%, или менее чем 0,1%).
Термин ʺсвободный человеческий сывороточный альбуминʺ или ʺсвободный ЧСАʺ, используемый в настоящем описании, относится к ЧСА, не включенному в наночастицы. Количество свободного ЧСА в наночастицах или их композициях может быть измерено посредством методов, известных в данной области, или метода, предоставленного в настоящем раскрытии, такого как в Примерах 11 и 17-19.
Термин "активный ингредиент", используемый в настоящем описании, относится к активному фармацевтическому ингредиенту. Конкретно, активный ингредиент относится к любому веществу или молекулярной основе, которые могут иметь терапевтическое значение (например, лечение, предотвращение, смягчение течения заболевания или подавление любого заболевания и/или расстройства).
Термин ʺкратность диализаʺ, используемый в настоящем описании, относится к объемному отношению потребляемого диализата к раствору образца в процессе диализа, где объем раствора образца поддерживается постоянным.
Термин ʺэксципиент лиофилизацииʺ, используемый в настоящем описании, относится к соединениям, добавленным к фармацевтической композиции, которая содержит очищенные терапевтические наночастицы, чтобы поддержать такие наночастицы во время процесса сублимационной сушки.
Термины, ʺлечитьʺ и ʺлечениеʺ относятся к медицинскому контролю заболевания, расстройства или состояния субъекта (т.е., пациента) (см., например, Stedmanʹs Medical Dictionary). ʺЛечение злокачественного новообразованияʺ относится к снижению числа симптомов злокачественного новообразования, уменьшению тяжести одного или более симптомов, или задержке прогрессирования злокачественного новообразования.
ʺТерапевтически эффективная дозаʺ конкретного терапевтического средства относится к такому количеству средства, достаточному для получения в результате снижения тяжести, устранения или задержки наступления или повторного наступления одного или более симптомов злокачественного новообразования статистически значимым образом.
В одном аспекте, настоящее раскрытие предоставляет очищенные терапевтические наночастицы, которые содержат активный ингредиент и ЧСА.
В нескольких вариантах осуществления массовое отношение человеческого сывороточного альбумина к активному ингредиенту в терапевтических наночастицах выбирают из группы, состоящей из 0,01:1, 0,02:1, 0,04:1, 0,05:1, 0,06:1, 0,07:1, 0,08:1, 0,09:1, 0,10:1, 0,11:1, 0,12:1, 0,13:1, 0,14:1, 0,15:1, 0,16:1, 0,17:1, 0,18:1, 0,19:1, 0,2:1, 0,21:1, 0,22:1, 0,23:1, 0,24:1, 0,25:1, 0,26:1, 0,27:1, 0,28:1, 0,29:1, 0,3:1, 0,31:1, 0,32:1, 0,33:1, 0,34:1, 0,35:1, 0,36:1, 0,37:1, 0,38:1, 0,39:1, 0,4:1, 0,41:1, 0,42:1, 0,43:1, 0,44:1, 0,45:1, 0,46:1, 0,47:1, 0,48:1, 0,49:1, 0,5:1, 0,51:1, 0,52:1, 0,53:1, 0,54:1, 0,55:1, 0,56:1, 0,57:1, 0,58:1, 0,59:1, 0,6:1, 0,65:1, 0,70:1, 0,75:1, 0,8:1, 0,85:1, 0,9:1, 0,95:1, 1:1, 1,5:1, 2:1, 2,5:1, 3:1, 3,5:1, 4:1, 4,5:1, 5:1, 5,5:1, 6:1, 6,5:1, 7:1, 7,5:1, 8:1, 8,5:1 или интервала между двумя отношениями, указанными выше.
В нескольких конкретных вариантах осуществления массовое отношение человеческого сывороточного альбумина к активному ингредиенту выбирают из группы, состоящей из 0,03:1, 0,04:1, 0,05:1, 0,06:1, 0,07:1, 0,08:1, 0,09:1, 0,10:1, 0,11:1, 0,12:1, 0,13:1, 0,14:1, 0,15:1, 0,16:1, 0,17:1, 0,18:1, 0,19:1, 0,2:1, 0,21:1, 0,22:1, 0,23:1, 0,24:1, 0,25:1, 0,26:1, 0,27:1, 0,28:1, 0,29:1, 0,3:1, 0,31:1, 0,32:1, 0,33:1, 0,34:1, 0,35:1, 0,36:1, 0,37:1, 0,38:1, 0,39:1, 0,4:1, 0,41:1, 0,42:1, 0,43:1, 0,44:1, 0,45:1, 0,46:1, 0,47:1, 0,48:1, 0,49:1, 0,5:1, 0,6:1, 0,7:1, 0,8:1, 0,9:1 или интервала между двумя отношениями, указанными выше, например, от 0,03:1 до 0,19:1, или от 0,21:1 до 0,9:1.
Более конкретно, в некоторых вариантах осуществления массовое отношение человеческого сывороточного альбумина к активному ингредиенту составляет 0,043:1, 0,071:1, 0,13:1, 0,15:1, 0,16:1, 0,17:1, 0,18:1, 0,24:1, или 0,57:1, или находится в интервале между двумя отношениями, указанными выше, например, от 0,043:1 до 0,071:1, от 0,043:1 до 0,13:1, от 0,043:1 до 0,14:1, от 0,043:1 до 0,15:1, от 0,043:1 до 0,16:1, от 0,043:1 до 0,17:1, от 0,043:1 до 0,18:1, от 0,043:1 до 0,24:1, от 0,043:1 до 0,57:1, от 0,071:1 до 0,13:1, от 0,071:1 до 0,15:1, от 0,071:1 до 0,16:1, от 0,071:1 до 0,17:1, от 0,071:1 до 0,18:1, от 0,071:1 до 0,24:1, от 0,071:1 до 0,57:1, от 0,13:1 до 0,15:1, от 0,13:1 до 0,16:1, от 0,13:1 до 0,17:1, от 0,13:1 до 0,18:1, от 0,13:1 до 0,24:1, от 0,13:1 до 0,57:1, от 0,15:1 до 0,16:1, от 0,15:1 до 0,17:1, от 0,15:1 до 0,18:1, от 0,15:1 до 0,24:1, от 0,15:1 до 0,57:1, от 0,16:1 до 0,17:1, от 0,16:1 до 0,18:1, от 0,16:1 до 0,24:1, от 0,16:1 до 0,57:1, от 0,17:1 до 0,18:1, от 0,17:1 до 0,24:1, от 0,17:1 до 0,57:1, от 0,18:1 до 0,24:1, от 0,18:1 до 0,57:1 или от 0,24:1 до 0,57:1.
В нескольких вариантах осуществления активный ингредиент, подходящий для инкапсулирования внутри человеческого сывороточного альбумина, является нерастворимым или малорастворимым в воде и растворимым или легкорастворимым в органическом растворителе. Органический растворитель может представлять собой чистый растворитель с низкой растворимостью в воде (т.е., растворимостью в воде менее чем 6%) и низкой температурой кипения (т.е., температурой кипения менее чем 80°C) или смесь указанного выше чистого растворителя с низкомолекулярными спиртами, включающими этанол, трет-бутанол, изопропанол и т.д. Конкретные органические растворители включают, но без ограничений, хлороформ, дихлорметан и т.д.
Активный ингредиент, подходящий для настоящего раскрытия, принадлежит к таксанам, включающим, но не ограниченным перечисленными, паклитаксел, доцетаксел, кабазитаксел, гидрофобные производные доцетаксела (например, 2ʹ-О-гексанолилдоцетаксел, и 2ʹ-бензоилдоцетаксел); или макролидам, включающим, но не ограниченным перечисленными, рапамицин и его производные (например, темсиролимус и эверолимус), эпотилон B и его производные, танеспимицин и его производные; или камптотецинам, включающим, но не ограниченным перечисленными, 10-гидроксикамптотецин, SN-38 и его производные; или антрациклиновым антибиотикам, включающим, но не ограниченным перечисленными, аклациномицин и пирарубицин; или другим активным ингредиентам, включающим колхицин и его производные, димер тиоколхицина, амиодарон, лиотиронин, циклоспорин, эксеместан, флутамид, фулвестрант, ромидепсин, семустин, ибупрофен, циклоспорин, пропофол, винбластин и т.д. В нескольких вариантах осуществления активный ингредиент выбирают из одного или нескольких из паклитаксела и доцетаксела. В конкретных вариантах осуществления активным ингредиентом является паклитаксел. В нескольких других вариантах осуществления активный ингредиент выбирают из доцетаксела, рапамицина и его производных, эксеместана, флутамида, фулвестранта и т.д.
В некоторых вариантах осуществления активный ингредиент включает, но не ограничен перечисленными: химиотерапевтические средства, радиотерапевтические средства, иммунотерапевтические средства и термические терапевтические средства и т.д. Например, аминоглутетимид, азатиоприн, блеомицина сульфат, бусульфан, кармустин, хлорамбуцил, цисплатин, циклофосфамид, циклоспорин, дакарбазин, дактиномицин, даунорубицин, амицин, паклитаксел, этопозид, фторурацил, интерферон-α, ломустин, меркаптопурин, метотрексат, митотан, прокарбазина гидрохлорид, тиогуанин, винбластина сульфат и винкристина сульфат.
В нескольких вариантах осуществления средний размер частицы терапевтических наночастиц выбирают из 30, 50, 70, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 165, 170, 175, 180, 185, 190, 195, 200 нм или интервала между двумя числовыми значениями, приведенными выше. Квалифицированный специалист в данной области должен понимать, что размер частиц может быть определен с использованием любого подходящего метода, который существует или появится в будущем, который включает, но не ограничен перечисленным, осадочный метод, метод ситового анализа, микроскопическое наблюдение или лазерный измеритель частиц. Следует также понимать, что когда терапевтические наночастицы, раскрытые в настоящем раскрытии, содержат множество частиц, не все терапевтические наночастицы должны иметь одинаковый размер частиц, и они также будут охватываться объемом настоящего раскрытия, поскольку их средний размер частиц (т.е., средний диаметр частиц) попадает в указанный выше интервал. В нескольких конкретных вариантах осуществления размер частиц определяют посредством лазерного измерителя частиц; и средний размер частицы терапевтических наночастиц выбирают из 50, 60, 70, 80, 90, 100, 110, 120, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160 нм или интервала между двумя числовыми значениями, приведенными выше.
В нескольких аспектах размеры частицы у наночастиц находятся в конкретном интервале, например, от 30 нм до 200 нм, предпочтительно, от 50 до 190 нм. Предпочтительно, размер частиц является по существу однородным.
В одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,043:1, и средний размер частицы терапевтических наночастиц равен 140 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,071:1, а средний размер частицы терапевтических наночастиц равен 134 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,13:1, а средний размер частицы терапевтических наночастиц равен 125 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,15:1, а средний размер частицы терапевтических наночастиц равен 136 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,16:1, и средний размер частицы терапевтических наночастиц равен 133 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,17:1, и средний размер частицы терапевтических наночастиц равен 136 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,18:1, и средний размер частицы терапевтических наночастиц равен 138 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,24:1, и средний размер частицы терапевтических наночастиц равен 141 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат паклитаксел и человеческий сывороточный альбумин, где паклитаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к паклитакселу составляет 0,57:1, и средний размер частицы терапевтических наночастиц равен 145 нм.
В еще одном конкретном варианте осуществления предоставлены очищенные терапевтические наночастицы, которые содержат доцетаксел и человеческий сывороточный альбумин, где доцетаксел инкапсулируют в человеческий сывороточный альбумин; и массовое отношение человеческого сывороточного альбумина к доцетакселу составляет 0,1:1, и размер частиц терапевтических наночастиц находится в интервале от 110 нм до 150 нм.
Очищенные терапевтические наночастицы настоящего раскрытия не содержат по существу свободного ЧСА. В некоторых вариантах осуществления очищенные терапевтические наночастицы содержат по большей мере 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5% или 0,1% масс. свободного ЧСА (т.е., по большей мере 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5% или 0,1% от общего ЧСА в очищенных терапевтических наночастицах представляют собой свободный ЧСА, который не связывается с активным ингредиентом и не включен в наночастицы.
В некоторых вариантах осуществления очищенные терапевтические наночастицы состоят по существу из активного ингредиента и ЧСА, где по существу весь ЧСА (т.е., более чем 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99,5% или 99,9% HSA) являются связанными с активным ингредиентом.
В некоторых вариантах осуществления очищенные терапевтические наночастицы содержат минимальное количество (например, менее чем 0,05, 0,04, 0,03, 0,02 или 0,01 мг/мл, или менее чем 5, 1, 0,5, 0,1, 0,05 или 0,01 мкг/мл) одного или нескольких органических растворителей, используемых при получении таких наночастиц.
В некоторых вариантах осуществления очищенные терапевтические наночастицы не содержат каких-либо поверхностно-активных веществ.
В некоторых вариантах осуществления очищенные наночастицы, предоставленные в настоящем описании, имеют относительно высокий электрокинетический потенциал, такой как от -20 до -45 или от -25 до -40.
В еще одном аспекте настоящее раскрытие предоставляет фармацевтическую композицию, которая содержит очищенные терапевтические наночастицы, предоставленные в настоящем описании, и по существу не содержит свободного ЧСА, который не включен в наночастицы. В некоторых вариантах осуществления фармацевтическая композиция содержит менее чем 10%, 9%, 8%, 7%, 6%, 5%, 4%, 3%, 2%, 1%, 0,5% или 0,1% свободного ЧСА.
В нескольких вариантах осуществления фармацевтическая композиция предоставлена в форме жидкости, включающей, но не ограниченной перечисленной, форму, подходящей для инъекции субъекту. В одном конкретном варианте осуществления фармацевтическую композицию получают внутри инъекции.
В нескольких других вариантах осуществления фармацевтическая композиция предоставлена в форме твердого вещества, включающей, но не ограниченной перечисленными, сухой порошок или лиофилизированный порошок.
В некоторых конкретных вариантах осуществления наночастица или фармацевтическая композиция настоящего раскрытия не содержит дефероксамин или его соль, например, дефероксамина мезилат.
В некоторых конкретных вариантах осуществления очищенные наночастицы или фармацевтические композиции настоящего раскрытия не содержат дополнительный стабилизатор.
При предоставлении в жидкой форме фармацевтическая композиция содержит терапевтические наночастицы настоящего раскрытия и фармацевтически приемлемый носитель. Терапевтические наночастицы суспендируют в фармацевтически приемлемом носителе. Фармацевтически приемлемый носитель включает, но не ограничен перечисленными, буферный раствор, консервант, воду для инъекции, нормальный физраствор и изотонический раствор. В нескольких конкретных вариантах осуществления, концентрация активного ингредиента терапевтических наночастиц составляет 1-10 мг/мл (например, 5 мг/мл) в фармацевтической композиции в жидкой форме.
При предоставлении в твердой форме фармацевтическая композиция содержит терапевтические наночастицы настоящего раскрытия и эксципиент лиофилизации, состоит по существу или состоит из них. В нескольких конкретных вариантах осуществления эксципиент лиофилизации выбирают из одного или нескольких из маннита, сахарозы, лактозы, мальтозы, трегалозы и декстрана. В некоторых конкретных вариантах осуществления терапевтические наночастицы суспендируют в растворе эксципиента лиофилизации с концентрацией от 5% до 10%, и, впоследствии, раствор лиофилизируют с получением фармацевтической композиции в форме лиофилизированного порошка. В конкретных вариантах осуществления раствор эксципиента лиофилизации выбирают из одного или нескольких из 5% раствора маннита, 10% раствора сахарозы, 5% раствора декстрана, 10% раствора лактозы, 10% раствора трегалозы и 10% раствора мальтозы. В нескольких конкретных вариантах осуществления содержание терапевтических наночастиц в фармацевтической композиции в твердой форме составляет от 4,8% до 10% масс.
Также предоставлен способ получения очищенных терапевтических наночастиц настоящего раскрытия, включающий в себя: (1) получение суспензии, содержащей терапевтические наночастицы, посредством смешивания активного ингредиента с человеческим сывороточным альбумином, и (2) очистку наночастиц из суспензии, чтобы получить по существу чистые терапевтические наночастицы.
В еще одном аспекте настоящее раскрытие также предоставляет терапевтические наночастицы, полученные посредством способа, предоставленного в настоящем описании.
В еще одном аспекте предоставлен способ получения очищенных терапевтических наночастиц. Способ включает в себя:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы, и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы;
3) удаление органического растворителя в эмульсии с получением суспензии, содержащей терапевтические наночастицы;
4) удаление свободного ЧСА, который не включен в наночастицы, из суспензии с получением очищенных терапевтических наночастиц.
Соответствующие органические растворитель (растворители) могут быть выбраны квалифицированным специалистом на основании свойств активного ингредиента. В нескольких конкретных вариантах осуществления подходящим органическим растворителем является хлороформ, этанол или смесь хлороформа и этанола, когда активными ингредиентами являются таксаны. Более конкретно, подходящим органическим растворителем является смесь хлороформа и этанола, когда активный ингредиент представляет собой паклитаксел или доцетаксел. В нескольких конкретных вариантах осуществления объемное отношение между хлороформом и этанолом находится в интервале от 1:1 до 20:1, и например, его выбирают из 1:1, 4:1, 9:1 и 11:1. В нескольких конкретных вариантах осуществления концентрация человеческого сывороточного альбумина в водной фазе находится в интервале от 2% до 10% (м./об.); например, ее выбирают из 2%, 4%, 5% и 10%. В нескольких вариантах осуществления отношение между активным ингредиентом и органическим растворителем в масляной фазе находится в интервале от 0,3-7,5 г/15-20 мл. В нескольких конкретных вариантах осуществления отношение между активным ингредиентом и органическим растворителем в масляной фазе выбирают из: 0,3 г/15 мл, 0,6 г/15 мл, 1 г/20 мл, 1,25 г/15 мл, 1,8 г/15 мл, 2 г/15 мл, 2,5 г/15 мл, 3 г/20 мл, 3 г/15 мл, 5 г/15 мл, 7,5 г/15 мл или интервала между двумя числовыми значениями, приведенными выше.
Когда образуют эмульсию типа масло-в-воде с масляной фазой и водной фазой, объемное отношение между двумя фазами выбирают из от 1:10 до 1:100. В нескольких вариантах осуществления отношение смешения масляной фазы и водной фазы равно 3:100 или 1:25. Эмульсию типа масло-в-воде образуют, используя метод, известный в данной области, который включает гомогенизацию, но не ограничивается лишь ей. В конкретных вариантах осуществления смесь масляной фазы и водной фазы эмульгируют, используя диспергатор с высоким сдвиговым усилием, и затем ее гомогенизируют, используя гомогенизатор высокого давления, таким образом, чтобы получить эмульсию типа масло-в-воде. В конкретных вариантах осуществления смесь масляной фазы и водной фазы эмульгируют в течение 2-10 минут, используя диспергатор с высоким сдвиговым усилием, и затем ее гомогенизируют, используя гомогенизатор высокого давления под давлением 10000-20000 фунтов/кв. дюйм, так, чтобы получить эмульсию типа масло-в-воде.
Органический растворитель может удаляться из эмульсии с использованием любого соответствующего метода. В нескольких вариантах осуществления органический растворитель удаляют из эмульсии с использованием испарения в вакууме при вращении. В конкретных вариантах осуществления органический растворитель удаляют из эмульсии с использованием роторного испарителя при 40°C под давлением 40 мбар. После удаления органического растворителя полученная в результате суспензия содержит терапевтические наночастицы настоящего раскрытия. Однако, избыточный альбумин, который не участвует в образовании наночастиц, также содержится в суспензии.
Избыточный свободный альбумин дополнительно удаляют из суспензии с получением очищенных терапевтических наночастиц настоящего раскрытия, которые не содержат по существу свободного ЧСА. В нескольких вариантах осуществления эту операцию проводят, используя центрифугирование, диализ или эксклюзионную хроматографию. В конкретных вариантах осуществления суспензию можно использовать непосредственно для удаления свободного ЧСА после удаления органического растворителя. Альтернативно, ее можно хранить для дополнительного применения.
Поскольку внутривенная инфузия является желательным путем введения для терапевтических наночастиц настоящего раскрытия, продукт должен быть стерильным. Стерилизация нагревом не является применимой в настоящем раскрытии, поскольку как наночастицы, так и человеческий сывороточный альбумин являются чувствительными к температуре. Таким образом, подходящие методы стерилизации включают получение в асептических условиях или стерилизующую фильтрацию. В таких случаях, после удаления органического растворителя, суспензию стерилизуют посредством пропускания через фильтр, и затем ее лиофилизируют с получением твердого вещества. Полученное твердое вещество ресуспендируют в растворе хлорида натрия. В нескольких вариантах осуществления твердое вещество ресуспендируют в 0,9% растворе хлорида натрия для достижения концентрации паклитаксела, равной приблизительно 5 мг/мл.
Когда терапевтические наночастицы очищают, используя центрифугирование, удаление свободного ЧСА проводят при 21000×g в течение 60 минут или в эквивалентных условиях.
Когда терапевтические наночастицы очищают посредством диализа, суспензию, содержащую наночастицы, полученную на стадии 3) диализируют, используя ультрафильтрационную мембрану, чтобы удалить свободный ЧСА. В конкретных вариантах осуществления жидкость, содержащую наночастицы согласно способу настоящего раскрытия, диализируют в равном объеме 5% раствора маннита, используя регенерированную целлюлозную ультрафильтрационную мембрану с пределом пропускания по молекулярной массе, равным 300 кДа, и кратностью диализа, равной 5.
Когда терапевтические наночастицы очищают, используя эксклюзионную хроматографию, терапевтические наночастицы отделяют от свободного ЧСА, используя эксклюзионную хроматографическую колонку. В конкретных вариантах осуществления суспензию, содержащую наночастицы, полученную на стадии 3), наносят на колонку с сефарозой, и элюируемый пик, соответствующий терапевтическим наночастицам, собирают.
В некоторых вариантах осуществления способ дополнительно включает в себя, между стадиями 3) и 4), стадию диализа суспензии стадии 3) с водным раствором (например, водой или 5% раствором маннита, 10% раствором сахарозы, 5% раствором декстрана, 10% раствором лактозы, 10% раствором трегалозы, и 10% раствором мальтозы и т.д) для удаления оставшегося органического растворителя из суспензии. Например, суспензия может быть диализирована в воде или водном растворе с использованием ультрафильтрационной мембраны, которая обеспечивает пропускание органического растворителя, но не ЧСА или наночастиц (например, целлюлозной ультрафильтрационной мембраны с пределом пропускания по молекулярной массе, равным 30 кДа). Не желая быть связанными какой-либо теорией, авторы полагают, что удаление или уменьшение оставшегося органического растворителя перед удалением свободного ЧСА из суспензии, содержащей наночастицы, может улучшить стабильность очищенных наночастиц или их композиции.
В еще одном аспекте также предоставлен способ получения фармацевтической композиции, содержащей терапевтические наночастицы. Способ включает в себя:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы, и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы;
3) удаление органического растворителя из эмульсии с получением суспензии, содержащей терапевтические наночастицы;
4) удаление свободного ЧСА, который не включен в наночастицы с получением очищенных терапевтических наночастиц;
5) повторное суспендирование очищенных терапевтических наночастиц в растворе, содержащем фармацевтически приемлемый носитель с получением фармацевтической композиции; и
6) необязательно, лиофилизацию повторно полученной суспензии очищенных терапевтических наночастиц, где фармацевтическая композиция находится в форме твердого вещества.
Стадии 1) и 4) данного способа являются такими же, как стадии, описанные по отношению к способу получения очищенных терапевтических наночастиц. Кроме того, в некоторых вариантах осуществления, способ включает в себя, между стадиями 3) и 4), стадию диализа суспензии стадии 3) с водным раствором для удаления оставшегося органического растворителя из суспензии, также как описано по отношению к способу получения очищенных терапевтических наночастиц.
В нескольких вариантах осуществления раствор, содержащий фармацевтически приемлемый носитель, представляет собой раствор, содержащий эксципиент лиофилизации. В нескольких конкретных вариантах осуществления эксципиент лиофилизации выбирают из одного или нескольких из маннита, сахарозы, лактозы, мальтозы, трегалозы и декстрана. В нескольких других конкретных вариантах осуществления эксципиент лиофилизации представляет собой ЧСА. В конкретных вариантах осуществления терапевтические наночастицы суспендируют в растворе эксципиента лиофилизации при концентрации от 5% до 10%.
Необязательно, фармацевтическую композицию получают в виде лиофилизированного порошка после лиофилизации. В конкретных вариантах осуществления раствор эксципиента лиофилизации выбирают из одного или нескольких из 5% раствора маннита, 10% раствора сахарозы, 5% раствора декстрана, 10% раствора лактозы, 10% раствора трегалозы, 10% раствора мальтозы и 10% раствора человеческого сывороточного альбумина.
В нескольких конкретных вариантах осуществления содержание терапевтических наночастиц в фармацевтической композиции, находящейся в жидкой форме, составляет от 0,1% до 30%, и предпочтительно, от 0,2% до 10%, и, более предпочтительно, от 0,5% до 5%, например, 1%. В нескольких конкретных вариантах осуществления содержание активного ингредиента (например, паклитаксела) в фармацевтической композиции, находящейся в жидкой форме, настоящего раскрытия составляет от 0,1 до 100 мг/мл, и предпочтительно, от 0,5 до 50 мг/мл, и, более предпочтительно, от 1 до 20 мг/мл, например, 5 мг/мл.
В некоторых вариантах осуществления содержание терапевтических наночастиц в фармацевтической композиции в твердой форме составляет от 0,1% до 80% масс., и, предпочтительно, от 0,5% до 50%, и, более предпочтительно, от 1% до 30%, например, от 2% до 10%.
В нескольких конкретных вариантах осуществления содержание активного фармацевтического ингредиента (например, паклитаксела) в фармацевтической композиции, находящейся в жидкой форме, составляет от 0,1% до 80% масс., и, предпочтительно, от 0,5% до 50%, и более предпочтительно, от 1% до 30%, например, от 2% до 10%.
Альтернативно, фармацевтическая композиция настоящего раскрытия может быть получена с использованием еще одной процедуры диализа. Способ включает в себя:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы, и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы, указанных выше;
3) удаление органического растворителя из эмульсии с получением суспензии, содержащей терапевтические наночастицы;
4) диализ суспензии, полученной после удаления органического растворителя, посредством раствора, содержащего фармацевтически приемлемый носитель для удаления свободного ЧСА, который не включен в наночастицы; и
5) необязательно лиофилизацию диализированной суспензии, когда фармацевтическую композицию получают в форме твердого вещества.
Стадии 1) и 4) данного способа являются аналогичными стадиям, описанным по отношению к способу получения очищенных терапевтических наночастиц, когда диализ применяют для удаления свободного ЧСА. Кроме того, в некоторых вариантах осуществления способ включает в себя, между стадиями 3) и 4), стадию диализа суспензии стадии 3) с водным раствором для удаления оставшегося органического растворителя из суспензии, так же, как описано по отношению к способу получения очищенных терапевтических наночастиц.
В нескольких вариантах осуществления в качестве диализата применяют раствор, содержащий фармацевтически приемлемый носитель, который представляет собой раствор, содержащий эксципиент лиофилизации. В нескольких конкретных вариантах осуществления эксципиент лиофилизации выбирают из одного или нескольких из маннита, сахарозы, лактозы, мальтозы, трегалозы и декстрана.
В конкретных вариантах осуществления терапевтические наночастицы суспендируют в растворе эксципиента лиофилизации с концентрацией от 5% до 10%. Необязательно, фармацевтическую композицию получают в виде лиофилизированного порошка после лиофилизации. В конкретных вариантах осуществления раствор эксципиента лиофилизации выбирают из одного или нескольких из 5% раствора маннита, 10% раствора сахарозы, 5% раствора декстрана, 10% раствора лактозы, 10% раствора трегалозы, 10% раствора мальтозы и 10% раствора человеческого сывороточного альбумина. В нескольких вариантах осуществления диализ осуществляют с использованием ультрафильтрационной мембраны с пределом пропускания по молекулярной массе, равным 300 кДа.
В еще одном аспекте настоящее раскрытие предоставляет способы применения очищенных терапевтических наночастиц или их композиций. Поскольку представленные очищенные наночастицы или композиции являются эффективным средством для доставки различных активных ингредиентов, они могут применяться для лечения любых заболеваний или расстройств, которые являются восприимчивыми к активным ингредиентам. Например, очищенные терапевтические наночастицы или их композиции могут применяться при лечении злокачественных новообразований, таких как рак печени, рак простаты и рак легких. Дополнительные заболевания или расстройства, которые могут подвергаться лечению, включают рак молочной железы, множественную миелому, отторжение трансплантата, рак толстой кишки, лимфому, жар и т.д.
В конкретном аспекте настоящее раскрытие предоставляет способ лечения злокачественного новообразования, который включает в себя введение терапевтически эффективного количества фармацевтической композиции, предоставленной в настоящем описании, субъекту, нуждающемуся в таком лечении. В конкретных вариантах осуществления субъект является млекопитающим, включающим, но не ограниченным перечисленными, человека, собаку, мышь и крысу.
Терапевтически эффективное количество фармацевтической композиции может определяться или подбираться в зависимости от различных факторов, включающих конкретные терапевтические средства или фармацевтические композиции, пути введения, состояние субъекта, то есть, стадию заболевания, тяжесть симптомов, вызываемых заболеванием, общее состояние здоровья, а также возраст, пол и массу тела, и другие факторы, очевидные для специалиста в области медицины. Аналогично, доза терапевтического средства для лечения заболевания или расстройства может определяться в соответствии с параметрами, понятными специалистам в области медицины. Оптимальные дозы могут обычно определяться с использованием экспериментальных моделей и/или клинических испытаний.
Фармацевтическая композиция может вводиться через любые подходящие пути, например, посредством перорального, назального, внутрикожного, подкожного, внутримышечного или внутривенного введения.
В еще одном аспекте в настоящем раскрытии также предоставлен фармацевтический набор, который содержит очищенные терапевтические наночастицы или их фармацевтическую композицию, предоставленные в настоящем описании. Если требуется, фармацевтический набор также содержит инструкцию, упаковку и контейнер, содержащий терапевтические наночастицы или фармацевтическую композицию.
Примеры
Примеры, приведенные ниже, были предназначены, чтобы лучше иллюстрировать терапевтические наночастицы и фармацевтическую композицию, раскрытую в настоящем описании, но не для ограничения любого аспекта настоящего раскрытия.
Пример 1
3 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 20 мл смеси хлороформ/этанол (9:1, об./об.), и добавляли в 500 мл раствора человеческого сывороточного альбумина (5% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Model F22E, Fluko Co., Ltd., Shanghai) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 136 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 2
0,32 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Model F22E, Fluko Co., Ltd., Shanghai) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 145 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 3
0,63 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), и добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 141 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 4
1,25 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Model F22Z, Fluko Co., Ltd., Shanghai) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 138 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 5
1,88 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 133 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 6
2,5 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 125 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 7
5 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 134 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 8
7,5 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 140 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 9
1 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 20 мл смеси хлороформ/этанол (4:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (2% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 136 нм, и суспензия была полупрозрачной.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе (Model LD-85S, Millrock, USA) с получением устойчивой лепешки почти белого цвета.
Пример 10. Методы определения содержания человеческого сывороточного альбумина и паклитаксела
Содержание человеческого сывороточного альбумина определяли посредством ВЭЖХ. Человеческий сывороточный альбумин определяли при длине волны, равной 228 нм на ВЭЖХ, оборудованном колонкой с гелем Tosohaas TSK G3000 SWXL и УФ-детектором (1260VWD G1314B, Agilent technologies), с подвижной фазой из 0,1 моль/л раствора дикалийгидрофосфата и объемом вводимой пробы, равным 10 мкл. Содержание альбумина рассчитывали, используя метод внешнего стандарта.
Получение растворов для тестирования: растворы для тестирования получали посредством разведения раствора для определения, используя 0,9% раствор хлорида натрия, до концентрации альбумина ниже, чем 3 мг/мл.
Содержание паклитаксела определяли посредством ВЭЖХ. Паклитаксел определяли при длине волны, равной 228 нм на ВЭЖХ, оборудованном колонкой с обращенной фазой C18 и УФ-детектором (1260VWD G1314B, Agilent technologies), с подвижной фазой в виде смеси ацетонитрил-вода (1:1, об./об.) и объемом пробы, равным 10 мкл. Содержание паклитаксела рассчитывали, используя метод внешнего стандарта.
Получение анализируемых растворов: анализируемые растворы получали посредством разбавления раствора для определения с использованием ацетонитрила до полного растворения паклитаксела, с концентрацией, равной 20-200 мкг/мл.
Пример 11
Продукт, полученный в Примере 1, восстанавливали в 0,9% растворе хлорида натрия с получением образца 1, с содержанием паклитаксела, равным 5 мг/мл. Затем образец 1 разбавляли, используя моделированную плазму крови, содержащую 5% альбумина, таким образом, что содержание паклитаксела могло достигать 20 мкг/мл с получением образца 2 (частицы были полностью дезинтегрированы в таких условиях, и никаких частиц со связанным с паклитакселом человеческим сывороточным альбумином не существовало). 1 мл образца 1 и 1 мл образца 2 центрифугировали при 21000×g в течение различной длительности, соответственно. Концентрации паклитаксела и альбумина в супернатанте определяли, используя методы, указанные в Примере 10, и результаты приведены в Таблице 1.
Таблица 1. Концентрации паклитаксела и альбумина в супернатанте при различной длительности центрифугирования
центрифугир.
(мин)
(мг/мл)
таксел
(мг/мл)
(мг/мл)
таксел
(мг/мл)
Замечание**: После восстановления концентрация альбумина увеличивалась, так как объем раствора снижался до 90% от его первоначального объема вследствие осаждения после центрифугирования суспензии
Авторы предположили, что никакого осаждения не происходило после центрифугирования для полностью дезинтегрированного образца (Образец 2). Никаких значительных вариаций концентраций паклитаксела и альбумина в супернатанте не наблюдали для различной длительности центрифугирования, т.е., в растворе не было кристаллов или тяжелых частиц паклитаксела.
Осадок выпадал на дно после центрифугирования для недезинтегрированного образца (Образец 1). Концентрация паклитаксела в супернатанте снижалась с течением времени центрифугирования и окончательно достигала равновесия. Концентрация альбумина немного увеличивалась с течением времени, и окончательно достигала равновесия (объем супернатанта составлял приблизительно 90% от общего объема). В завершении, наночастицы в образце могут быть выделены и очищены посредством центрифугирования.
После центрифугирования в течение 60 минут, концентрация паклитаксела в супернатанте достигала равновесия. Таким образом, частицы паклитаксела-альбумина могут быть выделены при 21000×g в течение 60 минут.
Пример 12. Получение наночастиц
С использованием метода центрифугирования, указанного в Примере 11, частицы выделяли из образцов, полученных в Примерах 1-9. После центрифугирования супернатанты отбрасывали, и осадки, полученные таким образом, представляли собой наночастицы паклитаксела-альбумина, которые получили наименование частицы 1, 2, 3, 4, 5, 6, 7, 8, и 9, в соответствии с Примерами 1-9.
Пример 13. Сканирующее электронное микроскопическое наблюдение морфологии частиц до и после разделения
Лиофилизированный порошок образца, полученный в Примере 5, и осадок из Примера 5, полученный в Примере 12 (частица 5) наблюдали под сканирующим электронным микроскопом (S-4800, Hitachi). В результате на ФИГ. 1 можно видеть, что небольшое количество частиц в образце из Примера 5 были погружены в средство подложки, образованное большим количеством альбумина. В то же время для Частицы 5, эти частицы существовали независимо (См. ФИГ. 2). Таким образом, можно подтвердить, что осадок, полученный с использованием метода разделения в Примере 11, представлял собой чистые или по существу чистые наночастицы.
Пример 14. Отношение между альбумином и паклитакселом в частице
Наночастицы паклитаксела-альбумина (соответствующие Примерам 1-9), полученные в Примере 12, ресуспендировали соответственно в 1 мл 0,9% растворе хлорида натрия. Каждое из значений содержания паклитаксела и альбумина в образцах было определено с использованием метода Примера 10, и результаты являются следующими:
Таблица 2. Отношение между альбумином и паклитакселом в различных очищенных наночастицах, полученных после центрифугирования
в масляной фазе
(мг/мл)
(мг/мл)
(мг/мл)
На основании результатов, приведенных выше, авторы предположили, что отношения между альбумином и паклитакселом в очищенных наночастицах из продуктов, полученных с использованием различных процессов составления были различными, и они имеют некоторую закономерность. Также можно видеть, что увеличенная концентрация паклитаксела в масляной фазе была обратно пропорциональной содержанию альбумина.
Пример 15. Получение очищенных наночастиц с использованием диализа
Диализ проводили в равном объеме для образцов, полученных в Примере 2, Примере 5 и Примере 8 после восстановления водой для инъекции против диализного раствора из 5% маннита с использованием регенерированной целлюлозной ультрафильтрационной мембраны (PXC300C50, Millipore) с пределом пропускания по молекулярной массе, равным 300 кДа, и кратность диализа была равна 5. Содержание паклитаксела и альбумина в образцах, определенное после диализа, было определено с использованием метода Примера 10, и результаты являются следующими:
Таблица 3. Отношения между альбумином и паклитакселом в очищенных наночастицах из различных составов, полученных посредством диализа
(мг/мл)
(мг/мл)
Приведенные выше результаты указывают на аналогичное по существу массовое отношение между альбумином и паклитакселом в частицах, полученных посредством диализа с использованием ультрафильтрационной мембраны, по сравнению с частицами, полученными посредством центрифугирования. В результате, диализ с использованием ультрафильтрационной мембраны также может применяться для отделения частиц в суспензии от избыточного альбумина, и для замены раствора, окружающего частицы.
Пример 16. Получение очищенных наночастиц на хроматографической колонке
20 мл геля Сефарозы 4B вносили в стеклянную колонку с диаметром 10 мм (Φ 10 мм*230 мм, Beijing Mancang Technology Ltd.). Колонку уравновешивали 0,9% раствором хлорида натрия, промывая трехкратным объемом колонки. По 1 мл образцов, полученных в Примере 2, Примере 5 и Примере 8 загружали в верхнюю часть колонки с гелем, и элюирование проводили, используя 0,9% раствор хлорида натрия, соответственно. Элюат регистрировали непрерывно, используя УФ-детектор при длине волны, равной 280 нм. Элюат, соответствующий первому пику на хроматограмме, представлял собой слегка мутную суспензию, которую собирали для определения паклитаксела и альбумина. Регистрацию продолжали до конца выхода второго пика. Оба пика хорошо разделялись, указывая на то, что частицы и свободный альбумин могут эффективно разделяться с использованием колонки с гелем Сефарозы 4B. Содержание паклитаксела и альбумина в образцах, определенное после диализа, было определено с использованием метода Примера 10, и результаты являются следующими:
Таблица 4. Отношения между альбумином и паклитакселом в очищенных наночастицах из различных составов, полученных посредством разделения на хроматографической колонке
(мг/мл)
(мг/мл)
Приведенные выше результаты указывают на аналогичное по существу массовое отношение между альбумином и паклитакселом в частицах, полученных посредством разделения на гелевой колонке, в сравнении с частицами, полученными посредством центрифугирования и диализа. В результате, разделение на колонке с гелем также может применяться для отделения частиц в суспензии от избыточного альбумина.
Пример 17. Определение содержания свободного альбумина в очищенных наночастицах, полученных посредством диализа, с использованием центрифугирования
Суспензию частиц, полученную посредством диализа в Примере 15, дополнительно разделяли центрифугированием в условиях Примера 11. После осаждения всех частиц на дне центрифужной пробирки определяли концентрацию альбумина в супернатанте, и результаты приведены в Таблице 5.
Таблица 5. Вариация концентрации альбумина в очищенных наночастицах из различных составов, полученных посредством диализа до и после центрифугирования
центрифугирования (мг/мл)
центрифугирования (мг/мл)
НО обозначает значение ниже предела определения, т.е., не обнаруженное.
Из приведенных выше результатов видно, что в суспензии наночастиц паклитаксела-альбумина, полученной посредством диализа, доля свободного альбумина была очень низкой, и большая часть альбумина была связана с паклитакселом с образованием наночастиц.
Пример 18. Определение содержания свободного альбумина в очищенных наночастицах, полученных посредством разделения на хроматографической колонке, с использованием центрифугирования
Суспензию частиц, полученную посредством разделения на хроматографической колонке в Примере 16, дополнительно подвергали центрифугированию в условиях Примера 11. После осаждения всех частиц на дне центрифужной пробирки определяли концентрацию альбумина в супернатанте, и результаты приведены в Таблице 6.
Таблица 6. Вариация концентрации альбумина в очищенных наночастицах из различных составов, полученных посредством разделения на хроматографической колонке, до и после центрифугирования
центрифугирования (мг/мл)
центрифугирования (мг/мл)
НО обозначает значение ниже предела определения, т.е., не обнаруженное.
Из приведенных выше результатов видно, что в суспензии наночастиц паклитаксела-альбумина, полученной посредством разделения на хроматографической колонке, почти не содержалось свободного альбумина, и большая часть альбумина была связана с паклитакселом с образованием наночастиц.
Пример 19. Определение содержания свободного альбумина в очищенных наночастицах, полученных посредством центрифугирования, с использованием разделения на хроматографической колонке
Суспензию частиц получали посредством повторного суспендирования наночастиц паклитаксела-альбумина, полученных в Примере 12 в 0,9% растворе хлорид натрия, и очищенные наночастицы и свободный альбумин (если содержался) отделяли друг от друга, используя колонку с Сефарозой 4B, как описано в Примере 16. Во время элюирования, регистрировали в непрерывном режиме кривую УФ-поглощения. После полного элюирования частиц, собирали последующий элюент, в котором определяли концентрацию альбумина. Результаты приведены в Таблице 7.
Таблица 7. Вариация концентрации альбумина в очищенных наночастицах из различных составов, полученных посредством центрифугирования до и после разделения на хроматографической колонке
разделения на колонке (мг/мл)
разделения на колонке (мг/мл)
НО обозначает значение ниже предела определения, т.е., не обнаруженное.
Из приведенных выше результатов видно, что в суспензии наночастиц паклитаксела-альбумина, полученной посредством центрифугирования, почти не содержалось свободного альбумина, и большая часть альбумина была связана с паклитакселом с образованием наночастиц. В Примерах 17, 18, и 19, может быть продемонстрировано очевидными результатами, наблюдаемыми при использовании трех методов разделения, что очищенные наночастицы могут быть получены посредством всех трех методов с почти полным отсутствием свободного альбумина.
Пример 20. Определение размера и потенциала частиц
Наночастицы паклитаксела-альбумина, полученные в Примере 12 (соответствующие Примерам 1-9), в каждом случае ресуспендировали в очищенной воде. Размер и электрокинетический потенциал (дзета-потенциал) очищенных наночастиц определяли, используя лазерный измеритель частиц Malven NANO-ZS, и результаты приведены ниже:
Таблица 8. Размер и потенциал частиц для очищенных наночастиц после центрифугирования
(нм)
частицы для
чистых частиц
(нм)
чистой наночастицы
(мВ)
По результатам можно видеть, что средний размер частицы для очищенных наночастиц, полученных после центрифугирования, был по существу таким же, как начальный средний размер частицы, и электрокинетический потенциал все еще был относительно высоким, таким образом, что заряд может использоваться для стабилизации частиц суспензии.
Пример 21. Дифракционная рентгенограмма
Кристаллическую форму определяли на рентгеновском дифракционном приборе в случае лекарственной субстанции паклитаксела и лиофилизированного порошка альбумина, и результаты показаны на ФИГ. 3 и ФИГ. 4. Как показано, лекарственная субстанция паклитаксела представляла собой кристаллический порошок, а лиофилизированный порошок альбумина являлся аморфным порошком. Представительные очищенные наночастицы, полученные в Примере 12 (соответствующие Примерам 2, 4, 6, и 8), лиофилизировали в лиофилизаторе с получением твердого порошка, кристаллическую форму которого анализировали на рентгеновском дифракционном приборе, и результаты показаны на ФИГ. 5, 6, 7, и 8. Из дифракционных рентгенограмм можно видеть, что отношение альбумина к паклитакселу в частицах находилось в интервале от 0,043:1 до 0,57:1, и как альбумин, так и паклитаксел находились в аморфной форме, которая существенно отличалась от формы лекарственной субстанции паклитаксела и слегка отличалась от формы лиофилизированного порошка альбумина.
Пример 22. Высвобождение паклитаксела из частиц
Используя метод разделения для частиц, определённый в Примере 11, свободный компонент может эффективно отделяться от компонента в форме наночастиц. Как следствие, высвобождение паклитаксела из частиц может быть обнаружено при различных концентрациях с использованием такого метода, и конкретная методика показана ниже:
Для наночастиц паклитаксела-альбумина, полученных в Примере 12 (соответствующих Примерам 1-9, соответственно), были выбраны представительные очищенные наночастицы (соответствующие Примерам 2, 4, 6, и 8, и именуемые как Частицы 2, 4, 6, и 8), и к ним добавляли 0,9% раствор хлорида натрия, исходя из отношения между альбумином и паклитакселом, с образованием исходного раствора с концентрацией паклитаксела, равной 5 мг/мл. Исходный раствор и раствор моделированной плазмы крови выдерживали при 37°C. Исходный раствор разбавляли, используя раствор моделированной плазмы крови, с получением ряда растворов паклитаксела с концентрациями, равными 5000, 1000, 200, 150, 100, 80, 50, 30, и 10 мкг/мл.
Определение степени высвобождения: определяли концентрации паклитаксела в полученных растворах образцов, и в то же самое время, также определяли концентрации паклитаксела в супернатантах после центрифугирования при 21000×g в течение 60 минут с использованием 1 мл каждого образца. Отношение высвобождения паклитаксела при каждой концентрации рассчитывали посредством деления концентрации паклитаксела в супернатанте на концентрацию перед центрифугированием, и кривые высвобождения, нанесенные на график, показаны на ФИГ. 9 и ФИГ. 10. По кривым высвобождения можно видеть, что отношение альбумина к паклитакселу в частицах находилось в интервале от 0,043:1 до 0,057:1, и характеристики высвобождения паклитаксела для частиц были по существу одинаковыми, все из которых коррелировали с концентрацией.
Пример 23. Получение композиций, содержащих терапевтические наночастицы
Способ 1 (Центрифугирование-Повторное суспендирование): терапевтические наночастицы, полученные в Примере 12, соответствующие Примерам 1-9 (именуемые как Частицы 1, 2, 3, 4, 5, 6, 7, 8, и 9), ресуспендировали в 5% растворе маннита, 10% растворе сахарозы, 5% растворе декстрана, 10% растворе лактозы, 10% растворе трегалозы, 10% растворе мальтозы, 10% растворе человеческого сывороточного альбумина, соответственно, чтобы установить концентрации паклитаксела, равные 5 мг/мл. Повторно полученные суспензии фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не наблюдали для частиц никакой значительной вариации размера частиц. Суспензии лиофилизировали в лиофилизаторе, соответственно, с получением композиций, содержащих фармацевтические наночастицы.
Способ 2 (Диализ): Каждый из образцов, полученных в Примерах 1-9, восстанавливали для получения суспензий с концентрацией паклитаксела, равной 5 мг/мл. Затем суспензии диализировали, используя ультрафильтрационную мембрану с 300 кДа, соответственно, используя 5% раствор маннита, 10% раствор сахарозы, 5% раствор декстрана, 10% раствор лактозы, 10% раствор мальтозы, 10% раствор трегалозы и 10% раствор человеческого сывороточного альбумина. После диализа до 5-кратного первоначального объема, весь свободный человеческий сывороточный альбумин в суспензиях заменяли. Суспензии фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате никакой значительной вариации размера частиц для частиц не наблюдали. Суспензии лиофилизировали в лиофилизаторе, соответственно с получением композиций, содержащих терапевтические наночастицы.
Пример 24
Каждую из композиций, содержащих терапевтические наночастицы, полученные в Способе 1 (Центрифугирование-Повторное суспендирование) и Способе 2 (Диализ) в Примере 23, восстанавливали в воде для инъекций для установления концентрации паклитаксела, равной 5 мг/мл. Для каждого раствора наблюдали стабильность, и определяли 12-часовую стабильность при комнатной температуре. Как показано, общепринятые лиопротекторы могут играть защитную роль для очищенных наночастиц паклитаксела-альбумина. Результаты показаны в Таблице 9 и Таблице 10:
Таблица 9. Стабильность композиций, полученных посредством Центрифугирования-Повторного суспендирования
композиц.
1
2
3
4
5
6
7
8
9
маннита
лактозы
Таблица 10. Стабильность композиций, полученных посредством Диализа
композиц.
1
2
3
4
5
6
7
8
9
маннита
лактозы
Как показано, стабильность частиц может поддерживаться посредством замены альбумина, окружающего частицы с использованием Центрифугирования-Повторного суспендирования (Способ 1) и Диализа (Способ 2).
Пример 25. Исследование сенсибилизации у морских свинок
В данном эксперименте продукт из Примера 6 и состав с 5% маннитом, содержащий Частицу 6 из Примера 24, были выбраны в качестве тестируемого лекарственного средства, где концентрации паклитаксела в составах приведены в таблице ниже. Были выбраны дозировки для сенсибилизации, равные 3 мг на морскую свинку и 1,25 мг на морскую свинку. Сенсибилизацию проводили посредством внутрибрюшинной инъекции один раз в два дня, всего за 3 инъекции. В то же время, устанавливали группу положительного контроля (0,2% овальбумина) и группу отрицательного контроля (инъекция 0,9% хлорида натрия). Детальный режим дозирования приведен в таблице ниже.
Таблица 11. Протокол для исследования сенсибилизации у морских свинок
животных
лек. средства
(мг/мл)
введения
(мл/морская свинка)
(мл/морская свинка)
введения
(мл/морская свинка)
(мг/морская свинка)
отриц.
контроля
полож.
контроля
продукта
из
прим. 6
продукта
из
прим. 6
Частицы 6
Частицы 6
Метод введения для сенсибилизации: спинку морской свинки плотно удерживали левой рукой, сложенной в форме чашечки, обеспечивая натяжение кожи на животе при фиксации морской свинки. Живот морской свинки поднимали, и голова была опущена вниз. После дезинфекции участка инъекции спиртовой салфеткой, Иглой одноразового шприца объемом 2 мл, удерживаемого правой рукой, протыкали кожу морской свинки. Иглу вводили на участке, находящемся на 1 мм слева от средней линии нижней части живота. При прохождении подкожной части, иглу вводили прямо на дополнительные от 5 мм до 10 мм, и последовательно проводили в брюшную полость под углом 45°. После фиксации иглы фармацевтический раствор вводили посредством медленной инъекции. После инъекции сухим ватным тампоном надавливали на точку укола, чтобы препятствовать вытеканию фармацевтического средства. После 3 раз сенсибилизации, морские свинки в группе состава из Примера 6 (3 мг/морскую свинку) становились истощенными и умирали. В других группах не наблюдали никаких аномалий.
Возбуждение аллергии: возбуждение проводили посредством внутривенной инъекции, и возбуждение проводили через 10 дней после последней сенсибилизации дозировками, равными 6 мг/животное и 2,5 мг/животное.
Метод введения для возбуждения: инъекцию проводили в латеральную метатарзальную вену морской свинки, фиксируемой ассистентом. Коленные суставы были зажаты оператором для фиксации тела животного. Его вену сжимали и лапы находились в вытянутом состоянии. Волосяной покров на участке инъекции сбривали (или срезали кожу на участке инъекции). После стерилизации спиртовыми салфетками можно было видеть толстую латеральную метатарзальную вену. Иглой одноразового шприца объемом 1 мл в правой руке протыкали кровеносный сосуд вдоль направления в сторону сердца. После инъекции сухим ватным тампоном надавливали на место прокола, чтобы предотвратить кровотечение.
Реакцию каждого животного и время появления или исчезновения симптомов аллергии наблюдали сразу же после возбуждения в течение 30 минут. Максимальная продолжительность наблюдения составляла 3 часа. Результаты аллергической реакции приведены в таблице 14.
Таблица 12. Симптомы аллергической реакции
Таблица 13. Критерии оценки для системной аллергической реакции
Таблица 14. Результаты для активной анафилаксии морских свинок
животных
оценка
отрицательного
контроля
контроля
На основании результатов активной анафилаксии у морских свинок авторы предположили, что состав наночастиц, содержащий избыточный альбумин, обладает более сильной сенсибилизацией, в то время как состав, содержащий очищенные наночастицы, может значительно уменьшить аллергическую реакцию.
Пример 26. Фармакокинетическое исследование на собаках
В данном эксперименте, продукт из Примера 6 и состав с 5% маннитом, содержащий Частицу 6 из Примера 24, были выбраны в качестве тестируемого лекарственного средства, где концентрация паклитаксела в составах составляла 5 мг/мл. Собак породы бигль использовали для in vivo фармакокинетических исследований обоих составов. В каждой группе 3-х собак породы бигль тестировали при вводимой дозировке, равной 5 мг/кг, и продолжительности введения, равной 30 мин. 2 мл крови собирали из головной вены передних конечностей собак породы бигль сразу же после введения (0 ч), 15 мин при процессе инфузии (0,25 ч после введения), на временной отметке вынимания иглы (0,5 ч после введения), и через 0,58, 0,75, 1,0, 1,5, 2,5, 3,5, 4,5, 6,5, 8,5, 24,5 ч после введения, и каждый из образцов крови помещали в пробирку с гепарином, встряхивали до гомогенности, и центрифугировали при 3000 об/мин в течение 10 мин для получения плазмы. Концентрацию паклитаксела в плазме определяли посредством ВЭЖХ/МС/МС, и концентрация лекарственного средства наносили на график зависимости от времени (См. ФИГ. 11). Из графика зависимости концентрации лекарственного средства от времени видно, что оба состава имели почти одинаковые характеристики in vivo, таким образом, не было резкого воздействия избыточного альбумина на характеристики частиц in vivo.
Пример 27. Аллергические симптомы во время фармакокинетического исследования у собак
При фармакокинетическом исследовании у собак, проводимом в Примере 26, неблагоприятные реакции наблюдали во время введения обоих составов. У образца из группы Примера 6, различная степень явных аллергических реакций проявилась у 3 собак, включая, в основном, яростное сопротивление, избыточное слюнотечение, эритему вокруг рта, и у индивидуальных экспериментальных животных во время введения имели место рвота и недержание мочи. В то же время для образца из группы Частицы 6, лишь небольшое сопротивление, слюнотечение и эритема вокруг рта проявились у части животных, и симптомы были слабо выраженными. Следовательно, состав очищенных наночастиц была способна значительно смягчать аллергические реакции лекарственного средства.
Пример 28
Следующие образцы исследовали для определения максимальной переносимой дозы, образцы, включающие композицию, содержащую терапевтические наночастицы, с использованием маннита в качестве лиопротектора (полученные Способом 1 Примера 24, соответствующие наночастицам Примера 2, 6, и 8, и именуемые ниже как Частица 2, Частица 6 и Частица 8), продукт из Примера 6, и Таксол®, полученный с использованием Кремофора.
На основании рекомендаций NCI США, были проведены модификации экспериментального метода определения максимальной переносимой дозы при острой токсичности посредством однократного введения дозы. Для Частицы 2, Частицы 6 и Частицы 8 и лиофилизированного порошка из Примера 6, доза 400 мг/кг была выбрана в качестве максимальной вводимой дозы, и вводимую дозу снижали при отношении 1,2, т.е., получали ряд доз, составляющих 400, 333, 278, 231 и 193 мг/кг. Для ТАКСОЛ® значение 48 мг/кг было выбрано в качестве максимальной вводимой дозы, и вводимую дозу также снижали при отношении 1,2, т.е., получали ряд доз, составляющих 48, 40, 33,3, 27,8, 23,1 и 19,3 мг/кг. В каждую группу дозирования включали самцов мышей 3 KM. Общее состояние и вариации массы тела животных наблюдали в течение 10 дней после введения. Если во время наблюдения ни одно животное не погибало, не происходило никакого необратимого токсического ответа или имела место потеря не более чем 15% массы тела, в течение 3 непрерывных дней в сравнении с массой тела до эксперимента, максимальную вводимую дозу считали максимальной переносимой дозой для однократного введения.
Результаты показали, что максимальная переносимая доза ТАКСОЛ® составляла 33,3 мг/кг. Во всех группах введения во время введения ТАКСОЛ® мыши яростно сопротивлялись, и после введения происходили кома и учащенное дыхание. В группах с дозами 40 мг/кг и 48 мг/кг, имели место летальные случаи. В остальных дозовых группах мыши постепенно восстанавливались после введения. Тяжесть симптомов токсичности и время, требуемое для восстановления, коррелировали с вводимой дозой. Все максимально переносимые дозы для продукта из Примера 6 и Частицы 2, Частицы 6 и Частицы 8 составляли 193 мг/кг. Неблагоприятные симптомы мышей в группах с дозами свыше 193 мг/кг в основном включали потерю массы тела, загрязнение вокруг ануса, слабость/паралич задних конечностей и смерть. Тяжесть неблагоприятных симптомов увеличивалась с увеличением вводимой дозы. Очевидного различия по токсичности для мышей, вызываемой Частицей 2, Частицей 6, Частицей 8 и продуктом из Примера 6, не наблюдали. Однако все указанные выше токсичности были ниже токсичности ТАКСОЛ®, полученного вместе с Кремофором.
Пример 29. Ингибирующий эффект на опухоль H22 рака печени у мышей
Частицу 2, Частицу 6, Частицу 8, продукт из Примера 6 и Таксол®, полученный вместе с Кремофором, исследовали на предмет ингибирующего эффекта на опухоль H22 рака печени у мышей.
Животных разделяли исходя из массы тела. Асцитические клетки H22 рака печени подкожно инокулировали в подкожную ткань подмышечной области передних конечностей самцов мышей KM при объеме инокуляции, равном 0,2 мл, который содержал приблизительно 1,0×106 Клеток. Животных поровну разделяли на 6 групп в соответствии со временем инокуляции.
Однократные внутривенные введения Частицы 2, Частицы 6, Частицы 8, продукта из Примера 6 и Таксол® проводили при максимально переносимой дозе для мышей через 24 часа после инокуляции, т.е., вводимые дозы Частицы 2, Частицы 6, Частицы 8, и продукта из Примера 6 составляли 193 мг/кг, и вводимая доза of Таксол® составляла 33,3 мг/кг. В холостой контрольной группе, инъекцию 0,9% хлорида натрия применяли для однократного внутривенного введения. На 12ый день после введения мышей умерщвляли посредством асфиксии CO2, и массу опухоли забирали и взвешивали. Степень ингибирования опухоли (%) может быть рассчитана по следующему уравнению: Степень ингибирования опухоли=(1 - средняя масса опухоли в тестируемых группах/средняя масса опухоли в контрольной группе) x 100%. Экспериментальные результаты статистически анализировали посредством однофакторного дисперсионного анализа ANOVA, используя программное обеспечение для статистики SPSS 19,0.
Экспериментальные результаты приведены в Таблице 15. Для однократного внутривенного введения при максимально переносимой дозе для мышей, рост опухоли H22 значительно ингибировался во всех группах. Межгруппового различия по ингибирующему эффекту на опухоль для Частицы 2, Частицы 6, Частицы 8 и продукта из Примера 6 на опухоль H22 не было. Степень ингибирования опухоли для Частицы 2, Частицы 8 и продукта из Примера 6 была значительно выше, чем для ТАКСОЛ®.
Таблица 15. Эффект на массу тела мышей, несущих опухоль H22 (n=10, χ±sd)
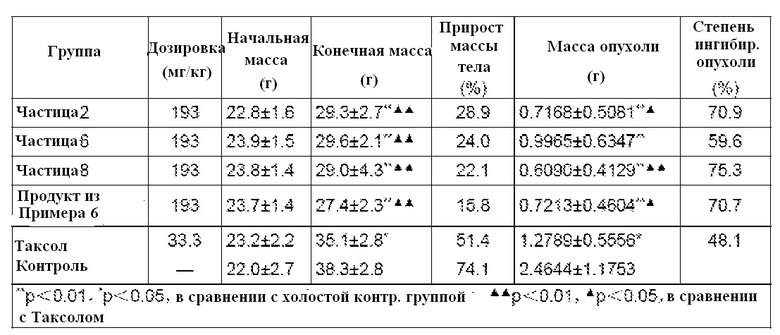
Пример 30. Ингибирующий эффект на опухоль RM-1 рака простаты мышей
Частицу 2, Частицу 6, Частицу 8, продукт из Примера 6 и ТАКСОЛ®, полученный вместе с Кремофором, исследовали на предмет ингибирующего эффекта на опухоль RM-1 рака простаты у мышей.
Опухолевые клетки RM-1 собирали на логарифмической фазе роста, и число клеток доводили до 2,5×106 клеток/мл. Суспензию опухолевых клеток инокулировали в подкожную ткань подмышечной области передних конечностей самцов мышей C75 с возрастом от 7 до 8 недель при объеме инокуляции, равном 0,2 мл, который содержал приблизительно 5×105 клеток. После инокуляции оставшуюся суспензию опухолевых клеток подсчитывали под оптическим микроскопом, причем число живых опухолевых клеток составляло > 95%. Мышей разделяли на 6 групп, исходя из времени инокуляции.
Однократные внутривенные введения Частицы 2, Частицы 6, Частицы 8, продукта из Примера 6 и Таксол® проводили при максимально переносимой дозе для мышей через 72 часа после инокуляции, т.е., вводимые дозы Частицы 2, Частицы 6, Частицы 8 и продукта из Примера 6 составляли 193 мг/кг, а вводимая доза Таксол® была равна 33,3 мг/кг. В холостой контрольной группе инъекцию 0,9% хлорида натрия применяли для однократного внутривенного введения. После разрастания опухоли диаметр измеряли используя штангенциркуль, и противоопухолевый эффект фармацевтического средства наблюдали в динамике. Объем опухоли (TV) может быть рассчитан следующим образом: V=1/2×a×b2, где a и b относятся к длине и ширине опухоли, соответственно. Объем опухоли может быть рассчитан на основании результатов. Степень ингибирования объема опухоли (%) может быть рассчитана по следующему уравнению: Степень ингибирования опухоли=(1 - средний объем опухоли в группах введения/средний объем опухоли в контрольной группе) х 100%. Экспериментальные результаты анализировали с использованием программного обеспечения для статистики SPSS 19,0. Вариацию объема опухоли между измерениями с течением времени анализировали, используя программу Repeated Measure Analysis, и межгрупповую вариацию объема опухоли для каждого измерения анализировали, используя Multivariate.
Экспериментальные результаты приведены в Таблице 16. Однократные внутривенные введения проводили при максимально переносимой дозе через 3 дня после инокуляции опухолевых клеток RM-1 рака простаты. В сравнении с холостой контрольной группой, значительный ингибирующий эффект на рост опухоли RM-1 рака простаты у мышей наблюдали для Частицы 2, Частицы 6, Частицы 8 и продукта из Примера 6, и никакого ингибирующего эффекта на опухоль не наблюдали для Таксол®, полученного вместе с Кремофором. Никакого значительного различия по ингибирующему эффекту на опухоль RM-1 мышей не наблюдали для Частицы 2, Частицы 6, Частицы 8 и продукта из Примера 6. Однако, в сравнении с Таксол®, все указанные выше составы имели значительный ингибирующий эффект на рост опухоли.
Таблица 16. Ингибирующий эффект на мышиную опухоль RM-1 рака простаты (n=10, χ±sd)
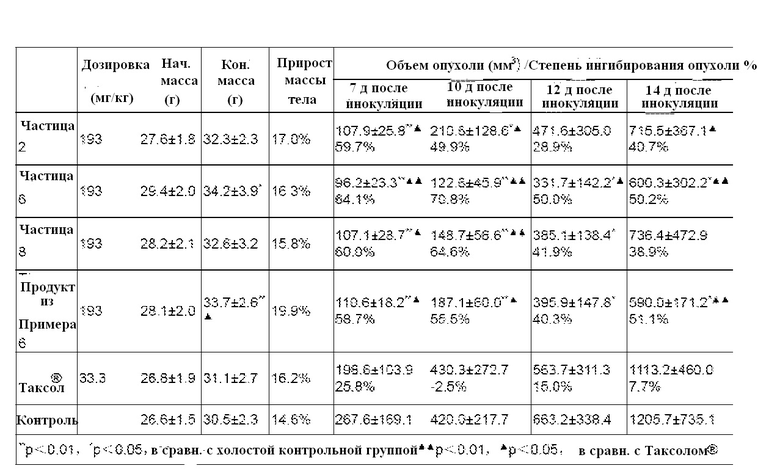
Пример 31:
Ингибирующий эффект на опухоль у мышей с раком легких Льюиса
Частицу 2, Частицу 6, Частицу 8, продукт Примера 6, и ТАКСОЛ®, полученные с использованием Кремофора, исследовали на предмет ингибирующего эффекта на опухоль у мышей с раком легких Льюиса.
Клетки опухоли Льюиса на логарифмической фазе роста собирали, и число клеток доводили до 2,5×106 клеток/мл. Суспензию опухолевых клеток инокулировали в подкожную ткань в подмышечную область передних конечностей самцам мышей C57 в возрасте от 7 до 8 недель при объеме инокуляции, равном 0,2 мл, который содержал приблизительно 5×105 клеток.
Распределение по группам, введение, наблюдение индикатора и статистический метод были такими же, как в Примере 18.
Экспериментальные результаты приведены в Таблице 17. Однократные внутривенные введения проводили при максимально переносимой дозе через 3 дня после инокуляции опухолевых клеток мышиного рака легких Льюиса. В сравнении с холостой контрольной группой, значительный ингибирующий эффект на рост опухоли мышиного рака легких Льюиса наблюдали для Частицы 2, Частицы 6, Частицы 8 и продукта из Примера 6, и никакой ингибирующий эффект на опухоль не был виден для Таксола®, полученного с использованием Кремофора. Никакого существенного различия по ингибирующему эффекту на опухоль мышиного рака легких Льюиса не наблюдали для Частицы 2, Частицы 6, Частицы 8 и продукта из Примера 6. Однако, в сравнении с Таксол®, все указанные выше составы имели значительный ингибирующий эффект на рост опухоли.
Таблица 17. Ингибирующий эффект на опухоль у мышей с раком легких Льюиса (n=10, χ±sd)
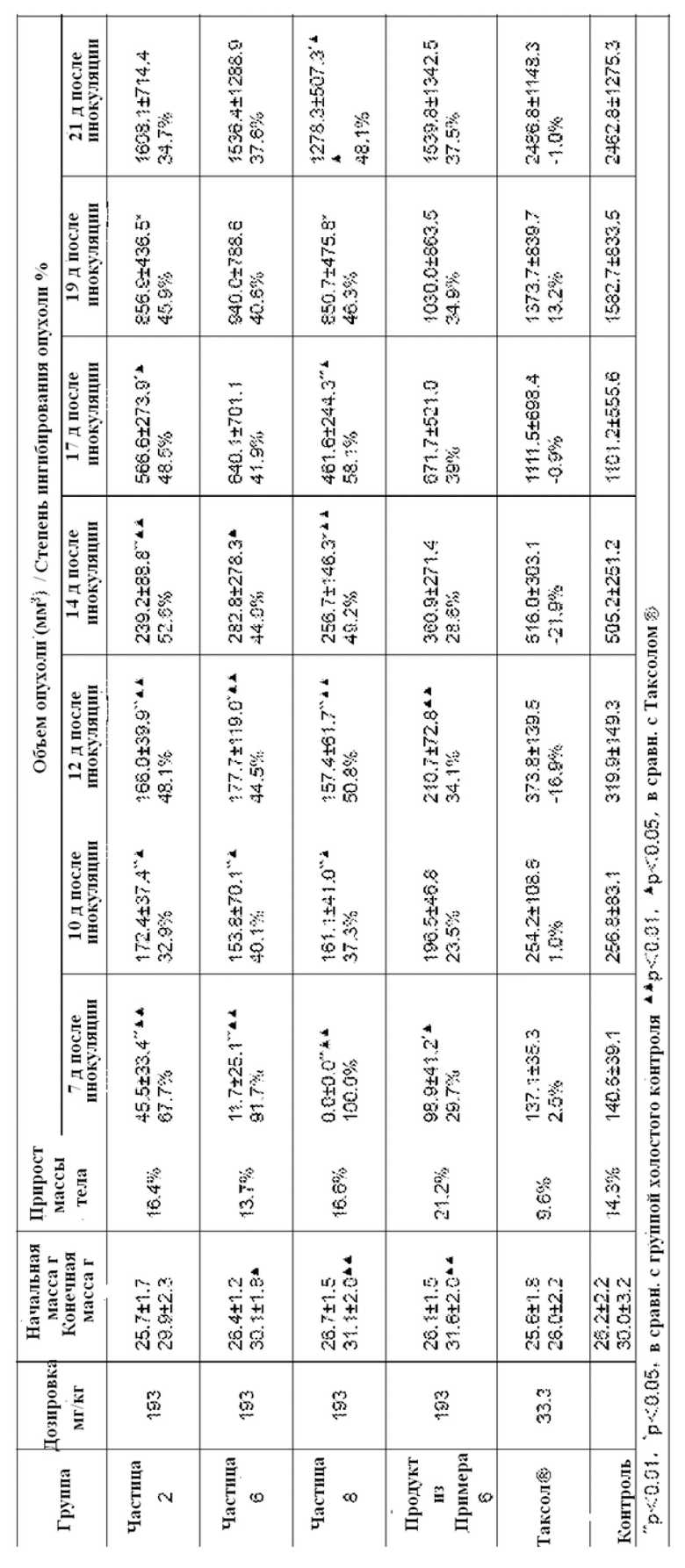
Пример 32
Как доступный для приобретения фармацевтический продукт, человеческий сывороточный альбумин должен содержать не более чем 5% полимера согласно его стандарту качества, так как полимер альбумина может индуцировать аллергическую реакцию. Метод определения альбумина в Примере 10 может различать мономер альбумина от полимера. Содержание полимера альбумина и паклитаксела было обнаружено в композициях из Примеров 1-9 (именуемых как первоначальный состав), и композициях полученных в результате Способа 1 Примера 24, соответственно, с использованием маннита и человеческого сывороточного альбумина в качестве лиопротектора (соответствующих Примерам 1-9), на основании метода определения альбумина и паклитаксела в Примере 10, и содержание полимера альбумина рассчитывали на 1 мг паклитаксела. Результаты приведены в Таблице 18.
Таблица 18. Относительное количество полимера альбумина и паклитаксела в различных составах
состав
(мг/мг)
качестве
лиопротектора
(мг/мг)
качестве
лиопротектора
(мг/мг)
На основании результатов авторы предположили, что содержание полимера альбумина в продукте, полученном непосредственно с использованием способа предшествующего уровня техники (Гомогенизации), было значительно выше, чем содержание в добавленном после альбумине композиции, полученной посредством способа настоящего раскрытия. Это являлось следствием вновь генерированного полимера альбумина в процессе гомогенизации или процессе испарения предшествующего уровня. Однако, содержание полимера альбумина в составе, содержащем протектор в виде маннита, было более низким, так как лишь очень малое количество альбумина содержалось в составе, и, таким образом, общее количество было ниже, чем количество полимера в первоначальном составе.
Пример 33. Извлечение альбумина для получения наночастиц
Диализат, полученный в результате диализа в Примере 15, собирали, и концентрировали до содержания альбумина, равного приблизительно 4%, используя регенерированную целлюлозную мембрану для ультрафильтрации с пределом пропускания по молекулярной массе, равным 10 кДа. Концентрат диализировали в равном объеме с очищенной водой. После диализа до 5-кратного объема, может быть извлечен 4% раствор альбумина.
Согласно методу в Примере 5 получали наночастицы паклитаксела-альбумина с использованием извлеченного раствора альбумина. Вследствие этого, средний диаметр полученных наночастиц паклитаксела-альбумина составлял 134 нм, и суспензия была полупрозрачной, по аналогии с продуктом, полученным в Примере 5.
Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм. Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали для суспензии после хранения в течение 48 часов при комнатной температуре. Суспензию разделяли на аликвоты и лиофилизировали в течение 24 часов в лиофилизаторе с получением устойчивой лепешки почти белого цвета.
Пример 34. Получение очищенных частиц доцетаксела-альбумина
3 г доцетаксела растворяли в 15 мл смеси хлороформ/этанол (1:1, об./об.), и добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы доцетаксела-альбумина, имеющие средний диаметр, равный 110-150 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц доцетаксел-альбумин методом центрифугирования, описанном в Примере 11 при 21000×g в течение 60 минут. Супернатант отбрасывали и очищенные наночастицы собирали. Очищенные наночастицы ресуспендировали в 5% растворе маннита. Повторно полученную суспензию фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание доцетаксела в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 48 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения человеческого сывороточного альбумина и доцетаксела в продукте, отношение между альбумином и доцетакселом может быть рассчитано как 0,1:1.
Пример 35. Получение очищенных частиц 2ʹ-O-гексаноилдоцетаксела-альбумина
695 мг 2ʹ-O-гексаноилдоцетаксела растворяли в 6 мл смеси хлороформ/этанол (10:1, об./об.), и добавляли в 102 мл раствора человеческого сывороточного альбумина (10% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы 2ʹ-O-гексаноилдоцетаксел-альбумина, имеющие средний диаметр, равный 75-100 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц 2ʹ-O-гексаноилдоцетаксела-альбумина методом диализа против 5% раствора маннита, описанным в Примере 15. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и не было никакой значительной вариации размера частиц в фильтрате. Содержание 2ʹ-O-гексаноилдоцетаксела в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 48 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и 2ʹ-O-гексаноилдоцетаксела в продукте, отношение между альбумином и 2ʹ-O-гексаноилдоцетакселом может быть рассчитано как 0,27:1.
Пример 36. Получение очищенных частиц 2ʹ-бензоилдоцетаксела-альбумина
226 мг 2ʹ-бензоилдоцетаксела растворяли в 2 мл смеси хлороформ/этанол (9:1, об./об.), и добавляли в 35 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 18000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы 2ʹ-бензоилдоцетаксела-альбумина, имеющие средний диаметр, равный 30-60 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц 2ʹ-бензоилдоцетаксела-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 10% раствор лактозы применяли в качестве диализата. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание 2ʹ-бензоилдоцетаксела в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 65 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и 2ʹ-бензоилдоцетаксела в продукте, отношение между альбумином и 2ʹ-бензоилдоцетакселом может быть рассчитано как 0,95:1.
Пример 37. Получение очищенных частиц рапамицина-альбумина
166 мг рапамицина растворяли в 1 мл смеси хлороформ/этанол (11:1, об./об.), и добавляли в 37 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы рапамицина, имеющие средний диаметр, равный 50-85 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц рапамицина-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 5% раствор декстрана применяли в качестве диализата. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание рапамицина в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 10 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 48 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и рапамицина в продукте, отношение между альбумином и рапамицином может быть рассчитано как 0,63:1.
Пример 38. Получение очищенных частиц темсиролимуса-альбумина
101 мг темсиролимуса растворяли в 0,5 мл смеси хлороформ/этанол (7:1, об./об.) и добавляли в 19,5 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы темсиролимуса, имеющие средний диаметр, равный 80-115 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц темсиролимуса-альбумина методом центрифугирования, описанным в Примере 11, при 21000×g в течение 60 минут. Супернатант отбрасывали и очищенные наночастицы собирали. Очищенные наночастицы ресуспендировали в 5% растворе маннита. Повторно полученную суспензию фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание темсиролимуса в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 10 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 48 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и темсиролимуса в продукте, отношение между альбумином и темсиролимусом может быть рассчитано как 0,16:1.
Пример 39. Получение очищенных частиц эверолимуса-альбумина
78 мг эверолимуса растворяли в 1 мл смеси хлороформ/этанол (9:1, об./об.), и добавляли в 22 мл раствора человеческого сывороточного альбумина (8% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы эверолимуса, имеющие средний диаметр, равный 80-110 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц эверолимуса-альбумина методом диализа, описанным в Примере 15 против 5% декстрана. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание эверолимуса в растворе определяли используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 10 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 60 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, Суспензия была полупрозрачной и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и эверолимуса в продукте, отношение между альбумином и эверолимусом может быть рассчитано как 0,32:1.
Пример 40. Получение очищенных частиц ромидепсина-альбумина
113 мг ромидепсина растворяли в 2 мл смеси хлороформ/этанол (8:1, об./об.) и добавляли в 35 мл раствора человеческого сывороточного альбумина (10% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы 2-ромидепсина, имеющие средний диаметр, равный 120-150 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц ромидепсина-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 10% раствор сахарозы применяли в качестве диализата. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание ромидепсина в растворе определяли используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 48 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и ромидепсина в продукте, отношение между альбумином и ромидепсином может быть рассчитано как 0,25:1.
Пример 41. Получение очищенных частиц пирарубицина-альбумина
147 мг пирарубицин растворяли в 0,75 мл смеси хлороформ/этанол (5:1, об./об.) и добавляли в 19,5 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы 2-пирарубицина, имеющие средний диаметр, равный 100-120 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц пирарубицина-альбумина методом диализа против 5% раствора маннита, описанным в Примере 15. Полученные очищенные наночастицы фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание пирарубицина в растворе определяли используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 10 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 48 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и пирарубицина в продукте, отношение между альбумином и пирарубицином может быть рассчитано как 0,47:1.
Пример 42. Получение очищенных частиц аклациномицина-альбумина
135 мг аклациномицина растворяли в 2 мл смеси хлороформ/этанол (8:1, об./об.) и добавляли в 25 мл раствора человеческого сывороточного альбумина (8% м./об.). Смесь эмульгировали в течение 2 минут используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы аклациномицина, имеющие средний диаметр, равный 80-150 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц аклациномицина-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16, 5% раствор маннита применяли в качестве диализата. Повторно полученную суспензию фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание аклациномицина в растворе определяли используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 10 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 48 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и аклациномицина в продукте, отношение между альбумином и аклациномицином может быть рассчитано как 0,13:1.
Пример 43 Получение очищенных частиц кабазитаксела-альбумина
289 мг кабазитаксела растворяли в 3 мл смеси хлороформ/этанол (10:1, об./об.) и добавляли в 67 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы кабазитаксела, имеющие средний диаметр, равный 65-85 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц кабазитаксела-альбумина равнообъемным диализом, описанным в Примере 15, против 5% маннита. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание кабазитаксела в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 55 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и кабазитаксела в продукте, отношение между альбумином и кабазитакселом может быть рассчитано как 0,27:1.
Пример 44 Получение очищенных частиц амиодарона-альбумина
90 мг амиодарона растворяли в 2 мл смеси хлороформ/этанол (11:1, об./об.) и добавляли в 58 мл раствора человеческого сывороточного альбумина (8% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы 2-амиодарона, имеющие средний диаметр, равный 70-105 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц амиодарона-альбумина методом центрифугирования, описанным в Примере 11, при 21000×g в течение 60 минут. Супернатант отбрасывали и очищенные наночастицы собирали. Очищенные наночастицы ресуспендировали в 5% растворе декстрана. Повторно полученную суспензию фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание амиодарона в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 10 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 57 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и амиодарона в продукте, отношение между альбумином и амиодароном может быть рассчитано как 0,43:1.
Пример 45 Получение очищенных частиц лиотиронина альбумина
93 мг лиотиронина растворяли в 2 мл смеси хлороформ/этанол (9:1, об./об.) и добавляли в 46 мл раствора человеческого сывороточного альбумина (8% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы 2-лиотиронина, имеющие средний диаметр, равный 85-125 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц лиотиронина-альбумина методом центрифугирования, описанным в Примере 11, при 21000×g в течение 60 минут. Супернатант отбрасывали и очищенные наночастицы собирали. Очищенные наночастицы ресуспендировали в 5% растворе маннита. Повторно полученную суспензию фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание лиотиронина в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 10 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 50 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и лиотиронина в продукте, отношение между альбумином и лиотиронином может быть рассчитано как 0,39:1.
Пример 46 Получение очищенных частиц Эпотилона B-альбумина
127 мг Эпотилона B растворяли в 2 мл смеси хлороформ/этанол (10:1, об./об.) и добавляли в 52 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы Эпотилона B, имеющие средний диаметр, равный 80-115 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц Эпотилона B-альбумина равнообъемным диализом против 5% маннита. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и не было никакой значительной вариации размера частиц в фильтрате. Содержание Эпотилона B в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 10 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 65 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и Эпотилона B в продукте, отношение между альбумином и Эпотилоном B может быть рассчитано как 0,14:1.
Пример 47 Получение очищенных частиц 10-гидроксикамптотецина-альбумина
93 мг 10-гидроксикамптотецина растворяли в 2 мл смеси хлороформ/этанол (10:1, об./об.) и добавляли в 48 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы 10-гидроксикамптотецина, имеющие средний диаметр, равный 100-135 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц 10-гидроксикамптотецина-альбумина методом центрифугирования, описанным в Примере 11 при 21000×g в течение 60 минут. Супернатант отбрасывали, и очищенные наночастицы собирали. Очищенные наночастицы ресуспендировали в 5% растворе маннита раствор. Повторно полученную суспензию фильтровали через стерильный фильтр 0,22 мкм, и не было никакой значительной вариации размера частиц в фильтрате. Содержание 10-гидроксикамптотецина в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 20 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 72 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и 10-гидроксикамптотецина в продукте, отношение между альбумином и 10-гидроксикамптотецином может быть рассчитано как 0,26:1.
Пример 48 Получение очищенных частиц циклоспорина-альбумина
148 мг циклоспорина растворяли в 1,5 мл смеси хлороформ/этанол (11:1, об./об.) и добавляли в 18,5 мл раствора человеческого сывороточного альбумина (4% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20), с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы циклоспорина, имеющие средний диаметр, равный 100-135 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц циклоспорина-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 10% лактозу использовали в качестве диализата. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание циклоспорина в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 72 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и циклоспорина в продукте, отношение между альбумином и циклоспорином может быть рассчитано как 0,26:1.
Пример 49 Получение очищенных частиц танеспимицина-альбумина
88 мг танеспимицина растворяли в 2 мл смеси хлороформ/этанол (8:1, об./об.) и добавляли в 18,5 мл раствора человеческого сывороточного альбумина (10% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм, с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы танеспимицина, имеющие средний диаметр, равный 85-105 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц танеспимицина-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 5% маннит использовали в качестве диализата. Суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание танеспимицина в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 72 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и танеспимицина в продукте, отношение между альбумином и танеспимицином может быть рассчитано как 0,62:1.
Пример 50 Получение очищенных частиц пропофола-альбумина
603 мг пропофола растворяли в 6 мл хлороформа, и добавляли в 73 мл раствора человеческого сывороточного альбумина (10% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы пропофола, имеющие средний диаметр, равный 70-115 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц пропофола-альбумина методом центрифугирования, описанным в Примере 11, при 21000×g в течение 60 минут. Супернатант отбрасывали и очищенные наночастицы собирали. Очищенные наночастицы ресуспендировали в 10% растворе лактозы. Повторно полученную суспензию фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание пропофола в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 72 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и пропофола в продукте, отношение между альбумином и пропофолом может быть рассчитано как 0,58:1.
Пример 51 Получение очищенных частиц винбластина сульфата-альбумина
1,25 г винбластина сульфата растворяли в 10 мл смеси хлороформ/изопропанол (10:1, об./об.) и добавляли в 202 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы винбластина сульфата, имеющие средний диаметр, равный 90-130 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц винбластина сульфата-альбумина равнообъемным диализом против 5% маннита. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание винбластина сульфата в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 72 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и винбластина сульфата в продукте, отношение между альбумином и винбластина сульфатом может быть рассчитано как 0,23:1.
Пример 52 Получение очищенных частиц эксеместана-альбумина
155 мг эксеместан растворяли в 3 мл смеси хлороформ/этанол (6:1, об./об.) и добавляли в 27 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы эксеместана, имеющие средний диаметр, равный 70-100 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц эксеместана-альбумина равнообъемным диализом против 5% маннита. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание эксеместана в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 72 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и эксеместана в продукте отношение между альбумином и эксеместаном может быть рассчитано как 0,19:1.
Пример 53 Получение очищенных частиц флутамида-альбумина
189 мг флутамида растворяли в 2 мл смеси хлороформ/трет-бутанол (6:1, об./об.) и добавляли в 38 мл раствора человеческого сывороточного альбумина (8% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы флутамида, имеющие средний диаметр, равный 100-120 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц флутамида-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 5% раствор декстрана применяли в качестве диализата. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание флутамида в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 50 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 60 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и флутамида в продукте отношение между альбумином и флутамидом может быть рассчитано как 0,35:1.
Пример 54 Получение очищенных частиц фулвестранта-альбумина
350 мг фулвестранта растворяли в 3 мл смеси дихлорметан/этанол (6:1, об./об.) и добавляли в 67 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы фулвестранта, имеющие средний диаметр, равный 105-150 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц фулвестранта-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 10% лактозу применяли в качестве диализата. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание фулвестранта в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 20 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 60 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и фулвестранта в продукте отношение между альбумином и фулвестрантом может быть рассчитано как 0,29:1.
Пример 55 Получение очищенных частиц семустина-альбумина
650 мг семустина растворяли в 5 мл смеси хлороформ/этанол (6:1, об./об.) и добавляли в 89 мл раствора человеческого сывороточного альбумина (4% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы семустина, имеющие средний диаметр, равный 85-120 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц семустина-альбумина методом центрифугирования, описанным в Примере 11 при 21000×g в течение 60 минут. Супернатант отбрасывали и очищенные наночастицы собирали. Очищенные наночастицы ресуспендировали в 5% растворе маннита. Повторно полученную суспензию фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание семустина в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 20 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 70 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и семустина в продукте, отношение между альбумином и семустином может быть рассчитано как 0,58:1.
Пример 56 Получение очищенных частиц димера тиоколхицина-альбумина
456 мг димера тиоколхицина растворяли в 5 мл смеси хлороформ/этанол (9:1, об./об.) и добавляли в 58 мл раствора человеческого сывороточного альбумина (8% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом, генерировали наночастицы димера тиоколхицина, имеющие средний диаметр, равный 65-90 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц димера тиоколхицина-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 10% лактозу применяли в качестве диализата. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание димера тиоколхицин в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 20 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 70 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и димера тиоколхицина в продукте, отношение между альбумином и димером тиоколхицина может быть рассчитано как 0,37:1.
Пример 57 Получение очищенных частиц ибупрофена-димера альбумина
348 мг ибупрофена растворяли в 3 мл смеси дихлорметан/этанол (11:1, об./об.) и добавляли в 62 мл раствора человеческого сывороточного альбумина (5% м./об.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы ибупрофена, имеющие средний диаметр, равный 65-95 нм, и суспензия была полупрозрачной.
Наночастицы выделяли из полученной в результате суспензии наночастиц ибупрофена-альбумина посредством метода разделения на хроматографической колонке, описанного в Примере 16. 5% декстран применяли в качестве диализата. Полученную суспензию очищенных наночастиц фильтровали через стерильный фильтр 0,22 мкм, и в фильтрате не было никакой значительной вариации размера частиц. Содержание ибупрофена в растворе определяли, используя ВЭЖХ. На основе содержания, раствор последовательно разделяли на аликвоты в пузырьки при количестве, равном 20 мг на пузырек. Пузырьки помещали в лиофилизатор и лиофилизировали в течение 50 часов.
Когда лиофилизированный продукт восстанавливали в воде для инъекции, полученную в результате лепешку быстро растворяли, суспензия была полупрозрачной, и никакой значительной вариации размера частиц не наблюдали. После определения содержания человеческого сывороточного альбумина и ибупрофена в продукте отношение между альбумином и ибупрофеном может быть рассчитано как 0,19:1.
Пример 58 Концентрация альбумина влияет на ингибирующий эффект лекарственных средств на человеческие опухолевые клетки
Человеческие клетки SPC-A-1 рака легких и клетки MCF-7 рака молочной железы высевали в 96-луночный планшет, культивировали в течение ночи и делали их прилипающими к стенкам. Затем, среду заменяли на бессывороточную среду, чтобы истощить клетки. Пример 6, частицы 6 и Абраксан добавляли к клеткам до конечных концентраций, равных 160, 320, и 640 мкг/мл (концентрация паклитаксела) в течение 24 часов при 37°С. Степень ингибирования клеток различных групп дозирования сравнивали, используя метод MTT, и результаты показаны в следующей таблице и на ФИГ. 12 и 13. Кроме того, также строили кривую зависимости степени ингибирования от концентрации лекарственного средства. Как показано на кривой зависимости степени ингибирования от концентрации лекарственного средства, степень ингибирования частиц 6 была значительно выше, чем для других двух групп, и отсутствовало существенное различие между Примером 6 и Абраксаном. Данное означает, что излишний альбумин будет уменьшать ингибирующий эффект паклитаксела на опухолевые клетки.
Таблица 19. Степень ингибирования клеток для различной обработки
Клетки MCF-7
ингиби
рования %
Клетки SPC-A-1
ингиби
рования %
Пример 59 Концентрация альбумина влияет на поглощение лекарственных средств человеческими васкулярными эндотелиальными клетками
Человеческие васкулярные эндотелиальные клетки пупочной вены EA.hy 926 засевали при 8×105 в 6-луночный планшет. Перед добавлением лекарственных средств среду заменяли на среду без сыворотки. Лекарственные средства разделяли на 5 групп, группа A представляла собой частицу 6, B являлась Примером 6, C представляла собой Абраксан, D являлась частицей 6+0,5% мономера ЧСА, E представляла собой частицу 6+0,5% полимера ЧСА. Различные группы лекарственных средств добавляли к клеткам при конечных концентрациях, равных 50, 100, и 200 мкг/мл (концентрация паклитаксела) в течение 2 часов при 37°С. Через 2 часа клетки промывали PBS в течение 3 раз, затем 500 мкл 5% Тритона X-100 добавляли в каждую лунку для лизиса клеток. Концентрацию паклитаксела при клеточном лизисе обнаруживали методом, указанным в Примере 10, и результаты показаны в следующей таблице и на ФИГ. 14. Результаты показывают, что клеточное поглощение паклитаксела для группы A было выше, чем для группы B и группы C, а группы B и C не имели значительного различия. Кроме того, показатели для группы D были выше, чем для группы E. Это означает, что более высокая концентрация ЧСА затрудняла поглощение лекарственного средства васкулярными эндотелиальными клетками, и, в сравнении с мономером, полимер ЧСА в большей степени мог бы затруднять клеточное поглощение.
Таблица 20. Концентрация лекарственного средства в клетках для различной обработки
Пример 60
Содержание альбумина и паклитаксела (PTX) и соотношение полимера в применямом человеческом сывороточном альбумине, продукте, полученном в Примере 6, и соответствующих наночастицах 6, определяли, используя метод Примера 10. Результаты приведены в следующей таблице.
Таблица 21. Содержание полимера альбумина (Поли-ЧСА)
(мг/мг)
(мг/мг)
Результаты продемонстрировали, что доля поли-ЧСА значительно увеличилась во время процесса получения наночастиц паклитаксела-альбумина. Однако, в продукте, полученном согласно способу настоящего раскрытия, количество поли-ЧСА в единице массы является значительно сниженным вследствие уменьшения общего содержание альбумина.
Количество полимера в растворе альбумин увеличивалось при диализе с использованием регенерированной целлюлозной ультрафильтрационной мембраны (PXC100C50, Millipore) с пределом пропускания по молекулярной массе, равным 100 кДа. Долю полимера альбумина определяли, используя метод Примера 10, а содержание альбумина определяли методом Кьельдаля, чтобы избежать отклонения, производимого различными ответными реакциями на потенциальные полимеры при методе ВЭЖХ.
Таблица 22. Содержание полимера альбумина в ЧСА до и после диализа
По результату можно видеть, что раствор альбумина с преобладающим количеством полимера может быть получен посредством процесса диализа.
Пример 61 Исследование сенсибилизации для поли-ЧСА у морских свинок
В данном эксперименте ЧСА и поли-ЧСА, полученные из Примера 60, были выбраны в качестве тестируемых лекарственных средств. Выбранные дозировки составляли 1 мг на морскую свинку и 0,3 мг на морскую свинку. Сенсибилизацию проводили посредством внутрибрюшинной инъекции один раз в два дня, всего за 3 инъекции. В то же время устанавливали группу положительного контроля (0,2% овальбумина) и группу отрицательного контроля (инъекция 0,9% хлорида натрия). Детальный режим дозирования приведен в таблице ниже.
Таблица 23. Протокол для исследования сенсибилизации у морских свинок
(мг/мл)
(мл/живот.)
(мг/живот.)
(мл/живот.)
(мг/живот.)
Метод введения для сенсибилизации: спинку морской свинки плотно удерживали левой рукой, сложенной в форме чашечки, обеспечивая натяжение кожи на животе при фиксации морской свинки. Живот морской свинки поднимали, и голова была опущена вниз. После дезинфекции участка инъекции спиртовой салфеткой, Иглой одноразового шприца объемом 2 мл, удерживаемого правой рукой, протыкали кожу морской свинки. Иглу вводили на участке, находящемся на 1 мм слева от средней линии нижней части живота. При прохождении подкожной части иглу вводили прямо на дополнительные от 5 мм до 10 мм, и последовательно проводили в брюшную полость под углом 45°. После фиксации иглы фармацевтический раствор вводили посредством медленной инъекции. После инъекции сухим ватным тампоном надавливали на точку укола, чтобы препятствовать вытеканию фармацевтического средства.
Возбуждение аллергии: возбуждение проводили посредством внутривенной инъекции, и возбуждение проводили через 10 дней после последней сенсибилизации с дозировками, равными 2 мг/животное и 0,6 мг/животное.
Метод введения для возбуждения: инъекцию проводили в латеральную метатарзальную вену морской свинки, фиксируемой ассистентом. Коленные суставы были зажаты оператором для фиксации тела животного. Его вену сжимали и лапы находились в вытянутом состоянии. Волосяной покров на участке инъекции сбривали (или срезали кожу на участке инъекции). После стерилизации спиртовыми салфетками можно было видеть толстую латеральную метатарзальную вену. Иглой одноразового шприца объемом 1 мл в правой руке протыкали кровеносный сосуд вдоль направления в сторону сердца. После инъекции сухим ватным тампоном надавливали на место прокола, чтобы предотвратить кровотечение.
Реакцию каждого животного и время появления или исчезновения симптомов аллергии наблюдали сразу же после возбуждения в течение 30 минут. Максимальная продолжительность наблюдения составляла 3 часа. Результаты аллергической реакции приведены в следующей таблице.
Таблица 24. Симптомы аллергической реакции
Таблица 25. Критерии оценки для системной аллергической реакции
Таблица 26. Результаты активной анафилаксии у морских свинок
оценка
Результаты показывают, что образцы, содержащие большее количество поли-ЧСА, обладали более сильной сенсибилизацией при одинаковом уровне общего содержания ЧСА.
Пример 62
2,5 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 129 нм, и суспензия была полупрозрачной. Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). Не было никакой значительной вариации размера частиц после фильтрации, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре.
Очищенные наночастицы выделяли соответственно центрифугированием, описанным в Примере 11, диализом, описанным в Примере 15, и разделением на хроматографической колонке, описанным в Примере 16. Результаты показали, что суспензия становилась мутной, и во время диализа происходило осаждение. Таким образом, очищенные наночастицы не могут быть получены посредством данного способа. В то время как очищенные наночастицы, полученные посредством центрифугирования и разделения на хроматографической колонке, были мутными и осаждались за 60 мин.
Пример 63
2,5 г паклитаксела (CAS: 33069-62-4, Yunnan Hande Bio-Tech Co., Ltd) растворяли в 15 мл смеси хлороформ/этанол (11:1, об./об.), которую добавляли в 500 мл раствора человеческого сывороточного альбумина (4% м./об.) (CAS: 70024-90-7, Guangdong Shuanglin Biopharmaceutical. Co., Ltd.). Смесь эмульгировали в течение 2 минут, используя диспергатор с высоким сдвиговым усилием (Fluko FZ-20) с получением первичной эмульсии. Первичную эмульсию затем гомогенизировали, используя гомогенизатор высокого давления (Model M110-EH30K, MFIC Company, USA) под давлением 10000-20000 фунт/кв. дюйм с получением наноэмульсии. Затем наноэмульсию переносили в роторный испаритель (Model R-210, Buchi Company, Switzerland) для удаления органического растворителя в растворе посредством испарения в вакууме при 40 мбар и при 40°C в водяной бане. Таким образом генерировали наночастицы паклитаксела-альбумина, имеющие средний диаметр, равный 128 нм, и суспензия была полупрозрачной. Суспензия может быть легко отфильтрована через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). После фильтрации не было никакой значительной вариации размера частиц, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре.
Диализ проводили в равном объеме по отношению к полученным образцам, против очищенной воды, используя регенерированную целлюлозную ультрафильтрационную мембрану (PXC300C50, Millipore) с пределом пропускания по молекулярной массе, равным 30 кДа, и кратность диализа составляла 5.
Очищенные наночастицы выделяли соответственно центрифугированием, описанным в Примере 11, диализом, описанным в Примере 15 и разделением на хроматографической колонке, описанном в Примере 16. Полученные в результате полупрозрачные суспензии от всех 3 методов были стабильными и могут быть легко отфильтрованы через стерильный фильтр 0,22 мкм (Sartorius AG, Germany). После фильтрации не было никакой значительной вариации размера частиц, и никакого значительного изменения не наблюдали после хранения в течение 48 часов при комнатной температуре
При сравнении Примеров 62 и 63 видно, что дополнительный процесс равнообъемного диализа против очищенной воды проводили в Примере 63 для удаления остаточных растворителей. Удаление остаточных растворителей улучшало стабильность суспензии очищенных наночастиц.
Обсуждение:
Составы предшествующего уровня техники имеют высокое отношение альбумина к активному ингредиенту (например, 9:1). Авторы настоящего изобретения неожиданно обнаружили, что в таком составе большая часть альбумина действует только как защитное средство или средство подложки в лиофилизированном продукте. Большая часть молекул лекарственного средства (например, паклитаксела) инкапсулирована в наночастицы, и альбумин, который не образует частицы, является по существу свободным от молекул лекарственного средства. В составах предшествующего уровня техники большая часть молекул человеческого сывороточного альбумина не является связанной с молекулами лекарственного средства.
После введения частицы составов предшествующего уровня техники быстро дезинтегрируются, и между молекулами лекарственного средства и эндогенным человеческим сывороточным альбумином образуются комплексы. Следовательно, избыточный человеческий сывороточный альбумин в составах предшествующего уровня техники не способствует эффективности действия составов (см. Пример 26), но вместо этого вызывает риски для безопасности вследствие его агрегации и иммуногенности (см. Примеры 32, 25 и 27). Кроме того, избыточный альбумин может конкурировать с комплексом лекарственное средство-альбумин за связывание с рецептором gp60, и, таким образом, уменьшать поглощение лекарственного средства васкулярными эндотелиальными клетками (см. Пример 59). Более того, после связывания с поли-ЧСА, образованным в процессе получения, рецептор gp60 может быть неспособен к эффективному трансцитозу, таким образом, имеет место предотвращение высвобождения рецептора. Это может дополнительно ингибировать поглощение лекарственного средства эндотелиальными клетками, и, таким образом, уменьшать эффективную концентрацию лекарственных средств.
Исходя из приведенных выше данных, очищенные наночастицы настоящего раскрытия имеют более низкое соотношение альбумин: активный ингредиент. Авторы настоящего изобретения подтвердили, что лишь небольшое количество человеческого сывороточного альбумина требовалось для образования стабильных наночастиц с молекулами лекарственного средства. Снижение содержания человеческого сывороточного альбумина в очищенных наночастицах и их композициях, предоставленных в настоящем описании, уменьшает неблагоприятные реакции, ассоциированные с агрегацией и иммуногенностью человеческого сывороточного альбумина.
После введения очищенные наночастицы, предоставленные в настоящем описании, обладали способностью к быстрой дезинтеграции, и связывались с эндогенным альбумином для циркуляции in vivo. При исследованиях на животных не было никакого значительного различия, наблюдаемого с позиций безопасности и эффективности действия, в сравнении с составами предшествующего уровня техники. Однако, очищенные наночастицы и их композиции, предоставленные в настоящем описании, улучшали поглощение человеческими васкулярными эндотелиальными клетками, а также доставку и эффективность активных ингредиентов в наночастицах, к человеческим целевым клеткам или в них.
Различные варианты осуществления, описанные выше, могут комбинироваться, чтобы предоставить дополнительные варианты осуществления. Все Патенты США, публикации заявок на Патент США, заявки на Патент США, патенты в иностранных государствах, заявки на патент в иностранных государствах и непатентные публикации, указанные в данном описании и/или приведенные в Информационном Листке Заявки полностью включены в данный документ посредством ссылки. Аспекты вариантов осуществления могут быть модифицированы, при необходимости, чтобы использовать концепции различных патентов, заявок и публикаций для предоставления еще дополнительных вариантов осуществления.
Эти и другие изменения могут быть проделаны с вариантами осуществления в свете подробного приведенного выше описания. В целом, в следующей формуле изобретения применяемые термины не должны рассматриваться, как ограничивающие формулу изобретения конкретными вариантами осуществления, раскрытыми в описании, и формуле изобретения, но должны рассматриваться, как включающие все возможные варианты осуществления наряду с полным объемом эквивалентов, права на которые такая формула изобретения предоставляет. Соответственно, формула изобретения не ограничивается раскрытием.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЭЛЕМЕН, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЕЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2838027C1 |
| СТАБИЛЬНАЯ КОМПОЗИЦИЯ НАНОЧАСТИЦ ДОЦЕТАКСЕЛА-АЛЬБУМИНА | 2022 |
|
RU2835478C1 |
| КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ СЛАБОРАСТВОРИМЫЕ В ВОДЕ ФАРМАЦЕВТИЧЕСКИЕ ВЕЩЕСТВА И ПРОТИВОМИКРОБНЫЕ ВЕЩЕСТВА | 2006 |
|
RU2433818C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ПРЕДОТВРАЩЕНИЯ ИЛИ МИНИМИЗАЦИИ ЭПИТЕЛИАЛЬНО-МЕЗЕНХИМАЛЬНОГО ПЕРЕХОДА | 2017 |
|
RU2764630C2 |
| КОМПОЗИЦИИ НАНОЧАСТИЦ АЛЬБУМИНА И ПАКЛИТАКСЕЛА | 2013 |
|
RU2663687C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ПОЛУЧЕНИЯ СЛАБОРАСТВОРИМЫХ В ВОДЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ С УВЕЛИЧЕННОЙ СТАБИЛЬНОСТЬЮ | 2006 |
|
RU2451510C2 |
| Способ получения препарата на основе магнитных наночастиц (МНЧ) оксида железа для МРТ-диагностики новообразований | 2017 |
|
RU2659949C1 |
| Способ получения радиомеченных частиц карбоната кальция с использованием дефероксамина в качестве хелатирующего вещества | 2022 |
|
RU2806148C1 |
| Способ получения препарата для диагностики новообразований методом магнитно-резонансной томографии | 2019 |
|
RU2723894C1 |
| Препарат для диагностики новообразований методом магнитно-резонансной томографии | 2019 |
|
RU2723932C1 |

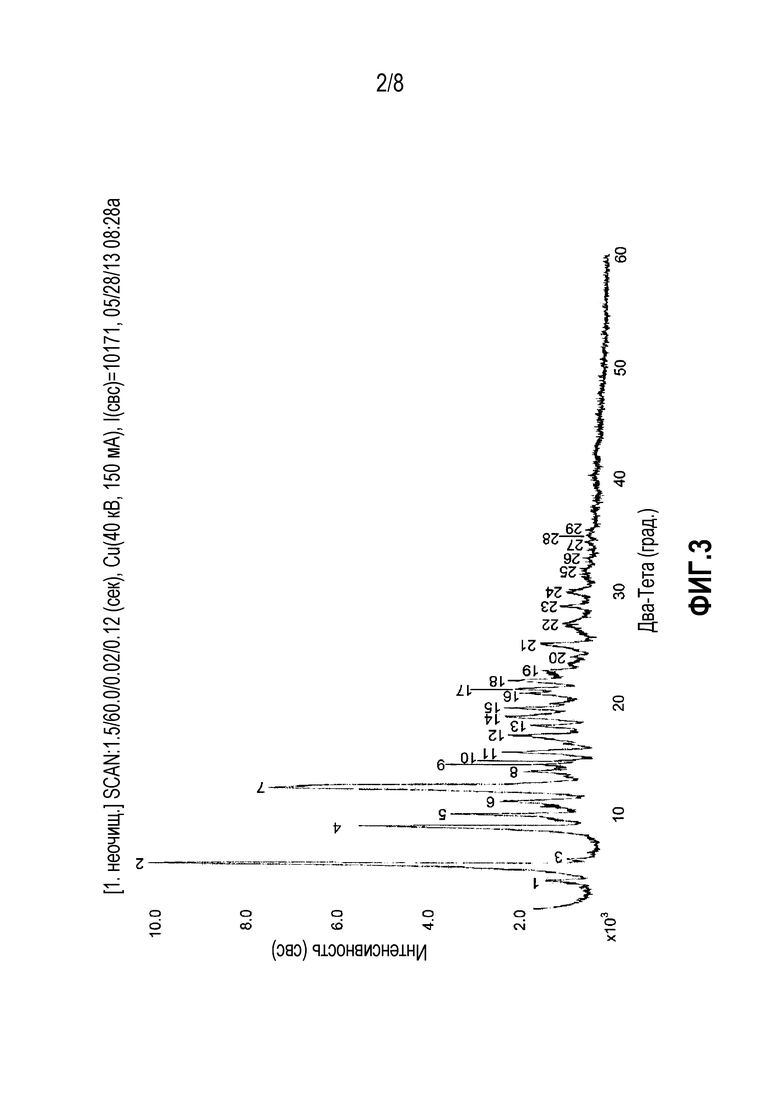
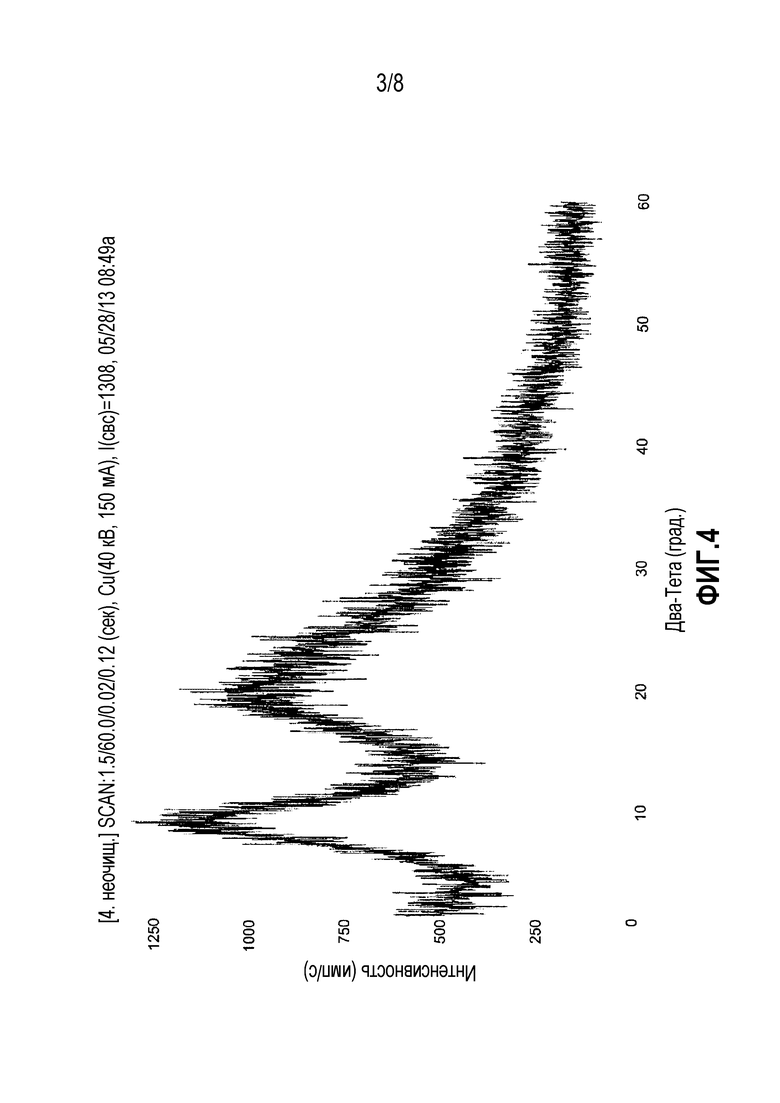
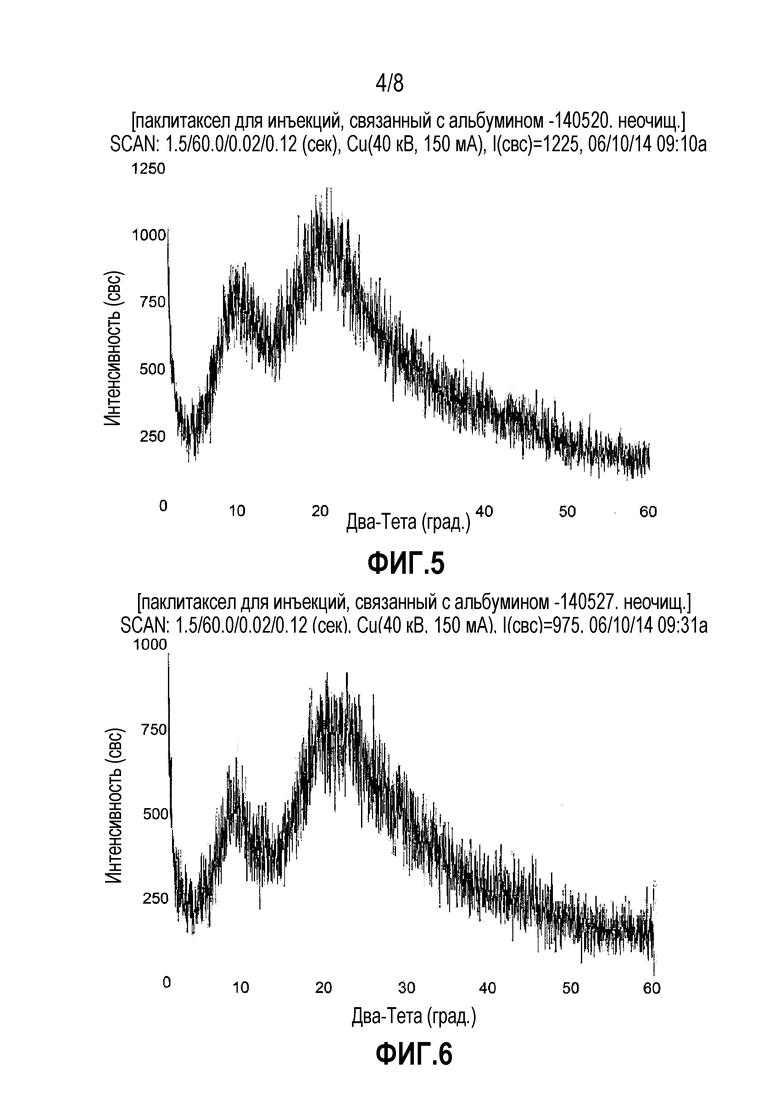
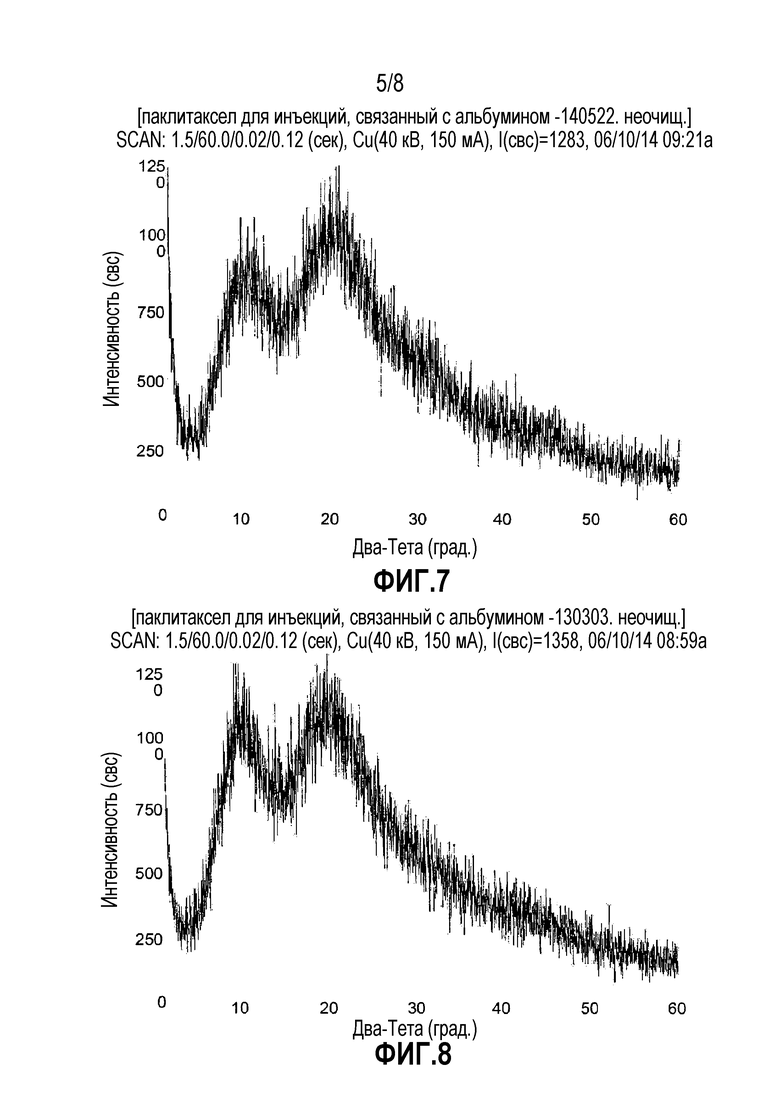
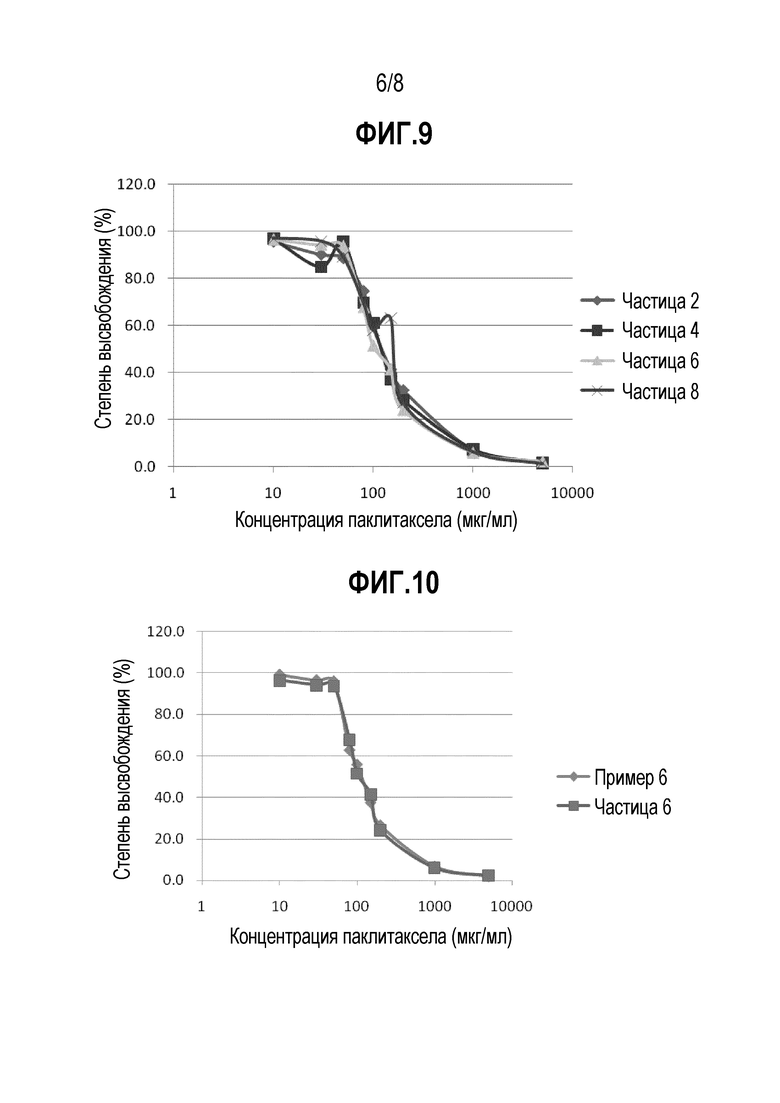
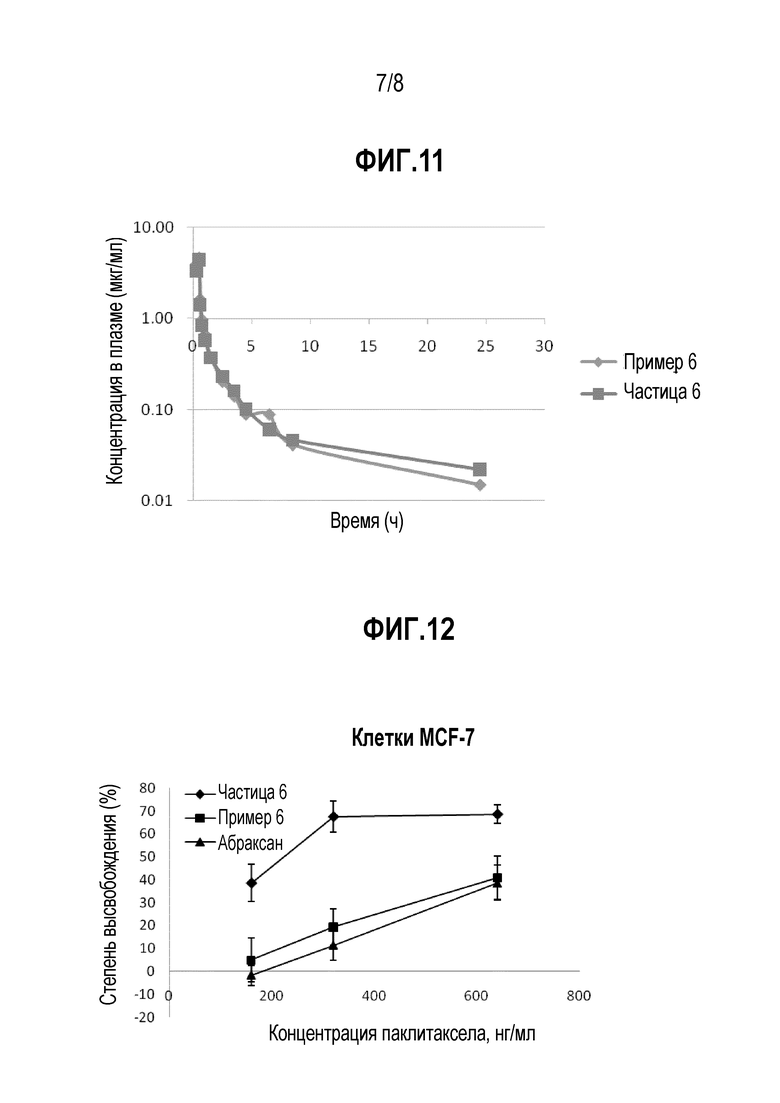
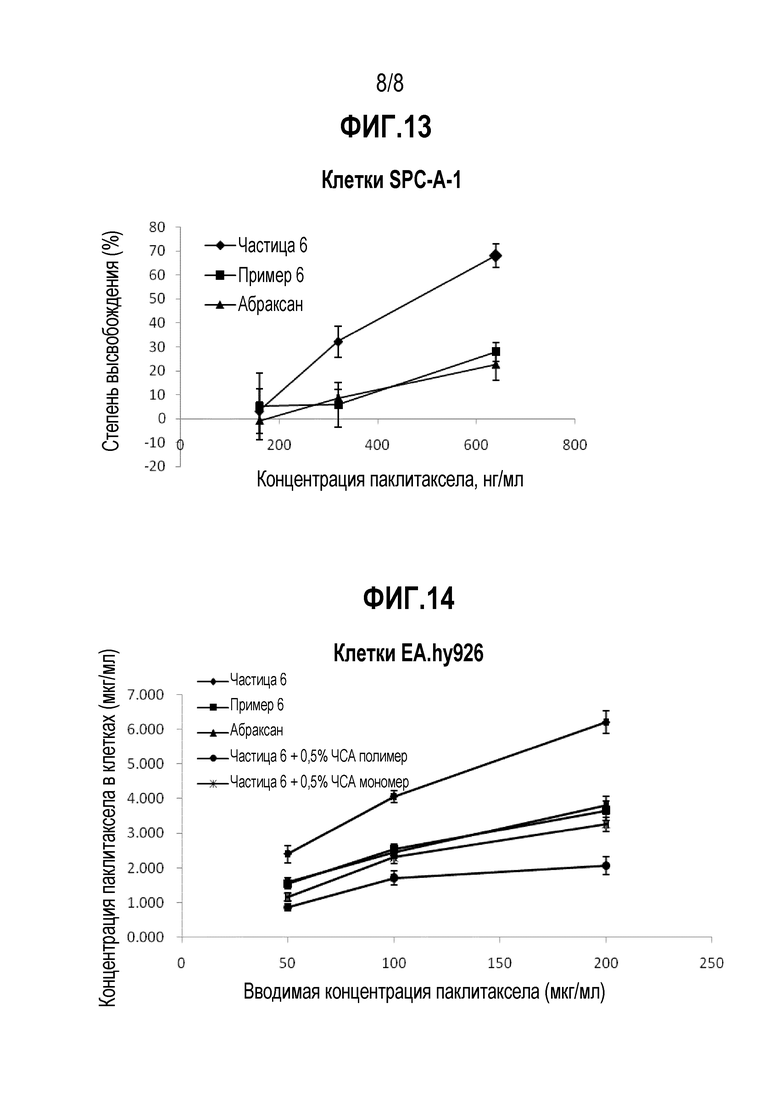
Изобретение относится к фармацевтической промышленности и представляет собой фармацевтическую композицию для лечения рака, содержащую очищенные терапевтические наночастицы, где указанные очищенные терапевтические наночастицы содержат активный ингредиент и человеческий сывороточный альбумин, где массовое отношение человеческого сывороточного альбумина (ЧСА) к активному ингредиенту в терапевтических наночастицах составляет от 0,03:1 до 0,95:1, активный ингредиент выбран из таксанов, камптотецинов, антрациклиновых антибиотиков, колхицина, димера тиоколхицина, лиотиронина, циклоспорина, эксеместана, флутамида, фулвестранта, ромидепсина и семустина и где указанная фармацевтическая композиция содержит, по большей мере, 5% свободного ЧСА (масс.). Изобретение обеспечивает стабилизацию наночастиц с молекулами лекарственного средства, а также уменьшение неблагоприятных реакций, ассоциированных с агрегацией и иммуногенностью человеческого сывороточного альбумина и снижение токсичности. 5 н. и 16 з.п. ф-лы, 14 ил., 26 табл., 63 пр.
1. Фармацевтическая композиция для лечения рака, содержащая очищенные терапевтические наночастицы, где указанные очищенные терапевтические наночастицы содержат активный ингредиент и человеческий сывороточный альбумин, где массовое отношение человеческого сывороточного альбумина (ЧСА) к активному ингредиенту в терапевтических наночастицах составляет от 0,03:1 до 0,95:1, активный ингредиент выбран из таксанов, камптотецинов, антрациклиновых антибиотиков, колхицина, димера тиоколхицина, лиотиронина, циклоспорина, эксеместана, флутамида, фулвестранта, ромидепсина и семустина и где указанная фармацевтическая композиция содержит, по большей мере, 5% свободного ЧСА (масс.).
2. Фармацевтическая композиция по п. 1, где активным ингредиентом является таксан.
3. Фармацевтическая композиция по п. 1, где таксан представляет собой паклитаксел или доцетаксел.
4. Фармацевтическая композиция по п. 1, где активный ингредиент инкапсулирован внутри человеческого сывороточного альбумина.
5. Фармацевтическая композиция по п. 1, где средний размер частицы терапевтических наночастиц выбран из: 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 140, 141, 142, 143, 144, 145, 146, 147, 148, 149, 150, 151, 152, 153, 154, 155, 156, 157, 158, 159, 160, 165, 170, 175, 180, 185, 190, 195, 200 нм или интервала между двумя числовыми значениями, приведенными выше.
6. Фармацевтическая композиция для лечения рака, содержащая очищенные терапевтические наночастицы, содержащие активный ингредиент и человеческий сывороточный альбумин, где активный ингредиент инкапсулирован внутри человеческого сывороточного альбумина; массовое отношение человеческого сывороточного альбумина к активному ингредиенту составляет от 0,03:1 до 0,95:1; активный ингредиент выбран из таксанов, камптотецинов, антрациклиновых антибиотиков, колхицина, димера тиоколхицина, лиотиронина, циклоспорина, эксеместана, флутамида, фулвестранта, ромидепсина и семустина; средний размер частицы терапевтических наночастиц составляет от 50 нм до 190 нм; и указанная фармацевтическая композиция содержит, по большей мере, 5% свободного ЧСА (масс.).
7. Фармацевтическая композиция по п. 6, где фармацевтическая композиция находится в форме жидкости или лиофилизированного порошка.
8. Фармацевтическая композиция по п. 6, где фармацевтическая композиция также содержит эксципиент лиофилизации, когда фармацевтическая композиция находится в форме лиофилизированного порошка.
9. Фармацевтическая композиция по п. 8, где эксципиент лиофилизации выбран из одного или нескольких из маннита, сахарозы, лактозы, мальтозы, трегалозы и декстрана.
10. Фармацевтическая композиция по п. 6, где активным ингредиентом является паклитаксел.
11. Способ получения фармацевтической композиции по п. 1, включающий:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы, указанных выше;
3) удаление органического растворителя в эмульсии с получением суспензии, содержащей терапевтические наночастицы; и
4) удаление свободного ЧСА, который не включен в наночастицы, из суспензии с получением очищенных терапевтических наночастиц.
12. Способ по п. 11, где органический растворитель выбран из одного или нескольких из хлороформа и этанола.
13. Способ по п. 11, дополнительно включающий в себя: между стадиями 3) и 4), стадию диализа суспензии стадии 3) с водным раствором для удаления оставшегося органического растворителя из суспензии.
14. Способ по п. 13, где водным раствором является вода.
15. Способ по п. 11, где указанное разделение на стадии 4) проводят с использованием метода, выбранного из центрифугирования, диализа и эксклюзионной хроматографии.
16. Способ получения фармацевтической композиции по п. 1, включающий:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы, указанных выше;
3) удаление органического растворителя из эмульсии с получением суспензии, содержащей терапевтические наночастицы;
4) удаление свободного ЧСА, который не включен в наночастицы с получением очищенных терапевтических наночастиц;
5) повторное суспендирование очищенных терапевтических наночастиц в растворе, содержащем эксципиент; и
6) необязательно лиофилизацию повторной суспензии очищенных терапевтических наночастиц с получением фармацевтической композиции.
17. Способ по п. 16, дополнительно включающий между стадиями 3) и 4) стадию диализа суспензии стадии 3) с водным раствором для удаления оставшегося органического растворителя из суспензии.
18. Способ по п. 17, где водным раствором является вода.
19. Способ получения фармацевтической композиции по п. 1, включающий:
1) растворение активного ингредиента в органическом растворителе с образованием масляной фазы, и растворение человеческого сывороточного альбумина в воде с образованием водной фазы;
2) образование эмульсии масло-в-воде с использованием масляной фазы и водной фазы, указанных выше;
3) удаление органического растворителя в эмульсии с получением суспензии, содержащей терапевтические наночастицы;
4) диализ суспензии, полученной после удаления органического растворителя посредством раствора, содержащего эксципиент, для удаления свободного ЧСА, который не включен в наночастицы; и
5) необязательно лиофилизацию диализированной суспензии с получением фармацевтической композиции.
20. Способ по п. 19, дополнительно включающий между стадиями 3) и 4) стадию диализа суспензии стадии 3) с водным раствором, чтобы удалить оставшийся органический растворитель из суспензии.
21. Способ по п. 20, где водным раствором является вода.
| US 5916596 A, 29.06.1999 | |||
| CN 103221042 A, 24.07.2013 | |||
| US 2005004002 A1, 06.01.2005 | |||
| СИСТЕМА-НОСИТЕЛЬ В ФОРМЕ НАНОЧАСТИЦ НА ОСНОВЕ ПРОТЕИНА ДЛЯ КЛЕТОЧНО-СПЕЦИФИЧЕСКОГО ОБОГАЩЕНИЯ ДЕЙСТВУЮЩИХ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ | 2005 |
|
RU2388463C2 |

