ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтических составов, в частности, к фармацевтической композиции, содержащей элемен, и способам ее получения и ее применению.
УРОВЕНЬ ТЕХНИКИ
Элемен представляет собой летучее маслянистое соединение, экстрагируемое из таких растений, как Curcuma Wenyujin Y. H. Chen et C. ling и Cymbopoqon citratus ((DC.)) Stapt. Указанное соединение выглядит как бледно-желтая или желтая прозрачная жидкость с резким запахом фенхеля. В настоящее время элемен применяют в клинической практике в виде смеси, содержащей α-элемен, β-элемен, δ-элемен и γ-элемен, которую широко используют при злокачественном плевральном выпоте, раке легкого, опухолях желудочно-кишечного тракта и других поверхностно расположенных опухолях. Поскольку доступный на рынке элемен и его составы содержат Cremophor EL и другие вспомогательные вещества, вызывающие сильное раздражение, после применения таких доступных на рынке лекарственных средств большинство пациентов страдают от серьезного флебита, лихорадки, местной боли, аллергической реакции или небольшой реакции желудочно-кишечного тракта, а также пирексии и других нежелательных реакций. В фактическом клиническом способе медикаментозной терапии, в связи с высокой частотой возникновения флебита и более высокой частотой его рецидивов, скорость внутривенной инфузии необходимо строго регулировать, что приводит к увеличению времени инфузии.
Элемен практически нерастворим в воде и обладает сильной растворимостью в липидах, особенно в органических растворителях, таких как петролейный эфир, диэтиловый эфир и т.п. Его молекулярная формула представляет собой C15H24 и состоит только из углеводородов. Вследствие его уникальной химической структуры и физико-химических свойств существуют большие трудности и препятствия при получении его фармацевтических составов. Для повышения растворимости элемена в процессе приготовления элемена для инъекций в доступные на рынке рецептурные лекарственные препараты добавляли Cremophor EL и другие вспомогательные вещества, что приводило к серьезным проблемам, связанным с гемолизом, и сильному раздражению. Кроме того, хотя маркетинг наносостава на основе паклитаксела+альбумина предоставляет новую форму фармацевтического состава для решения проблем, связанных с клиническими побочными эффектами, вследствие физико-химических свойств элемена, который является маслянистым при комнатной температуре, чрезвычайно сложно включить элемен в составы на основе альбумина. Таким образом, в настоящее время отсутствуют соответствующие исследования или разработки.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящей заявке предложена фармацевтическая композиция, содержащая элемен, масло для инъекций и белковый носитель, способный эффективно снижать один или более побочных эффектов фармацевтических композиций, вводимых в организм человека.
Согласно одному или более вариантам реализации настоящего изобретения белковый носитель, применяемый в фармацевтической композиции, включает белок, при этом можно использовать любой подходящий белок. Примеры подходящего белка включают, но не ограничиваются ими, альбумин, иммуноглобулин, в том числе, но не ограничиваясь ими, IgA, липопротеин, аполипопротеин B, альфа-кислый гликопротеин, бета-2-макроглобулин, тиреоглобулин, белок трансферрин, фибронектин, фактор VII, фактор VIII, фактор IX, фактор X и аналоги. Белковый носитель может быть природным или синтетическим. Согласно некоторым вариантам реализации белковый носитель представляет собой белок, не относящийся к крови, такой как казеин, альфа-лактальбумин и бета-лактоглобулин. Согласно некоторым вариантам реализации белковый носитель включает альбумин, такой как человеческий сывороточный альбумин (ЧСА), бычий сывороточный альбумин и т.п. Человеческий сывороточный альбумин представляет собой легкорастворимый глобулин, Mr65K, состоящий из 585 аминокислот. ЧСА является преобладающим белком в плазме и формирует от 70 до 80% коллоидного осмотического давления в плазме человека.
Согласно одному или более вариантам реализации настоящего изобретения «масло для инъекций», применяемое в предложенной фармацевтической композиции, включает, но не ограничивается ими, этилолеат, бензилбензоат, триглицерид средней цепи (MCT) и растительное масло, при этом растительное масло выбирают из одного или более масел из соевого масла, сафлорового масла, хлопкового масла, кукурузного масла, подсолнечного масла, арахисового масла и оливкового масла. Согласно одному или более вариантам реализации настоящего изобретения масло для инъекций представляет собой соевое масло.
Согласно одному или более вариантам реализации настоящего изобретения предложенную фармацевтическую композицию можно получить в виде частиц, размер которых составляет менее 180 нм, например, около от 70 до 170 нм, предпочтительно около от 70 до 150 нм.
Согласно одному или более вариантам реализации настоящего изобретения предложенная композиция содержит наночастицы любой формы (например, сферической или несферической формы), при этом согласно некоторым вариантам реализации средний или промежуточный диаметр частиц составляет не более около 180 нм. Согласно некоторым вариантам реализации средний или промежуточный диаметр частиц составляет от около 50 нм до около 180 нм. Согласно некоторым вариантам реализации средний или промежуточный диаметр частиц составляет от около 60 нм до около 180 нм. Согласно некоторым вариантам реализации средний или промежуточный диаметр частиц составляет от около 70 нм до около 170 нм. Согласно некоторым вариантам реализации средний или промежуточный диаметр частиц составляет от около 70 нм до около 170 нм. Согласно некоторым вариантам реализации средний или промежуточный диаметр частиц составляет от около 70 до около 150 нм. Согласно некоторым вариантам реализации средний или промежуточный диаметр частиц составляет около 60 нм, 70 нм, 80 нм, 90 нм, 100 нм, 110 нм, 120 нм, 130 нм, 140 нм или 150 нм. Согласно некоторым вариантам реализации частицы являются стерильными и поддаются фильтрованию.
Согласно одному или более вариантам реализации настоящего изобретения элемен в фармацевтической композиции выбирают из одного или более элеменов из α-элемена, β-элемена, γ-элемена и δ-элемена. Согласно одному или более вариантам реализации настоящего изобретения элемен в фармацевтической композиции представляет собой β-элемен.
Согласно одному или более вариантам реализации настоящего изобретения отношение альбуминового носителя, элемена и масла для инъекций в фармацевтической композиции представляет собой массовое отношение при дозировании.
Согласно одному или более вариантам реализации настоящего изобретения массовое отношение белкового носителя к элемену в фармацевтической композиции составляет 0,520; например, от 0,5 до 0.
Согласно одному или более вариантам реализации настоящего изобретения массовое отношение масла для инъекций к элемену в фармацевтической композиции составляет 0,3510; например, от 0,5 до 10.
Согласно одному или более вариантам реализации настоящего изобретения предложенная фармацевтическая композиция дополнительно содержит лиопротектор. Лиопротектор выбирают из одного или более лиопротекторов из глюкозы, сахарозы, мальтозы, лактозы, маннозы, трегалозы, глицина и декстрана, предпочтительно трегалозы или сахарозы. Согласно одному или более вариантам реализации отношение массы лиопротектора к объему раствора фармацевтической композиции составляет от 3:100 до 20:100 г/мл, предпочтительно от 5:100 до 10:100 г/мл.
Согласно одному или более вариантам реализации настоящего изобретения предложенная фармацевтическая композиция дополнительно содержит один или более компонентов из агента, регулирующего изоосмотические свойства, антиоксиданта, консерванта и регулятора рН. Агент, регулирующий изоосмотические свойства, представляет собой один или более компонентов из глицерола, сорбитола, маннитола или глюкозы; регулятор pH представляет собой один или более компонентов из гидроксида натрия, цитрата натрия, лимонной кислоты, фосфорной кислоты, уксусной кислоты или соляной кислоты; консервант представляет собой один или более компонентов из гидроксибензолалкиловых эфиров, бензойной кислоты, бензоата натрия, сорбиновой кислоты, ацетата хлоргексидина, бромида бензалкония; антиоксидант представляет собой один или более компонентов из сульфита натрия, бисульфита натрия, метабисульфита натрия, тиосульфата натрия, аскорбиновой кислоты, бутилированного гидроксианизола, 2,6-ди-трет-бутилированного гидрокситолуола и витамина Е.
Согласно одному или более вариантам реализации настоящего изобретения также предложена фармацевтическая композиция, в процессе получения которой не используют органические растворители.
Согласно одному или более вариантам реализации настоящего изобретения предложенную фармацевтическую композицию можно получить в виде частиц, размер которых составляет менее 180 нм, например, около от 70 до 150 нм.
Согласно одному или более вариантам реализации настоящего изобретения в предложенной фармацевтической композиции массовое отношение соевого масла к элемену составляет около от 0,35 до 3 и массовое отношение человеческого сывороточного альбумина к элемену составляет около от 0,5 до 20; предпочтительно массовое отношение соевого масла к элемену составляет около от 0,5 до 3, массовое отношение человеческого сывороточного альбумина к элемену составляет около от 0,5 до 2,5; или в предложенной фармацевтической композиции массовое отношение соевого масла к элемену составляет около от 0,35 до 1,5 и массовое отношение человеческого сывороточного альбумина к элемену составляет около от 1,5 до 10.
Согласно одному или более вариантам реализации настоящего изобретения массовое отношение соевого масла к элемену составляет около 0,5 и массовое отношение человеческого сывороточного альбумина к элемену составляет около 1,5.
Согласно одному или более вариантам реализации настоящего изобретения массовое отношение соевого масла к элемену составляет около 0,75 и массовое отношение человеческого сывороточного альбумина к элемену составляет около 5.
Согласно одному или более вариантам реализации настоящего изобретения массовое отношение соевого масла к элемену составляет около 1,5 и массовое отношение человеческого сывороточного альбумина к элемену составляет около 3-5.
Согласно одному или более вариантам реализации настоящего изобретения также предложена фармацевтическая композиция, которая в процессе получения содержит органический растворитель. При этом органический растворитель выбирают из одного или более растворителей из хлороформа, дихлорметана, третичного бутанола, изопропанола, этилацетата, этанола, тетрагидрофурана, диоксана, ацетонитрила, ацетона, диметилсульфоксида, диметилформамида и метилпирролидона. При этом липорастворимый органический растворитель представляет собой смешанный растворитель, состоящий из дихлорметана и этанола.
При этом объемное отношение дихлорметана к этанолу составляет 1:1-8, предпочтительно 1:4.
Фармацевтическую композицию можно получить в виде частиц, размер которых составляет около от 70 до 150 нм.
В предложенной фармацевтической композиции массовое отношение соевого масла к элемену составляет около от 2,4 до 10 и массовое отношение человеческого сывороточного альбумина к элемену составляет около от 0,5 до 10; в предложенной фармацевтической композиции предпочтительно массовое отношение соевого масла к элемену составляет около 2,4, массовое отношение человеческого сывороточного альбумина к элемену составляет около 3.
В одном или более вариантах реализации настоящего изобретения предложен способ получения описанной в настоящей заявке фармацевтической композиции, включающий следующие стадии:
(1) равномерное перемешивание элемена и масла для инъекций, необязательно с добавлением органического растворителя, с получением раствора элемена и масла для инъекций; растворение белкового носителя в воде с получением раствора белкового носителя;
(2) смешивание двух растворов, полученных на стадии (1), с получением эмульсии;
(3) гомогенизацию эмульсии на стадии (2) под высоким давлением и необязательно испарение эмульсии при пониженном давлении для удаления органического растворителя с получением раствора наночастиц.
В одном или более вариантах реализации настоящего изобретения предложен способ получения описанной в настоящей заявке фармацевтической композиции, включающий следующие стадии:
(1) равномерное перемешивание элемена и масла для инъекций;
(2) взятие альбумина и растворение альбумина в воде;
(3) смешивание обоих компонентов с получением эмульсии;
(4) гомогенизацию эмульсии на стадии (3) под высоким давлением с получением раствора наночастиц альбумина.
Согласно одному или более вариантам реализации настоящего изобретения на стадии (2) для смешивания используют способ с применением сдвига или способ с применением ультразвука; и на стадии (3) для гомогенизации используют способ гомогенизации высокого давления или микроструйной гомогенизации.
Согласно одному или более вариантам реализации настоящего изобретения стадия (3) может дополнительно включать: промывку трубопровода гомогенизатора с применением соответствующего количества водного раствора трегалозы для замены гомогенного раствора, оставшегося в гомогенизаторе, и затем промывку трубопровода гомогенизатора с применением соответствующего количества сверхчистой воды для замены остаточного водного раствора трегалозы, объединение водного раствора трегалозы с гомогенным раствором, аккуратное перемешивание и затем фильтрование.
Согласно одному или более вариантам реализации настоящего изобретения сдвиг можно осуществлять путем сдвига с помощью смесителя.
Согласно одному или более вариантам реализации настоящего изобретения скорость сдвига при применении смесителя составляет от 5000 об/мин до 10000 об/мин.
Согласно одному или более вариантам реализации настоящего изобретения время сдвига смесителя составляет от 1 минуты до 20 минут, предпочтительно, например, от 2 минут до 10 минут или более предпочтительно от 1 минуты до 10 минут.
Согласно одному или более вариантам реализации настоящего изобретения сдвиг с помощью смесителя предпочтительно осуществляют несколько раз, предпочтительно более 2 раз.
Согласно одному или более вариантам реализации настоящего изобретения в первый раз сдвиг предпочтительно осуществляют со скоростью 5000 об/мин в течение 1 минуты, и во второй раз сдвиг осуществляют со скоростью 10000 об/мин в течение 5 минут.
Согласно одному или более вариантам реализации настоящего изобретения давление гомогенизации высокого давления составляет от 200 до 1600 бар, предпочтительно, например, от 250 до 1550 бар.
Согласно одному или более вариантам реализации настоящего изобретения время гомогенизации высокого давления составляет от 1 до 10 минут, например, предпочтительно от 2 до 10 минут, например, еще более предпочтительно от 5 до 8 минут.
Согласно одному или более вариантам реализации настоящего изобретения гомогенизацию высокого давления предпочтительно проводят несколько раз, предпочтительно, например, более 2 раз.
Согласно одному или более вариантам реализации настоящего изобретения давление при микроструйной гомогенизации составляет 20000 psi (примерно 138 МПа).
Согласно одному или более вариантам реализации настоящего изобретения время микроструйной гомогенизации составляет 30 минут.
В одном или более вариантах реализации настоящего изобретения также предложен способ получения описанной в настоящей заявке фармацевтической композиции, в котором в процессе получения используют органический растворитель.
Согласно одному или более вариантам реализации настоящего изобретения способ получения предложенной в настоящей заявке фармацевтической композиции включает следующие стадии:
(1) растворение элемена и масла для инъекций в липорастворимом органическом растворителе;
(2) взятие альбумина и растворение альбумина в воде;
(3) смешивание двух растворов, полученных на стадии (1) и (2), с получением эмульсии;
(4) гомогенизацию эмульсии на стадии (3);
(5) выпаривание эмульсии на стадии (4) при пониженном давлении для удаления органического растворителя с получением раствора наночастиц альбумина.
Согласно одному или более вариантам реализации настоящего изобретения органический растворитель на стадии (1) выбирают из одного или более растворителей из хлороформа, дихлорметана, третичного бутанола, изопропанола, этилацетата, этанола, тетрагидрофурана, диоксана, ацетонитрила, ацетона, диметилсульфоксида, диметилформамида, метилпирролидона; например, смешанного растворителя, состоящего из дихлорметана и этанола, при этом объемное отношение этанола к дихлорметану в указанном смешанном растворителе составляет 1:1-8, например, 1:4.
Согласно одному или более вариантам реализации настоящего изобретения на стадии (3) для смешивания можно использовать способ с применением сдвига или способ с применением ультразвука; и на стадии (4) для гомогенизации можно использовать способ высокого давления или микроструйный способ.
Согласно одному или более вариантам реализации настоящего изобретения сдвиг можно осуществлять путем сдвига с помощью смесителя.
Согласно одному или более вариантам реализации настоящего изобретения скорость сдвига при применении смесителя составляет от 5000 до 10000 об/мин.
Согласно одному или более вариантам реализации настоящего изобретения время сдвига смесителя составляет от 1 минуты до 20 минут, например, от 2 до 10 минут.
Согласно одному или более вариантам реализации настоящего изобретения сдвиг с помощью смесителя предпочтительно осуществляют несколько раз, предпочтительно более 2 раз.
Согласно одному или более вариантам реализации настоящего изобретения, в первый раз сдвиг предпочтительно осуществляют со скоростью 5000 об/мин в течение 1 минуты, и во второй раз сдвиг осуществляют со скоростью 10000 об/мин в течение 5 минут.
Согласно одному или более вариантам реализации настоящего изобретения давление гомогенизации высокого давления составляет от 200 до 1600 бар, например, от 250 до 1550 бар.
Согласно одному или более вариантам реализации настоящего изобретения время гомогенизации высокого давления составляет от 1 до 10 минуты, например, от 2 до 10 минут, например, еще более предпочтительно от 5 до 8 минут.
Согласно одному или более вариантам реализации настоящего изобретения гомогенизацию высокого давления предпочтительно проводят несколько раз, например, более 2 раз.
Согласно одному или более вариантам реализации настоящего изобретения давление при микроструйной гомогенизации составляет 20000 psi (примерно 138 МПа).
Согласно одному или более вариантам реализации настоящего изобретения время микроструйной гомогенизации составляет 30 минут.
Согласно одному или более вариантам реализации настоящего изобретения способ получения предложенной в настоящей заявке фармацевтической композиции дополнительно включает стадию лиофилизации полученного раствора наночастиц альбумина.
В одном или более вариантах реализации настоящего изобретения дополнительно предложено применение описанной в настоящей заявке фармацевтической композиции в лекарственном препарате для предотвращения или лечения рака.
Согласно одному или более вариантам реализации настоящего изобретения рак, включенный в настоящую заявку, включает адренокортикальную карциному, криптогенную миелоидную метаплазию, рак, связанный со СПИДом, рак анального канала, рак червеобразного отростка, астроцитому, базально-клеточную карциному, холангиокарциному, рак мочевого пузыря, рак кости, глиому, эпендимому, олигодендроглиому, менингиому, краниофарингиому, гемангиобластому, медуллобластому, нейроэктодермальную опухоль, глиому и глиобластому зрительного пути и гипоталамическую глиому и глиобластому, аденому бронха, карциноидную опухоль, лимфому центральной нервной системы, рак шейки матки, рак толстой кишки, колоректальный рак, хроническое миелопролиферативное заболевание, рак эндометрия, эпендимому, семейство опухолей Юинга, рак глаза, рак желчного пузыря, рак желудка, карциноидную опухоль желудочно-кишечного тракта, стромальную опухоль желудочно-кишечного тракта, карциному половых клеток, гестационную трофобластную опухоль, рак головы и шеи, рак печени, рак гортани, лейкоз, рак губ и полости рта, рак легкого, лимфому, медуллобластому, меланому, мезотелиому, метастатический плоскоклеточный рак шеи, синдром множественной эндокринной неоплазии, миелодиспластический синдром, миелодиспластическое/миелопролиферативное заболевание, карциному полости носа и носовых пазух, нейробластому, нейроэндокринный рак, рак ротоглотки, рак носоглотки, опухоль головного мозга, рак костного метастаза, рак кишечника, рак пищевода, рак яичника, рак поджелудочной железы, рак кожи, карциному паращитовидной железы, карциному полового члена, перитонеальный рак, фарингеальную карциному, феохромоцитому, пинеобластому и супратенториальную примитивную нейроэктодермальную опухоль, опухоль гипофиза, плевропульмональную бластому, лимфому, первичную лимфому центральной нервной системы, легочный лимфангиомиоматоз, рак прямой кишки, рак почки, рак почечной лоханки и мочеточника, рабдомиосаркому, рак слюнных желез, рак тонкой кишки, карциному клеток плоского эпителия, рак яичка, рак горла, тимому и рак вилочковой железы, рак щитовидной железы, рак уретры, рак влагалища.
Согласно одному или более вариантам реализации настоящего изобретения рак, включенный в настоящую заявку, включает рак легкого, рак печени, рак пищевода, рак носоглотки, опухоль головного мозга, рак костного метастаза, рак желудка, рак кишечника, рак матки, рак шейки матки, карциному половых клеток, рак эндометрия, гестационную трофобластную опухоль, рак молочной железы, рак кожи, лимфому, лейкоз, злокачественную меланому.
Согласно одному или более вариантам реализации настоящего изобретения опухоль головного мозга включает глиому, глиому ствола головного мозга, мозжечковую или церебральную астроцитому, злокачественную глиому, эпендимому, олигодендроглиому, менингиому, краниофарингиому, гемангиобластому, медуллобластому, глиому и глиобластому зрительного пути и гипоталамическую глиому и глиобластому; мозжечковая или церебральная астроцитома представляет собой фиброзно-клеточную астроцитому, или диффузную астроцитому, или анапластическую (злокачественную) астроцитому.
Согласно одному или более вариантам реализации настоящего изобретения предложенную в настоящей заявке фармацевтическую композицию можно использовать в комбинации с лучевой терапией и химиотерапией для лечения рака легкого, рака печени, рака пищевода, рака носоглотки, опухоли головного мозга, костного метастаза и других злокачественных опухолей для усиления лечебного эффекта, уменьшения токсических и побочных эффектов лучевой терапии и химиотерапии, а также можно использовать для интервенционной, внутриполостной химиотерапии и лечения вызванных раком гидроторакса и асцита.
Согласно одному или более вариантам реализации настоящего изобретения предложенные в настоящей заявке соединения можно использовать в комбинации с другими лекарственными средствами, такими как противоопухолевые средства, при этом указанные противоопухолевые средства включают темозоломид, паклитаксел, доцетаксел, таксан, гемцитабин, лекарственные средства-ингибиторы ФРЭС, лекарственные средства на основе антител против PD-1; при этом лекарственные средства-ингибиторы ФРЭС включают АВАСТИН, ранибизумаб, афлиберцепт или конберцепт; при этом лекарственные средства на основе антител против PD-1 включают ниволумаб, пембролизумаб, торипалимаб, синтилимаб, цемиплимаб, атезолизумаб, авелумаб или дурвалумаб.
В одном или более вариантах реализации настоящего изобретения предложен способ лечения заболевания человека, включающий введение терапевтически эффективного количества описанной в настоящей заявке фармацевтической композиции. Указанную фармацевтическую композицию вводят парентерально, путем ингаляции, внутрибрюшинного, внутрипузырного, внутримышечного, внутривенного, внутритрахеального, подкожного, внутриглазного, интратекального, трансдермального, ректального или вагинального введения.
Согласно одному или более вариантам реализации настоящего изобретения предложенную в настоящей заявке фармацевтическую композицию можно получить в виде фармацевтического состава, подходящего для внутривенного введения, фармацевтического состава для парентерального введения, фармацевтического состава для гастроинтестинального введения, аэрозоля, фармацевтического состава для вагинального введения или другого подходящего фармацевтического состава, представляющего собой твердый состав, жидкий состав или газообразный состав.
Согласно одному или более вариантам реализации настоящего изобретения предложенную в настоящей заявке фармацевтическую композицию можно использовать в виде жидкого состава для инъекций, содержащего элемен и фармацевтически приемлемый носитель, при этом указанный фармацевтически приемлемый носитель включает альбуминовый носитель и масло для инъекций; при этом указанную композицию можно получить в виде частиц, размер которых составляет от 70 до 150 нм; и при этом в предложенной композиции массовое отношение альбуминового носителя к элемену составляет около от 0,5 до 20 и массовое отношение масла для инъекций и элемена составляет около от 0,35 до 10.
Согласно одному или более вариантам реализации настоящего изобретения предложенный в настоящей заявке жидкий состав может представлять собой стабильную водную суспензию, восстановленную из стерильного лиофилизированного порошка.
В одном или более вариантах реализации настоящего изобретения предложен герметичный контейнер, который может представлять собой однодозный контейнер или многодозный контейнер, содержащий описанную в настоящей заявке фармацевтическую композицию. Герметичный контейнер может представлять собой предварительно заполненный шприц. Предложенная фармацевтическая композиция представляет собой жидкую композицию или сухую композицию. Предложенная фармацевтическая композиция является лиофилизированной и стерильной.
Согласно одному или более вариантам реализации настоящего изобретения наносостав или наночастицы, описанные в настоящем документе, могут присутствовать в форме сухого состава (например, лиофилизированной композиции) или могут быть суспендированы в биосовместимой среде. Подходящая биосовместимая среда включает, но не ограничивается ими, воду, водную буферную среду, солевой раствор, буферный солевой раствор, необязательно буферный раствор аминокислоты, необязательно буферный раствор белка, необязательно буферный сахарный раствор, необязательно буферный витаминный раствор, необязательно буферный раствор синтетического полимера, липидосодержащую эмульсию и т.п.
Если прямо не указано иное, в настоящем документе «субъект» представляет собой млекопитающее, в том числе, но не ограничиваясь ими, примата, человека, крупный рогатый скот, представителя семейства лошадиных, представителя семейства кошачьих, представителя семейства псовых или грызуна.
В настоящей заявке «лечение» представляет собой способ получения благоприятных или желательных результатов, в том числе клинических результатов. Для целей настоящего изобретения благоприятные или желательные клинические результаты включают, но не ограничиваются ими, любой один или более из следующих результатов: уменьшение одного или более симптомов, вызванных заболеванием, уменьшение тяжести заболевания, стабилизацию заболевания (например, предотвращение или задержку обострения заболевания), предотвращение или задержку распространения (например, метастаза) заболевания, предотвращение или задержку возникновения или рецидива заболевания, предотвращение или замедление прогрессирования заболевания, улучшение состояния заболевания, обеспечение ремиссии (будь то частичной или полной) заболевания, уменьшение дозы одного или более других лекарственных средств, необходимых для лечения заболевания, задержку прогрессирования заболевания, повышение качества жизни и/или увеличение выживаемости. Согласно некоторым вариантам реализации предложенная композиция снижает тяжесть одного или более симптомов, связанных с раком, по сравнению с соответствующими симптомами у того же субъекта до лечения или по сравнению с соответствующими симптомами у других субъектов, которые не получают указанную композицию, по меньшей мере на около 10, 20, 30, 40, 50, 60, 70, 80, 90, 95 или 100%. «Лечение» также включает облегчение патологических последствий рака. Способы согласно настоящему изобретению предусматривают любой один или более из перечисленных терапевтических аспектов.
В настоящем документе «фармацевтически приемлемый» или «фармакологически совместимый» означает вещество, которое не является биологически или иным образом нежелательным, может быть включено в фармацевтическую композицию, вводимую пациенту, например, без причинения какого-либо значительного нежелательного биологического эффекта или без вредного взаимодействия с любым другим ингредиентом, содержащимся в композиции. Фармацевтически приемлемый носитель или вспомогательное вещество предпочтительно соответствует стандартам, необходимым для прохождения токсикологических и производственных испытаний.
В настоящем документе ссылка на «около» значение или параметр включает (и описывает) варианты реализации, которые относятся к самому значению или параметру. Например, описание, относящееся к «около X», включает описание «X».
Существует большее количество подходящих составов для фармацевтической композиции согласно настоящему изобретению. Приведенные ниже составы и способы являются только иллюстративными, а не ограничивающими.
Составы для перорального введения могут состоять из (а) жидкого раствора, например, эффективного количества активного ингредиента, растворенного в разбавителе, таком как вода, солевой раствор или апельсиновый сок, (b) капсул, саше или таблеток, каждые из которых содержит заданное количество активного ингредиента в твердой или гранулированной форме, (с) суспензии в соответствующей жидкости и (d) подходящей эмульсии. Таблетированные формы могут включать один или более компонентов из лактозы, маннитола, кукурузного крахмала, картофельного крахмала, микрокристаллической целлюлозы, арабской камеди, желатина, коллоидного диоксида кремния, кроскармеллозы натрия, талька, стеарата магния, стеариновой кислоты и других вспомогательных веществ, красителей, разбавителей, буферов, смачивающих агентов, консервантов, ароматизаторов и фармакологически совместимых вспомогательных веществ. Формы в виде таблеток для рассасывания могут содержать активный ингредиент в специях, обычно сахарозе и арабской камеди или трагаканте, при этом пастилки содержат активный ингредиент в инертной матрице, такой как желатин и глицерин, или сахарозе и арабской камеди, эмульсиях, гелях и т.д., в дополнение к активному ингредиенту, при этом они также содержат вспомогательные вещества, известные в данной области техники.
Составы для парентерального введения включают водный и неводный, изотонический стерильный раствор для инъекций, который может содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества для придания составу изотоничности с кровью предполагаемого реципиента, а также водную и неводную стерильную суспензию, которая может включать суспендирующие средства, солюбилизаторы, загустители, стабилизаторы и консерванты. Указанный состав может быть представлен в однодозовых или многодозовых герметичных контейнерах, таких как ампулы и флаконы, и может храниться в условиях вымораживания (лиофилизации) непосредственно перед применением, требуя только добавления стерильного жидкого вспомогательного вещества, такого как вода, для инъекции. Экстемпоральные инъекционные растворы и суспензии можно приготовить из некоторых видов стерильных порошков, гранул и таблеток, как описано выше.
Аэрозоли содержат фармацевтическую композицию согласно настоящему изобретению, при этом указанная фармацевтическая композиция включает водный и неводный, изотонический стерильный раствор, который может содержать антиоксиданты, буферы, бактериостатические агенты и растворенные вещества, а также водную и неводную стерильную суспензию, которая может содержать суспендирующие средства, солюбилизаторы, загустители, стабилизаторы и консерванты; при этом указанную фармацевтическую композицию можно получить в виде аэрозольного состава самого по себе или в комбинации с другими подходящими компонентами для введения путем ингаляции. Указанные аэрозольные составы можно поместить в приемлемые пропелленты под давлением, такие как дифторметан, пропан, азот и т.п. Их также можно приготовить в виде составов лекарственных средств не под давлением, например, в небулайзере или аэрозольном ингаляторе.
Также возможны и другие подходящие составы, например, путем применения различных основ, таких как эмульгированные основы или водорастворимые основы, можно приготовить суппозитории. Составы для вагинального введения могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или составов для распыления, содержащих, в дополнение к активному ингредиенту, подходящие носители, известные в данной области техники.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1 показано распределение диаметра гидратации наночастиц альбумина+β-элемен;
На фигуре 2 приведена диаграмма потенциала наночастиц альбумина+β-элемен;
На фигуре 3 показано полученное с помощью просвечивающего электронного микроскопа изображение наночастиц альбумина+β-элемен;
На фигуре 4 показаны результаты исследования стабильности размера частиц различных лиофилизированных образцов после восстановления;
На фигуре 5 показаны результаты исследования стабильности содержания различных лиофилизированных образцов после хранения;
На фигуре 6 показана выживаемость мышей-опухоленосителей в каждой группе;
На фигуре 7 показаны результаты гемолитического эксперимента;
На фигуре 8 показана кривая роста опухоли ортотопической модели опухоли у мышей U87R1;
На фигуре 9 показана кривая выживаемости ортотопической модели опухоли у мышей U87R1.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Приведенные ниже варианты реализации являются дополнительными иллюстрациями настоящего изобретения и не ограничивают его объем. Настоящее изобретение дополнительно подробно описано ниже со ссылкой на примеры, но специалистам в данной области техники следует понимать, что настоящее изобретение не ограничено указанными примерами и применяемыми способами получения. Кроме того, специалисты в данной области техники могут вносить в настоящее изобретение эквивалентные замены, комбинации, улучшения или модификации в соответствии с описанием настоящего изобретения, при этом все они будут включены в объем настоящего изобретения.
Соевое масло, применяемое в настоящем изобретении, получали из соевого масла (SIO, степень чистоты для инъекций, партия 19C4520/партия 18C4816); человеческий сывороточный альбумин (ЧСА), применяемый в настоящем изобретении, получали из человеческого сывороточного альбумина (20%, Baxter, партия A1U133A) или (20%, Grifols, партия ALAFD03042).
Широко применяемые способы обнаружения согласно настоящему изобретению включали определение размера частиц и распределения частиц по размеру, определение содержания, эксперимент по определению гемолиза и т. д., при этом подробности приведены ниже:
Размер частиц и распределение частиц по размеру
Размер частиц и распределение частиц по размеру определяли с помощью анализатора размера частиц (MALVERN ZETASIZER NANO ZS90). Отбирали 25 мкл образца, добавляли 1 мл сверхчистой воды для его разбавления для детектирования, угол детектирования: 90°; дисперсионная среда: вода; температура: 25°C.
Размер частиц представлял собой Z-среднее значение (средний размер частиц образца), измеренное с помощью анализатора размера частиц, при этом распределение частиц по размеру представляло собой значение PDI (коэффициент полидисперсности), измеренное с помощью анализатора размера частиц.
Определение содержания
Содержание определяли с помощью ВЭЖХ (общее правило 0512 четвертой части издания Китайской фармакопеи 2015 г.).
Хроматографическая колонка представляла собой CHIRALPAK® IG (4,6×150 мм, 5 мкм), в качестве подвижной фазы применяли смесь ацетонитрил-вода (55:45), при скорости потока 1,0 мл/мин. Объем введенной пробы составлял 20 мкл, температура колонки составляла 30°С, и длина волны детектирования составляла 210 нм.
Приготовление раствора эталонного вещества (0,2 мг/мл) (метод внешнего стандарта): около 10 мг β-элемена в качестве эталонного вещества точно взвешивали в мерной колбе объемом 50 мл с небольшим количеством добавленного ацетонитрила, добавляли ацетонитрил для растворения и разбавляли до объемной шкалы, затем смесь хорошо встряхивали с получением раствора эталонного вещества.
Приготовление исследуемого раствора: удаляли колпачок с 1 бутылки, содержащей данный продукт, и точно взвешивали (m1) указанный продукт, добавляли соответствующее количество воды (для обеспечения отношения активного фармацевтического ингредиента (АФИ) к воде 20 мг:1 мл), точно взвешивали (m2) и восстанавливали путем встряхивания. Около 500 мг восстановленного раствора (примерно эквивалентно 10 мг АФИ) (м3) точно взвешивали и помещали в мерную колбу объемом 50 мл, сначала добавляли около 1,5 мл воды, а затем медленно добавляли ацетонитрил до 80% от полного объема, обрабатывали ультразвуком в течение 10 минут, охлаждали и снова добавляли ацетонитрил для разбавления до объемной шкалы, смесь хорошо встряхивали и отфильтровывали через нейлоновый фильтр с размером пор 0,45 мкм с получением исследуемого раствора. После применения восстановленного раствора флакон промывали водой и высушивали, после чего точно взвешивали пустой флакон (m4).
Точно отмеряли 20 мкл, вводили в жидкостный хроматограф и записывали хроматограмму. Содержание β-элемена (C15H24) в исследуемом образце рассчитывали по площади пика в соответствии с методом внешнего стандарта.
Формула расчета была следующей:
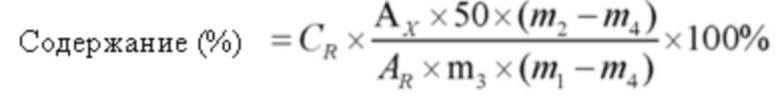
в формуле,
Ax: площадь пика АФИ в исследуемом растворе
AR: площадь пика эталонного вещества
CR: концентрация эталонного вещества
Метод определения величины гемолиза
Получение суспензии клеток крови: у кроликов (самцы новозеландских кроликов, выращивание в подходящих лабораторных условиях в течение 1 недели, масса тела от 2,5 кг до 3 кг, кровь брали из сердца) отбирали несколько миллилитров крови и помещали в стакан, кровь перемешивали стеклянным стержнем или бамбуковой палочкой для удаления фибриногена и получения дефибринированной крови. Добавляли 0,9% раствор натрия хлорида в примерно 10-кратном количестве, аккуратно перемешивали, центрифугировали со скоростью 1500 об/мин в течение 15 минут, отбрасывали супернатант и 2-3 раза промывали осажденные эритроциты 0,9% раствором натрия хлорида в соответствии с описанным выше способом до тех пор, пока супернатант не стал красным. Для последующего применения полученные эритроциты приготавливали в виде 2% суспензии эритроцитов, используя 0,9% раствор хлорида натрия.
Приготовление образца: в зависимости от содержания образца отбирали надлежащее количество и последовательно разбавляли физиологическим солевым раствором для получения образца с содержанием 1 мг/мл (фактическое содержание).
Наблюдение за реакцией: каждое лекарственное средство добавляли описанным ниже способом, при этом после смешивания смесь немедленно помещали на водяную баню при 37°С для инкубации и через 3 часа исследовали гемолиз.
Группа лекарственного средства: в пробирку последовательно добавляли 2,5 мл 2% суспензии клеток крови, 2,2 мл физиологического солевого раствора и 0,3 мл лекарственного средства.
Группа положительного контроля: в пробирку последовательно добавляли 2,5 мл 2% суспензии клеток крови и 2,5 мл дистиллированной воды.
Группа отрицательного контроля: в пробирку последовательно добавляли 2,5 мл 2% суспензии клеток крови и 2,5 мл физиологического солевого раствора.
Определение значения оптической плотности (ОП): 100 мкл супернатанта помещали в 96-луночный планшет с помощью пипетки и измеряли значение ОП при длине волны 540 нм.
Метод расчета величины гемолиза: (значение ОП образца - значение ОП группы отрицательного контроля) / (значение ОП группы положительного контроля - значение ОП группы отрицательного контроля).
Пример испытания 1
Получение наночастиц альбумина+β-элемен включало следующие стадии: 1) Взвешивали 200 мг элемена и 600 мг соевого масла и растворяли в 2 мл дихлорметана с получением масляной фазы. 2) 650 мг человеческого сывороточного альбумина растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3,6% (г/мл), то есть с получением водной фазы. 3) Под действием ультразвука водяной бани к водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. 4) Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1300 бар и гомогенизацию проводили 9 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен, после чего с помощью воды для инъекций доводили отмеряемый объем до 10 мг/мл. 5) К полученным наночастицам альбумина+элемен добавляли 3% сахарозу и лиофилизировали. Параметры лиофилизации были следующими: предварительное замораживание при -45°C в течение 5 часов; запуск вакуумного насоса и поддержание степени вакуумирования на уровне около 10 Па, программирование температуры, сушка при -40°C в течение 4 часов, сушка при -30°C в течение 10 часов и затем сушка при -20°C, -10°C, 0°C, 5°C и 10°C в течение 4 часов, соответственно. Лиофилизированные образцы, полученные, как описано выше, восстанавливали в чистой воде и исследовали размер частиц, потенциал и микроскопическую морфологию наночастиц с применением следующих способов.
Перед измерением анализатор размера частиц Malvern предварительно нагревали в течение 30 минут, отбирали 50 мкл водного раствора наночастиц альбумина+элемен, разбавляли водой до 1 мл, слегка встряхивали (чтобы избежать пузырьков) и помещали в чашку для измерения размера частиц Malvern Nano-ZS90 или потенциально подходящую чашку для измерения размера частиц и дзета-потенциала. Температура измерения составляла 25 °C, время установления равновесия составляло 120 секунд, при этом остальные параметры поддерживали на уровне установочных значений прибора, и полученные данные собирали и обрабатывали с помощью рабочей станции Malvern V2.2. Полученные диаграмма распределения частиц по размеру и дзета-потенциал наночастиц альбумина+элемен приведены на фигуре 1 и на фигуре 2. Как показано на фигурах 1 и 2, размер наночастиц альбумина+элемен составлял около 110 нм, PDI составлял 0,127 и потенциал составлял около -18 мВ.
Водный раствор наночастиц альбумина+элемен аспирировали с помощью пипетки и капали на медную сетку. После естественного испарения 1% раствор фосфорновольфрамовой кислоты капали на медную сетку таким же способом и испаряли естественным путем, аспирировали несколько капель чистой воды для тщательного промывания поверхности медной сетки. После испарения исследовали морфологию с помощью просвечивающего электронного микроскопа, как показано на фигуре 3, наночастицы были сферическими, правильной формы, относительно однородными по размеру, с диаметром около 100 нм.
Пример испытания 2
Наночастицы альбумина+элемен получали в соответствии со способом согласно контрольному эксперименту 1, при этом масло для инъекций применяли в том же количестве, что и масла, выбранные из соевого масла, кукурузного масла, масла камелии и триглицерида средней цепи (MCT), соответственно, с получением наночастиц альбумина+элемен при применении различных масел для инъекций в качестве сырья. Размер полученных наночастиц определяли с помощью лазерного анализатора размера частиц. После лиофилизации определяли содержание лекарственного средства в каждой группе составов до и после лиофилизации, соответственно, и рассчитывали уровень потери лекарственного средства при лиофилизации.
Получение наночастиц альбумина+элемен без соевого масла включало следующие стадии: 1) Взвешивали 200 мг элемена и растворяли в 2 мл дихлорметана с получением масляной фазы. 2) 650 мг человеческого сывороточного альбумина растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3,6% (г/мл), то есть с получением водной фазы. 3) Под действием ультразвука водяной бани к водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. 4) Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1300 бар и гомогенизацию проводили 9 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен, после чего с помощью воды для инъекций доводили отмеряемый объем до 10 мг/мл. 5) К полученным наночастицам альбумина+элемен добавляли 3% сахарозу и лиофилизировали в соответствии с параметрами лиофилизации примера испытания 1. Определяли содержание лекарственного средства в определенном объеме указанного состава до и после лиофилизации, соответственно, и рассчитывали уровень потери лекарственного средства в процессе лиофилизации.
Кроме того, согласно описанным выше способам были приготовлены группы, включающие соевое масло, кукурузное масло, масло камелии и только триглицерид средней цепи, не содержащие лиопротектор в виде 3% сахарозы, и определено содержание лекарственного средства в каждом определенном объеме указанных составов до и после лиофилизации, соответственно, после чего рассчитывали уровень потери лекарственного средства при лиофилизации. Результаты приведены в таблице 1.
Эксперименты показали, что уровни потери при лиофилизации наночастиц альбумина, полученных путем смешивания масла для инъекции с элеменом, уменьшились в разных степенях по сравнению с наночастицами, полученными без масла для инъекции.
Пример испытания 3
В соответствии со способом, описанном в примере испытания 1, количество соевого масла регулировали, наночастицы альбумина+элемен получали путем дозирования в соответствии с массовым отношением β-элемен/соевое масло 5:1, 1:1, 1: 3, 1:5, 1:10, 1:20 и без применения масел для инъекций.
Размер полученных наночастиц определяли с помощью лазерного анализатора размера частиц. После добавления 3% сахарозы и лиофилизации лиофилизированные образцы помещали в сушильную камеру при комнатной температуре. Образцы периодически отбирали и восстанавливали, затем определяли изменения размера частиц и содержания основного лекарственного средства в лиофилизированных образцах в течение 30 дней. Конкретные параметры детектирования были следующими: модель прибора: MALVERN ZETASIZER NANO ZS90, угол детектирования: 90°; режим детектирования: автоматический; время детектирования: 3; тип детектирования: наночастицы; показатель преломления образца, подлежащего определению: 1,340; дисперсионная среда: вода; температура: 25°C; вязкость: 0,8872 сП; показатель преломления дисперсионной среды: 1,330; время установления равновесия: 120 сек; тип чашки для определения размера частиц: DTS0012.
Полученные результаты приведены на фигуре 4 и фигуре 5. Размер частиц состава без соевого масла во время процесса приготовления составлял более 200 нм после лиофилизации. При увеличении времени хранения размер частиц образца после восстановления демонстрировал тенденцию к постепенному увеличению, при этом содержание лекарственного средства демонстрировало тенденцию к снижению. При постепенном увеличении доли соевого масла в исследуемом составе размер частиц всех лиофилизированных образцов после восстановления составлял менее 200 нм, стабильность размера частиц улучшалась, при этом стабильность содержания основного лекарственного средства также постепенно повышалась. Причем, когда массовое отношение соевого масло/элемен составляло от 1 до 10, полученный лиофилизированный образец наночастиц не проявлял явления агрегации в течение 30 дней при комнатной температуре, размер частиц составлял от 100 до 150 нм, PDI составлял менее 0,3, а содержание β-элемена в качестве основного лекарственного средства мало изменялось. Когда количество элемена составляло 1 часть, а количество соевого масла составляло менее 1 части, полученные наночастицы не демонстрировали достаточно хорошей стабильности.
Пример испытания 4
Для исследования терапевтического эффекта наночастиц альбумина+элемен на глиому головного мозга мыши применяли следующий экспериментальный метод.
Моделирование опухоли: хорошо выращенные клетки C6 в логарифмической фазе расщепляли с помощью трипсина, центрифугировали и отбрасывали верхний отработанный раствор. Клетки промывали с помощью фосфатно-солевого буфера (ФСБ), центрифугировали и отбрасывали верхний отработанный раствор, после чего клетки ресуспендировали в ФСБ с обеспечением конечной плотности клеток C6 около 1 × 106 клеток/5 мкл. Здоровых самцов мышей Kunming (от 28 до 30 г) анестезировали путем внутрибрюшинной инъекции соответствующего количества 4% хлоралгидрата. После сбривания волос на макушке применяли 75% этанол для дезинфекции, стерильными офтальмологическими ножницами делали небольшой разрез вдоль сагиттальной средней линии головы, быстро удаляли мембрану ткани с помощью ватного тампона, увлажненного 10% перекисью водорода, отделяли череп и смывали перекись водорода ватным тампоном, увлажненным ФСБ. Голову мыши закрепляли на стереотаксическом устройстве для исследования головного мозга, расположенном в положении 1,8 мм правее и 0,6 мм сзади относительно брегмы, в точке локализации просверливали небольшое отверстие с помощью иглы 2 мл шприца, которое не должно быть слишком глубоким, чтобы избежать кровотечения. 5 мкл вышеупомянутых клеток C6 с плотностью 1 × 106 клеток/5 мкл аспирировали с помощью иглы для микроинъекций. Иглу вводили на 4 мм и протягивали на 1 мм вдоль вертикального направления небольшого отверстия таким образом, чтобы оставить пространство для клеток, и медленно вводили клеточную суспензию в головной мозг. Микроиглу извлекали после пребывания в указанном положении в небольшом отверстии в течение 5 минут. Рану зашивали хирургической нитью и в заключении зашитую поверхность смазывали ватным тампоном, увлажненным двойным антителом.
На 10-е сутки после инокуляции опухоли мышей-опухоленосителей разделяли на группы, по 12 мышей в каждой группе, получавшие физиологический солевой раствор (NS), коммерчески доступные содержащие элемен липосомы (Lip, Dalian Jingang, номер официального утверждения: Guoyaozhunzi H10960114), наночастицы альбумина+элемен (NP), полученные в примере испытания 1. Дозировка при введении составляла 30 мг/кг, один раз в 2 дня, в общей сложности 4 раза. Регистрировали время выживаемости каждой мыши и составляли кривую выживания.
Результаты исследования выживаемости приведены на фигуре 6. Результаты эксперимента показали, что мыши-носители глиомы C6, которым вводили наночастицы альбумина+элемен, демонстрировали более длительную выживаемость. Средняя выживаемость группы, получавшей физиологический солевой раствор, составляла 21 день, средняя выживаемость группы, получавшей липосомы, содержащие элемен, составляла 28 дней, и средняя выживаемость группы, получавшей наночастицы альбумина+элемен, составляла 41 день. Таким образом, наночастицы альбумина+элемен, полученные в таком эксперименте, обладали эффектом увеличения выживаемости мышей-носителей глиомы C6.
Пример испытания 5
Эксперимент по определению гемолиза:
Новозеландских кроликов (самцы, содержащиеся в течение 1 недели в подходящих лабораторных условиях, масса тела от 2,5 кг до 3 кг) разделяли на две группы, получавшие коммерчески доступную инъекцию элемена (Ailineng, CSPC Yuanda Pharmaceutical Co., Ltd., номер партии 17050302) и наночастицы альбумина+элемен (NP), полученные в примере испытания 1, при этом в каждой группе было 2 мыши, дозировка при введении составляла 50 мг/кг. Соответственно, путем применения раствора глюкозы были получены два состава в форме лекарственных растворов с концентрацией 6 мг/мл, а затем кроликам вводили внутривенную инфузию.
В течение от 10 до 15 минут после введения из ушной вены отбирали около 1 мл венозной крови и помещали в центрифужную пробирку, покрытую гепарином натрия, центрифугировали со скоростью 3500 об/мин в течение 15 минут и отбирали супернатант для наблюдения за цветом.
Цвет полученной плазмы показан на фигуре 7. Результат показал, что новозеландские кролики, которым вводили наночастицы альбумина+элемен, не проявляли гемолиз, демонстрируя небольшое раздражение и высокую безопасность.
Пример испытания 6
1.1 Получение экспериментальных образцов
Образец 1
Взвешивали 3,33 г β-элемена и 8 г соевого масла и смешивали с получением масляной фазы. Отмеряли 50 мл (10 г) 20% человеческого сывороточного альбумина, аккуратно разбавляли путем добавления соответствующего количества сверхчистой воды и доводили отмеряемый объем до 333 мл с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3% (г/мл), а именно водной фазы. Водную фазу выливали в масляную фазу и применяли смеситель (FLUKO, FA30/30F) для обеспечения сдвига при скорости 10000 об/мин в течение 10 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в микроструйный гомогенизатор (Microfluidizer, M-110-EH) для гомогенизации при давлении гомогенизации 20000 psi (примерно 138 МПа) в течение 30 минут. Полученную эмульсию отфильтровывали с помощью пластинчатых фильтров из поливинилиденфторида (ПВДФ) с размером пор 0,45 мкм и 0,22 мкм и регулировали отмеряемый объем, после чего добавляли 3% (г/мл) сахарозу в качестве лиопротектора. Гомогенный раствор с добавлением лиопротектора лиофилизировали и после лиофилизации восстанавливали лиофилизированный порошок с применением стерилизованной сверхчистой воды. Размер частиц составлял 175,97 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,188.
Образец 2
Взвешивали 3,33 г β-элемена и 8 г соевого масла, растворяли в органическом растворителе и перемешивали с получением масляной фазы. Органический растворитель представлял собой смешанный растворитель, состоящий из 26,67 мл дихлорметана и 6,67 мл безводного этанола. Отмеряли 50 мл (10 г) 20% человеческого сывороточного альбумина, аккуратно разбавляли путем добавления соответствующего количества сверхчистой воды и доводили отмеряемый объем до 333 мл с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3% (г/мл), а именно водной фазы. Раствор водной фазы выливали в масляную фазу и использовали смеситель (FLUKO, FA30/30F) для обеспечения сдвига при скорости 10000 об/мин в течение 10 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в микроструйный гомогенизатор (Microfluidizer, M-110-EH) для гомогенизации высокого давления при давлении гомогенизации 20000 psi (примерно 138 МПа) в течение 30 минут. После гомогенизации органический растворитель удаляли из эмульсии, содержащей органический растворитель, с помощью тонкопленочного испарителя (Shanghai DEA), при этом параметры были следующими: температура 30°С, степень вакуумирования 4000 Па, скорость подачи жидкости 100 об/мин, скорость скребка 50 об/мин. Полученную эмульсию отфильтровывали с помощью пластинчатых фильтров из ПВДФ с размером пор 0,45 мкм и 0,22 мкм и регулировали отмеряемый объем, после чего добавляли 3% (г/мл) сахарозу в качестве лиопротектора. Гомогенный раствор с добавлением лиопротектора лиофилизировали и после лиофилизации восстанавливали лиофилизированный порошок с применением стерилизованной сверхчистой воды. Размер частиц составлял 78,34 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,207.
Образец 3 (инъекция элемена (торговое название: Ailineng)): CSPC Yuanda (Dalian) Pharmaceutical Co., Ltd., номер партии: 19061502; размер частиц составлял 14,74 нм, как было установлено с помощью анализатора размера частиц, PDI: 0,035.
1.2 Фармакокинетика и распределение наночастиц альбумина+элемен в тканях головного мозга мыши
Для эксперимента применяли мышей Kunming (самцы, категория SPF (свободные от патогенной микрофлоры), масса тела от 20 до 25 г). Дозировка при введении составляла 50 мг/кг, введение осуществляли через хвостовую вену. Образцы тканей головного мозга собирали в разные периоды времени и хранили при температуре -20°C для определения содержания лекарственного средства в ткани головного мозга. Для исследования трех образцов в общей сложности использовали 45 мышей, 3 мыши Kunming для каждой лекарственной формы в каждой временной точке. Глазные яблоки удаляли для сбора крови через 5 минут, 15 минут, 30 минут, 1 час и 2 часа после введения и собирали ткани головного мозга для определения содержания лекарственного средства в тканях головного мозга. Для количественного анализа β-элеменов в биологических образцах плазмы и ткани головного мозга мышей Kunming применяли ГХ-МС. Результаты исследования периода полувыведения в ткани головного мозга и воздействия на ткани головного мозга приведены в таблице 2.
Пример испытания 7
Получение и исследование стабильности образца 4
Взвешивали 1 г β-элемена и 3 г соевого масла и перемешивали с получением масляной фазы. Отмеряли 15 мл (3 г) 20% человеческого сывороточного альбумина, аккуратно разбавляли путем добавления 85 мл сверхчистой воды с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3% (г/мл), а именно водной фазы. Раствор водной фазы выливали в масляную фазу и применяли смеситель для обеспечения сдвига при скорости 5000 об/мин в течение 1 минуты и при скорости 10000 об/мин в течение 5 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1350 до 1400 бар в течение 9 минут. После гомогенизации общий объем гомогенного раствора измеряли с помощью мерного цилиндра, который составлял около 50 мл, добавляли 5 г трегалозы, хорошо встряхивали для растворения и отфильтровывали с помощью микропористой мембраны с размером пор 0,45 мкм с получением раствора наночастиц альбумина+элемен. Размер частиц составлял 141,0 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,162. Гомогенный раствор с добавлением трегалозы лиофилизировали и после лиофилизации восстанавливали лиофилизированный порошок с применением стерилизованной сверхчистой воды. Размер частиц составлял 144,07 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,123. Уровень потери содержания элемена до и после лиофилизации составлял 1,27%.
Получение и исследование стабильности образца 5
Взвешивали 4 г β-элемена и 2 г соевого масла и смешивали с получением масляной фазы. Отмеряли 30 мл (6 г) 20% человеческого сывороточного альбумина, аккуратно разбавляли путем добавления 170 мл сверхчистой воды с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3% (г/мл), а именно водной фазы. Раствор водной фазы выливали в масляную фазу и применяли смеситель для обеспечения сдвига при скорости 5000 об/мин в течение 1 минуты и при скорости 10000 об/мин в течение 10 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1350 до 1400 бар в течение 24 минут. После гомогенизации с помощью мерного цилиндра отмеряли 150 мл гомогенного раствора, добавляли 15 г трегалозы, хорошо встряхивали для растворения и отфильтровывали с помощью микропористой мембраны с размером пор 0,45 мкм с получением раствора наночастиц альбумина+элемен. Размер частиц составлял 132,4 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,133. Гомогенный раствор с добавлением трегалозы лиофилизировали и после лиофилизации восстанавливали лиофилизированный порошок с применением стерилизованной сверхчистой воды. Размер частиц составлял 123,68 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,144. Уровень потери содержания элемена после лиофилизации составлял 1,58%.
Получение и исследование стабильности образца 6
Взвешивали 1 г β-элемена и 2 г лецитина из яичного желтка, добавляли 50 мл сверхчистой воды и применяли смеситель для обеспечения сдвига при скорости 5000 об/мин в течение 1 минуты и обеспечения сдвига при скорости 10000 об/мин в течение 5 минут. Затем лецитин, прилипший к стенке бутылки, соскабливали в раствор и снова перемешивали со сдвигом со скоростью 15000 об/мин в течение 3 минут до обнаружения явного недиспергированного лецитина и получения первичной эмульсии. Первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1300 до 1350 бар в течение 12 минут. После гомогенизации гомогенный раствор отфильтровывали с помощью фильтрующей мембраны с размером пор 0,45 мкм и к отфильтрованному гомогенному раствору добавляли 50 мл 2% раствора ЧСА (содержащего 1 г ЧСА), после чего гомогенизировали при 280 бар в течение 1 цикла; затем выгружали гомогенный раствор, отмеряли с помощью мерного цилиндра 40 мл, добавляли 4 г трегалозы, хорошо встряхивали для растворения и отфильтровывали с помощью микропористой мембраны с размером пор 0,45 мкм с получением раствора наночастиц альбумина. Размер частиц составлял 150,86 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,114. Гомогенный раствор с добавлением трегалозы лиофилизировали и после лиофилизации восстанавливали лиофилизированный порошок с применением стерилизованной сверхчистой воды. Размер частиц составлял 181,56 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,251. Уровень потери содержания элемена после лиофилизации составлял 16,47%.
Получение и исследование стабильности образца 7
Взвешивали 1 г β-элемена, 2 г лецитина из яичного желтка и 0,5 г TPGS (токоферилполиэтиленгликольсукцинат), добавляли 50 мл сверхчистой воды и применяли смеситель для обеспечения сдвига при скорости 5000 об/мин в течение 1 минуты и для обеспечения сдвига при скорости 10000 об/мин в течение 5 минут. Затем лецитина, прилипший к стенке бутылки, соскабливали в раствор и снова перемешивали со сдвигом со скоростью 15000 об/мин в течение 3 минут до обнаружения явного недиспергированного лецитина и получения первичной эмульсии. Первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1300 до 1350 бар в течение 12 минут. После гомогенизации раствор отфильтровывали с помощью фильтрующей мембраны с размером пор 0,22 мкм и к отфильтрованному гомогенному раствору добавляли 50 мл 2% раствора ЧСА (содержащего 1 г ЧСА), гомогенизировали при 140 бар в течение 1 цикла; затем выгружали гомогенный раствор, отмеряли с помощью мерного цилиндра 40 мл, добавляли 4 г трегалозы, хорошо встряхивали для растворения и отфильтровывали с помощью микропористой мембраны с размером пор 0,45 мкм с получением раствора наночастиц альбумина. Размер частиц составлял 93,11 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,131. Гомогенный раствор с добавлением трегалозы лиофилизировали и после лиофилизации восстанавливали лиофилизированный порошок с применением стерилизованной сверхчистой воды. Размер частиц составлял 811,70 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,313. Уровень потери содержания элемена после лиофилизации составлял 82,64%.
Пример 1
Взвешивали 200 мг β-элемена и 600 мг соевого масла, растворяли в 2 мл органического растворителя и перемешивали с помощью вихревой мешалки с получением масляной фазы. Органический растворитель представлял собой дихлорметан или этилацетат или смесь дихлорметана и этанола. Отбирали 650 мг человеческого сывороточного альбумина и растворяли в 18 мл воды для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3,6% (г/мл), то есть с получением водной фазы. Под действием ультразвука водяной бани к водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 16 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен.
Экспериментальные результаты показали, что независимо от того, растворялись ли β-элемен и соевое масло в органических растворителях или нет в процессе приготовления, были получены составы альбумина+β-элемен с подходящим размером частиц. Среди различных органических растворителей наночастицы, полученные с применением дихлорметана или смешанного растворителя, состоящего из дихлорметана и этанола, имели меньший размер частиц.
Пример 2
Взвешивали 200 мг β-элемена и 600 мг соевого масла, растворяли в смешанном растворителе, состоящем из дихлорметана и этанола (дихлорметан:этанол=4:1, 2 мл), и перемешивали с помощью вихревой мешалки с получением масляной фазы объемом 2 мл. Отбирали 650 мг человеческого сывороточного альбумина и растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3,6% (г/мл), и затем добавляли 100 мг полиэтиленгликоль-12-гидроксистеарата с получением водной фазы. Под действием ультразвука водяной бани к водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 16 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Измеренный размер частиц составлял 116,4 нм, PDI составлял 0,118.
Пример 3
Взвешивали 200 мг β-элемена и 400 мг глицерида средне- и длинноцепочечной жирной кислоты, растворяли в смешанном растворителе, состоящем из дихлорметана и этанола (дихлорметан:этанол=4:1, 2 мл), и перемешивали с помощью вихревой мешалки с получением масляной фазы объемом 2 мл. Отбирали 900 мг человеческого сывороточного альбумина и растворяли в 18 мл воды для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом (г/мл) 5,0%, то есть с получением водной фазы. Под действием ультразвука водяной бани к водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 16 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Измеренный размер частиц составлял 105,3 нм, PDI составлял 0,116.
Пример 4
Взвешивали 800 мг β-элемена и 2400 мг кунжутного масла, растворяли в смешанном растворителе, состоящем из дихлорметана и этанола (дихлорметан:этанол=4:1, 10 мл), и перемешивали с помощью вихревой мешалки с получением масляной фазы. Отбирали 4000 мг человеческого сывороточного альбумина и растворяли в 100 мл воды для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом (г/мл) 4%, то есть с получением водной фазы. К водной фазе по каплям добавляли масляную фазу, и для получения первичной эмульсии применяли устройство для измельчения тканей при скорости 10000 об/мин для обеспечения сдвига в течение 10 минут. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 20 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Измеренный размер частиц составлял 121,3 нм, PDI составлял 0,133.
Пример 5
Взвешивали 800 мг β-элемена и 2400 мг оливкового масла, последовательно добавляли к смешанному растворителю дихлорметан/этанол (дихлорметан:этанол=4:1, 5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы. Отбирали 4000 мг человеческого сывороточного альбумина и растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 4% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу, и для приготовления первичной эмульсии применяли устройство для измельчения тканей при скорости 12000 об/мин для обеспечения сдвига в течение 10 минут. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1400 бар, и проводили процедуру гомогенизации 20 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Добавляли 3% сахарозу и лиофилизировали, после лиофилизации закрывали колпачком и хранили при 4°C. Отбирали лиофилизированные образцы и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 112,6 нм, PDI составлял 0,101.
Пример 6
Взвешивали 200 мг β-элемена и 600 мг соевого масла, растворяли в смешанном растворителе, состоящем из дихлорметана и этанола (дихлорметан:этанол=4:1, 10 мл), и перемешивали с помощью вихревой мешалки с получением масляной фазы. 4000 мг человеческого сывороточного альбумина растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом (г/мл) 10%, то есть с получением водной фазы. Под действием ультразвука водяной бани к водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 16 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Измеренный размер частиц составлял 140,3 нм, PDI составлял 0,164.
Пример 7
Взвешивали 200 мг β-элемена и 1000 мг соевого масла, растворяли в смешанном растворителе, состоящем из дихлорметана и этанола (дихлорметан:этанол=4:1, 5 мл), и перемешивали с помощью вихревой мешалки с получением масляной фазы. Отбирали 650 мг человеческого сывороточного альбумина и растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом (г/мл) 3,6%, то есть с получением водной фазы. Под действием ультразвука водяной бани к водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 16 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Измеренный размер частиц составлял 125,3 нм, PDI составлял 0,148.
Пример 8
Взвешивали 800 мг β-элемена и 2400 мг соевого масла, растворяли в смешанном растворителе, состоящем из дихлорметана и этанола (дихлорметан:этанол=4:1, 10 мл), и перемешивали с помощью вихревой мешалки с получением масляной фазы. 4000 мг человеческого сывороточного альбумина растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом (г/мл) 4%, то есть с получением водной фазы. К водной фазе по каплям добавляли масляную фазу, и для получения первичной эмульсии применяли устройство для измельчения тканей со скоростью 10000 об/мин для обеспечения сдвига в течение 10 минут. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 20 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Измеренный размер частиц составлял 119,3 нм, PDI составлял 0,143. Отбирали полученный раствор наночастиц и доводили до концентрации лекарственного средства 10 мг/мл, добавляли соответствующее количество лиопротектора, при этом количество лиопротектора составляло 3% (г/мл), и лиофилизировали. После лиофилизации для восстановления добавляли воду для инъекций и определяли размер частиц. Результаты приведены в следующей таблице.
Экспериментальные результаты показали, что в процессе лиофилизации наночастиц альбумина+элемен добавляли различные лиопротекторы для получения лиофилизированных порошков с рыхлой структурой и однородной текстурой, которые можно было восстановить путем добавления воды, при этом размер частиц после восстановления не превышал 200 нм. При этом наночастицы, полученные путем восстановления образца посредством лиофилизации с применением сахарозы в качестве лиопротектора, имели наименьший размер частиц.
Пример 9
Взвешивали 500 мг β-элемена и 1200 мг глицерида средне- и длинноцепочечной жирной кислоты, растворяли в смешанном растворителе, состоящем из дихлорметана и этанола (дихлорметан:этанол=4:1, 5 мл), и перемешивали с помощью вихревой мешалки с получением масляной фазы. Отбирали 1500 мг человеческого сывороточного альбумина и растворяли в 50 мл воды для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом (г/мл) 3% с получением водной фазы. К водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1400 бар, и проводили процедуру гомогенизации 10 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм, фильтрат разбавляли и доводили до концентрации лекарственного средства 15 мг/мл, добавляли 3% сахарозу, лиофилизировали, закрывали колпачком и хранили при 4°C. Отбирали лиофилизированный образец и восстанавливали в воде для инъекций, затем определяли размер частиц. Измеренный размер частиц составлял 103,5 нм, PDI составлял 0,123.
Пример 10
Взвешивали 800 мг β-элемена и 2400 мг кунжутного масла, последовательно добавляли к смешанному растворителю дихлорметан/изопропанол (дихлорметан:изопропанол=4:1, 5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы. 4000 мг человеческого сывороточного альбумина растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 4% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу, и для приготовления первичной эмульсии применяли устройство для измельчения тканей при скорости 10000 об/мин для обеспечения сдвига в течение 10 минут. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 20 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Водный раствор разбавляли и доводили до концентрации лекарственного средства 10 мг/мл, затем добавляли 3% сахарозу для лиофилизации, после лиофилизации закрывали колпачком и хранили при 4 °C. Отбирали лиофилизированный образец и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 115,2 нм, PDI составлял 0,102.
Пример 11
Взвешивали 800 мг β-элемена и 2400 мг оливкового масла, последовательно добавляли к смешанному растворителю дихлорметан/третичный бутанол (дихлорметан:третичный бутанол=4:1, 5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы. 4000 мг человеческого сывороточного альбумина растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 4% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу, и для приготовления первичной эмульсии применяли устройство для измельчения тканей при скорости 10000 об/мин для обеспечения сдвига в течение 10 минут. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 20 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Водный раствор разбавляли и доводили до концентрации лекарственного средства 10 мг/мл, затем добавляли 3% сахарозу для лиофилизации, после лиофилизации закрывали колпачком и хранили при 4°C. Отбирали лиофилизированный образец и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 109,6 нм, PDI составлял 0,113.
Пример 12
Взвешивали 800 мг β-элемена и 2400 мг кунжутного масла, последовательно добавляли к смешанному растворителю дихлорметан/тетрагидрофуран (дихлорметан:тетрагидрофуран=4:1, 5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы объемом 5 мл. Отбирали 4000 мг человеческого сывороточного альбумина и растворяли в воде для инъекций с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 4% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу, и для приготовления первичной эмульсии применяли устройство для измельчения тканей при скорости 10000 об/мин для обеспечения сдвига в течение 10 минут. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 20 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Для лиофилизации добавляли 3% трегалозу, после лиофилизации закрывали колпачком и хранили при 4°C. Отбирали лиофилизированный образец и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 112,6 нм, PDI составлял 0,101.
Пример 13
Взвешивали 500 мг β-элемена и 1200 мг соевого масла и последовательно добавляли к изопропанолу (5 мл), после чего перемешивали с помощью вихревой мешалки с получением масляной фазы. Отбирали 1500 мг бычьего сывороточного альбумина и растворяли в воде для инъекций с получением водного раствора бычьего сывороточного альбумина с массовым объемным процентом 3% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1400 бар, и проводили процедуру гомогенизации 10 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм. Фильтрат разбавляли и доводили до концентрации лекарственного средства 10 мг/мл, затем добавляли 3% сахарозу для лиофилизации, после лиофилизации закрывали колпачком и хранили при 4°C. Лиофилизированные образцы восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 128,1 нм, PDI составлял 0,132.
Пример 14
Взвешивали 500 мг β-элемена и 1200 мг соевого масла, последовательно добавляли к третичному бутанолу (5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы. Отбирали 1500 мг бычьего сывороточного альбумина и растворяли в воде для инъекций с получением водного раствора бычьего сывороточного альбумина с массовым объемным процентом 3% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1400 бар, и проводили процедуру гомогенизации 10 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм. Фильтрат разбавляли и доводили до концентрации лекарственного средства 5 мг/мл, затем добавляли 3% сахарозу для лиофилизации, после лиофилизации закрывали колпачком и хранили при 4°C. Отбирали лиофилизированный образец и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 116,8 нм, PDI составлял 0,113.
Пример 15
Взвешивали 500 мг β-элемена и 1200 мг кунжутного масла, последовательно добавляли к смешанному растворителю дихлорметан/изопропанол (дихлорметан:изопропанол=4:1, 5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы. 1500 мг бычьего сывороточного альбумина растворяли в воде для инъекций с получением водного раствора бычьего сывороточного альбумина с массовым объемным процентом 3% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1400 бар, и проводили процедуру гомогенизации 10 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм. Фильтрат разбавляли и доводили до концентрации лекарственного средства 1 мг/мл, затем добавляли 3% сахарозу для лиофилизации, после лиофилизации закрывали колпачком и хранили при 4°C. Лиофилизированный образец отбирали и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 121,8 нм, PDI составлял 0,111.
Пример 16
Взвешивали 500 мг β-элемена и 1200 мг соевого масла, последовательно добавляли к смешанному растворителю дихлорметан/третичный бутанол (дихлорметан:третичный бутанол=4:1, 5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы. 1500 мг бычьего сывороточного альбумина растворяли в воде для инъекций с получением водного раствора бычьего сывороточного альбумина с массовым объемным процентом 3% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1400 бар, и проводили процедуру гомогенизации 10 раз. После гомогенизации органический растворитель удаляли путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм. Фильтрат разбавляли и доводили до концентрации лекарственного средства 10 мг/мл, затем добавляли 3% сахарозу для лиофилизации, после лиофилизации закрывали колпачком и хранили при 4°C. Отбирали лиофилизированный образец и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 121,1 нм, PDI составлял 0,109.
Пример 17
Взвешивали 500 мг β-элемена и 1200 мг оливкового масла, последовательно добавляли к смешанному растворителю дихлорметан/этанол (дихлорметан:этанол=4:1, 5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы. 1500 мг бычьего сывороточного альбумина растворяли в воде для инъекций с получением водного раствора бычьего сывороточного альбумина с массовым объемным процентом 3% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1400 бар, и проводили процедуру гомогенизации 10 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм. Фильтрат разбавляли и доводили до концентрации лекарственного средства 12 мг/мл, затем добавляли 3% сахарозу для лиофилизации, после лиофилизации закрывали колпачком и хранили при 4°C. Отбирали лиофилизированный образец и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 109,7 нм, PDI составлял 0,105.
Пример 18
Взвешивали 500 мг β-элемена и 1200 мг соевого масла, последовательно добавляли к смешанному растворителю дихлорметан/тетрагидрофуран (дихлорметан:тетрагидрофуран=4:1, 5 мл) и перемешивали с помощью вихревой мешалки с получением масляной фазы. 1500 мг бычьего сывороточного альбумина растворяли в воде для инъекций с получением водного раствора бычьего сывороточного альбумина с массовым объемным процентом 3% (г/мл) с получением водной фазы. К водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1400 бар, и проводили процедуру гомогенизации 10 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания. Полученный путем ротационного выпаривания лекарственный раствор отфильтровывали и стерилизовали с помощью микропористой мембраны с размером пор 0,22 мкм. Фильтрат разбавляли и доводили до концентрации лекарственного средства 10 мг/мл, затем добавляли 3% сахарозу для лиофилизации, после лиофилизации закрывали колпачком и хранили при 4°C. Отбирали лиофилизированный образец и восстанавливали в воде для инъекций, при этом измеренный размер частиц составлял 112,7 нм, PDI составлял 0,112.
Пример 19
Взвешивали 200 мг элемена и 600 мг соевого масла, растворяли в смешанном растворителе, состоящем из дихлорметана и этанола (дихлорметан:этанол=4:1, 2 мл), и перемешивали с помощью вихревой мешалки с получением масляной фазы. 650 мг человеческого сывороточного альбумина растворяли в 18 мл 0,5% раствора натрия хлорида с получением раствора человеческого сывороточного альбумина с массовым объемным процентом (г/мл) 3,6%. Под действием ультразвука водяной бани к водной фазе по каплям добавляли масляную фазу и обрабатывали ультразвуком с помощью зонда в течение 8 минут (250 Вт) с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления для гомогенизации высокого давления, при этом давление гомогенизации составляло 1000 бар, и проводили процедуру гомогенизации 16 раз. Органический растворитель удаляли из гомогенизированной системы путем ротационного выпаривания и для фильтрации и стерилизации применяли микропористую мембрану с размером пор 0,22 мкм с получением водного раствора наночастиц альбумина+элемен. Измеренный размер частиц составлял 126,1 нм, PDI составлял 0,122.
Пример 20
Взвешивали 500 мг β-элемена и 1500 мг соевого масла, растворяли в 5 мл смешанного растворителя дихлорметан/этанол (дихлорметан:этанол=8:1) с получением масляной фазы; и 2500 мг человеческого сывороточного альбумина взвешивали и растворяли в 45 мл воды с получением водной фазы. Под действием ультразвука аккуратно перемешивали две фазы и переносили полученный раствор в гомогенизатор высокого давления для гомогенизации высокого давления при давлении от 400 до 500 бар 4-6 раз и при давлении от 1300 до 1400 бар 8-10 раз. Далее, после гомогенизации удаляли органический растворитель путем ротационного выпаривания, применяли микропористую мембрану для фильтрации и стерилизации, удаляли пузырьки в составе путем кратковременного отстаивания и получали раствор наночастиц альбумина+элемен. Измеренный размер наночастиц составлял 99,4 нм, PDI составлял 0,096. Полученный раствор наночастиц альбумина+элемен доводили до концентрации 10 мг/мл, добавляли 3% сахарозу, лиофилизировали и восстанавливали путем добавления сверхчистой воды, согласно измерениям размер частиц составлял 101,6 нм, и PDI составлял 0,113.
Пример 21
Соответствующие β-элемен, соевое масло, MCT (триглицерид средней цепи) и поверхностно-активные вещества (HS-15, а именно макрогол 15-гидроксистеарат; Твин 80; полоксамер P188) взвешивали в соответствии с количествами компонентов, перечисленными в таблице 5, и смешивали с получением масляной фазы. Отбирали 20% человеческий сывороточный альбумин (пересчитано по массе, как показано в таблице 5), разбавляли путем добавления сверхчистой воды до общего объема 50 мл и аккуратно перемешивали с получением водного раствора человеческого сывороточного альбумина, а именно водной фазы. К раствору водной фазы добавляли соответствующее количество поверхностно-активного вещества и выливали раствор водной фазы в масляную фазу, после чего перемешивали со сдвигом с помощью смесителя (IKA, T25) со скоростью 5000 об/мин в течение 1 минуты и со скоростью 10000 об/мин в течение 5 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления (ATS, AH-1500) для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1450 до 1550 бар в течение 6 минут. После гомогенизации общий объем гомогенного раствора измеряли с помощью мерного цилиндра, добавляли трегалозу 10% (г/мл), хорошо встряхивали для растворения и отфильтровывали с помощью микропористой мембраны с размером пор 0,45 мкм с получением раствора наночастиц альбумина+элемен. Устанавливали размер частиц и PDI и определяли величину гемолиза. Конкретные результаты приведены в таблице 5.
/г
/г
/г
Пример 22
Взвешивали β-элемен и соевое масло в соответствии с количествами компонентов, перечисленными в таблице 6, и аккуратно перемешивали с получением масляной фазы. Человеческий сывороточный альбумин (пересчитано по массе, как показано в таблице 6) измеряли, разбавляли до общего объема 50 мл путем добавления сверхчистой воды и аккуратно перемешивали с получением водного раствора человеческого сывороточного альбумина, а именно водной фазы. Водную фазу выливали в масляную фазу и перемешивали со сдвигом с помощью смесителя (IKA, T25) со скоростью 5000 об/мин в течение 1 минуты и со скоростью 10000 об/мин в течение 5 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления (ATS, AH-1500) для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1450 до 1550 бар в течение 6 минут. После гомогенизации общий объем гомогенного раствора измеряли с помощью мерного цилиндра, добавляли трегалозу в соотношении масса/объем 10% (г/мл), хорошо встряхивали для растворения и отфильтровывали с помощью микропористой мембраны с размером пор 0,45 мкм с получением раствора наночастиц альбумина+элемен. Устанавливали размер частиц и PDI и определяли величину гемолиза. Конкретные результаты приведены в таблице 6.
/г
/г
/г
Пример 23
Взвешивали 3,33 г β-элемена и 8 г соевого масла и смешивали с получением масляной фазы. Отмеряли 50 мл (10 г) 20% человеческого сывороточного альбумина (ЧСА), аккуратно разбавляли путем добавления соответствующего количества сверхчистой воды с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом (г/мл) 3%, а именно водной фазы. Раствор водной фазы выливали в масляную фазу и использовали смеситель (FLUKO, FA30/30F) для обеспечения сдвига при скорости 10000 об/мин в течение 10 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в микроструйный гомогенизатор (Microfluidizer, M-110-EH) для гомогенизации при давлении гомогенизации 20000 psi (примерно 138 МПа) в течение 30 минут. Гомогенизированный раствор отфильтровывали с помощью пластинчатых фильтров из ПВДФ с размером пор 0,45 мкм и 0,22 мкм, регулировали отмеряемый объем и добавляли различные лиопротекторы (г/мл) в соответствии с таблицей 7, соответственно. Гомогенный раствор с добавлением лиопротектора лиофилизировали и после лиофилизации восстанавливали лиофилизированный порошок с применением стерилизованной сверхчистой воды. Устанавливали размер частиц, PDI и величину гемолиза. Конкретные результаты приведены в таблице 7.
8 г соевого масла
10 г HSA
Пример 24
Взвешивали 3,33 г β-элемена и 8 г соевого масла, растворяли в органическом растворителе и перемешивали с получением масляной фазы. Органический растворитель представлял собой смешанный растворитель, состоящий из 26,67 мл дихлорметана и 6,67 мл безводного этанола. Отмеряли 50 мл (10 г) 20% человеческого сывороточного альбумина (ЧСА), аккуратно разбавляли путем добавления соответствующего количества сверхчистой воды с получением водного раствора человеческого сывороточного альбумина с массовым объемным процентом 3% (г/мл), а именно водной фазы. Раствор водной фазы выливали в масляную фазу и использовали смеситель (FLUKO, FA30/30F) для обеспечения сдвига при скорости 10000 об/мин в течение 10 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в микроструйный гомогенизатор (Microfluidizer, M-110-EH) для гомогенизации высокого давления при давлении гомогенизации 20000 psi (примерно 138 МПа) в течение 30 минут. После гомогенизации органический растворитель удаляли из эмульсии, содержащей органический растворитель, с помощью тонкопленочного испарителя, при этом параметры были следующими: температура 30 °С, степень вакуумирования 4000 Па, скорость подачи жидкости 100 об/мин, скорость скребка 50 об/мин. Полученную эмульсию отфильтровывали с помощью пластинчатых фильтров из ПВДФ с размером пор 0,45 мкм и 0,22 мкм и регулировали отмеряемый объем, после чего добавляли 10% (г/мл) трегалозы в качестве лиопротектора. Гомогенный раствор с добавлением лиопротектора лиофилизировали и после лиофилизации восстанавливали лиофилизированный порошок с применением стерилизованной сверхчистой воды. Установленный размер частиц составлял 76,28, PDI составлял 0,254 и величина гемолиза составляла 16,3.
Пример 25
Взвешивали β-элемен и соевое масло в соответствии с количествами компонентов, перечисленными в таблице 8, и растворяли в органическом растворителе, который представлял собой смешанный растворитель, состоящий из 1 мл этанола и 4 мл дихлорметана, и перемешивали с получением масляной фазы. Отмеряли соответствующее количество 20% человеческого сывороточного альбумина (ЧСА) (пересчитано по массе, как показано в таблице 8), разбавляли до общего объема 50 мл путем добавления сверхчистой воды, а именно водной фазы. Раствор водной фазы выливали в масляную фазу и перемешивали со сдвигом с помощью смесителя (IKA, T25) со скоростью 5000 об/мин в течение 1 минуты и со скоростью 10000 об/мин в течение 5 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления (ATS, AH-1500) для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1450 до 1550 бар в течение 6 минут. Органический растворитель удаляли с помощью роторного испарителя при 35 °C, 150 об/мин и вакууме 30 мбар. После завершения измеряли общий объем гомогенного раствора с помощью мерного цилиндра, добавляли трегалозу в соотношении масса/объем 10% (г/мл), хорошо встряхивали для растворения и отфильтровывали с помощью микропористой мембраны с размером пор 0,45 мкм с получением раствора наночастиц альбумина+элемен.
/г
/г
/г
Полученные образцы помещали в сушильный шкаф при 30°С и отбирали через 0 часов, 24 часа и 48 часов для определения размера частиц, PDI и содержания β-элемена. Результаты измерений приведены в таблице 9.
Пример 26
Взвешивали β-элемен и соевое масло в соответствии с количествами компонентов, перечисленными в таблице 10, с получением масляной фазы. Отмеряли соответствующее количество 20% человеческого сывороточного альбумина (ЧСА) (пересчитано по массе, как показано в таблице 10), разбавляли до общего объема 50 мл путем добавления сверхчистой воды, а именно водной фазы. Раствор водной фазы выливали в масляную фазу и перемешивали со сдвигом с помощью смесителя (IKA, T25) со скоростью 5000 об/мин в течение 1 минуты и со скоростью 10000 об/мин в течение 5 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления (ATS, AH-1500) для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1350 до 1450 бар в течение 6 минут. После завершения общий объем гомогенного раствора измеряли с помощью мерного цилиндра, добавляли трегалозу в соотношении масса/объем (г/мл), показанном в таблице 10, хорошо встряхивали для растворения и отфильтровывали с помощью микропористой мембраны с размером пор 0,45 мкм с получением раствора наночастиц альбумина+элемен.
/г
/г
Полученные образцы помещали в сушильный шкаф при 30°С и отбирали через 0 часов, 24 часа и 48 часов для определения размера частиц, PDI и содержания β-элемена. Результаты измерений приведены в таблице 11.
Пример 27
Взвешивали 2 г β-элемена и 1,5 г соевого масла и смешивали с получением масляной фазы. Отмеряли 45 мл (9 г) 20% человеческого сывороточного альбумина, а именно водную фазу. Раствор водной фазы выливали в масляную фазу и перемешивали со сдвигом с помощью смесителя (IKA, T25) со скоростью 5000 об/мин в течение 1 минуты и со скоростью 10000 об/мин в течение 5 минут с получением первичной эмульсии. Полученную первичную эмульсию переносили в гомогенизатор высокого давления (ATS, AH-1500) для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1450 до 1550 бар в течение 6 минут и собирали гомогенный раствор. Трубопровод гомогенизатора промывали водным раствором трегалозы (25 мл воды + 10 г трегалозы) для замены гомогенного раствора, оставшегося в гомогенизаторе, а затем трубопровод гомогенизатора промывали 30 мл сверхчистой воды для замены остаточного водного раствора трегалозы. Водный раствор трегалозы объединяли с гомогенным раствором, перемешивали и отфильтровывали через стерильную фильтрующую мембрану с размером пор 0,22 мкм. Размер частиц составлял 106,4 нм, как было установлено с помощью анализатора размера частиц, PDI составлял 0,08.
Пример 28
(г/г)
(г/г)
Взвешивали β-элемен и соевое масло, как показано в таблице 13, и смешивали с получением масляной фазы. Отмеряли 20% человеческий сывороточный альбумин (50 мл, 10 г), а именно водную фазу. Раствор водной фазы выливали в масляную фазу и перемешивали со сдвигом с помощью смесителя (IKA, T25) со скоростью 5000 об/мин в течение 1 минуты и со скоростью 10000 об/мин в течение 5 минут с получением первичной эмульсии. Полученный раствор переносили в гомогенизатор высокого давления (ATS, AH-1500) для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1550 до 1550 бар в течение 6 минут. Взвешивали 10 г трегалозы и растворяли в 50 мл путем добавления чистой воды (в которую добавляли образец № 5 с 15 г трегалозы и растворяли в 105 мл путем добавления чистой воды). Трубопровод гомогенизатора промывали водным раствором трегалозы для замены гомогенного раствора, оставшегося в гомогенизаторе, и остаточный гомогенный раствор объединяли с гомогенным раствором, перемешивали и отфильтровывали через стерильную фильтрующую мембрану с размером пор 0,45 мкм. Каждый образец исследовали для определения величины гемолиза. Образец разделяли и помещали в пробирки EP, хранили в термобаке при 30 °C и отбирали образцы через 0 часов, 24 часа и 48 часов для определения размера частиц, PDI и содержания β-элемена. Результаты приведены в таблице 14.
Пример 29
Взвешивали 2 г β-элемена и 1,5 г соевого масла и смешивали с получением масляной фазы. Отмеряли 50 мл (10 г) 20% человеческого сывороточного альбумина, а именно водную фазу. Раствор водной фазы выливали в масляную фазу и перемешивали со сдвигом с помощью смесителя (IKA, T25) со скоростью 5000 об/мин в течение 1 минуты и со скоростью 10000 об/мин в течение 5 минут с получением первичной эмульсии. Для очистки режущей головки использовали 5 мл воды для инъекций и объединяли с первичной эмульсией. Полученный раствор переносили в гомогенизатор высокого давления (ATS, AH-1500) для гомогенизации высокого давления при давлении гомогенизации от 250 до 300 бар в течение 2 минут и от 1500 до 1550 бар в течение 6 минут и собирали гомогенный раствор. Трубопровод гомогенизатора промывали 22,5 мл водного раствора трегалозы для замены гомогенного раствора, оставшегося в гомогенизаторе, а затем трубопровод гомогенизатора промывали 22,5 мл сверхчистой воды для замены остаточного водного раствора трегалозы. Раствор трегалозы объединяли с гомогенным раствором, перемешивали и отфильтровывали через стерильную фильтрующую мембрану с размером пор 0,22 мкм. Образец разделяли и помещали в пробирки EP, хранили в холодильнике при температуре от 2 до 8 °C и устанавливали размер частиц и PDI путем отбора образцов на 0-й день и второй месяц, соответственно. Результаты приведены в таблице 12.
Пример 30 Исследование эффективности лекарственного средства
Получение образца
Взвешивали 4,5 г β-элемена и 10,8 г соевого масла и смешивали с получением масляной фазы. Отмеряли 4 мл этанола и 16 мл дихлорметана и аккуратно перемешивали с получением органической фазы A. Масляную фазу добавляли к органической фазе и доводили отмеряемый объем до 36 мл с получением органической фазы B.
Взвешивали 13,5 г человеческого сывороточного альбумина и добавляли к 405 мл очищенной воды, перемешивали для растворения. Органическую фазу B добавляли к водной фазе и перемешивали с помощью высокоскоростного сдвигового устройства при передаче A в течение 2 минут, а затем при передаче B в течение 1 минуты с получением первичной эмульсии A. Первичную эмульсию A разделяли на 3 части и обрабатывали с применением клеточного дезинтегратора при 250 Вт в течение 8 минут с получением первичной эмульсии B. Первичную эмульсию B добавляли в гомогенизатор высокого давления, сначала при 200 бар в течение 3 циклов, а затем при 1300 бар в течение 5 циклов с получением наночастиц альбумина. Вышеуказанные наночастицы концентрировали при пониженном давлении при 37 °С в течение 1 часа, добавляли 12,30 г 3% сахарозы, отфильтровывали через 0,22 мкм и лиофилизировали. Размер частиц составлял 81,03 нм, PDI составлял 0,174.
Экспериментальные методы:
Создание мышиной модели ксенотрансплантата опухоли головного мозга человека: 50 бестимусных мышей подвергали анестезии с помощью 2,5% изофлурана, в черепе бестимусных мышей просверливали небольшое отверстие (<0,5 мм) и инъецировали в левую сторону лба (2,5 мм в латеральном направлении и 0,5 мм в переднем направлении от брегмы, глубина 2,5 мм) клетки, экспрессирующие люминесцирующий репортерный ген (5 × 104 U87MGR1 клеток глиомы человека).
Метод биолюминесцентной визуализации in vivo: бестимусные мыши получали однократную подкожную инъекцию раствора D-люциферина (120 мг/кг). Через 10 минут после инъекции флуоресцеина получали световые изображения животных при темном многоцветном освещении в течение времени экспозиции от 1 до 5 минут (для биолюминесцентной визуализации Gluc делали однократную внутривенную инъекцию коэлентеразина (20 мг/кг, при общем объеме 150 мкл)). Внутриопухолевую активность люциферазы оценивали путем регистрации количества фотонов с помощью камеры с зарядовой связью в течение 5 минут без освещения. Интенсивность изображения количественно анализировали с применением программного обеспечения Perkin Elmer 's Living Image 4.3.1.
Через неделю после инокуляции осуществляли биолюминесцентную визуализацию in vivo для оценки объема опухоли у бестимусных мышей. Отбирали 32 бестимусные мыши с аналогичным объемом опухоли и случайным образом разделяли по 8 мышей (половина самцов и половина самок)/на группу и вводили дозу путем инъекции в хвостовую вену в течение 3 недель, 3 раза в неделю:
1) модельная группа (контроль) (0,1% ДМСО, приготовленный с применением ФСБ), n=8;
2) 50 мг/кг темозоломида (TMZ), n=8;
3) 50 мг/кг Ailineng (ELE mic), n=8;
4) 50 мг/кг (Alb), n=8;
Еженедельно измеряли объем опухоли.
Во время эксперимента животных еженедельно взвешивали для контроля их состояния здоровья.
Прием лекарственного средства прекращали через две недели введения с последующим наблюдением в течение трех недель. Выживаемость и объем опухоли мышей в каждой группе регистрировали с применением программного обеспечения Graphpad для создания графиков Каплана-Мейера. Кривая роста опухоли мышиной ортотопической модели опухоли показана на фигуре 8 (бестимусные мыши-опухоленосители получали состав Alb, 3 раза/неделю × 2 недели (n=8, половина самцов и половина самок); сравнивали с темозоломидом (TMZ), p <0,01), кривая выживаемости мышиной ортотопической модели опухоли показана на фигуре 9 (бестимусные мыши-опухоленосители получали Alb, 3 раза/неделю × 2 недели (n=8, половина самцов и половина самок); сравнивали с темозоломидом (TMZ), p <0,01).
Пример 31 Исследование безопасности
Получение образца
Инъекция элемена (торговое наименование: Ailineng)): CSPC Yuanda (Dalian) Pharmaceutical Co., Ltd., номер партии: 17050302)
Состав в форме мицелл+β-элемен:
Взвешивали 17,08 г RH40 и нагревали при 60°C для растворения. Добавляли 2 мл β-элемена, нагревали и перемешивали при 60°С в течение 5 минут. Взвешивали 14,40 г пропиленгликоля и 38,72 г очищенной воды и перемешивали, затем нагревали при 60°C в течение 10 минут. Водный раствор пропиленгликоля добавляли к раствору RH40, содержащему β-элемен, и перемешивали на водяной бане при 60°C в течение 30 минут с получением мицелл+β-элемен. Вышеуказанные мицеллы отфильтровывали через 0,22 мкм и разделяли на части по 5 мл/на состав.
Состав в форме альбумина+β-элемен:
Взвешивали 2,5001 г β-элемена и 6,001 г соевого масла и смешивали для последующего применения. Отмеряли 2 мл этанола и 8 мл дихлорметана и аккуратно перемешивали для последующего применения. Объем масляной фазы доводили до 12 мл с помощью органической фазы для последующего применения.
Взвешивали 4,5015 г человеческого сывороточного альбумина и добавляли к 135 мл очищенной воды, перемешивали для растворения. К водной фазе добавляли органическую фазу и перемешивали с помощью высокоскоростного сдвигового устройства при передаче A в течение 2 минут, а затем при передаче B в течение 0,5 минуты с получением первичной эмульсии A. Первичную эмульсию A разделяли на 3 части и обрабатывали с применением клеточного дезинтегратора при 250 Вт в течение 8 минут с получением первичной эмульсии B. Первичную эмульсию B добавляли в гомогенизатор высокого давления, сначала при 200 бар в течение 3 циклов, а затем при от 1300 до 1400 бар в течение 5 циклов с получением наночастиц альбумина. Вышеуказанные наночастицы концентрировали при пониженном давлении при 37°С в течение 1 часа, добавляли 1,20 г сахарозы, отфильтровывали через 0,22 мкм и лиофилизировали.
Приготовление раствора:
Используя физиологический солевой раствор, приготавливали три состава в форме раствора с концентрацией 6 мг/мл.
Ailineng: отбирали 7,5 мл коммерчески доступного Ailineng с концентрацией 20 мг/мл и разбавляли с помощью 17,5 мл физиологического солевого раствора с получением 25 мл раствора образца с концентрацией 6 мг/мл.
Состав в форме мицелл+β-элемен: отбирали 5,91 мл состава в форме мицелл+β-элемен с концентрацией 25,41 мг/мл и разбавляли с помощью 19,09 мл физиологического солевого раствора с получением 25 мл раствора образца с концентрацией 6 мг/мл.
Состав в форме альбумина+β-элемен: отбирали 12 составов в форме мицелл+β-элемен, добавляли к каждому из них по 2,01 мл физиологического солевого раствора, затем перемешивали с получением 24 мл раствора образца с концентрацией 6 мг/мл.
Методы испытаний:
Самцов мышей KM, 20-25 г/мышь, случайным образом разделяли на 13 групп в соответствии с массой тела, по 7 мышей в каждой группе. Мышей группировали согласно предписанной дозе по массе. Дозировку каждого образца устанавливали равной 100 мг/кг, 130 мг/кг, 170 мг/кг и 220 мг/кг, и использовали еще одну холостую группу. Мышам вводили инъекцию через хвостовую вену и после введения отслеживали место введения, реакцию после введения и условия смерти. Результаты исследования приведены в таблице 15.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОЧИЩЕННЫЕ ТЕРАПЕВТИЧЕСКИЕ НАНОЧАСТИЦЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2015 |
|
RU2706735C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ПОЛУЧЕНИЯ СЛАБОРАСТВОРИМЫХ В ВОДЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ С УВЕЛИЧЕННОЙ СТАБИЛЬНОСТЬЮ | 2006 |
|
RU2451510C2 |
| НАНОЧАСТИЦА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ГЛАЗНЫХ ЗАБОЛЕВАНИЙ ИЛИ РАКА | 2018 |
|
RU2797114C2 |
| КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ СЛАБОРАСТВОРИМЫЕ В ВОДЕ ФАРМАЦЕВТИЧЕСКИЕ ВЕЩЕСТВА И ПРОТИВОМИКРОБНЫЕ ВЕЩЕСТВА | 2006 |
|
RU2433818C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ДОСТАВКИ ФАРМАКОЛОГИЧЕСКИХ АГЕНТОВ | 2009 |
|
RU2522977C2 |
| Фармацевтическая противоопухолевая композиция паклитаксела с гистоном Н1.3 в качестве носителя и способ получения композиции | 2017 |
|
RU2669937C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ДОСТАВКИ ФАРМАКОЛОГИЧЕСКИХ АГЕНТОВ | 2003 |
|
RU2361615C2 |
| КОМПОЗИЦИИ С ПАКЛИТАКСЕЛОМ, АЛЬБУМИНОМ И СВЯЗЫВАЮЩИМ СРЕДСТВОМ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ И ПОЛУЧЕНИЯ | 2017 |
|
RU2756892C2 |
| СТАБИЛЬНАЯ КОМПОЗИЦИЯ НАНОЧАСТИЦ ДОЦЕТАКСЕЛА-АЛЬБУМИНА | 2022 |
|
RU2835478C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПАРЕНТЕРАЛЬНОЙ ДОСТАВКИ В ФОРМЕ ЛИОФИЛИЗАТА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2007 |
|
RU2370258C2 |
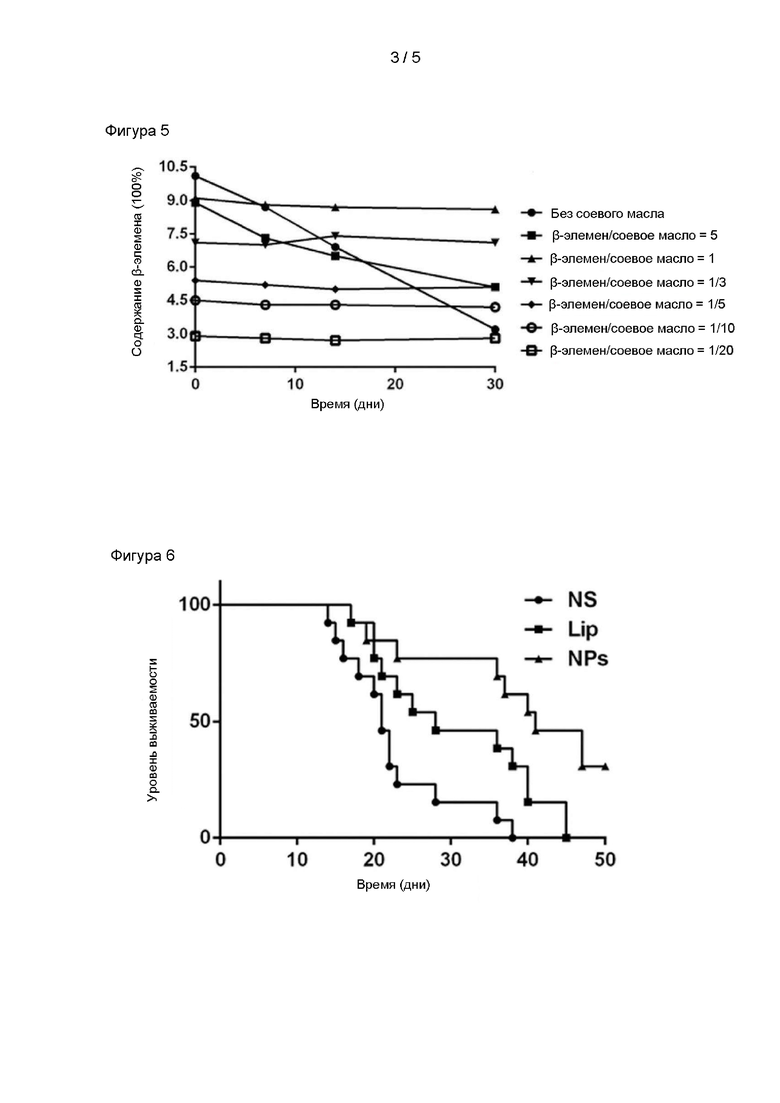
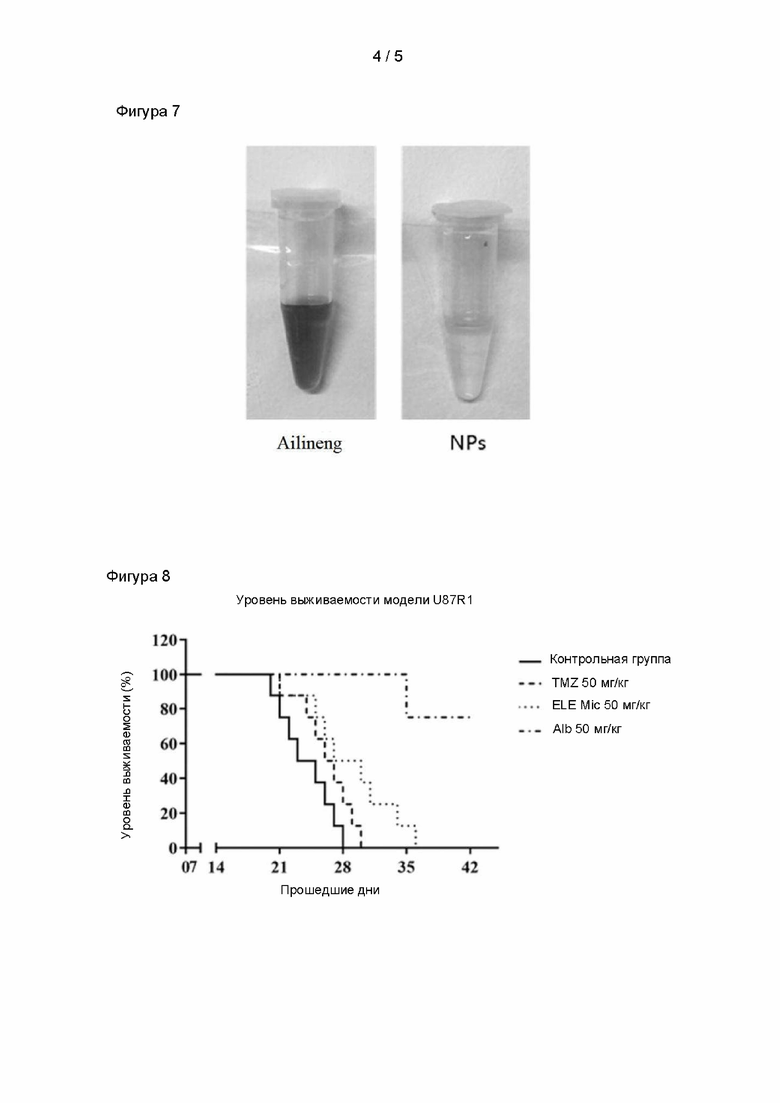
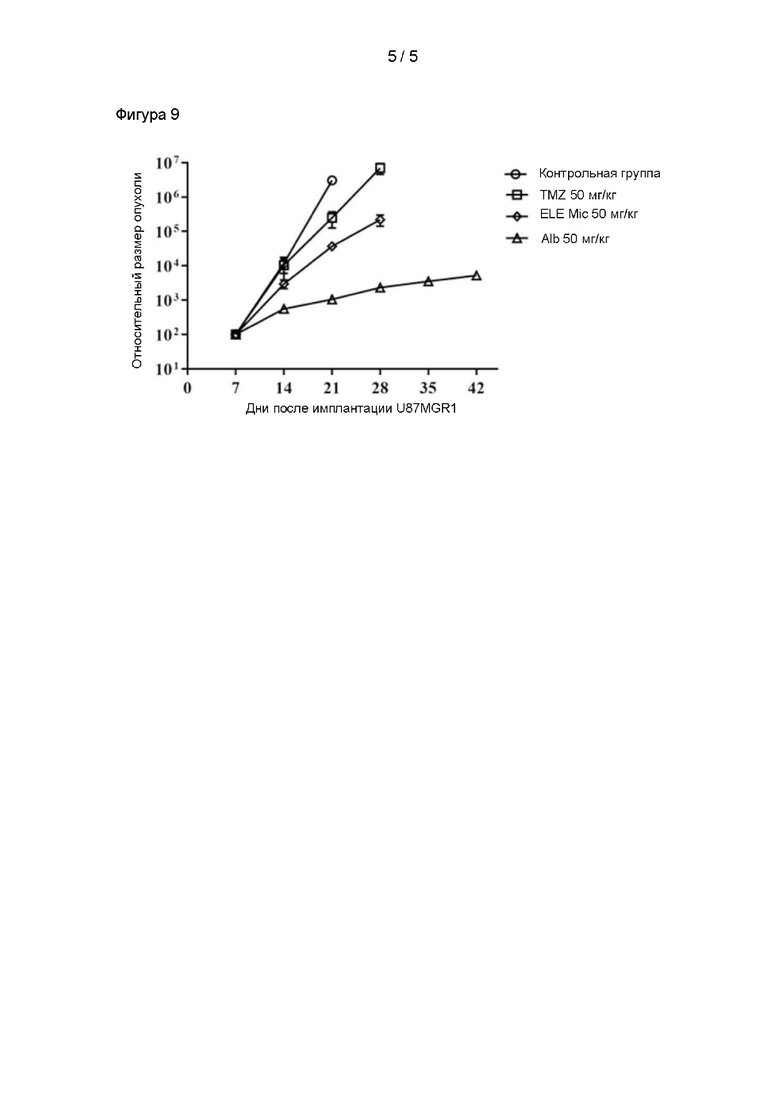
Группа изобретений относится к области фармацевтики, а именно к фармацевтической композиции для предотвращения или лечения рака, способам получения указанной композиции, применению указанной композиции для получения лекарственного препарата для предотвращения или лечения рака, способу лечения рака, фармацевтическому составу для предотвращения или лечения рака, герметичному контейнеру. Фармацевтическая композиция для предотвращения или лечения рака содержит элемен, масло для инъекций и белковый носитель, где указанное масло для инъекций представляет собой соевое масло, указанный белковый носитель представляет собой альбумин, причем массовое отношение соевого масла к элемену составляет около от 0,35 до 3, массовое отношение альбумина к элемену составляет около от 0,5 до 20. Способ получения указанной композиции включает стадии: (1) равномерного перемешивания элемена и масла для инъекций, добавления органического растворителя с получением раствора элемена и масла для инъекций; растворения белкового носителя в воде с получением раствора белкового носителя; (2) смешивания двух растворов, полученных на стадии (1), с получением эмульсии; (3) гомогенизации эмульсии на стадии (2) под высоким давлением и испарения эмульсии при пониженном давлении для удаления органического растворителя с получением раствора наночастиц, где указанное масло для инъекций представляет собой соевое масло, указанный белковый носитель представляет собой альбумин, причем массовое отношение соевого масла к элемену составляет около от 0,35 до 3, массовое отношение альбумина к элемену составляет около от 0,5 до 20. Способ получения указанной композиции включает стадии: (1) равномерного перемешивания элемена и масла для инъекций с получением раствора элемена и масла для инъекций; растворения белкового носителя в воде с получением раствора белкового носителя; (2) смешивания двух растворов, полученных на стадии (1), с получением эмульсии; (3) гомогенизации эмульсии на стадии (2) под высоким давлением с получением раствора наночастиц, где указанные масло для инъекций, белковый носитель и их массовое отношение к элемену характеризуются, как указано выше. Предложено применение указанной композиции для получения лекарственного препарата для предотвращения или лечения рака. Способ лечения рака включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества указанной композиции. Фармацевтический состав для предотвращения или лечения рака содержит указанную композицию. Герметичный контейнер содержит указанную композицию или указанный состав. Указанная группа изобретений позволяет создать эффективный и безопасный состав на основе альбумина, содержащий элемен. 7 н. и 50 з.п. ф-лы, 9 ил., 15 табл., 31 пр.
1. Фармацевтическая композиция для предотвращения или лечения рака, содержащая элемен, масло для инъекций и белковый носитель, где указанное масло для инъекций представляет собой соевое масло, указанный белковый носитель представляет собой альбумин, причем массовое отношение соевого масла к элемену составляет около от 0,35 до 3, массовое отношение альбумина к элемену составляет около от 0,5 до 20.
2. Фармацевтическая композиция по п. 1, в которой указанная фармацевтическая композиция имеет форму частиц, при этом размер указанных частиц составляет менее 180 нм, предпочтительно от 50 до 180 нм.
3. Фармацевтическая композиция по п. 2, в которой размер частиц составляет от 70 до 170 нм.
4. Фармацевтическая композиция по п. 3, в которой размер частиц составляет от 70 до 150 нм.
5. Фармацевтическая композиция по п. 1, в которой элемен выбран из одного или более элеменов из α-элемена, β-элемена, γ-элемена и δ-элемена.
6. Фармацевтическая композиция по п. 5, в которой элемен представляет собой β-элемен.
7. Фармацевтическая композиция по п. 1, в которой указанный альбумин выбран из одного или обоих из человеческого сывороточного альбумина и бычьего сывороточного альбумина.
8. Фармацевтическая композиция по п. 7, в которой белковый носитель представляет собой человеческий сывороточный альбумин.
9. Фармацевтическая композиция по любому из пп. 1-8, дополнительно содержащая лиопротектор.
10. Фармацевтическая композиция по п. 9, в которой лиопротектор выбран из одного или более лиопротекторов из глюкозы, сахарозы, мальтозы, лактозы, маннозы, трегалозы, глицина и декстрана.
11. Фармацевтическая композиция по п. 10, в которой лиопротектор представляет собой трегалозу или сахарозу.
12. Фармацевтическая композиция по пп. 9-11, в которой отношение массы лиопротектора к объему раствора фармацевтической композиции составляет от 3:100 до 20:100 г/мл.
13. Фармацевтическая композиция по любому из пп. 1-11, дополнительно содержащая один или более компонентов из агента, регулирующего изоосмотические свойства, антиоксиданта, консерванта и регулятора рН.
14. Фармацевтическая композиция по п. 13, в которой агент, регулирующий изоосмотические свойства, представляет собой один или более компонентов из глицерина, сорбитола, маннитола или глюкозы.
15. Фармацевтическая композиция по п. 13, в которой регулятор рН представляет собой один или более компонентов из гидроксида натрия, цитрата натрия, лимонной кислоты, фосфорной кислоты, уксусной кислоты или соляной кислоты.
16. Фармацевтическая композиция по п. 13, в которой консервант выбран из одного или более компонентов из гидроксибензолалкиловых эфиров, бензойной кислоты, бензоата натрия, сорбиновой кислоты, ацетата хлоргексидина, бромида бензалкония.
17. Фармацевтическая композиция по п. 13, в которой антиоксидант выбран из одного или более компонентов из сульфита натрия, бисульфита натрия, метабисульфита натрия, тиосульфата натрия, аскорбиновой кислоты, бутилированного гидроксианизола, 2,6-ди-трет-бутилированного гидрокситолуола и витамина Е.
18. Фармацевтическая композиция по любому из пп. 1-17, отличающаяся тем, что указанную фармацевтическую композицию получают в форме частиц, при этом размер указанных частиц составляет менее 180 нм.
19. Фармацевтическая композиция по п. 18, в которой размер частиц составляет от 70 до 150 нм.
20. Фармацевтическая композиция по любому из пп. 18, 19, отличающаяся тем, что в указанной фармацевтической композиции массовое отношение человеческого сывороточного альбумина к элемену составляет около от 0,5 до 20.
21. Фармацевтическая композиция по п. 20, отличающаяся тем, что в указанной фармацевтической композиции массовое отношение соевого масла к элемену составляет около от 0,5 до 3; массовое отношение человеческого сывороточного альбумина к элемену составляет около от 0,5 до 2,5.
22. Фармацевтическая композиция по п. 20, отличающаяся тем, что в указанной фармацевтической композиции массовое отношение соевого масла к элемену составляет около от 0,35 до 1,5, предпочтительно от 0,35 до 0,75; массовое отношение человеческого сывороточного альбумина к элемену составляет около от 1,5 до 10.
23. Фармацевтическая композиция по п. 22, отличающаяся тем, что в указанной фармацевтической композиции массовое отношение соевого масла к элемену составляет около 0,5; массовое отношение человеческого сывороточного альбумина к элемену составляет около 1,5.
24. Фармацевтическая композиция по п. 22, отличающаяся тем, что в указанной фармацевтической композиции массовое отношение соевого масла к элемену составляет около 0,75; массовое отношение человеческого сывороточного альбумина к элемену составляет около 5.
25. Фармацевтическая композиция по п. 22, отличающаяся тем, что в указанной фармацевтической композиции массовое отношение соевого масла к элемену составляет около 1,5; массовое отношение человеческого сывороточного альбумина к элемену составляет около от 3 до 5.
26. Фармацевтическая композиция по п. 20, отличающаяся тем, что в указанной фармацевтической композиции массовое отношение человеческого сывороточного альбумина к элемену составляет около от 0,5 до 10.
27. Фармацевтическая композиция по п. 26, отличающаяся тем, что в указанной фармацевтической композиции массовое отношение соевого масла к элемену составляет около 2,4; массовое отношение человеческого сывороточного альбумина к элемену составляет около 3.
28. Способ получения фармацевтической композиции по любому из пп. 1-27, включающий стадии:
(1) равномерного перемешивания элемена и масла для инъекций, добавления органического растворителя с получением раствора элемена и масла для инъекций; растворения белкового носителя в воде с получением раствора белкового носителя;
(2) смешивания двух растворов, полученных на стадии (1), с получением эмульсии;
(3) гомогенизации эмульсии на стадии (2) под высоким давлением и испарения эмульсии при пониженном давлении для удаления органического растворителя с получением раствора наночастиц;
где указанное масло для инъекций представляет собой соевое масло, указанный белковый носитель представляет собой альбумин, причем массовое отношение соевого масла к элемену составляет около от 0,35 до 3, массовое отношение альбумина к элемену составляет около от 0,5 до 20.
29. Способ получения фармацевтической композиции по любому из пп. 1-27, включающий стадии:
(1) равномерного перемешивания элемена и масла для инъекций с получением раствора элемена и масла для инъекций; растворения белкового носителя в воде с получением раствора белкового носителя;
(2) смешивания двух растворов, полученных на стадии (1), с получением эмульсии;
(3) гомогенизации эмульсии на стадии (2) под высоким давлением с получением раствора наночастиц;
где указанное масло для инъекций представляет собой соевое масло, указанный белковый носитель представляет собой альбумин, причем массовое отношение соевого масла к элемену составляет около от 0,35 до 3, массовое отношение альбумина к элемену составляет около от 0,5 до 20.
30. Способ по п. 28, в котором указанный органический растворитель используют на стадии (1) путем растворения элемена в указанном органическом растворителе и затем смешивания с маслом для инъекций; предпочтительно указанный органический растворитель выбран из одного или более из хлороформа, дихлорметана, третичного бутанола, изопропанола, этилацетата, этанола, тетрагидрофурана, диоксана, ацетонитрила, ацетона, диметилсульфоксида, диметилформамида и метилпирролидона.
31. Способ по п. 30, в котором указанный альбуминовый носитель представляет собой человеческий сывороточный альбумин; массовое отношение соевого масла к элемену составляет около от 2,4 до 10; массовое отношение указанного человеческого сывороточного альбумина к указанному элемену составляет около от 0,5 до 10.
32. Способ по п. 28 или 29, дополнительно включающий стадию лиофилизации полученного раствора наночастиц белкового носителя.
33. Способ по п. 28 или 29, в котором на стадии (2) для смешивания используют способ с применением сдвига или способ с применением ультразвука и на стадии (3) для гомогенизации используют способ гомогенизации высокого давления или способ микроструйной гомогенизации.
34. Применение фармацевтической композиции по любому из пп. 1-27 для получения лекарственного препарата для предотвращения или лечения рака.
35. Применение по п. 34, при котором рак представляет собой астроцитому, рак мочевого пузыря, рак кости, глиому, глиому и глиобластому зрительного пути и гипоталамическую глиому и глиобластому, рак молочной железы, рак шейки матки, рак толстой кишки, колоректальный рак, рак эндометрия, рак пищевода, семейство опухолей Юинга, рак желчного пузыря, рак печени, рак гортани, лейкоз, рак губ и полости рта, рак легкого, лимфому, меланому, мезотелиому, рак носоглотки, нейробластому, нейроэндокринный рак, опухоль головного мозга, рак костного метастаза, рак желудка, рак кишечника, рак яичника, рак поджелудочной железы, рак кожи, перитонеальный рак, опухоль гипофиза, рак прямой кишки, рак почки, карциному клеток плоского эпителия, рак горла, рак щитовидной железы.
36. Применение по п. 35, при котором рак представляет собой рак легкого, рак печени, рак пищевода, рак носоглотки, опухоль головного мозга, рак костного метастаза, рак желудка, рак кишечника, рак матки, рак шейки матки, рак эндометрия, рак молочной железы, рак кожи, лимфому, лейкоз или злокачественную меланому.
37. Применение по п. 36, при котором указанный рак представляет собой опухоль головного мозга.
38. Применение по п. 37, при котором указанный рак представляет собой глиому.
39. Способ лечения рака, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции по любому из пп. 1-27.
40. Способ по п. 39, в котором фармацевтическую композицию вводят путем парентерального введения, ингаляции, внутрибрюшинного, внутрипузырного, внутримышечного, внутривенного, внутритрахеального, подкожного, внутриглазного, интратекального, трансдермального, ректального или вагинального введения.
41. Способ по п. 40, в котором указанное введение представляет собой внутривенное введение.
42. Фармацевтический состав для предотвращения или лечения рака, содержащий фармацевтическую композицию по любому из пп. 1-27.
43. Фармацевтический состав по п. 42, отличающийся тем, что указанный фармацевтический состав представляет собой фармацевтический состав, предназначенный для внутривенного введения, фармацевтический состав, предназначенный для парентерального введения, фармацевтический состав, предназначенный для гастроинтестинального введения, фармацевтический состав, предназначенный для введения через органы дыхания, фармацевтический состав, предназначенный для вагинального введения, или другой подходящий фармацевтический состав; при этом указанный фармацевтический состав, предназначенный для введения через органы дыхания, представляет собой аэрозоль.
44. Фармацевтический состав по п. 42, отличающийся тем, что указанный фармацевтический состав представляет собой твердый состав, жидкий состав или газообразный состав.
45. Фармацевтический состав по п. 42, отличающийся тем, что указанный жидкий состав представляет собой жидкий состав для инъекций, содержащий элемен и фармацевтически приемлемый носитель, при этом фармацевтически приемлемый носитель содержит альбумин и соевое масло.
46. Фармацевтический состав по п. 44, представляющий собой состав в форме гранул, при этом размер частиц состава в форме гранул составляет от 70 до 150 нм, при этом в предложенной композиции массовое отношение альбумина к элемену составляет от 0,5 до 20 и массовое отношение масла для инъекций к элемену составляет от 0,35 до 10.
47. Фармацевтический состав по п. 44, отличающийся тем, что указанный жидкий состав представляет собой стабильную водную суспензию, восстановленную из стерильного лиофилизированного порошка.
48. Фармацевтический состав по п. 43, дополнительно содержащий другие лекарственные средства, в том числе противоопухолевые средства.
49. Фармацевтический состав по п. 48, отличающийся тем, что указанное противоопухолевое средство содержит темозоломид, паклитаксел, доцетаксел, таксан, гемцитабин, лекарственное средство, представляющее собой ингибитор ФРЭС, или лекарственное средство, представляющее собой антитело к PD-1.
50. Фармацевтический состав по п. 49, отличающийся тем, что указанное лекарственное средство, представляющее собой ингибитор ФРЭС, включает Авастин, ранибизумаб, афлиберцепт или конберцепт.
51. Фармацевтический состав по п. 45, в котором лекарственное средство, представляющее собой антитело к PD-1, включает ниволумаб, пембролизумаб, торипалимаб, синтилимаб, цемиплимаб, атезолизумаб, авелумаб или дурвалумаб.
52. Герметичный контейнер, содержащий фармацевтическую композицию по любому из пп. 1-27 или фармацевтический состав по любому из пп. 42-51.
53. Герметичный контейнер по п. 52, отличающийся тем, что указанный герметичный контейнер представляет собой однодозный контейнер или многодозный контейнер.
54. Герметичный контейнер по п. 52, в котором указанная фармацевтическая композиция представляет собой жидкую композицию или сухую твердую композицию.
55. Герметичный контейнер по п. 52, отличающийся тем, что указанная фармацевтическая композиция является лиофилизированной.
56. Герметичный контейнер по п. 52, отличающийся тем, что указанная фармацевтическая композиция является стерильной.
57. Герметичный контейнер по п. 52, отличающийся тем, что указанный герметичный контейнер представляет собой предварительно заполненный шприц.
| CN 100420445 C, 24.09.2008 | |||
| ZHAI, B., et al | |||
| Drug delivery systems for elemene, its main active ingredient β-elemene, and its derivatives in cancer therapy | |||
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| MU, L., et al | |||
| β-Elemene enhances the efficacy of gefitinib on glioblastoma multiforme cells through | |||

