ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к способу получения производных рапамицина, в частности, эверолимуса, с высоким выходом продукта и без значительного образования нежелательных побочных продуктов.
УРОВЕНЬ ТЕХНИКИ
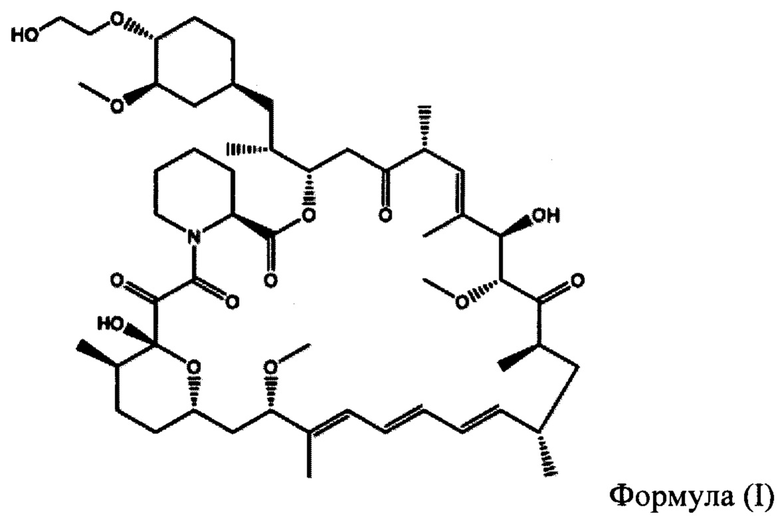
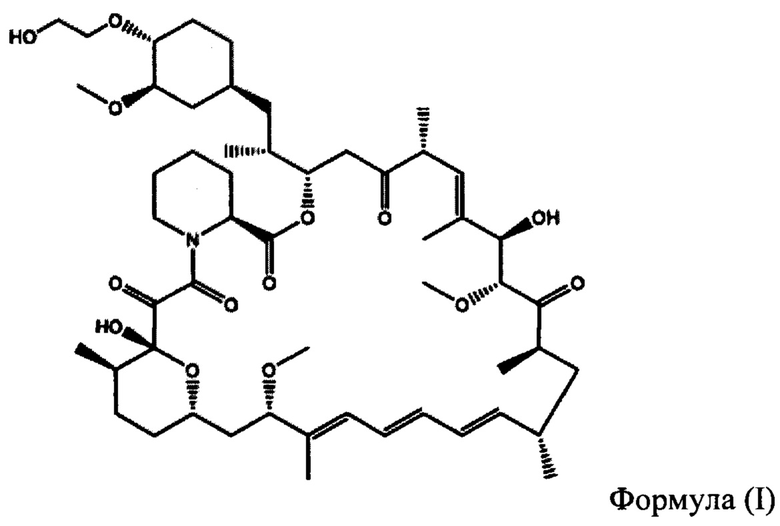
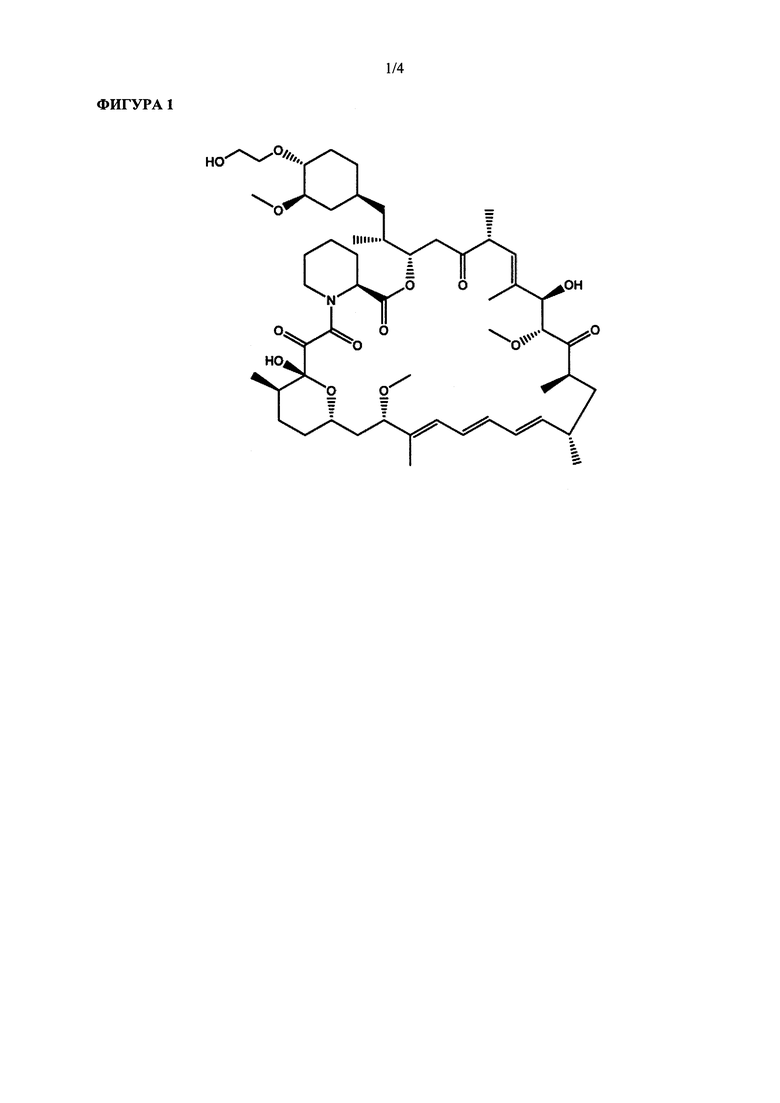
Эверолимус (40-О-(2-гидрокси)-этилрапамицин) (ФИГ. 1) представляет собой синтетическое производное сиролимуса (рапамицина), который впервые был выделен из Steptomycos hydroscopicus. Эверолимус относится к классу ингибиторов mTOR (мишень рапамицина в клетках млекопитающих, англ. - mammalian target of rapamycin,) и первоначально применялся в качестве иммунодепрессанта для предотвращения отторжения трансплантатов органов, а также для лечения некоторых видов рака, включая рак желудка, рак почки и лимфомы.
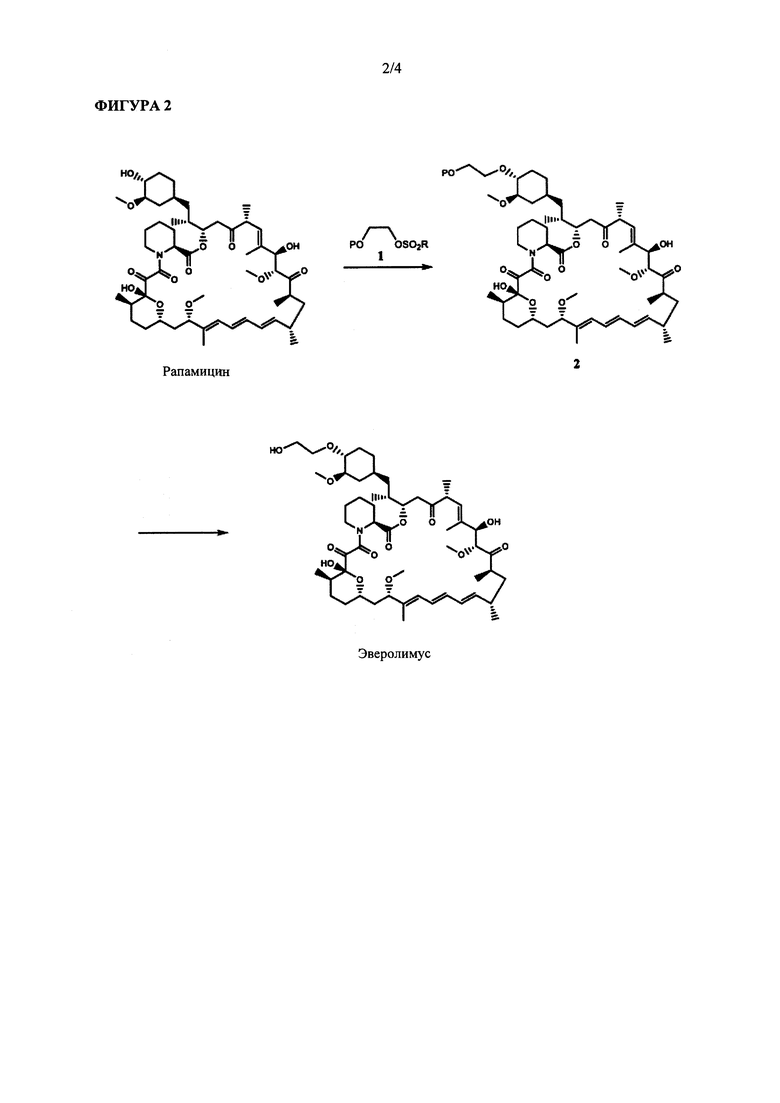
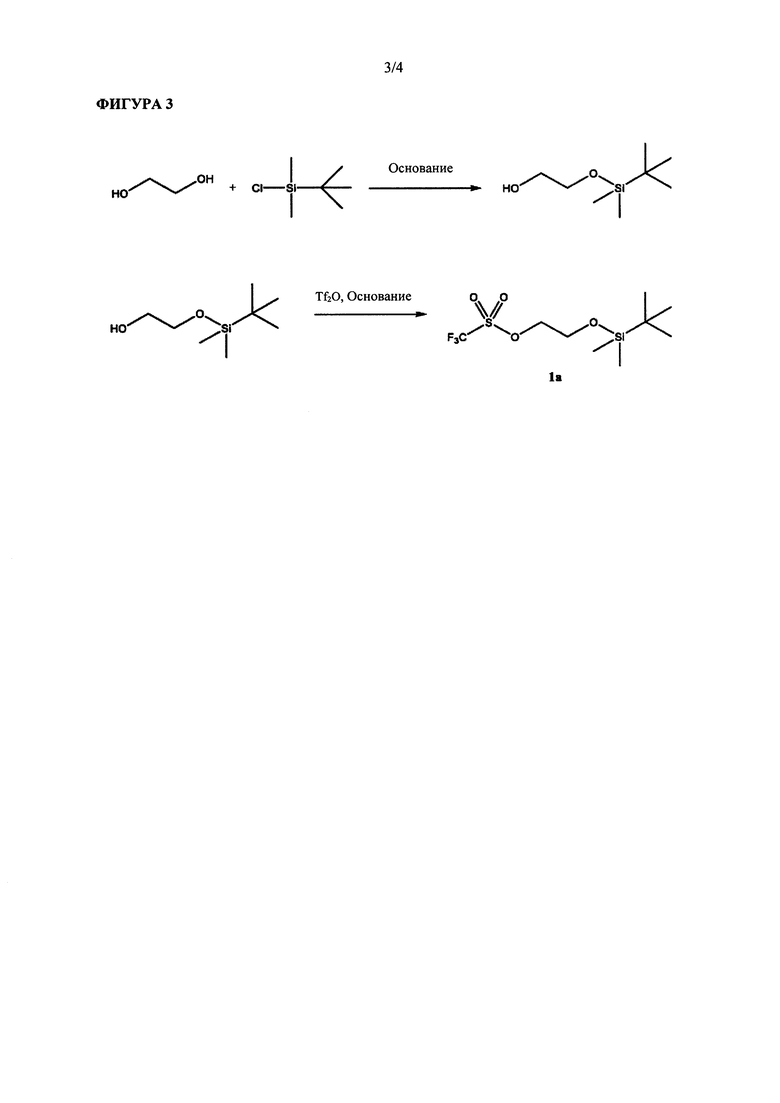
Известные способы синтеза эверолимуса основаны на алкилировании С40-гидроксильной группы рапамицина защищенным 2-гидроксиэтил-фторалкилсульфонатом 1 и последующем удалении защитной группы из полученного соединения 2 с получением эверолимуса (ФИГ. 2). Алкилирующий реагент, который обычно используют в данной схеме синтеза, представляет собой 2-((m-бутил-диметилсилил)окси)этил-трифторметансульфонат (1а на ФИГ. 3). Соединение 1а обычно получают достаточно сложным двухстадийным синтезом из этиленгликоля (ФИГ. 3). Вследствие его высокой нестабильности эверолимус необходимо применять сразу же после получения.
Фармакологическая активность эверолимуса, а также способ его получения, были первоначально описаны в WO 94/09010 А1. В указанной публикации синтез осуществляют посредством взаимодействия рапамицина с 4 эквивалентами 2-((m-бутилдиметилсилил)окси)этил-трифторметансульфоната в толуоле при температуре 60°С с применением 2,6-лутидина в качестве основания с получением 40-О-[2-(m-бутилдиметилсилил)окси]этил-рапамицина. Данный продукт очищают с помощью хроматографии и снимают защиту с применением 1N соляной кислоты в метаноле. Полученный неочищенный эверолимус далее снова очищают с помощью хроматографии.
Однако общий выход конечного продукта, как описано в WO 2012/103959 А1, составляет всего примерно 17%.
Применение защитной m-бутилдифенилсилильной группы для синтеза меченного тритием эверолимуса было предложено Моейиусом с соавторами (Moenius, Т. et al. (2000) J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)). В данном способе используют 2-(m-бутилдифенилсилил)оксиэтилтрифлат в смеси толуол-диметоксиэтан при температуре 50°С с применением N,N-диизопропилзтиламина в качестве основания. Однако общий выход эверолимуса, полученного после снятия защиты, был также очень низкий.
В WO 2012/066502 А1 предложен синтез эверолимуса посредством взаимодействия рапамицина с избытком в виде 4-8 эквивалентов 2-(m-бутилдиметилсилил)оксиэтилтрифлата с применением дихлорметана, этилацетата или толуола в качестве растворителя и 2,6-лутидина в качестве основания с последующим снятием защиты с полученного производного m-бутилдиметилсилил-эверолимуса. Общий выход конечного продукта, составляющий около 45%, был получен путем проведения реакции в дихлорметане с использованием 8 эквивалентов алкилирующего агента.
Способ, описанный в WO 2012/103959 А1, относится к алкилированию рапамицина более стабильным 2-(m-гексилдиметилсилил)оксиэтилтрифлатом. Реакцию проводят при температуре 70°С с 4 эквивалентами (m-гексилдиметилсилил)оксиэтил-трифлата в смеси толуол-диметоксиэтан с применением N,N-диизопропилэтиламина в качестве основания. Последующее снятие защиты с силильной группы с использованием 1N соляной кислоты в метаноле приводит к получению эверолимуса с немного большим общим выходом (около 52%) по сравнению с предыдущими способами.
Однако способ согласно WO 2012/103959 А1 затруднен сложным получением исходного 2-(m-гексилдиметилсилил)оксиэтилтрифлата. Требуется очистка полученного на первой стадии продукта, 2-((2,3-диметилбут-2-ил)диметилсилилокси)этанола, с применением вакуумной фракционной перегонки, проведение второй стадии реакции при низких температурах (-30°С) и дополнительная очистка сырого 2-(m-гексилдиметилсилил)оксиэтилтрифлата. Более того, данный способ необходимо осуществлять при высокой температуре (70°С, см. выше), что существенно увеличивает вероятность протекания нежелательных побочных реакций и вероятность наличия примесей в сыром продукте. Поэтому в качестве дополнительной стадии требуется хроматографическая очистка защищенного производного эверолимуса, после чего можно проводить стадию снятия защиты с очищенного продукта.
В WO 2014/203185 А1 предложено применение стерически затрудненных аминов в качестве оснований для синтеза эверолимуса, который включает взаимодействие рапамицина с соединением 1 (см. ФИГ. 2) и удаление защитной группы с получением эверолимуса. Применение аминов, таких как N,N-диизопропилпентан-3-амин, диизопропилнонан-5-амин и N,N-диизобутил-2,4-диметил-пентан-3-амин, в качестве основания в процессе алкилирования рапамицина увеличивает стабильность алкилирующего агента 1, что обеспечивает более высокий выход эверолимуса. Общий выход сырого эверолимуса, составляющий около 67%, был получен путем проведения реакции в толуоле при температуре 40°С с применением 2,5 эквивалентов 2-(m-бутилдифенилсилил)оксиэтилтрифлата и N,N-диизопропилпентан-3-амина в качестве оснований. Однако исходное вещество - 2-((m-бутилдифенилсилил)окси)этанол, используемое для получения трифлатного соединения, а также каждый из описанных выше стерически затрудненных аминов коммерчески недоступны и сложны в получении, что является главным недостатком данного способа.
Таким образом, по-прежнему существует потребность в улучшенных способах синтеза эверолимуса, которые позволили бы преодолеть недостатки известных способов.
В частности, существует потребность в менее трудоемком и при этом экономически выгодном способе получения 2-(тризамещенного силил)оксиэтилтрифлата, с увеличением тем самым общего выхода синтеза эверолимуса из рапамицина и с минимизацией при этом образования нежелательных побочных продуктов.
Соответственно, задачей настоящего изобретения является обеспечение улучшенного способа синтеза эверолимуса.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте настоящее изобретение относится к способу получения производного рапамицина формулы (I), включающему:
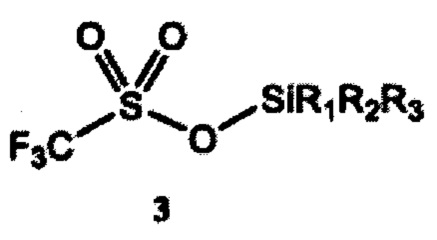
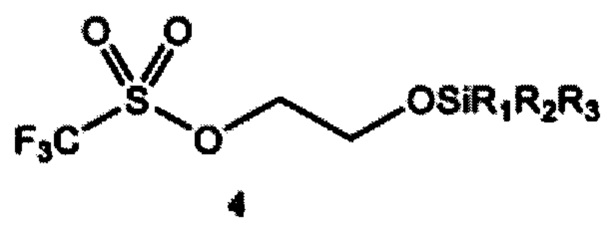
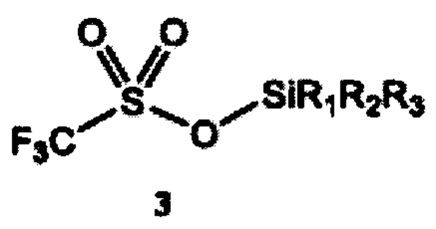
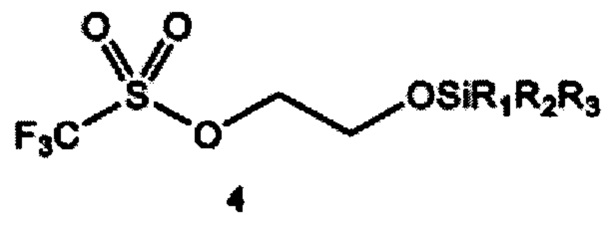
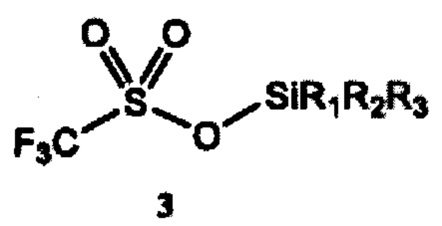
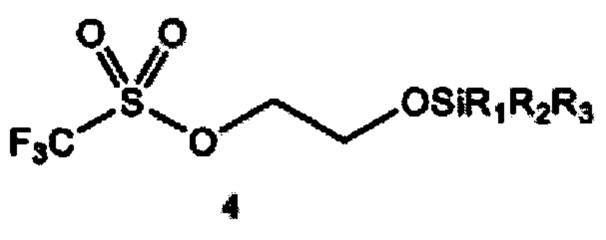
a) получение 2-(тризамещенного силил)оксиэтилтрифлата формулы 4 путем взаимодействия этиленоксида и тризамещенного силилтрифлата формулы 3
 ,
,
где R1, R2 и R3 независимо выбраны из группы, состоящей из С1-12алкила и C1-С12 арила;
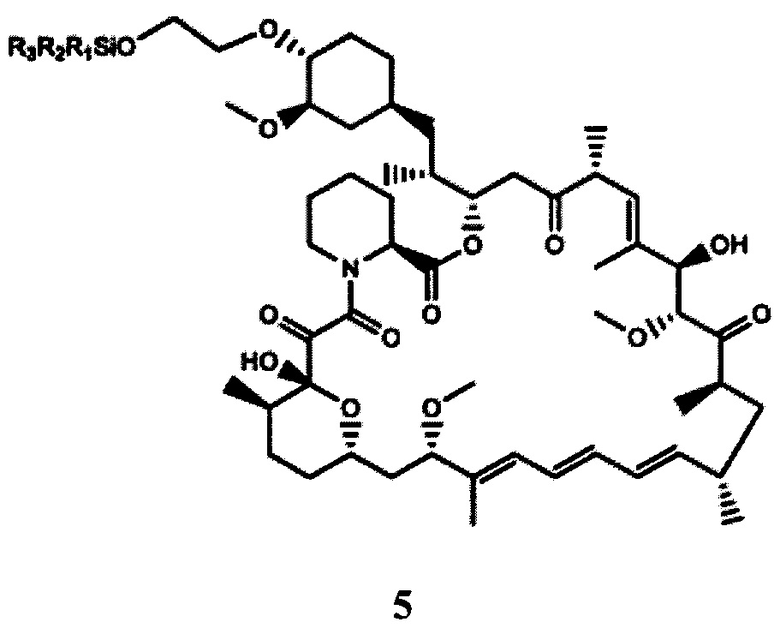
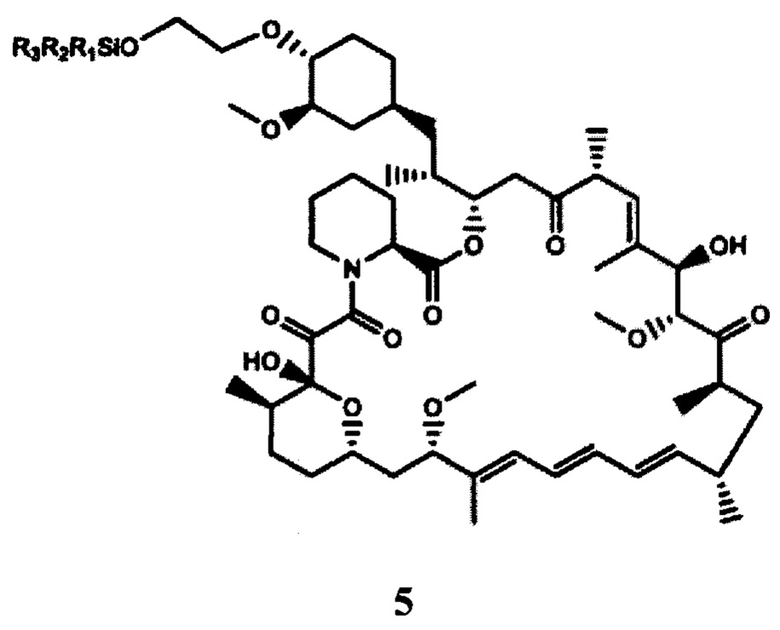
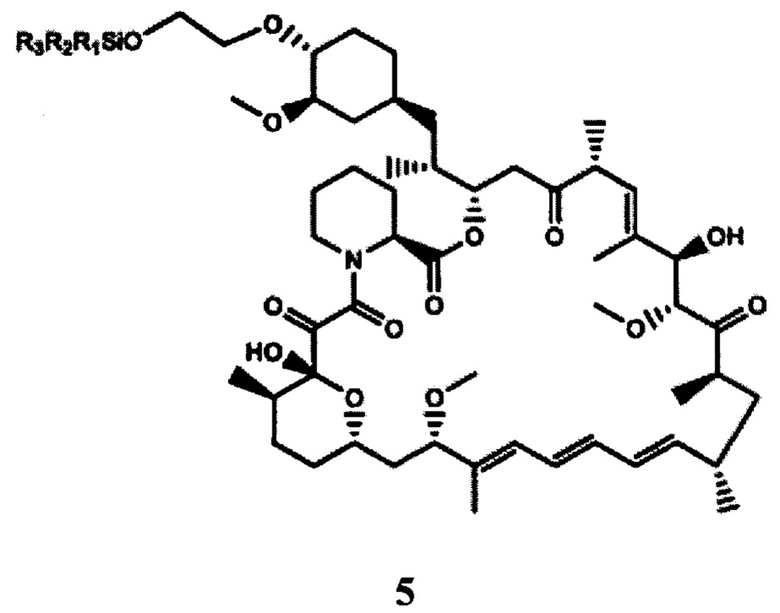
b) взаимодействие 2-(тризамещенного силил)оксиэтилтрифлата формулы 4, полученного на стадии (а), с рапамицином в присутствии молярного избытка органического основания с получением защищенного производного рапамицина формулы 5; и
c) снятие защиты с защищенного производного рапамицина формулы 5 с получением производного рапамицина формулы (I).
В частности, производное рапамицина формулы (I) представляет собой эверолимус.
Согласно конкретным вариантам реализации R1, R2 и R3 независимо выбраны из группы, состоящей из C1-С6 алкила и С6-С12 арила. Согласно предпочтительным вариантам реализации каждый из R1, R2 и R3 представляет собой изопропил; или каждый из R1 и R2 представляет собой метил, a R3 представляет собой m-бутил; или каждый из R1 и R2 представляет собой фенил, a R3 представляет собой m-бутил; или каждый из R1 и R2 представляет собой метил, a R3 представляет собой m-гексил.
Согласно другим предпочтительным вариантам реализации стадию (а) способа осуществляют в органическом растворителе при температуре реакции 15°С-45°С.
Особенно предпочтительно, органический растворитель представляет собой толуол.
Согласно другим конкретным вариантам реализации этиленоксид применяют в количестве 1,1-1,2 молярных эквивалентов по отношению к тризамещенному силилтрифлату формулы 3.
Согласно другим конкретным вариантам реализации 2-(тризамещенный силил)оксиэтилтрифлат формулы 4, полученный на стадии (а), применяют без дополнительной очистки.
Согласно другим предпочтительным вариантам реализации стадию (b) способа осуществляют в органическом растворителе при температуре реакции 40°С-55°С. Особенно предпочтительно, органический растворитель представляет собой смесь 85-95% (об./об.) толуола и 5-15% (об./об.) диметоксиэтана.
Согласно другим конкретным вариантам реализации 2-(тризамещенный силил)оксиэтилтрифлат формулы 4 применяют в количестве 4-12 молярных эквивалентов по отношению к рапамицину.
Согласно особенно предпочтительным вариантам реализации стадию (b) способа осуществляют в присутствии молярного избытка N,N-диизопропилэтиламина в качестве органического основания.
Согласно еще одним конкретным вариантам реализации защищенное производное рапамицина формулы 5, полученное на стадии (b), применяют без дополнительной очистки.
Согласно более предпочтительным вариантам реализации снятие защиты осуществляют посредством взаимодействия защищенного производного рапамицина формулы 5 с агентом, выбранным из группы, состоящей из соляной кислоты, уксусной кислоты, фторида тетрабутиламмония и гидрофторида пиридина.
Согласно другим конкретным вариантам реализации указанный способ дополнительно включает очистку производного рапамицина формулы (I), полученного на стадии (с).
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На ФИГУРЕ 1 приведена химическая структура 40-О-(2-гидрокси)этил-рапамицина (т.е. эверолимуса).
На ФИГУРЕ 2 приведена репрезентативная схема синтеза для получения эверолимуса, традиционно применяемая в данной области техники.
На ФИГУРЕ 3 приведена репрезентативная схема синтеза для получения 2-((m-бутилдиметилсилил)окси)этилтрифлата, традиционно применяемая в данной области техники.
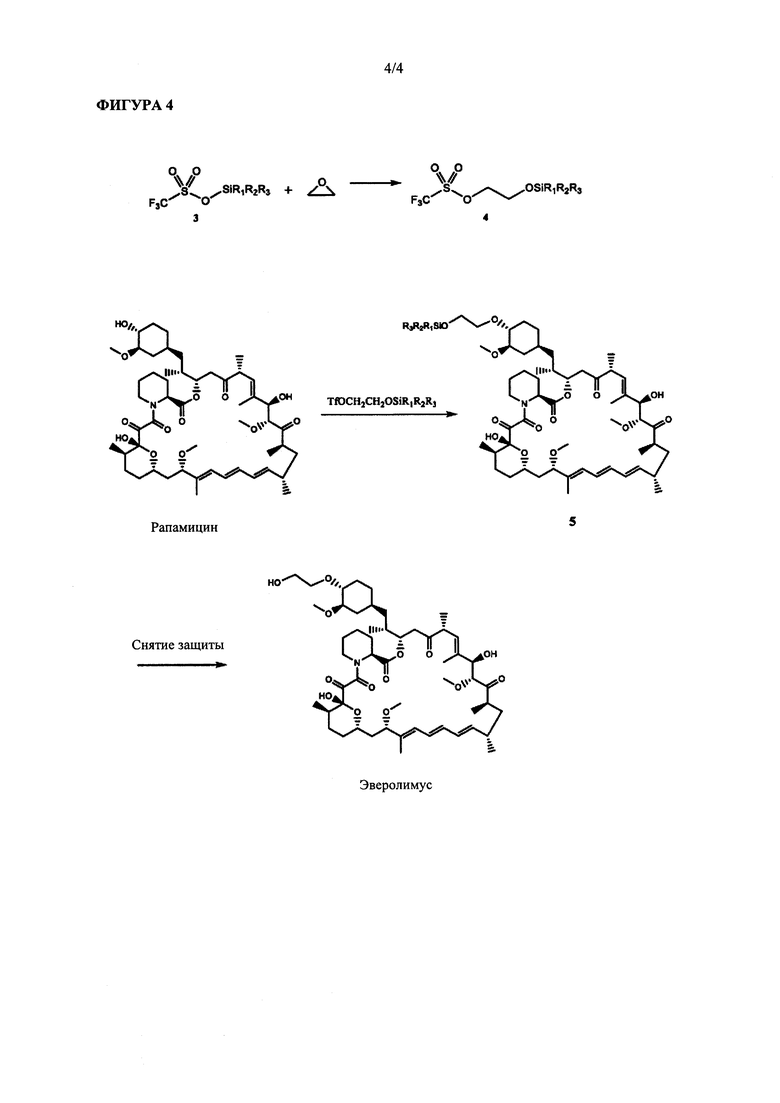
На ФИГУРЕ 4 приведена схема синтеза для получения эверолимуса согласно настоящему изобретению.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение основано на неожиданному обнаруженном факте, что взаимодействие этиленоксида с m-бутилдиметилсилил-трифторметансульфонатом в органическом растворителе приводит к получению высоко чистого 2-(m-бутилдиметилсилил)оксиэтилтрифлата, который можно непосредственно подвергнуть реакции с рапамицином в присутствии молярного избытка органического основания с получением силил-защищенного производного рапамицина (т.е. силил-защищенного эверолимуса) с высоким выходом и низким содержанием нежелательных побочных продуктов. Силил-защитная группа может быть удалена в слабокислой среде с получением чистого производного рапамицина (т.е. эверолимуса), с обеспечением тем самым более экономически выгодной и менее трудоемкой общей схемы синтеза, а также более высоких выходов конечного продукта.
Настоящее изобретение далее будет описано в соответствии с частными вариантами реализации и со ссылкой на конкретные графические материалы, но следует понимать, что изобретение не ограничено ими, а ограничено только прилагаемой формулой изобретения. Описанные графические материалы являются сугубо схематичными и не должны рассматриваться как ограничивающие настоящее изобретение.
Термин «включающий», используемый в настоящем описании и формуле изобретения, не исключает других элементов или стадий. Для целей настоящего изобретения термин "состоящий из" считается предпочтительным вариантом термина "включающий". Если в дальнейшем группа определена как включающая по меньшей мере некоторое количество вариантов реализации, это следует понимать как указание на группу, которая предпочтительно состоит только из этих вариантов реализации.
Если существительное упоминается в единственном числе с определенным или неопределенным артиклем, это включает и формы множественного числа для данного существительного, если специально не указано иное.
В случае, если в контексте настоящего изобретения используются числовые значения, специалисту в данной области техники будет понятно, что технический результат, обеспечиваемый конкретным признаком, обеспечивается в некотором интервале с допустимой погрешностью, которая обычно включает отклонения числовых значений в пределах ±10%, предпочтительно в пределах ±5%.
Кроме того, термины первый, второй, третий, (а), (b), (с) и т.п., используемые в настоящем описании и в формуле изобретения, применяются для различения сходных элементов и не обязательно описывают последовательность или хронологический порядок. Следует понимать, что используемые определения являются взаимозаменяемыми при соответствующих обстоятельствах, и что варианты реализации изобретения, описанные в настоящей заявке, могут быть реализованы в последовательности, отличной от описанной в настоящей заявке.
Дальнейшее определение терминов будет приведено ниже в контексте, в котором используются эти термины. Следующие термины и определения приведены исключительно для облегчения понимания изобретения. Указанные определения не следует истолковывать как имеющие более узкий смысл по сравнению со смыслом, очевидным среднему специалисту в данной области техники.
В одном аспекте настоящее изобретение относится к способу получения производного рапамицина формулы (I), включающему:
a) получение 2-(тризамещенного силил)оксиэтилтрифлата формулы 4 путем взаимодействия этиленоксида и тризамещенного силилтрифлата формулы 3
 ,
,
где R1, R2 и R3 независимо выбраны из группы, состоящей из С1-С12 алкила и C1-С12 арила;
b) взаимодействие 2-(тризамещенного силил)оксиэтилтрифлата формулы 4, полученного на стадии (а), с рапамицином в присутствии молярного избытка органического основания с получением защищенного производного рапамицина формулы 5; и
с) снятие защиты с защищенного производного рапамицина формулы 5 с получением производного рапамицина формулы (I).
В частности, производным рапамицина формулы (I), полученным в соответствии со способом согласно настоящему изобретению, является эверолимус. Аналогично, полученное в данных условиях защищенное производное рапамицина формулы 5 представляет собой защищенный эверолимус.
Способ согласно настоящему изобретению схематично представлен на ФИГ. 4.
Реакция между этиленоксидом и тризамещенным силилтрифлатом формулы 3 (т.е. способ согласно стадии (а)) может быть осуществлена при температуре реакции в диапазоне от -20°С до 70°С. Обычно данную реакцию осуществляют при температуре реакции в диапазоне от 0°С до 60°С или в диапазоне от 10°С до 50°С. Согласно предпочтительным вариантам реализации реакцию осуществляют при температуре в диапазоне от 15°С до 45°С или в диапазоне от 20°С до 40°С. Реакция может быть также осуществлена при температуре 15°С, 20°С, 25°С, 30°С, 35°С, 40°С и 45°С.
Реакцию можно проводить в органическом растворителе, в частности, в инертном органическом растворителе, то есть растворителе, который не вступает в реакцию с реагентами. Согласно конкретным вариантам реализации органический растворитель выбран из группы, состоящей из толуола, ацетонитрила, дихлорметана, гептана, пентана, гексана, бензола, ксилола и их смесей. Соответственно, применяемый органический растворитель может также представлять собой смесь двух или более отдельных растворителей, таких как толуол или дихлорметан. Особенно предпочтительно, применяемый органический растворитель представляет собой толуол.
Обычно этиленоксид можно применять в настоящей реакции в количестве 1,0-1,6 молярных эквивалентов по отношению к применяемому тризамещенному силилтрифлату формулы 3. Предпочтительно, этиленоксид применяют в количестве 1,0-1,3 молярных эквивалентов или 1,1-1,2 молярных эквивалентов по отношению к применяемому тризамещенному силилтрифлату формулы 3.
Заместители R1, R2 и R3 в тризамещенном силилтрифлате формулы 3 могут независимо представлять любую С1-С12 алкильную группу или С1-С12 арильную группу, тем самым включая линейные, разветвленные или циклические заместители, содержащие от 1 до 12 атомов углерода. Термины «алкил» и «арил» используются в данном документе в соответствии с их общепринятым значением в данной области техники. Например, линейные алкильные группы включают метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил, децил, ундецил и додецил. Разветвленные алкильные группы включают, в том числе, изо-бутил, втор-бутил, трет-бутил (m-бутил), изо-пропил, m-пропил, изо-гексил, m-гексил и т.д. Типичные арильные группы включают фенил и нафтил. Заместители могут также содержать гетероатомы или могут быть замещены, например, гидроксильной или аминной группой.
Согласно отдельным вариантам реализации R1, R2 и R3 независимо выбраны из группы, состоящей из C1-С6 алкила и С6-С12 арила. В предпочтительном варианте реализации изобретения каждый из R1, R2 и R3 представляет собой изопропил. В другом предпочтительном варианте реализации каждый из R1 и R2 представляет собой метил, a R3 представляет собой m-бутил. В дополнительном предпочтительном варианте реализации каждый из R1 и R2 представляет собой фенил, a R3 представляет собой m-бутил. В дополнительном предпочтительном варианте реализации каждый из R1 и R2 представляет собой метил, a R3 представляет собой m-гексил.
В отличие от известных способов с применением этиленгликоля в качестве исходного вещества, способ получения 2-(тризамещенного силил)оксиэтилтрифлата формулы 4 согласно настоящему изобретению представляет собой одностадийный синтез, который обеспечивает высокий выход продукта, а также высокую степень чистоты продукта, который, в свою очередь, не требует какой-либо дополнительной очистки перед применением на последующих стадиях реакции. При этом отсутствует необходимость в специальном оборудовании для осуществления обширного охлаждения или перегонки, что делает способ согласно настоящему изобретению легко применимым в промышленном производстве.
Согласно отдельным вариантам реализации 2-(тризамещенный силил)оксиэтилтрифлат формулы 4, полученный на стадии (а), соответственно применяют без дополнительной очистки.
Синтез защищенного эверолимуса (т.е. стадии (а) и (b) способа) может быть осуществлен в "одном сосуде" путем добавления рапамицина в присутствии молярного избытка органического основания к этиленоксиду и тризамещенному силилтрифлату формулы 3 и нагревания полученной реакционной смеси. В данных условиях 2-(тризамещенный силил)оксиэтилтрифлат формулы 4 представляет собой промежуточный продукт реакции, который непосредственно применяют для конденсации с рапамицином.
Способ согласно стадии (b) может быть осуществлен при температуре реакции в диапазоне от 20°С до 70°С. Обычно данную реакцию осуществляют при температуре реакции в диапазоне от 30°С до 60°С. Согласно предпочтительным вариантам реализации, реакцию осуществляют при температуре в диапазоне от 40°С до 55°, и особенно предпочтительно, в диапазоне от 48°С до 50°С. Реакция может быть также осуществлена при температуре 40°С, 42°С, 44°С, 46°С, 48°С, 50°С, 52°С и 55°С.
Реакцию можно также проводить в органическом растворителе, в частности, в апротонном органическом растворителе, то есть растворителе, который не может быть донором водорода. Согласно конкретным вариантам реализации органический растворитель выбран из группы, состоящей из толуола, ацетонитрила, дихлорметана, гептана, пентана, гексана, бензола, ксилола, диметоксиэтана и их смесей. Соответственно, применяемый органический растворитель может также представлять собой смесь двух или более отдельных растворителей, таких как толуол или диметоксиэтан. Особенно предпочтительно, применяемый органический растворитель представляет собой смесь 85-95% (об./об.) толуола и 5-15% (об./об.) диметоксиэтана.
Обычно 2-(тризамещенный силил)оксиэтилтрифлат формулы 4 можно применять в настоящей реакции в количестве от 1 до 30 молярных эквивалентов или от 2 до 20 молярных эквивалентов по отношению к используемому количеству рапамицина. Предпочтительно, 2-(тризамещенный силил)оксиэтилтрифлат формулы 4 применяют в количестве от 4 до 12 молярных эквивалентов или от 5 до 8 молярных эквивалентов по отношению к рапамицину, особенно предпочтительно, в количестве 6 молярных эквивалентов.
Стадию (b) способа проводят в присутствии молярного избытка органического основания для нейтрализации трифлатной кислоты, образующейся во время реакции. При этом было обнаружено, что добавление органического основания снижает протекание нежелательных побочных реакций, и таким образом способствует получению более чистого продукта и упрощает общую схему синтеза. Примеры подходящих органических оснований включают, помимо прочих, N,N-диизопропилэтиламин, 2,6-лутидин, 2,6-ди-трет-бутилпиридин и их смеси. Особенно предпочтительно, органическое основание представляет собой N,N-диизопропилэтиламин.
Согласно другим конкретным вариантам реализации защищенный эверолимус формулы 5, полученный на стадии (b), применяют без дополнительной очистки.
Стадия снятия защиты (т.е. стадия (с) способа) может быть осуществлена в подходящих для проведения реакции условиях, в частности, в слабокислой среде, известных в данной области техники, для удаления силил-защитной группы из защищенного эверолимуса формулы 5.
Согласно предпочтительным вариантам реализации снятие защиты проводят посредством взаимодействия защищенного эверолимуса формулы 5 с агентом, выбранным из группы, состоящей из соляной кислоты, уксусной кислоты, фторида тетрабутиламмония и гидрофторида пиридина.
Согласно другим конкретным вариантам реализации способ дополнительно включает очистку эверолимуса, полученного на стадии (с), например, с помощью аффинной хроматографии и/или последующей кристаллизацией. Подобные различные способы очистки хорошо известны в данной области техники.
Изобретение дополнительно описано с помощью фигур и следующих примеров, которые приведены исключительно с целью проиллюстрировать отдельные варианты реализации настоящего изобретения и не ограничивают настоящее изобретение каким-либо образом.
ПРИМЕРЫ
Пример 1: Синтез 40-О-[2-(т-бутилдиметилсилил)окси]этил-рапамицина
Смесь 7,9 мл 1М раствора этиленоксида в толуоле и 1,01 N,N-диизопропил-этиламина при перемешивании добавляли к 1,72 г m-бутилдиметилсилил-трифторметансульфоната. Полученную смесь далее перемешивали в течение 40 минут при температуре 20°С. Затем добавляли 0,66 мл 1,2-диметоксиэтана и 1,82 г N,N-диизопропилэтиламин-трифторметансульфоната. Полученную смесь перемешивали при температуре 20°С до полного растворения, после чего добавляли 1,0 г рапамицина. Реакционную смесь продували азотом в течение 10 минут, нагревали до 48-50°С и перемешивали при данной температуре в течение 18 часов. Далее реакционную смесь охлаждали до 20°С и добавляли 0,2 мл пиридина и 16 мл гептана. Полученную смесь перемешивали в течение 10 минут. Выпавший осадок отделяли фильтрованием и промывали смесью 4,0 мл толуола и 9,4 мл гептана. Затем к фильтрату добавляли 1 мл 0,2% раствора 2,6-ди-m-бутил-4-метилфенола в гептане, и полученный неочищенный раствор 40-О-[2-(m-бутилдиметилсилил)окси)этил-рапамицина подвергали стадии снятия зашиты. Анализ методом ВЭЖХ полученного в результате раствора 40-О-[2-(m-бутилдиметилсилил)окси]этил-рапамицина показал общий выход, равный 76% (895 мг).
Пример 2: Синтез 40-О-(2-гидрокси)этил-рапамицина (т.е. эверолимуса)
Неочищенный раствор 40-О-[2-(m-бутилдиметилсилил)окси]этил-рапамицина, полученный в Примере 1, упаривали до сухого остатка при пониженном давлении и температуре 25-30°С. Далее к остатку добавляли 66 мл гептана, и смесь упаривали до объема 40 мл. Затем добавляли 48 мл смеси ацетонитрила/воды (80:20) (об./об.) (рН=1,7, доведенный 75%-ой ортофосфорной кислотой), и значение рН полученной смеси доводили до значения 1,8 с применением 1N раствора соляной кислоты. Полученную в результате смесь перемешивали в течение одного часа при комнатной температуре и отделяли нижний слой, содержащий эверолимус. Общий выход эверолимуса, который определяли методом ВЭЖХ с применением внешнего стандарта, составил 68% (711 мг).
Пример 3: Синтез 40-О-[2-(триизопропилсилил)окси]этил-рапамицина
Смесь 7,9 мл 1М раствора этиленоксида в толуоле и 1,01 г N,N-диизопропил-этиламина при перемешивании добавляли к 1,99 г триизопропилсилил-трифторметансульфоната. Полученную смесь далее перемешивали в течение часа при температуре 40°С. Затем добавляли 0,66 мл 1,2-диметокси-этана и 1,82 г N,N-диизопропилэтиламин-трифторметансульфоната. Полученную смесь перемешивали при температуре 20°С до полного растворения, после чего добавляли 1,0 г рапамицина. Реакционную смесь продували азотом в течение 10 минут, нагревали до 48-50°С и перемешивали при данной температуре в течение 18 часов. Далее реакционную смесь охлаждали до 20°С и добавляли 0,2 мл пиридина и 16 мл гептана. Полученную смесь перемешивали в течение 10 минут. Выпавший осадок отделяли фильтрованием и промывали смесью 4,0 мл толуола и 9,4 мл гептана. Затем к фильтрату добавляли 1 мл 0,2% раствора 2,6-ди-m-бутил-4-метилфенола в гептане, и полученный неочищенный раствор 40-О-[2-(три-изопропилсилил)окси]этил-рапамицина подвергали стадии снятия зашиты. Анализ методом ВЭЖХ полученного в результате раствора 40-О-[2-(триизопропилсилил)окси]этил-рапамицина показал общий выход, равный 71% (864 мг).
Пример 4: Синтез 40-О-(2-гидрокси)этил-рапамицина (т.е. эверолимуса)
Неочищенный раствор 40-О-[2-(триизопропилсилил)окси]этил-рапамицина, полученный в Примере 3, упаривали досуха при пониженном давлении и температуре 25-30°С. Далее к остатку добавляли 66 мл гептана, и полученную смесь упаривали до объема 40 мл. Затем добавляли 48 мл смеси ацетонитрила/воды (80:20) (об./об.) (рН=1,7, доведенный 75%-ой ортофосфорной кислотой), и значение рН полученной смеси доводили до значения 1,8 с применением 1N раствора соляной кислоты. Полученную в результате смесь перемешивали в течение одного дня при комнатной температуре и отделяли нижний слой, содержащий эверолимус. Общий выход эверолимуса, который определяли методом ВЭЖХ с применением внешнего стандарта, составил 64% (668 мг).
Пример 5: Синтез 40-О-[2-(m-бутилдифенилсилил)окси]этил-рапамицина
Смесь 7,9 мл 1М раствора этиленоксида в толуоле и 1,01 г N,N-диизопропил-этиламина при перемешивании добавляли к 2,53 г m-бутилдифенилсилил-трифторметансульфоната. Полученную смесь далее перемешивали в течение часа при температуре 40°С. Затем добавляли 0,66 мл 1,2-диметоксиэтана и 1,82 г N,N-диизопропилэтиламин-трифторметансульфоната. Полученную смесь перемешивали при температуре 20°С до полного растворения, после чего добавляли 1,0 г рапамицина. Реакционную смесь продували азотом в течение 10 минут, нагревали до 48-50°С и перемешивали при данной температуре в течение 18 часов. Далее реакционную смесь охлаждали до 20°С и добавляли 0,2 мл пиридина и 16 мл гептана. Полученную смесь перемешивали в течение 10 минут. Выпавший осадок отделяли фильтрованием и промывали смесью 8,0 мл толуола и 8,0 мл гептана. Затем к фильтрату добавляли 1 мл 0,2% раствора 2,6-ди-m-бутил-4-метилфенола в гептане, и полученный неочищенный раствор 40-О-[2-(m-бутилдифенилсилил)окси]этил-рапамицина подвергали стадии снятия зашиты. Анализ методом ВЭЖХ полученного в результате раствора 40-О-[2-(m-бутилдифенилсилил,)окси]этил-рапамицина показал общий выход, равный 71% (932 мг).
Настоящее изобретение, иллюстративно описанное в данной заявке, может быть подходящим образом реализовано на практике в отсутствие любого элемента или элементов, ограничения или ограничений, конкретно не раскрытых в настоящей заявке. Так, например, термины «содержащий», «включающий» и т.п.следует понимать в широком смысле и без ограничений. Кроме того, применяемые в данной заявке термины и выражения были использованы в качестве терминов для описания, но не в качестве ограничения, и не следует применять данные термины и выражения, исключая любые эквиваленты представленных и описанных в данной заявке признаков или их частей, но при этом следует понимать, что в рамках заявленного объема настоящего изобретения возможны различные модификации. Таким образом, следует понимать, что хотя настоящее изобретение и описано конкретно посредством вариантов реализации и необязательных признаков, модификации и вариации изобретения, предложенного в качестве вариантов реализации, могут быть осуществлены специалистом в данной области техники, и такие модификации и вариации считаются находящимися в рамках настоящего изобретения.
В настоящей заявке изобретение описано широко и в общих чертах. Каждая из более узких интерпретаций и групп, входящих в общее описание изобретения, также является частью настоящего изобретения. Это включает общее описание изобретения с условием или отрицательным ограничением, исключающим любой объект из общего описания, независимо от того, упомянут ли данный исключенный материал конкретно в настоящей заявке.
Другие варианты реализации изобретения находятся в рамках нижеследующей формулы изобретения. Кроме того, когда признаки или аспекты настоящего изобретения описаны в виде групп Маркуша, специалистам в данной области будет понятно, что настоящее изобретение также тем самым описано в отношении каждого индивидуального представителя или подгруппы из групп Маркуша.
| название | год | авторы | номер документа |
|---|---|---|---|
| АЛКИЛИРОВАНИЕ СУЛЬФОНАТОМ АЛКИЛФТОРАЛКИЛА | 2014 |
|
RU2691728C2 |
| Новый способ получения эверолимуса | 2019 |
|
RU2716714C1 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ПРИМЕНЯЕМЫХ ДЛЯ СИНТЕЗА АНАЛОГОВ ВИТАМИНА D | 2005 |
|
RU2378252C2 |
| ПРОИЗВОДНОЕ 3'-ЭТИНИЛЦИТИДИНА | 2007 |
|
RU2441876C2 |
| ЭФИРЫ БОРОНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2325393C2 |
| 42-ОКСИМЫ И ГИДРОКСИЛАМИНЫ РАПАМИЦИНА, СПОСОБ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 1995 |
|
RU2172316C2 |
| АЛЬТЕРНАТИВНЫЕ СПОСОБЫ СИНТЕЗА ИНГИБИТОРОВ РЕНИНА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2005 |
|
RU2411230C2 |
| ГЕТЕРОТЕЛЕХЕЛАТНЫЙ БЛОК-СОПОЛИМЕР И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1996 |
|
RU2169742C2 |
| ЭФИРЫ КРЕМНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2375370C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ РАПАМИЦИНА | 2007 |
|
RU2448970C2 |
Изобретение относится к способу получения производного рапамицина формулы (I), который включает получение 2-(тризамещенного силил)оксиэтилтрифлата путем взаимодействия этиленоксида и тризамещенного силилтрифлата, взаимодействие полученного 2-(тризамещенного силил)оксиэтилтрифлата с рапамицином в присутствии молярного избытка органического основания и снятие защиты с получением производного рапамицина - соединения (I). Технический результат – высокий выход конечного продукта. 12 з.п. ф-лы, 4 ил., 5 пр.
1. Способ получения производного рапамицина формулы (I), включающий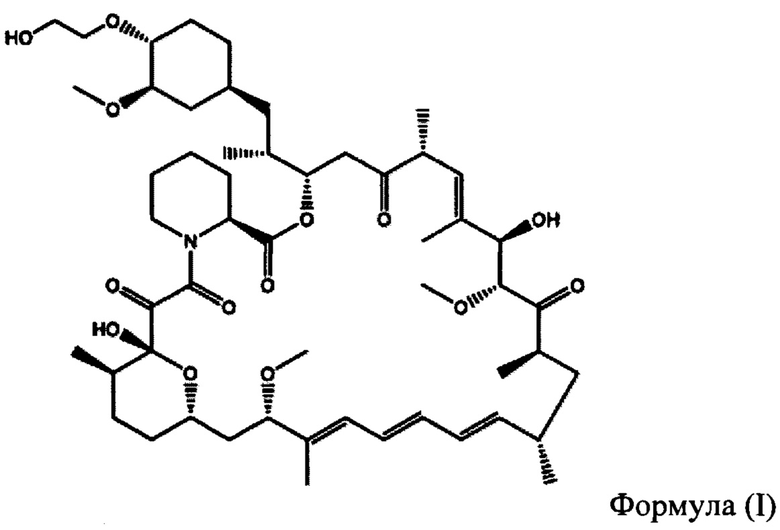
a) получение 2-(тризамещенного силил)оксиэтилтрифлата формулы 4 путем взаимодействия этиленоксида и тризамещенного силилтрифлата формулы 3
 ,
,
где R1, R2 и R3 независимо выбраны из группы, состоящей из С1-С12 алкила и C1-С12 арила;
b) взаимодействие 2-(тризамещенного силил)оксиэтилтрифлата формулы 4, полученного на стадии (а), с рапамицином в присутствии молярного избытка органического основания с получением защищенного производного рапамицина формулы 5; и
с) снятие защиты с защищенного производного рапамицина формулы 5 с получением производного рапамицина формулы (I),
при этом 2-(три-замещенный силил)оксиэтилтрифлат формулы 4, полученный на стадии (a), применяют без дополнительной очистки.
2. Способ по п. 1, отличающийся тем, что R1, R2 и R3 независимо выбраны из группы, состоящей из C1-С6 алкила и С6-С12 арила.
3. Способ по любому из пп. 1 или 2, отличающийся тем, что каждый из R1, R2 и R3 представляет собой изопропил; или каждый из R1 и R2 представляет собой метил, a R3 представляет собой m-бутил; или каждый из R1 и R2 представляет собой фенил, a R3 представляет собой m-бутил; или каждый из R1 и R2 представляет собой метил, a R3 представляет собой m-гексил.
4. Способ по любому из пп. 1-3, отличающийся тем, что стадию (а) проводят в органическом растворителе при температуре реакции 15-45°С.
5. Способ по п. 4, отличающийся тем, что органический растворитель представляет собой толуол.
6. Способ по любому из пп. 1-5, отличающийся тем, что этиленоксид применяют в количестве 1,1-1,2 молярных эквивалентов по отношению к тризамещенному силилтрифлату формулы 3.
7. Способ по любому из пп. 1-6, отличающийся тем, что стадию (b) проводят в органическом растворителе при температуре реакции 40-55°С.
8. Способ по п. 7, отличающийся тем, что органический растворитель представляет собой смесь 85-95% (об./об.) толуола и 5-15% (об./об.) диметоксиэтана.
9. Способ по любому из пп. 1-8, отличающийся тем, что 2-(тризамещенный силил)оксиэтилтрифлат формулы 4 применяют в количестве 4-12 молярных эквивалентов по отношению к рапамицину.
10. Способ по п. 1, отличающийся тем, что органическое основание, полученное на стадии (b), представляет собой N,N-диизопропилэтиламин.
11. Способ по любому из пп. 1-10, отличающийся тем, что защищенное производное рапамицина формулы 5, полученное на стадии (b), применяют без дополнительной очистки.
12. Способ по любому из пп. 1-11, отличающийся тем, что снятие защиты осуществляют посредством взаимодействия защищенного производного рапамицина формулы 5 с агентом, выбранным из группы, состоящей из соляной кислоты, уксусной кислоты, фторида тетра-н-бутиламмония и гидрофторида пиридина.
13. Способ по любому из пп. 1-12, дополнительно включающий очистку производного рапамицина формулы (I), полученного на стадии (с).
| CN 103848849 A, 11.06.2014 | |||
| Dilman, A | |||
| D | |||
| et al., Synthesis of C6F5-Substituted Aminoethanols via Acetate Ion Mediated C6F5-Group Transfer Reaction | |||
| Synthesis, n.3, 2006, p.489-495 | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| CN 102127092 A, 20.07.2011 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ РАПАМИЦИНА | 2007 |
|
RU2448970C2 |

