ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к производным 3'-этинилцитидина, обладающим отличными противоопухолевыми эффектами.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Рак, который характеризуется аномальной пролиферацией клеток, по-прежнему остается трудноизлечимым заболеванием, поэтому остро необходимо, чтобы в ближайшее время было разработано терапевтическое средство, эффективное для его лечения. Основываясь на знании о том, что для пролиферации клеток необходим биосинтез нуклеиновых кислот, были проведены активные исследования с целью получения антагониста метаболизма, способного ингибировать метаболизм нуклеиновых кислот. К настоящему времени были разработаны антагонисты метаболизма на основе цитидина, которые в настоящее время используются в клинике для лечения рака. Например, в качестве таких антагонистов могут быть упомянуты цитарабин (непатентный документ 1), анцитабин (непатентный документ 2) и гемцитабин (патентный документ 1), которые обладают противоопухолевой активностью посредством ингибирования синтеза ДНК. При этом содержащий 3'-этинилцитидин (ECyd) нуклеозид 3'-этинилпиримидин, который был разработан Matsuda et al., известен в качестве антагониста метаболизма нуклеиновых кислот, способного ингибировать синтез РНК (патентный документ 2 и непатентные документы 3 и 4).
Известно что ECyd обладает отличным противоопухолевым эффектом, более выраженным по сравнению с любым из лекарственных средств на основе фторпиримидина, в отношении 5 линий рака желудка, 3 линий рака толстой кишки, 2 линий рака поджелудочной железы, по 1 линии рака пищевода, рака желчного протока, рака легких, рака молочной железы и рака почки, что подтверждается способом применения, включающим его внутривенное введение (0,25 мг/кг непрерывно в течение 10 суток) «голым» мышам с подкожно трансплантированными опухолевыми клетками человека (непатентные документы 5 и 6).
Однако с такими внутривенно вводимыми лекарственными средствами существуют проблемы, такие как душевные и физические боли у больных раком и более высокая стоимость, связанная с амбулаторным лечением. Если было бы возможно заменить такие внутривенно вводимые лекарственные средства перорально вводимыми лекарственными средствами и обеспечить тем самым почти такой же терапевтический эффект, то считается, что качество жизни (QOL) пациентов значительно улучшится. Тем не менее при пероральном введении ECyd, скорее всего, обладает значительно менее высоким с противоопухолевым эффектом по сравнению с внутривенным введением. Поэтому существует повышенный спрос на разработку перорально вводимого лекарственного средства, обладающего противоопухолевой активностью, эквивалентной полученной при внутривенном введении ECyd.
[Патентный документ 1] Публикация патента Японии (kokoku) № 37394/1994
[Патентный документ 2] JP-B-3142874
[Непатентный документ 1] Evance, J. S. et al. Proc. Soc. Exp. Bio. Med., 106, 350 (1961)
[Непатентный документ 2] Hoshi, A. et al. Gann, 67, 725 (1972)
[Непатентный документ 3] Hattori, H. et al. J. Med. Chem. 39, 5005-5011 (1996)
[Непатентный документ 4] Hattori, H. et al. J. Med. Chem., 41, 2892-2902 (1998)
[Непатентный документ 5] Oncology Report Vol. 3, 1029 to 1034, 1996
[Непатентный документ 6] Motohiro Tanaka et al., Cancer & Chemotherapy Vol. 24-4, pp. 476 to 482, 1997
[Непатентный документ 7] Ludwig, P. S. et al. Synthesis (2002) 2387-2392
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
ПРОБЛЕМЫ, ПОДЛЕЖАЩИЕ РЕШЕНИЮ НАСТОЯЩИМ ИЗОБРЕТЕНИЕМ
Ввиду вышеизложенного задачей настоящего изобретения являлось создание производного ECyd, обладающего противоопухолевым эффектом, большим по сравнению с ECyd, при пероральном введении.
СРЕДСТВА РЕШЕНИЯ ПРОБЛЕМ
В попытке достичь упомянутую выше задачу авторы настоящего изобретения провели обширные исследования и обнаружили, что производное 3'-этинилцитидина, представленное следующей формулой (1), или его соль обладает отличной противоопухолевой активностью при пероральном введении. Настоящее изобретение было выполнено на основании этого открытия.
Соответственно, настоящее изобретение относится к производному 3'-этинилцитидина, представленному формулой (1):
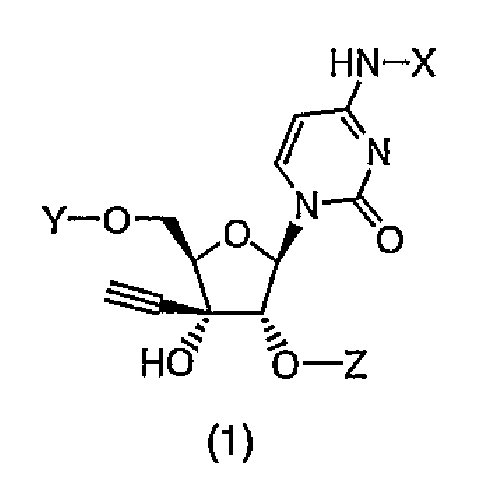
(в которой X представляет собой атом водорода, алкилкарбонильную группу, в которой алкильный фрагмент представляет собой неразветвленную или разветвленную C1-C6алкильную группу, которая может содержать заместитель, или алкоксикарбонильную группу, в которой алкокси-фрагмент представляет собой неразветвленную или разветвленную C1-C6алкоксигруппу, которая может содержать заместитель; один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляют собой неразветвленную или разветвленную C1-C10алкильную группу, которая может содержать заместитель, C3-C6циклоалкильную группу, которая может содержать заместитель, или C6-C14арильную группу, которая может содержать заместитель), или к его соли.
Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, представленное формулой (1), или его соль и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к противоопухолевому лекарственному средству, содержащему соединение, представленное формулой (1), или его соль и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к пероральному противоопухолевому лекарственному средству, содержащему соединение, представленное формулой (1), или его соль и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к применению соединения, представленного формулой (1), или его соли для производства лекарственного средства, в особенности противоопухолевого лекарственного средства.
Настоящее изобретение также относится к способу лечения опухоли, который включает введение соединения, представленного формулой (1), или его соли в эффективном количестве нуждающемуся в этом субъекту.
ЭФФЕКТЫ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Производное 3'-этинилцитидина или его соль по настоящему изобретению применимы в качестве противоопухолевого лекарственного средства, которое обладает отличной противоопухолевой активностью и прекрасной всасываемостью при пероральном введении.
НАИЛУЧШИЕ СПОСОБЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Производное 3'-этинилцитидина или его соль по настоящему изобретению представляет собой соединение, представленное формулой (1), причем соединение имеет химическую структуру, в которой силильную группу вводят по гидроксигруппе во 2'- и/или 5'-положении.
Одним из известных производных 3'-этинилцитидина, содержащих силильную группу во 2'- или 5'-положении, является 4-N-бензоил-2'-O-(трет-бутилдиметилсилил)-3'-C-триметилсилилэтинилцитидин (непатентный документ 7). Однако указанное соединение структурно отличается от производного 3'-этинилцитидина по настоящему изобретению тем, что содержит триметилсилильную группу в качестве заместителя в 3'-положении этинильной группы и содержит бензоильную группу в качестве заместителя в 4-N-положении. Кроме того, указанное соединение раскрыто исключительно в качестве промежуточного продукта синтеза, и о противоопухолевой активности данного соединения не сообщалось.
Примеры «неразветвленной или разветвленной C1-C6алкильной группы» в «алкилкарбонильной группе, в которой алкильный фрагмент представляет собой неразветвленную или разветвленную C1-C6алкильную группу, которая может содержать заместитель(и)», представленной X в формуле (1), включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил и н-гексил. Среди них предпочтительными являются неразветвленные C1-C6алкильные группы, а более предпочтительными являются метил и н-гексил. Примеры «заместителя» в «алкилкарбонильной группе, в которой алкильный фрагмент представляет собой неразветвленную или разветвленную C1-C6алкильную группу, которая может содержать заместитель(и)», представленной X в формуле (1), включают аминогруппу, в которой один или два атома водорода замещены неразветвленной или разветвленной C1-C6алкильной группой; например, метиламино, диметиламино и диэтиламино. Среди них предпочтительной является аминогруппа, в которой два атома водорода замещены неразветвленной или разветвленной C1-C6алкильной группой, а более предпочтительными являются диметиламино.
Примеры «неразветвленной или разветвленной C1-C6алкоксигруппы» в «алкоксикарбонильной группе, в которой алкокси-фрагмент представляет собой неразветвленную или разветвленную C1-C6алкоксигруппу, которая может содержать заместитель(и)», представленной X в формуле (1), включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси, н-пентилокси и н-гексилокси. Среди них предпочтительными являются неразветвленные или разветвленные C1-C4алкоксигруппы, а более предпочтительной является трет-бутокси. Примеры «заместителя» в «алкоксикарбонильной группе, в которой алкокси фрагмент представляет собой неразветвленную или разветвленную C1-C6алкокси группу, которая может содержать заместитель(и)», представленной X в формуле (1), включают неразветвленные или разветвленные C1-C6алкоксигруппы, например, метокси, а более предпочтительным является отсутствие заместителя.
Примеры «неразветвленной или разветвленной C1-C10алкильной группы» в «C1-C10алкилкарбонильной группе, которая может содержать заместитель(и)», представленной R1, R2, R3, R4, R5 или R6 в формуле (1), включают метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, н-гексил, тексил, н-октил и н-децил. Среди них предпочтительными являются неразветвленные C1-C8алкильные группы, такие как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, н-гексил, тексил и н-октил. Более предпочтительными примерами являются метил, изопропил, трет-бутил и тексил.
Примеры «заместителя» в «C1-C10алкилкарбонильной группе, которая может содержать заместитель(и)», представленной R1, R2, R3, R4, R5 или R6 в формуле (1), включают C3-C6циклоалкильные группы, такие как циклопропил, циклобутил, циклопентил и циклогексил; C1-C3алкоксигруппы, такие как метокси, этокси и изопропокси; C6-C14арильные группы, такие как фенил и нафтил; гидрокси; амино; атомы галогена, такого как хлор и бром; циано; и нитро.
Примеры «C3-C6циклоалкильной группы» в «C3-C6циклоалкильной группе, которая может содержать заместитель(и)», представленной R1, R2, R3, R4, R5 или R6 в формуле (1), включают циклопропильную, циклобутильную, циклопентильную и циклогексильную группы.
Примеры «заместителя» в «C3-C6циклоалкильной группе, которая может содержать заместитель(и)», представленной R1, R2, R3, R4, R5 или R6 в формуле (1), включают неразветвленные или разветвленные C1-C3алкильные группы, такие как метильная, этильная и изопропильная группы; C3-C6циклоалкильные группы, такие как циклопропил, циклобутил, циклопентил и циклогексил; C1-C3алкоксигруппы, такие как метокси, этокси и изопропокси; C6-C14арильные группы, такие как фенил и нафтил; гидрокси; амино; атомы галогена, такого как хлор и бром; циано; и нитро.
Примеры «C6-C14арильной группы» в «C6-C14арильной группе, которая может содержать заместитель(и)», представленной R1, R2, R3, R4, R5 или R6 в формуле (1), включают фенил и нафтил, причем предпочтительным является фенил.
Примеры «заместителя» в «C6-C14арильной группе, которая может содержать заместитель(и)», представленной R1, R2, R3, R4, R5 или R6 в формуле (1), включают неразветвленные или разветвленные C1-C3алкильные группы, такие как метильная, этильная и изопропильная группы; C3-C6циклоалкильные группы, такие как циклопропил, циклобутил, циклопентил и циклогексил; C1-C3алкоксигруппы, такие как метокси, этокси и изопропокси; C6-C14арильные группы, такие как фенил и нафтил; гидрокси; амино; атомы галогена, такого как хлор и бром; циано; и нитро.
Примеры групп (R1)(R2)(R3)Si- и (R4)(R5)(R6)Si-, которые идентичны друг другу или отличаются друг от друга и представлены Y и Z в формуле (1), включают трет-бутилдиметилсилил, триизопропилсилил, триизобутилсилил, диметил-н-октилсилил, диметилтексилсилил, триметилсилил, триэтилсилил, три-н-пропилсилил, три-н-бутилсилил, три-н-гексилсилил, н-пропилдиметилсилил, н-бутилдиметилсилил, изобутилдиметилсилил, н-пентилдиметилсилил, н-гексилдиметилсилил, н-децилдиметилсилил, (3,3-диметилбутил)диметилсилил, 1,2-диметилпропилдиметилсилил, ди-трет-бутилметилсилил, ди-н-бутилметилсилил, диэтилизопропилсилил, н-октилдиизопропилсилил, н-октилдиизобутилсилил, циклогексилдиметилсилил, дициклогексилметилсилил, изопропилдифенилсилил, трифенилсилил, диметилфенилсилил, трет-бутилдифенилсилил, метилдифенилсилил, дифенил(дифенилметил)силил, пара-толилдиметилсилил, бифенилдиметилсилил, бифенилдиизопропилсилил, три(2-бифенил)силил, три(орто-толил)силил, три(2-метоксифенил)силил, трибензилсилил, бензилдиметилсилил, фенэтилдиметилсилил, (3-фенилпропил)диметилсилил, пара-(трет-бутил)фенэтилдиметилсилил, фенэтилдиизопропилсилил, неофилдиметилсилил, бромметилдиметилсилил, хлорметилдиметилсилил, 4-хлорбутилдиметилсилил, (дихлорметил)диметилсилил, 3-хлорпропилдиметилсилил, 3,3,3-трифторпропилдиметилсилил, 1H,1H,2H,2H-перфтор-н-децилдиметилсилил, 1H,1H,2H,2H-перфтор-н-октилдиметилсилил, 3,3,4,4,5,5,6,6,6-нонафтор-н-гексилдиметилсилил, бис(хлорметил)метилсилил, пентафторфенилдиметилсилил, пентафторфенилпропилдиметилсилил, 3,5-бис(трифторметил)фенилдиметилсилил, 2-ацетоксиэтилдиметилсилил, 3-ацетоксипропилдиметилсилил, 3-метакрилоксипропилдиметилсилил, 3-цианопропилдиизопропилсилил, [3-(триметилсилокси)пропил]диметилсилил, н-бутилдиизопропилсилил, диизопропил-н-пропилсилил, диизопропил(2,2-диметилпропил)силил, (3-метилбутил)диизопропилсилил, (2-этилбутил)дициклопропилсилил, трет-амилдиэтилсилил, трет-бутилдиизобутилсилил, диэтил(3-метилпентан-3-ил)силил, изобутилдиизопропилсилил, диэтил(2-метилпентан-2-ил)силил, циклопропилдиизопропилсилил, дициклопропилизобутилсилил, диизопропил(3-метоксипропил)силил, (3-этоксипропил)диизопропилсилил, [3-(трет-бутилокси)пропил]диизопропилсилил, трет-бутилди(3-этоксипропил)силил и 3-феноксипропилдиметилсилил. В предпочтительных (R1)(R2)(R3)Si- и (R4)(R5)(R6)Si- одна или две группы из R1, R2 и R3, или из R4, R5 и R6, которые идентичны друг другу или отличаются друг от друга, представляют собой неразветвленные или разветвленные C1-C4алкильные группы, и оставшаяся одна или две группы, которые идентичны друг другу или отличаются друг от друга, представляют собой неразветвленные или разветвленные C2-C8алкильные группы или фенил. Примеры таких силилильных групп включают трет-бутилдиметилсилил, триэтилсилил, триизопропилсилил, диметил-н-октилсилил, диметилфенилсилил, диметилтексилсилил и трет-бутилдифенилсилил. В более предпочтительных (R1)(R2)(R3)Si- и (R4)(R5)(R6)Si- две группы из R1, R2 и R3, или из R4, R5 и R6, которые идентичны друг другу или отличаются друг от друга, представляют собой неразветвленные или разветвленные C1-C3алкильные группы, и оставшаяся одна группа представляет собой неразветвленную или разветвленную C2-C8алкильную группу. Примеры таких силилильных групп включают трет-бутилдиметилсилильную, триэтилсилильную, триизопропилсилильную, диметил-н-октилсилильную и диметилтексилсилильную группы. В особенно более предпочтительных (R1)(R2)(R3)Si- и (R4)(R5)(R6)Si- две группы из R1, R2 и R3, или из R4, R5 и R6, которые идентичны друг другу или отличаются друг от друга, представляют собой неразветвленные или разветвленные C1-C3алкильные группы, и оставшаяся одна группа представляет собой неразветвленную или разветвленную C3-C6алкильную группу. Примеры таких силилильных групп включают трет-бутилдиметилсилил, триизопропилсилил и диметилтексилсилил.
В формуле (1) один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-. Предпочтительно один из Y и Z представляет собой атом водорода, а другой представляет собой группу (R4)(R5)(R6)Si-.
Предпочтительно в соединении по настоящему изобретению, представленном формулой (1), X представляет собой атом водорода, алкилкарбонильную группу, в которой алкильной фрагмент представляет собой неразветвленную или разветвленную C1-C6алкильную группу, которая может содержать заместитель(и), или алкоксикарбонильную группу, в которой алкокси-фрагмент представляет собой неразветвленную или разветвленную C1-C6алкоксигруппу, которая может содержать заместитель(и); один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляет собой неразветвленную или разветвленную C1-C10алкильную группу, которая может содержать заместитель(и), C3-C6циклоалкильную группу, которая может содержать заместитель(и), или C6-C14арильную группу, которая может содержать заместитель(и).
Более предпочтительно в формуле (1) X представляет собой атом водорода, алкилкарбонильную группу, в которой алкильной фрагмент представляет собой неразветвленную или разветвленную C1-C6алкильную группу, которая в качестве заместителя(ей) может содержать моно- или дизамещенную неразветвленной или разветвленной C1-C6алкильной группой аминогруппу, или алкоксикарбонильную группу, в которой алкокси-фрагмент представляет собой неразветвленную или разветвленную C1-C6алкоксигруппу; один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляет собой неразветвленную или разветвленную C1-C10алкильную группу, C3-C6циклоалкильную группу или C6-C14арильную группу.
Еще более предпочтительно в формуле (1) X представляет собой атом водорода, алкилкарбонильную группу, в которой алкильной фрагмент представляет собой неразветвленную или разветвленную C1-C6алкильную группу, которая в качестве заместителя(ей) может содержать диметиламиногруппу, или алкоксикарбонильную группу, в которой алкокси-фрагмент представляет собой неразветвленную или разветвленную C1-C6алкоксигруппу; один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляет собой неразветвленную или разветвленную C1-C10алкильную группу, C3-C6циклоалкильную группу или C6-C14арильную группу.
Еще более предпочтительно в формуле (1) X представляет собой атом водорода; один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляет собой неразветвленную или разветвленную C1-C10алкильную группу, C3-C6циклоалкильную группу или C6-C14арильную группу.
Еще более предпочтительно в формуле (1) X представляет собой атом водорода; один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляет собой неразветвленную или разветвленную C1-C8алкильную группу или фенильную группу.
Еще более предпочтительно в формуле (1) X представляет собой атом водорода; один из Y и Z представляет собой атом водорода, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляет собой неразветвленную или разветвленную C1-C8алкильную группу или фенильную группу.
Еще более предпочтительно в формуле (1) X представляет собой атом водорода; один из Y и Z представляет собой атом водорода, а другой представляет собой трет-бутилдиметилсилильную группу, триэтилсилильную группу, триизопропилсилильную группу, диметил-н-октилсилильную группу, диметилфенилсилильную группу, диметилтексилсилильную группу или трет-бутилдифенилсилильную группу.
Конкретные примеры таких предпочтительных соединений включают следующие производные 3'-этинилцитидина (1)-(17) или их соли:
(1) 1-[5-O-(трет-бутилдиметилсилил)-3-C-этинил-β-D-рибофуранозил]цитозин,
(2) 1-[5-O-триэтилсилил-3-C-этинил-β-D-рибофуранозил]цитозин,
(3) 1-[5-O-триизопропилсилил-3-C-этинил-β-D-рибофуранозил]цитозин,
(4) 1-[5-O-(диметил-н-октилсилил)-3-C-этинил-β-D-рибофуранозил]цитозин,
(5) 1-[5-O-диметилфенилсилил-3-C-этинил-β-D-рибофуранозил]цитозин,
(6) 1-[5-O-диметилтексилсилил-3-C-этинил-β-D-рибофуранозил]цитозин,
(7) 1-[5-O-(трет-бутилдифенилсилил)-3-C-этинил-β-D-рибофуранозил]цитозин,
(8) 1-[2,5-бис-O-(трет-бутилдиметилсилил)-3-C-этинил-1-β-D-рибофуранозил]цитозин,
(9) 1-[2-O-(трет-бутилдиметилсилил)-3-C-этинил-1-β-D-рибофуранозил]цитозин,
(10) 1-(2,5-бис-O-триизопропилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин,
(11) 1-(2-O-триизопропилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин,
(12) 1-(2,5-бис-O-диметилтексилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин,
(13) 1-(2-O-диметилтексилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин,
(14) 1-[5-O-(трет-бутилдиметилсилил)-3-C-этинил-β-D-рибофуранозил]-4-N-гептаноилцитозин,
(15) 1-[5-O-(трет-бутилдиметилсилил)-3-C-этинил-β-D-рибофуранозил]-4-N-(трет-бутоксикарбонил)цитозин,
(16) 1-[5-O-(трет-бутилдиметилсилил)-3-C-этинил-β-D-рибофуранозил]-4-N-(N,N-диметилглицил)цитозин, и
(17) 1-[5-O-(триизопропилсилил)-3-C-этинил-β-рибофуранозил]-4-N-(N,N-диметилглицил)цитозин.
Конкретные примеры таких предпочтительных соединений включают следующие производные 3'-этинилцитидина или их соли:
(1) 1-[5-O-(трет-бутилдиметилсилил)-3-C-этинил-1-β-D-рибофуранозил]цитозин,
(3) 1-(5-O-триизопропилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин,
(6) 1-(5-O-диметилтексилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин,
(9) 1-[2-O-(трет-бутилдиметилсилил)-3-C-этинил-1-β-D-рибофуранозил]цитозин,
(11) 1-(2-O-триизопропилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин, и
(13) 1-(2-O-диметилтексилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин.
На соль производного 3'-этинилцитидина по настоящему изобретению не накладывается конкретного ограничения, поскольку соль является фармацевтически приемлемой. Примеры соли включают соли неорганических кислот, такие как гидрохлорид, гидробромид, сульфат, нитрат и фосфат; и соли органических кислот, такие как ацетат, пропионат, тартрат, фумарат, малеат, малат, цитрат, метансульфонат, пара-толуолсульфонат и трифторацетат. В зависимости от особенностей заместителя(ей) производное 3'-этинилцитидина по настоящему изобретению может образовывать оптические изомеры или геометрические изомеры, и настоящее изобретение охватывает такие оптические изомеры и геометрические изомеры при условии, что поддерживается стереоструктура скелета 3'-этинилцитидина, указанная в формуле (1). Такие изомеры могут быть использованы после разделения или использоваться в виде смеси. Производное 3'-этинилцитидина по настоящему изобретению также охватывает аморфные частицы, полиморфы и сольваты, такие как гидраты.
Производное 3'-этинилцитидина или его соль по настоящему изобретению может быть получено в соответствии со следующей схемой реакции, включающей стадии 1-7:
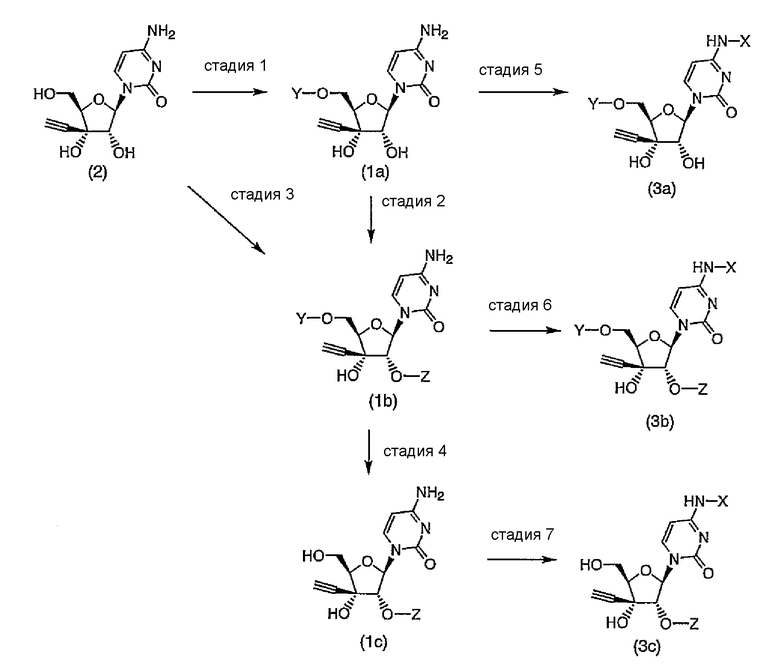
(где значения X, Y и Z определены выше).
(Стадия 1)
На стадии 1 осуществляют взаимодействие 3'-этинилцитидина, представленного формулой (2), или его соли с общеизвестным тризамещенным силилирующим агентом, таким как тризамещенный силилгалогенид, тризамещенный силилтрифлат или тризамещенный силилацетамид, представленным (R1)(R2)(R3)Si-W или (R4)(R5)(R6)Si-W (где W представляет собой атом галогена, трифторметансульфонилоксигруппу, ацетаминогруппу, и тому подобное, и значения R1-R6 определены выше), в результате чего может быть получено соединение, представленное формулой (1a).
Взаимодействие может быть осуществлено в соответствии с общеизвестным способом. На растворитель, используемый в реакции, конкретных ограничений не накладывается, и может быть использован любой растворитель при условии, что он инертен в реакции. Примеры растворителя включают дихлорметан, хлороформ, этилацетат, тетрагидрофуран, диоксан, диэтиловый эфир, бензол, толуол, N,N-диметилформамид и диметилсульфоксид. Указанные растворители могут использоваться по отдельности или в сочетании. При необходимости взаимодействие может быть осуществлено в присутствии основания. Примеры используемого основания включают органические амины, такие как имидазол, 1-метилимидазол, триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия и карбонат калия. Основание может быть использовано в качестве растворителя per se.
Тризамещенный силилгалогенид, который используется в реакции и представлен (R1)(R2)(R3)Si-W или (R4)(R5)(R6)Si-W, может быть получен общеизвестным способом. Например, осуществляют взаимодействие тригалогенсилана, моноалкилдигалогенсилана или диалкилмоногалогенсилана с алкиллитием или реактивом Гриньяра, представляющим интерес, с получением тем самым тризамещенного силана, представленного (R1)(R2)(R3)Si-H или (R4)(R5)(R6)Si-H, с которым затем осуществляют взаимодействие электрофильного реагента, такого как N-бромсукцинимид или N-хлорсукцинимид. При получении тризамещенного силана, представленного (R1)(R2)(R3)Si-H или (R4)(R5)(R6)Si-H, может быть использована добавка, такая как бромид меди. При необходимости тризамещенный силан, представленный (R1)(R2)(R3)Si-H или (R4)(R5)(R6)Si-H, и тризамещенный силилгалогенид, представленный (R1)(R2)(R3)Si-W или (R4)(R5)(R6)Si-W, могут быть выделены/очищены. В качестве альтернативы, полученный силан или силангалогенид могут использоваться на стадии 1 без проведения очистки.
В реакции упомянутый выше (R1)(R2)(R3)Si-W или (R4)(R5)(R6)Si-W используют в количестве приблизительно от 0,5 до приблизительно 10 моль, предпочтительно приблизительно от 1 до приблизительно 5 моль, а основание используют в количестве приблизительно от 0,5 до приблизительно 100 моль, предпочтительно приблизительно от 1 до приблизительно 10 моль, относительно 1 моля соединения, представленного формулой (2). Температура реакции составляет от -30 до 100°C, предпочтительно от 0 до 30°C, а продолжительность реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 20 часов. При необходимости полученное в ходе реакции соединение, представленное формулой (1a), может быть выделено/очищено. В качестве альтернативы, соединение может использоваться на следующей стадии без проведения очистки.
(Стадия 2)
На стадии 2 осуществляют взаимодействие производного 3'-этинилцитидина, представленного формулой (1a), с упомянутым выше тризамещенным силилирующим агентом, представленным (R1)(R2)(R3)Si-W или (R4)(R5)(R6)Si-W, в присутствии основания с получением соединения, представленного формулой (1b). Взаимодействие может осуществляться путем, сходным с описанным на стадии 1.
(Стадия 3)
По аналогии со стадией 1 на стадии 3 осуществляют взаимодействие 3'-этинилцитидина, представленного формулой (2), с упомянутым выше тризамещенным силилирующим агентом, представленным (R1)(R2)(R3)Si-W или (R4)(R5)(R6)Si-W, в присутствии основания с получением соединения, представленного формулой (1b). Температура реакции составляет от -30 до 150°C, предпочтительно от 0 до 100°C, а продолжительность реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 40 часов. При необходимости полученное в ходе реакции соединение, представленное формулой (1b), может быть выделено/очищено. В качестве альтернативы, соединение может использоваться на следующей стадии без проведения очистки.
(Стадия 4)
На стадии 4 соединение, представленное формулой (1c), получают из производного 3'-этинилцитидина, представленного формулой (1b), в кислой среде. В отношении кислот конкретных ограничений нет при условии, что она способна удалять заместитель Y. Примеры кислоты включают неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота; и органические кислоты, такие как трифторуксусная кислота, уксусная кислота, пропионовая кислота, муравьиная кислота, метансульфоновая кислота и пара-толуолсульфоновая кислота. Указанные кислоты могут быть смешаны с водой. При необходимости может использоваться растворитель. Примеры растворителя включают дихлорметан, хлороформ, этилацетат, тетрагидрофуран, диоксан, диэтиловый эфир, бензол, толуол, N,N-диметилформамид, диметилсульфоксид, метанол, этанол, н-пропанол, изопропанол и воду. Указанные растворители могут использоваться по отдельности или в сочетании. Температура реакции составляет от -30 до 150°C, предпочтительно от 0 до 100°C, а продолжительность реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 20 часов.
(Стадия 5)
На стадии 5 производное 3'-этинилцитидина, представленное формулой (1a), модифицируют группой X с получением соединения, представленного формулой (3a).
В том случае, если X представляет собой содержащую алкильную группу карбонильную группу, то модификацию проводят путем конденсации с галогенангидридом X-V (где V означает атом галогена), с ангидридом кислоты X-O-X или с карбоновой кислотой X-OH. Никаких ограничений на растворитель, используемый в реакции, нет и может быть использован любой растворитель при условии, что он инертен в реакции. Примеры растворителя включают дихлорметан, хлороформ, этилацетат, тетрагидрофуран, диоксан, диэтиловый эфир, бензол, толуол, N,N-диметилформамид, диметилсульфоксид и воду. Указанные растворители могут использоваться по отдельности или в сочетании. Если используются галогенангидрид X-V или ангидрид кислоты X-O-X, то может использоваться основание. Примеры используемого основания включают органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия и карбонат калия. Основание может быть использовано в качестве растворителя per se. В отношении реакции конденсации с карбоновой кислотой X-OH конкретных ограничений нет при условии, что в реакции происходит образование амида из карбоновой кислоты и амина. Например, может использоваться способ с использованием смешанных ангидридов, способ с использованием конденсирующего агента, и тому подобное. Примеры основания, используемого в способе с использованием смешанных ангидридов, включают органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия и карбонат калия. Основание может быть использовано в качестве растворителя per se. В качестве реагента для получения смешанного ангидрида с карбоновой кислотой X-OH могут использоваться изобутилхлоркарбонат, пивалоилхлорид, и тому подобное. Если используется конденсирующий агент, то могут использоваться карбодиимидные соединения, такие как дициклогексилкарбодиимид или гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида; или 1,1'-карбонилдиимидазол или сходное соединение. Примеры способствующих конденсации средств включают гидрат 1-гидроксибензотразола, N-гидроксисукцинимид, N-гидрокси-5-норборнен-2,3-дикарбоксимид и 4-(N,N-диметиламино)пиридин. При необходимости в реакции может использоваться основание. Примеры используемого основания включают органические амины, такие как триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия и карбонат калия. Основание может быть использовано в качестве растворителя per se.
Если в реакции используется галогенангидрид X-V или ангидрид кислоты X-O-X, то галогенангидрид X-V или ангидрид кислоты X-O-X используют в количестве приблизительно от 0,5 до приблизительно 20 моль, предпочтительно приблизительно от 1 до приблизительно 10 моль, относительно 1 моля соединения, представленного формулой (1a), а основание используют в количестве приблизительно от 0 до приблизительно 100 моль, предпочтительно приблизительно от 1 до приблизительно 20 моль. Температура реакции составляет от -30 до 100°C, предпочтительно от -10 до 30°C, а продолжительность реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 72 часов. Если конденсацию с карбоновой кислотой X-OH проводят по способу с использованием смешанного ангидрида, то карбоновую кислоту X-OH используют в количестве приблизительно от 0,5 до приблизительно 20 моль, предпочтительно приблизительно от 1 до приблизительно 10 моль; реагент для получения смешанного ангидрида используют в количестве приблизительно от 0,5 до приблизительно 20 моль, предпочтительно приблизительно от 1 до приблизительно 10 моль; и основание используют в количестве приблизительно от 0,5 до приблизительно 100 моль, предпочтительно приблизительно от 1 до приблизительно 20 моль, относительно 1 моля соединения, представленного формулой (1a). Температура реакции составляет от -30 до 100°C, предпочтительно от -10 до 30°C, а продолжительность реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 72 часов. Если используют конденсирующий агент, то карбоновую кислоту X-OH используют в количестве приблизительно от 0,5 до приблизительно 20 моль, предпочтительно приблизительно от 1 до приблизительно 10 моль; конденсирующий агент используют в количестве приблизительно от 0,5 до приблизительно 20 моль, предпочтительно приблизительно от 1 до приблизительно 10 моль; способствующее конденсации средство используют в количестве приблизительно от 0,1 до приблизительно 40 моль, предпочтительно приблизительно от 1 до приблизительно 10 моль; и основание используют в количестве от 0 до приблизительно 100 моль, предпочтительно от 0 до приблизительно 20 моль, относительно 1 моля соединения, представленного формулой (1a). Температура реакции составляет от -30 до 100°C, предпочтительно от -10 до 30°C, а продолжительность реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 72 часов. При необходимости полученное по любому из указанных способов соединение, представленное формулой (3a), может быть выделено/очищено. В качестве альтернативы, соединение может использоваться на следующей стадии без проведения очистки.
Если группа X представляет собой алкоксикарбонильную группу, то на реакцию на стадии 5 конкретное ограничение не накладывается при условии, что она представляет собой типовую реакцию. В одном типовом способе осуществляют взаимодействие производного 3'-этинилцитидина, представленного формулой (1a), или его соли с диалкилкарбонатом, алкилгалогенформиатом, алкил(пара-нитрофенил)карбонатом, сложным алкиловым эфиром 1H-имидазол-1-карбоновой кислоты, и тому подобное, которые представлены X-Q (где Q представляет собой алкоксикарбонилоксигруппу X-O, атом галогена, 4-нитрофенилоксигруппу, 1H-имидазол-1-ильную группу, и тому подобное). Взаимодействие может осуществляться в соответствии с общеизвестным способом. В отношении растворителя, используемого в реакции, конкретных ограничений нет и может быть использован любой растворитель при условии, что он инертен в реакции. Примеры растворителя включают дихлорметан, хлороформ, этилацетат, тетрагидрофуран, диоксан, диэтиловый эфир, бензол, толуол, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и воду. Указанные растворители могут использоваться по отдельности или в сочетании. При необходимости взаимодействие может быть осуществлено в присутствии основания. Примеры используемого основания включают органические амины, такие как имидазол, 1-метилимидазол, триметиламин, триэтиламин, трипропиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, лутидин и коллидин; и неорганические основания, такие как гидрокарбонат натрия, карбонат натрия и карбонат калия. Основание может быть использовано в качестве растворителя per se.
Алкилгалогенформиат X-Q, используемый в реакции, может быть получен общеизвестным способом. Например, галогенформиат может быть получен в реакции между трифосгеном и соответствующим алкиловым спиртом. По аналогии алкил(пара-нитрофенил)карбонат X-Q, используемый в реакции, может быть получен общеизвестным способом. Например, карбонат может быть получен в реакции между пара-нитрофенилхлороформиатом и соответствующим алкиловым спиртом. Кроме того, сложный алкиловый эфир 1H-имидазол-1-карбоновой кислоты X-Q, используемый в реакции, может быть получен общеизвестным способом. Например, сложный эфир может быть получен в реакции между 1,1'-карбонилдиимидазолом и соответствующим алкиловым спиртом. При необходимости алкилгалогенформиат, алкил(пара-нитрофенил)карбонат и сложный алкиловый эфир 1H-имидазол-1-карбоновой кислоты, представленные X-Q, могут быть выделены/очищены. В качестве альтернативы, указанные соединения могут использоваться на стадии 5 без проведения очистки.
В реакции упомянутое выше соединение X-Q используют в количестве приблизительно от 0,5 до приблизительно 20 моль, предпочтительно приблизительно от 1 до приблизительно 10 моль; а основание используют в количестве приблизительно от 0,5 до приблизительно 100 моль, предпочтительно приблизительно от 1 до приблизительно 20 моль, относительно 1 моля соединения, представленного формулой (1a). Температура реакции составляет от -30 до 100°C, предпочтительно от -10 до 30°C, а продолжительность реакции составляет от 0,1 до 100 часов, предпочтительно от 1 до 72 часов. При необходимости полученное в реакции соединение, представленное формулой (3a), может быть выделено/очищено. В качестве альтернативы, соединение может использоваться на следующей стадии без проведения очистки.
(Стадия 6)
По аналогии со стадией 5 на стадии 6 производное 3'-этинилцитидина, представленное формулой (1b), модифицируют группой X, с получением тем самым соединения, представленного формулой (3b).
(Стадия 7)
По аналогии со стадией 5 на стадии 7, производное 3'-этинилцитидина, представленное формулой (1c), модифицируют группой X, с получением тем самым соединения, представленного формулой (3c).
Полученное таким образом соединение по настоящему изобретению может быть преобразовано в его соли, в особенности фармацевтически приемлемые соли, общеизвестным способом.
Соединение или его соль по настоящему изобретению могут быть выделены и очищены общеизвестным способом разделения/очистки, таким как концентрирование, экстрагирование растворителем, фильтрация, перекристаллизация или хроматография.
Как описано посредством примеров, соединение или его соль по настоящему изобретению проявляет отличный противоопухолевый эффект при пероральном введении. Таким образом, оно представляет собой эффективное лекарственное средство, в особенности противоопухолевое лекарственное средство, для человека и млекопитающих.
При использовании соединения по настоящему изобретению в качестве лекарственного средства соединение смешивают с фармацевтически приемлемым носителем, и в соответствии с профилактическими и лечебными целями может быть выбран целый ряд вводимых форм. Могут использоваться различные вводимые формы, и примеры включают пероральные лекарственные средства, инъецируемые формы, суппозитории, мази и пластыри. Среди них предпочтительно используют пероральные формы. Указанные формы лекарственных средств могут быть получены любыми фармацевтическими методиками, известными в данной области техники.
Подлежащий использованию фармацевтически приемлемый носитель может представлять собой любые органические или неорганические носители, которые традиционно используются в качестве материалов для приготовления лекарственных средств. В твердых лекарственных формах носитель включают в состав в форме основы, смазки, связующего вещества, разрыхлителя или аналогичной добавки. В жидких лекарственных формах носитель включают в состав в виде растворителя, способствующего растворению средства, суспендирующего агента, придающего изотоничность агента, буфера, смягчающего агента или аналогичной добавки. При необходимости в состав могут быть также включены другие добавки, такие как консервант, антиоксидант, краситель и подсластитель.
При приготовлении твердой лекарственной формы для перорального введения соединение по настоящему изобретению смешивают с основой и необязательными добавками, такими как связующее вещество, разрыхлитель, смазка, краситель и подсластитель/ароматизатор, и преобразуют смесь традиционным способом в таблетки, покрытые оболочкой таблетки, гранулы, порошок, капсулы, и тому подобное. Указанные добавки могут быть обычно используемыми в данной области техники, и примеры включают лактозу, сахарозу, хлорид натрия, глюкозу, крахмал, карбонат кальция, каолин, микрокристаллическую целлюлозу и кремниевую кислоту (основы); воду, этанол, пропанол, простой сироп, жидкая глюкоза, жидкий крахмал, жидкий желатин, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилкрахмал, метилцеллюлозу, этилцеллюлозу, шеллак, фосфат кальция и поливинилпирролидон (связующие вещества); сухой крахмал, альгинат натрия, порошкообразный агар, гидрокарбонат натрия, карбонат кальция, лаурилсульфат натрия, моноглицерид стеариновой кислоты и лактозу (разрыхлители); очищенный тальк, соли стеариновой кислоты, боракс и полиэтиленгликоль (смазки); окись кремния и окись железа (красители); и сахарозу, апельсиновую цедру, лимонную кислоту и винную кислоту (подсластители/ароматизаторы).
При приготовлении жидкой лекарственной формы для перорального введения соединение по настоящему изобретению смешивают с добавками, такими как подсластитель, буфер, стабилизатор и ароматизатор, и преобразуют смесь традиционным способом в жидкую лекарственную форму для перорального введения, сироп, эликсир, и тому подобное. В этом случае подсластитель/ароматизатор может быть тем же, что и описанный выше. Примеры буфера включают цитрат натрия, а примеры стабилизатора включают трагакант, акацию и желатин.
При приготовлении инъецируемой формы соединение по настоящему изобретению смешивают с добавками, такими как регулятор pH, буфер, стабилизатор, придающий изотоничность агент и местный анестетик, и преобразуют смесь традиционным способом в инъецируемые формы для подкожного, внутримышечного и внутривенного введения. В этом случае примеры регулятора pH и буфера включают цитрат натрия, ацетат натрия и фосфат натрия, и примеры стабилизатора включают пиросульфит натрия, ЭДТА, тиогликолевую кислоту и тиомолочную кислоту. Примеры местного анестетика включают прокаина гидрохлорид и лидокаина гидрохлорид. Примеры придающего изотоничность агента включают хлорид натрия и глюкозу.
При приготовлении лекарственного средства в форме суппозитория соединение по настоящему изобретению смешивают с известным в данной области техники носителем для приготовления лекарственного средства, таким как полиэтиленгликоль, ланолин, какао-масло и триглицерид жирной кислоты, и необязательным поверхностно-активным веществом, таким как Tween™, и преобразуют смесь традиционным способом в суппозитории.
При приготовлении мази соединение по настоящему изобретению смешивают, в соответствии с требованиями, с обычно используемыми добавками, такими как основа, стабилизатор, увлажнитель, консервант, и тому подобное, смесь получают и преобразуют в лекарственное средство традиционным способом. Примеры основы мази включают вазелиновое масло, белый вазелин, осветленный пчелиный воск, октилдодециловый спирт и парафин. Примеры консерванта включают метил-пара-гидроксибензоат, этил-пара-гидроксибензоат и пропил-пара-гидроксибензоат.
При приготовлении лекарственного средства в форме пластыря упомянутую выше мазь, крем, гель, пасту или сходный материал традиционным способом наносят на стандартную подложку. Примеры подходящих подложек включают тканый и нетканый материал из хлопка, штапельное волокно или синтетическое волокно; и пленки и листовой пенопласт, сделанные из мягкого винилхлорида, полиэтилена или полиуретана.
Разовая доза соединения по настоящему изобретению, подлежащая включению в состав любой из упомянутых выше форм лекарственного средства, варьирует в зависимости от состояния пациентов, которым необходимо вводить соединение по настоящему изобретению, формы лекарственных средств или других факторов. Как правило, разовая доза предпочтительно составляет приблизительно от 0,05 до 1000 мг для пероральных лекарственных форм, приблизительно от 0,01 до 500 мг для инъецируемых форм и приблизительно от 1 до 1000 мг для суппозиториев. Суточная доза лекарственного средства в любой из упомянутых выше лекарственных форм варьирует в зависимости от состояния, массы тела, возраста, пола, и тому подобное, пациента, а потому она необязательно может быть определена немедленно. Тем не менее, как правило, суточная доза для взрослого человека составляет приблизительно от 0,05 до 5000 мг, предпочтительно от 0,1 до 1000 мг. Суточную дозу предпочтительно вводят один раз в сутки или дробно приблизительно от двух до четырех раз в сутки.
Примеры заболеваний (в случае злокачественных опухолей), которые могут лечиться посредством введения лекарственного средства, содержащего соединение по настоящему изобретению, включают рак шеи и головы, рак пищевода, рак желудка, рак толстой кишки, рак прямой кишки, рак печени, рак желчного пузыря/желчного протока, рак поджелудочной железы, рак легких, рак молочной железы, рак яичников, рак шейки матки, рак тела матки, рак почки, рак мочевого пузыря, рак предстательной железы, опухоль яичек, остеосаркому и саркому мягких тканей, лейкоз, злокачественную лимфому, множественную миелому, рак кожи и опухоль головного мозга.
ПРИМЕРЫ
Настоящее изобретение будет далее подробно описано со ссылкой на сравнительные примеры, примеры (демонстрационные примеры), примеры фармакологических тестов и подготовительные примеры. Однако ни один из не должен истолковываться как ограничивающий настоящее изобретение.
Пример 1
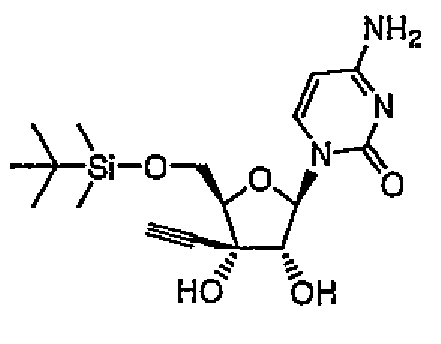
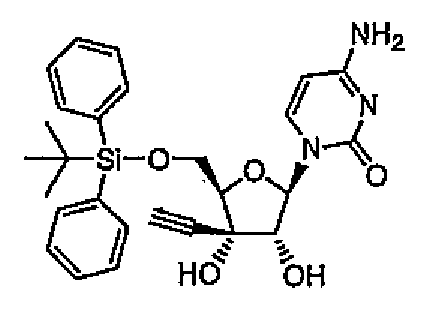
1-[5-O-(трет-Бутилдиметилсилил)-3-C-этинил-β-D-рибофуранозил]цитозин (соединение 1)
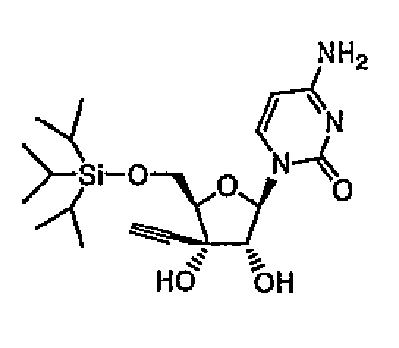
трет-Бутилдиметилсилилхлорид (12,5 г, 82,5 ммоль) постепенно добавляли при охлаждении на льду к раствору 3'-этинилцитидина (здесь и далее в этом документе называемого ECyd) (20 г, 75 ммоль) и имидазола (12,8 г, 188 ммоль) в N,N-диметилформамиде (здесь и далее в этом документе называемого DMF) (75 мл), и перемешивали смесь в течение 5 часов при комнатной температуре. После завершения реакции растворитель выпаривали при пониженном давлении, и добавляли к остатку этилацетат. Смесь промывали насыщенным водным гидрокарбонатом натрия (100 мл) и насыщенным солевым раствором (100 мл), и сушили над сульфатом магния. Сульфат магния удаляли путем фильтрации. Фильтрат перемешивали при комнатной температуре, и собирали выпавшие в осадок кристаллы путем фильтрации с получением соединения 1 (11,5 г, 52%).
Пример 2
1-[5-O-Триэтилсилил-3-C-этинил-β-D-рибофуранозил]цитозин (соединение 2)
Повторяли методику примера 1 за исключением того, что вместо использованного в примере 1 трет-бутилдиметилсилилхлорида использовали триэтилсилилхлорид, и синтезировали соединение 2.
Пример 3
1-[5-O-Триизопропилсилил-3-C-этинил-β-D-рибофуранозил]цитозин (соединение 3)
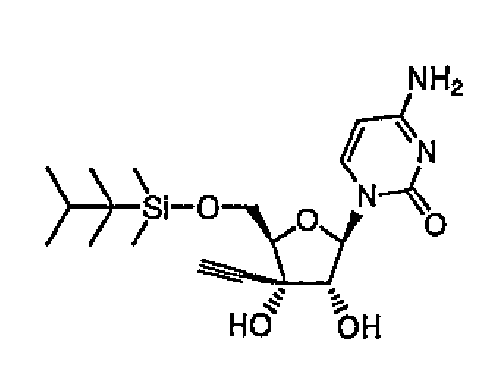
Повторяли методику примера 1 за исключением того, что вместо использованного в примере 1 трет-бутилдиметилсилилхлорида использовали триизопропилсилилхлорид, и синтезировали соединение 3.
Пример 4
1-[5-O-(Диметил-н-октилсилил)-3-C-этинил-β-D-рибофуранозил]цитозин (соединение 4)
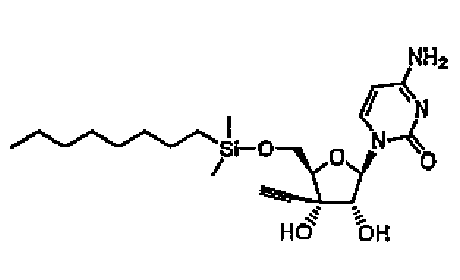
Повторяли методику примера 1 за исключением того, что вместо использованного в примере 1 трет-бутилдиметилсилилхлорида использовали диметил-н-октилсилилхлорид, и синтезировали соединение 4.
Пример 5
1-[5-O-Диметилфенилсилил-3-C-этинил-β-D-рибофуранозил]цитозин (соединение 5)
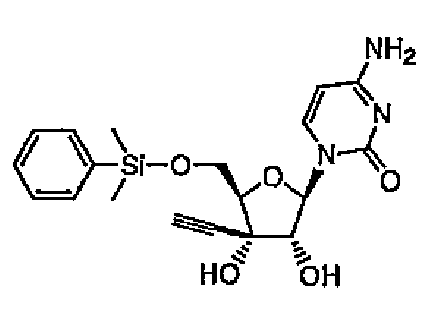
Повторяли методику примера 1 за исключением того, что вместо использованного в примере 1 трет-бутилдиметилсилилхлорида использовали диметилфенилсилилхлорид, и синтезировали соединение 5.
Пример 6
1-[5-O-Диметилтексилсилил-3-C-этинил-β-D-рибофуранозил]цитозин (соединение 6)
Повторяли методику примера 1 за исключением того, что вместо использованного в примере 1 трет-бутилдиметилсилилхлорида использовали диметилтексилсилилхлорид, и синтезировали соединение 6.
Пример 7
1-[5-O-(терт-Бутилдифенилсилил)-3-C-этинил-β-D-рибофуранозил]цитозин (соединение 7)
Повторяли методику примера 1 за исключением того, что вместо использованного в примере 1 трет-бутилдиметилсилилхлорида использовали трет-бутилдифенилсилилхлорид, и синтезировали соединение 7.
Пример 8
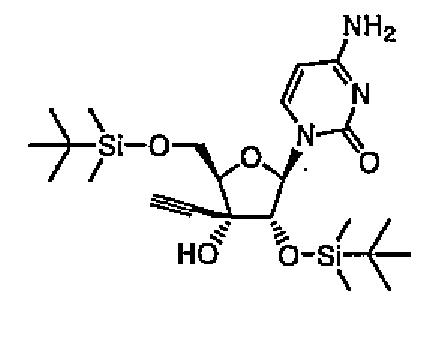
1-[2,5-бис-O-(трет-Бутилдиметилсилил)-3-C-этинил-1-β-D-рибофуранозил]цитозин (соединение 8)
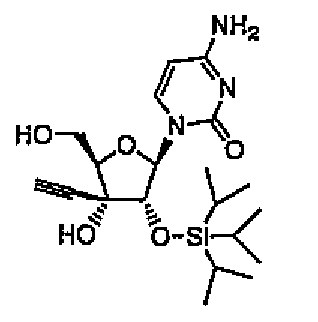
ECyd (5,00 г, 18,7 ммоль) растворяли в DMF (19 мл), добавляли к раствору имидазол (3,82 г, 56,1 ммоль) и трет-бутилдиметилсилилхлорид (6,20 г, 41,1 ммоль), а затем перемешивали в течение 4 часов при комнатной температуре под потоком азота. Растворитель выпаривали, и растворяли остаток в этилацетате. Раствор промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия, и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (0-5% метанол/хлороформ). Затем продукт кристаллизовали из гексана/эфира с получением соединения 8 (6,36 г, 12,8 ммоль, 69%) в виде белого твердого вещества.
Пример 9
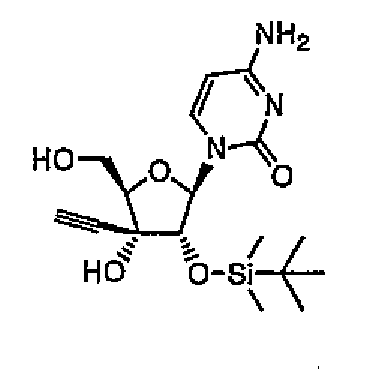
1-[2-O-(трет-Бутилдиметилсилил)-3-C-этинил-1-β-D-рибофуранозил]цитозин (соединение 9)
Соединение 8 (2,00 г, 4,03 ммоль) растворяли в тетрагидрофуране (здесь и далее в этом документе называемом THF) (20 мл), и добавляли к раствору 80% водную трифторуксусную кислоту (20 мл), а затем перемешивали в течение 1 часа при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и остаток трижды совместно кипятили с этанолом, и продукт распределяли между этилацетатом и водой. Органический слой промывали насыщенным водным гидрокарбонатом натрия и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель выпаривали, и очищали остаток колоночной хроматографией на силикагеле (0-6% метанол/хлороформ), а затем кристаллизовали из гексана/эфира с получением соединения 9 (645 мг, 1,69 ммоль, 42%) в виде белого твердого вещества.
Пример 10
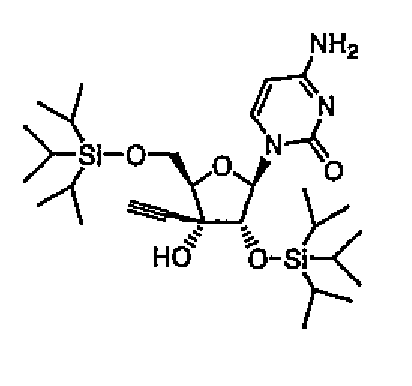
1-(2,5-бис-O-Триизопропилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин (соединение 10)
ECyd (5,00 г, 18,7 ммоль) растворяли в DMF (19 мл), добавляли к раствору имидазол (5,73 г, 84,2 ммоль) и триизопропилсилилхлорид (12,8 мл, 59,8 ммоль), а затем перемешивали в течение ночи при комнатной температуре под потоком азота. Растворитель выпаривали, и остаток растворяли в этилацетате. Раствор промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия, и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (0-6% метанол/хлороформ) с получением соединения 10 (5,05 г, 8,70 ммоль, 46%) в виде бесцветной пены.
Пример 11
1-(2-O-Триизопропилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин (соединение 11)
ECyd (4,01 г, 15 ммоль) растворяли в DMF (30 мл), добавляли к раствору имидазол (6,39 г, 93,9 ммоль) и триизопропилсилилхлорид (8,02 мл, 37,5 ммоль), а затем перемешивали под потоком азота в течение 3 часов при комнатной температуре, а затем в течение 24 часов при 50°C. Реакционную смесь распределяли между этилацетатом и водой, органический слой пять раз промывали водой, а затем сушили над безводным сульфатом натрия. Растворитель выпаривали, и получали 10,1 г остатка. 1,9 г полученного остатка растворяли в метаноле (5,2 мл), добавляли к раствору воду (0,58 мл) и метансульфоновую кислоту (347 мкл, 4,76 ммоль), а затем перемешивали в течение 1 часа при 40°C. Реакционную смесь распределяли между этилацетатом и насыщенным водным гидрокарбонатом натрия, органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель выпаривали, и остаток кристаллизовали из трет-бутилметилового эфира/воды/диизопропилового эфира. Полученные кристаллы снова кристаллизовали из метанола/воды/триэтиламина с получением соединения 11 (961 мг, 80%) в виде белого твердого вещества.
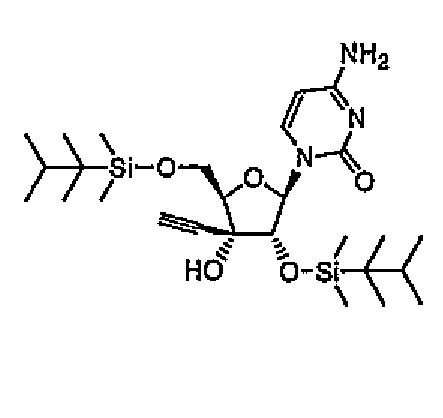
Пример 12
1-(2,5-бис-O-Диметилтексилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин (соединение 12)
ECyd (2,67 г, 10 ммоль) растворяли в DMF (100 мл), добавляли к раствору имидазол (4,50 г, 66 ммоль) и диметилтексилсилилхлорид (5,90 г, 33 ммоль), а затем перемешивали в течение 48 часов при комнатной температуре под потоком азота. Растворитель выпаривали, и растворяли остаток в этилацетате. Раствор промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом натрия, и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (0-3% метанол/хлороформ) с получением соединения 12 (4,34 г, 79%) в виде бесцветной пены.
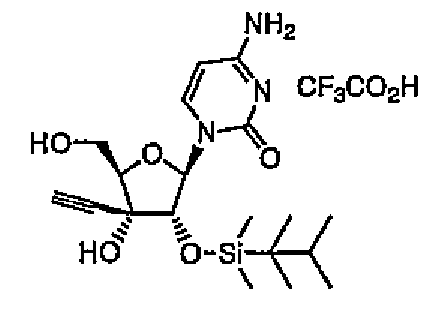
Пример 13
Трифторуксуснокислая соль 1-(2-O-диметилтексилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозина (соединение 13)
Соединение 12 (2,0 г, 3,6 ммоль) растворяли в THF (20 мл), добавляли к раствору 80% водную трифторуксусную кислоту (20 мл), а затем перемешивали в течение 4 часов при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении, и остаток трижды совместно кипятили с этанолом. К кипящему остатку добавляли хлороформ, и собирали выпавшее в осадок белое твердое вещество путем фильтрации с получением соединения 13 (1,54 г, 81%) в виде белого твердого вещества.
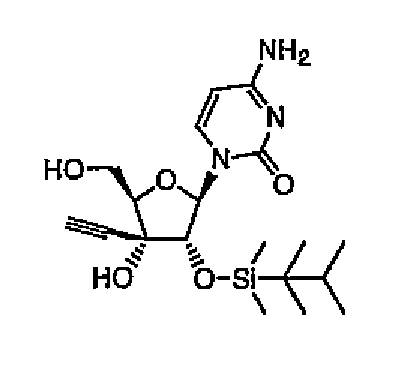
Пример 14
1-(2-O-Диметилтексилсилил-3-C-этинил-1-β-D-рибофуранозил)цитозин (соединение 14)
Соединение 13 (1,0 г, 1,9 ммоль) растворяли в смеси 5% метанола/хлороформа (100 мл), и промывали раствор насыщенным водным гидрокарбонатом натрия. Органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным сульфатом натрия. Растворитель выпаривали, и остаток кристаллизовали из гексана/эфира с получением соединения 14 (690 мг, 1,68 ммоль, 88%) в виде белого твердого вещества.
Пример 15
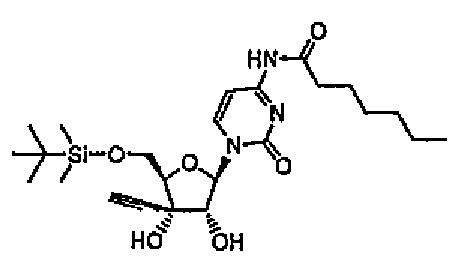
1-[5-O-(трет-Бутилдиметилсилил)-3-C-этинил-β-D-рибофуранозил]-4-N-гептаноилцитозин (соединение 15)
К смеси диоксана (14 мл) и воды (5 мл) добавляли соединение 1 (1,27 г, 3,3 ммоль) и ангидрид гептановой кислоты (1,8 мл, 6,8 ммоль), и перемешивали смесь в течение суток при 100°C. После завершения реакции реакционную смесь экстрагировали этилацетатом (50 мл), и нейтрализовали органический слой 1 н. водным гидроксидом натрия. Полученную смесь промывали насыщенным солевым раствором (50 мл), а затем сушили над сульфатом магния. Сульфат магния удаляли путем фильтрации, и выпаривали растворитель при пониженном давлении. Остаток очищали хроматографией на силикагеле (смесь 4% метанола/хлороформа). Элюат концентрировали, и перекристаллизовывали остаток из изопропанола/гексана с получением соединения 15 (0,55 г, 33%).
Пример 16
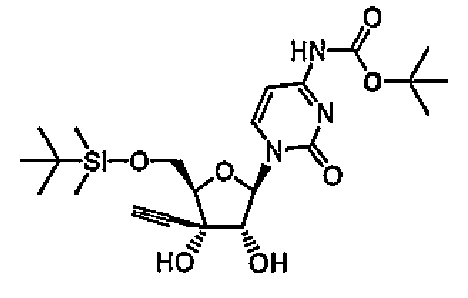
1-[5-O-(трет-Бутилдиметилсилил)-3-C-этинил-β-D-рибофуранозил]-4-N-(трет-бутоксикарбонил)цитозин (соединение 16)
При охлаждении на льду к тетрагидрофурану (20 мл) добавляли соединение 1 (1,65 г, 4,3 ммоль) и ди-трет-бутилдикарбонат (1,4 г, 6,5 ммоль), и перемешивали смесь в течение суток при 50°C. Реакционную смесь концентрировали, и очищали остаток хроматографией на силикагеле (смесь 4% метанола/хлороформа). Элюат концентрировали, и перекристаллизовывали остаток из гексана с получением соединения 16 (0,52 г, 26%).
Пример 17
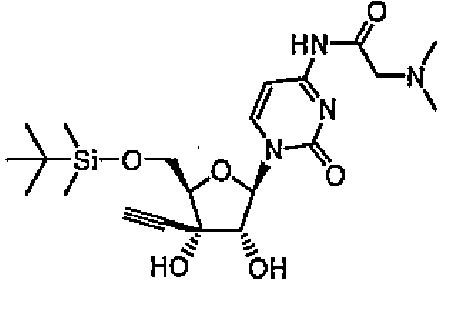
1-[5-O-(трет-Бутилдиметилсилил)-3-C-этинил-β-D-рибофуранозил]-4-N-(N,N-диметилглицил)цитозин (соединение 17)
К DMF (20 мл) при охлаждении на льду добавляли соединение 1 (1,9 г, 5 ммоль), N,N-диметилглицин (1,0 г, 10 ммоль), N,N-диметиламинопиридин (0,1 г, 0,8 ммоль) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (1,9 г, 10 ммоль), и перемешивали смесь в течение суток при 40°C. Реакционную смесь концентрировали, и добавляли к остатку этилацетат. Органический слой промывали насыщенным водным гидрокарбонатом натрия (50 мл) и насыщенным солевым раствором (50 мл), а затем сушили над сульфатом натрия. Сульфат магния удаляли путем фильтрации, и концентрировали фильтрат. Остаток очищали хроматографией на силикагеле (смесь 4% метанола/хлороформа). Элюат концентрировали, и перекристаллизовывали остаток из гексана с получением соединения 17 (0,46 г, 20%).
Пример 18
1-[5-O-(Триизопропилсилил)-3-C-этинил-β-рибофуранозил]-4-N-(N,N-диметилглицил)цитозин (соединение 18)
Повторяли методику примера 17 за исключением того, что вместо использованного в примере 17 соединения 1 использовали соединение 3, и синтезировали соединение 18 (выход: 47%).
Структура и физические свойства соединений, полученных в описанных выше примерах, представлены в таблицах 1-5.
(2H, с, исчезал в D2O), 5,93 (1H, д, J=7,1 Гц), 5,87 (1H, с, исчезал в D2O), 5,75 (1H, д, J=6,8 Гц, исчезал в D2O), 5,70 (1H, д, J=7,6 Гц), 4,03 (1H, дд, J=6,8 Гц, 7,1 Гц), 3,77-3,93 (3H, м), 3,55 (1H, с), 0,89 (9H, с), 0,09 (3H, с), 0,08 (3H, с)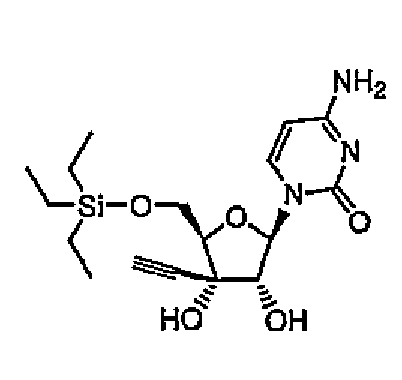
Фармакологический тест
Противоопухолевый эффект соединения по настоящему изобретению при пероральном введении крысам Donryu с подкожно имплантированными опухолевыми клетками
Клетки линии саркомы Йошиды (асцитная опухоль крыс), интраперитонеально субкультивированные в крысах Donryu (Charles River Laboratories Japan, Inc.), имплантировали под кожу спины крыс Donryu (возраст 5 недель) в количестве 2×104 клеток/0,2 мл. Через 4 суток после имплантации каждую крысу взвешивали, и делили крыс на группы таким образом, что средняя масса тела между группами была приблизительно равной (7 крыс в группе).
Каждое производное ECyd растворяли или суспендировали в 0,5% растворе гидроксипропилметилцеллюлозы, и перорально вводили полученный раствор или суспензию каждой крысе ежедневно с момента разбиения на группы один раз в сутки в течение 7 суток в дозе 12 мкмоль/кг/сутки. Производное ECyd оценивали трижды. В качестве контроля в каждом тесте использовали ECyd в количестве, эквимолярном по отношению к производному ECyd.
Через 7 суток после разбиения на группы у крыс из группы с введением фармацевтического средства измеряли массу опухоли. Массу опухоли также измеряли у крыс, которым фармацевтическое средство не вводили (группа без лечения). Рассчитывали среднюю массу опухоли в группе с введением фармацевтического средства и в группе без лечения. Степень ингибирования роста опухоли (IR) определяли с использованием следующего уравнения, оценивая тем самым противоопухолевый эффект.
IR(%)=[1-(TWtest)/(TWcont)]×100 (уравнение 1)
[где TWtest и TWcont представляют собой среднюю массу опухоли в группе с введением фармацевтического средства и в группе без лечения, соответственно].
Результаты теста представлены в таблице 6.
Как представлено в таблице 6, было обнаружено, что при пероральном введении соединение по настоящему изобретению обладает отличным противоопухолевым эффектом по сравнению с ECyd.
Отдельно соединение по настоящему изобретению перорально вводили самцам крыс Donryu, и измеряли содержание ECyd в сыворотке крови. Было обнаружено, что по сравнению с ECyd соединение по настоящему изобретению обнаруживается в крови в очень высоких концентрациях. Например, в очень высоких концентрациях в крови обнаруживались соединения 1, 3, 4, 9, 11, 12, 14, 15, 17 и 18.
Пример приготовления 1
Таблетки
Таблетки, каждая массой 250 мг и содержащая представленную выше композицию, получали обычным способом.
Пример приготовления 2
Гранулы
Гранулы (1000 мг/саше), содержащие представленную выше композицию, получали обычным способом.
Пример приготовления 3
Капсулы
Капсулы, каждая массой 193 мг и содержащая представленную выше композицию, получали обычным способом.
Пример приготовления 4
Инъекционная форма
для инъекций
(2 мл/ампула)
Инъецируемую жидкость, содержащую представленную выше композицию, получали обычным способом.
Пример приготовления 5
Сироп
Сироп, содержащий представленную выше композицию, получали обычным способом.
Пример приготовления 6
Суппозитории
Суппозитории, содержащие представленную выше композицию, получали обычным способом.
ПРИМЕНИМОСТЬ В ПРОМЫШЛЕННЫХ УСЛОВИЯХ
Производное 3'-этинилцитидина или его соль по настоящему изобретению представляет собой противоопухолевое лекарственное средство, которое обладает отличной противоопухолевой активностью и прекрасной всасываемостью при пероральном введении. Поэтому могут быть решены проблемы, связанные с внутривенным введением лекарственного средства пациентам, т.е. душевные и физические боли и чрезвычайно высокая стоимость амбулаторного лечения, и тем самым ожидается значительное улучшение качества жизни (QOL) пациентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| 3'-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ | 1995 |
|
RU2130029C1 |
| НОВЫЙ ПИРИМИДИНОВЫЙ НУКЛЕОЗИД ИЛИ ЕГО СОЛЬ | 2006 |
|
RU2395517C2 |
| КОМПЛЕКСЫ | 2015 |
|
RU2682680C2 |
| ФОСФОИНДОЛЫ КАК ИНГИБИТОРЫ ВИЧ | 2005 |
|
RU2393163C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ФТОР-2'-АЛКИЛЗАМЕЩЕННЫХ ИЛИ ДРУГИХ ЗАМЕЩЕННЫХ РИБОФУРАНОЗИЛПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2433124C2 |
| ПИРИМИДИН-НУКЛЕОЗИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2116306C1 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2466729C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2525392C2 |
| КОМПЛЕКСЫ | 2015 |
|
RU2684934C2 |
| ПРОИЗВОДНЫЕ 5'-ДЕЗОКСИЦИТИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1998 |
|
RU2238278C2 |
Изобретение относится к производным 3′-этинилцитидина, представленным формулой (1):
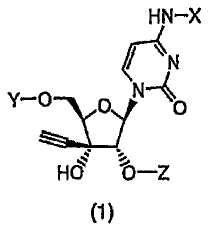
(в которой X представляет собой атом водорода, алкилкарбонильную группу, в которой алкильный фрагмент представляет собой неразветвленную или разветвленную C1-С6алкильную группу, которая в качестве заместителя(ей) может содержать моно- или дизамещенную неразветвленной или разветвленной С1-С6алкильной группой аминогруппу, или алкоксикарбонильную группу, в которой алкокси-фрагмент представляет собой неразветвленную или разветвленную C1-С6алкоксигруппу;
один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляют собой неразветвленную или разветвленную C1-С10алкильную группу или С6-С14арильную группу), или к их солям. Изобретение также относится к производному 3′-этинилцитидина, выбранному из соединений (1)-(17), к фармацевтической композиции, к противоопухолевому лекарственному средству, к пероральному противоопухолевому лекарственному средству, к применению производного 3′-этинилцитидина, а также к способу лечения опухоли. Технический результат - получение новых биологически активных соединений, обладающих противоопухолевой активностью. 7 н. и 7 з.п. ф-лы, 12 табл.
1. Производное 3′-этинилцитидина, представленное формулой (1):
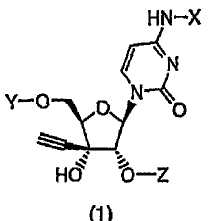
(в которой X представляет собой атом водорода, алкилкарбонильную группу, в которой алкильный фрагмент представляет собой неразветвленную или разветвленную С1-С6алкильную группу, которая в качестве заместителя(ей) может содержать моно- или дизамещенную неразветвленной или разветвленной С1-С6алкильной группой аминогруппу, или алкоксикарбонильную группу, в которой алкокси-фрагмент представляет собой неразветвленную или разветвленную С1-С6алкоксигруппу;
один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляют собой неразветвленную или разветвленную С1-С10алкильную группу или С6-С14арильную группу) или его соль.
2. Производное 3′-этинилцитидина или его соль по п.1, где в формуле (1) X представляет собой атом водорода, алкилкарбонильную группу, в которой алкильный фрагмент представляет собой неразветвленную или разветвленную С1-С6алкильную группу, которая в качестве заместителя может содержать диметиламиногруппу, или алкоксикарбонильную группу, в которой алкокси-фрагмент представляет собой неразветвленную или разветвленную С1-С6алкоксигруппу; один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляют собой неразветвленную или разветвленную С1-С10алкильную группу или С6-С14арильную группу.
3. Производное 3′-этинилцитидина или его соль по п.1, где в формуле (1) X представляет собой атом водорода; один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляют собой неразветвленную или разветвленную C1-С10алкильную группу или С6-С14арильную группу.
4. Производное 3′-этинилцитидина или его соль по п.1, где в формуле (1) X представляет собой атом водорода; один из Y и Z представляет собой атом водорода или группу (R1)(R2)(R3)Si-, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R1, R2, R3, R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляют собой неразветвленную или разветвленную С1-С8алкильную группу или фенильную группу.
5. Производное 3′-этинилцитидина или его соль по п.1, где в формуле (1) X представляет собой атом водорода; один из Y и Z представляет собой атом водорода, а другой представляет собой группу (R4)(R5)(R6)Si-; и каждый R4, R5 и R6, которые могут быть одинаковыми или отличаться друг от друга, представляют собой неразветвленную или разветвленную С1-С8алкильную группу или фенильную группу.
6. Производное 3′-этинилцитидина или его соль по п.1, где в формуле (1) X представляет собой атом водорода; один из Y и Z представляет собой атом водорода, а другой представляет собой трет-бутилдиметилсилильную группу, триэтилсилильную группу, триизопропилсилильную группу, диметил-н-октилсилильную группу, диметилфенилсилильную группу, диметилтексилсилильную группу или трет-бутилдифенилсилильную группу.
7. Производное 3′-этинилцитидина, выбранное из следующих соединений (1)-(17), или его соль:
(1) 1-[5-О-(трет-бутилдиметилсилил)-3-С-этинил-β-D-рибофуранозил]цитозин,
(2) 1-[5-О-триэтилсилил-3-С-этинил-β-D-рибофуранозил]цитозин,
(3) 1-[5-О-триизопропилсилил-3-С-этинил-β-D-рибофуранозил]цитозин,
(4) 1-[5-О-(диметил-н-октилсилил)-3-С-этинил-β-D-рибофуранозил]цитозин,
(5) 1-[5-O-диметилфенилсилил-3-С-этинил-β-D-рибофуранозил]цитозин,
(6) 1-[5-О-диметилтексилсилил-3-С-этинил-β-D-рибофуранозил]цитозин,
(7) 1-[5-О-(трет-бутилдифенилсилил)-3-С-этинил-β-D-рибофуранозил]цитозин,
(8) 1-[2,5-бис-О-(трет-бутилдиметилсилил)-3-С-этинил-1-β-D-рибофуранозил]цитозин,
(9) 1-[2-О-(трет-бутилдиметилсилил)-3-С-этинил-1-β-D-рибофуранозил] цитозин,
(10) 1-(2,5-бис-О-триизопропилсилил-3-С-этинил-1-β-D-рибофуранозил)цитозин,
(11) 1-(2-О-триизопропилсилил-3-С-этинил-1-β-D-рибофуранозил)цитозин,
(12) 1-(2,5-бис-О-диметилтексилсилил-3-С-этинил-1-β-D-рибофуранозил)цитозин,
(13) 1-(2-О-диметилтексилсилил-3-С-этинил-1-β-D-рибофуранозил)цитозин,
(14) 1-[5-О-(трет-бутилдиметилсилил)-3-С-этинил-β-D-рибофуранозил]-4-N-гептаноилцитозин,
(15) 1-[5-О-(трет-бутилдиметилсилил)-3-С-этинил-β-D-рибофуранозил]-4-N-(трет-бутоксикарбонил)цитозин,
(16) 1-[5-O-(трет-бутилдиметилсилил)-3-С-этинил-β-D-рибофуранозил]-4-N-(N,N-диметилглицил)цитозин и
(17) 1-[5-О-(триизопропилсилил)-3-С-этинил-β-рибофуранозил]-4-N-(N,N-диметилглицил)цитозин.
8. Фармацевтическая композиция, обладающая противоопухолевой активностью, содержащая производное 3′-этинилцитидина или его соль по любому из пп.1-7 и фармацевтически приемлемый носитель.
9. Противоопухолевое лекарственное средство, содержащее производное 3′-этинилцитидина или его соль по любому из пп.1-7 и фармацевтически приемлемый носитель.
10. Пероральное противоопухолевое лекарственное средство, содержащее производное 3′-этинилцитидина или его соль по любому из пп.1-7 и фармацевтически приемлемый носитель.
11. Применение производного 3′-этинилцитидина или его соли по любому из пп.1-7 для производства противоопухолевого лекарственного средства.
12. Применение по п.11, где лекарственное средство представляет собой пероральное противоопухолевое лекарственное средство.
13. Способ лечения опухоли, отличающийся тем, что включает введение производного 3′-этинилцитидина или его соли по любому из пп.1-8 в эффективном количестве нуждающемуся в этом субъекту.
14. Способ лечения опухоли по п.13, где введение означает пероральное введение.
| WO 9618636 A1, 20.06.1996 | |||
| JP 10298194 A, 10.11.1998 | |||
| ПРОИЗВОДНЫЕ 5'-ДЕЗОКСИЦИТИДИНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1998 |
|
RU2238278C2 |

