Изобретение относится к способу получения органических соединений, например, включая соли органических соединений и способы их получения.
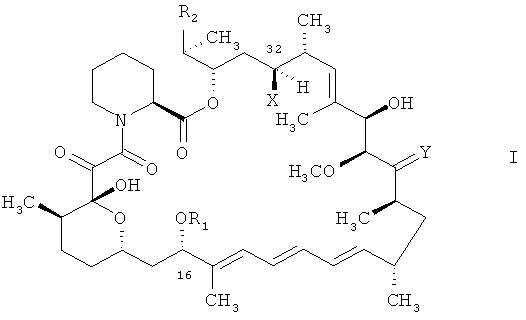
В международной заявке на изобретение WO 9641807 описаны, между прочим, соединения формулы
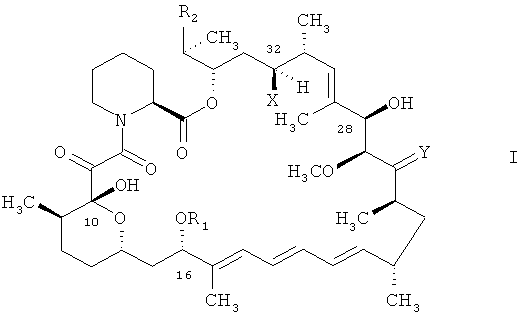
в которых
R1 является алкилом, алкенилом, алкинилом, гидроксиалкилом, гидроксиалкенилом, гидроксиалкинилом, бензилом, алкоксибензилом или хлорбензилом,
R2 выбран из группы формулы
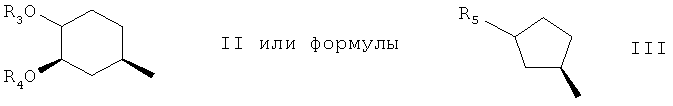
где R3 выбран из водорода, алкила, алкенила, алкинила, арила, арилалкила, гидроксиарилалкила, гидроксиарила, гидроксиалкила, дигидроксиалкила, гидроксиалкоксиалкила, гидроксиалкиларилалкила, дигидроксиалкиларилалкила, алкоксиалкила, алкилкарбонилоксиалкила, аминоалкила, алкиламиноалкила, алкоксикарбониламиноалкила, алкилкарбониламиноалкила, арилсульфамидоалкила, аллила, дигидроксиалкилаллила, диоксоланилаллила, карбалкоксиалкила и алкилсилила,
R4 является водородом, метилом или образует совместно с R3 (С2-С6)алкилен,
R5 является R6O-СН2-,
где R6 выбран из водорода, алкила, алкенила, алкинила, арила, алкилкарбонила, арилкарбонила, гетероарилкарбонила, гидроксиалкилкарбонила, аминоалкилкарбонила, формила, арилалкила, гидроксиарилалкила, гидроксиарила, гидроксиалкила, дигидроксиалкила, гидроксиалкоксиалкила, гидроксиалкиларилалкила, дигидроксиалкиларилалкила, алкоксиалкила, алкилкарбонилоксиалкила, аминоалкила, алкиламиноалкила, алкоксикарбониламиноалкила, алкилкарбониламиноалкила, арилсульфамидоалкила, аллила, дигидроксиалкилаллила, диоксоланилаллила, карбалкоксиалкила,
R7CO-,
где R7 выбран из водорода, алкила, гидроксигруппы, алкоксигруппы, арилоксигруппы, аминогруппы, алкиламиногруппы или N,N-дизамещенной аминогруппы, в которой заместители выбраны из алкила, арила или арилалкила,
R8NCH-,
где R8 является алкилом, арилом, аминогруппой, алкиламиногруппой, ариламиногруппой, гидроксигруппой, алкоксигруппой или арилсульфониламиногруппой, -O-СН-О- или замещенным диоксиметилином,
Y выбран из О, (Н, ОН) и (H, OR9),
где R9 выбран из (С1-С4)алкила, алкилкарбонила, арилкарбонила, гетероарилкарбонила, гидроксиалкилкарбонила, аминоалкилкарбонила, формила или арила и
Х является водородом или ОН.
В международной заявке на изобретение WO 9641807 также описаны способы получения соединений формулы I, в которых остатки отвечают вышеприведенным определениям. Ключевой стадией в получении соединений формулы I, в которых Х является водородом, является восстановление карбонильной группы в положении 32 в соединении формулы I, в котором карбонильная группа присоединена к атому углерода в положении 32 вместо Х и водорода.
Согласно международной заявке на изобретение WO 9641807 подобный способ может быть осуществлен согласно нескольким различным способам, например для получения соединения формулы I, в котором Х является водородом, посредством восстановления карбонильной функциональной группы в положении 32 соединения формулы
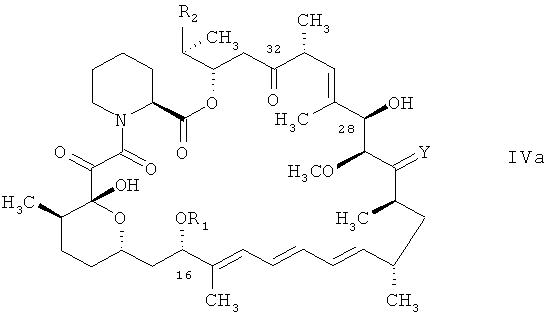
в котором R1, R2 и Y отвечают вышеприведенным определениям, в защищенной или незащищенной форме и, когда это требуется, удаления наличествующих защитных групп, а также, например, необязательного преобразования получаемого соединения формулы I, в котором R1 является алкилом, приводящего к соединению формулы I, в котором R1 является иной группой, нежели алкил. Например,
(а) восстановление до 32-дезоксо-соединения формулы I может быть легко осуществлено с помощью следующей цепочки взаимодействий, а именно
1) посредством взаимодействия соединения формулы IVa, предпочтительно в защищенной форме, с гидридом, например диизобутилалюминийгидридом или, предпочтительно, три(трет-бутокси)алюминийгидридом лития, с целью получения соответствующего 32-гидроксипроизводного (ОН-группа в положении 32 циклической структуры),
с последующей стадией 2), а именно преобразованием 32-гидроксипроизводного в соответствующее 32-галоидпроизводное, например, 32-бром- или (предпочтительно) 32-иодпроизводное, которое затем восстанавливают, например, с помощью гидрида, до искомого 32-дезоксопроизводного и, когда это требуется, удаляют в получаемом соединении защитные группы. Могут быть задействованы другие реагенты, такие как применяемые для восстановления галогенидов, включающие, например, металлы низкой валентности (т.е. литий, натрий, магний и цинк) и гидриды металлов (гидриды алюминия, боргидриды, силаны, гидриды меди) (см. Comprehensive Organic Transformations, R.C.Larock, VCH Publishers Inc., New York, 1989, стр.18-20, разделы 1.5.1. и 1.5.2.) (альтернативно, восстановление галогенидов может быть осуществлено с помощью водорода или источника водорода (например, муравьиной кислоты или ее соли) в присутствии подходящего металлического катализатора (т.е. никеля Ренея, металлического палладия или комплексов палладия, комплексов родия или рутения) (см. Comprehensive Organic Transformations, R.C.Larock, VCH Publishers Inc., New York, 1989, стр.20-24, раздел 1.5.3.)).
(б) могут также быть задействованы известные способы, применяемые для преобразования спирта в соответствующее дезокси-соединение. Эти способы включают, например, прямое восстановление или восстановление промежуточного фосфорсодержащего соединения, сульфоната, тиокарбоната, тиокарбамата или ксантата и описаны, например, в Comprehensive Organic Transformations, R.C.Larock, VCH Publishers Inc., New York, 1989, стр.27-31, разделы 1.9.1.-1.9.4); или
в) упомянутое восстановление может быть осуществлено через образование тозилгидразона с последующей обработкой бораном, например катехолбораном, или через образование дитиана с последующим восстановлением подходящим способом, например, с помощью никеля Ренея или гидрида, например гидрида трибутилолова. Могут быть задействованы и другие известные способы преобразования кетона в соответствующий алкан. Подобные способы включают, например, прямое восстановление (см. Comprehensive Organic Transformations, R.C.Larock, VCH Publishers Inc., New York, 1989, стр.35-37, раздел 1.12.1.) или восстановление через гидразоны (Comprehensive Organic Transformations, R.C.Larock, VCH Publishers Inc., New York, 1989, стр.37-38, раздел 1.12.2) и через серо- и селеносодержащие производные (Comprehensive Organic Transformations, R.C.Larock, VCH Publishers Inc., New York, 1989, стр.34-35, разделы 1.10. и 1.1L).
Проиллюстрированный в международной заявке на изобретение WO 9641807 способ получения 32-дезоксорапамицина состоит в получении 32-гидроксипроизводного рапамицина в защищенной форме, преобразовании гидроксигруппы в положении 32 в мезилатную группу, преобразовании таковой мезилатной группы в иодный заместитель, обработке получаемого защищенного соединения формулы I, в котором Х является иодом, гидридом трибутилолова и последующей обработке раствором триэтилборана в гексане, причем получаемое защищенное 32-дезоксо-производное очищают с помощью колоночной хроматографии с целью получения защищенного 32-дезоксо-производного в твердом виде и обрабатывают получаемое защищенное 32-дезоксо-производное серной кислотой в метаноле, а затем NaHCO3 с целью получения незащищенного 32-дезоксо-производного, каковое незащищенное 32-дезоксо-производное может быть получено в кристаллической форме.
Согласно настоящему изобретению был неожиданно обнаружен усовершенствованный способ получения соединения формулы I, в котором Х является водородом, a R1, R2 и Y отвечают вышеприведенным определениям, например, способ, применимый в техническом масштабе.
В одном из вариантов осуществления настоящего изобретения его объектом является способ получения соединения формулы I, в котором Х является водородом, a R1, R2 и Y отвечают вышеприведенным определениям, включающий
А) обработку или
а) соединения формулы
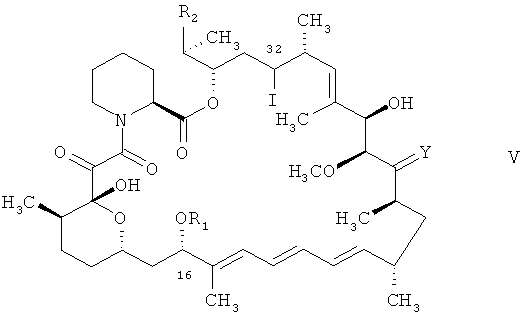
в котором R1, R2 и Y отвечают определениям для соединения формулы I и в котором наличествующие реакционноспособные группы находятся в незащищенной или в защищенной форме, предпочтительно в защищенной форме, трис(триметилсилил)силаном, (С6-С18)алкилмеркаптаном, например трет-додецилмеркаптаном и α,α'-азоизобутиронитрилом в органическом растворителе или
б) соединения формулы
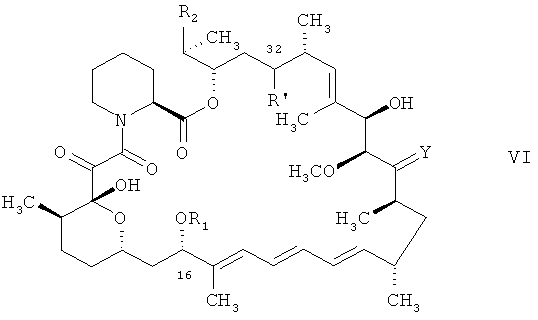
в котором R1, R2 и Y отвечают определениям для соединения формулы I и в котором R' является остатком арилтионокарбоната или арилтионокарбамата, каковой арилтионокарбонат или арилтионокарбамат присоединен к атому углерода в положении 32 циклической структуры через атом кислорода, а также в котором наличествующие реакционноспособные группы находятся в незащищенной или в защищенной форме, предпочтительно в защищенной форме, трис(триметилсилил)силаном и α,α'-азоизобутиронитрилом в органическом растворителе,
Б) расщепление защитных групп, если таковые наличествуют,
В) выделение соединения формулы I, в котором R1, R2 и Y отвечают вышеприведенным определениям, и
Г) необязательное преобразование получаемого соединения формулы I в другое соединение формулы I, например преобразование соединения формулы I, в котором R1 является алкилом, в другое соединение формулы I, в котором R1 отвечает вышеприведенным определениям, но является иной группой, нежели алкил.
В соединении формулы VI арилтионокарбонат или арилтионокарбамат, присоединенный к атому углерода в положении 32 циклической структуры через атом кислорода, включает группу формулы
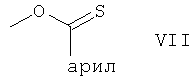
в которой арил является (С6-С18)арилоксигруппой (остаток арилтионокарбоната) или арил является арильным 5- или 6-членным гетероциклилом, включающим от 1 до 4 гетероатомов, выбранных из азота, кислорода и серы, при том условии, что гетероциклил содержит хотя бы один атом азота, каковой гетероциклил присоединен к группе C=S в группе формулы VII через гетероциклический атом азота (остаток арилтионокарбамата).
(С6-С18)арил включает фенил, например незамещенный фенил или замещенный фенил, предпочтительно незамещенный фенил или фенил, замещенный группами, остающимися инертными в условиях реакции, например фенил, замещенный галоидом, например фтором.
Арильный 5- или 6-членным гетероциклил, включающий от 1 до 4 гетероатомов, выбранных из азота, кислорода и серы, при том условии, что гетероциклил содержит хотя бы один атом азота, может быть необязательно конденсирован с другим циклом (системой). Арильный гетероциклил предпочтительно содержит 5 кольцевых атомов и предпочтительно является имидазолилом.
В соединении формулы I упоминание заместителя, включающего «алк» или «алкил» относится к C1-С10 алифатическому заместителю, необязательно прерываемому кислородным мостиком, а упоминание заместителя, включающего «ар» или «арил», относятся к моноциклическому С4-С14 ароматическому заместителю, необязательно являющемуся гетероциклическим и необязательно замещенному.
Примеры вышеупомянутых необязательно замещенных остатков «ар» или «арил» могут включать, например, фенил, бензил, толил, пиридил и им подобные.
В тех случаях, когда R1 является хлорбензилом или алкоксибензилом, заместитель предпочтительно находится в орто-положении.
В тех случаях, когда R7CO- является N,N-дизамещенным карбамоилом, он может являться, например, N-метил-N-(2-пиридин-2-илэтил)карбамоилом, (4-метилпиперазин-1-ил)карбонилом или (морфолин-4-ил)карбонилом.
В тех случаях, когда R8 является замещенным диоксиметилином, он может являться, например, О,О-(алкилен)диоксиметилином, т.е. где оба атома кислорода 2 связаны с алкиленовой группой.
В соединении формулы I следующие значения являются предпочтительными как по отдельности, так и в любом сочетании или частичном сочетании.
1. R1 является (С1-С10)алкилом, (С3-С10)алк-2-енилом, гидрокси(С3-С10)алк-2-енилом, (С3-С10)алк-2-инилом, гидрокси(С3-С10)алк-2-инилом или (С1-С10)алкокси(С1-С10)алкилом, предпочтительно, (С1-С6)алкилом или (С3-С6)алк-2-инилом, более предпочтительно, (С1-С4)алкилом, наиболее предпочтительно, метилом,
2. R1 является (С3-С6)алк-2-инилом, как, например, R1 является 2-пропинилом или пент-2-иниилом, предпочтительно, пент-2-инилом,
3. Y является кислородом, (Н, ОН) или (H, (С1-С4)алкоксигруппой), предпочтительно, кислородом,
4. R2 является группой формулы II;
5. В группе формулы II, R3 является водородом, гидрокси(С1-С6)алкилом, гидрокси(С1-С6)алкокси(С1-С6)алкилом, ((С1-С6)алкил)карбониламино-(С1-С6)алкилом, (С1-С6)алкокси(С1-С6)алкоксигруппой или амино(С1-С6)алкилом, предпочтительно, водородом, гидроксиэтилом, гидроксипропилом, гидроксиэтоксиэтилом, метоксиэтилом или ацетиламиноэтилом, в особенности, водородом, когда R1 является алкинилом,
6. В группе формулы II R4 является метилом,
7. R2 является остатком формулы III, в котором R5 является
-R6OCH2-, где R6 выбран из водорода, (С1-С6)алкила, (С3-С6)алк-2-енила, (С3-С6)алк-2-инила, арила, (С1-С6)алкилкарбонила, арилкарбонила, гидрокси(С1-С6)алкила, (С1-С6)алкокси(С1-С6)алкила или амино(С1-С6)алкила,
-R7CO-, где R7 выбран из водорода, гидроксигруппы, (С1-С6)алкоксигруппы, аминогруппы, (С1-С6)алкиламиногруппы, остатка аминокислоты или N,N-дизамещенной аминогруппы, в которой заместители выбраны
(а) из (С1-С6)алкила или арила или
(б) из гетероциклической структуры
-R8NCH-, где R8 является алкилом, арилом, аминогруппой, алкиламиногруппой, ариламиногруппой, гидроксигруппой, алкоксигруппой или арилсульфониламиногруппой, -O-СН-О- или замещенным диоксиметилином.
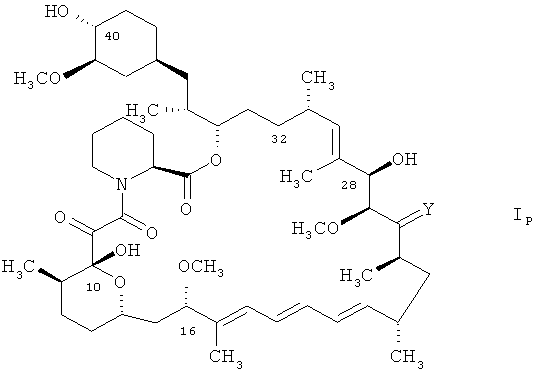
Особенно предпочтительные соединения формулы I включают, например, 16-O-пент-2-инил-32-дезоксорапамицин, 16-O-пент-2-инил-32-дезоксо-40-O-(2-гидроксиэтил)рапамицин и 32-дезоксорапамицин, как то 32-дезоксорапамицин формулы
Соединения формулы I могут проявлять изомерию и, соответственно, будут существовать другие их изомерные формы. Следует понимать, что настоящее изобретение включает в свой объем соединение формулы I в любой индивидуальной изомерной форме и любую изомерную смесь, например соединение формулы I в форме индивидуальных изомеров и индивидуальные изомеры формулы
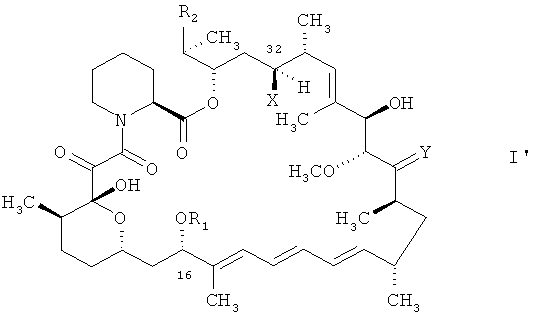
в которой R1, R2, Y и Х отвечают вышеприведенным определениям, равно как и их изомерные смеси.
Индивидуальные изомеры могут быть разделены подходящим способом, например общепринятым способом.
Термин «соединение в защищенной форме», как он употребляется в контексте, относится к описанному в контексте соединению указанной формулы, такому как соединение формулы I, I', Ip, IVa, V и VI, в котором реакционноспособные группы снабжены защитой. Подходящие защитные группы включают применимые в соответствующем случае защитные группы, как то, например, общепринятые группы. Реакционноспособные группы включают, например, гидроксигруппы, такие как гидроксигруппа в положении 28, и, в тех случаях, когда R2 является соединением формулы II, а R3 является водородом, гидроксигруппа, присоединенная к циклической системе в положении 40. Было обнаружено, что гидроксигруппа в положении 10 циклической структуры в соединении формулы V не вступает во взаимодействия в реакционных условиях и не нуждается в защите.
Защитные группы для гидроксигрупп и способы защиты и снятия защитной группы описаны, например, в монографии «Protective Groups in Organic Synthesis», второе издание, T.W.Greene и P.G.M.Wuts, John Wiley & Sons, New York, 1991, глава 2, и приведенных там ссылках. Предпочтительными защитными группами для гидроксигрупп являются, например, триорганосилильные группы, такие как три(С1-С6)алкилсилил (например, триметилсилил, триэтилсилил), триизопропилсилил, изопропилдиметилсилил, трет-бутилдиметилсилил, триарилсилил (например, трифенилсилил) или триарилалкилсилил (например, трибензилсилил). Снятие защиты может быть осуществлено в мягких кислотных условиях.
Предпочтительно, гидроксигруппы в положениях 28 и 40 циклической структуры в соединении формулы I, I', Ip, IVa, V и VI защищены триорганосилильными группами, более предпочтительно, триэтилсилилом.
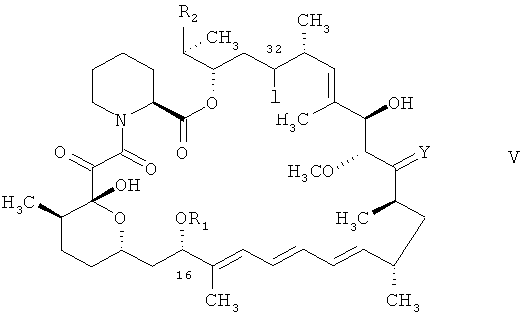
Соединения формулы V, например, в защищенной форме, применяемые в качестве исходного вещества согласно настоящему изобретению, могут быть получены соответствующим способом, например, согласно общепринятому способу, например, аналогично ему. Соединения формулы V, например, в защищенной форме и их получение известны, например, из международной заявки на изобретение WO 9641807.
Соединения формулы VI, например, в защищенной форме, применяемые в качестве исходного вещества согласно настоящему изобретению, могут быть получены соответствующим способом, например, согласно общепринятому способу, например, аналогично ему, или же как это описано в контексте.
В другом варианте осуществления настоящего изобретения его объектом является способ получения соединения формулы VI, в котором R', R1, R2 и Y отвечают вышеприведенным определениям, например, соединения формулы VI в защищенной форме, каковой способ включает обработку соединения формулы I, в котором Х является гидроксигруппой, a R1, R2 и Y отвечают вышеприведенным определениям, например, в защищенной форме, хлорангидридом арилтиономуравьиной кислоты или арилтионокарбаматом в реакционноспособной форме, например хлорангидридом арилтиономуравьиной кислоты формулы
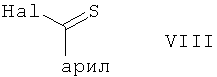
в которой Hal является галоидом, например, бромом, хлором, а арил отвечает вышеприведенному определению, или, в случае арилтионокарбамата, обработку соединением формулы
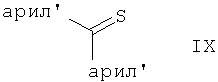
где оба арила независимо друг от друга являются арильными гетероциклилами, как они определены выше для арила, в органическом растворителе в присутствии основания и в необязательном присутствии агента для сочетания, такого как сукцинимид, например N-гидроксисукцинимид, а также включает выделение получаемого соединения формулы VI из реакционной смеси.
В предпочтительном варианте осуществления настоящего изобретения его объектом является способ согласно настоящему изобретению согласно стадии а) в стадиях А)-Г), как они описаны в контексте (также обозначаемый в контексте как способ А)а)). В качестве исходного вещества используют соединение формулы V, реакционноспособные группы в котором предпочтительно защищены.
Взаимодействие А)а) согласно настоящему изобретению может быть осуществлено следующим образом. Взаимодействие осуществляют в органическом растворителе. Подходящий органический растворитель включает растворитель, являющийся в реакционных условиях инертным, например углеводороды, такие как алифатические необязательно галогенированные углеводороды, ароматические или циклоалифатические углеводороды, простые эфиры, эфиры уксусной кислоты или индивидуальные смеси указанных растворителей. Предпочтительно выбирают такой растворитель, который при контакте с водой может образовывать двухфазную систему, включающую органический слой и водный слой.
Предпочтительный растворитель включает углеводороды, например циклические углеводороды, такие как циклогексан, и эфиры уксусной кислоты, такие как этилацетат, пропилацетат, изопропилацетат, например изопропилацетат. Предпочтительно используют смесь растворителей, например смесь углеводородов и эфиров уксусной кислоты, такую как смесь изопропилацетата и циклогексана.
Для осуществления взаимодействия соединение формулы V, предпочтительно в защищенной форме, смешивают с трис(триметилсилил)силаном, (С6-18)алкилмеркаптаном, например трет-додецилмеркаптаном, и α,α'-азоизобутиронитрилом в органическом растворителе и приводят во взаимодействие при подходящей температуре. Предпочтительно, трис(триметилсилил)силан в органическом растворителе, например углеводороде, обрабатывают соединением формулы V в органическом растворителе, например углеводороде, добавляют (С6-С18)алкилмеркаптан и нагревают получаемую таким образом смесь до соответствующей температуры, например до температурного диапазона (приблизительно) от 50°С до 80°С, более предпочтительно, до температурного диапазона (приблизительно) от 60°С до 70°С. К получаемой смеси добавляют α,α'-азоизобутиронитрил в органическом растворителе, например в эфире уксусной кислоты, например, порциями, и ввергают получаемую смесь во взаимодействие до израсходования исходных веществ вплоть до уровня ниже заранее заданного диапазона концентраций (контроль с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ)) (приблизительно, от 1 до 3 часов). Получают соединение V, необязательно в защищенной форме, в котором Х является водородом, a R1, R2 и Y отвечают вышеприведенным определениям, например, в котором гидроксигруппы в положении 28 и, если это применимо, в положении 40 циклической системы защищены триорганосилильными группами.
Снятие защитных групп может быть осуществлено следующим способом. Получаемую смесь охлаждают до температуры -10°С или ниже, например, в температурный диапазон (приблизительно) от -30°С до 0°С, такой как (приблизительно) от -20°С до -10°С, и добавляют предварительно охлажденный полярный органический растворитель. Полярный органический растворитель включает, например, спирты, такие как метанол. Получаемую таким образом смесь перемешивают при температуре от -10°С до -20°С до израсходования исходных веществ вплоть до уровня ниже заранее заданного диапазона концентраций (например (приблизительно), от 0,25 до 2 часов). Получают соединение формулы I, в котором R1, R2 и Y отвечают вышеприведенным определениям, такое как соединение формулы Ip, в незащищенной форме.
Завершающая обработка реакционной смеси может быть осуществлена следующим образом. К получаемой смеси добавляют основание с целью достижения pH смеси приблизительно 7. Основание включает неорганические основания, например соль натрия или калия, например гидрокарбонат натрия или калия. Основание предпочтительно добавляют в растворе, более предпочтительно, в водном растворе, например, таким образом, что температура не превосходит диапазона от -20°С до -10°С. К получаемой таким образом смеси добавляют органический растворитель, который при контакте с водой может образовывать двухфазную систему, содержащую органический слой и водный слой, например, так, как это описано выше для реакции получения соединения формулы I в защищенной форме, а также воду. Подобный органический растворитель предпочтительно является эфиром уксусной кислоты. Получаемую смесь перемешивают и разделяют получаемые фазы. Если это требуется, водную фазу дополнительно экстрагируют органическим растворителем и объединяют получаемые таким образом органические слои. Из органического слоя выпаривают раствор при пониженном давлении. Получают соединение формулы I, в котором R1, R2 и Y отвечают вышеприведенным определениям, например соединение формулы Ip, например в форме масла.
В еще одном предпочтительном варианте осуществления настоящего изобретения его объектом является способ согласно настоящему изобретению согласно стадии а) в стадиях А)-Г), как они описаны в контексте (также обозначаемый в контексте как способ А)б)). В качестве исходного вещества используют соединение формулы VI, в котором реакционноспособные группы предпочтительно находятся в защищенной форме.
Способ А)б) согласно настоящему изобретению может быть осуществлен следующим образом. Взаимодействие осуществляют в органическом растворителе. Подходящий органический растворитель включает растворитель, как он описан выше для реакции А)а), такой как ароматические углеводороды, например толуол.
Для осуществления взаимодействия соединение формулы VI, предпочтительно, в защищенной форме смешивают с трис(триметилсилил)силаном, (С6-С18)алкилмеркаптаном, например трет-додецилмеркаптаном и α,α'-азоизобутиронитрилом в органическом растворителе и вводят их во взаимодействие при соответствующей температуре.
Предпочтительно, соединение формулы VI в смеси органического растворителя с (С6-С18)алкилмеркаптаном нагревают, например, до диапазона температур (приблизительно) от 80°С до 110°С, такого как (приблизительно) от 95°С до 105°С, добавляют трис(триметилсилил)силан и α,α'-азоизобутиронитрил в органическом растворителе и вводят получаемую таким образом смесь во взаимодействие до израсходования исходных веществ вплоть до уровня ниже заранее заданного диапазона концентраций (контроль с помощью ВЭЖХ) (приблизительно до 1 часа). Получают соединение формулы VI, необязательно в защищенной форме, в котором Х является водородом, a R1, R2 и Y, отвечают вышеприведенным определениям, например, в котором гидроксигруппы в положении 28 и, если это применимо, в положении 40 циклической системы защищены триорганосилильными группами, например триэтилсилилом.
Снятие защитных групп может быть осуществлено так, как это писано в международной заявке на изобретение WO 9641807. Получают соединение формулы I в незащищенной форме, в котором R1, R2 и Y отвечают вышеприведенным определениям.
Заключительная обработка реакционной смеси может быть осуществлена так, как это описано выше для способа А)а), но с применением в качестве предпочтительного растворителя ароматического углеводорода, такого как толуол.
Получают соединение формулы I, в котором R1, R2 и Y отвечают вышеприведенным определениям, например в форме масла.
Соединение формулы I, получаемое согласно любому из способов А)а) или А)б) может быть в дальнейшем очищено с помощью подходящего способа, например с помощью хроматографии, такой как колоночная хроматография.
Хроматографическая очистка может быть осуществлена посредством колоночной хроматографии на силикагеле 60, с использованием подходящей смеси растворителей в качестве подвижной фазы, такой как смесь эфира уксусной кислоты и углеводорода, например смеси этилацетата и н-гексана, например смеси этилацетата и н-гексана 2:1. Соединение формулы I может быть получено в твердом виде, например посредством добавления осадителя к фракциям, содержащим соединения формулы I.
Помимо вышеизложенного, было обнаружено, что соединение формулы Ip может также быть получено в кристаллической форме, например, посредством добавления н-гептана к фракциям, содержащим соединение формулы Ip, полученное в результате хроматографической очистки.
Согласно настоящему изобретению было, кроме того, неожиданно обнаружено, что соединение формулы Ip может также быть получено в форме сольвата с органическим растворителем, таким как сольват с ацетоном, или сольват с пропиленгилколем, или сольват с водой, например гидрата, такого как моногидрат, если в смеси растворителей для кристаллизации присутствуют ацетон, пропиленгликоль или вода, соответственно.
Кристаллическая форма соединения формулы Ip, получаемая в тех случаях, когда в смеси растворителей для кристаллизации присутствует ацетон, именуется в контексте формой А (сольват с ацетоном), а кристаллическая форма гидрата соединения формулы Ip, получаемая в тех случаях, когда в смеси растворителей для кристаллизации присутствует вода - формой Б. Было, кроме того, обнаружено, что может быть получена другая моногидратная форма соединения формулы Ip, именуемая формой 2, если в смеси растворителей для кристаллизации присутствуют вода и метанол. При нагревании форма 2 обратимо переходит в безводную форму 2'.
Было обнаружено, что форма Б может быть получена в тех случаях, когда в смеси растворителей для кристаллизации присутствует спирт, например метанол или этанол, а вода используется в качестве осадителя. Было обнаружено, что форма А может быть получена в тех случаях, когда в смеси растворителей для кристаллизации присутствует ацетон и когда в смеси растворителей для кристаллизации присутствует органический осадитель, например углеводород, такой как гексан.
Было также обнаружено, что из обеих модификаций, формы А и формы Б, при нагревании может быть получена безводная форма 1А'.
В еще одном варианте осуществления настоящего изобретения его объектом является соединение формулы Ip в кристаллической форме в форме сольвата, например в форме сольвата с органическим растворителем, такого как сольват с ацетоном или сольват с пропиленгликолем, или, например, в форме сольвата с водой, такого как гидрат, например моногидрат.
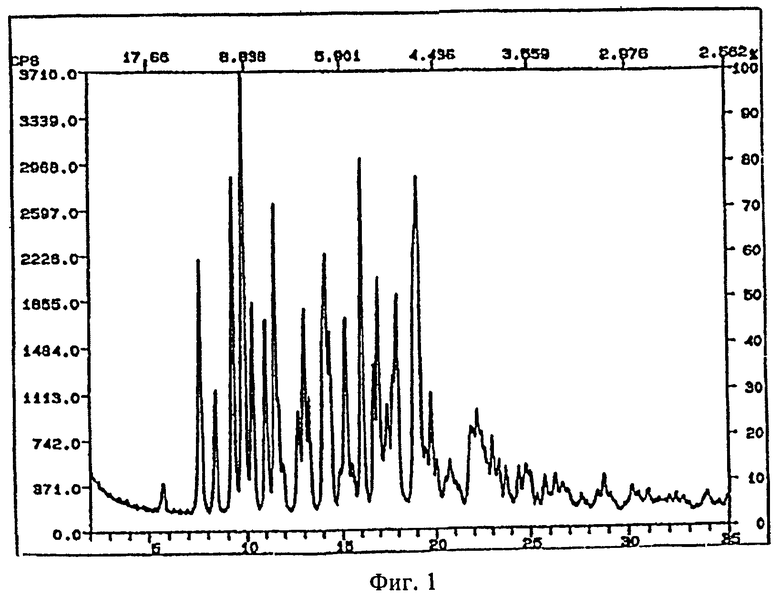
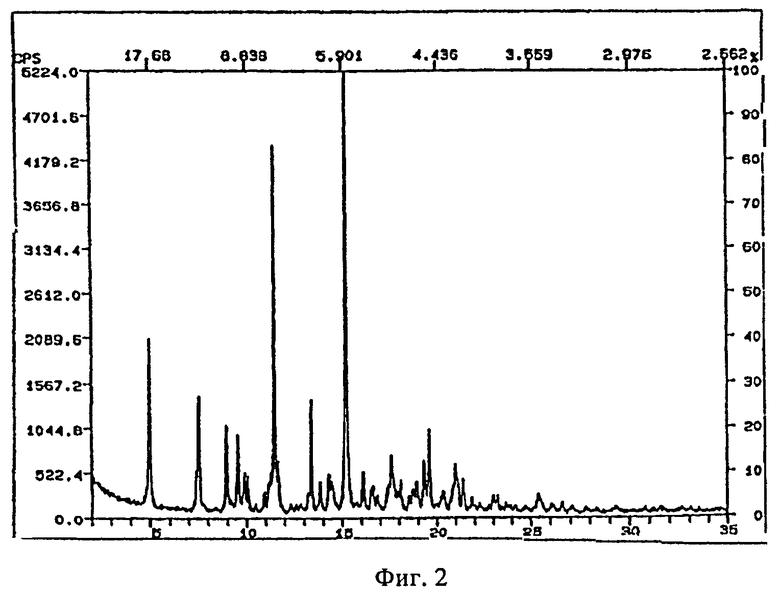
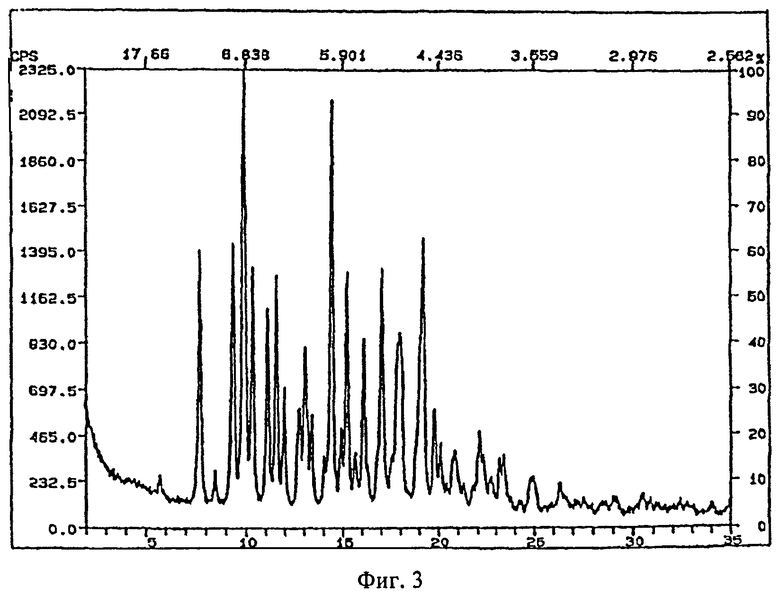
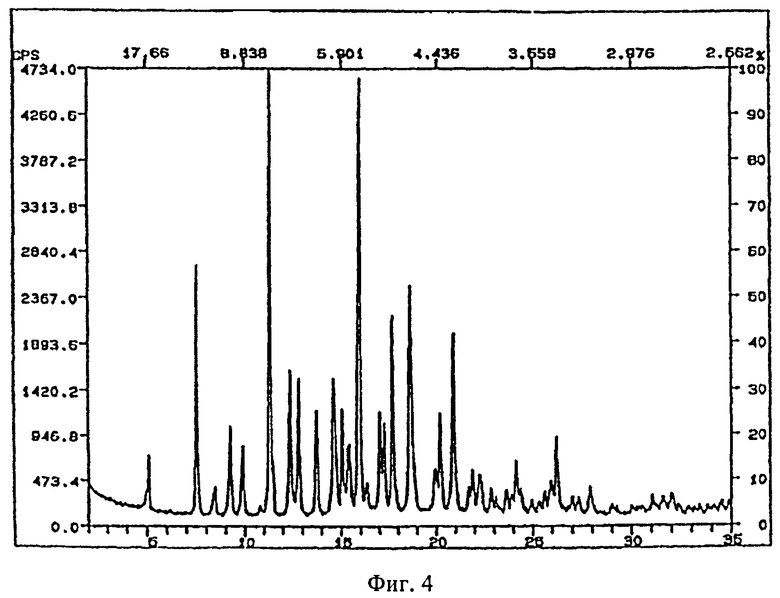
Рентгеновские порошковые дифрактограммы кристаллических соединений, являющихся объектом настоящего изобретения, представлены на
фигуре 1 (сольват с ацетоном),
фигуре 2 (сольват с пропиленгилколем),
фигуре 3 (гидрат, форма 1)
фигуре 4 (гидрат, форма 2).
На фигурах 1-4 ось абсцисс обозначает угол рассеяния, ось ординат обозначает измеренную интенсивность. Длина волны: 1,54060.
В нижеследующих примерах все температуры приведены в градусах Цельсия.
32-Дезоксорапамицин является соединением формулы Ip.
Соединение 28,40-O-бис(триэтилсилил)-32-иодрапамицин является соединением формулы
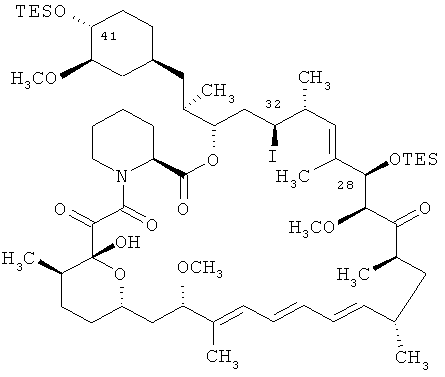
Соединение 28,40-O-бис(триэтилсилил)-32-гидроксирапамицин является соединением формулы
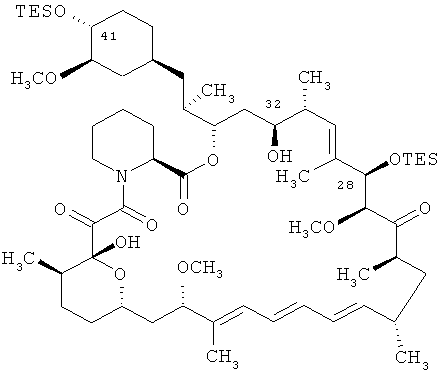
TES является триэтилсилильной группой.
Пример 1
32-Дезоксорапамицин в кристаллической форме
А) 28,40-O-Бис(триэтилсилил)-32-дезоксорапамицин
К смеси 1,74 г трис(триметилсилил)силана в 5 г циклогексана добавляют в атмосфере аргона раствор 8,5 г 28,40-бис-O-(триэтилсилил)-32-иодрапамицина в 70,6 г циклогексана и 14,44 г трет-додецилмеркаптана и нагревают получаемую таким образом смесь до температуры около 65°С. К получаемой смеси порциями добавляют 0,1135 г α,α'-азоизобутиронитрила (АИБН) в 7,4 г изопропилацетата, поддерживая при этом температуру близко к 65°С. Получают 28,40-O-бис(триэтилсилил)-32-дезоксорапамицин.
Б) 32-Дезоксорапамицин
Смесь, получаемую на стадии А, охлаждают до температуры ниже -20°С и выливают в 90 г метанола, предварительно охлажденного до температуры -20°С, не превышая при этом температуры приблизительно -15°С. Получаемую таким образом смесь перемешивают в течение приблизительно одного часа и обрабатывают 44,4 г насыщенного водного раствора NaHCO3, не превышая при этом температуры приблизительно -15°С. Получаемую смесь перемешивают в течение приблизительно 15 минут и обрабатывают 68 г изопропилацетата и 84,5 г воды. Получаемую смесь интенсивно перемешивают и нагревают до комнатной температуры. Фазы разделяют, водную фазу экстрагируют изопропилацетатом и промывают объединенные органические фазы водой. Отгоняют из органической фазы изопропилацетат при пониженном давлении. Получают масло, которое обрабатывают 116 г н-гептана. Осаждающийся 32-дезоксорапамицин отфильтровывают. Получают 4,28 г твердого 32-дезоксорапамицина во влажном виде и дополнительно 0,435 г 32-дезоксорапамицина во влажном виде из фильтрата. Всего получают 4,715 г 32-дезоксорапамицина во влажном виде, содержащего 3,080 г 32-дезоксорапамицина.
В) Очистка 32-дезоксорапамицина
Колонку, заполненную 118 г силикагеля, обрабатывают 3,98 г 32-дезоксорапамицина, полученного согласно способу, описанному на стадии Б), растворенного в смеси этилацетата и гексана 2:1. Собирают фракции, содержащие 32-дезоксорапамицин и выпаривают растворитель до достижения объема приблизительно 10 мл.
Г) Кристаллизация 32-дезоксорапамицина
К остатку от выпаривания, получаемому на стадии В), добавляют приблизительно 7 мл этилацетата и добавляют по каплям к получаемой таким образом смеси приблизительно 20 мл н-гептана. В получаемую смесь добавляют в качестве зародышей кристаллизации заранее приготовленные кристаллы 32-дезоксорапамицина, охлаждают получаемую смесь до температуры приблизительно от 0°С до 5°С и перемешивают в течение приблизительно 1 часа. Кристаллизованный 32-дезоксорапамицин выделяют с помощью фильтрования и сушат. Выход: 1,69 г (около 74% от теоретического), чистота более 98%. Температура плавления по данным дифференциальной сканирующей калориметрии (ДСК): 127°С (начало процесса). Рентгенодифракционный анализ кристаллов показывает, что получаемый таким образом 32-дезоксорапамицин находится в гидратной форме 1. Из получаемого фильтрата может быть получено дополнительное количество выкристаллизовавшегося 32-дезоксорапамицина.
Согласно методике, описанной на стадиях А)-Г), но с использованием подходящих количеств исходных веществ на опытной установке получают несколько килограммов кристаллического 32-дезоксорапамицина в гидратной форме 1.
Д) Кристаллизация 32-дезоксорапамицина в форме сольвата с ацетоном
Данный сольват получают посредством охлаждения и осаждения из фракции в диэтиловом эфире и гексане при добавлении ацетона или посредством кристаллизации из ацетона.
Е) Кристаллизация 32-дезоксорапамицина в форме сольвата с пропиленгликолем
Данный сольват получают посредством охлаждения и осаждения из фракции в диэтиловом эфире и гексане при добавлении пропиленгликоля или посредством кристаллизации из пропиленгликоля.
Ж) Кристаллизация 32-дезоксорапамицина в форме моногидрата
Данный гидрат получают из растворителя, например метанола, содержащего воду (форма 2). Его также получают после выдерживания сольвата с ацетоном в условиях с высокой влажностью. Форма 1 может быть получена из растворителя, иного, нежели метанол, содержащего воду.
Пример 2
Получение 32-дезоксорапамицина
1 г соединения формулы
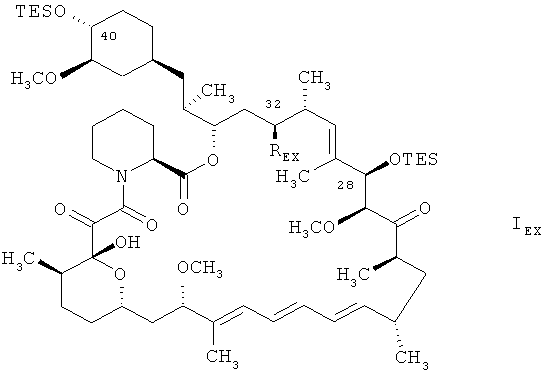
в котором REX является группой формулы
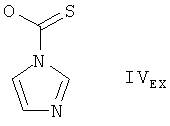
растворяют в смеси 10 мл додецилмеркаптана и 10 мл толуола. Получаемую таким образом смесь нагревают до температуры 100°С и добавляют при температуре 100°С 0,407 г трис(триметилсилил)силана, а затем, при той же температуре, раствор 0,0262 г АИБН в 1 мл толуола. Получаемую смесь перемешивают в течение приблизительно 15 минут при температуре 100°С и охлаждают до температуры 5-10°С. Смесь медленно добавляют к 20 мл предварительно охлажденного метанола, поддерживая в ходе добавления температуру в пределах между -10 и -20°С. К получаемой таким образом смеси добавляют 25 мл толуола и дают получаемой смеси нагреться до комнатной температуры. Экстрагируют получаемую смесь насыщенным водным раствором NaHCO3 и водой. Получаемый таким образом водный слой экстрагируют толуолом, органические слои объединяют, сушат над сульфатом натрия и выпаривают растворитель. Получают неочищенный 28,40-O-бис(триэтилсилил)-32-дезоксорапамицин в форме желтоватого раствора. Поучаемый раствор подвергают очистке с помощью флэш-хроматографии на силикагеле, используя в качестве элюента сначала гексан, а затем смесь гексана и трет-бутилметилового эфира 3:1. Получают 0,573 г 28,40-O-бис(триэтилсилил)-32-дезоксорапамицина в форме пены белого цвета, с высокой чистотой (приблизительно 98%). Получаемый таким образом 28,40-O-бис(триэтилсилил)-32-дезоксорапамицин обрабатывают 2 н. водным раствором серной кислоты в метаноле (согласно методике, описанной в международной заявке на изобретение WO 9641807). Получают с высокой чистотой 32-дезоксорапамицин.
Получение исходных веществ
Пример А
Получение соединения формулы IEX, в котором REX является группой формулы
При комнатной температуре обрабатывают 4,0 г 28,40-O-бис(триэтилсилил)-32-гидроксирапамицина в 40 мл CH2Cl2 2,75 г пиридина и 43 мг 4-диметиламинопиридина. К получаемой таким образом смеси добавляют 1,275 г хлорангидрида фенилтиономуравьиной кислоты и перемешивают получаемую смесь в течение 7 часов при комнатной температуре. Получаемую смесь подвергают хроматографической очистке на силикагеле (элюируя метил-трет-бутиловым эфиром). Получают соединение формулы IEX, в котором REX является группой формулы IIEX, в форме пены белого цвета. Масс-спектрометрия (МС) (ионизация распылением в электрическом поле в режиме отрицательных ионов): 1324,8 (M+HCOO)-, 1278,9 (М-Н)-. Ядерный магнитный резонанс 1Н-ЯМР подтверждает предложенную структуру.
Пример Б
Получение соединения формулы IEX, в котором REX является группой формулы
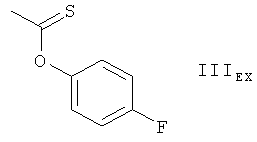
Обрабатывают 5,0 г 28,40-O-бис(триэтилсилил)-32-гидроксирапамицина в 45 мл CH2Cl2 0,052 г N-гидроксисукцинимида и 1,04 г пиридина. К получаемой таким образом смеси добавляют по каплям 1,70 г хлорангидрида 4-фторфенилтиономуравьиной кислоты так, чтобы температура не превысила 30°С. Получаемую смесь перемешивают в течение 3,5 часов при комнатной температуре, выливают в 80 мл хлористого метилена и экстрагируют получаемую таким образом смесь водой. Получаемый органический слой промывают насыщенным водным раствором NaHCO3 и водой, сушат его и выпаривают растворитель с целью получения концентрированного раствора в CH2Cl2 (приблизительно 40 мл). К получаемому таким образом раствору добавляют 60 мл трет-бутилметилового эфира и выпаривают растворитель до достижения конечного объема приблизительно 25 мл. Происходит выпадение осадка. Получаемую таким образом суспензию охлаждают до температуры 0-5°С в течение 45 минут и удаляют осадок посредством фильтрования. Получаемый таким образом фильтрат концентрируют посредством выпаривания и подвергают остаток от концентрирования хроматографической очистке на силикагеле. Получают соединение формулы IEX, в котором REX является группой формулы IIIEX, в форме пены. MS (бомбардировка быстрыми атомами): 1304 (M+Li)+. ИК-спектр (KBr): 3420 (широкая полоса), 2956, 2936, 2876, 1746, 1627, 1504, 1458, 1376, 1294, 1242, 1191, 1145, 1107, 1007, 988, 742 cm-1. ЯМР-спектры подтверждают предложенную структуру.
Пример В
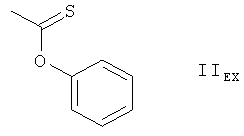
Получение соединения формулы IEX, в котором REX является группой формулы IVEX
Обрабатывают 50 г 28,40-O-бис(триэтилсилил)-32-гидроксирапамицина в 500 мл толуола 11,7 г 1,1-тиокарбонилдиимидазола и 533,6 мг 4-диметиламинопириидна, нагревают получаемый таким образом раствор до температуры 40°С и перемешивают в течение 20 часов при данной температуре. К получаемой смеси добавляют еще 0,778 г 1,1-тиокарбонилдиимидазола и перемешивают получаемую смесь в течение 2 часов при температуре 40°С. Получаемую таким образом смесь охлаждают до температуры 0°С и обрабатывают насыщенным водным раствором NaHCO3. Получаемые таким образом слои разделяют и экстрагируют органический слой насыщенным водным раствором NaHCO3 и водой. Получаемый таким образом органический слой подвергают хроматографической очистке на силикагеле, используя в качестве элюента сначала толуол, а затем смесь трет-бутилметилового эфира и гексана 1:1. Выпаривают растворитель и получают 46,1 г соединения формулы IEX, в котором REX является группой формулы IVEX, в форме пены. МС (ионизация распылением в электрическом поле в режиме положительных ионов): 1254.8 (М+Н)+, 1222.8 (М-ОСН3)+. ИК-спектр (KBr): 3428 (широкая полоса), 3133, 2936, 2876, 2824, 1746, 1730, 1650, 1626, 1532, 1460, 1415, 1387, 1326, 1285, 1232, 1192, 1167, 1142, 1107, 1075, 1005, 988, 890, 867, 824, 742, 656, 642, 613 и 560 cm-1. ЯМР-спектры продукта подтверждают предложенную структуру.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МАКРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2456296C2 |
| ПРОИЗВОДНЫЕ ОКСАДИАЗОЛА | 1997 |
|
RU2182905C2 |
| СПОСОБ ПОЛУЧЕНИЯ БИАРИЛЗАМЕЩЕННОЙ 4-АМИНОМАСЛЯНОЙ КИСЛОТЫ ИЛИ ЕЕ ПРОИЗВОДНЫХ И ИХ ПРИМЕНЕНИЕ В ИЗГОТОВЛЕНИИ ИНГИБИТОРОВ НЭП | 2007 |
|
RU2469019C2 |
| β-ЛАКТАМ ИЛИ ЕГО ЭНАНТИОМЕР ДЛЯ СИНТЕЗА ТАКСОЛОВ И СПОСОБ ПОЛУЧЕНИЯ ТАКСОЛОВ | 1990 |
|
RU2097374C1 |
| АМИНОАЛКИЛАМИДОМЕТИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 2-(4-СУЛЬФОНИЛАМИНО)-3-ГИДРОКСИ-3,4-ДИГИДРО-2Н-ХРОМАН-6-ИЛА И ЛЕКАРСТВЕННЫЕ СРЕДСТВА, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ | 2006 |
|
RU2413725C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО-ИМИДАЗОПИРИМИДИНА, ОБЛАДАЮЩИЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ КОРТИКОТРОПИН-РИЛИЗИНГ ФАКТОРА (CRF) | 2004 |
|
RU2377241C2 |
| ИНГИБИТОРЫ MIF | 2005 |
|
RU2383541C2 |
| ПОЛУЧЕНИЕ ДИГИДРОТИЕНО[3,2-d]ПИРИМИДИНОВ И ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ, ПРИМЕНЯЮЩИХСЯ ДЛЯ ИХ СИНТЕЗА | 2008 |
|
RU2528340C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2678305C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2696270C1 |
В настоящем изобретении описаны способы получения 32-дезоксорапамицина из 32-иод- или 32-гидроксирапамицина, в котором гидроксигруппа замещена остатком арилтиокарбоната или арилтиокарбамата, в присутствии трис(триметилсилил)силана и α,α'-азоизобутиронитрила в органическом растворителе. 4 з.п. ф-лы, 4 ил., 2 пр.
1. Способ получения соединения формулы
в котором R1 является метилом,
R2 представляет собой
где R3 представляет собой водород,
R4 представляет собой водород,
Y представляет собой О, и
Х представляет собой водород,
способ включает
А) обработку или
а) соединения формулы
в котором R1, R2 и Y отвечают вышеприведенным определениям, и в котором наличествующие гидроксигруппы находятся в незащищенной или в защищенной форме, предпочтительно в защищенной форме, в виде эфиров триэтилсилила, трис(триметилсилил)силаном, (С6-С18)алкилмеркаптаном, например трет-додецилмеркаптаном и α,α'-азоизобутиронитрилом в органическом растворителе, или
б) соединения формулы
в котором R1, R2 и Y отвечают вышеприведенным определениям, и в котором R' является остатком формулы IVEX
, который присоединен к атому углерода в положении 32 циклической структуры через атом кислорода, а также в котором наличествующие гидроксигруппы находятся в незащищенной или в защищенной форме, предпочтительно в защищенной форме, в виде эфиров триэтилсилила, трис(триметилсилил)силаном и α,α'-азоизобутиронитрилом в органическом растворителе,
Б) отщепление защитных групп, если таковые наличествуют,
В) выделение соединения формулы I, в котором R1, R2 и Y отвечают вышеприведенным определениям.
2. Способ получения соединения формулы I по п.1, в котором задействованы стадии способа А) а), необязательно Б), и В).
3. Способ по п.1, в котором гидроксигруппы соединения формулы V находятся в защищенной форме, предпочтительно в защищенной форме, в виде эфиров триэтилсилила.
4. Способ получения соединения формулы I по п.1, в котором задействованы стадии способа А) б), необязательно Б) и В).
5. Способ по п.1, в котором гидроксигруппы соединения формулы VI находятся в защищенной форме, предпочтительно в защищенной форме, в виде эфиров триэтилсилила.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| US 5256790 A, 26.10.1993 | |||
| ПРОИЗВОДНЫЕ РАПАМИЦИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2143434C1 |

