ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому производному хинолина, пригодному для применения в лечении или профилактике вирусных инфекций и связанных с вирусом состояний, в частности ВИЧ-инфекций.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Изобретение относится к новому соединению, пригодному для получения композиций, полезных для лечения заболеваний, вызванных изменениями процессов сплайсинга.
Некоторые производные индола, такие как производные эллиптицина и производные аза-эллиптицина, уже известны как интеркалирующие молекулы для коррекции дисфункций в экспрессии генов, особенно в репликации ДНК. Они были более конкретно описаны как полезные для лечения таких заболеваний, как рак, лейкемия или СПИД (см., в частности, патенты FR 2627493, FR 2645861, FR 2436786).
Одна из стратегий борьбы с вирусными инфекциями и/или связанными с вирусом состояниями, и в особенности ВИЧ/СПИД, заключается в применении производных, способных избирательно ингибировать некоторые дефекты сплайсинга.
В международной патентной заявке WO 05023255, поданной Заявителем, описано применение производных индола для лечения заболеваний, связанных с процессом сплайсинга предшественника матричной РНК в клетке.
Затем было показано, что некоторые производные индола оказываются особенно эффективными при лечении метастазирующего рака и при лечении СПИДа (BAKKOUR et al., PLoS Pathogens, vol. 3, p. 1530-1539, 2007).
Однако остается потребность в новых соединениях, пригодных для лечения или профилактики вирусной инфекции или связанного с вирусом состояния у пациента, включая ВИЧ и СПИД.
Среди связанных с вирусом состояний СПИД развился во всемирную пандемию. Более 30 миллионов человек инфицированы вирусом иммунодефицита человека (ВИЧ). Современные методы лечения позволили контролировать заболевание, но долгосрочное применение антиретровирусной терапии (APT) ограничено проблемами устойчивости к лекарственным средствам и побочных эффектов.
Таким образом, были предложены альтернативы APT, включая, например, комбинацию 3ТС-Тенофовир-Ралтегравир и AZT (ВААРТ).
Доступ к высокоактивной антиретровирусной терапии (ВААРТ), основанный на комбинации ВИЧ-протеазы и ингибиторов обратной транскриптазы, резко изменил прогноз течения ВИЧ-инфекции. В результате в развитых странах ВИЧ рассматривается как хроническое заболевание. Однако долгосрочное применение ВААРТ ограничено проблемами устойчивости к лекарствам и побочных эффектов.
Существует постоянная потребность в новых лекарствах, в частности тех, которые имеют новые и пока еще не исследованные механизмы действия в отношении лечения и/или профилактики вирусных инфекций и связанных с вирусом состояний и, в частности, для достижения контроля или лечения ВИЧ-инфекции.
Также существует потребность в лекарственных средствах и их. композициях, которые пригодны для введения с более низкой частотой и/или характеризуются долговременной эффективностью и/или устойчивым воздействием лекарственного средства.
Недавно некоторые производные хинолина были описаны в следующих патентных заявках: WO 02010/143169, WO 2012080953, ЕР 14306164 и ЕР 14306166, как полезные при лечении ВИЧ/СПИДа или воспалительных заболеваний.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
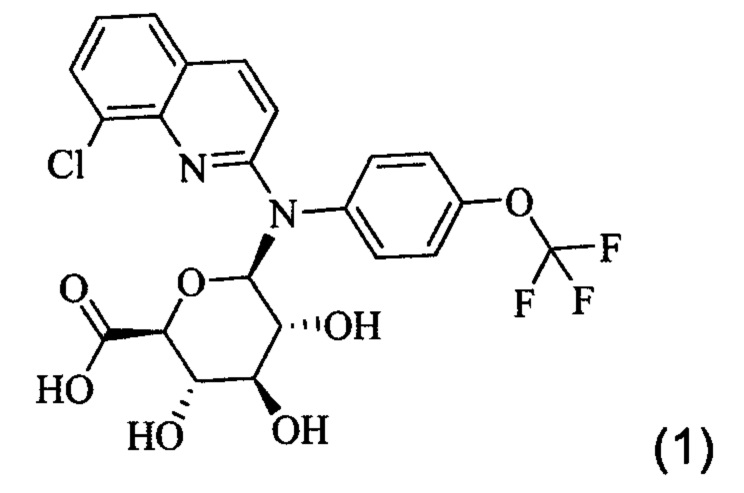
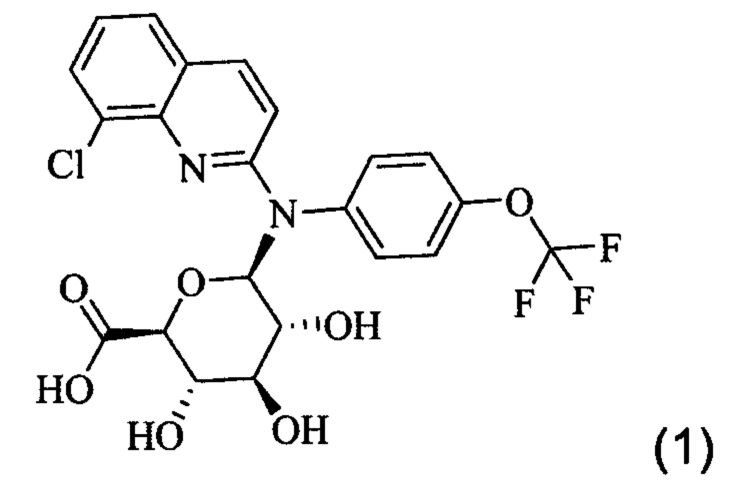
Настоящее изобретение относится к соединению формулы (1)
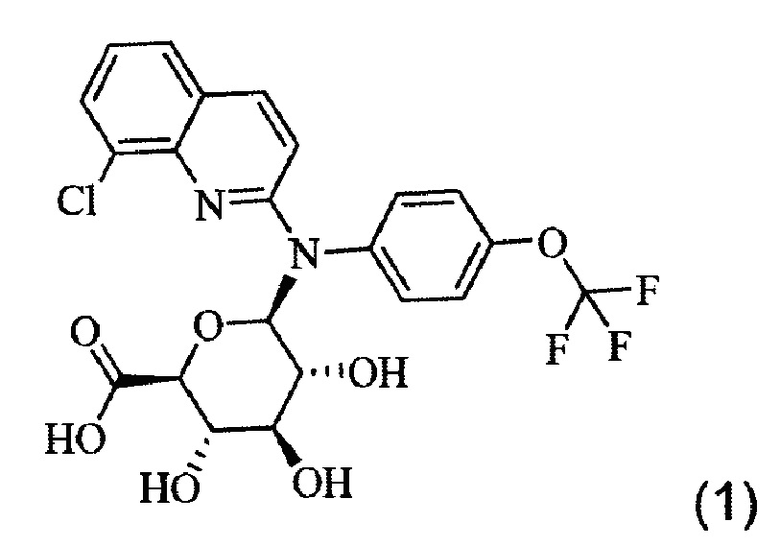
или одной из его фармацевтически приемлемых солей, и к его фармацевтической композиции. Это соединение может применяться для лечения или профилактики вирусной или ретровирусной инфекции и связанных с вирусом состояний, в частности СПИДа или состояния связанного со СПИДом или вирусом иммунодефицита человека (ВИЧ).
Изобретение также относится к соединению формулы (1) или одной из его фармацевтически приемлемых солей в лекарственном средстве.
Изобретение также относится к способу получения соединения формулы (1), а также к одному промежуточному соединению в указанном способе.
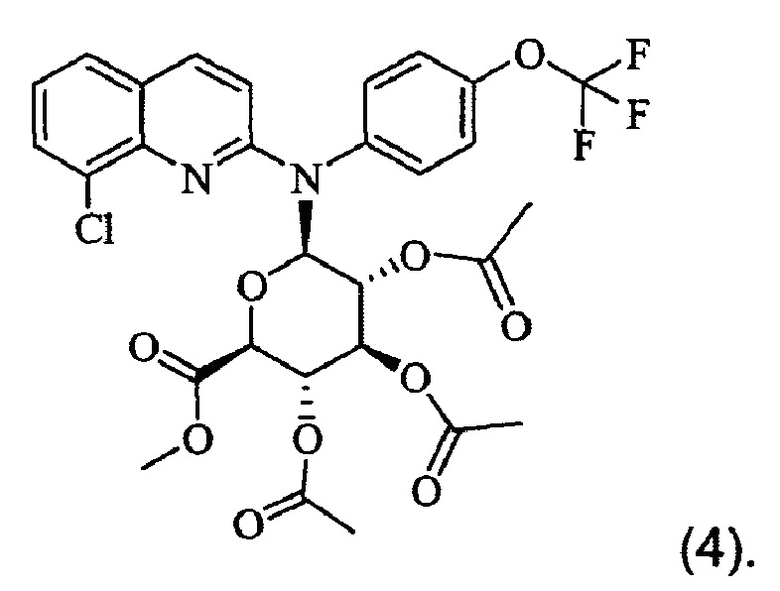
Изобретение также относится к промежуточному соединению формулы (4)
КРАТКОЕ ОПИСАНИЕ ФИГУР
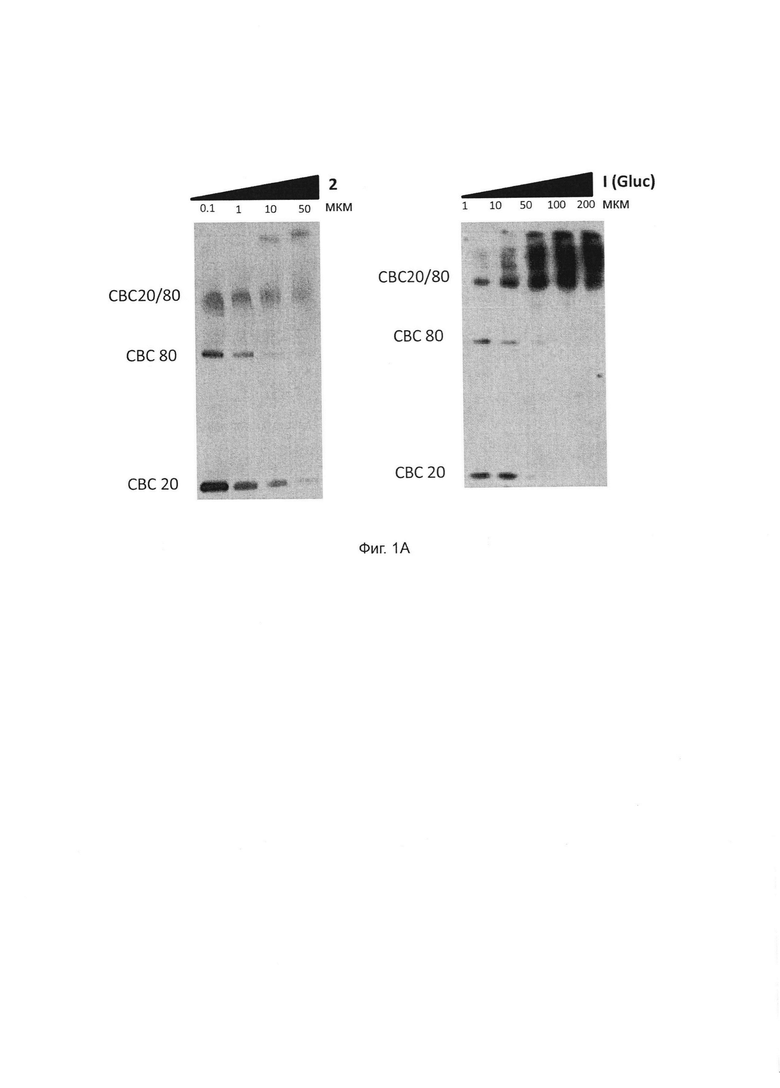
Фиг. 1А. Соединения 1 и 2 взаимодействуют с комплексом СВС. Очищенные рекомбинантные белки СВР20 и СВР80 инкубировали с увеличением концентрации соединения 2 (левая панель) или соединения 1 (правая панель, Gluc) и обрабатывали в течение 30 минут (соединение 1) или 15 минут (соединение 2) УФ-излучением. Белки были обнаружены с помощью Вестерн-блоттинга с применением антител СВР20 и СВР80. Левая панель: слева направо, инкубация с 0,1 (дорожка 1), 1 (дорожка 2), 10 (дорожка 3) и 50 мкМ (дорожка 4) соединения 1. Правая панель: слева направо, инкубация с 1 (дорожка 1), 10 (дорожка 2), 50 (дорожка 3), 100 мкМ (дорожка 4) и 200 мкМ (дорожка 5) соединения 1.
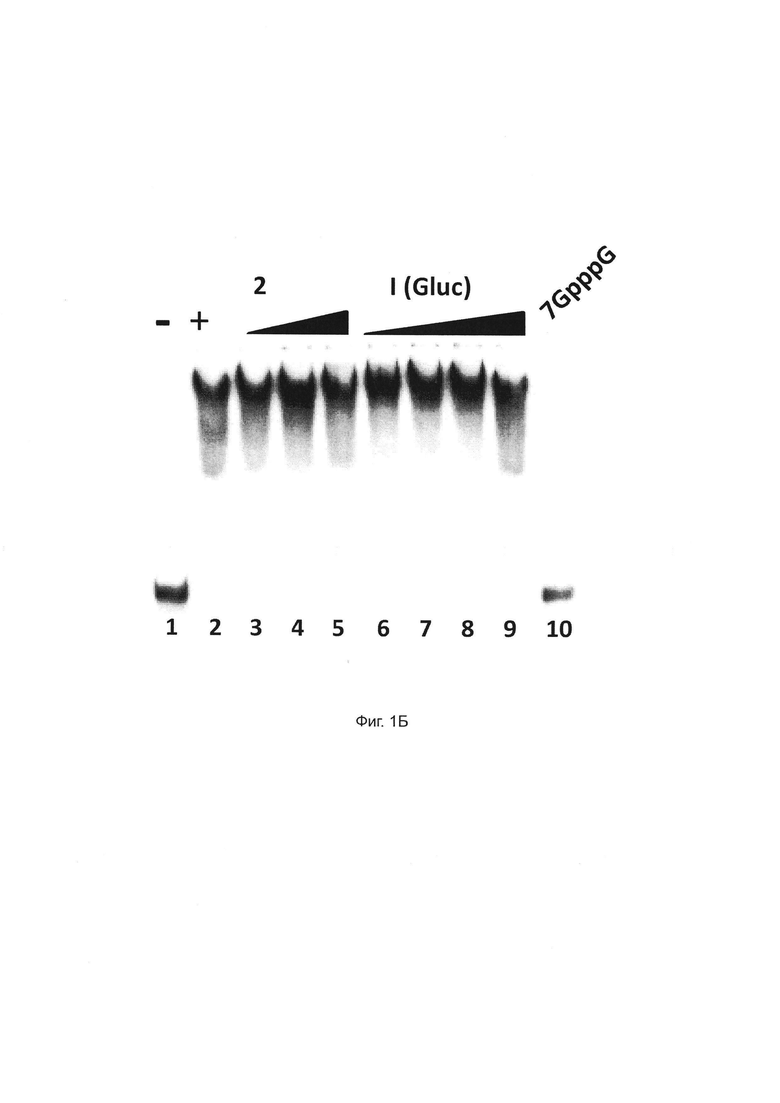
Фиг. 1Б. В отличие от структуры кэпа m7GpppG, соединения 1 и 2 не мешают связыванию РНК с кэпом с комплексом СВС. Рекомбинантный СВС человека инкубировали с РНК-субстратом с кэпом и анализировали с помощью нативного гель-электрофореза для того, чтобы разрешить различные комплексы РНК и РНК-белка: свободную РНК (дорожка 1) и комплексы СВС-РНК (полосы 2-10) в присутствии 12 мМ m7GppG (дорожки 10) или 5 мкМ, 10 мкМ или 50 мкМ соединения 2 (полосы 2-5 соответственно) и 5 мкМ, 10 мкМ, 50 мкМ или 100 мкМ соединения 1 (полосы 6-9, соответственно).
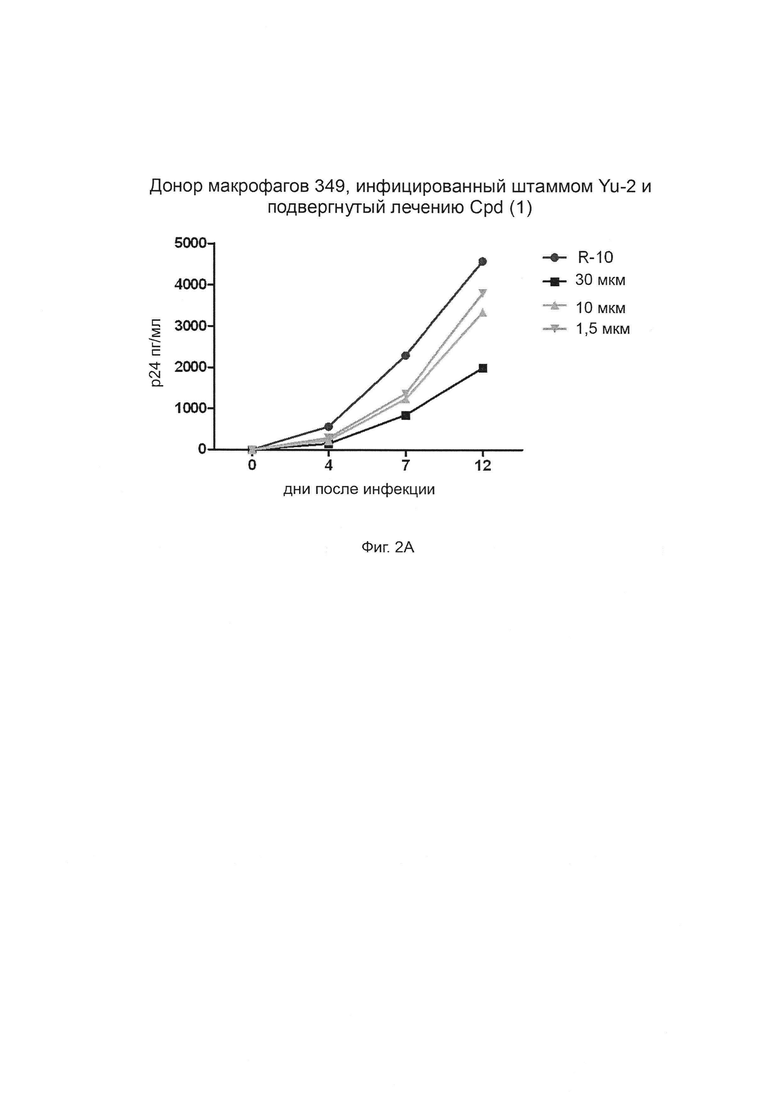
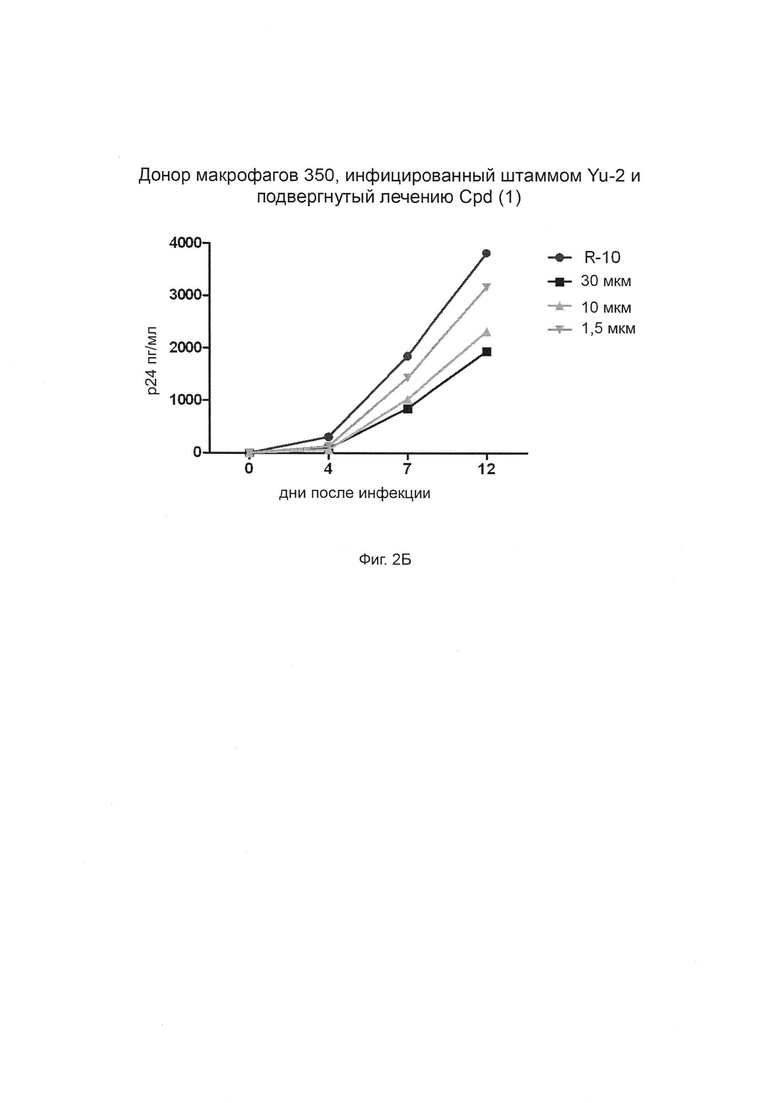
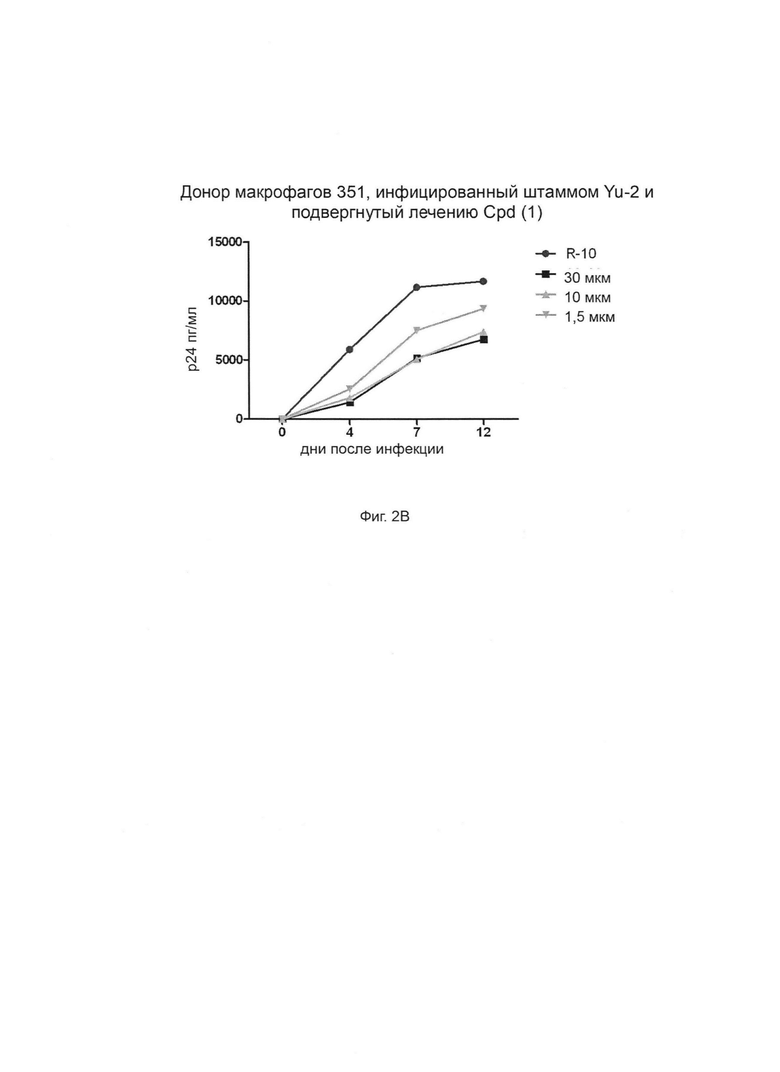
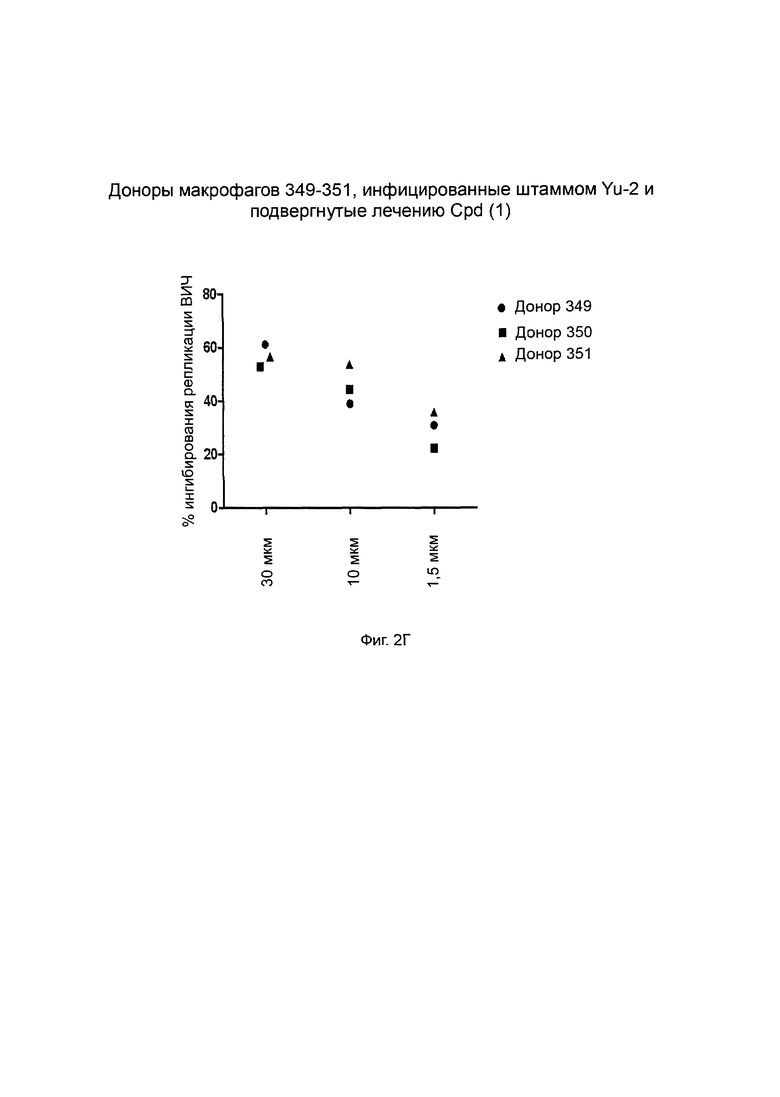
Фиг. 2А-Г. Способность соединения 1 ингибировать продуцирование ВИЧ-1 в инфицированных клетках макрофагов. Штамм ВИЧ-1 Yu-2 применяли для инфицирования моноцитарных макрофагов от трех разных доноров (доноры 349, 350 и 351, соответственно, Фиг. 2А, 2Б и 2В) в присутствии возрастающих концентраций (глюкуронидированного) соединения 1 при 1,5 мкМ, 10 мкМ и 30 мкМ. Белок р24 вирусного капсида определяли количественно с применением стандартного протокола ELISA (выраженного в пг/мл по оси Y). R-10 соответствует необработанным клеткам. Фиг. 2Г: % ингибирования репликации ВИЧ дополнительно оценивается при трех концентрациях и сравнивается для каждой концентрации между донорами.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение имеет целью удовлетворить вышеупомянутые потребности.
Настоящее изобретение относится к соединению формулы (1)
или одной из его фармацевтически приемлемых солей. Это соединение также упоминается здесь как Соединение 1, cpd (1) или Gluc.
Указанное соединение может существовать в форме основания или аддукта соли с кислотой, в частности, фармацевтически приемлемой кислотой.
Подходящие физиологически приемлемые кислотно-аддитивные соли соединения формулы (1) включают гидробромид, тартрат, цитрат, трифторацетат, аскорбат, гидрохлорид, тартрат, трифлат, малеат, мезилат, формиат, ацетат и фумарат.
Соединение формулы (1) и его соли могут образовывать сольваты или гидраты, и изобретение включает все такие сольваты и гидраты.
Термины «гидраты» и «сольваты» просто означают, что соединение формулы (1) в соответствии с изобретением может быть в форме гидрата или сольвата, то есть объединено или связано с одной или более молекулами воды или растворителя.
Соединение формулы (1), как показано выше, представляет собой неожиданный метаболит печени человека и, более конкретно, метаболит N-глюкуронида, соединения формулы (2)
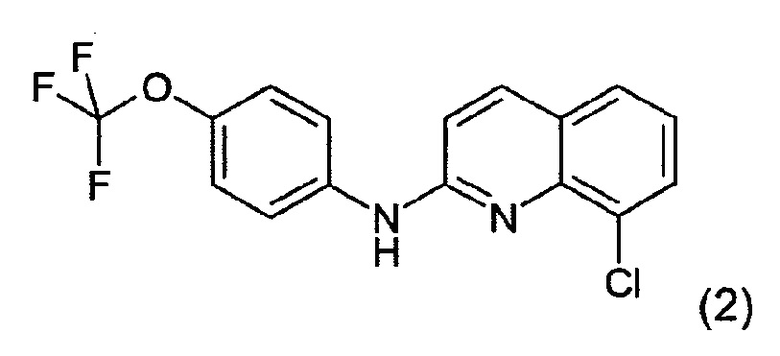
которое как таковое является активным соединением, пригодным для лечения вирусной инфекции или связанного с вирусом состояния у пациента, в частности инфекции вирусом иммунодефицита человека (ВИЧ) и связанного с вирусом состояния, такого как СПИД, как описано в WO 2010/143169.
Соединение (1) имеет следующее химическое название: N-β-глюкуронид 8-хлор-N-(4-(трифторметокси)фенил)хинолин-2-амина.
Соединение (1) может быть дополнительно охарактеризовано устойчивым воздействием лекарственного средства, которое транслируется в чрезвычайно длительный период полувыведения примерно 100 ч (от примерно 90 до примерно 110 ч). Это удивительно, потому что глюкуронидирование также было сообщено в предшествующем уровне техники как механизм клиренса для многих лекарств (см., например, Williams et al. Drugabalism and Disposition, Drug.32, No. 11, 2004).
Высокий клиренс обычно связан с коротким периодом полувыведения и коротким воздействием лекарственного средства. Напротив, низкий клиренс обычно связан с длительным периодом полувыведения и продолжительным воздействием лекарственного средства.
Это соединение формулы (2) также упоминается здесь как соединение 2.
Таким образом, согласно другому аспекту, предмет настоящего изобретения относится к соединению формулы (1) или его фармацевтически приемлемым солям для прменения в качестве лекарственного средства.
Согласно другой своей цели изобретение относится к фармацевтической композиции, содержащей соединение формулы (1) или его фармацевтически приемлемые соли и по меньшей мере один фармацевтически приемлемый эксципиент, и к лекарственному средству, содержащему соединение формулы (1), или одну из его фармацевтически приемлемых солей.
Соединение формулы (1) или одну из его фармацевтически приемлемых солей можно применять для лечения или профилактики вирусных или ретровирусных инфекций и связанных с вирусом состояний, в частности СПИДа или состояния, связанного со СПИДом или вирусом иммунодефицита человека (ВИЧ). Более конкретно, здесь показано, что такое соединение (I) снижает вирусную нагрузку ВИЧ-1 у ВИЧ-инфицированных млекопитающих и (II) поддерживает или восстанавливает высокий уровень количества лимфоцитов CD4+у ВИЧ-инфицированных млекопитающих.
Изобретатели далее предоставляют доказательства того, что это соединение (1) оказывает долгосрочное лечебное действие на пациентов и подходит для лечения или профилактики вирусной инфекции или связанного с вирусом состояния у пациента.
Изобретатели далее предоставляют доказательства того, что указанное соединение (1) является особенно подходящим в качестве лекарственного средства из-за его улучшенных фармакокинетических свойств.
Соединение формулы (1), которое подходит для изобретения, может быть получено в соответствии со схемой I ниже:
Схема 1
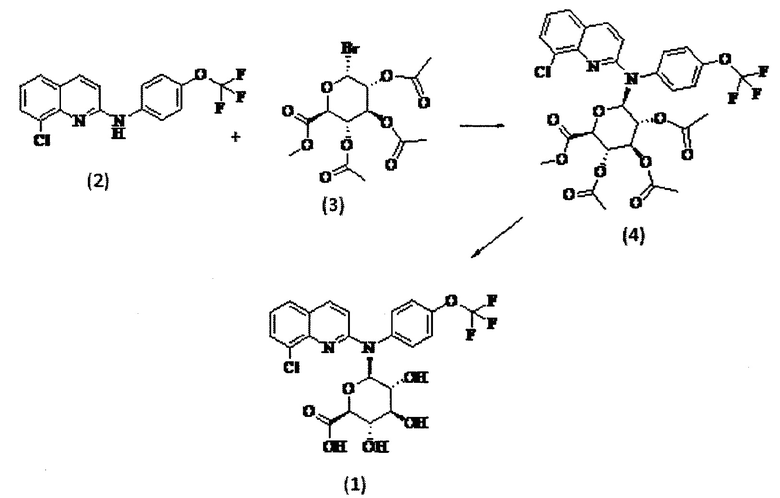
Соединение (2) можно синтезировать в соответствии со способом, описанным в WO 2010/143169.
Соединение (3) можно синтезировать в два этапа в соответствии со схемой 2 ниже.
Схема 2
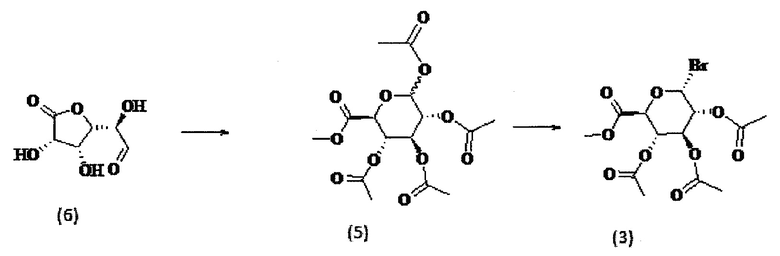
Соединение (6), которое является коммерчески доступным, может быть помещено в безводный метанол в присутствии металла, такого как натрий, при температуре в пределах от -20°C до 10°C, например, при 0°C, в течение некоторого времени от 1 до 7 часов, например, в течение 5 часов. Реакционную смесь можно обрабатывать смолой, например, смолой Amberlite ® IR-120 (Ir), например, до тех пор, пока рН не достигнет 3, и затем фильтровать. Смолу, полученную после фильтрации и удаления растворителя, можно растворить в уксусном ангидриде в присутствии хлорной кислоты. Реакционную смесь можно перемешивать в течение некоторого времени, например, в течение от 1 до 16 часов, в частности в течение 12 часов в атмосфере инертного газа, затем промывать и сушить с получением соединения (5). Указанные условия стадии способа более подробно описаны у Bollenback, G.N., Long, J.W., Benjamin, D.G., Lindquist, J.A., J. Am. Chem. Soc., 1955, 77, 3310. Иллюстрация указанной стадии способа приведена в примере 1 настоящего документа.
К соединению (5), полученному выше, бромистоводородную кислоту в уксусной кислоте можно добавлять в атмосфере инертного газа, например, аргона или азота, при температуре от -20 до 10°C, например, при 0°C, и перемешивать в течение некоторого времени, например, в течение от 1 до 5 дней, в частности, в эксикаторе, например, в течение 2 дней при 4°C. Полученную смесь можно разбавить этилацетатом и вылить в лед, затем промыть, высушить и необязательно очистить с получением соединения (3). Иллюстрация указанной стадии способа приведена в примере 1 настоящего документа.
Соединение (2) можно активировать, помещая его в растворитель, такой как безводный толуол, в присутствии соли тяжелого металла, такой как соль кадмия, и, например, карбонат кадмия. Реакцию между соединениями (2) и (3) можно проводить в соответствии с синтезом Koenigs-Knorr, который хорошо известен специалисту в данной области, подходящим образом в растворителе, таком как нитрометан, обычно при температуре кипения растворителя. После дефлегмации и, необязательно, стадии фильтрации и/или промывки и/или очистки получают соединение (4). Пример 2 настоящего документа иллюстрирует эту стадию.
Затем соединение (4) может быть обработано с применением гидропероксидных солей, например, путем добавления перекиси водорода к моногидрату гидроксида лития в воде с получением раствора гидропероксида лития. Таким образом, соединение (4) можно помещать в растворитель, такой как тетрагидрофуран или диоксан, в присутствии предыдущего полученного раствора и перемешивать, например, в течение 0,5-1,5 часов. Полученный осадок может быть очищен с получением соединения (1). Пример 3 настоящего документа иллюстрирует эту стадию синтеза.
Следовательно, изобретение также относится к способу получения соединения формулы (1), включающему стадию обработки соединения (4) в растворе гидропероксида лития, например, в растворителе, таком как тетрагидрофуран или диоксин, которой необязательно предшествует стадия получения соединения (4), состоящая во взаимодействии соединения формулы (2) с соединением формулы (3), как определено выше, в присутствии соли тяжелого металла, такой как соль кадмия, например карбонат кадмия, в частности в растворителе, таком как нитрометан, обычно при температуре кипения растворителя.
Изобретение также распространяется на соединение формулы (4), которое представляет собой промежуточное соединение:
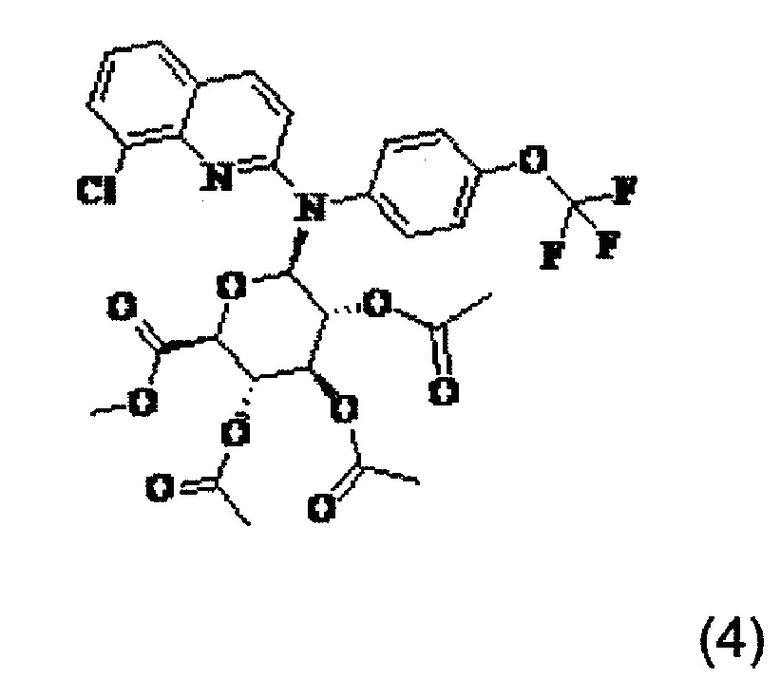
или одну из его фармацевтически приемлемых солей.
Вирусы
Производное хинолина согласно настоящему изобретению подходит для лечения или профилактики вирусных инфекций и, в частности, ВИЧ-инфекции или связанного с вирусом состояния, и в частности СПИДа.
Примеры вирусов, которые рассматриваются в настоящем изобретении, включают в себя без ограничений вирусы с оболочкой и без оболочки, которые включают ДНК-содержащие вирусы, РНК-содержащие вирусы и ретровирусы, которые включают вирусы с двухцепочечной ДНК, вирусы с одноцепочечной ДНК, вирусы с двухцепочечной РНК, вирусы с одноцепочечной (+) РНК, вирусы с одноцепочечной (-) РНК, вирусы с одноцепочечной РНК-зависимой ДНК-полимеразой и вирусы с двуцепочечной ДНК-зависимой ДНК-полимеразой, которые включают онковирусы, лентивирусы и спумавирусы.
Таким образом, онковирусы называются так потому, что они могут быть связаны с раковыми заболеваниями и злокачественными инфекциями. Можно упомянуть, например, вирусы-возбудители лейкоза (такие как вирус птичьего лейкоза (ALV), вирус мышиного лейкоза (MULV), также называемый вирусом Молони, вирус кошачьей лейкемии (FELV), вирусы лейкемии человека (HTLV), такие как HTLV1 и HTLV2, вирус обезьяньей лейкемии или STLV, вирус бычьей лейкемии или BLV, онковирусы приматов типа D, онковирусы типа В, которые являются индукторами опухолей молочной железы, или онковирусы, которые вызывают быстротекущий рак (например, вирус саркомы Рауса или RSV).
Спумавирусы проявляют достаточно низкую специфичность для данного типа клеток или данного вида, и они иногда связаны с явлением иммуносупрессии; как, например, в случае вируса пенистости обезьян (или SFV).
Таким образом, лентивирусы, такие как ВИЧ, названы так потому, что они отвечают за медленно прогрессирующие патологические состояния, которые очень часто включают явление иммуносупрессии, в том числе СПИД.
Известно, что вирусы и, в частности, ретровирусы, такие как ВИЧ, HTLV-I и HTLV-II, полагаются на регуляцию сплайсинга и сплайсинг РНК для распространения и расселения внутри клеток и тканей инфицированного человека. Другие вирусы, представляющие интерес, являются вирусами, патогенными для человека, включая, но не ограничиваясь перечисленными, вирусы семейства HSV (включая 1, 2, 6), CMV, VZV, HBV, HCV, вирус гепатита Е, вирусы папилломы, RSV, риновирусы, вирусы гриппа, аденовирусы, вирус герпеса человека 4-го типа, вирус Эболы, Нипах и другие арбовирусы, вирусы денге, чикунгунья, западного Нила, вирус долины Рифта, вирус японского энцефалита, другие коронавирусы SRAS, парвовирус, энтеровирусы.
Другие вирусы, представляющие интерес, являются вирусами, патогенными для животных, включая, но не ограничиваясь ими, грипп, FLV, пестивирус, хантавирус и лиссавирус.
В частности, вирусы и связанные с вирусом состояния, которые рассматриваются, включают вирусы, у которых репликация вируса требует сплайсинга РНК и/или экспорта вирусной РНК из ядра в цитоплазму.
Примеры вирусов включают латентные вирусы и/или ретровирусы и/или вирусы, которые связаны с хроническими вирусными инфекциями.
Вирусы, которые более конкретно рассматриваются, представляют собой РНК-содержащие вирусы и ретровирусы, включая лентивирусы и предпочтительно ВИЧ. Соответственно, связанные с вирусом состояния, которые более подробно рассматриваются, связаны с РНК-содержащим вирусом или ретровирусом, предпочтительно с ВИЧ.
ВИЧ может включать ВИЧ-1, ВИЧ-2 и все его подтипы, которые включают штаммы ВИЧ-I, относящиеся к подтипу ВИЧ-I В, подтипу ВИЧ-I С и рекомбинантам ВИЧ-I. Примеры включают штаммы ВИЧ-I, выбранные из Ad8, AdaM, изолята В, изолята С, CRF01, CRF02 и CRF06.
В соответствии с конкретным вариантом осуществления вирусы могут включать штаммы ВИЧ, у которых развились резистентность к текущим методам лечения.
Согласно предпочтительному варианту осуществления, связанным с вирусом состоянием является СПИД.
Терапевтическое применение
Вышеупомянутое соединение особенно подходит для лечения или профилактики вирусной инфекции и, более конкретно, ВИЧ-инфекции или состояния, связанного с ВИЧ. Кроме того, вышеупомянутое соединение особенно подходит для лечения латентной ВИЧ-инфекции у индивидуума, для искоренения ВИЧ-инфекции или связанного с ВИЧ состояния у индивидуума, включая искоренение ВИЧ и/или для применения в качестве лекарственного средства для лечения ВИЧ и связанных с ВИЧ состояний.
Изобретение также относится к применению соединения формулы (1) для получения композиции, такой как лекарственное средство, для лечения или профилактики вирусной инфекции или связанного с вирусом состояния, и в особенности СПИДа, связанного со СПИДом или ВИЧ состояния.
Изобретение также относится к способу лечения или профилактики вирусной инфекции или связанного с вирусом состояния и, в частности, для лечения или профилактики ВИЧ-инфекции у пациента, заключающегося в введении пациенту, нуждающемуся в этом, эффективного количества производного хинолина формулы (1), как описано выше, или его фармацевтической композиции.
Кроме того, изобретение относится к производному хинолина формулы (1), как определено здесь выше, или любой из его фармацевтически приемлемых солей для применения для снижения вирусной нагрузки и/или для увеличения или восстановления уровня количества клеток CD4+ у ВИЧ-положительных пациентов.
В частности, изобретение относится к производному хинолина формулы (1), как определено здесь выше, или любой из его фармацевтически приемлемых солей для применения в лечении или профилактике ВИЧ-инфекции или связанного с ВИЧ состояния у пациента; а затем прекращения лечения, когда вирусная нагрузка становится низкой или неопределяемой; и/или уровень количества клеток CD4+ сохраняется или восстанавливается.
Для справки и, как дополнительно описано ниже, низкая вирусная нагрузка обычно ниже 500 копий/мл плазмы, и неопределяемая вирусная нагрузка обычно составляет ниже 40 копий/мл.
Для справки и, как дополнительно описано ниже, восстановленное количество клеток CD4+ может соответствовать физиологическому (или «нормальному») количеству CD4 + клеток, которое обычно равно или превосходит 500 CD4 + клеток/мм3 плазмы и обычно варьирует между 500 и 1500 CD4+ клеток/мм3 плазмы, хотя для отдельных индивидов она может быть ниже.
Альтернативно, восстановленное количество клеток CD4+ может соответствовать увеличению количества клеток CD4+ по сравнению с количеством клеток CD4+ у указанного пациента до указанного лечения.
Изобретение также относится к производному хинолина формулы (1), как определено здесь выше, или любой из его фармацевтически приемлемых солей для применения в лечение или профилактике ВИЧ-инфекции или связанного с ВИЧ состояния у пациентов, для которых показаны неэффективность предшествующей антиретровирусной терапии или снижение эффективности предшествующей антиретровирусной терапии.
Изобретение также относится к производному хинолина формулы (1), как определено здесь выше, или любой из его фармацевтически приемлемых солей для применения в лечение или профилактике ВИЧ-инфекции или связанного с ВИЧ состояния у пациента в случае, если пациент инфицирован устойчивым к лекарствам штаммом ВИЧ.
Используемый здесь термин «пациент» может распространяться на людей или млекопитающих, таких как кошки или собаки. Используемый здесь термин «профилактика» также включает «уменьшение вероятности возникновения» или «снижение вероятности повторения».
В приведенных в настоящем документе примерах показано, что производное хинолина формулы (1) снижает репликацию ВИЧ у инфицированных ВИЧ млекопитающих.
Согласно конкретному варианту осуществления изобретения изобретатели предоставляют доказательства того, что указанное производное хинолина формулы (1) обладает долгосрочным лечебным эффектом и обеспечивает значительно сниженное возобновление симптомов, в частности, по сравнению с классическими антиретровирусными лекарственными средствами.
Не желая связывать себя какой-либо конкретной теорией, изобретатели считают, что производное хинолина согласно изобретению способно модулировать активность вирусного белка Rev и, в частности, модулировать Rev-опосредованный экспорт вирусных РНК.
Не желая связывать себя какой-либо конкретной теорией, изобретатели также считают, что такое производное хинолина имеет неожиданные свойства при направленном взаимодействии со скрытыми резервуарами ВИЧ.
Причины, объясняющие возобновление симптомов вирусных инфекций у пациентов, ранее подлежавших лечению, включают:
(I) тот факт, что многие вирусы, включая ретровирусы, такие как ВИЧ или ДНК-содержащие вирусы семейства Herpesviridae, характеризуются латентностью вируса, которая является способностью вируса находиться бездействующим в клетке, таким образом определяя лизогенную часть жизненного цикла вируса. Латентная фаза представляет собой фазу цикла репликации вируса, при которой после первичной инфекции пролиферация вирусных частиц прекращается без полной ликвидации. Явление латентности вируса связано с появлением так называемых «резервуаров» внутри хозяина, которые, как правило, труднодоступны, и которые также являются одной из основных причин трудности лечения ВИЧ;
(II) появление устойчивых к лекарствам штаммов, особенно для вирусных инфекций, требующих длительного лечения. Вероятность появления мутантных штаммов особенно важна для ретровирусов, включая ВИЧ. Действительно, устойчивость к лекарствам против ВИЧ можно объяснить на биологическом уровне следующим образом. Как ретровирус ВИЧ использует фермент обратную транскриптазу для синтеза ДНК из его РНК генома и не имеет механизма для исправления ошибок, возникающих при воспроизведении его генома. В результате ВИЧ реплицирует свой геном с наибольшей скоростью мутаций, известной для любого «живого» организма. Это создает идеальную ситуацию для воздействия естественного отбора на популяцию ВИЧ, поскольку генетические вариации являются сырьем для естественного отбора.
Эти мутации накапливаются в течение поколений и в популяциях, что приводит к большой генетической вариации среди популяций ВИЧ и увеличивает вероятность того, что вирион развивает эволюционное избирательное преимущество перед другими вирионами. Естественный отбор затем действует на ВИЧ, выбирая вирионы с более высокой приспособленностью, так как все остальные в конечном итоге уничтожаются при лечении лекарственными средствами. Вирионы, способные избежать вредного воздействия лекарственного средства, создают совершенно новую, устойчивую к лекарствам популяцию.
Следствием снижения эффективности предшествующего лечения является то, что вирионы воспроизводятся до тех пор, пока у пациента не будет увеличенной, обнаруживаемой вирусной нагрузки, столь же большой, как изначально, до тех пор, пока лечение не уменьшило это число. Это создает цикл, в котором пациенты, особенно ВИЧ-инфицированные, впервые испытывают успех в лечении, поскольку:
- их вирусная нагрузка находится под контролем или даже уменьшается;
- уровень количества CD4+ клеток поддерживается или даже восстанавливается; и/или
- клинические признаки, которые обычно связаны со связанным с вирусом состоянием, таким как СПИД, стабилизируются или даже исчезают. Клинические признаки СПИДа различаются в зависимости от стадии заражения.
Затем со временем у этих пациентов может наблюдаться снижение эффективности лечения, так как вирус развивает устойчивость и восстанавливает популяцию вирусных частиц.
В частности, это явление усиливается для терапий против ВИЧ, по меньшей мере по трем причинам, которые включают:
(I) тот факт, что ВИЧ является ретровирусом, и появление новых мутантных штаммов особенно важно для этого класса вирусов, как указано ранее;
(II) тот факт, что ВИЧ имеет способность переходить в латентную фазу и, таким образом, формировать «скрытые» резервуары, на которые доступные в настоящее время методы лечения не имеют эффективного направленного взаимодействия;
(III) тот факт, что имеющиеся в настоящее время методы лечения также склонны отбирать мутантные штаммы ВИЧ с течением времени, что в долгосрочной перспективе играет важную роль в возникновении устойчивости к лекарственным средствам.
Используемый здесь термин «агент против ВИЧ» означает классическое лекарственное средство или комбинацию лекарств, вводимых для борьбы с ВИЧ-инфекцией. Это может быть, в частности, APT (антиретровирусная терапия) или ВААРТ (высокоактивная антиретровирусная терапия).
«АРТ» и «ВААРТ» обычно относятся к комбинациям из двух, трех или более антиретровирусных лекарственных средств. Такие антиретровирусные лекарственные средства включают:
(I) ингибиторы обратной транскриптазы нуклеозидов/нуклеотидов, также называемые нуклеозидными аналогами, такими как абакавир, эмтрицитабин и тенофовир;
(II) ненуклеозидные ингибиторы обратной транскриптазы (ННИОТ), такие как эфавиренц, этравирин и невирапин;
(III) ингибиторы протеазы (ИП), такие как атазанавир, дарунавир и ритонавир;
(IV) ингибиторы входа, такие как энфувиртид и маравирок;
(V) ингибиторы интегразы, такие как долутегравир и ралтегравир.
Используемый здесь термин «лечение ВИЧ» включает, в частности:
- действие агента против ВИЧ по снижению вирусной нагрузки в течение определенного периода времени, но не обязательно демонстрирующее длительное снижение указанной вирусной нагрузки после прекращения указанного лечения; и/или
- действие агента против ВИЧ по увеличению уровня количества CD4+ клеток у ВИЧ-инфицированных пациентов.
Согласно одному варианту осуществления изобретения настоящее изобретение относится к производному хинолина формулы (1), как описано выше, или к одной из его фармацевтически приемлемых солей, пригодных для применения при лечении или профилактике вирусной инфекции или связанного с вирусом состояния у пациентов, в частности ВИЧ-инфекции или СПИДа, или, особенно, в тех случаях, когда их применение поддерживает низкую вирусную нагрузку после прекращения лечения.
В соответствии с указанным аспектом изобретение относится к производному хинолина формулы (1), как определено здесь, или к одной из его фармацевтически приемлемых солей, пригодных для применения при лечении или профилактике вирусной инфекции или связанных с вирусом состояний у пациента, в частности ВИЧ-инфекции или состояния, связанного с ВИЧ, при этом сохраняется низкая или неопределяемая вирусная нагрузка; и/или количество CD4+ клеток стабильно или увеличивается после прекращения лечения.
Тем не менее, в соответствии с указанным аспектом изобретение относится к производному хинолина формулы (1), как определено здесь, или к одной из его фармацевтически приемлемых солей для применения при лечении или профилактике вирусной инфекции или связанных с вирусом состояний у пациента, в частности ВИЧ-инфекции или состояния, связанного с ВИЧ, и затем прекращения лечения, при этом низкая или неопределяемая вирусная нагрузка сохраняется; и/или количество лимфоцитов CD4+ стабильно или увеличено после прекращения лечения.
В рамках настоящего изобретения «поддержание низкой вирусной нагрузки после окончания лечения» означает поддержание вируса не уровне менее обнаружимого или увеличение времени возобновления симптомов до 2 недель по сравнению с APT и ВААРТ.
Используемый здесь термин «вирусная нагрузка» также относится к «вирусному титру», и его можно определить прямо или косвенно. Для справки, вирусная нагрузка обычно относится к:
- количеству копий вирусной РНК или ДНК на мл образца плазмы;
- количеству вирусных частиц на мл образца плазмы; и/или
- активности вирусного белка в образце плазмы.
Используемый здесь термин «вирусная нагрузка ВИЧ» также относится к «титру ВИЧ-инфекции», и его можно определить прямо или косвенно. Для справки, вирусная нагрузка обычно относится к:
- количеству копий РНК ВИЧ на мл образца плазмы; и/или
- количеству частиц ВИЧ на мл образца плазмы; и/или
- активности белка, связанного с ВИЧ, в образце плазмы, которая может, например, включать определение активности обратной транскриптазы (RT) в указанном образце плазмы.
Для справки, методы определения вирусной нагрузки ВИЧ в образце включают:
- определение количества копий РНК ВИЧ на мл образца; и/или
- определение количества частиц ВИЧ на мл образца; и/или
- определение активности белка, связанного с ВИЧ, в образце.
Предпочтительно «вирусная нагрузка ВИЧ» относится к числу копий РНК ВИЧ на мл образца плазмы.
Низкая вирусная нагрузка обычно составляет менее 500 копий/мл плазмы; что включает от 20 до 500 копий/мл плазмы, или от 40 до 500 копий/мл плазмы, в зависимости от типа и чувствительности используемого теста. Этот результат указывает на то, что ВИЧ не воспроизводится активно и что риск прогрессирования заболевания низкий.
Низкая вирусная нагрузка может состоять из вирусной нагрузки ниже 500 копий/мл и включает уровни ниже 450, 400, 350, 300, 250, 200, 150, 100, 50, 40, 30, 20, 10, 9, 8, 7, 6, 5, 4, 3, 2 и 1 копий/мл плазмы.
Неопределяемая для обычных методов вирусная нагрузка обычно ниже 40 копий/мл плазмы, и включает 20 копий/мл плазмы, в частности, при измерении способом и/или наборами, выбранными из: COBAS® AmpliPrep/COBAS® TaqMan® HIV-1 Test и COBAS® AMPLICOR HIV-1 MONITOR Test, продаваемые Roche Molecular Diagnostic или NucliSENS EasyQ®HIV-1, продаваемый Biomerieux Diagnostics.
Более конкретно, в соответствии с этим аспектом изобретение относится к дозам и схемам производного хинолина формулы (1) или одной из его фармацевтически приемлемых солей при лечении или профилактике вирусной или ретровирусной инфекции и, в частности, ВИЧ-инфекции, при котором вирусная нагрузка после окончания лечения поддерживается низкой.
Другими словами, вирусная нагрузка остается предпочтительно на неопределяемом уровне по меньшей мере через две недели после окончания лечения по сравнению с терапией APT или ВААРТ, которая включает по меньшей мере три, четыре или пять недель после окончания лечения.
Это означает, что производное хинолина формулы (1) или одна из его фармацевтически приемлемых солей предоставляет удивительно длительный терапевтический эффект и отсутствие резистентности.
В частности, период полувыведения был определен в примерах и составляет около 100 ч (варьируя от примерно 90 ч до примерно 110 ч).
Согласно конкретному варианту осуществления производное хинолина формулы (1) в соответствии с настоящим изобретением может вводиться при различных дозировках и режимах и, в частности, один раз в день, один раз в три дня, один раз в неделю, раз в две недели или один раз в месяц, в дозах от 25 до 1000 мг, в частности от 25 до 700 мг, например, от 25 до 500 мг, а более конкретно от 25 до 300 мг, в течение периода лечения или в виде непрерывного лечения.
Согласно одному варианту осуществления производное хинолина формулы (1) в соответствии с настоящим изобретением можно таким образом вводить в дозах от примерно 50 до примерно 200 мг, от примерно 50 мг до примерно 300 мг или от примерно 50 до примерно 400 мг.
Из-за длительного периода его полувыведения, производное хинолина можно вводить, в частности, один раз каждые три дня, один раз в четыре дня, один раз в пять дней, один раз в шесть дней, раз в неделю, раз в две недели или даже один раз в месяц.
«Непрерывное лечение» означает долгосрочное лечение, которое может быть реализовано с различными частотами введения, например, один раз в три дня или раз в неделю или раз в две недели или раз в месяц.
Период лечения, т.е. когда лечение не является непрерывным, может варьировать от 2 недель до 8 недель, что включает 2, 3, 4, 5, 6, 7 и 8 недель.
Согласно другому варианту осуществления изобретение относится к производному хинолина формулы (1), как определено здесь, или к одной из его фармацевтически приемлемых солей, пригодных для применения при лечении или профилактике вирусной инфекции или связанных с вирусом состояний у пациента, в частности ВИЧ-инфекции или состояния, связанного с ВИЧ, для которых было указано снижение эффективности предшествующего противовирусного или антиретровирусного лечения.
Как использовано здесь «было указано снижение эффективности предшествующего лечения» может указывать на то, что устойчивые штаммы вируса появляются во время указанного предшествующего лечения, причем такие штаммы не подвержены воздействию агента против ВИЧ.
Не ограничивающим образом неэффективность или снижение эффективности предшествующего лечения у пациента могут возникать, например, потому что:
- пациент инфицирован штаммом вируса, в частности штаммом ВИЧ, для которого считается, что репликация и/или инфекционность были стабилизированы или даже уменьшены, но он больше не реагирует на лечение, которое включает лечение APT и ВААРТ; и/или
- пациент инфицирован устойчивым к лекарствам штаммом.
В частности, определение включает ранее подвергавшихся лечению пациентов, у которых вирусная нагрузка ВИЧ и/или уровень количества клеток CD4+ и/или низкая, устанавливающая таким образом контрольное значение, и которые во время или после лечения демонстрируют, по меньшей мере, один из следующих фактов:
- увеличение вирусной нагрузки ВИЧ; и/или
- снижение уровня количества клеток CD4+; и/или
где вирусная нагрузка ВИЧ и/или уровень количества клеток CD4+ установлены предпочтительно в образце плазмы.
В таких случаях утверждение о неэффективности или снижении эффективности упомянутого предшествующего лечения может быть оценено путем измерения вирусной нагрузки, которая увеличилась выше определяемого уровня, в частности в течение нескольких последовательных недель, например, в течение по меньшей мере одной или двух недель, в частности, по меньшей мере, 3 недель или 4 недель лечения антиретровирусным агентом, включая агент против ВИЧ, причем вирусная нагрузка является такой, как определено здесь выше.
Альтернативно, утверждение о неэффективности или снижении эффективности указанного предшествующего лечения может быть оценено путем измерения количества клеток CD4+ в плазме крови, которое уменьшилось (снова) до уровня ниже 500/мм3, в частности в течение нескольких последовательных недель, например, при по меньшей мере, одной или двух недель, в частности, по меньшей мере, трех недель или четырех недель лечения антиретровирусным агентом, включая агент против ВИЧ, причем количество клеток CD4+ определено более подробно здесь выше.
Для справки, восстановленное количество клеток CD4+ может соответствовать физиологическому (или «нормальному») количеству клеток CD4+, которое обычно равно или превосходит 500 клеток CD4+/мм3 плазмы, которое обычно варьирует между 500 и 1500 клеток CD4+/мм3 плазмы, хотя для отдельных индивидуумов оно может быть ниже.
Соответственно, низкое количество клеток CD4+ включает в себя количество клеток CD4+ ниже 500/мм3 в плазме крови, которое составляет менее 450, 350, 300; 250; 200; 150 и 100/мм3.
Согласно еще одному варианту осуществления изобретение относится к производному хинолина формулы (1), как определено здесь, или к одной из его фармацевтически приемлемых солей, пригодных для применения при лечении или профилактике вирусной инфекции или связанного с вирусом состояния у пациента, в особенности ВИЧ-инфекции или связанного с ВИЧ состояния, при котором пациент инфицирован устойчивым к лекарственным средствам штаммом.
Возникновение устойчивого к лекарственным средствам штамма у пациента может быть следствием:
- отбора штамма на устойчивость к лекарству у указанного пациента после предшествующего лечения, как описано выше; и/или
- первичного инфицирования пациента устойчивым к лекарству штаммом.
В частности, вышеупомянутые способы пригодны для лечения или профилактики вирусной инфекции или связанного с вирусом состояния, например, у пациентов со штаммами, устойчивыми к ламивудину (3ТС), устойчивыми к тенофовиру, устойчивыми к ралтегравиру и азидотимидину (AZT).
Производное хинолина в соответствии с изобретением также особенно подходит для лечения или профилактики вирусной инфекции или связанного с вирусом состояния у устойчивых к лечению индивидуумов, включая людей, инфицированных устойчивым штаммом ВИЧ, включая индивидуумов со штаммами, устойчивыми к ВААРТ и устойчивыми к APT.
Из-за широкой эффективности хинолиновых производных по изобретению теперь можно предлагать новые стратегии лечения, даже для пациентов с первичной инфицекцией, у которых в противном случае остаются неизлечимые штаммы.
Используемый здесь термин «устойчивость к лекарственным средствам против ВИЧ» относится к способности ВИЧ мутировать и воспроизводить себя в присутствии антиретровирусных лекарственных средств.
Для справки, «устойчивый к лекарственным средствам штамм ВИЧ» может быть определен путем измерения активности обратной транскриптазы (RT) в мононуклеарных клетках периферической крови человека (РВМС), инфицированных тестируемым штаммом, и затем обработки соединением или комбинацией соединения, в отношении которого подозревается резистентность.
Соответственно, пациент не обязательно ранее получал противовирусное лечение, включая антиретровирусное лечение или даже лечение против ВИЧ, отличное от указанного производного хинолина.
Соответственно, изобретение также относится к производному хинолина формулы (1), как определено выше, или любому из его метаболитов для применеия при лечении или профилактике ВИЧ-инфекции или связанного с ВИЧ состояния у пациента, при котором низкая или неопределяемая вирусная нагрузка сохраняется и/или количество клеток CD4+ сохраняется или восстанавливается после окончания лечения и для которого пациент ранее не получал антиретровирусного лечения, включая лечение против ВИЧ.
Соответственно, изобретение также относится к производному хинолина формулы (1), как определено выше, или любому из его метаболитов для использования при лечении или профилактике ВИЧ-инфекции или связанного с ВИЧ состояния у пациента, при этом пациент инфицирован штаммом ВИЧ, устойчивым к лекарственным средствам, причем пациент ранее не получал антиретровирусного лечения.
Примеры устойчивых к лекарственным средствам штаммов ВИЧ выбирают из: мутантов штамма NL4.3, K103N (устойчивых к эфавиренцу), K65R (устойчивых к тенофовиру и 3ТС) и мутантов M184V (устойчивых к 3ТС), штаммов ВИЧ-1 В и отобранных из Ad8 и AdaM; и клинических изолятов, выбранных из CRF01, CRF02 и CRF06.
Типичные устойчивые штаммы более подробно описаны у Pinar lyodogan et al. ("Current Perspectives on HIV-1 Antiretroviral Drug Resistance", Viruses 2014, 6,4095-4139; doi 10.3390/4095) и далее описаны ниже.
В частности, штамм вируса может быть штаммом, устойчивым к лекарственному средству или лечению, включающему введение лекарственного средства, выбранного из APT и/или терапии ВААРТ, и/или включая
(I) нуклеозидные/нуклеотидные ингибиторы обратной транскриптазы (НИОТ), также называемые нуклеозидными аналогами, такими как абакавир, эмтрицитабин и тенофовир;
(II) ненуклеозидные ингибиторы обратной транскриптазы (ННИОТ), такие как эфавиренц, этравирин и невирапин;
(III) ингибиторы протеазы (ИП), такие как атазанавир, дарунавир и ритонавир;
(IV) ингибиторы входа, такие как энфувиртид и маравирок;
(V) ингибиторы интегразы, такие как долутегравир и ралтегравир;
и их комбинации.
Соответственно, устойчивый к лекарственным средствам штамм ВИЧ охватывает устойчивые к НИОТ, ННИОТ, ИП, ингибиторам входа и ингибиторам интегразы штаммы ВИЧ.
Устойчивые штаммы известны в области техники и включают в себя не ограничивающим образом штаммы, несущие мутацию устойчивости, как описано в базе данных Международного общества по борьбе с вирусными заболеваниями (IAS-USA) и базе данных лекарственных средств от ВИЧ/СПИДа в Стенфорде.
Типичные устойчивые штаммы ВИЧ включают штаммы, несущие мутацию устойчивости, выбранные из:
- М41; К65; D67; К70; L74; Y115; М184 (включая М184 V/I); L210; Т215; К219; как основные мутации устойчивости к НИОТ;
- М41; А62; D67; Т69; К70; V75; F77; F116; Q151; L210; Т215; К219; как мутации с множественной устойчивостью к НИОТ;
- V90; А98; L100; К101; К103; V106; V108; Е138; V179; Y181; Y188; G190; Н221; Р225; F227; М230; как основные мутации устойчивости ННИОТ;
- L10; V11; G16; К20; L24; D30; V32; L33; Е34; М36; К43; М46; I47; G48; I50; F53; I54; Q58; D60; I62; L63; I64; Н69; А71; G73; L74; L76; V77; V82; N83; I84; I85; N88; L89; L90; I93 как основные мутации устойчивости к ингибитору протеазы;
- Т66; L74; Е92; Т97; Е138; G140, Y143; S147; Q148; N155 как основные мутации устойчивости к ингибиторам интегразы;
- G36; I37; V38; Q39; Q40; N42; N43 как основные мутации устойчивости к ингибитору входа; и их комбинации.
Следует отметить, что конкретные подкатегории мутантных/устойчивых штаммов, включая точечные мутации, такие как замены одного нуклеотида на другой, известны в данной области техники и также рассматриваются в изобретении.
Примеры лекарственных средств, для которых были найдены устойчивые к лекарственным средствам штаммы ВИЧ, включают: Зидовудин, Ламивудин, Эмтрицитабин, Диданозин, Ставудин, Абакавир, Залцитабин, Тенофивир, Рацивир, Амдоксовир, Априцитабин, Эльвуцитабин, Эфавиренц, Невирапин, Этравирин, Делавирдин, Рилпивирин, Тенофовир, Фосальвудин, Ампренавир, Типранавир, Индинавир, Саквинавир, Фосампренавир, Ритонавир, Дарунавир, Атазанавир, Нелфинавир, Лопинавир, Ралтегравир, Эльвитегравир, Долутегравир, Энфувиртид, Маравирок, Викривирок и их комбинации.
В частности, штамм ВИЧ, который подвергается лечению, может быть устойчивым к ламивудину (3ТС), тенофовиру, ралтегравиру, зидовудину (AZT), невирапину (NVP), эфавиренцу (EFV) и их комбинациям.
В качестве изобретения рассматриваются и применение, и способы.
Таким образом, изобретение также относится к способу лечения или профилактики вирусной инфекции или связанного с вирусом состояния у пациента, включая ВИЧ-инфекцию, состоящем во введении пациенту, нуждающемуся в этом, эффективного количества производного хинолина формулы (1), как описано выше, где указанный способ позволяет поддерживать низкую вирусную нагрузку после окончания лечения.
Таким образом, изобретение также относится к способу лечения или профилактики вирусной инфекции или связанного с вирусом состояния у пациента, включая ВИЧ-инфекцию, состоящему в введении пациенту, нуждающемуся в этом, эффективного количества производного хинолина формулы (1), как описано выше; а затем прекращении лечения, причем вирусная нагрузка низкая или неопределяемая; и/или уровень количества клеток CD4+ сохраняется или восстанавливается.
Таким образом, изобретение также относится к способу лечения или профилактики вирусной инфекции, в частности ВИЧ-инфекции, заключающемуся во введении пациенту, для которого были показаны неэффективность или снижение эффективности предшествующего антивирусного (или антиретровирусного) лечения, эффективного количества производного хинолина формулы (1), как описано выше.
Таким образом, изобретение также относится к способу лечения или профилактики вирусной инфекции, в частности ВИЧ-инфекции, заключающемуся во введении пациенту, инфицированному устойчивым к лекарственным средствам штаммом, эффективного количества производного хинолина формулы (1) как описано выше.
Согласно некоторым вариантам осуществления изобретение также относится к способу лечения ВИЧ-инфекции или связанного с ВИЧ состояния у пациента, состоящему из:
(I) введения пациенту, нуждающемуся в этом, эффективного количества производного хинолина формулы (1), таким образом, лечения пациента;
(II) прекращения лечения;
(III) необязательно измерения вирусной нагрузки и/или количества клеток CD4+ у указанного пациента после прекращения лечения; где предпочтительно:
- сохраняется низкая или неопределяемая вирусная нагрузка; и/или
- количество клеток CD4+ стабильно или увеличивается после окончания лечения;
(IV) необязательно введения снова указанному пациенту, нуждающемуся в этом, эффективного количества производного хинолина формулы (1), если вирусная нагрузка не является низкой или неопределяемой и/или количество клеток CD4+ уменьшается.
Соединение в соответствии с настоящим изобретением может быть реализовано в фармацевтической композиции, которая может содержать эффективное количество указанного соединения и один или более фармацевтических эксципиентов.
Вышеупомянутые эксципиенты выбирают в соответствии с лекарственной формой и желаемым способом введения.
В этом контексте они могут присутствовать в любой фармацевтической форме, пригодной для энтерального или парентерального введения, в сочетании с соответствующими эксципиентами, например, в виде обычных таблеток или таблеток покрытых оболочкой, твердого желатина, капсул с мягкой оболочкой и других капсул, суппозиториев или пригодных для питья, таких как суспензии, сиропы или инъекционных растворов или суспензий.
Может использоваться любой способ введения. Например, соединение формулы (1) можно вводить пероральным, парентеральным, внутривенным, трансдермальным, внутримышечным, ректальным, подъязычным, слизистым, назальным способом или другими средствами. Кроме того, соединение формулы (1) можно вводить в форме фармацевтической композиции и/или стандартной лекарственной формы.
В частности, фармацевтические композиции по изобретению могут вводиться перорально и/или парентерально.
Согласно одному иллюстративному варианту осуществления фармацевтические композиции согласно изобретению могут вводиться перорально.
Подходящие лекарственные формы включают, но не ограничиваются перечисленными, капсулы, таблетки (включая таблетки с быстрым растворением и с замедленным высвобождением), порошок, сиропы, пероральные суспензии и растворы для парентерального введения и, более конкретно, капсулы.
Фармацевтическая композиция может также содержать другое лекарственное средство для лечения ВИЧ, хорошо известное специалисту в данной области, в сочетании с соединением в соответствии с настоящим изобретением.
Следующие примеры представлены в качестве иллюстраций и никоим образом не ограничивают объем настоящего изобретения.
ПРИМЕРЫ
Пример 1: Синтез метил-2,3,4-три-O-ацетил-D-глюкопиранозилуронат-бромида - соединения (3)
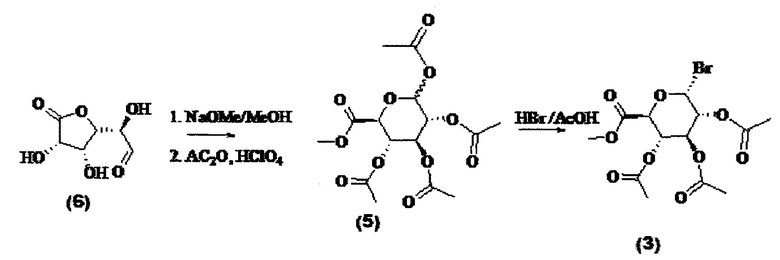
К коммерчески доступному соединению D-глюкуроно-6,3-лактону (6) (48,6 г, 276 ммоль) добавляли безводный метанол (500 мл) и металлический Na (200 мг) при 0°C. Смесь перемешивали в атмосфере азота в течение 5 часов. Раствор обрабатывали смолой Amberlite® IR-120 (Ir) до рН 3. После фильтрации растворитель удаляли в вакууме с получением желтой смолы. Остаток частично растворяли в Ac2O (100 мл) и к реакционной смеси по каплям добавляли раствор HClO4 (0,1 мл) в Ac2O (1 мл) с такой скоростью, чтобы температура раствора не превышала 40°C. Затем реакционную смесь перемешивали в течение ночи при комнатной температуре в атмосфере N2. Затем продукт растворяли в этилацетате, промывали 1Н HCl, H2O и соляным раствором, органическую фазу сушили над Na2SO4.
Растворитель удаляли вакуумной сушкой с получением промежуточного соединения (5) пер-О-ацетата (96,5 г, 93%) в виде белой смолы с соотношением α/β, состовляющим 75:25. Спектроскопические данные согласуются с ранее полученными спектроскопическими данными (1)
1Н ЯМР (400 MHz, CDCl3): α-аномер 6,42 (d, 1 Н, J=3,9 Hz, 1Н); 5,54 (dd, 1 Н, J1=10,0, J2=9,7 Hz, 1H); 5,24 (dd, 1 H, J1=10,2, J2=9,7 Hz,1 H); 5,14 (dd, 1 H,=10,0, J2=3,9 Hz, 1H); 4,43 (d, 1 H, J=10,2 Hz, 1H); 3,77 (s, 3 H, CO2Me); 2,21, 2,06, 2,03 (3 s, 12 H, 4 OAc).
1Н ЯМР (400 MHz, CDCl3): β-аномер 5,75 (d, 1 Н, J=7,8 Hz, 1 Н); 5,32-5,09 (m, 3 Н); 4,16 (d, 1 Н, J=9,3 Hz, H-5); 3,73 (s, 3 H, CO2Me); 2,10, 2,02, 2,01 (3 s, 12Н,4 OAc).
В полученное выше соединение (5) пер-О-ацетата (7,73 г, 20,54 ммоль) в атмосфере N2 при 0°C по каплям добавляли 45% HBr в уксусной кислоте (25 мл). Круглодонную колбу помещали внутрь эксикатора и перемешивали содержимое при 4°C в течение 48 часов. Смесь разбавляли этилацетатом (100 мл), выливали на лед (50 г). Раствор промывали насыщ. водн. NaHCO3 (50 мл), солевым раствором (50 мл), H2O (100 мл), органическую фазу сушили над Na2SO4, фильтровали и растворитель удаляли вакуумной сушкой. Продукт очищали флэш-хроматографией (гексан/этилацетат 2:1) с получением соединения глюкуронозилбромида (3) (3,50 г, 43%) в виде розовой смолы. Спектры ЯМР 1Н и 13С согласуются с литературными данными (Bollenback, G.N., Long, J.W., Benjamin, D.G., Lindquist, J.A., J.Am. Chem. Soc, 1955, 77, 3310).
1H ЯМР (400 MHz, CDCl3): 6,64 (d, J=4,1 Hz, 1H); 5,62 (dd, J1=9,9, J2=9,9 Hz, 1H); 5,25 (dd, J1=10,2, J2=9,9 Hz, 1H); 4,86 (dd, J1=9,9, J2=4,1 Hz, 1 H); 4,59 (d, J=10,2 Hz, 1H); 3,77 (s, 3 H, CO2Me); 2,11, 2,06, 2,05 (3 s, 9 H, 3 OAc).
Пример 2. Получение защищенных β-глюкуронидов соединения (4)
К раствору соединения (2) (10 г, 25,72 ммоль) в безводном толуоле (300 мл) добавляли карбонат кадмия (2,6 г, 15,12 ммоль) и весь раствор подвергали кипячению с обратным охлаждением в аппарате с ловушкой Дина-Старка в течение 12 часов. После охлаждения добавляли метил-α-ацетобромглюкуронат III (10,3 г, 25,94 ммоль) и все дополнительно кипятили с обратным холодильником в течение 24 часов. Осадок удаляли фильтрованием и промывали смесью CH2Cl2/СН3ОН: 95/05. Фильтрат и промывочный раствор объединяли и выпаривали. Маслянистый остаток очищали хроматографией на колонке на силикагеле, элюируя смесью CH2Cl2/MeOH: 98/02 с получением соединения (4) защищенного глюкуронида (3,5 г, 5,35 ммоль) в виде белой пены.
1Н ЯМР (400 MHz, CDCl3): 7,82 (d, J=9,2 Hz, 1H); 7,75 (dd, J1=7,53 Hz, J2=1,18 Hz, 1H); 7,56 (dd, J1=8 Hz, J2=1,18 Hz, 1H); 7,39 (d, J=8,43 Hz, 2H); 7,33 (d, J=8,43 Hz, 2H); 6,82 (d, J=8,1 Hz, 1H); 6,50 (d, J=8,65 Hz,1H); 5,47 (t, J=9,44 Hz, 1H); 5,18 (t, J=9,44 Hz, 1H); 4,86 (m, 1H); 4,35 (d, J=10,20 Hz, 1H); 3,71 (s, 3 H, CO2Me); 2,05, 1,95, 1,94 (3 s, 9 H, 3 OAc).
Пример 3. Получение N-β-глюкуронида 8-хлор-N-(4-(трифторметокси) фенил)хинолин-2-амина соединения (1)
Перекись водорода (30%, 10,4 мл) добавляли к перемешиваемой суспензии моногидрата гидроксида лития (4,73 г, 112 ммоль) в воде (44 мл) с образованием раствора в течение 3-4 минут. Смесь перемешивали еще 10 мин и затем добавляли к перемешиваемому раствору соединение (4) (3,4 г, 5,2 ммоль) в ТГФ (140 мл). Осадок образовывался в течение 15 мин, и смесь гасили добавлением тиосульфата натрия через 75 мин. О завершении реакции судили по тонкослойной хроматографии через 45 мин. Смесь подкисляли до рН 2,5 с помощью 1М соляной кислоты, и неочищенный продукт три раза экстрагировали этилацетатом. Объединенные экстракты сушили (Na2SO4), и растворитель удаляли при пониженном давлении, оставляя хроматографически чистый глюкуронид 2 (2,5 г, 4,86 ммоль, 93%), который подвергали хроматографии с получением желтого твердого вещества.
1Н ЯМР (400 MHz, ДМСО-D6): 8,07 (d, J=9,2 Hz, 1H); 7,81 (dd, J1=8 Hz, J2=0,9 Hz, 1H); 7,74 (dd, J1=8 Hz, J2=0,9 Hz, 1H); 7,56 (d, J=8,77 Hz, 2H); 7,52 (d, J=8,97 Hz, 2H); 7,28 (d, J=7,75 Hz, 1H); 6,50 (d, 1 H, J=9,2 Hz,1H); 6,34 (d, 1 H, J=8,2 Hz, 1H); 5,17 (broad, 2H); 3,78 (d, 1 H, J=9,4 Hz, 1H); 3,14-3,48 (m, 4H).
13C ЯМР (133 MHz, ДМСО-D6): 171,25; 157,07; 148,08; 142,86; 138,91; 133,11; 130,27; 130,02; 127,38; 125,37; 123,66; 122,58; 121,84; 119,29; 112,47; 84,27; 78,32; 72,35; 70,18
Пример 4: Соединения формул (1) и (2) взаимодействуют с комплексом СВС и способствуют взаимодействию СВР20 с СВР80.
1. Материал и методы
А. Подготовка рекомбинантного комплекса СВС для исследований in vitro.
Рекомбинантный комплекс СВС, содержащий СВР20 и СВР80, получают в соответствии с протоколом, который был описан у Worch, R. et al. (Specificity of recognition of mRNA 5' cap by human nuclear cap-binding complex. RNA 11, 1355-1363 (2005)).
Б. Маркировка соединения 1 фотоактивным фрагментом и индукция дозозависимого образования ковалентных мостиков.
Соединение 1 может производить ковалентное связывание с очищенным комплексом СВС после 15-минутного облучения ультрафиолетовым светом при длине волны 365 нм.
В. Анализ сдвига гель-подвижности
Рекомбинантный СВС человека инкубировали с РНК с кэпом в присутствии возрастающих концентраций соединения 1 или соединения 2 или m(7)GpppG-кеппированного аналога и анализировали с помощью нативного гель-электрофореза для того, чтобы разрешить различные комплексы РНК и РНК-белка в соответствии с Mazza et al. (Large-scale induced fit recognition of an m(7)GpppG cap analogue by the human nuclear cap-binding complex. EMBO J. 21, 5548-5557 (2002).
Г. Ограниченный протеолиз на комплексе СВС.
Ограниченный протеолиз комплекса СВС был установлен в соответствии с протоколом, описанным в Mazza et al. (Large-scale induced fit recognition of an m(7)GpppG cap analogue by the human nuclear cap-binding complex. EMBO J. 21, 5548-5557 (2002)).
Д. Масс-спектрометрический анализ.
Масс-спектрометрический анализ комплекса СВС был проведен в соответствии с протоколом, описанным в Schirie et al. (Mass spectrometry-based proteomics in preclinical drug discovery. Chem Biol., 19:72-84 (2012)).
Белки отделяли в гелях SDS-PAGE (4-15% полиакриламида, мини-PROTEAN® TGX™ Precast Gels, Bio-Rad, Hercules USA) и окрашивали с помощью Page Blue Stain (Fermentas). Полосы геля разрезали на 3 части и сушили с тремя промывками в 50% ацетонитриле и 50 мМ ТЕАВС (бикарбонат триэтиламмония). После восстановления белка (с 10 мМ дитиотреитола в 50 мМ ТЕАВС при 56°C в течение 45 мин) и алкилирования (55 мМ йодацетамида ТЕАВС при комнатной температуре в течение 30 мин) белки расщепляли в геле с применением трипсина (1 мкг/полоса, Gold, Promega, г. Мэдисон, США), как описано ранее у Shevchenko et al. (Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68 (5), 850-8; 1996). Обработанные продукты дегидратировали в вакуумной центрифуге и уменьшали объем до 4 мкл.
Сгенерированные пептиды анализировали в режиме реального времени с применением нанофлюидной ВЭЖХ-наноэлектрораспылительной ионизацией на масс-спектрометре Q-Exactive (ThermoScientific, Уолтем, США) в сочетании с аппаратом Ultimate 3000 RSLC (Thermo Fisher Scientific). Осаждение и предварительную концентрацию образцов проводили в режиме онлайн на предварительной колонке Рертар® (0,3 мм × 10 мм). Градиент, состоящий из 0-55% В в течение 35 мин и 90% В в течение 10 мин (А=0,1% муравьиной кислоты в воде, В=0,1% муравьиной кислоты, 80% ацетонитрила в воде) при 300 нл/мин использовали для элюирования пептидов из капиллярной (0,075 мм × 150 мм) колонки с обращенной фазой (Acclaim РерМар® RSLC, Thermo Fisher Scientific), оснащенной непокрытым силиконовым PicoTip Emitter (NewOjective, Уоборн, США). Элюированные пептиды подвергали электрораспылению онлайн при напряжении 1,9 кВ на масс-спектрометре Q-Exactive. Спектры MS (m/z, 400-2000) были получены с применением программного обеспечения Xcalibur (v 3.0, Thermo Fisher Scientific) в режиме положительного иона с разрешением 70000 для сканирования ионов-предшественников. Для всех полных измерений сканирования с помощью детектора Orbitrap в качестве внутреннего калибратора использовался ион блокирующей массы из окружающего воздуха (м/з 445,120024), как описано у Olsen et al. Olsen et al. (Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010-2021; 2005). Спектры MS/MS были получены в режиме зависимого от процесса сбора данных, в котором ТОР10 наиболее распространенных ионов-предшественников с максимальным временем интегрирования 250 мсек и целевым значением ионов 3*106. Фрагментацию пептида осуществляли путем столкновительной диссоциации более высокой энергии, установленной при 26 В нормализованной энергии столкновения. Спектры MS/MS были получены с разрешением 17500 с целевым значением ионов 1*105 и максимальным временем интегрирования 120 мсек.
Все спектры MS/MS были исследованы по базе данных CPS Homo sapiens (85895 последовательностей и специфических последовательностей СВР80-СВС20, выпуск от сентября 2014 г., http://www.uniprot.org/) с применением программного обеспечения Proteome Discoverer v1.4 (Thermo Fisher Scientific) и алгоритма Mascot v2.5 (http://www.matrixscience.com/) с трипсиновой ферментативной специфичностью и одним пропущенным расщеплением трипсином. Карбамидометилирование устанавливали как фиксированную модификацию цистеина, а окисление устанавливали как переменную модификацию метионина для поиска. Массовые допуски в MS и MS/MS устанавливали соответственно 5 миллионных долей и 0,5 Да. Управление и валидация данных масс-спектрометрии проводили с применением программного обеспечения Proteome Discoverer (порог значимости Mascot р<0,05 с минимум одним пептидом на белок).
Помимо идентификации белка/пептида, программное обеспечение Skyline v2.6. (http://proteome.gs.washington.edu/software/skyline) использовали для обработки хроматограмм ионной интенсивности конкретных пептидов из полномасштабных масс-спектральных данных (MS1), полученных в ходе протеомических экспериментов HPLC MS/MS, как описано у MacLean et al. (Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal Chem 82, 10116-10124; 2010).
2. Результаты
Используя производное соединения 2, которое имеет фотоактивируемую часть, и конкуренцию с соединением 2 на очищенных рекомбинантных СВР20 и СВР80 (СВС), мы обнаружили, что само соединение 2 может индуцировать дозозависимое образование ковалентных мостиков между СВР20 и СВР80 после облучения ультрафиолетом, и этот комплекс может быть разрешен с помощью SDS-PAGE. Те же результаты были получены с глюкуронидированным соединением 1, более растворимым производным соединения 2, которое получают в виде метаболита с применением гепатоцитов человека.
Масс-спектрометрический анализ очищенных гелем СВР20, СВР80 и комплекса СВС (80 и 20) показал, что расщепление трипсином комплекса СВС (СВР80 и СВР20) привело к росту всех предсказанных пептидов, за исключением пептида в положении 37-66 СВР20, который воспроизводимо недопредставлен или отсутствует. Однако индивидуальное расщепление трипсином или СВР20 или СВР80 из того же образца привело к росту всех предсказанных пептидов. Примечательно, что пептид в положении 37-66 в кристаллической структуре СВС (Mazza et al.] Crystal structure of the human nuclear cap binding complex. Mol. Cell 8, 383-396 (2001)), соответствует интерфейсу между CBP20 и СВР80, который может быть местом взаимодействия между соединением 2 и СВС.
Однако ни соединение 2, ни его метаболит соединение 1 не влияло на связывание комплекса СВС с РНК с кэпом в анализе сдвига подвижности геля. В то время как комплекс СВС и РНК с кэпом конкурировал с m7GpppG, никакой конкуренции не наблюдалось в отношении соединений 1 или 2 при любой проверенной концентрации, что подтверждает, что оба соединения не взаимодействуют с сайтом связывания кэпа с СВР20 (Фиг. 1А и 1Б).
Эти результаты, таким образом, полностью подтверждают общий механизм взаимодействия между соединениями 1 и 2. Эти результаты также подтверждают, что оба соединения напрямую связываются с комплексом СВС, но не препятствуют связыванию с кепом и не экспортируют объемные транскрипты pol II, одновременно предотвращая экспорт вирусных РНК, включая Rev-опосредованный экспорт вирусной РНК.
Пример 5: Способность соединений 1 ингибировать продукцию ВИЧ-1 в клетках, инфицированных макрофагами.
1. Материал и способы
А. Клеточная культура и инфекция
Лейкоцитарную фракцию от ВИЧ-отрицательных индивидуумов получали из местного пункта взятия крови в Цюрихе, Швейцария (http://www.blutspendezurich.ch/) и Центра переливания крови, г. Монпелье. Мононуклеарные клетки периферической крови человека (РВМС) выделяли центрифугированием в градиенте Ficoll (Axis-Shield РоС AS). Затем клетки культивировали при 37°C, 5% СО2 до плотности 1×106 клеток/мл в среде RPMI Glutamax (Life Technologies Ref. 61870-010), дополненной 10% фетальной бычьей сывороткой (FCS) (Thermo Fischer Ref SV30160. 03), 1000 ед/мл IL2 (Peprotech Ref 200-02) и 5 мкг/мл ФГА (Roche Ref 1249738) для активации. Через три дня клетки объединяли и ресуспендировали до плотности 1×106 клеток/мл в среде RPMI Glutamax, дополненной 10% фетальной бычьей сывороткой (FCS) 1000 U/мл IL-2 для инфекции. Изучение ВИЧ-1 инфекции проводили на 10 мкг штамма ВИЧ-инфекции Ada-M R5 на мл клеток в течение 4 часов. Затем клетки центрифугировали и ресуспендировали до плотности 1×106 клеток/мл в среде, дополненной разбавленным ДМСО солюбилизированным лекарственным средством (Sigma Ref D4818) в соответствии с конечной концентрацией 0,05% ДМСО. Клетки обрабатывали в течение 6 дней с частичным изменением среды на 3-й день. Супернатант для культивирования клеток с ВИЧ р24 проводили с помощью ELISA с помощью набора Ingen Innotest (Ingen Ref 80564) в соответствии с инструкциями производителя.
Для получения макрофагов, полученных из моноцитов (МДМ), моноциты выделяли с применением микробидов CD14 (№ по каталогу 130-050-201, Miltenyi) и культивировали в среде X-VIVO10 (Lonza), дополненной GM-CSF 1000U/мл и М-CSF 100 нг/мл в течение 6 дней. Моноциты высевали в количестве клеток 50000 клеток на лунку в 96-луночном планшете. Через 6 дней среду заменяли на X-VIVO10 без цитокинов. Через 2 дня макрофаги обрабатывали соединением (1) ночью и на следующий день заражали вирусом Yu-2 в течение 6 часов, промывали PBS и культивировали в среде, содержащей эти соединения в течение 12 дней. Супернатант для р24 ELISA собирали 2 раза в неделю.
2. Результаты
На Фиг. 2 показаны результаты. График внизу и справа объединяет результаты трех других графиков. «Cpd (1)» означает соединение формулы (1).
Клетки обрабатывали от 1,5 мкМ до 30 мкМ, а уровни антигена р24 контролировали в супернатанте культуры в течение 12-дневного периода (R-10 соответствует необработанным клеткам) (Фиг. 2). Интересно, что соединение (1) эффективно блокирует репликацию вируса и эффект зависит от дозы, достигая уровней ингибирования до 60% в первичных макрофагах при 30 мкМ.
Эти результаты свидетельствуют о том, что соединение (1) согласно изобретению обладает низкой токсичностью, но остается пригодным для ингибирования репликации ВИЧ-1 в макрофагах.
Пример 6: Фармакокинетические (ФК) параметры соединения (1) после однократного перорального введения соединения (2).
1. Материал и способы
1.1 Группа пациентов
В настоящем документе описываются результаты, касающиеся фармакокинетики (ФК), полученные в первом исследовании на людях соединения (2), включающем введение одной пероральной восходящей дозы здоровым мужчинам. Было исследовано четыре (4) уровня дозировки (50, 100, 150 и 200 мг).
На каждом уровне дозы в исследование были включены 6 субъектов, и они получили одну дозу соединения (2) перорально, так что в исследование было включены и завершили его 24 субъекта. При анализе ФК не было выявлено отклонения от исследования, и все испытуемые были включены в анализ ФК.
В этом исследовании измеряли уровни соединения (1) в плазме.
Первоначальный забор крови для анализа на ФК проводили до 48 ч после введения дозы. В дополнение к первым результатам после однократного перорального введения 50 мг соединения (2) показано, что соединение (1) проявило длительный период полувыведения (t1/2), было принято решение увеличить повторность образцов для анализа ФК, добавив 3 собранных образца крови до 45 дней после введения соединения (2).
Сбор образцов крови для оценки уровней соединения (1) в плазме запланирован в следующие моменты времени:
- День 1. Перед приемом дозы, через 0,33, 0,66, 1,00, 1,50, 2,00, 2,50, 3,00, 4,00, 6,00, 8,00, 10,00 и 12,00 ч после приема;
- День 2. Через 24,00 и 36,00 ч после приема;
- День 3. Через 48,00 ч после приема;
- День 10. Через 240 ч после приема;
- День 24. Через 576 ч после приема;
- День 45. Через 1080 ч после приема.
1.2 Фармакокинетика
Концентрации в плазме обрабатывались всем программным обеспечением для ФК анализа для генерации данных об ФК. Параметры ФК были рассчитаны с помощью анализа без учета компартнемтов (NCA) с применением Phoenix® WinNonlin® (Pharsight Corporation), работающего на персональном компьютере.
Для расчета параметров и характеристик ФК были применены следующие правила:
- Все подтвержденные концентрации в плазме, предоставленные исследователю фармакокинетики, использовались для анализа ФК.
- Использовались фактические моменты времени отбора проб крови, связанные с предшествующим введением лекарственного средства.
- В моменты времени в промежутке времени между нулевым временем и первой концентрацией, равной или превышающей предельную величину квантования (LOQ), концентрации ниже LOQ устанавливали на ноль (0). Концентрации ниже предела количественной оценки (BLOQ) между 2 концентрациями, равными или превышающими LOQ, считались недостающими данными. В расчетах использовались замыкающие концентрации BLOQ.
- Если концентрация перед введением дозы лекарственного средства была отсутствующей, она была произвольно принята за ноль (0), что предполагало, что ожидаемыми результатами были бы BLOQ.
Для каждого субъекта, получающего активное лечение в каждой группе, должны были быть получены следующие параметры ФК (1):
Cmax Наблюдаемая максимальная концентрация, измеренная в плазме, была получена непосредственно из данных о времени после приема и концентрации.
tmax Время, в течение которого Cmax была очевидной, определенное путем проверки концентрации лекарственного средства в плазме в сравнении с данными по времени с помощью Phoenix® WinNonlin®.
AUC0-t Площадь под кривой концентрации-времени от нулевого времени (до приема дозы) до времени последней измеряемой концентрации рассчитывалась с применением линейного метода трапеций.
ke Константа скорости конечной элиминации из плазмы оценивалась посредством анализа логарифмической регрессии конечной фазы профиля времени и концентрации в плазме. Количество точек, включенных в конечную фазу, определялось визуальным осмотром полулогарифмических графиков концентрации в плазме в зависимости от времени (не менее 3).
AUC0-∞ AUC от времени 0 до бесконечности рассчитывали по формулам AUC0-∞=AUC0-t+AUCt-∞, где AUC0-t представляет собой площадь под кривой концентрации-времени от нулевого времени (до приема дозы) до времени последней измеряемой концентрации, рассчитанную с применением логарифмически-линейного метода трапеций, и AUCt-∞=Ct/ke, где Ct представляет собой измеренную концентрацию во время последней измеренной концентрации t. Экстраполированная часть AUC0-∞ должна составлять <20% для того, чтобы значение считалось надежным.
t1/2 Период полувраспада видимой конечной элиминации рассчитывали как In2/ke, где ke представляет собой константу скорости элиминации во время конечной фазы, определяемую логарифмической регрессией, полученной по меньшей мере по 3 последним измеренным концентрациям и с применением фактического времени забора крови. Коэффициент корреляции для соответствия подгонки линии регрессии через точки данных должен составлять 0,8500 или выше, чтобы значение считалось надежным.
2. Результаты
После однократного перорального введения соединения (2), независимо от дозы, биотрансформация соединения (2) в соединение (1) происходит быстро и оказывается основным путем метаболизма лекарственного средства, поскольку концентрация соединения (1) в плазме значительно выше, чем в исходном лекарственном средстве. Концентрация первого количественного оцененного соединения (1) обычно наблюдается через 40 минут после введения дозы (у 20 субъектов из 24), ранее в одном случае (20 минут после дозы для субъекта №306), а затем в 3 случаях (1 час после приема для субъектов №102, №106 и №406).
Затем концентрации в плазме возрастают до Cmax, как правило, через 4 часа после приема дозы (2/3 испытуемых), но в пределах от 3 часов после приема дозы (2 случая) до 6 часов после приема дозы (6 случаев). Снижение концентрации соединения (1) в плазме происходит медленно и может наблюдаться до 24-45 дней после введения лекарственного средства.
Во всех случаях значения ФК определяется с очень хорошей точностью, а экстраполированная область под кривой (AUC) была очень ограниченной, поэтому все параметры ФК всех субъектов являются надежными и используются для дальнейшей статистической оценки.
Индивидуальные концентрации в плазме представлены уровнем дозы. Описательная статистика для концентраций в плазме представлена как среднее и стандартное отклонение (SD) и рассчитывается, если по меньшей мере 2/3 (т.е. 4 из 6) значений в плазме в момент времени были выше предела количественной оценки (LOQ).
Описательная статистика параметров ФК была представлена как среднее арифметическое, SD, коэффициент вариации (CV%), медианное, минимальное (Min) и максимальное (Мах) значения и среднее геометрическое (GM).
Описательная статистика полученных фармакокинетических (ФК) параметров соединения (1) после однократного перорального введения 50-200 мг соединения (2) приведена ниже в Таблице 1.
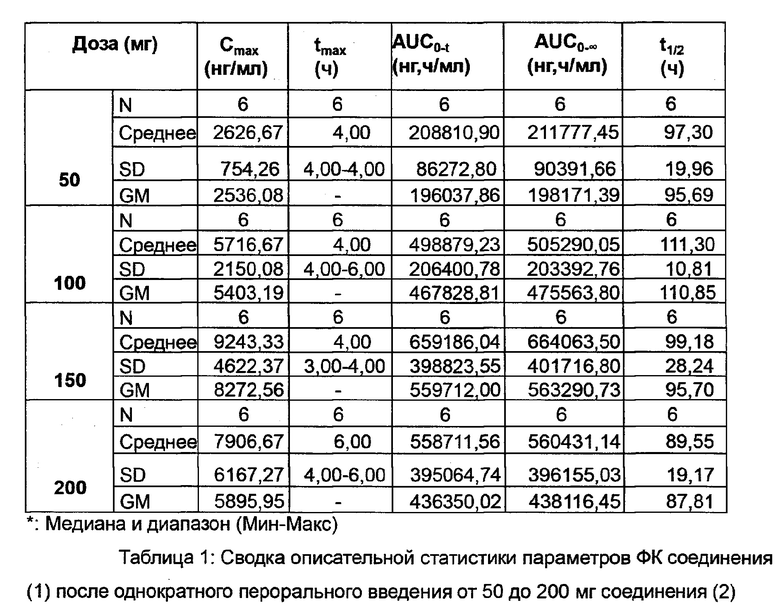
Медиана tmax была сопоставима по уровням дозы, варьирующим от 4 до 6 ч после приема дозы с очень низкой индивидуальной изменчивостью.
Индивидуальная изменчивость Cmax и AUC, как правило, увеличивалась с дозами соединения (2), составляя менее 50% для 2 более низких уровней доз и достигая 70-80% при 200 мг. Можно отметить, что значения Cmax и AUC были в пределах одного и того же диапазона для субъектов из группы 200 мг.
t1/2 соединения (1) было приблизительно сопоставимо со средними значениями, а изменчивость от 50 до 200 мг составляла от 90 до 110 ч.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ХИНОЛИНА ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ИЛИ ПРОФИЛАКТИКЕ ВИРУСНОЙ ИНФЕКЦИИ | 2016 |
|
RU2723016C2 |
| МикроРНК-124 В КАЧЕСТВЕ БИОМАРКЕРА | 2014 |
|
RU2687366C2 |
| ПРОИЗВОДНЫЕ, РОДСТВЕННЫЕ ЛИЗИНУ, КАК ИНГИБИТОРЫ АСПАРТИЛПРОТЕАЗЫ ВИЧ | 2007 |
|
RU2458916C2 |
| СПОСОБ ИНГИБИРОВАНИЯ РЕПЛИКАЦИИ ВИЧ В КЛЕТКАХ МЛЕКОПИТАЮЩИХ И У ЛЮДЕЙ | 2011 |
|
RU2593948C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОПТИМАЛЬНОЙ ХИМИОТЕРАПИИ ПАЦИЕНТОВ, СЕРОПОЗИТИВНЫХ ПО ВИЧ, ОСНОВАННЫЙ НА ФЕНОТИПИЧЕСКОЙ ЛЕКАРСТВЕННОЙ ЧУВСТВИТЕЛЬНОСТИ ЧЕЛОВЕЧЕСКИХ ШТАММОВ ВИЧ | 1997 |
|
RU2174014C2 |
| КОМПОЗИЦИИ ДЛЯ ПРЕДУПРЕЖДЕНИЯ И/ИЛИ ЛЕЧЕНИЯ ИНФЕКЦИИ, ВЫЗВАННОЙ ВИРУСОМ ВИЧ-1 | 2012 |
|
RU2603262C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ И(ИЛИ) ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ВИЧ У ЛЮДЕЙ | 2012 |
|
RU2609769C2 |
| ПРОИЗВОДНЫЕ ИНГЕНОЛА ДЛЯ РЕАКТИВАЦИИ ЛАТЕНТНОГО ВИРУСА ВИЧ | 2013 |
|
RU2609512C2 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ИНФЕКЦИОННЫХ ЗАБОЛЕВАНИЙ | 2014 |
|
RU2579262C1 |
| Циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноаты, способ их получения и применения | 2017 |
|
RU2647576C1 |
Изобретение относится к производному хинолина формулы (1) или к одной из его фармацевтически приемлемых солей, выбранных из гидробромида, тартрата, цитрата, трифторацетата, аскорбата, гидрохлорида, трифталата, малеата, мезилата, формиата, ацетата и фумарата. Изобретение также относится к фармацевтической композиции, лекарственному средству, способу получения, соединению формулы (4). Технический результат: получено новое производное хинолина формулы (1), которое может применяться для лечения или профилактики ВИЧ-инфекции или связанного с ВИЧ состояния. 5 н. и 5 з.п. ф-лы, 6 ил., 1 табл., 6 пр.
1. Производное хинолина формулы (1)
или одна из его фармацевтически приемлемых солей, выбранных из гидробромида, тартрата, цитрата, трифторацетата, аскорбата, гидрохлорида, трифталата, малеата, мезилата, формиата, ацетата и фумарата.
2. Производное хинолина по п. 1 или одна из его фармацевтически приемлемых солей, выбранных из гидробромида, тартрата, цитрата, трифторацетата, аскорбата, гидрохлорида, трифталата, малеата, мезилата, формиата, ацетата и фумарата, для применения при лечении или профилактике ВИЧ-инфекции или связанного с ВИЧ состояния.
3. Фармацевтическая композиция для лечения или профилактики ВИЧ-инфекции или связанного с ВИЧ состояния, содержащая производное хинолина по п. 1 в эффективном количестве или одну из его фармацевтически приемлемых солей, выбранных из гидробромида, тартрата, цитрата, трифторацетата, аскорбата, гидрохлорида, трифталата, малеата, мезилата, формиата, ацетата и фумарата, и по меньшей мере один фармацевтически приемлемый эксципиент.
4. Лекарственное средство для лечения или профилактики ВИЧ-инфекции или связанного с ВИЧ состояния, содержащее производное хинолина по п. 1 в эффективном количестве или одну из его фармацевтически приемлемых солей, выбранных из гидробромида, тартрата, цитрата, трифторацетата, аскорбата, гидрохлорида, трифталата, малеата, мезилата, формиата, ацетата и фумарата.
5. Способ получения производного хинолина по п. 1, включающий стадию обработки соединения (4)
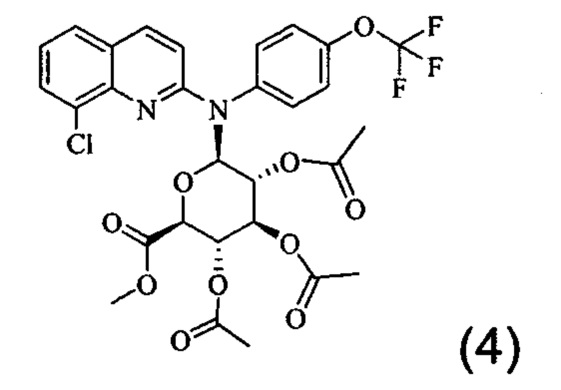
в растворе гидропероксида лития с получением таким образом производного хинолина по п. 1.
6. Способ по п. 5, где стадию обработки соединения (4) в растворе гидропероксида лития проводят в растворителе, таком как тетрагидрофуран или диоксин.
7. Способ по п. 5 или 6, включающий стадию взаимодействия соединения формулы (2)
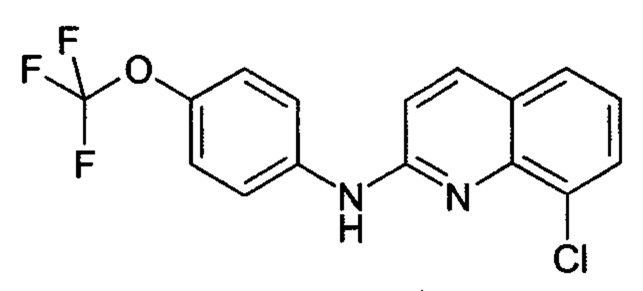
с соединением формулы (3)
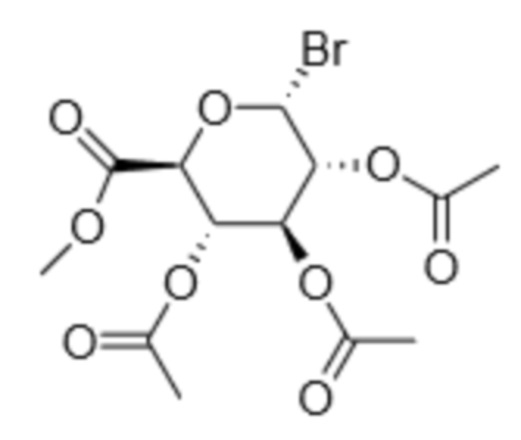 (3)
(3)
в присутствии соли тяжелого металла с получением таким образом соединения (4).
8. Способ по п. 7, где соль тяжелого металла представляет собой соль кадмия.
9. Способ по п. 7 или 8, где стадию реакции соединения формулы (2) осуществляют в растворителе, представляющем собой нитрометан.
10. Соединение формулы (4)
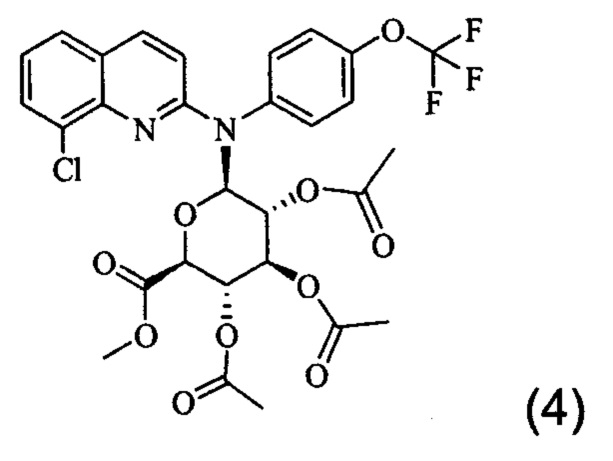
или одна из его фармацевтически приемлемых солей, выбранных из гидробромида, тартрата, цитрата, трифторацетата, аскорбата, гидрохлорида, трифталата, малеата, мезилата, формиата, ацетата и фумарата.
| WO 2010143169 A2, 16.12.2010 | |||
| WO 2015001518 A1, 08.01.2015 | |||
| RU 2011149571 A, 20.07.2013 | |||
| Lai-Xi Wang et al | |||
| "Resveratrol glucuronides as the metabolites of resveratrol in humans: Characterization, synthesis, and anti-HIV activity" Journal of Pharmaceutical Sciences, vol.93, N10, 2004, 2448-2457 | |||
| M | |||
| T | |||
| Bilodeau et al | |||
| "Potent |

