Настоящее изобретение относится к химиотерапевтическим средствам для лечения вирусных и раковых заболеваний. Эти соединения являются пролекарствами ингибиторов вируса имунодефицита человека (ВИЧ), вируса гепатита В (ВГВ, HBV) ДНК-полимеразы, вируса гепатита С полимеразы (ВГС, HCV) и предназначены для лечения вируса иммунодефицита человека, гепатита С, гепатита В, а также ко-инфекций ВИЧ/ВГС, ВИЧ/ВГВ, ВИЧ/ВГС/ВГВ и ВГС/ВГВ.
Вирус иммунодефицита человека (ВИЧ) относится к лентивирусам приматов, которые являются этиологическими агентами синдрома приобретенного иммунодефицита (СПИД). Болезнь была впервые описана в 1981 году, а ВИЧ-1 был выделен в конце 1983 г. С тех пор СПИД стал всемирной эпидемией, расширяется по масштабам и величине, такие инфекции как ВИЧ-инфекции повлияли на различные группы населения и географические регионы. Миллионы людей заражены во всем мире; в случае заражения люди остаются инфицированными всю жизнь. В течение десяти лет, при отсутствии антиретровирусной терапии, у подавляющего большинства ВИЧ-инфицированных индивидуумов болезнь развивается со смертельным исходом под влиянием инфекций, вызванных в результате ВИЧ-индуцированных недостатков в иммунной системе. СПИД является одной из самых важных проблем общественного здравоохранения во всем мире в начале 21-го века. Развитие высокоактивной антиретровирусной терапии (ВААРТ) при хроническом подавлении репликации и профилактики СПИДа ВИЧ является крупным достижением в области медицины ВИЧ [http://basicmedicalkey.com/aids-and-lentiviruses/].
ВИЧ продолжает оставаться одной из основных глобальных проблем общественного здравоохранения. В 2015 году, с ВИЧ живут 36,7 миллиона человек (в том числе 1,8 миллиона детей) - глобальная распространенность ВИЧ на уровне 0,8%. Подавляющее большинство из этого числа живут в странах с низким и средним уровнем доходов. В том же году, 1,1 миллиона человек умерли от заболеваний, связанных со СПИДом. С начала эпидемии, по оценкам экспертов, 78 миллионов человек заразились ВИЧ и 35 миллионов человек умерли от связанных со СПИДом заболеваний. По оценкам, 25,5 миллиона человек, зараженных ВИЧ, живут в странах Африки к югу от Сахары. Подавляющее большинство из них (по оценкам, 19 миллионов) живут на востоке и на юге Африки, что составило 46% новых случаев ВИЧ-инфекции во всем мире в 2015. Около 40% всех людей, живущих с ВИЧ, не знают, что у них есть вирус [http://www.avert.org/global-hiv-and-aids-statistics].
Развитие антиретровирусных препаратов существенно изменило восприятие ВИЧ / СПИДа от фатального до хронического и потенциально управляемого заболевания, а также наличие и введение антиретровирусной терапии (APT) привело к значительному снижению смертности и заболеваемости, связанной с ВИЧ и СПИДом. Существует взаимосвязь между антиретровирусной терапией (APT) и качеством жизни людей, живущих с ВИЧ и СПИДом, а также несколько исследований отметили сильную положительную связь между APT и улучшением качества жизни в различных областях среди людей, живущих с ВИЧ и СПИДом как в развитых, так и развивающихся странах [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3418767/].
ВИЧ-инфицированные пациенты живут дольше со времени введения высокоактивной антиретровирусной терапии (ВААРТ). При проведении антиретровирусной терапии продолжительность жизни пациента может быть продлена до 70-80 лет. Однако сопутствующая инфекция с ВГВ или/и ВГС приводит к повышенной заболеваемости и смертности от болезней печени. Неконтролируемая ВИЧ-инфекция ускоряет прогрессирование ВГС-индуцированного склероза печени.
Сопутствующая инфекция ВИЧ/ВГВ является обычным явлением. Хроническая инфекция ВГВ встречается у 5-10% ВИЧ-инфицированных лиц, которые подвергаются ВГВ, со скоростью в 10 раз выше, чем для населения в целом [http://hivinsite.ucsf.edu/InSite?page=kb-05-03-04#S1X].
ВИЧ/ВГС-инфицированные больные имеют в три раза больший риск развития цирроза или декомпенсации заболевания печени, чем пациенты с ВГС - моноинфекцией [https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv-guidelines/26/hcv-hiv]. В 2006 году в многонациональной когорте, включающей более 25000 ВИЧ-инфицированных людей в США и Европе, 14% смертей были связаны с заболеваниями печени и, из них, 66% произошло у лиц с сопутствующей инфекцией ВИЧ/ВГС [http://hivinsite.ucsf.edu/InSite?page=kb-00&doc=kb-05-03-05].
Моноинфекция ВГС или ВГВ представляет собой одну из основных причин хронических заболеваний печени во всем мире. Тем не менее, в эндемичных районах, значительное число пациентов инфицированы обоими вирусами в основном в результате общих путей передачи. Многочисленные исследования показали, что ВГС/ВГВ - инфицированные пациенты подвержены большему риску заболевания циррозом печени и гепатоцеллюлярной карциномой по сравнению с моноинфицированными пациентами. Поразительно, что примерно у 60% пациентов с неактивной формой инфекции ВГВ до начала лечения гепатита С может происходить ВГВ реактивация, в то время как другие пациенты, инфицированные ВГВ испытывают проявление сероконверсии антигена гепатита В [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4367211/].
Нуклеозиды (и нуклеотиды уже используются в клинической практике в течение почти 50 лет и стали краеугольным камнем при лечении пациентов с вирусными инфекциями или раком. Разработка нескольких новых препаратов за последнее десятилетие показывает, что этот класс соединений все еще обладает большим потенциалом. Нуклеоз(т)иды сыграли важную роль в лечении вирусных заболеваний. Для пациентов с вирусом иммунодефицита человека (ВИЧ) они оказались основой в ряде комбинированных схем лечения. В настоящее время нуклеоз(т)иды являются предпочтительным вариантом и стандартом лечения пациентов, инфицированных вирусом гепатита В (HBV).
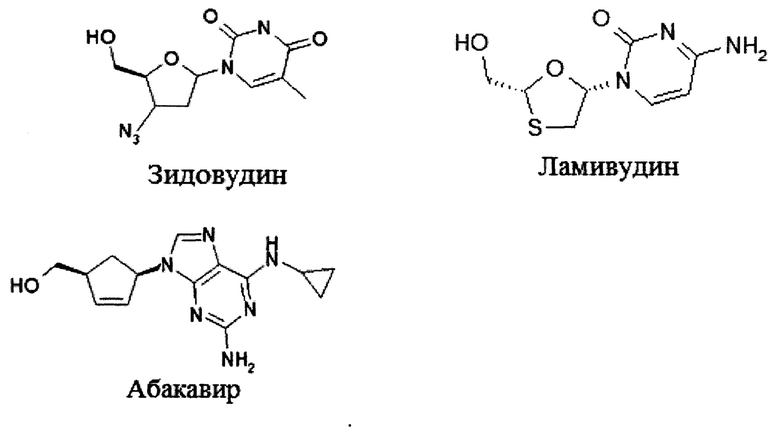
Ряд нуклеоз(т)идных ингибиторов RT был одобрен для лечения ВИЧ-инфекции [R.F. Shinazi et al. Pharmacology of current and promising nucleosides for the treatment of human immunodeficiency viruses. J. Antiviral Res. 2006, 71, 322-334. E. De Clereq. The nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, and protease inhibitors in the treatment of HIV infections (AIDS). Adva Pharmacol. 2013, 67, 317-358]. Некоторые из них используют в адъювантной терапии совместно с другими ингибиторами репликации ВИЧ, обеспечивающими удобные терапевтические режимы, которые стали стандартом в высокоактивной антиретровирусной терапии (ВААРТ). Эти комбинации включают Комбивир (Combivir®), Тризивир (Trizivir®), Эпзиком (Epzicom®), Трувадас (Truvadas), Атрипл (Atriple), Стрибил (Stribile), и Комплерас (Compleras). Трувадас, Атрипл, Комплерас и Стрибил включают нуклеозид эмтрицитабин и ациклический нуклеотид тенофовир диизопропилфумарат (TDF), в то время как Комбивир (CombivirTM), Тризивир (Trizivir) и Эпзиком (Epzicom), включают в себя комбинацию из двух или трех лекарств, содержащих нуклеозиды Зидовудин (Zidovudine (AZT)), Ламивудин (Lamivudine, 3ТС, Nuc21), и/или Абакавир (Abacavir (ABC)) [R.F. Shinazi et al. Pharmacology of current and promising nucleosides for the treatment of human immunodeficiency viruses. J. Antiviral Res. 2006, 71, 322-334]. Успех антиретровирусной терапии ВИЧ привел к резкому увеличению продолжительности жизни людей, инфицированных этой когда-то неизлечимой болезнью. Несмотря на эти успехи, продолжается поиск новых агентов, направленных на лечение хронически инфицированного населения, снижение резистенции и побочных эффектов, связанных с долгосрочным использованием лекарственных средств.
Одним из достаточно новых пролекарств противовирусного ациклического нуклеозида является фосфонат тенофовир (TFV). Улучшенные свойства по сравнению с ним показал дизопроксилфумарат тенофовир (TDF), а так же тенофовир алафенамид и его соли (TAF, GS-7340, Vemlidy). Это пролекарство является противовирусным препаратом и было предназначено именно для комбинированного лечения пациентов, нуждающихся в этом [WO 2002008241. US 7390791. A.S. Ray, M.W. Fordyce, M.J.M. Hitchcock. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus. Antiviral Research Volume 125, January 2016, Pages 63-70. WO 2002008241. US 7390791.
WO 2013025788, WO 2013116720, US 9296769.
http://www.gilead.ca/pdf/ca/genvoya_pm_english.pdf.
http://www.accessdata.fda.gov/drugsatfdadocs/nda/2015/207561_Origls000PharmR.pdf.].
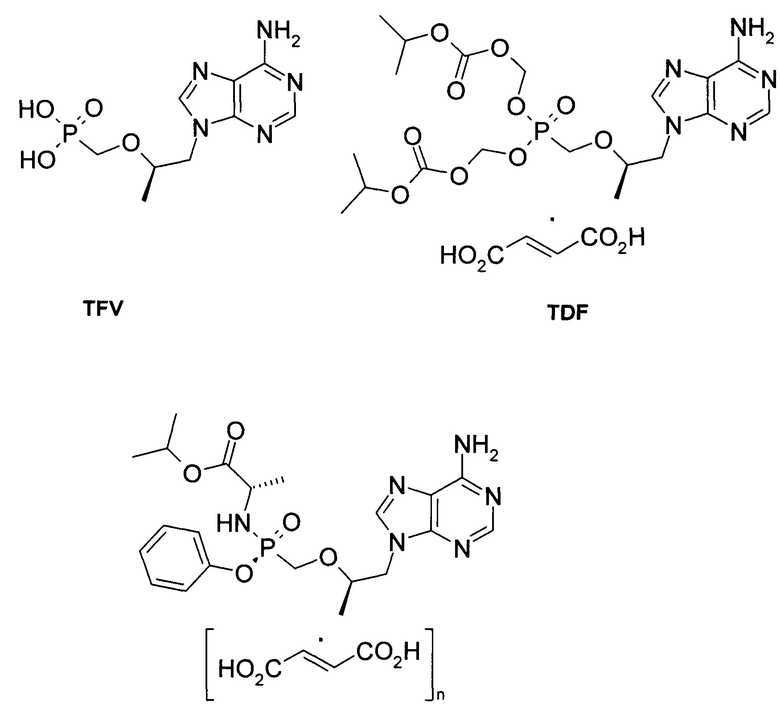
n=0: TAF (Тенофовир алафенамид, GS-7340, Vemlidy).
n=1: TAF фумарат.
n=0,5: TAF полуфумарат.
В 2015 году FDA одобрило первую композицию на основе TAF для комбинированного лечения ВИЧ [http://www.dailykos.com/story/2013/4/l0/1200735/-Gilead-s-New-FDA-Approved-HIV-Drug-Improves-Nothing-So-Naturally-It-Costs-A-Lot], а в 2016 году TAF одобрен FDA в качестве препарата для лечения ВГВ [https://www.hepmag.com/article/fda-approves-vemlidy-tenofovir-alafenamide-taf-hepatitis-b].
TAF является мощным пролекарством против вируса гепатита В (HBV). По сравнению с TDF он имеет меньше побочных воздействий на кости и почки [http://www.aidsmap.com/Tenofovir-alafenamide-works-well-against-hepatitis-B-with-less-effect-on-bones-and-kidneys/page/3051008/].
Несмотря на значительный прогресс в разработке схем комбинированной антиретровирусной терапии, которые подавляют долговечные ВИЧ и ВГВ, необходимы новые, безопасные и способные поддерживать высокую эффективность на протяжении всей жизни пациентов препараты.
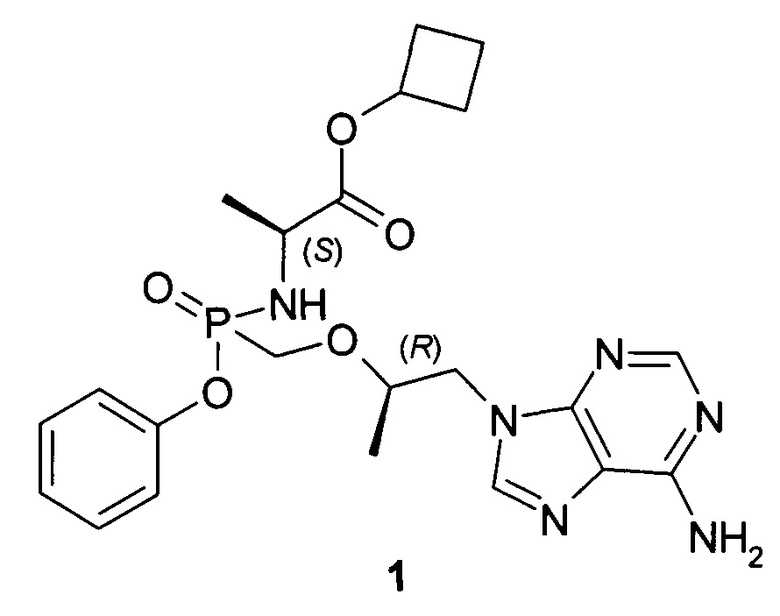
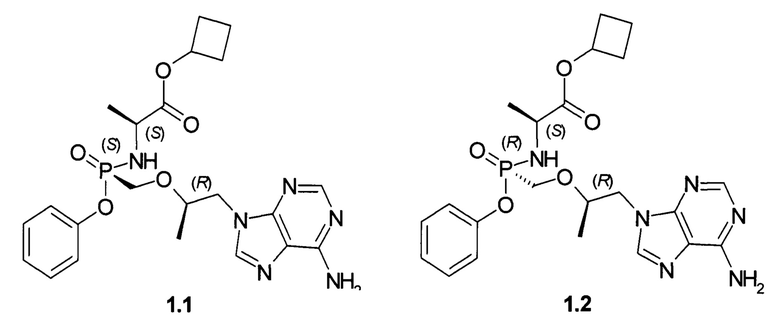
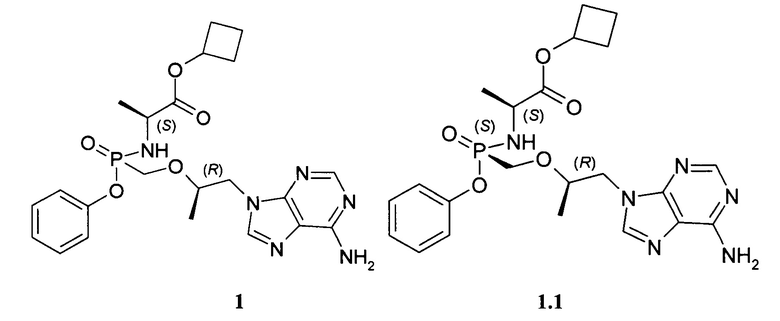
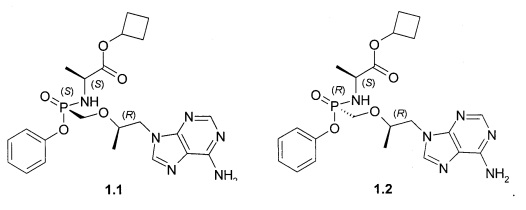
Авторы неожиданно обнаружили, что неизвестные ранее циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат общей формулы 1, его стереоизомер (циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат формулы 1.1 и циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат формулы 1.2, их изотопно-обогащенные аналоги, фармацевтически приемлемые соли, гидраты, сольваты, кристаллические или поликристаллические формы являются более эффективными пролекарствами TFV, представляющими интерес для комбинированного лечения вирусных заболеваний и прежде всего для лечения ВИЧ и гепатита В (ВГВ).
Ниже приведены определения различных терминов, используемых для описания данного изобретения. Эти определения применимы к терминам, как они использованы в данном описании и формуле изобретения, если не ограничены иным в конкретных случаях, либо по отдельности, либо как часть большей группы.
Термин «пролекарство» относится к соединениям согласно настоящему изобретению, которые расщепляются химически или метаболически и становятся, путем сольволиза или в физиологических условиях, соединением по настоящему изобретению, которое фармацевтически активно в естественных условиях. Пролекарства часто имеют более высокую растворимость, тканевую совместимость, доставку или замедленное высвобождение у млекопитающих (see, Bungard, Н., Desing of products, pp. 7-9, 21-24, Elsevier, Amsterdam 1985). Пролекарства включают кислотные производные, хорошо известные специалистам в данной области техники, такие как, например, сложные эфиры, полученные реакцией исходного кислотного соединения с подходящим спиртом, или амиды, полученные реакцией соединения исходной кислоты с подходящим амином. Примеры пролекарств включают, но не ограничиваются ими, ацетат, формиат, бензоат или другие ацилированные производные спиртов или аминов функциональных групп в соединениях по настоящему изобретению.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного (биотехнологического, растительного, животного, бактериального и так далее) происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
Термин «кристаллическая форма» - структура вещества, характеризующаяся упаковкой образующих их молекул в один из видов кристаллической решетки.
Термин «поликристаллическая форма» - структура вещества, имеющая поликристаллическое строение, т.е. состоящая из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «лекарственный препарат» означает вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Термин «терапевтический коктейль» представляет одновременно администрируемую комбинацию двух и более лекарственных препаратов, обладающих различным механизмом фармакологического действия и направленных на различные биомишени, участвующие в патогенезе заболевания.
Термин «фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
Термин «инертный наполнитель», используемый в данном описании, относится к соединению, которое используют для получения фармацевтической композиции, и, как правило, безопасному, нетоксичному, ни биологически, ни иным образом нежелательному, и включает в себя вспомогательные вещества, которые являются приемлемыми для применения в ветеринарии, а также фармакологически приемлемыми для человеческого использования. Соединения по данному изобретению могут быть введены отдельно, но обычно их вводят в смеси с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого пути введения и стандартной фармацевтической практики.
Термин «фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, дихлорацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в (Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как, холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Термин «терапевтически эффективное количество», используемый здесь, означает количество субстанции, пролекарства или лекарства, необходимое для уменьшения симптомов заболевания у субъекта. Доза субстанции, пролекарства или лекарства будет соответствовать индивидуальным требованиям в каждом конкретном случае. Эта доза может варьироваться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, с помощью которых пациент проходит лечение, способа и формы введения и опыта лечащего врача. Для перорального введения суточная доза составляет приблизительно от 0,01 до 10 г, включая все значения между ними, в день в монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет примерно от 0,1 до 7 г в день. Как правило, лечение начинают с большей начальной «нагрузочной дозы», чтобы быстро уменьшить или устранить вирус, сопровождающей убывающую дозу до уровня, достаточного для предотвращения всплеска инфекции.
Термин «субъект» означает млекопитающее, которое включает, но не ограничивается ими, крупный рогатый скот, свиней, овец, куриц, индеек, буйволов, лам, страусов, собак, кошек и человека, предпочтительным субъектом является человек. Предполагается, что в способе лечения субъекта может быть использовано любое из пролекарств общей формулы 1, его стерео изомер, изотопно-обогащенный аналог, его фармацевтически приемлемая сол, гидрат, сольват, кристаллическая и полиморфная форма, либо в их сочетании с другим соединением, в том числе с ингибитором NS5A HCV.
Термин «сольват» означает комплекс или агрегат, образуемый одной или более молекулами растворенного вещества, т.е. соединением согласно настоящему изобретению или его фармацевтически приемлемой солью и одной или более молекулами растворителя. Такие сольваты являются типичными твердыми кристаллами, имеющими, фиксированное молярное отношение растворенного вещества и растворителя. Репрезентативные растворители включают в себя, но не ограничиваются перечисленными, воду, этанол, изопропанол, уксусную кислоту и пр. Когда растворителем является вода, образуемый сольват представляет собой гидрат.
Настоящее изобретение относится к новому пролекарству TFV, представляющему собой циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат общей формулы 1, его стереоизомеру (циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноату формулы 1.1 и циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноату формулы 1.2) или их фармацевтически приемлемой соли,
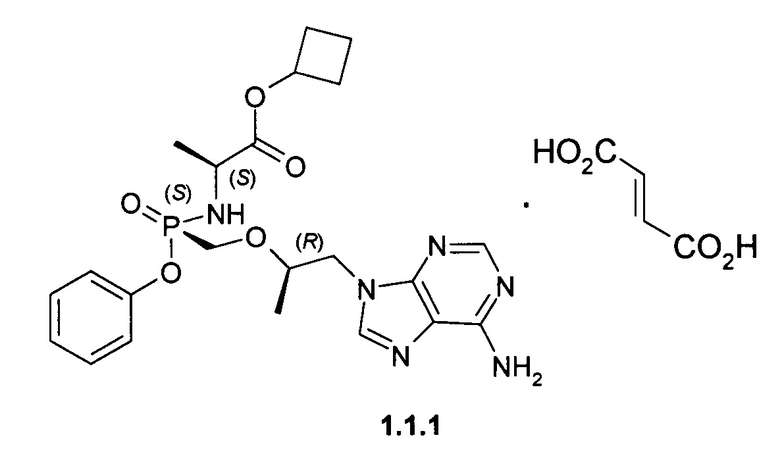
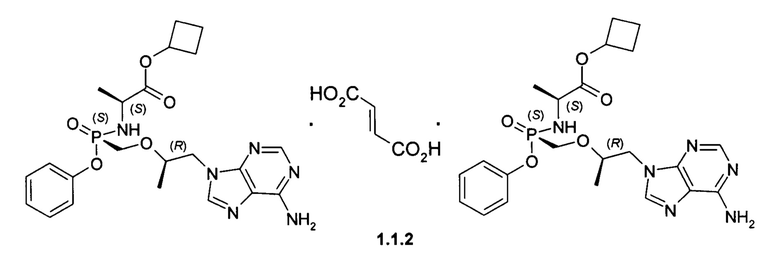
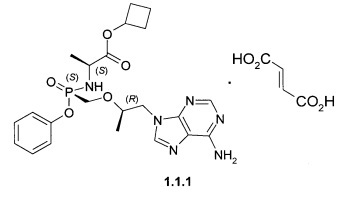
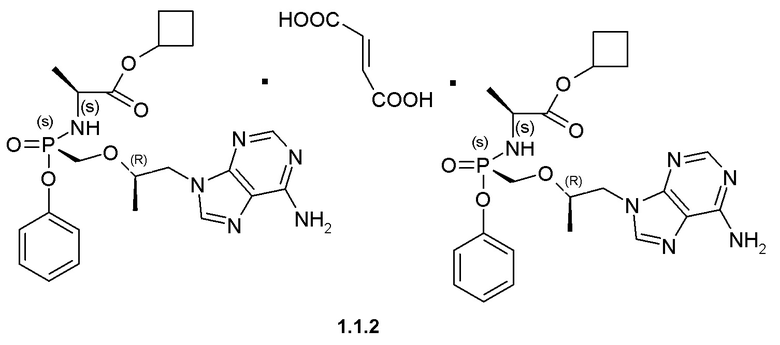
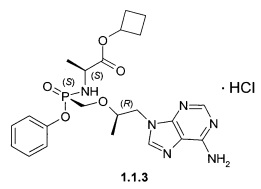
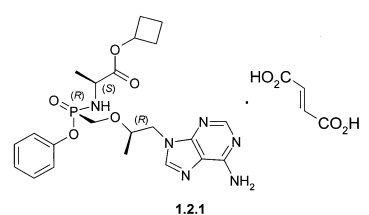
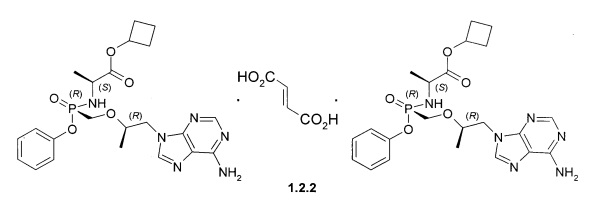
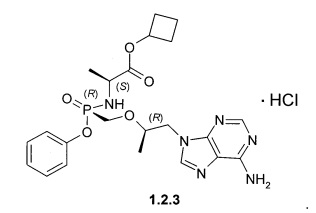
Предпочтительными солями являются фумараты формулы 1.1.1 и 1.2.1 полуфумараты формулы 1.1.2 и 1.2.2 и гидрохлориды формулы 1.1.3 и 1.2.3.
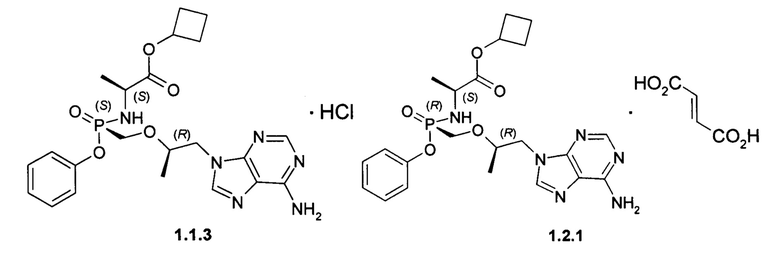
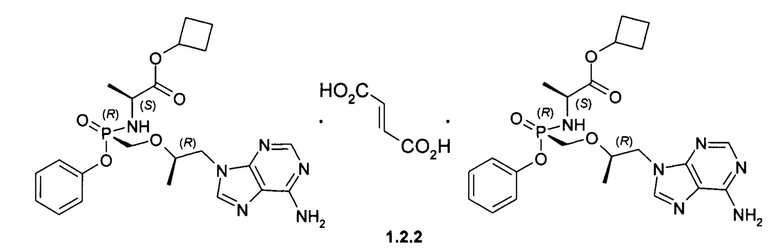
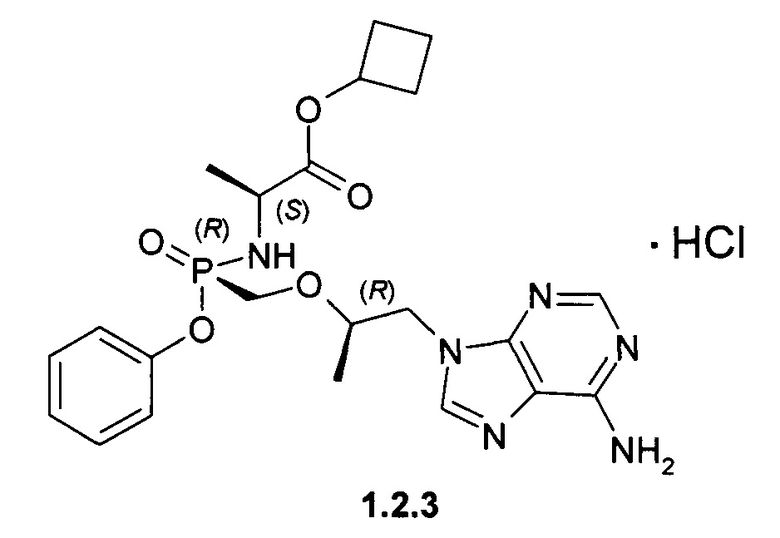
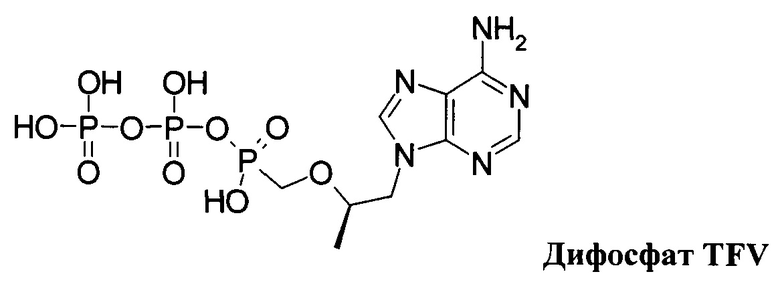
Неожиданно, новые пролекарства формулы 1.1 и формулы 1.2 оказались более эффективными, чем известное пролекарство TAF и его соли, использующееся в настоящее время в терапии ВИЧ и ВГВ инфицированных субъектов. Действительно фумараты соединений формулы 1.1 и 1.2 метаболизируют в TVF и дифосфат TVF в периферических мононуклеарных клетках крови (РВМС) с образованием более высокой концентрации и AUClast метаболитов, чем с образованием этих метаболитов в сопоставимых условиях при метаболизме TAF. Как видно из Таблицы 1, скорость образования метаболита TVF пролекарства формулы 1.1 Сmах и AUClast дифосфата TVF (лекарства) почти в 2 раза выше, чем значения этих параметров наблюдаемых при метаболизме TAF. Более высокие значения Сmах и AUClast (Таблица 1) наблюдаются и при метаболизме пролекарства формулы 1.2 по сравнению с значениями этих параметров, наблюдаемых при метаболизме TAF.
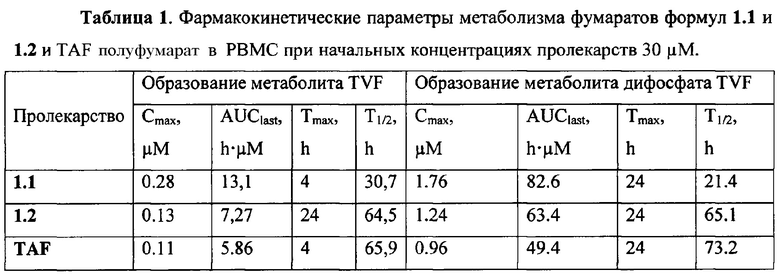
Оценка антивирусной активности фумаратов соединений формулы 1.1, 1.2 и TAF в ВИЧ-тесте с использованием клеток SupT1 зараженых штаммом NL4.3 ВИЧ, несущей вирус GFP-репортер (NL4.3GFP), показала, что фумарат соединений формулы 1.1 оказался наиболее эффективным (Таблица 2). Его активность и селективность в 1,4 раза выше активности и селективности TAF.

Предметом данного изобретения является фармацевтическая композиция для комбинированного лечения и профилактики вирусных инфекций в форме таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, включающая соединение общей формулы 1, или его стереоизомер, или их изотопно-обогащенный аналог, фармацевтически приемлемую соль, гидрат, сольват, кристаллическую или полиморфную форму.
Предпочтительной является фармацевтическая композиция, содержащая фумарат, полуфумарат, дихлорацетат или гидрохлорид пролекарства формулы 1.1 или его изотопно-обогащенный аналог, гидрат, сольват, кристаллическую или поликристаллическую форму.
Согласно изобретению фармацевтическая композиция может включать фармацевтически приемлемый наполнитель и дополнительный терапевтический агент, выбранный из группы, состоящей из вируса иммунодефицита человека (ВИЧ), протеазы ингибирующих соединений, ненуклеозидных ВИЧ ингибиторов обратной транскриптазы, нуклеозидных ВИЧ ингибиторов - обратной транскриптазы, ингибиторов ВИЧ-нуклеотидного реверса транскриптазы, ингибиторов ВИЧ-интегразы и ингибиторов CCR5.
Предметом изобретения является также способ лечения вируса иммунодефицита человека (ВИЧ), который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения общей формулы 1, или его стереоизомера, или их изотопно-обогащенного аналога, фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы.
Предпочтительным является также способ лечения вируса иммунодефицита человека (ВИЧ), который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения общей формулы 1.1, или его изотопно-обогащенного аналога, фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы.
Предпочтительным является также способ лечения вируса иммунодефицита человека (ВИЧ), который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества фумарата, или полуфумарата, или дихлорацетата или гидрохлорида пролекарства формулы 1.1 или его изотопно-обогащенного аналога, гидрата, сольвата, кристаллической или поликристаллической форм.
Предпочтительным является также способ лечения вируса иммунодефицита человека (ВИЧ), который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества приведенной выше фармацевтической композиции.
Предметом изобретения является также способ лечения вируса иммунодефицита человека (ВИЧ), который дополнительно включает введение субъекту одного или более дополнительных терапевтических агентов, выбранных из группы, состоящей из ингибиторов протеазы вируса иммунодефицита человека (ВИЧ), ингибирующих соединений, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеотидных ингибиторов обратной транскриптазы, ингибиторов ВИЧ-интегразы и ингибиторов CCR5.
Предметом изобретения является также способ лечения инфекции ВГВ, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения общей формулы 1, или его стереоизомера, или их изотопно-обогащенного аналога, фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы.
Предпочтительным является способ лечения инфекции ВГВ, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества фумарата, или полуфумарата, или дихлорацетата или гидрохлорида пролекарства формулы 1.1 или его изотопно-обогащенного аналога, гидрата, сольвата, кристаллической или поликристаллической формы.
Предметом изобретения является также способ лечения инфекции ВГВ, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективное количество одного или более дополнительных терапевтических агентов, выбранных из группы, состоящей из ингибитора протеазы вируса иммунодефицита человека (ВИЧ), ингибирующих соединений, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеотидных ингибиторов обратной транскриптазы, ингибиторов ВИЧ-интегразы и ингибиторов CCR5.
Предметом изобретения является также способ лечения вируса иммунодефицита человека (ВИЧ), который включает введение субъекту, нуждающемуся в этом, одной или нескольких доз терапевтически эффективного количества приведенной выше фармацевтической композиции.
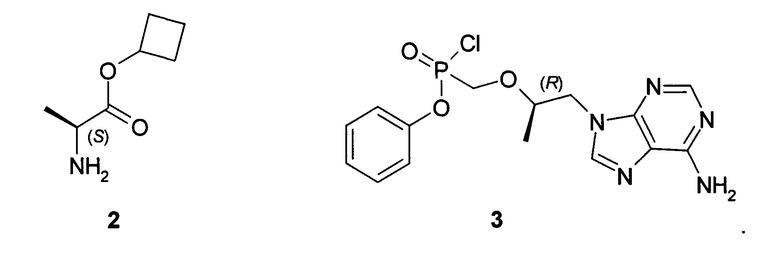
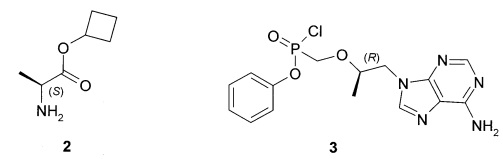
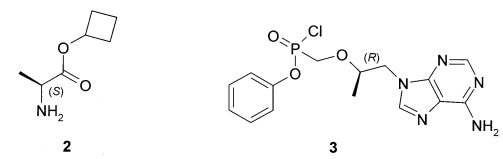
Предметом изобретения является также способ получения циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат общей формулы 1, его стереоизомера (циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.1 и циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноата формулы 1.2, и их фармацевтически приемлемых солей, взаимодействием циклобутилового эфира L-аланина формулы 2 с соединением общей формулы 3, с последующим при необходимости разделением соединения формулы 1 на соответствующие стереоизомеры 1.1 и 1.2 и необязательной обработкой полученных соединений 1.1 или 1.2 кислотой. Предпочтительными являются фумаровая кислота или соляная кислота.
Настоящее изобретение далее будет описано в связи с определенными вариантами осуществления, но не ограничивается ими. Напротив, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем формулы изобретения. Таким образом, следующие примеры, которые включают в себя конкретные варианты осуществления, иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Общий протокол синтеза пролекарств общей формулы 1 (Схема 1).
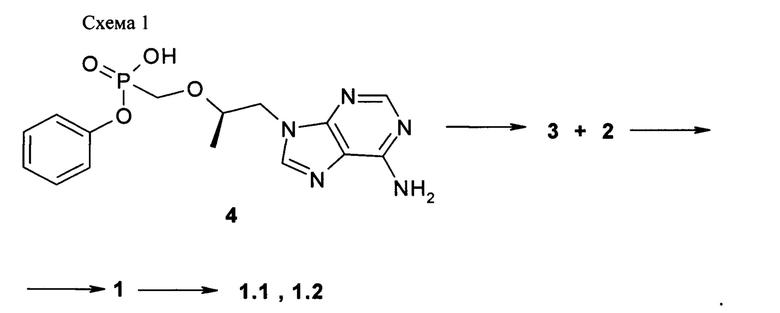
К суспензии 3,63 г (10 ммоль) [(R)-2-(6-амино-пурин-9-ил)-1-метил-этоксиметил]фосфоновой кислоты монофенилового эфира (4) [WO 2013116720] в 14 мл сульфолана и 12 мл дихлорметана прикапывали при перемешивании 3 мл (40 ммоль) тионилхлорида. Затем смесь перемешивали с обратным холодильником при температуре 50-55°С под слабым током аргона в течение 15 ч. После этого к колбе подключали вакуум (мембранный насос) и в течение 2 часов удаляли летучие компоненты при 50-55°С. Реакционной массе давали охладиться до 30°С и при перемешивании вливали смесь 10 мл дихлорметана и 40 мл сухого ацетонитрила. Реакционную массу, содержащую хлорид (3) охлаждали до (-60)-(-50)°С и приливали раствор 1,546 г (12 ммоль) циклобутилового эфира L-аланина (2) и 4,172 мл (30 ммоль) триэтиламина в 6 мл ацетонитрила. Смеси давали медленно нагреться до комнатной температуры, затем разбавляли 100 мл дихлорметана и наносили раствор на ~100 мл силикагеля на стеклянном фильтре. Далее проводили выделение продукта методом сухой флэш-хроматографии, элюируя сначала дихлорметаном, 30% раствором ацетона в дихлорметане и затем чистым тетрагидрофураном. Получали 2 г соединения общей формулы 1 с соотношением стереоизомеров формулы 1.1 и 1.2 в соотношении 2:3. Разделение стереоизомеров формулы 1.1 и 1.2 проводили методом ВЭЖХ на оптической колонке Phenomenex Amylose-2 AXIA-Pac 250×21.20 мм в изократической системе AcCN:EtOH:HCOOH 200:20:0.5 со скоростью потока 20 мл/мин с УФ-детектором 254 нм. Циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат формулы 1.1, LC-MS (ESI) 489 (М+Н)+. 1Н NMR (400 MHz, DMSO) δ 1.06 (d, J=5.8 Hz, 3Н), 1.13 (d, J=7.0 Hz, 3H), 1.57 (m, 1H), 1.71 (m, 1H), 1.93 (m, 2H), 2.22 (m, 2H), 3.81 (m, 4H), 4.13 (dd, J=14.3, 6.0 Hz, 1H), 4.27 (m, 1H), 4.92-4.79 (m, 1H), 5.66 (m, 1H), 7.05 (d, J=7.7 Hz, 2H), 7.35-7.09 (m, 5H), 8.11 (d, J=16.6 Hz, 2H). Циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат формулы 1.2, LC-MS (ESI) 489 (M+H)+. 1H NMR (400 MHz, DMSO) δ 1.05 (d, J=6.1 Hz, 3H), 1.13 (d, J=7.0 Hz, 3H), 1.57 (m, 1H), 1.70 (m, 1H), 1.91 (m, 2H), 2.21 (m, 2H), 3.86 (m, 4H), 3.98 (m, 1H), 4.18-4.22 (m, 1H), 4.86-4.95 (m, 1H), 5.55-5.60 (m, 1H), 7.02-7.08 (m, 2H), 7.1-7.4 (m, 5H), 8.13 (d, J=11.2 Hz, 2H). Фумараты 1.1.1 и 1.2.1 получали кристаллизацией стереоизомеров 1.1 и 1.2 с эквимолярным количеством фумаровой кислоты из 100 мл ацетонитрила. Фумарат (S)-циклобутил 2-((S)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)-фосфориламино)-пропаноата (1.1.1), LC-MS (ESI) 489 (М+Н)+. 1Н NMR (DMSO-d6, 300 MHz) δ 8.14 (s, 1Н), 8.10 (s, 1H), 7.30 (m, 2H), 7.19 (s, 2H), 7.14 (m, 1H), 7.06 (m, 2H), 6.63 (s, 2H), 5.64 (t, J=11.1 Hz, 1H), 4.86 (p, J=7.2 Hz, 1H), 4.27 (dd, J1=14.4 Hz, J2=3.0 Hz, 1H), 4.14 (dd, J1=14.4 Hz, J2=6.6 Hz, 1H), 3.85 (m, 4H), 2.23 (m, 2H), 1.94 (m, 2H), 1.72 (m, 1H), 1.59 (m, 1H), 1.13 (d, J=6.9 Hz, 3H), 1.07 (d, J=6.6 Hz, 3H). 31P NMR (DMSO-d6, 121.5 MHz) δ 22.056. Фумарат (S)-циклобутил 2-((R)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)пропаноата (1.2.1), LC-MS (ESI) 489 (M+H)+. 1H NMR (DMSO-d6, 300 MHz) δ 8.14 (s, 1H), 8.12 (s, 1H), 7.34 (m, 2H), 7.21 (s, 2H), 7.15 (m, 1H), 7.11 (m, 2H), 6.63 (s, 2H), 5.53 (dd, J1=12.0 Hz, J2=10.5 Hz, 1H), 4.82 (p, J=7.5 Hz, 1H), 4.29 (dd, J1=14.4 Hz, J2=3.6 Hz, 1H), 4.20 (dd, J1=14.4 Hz, J2=5.7 Hz, 1H), 3.98 (m, 1H), 3.86 (m, 3H), 2.21 (m, 2H), 1.91 (m, 2H), 1.69 (m, 1H), 1.57 (m, 1H), 1.13 (d, J=6.9 Hz, 3H), 1.05 (d, J=6.3 Hz, 3H). 31P NMR (DMSO-d6, 121.5 MHz) δ 22.86.
Полуфумараты 1.1.2 и 1.2.2 получали двойной кристаллизацией стереоизомеров 1.1 и 1.2 с 0,5 экв фумаровой кислоты из ацетонитрила. Смесь нагревали до растворения твердых веществ, охлаждали до комнатной температуры и снова нагревали с обратным холодильником, чтобы вызвать диссоциацию второй ступени. Полуфумарат циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноата (1.1.2), LC-MS (ESI) 489 (М+Н)+. 1Н NMR 1Н NMR (400 MHz, DMSO) δ 8.14 (s, 1H), 8.10 (s, 1H), 7.29 (t, J=7.6 Hz, 2H), 7.15 (dd, J=19.6, 12.1 Hz, 3H), 7.05 (d, J=7.6 Hz, 2H), 6.63 (s, 1H), 5.64 (t, J=11.0 Hz, 1H), 4.93-4.79 (m, 1H), 4.27 (d, J=11.1 Hz, 1H), 4.14 (dd, J=14.4, 6.5 Hz, 1H), 3.98-3.71 (m, 4H), 2.23 (s, 2H), 1.94 (d, J=10.3 Hz, 2H), 1.71 (d, J=9.9 Hz, 1H), 1.65-1.50 (m, 1H), 1.13 (d, J=7.1 Hz, 3H), 1.07 (d, J=6.1 Hz, 3H). (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата (1.2.2), LC-MS (ESI) 489 (M+H)+. 1H NMR (DMSO-d6, 400 MHz) δ 8.13 (s, 1H), 8.11 (s, 1H), 7.34 (m, 2H), 7.19 (s, 2H), 7.15 (m, 1H), 7.11 (m, 2H), 6.63 (s, 1H), 5.54 (dd, J1=12.0 Hz, J2=10.4 Hz, 1H), 4.82 (p, J=7.6 Hz, 1H), 4.28 (dd, J1=14.4 Hz, J2=3.6 Hz, 1H), 4.20 (dd, J1=14.4 Hz, J2=6.0 Hz, 1H), 3.97 (m, 1H), 3.85 (m, 3H), 2.21 (m, 2H), 1.91 (m, 2H), 1.69 (m, 1H), 1.55 (m, 1H), 1.12 (d, J=7.2 Hz, 3H), 1.05 (d, J=6.0 Hz, 3H).
Гидрохлорды 1.1.3 и 1.2.3 получали кристаллизацией стереоизомеров 1.1 и 1.2 с эквимолярным количеством 12 M НСl из ацетонитрила. Гидрохлорд циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноата (1.1.3), LC-MS (ESI) 489 (М+Н)+. 1Н NMR (DMSO-d6, 400 MHz) δ 9.37 (brs, 1Н), 8.84 (brs, 1H), 8.50 (s, 1H), 8.45 (s, 1H), 7.32 (m, 2H), 7.15 (m, 1H), 7.07 (m, 2H), 5.64 (dd, J1=11.6 Hz, J2=10.4 Hz, 1H), 4.84 (p, J=7.2 Hz, 1H), 4.41 (dd, J1=14.4 Hz, J2=3.2 Hz, 1H), 4.24 (dd, J1=14.4 Hz, J2=6.8 Hz, 1H), 4.00 (m, 1H), 3.84 (m, 3H), 2.22 (m, 2H), 1.92 (m, 2H), 1.71 (m, 1H), 1.57 (m, 1H), 1.12 (d, J=7.2 Hz, 3H), 1.10 (d, J=6.8 Hz, 3Н). Гидрохлорд циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метилфенокси-фосфорил]амино]-пропаноата (1.2.2), LC-MS (ESI) 489 (M+H)+. 1H NMR (DMSO-d6, 400 MHz) δ 9.25 (brs, 1H), 8.73 (brs, 1H), 8.46 (s, 1H), 8.45 (s, 1H), 7.34 (m, 2H), 7.15 (m, 1H), 7.09 (m, 2H), 5.51 (dd, J1=12.0 Hz, J2=10.0 Hz, 1H), 4.81 (p, J=7.2 Hz, 1H), 4.41 (dd, J1=14.4 Hz, J2=2.8 Hz, 1H), 4.28 (dd, J1=14.4 Hz, J2=6.8 Hz, 1H), 4.01 (m, 1H), 3.86 (m, 3H), 2.21 (m, 2H), 1.91 (m, 2H), 1.70 (m, 1H), 1.56 (m, 1H), 1.10 (m, 6H).
Пример 2. Получение фармацевтической композиции в виде таблетки. Крахмал (1600 мг), молотую лактозу (1600 мг), тальк (400 мг), и 1000 мг соли 1.1.1, 1.1.2 или 1.1.3 смешивали и прессовали в бар. Полученный брусок измельчали в гранулы и просеивали через сито, чтобы собрать гранулы размером 14-16 меш. Полученные таким образом гранулы были сформированы в таблетки подходящей формы весом 200 или 400 мг каждая.
Пример 3. Получение фармацевтической композиции в виде капсул. Соль 1.1.1, 1.1.2 или 1.1.3 тщательно смешивали с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывали в желатиновые капсулы подходящего размера по 150 или 300 мг в каждой капсуле.
Пример 4. Получение фармацевтической композиции в форме композиций для внутримышечных, внутрибрюшинных или подкожных инъекций. Смешивали 500 мг соли 1.1.1, 1.1.2 или 1.1.3, 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл воды для инъекций. Полученный раствор фильтровали, помещали в ампулы по 5 мл и запечатывали.
Пример 5. Определение параметров метаболизма соединений общей формулы формул 1.1 и 1.2 и прототипа TAF в периферических мононуклеарных клетках крови (РВМС) человека.
Исходные растворы тестируемых соединений формул 1.1.1, 1.2.1, 1.1.2, 1.2.2, 1.1.3, 1.2.3 и TAF были приготовлены в DMSO (Sigma) и хранились при -20°С. РВМС были выделены из крови человека центрифугированием в градиенте Ficoll-Paque Premium (GE Healthcare), до использования хранились в жидком азоте. РВМС сажали в 24-луночные планшеты (Greiner Bio-one) по 1,5 млн. клеток в лунку (4,2 млн/мл) в среде RPMI-1640, содержащей 2 мМ L-глутамина, 0,11 мг/мл пирувата натрия, заменимые и незаменимые аминокислоты, 50 Ед/мл пенициллина, 50 мкг/мл стрептомицина (все реактивы ПанЭко) и 5% HI (Heat Inactivated) фетальной бычьей сыворотки (HyClone). Инкубировали в течение ночи при 37°С и 5% СО2. На следующий день к клеткам добавляли тестируемые и референсные соединения в финальной концентрации 30 мкМ. Инкубировали клетки с соединениями при 37°С и 5% СО2. После 2, 4, 8, 24, 48 и 72 часов инкубации неприкрепившиеся клетки со средой переносили в 1,5 мл пробирки (Eppendorf), центрифугировали 5 минут при 1000 g и удаляли среду. Клетки промывали 1 мл фосфатным буфером (Gibco), лизировали 200 мкл охлажденного до -20°С 70% метанола. Клетки, прикрепившиеся к лункам планшета, промывали 1 мл PBS (Gibco) и лизировали 200 мкл охлажденного до -20°С 70% метанола. Лизаты прикрепившихся и неприкрепившихся клеток из соответствующих лунок объединяли и перемешивали.
Анализ клеточных лизатов на содержание тенофовира (TVF) и дифосфата тенофовира (DP-TVF) проводили методом UPLC-MS/MS с использованием хроматографа 1290 UPLC System (Agilent) и масс-спектрометра с тройным квадруполем QTrap5500 System (АВ Sciex). Разделение аналитов проводили на колонке Thermo Hypercarb (50×3.0 мм, 5 мкм, Thermo Scientific) в подвижной фазе следующего состава: А - 0,5% аммиак в 25 мМ ацетата аммония и В - 0,5% аммиак в 25 мМ ацетата аммония: 2-пропанол: метанол (1:1:3) при скорости потока 0,8 мл/мин. В качестве источника ионов использовали электрораспыление (TurboIonSpray) в режиме регистрации отрицательных ионов. Детектирование аналитов проводили в режиме MRM по переходам для TVF 286>107, 286>79, 286>63 m/z, для DP-TVF 446>348, 446>176, 446>158, 446>79 m/z. Обработку хроматограмм проводили в Analyst 1.5.2 Software (АВ Sciex). Концентрацию TVF и дифосфата TVF в клеточных лизатах оценивали по калибровочным кривым, полученным с помощью стандартных образцов TVF и DP-TVF в 70% метаноле. Полученные результаты представлены в Таблице 1.
Пример 6. Определение анти-ВИЧ активности соединений общей формулы 1 и прототипа TAF полуфумарата. Определение антивирусной активности тестируемых соединений проводили на линии Т-лимфоцитов, SupT1. Клетки были инфицированы ВИЧ штаммом NL4.3, несущим ген зеленого флуоресцентного белка (NL4.3-GFP). Препарат вируса получали путем трансфекции клеток 293Т провирусной ДНК. Через 48 часов после трансфекции препарат замораживали и хранили до использования. Для повышения эффективности инфекции, суспензию клеток SupT1 осаждали из инфекционной смеси центрифугированием. Тестируемые вещества добавляли к клеткам непосредственно перед добавлением вируса. Через 2 часа инкубации инфекционную смесь заменяли свежей культуральной средой с тестируемыми препаратами. Эффективность инфекции определяли через 45 часов путем подсчета процента флуоресцирующих клеток, по сравнению с неинфицированными клеточными культурами. Цитотоксичность тестируемых соединений определяли параллельно на той же, но не инфицированной клеточной линии SupT1, используя реагент ХТТ. Использовали серийные десятикратные разведения препаратов (начиная от 10 мкМ для определения антивирусной активности, или от 100 мкМ - для цитотоксичности). В качестве негативного контроля использовали 0,1% ДМСО. Были рассчитаны значения ЕС50, СС50 и СИ (селективный индекс). Качество тестов определяли при помощи следующих контролей: отношение сигнала к фону, интегразный ингибитор ралтегравир (1 мкМ), а также воспроизводимость теста. Контролем для определения цитотоксичности служил препарат эметин (0,03, 0,09 и 0,2 мкМ). Полученные результаты представлены в Таблице 2.
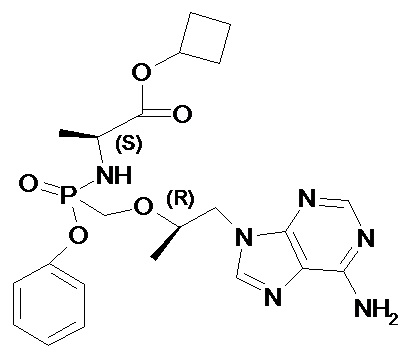
Изобретение относится к новым изомерным формам циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1 и его фармацевтически приемлемым солям, которые обладают противовирусным действием и могут быть использованы при лечении гепатита В, ВИЧ-инфекции. Соединения обладают высокими фармакокинетическими параметрами метаболизма, высокой противовирусной активностью и селективностью, превосходящей действие известных используемых в настоящее время аналогов. Циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат соответствует формуле 1
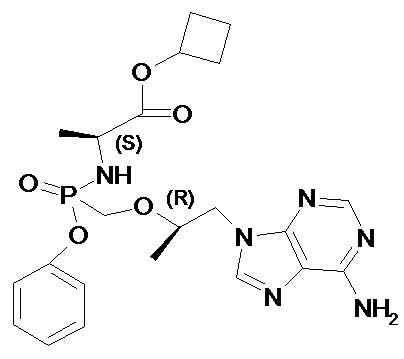
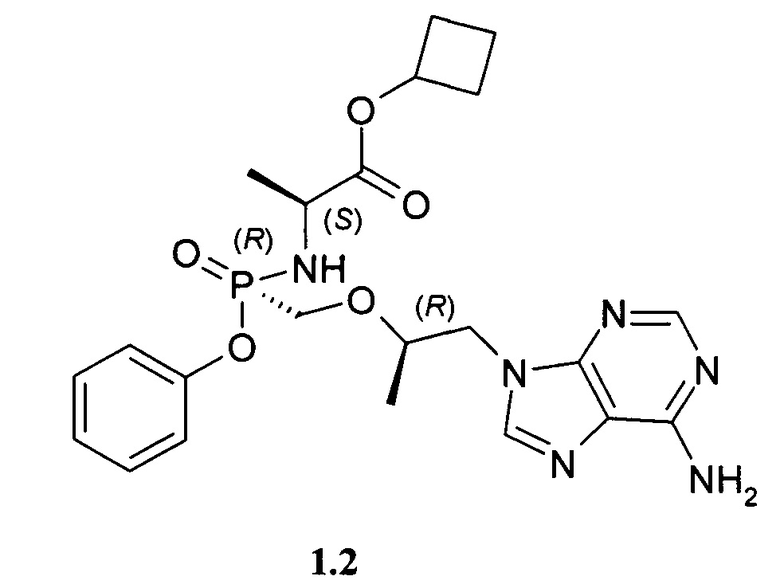
Предпочтительно указанное соединение представляет собой циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат формулы 1.1 или циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат формулы 1.2 и их фармацевтически приемлемые соли
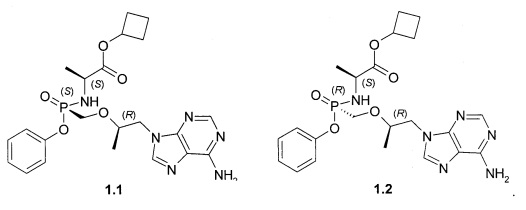
Предпочтительные соли собой представляют собой фумарат циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.1.1; полуфумарат циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.1.2; гидрохлорид циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.1.3; фумарат циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.2.1; полуфумарат; циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.2.2 или гидрохлорид циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.2.3. Структурные формулы указанных соединений указаны в формуле изобретения. Изобретение также относится к способу получения соединений формулы 1, 1.1 и 1.2 и их фумаратов, полуфумаратов, гидрохлоридов, взаимодействием циклобутилового эфира L-аланина формулы (2) и соединения формулы 3
При необходимости проводят разделение соединения формулы 1 на соответствующие стереоизомеры 1.1 и 1.2 и необязательной обработкой полученных соединений 1.1 или 1.2 соответствующей кислотой. 4 н. и 6 з.п. ф-лы, 2 табл., 6 пр.
1. Циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат общей формулы 1 и их фармацевтически приемлемые соли
2. Соединения по п. 1, представляющие собой циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат формулы 1.1 или циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноат формулы 1.2, и их фармацевтически приемлемые соли
3. Соединение по п. 1 или 2, представляющее собой фумарат циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.1.1, полуфумарат циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.1.2, гидрохлорид
циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.1.3, фумарат циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.2.1, полуфумарат циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.2.2 или гидрохлорид циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.2.3
4. Фармацевтическая композиция для лечения и профилактики вирусных инфекций в форме таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, включающая соединение по любому из пп. 1, 2 или 3 в терапевтически эффективном количестве и один или несколько фармацевтически приемлемых наполнителей.
5. Способ лечения вируса иммунодефицита человека, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1, 2, 3 или фармацевтической композиции по п. 4.
6. Способ лечения по п. 5, который включает введение субъекту одной или нескольких доз соединения по любому из пп. 1, 2, 3 или фармацевтической композиции по п. 4.
7. Способ лечения вируса гепатита В, который включает введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1, 2, 3 или фармацевтической композиции по п. 4.
8. Способ лечения по п. 7, который включает введение субъекту одной или нескольких доз соединения по любому из пп. 1, 2, 3 или фармацевтической композиции по п. 4.
9. Способ получения циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата общей формулы 1, циклобутил (S)-2-[(S)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]пропаноата формулы 1.1 и циклобутил (S)-2-[(R)-[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноата формулы 1.2 и их фумаратов, полуфумаратов, гидрохлоридов, взаимодействием циклобутилового эфира L-аланина (2) и соединения общей формулы 3
с последующим, при необходимости, разделением соединения формулы 1 на соответствующие стереоизомеры 1.1 и 1.2 и необязательной обработкой полученных соединений 1.1 или 1.2 кислотой.
10. Способ по п. 9, в котором в качестве кислоты используют фумаровую кислоту, хлористоводородную или соляную кислоту.
| CN 106317116 A, 11.01.2017 | |||
| СЕНСОРНЫЙ ПЕРЕКЛЮЧАТЕЛЬ | 1990 |
|
RU2067354C1 |
| WO 2013116720 A1, 08.08.2013 & EA 026138 B1, 31.03.2017 | |||
| Подогреватель питательной воды для паровозов | 1926 |
|
SU4926A1 |

