Уровень техники
Сердечно-сосудистое заболевание представляет собой одно из основных заболеваний, приводящих к смерти в США и в большинстве европейских стран. Установлено, что более 70 миллионов человек только в США страдают от сердечно-сосудистого заболевания или нарушения, включая, но не ограничиваясь ими, высокое артериальное давление, коронарную болезнь сердца, дислипидемию, застойную сердечную недостаточность и инсульт. Существует потребность в улучшенных способах лечения заболеваний и нарушений связанных с сердечно-сосудистой системой.
Сущность изобретения
В различных вариантах осуществления данное изобретение относится к фармацевтическим композициям и способам использования таких композиции для повышения уровней EPA в плазме, сыворотке и/или красных кровяных клетках (RBC) и/или для лечения или предотвращения связанных с сердечно-сосудистой системой заболеваний.
В одном варианте осуществления изобретение относится к фармацевтической композиции, включающей, состоящей из или состоящей в основном из по меньшей мере 95% по массе этилэйкозапентаеноата (EPA-E), от приблизительно 0,2% до приблизительно 0,5% по массе этилоктадекатетраеноата (ODTA-E), от приблизительно 0,05% до приблизительно 0,25% по массе этилнонаэкапентаеноата (NDPA-E), от приблизительно 0,2% до приблизительно 0,45% по массе этиларахидоната (AA-E), от приблизительно 0,3% до приблизительно 0,5% по массе этилэйкозатетраеноата (ETA-E) и от приблизительно 0,05% до приблизительно 0,32% этилгенэйкозапентаеноата (HPA-E). В еще одном варианте осуществления композиция представлена в капсуле с оболочкой. В еще одном варианте осуществления композиция практически не содержит или не содержит докозагексаеновую кислоту (DHA) или ее производное, такое как этил-DHA (DHA-E), например, не больше чем приблизительно 0,06%, приблизительно 0,05% или приблизительно 0,04% по массе.
В еще одном варианте осуществления изобретение относится к способу повышения уровней EPA в сыворотке, плазме и/или красных кровяных клетках (RBC), включающему введение композиции, описанной в данном документе, пациенту, нуждающемуся в повышенных уровнях EPA в сыворотке, плазме и/или RBC. В родственном варианте осуществления начальный уровень EPA у пациента в плазме, сыворотке и/или RBC имеет значение не выше чем приблизительно 50 мкг/г, и после введения композиции пациенту в течение по меньшей мере приблизительно 6 недель, у пациента проявляется по меньшей мере 100%, по меньшей мере 150%, по меньшей мере 200%, по меньшей мере 250%, по меньшей мере 300%, по меньшей мере 350% или по меньшей мере 400% повышение уровней EPA (изменение уровня EPA, деленное на начальный уровень EPA) в плазме, сыворотке и/или RBC по сравнению с начальным. В родственном варианте осуществления начальный уровень EPA в плазме, сыворотке и/или RBC пациента имеет значение не выше чем приблизительно 50 мкг/г. В еще одном варианте осуществления пациенту предоставляется количество указанной композиции, эффективное для достижения указанных повышений уровней EPA. В еще одном варианте осуществления пациенту предоставляется от приблизительно 2 г до приблизительно 4 г в сутки указанной композиции.
В еще одном варианте осуществления изобретение относится к способу лечения связанного с сердечно-сосудистой системой заболевания у пациента, нуждающегося в этом, включающему введение композиции пациенту, как описано в данном документе. В родственном варианте осуществления начальный уровень EPA в плазме, сыворотке и/или RBC пациента имеет значение не выше чем приблизительно 50 мкг/кг и после введения композиции пациенту в течение по меньшей мере приблизительно 6 недель у пациента проявляется по меньшей мере приблизительно 100%, по меньшей мере приблизительно 150%, по меньшей мере приблизительно 200%, по меньшей мере приблизительно 250%, по меньшей мере приблизительно 300%, по меньшей мере приблизительно 350% или по меньшей мере приблизительно 400% повышение уровней EPA в плазме, сыворотке и/или RBC по сравнению с начальным. В родственном варианте осуществления начальный уровень EPA в плазме, сыворотке и/или RBC пациента имеет значение не выше чем приблизительно 50 мкг/г. В еще одном варианте осуществления пациенту предоставляется от приблизительно 2 г до приблизительно 4 г в сутки указанной композиции.
Эти и другие варианты осуществления данного изобретения будут раскрыты в данном документе в дополнительных подробностях далее. На фиг. 1 показаны уровни EPA в крови после различных введений EPA.
На фиг. 2 показано повышение EPA по сравнению с начальным после различных введений EPA.
Подробное описание
Несмотря на то что данное изобретение может быть воплощено в различных формах далее предоставлено описание нескольких вариантов осуществления, но необходимо понимать, что данное описание должно рассматриваться, как иллюстративный пример изобретения, и не подразумевается, что оно ограничивает изобретение конкретными проиллюстрированными вариантами осуществления. Заголовки представлены только для удобства и не должны истолковываться как ограничивающие изобретение каким-либо образом. Варианты осуществления, проиллюстрированные под любым заголовком, могут быть комбинированы с вариантами осуществления, проиллюстрированными под любым другим заголовком.
Использование числовых значений в различных количественных значениях, установленных в данной заявке, если особым образом не указано иное, устанавливаются в виде приближений как если бы оба минимальное и максимальное значение в установленных пределах предшествовались словом "приблизительно". Кроме того, под раскрытием пределов подразумевается непрерывный диапазон, включая каждое значение между минимальным и максимальным приведенными значениями, а также любые пределы, которые могут быть образованы такими значениями. Также в данном документе раскрыты любые и все соотношения (и пределы любых таких соотношений), которые могут быть образованы делением раскрытого числового значения на любое другое раскрытое числовое значение. Соответственно, специалист в данной области примет во внимание то, что многие такие соотношения, пределы и пределы соотношений могут быть однозначно получены из числовых значений, представленных в данном документе, и во всех примерах такие соотношения, пределы и пределы соотношений отображают различные варианты осуществления данного изобретения.
В одном варианте осуществления изобретение относится к фармацевтической композиции, включающей эйкозапентаеновую кислоту или ее производное. В одном варианте осуществления такие композиции содержат эйкозапентаеновую кислоту или фармацевтически приемлимый эфир, производное, конъюгат или их соль или смеси любого из вышеупомянутого, собирательно называемые в данном документе "EPA". Термин "фармацевтически приемлемый" в контексте данного документа означает, что указанное вещество не вырабатывает неприемлемую токсичность для пациента или взаимодействие с другими компонентами композиции.
В одном варианте осуществления EPA содержит полностью цис-эйкоза-5,8,11,14,17-пентаеновую кислоту. В еще одном варианте осуществления EPA содержит эфир эйкозапентаеновой кислоты. В еще одном варианте осуществления EPA содержит C1-C5 алкиловый эфир эйкозапентаеновой кислоты. В еще одном варианте осуществления EPA содержит этиловый эфир эйкозапентаеновой кислоты, метиловый эфир эйкозапентаеновой кислоты, пропиловый эфир эйкозапентаеновой кислоты или бутиловый эфир эйкозапентаеновой кислоты. В еще одном варианте осуществления EPA содержит этиловый эфир полностью цис-эйкоза-5,8,11,14,17-пентаеновой кислоты.
В еще одном варианте осуществления EPA в форме этил-EPA, литий EPA, моно-, ди- или триглицерида EPA или любого другого эфира или соли EPA или формы свободной кислоты EPA. EPA может также быть в форме 2-замещенного производного или другого производного, которое замедляет скорость его окисления, но не изменяет в значительной степени иным образом его биологическое действие.
В еще одном варианте осуществления композиция представлена в единице дозирования (например, капсуле) в количестве от приблизительно 50 мг до приблизительно 5000 мг, от приблизительно 75 мг до приблизительно 2500 мг или от приблизительно 100 мг до приблизительно 1000 мг, например, приблизительно 75 мг, приблизительно 100 мг, приблизительно 125 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг, приблизительно 325 мг, приблизительно 350 мг, приблизительно 375 мг, приблизительно 400 мг, приблизительно 425 мг, приблизительно 450 мг, приблизительно 475 мг, приблизительно 500 мг, приблизительно 525 мг, приблизительно 550 мг, приблизительно 575 мг, приблизительно 600 мг, приблизительно 625 мг, приблизительно 650 мг, приблизительно 675 мг, приблизительно 700 мг, приблизительно 725 мг, приблизительно 750 мг, приблизительно 775 мг, приблизительно 800 мг, приблизительно 825 мг, приблизительно 850 мг, приблизительно 875 мг, приблизительно 900 мг, приблизительно 925 мг, приблизительно 950 мг, приблизительно 975 мг, приблизительно 1000 мг, приблизительно 1025 мг, приблизительно 1050 мг, приблизительно 1075 мг, приблизительно 1100 мг, приблизительно 1025 мг, приблизительно 1050 мг, приблизительно 1075 мг, приблизительно 1200 мг, приблизительно 1225 мг, приблизительно 1250 мг, приблизительно 1275 мг, приблизительно 1300 мг, приблизительно 1325 мг, приблизительно 1350 мг, приблизительно 1375 мг, приблизительно 1400 мг, приблизительно 1425 мг, приблизительно 1450 мг, приблизительно 1475 мг, приблизительно 1500 мг, приблизительно 1525 мг, приблизительно 1550 мг, приблизительно 1575 мг, приблизительно 1600 мг, приблизительно 1625 мг, приблизительно 1650 мг, приблизительно 1675 мг, приблизительно 1700 мг, приблизительно 1725 мг, приблизительно 1750 мг, приблизительно 1775 мг, приблизительно 1800 мг, приблизительно 1825 мг, приблизительно 1850 мг, приблизительно 1875 мг, приблизительно 1900 мг, приблизительно 1925 мг, приблизительно 1950 мг, приблизительно 1975 мг, приблизительно 2000 мг, приблизительно 2025 мг, приблизительно 2050 мг, приблизительно 2075 мг, приблизительно 2100 мг, приблизительно 2125 мг, приблизительно 2150 мг, приблизительно 2175 мг, приблизительно 2200 мг, приблизительно 2225 мг, приблизительно 2250 мг, приблизительно 2275 мг, приблизительно 2300 мг, приблизительно 2325 мг, приблизительно 2350 мг, приблизительно 2375 мг, приблизительно 2400 мг, приблизительно 2425 мг, приблизительно 2450 мг, приблизительно 2475 мг или приблизительно 2500 мг.
В еще одном варианте осуществления подходящая композиция в соответствии с изобретением содержит не больше чем приблизительно 10%, не больше чем приблизительно 9%, не больше чем приблизительно 8%, не больше чем приблизительно 7%, не больше чем приблизительно 6%, не больше чем приблизительно 5%, не больше чем приблизительно 4%, не больше чем приблизительно 3%, не больше чем приблизительно 2%, не больше чем приблизительно 1% или, если содержит, не больше чем от приблизительно 0,5% по массе докозагексаеновой кислоты (DHA) или ее производного, такого как этил-DHA. В еще одном варианте осуществления композиция по изобретению практически не содержит DHA или этил-DHA. В еще одном варианте осуществления подходящая композиция по данному изобретению не содержит DHA или ее производное, такое как DHA-E.
В еще одном варианте осуществления EPA содержит по меньшей мере 70%, по меньшей мере 80%, по меньшей мере 90%, по меньшей мере 95%, по меньшей мере 96%, по меньшей мере 97%, по меньшей мере 98%, по меньшей мере 99% или 100% по массе от всех жирных кислот, представленных в композиции, в соответствии с изобретением.
В еще одном варианте осуществления подходящая композиция в соответствии с изобретением содержит менее 10%, менее 9%, менее 8%, менее 7%, менее 6%, менее 5%, менее 4%, менее 3%, менее 2%, менее 1%, менее 0,5% или менее 0,25% по массе от всей композиции или по массе от всего содержания жирных кислот, любой жирной кислоты или ее производного, отличного от EPA. Иллюстративные примеры "жирной кислоты, отличной от EPA," включают линоленовую кислоту (LA), арахидоновую кислоту (AA), докозагексаеновую кислоту (DHA), альфа-линоленовую кислоту (ALA), стеарадоновую кислоту (STA), эйкозатриеновую кислоту (ETA) и/или докозапентаеновую кислоту (DPA). В еще одном варианте осуществления подходящая композиция в соответствии с изобретением содержит от приблизительно 0,1% до приблизительно 4%, от приблизительно 0,5% до приблизительно 3% или от приблизительно 1% до приблизительно 2% по массе всех жирных кислот, отличных от EPA и/или DHA.
В еще одном варианте осуществления композиция в соответствии с изобретением обладает одним или несколькими из следующих свойств: (a) этиловый эфир эйкозапентаеновой кислоты составляет по меньшей мере приблизительно 96%, по меньшей мере приблизительно 97% или по меньшей мере приблизительно 98% по массе от всех жирных кислот, представленных в композиции; (b) композиция содержит не больше чем приблизительно 4%, не больше чем приблизительно 3% или не больше чем приблизительно 2% по массе от всего количества жирных кислот, отличных от этилового эфира эйкозапентаеновой кислоты; (c) композиция содержит не больше чем приблизительно 0,6%, не больше чем приблизительно 0,5% или не больше чем приблизительно 0,4% любой отдельной жирной кислоты, отличной от этилового эфира эйкозапентаеновой кислоты; (d) композиция имеет показатель преломления (20 °C) от приблизительно 1 до приблизительно 2, от приблизительно 1,2 до приблизительно 1,8 или от приблизительно 1,4 до приблизительно 1,5; (e) композиция имеет удельную плотность (при 20 °C) от приблизительно 0,8 до приблизительно 1,0, от приблизительно 0,85 до приблизительно 0,95 или от приблизительно 0,9 до приблизительно 0,92; (e) композиция содержит не больше чем приблизительно 20 ppm, не больше чем приблизительно 15 ppm или не больше чем приблизительно 10 ppm тяжелых металлов, (f) композиция содержит не больше чем приблизительно 5 ppm, не больше чем приблизительно 4 ppm, не больше чем приблизительно 3 ppm, или не больше чем приблизительно 2 ppm мышьяка и/или (g) композиция имеет пероксидное число не больше чем приблизительно 5 мэкв/л, не больше чем приблизительно 4 мэкв/л, не больше чем приблизительно 3 мэкв/л или не больше чем приблизительно 2 мэкв/л.
В еще одном варианте осуществления изобретение относится к композиции, включающей, состоящей в основном из или состоящей из по меньшей мере 95%, 96% или 97% по массе этилэйкозапентаноата, от приблизительно 0,2% до приблизительно 0,5% по массе этилоктадекатетраеноата, от приблизительно 0,05% до приблизительно 0,25% по массе этилнонаэкапентаеноата, от приблизительно 0,2% до приблизительно 0,45% по массе этиларахидоната, от приблизительно 0,3% до приблизительно 0,5% по массе этилэйкозатетраеноата и от приблизительно 0,05% до приблизительно 0,32% этилгенэйкозапентаеноата. При желании композиция содержит не больше чем приблизительно 0,06%, приблизительно 0,05% или приблизительно 0,04% по массе DHA или ее производного, такого как этил-DHA. В одном варианте осуществления композиция практически не содержит или не содержит DHA или ее производное, такое как этил-DHA. Композиция дополнительно при желании содержит один или более антиоксидантов (например, токоферол) или другие примеси в количестве не более чем приблизительно 0,5% или не более чем 0,05%. В еще одном варианте осуществления композиция содержит от приблизительно 0,05% до приблизительно 0,4%, например, от приблизительно 0,2% по массе токоферола. В еще одном варианте осуществления от приблизительно 500 мг до приблизительно 1 г композиции предоставляется в капсульной оболочке.
В еще одном варианте осуществления изобретение относится к композиции, включающей, состоящей из или состоящей в основном из по меньшей мере 96% по массе этилэйкозапентаеноата, от приблизительно 0,22% до приблизительно 0,4% по массе этилоктадекатетраеноата, от приблизительно 0,075% до приблизительно 0,20% по массе этилнонаэкапентаеноата, от приблизительно 0,25% до приблизительно 0,40% по массе этиларахидоната, от приблизительно 0,3% до приблизительно 0,4% по массе этилэйкозатетраеноата и от приблизительно 0,075% до приблизительно 0,25% этилгенэйкозапентаеноата. При желании композиция содержит не больше чем приблизительно 0,06%, приблизительно 0,05% или приблизительно 0,04% по массе DHA или ее производного, такого как этил-DHA. В одном варианте осуществления композиция практически не содержит или не содержит DHA или ее производное, такое как этил-DHA. Композиция дополнительно при желании содержит один или более антиоксидантов (например, токоферол) или другие примеси в количестве не больше чем приблизительно 0,5% или не больше чем 0,05%. В еще одном варианте осуществления композиция содержит от приблизительно 0,05% до приблизительно 0,4%, например, приблизительно 0,2% по массе токоферола. В еще одном варианте осуществления изобретение относится к лекарственной форме, включающей от приблизительно 500 мг до приблизительно 1 г вышеупомянутой композиции в капсульной оболочке.
В еще одном варианте осуществления изобретение относится к композиции включающей, состоящей из или состоящей в основном из по меньшей мере 96%, 97% или 98% по массе этилэйкозапентаеноата, от приблизительно 0,25% до приблизительно 0,38% по массе этилоктадекатетраеноата, от приблизительно 0,10% до приблизительно 0,15% по массе этилнонаэкапентаеноата, от приблизительно 0,25% до приблизительно 0,35% по массе этиларахидоната, от приблизительно 0,31% до приблизительно 0,38% по массе этилэйкозатетраеноата и от приблизительно 0,08% до приблизительно 0,20% этилгенэйкозапентаеноата. При желании композиция содержит не больше чем приблизительно 0,06%, приблизительно 0,05% или приблизительно 0,04% по массе DHA или ее производного, такого как этил-DHA. В одном варианте осуществления композиция практически не содержит или не содержит DHA или ее производное, такое как этил-DHA. Композиция при желании дополнительно содержит один или более антиоксидантов (например, токоферол) или другие примеси в количестве не больше чем приблизительно 0,5% или не больше чем 0,05%. В еще одном варианте осуществления композиция содержит от приблизительно 0,05% до приблизительно 0,4%, например, приблизительно 0,2% по массе токоферола. В еще одном варианте осуществления изобретение относится к лекарственной форме, включающей от приблизительно 500 мг до приблизительно 1 г вышеупомянутой композиции в капсульной оболочке.
В еще одном варианте осуществления изобретение относится к способу повышения уровней EPA в сыворотке, плазме и/или красных кровяных клетках (RBC), содержащему введение композиции, описанной в данном документе, пациенту, нуждающемуся в таком лечении. В одном варианте осуществления при оральном введении композиции пациенту как установлено далее в данном документе в течение по меньшей мере приблизительно 5, приблизительно 10, приблизительно 15, приблизительно 20, приблизительно 25, приблизительно 30, приблизительно 35, приблизительно 40, приблизительно 42, приблизительно 45 или приблизительно 50 дней, у пациента проявляется по меньшей мере приблизительно 2-кратное, по меньшей мере приблизительно 3-кратное, по меньшей мере приблизительно 3,5-кратное, по меньшей мере приблизительно 3,75-кратное или по меньшей мере приблизительно 4-кратное изменение уровня EPA (конечный абсолютный уровень EPA, деленный на начальный уровень EPA) в сыворотке, плазме и/или RBC. В одном варианте осуществления способ содержит этап идентификации пациента, нуждающегося в повышении EPA в сыворотке, плазме и/или красных кровяных клетках (RBC) перед упомянутым этапом введения. В родственном варианте осуществления начальный уровень EPA в плазме, сыворотке и/или RBC пациента имеет значение не выше чем приблизительно 50 мкг/г. В еще одном варианте осуществления пациенту предоставляется от приблизительно 2 г до приблизительно 4 г в сутки указанной композиции. В еще одном варианте осуществления после введения композиции пациенту, как указано выше, у пациента проявляется снижение уровней DHA, AA и/или DGLA в плазме, сыворотке и/или RBC. В еще одном варианте осуществления после введения композиции пациенту, как указано выше, у пациента проявляется повышение уровней DPA в плазме, сыворотке и/или RBC. В еще одном варианте осуществления после введения композиции пациенту, как указано выше, уровни DHA в плазме, сыворотке и/или RBC снижаются по меньшей мере на 16%, уровни DGLA в плазме, сыворотке и/или RBC снижаются по меньшей мере на 31%, уровни AA в плазме, сыворотке и/или RBC снижаются по меньшей мере на 20%, и/или уровни DPA в плазме, сыворотке и/или RBC уровни повышаются на более чем 130%.
В еще одном варианте осуществления изобретение относится к способу повышения уровней EPA в сыворотке, плазме и/или красных кровяных клетках (RBC), содержащему введение композиции, описанной в данном документе, пациенту, нуждающемуся в повышенных уровнях EPA в сыворотке, плазме и/или RBC. В родственном варианте осуществления после введения композиции пациенту в течение по меньшей мере приблизительно 5, приблизительно 10, приблизительно 15, приблизительно 20, приблизительно 25, приблизительно 30, приблизительно 35, приблизительно 40, приблизительно 42, приблизительно 45 или приблизительно 50 дней у пациента проявляется по меньшей мере приблизительно 100%, по меньшей мере приблизительно 150%, по меньшей мере приблизительно 200%, по меньшей мере приблизительно 250%, по меньшей мере приблизительно 300%, по меньшей мере приблизительно 350% или по меньшей мере приблизительно 400% повышение (изменение уровня EPA по сравнению с начальным, деленное на начальный уровень EPA) уровней EPA в плазме, сыворотке и/или RBC по сравнению с начальным. В родственном варианте осуществления начальный уровень EPA в плазме, сыворотке и/или RBC пациента имеет значение не выше чем приблизительно 50 мкг/г. В еще одном варианте осуществления пациенту предоставляется от приблизительно 2 г до приблизительно 4 г в сутки указанной композиции. В еще одном варианте осуществления после введения композиции пациенту, как указано выше, у пациента проявляется снижение уровней DHA, AA и/или DGLA в плазме, сыворотке и/или RBC. В еще одном варианте осуществления после введения композиции пациенту, как указано выше, у пациента проявляется повышение уровня DPA в плазме, сыворотке и/или RBC. В еще одном варианте осуществления после введения композиции пациенту как указано выше, уровни DHA в плазме, сыворотке и/или RBC снижаются по меньшей мере на 16%, уровни DGLA в плазме, сыворотке и/или RBC снижаются по меньшей мере на 31%, уровни AA в плазме, сыворотке и/или RBC снижаются по меньшей мере на 20% и/или уровни DPA в плазме, сыворотке и/или RBC повышаются на более чем 130%.
В родственном варианте осуществления при оральном введении от приблизительно 2 до приблизительно 4 г в сутки композиции как установлено в данном документе пациенту в течение по меньшей мере приблизительно 5, приблизительно 10, приблизительно 15, приблизительно 20, приблизительно 25, приблизительно 30, приблизительно 35, приблизительно 40, приблизительно 45 или приблизительно 50 дней у пациента проявляется по меньшей мере приблизительно 10 мкг/кг увеличение, по меньшей мере приблизительно 15 мкг/г увеличение, по меньшей мере приблизительно 20 мкг/г увеличение, по меньшей мере приблизительно 25 мкг/г увеличение, по меньшей мере приблизительно 30 мкг/г увеличение, по меньшей мере приблизительно 35 мкг/г увеличение, по меньшей мере приблизительно 40 мкг/г увеличение, по меньшей мере приблизительно 45 мкг/г увеличение, по меньшей мере приблизительно 50 мкг/г увеличение, по меньшей мере приблизительно 75 мкг/г увеличение, по меньшей мере приблизительно 100 мкг/г увеличение или по меньшей мере приблизительно 150 мкг/г увеличение уровня EPA в сыворотке, плазме и/или RBC по сравнению с начальным. В еще одном варианте осуществления после введения композиции пациенту как указано выше у пациента проявляется снижение уровней DHA, AA и/или DGLA в плазме, сыворотке и/или RBC. В еще одном варианте осуществления после введения композиции пациенту как указано выше у пациента проявляется повышение уровней DPA в плазме, сыворотке и/или RBC. В еще одном варианте осуществления после введения композиции пациенту как указано выше DHA уровни в плазме, сыворотке и/или RBC снижаются по меньшей мере на 16%, уровни DGLA в плазме, сыворотке и/или RBC снижаются по меньшей мере на 31%, уровни AA в плазме, сыворотке и/или RBC снижаются по меньшей мере на 20% и/или уровни DPA в плазме, сыворотке и/или RBC повышаются на более чем 130%.
В еще одном варианте осуществления пациент не получал терапию или добавку омега-3 жирной кислоты в течение по меньшей мере 2 недель, 3 недель, 4 недель, 6 недель или 12 недель до начала лечения, описанного в данном документе.
В одном варианте осуществления изобретение относится к способу лечения и/или профилактики связанного с сердечно-сосудистой системой заболевания, включающему введение пациенту, нуждающемуся в таком лечении или профилактике, композиции как установлено в данном документе. Термин "связанное с сердечно-сосудистой системой заболевание" в данном документе относится к любому заболеванию или нарушению сердца или кровеносных сосудов (т.е. артерии и вены) или их любым симптомам. Неограничивающие примеры связанных с сердечно-сосудистой системой заболеваний и нарушений включают гипертриглицеридемию, гиперхолестеринемию, смешанную дислипидемию, коронарную болезнь сердца, сосудистое заболевание, инсульт, атеросклероз, аритмию, гипертензию, инфаркт миокарда и другие сердечно-сосудистые патологии.
Термин "лечение" в отношении данного заболевания или нарушения, включает, но не ограничивается ими, замедление заболевания или нарушения, например, подавление развития заболевания или нарушения; облегчение заболевания или нарушения, например, приводящее к регрессии заболевания или нарушения или облегчение состояния, вызванного и возникшего вследствии заболевания или нарушения, например, облегчение, предотвращение или лечение симптомов заболевания или нарушения. Термин "профилактика" в отношении данного заболевания или нарушения означает: предотвращение начала развития заболевания, если оно не имело место, предотвращение возникновения заболевания или нарушения у пациента, который может быть предрасположен к нарушению или заболеванию, но у которого еще не было диагностировано нарушение или заболевание, и/или предотвращение дальнейшего развития заболевания/нарушения, если оно уже имеет место.
В одном варианте осуществления данное изобретение относится к способу терапии липидов крови, включающему введение пациенту или группе пациентов, нуждающихся в этом, фармацевтической композиции, описанной в данном документе. В еще одном варианте осуществления пациент или группа пациентов страдают от гипертриглицеридемии, гиперхолестеринемии, смешанной дислипидемии и/или очень высокого уровня триглицеридов.
В еще одном варианте осуществления начальный уровень триглицеридов у пациента или группы пациентов (или средний или срединный начальный уровень триглицеридов в случае группы пациентов), подвергающихся лечению, сохраняется после приема пищи или натощак от приблизительно 200 мг/дл до приблизительно 500 мг/дл. В еще одном варианте осуществления начальный уровень ХС-ЛПНП (или средний или срединный начальный ХС-ЛПНП уровень) у пациента или группы пациентов, несмотря на статиновую терапию, составляет от приблизительно 40 мг/дл до приблизительно 100 мг/дл.
В одном варианте осуществления пациент или группа пациентов, подвергающихся лечению в соответствии со способами изобретения, проходят сопутствующую статиновую терапию, например, аторвастатиновую, розувастатиновую или симвастатиновую терапию (с или без эзетимиба). В еще одном варианте осуществления пациент проходит сопутствующую постоянную статиновую терапию во время начала терапии EPA с высоким уровнем очистки.
В еще одном варианте осуществления пациент или группа пациентов, подвергающихся лечению в соответствии со способами изобретения, имеют индекс массы тела (ИМТ или средний ИМТ) не больше чем приблизительно 45 кг/м2.
В еще одном варианте осуществления изобретение относится к способу поддержания уровня ЛПНП у пациента, который проходит постоянную статиновую терапию и для которого требуется понижающая уровень триглицеридов терапия, при этом способ содержит идентификацию пациента, который проходит постоянную статиновую терапию и для которого требуется понижающая уровень триглицеридов терапия, введение пациенту фармацевтически приемлемой композиции, включающей от приблизительно 1 г до приблизительно 4 г EPA в сутки (например, EPA с высоким уровнем очистки), где после введения композиции пациенту, у пациента проявляется клинически значимое снижение уровня триглицеридов натощак по сравнению с контролем. В контексте данного документа термин "клинически значимое снижение уровня триглицеридов натощак" означает снижение уровней триглицеридов в степени, соответствующей снижению риска сердечно-сосудистой патологии. Как правило, каждое снижение уровня триглицеридов на 10 мг/дл приводит к на 1,6% более низкому риску смерти, инфаркта миокарда и рецидивного острого коронарного синдрома. Например, Miller et al., Impact of triglyceride level beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. JACC Vol. 51, No. 7 (2008), включенный в данный документ путем ссылки. Вследствие этого, в одном варианте осуществления "клинически значимое снижение уровня триглицеридов натощак" означает снижение на 10 мг/дл. В контексте данного документа термин "регулирования уровня ЛПНП" означает отсутствие клинически значимых побочных изменений уровней ЛПНП во время терапии.
В одном варианте осуществления изобретение относится к способу понижения уровня триглицеридов у пациента, проходящего постоянную статиновую терапию, имеющего начальный уровень триглицеридов натощак от приблизительно 200 мг/дл до приблизительно 500 мг/дл, при этом способ включает введение пациенту фармацевтической композиции, включающей от приблизительно 1 г до приблизительно 4 г EPA (например, EPA с высоким уровнем очистки), где после введения композиции пациенту ежедневно в течение приблизительно 12 недель уровень триглицеридов натощак у пациента имеет по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 45%, по меньшей мере на 50%, по меньшей мере на 55%, по меньшей мере на 60%, по меньшей мере на 65%, по меньшей мере на 70% или по меньшей мере на 75% более низкое значение, чем у контрольного пациента, проходящего постоянную статиновую терапию без сопутствующей EPA с высоким уровнем очистки в течение приблизительно 12 недель, где начальный уровень триглицеридов натощак у контрольного пациента также составляет от приблизительно 200 мг/дл до приблизительно 500 мг/дл. Термин "постоянная статиновая терапия" в данном документе означает, что исследуемые пациент, группа пациентов, контрольный пациент или контрольная группа пациентов получали постоянную ежедневную дозу статина (например, аторвастатин, розувастатин или симвастатин) в течение по меньшей мере 4 недель перед начальным измерением уровня триглицеридов натощак ("испытательный период"). Например, пациент или контрольный пациент, проходящий постоянную статиновую терапию, будет получать на постоянной основе ежедневно (т.е. одинаковая доза каждый день) дозу статина в течение по меньшей мере 4 недель непосредственно перед измерением начального уровня триглицерида натощак. В одном варианте осуществления ХС-ЛПНП пациента и контрольного пациента поддерживается между приблизительно 40 мг/дл и приблизительно 100 мг/дл во время испытательного периода. Затем пациент и контрольный пациент продолжают получать постоянную дозу статина в течение 12-недельного периода после начала.
В одном варианте осуществления статин вводится пациенту и контрольному пациенту в количестве от приблизительно 1 мг до приблизительно 500 мг, от приблизительно 5 мг до от приблизительно 200 мг или от приблизительно 10 мг до приблизительно 100 мг, например, приблизительно 1 мг, приблизительно 2 мг, приблизительно 3 мг, приблизительно 4 мг, приблизительно 5 мг, приблизительно 6 мг, приблизительно 7 мг, приблизительно 8 мг, приблизительно 9 мг или приблизительно 10 мг; приблизительно 15 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 30 мг, приблизительно 35 мг, приблизительно 40 мг, приблизительно 45 мг, приблизительно 50 мг, приблизительно 55 мг, приблизительно 60 мг, приблизительно 65 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг, приблизительно 90 мг, приблизительно 100 мг, приблизительно 125 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг, приблизительно 325 мг, приблизительно 350 мг, приблизительно 375 мг, приблизительно 400 мг, приблизительно 425 мг, приблизительно 450 мг, приблизительно 475 мг или приблизительно 500 мг. В еще одном варианте осуществления начальный уровень ХС-ЛПНП у пациента (и при желании у контрольного пациента) имеет высокое значение, несмотря на постоянную статиновую терапию, от приблизительно 40 мг/дл до приблизительно 100 мг/дл. В еще одном варианте осуществления пациент и/или контрольный пациент имеет индекс массы тела (ИМТ или средний ИМТ) не больше чем приблизительно 45 кг/м2.
В еще одном варианте осуществления изобретение относится к способу снижения уровня триглицеридов в группе пациентов, проходящих постоянную статиновую терапию, имеющих средний начальный уровень триглицеридов натощак от приблизительно 200 мг/дл до приблизительно 500 мг/дл, при этом способ включает введение членам группы пациентов фармацевтической композиции, включающей от приблизительно 1 г до приблизительно 4 г в сутки EPA с высоким уровнем очистки, где после введения композиции членам группы пациентов ежедневно в течение периода приблизительно 12 недель у группы пациентов проявляется по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 45%, по меньшей мере на 50%, по меньшей мере на 55%, по меньшей мере на 60%, по меньшей мере на 65%, по меньшей мере на 70%, по меньшей мере на 75% более низкий средний уровень триглицеридов натощак, чем у контрольной группы пациентов, подвергающихся постоянной статиновой терапии без сопутствующей EPA с высоким уровнем очистки в течение приблизительно 12 недель, где средний начальный уровень триглицеридов натощак у контрольной группы пациентов также составляет от приблизительно 200 мг/дл до приблизительно 500 мг/дл. В родственном варианте осуществления постоянной статиновой терапии будет достатачно, если средний уровень холестерина-ЛПНП у группы пациентов составляет приблизительно по меньшей мере приблизительно 40 мг/дл и не больше чем приблизительно 100 мг/дл в течение 4 недель непосредственно перед начальным измерением уровня триглицеридов натощак.
В еще одном варианте осуществления изобретение относится к способу снижения уровня триглицеридов у группы пациентов, проходящих постоянную статиновую терапию и имеющих средний начальный уровень триглицеридов натощак от приблизительно 200 мг/дл до приблизительно 500 мг/дл, при этом способ включает введение членам группы пациентов фармацевтической композиции, включающей от приблизительно 1 г до приблизительно 4 г EPA с высоким уровнем очистки, где после введения композиции членам группы пациентов ежедневно в течение приблизительно 12 недель у группы пациентов проявляется (a) по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 45%, по меньшей мере на 50%, по меньшей мере на 55%, по меньшей мере на 60%, по меньшей мере на 65%, по меньшей мере на 70%, по меньшей мере на 75% более низкий средний уровень триглицеридов натощак по сравнению с контрольной группой пациентов, подверженных постоянной статиновой терапии без сопутствующей EPA с высоким уровнем очистки в течение приблизительно 12 недель, и (b) отсутствие повышения средних уровней ХС-ЛПНП в сыворотке по сравнению с начальным, где средний начальный уровень триглицеридов натощак у контрольного пациента также составляет от приблизительно 200 мг/дл до приблизительно 500 мг/дл.
В еще одном варианте осуществления изобретение относится к способу снижения уровня триглицеридов у пациента, проходящего постоянную статиновую терапию и имеющего средний начальный уровень триглицеридов натощак от приблизительно 200 мг/дл до приблизительно 500 мг/дл, при этом способ включает введение пациенту фармацевтической композиции, включающей от приблизительно 1 г до приблизительно 4 г EPA с высоким уровнем очистки, где после введения композиции пациенту ежедневно в течение приблизительно 12 недель у пациента проявляется (a) по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 45%, по меньшей мере на 50%, по меньшей мере на 55%, по меньшей мере на 60%, по меньшей мере на 65%, по меньшей мере на 70% или по меньшей мере на 75% более низкий уровень триглицеридов натощак по сравнению с контрольным пациентом, подверженным постоянной статиновой терапии без сопутствующей EPA с высоким уровнем очистки в течение приблизительно 12 недель, и (b) отсутствие повышения в сыворотке уровней холестерина-ЛПНП по сравнению с начальным, где контрольный начальный уровень триглицеридов натощак у пациента также составляет от приблизительно 200 мг/дл до приблизительно 500 мг/дл.
В еще одном варианте осуществления изобретение относится к способу снижения уровня триглицеридов у группы пациентов, проходящих постоянную статиновую терапию и имеющих средний начальный уровень триглицеридов натощак от приблизительно 200 мг/дл до приблизительно 500 мг/дл, при этом способ включает введение членам группы пациентов фармацевтической композиции включающей от приблизительно 1 г до приблизительно 4 г EPA с высоким уровнем очистки, где после введения композиции членам группы пациентов ежедневно в течение приблизительно 12 недель у группы пациентов проявляется (a) по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 45%, по меньшей мере на 50%, по меньшей мере на 55%, по меньшей мере на 60%, по меньшей мере на 65%, по меньшей мере на 70%, по меньшей мере на 75% более низкий средний уровень триглицеридов натощак и (b) по меньшей мере на 5%, по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на45% или по меньшей мере на 50% более низкие средние уровни ХС-ЛПНП в плазме или сыворотке по сравнению с контрольной группой пациентов, подверженных постоянной статиновой терапии без сопутствующей EPA с высоким уровнем очистки в течение приблизительно 12 недель, где средний начальный уровень триглицеридов натощак у контрольного пациента также составляет от приблизительно 200 мг/дл до приблизительно 500 мг/дл.
В еще одном варианте осуществления изобретение относится к способу снижения уровня триглицеридов у группы пациентов, проходящих постоянную статиновую терапию и имеющих средний начальный уровень триглицеридов натощак от приблизительно 200 мг/дл до приблизительно 500 мг/дл, при этом способ включает введение членам группы пациентов фармацевтической композиции, включающей от приблизительно 1 г до приблизительно 4 г EPA с высоким уровнем очистки, где после введения композиции членам группы пациентов ежедневно в течение приблизительно 12 недель у группы пациентов проявляется (a) по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 45%, по меньшей мере на 50%, по меньшей мере на 55%, по меньшей мере на 60%, по меньшей мере на 65%, по меньшей мере на 70%, по меньшей мере на 75% более низкий средний уровень триглицеридов натощак и (b) по меньшей мере на 5%, по меньшей мере на 10%, по меньшей мере на 15%, по меньшей мере на 20%, по меньшей мере на 25%, по меньшей мере на 30%, по меньшей мере на 35%, по меньшей мере на 40%, по меньшей мере на 45% или по меньшей мере на 50% более низкие средние уровни холестерина-ЛПНП в плазме или сыворотке по сравнению с контрольной группой пациентов, подверженных постоянной статиновой терапии без сопутствующей EPA с высоким уровнем очистки в течение приблизительно 12 недель, где средний начальный уровень триглицеридов натощак у контрольной группы пациентов также составляет от приблизительно 200 мг/дл до приблизительно 500 мг/дл.
В еще одном варианте осуществления у пациента или группы пациентов, подвергающихся лечению в соответствии со способами изобретения, проявляется начальный абсолютный уровень в плазме натощак общего количества свободной жирной кислоты (или его среднее значение) не выше чем приблизительно 300 нмоль/мл, не выше чем приблизительно 250 нмоль/мл, не выше чем приблизительно 200 нмоль/мл, не выше чем приблизительно 150 нмоль/мл, не выше чем приблизительно 100 нмоль/мл или не выше чем приблизительно 50 нмоль/мл.
В еще одном варианте осуществления у пациента или группы пациентов, подвергающихся лечению в соответствии со способами изобретения, проявляется начальный абсолютный уровень в плазме свободной EPA натощак (или его среднее значение в случае группы пациентов) не выше чем от приблизительно 0,70 нмоль/мл, не выше чем от приблизительно 0,65 нмоль/мл, не выше чем от приблизительно 0,60 нмоль/мл, не выше чем от приблизительно 0,55 нмоль/мл, не выше чем от приблизительно 0,50 нмоль/мл, не выше чем от приблизительно 0,45 нмоль/мл, или не выше чем от приблизительно 0,40 нмоль/мл. В еще одном варианте осуществления у пациента или группы пациентов, подвергающихся лечению в соответствии со способами изобретения, проявляется начальный уровень натощак свободной EPA в плазме (или его среднее значение), выраженный в виде процента от общего количества свободной жирной кислоты, не больше чем приблизительно 3%, не больше чем приблизительно 2,5%, не больше чем приблизительно 2%, не больше чем приблизительно 1,5%, не больше чем приблизительно 1%, не больше чем приблизительно 0,75%, не больше чем приблизительно 0,5%, не больше чем приблизительно 0,25%, не больше чем приблизительно 0,2% или не больше чем приблизительно 0,15%. В одном из таких вариантов осуществления перед началом терапии определяются уровни свободной EPA в плазме и/или общего количества жирной кислоты.
В еще одном варианте осуществления у пациента или группы пациентов, подвергающихся лечению в соответствии со способами изобретения, проявляется начальный абсолютный свободный уровень EPA в плазме натощак (или его среднее значение) не выше чем приблизительно 1 нмоль/мл, не выше чем приблизительно 0,75 нмоль/мл, не выше чем приблизительно 0,50 нмоль/мл, не выше чем приблизительно 0,4 нмоль/мл, не выше чем приблизительно 0,35 нмоль/мл или не выше чем приблизительно 0,30 нмоль/мл. В еще одном варианте осуществления у пациента или группы пациентов, подвергающихся лечению в соответствии со способами изобретения, проявляется начальный уровень EPA натощак в плазме, сыворотке или мембранах красных кровяных клеток не выше чем приблизительно 150 мкг/мл, не выше чем приблизительно 125 мкг/мл, не выше чем приблизительно 100 мкг/мл, не выше чем приблизительно 95 нг/мл, не выше чем приблизительно 75 мкг/мл, не выше чем приблизительно 60 мкг/мл, не выше чем приблизительно 50 мкг/мл, не выше чем приблизительно 40 мкг/мл, не выше чем приблизительно 30 мкг/мл или не выше чем приблизительно 25 мкг/мл.
В еще одном варианте осуществления способы по данному изобретению содержат этап измерения начального липидного профиля пациента (или среднего для группы пациентов) перед началом терапии. В еще одном варианте осуществления способы по изобретению содержат этап идентификации пациента или группы пациентов, характеризующихся наличием одним или более из следующего: начальная величина уровня ХС-ЛПНВП от приблизительно 200 мг/дл до приблизительно 400 мг/дл, например, по меньшей мере приблизительно 210 мг/дл, по меньшей мере приблизительно 220 мг/дл, по меньшей мере приблизительно 230 мг/дл, по меньшей мере приблизительно 240 мг/дл, по меньшей мере приблизительно 250 мг/дл, по меньшей мере приблизительно 260 мг/дл, по меньшей мере приблизительно 270 мг/дл, по меньшей мере приблизительно 280 мг/дл, по меньшей мере приблизительно 290 мг/дл или по меньшей мере приблизительно 300 мг/дл; начальное общее количество холестерина от приблизительно 250 мг/дл до приблизительно 400 мг/дл, например, по меньшей мере приблизительно 260 мг/дл, по меньшей мере приблизительно 270 мг/дл, по меньшей мере приблизительно 280 мг/дл или по меньшей мере приблизительно 290 мг/дл; начальный уровень ХС-ЛПОНП от приблизительно 140 мг/дл до приблизительно 200 мг/дл, например, по меньшей мере приблизительно 150 мг/дл, по меньшей мере приблизительно 160 мг/дл, по меньшей мере приблизительно 170 мг/дл, по меньшей мере приблизительно 180 мг/дл или по меньшей мере приблизительно 190 мг/дл; начальный уровень ХС-ЛПВП от приблизительно 10 до приблизительно 100 мг/дл, например, не больше чем приблизительно 90 мг/дл, не больше чем приблизительно 80 мг/дл, не больше чем приблизительно 70 мг/дл, не больше чем приблизительно 60 мг/дл, не больше чем приблизительно 60 мг/дл, не больше чем приблизительно 50 мг/дл, не больше чем приблизительно 40 мг/дл, не больше чем приблизительно 35 мг/дл, не больше чем приблизительно 30 мг/дл, не больше чем приблизительно 25 мг/дл, не больше чем приблизительно 20 мг/дл или не больше чем приблизительно 15 мг/дл; и/или начальный уровень ХС-ЛПНП от приблизительно 30 до приблизительно 300 мг/дл, например, не меньше чем приблизительно 40 мг/дл, не меньше чем приблизительно 50 мг/дл, не меньше чем приблизительно 60 мг/дл, не меньше чем приблизительно 70 мг/дл, не меньше чем приблизительно 90 мг/дл или не меньше чем приблизительно 90 мг/дл.
В родственном варианте осуществления при лечении в соответствии с данным изобретением, например, в течение периода от приблизительно 1 до приблизительно 200 недель, от приблизительно 1 до приблизительно 100 недель, от приблизительно 1 до приблизительно 80 недель, от приблизительно 1 до приблизительно 50 недель, от приблизительно 1 до приблизительно 40 недель, от приблизительно 1 до приблизительно 20 недель, от приблизительно 1 до приблизительно 15 недель, от приблизительно 1 до приблизительно 12 недель, от приблизительно 1 до приблизительно 10 недель, от приблизительно 1 до приблизительно 5 недель, от приблизительно 1 до приблизительно 2 недель или приблизительно 1 недели у пациента или у группы пациентов проявляется одно или более из следующих последствий:
сниженные уровни триглицеридов по сравнению с начальным;
сниженные уровни апо-B по сравнению с начальным;
сниженные апо-B уровни по сравнению с начальным;
d) отсутствие повышения уровней холестерина-ЛПНП по сравнению с начальным;
e) снижение уровней холестерина-ЛПНП по сравнению с начальным;
f) снижение уровней ХС-ЛПНВП по сравнению с начальным;
g) снижение уровней ЛПОНП по сравнению с начальным;
h) повышение уровней апо A-I по сравнению с начальным;
i) повышение соотношения апо A-I/апо-B по сравнению с начальным;
j) снижение уровней липопротеина по сравнению с начальным;
k) снижение количества частиц ЛПНП по сравнению с начальным;
l) снижение размера ЛПНП по сравнению с начальным;
m) снижение количества частиц холестерина, подобных остаточным, по сравнению с начальным;
n) снижение количества окисленных ЛПНП по сравнению с начальным;
o) снижение уровня глюкозы в плазме натощак (FPG) по сравнению с начальным;
p) снижение уровня гемоглобина A1с (HbA1с) по сравнению с начальным;
q) снижение гомеостатической модели резистентности к инсулину по сравнению с начальной;
r) снижение уровня липопротеин-ассоциированной фосфолипазы А2 по сравнению с начальным;
s) снижение уровня молекул внутриклеточной адгезии 1 типа по сравнению с начальным;
t) снижение уровня интерлейкина-2 по сравнению с начальным;
u) снижение уровня ингибитора активатора плазминогена типа 1 по сравнению с начальным;
v) снижение уровня высокочувствительного C-реактивного белка (hsCRP) по сравнению с начальным;
w) повышение уровня EPA фосфолипида в плазме или сыворотке по сравнению с начальным;
x) повышение уровня личества EPA в мембране красных кровяных клеток по сравнению с начальным и/или
y) снижение или повышение содержания одного или более из следующего: докозагексаеновая кислота (DHA), докозапентаеновая кислота (DPA), арахидоновая кислота (AA), пальмитиновая кислота (PA), стеаридоновая кислота (SA) или олеиновая кислота (OA) в плазме, сывороточных фосфолипидах и/или красных кровяных клетках по сравнению с начальным.
В одном варианте осуществления способы по данному изобретению содержат измерение начальных уровней одного или более маркеров, изложенных в в (a)-(y) выше, перед введением доз пациенту или группе пациентов. В еще одном варианте осуществления способы содержат введение композиции, раскрытой в данном документе, пациенту после того как определены начальные уровни одного или более маркеров, изложенных в (a)-(y), и с проведением впоследствии дополнительных измерений упомянутых одного или более маркеров.
В еще одном варианте осуществления при лечении композицией по данному изобретению, например, в течение периода от приблизительно 1 до приблизительно 200 недель, от приблизительно 1 до приблизительно 100 недель, от приблизительно 1 до приблизительно 80 недель, от приблизительно 1 до приблизительно 50 недель, от приблизительно 1 до приблизительно 40 недель, от приблизительно 1 до приблизительно 20 недель, от приблизительно 1 до приблизительно 15 недель, от приблизительно 1 до приблизительно 12 недель, от приблизительно 1 до приблизительно 10 недель, от приблизительно 1 до приблизительно 5 недель, от приблизительно 1 до приблизительно 2 недель или приблизительно 1 недели у пациента или у группы пациентов проявляются любые 2 или более, любые 3 или более, любые 4 или более, любые 5 или более, любые 6 или более, любые 7 или более, любые 8 или более, любые 9 или более, любые 10 или более, любые 11 или более, любые 12 или более, любые 13 или более, любые 14 или более, любые 15 или более, любые 16 или более, любые 17 или более, любые 18 или более, любые 19 или более, любые 20 или более, любые 21 или более, любые 22 или более, любые 23 или более, любые 24 или более или все 25 последствий (a)-(y), непосредственно описанных выше.
В еще одном варианте осуществления при лечении композицией по данному изобретению, у пациента или у группы пациентов проявляется одно или более из следующих последствий:
(a) снижение уровня триглицерида по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 55% или по меньшей мере на приблизительно 75% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(b) менее чем 30% увеличение, менее чем 20% увеличение, менее чем 10% увеличение, менее чем 5% повышение или отсутствие повышения уровней ХС-ЛПНВП или снижение уровней ХС-ЛПНВП по меньшей мере на приблизительно 1%, по меньшей мере на приблизительно 3%, по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 55% или по меньшей мере на приблизительно 75% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(c) повышение уровней ХС-ЛПВП по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 55% или по меньшей мере на приблизительно 75% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(d) менее чем 30% увеличение, менее чем 20% увеличение, менее чем 10% увеличение, менее чем 5% увеличение или отсутствие увеличения уровней ХС-ЛПНП или снижение уровней ХС-ЛПНП по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 55%, по меньшей мере на приблизительно 55% или по меньшей мере на приблизительно 75% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(e) снижение уровней апо-B по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 55% или по меньшей мере на приблизительно 75% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(f) снижение уровней ЛПОНП по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным; (g) повышение уровней апо A-I по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(h) повышение соотношения апо A-I/апо-B по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(i) снижение уровней липопротеина по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(j) снижение среднего количества частиц ЛПНП по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(k) увеличение среднего размера частиц ЛПНП по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(l) снижение количества холестерина в остаточных частицах по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(m) снижение уровня окисленных ЛПНП по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(n) снижение уровня глюкозы в плазме натощак (FPG) по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(o) снижение уровня гемоглобина A1с (HbA1с) по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45% или по меньшей мере на приблизительно 50% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(p) снижение показателя гомеостатической модели резистентности к инсулину по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(q) снижение уровня липопротеин-ассоциированной фосфолипазы A2 по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(r) снижение уровня внутриклеточных молекул внутриклеточной адгезии типа 1 по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(s) снижение уровня интерлейкина-2 по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(t) снижение уровня ингибитора активатора плазминогена типа 1 по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(u) снижение уровня высокочувствительного С-реактивного белка (hsCRP) по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50% или по меньшей мере на приблизительно 100%) (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(v) повышение уровня EPA в плазме, сывороточных фосфолипидах или RBC по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 100%, по меньшей мере приблизительно 200% или по меньшей мере приблизительно 400% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(w) повышение уровня EPA в плазме, сывороточных фосфолипидах и/или в мембранах RBC по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 100%, по меньшей мере на приблизительно 200% или по меньшей мере на приблизительно 400% (конкретный % изменения или срединный % изменения) по сравнению с начальным;
(x) снижение или повышение в плазме, сывороточных фосфолипидах и/или RBC одного или более из DHA, DPA, AA, PA и/или OA по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 55%) или по меньшей мере на приблизительно 75% (конкретный % изменения или срединный % изменения) по сравнению с начальным и/или
(y) снижение общего уровня холестерина по меньшей мере на приблизительно 5%, по меньшей мере на приблизительно 10%, по меньшей мере на приблизительно 15%, по меньшей мере на приблизительно 20%, по меньшей мере на приблизительно 25%, по меньшей мере на приблизительно 30%, по меньшей мере на приблизительно 35%, по меньшей мере на приблизительно 40%, по меньшей мере на приблизительно 45%, по меньшей мере на приблизительно 50%, по меньшей мере на приблизительно 55% или по меньшей мере на приблизительно 75% (конкретный % изменения или срединный % изменения) по сравнению с начальным.
В одном варианте осуществления способы по данному изобретению содержат измерение начальных уровней одного или более маркеров, изложенных в (a)-(y), перед введением доз пациенту или группе пациентов. В еще одном варианте осуществления способы содержат введение композиции, раскрытой в данном документе, пациенту после того как определены начальные уровни одного или более маркеров, изложенных в (a)-(y), и последующее проведение второго измерения одного или более маркеров как это было измерено в начале для сравнения.
В еще одном варианте осуществления при лечении композицией по данному изобретению, например, в течение периода от приблизительно 1 до приблизительно 200 недель, от приблизительно 1 до приблизительно 100 недель, от приблизительно 1 до приблизительно 80 недель, от приблизительно 1 до приблизительно 50 недель, от приблизительно 1 до приблизительно 40 недель, от приблизительно 1 до приблизительно 20 недель, от приблизительно 1 до приблизительно 15 недель, от приблизительно 1 до приблизительно 12 недель, от приблизительно 1 до приблизительно 10 недель, от приблизительно 1 до приблизительно 5 недель, от приблизительно 1 до приблизительно 2 недель или приблизительно 1 недели у пациента или у группы пациентов проявляется любые 2 или более, любые 3 или более, любые 4 или более, любые 5 или более, любые 6 или более, любые 7 или более, любые 8 или более, любые 9 или более, любые 10 или более, любые 11 или более, любые 12 или более, любые 13 или более, любые 14 или более, любые 15 или более, любые 16 или более, любые 17 или более, любые 18 или более, любые 19 или более, любые 20 или более, любые 21 или более, любые 22 или более, любые 23 или более, любые 24 или более или все 26 или более последствий (a)-(y), непосредственно описанных выше.
Параметры (a)-(y) могут быть определены в соответствии с любой клинически приемлемой методикой. Например, триглицериды, общий холестерин, ХС-ЛПВП и сахар, содержащиеся в крови натощак могут быть в виде образца из сыворотки и проанализированы с использованием стандартных фотометрических технологий. Содержание ТГ-ЛПОНП, ХС-ЛПНП и ХС-ЛПОНП может быть рассчитано или определено с использованием фракционирования липопротеинов сыворотки посредством препаративного ультрацентрифугирования и последующего количественного анализа посредством рефрактометрииили посредством аналитических методик ультрацентрифугирования. Апо A1, апо-B и hsCRP могут быть определены в сыворотке с использованием стандартных технологий нефелометрии. Уровень липопротеина (a) может быть определен в сыворотке с использованием стандартных технологий турбидиметрического иммуноанализа. Количество частиц ЛПНП и размер частиц могут быть определены с использованием спектрометрии ядерно-магнитного резонанса (ЯМР). Остаточные липопротеины и ЛПНП-фосфолипаза A2 могут быть определены в ЭДТА плазме или сыворотке и сыворотке, соответственно, с использованием технологий ферментативного иммуноразделения. Уровни окисленного ЛПНП, молекул межклеточной адгезии типа 1 и интерлейкина-2 могут быть определены в сыворотке с использованием стандартных технологий иммуноферментного анализа. Такие технологии подробно описаны в стандартных руководствах, например, Tietz Fundamentals of Сlinical Сhemistry, 6th Ed. (Burtis, Ashwood и Borter Eds.), WB Saunders Company.
В одном варианте осуществления пациенты не получают пищу в течение до 12 часов перед отбором проба крови, например, приблизительно в течение 10 часов.
В еще одном варианте осуществления данное изобретение относится к способу лечения или предотвращения первичной гиперхолестеринемии и/или смешанной дислипидемии (типы IIa и IIb по Фредриксону) у пациента, нуждающегося в этом, включающему введение пациенту одной или более композиций, раскрытых в данном документе. В родственном варианте осуществления данное изобретение относится к способу понижения уровней триглицеридов у пациента или пациентов, когда лечение статином или монотерапия с продленным высвобождением ниацина считаются неподходящими (гиперлипидемия типа IV по Фредриксону).
В еще одном варианте осуществления данное изобретение относится к способу лечения или предотвращения риска рецидивного несмертельного инфаркта миокарда у пациента, уже имевшего инфаркт миокарда, включающему введение пациенту одной или более композиций, раскрытых в данном документе.
В еще одном варианте осуществления данное изобретение относится к способу замедления развития или стимуляции регрессии атеросклеротического заболевания у пациента, нуждающегося в этом, включающему введение пациенту, нуждающемуся в этом, одной или более композиций, раскрытых в данном документе.
В еще одном варианте осуществления данное изобретение относится к способу лечения или предотвращения очень высоких уровней триглицеридов в сыворотке (например, гиперлипидемия типов IV и V) у пациента, нуждающегося в этом, включающему введение пациенту одной или более композиций, раскрытых в данном документе.
В одном варианте осуществления композиция по изобретению вводится пациенту в количестве, достаточном для обеспечения ежедневно дозы этилэйкозапентаеновой кислоты от приблизительно 1 мг до приблизительно 10000 мг, от приблизительно 25 до приблизительно 5000 мг, от приблизительно 50 до приблизительно 3000 мг, от приблизительно 75 мг до приблизительно 2500 мг или от приблизительно 100 мг до приблизительно 1000 мг, например, приблизительно 75 мг, приблизительно 100 мг, приблизительно 125 мг, приблизительно 150 мг, приблизительно 175 мг, приблизительно 200 мг, приблизительно 225 мг, приблизительно 250 мг, приблизительно 275 мг, приблизительно 300 мг, приблизительно 325 мг, приблизительно 350 мг, приблизительно 375 мг, приблизительно 400 мг, приблизительно 425 мг, приблизительно 450 мг, приблизительно 475 мг, приблизительно 500 мг, приблизительно 525 мг, приблизительно 550 мг, приблизительно 575 мг, приблизительно 600 мг, приблизительно 625 мг, приблизительно 650 мг, приблизительно 675 мг, приблизительно 700 мг, приблизительно 725 мг, приблизительно 750 мг, приблизительно 775 мг, приблизительно 800 мг, приблизительно 825 мг, приблизительно 850 мг, приблизительно 875 мг, приблизительно 900 мг, приблизительно 925 мг, приблизительно 950 мг, приблизительно 975 мг, приблизительно 1000 мг, приблизительно 1025 мг, приблизительно 1050 мг, приблизительно 1075 мг, приблизительно 1100 мг, приблизительно 1025 мг, приблизительно 1050 мг, приблизительно 1075 мг, приблизительно 1200 мг, приблизительно 1225 мг, приблизительно 1250 мг, приблизительно 1275 мг, приблизительно 1300 мг, приблизительно 1325 мг, приблизительно 1350 мг, приблизительно 1375 мг, приблизительно 1400 мг, приблизительно 1425 мг, приблизительно 1450 мг, приблизительно 1475 мг, приблизительно 1500 мг, приблизительно 1525 мг, приблизительно 1550 мг, приблизительно 1575 мг, приблизительно 1600 мг, приблизительно 1625 мг, приблизительно 1650 мг, приблизительно 1675 мг, приблизительно 1700 мг, приблизительно 1725 мг, приблизительно 1750 мг, приблизительно 1775 мг, приблизительно 1800 мг, приблизительно 1825 мг, приблизительно 1850 мг, приблизительно 1875 мг, приблизительно 1900 мг, приблизительно 1925 мг, приблизительно 1950 мг, приблизительно 1975 мг, приблизительно 2000 мг, приблизительно 2025 мг, приблизительно 2050 мг, приблизительно 2075 мг, приблизительно 2100 мг, приблизительно 2125 мг, приблизительно 2150 мг, приблизительно 2175 мг, приблизительно 2200 мг, приблизительно 2225 мг, приблизительно 2250 мг, приблизительно 2275 мг, приблизительно 2300 мг, приблизительно 2325 мг, приблизительно 2350 мг, приблизительно 2375 мг, приблизительно 2400 мг, приблизительно 2425 мг, приблизительно 2450 мг, приблизительно 2475 мг или приблизительно 2500 мг.
В еще одном варианте осуществления любые из способов, раскрытых в данном документе, используются при лечении пациента или пациентов, которые потребляют традиционное западное питание. В одном варианте осуществления способы по изобретению включают этап идентификации пациента как потребителя западного питания или потребителя целесообразного питания и затем лечение пациента, если пациент признается потребителем западного питания. Термин "западное питание" в данном документе в целом относится к типичному питанию, состоящему из (по проценту от общего количества калорий) от приблизительно 45% до приблизительно 50% углеводов, от приблизительно 35% до приблизительно 40% жиров и от приблизительно 10% до приблизительно 15% белков. Западное питание может с другой стороны или дополнительно характеризоваться относительно высоким потреблением красного мяса и готовых мясных блюд, сладостей, очищенных зерновых продуктов и десертов, например, более 50%, более 60% или более или 70% от общего количества калорий поступают из этих источников.
В еще одном варианте осуществления любые из способов, раскрытых в данном документе, используются при лечении пациента или пациентов, которые потребляют менее чем (фактически или в среднем) приблизительно 150 г, менее чем приблизительно 125 г, менее чем приблизительно 100 г, менее чем приблизительно 75 г, менее чем приблизительно 50 г, менее чем приблизительно 45 г, менее чем приблизительно 40 г, менее чем приблизительно 35 г, менее чем приблизительно 30 г, менее чем приблизительно 25 г, менее чем приблизительно 20 г или менее чем приблизительно 15 г рыбы в сутки.
В еще одном варианте осуществления любые из способов, раскрытых в данном документе, используются при лечении пациента или пациентов, которые потребляют менее чем (фактически или в среднем) приблизительно 10 г, менее чем приблизительно 9 г, менее чем приблизительно 8 г, менее чем приблизительно 7 г, менее чем приблизительно 6 г, менее чем приблизительно 5 г, менее чем приблизительно 4 г, менее чем приблизительно 3 г, менее чем приблизительно 2 г в сутки омега-3 жирных кислот из пищевых источников.
В еще одном варианте осуществления любые из способов, раскрытых в данном документе, используются при лечении пациента или пациентов, которые потребляют менее чем (фактически или в среднем) приблизительно 2,5 г, менее чем приблизительно 2 г, менее чем приблизительно 1,5 г, менее чем приблизительно 1 г, менее чем приблизительно 0,5 г, менее чем приблизительно 0,25 г или менее чем приблизительно 0,2 г в сутки EPA и DHA (вместе взятых) из пищевых источников.
В одном варианте осуществления композиция, описанная в данном документе, вводится пациенту один раз или два раза в сутки. В еще одном варианте осуществления 1, 2, 3 или 4 капсулы, каждая из которых содержит от приблизительно 500 мг до приблизительно 1 г композиции, описанной в данном документе, вводятся пациенту ежедневно. В еще одном варианте осуществления 1 или 2 капсулы, каждая из которых содержит приблизительно 1 г композиции, описанной в данном документе, вводятся пациенту утром, например, между приблизительно 5 и приблизительно 11 утра, и 1 или 2 капсулы, каждая из которых содержит приблизительно 1 г композиции, описанной в данном документе, вводятся пациенту вечером, например, между приблизительно 5 и приблизительно 11 вечера.
В одном варианте осуществления пациент, подвергающийся лечению в соответствии со способами изобретения, не проходит фибратную или нитратную терапию.
В еще одном варианте осуществления композиции, пригодные в соответствии со способами изобретения, являются доставляемыми орально. Термины "орально доставляемые" или "орально вводимые" в данном документе включают любой вид доставки терапевтического агента или его композиции пациенту, где агент или композиция помещаются в рот пациента, независимо от того, проглатывается композиция или нет. Таким образом "оральное введение" включает буккальное и сублингвальное, а также введение через пищевод. В одном варианте осуществления композиция входит в состав капсулы, например, мягкой желатиновой капсулы.
Композиция для применения в соответствии с изобретением может быть приготовлена в виде одной или более единиц дозирования. Термины "единица дозы" и "единица дозирования" в данном документе относятся к порции фармацевтической композиции, которая содержит количество терапевтического агента, пригодное для однократного введения для обеспечения терапевтического эффекта. Такие единицы дозирования могут вводиться от одного до множества раз (т.е. от 1 до приблизительно 10, от 1 до 8, от 1 до 6, 1 до 4 или от 1 до 2) в сутки или столько раз, сколько требуется для достижения терапевтического ответа.
В еще одном варианте осуществления изобретение относится к применению любой композиции, описанной в данном документе, для лечения от умеренной до острой гипертриглицеридемии у пациента, нуждающегося в этом, включающему: обеспечение пациента, имеющего начальный уровень триглицеридов натощак от приблизительно 500 мг/дл до приблизительно 1500 мг/дл, и введение пациенту фармацевтической композиции, описанной в данном документе. В одном варианте осуществления композиция содержит от приблизительно 1 г до приблизительно 4 г этилового эфира эйкозапентаеновой кислоты, при этом композиция практически не содержит докозагексаеновую кислоту.
В еще одном варианте осуществления пациент, подвергающийся лечению, страдает от диабета.
ПРИМЕРЫ
Следующие примеры представлены только в иллюстративных целях и не должны рассматриваться как ограничивающие изобретение каким-либо образом.
Пример 1
Выполняли одноцентровое двойное слепое рандомизированное плацебо-контролируемое исследование в параллельных группах с целью определения оптимальной дозы E-EPA у пациентов со связанными с возрастом нарушениями (AAMI). Первостепенной целью являлось исследование эффекта этил-EPA в сравнении с плацебо на когнитивное функционирование у пациентов с AAMI, посредством измерения способности выполнять задания на внимание в компьютеризированной тестовой батарее в течение периода 6 недель. Второстепенными задачами являлось:
(1) исследовать эффект E-EPA в сравнении с плацебо в течение 6 недель в следующих тестах в компьютеризированной когнитивной батерее: задания на продолжительность внимания; задания на качество кратковременной памяти; задания на качество эпизодической памяти; задания на скорость внимания;
(2) оценить безопасность и переносимость E-EPA в сравнении с плацебо в общепринятых клинических лабораторных тестах, контроль нежелательных явлений (НЯ) и показателей жизнедеятельности и
(3) оценить потенциальную зависимость доза-эффект E-EPA с конечными когнитивными показателями посредством измерения уровня незаменимых жирных кислот в плазме и мембранах красных кровяных клеток. 94 пациента были рандомизированы.
План исследования заключался в задействовании 96 пациентов, которые должны были рандомизированно распределены с 1 по 4 возможные группы лечение в течение 6 недель, с равными группами (24 пациента на группу), следующим образом:
1 г этил-EPA ежедневно
2 г этил-EPA ежедневно
4 г этил-EPA ежедневно
Плацебо (парафиновое масло) ежедневно
Этил-EPA предоставлялся по 500 мг в мягких желатиновых капсулах, обеспечивающих этил-EPA >96% чистоты, от 0,25% до 0,38% по массе этилоктадекатетраеноата, от 0,075% до 0,15% по массе этилнонаэкапентаеноата, от 0,25% до 0,35% по массе этиларахидоната, от 0,3% до 0,4% по массе этилэйкозатетраеноата (ETA-E), от 0,075% до 0,15% этилгенэйкозапентаеноата и 0,2% dl-α-токоферола в качестве антиоксиданта. Наполнение капсул плацебо включало 467 г жидкого парафина и 0,2% dl- α-токоферола. Плацебо группу дополнительно рандомизировали, так что одинаковое количество пациентов (8) получало 1 г, 2 г или 4 г плацебо. Исследуемое лекарственное средство принималось по два раза в сутки (BID) в виде разделенной дозы (например, для 1 г дозы, 500 мг давалось утром и дополнительно 500 мг давалось вечером) с легкой закуской или едой.
Исследование состояло из скринингового визита, тренировочного визита и 4 экспериментальных визитов. Во время скринингового визита определялось соответствие пациентов требованиям посредством когнитивных тестов: вербальное заучивание парных ассоциаций (verbal paired associated learning [PAL] subscale), словарный субтест(vocabulary subtest), Опросный лист оценки памяти для клиник (Memory Assessment Clinics Questionnaire [MAC-Q]), Мини-исследование психического статуса (mini mental state evaluation [MMSE]) и Краткий международный нейропсихиатрический опросник (MINI [mini international neuropsychiatric interview; sections 1 and 2 of Diagnostic and Statistical Manual of Mental Disorders, 4th Ed. (DSM-IV) plus dysthymia]), гематологии, клинической биохимии и электрокардиограммы в 12 отведениях (ECG). Во время тренировочного визита пациентам было показано, как использовать CDR компьютеризированную систему. Пациенты принимали исследуемое лекарственное средство в течение 6 недель и в дни 0, 14, 28 и 42 пациенты подвергались CDR батарее когнитивных тестов.
Критерии включения
1.Письменное информированное согласие.
2. Добровольцы мужского и женского пола возрастом между 50 и 70 годами.
3. Собственные жалобы на потерю памяти, отражаемые в таких ежедневных проблемах, как сложности с запоминанием имен отдельных людей после представления, размещение предметов не на их место, сложности с запоминанием многочисленных объектов, которые должны быть приобретены, или множественных задач, которые должны быть выполнены, проблемы с запоминанием телефонных номеров или почтовых кодов и сложности с быстрым вспоминанием информации или последующим отвлечением внимания, что установлено оценкой 25 или выше в опросном листе MAC-Q. Появление потери памяти должно было быть описано как постепенное без резкого ухудшения в последние месяцы.
4. Обладание субъективным и объективным когнитивным нарушением с оценкой по меньшей мере на 1 стандартное отклонение (SD) ниже чем среднее для выровненного по возрасту населения старших возрастов, что установлено общей оценкой между 13 и 20 в субтестах PAL шкалы памяти Векслера.
5. Проявление удовлетворительной интеллектуальной функции, что установлено вычисленной оценкой по меньшей мере 9 (исходная оценка по меньшей мере 32) словарного субтеста шкалы интеллекта взрослых Векслера (WAIS).
6. Отсутствие слабоумия, что установлено оценкой 24 или выше в MMSE.
7. Некурящие или некурящие в течение более 3 месяцев.
8. Были способны приехать в центр и были оценены исследователем как вероятно способные продолжать приезжать в течение срока исследования и соответствующие логистическим аспектам исследования.
9. Индекс массы тела (ИМТ)<29,5 кг/м2.
Критерии невключения
Маловероятность или неспособность выполнять требования исследуемого введения лекарственных доз.
2. Диагноз тяжелое депрессивное расстройство, болезнь Альцгеймера или сосудистая деменция, определенные в соответствии с критериями MINI/DSM-IV редактирование текста (TR).
3. Предшествующий или текущий анамнез неврологического или психиатрического нарушения, которое могло повлиять на когнитивную функцию.
4. Предшествующий или текущий анамнез воспалительного заболевания желудочно-кишечного тракта, такого как болезнь Крона или язвенный колит.
5. Запор, который требовал активное лечение.
6. Текущий или предшествующий анамнез рака за исключением диагноза базально-клеточная карцинома.
7. Любая история или признаки клинически значимых нарушений сердечной деятельности, определенные ЭКГ в 12 отведениях.
8. Любое другое медицинское состояние или интеркуррентное заболевание, не контролируемое соответствующим образом, которое, по мнению исследователя, может подвергнуть пациента риску при участии в исследовании или может повлиять на результаты исследования или повлиять на способность пациента принимать участие в исследовании.
9. Клинически значимые аномальные результаты лабораторного скрининга (гематология, биохимия) при скрининге или показатели жизнедеятельности, которые не попадают в нормальные пределы для этой популяции, которые, по мнению исследователя, могут повлиять на соответствие пациента требованиям исследования.
10. Любые изменения по отношению к прописанному лечению медицинского состояния в течение 4 недель от начального визита.
11. Добавки омега-3 в течение 4 недель от начального визита или во время исследуемого периода лечения.
12. В настоящее время принимает антикоагулянты или ежедневную дозу аспирина >325 мг.
13. Средства против кашля или простуды, содержащие опиаты или антигистаминные средства, в течение 2 недель от начального визита или во время 6-недельного периода лечения.
14. Известная аллергия к любому из ингредиентов исследуемого лекарственного средства или плацебо.
Любой пациент мог отказаться от исследования в любое время по своему требованию или по требованию его официального опекуна, или по выбору исследователя, если задействование, не представляло интереса для пациентов, или в случае серьезного или неожиданного НЯ. Все разумные усилия были предприняты для документирования результатов пациента и причин прекращения участия. За любыми существующими НЯ наблюдали до тех пор, пока явление не было устранено, стабилизировано или объяснено другим образом. Пациенты, участие которых было прекращено, не были заменены. За пациентами были закреплены индивидуальные идентификационные номера в соответствии с предопределенным рандомизационным списком, генерированным Catalent Pharma Solutions, и использовались при упаковывании лекарственного средства.
Исследуемое лекарственное средство вводили орально BID в виде разделенной дозы с едой в течение 6 недель. Пациентов рандомизировали на 1 из 6 возможных групп лечения (Таблица 1).
Группы Лечения
Исследуемое лекарственное средство было распределено в визиты 3, 4 и 5; максимальный период между визитом 3 и каждым следующим визитом составлял:
от визита 3 до визита 4 (2 недели ±2 дня от визита 3),
от визита 3 до визита 5 (4 недель ±2 дня от визита 3),
от визита 3 до визита 6 (6 недель ±2 дня от визита 3).
Все лечебные упаковки были внешне одинаковыми, для того чтобы пациент и исследователь оставались в неведении на протяжении всего исследования. Исследователь, спонсор/персонал клинической экспериментальной организации и пациенты оставались в неведении на протяжении всего этого исследования. Исследователю было позволено демаскировать отдельных пациентов, если это считалось обоснованным с медицинской точки зрения. Процесс демаскирования изложен ниже.
Прием омега-3 добавки должен быть прекращен по меньшей мере за 4 недели перед начальным визитом (визит 3). Прием лекарственных средств от кашля и гриппа, содержащих опиаты или антигистаминные средства, должен быть прекращен за 2 недели перед начальным визитом (визит 3) и не был разрешен во время исследования.
Действующее лекарственное лечение должно было быть стабильным в течение 4 недель перед начальным визитом (визит 3) и дозу сохраняли во время исследования. Когда изменение дозы было определенно необходимо, это записывалось в электронную индивидуальную регистрационную карту (eCRF).
Пациенты, которым требовалось противокоагулирующее лечение во время исследования, должны были быть исключены. Психологическое консультирование или терапия не разрешались во время исследования, так как на это могло повлиять на последствия исследования. Неиспользованное исследуемое лекарственное средство было возвращено в экспериментальный центр. Пациенты, которые использовали менее 80% предписанной дозы, считались не соблюдающими режим лечения.
Были проведены скрининговое когнитивное тестирование и проверка соответствия требованиям исследования с использованием вербальных спаренных ассоциативных связей (Verbal Paired Associates 1) (Шкала памяти Векслера), словарного субтеста WAIS, MAC-Q, MMSE и MINI (DSM-IV разделы 1 и 2 и дистимия).
Набор заданий из компьютеризированной когнитивной системы оценки CDR был осуществлен (приложение 8 протокола) в визит 2 (тренировочный визит), визит 3 (начальный), визит 4 (день 14), визит 5 (день 28) и визит 6 (день 42). В каждом сеансе тестирования присутствовали параллельные формы тестов. Все задания контролировались компьютером, информация предоставлялась на мониторах с высоким разрешением, и ответы записывались с помощью модели откликов, содержащей 2 кнопки: 1 - обозначенная 'нет', другая - 'да'. Пять суммарных баллов CDR использовали в качестве первичных/вторичных конечных величин.
Задания имели следующие названия:
Словесное представление (Word presentation)
Непосредственное словесное воспроизведение (Immediate Word Recall)
Представление изображений (Picture Presentation)
Время простой реакции (Simple Reaction Time)
Цифровая внимательность (Digit Vigilance)
Время реакции выбора (Choice Reaction Time)
Пространственная память (Spatial Working Memory)
Числовая память (Numeric Working Memory)
Замедленное вспоминание слов (Delayed Word Recall)
Распознавание слов (Word Recognition)
Распознавание изображений (Picture Recognition)
Визуальный Бонд-Ладера (Bond-Lader Visual)
Аналоговые шкалы настроения и восприимчивости (Analogue Scales of Mood and Alertness)
Скрининг с использованием компьютерной мыши (Screen, Using the Computer Mouse).
Для обеспечения систематичности подхода, все обучение когнитивным тестам и батарее тестов CDR было предоставлено исследовательскому персоналу на местах и пациентам. Результаты каждой величины были автоматически записаны с использованием машинного интерфейса, разработанного CDR.
НЯ определяли как любое неблагоприятное медицинское явление, временно относящееся к использованию лекарственного средства, независимо от того связано оно с лекарственным средством или нет. Исследователь был ответственен за обнаружение и документирование НЯ. Во время каждого визита пациента спрашивали об НЯ посредством ненаводящих вопросов. НЯ записывали начиная со времени, когда пациент предоставлял письменное информированное согласие и считался подходящим для участия до завершения периода лечения. За НЯ, продолжающимися в конце периода лечения, наблюдали до прекращения или возвращения к начальному или обычному значению или до того, как явление признавали несвязанным с исследуемым лекарственным средством.
Серьезное побочное явление (СНЯ) было определено как любое НЯ при любой дозе, которое:
приводило к смерти;
было опасно для жизни;
требовало госпитализацию или продление имеющейся госпитализации;
приводило к инвалидности или нетрудоспособности или
приводило к врожденной аномалии/пороку развития.
Другие явления считались СНЯ, если они подвергали пациента опасности или требовали медицинское или хирургическое вмешательство для предотвращения приведенных выше последствий.
Независимо от приведенных выше критериев, о любом НЯ, которое спонсор или исследователь считали серьезным, должно было быть непосредственно сообщено как о СНЯ. О смерти или любом СНЯ, испытываемом пациентом во время получения или в течение 30 дней после последней дозы исследуемого лекарственного средства, должно было быть немедленно сообщено в фармакологический надзор (в течение 24 часов после получения информации о явлении). Все НЯ (включая СНЯ) должны были быть точно записаны на листе побочных явлений eCRF пациента, начиная с первого введения исследуемого лекарственного средства до 30 дней после последней дозы.
Образцы крови для гематологических лабораторных исследований (5 мл образец крови) и клиническая биохимия (10 мл образец крови), приведенные в таблице 2, были отобраны во время скринингового визита (визит 1). Образцы обрабатывали и анализировали с помощью Simbec Laboratories Ltd.
Лабораторные исследования
Образцы крови (10 мл) отбирали во время визита 1 (скрининг) и во время визитов 4, 5 и 6. Анализ выполняли с помощью липидного анализа MSR, Scottish Crop Research Institute, Dundee, UK. Скрининговый образец считался начальным во время измерений EFA.
Липид экстрагировали из плазмы, сыворотки и суспензий RBC и превращали в метиловые эфиры жирных кислот, которые анализировали с помощью газовой хроматографии с получением профилей жирных кислот в микрограммах жирной кислоты на грамм образца (мкгЖК/г) и нормализованного процента площади. Использовали компьютеризированную систему CDR для измерения эффектов лекарственных препаратов на когнитивную функцию в ряде клинических испытаний. Эффективность достигалась посредством батареи когнитивных тестов, разработанных CDR. Показатели безопасности анализировались с помощью Quanticate.
Анализируемое население включало следующие категории:
Популяция пациентов, "намеренных лечиться" (Intent to Treat (ITT)): все рандомизированные пациенты с по меньшей мере с 1 визитом после начального были включены в эту популяцию, независимо от того, в действительности ли они получили лечение.
Популяция по протоколу (PP): все рандомизированные пациенты, которые завершили исследование, за исключением существенного отклонения от протокола, были определены как Безопасная популяция PP. Эффективная популяция PP была основана на эффективно завершивших курс лечения. Перекрывание Безопасной и Эффективной PP популяций определяло Исследовательскую популяцию PP.
Безопасная популяция: все рандомизированные пациенты, которые получили по меньшей мере 1 дозу исследуемого лекарственного средства.
Суммарная статистика была обеспечена для ITT и Исследовательской PP популяции отдельно для всех суммарных баллов, главных и вспомогательных величин. Суммарная статистика была выполнена как для нескорректированных, так для отличия от начальных данных (т.е. отличие от согласованных во времени оценок перед дозой в день 0). Суммарная статистика была рассчитана по лечению, дням и моментам времени. Суммарная статистика содержала n, среднее, срединное, SD, стандартную ошибку среднего (SEM), минимальное и максимальное значения.
Отличие от начальных данных для каждой главной величины оценивалось с помощью ковариационного анализа (ANCOVA) с использованием SAS® PROC MIXED версия 8.2.
Были установлены фиксированные эффекты для лечения, дней, моментов времени, лечения по дням, лечения по моментам времени, лечения по дням по моментам времени. Пациент в лечении был установлен в качестве итеративного воздействия с использованием итеративного выражения. Применялась ковариационная структура со сложной симметрией. Согласованные во времени оценки пациента перед дозой в день 0 были использованы в анализе в качестве ковариаты. Пределы среднего (LS means) были рассчитаны для лечения по дням, лечения по моментам времени и лечения при взаимосвязи по дням и моментам времени. Этот формальный анализ был проведен отдельно для ITT и Исследовательской популяции PP.
Оценки безопасности были основаны на безопасной популяции. Безопасность и переносимость оценивали при помощи НЯ, показателей жизнедеятельности, ЭКГ в 12 отведениях, клинико-лабораторных данных, медицинского анамнеза и комплаентности к исследуемому лекарственному средству. Данные о безопасности и переносимости были представлены по группам лечения. Все данные о безопасности были приведены для пациента отдельно.
Данные о EFA в RBC и плазме отбирали в начале, дни 14, 28 и 42 и обобщали по визитам для каждой группы лечения. Изменение по сравнению с начальным и процент изменения по сравнению с начальным обобщали. Выполняли сопоставление ANCOVA групп доз этил-EPA и этил-EPA в сравнении с плацебо.
Расчет размера выборки был основан на Способности к вниманию(Power of Attention). Испрониклин (50 мг), частичный агонист нейронального никотинового холинорецептора, у пациентов с AAMI на день 21 повторного введения доз в более раннем исследовании продемонстрировал улучшение на 61 мс (50 мг среднее=-32,54, SD=61,22; плацебо среднее=28,25, SD=49,64) Способности к вниманию. С использованием объединенных SD, размер выборки из 15 пациентов на группу лечения считался достаточным для детектирования 61 мс, с 80% способностью и 5% уровнем значимости (без поправки для множественных сравнений). Так как предшествующего опыта с соединением или механизмом действия таких когнитивных критериев, то в качестве достаточного был выбран размер выборки 24 пациента на группу лечения, для того чтобы иметь возможность более раннего прекращения участия.
Для проведения исследования не было внесено никаких изменений. Следующие изменения были внесены в планируемые анализы: формула для расчета скорости запоминания (Speed of Memory) была изменена на SPEEDMEM (скорость запоминания) = SPMRT (пространственная скорость запоминания [spatial working memory speed])+NWMRT (числовая скорость запоминания [numeric working speed])+DRECRT (скорость распознавания слов [word recognition speed])+DPICRT (скорость распознавания изображений [picture recognition speed]).
Согласованные во времени оценки пациента перед дозой в день 0 использовали в анализе в качестве ковариаты.
День 0 был удален из значений дней в списке значений параметров ANCOVA. Ковариата = Начальный уровень была изменена на Ковариата = Согласованные во времени оценки перед введением доз в день 0 в списке значений параметров ANCOVA.
День по моменту времени был добавлен в список модельных эффектов в SAS® code модели ANCOVA.
Таблица F тестов и таблица эффектов лечения были добавлены список сводных таблиц ANCOVA.
Сводные таблицы ANCOVA были перенумерованы в соответствии с необработанными результатами ANCOVA.
Были включены фигуры для лечения, лечения по дням, лечения по моментам времени, эффекты лечения по дням по моментам времени для предела среднего ANCOVA.
Были добавлены фигуры для различий пределов среднего ANCOVA от плацебо (95% доверительный интервал [ДИ]).
Выполняли анализ по полученным результатам, в котором сравнивали отдельные группы плацебо (1 г, 2 г и 4 г парафинового масла) с соответствующей дозой этил-EPA, а не с группой без плацебо.
Девяносто один пациент завершил исследование, три пациента прервали; 2 пациента из группы лечения этил-EPA 2 г (1 пациент из-за СНЯ, которое оказалось несвязанным с исследуемым лекарственным средством, и 1 из-за нарушения протокола и 1 пациент из группы плацебо 2 г из-за НЯ.
Для Способности к вниманию не наблюдалось статистически значимого эффекта от лечения, ни при лечении по дням, ни при лечении по моментам времени или при сочетании лечения по дням и по моментам времени. Не имело место отличие предела среднего между активным лечением и плацебо в любой момент времени. Для времени реакции выбора (Choice Reaction Time) имели место статистически значимые преимущества для этил-EP 1 г и 2 г в день 28 и некоторые признаки преимущества для 1 и 4 г этил-EPA в день 42 по сравнению с плацебо; тем не менее не наблюдали однозначной связанной с проводимой терапией модели.
Продолжительность внимания (Continuity of Attention) не демонстрировала отличия между плацебо и этил-EPA, за исключением общего снижения для 2 г этил-EPA, которое было заметно только в ITT популяции. Элемент задания обнаруженные мишени для внимания к цифрам (Digit Vigilance Targets Detected) продемонстрировал отдельные снижения для активного лечения по сравнению с плацебо, но не имело места наглядная связанная с проводимой терапией модель.
Качество кратковременной памяти (Quality of working memory) являлось только суммарной оценкой, которая демонстрировала статистически значимое лечение во взаимосвязи по дням по статистике Фишера. Тем не менее, были выделены только статистически значимые снижения для этил-EP 1 г и 2 г в сравнении с плацебо на дни 14 и 28, и они наиболее вероятно являлись случайными и не связанными с лечением.
Качество эпизодической вторичной памяти (Episodic Secondary Memory) продемонстрировало статистически значимые снижения для этил-EPA в сравнении с плацебо в различные моменты времени. Тем не менее, это маловероятно являлось эффектом активного лечения, так как несогласованные данные продемонстрировали изначально присутствующие различия между группами лечения, что было наиболее заметно в день 0 в первом периоде оценки. В отличие от начальных данных, которые были рассчитаны перед анализом ANCOVA, эти различия больше не были выражены. Это предполагает, что модель ANCOVA устанавливала выраженную обратную корреляцию с начальными значениями. Это часто имеет место, когда вариабельность у пациентов накладывается на вариабельность между пациентами.
Скорость памяти и элементы задания Скорости пространственной и числовой памяти (Spatial and Numeric Working Memory Speeds) и Скорость распознавания слов и изображений (Word and Picture Recognition Speed) не продемонстрировали различия в производительности между этил-EPA и плацебо в статистике Фишера.
Для оценки собственной настороженности (Self-rated Alertness) не имело место явное отличие в оценках между этил-EPA и плацебо. Были получены снижения оценок для активного лечения по сравнению с плацебо, что вероятно не было связано с соединением.
Оценка собственной удовлетворенности (Self-rated Contentment) продемонстрировала статистически значимые снижения оценок для этил-EPA 2 г в день 28. Тем не менее, эти отдельные снижения не были статистически значимыми. Маловероятно, что это было связано с эффектом проводимой терапии, так как это было ограничено единственным днем, и никакой другой уровень дозы не продемонстрировал подобную модель в любой другой день. Для оценки собственного спокойствия (Self-rated Calmness) не наблюдалось отличия в оценках между активным лечением и плацебо.
Когда осуществили сравнение результатов каждой дозы этил-EPA и соответствующего ей плацебо (по полученным результатам анализа), оказалось, что этил-EPA 4 г улучшал время реакции пациента в заданиях на внимательность (способность к вниманию (Power of Attention), время простой реакции (Simple Reaction Time) и время реакции выбора (Choice Reaction Time)). Наиболее очевидно это было видно из времени реакции выбора, где наблюдалась модель постепенного улучшения в течение оцениваемого дня для 4 г. Возможно, что более длительный период введения прояснил бы эффекты этил-EPA на эти параметры.
Значения EPA (приведено в таблице 3), DPAn-3 и соотношение EPA/AA (данные не приведены) в плазме и RBC повышались практически от начала до дня 42 для AMR-101 групп лечения 1,2, и 4 г. Уровни AA, DHA и DGLA снижались практически от начала (данные не приведены).
Среднее (SD) EPA (В плазме и RBC (мкг/г)) изменение от начального
(24,69)
(8,91)
(9,15)
(7,06)
(8,01)
(6,97)
Как видно из таблицы 3, при дозе AMR101 2 г в сутки уровни EPA в плазме повышались на 297% после 42 дней и при дозе AMR101 4 г в сутки уровни EPA в плазме повышались на 417% по сравнению с начальным.
[0167] Grimsgaard et al. ранее опубликовали статью, описывающую уровни фосфолипидов в сыворотке в начале и после 7 недель добавки 4 г в сутки 90% этил-DHA, 4 г в сутки 95% этил-EPA с некоторым количеством DHA или кукурузным маслом. Am. J. Clin. Nutr. 1997; 66:649-59 (1997). Полный профиль дополнительных жирных кислот и ингредиентов, представленных в композиции, неизвестен. После добавки в течение периода 7 недель у пациентов проявлялось только 297% повышение фосфолипид EPA в сыворотке по сравнению с повышением 417%, продемонстрированным выше для композиции по изобретению. Сопоставление других изменений жирных кислот в плазме/сыворотке приведено в таблице 4.
Процент изменения уровня жирных кислот по сравнению с начальным после введения дозы 4 г
Кроме того, в Japanese Eicosapentaenoic Acid (EPA) Lipid Intervention Study (JELIS), Yokoyama et al. Yokoyama et al. сообщили о том, что они наблюдали за более чем 18000 пациентов, рандомизированно получавших или 1800 мг композиции EPA (Epadel) со статином, или только статин с наблюдением в течение 5-лет. Lancet 2007; 369: 1090-98. После 5 лет лечения у пациентов проявлялось повышение EPA в плазме только на 70% (от начального 93 мг/л до 169 мг/л).
Фигуры 1 и 2 и демонстрируют сопоставление изменения уровней EPA в плазме/сыворотке, наблюдаемое для лечения AMR101 в данном исследовании, по сравнению с изменениями, наблюдаемыми для других композиций EPA в исследованиях JELIS и Grimsgaard. Как будет отмечено, при ~2 г в сутки для AMR101 достигалось гораздо более высокое повышение EPA в плазме по сравнению с начальным (~4-кратное) после только 6 недель, чем для исследования JELIS (< 2- кратного) после 5 лет лечения. Кроме того, при дозе 4 г в сутки при лечении AMR101 в течение 6 недель достигались намного более высокие (>250 мкг/г) уровни EPA в плазме, чем те, о которых сообщал Grimsgaard после 7 недель лечения (87,66 мкг/кг сыворотка). В общем, доза AMR101 4 г в сутки приводила к более чем 5-кратному повышению EPA в плазме по сравнению с начальным, тогда как доза 4 г в сутки композиции Grimsgaard приводила к менее чем 3-кратному повышению EPA в сыворотке. Эти результаты оказались неожиданными.
Пример 2
Проводили многоцентровое двойное слепое рандомизированное плацебо-контролируемое исследование в параллельных группах в Северной Америке с целью для определения того, улучшает ли 1 грамм EPA два раза в сутки в течение 6 месяцев двигательную активность у пациентов с заболеванием Хантингтона(HD). Выполняли анализ по полученным результатам для установления эффекта EPA на уровень триацилглицеринов после приема пищи.
В исследовании эффектов этил-EPA на развитие заболевания Хантингтона были задействованы участники исследования в 41 месте в Канаде и США. На основании результатов более раннего исследования были разработаны критерии включения в исследование для достижения задействования пациентов с заболеванием Хантингтона с CAG повтором менее чем 45, не требующих генетический анализ для выявления длины умножений для экспериментальных участников или исследователей. Для участия в исследовании пациенты должны были иметь клинические признаки HD и или подтверждающий семейный анамнез, или известное умножение CAG. Критерии отбора включали минимальный возраст 35, общую функциональную способность по меньшей мере 7, минимальную дистонию (не превышающую 2 в UHDRS или в стволе или в концевых частях), минимальную брадикинезию (не превышающую 2 в разделе UHDRS для брадикинезии), использование приемлемого предупреждения беременности, способность принимать оральные лекарственные средства и готовность и способность выполнять требования исследования. Пациенты считались непригодными для участия, если в течение 60 дней до начального визита они использовали добавки омега-3 жирных кислот, тетрабеназин или резерпин, высокие или переменные дозы оральных антипсихотических лекарственных средств (например, галоперидол), стероиды, отличные от средств местного применения, высокие дозы добавок селена, лития, высокие дозы бензодиазепинов, антикоагуляционные лекарственные средства (например, кумадин), высокие дозы (больше чем 325 мг в сутки) ампирина, неустойчивые дозы антагонистов NMDA-рецептора (например, мемантин), неустойчивые дозы противоэпилептических лекарственных средств или если они принимали участие в других экспериментальных исследованиях лекарственных средств. Дополнительными критериями невключения являлось применение базовых нейролептиков в течение 6 месяцев до начального визита, анамнез поздней дискинезии, неустойчивое медицинское или психиатрическое расстройство, глубокая депрессия (определенная оценкой больше 20 в шкале оценки депрессий Бека II), суицидальное мышление, клинически значимая химическая зависимость в течение 12 месяцев до начального визита, беременность или лактация для женщин, известная аллергия к этил-EPA или плацебо или предшествующее участие в экспериментальном исследовании EPA.
Исследование EPA (1 грамм два раза в сутки) являлось двойным слепым рандомизированным плацебо-контролируемым в параллельных группах. Экспертный совет организации в каждом участвующем месте одобрил план исследований и разрешительные документы. Пригодные к исследованию участники предоставили письменное согласие. Во время начального визита участники были рандомизированы в соответствии со сбалансированным по блокам генерируемым компьютером планом рандомизации, который был разделен по местам и генерирован Центре статистики университета Рочестера. Пациенты были рандомизированы в соотношении 1:1, чтобы получать или активное лекарственное средство (n=158) в форме двух капсул по 500 мг AMR101 орально или плацебо (n=154) в форме двух капсул по 500 мг, содержащих легкое парафиновое масло и 0,2% dl-альфа-токоферол два раза в сутки орально в течение 6 месяцев. После 6 месяцев, все участники TREND-HD подвергались лечению с AMR101 в течение 6 месяцев в форме открытого исследования. Данные только за первые 6 месяцев использовали для оценки эффектов AMR101 на липиды.
Критерием эффективности этого исследования было изменение уровней триацилглицерина (TG) после приема пищи для AMR101 по сравнению с плацебо.
Оценивали безопасность всех экспериментальных визитов, включая определение и оценку нежелательных явлений и серьезных нежелательных явлений и обзор клинических лабораторных тестов (полный анализ крови, биохимический анализ сыворотки и тест мочи на беременность). За безопасностью участников наблюдали анонимно посредством медицинских наблюдателей от спонсора и из группы исследования заболевания Хантингтона. Кроме того, независимый Комитет по мониторингу данных о безопасности, который имел доступ к распределению в группы лечения, проверял данные по безопасности на протяжении всего исследования для определения того, нужно ли было вносить какие-либо изменения в проведение испытаний.
Сравнивали изменения уровней липидов с использованием ковариационного анализа (ANCOVA) с группой лечения в качестве целевого фактора, места в качестве фактора разделения и начального значения в качестве ковариаты. Все пациенты, которые получали исследуемое лекарственное средство, были включены в анализ безопасности. Для каждого типа побочных явлений, группы лечения сравнивали в соответствии с встречаемостью по меньшей мере одного явления с использованием точного критерия Фишера. Непрерывные измерения безопасности, такие как результаты лабораторных тестов и показатели жизнедеятельности анализировали с использованием способов, сходных с описанными выше для величин первичных последствий (ANCOVA). Не было внесено никаких изменений для множественных сопоставлений при оценке показателей безопасности.
У сто сорока пяти пациентов, принимавших AMR101 (92% включенных в состав), и 141 пациента, принимавшего плацебо (92% включенных в состав), определяли содержание EPA в красных кровяных клетках в начале и через 6 месяцев. Начальное содержание 20:5n3 (EPA) в красных кровяных клетках значительно повышалось после 6 месяцев применения AMR101 (от среднего 0,52% до 3,07%), но снижалось для пациентов, принимавших плацебо (от среднего 0,61% до 0,55%); p<0,0001). После 6 месяцев, у пациентов, принимавших AMR101, имело место снижение уровней TG на 26 мг/дл от начального 171, сравниваемое со снижением на 11 мг/дл от начального 187 мг/дл для пациентов, принимавших плацебо; p=0,007. Общий холестерин был значительно больше снижен у пациентов, принимавших AMR101, (на 9,5 мг/дл) от начального 204 мг/дл, чем у пациентов, принимавших плацебо (на 2,5 мг/дл) от начального 208 мг/дл; p=0,009. Липидные показатели и показатели двигательной активности приведены в таблицах 5 и 6, соответственно.
Результаты оценки двигательной активности
n=316
n=221
EPA
величина
EPA
величина
[средние (SD)]
(8,1)
(8,3)
(7,7)
Результаты измерения липидных параметров.
По сравнению с этими данными для AMR101 Grimsgaard продемонстрировал снижение (от начального) уровня триглицеридов в сыворотке только на 12% в группе EPA после 7 недель лечения. Кроме того, добавление композиции Epadel EPA к действующей статиновой терапии в исследовании JELIS привело только к 9% снижению уровня триглицеридов после 5 лет лечения.
Пример 3
Выполняли исследование для оценки и сравнения содержимого капсул Epadel и капсул AMR101. Для анализа отбирали шесть капсул каждой композиции посредством газовой хромотографии. Средние величины для шести капсул для каждой из двух композиций приведены в таблице 7. Таблица 7. Измеренные и идентифицированные компоненты AMR101 и Epadel.
Пример 4
В фазе I было проведено одноцентровое фармакокинетическое исследование множественных доз на здоровых добровольцах мужского пола. Двадцать четыре пациента были разделены на две группы лечения по 12 пациентов в каждой (группы A и B). Обе группы получали ежедневно одинаковое общее количество дозы AMR101, но режимы введения были различными. Все пациенты получали однократную оральную дозу 2 г AMR101 в день 1. Группа лечения A получала ежедневно 28 доз по 2 г AMR101. Группа лечения В получала 27 доз два раза в сутки 1 г AMR101 и однократную дозу 2 г AMR101 в день 30.
Определяли уровни EPA и других незаменимых жирных кислот в плазме и красных кровяных клетках. Образцы крови для фармакокинетического анализа отбирали в следующие моменты времени для групп лечения A и B:
Дни 1 и 30: до приема препарата, 1, 2, 3, 4, 5, 6, 8, 20, 12, 24, 36 и 48 ч после приема препарата;
Дни 9, 16, 23: предутренняя доза;
Дни 37, 44, 58: после последнего приема препарата.
Первый промежуточный отчет представляет следующие фармакокинетические результаты для группы лечения B:
Плазма - День 1 (до приема препарата, 1, 2, 3, 4, 5, 6, 8, 20, 12, 24, 36 и 48 ч после приема препарата);
Красные кровяные клетки -День 1 (до приема препарата и 36 ч), день 30 (1 ч после приема препарата), день 37, день 44, день 58.
С использованием скорректированного значения, полученного вычитанием концентрации перед введением из концентраций при каждом отборе, однократная оральная доза 2 г AMR101 приводила к быстрому возрастанию в плазме EPA-липидов. Максимальные значения наблюдались через 5 часов после введения, при этом уровни EPA оставались выше начальных через 48 часов после введения. Полупериод выведения EPA из липидов плазмы составлял 87±65 ч (без вычета начального) и 42±31 ч (с вычетом начального). Суммарные фармакокинетические данные приведены в таблице 8.
Некомпартментный анализ - арифметическое среднее и SD.
В популяции по протоколу оральное введение AMR101 приводило к уровням EPA в RBC, возрастающим от среднего значения 190,4 мг/г перед введением доз в день 1 на 40,3 мг/г через один час после последней дозы в день 30.
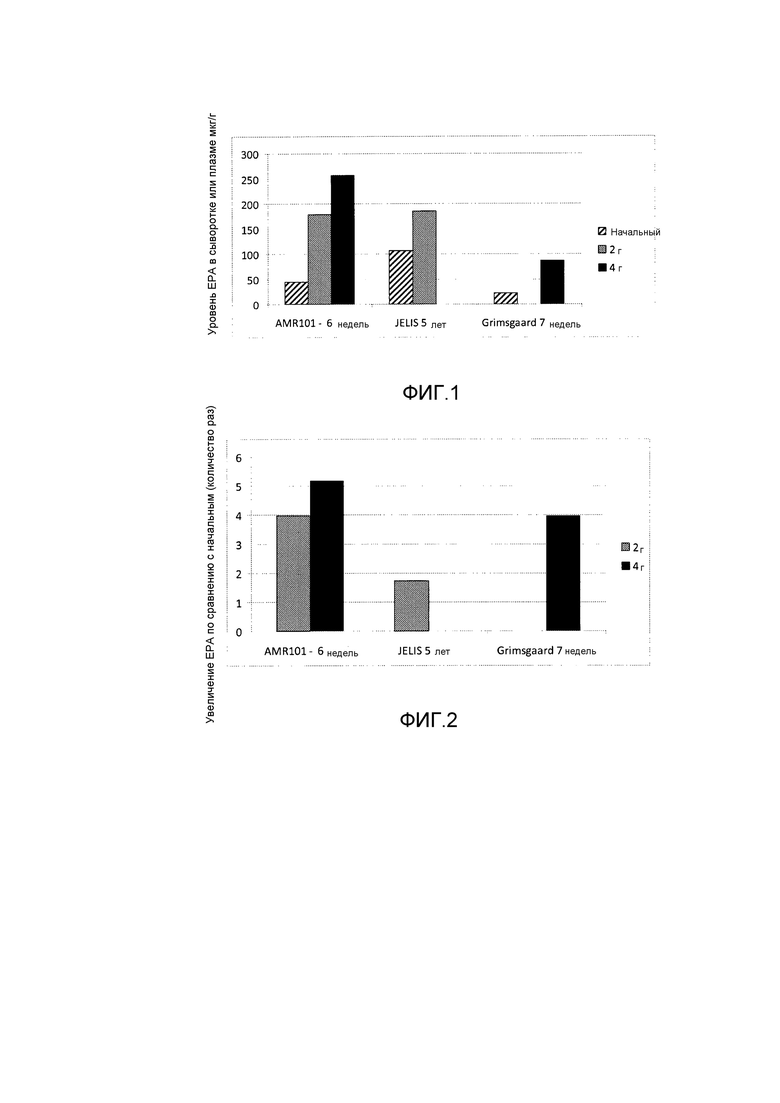
Группа изобретений относится к области медицины, а именно к способу повышения уровня этилэйкозапентаеновой кислоты (EPA)в плазме и/или сыворотке у пациента, нуждающегося в этом, включающему введение пациенту ежедневно фармацевтической композиции, включающей по меньшей мере 96% по массе этилэйкозапентаеноата, от 0,2% до 0,5% по массе этилоктадекатетраеноата, от 0,05% до 0,25% по массе этилнонаэкапентаеноата, от 0,2% до 0,45% по массе этиларахидоната, от 0,3% до 0,5% по массе этилэйкозатетраеноата, от 0,05% до 0,32% этилгенэйкозапентаеноата и не больше чем 0,05% этилдокозагексаеновой кислоты (этил-DHA), если она содержится, в количестве, достаточном для повышения уровней EPA у пациента в плазме и/или сыворотке на по меньшей мере 200% по сравнению с начальным, относится к способу повышения уровней EPA в плазме и/или сыворотке у пациента, нуждающегося в этом, включающему введение пациенту фармацевтической композиции, включающей по меньшей мере 96% по массе этилэйкозапентаеноата, от 0,2% до 0,5% по массе этилоктадекатетраеноата, от 0,05% до 0,25% по массе этилнонаэкапентаеноата, от 0,2% до 0,45% по массе этиларахидоната, от 0,3% до 0,5% по массе этилэйкозатетраеноата, от 0,05% до 0,32% этилгенэйкозапентаеноата и не больше чем 0,05% этил-DHA, где при ежедневном введении 2 г композиции пациенту в течение по меньшей мере 6 недель, у пациента проявляется по меньшей мере 200% повышение уровня EPA в плазме и/или сыворотке по сравнению с начальным, и относится к способу повышения уровня EPA в плазме или сыворотке пациента, нуждающегося в этом, включающему введение пациенту фармацевтической композиции, включающей по меньшей мере 96% по массе этилэйкозапентаеноата, от 0,2% до 0,5% по массе этилоктадекатетраеноата, от 0,05% до 0,25% по массе этилнонаэкапентаеноата, от 0,2% до 0,45% по массе этиларахидоната, от 0,3% до 0,5% по массе этилэйкозатетраеноата, от 0,05% до 0,32% этилгенэйкозапентаеноата и не больше чем 0,05% этил-DHA, где при ежедневном введении 4 г композиции пациенту в течение по меньшей мере приблизительно 6 недель, у пациента проявляется по меньшей мере приблизительно 300% повышение уровня EPA в плазме и/или сыворотке по сравнению с начальным. Группа изобретений обеспечивает улучшенные способы лечения заболеваний и нарушений связанных с сердечно-сосудистой системой. 3 н. и 16 з.п. ф-лы, 2 ил., 8 табл., 4 пр.
1. Способ повышения уровня этилэйкозапентаеновой кислоты (EPA)в плазме и/или сыворотке у пациента, нуждающегося в этом, включающий введение пациенту ежедневно фармацевтической композиции, включающей по меньшей мере 96% по массе этилэйкозапентаеноата, от 0,2% до 0,5% по массе этилоктадекатетраеноата, от 0,05% до 0,25% по массе этилнонаэкапентаеноата, от 0,2% до 0,45% по массе этиларахидоната, от 0,3% до 0,5% по массе этилэйкозатетраеноата, от 0,05% до 0,32% этилгенэйкозапентаеноата и не больше чем 0,05% этилдокозагексаеновой кислоты (этил-DHA), если она содержится, в количестве, достаточном для повышения уровней EPA у пациента в плазме и/или сыворотке на по меньшей мере 200% по сравнению с начальным.
2. Способ по п.1, где композиция вводится в количестве, достаточном для повышения уровней EPA у пациента в плазме и/или сыворотке на по меньшей мере 300% по сравнению с начальным.
3. Способ по п.1, где композиция вводится в количестве, достаточном для повышения уровней EPA у пациента в плазме и/или сыворотке на по меньшей мере 400% по сравнению с начальным.
4. Способ по п.3, где композиция вводится пациенту ежедневно в течение по меньшей мере 6 недель.
5. Способ по п.4, где начальный уровень EPA в плазме и/или сыворотке пациента составляет не больше чем 50 мкг/мл.
6. Способ по п.1, где при введении 2 г композиции пациенту ежедневно в течение по меньшей мере 6 недель у пациента проявляется по меньшей мере 200% повышение уровня EPA в плазме и/или сыворотке по сравнению с начальным.
7. Способ по п.1, где при введении 4 г композиции пациенту ежедневно в течение по меньшей мере 6 недель у пациента проявляется по меньшей мере 300% повышение уровня EPA в плазме и/или сыворотке по сравнению с начальным.
8. Способ по п.1, где при введении ежедневно 4 г композиции пациенту в течение по меньшей мере 6 недель у пациента проявляется по меньшей мере 400% повышение уровня EPA в плазме и/или сыворотке по сравнению с начальным.
9. Способ по п.1, где при ежедневном введении композиции пациенту у пациента проявляется повышение уровней докозапентаеновой кислоты (DPA) в сыворотке и/или плазме и снижение уровней арахидоновой кислоты (AA), докозагексаеновой кислоты (DHA) и/или DGLA в сыворотке и/или плазме.
10. Способ по п.1, где при ежедневном введении композиции пациенту в течение по меньшей мере 6 недель у пациента проявляется повышение уровней DPA в сыворотке и/или плазме и снижение уровней AA, DHA и/или DGLA в сыворотке и/или плазме по сравнению с начальными.
11. Способ по п.1, где при ежедневном введении композиции пациенту в течение по меньшей мере 6 недель у пациента проявляется снижение уровней DHA в плазме и/или сыворотке по меньшей мере на 16% по сравнению с начальным, снижение уровней DGLA в плазме и/или сыворотке по меньшей мере на 31 % по сравнению с начальным, снижение уровней AA в плазме и/или сыворотке по меньшей мере на 20% по сравнению с начальным и/или повышение уровней DPA в плазме и/или сыворотке на по меньшей мере на 130% по сравнению с начальными.
12. Способ по п.1, где композиция содержит по меньшей мере 96% по массе этилэйкозапентаеноата, от 0,22% до 0,4% по массе этилоктадекатетраеноата, от 0,075% до 0,20% по массе этилнонаэкапентаеноата, от 0,25% до 0,40% по массе этиларахидоната, от 0,3% до 0,4% по массе этилэйкозатетраеноата и от 0,075% до 0,25% этилгенэйкозапентаеноата.
13. Способ по п.1, где композиция содержит по меньшей мере 98% по массе этилэйкозапентаеноата, от 0,25% до 0,38% по массе этилоктадекатетраеноата, от 0,10% до 0,15% по массе этилнонаэкапентаеноата, от 0,25% до 0,35% по массе этиларахидоната, от 0,31% до 0,38% по массе этилэйкозатетраеноата и от 0,08% до 0,20% этилгенэйкозапентаеноата.
14. Способ по п.1, где композиция дополнительно содержит токоферол в количестве от 0,1% до 0,3% по массе.
15. Способ по п.1, где композиция представлена в капсульной оболочке.
16. Способ повышения уровней EPA в плазме и/или сыворотке у пациента, нуждающегося в этом, включающий введение пациенту фармацевтической композиции, включающей по меньшей мере 96% по массе этилэйкозапентаеноата, от 0,2% до 0,5% по массе этилоктадекатетраеноата, от 0,05% до 0,25% по массе этилнонаэкапентаеноата, от 0,2% до 0,45% по массе этиларахидоната, от 0,3% до 0,5% по массе этилэйкозатетраеноата, от 0,05% до 0,32% этилгенэйкозапентаеноата и не больше чем 0,05% этил-DHA, где при ежедневном введении 2 г композиции пациенту в течение по меньшей мере 6 недель у пациента проявляется по меньшей мере 200% повышение уровня EPA в плазме и/или сыворотке по сравнению с начальным.
17. Способ по п.16, где начальный уровень EPA в плазме или сыворотке пациента составляет не больше чем приблизительно 50 мкг/мл.
18. Способ повышения уровня EPA в плазме или сыворотке пациента, нуждающегося в этом, включающий введение пациенту фармацевтической композиции, включающей по меньшей мере 96% по массе этилэйкозапентаеноата, от 0,2% до 0,5% по массе этилоктадекатетраеноата, от 0,05% до 0,25% по массе этилнонаэкапентаеноата, от 0,2% до 0,45% по массе этиларахидоната, от 0,3% до 0,5% по массе этилэйкозатетраеноата, от 0,05% до 0,32% этилгенэйкозапентаеноата и не больше чем 0,05% этил-DHA, где при ежедневном введении 4 г композиции пациенту в течение по меньшей мере приблизительно 6 недель у пациента проявляется по меньшей мере приблизительно 300% повышение уровня EPA в плазме и/или сыворотке по сравнению с начальным.
19. Способ по п.18, где начальный уровень EPA в плазме или сыворотке пациента составляет не больше чем 50 мкг/мл.
| ГРОМОВА О.А | |||
| и др | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Трудный пациент | |||
| Колосоуборка | 1923 |
|
SU2009A1 |
| Установка для осушки сжатого воздуха | 1982 |
|
SU1047501A1 |
| Приспособление для записи звуковых явлений на светочувствительной поверхности | 1919 |
|
SU101A1 |

