Область техники
Настоящее изобретение относится к области органической и медицинской химии, а также молекулярной биологии и касается новых соединений биологически активных веществ с улучшенными значениями цитотоксичности, которые являются ингибиторами белок-белкового взаимодействия MDM2/p53 и способа их получения, которые могут быть использованы в качестве активных компонентов для фармацевтических композиций и лекарственных средств для лечения онкологических заболеваний.
Уровень техники
Последние несколько лет наблюдается повышенный интерес к поиску непептидных низкомолекулярных соединений способных препятствовать взаимодействию клеточных белков p53-MDM2. Белок р53 является опухолевым супрессором, играющим важную роль в жизненном цикле клетки и апоптозе, a MDM2 - его эндогенный ингибитор [Wu X., Bayle J.Н., Olson D., Levine A.J., Genes DeV., 1993, 7, 1126; Vogelstein В., Lane D., Levine A.J., Nature, 2000, 408, 307]. Если блокировать взаимодействие этих белков, то высвобождающийся р53 активирует процесс разрушения опухоли, что является новым направлением в терапии раковых заболеваний. Как известно, нарушение взаимодействия p53-MDM2 сопутствует большинству онкологических заболеваний [Hock, А. & Vousden, K.Н. Regulation of the р53 pathway by ubiquitin and related proteins. Int. J. Biochem. Cell Biol. 42, 1618-1621 (2010)], а повышенная концентрация белка MDM2 присутствует в 80% раковых клеток [Vogelstein, В., Lane, D. & Levine, A.J. Surfing the p53 network. Nature 408, 307-310(2000)].
Соединения на основе тиазолидинов и роданинов являются перспективными мишенями для исследования в органической и медицинской химии в связи с их широким спектром биологической активности. Также, известно некоторое количество медицинских препаратов на основе роданиновых гетероциклических производных, обладающих противодиабетической [Momosw Y, Maekawa Т, Yamano Т, Kawada М, Odaka Н, Ikeda Н, Sohda Т (2002) Novel5-substituted-2,4-thiazolidinedione and oxazolidinedione derivatives as insulin sensitizers with antidiabetic activity. J. Med. Chem. 45:1518-1534], противовоспалительной [Unangst PC, Connor DT, Cetenko WA, Sorenson RJ, Sircar JC, Wright CD, Schrier DJ, Dyer RD (1993) Oxazole, thiazole, and imidazole derivatives of 2,6-di-tert-butylphenol as dual 5-lipoxygenase and cyclooxygenase inhibitors. Bioorg. Med. Chem. Lett. 3:1729-1734], противовирусной [Grant EB, Guiadeen D, Baum EZ, Foleno BD, Jin H, Montenegro DA, Nelson EA, Bush K, Hlasta DJ (2000) The synthesis and SAR of rhodanines as novel class β-lactamase inhibitors. Bioorg. Med. Chem. Lett., 10:2179-2182], противогрибковой [Jiang C, Ting AT, Seed В (1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature, 391:82-86] и противоопухолевой активностью [Kubota T, Koshizuka K, Williamson EA, Asou H, Said JW, Holden S, Miyoshi I, Koeffler HP (1998) Ligand for peroxisome proliferator-activated receptor γ (troglitazone) has potent antitumor effects against human prostate cancer both in vitro and in vivo. Cancer Res., 58:3344-3352]. Сочетание данных двух подходов по созданию молекулы, содержащей спироиндолиноновый фрагмента, для ингибирования p53-MDM2 белок-белкового взаимодействия и 3-арилзамещенного роданинового гетероцикла, обладающим противоопухолевой актиностью, позволило создать серию диспироиндолинонов на основе 3-арилроданинов в качетвте ингибиторов p53-MDM2 белок-белкового взаимодействия.
Алкалоиды спиро-оксиндольного типа были впервые выделены из растений семейств Apocynaceae и Rubiacae [Galliford, С.V.; Scheidt, K.A. Angew. Chem. Int. Ed. 2007, 46, 8748]. Ключевой особенностью этих соединений является наличие индолинонового спиросодержащего фрагмента с различными заместителями в фрагментах пирролидина и индолинона.
Так, известны соединения и способы их получения такие как, A1 (Sanofi) [Allen, J.G. et al. Discovery and optimization of chromenotriazolopyrimidines as potent inhibitors of the mouse double minute 2-tumor protein 53 protein-protein interaction. J. Med. Chem. 52, 7044-7053 (2009)], RD-37 (University of California) [Wassman, C.D. et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nature Commun. 4, 1407 (2013)] со значением IC50=124 нМ на клетках LNCaP, А2 (University of Debrecen) [Noguchi, T. et al. Affinity-based screening of MDM2/ MDMX-p53 interaction inhibitors by chemical array: identification of novel peptidic inhibitors. Bioorg. Med. Chem. Lett. 23, 3802-3805 (2013)] со значением IC50<1 мМ на клетках Hep G2, A3 (Johnson & Johnson) [Vu, B. et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 4, 466-469 (2013)]. Получение данных производных, на примере соединения A3, возможно из ацилированных 1-дезокси-β-D-глюкопиразонил цианидов путем первоначального радикального бромирования с получением соответствующих бром-производных, в последующим их восстановлением до амидов с использованием TiCl4 в системе вода-уксусная кислота. После хроматографической очистки продукт вводили в реакцию с незамещенным тиогидантоином в присутствии 4-х эквивалентов цианата серебра или калия, в нитрометане при температуре 80°С. Побочно наблюдали образование гидроксиамида в соотношении с целевым продуктом 1:2.
Основными недостатками способа получения данных соединений является трудоемкий способ синтеза конечных соединений, использование больших количеств высокотоксичных реагентов и элюэнтов при синтезе и очистке, инертных атмосфер, в некоторых случаях при синтезе оптически-активных соединений необходимо использование дорогостоящих неорганических катализаторов, также являющихся токсичными. На некоторых из стадий авторам удавалось выделить целевые продукты с выходами 4-20%. Также нежелательным является и использование в синтезе радикальных реакций, поскольку в этом случае в смеси наблюдаются не только продукты монобромирования, но и полибромирования, что значительно понижает общий выход и эффективность реакции.
Из уровня техники известен способ получения ряда спиро-индолинонов с перспективной противоопухолевой активностью (например, B1-В6). Эти соединения были представлены как высокоактивные ингибиторы белок-белкового взаимодействия (PPI) MDM2/p53 [Liu, Jin-Jun; et al World Intellectual Property Organization, WO 2008055812 A1 2008-05-15]. Получение данных производных, на примере соединения В1, возможно из соответствующего рацемического (1R,2S)-6'-хлор-2-(3-хлорфенил)спиро[5-циклогексен-1,3'-[3Н]индол]-2',4(1'Н)-диона, CuCl, Ph3P и фенилмагний бромида в атмосфере аргона в абсолютном ТГФ при 0°С. После экстракции и отделения органической фазы из смеси были выделены два изомера, которые далее были разделены на хиральной колонке. Основными недостатками получаемых производных являются сложный путь синтеза исходного рацемического (1R,2S)-6'-хлор-2-(3-хлорфенил)спиро[5-циклогексен-1,3'-[3Н]индол]-2',4(1'Н)-диона, использование дорогостоящих хиральных колонок и токсичных органических растворителей при очистке.
Из уровня техники известен способ получения соединений, содержащих в своей структуре один спиро фрагмент, например, серия MI-43 или ISA-27, которые в настоящее время проходят доклинические испытания и, заключающийся в первоначальной конденсации ароматического альдегида с бензилцианидами, дальнейшей циклизацией совместно с третбутиловым эфиром енамина в присутствии фторида серебра и триметиламина, гидролизом сложного эфира в трифторуксусной кислоте, дальнейшим разделением энантиомеров на хиральной колонке и конденсацией с 2-метокси-4-карбоксианилином совместно с дифенилфосфинил хлоридом. В ходе дальнейших реакций оптимизации стереохимии, замещения бензольных колец на алкильные цепочки, введения амидных связей с подбором алициклического фрагмента была получена целевая структура соединения MI-43 или ISA-27 [Gomez-Monterrey, I.; Bertamino, A.; Porta, A.; et al. J. Med. Chem. 2010, 53, 8319]. MI-43 в настоящее время находится в Фазе I клинических испытаний против рака [http://clinicaltrials.gov/show/NCT01636479]. Основными недостатками способа получения данных соединений является длительный и трудоемкий способ синтеза конечных соединений, включающий более 15 стадий, а также необходимость разделения хиральных препаратов на дорогостоящих оптически-активных препаративных колонках и использование при этом больших объемов токсичных элюэнтов и растворителей.
Также, Daiichi Sankyo сообщает о высоко активных ингибиторах белок-белкового взаимодействия MDM2/p53 со значениями IC50 в диапазоне от 24.1 нМ до 181 мкМ [Zhao, Y.; Liu, L.; Sun, W.; et al. J. Am. Chem. Soc. 2013, 135, 7223]. Диспиро аналоги среди данных соединений описаны более слабо и включают арил-замещенный (3''R)-4,4-диметилдиспиро[циклогексан-1,2'-пирролидин-3',3''-индол]-2''(1''H)-он В6 [Sugimoto, Y.U.S. 20130165424 А9, 2012]. Эти агенты ингибируют белок-белковое взаимодействие MDM2/p53 со значениями IC50 в диапазоне 0.001-0.05 мкМ и являются ингибиторами р53-MDM2 взаимодействия. На первом этапе синтеза целевого соединения была проведена конденсация 5-замещенного изатина с этиловым эфиром цистеина при помощи микроволнового излучения, на следующем этапе с использованием тиофосгена и триэтиламина и последующим введением производного анилина. Далее, полученное диспиропроизводное ацилировали по атому азота изатина и выделяли нужный изомер на препаративной хиральной колонке. Основными недостатками метода получения данных соединений являются сложный и длительный путь синтеза конечных препаратов, необходимость дополнительных очисток и перекристаллизаций с использованием токсичных растворителей, необходимость использования дорогостоящей препаративной колонки, а также ряд побочных эффектов самих соединений.
Наиболее близкими к заявляемому являются диспиропроизводные соединений на основе 2-тиогидантоинов, являющиеся ингибиторами p53-MDM2 взаимодействия и способ их получения, заключающийся в трехкомпонентной реакции 1,3-диполярного циклоприсоединения с изатином, саркозином и 2-тиогидантоином [(1)Yan A. Ivanenkov, et al. Bioorganic & Medicinal Chemistry Letters, 25, 2, 2015, 404; (2) Белоглазкина E.К., Белоглазкина A.A. Кукушкин M.E., Мажуга А.Г, Иваненков Я.А. Номер заявки ФИПС 2015113026/04(020403) от 09.04.2015]. Согласно данному способу в реакции 1,3-диполярного циклоприсоединения все компоненты загружают в реактор и кипятят с обратным холодильником в течение 6-10 часов, в качестве растворителя используют токсичный метанол. Таким образом известным способом получают соединения с низкими выходами (30-60%), низкой растворимостью (0.0015 г / в 100 г воды), низкой цитотоксичностью (около 20 мкМ), высокой общей токсичностью и селективностью по отношению к клеткам рака предстательной железы, для подобных соединений отсутствует селективность по отношению к клеткам рака почек.
По сравнению с близкими прототипами, получение диспироиндолинонов на основе 3-замещенных роданинов возможно путем кипячения в толуоле в течение 4-8 часов. Полученные диспиропроизводные обладают более высокими выходами (более 62%), лучшей оастворимостью (0.01 г / в 100 г воды), высокой цитотоксичностью (до 10 мкМ), низкой общей токсичностью и выоской селективностью по отношению к опухолевым клеточным линиям почек SN12C.
Раскрытие изобретения
Технической проблемой, на решение которой направлено настоящее изобретение является разработка синтез новых низкомолекулярных ингибиторов белок-белкового взаимодействия MDM2/p53.
Техническим результатом заявляемого изобретения является способ получения диспиропроизводных на основе 2-сульфанилиден-1,3-тиазолидин-4-онов, который позволяет получить соединения с более выходами не менее 62%.
Также полученные диспиропроизводные на основе 2-сульфанилиден-1,3-тиазолидин-4-онов, являющихся ингибиторами белок-белкового взаимодействия MDM2/p53 с высокой активностью и селективностью действия в отношении клеток экспрессирующих белок р53 (А549 и MCF7), так и нераковых клеточных линий (HEK293T и VA13). Данное свойство позволяет использовать активный компонент для лечения заболеваний, связанных с нарушением активности белка р53, с меньшей дозировкой при избирательном действии на раковые клетки, не затрагивая при этом здоровые клетки. Полученные соединения обладают низкой общей токсичностью, о чем свидетельствует отсутствие активности на клеточной линии здоровых клеток фибробластов легких человека VA13.
Техническая проблема решается новыми соединениями на основе диспироиндолинонов общей формулы 1, или их фармацевтически приемлемыми солями, или оптическими изомерами.
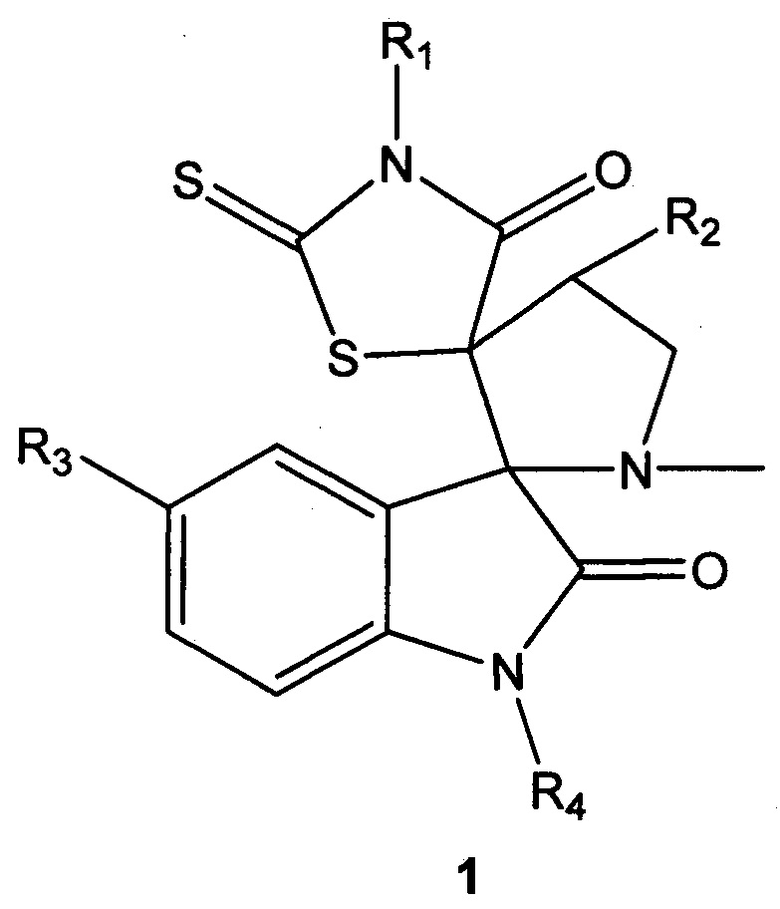
где R1 выбран из группы, включающей фенил, возможно замещенный одним заместителем, выбранными из атома галогена (хлор или бром), низшей алкокси-группы (метокси или этокси) или C1-С6 алкила (метил, этил, пропил, аллил). Более предпочтительным является соединение, отличающееся тем, что заместитель R1 выбран из группы, включающей аллил, фенил С6Н5-, 4-этоксифенил 4-С2Н5О-С6Н4-, 4-хлорфенил 4-Cl-С6Н4-, 4-бромфенил 4-Br-С6Н4-.
R2 выбран из группы, включающей фенил, замещенный 1-2 заместителями, выбранными из атома галогена (фтор, хлор, бром), низшей алкокси-группы (метокси, этокси), или С1-С2 алкила (метил, этил); Более предпочтительным является соединение, отличающееся тем, что заместитель R2 фенил С6Н5-, 4-этоксифенил 4-С2Н5О-С6Н4-, 2-хлорфенил 2-Cl-С6Н4-, 3-хлорфенил 3-Cl-С6Н4-, 4-хлорфенил 4-Cl-С6Н4-, 4-этилфенил 4-С2Н5-С6Н4-, 3,4-диметоксифенил 3,4-ОМе-С6Н3-, 4-метоксифенил 4-СН3О-С6Н4-, 4-фторфенил 4-F-C6H4-, 2-метилфенил 2-СН3-С6Н4-.
R3 выбран из группы, включающей атом галогена, водорода. Более предпочтительным является соединение, отличающееся тем, что заместитель R3 выбран из группы включающей атом хлора, брома или водорода.
R4 представляет собой атом водорода или алкил С1-С3 (метил, этил, пропил); R4 выбран из группы, включающей атом водорода или пропаргил НС≡С-СН2-.
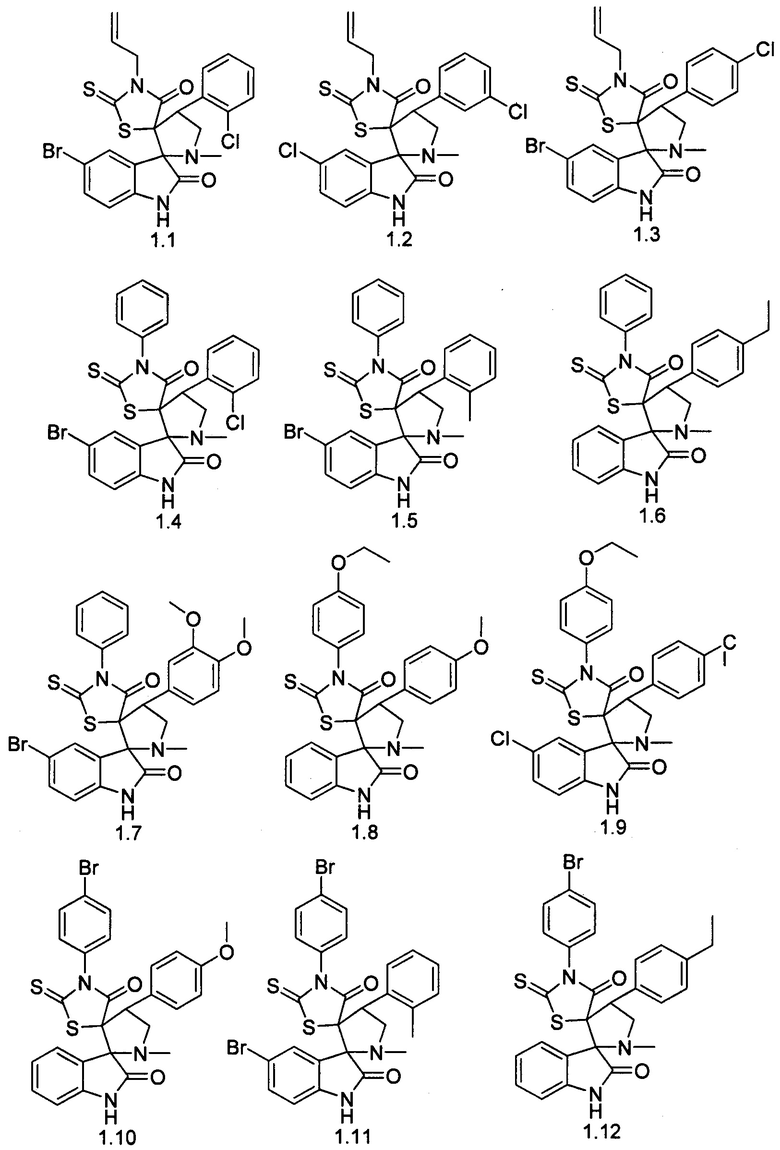
Более предпочтительным является соединение, выбранное из группы, включающей:
4'-(2-хлорфенил)-1'-метил-4''-(проп-2-ен-1-ил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.1);
5-бром-4'-(3-хлорфенил)-1'-метил-4''-(проп-2-ен-1-ил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.2);
5-хлор-4'-(4-хлорфенил)-1'-метил-4''-(проп-2-ен-1-ил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.3);
5-бром-4'-(2-хлорфенил)-1'-метил-4''-фенил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.4);
5-бром-1'-метил-4'-(2-метилфенил)-4''-фенил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.5);
4'-(4-этилфенил)-1'-метил-4''-фенил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.6);
5-бром-4'-(3,4-диметоксифенил)-1'-метил-4''-фенил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.7);
4''-(4-этоксифенил)-4'-(4-метоксифенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.8);
5-хлор-4'-(4-хлорфенил)-4''-(4-этоксифенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.9);
4''-(4-бромфенил)-4'-(4-метоксифенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.10);
5-бром-4''-(4-бромфенил)-1'-метил-4'-(2-метилфенил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.11);
4''-(4-бромфенил)-4'-(4-этилфенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.12);
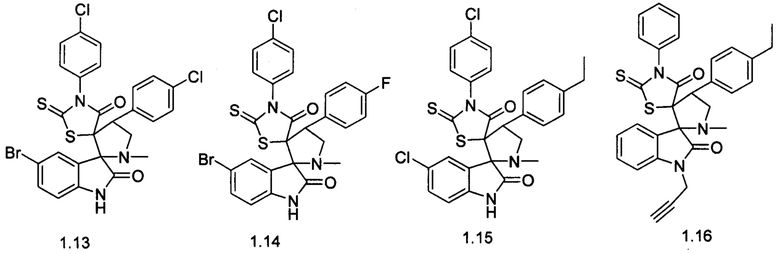
5-бром-4',4''-бис(4-хлорфенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.13);
5-бром-4''-(4-хлорфенил)-4'-(4-фторфенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.14);
5-хлор-4''-(4-хлорфенил)-4'-(4-этилфенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.15);
4'-(4-этилфенил)-1'-метил-4''-фенил-1-(проп-2-ин-1-ил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-дион (1.16).
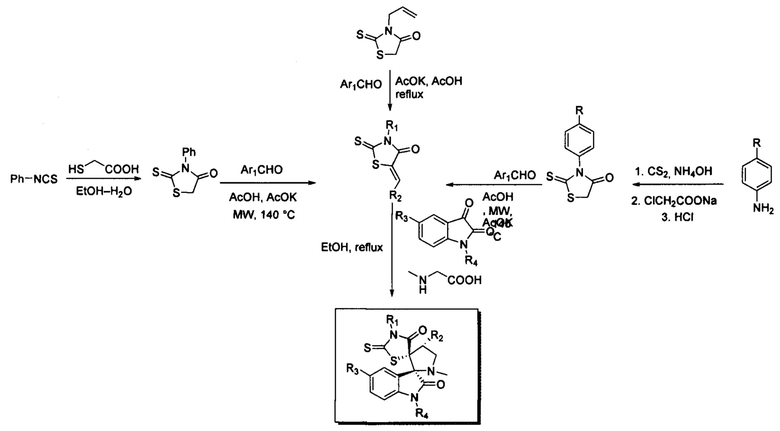
Также техническая проблема решается способом получения диспироиндолинонов на основе различных классов гетероциклов, заключающийся в трехкомпонентной реакции между соответствующим 1,3-тиазолидин-4-оном, замещенным по 3-му или 5-му положению или незамещенным или 5-галогензамещенным изатином и сакозином в одноатомном ароматическом спирте. При этом способ получения диспиро-индолинонов общей формулы 1, заключается в том, что 1±0.1 мольных экв. соответствующего имидазолидин-4-она растворяют в толуоле, взятого в количестве 10 мл на 0.5±0.1 ммоль и кипятят при температуре 100±10°С в течение 10±2 минут после закипания образовавшегося раствора, затем в кипящую смесь добавляют саркозин, взятый из расчета 2±0.2 экв. и соответствующего изатина, взятого в количестве 2±0.2 экв по отношению к соответствующему имидазолидин-4-ону, полученную смесь кипят до окончания реакции, затем смесь охлаждают до комнатной температуры, выпавший осадок отфильтровывают на вакууме со стеклянным пористым фильтром и колбой Бунзена.
Техническая проблема решается также активным компонентом, обладающим свойством ингибитора белок-белкового взаимодействия MDM2/p53, представляющим собой соединение общей формулы 1.
Также техническая проблема решается фармацевтической композицией для профилактики и лечения онкологического заболевания, связанного с нарушением белок-белкового взаимодействием MDM2/p53, включающей в качестве лекарственного начала соединения общей формулы 1, или их фармацевтически приемлемые соли и/или гидраты, в терапевтически эффективном количестве или содержащая в эффективном количестве активный компонент по настоящему изобретению. Более предпочтительной является фармацевтическая композиция, где онкологическое заболевание представляет собой рак легких или почек.
Также техническая проблема решается лекарственным средством для профилактики и лечения пролиферативных заболеваний, связанных с белок-белковым взаимодействием MDM2/p53, выполненным в виде таблеток, капсул или инъекций,помещенных в фармацевтически приемлемую упаковку, содержащее в эффективном количестве активный компонент, представляющий собой соединение общей формулы 1 или фармацевтическую композицию, включающую в качестве лекарственного начала соединение общей формулы 1.
Техническая проблема также решается способом ингибирования белок-белкового взаимодействия MDM2/p53, включающим стадию введения в клетку соединения общей формулы 1 по настоящему изобретению.
Осуществление изобретения
Ниже приведены определения терминов, которые используются в описании настоящего изобретения.
«IC50» (ПК50) - концентрация соединения, при которой наблюдается 50% ингибирование ферментативной активности
«Алифатический заместитель» (алкил, алкенил, алкинил) означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи, необязательно содержащий двойную или тройную связь. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» заместителей. Алкил, алкенил, алкинил могут иметь один или несколько одинаковых или различных заместителей («алкильных заместителей») включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбнил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонилгетероаралкилокси, аннелированный гетероарилциклоалкенил, аннелированный гетероарилциклоалкил, аннелированный гетероарилгетероцикленил, аннелированный гетероарилгетероциклил, аннелированный арилциклоалкенил, аннелированный арилциклоалкил, аннелированный арилгетероцикленил, аннелированный арилгетероциклил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или RkaRk+1aN-, RkaRk+1aNC(=O)-, RkaRk+1aNC(=S)-, RkaRk+1aNSO2-, где Rka и Rk+1a независимо друг от друга представляют собой «заместители амино группы», например, атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rka и Rk+1a вместе с атомом N, с которым они связаны, образуют через Rka и Rk+1a 4-7 членный гетероциклил или гетероцикленил. Предпочтительными алкильными группами являются метил, трифторметил, циклопропилметил, циклопентилметил, этил, н-пропил, изо-пропил, н-бутил, трет-бутил, н-пентил, 3-пентил, метоксиэтил, карбоксиметил, метоксикарбонилметил, этоксикарбонилметил, бензилоксикарбонилметил метоксикарбонилметил и пиридилметилоксикарбнилметил. Предпочтительными «алкильными заместителями» являются циклоалкил, арил, гетероарил, гетероциклил, гидрокси, алкокси, алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или RkaRk+1aN-, RkaRk+1aNC(=O)-, аннелированный арилгетероцикленил, аннелированный арилгетероциклил.
Предпочтительными алкильными группами в данном изобретении являются аллил, циклопропил, циклогексил и 4-этилморфолин.
«Арил» означает ароматическую моноциклическую или полициклическую систему, включающую от 6 до 14 атомов углерода, преимуществено от 6 до 10 атомов углерода. Арил может содержать один или более «заместителей циклической системы», которые могут быть одинаковыми или разными. Представителями арильных групп являются фенил или нафтил, замещенный фенил или замещенный нафтил. Арил может быть аннелирован с ароматической и неароматической циклической системой или гетероциклом, включая арил, ароил, аралкокси, арилгидрокси, арилокси, арилоксикарбонил, аралкилтио, арилсульфонил, арилгалоген, гетероарил гетероциклил, гетероаралкилокси, гетероарилтио, аннелированный гетероарилциклоалкенил, аннелированный гетероарилциклоалкил, аннелированный гетероарилгетероцикленил, аннелированный гетероарилгетероциклил, аннелированный арилциклоалкенил, аннелированный арилциклоалкил, аннелированный арилгетероцикленил, аннелированный арилгетероциклил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или RkaRk+1aN-, RkaRk+1aNC(=O)-, RkaRk+1aNC(=S)-, RkaRk+1aNSO2-, где Rka и Rk+1a независимо друг от друга представляют собой «заместители амино группы», значение которых определено в данном разделе.
Предпочтительными арильными группами в данном изобретении являются фенил С6Н5-, 3-хлор-4-фторфенил 3-Cl-4-F-С6Н3-, 4-этоксифенил 4-С2Н5О-С6Н4-, 3-хлор-4-этоксифенил 3-Cl-4- С2Н5О-С6Н3-, 2-хлорфенил 2-Cl-С6Н4-, 4-хлорфенил 4-Cl-С6Н4-, 4-этилфенил 4-С2Н5-С6Н4-, 3,4-диметоксифенил 3,4-ОМе-С6Н3-,4-метоксифенил 4-СН3О-С6Н4-, 4-фторфенил 4-F-C6H4-, 4-бромфенил 4-Br-С6Н4-, 4-пропилоксифенил 4-O(iC3H7)С6Н4, циклопентоксибензил.
«Белок-белковое взаимодействие» означает установление связей между белками р53 и MDM2 в результате их взаимодействий за счет электростатических, гидрофобных или гидрофильных контактов.
«Галоген» означает фтор, хлор, бром и йод. Предпочтительными являются фтор, хлор и бром.
«Гетероциклил» (гетероциклический заместитель) означает ароматическую или неароматическую насыщенную моноциклическую или полициклическую систему, включающую от 3 до 10 атомов углерода, преимущественно от 5 до 6 атомов углерода, в которой один или несколько атомов углерода заменены на гетероатом, такой как азот, кислород, сера. Приставка «аза», «окса» или «тиа» перед гетероциклилом означает наличие в циклической системе, атома азота, атома кислорода или атома серы, соответственно. Гетероциклил может иметь один или несколько «заместителей циклической системы», которые могут быть одинаковыми или разными. Атомы азота и серы, находящиеся в гетероциклиле могут быть окислены до N-оксида, S-оксида или S-диоксида. Представителями гетероциклилов являются пиперидин, пирролидин, пиперазин, морфолин, тиоморфолин, тиазолидин, 1,4-диоксан, тетрагидрофуран, тетрагидротиофен, гетероаралкил, гетероциклил или гетероарил, или Rka и Rk+1a вместе с атомом N, с которым они связаны, образуют через Rka и Rk+1a 4-7 членный гетероцикл.
Предпочтительной гетероциклической группой в данном изобретении является пиридин.
«Гидрат» означает сольват, в котором вода является молекулой или молекулами растворителя.
«Заместитель» означает химический радикал, который присоединяется к скэффолду (фрагменту), например, «заместитель алкильный», «заместитель амино группы», «заместитель карбамоильный», «заместитель циклической системы», значения которых определен в данном разделе.
«Лекарственное начало» (активный компонент, лекарственная субстанция лекарственное вещество, drag-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции), в виде таблеток капсул инъекций, мазей и др. готовых форм предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлимых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений, или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как, холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
«Циклоалкил» означает не ароматическую моно- или полициклическую систему, включающую от 3 до 10 атомов углерода. Циклоалкил может иметь один или несколько «заместителей циклической системы», которые могут быть одинаковыми или разными. Представителями циклоалкильных групп являются циклопропил, циклобутил, циклопентил, циклогексил, декалин, адамант-1-ил и т.п. Циклоалкил может быть аннелирован с ароматическим циклом или гетероциклом.
Ранее был проведен синтез разнообразных гетероциклических диспиро соединений [Yan A. Ivanenkov, et al. Bioorganic & Medicinal Chemistry Letters, 25, 2, 2015, 404.], а также биологические испытания этих соединений в экспериментах с клеточными линиями HepG2, Hek, MCF-7, SiHa, HCT p53(+,+) и HCT р53(-,-), РС3 и LNCap. Заявляемые соединения обладают выраженной активностью по отношению к клеточным линиям, экспрессирующим р53. Были найдены новые производные 2-сульфанилиден-1,3-тиазолидин-4-онов. Отсутствие в целевой молекуле кислого NH протона в роданиновом цикле по сравнению с известным тиогидантоиновым аналогом, делает целевую молекулу болеее устройчивой в щелочной и кислой среде. Кроме того, меньшая электроотрицательность атома серы по сравнению с азотом препятствует протеканию гидролиза тионного фрагмента до карбонильного в водных растворах в кислых и щелочных условиях, что осложняет использование близких соединений-прототипов в условиях, близких к физиологическим. Также была проведена оптимизация способа получения 2-сульфанилиден-1,3-тиазолидин-4-онов в виде оснований и фармакологически приемлемых солей и гидратов, обладающие свойствами низкомолекулярных ингибиторов белок-белкового взаимодействия MDM2/p53. Получение данных соединений является экономичным и включает в себя две-пять стадий в зависимости от заместителей, не требует применения больших объемов растворителей, в ходе реакции сразу образуется оптически-активная молекула, которая не требует применения дорогостоящих хиральных катализаторов и дальнейшего разделения на хиральных колонках. Эффективность по отношению к белку р53 подтверждается активностью как активностью на опухолевых клеточных линиях, экспрессирующих данный белок (MCF7, А549 и SN12C), так и полным отсутствием цитотоксического эффекта на нераковых клеточных линиях HEK293T и VA13. Дополнительно для соединений данного типа была проведена оценка цитотоксичности и селективности по отношению к 60 различным опухолевым клеточным линиям (National Cancer Institute, USA), которая продемонстировала селективность диспироиндолинонов по отноешнию к клеточным линиям рака почек SN12C.
Известно, что применяемые ингибиторы белок-белкового взаимодействия р53-MDM2 в экспериментах на аналогичных клеточных линиях показывают худшую активность [Zhao, Y.; Liu, L.; Sun, W.; Lu, J.; McEachern, D.; Li, X.; Yu, S.; Bernard, D.; Ochsenbein, P.; Ferey, V.; Carry, J.-C; Deschamps, J.R.; Sun, D.; Wang, S. Diastereomeric spirooxindoles as highly potent and efficacious MDM2 inhibitors. J. Am. Chem. Soc. 2013, 135, 7223- 7734], однако в настоящее время они проходят дальнейшие клинические испытания в терапии онкологических заболеваний, что свидетельствует о перспективности исследуемых в настоящей заявке ингибиторов. В настоящий момент, на основании предложенной ранее методики получения диспиропроизводных на основе 2-арил-5-арилметилидензамещенных тетрагидро-4Н-имидазол-4-онов [Yan A. Ivanenkov, et al. Bioorganic & Medicinal Chemistry Letters, 25, 2, 2015, 404] была произведена их оптимизация и получены новые ингибиторы p53-MDM2 взаимодействия на основе 2-сульфанилиден-1,3-тиазолидин-4-онов. В ходе оптимизации было подобрано соотношение реагентов на конечной стадии образования диспиропрозводного, заключающееся в двукратном избытке изатина и саркозина по отношению к исходному 2-сульфанилиден-1,3-тиазолидин-4-онов; был оптимизирован порядок добавления реагентов (первоначальное добавлениие саркозина в кипящий раствор с последующим добавлением изатина, а также улучшение биодоступности и цитотоксичности путем варьирования заместителей в молекуле. Полученные параметры позволили улучить выходы конечных ингибиторов в 1,5-3 раза, а также улучшить значения цитотоксичности на два порядка.
Для получения заявляемых соединений 1±0.1 экв. соответствующего тиазолидин-4-она растворяют в толуоле, взятом в количестве 10 мл на 0.5±0.1 ммоль и кипятят при температуре 100±10°С в течение 10±2 минут после закипания образовавшегося раствора, затем в кипящую смесь добавляют саркозин, взятый из расчета 2±0.2 экв. и соответствующего изатина, взятого в количестве 2±0.2 экв по отношению к соответствующему тиазолидин-4-ону. Полученную смесь кипят до окончания реакции, окончание реакции определяют по возникновению пятна нового продукта на ТСХ в системе петролейный эфир : этилацетат = 3:1. В среднем реакция протекает в течение 6±2 часов. После охлаждения смеси до комнатной температуры на воздухе наблюдают выпадение белого осадка. Затем выпавший осадок отфильтровывают на вакууме со стеклянным пористым фильтром и колбой Бунзена.
Общая схема синтеза получаемых соединений исходя из 3-замещенных тиазолидин-4-онов представлена на следующей синтетической схеме:
Таким образом, за счет использования двукратного избытка компонентов на конечной стадии, последовательности введения реагентов, экологичной температуры 100±10°С, были синтезированы диспиропроизводные на основе 2-сульфанилиден-1,3-тиазолидин-4-онов. Цитотоксичность получаемых соединений была оценена не только на клеточных линиях экспрессирующих белок р53, в частности линия раковых клеток молочной железы MCF-7, линию аденокарциномы легких человека А549 и линию клеток рака почек SN12C, но и на нераковых клеточных линиях почек VA13 и эмбриональных трансфицированных клеток почек НЕК293Т.
Для получения солей была использована методика, разработанная авторами ранее [Белоглазкина Е.К., Белоглазкина А.А. Кукушкин М.Е., Мажуга А.Г, Иваненков Я.А., заявка РФ №2015113026/04(020403) от 9.04.2015].
Фармацевтические композиции могут включать фармацевтически приемлемые эксципиенты. Под фармацевтически приемлемым эксципиентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители. Фармацевтическая композиция наряду с соединением общей формулы 1, или его фармацевтически приемлемой солью и/или гидратом по настоящему изобретению, может включать и другие активные субстанции, в том числе обладающие противоопухолевой активностью, при условии, что они не вызывают нежелательных эффектов.
При необходимости использования фармацевтической композиции по настоящему изобретению в клинической практике она может смешиваться с традиционными фармацевтическими носителями.
Носители, используемые в фармацевтических композиций по настоящему изобретению, представляют собой носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: в пероральных формах используются связующие вещества, смазывающие агенты, дезинтеграторы, растворители, разбавители, стабилизаторы, суспендирующие агенты, бесцветные агенты, корригенты вкуса; в формах для инъекций используются антисептические агенты, солюбилизаторы, стабилизаторы; в местных формах используются основы, разбавители, смазывающие агенты, антисептические агенты.
Лекарственное средство для профилактики и лечения пролиферативных заболеваний, связанных с белок-белковым взаимодействием MDM2/p53, а именно рак простаты и колоректальный рак, являющиеся наиболее распространенными заболеваниями данного типа, в виде таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, содержащее в эффективном количестве активный компонент или фармацевтическую композицию по настоящему изобретению.
В соответствии с данным изобретением способ профилактики или лечения пролиферативных заболеваний, связанных с белок-белковым взаимодействием MDM2/p53 у животных и людей, заключается во введении пациенту нового лекарственного средства или новой фармацевтической композиции.
Лекарственные средства могут вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно или местно). Клиническая дозировка средства, содержащего соединение общей формулы 1, у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента, при этом суточная доза у взрослых обычно составляет 10~500 мг, предпочтительно - 50~300 мг. Поэтому во время приготовления из фармацевтической композиции лекарственного средства по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку, при этом каждая единица дозировки препарата должна содержать 10~500 мг средства общей формулы 1, предпочтительно - 50~300 мг. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно - от одного до шести раз) до достижения терапевтического эффекта.
Представленные ниже примеры иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Получение 4'-(2-хлорфенил)-1'-метил-4''-(проп-2-ен-1-ил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.1).
Смесь 0.1 г (1.0 ммоль, 1±0.1 экв.) соответствующего (Z)-3-аллил-5-(2-хлорбензилиден)-2-тиоксотиазолидин-4-она растворяли в 10 мл толуола и кипятили в течение 10 минут при температуре 80°С, после чего добавляли 0.06 г (2.0 ммоль, 2±0.2 экв.) саркозина и 0.15 г (2.0 ммоль, 2±0.2 экв.) 5-бромизатина и кипятили в течение 7 часов (окончание реакции определяют по ТСХ). Затем выпавший осадок отфильтровывали и промывали при необходимости спиртом. Выход составил 59%. Тпл=256°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.05 (с, 1Н, NH-индолинон), 7.78 (д, J=7.8 Гц, 1Н, Ar), 7.42-7.53 (м, 3Н, Ar), 7.35 (т, J=7.8 Гц, 1Н, Ar), 7.19 (s, 1Н, Ar), 6.82 (д, J=8.2 Гц, 1Н, Ar), 5.45-5.57 (м, 1Н, allyl), 5.05 (д, J=10.4 Гц, 1Н, allyl), 4.66 (т, J=8.2 Гц, 1Н, пирролидин-Н3), 4.76 (д, J=17.4 Гц, 1H, allyl), 4.37 (д, J=4.3 Гц, 2Н, allyl), 4.08 (т, J=9.0 Гц, 1H, allyl, пирролидин-Н2), 3.56 (т, J=7.8 Гц, 1Н, СН, пирролидин-Н1), 2.14 (с, 3Н, NCH3). Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C23H19BrClN3O2S2, М+Н: 546.9831, масса найденная (М+Н): 546.9837.
Пример 2. Получение 5-бром-4'-(3-хлорфенил)-1'-метил-4''-(проп-2-ен-1-ил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.2).
В результате реакции 0.1 г (1 ммоль) соответствующего роданина, 0.12 г (2 ммоль) 5-бромизатина и 0.06 г (2 ммоль) саркозина получили 0.13 г (81%) соединения в виде белого порошка. Тпл=210°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.07 (с, 1H, NH-индолинон), 7.55 (с, 1Н, Ar), 7.36-7.42 (м, 4Н, Ar), 7.06 (д, J=2.2 Гц, 1Н, Ar), 6.88 (д, J=8.3 Гц, 1Н, Ar), 5.38-5.48 (м, 1Н, Allyl), 4.96 (д, J=10.4 Гц, 1H, Allyl), 4.58 (д, J=17.2 Гц, 1Н, Allyl), 4.45 (т, J=8.7 Гц, 1Н, пирролидин-Н3), 4.37 (м, 2Н, Allyl), 3.80 (т, J=9.6 Гц, 1Н, пирролидин-Н2), 3.58 (т, J=8.4 Гц, 1Н, пирролидин-Н1), 2.11 (с, 3Н, NCH3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C23H19Cl2N3O2S2, М+Н: 503.0347, масса найденная (М+Н): 503.0343.
Пример 3. Получение 5-хлор-4'-(4-хлорфенил)-1'-метил-4''-(проп-2-ен-1-ил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.3).
В результате реакции 0.1 г (1 ммоль) соответствующего роданина, 0.15 г (2 ммоль) 5-бромизатина и 0.06 г (2 ммоль) саркозина получили 0.13 г (78%) соединения в виде белого порошка. Тпл=245°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.08 (с, 1H, NH-индолинон), 7.49 (дд, J1=2.2 Гц, J2=8.3 Гц, 1H, Ar), 7.45 (м, 4Н, Ar), 7.19 (д, J=2.0 Гц, 1Н, Ar), 6.82 (д, J=8.3 Гц, 1H, Ar), 5.40-5.50 (м, 1Н, Allyl), 4.98 (д, J=10.4 Гц, 1Н, Allyl), 4.58 (д, J=17.2 Гц, 1Н, Allyl), 4.45 (т, J=8.9 Гц, 1Н, пирролидин-Н3), 4.37 (д, J=5.1 Гц, 2Н, Allyl), 3.82 (т, J=9.6 Гц, 1Н, пирролидин-Н2), 3.56 (т, J=8.5 Гц, 1Н, пирролидин-Н1), 2.11 (с, 3Н, NCH3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C23H19BrClN3O2S2, М+Н: 546.9831, масса найденная (М+Н): 546.9831.
Пример 4. Получение 5-бром-4'-(2-хлорфенил)-1'-метил-4''-фенил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.4).
Смесь 0.06 г (0.5 ммоль, 1±0.1 экв.) соответствующего (Z)-3-аллил-5-(2-хлорбензилиден)-2-тиоксотиазолидин-4-она растворяли в 10 мл толуола и кипятили в течение 10 минут при температуре 80°С, после чего добавляли 0.03 г (1.0 ммоль, 2±0.2 экв.) саркозина и 0.08 г (1.0 ммоль, 2±0.2 экв.) 5-бромизатина и кипятили в течение 7 часов (окончание реакции определяют по ТСХ). Затем выпавший осадок отфильтровывали и промывали при необходимости спиртом. Выход составил 84%. Тпл=258°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.12 (с, 1Н, NH-индолинон), 7.82 (д, J=7.8 Гц, 1Н, Ar), 7.62 (дд, J1=1.8 Гц, J2=8.3 Гц, 1Н, Ar), 7.56-7.53 (м, 5Н, Ar), 7.48 (т, J=7.7 Гц, 1Н, Ar), 7.39 (т, J=7.7 Гц, 1H, Ar), 7.12 (д, J=1.7 Гц, 1Н, Ar), 6.91 (д, J=8.3 Гц, 2Н, Ar), 4.88 (т, J=8.5 Гц, 1H, пирролидин-Н3), 4.14 (т, J=8.7 Гц, 1Н, пирролидин-Н2), 3.58 (т, J=8.7 Гц, 1Н, пирролидин-Н1), 2.17 (с, 3Н, NCH3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C26H19BrClN3O2S2, М+Н: 582.9783, масса найденная (М+Н): 582.9785.
Пример 5. Получение 5-бром-1'-метил-4'-(2-метилфенил)-4''-фенил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.5).
В результате реакции 0.1 г (0.7 ммоль) соответствующего роданина, 0.145 г (1.4 ммоль) 5-бромизатина и 0.057 г (1.4 ммоль) саркозина получили 0.18 г (65%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе ПЭ:ЭА=20:1-5:1.Тпл=271°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.12 (с, 1H, NH-индолинон), 7.74 (д, J=8.0 Гц, 1Н, Ar), 7.62 (д, J=8.0 Гц, 1H, Ar), 7.50 (с, 3Н, Ar), 7.29-7.32 (м, 1H, Ar), 7.21-7.23 (м, 4Н, Ar), 7.08 (с, 1Н, Ar), 6,91 (д, J=8.1 Гц, 1Н, Ar), 4.82 (т, J=8.2 Гц, 1H, пирролидин-Н3), 4.00 (т, J=8.1 Гц, 1Н, пирролидин-Н2), 3.52 (т, J=8.5 Гц, 2Н, пирролидин-Н1), 2.23 (с, 3Н, CH3),2.15 (c, 3H, NCH3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C27H22BrN3O2S2, М+Н: 563.0325, масса найденная (М+Н): 563.0327.
Пример 6. Получение 4'-(4-этилфенил)-1'-метил-4''-фенил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.6).
В результате реакции 0.1 г (0.7 ммоль) соответствующего роданина, 0.145 г (1.4 ммоль) 5-бромизатина и 0.057 г (1.4 ммоль) саркозина получили 0.11 г (64%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:ЭА=40:1. Тпл=246°С.
Спектр 1H ЯМР (DMSO-d6,400 МГц, δ, м.д): 11.13 (с, 1Н, NH-индолинон), 7.60 (дд, J1=8.2 Гц, J2=1.9 Гц, 1Н, Ar), 7.42-7.48 (м, 7Н, Ar), 7.24 (д, J=7.9 Гц, 2Н, Ar), 7.06 (д, J=1.9 Гц, 1Н, Ar), 6,91 (д, J=8.3 Гц, 1Н, Ar), 4.51 (т, J=8.8 Гц, 1H, пирролидин-Н3), 3.87 (т, J=9.4 Гц, 1Н, пирролидин-Н2), 3.54 (т, J=8.4 Гц, 1Н, пирролидин-Н1), 2.61 (кв, J=7.5 Гц, 2Н, СН2), 2.12 (с, 3Н, NCH3), 1.18 (т, J=7.6 Гц, 3Н, СН3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C28H24BrN3O2S2, М+Н: 577.0549, масса найденная (М+Н): 577.0545.
Пример 7. Получение 5-бром-4'-(3,4-диметоксифенил)-1'-метил-4''-фенил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.7).
В результате реакции 0.19 г (1.1 ммоль) соответствующего роданина, 0.37 г (2.2 ммоль) 5-бромизатина и 0.146 г (2.2 ммоль) саркозина получили 0.18 г (54%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:МеОН=40:1. Тпл=229°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.15 (с, 1H, NH-индолинон), 7.60 (д, J=8.3 Гц, 1Н, Ar), 7.48 (с, 5Н, Ar), 7.25 (с, 1Н, Ar), 7.06 (с, 1Н, Ar), 7.03 (д, J=8.3 Гц, 1Н, Ar), 6.90-6.96 (м, 2Н, Ar), 4.47 (т, J=8.7 Гц, 1H, пирролидин-Н3), 3.85 (т, J=9.4 Гц, 1Н, пирролидин-Н2), 3.78 (с, 3Н, ОСН3), 3.75 (с, 3Н, ОСН3), 3.56 (т, J=8.4 Гц, 1Н, пирролидин-Н1), 2.17 (с, 3H, NCH3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C28H24BrN3O4S2, М+Н: 609.0427, масса найденная (М+Н): 609.0429.
Пример 8. Получение 4''-(4-этоксифенил)-4'-(4-метоксифенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.8).
В результате реакции 0.2 г (1 ммоль) соответствующего роданина, 0.18 г (1 ммоль) 5-бромизатина и 0.09 г (1 ммоль) саркозина получили 0.22 г (74%) соединения в виде белого порошка. Тпл=267°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.14 (ус, 1Н, NH-индолинон), 7.58 (д, J=8.2 Гц, 4Н, Ar), 7.43-7.51 (м, 3Н, Ar), 6.89-7.01 (м, 4Н, Ar), 4.56 (т, J=8.4 Гц, 1H, пирролидин-Н3), 4.37 (т, J=8.4 Гц, 1Н, пирролидин-Н2), 4.05 (кв, J=6.9 Гц, 2Н, ОСН2), 3.58 (т, J=9.4 Гц, 1Н, пирролидин-Н1), 2.13 (с, 3Н, NCH3), 1.33 (т, J=7.0 Гц, 3Н, СН3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C28H23Cl2N3O3S2, М+Н: 583.0631, масса найденная (М+Н): 583.0633.
Пример 9. Получение 5-хлор-4'-(4-хлорфенил)-4''-(4-этоксифенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.9).
В результате реакции 0.2 г (1 ммоль) роданина, 0.27 г (1 ммоль) 5-бромизатина и 0.09 г (1 ммоль) саркозина получили 0.1 г (30%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:ЭА=40:1. Тпл=239°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.12 (ус, 1Н, NH-индолинон), 7.70 (д, J=8.8 Гц, 2Н, Ar), 7.58 (дд, J1=6.3 Гц, J2=2.1 Гц, 1H, Ar), 7.46 (д, J=8.8 Гц, 2Н, Ar), 7.01 (с, 2Н, Ar), 6.95 (д, J=8.8 Гц, 2Н, Ar), 6.90 (д, J=8.3 Гц, 2Н, Ar), 4.48 (т, J=9.3 Гц, 1Н, пирролидин-Н3), 3.82 (т, J=9.3 Гц, 1H, пирролидин-Н2), 3.64 (с, 3Н, СН3), 3.54 (т, J=8.5 Гц, 1Н, пирролидин-Н1), 2.11 (с, 3Н, NCH3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C27H21Br2N3O3S2, М+Н: 656.9397, масса найденная (М+Н): 656.9393.
Пример 10. Получение 4''-(4-бромфенил)-4'-(4-метоксифенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.10).
В результате реакции 0.2 г (1 ммоль) роданина, 0.15 г (1 ммоль) изатина и 0.09 г (1 ммоль) саркозина получили 0.15 г (52%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:МеОН=40:1. Тпл=246°C.
Спектр 1H ЯМР (DMSO-d6, 400 МГц, δ, м.д): 10.94 (ус, 1Н, NH-индолинон), 7.67 (д, J=8.8 Гц, 2Н, Ar), 7.61(д, J=8.1 Гц, 2Н, Ar), 7.45 (д, J=8.8 Гц, 2Н, Ar), 7.35-7.39 (м, 1H, Ar), 6.90-7.02 (м, 5Н, Ar), 4.50 (т, J=9.3 Гц, 1Н, пирролидин-Н3), 3.87 (т, J=9.4 Гц, 1Н, пирролидин-Н2), 3.76 (с, 3Н, СН3), 3.52 (т, J=8.3 Гц, 1Н, пирролидин-Н1), 2.09 (с, 3Н, NCH3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C27H22BrN3O3S2 М+Н: 579.0347, масса найденная (М+Н): 579.0349.
Пример 11. Получение 5-бром-4''-(4-бромфенил)-1'-метил-4'-(2-метилфенил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона(1.11).
В результате реакции 0.15 г (0.8 ммоль) роданина, 0.18 г (1.6 ммоль) 5-бромизатина и 0.08 г (1.6 ммоль) саркозина получили 0.15 г (58%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:ЭА=40:1. Тпл=207°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.12 (ус, 1Н, NH-индолинон), 7.72 (д, J=9.1 Гц, 3Н, Ar), 7.60 (дд, J1=6.2 Гц, J2=2.0 Гц, 1Н, Ar), 7.30 (д, J=7.7 Гц, 1Н, Ar), 7.19-7.25 (м, 3Н, Ar), 7.03 (с, 1Н, Ar), 6.91 (д, J=8.3 Гц, 2Н, Ar), 4.38 (т, J=8.0 Гц, 1Н, пирролидин-Н3), 3.97 (т, J=9.3 Гц, 1Н, пирролидин-Н2), 3.54 (т, J=8.4 Гц, 1Н, пирролидин-Н1), 2.24 (с, 3Н, СН3), 2.14 (с, 3Н, NCH3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C27H21Br2N3O2S2, М+Н: 640.9405, масса найденная (М+Н): 640.9407.
Пример 12. Получение 4''-(4-бромфенил)-4'-(4-этилфенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.12).
В результате реакции 0.15 г (0.8 ммоль) роданина, 0.12 г (1.6 ммоль) изатина и 0.08 г (1.6 ммоль) саркозина получили 0.16 г (69%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:ЭА=40:1. Тпл=254°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 10.93 (ус, 1Н, NH-индолинон), 7.67 (д, J=8.8 Гц, 1Н, Ar), 7.43 (д, J=8.0 Гц, 2Н, Ar), 7.37 (д, J=8.0 Гц, 1Н, Ar),7.23 (д, J=7.9 Гц, 2Н, Ar), 6.97-7.02 (м, 4Н, Ar), 6.92 (д, J=7.7 Гц, 1Н, Ar), 6.92 (д, J=7.8 Гц, 1Н, Ar), 4.51 (т, J=9.1 Гц, 1Н, пирролидин-Н3), 3.89 (т, J=9.4 Гц, 1Н, пирролидин-Н2), 3.52 (т, J=8.4 Гц, 1Н, пирролидин-Н1), 2.60 (м, 2Н, СН2), 2.10(с, 3Н, NCH3), 1.18 (с, 3Н, СН3).
Масс-спектр высокого разрешения (ESI, m/Z): масса рассчитанная для C28H24BrN3O2S2, М+Н: 577.0541, масса найденная (М+Н): 577.0545.
Пример 13. Получение 5-бром-4',4''-бис(4-хлорфенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.13).
В результате реакции 0.15 г (0.8 ммоль) роданина, 0.17 г (1.6 ммоль) 5-бромизатина и 0.08 г (1.6 ммоль) саркозина получили 0.25 г (97%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:ЭА=40:1. Тпл=232°С.
Спектр 1Н ЯМР (DMSO-d6,400 МГц, δ, м.д): 11.03 (ус, 1Н, NH-индолинон), 7.50-7.63 (м, 7Н, Ar), 7.39-7.48 (м, 2Н, Ar), 6.99 (д, J=7.2 Гц, 1Н, Ar), 7.89 (д, J=7.2 Гц, 1Н, Ar), 4.61 (т, J=9.1 Гц, 1H, пирролидин-Н3), 3.79 (т, J=9.4 Гц, 1H, пирролидин-Н2), 3.56 (т, J=8.5 Гц, 1H, пирролидин-Н1), 2.16 (с, 3Н, NCH3).
Пример 14. Получение 5-бром-4''-(4-хлорфенил)-4'-(4-фторфенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.14).
В результате реакции 0.14 г (0.8 ммоль) роданина, 0.17 г (1.6 ммоль) 5-бромизатина и 0.08 г (1.6 ммоль) саркозина получили 0.19 г (87%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:ЭА=40:1. Тпл=237°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.13 (ус, 1Н, NH-индолинон), 7.47-7.63 (м, 7Н, Ar), 7.17-7.25 (м, 2Н, Ar), 6.98 (д, J=7.1 Гц, 1H, Ar), 6.89 (д, J=7.1 Гц, 1H, Ar), 4.53 (т, J=9.6 Гц, 1H, пирролидин-Н3), 3.79 (т, J=9.4 Гц, 1H, пирролидин-Н2), 3.56 (т, J=8.5 Гц, 1Н, пирролидин-Н1), 2.17 (с, 3Н, NCH3).
Пример 15. Получение 5-хлор-4''-(4-хлорфенил)-4'-(4-этилфенил)-1'-метил-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.15).
В результате реакции 0.16 г (0.8 ммоль) роданина, 0.15 г (1.6 ммоль) 5-хлоризатина и 0.08 г (1.6 ммоль) саркозина получили 0.21 г (86%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:ЭА=40:1. Тпл=242°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 11.11 (ус, 1Н, NH-индолинон), 7.55 (д, J=8.5 Гц, 2Н, Ar), 7.46 (д, J=8.5 Гц, 1Н, Ar), 7.43 (д, J=7.7 Гц, 2Н, Ar),7.20-7.26 (м, 4Н, Ar), 6.94 (д, J=8.3 Гц, 1Н, Ar), 6.88 (д, J=8.3 Гц, 1Н, Ar), 4.50 (т, J=9.8 Гц, 1Н, пирролидин-Н3), 3.84 (т, J=9.4 Гц, 1Н, пирролидин-Н2), 3.53 (т, J=9.0 Гц, 1Н, пирролидин-Н1), 2.59 (кв, J=7.6 Гц, 2Н, СН2), 2.11 (с, 3Н, NCH3), 1.12-1.22 (м, 3Н, СН3).
Пример 16. Получение 4'-(4-этилфенил)-1'-метил-4''-фенил-1-(проп-2-ин-1-ил)-5''-сульфанилиден-1,2-дигидродиспиро[индол-3,2'-пирролидин-3',2''-[1,4]тиазолидин]-2,3''-диона (1.16).
В результате реакции 0.17 г (0.8 ммоль) роданина, 0.19 г (1.6 ммоль) N-пропаргилизатина и 0.08 г (1.6 ммоль) саркозина получили 0.14 г (68%) соединения в виде белого порошка. Продукт очищали колоночной хроматографией в системе CHCl3:ЭА=40:1. Тпл=219°С.
Спектр 1Н ЯМР (DMSO-d6, 400 МГц, δ, м.д): 7.77 (д, J=8.4 Гц, 1Н, Ar), 7.46-7.34 (м, 6Н, Ar), 7.23-7.01 (м, 4Н, Ar), 4.57-4.63 (м, 2Н), 4.46 (с, 1Н), 3.86 (т, J=9.5 Гц, 1H, пирролидин-Н3), 3.58 (т, J=8.5 Гц, 1Н, пирролидин-Н2), 3.51 (т, J=8.5 Гц, 1H, пирролидин-Н1), 2.60 (м, 2Н, СН2), 2.08 (с, 3Н, NCH3), 1.17 (с, 3Н, СН3).
Пример 31. Получение солей (в виде гидрохлоридов)
Для получения солей диспиро-индолинонов формулы 1 полученные соединения 1.1-1.30 растворяют в спирте, добавляют к ним насыщенный спиртовой раствор хлороводорода. После окончания реакции выпавший осадок отфильтровывают. Получают кристаллы гидрохлоридов соединений 1.1-1.16.
Пример 32. Биологические испытания.
Все представленные диспиро-индолиноны формулы 1 были протестированы на их способность блокировать клеточный рост и вызывать апоптоз на различных штаммах опухолевых клеточных линий.
Эти модели включают дифференцированные клетки эмбриональные трансфицированные клетки почек (НЕК293Т), линию раковых клеток молочной железы (MCF-7), а также линии аденокарциномы легких человека А549, линию клеток рака почек SN12C и нормальный клеточной линии почек VA13. Цитотоксичность этих вещества оценивали с помощью стандартного МТТ-теста с (3- (4,5-диметилтиазол-2-ил)-2,5-дифенил тетразолий бромидом. Результаты биологических испытаний представлены ниже в Таблице 1.

Известно, что в клеточной линии LNCAp белок р53 экспрессирован в большей концентрации по сравнению с клеточной линией РС3 [Wassman, C.D. et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nature Commun. 4, 1407 (2013)]. Таким образом, имея различия в значениях цитотоксичности соединений на этих двух линиях более, чем на один порядок, мы можем судить о эффективности полученных соединений как ингибиторов p53-MDM2 для терапии рака простаты. Как видно из данных Таблицы 1, соединение 1.2 обладает значительным цитототоксическим эффектом на клеточных линиях всех типов, проявляя селективность пот отношению к линии А549 со значением IC50 5.6±0.4 мкМ, при этом показывая отсутствие цитотоксичности на нераковой клеточной линии VA13; соединение 1.6, также проявляет селективность по отношению к линии А549 со значением IC50 4.2±0.5 мкМ и также имеет слабовыраженный цитотоксический эффект на клеточной линии VA13. Соединение 1.10 проявляет цитотоксический эффект по отношению к линии А549 со значением IC50 6.2±0.5 мкМ, а также соединение 1.16 имеет значение IC50 5.0±0.4 мкМ на той же клеточной линии. Данные соединения имеют высокие значения цитотоксичности на нераковых клеточных линиях VA13 и могут быть использованы как противоопухолевые препараты для терапии рака легкого.
Дополнительно было проведено исследование цитотоксичности на клеточных линиях рака почек SN12C, экспрессирующей белок р53. Как видно из Таблицы 1, соединения 1.1, 1.7, 1.8 и 1.15 имеют средние значения цитоксичности IC50 на клеточной линии рака почек SN12C 6.5±1.2, 5.2±1.4, 6.1±1.3 и 3.4±0.5 мкМ соответственно. Наилучший цитотоксический проявляют соединения 1.3, 1.12 и 1.14 которые имеют IC50 1.3±0.2, 2.1±0.4 и 2.1±0.6 мкМ на опухолевой клеточной линии SN12C. Данные соединения могут быть использованы как противоопухолевые препараты для терапии рака почек.
Пример 33. Протокол исследования цитотоксичности.
Цитотоксичность тестируемых веществ оценивали с помощью стандартного МТТ-теста с использованием 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил тетразолий бромида[Ciapetti G, Cenni Е, Pratelli L, Pizzoferrato А. 1993. In vitro evaluation of cell/biomaterial interaction by MTT assay. Biomaterials 14:359-364]. Для этого рассевают 4000 клеток на лунку в 130 мкл среды DMEM в 96-луночном планшете и выдерживают в инкубаторе при температуре 37°С с 5% СО2 в течение первых 24 ч без обработки. Затем в плашки добавляют 15 мкл растворов вода-ДМСО испытуемых веществ к клеткам (восемь разведений от 50 нМ до 100 мкМ) и инкубируют клетки 72 ч, используя в качестве контроля доксорубицин (восемь разведений от 3 нМ до 6 мкмоль). После этого добавляют МТТ с концентрацией 0,5 мг/мл в среде, инкубируют клетки 2 ч с последующим удалением среды и добавлением 100 мкл ДМСО и измеряют пропускание при 565 нм с использованием планшетного ридера. IC50 рассчитывают с использованием "GraphPad Prism 6" software (GraphPad Software, Inc., San Diego, CA).
Настоящее изобретение может быть использовано для синтеза соединений для терапии онкологических заболеваний.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ДИСПИРО-ИНДОЛИНОНЫ, ИНГИБИТОРЫ MDM2/p53 ВЗАИМОДЕЙСТВИЯ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2015 |
|
RU2629750C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИСПИРОИНДОЛИНОНОВ | 2018 |
|
RU2682678C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИСПИРОИНДОЛИНОНОВ НА ОСНОВЕ 5-ИНДОЛИДЕН-2-ТИОГИДАНТОИНОВ | 2020 |
|
RU2756463C1 |
| НОВЫЕ 2',5'-ДИАРИЛСПИРО[ИНДОЛ-3,3'-ПИРРОЛИДИН]-2(1Н)-ОНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2730287C1 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-R-6-R-5-АРИЛ-ПИРРОЛО[3,4-с]КАРБАЗОЛ-1,3(2Н,6Н)-ДИОНОВ | 2009 |
|
RU2404983C1 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
| ЗАМЕЩЕННЫЕ N-(ИНДОЛ-2-КАРБОНИЛ)-ГЛИЦИНАМИДЫ И ИХ ПРОИЗВОДНЫЕ, СПОСОБЫ ЛЕЧЕНИЯ И ФАРМКОМПОЗИЦИЯ | 1996 |
|
RU2143424C1 |
| ЗАМЕЩЕННЫЕ 1,3-ДИЭТИЛ-8-ВИНИЛ-7-МЕТИЛ-3,7-ДИГИДРО-ПУРИН-2,6-ДИОНЫ-АНТАГОНИСТЫ АДЕНОЗИНОВОГО A РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2480469C1 |
| Способ получения α-диазокарбонильных соединений в водной среде | 2018 |
|
RU2686489C1 |
Изобретение относится к соединениям формулы 1, где R1 выбран из группы, включающей 4-этоксифенил, 4-хлорфенил, 4-бромфенил; R2 выбран из группы, включающей фенил, 4-этоксифенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 4-этилфенил, 3,4-диметоксифенил, 4-метоксифенил, 4-фторфенил, 2-метилфенил; R3 выбран из группы, включающей атом хлора, брома или водорода; R4 представляет собой атом водорода или пропаргил НС≡С-СН2-. Также изобретение относится к способу получения соединений формулы 1, применению в качестве ингибитора белок-белкового взаимодействия MDM2/p53, фармацевтической композиции и лекарственному средству для лечения онкологического заболевания, связанного с белок-белковым взаимодействием MDM2/p53 и способу ингибирования. Технический результат – соединения формулы 1 в качестве ингибиторов белок-белкового взаимодействия MDM2/p53. 6 н. и 1 з.п. ф-лы, 1 табл., 33 пр.
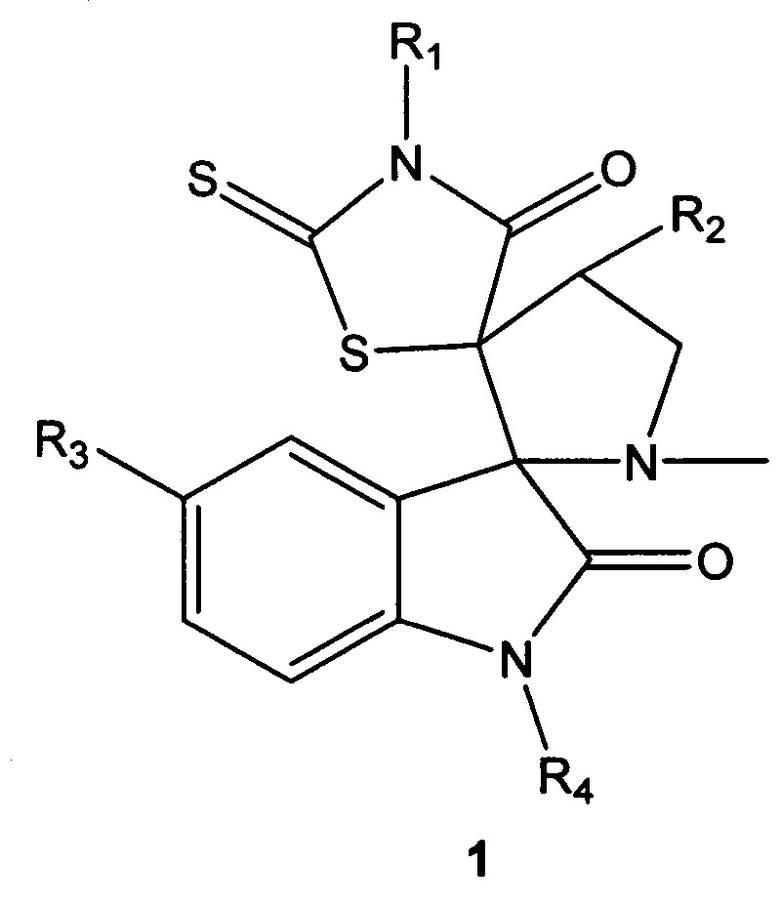
1. Диспироиндолиноны общей формулы 1
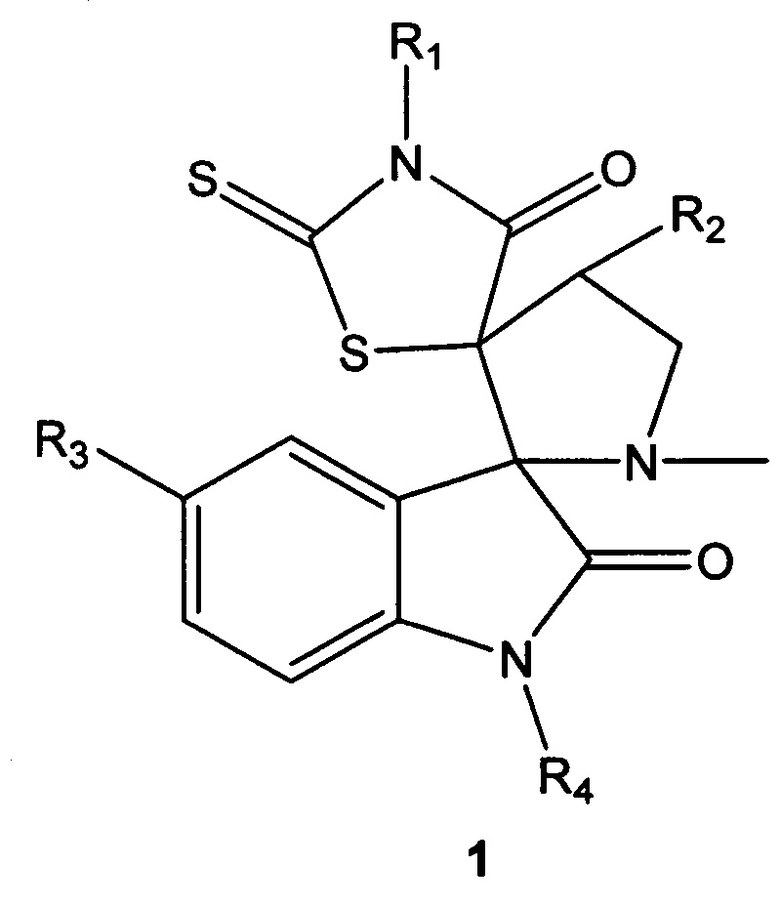 , где
, где
R1 выбран из группы, включающей 4-этоксифенил 4-С2Н5О-С6Н4-, 4-хлорфенил 4-Cl-C6H4-, 4-бромфенил 4-Br-C6H4-;
R2 выбран из группы, включающей фенил С6Н5-, 4-этоксифенил 4-С2Н5О-С6Н4-, 2-хлорфенил 2-Cl-C6H4-, 3-хлорфенил 3-Cl-C6H4-, 4-хлорфенил 4-Cl-C6H4-, 4-этилфенил 4-С2Н5-C6H4-, 3,4-диметоксифенил 3,4-ОМе-C6H3-, 4-метоксифенил 4-CН3О-C6H4-, 4-фторфенил 4-F-C6H4-, 2-метилфенил 2- CН3-C6H4-;
R3 выбран из группы, включающей атом хлора, брома или водорода;
R4 представляет собой атом водорода или пропаргил НС≡С-СН2-.
2. Способ получения диспироиндолинонов общей формулы 1 по п. 1, характеризующийся тем, что 1±0.1 мольных экв. соответствующего имидазолидин-4-она растворяют в толуоле, взятом в количестве 10 мл на 0.5±0.1 ммоль, и кипятят при температуре 100±10°С в течение 10±2 минут после закипания образовавшегося раствора, затем в кипящую смесь добавляют саркозин, взятый из расчета 2±0.2 экв. и соответствующего изатина, взятого в количестве 2±0.2 экв. по отношению к соответствующему имидазолидин-4-ону, полученную смесь кипят до окончания реакции, затем смесь охлаждают до комнатной температуры, выпавший осадок отфильтровывают на вакууме со стеклянным пористым фильтром и колбой Бунзена.
3. Применение соединения общей формулы 1 по п. 1 в качестве ингибитора белок-белкового взаимодействия MDM2/p53.
4. Фармацевтическая композиция для профилактики и лечения онкологического заболевания, связанного с белок-белковым взаимодействием MDM2/p53, содержащая в эффективном количестве соединение по п. 1.
5. Фармацевтическая композиция по п. 4, где онкологическое заболевание представляет собой рак легких или почек.
6. Лекарственное средство для профилактики и лечения пролиферативных заболеваний, связанных с белок-белковым взаимодействием MDM2/p53, в виде таблеток, капсул или инъекций, помещенных в фармацевтически приемлемую упаковку, содержащее в эффективном количестве соединение по п. 1 или фармацевтическую композицию по п. 4.
7. Способ ингибирования белок-белкового взаимодействия MDM2/p53, включающий введение в клетку соединения по п. 1.
| GUO-LIANG FENG et al., "A facile regioselective 1,3-dipolar cycloaddition reaction for the synthesis of dispiro[indole-3,2-pyrrolidine-3',5'-[1,3]thiazolidine] derivatives", Journal of Chemical Research, 2015, vol.39, no.3, pp.134-137 | |||
| НОВЫЕ ДИСПИРО-ИНДОЛИНОНЫ, ИНГИБИТОРЫ MDM2/p53 ВЗАИМОДЕЙСТВИЯ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2015 |
|
RU2629750C2 |
| HAI LIU et al., "An efficient one-pot synthesis of dispiropyrrolidine derivatives | |||

