Изобретение относится к ингибиторам гликогенфосфорилазы, фармацевтическим композициям, содержащим такие ингибиторы, и применению подобных ингибиторов для лечения диабета, гипергликемии, гиперхолестеринемии, гипертензии, гиперинсулинемии, гиперлипидемии, атеросклероза и ишемии миокарда у млекопитающих.
Несмотря на раннее открытие инсулина и его последующее широкое использование при лечении диабета и более позднее открытие и использование сульфонилмочевин (например, хлорпропамидаТМ (Pfizer), толбутамидТМ (Upjohn), ацетогексамидТМ (E.I.Lilly), толазамидТМ (Upjohn)) и бигуанидов (например, фенформинТМ (Ciba Geigy), метформинТМ (G.D.Searle)) в качестве пероральных гипогликемических средств, лечение диабета остается менее чем удовлетворительным. Применение инсулина необходимо примерно у 10% больных диабетом, для которых синтетические гипогликемические средства неэффективны (диабет типа I, инсулинозависимый сахарный диабет), требует многократного ежедневного введения, обычно путем самостоятельных инъекций. Определение соответствующей дозировки инсулина требует частого определения сахара в моче или крови. Введение избыточной дозы инсулина вызывает гипогликемию с эффектами, изменяющимися от легких отклонений от нормы содержания глюкозы в крови до комы или даже смерти. Лечение инсулин-независимого сахарного диабета (диабет типа II, ИНСД) обычно состоит из комбинации диеты, упражнений, приема пероральных средств, например сульфонилмочевин, и в более тяжелых случаях, введения инсулина. Однако, клинически доступные гипогликемические средства могут обладать другими побочными действиями, которые ограничивают их применение. В любом случае, когда один из этих препаратов может быть неэффективным в отдельном конкретном случае, другой может обеспечить успех. Остающаяся потребность в гипогликемических средствах, которые обладают меньшими побочными действиями или дают успех, когда другие неэффективны, очевидна.
Атеросклероз, заболевание артерий, как установлено, является главной причиной смертности в Соединенных Штатах и Западной Европе. Патологическая цепь, приводящая к атеросклерозу и окклюзивному заболеванию сердца, хорошо известна. Самой ранней стадией этой цепи является образование "жировых полосок" в сонных артериях, коронарных и церебральных артериях и в аорте. Эти поражения имеют желтый цвет из-за наличия жировых отложений, обнаруживаемых в основном в гладкомышечных клетках и в макрофагах внутреннего слоя артерий и аорты. Кроме того, установлено, что большая часть холестерина, обнаруживаемого в жировых полосках, в свою очередь вызывает развитие "фиброзных бляшек", которые состоят из скоплений внутренних гладкомышечных клеток, нагруженных липидом и окруженных внеклеточным жиром, коллагеном, эластином и протеогликанами. Клетки плюс матрица образуют фиброзное утолщение, которое покрывает более глубокое отложение из клеточных остатков и дополнительными экстраклеточными липидами. Липид представлен главным образом свободным или этерифицированным холестерином. Фиброзная бляшка образуется медленно и, вероятно, в свое время становится кальцифицированной и некротической, что приводит к "осложненному поражению", чем объясняется артериальная окклюзия и тенденция к интрамуральному тромбозу и спазму мышц артерий, которые являются характерными особенностями развитого атеросклероза.
Эпидемиологические данные позволили твердо определить гиперлипидемию в качестве главного фактора риска в причинности сердечно-сосудистого заболевания (ССЗ), связанного с атеросклерозом. В последние годы ведущие специалисты в медицине придают новое значение снижению уровней холестерина в плазме крови и особенно липопротеинхолестерина низкой плотности, как существенному этапу в профилактике ССЗ. Верхние пределы нормы, как теперь известно, значительно ниже, чем считалось до сих пор. В результате, как теперь стало ясно, большая часть населения Запада подвержены чрезвычайно высокому риску заболевания. Такие независимые факторы риска включают непереносимость глюкозы, гипертрофию левого желудочка, гипертензию и принадлежность к мужскому полу. Сердечно-сосудистые заболевания особенно преобладают среди лиц с диабетом, по крайней мере частично из-за существования независимых факторов риска у этой части населения. Успешное лечение гиперлипидемии у основного населения и у лиц с диабетом в частности, имеет поэтому исключительное медицинское значение.
Гипертензия (или высокое кровяное давление) является состоянием, которое встречается у людей в качестве вторичного симптома при различных других заболеваниях, таких как стеноз почечной артерии, феохромоцитома или эндокринные расстройства. Однако гипертензия наблюдается у многих пациентов, у которых причина заболевания неизвестна. В то время как такая гипертоническая болезнь часто связана с заболеваниями, такими как ожирение, диабет и гипертриглицеридемия, связь между этими заболеваниями не выяснена. К тому же у многих больных проявляются симптомы высокого кровяного давления при полном отсутствии любых других признаков заболевания или нарушений.
Известно, что гипертензия может прямо привести к сердечной недостаточности, почечной недостаточности и "удару" (кровоизлиянию в мозг). Эти состояния могут быть причиной скоропостижной смерти больного. Гипертензия может также способствовать развитию атеросклероза и коронарного заболевания. Эти состояния постепенно ослабляют больного и могут привести к смерти после длительной болезни.
Точная причина гипертонической болезни неизвестна, хотя ряд факторов, как считают, способствует началу заболевания. Среди таких факторов присутствуют стресс, неконтролируемые эмоции, нерегулируемое выделение гормонов (ренина, ангиотензина, альдостероновой системы), избыток соли и воды из-за плохой функции почек, утолщение стенки сосудов и гипертрофия сосудистой сети, приводящих к сужению кровеносных сосудов, и генетические факторы.
Лечение гипертонической болезни производилось с учетом вышеприведенных факторов. Так разработаны и представлены на рынке лекарственных препаратов широкий ряд бетаблокаторов, сосудосуживающих препаратов, ингибиторов ферментов, превращающих ангиотензин, и тому подобные, в качестве антигипертензивных средств. Лечение гипертензии с использованием этих соединений, как было доказано, благоприятно для предотвращения скоропостижной смерти в результате таких состояний, как сердечная недостаточность, почечная недостаточность и кровоизлияния в мозг. Однако развитие атеросклероза или сердечного заболевания в течение длительного периода времени остается нерешенной проблемой. Это означает, что хотя высокое кровяное давление снижается, основная причина гипертонической болезни не реагирует на это лечение.
Была установлена связь гипертензии с повышенным уровнем инсулина в крови, состоянием, известным как гиперинсулинемия. Инсулин, пептидный гормон, чье основное действие состоит в стимуляции утилизации глюкозы, протеинового синтеза и образования и накопления нейтральных жиров, действует также в качестве стимулятора роста клеток сосудов и повышает задержку натрия почками, среди прочего. Эти последние функции могут осуществляться без влияния на уровни глюкозы и, как известно, вызывают гипертензию. Рост периферийной сосудистой сети, например, может вызывать сужение периферийных капилляров; в то время как задержка натрия увеличивает объем крови. Таким образом, снижение уровней инсулина при гиперинсулинемии может предупреждать ненормальный рост сети сосудов и задержку натрия почками, вызываемую высокими уровнями инсулина, и тем самым облегчать гипертензию.
Кардиальная гипертрофия является значительным фактором риска в явлении внезапной смерти, инфаркта миокарда и сердечной недостаточности при перегрузке. Эти сердечные явления связаны, по крайней мере, частично с повышенной чувствительностью к повреждению миокарда после ишемии и возобновленной перфузии, которые могут происходить у поликлинического больного, так же как и в условиях операционной. Существует неудовлетворенная потребность в медицине в предотвращении или сведении к минимуму неблагоприятных исходов операций для миокарда, особенно инфаркта миокарда до и после операции. Как некардиальная, так и кардиальная хирургия связаны с существенным риском инфаркта миокарда или смерти. Считается, что риску подвергаются примерно 7 миллионов пациентов при некардиальной хирургии со случаями смерти до или после операции и серьезными осложнениями для сердца на таком уровне, как 20-25% в отдельных сериях. Кроме того, у 400000 больных, подвергающихся ежегодно операции коронарного шунтирования, возникновение инфаркта миокарда до и после операции оценивается на уровне 5% и смертность - на уровне 1-2%. В настоящее время не существует лекарственной терапии в этой области, которая снижает повреждение ткани сердца в результате ишемии миокарда при оперативном вмешательстве или повышает кардиальную устойчивость к эпизодам ишемии. Такая терапия, как предполагают, является жизнесберегающей и снижает количество госпитализаций, повышает качество жизни и снижает общие затраты на лечение больных с высоким риском осложнений.
Продукция глюкозы в печени является важной мишенью для терапии ИНСД. Печень является главным регулятором уровней глюкозы в плазме в состоянии после всасывания (устойчивом), и степень продукции глюкозы в печени у больных ИНСД значительно повышена по сравнению с нормальными индивидуумами. Подобным же образом в послеобеденном (приема пищи) состоянии, когда печень играет пропорционально меньшую роль в общем поступлении глюкозы в плазму, продукция глюкозы в печени остается ненормально высокой у больных ИНСД.
Гликогенолиз является важной мишенью для прерывания продукции глюкозы в печени. Печень продуцирует глюкозу путем гликогенолиза (расщепления полимера глюкозы гликогена) и глюконеогенез (синтеза глюкозы из предшественников из 2-х и 3-х углеродных атомов). Данные нескольких направлений показывают, что гликогенолиз может осуществлять важный вклад в выход глюкозы из печени при ИНСД. Во-первых, установлено, что у нормального человека после завершения всасывания до 75% продукции глюкозы в печени происходит в результате гликогенолиза. Во-вторых, у больных, страдающих болезнями с накоплением гликогена в печени, включая болезнь Герса (гликогенфосфорилазная недостаточность), проявляется эпизодическая гипогликемия. Эти наблюдения наводят на мысль о том, что гликогенолиз может быть важным процессом для продукции глюкозы в печени.
Гликогенолиз катализируется в печени, мышцах и мозгу тканеспецифичными изоформами фермента гликогенфосфорилазы. Этот фермент расщепляет гликогеновую макромолекулу с выделением глюкозо-1-фосфата и новой укороченной гликогеновой макромолекулы. К настоящему времени сообщено о двух типах ингибиторов гликогенфосфорилазы: глюкозе и аналогах глюкозы (Martin, J.L. et al. Biochemistry 1991, 30, 10101) и кофеин и другие пуриновые аналоги (Kasvinski, P. J. et al. J. Biol. Chem. 1978, 253, 3343-3351 и 9102-9106). Эти соединения и ингибиторы гликогенфосфорилазы, как утверждалось, обладают потенциалом для использования при лечении ИНСД путем снижения продукции глюкозы в печени и снижения гликемии (Blundell, T.B. et al. Diabetologia, 1992, 35, Suppl. 2, 569-576 и Martin et al. Biochemistry, 1991, 30, 10101).
Механизмы, ответственные за повреждение миокарда, наблюдаемое после ишемии и возобновления перфузии, еще полностью не поняты. Сообщалось, что (M. F. Allard, et al. Am J. Physiol. 267, Н66-Н74, 1994) "предишемическое снижение гликогена. .. связано с улучшенным послеишемическим функциональным восстановлением левого желудочка у крыс с гипертрофией сердца".
Таким образом, хотя существует множество способов терапии гипергликемии, гиперхолестеринемии, гипертензии, гиперинсулинемии, гиперлипидемии, атеросклероза и ишемии миокарда сохраняется потребность и продолжаются исследования в этой области по поиску альтернативной терапии.
Это изобретение направлено на получение ингибиторов гликогенфосфорилазы - соединений формулы I, применяемых для лечения диабета, гипергликемии, гиперхолестеринемии, гиперинсулинемии, гипертензии, гиперлипидемии, атеросклероза и ишемии миокарда.
Соединения этого изобретения имеют формулу I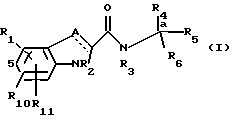
и их фармацевтически приемлемые соли и пролекарства,
где (прерывистая линия (---) является необязательной связью;
A представляет -C(H)=, -C((C1-C4)алкил) =, -C(галоген)= или -N=, когда прерывистая линия (---) является связью, или A является метиленом или представляет -CH((C1-C4)алкил)-, когда прерывистая линия (---) не представлена связью;
R1, R10 или R11 каждый независимо являются H, галогеном, циано-, 4-, 6- или 7-нитро-, (C1-C4)алкилом, (C1-C4)алкоксилом, фторметилом, дифторметилом или трифторметилом;
R2 является H;
R3 является H или (C1-C5)алкилом;
R4 является H, метилом, этилом, н-пропилом, гидрокси (C1-C3)-алкилом, (C1-C3)алкокси (C1-C3)алкилом, фенил(C1-C4)алкилом, фенилгидрокси(C1-C4)алкилом, (фенил) ((C1-C4)-алкилом, -алкокси) (C1-C4)тиен-2- или -3-ил(C1-C4)алкилом или фур-2- или -3-ил (C1-C4) алкилом, причем указанные кольца R4 являются моно-, ди- или тризамещенными, независимо, у атома углерода, водородом, галогеном, (C1-C4)алкилом, (C1-C4)алкоксилом, трифторметилом, гидроксилом, амино- циано- или 4,5-дигидро-1Н-имидазол-2-илом; или
R4 является пирид- -2-, -3- или -4-ил (C1-C4)алкилом, тиазол-2-, -4- или -5-ил (C1-C4)алкилом, имидазол-2-, -4- или -5-ил(C1-C4)алкилом, пиррол-2- или -3-ил (C1-C4)алкилом, оксазол-2-, -4- или -5-ил (C1-C4)алкилом, пиразол-3-, -4- или -5-ил (C1-C4) алкилом, изоксазол-3-, -4- или -5-ил (C1-C4) алкилом, изотиазол-3-, -4- или -5-ил-(C1-C4) алкилом, пиридазин-3- или -4-ил (C1-C4)алкилом, пиримидин-2-4-, -5- или -6-ил(C1-C4)алкилом, пиразин-2- или -3-ил(C1-C4)алкилом, 1,3,5-триазин-2-ил(C1-C4) алкилом или индол-2--(C1-C4)алкилом, причем указанные предыдущие гетероциклы R4 являются произвольно моно- или ди-замещенными независимо галогеном, трифторметилом, (C1-C4)алкилом, (C1-C4)алкоксилом, амино-, гидроксилом или циано-, и указанные заместители связаны с углеродом; или
R4 является R15-карбонилоксиметилом, где R15 является фенилом, тиазолилом, имидазолилом, 1H-индолилом, фурилом, пирролилом, оксазолилом, пиразолилом, изоксазолилом, изотиазолилом, пиридилом, пиридазинилом, пиримидинилом, пиразинилом или 1,3,5-триазинилом и причем указанные предыдущие кольца R15 являются произвольно моно- или ди-замещенными независимо галогеном, амино-, гидроксилом, (C1-C4)алкилом, (C1-C4)алкоксилом или трифторметилом, и указанные моно- или ди-заместители связаны с углеродом;
R5 является метилом, этилом, н-пропилом, гидроксиметилом или гидроксиэтилом;
R6 является карбоксилом, (C1-C8)алкоксикарбонилом, бензилоксикарбонилом, C(O)NR8R9 или C(O)R12, где
R8 является H, (C1-C6)алкилом, цикло(C3-C6)алкилом, цикло(C3-C6)алкил(C1-C5)алкилом, гидроксилом или (C1-C8)алкоксилом; и
R9 является H, цикло(C3-C8)алкилом, цикло(C3-C8)алкил (C1-C5)-алкилом, цикло(C4-C7)алкенилом, цикло(C3-C7)алкил(C1-C5)алкоксилом, цикло(C3-C7)алкилокси, гидроксилом, метилен-перфторированным (C1-C8)-алкилом, фенилом или гетероциклом, причем указанный гетероцикл является пиридилом, фурилом, пирролилом, пирролидинилом, оксазолилом, тиазолилом, имидазолилом, пиразолилом, пиразолинилом, пиразолидинилом, изоксазолилом, изотиазолилом, пиранилом, пиридинилом, пиперидинилом, морфолинилом, пиридазинилом, пиримидинилом, пиразинилом, пиперазинилом, 1,3,5-триазинилом, бензотиазолилом, бензоксазолилом, бензимидазолилом, тиохроманилом или тетрагидробензотиазолилом, причем указанные гетероциклические кольца являются связанными через углерод-азот; или
R9 является (C1-C6)алкилом или (C1-C8)алкоксилом, причем указанный (C1-C6)алкил или (C1-C8)алкоксил является произвольно монозамещенным цикло(C4-C7)алкен-1-илом, фенилом, тиенилом, пиридилом, фурилом, пирролилом, пирролидинилом, оксазолилом, тиазолилом, имидазолилом, пиразолилом, пиразолинилом, пиразолидинилом, изоксазолилом, изотиазолилом, пиранилом, пиперидинилом, морфолинилом, тиоморфолинилом, 1-оксотиоморфолинилом, 1,1-диоксотиоморфолинилом, пиридазинилом, пиримидинилом, пиразинилом, пиперазинилом, 1,3,5-триазинилом или индолилом, и где указанный (C1-C6)алкил или (C1-C8)алкоксил являются произвольно дополнительно независимо моно- или дизамещенными галогеном, гидроксилом, (C1-C5)алкоксилом, амино-, моно-N- или ди-N,N-(C1-C5)алкиламино-, циано-, карбоксилом или (C1-C4)-алкоксикарбонилом; и
причем кольца R9 являются произвольно моно- или ди-замещенными независимо у атома углерода галогеном, (C1-C4)алкилом, (C1-C4)-алкоксилом, гидроксилом, гидрокси(C1-C4)алкилом, амино(C1-C4)алкилом, моно-N- или ди-N,N-(C1-C4)алкиламино(C1-C4)алкилом, (C1-C4)-алкокси(C1-C4)алкилом, амино-, моно-N- или ди-N,N-(C1-C4)алкиламино-, циано-, карбоксилом, (C1-C5)алкоксикарбонилом, карбамоилом, формилом или трифторметилом, и указанные кольца R9 могут быть произвольно дополнительно моно- или ди-замещенными независимо (C1-C5)-алкилом или галогеном;
при условии, что не включен ни один четвертичный азот в любой гетероцикл R9;
R12 является морфолино-, тиоморфолино-, 1-оксотиоморфолино-, 1,1-диоксотиоморфолино-, тиазолидин-3-илом, 1-оксотиазолидин-3-илом, 1,1-диоксотиазолидин-3-илом, пирролидин-1-илом, пиперидин-1-илом, пиперазин-1-илом, пиперазин-4-илом, азетидин-1-илом, 1,2-оксазинан-2-илом, пиразолидин-1-илом, изоксазолидин-2-илом, изотиазолидин-2-илом, 1,2-оксазетидин-2-илом, оксазолидин-3-илом, 3,4-дигидроизохинолин-2-илом, 1,3-дигидроизоиндол-2-илом, 3,4-дигидро-2Н-хинол-1-илом, 2,3-дигидробензо[1,4]оксазин-4-илом, 2,3-дигидробензо[1,4] -тиазин-4-илом, 3,4-дигидро-2Н-хиноксалин-1-илом, 3,4-дигидробензо[c][1,2]оксазин-1-илом, 1,4-дигидробензо[d][1,2]оксазин-3-илом, 3,4-дигидробензо[e] [1,2] -оксазин-2-илом, 3H-бензо[d]-изоксазол-2-илом, 3H-бензо[c] изоксазол-1-илом или азепан-1-илом,
причем указанные кольца R12 являются произвольно моно-, ди- или три-замещенными независимо галогеном, (C1-C5)алкилом, (C1-C5)-алкоксилом, гидроксилом, амино-, моно-N- или ди-N,N-(C1-C5)алкиламино-, формилом, карбоксилом, карбамоилом, моно-N- или ди-N,N-(C1-C5)алкилкарбамоилом, (C1-C6)алкокси(C1-C3)алкоксилом, (C1-C5)алкоксикарбонилом, бензилоксикарбонилом, (C1-C5)алкоксикарбонил(C1-C5)алкилом, (C1-C4)алкоксикарбониламино,
карбокси(C1-C5)алкилом, карбамоил(C1-C5)алкилом, моно-N- или ди-N,N-(C1-C5)алкилкарбамоил(C1-C5)алкилом, гидрокси(C1-C5)алкилом, (C1-C4)алкокси(C1-C4)алкилом, амино(C1-C4)алкилом, моно-N- или ди-N,N-(C1-C4)алкиламино(C1-C4)алкилом, оксо-, гидроксиимино- или (C1-C6)алкоксиимино- и причем не более, чем два заместителя, выбираемые из оксо-, гидроксиимино- или (C1-C6)алкоксиимино- и оксо-, гидроксиимино- или (C1-C6)алкоксиимино-, находятся у неароматического углерода; и
причем кольца R12 являются произвольно дополнительно моно- или ди-замещенными независимо (C1-C5)алкилом или галогеном;
при условии, что когда R6 является (C1-C5)алкоксикарбонилом или бензилоксикарбонилом, тогда R1 является 5-галогеном, 5-(C1-C4)-алкилом или 5-циано- и R4 является (фенил) (гидрокси) (C1-C4)алкилом, (фенил) ((C1-C4)алкокси)(C1-C4)алкилом, гидроксиметилом или Ar(C1-C2)алкилом, где Ar является тиен-2- или -3-илом, фур-2- или -3-илом или фенилом, причем указанный Ar является произвольно моно- или ди-замещенным независимо галогеном; при условии, что когда R4 является бензилом и R5 является метилом, R12 не является 4-гидроксипиперидин-1-илом или когда R4 является бензилом и R5 является метилом, R6 - не C(O)N(CH3)2;
при условии, что когда R1 и R10 и R11 являются H, R4 - не имидазол-4-илметил, 2-фенилэтил или 2-гидрокси-2-фенилэтил;
при условии, что когда оба R8 и R9 являются н-пентилом, ни один из R1 не является 5-хлор-, 5-бром-, 5-циано-, 5(C1-C5)алкилом, 5(C1-C5)алкоксилом или трифторметилом;
при условии, что когда R12 является 3,4-дигидроизохинол-2-илом, указанный 3,4-дигидроизохинол-2-ил не замещен карбокси((C1-C4)алкилом;
при условии, что когда R8 является H и R9 является (C1-C6)-алкилом, R9 не замещен карбоксилом или (C1-C4)алкоксикарбонилом у углерода, который связан с атомом азота N из NHR9; и
при условии, что когда R6 является карбоксилом и R1, R10, R11 и R5 являются все H, тогда R4 не является бензилом, H, (фенил) (гидрокси)метилом, метилом, этилом или н-пропилом.
Первая группа предпочтительных соединении с формулой I состоит из таких соединений, где
R1 является 5-Н, 5-галогеном, 5-метилом, 5-циано- или 5-трифторметилом;
R10 и R11 являются каждый независимо H или галогеном;
A является -C(H)=;
R2 и R3 являются H;
R4 является H, метилом, фенил (C1-C2)алкилом, где указанные фенильные группы являются моно- или ди-замещенными независимо H, галогеном, (C1-C4)алкилом, (C1-C4)алкоксилом, трифторметилом, гидроксилом, амино- или циано- и где указанные R4 группы произвольно дополнительно монозамещены галогеном; или
R4 является тиен-2- или -3-ил(C1-C2)алкилом, пирид-2-, -3- или -4-ил(C1-C2)алкилом, тиазол-2-, -4- или -5-ил(C1-C2)алкилом, имидазол-2-, -4- или -5-ил(C1-C2)алкилом, фур-2- или -3-ил(C1-C2)-алкилом, пиррол-2- или -3-ил(C1-C2)алкилом, оксазол-2-, -4- или -5-ил(C1-C2)алкилом, пиразол-3-, -4- или -5-ил(C1-C2)алкилом, изоксазол-3-, -4- или -5-ил(C1-C2)алкилом, изотиазол-3-, -4- или -5-ил(C1-C2)алкилом, пиридазин-3- или -4-ил(C1-C2)алкилом, пиримидин-2-, -4-, -5- или -6-ил(C1-C2)алкилом, пиразин-2- или -3-ил(C1-C2)-алкилом или 1,3,5-триазин-2-ил(C1-C2)алкилом, где указанные предшествующие R4 гетероциклы являются произвольно моно- или ди-замещенными независимо галогеном, трифторметилом, (C1-C4)алкилом, (C1-C4)алкоксилом, амино- или гидроксилом, и указанные моно- или ди-заместители связаны с атомом углерода;
R5 является H; и
R6 является C(O)NR8R9 или C(O)R12.
Внутри вышеприведенной первой группы предпочтительных соединений с формулой I существует группа особенно предпочтительных соединений, где
R4 является H, фенил(C1-C2)алкилом, тиен-2- или -3-ил(C1-C2)-алкилом, фур-2- или -3-ил(C1-C2)алкилом, где R4 кольца являются моно- или ди-замещенными независимо H или фтором;
R6 является C(O)R12; и
R12 является морфолино-, тиоморфолино-, 1-оксотиоморфолино-, 1,1-диоксотиоморфолино, тиазолидин-3-илом, 1-оксотиазолидин-3-илом, 1,1-диоксотиазолидин-3-илом, пирролидин-1-илом, пиперидин-1-илом, пиперазин-1-илом, пиперазин-4-илом, азетидин-1-илом, 1,2-оксазинан-2-илом, изоксазолидин-2-илом, изотиазолидин-2-илом, 1,2-оксазетидин-2-илом, оксазолидин-3-илом, 1,3-дигидроизоиндол-2-илом или азепан-1-илом,
где указанный R12 кольца являются произвольно моно- или ди-замещенными независимо галогеном, (C1-C5)алкилом, (C1-C5)алкоксилом, гидроксилом, амино-, моно-N- или ди-N,N-(C1-C5)алкиламино-, формилом, карбоксилом, карбамоилом, моно-N- или ди-N,N-(C1-C5)алкилкарбамоилом, (C1-C5)алкоксикарбонилом, гидрокси(C1-C5)алкилом, амино(C1-C4)алкилом, моно-N- или ди-N,N-(C1-C4)алкиламино(C1-C4)алкилом, оксо-, гидроксиимино- или (C1-C6)алкоксиимино- при условии, что только R12 гетероциклы тиазолидин-3-ил, пирролидин-1-ил, пиперидин-1-ил, пиперазин-4-ил, азетидин-1-ил, 1,2-оксазинан-2-ил, изоксазолидин-2-ил, изоксазолидин-2-ил или оксазопидин-3-ил являются произвольно моно- или ди-замещенными оксо-, гидроксиимино- или (C1-C6)-алкоксиимино; и
где указанные R12 кольца являются произвольно дополнительно моно- или ди-замещенными независимо (C1-C5)алкилом.
В вышеприведенной группе особенно предпочтительными соединениями являются соединения:
5-хлор-1H-индол-2-карбоновой кислоты [(1S)-бензил-2-(3- гидроксииминопирролидин-1-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [2-(цис-3,4- дигидроксипирролидин-1-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [2-((3S, 4S)- дигидроксипирролидин-1-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [2-(1,1- диоксотиазолидин-3-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [2-оксо-2-тиазолидин-3-ил- этил)-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [(1S)-(4-фторбензил)- 2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-((3RS)- гидроксипиперидин-1-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [2-оксо-2-((1RS)-оксо-1- тиазолидин-3-ил)этил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [(1S)-(2-фторбензил) -2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-((3S, 4S)-дигидроксипирролидин-1-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(3- гидроксиазетидин-1-ил)-2-оксоэтил]-амид,
5-хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(3- гидроксииминоазетидин-1-ил)-2-оксоэтил]амид или
5-хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(4- гидроксииминопиперидин-1-ил)-2-оксоэтил]-амид.
В вышеприведенной группе особенно предпочтительных соединений существует ведущая группа особенно предпочтительных соединений, в которых
R4 является H; и
R12 является тиазолидин-3-илом, 1-оксотиазолидин-3-илом, 1,1-диоксотиазолидин-3-илом или оксазолидин-3-илом или указанные R12 заместители произвольно являются моно- или ди-замещенными независимо карбоксилом, (C1-C5)алкоксикарбонилом, гидрокси (C1-C3)алкилом, амино(C1-C3)алкилом, моно-N- или ди-N,N-(C1-C3)алкиламино(C1-C3)алкилом или
R12 является моно- или ди-замещенным пирролидин-1-илом, причем указанные заместители являются независимо карбоксилом, (C1-C5)алкоксикарбонилом, (C1-C5)алкоксилом, гидроксилом, гидрокси(C1-C3)алкилом, амино, амино(C1-C3)алкилом, моно-N- или ди-N, N-(C1-C3)алкиламино(C1-C3)алкилом или моно-N- или ди-N,N-(C1-C4)алкиламино; и
кольца R12 произвольно дополнительно независимо ди-замещены (C1-C5)алкилом.
Предпочтительными соединениями в непосредственно предшествующей группе особенно предпочтительных соединений являются соединения, в которых
a. R1 является 5-хлором;
R10 и R11 являются H; и
R12 является цис-3,4-дигидроксипирролидин-1-илом;
b. R1 является 5-хлором;
R10 и R11 являются H; и
R12 является (3S, 4S)-дигидроксипирролидин-1-илом;
c. R1 является 5-хлором;
R10 и R11 являются H; и
R12 является 1,1-диоксотиазолидин-3-илом;
d. R1 является 5-хлором;
R10 и R11 являются H; и
R12 является тиазолидин-3-илом; и
е. R1 является 5-хлором;
R10 и R11 является H; и
R12 является 1-оксотиазолидин-3-илом.
В пределах вышеуказанной группы особенно предпочтительных соединений находится вторая группа особенно предпочтительных соединений, в которых
R4 является фенилметилом, тиен-2- или -3-илметилом, причем R4 кольца произвольно моно- или ди-замещены фтором; и
R12 является тиазолидин-3-илом, 1-оксотиазолидин-3-илом, 1,1-диоксотиазолидин-3-илом или оксазолидин-3-илом или указанные заместители R12 произвольно моно- и ди-замещены независимо карбоксилом, или (C1-C5)алкоксикарбонилом, гидрокси(C1-C3)алкилом, амино(C1-C3)-алкилом или моно-N- или ди-N,N-(C1-C3)алкиламино(C1-C3)алкилом,
или R12 является моно- или ди-замещенным азетидин-1-илом или моно- или ди-замещенным пирролидин-1-илом или моно- или ди-замещенным пиперидин-1-илом, причем указанные заместители являются независимо карбоксилом, (C1-C5)алкоксикарбонилом, гидрокси(C1-C3)алкилом, амино(C1-C3)алкилом,
моно-N- или ди-N, N-(C1-C3)алкиламино -(C1-C3)-алкилом, гидроксилом, (C1-C3)алкоксилом, амино-, моно-N- или ди-N,N-(C1-C5)алкиламино-, оксо-, гидроксиимино- или (C1-C5)алкоксиимино-; и R12 кольца являются произвольно дополнительно моно- или ди-замещенными независимо (C1-C5)алкилом.
Предпочтительными соединениями в непосредственно предшествующей группе особенно предпочтительных соединений являются соединения, в которых
а. R1 является 5-хлором;
R10 и R11 являются H;
R4 является 4-фторбензилом;
R12 является 4-гидроксипиперидин-1-илом; и
стереохимия углерода (а) имеет (S) форму;
b. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R12 является 3-гидроксипиперидин-1-илом; и
стереохимия углерода (а) имеет (S) форму;
c. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R12 является цис-3,4-дигидроксипирролидин-1-илом; и
стереохимия углерода (а) имеет (S) форму;
d. R1 является 5-хлором;
R10 и R11 являются H; R4 является бензилом;
R12 является 3-гидроксииминопирролидин-1-илом; и
стереохимия углерода (а) представляет собой (S ) форму;
e. R1 является 5-хлором;
R10 и R11 являются H;
R4 является 2-фторбензилом;
R12 является 4-гидроксипиперидин-1-илом; и
стереохимия углерода (а) имеет (-S) форму;
f. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R12 является (3,4)-дигидроксипирролидин-1-илом; и
стереохимия углерода (а) имеет форму (S);
g. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R12 является 3-гидроксиазетидин-1-илом; и
стереохимия углерода (а) имеет форму (S);
h. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R12 является 3-гидроксииминоазетидин-1-илом; и
стереохимия углерода (а) представляет собой (S) форму; и
i. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R12 является 4-гидроксииминопиперидин-1-илом; и
стереохимия углерода (а) имеет (S) форму.
Второй группой особенно предпочтительных соединений в ведущей группе предпочтительных соединений являются соединения, в которых
R4 является H, фенил(C1-C2)алкилом, тиен-2- или -3-ил-(C1-C2)-алкилом, фур-2- или -3-ил(C1-C2)алкилом, причем указанные кольца R4 являются моно- или ди-замещенными независимо H или фтором;
R6 является C(O)NR8R9; и
R8 является H, (C1-C2)алкилом, гидроксилом или (C1-C4)алкоксилом; и
R9 является H, цикло(C4-C6)алкилом, цикло(C3-C6)алкил(C1-C5)-алкилом, метиленперфторированным(C1-C3)алкилом, пиридилом, пирролидинилом, оксазолилом, тиазолилом, имидазолилом, пиперидинилом, бензотиазолилом или тиохроманилом; или
R9 является (C1-C5)алкилом, причем указанный (C1-C5)алкил произвольно замещен цикло(C4-C6)алкенилом, фенилом, тиенилом, пиридилом, пирролидинилом, оксазолилом, тиазолилом, имидазолилом, пиразолилом, пиперидинилом, морфолинилом, тиоморфолинилом, 1-оксотиоморфолинилом или 1,1-диоксотиоморфолинилом и где указанные (C1-C5)-алкил или (C1-C4)алкоксил произвольно дополнительно независимо замещены моно- или ди-галогеном, гидроксилом, (C1-C5)алкоксилом, амино-, моно-N- или ди-N,N-(C1-C5)алкиламино-, циано-, карбоксилом или (C1-C4)алкоксикарбонилом; и
причем кольца R9 являются произвольно моно- или ди-замещенными независимо у атома углерода галогеном, (C1-C4)алкилом, (C1-C4)алкоксилом, гидроксилом, амино-, моно-N- или ди-N,N-(C1-C4)алкиламино-, карбамоилом, (C1-C5)алкоксикарбонилом или карбамоилом.
В непосредственно предшествующей второй группе особенно предпочтительных соединений находятся соединения, в которых:
a. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R8 является метилом; и
R9 является 3-(диметиламино)пропилом;
стереохимия углерода (а) имеет (S) форму;
b. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R8 является метилом; и
R9 является 3-пиридилом;
стереохимия углерода (а) имеет (S) форму;
c. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R8 является метилом; и
R9 является 2-гидроксиэтилом; и
стереохимия углерода (а) представляет собой (S) форму;
d. R1 является 5-фтором;
R10 и R11 являются H;
R4 является 4-фторфенилметилом;
R8 является метилом; и
R9 является 2-морфолиноэтилом.
Третьей группой особенно предпочтительных соединений в ведущей группе предпочтительных соединений являются соединения, в которых
R4 является H, фенил(C1-C2)алкилом, тиен-2- или -3-ил-(C1-C2)алкилом, фур-2- или -3-ил(C1-C2)алкилом, причем указанные кольца R4 являются моно- или ди-замещенными независимо H или фтором;
R6 является C(O)NR8R9; и
R8 является H, (C1-C5)алкилом, гидроксилом или (C1-C5)алкоксилом; и
R9 является (C1-C4)алкоксилом, причем указанный алкоксил является произвольно замещенным цикло(C4-C6)алкенилом, фенилом, тиенилом, пиридилом, пирролидинилом, оксазолилом, тиазолилом, имидазолилом, пиразолилом, пиперидинилом, морфолинилом, тиоморфолинилом, 1-оксотиоморфолинилом или 1,1-диоксотиоморфолинилом и причем указанный (C1-C5)алкил или (C1-C4)алкоксил является произвольно дополнительно независимо моно- или ди-замещенным галогеном, гидроксилом, (C1-C5)алкоксилом, амино-, моно-N- или ди-N, N-(C1-C5)алкиламино-, циано-, карбоксилом или (C1-C4)алкоксикарбонилом; и
причем кольца R9 являются произвольно моно- или ди-замещенными независимо у углеродного атома галогеном, (C1-C4)-алкоксилом, гидроксилом, амино-, моно-N- или ди-N,N-(C1-C4)алкиламино-, карбамоилом, (C1-C5)алкоксикарбонилом или карбамоилом.
В непосредственно предшествующей третьей группе особенно предпочтительных соединений находятся соединения, в которых:
a. R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R8 является метилом; и
R9 является 2-гидроксиэтоксилом;
стереохимия углерода (а) представляет собой (S) форму;
b. R1 является 5-хлором;
R10 и R11 являются H;
R4 является 4-фторфенилметилом;
R8 является метилом; и
R9 является метоксилом;
c. Стереохимия углерода (а) представляет собой (S) форму;
R1 является 5-хлором;
R10 и R11 являются H;
R4 является бензилом;
R8 является метилом; и
R9 является метоксилом;
Вторую группу предпочтительных соединений с формулой I составляют те соединения, в которых;
R1 является 5-галогеном, 5-метилом, 5-циано- или трифторметилом;
R10 и R11 являются, каждый независимо, H или галогеном;
A является -C(H)=;
R2 и R3 являются H,
R4 является H, фенил(C1-C2)алкилом, тиен-2- или -3-ил-(C1-C2)-алкилом, фур-2- или -3-ил(C1-C2)алкилом, причем указанные кольца являются моно- или ди-замещенными независимо H или фтором;
R5 является H; и
R6, является (C1-C5)алкоксикарбонилом.
Третья группа предпочтительных соединений с формулой I представляет те соединения, в которых
R1 является 5-галогеном, 5-метилом, 5-циано- или трифторметилом;
R10 и R11 являются каждый независимо H или галогеном;
A является -C(H)=;
R2 и R3 являются H;
R4 является H, метилом или фенил(C1-C2)алкилом, причем указанные фенильные группы являются моно- или ди-замещенными независимо H, галогеном, (C1-C4)алкилом, (C1-C4)алкоксилом, трифторметилом, гидроксилом, амино- или циано- и причем указанные фенильные группы являются дополнительно моно- или ди-эамещенными независимо H или галогеном; или
R4 является тиен-2- или -3-ил(C1-C2)алкилом, пирид-2-, -3- или -4-ил(C1-C2)алкилом, тиазол-2-, -4- или -5-ил(C1-C2)алкилом, имидазол-2-, -4- или -5-ил(C1-C2)алкилом, фур-2- или -3-ил(C1-C2)алкилом, пиррол-2- или -3-ил(C1-C2)алкилом, оксазол-2-, -4- или -5-ил-(C1-C2)алкилом, пиразол-3-, -4- или -5-ил-(C1-C2)алкилом, изоксазол-3-, -4- или -5-ил(C1-C2)алкилом, изотиазол-3-, -4- или -5-ил(C1-C2)алкилом, пиридазин-3- или -4-ил-(C1-C2)алкилом, пиримидин-2-, -4-, -5- или -6-ил(C1-C2)алкилом, пиразин-2- или -3-ил(C1-C2)алкилом или 1,3,5-триазин-2-ил-(C1-C2)алкилом, причем указанные предшествующие R4 гетероциклы произвольно моно- или ди-замещены независимо галогеном, трифторметилом, (C1-C4)алкилом, (C1-C4)алкоксилом, амино- или гидроксилом, и указанные моно- или ди-заместители связаны с углеродом;
R5 является H; и
R6 является карбоксилом.
В третьей группе предпочтительных соединений есть ведущая группа особенно предпочтительных соединений, в которых
R10 и R11 являются H; и
R4 является H.
Особенно предпочтительно в непосредственно предшествующей особенно предпочтительной группе соединение, в котором
R1 является 5-хлором.
В другом аспекте это изобретение направлено на получение промежуточных соединений, применимых для получения некоторых соединений с формулой I. Промежуточные соединения имеют формулу QZ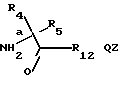
где R5 является H;
R4 является H, фенилметилом, тиен-2- или -3-илметилом, фур-2- или -3-илметилом, причем указанные кольца произвольно моно- или ди-замещены фтором; и
R12 является тиазолидин-3-илом, 1-оксотиазолидин-3-илом, 1,1-диоксотиазолидин-3-илом, пирролидин-1-илом, пиперидин-1-илом, азетидин-1-илом, 1,2-оксазинан-2-илом, изоксазолидин-2-илом, изотиазолидин-2-илом, 1,2-оксазетидин-2-илом или оксазолидин-3-илом,
причем указанные R12 кольца являются произвольно моно- или ди-замещенными независимо галогеном, (C1-C5)алкилом, (C1-C5)алкоксилом, гидроксилом, амино-, моно-N- или ди-N,N-(C1-C5)алкиламино-, формилом, карбоксилом, карбамоилом, моно-N- или ди-N,N-(C1-C5)алкилкарбамоилом, (C1-C5) алкоксикарбонилом, гидрокси(C1-C5)алкилом, амино(C1-C4)алкилом, моно-N- или ди-N,N-(C1-C4)алкиламино(C1-C4)алкилом, оксо-, гидроксиимино или (C1-C6)алкоксиимино- при условии, что только R12 гетероциклы тиазолидин-3-ил, пирролидин-1-ил, пиперидин-1-ил, азетидин-1- ил, 1,2-оксазинан-2-ил, оксазолидин-2-ил или оксазолидин-3-ил являются произвольно моно- или ди-замещенными независимо оксо-, гидроксиимино-, или (C1-C6)алкоксиимино-группами; и
причем указанные R12 кольца являются произвольно дополнительно моно- или ди-замещенными независимо (C1-C5)алкилом и
при условии, что R12 не 2-карбокси-4-гидрокси-пирролидин-1-ил, 2-((C1-C5)алкоксикарбонил)-4-гидроксипирролидин-1-ил, 2-карбоксипиперидин-1-ил или 2-((C1-C5)алкоксикарбонил)-пиперидин-1-ил.
Конкретные соединения в вышеприведенной группе промежуточных соединений представляют соединения, в которых
а. R4 является H; и
R12 является тиазолидин-3-илом;
b. R4 является H; и
R12 является 1,1-диоксотиазолидин-3-ил; и
c. R4 является H; и
R12 является 1-оксотиазолидин-3-илом.
Ведущей группой предпочтительных соединении с формулой QZ являются соединения, в которых
R4 является фенилметилом, и указанный фенил произвольно моно- или ди-замещен фтором; и
R12 является 3-моно-замещенным азетидин-1-илом, 3-моно- или 3,4-дизамещенным пирролидин-1-илом, 3-, 4- или 5-моно- или дизамещенным пиперидин-1-илом, тиазолидин-3-илом, 1-оксотиазолидин-3-илом или 1,1-диоксотиазолидин-3-илом, причем указанный пирролидин-1-ил или пиперидин-1-ил являются моно- или дизамещенными независимо гидроксилом, оксо-, гидроксиимино, амино, моно-N- или ди-N, N-(C1-C4)-алкиламино, (C1-C5)алкоксикарбонилом или карбоксилом
и указанные R12 кольца являются произвольно дополнительно моно- или ди-замещенными независимо (C1-C4)алкилом.
Конкретными соединениями в вышеприведенной непосредственно предшествующей группе предпочтительных соединений являются соединения, в которых
а. R4 является бензилом;
R12 является 3-гидроксипирролидин-3-илом; и
стереохимия углерода (а) имеет (S)-форму;
b. R4 является бензилом;
R12 является 3-гидроксиазетидин-1-илом; и
стереохимия углерода (а) представляет собой (S) форму;
c. R4 является бензилом;
R12 является 3,4-дигидроксипирролидин-1-илом; и
стереохимия углерода (а) представлена (S)-формой;
d. R4 является бензилом;
R12 является 4-гидроксипиперидин-1-илом;
стереохимия углерода (а) представляет собой (S)-форму;
e. R4 является 4-фторфенилметилом;
R12 является 4-гидроксипиперидин-1-илом; и
стереохимия углерода (а) имеет (S)-форму; и
f. R4 является бензилом;
R12 является 4-гидроксииминоазетидин-1-илом; и
стереохимия углерода (а) имеет (S)-форму.
Еще один аспект этого изобретения направлен на способ лечения заболевания или состояния, связанного с гликогенфосфорилазой, у млекопитающих путем введения млекопитающему, страдающему заболеванием или состоянием, зависимым от гликогенфосфорилазы, количества соединения с формулой I, излечивающего заболевание или состояние, зависимое от гликогенфосфорилазы.
Еще один аспект этого изобретения направлен на способ лечения гипергликемии у млекопитающих путем введения млекопитающему, страдающему от гипергликемии, количества соединения с формулой I, лечащего гипергликемию.
Еще один аспект этого изобретения направлен на способ лечения диабета у млекопитающих путем введения млекопитающему, страдающему от диабета, количества соединения по формуле I, излечивающего диабет. В лечение диабета включается предупреждение или ослабление отсроченных осложнений, таких как нейропатия, нефропатия, ретинопатия или катаракты.
Еще один аспект этого изобретения направлен на способ лечения гиперхолестеринемии у млекопитающих путем введения млекопитающему, страдающему гиперхолестеринемией, количества соединения по формуле I, излечивающего гиперхолестеринемию.
Еще один аспект этого изобретения направлен на способ лечения атеросклероза у млекопитающих путем введения млекопитающему, страдающему атеросклерозом, количества соединения по формуле I, излечивающего атеросклероз.
Еще один аспект этого изобретения направлен на способ
лeчения гиперинсулинемии у млекопитающих путем введения млекопитающему, страдающему гиперинсулинемией, количества соединения по формуле I, излечивающего гиперинсулинемию.
Еще один аспект этого изобретения направлен на способ лечения гипертензии у млекопитающих путем введения млекопитающему, страдающему гипертензией, количества соединения по формуле I, излечивающего гипертензию.
Еще один аспект этого изобретения направлен на способ лечения гиперлипидемии у млекопитающих путем введения млекопитающему, страдающему гиперлипидемией, количества соединения по формуле I, излечивающего гиперлипидемию.
Еще один аспект этого изобретения направлен на способ предупреждения ишемического повреждения миокарда у млекопитающих путем введения млекопитающему при риске ишемического повреждения миокарда в условиях до и после операции количества соединения по формуле I, предотвращающего ишемическое повреждение миокарда.
Еще один аспект этого изобретения направлен на способ предупреждения ишемического поражения миокарда у млекопитающих путем введения млекопитающему при риске ишемического повреждения миокарда в условиях до и после операции количества ингибитора гликогенфосфорилазы, предупреждающего ишемическое повреждение миокарда в условиях операции.
Это изобретение также направлено на создание фармацевтических композиций, которые включают терапевтически эффективное количество соединения по формуле I и фармацевтически приемлемый носитель.
Предпочтительные композиции включают фармацевтические композиции для лечения заболеваний и состояний, зависимых от гликогенфосфорилазы, у млекопитающих, которые содержат количество соединения по формуле I, излечивающее заболевание или состояние, зависимое от гликогенфосфорилазы и фармацевтически приемлемый носитель.
Еще один аспект этого изобретения направлен на создание фармацевтических композиций для лечения диабета, которые включают терапевтически эффективное количество ингибитора гликогенфосфорилазы;
одно или более из противодиабетических средств, таких как инсулин и аналоги инсулина (например, Lys-Pro-инсулин); GLP-1 (7-37) (инсулинотропин) и GLP-1 (7-36)-NH2; сульфонмочевины и аналоги: хлорпропамид, глибенкламид, толбутамид, толазамид, ацетогексамид, глипизид®, глимепирид, репаглинид, меглитинид; бигуаниды: метформин, фенформин, буформин; α2-aнтагонисты и имидазолины: мидаглизол, изаглидол, дериглидол, идазокса, эфароксан, флупароксан; другие усиливающие секрецию инсулина вещества: линоглирид, A-4166; глитазоны: циглитазон, пиоглитазон, энглитазон, троглитазон, дарглитазон, BRL 49653; ингибиторы окисления жирных кислот: кломоксир, этомоксир; ингибиторы α-глюкозидазы: акарбоза, миглитол, эмиглитат, воглибоза, MDL-25637, камиглибоза, MDL-73945; β-агонисты: BRL 35135, BRL 37344, Ro 16-8714, ICID7114, CL 316243; ингибиторы фосфодиэстеразы: L-386398; липидопонижающие средства: бенфлуорекс; средства против ожирения: фенфлурамин; ванадат и ванадиевые комплексы (например, нагливан®) и пероксованадиевые комплексы; амилиновые антагонисты; глюкагоновые антагонисты; ингибиторы глюконеогенеза; аналоги соматостатина; антилиполитические средства: никотиновая кислота, аципимокс, WAG 994; и
произвольно, фармацевтически приемлемый носитель.
Предпочтительные фармацевтические композиции в непосредственно предшествующей группе - это те композиции, в которых ингибитором гликогенфосфорилазы является соединение по формуле I.
Еще одним аспектом этого изобретения является способ лечения диабета у млекопитающих с помощью вышеописанных композиций с комбинациями препаратов.
Заболевания или состояния, зависимые от гликогенфосфорилазы, относятся к заболеваниям, которые опосредуются, вызываются или поддерживаются, в целом или частично, расщеплением макромолекулы гликогена с помощью гликогенфосфорилазных ферментов с выделением глюкозо-1-фосфата и новой укороченной молекулы гликогена. Состояние при этих заболеваниях улучшается путем снижения активности гликогенфосфорилазы и характеризуется повышенной активностью гликогенфосфорилазы. Примеры включают диабет, гипергликемию, гиперхолестеринемию, гипертензию, гиперинсулинемию, гиперлипидемию, атеросклероз и ишемию миокарда.
Термин ингибитор гликогенфосфорилазы относится к любому веществу или средству или любой комбинации веществ и/или средств, которые снижают, замедляют или устраняют ферментативное действие гликогенфосфорилазы. Известное к настоящему времени действие гликогенфосфорилазы состоит в разрушении гликогена путем катализа обратимой реакции макромолекулы гликогена с неорганическим фосфатом с образованием глюкозо-1-фосфата и макромолекулы гликогена, которая на один гликозильный остаток короче, чем первоначальная макромолекула гликогена (в прямом направлении гликогенолиза).
Термин "лечение", как он использован здесь, включает предотвращающее (например, профилактическое) и паллиативное лечение.
Под галогеном подразумевается хлор, бром, иод или фтор.
Под алкилом подразумевается насыщенный углеводород с прямой или с разветвленной цепью. Примерами таких алкильных групп (принимая во внимание обозначенную длину, включает конкретный пример) являются метил, этил, пропил, изопропил, бутил, сек-бутил, третичный бутил, пентил, изопентил, гексил и изогексил.
Под алкоксилом подразумевается насыщенный алкил с прямой или разветвленной цепью, связанный через окси-группу. Примерами таких алкоксильных групп (принимая во внимание обозначенную длину, охватывает конкретный пример) являются метоксил, этоксил, пропоксил, изопропоксил, бутоксил, изобутоксил, третичный бутоксил, пентоксил, изопентоксил, гексоксил и изогексоксил.
Выражение "фармацевтически приемлемая анионная соль" относится к нетоксичным анионным солям, содержащим анионы, такие как (но не ограничиваются ими) хлорид, бромид, иодид, сульфат, бисульфат, фосфат, ацетат, малеат, фумарат, оксалат, лактат, тартрат, цитрат, глюконат, метансульфонат и 4-толуолсульфонат.
Выражение "фармацевтически приемлемая катионная соль" относится к нетоксическим катионным солям, таким как (но не ограничивается ими) соли натрия, калия, кальция, магния, аммония или протонированных бензатина (N,N'-дибензилэтилендиамин), холина, этаноламина, диэтаноламина, этилендиамина, мегламина (N-метилглюкамин), бенетамина (N-бензилфенетиламин), пиперазина или трометамина (2-амино-2-гидроксиметил-1,3-пропандиол).
Выражение "пролекарство" относится к соединениям, являющимся предшественниками лекарства, которые после введения выделяют лекарство in vivo при помощи какого-то химического или физиологического процесса (например, пролекарство при доведении до физиологического pH превращается в желаемую форму лекарства). Некоторые приведенные в качестве примеров пролекарства после расщепления выделяют соответствующую свободную кислоту и такие гидролизуемые образующие эфир остатки соединений этого изобретения включают, но не ограничиваются ими, заместители карбоновых кислот (например, R6 является карбоксилом, или R8, R9 или R12 содержит карбоксил), в которых свободный водород замещен (C1-C4)алкилом, (C2-C12)алканоилоксиметилом, 1-(алканоилокси)этилом, имеющими от 4 до 9 атомов углерода, 1-метил-1-(алканоилокси)-этилом, имеющим от 5 до 10 атомов углерода, алкоксикарбонилоксиметилом, имеющим от 3 до 6 атомов углерода, 1-(алкоксикарбонилокси)этилом, имеющим от 4 до 7 атомов углерода, 1-метил-1-(алкоксикарбонилокси)этилом, имеющим от 5 до 8 атомов углерода, N-(алкоксикарбонил)аминометилом, имеющим от 3 до 9 атомов углерода, 1-(N-(алкоксикарбонил)амино)этилом, имеющим от 4 до 10 атомов углерода, 3-фталидилом, 4-кротонолактонилом, гамма-бутиролактон-4-илом, ди-N,N-(C1-C2)алкиламино(C2-C3)алкилом (таким как β -диметиламиноэтил), карбамоил-(C1-C2)алкилом, N,N-ди(C1-C2)алкилкарбамоил-(C1-C2)-алкилом и пиперидино-, пирролидино- или морфолино(C2-C3)алкилом.
Другие приводимые в качестве примеров пролекарства выделяют спирт с формулой I, в котором свободный водород гидроксильного заместителя (например, R8, R9 или R12 содержат гидроксил) замещается (C1-C6)алканоилоксиметилом, 1-((C1-C6)алканоилокси)этилом, 1-метил-1((C1-C6)алканоилокси)этилом, (C1-C6)-алкоксикарбонилоксиметилом, N-(C1-C6)алкоксикарбониламинометилом, сукциноилом, (C1-C6)алканоилом, α-амино (C1-C4)алканоилом, арилацилом и α- аминоацилом, или α-аминоацил -α аминоацилом, причем указанные α-аминоацильные составляющие независимо являются любыми из встречающихся в природе L-аминокислот, обнаруженных в белках, P(O)(OH)2, -P(O) (O(C1-C6)алкил)2 или гликозилом (радикал, получающийся в результате отщепления гидроксила гемиацеталя углевода).
Другие примеры пролекарств включают, но не ограничиваются ими, производные по формуле I, в которых R2 является свободным водородом, который замещается R-карбонилом, RO-карбонилом, NRR'-карбонилом, где R и R' являются каждый независимо (C1-C10)алкилом, (C3-C7)циклоалкилом, бензилом, или R-карбонил является природным α- аминоацилом или природным α- -аминоацил-природный -α аминоацилом, -C(OH)C(O)OY, где Y является H, (C1-C6)алкилом или бензилом; -C(OY0)Y1, где Y0 является (C1-C4))алкилом, и Y1 является ((C1-C6))алкилом,
карбокси-(C1-C6)алкилом, амино(C1-C4)алкилом или моно-N- или ди-N,N-(C1-C6)алкиламиноалкилом, -C(Y2)Y3, где Y2 является H или метилом и Y3 является моно-N- или ди-N,N-(C1-C6)алкиламино, морфолино, пиперидин-1-илом или пирролидин-1-илом.
Другие примеры пролекарств включают, но не ограничиваются ими, производные по формуле I, несущие гидролизуемые заместители при R3 из которых выделяется соединение по формуле I, где R3 является свободный водород после гидролиза. Такие гидролизуемые заместители при R3 представляют собой/включают 1-гидрокси(C1-C6)алкил или 1-гидрокси-1-фенилметил.
Другие примеры пролекарств включают циклические структуры, такие, как соединения по формуле I, в которых R2 и R3 являются общим углеродом, образуя таким образом пятичленные кольца. Связывающий углерод может быть моно или ди-замещенным независимо H, (C1-C6)алкилом, (C3-C6)циклоалкилом или фенилом.
Как они использованы здесь, выражения "реакционно-инертный растворитель" и "инертный растворитель" относятся к растворителю, который не взаимодействует с исходными материалами, реагентами, промежуточными соединениями или продуктами таким образом, который влияет на выход желаемого продукта.
Химик обычной квалификации поймет, что некоторые соединения этого изобретения будут содержать один или более атомов, которые могут находиться в определенной стереохимической или геометрической конфигурации, что приводит к образованию стереоизомеров и конфигурационных изомеров. Все такие изомеры и их смеси включаются в это изобретение. Гидраты соединений этого изобретения также включаются в качестве одного из аспектов этого изобретения.
Химик обычной квалификации поймет, что некоторые комбинации содержащих гетероатомы заместителей, перечисленных в этом изобретении, определяют соединения, которые будут менее стабильны в физиологических условиях (например, те, которые содержат ацетальные или аминальные связи). Соответственно, такие соединения менее предпочтительны.
Термин "Rx кольцо (кольцо Rx)", где x является показателем, целым числом, например "R9 кольцо", "R12 кольцо" или "R4 кольцо", которые использованы здесь в отношении замещения на кольце, относится к заместителям, в которых кольцо является Rx, а также где кольцо содержится в пределах Rx.
Как он использован здесь, термин моно-N- или ди-N,N-(C1-Cx)алкил... относится к (C1-Cx)алкильному заместителю, взятому независимо, когда он является ди-N,N-(C1-Cx)алкилом... (x относится к показателю, целому числу).
Другие особенности и преимущества будут очевидны из описания и пунктов формулы изобретения, в которых описано это изобретение.
В основном соединения формулы I могут быть получены с помощью процессов, которые включают процессы, известные в химической отрасли, конкретно, в свете содержащегося здесь описания. Некоторые процессы производства соединений по формуле I представлены в качестве дополнительных характерных черт изобретения и иллюстрируются следующими схемами реакций (см. в конце описания).
По схеме реакций 1 соединения формулы I, где R1, R10, R11, A, R2, R3, R4, R5 и R6 дано определение выше, могут быть получены с помощью любого из двух основных процессов. При первом процессе желаемое соединение по формуле I может быть получено путем взаимодействия соответствующей индол-2-карбоновой кислоты с формулой II, индолин-2-карбоновой кислоты или бензимидазол-2-карбоновой кислоты с соответствующим амином с формулой III (т.е. ацилируя амин). Во втором процессе желаемое соединение с формулой I может быть получено путем взаимодействия соответствующего соединения с формулой IV (т. е. соединения с формулой I, где R6, является карбоксилом) с соответствующим спиртом или амином с формулой R8R9NH или R12H, где R8, R9 и R12 дано определение выше (т.е. ацилируя амин или спирт). Первый процесс (взаимодействие соединений с формулой II с соединениями с формулой III) обычно предпочтителен, когда R4 не H, а R5 является H.
Обычно соединение формулы II соединяется с соединением по формуле III (или соединение формулы IV соединяется с соответствующим амином (например, R12H или R8R9NH) или спиртом в присутствии подходящего агента сочетания. Подходящим агентом сочетания (связывающим средством) является такой, который трансформирует карбоновую кислоту в реакционноспособную разновидность, которая образует амидную или эфирную связь в реакции с амином или спиртом, соответственно.
В качестве агента сочетания может быть реагент, который производит конденсацию в одновременном процессе, когда смешан вместе с карбоновой кислотой и амином или спиртом. Если кислота должна конденсироваться со спиртом, предпочтительно использовать большой избыток спирта в качестве растворителя для реакции с добавлением 1,0-1,5 эквивалентов диметиламинопиридина или без него. Примерами соединяющих реагентов являются 1-(3-диметиламинопропил)- 3-этилкарбодиимида гидрохлорид-гидроксибензотриазол (ДЭК/ГБТ), карбонилдиимидазол, дициклогексилкарбодиимид/гидроксибензотриазол (ГБТ), 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (ЭЭДХ), карбонилдиимидазол/ГБТ, пропанфосфоновый ангидрид (пропанфосфоновой кислоты ангидрид, ПФА) и диэтилфосфорилцианид. Взаимодействие осуществляется в инертном растворителе, предпочтительно апротонном растворителе при температуре, равной от примерно -20oC до примерно 50oC в течение от примерно 1 до примерно 48 часов при произвольном присутствии основания в виде третичного амина, такого, как триэтиламин. Примеры растворителей включают ацетонитрил, дихлорметан, этилацетат, диметилформамид и хлороформ или их смеси. Примером подходящей процедуры соединения является процедура A, содержащаяся здесь (непосредственно перед примерами).
Агентом сочетания может быть также вещество, которое превращает карбоновую кислоту в активированное промежуточное соединение, которое выделяется и/или образуется на первой стадии и которому дают прореагировать с амином или спиртом на второй стадии. Примерами таких соединяющих средств и активированных промежуточных соединений являются тионилхлорид или оксалилхлорид для образования хлорида кислоты, цианурфторид для образования фторида кислоты или алкилхлороформат, такой как изобутил или изопропенилхлороформат (с третичноаминным основанием) для образования смешанного ангидрида карбоновой кислоты. Если связывающим средством является оксалилхлорид, целесообразно использовать небольшое количество диметилформамида в качестве сорастворителя вместе с другим растворителем (таким, как дихлорметан) для катализа образования хлорида кислоты. Этот хлорид кислоты может соединяться путем смешивания с промежуточным соединением с формулой III в соответствующем растворителе вместе с подходящим основанием. Подходящими комбинациями растворителя/основания являются, например, дихлорметан, диметилформамид или ацетонитрил или их смеси в присутствии третичноаминного основания, например, триэтиламина. Другие подходящие комбинации растворителя/основания включают воду или (C1-C5)спирт или их смесь вместе с сорастворителем, таким как дихлорметан, тетрагидрофуран или диоксан и основанием, таким, как карбонат натрия или калия, натрия, калия или лития гидроксид или натрия бикарбонат в достаточном количестве для поглощения освобождаемой кислоты. Использование межфазного катализатора (обычно от 1 до 10 моль.%), такого как галогенид четвертичного аммония (например, тетрабутиламмония бромид или метилтриоктиламмония хлорид) целесообразно, когда используется смесь только частично смешиваемых растворителей (например дихлорметана-воды или дихлорметана-метанола). Использование таких соединяющих средств и соответствующий выбор растворителей и температур известны специалистам и могут быть легко определены по литературным данным. Эти и другие примеры условий, применимых для реакций соединения с карбоновыми кислотами, описаны у Houben - Weyl, Vol XV, part II, E. Wunsch, Ed. , G. Theime Veriag, 1974, Stuttgart, and M. Bodansky, Principles of Peptide Synthesis, Springer - Verlag Berlin 1984 and The Peptides, Analysis, Synthesis and Biology (ed. E. Gross and J. Meienhofer), vols. 1-5 (Academic Press NY 1979-1983).
Соединения формулы IV, где R1, R10, R11, A, R2, R3, R4 и R5 дано определение выше, могут быть получены из соответствующих эфиров формулы V (т.е. соединений формулы I, где R6 является (C1-C5)алкоксикарбонилом или бензилоксикарбонилом) путем гидролиза водным раствором щелочи при температуре, равной от примерно -20oC до примерно 100oC, обычно при примерно 20oC в течение от примерно 30 минут до примерно 24 часов.
Или иначе соединения формулы IV получают путем активации индолкарбоновой кислоты формулы II при помощи агента сочетания (который описан выше), который дает активированное промежуточное соединение (такое как хлорангидрид кислоты, фторангидрид кислоты или смешанный ангидрид), которому затем дают прореагировать с соединением с формулой III, где R3, R4 и R5 дано определение выше, и R6 является карбоксилом, в подходящем растворителе в присутствии подходящего основания. Подходящие растворители включают воду или метанол или их смесь вместе с сорастворителем, таким как дихлорметан, тетрагидрофуран или диоксан. Подходящие основания включают гидроксиды натрия, калия или лития, бикарбонаты натрия или калия, карбонаты натрия или калия, или карбонат калия вместе с тетрабутиламмония бромидом (1 эквивалент) в достаточном количестве для связывания освобождаемой при реакции кислоты (в основном это количество, достаточное для поддержания pH реакции на уровне более 8). Основание может добавляться в возрастающем количестве вместе с активированным промежуточным соединением для осуществления соответствующего регулирования pH реакционной смеси. Реакция проводится в основном при температуре от -20oC до 50oC. Процедуры выделения подбираются специалистом так, чтобы удалить примеси, но обычно состоят из удаления смешиваемых с водой растворителей путем выпаривания, экстрагирования примесей при высоком pH с помощью органического растворителя, подкисления до низкого pH (1-2) и фильтрации или экстракции желаемого продукта подходящим растворителем, таким как этилацетат или дихлорметан.
Соединения формулы V могут быть получены путем связывания соответствующего соединения формулы III, где R6 является алкоксикарбонилом, и соответствующего соединения формулы II при процедуре, аналогичной описанной выше (например, процедура A).
Или иначе, соединения формулы I, которые содержат атомы серы в состоянии окисления до сульфоксида или сульфона, могут быть получены из соответствующих соединений формулы I, имеющих атом серы в неокисленной форме, путем обработки подходящим окисляющим средством, таким как м-хлорпероксибензойная кислота в дихлорметане при температуре, равной от примерно 0oC до примерно 25oC в течение от примерно 1 до примерно 48 часов с использованием от примерно 1 до примерно 1,3 эквивалента для превращения в состояние окисления до сульфоксида и более 2 эквивалентов для превращения в состояние окисления до сульфона.
Некоторые из методов получения, описанных здесь, могут требовать защиты отдельных функциональных групп (т. е. первичных аминов, вторичных аминов, карбоксилов в предшественниках соединений с формулой I). Необходимость такой защиты будет меняться в зависимости от природы отдельных функциональных групп и условий методов получения. Необходимость такой защиты легко определяется специалистом. Использование таких методов защиты/удаления защиты также находится в пределах компетенции специалиста. В отношении общего описания защитных групп и их использования смотрите T.W. Greene, Protective Groups in Organic Synthesis, John Wiley &, Sons, New York, 1991.
Например, в схеме реакций 1 некоторые соединения формулы I содержат функциональные группы первичного амина, вторичного амина или карбоновой кислоты в части молекулы, определяемой R6, которые могут мешать преднамеченной реакции соединения по схеме реакций 1, если промежуточное соединение формулы III или R12H или R8R9NH-амин остается незащищенным. Соответственно функциональные группы первичного амина, вторичного амина или карбоновой кислоты могут быть защищенными, там, где они присутствуют в R6-составляющих по формуле III промежуточного соединения R8R9NH или R12H амина, соответствующими защитными группами во время реакции соединения по схеме реакций 1. Продуктом такой реакции присоединения в подобном случае является соединение по формуле I, содержащее защищающую группу. Эта защищающая группа удаляется на последующей стадии с получением соединения по формуле I. Подходящие защищающие группы для защиты амина и карбоновой кислоты включают те защитные группы, которые обычно используются в пептидном синтезе (такие, как N-т-бутоксикарбонил, N-карбобензилокси- и 9-фторенилметиленоксикарбонил для аминов и сложные эфиры низших алкилов или бензила для карбоновых кислот), которые химически не реактивны в условиях реакции присоединения, описанных выше (и непосредственно предшествуя примерам здесь в виде описания процедуры A), и могут удаляться без химического изменения других функциональных группировок в соединении по формуле I.
Исходные индол-2-карбоновые кислоты и индолин-2-карбоновые кислоты, использованные в схеме реакции 1, когда они коммерчески недоступны или неизвестны из предыдущего опыта (такие работы широко публикуются), могут быть получены общепринятыми методами синтеза. Например, по схеме реакций II индольный эфир с формулой VII (где A не является азотом) может быть получен из соединения по формуле VI (где O выбирается так, чтобы достичь желаемого A, которому дано определение выше, за исключением) путем синтеза индолов по Фишеру (смотрите The Fischer indoll Synthesis Robinson. B. (Wiley, New York, 1982) с последующим омылением полученного в результате сложного эфира индола с формулой VII с получением соответствующей кислоты с формулой VIII. Исходный арилгидразон может быть получен путем конденсации легкодоступного гидразина с соответствующим карбонильным производным или путем реакции Яппа-Клингемана (смотрите Organic Reactions, Phillips, R.R., 1959, 10, 143).
Или иначе, индол-2-карбоновая кислота по формуле VIIIA может быть получена путем конденсации орто-метилнитросоединения по формуле IX с оксалатным сложным эфиром с получением индольного сложного эфира с формулой X с последующим восстановлением нитро-группы и последующего гидролиза.
Этот трехстадийный процесс известен как синтез индолов Райссерта (Reissert, Chemische Berichte, 1987, 30, 1030). Условия выполнения этой последовательности и рекомендации по ней описаны в литературе (Kermark, et al. , J. Chem. Soc. 1921, 119, 1602; Cannon et al., J. Med. Chem. 1981, 24, 238; Julian, et al. in Heterocyclic Compounds, vol. 3 (Wiley, New York, NY, 1962, R. C. Elderfield, ed.) p. 18). Пример конкретного осуществления этой последовательности изложен здесь в примерах 10A-10C.
3-Галоген-5-хлор-1H-индол-2-карбоновые кислоты могут быть также получены путем галогенирования 5-хлор-1H-индол-2-карбоновых кислот.
Согласно схеме реакций III промежуточные соединения бензимидазол-2-карбоновой кислоты по формуле XI могут быть получены путем конденсации ортодиамино-соединения по формуле XIII с гликолевой кислотой с последующим окислением полученного в результате бензимидазол-2-метанола с формулой XII (Bistrzycki, A. and Przeworski, G. Ber. 1912, 45, 3483).
Или же иначе (к схеме реакций II) замещенные индолины с формулой XIV могут быть получены путем восстановления соответствующих индолов с формулой XV при помощи восстанавливающего средства, такого как магний в метаноле при температуре от примерно 25oC до примерно 65oC в течение от примерно 1 до примерно 48 часов (схема реакций III).
Индолинкарбоновые кислоты по формуле XVI получают путем омыления соответствующего эфира с формулой XVII (схема реакций III). Сложный эфир с формулой XVII получают путем восстановления соответствующего эфира индола с формулой VII при помощи восстанавливающего средства, такого как магний в метаноле, как описано для превращения соединения с формулой XV в соединение с формулой XIV, выше.
В следующих разделах описываются пути получения различных аминов, которые используются в вышеприведенных схемах реакций.
В соответствии со схемой реакции IV альфа-аминокислота с формулой XXIII может быть защищена по азоту соответствующей защищающей группой (Pt) (например, t-Boc) с образованием соединения с формулой XXIV. Специалист может легко выбрать соответствующую защищающую группу и метод ее введения. Например, двумя обычными защитными группами являются t-Boc (вводимая путем обработки аминокислоты ди-т-бутилдикарбонатом в, предпочтительно, подходящем протонном растворителе или смеси растворителей при высоком pH) и CBZ (вводимая путем обработки аминокислоты бензилхлорформиатом в подходящем, предпочтительно, протонном растворителе или смеси растворителей и основанием). Соединение с формулой XXIV соединяется (при процедуре, аналогичной процессу присоединения, описанному в схеме реакций 1) с соответствующим R8R9NH или HR12 амином с образованием соединения с формулой XXV, у которого затем удаляются защищающие группы, что дает в результате соединение с формулой IIIb (т. е. соединение с формулой III, где R6 является C(O)R12 или C(O)NR8R9). Если защищающими группами являются t-Boc, удаление производится путем обработки соединения с формулой XXV кислотой в подходящем, предпочтительно апротонном растворителе. Кислоты для этого удаления защиты включают HCl, MeSO3H или трифторуксусную кислоту.
В соответствии со схемой реакций V соединение с формулой XXXI (N-защищенный амин с формулой III, где R6 является (C1-C8)-алкоксикарбонилом или бензилоксикарбонилом) может быть получено из соответствующей незащищенной аминокислоты с формулой XXX путем защиты N-(с получением защищенной аминокислоты с формулой XXXIII) с последующей этерификацией. Например, соединение с формулой XXXIII может быть этерифицировано с помощью соответствующего спирта и кислотного катализатора, такого как хлористый водород или тионилхлорид, в случае трет-бутанола путем обработки аминокислоты изобутиленом и кислотным катализатором, таким как концентрированная серная кислота, или путем обработки алкилгалогенидом (например, метилиодидом) и основанием (например, карбонатом калия). Или же иначе, этерификация может предшествовать стадии защиты.
В соответствии со схемой реакции VI соединения по формуле XXX, где R3 не H, используемые в схеме реакций V, могут быть получены следующим образом. Аминокислоты с формулой XLI могут быть получены путем N-алкилирования защищенных (Pt) аминокислот путем обработки соответствующим основанием и алкилирующим средством. Конкретные процедуры для этого алкилирования описаны. Benoiton, Can. J. Chem. 1977, 58, 906-910 и Hansen, J. Org. Chem. 1985, 50, 945-950.
Например, когда R3 является метилом, а Pt - это Boc, используются гидрид натрия и метилиодид в тетрагидрофуране. Удаление защитных групп у соединения с формулой XII дает желаемое соединение с формулой XXX.
Или же иначе, аминокислота с формулой XLII может быть N-алкилирована с помощью последовательности из трех стадий, включающей восстановительное бензилирование (такое, как с помощью бензальдегида, катализируемой Pd/C гидрогенизации) с получением моно-N-бензильного производного, и восстановительное аминирование с помощью соответствующего карбонильного соединения (например, с помощью формальдегида и цианоборогидрида натрия для введения R3 в виде метила) для получения N-бензил, N-R3-замещенной аминокислоты, N-бензильная защищающая группа удобно удаляется (например, путем гидрогенизации с помощью подходящего катализатора) с получением соединения с формулой XXX. Конкретные условия для этой трехстадийной процедуры алкилирования описаны Reinhold et al., J. Med. Chem., 1968, 11, 258-260.
Непосредственно предшествующая процедура получения может также использоваться для введения заместителя R3 в промежуточное соединение с формулой IIIa (которое является промежуточным соединением с формулой III, где R3 является H).
Аминокислоты, использованные здесь в схемах (например, XL, XLII), если они коммерчески недоступны или о них нет публикаций в литературе, могут быть получены с помощью ряда методов, известных специалистам. Например, может использоваться синтез Стрекера или его разновидности. Соответственно, альдегид (R4CHO), цианид натрия или калия и хлорид аммония реагируют с образованием соответствующего аминонитрила. Аминонитрил гидролизуется минеральной кислотой с образованием желаемой аминокислоты с формулой XLII R4C(NH2)COOH. Или иначе, может использоваться метод Бухерера-Бергса, при котором образуется гидантоин путем нагревания альдегида (R4CHO) с карбонатом аммония и цианидом калия с последующим гидролизом (например, с помощью гидроксида бария при нагревании в колбе с обратным холодильником в кипящем диоксане) кислотой или основанием с образованием желаемой аминокислоты с формулой XLII R4C(NH2)COOH.
В литературе сообщается также и о других методах синтеза α- аминокислот, которые должны позволить специалисту получить желаемое промежуточное соединение с формулой XLII R4C(NH2)COOH, необходимое для синтеза соединений по формуле I.
Подходящие методы синтеза и/или разделения соединений по формуле XLIi можно найти в обзорах Duthaler (Tetrahedron 1994, 50, 1539-1650) или Williams (R. M. Williams, Synthesis of optically active amino acids. Pergamon: Oxford, U.K., 1989).
Конкретный метод синтеза промежуточного соединения с формулой XLII в любой энантиомерной форме из соответствующего промежуточного соединения R4X (X= Cl, Br или I) - это способ Пиррунга и Кришнамурти (J. Org. Chem. 1993, 58, 957-958) или по способу O'Donnell, et al. (J. Am. Chem. Soc, 1989, 111, 2353-2355). Необходимые промежуточные соединения R4X легко получаются с помощью многих методов, знакомых квалифицированному химику. Например, эти соединения, когда R4X является ArCH2X, могут быть получены путем радикального галогенирования соединения ArCH3 или путем формилироваиия арена Ar-H и превращения спирта в бромид.
Другим конкретным методом синтеза промежуточных соединений по формуле XLII в любой энантиомерной форме является метод Кори и Линк (Corey and Link, J. Am. Chew, Soc. 1992, 114, 1906-1908). Так, промежуточное соединение по формуле R4COCCl3 восстанавливается энантиоспецифически до промежуточного соединения R4CH(ОН)CCl3, которое превращается при обработке азидом и основанием в промежуточное соединение R4CH(N3)COOH, которое восстанавливается путем каталитической гидрогенизации в желаемое соединение с формулой XLII. Необходимый трихлорметилкетон R4COCCl3 получается путем реакции альдегида R4CHO с трихлорметильным анионом с последующим окислением (Gallina and Gicrdano, Synthesis, 1989, 466-468).
Соединение по формуле R8NH2 или R9NH2 моноалкилируется карбонильным соединением, соответствующим R8 или R9, соответственно, в необходимых условиях восстановительного аминирования с получением амина с формулой R8R9NH. Чтобы избежать диалкилирования, может быть предпочтительно защитить амины (R8NH2 или R9NH2) подходящими защищающими группами Pt с получением R8(Pt)NH или R9(Pt)NH, например путем реакции с бензальдегидом и восстанавливающим средством. Защищенные амины моноалкилируются с помощью карбонильного соединения, соответствующего R9 или R8, соответственно, в нужных условиях восстановительного аминирования, с получением R8R9N(Pt). Защищающая группа (Pt) удаляется (например, путем исчерпывающей каталитической гидрогенизации, когда Pt является бензилом) с получением соединения по формуле R8R9NH. Соответствующие условия восстановительного аминирования специалист может получить из литературы. Эти условия включают изложенные Borch et al. (J. Am. Chem. Soc 1971, 2897-2904) и рассматриваемые Emerson (Orhanic Reactions, Wiley; Ney York, 1948(14), 174), Hutchins et al. (Org Prep. Proced Int 1979 (II), 20 and Lane et al. (Synthesis) 1975, 135). Условия восстановительного аминирования, способствующие N-моноалкилированию, включают сообщенные Motales et al. (Synthetic Communications 1984, 1213-1220) and Verardo et al. (Synthesis 1992, 121-125). Амины R8NH2 или R9NH2 могут быть также моноалкилированы R9X или R8X, соответственно, где X является хлоридом, бромидом, тосилатом или мезилатом. Или же промежуточное соединение с формулой к R8(Pt)NH или R9(Pt)NH может быть алкилировано R9X или R8X, и защищающая группа удаляется с получением соединения по формуле R8R9NH.
Для получения аминов по формуле R8R9NH, где R8-NH или R9-NH связаны через кислород-азот, могут использоваться дополнительные методы. Так легкодоступное соединение с формулой (C1-C4)алкоксикарбонил-NHOH или NH2CONHOH диалкилируется по азоту и кислороду путем обработки основанием и избытком подходящего алкилирующего средства (R-X) с получением соответствующего (C1-C4)алкоксикарбонил-N(R)OR, которое затем гидролизуется с получением соединения с формулой R8R9NH (где R8=R9=R). Подходящие условия, основание и алкилирующие средства включают описанные Goel and Krolls (Orh. Prep. Proced. Int. 1987, 19, 75-78) and Majorand Fleck (J.Am. Chem. Soc. 1928, 50, 1479). Или иначе, N-гидроксимочевину (NH2CONH(OH)) можно последовательно алкилировать, сначала по кислороду с получением NH2CONH(OR'), затем по азоту с получением NH2CON(R'') (OR') путем последовательной обработки алкилирующими веществами R'X и R''X, соответственно, в присутствии подходящего основания. Подходящее основание и алкилирующие вещества включают описанные Kreutzkamp and Messinger (Chem. Ber. 100, 3463-3465 (1967) and Danen et al. (J. Am. Chem. Soc. 1973, 95, 5716-5724). Гидролиз этих алкилированных производных гидроксимочевины дает амины R'ONH2, и R'ONHR'', которые соответствуют определенным аминам по формуле R8R9NH. Квалифицированный химик может адаптировать процедуры, описанные в этом разделе для других алкилирующих средств R, R' и R''-X для получения других аминов с формулой R8R9NH, где R8-N или R9-N связаны через кислород-азот. Uno et al. (Syn Lett 1991, 559-560) описывают катализируемое BF3 присоединение металлоорганического реагента R-Li; к O-алкилоксиму с формулой R'CH= N-OR'', с получением соединений по формуле R'RCH-NH(OR''). Этот путь может также использоваться для получения соединений с формулой R8R9NH, где один из R8-NH или R9-NH связаны через кислород-азот.
Пролекарства этого изобретения, где карбоксильная группа в карбоновой кислоте с формулой I замещается эфиром, могут быть получены путем соединения карбоновой кислоты с соответствующим алкилгалогенидом в присутствии основания, такого как карбонат калия в инертном растворителе, таком как диметилформамид при температуре от примерно 0 до 100oC в течение от примерно 1 до примерно 24 часов. Или же кислота соединяется с соответствующим спиртом в присутствии каталитического количества кислоты, такой как концентрированная серная кислота, при температуре от примерно 20 до 120oC, предпочтительно при температуре кипения в колбе с обратным холодильником в течение от примерно 1 до примерно 24 часов. Другим способом является реакция кислоты со стехиометрическим количеством спирта в присутствии каталитического количества кислоты в инертном растворителе, таком как тетрагидрофуран, с сопутствующим удалением воды, что производится с помощью физических (например, сепаратор Дин-Старк) или химических (например, молекулярные сита) средств.
Пролекарства этого изобретения, где функциональная спиртовая группа превращена в производное, такое как эфир, могут быть получены путем соединения спирта с соответствующим алкилбромидом или иодидом в присутствии основания, такого как карбонат калия в инертном растворителе, таком как диметилформамид при температуре от примерно 0 до 100oC в течение от примерно 1 до примерно 24 часов. Алканоиламинометиловые эфиры могут быть получены путем реакции спирта с бис-(алканоиламино)метаном в присутствии каталитического количества кислоты в инертном растворителе, таком как тетрагидрофуран, по методу, описанному в US 4997984. Или же эти соединения могут быть получены методами, описанными Hoffman et al. in J. Org. Chem. 1994, 59, 3530.
Диалкилфосфатные эфиры могут быть получены с помощью реакции спирта с диалкилхлорфосфатом в присутствии основания в инертном растворителе, таком как тетрагидрофуран. Диводородфосфаты могут быть получены путем реакции спирта с диарил- или дибензилхлорфосфатом, как описано выше, с последующим гидролизом или гидрогенизацией в присутствии катализатора из благородных металлов, соответственно.
Гликозиды получают путем реакции спирта и углевода в инертном растворителе, таком как толуол в присутствии кислоты. Обычно вода, образующаяся при реакции, удаляется, как только образуется, как описано выше. Альтернативной процедурой является реакция спирта с соответственно защищенным гликозилгалогенидом в присутствии основания с последующим удалением защитных групп.
N-(1-Гидроксиалкил)амиды, N-(1-гидрокси-1-(алкоксикарбонил)-метил)амиды или соединения, где R2 были замещены C(OH)C(O)OY могут быть получены путем реакции исходного амида или индола с соответствующим альдегидом в нейтральных или щелочных условиях (например, этоксид натрия в этаноле) при температурах от 25 до 70oC. N-алкоксиметилиндолы или N-1-(алкокси)-алкилиндолы могут быть получены путем реакции N-незамещенного индола с необходимым алкилгалогенидом в присутствии основания в инертном растворителе. 1-(N,N-диалкиламинометил)индол, 1-(1-(N,N-диалкиламино)этил)индол и N,N-диалкиламинометиламиды (например, R3= CH2N(CH3)2) могут быть получены путем реакции исходного N-H соединения с соответствующим альдегидом и амином в спиртовом растворителе при 25-70oC.
Пролекарства этого изобретения, где R2 и R3 являются обычным углеродом, могут быть получены путем реакции исходного соединения (лекарства) с бензальдегидом или кетоном или его диметилацеталем в инертном растворителе в присутствии каталитического количества кислоты с одновременным удалением воды или метанола.
Исходные материалы и реагенты для вышеописанных схем реакций (например, амины, замещенные индолкарбоновые кислоты, замещенные индолинкарбоновые кислоты, аминокислоты), хотя получение большинства из них описано выше, к тому же легко доступны и могут быть легко синтезированы квалифицированными специалистами общепринятыми методами органического синтеза. Например, многие из промежуточных соединений, использованных здесь для получения соединений по формуле I, являются, связаны с аминокислотами или являются производными аминокислот, обнаруживаемых в природе, к которым существуют большой научный интерес и коммерческая потребность, и соответственно многие такие промежуточные соединения коммерчески доступны и о них есть публикации в литературе, и их легко получить из других обычно доступных соединений методами, о которых сообщается в литературе. Такие промежуточные соединения включают, например, соединения с формулой XXX, формулой XLII, формулой XXXII и формулой XXXIII.
Некоторые соединения формулы I имеют асимметрические атомы углерода, и поэтому являются энантиомерами или диастереомерами. Смеси диастереомеров могут быть разделены на отдельные диастереомеры на основе их физических и химических различий методами, известными per se, например, с помощью хроматографии и/или фракционной кристаллизации. Энантиомеры (например, с формулами III, VIII или IX) могут быть разделены путем превращения смеси энантиомеров в смесь диастереомеров путем реакции с соответствующим оптически активным соединением (например, спиртом), разделения диастереомеров и превращения (например, гидролизом) отдельных диастереомеров в соответствующие чистые энантиомеры. Все такие изомеры, включая диастереомеры, энантиомеры и их смеси считаются частью этого изобретения.
Хотя многие соединения этого изобретения не ионизируются в физиологических условиях, некоторые соединения этого изобретения являются ионизируемыми в физиологических условиях. Так, например, некоторые из соединений этого изобретения являются кислотными и они образуют соль с фармацевтически приемлемым катионом. Все такие соли охватываются объемом этого изобретения и они могут быть получены общепринятыми методами. Например, они могут быть получены просто путем связывания кислотных и основных веществ, обычно в стехиометрическом соотношении или в водной, или в неводной или частично водной среде, как необходимо. Соли выделяют или путем фильтрования, или путем осаждения с помощью жидкости, не являющейся растворителем, с последующим фильтрованием, или путем выпаривания растворителя, или, в случае водных растворов, путем лиофилизации, по необходимости.
Кроме того, некоторые соединения этого изобретения являются основными, и они образуют соль с фармацевтически приемлемым анионом. Все такие соли входят в объем этого изобретения, и они могут быть получены общепринятыми методами. Например, они могут быть получены просто путем связывания кислотных и основных веществ обычно в стехиометрических соотношениях или в водной или неводной, или частично водной среде, как необходимо. Соли выделяются или путем отфильтровывания, или осаждением с помощью нерастворяющей жидкости с последующим фильтрованием, или путем выпаривания растворителя, или, в случае водных растворов, путем лиофилизации, как необходимо.
Кроме того, когда соединения этого изобретения образуют гидраты или сольваты, они также входят в объем изобретения.
Применимость соединений данного изобретения в качестве медикаментов при лечении метаболических заболеваний (таких, которые детально охарактеризованы здесь) у млекопитающих (например, людей) демонстрируется активностью соединений этого изобретения при общепринятых исследованиях и исследованиях in vitro и in vivo, описанных ниже. Такие исследования также дают средства, с помощью которых активность соединений этого изобретения можно сравнить с активностью других известных соединений. Результаты этого сравнения полезны для определения уровней дозировки у млекопитающих, включая людей, для лечения таких заболеваний.
Очищенная человеческая печеночная гликогенфосфорилаза (ЧПГФ) получена с помощью следующей процедуры.
Экспрессия и ферментация
кДНК ЧПГФ экспрессируется с помощью плазмиды рКК233-2 (Pharmacia Biotech. Inc. , Piscataway, New Jersey) в E.coli, штамм XL-1 Blue (Stratagene Cloning Sistems, La Jolla, CA). Штамм засевается в среду LB (состоящую из 10 г триптона, 5 г дрожжевого экстракта, 5 г NaCl и 1 мл 1N NaCl на литр) плюс 100 мг/л ампициллина, 100 мг/л пиридоксина и 600 мг/л MnCl2 и выращивается при 37oC до плотности клеток по OD550=1,0. В этот момент клетки индуцируются 1 мМ изопропил-1-тио -β-D-галактозидом (ИПТГ). Через три часа после индукции клетки собирают путем центрифугирования и клеточные осадки замораживаются при -70oC до тех пор, пока не требуются для очистки.
Очистка гликогенфосфорилазы
Клетки в осадках, описанные выше, снова суспендируют в 25 мМ β-глицерофосфате (pH 7,0) с 0,2 мМ ДТТ, 1 мM MqCl2 плюс следующие ингибиторы протеазы: 0,7 мкг/мл - пепстатин A; 0,5 мкг/мл - леупептин; 0,2 мМ - фенилметилсульфонилфторид (ФМСФ) и 0,5 мМ - ЭДТУ, лизируют путем предварительной обработки 200 мкг/мл лизоцима и 3 мкг/мл ДНКазы с последующим разрушением ультразвуком порциями по 250 мл в течение 5 х 1,5 минут на льду с использованием ультразвукового прибора для разрушения клеток Branson модель 450 (Brancon Sonic Power Co., Danbury CT). Лизаты осветляли центрифугированием при 35000 xq в течение одного часа с последующим фильтрованием через 0,45-микронные фильтры. ЧПГФ в растворенной фракции лизатов (оценная, как составляющая менее 1% общего белка) очищается путем контроля ферментативной активности (как описано в разделе Определение активности ЧПГФ, ниже) в ряде этапов хроматографии, детализированных ниже.
Аффинная хроматография на иммобилизированном носителе с металлом (IMAC)
Этот этап основан на методе Luocng et al. (Luong et at. Journal of Chromatography (1992) 584, 77-84). 500 мл профильтрованной растворенной фракции клеточных лизатов (полученных из примерно 160 г первоначального осадка клеток) загружают на 130 мл колонку IMAC хелатирующей-сефарозы (Pharmacia LKB Biotechnology Piscataway, New Jersey), которая была нагружена 50 мM CuCl2 и 25 мМ β-глицерофосфата, 250 мM NaCl и 1 мМ имидазола в уравновешивающем буфере pH 7. Колонку промывают уравновешивающим буфером до тех пор, пока A280 не возвращается к исходной линии. Затем образец элюируют с колонки тем же буфером, содержащим 100 мМ имидазола для удаления связанной ЧПГФ и других связанных белков. Фракции, содержащие активную ЧПГФ, объединяют (примерно 600 мл) и добавляют этилендиаминтетрауксусную кислоту (ЭДТУ), DL -дитиотреитол (ДТТ), фенилметилсульфонилфторид (ФМСФ), леупептин и пестатин A с получением концентраций 0,3 мМ, 0,2 мM, 0,2 мМ, 0,5 мкг/мл и 0,7 мкг/мл, соответственно. Объединенную ЧПГФ обессоливали на колонке сефадекса G-25 (Sigma. Chemical Co., St. Louis Missouri), уравновешенной 25 мМ Трис-HCl (pH 7,3), 3 мМ буфером ДТТ (буфер A) для удаления имидазола и хранили на льду до второго этапа хроматографии.
Хроматография на 5'-АМФ сефарозе
Обессоленный объединенный образец ЧПГФ затем смешивали с 70 мл 5'-АМФ сефарозы (Pharmacia LKB Biotechnology, Piscataway, New Jersey), которая была уравновешена буфером A (см. выше). Смесь слегка взбалтывали в течение одного часа при 22oC, затем упаковывали в колонку и промывали буфером A до тех пор, пока A280 не возвращался к исходному уровню. ЧПГФ и другие белки элюировали с колонки с помощью 25 мМ Трис-HCl, 0,2 мМ ДТТ и 10 мМ аденозин-5'-монофосфата (АМФ) при pH 7,3 (буфер B). Фракции, содержащие ЧПГФ, объединяли после идентификации путем определения ферментативной активности (описанного ниже) и визаулизации М, примерно 97 кД полосы белка ЧПГФ с помощью электрофореза в полиакриламидном геле с додецилсульфатом натрия (SDS-PAGE) с последующим окрашиванием серебром (2D-серебряная краска 11 "Daiichi" Kit; Daiichi Pure Chemicals Co., LTD., Tokyo, Lapan). Объединенную ЧПГФ диализировали в 25 мМ β- глицерофосфат, 0,2 мМ ДТТ, 0,3 мМ ЭДТУ, 200 мМ NaCl, pH 7,0 буфер (буфер C) и хранили на льду до использования.
Определение ферментативной активности ЧПГФ
А) Активация ЧПГФ: Превращение ЧПГФb в ЧПГФa
Перед определением ферментативной активности ЧПГФ фермент превращали из неактивной формы, которая экспрессировалась в E. coli, штамм ХL-1 Blue (обозначаемой ЧПГФb) (Stragene Cloning Sistems, LaJolla, California) в активную форму (обозначаемую ЧПГФа) путем фосфорилирования ЧПГФ, используя фосфорилазокиназу следующим образом:
ЧПГФb реакция с иммобилизованной фосфорилазокиназой
Фосфорилазокиназу (Siqma Chemical Company, St. Louis, МО) иммобилизировали на Affi-Gel 10 (BioRad Corp., Melvile, NY) по инструкциям производителя. Вкратце, фермент фосфорилазокиназу (10 мг) инкубировали с промытыми гранулами аффи-геля (1 мл) в 2,5 мл 100 мМ HEPES и 80 мМ CaCl2 при pH 7,4 в течение 4 часов при 4oC. Гранулы аффи-геля затем промывали один раз тем же буфером перед блокированием 50 мМ HEPES и 1 М глицинметиловым эфиром при pH 8,0 в течение одного часа при комнатной температуре. Блокирующий буфер удаляли и заменяли 50 мМ HEPES (pH 7,4), 1 мМ β- меркаптоэтанолом и 0,2% NaN3 для сохранения. Перед использованием для превращения ЧПГФb в ЧПГФa, гранулы аффи-геля с иммобилизованной фосфорилазокиназой уравновешивали путем промывания в буфере, используемом для выполнения киназной реакции, состоящем из 25 мМ β- глицерофосфата, 0,3 мМ ДТТ и 0,3 мМ ЭДТУ при pH 7,8 (буфер для исследований с киназой).
Частично очищенную неактивную ЧПГФb, полученную после вышеуказанной хроматографии на 5'-АМФ-сефарозе, разводили 1:10 буфером для исследования киназы, затем смешивали с вышеупомянутым ферментом фосфорилазокиназой, иммобилизированной на гранулах аффи-геля. Добавляли NaАТФ до 5 мМ и MgCl2 до 6 мМ. Полученную в результате смесь мягко перемешивали при 25oC в течение от 30 до 60 минут. Образец отделяли от гранул и устанавливали процент активации ЧПГФb путем превращения в ЧПГФa с помощью определения ферментативной активности ЧПГФ в присутствии или в отсутствие 3,3 мМ АМФ. Процент общей ферментативной активности ЧПГФ, связанной с ферментативной активностью ЧПГФa (АМФ-независимой), затем рассчитывается следующим образом:
B) Определение активности ЧПГФ
Гипогликемическая активность (так же как и активность по лечению/предупреждению других заболеваний/состояний) соединений этого изобретения может быть не напрямую определена путем оценки действия соединений этого изобретения на активность активированной формы гликогенфосфорилазы (ГФа) с помощью одного из двух методов; активность гликогенфосфорилазы а измеряли в прямом направлении путем контроля продукции глюкозо-1-фосфата из гликогена или следуя обратной реакции, измеряя синтез гликогена из глюкозо-1-фосфата путем выделения неорганического фосфата. Все реакции проводили в трехкратных повторностях в 96-ячеечных платах и изменение в поглощении, связанное с образованием продукта реакции, измеряется при длине волны, указанной ниже, в считывающем устройстве Elisa MCC/340 MK11 (Lab Systems, Finland), соединенном с приемником микроплат (ISN Biomedical Co, Huntsville, Alabama).
Для измерения ферментативной активности ЧПГФ в прямом направлении, продукцию глюкозо-1-фосфата из гликогена отслеживали с помощью полиферментного сочетанного общего метода Pesce et al. (Pesce, M.A. Bodourian, S.H. Harris R. C. and Nicholson. J.F. (1977) Clinical Chemistry 23, 1711-1717), модифицированного следующим образом: от 1 до 100 мкг фосфорилазы а, 10 единиц фосфоглюкомутазы и 15 единиц глюкозо-6-фосфатдегидрогеназы (Boehringer Mannheim Biochemicals, Indianapolis, IN) разводят до 1 мл в буфере A (описанном здесь далее). Буфер A с pH 7,2 содержит 50 мМ HEPES, 100 мM KCl, 2,5 мМ этиленгликольтетрауксусной кислоты (ЭГТУ), 2,5 мМ MgCl2, 3,5 мМ KH2PO4 и 0,5 мМ дитиотреитола. 20 мкл этого исходного, стандартного раствора добавляют к 80 мкл буфера A, содержащего 0,47 мг/мл гликогена, 9,4 мМ глюкозы, 0,63 мМ окисленной формы никотинамидадениндинуклеотидфосфата (НАДФ+). Соединения, которые необходимо испытать, добавляют в виде 5 мкл раствора в 14% диметилсульфоксиде (ДМСО) перед добавлением ферментов. Исходную степень ферментативной активности ЧПГФа в отсутствие ингибиторов определяют путем добавления 5 мкл 14% ДМСО, а ферментативную активность при полностью подавленной ЧПГФа получают путем добавления 20 мкл 50 мМ положительного контроля испытуемого вещества, кофеина. Реакция прослеживается при комнатной температуре путем измерения превращения окисленной НАДФ+ в восстановленной НАДФН при 340 нм.
Для измерения ферментативной активности в обратном направлении превращение глюкозо-1-фосфата в гликоген плюс неорганический фосфат измеряется с помощью общего метода, описанного Engers et al. {Engers, H.D. Shechosky, S. and Madsen, N. B. (1970) Can. J.Biochem. 48, 746-754}, модифицированного следующим образом: 1-100 мкг ЧПГФa разводится до 1 мл в буфере B (описанном здесь далее). Буфер B с pH 7,2 содержит 50 мМ HEPES, 100 мМ KCl2, 2,5 мМ ЭГТУ, 2,5 мМ MgCl2 и 0,5 мМ дитиотреитола. 20 мкл этого исходного раствора добавляется к 80 мкл буфера B с 1,25 мг/мл гликогена, 9,4 мМ глюкозы и 0,63 мМ глюкозо-1-фосфата. Соединения, подлежащие испытанию, добавляются в виде 5 мкл раствора в 14% ДМСО перед добавлением фермента. Исходная степень ферментативной активности ЧПГФа в отсутствие добавляемых ингибиторов определяется путем добавления 5 мкл 14% ДМСО и полностью подавленную ферментативную активность ЧПГФа получали путем добавления 20 мкл 50 мМ кофеина. Эту смесь инкубировали при комнатной температуре в течение 1 часа, неорганический фосфат, выделившийся из глюкозо-1-фосфата, определяли количественно по общему методу Lanzetta et al. {Lanzetta, P.A., Alvarez, L.J., Reinach, P.S. and Candia, O. A. (1979) Anal. Biochem. 100, 95-97}, модифицированного следующим образом: 150 мкл раствора молибдата аммония 10 мг/мл, 0,38 мг/мл малахитового зеленого в 1N HCl добавляют к 100 мкл ферментной смеси. После 20 минут инкубации при комнатной температуре измеряли поглощение при 620 нм.
Соединения этого изобретения легко адаптируются для клинического применения в качестве гипогликемических средств. Гипогликемическую активность соединений этого изобретения можно определить по количеству испытуемого соединения, которое снижает уровни глюкозы в сравнении с носителем без испытуемого соединения у самцов мышей ob/ob. Испытание также дает возможность определения примерного значения минимальной эффективной дозы (МЭД) по снижению in vivo концентрации глюкозы в плазме у таких мышей для подобных испытуемых соединений.
Так как концентрация глюкозы в крови тесно связана с развитием диабетических нарушений, эти соединения благодаря своему гипогликемическому действию предупреждают, останавливают развитие и/или обеспечивают регрессию диабетических нарушений.
Пяти-восьминедельных самцов мышей C57BL/6J-ob/ob (полученных из Jackson, Laboratory, Bar Harbor, ME) помещали по пять в клетку при стандартной практике ухода за животными. После однонедельного периода акклиматизации животных взвешивали и отбирали 25 мкл крови из ретро-орбитального синуса перед каждым курсом терапии. Образец крови сразу же разводили 1:5 физиологическим раствором, содержащим 0,025% гепарина-натрия, и держали на льду до метаболитного анализа. Животных распределяли по группам терапевтического воздействия, так что каждая группа имела сходную среднюю концентрацию глюкозы в плазме. После распределения на группы животным перорально ежедневно в течение четырех дней вводили дозу носителя, содержащего одно из: 1) 0,25% вес/объем метилцеллюлозы в воде без установки pH; или 2) 0,1% Pluronic® P105 блокирующего сополимерного сурфактанта (BASE Corporation, Parsippanay, NJ) в 0,1% растворе соли без установления pH. На 5-й день животных снова взвешивали и затем перорально вводили дозу испытуемого соединения или одного носителя. Все лекарственные препараты вводили в носителе, состоящем из или: 1) 0,25% вес/объем метилцеллюлозы в воде без установления pH; или 2) 10% ДМСО/0,1% Pluronic® P105 (BASF Corporation, Parsippany, NJ) в 0,1% растворе соли без установления pH. Затем у животных через три часа из ретро-орбитального синуса отбирали кровь для определения уровней метаболитов в крови. Свежеотобранные образцы центрифугировали в течение двух минут при 10000xq при комнатной температуре. Супернатант анализировали на глюкозу, например, с помощью Abbott VPТМ (Abbott Laboratories, Diagnostics Division, Irving, TX) и автоанализатора VP Syper System® (Abbott Laboratories, Irving, TX), используя систему реагентов для УФ определения глюкозы A-GentТМ Glucose-UN test (Abbott Laboratories, Irving, TX) (модификация метода Richterich and Dauwalder, Schweizerische Medizinische Wochenschrift, 101, 860 (1971)) (гексокиназный метод) с использованием 100 мг/дл стандарта.
Глюкозу плазмы рассчитывали по равенству:
Глюкоза плазмы (мг/дл) = значение для образца х 5 х 1,784 = 8,92 х значение для образца,
где 5 - это показатель разведения и 1,784 - это поправка на гематокрит плазмы (принимая, что гематокрит плазмы составляет 44%).
У животных, которым вводили носитель, сохранялись по существу неизменные гипергликемические уровни глюкозы (например, больше или равные 250 мг/дл), животные, которых лечили испытуемыми соединениями в соответствующей дозировке, имели значительно сниженные уровни глюкозы. Гипогликемическая активность испытуемых соединений определяется путем статистического анализа (непарный т-тест) средней концентрации глюкозы в плазме между группой, получавшей испытуемое соединение, и группой, получавшей носитель, на пятый день. Вышеприведенное исследование, проведенное с рядом доз испытуемых соединений, дает возможность определения примерного значения минимальной эффективной дозы (МЭД) для снижения концентрации глюкозы в плазме in vivo.
Соединения этого изобретения легко адаптируются для клинического использования в качестве средств, изменяющих гиперинсулинемию, средств, снижающих уровень триглицеридов и гипохолестеринемических средств. Такая активность может определяться количеством испытуемого соединения, которое снижает уровень инсулина, триглицеридов или холестерина по сравнению с контролем с носителем без испытуемого соединения на самцах мышей ob/ob.
Так как концентрация холестерина в крови тесно связана с развитием сердечно-сосудистых заболеваний и болезней сосудов мозга и периферических сосудов, соединения этого изобретения благодаря своему гипохолестеринемическому действию предупреждают, останавливают развитие и/или вызывают регрессию атеросклероза.
Так как концентрация инсулина в крови связана со стимуляцией роста клеток сосудов и повышенную задержку натрия почками (в дополнение к другим действиям, например, стимуляции утилизации глюкозы) и эти действия, как известно, вызывают гипертензию, соединения этого изобретения благодаря их гиперинсулинемическому действию, предупреждают, останавливают развитие и/или вызывают регрессию гипертензии.
Так как концентрация триглицеридов в крови способствует общему повышению уровней липидов в крови, соединения этого изобретения, благодаря их снижающий уровень триглицеридов активности, предупреждают, останавливают развитие и/или вызывают регрессию гиперлипидемии.
Пяти-восьминедельных самцов мышей C57BL/6j-ob/ob (полученных из Jackson Laboratory, Bar Harbor, ME) помещали по пять на клетку при стандартной практике ухода за животными и стандартной диете питания для грызунов по потребности. После однонедельного периода акклиматизации животных взвешивали и из ретро-орбитального синуса перед любым воздействием отбирали 25 микролитров крови. Образец крови сразу же разводили 1:5 физиологическим раствором, содержащим 0,025% гепарина натрия, и держали на льду для анализа на глюкозу плазмы. Животных распределяли на группы по виду производимого воздействия, так что каждая группа имела аналогичную среднюю концентрацию глюкозы в плазме. Соединение, подлежащее испытанию, вводили перорально с помощью зонда в виде примерно от 0,02% до 2,0% растворов (вес/объем (в/о)) в любом 1) 10% ДМСО/0,1% блокирующем сополимерном сурфактанте Pluronic® Р105 (BASE Corporation, Parsippany, NJ) в 0,1% солевом растворе без установления pH, или 2) 0,25% в/о растворе метилцеллюлозы в воде без установления pH. Однократное введение в сутки (о.с.в.) или двукратное введение в сутки (д.с.в.) производили в течение от 1 до 15 дней. Контрольные мыши получали 10% ДМСО/0,1% Pluronic® P105 в 0,1% солевом растворе без установления pH или только в 0,25% в/о метилцеллюлозе в воде без установления pH.
Через три часа после последнего введения животных забивали отсечением головы и кровь из центрального кровообращения собирали в 0,5 мл пробирки для отделения плазмы, содержащие 3,6 мг 1:1 вес/вес смеси фторида натрия: оксалата калия. Свежесобранные образцы центрифугировали и течение двух минут при 10000 х g при комнатной температуре, и сывороточный супернатант переносили и разводили 1:1 объем/объем раствором апротинина 1 ТМЕ/мл в 0,1% солевом растворе без установления pH.
Разведенные образцы сыворотки затем хранили при -80oC до анализа. Оттаявшие образцы разведенной сыворотки анализировали на инсулин, триглицериды и уровень холестерина. Концентрацию инсулина определяли, используя наборы Equate® RIA INSULIN (двойной антительный метод; как указано производителем), закупленные у Binax, South Portland, ME. Коэффициент вариации между определениями составляет ≤ 10%. Сывороточные триглицериды определяли, используя автоанализаторы Abbott VMТМ и VP Super System® (Abbott Laboratories, Irving, TX), используя систему реагентов для определения триглицеридов A-GentТМ Triglycerides Test (Abbott Laboratories, Diagnostics Division, Irving, TX) (ферментативный метод со связанной липазой; модификация метода Sampson, et al. , Clinical Chemistry 21, 1983 (1975)). Уровни общего холестерина сыворотки определяли, используя автоанализатор Abbott VMТМ и VP Super System® (Abbott Laboratories, Irving, TX) и систему реагентов для определения холестерина A GentТМ (ферментативный метод со связанной холестеринэстеразой; модификация метода Allain. et al., Clinical Chemictry 20, 470 (1974)) с использованием стандартов 100 и 300 мг/дл. Сывороточные уровни инсулина, триглицеридов и общего холестерина затем рассчитывали по уравнениям.
Сывороточный инсулин (мкЕд/мл) = Значение для образца х 2.
Сывороточные триглицериды (мг/дл) = Значение для образца х 2. Сывороточный общий холестерин (мг/дл) = Значение для образца х 2, где 2 является показателем разведения.
У животных, которым вводили носитель, сохранялся почти неизменным повышенный уровень сывороточного инсулина (например, 225 мкЕд/мл), сывороточных триглицеридов (например, 225 мг/дл) и общего сывороточного холестерина (например, 160 мг/дл), в то время как у животных, которых лечили испытуемыми соединениями этого изобретения, в основном выявлялся сниженный уровень сывороточного инсулина, триглицеридов и общего холестерина. Активность испытуемых соединений по снижению сывороточного инсулина, триглицеридов и общего холестерина определяли с помощью статистического анализа (непарного t-теста) сравнения средних концентраций инсулина, триглицеридов или общего холестерина между группами, получавшими испытуемое вещество, и контрольной группой, получавшей носитель.
Активность по обеспечению защиты от повреждения ткани сердца для соединений этого изобретения может быть продемонстрирована in vitro в соответствии с представленным у Butwell et al., Am. J. Physiol, 264, H1884-H1889, 1993 and Allard et al., Am. J. Physiol, 1994, 267, H66-H74. Эксперименты выполнялись с использованием изообъемных препаратов изолированного сердца крысы по существу так же, как описано в приведенной выше в виде ссылки статье. Нормальным самцам крыс Sprague-Dawley, подготовленным так, чтобы у них была гипертрофия сердца, путем операции перевязывания аорты, самцам крыс BB/W с острым диабетом или подходящим по возрасту контрольным крысам BB/W без диабета предварительно вводили гепарин (1000 ед, в/б), с последующим введением фенобарбитала (65 мг/кг в/б). После того, как достигалась глубокая анестезия, что определяли по отсутствию подошвенного рефлекса, сердце быстро иссекали и помещали в ледяной физиологический раствор. Сердце ретроградно перфузировали через аорту в течение 2 минут. Частоту сердцебиений и давление в желудочке определяли, используя латексный баллончик, помещенный в левый желудочек, с трубочкой для высокого давления, соединенной с датчиком давления. Сердце перфузировали раствором для перфузии, состоящим из (мМ) NaCl - 118, KCl - 4,7, CaCl2 - 1,2, MgCl2 - 1,2, NaHCO3 - 25, глюкозы - 11. В аппарате для перфузии строго регулировали температуру с помощью подогреваемых бань, используемых для перфузата и для водяной рубашки вокруг трубопровода для перфузии так, чтобы поддерживать температуру сердца на уровне 37oC. Оксигенация перфузата обеспечивалась с помощью педиатрического полого волоконного оксигенератора (Capiax, Terumo, Corp., Tokyo, Japan) непосредственно проксимально к сердцу. Сердца выдерживали с перфузионным раствором ± испытуемое соединение в течение 10 минут или более с последующими 20 минутами полной ишемии и 60 минутами возобновленной перфузии в отсутствие испытуемого вещества. Сердцебиение контрольного и подвергаемого действию испытуемых соединений сердца сравнивали в период после ишемии. Давление в левом желудочке контрольного и подвергаемого воздействию испытуемого вещества сердца сравнивали в период после ишемии. В конце эксперимента сердца также перфузировали и окрашивали для определения отношения площади инфаркта к площади подвергавшейся отрицательному воздействию (%ПИ/ПОВ), как описано ниже.
Терапевтические эффекты соединений этого изобретения по предупреждению поражения ткани сердца, в противном случае являющегося следствием ишемического инсульта, можно также продемонстрировать in vivo в соответствии с представленным у Liu et al., Circulation, Vol. 84, N 1 (July 1991), как, в частности, описано здесь. В исследовании in vivo проверяется кардиозащита испытуемых соединений в сравнении с контрольной группой, которая получает носитель в виде физраствора. В качестве исходной информации отмечено, что короткие периоды миокардиальной ишемии с последующим возобновлением перфузии коронарных артерий защищает сердце от последующей тяжелой миокардиальной ишемии (Murry et al., Circulation, 74; 1124-1136, 1986). Кардиозащита, как показано восстановлением пораженного инфарктом миокарда, может быть создана фармакологически с использованием внутривенного введения агонистов аденозиновых рецепторов интактным анестезированным кроликам, изучаемым в качестве модели in situ предварительно созданной ишемии миокарда (Liu et al., Circulation, 84: 350-356, 1991). При исследовании in vivo определяется, могут ли вещества фармакологически индуцировать кардиозащиту, т.е. обеспечивать уменьшенный размер инфаркта миокарда при парентеральном введении анестезированным кроликам. Эффекты соединений этого изобретения на предварительно созданную ишемию можно сравнить с использованием агониста аденозина A1, N6-1-(фенил-2R-изопропил)аденозина (ФИА), который, как было показано, фармакологически вызывает кардиозащиту у интактных анестезированных кроликов, использованных для изучения in situ (Liu et al., Circulation 84: 350-356, 1991). Точная методика описывается ниже.
Хирургия: самцов новозеландских белых кроликов (3-4 кг) анестезировали пентобарбиталом (30 мг/кг, в/в). Трахеотомию выполняли путем вентрального срединного сервикального сечения, и кроликам делали искусственную вентиляцию 100% кислородом с использованием аппарата искусственного дыхания с созданием положительного давления. Катетеры устанавливали в левой яремной вене для введения лекарства и в левой сонной артерии для измерения давления крови. Затем выводили сердца через левую торакотомию и вокруг выделенной веточки левой коронарной артерии делали петлю (шелк 00). Ишемию вызывали путем тугого затягивания петли и фиксирования ее на месте. Отпуская петлю, давали возобновиться кровообращению в пораженной области. Об ишемии миокарда свидетельствовал региональный цианоз; о возобновлении кровообращения свидетельствовала реактивная гиперемия.
Методика проведения исследования: если артериальное давление и частота сердцебиений оставались стабильными в течение по крайней мере 30 минут, эксперимент начинают. Предварительное состояние ишемии вызывается с помощью двукратной окклюзии коронарной артерии в течение 5 минут с последующим 10 мин возобновления перфузии. Фармакологическое предупредительное лечение осуществлялось путем двукратного вливания испытуемого вещества в течение, например, 5 минут и, давая 10 минут перед дальнейшим вмешательством, или путем инфузии агониста аденозина, ФИА (0,25 мг/кг). После предварительного создания ишемического фона, фармакологического фона или без предварительного вмешательства (без создания предварительного состояния, контроль с введением носителя) производили окклюзию артерии на 30 минут и затем возобновляли кровообращение на два часа, чтобы вызвать инфаркт миокарда. Испытуемое соединение и ФИА растворяли в физрастворе или другом подходящем носителе и вводили в количестве 1-5 мл/кг, соответственно.
Окрашивание (Liu et al., Circulation 84:350-356, 1991): В конце 2 часа периода возобновления кровообращения сердца быстро удаляли, подвешивали в аппарате Лангендорфа и заполняли на 1 минуту нормальным физраствором, нагретым до температуры тела (38oC). Шовный шелк, использованный в качестве петли, затем туго завязывали, чтобы снова вызвать окклюзию артерии, и производили вливание 0,5% суспензии флюоресцентных частиц (1-10 мкм) с перфузатом для окрашивания всего миокарда за исключением области, на которую производилось отрицательное воздействие (нефлюоресцирующий желудочек). Сердца затем быстро замораживали и хранили в течение ночи при -20oC. На следующий день сердца разрезали на слои 2 мм и окрашивали 1% трифенилтеразолийхлоридом (ТТХ). Так как ТТХ реагирует с живой тканью, эта краска дифференцирует живую (красное окрашивание) ткань и мертвую ткань (неокрашенная инфарктная ткань). Область инфаркта (неокрашенная) и область отрицательного воздействия (нет флюоресцентных частиц) обсчитываются для каждого среза левого желудочка с использованием предварительного откалиброванного анализатора изображения. Чтобы упорядочить ишемическое поражение по различиям в площади отрицательного воздействия у разных сердец, данные выражали в виде отношения площади инфаркта к площади отрицательного воздействия (%ПИ/ПОВ). Все данные выражаются в виде среднего значения ± средняя квадратичная ошибка (СКО) и сравниваются статистически с использованием однофакторного вариационного анализа (ANOVA) или непарного т-теста. Значение принимается во внимание, когда p < 0,05.
Введение соединений этого изобретения может происходить любым путем, который доставляет соединение этого изобретения предпочтительно в печеночную и/или сердечную ткани. Эти способы введения включают пероральный путь, парентеральный, интрадуоденальный пути введения и т.д. В основном, соединения данного изобретения вводятся в виде однократного (например, один раз в день) или многократного введения.
Однако, количество и режим введения соединений будут, конечно, зависеть от конкретного заболевания/состояния, которое нужно лечить, субъекта, которого нужно лечить, тяжести заболевания, способа введения и суждения прописывающего врача. Таким образом, из-за разнообразия состояний больных дозировка, даваемая ниже, является направляющим указанием и врач может подбирать дозы лекарства для достижения действия (например, снижающего уровень глюкозы действия), которое врач считает адекватным для данного больного. Рассматривая степень желаемой активности, врач должен сбалансировать ряд факторов, таких как исходный уровень, другие факторы отрицательного действия (сердечно-сосудистые), наличие предшествующих заболеваний, возраст больного и мотивацию больного.
В основном, эффективная дозировка для действия соединений этого изобретения, например, действия по снижению в крови глюкозы, триглицеридов и холестерина и действия по обратному развитию гиперинсулинемии, находится в интервале от 0,005 до 50 мг/кг/сут, предпочтительно от 0,01 до 25 мг/кг/сут и наиболее предпочтительно от 0,1 до 15 мг/кг/сут.
В основном, соединения этого изобретения вводятся перорально, но может применяться парентеральное введение (например, внутривенное, внутримышечное, подкожное или интрамедуллярно), например, когда пероральное введение не подходит по экстренности цели применения, или когда больной неспособен проглотить лекарство. Может быть также показано местное введение, например, когда больной страдает желудочно-кишечными расстройствами или когда медикаментозное лечение лучше всего применять на поверхности ткани или органа, что определяется лечащим врачом.
Соединения данного изобретения в основном вводятся в форме фармацевтической композиции, содержащей по крайней мере одно из соединений этого изобретения вместе с фармацевтически приемлемым носителем или растворителем. Таким образом, соединения этого изобретения могут вводиться отдельно или вместе в любой общепринятой пероральной, парентеральной или трансдермальной дозированной форме.
Для перорального введения фармацевтическая композиция может принимать форму растворов, суспензий, таблеток, пилюль, капсул, порошков и тому подобного. Таблетки, содержащие различные наполнители, такие как цитрат натрия, карбонат кальция и фосфат кальция, применяются вместе с различными дезинтеграторами, такими как крахмал и предпочтительно картофельный крахмал или тапиоки и некоторые сложные силикаты, вместе со связывающими веществами, такими как поливинилпирролидон, сахароза, желатин и аравийская камедь. Кроме того, часто применяются скользящие вещества, такие как стеарат магния, лаурилсульфат натрия и тальк для таблетирования. Твердые композиции подобного типа также применяются в качестве наполнителей в мягких и твердых заполненных желатиновых капсулах; предпочтительные в этой связи материалы включают также лактозу или молочный сахар, а также полиэтиленгликоли с высоким молекулярным весом. Если для перорального введения желательны водные суспензии и/или эликсиры, соединения этого изобретения могут сочетаться с различными подслащивающими средствами, улучшающими вкус и запах, подкрашивающими веществами, эмульгирующими веществами и/или суспендирующими средствами, а также растворителями, такими как вода, этанол, пропиленгликоль, глицерин и различные подобные их комбинации.
Для парентерального введения могут использоваться растворы в кунжутном или арахисовом масле или в водном пропиленгликоле, так же как и стерильные водные растворы соответствующих водорастворимых солей. Такие водные растворы могут быть соответственно буферными, если необходимо, и водный растворитель сначала изотонируют с помощью достаточного количества раствора соли или глюкозы. Эти водные растворы особенно подходят для внутривенного, внутримышечного, подкожного и внутрибрюшинного введения. В этой связи используемые водные среды легко можно получить с помощью стандартных методик, хорошо известных специалистам.
Для трансдермального введения (например, местного) готовятся разбавленные стерильные, водные или частично водные растворы (обычно от 0,1% до 5% концентрации), или же подобные вышеописанным растворам для парентерального введения.
Методы получения различных фармацевтических композиций с определенным количеством активного ингредиента известны или будут ясны в свете этого описания для специалистов. Например, смотрите Remington's Pharmaceutical Sciences, Mack Publishing Company, Easter, Pa, 15th Edition (1975).
Фармацевтические композиции по этому изобретению могут содержать 0,1-95% соединения(й) этого изобретения, предпочтительно 1-70%. В любом случае композиция или лекарственная форма для введения будет содержать количество соединения(й) по этому изобретению, эффективное для лечения заболевания/состояния субъекта, которого нужно лечить, т.е. заболевания/состояния, зависимого от гликогенфосфорилазы.
Общие процедуры эксперимента для примеров 1-99 и 166-172
ЯМР спектры регистрировались на спектрометре Varian XL-300 (Varian Co., Palo Alto, California) или Bruker АМ-300 (Bruker Co., Billerica, Massachusetts) при примерно 23oC и 300 МГц для протонного и 75,4 МГц для углеродных ядер. Химический сдвиг выражается в частях на миллион вниз от поля триметилсилана. Резонансы, обозначенные как обменные, не появляются в отдельном эксперименте ЯМР, где образец встряхивали с несколькими каплями D2O в том же растворителе. FAB-MS спектры получали на спектрометре VG70-2505 (V4 analitical LTD, Wythanshaw, Manchester, U.K.) с использованием жидкой матрицы, состоящей из дитиотреитола/дитиоэритритола 3:1. МС при термораспылении (ТР МС были получены на спектрометре Fisons Trio-1000 (Fisons, Co., Valencia, California) с использованием ионизации аммиаком. Масс-спектры при химической ионизации получали на приборе Hewlett-Packard 5989 (Hewlett-Packard Co., Palo Alto, California) (аммиачная ионизация, PBMS). Там где описывается интенсивность ионов, содержащих хлор или бром, наблюдалось ожидаемое отношение интенсивности (примерно 3: 1 для 35Cl/37Cl-coдеpжащиx ионов и 1:1 для 79Br/81Br-содержащих ионов) и дается интенсивность только для иона более низкой массы.
ВЭЖХ выполняли с детекцией при 214 нм на 250х4,6 мм колонке Rainin Microsorb C-18 (Rainin Co. , Woburn, Massachusetts), которую элюировали изократно с помощью системы из двух насосов/смешивающего устройства, подающей указанную смесь ацетонитрила и водного 0,1 М KH2PO4 pH 2,1 (с помощью H3PO4), соответственно со скоростью 1,5 мл/мин. Образцы вводили в смеси 1:1 ацетонитрила и фосфатного буфера pH 7,0 (0,025 М каждого Na2HPO4 и KH2PO4). Процент чистоты относится к проценту общей интегрированной площади обычно с ходом (протеканием) свыше 10-15 минут. Температуры плавления неоткорректированы и их определяли в аппарате для определения температуры плавления Buchi 510 (Buchi Laboratorums-Technik Ag, Flawil, Switzerland), где были получены температуры плавления, равные 120,5-122oC для бензойной кислоты и 237,5-240,5oC для п-хлорбензойной кислоты (Aldrich 99+% градусов). Колоночную хроматографию выполняли с помощью силикагеля Amicon (30 мкМ, размер пор 60А) (Amicon D Vision, W.R. Grace & Co., Beverly, Mass.) в стеклянных колонках под низким давлением азота. Если не указано иначе, использовались реагенты из коммерческих источников. Диметилформамид, 2-пропанол, тетрагидрофуран и дихлорметан, используемые в качестве растворителей при реакциях, были марки "безводный" и поставлялись Aldrich Chemical Company (Milwaukee, Misconsin). Микроанализы выполняли с помощью микроаналитической лаборатории Шварцкопфа (Schwarzkopf Microanalytical Laboratory), Woodside, NY. Термины "концентрированный" или совыпаренный относятся к удалению растворителя на роторном испарителе с водным отсасывающим устройством на бане с температурой менее 45oC. Реакции, проводимые при "0-20oC" или "0-25oC", проводили с первоначальным охлаждением сосуда в изолированной ледяной бане, которой давали нагреться до комнатной температуры в течение нескольких часов. Сокращения "мин" и "ч" обозначают "минуты" и "часы", соответственно.
Процедура А (связывание пептида с использованием ДЭК)
0,1-0,7 М раствор первичного амина (1,0 эквив., или первичного амина гидрохлорида и от 1,0 до 1,3 эквивалентов триэтиламина на эквив. HCl) в дихлорметане (если не указан определенный растворитель), обрабатывается последовательно при 25oC 0,95-1,2 эквивалента указанной карбоновой кислоты, 1,2-1,8 эквивалента гидроксибензотриазолгидрата (обычно 1,5 эквивалента относительно карбоновой кислоты) и 0,95-1,2 эквивалента (соответствующим в мольном отношении количеству карбоновой кислоты) 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорида (ДЭК), и смесь перемешивают в течение 14-20 часов (смотрите примечание 1, ниже). Смесь разводят этилацетатом, промывают 2-3 раза 1 или 2N NaOH, 2-3 раза 1 или 2N HCl (примечание 2), органический слой сушили над MgSO4 и концентрировали с получением продукта-сырца, который очищали с помощью хроматографии на силикагеле, растирания или перекристаллизации, как указано с использованием указанных растворителей. Очищенные продукты анализировали с помощью ВЭЖХ с обращенной фазой и обнаруживали, что чистота составляла более 95%, если не указано иначе. Реакции, проводимые при температуре 0-25oC, проводили с первоначальным охлаждением сосуда в изолированной ледяной бане, которой давали нагреться до комнатной температуры в течение нескольких часов.
Примечание 1: при реакциях соединения большего масштаба (> 50 мл растворителя) смесь в этот момент концентрировали и остаток растворяли в этилацетате.
Примечание 2: если продукт содержал ионизируемую функциональную аминную группу, промывание кислотой не включалось. Исключения при применении процедуры А отмечаются отдельно (где необходимо, ниже), обычно в скобках, непосредственно после упоминания процедуры А.
Пример 1.
Метиловый эфир (2S)-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3- фенилпропионовой кислоты
Гидрохлорид метилового эфира L-Фенилаланина (77,0 ммолей) и 5-хлор-1Н-индол-2-карбоновую кислоту (77 ммолей) связывали по процедуре A (0-25oC), и продукт очищали с помощью хроматографии на силикагеле в 10 и 20% этилацетате-гексанах с получением соединения заголовка в виде не совсем белого твердого вещества (22,12 г, 81%): т.пл. 156-157oC; ВЭЖХ (60/40) 9,5 минут (98%); PBMS 357/359 (MH+ 100%).
1H ЯМР (CDCl2) δ 9,40 (шир, 1Н), 7,60 (д, 1Н, J =  1 Гц), 7,35 (д, 1Н, J= 8,9 Гц), 7,3-7,2 (м, 4Н), 7,13 (м, 2Н), 6,74 (д, 1Н, J=1,7 Гц), 6,62 (д, 1Н, J=7,5 Гц), 5,11 (м, 1Н), 3,77 (с, 3Н), 3,26 (м, 2Н);
1 Гц), 7,35 (д, 1Н, J= 8,9 Гц), 7,3-7,2 (м, 4Н), 7,13 (м, 2Н), 6,74 (д, 1Н, J=1,7 Гц), 6,62 (д, 1Н, J=7,5 Гц), 5,11 (м, 1Н), 3,77 (с, 3Н), 3,26 (м, 2Н);
Анал. Расчетное для C19H17ClN2O3: C 63,96; H, 4,80; N, 7,85. Обнаруженное: C, 64,24; H, 4,84; N, 8,02.
Пример 2.
2-[(5-Хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовая кислота
Водный 2 М LiOH (33,10 мл) добавляли к раствору (2S)-[(5-хлор-1Н-индол-2-карбонил)амино] -3-фенилпропионовой кислоты метилового эфира (21,47 г, 60 ммолей) в ТГФ (140 мл) при 0-5oC. Через 0,5 часа смесь частично концентрировали, подкисляли до pH 1-2 6N HCl, концентрировали до сухости и твердые вещества промывали водой и затем эфиром с получением бесцветного твердого вещества (18,78 г, 91%): т.пл. 248-255oC; ВЭЖХ (60/40) 5,21 минуты (98%); ТРМС 343/345 (MH+, 100%).
1H ЯМР (ДМСО-d6) δ 12,85 (шир. 1Н), 11,75 (д, 1Н, J ≤ 1 Гц), 8,84 (д, 1H, J= 8,4 Гц), 7,35-7,14 (м, 7Н), 4,65 (м, 1Н), 3,30 (A из AB, 1Н, J=4,5, 13,9 Гц), 3,07 (B из AB, 1Н, J =10,8, 13,8 Гц);
Анал. Расчетное для C18H15ClN2O3: C, 63,07, H,4,41; N, 8,17.
Обнаруженное: C, 62,90; H,4,60; N, 8,04.
Пример 3.
Метиловый эфир [(5-Хлор-1Н-индод-2-карбонил)-амино]-уксусной кислоты
Гидрохлорид глицинметилового эфира (50 ммолей) и 5-хлор-1Н-индол-2-карбоновую кислоту (50 ммолей) соединяли по процедуре А, изменяя следующую обработку: реакционную смесь перемешивали с этилацетатом (250 мл), гексанами (50 мл) и 1N NaOH (50 мл) и суспензию фильтровали. Твердое вещество промывали 1N NaOH, 1N HCl, водой, этилацетатом и сушили: выход 11,5 г, 86%; т.пл. 252-254oC с разложением;
1H ЯМР (ДМСО-d6) δ 11,87 (шир, 1Н), 9,05 (т, 1H, J=6,0 Гц), 7,72 (д, 1Н, J= 2,0 Гц), 7,45 (д, 1Н, J=8,7 Гц), 7,19 (дд, 1Н, J=2,0, 8,7 Гц), 4,05 (д, 2H, J=6,0 Гц), 3,91 (с, 3Н);
Анал. Расчетное для C12H11ClN2O3: C, 54,05; H, 4,16; N, 10,50.
Обнаруженное: C, 54,11; H, 4,23; N, 10,56.
Пример 4.
[(5-Хлор-1Н-индол-2-карбонил)амино]-уксусная кислота
1N NaOH (35 мл) добавляли к суспензии [(5-хлор-1Н-индол-2-карбонил)-амино] -уксусной кислоты метилового эфира (8,0 г, 30 ммолей) в ТГФ (100 мл) и полученную в результате смесь перемешивали в течение 18 часов при 25oC. Раствор подкисляли 6N HCl (7 мл), смесь концентрировали, твердые вещества суспендировали в воде, фильтровали и промывали водой (7,42 г, 98%): ВЭЖХ (60/40) 2,89 минуты (100%);
1H ЯМР (300 МГц, ДМСО-d6) δ 12,68 (шир, 1Н), 11,85 (шир, 1Н), 8,95 (т, 1Н, J=5,9 Гц), 7,72 (д, 1Н, J=2,0 Гц), 7,44 (д, 1Н, J=8,7 Гц), 7,19 (дд, 1H, J=2,0, 8,7 Гц), 7,14 (д, 1Н, J= < 2 Гц), 3,96 (д, 2Н, J=5,9 Гц).
Анал. Расчетное для C11H9N2O3Cl: C, 52,29; H, 3,59; N, 11,09.
Обнаруженное: C, 52,26; H, 3,73; N, 11,20.
Пример 5.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-((3RS)гидроксипирролидин-1-ил)-2-оксоэтил]-амид
3-Пирролидинол (1,25 ммоля) и [(5-хлор-1Н-индол-2-карбонил)-амино]-уксусную кислоту (1,19 ммоля) соединяли по процедуре А со следующей обработкой: реакционную смесь разводили этилацетатом и 2N HCl, перемешивали 1 час, смесь фильтровали и полученные в результате твердые вещества промывали последовательно 2N HCl, 2N NaOH, 2N HCl, сушили, растирали с эфиром/гексанами 1: 1 и сушили с получением не совсем белого твердого вещества: выход 280 мг, 73%; ВЭЖХ (60/40) 4,66 минуты (96%);
PBMS 322/324 (MH+, 100%).
1H ЯМР (ДМСО-d6) δ 11,87 (шир, 1Н), 8,71 (кв, 1Н), 7,71 (д, 1Н, J=2,1 Гц), 7,45 (д, 1Н, J=8,8 Гц), 7,19 (дд, 1Н, J=3,1, 8,8 Гц), 7,16 (s, 1Н), 5,07 (д, 0,5H, J=3,6 Гц), 4,97 (д, 0,5H, J=3,1 Гц), 4,35 (м, 1Н), 4,27 (м, 1Н), 4,10 (т, 1Н), 4,03 (д, 1Н), 3,59 (м, 1Н), 3,49-3,27 (м, 2Н), 2,04-1,79 (м, 2Н).
Пример 6.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-(цис-3,4-дигидроксипирролидин-1-ил)-2-оксоэтил]-амид
(3R, 4S)-3,4-Дигидроксипирролидин-гидрохлорид (цис или мезоизомер, 1,79 ммоля) и [(5-хлор-1Н-индол-2-карбонил)-амино]- уксусную кислоту (0,85 ммоля) соединяли по процедуре А (1:1, CH2Cl2/ДМФ в качестве растворителя при реакции) со следующей обработкой: реакционную смесь концентрировали, остаток суспендировали в 10 мл EtOAc и 10 мл 2N NaOH, твердые вещества отфильтровывали и промывали последовательно водным 1N NaOH, EtOAc, водной 1N HCl, H2O и эфиром. Последовательность промывания повторяли и полученные в результате твердые вещества суспендировали в EtOAc, перемешивали в течение 1 часа, фильтровали и сушили: выход 252 мг, 88%; ВЭЖХ (60/40) 2,33 минуты (93%); ТРМС 338/340 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,82 (с, 1H) , 8,72 (т, 1Н), 7,73 (д, 1H), 7,45 (д, 1H), 7,20 (дд, 1H), 7,15 (с, 1H), 5,05 (д, 1H), 4,98 (д, 1H), 4,10 (м, 1H), 4,03 (м, 3Н), 3,68 (дд, 1Н), 3,42 (дд, 1H), 3,33 (дд, 1H), 3,23 (дд, 1H).
Пример 7.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
4-Гидроксипиперидин (0,83 ммоля) и [(5-хлор-1Н-индол-2-карбонил)-амино] -уксусную кислоту (0,8 ммоля) соединяли по процедуре А (диметилформамиддихлорметан в качестве растворителя для реакции) со следующей обработкой: реакционную смесь перемешивали с этилацетатом и водной 2N HCl, полученную в результате суспензию фильтровали и собранное твердое вещество промывали последовательно водным 2N HCl, водным 2N NaOH, эфиром и сушили: выход 180 мг, 68%; ТРМС 336/338
1H ЯМР (ДМСО-d6) δ 11,84 (шир, 1Н), 8,68 (шир, 1Н), 7,71 (d, 1Н), 7,43 (д, 1Н), 7,17 (дд, 1Н), 7,14 (с, 1Н), 4,80 (шир, 1Н), 4,15 (м, 2Н), 3,91 (м, 1Н), 3,72 (м, 2Н), 3,20 (м, 1Н), 3,05 (м, 1Н), 1,75 (м, 2Н), 1,48 (м, 1Н), 1,38 (м, 1Н).
Пример 8.
5-хлор-1Н-индол-2-карбоновой кислоты [1-бензил-2-(3-гидроксипирролидин-1-ил)-2-оксоэтил]-амид
Рацемический 3-пирролидинол (2,0 ммоля) и 2-[(5-xлop-1H- индол-2-карбонил)-амино] -3-фенилпропионовую кислоту (1 ммоль) соединяли по процедуре А (0-25oC - температура реакции, промывание сначала кислотой, затем основанием), и продукт очищали путем колоночной хроматографии на силикагеле с элюированием 0,5-16% этанолом в дихлорметане с получением бесцветной пены: выход 260 мг, 63%; ВЭЖХ (60/40) 100%, 3,86 минуты; PBMS 412/414 (NH+, 100%);
Анал. Расчетное для C22H22ClN3O3 + 0,2H2O: C, 63,60; H, 5,43, N, 10,11.
Обнаруженное: C, 63,90; H, 5,93; N, 10,11.
Пример 9,
5-хлор-1Н-индол-2-карбоновой кислоты (1-диэтилкарбамоил-2-фенил-этил)-амид
Диэтиламин (1,2 ммоля) и 2-[(5-хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовую кислоту (0,6 ммоля) соединяли по процедуре А (0-25oC в течение 5 дней) с изменением следующей обработки: неочищенный продукт суспендировали в хлороформе/дихлорметане 1:1, обрабатывали ультразвуком, отфильтровывали для удаления твердых веществ, концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле с элюированием 10, 20 и 30% этилацетатом в гексанах: выход 14 мг, 6%; ВЭЖХ (60/40) 8,88 минут (98%); PBMS 398/400 (МН+, 100%);
1H ЯМР (CDCl3) δ 9,31 (шир, 1Н), 7,61 (д, 1Н), 7,32 (д, 1Н, J= 8,7 Гц), 7,28-7,18 (м, 7Н), 6,87 (д, 1H, J=1,4 Гц), 5,26 (м, 1Н), 3,6 (м, 1-1, 5Н), 3,2-2,9 (м, 4,5-5Н), 1,07 (т, 3Н, J =7,2 Гц), 1,02 (т, 3Н, J=7,2 Гц).
Анал. Расчетное для C22H24ClN3O2 + 0,25 H2O: C 65,66; H, 6,14; N 10,44. Обнаруженное: C, 65,61; H, 6,20; N, 10,11.
Пример 10.
4-{ 2-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3-фенилпропионил} - пиперазин-1-карбоновой кислоты терт-бутиловый эфир
т-Бутиловый эфир 1-Пиперазинкарбоновой кислоты (1,2 ммоля) и 2 [(5-хлор-1Н-индол-2-карбонил)-амино] -3-фенилпропионовую кислоту (0,6 ммоля) соединяли по процедуре А (0-25oC - температура реакции, время реакции - 4 дня, экстрагирование сначала кислотой, затем основанием) и продукт-сырец очищали с помощью колоночной хроматографии на силикагеле с элюированием 30% этилацетатом в гексанах с получением бесцветной пены: выход 290 мг, 95%; ВЭЖХ (70/30) 6,23 мин (99%); PBMS 512/514 (МН+, 100%);
1H ЯМР (CDCl3) δ 9,32 (шир, 1Н), 7,60 (д, 1H, J=1,9 Гц), 7,32 (д, 1Н, J= 8,7 Гц), 7,3-7,15 (м,  7Н), 6,87 (д, 1Н, J=1,5 Гц), 5,33 (м, 1Н), 3,65-2,9 (перекрывание м, 9Н), 2,70 (м, 1Н), 1,43 (с, 9Н).
7Н), 6,87 (д, 1Н, J=1,5 Гц), 5,33 (м, 1Н), 3,65-2,9 (перекрывание м, 9Н), 2,70 (м, 1Н), 1,43 (с, 9Н).
Анал. Расчетное для C27H31ClN4O4: C, 63,46; H,6,11; N, 10,96.
Обнаруженное: C, 63,33; H, 5,97; N, 10,97.
Пример 11.
5-Хлор-1Н-индол-2-карбоновой кислоты [1-бензил-2-(4-метиламинопиперидин-1-ил)-2-оксоэтил]амид
Диметиламина гидрохлорид (1,1 ммоля), ацетат натрия (2,1 ммоля), активированное 3  молекулярное сито и цианоборогидрид натрия (0,25 ммоля) добавляли в этом порядке к 5-хлор-1Н-индол-2-карбоновой кислоты [1-бензил-2-оксо-2-(4-оксопиперидин-1-ил)-этил] амиду (0,21 ммоля) в метаноле (2 мл) при 0oC. Через 18 часов смесь концентрировали, остаток собирали в этилацетат, полученный в результате раствор промывали 2N NaOH и солевым раствором, сушили Na2SO4 и концентрировали. Продукт очищали хроматографией на силикагеле с элюированием 1-8% этанолом в дихлорметане, содержащем 0,5% NH4OH, с последующим растиранием в порошок с эфиром: выход 82%; ВЭЖХ (60/40) 2,79 минуты (98%); PBMS 439/441 (МН+, 100%);
молекулярное сито и цианоборогидрид натрия (0,25 ммоля) добавляли в этом порядке к 5-хлор-1Н-индол-2-карбоновой кислоты [1-бензил-2-оксо-2-(4-оксопиперидин-1-ил)-этил] амиду (0,21 ммоля) в метаноле (2 мл) при 0oC. Через 18 часов смесь концентрировали, остаток собирали в этилацетат, полученный в результате раствор промывали 2N NaOH и солевым раствором, сушили Na2SO4 и концентрировали. Продукт очищали хроматографией на силикагеле с элюированием 1-8% этанолом в дихлорметане, содержащем 0,5% NH4OH, с последующим растиранием в порошок с эфиром: выход 82%; ВЭЖХ (60/40) 2,79 минуты (98%); PBMS 439/441 (МН+, 100%);
1H ЯМР (ДМСО-d6) δ 11,75 (шир, 1Н), 8,94 (д, 0,5Н), J=8,8 Гц), 8,90 (д, 0,5Н), 7,71 (д, 1Н, J=1,8 Гц), 7,40 (д, 1Н, J=8,7 Гц), 7,31-7,20 (м, 6-7Н), 7,17 (дд, 1Н, J=2,1, 8,7 Гц), 5,15 (м, 1H), 4,22 (м, 0,5Н), 4,08 (м, 0,5Н), 3,96 (м, 0,5Н), 3,85 (м, 0,5Н), 3,2-2,9 (м, 4Н), 2,78 (м, 0,5Н), 2,72 (м, 0,5Н), 2,25 (с, 1,5Н), 2,24 (с, 1,5Н), 1,75 (м, 2Н), 1,3-0,8 (м, 2Н).
Анал. Расчетное для C24H27ClN4O2 + 1,0 H2О: C, 63,08; H, 6,40; N, 12,26.
Обнаруженное: C, 63,18; H, 6,16; N, 12,46.
Пример 12.
5-Хлор-1Н-индол-2-карбоновой кислоты (1-бензил-2-морфолин-4-ил-2-оксоэтил)-амид
Морфолин (0,33 ммоля) и 2-[(5-хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовую кислоту (0,30 ммоля) соединяли по процедуре А (температура реакции 0-25oC, 48 часов - время реакции). Продукт-сырец хроматографировали на силикагеле с элюированием этилацетатом/гексанами 1:1, желаемые фракции концентрировали и остаток растворяли в хлороформе и метаноле, и полученный в результате раствор перемешивали 18 часов с примерно 128 мг диметиламинопиридинполистирольной смолы (Fluka Chemical Co.). Раствор фильтровали, концентрировали и остаток растирали с эфиром: выход 51%; ВЭЖХ (60/40) 5,92 мин (98%).
PBMS 412/414 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,75 (шир, 1H), 8,95 (д, 1Н), 7,72 (д, 1H), 7,39 (д, 1H, J=8,7 Гц), 7,35-7,15 (м, 7Н), 5,13 (м, 1H), 3,65-3,10 (м, 8Н), 3,05 (м, 2Н).
Анал. Расчетное для C22H22ClN3O3 + 0,33 H2O: C, 63,23; H, 5,47; N, 10,06. Обнаруженное: C, 63,28; H, 5,32; N, 10,10.
Пример 13.
5-хлор-1Н-индол-2-карбоновой кислоты (1-бутилкарбамоил-2- фенилэтил)-амид
н-Бутиламин (0,66 ммоля) и 2-[(5-хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовую кислоту (0,60 ммоля) соединяли в соответствии с процедурой А (температура реакции 0-25oC). Продукт-сырец растворяли в хлороформе и метаноле, и полученный в результате раствор перемешивали 18 часов с 50 мг диметиламинопиридинполистирольной смолы (Fluka Chemical Co.), раствор фильтровали, концентрировали и твердые вещества растирали в порошок с эфиром: выход 83%; ВЭЖХ (60/40) 8,88 минут (92%); т. пл. 192-193oC; ТРМС 398/400 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,71 (шир, 1Н), 8,70 (д, 1H, J=8,3 Гц), 8,10 (т, 1Н), 7,72 (д, 1Н, J=2,0 Гц), 7,39 (д, 1Н, J =8,7 Гц), 7,35-7,15 (м, 7Н), 4,70 (м, 1Н), 3,13-2,93 (м, 4Н), 1,38 (м, 2Н), 0,86 (т, 3Н, J=7,2 Гц);
Анал. Расчетное для C22H24ClN3N3O2: C, 66,41; H, 6,08; N, 10,56.
Обнаруженное: C, 65,15; H, 6,04; N, 10,52.
Пример 14.
5-Хлор-1Н-индол-2-карбоновой кислоты [1-бензил-2-оксо-2- (4-оксопиперидин-1-ил)-этил]-амид
4-Пиперидон моногидрат (2,0 ммоля) и 2-[(5-хлор-1Н-индол- 2-карбонил)-амино] -3-фенилпропионовую кислоту (1,0 ммоль) соединяли в соответствии с процедурой А (температура реакции 0-25oC) с изменением следующей обработки: реакционную смесь разводили этилацетатом, полученный в результате раствор промывали 2N NaOH и 2N HCl, суспензию фильтровали и твердое вещество высушивали: выход 111 мг, 26%; ВЭЖХ (60/40) 8,88 минут (92%); PBMS 424/426 (MH+, 100%); т.пл. 258-261oC; PBMS 424/426 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,75 (шир, 1Н), 9,03 (д, 1H, J=8,1 Гц), 7,72 (д, 1H, J=1,9 Гц), 7,39 (д, 1H, J=8,7 Гц), 7,4-7,15 (м, 7Н), 5,20 (м, 1H, J=8,2 Гц), 3,88 (м, 1H), 3,73 (м, 3Н), 3,1 (м, 3Н), 2,5-2,22 (м, 3Н), 2,05 (м, 1H).
Анал. Расчетное для C23H22ClN3O3 + 0,75H2O: C 63,16; H, 5,42; N 9,61.
Обнаруженное: C, 63,11; H, 5,15; N, 9,53.
Пример 15.
5-Хлор-1Н-индол-2-карбоновой кислоты (1-бензил-2-оксо-2- пирролидин-1-илэтил)-амид
Пирролидин (0,35 ммоля) и 2-[(5-хлор-1Н-индол-2-карбонил)- амино]-3-фенилпропионовую кислоту (0,31 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 140 часов) и продукт-сырец растирали с эфиром: выход 89 мг, 71%; ВЭЖХ (70/30) 7,57 минут (98%); PBMS 396/398 (MH+, 100/80%);
Анал. Расчетное для C22H22ClN3O2 + 0,33H2O: C, 65,75; H, 5,68; N, 10,48.
Обнаруженное: C, 65,56; H, 5,81; N, 10,44.
Пример 16.
5-Хлор-1Н-индол-2-карбоновой кислоты [1-[(3-диметиламинопропил) метилкарбамоил]-2-фенилэтил}-амид
N, N,N'-Триметил-1,3-диаминопропан (0,31 ммоля) и 2-[(5-хлор- 1Н-индол-2-карбонил)амино] -3-фенилпропионовую кислоту (0,28 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 120 часов) и продукт очищали с помощью хроматографии на силикагеле с элюированием 1-8% этанолом в дихлорметане, содержащем 0,5% гидроксида аммония: выход 86 мг, 69%; ВЭЖХ (40/60) 7,57 минут (> 99%); т.пл. 187-190,5oC; ТРМС 441/443 (MH+, 100%);
Анал. Расчетное для C24H29ClN4O2 + 0,25 H2O: C, 64,71, H, 6,68; N, 12,58.
Обнаруженное: C, 64,73; H, 6,94; N, 12,86.
Пример 17.
5-Хлор-1Н-индол-2-карбоновой кислоты [1-(3-морфолин-4- ил-пропилкарбамоил)-2-фенилэтил]-амид
4-(3-Аминопропил)морфолин (0,34 ммоля) и 2-[(5-хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовую кислоту (0,30 ммоля) соединяли по процедуре А (температура реакции 0-25oC) с изменением следующей обработки: реакционную смесь разводили этилацетатом, полученный в результате раствор промывали три раза 2 N NaOH и один раз солевым раствором, сушили над Na2SO4 и концентрировали. Остаток перемешивали в атмосфере эфира в течение 1 часа, твердое вещество отфильтровывали и сушили: выход 125 мг, 87%; ВЭЖХ (60/40) 2,85 минуты (98%); PBMS 469/471 (MH+, 100/90%);
Анал. Расчетное для C25H29ClN4O3 + 0,25 H2O: C, 63,42, H, 6,28, N, 11,83.
Обнаруженное: C, 63,31; H, 6,57; N, 12,04.
Пример 18.
5-Хлор-1Н-индол-2-карбоновой кислоты (1-диметилкарбамоил-2-фенилэтил)-амид
Гидрохлорид диметиламина (0,96 ммоля) и 2 [(5-хлор-1Н- индол-2-карбонил)амино] -3-фенилпропионовую кислоту (0,90 ммоля) соединяли по процедуре А (температура реакции 0-25oC, 60 часов - время реакции, промывали сначала кислотой, затем основанием) и полученное в результате твердое вещество растирали с эфиром: выход 320 мг, 99%; ВЭЖХ (60/40), 5,87 минут (100%);
Т.пл. 224-225oC; PBMS 370/372 (M+, 100%).
Образец перекристаллизовывали из этилацетата для анализа (т.пл. 224-225oC).
Анал. Расчетное для C20H20ClN3O2 + 0,5 C4H8O2: C, 63,80; H, 5,84; N, 10,15.
Обнаруженное: C, 63,81; H, 5,80; N 10,21
Пример 19.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-((3R,4R)-дигидроксипирролидин-1-ил)-2-оксоэтил]-амид
(3R, 4R)-3,4-гидроксипирролидин (из неприродной винной кислоты путем процедуры, описанной в патенте США N"4634775, 1,0 ммоль) и [(5-хлор-1Н-индол-2-карбонил)-амино] уксусную кислоту (1,1 ммоля) соединяли по процедуре А (диметилформамид в качестве растворителя при реакции) с изменением следующей обработки: реакционную смесь концентрировали, разводили 20 мл этилацетата и 20 мл 2N NaOH, суспензию перемешивали 0,5 часа, фильтровали и полученные в результате твердые вещества промывали последовательно 2 N NaOH, водой, 1N HCl и этилацетатом: выход 77%; ВЭЖХ (40/60, 10,7 минут (99%); ТРМС 338/340 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,84 (шир, 1Н, обмены), 8,72 (т, 1Н, обмены), 7,72 (д, 1Н, J= 1,9 Гц), 7,44 (д, 1Н, J=8,7 Гц), 7,19 (дд, 1Н, J=2,1, 8,8 Гц), 7,16 (с, 1Н), 5,26 (д, 1Н, J=4,4 Гц, обмены), 5,17 (д, 1Н, J=3,2 Гц, обмены), 4,04 (м, 3Н), 3,92 (м, 1Н), 3,66 (дд, 1Н, 1=4,0, 10,8 Гц), 3,42-3,28 (м, 3Н);
Анал. Расчетное для C15H16ClN3O4 + 0,25 H2O: C, 52,64; H, 4,86; N, 12,28.
Обнаруженное: C, 52,61; H, 4,85; N, 12,23.
Пример 20.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-((3S, 4S,)-дигидроксипирролидин-1-ил)-2-оксоэтил]амид
(3S, 4S)-3,4-Дигидроксипирролидин (из встречающейся в природе винной кислоты путем процедуры, описанной в патенте США N 4634775, 1,0 ммоль) и [(5-хлор-1Н-индол-2-карбонил)-амино] -уксусную кислоту (1,0 ммоль) соединяли по процедуре А (растворитель для реакции - диметилформамид), заменяя следующей обработкой: реакционную смесь разводили этилацетатом и 2N NaOH, полученную в результате суспензию фильтровали и твердые вещества промывали этилацетатом, водой и сушили: выход 135 мг, 40%; ВЭЖХ (40/60) 7,29 минут (98%); ТРМС 338/340 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 12,1 (шир, 1Н), 8,86 (шир, 1Н), 7,71 (д, 1Н, J=2 Гц), 7,43 (д, 1Н, J=8,8 Гц), 7,17 (дд, 1Н, J=2, 8,8 Гц), 7,13 (с, 1Н), 5,35 (шир, 1Н, обмены с D2O), 5,28 (шир, 1Н, обмены с D2O), 4,03 (м, 3Н), 3,92 (с, 1Н), 3,66 (дд, 1Н, J=4,11 Гц), 3,4-3,2 (м, 3Н).
Анал. Расчетное для C15H16ClN3O4 + 1,5 H2O: C, 49,39; H, 5,25, N, 11,52.
Обнаруженное: C, 49,50; H, 5,04; N, 11,27.
Пример 21.
5-Хлор-1Н-индол-2-карбоновой кислоты [1-бензил-2- (4-метоксиметоксипиперидин-1-ил)-2-оксоэтил]-амид
4-Метоксиметоксипиперидин (1,0 ммоль) и 2-[(5-хлор-1Н-индол-2-карбонил)-амино] -3-фенилпропионовую кислоту (1,0 ммоль) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием этилацетатом-гексанами 1: 1; выход 241 мг, 50%; ВЭЖХ (60/40) 7,67 минуты (94%); PBMS 470/472 (MH+, 100%);
Анал. Расчетное для C25H28ClN3O4: C, 63,89; H, 6,01; N, 8,94.
Обнаруженное: C, 63,91; H, 6,00; N, 8,95.
Пример 22.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-фенил-1-(2,2,6,6-тетраметилпиперидин-4-илкарбамоил)-этил]-амид
2,2,6,6-Тетраметилпиперидин (1,0 ммоль) и 2-[(5-хлор-1Н- индол-2-карбонил)-амино] -3-фенилпропионовую кислоту (1,0 ммоль) соединяли по процедуре А. Полученную в результате желтую пену растирали с эфиром, дихлорметаном-эфиром 1:5. Полученное в результате твердое вещество растворяли в дихлорметане и полученный раствор обрабатывали 0,20 мл 4N HCl в диоксане. Образовавшийся осадок, который отфильтровывали, промывали дихлорметаном и сушили: выход 220 мг, 42%; ВЭЖХ (60/40), 3,19 минуты (96%); PBMS 481/483 (MH+, 100%);
Анал. Расчетное для C27H33ClN4O2 + HCl + 1,5 H2O: C, 59,56; H, 6,85; N, 10,29. Обнаруженное: C, 59,30; H, 6,90; N, 10,22.
Пример 23.
(1-{ 2-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3-фенилпропионил}- пирролидин-(3RS)-ил)карбаминовой кислоты трет-бутиловый эфир
Рацемический трет-бутиловый эфир пирролидин-3-карбаминовой кислоты (1,0 ммоль) и 2-[(5-хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовую кислоту (1,0 ммоль) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием этил-ацетатом-гексанами 1:1: выход 302 мг, 59%; PBMS 511/513 (MH+, 100%);
Анал. Расчетное для C27H31ClN4O4: C, 63,46; H, 6,11; N, 10,96.
Обнаруженное: C, 63,32; H, 6,26; N, 10,89.
Пример 24.
5-Хлор-1Н-индол-2-карбоновой кислоты (2-морфолин-4-ил-2-оксоэтил)-амид
Морфолин (1,0 ммоль) и [(5-хлор-1Н-индол-2-карбонил)-амино]-уксусную кислоту (1,0 ммоль) соединяли по процедуре А. Полученное в результате твердое вещество суспендировали в эфире, фильтровали и сушили с получением бежевого твердого вещества: выход 264 мг, 71%; ВЭЖХ (60/40) 3,28 минуты (100%); ТРМС 322/324 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,85 (с, 1Н), 8,68 (т, 1Н), 7,72 (д, 1Н, J=2,0 Гц), 7,43 (д, 1Н, J=8,8 Гц), 7,19 (дд, 1Н, J=2,1, 8,8 Гц), 7,16 (с, 1Н), 4,17 (д, 2Н, J=5,7 Гц), 3,65-3,45 (м, 8Н).
Анал. Расчетное для C15H16ClN3O3 + 0,25 H2O: C, 55,22; H, 5,10; N, 12,88.
Обнаруженное: C, 55,22; H, 5,08; N, 12,82.
Пример 25.
5-Хлор-1Н-индол-2-карбоновой кислоты [(метоксиметилкарбамоил) метил]-амид
Метоксиметиламин гидрохлорид (1,0 ммоль) и [(5-хлор-1Н-индол-2-карбонил)-амино] -уксусную кислоту (1,0 ммоль) соединяли по процедуре А. Полученное в результате твердое вещество суспендировали в эфире, отфильтровывали и сушили: выход 158 мг, 53%; PBMS 296/298 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,82 (с, 1Н), 8,77 (т, 1Н, J =6 Гц), 7,73 (д, 1H, J= 2,0 Гц), 7,43 (д, 1H, J=8,7 Гц), 7,19 (дд, 1Н, J=2,0, 8,7 Гц), 7,16 (s, 1Н), 4,22 (д, 2H, J=5,7 Гц), 3,76 (с, 3Н), 3,14 (с, 3Н).
Анал. Расчетное для C13H14ClN3O3: C, 52,80; H, 4,77; N, 14,21.
Обнаруженное: C, 52,51; H, 4,82; N, 14,01.
Пример 26.
5-Хлор-1Н-индол-2-карбоновой кислоты [1-бензил-2- (4-диметиламинопиперидин-1-ил)-2-оксоэтил]-амид
4-Диметиламинопиперидин (1,0 ммоль) и 2-[(5-хлор-1Н-индол-2-карбонил)-амино] -3-фенилпропионовую кислоту соединяли по процедуре А. Остаток очищали с помощью хроматографии на силикагеле с элюированием 5-30% этанолом в дихлорметане, содержащем 0,5% гидроксида аммония, с последующим растиранием с эфиром: выход 21 мг, 5%; PBMS 453/455 (MH+, 100%).
1H ЯМР (ДМСО-d6, частичный) δ 11,75 (шир, 1Н), 8,94 (м, 1Н), 7,72 (д, 1Н, J=2 Гц), 7,45-7,10 (м, 8Н), 5,17 (м, 1Н), 4,63 (м), 4,38 (м), 4,03 (м), 3,50 (м), 3,15-2,8 (м), 2,51 (с, 3Н), 2,50 (с, Н).
Пример 27.
5-Хлор-1Н-индол-2-карбоновой кислоты (1-бензил-2-оксо-2-пиперазин-1-илэтил)-амид
Трифторуксусную кислоту (4 мл) добавляли к 4-{2-[(5-Хлор- 1Н-индол-2-карбонил)-амино] -3-фенилпропионил} пиперазин-1-карбоновой кислоты трет-бутиловому эфиру (0,6 ммоля) при 0oC и полученный в результате раствор перемешивали в течение 0,3 часа и концентрировали. Остаток разделяли между этилацетатом и 2 N NaOH, органический слой отделяли и промывали солевым раствором, сушили Na2SO4, концентрировали и полученное в результате твердое вещество растирали с эфиром: выход 189 мг, 77%; ВЭЖХ (60/40) 2,63 минуты (99%); т.пл. 166,5-168oC; ТРМС 411/413 (MH+, 100%);
Анал. Расчетное для C22H23ClN4O2 + 0,5 H2O: C, 62,93; H, 5,76; N, 13,34.
Обнаруженное: C, 62,64; H, 5,52; N, 13,34.
Пример 28
5-Хлор-1Н-индол-2-карбоновой кислоты [2-((3RS)-аминопирролидин-1-ил)-1-бензил-2-оксоэтил]-амид
4N HCl в 1,4-диоксане (5 мл) добавляли к (1-{2-[(5-хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропионил} -пирролидин-(3RS-ил)-карбаминовой кислоты трет-бутиловому эфиру (0,5 ммоля). Полученный в результате раствор перемешивали при 25oC в течение 0,5 часа, концентрировали и остаток растирали с эфиром: выход 190 мг, 85%; ВЭЖХ (60/40) 2,62 минуты (98%); PBMS 411/413 (МН+, 100%);
Анал. Расчетное для C22H23ClN4O2 + HCl + 1,7 H2О: C, 55,28; H, 5,78; N, 11,72.
Обнаруженное: C, 55,14; H, 5,86; N, 11,45.
Пример 29.
1-{ (2RS)-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3-фенилпропионил} - пирролидин-(2S)-карбоновая кислота
Трифторуксусную кислоту добавляли к 1-{2-(RS)-[(5-хлор-1Н- индол-2-карбонил)-амино] -3-фенилпропионил} -пирролидин-(2S)-карбоновой кислоты трет-бутиловому эфиру (1,0 ммоль) при 25oC. Через 1,5 часа реакционную смесь концентрировали и остаток растирали сначала с эфиром, затем со смесью эфира и гексанов. Выход 360 мг, 82%; ВЭЖХ (60/40) 4,84 минуты (99%); PBMS 440/442 (MH+, 40%), 396/398 (МН-44, 100%);
Анал. Расчетное для C23H22ClN3O4 + 0,8 H2O: C, 60,81; H, 5,24; N, 9,25.
Обнаруженное: C, 60,74; H, 5,42; N, 8,96.
Пример 29а.
1-{ 2(R, S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропионил}- пирролидин-(2S)-карбоновой кислоты трет-бутиловый эфир
L-Пролин-т-бутиловый эфир (2,0 ммоля) и 2-[(5-хлор-1Н-индол-2- карбонил)-амино] -3-фенилпропионовую кислоту (2,0 ммоля) соединяли по процедуре А и продукт-сырец очищали с помощью хроматографии на силикагеле с элюированием этилацетатом-гексанами 1: 2: выход 611 мг, 62%; ВЭЖХ (60/40) 13,45 минуты (57%) и 14,46 минуты (41%).
Пример 30.
5-Хлор-1Н-индол-2-карбоновой кислоты ((1S)-метилкарбамоил-2-тиазол-4-илэтил)-амид
(S)-2-Амино-N-метил-3-тиазол-4-илпропионамида гидрохлорид (0,6 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,51 ммоля) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции - диметилформамид). Продукт-сырец перемешивали в эфире в течение 0,5 часа, затем фильтровали с получением бежевого твердого вещества: 182 мг, 98%; ВЭЖХ (60/40) 3,41 минуты (98%); т.пл. 260oC (разл.); ТРМС 363/365 (MH+, 100%);
1H ЯМР (ДМСО-D6) δ 11,82 (шир, 1H), 9,0 (д, 1H), 8,82 (шир, 1H), 8,10 (шир, 1H), 7,70 (м, 1Н), 7,44-7,38 (м, 2Н), 7,21-7,15 (м, 2Н), 4,80 (м, 1H), 3,24 (м, 1H), 3,05 (м, 1H), 2,60 (д, 3Н).
Пример 30а.
(S)-2-Амино-N-метил-3-тиазол-4-илпропионамида гидрохлорид
(S)-(1-Метилкарбамоил-2-тиазол-4-ил-этил)карбаминовой кислоты трет-бутиловый эфир (248 мг, 0,87 ммоля) растворяли в 4М HCl-диоксане при 0oC. Полученную в результате смесь перемешивали в течение 1 часа при 25oC, концентрировали и остаток растирали с эфиром. Выход 202 г, 102%; ВЭЖХ (70/30) 2,41 минуты (96%).
Пример 30b.
(S)-2-(N-т-Бутоксикарбониламино)-N-метил-3-тиазол-4-ил-пропионамид
Метиламина гидрохлорид (1,2 ммоля) и Boc-L-3-(4-тиазолил)-аланин (1,0 ммоля) соединяли по процедуре А (0-25oC - температура реакции, промывание кислотой исключается) и продукт использовали без дальнейшей очистки. Выход 250 мг, 88%.
Пример 31.
(±)-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3-гидроксипропионовой кислоты метиловый эфир
D, L-Серин-метилового эфира гидрохлорид (2,1 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (2,0 ммоля) соединяли по процедуре А (температура реакции 0-25oC, промывали сначала кислотой, затем насыщенным NaHCO3) и продукт очищали с помощью хроматографии на силикагеле с элюированием 10, 20, 40 и 60% этилацетатом в гексанах: выход 565 мг, 95%; ВЭЖХ (60/40) 3,46 минуты (98%); т.пл. 153-155oC; ТРМС 297/299 (MH+, 100/40%);
Анал. Рассчитано для C13H13ClN2O4: C, 52,62; H, 4,42; N, 9,44.
Обнаруженное: C, 52,62; H, 4,54; N, 9,53.
Пример 32
5-Хлор-1Н-индол-2-карбоновой кислоты ((1S)-диметилкарбамоил-2- тиазол-4-ил-этил)-амид
(S)-2-Амино-N,N-диметил-3-тиазол-4-илпропионамида гидрохлорид (0,43 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,40 ммоля) соединяли по процедуре А (температура реакции 0-25oC) и продукт-сырец растирали сначала с эфиром-гексанами 1: 1, затем с гексанами. Выход 115 мг, 75%; ВЭЖХ (60/40) 3,72 минуты (99%); т.пл. 198-202oC (сокращения на вставке при 192oC); PBMS 377/379 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,75 (с, 1Н), 9,02 (д, 1H, J=2 Гц), 8,9 (д, 1H, J= 8,2 Гц), 7,7 (д, 1H, J=1,8 Гц), 7,41 (д, 1Н, J=6,7 Гц), 7,39 (с, 1Н), 7,22 (с, 1Н), 7,17 (дд, 1H, J=2,2, 8,7 Гц), 5,30 (м, 1Н), 3,24 (дд, A из AB, 1H, J= 7,13 Гц), 3,16 (дд, B из AB, 1Н, J=8,5, 16 Гц), 3,07 (с, 3Н), 2,84 (с, 3Н).
Анал. Расчетное для C13H17ClN4O2S + 0,125 H2O: C, 53,86; H, 4,59; N, 14,78.
Обнаруженное: C, 53,92; H, 4,47; N, 14,42.
Пример 32а.
(S)-2-Амино-N,N-диметил-3-тиазолил-4-пропионамида гидрохлорид
(S)-(1-Диметилкарбамоил-2-тиазол-4-илэтил)-карбаминовой кислоты трет-бутиловый эфир растворяли в 4М HCl-диоксанах при 0oC и перемешивали при 25oC в течение 2 часов. Смесь концентрировали и остаток растирали с эфиром. Выход 3,06 г, 105%; ВЭЖХ (70/30), 2,12 минуты (97%); PBMS 200 (MH+, 100%).
Пример 32b.
(S)-(1-Диметилкарбамоил-2-тиазол-4-илэтил)-карбаминовой кислоты трет-бутиловый эфир
Диметиламина гидрохлорид (1,2 ммоля) и Вос-L-3-(4-тиазолил)-аланин (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC) и продукт очищали с помощью хроматографии на силикагеле с элюированием 1-16% этанолом в дихлорметане, содержащем 0,5% гидроксида аммония. Выход 124 мг, 41%.
Пример 33.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-((3R, 4S) -дигидроксипирролидин-1-ил)-2-оксоэтил]-амид
(3R, 4S)-Дигидроксипирролидина гидрохлорид (0,5 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,5 ммоля) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием 2-10% этанолом в дихлорметане. Выход 180 мг, 86%; ВЭЖХ (60/40) 3,14 минуты (98%); ТРМС 428/430 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,75 (шир, 1Н), 8,94 (д, 1Н, J=8 Гц), 7,72 (с, 1Н), 7,4-7,1 (м, 8Н), 5,03 (д, 0,5H, J=5 Гц), 4,95 (д, 0,5H, J=5 Гц), 4,90 (д, 1Н, J =5 Гц), 4,87 (м, 1Н), 4,08 (м, 0,5Н), 4,00 (м, 0,5Н), 3,88 (м, 1,5Н), 3,5-3,3 (м, 2,5Н), 3,2 (м, 0,5Н), 3,0 (м, 2Н).
Анал. Расчетное для C22H22ClN3O4 + 0,25 H2O: C, 61,11; H, 5,25; N, 9,72.
Обнаруженное: C, 60,91; H, 5,46; N, 9,43.
Пример 33а.
(цис-3,4)-Дигидроксипирролидина гидрохлорид
цис-3,4-Дигидроксипирролидин-1-карбоновой кислоты трет-бутиловый эфир (1,99 г, 9,8 ммоля) растворяли в 4М HCl-диоксане при 5oC и полученную в результате суспензию перемешивали при 25oC в течение 1 часа. Смесь концентрировали и остаток растирали с эфиром с получением легкого пурпурного порошка (1,30 г, 95%).
Пример 33b.
цис-3,4-Дигидроксипирролидин-1-карбоновой кислоты трет-бутиловый эфир
Раствор неочищенного 2,5-дигидропиррол-1-карбоновой кислоты трет-бутилового эфира (10,5 г, 62,1 ммоля) в тетрагидрофуране (300 мл) обрабатывали последовательно тетроксидом осмия (2,5% в т-бутаноле, 6 мл) и N-метилморфолин-N-оксидом при 25oC. Через 48 часов добавляли водный 10% раствор тиосульфата натрия и смесь перемешивали 30 минут, частично концентрировали для удаления тетрагидрофурана и полученную в результате водную смесь экстрагировали дважды эфиром. Эфирные экстракты промывали 10% тиосульфатом натрия, 0,1N HCl, сушили и концентрировали с получением темно-оранжевого масла, которое хроматографировали на двуокиси кремния с элюированием 1%, 2%, 4%, 8% и 10% этанолом-дихлорметаном с получением янтарного сиропа (4,09 г).
Пример 33с
2,5-Дигидропиррол-1-карбоновой кислоты трет-бутиловый эфир
ди-т-Бутилдикарбонат (83 г, 380 ммолей) добавляли к раствору 3-пирролина (содержащего 35% пирролидина, 25 г, 362 ммоля) в тетрагидрофуране (500 мл) при 0oC. Смесь перемешивали при 25oC в течение 1 часа и концентрировали с получением 76,2 г желтого масла, которое использовали без очистки.
Пример 34.
(3S)-[(5-Хлор-1Н-индол-2-карбонил)амино] -4-(4-гидроксипиперидин- 1-ил)-4-оксомасляной кислоты трет-бутиловый эфир
(S)-3-Амино-4-(4-гидроксипиперидин-1-ил)-4-оксомасляной кислоты трет-бутиловый эфир (0,8 ммоля) и 5-хлор-1Н-индол-2-карбоновой кислоты (0,8 ммоля) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием 25, 40, 50, 75 и 100% этилацетатом в гексанах. Выход 330 мг, 94%; ВЭЖХ (60/40), 4,18 минуты (97%); ТРМС 450/452 (MH+, 100%).
Пример 34а.
(S)-3-Амин-4-(4-гидроксипиперидин-1-ил)-4-оксомасляной кислоты трет-бутиловый эфир
Диэтиламин (1,0 ммоль) добавляли к (S)-3-(9Н-флюорен-9- илметоксикарбониламино)-4-(4-гидроксипиперидин-1-ил)-4-оксомасляной кислоты трет-бутиловому эфиру в диметилформамиде (5 мл) при 25oC. Через 1 час реакционную смесь концентрировали, остаток суспендировали в эфире/дихлорметане 1:1, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле с элюированием 1-50% этанолом в дихлорметане, содержащем 0,5% гидроксида аммония. Выход 217 мг, 80%.
Пример 34b.
(S)-3-(9Н-Флуорен-9-илметоксикарбониламино)-4-(4- гидроксипиперидин-1-ил)-4-оксомасляная кислота
4-Гидроксипиперидин (2,1 ммоля) и N-FMOC-L-аспарагиновой кислоты β- т-бутиловый эфир (2,0 ммоля) соединяли по процедуре А (время реакции 96 часов, промывали только кислотой) и продукт очищали с помощью хроматографии на силикагеле с элюированием 1-4% этанолом в дихлорметане. Выход 516 мг, 52%: ВЭЖХ (60/40) 5,33 минуты (93%).
Пример 35.
5-Хлор-1Н-индод-2-карбоновой кислоты [(1R)-бензил-2-(4- гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(R)-2-Амино-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-она гидрохлорид (3,1 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (3,4 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции 60 часов) и продукт очищали с помощью хроматографии на силикагеле с элюированием 50,75 и 100% этилацетатом в гексанах с последующим растиранием с эфиром-гексанами 1:1. Выход 1,1 г, 84%; ВЭЖХ (60/40) 4,06 минуты (99%); PBMS 426/428 (MH+, 100%);
Анал. Расчетное для C23H24ClN3O3 + 0,25 H2O: C, 64,18; H, 5,74; N, 9,76.
Обнаруженное: C, 64,28; H, 5,94; N, 9,41.
Пример 35а.
(R)-2-Амино-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-она гидрохлорид
(R)-2-(N-т-Бутоксикарбониламино)-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-он (12,5 ммолей) растворяли в 4М HCl-диоксане при 0oC и полученную суспензию перемешивали при 25oC в течение 1 часа. Смесь концентрировали и остаток растирали с эфиром. Выход 3,44 г, 97%.
Пример 35b.
(R)-2-(N-т-Бутоксикарбониламино)-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-он
(R)-2-(N-т-Бутоксикарбокиламино)-3-фенилпропан-1-он (14 ммолей) и 4-гидроксипиперидин (21,5 ммоль) соединяли по процедуре А (температура реакции 0-25oC, промывали сначала кислотой, затем основанием) и продукт использовали без дополнительной очистки. Выход 4,7 г, 94%; ВЭЖХ (60/40) 3,52 минуты (98%).
Пример 36.
1Н-Индол-2-карбоновой кислоты [2-(1,1-диоксо-1-тиазолидин-3-ил)- 2-оксоэтил]-амид
2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-этанона гидрохлорид (1,0 ммоль) и 1Н-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC, время реакции 60 часов) со следующей обработкой: реакционную смесь разводили этилацетатом и 2N NaOH, полученный в результате осадок собирали и промывали 2N NaOH, 1N HCl и водой. Выход 135 мг, 42%; ВЭЖХ (60/40) 2,97 минуты (97%); PBMS 322 (MH+, 100%);
Анал. Расчетное для C14H15N3O4 + 0,25 H2O: C, 51,60; H, 4,79; N, 12,90.
Обнаруженное: C, 51,31; H,4,66; N, 12,88.
Пример З6а.
2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-этанона гидрохлорид
[2-(1,1-диоксо-1-Тиазолидин-3-ил)-2-оксоэтил] карбаминовой кислоты трет-бутиловый эфир (11 ммолей) растворяли в 4М HCl-диоксане при 0oC и полученную в результате суспензию перемешивали при 25oC в течение 1 часа. Смесь концентрировали и остаток растирали с эфиром. Выход 2,3 г, 100%.
Пример 36.
[2-(1,1-Диоксо-1-тиазолидин-3-ил)-2-оксоэтил]-карбаминовой кислоты трет-бутиловый эфир
м-Хлорпероксибензойную кислоту (35 ммолей) добавляли медленно к (2-оксо-2-тиазолидин-3-илэтил)-карбаминовой кислоты трет-бутиловому эфиру (14 ммолей) в дихлорметане (35 мл) при 0oC. После того как вспенивание стихало, смесь перемешивали дополнительно еще 2,5 часа при 25oC. Смесь разбавляли этилацетатом, полученный в результате раствор промывали три раза смесью 1:1 насыщенного водного NaHCO3 и 10% водного раствора NaS2O3, один раз насыщенным NaHCO3, сушили, концентрировали и остаток растирали с эфиром-гексанами 1:1. Выход 3,6 г, 92%.
Пример 36с.
(2-оксо-2-Тиазолидин-3-илэтил)-карбаминовой кислоты трет-бутиловый эфир
Тиазолидин (85 ммолей) и Boc-глицин (57 ммолей) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 60 ч) и продукт использовали без очистки. Выход 12,7 г, 90%.
Пример 37г.
5-Фтор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(4- гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(S)-2-Амино-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-она гидрохлорид (0,65 ммоля) и 5-фтор-1Н-индол-2-карбоновую кислоту (0,73 ммоля) соединяли по процедуре А (температура реакции 0-25oC) и продукт очищали с помощью хроматографии на силикагеле с элюированием 20, 30, 40, 50, 75 и 100% этилацетатом в гексанах. Выход 228 мг, 84%; ВЭЖХ (60/40) 3,57 минуты (98%); PBMS 410 (MH+, 100%);
Анал. Расчетное для C23H24FN3O3 + 0,25 H2O: C, 66,73; H, 5,97; N, 10,15. Обнаруженное: C, 66,68; H, 6,19; N, 9,94.
Пример 38.
1Н-Индол-2-карбоновой кислоты [(1S)-бензил-2-(4- гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(S)-2-Амино-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-она гидрохлорид (3,4 ммоля) и 1Н-индол-2-карбоновую кислоту (3,7 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 48 часов). Продукт очищали с помощью хроматографии на силикагеле с элюированием 50, 75 и 100% этилацетатом в гексанах с последующим растиранием с эфиром-гексанами 1:1. Выход 1,14 г, 86%; ВЭЖХ (60/40) 3,52 минуты (98%); PBMS (MH+, 100%);
Анал. Расчетное для C23H25N3O3 + 0,25 H2O: C, 69,77; H, 6,49; N, 10,61.
Обнаруженное: C, 69,99; H, 6,72; N, 10,47.
Пример 39.
5-Фтор-1Н-индол-2-карбоновой кислоты [(1S)-(4-фторбензил)-2-морфолин-4-ил-2-оксоэтил]-амид
(S)-2-Амино-3-(4-фторфенил)-1-морфолин-4-илпропан-1-она гидрохлорид (0,48 ммоля) и 5-фтор-1Н-индол-2-карбоновую кислоту (0,48 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 48 часов, промывали сначала кислотой, затем основанием) и продукт очищали путем хроматографии на силикагеле с элюированием 20, 30, 40, 50 и 75% этилацетатом в гексанах. Выход 189 мг, 95%; ВЭЖХ (60/40) 4,76 минуты (97%); PBMS 414 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,23 (шир, 1Н), 7,4-7,1 (м, 5Н), 7,1-6,94 (м, 3Н), 6,9 (д, 1H, J= 2 Гц), 5,30 (м, 1Н), 3,72-3,48 (м, 5Н), 3,42 (м, 1Н), 3,03 (м, 4Н).
Анал. Расчетное для C22H21F2N3O3: C, 63,92; H, 5,12; N, 10,16.
Обнаруженное: C, 64,30; H, 5,34; N, 9,82.
Пример 39а.
(S)-2-Амино-3-(4-фторфенил)-1-морфолин-4-илпропан-1-она гидрохлорид
(S)-2-(N-т-Бутоксикарбониламино)-3-(4-фторфенил)-1-морфолин-4- илпропан-1-он (3,1 ммоля) растворяли в 4М HCl-диоксане при 0oC и полученную в результате суспензию перемешивали при 25oC в течение 1 часа. Смесь концентрировали и остаток растирали с эфиром. Выход 776 мг, 88%; ВЭЖХ (60/40) 2,31 минуты (99%).
Пример 39b.
(S)-2-(N-т-Бутоксикарбониламино)-3-(4-фторфенил)-1-морфолин-4-ил-пропан- 1-он
Морфолин (3,7 ммоля) и (S)-Boc-4-фторфенилаланин (3,5 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции 60 часов, промывали сначала кислотой, затем основанием) и продукт очищали с помощью хроматографии на силикагеле с элюированием 20, 30 и 40% этилацетатом в гексанах. Выход 1,08 г масла, 87%.
Пример 40.
5-Фтор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2- (1,1-диоксо-1-тиазолидин-3-ил)-2-оксоэтил]-амид
(S)-2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-3-фенилпропан-1-она гидрохлорид (1,0 ммоль) и 5-фтор-1Н-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC, время реакции 48 часов, промывали сначала кислотой, затем основанием) и продукт очищали с помощью хроматографии на силикагеле с элюированием 20, 30, 40 и 50% этилацетатом в гексанах. Выход 404 мг, 94%; ВЭЖХ (60/40) 4,74 мин (98%); PBMS 430 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,53 (шир, 0,5Н), 9,44 (шир, 0,5Н), 7,44 (д, 0,5H, J=9 Гц), 7,4-7,1 (м, 7Н), 7,02 (м, 1Н), 6,84 (с, 0,5Н), 6,81 (с, 0,5Н), 5,20 (м, 0,5Н), 4,96 (м, 0,5Н), 4,68 (д, 0,5H, J=11 Гц), 4,52 (д, A из AB, 0,5H, J= 11,5 Гц), 4,37 (д, B из AB, 0,5H, J=11,5 Гц), 4,20 (м, 0,5Н), 4,03 (м, 0,5Н), 3,80 (м, 0,5Н), 3,50 (д, 0,5H, J=11 Гц), 3,3-3,0 (м, 4Н), 2,69 (м, 0,5Н).
Пример 40а.
(S)-2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-3-фенилпропан-1-она гидрохлорид
(S)-2-(N-т-Бутоксикарбониламино)-1-(1,1-диоксо-1-тиазолидин-3-ил)-3- фенилпропан-1-он растворяли в 4М HCl-диоксанах при 0oC. Раствор перемешивали при 25oC в течение 1 часа, концентрировали и остаток растирали с эфиром. Выход 8 мг, 84%.
Пример 40b.
(S)-2-(N-т-Бутоксикарбониламино)-1-(1,1-диоксо-1-тиазолидин-3-ил)-3- фенилпропан-1-он
Раствор м-хлорпероксибензойной кислоты (9 ммолей) и (S)-(1-бензил-2-оксо-2-тиазолидин-3-ил-этил)-карбаминовой кислоты трет-бутилового эфира (3 ммоля) в дихлорметане (9 мл) нагревали в колбе с обратным холодильником в течение 6 часов. Смесь разбавляли этилацетатом, полученный в результате раствор промывали три раза смесью 1: 1 10% водного NaS2O3 и насыщенного водного NaHCO3, сушили и концентрировали. Полученную в результате пену очищали с помощью хроматографии на силикагеле с элюированием 20, 30 и 40% этилацетатом в гексанах, с получением бесцветной пены (979 мг, 89% выход).
Пример 40с.
(S)-(1-Бензил-2-оксо-2-тиазолидин-3-ил-этил)-карбаминовой кислоты трет-бутиловый эфир
Тиазолидин (38 ммолей) и Boc-L-фенилаланин (19 ммолей) соединяли по процедуре А (температура реакции 0-25oC, промывали кислотой, затем основанием) и продукт использовали без дополнительной очистки. Выход 5,5 г, 86%.
Пример 41.
5-Фтор-1Н-индол-2-карбоновой кислоты [2-(1,1-диоксо-1- тиазолидин-3-ил)-2-оксоэтил]-амид
2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-этанона гидрохлорид (1,0 ммоль) и 5-фтор-1Н-индол-2-карбоновую кислоту соединяли по процедуре А (температура реакции 0-25oC, время реакции 48 часов) со следующей обработкой. Реакционную смесь разводили этилацетатом и 1N HCl, полученную в результате суспензию фильтровали и собранное твердое вещество промывали 2N HCl, 2N NaOH и водой. Отфильтрованное твердое вещество кипятили в ацетоне, отфильтровывали и сушили. Выход 134 мг, 40%; ВЭЖХ (60/40) 3,06 минуты (97%); т.пл. 239-241oC (с разложением); PBMS 340 (М+, 70%), 357 (100%)
1H ЯМР (ДМСО-d6) δ 11,74 (с, 1Н), 8,82 (м, 1Н), 7,43 (м, 2Н), 7,17 (с, 1Н), 7,05 (д, 1H, J=3,9 Гц), 4,86 (с, 1,2Н), 4,52 (с, 0,8Н), 4,27 (д, 0,8H, J= 5,5 Гц), 4,13 (д, 1,2H, J=6 Гц, наложение на м, 1,2Н), 3,86 (т, 1,2H, J= 7,4 Гц), 3,58 (т, 0,8H, J=7 Гц), 3,46 (т, 1,2H, J=7,2 Гц).
Анал. Расчетное для C14H14FN3О4 + 0,6 H2O: C, 48,02; H, 4,38; N, 12,00.
Обнаруженное: C, 47,99; H, 4,04; N, 12,00.
Пример 42.
5-Циано-1H-индол-2-карбоновой кислоты ((1S)-бензил-2-оксо-2- тиазолидин-3-ил-этил)-амид
(S)-2-Амино-3-фенил-1-тиазолидин-3-илпропан-1-она гидрохлорид (4,0 ммоля) и 5-циано-1Н-индол-2-карбоновую кислоту (4 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 48 часов) со следующей обработкой; реакционную смесь разводили этилацетатом и 2N HCl, полученный в результате осадок собирали фильтрованием, промывали 2N HCl и 2N NaOH. Продукт-сырец очищали с помощью хроматографии на силикагеле с элюированием 30, 40 и 50% этилацетатом в гексанах. Выход 1,22 г, 75%. ВЭЖХ (60/40) 4,74 минуты (97%); PBMS 405 (NH+, 100%);
Анал. Расчетное для C22H20N4O2S + 0,5 H2O: C, 63,90; H, 5,12; N, 13,55.
Обнаруженное: C, 64,18; H, 5,04; N, 13,47.
Пример 42а.
(S)-2-Акино-3-фенил-1-тиазолидин-3-илпропан-1-она гидрохлорид
(S)-(1-Бензил-2-оксо-2-тиазолидин-3-илэтил)-карбаминовой кислоты трет-бутиловый эфир (16 ммолей) растворяли в 4М HCl-диоксанах при 0oC, раствор перемешивали при 25oC в течение 1 часа, реакционную смесь концентрировали и остаток растирали с эфиром. Выход 4,2 г, 95%.
Пример 42b.
5-Циано-1Н-индол-2-карбоновая кислота
5-Циано-1Н-индол-2-карбоновой кислоты этиловый эфир (1,71 г, 8,0 ммолей) добавляли к раствору этанола (10 мл) и гидроксида калия (2 г) и полученную в результате смесь нагревали в колбе с обратным холодильником в течение 1 часа. Для растворения осадка добавляли воду и 6N HCl добавляли для доведения pH до 1. Образовывался осадок. Смесь охлаждали на ледяной бане, фильтровали и полученное в результате бесцветное твердое вещество промывали холодной водой и сушили (1,51 г). Часть (1,4 г) суспендировали в горячей уксусной кислоте (40 мл) и охлаждали с получением твердого вещества, которое отфильтровывали, промывали холодным этилацетатом и сушили: выход 980 мг, (70%); ВЭЖХ (60/40) 3,09 минуты (97%).
Пример 43.
1Н-Индол-2-карбоновой кислоты [(1S)-бензил-2-(1,1-диоксо-1- тиазолидин-3-ил)-2-оксоэтил]-амид
(S)-2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-3-фенилпропан- 1-она гидрохлорид (0,56 ммоля) и 1Н-индол-2-карбоновую кислоту (0,56 ммоля) соединяли по процедуре А (температура реакции 0-25oC) и продукт растирали с эфиром-гексанами 1: 1. Выход 213 мг, 92%; ВЭЖХ (60/40) 4,15 минуты (99%); PBMS 412 (MH+, 100%);
Анал. Расчетное для C21H21N3O4O + 0,5 H2O: C, 59,99; H, 5,27; N, 9,99.
Обнаруженное: C, 60,25; H, 5,27; N, 9,98.
Пример 44.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-(1,1-диоксо-1-тиазолидин-3-ил)-2-оксоэтил]-амид
2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-этанона гидрохлорид (0,6 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,6 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции 120 часов) со следующей обработкой: реакционную смесь разводили этилацетатом и 2N HCl, полученный в результате осадок собирали фильтрованием с последующим промыванием 2N HCl, 2N NaOH, водой и эфиром. Выход 110 мг, 52%; ВЭЖХ (60/40) 3,37 минуты (99%); т. пл. 236-239oC (разл.); PBMS 356/358 (MH+, 100%);
1H ЯМР (ацетон-d6) δ 11,0 (шир, 1Н), 8,0 (шир, 1Н), 7,66 (д, 1Н, J=2 Гц), 7,55 (д, 1H, J=8,7 Гц), 7,21 (дд, 1Н, J=2,0, 8,7 Гц), 7,15 (д, 1Н, J=2 Гц), 4,77 (с, 1,1Н), 4,49 (с, 0,9Н), 4,37 (д, 0,9H, J=5,3 Гц), 4,27 (д,  1H, J= 5,3 Гц, наложенный на м,
1H, J= 5,3 Гц, наложенный на м,  1Н), 4,04 (т, 1,1H, J=7 Гц), 3,54 (т, 0,9H, J=7 Гц), 3,40 (т, 1,1H, J=7 Гц).
1Н), 4,04 (т, 1,1H, J=7 Гц), 3,54 (т, 0,9H, J=7 Гц), 3,40 (т, 1,1H, J=7 Гц).
Анал. Расчетное для C14H14ClN3O4S + 1,6 H2O : C, 43,72; H, 4,51; N, 10,93.
Обнаруженное: C, 44,05; H, 3,88; N, 10,99.
Пример 45.
5-Хлор-1Н-индол-2-карбоновой кислоты (2-оксо-2-тиазолидин-3-ил-этил)-амид
2-Амино-1-тиазолидин-3-илэтанона гидрохлорид (3,1 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (3,4 ммоля) соединяли по процедуре А (0-25oC, время реакции 120 часов), заменяя следующую обработку: реакционную смесь перемешивали с этилацетатом и 2N HCl, фильтровали, и отфильтрованное твердое вещество промывали 2N HCl, 2N NaOH и эфиром. Выход 988 мг, 98%; ВЭЖХ (70/30) 3,25 минуты (99%); т. пл. 253-255oC (разл., потемнение при 243oC); PBMS 324/326 (MH+, 100%);
1H ЯМР (ацетон-d6) δ 11,03 (шир, 1Н), 7,88 (шир, 1Н), 7,66 (д, 1H, J=2 Гц), 7,54 (д, 1Н, J=8,3 Гц), 7,21 (дд, 1Н, J=2, 8, 3 Гц), 4,67 (с, 0,8Н), 4,53 (с, 1,2Н), 4,24 (м, 2Н), 3,87 (т, 1,2H, J=7 Гц), 3,78 (т, 0,8H, J=7 Гц), 3,18 (т, 1,2H, J=7 Гц), 3,05 (т, 0,8H, J=7 Гц).
Образец перекристаллизовывали из уксусной кислоты для анализа (т.пл. 262-264oC):
Анал. Расчетное для C14H14ClN3O2S: C, 51,93; H, 4,36; N, 12,98.
Обнаруженное: C, 52,78, H, 4,38; N, 12,95.
Пример 45а.
2-Амино-1-тиазолидин-3-илэтанона гидрохлорид
(2-Оксо-2-тиазолидин-3-ил-этил)карбаминовой кислоты трет-бутиловый эфир (5,41 г, 22 ммоля) растворяли в 4М HCl-диоксане (80 мл) при 0oC. Полученный в результате раствор перемешивали при 25oC в течение 2 часов, концентрировали и остаток растирали с эфиром. Выход 3,9 г, 97%.
Пример 46.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(4- гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(S)-2-Амино-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-она гидрохлорид (0,8 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,9 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции 48 часов) и продукт очищали с помощью хроматографии на силикагеле с элюированием 50, 75 и 100% этилацетатом в гексанах с последующим растиранием с эфиром-гексанами 1:1. Выход 266 мг, 76%; ВЭЖХ (60/40) 4,09 минуты (99%); PBMS 426/428 (MH+, 100%);
Анал. Расчетное для C23H24ClN3O3 + 0,33 H2O: C, 63,96; H, 5,76; N 9,73.
Обнаруженное: C, 63,90; H, 5,74; N, 9,58.
Пример 46а.
(S)-2-Амино-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-она гидрохлорид
(S)-[1-Бензил-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил] -карбаминовой кислоты трет-бутиловый эфир (3,66 г, 10,5 ммоля) растворяли в 4М HCl-диоксане (39 мл) при 0oC. Смесь перемешивали при 25oC в течение 1 часа, концентрировали и остаток растирали с эфиром.
Выход 3,06 г, 102%.
Пример 46b.
(S)-[1-Бензил-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-карбаминовой кислоты трет-бутиловый эфир
4-Гидроксипиперидин (75 ммолей) и Вос-L-фенилаланин (38 ммолей) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 144 часа) и продукт использовали без дальнейшей очистки. Выход 12,2 г, 96%; ВЭЖХ (60/40) 3,45 минуты (97%).
Пример 47.
5-Бром-1Н-индод-2-карбоновой кислоты [(1S)-бензил-2- (1,1-диоксо-1-тиазолидин-3-ил)-2-оксоэтил]-амид
(S)-2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-3-фенилпропан-1-он гидрохлорид (0,3 ммоля) и 5-бром-1Н-индол-2-карбоновую кислоту (0,3 ммоля) соединяли по процедуре А (температура реакции 0-25oC, промывали сначала кислотой, затем основанием) и продукт очищали с помощью хроматографии на силикагеле с элюированием 30, 40 и 50% этилацетатом в гексанах. Продукт собирали в виде не совсем белой пены и растирали с эфиром-гексанами 1:1 с получением 107 мг, 73%; ВЭЖХ (60/40) 6,21 минуты (99%); PBMS 490/492 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,53 (шир, 0,5Н), 9,44 (шир, 0,5Н), 7,78 (д, 0,5H, J=2 Гц), 7,76 (д, 0,5H, J=2 Гц), 7,4-7,2 (м, 7Н), 7,10 (д, 0,5H, J=9 Гц), 7,02 (д, 0,5H, J=9 Гц), 6,86 (с, 0,5Н), 6,81 (с, 0,5Н), 5,21 (м, 0,5Н), 4,95 (м, 0,5Н), 4,62 (д, 0,5H, J=11 Гц), 4,47 (д, A из AB, 0,5H, J=13 Гц), 4,38 (д, B из AB, 0,5H, J=13 Гц), 4,20 (м, 0,5Н), 4,03 (м, 0,5Н), 3,82 (м, 0,5Н), 3,44 (д, 0,5H, J=11 Гц), 3,33-3,0 (м, 4Н), 2,70 (м, 0,5Н).
Анал. Расчетное для C21H20BrN3O4S + 0,2 H2O: C, 51,06; H, 4,16; N, 8,51.
Обнаруженное: C, 51,44; H, 4,36; N, 7,93.
Пример 48.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-оксо-2- (3-оксопирролидин-1-ил)этил]-амид
(S)-1-(2-Амино-3-фенилпропионил)-пирролидин-3-он гидрохлорид (0,06 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,06 ммоля) соединяли по процедуре А (температура реакции 0-25oC, промывали сначала кислотой, затем основанием) и продукт очищали с помощью хроматографии на силикагеле с элюированием 40 и 50% этилацетатом в гексанах с последующим растиранием полученной в результате пены с эфиром. Выход 112 мг, 45%; ВЭЖХ (60/40) 5,13 минуты (> 99%); PBMS 410/412 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,19 (м, 1Н), 7,60 (м, 1Н), 7,3-7,15 (м, 8Н), 6,86 (м, 1Н), 4,23 (м, 0,5Н), 4,95 (м, 0,5Н), 4,0-3,7 (м, 3Н), 3,27 (м, 1Н), 3,15 (м, 1Н), 3,05 (м, 0,5Н), 2,85 (д, 0,5H, J=28 Гц), 2,45 (м, 1,5Н), 2,15 (м, 0,5Н).
Анал. Расчетное для C22H20ClN3O3 + 0,55 H2O: C, 62,95; H, 5,07; N, 10,01.
Обнаруженное: C, 63,31; H, 5,09; N, 9,61.
Пример 48а.
(S)-1-(2-Амино-3-фенилпропионил)-пирролидин-3-она-гидрохлорид
(S)-[1-Бензил-2-оксо-2-(3-оксопирролидин-1-ил)-этил]-карбаминовой кислоты трет-бутиловый эфир (552 мг, 1,7 ммоля) растворяли в 4М HCl-диоксане (6,2 мл) при 0oC. Смесь перемешивали при 25oC в течение 1 часа, концентрировали и остаток растирали с эфиром с получением светящегося коричневого твердого вещества. Выход 482 мг, 108%.
Пример 48b.
(S)-[1-Бензил-2-оксо-2-(3-оксопирролидин-1-ил)-этил] - карбаминовой кислоты трет-бутиловый эфир
Раствор диметилсульфоксида (4,07 г, 52 ммоля) и оксалилхлорида (3,61 г, 28 ммолей) медленно добавляли в этом порядке к дихлорметану (50 мл) при -78oC. К вышеуказанному раствору через канюлю добавляли (S)-[1-бензил-2-(3-гидроксипирролидин-1-ил)-2- оксоэтил] -карбаминовой кислоты трет-бутиловый эфир (24 ммоля) в дихлорметане (30 мл), температуру реакции доводили до -30oC в течение 0,5 часа, затем понижали до -78oC с последующим добавлением триэтиламина (118 ммолей). Затем реакционную смесь нагревали до 25oC, разводили этилацетатом, промывали три раза насыщенным NaHCO3/солевым раствором, органические вещества сушили MgSO4 и концентрировали. Полученную в результате пену очищали с помощью хроматографии на силикагеле с элюированием 30, 40 и 50% этилацетатом в гексанах с получением легкой желтой пены (7,5 г, 95%).
Пример 48с.
(S)-[1-Бензил-2-(3R,S)-гидроксипирролидин-1-ил)-2-оксоэтил]- карбаминовой кислоты трет-бутиловый эфир
(±)-3-Пирролидинол (75 ммолей) и Boc-L-фенилаланин (38 ммолей) соединяли по процедуре А (температура реакции 0-25oC, промывали сначала кислотой, затем основанием) и продукт использовали без дополнительной очистки. Выход 12,2 г, 96%; ВЭЖХ (60/40) 3,45 минуты (96%).
Пример 49.
5-Хлор-1Н-индол-2-карбоновой кислоты ((1S)-бензил-2-оксо- 2-тиазолидин-3-ил-этил)-амид
(S)-2-Амино-3-фенил-1-тиазолидин-3-илпропан-1-она гидрохлорид (2,6 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту соединяли по процедуре А (температура реакции 0-25oC, время реакции 96 часов, промывали сначала кислотой, затем основанием). Неочищенный продукт растирали с эфиром-гексанами 1: 1 и сушили. Выход 966 мг, 91%; ВЭЖХ (60/40) 7,99 минуты (97%); PBMS 414/416 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,26 (шир,1Н), 7,59 (м, 1Н), 7,35-7,20 (м, 8Н), 6,84 (м, 1Н), 5,14 (м, 1Н), 4,61 (д, A из AB, 0,6Н, J=10,3 Гц), 4,52 (д, 0,4H, J= 11,6 Гц), 4,42 (д, B из AB, 0,6H, J=10,3 Гц), 3,80-3,65 (м,  1,5Н), 3,2 (м,
1,5Н), 3,2 (м,  2,5Н), 3,04 (м, 0,4Н), 2,95-2,8 (м, 1,2Н), 2,63 (м, 0,6Н).
2,5Н), 3,04 (м, 0,4Н), 2,95-2,8 (м, 1,2Н), 2,63 (м, 0,6Н).
Анал. Расчетное для C21H20ClN3O2S + 0,6 H2O: C, 59,39; H, 5,03; N, 9,89.
Обнаруженное: C, 59,39; H, 4,96; N, 9,52.
Пример 50.
5-Хлор-1Н-индол-2-карбоновой кислоты ((1S)-бензил-2-оксо-2- тиоморфолин-4-ил-этил)-амид
(S)-2-Амино-3-фенил-1-тиоморфолин-4-илпропан-1-она гидрохлорид (2,6 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (2,6 ммоля) соединяли по процедуре А (температура реакции 0-25oC, промывали сначала кислотой, затем основанием). Неочищенный продукт затем растирали с эфиром-гексанами 1:1 и сушили. Выход 1,03 г, 94%; ВЭЖХ (60/40) 8,74 минуты (99%); PBMS 428/430 (MH+, 100%);
Анал. Расчетное для C22H22ClN3O2S: C, 61,75; H, 5,18; N, 9,82.
Обнаруженное: C, 62,04; H, 5,58; N, 9,72.
Пример 50а.
(S)-2-Амино-3-фенил-1-тиоморфолин-4-илпропан-1-она гидрохлорид
(S)-(1-Бензил-2-оксо-2-тиоморфолин-4-ил-этил)-карбаминовой кислоты трет-бутиловый эфир (17,8 ммоля) растворяли в 4М HCl-диоксане (67 мл) при 0oC, раствор перемешивали при 25oC в течение 1 часа, реакционную смесь концентрировали и остаток растирали с эфиром. Выход 5,0 г, 98%; PBMS 251 (MH+, 100%).
Пример 50b
(S)-(1-Бензил-2-оксо-2-тиоморфолин-4-ил-этил)-карбаминовой кислоты трет-бутиловый эфир
Тиоморфолин (38 ммолей) и Boc-L-фенилаланин (19 ммолей) соединяли по процедуре А (температура реакции 0-25oC) со следующей обработкой: реакционную смесь концентрировали, разбавляли этилацетатом, затем промывали сначала 1N HCl три раза, затем 2N NaOH, органический слой сушили MgSO4, и концентрировали. Полученную в результате пену использовали без дополнительной очистки. Выход 6,3 г, 95%.
Пример 51.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2- (1,1-диоксо-1-тиазолин-3-ил)-2-оксоэтил]-амид
(S)-2-Амино-1-(1,1-диоксо-1-тиазолидин-3-ил)-3-фенилпропан-1-она гидрохлорид (0,8 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,8 ммоля) соединяли по процедуре А (температура реакции 0-25oC, промывали сначала кислотой, затем основанием). Продукт очищали с помощью хроматографии на силикагеле с элюированием 30, 40 и 50% этилацетатом в гексанах с последующим растиранием с эфиром-гексанами 1:1. Выход 266 мг, 75%; ВЭЖХ (60/40), 5,52 минуты (>99%); PBMS 446/448 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,21 (шир, 0,5Н), 9,15 (шир, 0,5Н), 7,62 (шир, 0,5H, J= 2 Гц), 7,60 (д, 0,5H, J=2 Гц), 7,35-7,20 (м, 7Н), 7,10 (д, 0,5H, J=8,5 Гц), 7,02 (д, 0,5H, J=8,5 Гц), 6,84 (д, 0,5Н, J=2 Гц), 6,81 (д, 0,5H, J=2 Гц), 5,21 (м, 0,5Н), 4,93 (м, 0,5H), 4,62 (д, 0,5H, J=11 Гц), 4,47 (д, A из AB, 0,5H, J= 13 Гц), 4,39 (д, B из AB, 0,5H, J=13 Гц), 4,22 (м, 0,5H), 4,03 (м, 0,5H), 3,83 (м, 0,5Н), 3,44 (д, 0,5H, J=11 Гц), 3,3-3,0 (м, 4Н), 2,67 (м, 0,5Н).
Пример 52.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-(4-хлорбензил)- 2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(S)-2-Амино-3-(4-хлорфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид (0,98 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,92 ммоля) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием 50, 75 и 100% этилацетатом в гексанах. Выход 362 мг, 86%; ВЭЖХ (60/40) 5,06 минуты (97%); т. пл. 227-229oC; ТРМС 460/462 (NH+, 100%);
Анал. Рассчитанное для C23H23Cl2N3O3: C, 60,01; H, 5,04; N, 9,13.
Обнаруженное: C, 59,83; H, 5,18; N, 9,16.
Пример 52а.
(S)-2-Амино-3-(4-хлорфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид
(S)-[1-(4-Хлорбензил)-2-(4-гидроксипиперидин-1-ил)-2- оксоэтил]-карбаминовой кислоты трет-бутиловый эфир (475 мг, 1,2 ммоля) растворяли в 4М HCl-диоксане (5 мл) при 0oC. Смесь перемешивали в течение 1,5 часов при 25oC, концентрировали и остаток растирали с эфиром. Выход 422 мг, 105%; ТРМС 283 (MH+, 100%).
Пример 52b.
(S)-[1-(4-Хлорбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир
4-Гидроксипиперидин (2,6 ммоля) и Boc-L-п-хлорфенилаланин (2,5 ммоля) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием этилацетатом-гексанами 1:1 и 3:1. Выход 662 мг, 69%.
Пример 53.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-(4-гидроксипиперидин-1-ил)-(1S)-(1Н-имидазол-4-илметил)-2-оксоэтил]-амид
(S)-2-Амино-1-(4-гидроксипиперидин-1-ил)-3-(1H-имидазол-4-ил)- пропан-1-она гидрохлорид (0,7 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,7 ммоля) соединяли по процедуре А (время реакции 120 часов, промывание кислотой исключалось). Неочищенный продукт дважды растирали с эфиром, с эфиром-гексанами 1:1 и остаток очищали с помощью хроматографии на силикагеле с элюированием 5-20% этанолом в дихлорметане, содержащем 0,5% гидроксида аммония. Выход 232 мг, 81%; ВЭЖХ (40/60) 2,57 минуты (98%); PBMS 416/418 (MH+, 100%);
Анал. Расчетное для C20H22ClN5O3 + 0,55 H2O: C, 56,42; H, 5,47; N, 16,45.
Обнаруженное: C, 6,07; H, 5,65; N, 16,08.
Пример 53а.
(S)-2-Амино-1-(4-гидроксипиперидин-12-ил)-3-(1H-имидазол-4-ил)-пропан- 1-она гидрохлорид
(S)-{ 2-(4-Гидроксипиперидин-1-ил)-2-оксо-1-[l-(толуол-4- сульфонил)-1H-имидазол-4-илметил] -этил} -карбаминовой кислоты трет-бутиловый эфир (512 мг, 1,0 ммоль) растворяли в 4M HCl-диоксане (3 мл) при 0oC. Смесь перемешивали при 25oC в течение 1,5 часов, концентрировали и остаток растирали с эфиром. Выход 422 мг, 105%; ТРМС 283 (MH+, 100%).
Пример 53b.
(S)-{ 2-(4-Гидроксипиперидин-1-ил)-2-оксо-1-[1-(толуол-4- сульфонил)-1Н-имидазол-4-илметил]-этил}-карбаминовой кислоты трет-бутиловый эфир
4-Гидроксипиперидин (303 мг, 3,0 ммоля), триэтиламин (394 мг, 3,9 ммоля) и диэтилцианофосфонат (636 мг, 3,9 ммоля) добавляли в этом порядке к Boc-NiM-тосил-L-гистидину (J. Med. Chem. 30 536 (1987); 1,32 г, 3,9 ммоля) в дихлорметане (10 мл) при 25oC. Через 120 часов раствор разбавляли этилацетатом, промывали дважды насыщенным NaHCO3, сушили и концентрировали. Остаток очищали с помощью хроматографии на силикагеле с элюированием 1-8% этанолом в дихлорметане. Выход 517 мг, 35%; ВЭЖХ (50/50) 4,75 минуты (97%).
Пример 54
5-Хлор-1Н-индол-2-карбоновой кислоты (2S)-[(5-хлор-1Н- индол-2-карбонил)-амино]-3-(4-гидроксипиперидин-1-ил)-3-оксопропиловый эфир
(S)-2-Амино-3-гидрокси-1-(4-Гидроксипиперидин-1-ил)пропан- 1-она гидрохлорид (0,89 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,85 ммоля) соединяли по процедуре А и продукт выделяли с помощью хроматографии на силикагеле вместе с более полярным сериновым аналогом (40%) при элюировании 1-16% этанолом в дихлорметане. Выход 51 мг, 16%; ВЭЖХ (60/40) 7,06 минуты (96%); PBMS 348/350 (100%), 543/545 (MH+, 5%).
Анал. Расчетное для C26H24Cl2N4O5 + 0,57 H2O: C, 56,40; H, 4,58; N, 10,12.
Обнаруженное: C, 56,79; H, 4,90; N 9,65.
Пример 54а.
(S)-2-Амино-3-гидрокси-1-(4-гидроксипиперидин-1-ил)-пропан-1-она гидрохлорид
(S)-[1-Гидроксиметил-2-(4-гидроксипиперидин-1-ил)-2- оксоэтил] - карбаминовой кислоты трет-бутиловый эфир (595 мг, 2,0 ммоля) растворяли в 4М HCl-диоксанах (2 мл) при 0oC. Смесь перемешивали при 25oC в течение 1 часа, концентрировали и остаток растирали с эфиром. Выход 506 мг, 105%; МС 189 (MH+ 100%).
Пример 54b
(S)-[1-гидроксиметил-2-(4-гидроксипиперидин-1-ил)-2- оксоэтил]-карбаминовой кислоты трет-бутиловый эфир
4-Гидроксипиперидин (6,07) и Boc-L-серин (6,04 ммоля) соединяли по процедуре А (время реакции 60 часов) со следующей обработкой: реакционную смесь концентрировали, остаток растворяли в хлороформе и 1N NaOH (6 мл) и полученный в результате раствор несколько раз (десять или более) экстрагировали хлороформом. Хлороформные экстракты концентрировали и остаток очищали с помощью хроматографии на силикагеле с элюированием 1-16% этанолом в дихлорметане. Выход 751 мг, 41%; ВЭЖХ (40/60) 2,72 минуты (96%).
Пример 55.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-(4-гидроксибензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(S)-2-Амино-3-(4-гидроксифенил)-1-(4-гидроксипиперидин-1- ил)-пропан-1-она гидрохлорид (0,68 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,65 ммоля) соединяли по процедуре А (температуре реакции 0-25oC) со следующей обработкой: реакционную смесь разбавляли этилацетатом, полученный в результате раствор промывали 1N NaOH (2 мл), водный слой экстрагировали три раза этилацетатом, объединенные органические экстракты промывали 1N HCl, сушили и концентрировали. Остаток очищали с помощью хроматографии на силикагеле с элюированием 1-16% этанолом в дихлорметане. Выход 150 мг, 52%; ВЭЖХ (60/40) 3,53 минуты (99%); PBMS 442/444 (NH+, 100%);
Анал. Расчетное для C23H24ClN3O4 + 0,5 H2O: C, 61,26; H, 5,59; N, 9,32.
Обнаруженное: C, 61,52; H, 5,89; N, 8,98.
Пример 55а.
(S)-2-Амино-3-(4-гидроксифенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид
(S)-[1-(4-Гидроксибензил)-2-(4-гидроксипиперидин-1-ил)-2- оксоэтил]-карбаминовой кислоты трет-бутиловый эфир (450 мг, 1,2 ммоля) растворяли в 4М HCl-диоксане (2 мл) при 0oC. Смесь перемешивали при 25oC в течение 1 часа, концентрировали и остаток растирали с эфиром. Выход 400 мг, 107%; МС 265 (MH+, 100%).
Пример 55b.
(S)-[1-(4-Гидроксибензил)-2-(4-гидроксипиперидин-1-ил)- 2-оксоэтил]-карбаминовой кислоты трет-бутиловый эфир
4-Гидроксипиперидин (3,9 ммоля) и Boc-L-тирозин (3,7 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 60 часов) со следующей обработкой: реакционную смесь разводили этилацетатом и промывали один раз основанием, щелочной слой подкисляли 2N HCl и экстрагировали три раза хлороформом, и хлороформные экстракты концентрировали. Полученную в результате пену очищали с помощью хроматографии на силикагеле с элюированием 1-8% этанолом в дихлорметане, содержащим 0,5% NH4OH. Выход 550 мг, 41%; ВЭЖХ (40/60) 5,02 минуты (87%).
Пример 56.
5-Хлор-1Н-индол-2-карбоновой кислоты [2(4-гидроксипиперидин-1-ил)- 2-оксо-(1S)-пиридин-3-илметилэтил]-амид
(S)-2-Амино-1-(4-гидроксипиперидин-1-ил)-3-пиридин-3-ил- пропан-1-она дигидрохлорид (0,8 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту соединяли по процедуре А (температура реакции 0-25oC) и продукт очищали с помощью хроматографии на силикагеле с элюированием 1-16% этанолом в дихлорметане. Выход 26 мг, 8%; ВЭЖХ (50/50) 5,02 минуты (99%); PBMS 427/429 (MH+, 100%);
Анал. Расчетное для C22H23ClN4O3 + 0,5 H2O: C, 60,62; H, 5,55; N, 12,85.
Обнаруженное: C, 60,57; H, 5,74; N, 12,53.
Пример 56а.
(S)-2-Амино-1-(4-Гидроксипиперидин-1-ил)-3-пиридин-3- ил-пропан-1-она дигидрохлорид
(S)-[2-(4-Гидроксипиперидин-1-ил)-2-оксо-1-пиридин-3-ил- метилэтил] -карбаминовой кислоты трет-бутиловый эфир (367 мг, 1,05 ммоля) растворяли в 4М HCl-диоксане при 0oC. Полученную в результате суспензию перемешивали в течение 1,5 часов при 25oC, концентрировали и остаток растирали с эфиром. Выход 450 мг, 100%.
Пример 56b.
(S)-[2-(4-Гидроксипиперидин-1-ил)-2-оксо-1-пиридин-3-ил-метилэтил] - карбаминовой кислоты трет-бутиловый эфир
4-Гидроксипиперидин (2,9 ммоля) и N-т-Boc-L-3-(3-пиридил) аланин (2,8 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции 96 часов, промывание кислотой исключалось) и продукт очищали с помощью хроматографии на силикагеле с элюированием 1-8% этанолом в дихлорметане. Выход 454 мг, 46%; МС 350 (MH+, 100%).
Пример 57.
1Н-Индол-2-карбоновой кислоты [(1R)-(4-фторбензил)-2- )4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(R)-2-Амино-3-(4-фторфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид (0,5 ммоля) и 1Н-индол-2-карбоновую кислоту (0,5 ммоля) соединяли по процедуре А (температура реакции 0-25oC) и продукт очищали с помощью хроматографии на силикагеле с элюированием 25, 30, 50, 75 и 80% этилацетатом в гексанах. Выход 150 мг, 60%; ВЭЖХ (60/40) 3,66 минуты (97%); т.пл. 204-207oC; PBMS 410 (MH+, 100%);
Анал. Расчетное для C23H24FN3O3: C, 67,47; H, 5,91; N, 10,26.
Обнаруженное: C, 67,18; H, 6,03; N, 10,21.
Пример 57а.
(R)-2-Амино-3-(4-фторфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид
(R)-[1-(4-Фторбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир (2,6 ммоля) растворяли в 4М HCl-диоксане (2 мл) при 0oC. Раствор перемешивали 2 часа при 25oC, концентрировали и остаток растирали с эфиром. Выход 920 мг, 124%; ВЭЖХ (60/40) 2,23 минуты (98%).
Пример 57.
(R)-[1-(4-Фторбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир
4-Гидроксипиперидин (3,7 ммоля) и (R)-N-т-Вос-п-фторфенилаланин (3,5 ммоля) соединяли по процедуре А с получением пенообразного вещества, которое использовали без дальнейшей очистки. Выход 940 мг, 73%; ВЭЖХ (60/40) 3,64 минуты (95%); МС 367 (MH+, 100%).
Пример 58.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1R)-(4-фторбензил)- 2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(R)-2-Амино-3-(4-фторфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид (0,6 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,6 ммоля) соединяли по процедуре А и неочищенный продукт очищали с помощью хроматографии на силикагеле с элюированием 50, 75 и 100% этилацетатом в гексанах. Выход 171 мг, 765%; ВЭЖХ (60/40) 4,23 минуты (97%); МС 444/446 (MH+, 200%); ТРМС 444/446 (MH+, 100%);
1H ЯМР (CDCl)3 δ 9,20 (шир, 1Н), 7,57 (д, 1Н, J=2 Гц), 7,33 (д, 1Н, J=8 Гц), 7,3-7,2 (м, 2Н), 7,14 (м, 2Н), 6,97 (м, 2Н), 6,85 (м, 1Н), 5,34 (м, 1Н), 4,05-3,80 (м, 2Н), 3,7-3,3 (м, 1,5H,), 3,25 (м, 1H), 3,10 (м, 2Н), 2,93 (м, 0,5Н), 1,9-1,7 (м, 2,5Н), 1,45 (м, 2Н), 1,15 (м, 0,5Н).
Анал. Расчетное для C23H23ClFN3O3 + 0,05 H2O: C, 62,11; H, 5,23; N, 9,45.
Обнаруженное: C, 62,51; H, 5,66; N, 9,19.
Пример 59.
5-Фтор-1Н-индол-2-карбоновой кислоты [(1S)-(4-фторбензил)- 2-(4-гидроксипиперидин-12-ил)-2-оксоэтил]-амид
(S)-2-Амино-3-(4-фторфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид (0,5 ммоля) и 5-фтор-1Н-индол-2-карбоновую кислоту (0,5 ммоля) соединяли по процедуре А. Неочищенный продукт растирали один раз с эфиром-гексанами 1: 1 и один раз с гексанами. Полученное в результате твердое вещество кипятили в этилацетате, полученную суспензию фильтровали и собранное твердое вещество сушили. Выход 103 мг, 48%; ВЭЖХ (60/40) 3,69 минуты (95%); PBMS 428 (MH+, 100%);
Анал. Расчетное для C23H23F2N3O3 + 0,25 H2O: C, 63,95; H, 5,48; N, 9,73.
Обнаруженное: C, 63,93; H, 5,66; N, 9,87.
Пример 59а.
(S)-2-Амино-3-(4-фторфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид
[(S)-1-(4-Фторбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир (20,2 г, 55 ммолей) растворяли в 4М HCl-диоксане (25 мл) при 25oC. Через 3 часа осаждался густой сироп и добавляли дополнительно 4М HCl-диоксаны (10 мл). Смесь перемешивали в течение 2 часов, концентрировали и твердый остаток суспендировали в 4М HCl-диоксанах. После 2 часов при 25oC смесь концентрировали и остаток дважды выпаривали вместе с эфиром. Полученное в результате твердое вещество перемешивали в смеси эфира (75 мл) и гексанов (10 мл) при 25oC в течение 18 часов, смесь фильтровали и отфильтрованное твердое вещество промывали эфиром-гексанами 1: 1 и сушили с получением гигроскопичного твердого вещества (16,3 г, 97%).
Пример 59b.
[(S)-1-(4-Фторбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир
4-Гидроксипиперидин (0,29 ммоля) и (S)-N-т-Boc-п-фторфенилаланин (0,28 ммоля) соединяли по процедуре А с получением неочищенного продукта в виде пены с выходом 84%. Часть этого вещества (81,6 г) растворяли в горячем этилацетате (400 мл) и добавляли гексаны (25oC) к полученному раствору до тех пор, пока не появлялось легкое помутнение. Смесь нагревали до кипения и полученному в результате прозрачному раствору давали остыть до 25oC в течение ночи. Полученную суспензию фильтровали и собранное твердое вещество промывали этилацетатом-гексанами и сушили (68,1 г, 67%) .
Пример 60.
1-{ (2S)-[(5-Хлор-1Н-индол-2-карбонил)-амино]-3-фенил- пропионил}-(4R)-гидроксипирролидин-(2S)-карбоновой кислоты бензиловый эфир
1-((2S)-Амино-3-фенилпропионил)-(4R)-гидроксипирролидин-(2R)- карбоновой кислоты бензилового эфира гидрохлорид (0,56 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,53 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции 60 часов) и продукт очищали с помощью хроматографии на силикагеле с элюированием 20, 30 и 50% этилацетатом в гексанах. Выход 26 мг, 8%; ВЭЖХ (60/40) 8,14 минут (98%); PBMS 546/548 (MH+, 100%);
Анал. Расчетное для C30H28ClN3O5: C, 65,99; H, 5,17; N, 7,70.
Обнаруженное: C, 66,14; H, 5,37; N, 7,60.
Пример 60а.
1-((2S)-Амино-3-фенилпропионил)-(4R)-гидроксипирролидин- (2S)-карбоновой кислоты бензилового эфира гидрохлорид
1-((2S)-трет-Бутоксикарбониламино-3-фенилпропионил)-(4R)- гидроксипирролидин-(2S)-карбоновой кислоты бензиловый эфир (3,0 ммоля) растворяли в 4М HCl-диоксане при 0oC. Смесь перемешивали при 25oC в течение 1 часа, концентрировали и остаток растирали с эфиром. Выход 1,16 г, 96%.
Пример 60b.
1-((2S)-трет-Бутоксикарбониламино-3-фенилпропионил)-(4R)- гидроксипирролидин-(2S)-карбоновой кислоты бензиловый эфир
Транс-1-гидроксипролина бензиловый эфир (3,15 ммоля) и 1-Boc-фенилаланин (3,0 ммоля) соединяли по процедуре А (температура реакции 0-2oC, дихлорметан/диметилформамид 1:1) и продукт использовали без дополнительной очистки. Выход 1,31 г, 99%; ВЭЖХ (60/40) 6,1 минуты (95%).
Пример 61.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-(4-фторбензил)- 2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
(S)-2-Амино-3-(4-фторфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид (0,051 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,051 ммоля) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием 50, 75, 80 и 100% этилацетатом-гексанами с получением пенообразного вещества (выход 78%), ВЭЖХ (60/40) 4,21 минуты (99%). Часть этого материала перекристаллизовывали путем растворения в горячем этилацетате (примерно 5-7 мл/г) и добавления примерно равного объема гексанов при нагревании в колбе с обратным холодильником с последующим медленным охлаждением раствора до 25oC. Твердое вещество отфильтровывали и промывали этилацетатом-гексанами 1:4 и сушили (70-90% извлечения): т.пл. 175-177oC.
1H ЯМР (CDCl3) δ 9,41 (м, 0,5Н), 9,36 (м, 0,5Н), 7,59 (д, 1Н, J=2 Гц), 7,37 (д, 1H, J=8 Гц), 7,29 (дд, 1H, J=2,9 Гц), 7,20 (дд, 1Н, J=2,0, 8,9 Гц), 7,14 (м, 2Н), 6,95 (м, 2Н), 6,86 (м, 1H), 5,34 (м, 1H), 4,05 (м, 0,5Н), 3,90 (м, 1,5Н), 3,65 (м, 0,5Н), 3,45 (м,1Н), 3,25 (м, 1H), 3,10 (м, 2Н), 2,93 (м, 0,5Н), 1,88 (шир, 1H, обмены с D2O), 1,80 (м, 1,5Н), 1,45 (м, 2Н), 1,12 (м, 0,5Н).
ФБМС 444/446 (MH+, 100%);
Анал. Расчетное для C23H23ClFN3O3 + 0,2 H2O: C, 61,73; H, 5,27; N, 9,39.
Обнаруженное: C, 61,40; H, 5,37; N, 9,11.
Пример 62.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-(метоксиметилкарбамоил)-2-пиридин-3-ил-этил]-амид
(2S)-Амино-N-метокси-N-метил-3-пиридин-3-ил-пропионамида гидрохлорид (1,3 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (1,25 ммоля) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции - дихлорметан/ДМФ 1:1) и продукт очищали с помощью хроматографии на силикагеле с элюированием этилацетатом. Выход 313 мг, 65%. ВЭЖХ (60/40) 2,84 минуты (99%); ТРМС 387/389 (MH+, 100%).
1H ЯМР (CDCl3) δ 9,1 (шир, 1H), 8,48 (дд, 1H), 8,43 (м, 1H), 7,60 (д, 1H), 7,50 (м, 1H, J=ca, 8 Гц), 7,37 (д, 1H, J=ca, 8 Гц), 7,23 (д, 1H), 7,18 (дд, 1H, J=ca, 8 Гц), 7,10 (д, 1Н, J=са, 8 Гц), 6,82 (д, 1H), 5,42 (м, 1H), 3,78 (с, 3Н), 3,25 (с, 3Н), 3,32 (дд, A из AB, 1H, J=ca, 7,14 Гц), 3,10 (дд, B из AB, 1H, J=са.7, 14 Гц).
Анал. Расчетное для C19H19ClN4O3 + 0,4 H2O: C, 57,91; H, 5,07; N, 14,22.
Обнаруженное: C, 58,19; H, 5,23; N, 13,82.
Пример 62а.
(2S)-Амино-N-метокси-N-метил-3-пиридин-3-илпропионамида дигидрохлорид
[(1S)-(Метоксиметилкарбамоил)-2-пиридин-3-ил-этил] -карбаминовой кислоты трет-бутиловый эфир (1,5 ммоля) растворяли в 4М HCl-диоксане при 0oC. Полученный в результате раствор перемешивали в течение 2 часов при 25oC, концентрировали и остаток растирали с эфиром. Выход 390 мг, 95%.
Пример 62b.
(S)-[1-(Метоксиметилкарбамоил)-2-пиридин-3-ил-этил] - карбаминовой кислоты трет-бутиловый эфир
N, O-Диметилгидроксиламина гидрохлорид (1,7 ммоля) и Boc-3-пиридил-L-аланин (1,6 ммоля) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции - дихлорметан/диметилформамид, промывание кислотой исключалось, для высушивания использовали Na2SO4). Остаток растирали с эфиром с получением 428 мг (86% выход) желтого твердого вещества.
Пример 63.
(R, S)-2-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3-(3- фторфенил)-пропионовой кислоты метиловый эфир
(R, S)-2-Амино-3-(3-фторфенил)-пропионовой кислоты метиловый эфир (2,05 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (2,03 ммоля) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции дихлорметан/ДМФ 1: 1) и продукт очищали с помощью хроматографии на силикагеле с элюированием 10, 20 и 40% этилацетатом в гексанах. Остаток растирали с эфиром-гексанами 1:1 и гексанами с получением не совсем белого твердого вещества (484 мг, 63%): ВЭЖХ (60/40) 8,13 минуты (95%); ТРМС 375/377 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,26 (шир, 1Н), 7,60 (д, 1Н, J =  1 Гц), 7,35 (д, 1Н, J=8,7 Гц), 7,25 (м, 2Н), 6,95 (м, 1Н), 6,91 (м, 1Н), 6,84 (м, 1Н), 6,77 (д, 1H, J=1,5 Гц), 6,63 (д, 1Н, J=7,7 Гц), 5,08 (м, 1Н), 3,78 (с, 3Н), 3,28 (дд, 1Н, A из AB, J=5,7,14 Гц), 3,21 (дд, 1Н, B из AB, J=5,5,14 Гц).
1 Гц), 7,35 (д, 1Н, J=8,7 Гц), 7,25 (м, 2Н), 6,95 (м, 1Н), 6,91 (м, 1Н), 6,84 (м, 1Н), 6,77 (д, 1H, J=1,5 Гц), 6,63 (д, 1Н, J=7,7 Гц), 5,08 (м, 1Н), 3,78 (с, 3Н), 3,28 (дд, 1Н, A из AB, J=5,7,14 Гц), 3,21 (дд, 1Н, B из AB, J=5,5,14 Гц).
Анал. Расчетное для C19H16ClFN2O3: C, 60,89; H, 4,30; N, 7,47.
Обнаруженное: C, 60,79; H, 4,58; N, 7,18.
Пример 63а.
(R, S)-2-Амино-3-(3-фторфенил)-пропионовой кислоты метилового эфира гидрохлорид
Триметилсилилхлорид (1,07 г, 9,9 ммоля) добавляли к суспензии м-фтор-DL-фенилаланина (0,404 г, 2,2 ммоля) в метаноле (4 мл) при 25oC. Полученный в результате раствор кипятили в колбе с обратным холодильником в течение 1 часа, охлаждали и концентрировали. Остаток растирали с эфиром. Выход 515 мг, 100%; ВЭЖХ (60/40) 2,31 минуты (95%).
Пример 64.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-(метоксиметилкарбамоил)- 2-тиофен-2-ил-этил]-амид
(S)-2-Амино-N-метокси-N-метил-3-тиофен-2-ил-пропионамида гидрохлорид (1,2 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (1,2 ммоля) соединяли по процедуре А (0-25oC - температура реакции, растворитель для реакции - дихлорметан/диметилформамид 2:1). Продукт-сырец очищали с помощью хроматографии на силикагеле с элюированием 10, 20, 30 и 40% этилацетатом в гексанах. Выход 375 мг, 80%; ВЭЖХ (60/40) 6,36 минуты (99%); PBMS 392/394 (MH+, 100%).
1H ЯМР (CDCl3) δ 9,33 (шир, 1Н), 7,60 (д, 1H, J=4  1 Гц), 7,30 (д, 1Н, J= 8,8 Гц), 7,20 (дд, 1H, J=2,0, 8,7 Гц), 7,15 (дд, 1H, J=1, 5,0 Гц), 6,91 (дд, 1H, J=3,4, 5,1 Гц), 6,86 (д, 1Н, J=1,6 Гц), 6,84 (д, 1Н, J=
1 Гц), 7,30 (д, 1Н, J= 8,8 Гц), 7,20 (дд, 1H, J=2,0, 8,7 Гц), 7,15 (дд, 1H, J=1, 5,0 Гц), 6,91 (дд, 1H, J=3,4, 5,1 Гц), 6,86 (д, 1Н, J=1,6 Гц), 6,84 (д, 1Н, J=  2 Гц), 5,40 (м, 1Н), 3,77 (с, 3Н), 3,46 (дд, 1Н, A из AB, J=6,2,
2 Гц), 5,40 (м, 1Н), 3,77 (с, 3Н), 3,46 (дд, 1Н, A из AB, J=6,2,  14 Гц), 3,37 (дд, 1Н, B из AB, J=6,2
14 Гц), 3,37 (дд, 1Н, B из AB, J=6,2  14,2 Гц), 3,25 (с, 3Н).
14,2 Гц), 3,25 (с, 3Н).
Анал. Расчетное для C18H18ClN3O3S + 0,25 C4H8O2: C, 55,14, H, 4,87, N, 10,15.
Обнаруженное: C, 55,41; H, 4,79; N, 10,17.
Пример 64а.
(S)-2-Амино-N-метокси-N-метил-3-тиофен-2-ил-пропионамида гидрохлорид
(S)-[1-(Метоксиметилкарбамоил)-2-тиофен-2-ил-этил] -карбаминовой кислоты трет-бутиловый эфир (1,3 ммоля) растворяли в 4М HCl-диоксане (1 мл) при 0oC и полученный в результате раствор перемешивали при 25oC в течение 2 часов. Смесь концентрировали и остаток растирали с эфиром с получением желтого твердого вещества (321 мг, 96%; ВЭЖХ (60/40) 2,24 минуты (98%); МС 215 (M+, 100%).
Пример 64b.
(S)-[1-(Метоксиметилкарбамоил)-2-тиофен-2-ил-этил]- карбаминовой кислоты трет-бутиловый эфир
N, O-диметилгидроксиламина гидрохлорид (1,4 ммоля) и Boc-(2-тиенил)-L-аланин (1,3 ммоля) соединяли по процедуре А (температура реакции 0-25oC) с получением продукта, который использовали без дополнительной очистки. Выход 426 мг, 104%.
Пример 65.
(RS)-2-[(5-Хлор-1Н-индол-2-карбонил)-амино]-3-(4-фторфенил)- пропионовой кислоты метиловый эфир
(R, S)-2-Амино-3-(3-фторфенил)-пропионовой кислоты метиловый эфир (3,0 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (2,9 ммоля) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции - дихлорметан/диметилформамид 3:2) и полученный продукт-сырец растирали с эфиром-гексанами 1:1. Выход 1,03 г, 92%; ВЭЖХ (60/40) 7,95 минуты (96%); PBMS 375/377 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,30 (шир, 1Н), 7,60 (д, 1H, J=  1 Гц), 7,35 (д, 1H, J= 8,8 Гц), 7,25 (дд, 1H, J=2,0, 8,7 Гц), 7,10 (м, 2Н), 6,97 (м, 2Н), 6,77 (д, 1H, J= 2 Гц), 6,62 (д, 1Н, J=7,8 Гц), 5,06 (м, 1Н), 3,78 (s, 3Н), 3,27 (дд, 1Н, A из AB, J=7,14 Гц), 3,19 (дд, 1Н, B из АВ, J=7,14 Гц).
1 Гц), 7,35 (д, 1H, J= 8,8 Гц), 7,25 (дд, 1H, J=2,0, 8,7 Гц), 7,10 (м, 2Н), 6,97 (м, 2Н), 6,77 (д, 1H, J= 2 Гц), 6,62 (д, 1Н, J=7,8 Гц), 5,06 (м, 1Н), 3,78 (s, 3Н), 3,27 (дд, 1Н, A из AB, J=7,14 Гц), 3,19 (дд, 1Н, B из АВ, J=7,14 Гц).
Анал. Расчетное для C19H16ClFN2O3: C, 60,89; H, 4,30; N, 7,47.
Обнаруженное: C, 60,74; H, 4,36; N, 7,55.
Пример 66.
5-Хлор-1Н-индол-2-карбоновой кислоты [2-(4-аминофенил)- (1S)-диметилкарбамоилэтил]-амида гидрохлорид
(S)-2-Амино-3-(4-аминофенил)-N, N-диметилпропионамида дигидрохлорид (0,7 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,7 ммоля) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции дихлорметан/ДМФ 3:1, промывали только основанием) и продукт очищали с помощью хроматографии на силикагеле с элюированием 1-16% этанолом в дихлорметане с 0,5% NH4OH. Объединенные фракции концентрировали, растворяли в метаноле при 0oC, полученный в результате раствор обрабатывали 1,01 N HCl (1,05 экв.). Через 5 минут реакционную смесь концентрировали и остаток растирали с эфиром с получением оранжевого твердого вещества (79 мг, выход 29%); ТРМС 385/387 (MH+, 100%);
Анал. Расчетное для C20H21ClN4O2+ 1,5 HCl: C, 54,65; H, 5,16; N, 12,75. Обнаруженное: C, 54,96; H, 5,53; N, 12,53.
Пример 66а.
(S)-2-Амино-3-(4-аминофенил)-N,N-диметилпропионамида дигидрохлорид
(S)-[2-(4-Аминофенил)-1-диметилкарбамоил-этил] -карбаминовой кислоты трет-бутиловый эфир (214 мг, 0,7 ммоля) растворяли в 4М HCl-диоксане (2 мл) при 0oC и раствор перемешивали в течение 2 часов при 25oC. Смесь концентрировали и остаток растирали с эфиром. Выход 294 мг, 102%; PBMS 208 (MH+, 100%).
Пример 66b.
(S)-[2-(4-Аминофенил)-1-диметилкарбамоил-этил]-карбаминовой кислоты трет-бутиловый эфир
Диметиламина гидрохлорид (2,04 ммоля) и Boc-п-амино-L-фенилаланин (1,7 ммоля) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции - дихлорметан/диметилформамид 4:1, промывали только основанием). Продукт очищали с помощью хроматографии на силикагеле с элюированием 50, 60, 70 и 100% этилацетатом в гексанах. Выход 226 мг, 42%; ВЭЖХ (70/30) 2,45 минуты (100%).
Пример 67.
5-Хлор-1Н-индол-2-карбоновой кислоты ((1S)-диметилкарбамоил-3- фенилпропил)-амид
(S)-2-Амино-N, N-диметил-4-фенилбутирамида гидрохлорид (0,76 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,76 ммоля) соединяли по процедуре А (температура реакции 0-25oC; растворитель для реакции - дихлорметан/ДМФ 3:1) и продукт очищали с помощью хроматографии на силикагеле с элюированием 10, 20, 30, 40, 50 и 60% этилацетатом в гексанах. Выход 263 мг, 90%; ВЭЖХ (60/40) 7,12 минуты (99%); ТРМС 384/386 (MH+ 100%);
Анал. Расчетное для C21H22ClN3O2: C, 65,71; H, 5,78; N, 10,95.
Обнаруженное: C, 65,34; H, 5,93; N, 10,91.
Пример 67а.
(S)-2-Амино-N,N-диметил-4-фенилбутирамида гидрохлорид
(S)-(1-Диметилкарбамоил-3-фенилпропил)-карбаминовой кислоты трет-бутиловый эфир (235 мг, 0,8 ммоля) растворяли в 4М HCl-диоксане (2 мл) при 0oC. Смесь перемешивали при 25oC в течение 1,5 часов, концентрировали и остаток растирали с эфиром. Выход 187 мг, 100%; ВЭЖХ (60/40) 2,31 минуты (99%).
Пример 67b.
(S)-(1-Диметилкарбамоил-3-фенилпропил)-карбаминовой кислоты трет-бутиловый эфир
Диметиламина гидрохлорид (1,0 ммоль) и (S)-N-т-бутоксикарбонил-2- амино-4-фенилмасляную кислоту (0,84 ммоля) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции дихлорметан/ДМФ 3:1) с получением продукта, который использовали без дополнительной очистки. Выход 238 мг, 93%; ВЭЖХ (60/40) 5,98 минуты (97%).
Пример 68.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-диметилкарбамоил-2- (4-гидроксифенил)-этил]-амид
(S)-2-Амино-3-(4-гидроксифенил)-N, N-диметилпропионамида гидрохлорид (1,05 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции дихлорметан/ДМФ 2: 1, промывали только кислотой) и продукт очищали с помощью хроматографии на силикагеле с элюированием 20, 40, 50 и 75% этилацетатом в гексанах с последующим растиранием с эфиром. Выход 400 мг, 104%; ВЭЖХ (60/40) 3,93 минуты, (98%); т. пл. 228-231oC (разложение, желтел при 210oC); ТРМС 386/388 (М+, 100%);
Анал. Расчетное для C20H20ClN3O3 + 0,9 H2O: C, 59,75; H, 5,47; N, 10,45.
Обнаруженное: C, 61,05, H, 5,79; N, 10,08.
Пример 68а.
(S)-2-Амино-3-(4-гидроксифенил)-N,N-диметилпропионамида гидрохлорид
(S)-[1-Диметилкарбамоил-2-(4-гидроксифенил)-этил] -карбаминовой кислоты трет-бутиловый эфир (5,7 г, 18,5 ммоля) растворяли в 4М HCl-диоксане (7 мл) при 0oC. Смесь перемешивали при 25oC в течение 3 часов, концентрировали и остаток растирали с эфиром. Выход, 5,23 г; ВЭЖХ (60/40) 3,32 минуты (98%).
Пример 68b.
(S)-[1-Диметилкарбамоил-2-(4-гидроксифенил)-этил] -карбаминовой кислоты трет-бутиловый эфир
Диметиламина гидрохлорид (79 ммолей) и Boc-L-тирозин (66 ммолей) соединяли по процедуре А (температура реакции 0-25oC, растворитель для реакции - дихлорметан/ДМФ 12: 1, время реакции - 60 часов) и продукт очищали с помощью хроматографии на силикагеле с элюированием 10, 20, 30, 50 и 70% этилацетатом в гексанах.
Пример 69.
5-Хлор-1Н-индол-2-карбоновой кислоты ((1S)-метоксикарбамоил-2-фенилэтил)-амид
(2S)-Амино-N-метокси-3-фенилпропионамида гидрохлорид (1,02 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (1,02 ммоля) соединяли по процедуре А. Остаток растирали с эфиром с получением легкого желтого твердого вещества. Выход 160 мг, 36%; т.пл. 210-213oC (разлож.); PBMS 372/374 (MH+, 100%);
Анал. Расчетное для C19H18ClN3O3 + 1,75 H2O: C, 56,58; H, 5,37; N, 10,42.
Обнаруженное: C, 56,88; H, 5,09; N, 10,03.
Пример 69а.
(2S)-Амино-N-метокси-3-фенилпропионамида гидрохлорид
[(1S)-(Метоксикарбамоил)-2-фенилэтил] -карбаминовой кислоты трет-бутиловый эфир (200 мг, 0,68 ммоля) растворяли в 4М HCl-диоксане при 0oC и смесь перемешивали при 25oC. Через 0,5 часа смесь концентрировали и остаток растирали с эфиром.
Пример 69b.
[(1S)-(Метоксикарбамоил)-2-фенилэтил] -карбаминовой кислоты трет-бутиловый эфир
Метоксиамина гидрохлорид (83,5 ммоля) и Boc-L-фенилаланин (20 ммолей) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием этилацетатом/гексанами 1:1 и 2:1 с последующим растиранием с эфиром. Выход 1,80 г, 31%.
Пример 70.
5-Хлор-1Н-индол-2-карбоновой кислоты ((1R)-метилкарбамоил-2- фенилэтил)-амид
(R)-2-Амино-N-метил-3-фенилпропионамида гидрохлорид (0,84 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,84 ммоля) соединяли по процедуре А (температура реакции 0-25oC). Продукт-сырец растирали с дихлорметаном и затем с эфиром и сушили. Выход 236 мг, 79%: ВЭЖХ (60/40) 4,63 минуты (97%); PBMS 356/358 (MH+, 100%);
Анал. Расчетное для C19H18ClN3O2 + 0,25 H2O: C, 63,33, H, 5,18; N, 11,66.
Обнаруженное: C, 63,37; H, 5,50; N, 12,06.
Пример 70а.
(R)-2-Амино-N-метил-3-фенилпропионамида гидрохлорид
(R)-(1-Метилкарбамоил-2-фенилэтил)-карбаминовой кислоты трет-бутиловый эфир (722 мг, 2,6 ммоля) растворяли в 4М HCl-диоксане (10 мл) при 0oC. Смесь перемешивали в течение 1 часа при 25oC, концентрировали и остаток растирали с эфиром. Выход 517 мг, 93%.
Пример 70b.
(R)-(1-Метилкарбамоил-2-фенилэтил)-карбаминовой кислоты трет-бутиловый эфир
Метиламина гидрохлорид (3,1 ммоля) и Boc-фенилаланин (2,8 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции 144 часа, промывали сначала кислотой, затем основанием) с получением продукта, который использовали без дополнительной очистки. Выход 760 мг, 96%.
Пример 71.
5,6-Дихдор-1Н-индол-2-карбоновой кислоты ((1S)-диметилкарбамоил-2-фенилэтил)-амид
(S)-2-Амино-N, N-диметил-3-фенилпропионамида гидрохлорид (0,06 ммоля) и 5,6-дихлор-1Н-индол-2-карбоновую кислоту (0,06 ммоля) соединяли по процедуре А (температура реакции 0-25oC, время реакции 96 часов). Неочищенный продукт растирали с эфиром-гексанами 1:1 и сушили. Выход 24 мг, 96%. ВЭЖХ (60/40) 8,05 минуты (97%); PBMS 405/407 (MH+, 100%).
Анал. Расчетное для C20H19Cl2N3O2 + 0,25 H2O: C, 58,76; H, 4,81; N, 10,28.
Обнаруженное: C, 58,95; H, 4,89; N, 9,90.
Пример 71а.
(S)-2-Амино-N,N-диметил-3-фенилпропионамида гидрохлорид
(1-Диметилкарбамоил-2-фенилэтил)-карбаминовой кислоты трет-бутиловый эфир (8,6 г, 29 ммолей) растворяли в 4М HCl-диоксане (110 мл) при 0oC и смесь перемешивали при 25oC в течение 1 часа. Смесь концентрировали и твердые вещества растирали с эфиром. Выход 6,2 г, 92%; PBMS 193 (MH+, 100%).
Пример 71b.
5,6-Дихлор-1Н-индол-2-карбоновая кислота
Цинковую пыль (3,52 г, 54 ммоля) медленно добавляли к теплому раствору 3,4-дихлор-5-нитрофенилпировиноградной кислоты (1,5 г, 5,4 ммоля) в уксусной кислоте (15 мл). Через несколько минут происходила бурная реакция (экзотермическая). Полученный в результате раствор нагревали до 80oC и реакция, по-видимому, заканчивалась (ТСХ). Смесь фильтровали, твердые отфильтрованные вещества промывали уксусной кислотой и фильтрат концентрировали. Остаток растворяли в 2N NaOH, полученный в результате раствор промывали эфиром (3х), дихлорметаном (2х) и подкисляли до pH 6N HCl и экстрагировали этилацетатом. Экстракты сушили и концентрировали с получением легкого коричневого твердого вещества (458 мг, 34%): ВЭЖХ (60/40) 5,31 (93%).
Пример 71с.
3,4-Дихлор-5-нитрофенилпировиноградная кислота
Абсолютный спирт (25 мл) добавляли при 3-15oC к перемешиваемой смеси металлического калия (2,67 г, 68 ммолей) в эфире (100 мл). Полученный в результате раствор обрабатывали при 3oC раствором диэтилоксалата (10,0 г, 62 ммоля) в течение 5-10 минут, и полученный раствор перемешивали в течение 30 минут при 3oC и 25oC в течение 18 часов. Смесь фильтровали и полученное твердое вещество промывали эфиром и сушили (13,7 г). Этот материал (12,7 г) растворяли в 400 мл горячей воды, раствор охлаждали и экстрагировали эфиром. Полученный в результате водный слой подкисляли до pH 2 концентрированной HCl и эфирный слой отделяли, сушили и концентрировали с получением 7,5 г твердого вещества, которое растирали с гексанами, получая вещество заголовка в виде желтого твердого вещества (7,01 г, 41%).
Пример 72.
5-Бром-1Н-индол-2-карбоновой кислоты ((1S)-диметилкарбамоил-2- фенилэтил)-амид
(S)-2-Амино-N, N-диметил-3-фенилпропионамида гидрохлорид (1,0 ммоль) и 5-бром-1H-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC) и полученное пенообразное вещество растирали с эфиром-гексанами 1:1 и сушили.
Выход 374 мг, 90%; БЭЖХ (60/40) 6,17 минуты (98%); т.пл. 199-201oC; PBMS 414/416 (MH+, 100%);
Анал. Расчетное для C20H20BrN3O2: C, 57,98; H, 4,82; N, 10,14.
Обнаруженное: C, 58,07; H, 5,12; N, 10,08.
Пример 73.
5-Метил-1Н-индол-2-карбоновой кислоты ((1S)-диметилкарбамоил-2-фенилэтил)-амид
(S)-2-Амино-N, N-диметил-3-фенилпропионамида гидрохлорид (1,0 ммоль) и 5-метил-1Н-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC). Неочищенный продукт растирали с эфиром-гексанами 1: 1 и сушили. Выход 302 мг, 87%, ВЭЖХ (60/40) 5,46 минуты (99%); т.пл. 198,5-200oC; PBMS 350 (MH+, 100%).
Анал. Расчетное для C21H23N3O2: C, 72,18; H, 6,63; N, 12,04.
Обнаруженное: C, 72,14; H, 6,90; N, 12,11.
Пример 74.
5-Метокси-1Н-индол-2-карбоновой кислоты ((1S)-диметилкарбамоил-2-фенилэтил)-амид
(S)-2-Амино-N, N-диметил-3-фенилпропионамида гидрохлорид (1,0 ммоль) и 5-метокси-1Н-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC, время реакции 60 часов) и полученное в результате пенообразное вещество растирали с эфиром. Выход 329 мг, 90%; ВЭЖХ (60/40) 4,27 минуты (99%); PBMS 366 (MH+, 100%);
Анал. Расчетное для C21H23N3O3 + 0,125 H2O: C, 68,60; H, 6,37; N, 11,43.
Обнаруженное: C, 68,50; H, 6,34; N, 11,45.
Пример 75.
5-Фтор-1Н-индол-2-карбоновой кислоты ((1S)-диметилкарбамоил- 2-фенилэтил)-амид
(S)-2-Амино-N, N-диметил-3-фенилпропионамида гидрохлорид (1,0 ммоль) и 5-фтор-1Н-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC, время реакции - 60 часов) и полученное твердое вещество растирали с эфиром. Выход 320 мг, 91%; ВЭЖХ (60/40) 4,74 минуты (100%); т.пл. 229,5-232oC; PBMS 354 (MH+, 100%).
Анал. Расчетное для C20H20FN3O2: C, 67,97; H, 5,70; N, 11,89.
Обнаруженное: C, 67,88; H, 5,74; N, 11,71.
Пример 76.
5-Циано-1Н-индол-2-карбоновой кислоты ((1S)-диметилкарбамоил-2- фенилэтил)-амид
(S)-2-Амино-N, N-диметил-3-фенилпропионамида гидрохлорид (0,16 ммоля) и 5-циано-1Н-индол-2-карбоновую кислоту (0,16 ммоля) соединяли по процедуре А (температура реакции 0-25oC) и продукт очищали с помощью хроматографии на силикагеле с элюированием этилацетатом/гексанами 1:1. Выход 38 мг, 66%; ВЭЖХ (60/40) 4,08 минуты (97%); PBMS 361 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 12,1 (шир, 1Н), 9,04 (д, 1Н, J=8,1 Гц), 8,27 (с, 1Н), 7,52 (м, 2Н), 7,43 (м, 1Н), 7,33 (м, 2Н), 7,25 (м, 2Н), 7,18 (м, 1Н), 5,10 (м, 1Н), 3,03 (м, 2Н), 3,00 (с, 3Н), 2,83 (с, 3Н).
Анал. Расчетное для C21H20N4O2 + 0,5 H2O: C, 68,28; H, 5,73; N, 15,17.
Обнаруженное: C, 68,51; H, 5,66; N, 14,85.
Пример 76а.
5-Циано-1Н-индол-2-карбоновая кислота
5-Циано-1Н-индол-2-карбоновой кислоты этиловый эфир (1,71 г, 8 ммолей) добавляли к раствору этанола (10 мл) и гидроксида калия (2 г) и полученную смесь нагревали в колбе с обратным холодильником в течение 1 часа. Добавляли воду для растворения осадка и 6N HCl добавляли для доведения pH до 1. Смесь охлаждали на ледяной бане, фильтровали и полученное в результате бесцветное твердое вещество промывали холодной водой и сушили (1,51 г). Часть (1,4 г) суспендировали в горячей уксусной кислоте (40 мл) и охлаждали с получением твердого вещества, которое отфильтровывали, промывали холодным ацетатом и сушили: выход 980 мг, 70%; ВЭЖХ (60/40) 3,09 минуты (97%).
Пример 76b.
5-Циано-1Н-индол-2-карбоновой кислоты этиловый эфир
Цинковую пыль (57,8 г, 887 ммолей) добавляли в горячую суспензию 3-циано-5-нитрофенилпировиноградной кислоты этилового эфира (23,2 г, 88 ммолей) в уксусной кислоте (225 мл) и воде (225 мл, Осторожно! первоначально бурно экзотермичная) такими порциями, чтобы поддерживать кипение в колбе с обратным холодильником и реакция с сохранением кипения продолжалась в течение 0,5 часа. Смесь фильтровали, отфильтрованные соли промывали горячей уксусной кислотой (150 мл), и фильтрат охлаждался в течение ночи с получением кристаллов, которые отфильтровывали, промывали холодной уксусной кислотой - водой 1: 1, водой и сушили (10,11 г, 53%). Фильтрат концентрировали, остаток растворяли в этилацетате, и полученный раствор промывали насыщенным водным раствором бикарбоната натрия, солевым раствором, сушили и концентрировали с получением второй порции (5,05 г). Основная масса использовалась при последующих превращениях.
Пример 76с.
3-Циано-5-нитрофенилпировиноградной кислоты этиловый эфир
Раствор этоксида натрия в этаноле (из 2,2 г, 400 ммолей металлического в 400 мл этанола) добавляли при 0oC к смеси перегнанного диэтилоксалата (120 г, 821 ммоля) и 3-метил-4-нитро-бензонитрила (32 г, 197 ммолей). Полученный в результате красный раствор нагревали при 40oC в течение 18 часов. Охлажденную смесь разводили водой (600 мл) и подкисляли концентрированной HCl до pH 2,1. Осадок, который образовался, собирали фильтрованием смеси с температурой 13oC, сушили и очищали с помощью хроматографии на силикагеле с элюированием 15, 30 и 50% ацетоном-гексанами с получением оранжевого твердого вещества, которое использовали без очистки (23,6 г, 31%). Образец перекристаллизовывали из этилацетата для определения характеристик.
Пример 77.
1Н-Индол-2-карбоновой кислоты ((1S)-диметилкарбамоил-2-фенилэтил)-амид
(S)-2-Амино-N, N-диметил-3-фенилпропионамида гидрохлорид (1,0 ммоль) и 1Н-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (температура реакции 0-25oC). Полученное в результате твердое вещество растирали с гексанами, затем с эфиром. Выход 272 мг, 81%; ВЭЖХ (70/30), 3,49 минуты (99%); т.пл. 199-200oC; PBMS 336 (MH+, 1000).
Анал. Расчетное для C20H21N3O2: C, 71,62; H, 6,31; N, 12,53.
Обнаруженное: C, 71,45; H, 6,39; N, 12,50.
Пример 78.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-((3S, 4S) -дигидроксипирролидин-1-ил)-2-оксоэтил]-амид
(3S, 4S)-2-Амино-1-(3,4-дигидроксипирролидин-1-ил)-3-фенилпропан-1-она гидрохлорид (0,94 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (1,03 ммоля) соединяли по процедуре А (время реакции 170 часов) и продукт-сырец очищали с помощью колоночной хроматографии на силикагеле с элюированием этилацетатом. Выход 150 мг, 37%; ВЭЖХ (60/40) 3,08 минуты (96%);
1H ЯМР (ДМСО-d6) δ 11,73 (с, 1Н), 8,90 (д, 1Н, J=8,5 Гц), 7,72 (д, 1Н, J= 1,5 Гц), 7,39 (д, 1H, J=8,7 Гц), 7,30 (м, 2Н), 7,30-7,1 (м, 5Н), 5,22 (м, 1Н), 5,13 (м, 1Н), 4,91 (м, 1Н), 3,97 (м, 1Н), 3,91 (м, 1Н), 3,60 (м, 2Н), 3,5-3,2 (м, 2Н), 3,00 (м, 2Н).
Анал. Расчетное для C22H22ClN3O4: C, 61,75; H, 5,18; N, 9,82.
Обнаруженное: C, 61,65; H, 5,45; N, 9,17.
Пример 78а.
(3S, 4S)-2-Амино-1-(3,4-дигидроксипирролидин-1-ил)-3- фенилпропан-1-она гидрохлорид
(3S, 4S)-[1-Бензил-2-(3,4-дигидроксипирролидин-1-ил)-2- оксоэтил]-карбаминовой кислоты трет-бутиловый эфир (360 мг, 1,00 ммоль) растворяли в 4М HCl-диоксане (4 мл) при 25oC в течение 3 часов. Смесь концентрировали и полученное в результате желтое твердое вещество растирали с эфиром и сушили. Выход 304 мг, 103%.
Пример 78b.
[1-бензил-2-(3,4-дигидроксипирролидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир
Boc-L-фенилаланин (2,2 ммоля) и (3S,4S)-дигидроксипирролидин (патент США N 4634775, пример 1С, 206 мг, 2,0 ммоля) соединяли по процедуре А (температура реакции 0-25oC) с получением бесцветного твердого вещества, которое использовали без дополнительной очистки. Выход 431 мг, 61%.
Пример 79.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-((3RS) - гидроксипиперидин-1-ил)-2-оксоэтил]-амид
2(S)-Амино-1-((3RS)-гидроксипиперидин-1-ил)-3-фенилпропан-1-она гидрохлорид (570 мг, 2,0 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (429 мг, 2,2 ммоля) соединяли по процедуре А (растворитель - дихлорметан-диметилформамид 5:2) и продукт-сырец растирали с эфиром-гексанами 1:1. Полученное в результате твердое вещество очищали с помощью колоночной хроматографии на силикагеле с элюированием этилацетатом/гексанами 3:2 и 2:1 с последующим растиранием с эфиром-гексанами 1: 1. Выход 430 мг, 51%: ВЭЖХ (60/40) 3,45 минуты (95%);
Анал. Расчетное для C23H24ClN3O3 + 0,125 C6H14: C, 65,32; H, 5,94; N, 9,62.
Обнаруженное: C, 65,01; H, 6,19; N, 9,22.
Пример 79а.
(2S)-Амино-1-(3-гидроксипиперидин-1-ил)-3-фенилпропан-1-она гидрохлорид
[(1S)-Бензил-2-((3RS)-гидроксипиперидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир (7,13 г, 20 ммолей) растворяли в 4М HCl-диоксане (40 мл) при 25oC в течение 3 часов. Смесь концентрировали и полученное в результате масло перемешивали в атмосфере эфира в течение 72 часов. Полученную в результате суспензию фильтровали и твердое вещество промывали эфиром и сушили. Выход 5,64 г, 99%.
Пример 79b.
[(1S)-Бензил-2-((3RS)-гидроксипиперидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир
Boc-L-фенилаланин (8,17 г, 30,8 ммоля) и 3-гидроксипиперидина гидрохлорид (4,24 г, 30,8 ммоля) соединяли по процедуре А с получением соединения заголовка в виде масла, которое использовали без дополнительной очистки. Выход 7,79 г, 73%.
Пример 80.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-оксо-2-(3-оксопиперазин-1-ил)-этил]-амид
4-((2S)-Амино-3-фенилпропионил)-пиперазин-2-она гидрохлорид (140 мг, 0,5 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (98 мг, 0,5 ммоля) соединяли по процедуре А и продукт-сырец очищали с помощью колоночной хроматографии на силикагеле с элюированием этилацетатом и 2% этанолом в этилацетате с последующим растиранием с эфиром. Выход 71 мг, 33%; ВЭЖХ
(60/40) 3,53 минуты (100%); PBMS 425/427 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,78 (шир, 0,5Н), 11,76 (шир, 0,5Н), 9,03 (м, 0,5Н), 9,02 (м, 0,5Н), 8,06 (м, 0,5Н), 8,04 (м. 0,5Н), 7,73 (д, 1Н, J=2 Гц), 7,38 (д, 1Н, J= 8,7 Гц), 7,32 (м, 2Н), 7,20 (м, 2Н), 7,2-7,1 (м, 2Н), 5,15 (м, 0,5Н), 5,05 (м, 0,5Н), 4,20 (д, 0,5H, J=17 Гц), 4,08 (д, 0,5H, J=17 Гц), 3,85 (д, 0,5H, J=17 Гц), 3,9 (м, 0,5Н), 3,6 (м, 2Н), 3,2-2,9 (м, 4Н).
Пример 80а.
4-((2S)-Амино-3-фенилпропионил)-пиперазин-2-она гидрохлорид
[(1S)-Бензил-2-оксо-2-(3-оксопиперазин-1-ил)-этил] -карбаминовой кислоты трет-бутиловый эфир (400 мг, 1,2 ммоля) растворяли в 4М HCl-диоксане (10 мл) при 25oC в течение 0,5 часа. Смесь концентрировали и остаток выпаривали вместе с дихлорметаном, растирали с эфиром и сушили. Выход 340 мг, 103%.
Пример 80b.
[(1S) -Бензил-2-оксо-2-(3-оксопиперазин-1-ил)-этил] - карбаминовой кислоты трет-бутиловый эфир
Boc-L-фенилаланин (530 мг, 2 ммоля) и пиперазин-2-он (J. Am. Chem. Soc., 62, 1202 (1940), 200 мг, 2 ммоля) соединяли по процедуре А (растворитель для реакции дихлорметан/диметилформамид 2:1, промывали 1N NaOH после промываний кислотой) и продукт использовали без дополнительной очистки. Выход 404 мг, 58%.
Пример 81.
5-Хлор-1H-индол-2-карбоновой кислоты ((1S)-метил-2-морфолин-4-ил-2-оксоэтил)-амид
(2S)-Амино-1-морфолин-4-ил-пропан-1-она гидрохлорид (195 мг, 1,0 ммоль) и 5-хлор-1Н-индол-2-карбоновую кислоту (195 мг, 1,0 ммоль) соединяли по процедуре А (промывали 1N NaOH после промываний кислотой) с получением продукта-сырца, который растирали с эфиром и сушили. Выход 150 мг, 45%: ВЭЖХ (60/40) 3,61 минуты (100%); PBMS 336/338 (MH+, 100%);
Анал. Расчетное для C16H18ClN3O3: C, 57,23; H, 5,40; N, 12,51.
Обнаруженное: C, 57,01; H, 5,49; N, 12,24.
Пример 81а.
(2S)-Амино-1-морфолин-4-ил-пропан-1-она гидрохлорид
((1S)-Метил-2-морфолин-4-ил-2-оксоэтил)-карбаминовой кислоты трет-бутиловый эфир (3,88 г, 15 ммолей) растворяли в 4М HCl-диоксане (20 мл) при 25oC в течение 1,25 часа. Смесь концентрировали и остаток растирали с эфиром и сушили: выход 2,51 г, 86%.
Пример 81b.
((1S)-Метил-2-морфолин-4-ил-2-оксоэтил)-карбоновой кислоты трет-бутиловый эфир
Boc-L-Аланин (3,50 мг, 20 ммолей) и морфолин (1,74 г, 20 ммолей) соединяли по процедуре А (промывали 1N NaOH после промывания кислотой) с получением бесцветного масла, которое использовали без дополнительной очистки. Выход 3,94 г, 76%.
Пример 82.
5-Хлор-1Н-индол-2-карбоновой кислоты ((1S)-метилкарбамоил-2-фенилэтил)-амид
(2S)-Амино-N-метил-3-фенилпропионамида гидрохлорид (214 мг, 1,0 ммоль) и 5-хлор-1Н-индол-2-карбоновую кислоту (195 мг, 1,0 ммоль) соединяли по процедуре А и продукт-сырец растирали с эфиром и сушили. Выход 160 мг, 45%: ВЭЖХ (60/40) 4,60 минуты (100%);
1H ЯМР (ДМСО-d6) δ 11,70 (шир, 1Н), 8,73 (д, 1Н, J=8,5 Гц), 8,08 (чет, 1H, J=4,6 Гц), 7,72 (д, 1H, J=1,9 Гц), 7,39 (д, 1Н, J=8,8 Гц), 7,32 (м, 2Н), 7,25-7,10 (м, 5Н), 4,68 (м, 1Н), 3,10 (д, A из AB, 1Н, J=4,2, 13 Гц), 2,96 (дд, 1Н, J=10,7, 13 Гц), 2,62 (д, 3Н, J=4,6 Гц).
Пример 82а.
(2S)-Амино-N-метил-3-фенил-пропионамида гидрохлорид
[((1S)-1-Метилкарбамоил-2-фенилэтил)-карбаминовой кислоты трет-бутиловый эфир (2,35 г, 8,45 ммоля) растворяли в 4М HCl-диоксане (20 мл) при 25oC в течение 2 часов. Смесь концентрировали и остаток растирали с эфиром и сушили. Выход 1,70 г, 94%.
Пример 82b.
((1S)-1-Метилкарбамоил-2-фенилэтил)-карбаминовой кислоты трет-бутиловый эфир
Boc-N-фенилаланин (2,65 г, 10 ммолей) и метиламина гидрохлорид (675 мг, 10 ммолей) соединяли по процедуре А (промывали 1N NaOH после промываний кислотой) с получением вещества заголовка в виде бесцветного твердого вещества, которое использовали без дополнительной очистки. Выход 2,41 г, 87%; ВЭЖХ (60/40) 3,83 минуты (100%).
Пример 83.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-(метоксиметилкарбамоил)-этил] -амид
(2S)-Амино-N-метокси-N-метилпропионамида гидрохлорид (169 мг, 1,0 ммоль) и 5-хлор-1Н-индол-2-карбоновую кислоту соединяли по процедуре А (промывали 1N NaOH после промываний кислотой) с получением продукта (290 мг, 94%): ВЭЖХ (60/40) 4,03 минуты (94%); PBMS 310/312 (MH+, 100%);
Анал. Расчетное для C14H16ClN3O3: C, 54,29; H, 5,21; N, 13,57.
Обнаруженное: C, 54,17; H, 5,26; N, 13,31.
Пример 83а.
(2S)-Амино-N-метокси-N-метилпропионамида гидрохлорид
[(1S)-(Метоксиметилкарбамоил)-этил] -карбаминовой кислоты трет-бутиловый эфир (3,55 г, 15,3 ммоля) растворяли в 4М HCl-диоксане (20 мл) при 25oC в течение 0,75 часа. Смесь концентрировали и остаток выпаривали вместе с эфиром и дихлорметаном и сушили. Выход 2,2 г (86%).
Пример 83b.
[1-(Метоксиметилкарбамоил)-этил] -карбаминовой кислоты трет-бутиловый эфир
L-Boc-Аланин (3,50 г, 20 ммолей) и O,N-диметилгидроксиамина гидрохлорид (1,94 г, 20 ммолей) соединяли по процедуре А (промывали 1N NaOH после промываний кислотой) и полученное в результате бесцветное твердое вещество использовали без дополнительной очистки. Выход 3,71 г (80%).
Пример 84.
5-Бром-1H-индол-2-карбоновой кислоты ((1S)-карбамоил-2- фенилэтил)-амид
L-фенилаланинамида гидрохлорид (835 мг, 4,17 ммоля) и 5-бром-1Н-индол-2-карбоновую кислоту (1,0 г, 4,17 ммоля) соединяли по процедуре А с заменой следующей обработкой: реакционную смесь разводили этилацетатом и 2N NaOH. Полученную суспензию фильтровали и собранное твердое вещество промывали этилацетатом, 2N NaOH, 2N HCl, эфиром и сушили. Выход 890 мг, PBMS 386/388 (MH+, 100%);
Анал. Расчетное для C18H16BrN3O2: C, 55,97; H, 4,18; N, 10,88.
Обнаруженное: C, 55,69; H, 4,48; N, 10,48.
Пример 85.
5-Хлор-1Н-индол-2-карбоновой кислоты ((1S)-(метоксиметилкарбамоил)-2-фенилэтил)-амид
(2S)-Амино-N-метокси-N-метил-3-фенилпропионамида гидрохлорид (317 мг, 1,3 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (253 мг, 1,3 ммоля) соединяли по процедуре А (0-25oC, промывали сначала кислотой, затем основанием). Продукт-сырец очищали с помощью колоночной хроматографии на силикагеле с элюированием 30 и 40% этилацетатом в гексанах. Полученное пенообразное вещество растирали с изопропиловым эфиром с получением не совсем белого твердого вещества (356 мг, 71%): ВЭЖХ (60/40) 8,28 минуты (98%);
Анал. Расчетное для C20H20ClN3O3: C, 62,26; H, 5,22; N, 10,89.
Обнаруженное: C, 62,22; H, 5,60; N, 10,73.
Пример 85а.
(2S)-Амино-N-метокси-N-метил-3-фенилпропионамида-гидрохлорид
[(1S)-(Метоксиметилкарбамоил)-2-фенилэтил] -карбаминовой кислоты трет-бутиловый эфир (2,97 г, 9,6 ммоля) растворяли в 4М HCl-диоксане (36 мл) при 0oC. Полученную смесь перемешивали при 25oC в течение 1 часа, концентрировали и остаток растирали с эфиром и сушили. Выход 2,27 г, 96%.
Пример 85b.
[(1S)-(Метоксиметилкарбамоил)-2-фенилэтил]-карбаминовой кислоты трет-бутиловый эфир
Boc-L-фенилаланин (4,0 г, 15,1 ммоля) и N,O-диметилгидроксиламина гидрохлорид (3,82 г, 15,1 ммоля) соединяли по процедуре А (0-25oC, промывали сначала кислотой, затем основанием). Полученное бесцветное масло использовали без очистки (3,22 г, 69%).
Пример 86.
(2RS)-[(5-Хлор-1Н-индол-2-карбонил)-амино] -2-метил-3- фенилпропионовой кислоты метиловый эфир
Рацемический 2-амино-2-метил-3-фенилпропионовой кислоты метиловый эфир (200 мг, 0,87 ммоля) и 5-хлор-1H-индол-2-карбоновую кислоту (170 мг, 0,87 ммоля) соединяли по процедуре А (растворитель - дихлорметан/диметилформамид 2: 1) и продукт очищали с помощью хроматографии на силикагеле с элюированием 10% этилацетатом в гексанах. Выход 286 мг, 89%; ВЭЖХ (60/40) 9,63 минуты (85%); ТРМС 371/373 (MH+, 100>);
1H ЯМР (CDCl3) δ 9,31 (с, 1Н), 7,57 (д, 1H J ≤ Гц), 7,37 (д, 1Н, J=8,8 Гц), 7,20 (м, 4Н), 7,04 (м, 2Н), 6,84 (с, 1Н), 6,66 (с, 1Н), 3,81 (с, 3Н), 3,67 (A из AB, 1Н, J=13,5 Гц), 3,28 (B из AB, 1H, J=13,5 Гц), 1,80 (с, 3Н).
Пример 87.
(2RS)-[(5-Хлор-1Н-индол-2-карбонил)-амино] -2-метил-3- фенилпропионовая кислота
Водный 2N LiOH (0,10 мл, 0,50 ммоля) добавляли к раствору (2RS)-[(5-хлор-1Н-индол-2-карбонил)-амино)-2-метил-3-фенил-пропионовой кислоты метилового эфира (132 мг, 0,36 ммоля) в тетрагидрофуране (8 мл) при 25oC. Полученный в результате раствор перемешивали в течение 1 часа, концентрировали и остаток растворяли в этилацетате и воде (15 мл). pH доводили до 1 с 2N HCl при 0oC. Органический слой отделяли, промывали водой, солевым раствором и сушили с получением пенообразного вещества, которое использовали без дополнительной очистки (129 мг, 102%): ВЭЖХ (60/40) 4,42 минуты (99%); ТРМС 357/359 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,88 (с, 1Н), 7,57 (с, 1Н), 7,35 (д, 1Н, J=8,8 Гц), 7,3-7,2 (м, 5Н), 7,16 (м, 2Н), 6,75 (м, 1Н), 6,67 (м, 1Н), 3,57 (A из AB, 1Н, J=13,7 Гц), 3,42 (B из AB, 1Н, J=13,7 Гц), 1,80 (с, 3Н).
Анал. Расчетное для C19H17ClN2O3 + 0,3 H2O: C, 63,00; H, 4,90; N, 7,73.
Обнаруженное: C, 63,38; H, 5,31; N, 7,42.
Пример 88.
5-Хлор-1Н-индол-2-карбоновой кислоты [[1S)-бензил-2-оксо-2- (1-оксо-1-тиоморфолин-4-ил)-этил]-амид
м-Хлорпероксибензойную кислоту (80 мг, равные 50%, 0,23 ммоля) добавляли при 25oC к раствору 5-хлор-1Н-индол-2-карбоновой кислоты ((1S)-бензил-2-оксо-2-тиоморфолин-4-илэтил)амида (100 мг, 0,23 ммоля) в дихлорметане (2 мл). Через 1 час смесь разводили этилацетатом и промывали три раза смесью 50/50 насыщенного водного раствора бикарбоната натрия и 10% водного раствора тиосульфата натрия, один раз насыщенным водным бикарбонатом натрия, солевым раствором и сушили. Продукт-сырец очищали с помощью колоночной хроматографии на силикагеле с элюированием 0,5-8% этанолом в дихлорметане с получением соединения заголовка. Выход 76%; ВЭЖХ (60/40) 3,97 минуты (97%); т.пл. 230-234oC; ТРМС 444/446 (MH+, 100%);
Анал. Расчетное для C22H22ClN3O3S + 0,5 H2O: C, 58,34; H, 5,12; N, 9,28.
Обнаруженное: C, 58,41; H, 5,37; N, 8,90.
Пример 89.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(1,1- диоксо-1-тиоморфолин-4-ил)-2-оксоэтил]-амид
м-Хлорпероксибензойную кислоту (202 мг, равные 50%, 0,58 моля) добавляли при 25oC к раствору 5-хлор-1Н-индол-2-карбоновой кислоты ((1S)-бензил-2-оксо-2-тиоморфолин-4-илэтил)-амида (100 мг, 0,23 ммоля) в дихлорметане (2 мл). Через 1 час смесь разводили этилацетатом и полученный раствор промывали три раза смесью 50/50 насыщенного водного бикарбоната натрия и 10% водного тиосульфата натрия, один раз насыщенным водным бикарбонатом натрия, солевым раствором и сушили. Продукт-сырец очищали с помощью колоночной хроматографии на силикагеле с элюированием 30, 40 и 50% этилацетатом в гексанах с получением соединения заголовка. Выход 60%: ВЭЖХ (60/40) 5,69 минуты (98%); PBMS 460/462 (MH+, 100%);
Анал. Расчетное для C22H22ClN3O4S + 0,4 H2O: C, 56,56; H, 4,92; N, 8,99.
Обнаруженное: C, 56,77; H, 5,15; N, 8,60.
Пример 90.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-оксо-2- (1-оксо-1-тиазолидин-3-ил)-этил]-амид
м-Хлорпероксибензойную кислоту (167 мг, равное 50%, 0,48 ммоля) добавляли при 25oC к раствору 5-хлор-1Н-индол-2-карбоновой кислоты ((1S)-бензил-2-оксо-2-тиазолидин-3-илэтил)-амида (200 мг, 0,48 ммоля) в дихлорметане (4 мл). Через 0,5 часа смесь разводили этилацетатом и промывали три раза смесью 50/50 насыщенного водного бикарбоната натрия и 10% водного тиосульфата натрия, один раз насыщенным водным бикарбонатом натрия, солевым раствором и сушили. Продукт-сырец концентрировали до желтого твердого вещества и затем очищали с помощью колоночной хроматографии на силикагеле с элюированием 1-8% этанолом в дихлорметане и затем растирали с эфиром с получением вещества заголовка. Выход 151 мг (73%); ВЭЖХ (60/40) 3,64 минуты (98%); PBMS 430/432 (MH+, 100%);
Анал. Расчетное для C21H20ClN3O3S + 0,6 H2O: C,57,23; H, 4,85; N, 9,53.
Обнаруженное: C, 57,00; H, 4,85; N, 9,25.
Пример 91.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2- (3-гидроксииминопирролидин-1-ил)-2-оксоэтил]-амид
Гидроксиламина гидрохлорид (68 мг, 0,82 ммоля) и карбонат калия (136 мг, 0,98 ммоля) добавляли к раствору 5-хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-оксо-2- (3-оксопирролидин-1-ил)-этил]-амида в этаноле (5 мл) и воде (1 мл) при 25oC. Через 48 часов реакционную смесь концентрировали и остаток растворяли в этилацетате. Полученный в результате раствор промывали два раза водой и один раз солевым раствором, сушили Na2SO4 и концентрировали. Два вещества, по-видимому, являются син/анти изомерами оксима, разделяемыми с помощью хроматографии на силикагеле с элюированием 2,5%, 5% и 10% этанолом в дихлорметане.
Пример 91 (I)
Для менее полярного изомера:
Выход 48 мг (14%); ВЭЖХ (60/40) 4,69 минуты (97%); т.пл. 216-220oC (темнел при 210oC); PBMS 425/427 (MH+, 100%).
1H ЯМР (ДМSO-d6) δ 11,75 (шир, 1Н), 10,87 (с, 0,5H), 10,86 (с, 0,5Н), 9,02 (м, 1Н), 7,72 (д, 1H, J=2,0 Гц), 7,4-7,1 (м, 8Н), 4,95 (м, 0,5Н), 4,85 (м, 0,5Н), 4,40 (д, 0,5H, J=15 Гц), 4,0 (м, 1,5H), 3,9 (м, 0,5H), 3,61 (м, 1Н), 3,5 (м, 0,5H), 3,10 (м, 2Н), 2,8-2,5 (м, 2Н);
Анал. Расчетное для C22H21ClN4O3: C, 62,19; H, 4,98; N, 13,19.
Обнаруженное: C, 61,82; H, 5,07; N, 12,95.
Пример 91 (II)
Для более полярного изомера:
Выход 69 мг (20%); ВЭЖХ (60/40) 6,78 минуты (> 99%); т.пл. 223-224oC (разл., смола); PBMS 425/427 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,74 (шир, 1Н), 10,87 (с, 1Н), 10,84 (с, 1Н), 9,05 (д, 0,5H, J= 8,1 Гц), 8,99 (д, 1H, J=8,0 Гц), 7,73 (д, 1Н, J=2Гц), 7,4-7,1 (м, 8Н), 4,97 (м, 1Н), 4,85 (м, 1Н), 4,47 (д, 0,5H, J=17 Гц), 3,95 (м, 1,5Н), 3,87 (м, 0,5Н), 3,65-3,4 (м, 1,5Н), 3,10 (м, 2Н), 2,7-2,5 (м, 2Н).
Анал. Расчетное для C22H21ClN4O3: C, 62,19; H, 4,98; N, 13,19.
Обнаруженное: C, 61,85; H, 5,17; N, 13,16.
Пример 92.
5-Хлор-1Н-индол-2-карбоновой кислоты (1-бензил-2-оксо-2-пиперидин-1-илэтил)-амид
Пиперидина гидрохлорид (0,34 ммоля) и 2[(5-хлор-1Н-индол-2- карбонил)-амино] -3-фенилпропионовую кислоту (0,30 ммоля) соединяли по процедуре А (температура реакции 0-25oC). Продукт-сырец хроматографировали па силикагеле с элюированием 20%, 30%, 40%, 50%, 75% и 100% этилацетатом в гексане с получением частичного разделения. Чистые фракции объединяли, получая 31 мг (25%) вещества заголовка: ВЭЖХ (60/40) 9,38 минуты (94%); PBMS 410/412 (MH+, 100%);
Анал. Расчетное для C23H24N3O2Cl + 0,5 H2O: C, 65,94; H, 6,02; N, 10,03.
Обнаруженное: C, 65,70; H, 6,19; N, 9,66.
Пример 93.
5-Хлор-1Н-индол-2-карбоновой кислоты карбамоилметиламид
[(5-Хлор-1Н-индол-2-карбонил)-амино] -уксусной кислоты метиловый эфир (100 мг, 0,40 ммоля) добавляли к насыщенному раствору аммиака в метаноле ( 3 мл) при 25oC. Суспензию обрабатывали ультразвуком в течение 1 часа и полученный в результате раствор концентрировали. Остаток растирали с эфиром/гексанами и сушили. Выход 77 мг, 77%; ВЭЖХ (60/90) 2,78 минуты (98%); PBMS 252/254 (MH+, 100%);
3 мл) при 25oC. Суспензию обрабатывали ультразвуком в течение 1 часа и полученный в результате раствор концентрировали. Остаток растирали с эфиром/гексанами и сушили. Выход 77 мг, 77%; ВЭЖХ (60/90) 2,78 минуты (98%); PBMS 252/254 (MH+, 100%);
1H ЯМР (ДМСО-d6) δ 11,82 (шир, 1Н), 8,80 (т, 1Н), 7,71 (д, 1H, J=  1 Гц), 7,43 (д, 1H, J=7-8 Гц), 7,42 (шир, 1Н), 7,18 (дд, 1H, J=7-8,
1 Гц), 7,43 (д, 1H, J=7-8 Гц), 7,42 (шир, 1Н), 7,18 (дд, 1H, J=7-8,  2 Гц), 7,14 (с, 1Н), 7,08 (шир, 1Н), 3,82 (м, 2Н).
2 Гц), 7,14 (с, 1Н), 7,08 (шир, 1Н), 3,82 (м, 2Н).
Анал. Расчетное для C11H10ClN3O3 + 0,125 H2O: C, 52,03; H, 4,07; N, 16,55.
Обнаруженное: C, 52,05; H, 4,08; N, 16,63.
Пример 94.
1-{ (2)-[(5-Бром-1Н-индол-2-карбонил)-амино] -3-фенилпропионил} - пирролидин-(2S)-карбоновая кислота
Трифторуксусную кислоту добавляли в раствор 1-{(2S)-[(5-бром-1Н-индол-2-карбонил)-амино] -3-фенилпропионил} - пирролидин-(2S)-карбоновой кислоты трет-бутилового эфира (345 кг, 0,64 ммоля) в дихлорметане (2 мл) при 0oC. Через 1 час при 25oC реакционную смесь концентрировали, растирали с эфиром и сушили с получением желтого твердого вещества.
Выход 273 мг, 88%; ВЭЖХ (70/30) 4,75 минуты (98%); ТРМС 484/486 (MH+, 100%);
Анал. Расчетное для C23H22BrN3O4 + 0,25 H2O: C, 56,51; H, 4,64; N, 8,60.
Обнаруженное: C, 56,28; H, 4,78; N, 8,26.
Пример 94а.
1-{ (2S)-[(5-Бром-1Н-индол-2-карбонил)-амино] -3-фенилпропионил}- пирролидин-(2S)-карбоновой кислоты трет-бутиловый эфир
L-Фенилаланин-L-пролина трет-бутиловый эфир (333 мг, 1,0 ммоль) и 5-бром-1Н-индол-2-карбоновую кислоту соединяли по процедуре А (72 часа - время реакции). Продукт очищали с помощью колоночной хроматографии на силикагеле с элюированием 15%, 20% и 30% этилацетатом с получением бледно-желтой пены. Выход 428 мг (79%); ВЭЖХ (70/30) 5,84 минуты (81%).
Пример 95.
5-Хлор-1H-индол-2-карбоновой кислоты [2-окco-2-(1RS)-оксо-1- тиазолидин-3-ил)-этил]-амид
м-Хлорпероксибензойную кислоту (426 мг, 50%, 1,2 ммоля) добавляли при 25oC к раствору 5-хлор-1Н-индол-2-карбоновой кислоты (2-оксо-2-тиазолидин-3-ил-этил)-амида (400 мг, 1,2 ммоля) в дихлорметане (8 мл) при 25oC. Через 1 час смесь разводили этилацетатом (ca 80 мл) и полученный в результате раствор промывали три раза смесью 1:1 насыщенного водного NaHCO3/10% водного Na2S2O3 насыщенным водным NaHCO3 и солевым раствором. Полученную в результате суспензию фильтровали и отфильтрованное твердое вещество промывали водой и сушили с получением кристаллического твердого вещества. ВЭЖХ (60/40) 2,52 минуты (98,5%); ТРМС 340/342 (MH+, 70%), 357 (100%);
1H ЯМР (ДМСО-d6) δ 11,82 (шир, 1Н), 3,84 (м, 1Н), 7,73 (д, 1Н, J=2,0 Гц), 7,43 (д, 1H, J=8,7 Гц), 7,19 (дд, 1Н, J=2,0, 8,7 Гц), 7,18 (с, 1Н), 4,92 (дд, 0,5H, J=12,1 Гц), 4,71 (дд, 0,5H, J=2,2, 13 Гц), 4,47 (д, 1H, J= 12,1 Гц), 4,4-3,9 (м, 4,5Н), 3,3 (м, 0,5Н), 3,13 (м, 1Н), 3,0 (м, 0,5Н).
Анал. Расчетное для C14H14ClN3O3S + 0,8 H2O: C, 47,47; H, 4,44; N, 11,86.
Обнаруженное: C, 47,46; H, 4,07; N, 11,83.
Пример 96.
1-{ (2S)-[(5-Хдор-1Н-индол-2-карбонил)-амино] -3-фенилпропионил}- (4R)-гидроксипирролидин-(2S)-карбоновая кислота
Избыток водного 2М LiOH добавляли к раствору 1-{(2S)- [(5-хлор-1Н-индол-2-карбонил)-амино] -3-фенилпропионил} -(4R)- гидроксипирролидин-(2S)-карбоновой кислоты бензилового эфира (215 мг, 0,40 ммоля) в тетрагидрофуране при 25oC. Через 2 часа смесь разводили этилацетатом и льдом и смесь подкисляли до pH 1-2 6N HCl. Кислотный слой экстрагировали три раза этилацетатом и органические слои объединяли и сушили. Остаток растирали с эфиром и сушили с получением бесцветного твердого вещества (190 мг, 106%): ВЭЖХ (60/40) 3,43 минуты (94%); ТРМС 456/458 (MH+, 100%);
Анал. Расчетное для C23H22ClN3O5 + 0,5 C4H8O2: C, 60,06; H, 5,24; N, 8,40.
Обнаруженное: C, 60,27; H, 5,33; N, 8,13.
Пример 97.
(S)-2-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3-(1H-индол-3-ил)- пропионовой кислоты метиловый эфир
L-Триптофана метилового эфира гидрохлорид (1,05 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (1,0 ммоль) соединяли по процедуре А (0-25oC, растворитель для реакции - диметилформамид) и продукт очищали с помощью хроматографии на силикагеле с элюированием 10%, 20%, 30%, 40%, 50% и 60% этилацетатом в гексанах с получением желтого пенообразного вещества.
ВЭЖХ (60/40) 7,43 минуты (96%);
1H ЯМР (CDCl3) δ 11,78 (шир, 1Н), 10,85 (шир,1Н), 8,93 (д, 1Н, J=7,7 Гц), 7,73 (д, 1H, J=1,9 Гц), 7,57 (д, 1H, J=7,7 Гц), 7,41 (д, 1Н, J=8,7 Гц), 7,32 (д, 1H, J=8,0 Гц), 7,22 (м, 2Н), 7,18 (дд, 1Н, J=2,1, 8,8 Гц), 7,06 (м, 1Н), 6,99 (м, 1Н), 4,74 (м, 1Н), 3,65 (с, 3H), 3,35-3,2 (м, 2Н).
Пример 98.
(±)-3-{ [(5-Хлор-1Н-индол-2-карбонил)-амино] -ацетил}- тиазолидин-2-карбоновой кислоты метиловый эфир
(±)-Тиазолидин-2-карбоновой кислоты метилового эфира гидрохлорид (1,02 ммоля) и [(5-хлор-111-индол-2-карбонил)-амино]- уксусную кислоту (1,02 ммоля) соединяли по процедуре А (растворитель - дихлорметан-диметилформамид 1: 1) и неочищенный продукт растирали с эфиром-гексанами 1:1 с получением легкого желтого твердого вещества. Выход 79%; ВЭЖХ (60/40) 4,47 минуты (95%) TPMC 382/384 (MH+, 100%).
1H ЯМР (ДМСО-d6) δ 11,82 (с, 1Н), 8,85 (т, 1H, J=7 Гц), 7,73 (д, 1H, J=2 Гц), 7,43 (д, 1H, J=8,8 Гц), 7,18 (дд, 1Н, J=8,8,2 Гц), 7,17 (с, 1Н), 5,44 (с, 1Н), 4,25 (м, 1Н), 4,1 (м, 1H), 3,95 (м, 1Н), 3,34 (с, 3Н), 3,3 (м, 2Н).
Анал. Расчетное для C16H16ClN3O4S: C, 50,33; H, 4,22; N, 11,00.
Обнаруженное: C, 50,56; H, 4,46; N, 10,89.
Пример 99.
(±)-3-{ [(5-Хлор-1Н-индол-2-карбонил)-амино] -ацетил}- тиазолидин-2-карбоновая кислота
Раствор 3-{[(5-хлор-1Н-индол-2-карбонил)-амино]-ацетил}- тиазолидин-2-карбоновой кислоты метилового эфира (196 мг, 0,5 ммоля) в метаноле (10 мл) обрабатывали водным 1N NaOH (0,5 мл) при 25oC. Через 3 часа добавляли еще 1N NaOH (0,25 мл). Смесь перемешивали при 25oC в течение ночи, концентрировали, остаток смешивали с этилацетатом (30 мл) и 1N HCl (5 мл), и полученную смесь подкисляли до pH 1,8 водным 6N HCl. Водный слой отделяли и экстрагировали этилацетатом. Органические слои объединяли, сушили и концентрировали с получением твердого вещества, которое растирали с эфиром-гексаном 1:1 и сушили. Выход 186 мг, 99%; ВЭЖХ (60/40) 3,13 минуты (98%); ТРМС 368/370 (MH+), 70%) , 339 (100%).
1H ЯМР (ДМСО-d6) δ 11,80 (с, 1Н), 8,84 (шир, 1Н), 7,23 (с, 1Н), 7,44 (д, 1H, J=8,8 Гц), 7,18 (дд, 1Н), 7,17 (с, 2Н), 4,32 (с, 1Н), 4,25 (м, 2Н), 4,0 (м, 2Н), 3,3 (м, 2Н).
Пример 99а.
(±)-Тиазолидин-2-карбоновой кислоты метиловый эфир
Смесь (±)-тиазолидин-2-карбоновой кислоты (1,58 г, 11,9 ммоля) и хлортриметилсилана (5,1 г, 47 ммолей) в метаноле (22 мл) нагревали в колбе с обратным холодильником в течение 5 часов, охлаждали и концентрировали с получением твердого вещества (2,19 г, 100%).
Пример 100.
S-трет-Бутил-2-[(5-хлор-1Н-индол-2-карбонил)-амино]-3- фенил-пропионат
Процедура B
К раствору 5-хлор-1Н-индол-2-карбоновой кислоты (0,50 г, 2,6 ммоля), L-фенилаланина трет-бутилового эфира гидрохлорида (0,66 г, 2,6 ммоля), триэтиламина (0,36 мл, 2,6 ммоля) и 4-диметиламинопиридина (0,16 г, 1,3 ммоля) в дихлорметане (20 мл) добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимид (0,73 г, 3,8 ммоля). Смесь перемешивали при комнатной температуре в течение ночи, разбавляли хлороформом, промывали 2N HCl, водой и солевым раствором, сушили над сульфатом магния и концентрировали. Продукт очищали с помощью тонкослойной хроматографии (30% ацетон в гексанах) и получали в виде бледно-желтой пены (0,86 г, 85%).
Анал., расч.: C, 66,25; H, 5,81, N, 7,03;
Обнаруженное: C, 66,57; H, 6,11, N, 6,86.
Следующие примеры соединений (101-122) были получены с помощью методов, аналогичных процедуре B.
Пример 101.
R-Метил-2-[(5-фтор-1H-индол-2-карбонил)-амино]-3-фенил-пропионат
Из 5-фтор-1Н-индол-2-карбоновой кислоты и D-фенилаланина метилового эфира.
1H ЯМР (300 МГц, CDCl3) δ 3,22 (м, 2Н), 3,80 (с, 3Н), 5,10 (м, 1Н), 6,62 (д, 6 Гц, 1Н), 6,75 (д, 2 Гц, 1Н), 7,05 (д, 2 Гц, 8 Гц, 1Н), 7,10-7,15 (м, 2Н), 7,25-7,40 (м, 4Н), 7,73 (д, 2,1 Гц, 1Н), 9,50 (шир, 1Н).
Пример 102.
R-Метил-2-[(5-7-дихлор-1Н-индол-2-карбонил)-амино]-3- фенил-пропионат
Из 5,7-дихлор-1Н-индол-2-карбоновой кислоты и D-фенилаланина метилового эфира.
1H ЯМР (300 МГц, CDC13) δ 3,25 (м, 2Н), 3,80-3,95 (с, 3Н), 5,10 (м, 1Н), 6,62 (д, 6 Гц, 1Н), 6,69 (д, 2 Гц, 1Н), 7,10-7,15 (м, 2Н), 7,25-7,35 (м, 3Н), 7,50-7,56 (с, 1Н), 9,35 (шир, 1Н).
Пример 102а.
5,7-Дихлор-1Н-индол-2-карбоновая кислота
A. Этил-2-оксопропионат-2,4-дихлорфенилгидразон
Смесь 2,4-дихлорфенилгидразина (1,0 г, 4,7 ммоля), этилпирувата (0,53 мл, 4,7 ммоля), триэтиламина (0,65 мл, 4,7 ммоля) и этанола (5 мл) нагревали в колбе с обратным холодильником в течение ночи. Растворитель выпаривали и остаток собирали в хлороформ. Раствор промывали водой и солевым раствором и сушили над сульфатом магния и концентрировали, с получением остатка в виде масла (1,1 г, 98%).
B. Этил-5,7-дихлор-1H-индол-2-карбоксилат
Раствор этил-2-оксопропионат-2,4-дихлорфенилгидразона (1,1 г, 4,6 ммоля) и безводного хлорида цинка (10 г, 74 ммоля) в ледяной уксусной кислоте (12 мл) нагревали в колбе с обратным холодильником в течение 1/2 часа. Реакционную смесь выливали в воду и экстрагировали дважды эфиром. Объединенные органические слои промывали водой и солевым раствором, сушили сульфатом магния и концентрировали. Продукт очищали с помощью тонкослойной хроматографии (30% этилацетат в гексанах) и получали в виде масла (0,80 г, 67%).
C. 5,7-Дихлор-1Н-индол-2-карбоновая кислота
Раствор этил-5,7-дихлор-1Н-индол-2-карбоксилата (0,80 г, 3,1 ммоля) в 1N NaOH (40 мл) и метаноле (50 мл) нагревали в колбе с обратным холодильником в течение 3 часов. Метанол удаляли под вакуумом и водный остаток подкисляли 1N HCl и экстрагировали дважды хлороформом. Объединенные экстракты промывали водой и солевым раствором, сушили сульфатом магния и концентрировали до образования твердого вещества (0,58 г, 76%).
Следующие индол-карбоновые кислоты получали с помощью той же самой последовательности: 4-хлор-5-фтор-1H-индол-2-карбоновую кислоту и 6-хлор-5-фтор-1Н-индол-2-карбоновую кислоту (в виде смеси) из 3-хлор-4-фторфенилгидразина; 5,7-дифтор-1Н-индол-2-карбоновую кислоту из 2,4-дифторфенилгидразина.
Пример 103.
(±)-Этил-2-[(5-хлор-1Н-индол-2-карбонил-амино]-3- фенилпропионат
Из 5-хлор-1Н-индол-2-карбоновой кислоты и D,L -фенилаланина этилового эфира. Т.пл. 146-147oC.
Анал. Расч.: C 64,61, H 5,42; N 7,54.
Обнаруженное: C 64,73, H 5,26, N 7,57.
Пример 104.
Из 3-бром-5-хлор-1Н-индол-2-карбоновой кислоты (1-диметилкарбамоил-2-фенилэтил)-амид
Из 3-бром-5-хлор-1Н-индол-2-карбоновой кислоты и S-2-амино-N,N-диметил-3-фенилпропионамида.
Анал.Расч.: C 53,53; H 4,27; N 9,36.
Обнаруженное: C 53,51, H 4,46, N 9,38.
Пример 104а.
3-Бром-5-хлор-1H-индол-2-карбоновая кислота
К раствору 5-хлор-1Н-индол-2-карбоновой кислоты (2,0 г, 10,2 ммоля) в уксусной кислоте (24 мл) добавляли раствор брома (0,53 мл, 10,2 ммоля) в уксусной кислоте (16 мл). Через 20 минут смесь выливали в воду и дважды экстрагировали хлороформом. Объединенные экстракты дважды промывали водой и солевым раствором, сушили сульфатом магния и концентрировали. Продукт получали в виде твердого вещества (2,5 г, 89%).
Пример 104.
(S)-2-Амино-N,N-диметил-3-фенилпропионамида гидрохлорид
A. (S)-(1-Диметилкарбамоил-2-фенилэтил)-карбаминовой кислоты трет-бутиловый эфир
К раствору трет-Boc-фенилаланина (10 г, 38 ммолей), диметиламина гидрохлорида (3,4 г, 41 ммоль), триэтиламина (5,8 мл, 42 ммоля) и гидроксибензотриазола (6,6 г, 49 ммолей) в дихлорметане (300 мл) добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимид (9,4 г, 49 ммолей). Смесь перемешивали в течение ночи, затем гасили 2N HCl и концентрировали. Остаток собирали в этилацетат и этот раствор промывали водой и солевым раствором, сушили сульфатом магния и концентрировали. Остаток растирали с хлороформом, твердое вещество отфильтровывали и фильтрат концентрировали до масла (11 г, 100%).
B. (S)-2-Амино-N,N-диметил-3-фенилпропионамида гидрохлорид
(S)-(1-Диметилкарбамоил-2-фенилэтил)-карбаминовой кислоты трет-бутиловый эфир (11,0 г, 38 ммолей) растворяли в этилацетате (125 мл) и через раствор пропускали газообразный HCl в течение 10 мин. Раствор перемешивали в течение 1 часа при комнатной температуре, затем концентрировали. Остаток растирали с эфиром, твердое вещество отфильтровывали и сушили под высоким вакуумом (8,6 г, 100%).
Пример 105.
S-5-Хлор-4-нитро-1Н-индол-2-карбоновой кислоты (1-диметилкарбамоил-2-фенилэтил)-амид
Из 4-нитро-5-хлор-1Н-индол-2-карбоновой кислоты и N-2-амино-N,N-диметил-3-фенилпропионамида.
1H ЯМР (300 МГц, CDCl3) δ 2,75 (с, 3Н), 2,97 (с, 3Н), 3,20 (м, 2Н), 5,30 (м, 1Н), 7,07 (д, 2 Гц, 1H), 7,24-7,32 (м, 5Н), 7,40 (д, 7 Гц, 1H), 8,12 (шир, д, 7 Гц, 1H), 9,85 (шир, 1Н).
Пример 105а.
4-Нитpo-5-хлop-1H-индoл-2-кapбoнoвaя кислота
A. 2-[(4-Хлор-3-нитрофенил)-гидразоно]-пропионовой кислоты этиловый эфир
К раствору нитрита натрия (2,17 г, 31 ммоль) в воде (60 мл) и конц. HCl (12 мл) при 0oC добавляли 4-хлор-3-нитроанилин (5,0 г, 29 ммолей). Через 5 минут добавляли раствор этилметилацетоацетата (4,5 мл, 29 ммолей) в воде (60 мл), этанол (30 мл) и 50% гидроксид калия (10 мл) и реакционную смесь перемешивали в течение ночи. Осадок собирали (7,0 г, 91%).
B. Этил-5-хлор-4-нитро-1Н-индол-2-карбоксилат
Смесь 2-[(4-хлор-3-нитрофенил)-гидразоно]-пропионовой кислоты этилового эфира (2,0 г, 6,7 ммоля) и полифосфорной кислоты (7 г) нагревали до 90-110oC в течение 2 часов. Смесь охлаждали, выливали в смесь льда/воды и твердое вещество собирали. Тонкослойная хроматография (1% метанол в хлороформе) давала соединение заголовка (0,58 г, 32%) и 5-хлор-6-нитро-1Н-индол-2-карбоксилат (0,31 г, 17%).
C. 4-Нитро-5-хлор-1Н-индол-2-карбоновая кислота
Соединение заголовка получали путем гидролиза этил-5-хлор-4-нитро-1H-индол-2-карбоксилата, как описано для получения 5,7-дихлор-1H-индол-2-карбоновой кислоты.
Пример 106.
S-7-Нитро-1H-индол-карбоновой кислоты (1-диметил-карбамоил-2-фенилэтил)-амид
Из 7-нитро-1Н-индол-2-карбоновой кислоты и N-2-амино-N,N-диметил-3-фенилпропионамида.
1H ЯМР δ 2,8 (с, 3Н), 3,0 (с, 3Н), 3,1-3,3 (м, 2Н), 5,35 (кВ, 7 Гц, 1Н), 6,95 (с, 1Н), 7,15-7,3 (м, 6Н), 7,9 (д, 8 Гц, 1H), 8,2 (д, 8 Гц, 1Н) , 10,3 (шир, 1Н).
Пример 107.
(±)-Метил-2-[(5-хлор-1Н-индол-2-карбонил)-амино]-3- фенил-бутират
Из 5-хлор-1Н-индол-2-карбоновой кислоты и DL-в-метилфенилаланина метилового эфира.
Т.пл. 135-136oC.
Анал. Расч.: C 64,78, H 5,17, N 7,56;
Обнаруженное: C 64,76, H, 5,26, N 7,64.
Пример 108.
(±)-5-Хлор-1Н-индол-2-карбоновой кислоты [1-(2-фторбензил)-2-oкco-2-тиaзoлидин-3-илэтил]-амид
Из 5-хлор-1Н-индол-2-карбоновой кислоты и (±)-2-амино-3-(2-фторфенил)-1-тиазолидин-3-илпропан-1-она.
Т.пл. 216-217oC.
Анал. Расч.: C 58,40, H 4,43, N 9,73.
Обнаруженное: C 58,45, H 4,53, N 9,71.
Пример 108а.
(±)-2-Амино-3-(2-фторфенил)-1-тиазолидин-3-илпропан-1-она гидрохлорид
A. (±)-2-трет-Бутоксикарбониламино-3-(2-фторфенил)-пропионовая кислота
К смеси DL-3-фторфенилаланина (1,0 г, 5,5 ммоля) и триэтиламина (1,14 мл, 8,2 ммоля) в дихлорметане (20 мл) добавляли ди-трет-бутил-дикарбонат (1,4 г, 6,55 ммоля). Смесь перемешивали при комнатной температуре в течение ночи, затем выливали в воду, подкисляли 1N HCl и экстрагировали хлороформом
Объединенные экстракты промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Продукт очищали с помощью тонкослойной хроматографии (хлороформ/метанол/уксусная кислота, 89: 10:1) и получали в виде твердого вещества (1,28 г, 83%, т.пл. 118-119oC).
B. (±)-[1-(2-Фторбензил)-2-оксо-2-тиазолиди-3-илэтил] карбаминовой кислоты трет-бутиловый эфир
К смеси 2-трет-бутоксикарбониламино-3-(2-фторфенил)пропионовой кислоты (0,50 г, 1,77 ммоля), тиазолидина (0,15 мл, 1,94 ммоля) и 4-диметиламинопиридина (0,21 г, 1,77 ммоля) в дихлорметане (15 мл) добавляли ЭДХ (0,44 г, 2,31 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение ночи, разводили хлороформом, промывали 2N HCl, водой и солевым раствором, сушили сульфатом магния и концентрировали. Продукт очищали с помощью тонкослойной хроматографии (30% ацетон в гексанах) и получали в виде бесцветного твердого вещества (0,39 г, 62%, т.пл. 133-134oC).
C. (±)-2-Амино-3-(2-фторфенил)-1-тиазолидин-3-илпропан-1-она гидрохлорид
Газообразный HCl пропускали в раствор [1-(2-фторбензил)-2-оксо-2- тиазолидин-3-илэтил] -карбаминовой кислоты трет-бутилового эфира (0,39 г, 1,1 ммоля) в этилацетате (15 мл). Раствор концентрировали, остаток растирали с эфиром, твердое вещество отфильтровывали и сушили (0,27 г, 84%, т.пл. 217-218oC).
Следующие амины были получены аналогичными методами и в той же последовательности:
(±)-2-амино-3-(2-хлорфенил)-1-тиазолидин-3-илпропан-1-он из DL-2-хлорфенилаланина,
(±)-2-амино-3-(3-цианофенил)-1-тиазолидин-3-илпропан-1-он из DL-3-цианофенилаланина,
(±)-2-амино-3-(3-хлорфенил)-1-тиазолидин-3-илпропан-1-он из DL-3-хлорфенилаланина,
(±)-2-амино-3-(3-трифторметилфенил)-1-тиазолидин-3-илпропан- 1-он из DL-3-трифторметилфенилаланина,
(S)-2-амино-1-(4-гидроксипиперидин-1-ил)-3-(4-метоксифенил)- пропан-1-он из L-4-метоксифенилаланина.
Пример 109.
(±)-5-Хлор-1Н-индол-2-карбоновой кислоты [1-(2-хлорбензил)- 2-оксо-2-тиазолидин-3-илэтил]-амид
Из 5-Хлор-1Н-индол-2-карбоновой кислоты и (±)-2-амино-3-(2-хлорфенил)-1-тиазолидин-3-илпропан-1-она.
Т.пл. 214-216oC.
Анал. Расч.: C 56,26, H 4,58, N 9,37.
Обнаруженное: C 56,27, H 4,54, N 9,36.
Пример 110.
(±)-5-Хлор-1H-индол-2-карбоновой кислоты [2-(3-цианофенил)-1-(тиазолидин-3-карбонил)-этил]-амид
Из 5-Хлор-1Н-индол-2-карбоновой кислоты и (±)-2-амино-3- (3-цианофенил)-1-тиазолидин-3-илпропан-1-она.
Т.пл. 183-184oC.
Анал. Расч.: С, 60,20, H 4,36, N 12,77.
Обнаруженное: C 60,11, H 4,84, N 12,43.
Пример 111.
(±)-5-Хлор-1Н-индол-2-карбоновой кислоты [1-(3-хлорбензил) 2-оксо-2-тиазолидин-3-илэтил]-амид
Из 5-хлор-1Н-индол-2-карбоновой кислоты и (±)-2-амино-3-(3-хлорфенил)-1-тиазолидин-3-илпропан-1-она.
Т.пл. 188-190oC.
Анал. Расч.: C 56,26, H 4,27, N 9,37.
Обнаруженное: C 56,38, H 5,04, N 9,04.
Пример 112.
(±)-5-Хлор-1Н-индол-2-карбоновой кислоты [2-оксо-2-тиазолидин-3-ил-1-(3-трифторметилбензил)-этил]-амид
Из 5-хлор-1Н-индол-2-карбоновой кислоты и (±)-2-амино-3-(3-трифторметилфенил)-1-тиазолидин-3-илпропан-1-она.
Т.пл. 205-207oC.
Анал.Расч.: C 54,83, H 3,97, N 8,72.
Обнаруженное: C 54,44, H 4,14, N 8,88.
Пример 113.
S-5-Хлор-1Н-индол-2-карбоновой кислоты [1-(4-метоксибензил)-2-оксо-2-тиaзoлидин-3-илэтил]-aмид
Из 5-хлор-1Н-индол-2-карбоновой кислоты и S-2-амино-3-(4-метоксифенил)-1-тиазолидин-3-илпропан-1-она.
Анал.Расч.: C, 59,52, H 5,00, N 9,47.
Обнаруженное: C 60,00, H 5,55, N 8,90.
Масс-спектр м/э 444 (M++1).
Пример 114.
(±)-5-Хлор-1Н-индол-2-карбоновой кислоты [1-(3-хлорбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
Из 5-хлор-1Н-индол-2-карбоновой кислоты и (±)-2-амино-1-(4-гидроксипиперидин-1-ил)-3-(3-хлорфенил)- пропан-1-она.
Пример 115.
S-5-Xлop-4-фтop-1H-индoл-2-кapбонoвoй кислоты (1-бензил-2-оксо-2-тиазолидин-3-илэтил)-амид и S-6-хлор-4-фтор-1Н-индол-2-карбоновой кислоты (1-бензил-2-оксо-2-тиазолидин-3-илэтил)-амид
Из смеси 5-хлор-4-фтор-1Н-индол-2-карбоновой кислоты и 6-хлор-4-фтор-1Н-индол-2-карбоновой кислоты и 5-2-амино-3-фенил-1-тиазолидин-3-илпропан-1-она.
Т.пл. 105-125oC, разл.
Анал.расчеты: C 58,40, H 4,43, N 9,73.
Обнаруженное: C 58,54, H 4,59, N 9,58.
Пример 116.
(+)-2-[(5-Хлор-1Н-индол-2-карбонил)-амино]-пропионовой кислоты метиловый эфир
Из 5-хлор-1Н-индол-2-карбоновой кислоты гидрохлорида метилового метила и DL-аланина.
Т.пл. 199-201oC.
Анал.расчетн. : C 55,63, H 4,67, N 9,98.
Обнаруженное: C 55,70, H 4,75, N 10,06.
Пример 117.
(±)-2-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3-[4-(4,5- дигидро-1Н-имидазол-2-ил)-фенил]-пропионовой кислоты метиловый эфир
Из 5-хлор-1Н-индол-2-карбоновой кислоты и (±)-2-амино-3-[4-(4,5-дигидро-1H-имидазол-2-ил)-фенил]-пропионовой кислоты метилового эфира.
1H ЯМР (300 МГц, CDC13,) δ 3,1-3,3 (м, 2Н), 3,70 (с, 3Н), 3,95 (с, 4Н), 4,85 (м, 1Н), 7,15 (с, 1Н), 7,17 (д, 8 Гц, 1Н), 7,40 (д, 8 Гц, 1Н), 7,65 (д, 7 Гц, 1Н), 7,75 (с, 1Н), 7,88 (д, 8 Гц, 1Н), 9,10 (шир, д, 9 Гц, 1Н), 10,5 (с, 1Н), 11,8 (шир, с, 1Н).
Пример 117а.
(±)-2-Амино-2-[4-(4,5-дигидро-1Н-имидазол-2-ил)-фенил] - пропионовой кислоты метиловый эфир
A. 2-Ацетиламино-2-[4-(4,5-дигидро-1Н-имидазол-2-ил)-бензил]- малоновой кислоты диэтиловый эфир
Раствор 2-ацетиламино-2-[(4-метоксиуглерод-имидоил)-бензил] - малоновой кислоты диэтилового эфира (G.Wagner et al. Pharmazia 1994, 29, 12) (5,3 г, 13 ммолей) и этилендиамина (4,8 г, 80 ммолей) в этаноле (100 мл) перемешивали при 60oC в течение 5 часов. После охлаждения растворитель выпаривали, к остатку добавляли воду и твердое вещество отфильтровывали и растворяли в горячей 1N HCl. После охлаждения остаток отфильтровывали и сушили (3,1 г).
B. (±)-2-Амино-3-[4-(4,5-дигидро-1Н-имидазол-2-ил)- фенил]-пропионовой кислоты дигидрохлорид
К 2-ацетиламино-2-[4-(4,5-дигидро-1Н-имидазол-2-ил)-бензил] - малоновой кислоты диэтиловому эфиру (3,0 г, 7,3 ммоля) добавляли ледяную уксусную кислоту (50 мл) и 3N HCl (100 мл).
Раствор нагревали в колбе с обратным холодильником в течение 3 часов, охлаждали и концентрировали до образования твердого белого вещества, которое перекристаллизовывали из метанола/эфира (2,0 г, т.пл. 270-272oC, разл).
C. (±)-2-Амино-3-[4-(4,5-дигидро-1Н-имидазол-2-ил)- фенил]-пропионовой кислоты метиловый эфир
(±)-2-Амино-3-[4-(4,5-дигидро-1Н-имидазол-2-ил)- фенил]-пропионовой кислоты дигидрохлорид (0,50 г, 1,6 ммоля) помещали в тионилхлорид (1 мл) и метанол (25 мл). Смесь нагревали в колбе с обратным холодильником в течение 30 минут, через этот срок добавляли еще тионилхлорида (3 мл) и метанола (75 мл). После еще 3 часов нагревания в колбе с обратным холодильником раствор концентрировали, остаток растворяли в небольшом количестве метанола и, чтобы вызвать осаждение, добавляли этилацетат. Твердое вещество собирали и сушили (0,40 г, т.пл. 230oC, разл.).
Пример 118.
(S)-5,7-Дифтор-1Н-индол-2-карбоновой кислоты [1-бензил-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
Из 5,7-дифтор-1Н-индол-2-карбоновой кислоты и (S)-2-амино-1-(4-гидроксипиперидин-1-ил)-3-фенилпропан-1-она.
Т.пл. 95-110oC.
Пример 119.
S-4-хлор-5-фтор-1Н-индол-2-карбоновой кислоты (1-диметилкарбамоил-2-фенилэтил)-амид и S-6-хлор-5-фтор-1Н-индол-2-карбоновой кислоты (1-диметилкарбамоил-2-фенилэтил)-амид
Из смеси 5-хлор-4-фтор-1Н-индол-2-карбоновои кислоты и 6-хлор-4-фтор-1Н-индол-2-карбоновой кислоты и (S)-2-амино-N,N- диметил-3-фенилпропионамида.
Т.пл. 200-210oC.
Анал.Расчет.: C 61,94, H 4,94, N 10,83.
Обнаруженное: C 62,21, H 4,99, N 10,84.
Пример 120.
(S)-5,7-Дифтор-1Н-индол-2-карбоновой кислоты (1-бензил-2-оксо-2-тиазолидин-3-илэтил)-амид
Из 5,7-дифтор-1Н-индол-2-карбоновой кислоты и (S)-2-амино-3-фенил-1-тиазолидин-3-илпропан-1-она.
Т.пл.: 175-185oC.
Анал.Расчетн. : C 60,71, H 4,61, N 10,11.
Обнаруженное: C 60,79, H 4,66, N 9,93.
Пример 121
(S)-5,7-дифтор-1Н-индол-2-карбоновой кислоты [1-бензил-2-(1,1- диоксо-1-тиазолидин-3-ил)-2-оксоэтил]-амид
Из 5,7-дифтор-1Н-индол-2-карбоновой кислоты и (S)-2-амино-1- (1,1-диоксо-1-тиазолидин-3-ил)-3-фенилпропан-1-она гидрохлорида.
Т.пл. 95-110oC.
MC 448 (MH+).
Пример 122.
S-5-Хлор-1Н-индол-2-карбоновой кислоты [1-(2-фторбензил)- 2-(4-гидроксипиперидин-1-ил)-2-оксоэтил-амид
Процедура C
К раствору 5-хлор-1Н-индол-2-карбоновой кислоты (0,49 г, 2,5 ммоля), S-2-амино-3-(2-фторфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она (0,76 г, 2,5 ммоля), триэтиламина (0,35 мл, 2,5 ммоля) и гидроксибензотриазол (0,34 г, 2,5 ммоля) в дихлорметане (6 мл) добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимид (0,53 г, 2,8 ммоля). Смесь перемешивали при комнатной температуре в течение ночи, разводили дихлорметаном, промывали водой, 1N HCl и насыщенным бикарбонатом натрия, сушили над сульфатом магния и концентрировали. Продукт очищали с помощью тонкослойной хроматографии (хлороформ/метанол, 8:1) и получали в виде не совсем белого твердого вещества (0,82 г, 73%). Т.пл. 120-122oC.
Пример 122а.
(S)-2-Амино-3-(2-фторфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-она гидрохлорид
A. (S)-трет-Бутоксикарбониламино-3-(2-фторфенил)-1-тиазолидин-3-ил-пропан-1-он
Из L-Boc-2-фторфенилаланина и 4-гидроксипиперидина с помощью метода, аналогичного процедуре C.
B. (S)-2-Амино-3-(2-фторфенил)-1-(4-гидроксипиперидин-1-ил) пропан-1-она гидрохлорид
Соединение заголовка получали с помощью реакции L-2-трет- бутоксикарбониламино-3-(2-фторфенил)-1-тиазолидин-3-ил-пропан-1-она с HCl по методу, аналогичному описанному в примере 108а, стадия C.
Следующие амины были получены с помощью аналогичных методов в той же последовательности:
S-2-амино-3-(4-метоксифенил)-1-тиазолидин-3-илпропан-1-он;
S-2-амино-3-(2-фторфенил)-1-(4-гидроксипиперидин-1-ил)-пропан-1-он,
S-2-амино-1-(4-гидроксипиперидин-1-ил)-3-(4-метоксифенил)- пропан-1-он,
S-2-амино-3-(2-хлорфенил)-1-(4-гидроксипиперидин-1-ил)- пропан-1-он,
S- 2-амино-3-(4-метоксифенил)-1-морфолин-4-ил-пропан-1-он,
S-2-амино-3-(4-метоксифенил)-1-(4-ацетилпиперазинил)-пропан-1-он.
С помощью процесса, аналогичного процессу (процедуры C), были получены следующие примеры соединений (122-138).
Пример 123.
(2SR), (3RS)-2-[(5-Хлор-1Н-индол-2-карбонил)-амино]-3- гидрокси-3-фенилпропионовой кислоты метиловый эфир
Из 5-хлор-1Н-индол-2-карбоновой кислоты и (±)- трет-b-фенилсерина метилового эфира.
Т.пл. 196-197oC.
Пример 124.
S-5-Фтор-1Н-индол-2-карбоновой кислоты [1-(4-метоксибензил) 2-оксо-2-тиазолидин-3-илэтил]амид
Из 5-Фтор-1Н-индол-2-карбоновой кислоты и S-2-амино-3-(4- метоксифенил)-1-тиазолидин-3-илпропан-1-она.
Т.пл. 90-115oC.
Анал.Расчитан.: C 61,81, H 5,19, N 9,83.
Обнаруженное: C 60,94, H 5,33, N 10,01.
Пример 125.
S-5-Хлор-1Н-индол-2-карбоновой кислоты [1-(2-хлорбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил]-амид
Из 5-хлор-1Н-индол-2-карбоновой кислоты и S-2-амино-3-(2- хлорфенил)-1-(4-гидроксипиперидин-1-ил)-пропан-1-она.
Т.пл. 127-129oC.
Пример 126.
S-5-Хлор-1Н-индол-2-карбоновой кислоты [1-(4-метоксибензил)- 2-морфолин-4-ил-2-оксоэтил]-амид
Из 5-хлор-1Н-индол-2-карбоновой кислоты S-2-амино-3-(4- метоксифенил)-1-морфолин-4-илпропан-1-она.
Т.пл. 95-105oC.
Анал.расчетн. : C 62,51, H 5,47, N 9,51.
Обнаруженное: C 61,82, H 6,05, N 8,97.
Пример 127.
S-5-Хлор-1Н-индол-2-карбоновой кислоты [2-(4-ацетилпиперазин- 1-ил)-1-(4-метоксибензил)-2-оксоэтил]-амид
Из 5-хлор-1Н-индол-2-карбоновой кислоты и S-2-амино-3-(4-метоксифенил)-1-(4-ацетилпиперазинил)-пропан-1-она.
Т.пл. 120-135oC.
Анал. Расчетн.: C 62,17, H 5,64, N 11,60.
Обнаруженное: C 62,76, H 6,20, N 10,44.
Пример 128.
S-5-фтор-1H-индол-2-карбоновой кислоты [1-(бензотиазол-2 ил-карбамоил)-2-фенилэтил]-амид
Из S-2-[(5-фтор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и 2-амино-1,3-бензотиазола.
Т.пл. 139-141oC.
Пример 129.
S-5-фтор-1Н-индол-2-карбоновой кислоты (1-бензил-2-морфолин-4-ил-2-оксоэтил)-амид
Из S-2-[(5-фтор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и морфолина.
Т.пл. 234-236oC.
Пример 130.
5-фтор-1Н-индол-2-карбоновой кислоты [1S-бензил-2-оксо-2- (3,3,5RS-триметилазепан-1-ил)-этил]-амид
Из 2-[(5-фтор-1Н-индол-2-карбонил)-амино] -3-фенилпропионовой кислоты и (±)-3,3,5-триметилазепан.
Т.пл. 125-127oC.
Анал.расчетн.: C 72,14, H 7,18, N 9,35.
Обнаруженное: C 72,00, H 7,58, N 9,10.
Пример 131.
5-фтор-1Н-индол-2-карбоновой кислоты [1S-бензил-2-(3RS- карбамоилпиперидин-1-ил)-2-оксоэтил]-амид
Из S-2-[(5-фтор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и 3-карбамоилпиперидина.
Т.пл. 234-236oC.
Пример 132.
5-Фтор-1Н-индол-2-карбоновой кислоты [2-фенил-1S- (тиохроман-4RS-илкарбамоил)-этил]-амид
Из S-2-[(5-фтор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и (±)-тиохроман-4-иламина.
Т.пл. 225-226oC.
Анал.рассчит.: C 68,48, H 5,11, N 8,88.
Обнаруженное: C 68,40, H 5,64, N 8,61.
Пример 133.
S-5-Фтор-1Н-индол-2-карбоновой кислоты [1-(5-метил-изоксазол-3- илкарбамоил)-2-фенилэтил]-амид
Из S-2-[(5-фтор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и 5-метилизоксазол-3-иламина.
Т.пл. 219-221oC.
Пример 134.
S-5-Фтор-1Н-индол-2-карбоновой кислоты [2-фенил-1-(4,5,6,7-тетрагидробензотиазол-2-илкарбамоил)-этил]-амид
Из S-2-[(5-фтор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и 4,5,6,7-тетрагидробензотиазол-2-иламина.
Т.пл. 162-165oC.
Пример 135.
S-5-фтор-1H-индол-2-карбоновой кислоты [1-(5-метилтиазол 2-илкарбамоил)-2-фенилэтил]-амид
Из S-2-[(5-фтор-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и 4-метилтиазол-2-иламин.
Т.пл. 211-213oC.
Пример 136.
S-5-Метил-1Н-индол-2-карбоновой кислоты [1-(5-метилизоксазол- 3-илкарбамоил)-2-фенилэтил]-амид
Из S-2[(5-метил-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и 5-метил-изоксазол-3-иламин.
Т.пл. 243-245oC.
Анал.расчетн.: C 68,64, H 5,51, N 13,93.
Обнаруженное: C 68,29, H 5,81, N 14,05.
Пример 137.
S-5-Метил-1Н-индол-2-карбоновой кислоты [2-(4-ацетилпиперазин- 1-ил)-1-бензил-2-оксоэтил]-амид
Из S-2-[(5-метил-1Н-индол-2-карбонил)-амино]-3-фенилпропионовой кислоты и 1-пиперазин-1-илэтанона.
Т.пл. 221-223oC.
Пример 138.
S-5-Хлор-1Н-индол-2-карбоновой кислоты (1-карбамоил-2-фенилэтил)-амид
Из 5-хлор-1Н-индолкарбоновой кислоты и S-2-амино-3-фенилпропионамида.
Т.пл. 257-258oC.
Пример 139 и 140.
(2RS)-2,3-Дигидро-1Н-индол-2-карбоновой кислоты
(R-1-диметилкарбамоил-2-фенилэтил)-амид
К смеси DL-индолин-2-карбоновой кислоты (0,38 г, 2,3 ммоля), (R)-2-амино-N, N-диметил-3-фенилпропионамида гидрохлорида (0,53 г, 2,3 ммоля), гидроксибензотриазола (0,66 г, 4,2 ммоля) и триэтиламина (0,32 мл, 2,3 ммоля) в дихлорметане (5 мл) добавляли ЭДХ (EDC) (0,64 г, 2,7 ммоля). Раствор перемешивали в течение ночи, разводили дихлорметаном, промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Два изомерных продукта разделяли с помощью тонкослойной хроматографии (EtOAc, затем EtOAc/MeOH, 20:1).
Пример 139.
Менее полярный изомер (масло, 0,23 г, 30%):
1H ЯМР (300 МГц, CDCl3) δ 2,68 (с, 3Н), 2,87 (с, 3Н), 3,02-3,09 (м, 3Н), 3,55 (дд, J=10 Гц, 6 Гц, 1Н), 4,61 (м, 1Н), 5,10 (кB, J=8 Гц, 1Н), 6,95 (д, J=8 Гц, 1Н), 7,11-7,30 (м, 8Н), 8,12 (шир, 1Н). МС (Cl, NH3), 394 (M+ +17).
Пример 140.
Более полярный изомер (0,11 г, 14%): т.пл. 136-140oC.
Примеры 141 и 142.
(2RS)-5-Хлор-2,3-дигидро-1Н-индол-2-карбоновой кислоты (1-S-диметилкарбамоил-2-фенилэтил)-амид
К раствору S-5-хлор-1Н-индол-2-карбоновой кислоты (1-диметилкарбамоил-2-фенилэтил)-амида (2,60 г, 7,0 ммолей) в ТГФ (20 мл) и метаноле (20 мл) добавляли магний (1,75 г, 73 ммоля) порциями с такой скоростью, чтобы поддерживать течение реакции без избыточного выделения тепла. После того, как реакция прекращалась, реакционную смесь концентрировали до малого объема, остаток разделяли между 1N HCl и этилацетатом, объединенные этилацетатные слои промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Продукты разделяли с помощью тонкослойной хроматографии (1% метанол в хлороформе).
Пример 141.
Менее полярный изомер:
1H ЯМР (300 МГц, CDCl3) δ 2,61 (с, 3Н), 2,83 (с, 3Н), 3,00-3,02 (м, 3Н), 3,47 (дд, J= 9,9 Гц, 6,4 Гц, 1Н), 4,43 (м, 1Н), 5,10 (кв, J=7,5 Гц, 1Н), 6,64 (д, J=8,8 Гц, 1Н), 7,00 (с, 1Н), 7,16-7,29 (м, 7Н), 7,70 (шир, 1Н). МС (Cl, NH3) 372 (M+ +1).
Пример 142.
Более полярный изомер: т.пл. 125oC, разл.
Примеры 143 и 144.
2RS-5-хлор-2,3-дигидро-1Н-индол-2-карбоновой кислоты (1R-диметилкарбамоил-2-фенилэтил)-амид
С помощью метода, аналогичного методу из примера 141 и 142 с использованием R-5-хлор-1Н-индол-2-карбоновой кислоты (1-диметилкарбамоил-2-фенилэтил)-амида, были получены два диастереомера.
Пример 143.
Менее полярный изомер: т.пл. 122-124oC, разл.
Пример 144.
Более полярный изомер:
1H ЯМР (300 МГц, CDCl3) δ 2,70 (с, 3Н), 2,83 (с, 3Н), 2,77-2,97 (м, 3Н), 3,40 (дд, J= 16,6 Гц, 10,8 Гц, 1Н), 4,28 (м, 1Н), 4,40 (д, J=5,2 Гц, 1Н), 5,12 (кB, J= 7,8 Гц, 1Н), 6,56 (д, J=9,0 Гц, 1Н), 6,96 (д, J=7,8 Гц, 1Н), 6,99 (с, 1Н), 7,03-7,07 (м, 2Н), 7,11-7,18 (м, 3Н), 7,74 (д, J=8,8 Гц, 1Н).
Пример 145.
3-Хлор-1Н-индол-2-карбоновой кислоты (1R-диметилкарбамоил- 2-фенилэтил)-амид
К раствору 2,3-дигидро-1Н-индол-2-карбоновой кислоты (1R-диметилкарбамоил-2-фенилэтил)-амида (менее полярный изомер, 0,50 г, 1,42 ммоля) в ДМФ (7,5 мл) добавляли N-хлорсукцинимид (0,55 г, 1,42 ммоля). После перемешивания в течение ночи растворитель выпаривали и продукт очищали с помощью тонкослойной хроматографии (гексаны/этилацетат, 1:1).
1H ЯМР (300 МГц, CDCl3) δ 2,55 (с, 3Н), 2,80 (с, 3Н), 3,05-3,20 (м, 2Н), 5,32 (м, 1Н), 7,10-7,25 (м, 6Н), 7,30 (д, 7 Гц, 1Н), 7,58 (д, 7 Гц, 1Н), 8,11 (шир, д, 7 Гц, 1Н), 10,20 (шир, 1Н). МС м/э 370 (M++1).
Пример 146.
3-Хлор-1Н-индол-2-карбоновой кислоты (1S-диметилкарбамоил 2-фенилэтил)-амид
Соединение заголовка получали методом, аналогичным методу из примера 145 из более полярного изомера 2,3-дигидро-1H-индол-2-карбоновой кислоты (1R-диметилкарбамоил-2-фенилэтил)-амида.
1H ЯМР (300 МГц, CDCl3,) δ 2,55 (с, 3Н), 2,85 (с, 3Н), 3,05-3,20 (м, 2Н), 5,32 (м, 1Н), 7,10-7,25 (м, 6Н), 7,35 (д, 7 Гц, 1Н), 7,58 (д, 7 Гц, 1Н), 8,11 (шир д, 7 Гц, 1Н), 10,30 (шир, 1Н).
МС м/э 370 (М+ +1).
Условия ВЭЖХ для примеров 147-165: детекторная длина волны - 215 нм. Время удерживания при ВЭЖХ (в минутах) - по колонке Water's Novapac C18 3,9 х 150 мм. Элюент A= 50 мМ KH2PO3, pH 3; элюент B = ацетонитрил; скорость потока 1,5 мл/минуту; градиент 90% A/10% B (5 минут) до 40% A/60% B (5 минут задержки). Время удерживания при ВЭЖХ (RT) представлено в минутах. Процентное значение дано в процентах от общего целого, в связи с конкретным пиком. По ВЭЖХ исходная кислота присутствовала в количестве менее 5% от общего целого, если не указано иначе.
Пример 147.
(S)-2-[(5-Фтор-1Н-индол-2-карбонил)-амино] -3-фенилпропионовая кислота
К раствору 5-фториндол-2-карбоновой кислоты (5,0 г, 28 ммолей), L-фенилаланина т-бутилового эфира гидрохлорид (6,54 г, 27,9 ммоля) и метиленхлорида (250 мл) добавляли 1-(3-диметиламинопропил)-3- этилкарбодиимида гидрохлорид (5,53 г, 27,9 ммоля) и триэтиламин (7,1 мл, 5,13 г, 51 ммоль). После перемешивания в течение 40 часов при комнатной температуре реакционную смесь промывали равным объемом воды и затем равным объемом 1N HCl. Водный раствор кислоты экстрагировали метиленхлоридом и объединенные органические слои последовательно промывали равными объемами воды (дважды) и солевого раствора. Органический раствор сушили (MgSO4), фильтровали и концентрировали с получением S-т-бутил-2-[(5-фтор-1Н-индол-2-карбонил)-амино] -3-фенилпропионата (2,97 г, 31%). Его затем разводили метиленхлоридом (75 мл) и охлаждали до 0oC. Добавляли трифторуксусную кислоту (8 мл) и реакционную смесь затем перемешивали при комнатной температуре в течение 2 дней и затем нагревали в колбе с обратным холодильником в течение 6,5 часов. Затем смеси давали дойти до комнатной температуры в течение ночи и раствор концентрировали до сухости с получением коричневого твердого вещества. Это вещество затем растворяли в небольшом количестве эфира и пентанов, фильтровали для удаления частиц и концентрировали с получением соединения заголовка в виде коричневой пены (2,65 г, количественный выход): т. пл. 125-127oC; ВЭЖХ RT 5,72; ТРМС-ион (ожидаемое) 327(326);
1H ЯМР (CDCl3) δ 9,0 (шир, с, 2Н), 7,4-7,2 (м, 6Н), 7,02 (дт, J=2,4,9,1 Гц, 1Н), 6,80 (д, J=7,7 Гц, 1Н), 6,75 (д, J=1,6 Гц, 1Н), 5,09 (кB, J=7,6 Гц, 1Н), 3,35 (дд, J=5,8, 7,6 Гц, 1Н), 3,26 (дд, J=5,8, 7,6 Гц, 1Н).
Пример 148.
(S)-2-[(5-Метил-1Н-индол-2-карбонил)-амино)-3-фенилпропионовая кислота
Повторение вышеприведенной процедуры с 5-метилиндол-2-карбоновой кислотой (3,0 г, 17 ммолей), метиленхлоридом (185 мл), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлоридом (3,28 г, 17,1 ммоля), L-фенилаланина т-бутилового эфира гидрохлоридом (4,01 г, 15,6 ммоля) и триэтиламином (4,5 мл, 3,31 г, 32,7 ммоля) давало аналогичный бутиловый эфир (2,42 г, 41%). После разведения метиленхлоридом (60 мл) и трифторуксусной кислотой (6,6 мл) реакционную смесь нагревали в колбе с обратным холодильником в течение 3 часов, давали дойти до комнатной температуры в течение ночи и концентрировали. Продукт-сырец суспендировали в этилацетате, фильтровали для удаления нерастворимого материала и концентрировали (дважды) с получением соединения заголовка в виде коричневой пены (2,54 г, количественный выход).
ВЭЖХ RT 5,98; ТРМС-ион (ожидаемое) 323(322);
1H ЯМР (CDCl3) δ 9,9 (шир, с, 1Н), 8,5 (шир,с, 2Н), 7,38 (с, 1Н), 7,3-7,1 (м, 6Н), 6,77 (м, 2Н), 5,09 (кВ, J=7,6 Гц, 1Н), 3,35 (дд, J=5,6, 7,6 Гц, 1Н), 3,26 (дд, J=5,6, 7,6 Гц, 1Н), 2,43 (с, 3Н).
Пример 149.
5-Фтор-1Н-индол-2-карбоновой кислоты {1-[2-(5-метокси-1Н-индол-3-ил)-этилкарбамоил]-2-фенилэтил]-амид
К 5,0 мкмолям 2-[(5-фтор-1Н-индол-2-карбонил)-амино]-3- фенилпропионовой кислоты (50 мкл 0,1 мM раствора в диметилформамиде) добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (50 мкл 0,11 мМ раствора в ДМФ, 5,5 мкмоля) с последующим добавлением 5-метокситриптамина (50 мкл 0,11 мМ раствора в ДМФ, 5,5 мкмоля). Реакционную смесь взбалтывали в течение 3 дней и затем концентрировали до сухости. Продукт-сырец разделяли между хлороформом (0,5 мл) и водой (0,25 мл), и органический слой затем концентрировали с получением соединения заголовка.
ТРМС ион (ожидаемое) 499(499); ВЭЖХ RT 6,78 (25%).
В отношении следующих примеров соединений, полученных аналогично примеру 149, в примерах 150-156 использовали 2-[(5-фтор-1Н-индол-2-карбонил)-амино] -3-фенилпропионовую кислоту и в примерах 157-163 использовали 2-[(5-метил-1Н-индол-2-карбонил)-амино]- 3-фенилпропионовую кислоту.
Пример 150.
5-Фтор-1Н-индол-2-карбоновой кислоты {1-[2-(1H-индол-3-ил)-1- метилэтилкарбамоил]-2-фенилэтил}-амид
ТРМС-ион (ожидаемое) 482 (483); ВЭЖХ RT неизвестно, отмечено несколько небольших пиков; дост.чистота < 10%; % SM (ВЭЖХ) не опр.
Пример 151.
5-Фтор-1Н-индол-2-карбоновой кислоты [1-бензил-2-(2- этилпиперидин-1-ил)-2-оксоэтил]-амид
ТРМС-ион (ожидаемое) 420(421); ВЭЖХ RT 6,61 (40%).
Пример 152.
5-Фтор-1Н-индол-2-карбоновой кислоты (1-циклогексилкарбамоил-2-фенил)-амид
ТРМС-ион (ожидаемое) 408 (407); ВЭЖХ RT 6,60/7,11 (два самых больших пика были примерно равной концентрации); дост. чистота (25%).
Пример 153.
5-Фтор-1Н-индол-2-карбоновой кислоты {2-фенил-1-[(тиофен-2- илметил)-карбамоил]-этил}-амид
ТРМС-ион (ожидаемое) 422 (421); ВЭЖХ RT 7,50 (50%).
Пример 154.
5-Фтор-1Н-индол-2-карбоновой кислоты [1-бензил-2-(3,4-дигидро- 1Н-изохинолин-2-ил)-2-оксоэтил]-амид
ТРМС-ион (ожидаемое) 442 (441); ВЭЖХ RT 6,78 (35%), 5% ИМ.
Пример 155.
5-Фтор-1Н-индол-2-карбоновой кислоты [1-(2-циклогексан-1-ил-этилкарбамоил)-2-фенилэтил]-aмид
ТРМС-ион (ожидаемое) 434 (433); ВЭЖХ RT 6,27/6,60 (два самых больших пика примерно равной концентрации); дост.чистота (35%), 5% ИМ.
Пример 156.
5-Фтор-1Н-индол-2-карбоновой кислоты [1-(5-цианопентилкарбамоил)-2-фенилэтил]-амид
ТРМС-ион (ожидаемое) 421 (420); ВЭЖХ RT 6,61/7,71 (два самых больших пика примерно равной концентрации) (40%).
Пример 157.
5-Метил-1Н-индол-2-карбоновой кислоты [2-фенил-1-(тиохроман- 4-илкарбамоил)-этил]-амид
ТРМС-ион (ожидаемое) 470 (470).
Пример 158.
5-Метил-1Н-индол-2-карбоновой кислоты (1-циклогексилкарбамоил-2-фенилэтил)-амид
ТРМС-ион (ожидаемое) 404 (404); ВЭЖХ RT 6,21 (70%).
Пример 159.
5-Метил-1Н-индол-2-карбоновой кислоты (1-бензил-2-морфолин-4-ил-2-оксоэтил)-амид
ТРМС-ион (ожидаемое) 392 (391); ВЭЖХ RT 6,86 (50%)
Пример 160.
5-Метил-1Н-индол-2-карбоновой кислоты (1-бензил-2-оксо-2-пирролидин-1-илэтил)-амид
ТРМС-ион (ожидаемое) 376 (375); ВЭЖХ RT 6,50 (40%).
Пример 161.
5-Метил-1Н-индол-2-карбоновой кислоты {2-фенил-1-[(тиофен- 2-илметил)-карбамоил]-этил}-амид
ТРМС-ион (ожидаемое) 418 (417); ВЭЖХ RT 7,89 (70%).
Пример 162.
5-Метил-1Н-индол-2-карбоновой кислоты [1-(5-цианопентилкарбамоил)-2-фенилэтил]-амид
ТРМС-ион (ожидаемое) 417 (417); ВЭЖХ RT 6,49/6,88 (два самых больших пика примерно равной концентрации); (40%).
Пример 163.
5-Метил-1Н-индол-2-карбоновой кислоты (1-циклопентилкарбамоил-2-фенилэтил)-амид
ТРМС-ион (ожидаемое) 390 (389); ВЭЖХ RT 6,96 (55%).
Пример 164.
{2-[(5-Хлор-1Н-индол-2-карбонил)-амино]-3-фенилпропиониламино}- уксусной кислоты метиловый эфир
К 5 ммолям 5-хлор-1Н-индол-2-карбоновой кислоты (50 мл 0,1 М раствора в ацетонитриле) добавляли 1-(3-диметиламинопропил)- 3-этилкарбодиимида гидрохлорид (50 мл 0,1 М раствора в ацетонитриле, 5 ммолей), 1-гидроксибензотриаэола (50 мл 0,1 M раствора в ацетонитриле, 5,0 ммолей) с последующим добавлением (2-амино-3-фенилпропиониламино)-уксусной кислоты метилового эфира (50 мл 0,1 М раствора в ацетонитриле, 5,0 ммоля). Реакционную смесь перемешивали в течение ночи при 80oC и затем концентрировали до сухости с получением соединения заголовка.
ВЭЖХ RT 8,15 (65%).
Пример 165.
2-(S)-[(5-Хлор-1Н-индол-2-карбонил)-амино] -3-фенилпропионовой кислоты бензиловый эфир
Соединение заголовка получали путем замещения L-фенилаланина бензилового эфира (2-амино-3-фенилпропиониламино)-уксусной кислоты метилового эфира по методу, аналогичному методу из процедуры примера 164.
ВЭЖХ RT 8,13 (40%).
Пример 166.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(3- гидроксиазетидин-1-ил)-2-оксоэтил]-амид
2-Амино-1-(3-гидроксиазетидин-1-ил)-3-фенилпропан-1-она гидрохлорид (1,18 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (1,18 ммоля) соединяли по процедуре А (растворитель для реакции дихлорметан-диметилформамид 4:1) и продукт очищали с помощью хроматографии на силикагеле с элюированием 25%, 50%, 75% и 100% этилацетатом-гексанами с получением вещества заголовка в виде бесцветной пены (104 мг, 22%), Была также выделена смесь менее полярных продуктов.
Вещество заголовка: ВЭЖХ (60/40) 4,18 минуты (97%); ТРМС 398/400 (MH+, 100%).
Пример 166а.
(2S)-Амино-1-(3-гидроксиазетидин-1-ил)-3-фенилпропан-1-она гидрохлорид
[(1S)-Бензил-2-(3-гидроксиазетидин-1-ил)-2-оксоэтил] -карбаминовой кислоты трет-бутиловый эфир (515 мг, 1,6 ммоля) растворяли в холодной 4N HCl-диоксане, смесь перемешивали 2 часа при 25oC, концентрировали и остаток выпаривали с эфиром с получением бесцветного твердого вещества (415 мг, 100%).
Пример 167.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(3-гидроксииминоазетидин-1-ил)-2-оксоэтил]-амид
Раствор 5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(3-оксоазетидин-1-ил)-2-оксоэтил]-амида (продукт из примера 170, 50 мг, 0,13 ммоля), ацетата натрия тригидрата (43 мг, 0,32 ммоля) и гидроксиламина гидрохлорида (18 мг, 0,25 ммоля) в метаноле (2 мл) нагревали в колбе с обратным холодильником в течение 8 часов и концентрировали. Остаток разделяли между дихлорметаном и насыщенным водным NaHCO3. Органический слой отделяли и сушили с получением бесцветного твердого вещества, которое растирали с эфиром-гексанами и сушили (выход 36 мг, 69%): ВЭЖХ (50/50) 6,74 мин (99%), ТРМС 411/413 (MH+, 10%), 180 (100%);
1H ЯМР (ДМСО-d6) δ 11,75 (шир,1Н), 11,10 (с, 0,5H), 11,08 (с, 0,5H), 8,99 (д, 1H, J=9 Гц), 7,73 (д, 1H, J=2 Гц), 7,4-7,1 (м, 8Н), 5,0 (м, 1Н), 4,8-4,5 (м, 4Н), 3,1 (м, 2Н).
Пример 168.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2- (4-гидроксииминопиперидин-1-ил)-2-оксоэтил]-амид
Смесь 5-Хлор-1Н-индол-2-карбоновой кислоты [1(S)-бензил- 2-оксо-2-(4-оксопиперидин-1-ил)-этил] амида (406 мг, 0,96 ммоля), гидроксиламина гидрохлорида (80 мг, 1,15 ммоля) и карбоната калия (159 мг, 1,15 ммоля) в этаноле (6 мл) и воде (1 мл) перемешивали при 25oC в течение 18 часов и концентрировали. Остаток растворяли в этилацетате и полученный в результате раствор промывали водой и сушили (411 мг, 98%); ВЭЖХ (60/40) 5,13 минуты (97%); ТРМС 439/441 (MH+, 100%);
1H ЯМР (ДМСР-d6) δ 11,75 (шир, 1Н), 10,45 (с, 0,5H), 10,44 (с, 0,5H), 9,00 (м, 1Н), 7,72 (д, 1Н, J=2 Гц), 7,40 (д, 1Н, J=8,8 Гц), 7,35-7,15 (м, 7Н), 5,17 (м, 1Н), 5,17 (м, 1Н), 3,8-3,5 (м, 4Н), 3,1 (м, 2Н), 2,45 (м, 2Н), 2,25 (м, 2Н).
Пример 168а.
5-Хлор-1Н-индол-2-карбоновой кислоты [1(S)-бензил-2- оксо-2-(4-оксопиперидин-1-ил)-этил]-амид
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(4- гидроксипиперидин-1-ил)-2-оксоэтил] -амид (пример 46, 669 мг) добавляли одной порцией при 0oC к смеси 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (ДЭК, 1,80 г, 9,4 ммоля) и дихлоруксусной кислоты (307 мг, 1,5 ммоля) в безводном толуоле (3 мл? ) и безводном диметилсульфоксиде (3 мл). Смесь перемешивали при 0-20oC в течение 2 ч, разводили этилацетатом и полученный раствор промывали дважды 1N HCl, дважды насыщенным водным раствором NaHCO3, сушили, концентрировали и остаток очищали с помощью хроматографии на силикагеле с элюированием 25%, 50% и 75% этилацетатом-гексанами с получением пенообразного вещества (424 мг, 64%).
Пример 169.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(1,3-дигидроизоиндол-2-ил)-2-оксоэтил]-амида
2-Амино-1-(1,3-дигидроизоиндол-2-ил)-3-фенилпропан-1-она гидрохлорид (0,20 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,20 ммоля) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием 5%, 10%, 20% и 50% этилацетатом-гексанами (55 мг, 62% выход): ВЭЖХ (70/30) 6,58 минуты;
ТРМС 444/446 (MH+, 50%), 180 (1000).
1H ЯМР (CDCl3) δ 9,25 (шир, 1Н), 7,60 (с, 1H), 7,45 (м,  1Н), 7,3-7,1 (м,
1Н), 7,3-7,1 (м,  ), 6,90 (5,25 (м, 1H), 5,0 (д, 1H,
), 6,90 (5,25 (м, 1H), 5,0 (д, 1H,  16 Гц), 4,85 (д, 1H, J=
16 Гц), 4,85 (д, 1H, J=  16 Гц), 4,70 (д, 1H, J=
16 Гц), 4,70 (д, 1H, J=  16 Гц), 4,20 (д, 1H, J=16 Гц).
16 Гц), 4,20 (д, 1H, J=16 Гц).
Пример 169а.
(2S)-Амино-1-(1,3-дигидро-изоиндол-2-ил)-3-фенил-пропан-1-она гидрохлорид
[(1S)-Бензил-2-(1,3-дигидро-изоиндол-2-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутиловый эфир (88 мг) растворяли в холодной 4N HCl-диоксане (1,5 мл), перемешивали 2 ч при 25oC и смесь концентрировали. Остаток растирали с эфиром и сушили (65 мг, 91%). ТРМС 267 (MH+, 100%).
Пример 169b.
[(1S)-Бензил-2-(1,3-дигидроизоиндол-2-ил)-2-оксоэтил]- карбаминовой кислоты трет-бутиловый эфир
N-t-Boc-L-фенилаланин (1 ммоль) и изоиндолин (J. Org. Chem., 1988, 53, р. 5382, 70-80% чистоты, 1 ммоль) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием 20% и 50% этилацетатом-гексанами, получая янтарное масло (88 мг, 23%): ТРМС 367 (MH+, 100%).
Пример 170.
5-Хлор-1Н-индол-2-карбоновой кислоты [(1S)-бензил-2-(3-оксоазетидин-1-ил)-2-оксоэтил]-амид
1-((2S)-Амино-3-фенилпропионил)-азетидин-3-она гидрохлорид (3,2 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (3,2 ммоля) соединяли по процедуре А (0-25oC - температура реакции) и полученную желтую пену очищали с помощью хроматографии на силикагеле с элюированием 20%, 30%, 40% и 50% этилацетатом в гексане, получая вещество заголовка в виде бесцветной пены (600 мг, 47%): ВЭЖХ (60/40) 5,09 минуты (98%); ТРМС 396 (MH+, 100%);
1H ЯМР (CDCl3) δ 9,14 (шир, 1Н), 7,62 (д, 1H, J=3 Гц), 7,4-7,2 (м, 7Н), 7,11 (д, 1H, J=8,0 Гц), 6,85 (м, 1Н), 4,90 (м, 1Н), 4,78 (м, 2Н), 4,63 (м, 1Н), 3,65 (м, 1Н), 3,25 (дд, 1Н, A из AB, J=5,1, 12,9 Гц), 3,10 (дд, 1Н, B из AB, J=10, 12,9 Гц).
Пример 170а.
1-((2S)-Амино-3-фенилпропионил)-азетидин-3-она гидрохлорид
[(1S)-Бензил-2-оксо-2-(3-оксоазетидин-1-ил)-этил] -карбаминовой кислоты трет-бутиловый эфир (297 мг, 0,9 ммоля) растворяли в 4N HCl-диоксане (3 мл). Полученный раствор перемешивали при 25oC в течение 2 ч, концентрировали и остаток растирали с эфиром и сушили (196 мг, 82%).
Пример 170b.
[(1S)-Бензил-2-оксо-2-(3-оксоазетидин-1-ил)-этил] -карбаминовой кислоты трет-бутиловый эфир
[(1S)-Бензил-2-(3-гидроксиазетидин-1-ил)-2-оксоэтил] -карбаминовой кислоты трет-бутиловый эфир (320 мг, 1 ммоль) добавляли одной порцией к смеси 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (ДЭК, 575 мг, 3 ммоля) и дихлоруксусной кислоты (192 мг, 1,5 ммоля) в безводном толуоле (2 мл) и безводном диметилсульфоксиде (2 мл). Смесь перемешивали при 0-20oC в течение 1 ч, разводили этилацетатом, полученный раствор промывали дважды 1N HCl, дважды насыщенным водным NaHCO3, сушили и концентрировали с получением бесцветного твердого вещества (304 мг, 96%).
Пример 170c.
[(1S)-Бензил-2-(3-гидроксиазетидин-1-ил)-2-оксоэтил] - карбаминовой кислоты трет-бутилового эфира
3-Гидроксиазетидина гидрохлорид (J. Chem. Soc., Chem. Commun., 1968, р. 93, 27 ммолей) и N-t-Boc-L-фенилаланин (27 ммолей) соединяли по процедуре А с получением вещества заголовка в виде бесцветной пены (8,15 г, 93%).
Пример 171.
5-Хлор-1Н-бензоимидазол-2-карбоновой кислоты (1-диметилкарбамоил-2-фенилэтил)-амид
(S)-2-Амино-N, N-диметил-3-фенилпропионамида гидрохлорид (2,0 ммоля) и 5-хлор-1Н-бензоимидазол-2-карбоновую кислоту (Crowther et al., J. Chem. Soc. , 1949, р.1268, 2 ммоля) соединяли по процедуре А и продукт очищали с помощью хроматографии на силикагеле с элюированием этилацетатом-гексанами 1:1 (235 мг, 63%): ВЭЖХ (60/40) 4,92 мин (91%); PBMS (MH+, 100%);
1H ЯМР (CDCl3) δ 11,25 (шир, 0,6Н), 10,9 (шир, 0,4Н), 8,36 (м, 1Н), 7,78 (д, 0,4H, J=7,72 (d, 0,6H, J=8,8 Гц), 7,52 (д, О,6Н, J=2 Гц), 7,41 (д, 0,4H, J= 8,4 Гц), 7,35-7,1 (м, 6Н), 7,35 (м, 1Н), 3,16 (м, 2Н), 2,90 (с, 3Н), 2,68 (с,  2Н), 2,67 (с.
2Н), 2,67 (с.  1H).
1H).
Пример 172.
5-Хлор-1Н-индол-2-карбоновой кислоты [1-бензил-2-оксо-2- (2-оксо-оксазолидин-3-ил)-этил]-амид
3-((2S)-Амино-3-фенилпропионил)-оксазолидин-2-она гидрохлорид (0,50 ммоля) и 5-хлор-1Н-индол-2-карбоновую кислоту (0,50 ммоля) соединяли по процедуре А (растворитель для реакции - диметилформамид-дихлорметан 3:1) и продукт растирали с эфиром-гексанами 2:1 и сушили (130 мг, 63%): ВЭЖХ (60/40) 6,22 минуты (95%); ТРМС 429/431 (45%, MH+ NH3), 412/414 (30%, MH+, 325/327 (100%).
1H ЯМР (ДМСО-d6) δ 11,68 (шир, 1Н), 8,92 (д, 1Н, J=8,5 Гц), 7,75 (с, 1Н), 7,42 (м, 3Н), 7,26 (м, 3Н), 7,18 (м, 2Н), 5,83 (м, 1Н), 4,50 (м, 2Н), 4,0 (м, 1Н), 3,25 (м, 1Н), 2,95 (м, 1Н).
Пример 172а.
3-((2S)-Амино-3-фенилпропионил)-оксазолидин-2-она гидрохлорид
[(1S)-Бензил-2-оксо-2-(2-оксо-оксазолидин-3-ил)-этил] -карбаминовой кислоты трет-бутиловый эфир (2,29 г, 6,68 ммоля) растворяли в 4М HCl-диоксане (10 мл) при 0oC. Полученный раствор перемешивали при 25oC в течение 2 ч, концентрировали и остаток растирали с эфиром и сушили (1,98 г, 107%).
Пример 172b.
[(1S)-Бензил-2-оксо-2-(2-оксо-оксазолидин-3-ил)-этил]- карбаминовой кислоты трет-бутиловый эфир
N-Бутиллитий (2,35 М в гексанах, 11,5 мл) добавляли при 78oC к раствору 2-оксазолидинона (2,04 г, 23,4 ммоля) в тетрагидрофуране (25 мл). После 30 минут при 78oC раствор обрабатывали N-t-Boc-L-фенилаланина N-гидроксисуцинимидным эфиром (9,31 г, 25,7 ммоля) в тетрагидрофуране (10 мл), и перемешиваемой смеси давали нагреться до 25oC в течение ночи. Добавляли воду (10 мл) и полученную смесь концентрировали, остаток растворяли в этилацетате, и полученный раствор промывали дважды 1N NaOH, один раз водой, один раз солевым раствором, сушили и концентрировали. Остаток хроматографировали на силикагеле, элюировали 25% и 50% этилацетатом в гексанах с получением бесцветного твердого вещества (3,42 г, 44%).
Должно быть понятно, что изобретение не ограничивается конкретными осуществлениями, описанными здесь, но что различные изменения и модификации могут быть произведены, не выходя за сущность и объем этой новой идеи, что определено следующими пунктами формулы изобретения.
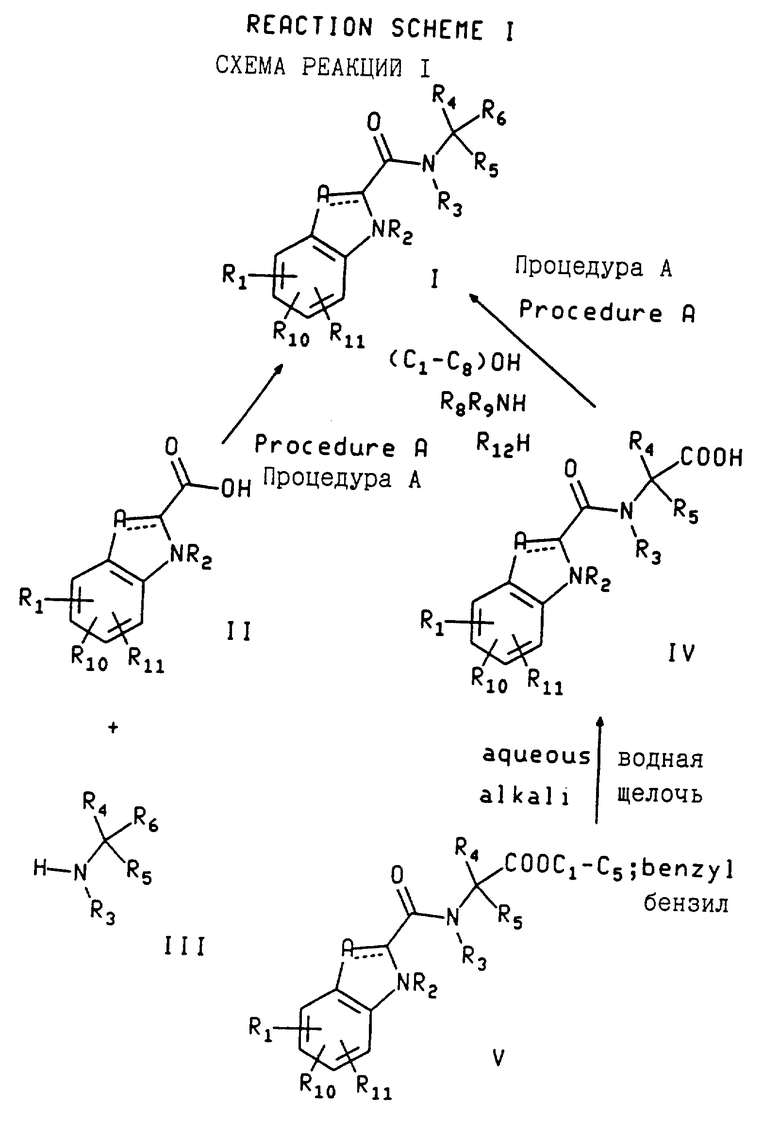
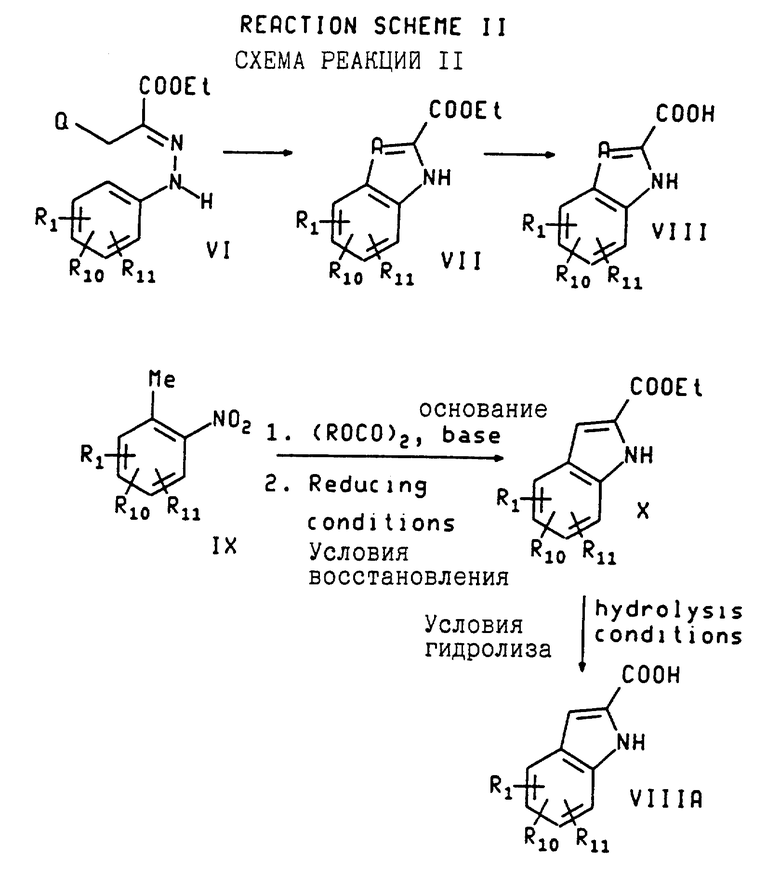

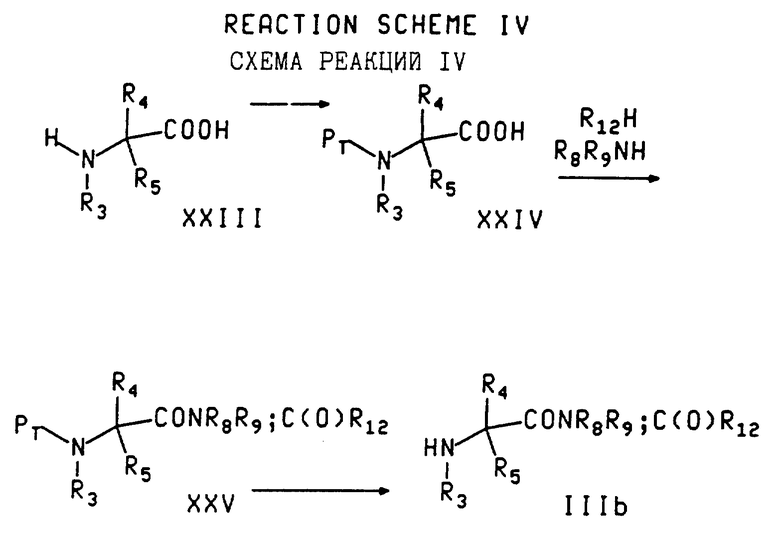
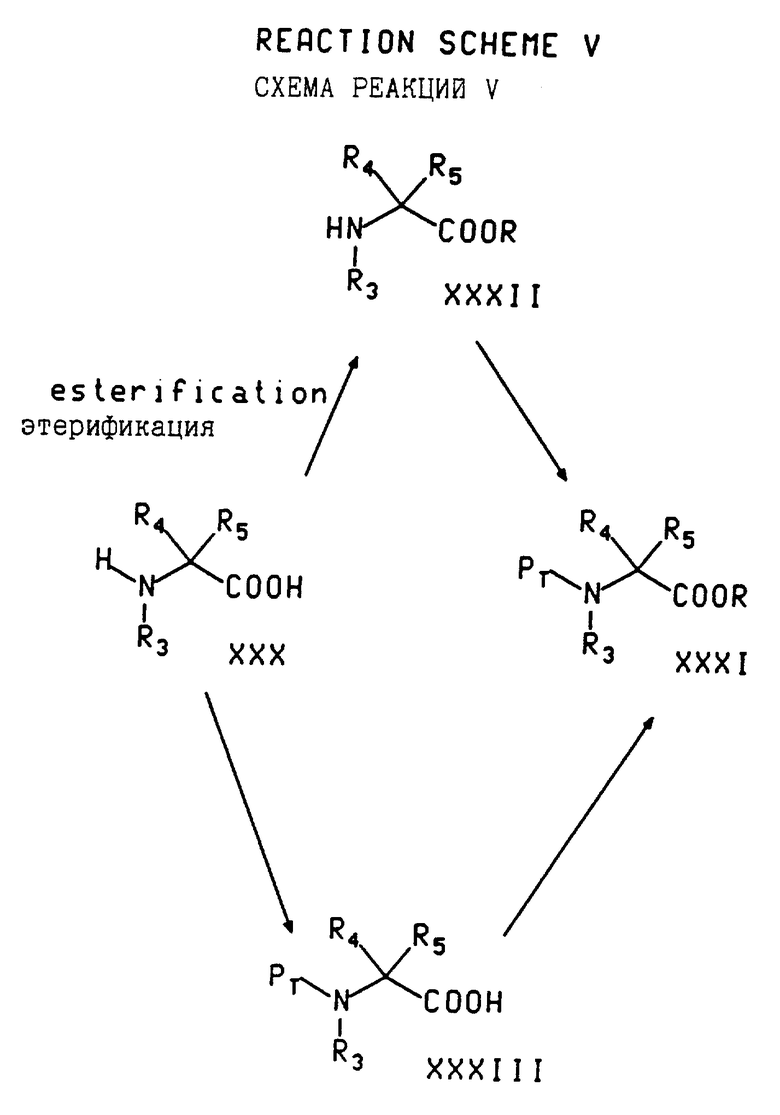
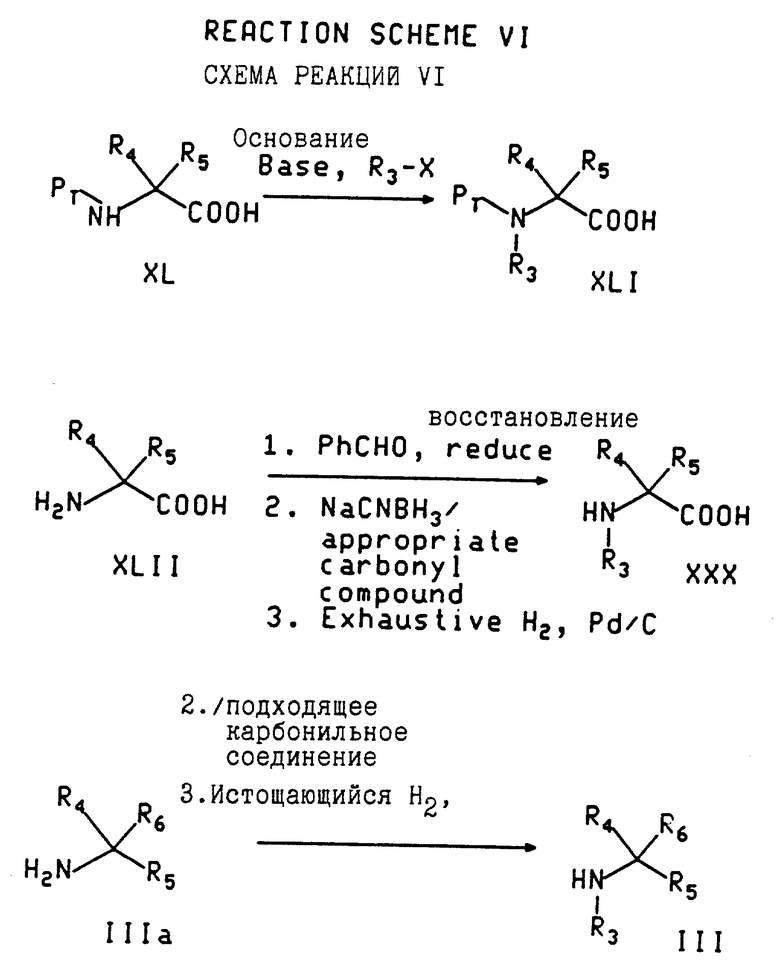
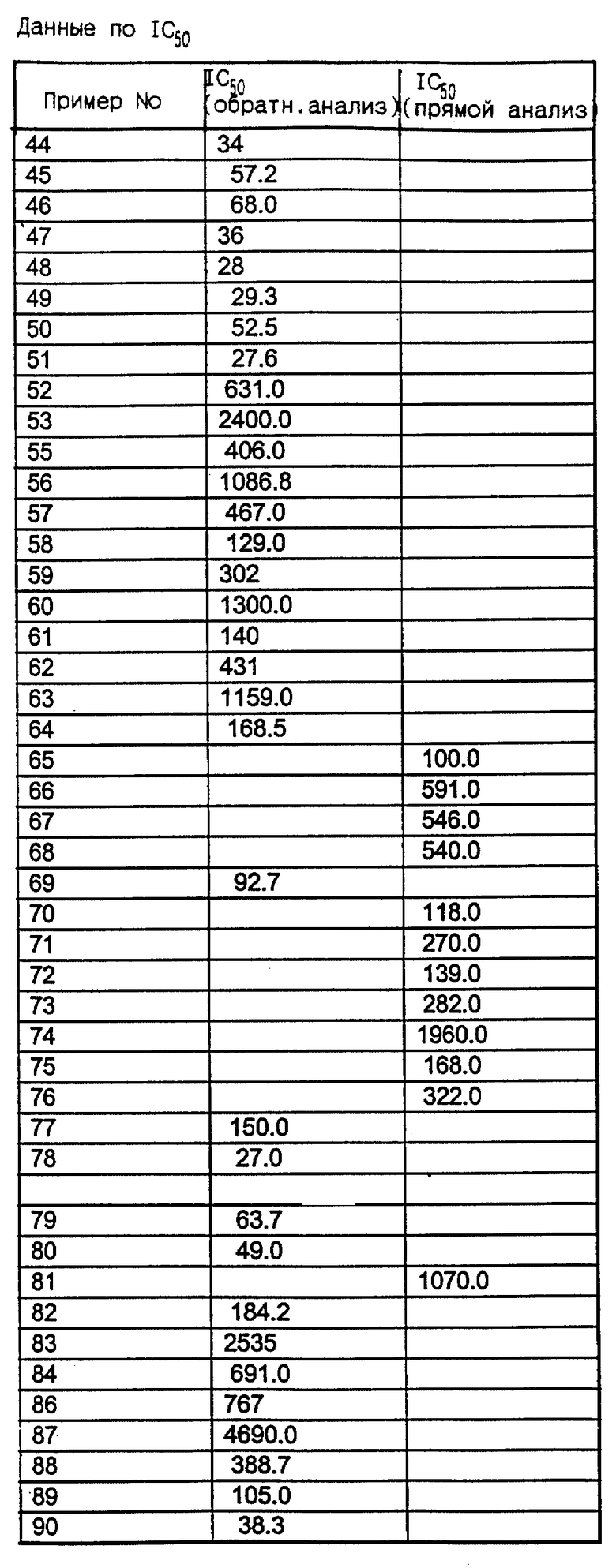
Описываются замещенные N-(индол-2-карбонил)-глицинамиды и их производные общей формулы (I), где значения R1 - R6, R10, R11 и А указаны в п.1 формулы изобретения, которые проявляют свойства ингибиторов гликогенфосфорилазы. Описывается также способ лечения и фармкомпозиция на основе соединений формулы (I) для снижения активности гликогенфосфорилазы у млекопитающих. 4 с. и 51 з.п.ф-лы, 1 табл.
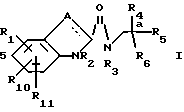
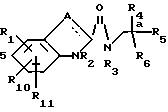
и их фармацевтически приемлемые соли,
где прерывистая линия (---) является необязательной связью;
A представляет -C(H)=, -C((C1 - C4)алкил)=, -C(галоген)= или -N=, когда прерывистая линия (---) является связью, или A является метиленом или представляет -CH((C1 - C4)алкил)-, когда прерывистая линия (---) не представлена связью;
R1, R10 или R11 каждый независимо является H, галогеном, циано-, 4-, 6- или 7-нитро-, (C1 - C4)алкилом, (C1 - C4)алкокси, фторметилом, дифторметилом или трифторметилом;
R2 - H;
R3 - H или (C1 - C5)алкил;
R4 является H, метилом, этилом, н-пропилом, гидрокси(C1 - C3)алкилом, (C1 - C3)алкокси(C1 - C3)алкилом, фенил(C1 - C4)алкилом, фенилгидрокси(C1 - C4)алкилом, (фенил)((C1 - C4)алкокси)(C1 - C4)алкилом, фур-2 или 3(C1 - C4)алкилом, тиен-2- или -3-ил(C1 - C4)алкилом, причем указанные кольца R4 являются независимо моно-, ди- или тризамещенными у атома углерода водородом, галогеном, (C1 - C4)алкилом, (C1 - C4)алкокси, трифторметилом, гидроксилом, аминогруппой, цианогруппой или 4,5-дигидро-1Н-имидазол-2-илом; или R4 является пирид-2-, -3- или -4-ил(C1 - C4)алкилом, тиазол-2-, -4- или -5-ил(C1 - C4)алкилом, имидазол-2-, -4- или -5-ил(C1 - C4)алкилом или индол-2-(C1 - C4)алкилом, причем указанные предыдущие гетероциклы R4 являются необязательно моно- или дизамещенными независимо галогеном, трифторметилом, (C1 - C4)алкилом, (C1 - C4)алкокси, амино, гидроксилом или циано, и указанные заместители связаны с углеродом;
R5 - водород;
R6 является карбоксилом, (C1 - C8)алкоксикарбонилом, бензилоксикарбонилом, C(O)NR8R9 или C(O)R12, где R8 - H, (C1 - C6)алкил, цикло(C3 - C6)алкил или (C1 - C8)алкокси, R9 - H, цикло(C3 - C8)алкил, цикло(C3 - C8)алкил(C1 - C5)алкил, цикло(C4 - C7)алкенил, цикло(C3 - C7)алкил(C1 - C5)алкокси, цикло(C3 - C7)алкилокси, гидроксил, метиленперфторированный (C1 -C8)алкил, фенил или гетероцикл, причем указанный гетероцикл является тиазолилом, изоксазолилом, изотиазолилом, морфолинилом, бензотиазолилом, тиохроманилом или тетрагидробензотиазолилом, где указанные гетероциклические кольца являются связанными через углерод-азот, или R9 является (C1 - C6)алкилом или (C1 - C8)алкокси, где указанный (C1 - C6)алкил или (C1 - C8)алкокси является необязательно монозамещенным цикло(C4 - C7)алкен-1-илом, фенилом, тиенилом, морфолинилом или индолилом, и где кольца R9 являются независимо необязательно моно- или дизамещенными у атома углерода (C1 - C4)алкилом, (C1 - C4)алкокси, при условии, что не включен ни один четвертичный азот в любой гетероцикл R9;
R12 - морфолино, тиоморфолино, 1-оксотиоморфолино, 1,1-диоксотиоморфолино, тиазолидин-3-ил, 1-оксотиазолидин-3-ил, 1,1-диоксотиазолидин-3-ил, пирролидин-1-ил, пиперидин-1-ил, пиперазин-1-ил, пиперазин-4-ил, азетидин-1-ил, оксазолидин-3-ил, 3,4-дигидроизохинолин-2-ил, 1,3-дигидроизоиндол-2-ил, где указанные кольца R12 являются независимо необязательно моно-, ди- или тризамещенными галогеном, (C1 - C5)алкилом, (C1 - C5)алкокси, гидроксилом, амино, моно-N- или ди-N,N-(C1 - C5)алкиламино, формилом, карбоксилом, карбамоилом, (C1 - C6)алкокси(C1 - C3)алкокси, (C1 - C5)алкоксикарбонилом, бензилоксикарбонилом, (C1 - C4)алкоксикарбониламино, карбокси(C1 - C5)алкилом, оксо, гидроксиимино или (C1 - C6)алкоксиимино, и где не более чем два заместителя, выбираемые из оксо, гидроксиимино или (C1 - C6)алкоксиимино и оксо, гидроксиимино или (C1 - C6)алкоксиимино, находятся у неароматического углерода и
при условии, что когда R6 является (C1 - C5)алкоксикарбонилом или бензилоксикарбонилом, тогда R1 является 5-галогеном, 5-(C1 - C4)алкилом или 5-циано и R4 является (фенил)(гидрокси)(C1 - C4)алкилом, (фенил)((C1 - C4)алкокси)(C1 - C4)алкилом, гидроксиметилом или Ar(C1 - C2)алкилом, где Ar - тиен-2- или -3-ил, фур-2- или -3-ил или фенил, где указанный Ar является необязательно моно- или дизамещенным независимо галогеном; при условии, что когда R4 - бензил и R5 - метил, R12 не является 4-гидроксипиперидин-1-илом, или когда R4 - бензил и R5 - метил, R6 - не C(O)N(CH3)2;
при условии, что когда R1 и R10 и R11 - H, R4 - не имидазол-4-илметил, 2-фенэтил или 2-гидрокси-2-фенилэтил;
при условии, когда R8 и R9 оба - н-пентил, R1 - 5-хлор, 5-бром, 5-циано, 5(C1 - C5)алкил, 5(C1 - C5)алкокси или трифторметил;
при условии, что когда R12 - 3,4-дигидроизохинол-2-ил, указанный 3,4-дигидроизохинол-2-ил не замещен карбокси(C1 - C4)алкилом; и
при условии, что когда R6 - карбоксил и R1, R10, R11 и R5 все - H, тогда R4 не является бензилом, H, (фенил)(гидрокси)метилом, метилом, этилом или н-пропилом.
[(1S)-бензил-2-(3-гидроксииминопирролидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[2-(цис-3,4-дигидроксипирролидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[2-((3S, 4S)-дигидроксипирролидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-(цис-3,4-дигидроксипирролидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[2-(1,1-диоксотиазолидин-3-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
(2-оксо-2-тиазолидин-3-илэтил)амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-(4-фторбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-((3RS)-гидроксипиперидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[2-оксо-2-((1RS)-оксо-1-тиазолидин-3-ил)этил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-(2-фторбензил)-2-(4-гидроксипиперидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-((3S, 4S)-дигидроксипирролидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-(3-гидроксиазетидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-(3-гидроксииминоазетидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты или
[(1S)-бензил-2-(4-гидроксииминопиперидин-1-ил)-2-оксоэтил] амид 5-хлор-1Н-индол-2-карбоновой кислоты.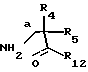
где R5 - водород;
R4 является H, фенилметилом, тиен-2- или -3-илметилом, причем указанные кольца являются необязательно моно- или дизамещенными фтором;
R12 является тиазолидин-3-илом, 1-оксотиазолидин-3-илом, 1,1-диоксотиазолидин-3-илом, пирролидин-1-илом, пиперидин-1-илом, азетидин-1-илом или оксазолидин-3-илом, где указанные кольца R12 являются необязательно моно- или дизамещенными независимо галогеном, (C1 - C5)алкилом, (C1 - C5)алкокси, гидроксилом, амино, моно-N- или ди-N,N-(C1 - C5)алкиламино, карбоксилом, карбамоилом, (C1 - C5)алкоксикарбонилом, амино(C1 - C4)алкилом, оксо, гидроксиимино или (C1 - C6)алкоксиимино при условии, что только R12 гетероциклы тиазолидин-3-ил, пирролидин-1-ил, пиперидин-1-ил, азетидин-1-ил или оксазолидин-3-ил являются необязательно моно- или дизамещенными независимо оксо, гидроксиимино или (C1 - C6)алкоксиимино, при условии, что R12 не является 2-карбокси-4-гидроксипирролидин-1-илом, 2-((C1 - C5)алкоксикарбонил)-4-гидроксипирролидин-1-илом, 2-карбоксипиперидин-1-илом или 2-((C1 - C5)алкоксикарбонил)пиперидин-1-илом.
| Железобетонные блоки для крепления вертикальных стволов шахт | 1955 |
|
SU105763A1 |
| Способ получения производных пиридо (2,3- )пиримидина | 1974 |
|
SU624575A3 |
| Kasvinsky P.J | |||
| et al | |||
| J.Biol | |||
| Чугунный экономайзер с вертикально-расположенными трубами с поперечными ребрами | 1911 |
|
SU1978A1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЙ, НАДПИСЕЙ И Т.П. ОТБРАСЫВАЕМЫХ НА ЭКРАН | 1926 |
|
SU3343A1 |
| Кон Р.И., Рот К.С | |||
| Ранняя диагностика болезней обмена веществ | |||
| - М.: Медицина, 1986, с | |||
| Подвижная хлебопекарная печь | 1925 |
|
SU433A1 |
