Настоящее изобретение относится к области фармацевтики и медицины, конкретнее к фармацевтической композиции, обладающей превосходной противовирусной активностью, предназначенной для облегчения клинических симптомов, течения и излечения широкого спектра заболеваний, вызванных воздействием вирусов, геном которых закодирован одноцепочечной нитью РНК и которые используют вирусную РНК-зависимую-РНК-полимеразу для своей репликации, включая вирусы гриппа, как высоко, так и низко вирулентных штаммов, коронавирусы, пикорнавирусы, вирусы семейств арена-, флави-, бунья-, тога-, кальци и норавирусов, вызывающие тяжелейшие и, в основном, летальные лихорадки.
Одноцепочечные РНК вирусы составляют широкую и разнообразную группу патогенов, представляющих серьезную опасность для здоровья человека. Ежегодно они поражают миллионы людей во всем мире и, в зависимости от семейства вируса, вызывают болезни от простого ОРВИ, поражающего верхние дыхательные пути, до пневмонии и острого дистресс синдрома нижних дыхательных путей, таких, как SARS и MERS [70], а в регионах Азии и Африки - неизлечимые тропические гемморагические лихорадки, такие как вирус Денге (DENV) и вновь возобновившиеся вспышки вирусов Чикугунья (CHIKV) и Зика (ZIKV). Для большинства данных вирусов, независимо от семейства, не существует специфичной противовирусной терапии, приводящей к полному излечению пациентов и не приводящей к значительным осложнениям, а также предотвращающей летальный исход [68, 69]. Одноцепочечные вирусы со смысловым РНК-геномом используют репликативный и трансляционный аппарат самой клетки, поэтому любые препараты, действующие на вирус, будут априори довольно токсичны для клеток самого человека.
В частности, многие терапевтические агенты, подавляющие синтез ДНК и РНК и доказано блокирующие репликацию а и b - коронавирусов, относятся к цитостатическим средствам, вызывающим серьезные токсичные побочные эффекты [71, 76]. Дополнительная угроза здоровью от данных вирусов состоит в том, что многие РНК-вирусы помимо сродства к тропному органу, проявляют высокую нейроинвазивность, что приводит к более значимым поражениям организма. Таковы, в частности, вирусы гриппа, большинство коронавирусов, флавивирусы, энтеровирусы, Чикугунья [72]. Они проникают в ЦНС различными путями: через ольфакторные нейроны полости носа, чувствительные волокна вагуса, иннервирующие органы дыхательных путей, периферические нейроны и лицевой нерв [73, 74]. Большинство РНК-вирусов научились «обманывать» иммунную систему, проскакивая незаметно мимо немедленной системы узнавания и интерферонового ответа организма, вызывая острые течения заболевания, когда вирусные титры уже колоссальны, а поражение тропного органа обширно и иммунный ответ либо отсутствует, либо, напротив, зашкаливает. Флави-, бунья-, вирус Денге, коронавирусы научились обходить немедленный иммунный ответ, совершенно различными способами выключая, как экспрессию генов интерферона, так и способность РНКаз узнавать чужеродную одноцепочечную РНК, а также NFκB путь [75]. РНК-вирусам свойственно очень быстро мутировать, таким образом, адаптируясь к целому спектру зоонотических хозяев, а далее «перескакивая» к человеку и вызывая жизнеугрожающие состояния, требующие госпитализации, а также периодически вызывая новые неожиданные вспышки в разных эндемичных регионах [72].
Ясно, что опасные одноцепочечные РНК вирусы требуют разработки новых подходов и применения новых улучшенных схем с мультифакторным действием на заболевания. И даже хорошо изученные вирусы гриппа, вызывающие ежегодные пандемии с осложнениями, а также вездесущие коронавирусы, приводящие к разнообразным острым респираторным синдромам и вирусным ринитам, фарингитам, ларингитам и т.п., бывают возбудителями гораздо более опасных летальным исходом состояний.
Одним из подходов к разработке высокоспецифичных противовирусных препаратов остается направленное таргетирование вирус-специфических функциональных молекул, таких как структурные и неструктурные белки, вирусные протеазы и отсутствующая в животных клетках РНК-зависимая-РНК-полимераза. Последняя поэтому представляет особый интерес в качестве мишени ингибиторов вирусной репликации.
Общественная потребность в создании композиции, обладающей активностью в отношении вышеуказанных вирусов, в настоящее время является очень актуальной.
Так, например, вспыхнув в Ухани (Китай) в декабре 2019 года, новый вирус из семейства коронавирусов (SARS-CoV-2), вызывающий атипичную пневмонию, переходящую в острый респираторный дистресс синдром (ОРДС) с системной гипоксемией и воспалительным эндоваскулитом, стремительно распространился по разным уголкам планеты, окунув ее в давно невиданную пандемию. Менее, чем за 2 месяца, на момент 10 февраля 2020 г., вирус достиг 25 стран, 4х континентов, и количество подтвержденных случаев заболевания перевалило за 400,000. На тот момент риск смертности составил около 2%. К 15 апреля 2020 г. количество подтвержденных случаев заболевания по всему миру составило 1 991 562 человек, унесло оно 130 885 жизней [1]. Данные показатели сводят уровень смертности до 0,066%, однако, как показала клиническая практика последних 3,5 месяцев, она варьирует в разных группах населения, в зависимости от возраста, представляя наибольшую опасность своим агрессивным течением для людей от 70 и выше лет с сопутствующими ко-морбидными состояниями.
К сожалению, на настоящий момент лекарств, противодействующих инфекции нет, также нет точных установленных клинических предписаний, поскольку течение вируса неоднородно по мере развития заболевания у индивидуума и различно в разных группах населения, как возрастных, так и этнических. Механизм возникновения симптомов COVID-19 изучается многими научными и клиническими лабораториями в авральном режиме, непосредственно в процессе пандемии, снабжая врачей и ученых лишь обрывочными знаниями о природе вируса и физиологии течения заболевания, в особенности сценария, приводящего к развитию острого респираторного дистресс синдрома, который не всегда успешно снимается поддержкой больного на аппарате искусственной вентиляции легких (ИВЛ).
Однако, принадлежность SARS-CoV-2 довольно хорошо изученному семейству b-коронавирусов, а значит понимание, в целом, жизненного цикла вируса в человеческом организме, позволяет сделать некоторые клинические предположения и выбрать селективные и эффективные терапевтические решения из арсенала лекарств, используемых для подтипов того же семейства - коронавирусов, исходя из опыта вспышек SARS и MERS в 2002 и 2012 гг., соответственно, протекавших с похожими клиническими симптомами и гораздо большей смертностью.
2019-nCoV или SARS-CoV-2 - капсидный коронавирус семейства b-coronaviridae, несущий смысловую нить одноцепочечной РНК. По аналогии с SARS и MERS, геном 2019-nCoV кодирует неструктурные белки (3-химотрипсин-подобную протеазу, папаин-подобную протеазу, хеликазу, РНК-зависимую-РНК-полимеразу (РНКзРНКпол / РзРп)), обеспечивающие репликацию и сборку вирусных частиц, структурные белки, включая S-гликопротеин, необходимый для узнавания клеточного рецептора и осуществления входа в клетку, М (мембранный белок), Е (белок оболочки), N (белок нуклеокапсида), и вспомогательные белки, играющие роль в поддержании жизнедеятельности вируса, его репликации и геномной стабильности [2,3]. Все 5 неструктурных белков и РНК-з-РНК-пол являются прямыми мишенями для потенциального лекарства против коронавируса 2019-nCoV.
Анализ генома 2019-nCoV (Фиг. 1) показал, что каталитические протеазы достаточно консервативны в семействе b-коронавирусов, будучи гомологичными соответствующим ферментам вирусов SARS и MERS [4]. Поэтому в настоящий момент ведутся множественные международные исследования по репозиционрованию успешно работающих препаратов, используемых от SARS и MERS.
Таким образом, имеются обоснованные предположения в отношении веществ-кандидатов для потенциального фармацевтического использования в вышеуказанных целях.
Потенциальные кандидаты на репозиционирование для терапии 2019-nCoV.
Лекарства, взаимодействующие с вирусным аппаратом репликации.
Одобренные аналоги нуклеозидов, в частности фавипиравир, обладают хорошим клиническим потенциалом против вирусов, использующих в репликации РНК-зависимую-РНК-полимеразу, а, следовательно, в частности, против 2019-nCoV. Аналоги нуклеозидов - как адениновые, так и гуаниновые производные - напрямую нацелены на блокировку активности РНК-зависимой-РНК-полимеразы и блокируют синтез вирусной цепи РНК для широкого спектра РНК-вирусов, включая семейство человеческих коронавирусов [5]. Фавипиравир (Т-705), гуаниновый аналог, одобренный в клинической практике для лечения гриппа, доказано эффективно блокирует РНК-зависимую-РНК-полимеразу вирусов гриппа (разных типов), вируса Эбола, желтой лихорадки, чикунгунья, норовирусов, энтеровирусов [5]. Недавнее исследование показало его эффективность против 2019-nCoV (ЭК50=61.88 μM в культуре клеток Vero Е6) [6, 33]. Исследование проверяло сочетанное применение фавипиравира с интерфероном-α (ChiCTR2000029600) и фавипиравира с балоквавир марбоксилом (одобренным в показании для гриппа ингибитором кэп-зависимой эндонуклеазы)(ChiCTR2000029544) у пациентов с SARS-CoV-2. Фавипиравир проявляет доказанное направленное действие против SARS-CoV-2. Это легко объяснить его свойствами ингибитора РНК-зависимой-РНК-полимеразы, доказанной на множестве исследований РНК-вирусов разных семейств, всех использующих этот фермент, приведенных ниже.
Механизм действия Фавипиравира
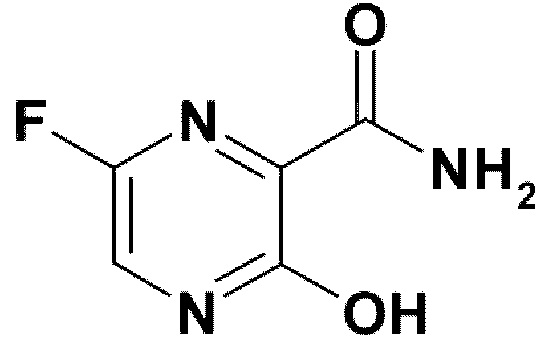
Соединение 1
Изначально фавипиравир (Соединение 1) был открыт как мощный ингибитор вирусной репликации в исследованиях на вирусе гриппа [7]. В исследованиях in vitro на модели клеточных линий Мадин-Дарби (Madin Darby Canine Kidney), зараженных вирусом гриппа, действие фавипиравира ослаблялось в присутствии пуриновых нуклеозидов или пуриновых оснований, указывая на избирательное соревнование вещества с пуриновыми основаниями при синтезе новых цепей РНК в процессе репликации. К клеткам MDCK добавляли фавипиравир, и далее клеточные метаболиты анализировали с помощью жидкостной хроматографии высокого давления. В результате были выявлены фавипиравир рибофуранозил- 5'-трифосфат (фавипиравир - РТФ), фавипиравир-рибофураноза (фавипиравир Р) и фавипиравир рибофуранозил 5' монофосфат (фавипиравир РМФ), указывая на то, что действие фавипиравира происходит при его попадании в клетку. Химически синтезированный фавипиравир - РТФ ингибирует вирусную РНК-зависимую-РНК-полимеразу в нано- и микро-молярных концентрациях (Фиг. 2, 3) [7]. Эти данные показали, что фавипиравир как субстанция является про-лекарством и, будучи фосфо-рибозилированным в клетке до фавипиравира-РТФ, ингибирует вирусную РНК-зависимую-РНК-полимеразу.
В настоящий момент считается, что фавипиравир встраивается в растущую цепь вирусной РНК, прерывая ее, а также одновременно структурно взаимодействует с доменами вирусной РзРп, таким образом, ингибируя как транскрипцию, так и трансляцию вирусного генома [8]. Также в исследованиях на вирусе гриппа было показано, что фавипиравир приводит к летальному мутагенезу вирусного генома, резко снижая вирусные титры в культуре клеток [9]. Значимо, что за все время многолетних исследований данного вещества не удалось выявить ни одного вирусного штамма, резистентного к фавипиравиру. Исходя из описанного механизма действия, мутагенез, вызываемый фавипиравиром, может происходить и в других РНК-вирусах, также, как и ингибирование РзРп, что делает его мощным, эффективным и универсальным ингибитором целой группы эпидемиологически значимых вирусов различных семейств, вызывающих серьезные, часто смертельные заболевания [10-12].
Синтез РНК является неотъемлемой частью жизни человеческой клетки. В отличие от вирусов у людей нет РзРп, однако есть ДНК-зависимая-РНК-полимераза (ДзРп), а также ДНК-зависимая-ДНК-полимераза. Фавипиравир-РТФ способен ингибировать эти два человеческих фермента в разных концентрациях активного лекарственного вещества. Было показано, что фавипиравир-РТФ блокирует РзРп гриппа при IC50 0.341 μмоль/литр, но не оказывал никакого ингибирующего действия на ДНК-полимеразу α, β, γ в концентрациях до 1000 μмоль/литр. Фавипиравир незначительно угнетал человеческую РНК-полимеразу II, относящуюся к ДзРп, но при высоких концентрациях - IC50 905 μмоль/литр [13]. В концентрациях 637 μмоль/литр in vitro в клетках MDCK фавипиравир не блокировал ни синтез клеточной ДНК, ни синтез клеточной РНК.
Такой универсальный механизм действия фавипиравира специфично на основополагающий фермент репликативного аппарата вируса предполагает широкий спектр противовирусной активности данного вещества, что было продемонстрированно во многих исследованиях, указанных ниже.
Противовирусная активность фавипиравира
Использование фавипиравира для лечения пациентов с COVID-19.
Согласно глобальному отчету по фарм-субстанциям, в настоящее время фавипиравир вошел в клинические испытания против заболевания COVID-19, исходя из доказанного механизма его действия против вирусной РзРп (https://www.clinicaltrialsarena.com/comment/influenza-favipiravir-covid-19/). В клиническом исследовании Национального Центра Клинических Исследований Инфекционных Заболеваний в Шеньжене, фавипиравир вводился 340 пациентам (возрастные группы и ко-морбидности не уточняются) в два приема (2 дозы) по 1600 мг в первый день и две дозы по 600 мг последующие 13 дней в дополнение к ингаляционному аэрозолю интерферона-альфа (5 млн. единиц* 2/день). Эта дозировка приводила к более быстрому исчезновению вируса до недетектируемых в крови значений), чем в группе пациентов, принимавших комбинацию анти-ВИЧ протеаз - лопинавира / ритонавира, с медианой вывода вирусных частиц в 4 дня, против 11, соответственно, и оценивалась по контролю КТ грудного отдела. По данным текущего исследования применение фавипиравира снижает количество детектируемых вирусных частиц в крови, а значит, доказательно ингибирует вирусную репликацию, отсрочивает развитие агрессивного сценария течения COVID-19 или вовсе предотвращает его.
С учетом универсальности механизма действия фавипиравира, данное лекарство может позиционироваться в качестве ингибитора РзРп широкого спектра действия и успешно применяться для борьбы с другими заболеваниями, вызванными РНК-вирусами, оснащенными РзРп репликативным аппаратом, как описано далее. Это особенно актуально, в свете участившихся случаев зоонотического переноса вирусов во всем мире. Так вспышки вируса Эбола и Ласса в Западной Африке в 2014 г. вызвало большую волну социальной озабоченности возможными последующими вспышками смертоносных вирусов в странах Азии и Африки [15, 16].
Эффект применения фавипиравира против вируса гриппа
Эпидемия гриппа случается во всем мире ежегодно. Заболевание вызвано штаммами вируса гриппа различной вирулентности. Высоко патогенный вирус птичьего гриппа A(H5N1) вызвал первую вспышку заболевания в Гонг-Конге в 1997 г. и продолжает каждый год вызывать локальные вспышки данного вида гриппа. Эпидемия птичьего гриппа A(H7N9) в Китае в 2013 г. и пандемия гриппа A(H1N1) в 2009 привела к 17,700 смертям, и грипп до сих пор грипп является одной из серьезных проблем для здравоохранения во всем мире, не только по масштабу заболеваемости, но и по критическим для здоровья осложнениям, которые он вызывает [14]. Эпидемия A(H1N1) показала, что данный штамм устойчив к оцетальмивиру (тамифлю) - ингибитору нейрамидазы и к амантадину - ингибитору неструктурного белка М2. В связи с этим в медицинской практике остро необходимо лекарство другого механизма действия.
Фавипиравир (Т-705; 6-флуоро-3-гидрокси-2-пиразинкарбоксамид) эффективен в отношении широкого диапазона штаммов вируса гриппа, включая A(H1N1) (пандемия 2009), A(H5N1) и A(H7N9), за счет того, что вирусная РзРп ошибочно принимает метаболит фавипировир-РТФ за пуриновый нуклеотид. Фавипировир прошел исследования III фазы в Японии и Пой - в США по лечению гриппа.
В дополнение к антивирусной активности против гриппа фавипиравир ингибирует репликацию аренавирусов (Юнин, Мачупо и Пичиндэ), флебовирусов (лихорадка Рифт-Валле, вирус флеботомной лихорадки и лихорадки Пунта Торо), хантавирусы (Мапорал, Добрава и Проспект Хилл); флавивирусы (желтая лихорадка и лихорадка Западного Нила); энтеровирусы (полно- и ринофирусы); альфавирус Западного энцефалита, парамиксовирус респираторного синцития и норовирус [8].
Использование фавипиравира в in vitro моделях гриппа
В исследованиях in vitro фавипиравир показал высокую противовирусную активность в отношении всех штаммов вируса гриппа, А, В и С. Основываясь на подсчете бляшкообразующих единиц (БОЕ) в культуре клеток MDCK, показатель эффективной концентрации (далее ЭК50) был в пределах от 0.014 to 0.55 μг/мл [17]. Лекарство не является цитотоксичным для клеточной культуры MDCK. В таблице 1 приведены измеренные активности фавипиравира против 53 штаммов вируса гриппа, включая сезонные штаммы A(H1N1), A(H3N2), и штаммы вируса гриппа типа В; A(H1N1)pdm09 пандемичный вирус, высокопатогенный птичий грипп A(H5N1), выделенный от человека, штаммы А(H1N1) и А(H1N2), выделенные из свиней, и A(H2N2), A(H4N2), A(H7N2) [78]. В это исследование вошло большое количество штаммов, резистентных к существующим противогриппозным препаратам, таким как амантадин, римантадин, оцетальмивир, занамивир [77].


50%-ная цитотоксическая концентрация фавипиравира (СС50) в MDCK клетках была более 2,000 μg/ml, демонстрируя высокоселективное ингибирование репликации вируса гриппа (селективный индекс более 3000). Единственным ограничением в применении фавипиравира является его эмбрио- и тератогенность [18], таким образом, не позволяя применять для беременных и кормящих женщин.
Использование фавипиравира в in vivo моделях гриппа
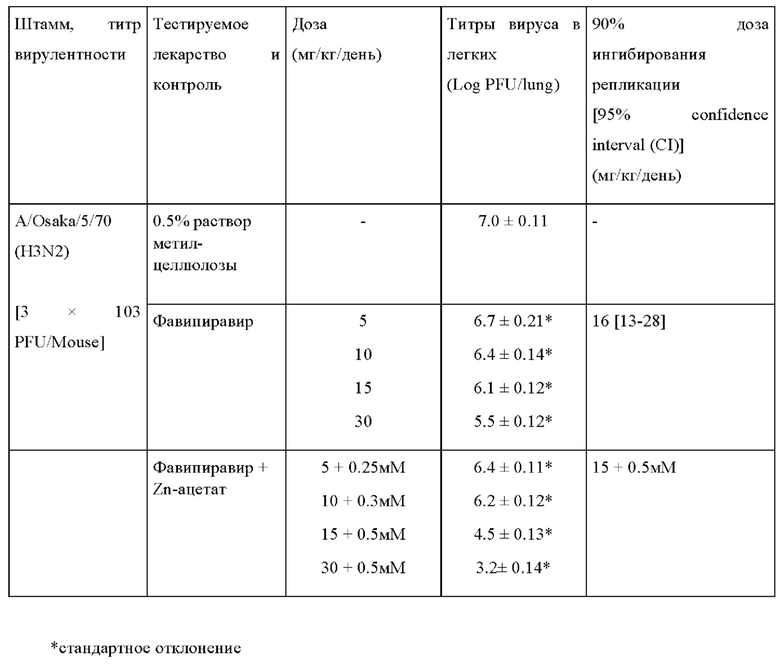
In vivo на мышиных моделях вирусной инфекции летальными дозами штаммов H3N2 (A/Victoria/3/75), H3N2 (A/Osaka/5/70) или H5N1 (A/Duck/MN/1525/81) фавипиравир применялся час спустя после заражения. Выживаемость мышей при дозах от 30 мг/кг/день 2 или 4 раза в день была значительна, тогда как все зараженные мыши контрольной группы погибли (Фиг. 4).
При применении от 60 до 300 мг/кг/день фавипиравир продемонстрировал свою эффективность в снижении вирусной нагрузки в легких мышей, инфицированных H1N1 (A/California/04/09), а также при отсроченном применении, вплоть до 96 часов после инфицирования [19, 20].
Фавипиравир показал значительный терапевтический эффект в сравнении с оцетальмивиром на мышах, которым была введена доза вируса в 100 раз большая, а само лечение было отсрочено на 96 часов пост-инфекции [79].
Эффективность фавипиравира для других семейств РНК-вирусов в исследованиях in vitro и in vivo.
Arenaviridae
Аренавирусы вызывают фатальные заболевания человека [21], от которых не существует противовирусных препаратов, кроме рибавирина, обладающего выраженным токсичным действием.
Использование фавипиравира в in vivo моделях аренавирусов
In vitro фавипиравир показал большую селективность, чем рибавирин. При измерении цитопатического эффекта в культуре клеток значения ЭК50 для препарата составили 0.79-0.94 μг/мл для вирусов Юнин, Пичинде и Такарибэ. Также вирусная нагрузка при применении фавипиравира значительно уменьшалась уже к третьему дню. При исследовании методом подсчета очагов гемолиза ЭК90s против высокопатогенного штамма Ромеро вируса Гуанарито JUNV (Romero) и вируса Мачупо составила 3.3-8.4 μг/мл (21-53 μМ) (Таблица 2) [80].

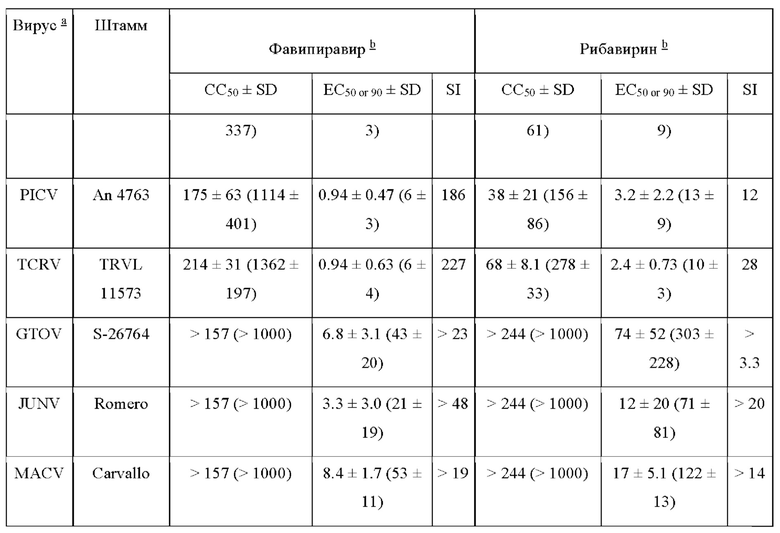
При пероральном применении фавипиравира на модельной линии хомяков, зараженных вирусом Пичинде, препарат предотвращал смертельный исход, уменьшал количество вирусных титров в крови и тканях, при дозировках 60 мг/кг/день двукратно предотвращал разрушение печени при применении в течение 7 дней, начиная с 4 часа пост-инфицирования [22] Вирусная нагрузка значительно уменьшалась также при начале лечения от 4, 5 и 6 дня пост-инфекции. При применении в дозировках от 100 мг/кг/день значительно увеличивалась выживаемость животных [23].
На модели заражения морских свинок фавипиравир демонстрировал свою эффективность уже даже после возникновения острых симптомов болезни [24].
При применении фавпиравира в дозе 300 мг/кг/день он показал значимые эффекты для выживаемости: 100% животных, участвующих в эксперименте, выжило, при дозе 150 мг/кг/день показатели снижались до 50 и 25%, животные сохраняли массу тела, у них падала температура до нормы и все показатели в целом были лучше, чем таковые для рибавирина в дозировке 50 мг/кг/день. Значительно дозозависимо понижалась концентрация прогностического маркера тяжести заболевания лихорадкой Ласса в крови -аспартат аминотрансферазы (AST) к десятому дню заболевания при лечении фавипиравиром. Виремия средней инфекционной дозы на 1 мл элюата снизилась в среднем до 2.1, 1.3, и 1.6 log10 CCID50/мл в группах, пролеченных высокой и средней дозой фавипиравира и рибавирином, соответственно.
Эффективность перорального применения фавипиравира была также показана на моделях, зараженных летальным Ласса вирусом морских свинок и мышей [25]. Терапевтический эффект наблюдался на 2ой день после заражения. Подкожное применение фавипиравира в дозировках 300 мг/кг/день 1 раз в день снижало температуру, предотвращало уменьшение массы тела и увеличивало выживаемость животных. Эффекты от применения фавипиравира во много раз превосходили терапевтические результаты от применения рибавирина в дозировке 50 мг/кг/день. Значимое улучшение выживаемости зараженных Ласса-вирусом морских свинок наблюдалось даже на 5,7 и 9 день постинфицирования [25].
Bunyaviridae
Вирусы семейства Буньявирида, включая вирус Ла Кроче (LACV), вирус лихорадки Рифт-Валле (RVFV), вирус конго-крымской геморрагической лихорадки (CCHFV), вирус острой лихорадки с синдромом тромбоцитопении (SFTSV) и хантавирус, вызывают тяжелые геморрагические лихорадки с сопутствующими легочными и почечными осложнениями.
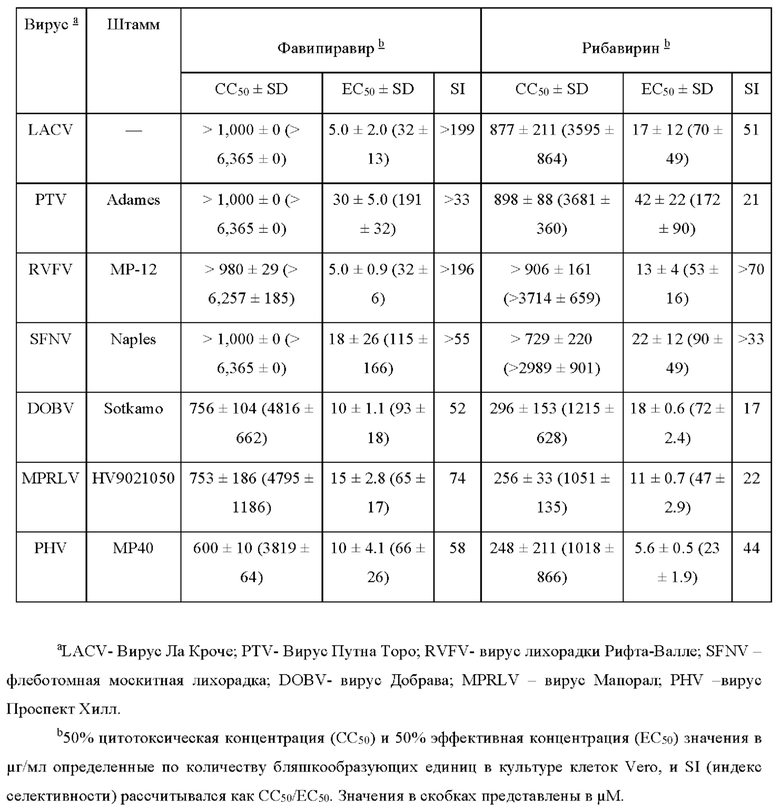
In vitro исследования показали превосходство применения фавипиравира над другими препаратами в направленности, эффективности и быстроте действия против целого ряда подобных вирусов (Таблица 3 [8, 26]). Значения ЭК50 в исследованиях бляшкообразующих единиц (БОЕ) в клеточной культуре начинались в пределах 0.9-30 μг/мл препарата для вирусов Ласса, Пунта Торо, Рифа-Валле, острой лихорадки (SFTSV), флеботомной лихорадки, и хантавирусов Добрава, Мапорал и Проспект Хилл [27].

Использование фавипиравира в in vitro и in vivo моделях буньявирусов
Вирус острой лихорадки с тромбоцитопенией (SFTSV) возник несколько лет назад как сезонное заболевание в Китае, Корее и Японии [28, 29]. Фавипиравир ингибировал репликацию SFTSV в культуре клеток с показателями ЭК50 в 0.71-1.3 μг/мл.
Терапевтический эффект фавипиравира был продемонстрирован на мышиных моделях с нокаутом рецепторов к интерферону-a (IFNAR-/-), которые не развивают немедленный иммунный ответ.
При пероральном применении фавипиравира в дозе от 300 мг/кг/день в течение 5 дней, начиная с 3 дня пост-заражения, все экспериментальные мыши выжили (Р<0.001), а начиная с 4 и 5 дня постинфекции значительно улучшились показатели выживания в группе. На базе данных исследований in vivo в Японии препарат вошел в клинические испытания и успешно их закончил к настоящему моменту [30]. Оно показало эффективность применения фавипиравира даже после начала заболевания и наступления клинических симптомов (Таблица 4) [8].

Flaviviridae
Фавипиравир блокирует репликацию вирусов семейства флавивирида, включая вирус желтой лихорадки (YFV) и вирус Западного Нила (WNV) [31,32], однако, в более высоких концентрациях, чем необходимы для блокировки активности вируса гриппа.
ЭК90 фавипиравира в отношении YFV составляет 51.8 μг/мл в in vitro исследованиях по определению выделения активных вирусных частиц на культуре клеток Vero.
Использование фавипиравира в in vivo моделях флавивирусов
Инфицированных YFV-хомяков пролечивали перорально фавипиравиром в дозах от 200 до 400 мг/кг/день в течение 8 дней, начав лечение за 4 часа до инфицирования. Эта терапия серьезно снизила уровень смертности животных при начале лечения [31].
Полное выздоровление было достигнуто при введении 400 мг/кг/день, начиная со 2ого дня пост-инфицирования.
In vitro и in vivo противовирусная эффективность фавипиравира в отношении вируса Западного Нила достигалась в концентрациях ЭК50 53 μг/мл в культуре клеток Vero.
Пероральное применение фавипиравира в дозах от 400 мг/кг/день 2 раза в день, спустя 4 часа после заражения спасло 90% мышей от смертельного исхода, а также значимо уменьшило экспрессию вирусных белков и вирусной РНК в тканях мозга. Такая же эффективность была показана и на линиях, зараженных WNV, хомяков в таких же дозах. Белки оболочки вируса WNV не детектировались в мозге пролеченных животных.
Вирус Zika (ZIKV) - недавно возникший вирус вида arbovirus семейства Flaviviridae, передается в основном укусами комара. Данные указывают на связь между инфекцией развивающегося плода и микроцефалией. Фавипиравир блокировал репликацию ZIKV в культуре клеток Vero при ЭК50 от 3.5-3.8 μг/мл [34].
Togaviridae
Фавипиравир проявляет противовирусную активность против Западного вируса лошадиного энцефалита (WEEV) в культуре клеток Vero, достигая ЭК90 при 49 μг/мл [35].
В мышах, инфицированных WEEV оральное применение фавипиравира значительно увеличивало выживаемость и продолжительность жизни зараженных животных при двукратном применении в дозе 400 мг/кг/день в течение 7 дней, начиная с 4ого часа пост-инфекции. Вирусные титры в тканях мозга были снижены на 4ой день пост-инфицирования, однако, полного излечения от инфекции при применении фавипиравира не произошло.
Фавипиравир показал противовирусную активность против вируса Чикунгунья (CHIKV) в культуре клеток Vero, достигая ЭК50 при 0.3-9.4 μг/мл. На мышах, инфицированных CHIKV, оральное применение фавипиравира улучшало показатели выживаемости при двукратных дозах от 300 мг/кг/день, начиная за сутки до или 4 часа после инфицирования [36].
Picornaviridae
Использование фавипиравира в in vitro моделях пикорнавирусов
Репликация энтеровируса везикулярного стоматита ингибировалась фавипировиром в исследованиях in vitro с ЭК50 в 14 μг/мл [37, 38].
Фавипиравир также блокировал репликацию вируса полиомиелита в культуре клеток Vero и риновируса в культуре клеток HeLa при ЭК50s в 4.8 и 23 μг/мл, и с индексом селективности в значениях 29 и >43, соответственно [39]. Фавипиравир ингибировал репликацию Энтеровируса при ЭК50 в 23 μг/мл [40].
Caliciviridae
Фавипиравир проявляет активность против мышиного норавируса с показателями ЭК50 от 39 μг/мл в исследованиях по подсчету вирусных бляшек в клеточной линии мышиных макрофагов RAW 264.7. ПЦР реального времени выявил блокирование синтеза РНК с помощью фавипиравира с ЭК50 от 19 μг/мл [41].
Использование фавипиравира в in vivo моделях рода норавирусов из семейства Калицивирусов
В мышиной модели персистирующей инфекции оральное применение фавпиравира в дозировке 600 мг/кг/день двукратно в течение 8 недель, спустя 4 недели пост-инфицирования, привело к значительному снижению вирусных титров в испражнениях мышей и количестве норовирус-положительных мышей. Научные данные также подтверждают, что фавипиравир-РТФ ингибирует РНК-полимеразную активность Норавирусов человека [42].
Filoviridae
Фавипиравир показал антивирусную активность против вируса Zaire Ebola (штамм Mayinga 1976) в культуре клеток Vero Е6 с ЭК50 в 10.5 цг/мл. В линии мышей, зараженных штаммом Mayinga 1976 с отсутствием рецептора к интерферону-альфа (IFNAR-/- C57BL/6), оральное применение фавипиравира позволило избежать летального исхода и снижало вирусные титры в крови при двукратном применении от 300 мг/кг/день в течение 8 дней с 6 дня пост-инфекции, тогда как в плацебо-группе все мыши умерли [43]. Сходным образом, в линии А129, нокаутной по рецептору интерферона IFNAR-/-, зараженной шитаммом Эбола Е718, лечение фавипиравиром орально полностью спасло всех зараженных мышей от смерти при двукратном применении лекарства в дозе от 300 мг/кг/день в течение 14 дней, начиная с 1ого часа после заражения [44].
При вспышке Эбола в западной Африке в 2014 году Французский институт Здоровья и Медицинских Исследований (INSERM) и правительство Гвинеи провели клиническое исследование фавипиравира на больных [45]. Фавипиравир хорошо переносился пациентами и снижал количество смертельных исходов у пациентов с низкими вирусными титрами [46]. Группа китайских исследователей также отметила увеличение выживаемости пациентов с Эбола при применении фавипиравира в Сьерра Леоне [47].
Rhabdoviridae
Активность фавипиравира против вируса бешенства (RABV) была выявлена на клеточной линии мышиной нейробластомы Neuro-2a с ЭК50s при 5.1-7.0 μг/мл [48]. Фавипиравир значительно снизил показатели заболеваемости и смертности у мышей, зараженных RABV, при пероральном применении в дозах от 300 мг/кг/день двукратно в течение 7 дней, начиная с 1ого часа пост-инфекции.
Все описанные выше обобщенные данные по неоспоримой эффективности фавипиравира против очень широкого спектра РНК-вирусов показывают, что данное лекарство прекрасно «закрывает» до сих пор эффективно неохваченную нишу неизлечимых, острых и смертельных вирусных недугов, включая большой спектр тропических лихорадок. До исследований на фавипиравире, против вышеозначенных РНК-инфекций применялись рибавирин и/или альфа-интерферон, однако первый обладает значимо меньшей противовирусной активностью и эффективностью против РНК-вирусов, и оба препарата при длительном применении приводят к дебилитирующим побочным эффектам. Фавипиравир, в отличие от других препаратов, блокирующих РНК-полимеразу, хорошо переносится в клинических исследованиях и заслуживает широкого применения за счет изученности и универсальности механизма.
Ясно, что опасные одноцепочечные «сенс»-РНК вирусы требуют разработки новых подходов и применения новых улучшенных схем с мультифакторным действием на заболевания. И даже хорошо изученные вирусы гриппа, вызывающие ежегодные пандемии с осложнениями, а также вездесущие коронавирусы, приводящие к разнообразным острым респираторным синдромам и вирусным ринитам, фарингитам, ларингитам и т.п., бывают возбудителями гораздо более опасных летальным исходом состояний.
Одним из наиболее эффективных подходов к разработке высокоспецифичных противовирусных препаратов остается направленное таргетирование вирус-специфических функциональных молекул, таких как структурные и неструктурные белки, вирусные протеазы и отсутствующая в животных клетках РНК-зависимая-РНК-полимераза. Последняя поэтому представляет особый интерес в качестве мишени ингибиторов вирусной репликации.
Фавипиравир, опираясь на универсальный и доказанный механизм его действия в отношении РзРп, также может облегчить течение вирусных заболеваний, при своевременном начале приема, а также значимо снизить вирусную нагрузку при течении заболеваний с осложнениями.
Однако, учитывая многокомпонентность и многосложность клинической картины вирусных заболеваний, в частности, демонстрируемую сегодня COVID-19, способность вирусов к мутации, а также перестройки физиологических механизмов борьбы с вирусом внутри организма, существует необходимость в создании эффективных противовирусных комбинаций фавипиравира с другими препаратами, опираясь, как на клеточный цикл вируса в организме, так и на физиологический ответ для эффективной борьбы.
Ранее было выдвинуто предположение, что клинически одобренные ингибиторы ретровирусных протеаз: ритонавир и лопинавир, активны против вирусов SARS и MERS, гипотетически, воздействуя на химотрипсин-подобные протеазы обоих коронавирусов. В клинических исследованиях было достигнуто улучшение клинических показателей пациентов с SARS, пролеченных комбинацией препаратов [2], что послужило началом исследований комбинации лопинавира с ритонавиром (ChiCTR2000029539) для пациентов с 2019-nCoV. В рамках текущей пандемии предпринималось несколько попыток воздействовать на репликацию вируса путем репозиционирования анти-ВИЧ протеаз: ритонавира, усиленного действием лопинавира, однако, очевидно, что эти попытки не могли дать значимых клинических результатов, поскольку сайт узнавания и разрезания полипептидной цепи ВИЧ-протеазами не совпадает с папаин- и химотрипсин-подобными протеазами, а соответственно, структура их ингибиторов не совпадает, и ритонавир не проявил ингибирующего эффекта в отношении других протеаз.
Cai et al провели открытое контролируемое исследование по сравнению эффектов фавипиравира (1600 мг 2 раза в день в 1ый день и далее 600 мг 2 раза в день со 2ого по 14 день) против сочетания лопинавира с ритонавира (400 мг/100 мг двукратно) в сочетании с интерфероном- a1b в дозировке 60 мг 2 раза день путем ингалирования для терапии COVID-19 [66]. Предварительные результаты показывают значительные клинические различия между применением фавипиравира (35 пациентов) и комбинации лопинавир/ритонавир (45 пациентов) с медианой очищения вирусной нагрузки по времени 4 дня против 11 дней, оцениваемой по улучшению результатов КТ грудной клетки (91.43% vs. 62.22%). Слабые клинические результаты данного исследования в отношении применения антиретровирусных протеаз четко указывают на необходимость использования строго-специфичных ингибиторов химотрипсин-подобных и папаин-подобных протеаз.
Лопинавир, ингибирующий химотрипсин-подобные протеазы типа 3CLpro, в теории, мог бы выступить успешным терапевтическим агентом, однако, показано, что протеазы коронавируса SARS-CoV-2 содержат С2-симметричный сайт взаимодействия, куда лопинавир зайти не может. Компьютерное моделирование in silico по поиску специфических структурных блокаторов химотрипсин-подобных протеаз на основе имеющихся одобренных лекарств показало, что среди ретровирусных протеаз дарунавир может взаимодействовать с карманом 3CLpro, специфичным для протеаз SARS-CoV-2 [67].
Однако введение большого количества дополнительных активных компонентов не может быть решением проблемы, поскольку нередко оно бывает сопряжено с возникновением у пациентов осложнений, обусловленных побочными эффектами каждого из компонентов комбинации. Кроме того, в этом случае необходимо проверить безопасность и эффективность взаимного действия каждого из активных компонентов, что для многокомпонентных композиций объективно затруднено.
Задачей настоящего изобретения является создание композиции, проявляющей профилактическую и/или терапевтическую эффективность в отношении широкого спектра заболеваний, вызванных воздействием вирусов, геном которых закодирован одноцепочечной нитью РНК и которые используют вирусную РНК-зависимую-РНК -полимеразу для своей репликации, включающей в своем составе эффективное количество фавипиравира в комбинации с эффективным количеством иного средства,. Причем новая композиция должна действовать эффективнее чем фавипиравир отдельно.
Опираясь на приведенные выше данные, авторами предлагается применение комбинации фавипиравира с, по меньшей мере, одним соединением цинка для терапевтических подходов к лечению вирусов, использующих РзРп для репликации и химотрипсин-подобные протеазы для трансляции, подтверждая их эффективность в исследованиях in vitro и in vivo.
Было обнаружено, что совместное применение, по меньшей мере, одного соединения цинка и фавипиравира в эффективных количествах приводит к значительному усилению взаимного эффекта в отношении вирусов, геном которых закодирован одноцепочечной нитью РНК и которые используют вирусную РНК-зависимую-РНК-полимеразу для своей репликации, при отсутствии дополнительных побочных эффектов. Терапевтический эффект комбинации может быть обусловлен разнонаправленным действием компонентов комбинации на вирус. Такое разнонаправленное действие позволяет сохранить необходимо высокую активность даже в случаях мутации вируса-мишени, что особенно важно с вирусами, относительно недавно преодолевшими межвидовой барьер. Вывод об усилении противовирусного терапевтического эффекта введения в композицию в эффективном количестве цинка и фавипиравира сделан на основе описанных примеров и наблюдений. Точное объяснение механизмов такого неожиданно эффективного совместного действия требует дальнейших углубленных исследований.
Вышеуказанные свойства могут быть расценены как технический результат, проявляющийся при использовании настоящего изобретения - фармацевтической композиции для облегчения клинических симптомов, течения и излечения заболевания, вызванного воздействием вируса, геном которого закодирован одноцепочечной нитью РНК и который использует вирусную РНК-зависимую-РНК -полимеразу для своей репликации, содержащей эффективное количество фавипиравира и, по меньшей мере, одного соединения цинка.
Ранее было показано, что сочетанное применение Zn2+ и ионофоров Zn2+, таких, как пиритион (ПТ) в концентрациях (2 μМ Zn2+ и 2 μМ ПТ) эффективно снижает скорость репликации вируса, а также влияет на процессирование вирусных полипротеиновых цепей для РНК-вирусов, несущих смысловую РНК-цепь в геноме и использующих РзРп [50].
Достоверно известно, что цинк играет одну из ключевых ролей в гомеостазе иммунной системы [83]. Дефицит цинка ассоциирован с ограничением и ухудшением эффективности клеточного иммунитета, сопровождаемым дисбалансом между Th1 и Th2 [84, 85]. Дефицит цинка приводит также к пониженному синтезу интерлейкинов и гамма-интерферона [86].
На уровне клеточных механизмов цинк необходим для поддержания каталитической активности многих клеточных ферментов, а также связывается с белками, содержащими zinc-finger последовательности и играющими важные роли в поддержании метаболического и иммунного гомеостаза [87]. Поэтому неудивительно, что соли цинка обладают способностью блокировать пролиферацию широкого спектра вирусов, включая ВИЧ, вирус гастроэнтерита, вирус простого герпеса, вакциния-вирус, вирус острого респираторного синдрома (SARS-CoV), лошадиного энцефалита, риновирус, вирус респираторного синцития [88-94]. Антивирусный эффект достигается за счет прямого ингибирования вирусной РНК-зависимой-РНК-полимеразы и непрямого блокирования каталитического процессирования полипептидных вирусных цепей [94].
Прямое ингибирование ионами Zn2+ РзРп корона и артери-вирусов в in vitro модели.
Добавление ZnOAc2 к активной РНК-полимеразе вирусов лошадиного энцефалита (EAV) и SARS-CoV приводило к сильному доза-зависимому нигибированию энзиматической активности фермента обоих вирусов, аналогичному тому, что наблюдался в исследованиях по скорости синтеза РНК для данных вирусов. В сравнении с активностью других дивалентных атомов, в частности, Со2+ и Са2+, обычно связывающих полипептидные цепи по атомам кислорода, а не по серосодержащим радикалам, Zn2+ являлся наиболее эффективным ингибитором SARS-CoV nsp12-белка РзРп. В концентрации от 2mM до 6 mM Zn2+ достигался эффект полного блокирования синтеза цепей РНК, а также производства sgRNA in vitro [92].
Ингибирование ионами Zn2+ РзРп коронавируса и вируса респираторного синцития (RSV) в in vitro модели.
Для приготовления растворов цинка использовался USP-grade ацетат цинка (AMEND Chemical, Irvington, N.J.), или лактат или сульфат цинка (Sigma Chemical, St. Louis, Mo.). Стоковые растворы использовались в концентрации 250 mM солей в деионизированной воде, стерилизованные путем фильтрования сквозь 0.22-μm-поровый шприцевый фильтр.
(а) Выход вирусных частиц. При добавлении цинка в процессе абсорбции вируса клетками, проникновения и выхода вирусных частиц, наблюдалась существенное снижение выхода вириомов, даже в присутствии самых низких концентраций 10 μМ раствора, в сравнении с «пустым» - бессолевым контролем и контролем солями других дивалентных металлов - Mg2+ и Со2+. Среднее значение вирусных титров составило 5.3 log10PFU/мл (стандартная средняя ошибка [SEM]=0.7 log10 PFU/мл) в сравнении с 2.8 log10PFU/мл (SEM=0.7 log10 PFU/мл) для любой соли цинка (Р<0.01). Снижение вирусных титров в 10.000 и более раз было достигнуто при концентрациях соли цинка 100 μМ. Напротив, ацетат кальция, сульфат магния и сульфат марганца не показали подобных ингибирующих свойств даже при высочайшей концентрации в 10 mM.
(b) Полное ингибирование формирования вирусных бляшек RSV наблюдалось при концентрациях солей цинка в 10 mM, тогда как 88% ингибирование уже достигалось при 1 mM концентрации (SEM, 4%; Р<0.001) [94].
(c) Эффект цинка на вирусную пенетрацию.
Значимый блокирующий эффект солей цинка на вход вируса в клетку был обнаружен при концентрациях в 100 μM, 1 mM, и 10 mM по сравнению с эффектом от контрольных солей и контрольной культуры, где средний вирусный титр составил 6.6 log10 PFU/мл (SEM, 0.2 log10 PFU/мл).
Соединения цинка также блокируют другие РНК-вирусы, в частности ВИЧ, что обусловлено ингибированием РНК транскрипции на матрице ДНК [95].
Ингибирование ионами Zn2+ РзРп РНК вируса везикулярно-инвазивного ящура.
Цинк в концентрациях 0.5 мМ добавлялся к культуре зараженных ящуром клеток на протяжении некоторого времени с интервалами. Он блокировал продуцирование новых вирусных частиц практически моментально при условии добавления его к клеткам не позже, чем через 90 минут постинфекции и значительно уменьшал количество и выход вирусных частиц при добавлении через 120 и 150 минут пост-инфекции [96]. 0.5 mM концентрации Zn2+ показанные достаточными для полного ингибирования репликации везикулярно-пузырькового ящура, сравнимы с результатами по блокированию репликации человеческого риновируса, энцефаломиокардита и полиомиелита [97]. Также как и для данных вирусов, при повышении внутриклеточной концентрации ионов цинка в клутках повышалось количество непроцессированных полипептидных цепей предшественников вирусных белков. Основные структурные белки капсида ящура, весом 30.000 Дальтон уже не производились в нужных количествах при концентрации цинка 0.5 mM, очевидно, из-за влияния цинка на протеолитический процессинг белков-предшественников [96].
Однако, при поступлении цинка в организм может возникнуть проблема его избыточного накопления локально в определенном органе, а также проблема недостатка в той внуткриклеточной системе, где он нужен.
В связи с этим рекомендовано его употребление вместе с «ионофорами» улучшающими его трансмембранную доставку, такими, как дитиокарбаматы в эквимолярных концентрациях и цинк - диэтил-бис(N4-метилтиосемикарбазон) - Zn-DTSM. Zn-DTSM значительно увеличивал внутриклеточное содержание цинка (р<0.0001; +287% в сравнении с контролем), и при этом значимо не изменял уровень содержания других металлов (Fe и Cu).
Раскрытые выше эффекты возможно объяснить тем, что ионы цинка вовлечены в большое количество различных клеточных процессов и совершенно необходимы для правильного фолдинга клеточных ферментов и факторов транскрипции. Отсюда возможно предположить, что Zn2+ также может выступать ко-фактором для вирусных белков. Внутриклеточная концентрация ионов Zn2+ в норме поддерживается в низких концентрациях с помощью металлотеонеинов, вероятнее всего потому, что Zn2+ может выступать вторичным мессенджером в качестве триггера апоптоза и вещества снижающего повышенный уровень белкового синтеза [51, 52]. Во многих исследованиях в культуре клеток было показано, что повышенные концентрации ионов Zn2+, совместно с другими компонентами, стимулирующими повышение его внутриклеточной концентрации, в частности, с хинокитолом, пирролидин дитиокарбаматом и пиритионом, ингибируют репликацию РНК-вирусов, включая вирус гриппа [53], вирус респираторного синцития [54] и несколько видов пикорнавирусов [55-58]. Несмотря на то, что эти более ранние исследования не отражают конкретного механизма действия Zn2+, тем не менее, они доказательно описывают, что ионы цинка участвуют в вирусной репликации [50].
Для корона- и артеривирусной инфекции характерной особенностью цикла репликации является транскрипция 5'- и 3'- концевых небольших фрагментов субгеномной информационной РНК (sg mRNA), с которых далее происходит трансляция структурных и неструктурных вспомогательных вирусных белков [59, 60]. В исследованиях на культуре клеток in vitro, также, как и в случае для семейства пикорнавирусов, было показано, что ионы цинка ингибируют определенные сайты протеолитического процессинга полипротеинов коронавируснго репликазного комплекса в инфицированных клетках и бесклеточных системах [61, 62]. В частности, пиритион ионофор цинка в комбинации с ионами цинка проявлял себя мощным ингибитором репликации SARS-коронавируса (SARS-CoV) и вируса лошадиного артерита (EAV). В дополнение, данный эффект наблюдался в исследованиях на рекомбинантной РзРп SARS-CoV и EAV [63].
При исследовании на коронавирусах, в частности на близком по структуре к SARS-CoV-2 вирусе SARS-CoV было показано, что повышение концентрации Zn2+ до микромолярных концентраций в культуре зараженных клеток резко снижает эффективность связывания РзРп с матрицей геномной и информационной вирусной РНК [50] (Фиг. 5).
Большинство семейств РНК-вирусов экспрессируют свой геном в виде длинных полипептидных цепей - предшественников функциональных белков, которые претерпевают процессинг вирусными и клеточными протеазами. Такая стратегия совмещенных транскрипции и трансляции позволяет одновременно и с минимальными энергетическими и биохимическими затратами активизировать большое количество разнофункциональных белков из одного предшественника. Вирусные протеазы, хотя и разные по структуре, высоко специфичны в отношении своих полипептидных субстратов и направленного узнавания отдельных сайтов «разрезания». Каскад процессирования - жестко регулируемый процесс, который в некоторых случаях подразумевает участие ко-факторных белков - модификаторов действия отдельных протеиназ [64]. Многие семейства одноцепочечных «сенс»-РНК вирусов имеют сходные химотрипсин-подобные и папин-подобные протеазы, в частности, Picorna-, Flavi- и Coronaviridae [65].
Мы предположили, что ингибирование данных, а также других видов протеаз, специфичных для РНК(+)-вирусов является успешной антивирусной терапевтической стратегией, параллельно с описанной выше блокировкой вирусной репликации. Опираясь на исследования ингибирования сайтов полипептидного процессинга коронавирусов с помощью цинка, а также на необходимость производить полипептидный процессинг для жизнедеятельности вируса, в терапии РНК-вирусов должны эффективно срабатывать ингибиторы папаин-подобных и химотрипсин-подобных протеаз, в частности, закодированных в геноме SARS-CoV-2. А их сочетанное применение с фавипиравиром приведет к еще более эффективному и быстрому ингибированию вируса, причем не только коронавируса, но и большинства обсуждаемых выше РНК(+)-вирусов.
Таким образом установлено, что добавление к фавипиравиру по меньшей мере одного соединения цинка в заявленных пределах усиливает требуемую фармацевтическую активность, при этом не вызывает побочных эффектов при проведении противовирусной терапии.
Изобретение также относится к способу лечения, который предусматривает введение пациенту, нуждающемся в таком противовирусном лечении, например, пероральное или инъекционное эффективного количества предлагаемой композиции.
Изображения, поясняющие сущность изобретения:
Фиг. 1: (а) геномная структура 2019-nCoV с указанием протеаз и РНК-зависимой-РНК-полимеразы, (b) нуклеозидные блокаторы РНК-зависимой-РНК-полимеразы.
Фиг. 2: Эффект действия фавипиравира-РТФ, фавипиравира и фавипиравира-МТФ на активность РНК-зависимой-РНК-полимеразы (РзРп) вируса гриппа [7]. Активность РзРп измерялась по включению 32P-GTP в нативную нить РНК. Р<0.01; результаты значительно отличны от контроля, согласно критерию Тьюки [8].
Фиг. 3: Схематичное изображение механизма действия фавипировира и его превращений при попадании в клетку [8].
Фиг. 4: Терапевтический эффект фавипиравира на модели мышиной инфекции гриппом [8]. 10 самок инфицировали 100% летальной дозой вируса гриппа одного из штаммов: A/Victoria/3/75 (H3N2), A/Osaka/5/70 (H3N2) или A/Duck/MN/1525/81 (H5N1) и следили за выживаемостью в течение 21 дня пост-инфекции. Фавипиравир применялся перорально, спустя 1 час после заражения 2 раза в день в группе Osaka и Duck или 4 раза в группе Victoria в течение 5 дней. **, р<0.001 в сравнении с контролем (Chi-square test). ++, р<0.001 в сравнении с контролем (метод Каплана-Майера).
Фиг. 5: Эффект присутствия растущих концентраций ионов цинка на связывание РзРп вируса SARS-CoV nsp12 на связывание с субстратом.
Осуществление изобретения
Фармацевтическая композиция согласно изобретению содержит эффективное количество фавипиравира.
Эффективное количество фавипиравира составляет от 50 мг до 800 мг. Предпочтительно эффективное количество фавипиравира составляет от 150 мг до 600 мг. Предпочтительно эффективное количество фавипиравира составляет от 200 мг до 400 мг. Предпочтительно эффективное количество фавипиравира составляет от 100 мг до 800 мг.
Фармацевтическая композиция согласно изобретению содержит эффективное количество, по меньшей мере, одно соединение цинка.
При этом соединение цинка может быть выбрано из ацетата цинка, лактата цинка, аспартата цинка, сульфата цинка, ундецилената цинка, но не ограничиваться ими.
При этом соединение цинка может быть представлено, как солью, так и йонофорной формой, например, пиритиона цинка, дитиокарбаматы, цинк-диэтил-бис(N4 метилтиосемикарбазон) цинк-DTSM, хинокитол, пирролидин дитиокарбамат цинка, но не ограничивается ими.
Эффективное количество солей цинка в случае их использования составляет от 15 до 250 мг. Максимальная концентрация составляет до 900 мг. Ионофоры в случае их применения используются в эквимолярном количестве. Предпочтительно, в композиции можно использовать указанных солей цинка в количестве от 15 до 250 мг, а также предпочтительно от 100 до 150 мг.
Массовое соотношение компонентов фавипиравир : цинк / цинк-ионофор (в случае его использования) может составлять 1:1 - 10:1, более предпочтительно 1:1; 1,5:1; 2:1; 2,5:1.
Фармацевтическая композиция согласно изобретению, может дополнительно включать фармацевтически приемлемые вещества.
Фармацевтическая композиция согласно изобретению, может содержать один или более эксципиентов, выбранных из растворителей, диспергаторов, наполнителей, поверхностно-активных веществ, солюбилизаторов, эмульгаторов, стабилизаторов, лубрикантов, консервантов, антиоксидантов, буферных соединений.
Фармацевтическая композиция согласно изобретению, может содержать один или более растворителей, выбранных из воды, например, воды для инъекций, дистиллированной воды, апирогенной воды, буферного раствора, но не ограничивается ими.
Фармацевтическая композиция согласно изобретению может содержать один или более диспергаторов, выбранных из крахмала, агара, альгиновой кислоты или ее солей, но не ограничивается ими.
Фармацевтическая композиция согласно изобретению, может содержать один или более поверхностно-активных веществ, выбранных из полиэтиленгликоля, глицерина, глицерида органической кислоты, полисорбата, Твина-80 но не ограничивается ими.
Фармацевтическая композиция согласно изобретению, может содержать один или более солюбилизаторов, выбранных из Твина-80, ПЭГ, бензола, глицерина, сорбита и его производных, ксилита, альгиновой кислоты, но не ограничивается ими.
Фармацевтическая композиция согласно изобретению, может содержать один или более стабилизаторов, выбранных из лимонной кислоты, аскорбиновой кислоты, натрия гидрокарбоната, натрия сульфита, унитиол, токоферола, но не ограничивается ими.
Фармацевтическая композиция согласно изобретению, может содержать один или более консервантов, выбранных из сорбиновой кислоты, бензойной кислоты, метабисульфита натрия, бензилового спирта, но не ограничивается ими.
Фармацевтическая композиция согласно изобретению, может содержать один или более лубрикантов, выбранных из стеариновой кислоты и ее производных, но не ограничивается ими.
Фармацевтическая композиция согласно изобретению, может содержать один или более наполнителей, выбранных из сахарозы, карбоната магния, диоксида кремния, крахмала, маннита, поливинилпирролидона, повидона, моногидрата лактозы, различных производных целлюлозы, например, микрокристаллической целлюлозы, кроскамеллозы, но не ограничивается ими.
Фармацевтические композиции могут быть получены любыми способами, известными в фармацевтической области, например, смешиванием активных ингредиентов с фармацевтически приемлемыми веществами в стерильных условиях.
Фармацевтические композиции могут быть выполнены в форме таблетки или капсулы, в лиофилизированной форме или в жидкой лекарственной форме в виде инъекционного раствора.
Таблетка может быть покрыта пленочной оболочкой.
Изготовление композиций согласно настоящему изобретению можно производить различными способами, включающими в себя экструзию, распылительную сушку и выпаривание из растворителя, но не ограниченными ими.
Фармацевтические композиции по настоящему изобретению обладают превосходной противовирусной активностью и направлены на профилактику и/или терапию заболеваний, вызванных воздействием вирусов, геном которых закодирован одноцепочечной нитью РНК и которые используют вирусную РНК-зависимую-РНК-полимеразу для своей репликации.
Фармацевтические композиции по настоящему изобретению направлены на значимое облегчение клинических симптомов, течения и излечения широкого спектра заболеваний, вызванных воздействием вирусов, геном которых закодирован одноцепочечной смысловой (+)-нитью, а также антисмысловой (-)-нитью РНК и которые используют вирусную РНК-зависимую-РНК-полимеразу для своей репликации.
Вирусы, в отношении которых проявляют активность композиции по настоящему изобретению, представляют собой вирусы гриппа штаммов как высокой, так инизкой вирулентности, вирусы семейств Coronaviridae, Picornaviridae, Arenaviridae, Flaviviridae, Bunyaviridae, Togaviridae, Orthomyxoviridae.
Назначение композиции, а именно, облегчение симптомов, вплоть до выздоровления, у пациента, нуждающемся в лечении может быть связано с инфицированием пациента вирусом, который представляет собой калицивирус или норавирус.
Фармацевтические композиции по настоящему изобретению применяются в лечении или профилактике курсом.
Длительность курса и суточная доза определяется задачей, сложностью заболевания и индивидуальными особенностями пациента и может быть скорректирована в ходе терапии и/или профилактики.
Длительность профилактического курса может составлять от одной до четырех недель и включать прием от одной до тридцати лекарственных форм в сутки.
Длительность терапевтического курса может составлять от одной до четырех недель и включать прием от одной до тридцати лекарственных форм в сутки.
Курс для терапии заболеваний может включать прием от 3-х доз композиции в день в первые 2 дня, далее по 2 дозы.
Курс для терапии заболеваний может включать прием от 2 доз в сутки.
Предпочтительно, одна доза включает фавипиравир от 200 до 6000 мг, соединение Zn2+ в количестве от 10 до 100 мг (в пересчете на цинк).
Предпочтительно, одна доза включает фавипиравир от 200 до 1600 мг, соединение Zn2+ от 10 до до 60 мг (в пересчете на цинк).
Примеры
Настоящее изобретение и способы его осуществления будет понятно специалисту из следующих примеров.
Данные примеры, относящиеся к композициям фавипиравира - по меньшей мере одного соединения цинка, не являются ограничивающими изобретение и относятся лишь его частным вариантам реализации.
Пример 1. Изготовление вариантов готовых лекарственных препаратов.
1. Таблетка, покрытая пленочной оболочкой
Состав:
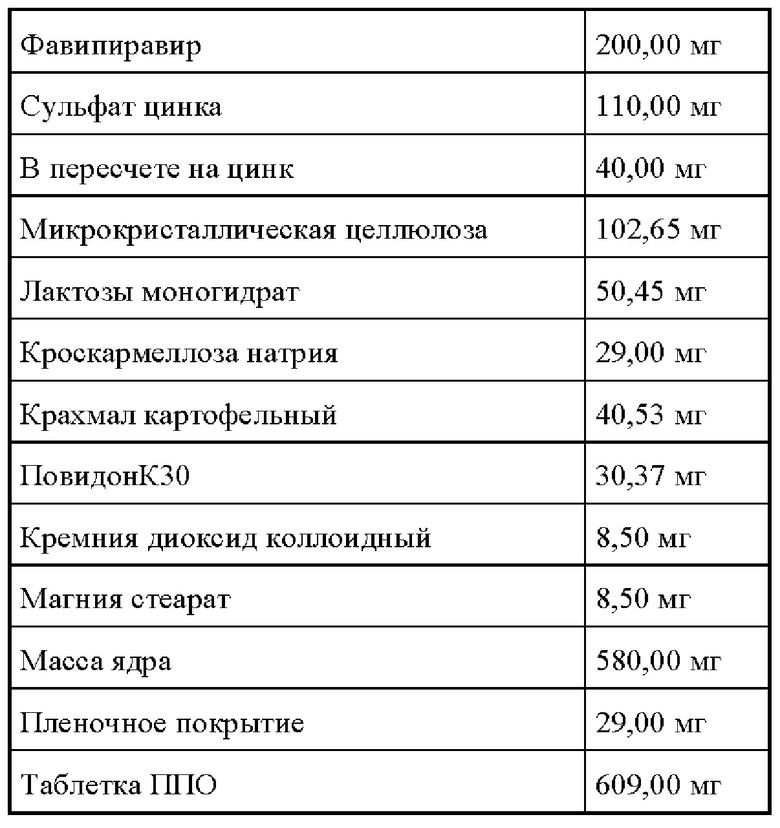
В смеситель с высоким усилием сдвига загружают лактозы моногидрат, фавипиравир и кроскармеллозу натрия, перемешивают в течение 5 минут на скорости 100 об/ мин на нижней мешалке. Добавляют с равномерной скоростью, при постоянном перемешивании мешалки 150 об/мин и чоппера 500 об/ мин, увлажнитель, 10% раствор повидона КЗО, увлажняют до полного внесения увлажнителя. Увлажненную массу гранулируют при перемешивании мешалки 200 об/ мин и чоппера 650 об/ мин.
Полученные таким путем гранулы пропускают через сетку с размером ячеек 1 мм и сушат в установке псевдоожиженного слоя при температуре входящего воздуха 50 - 60°С, и температуры продукта 30 - 45°С до остаточной влажности не более 2%.
В бин гравиметрического смесителя загружают полученные гранулы фавипиравира, микрокристаллическую целлюлозу, крахмал картофельный, кремния диоксид коллоидного смешенного с сульфатом цинка, перемешивают в течении 30 минут (по 15 минут на каждую сторону).
Загружают магния стеарат и опудривают 2 минуты (по 1 минуте на каждую сторону).
Полученную массу таблетируют на роторном таблетпрессе с усилием 120 -150 Н, и средней массой ядра 5 80 мг.
Таблетки покрывают готовой пленочной оболочкой в коутере с перфорированным барабаном до прироста массы на 5%.
2. Капсула твердая желатиновая
Состав:
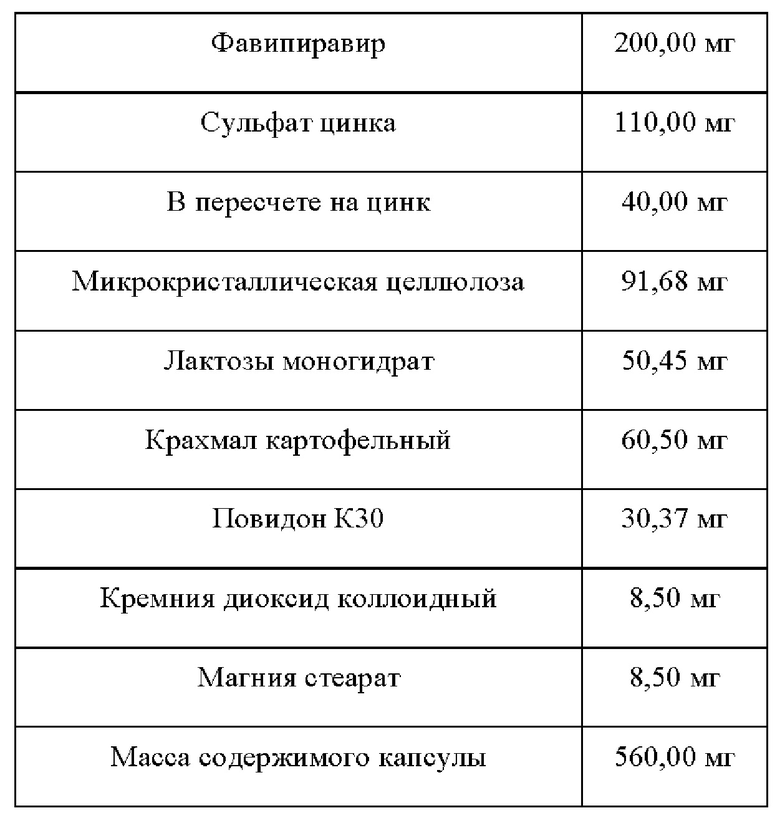
В смеситель с высоким усилием сдвига загружают лактозы моногидрат, фавипиравир половину от состава картофельного крахмала, перемешивают в течении 5 минут на скорости 100 об/ мин на нижней мешалке. Добавляют с равномерной скоростью, при постоянном перемешивании мешалки 150 об/мин и чоппера 500 об/ мин, увлажнитель, 10% раствор повидона К30, увлажняют до полного внесения увлажнителя. Увлажненную массу гранулируют при перемешивании мешалки 200 об/ мин и чоппера 650 об/ мин.
Полученные таким путем гранулы пропускают через сетку с размером ячеек 1 мм и сушат в установке псевдоожиженного слоя при температуре входящего воздуха 50 - 60°С, и температуры продукта 30 - 45°С до остаточной влажности не более 2%.
В бин гравиметрического смесителя загружают полученные гранулы фавипиравира, микрокристаллическую целлюлозу, оставшеюся часть крахмала картофельного, кремния диоксид коллоидного смешенного с сульфатом цинка, перемешивают в течении 30 минут (по 15 минут на каждую сторону).
3. Инъекционный раствор
Состав композиции на 100 мл водного раствора:

Заявленный раствор получали следующим способом: в снабженный примешивающим устройством и термостатирующей рубашкой стеклянный или нержавстальной аппарат для приготовления растворов помещают отмеренные количества фавипирафира, пропиленгликоля, сорбитола и повидона К15 и нагревают при непрерывном перемешивании до температуры 40-45°С. Полученную смесь перемешивают при данной температуре до полного растворения компонентов. Далее к раствору прибавляют заранее отмеренную и нагретую до температуры 40°С воду для инъекций и перемешивают до получения однородного раствора.
Затем раствор охлаждают до комнатной температуры, прибавляют расчетное количество сульфата цинка и перемешивают до его полного растворения. Полученный раствор фильтруют через мембранные пластины типа «Миллипор» с диаметром пор 0,22 мкм и разливают в ампулы или флаконы.
Представленные примеры демонстрируют варианты готовых лекарственных форм по изобретению и возможные способы их изготовления.
Пример 2. Измерение ингибирования репликации вируса гриппа в легких инфицированных мышей фавипиравиром в комбинации с солями цинка.
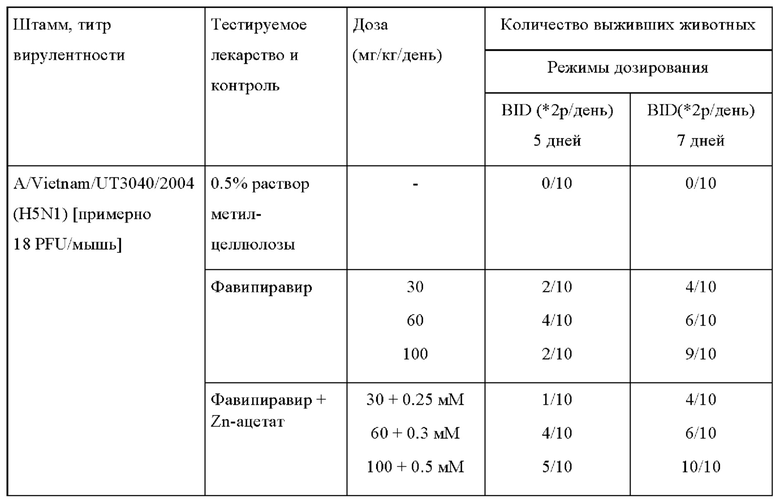
Пример 3. Терапевтический эффект фавипиравира и фавипиравира в комбинации с солями цинка на модели мышей инфицированных высокопатогенным вирусом птичьего гриппа.
Пример 4. Терапевтический эффект на инфицированных гриппом иммунодефицитных мышах


Проведенные примеры позволяют сделать вывод о наличии у заявленных комбинаций повышенной терапевтической активности в отношении различных видов возбудителей по сравнению с отдельными компонентами.
Список используемой литературы
[1] https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200416-sitrep-87-covid-19.pdf?sfvrsn=9523115a_2
[2] Zumla, A., Chan, J.F., Azhar, E.I., Hui, D.S. & Yuen, K.Y. Coronaviruses - drug discovery and therapeutic options. Nat. Rev. Drug Discov. 15, 327-347 (2016).
[3] S. van Boheemen, M. de Graaf, C. Lauber, T.M. Bestebroer, V.S. Raj, A.M. Zaki, et al.Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio, 3 (6) (2012) e00473-12.
[4] Morse, J. S., Lalonde, Т., Shiqing, X. & Liu, W. R. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019 - nCoV. ChemBioChem https://doi.org/10.1002/cbic.202000047 (2020).
[5] De Clercq, E. New nucleoside analogues for the treatment of hemorrhagic fever virus infections. Chem.AsianJ. 14, 3962-3968 (2019).
[6] Wang, M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. https://doi.org/10.1038/s41422-020-0282-0 (2020).
[7] Furuta Y., Takahashi K., Kuno-Maekawa M., Sangawa H., Uehara S., Kozaki K., Nomura N., Egawa H., Shiraki K. (2005) Mechanism of action of T-705 against influenza virus. Antimicrob. Agents Chemother. 49, 981-986.
[8] Yousuke Furuta, Brian B.Gowen, KazumiTakahashi, KimiyasuShiraki, Donald F. Smee, Dale L. Barnard. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Research
Volume 100, Issue 2, November 2013, Pages 446-454
[9] Baranovich Т., Wong S.S., Armstrong J., Marjuki H., Webby R.J., Webster R.G., Govorkova E.A. (2013) T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 87, 3741-3751.
[10] Vanderlinden E., Vrancken В., Van Houdt J., Rajwanshi V.K., Gillemot S., Andrei G., Lemey P., Naesens L. (2016) Distinct effects of T-705 (favipiravir) and ribavirin on influenza virus replication and viral RNA synthesis. Antimicrob. Agents Chemother. 60, 6679-6691.
[11] e  Gallego L, Soria M.E., Gregori J., Quer J., Esteban J.I., Rice С.М., Domingo E., Perales C. (2016) Lethal mutagenesis of Hepatitis С virus induced by Favipiravir. PLoS One 11, e0164691.
Gallego L, Soria M.E., Gregori J., Quer J., Esteban J.I., Rice С.М., Domingo E., Perales C. (2016) Lethal mutagenesis of Hepatitis С virus induced by Favipiravir. PLoS One 11, e0164691.
[12] Arias A., Thorne L., Goodfellow I. (2014) Favipiravir elicits antiviral mutagenesis during virus replication in vivo. eLife 3, e03679.
[13] Kiso M., Takahashi K., Sakai-Tagawa Y., Shinya K., Sakabe S., Le Q.M., Ozawa M., Furuta Y., Kawaoka Y. (2010) T-705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc. Natl. Acad. Sci. U.S.A. 107, 882-887.
[14] Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza (2010) Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N. Engl. J. Med. 362, 1708-1719.
[15] WHO Ebola Response Team (2014) Ebola virus disease in West Africa-the first 9 months of the epidemic and forward projections. N. Engl. J. Med. 371, 1481-1495.
[16] Mylne A.Q., Pigott D.M., Longbottom J., Shearer F., Duda K.A., Messina J.P., Weiss D.J., Moyes C.L., Golding N., Hay S.I. (2015) Mapping the zoonotic niche of Lassa fever in Africa. Trans. R. Soc. Trop.Med. Hyg. 109, 483-492.
[17] Furuta Y., Takahashi K., Fukuda Y., Kuno M., Kamiyama Т., Kozaki K., Nomura N., Egawa H., Minami S., Watanabe Y., Narita H., Shiraki K. (2002) In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 46, 977-981.
[18] Nagata Т., Lefor A.K., Hasegawa M., Ishii M. (2015) Favipiravir: a new medication for the Ebola virus disease pandemic. Disaster Med. Public Health Prep.9, 79 81.
[19] Takahashi K., Furuta Y., Fukuda Y., Kuno M., Kamiyama Т., Kozaki K., Nomura N., Egawa H., Minami S., Shiraki K. (2003) In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antivir. Chem. Chemother. 14, 235-241.
[20] Sidwell R.W., Barnard D.L., Day C.W., Smee D.F., Bailey K.W., Wong M.H., Morrey J.D., Furuta Y. (2007) Efficacy of orally administered T-705 on lethal avian influenza A (H5N1) virus infections in mice. Antimicrob. Agents Chemother. 51, 845-851.
[21] Moraz M.L., Kunz S. (2011) Pathogenesis of arenavirus hemorrhagic fevers. Expert Rev. Anti Infect. Ther. 9, 49-59.
[22] Gowen B.B., Wong M.H., Jung K.H., Sanders A.B., Mendenhall M., Bailey K.W., Furuta Y., Sidwell R.W. (2007) In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob. Agents Chemother. 51, 3168-3176.
[23] Gowen B.B., Smee D.F., Wong M.H., Hall J.O., Jung K.H., Bailey K.W., Stevens J.R., Furuta Y., Morrey J.D. (2008) Treatment of late stage disease in a model of arenaviral hemorrhagic fever: T-705 efficacy and reduced toxicity suggests an alternative to ribavirin. PLoS One 3, e3725.
[24] Mendenhall M., Russell A., Smee D.F., Hall J.O., Skirpstunas R., Furuta Y., Gowen B.B. (2011) Effective oral favipiravir (T-705) therapy initiated after the onset of clinical disease in a model of arenavirus hemorrhagic fever. PLoS Negl. Trop.Dis. 5, el342.
[25] Safronetz D., Rosenke K., Westover J.B., Martellaro C, Okumura A., Furuta Y., Geisbert J., Saturday G., Komeno Т., Geisbert T.W., Feldmann H., Gowen B.B. (2015) The broad-spectrum antiviral favipiravir protects guinea pigs from lethal Lassa virus infection post-disease onset. Sci. Rep.5, 14775.
[26] Gowen B.B., Wong M.H., Jung K.H., Smee D.F., Morrey J.D., Furuta Y. (2010) Efficacy of favipiravir (T-705) and T-1106 pyrazine derivatives in phlebovirus disease models. Antiviral Res. 86, 121-127.
[27] Tani H., Fukuma A., Fukushi S., Taniguchi S., Yoshikawa Т., Iwata-Yoshikawa N., Sato Y., Suzuki Т., Nagata N., Hasegawa H., Kawai Y., Uda A., Morikawa S., Shimojima M., Watanabe H., Saijo M. (2016) Efficacy of T-705 (Favipiravir) in the treatment of infections with lethal severe fever with thrombocytopenia syndrome virus. mSphere 1, e00061-15.
[28] Yu X., Liang M., Zhang S., Liu Y., Li J., Sun Y., Zhang L., Zhang Q., Popov V.L., Li C, Qu J., Li Q., Zhang Y., Hai R., Wu W., Wang Q., Zhan F., Wang X., Kan В., Wang S., Wan K.L., Jing H.Q., Lu J.X., Yin W.W., Zhou H., Guan X.H., Liu J.F., Bi Z.Q., Liu G.H., Ren J., Wang H., Zhao Z., Song J.D., He J.R., Wan Т., Zhang J.S., Fu X.P., Sun L.N., Dong X.P., Feng Z.J., Yang W.Z., Hong Т., Zhang Y., Walker D.H., Wang Y., Li D.X. (2011) Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 364, 1523-1532.
[29] Kim К., Yi J., Kim G., Choi S.J., Jun K.I., Kim N., Choe P.G., Kim N., Lee J., Oh M. (2013) Severe fever with thrombocytopenia syndrome, South Korea, 2012. Em erg. Infect. Dis. 19, 1892-1894.
[30] Clinical study of favipiravir for patients with severe fever with thrombocytopenia syndrome. UMIN-CTR Clinical Trial https://upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000025710.
[31] Julander J.G., Shafer K., Smee D.F., Morrey J.D., Furuta Y. (2009) Activity of T-705 in a hamster model of yellow fever virus infection in comparison with a chemically related compound T-1106. Antimicrob. Agents Chemother. 53, 202-209.
[32] Morrey J.D., Taro B.S., Siddharthan V., Wang H., Smee D.F., Christensen A. J., Furuta Y. (2008) Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res. 80, 377-379.
[33] Rebecca Sheets History and Characterization of the Vero Cell Line. A Report for the Vaccines and Related Biological Products Advisory Committee Meeting.
[34] Zmurko J., Marques R.E., Schols D., Verbeken E., Kaptein S.J., Neyts J. (2016) The viral polymerase inhibitor 7-deaza-2'-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl. Trop.Dis. 10, e0004695.
[35] Julander J.G., Smee D.F., Morrey J.D., Furuta Y. (2009) Effect of T-705 treatment on western equine encephalitis in a mouse model. Antiviral Res. 82, 169-171.
[36] Delang L., Segura Guerrero N., Tas A., Querat G., Pastorino В., Froeyen M., Dallmeier K., Jochmans D., Herdewijn P., Bello F., Snijder E.J., de Lamballerie X., Martina В., Neyts J., van Hemert M.J., Leyssen P. (2014) Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. J. Antimicrob. Chemother. 69, 2770 2784.
[37] Sakamoto K., Ohashi S., Yamazoe R., Takahashi K., Furuta Y. (2006) The inhibition of FMD virus excretion from the infected pigs by an antiviral agent, T-1105. FAO report of the research group of the standing technical committee of european commission for the control of Foot-and-Mouth Disease, Paphos, Cyprus. FAO Appendix 64, 418-424.
[38] Furuta Y., Takahashi K., Shiraki K., Sakamoto K., Smee D.F., Barnard D.L., Gowen B.B., Julander J.G., Morrey J.D. (2009) T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 82, 95 102.
[39] Furuta Y., Takahashi K., Fukuda Y., Kuno M., Kamiyama Т., Kozaki K., Nomura N., Egawa FL, Minami S., Watanabe Y., Narita H., Shiraki K. (2002) In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 46, 977-981.
[40] Wang Y., Li G., Yuan S., Gao Q., Lan K., Altmeyer R., Zou G. (2016) In vitro assessment of combinations of enterovirus inhibitors against enterovirus 71. Antimicrob. Agents Chemother. 60, 5357-5367.
[41] Rocha-Pereira J., Jochmans D., Dallmeier K., Leyssen P., Nascimento M.S., Neyts J. (2012) Favipiravir (T-705) inhibits in vitro norovirus replication. Biochem. Biophys. Res. Commun. 10 (424), 777-780.
[42] Jin Z., Tucker K., Lin X., Kao C.C., Shaw K., Tan H., Symons J., Behera I., Rajwanshi V.K., Dyatkina N., Wang G., Beigelman L., Deval J. (2015) Biochemical evaluation of the inhibition properties of Favipiravir and 2'-C-methyl-cytidine triphosphates against human and mouse norovirus RNA polymerases. Antimicrob. Agents Chemother. 59, 7504-7516.
[43] Oestereich L., Ludtke A., Wurr S., Rieger Т., Munoz-Fontela C, Gunther S. (2014) Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res. 105, 17-21.
[44] Smither S.J., Eastaugh L.S., Steward J.A., Nelson M., Lenk R.P., Lever M.S. (2014) Post-exposure efficacy of oral T-705 (Favipiravir) against inhalational Ebola virus infection in a mouse model. Antiviral Res. 104, 153-155.
[45] Mentre F., Taburet A.M., Guedj J., Anglaret X., Keita S., de Lamballerie X., Malvy D. (2015) Dose regimen of favipiravir for Ebola virus disease. Lancet Infect. Dis. 15, 150-151.
[46] Sissoko D., Laouenan C, Folkesson E., M'Lebing A.B., Beavogui A.H., Baize S., Camara A.M., Maes P., Shepherd S., Danel C, Carazo S., Conde M.N., Gala J.L., Colin G., Savini H., Bore J.A., Le Marcis F., Koundouno F.R., Petitjean F., Lamah M.C., Diederich S., Tounkara A., Poelart G., Berbain E., Din dart J.M., Duraffour S., Lefevre A., Leno Т., Peyrouset O., Irenge L., Bangoura N., Palich R., Hinzmann J., Kraus A., Barry T.S., Berette S., Bongono A., Camara M.S., Munoz V.C., Doumbouya L., Harouna S., Kighoma P.M., Koundouno F.R., Lolamou R., Loua С.М., Massala V., Moumouni K., Provost C, Samake N., Sekou C, Soumah A., Arnould I., Komano M.S., Gustin L., Berutto C, Camara D., Camara F.S., Colpaert J., Delamou L., Jansson L., Kourouma E., Loua M., Malme K., Manfrin E., Maomou A., Milinouno A., Ombelet S., Sidiboun A.Y., Verreckt I., Yombouno P., Bocquin A., Carbonnelle C, Carmoi Т., Frange P., Mely S., Nguyen V.K., Pannetier D., Taburet A.M., Treluyer J.M., Kolie J., Moh R., Gonzalez M.C, Kuisma E., Liedigk В., Ngabo D., Rudolf M., Thorn R., Kerber R., Gabriel M., Di Caro A., Wolfel R., Badir J., Bentahir M., Deccache Y., Dumont C, Durant J.F., El Bakkouri K., Uwamahoro M.G., Smits В., Toufik N., Van Cauwenberghe S., Ezzedine K., Dortenzio E., Pizarro L., Etienne A., Guedj J., Fizet A., de Sainte Fare E.B., Murgue В., Tran-Minh Т., Rapp C, Piguet P., Poncin M., Draguez В., Allaford Duverger Т., Barbe S., Baret G., Defourny I., Carroll M., Raoul FL, Augier A., Eholie S.P., Yazdanpanah Y., Levy-Marchal C, Antierrens A., Van Негр M.,  de, Lamballerie X., Keita S., Mentre F., Anglaret X., Malvy D., JIKI Study Group (2016) Experimental treatment with Favipiravir for Ebola virus disease (the JIKI Trial): A historically controlled, single-arm proof-of-concept trial in Guinea. PLoS Med. 13, el001967.
de, Lamballerie X., Keita S., Mentre F., Anglaret X., Malvy D., JIKI Study Group (2016) Experimental treatment with Favipiravir for Ebola virus disease (the JIKI Trial): A historically controlled, single-arm proof-of-concept trial in Guinea. PLoS Med. 13, el001967.
[47] Bai C.Q., Mu J.S., Kargbo D., Song Y.B., Niu W.K., Nie W.M., Kanu A., Liu W.W., Wang Y.P., Dafae F., Yan Т., Hu Y., Deng Y.Q., Lu H.J., Yang F., Zhang X.G., Sun Y., Cao Y.X., Su H.X., Sun Y., Liu W.S., Wang C.Y., Qian J., Liu L., Wang FL, Tong Y.G., Liu Z.Y., Chen Y.S., Wang H.Q., Kargbo В., Gao G.F., Jiang J.F. (2016) Clinical and virological characteristics of Ebola virus disease patients treated with favipiravir (T-705)-Sierra Leone, 2014. Clin. Infect. Dis. 63, 1288-1294.
[48] Yamada K., Noguchi K., Komeno Т., Furuta Y., Nishizono A. (2016) Efficacy of Favipiravir (T-705) in rabies postexposure prophylaxis. J. Infect. Dis. 213, 1253-1261.
[49] https://www.cebm.net/covid-19/lopinavir-ritonavir-a-rapid-review-of-the-evidence-for-effectiveness-in-treating-covid/
[50] Aartjan J. W. te Velthuis, Sjoerd H. E. van den Worm, Amy C. Sims, Ralph S. Baric, Eric J. Snijder, Martijn J. van Hemert. Zn2+Inhibits Coronavirus and Arterivirus RNA Polymerase Activity In Vitro and Zinc Ionophores Block the Replication of These Viruses in Cell Culture. PLOS Pathogens November 4, 2010 (https://doi.org/10.1371/journal.ppat.1001176)
[51] Lazarczyk M, Favre M (2008) Role of Zn2+ions in host-virus interactions. J Virol 82: 11486-11494.
[52] Frederickson CJ, Koh JY, Bush Al (2005) Neurobiology of zinc in health and disease. Nat Rev Neurosci 6: 449-462.
[53] Uchide N, Ohyama K, Bessho T, Yuan B, Yamakawa T (2002) Effect of antioxidants on apoptosis induced by influenza virus infection: inhibition of viral gene replication and transcription with pyrrolidine dithiocarbamate. Antiviral Res 56: 207-217.
[54] Suara RO, Crowe JEJ (2004) Effect of zinc salts on respiratory syncytial virus replication. Antimicrob Agents Chemother 48: 783-790.
[55] Korant BD, Kauer JC, Butterworth BE (1974) Zinc ions inhibit replication of rhinoviruses. Nature 248: 588-590.
[56] Polatnick J, Bachrach HL (1978) Effect of zinc and other chemical agents on foot-and-mouth-disease virus replication. Antimicrob Agents Chemother 13: 731-734.
[57] Lanke K, Krenn BM, Melchers WJG, Seipelt J, van Kuppeveld FJM (2007) PDTC inhibits picornavirus polyprotein processing and RNA replication by transporting zinc ions into cells. J Gen Virol 88: 1206-1217.
[58] Krenn BM, Gaudernak E, Holzer B, Lanke K, Van Kuppeveld FJM, et al. (2009) Antiviral Activity of the Zinc Ionophores Pyrithione and Hinokitiol against Picornavirus Infections. J Virol 83: 58 64.
[59] Pasternak AO, Spaan WJ, Snijder EJ (2006) Nidovirus transcription: how to make sense…? J Gen Virol 80: 1403-1421.
[60] Sawicki SG, Sawicki DL, Siddell SG (2007) A Contemporary View of Coronavirus Transcription. J Virol 81: 20-29.
[61] Denison MR, Perlman S (1986) Translation and processing of mouse hepatitis virus virion RNA in a cell-free system. J Virol 60: 12-18.
[62] Denison MR, Zoltick PW, Hughes SA, Giangreco B, Olson AL, et al. (1992) Intracellular processing of the N-terminal ORF la proteins of the coronavirus MHV-A59 requires multiple proteolytic events. Virology 189: 274-284.
[63] te Velthuis AJ, Arnold JJ, Cameron CE, van den Worm SH, Snijder EJ (2009) The RNA polymerase activity of SARS-coronavirus nspl2 is primer dependent. Nucleic Acids Res 38: 203-214.
[64] Valerie E.SpallM.Shanks, G.P.Lomonossoff. Polyprotein Processing as a Strategy for Gene Expression in RNA Viruses. Seminars in Virology Volume 8, Issue 1, February 1997, Pages 15-23.
[65] Jian Lei, Rolf Hilgenfeld. RNA - virus proteases counteracting host innate immunity. FEBS Lett. 2017 Oct; 591(20): 3190-3210.
[66] Chih-Chia Lua, Mei-Yu Chena, Yuh-Lih Chang. Potential therapeutic agents against COVID-19: What we know so far. Journal of the Chinese Medical Association Publish Ahead of Print DOI: 10.1097/JCMA.0000000000000318
[67] Salman Ali et al. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. Journal of Biomolecular Structure and Dynamics. Published online: 13 Apr 2020.
[68] Tessa Nelemans, Marjolein Kikkert. Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses. 2019 Oct; 11(10): 961.
[69] Brechot C., Bryant J., Endtz H., Garry R.F., Griffin D.E., Lewin S.R., Mercer N., Osterhaus A., Picot V., Vahlne A., et al. 2018 International Meeting of the Global Virus Network. Antiviral Res. 2019;163:140-148. doi: 10.1016/j.antiviral.2019.01.013.
[70] WHO Middle East respiratory syndrome coronavirus (MERS-CoV) [(accessed on 16 September 2019)]; Available online: https://www.who.int/news-room/fact-sheets/detail/middle-east-respiratory-syndrome-coronavirus-(MERS-CoV)
[71] Shen. High-throughput screening and identification of potent broad-spectrum inhibitors of coronaviruses. J. Virol. 2019;93(12):e00023 19.
[72] Marc Desforges, Alain Le Coupanec, Philippe Dubeau, Andreanne Bourgouin, Louise Lajoie, Mathieu Dube, Pierre J. Talbot. Human Coronaviruses and Other Respiratory Viruses: Underestimated Opportunistic Pathogens of the Central Nervous System? Viruses. 2020 Jan; 12(1): 14.
[73] Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host Microbe. 2013 Apr 17; 13(4):379-93.
[74] Berth SH, Leopold PL, Morfini GN. Virus-induced neuronal dysfunction and degeneration. Front Biosci (Landmark Ed). 2009 Jun 1; 14():5239-59.
[75] Daphne Y Ma, Mehul S Suthar Mechanisms of innate immune evasion in re-emerging RNA viruses Curr Opin Virol. 2015 Jun; 12: 26-37.
[76] Thanigaimalai Pillaiyar, Sangeetha Meenakshisundaram, Manoj Manickam. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov Today. 2020 Jan 30
[77] Furuta Y, Takahashi K, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Watanabe Y, Narita H, Shiraki K. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob Agents Chemother. 2002 Apr; 46(4):977-81.
[78] Sleeman К, Mishin VP, Deyde VM, Furuta Y, Klimov AI, Gubareva LV. In vitro antiviral activity of favipiravir (T-705) against drug-resistant influenza and 2009 A(H1N1) viruses. Antimicrob Agents Chemother. 2010 Jun; 54(6):2517-24.
[79] Takahashi K, Furuta Y, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Shiraki K
Antivir Chem Chemother. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. 2003 Sep; 14(5):235-41.
[80] Mendenhall M, Russell A, Juelich T, Messina EL, Smee DF, Freiberg AN, Holbrook MR, Furuta Y, de la Torre JC, Nunberg JH, Gowen BB. Antimicrob Agents Chemother. T-705 (favipiravir) inhibition of arenavirus replication in cell culture. 2011 Feb; 55(2):782-7.
[81] Gowen BB, Wong MH, Jung KH, Sanders AB, Mendenhall M, Bailey KW, Furuta Y, Sidwell RW. In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob Agents Chemother. 2007 Sep; 51(9):3168-76.
[83] Shankar AH, Prasad AS. Zinc and immune function: the biological basis of altered resistance to infection. Am J Clin Nutr. 1998 Aug;68(2 Suppl):447S-463S
[84] Prasad, A. S., J. T. Fitzgerald, B. Bao, F. W. J. Beck, and H. Chandrasekar. 2000. Duration of symptoms and plasma cytokine levels in patients with the common cold treated with zinc acetate. Ann. Intern. Med. 133:245-252.
[85] Mulholland, E.K., О.O. Ogunlesi, R.A. Adegbola, M. Weber, В.E. Sam, A. Palmer, M.J. A. Manary, O. Secka, M. Aidoo, D. Hazlett, H. Whittle, and В. M. Greenwood. 1999. Etiology of serious infections in young Gambian infants. Pediatr. mfect. Dis. J. 18(10 Suppl.):S35-S41
[86] Beck, F.W. J., A.S. Prasad, J. Kaplan, J.T. Fitzgerald, and G.J. Brewe. 1997. Changes in cytokine production and T cell subpopulations in experimentally induced zinc-deficient humans. Am. J. Physiol. 272: E1002-E1007.
[87] Nidhi Kaushik, Chandru Subramani, Saumya Anang, Rajagopalan Muthumohan, Shalimar, Baibaswata Nayak, С.T. Ranjith-Kumar,Milan Surjit. Zinc Salts Block Hepatitis E Virus Replication by Inhibiting the Activity of Viral RNA-Dependent RNA Polymerase. J Virol. 2017 Nov 1; 91(21): e00754-17.
[88] Haraguchi Y, Sakurai H, Hussain S, AnnerBM, Hoshino H. 1999. Inhibition of HIV-1 infection by zinc group metal compounds. Antiviral Res 43:123-133
[89] Wei Z, Burwinkel M, Palissa C, Ephraim E, Schmidt MFG. 2012. Antiviral activity of zinc salts against transmissible gastroenteritis virus in vitro. Vet Microbiol 160:468-472.
[90] Wahba A. 1980. Topical application of zinc-solutions: a new treatment for herpes simplex infections of the skin? Acta Derm Venereol 60:175-177.
[91] Katz E, Margalith E. 1981. Inhibition of vaccinia virus maturation by zinc chloride. Antimicrob Agents Chemother 19:213-217.
[92] te Velthuis AJW, van den Worm SHE, Sims AC, Baric RS, Snijder EJ, van Hemert MJ. 2010. Zn2+ inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathog 6:e1001176.
[93] Korant BD, Kauer JC, Butterworth BE. 1974. Zinc ions inhibit replication of rhinoviruses. Nature 248:588-590.
[94] Suara RO, Crowe JE Jr. 2004. Effect of zinc salts on respiratory syncytial virus replication. Antimicrob Agents Chemother 48:783-790.
[95] Haraguchi, Y., H. Sakurai, S. Hussain, В. M. Anner, and H. Hoshino. 1999. Inhibition of HIV-1 infection by zinc group metal compounds. Antivir. Res. 43:123-133.
[96] Polatnick J, Bachrach H.L. Effect of Zinc and Other Chemical Agents on Foot-and-Mouth Disease Virus Replication. Antimicrobial Agents and Chemotherapy, May 1978, p. 731-734, Vol. 13, No. 5
[97] Butterworth, В. E., and B. D. Korant. 1974. Characterization of the large picomaviral polypeptides produced in the presence of zinc ion. J. Virol. 14:282-291.
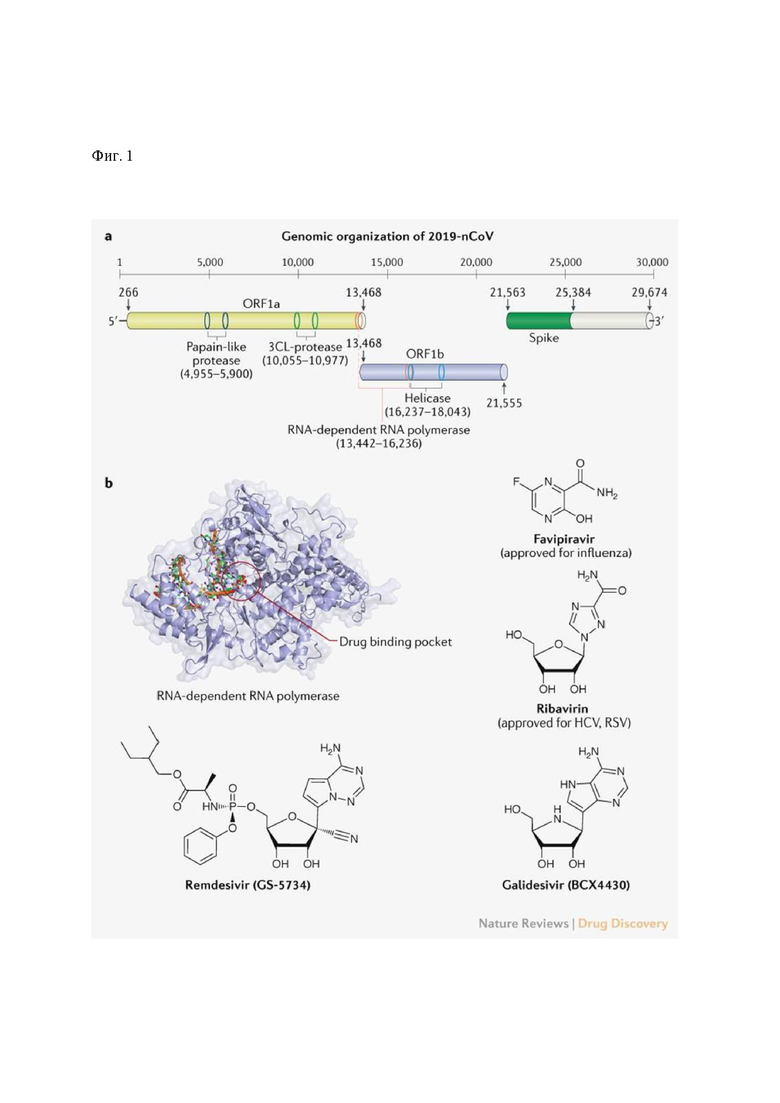
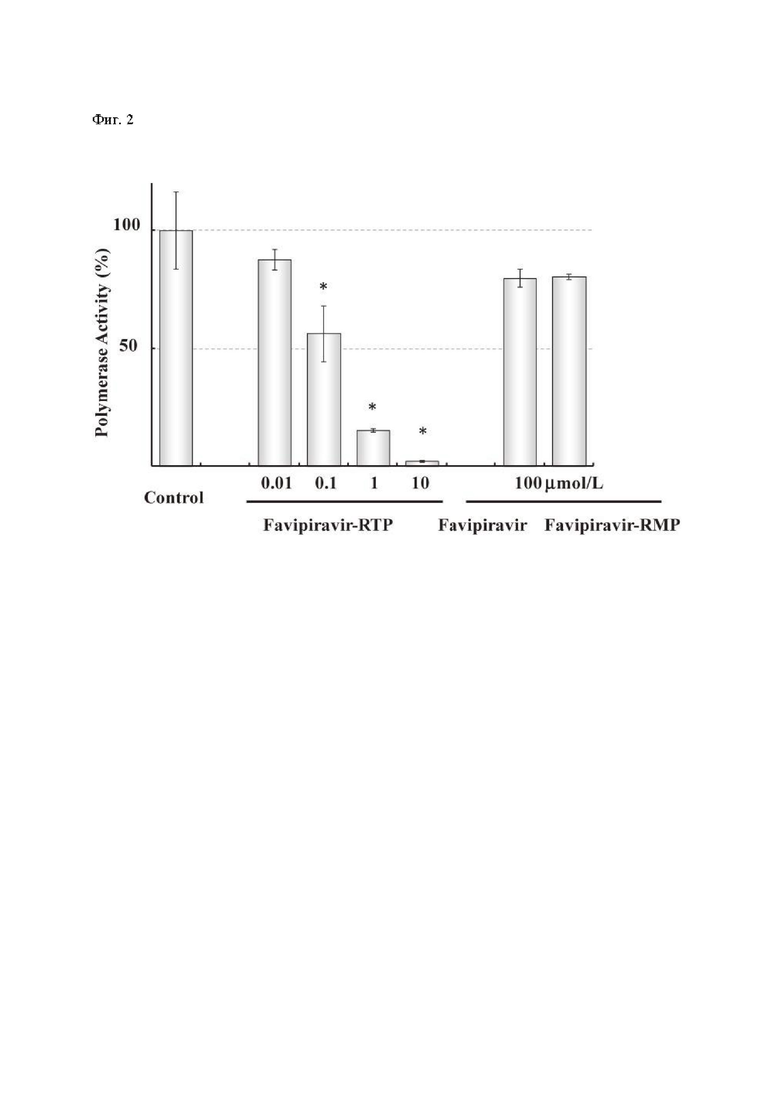
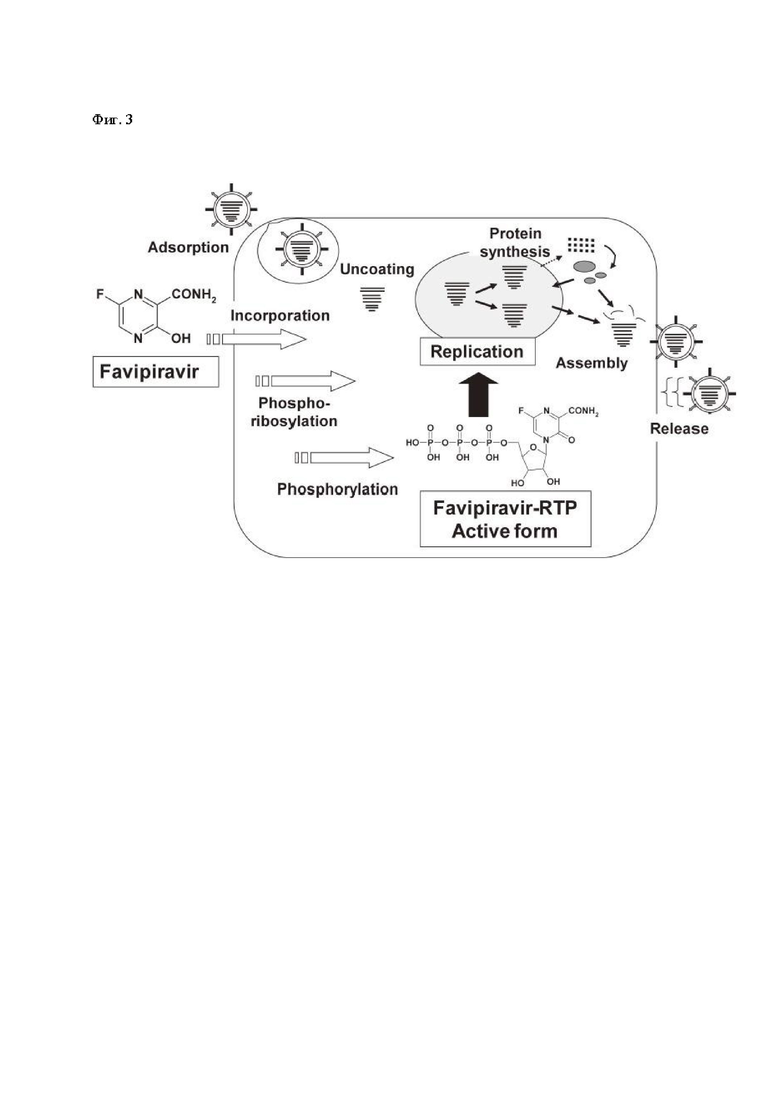
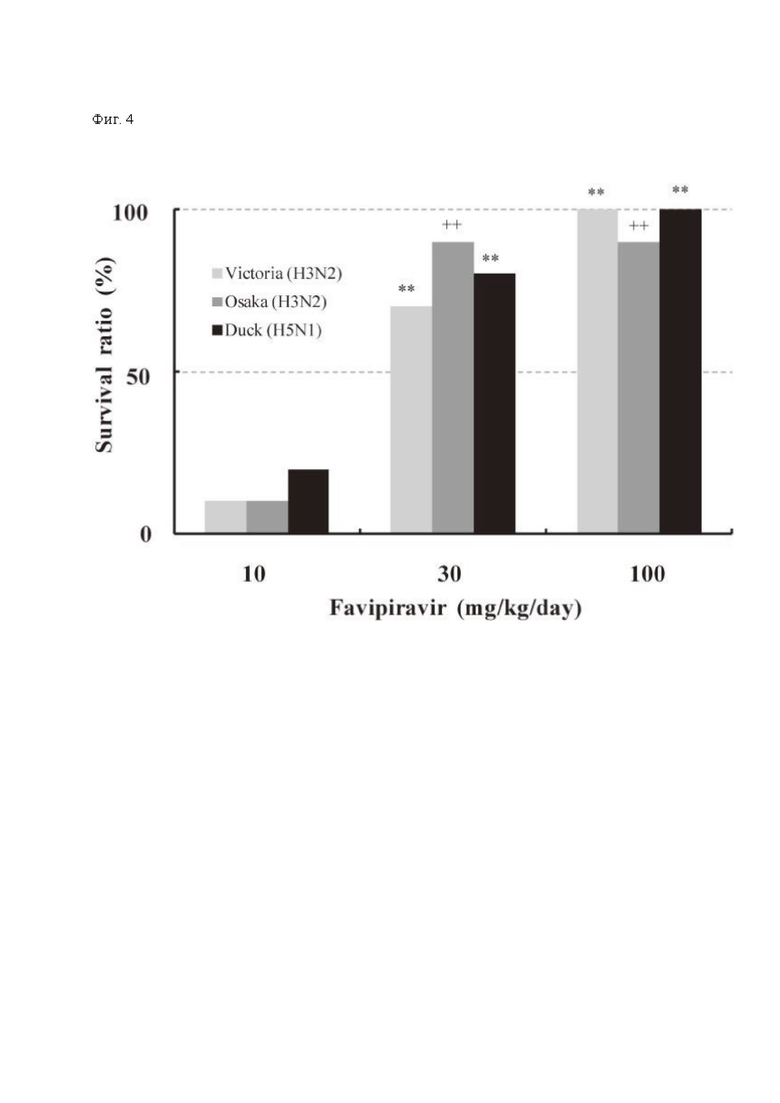
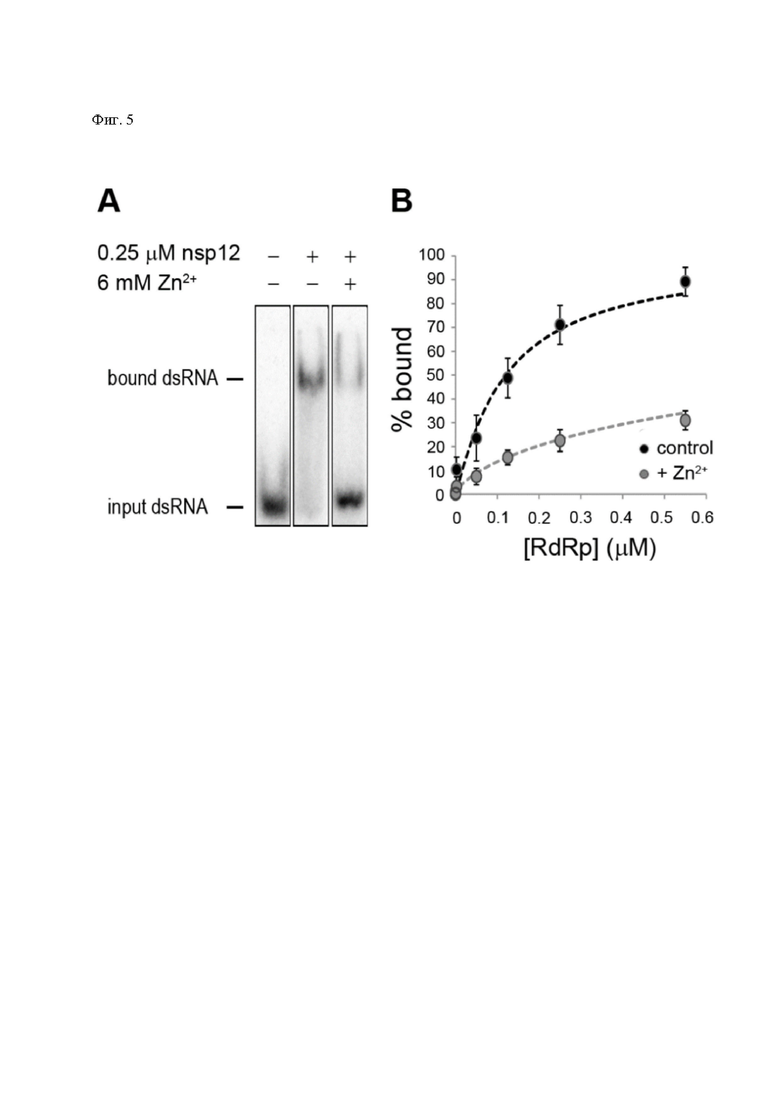
Группа изобретений относится к фармацевтической промышленности, а именно к композиции для облегчения клинических симптомов, течения и/или излечения заболевания, вызванного воздействием вируса, геном которого закодирован одноцепочечной нитью РНК и который использует вирусную РНК-зависимую-РНК-полимеразу для своей репликации. Фармацевтическая композиция для облегчения клинических симптомов, течения и/или излечения заболевания, вызванного воздействием вируса, геном которого закодирован одноцепочечной нитью РНК и который использует вирусную РНК-зависимую-РНК-полимеразу для своей репликации, содержащая эффективное количество фавипиравира и эффективное количество соединения цинка, выбранного из сульфата цинка, ацетата цинка, лактата цинка, цинка-диэтил-бис(N-4-метилтиосемикарбазона), дитиокарбамата цинка, в массовом соотношении фавипиравир к соли цинка 1:1-10:1, где эффективное количество фавипиравира составляет 50-800 мг, эффективное количество соли цинка составляет 15-250 мг. Способ лечения и/или профилактики заболевания, вызванного воздействием вируса, геном которого закодирован одноцепочечной нитью РНК и который использует вирусную РНК-зависимую-РНК-полимеразу для своей репликации, включающий введение нуждающемуся субъекту вышеописанной фармацевтической композиции. Вышеописанная композиция обладает выраженным действием для облегчения клинических симптомов, течения и/или излечения заболевания, вызванного воздействием вируса, геном которого закодирован одноцепочечной нитью РНК и который использует вирусную РНК-зависимую-РНК-полимеразу для своей репликации. 2 н. и 8 з.п. ф-лы, 5 ил., 4 табл., 4 пр.
1. Фармацевтическая композиция для облегчения клинических симптомов, течения и/или излечения заболевания, вызванного воздействием вируса, геном которого закодирован одноцепочечной нитью РНК и который использует вирусную РНК-зависимую-РНК-полимеразу для своей репликации, содержащая эффективное количество фавипиравира и эффективное количество соединения цинка, выбранного из сульфата цинка, ацетата цинка, лактата цинка, цинка-диэтил-бис(N-4-метилтиосемикарбазона), дитиокарбамата цинка, в массовом соотношении фавипиравир к соли цинка 1:1-10:1, где эффективное количество фавипиравира составляет 50-800 мг, эффективное количество соли цинка составляет 15-250 мг.
2. Фармацевтическая композиция по п. 1, в которой вирус представляет собой вирус гриппа, коронавирус, пикорнавирус, аренавирус, флавивирус, буньявирус, тогавирус, калицивирус или норавирус.
3. Фармацевтическая композиция по п. 2, в которой вирус гриппа представляет собой высоковирулентный вирус или низковирулентный вирус.
4. Фармацевтическая композиция по пп. 1-3, в которой количество фавипиравира составляет от 100 до 800 мг.
5. Фармацевтическая композиция по пп. 1-3, в которой количество фавипиравира составляет от 200 до 400 мг.
6. Фармацевтическая композиция по п. 1, в которой количество соединения цинка составляет от 50 до 250 мг.
7. Фармацевтическая композиция по п. 1, в которой количество соединения цинка составляет от 100 до 150 мг.
8. Фармацевтическая композиция по п. 1, выполненная в твердой или жидкой форме.
9. Фармацевтическая композиция по п. 8, в которой твердая форма представляет собой таблетку, покрытую пленочной оболочкой или полученную методом прессования.
10. Способ лечения и/или профилактики заболевания, вызванного воздействием вируса, геном которого закодирован одноцепочечной нитью РНК и который использует вирусную РНК-зависимую-РНК-полимеразу для своей репликации, включающий введение нуждающемуся субъекту фармацевтической композиции по пп. 1-9.
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |

