Изобретение относится к области химико-фармацевтической промышленности и медицины и представляет собой новые производные фавипиравира, а именно новые соли фавипиравира с аминокислотами, а также фармацевтическую композицию и лекарственное средство, содержащие новые соли фавипиравира.
Одна из наиболее актуальных проблем современной медицины - это борьба с вирусными инфекциями. По данным ВОЗ, ежегодно в мире от инфекционных болезней страдают 2 млрд человек и для 17 млн из них это причина смерти. Ежедневно в мире 50 тыс. летальных исходов обусловлены инфекционными болезнями, которые по-прежнему остаются в числе ведущих причин смертности и первой причиной преждевременной смерти [Брико Н.И., Миндлина А.Я., Полибин Р.В. Универсальность изменений в проявлениях эпидемического процесса антропонозных инфекций за последние десятилетия. Журнал микробиологии. 2015; 5: 12-20].
Большую часть вирусов представляют собой вирусы, геном которых закодирован одноцепочечной нитью РНК и которые используют вирусную РНК-зависимую-РНК- полимеразу для своей репликации. Примерами таких вирусов являются вирус гриппа, коронавирус, пикорнавирус, аренавирус, флавивирус, буньявирус, филовирус, флебовирус, хантавирус, энтеровирус, тогавирус, калицивирус, респираторно-синцитиальный вирус, вирус парагриппа, риновирус, метапневмовирус, ротавирус или норавирус.
Препараты - аналоги нуклеозидов напрямую нацелены на блокировку активности РНК-зависимой-РНК-полимеразы и блокируют синтез вирусной цепи РНК для широкого спектра РНК-вирусов, включая семейство человеческих коронавирусов. Например, фавипиравир (6-фторо-3-гидрокси-2-пиразинкарбоксамид), гуаниновый аналог, одобренный в клинической практике для лечения гриппа, эффективно блокирует РНК-зависимую-РНК- полимеразу вирусов гриппа (разных типов), вируса Эбола, желтой лихорадки, чикунгунья, норовирусов, энтеровирусов и др. [De Clercq, E. New nucleoside analogues for the treatment of hemorrhagic fever virus infections. Chem. Asian J. 14, 3962-3968, 2019].
Универсальный механизм действия фавипиравира специфично действующий на основополагающий фермент репликативного аппарата вируса предполагает широкий спектр противовирусной активности данного вещества, что было продемонстрировано во многих исследованиях, указанных ниже.
Противовирусная активность фавипиравира
Эффект применения фавипиравира против вируса гриппа
Эпидемия гриппа случается во всем мире ежегодно. Заболевание вызвано штаммами вируса гриппа различной вирулентности. Высоко патогенный вирус птичьего гриппа A(H5N1) вызвал первую вспышку заболевания в Гонг-Конге в 1997 г. и продолжает каждый год вызывать локальные вспышки данного вида гриппа. Эпидемия птичьего гриппа A(H7N9) в Китае в 2013 г. и пандемия гриппа A(H1N1) в 2009 привела к 17,700 смертям, и до сих пор грипп является одной из серьезных проблем для здравоохранения во всем мире, не только по масштабу заболеваемости, но и по критическим для здоровья осложнениям, которые он вызывает [Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. - 2010. - N. Engl. J. Med.]. Эпидемия A(H1N1) показала, что данный штамм устойчив к оцетальмивиру (тамифлю) - ингибитору нейрамидазы и к амантадину- ингибитору неструктурного белка М2. В связи с этим в медицинской практике остро необходимо лекарство другого механизма действия.
Фавипиравир (T-705; 6-флуоро-3-гидрокси-2-пиразинкарбоксамид) эффективен в отношении широкого диапазона штаммов вируса гриппа, включая A(H1N1), A(H5N1) и A(H7N9), за счет того, что вирусная РНК-зависимая РНК-полимераза (РзРп) ошибочно принимает метаболит фавипировир-РТФ за пуриновый нуклеотид. Фавипиравир прошел исследования III фазы в Японии и II-ой - в США по лечению гриппа.
В дополнение к антивирусной активности против гриппа фавипиравир ингибирует репликацию аренавирусов, флебовирусов (лихорадка Рифт-Валле, вирус флеботомной лихорадки и лихорадки Пунта Торо), хантавирусы (Мапорал, Добрава и Проспект Хилл); флавивирусы (желтая лихорадка и лихорадка Западного Нила); энтеровирусы (полио- и ринофирусы); парамиксовирус респираторного синцития и норовирус [Yousuke Furuta, Brian B.Gowen, KazumiTakahashi, KimiyasuShiraki, Donald F.Smee, Dale L.Barnard. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Research, 2013, Volume 100, Issue 2].
Использование фавипиравира в in vitro моделях гриппа
В исследованиях in vitro фавипиравир показал высокую противовирусную активность в отношении всех штаммов вируса гриппа, A, B и C. Основываясь на подсчете бляшкообразующих единиц (БОЕ) в культуре клеток MDCK, показатель эффективной концентрации (далее ЭК50) был в пределах от 0.014 to 0.55μг/мл [Furuta Y., Takahashi K., Fukuda Y., Kuno M., Kamiyama T., Kozaki K., Nomura N., Egawa H., Minami S., Watanabe Y., Narita H., Shiraki K. In vitro and in vivo activities of anti-influenza virus compound T-705. -2002. - Antimicrob Agents Chemother; 46(4), p. 977-981].
Известна активность фавипиравира против 53 штаммов вируса гриппа, включая сезонные штаммы A(H1N1), A(H3N2), и штаммы вируса гриппа типа В; A(H1N1)pdm09 пандемичный вирус, высокопатогенный птичий грипп A(H5N1), выделенный от человека, штаммы A(H1N1) и A(H1N2), выделенные из свиней, и A(H2N2), A(H4N2), A(H7N2).
Использование фавипиравира в in vivo моделях гриппа
In vivo на мышиных моделях вирусной инфекции летальными дозами штаммов H3N2 (A/Victoria/3/75), H3N2 (A/Osaka/5/70) или H5N1 (A/Duck/MN/1525/81) фавипиравир применялся час спустя после заражения. Выживаемость мышей при дозах от 30 мг/кг/день 2 или 4 раза в день была значительна, тогда как все зараженные мыши контрольной группы погибли.
При применении от 60 до 300 мг/кг/день фавипиравир продемонстрировал свою эффективность в снижении вирусной нагрузки в легких мышей, инфицированных H1N1 (A/ California/04/09), а также при отсроченном применении, вплоть до 96 часов после инфицирования [Takahashi K., Furuta Y., Fukuda Y., Kuno M., Kamiyama T., Kozaki K., Nomura N., Egawa H., Minami S., Shiraki K. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. Antivir. Chem. Chemother. - 2003; Sidwell R.W., Barnard D.L., Day C.W., Smee D.F., Bailey K.W., Wong M.H., Morrey J.D., Furuta Y. Efficacy of orally administered T-705 on lethal avian influenza A (H5N1) virus infections in mice. - 2007. - Antimicrob Agents Chemother]. Фавипиравир показал значительный терапевтический эффект в сравнении с оцетальмивиром на мышах, которым была введена доза вируса в 100 раз больше, а само лечение было отсрочено на 96 часов пост-инфекции [Takahashi K, Furuta Y, Fukuda Y, Kuno M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Shiraki K. Antivir Chem Chemother. In vitro and in vivo activities of T-705 and oseltamivir against influenza virus. 2003].
Эффективность фавипиравира для других семейств РНК-вирусов в исследованиях in vitro и in vivo
Arenaviridae
Аренавирусы вызывают фатальные заболевания человека [Moraz M.L., Kunz S. Pathogenesis of arenavirus hemorrhagic fevers. - 2011. - Expert Rev. Anti Infect. Ther. - 9(1), p. 49-59], от которых не существует противовирусных препаратов, кроме рибавирина, обладающего выраженным токсичным действием.
In vitro фавипиравир показал большую селективность, чем рибавирин. При измерении цитопатического эффекта в культуре клеток значения ЭК50 для препарата составили 0.79-0.94 μг/мл для вирусов Юнин, Пичинде и Такарибэ. Также вирусная нагрузка при применении фавипиравира значительно уменьшалась уже к третьему дню. При исследовании методом подсчета очагов гемолиза ЭК90s против высокопатогенного штамма Ромеро вируса Гуанарито JUNV (Romero) и вируса Мачупо составила 3.3-8.4 μг/мл (21-53 μM) [Mendenhall M, Russell A, Juelich T, Messina EL, Smee DF, Freiberg AN, Holbrook MR, Furuta Y, de la Torre JC, Nunberg JH, Gowen BB. Antimicrob Agents Chemother. T-705 (favipiravir) inhibition of arenavirus replication in cell culture. - 2011. - Antimicrob Agents Chemother. - 55(2), p. 782-787].
При пероральном применении фавипиравира на модельной линии хомяков, зараженных вирусом Пичинде, препарат предотвращал смертельный исход, уменьшал количество вирусных титров в крови и тканях, при дозировках 60 мг/кг/день двукратно предотвращал разрушение печени при применении в течение 7 дней, начиная с 4 часа пост- инфицирования [Gowen B.B., Wong M.H., Jung K.H., Sanders A.B., Mendenhall M., Bailey K.W., Furuta Y., Sidwell R.W. In vitro and in vivo activities of T-705 against arenavirus and bunyavirus infections. Antimicrob. Agents Chemother. - 2007. - 51(9), p. 3168-3176]. Вирусная нагрузка значительно уменьшалась также при начале лечения от 4, 5 и 6 дня пост-инфекции. При применении в дозировках от 100 мг/кг/день значительно увеличивалась выживаемость животных [Gowen B.B., Smee D.F., Wong M.H., Hall J.O., Jung K.H., Bailey K.W., Stevens J.R., Furuta Y., Morrey J.D. Treatment of late stage disease in a model of arenaviral hemorrhagic fever: T-705 efficacy and reduced toxicity suggests an alternative to ribavirin. - 2008. - PLoS One 3, e3725].
На модели заражения морских свинок фавипиравир демонстрировал свою эффективность уже даже после возникновения острых симптомов болезни [Mendenhall M., Russell A., Smee D.F., Hall J.O., Skirpstunas R., Furuta Y., Gowen B.B. Effective oral favipiravir (T-705) therapy initiated after the onset of clinical disease in a model of arenavirus hemorrhagic fever. - 2011. - PLoS Negl. Trop. Dis. 5, e1342].
При применении фавипиравира в дозе 300 мг/кг/день он показал значимые эффекты для выживаемости: 100% животных, участвующих в эксперименте, выжило, при дозе 150 мг/кг/ день показатели снижались до 50 и 25%, животные сохраняли массу тела, у них падала температура до нормы и все показатели в целом были лучше, чем таковые для рибавирина в дозировке 50 мг/кг/день. Значительно дозозависимо понижалась концентрация прогностического маркера тяжести заболевания лихорадкой Ласса в крови - аспартат аминотрансферазы (AST) к десятому дню заболевания при лечении фавипиравиром. Виремия средней инфекционной дозы на 1 мл элюата снизилась в среднем до 2.1, 1.3, и 1.6 log10 CCID50/мл в группах, пролеченных высокой и средней дозой фавипиравира и рибавирином, соответственно.
Эффективность перорального применения фавипиравира была также показана на моделях, зараженных летальным Ласса вирусом морских свинок и мышей [Safronetz D., et al. The broad-spectrum antiviral favipiravir protects guinea pigs from lethal Lassa virus infection post-disease onset. - 2015. - Sci. Rep. 5, 14775]. Терапевтический эффект наблюдался на 2-ой день после заражения. Подкожное применение фавипиравира в дозировках 300 мг/кг/день 1 раз в день снижало температуру, предотвращало уменьшение массы тела и увеличивало выживаемость животных. Эффекты от применения фавипиравира во много раз превосходили терапевтические результаты от применения рибавирина в дозировке 50 мг/кг/день. Значимое улучшение выживаемости зараженных Ласса-вирусом морских свинок наблюдалось даже на 5, 7 и 9 день пост- инфицирования [Safronetz D.,et al. The broad-spectrum antiviral favipiravir protects guinea pigs from lethal Lassa virus infection post-disease onset. - 2015. - Sci. Rep. 5, 14775].
Bunyaviridae
Вирусы семейства Буньявирида, включая вирус Ла Кроче (LACV), вирус лихорадки Рифт-Валле (RVFV), вирус конго-крымской геморрагической лихорадки (CCHFV), вирус острой лихорадки с синдромом тромбоцитопении (SFTSV) и хантавирус, вызывают тяжелые геморрагические лихорадки с сопутствующими легочными и почечными осложнениями.
In vitro исследования показали превосходство применения фавипиравира над другими препаратами в направленности, эффективности и быстроте действия против целого ряда подобных вирусов [Yousuke Furuta, et al. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. - 2013. - Antiviral Research. Volume 100; Gowen B.B.,et al. Efficacy of favipiravir (T-705) and T-1106 pyrazine derivatives in phlebovirus disease models. - 2010. - Antiviral Res., Volume 86, Issue 2]. Значения ЭК50 в исследованиях бляшкообразующих единиц (БОЕ) в клеточной культуре начинались в пределах 0.9-30 μг/мл препарата для вирусов Ласса, Пунта Торо, Рифа-Валле, острой лихорадки (SFTSV), флеботомной лихорадки, и хантавирусов Добрава, Мапорал и Проспект Хилл [Tani H., et al. Efficacy of T-705 (Favipiravir) in the treatment of infections with lethal severe fever with thrombocytopenia syndrome virus. - 2016. - mSphere 1].
Использование фавипиравира в in vitro и in vivo моделях буньявирусов.
Вирус острой лихорадки с тромбоцитопенией (SFTSV) возник несколько лет назад как сезонное заболевание в Китае, Корее и Японии [Yu X., Liang M., et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. - 2011. - N. Engl. J. Med, 364. P. 523-1532]. Фавипиравир ингибировал репликацию SFTSV в культуре клеток с показателями ЭК50 в 0.71-1.3 μг/мл.
Терапевтический эффект фавипиравира был продемонстрирован на мышиных моделях с нокаутом рецепторов к интерферону-a (IFNAR-/-), которые не развивают немедленный иммунный ответ.
При пероральном применении фавипиравира в дозе от 300 мг/кг/день в течение 5 дней, начиная с 3 дня пост-заражения, все экспериментальные мыши выжили (P < 0.001), а начиная с 4 и 5 дня пост-инфекции - значительно улучшились показатели выживания в группе. На базе данных исследований in vivo в Японии препарат вошел в клинические испытания и успешно их закончил к настоящему моменту. Исследования показали эффективность применения фавипиравира даже после начала заболевания и наступления клинических симптомов [Yousuke Furuta,et al. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. - 2013. - Antiviral Research. Volume 100, Issue 2].
Flaviviridae.
Фавипиравир блокирует репликацию вирусов семейства флавивирида, включая вирус желтой лихорадки (YFV) и вирус Западного Нила (WNV) [Julander J.G.,et al.Activity of T-705 in a hamster model of yellow fever virus infection in comparison with a chemically related compound T-1106. - 2009. - Antimicrob. Agents Chemother, 53(1), p. - 202-209; Morrey J.D., et al. Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. - 2008. - Antiviral Res., 80(3), p. 377-379], однако, в более высоких концентрациях, чем необходимы для блокировки активности вируса гриппа.
ЭК90 фавипиравира в отношении YFV составляет 51.8 μг/мл в in vitro исследованиях по определению выделения активных вирусных частиц на культуре клеток Vero.
Инфицированных YFV-хомяков пролечивали перорально фавипиравиром в дозах от 200 до 400 мг/кг/день в течение 8 дней, начав лечение за 4 часа до инфицирования. Эта терапия серьезно снизила уровень смертности животных при начале лечения [Julander J.G., et al. Activity of T-705 in a hamster model of yellow fever virus infection in comparison with a chemically related compound T-1106. - 2009. - Antimicrob. Agents Chemother, 53(1), p. 202-209].
Полное выздоровление было достигнуто при введении 400 мг/кг/день, начиная со 2-ого дня пост-инфицирования.
In vitro и in vivo противовирусная эффективность фавипиравира в отношении вируса Западного Нила достигалась в концентрациях ЭК50 53 μг/мл в культуре клеток Vero.
Пероральное применение фавипиравира в дозах от 400 мг/кг/день 2 раза в день, спустя 4 часа после заражения спасло 90% мышей от смертельного исхода, а также значимо уменьшило экспрессию вирусных белков и вирусной РНК в тканях мозга. Такая же эффективность была показана и на линиях, зараженных WNV, хомяков в таких же дозах. Белки оболочки вируса WNV не детектировались в мозге пролеченных животных.
Togaviridae
Фавипиравир проявляет противовирусную активность против Западного вируса лошадиного энцефалита (WEEV) в культуре клеток Vero, достигая ЭК90 при 49 μг/мл [Julander J.G., et al. Effect of T-705 treatment on western equine encephalitis in a mouse model. Antiviral Res, 82(3), p. 169-171].
В мышах, инфицированных WEEV оральное применение фавипиравира значительно увеличивало выживаемость и продолжительность жизни зараженных животных при двукратном применении в дозе 400 мг/кг/день в течение 7 дней, начиная с 4-ого часа пост- инфекции. Вирусные титры в тканях мозга были снижены на 4-ый день пост-инфицирования.
Фавипиравир показал противовирусную активность против вируса Чикунгунья (CHIKV) в культуре клеток Vero, достигая ЭК50 при 0.3-9.4 μг/мл. На мышах, инфицированных CHIKV, оральное применение фавипиравира улучшало показатели выживаемости при двукратных дозах от 300 мг/кг/день, начиная за сутки до или 4 часа после инфицирования [Delang L., et al. Mutations in the chikungunya virus non-structural proteins cause resistance to favipiravir (T-705), a broad-spectrum antiviral. - 2014. - J. Antimicrob. Chemother, 69(10), p. 2770-2784].
Picornaviridae
Репликация энтеровируса везикулярного стоматита ингибировалась фавипировиром в исследованиях in vitro с ЭК50 в 14 μг/мл [Sakamoto K., et al. The inhibition of FMD virus excretion from the infected pigs by an antiviral agent, T-1105. FAO report of the research group of the standing technical committee of european commission for the control of Foot-and-Mouth Disease. - 2006. - Paphos, Cyprus. FAO Appendix 64; Furuta Y., et al. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. - 2009. - Antiviral Res, ;82(3), p. 95-102].
Фавипиравир также блокировал репликацию вируса полиомиелита в культуре клеток Vero и риновируса в культуре клеток HeLa при ЭК50s в 4.8 и 23 μг/мл, и с индексом селективности в значениях 29 и >43, соответственно [Furuta Y., et al. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. 2002. - Agents Chemother, 46(4), p. 977-981]. Фавипиравир ингибировал репликацию Энтеровируса при ЭК50 в 23 μг/мл [Wang Y., et al. In vitro assessment of combinations of enterovirus inhibitors against enterovirus 71. - 2016. - Antimicrob. Agents Chemother, 60(9), p. 5357-5367].
Caliciviridae.
Фавипиравир проявляет активность против мышиного норавируса с показателями ЭК50 от 39 μг/мл в исследованиях по подсчету вирусных бляшек в клеточной линии мышиных макрофагов RAW 264.7. ПЦР реального времени выявил блокирование синтеза РНК с помощью фавипиравира с ЭК50 от 19 μг/мл [Rocha-Pereira J., et al. Favipiravir (T-705) inhibits in vitro norovirus replication. - 2012. - Biochem. Biophys. Res. Commun, 424(4), p. 777-780].
Использование фавипиравира в in vivo моделях рода норавирусов из семейства Калицивирусов
В мышиной модели персистирующей инфекции оральное применение фавпиравира в дозировке 600 мг/кг/день двукратно в течение 8 недель, спустя 4 недели пост-инфицирования, привело к значительному снижению вирусных титров в испражнениях мышей и количестве норовирус-положительных мышей. Научные данные также подтверждают, что фавипиравир- РТФ ингибирует РНК-полимеразную активность Норавирусов человека [Jin Z., et al. Biochemical evaluation of the inhibition properties of Favipiravir and 2′-C-methyl-cytidine triphosphates against human and mouse norovirus RNA polymerases. - 2015 Antimicrob. Agents Chemother].
Filoviridae
Фавипиравир показал антивирусную активность против вируса Zaire Ebola (штамм Mayinga 1976) в культуре клеток Vero E6 с ЭК50 в 10.5 μг/мл. В линии мышей, зараженных штаммом Mayinga 1976 с отсутствием рецептора к интерферону-альфа (IFNAR-/- C57BL/6), оральное применение фавипиравира позволило избежать летального исхода и снижало вирусные титры в крови при двукратном применении от 300 мг/кг/день в течение 8 дней с 6 дня пост-инфекции, тогда как в плацебо-группе все мыши умерли [Oestereich L., et al. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. - 2014. - Antiviral Res, 105, p. 17-21]. Сходным образом, в линии A129, нокаутной по рецептору интерферона IFNAR-/-, зараженной штаммом Эбола E718, лечение фавипиравиром орально полностью спасло всех зараженных мышей от смерти при двукратном применении лекарства в дозе от 300 мг/кг/день в течение 14 дней, начиная с 1-ого часа после заражения [Smither S.J., et al. - Post-exposure efficacy of oral T-705 (Favipiravir) against inhalational Ebola virus infection in a mouse model. - 2014. - Antiviral Res, Volume 104, Pages 153-155].
При вспышке Эбола в западной Африке в 2014 году Французский институт Здоровья и Медицинских Исследований и правительство Гвинеи провели клиническое исследование фавипиравира на больных [Mentré F., Taburet A.M. Dose regimen of favipiravir for Ebola virus disease. 2015. - Lancet Infect. Dis, VOLUME 15, ISSUE 2, P.150-151]. Фавипиравир хорошо переносился пациентами и снижал количество смертельных исходов у пациентов с низкими вирусными титрами. Группа китайских исследователей также отметила увеличение выживаемости пациентов с Эбола при применении фавипиравира в Сьерра Леоне [Bai C.Q., Clinical and virological characteristics of Ebola virus disease patients treated with favipiravir (T-705)-Sierra Leone, 2016. Clin. Infect. Dis, 63(10), p.1288-1294].
Rhabdoviridae
Активность фавипиравира против вируса бешенства была выявлена на клеточной линии мышиной нейробластомы Neuro-2a с ЭК50s при 5.1-7.0 μг/мл [Yamada K., et al. Efficacy of Favipiravir (T-705) in rabies postexposure prophylaxis. - 2016. - J. Infect. Dis, 213(8), p. 1253-1261]. Фавипиравир значительно снизил показатели заболеваемости и смертности у мышей, зараженных RABV, при пероральном применении в дозах от 300 мг/кг/день двукратно в течение 7 дней, начиная с 1-ого часа пост-инфекции.
Coronavirus
Согласно глобальному отчету по фармсубстанциям, в настоящее время фавипиравир вошел в клинические испытания против заболевания COVID-19, исходя из доказанного механизма его действия против вирусной РзРп. В клиническом исследовании Национального Центра Клинических Исследований Инфекционных Заболеваний в Шеньжене, фавипиравир вводился 340 пациентам (возрастные группы и ко-морбидности не уточняются) в два приема (2 дозы) по 1600 мг в первый день и две дозы по 600 мг последующие 13 дней в дополнение к ингаляционному аэрозолю интерферона-альфа (5 млн.единиц* 2/день). Эта дозировка приводила к более быстрому исчезновению вируса до недетектируемых в крови значений), чем в группе пациентов, принимавших комбинацию анти-ВИЧ протеаз - лопинавира / ритонавира, с медианой вывода вирусных частиц в 4 дня, против 11, соответственно, и оценивалась по контролю КТ грудного отдела. По данным текущего исследования применение фавипиравира снижает количество детектируемых вирусных частиц в крови, а значит, доказательно ингибирует вирусную репликацию, отсрочивает развитие агрессивного сценария течения COVID-19 или вовсе предотвращает его.
Недавнее исследование показало эффективность фавипиравира против SARS-CoV-2 (ЭК50 = 61.88 μM в культуре клеток Vero E6) [Wang, M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. - 2020. - 30, p. 269-271]. Исследование проверяло сочетанное применение фавипиравира с интерфероном-α и фавипиравира с балоквавир марбоксилом (одобренным в показании для гриппа ингибитором кэп-зависимой эндонуклеазы) у пациентов с SARS-CoV-2.
Все описанные выше обобщенные данные по неоспоримой эффективности фавипиравира против очень широкого спектра РНК-вирусов показывают, что данное лекарственное средство прекрасно «закрывает» до сих пор эффективно неохваченную нишу неизлечимых, острых и смертельных вирусных недугов, включая большой спектр тропических лихорадок. До исследований на фавипиравире, против вышеозначенных РНК-инфекций применялись рибавирин и/или альфа-интерферон, однако первый обладает значимо меньшей противовирусной активностью и эффективностью против РНК-вирусов, и оба препарата при длительном применении приводят к дебилитирующим побочным эффектам.
Ввиду универсальности и высокой эффективности, продемонстрированной препаратами фавипиравира, представляется необходимым использовать их в периоды эпидемий.
Вспышка SARS-CoV-2 по всему миру и связанные с ней последствия являются угрозой для общественного здравоохранения и экономики многих стран. Отсутствие специальной терапии против нового вируса и его высокая изменчивость требует создания новых лекарственных средств.
В рамках борьбы с новой инфекцией был разработан ряд профилактических вакцин, а также средств неспецифической терапии, облегчающих течение заболевания. Однако в дополнение к существующим средствам терапии необходимы специфические препараты, оказывающие непосредственное действие на вирус, приводящие к облегчению симптомов заболевания, ускоренному разрешению заболевания, блокированию передачи инфекции и уменьшению риска развития клинических осложнений.
Фавипиравир, опираясь на широкий спектр и доказанный механизм его действия, также может облегчить течение вирусных заболеваний, при своевременном начале приема, а также значимо снизить вирусную нагрузку при течении заболеваний с осложнениями. Несмотря на высокую эффективность фавипиравира против вирусных заболеваний существует ряд трудностей, связанных с физико- химическими, технологическими и фармакологическими характеристиками лекарственных препаратов на основе фавипиравира в свободной форме, в частности, растворимость фавипиравира в свободной форме в воде крайне низкая, что осложняет производство новый лекарственных форм, при этом субстанция фавипиравира в свободной форме остается стабильной и проявляет приемлемые показатели, в том числе гигроскопичности и сыпучести, всего лишь около трех лет.
Также одной из проблем применения противовирусных препаратов, в том числе фавипиравира, является повышенный риск проявления гепатотоксических эффектов при терапии (Barbara Styrt MD, Joel P. Freiman MD. Hepatotoxicity of antiviral agents // Gastroenterology Clinics of North America. - 1995. - V. 24, I. 4, P. 839-852).
Именно поэтому на сегодняшний день весьма актуальна проблема поиска новых соединений эффективных против вирусных заболеваний.
Исходя из вышесказанного, представляется необходимым создание новых противовирусных соединений, обладающих улучшенным профилем безопасности, а также улученными физико-химическими, технологическими и фармакологическими свойствами.
Ранее исследователями были предприняты попытки получения соединений фавипиравира с различными веществами основной природы с целью улучшения их физико- химических свойств.
Из уровня техники известны производные фавипиравира с различными веществами основной природы, а именно, соединения фавипиравира с меглумином (CN 103209967, Toyama Chemical Co LTD) и натриевая соль фавипиравира (TW 201934539, Fujifilm Toyama Chemical Co LTD).
В качестве ближайшего аналога настоящего изобретения авторы рассматривают соль фавипиравира с аргинином (CN 111214446, Reyoung Pharmaceutical Co LTD et al.).
Задачей настоящего изобретения является создание новых противовирусных соединений, а именно производных фавипиравира, субстанции и лекарственной формы, которые обладают улучшенным профилем безопасности, а также улучшенными физико-химическими, технологическими и фармакологическими свойствами. Поставленная задача решается путем создания новых производных фавипиравира, в частности солей фавипиравира с аминокислотами, а именно, соединений формулы I:
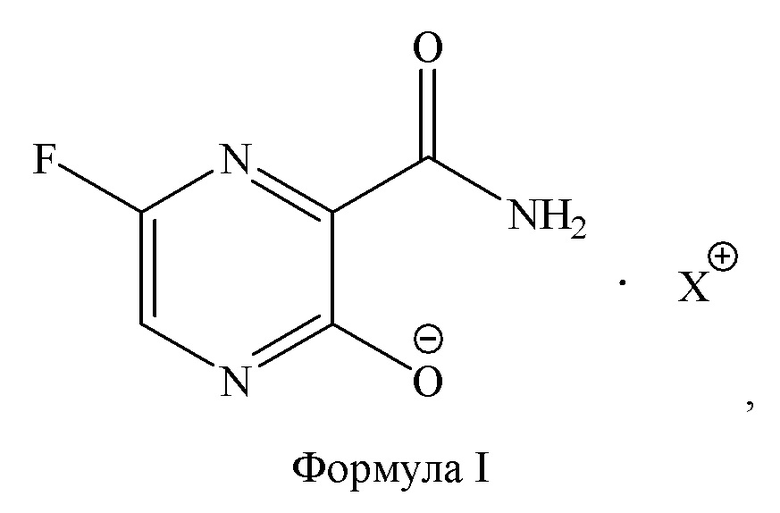
где X представляет собой остаток аминокислоты, выбранной из группы: лизин, орнитин или гистидин.
Техническими результатами настоящего изобретения являются:
- уменьшение риска повышения уровня печеночных трансаминаз во время терапии, ассоциированного с токсическим поражением печени;
- улучшение физико-химических и технологических свойств субстанции в виде новых производных фавипиравира по настоящему изобретению, фармацевтических композиций и лекарственных форм, которые их содержат.
Ниже приведены термины, которые используются в описании настоящего изобретения. Если не указано иное, все технические и специальные термины, использованные в описании, имеют общепринятое в данной области техники значение.
Термин "фармацевтическая композиция" обозначает композицию, включающую соль фавипиравира с аминокислотой в эффективном количестве, а также фармацевтическая композиция в контексте настоящего изобретения может дополнительно содержать один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых эксципиентов, таких как, не ограничиваясь указанным, наполнителей, солюбилизаторов, растворителей, со-растворителей, антиоксидантов, буферных агентов, криопротекторов, разбавителей, консервантов, стабилизаторов, увлажнителей, эмульгаторов, лубрикантов, скользящих веществ, суспендирующих агентов, загустителей, подсластителей, отдушек, ароматизаторов, антибактериальных агентов, фунгицидов, регуляторов пролонгированной доставки, изотонических агентов, агентов, регулирующих рН, выбор и соотношение которых зависит от природы, назначения и дозировки лекарственного средства.
Неограничивающими (иллюстративными) примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, а также смеси этих веществ.
Защита фармацевтической композиции от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, не ограничиваясь указанным, сорбиновая кислота, парабены (метилпарабен, пропилпарабен, этилпарабен, бутилпарабен, изобутилпарабен, изопропилпарабен, бензилпарабен, гептилпарабен и их смеси), хлорбутанол и подобные им соединения.
В качестве изотонических агентов фармацевтическая композиция может включать, не ограничиваясь указанным, сахара, хлорид натрия, гидрокарбонат натрия и др. Пролонгированное действие фармацевтической композиции может быть обеспечено, не ограничиваясь указанным, с помощью агентов, замедляющих абсорбцию активного агента (например, гидрофильные и гидрофобные полимерные замедлители высвобождения).
Неограничивающими (иллюстративными) примерами подходящих наполнителей являются лактоза, различные типы крахмала, микрокристаллическая целлюлоза, карбонат и фосфат кальция и др.
В качестве растворителей и разбавителей могут быть использованы, не ограничиваясь указанным, вода, пригодные для парентеральных форм органические сложные эфиры, этанол, полиспирты, а также их смеси. Примерами лубрикантов могут быть стеарат магния или кальция, тальк, лаурилсульфат натрия, стеарилфумарат натрия и др. В качестве скользящих веществ могут выступать диоксид кремния, тальк, каолин, бентониты и др. Для регулирования рН могут быть использованы различные органические и неорганические кислоты или щелочи, такие как, не ограничиваясь указанным, соляная кислота, яблочная, аскорбиновая, лимонная, уксусная, янтарная, винная, фумаровая, молочная, аспарагиновая, глутаровая, глутаминовая, сорбиновая кислоты, гидроксид натрия и т.д. Примерами диспергирующих агентов и распределяющих средств являются, не ограничиваясь указанным, крахмал, альгиновая кислота и ее соли, силикаты.
Фармацевтическая композиция может быть введена животным и людям перорально, парентерально (внутрикожно, подкожно, внутримышечно, внутривенно, внутриартериально, в полости), сублингвально, местно, в том числе не ограничиваясь указанным, глазное, назальное введение и др., ректально в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают (без ограничения) пероральные формы: таблетки, капсулы, пеллеты, гранулы, порошки, растворы, растворы для распыления в полости рта и носа, сиропы, суспензии и др., пероральные: растворы, суспензии, эмульсии, концентраты для приготовления инъекционных и инфузионных лекарственных форм, аэрозоли и порошки для ингаляционного введения, порошки и лиофилизаты для приготовления инъекционных и инфузионных лекарственных форм; ректальные: суппозитории, капсулы и др.; а также глазные капли.
Термин «эксципиент» в контексте настоящего изобретения характеризует вещества неорганического или органического происхождения, используемые в процессе производства, изготовления лекарственных препаратов для придания им необходимых физико-химических свойств.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении.
Термин «фармацевтически приемлемый» в контексте настоящего изобретения означает, что данное вещество или композиция, в отношении которых применяется этот термин, должны быть совместимы с точки зрения химии и/или токсикологии с другими ингредиентами, входящими в состав препарата, и безопасны для того, кого лечат этим веществом или композицией.
Термин «аминокислоты» в контексте настоящего изобретения означает органические вещества, молекулы которых одновременно содержат карбоксильные и аминные группы, которые могут существовать в виде изомеров и их смесей.
Термин «субстанция» в контексте настоящего изобретения означает продукт синтеза, представляющий собой любое вещество или смесь веществ синтетического или иного (биологического, биохимического, минерального, растительного, животного, микробного и прочего) происхождения, предназначенный для получения фармацевтических композиций и лекарственных средств, который в процессе производства лекарственного средства (препарата) становится активным ингредиентом этого лекарственного средства и определяет его эффективность. Такие вещества предназначены для проявления фармакологической активности или другого прямого эффекта при диагностике, лечении, облегчении симптомов, или профилактики болезни и облегчения симптомов, или для воздействия на структуру или функцию организма.
Термин «лекарственное средство» в контексте настоящего изобретения характеризует вещества или их комбинации, вступающие в контакт с организмом человека или животного, проникающие в органы, ткани организма человека или животного, применяемые для профилактики, лечения заболевания, полученные методами синтеза. К лекарственным средствам относятся лекарственные препараты. Лекарственное средство может быть представлено в виде различных готовых форм, предназначенных для введения в организм животного или человека различными способами, например, не ограничиваясь указанным, перорально, сублингвально, местно, ректально, парентерально.
Также для целей настоящего изобретения термины «содержащий», «содержит», «включающий» означают, что указанные комбинации, композиции и концентрат включают перечисленные компоненты, но не исключают включение других компонентов.
Термин «терапевтически эффективное количество» в контексте настоящего изобретения означает количество вещества (а также комбинации веществ), которое при ведении субъекту для лечения или предотвращения заболевания, или по меньшей мере одного из клинических симптомов заболевания, или нарушения является достаточным для воздействия такого лечения (профилактики) на заболевание, нарушение или симптом. «Терапевтически эффективное количество» может меняться в зависимости от формы вещества (например, полиморфная форма, соль, сольват, гидрат и т.д.), заболевания, нарушения и/или симптомов заболевания или нарушения, тяжести заболевания, нарушения и/или симптомов заболевания или нарушения, а также от возраста и/или веса субъекта, которому необходимо такое лечение (профилактика).
Термины «примерно», «приблизительно», «около» характеризуют плюс минус десять процентов от указанной величины.
Термин «свободная форма» вещества в контексте настоящего изобретения означает форму вещества, в которой молекулы данного вещества не взаимодействуют с другими молекулами и/или частицами, в частности катионами или анионами, лигандами или комплексообразователями посредством ионных или Ван-дер-Вальсовых взаимодействий, при этом атомы в данной молекуле связаны только ковалентными связями.
Термин «связанная форма» вещества в контексте настоящего изобретения означает форму вещества, в которой молекулы данного вещества взаимодействуют с другими молекулами и/или частицами, в частности катионами или анионами, лигандами или комплексообразователями посредством ионных или Ван-дер-Вальсовых взаимодействий. Примерами связанных соединений являются органические и неорганические соли и комплексы.
Термин «производные органических соединений» в контексте настоящего изобретения означает соединение, полученное из соединения аналогичного строения (структурного аналога) посредством химической реакции, если один или несколько атомов замещаются другими атомами (или группой атомов), в том числе, функциональными группами; или происходит присоединение к исходной молекуле остатков кислот или оснований, молекул растворителя или иных малых молекул посредством химических связей или межмолекулярных взаимодействий; или отщепление атомов или групп атомов с образованием кратных связей.
Термин «физические свойства лекарственных веществ» в контексте настоящего изобретения характеризует плотность, форму, размер и характер поверхности частиц, удельную поверхность частиц, силы адгезии (слипание на поверхности) и когезии (слипание частиц внутри тела), поверхностную активность, температуру плавления лекарственных веществ и т.д.
Термин «химические свойства лекарственных веществ» в контексте настоящего изобретения характеризует растворимость, стабильность, реакционную способность лекарственных веществ и др.
Термин «технологические свойства лекарственных веществ» в контексте настоящего изобретения характеризует объемную плотность, степень уплотнения, сыпучесть, влажность, фракционный состав, дисперсность, пористость, прессуемость лекарственных веществ и др.
краткое описание чертежей
Настоящее изобретение дополнительно проиллюстрировано посредством Фиг. 1 и Фиг. 2.
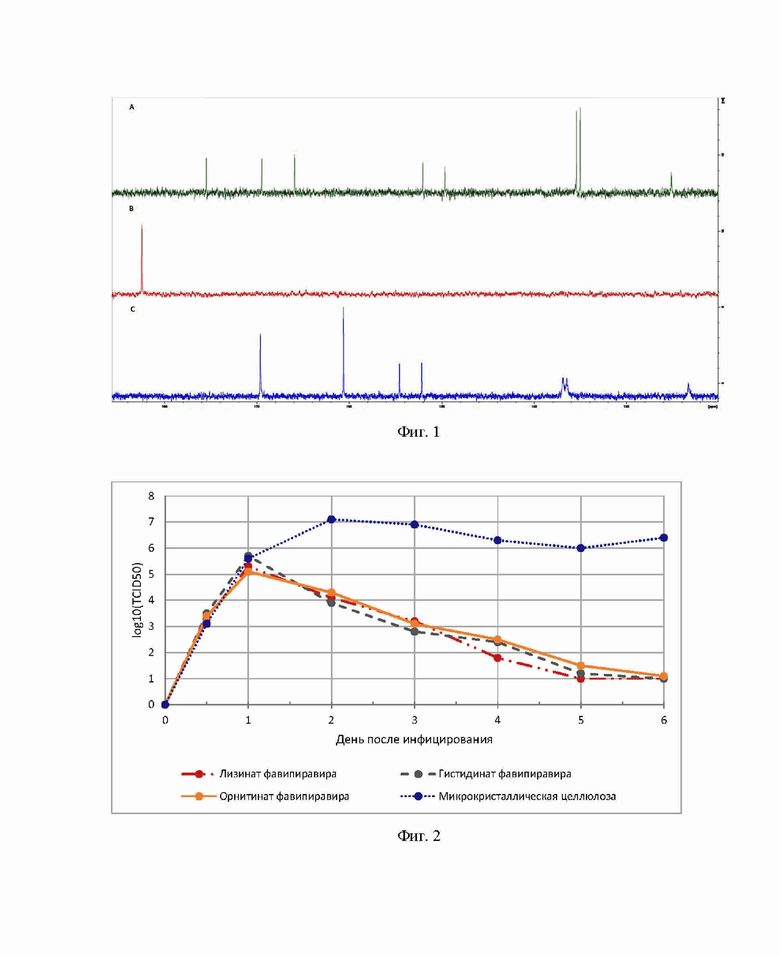
На Фиг. 1 изображены 13С ЯМР спектры лизината фавипиравира (А), лизина (B) и фавипиравира (C).
На Фиг. 2 показан титр вируса из назального лаважа мышей (введение препаратов осуществляли через 24 ч после инфицирования).
осуществление изобретения
Поставленная задача решается, а заявленный технический результат достигается получением новых солей фавипиравира с аминокислотами, в частности, лизината фавипиравира, орнитината фавипиравира и гистидината фавипиравира.
Более предпочтительно аминокислоты по настоящему изобретению представляют собой аминокислоты в L- или D- конфигурации.
Более предпочтительно аминокислота по настоящему изобретению представляет собой L-лизинат фавипиравира.
Более предпочтительно аминокислота по настоящему изобретению представляет собой L-орнитинат фавипиравира.
Более предпочтительно аминокислота по настоящему изобретению представляет собой L-гистидинат фавипиравира.
Более предпочтительно аминокислота по настоящему изобретению представляет собой D-лизинат фавипиравира.
Более предпочтительно аминокислота по настоящему изобретению представляет собой D-орнитинат фавипиравира.
Более предпочтительно аминокислота по настоящему изобретению представляет собой D-гистидинат фавипиравира.
Новые соединения по настоящему изобретению могут быть введены в форме пролекарств. Пролекарство может включать ковалентно связанный носитель, который высвобождает активное исходное лекарственное средство при введении субъекту-млекопитающему. Пролекарства могут быть получены путем модификации функциональных групп, присутствующих в соединениях, таким образом, что модификации расщепляются, либо в ходе обычных манипуляций, либо in vivo, до исходных соединений. Пролекарства включают, например, соединения, в которых гидроксильная группа связана с любой группой, которая при введении субъекту расщепляется с образованием свободной гидроксильной группы. Примеры пролекарств включают, без ограничений, ацетатные, формиатные и бензоатные производные спиртовых функциональных групп в соединениях.
Типичные пролекарства образуют активный метаболит путем превращения пролекарства гидролитическими ферментами, гидролиза амидов, лактамов, пептидов, сложных эфиров карбоновых кислот, эпоксидов или расщепления сложных эфиров неорганических кислот.
Для получения новых производных фавипиравира с аминокислотами по настоящему изобретению в качестве растворителей фавипиравира можно использовать диоксан, диметилсульфоксид, этиленгликоль, тетрагидрофуран, спирты (например, не ограничиваясь указанным, метанол, этанол, изопропанол), диметилацетамид, диметилформамид и др., а также их смеси в различных соотношениях.
Поставленная задача решается, а заявленный технический результат достигается получением фармацевтической композиции для лечения и/или профилактики вирусных заболеваний, содержащей в терапевтически эффективном количестве соединение, представляющее собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира.
Поставленная задача решается, а заявленный технический результат достигается получением фармацевтической композиции для лечения и/или профилактики вирусных заболеваний, содержащей в терапевтически эффективном количестве соединение, представляющее собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, и по меньшей мере один фармацевтически приемлемый эксципиент.
Одним из вариантов воплощения изобретения является фармацевтическая композиция, в которой количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 100 до 4000 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 100 до 3700 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 110 до 3300 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 120 до 3000 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 130 до 2700 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 140 до 2400 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 150 до 2100 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 160 до 1800 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 170 до 1500 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 180 до 1200 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 190 до 900 мг.
Более предпочтительно количество соединения, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, составляет от 200 до 600 мг.
Одним из вариантов воплощения изобретения является фармацевтическая композиция, в которой фармацевтически приемлемые эксципиенты выбраны из группы, включающей, не ограничиваясь указанным, формообразующие вещества, стабилизаторы, солюбилизаторы, растворители, увлажнители, лубриканты, скользящие вещества, загустители, подсластители, отдушки, ароматизаторы, фунгициды, регуляторы пролонгированной доставки, сорастворители, разбавители, наполнители, эмульгаторы, консерванты, антиоксиданты, буферные агенты, криопротекторы, регулирующие рН агенты, вещества для поддержания изотоничности или корригирующие вещества.
Более предпочтительно растворитель по настоящему изобретению представляет собой, не ограничиваясь указанным, физиологический раствор, инъекционную воду, апирогенную воду, дистиллированную воду, раствор Рингера, раствор Хартмана или раствор глюкозы.
Одним из вариантов воплощения изобретения является фармацевтическая композиция для лечения и/или профилактики вирусных заболеваний, где вирус представляет собой РНК- содержащий вирус.
Одним из вариантов воплощения изобретения является фармацевтическая композиция для лечения и/или профилактики вирусных заболеваний, где вирус представляет собой вирус, геном которого закодирован одноцепочечной смысловой (+)-нитью, а также антисмысловой (-)-нитью РНК и который использует вирусную РНК-зависимую-РНК-полимеразу для своей репликации.
Более предпочтительно вирус по настоящему изобретению представляет собой вирус гриппа, коронавирус, пикорнавирус, аренавирус, флавивирус, буньявирус, филовирус, флебовирус, хантавирус, энтеровирус, тогавирус, калицивирус, респираторно-синцитиальный вирус, вирус парагриппа, риновирусы, метапневмовирусы, ротавирус или норавирус.
Более предпочтительно вирус по настоящему изобретению представляет собой высоковирулентный или низковирулентный вирус.
Более предпочтительно вирус по настоящему изобретению представляет собой SARS-CoV, SARS-CoV-2, MERS-CoV или Influenza A, B, С.
Более предпочтительно вирус гриппа по настоящему изобретению представляет собой, не ограничиваясь указанным, вирус гриппа А, включая штаммы А (H1N1), А (H1N1) pdm09, А (H1N2), А (H3N2), A (H2N2), A (H4N2), A (H7N2), свиной грипп типа А, птичий грипп типа А, включая высокопатогенные штаммы (в том числе, H5N1 и H7N9).
Поставленная задача решается, а заявленный технический результат достигается за счет получения лекарственного средства для лечения и/или профилактики вирусных заболеваний, содержащего фармацевтическую композицию по настоящему изобретению.
Поставленная задача решается, а заявленный технический результат достигается за счет получения лекарственного средства для лечения и/или профилактики вирусных заболеваний, содержащего фармацевтическую композицию по настоящему изобретению и по меньшей мере один фармацевтически приемлемый эксципиент.
Поставленная задача решается, а заявленный технический результат достигается за счет получения лекарственного средства для лечения и/или профилактики вирусных заболеваний, содержащего фармацевтическую композицию по настоящему изобретению в терапевтически эффективном количестве.
Одним из вариантов воплощения изобретения является лекарственное средство, представляющее собой твердое лекарственное средство.
Более предпочтительно твердое лекарственное средство по настоящему изобретению представляет собой, не ограничиваясь указанным, таблетку, капсулу, пеллету, саше, драже, порошок или лиофилизат.
Лиофилизация (лиофильная сушка) представляет собой процесс удаления растворителя из замороженного материала путем возгонки (сублимации) кристаллов растворителя в условиях вакуума, т.е. превращения его в пар, минуя жидкую фазу. Лекарственное средство в форме лиофилизата полностью сохраняет свою фармакологическую активность.
Удаление растворителя при лиофилизационной сушке осуществляется главным образом за счет сублимации. Сублимация - это удаление растворителя из замороженного объекта без образования жидкой фазы, она проводится под вакуумом или значительно реже в инертном газе. Стадия замораживания является одной из определяющих стадий для получения качественного лекарственного средства в форме лиофилизата.
К вспомогательным эксципиентам, используемым в лиофильно высушенных лекарственных препаратах по настоящему изобретению, относятся: растворители, со-любилизаторы (ЭДТА, α-циклодекстрин и др.), наполнители (маннит, гли-цин, глюкоза, сахароза, лактоза, молоко и др.), консерванты (бензиловый спирт, этил- и метилпарагидроксибензоат и др.), регуляторы рН (буферные растворы, натрия гидроксид, хлористоводородная кислота), стабилизаторы, криопротекторы (декстран, желатин, гидроксиэтилкрахмал и др.).
В форме лиофилизатов по настоящему изобретению могут быть представлены как индивидуальные лекарственные вещества, так и их смеси с вспомогательными веществами.
Одним из вариантов воплощения изобретения является лекарственное средство, представляющее собой пероральное лекарственное средство.
Более предпочтительно пероральное лекарственное средство по настоящему изобретению представляет собой, не ограничиваясь указанным, таблетку, капсулу, пеллету, драже, порошок, суспензию, сироп или раствор.
Одним из вариантов воплощения изобретения является лекарственное средство, представляющее собой жидкое лекарственное средство.
Более предпочтительно жидкое лекарственное средство по настоящему изобретению представляет собой, не ограничиваясь указанным, раствор, концентрат, сироп, суспензию.
Более предпочтительно жидкое лекарственное средство по настоящему изобретению представляет собой парентеральное лекарственное средство.
Парентеральное введение лекарственных средств - это такие пути введения лекарственных средств в организм, при которых они минуют желудочно-кишечный тракт, в отличие от перорального способа применения лекарств. Парентеральные лекарственные средства- это стерильные препараты, предназначенные для введения путем инъекций, инфузий, ингаляций или имплантаций в организм человека или животного. К ним относятся растворы, эмульсии, суспензии, аэрозоли, порошки и таблетки для получения растворов и имплантации, порошки для ингаляций, лиофилизированные препараты для получения лекарственных форм, вводимых в организм парентерально (подкожно, внутримышечно, внутривенно, внутриартериально, в различные полости).
Парентеральные пути введения включают введение в ткани (внутрикожно, подкожно, внутримышечно, внутрикостно), в сосуды (внутривенно, внутриартериально, в лимфатические сосуды), в полости (в плевральную, брюшную, сердечную и суставную полости), в субарахноидальное пространство, а также ингаляционное, интраназальное и субконъюнктивальное введение.
Более предпочтительно парентеральное лекарственное средство представляет собой инфузионный раствор.
Более предпочтительно парентеральное лекарственное средство представляет собой инъекционный раствор.
Одним из вариантов воплощения изобретения является лекарственное средство, представляющее собой ингаляционное лекарственное средство.
Более предпочтительно ингаляционное лекарственное средство по настоящему изобретению представляет собой, не ограничиваясь указанным, аэрозоль, порошок или пульмопорошок.
Одним из вариантов воплощения изобретения является лекарственное средство, представляющее собой лекарственное средство для местного введения.
Под местным введением лекарственных средств понимается нанесение лекарственного препарата на слизистые оболочки (в т.ч. глазное, назальное, ректальное, вагинальное применение, нанесение на десны, слизистую оболочку полости рта и др.), а также введение в наружный слуховой проход.
Более предпочтительно местное лекарственное средство по настоящему изобретению представляет собой лекарственное средство для глазного, назального и ректального введения.
Одним из вариантов воплощения изобретения является лекарственное средство, представляющее собой ректальное лекарственное средство.
Более предпочтительно ректальное лекарственное средство по настоящему изобретению представляет собой, не ограничиваясь указанным, суппозитории или капсулы.
Одним из вариантов воплощения изобретения является лекарственное средство, в котором фармацевтически приемлемые эксципиенты выбраны из группы, включающей, не ограничиваясь указанным, формообразующие вещества, стабилизаторы, солюбилизаторы, растворители, увлажнители, лубриканты, скользящие вещества, загустители, подсластители, отдушки, ароматизаторы, фунгициды, регуляторы пролонгированной доставки, сорастворители, разбавители, наполнители, эмульгаторы, консерванты, антиоксиданты, буферные агенты, криопротекторы, регулирующие рН агенты, вещества для поддержания изотоничности или корригирующие вещества.
Более предпочтительно растворитель по настоящему изобретению представляет собой, не ограничиваясь указанным, физиологический раствор, инъекционную воду, апирогенную воду, дистиллированную воду, раствор Рингера или раствор глюкозы.
Поставленная задача решается, а заявленный технический результат достигается за счет применения новых соединений по настоящему изобретению, представляющих собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, для получения фармацевтической композиции для лечения и/или профилактики вирусных заболеваний.
Поставленная задача решается, а заявленный технический результат достигается за счет применения фармацевтической композиции по настоящему изобретению для получения лекарственного средства для лечения и/или профилактики вирусных заболеваний.
Поставленная задача решается, а заявленный технический результат достигается за счет применения новых соединений по настоящему изобретению, представляющих собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, для лечения и/или профилактики вирусных инфекций.
Поставленная задача решается, а заявленный технический результат достигается за счет применения фармацевтической композиции или лекарственного средства для лечения и/или профилактики вирусных инфекций.
Более предпочтительно вирусная инфекция по настоящему изобретению представляет собой SARS-CoV, SARS-CoV-2, MERS-CoV или Influenza A, B С.
Поставленная задача решается, а заявленный технический результат достигается за счет способа лечения и/или профилактики вирусных заболеваний, включающего введение пациенту, нуждающемуся в таком лечении, нового соединения по настоящему изобретению, представляющего собой лизинат фавипиравира, орнитинат фавипиравира или гистидинат фавипиравира, в терапевтически эффективном количестве.
Поставленная задача решается, а заявленный технический результат достигается за счет способа лечения и/или профилактики вирусных заболеваний, включающего введение пациенту, нуждающемуся в таком лечении, фармацевтической композиции по настоящему изобретению, в терапевтически эффективном количестве.
Поставленная задача решается, а заявленный технический результат достигается за счет способа лечения и/или профилактики вирусных заболеваний, включающего введение пациенту, нуждающемуся в таком лечении, лекарственного средства по настоящему изобретению, в терапевтически эффективном количестве.
Далее приводятся примеры осуществления изобретения, которые иллюстрируют изобретение, но не охватывают все возможные варианты его осуществления и не ограничивают изобретение.
Специалисту в данной области понятно, что возможны и другие частные варианты осуществления изобретения.
Примеры
Пример 1. Получение новых соединений по настоящему изобретению
Общая методика получения новых соединений по настоящему изобретению, а именно, соединений общей формулы I:
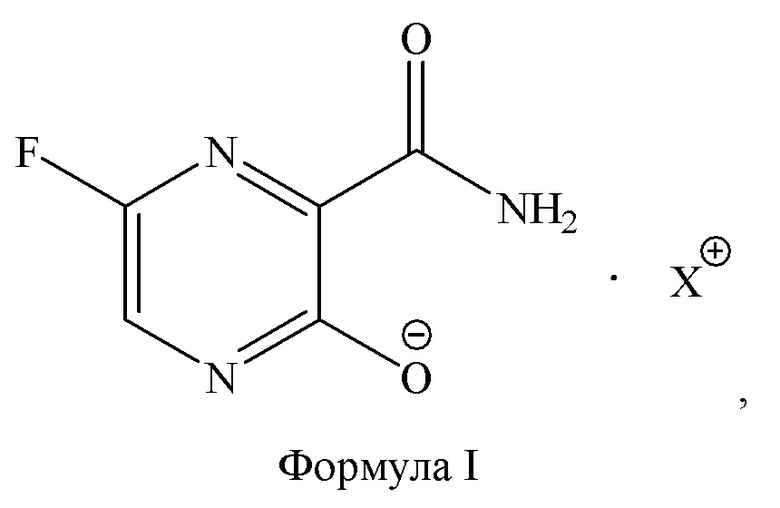
где X представляет собой остаток аминокислоты, выбранной из: лизина, орнитина гистидина, триптофана, гидроксилизина или лейцина.
Реакционную емкость заполняли дистиллированной водой и термостатировали при температуре 40-45°C. Далее при перемешивании в воду постепенно добавляли рассчитанное количество аминокислоты. Температуру реакционной среды повышали до 60-70°C и добавляли в раствор рассчитанное количество смеси фавипиравира с диметилформамидом при условии, что общий объем указанной смеси не превышал 5 - 15 % от объема раствора аминокислоты. Добавление фавипиравира происходило поэтапно, а именно четыре порции в массовом соотношении 4:2:1:0,5. При этом после добавления третьей порции смеси фавипиравира с диметилформамидом реакционную емкость выдерживали в ультразвуковой бане на протяжении 5-10 минут, после чего добавляли четвертую порцию смеси. Затем температуру реакционной среды повышали до 75-80°С. Раствор выдерживали при перемешивании и постоянной температуре, затем удаляли растворитель из реакционной массы выпариванием при пониженном давлении. Температура в процессе выпаривания не превышала 50°C. В результате был получен мелкодисперсный порошок соли фавипиравира с указанными аминокислотами.
Идентификация новых соединений по настоящему изобретению:
Лизинат фавипиравира
ЯМР 1H (D2O, 300 МГц) δ (м.д.): 1,56 - 1,34 (m, 2H), 1,69 (p, J = 7,62 Гц, 2H), 1,95 - 1,80 (m, 2H), 3,00 (t, J = 7,60 Гц, 1H), 3,73 (t, J = 6,08 Гц, 2H), 8,01 (d, J = 8,08 Гц). ЯМР 13C δ (м.д.): 22,10, 27,06, 30,64, 39,71, 55,18, 125,16, 135,23 (d, J = 41,18 Гц), 150,85 (d, J = 235,52 Гц), 165, 94, 169,49, 175,50. Погрешность измерения химических сдвигов составляет 0,1 м.д., констант спин-спинового взаимодействия 0,3 Гц.
Элементный анализ C11H18FN5O4: рассчитано C 43,56%, H 5,98 %, F 6,26%, N 23,09%, O 21,10%; найдено C 43,68%, H 6,09 %, F 6,41%, N 22,96%, O 21,37%.
Сопоставление 13С ЯМР спектров лизината фавипиравира (А), лизина (B) и фавипиравира (C) представлено на Фиг.1.
В результате анализа проведенных экспериментов 13C ЯМР-спектроскопии лизина, фавипиравира и лизината фавипиравира была подтверждена структура лизината фавипиравира. В частности, образование лизиновой соли подтверждалось слабопольными сдвигами сигналов С(4) и C(5) фавипиравира с 122,8 на 125,1 м.д., с 160,2 на 165,9 м.д. соответственно, и сильнопольными сдвигами сигналов фавипиравира при C(2) и C(1) с 136,2 м.д. на 135,2 м.д., с 152,9 на 150,1 м.д соответственно. Также было обнаружено смещение всех сигналов в 13C ЯМР спектре лизина в сильное поле.
Орнитинат фавипиравира
ЯМР 1H (D2O, 300 МГц) δ (м.д.): 1,54 - 1,81 (m, 2H), 1,85 - 2,03 (m, 2H), 2,54 - 2,94 (m, 2H), 3,74 - 3,87 (m, 2H), 7,80 (d, J = 7,7 Гц, 1H), 7,90 (t, J = 7.2 Hz, 3H). ЯМР 13C δ (м.д.): 22,3, 27,4, 30,9, 40,0, 55,4, 137,9, 140,5, 143,1, 159,1, 161,9, 178,0. Погрешность измерения химических сдвигов составляет 0,1 м.д., констант спин-спинового взаимодействия 0,3 Гц.
Элементный анализ C10H18FN5O4: рассчитано C 41,52%, H 5,58 %, F 6,57%, N 24,21%, O 22,12%; найдено C 41,40%, H 5,64 %, F 6,71%, N 24,39%, O 21,97%.
Гистидинат фавипиравира:
ЯМР 1H (D2O, 300 МГц) δ (м.д.): 3.53 - 3.71 (m, 2H), 4,06 - 4,23 (m, 1H), 4,51 (d, J = 15,8 Гц, 2H), 7.80 (s, 1H), 8.79 - 8.94 (m, 1H). ЯМР 13C δ (м.д.): 38,1, 50,3, 50,1, 53,8, 138,4, 140,6, 143,1, 158,9, 161,3, 164,5, 168,5, 171,8. Погрешность измерения химических сдвигов составляет 0,1 м.д., констант спин-спинового взаимодействия 0,3 Гц.
Элементный анализ C11H13FN6O4: рассчитано C 42,31%, H 4,20 %, F 6,08%, N 26,91%, O 20,49%; найдено C 42,04%, H 4,51 %, F 5,90%, N 26,58%, O 20,75%.
Гидроксилизинат фавипиравира:
ЯМР 1H (D2O, 300 МГц) δ (м.д.): 1,55 - 2,07 (m, 4H), 2,80 - 3,04 (m, 1H), 3,58 - 4,02 (m, 2H), 5,28 (d, J = 7,3 Гц, 1H), 7,71 - 7,95 (m, 4H), 8,03 (t, J = 6,9 Гц, 1H). ЯМР 13C δ (м.д.): 26,1, 32,8, 50,1, 53,8, 74,2, 135,9, 142,7, 146,6, 163,0, 165,3, 179,1.
Элементный анализ C16H16FN5O4: рассчитано C 41,38%, H 5,68 %, F 5,95%, N 21,93%, O 25,05%; найдено C 41,44%, H 5,61 %, F 5,94%, N 21,87%, O 25,12%.
Лейцинат фавипиравира:
ЯМР 1H (D2O, 300 МГц) δ (м.д.): 0,90 (d, J = 6,6 Гц, 6H), 1,67 - 2,24 (m, 3H), 3,83 - 4,09 (m, 1H), 7,59 - 7,97 (m, 3H), 10,2 (s, 1H). ЯМР 13C δ (м.д.): 22,0, 22,3, 40,5, 58,1, 136,5, 142,7, 145,2, 160,9, 163,8, 172,4.
Элементный анализ C16H16FN5O4: рассчитано C 45,83%, H 5,94 %, F 6,59%, N 19,44%, O 22,20%; найдено C 45,91%, H 5,86 %, F 6,64%, N 19,50%, O 22,16%.
Триптофанат фавипиравира:
Анализ ЯМР 1H, 13C и элементный анализ не показали сигналов продукта триптофаната фавипиравира.
При этом в ряде экспериментов отмечались улучшенные характеристики новых субстанций в виде производных фавипиравира по настоящему изобретению, а именно, улучшение растворимости, стабильности (в том числе фотостабильности, ускоренной и долгосрочной стабильности), уменьшение гигроскопичности, улучшение прессуемости с сохранением хороших показателей сыпучести без заметных электростатических явлений.
Пример 2. Получение лекарственных форм новых производных фавипиравира
Инъекции
Для приготовления раствора для инъекций новых производных фавипиравира по настоящему изобретению предварительно в стерильных условиях в емкость для приготовления растворов помещали воду для инъекций. Далее в емкость добавляли рассчитанное количество соли фавипиравира по настоящему изобретению и перемешивали до полного растворения активного агента при температуре 50-70°С до получения прозрачной фармацевтической композиции в виде раствора. Далее, при необходимости, корректировали значение рН, фильтровали через мембранный фильтр и фильтрат помещали в стерильные емкости и плотно укупоривали.
Таблетки
Для получения твердого лекарственного средства в форме таблетки, содержащего новые производные фавипиравира в качестве активного агента, первоначально приготовили 10 % раствор увлажнителя. Далее в миксер гранулятор загрузили последовательно низкозамещенную гипролозу, соль фавипиравира по настоящему изобретению и половину микрокристаллической целлюлозы. Полученную смесь перемешивали мешалкой в течение 5 минут с последующим добавлением увлажнителя. Процесс перемешивания продолжали до полного внесения увлажнителя.
Полученную фармацевтическую композицию гранулировали с последующей сушкой гранул в установке кипящий слой. Далее гранулы калибровали через сито с размером ячеек 1 мм.
Для получения готовой фармацевтической композиции для таблетирования, полученный гранулят смешивали с оставшейся половиной микрокристаллической целлюлозы, кросповидоном и кремнием диоксидом коллоидным в гравиметрическом смесителе. Полученную фармацевтическую композицию опудривали стеариновой кислотой с последующим таблетированием на роторном таблетпрессе.
Пример 3. Исследование биохимических показателей крови, связанных с поражением печени
Для подтверждения эффекта снижения риска повышения уровня печеночных трансаминаз посредством использования новых производных фавипиравира, а именно солей фавипиравира с аминокислотами, выбранными из группы: лизин, гидроксилизин, орнитин, лейцин или гистидин, был проведен эксперимент на лабораторных животных. В качестве критерия для оценки положительного влияния представленной композиции были исследованы биохимические показатели крови, такие как уровни аланин-и аспартат-аминотрансфераз, щелочной фосфатазы и билирубина, повышение которых в большинстве случаев связано с поражением печени.
Исследования проводили на 60 половозрелых мышах, которые были разделены на четыре группы по десять особей в каждой. Животных распределяли по группам, используя в качестве критерия массу тела, так, чтобы индивидуальная масса животных не отличалась более чем на 10% от средней массы животных.
Вводимые препараты:
- 1-я группа: препарат лизината фавипиравира,
- 2-я группа: препарат орнитината фавипиравира;
- 3-я группа: препарат гистидината фавипиравира;
- 4-я группа: препарат гидроксилизината фавипиравира;
- 5-я группа: препарат лейцината фавипиравира;
- 6-я группа: препарат аргинината фавипиравира, полученный в соответствии со способом, описанном в источнике CN111214446 (ближайший аналог).
Дозирование препарата животным осуществляли по следующей схеме: ежедневно особям из 1, 2, 3, 4, 5 и 6 групп перорально вводили 1500 мг препарата в пересчете на фавипиравир в свободном виде (дозировка приведена в пересчете на человека) при помощи зонда в течение 10 дней.
На 11-е сутки эксперимента животных подвергали одномоментной декапитации с учетом требований этического комитета.
Биохимическому исследованию подвергалась сыворотка, отделяемая центрифугированием цельной крови, собираемой в момент декапитации животных. В ней определяли уровень ферментов аспарагиновой (АсАТ) и аланиновой (АлАТ) аминотрансфераз, щелочной фосфотазы (ЩФ) и билирубина.
Полученные результаты представлены в Табл. 1.
Как видно из приведенных данных биохимических показателей крови мышей, при введении лизината фавипиравира, орнитината фавипиравира и гистидината фавипиравира не наблюдается статистически значимых ухудшений биохимических показателей крови, вызванных токсическим поражением клеток печени, в отличии от новых производных фавипиравира, таких как гидроксилизинат фавипиравира и лейцинат фавипиравира, а также аргинината фавипиравира по прототипу.
Таким образом новые соли фавипиравира с аминокислотами, а именно, лизинат фавипиравира, орнитинат фавипиравира и гистидинат фавипиравира, неожиданно имеют преимущество перед впервые синтезированными новыми солями фавипиравира, а именно гидроксилизинатом фавипиравира и лейцинатом фавипиравира, а также известным в уровне техники аргининатом фавипиравира с точки зрения безопасности использования.
Пример 4. Изучение терапевтического эффекта новых производных фавипиравира против вируса гриппа
Моделирование вирусной инфекции у мышей проводили путем интраназального введения единиц высокопатогенного вируса гриппа H1N1. Эксперимент проводили на 40 половозрелых мышах. Животных содержали в стандартных условиях вивария (они получали, в том числе, стерильный корм для грызунов и стерильную воду).
В ходе эксперимента ежедневно оценивали интегральные показатели (изменение веса животных) и оценивали клиническую симптоматику (лихорадка, слизистые выделения, уровень активности, дыхание).
Животных разделили на четыре группы по 10 особей.
Вводимые препараты:
- 1-я группа: препарат лизината фавипиравира;
- 2-я группа: препарат орнитината фавипиравира;
- 3-я группа: препарат гистидината фавипиравира;
- 4-я группа: микрокристаллическая целлюлоза (плацебо).
Лечение начинали спустя 24 ч после инфицирования. Препараты вводили перорально в виде суспензии через зонд в дозировке 400 мг 4 раза в день в течении 5 дней (дозировки приведены в пересчете на фавипиравир в свободном виде в пересчете на человека). Аналогично четвертая группа получала микрокристаллическую целлюлозу в виде суспензии через зонд 4 раза в день.
На 6 день животных умерщвляли. Для титрования вирусов использовали монослойную культуру чувствительных клеток Vero E6 выращенную в 96-луночных планшетах. Предварительно были приготовлены 10-кратные разведения плазмы на питательной среде. Инфицированные клетки инкубировали в течение 7 суток. Оценку наличия вируса проводили по характерным вирусиндуцированным изменениям клеточной морфологии. Титр вируса определяли в тканевых цитопатических дозах (ТЦПД/50).
Титры вируса в назальном лаваже оценивали ежедневно на протяжении всего эксперимента. Усредненные результаты представлены Фиг .2.
Результаты эксперимента наглядно демонстрируют значительное снижение титра вируса уже на 2 день эксперимента. При этом за время лечения у особей в группах 1-3 не наблюдалось существенного изменения в весе, при этом смертность на протяжении всего эксперимента в группах 1-3 была нулевая. Напротив, в группе 4 (группа контроля) к концу эксперимента смертность составляла 80 %.
Таким образом, новые производные фавипиравира в виде фармацевтически приемлемых солей фавипиравира с аминокислотами, а именно, лизинат фавипиравира, орнитинат фавипиравира и гистидинат фавипиравира, проявляют высокоэффективную противовирусную активность.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО, СОДЕРЖАЩИЕ НОВЫЕ ПРОИЗВОДНЫЕ ФАВИПИРАВИРА | 2023 |
|
RU2832326C1 |
| ПРОТИВОВИРУСНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ФАВИПИРАВИР И АМИНОКИСЛОТУ, ЛЕКАРСТВЕННОЕ СРЕДСТВО, ЕЕ СОДЕРЖАЩЕЕ, А ТАКЖЕ ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ВИРУСНЫХ ИНФЕКЦИЙ | 2021 |
|
RU2783282C1 |
| ПРОТИВОВИРУСНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ФАВИПИРАВИР И АМИНОКИСЛОТУ, ЛЕКАРСТВЕННОЕ СРЕДСТВО, ЕЕ СОДЕРЖАЩЕЕ, А ТАКЖЕ ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ВИРУСНЫХ ИНФЕКЦИЙ | 2022 |
|
RU2839470C2 |
| ПРОТИВОВИРУСНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ФАВИПИРАВИР И АМИНОКИСЛОТУ, ЛЕКАРСТВЕННОЕ СРЕДСТВО, ЕЕ СОДЕРЖАЩЕЕ, А ТАКЖЕ ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ВИРУСНЫХ ИНФЕКЦИЙ | 2022 |
|
RU2814927C1 |
| ПРОТИВОВИРУСНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ФАВИПИРАВИР И ГИДРАТ АМИНОКИСЛОТЫ И/ИЛИ ЕЕ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ СОЛИ | 2021 |
|
RU2789612C1 |
| Противовирусная композиция | 2020 |
|
RU2751108C1 |
| ПРОТИВОВИРУСНАЯ КОМПОЗИЦИЯ | 2020 |
|
RU2740660C1 |
| ПРОТИВОВИРУСНАЯ КОМПОЗИЦИЯ | 2020 |
|
RU2740657C1 |
| Противо-COVID-19 (SARS-CoV-2) вирусная фармацевтическая композиция | 2020 |
|
RU2731932C1 |
| Противо-РНК вирусное, в том числе противокоронавирусное средство - замещенный хиноксалин, фармацевтическая композиция и применения | 2020 |
|
RU2744429C1 |
Изобретение относится к области химико-фармацевтической промышленности и медицины, а именно к способу получения соли фавипиравира с аминокислотами. Способ включает взаимодействие аминокислоты, выбранной из группы, включающей лизин, орнитин, гистидин, гидроксилизин и лейцин, с фавипиравиром, при условии, что в водный раствор аминокислоты, нагретый до 60-70°С, добавляют смесь фавипиравира с диметилформамидом в количестве 5-15 % от объема раствора аминокислоты поэтапно четырьмя порциями в массовом соотношении 4:2:1:0,5, с дальнейшим выпариванием реакционной смеси при пониженном давлении при температуре не выше 50°С. Технический результат изобретения заключается в получении новых противовирусных соединений, а именно соли фавипиравира, которые обладают улучшенным профилем безопасности, а также улучшенными физико-химическими, технологическими и фармакологическими свойствами. 3 з.п. ф-лы, 2 ил., 1 табл., 4 пр.
1. Способ получения соли фавипиравира с аминокислотами, который включает взаимодействие аминокислоты, выбранной из группы, включающей лизин, орнитин, гистидин, гидроксилизин и лейцин, с фавипиравиром, при условии, что в водный раствор аминокислоты, нагретый до 60-70°С, добавляют смесь фавипиравира с диметилформамидом в количестве 5-15 % от объема раствора аминокислоты поэтапно четырьмя порциями в массовом соотношении 4:2:1:0,5, с дальнейшим выпариванием реакционной смеси при пониженном давлении при температуре не выше 50°С.
2. Способ по п. 1, в котором аминокислота представляет собой аминокислоту в L- или D-конфигурации.
3. Способ по п. 1, в котором после добавления третьей порции смеси фавипиравира с диметилформамидом реакционную емкость выдерживают в ультразвуковой бане на протяжении 5-10 минут.
4. Способ по п. 1, в котором температуру реакционной среды после внесения всего объема смеси фавипиравира с диметилформамидом повышают до 75-80°С.
| CN 111214446 A, 02.06.2020 | |||
| TILBORG A | |||
| et al., Pharmaceutical salts and cocrystals involving amino acids: A brief structural overview of the state-of-art, European Journal of Medicinal Chemistry, 2014, vol.74, p.411-426 | |||
| CN 103209967 B, 08.04.2015 | |||
| TW 201934539 A, 01.09.2019 | |||
| CN 112574130 A, 30.03.2021 | |||
| CN 111875550 A, 03.11.2020 | |||
| ПРОТИВОВИРУСНАЯ КОМПОЗИЦИЯ | 2020 |
|
RU2740660C1 |

