ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка испрашивает приоритет на основании заявки на патент Кореи № 10-2018-0100359, поданной 27 августа 2018 года, и заявки на патент Кореи № 10-2019-0104641, поданной 26 августа 2019 года в Корейское агентство по интеллектуальной собственности, каждая из которых включена в настоящий документ посредством ссылки в полном объеме.
Настоящее изобретение относится к новому производному гетероциклического амина, подходящему в качестве ингибитора BTK (тирозинкиназы Брутона), и к содержащей его фармацевтической композиции.
УРОВЕНЬ ТЕХНИКИ
ITK (интерлейкин-2-индуцибельная тирозинкиназа) и BTK (тирозинкиназа Брутона) и представляют собой тип тирозинкиназ, наряду с Tec (тирозинкиназой, экспрессируемой при гепатоцеллюлярной карциноме), RLK (киназой покоящихся лимфоцитов) и BMX (ген тирозинкиназы костного мозга на X-хромосоме), который не имеет рецептора TEC-семейства и действует на различные иммунные ответы.
ITK экспрессируется не только в T-клетках, но также в NK-клетках и тучных клетках и играет важную роль в пролиферации T-клеток и продуцировании важных цитокинов, таких как IL-2, IL-4, IL-5, IL-10, IL-13 и IL-17 (Schaeffer et al. Nat. Immune 2001,2, 1183; Fowell et al. Immunity, 1999, 11, 399). Т-клетки активируются посредством TCR-сигналинга, и активированные Т-клетки продуцируют воспалительный цитокин и активируют В-клетки и макрофаги, вызывая аутоиммунные заболевания, такие как RA (Sahu N. et al. Curr Top Med Chem. 2009, 9, 690). Ранее было известно, что Т-клетки активируются в Th1-клетки, индуцируя RA заболевания, но недавно было показано, что не только Th17/Treg, но также и Th1-клетки играют роль в патогенезе RA (J Leipe J. et al. Arthritis Rheum. 2010, 62, 2876). Кроме того, ITK ранее была разработана в качестве иммунотерапевтической лекарственной мишени, например, при астме, но ITK не была разработана в качестве терапевтического средства для RA (Lo H. Y Expert Opin Ther Pat. 2010, 20, 459). Однако недавно сообщалось, что она регулирует развитие Th17- и Treg-клеток через ITK−/− у мышей и обладает достаточным потенциалом в качестве терапевтической мишени при RA (Gomez-Rodriguez J. et al. J. Exp. Med. 2014, 211, 529).
В исследовании с использованием ингибитора ITK PRN694 сообщалось о снижении уровня TNF-α, который является типичным воспалительным цитокином при заболеваниях RA, что подтверждает возможность разработки в качестве терапевтического агента путем регулирования экспрессии Th17 через ингибирование ITK (Zhong Y. et al. THE JOURNAL OF BIOLOGICAL CHEMISTRY 2015, 290, 5960).
BTK действует как регулятор развития ранних B-клеток, а также активации, передачи сигналов и выживания зрелых B-клеток. B-клетка передает сигнал с помощью B-клеточного рецептора (BCR), который распознает антиген, присоединенный к поверхности антиген-презентирующей клетки, и активируется в зрелую антитело-продуцирующую клетку. Однако аберрантная передача сигналов через BCR приводит к аномальной пролиферации B-клеток и образованию патологических аутоантител и, тем самым, может вызывать рак, аутоиммунные и/или воспалительные заболевания. Таким образом, при аномальной пролиферации B-клеток передача сигналов через BCR может блокироваться при дефиците BTK. В связи с этим, ингибирование BTK может блокировать опосредованные B-клетками процессы заболевания, и применение ингибиторов BTK может быть полезным подходом для лечения заболеваний, опосредованных B-клетками.
Кроме того, BTK может экспрессироваться другими клетками, которые могут быть связаны с заболеванием, кроме B-клеток. Например, BTK является важным компонентом для передачи сигналов Fc-гамма в клетках костного мозга и экспрессируется тучными клетками. В частности, индуцированные тучные клетки костного мозга с дефицитом BTK демонстрируют нарушенную антиген-индуцированную дегрануляцию, и известно, что ингибирование активности BTK полезно для лечения патологических откликов тучных клеток, таких как аллергия и астма (Iwaki et al. J. Biol Chem. 2005 280: 40261). Кроме того, известно, что моноциты у пациентов XLA, у которых отсутствует активность BTK, снижают продуцирование TNF-альфа после стимулирования и, следовательно, опосредованное TNF-альфа воспаление может быть ингибировано ингибиторами BTK (см., Horwood et al., J. Exp. Med. 197: 1603, 2003).
В настоящее время отсутствуют случаи его разработок в качестве вещества, которое двойственно ингибирует BTK и ITK. Однако, в качестве ингибитора BTK в WO 2008/039218 описаны производные 4-аминопиразоло[3,4-d]пиримидинилпиперидина, и в WO 2015/061247 описаны гетеросоединения, такие как пиридин, пиримидин, пиразин и пиридазин, и в WO 2014/055934 описаны производные пиримидинилфенилакриламида. В качестве ингибитора ITK в WO 2005/066335 описаны аминобензимидазолы, в WO 2005/056785 описаны пиридоны, в WO 2002/050071 описаны производные аминотиазола, и недавно в WO 2014/036016 описаны производные бензимидазола.
Учитывая вышесказанное, в результате изучения новых соединений авторы настоящего изобретения обнаружили, что соединение, имеющее химическую структуру, отличную от ингибиторов BTK, ITK, о которых до сих пор сообщалось, обладает превосходным двойным ингибирующим действием активности BTK и ITK, в чем и заключается настоящее изобретение. Соединения, относящиеся к настоящему изобретению, в основном обладают собственной ингибирующей активностью в отношении BTK и ITK, но не исключают возможности проявления фармакологического действия в качестве эффективного агента со стороны специальной среды организма или продуктов метаболического процесса, после поглощения в организме.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Техническая задача
Задачей настоящего изобретения является предложить новое производное гетероциклического амина, подходящее в качестве ингибитора BTK, и содержащую его фармацевтическую композицию.
Техническое решение
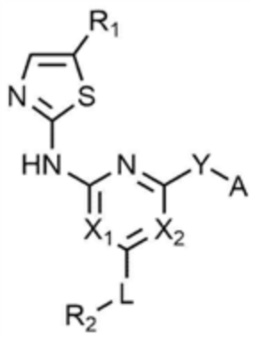
Для достижения указанных выше целей предлагается соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль:
[Химическая формула 1]
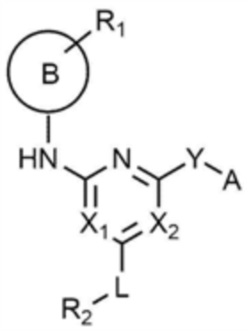
при этом, в химической формуле 1,
B представляет собой 5-членный или 6-членный гетероцикл, содержащий от 1 до 3 гетероатомов, каждый из которых независимо выбран из группы, состоящей из N, O и S, при условии, что 5-членный или 6-членный гетероцикл включает по меньшей мере один N,
R1 представляет собой водород, галоген, C1-4 алкил или C1-4 галогеналкил,
каждый из X1 и X2 независимо представляет собой N или CR',
где R' представляет собой водород или галоген,
L представляет собой связь, C1-4 алкилен или -O-,
R2 представляет собой циано; C1-4 алкил; C6-10 арил; пиридинил; морфолино; пиперазинил; или пиперидинил,
при этом каждый из пиперазинила и пиперидинила независимо является незамещенным, или замещенным C1-4 алкилом, C1-4 алкилом, замещенным циано, C1-4 алкилом, замещенным амино, C1-4 алкилом, замещенным C1-4 алкокси, или -CO-(C1-4 алкилом),
Y представляет собой связь, -O-, -NH- или -N(C1-4 алкил)-,
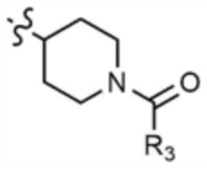
A представляет собой C1-4 алкил, 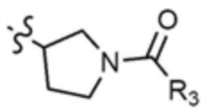 ,
, 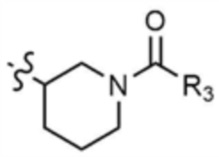 ,
,  ,
, 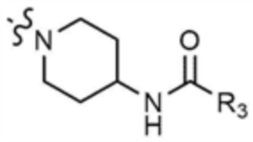 ,
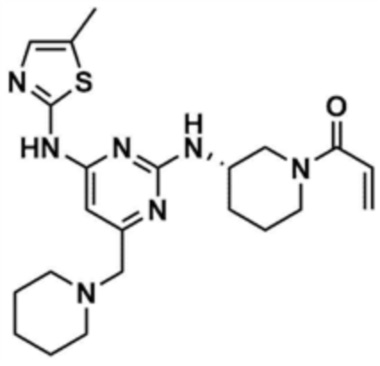
, 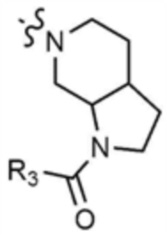 , или
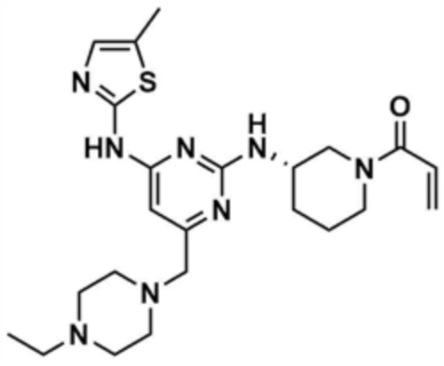
, или 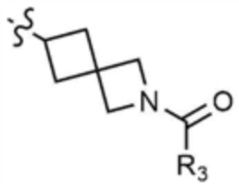 ,
,
где R3 представляет собой C1-4 алкил, C1-4 галогеналкил, C2-4 алкенил, C2-4 галогеналкенил, C2-4 алкинил или C2-4 галогеналкинил.
Предпочтительно B представляет собой тиазольное, пиразольное, пиридиновое или пиримидиновое кольцо, и
R1 представляет собой водород, хлор, метил или трифторметил.
Предпочтительно,
и X1 и X2 представляют собой CH;
один из X1 и X2 является CF, и другой является CH; или
один из X1 и X2 является N, и другой является CH.
Более предпочтительно,
и X1 и X2 представляют собой CH, или
X1 представляет собой CH, и X2 представляет собой CF; или
X1 представляет собой CH, и X2 представляет собой N; или
X1 представляет собой N, и X2 представляет собой CH.
Предпочтительно, L представляет собой связь, метилен или -O-.
Предпочтительно, R2 представляет собой циано; метил; фенил; пиридинил; морфолино; пиперазинил, замещенный метилом; пиперазинил, замещенный этилом; пиперазинил, замещенный 2-цианоэтилом; пиперазинил, замещенный 3-аминопропилом; пиперазинил, замещенный 2-метоксиэтилом; пиперазинил, замещенный -CO-(метилом); незамещенный пиперидинил; или пиперидинил, замещенный метилом.
Предпочтительно, Y представляет собой связь, -O-, -NH- или -N(метил)-.
Предпочтительно, R3 представляет собой -CH2CH2Cl, -CH=CH2, -CH=CHCH3, -CH=CHCl, -C≡CH или -C≡CCH3.
Предпочтительно, соединение, представленное химической формулой 1, представлено следующей химической формулой 1-1:
[Химическая формула 1-1]
при этом, в химической формуле 1-1,
R1 представляет собой водород, C1-4 алкил или C1-4 галогеналкил,
каждый из X1 и X2 независимо представляет собой N или CR',
где R' представляет собой водород или галоген,
L представляет собой связь, C1-4 алкилен или -O-,
R2 представляет собой циано; C1-4 алкил; C6-10 арил; пиридинил; морфолино; пиперазинил; или пиперидинил,
при этом каждый из пиперазинила и пиперидинила независимо является незамещенным, или замещенным C1-4 алкилом, C1-4 алкилом, замещенным циано, C1-4 алкилом, замещенным амино, C1-4 алкилом, замещенным C1-4 алкокси, или -CO-(C1-4 алкилом),
Y представляет собой связь, -O-, -NH- или -N(C1-4 алкил)-,
A представляет собой C1-4 алкил, , , , , , или ,
где R3 представляет собой C1-4 галогеналкил, C2-4 алкенил, C2-4 галогеналкенил или C2-4 алкинил.
Предпочтительно, в химической формуле 1-1,
R1 представляет собой C1-4 алкил,
каждый из X1 и X2 независимо представляет собой N или CH,
L представляет собой связь, C1-4 алкилен или -O-,
R2 представляет собой циано; C1-4 алкил; C6-10 арил; пиридинил; морфолино; пиперазинил; или пиперидинил,
при этом каждый из пиперазинила и пиперидинила независимо является незамещенным, или замещенным C1-4 алкилом, C1-4 алкилом, замещенным циано, C1-4 алкилом, замещенным C1-4 алкокси, или -CO-(C1-4 алкилом),
Y представляет собой связь, -O-, -NH- или -N(C1-4 алкил)-,
A представляет собой , , , , , или ,
где R3 представляет собой C2-4 алкенил или C2-4 алкинил.
Также предпочтительно, соединение, представленное химической формулой 1, представлено следующей химической формулой 1-2:
[Химическая формула 1-2]
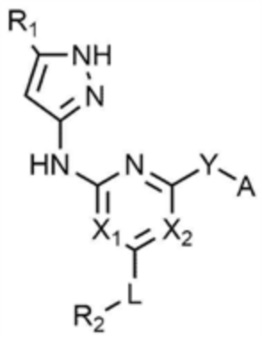
при этом, в химической формуле 1-2,
R1 представляет собой водород или галоген,
каждый из X1 и X2 независимо представляет собой N или CH,
L представляет собой C1-4 алкилен,
R2 представляет собой морфолино,
Y представляет собой -NH-,
A представляет собой ,
где R3 представляет собой C2-4 алкенил.
Кроме того, предпочтительно, соединение, представленное химической формулой 1, представлено следующей химической формулой 1-3:
[Химическая формула 1-3]
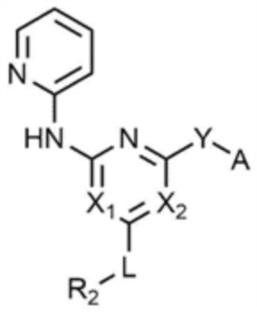
при этом, в химической формуле 1-3,
каждый из X1 и X2 независимо представляет собой N или CH,
L представляет собой C1-4 алкилен,
R2 представляет собой морфолино,
Y представляет собой -NH-,
A представляет собой ,
где R3 представляет собой C2-4 алкенил.
Кроме того, предпочтительно, соединение, представленное химической формулой 1, представлено следующей химической формулой 1-4:
[Химическая формула 1-4]
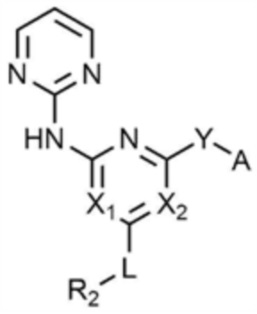
при этом, в химической формуле 1-4,
каждый из X1 и X2 независимо представляет собой N или CH,
L представляет собой C1-4 алкилен,
R2 представляет собой морфолино,
Y представляет собой -NH-,
A представляет собой ,
где R3 представляет собой C2-4 алкенил.
Кроме того, соединения настоящего изобретения могут существовать в форме солей, особенно фармацевтически приемлемых солей. В качестве солей без ограничения можно использовать соли, обычно используемые в данной области, такие как кислотно-аддитивные соли, образованные фармацевтически приемлемыми свободными кислотами. Используемый здесь термин «фармацевтически приемлемая соль» относится к любой органической или неорганической аддитивной соли соединения, представленного химической формулой 1, чья концентрация относительно нетоксична и безвредна для пациента и эффективно активирует, и чьи побочные эффекты не снижают полезную эффективность вышеуказанного соединения.
В качестве свободной кислоты можно использовать органическую кислоту и неорганическую кислоту. Примеры неорганических кислот включают соляную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, винную кислоту и тому подобное. Примеры органических кислот включают метансульфоновую кислоту, п-толуолсульфоновую кислоту, уксусную кислоту, трифторуксусную кислоту, малеиновую кислоту, янтарную кислоту, щавелевую кислоту, бензойную кислоту, винную кислоту, фумаровую кислоту, миндальную кислоту, пропионовую кислоту, лимонную кислоту, молочную кислоту, гликолевую кислоту, глюконовую кислоту, галактуроновую кислоту, глутаминовую кислоту, глутаровую кислоту, глюкуроновую кислоту, аспарагиновую кислоту, аскорбиновую кислоту, угольную кислоту, ванилиновую кислоту, иодистоводородную кислоту и тому подобное, но не ограничиваются ими. Предпочтительно соль может быть гидрохлоридной солью.
Кроме того, фармацевтически приемлемая соль металла может быть получена обычным способом с использованием основания. Например, соединение, представленное химической формулой 1, растворяют в избыточном количестве раствора гидроксида щелочного металла или раствора гидроксида щелочноземельного металла, нерастворимую соль отфильтровывают и затем фильтрат выпаривают и сушат с получением фармацевтически приемлемой соли металла. При этом особенно предпочтительно получить натриевую соль, калиевую соль или кальциевую соль в качестве соли металла.
В дополнение к этому, фармацевтически неприемлемая соль или сольват соединения химической формулы 1 могут быть использованы в качестве промежуточного соединения при получении соединения химической формулы 1 или его фармацевтически приемлемой соли или сольвата.
Кроме того, соединение, представленное химической формулой 1 согласно настоящему изобретению, включает не только его фармацевтически приемлемые соли, но также и сольваты, такие как гидраты, которые могут быть получены из него, и включает все возможные стереоизомеры, но не ограничивается ими. Сольват и стереоизомер соединения, представленного химической формулой 1, могут быть получены из соединения химической формулы 1 обычными способами, известными в данной области.
Кроме того, соединение, представленное химической формулой 1, согласно настоящему изобретению может быть получено либо в кристаллической форме, либо в некристаллической форме, и когда соединение химической формулы 1 получают в кристаллической форме, оно может быть необязательно гидратировано или сольватировано. В настоящем изобретении соединение, представленное химической формулой 1, может не только включать стехиометрический гидрат, но также может включать соединение, содержащее различные количества воды. Сольват соединения, представленного химической формулой 1, согласно настоящему изобретению включает как стехиометрические сольваты, так и нестехиометрические сольваты.
Типичными примерами соединения, представленного химической формулой 1, или его фармацевтически приемлемой соли являются следующие:
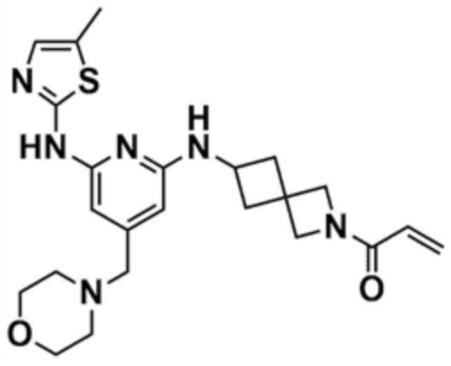
1) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
2) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
3) 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
4) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
5) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
6) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
7) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
8) 1-(3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-4-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
9) (E)-1-(3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-4-ил)амино)пирролидин-1-ил)бут-2-ен-1-он,
10) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пирролидин-1-ил)проп-2-ен-1-он,
11) 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он,
12) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он,
13) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
14) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)бут-2-ин-1-он,
15) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он,
16) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он,
17) 1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
18) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
19) 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
20) (S)-1-(3-((4-((4-ацетилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
21) (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
22) (S)-1-(3-((4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
23) (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
24) (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
25) (R)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
26) (S)-1-(3-(метил(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
27) N-(1-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)пиперидин-4-ил)акриламид,
28) 1-(6-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)октагидро-1H-пирроло[2,3-c]пиридин-1-ил)проп-2-ен-1-он,
29) 1-(6-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)октагидро-1H-пирроло[2,3-c]пиридин-1-ил)бут-2-ин-1-он,
30) 1-(6-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)-2-азаспиро[3,3]гептан-2-ил)проп-2-ен-1-он,
31) (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(пиперидин-1-илметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
32) (S)-1-(3-((4-((4-этилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
33) (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
34) (S)-3-(4-((2-((1-акрилоилпиперидин-3-ил)амино)-6-((5-метилтиазол-2-ил)амино)пиридин-4-ил)метил)пиперазин-1-ил)пропаннитрил,
35) (S)-1-(3-((4-метил-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
36) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-3-илметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
37) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-2-илметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
38) (S)-2-((1-акрилоилпиперидин-3-ил)амино)-6-(5-метилтиазол-2-ил)амино)изоникотинoнитрил,
39) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-фенилпиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
40) (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ин-1-он,
41) (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
42) (S)-1-(3-((4-((4-этилпиперазин-1-ил)метил)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
43) (S)-1-(3-((4-((4-метилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
44) 5-метил-N-(6-метил-4-(морфолинoметил)пиридин-2-ил)тиазол-2-амин,
45) (S)-3-хлор-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)пропaн-1-он,
46) (S, E)-3-хлор-1-(3-(6-(5-метилтиазол-2-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
47) (S)-1-(3-(4-((4-(3-аминопропил)пиперазин-1-ил)метил)-6-(5-метилтиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
48) (S)-1-(3-(6-(1H-пиразол-3-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
49) (S)-1-(3-(4-(морфолинoметил)-6-(5-(трифторметил)тиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
50) (S)-1-(3-(6-(5-хлор-1H-пиразол-3-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
51) (S)-1-(3-(4-(морфолинoметил)-6-(тиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
52) (S)-1-(3-(3-фтор-6-(5-метилтиазол-2-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
53) (S)-1-(3-(4-(морфолинoметил)-6-(пиридин-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он, и
54) (S)-1-(3-(4-(морфолинoметил)-6-(пиримидин-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он.
Кроме того, согласно настоящему изобретению, когда A не является C1-4 алкилом, соединение, представленное химической формулой 1, может быть получено, например, по схеме реакции 1 ниже.
[Схема реакции 1]
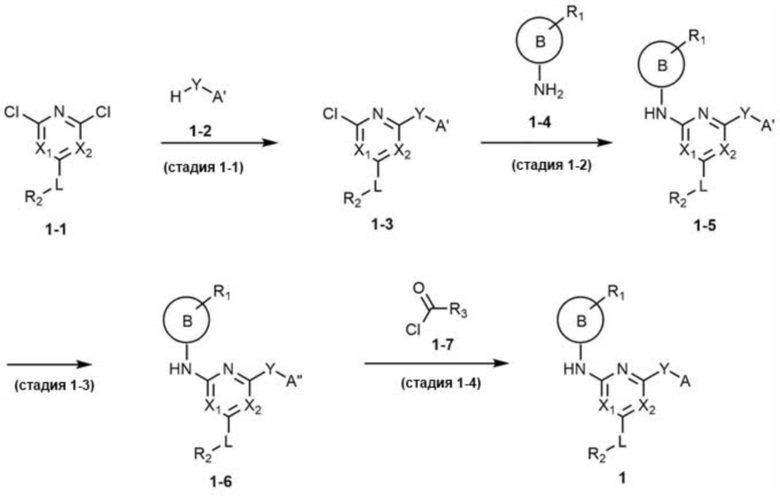
В схеме реакции 1, A' представляет собой 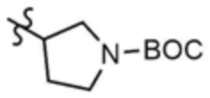 ,
,  ,
,  ,
,  ,
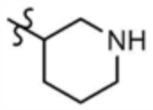
, 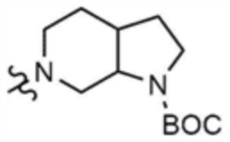 , или
, или 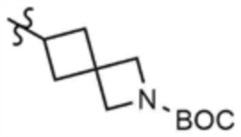 , A" представляет собой
, A" представляет собой 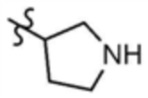 ,
,  ,
, 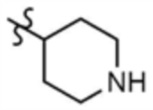 ,
, 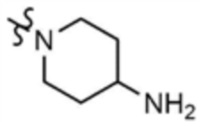 ,
, 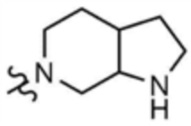 , или
, или 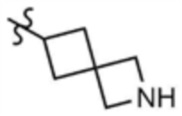 и остальное является таким, как определено выше.
и остальное является таким, как определено выше.
Стадия 1-1 представляет собой стадию реакции соединения, представленного химической формулой 1-1, и соединения, представленного химической формулой 1-2, с получением соединения, представленного химической формулой 1-3. Когда реакция представляет собой реакцию аминного замещения, - реакция предпочтительно проводится в присутствии палладиевого катализатора и основания, и когда реакция представляет собой реакцию сольволиза алкилхлорида из-за вторичного спирта, реакцию предпочтительно проводят в присутствии основания.
Стадия 1-2 представляет собой стадию реакции соединения, представленного химической формулой 1-3, и соединения, представленного химической формулой 1-4, с получением соединения, представленного химической формулой 1-5. Реакция представляет собой реакцию аминного замещения, которая предпочтительно проводится в присутствии палладиевого катализатора и основания.
Стадия 1-3 представляет собой стадию удаления защитной группы (ВОС; трет-бутилоксикарбонильная защитная группа) из соединения, представленного химической формулой 1-5, с получением соединения, представленного химической формулой 1-6. Реакцию предпочтительно проводят в кислых условиях, способных удалять защитную группу.
Стадия 1-4 представляет собой стадию реакции соединения, представленного химической формулой 1-6, с соединением, представленным химической формулой 1-7, с получением соединения, представленного химической формулой 1. Реакция представляет собой реакцию амидирования, которая предпочтительно проводится в присутствии основания.
Кроме того, на схеме реакции 1 может быть добавлена реакция защиты защитной группой и реакция удаления защитной группы в зависимости от каждого заместителя.
В соответствии с другим вариантом осуществления настоящего изобретения, в соединении, представленном химической формулой 1, когда L представляет собой метилен, и A не является C1-4 алкилом, соединение, представленное химической формулой 1, может быть получено, например, по схеме реакции 2 ниже.
[Схема реакции 2]
В схеме реакции 2 определение каждого заместителя является таким же, как определено выше.
Стадия 2-1 представляет собой стадию реакции соединения, представленного химической формулой 2-1, и соединения, представленного химической формулой 1-2, с получением соединения, представленного химической формулой 2-2. Когда реакция представляет собой реакцию аминного замещения, - реакция предпочтительно проводится в присутствии палладиевого катализатора и основания, и когда реакция представляет собой реакцию сольволиза алкилхлорида из-за вторичного спирта, реакцию предпочтительно проводят в присутствии основания.
Стадия 2-2 представляет собой стадию реакции соединения, представленного химической формулой 2-2, и соединения, представленного химической формулой 2-3, с получением соединения, представленного химической формулой 2-4. Реакция представляет собой реакцию амидирования, которую предпочтительно проводят в присутствии амидирующего реагента.
Стадия 2-3 представляет собой стадию реакции соединения, представленного химической формулой 2-4, и соединения, представленного химической формулой 1-4, с получением соединения, представленного химической формулой 2-5. Реакция представляет собой реакцию аминного замещения, которая предпочтительно проводится в присутствии палладиевого катализатора и основания.
Стадия 2-4 представляет собой стадию удаления защитной группы (ВОС) из соединения, представленного химической формулой 2-5, с получением соединения, представленного химической формулой 2-6. Реакцию предпочтительно проводят в кислых условиях.
Стадия 2-5 представляет собой стадию реакции соединения, представленного химической формулой 2-6, с соединением, представленным химической формулой 1-7, с получением соединения, представленного химической формулой 1. Реакция представляет собой реакцию амидирования, которая предпочтительно проводится в присутствии основания.
Кроме того, на схеме реакции 2 может быть добавлена реакция защиты защитной группой и реакция удаления защитной группы в зависимости от каждого заместителя.
Кроме того, согласно настоящему изобретению, в соединении, представленном химической формулой 1, соединение, в котором A представляет собой C1-4 алкил, может быть получено с использованием того же способа, что и на стадии 1-1 и стадии 1-2 схемы реакции 1, за исключением того, что на стадии 1-1 на схеме реакции 1 соединение, представленное  , используется вместо соединения, представленного химической формулой 1-2.
, используется вместо соединения, представленного химической формулой 1-2.
Способ получения каждой описанной выше стадии может быть более подробно представлен в примерах, описанных ниже.
В соответствии с дополнительным вариантом осуществления настоящего изобретения, предложена фармацевтическая композиция для профилактики или лечения аутоиммунных заболеваний или раковых заболеваний, которая эффективна для ингибирования ITK и BTK, содержащая соединение, представленное химической формулой 1, или его фармацевтически приемлемую соль, гидрат, сольват или изомер в качестве активного ингредиента.
В данном случае аутоиммунные заболевания включают следующее: ревматоидный артрит, системная красная волчанка, сахарный диабет у детей, псориаз, афтозный стоматит, хронический тиреоидит, приобретенная апластическая анемия, первичный цирроз, язвенный колит, болезнь Бехчета, болезнь Крона, силикоз, асбестоз, синдром Шегрена, синдром Гийена-Барре, дерматомиозит, полимиозит, рассеянный склероз, аутоиммунная гемолитическая анемия, аутоиммунный энцефаломиелит, миастения гравис, гиперплазия щитовидной железы Грейвса, узелковый полиартериит, анкилозирующий спондилит, фиброзит, височный артериит, болезнь Вильсона или синдром Фанкони, и
рак может представлять собой рак крови, экстранодальную В-клеточную лимфому маргинальной зоны, глиобластому, лимфоплазмацитарную лимфому, острый миелогенный лейкоз, макроглобулинемию, B-клеточную лимфому, хронический лимфоцитарный лейкоз, фолликулярную лимфому, неходжкинскую лимфому, диффузную B-крупноклеточную лимфому, волосатоклеточный лейкоз, мантийноклеточную лимфому, глиобластому, рак мочевого пузыря, рак поджелудочной железы, рак яичников, колоректальный рак, рак почки, рак желудка, переходно-клеточную карциному, карциноидную опухоль, рак молочной железы, немелкоклеточный рак легкого или множественную миелому.
Используемый здесь термин «профилактика» относится к любому действию, направленному на задержку или ингибирование возникновения, распространения или рецидива указанных выше заболеваний путем введения композиции настоящего изобретения, и «лечение» относится к любому действию, направленному на улучшение или изменение симптомов указанных выше заболеваний в лучшую сторону путем введения композиции настоящего изобретения.
Фармацевтическая композиция по настоящему изобретению может быть приготовлена в виде для перорального или парентерального введения в соответствии со стандартной фармацевтической практикой. Эти композиции могут содержать добавки, такие как фармацевтически приемлемый носитель, адъювант или разбавитель, в дополнение к активному ингредиенту.
Подходящие носители включают, например, физиологический раствор, полиэтиленгликоль, этанол, растительное масло и изопропилмиристат, и тому подобное. Разбавители включают, например, лактозу, декстрозу, сахарозу, маннит, сорбит, целлюлозу и/или глицин и тому подобное, но не ограничиваются этим. Кроме того, соединения настоящего изобретения могут быть растворены в маслах, пропиленгликоле или других растворителях, обычно используемых при приготовлении инъекционных растворов. Кроме того, соединения настоящего изобретения могут быть приготовлены в виде мазей или кремов для местного применения.
Предпочтительная доза соединения настоящего изобретения может варьировать в зависимости от состояния и веса пациента, тяжести заболевания, типа лекарственного средства, а также пути и продолжительности введения, при этом она может быть соответствующим образом выбрана специалистами в данной области. Однако для достижения желаемых эффектов соединение настоящего изобретения можно вводить ежедневно в дозе от 0,0001 до 100 мг/кг (массы тела) и предпочтительно от 0,001 до 100 мг/кг (массы тела). Введение препарата может осуществляться один раз в день или в разделенных дозах каждый день перорально или парентерально.
В зависимости от способа введения фармацевтическая композиция может содержать соединение настоящего изобретения в количестве 0,001-99% масс., предпочтительно 0,01-60% масс.
Фармацевтическую композицию настоящего изобретения можно вводить млекопитающим, таким как крыса, мышь, домашнее животное, человек, различными путями. Введение может осуществляться всеми возможными способами, например, пероральным, ректальным, внутривенным, внутримышечным, подкожным, внутриматочным, интрацеребровентрикулярным.
ПОЛЕЗНЫЕ ЭФФЕКТЫ
Соединение, представленное химической формулой 1 в соответствии с настоящим изобретением, или его фармацевтически приемлемая соль, гидрат, сольват или изомер могут быть эффективно использованы для профилактики или лечения аутоиммунных заболеваний или раковых заболеваний.
ПОДРОБНОЕ ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Ниже настоящее изобретение будет описано более подробно с помощью примеров. В то же время, эти примеры приводятся только для иллюстративных целей и не должны рассматриваться как ограничивающие объем настоящего изобретения.
Пример 1: Получение 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 1-1: Получение (2,6-дихлорпиридин-4-ил)(морфолино)метанона
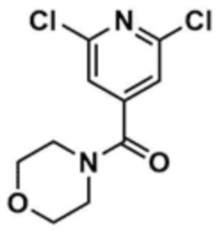
2,6-Дихлоризоникотиновую кислоту (10,0 г, 1,0 экв.) растворяли в диметилформамиде (100,0 мл) и затем к раствору добавляли 1,1-карбонилдиимидазол (1,0 г, 1,2 экв.). Смесь перемешивали при комнатной температуре (25 ~ 30 °С) в течение 1 ч в атмосфере азота, и затем добавляли морфолин (5,4 мг, 1,2 экв.) и перемешивали при той же температуре в течение 2 ч для завершения реакции. К смеси добавляли этилацетат (200,0 мл) и воду (200,0 мл) с последующей экстракцией, и водный слой реэкстрагировали трижды этилацетатом (200,0 мл). Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:5) с получением указанного в заголовке соединения (13,0 г, выход: 93,0%).
Стадия 1-2: Получение 4-((2,6-дихлорпиридин-4-ил)метил)морфолина
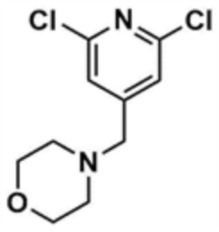
Промежуточное соединение (10,0 г, 1,0 экв.), полученное на стадии 1-1, растворяли в дихлорметане (100,0 мл) и затем охлаждали до 0-10 °С в атмосфере азота. Медленно по каплям добавляли 1 М боран-тетрагидрофуран (115,0 мл, 3,0 экв.). Смесь перемешивали при комнатной температуре в течение 12 ч для завершения реакции. Реакционный раствор охлаждали до 0-10 °С, и далее медленно по каплям добавляли 6 н. водный раствор соляной кислоты (256,0 мл, 20,0 экв.), и затем перемешивали при той же температуре в течение 1 ч. После доведения рН до 9~12 с использованием 10 н. водного раствора гидроксида натрия, экстракцию проводили дважды дихлорметаном. Дихлорметановый слой отделяли, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:1) с получением указанного в заголовке соединения (8,1 г, выход: 90,0%).
Стадия 1-3: Получение трет-бутил-3-((6-хлор-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-карбоксилата
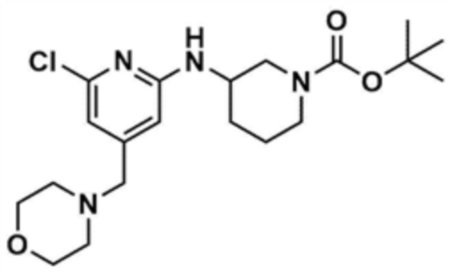
После добавления 1,4-диоксана (10,0 мл) к промежуточному соединению (1,0 г, 1,0 экв.), полученному на стадии 1-2, и растворения, к нему добавляли трис(дибензилиденацетон)дипалладий (0) (465,8 мг, 0,2 экв.) и Ксантфос (1,5 г, 0,4 экв.). Добавляли трет-бутил-3-аминопиперидин-1-карбоксилат (780,0 мкл, 1,0 экв.) и затем добавляли карбонат натрия (1,3 г, 3,0 экв.), и смесь кипятили с обратным холодильником в течение 12 ч для завершения реакции. После охлаждения до 30°C или менее добавляли воду (20,0 мл) и этилацетат (20,0 мл) и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:1) с получением указанного в заголовке соединения (900,0 мг, выход: 54,1%).
Стадия 1-4: Получение трет-бутил-3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-карбоксилата
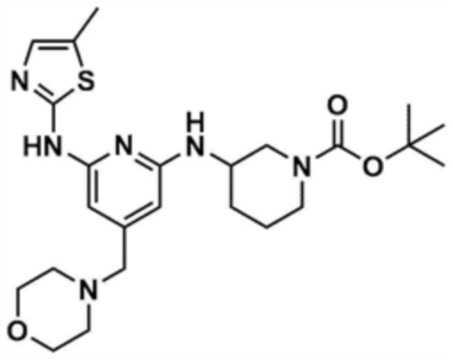
Промежуточное соединение (730,0 мг, 1.0 экв.), полученное на стадии 1-3, растворяли в 1,4-диоксане (14,0 мл). Последовательно добавляли ацетат палладия (40,0 мг, 0,1 экв.), Ксантфос (204,7 мг, 0,2 экв.), 5-метилтио-2-амин (203,6 мг, 1,0 экв.) и карбонат цезия (1,7 г, 3,0 экв.). Смесь подвергали реакции в микроволновом реакторе при 150°С в течение 30 мин. Добавляли этилацетат (10,0 мл) и воду (10,0 мл) и полученное твердое вещество фильтровали с получением указанного в заголовке соединения (424,9 мг, выход: 65,4%). После охлаждения до 30°C или менее к смеси добавляли воду (15,0 мл) и этилацетат (15,0 мл), с последующим разделением слоев. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (EA 100%) с получением указанного в заголовке соединения (564,0 мг, выход: 65,0%).
Стадия 1-5: Получение N2-(5-метилтиазол-2-ил)-4-(морфолинoметил)-N6-(пиперидин-3-ил)пиридин-2,6-диамина
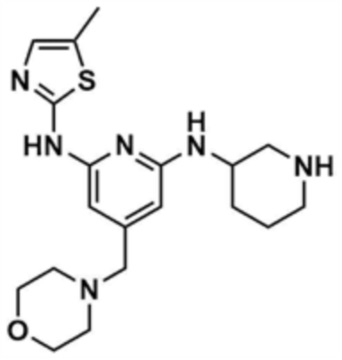
Промежуточное соединение (500,0 мг, 1,0 экв.), полученное на стадии 1-4, растворяли в дихлорметане (10,0 мл) и затем охлаждали до 0-10 °С. Медленно по каплям добавляли трифторуксусную кислоту (1,6 мл, 20,0 экв.) и затем перемешивали в течение 1 ч. После доведения pH до 9-12 с использованием 12 н. водного раствора гидроксида натрия, отделенный дихлорметановый слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат (10,0 мл) для образования кристаллов в течение 30 мин. Кристаллы отфильтровывали и сушили с получением указанного в заголовке соединения (357,5 мг, выход: 90,0%).
Стадия 1-6: Получение 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
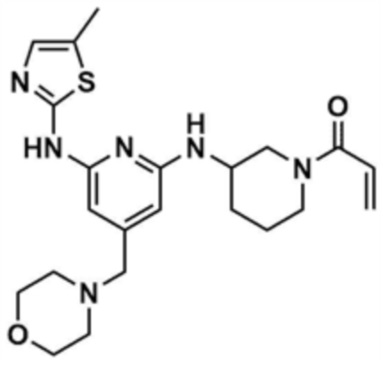
После растворения промежуточного соединения (350,0 мг, 1,0 экв.), полученного на стадии 1-5, в тетрагидрофуране (7,0 мл) добавляли воду (7,0 мл) и добавляли бикарбонат натрия (226,8 мг, 3,0 экв.), и затем охлаждали до 0-10 °С. Медленно по каплям добавляли акрилоилхлорид (73,1 мкл, 1,0 экв.) и затем перемешивали в течение 30 мин для завершения реакции. Слои разделяли дихлорметаном, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=15:1) с получением указанного в заголовке соединения (318,0 мг, выход: 80,0%).
1H ЯМР (500 МГц, ДМСО): 10,5 (c, 1H), 6,94 (c, 1H), 6,86-6,80 (кв, 1H), 6,50-6,49 (д, 1H), 6,10-6,07 (д, 1H), 6,04 (c, 1H), 5,94 (c, 1H), 5,66-5,64 (д, 1H), 4,38-4,36 (м, 1H), 4,18-4,16 (м, 1H), 4,08-4,06 (м, 1H), 3,55 (м, 4H), 3,21 (c, 3H), 2,88-2,83 (м, 2H), 2,32 (м, 4H), 2,28 (c, 3H), 2,03 (м, 2H), 1,30 (м, 2H)
Пример 2: Получение 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она
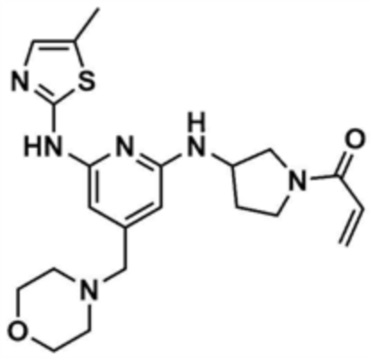
Указанное в заголовке соединение (15,0 мг, выход: 23,0%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 использовали трет-бутил-3-аминопирролидин-1-карбоксилат вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,58-10,57 (м, 1H), 6,94 (c, 1H), 6,83-6,75 (м, 1H), 6,63-6,48 (м, 1H), 6,14-6,10 (м, 1H), 6,08 (c, 1H), 5,99-5,98 (м, 1H), 5,67-5,59 (м, 1H), 4,65-4,50 (м, 1H), 3,99-3,97 (м, 0,5H), 3,70-3,66 (м, 1,5H), 3,55 (м, 4H), 3,48 (м, 1H), 3,35 (м, 1H), 3,22 (c, 2H), 2,34-2,32 (м, 4H), 2,26-2,24 (д, 3H), 2,25 (м, 1H), 1,95-1,85 (м, 1H)
Пример 3: Получение 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
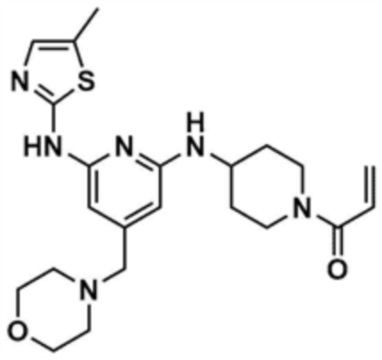
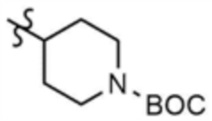
Указанное в заголовке соединение (8,0 мг, выход: 53,0%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 использовали трет-бутил-4-аминопиперидин-1-карбоксилат вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,54 (c, 1H), 6,94 (c, 1H), 6,93-6,83 (м, 1H), 6,51-6,49 (д, 1H), 6,10 (д, 1H), 6,07 (c, 1H), 5,94 (c, 1H), 5,66-5,64 (д, 1H), 4,38-4,36 (м, 1H), 4,16 (м, 1H), 4,08-4,02 (м, 1H), 3,56 (м, 4H), 3,21 (c, 2H), 2,85 (м, 1H), 2,61 (м, 1H), 2,34-2,33 (м, 4H), 2,28 (c, 3H), 2,0 (м, 2H), 1,30-1,21 (м, 2H)
Пример 4: Получение (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
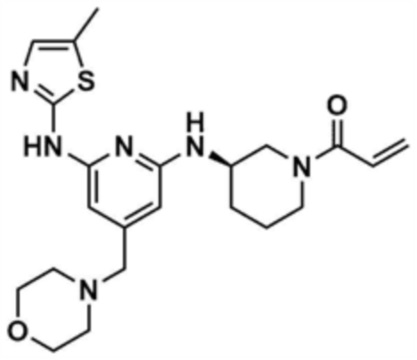
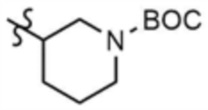
Указанное в заголовке соединение (10,0 мг, выход: 53,0%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 использовали трет-бутил-(R)-3-аминопиперидин-1-карбоксилат вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,55-10,50 (м, 1H), 6,91-6,90 (м, 1H), 6,90-6,78 (м, 0,5H), 6,47-6,56 (м, 1,5H), 6,06-5,96 (м, 3H), 5,65-5,67 (м, 0,5H), 5,42-5,40 (м, 0,5H), 4,42-4,40 (м, 0,5H), 4,10-4,0 (м, 1H), 3,90-3,87 (м, 1,5H), 3,56 (м, 4H), 3,20 (c, 2H), 3,14-3,10 (м, 1H), 2,68-2,63 (м, 0,5H), 2,32 (м, 4H), 2,19 (c, 3H), 1,90-2,0 (м, 1H), 1,80 (м, 1H), 1,50-1,40 (м, 2,5H)
Пример 5: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
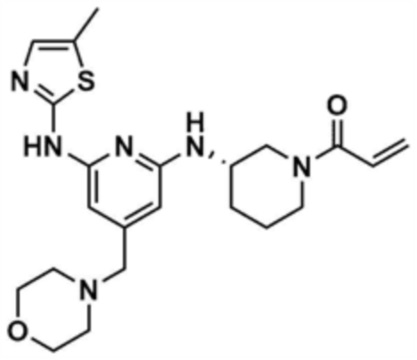
Указанное в заголовке соединение (13,0 мг, выход: 63,0%) получали таким же образом, как и в примере 1, за исключением того, что на стадиях 1-3 примера 1 использовали трет-бутил-(S)-3-аминопиперидин-1-карбоксилат вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,57 (м, 1H), 6,91-6,90 (м, 1H), 6,80-6,85 (м, 0,5H), 6,70-6,40 (м, 1,5H), 6,10-5,96 (м, 3H), 5,65-5,63 (д, 0,5H), 5,42-5,40 (д, 0,5H), 4,42-4,40 (м, 0,5H), 4,10-4,0 (м, 1H), 3,90-3,87 (м, 1,5H), 3,56 (м, 4H), 3,20 (c, 2H), 3,14-3,10 (м, 1H), 2,68-2,63 (м, 0,5H), 2,32 (м, 4H), 2,19 (c, 3H), 1,90-2,0 (м, 1H), 1,80 (м, 1H), 1,50-1,40 (м, 2,5H)
Пример 6: Получение (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она
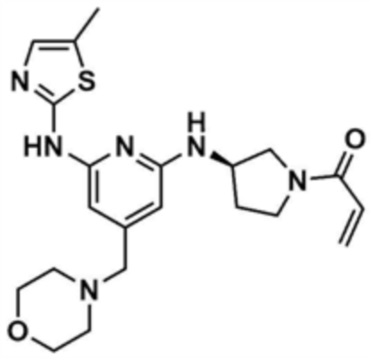
Указанное в заголовке соединение (10,0 мг, выход: 58,0%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 использовали трет-бутил-(R)-3-аминопирролидин-1-карбоксилат вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,7 (м, 1H), 6,94 (c, 1H), 6,83-6,75 (м, 1H), 6,63-6,47 (м, 1H), 6,14-6,10 (м, 2H), 6,09-3,08 (м, 1H), 5,67-5,59 (м, 1H), 4,67-4,50 (1H), 3,97-3,96 (м, 0,5H), 3,70 (м, 1,5H), 3,55-3,54 (м, 4H), 3,40 (м, 1H), 3,38 (м, 1H), 3,22 (c, 2H), 2,32 (м, 4H), 2,26-2,24 (д, 3H), 2,20 (м, 1H), 1,95-1,80 (м, 1H)
Пример 7: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она
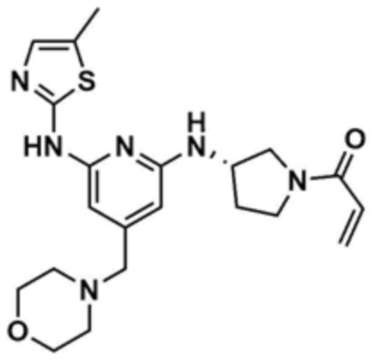
Указанное в заголовке соединение (15,0 мг, выход: 63,0%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 использовали трет-бутил-(S)-3-аминопирролидин-1-карбоксилат вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,7 (м, 1H), 6,94 (c, 1H), 6,83-6,75 (м, 1H), 6,63-6,47 (м, 1H), 6,14-6,10 (м, 2H), 6,09-3,08 (м, 1H), 5,67-5,59 (м, 1H), 4,67-4,50 (1H), 3,97-3,96 (м, 0,5H), 3,70 (м, 1,5H), 3,55-3,54 (м, 4H), 3,40 (м, 1H), 3,38 (м, 1H), 3,22 (c, 2H), 2,32 (м, 4H), 2,26-2,24 (д, 3H), 2,20 (м, 1H), 1,95-1,80 (м, 1H)
Пример 8: Получение 1-(3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-4-ил)амино)пирролидин-1-ил)проп-2-ен-1-она
Стадия 8-1: Получение метил-6-((1-(трет-бутоксикарбонил)пирролидин-3-ил)амино)-2-хлорпиримидин-4-карбоксилата
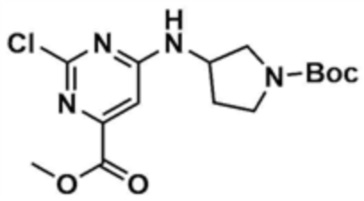
После растворения метил-2,4-дихлорпиримидин-6-карбоксилата (500 мг, 1,0 экв.) в тетрагидрофуране (10,0 мл) к нему добавляли диизопропилэтиламин (1,5 экв.) и трет-бутил-3-аминопирролидин-1-карбоксилат (1,5 экв.) и затем перемешивали при 80°C в течение 1 ч. По завершении реакции смесь охлаждали до 30°C или ниже, к ней добавляли воду (100,0 мл) и дихлорметан (100,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:1) с получением указанного в заголовке соединения (640,0 мг, выход: 74,0%).
Стадия 8-2: Получение трет-бутил-3-((2-хлор-6-(морфолин-4-карбонил)пиримидин-4-ил)амино)пирролидин-1-карбоксилата
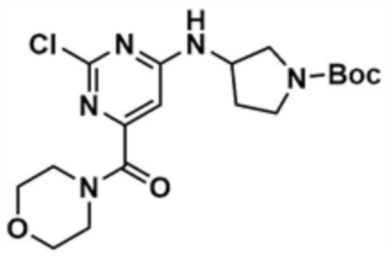
После того, как метил-6-((1-трет-бутоксикарбонил)пирролидин-3-ил)амино)-2-хлорпиримидин-4-карбоксилат (640,0 мг, 1,0 экв.), полученный на стадии 8-1, растворяли в тетрагидрофуране (10,0 мл), в раствор добавляли 1,5,7-триазабицикло[4,4,0]дец-5-ен (0,3 экв.) и морфолин (1,2 экв.) и перемешивали при 60°C в течение 3 ч. После завершения реакции к смеси добавляли воду (200,0 мл) и дихлорметан (200,0 мл), с последующей экстракцией. Отделенный органический слой сушили над сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=9:1) с получением указанного в заголовке соединения (470,0 мг, выход: 63,7%).
Стадия 8-3: Получение трет-бутил-3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолин-4-карбонил)пиримидин-4-ил)амино)пирролидин-1- карбоксилата
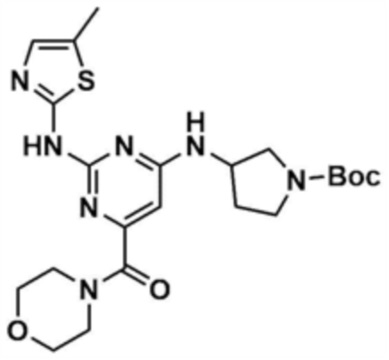
После того, как трет-бутил-3-((2-хлор-6-(морфолин-4-карбонил)пиримидин-4-ил)амино)пирролидин-1-карбоксилат (450,0 мг, 1,0 экв.), полученный на стадии 8-2, растворяли в 1,4-диоксане (10,0 мл), к ним добавляли ацетат палладия (0,1 экв), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (0,2 экв), карбонат цезия (3,0 экв), 2-амино-5-метилтиазол (1,2 экв), и подвергали реакции в микроволновом реакторе (160°С, 30 мин). После завершения реакции к смеси добавляли воду (200,0 мл) и этилацетат (200,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=9:1) с получением указанного в заголовке соединения (410,0 мг, выход: 76,6%).
Стадия 8-4: Получение N2-(5-метилтиазол-2-ил)-6-(морфолинoметил)-N4-(пирролидин-3-ил)пиримидин-2,4-диамина
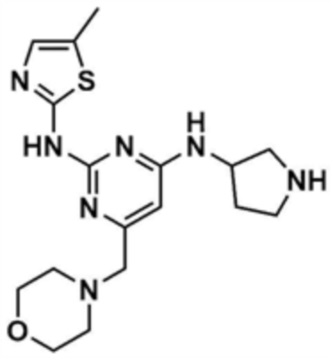
После того, как трет-бутил-3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолин-4-карбонил)пиримидин-4-ил)амино)пирролидин-1-карбоксилат (250,0 мг, 1,0 экв.), полученный на стадии 8-3, растворяли в тетрагидрофуране (10,0 мл), туда добавляли 0,9 М раствор боран-тетрагидрофурана (5,0 экв.), и перемешивали при 50°C в течение 5 ч. Реакционный раствор охлаждали до 0°C и затем добавляли 6 н. водный раствор соляной кислоты (5,0 экв.), и затем перемешивали при 50°C в течение 12 ч. Реакционный раствор снова охлаждали до 0°C, и затем рН доводили до 12 с использованием 12 н. водного раствора гидроксида натрия, и затем экстрагировали дихлорметаном (200,0 мл) и водой (200,0 мл). Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении, с получением указанного в заголовке соединения (40,0 мг, выход: 20,9%).
Стадия 8-5: Получение 1-(3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-4-ил)амино)пирролидин-1-ил)проп-2-ен-1-она
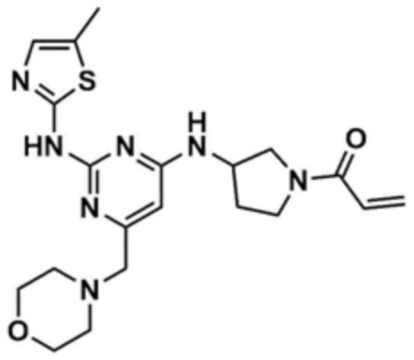
После того, как N2-(5-метилтиазол-2-ил)-6-(морфолинoметил)-N4-(пирролидин-3-ил)пиримидин-2,4-диамин (50,0 мг, 1,0 экв.), полученный на стадии 8-4, растворяли в тетрагидрофуране (1,6 мл) и воде (0,4 мл), в раствор добавляли бикарбонат натрия (3,0 экв.) и акрилоилхлорид (1,1 экв.), и перемешивали при 0°C в течение 30 мин. После завершения реакции к смеси добавляли воду (50,0 мл) и этилацетат (50,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=3:1) с получением указанного в заголовке соединения (7,0 мг, выход: 12,2%).
1H ЯМР (500 МГц, CDCl3): 6,95 (c, 1H), 6,40-6,46 (м, 3H), 6,18 (c, 1H), 5,69-5,74 (м, 1H), 3,91-4,10 (м, 1H), 3,76 (c, 2H), 3,50-3,75 (м, 8H), 2,46-2,56 (м, 6H), 2,36 (c, 3H)
Пример 9: Получение (E)-1-(3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-4-ил)амино)пирролидин-1-ил)бут-2-ен-1-она
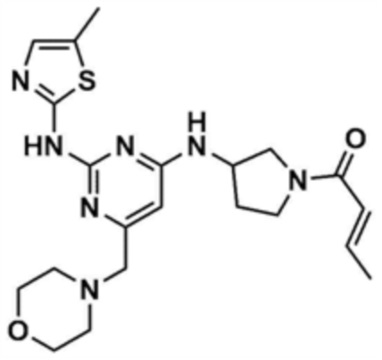
Указанное в заголовке соединение (5,0 мг, выход: 8,5%) получали таким же образом, как и в примере 8, за исключением того, что на стадии 8-5 примера 8 кротонилхлорид использовали вместо акрилоилхлорида.
1H ЯМР (500 МГц, CDCl3): 6,94-7,00 (м, 3H), 6,18 (c, 1H), 6,13-6,16 (м, 2H), 4,3 (c, 1H), 3,75 (c, 2H), 3,61-3,73 (м, 8H), 2,56 (c, 4H), 2,29-2,36 (м, 5H), 1,86-1,91 (м, 3H)
Пример 10: Получение 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пирролидин-1-ил)проп-2-ен-1-она
Стадия 10-1: Получение трет-бутил-3-((6-хлор-4-(морфолинoметил)пиридин-2-ил)окси)пирролидин-1-карбоксилата
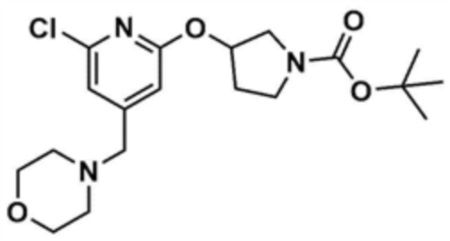
После растворения трет-бутил-3-гидроксипирролидин-1-карбоксилата (2,0 г, 1,0 экв.) в диметилформамиде (10,0 мл) к нему добавляли т-бутоксид калия (1,4 г, 1,5 экв) и перемешивали в течение 30 мин. Добавляли промежуточное соединение (2,0 г, 1,0 экв.), полученное на стадии 1-2 примера 1, и затем смесь перемешивали при 60-80 °С в течение 4 ч. После охлаждения до 30°C или менее добавляли воду (40,0 мл) и этилацетат (40,0 мл), и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:5) с получением указанного в заголовке соединения (1,8 г, выход: 55,3%).
Стадия 10-2: Получение (1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пирролидин-1-ил)проп-2-ен-1-она
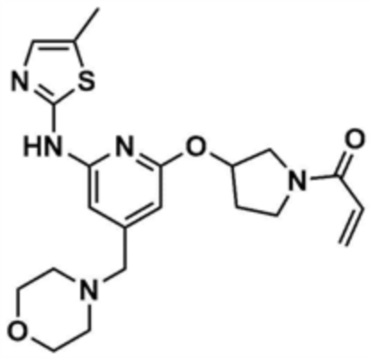
Указанное в заголовке соединение (13,2 мг, выход 65,5%) получали таким же образом, как и в примере 1, за исключением того, что использовали промежуточное соединение, полученное на стадии 10-1, вместо промежуточного соединения, полученного на стадии 1-3 примера 1.
1H ЯМР (500 МГц, ДМСО): 10,18 (c, 1H), 6,62 (м, 1H), 6,53 (c, 1H), 6,04 (м, 1H), 5,65 (c, 1H), 5,58 (м, 1H), 5,20 (c, 1H), 4,44 (c, 2H), 3,99 (м, 1H), 3,71 (м, 2H), 3,57 (м, 4H), 3,50-3,49 (м, 2H), 2,42 (м, 4H), 2,04 (м, 2H)
Пример 11: Получение 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-она
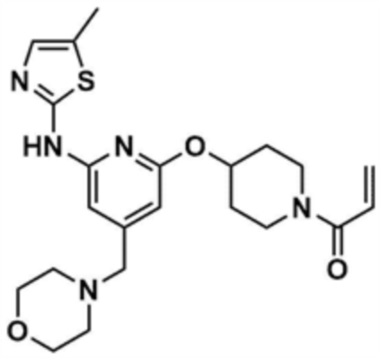
Указанное в заголовке соединение (8,5 мг, выход: 65,0%) получали таким же образом, как и в примере 10, за исключением того, что на стадии 10-1 примера 10 трет-бутил-4-гидроксипиперидин-1-карбоксилат использовали вместо трет-бутил-3-гидроксипирролидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,9 (c, 1H), 7,00 (c, 1H), 6,86-6,80 (м, 1H), 6,53 (c, 1H), 6,20 (c, 1H), 6,11-6,08 (д, 1H), 5,68-5,65 (д, 1H), 5,36-5,32 (м, 1H), 4,03 (м, 1,5H), 3,90 (м, 1,5H), 3,50 (м, 4H), 3,45 (м, 2H), 3,40 (c, 2H), 2,34 (м, 4H), 2,31 (c, 3H), 2,08-2,06 (м, 2H), 1,61-1,59 (м, 2H)
Пример 12: Получение 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-она
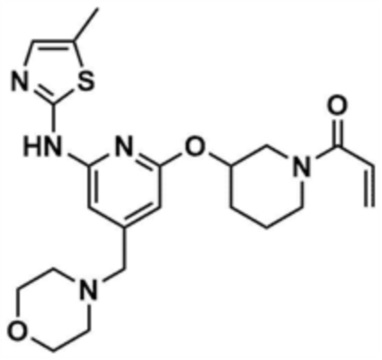
Указанное в заголовке соединение (9,5 мг, выход: 63,0%) получали таким же образом, как и в примере 10, за исключением того, что на стадии 10-1 примера 10 трет-бутил-3-гидроксипиперидин-1-карбоксилат использовали вместо трет-бутил-3-гидроксипирролидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,994-10,92 (м, 1H), 7,00-6,99 (м, 1H), 6,95-6,85 (м, 0,5H), 6,53 (c, 1H), 6,60-6,5 (м, 0,5H), 6,15-6,13 (м, 1H), 6,10-5,96 (м, 1H), 5,74 (д, 0,5H), 5,43-5,45 (д, 0,5H), 5,25-5,15 (м, 1H), 4,01-3,95 (м, 0,5H), 3,90-3,75 (м, 2H), 3,70 (м, 0,5H), 3,55 (м, 4H), 3,40 (c, 2H), 2,34 (м, 4H), 2,37-2,20 (c, 3H), 2,09-2,04 (м, 1,5H), 1,97-1,78 (м, 2,5H), 1,50 (м, 1H)
Пример 13: Получение (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-она
Стадия 13-1: Получение (R)-N2-(5-метилтиазол-2-ил)-4-(морфолинoметил)-N6-(пиперидин-3-ил)пиридин-2,6-диамина
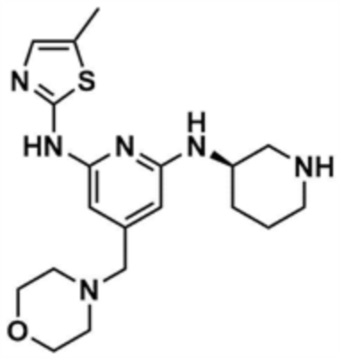
Указанное в заголовке соединение (150,0 мг, выход: 75,0%) получали таким же образом, как и на стадиях 1-3, 1-4 и 1-5 примера 1, за исключением того, что на стадии 1-3 примера 1 трет-бутил-(R)-3-аминопиперидин-1-карбоксилат использовали вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
Стадия 13-2: Получение (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-она
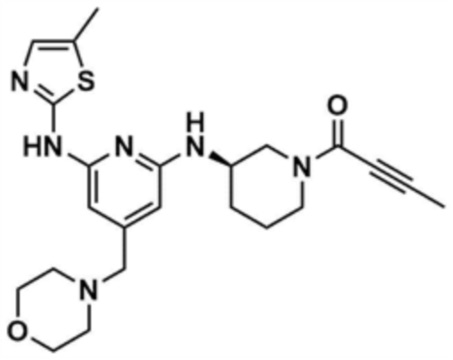
После растворения 2-бутиновой кислоты (21,6 мг, 1,0 экв.) в диметиламиде (1,0 мл) добавляли 1-[бис(диметиламино)метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксидгексафторфосфат (97,3 мг, 1,0 экв.), и затем перемешивали в течение 30 мин. Добавляли промежуточное соединение (100,0 мг, 1,0 экв.), полученное на стадии 13-1, и триэтиламин (53,5 мкл, 1,5 экв.), и затем перемешивали в течение 1 ч. Добавляли воду (1,0 мл) и этилацетат (1,0 мл) и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (64,0 мг, выход: 55,0%).
1H ЯМР (500 МГц, ДМСО): 10,59-10,58 (д, 1H), 6,95-6,94 (м, 1H), 6,84-6,79 (м, 1H), 6,10 (м, 1H), 5,99-5,97 (м, 1H), 4,57-4,56 (м, 1H), 3,85-3,65 (м, 2H), 3,55 (м, 4H), 3,45-3,35 (м, 4H), 3,20 (c, 2H), 3,30 (м, 4H), 2,26 (c, 3H), 2,25-2,15 (м, 2H), 2,0 (д, 3H)
Пример 14: Получение 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)бут-2-ин-1-она
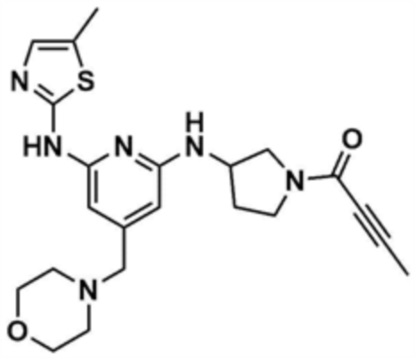
Указанное в заголовке соединение (58,6 мг, выход: 50,0%) получали таким же образом, как и в примере 13, за исключением того, что на стадии 13-1 примера 13 трет-бутил-3-аминопирролидин-1-карбоксилат использовали вместо трет-бутил-(R)-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, CDCl3): 7,03 (c, 1H), 6,11 (c, 1H), 5,96 (c, 1H), 4,56-4,54 (д, 1H), 4,42-4,40 (д, 1H), 3,73-3,71 (м, 4H), 3,33 (c, 2H), 3,30 (м, 1H), 2,93 (м, 1H), 2,45 (м, 4H), 2,38 (c, 3H), 1,47-1,40 (м, 1H)
Пример 15: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-она
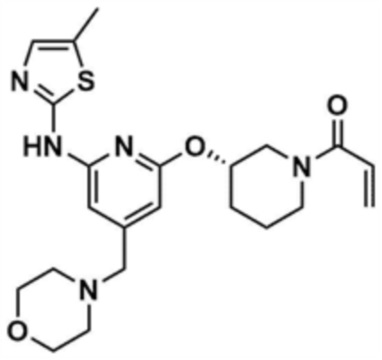
Указанное в заголовке соединение (496,0 мг, выход: 50,0%) получали таким же образом, как и в примере 10, за исключением того, что на стадии 10-1 примера 10 трет-бутил-(S)-3-гидроксипиперидин-1-карбоксилат использовали вместо трет-бутил-3-гидроксипирролидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,95 (м, 1H), 6,99-6,98 (м, 1H), 6,75-6,85 (м, 0,5H), 6,50 (c, 1H), 6,4-6,5 (м, 0,5H), 5,74-5,65 (д, 1H), 6,45-6,43 (д, 1H), 5,24-5,15 (м, 1H), 4,02-4,00 (м, 0,5H), 3,82-3,81 (м, 2H), 3,78 (м, 0,5H), 3,55 (м, 4H), 3,50 (м, 0,5H), 3,15-3,14 (д, 2H), 2,32 (м, 4H), 2,27-2,24 (д, 3H), 2,06-1,96 (м, 1,5H), 1,78-1,72 (м, 2,5H), 1,51 (м, 1H)
Пример 16: Получение (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-она
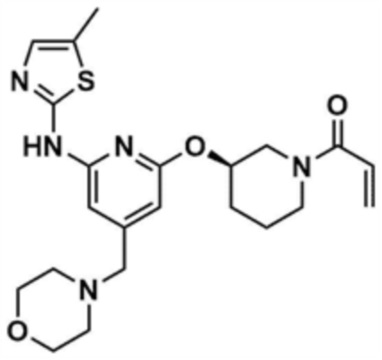
Указанное в заголовке соединение (15,0 мг, выход: 55,0%) получали таким же образом, как и в примере 10, за исключением того, что на стадии 10-1 примера 10 трет-бутил-(R)-3-гидроксипиперидин-1-карбоксилат использовали вместо трет-бутил-3-гидроксипирролидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,95 (м, 1H), 6,99-6,98 (м, 1H), 6,75-6,85 (м, 0,5H), 6,50 (c, 1H), 6,4-6,5 (м, 0,5H), 5,74-5,65 (д, 1H), 6,45-6,43 (д, 1H), 5,24-5,15 (м, 1H), 4,09-4,00 (м, 0,5H), 3,82-3,81 (м, 2H), 3,78 (м, 0,5H), 3,56 (м, 4H), 3,50 (м, 0,5H), 3,15-3,14 (д, 2H), 2,32 (м, 4H), 2,27-2,25 (д, 3H), 2,04 (м, 1,5H), 1,87-1,72 (м, 2,5H), 1,51 (м, 1H)
Пример 17: Получение 1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
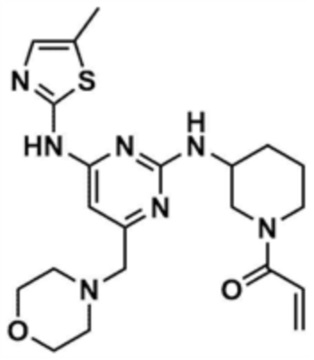
Указанное в заголовке соединение (10,0 мг, выход: 17,5%) получали таким же образом, как и в примере 8, за исключением того, что на стадии 8-1 примера 8 3-амино-1-трет-бутокси-карбонилпиперидин использовали вместо трет-бутил-3-аминопирролидин-1-карбоксилата.
1H ЯМР (500 МГц, CDCl3): 7,25 (c, 1H), 6,56-6,65 (м, 1H), 6,25-6,39 (м, 2H), 5,53 (c, 0,5H), 5,22 (c, 0,5H), 4,26 (c, 1H), 4,09 (c, 1H), 3,77 (c, 4H), 3,72 (c, 0,5H), 3,27-3,37 (м, 4,5H), 2,54 (c, 4H), 2,40 (c, 3H), 2,14 (c, 1H), 1,85 (c, 1H), 1,66-1,67 (м, 2H)
Пример 18: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-она
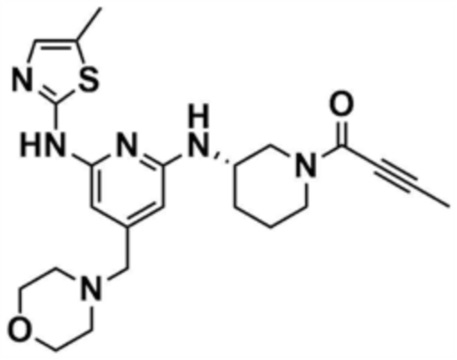
Указанное в заголовке соединение (12,1 мг, выход: 68,0%) получали таким же образом, как и в примере 13, за исключением того, что на стадии 13-1 примера 13 трет-бутил-(S)-3-аминопиперидин-1-карбоксилат использовали вместо трет-бутил-(R)-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,18 (c, 1H), 6,53 (c, 1H), 5,87 (c, 1H), 5,85 (c, 1H), 4,44 (c, 2H), 3,71-3,47 (м, 8H), 2,78 (м, 1H), 2,42 (м, 4H), 2,30 (c, 3H), 1,87-1,58 (м, 4H), 1,80 (c, 3H)
Пример 19: Получение 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-она
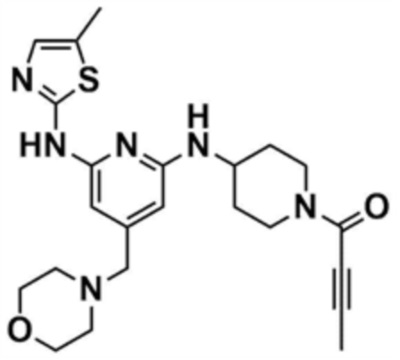
Указанное в заголовке соединение (8,5 мг, выход: 53,0%) получали таким же образом, как и в примере 13, за исключением того, что на стадии 13-1 примера 13 трет-бутил-4-аминопиперидин-1-карбоксилат использовали вместо трет-бутил-(R)-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,18 (c, 1H), 6,53 (c, 1H), 5,87 (c, 1H), 5,80 (c, 1H), 4,44 (c, 2H), 3,59-3,49 (м, 8H), 2,68 (м, 1H), 2,42 (м, 4H), 2,30 (c, 3H), 1,97-1,72 (м, 4H), 1,80 (c, 3H)
Пример 20: Получение (S)-1-(3-((4-((4-ацетилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 20-1: Получение 1-((2,6-дихлорпиридин-4-ил)метил)пиперазина
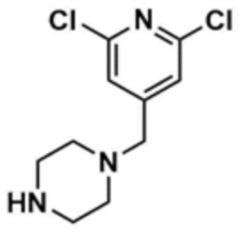
Указанное в заголовке соединение (11,0 мг, выход: 86,0%) получали таким же образом, как и на стадиях 1-1 и 1-2 примера 1, за исключением того, что на стадии 1-1 примера 1 трет-бутилпиперазин-1-карбоксилат использовали вместо морфолина.
Стадия 20-2: Получение 1-(4-((2,6-дихлорпиридин-4-ил)метил)пиперазин-1-ил)этан-1-она
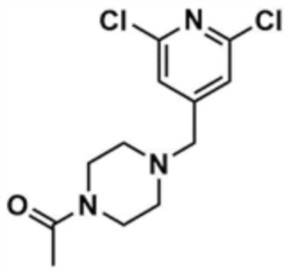
Промежуточное соединение (1,0 г, 1,0 экв.), полученное на стадии 20-1, растворяли в тетрагидрофуране (10,0 мл), и затем к нему добавляли триэтиламин (1,1 мл, 2,0 экв.). К этому добавляли ацетилхлорид (434,6 мкл, 1,5 экв.), и перемешивали в течение 6 ч. После концентрирования добавляли этилацетат (10,0 мл) и воду (10,0 мл), и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (1,0 г, выход: 85,5%).
Стадия 20-3: Получение (S)-1-(3-((4-((4-ацетилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
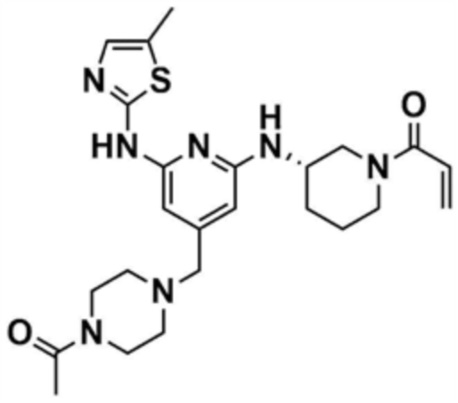
Указанное в заголовке соединение (50,0 мг, выход 45,0%) получали таким же образом, как и в примере 1, за исключением того, что использовали промежуточное соединение, полученное на стадии 20-2 примера 20, вместо промежуточного соединения, полученного на стадии 1-2 примера 1, и использовали трет-бутил-(S)-3-аминопиперидин-1-карбоксилат вместо трет-бутил-3-аминопиперидин-1-карбоксилата на стадиях 1-3.
1H ЯМР (500 МГц, ДМСО): 10,55 (м, 1H), 6,91-6,90 (м, 1H), 6,85-6,75 (м, 0,5H), 6,57-6,48 (м, 1,5H), 6,06-6,04 (м, 1,5H), 5,99-5,96 (м, 1,5H), 5,65-5,63 (м, 0,5H), 5,43-5,41 (м, 0,5H), 4,43-4,40 (м, 0,5H), 4,14-4,10 (м, 1H), 3,98-3,88 (1,5H), 3,40 (м, 4H), 3,24 (c, 2H), 3,15-3,11 (м, 2H), 2,67 (м, 0,5H), 2,33-2,27 (м, 4H), 2,19 (c, 3H), 1,96 (д, 3H), 1,79 (м, 1H), 1,54-1,44 (м, 2,5H)
Пример 21: Получение (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 21-1: Получение метил-(S)-6-((1-(трет-бутоксикарбонил)пиперидин-3-ил)амино)-2-хлорпиримидин-4-карбоксилата
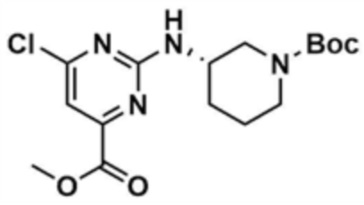
После растворения метил-2,4-дихлорпиримидин-6-карбоксилата (5,0 г, 1,0 экв.) в тетрагидрофуране (100,0 мл) добавляли диизопропилэтиламин (1,2 экв.) и трет-бутил-(S)-3-аминопиперидин-1-карбоксилат (1,2 экв.) и перемешивали при 80°С в течение 1 ч. По завершении реакции смесь охлаждали до 30°C или ниже, к ней добавляли воду (500,0 мл) и дихлорметан (500,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:3) с получением указанного в заголовке соединения (1,7 г, выход: 19,3%).
Стадия 21-2: Получение трет-бутил-(S)-3-((2-хлор-6-(морфолин-4-карбонил)пиримидин-4-ил)амино)пиперидин-1-карбоксилата
После того, как метил-(S)-6-((1-(трет-бутоксикарбонил)пиперидин-3-ил)амино)-2-хлорпиримидин-4-карбоксилат (1,7 г, 1,0 экв.), полученный на стадии 21-1, растворяли в тетрагидрофуране (20,0 мл), к раствору добавляли 1,5,7-триазабицикло[4,4,0]дец-5-ен (0,3 экв.) и морфолин (1,2 экв.) и перемешивали при комнатной температуре в течение 3 ч. После завершения реакции к смеси добавляли воду (200,0 мл) и дихлорметан (200,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=19:1) с получением указанного в заголовке соединения (970,0 мг, выход: 55,7%).
Стадия 21-3: Получение трет-бутил-3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолин-4-карбонил)пиримидин-4-ил)амино)пиперидин-1-карбоксилата
После того, как трет-бутил-(S)-3-((2-хлор-6-(морфолин-4-карбонил)пиримидин-4-ил)амино)пиперидин-1-карбоксилат (950,0 мг, 1,0 экв.), полученный на стадии 21-2, растворяли в 1,4-диоксане (10,0 мл), в раствор добавляли ацетат палладия (0,1 экв), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (0,2 экв), карбонат цезия (3,0 экв) и 2-амино-5-метилтиазол (1,2 экв), и подвергали реакции в микроволновом реакторе (160°С, 30 мин). После завершения реакции к смеси добавляли воду (100,0 мл) и этилацетат (100,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=19:1) с получением указанного в заголовке соединения (900,0 мг, выход: 80,0%).
Стадия 21-4: Получение (S)-N2-(5-метилтиазол-2-ил)-6-(морфолинoметил)-N4-(пиперидин-3-ил)пиримидин-2,4-диамина
После того, как трет-бутил-3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолин-4-карбонил)пиримидин-4-ил)амино)пиперидин-1-карбоксилат (500,0 мг, 1,0 экв.), полученный на стадии 21-3, растворяли в тетрагидрофуране (10,0 мл), к нему добавляли 0,9 М раствор боран-тетрагидрофурана (3,0 экв.), и перемешивали при 50°C в течение 5 ч. Реакционный раствор охлаждали до 0°C и затем добавляли 6 н. водный раствор соляной кислоты (5,0 экв.), и затем перемешивали при 50°C в течение 12 ч. Реакционный раствор снова охлаждали до 0°C, и затем рН доводили до 12 с использованием 12 н. водного раствора гидроксида натрия, и экстрагировали дихлорметаном (200,0 мл) и водой (200,0 мл). Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении, с получением указанного в заголовке соединения (270,0 мг, выход: 69,8%).
Стадия 21-5: Получение (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
После того, как (S)-N2-(5-метилтиазол-2-ил)-6-(морфолинoметил)-N4-(пиперидин-3-ил)пиримидин-2,4-диамин (270,0 мг, 1,0 экв.), полученный на стадии 21-4, растворяли в тетрагидрофуране (4,0 мл) и воде (1,0 мл), к ним добавляли бикарбонат натрия (3,0 экв.) и акрилоилхлорид (1,2 экв.), и перемешивали при 0°C в течение 30 мин. После завершения реакции к смеси добавляли воду (100,0 мл) и дихлорметан (100,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=5:1) с получением указанного в заголовке соединения (45,0 мг, выход: 14,6%).
1H ЯМР (500 МГц, CDCl3): 7,17 (c, 1H), 6,57-6,61 (м, 1H), 6,25-6,42 (м, 2H), 5,53 (c, 1H), 4,25 (c, 1H), 4,10 (c, 1H), 3,77 (c, 4H), 3,72 (c, 0,5H), 3,30-3,37 (м, 4,5H), 2,54 (c, 4H), 2,40 (c, 3H), 2,15 (c, 1H), 1,85 (c, 1H), 1,66-1,67 (м, 2H)
Пример 22: Получение (S)-1-(3-((4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 22-1: Получение 1-((2,6-дихлорпиридин-4-ил)метил)-4-(2-метоксиэтил)пиперазина
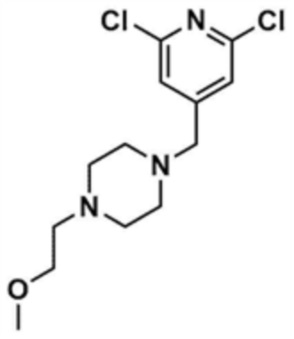
После того, как промежуточное соединение (1,0 г, 1,0 экв.), полученное на стадии 20-1 примера 20, растворяли в тетрагидрофуране (10,0 мл), к нему добавляли триэтиламин (1,1 мл, 2,0 экв.). К этому добавляли 1-бром-2-метоксиэтан (572,0 мкл, 1,5 экв.) и перемешивали в течение 6 ч. После концентрирования добавляли этилацетат (10,0 мл) и воду (10,0 мл), и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (711,1 мг, выход: 60,0%).
Стадия 22-2: Получение (S)-1-(3-((4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
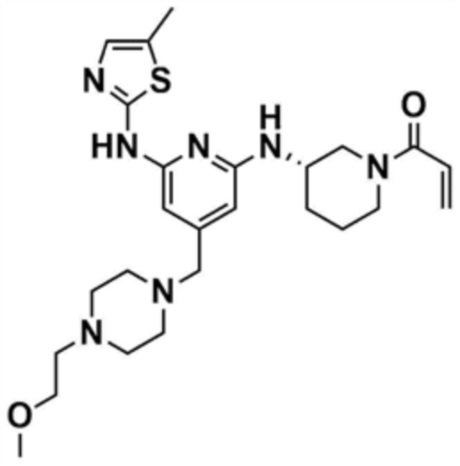
Указанное в заголовке соединение (15,0 мг, выход 68,0%) получали таким же образом, как и в примере 1, за исключением того, что использовали промежуточное соединение, полученное на стадии 22-1 примера 22, вместо промежуточного соединения, полученного на стадии 1-2 примера 1, и использовали трет-бутил-(S)-3-аминопиперидин-1-карбоксилат вместо трет-бутил-3-аминопиперидин-1-карбоксилата на стадии 1-3.
1H ЯМР (500 МГц, CDCl3): 10,50 (м, 1H), 7,04-7,02 (м, 1,5H), 6,60 (м, 0,5H), 6,44-6,49 (м, 1H), 6,33-3,18 (м, 3H), 5,50-5,48 (м, 0,5H), 5,80 (м, 0,5H), 4,52-4,05 (м, 2H), 4,0-3,70 (м, 2H), 3,47 (c, 2H), 3,36 (c, 3H), 2,74-2,52 (м, 13H), 2,35 (c, 3H), 2,12-2,01 (м, 1H), 1,83 (м, 1H), 1,67 (м, 2H)
Пример 23: Получение (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-она
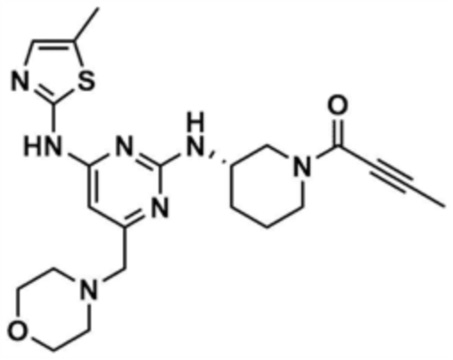
Указанное в заголовке соединение (25,0 мг, выход 10,7%) получали таким же образом, как и в примере 21, за исключением того, что на стадии 21-5 примера 21 использовали бут-2-инoилхлорид вместо акрилоилхлорида.
1H ЯМР (500 МГц, CDCl3): 7,17 (д, 1H), 6,34 (д, 1H), 5,30 (c, 1H), 4,32-4,36 (м, 1H), 4,22 (c, 1H), 4,01 (c, 1H), 3,78 (c, 4H), 3,41-3,45 (м, 1H), 3,39 (д, 2H), 3,36 (c, 1H), 2,54 (c, 4H), 2,45 (c, 3H), 2,17 (c, 1H), 2,04 (c, 3H), 1,62-1,72 (м, 3H)
Пример 24: Получение (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она
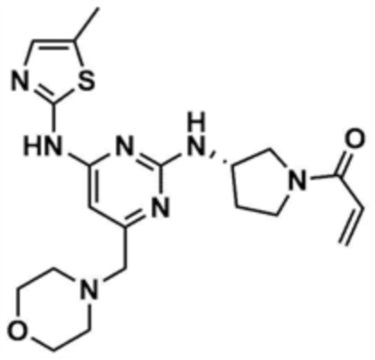
Указанное в заголовке соединение (15,0 мг, выход: 10,1%) получали таким же образом, как и в примере 21, за исключением того, что на стадии 21-1 примера 21 трет-бутил-(S)-3-аминопирролидин-1-карбоксилат использовали вместо трет-бутил-(S)-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, CDCl3): 11,4 (c, 1H), 7,21 (c, 1H), 6,38-6,47 (м, 2H), 5,66-5,72 (м, 1H), 5,34 (c, 1H), 4,75-4,80 (м, 1H), 3,91 (д, 0,5H), 3,68-3,77 (м, 8H), 3,51 (д, 0,5H), 3,38 (д, 2H), 2,54 (c, 4H), 2,43 (д, 3H), 2,17-2,26 (м, 1H), 1,96-2,03 (м, 1H).
Пример 25: Получение (R)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
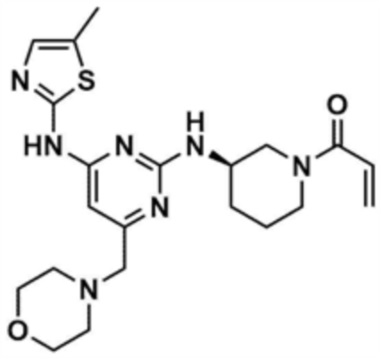
Указанное в заголовке соединение (7,0 мг, выход: 6,2%) получали таким же образом, как и в примере 21, за исключением того, что на стадии 21-1 примера 21 трет-бутил-(R)-3-аминопиперидин-1-карбоксилат использовали вместо трет-бутил-(S)-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, CDCl3): 7,10 (c, 1H), 6,50-6,61 (м, 2H), 6,28-6,31 (м, 3H), 4,40 (c, 1H), 4,20 (c, 1H), 4,01 (c, 1H), 3,77 (c, 4H), 3,30-3,36 (м, 4H), 2,53 (c, 2H), 2,37 (c, 3H), 1,84 (c, 2H), 1,70 (м, 4H)
Пример 26: Получение (S)-1-(3-(метил(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
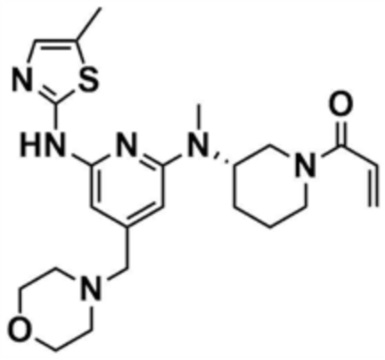
Указанное в заголовке соединение (145,0 мг, выход: 60,0%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 трет-бутил-(S)-3-(метиламино)пиперидин-1-карбоксилат использовали вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, ДМСО): 10,62-10,60 (м, 1H), 6,92 (c, 1H), 6,91-6,81 (м, 0,5H), 6,66 (м, 0,5H), 6,22 (c, 1H), 6,09-5,99 (м, 2H), 5,66-5,64 (м, 0,5H), 5,50-5,48 (м, 0,5H), 4,9 (м, 0,5H), 4,8 (м, 0,5H), 4,49-4,40 (м, 1H), 4,08-3,92 (м, 1H), 3,56 (м, 4H), 3,30 (c, 2H), 3,25-3,22 (м, 0,5H), 2,86 (c, 3H), 2,99-2,96 (м, 0,5H), 2,80-2,77 (м, 0,5H), 2,59-2,54 (м, 0,5H), 2,34 (м, 4H), 2,18 (c, 3H), 1,83-1,77 (м, 3H), 1,50 (м, 1H)
Пример 27: Получение N-(1-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)пиперидин-4-ил)акриламида
Стадия 27-1: Получение трет-бутил-(1-(6-хлор-4-(морфолинoметил)пиридин-2-ил)пиперидин-4-ил)карбамата
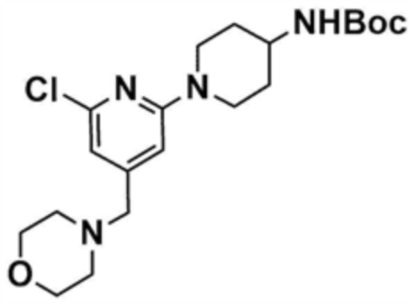
После растворения 4-((2,6-дихлорпиридин-4-ил)метил)морфолина (1,0 г, 3,8 ммоль) в N, N-диметилформамиде (8 мл) в раствор добавляли трет-бутилпиперидин-4-илкарбамат (0,9 г, 3,8 ммоль) и карбонат цезия (1,3 г, 3,8 ммоль) и затем смесь перемешивали при кипячении с обратным холодильником при 80°C в течение 12 ч. После завершения реакции реакционную смесь разбавляли этилацетатом и промывали насыщенным солевым раствором. Органический слой собирали, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали колоночной хроматографией (гексан/этилацетат=1/1) с получением указанного в заголовке соединения (580 мг, выход: 35%).
Стадия 27-2: Получение трет-бутил-(1-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)пиперидин-4-ил)карбамата
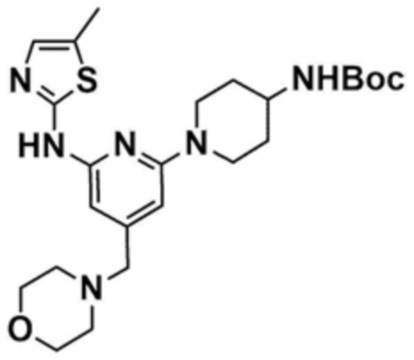
После того, как трет-бутил-(1-(6-хлор-4-(морфолинoметил)пиридин-2-ил)пиперидин-4-ил)карбамат (0,5 г, 1,3 ммоль), полученный на стадии 27-1, растворяли в 1,4-диоксане (9 мл), в раствор последовательно добавляли 5-метилтиазол-2-амин (0,2 г, 1,4 ммоль), ацетат палладия (0,06 г, 0,3 ммоль), Ксантфос (0,3 г, 0,5 ммоль) и карбонат цезия (1,2 г, 3,8 ммоль) и затем подвергали реакции в микроволновом реакторе при 150°С в течение 1 ч. После завершения реакции реакционную смесь фильтровали через целит, концентрировали при пониженном давлении и затем очищали колоночной хроматографией (дихлорметан/метанол=9/1) с получением указанного в заголовке соединения (106 мг, выход: 17%).
Стадия 27-3: Получение N-(6-(4-аминопиперидин-1-ил)-4-(морфолинoметил)пиридин-2-ил)-5-метилтиазол-2-амина
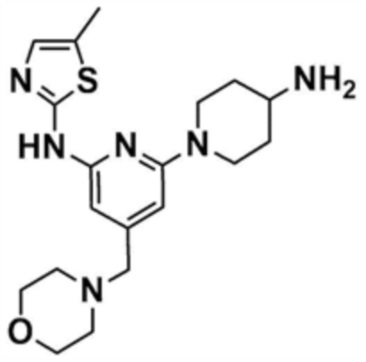
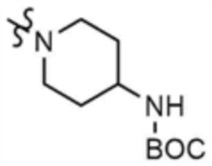
После того, как трет-бутил-(1-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)пиперидин-4-ил)карбамат (0,1 г, 0,2 ммоль), полученный на стадии 27-2, растворяли в дихлорметане (11 мл), в раствор добавляли трифторуксусную кислоту (229 мг, 3,0 ммоль) и затем перемешивали при 20°C в течение 2 ч. После завершения реакции добавляли 1 н. раствор гидроксида натрия для доведения рН до 7, разбавляли этилацетатом и промывали насыщенным солевым раствором. Органические слои собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (195 мг, выход: 100%).
Стадия 27-4: Получение N-(1-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)пиперидин-4-ил)акриламида
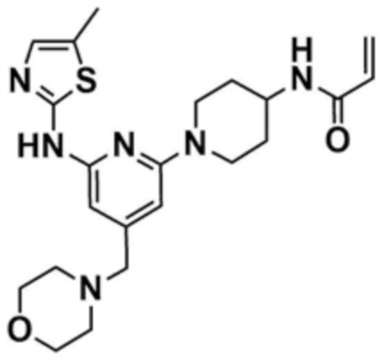
N-(6-(4-аминопиперидин-1-ил)-4-(морфолинoметил)пиридин-2-ил)-5-метилтиазол-2-амин (0,1 г, 0,2 ммоль), полученный на стадии 27-3, растворяли в тетрагидрофуране (6 мл) и воде (2 мл), и затем охлаждали до 0°С, и к нему добавляли бикарбонат натрия (0,08 г, 1,0 ммоль). Акрилоилхлорид (0,02 мл, 0,3 ммоль) медленно добавляли в реакционный раствор и затем перемешивали при 0°С в течение 10 мин. После завершения реакции реакционную смесь разбавляли этилацетатом, промывали насыщенным солевым раствором, концентрировали при пониженном давлении, и очищали колоночной хроматографией (дихлорметан/метанол=9/1) с получением указанного в заголовке соединения (33 мг, выход: 37%).
1H ЯМР (500 МГц, CDCl3): 7,02 (c, 1H), 6,30-6,32 (м, 1H), 6,15-6,20 (м, 1H), 6,01-6,10 (м, 1H), 5,60-5,62 (м, 1H), 5,41-5,45 (м, 1H),4,35-4,38 (м, 2H), 4,15-4,20 (м, 1H),3,78 (c, 4H), 3,38 (c, 2H), 3,07-3,12 (м, 2H), 2,45 (c, 4H), 2,37 (c, 3H), 2,10-2,12 (м, 2H), 1,50-1,60 (м, 2H).
Пример 28: Получение 1-(6-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)октагидро-1H-пирроло[2,3-c]пиридин-1-ил)проп-2-ен-1-она
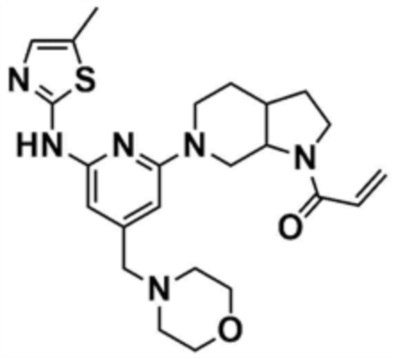
Указанное в заголовке соединение (7 мг, выход 19%) получали таким же образом, как и в примере 27, за исключением того, что на стадии 27-1 примера 27 трет-бутилоктагидро-1H-пирроло-[2,3-c]пиридин-1-карбоксилат использовали вместо трет-бутилпиперидин-4-илкарбамата.
1H ЯМР (500 МГц, CDCl3): 6,97 (c, 1H), 6,40-6,47 (м, 2H), 6,07-6,20 (м, 2H), 5,67-5,71 (м, 1H), 4,32-4,42 (м, 2H), 4,05-4,15 (м, 2H), 3,70 (c, 4H), 3,58-3,62 (м, 2H), 3,50 (c, 2H), 3,32 (c, 2H), 2,95-3,05 (м, 2H), 2,50-2,52 (м, 1H), 2,47 (c, 4H),2,37 (c, 3H), 1,85-1,90 (м, 1H).
Пример 29: Получение 1-(6-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)октагидро-1H-пирроло[2,3-c]пиридин-1-ил)бут-2-ин-1-она
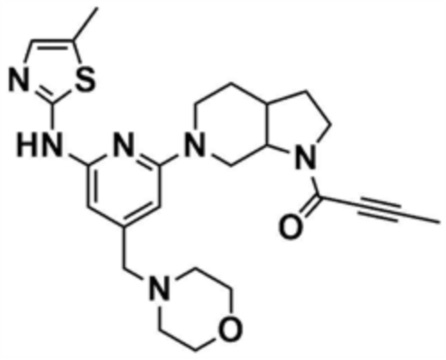
Указанное в заголовке соединение (12 мг, выход: 15%) получали таким же образом, как и в примере 27, за исключением того, что на стадии 27-1 примера 27 трет-бутилоктагидрокси-1H-пирроло-[2,3-c]пиридин-1-карбоксилат использовали вместо трет-бутилпиперидин-4-илкарбамата, и на стадии 27-4 бут-2-иноилхлорид использовали вместо акрилоилхлорида.
1H ЯМР (500 МГц, CDCl3): 6,98 (c, 1H), 6,06-6,10 (м, 2H), 4,20-4,25 (м, 2H), 3,67 (c, 3H), 3,60-3,65 (м, 2H), 3,41-3,50 (м, 2H), 3,31-3,37 (м, 2H), 2,95-3,05 (м, 2H), 2,39-2,50 (м, 4H), 2,38 (c, 3H), 2,27-2,35 (м, 2H), 1,87-1,95 (м, 4H), 1,75-1,87 (м, 2H).
Пример 30: Получение 1-(6-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)-2-азаспиро[3,3]гептан-2-ил)проп-2-ен-1-она
Указанное в заголовке соединение (5 мг, выход 11%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 трет-бутил-6-амино-2-азаспиро[3.3]гептан-2-карбоксилат использовали вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
1H ЯМР (500 МГц, CDCl3): 6,95 (c, 1H), 6,40-6,46 (м, 2H), 6,25-6,30 (м, 1H), 6,18 (м, 1H), 5,69-5,74 (м, 1H), 3,82 (c, 2H), 3,72-3,77 (м, 2H), 3,65-3,70 (м, 2H), 3,10-3,12 (м, 1H), 2,39-2,50 (м, 4H), 2,36 (c, 3H), 1,87-1,95 (м, 4H), 1,85-1,87 (м, 2H), 1,60-1,62 (м, 2H).
Пример 31: Получение (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(пиперидин-1-илметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
Указанное в заголовке соединение (1,0 мг, выход: 1,5%) получали таким же образом, как и в примере 21, за исключением того, что на стадии 21-2 примера 21 пиперидин использовали вместо морфолина.
1H ЯМР (500 МГц, CDCl3): 7,05 (c, 1H), 6,50-6,63 (м, 2H), 6,21-6,28 (м, 2H), 5,70 (c, 0,5H), 5,54 (c, 0,5H), 4,50-4,70 (м, 1H), 4,20-4,40 (м, 4H), 3,31 (c, 2H), 3,20-3,30 (м, 4H), 2,36 (c, 3H), 1,33-1,72 (м, 10H)
Пример 32: Получение (S)-1-(3-((4-((4-этилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
Указанное в заголовке соединение (0,5 мг, выход: 1,1%) получали таким же образом, как и в примере 21, за исключением того, что на стадии 21-2 примера 21 1-этилпиперазин использовали вместо морфолина.
1H ЯМР (500 МГц, CDCl3): 7,06 (c, 1H), 6,50-6,63 (м, 1H), 6,19-6,28 (м, 2H), 5,71 (c, 0,5H), 5,54 (c, 0,5H), 4,20-4,44 (м, 1H), 3,51-3,60 (м, 1H), 3,37 (c, 2H), 3,27-3,32 (м, 1H), 2,52-2,57 (м, 4H), 2,41-2,46 (м, 4H), 2,36 (c, 3H), 1,66-1,84 (м, 8H), 1,08 (т, 3H)
Пример 33: Получение (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 33-1: Получение трет-бутил-4-((2,6-дихлорпиридин-4-ил)окси)пиперидин-1-карбоксилата
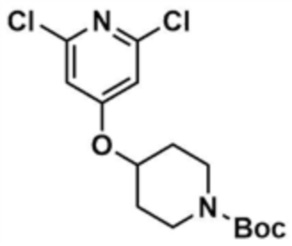
После растворения 1-(трет-бутоксикарбонил)-4-гидроксипиперидина (5,5 г, 1,0 экв.) в диметилформамиде (50,0 мл) добавляли 60% гидрид натрия (3,0 экв) и проводили реакцию при 0°С в течение 10 мин, затем добавляли 2,4,6-трихлорпиридин (1,0 экв) и проводили реакцию в течение 30 мин. После завершения реакции к смеси добавляли воду (500,0 мл) и этилацетат (500,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:5) с получением указанного в заголовке соединения (5,1 г, выход: 53,7%).
Стадия 33-2: Получение 2,6-дихлор-4-(пиперидин-4-илoкси)пиридина
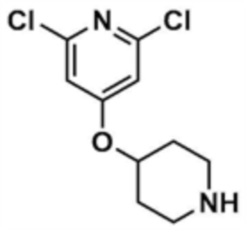
К трет-бутил-4-((2,6-дихлорпиридин-4-ил)окси)пиперидин-1-карбоксилату (5,0 г, 1,0 экв.), полученному на стадии 33-1, добавляли 4M раствор соляной кислоты в диоксане (50,0 мл), и реакцию проводили при комнатной температуре в течение 30 мин. После завершения реакции реакционный раствор охлаждали до 0°C и затем рН доводили до 12 с использованием 12 н. водного раствора гидроксида натрия, и к смеси добавляли воду (250 мл) и этилацетат (500,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении, с получением указанного в заголовке соединения (2,4 г, выход: 68,2%).
Стадия 33-3: Получение 2,6-дихлор-4-((1-метилпиперидин-4-ил)окси)пиридина
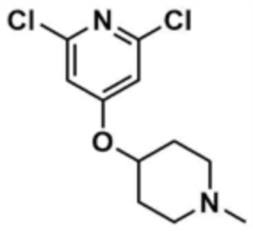
После того, как 2,6-дихлор-4-(пиперидин-4-илoкси)пиридин (2,4 г, 1,0 экв.), полученный на стадии 33-2, растворяли в метаноле (50,0 мл) и дихлорметане (50,0 мл), к нему добавляли раствор формальдегида (1,0 экв), уксусную кислоту (0,1 экв) и триацетоксиборгидрид натрия (2,0 экв), и реакцию проводили при комнатной температуре в течение 30 мин. После завершения реакции к смеси добавляли воду (500,0 мл) и дихлорметан (500,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении, с получением указанного в заголовке соединения (5,1 г, выход: 89,8%).
Стадия 33-4: Получение трет-бутил-(S)-3-((6-хлор-4-((1-метилпиперидин-4-ил)окси)пиридин-2-ил)амино)пиперидин-1-карбоксилата
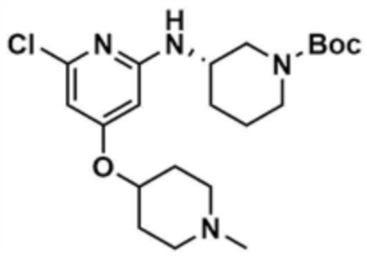
После того, как 2,6-дихлор-4-((1-метилпиперидин-4-ил)окси)пиридин (2,0 г, 1,0 экв.), полученный на стадии 33-3, растворяли в 1,4-диоксане (20,0 мл), в раствор добавляли ацетат палладия (0,1 экв), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (0,2 экв), карбонат цезия (3,0 экв) и 2-амино-5-метилтиазол (1,2 экв), и подвергали реакции в микроволновом реакторе (150°С, 30 мин). После завершения реакции к смеси добавляли воду (250,0 мл) и дихлорметан (250,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=1:1) с получением указанного в заголовке соединения (900,0 мг, выход: 27,7%).
Стадия 33-5: Получение трет-бутил-(S)-3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-карбоксилата
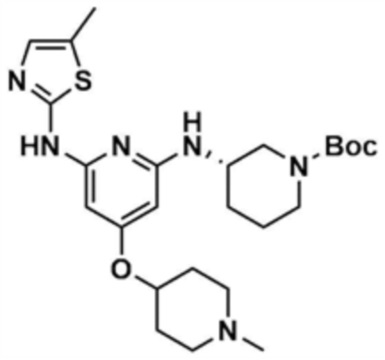
После того, как трет-бутил-(S)-3-((6-хлор-4-((1-метилпиперидин-4-ил)окси)пиридин-2-ил)амино)пиперидин-1-карбоксилат (900,0 мг, 1,0 экв.), полученный на стадии 33-4, растворяли в 1,4-диоксане (20,0 мл), в раствор добавляли ацетат палладия (0,1 экв), 4,5-бис(дифенилфосфино)-9,9-диметилксантен (0,2 экв), карбонат цезия (3,0 экв) и 2-амино-5-метилтиазол (1,1 экв), и подвергали реакции в микроволновом реакторе (160°С, 2 ч). После завершения реакции к смеси добавляли воду (250,0 мл) и этилацетат (250,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=1:1) с получением указанного в заголовке соединения (300,0 мг, выход: 28,3%).
Стадия 33-6: Получение (S)-4-((1-метилпиперидин-4-ил)окси)-N2-(5-метилтиазол-2-ил)-N6-(пиперидин-3-ил)пиридин-2,6-диамина
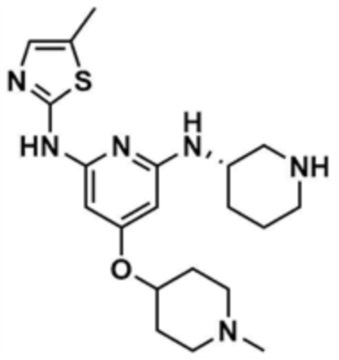
В трет-бутил-(S)-3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-карбоксилат (300,0 мг, 1,0 экв.), полученный на стадии 33-5, добавляли 1,25 М раствор хлористоводородной кислоты в метаноле (5,0 мл) и проводили реакцию при 50°С в течение 12 ч. После завершения реакции реакционный раствор охлаждали при 0°С, и затем pH доводили до 8~9 с использованием насыщенного водного раствора бикарбоната натрия, и к смеси добавляли воду (100,0 мл) и этилацетат (100,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении, с получением указанного в заголовке соединения (150,0 мг, выход: 62,5%).
Стадия 33-7: Получение (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
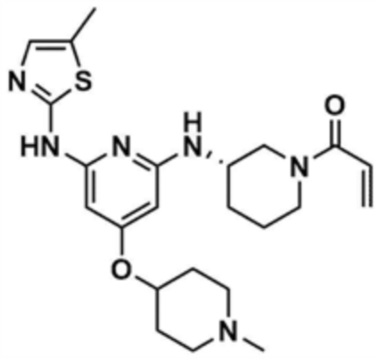
После того, как (S)-4-((1-метилпиперидин-4-ил)окси)-N2-(5-метилтиазол-2-ил)-N6-(пиперидин-3-ил)пиридин-2,6-диамин (130,0 мг, 1,0 экв.), полученный на стадии 33-6, растворяли в тетрагидрофуране (2,4 мл) и воде (0,6 мл), в раствор добавляли бикарбонат натрия (3,0 экв.) и акрилоилхлорид (1,2 экв.), и проводили реакцию при комнатной температуре в течение 1 ч. После завершения реакции к смеси добавляли воду (100,0 мл) и дихлорметан (100,0 мл), с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:метанол=1:1) с получением указанного в заголовке соединения (1,0 мг, выход: 0,7%).
1H ЯМР (500 МГц, CDCl3): 6,99 (c, 1H), 6,60-6,65 (м, 0,5H), 6,46-6,51 (м, 0,5H), 6,32 (д, 0,5H), 6,24 (д, 0,5H), 5,71 (c, 1H), 5,53-5,59 (м, 1H), 5,40 (д, 2H), 4,46 (c, 0,5H), 4,31 (c, 2H), 4,29 (c, 0,5H), 3,93 (д, 1H), 3,86 (c, 0,5H), 3,71 (c, 0,5H), 3,48 (c, 1H), 3,37-3,39 (м, 1H), 2,69 (c, 2H), 2,34 (c, 3H), 2,30 (c, 5H), 2,11 (c, 1H), 2,00 (c, 2H), 1,81 (c, 3H), 1,65 (c, 2H)
Пример 34: Получение (S)-3-(4-((2-((1-акрилоилпиперидин-3-ил)амино)-6-((5-метилтиазол-2-ил)амино)пиридин-4-ил)метил)пиперазин-1-ил)пропаннитрила
Стадия 34-1: Получение 3-(4-((2,6-дихлорпиридин-4-ил)метил)пиперазин-1-ил)пропаннитрила
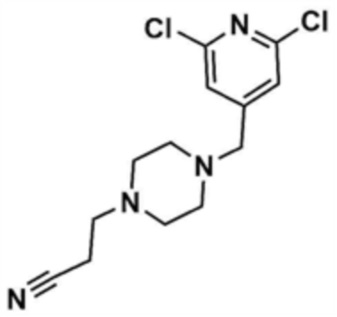
Указанное в заголовке соединение (7,1 г, выход: 57,2%) получали таким же образом, как и на стадиях 1-1 и 1-2 примера 1, за исключением того, что на стадии 1-1 примера 1 3-(пиперазин-1-ил)пропаннитрил использовали вместо морфолина.
Стадия 34-2: Получение трет-бутил-(S)-3-((4-((4-(2-цианоэтил)пиперазин-1-ил)метил)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-карбоксилата
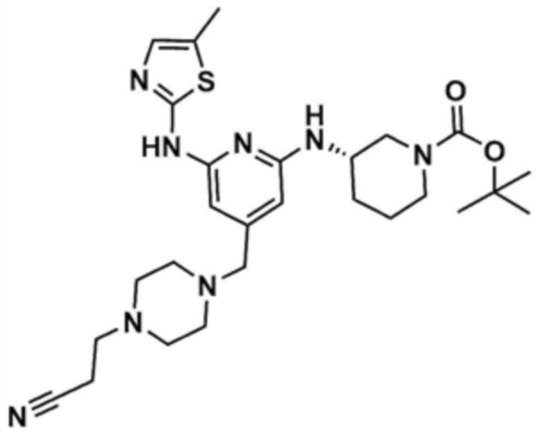
Указанное в заголовке соединение (400 мг, выход: 76,0%) получали таким же образом, как и на стадиях 1-3 и 1-4 примера 1, за исключением того, что промежуточное соединение, полученное на стадии 34-1, использовали вместо промежуточного соединения, полученного на стадии 1-3 примера 1, и трет-бутил-(S)-3-аминопиперидин-1-карбоксилат использовали вместо трет-бутил-3-аминопиперидин-1-карбоксилата.
Стадия 34-3: Получение (S)-3-(4-((2-((5-метилтиазол-2-ил)амино)-6-(пиперидин-3-иламино)пиридин-4-ил)метил)пиперазин-1-ил)пропаннитрила
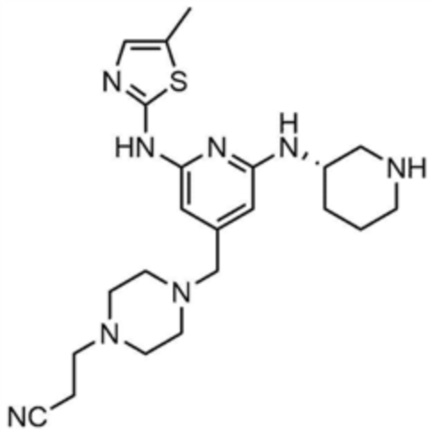
После того, как трет-бутил-(S)-3-((4-((4-(2-цианоэтил)пиперазин-1-ил)метил)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-карбоксилат (100,0 мг, 1,0 экв.), полученный на стадии 34-2, растворяли в дихлорметане (10,0 мл), в раствор добавляли трифторуксусную кислоту (141,5 мкл, 10,0 экв.) при комнатной температуре, и реакционной смеси давали возможность реагировать при комнатной температуре в течение 2 ч. Реакционную смесь нейтрализовали 2,0 М водным раствором гидроксида натрия и затем экстрагировали дихлорметаном. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией с получением 65,0 мг (выход: 77,6%) указанного в заголовке соединения в виде коричневого твердого вещества.
Стадия 34-4: Получение (S)-3-(4-((2-((1-акрилоилпиперидин-3-ил)амино)-6-((5-метилтиазол-2-ил)амино)пиридин-4-ил)метил)пиперазин-1-ил)пропаннитрила
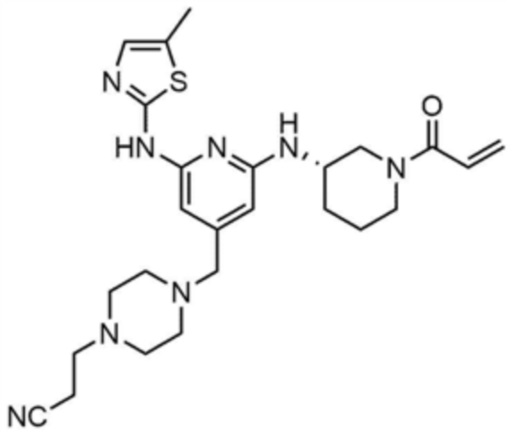
После того, как (S)-3-(4-((2-((5-метилтиазол-2-ил)амино)-6-(пиперидин-3-иламино)пиридин-4-ил)метил)пиперазин-1-ил)пропаннитрил (65,0 мг, 1,0 экв.), полученный на стадии 34-3, растворяли в тетрагидрофуране (5,0 мл) и воде (1,0 мл), гидрокарбонат натрия (24,8 мг, 2,0 экв) добавляли при комнатной температуре и проводили реакцию в течение 30 мин. Акрилоилхлорид (24,0 мкл, 2,0 экв.) добавляли к смеси при комнатной температуре. Реакционной смеси давали возможность прореагировать при комнатной температуре в течение 10 мин, затем добавляли метанол, а также добавляли воду и этилацетат с последующей экстракцией. Отделенный органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией с получением 25,0 мг (выход: 34,1%) указанного в заголовке соединения в виде коричневого твердого вещества.
1H ЯМР (500 МГц, MeOD): 6,89 (д, 1H), 6,84-6,79 (м, 1H), 6,55-6,50 (м, 1H), 6,20-6,03 (м, 2H), 5,47 (д, 1H), 4,61 (д, 1H), 4,38-4,30 (м, 1H), 4,29-4,18 (м, 1H), 4,09-3,97 (м, 2H), 3,34 (с, 3H), 2,83 (т, 1H), 2,61-4,24 (м, 14H), 2,19-2,11 (м, 1H), 1,97-1,88 (м, 1H), 1,71-1,55 (м, 1H).
Пример 35: Получение (S)-1-(3-((4-метил-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
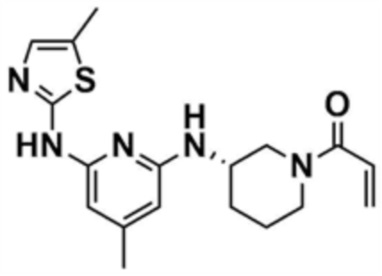
Указанное в заголовке соединение (15 мг, выход: 21%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 использовали 2,6-дихлор-4-метилпиридин вместо 4-((2,6-дихлорпиридин-4-ил)метил)морфолина.
1H ЯМР (500 МГц, CDCl3): 6,94-6,98 (м, 1H), 6,45-6,50 (м, 1H), 6,20-6,25 (м, 1H), 5,97-6,00 (м, 1H), 5,75-5,85 (м, 1H), 5,47-5,51 (м, 1H), 4,22-4,40 (м, 2H), 3,78-3,96 (м, 2H), 3,70-3,72 (м, 1H), 3,27-3,40 (м, 2H), 2,36 (c, 3H), 2,21 (c, 3H), 1,85-1,90 (м, 1H), 1,75-1,80 (м, 1H).
Пример 36: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-3-илметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 36-1: Получение 2,6-дихлор-4-(пиридин-3-илметил)пиридина
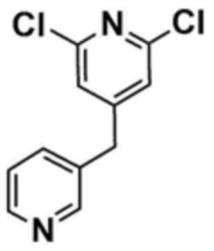
После того, как (2,6-дихлорпиридин-4-ил)бороновую кислоту (0,5 г, 2,6 ммоль) растворяли в 1,4-диоксане (13 мл) и воде (1,6 мл), в раствор последовательно добавляли 3-(бромметил)пиридингидробромид (0,7 г, 1,6 ммоль), карбонат калия (1,8 г, 13,0 ммоль), [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,1 г, 0,2 ммоль), и затем смесь перемешивали при кипячении с обратным холодильником при 110°C в течение 2 ч. После завершения реакции реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией (гексан/этилацетат=1/1) с получением указанного в заголовке соединения (420 мг, выход: 67%).
Стадия 36-2: Получение трет-бутил-(S)-3-((6-хлор-4-(пиридин-3-илметил)пиридин-2-ил)амино)пиперидин-1-карбоксилата
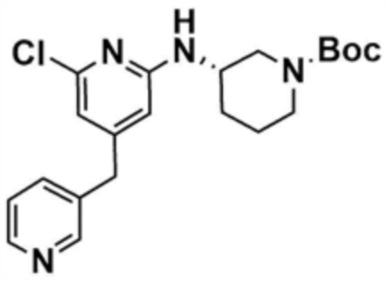
После того, как 2,6-дихлор-4-(пиридин-3-илметил)пиридин (1,7 г, 7,1 ммоль), полученный на стадии 36-1, растворяли в 1,4-диоксане (24 мл), в раствор последовательно добавляли трет-бутил-(S)-3-аминопиперидин-1-карбоксилат (1,6 г, 7,8 ммоль), ацетат палладия (0,2 г, 0,7 ммоль), Ксантфос (0,8 г, 1,4 ммоль) и карбонат натрия (2,3 г, 21,3 ммоль) и затем перемешивали при кипячении с обратным холодильником при 100°C в течение 12 ч. После завершения реакции реакционную смесь фильтровали через целит, концентрировали при пониженном давлении и очищали колоночной хроматографией (этилацетат=100%) с получением указанного в заголовке соединения (280 мг, выход: 15%).
Стадия 36-3: Получение трет-бутил-3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-3-илметил)пиридин-2-ил)амино)пиперидин-1-карбоксилата
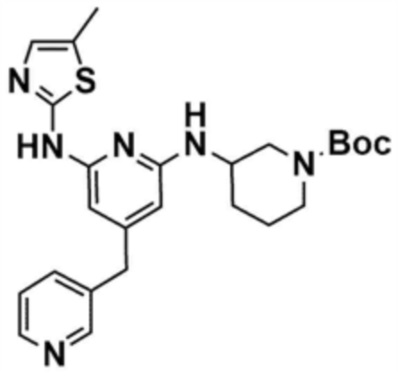
После того, как трет-бутил-(S)-3-((6-хлор-4-(пиридин-3-илметил)пиридин-2-ил)амино)пиперидин-1-карбоксилат (0,4 г, 2,3 ммоль), полученный на стадии 36-2, растворяли в 1,4-диоксане (6 мл), в раствор последовательно добавляли 5-метилтиазол-2-амин (0,1 г, 2,6 ммоль), ацетат палладия (0,02 г, 0,2 ммоль), Ксантфос (0,1 г, 0,5 ммоль) и карбонат цезия (0,9 г, 7,0 ммоль), и подвергали реакции в микроволновом реакторе при 150°С в течение 1 ч. После завершения реакции реакционную смесь фильтровали через целит, концентрировали при пониженном давлении и очищали колоночной хроматографией (дихлорметан/метанол=9/1) с получением указанного в заголовке соединения (360 мг, выход: 84%).
Стадия 36-4: Получение N2-(5-метилтиазол-2-ил)-N6-(пиперидин-3-ил)-4-(пиридин-3-илметил)пиридин-2,6-диамина
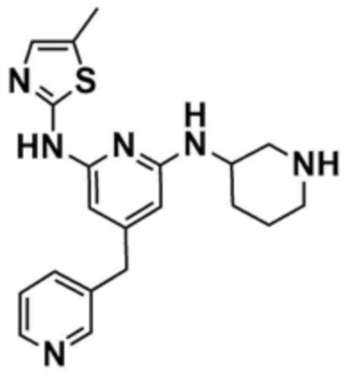
После того, как трет-бутил-3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-3-илметил)пиридин-2-ил)амино)пиперидин-1-карбоксилат (0,2 г, 0,3 ммоль), полученный на стадии 36-3, растворяли в дихлорметане (2 мл), в раствор добавляли трифторуксусную кислоту (0,5 мл, 6,6 ммоль) и затем перемешивали при 20°C в течение 2 ч. После завершения реакции добавляли 1 н. раствор гидроксида натрия для доведения рН до 7, разбавляли этилацетатом и промывали насыщенным солевым раствором. Органические слои собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (180 мг, выход: 100%).
Стадия 36-5: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-3-илметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
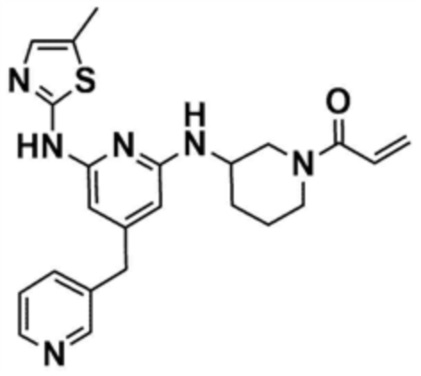
N2-(5-метилтиазол-2-ил)-N6-(пиперидин-3-ил)-4-(пиридин-3-илметил)пиридин-2,6-диамин (0,2 г, 0,4 ммоль), полученный на стадии 36-4, растворяли в тетрагидрофуране (6 мл) и воде (2 мл), и затем охлаждали до 0°С, и в раствор добавляли бикарбонат натрия (0,1 г, 1,1 ммоль). Акрилоилхлорид (0,05 мл, 0,6 ммоль) медленно добавляли в реакционный раствор и затем перемешивали при 0°С в течение 10 мин. После завершения реакции реакционную смесь разбавляли этилацетатом, промывали насыщенным солевым раствором, концентрировали при пониженном давлении, и очищали колоночной хроматографией (дихлорметан/метанол=9/1) с получением указанного в заголовке соединения (19 мг, выход: 25%).
1H ЯМР (500 МГц, CDCl3): 8,49 (м, 2H), 7,45-7,48 (м, 1H), 7,20-7,25 (м, 1H), 6,78 (c, 1H), 6,40-6,59 (м, 1H), 6,18-6,30 (м, 1H), 5,50-5,85 (м, 3H), 4,34-4,48 (м, 1H), 4,15-4,30 (м, 2H), 4,85-4,89 (м, 1H), 3,83 (s 2H), 3,31-3,70 (м, 3H), 2,32 (c, 3H), 2,10-2,15 (м, 1H), 1,80-1,85 (м, 1H).
Пример 37: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-2-илметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
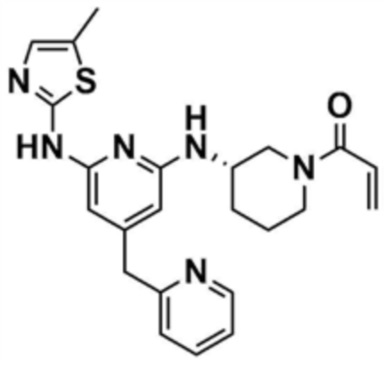
Указанное в заголовке соединение (11 мг, выход: 22%) получали таким же образом, как и в примере 36, за исключением того, что на стадии 36-1 примера 36 использовали 2-(бромметил)пиридингидробромид вместо 3-(бромметил)пиридингидробромида.
1H ЯМР (500 МГц, CDCl3): 8,55 (м, 1H), 7,59-7,61 (м, 1H), 7,27-7,35 (м, 2H), 6,90-6,95 (c, 1H), 6,45-6,55 (м, 1H), 6,20-6,25 (м, 1H), 5,95-6,00 (м, 1H), 5,85-5,90 (м, 1H), 5,42-5,48 (м, 1H), 4,18-4,30 (м, 2H), 3,92 (c, 2H), 3,85-3,90 (м, 1H), 3,35-3,40 (м, 2H), 2,37 (c, 3H), 2,01-2,10 (м, 1H), 1,85-1,90 (м, 1H).
Пример 38: Получение (S)-2-((1-акрилоилпиперидин-3-ил)амино)-6-(5-метилтиазол-2-ил)амино)изоникотинoнитрила
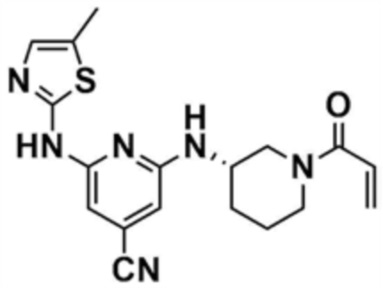
Указанное в заголовке соединение (11 мг, выход: 22%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 использовали 2,6-дихлоризоникотинoнитрил вместо 4-((2,6-дихлорпиридин-4-ил)метил)морфолина.
1H ЯМР (500 МГц, CDCl3): 6,95-7,02 (м, 1H), 6,20-6,45 (м, 3H), 5,50-5,75 (м, 1H), 4,95-5,05 (м, 1H), 4,17-4,28 (м, 1H), 3,80-4,10 (м, 1H), 3,50-3,78 (м, 3H), 2,37 (c, 3H), 2,10-2,15 (м, 1H), 1,80-1,87 (м, 1H).
Пример 39: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-фенилпиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
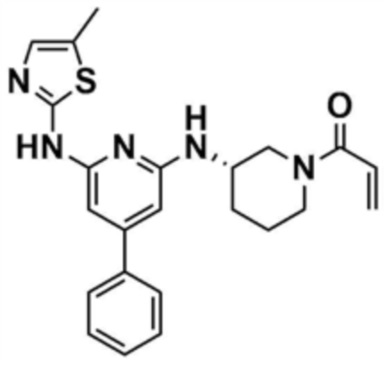
Указанное в заголовке соединение (10 мг, выход: 22%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 1-3 примера 1 использовали 2,6-дихлор-4-фенилпиридин вместо 4-((2,6-дихлорпиридин-4-ил)метил)морфолина.
1H ЯМР (500 МГц, CDCl3): 7,52-7,60 (м, 2H), 7,35-7,50 (м, 3H), 6,95-7,02 (c,1H), 6,42-6,50 (м, 1H), 6,17-6,37 (м, 3H), 5,42-5,50 (м, 1H), 4,42-4,50 (м, 1H), 4,28-4,33 (м, 1H), 3,89-3,95 (м, 1H), 3,33-3,50 (м, 2H), 2,30 (c, 3H), 2,05-2,15 (м, 2H),1,81-1,90 (м, 2H).
Пример 40: Получение (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ин-1-она
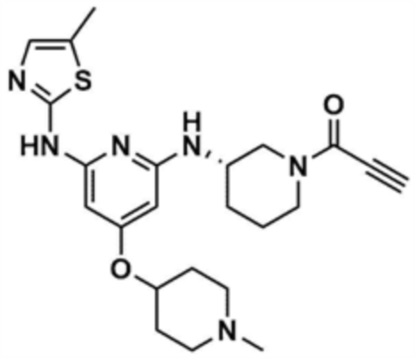
Указанное в заголовке соединение (10,0 мг, выход: 8,9%) получали таким же образом, как и в примере 33, за исключением того, что на стадии 33-7 примера 33 пропиолоилхлорид использовали вместо акрилоилхлорида.
1H ЯМР (500 МГц, CDCl3): 7,40 (c, 1H), 6,97 (c, 1H), 5,74 (д, 1H), 5,55 (д, 1H), 4,31-4,40 (м, 2H), 4,16-4,21 (м, 1H), 4,24 (д, 1H), 3,82-3,87 (м, 1H), 2,67 (c, 2H), 2,35 (т, 3H), 2,30 (c, 6H), 1,94-1,99 (м, 3H), 1,83-1,89 (м, 4H), 1,65-1,74 (м, 3H)
Пример 41: Получение (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-она
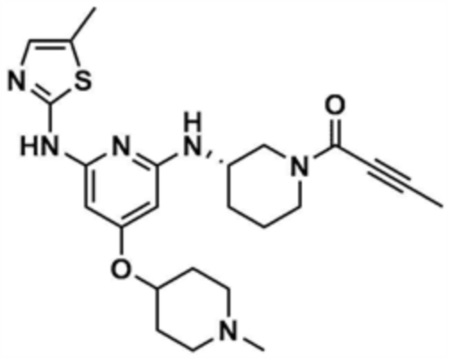
Указанное в заголовке соединение (40,0 мг, выход 34,4%) получали таким же образом, как и в примере 33, за исключением того, что на стадии 33-7 примера 33 использовали бут-2-инoилхлорид вместо акрилоилхлорида.
1H ЯМР (500 МГц, CDCl3): 10,4 (c, 1H), 6,99 (c, 1H), 5,73 (c, 0,5H), 5,71 (c, 0,5H), 5,55 (д, 0,5H), 5,53 (д, 0,5H), 4,35-4,49 (м, 1H), 4,28-4,29 (м, 1H), 4,24 (д, 1H), 3,82-3,86 (м, 1H), 3,33-3,47 (м, 2H), 2,70 (c, 2H), 2,38 (д, 3H), 2,30 (c, 3H), 2,21-2,26 (м, 3H), 2,09-2,13 (м, 1H), 2,00 (c, 2H), 1,80-1,83 (м, 2H), 1,78 (c, 3H), 1,61-1,67 (м, 2H)
Пример 42: Получение (S)-1-(3-((4-((4-этилпиперазин-1-ил)метил)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
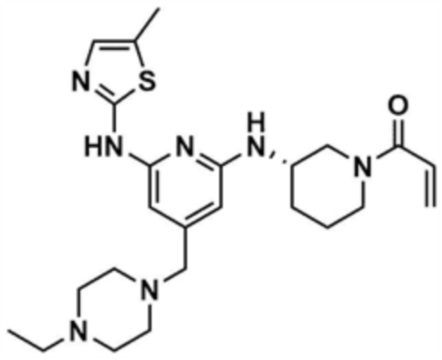
Указанное в заголовке соединение (50,0 мг, выход: 25,0%) получали таким же образом, как и в примере 22, за исключением того, что на стадии 22-1 примера 22 йодэтан использовали вместо 1-бром-2-метоксиэтана.
1H ЯМР (500 МГц, MeOH): 6,89-6,88 (м, 1H), 6,85-6,45 (м, 1H), 6,20-6,02 (м, 3H), 5,80 (м, 0,5H), 5,45 (м, 0,5H), 4,60 (м, 0,5H), 4,4-4,2 (м, 1H), 4,15-4,00 (1,5H), 3,40 (c, 2H), 2,82-2,80 (м, 2H), 2,50 (м, 7H), 2,40 (м, 2H), 2,27 (c, 3H), 2,20-2,10 (м, 1H), 1,92-1,80 (м, 1H), 1,66-1,59 (м, 3H), 1,10-1,07 (т, 3H)
Пример 43: Получение (S)-1-(3-((4-((4-метилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
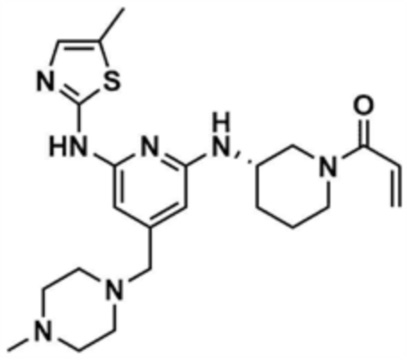
Указанное в заголовке соединение (61,0 мг, выход: 21,0%) получали таким же образом, как и в примере 22, за исключением того, что на стадии 22-1 примера 22 йодметан использовали вместо 1-бром-2-метоксиэтана.
1H ЯМР (500 МГц, ДМСО): 10,53-10,49 (м, 1H), 6,91 (м, 1H), 6,80-6,70 (м, 0,5H), 6,54-6,46 (м, 1,5H), 6,07-5,95 (м, 3H), 5,65-5,63 (м, 0,5H), 5,42-5,40 (м, 0,5H), 4,40-4,50 (м, 0,5H), 4,20-4,00 (м, 1H), 3,90-3,87 (м, 1,5H), 3,26 (c, 2H), 3,12-3,08 (м, 1H), 2,66-2,61 (м, 0,5H), 2,41-2,31 (м, 4H), 2,19 (c, 3H), 2,13 (c, 3H), 2,06-1,97 (м, 3H), 1,81 (м, 1,5H) 1,81 (м, 1H), 1,54-1,44 (м, 2H)
Пример 44: Получение 5-метил-N-(6-метил-4-(морфолинoметил)пиридин-2-ил)тиазол-2-амина
Стадия 44-1: Получение 4-((2-хлор-6-метилпиридин-4-ил)метил)морфолина
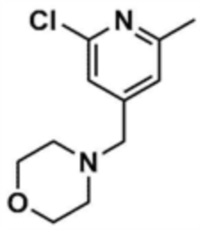
Указанное в заголовке соединение (112,0 мг, выход: 46%) получали таким же образом, как и на стадии 1-1, за исключением того, что на стадии 1-1 примера 1 использовали 2-хлор-6-метилизоникотиновую кислоту вместо 2,6-дихлоризоникотиновой кислоты.
Стадия 44-2: Получение 5-метил-N-(6-метил-4-(морфолинoметил)пиридин-2-ил)тиазол-2-амина
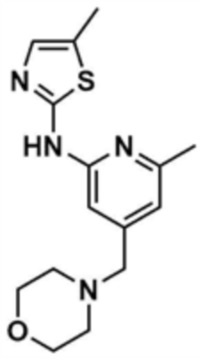
Указанное в заголовке соединение (55,0 мг, выход: 54%) получали таким же образом, как и на стадии 1-2 примера 1, используя промежуточное соединение, полученное на стадии 44-1.
1H ЯМР (500 МГц, CDCl3): 7,10 (c, 1H), 6,72 (c, 1H), 6,67 (c, 1H), 3,74-3,73 (м, 4H), 3,43 (c, 2H), 2,51 (c, 3H), 2,46 (м, 4H), 2,41 (c, 3H)
Пример 45: Получение (S)-3-хлор-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)пропaн-1-она
Стадия 45-1: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он∙гидрохлорида
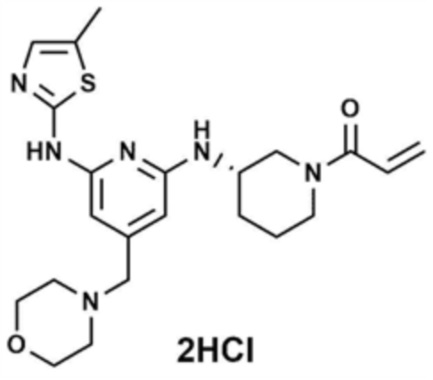
10,0 г (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она в качестве вещества примера 5 растворяли в 200,0 мл этилацетата, к этому добавляли 3 экв. 1 н. соляной кислоты, растворенной в этилацетате. Смесь перемешивали при комнатной температуре в течение 1 ч, отфильтровывали и затем сушили при пониженном давлении в течение 12 ч при комнатной температуре, с получением (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он∙гидрохлорида (11,6 г, выход: 85%).
Стадия 45-2: Получение (S)-3-хлор-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)пропaн-1-она
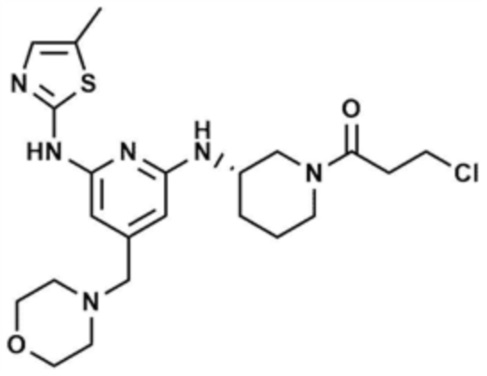
Вещество, полученное на стадии 45-1, хранили при -20°С в течение 7 месяцев. Вещество, полученное во время хранения, разделяли колоночной хроматографией с использованием смешанного растворителя метиленхлорида и метанола в соотношении 15:1 с получением указанного в заголовке соединения (30,0 мг, выход: 5%).
1H ЯМР (500 МГц, CDCl3): 7,03-7,0 (д, 1H), 6,17-6,12 (д, 1H), 5,98 (c, 1H), 4,38-4,30 (м, 1H), 4,20-4,11 (м, 2H), 3,88-3,86 (м, 1H), 3,75-3,70 (м, 4H), 3,68-3,56 (м, 1H), 3,43-3,39 (м, 1H), 3,34 (c, 2H), 2,91-2,80 (м, 1H), 2,80-2,71 (м, 1H), 2,62-2,45 (м, 1H), 2,37 (м, 4H), 2,12 (c, 3H), 2,12-2,11 (м, 1H), 2,10-2,12 (м, 1H), 1,90-1,80 (м, 2H)
Пример 46: Получение (S, E)-3-хлор-1-(3-(6-(5-метилтиазол-2-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
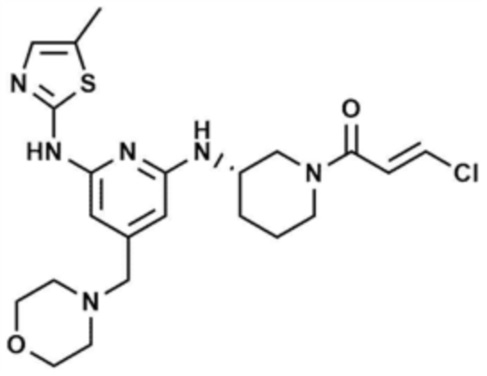
Вещество, полученное на стадии 45-1, хранили при -20°С в течение 7 месяцев. Вещество, полученное во время хранения, разделяли колоночной хроматографией с использованием смешанного растворителя метиленхлорида и метанола в соотношении 15:1 с получением указанного в заголовке соединения (3,0 мг, выход: 0,5%).
1H ЯМР (500 МГц, CDCl3): 6,99 (c, 1H), 6,70-6,60 (м, 1H), 6,4-6,2 (м, 2H), 5,8-5,5 (м, 1H), 3,9-3,8 (м, 2H), 3,8-3,7 (м, 4H), 3,6-3,7 (м, 1H), 3,49 (c, 2H), 3,39-3,41 (м, 2H), 2,52 (м, 4H), 2,33 (c, 3H), 1,85-1,82 (м, 2H), 1,68-1,60 (м, 2H)
Пример 47: Получение (S)-1-(3-(4-((4-(3-аминопропил)пиперазин-1-ил)метил)-6-(5-метилтиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
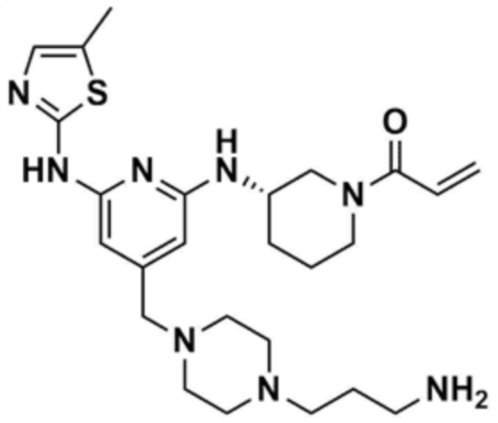
После того, как (S)-3-(4-((2-((1-акрилоилпиперидин-3-ил)амино)-6-((5-метилтиазол-2-ил)амино)пиридин-4-ил)метил)пиперазин-1-ил)пропаннитрил (35,0 мг, 1,0 экв.) в качестве вещества примера 34 растворяли в метаноле (5,0 мл), в раствор добавляли 10% палладий на угле при комнатной температуре и оставляли реагировать в течение 5 мин. Затем реакционную смесь фильтровали с метанолом, используя целит. Отделенный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией с получением указанного в заголовке соединения (4,0 мг, выход: 12,1%) в виде коричневого твердого вещества.
1H ЯМР (500 МГц, MeOD): 6,57 (c, 1H), 6,53-6,50 (м, 1,5H), 6,08-5,98 (м, 2,5H), 5,67-5,61 (м, 0,5H), 5,42-5,38 (м, 0,5H), 4,48-4,29 (м, 0,5H), 4,20-4,06 (м, 1H), 3,98-3,90 (м, 1,5H), 3,29 (c, 2H), 3,17-3,11 (м, 1H), 2,65-2,62 (м, 2,5H), 2,40-2,36 (м, 6H), 2,23 (c, 3H), 2,06-1,99 (м, 3H), 1,80-1,79 (м, 2,5H), 1,77 (м, 2H) 1,54-1,40 (м, 2H)
Пример 48: Получение (S)-1-(3-(6-(1H-пиразол-3-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 48-1: Получение трет-бутил-(S)-3-((6-((1H-пиразол-3-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-карбоксилата
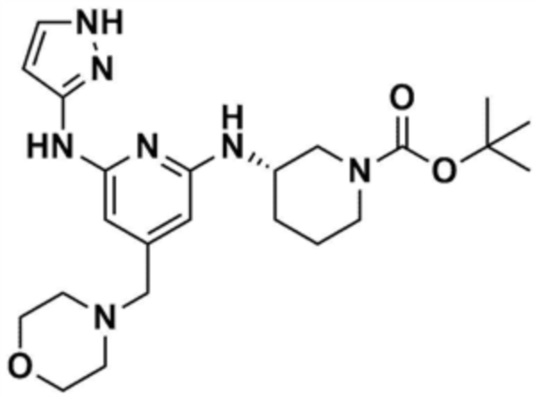
После того, как (S)-трет-бутил-3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-карбоксилат (41,0 мг, 1,0 экв.) растворяли в 1,4-диоксане (2,0 мл), к нему добавляли трис(дибензилиденацетон)дипалладий(0) (9,2 мг, 0,1 экв.) и (±)-2,2'-бис(дифенилфосфино)-1,1'-бинафталин (12,5 мг, 0,2 экв.). Добавляли 1H-пиразол-3-амин (8,3 мг, 1,0 экв.) и затем последовательно добавляли карбонат цезия (97,7 мг, 3,0 экв.). Смесь подвергали реакции в микроволновом реакторе при 130°С в течение 30 мин. После охлаждения до 30°C или менее добавляли воду (10,0 мл) и этилацетат (10,0 мл) и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:1) с получением указанного в заголовке соединения (50,2 мг, выход: 99,8%).
Стадия 48-2: Получение (S)-4-(морфолинoметил)-N2-(пиперидин-3-ил)-N6-(1H-пиразол-3-ил)пиридин-2,6-диамина
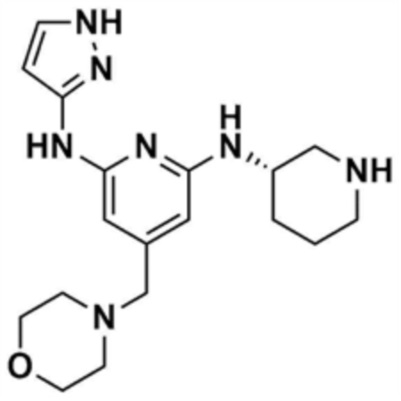
После того, как промежуточное соединение (50,0 мг, 1,0 экв.), полученное на стадии 48-1, растворяли в этилацетате (10,0 мл), водный раствор 6 н. соляной кислоты (0,4 мл, 20,0 экв.) медленно добавляли по каплям и затем перемешивали в течение 2 ч. После доведения pH до 9~12 с использованием 12 н. водного раствора гидроксида натрия, отделенный дихлорметановый слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (34,2 мг, выход: 87,9%).
Стадия 48-3: Получение (S)-1-(3-(6-(1H-пиразол-3-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
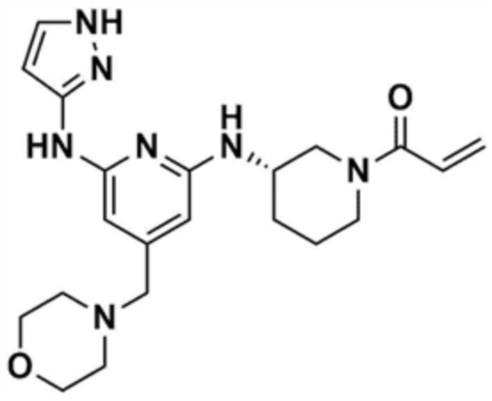
После растворения промежуточного соединения (20,0 мг, 1,0 экв.), полученного на стадии 48-2, в тетрагидрофуране (2,0 мл) добавляли воду (1,0 мл) и добавляли бикарбонат натрия (14,1 мг, 3,0 экв.), и затем охлаждали до 0-10 °С. Медленно по каплям добавляли акрилоилхлорид (5,6 мкл, 1,0 экв.) и затем перемешивали в течение 30 мин для завершения реакции. Слои разделяли дихлорметаном и далее сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=15:1) с получением указанного в заголовке соединения (5,7 мг, выход: 15,4%).
1H ЯМР (500 МГц, MeOD): 7,76 (д, 1H), 6,54 (д, 1H), 6,17-6,13 (м, 1H), 6,06 (c, 2H), 6,02-5,92 (м, 1H), 5,50-5,25 (м, 1H), 3,83-3,74 (м, 4H), 3,64 (c, 2H), 2,65-2,27 (м, 4H), 2,12-2,07 (м, 1H), 1,73 (м, 1H), 1,65-1,48 (м, 2H), 1,43-1,20 (м, 5H)
Пример 49: Получение (S)-1-(3-(4-(морфолинoметил)-6-(5-(трифторметил)тиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 49-1: Получение трет-бутил-(S)-3-((4-(морфолинoметил)-6-((5-(трифторметил)тиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-карбоксилата
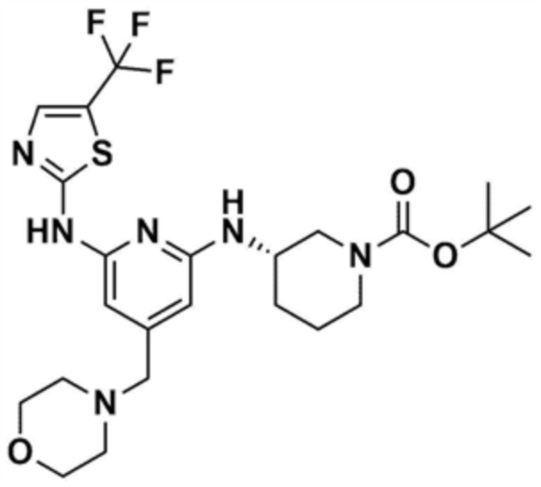
После того, как (S)-трет-бутил-3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-карбоксилат (200,0 мг, 1,0 экв.) растворяли в 1,4-диоксане (2,0 мл), в раствор добавляли ацетат палладия (11,9 мг, 0,1 экв.) и Ксантфос (56,7 мг, 0,2 экв.). Добавляли 5-(трифторметил)тиазол-2-амин (81,8 мг, 1,0 экв.) и затем последовательно добавляли карбонат цезия (476,0 мг, 3,0 экв.). Реакционной смеси давали возможность прореагировать в микроволновом реакторе при 150°С в течение 1 ч. После охлаждения до 30°C или менее добавляли воду (10,0 мл) и этилацетат (10,0 мл) и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:1) с получением указанного в заголовке соединения (68,2 мг, выход: 25,8%).
Стадия 49-2: Получение (S)-4-(морфолинoметил)-N2-(пиперидин-3-ил)-N6-(5-(трифторметил)тиазол-2-ил)пиридин-2,6-диамина
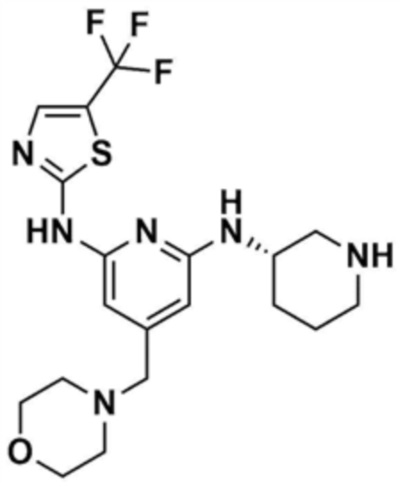
После того, как промежуточное соединение (50,0 мг, 1,0 экв.), полученное на стадии 48-1, растворяли в ацетате (10,0 мл), водный раствор 6 н. соляной кислоты (0,4 мл, 20,0 экв.) медленно добавляли по каплям и затем перемешивали в течение 2 ч. После доведения pH до 9~12 с использованием 12 н. водного раствора гидроксида натрия, отделенный дихлорметановый слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=10:1) с получением указанного в заголовке соединения (27,0 мг, выход: 44,3%).
Стадия 49-3: Получение (S)-1-(3-(4-(морфолинoметил)-6-(5-(трифторметил)тиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
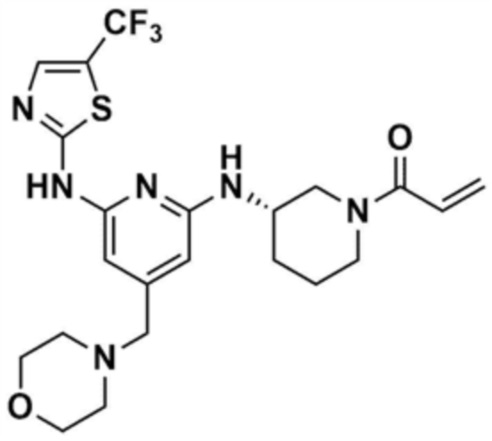
После растворения промежуточного соединения (20,0 мг, 1,0 экв.), полученного на стадии 49-2, в тетрагидрофуране (2,0 мл) добавляли воду (1,0 мл) и добавляли бикарбонат натрия (17,2 мг, 3,0 экв.), и затем охлаждали до 0-10 °С. Медленно по каплям добавляли акрилоилхлорид (4,8 мкл, 1,0 экв.) и затем перемешивали в течение 30 мин для завершения реакции. Слои разделяли и затем сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=15:1) с получением указанного в заголовке соединения (3,6 мг, выход: 16,0%).
1H ЯМР (500 МГц, MeOD): 7,66 (д, 1H), 6,83-6,72 (м, 0,5H), 6,55-6,44 (м, 0,5H), 6,23 (д, 1H), 6,16 (д, 1H), 6,05 (д, 0,5H), 5,73 (д, 0,5H), 5,48 (д, 0,5H), 4,54 (д, 0,5H), 4,43-4,14 (м, 1H), 4,03-3,93 (м, 1,5H), 3,75-3,62 (м, 4H), 3,38 (c, 2H), 3,27-3,18 (м, 1H), 2,86 (т, 0,5H), 2,53-2,38 (м, 4H), 2,28-2,12 (м, 1H), 1,96-1,83 (м, 1H), 1,72-1,49 (м, 2,5H), 1,38-1,23 (м, 1,5H)
Пример 50: Получение (S)-1-(3-(6-(5-хлор-1H-пиразол-3-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
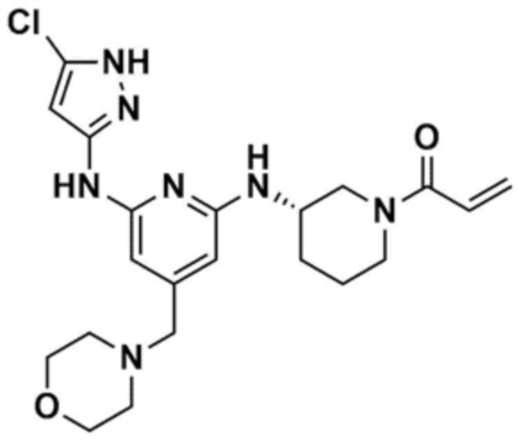
Указанное в заголовке соединение (14,0 мг, выход: 7,7%) получали таким же образом, как и в примере 48, за исключением того, что на стадии 48-1 использовали 5-хлор-1H-пиразол-3-амин вместо 1H-пиразол-3-амина.
1H ЯМР (500 МГц, MeOD): 6,16-6,14 (м, 1H), 6,13-6,11 (м, 1H), 6,06 (c, 2H), 6,03-6,02 (м, 1H), 5,52-5,50 (м, 1H), 3,73-3,70 (м, 4H), 3,62 (с, 2H), 2,60-2,15 (м, 4H), 2,32-2,06 (м, 1H), 1,72-1,65 (м, 1H), 1,65-1,50 (м, 2H), 1,44-1,25 (м, 5H)
Пример 51: Получение (S)-1-(3-(4-(морфолинoметил)-6-(тиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
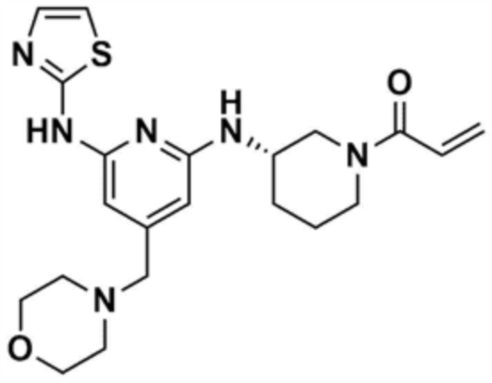
Указанное в заголовке соединение (3,2 мг, выход: 21,1%) получали таким же образом, как и в примере 49, за исключением того, что на стадии 49-1 тиазол-2-амин использовали вместо 5-(трифторметил)тиазол-2-амина.
1H ЯМР (500 МГц, MeOD): 6,68-6,53 (д, 1H), 6,38-6,30 (д, 2H), 6,17-6,05 (м, 2H), 5,89-5,83 (д, 1H), 5,47-5,43 (м, 0,5H), 5,37-5,32 (м, 0,5H), 3,94-3,87 (м, 1H), 3,80-3,66 (м, 2H), 3,63-3,48 (м, 2H), 2,58-2,16 (м, 4H), 2,08-1,73 (м, 4H), 1,65-1,55 (м, 2H), 1,41-1,33 (м, 4H)
Пример 52: Получение (S)-1-(3-(3-фтор-6-(5-метилтиазол-2-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
Стадия 52-1: Получение (2,6-дихлор-3-фторпиридин-4-ил)(морфолино)метанона
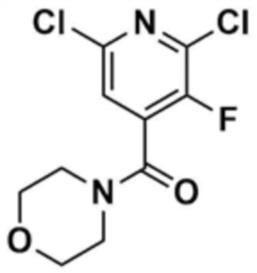
После растворения 2,6-дихлор-3-фторникотиновой кислоты (500,0 мг, 1,0 экв.) в тетрагидрофуране (15,0 мл) в раствор добавляли 1,1-карбонилдиимидазол (463,3 мг, 1,2 экв). Смесь перемешивали при комнатной температуре (25 ~ 30 °С) в течение 1 ч в атмосфере азота, и затем добавляли морфолин (0,2 мл, 1,2 экв.) и перемешивали при той же температуре в течение 2 ч для завершения реакции. К смеси добавляли этилацетат (50,0 мл) и воду (50,0 мл) с последующей экстракцией, и водный слой реэкстрагировали трижды этилацетатом (50,0 мл). Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:5) с получением указанного в заголовке соединения (581,0 мг, выход: 87,5%).
Стадия 52-2: Получение 4-((2,6-дихлор-3-фторпиридин-4-ил)метил)морфолина
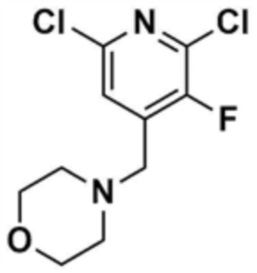
Промежуточное соединение (500,0 мг, 1,0 экв.), полученное на стадии 52-1, растворяли в дихлорметане (20,0 мл) и затем перемешивали при комнатной температуре (25-30 °С). Медленно по каплям добавляли 0,9 М боран-тетрагидрофуран (6,0 мл, 3,0 экв.). Смесь перемешивали при комнатной температуре в течение 12 ч для завершения реакции. Реакционный раствор охлаждали до 0-10 °С, и далее медленно по каплям добавляли 6 н. водный раствор соляной кислоты (39,0 мл, 20,0 экв.), и затем перемешивали при той же температуре в течение 1 ч. После доведения pH до 9-12 с использованием 6 н. водного раствора гидроксида натрия, смесь дважды экстрагировали дихлорметаном. Дихлорметановый слой отделяли, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:1) с получением указанного в заголовке соединения (430,2 мг, выход: 85,9%).
Стадия 52-3: Получение трет-бутил-(S)-3-((6-хлор-3-фтор-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-карбоксилата
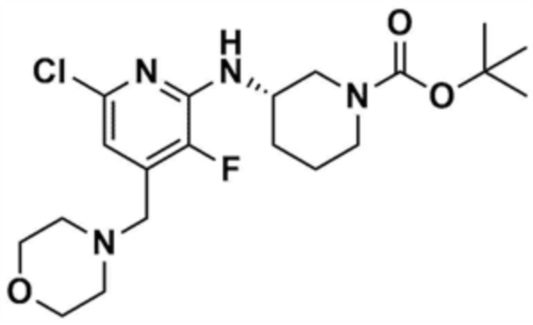
К промежуточному соединению (100,0 мг, 1,0 экв.), полученному на стадии 52-2, добавляли 1,4-диоксан (2,0 мл) и растворяли, и затем к этому добавляли ацетат палладия (9,3 мг, 0,1 экв.) и Ксантфос (43,4 мг, 0,2 экв.). Добавляли трет-бутил-(S)-3-аминопиперидин-1-карбоксилат (75,5 мг, 1,0 экв.) и далее вслед за этим добавляли карбонат цезия (325,8 мг, 3,0 экв.). Смесь подвергали реакции в микроволновом реакторе при 140°С в течение 30 мин. После охлаждения до 30°C или менее добавляли воду (10,0 мл) и этилацетат (10,0 мл) и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:1) с получением указанного в заголовке соединения (144,0 мг, выход: 89,4%).
Стадия 52-4: Получение трет-бутил-(S)-3-((3-фтор-6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-карбоксилата
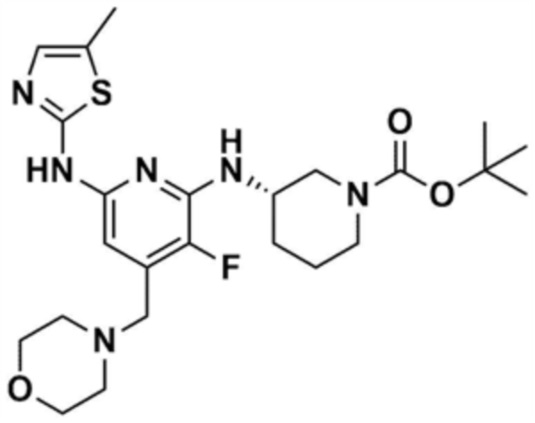
Промежуточное соединение (100,0 мг, 1,0 экв.), полученное на стадии 52-3, растворяли в 1,4-диоксане (2,0 мл). Последовательно добавляли ацетат палладия (5,1 мг, 0,1 экв.), Ксантфос (24,3 мг, 0,2 экв.), 5-метилтиано-2-амин (24,0 мг, 1,0 экв.) и карбонат цезия (205,8 г, 3,0 экв.). Смесь подвергали реакции в микроволновом реакторе при 150°С в течение 30 мин. После охлаждения до 30°C или менее добавляли воду (10,0 мл) и этилацетат (10,0 мл) и затем слои разделяли. Этилацетатный слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (этилацетат:гексан=1:1) с получением указанного в заголовке соединения (86,2 мг, выход: 81,8%).
Стадия 52-5: Получение (S)-3-фтор-N6-(5-метилтиазол-2-ил)-4-(морфолинoметил)-N2-(пиперидин-3-ил)пиридин-2,6-диамина
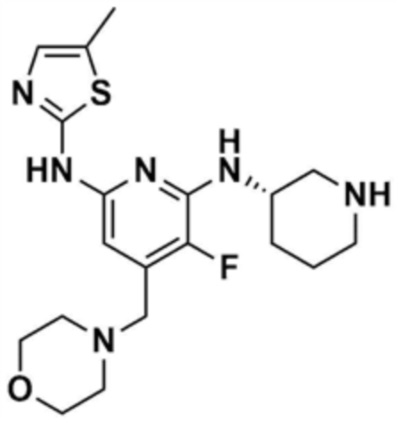
После того, как промежуточное соединение (80,0 мг, 1,0 экв.), полученное на стадии 52-4, растворяли в этилацетате (10,0 мл), водный раствор 6 н. соляной кислоты (0,6 мл, 20,0 экв.) медленно добавляли по каплям и затем перемешивали в течение 2 ч. После доведения pH до 9~12 с использованием 12 н. водного раствора гидроксида натрия, отделенный дихлорметановый слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат (10,0 мл) и получали кристаллы в течение 30 мин. Кристаллы отфильтровывали и затем сушили с получением указанного в заголовке соединения (60,5 мг, выход: 99,9%).
Стадия 52-6: Получение (S)-1-(3-(3-фтор-6-(5-метилтиазол-2-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
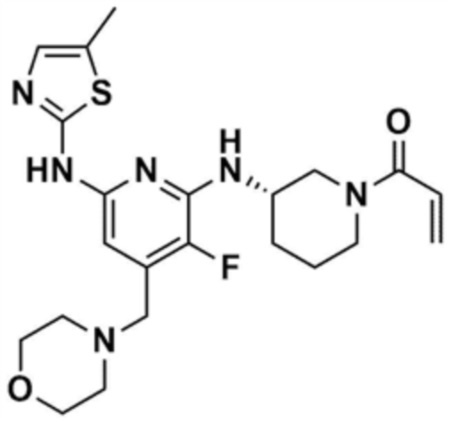
После растворения промежуточного соединения (50,0 мг, 1,0 экв.), полученного на стадии 52-5, в тетрагидрофуране (4,0 мл) добавляли воду (1,0 мл) и добавляли бикарбонат натрия (31,0 мг, 3,0 экв.), и затем охлаждали до 0-10 °С. Медленно по каплям добавляли акрилоилхлорид (9,9 мкл, 1,0 экв.) и затем перемешивали в течение 30 мин для завершения реакции. Слои разделяли дихлорметаном, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией (дихлорметан:метанол=15:1) с получением указанного в заголовке соединения (26,5 мг, выход: 46,8%).
1H ЯМР (500 МГц, MeOD): 6,88-6,80 (м, 1H), 6,65 (м, 1H), 6,21-6,15 (м, 1 H), 5,68 (д, 1H), 5,37-5,28 (м, 1H), 3,72-3,65 (м, 4H), 3,53-3,48 (c, 3H), 2,76-2,69 (м, 2H), 2,52-2,42 (м, 4H), 2,28-2,14 (м, 3H), 2,12-1,98 (м, 2H), 1,66-1,53 (м, 4H)
Пример 53: Получение (S)-1-(3-(4-(морфолинoметил)-6-(пиридин-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
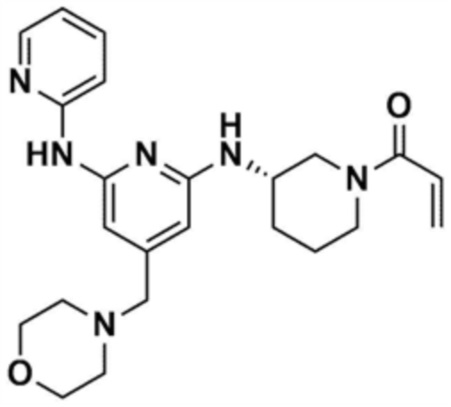
Указанное в заголовке соединение (11,0 мг, выход: 31,0%) получали таким же образом, как и в примере 48, за исключением того, что на стадии 48-1 пиридин-2-амин использовали вместо 1H-пиразол-3-амина.
1H ЯМР (500 МГц, MeOD): 8,50-8,45 (т, 2H), 7,51 (д, 1H), 6,80-6,75 (м, 1H), 6,70-6,62 (м, 0,5H), 6,24-6,16 (м, 1,5H), 6,14-6,08 (д, 0,5H), 5,77-5,51 (м, 0,5H), 4,03-3,92 (м, 1H), 3,91-3,77 (м, 2H), 3,74-3,65 (м, 4H), 3,42-3,37 (м, 3H), 3,26-3,18 (м, 0,5H), 2,81-2,74 (м, 0,5H), 2,54-2,40 (м, 4H), 2,12-1,98 (м, 1H), 1,89-1,81 (м, 1H)
Пример 54: Получение (S)-1-(3-(4-(морфолинoметил)-6-(пиримидин-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-она
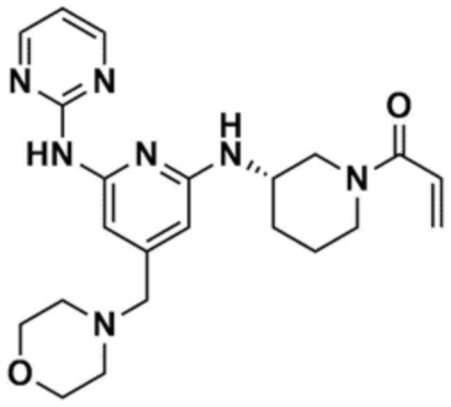
Указанное в заголовке соединение (24,2 мг, 24,3%) получали таким же образом, как и в примере 1, за исключением того, что на стадии 48-1 пиримидин-2-амин использовали вместо 1H-пиразол-3-амина.
1H ЯМР (500 МГц, MeOD): 8,15-8,10 (м, 1H), 7,80 (д, 1H), 7,62-7,56 (м, 1H), 6,86-6,80 (м, 1H), 6,58-6,45 (м, 1,5H), 6,24-6,19 (м, 0,5H), 6,09 (м, 1H), 6,06-6,00 (м, 0,5H), 5,79-5,73 (м, 0,5H), 4,01-3,80 (м, 4H) 3,73-3,65 (м, 4H), 3,36 (c, 2H), 2,52-2,41 (м, 4H), 2,14-2,304 (м, 1H), 1,94-1,86 (м, 2H), 1,73-1,56 (м, 2H)
Экспериментальный пример: Ингибирующая активность в отношении BTK и ITK
Ингибирующую активность в отношении BTK и ITK измеряли для соединений, полученных в приведенных выше примерах, следующим образом.
Ингибирующую активность в отношении BTK оценивали с использованием набора «ADP-Glo™+BTK Kinase enzyme system» (Promega Corporation). В белом 96-луночном планшете 10 мкл фермента BTK, приготовленного таким образом, чтобы иметь конечную концентрацию 1 нг/мкл, смешивали с 5 мкл соединений, имеющих конечную концентрацию 1 мкл, в случае оценки единственной концентрации соединения, и концентрацию 1000, 300, 100, 30, 10, 3, 1, 0,3, 0,1 и 0,03 нМ в случае оценки IC50, и затем проводили реакцию при комнатной температуре в течение 15 мин. 5 мкл субстрата и 5 мкл АТФ, полученных таким образом, чтобы иметь конечную концентрацию 10 мкМ, добавляли в планшет, на котором реакции были завершены, и затем давали возможность прореагировать при температуре 30°С в течение 1 ч. Все лунки планшета обрабатывали 25 мкл реагента ADP-Glo™ и давали возможность прореагировать при 30°С в течение 40 мин. После этого все лунки обрабатывали 50 мкл буфера для детектирования киназы, и затем оставляли реагировать при 30°С в течение 30 мин в защищенных от света условиях. Для планшета, на котором все реакции были завершены, измеряли люминесценцию и рассчитывали результаты. Оценку проводили дважды, и негативный контроль и позитивный контроль рассчитывали в зависимости от того, был ли фермент добавлен без обработки соединения. IC50 рассчитывали на основании вычисленных значений.
Ингибирующую активность в отношении ITK оценивали с использованием набора «ADP-Glo™+ITK Kinase enzyme system» (Promega Corporation). В белом 96-луночном планшете 10 мкл фермента ITK, приготовленного таким образом, чтобы иметь конечную концентрацию 0,4 нг/мкл, смешивали с 5 мкл соединений, имеющих конечную концентрацию 1 мкл, в случае оценки единственной концентрации соединения, и концентрацию 1000, 300, 100, 30, 10, 3, 1, 0,3, 0,1 и 0,03 нМ в случае оценки IC50, и затем проводили реакцию при комнатной температуре в течение 15 мин. В планшет, на котором реакции были завершены, добавляли 5 мкл субстрата и 5 мкл АТФ, полученных таким образом, чтобы иметь конечную концентрацию 25 мкМ, и затем давали возможность прореагировать при температуре 30°С в течение 1 ч. Все лунки планшета обрабатывали 25 мкл реагента ADP-Glo™ и затем давали возможность прореагировать при 30°С в течение 40 мин. После этого все лунки обрабатывали 50 мкл буфера для детектирования киназы, и затем давали возможность реагировать при 30°С в течение 30 мин в защищенных от света условиях. Для планшета, на котором все реакции были завершены, измеряли люминесценцию и рассчитывали результаты. Оценку проводили дважды, и негативный контроль и позитивный контроль рассчитывали в зависимости от того, был ли фермент добавлен без обработки соединения. IC50 рассчитывали на основании вычисленных значений. Результаты показаны в таблице 1 ниже.
[Таблица 1]
(нМ)
(нМ)
(нМ)
(нМ)
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПРОИЗВОДНОЕ ФЕНИЛПИРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2748945C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОПИРИДИНА И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2017 |
|
RU2722594C1 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2733384C1 |
| 3,5-ДИЗАМЕЩЕННОЕ АЛКИНИЛБЕНЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО СОЛЬ | 2013 |
|
RU2576384C1 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| ПРОТИВООПУХОЛЕВОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ПРЕРЫВИСТОГО ВВЕДЕНИЯ ИНГИБИТОРА FGFR | 2014 |
|
RU2664118C2 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2014 |
|
RU2719480C2 |
| НОВОЕ АМИНОАРИЛЬНОЕ ПРОИЗВОДНОЕ, ПРИГОДНОЕ В КАЧЕСТВЕ ИНГИБИТОРА ДИАЦИЛГЛИЦЕРОЛАЦИЛТРАНСФЕРАЗЫ 2, И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2799819C1 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2829484C2 |
Изобретение отсносится к соединению, представленному химической формулой 1, или к его фармацевтически приемлемой соли, где B представляет собой тиазольное, пиразольное, пиридиновое или пиримидиновое кольцо, R1 представляет собой водород, галоген, C1-4 алкил или C1-4 галогеналкил, каждый из X1 и X2 независимо представляет собой N или CR', где R' представляет собой водород или галоген, L представляет собой C1-4 алкилен или -O-, R2 представляет собой циано, C1-4 алкил, фенил, пиридинил, морфолино, пиперазинил или пиперидинил, при этом каждый из пиперазинила и пиперидинила независимо является незамещенным или замещенным C1-4 алкилом, C1-4 алкилом, замещенным циано, C1-4 алкилом, замещенным амино, C1-4 алкилом, замещенным C1-4 алкокси или -CO-(C1-4 алкилом), Y представляет собой связь, -O-, -NH- или -N(C1-4 алкил)-, A представляет собой 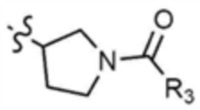 ,
, 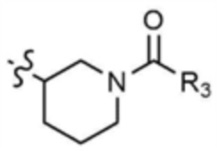 ,
, 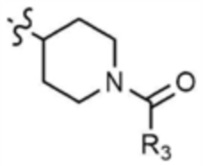 ,
, 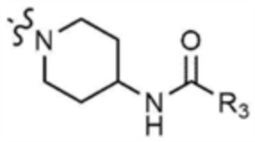 ,
, 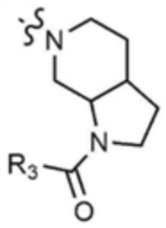 или
или 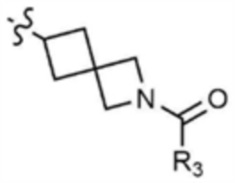 , где R3 представляет собой C1-4 галогеналкил, C2-4 алкенил, C2-4 галогеналкенил или C2-4 алкинил. Изобретение также относится к фармацевтической композиции на основе соединения химической формулы 1. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для профилактики или лечения аутоиммунных заболеваний или раковых заболеваний. 4 н. и 11 з.п. ф-лы, 1 табл., 54 пр.
, где R3 представляет собой C1-4 галогеналкил, C2-4 алкенил, C2-4 галогеналкенил или C2-4 алкинил. Изобретение также относится к фармацевтической композиции на основе соединения химической формулы 1. Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для профилактики или лечения аутоиммунных заболеваний или раковых заболеваний. 4 н. и 11 з.п. ф-лы, 1 табл., 54 пр.
[Химическая формула 1]
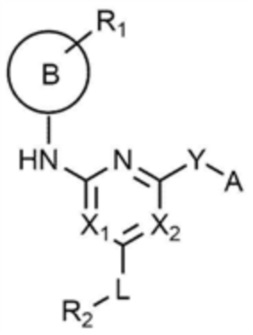
1. Соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль:
[Химическая формула 1]
 ,
,
при этом в химической формуле 1
B представляет собой тиазольное, пиразольное, пиридиновое или пиримидиновое кольцо,
R1 представляет собой водород, галоген, C1-4 алкил или C1-4 галогеналкил,
каждый из X1 и X2 независимо представляет собой N или CR',
где R' представляет собой водород или галоген,
L представляет собой C1-4 алкилен или -O-,
R2 представляет собой пиридинил, морфолино, пиперазинил или пиперидинил,
при этом каждый из пиперазинила и пиперидинила независимо является незамещенным или замещенным C1-4 алкилом, C1-4 алкилом, замещенным циано, C1-4 алкилом, замещенным амино, C1-4 алкилом, замещенным C1-4 алкокси или -CO-(C1-4 алкилом),
Y представляет собой связь, -O-, -NH- или -N(C1-4 алкил)-,
A представляет собой 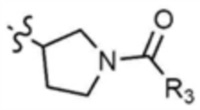 ,
, 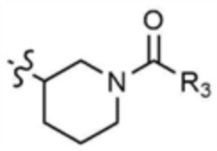 ,
, 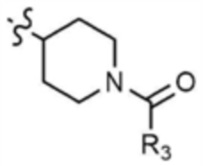 ,
, 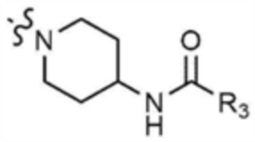 ,
, 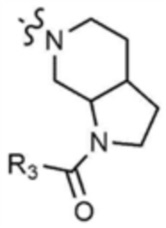 или
или 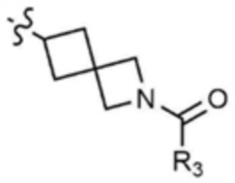 ,
,
где R3 представляет собой C1-4 галогеналкил, C2-4 алкенил, C2-4 галогеналкенил или C2-4 алкинил.
2. Соединение или его фармацевтически приемлемая соль по п.1, где
R1 представляет собой водород, хлор, метил или трифторметил.
3. Соединение или его фармацевтически приемлемая соль по п.1, где
и X1 и X2 представляют собой CH; или
один из X1 и X2 является CF и другой является CH; или
один из X1 и X2 является N и другой является CH.
4. Соединение или его фармацевтически приемлемая соль по п.1, где
L представляет собой метилен или -O-.
5. Соединение или его фармацевтически приемлемая соль по п.1, где
R2 представляет собой пиридинил; морфолино; пиперазинил, замещенный метилом; пиперазинил, замещенный этилом; пиперазинил, замещенный 2-цианоэтилом; пиперазинил, замещенный 3-аминопропилом; пиперазинил, замещенный 2-метоксиэтилом; пиперазинил, замещенный -CO-(метилом); незамещенный пиперидинил; или пиперидинил, замещенный метилом.
6. Соединение или его фармацевтически приемлемая соль по п.1, где
Y представляет собой связь, -O-, -NH- или -N(метил)-.
7. Соединение или его фармацевтически приемлемая соль по п.1, где
R3 представляет собой -CH2CH2Cl, -CH=CH2, -CH=CHCH3, -CH=CHCl, -C≡CH или -C≡CCH3.
8. Соединение или его фармацевтически приемлемая соль по п.1, где
соединение, представленное химической формулой 1, представлено следующей химической формулой 1-1:
[Химическая формула 1-1]
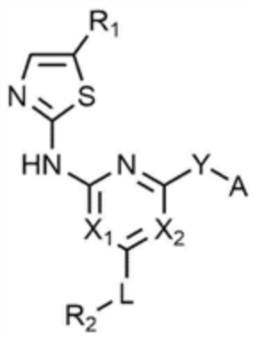 ,
,
при этом в химической формуле 1-1
R1 представляет собой водород, C1-4 алкил или C1-4 галогеналкил,
каждый из X1 и X2 независимо представляет собой N или CR',
где R' представляет собой водород или галоген,
L представляет собой C1-4 алкилен или -O-,
R2 представляет собой пиридинил, морфолино, пиперазинил или пиперидинил,
при этом каждый из пиперазинила и пиперидинила независимо является незамещенным или замещенным C1-4 алкилом, C1-4 алкилом, замещенным циано, C1-4 алкилом, замещенным амино, C1-4 алкилом, замещенным C1-4 алкокси или -CO-(C1-4 алкилом),
Y представляет собой связь, -O-, -NH- или -N(C1-4 алкил)-,
A представляет собой, , 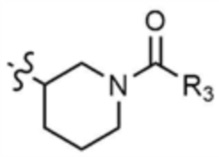 ,
, 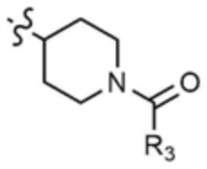 ,
, 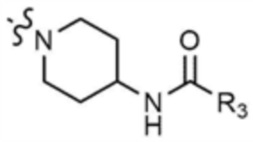 ,
, 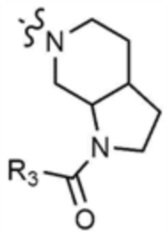 или
или 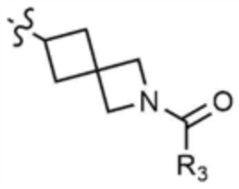 ,
,
где R3 представляет собой C1-4 галогеналкил, C2-4 алкенил, C2-4 галогеналкенил или C2-4 алкинил.
9. Соединение или его фармацевтически приемлемая соль по п.8, где
R1 представляет собой C1-4 алкил,
каждый из X1 и X2 независимо представляет собой N или CH,
L представляет собой C1-4 алкилен или -O-,
R2 представляет собой пиридинил, морфолино, пиперазинил или пиперидинил,
при этом каждый из пиперазинила и пиперидинила независимо является незамещенным или замещенным C1-4 алкилом, C1-4 алкилом, замещенным циано, C1-4 алкилом, замещенным C1-4 алкокси или -CO-(C1-4 алкилом),
Y представляет собой связь, -O-, -NH- или -N(C1-4 алкил)-,
A представляет собой  , , , , или ,
, , , , или ,
где R3 представляет собой C2-4 алкенил или C2-4 алкинил.
10. Соединение или его фармацевтически приемлемая соль по п.1, где
соединение, представленное химической формулой 1, представлено следующей химической формулой 1-2:
[Химическая формула 1-2]
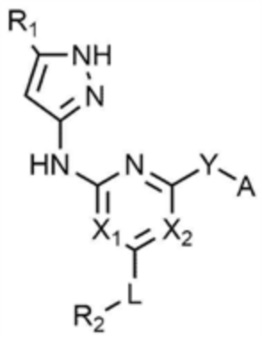
при этом в химической формуле 1-2
R1 представляет собой водород или галоген,
каждый из X1 и X2 независимо представляет собой N или CH,
L представляет собой C1-4 алкилен,
R2 представляет собой морфолино,
Y представляет собой -NH-,
A представляет собой ,
где R3 представляет собой C2-4 алкенил.
11. Соединение или его фармацевтически приемлемая соль по п.1, где
соединение, представленное химической формулой 1, представлено следующей химической формулой 1-3:
[Химическая формула 1-3]
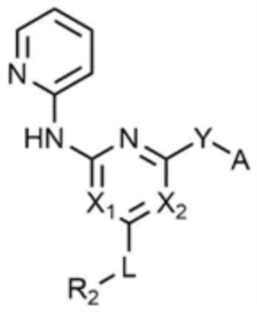 ,
,
при этом в химической формуле 1-3
каждый из X1 и X2 независимо представляет собой N или CH,
L представляет собой C1-4 алкилен,
R2 представляет собой морфолино,
Y представляет собой -NH-,
A представляет собой ,
где R3 представляет собой C2-4 алкенил.
12. Соединение или его фармацевтически приемлемая соль по п.1, где
соединение, представленное химической формулой 1, представлено следующей химической формулой 1-4:
[Химическая формула 1-4]
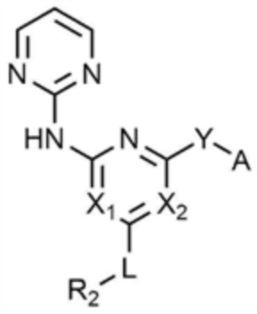 ,
,
при этом в химической формуле 1-4
каждый из X1 и X2 независимо представляет собой N или CH,
L представляет собой C1-4 алкилен,
R2 представляет собой морфолино,
Y представляет собой -NH-,
A представляет собой ,
где R3 представляет собой C2-4 алкенил.
13. Соединение, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль:
[Химическая формула 1]
,
при этом в химической формуле 1
B представляет собой тиазольное кольцо,
R1 представляет собой C1-4 алкил,
X1 и X2 представляет собой CH,
L представляет собой связь,
R2 представляет собой циано, C1-4 алкил или фенил,
Y представляет собой -NH-,
A представляет собой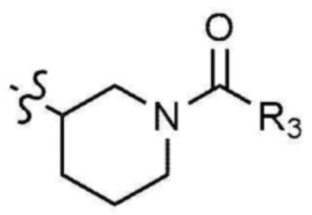 ,
,
где R3 представляет собой C2-4 алкенил.
14. Соединение или его фармацевтически приемлемая соль, где
соединение представляет собой любое соединение, выбранное из группы, состоящей из следующего:
1) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
2) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
3) 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
4) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
5) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
6) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
7) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
8) 1-(3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-4-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
9) (E)-1-(3-((2-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-4-ил)амино)пирролидин-1-ил)бут-2-ен-1-он,
10) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пирролидин-1-ил)проп-2-ен-1-он,
11) 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он,
12) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он,
13) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
14) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пирролидин-1-ил)бут-2-ин-1-он,
15) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он,
16) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)окси)пиперидин-1-ил)проп-2-ен-1-он,
17) 1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
18) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
19) 1-(4-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
20) (S)-1-(3-((4-((4-ацетилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
21) (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
22) (S)-1-(3-((4-((4-(2-метоксиэтил)пиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
23) (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
24) (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-он,
25) (R)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(морфолинoметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
26) (S)-1-(3-(метил(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
27) N-(1-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)пиперидин-4-ил)акриламид,
28) 1-(6-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)октагидро-1H-пирроло[2,3-c]пиридин-1-ил)проп-2-ен-1-он,
29) 1-(6-(6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)октагидро-1H-пирроло[2,3-c]пиридин-1-ил)бут-2-ин-1-он,
30) 1-(6-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)-2-азаспиро[3,3]гептан-2-ил)проп-2-ен-1-он,
31) (S)-1-(3-((4-((5-метилтиазол-2-ил)амино)-6-(пиперидин-1-илметил)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
32) (S)-1-(3-((4-((4-этилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиримидин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
33) (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
34) (S)-3-(4-((2-((1-акрилоилпиперидин-3-ил)амино)-6-((5-метилтиазол-2-ил)амино)пиридин-4-ил)метил)пиперазин-1-ил)пропаннитрил,
35) (S)-1-(3-((4-метил-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
36) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-3-илметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
37) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(пиридин-2-илметил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
38) (S)-2-((1-акрилоилпиперидин-3-ил)амино)-6-(5-метилтиазол-2-ил)амино)изоникотинoнитрил,
39) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-фенилпиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
40) (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ин-1-он,
41) (S)-1-(3-((4-((1-метилпиперидин-4-ил)окси)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)бут-2-ин-1-он,
42) (S)-1-(3-((4-((4-этилпиперазин-1-ил)метил)-6-((5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
43) (S)-1-(3-((4-((4-метилпиперазин-1-ил)метил)-6-(5-метилтиазол-2-ил)амино)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он,
44) 5-метил-N-(6-метил-4-(морфолинoметил)пиридин-2-ил)тиазол-2-амин,
45) (S)-3-хлор-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинoметил)пиридин-2-ил)амино)пиперидин-1-ил)пропaн-1-он,
46) (S, E)-3-хлор-1-(3-(6-(5-метилтиазол-2-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
47) (S)-1-(3-(4-((4-(3-аминопропил)пиперазин-1-ил)метил)-6-(5-метилтиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
48) (S)-1-(3-(6-(1H-пиразол-3-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
49) (S)-1-(3-(4-(морфолинoметил)-6-(5-(трифторметил)тиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
50) (S)-1-(3-(6-(5-хлор-1H-пиразол-3-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
51) (S)-1-(3-(4-(морфолинoметил)-6-(тиазол-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
52) (S)-1-(3-(3-фтор-6-(5-метилтиазол-2-иламино)-4-(морфолинoметил)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он,
53) (S)-1-(3-(4-(морфолинoметил)-6-(пиридин-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он и
54) (S)-1-(3-(4-(морфолинoметил)-6-(пиримидин-2-иламино)пиридин-2-иламино)пиперидин-1-ил)проп-2-ен-1-он.
15. Фармацевтическая композиция для профилактики или лечения аутоиммунных заболеваний или раковых заболеваний, опосредованных киназой BTK (тирозинкиназа Брутона) или ITK (интерлейкин-2-индуцибельная тирозинкиназа), содержащая соединение по любому из пп.1-14 или его фармацевтически приемлемую соль в качестве активного ингредиента.
| WO 2015108861 A1, 23.07.2015 | |||
| WO 2011034907 A2, 24.03.2011 | |||
| WO 2010144359 A1, 16.12.2010 | |||
| WO 2002022602 A2, 21.03.2002 | |||
| WO 2015061247 A2, 30.04.2015 | |||
| WO 2014036016 A1, 06.03.2014 | |||
| Разрядная трубка | 1929 |
|
SU23263A1 |

