Изобретение относится к преимплантационному генетическому тестированию моногенных заболеваний. В настоящее время в мире насчитывается более 350 миллионов людей, страдающих редким заболеванием (по данным RARE Project). Общее количество таких заболеваний по подсчетам European Organization for Rare Diseases (EURORDIS) варьируется от 5 до 7 тысяч. При этом около 80% редких заболеваний имеют генетическую причину. Известная генетическая основа заболевания позволяет с высокой точностью предсказать не только здоровье уже родившегося ребенка, но и оценить риск рождения такого ребенка при анализе генотипов родителей, а также провести генетическую диагностику на самых ранних этапах. Преимплантационное генетическое тестирование (ПГТ) моногенного заболевания становится мощным инструментом для профилактики таких заболеваний.
Настоящее изобретение относится к способу преимплантационного генетического тестирования синдрома Луи-Бара (также известного как атаксия-телеангиэктазия). Синдром Луи-Бара - это наследственное заболевание, характеризующееся прогрессирующей церебральной атаксией. Синдром Луи-Бара встречается с частотой 1 на 40,000-100,000 новорожденных в США. При этом частота зависит от количества близкородственных браков в популяции. Это заболевание приводит к церебральной атаксии, начинающейся в возрасте от 1 до 4 лет, глазодвигательной апраксии, иммунодефициту и повышенному риску развития злокачественных новообразований, особенно лейкемии и лимфомы. При аутосомно-рецессивном типе наследования заболевания вероятность рождения ребенка с этим заболеванием в семье составляет 25%.
К заболеванию синдром Луи-Бара могут приводить патогенные генетические варианты в гене ATM, располагающемся на хромосоме 11 [Amirifar P. Pediatr Allergy Immunol. 2019 May;30(3):277-288]. Этот ген кодирует белок ATM, который участвует в контроле клеточного деления и вовлечен в репарацию ДНК.
ПГТ синдрома Луи-Бара проводится для семей, имеющих подтвержденную молекулярно-генетическую природу заболевания. Важно отметить, что обоснование патогенности и каузативности генетических вариантов происходит до проведения ПГТ моногенного заболевания и не входит ни цели и задачи ПГТ моногенного заболевания, ни в комплекс мероприятий по проведению ПГТ моногенного заболевания. Оценку патогенности проводят по международному стандарту - по критериям, описанным в 2015 году Американским Колледжем Медицинской генетики и Геномики (American College of Medical Genetics and Genomics-Association for Molecular Pathology (ACMG-AMP)) в ходе поиска молекулярно-генетической причины заболевания. ПГТ рекомендуется семьям с высоким риском рождения ребенка с тяжелым (неизлечимым) наследственным заболеванием с установленными патогенными вариантами, обуславливающими этот риск. ПГТ позволяет выбрать из всех эмбрионов, полученных при ЭКО (экстракорпоральном оплодотворении), эмбрионы без патогенных вариантов в компаунд-гетерозиготе и, следовательно, без риска развития заболевания.
Основной проблемой при генетической диагностике эмбрионов является малое исходное количество биоматериала, так как биоптат содержит от одной до трех клеток. В этом случае для повышения эффективности и точности анализа важно как полностью исключить возможность контаминации, так и нивелировать возможный эффект неравномерной и/или неполной амплификации, а также деградации биоматериала. Это требует разработки тест-системы с особыми характеристиками. При этом тест-система разрабатывается с учетом возможности использовать биоматериал разного типа - тотальную дезоксирибонуклеиновую кислоту (ДНК), выделенную из разных тканей, продукт полногеномной амплификации (Whole Genome Amplification, WGA), а также единичные клетки. Сочетание универсальности по отношению к биоматериалу с поэтапной амплификацией целевых фрагментов позволяет проводить анализ нескольких патогенных вариантов для одного образца, в том числе на единичных клетках, а также выявить ситуацию неполной амплификации, контаминации или деградации образца. Еще одной особенностью ПГТ является отсутствие информации о биологических особенностях эмбриона: в отличие от взрослого человека, у эмбриона могут быть любые хромосомные аномалии, которые усложняют задачу оценки статуса эмбриона по конкретному генетическому варианту. Поэтому тест-система для ПГТ моногенного заболевания должна давать возможность выявить такие случаи и оценить их влияние на достоверность результата диагностики.
Наиболее близким техническим решением является проведение ПГТ синдрома Луи-Бара, предложенное Ali Hellani и коллегами [Hellani A. Mol Hum Reprod. 2002 Aug;8(8):785-8] для детекции делеции экзона 19 гена ATM с помощью мультплексной гнездовой полимеразной цепной реакции (ПЦР) в 2 раунда. Важным преимуществом предлагаемого нами подхода является использование не только прямой диагностики, но и использование косвенной диагностики с помощью анализа наследования аллелей полиморфных маркеров, сцепленных с патогенным вариантом. Такой подход позволяет повысить достоверность результата ПГТ моногенных заболеваний (ПГТ-М) за счет увеличения вероятности выявления случая выпадения аллеля (allele drop out, ADO) при непосредственном тесте на наличие патогенного варианта у эмбриона. Также использование косвенной диагностики является основой универсальности предлагаемой нами тест-системы, так как позволяет проводить ПГТ-М для любого патогенного варианта гене ATM.
Представленный нами метод ПГТ синдрома Луи-Бара решает задачу разработки более точного способа преимплантационного генетического тестирования этого моногенного заболевания без использования дорогостоящих приборов и реагентов, который можно было бы применять на биоматериале различного типа: ДНК, выделенную из разных тканей, продукт полногеномной амплификации (WGA), единичные клетки.
Техническим результатом стало создание тест-системы для диагностики патогенного варианта (номер нуклеотида в референсной последовательности геномной ДНК обозначен префиксом NC, номер нуклеотида в референсной последовательности кодирующего транскрипта обозначен префиксом NM): NC_000011.9:g.chr11:108198481delA (NM_000051:c.7085delA, p.Lys2363ArgFS*3), в гене ATM с двойной системой детекции - прямой и косвенной. Такая система необходима при работе с малым количеством биоматериала, так как нестабильная амплификация может привести к потере информации или сниженной точности анализа. Данный вариант не упоминался ранее в литературе как причина синдрома Луи-Бара. Однако, так как он удовлетворял критерии ACMG-AMP - в частности то, что данный вариант находится в аминокислотной позиции с высокой степенью консервативности по данным алгоритмов PhastCons и PhyloP, не встречается у здоровых людей, приводит к сдвигу рамки считывания - он был расценен как патогенный. Прямая диагностика подразумевает анализ непосредственно наличия-отсутствия патогенного варианта. В данном случае для генетического варианта NC_000011.9:g.chr11:108198481delA (NM_000051:c.7085delA, p.Lys2363ArgFS*3) типа делеция (отсутствие нескольких нуклеотидов, англ. Deletion, del) были подобраны праймеры, которые позволяют произвести детекцию мутации по разнице длин фрагментов с помощью капиллярного электрофореза. Косвенная диагностика заключается в анализе наследования молекулярно-генетических маркеров, сцепленных с мутацией, т.е. наследуемых вместе с ней. Для этого на расстоянии не более 3 мБ (что соответствует 3% кроссинговера в среднем) от гена ATM в каждую сторону были выбраны полиморфные локусы, называемые STR (short tandem repeat - короткий тандемный повтор), с гетерозиготностью не менее 0,70 для обеспечения максимальной информативности косвенной диагностики. STR представляют собой повторы 2х и более нуклеотидов расположенные друг за другом (например пара аденин-цитозин (АЦ), повторяющаяся несколько раз подряд: АЦАЦАЦАЦ) и в большом количестве присутствуют в геноме человека. Количество повторов в каждом из них может варьироваться от индивидуума к индивидууму, а также может быть разным у одного и того же человека на 2х гомологичных хромосомах. Гетерозиготность выше 0,70 означает, что высока вероятность того, что у одного и того же человека количество повторов нуклеотидов в данном STR на одной хромосоме будет отличаться от количества повторов в этом же STR на гомологичной хромосоме, другими словами, аллели данного маркера у этого человека будут отличаться между собой по длине. При амплификации фрагмента, содержащего такой маркер, будут получены ампликоны двух разных длин. Проанализировав количество повторов в нескольких маркерах, окружающих патогенный вариант, и изучив то, как они наследуются в тестируемой семье, можно установить сцепление между аллелями маркеров и патогенным вариантом. Диагностическая ценность исследования количества повторов в данных маркерах у эмбрионов состоит в том, что по тому, какой аллель каждого из маркеров был унаследован эмбрионом, можно судить о том, унаследовал ли эмбрион ген ATM, несущий патогенный вариант, или же он унаследовал ген ATM с другой, гомологичной хромосомы, не содержащий патогенный вариант. Для каждого из этих локусов были подобраны праймеры для амплификации по типу гнездовой или полугнездовой ПЦР в 2 раунда, позволяющей повысить точность и эффективность амплификации. В тест-систему были включены 9 STR локусов для гена ATM: D11S107.4, D11S107.41, D11S107.5, D11S107.67, D11S108.2, D11S1778, D11S108.3, D11S108.91, D11S109. Праймеры для амплификации находятся на 11 хромосоме в районе координат 107401327-109080729 (в соответствии с hg19). Важно отметить, что при подборе праймеров соблюдали ряд особенных требований: длина продукта с внешними праймерами для первого раунда ПЦР не должна превышать 500 п.н. (для наработки с фрагментов, получаемых при полногеномной амплификации), длина продукта с внутренних праймеров для второго раунда ПЦР от 120 до 350 п.н., высокая специфичность внешних праймеров, температура отжига не отличается более чем на 1°С.
Подготовительный этап ПГТ
На подготовительном этапе проводится отработка тест-системы: подбор условий амплификации, оптимальных для работы праймеров, анализ эффективности и специфичности ПЦР-амплификации в обоих раундах, оценки универсальности тест-системы для биообразцов различного типа (ДНК, продукт WGA, единичные клетки). При отработке тест-системы были приготовлены стоковые разведения праймеров с концентрацией 10 mM, и рабочие разведения комбинаций праймеров (комбинация пар праймеров для 1 и 2 раунда ПЦР) с концентрацией 10 mM каждого праймера в растворе. Так как в рамках диагностики клинического материала могут быть использованы различные типы матриц, при отработке тест-системы были использованы две биопсии единичных клеток, находящихся в специальном лизирующем буфере (1×PCR Buffer, 0,1% Tween-20, 0,1% Triton X-100, 1 мкг Proteinase K), два образца продуктов полногеномной амплификации биопсиий эмбриона (WGA), а также тотальной ДНК членов семьи, выделенной из крови, для составления родословной и выявления сцепления патогенного варианта с аллелями полиморфных маркеров.
В рамках гнездовой и полугнездовой ПЦР амплификация проводится в два этапа. На первом этапе проводится мультиплексная ПЦР со всеми внешними праймерами для всех локусов, входящих в тест-систему, для обогащения образца всеми целевыми фрагментами. На втором этапе проводится индивидуальная амплификация каждого фрагмента с внутренними праймерами.
Полугнездовая ПЦР
Для первого этапа были подобраны внешние высокоспецифичные праймеры для амплификации фрагментов от 300 до 500 п.н. Для второго этапа были подобраны праймеры для амплификации фрагментов длиной не более 350 пар оснований, а также были введены метки для детекции методом фрагментного анализа. ПЦР-смесь для первого раунда амплификации содержала 1×PCR Buffer with Mg2+ (Евроген, Россия), 0.1 mM каждого деоксинуклеотида, 0.15 μМ каждого праймера, 2,5 U/μl of HsTaq DNA-polymerase (Евроген, Россия), 6% of DMSO и 1 мкл тотальной ДНК или 2,5 мкл WGA или 5 мкл лизирующего буфера с образцом в качестве матрицы. Первый этап амплификации проводился по следующему протоколу: этап денатурации 94°С в течение 2 минут, 30 циклов с понижением температуры отжига праймеров с 62 до 45°С в каждом, этап достройки всех матриц 72°С 10 минут. Далее продукты 1-ого этапа были разнесены в индивидуальные пробирки с одной парой праймеров на определенный локус.
В состав ПЦР смеси для второго этапа входили 1×PCR буфер с Mg2+ (Евроген, Россия), 0.5×RediLoad™ загрузочный буфер (Thermo Fisher Scientific, USA), 0.2 mM каждого деоксинуклеотида, 0.2 μМ каждого праймера, 1U/μl ДНК полимеразы HsTaq (Евроген, Россия), 6% диметилсульфоксида (DMSO) и 1 μl ПЦР-продукта первого этапа амплификации в качестве матрицы. Второй этап амплификации проводился по следующему протоколу: этап денатурации 95°С в течение 2 минут, 35 циклов: денатурация 95°С 30 секунд, отжиг праймеров - 57°С 30 секунд, синтез матрицы - 72°С 1 минута, этап достройки всех матриц 72°С 5 минут. Оценку эффективности и специфичности амплификации проводили с помощью электрофореза в 2% агарозном геле. Результат электрофореза в агарозном геле позволяет определить необходимую степень разведения продуктов амплификации для нанесения на фрагментный анализ (продукты амплификации ДНК членов семьи).
Фрагментный анализ продуктов амплификации проводили с помощью капиллярного электрофореза на приборе 3130×l Genetic Analyzer (Applied Biosystems, USA). По результатам фрагментного анализа составляется родословная и отмечаются информативные полиморфные STR-локусы для каждой семьи, которые в дальнейшем будут использованы в клинической диагностике. Локусы делятся на неинформативные (носитель патогенного варианта гомозиготен по этому локусу), полуинформативные (на некоторых и родительских хромосом аллели по этому маркеру совпадают), информативные (на всех хромосомах родителей аллели этого маркера разные, что дает возможность отличить каждую из них при анализе генотипа эмбриона). Детекция патогенного варианта NC_000011.9:g.chr11:108198481delA (NM_000051:c.7085delA, p.Lys2363ArgFS*3), проводилась с помощью праймеров для амплификации:
Внешние праймеры:
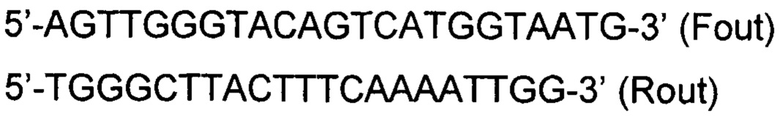
Внутренние праймеры:

Длины продуктов амплификации так же оценивались с помощью фрагментного анализа.
Пример 1
Пациенты А
В ЦГРМ Генетико обратилась семья А, в которой родился ребенок с заболеванием синдром Луи-Бара с компаунд-гетерозиготным носительством патогенных вариантов NC_000011.9:g.chr11:108198481delA (NM_000051:c.7085delA, p.Lys2363ArgFS*3), в гене ATM. Паре было рекомендовано проведение ПГТ заболевания синдром Луи-Бара в рамках ЭКО для отбора эмбрионов, не унаследовавших заболевание.
Гаплотипирование семьи и преимплантационное генетическое тестирование
Был получен биоматериал (периферическая кровь) пациентки и ее партнера для детекции патогенного варианта и выявления информативных полиморфных маркеров. Было проанализировано 9 STR локусов. Из них 8 оказались информативными по матери и/или отцу. Образцы эмбрионов тестировались только на информативные маркеры.
Гаплотипирование проводилось путем анализа наследования аллелей полиморфных маркеров у эмбрионов. В цикле ЭКО было получено 11 эмбрионов, проведена биопсия на 5 день развития (в клинике ЭКО), биоптат в буфере для WGA (1×PBS (Invitrogen, США), 1% поливинилпирролидона (PVP) (Fertipro, Бельгия)) направлен в лабораторию «Генетико». Для контроля контаминации на разных этапах работы с образцом в лаборатории разработана система контролей: контроль контаминации буфера для биопсии, контроль контаминации при транспортировке (одна пробирка с буфером не открывается эмбриологом), контроль контаминации каждого образца (проба среды из последней отмывочной капли биопсиийного материала). Все эти контроли вместе с образцами проходят этап полногеномной амплификации, после которого будет заметно малейшее количество ДНК, контаминировавшей контроли. Полногеномную амплификацию проводили с помощью коммерческого набора SurePlex (lllumina, США).
Продукт полногеномной амплификации, а также ДНК всех членов семьи амплифицировали на 1 этапе в мультиплексной ПЦР с праймерами для детекции патогенного варианта и праймерами для информативных для семьи А полиморфных маркеров в соответствии с разработанным в рамках подготовительного этапа протоколом для тест-системы. На 2 этапе амплификацию проводили для каждого маркера отдельно в соответствии с разработанным протоколом для тест-системы. Таким образом были установлены группы сцепления, унаследованные каждым эмбрионом
Полученные результаты по информативным маркерам (аллели, указанные с одной стороны от символа «/», для разных маркеров располагаются на одной хромосоме, то есть составляют группу сцепления): эмбрионы 1, 6, 7, 8: D11S107.4 - 204, D11S107.41 - 302, D11S107.5 - 273, D11S107.67 - 170, ATM_c.7085delA - c.7085delA, D11S108.2 - 322D11S1778 - 213, D11S108.91 - 280, D11S109 - 266; эмбрион2: D11S107.4 - 215/204, D11S107.41 - ADO/302, D11S107.5 - 264/273, D11S107.67 - 170, ATM_c.7085delA - N/c.7085delA, D11S108.2 - 318/322, D11S1778 - 194/213, D11S108.91 - 264/280, D11S109 - 270/ADO; эмбрион3: D11S107.4 - 215/204, D11S107.41 - 304/302, D11S107.5 - 264/273, D11S107.67 - 170/181, ATM_c.7085 delA - N, D11S108.2 - 318, D11S1778 - 194/196, D11S108.91 - 264/280, D11S109 - 270; эмбрион4: D11S107.4 - 204, D11S107.41 - 302, D11S107.5 - 273, D11S107.67 - 170/181, ATM_c.7085delA - c.7085delA/N, D11S108.2 - 322/318, D11S1778 - 213/196, D11S108.91 - 280, D11S109 - 266/270; эмбрион 5: D11S107.4 - 215/204, D11S107.41 - 304/302, D11S107.5 - 264/273, D11S107.67 - 170, ATM_c.7085delA - N/c.7085delA, D11S108.2 - 318/322, D11S1778 - 194/213, D11S108.91 - 264/280, D11S109 - 270/ADO; эмбрион 9: D11S107.4 - 204, D11S107.41 - 302, D11S107.5 - 273, D11S107.67 - 170/181, ATM_c.7085delA - c.7085delA/N, D11S108.2 - 322/318, D11S1778 - ADO/196, D11S108.91 - 280, D11S109 - 266/270; эмбрион 10: D11S107.4 - 215/204, D11S107.41 - 304/302, D11S107.5 - 264/273, D11S107.67 - 170/181, ATM_c.7085 delA - N, D11S108.2 - 318, D11S1778 - 194/196, D11S108.91 - 264/280, D11S109 - 270/266; эмбрион 11: D11S107.4 - 215/204, D11S107.41 - 304/302, D11S107.5 - 264/273, D11S107.67 - 170, ATM_c.7085delA - N/c.7085delA, D11S108.2 - 318/322, D11S1778 - 194/213, D11S108.91 - 264/280, D11S109 - 270/266; партнер пациентки (носитель патогенного варианта c.7085delA): D11S107.4 - 204/215, D11S107.41 - 302/304, D11S107.5 - 273/264, D11S107.67 - 170/170, ATM_c.7085delA - c.7085delA/N, D11S108.2 - 322/318, D11S1778 - 213/194, D11S108.91 - 280/264, D11S109 -266/270; пациентка (носитель патогенного варианта c.7085delA): D11S107.4 -204/204, D11S107.41 - 302/302, D11S107.5 - 273/273, D11S107.67 - 170/181, ATM_c.7085delA - c.7085delA/N, D11S108.2 - 322/318, D11S1778 - 213/196, D11S108.91 -280/280, D11S109 - 266/270.
В результате гаплотипирования был сделан вывод, что, как у партнера пациентки, так и у самой пациентки, с патогенным вариантом были связаны следующие аллели STR-маркеров: D11S107.4 - 204, D11S107.41 - 302, D11S107.5 - 273, D11S107.67 - 170, D11S108.2 - 322, D11S1778 - 213, D11S108.91 - 280, D11S109-266;
По результатам прямой и косвенной диагностики 7 эмбрионов (эмбрионы 2, 3, 4, 5, 9, 10, 11) не унаследовали заболевание, у 4 эмбрионов (эмбрионы 1, 6, 7, 8) выявлен гаплотип, соответствующий унаследованному заболеванию, т.е. они унаследовали и мутантный аллель гена ATM от обоих родителей. При этом, эмбрионы 3 и 10 унаследовали здоровые аллели гена ATM от обоих родителей, эмбрионы 2, 5, 11 унаследовали патогенный вариант от пациентки и нормальный ген ATM от партнера пациентки, а эмбрионы 4 и 9 унаследовали патогенный вариант от партнера пациентки и нормальный ген ATM от пациентки. С согласия родителей для эмбрионов, не унаследовавших заболевание, провели преимплантационный генетический скрининг хромосомных аномалий. У эмбрионов 3, 5, 9, 10, 11 были выявлены хромосомные аномалии, в связи с чем они не были рекомендованы к переносу. Заключения по скринингу хромосомных аномалий для эмбриона 2 получить не удалось из-за выраженной деградации биоматериала. По результатам всех проведенных анализов эмбрион 4 был рекомендован к переносу.
--->
Перечень последовательностей праймеров
Перечень последовательностей праймеров с указанием маркера или
патогенного варианта, для которых использованы указанные
праймеры (внешние праймеры обозначены как Fout (прямой праймер)
и Rout (обратный праймер), а внутренние - как Fin (прямой праймер)
и Rin (обратный праймер)):
D11S107.4:
SEQ ID NO 1: 5’-CAATCCATGTCCCTCTACAATG-3’ (Fout);
SEQ ID NO 2: 5’-TTGAAGGCTGCCATTGATAC-3’ (Rout);
SEQ ID NO 3: 5’-CACCCAAAGATTCTGATAAACATC-3’ (Fin);
D11S107.41:
SEQ ID NO 4: 5’-AGAGCCATACAGAAGTCACCAT-3’ (Fout);
SEQ ID NO 5: 5’-TCCAACCTCAAATTCAGCC-3’ (Rout);
SEQ ID NO 6: 5’-GATTCCAGGCCCCATACT-3’ (Rin);
D11S107.5:
SEQ ID NO 7: 5’-CCCAGAACAGGGCTTCAT-3’ (Fout);
SEQ ID NO 8: 5’-ATAGTCCTCAATGCGTTGTCC-3’ (Rout);
SEQ ID NO 9: 5’-ATGTACTCACAGACTTGTGGCTC-3’ (Rin);
D11S107.67:
SEQ ID NO 10: 5’-TGCTTTTGTTTGGTTGGACA-3’ (Fout);
SEQ ID NO 11: 5’-CAACAGTGCGAGACTCCG-3’ (Rout);
SEQ ID NO 12: 5’-GGACAGTGCTTGCCACG-3’ (Fin);
D11S108.2:
SEQ ID NO 13: 5’-TACGGGTGTTGAAGGTGTCTT-3’ (Fout);
SEQ ID NO 14: 5’-ATGTATGTTACATGGGCTTAGGTC-3’ (Rout);
SEQ ID NO 15: 5’-TATTGATAAGGCACTGGAATACGA-3’ (Rin);
D11S1778:
SEQ ID NO 16: 5’-AATGGCAAACCAATTCCCT-3’ (Fout);
SEQ ID NO 17: 5’-AGCCACACACCTTTGACTAGC-3’ (Rout);
SEQ ID NO 18: 5’-CAATGGGGTTCTGGAGAGATAG-3’ (Fin);
D11S108.3:
SEQ ID NO 19: 5’-GCTGGCTGTTGATTGCC-3’ (Fout);
SEQ ID NO 20: 5’-GACCCTTTCATCGTCCCA-3’ (Rout);
SEQ ID NO 21: 5’-CTGTAGGCACACGACACCA-3’ (Fin);
SEQ ID NO 22: 5’-ACAATCACCACCGGGTCTAC-3’ (Rin);
D11S108.91:
SEQ ID NO 23: 5’-CAGCCAATGCACTGAAAGT-3’ (Fout);
SEQ ID NO 24: 5’-GTACACAACTTTGGCGGG-3’ (Rout);
SEQ ID NO 25: 5’-ATTCCTGGGATGCTTCCA-3’ (Rin);
D11S109:
SEQ ID NO 26: 5’-ACTTGTACTCTTTGACTTCCCCA-3’ (Fout);
SEQ ID NO 27: 5’-GCATTAACCCAGACAGCCA-3’ (Rout);
SEQ ID NO 28: 5’-CACAGGTAAGAGGGATCATGTAGA-3’ (Fin).
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ преимплантационного генетического тестирования семейной гипертрофической кардиомиопатии | 2021 |
|
RU2772938C1 |
| Способ преимплантационного генетического тестирования семейной фокальной эпилепсии 1 типа | 2021 |
|
RU2777084C1 |
| Способ преимплантационного генетического тестирования буллезного эпидермолиза | 2021 |
|
RU2777086C1 |
| Способ преимплантационного генетического тестирования анемии Фанкони | 2022 |
|
RU2792147C1 |
| Способ преимплантационного генетического тестирования несиндромальной нейросенсорной тугоухости | 2022 |
|
RU2791878C1 |
| Способ преимплантационного генетического тестирования муковисцидоза | 2021 |
|
RU2777078C1 |
| Способ преимплантационного генетического тестирования болезни Канаван | 2020 |
|
RU2748289C1 |
| Способ преимплантационного генетического тестирования синдрома Смита-Лемли-Опица | 2023 |
|
RU2816650C1 |
| Способ преимплантационного генетического тестирования синдрома Альпорта | 2022 |
|
RU2795481C1 |
| Способ преимплантационного генетического тестирования наследственных множественных остеохондром типа 1 | 2022 |
|
RU2795824C1 |
Изобретение относится к биотехнологии. Описан способ диагностики моногенного заболевания синдром Луи-Бара в условиях преимплантационного генетического тестирования (ПГТ). Для этого проводят диагностику патогенных вариантов NC_000011.9:g.chr11:108198481delA (NM_000051:c.7085delA, p.Lys2363ArgFS*3), в гене ATM. В рамках способа были показаны высокая специфичность и эффективность, а также универсальность в отношении биообразцов различного типа: единичные клетки, продукт полногеномной амплификации, тотальной ДНК, выделенной из разных тканей. С помощью разработанной тест-системы было проведено преимплантационное генетическое тестирование заболевания синдром Луи-Бара для семей с высоким риском развития этого заболевания у будущих детей. 1 пр.
Способ преимплантационного генетического тестирования синдрома Луи-Бара, предусматривающий выявление наследования патогенных вариантов NC_000011.9:g.chr11:108198481delA (NM_000051:c.7085delA, p.Lys2363ArgFS*3) в гене ATM, включающий двойную систему детекции – прямую и косвенную, где прямую детекцию осуществляют с помощью праймеров
SEQ ID NO 29: 5’-AGTTGGGTACATGGTAATG-3’ (Fout),
SEQ ID NO 30: 5’-TGGGCTTACTTTCAAAATTGG-3’ (Rout),
SEQ ID NO 31: 5’-CTGGTTAGCAGAAACGTGTGCT-3’ (Fin),
а косвенную детекцию осуществляют с помощью праймеров для анализа наследования молекулярно-генетических полиморфных маркеров типа (STR), сцепленных с патогенным вариантом, выбранных из SEQ ID №1-28, при этом используют праймеры, направленные на те STR, аллели которых разные на всех хромосомах родителей, где внешние праймеры обозначены как Fout (прямой праймер) и Rout (обратный праймер), а внутренние праймеры как Fin (прямой праймер) и Rin (обратный праймер), при этом диагностику проводят в два этапа полугнездовой ПЦР: на первом этапе проводят мультиплексную ПЦР с внешними праймерами для STR и гена ATM, на втором этапе проводят индивидуальную ПЦР каждого фрагмента с внутренними праймерами для STR, а также методом ПЦР-ПДРФ для определения патогенного варианта гена ATM.
| Hellani A | |||
| и др., Pregnancy after preimplantation genetic diagnosis for Ataxia Telangiectasia, Mol Hum Reprod | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| Гильза для удерживания скрепляющих подошвенные части винтов | 1916 |
|
SU785A1 |
| И.Ю | |||
| Коган и др., Преимплантационное генетическое тестирование моногенных заболеваний | |||
| Описание клинического случая | |||
| Журнал акушерства и женских болезней, 2018, том 67, выпуск 1, стр | |||
| Автоматический огнетушитель | 0 |
|
SU92A1 |
| Соловьева | |||

