Настоящее изобретение относится к CH-кислотным сложным метакриловым эфирам, а также к сополимерам, получаемым из CH-кислотных сложных метакриловых эфиров. Кроме того, настоящее изобретение относится к композициям для нанесения покрытия, содержащим такие сополимеры.
Смолы, сшиваемые при комнатной температуре, механизм сшивания которых основан на реакциях с участием карбонильных групп, являются известными в уровне техники и описаны, например, в EP 0016518, DE 4237030, WO 2009/146995 A1 или EP 2246403 для областей применения, связанных с составами красок или клеящими веществами. В целом, такие системы смол обычно содержат сополимеры – в более поздних вариантах реализации обычно полимеры, полученные в результате полимеризации в водной эмульсии, не содержащей VOC, образованные из сомономеров с боковой карбонильной функциональной группой. Наиболее часто применяемыми для данной цели являются диацетонакриламид (DAAM) или ацетоацетоксиэтилметакрилат (AEEMA).
Однако данные два мономера с карбонильными функциональными группами обладают критическими недостатками: карбонильная функциональная группа в относительно высокой степени является стерически затрудненной и, следовательно, труднодоступной, при этом сложноэфирная группа характеризуется значительной чувствительностью к гидролизу. На практике это приводит к получению композиций для нанесения покрытия на основе таких сомономеров, которые характеризуются устойчивостью при хранении ниже среднего уровня. Кроме того, при гидролизе таких сомономеров образуется диоксид углерода, что тем самым приводит к значительному повышению давления в сосуде, который вследствие этого должен характеризоваться особенно высокой устойчивостью к давлению.
Кроме этого, мономеры, такие как AAEMA и DAAM, можно получать только со значительными затратами. Для получения AAEMA требуется дикетен в качестве исходного материала, который является токсичным и обладает только ограниченной устойчивостью при хранении. Для получения DAAM требуются токсичный акрилонитрил и олеум, обращение с которым сопряжено с трудностями. Кроме того, данный мономер представляет собой твердое вещество, что, как правило, в условиях крупного масштаба приводит к трудностям при обращении с ним.
Кроме того, сополимеры на основе ацетоацетамидов или их сложных эфиров образуют слабоокрашенные комплексы с ионами металлов, даже если последние присутствуют только в следовых количествах. По этой причине, такие сополимеры являются непригодными для широкого ряда вариантов применений, связанных с прозрачными покрытиями.
Следовательно, целью настоящего изобретения является преодоление или по меньшей мере сведение к минимуму указанных недостатков в отношении профиля свойств сополимеров на основе AAEMA и DAAM, а также трудностей в отношении их получения. В данном случае необходимым являлось обеспечение того, чтобы разработанный CH-кислотный мономер обладал достаточно высокой растворимостью в воде, и в результате мог быть подвергнут полимеризации в эмульсии даже при отсутствии органических растворителей, и, таким образом, мог обеспечивать получение по сути не содержащих растворителя водных дисперсий.
Кроме того, наряду с этим целью настоящего изобретения также являлось обеспечение композиции для нанесения покрытия, которая характеризуется особенно длительными сохраняемостью и сроком хранения. Кроме того, предусматривается, что твердость покрытий, получаемых из композиций для нанесения покрытия, может варьироваться в широком диапазоне. В частности, предусматривается, что можно получать особенно твердые, устойчивые к царапанию покрытия.
Дополнительной целью являлось обеспечение сополимеров, применение которых обеспечивает получение композиций для нанесения покрытия без летучих органических растворителей.
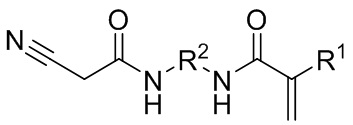
Данные цели достигались путем обеспечения CH-кислотных (мет)акрилатов общей формулы (I):
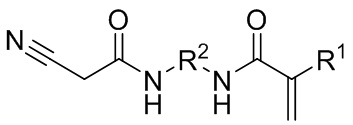 (I),
(I),
где R1 выбран из атома водорода и метильной группы, и
R2 представляет собой необязательно замещенную алкиленовую группу, имеющую состав
CnHmOxNy, при этом
n = 2–5,
m = 2–10,
x = 0–4,
y = 0–4.
CH-кислотные (мет)акрилаты общей формулы (I) содержат CH-кислотную функциональную группу, которая является легкодоступной для сшивания, и они являются легко получаемыми с помощью способа, описанного ниже. Их можно применять для дополнительной обработки; причем обычно это полимеризация в водной эмульсии или суспензии, без дополнительного выделения. Соответствующие сополимеры характеризуются неожиданно высокой стабильностью в отношении гидролиза. Таким образом, продукты на основе таких сополимеров являются особенно стабильными при хранении. Кроме того, даже если во время хранения в небольшой степени произойдет гидролиз таких сополимеров, в ходе этого не будут образовываться какие-либо газообразные соединения, приводящие к нежелательному повышению давления в сосуде.
Кроме того, можно достичь следующих преимуществ путем применения CH-кислотных сложных метакриловых эфиров формулы (I) по настоящему изобретению.
CH-кислотные сложные метакриловые эфиры по настоящему изобретению можно обрабатывать с получением сополимеров, композиций для нанесения покрытия и покрытий, характеризующихся очень низким остаточным содержанием мономеров.
Твердость покрытий, получаемых с помощью композиций для нанесения покрытия по настоящему изобретению, которые, в свою очередь, основаны на сополимерах или CH-кислотных сложных метакриловых эфирах, может варьироваться в широком диапазоне. В соответствии с предпочтительной модификацией в соответствии с настоящим изобретением можно получать особенно твердые, устойчивые к царапанию покрытия. Покрытия, получаемые с помощью композиций для нанесения покрытия по настоящему изобретения, демонстрируют неожиданно высокую устойчивость к действию растворителей, что особенно заметно в экспериментах с метилизобутилкетоном (MIBK), растворами аммиака или этанолом. Таким образом, полученные покрытия обладают исключительными параметрами, особенно в экспериментах в соответствии тестом мебельных поверхностей DIN 68861-1.
Для композиций для нанесения покрытия, получаемых с применением CH-кислотных сложных метакриловых эфиров формулы (I) по настоящему изобретению, обычно не требуются какие-либо летучие органические растворители. Более того, композиции для нанесения покрытия по настоящему изобретению демонстрируют особенно высокую устойчивость при хранении, длительный срок хранения и хорошую сохраняемость. В частности, по сути отсутствует образование агрегатов.
Покрытия, получаемые с помощью композиций для нанесения покрытия по настоящему изобретению, также демонстрируют высокую устойчивость к атмосферным воздействиям, особенно высокую устойчивость к УФ-излучению. Кроме того, пленки, получаемые с помощью композиций для нанесения покрытия, характеризуются низкой липкостью по прошествии короткого периода времени.
CH-кислотные сложные метакриловые эфиры формулы (I), сополимеры и композиции для нанесения покрытия по настоящему изобретению можно получать в крупном масштабе эффективным с точки зрения затрат способом. Композиции для нанесения покрытия по настоящему изобретению являются безвредными для окружающей среды, и их можно обрабатывать и получать безопасно и без значительных затрат. В данном случае композиции для нанесения покрытия по настоящему изобретению обладают исключительной прочностью на сдвиг.
В соответствии с настоящим изобретением радикал R1 в общей формуле (I) может представлять собой атом водорода или метильную группу, при этом R1 предпочтительно представляет собой метильную группу.
В соответствии с настоящим изобретением радикал R2 представляет собой необязательно замещенную алкиленовую группу, имеющую состав
CnHmOxNy, при этом
n = 2–5,
m = 2–10,
x = 0–4,
y = 0–4.
Радикал R2 может быть разветвленным или неразветвленным. Кроме того, R2 может содержать один или более заместителей, в частности атомов галогена или гидроксигрупп. Авторы настоящего изобретения неожиданно установили, что количество атомов углерода в радикале R2 оказывает определяющее влияние на растворимость CH-кислотных сложных метакриловых эфиров формулы (I) в воде. Так, соответствующие соединения, содержащие 2–5 атомов углерода в радикале R2, характеризуются исключительной растворимостью в воде. Следовательно, такие соединения можно применять даже при отсутствии органического растворителя при полимеризации в эмульсии для получения водных дисперсий. Это обеспечивает возможность получения водных дисперсий, по сути не содержащих растворителя, которые являются предпочтительными с токсикологической точки зрения.
С другой стороны, аналогичные CH-кислотные сложные метакриловые эфиры с более чем 6 атомами углерода в радикале R2 являются по сути нерастворимыми в воде. В результате этого для полимеризации в эмульсии таких соединений обычно требуется добавление растворимого в воде органического растворителя, например этанола. Следовательно, получение водных дисперсий, не содержащих растворителя, является невозможным.
Предпочтительно радикал R2 представляет собой необязательно замещенную алкиленовую группу, имеющую состав
CnHm, при этом
n = 2–4,
m = 2–8.
Особенно предпочтительные алкиленовые группы R2 могут представлять собой, например, одну из следующих групп: 1,2-этиленовую группу, 1,3-пропиленовую группу, 1,2-пропиленовую группу, 1,4-бутиленовую группу, 1,3-(2-гидрокси)пропиленовую группу, 1,3-(2,2-диметил)пропиленовую группу, при этом, как было показано, особенно предпочтительно, если алкиленовая группа R2 представляет собой 1,2-этиленовую группу или 1,4-бутиленовую группу.
Авторы настоящего изобретения неожиданно определили, что (мет)акрилаты общей формулы (I) по настоящему изобретению могут быть получены с помощью способа, в котором сложный эфир циануксусной кислоты (A) вводят в реакцию с диамином (B) с получением соответствующего цианоацетамида (IP), и цианоацетамид (IP) на следующей стадии способа вводят в реакцию с (мет)акриловым производным (C) с получением (мет)акрилата общей формулы (I) по настоящему изобретению. Таким образом, соединение общей формулы (I) по настоящему изобретению можно выделять с особенно высоким выходом продукта и высокой степенью чистоты.
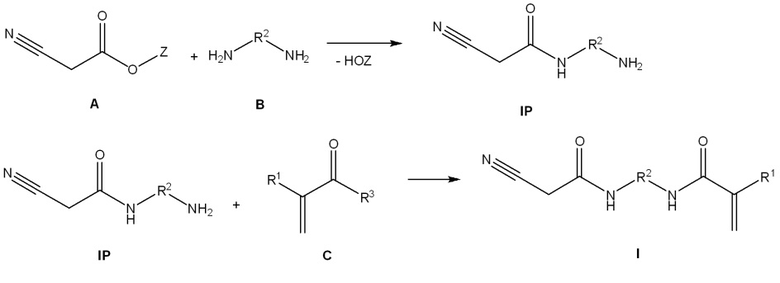
В предпочтительном варианте осуществления способ получения соединения общей формулы (I) по настоящему изобретению предусматривает следующие стадии способа:
a) осуществление реакции сложного эфира циануксусной кислоты (A) с диамином (B), который присутствует в избытке, с образованием цианоацетамида (IP);
b) удаление непрореагировавшего диамина (B),
c) осуществление реакции цианоацетамида (IP) со стадии b) способа либо
c1) с (мет)акриловым сложным эфиром (C1), либо
c2) с (мет)акриловым ангидридом (C2), либо
c3) с (мет)акрилоилгалогенидом (C3), и
d) необязательно выделение соединения общей формулы (I) с применением экстракции или кристаллизации.
В качестве сложных эфиров циануксусной кислоты (A) предпочтительно применять сложные метиловые и этиловые эфиры (Z = CH3, C2H5) циануксусной кислоты, поскольку они обеспечивают получение требуемого цианоацетамида (IP) с особенно высокими значениями выхода.
Кроме того, было показано, что получение цианоацетамида (IP) предпочтительно осуществлять в условиях избытка диамина (B) с целью подавления образования нежелательных побочных продуктов. Диамин (B) обычно характеризуется более низкой точкой кипения по сравнению с цианоацетамидом (IP), и, следовательно, его можно удалять путем перегонки на стадии b) способа без значительных сложностей. Неожиданным является то, что образование нежелательных побочных продуктов можно эффективно подавлять с помощью даже небольших уровней избытка диамина (B), например, при молярном соотношении B и A, составляющим по меньшей мере 1,001:1. При молярном соотношении B и A, составляющем по меньшей мере 10:1, особенно предпочтительно составляющем по меньшей мере 4:1, обычно получают цианоацетамид (IP), который даже в виде неочищенного продукта после удаления избытка диамина (B) выделяют со степенью чистоты, составляющей более 99%. Отделение диамина (B) в данном случае можно проводить посредством экстракции или кристаллизации, но особенно посредством перегонки, предпочтительно при пониженном давлении.
Подходящие диамины (B) для получения соединения общей формулы (I) можно выбирать по сути без ограничения из группы алифатических, линейных, или разветвленных, или циклических замещенных и незамещенных диаминов. Особенно предпочтительные диамины выбраны из группы, содержащей 1,2-этилендиамин, 1,3-диаминопропан, 1,4-диаминобутан, 1,5-диаминопентан, 1,2-диаминопропан, 1,3-диамино-2-гидроксипропан и 2,2-диметил-1,3-пропандиамин.
В особенно предпочтительном варианте осуществления цианоацетамид (IP) вводят в реакцию с получением соединения общей формулы (I) с применением (мет)акрилового ангидрида (C2) или (мет)акрилоилгалогенида (C3). В данном случае было показано, что особенно предпочтительным является применение метакрилового ангидрида и акрилового ангидрида.
В данной заявке на патент выражение «(мет)акрилат» в данном случае означает как метакрилат, например, метилметакрилат, этилметакрилат и т. д., так и акрилат, например, метилакрилат, этилакрилат и т. д., а также смеси обоих из них.
Особенно высокие значения выхода соединения общей формулы (I) можно получать, в частности, при молярном соотношении C и IP на стадии c) способа, которое находится в диапазоне от 0,2:1 до 5:1, предпочтительно в диапазоне от 0,8:1 до 2:1, особенно предпочтительно в диапазоне от 0,9:1 до 1,5:1.
Реакцию на стадии a) способа обычно проводят при значениях температуры от 0°C до 120°C, предпочтительно от 10°C до 40°C во время дозирования и при значениях температуры, составляющих не более 100°C, во время фазы после реакции и при подготовке к выделению.
Было показано, что на стадии c1) способа предпочтительно проводить реакцию при температуре, находящейся в диапазоне от 60°C до 140°C, предпочтительно от 100°C до 120°C. На стадии c2) способа, с другой стороны, реакцию предпочтительно проводят при температуре реакции, составляющей от примерно 0°C до 40°C; необязательно после реакции при температуре составляющей не более 100°C. Чтобы избежать образования нежелательных побочных продуктов, температуру реакции поддерживают как можно более низкой.
Реакция на стадии c) способа может проходить в присутствии катализатора. Соединения металлов и/или амины катализируют реакции (мет)акрилового ангидрида с гидрокси- или аминными группами несколько раз. Металлосодержащие соединения и амины являются известными из уровня техники и изложены, например, в Ullmann‘s Encyclopedia of Industrial Chemistry (6th edition), Wiley-VCH publishing, Вайнхайм 2003, или Römpp Chemielexikon, 2nd edition, на CD-ROM. В частности, к металлосодержащим соединениям относятся соли, такие как, например, галогениды, гидроксиды или оксиды щелочных металлов, такие как LiOH, KOH, или соединения циркония. К аминам относятся, например, аммиак, триэтиламин, трибутиламин и другие.
Предпочтительно в реакции применяют ингибиторы полимеризации. Такие соединения, например, гидрохиноны, простые эфиры гидрохинонов, такие как простой монометиловый эфир гидрохинона или ди-трет-бутилкатехин, фенотиазин, 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксил, метиленовый синий или стерически затрудненные фенолы, например, 2,4-диметил-6-трет-бутилфенол, являются широко известными в уровне техники. Данные соединения можно применять по отдельности или в виде смесей, и они, как правило, являются коммерчески доступными. Дополнительные подробности можно найти в соответствующей специализированной литературе, в частности, Römpp-Lexikon Chemie; редакторы: J. Falbe, M. Regitz; Штутгарт, Нью-Йорк; 10th edition (1996); ключевое слово «Antioxidantien», а также в литературных источниках, приведенных в нем.
Особое предпочтение отдается применению фенолов в качестве ингибитора полимеризации. Особенно неожиданные преимущества можно получить с применением смесей, которые содержат простой монометиловый эфир гидрохинона и/или 2,4-диметил-6-трет-бутилфенол. Молярное соотношение простого монометилового эфира гидрохинона и 2,4-диметил-6-трет-бутилфенола особенно предпочтительно находится в диапазоне от 2:1 до 1:2. На основании веса всей реакционной смеси доля ингибиторов, представленных либо по отдельности, либо в виде смеси, обычно может составлять от 0,01 до 0,5% (вес/вес).
Такие ингибиторы полимеризации можно добавлять в реакционную смесь до или во время начала реакции. Более того, некоторые из добавляемых ингибиторов полимеризации также можно добавлять во время проведения реакции.
В предпочтительном варианте осуществления реакцию можно осуществлять в присутствии кислорода, в частности атмосферного кислорода. Более того, реакция также может проходить в атмосфере с недостатком кислорода, в частности в атмосфере азота.
Продолжительность реакции на стадиях a) и c) способа обычно составляет от 15 мин до 10 часов, предпочтительно от 1 часа до 5 часов. Реакция может проходить при повышенном давлении или пониженном давлении. В соответствии с особенно выгодной модификацией настоящего изобретения реакции можно проводить при давлении в диапазоне от 200 мбар до 2000 мбар, особенно предпочтительно в диапазоне от 500 мбар до 1300 мбар.
Цианоацетамид (IP) можно дополнительно применять без водного выделения. Цианоацетамид (IP) со стадии b) способа предпочтительно поглощают растворителем в горячем состоянии, поскольку в ином случае он будет подвергаться затвердеванию с образованием стекловидной массы, а также за счет этого он вступает в реакцию со значительно большей легкостью. Подходящими растворителями для данной цели являются вода, метил-трет-бутиловый эфир (MTBE), тетрагидрофуран (THF), ацетонитрил, диоксан и спирты. Выбор является очевидным для специалиста в данной области техники исходя из соответствующей цели реакции.
На стадии c2) способа предпочтительно применяют растворитель, такой как вода, MTBE, THF, ацетонитрил, диоксан и спирты, или их смеси. (Мет)акриловая кислота или ее соли, содержащиеся в реакционной смеси, могут оставаться в полученном неочищенном продукте, что не оказывает отрицательного влияния на получаемые из него сополимеры. Однако, в зависимости от предусмотренного применения сополимеров, полученную (мет)акриловую кислоту можно удалять из реакционной смеси с помощью способов экстракции или перегонки.
Соединение общей формулы (I) по настоящему изобретению можно применять без дополнительного выделения. При необходимости его также можно высушивать при пониженном давлении, подвергать перекристаллизации путем добавления полярного растворителя или экстрагировать путем добавления несмешивающегося растворителя.
В соответствии с одной конфигурацией способа по настоящему изобретению полученную смесь продукта можно очищать с помощью способов фильтрации. Такие способы являются известными из уровня техники (W. Gösele, Chr. Alt, в Ullmann's Encyclopedia of Industrial Chemistry, (6th edition), Wiley-VCH publishing, Вайнхайм, 2003, том 13, страницы 731 и 746), при этом можно применять стандартные добавки для фильтрации, например, отбеливающую землю и силикаты алюминия (перлит). Например, среди прочего, можно применять фильтры для фильтрования с предварительно нанесенным слоем вспомогательного вещества, работающие в непрерывном режиме.
Сополимеры на основе CH-кислотных сложных метакриловых эфиров
Дополнительный аспект настоящего изобретения относится к сополимерам на основе CH-кислотных сложных метакриловых эфиров. Соответствующие сополимеры могут быть получены путем полимеризации смеси мономеров, которая обычно содержит по меньшей мере 0,5 вес. %, предпочтительно по меньшей мере 1 вес. % CH-кислотного (мет)акрилата общей формулы (I) в пересчете на вес смеси мономеров.
Вместе с по меньшей мере одним (мет)акрилатным мономером в соответствии с формулой (I) смесь мономеров содержит по меньшей мере один дополнительный мономер, который способен к сополимеризации с (мет)акрилатным мономером в соответствии с формулой (I). Такие способные к сополимеризации мономеры включают мономеры с кислотной группой, мономеры, содержащие сложноэфирные группы, отличные от (мет)акрилатного мономера в соответствии с формулой (I), и стироловые мономеры.
Мономеры, содержащие кислотные группы, представляют собой соединения, которые предпочтительно можно подвергать радикальной сополимеризации с вышеуказанными (мет)акрилатными мономерами в соответствии с формулой (I). Они включают, например, мономеры с группой сульфоновой кислоты, например, винилсульфоновой кислоты; мономеры с группой фосфоновой кислоты, например, винилфосфоновой кислоты и ненасыщенных карбоновых кислот, например, метакриловой кислоты, акриловой кислоты, фумаровой кислоты и малеиновой кислоты. Особенно предпочтительными являются метакриловая кислота и акриловая кислота. Мономеры, содержащие кислотные группы, можно применять по отдельности или в виде смеси двух, трех или более мономеров, содержащих кислотные группы.
Предпочтительные мономеры, содержащие сложноэфирные группы, включают, в частности, (мет)акрилаты, отличные от мономеров в соответствии с формулой (I), фумараты, малеаты и/или винилацетат. Выражение «(мет)акрилаты» охватывает метакрилаты и акрилаты, а также их смеси. Такие мономеры являются хорошо известными.
Они включают, в частности, (мет)акрилаты, содержащие от 1 до 6 атомов углерода в алкильном радикале и полученные из насыщенных спиртов, такие как метил(мет)акрилат, этил(мет)акрилат, н-пропил(мет)акрилат, изопропил(мет)акрилат, н-бутил(мет)акрилат, трет-бутил(мет)акрилат и пентил(мет)акрилат, гексил(мет)акрилат; циклоалкил(мет)акрилаты, такие как циклопентил(мет)акрилат, циклогексил(мет)акрилат; и (мет)акрилаты, полученные из ненасыщенных спиртов, такие как 2-пропинил(мет)акрилат, аллил(мет)акрилат и винил(мет)акрилат.
Для получения сополимеров по настоящему изобретению особенно предпочтительно применять смеси, которые содержат метакрилаты и акрилаты. Таким образом, в частности, можно применять смеси метилметакрилата и акрилатов, содержащих от 2 до 6 атомов углерода, таких как этилакрилат, бутилакрилат и гексилакрилат.
Более того, сомономеры включают, например, (мет)акрилаты, содержащие по меньшей мере 7 атомов углерода в алкильном радикале и полученные из насыщенных спиртов, например, 2-этилгексил(мет)акрилат, гептил(мет)акрилат, 2-трет-бутилгептил(мет)акрилат, октил(мет)акрилат, 3-изопропилгептил(мет)акрилат, нонил(мет)акрилат, децил(мет)акрилат, ундецил(мет)акрилат, 5-метил-ундецил(мет)акрилат, додецил(мет)акрилат, 2-метилдодецил(мет)акрилат, тридецил(мет)акрилат, 5-метилтридецил(мет)акрилат, тетрадецил(мет)акрилат, пентадецил(мет)акрилат, гексадецил(мет)акрилат, 2-метилгексадецил(мет)акрилат, гептадецил(мет)акрилат, 5-изопропилгептадецил(мет)акрилат, 4-трет-бутилоктадецил(мет)акрилат, 5-этилоктадецил(мет)акрилат, 3-изопропилоктадецил(мет)акрилат, октадецил(мет)акрилат, нонадецил(мет)акрилат, эйкозил(мет)акрилат, цетилэйкозил(мет)акрилат, стеарилэйкозил(мет)акрилат, докозил(мет)акрилат и/или эйкозилтетратриаконтил(мет)акрилат; циклоалкил(мет)акрилаты, такие как 3-винилциклогексил(мет)акрилат, изоборнил(мет)акрилат, циклоалкил(мет)акрилаты, такие как 2,4,5-три-трет-бутил-3-винилциклогексил(мет)акрилат, 2,3,4,5-тетра-трет-бутилциклогексил(мет)акрилат; гетероциклические (мет)акрилаты, такие как 2-(1-имидазолил)этил(мет)акрилат, 2-(4-морфолинил)этил(мет)акрилат и 1-(2-метакрилоилоксиэтил)-2-пирролидон; нитрилы (мет)акриловой кислоты и другие азотсодержащие метакрилаты, такие как N-(метакрилоилоксиэтил)диизобутилкетимин, N-(метакрилоилоксиэтил)дигексадецилкетимин, метакрилоиламидоацетонитрил, 2-метакрилоилоксиэтилметилцианамид, цианометилметакрилат; арил(мет)акрилаты, такие как бензил(мет)акрилат или фенил(мет)акрилат, где арильные радикалы в каждом случае могут быть незамещенными или не более чем четырежды замещенными; (мет)акрилаты, которые содержать две или более (мет)акрилоильных групп, гликольди(мет)акрилаты, такие как этиленгликольди(мет)акрилат, диэтиленгликольди(мет)акрилат, триэтиленгликольди(мет)акрилат, тетра- и полиэтиленгликольди(мет)акрилат, 1,3-бутандиол(мет)акрилат, 1,4-бутандиол(мет)акрилат, 1,6-гександиолди(мет)акрилат, глицеринди(мет)акрилат; диметакрилаты этоксилированного бисфенола A; (мет)акрилаты, содержащие три или более двойных связей, например, глицеринтри(мет)акрилат, триметилолпропантри(мет)акрилат, пентаэритриттетра(мет)акрилат и дипентаэритритпента(мет)акрилат; (мет)акрилаты, функциональные метакрилаты с простыми эфирными или аминовыми группами в боковой цепи.
Более того, мономеры, содержащие сложноэфирные группы, включают сложные виниловые эфиры, такие как винилацетат; производные малеиновой кислоты, например, малеиновый ангидрид, сложные эфиры малеиновой кислоты, например, диметилмалеат, метилмалеиновый ангидрид; и производные фумаровой кислоты, такие как диметилфумарат.
Дополнительной предпочтительной группой сомономеров являются стироловые мономеры, например, стирол, замещенные стиролы, содержащие алкильный заместитель в боковой цепи, например, α-метилстирол и α-этилстирол, замещенные стиролы, содержащие алкильный заместитель в кольце, такие как винилтолуол и п-метилстирол, галогенированные стиролы, например, монохлорстиролы, дихлорстиролы, трибромстиролы и тетрабромстиролы.
Наряду с вышеуказанными мономерами полимеры в соответствии с настоящим изобретением, полученные с помощью полимеризации смесей мономеров, также могут содержать дополнительные мономеры. Они включают, например, гетероциклические виниловые соединения, такие как 2-винилпиридин, 3-винилпиридин, 2-метил-5-винилпиридин, 3-этил-4-винилпиридин, 2,3-диметил-5-винилпиридин, винилпиримидин, винилпиперидин, 9-винилкарбазол, 3-винилкарбазол, 4-винилкарбазол, 1-винилимидазол, 2-метил-1-винилимидазол, N-винилпирролидон, 2-винилпирролидон, N-винилпирролидин, 3-винилпирролидин, N-винилкапролактам, N-винилбутиролактам, винилоксолан, винилфуран, винилтиофен, винилтиолан, винилтиазолы и гидрогенизированные винилтиазолы, винилоксазолы и гидрогенизированные винилоксазолы; малеимид, метилмалеимид; виниловый и изопрениловый эфир; и виниловые галогениды, например, винилхлорид, винилфторид, винилиденхлорид и винилиденфторид.
Смеси мономеров, предпочтительно применяемые в соответствии с настоящим изобретением, содержат
от 0,1 до 90 вес. %, предпочтительно от 0,5 до 30 вес. % (мет)акрилатного мономера в соответствии с формулой (I);
от 10 до 95 вес. %, предпочтительно от 40 до 90 вес. % мономеров со сложноэфирными группами;
от 0 до 20 вес. %, предпочтительно от 1 до 8 вес. %, в частности от 1 до 3 вес. %, мономера с кислотной группой и
от 0 до 70 вес. %, предпочтительно от 0 до 50 вес. %, в частности от 0 до 30 вес. %, стироловых мономеров, при этом числа в каждом случае означают общий вес мономеров.
Смеси мономеров, применяемые в соответствии с настоящим изобретением, которые характеризуются высокой долей (мет)акрилатного мономера в соответствии с формулой (I), обычно обеспечивают получение полимеров или композиций для нанесения покрытия, которые обеспечивают возможность получения особенно устойчивых к атмосферным воздействиям, устойчивых к действию растворителей и твердых покрытий.
Такие смеси мономеров предпочтительно содержат
от 10 до 90 вес. %, предпочтительно от 15 до 40 вес. % (мет)акрилатного мономера в соответствии с формулой (I);
от 10 до 90 вес. %, предпочтительно от 40 до 85 вес. % мономеров со сложноэфирными группами;
от 0 до 10 вес. %, предпочтительно от 1 до 8 вес. % мономера с кислотной группой и
от 0 до 50 вес. %, предпочтительно от 0 до 30 вес. % стироловых мономеров, при это числа в каждом случае означают общий вес мономеров в смеси мономеров.
CH-кислотные (мет)акриловые сложные эфиры формулы (I) по настоящему изобретению, в частности, обеспечивают получение или модификацию сополимеров. Полимеризацию можно проводить любым известным способом. Она включает, в частности, радикальную, катионную или анионную полимеризацию, при этом также можно применять варианты таких способов полимеризации, например, ATRP (= радикальная полимеризация с переносом атома), NMP (нитроксид-опосредованная полимеризация) или RAFT (= полимеризация с обратимой передачей цепи по механизму присоединения- фрагментации).
Вышеуказанные мономеры или CH-кислотные сложные метакриловые эфиры формулы (I) можно превращать, например, с помощью видов полимеризации в растворе, видов полимеризации в массе или видов полимеризации в эмульсии, при этом можно получить неожиданные преимущества с помощью радикальной полимеризации в эмульсии.
Дополнительный аспект настоящего изобретения относится к водной дисперсии, которая содержит сополимер по настоящему изобретению. Содержание сополимера в дисперсии обычно составляет от 0,1 до 90 вес. %, предпочтительно от 20 до 80 вес. %, особенно предпочтительно от 30 до 60 вес. %, в пересчете на вес водной дисперсии. Такие водные дисперсии можно получать, в частности, с помощью полимеризации в эмульсии.
Способы полимеризации в эмульсии, среди прочего, описаны в Ullmann's Encyclopedia of Industrial Chemistry, пятое издание. Обычно для данной цели получают водную фазу, которая наряду с водой может содержать стандартные добавки, в частности эмульгаторы и защитные коллоиды для стабилизации эмульсии.
Затем в данную водную фазу добавляют мономеры и их подвергают полимеризации в водной фазе. В данном случае при получении однородных частиц полимеров CH-кислотный сложный метакриловый эфир можно добавлять непрерывно или партиями в течение определенного периода времени.
Например, полимеризацию в эмульсии можно осуществлять в виде мини- или микроэмульсии, которые описаны более подробно в Chemistry and Technology of Emulsion Polymerisation, A.M. van Herk (редактор), Blackwell Publishing, Оксфорд, 2005, и J. O'Donnell, E.W. Kaler, Macromolecular Rapid Communications 2007, 28(14), 1445-1454. Миниэмульсия обычно характеризуется применением совместных стабилизаторов или средств, способствующих набуханию, при этом часто применяют алканы или алканолы с длинной цепью. Размер капель в миниэмульсиях предпочтительно находится в диапазоне от 0,05 мкм до 20 мкм. Размер капель в микроэмульсиях предпочтительно находится в диапазоне ниже 1 мкм, в результате чего можно получать частицы с размером менее 50 нм. В микроэмульсиях часто применяют дополнительные поверхностно-активные вещества, например, гексанол или подобные соединения.
Диспергирование мономер-содержащей фазы в водной фазе можно осуществлять с применением известных способов. Они включают, в частности, механические способы, а также применение ультразвука.
При получении однородных полимеров, получаемых в результате полимеризации в эмульсии, предпочтительно можно применять смеси мономеров, которые содержат от 0,5 до 30 вес. % CH-кислотного сложного метакрилового эфира формулы (I).
При получении полимеров типа «ядро-оболочка» состав смеси мономеров можно модифицировать поэтапно, при этом до модификации композиции полимеризацию предпочтительно проводят до степени превращения, составляющей по меньшей мере 80 вес. %, особенно предпочтительно по меньшей мере 95 вес. %, в каждом случае в пересчете на общий вес применяемой смеси мономеров. Ход реакции полимеризации на каждой стадии способа можно контролировать известным способом, например, посредством гравиметрических способов или посредством газовой хроматографии.
Полимеризацию в эмульсии предпочтительно проводят при температуре в диапазоне от 0°C до 120°C, особенно предпочтительно в диапазоне от 30°C до 100°C. В данном случае было показано, что особенно предпочтительными являются значения температуры полимеризации в диапазоне от более 60°C до менее 90°C, целесообразно в диапазоне от более 70°C до менее 85°C, предпочтительно в диапазоне от более 75°C до менее 85°C.
Инициацию полимеризации осуществляют с помощью инициаторов, обычно применяемых для полимеризации в эмульсии. Пригодными органическими инициаторами являются, например, гидропероксиды, такие как трет-бутилгидропероксид или гидропероксид кумола. Подходящими неорганическими инициаторами являются пероксид водорода и соли щелочного металла и аммония с пероксодисерной кислотой, в частности, пероксодисульфаты аммония, натрия и калия. Подходящими окислительно-восстановительными системами инициаторов являются, например, комбинации третичных аминов с пероксидами или дисульфитом натрия и соли щелочного металла и аммония с пероксодисерной кислотой, особенно пероксодисульфаты натрия и калия. Дополнительные подробности можно найти в специализированной литературе, в частности H. Rauch-Puntigam, Th. Völker, "Acryl- und Methacrylverbindungen” [акриловые и метакриловые соединения], Springer, Heidelberg, 1967 или Kirk-Othmer, Encyclopedia of Chemical Technology, том 1, страницы 386ff, J. Wiley, New York, 1978. В контексте настоящего изобретения особенно предпочтительным является применение органических и/или неорганических инициаторов.
Указанные инициаторы можно применять по отдельности или в виде смеси. Предпочтительно их применяют в количестве от 0,05 до 3,0 вес. % в пересчете на общий вес мономеров на конкретной стадии. Предпочтительной также является возможность проведения полимеризации с помощью смеси различных инициаторов полимеризации с различными значениями периода полураспада с целью поддержания постоянной подачи радикалов на протяжении полимеризации, а также при различных значениях температуры полимеризации.
Смесь предпочтительно стабилизируют с помощью эмульгаторов и/или защитных коллоидов. Эмульсию предпочтительно стабилизируют с помощью эмульгаторов с целью получения низкой вязкости дисперсии. Общее количество эмульгатора предпочтительно составляет от 0,1 до 15 вес. %, в частности от 1 до 10 вес. %, а особенно предпочтительно от 2 до 5 вес. %, в пересчете на общий вес применяемых мономеров. В соответствии с дополнительным аспектом настоящего изобретения во время полимеризации можно добавлять порцию эмульгаторов.
Особенно подходящими эмульгаторами являются анионные или неионогенные эмульгаторы или их смеси, в частности:
- алкилсульфаты, предпочтительно содержащие от 8 до 18 атомов углерода в алкильном радикале, сульфаты простых алкиловых и алкилариловых эфиров, содержащие от 8 до 18 атомов углерода в алкильном радикале и от 1 до 50 звеньев этиленоксида;
- сульфонаты, предпочтительно алкилсульфонаты, содержащие от 8 до 18 атомов углерода в алкильном радикале, алкиларилсульфонаты, содержащие от 8 до 18 атомов углерода в алкильном радикале, сложные эфиры сульфоянтарной кислоты с одноатомными спиртами или алкилфенолами, содержащими от 4 до 15 атомов углерода в алкильном радикале; причем такие спирты или алкилфенолы также необязательно могут быть этоксилированы 1–40 звеньями этиленоксида;
- неполные сложные эфиры фосфорной кислоты и их соли щелочного металла и аммония, предпочтительно алкил- и алкиларилфосфаты, содержащие от 8 до 20 атомов углерода в алкильном или алкиларильном радикале и от 1 до 5 этиленоксидных звеньев;
- простые алкилполигликолевые эфиры, предпочтительно содержащие от 8 от 20 атомов углерода в алкильном радикале и от 8 до 40 этиленоксидных звеньев;
- простые алкиларилполигликолевые эфиры, предпочтительно содержащие от 8 до 20 атомов углерода в алкильном или алкиларильном радикале и от 8 до 40 этиленоксидных звеньев;
- сополимеры этиленоксида и пропиленоксида, предпочтительно блок-сополимеры, предпочтительно содержащие от 8 до 40 этиленоксидных и/или пропиленоксидных звеньев.
Особенно предпочтительные анионные эмульгаторы включают, в частности, сульфаты простых эфиров жирного спирта, диизооктилсульфосукцинат, лаурилсульфат, сульфонат C15-парафина, при этом такие соединения обычно можно применять в виде соли щелочного металла, в частности, в виде соли натрия. Такие соединения являются коммерчески доступными, в частности, под торговыми названиями Disponil® FES 32, Aerosol® OT 75, Texapon® K1296 и Statexan® K1 от компаний Cognis GmbH, Cytec Industries, Inc. и Bayer AG.
Целесообразным является, если неионогенные эмульгаторы, среди прочего, представляют собой трет-октилфенолэтоксилат с 30 звеньями этиленоксида и простые эфиры жирного спирта с полиэтиленгликолем, которые предпочтительно содержат от 8 до 20 атомов углерода в алкильном радикале и от 8 до 40 звеньев этиленоксида. Такие эмульгаторы являются коммерчески доступными под торговыми названиями Triton® X 305 (Fluka), Tergitol® 15-S-7 (Sigma-Aldrich Co.), Marlipal® 1618/25 (Sasol Germany) и Marlipal® O 13/400 (Sasol Germany).
Предпочтительно можно применять смеси анионного эмульгатора и неионогенного эмульгатора. Целесообразным является, если весовое соотношение анионного эмульгатора и неионогенного эмульгатора может находиться в диапазоне от 20:1 до 1:20, предпочтительно от 2:1 до 1:10 и более предпочтительно от 1:1 до 1:5. В данном случае было показано, что смеси, содержащие сульфат, в частности сульфат простого эфира жирного спирта, лаурилсульфат или сульфонат, в частности диизооктилсульфосукцинат или парафинсульфонат, в качестве анионного эмульгатора и алкилфенолэтоксилат или простой эфир жирного спирта с полиэтиленгликолем, каждый из которых предпочтительно содержит от 8 до 20 атомов углерода в алкильном радикале и от 8 до 40 звеньев этиленоксида, являются особенно эффективными в качестве неионогенных эмульгаторов.
Необязательно эмульгаторы можно также применять в смеси с защитными коллоидами. Подходящие защитные коллоиды включают частично гидролизованные поливинилацетаты, поливинилпирролидоны, карбоксиметил-, метил-, гидроксиэтил- и гидроксипропилцеллюлозу, разновидности крахмала, белки, поли(мет)акриловую кислоту, поли(мет)акриламид, поливинилсульфоновые кислоты, меламинформальдегидные сульфонаты, нафталинформальдегидные сульфонаты, сополимеры стирола и малеиновой кислоты и простого винилового эфира и малеиновой кислоты. В случае применения защитных коллоидов их предпочтительно применяют в количестве от 0,01 до 1,0 вес. % в пересчете на общее количество мономеров. Защитные коллоиды можно загружать сразу или вводить дозированно перед началом полимеризации. Инициатор можно загрузить сразу или вводить дозировано. Кроме того, также можно сначала загрузить часть инициатора и дозировано загрузить остаток.
Предпочтительно полимеризацию начитают путем нагревания смеси до температуры полимеризации и дозированного введения инициатора, предпочтительно в водном растворе. Дозированное добавление эмульгатора и мономеров можно проводить по отдельности или в виде смеси. В случае дозированного добавления смесей эмульгатора и мономера процедура представляет собой предварительное смешивание эмульгатора и мономера в смесителе, присоединенном выше по потоку относительно реактора полимеризации. Предпочтительно остатки эмульгатора и мономера, которые не загрузили первоначально, дозированно вводят по отдельности после начала полимеризации. Предпочтительно дозированное добавление можно начинать через 15–35 минут после начала полимеризации.
Предпочтительные полимеры, получаемые в результате полимеризации в эмульсии, которые характеризуются высоким содержанием нерастворимых полимеров, можно получать описанным выше способом, при этом параметры реакции для обеспечения высокого молекулярного веса являются известными. Таким образом, в таком случае можно, в частности, исключить применение регуляторов молекулярной массы.
На регулирование радиуса частиц может влиять, среди прочего, доля эмульгаторов. Чем больше эта доля, особенно в начале полимеризации, тем меньше получаемые частицы.
Полимеры, получаемые в соответствии с вышеописанным способом, особенно предпочтительно полимеры, получаемые в результате полимеризации в эмульсии, являются дополнительным субъектом настоящего изобретения.
В соответствии с предпочтительной модификацией настоящего изобретения полимер, получаемый в результате полимеризации в эмульсии, может характеризоваться набуханием, составляющим по меньшей мере 1000%, особенно предпочтительно по меньшей мере 1400% и в наибольшей степени предпочтительно по меньшей мере 1600%, в тетрагидрофуране (THF) при 20°C. Верхний предел значения набухания не является критичным per se, при этом набухание предпочтительно составляет не более 5000%, особенно предпочтительно не более 3000% и в наибольшей степени предпочтительно не более 2500%. С целью определения набухания образец полимера, полученного в результате полимеризации в эмульсии, высушенный без доступа кислорода, хранят в 200-кратном количестве THF при 20°C в течение 4 часов. В результате образец набухает. Образец, набухший таким образом, отделяют от оставшегося растворителя. Затем растворитель удаляют из образца. Например, можно выпаривать значительную часть растворителя при комнатной температуре (20°C). Остатки растворителя можно удалять в сушильном шкафу (140°C), чего обычно достигают в течение 1 часа. Набухание приведено относительно веса растворителя, поглощенного образцом, и веса сухого образца. Более того, растворимая фракция полимера, полученного путем полимеризации в эмульсии, приведена относительно различия в весе образца перед экспериментом с набуханием и веса высушенного образца после эксперимента с набуханием.
Радиус частиц полимеров, полученных в результате полимеризации в эмульсии, может находиться в широком диапазоне. Таким образом, в частности, можно применять полимеры, полученные в результате полимеризации в эмульсии, с радиусом частиц в диапазоне от 1 до 100 нм, предпочтительно от 5 до 59 нм. В соответствии с дополнительным аспектом настоящего изобретения, радиус частиц предпочтительно находится в диапазоне от 60 нм до 500 нм, особенно предпочтительно от 70 до 150 нм и в наибольшей степени предпочтительно от 75 до 100 нм. Радиус частиц можно определять с помощью PCS (фотонной корреляционной спектроскопии), при этом приводимые данные относятся к значению d50 (50% частиц меньше такого значения, 50% – больше). Для данной цели, например, можно применять анализатор размера субмикронных частиц N5 от Beckman Coulter.
Неожиданные преимущества демонстрируются, в частности, полимерами, полученными в результате полимеризации в эмульсии, с высокой набухаемостью. Предпочтительные полимеры, полученные в результате полимеризации в эмульсии, характеризуются набухаемостью, составляющей по меньшей мере 2, в частности по меньшей мере 4, особенно предпочтительно по меньшей мере 6 и в наибольшей степени предпочтительно по меньшей мере 8. С целью определения набухаемости сначала радиус частиц полимеров, полученных в результате полимеризации в эмульсии, измеряют в воде вышеописанным способом. Затем обеспечивают набухание полимеров, полученных в результате полимеризации в эмульсии, в смеси растворитель/вода (THF/вода = 90:10) и количественно оценивают размер частиц (микрогелей) путем измерения с помощью Nanosizer N5 от Coulter (r раств.). Обычно для данной цели в дисперсию добавляют соответствующее количество тетрагидрофурана (THF) с целью регулирования объемного соотношения THF/вода в дисперсии до 90:10. Измерение проводят при 20°C, при этом обеспечивают набухание дисперсии в течение 5 минут после добавления растворителя (THF). Отношение объемов частиц, рассчитанных на основании полученных радиусов частиц (r раств. и r воды), определяется как набухаемость (SF):
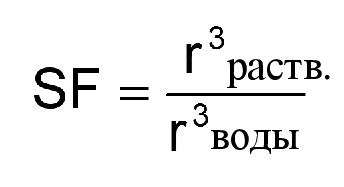
Полимерами, полученными в результате полимеризации в эмульсии, с высокими значениями набухаемости являются полимеры, характеризующиеся низким предварительным сшиванием. Предварительное сшивание полимера, полученного в результате полимеризации в эмульсии, происходит при полимеризации в присутствии мономеров, содержащих более чем одну способную к полимеризации двойную связь (например, дополнительную метакрилатную группу). Такие мономеры иногда называют сшивающими средствами, поскольку они обеспечивают возможность соединения двух полимерных цепей друг с другом. Формально это соответствует обычному сшиванию при нанесении покрытия на изделия, однако нежелательным является время сшивания, которое происходит до применения. Полимеры, полученные в результате полимеризации в эмульсии, которые получали из CH-кислотных сложных метакриловых эфиров с низкой долей сшивающих средств, соответственно демонстрируют высокую набухаемость.
Температура стеклования полимера по настоящему изобретению предпочтительно находится в диапазоне от −30°C до 70°C, особенно предпочтительно в диапазоне от −20 до 40°C и в наибольшей степени предпочтительно в диапазоне от 0 до 25°C. Температура стеклования может зависеть от типа и доли мономеров, применяемых для получения полимера. Температуру стеклования Tg полимера в данном случае можно определять известным способом с помощью дифференциальной сканирующей калориметрии (DSC). Кроме того, температуру стеклования Tg также можно предварительно примерно рассчитать с помощью уравнения Фокса. Согласно Fox T. G., Bull. Am. Physics Soc. 1, 3, стр. 123 (1956):
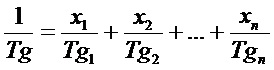
где xn представляет собой массовую долю (вес. %/100) мономера n, и Tgn представляет собой температуру стеклования гомополимера, состоящего из мономеров n, в кельвинах. Дополнительные полезные рекомендации могут быть найдены специалистом в данной области техники в Polymer Handbook 2nd Edition, J. Wiley & Sons, New York (1975), в котором приведены значения Tg для наиболее распространенных гомополимеров. В данном контексте полимер может характеризоваться одним или более разными значениями температуры стеклования. Следовательно, такие числа относятся к одному сегменту, получаемому путем полимеризации CH-кислотного сложного метакрилового эфира по настоящему изобретению.
Архитектура сополимера не является критичной для многих вариантов применения и свойств. Соответственно, сополимеры, особенно сополимеры, полученные в результате полимеризации в эмульсии, могут представлять собой статистические сополимеры, градиентные сополимеры, блок-сополимеры и/или привитые сополимеры. Блок-сополимеры или градиентные сополимеры, например, можно получать путем ступенчатого изменения состава мономеров во время роста цепи. В соответствии с предпочтительным аспектом настоящего изобретения, полимер, полученный в результате полимеризации в эмульсии, представляет собой статистический сополимер, в котором состав мономеров является по сути постоянным в ходе полимеризации. Однако, поскольку мономеры могут характеризоваться различными параметрами сополимеризации, точный состав может изменяться на протяжении полимерной цепи полимера.
Полимер может представлять собой однородный полимер, который, например, в водной дисперсии образует частицы с постоянным составом. В данном случае полимер, который предпочтительно представляет собой полимер, полученный в результате полимеризации в эмульсии, может состоять из одного или более сегментов, получаемых путем полимеризации CH-кислотных метакриловых сложных эфиров формулы (I) по настоящему изобретению.
В соответствии с дополнительным вариантом осуществления полимер, полученный в результате полимеризации в эмульсии, может представлять собой полимер типа «ядро-оболочка», который имеет одну, две, три или более оболочек. В данном случае сегмент, получаемый путем полимеризации CH-кислотных сложных метакриловых эфиров формулы (I) по настоящему изобретению, предпочтительно образует самую внешнюю оболочку полимера типа «ядро-оболочка». Оболочка может быть присоединена к ядру или внутренним оболочкам посредством ковалентных связей. Кроме того, оболочку также можно подвергать полимеризации на ядре или внутренней оболочке. В данном варианте осуществления сегмент, получаемый путем полимеризации CH-кислотных сложных метакриловых эфиров формулы (I) по настоящему изобретению, можно отделять и выделять из ядра множеством способов с помощью подходящих растворителей.
Предпочтительно весовое соотношение сегмента, получаемого путем полимеризации CH-кислотных сложных метакриловых эфиров формулы (I) по настоящему изобретению, и ядра находится в диапазоне от 2:1 до 1:6, особенно предпочтительно от 1:1 до 1:3.
Предпочтительно ядро можно образовывать из полимеров, которые содержат от 50 до 100 вес. %, предпочтительно от 60 до 90 вес. % звеньев, полученных из (мет)акрилатов. В данном контексте предпочтение отдается сложным эфирам (мет)акриловой кислоты, спиртовой радикал которых предпочтительно содержит от 1 до 30 атомов углерода, особенно предпочтительно от 1 до 20 атомов углерода и в наибольшей степени предпочтительно от 1 до 10 атомов углерода. Они включают, в частности, (мет)акрилаты, полученные из насыщенных спиртов, такие как метил(мет)акрилат, этил(мет)акрилат, н-пропил(мет)акрилат, изопропил(мет)акрилат, н-бутил(мет)акрилат, трет-бутил(мет)акрилат и пентил(мет)акрилат, гексил(мет)акрилат.
В соответствии с конкретной конфигурацией настоящего изобретения для получения ядра можно применять смесь, содержащую метакрилаты и акрилаты. Таким образом, в частности, можно применять смеси метилметакрилата и акрилатов, содержащих от 2 до 6 атомов углерода, таких как этилакрилат, бутилакрилат и гексилакрилат.
Более того, полимеры ядра могут содержать вышеописанные сомономеры. В соответствии с предпочтительной модификацией ядро может быть сшитым. Такое сшивание можно получать путем применения мономеров с двумя, тремя или более способными к радикальной полимеризации двойными связями.
В соответствии с конкретным аспектом ядро предпочтительно может характеризоваться температурой стеклования в диапазоне от −30°C до 200°C, особенно предпочтительно в диапазоне от −20°C до 150°C. Оболочка полимера, полученного в результате полимеризации в эмульсии, по настоящему изобретению, предпочтительно получаемая путем полимеризации CH-кислотных сложных метакриловых эфиров формулы (I) по настоящему изобретению, предпочтительно может характеризоваться температурой стеклования в диапазоне от −30°C до 70°C, особенно предпочтительно в диапазоне от −20°C до 40°C и в наибольшей степени предпочтительно в диапазоне от 0°C до 25°C. В соответствии с конкретным аспектом настоящего изобретения температура стеклования ядра может быть выше температуры стеклования оболочки. Целесообразным является, если температура стеклования ядра может быть на по меньшей мере 10°C, предпочтительно на по меньшей мере 20°C, выше температуры стеклования оболочки.
Сополимеры, получаемые путем полимеризации CH-кислотных сложных метакриловых эфиров формулы (I) по настоящему изобретению, можно выделять. В соответствии с конкретной конфигурацией настоящего изобретения, дисперсии, получаемые путем полимеризации в эмульсии, можно применять как таковые в качестве композиции для нанесения покрытия.
Соответственно, водные дисперсии являются дополнительным субъектом настоящего изобретения. Предпочтительно водные дисперсии характеризуются содержанием твердых веществ в диапазоне от 10 до 70 вес. %, особенно предпочтительно от 20 до 60 вес. %. Целесообразным является, если дисперсия может характеризоваться динамической вязкостью в диапазоне от 0,1 до 180 мПа·с, предпочтительно от 1 до 80 мПа·с и в наибольшей степени предпочтительно от 10 до 50 мПа·с, что измеряли в соответствии с DIN EN ISO 2555 при 25°C (Brookfield).
Кроме того, водные дисперсии по настоящему изобретению можно известными способами снабжать добавками или дополнительными компонентами с целью адаптирования свойств композиции для нанесения покрытия к конкретным требованиям. Такие добавки, в частности, включают средства, способствующие высушиванию, которые называют высушивающими веществами, вещества, улучшающие текучесть, пигменты и красители.
Предпочтительно композиции для нанесения покрытия по настоящему изобретению характеризуются минимальной температурой пленкообразования, составляющей не более 50°C, особенно предпочтительно не более 35°C и в наибольшей степени предпочтительно не более 25°C, что можно измерять в соответствии с DIN ISO 2115.
Водные дисперсии по настоящему изобретению, в частности, можно применять в качестве композиции для нанесения покрытия или в качестве добавки. Они включают, в частности, краски, пропитывающие средства, клеи и/или грунтовки. Водные дисперсии особенно предпочтительно могут обеспечивать получение красок или пропитывающих средств для вариантов применения в отношении дерева и/или металла.
Дополнительный аспект настоящего изобретения относится к покрытию, получаемому путем нанесения водной дисперсии по настоящему изобретению на изделие, подлежащее нанесению покрытия, и ее последовательного отверждения и высушивания. При высушивании и отверждении обычно происходят реакции сшивания с дополнительными компонентами водных дисперсий, и, таким образом, покрытие по настоящему изобретению содержит сшитый сополимер. Тип реакций сшивания зависит от состава водной дисперсии.
Если водная дисперсия по настоящему изобретению содержит кетон или альдегид, будет происходить вторичное сшивание путем реакции конденсации с кетоном или альдегидом при отверждении сополимера по настоящему изобретению.
В дополнительном варианте осуществления сополимер по настоящему изобретению также можно подвергать вторичному сшиванию путем присоединения по Михаэлю с участием ненасыщенной связи.
В системах для нанесения покрытия, содержащих изоцианат, сополимер по настоящему изобретению также можно подвергать вторичному сшиванию путем присоединения с участием изоцианата.
Покрытия, получаемые с помощью композиций для нанесения покрытия по настоящему изобретению, демонстрируют высокую устойчивость к действию растворителей, при этом, в частности, только небольшие части покрытия подвергаются растворению растворителями. Предпочтительные покрытия демонстрируют, в частности, высокую устойчивость к действию метилизобутилкетона (MIBK). Таким образом, потеря веса после обработки с помощью MIBK предпочтительно составляет не более 50 вес. %, предпочтительно не более 35 вес. %. Поглощение MIBK предпочтительно составляет не более 300 вес. %, особенно предпочтительно не более 250 вес. %, в пересчете на вес применяемого покрытия. Такие значения измеряют при температуре, составляющей примерно 25°C, и времени контакта, составляющем по меньшей мере 4 часа, при этом измерение проводят в отношении полностью высушенного покрытия. В данном случае высушивание происходит в присутствии кислорода, например, воздуха, с целью обеспечения возможности сшивания.
Покрытия, полученные с помощью композиций для нанесения покрытия по настоящему изобретению, демонстрируют высокую механическую прочность. Твердость, определенная с помощью маятникового прибора, предпочтительно составляет по меньшей мере 15 с, предпочтительно по меньшей мере 25 с, что измеряли в соответствии с DIN ISO 1522.
Дисперсии по настоящему изобретению также могут содержать дополнительные составляющие, а также полимеры, полученные в результате полимеризации в эмульсии.
Ниже предусматривается более подробное описание настоящего изобретения с использованием примеров и сравнительных примеров, которые не являются ограничивающими.
ПРИМЕРЫ
Анализ
Газовая хроматография (GC).
Прибор: 7820A от Agilent Technologies.
Колонка: DB5, 30 м, диаметр 0,250 мм, пленка 0,25 мкм.
Температурный режим
Введение при 60°C, затем удерживание в течение 2 мин. Затем нагревание до 240°C при 20°C/мин и после достижения данной температуры удерживание в течение 240°C в течение 8 мин.
Пример 1. Получение N-(2-этиламино)-2-цианоацетамида
Изначально загружали 600 г (10,0 моль) этилендиамина в четырехгорлую круглодонную колбу объемом 2 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 500 мл. В нее дозировали по каплям 248 г (2,5 моль) метилцианоацетата в течение 60 минут, с тем чтобы температура реакции не превышала 30°C. В течение данного времени четырехгорлую круглодонную колбу охлаждали на бане с ледяной водой. В ходе добавления метилцианоацетата реакционная смесь все больше окрашивалась в розовый цвет, а затем – в сиреневый. Для завершения реакции реакционную смесь перемешивали дополнительно в течение 90 минут при комнатной температуре.
Затем избыток этилендиамина удаляли при пониженном давлении. Для данной цели реакционную смесь нагревали до 100°C (температура масляной бани) и летучие составляющие отгоняли в течение периода времени, составляющего 2 часа, при давлении, составляющем не более 5 мбар.
Продукт получали в виде темного стекловидного твердого вещества с чистотой, составляющей 97,9% площади (определяли с применением GC-RV). Выход продукта составил 309 г (95%).
Сравнительный пример 1. Получение N,N’-этилен-бис-метакриламида
Изначально загружали 40% водный раствор этилендиамина (25,5 г, 0,17 моль) в четырехгорлую круглодонную колбу объемом 250 мл с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 100 мл. В нее дозировали по каплям 26 г (0,17 моль) метакрилового ангидрида в течение 60 минут, с тем чтобы температура реакции не превышала 30°C. В течение данного времени четырехгорлую круглодонную колбу охлаждали на бане с ледяной водой. В ходе добавления метакрилового ангидрида образовывалось белое твердое вещество.
Белое твердое вещество отделяли путем фильтрации и высушивали. Оно представляло собой N,N´-этилен-бис-метакриламид с чистотой 74,8% площади (определяли с применением GC-RV). Выход продукта составил 20 г (60%).
Пример 2. Получение N-(2-цианоэтиламидоэтил)метакриламида
Изначально загружали смесь 147 г (1,2 моль) N-(2-этиламино)-2-цианоацетамида и 600 г (6,0 моль) метилметакрилата в четырехгорлую круглодонную колбу объемом 1 л со впуском для воздуха, сабельной мешалкой, электродвигателем для мешалки и колонкой из зеркального материала длиной 50 см и толщиной 29 мм с неупорядоченной насадкой, заполненной кольцами Рашига 6 × 6. В нее добавляли 7 мг (10 ppm) 4-гидрокси-2,2,6,6-тетраметилпиперидинооксила и 0,15 г (200 ppm) простого гидрохинон-монометилового эфира с последующим добавлением 7,4 г смеси, содержащей 65,6 вес. % оксида диоктилолова и 34,4 вес. % тетраизопропилтитаната.
Реакционную смесь нагревали с обратным холодильником, при этом образующийся метанол отгоняли в виде азеотропной смеси с помощью колонки с неупорядоченной насадкой. Через примерно 3,5 часа степень превращения, определенная с помощью GC, составляла 58%.
Пример 3. Получение N-(2-цианоэтиламидоэтил)метакриламида
Растворяли 312 г (2,4 моль) N-(2-этиламино)-2-цианоацетамида из примера 1 в 468 г воды в трехгорлой круглодонной колбе объемом 2 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 500 мл. В нее медленно добавляли по каплям 370 г (2,4 моль) метакрилового ангидрида, при этом образовывался светло-коричневый осадок. Затем реакционную смесь перемешивали дополнительно в течение 1,5 часа при 80°C.
Из полученной прозрачной темно-красной реакционной смеси удаляли низкокипящие вещества при пониженном давлении, ее концентрировали до 646 г и к ней добавляли 400 г изопропанола. Это приводило к образованию осадка, который отделяли путем фильтрации.
Продукт получали в виде коричневого кристаллического твердого вещества с чистотой, составляющей 95,0% площади (определяли с применением GC-RV). Выход продукта составил 346 г (73,9%).
Растворимость продукта в воде определяли следующим образом.
Продукт перемешивали с водой в течение трех дней при комнатной температуре, и состав водной фазы, полученной таким образом, определяли с применением HPLC. Водная фаза содержала 6,7 вес. % продукта.
Дополнительный образец продукта перемешивали с водой в течение трех часов при 80°C и охлаждали обратно до комнатной температуры. Затем определяли состав водной фазы, полученной таким образом, с применением HPLC. Водная фаза содержала более 10 вес. % продукта.
Продукт характеризовался высокой растворимостью в воде. Следовательно, его можно применять при полимеризации в эмульсии даже при отсутствии органических растворителей.
Пример 4. Получение N-(2-бутиламино)-2-цианоацетамида
Плавили 353 г (4,0 моль) 1,4-диаминобутана при примерно 30°C в четырехгорлой круглодонной колбе объемом 1 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 250 мл. В нее дозировали по каплям 99 г (1,0 моль) метилцианоацетата в течение 30 минут, с тем чтобы температура реакции оставалась на уровне от примерно 30°C до 40°C. В течение данного времени круглодонную колбу охлаждали на бане с ледяной водой. В ходе добавления метилцианоацетата реакционная смесь все больше окрашивалась в интенсивный желтый цвет. Для завершения реакции реакционную смесь перемешивали дополнительно в течение 90 минут при комнатной температуре, при этом реакционная смесь окрашивалась в красный цвет.
Затем удаляли избыток 1,4-диаминобутана при пониженном давлении. Для данной цели реакционную смесь нагревали до 100°C (температура масляной бани) и летучие составляющие отгоняли в течение периода времени, составляющего 2,5 часа, при давлении, составляющем не более 2 мбар.
Продукт получали в виде темного стекловидного твердого вещества с чистотой, составляющей 89,1% площади (определяли с применением GC-RV). Выход продукта составил 146 г (84%).
Пример 5. Получение N-(2-цианоэтиламидобутил)метакриламида
Растворяли 360 г (0,93 моль) N-(2-бутиламино)-2-цианоацетамида из примера 3 в 540 г воды в четырехгорлой круглодонной колбе объемом 1 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 500 мл и охлаждали до 0°C на бане с ледяной водой. В нее медленно добавляли по каплям 143 г (0,93 моль) метакрилового ангидрида, растворенного в 300 мл метанола. Затем реакционную смесь перемешивали в течение ночи при комнатной температуре, при этом реакционная смесь окрашивалась в зеленый цвет.
Реакционную смесь концентрировали при пониженном давлении при 80°C и 35 мбар до 263 г. Остаток растворяли в 160 г изопропанола, и полученный раствор хранили при комнатной температуре. Это приводило к образованию осадка, который отделяли путем фильтрации.
Продукт получали в виде желтого кристаллического твердого вещества. Чистота составляла примерно 94,0% площади (определяли с применением GC-RV).
Растворимость продукта в воде определяли следующим образом.
Продукт перемешивали с водой в течение двух дней при комнатной температуре, и состав водной фазы, полученной таким образом, определяли с применением HPLC. Водная фаза содержала 6,2 вес. % продукта.
Дополнительный образец продукта перемешивали с водой в течение одного часа при 60°C и охлаждали обратно до комнатной температуры. Затем определяли состав водной фазы, полученной таким образом, с применением HPLC. Водная фаза содержала 8,1 вес. % продукта.
Продукт характеризовался высокой растворимостью в воде. Следовательно, его можно применять при полимеризации в эмульсии даже при отсутствии органических растворителей.
Сравнительный пример 2. Получение N-(2-гексиламино)-2-цианоацетамида
Плавили 465 г (4,0 моль) 1,6-диаминогексана при примерно 41°C в четырехгорлой круглодонной колбе объемом 1 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 250 мл. В нее дозировали по каплям 99 г (1,0 моль) метилцианоацетата в течение 30 минут, с тем чтобы температура реакции оставалась на уровне от примерно 50°C до 75°C. В ходе добавления метилцианоацетата реакционная смесь все больше окрашивалась в интенсивный желтый цвет. Для завершения реакции реакционную смесь перемешивали дополнительно в течение 90 минут при температуре от примерно 50°C до 75°C, при этом реакционная смесь окрашивалась в красный цвет.
Затем удаляли избыток 1,6-диаминогексана при пониженном давлении. Для данной цели реакционную смесь нагревали до 120°C (температура масляной бани) и летучие составляющие отгоняли в течение периода времени, составляющего 4 часа, при давлении, составляющем не более 2 мбар.
Продукт получали в виде темного стекловидного твердого вещества с чистотой, составляющей примерно 100% площади (определяли с применением GC-RV). Выход продукта составил 172 г (94%).
Сравнительный пример 3. Получение N-(2-цианоэтиламидогексил)метакриламида
Изначально загружали 31 г (0,2 моль) метакрилового ангидрида и 150 г воды в четырехгорлую круглодонную колбу объемом 1 л с сабельной мешалкой, электродвигателем мешалки, термометром и капельной воронкой объемом 500 мл и охлаждали до 0°C на бане с ледяной водой.
Растворяли 360 г (0,93 моль) N-(2-гексиламино)-2-цианоацетамида из сравнительного примера 2 в 3240 г метанола при 60°C, и его охлаждали до комнатной температуры. Данный раствор добавляли в течение периода времени, составляющего 30 минут, с помощью капельной воронки к метакриловому ангидриду. Температуру реакции поддерживали на уровне ниже 20°C. Затем реакционную смесь перемешивали дополнительно в течение 3 часов при комнатной температуре.
Образованный продукт обнаруживали в реакционной смеси с применением GC-RV, а его выделение можно проводить путем кристаллизации из изопропанола.
Растворимость продукта в воде определяли следующим образом.
Продукт перемешивали с водой в течение трех дней при комнатной температуре, и состав водной фазы, полученной таким образом, определяли с применением HPLC. Продукт был по сути нерастворимым в воде.
Дополнительный образец продукта перемешивали с водой в течение трех часов при 80°C и охлаждали до комнатной температуры. Затем определяли состав водной фазы, полученной таким образом, с применением HPLC. Продукт был по сути нерастворимым в воде; причем только имеющиеся в продукте примеси подвергались растворению и обнаружению.
Продукт характеризовался очень низкой растворимостью в воде. Следовательно, его нельзя применять при полимеризации в эмульсии при отсутствии органических растворителей.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВОДНОЕ СВЯЗУЮЩЕЕ СРЕДСТВО ДЛЯ ПРИМЕНЕНИЯ В ТЕРМИЧЕСКОЙ СВАРКЕ | 2013 |
|
RU2639157C2 |
| ФУНКЦИОНАЛИЗОВАННЫЙ (МЕТ) АКРИЛАТНЫЙ МОНОМЕР, ПОЛИМЕР, ПОКРОВНОЕ СРЕДСТВО, СПОСОБ ПОЛУЧЕНИЯ И СПОСОБ СШИВАНИЯ | 2009 |
|
RU2523549C9 |
| ВОДНЫЕ ПЕРВИЧНЫЕ ДИСПЕРСИИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, А ТАКЖЕ ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2662226C1 |
| ПРИМЕНЕНИЕ МНОГОСТАДИЙНЫХ ПОЛИМЕРНЫХ ДИСПЕРСИЙ ДЛЯ НАНЕСЕНИЯ ПОКРЫТИЙ НА МЕТАЛЛИЧЕСКИЕ ЛИСТЫ | 2013 |
|
RU2674410C2 |
| БЫСТРООТВЕРЖДАЮЩАЯСЯ ВОДНАЯ КОМПОЗИЦИЯ ДЛЯ ПОКРЫТИЯ И КРАСКА | 1996 |
|
RU2158744C2 |
| ВОДНЫЕ ДИСПЕРСИИ, СОДЕРЖАЩИЕ ПО МЕНЬШЕЙ МЕРЕ ОДНУ АЛКИДНУЮ СМОЛУ И ПО КРАЙНЕЙ МЕРЕ ОДИН ПОЛИМЕРИЗАТ ПО МЕНЬШЕЙ МЕРЕ С ОДНИМ (МЕТ)АКРИЛАТНЫМ СЕГМЕНТОМ | 2008 |
|
RU2478677C2 |
| МНОГОСТАДИЙНЫЕ ПОЛИМЕРНЫЕ ДИСПЕРСИИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2588130C2 |
| ВОДНЫЕ ЭМУЛЬСИОННЫЕ ПОЛИМЕРИЗАТЫ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2011 |
|
RU2583807C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНЫХ ДИСПЕРСИЙ | 2017 |
|
RU2754822C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНЫХ ЛАТЕКСОВ КАК СВЯЗУЮЩИХ | 2006 |
|
RU2407755C2 |
Изобретение относится к соединениям общей формулы (I), которые могут найти применение в качестве мономеров для синтеза сополимеров, обеспечивающих получение композиций для нанесения покрытия. В формуле (I) R1 выбран из метильной группы и R2 представляет собой алкиленовую группу, имеющую состав CnHm, при этом n = 2-4, m = 4-8. 1 з.п. ф-лы, 5 пр.
 (I)
(I)
1. Соединение общей формулы (I)
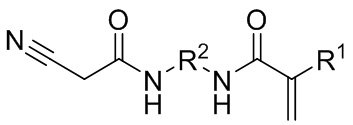 (I),
(I),
где R1 выбран из метильной группы и
R2 представляет собой алкиленовую группу, имеющую состав CnHm, при этом
n = 2-4,
m = 4-8.
2. Соединение по п. 1, где алкиленовая группа R2 представляет собой одну из следующих групп: 1,2-этиленовую группу, 1,3-пропиленовую группу, 1,2-пропиленовую группу, 1,4-бутиленовую группу.
| Колосоуборка | 1923 |
|
SU2009A1 |
| US 4215195 A, 29.07.1980 | |||
| МНОГОСТАДИЙНЫЕ ПОЛИМЕРНЫЕ ДИСПЕРСИИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2588130C2 |

