Изобретение относится к бензимидазольным производным общей формулы I
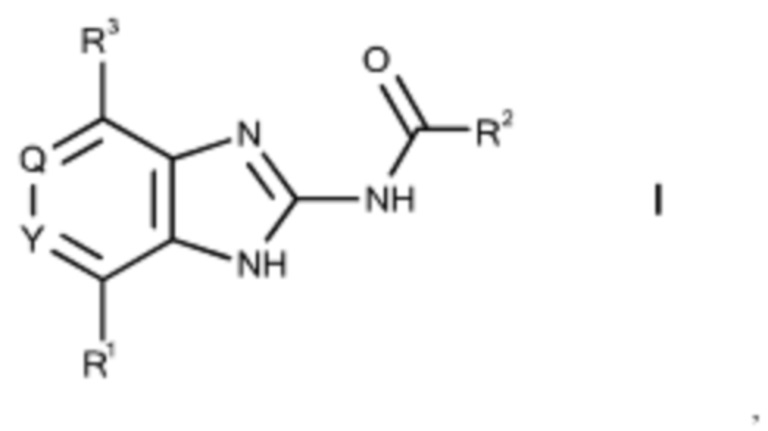
и применению соединений настоящего изобретения для лечения и/или предотвращения гиперпролиферативных или инфекционных заболеваний и нарушений у млекопитающих, в особенности людей, и фармацевтическим композициям, содержащим такие соединения.
Предпосылки создания изобретения
Аденозин является распространенным модулятором многочисленных физиологических действий, особенно в сердечно-сосудистой, нервной и иммунной системах. Аденозин связан как структурно, так и метаболически, с биоактивными нуклеотидами: аденозинтрифосфатом (АТФ), аденозиндифосфатом (АДФ), аденозинмонофосфатом (АМФ) и циклическим аденозинмонофосфатом (цАМФ), с биохимическим метилирующим агентом S-аденозил-L-метионом (SAM) и структурно с коферментами NAD, FAD и коферментом А и с РНК.
Через рецепторы клеточной поверхности аденозин модулирует различные физиологические функции, включая индукцию седации, вазодилатацию, подавление сердечного ритма и сократительной способности, ингибирование агрегации тромбоцитов, стимуляцию глюконеогенеза и ингибирование липолиза. Исследования показывают, что аденозин способен активировать аденилатциклазы, открывать калиевые каналы, уменьшать поток через кальциевые каналы и ингибировать или стимулировать обмен фосфоинозитидов с помощью рецептор-опосредованных механизмов (Muller СЕ и Stein В., Current Pharmaceutical Design, 2: 501, 1996; Muller СЕ, Exp. Opin. Ther. Patents, 7 (5): 419, 1997).
Аденозиновые рецепторы принадлежат к суперсемейству рецепторов, сопряженных с G-белками (GPCR). Четыре основных подтипа аденозиновых рецепторов были фармакологически, структурно и функционально охарактеризованы (Fredholm и др., Pharm. Rev., 46: 143-156, 1994) и названы как A1, A2A, A2B и А3. Хотя один и тот же аденозиновый рецептор может связываться с разными G-белками, аденозиновые рецепторы A1 и А3 обычно связываются с ингибирующими G-белками, называемыми Gi и G0, которые ингибируют аденилатциклазу и снижают уровень цАМФ в клетках. В отличие от этого, аденозиновые рецепторы A2A и A2B сопряжены со стимулирующими G-белками, называемыми GS, которые активируют аденилатциклазу и повышают уровни цАМФ в клетках (Linden J., Annu. Rev. Pharmacol. Toxicol., 41: 775-87 2001).
В соответствии с изобретением, «лиганды, селективные к аденозиновым рецепторам» представляют собой вещества, которые селективно связываются с одним или несколькими подтипами аденозиновых рецепторов, таким образом либо имитируя действие аденозина (агонисты аденозина), либо блокируя его действие (антагонисты аденозина). В соответствии с их рецепторной селективностью, лиганды, селективные к аденозиновым рецепторам, можно разделить на различные категории, например, лиганды, которые селективно связываются с рецепторами A1 или А2, а в случае последних также, например, те, которые селективно связываются с рецепторами A2A или A2B. Также возможны лиганды аденозиновых рецепторов, которые селективно связываются с множеством подтипов аденозиновых рецепторов, например, лиганды, которые селективно связываются с A1 и А2, но не с рецепторами А3. Селективность вышеупомянутых рецепторов может быть определена по воздействию веществ на клеточные линии, которые после стабильной трансфекции соответствующей кДНК, экспрессируют рассматриваемые подтипы рецептора (Olah, М.Е. и др., J. Biol. Chem., 267: 10764-10770, 1992). Влияние веществ на такие клеточные линии можно контролировать с помощью биохимического измерения внутриклеточного мессенджера цАМФ (Klotz, K.N. и др., Naunyn Schmiedebergs Arch. Pharmacol. 357: 1-9, 1998).
Известно, что система рецептора A1 включает активацию фосфолипазы С и модуляцию как калиевых, так и кальциевых ионных каналов. Подтип А3, помимо своей ассоциации с аденилатциклазой, также стимулирует фосфолипазу С и, таким образом, активирует кальциевые ионные каналы.
Рецептор A1 (326-328 аминокислот) был клонирован из различных видов (псовые, человек, крысы, собака, цыпленок, крупный рогатый скот, морская свинка) с последовательностью 90-95%, идентифицированной среди видов млекопитающих. Рецептор A2A (409-412 аминокислот) был клонирован из псовых, крыс, человека, морской свинки и мыши. Рецептор A2B (332 аминокислот) был клонирован из человека и мыши с 45% гомологией A2B человека с человеческими рецепторами A1 и A2A. Рецептор А3 (317-320 аминокислот) был клонирован из человека, крысы, собаки, кролика и овцы.
Предполагается, что подтипы рецепторов A1 и A2A играют взаимодополняющие роли в регуляции аденозинового энергоснабжения. Аденозин, который является продуктом метаболизма АТФ, диффузирует из клетки и действует локально, активируя аденозиновые рецепторы, уменьшая потребность в кислороде (A1 и А3) или увеличивая снабжение кислородом (A2A), и таким образом восстанавливая баланс энергообеспечения / энергопотребления в тканях. Действием обоих подтипов является увеличение количества кислорода, доступного ткани, и защита клеток от повреждения, вызванного кратковременным дисбалансом кислорода. Одной из важных функций эндогенного аденозина является предотвращение повреждений при травмах, таких как гипоксия, ишемия, гипотензия и судорожная активность. Кроме того, известно, что связывание агониста аденозинового рецептора с тучными клетками, экспрессирующими рецептор А3 крысы, приводило к повышению концентраций инозитолтрифосфата и внутриклеточного кальция, что усиливало антиген-индуцированную секрецию медиаторов воспаления. Следовательно, рецептор А3 играет роль в опосредовании приступов астмы и других аллергических реакций.
Эти аденозиновые рецепторы кодируются различными генами и классифицируются по их сродству с аналогами аденозина и антагонистами метилксантина (Klinger и др., Cell Signal., 14 (2): 99-108, 2002).
Касательно роли аденозина в нервной системе, то были сделаны первые наблюдения о влиянии кофеина, который является наиболее широко используемым из всех психоактивных лекарственных веществ. На самом деле кофеин - это известный антагонист аденозинового рецептора, который способен повысить восприятие и способность к обучению млекопитающих. Путь аденозинового рецептора A2A отвечает за эти эффекты (Fredholm и др., Pharmacol. Rev., 51 (1): 83-133, 1999; Huang и др., Nat Neurosci., 8 (7): 858-9, 2005), и влияние кофеина на сигнальный путь аденозинового рецептора A2A стимулировало исследование высокоспецифических и сильнодействующих антагонистов аденозинового A2A.
У млекопитающих аденозиновые рецепторы A2A имеют ограниченное распределение в мозге и обнаруживаются в стриатуме, обонятельном бугорке и прилежащем ядре (Dixon и др., Br. J. Pharmacol., 118 (6): 1461-8, 1996). Высокие и промежуточные уровни экспрессии могут наблюдаться в иммунных клетках, сердце, легких и кровеносных сосудах. В периферической системе G3, по-видимому, является основным G-белком, связанным с аденозиновым рецептором A2A, но в стриатуме было показано, что стриарные аденозиновые рецепторы A2A опосредуют свое действие посредством активации G-белка, называемого Goif (Kull и др., Mol. Pharmacol., 58 (4): 772-7, 2000), который подобен G3 и также связан с аденилатциклазой.
На сегодняшний день исследования генетически модифицированных мышей и фармакологический анализ показывают, что рецептор A2A является многообещающей терапевтической мишенью для лечения нарушений центральной нервной системы (ЦНС) и таких заболеваний, как болезнь Паркинсона, болезнь Хантингтона, синдром дефицита внимания и гиперактивности (СДВГ), удар (ишемическая черепно-мозговая травма) и болезнь Альцгеймера (Fredholm и др., Annu. Rev. Pharmacol. Toxicol., 45: 385-412, 2005; Higgins и др.; Behav. Brain Res. 185: 32-42, 2007; DaIl' Igna и др., Exp. Neurol., 203 (1): 241-5, 2007; Arendash и др., Neuroscience, 142 (4): 941-52, 2006; Trends in Neurosci., 29 (11), 647-654, 2006; Expert Opinion Ther. Patents, 17, 979-991, 2007; Exp. Neurol., 184 (1), 285-284, 2003; Prog. Brain Res, 183, 183-208, 2010; J. Alzheimer Dis., Suppl 1, 1 17-126, 2010; J. Neurosci., 29 (47), 14741-14751, 2009; Neuroscience, 166 (2), 590-603, 2010; J. Pharmacol. Exp. Ther., 330 (1), 294-303, 2009; Frontiers Biosci., 13, 2614-2632, 2008), а также для лечения различных психозов органического происхождения (Weiss и др., Neurology, 61 (11 Suppl 6): 88-93, 2003).
Применение аденозиновых рецепторов A2A у нокаутных мышей показало, что инактивация аденозинового рецептора A2A защищает от гибели нейрональных клеток, вызванной ишемией (Chen и др., J. Neurosci., 19 (21): 9192-200, 1999 и Monopoli и др., Neuroreport, 9 (17): 3955-9, 1998) и митохондриальным токсином 3-NP (Blum и др., J. Neurosci., 23 (12): 5361-9, 2003). Эти результаты послужили основой для лечения ишемии и болезни Хантингтона с помощью антагонистов аденозинового A2A. Блокада аденозиновых рецепторов A2A оказывает также антидепрессивное действие (El Yacoubi и др., Neuropharmacology, 40 (3): 424-32, 2001). В заключение, эта блокада предотвращает дисфункцию памяти (Cunha и др., Exp. Neurol., 210 (2): 776-81, 2008; Takahashi и др., Front. Biosci., 13: 2614-32, 2008), и это может быть перспективным терапевтическим путем для лечения и/или предотвращения болезни Альцгеймера.
Для обзора относительно аденозиновых рецепторов A2A см., например, Moreau и др. (Brain Res. Reviews 31: 65-82, 1999) и Svenningsson и др. (Progress in Neurobiology 59: 355-396, 1999).
На сегодняшний день несколько антагонистов аденозиновых рецепторов A2A продемонстрировали многообещающий потенциал для лечения болезни Паркинсона. В качестве примера, KW-6002 (Истрадефиллин) завершил клиническое испытание III фазы в США после того, как исследования продемонстрировали его эффективность в смягчении симптомов заболевания (Bara-Himenez и др., Neurology, 61 (3): 293-6, 2003 и Hauser и др., Neurology, 61 (3): 297-303, 2003). SCH420814 (Преладенант) в настоящее время находится на этапе II клинического испытания в США и обеспечивает улучшение моторной функции на животных моделях болезни Паркинсона (Neustadt и др., Bioorg. Med. Chem. Lett., 17 (5): 1376-80, 2001), а также у пациентов-людей (Hunter J.С, poster Boston 2006 - http://www.a2apd.org/Speaker abstracts/Hunter.pdf).
Помимо полезности антагонистов рецепторов A2A для лечения нейродегенеративных заболеваний, эти соединения были рассмотрены в качестве полезных для применения при дополнительных симптоматических показаниях. Это основано на доказательствах того, что активация рецептора A2A может способствовать патофизиологии ряда психоневрологических нарушений и дисфункций, таких как депрессия, чрезмерная дневная сонливость, синдром беспокойных ног, синдром дефицита внимания и гиперактивности и когнитивная усталость (Neurology, 61 (Suppl 6), 82-87, 2003; Behav. Pharmacol., 20 (2), 134-145, 2009; CNS Drug Discov., 2 (1), 1-21, 2007).
Некоторые авторы предлагают применение антагонистов A2A для лечения диабета (WO 1999035147; WO 2001002400). Другие исследования предполагают участие аденозиновых рецепторов A2A в заживлении ран или мерцательной аритмии (Am. J. Path., 6, 1774-1778, 2007; Arthritis & Rheumatism, 54 (8), 2632-2642, 2006).
Некоторые из сильнодействующих антагонистов аденозиновых A2A, обнаруженных в прошлом фармацевтическими компаниями, перешли в клинические испытания, показывающие положительные результаты и демонстрирующие потенциал этого класса соединений для лечения нейродегенеративных нарушений, таких как болезнь Паркинсона, Хантингтона или Альцгеймера, а также при других заболеваниях, связанных с ЦНС, таких как депрессия, синдром беспокойства, нарушение сна и тревожное расстройство (Clin. Neuropharmacol., 33, 55-60, 2010; J. Neurosci., 30 (48), 2010), 16284-16292; Parkinson Relat. Disord., 16 (6), 423-426, 2010; Expert Opinion Ther. Patents, 20(8), 987-1005, 2010; Current Opinion in Drug Discovery & Development, 13 (4), 466-480, 2010 и ссылки в данном документе; Mov. Disorders, 25 (2), S305, 2010).
Известными ингибиторами A2A являются истрадефиллин (KW-6002), преладенант (SCH420814), SCH58261, CGS15943, тозаденант, випаденант (V-2006), V-81444 (CPI-444, HTL-1071, PBF-509, Medi-9447, PNQ-370, ZM-241385, ASO-5854, ST-1535, ST-4206, DT1133 и DT-0926, которые в большинстве случаев разработаны для лечения болезни Паркинсона.
Аденозиновые рецепторы A2B были клонированы из гипоталамуса крысы (Rivkees и Reppert, 1992), гиппокампа человека (Pierce и др., 1992) и тучных клеток мыши (Marquardt и др., 1994) с использованием стандартных методов полимеразной цепной реакции с вырожденными олигонуклеотидными праймерами, предназначенными для распознавания консервативных областей большинства рецепторов, сопряженных с G-белком. Человеческий рецептор A2B имеет от 86 до 87% гомологии аминокислотной последовательности с рецепторами A2B крысы и мыши (Rivkees and Reppert, 1992; Pierce и др., 1992; Marquardt и др., 1994) и 45% гомологии аминокислотной последовательности с человеческими рецепторами A1 и A2A. Как и ожидалось для близкородственных видов, крысиные и мышиные рецепторы A2B имеют 96% гомологию аминокислотной последовательности. Для сравнения, общая аминокислотная идентичность между рецепторами A1 различных видов составляет 87% (Palmer и Stiles, 1995). Рецепторы A2A имеют 90% гомологии между видами (Ongini и Fredholm, 1996), при этом большинство различий наблюдается во 2-й внеклеточной петле и длинном С-концевом домене (Palmer и Stiles, 1995). Самая низкая (72%) степень идентичности между видами наблюдается для последовательностей рецептора А3 (Palmer и Stiles, 1995).
Аналог аденозина NECA остается наиболее мощным агонистом A2B (Bruns, 1981; Feoktistov and Biaggioni, 1993, 1997; Brackett и Daly, 1994), с концентрацией, вызывающей полумаксимальный эффект (ЕС50) для стимуляции аденилциклазы приблизительно 2 мкМ. Это, однако, неселективно и активирует другие аденозиновые рецепторы с еще большей аффинностью, со значением ЕС50 в низком наномолярном (A1 и A2A) или высоком наномолярном (A3) диапазоне. Поэтому определение характеристик рецепторов A2B часто зависит от недостаточной эффективности соединений, которые являются сильными и селективными агонистами других типов рецепторов. Рецепторы A2B были охарактеризованы методом исключения, то есть отсутствием эффективности агонистов, специфичных для других рецепторов. Селективный агонист A2A CGS-21680 (Webb и др., 1992), например, был полезен для дифференциации между аденозиновыми рецепторами A2A и A2B (Hide и др., 1992; Chern и др., 1993; Feoktistov and Biaggioni, 1995; van der Ploeg и др., 1996). Оба рецептора положительно связаны с аденилциклазой и активируются неселективным агонистом NECA. CGS-21680 практически неэффективен в отношении рецепторов A2B, но все же эффективен, как NECA, в активации рецепторов A2A, со значением ЕС50 в низком наномолярном диапазоне для обоих агонистов (Jarvis и др., 1989; Nakane и Chiba, 1990; Webb и др., 1992; Hide и др., 1992; Feoktistov и Biaggioni, 1993; Alexander и др., 1996). Рецепторы A2B также имеют очень низкое сродство с A1-селективным агонистом R-PIA (Feoktistov and Biaggioni, 1993; Brackett и Daly, 1994), а также с A3-селективным агонистом N6-(3-йодбензил)-N-метил-5'-карбамоиладенозином (IB-MECA) (Feoktistov и Biaggioni, 1997). Профиль агонистов NECA > R-PIA=IB-MECA > CGS-21680 определяли в клетках эритролейкемии человека (HEL) для A2B-опосредованного накопления цАМФ. Разница между ЕС50 для NECA и остальных агонистов составляет приблизительно 2 порядка величин. Поэтому ответы, вызванные NECA при концентрациях в низком микромолярном диапазоне (1-10 мкМ), но не R-PIA, IB-MECA или CGS-21680, характерны для рецепторов A2B. В то время как рецепторы A2B в общем имеют более низкое сродство с агонистами по сравнению с другими подтипами рецепторов, это не относится к антагонистам. Отношение структурной активности аденозиновых антагонистов на рецепторы A2B не были полностью охарактеризованы, но по крайней мере некоторые ксантины являются такими же или более сильными антагонистами подтипов рецепторов A2B, чем других подтипов. В частности, DPSPX (1,3-дипропил-8-сульфофенилксантин), DPCPX (1,3-дипропил-8-циклопентилксантин), DPX (1,3-диэтилфенилксантин), противоастматическое лекарственное вещество энпрофиллин (3-н-пропилксантин) и нексантиновое соединение 2,4-диоксобензоптеридин (аллоксазин) имеют сродство в нМ диапазоне от средних до высоких значений.
Другими известными ингибиторами A2B являются ATL801, PSB-605, PSB-1115, ISAM-140, GS6201, MRS1706 и MRS1754.
В данном документе раскрыто, что аденозиновые рецепторы играют статически неопределимую роль в понижающей регуляции воспаления in vivo, действуя как физиологический «СТОП» (механизм прекращения), который может ограничивать иммунный ответ и тем самым защищать нормальные ткани от чрезмерного иммунного повреждения во время патогенеза различных заболеваний.
Антагонисты рецепторов A2A обеспечивают долгосрочное усиление иммунных ответов путем уменьшения опосредованной Т-клетками переносимости антигенных стимулов, усиления индукции Т-клеток памяти и повышения эффективности введения пассивных антител для лечения злокачественного новообразования и инфекционных заболеваний, в то время как агонисты рецепторов A2A обеспечивают длительное снижение иммунных ответов путем повышения переносимости, опосредованной Т-клетками, антигенных стимулов, в частности, для снижения использования иммунодепрессантов в определенных условиях.
Иммуномодуляция является критическим аспектом лечения ряда заболеваний и нарушений. Т-клетки в особенности играют жизненно важную роль в борьбе с инфекциями и обладают способностью распознавать и уничтожать раковые клетки. Усиление опосредованных Т-клетками ответов является ключевым компонентом для усиления ответов на терапевтические средства. Однако для иммуномодуляции крайне важно, чтобы любое усиление иммунного ответа было сбалансировано с необходимостью предотвращения аутоиммунитета, а также хронического воспаления. Хроническое воспаление и самораспознавание Т-клетками является основной причиной патогенеза системных нарушений, таких как ревматоидный артрит, рассеянный склероз и системная красная волчанка. Кроме того, для предотвращения отторжения пересаженных органов или трансплантатов требуется длительная иммуносупрессия.
Иммуносупрессия, индуцированная опухолью, является основным препятствием для эффективности современных методов лечения злокачественных новообразований. Из-за их замечательной клинической эффективности против более широкого спектра злокачественных новообразований, недавние успехи с ингибиторами блокады иммунных контрольных точек, такими как анти-CTLA-4 и анти-PD-1/PDL1, представляют собой революционное лечение злокачественных заболеваний.
Аденозин является одной из новых перспективных иммуносупрессивных мишеней, выявленных в доклинических исследованиях. Этот метаболит вырабатывается эктоэнзимом - CD73, экспрессируемым на клетках-супрессорах хозяина и опухолевых клетках. Повышенная экспрессия CD73 коррелирует с плохим прогнозом у пациентов с рядом видов злокачественных новообразований, включая колоректальный рак (Liu и др., J. Surgical Oncol, 2012), рак желудка (Lu и др., World J. Gastroenterol., 2013), рак желчного пузыря (Xiong и др., Cell and Tissue Res., 2014). Доклинические исследования продемонстрировали, что проопухолевые эффекты CD73 могут быть вызваны (по крайней мере частично) аденозин-опосредованной иммуносупрессией. Как раскрыто выше, аденозин связывается с четырьмя известными рецепторами A1, A2A, A2B и А3, с активацией рецепторов A2A и A2B, которые, как известно, подавляют эффекторные функции многих иммунных клеток, то есть, рецепторы A2A и A2B индуцируют аденилат-циклаз-зависимое накопление цАМФ, что приводит к иммуносупрессии. Поскольку антагонизирование A1 и А3 будет противодействовать желаемому эффекту, а агонисты A1 и А3 служат потенциальными кардиозащитными агентами, необходимо достичь селективности по отношению к A1 и A3 (Antonioli и др., Nat. rev. Cancer, 2013, Thiel и др., Microbes и Infection, 2003). Было продемонстрировано, что в микроокружении опухоли активация рецепторов как A2A, так и A2B, подавляет противоопухолевый иммунитет и увеличивает распространение опухолей CD73. Кроме того, блокада A2A или A2B низкомолекулярными антагонистами может уменьшить метастазирование опухоли. Было обнаружено, что блокирование рецептора A2A может преодолеть механизмы ускользания опухоли, включая как анергию, так и регуляторную индукцию Т-клеток, вызванную опухолевыми клетками, и вызывать долговременную восприимчивость опухоли к лечению. Ohta и др. продемонстрировали отторжение приблизительно 60% установленных опухолей меланомы CL8-1 у мышей с дефицитом рецептора A2A по сравнению с отсутствием отторжения у нормальных мышей (Ohta, и др.; PNAS 103 (35): 13132-7, 2006). В соответствии с этим, исследователи также показали улучшенное ингибирование роста опухоли, разрушение метастазов и предотвращение неоваскуляризации противоопухолевыми Т-клетками после лечения антагонистом рецептора A2A.
Было продемонстрировано, что опухоли избегают повреждения иммунной системой, препятствуя активации Т-клеток за счет ингибирования костимуляторных факторов в семействах B7-CD28 и TNF, а также путем привлечения регуляторных Т-клеток, которые ингибируют противоопухолевые Т-клеточные ответы (Wang, Cancer. Semin. Cancer. Biol. 16: 73-79, 2006; Greenwald и др., Ann. Rev. Immunol. 23: 515-48, 2005; Watts, Ann. Rev. Immunol. 23: 23-68, 2005; Sadum и др., Clin. Cane. Res. 13 (13): 4016-4025, 2007). Поскольку экспрессия рецептора A2A увеличивается в лимфоцитах после активации, методы терапии, которые высвобождают ответы эффекторных лимфоцитов, такие как терапия анти-CTLA-4 и анти-PD-1, могут также усиливать эффекты A2A-опосредованной иммуносупрессии. Иммунная блокада контрольных точек в сочетании с антагонистами A2A или антагонистами двойных A2A/2B увеличивает величину иммунных ответов на опухоли и метастазы. Соответственно, комбинация ингибирования A2A с терапией анти-PD-1 усиливает продуцирование IFN-γ Т-клетками в совместной культуре с опухолевыми клетками МС38, улучшает выживаемость мышей в модели опухоли молочной железы 4Т1 и уменьшает рост опухоли в опухолях AT-3ovadim CD73+. (Beavis и др., Cancer Immunol. Res., 2015; Mittal и др., Cancer Res., 2014).
Кроме того, доклинические исследования показали, что ингибирование A2B приводит к снижению роста опухоли и увеличению выживаемости мышей в моделях карциномы легких Льюиса, карциномы мочевого пузыря МВ49, карциномы молочной железы орто-4Т1 (Ryzhov и др., 2009, Cekic и др., 2012), и комбинация ингибирования A2B с анти-PD-1 терапией уменьшает метастазы в легкие опухолей B16-F10 меланомы и улучшает выживаемость мышей в модели опухоли молочной железы 4Т1.
В WO 03/050241 описаны способы усиления иммунного ответа на антиген, повышения эффективности вакцины или усиления иммунного ответа на опухолевый антиген, или разрушения опухоли, опосредованного иммунными клетками, путем введения агента, который ингибирует внеклеточный аденозин или ингибирует аденозиновые рецепторы.
WO 2004/089942, WO 2005/000842 и WO 2006/008041 раскрывают бензотиазольные производные, включая тозаденант, в качестве ингибиторов A2A для лечения болезни Паркинсона. WO 2004/092171 и WO 2005/028484 раскрывают подобные производные тиазолопиридина и пиразолопиримидина также в качестве ингибиторов A2A для лечения болезни Паркинсона. Однако эти соединения не проявляют значительной ингибирующей активности на A2B и показывают только хорошие фармакокинетические свойства у крыс, животных моделях болезни Паркинсона, но не у мышей, и животных моделях злокачественного новообразования. Кроме того, соединения не показывают, что они способны предотвращать иммуносупрессию и, следовательно, способны поддерживать индуцированное противораковыми Т-клетками ингибирование роста опухоли, уменьшение или разрушение метастазов и предотвращение неоваскуляризации.
Таким образом, остается потребность в способах лечения, которые обеспечивают долгосрочное усиление иммунных ответов на специфические антигены, особенно для лечения и предотвращения гиперпролиферативных и инфекционных заболеваний и нарушений, и, таким образом, целью настоящего изобретения было обеспечить способы лечения, которые позволили бы упростить протоколы лечения и улучшить иммунные ответы в отношении определенных антигенов. Конкретной задачей изобретения было обеспечить улучшенные способы предотвращения или лечения гиперпролиферативных и инфекционных заболеваний и нарушений у хозяина, в особенности, обеспечить эффективные антагонисты A2A или двойные антагонисты A2A/2B для лечения и предотвращения таких заболеваний.
Краткое описание изобретения
Неожиданно было обнаружено, что бензимидазольные производные в соответствии с изобретением являются высокоэффективными ингибиторами аденозинового рецептора A2A или аденозиновых рецепторов A2A и A2B, и в то же время имеют высокую селективность по сравнению с аденозиновыми рецепторами A1 и А3, и, таким образом, соединения настоящего изобретения могут применяться для лечения гиперпролиферативных заболеваний и нарушений, таких как злокачественное новообразование и инфекционные заболевания и нарушения.
В частности, в отличие от известного антагониста аденозинового рецептора A2A тозаденанта и подобных бензотиазольных производных, соединения настоящего изобретения неожиданно проявляют двойную активность A2A/A2B, которая является предпочтительной для лечения и/или предотвращения гиперпролиферативных и инфекционных заболеваний и нарушений, как раскрыто выше, или соединения настоящего изобретения демонстрируют, по меньшей мере, высокую ингибирующую активность на A2A вместе с другими раскрытыми в данном документе неожиданными преимуществами, которые приводят к высокой эффективности в лечении и/или предотвращении гиперпролиферативных и инфекционных заболеваний и нарушений.
Кроме того, по сравнению с известным антагонистом аденозинового рецептора A2A тозаденантом и подобными бензотиазольными производными, соединения настоящего изобретения неожиданно демонстрируют лучшие фармакокинетические свойства у мыши как у животной модели, актуальной для злокачественного новообразования, что является предпочтительным для лечения и/или предотвращения гиперпролиферативных и инфекционных заболеваний и нарушений, как раскрыто выше.
Кроме того, как отмечалось выше, аденозин в микроокружении опухоли может ингибировать активность Т-клеток путем передачи сигналов через рецепторы A2A и подавлять секрецию цитокинов Т-клетками. A2A-специфические агонисты, такие как CGS-21680, как и аденозин, ингибируют секрецию цитокинов Т-клетками in vitro и in vivo. В отличие от этого, потенциальные антагонисты A2A или двойные антагонисты A2A/A2B могут спасти Т-клетки от этого ингибирования. В отличие от известного антагониста аденозинового рецептора A2A тозаденанта, соединения настоящего изобретения демонстрируют, что они способны спасти Т-клетки от ингибирования и способны предотвратить подавление секреции цитокинов, индуцированной аденозином или A2A-специфическими агонистами, такими как CGS-2168, что является предпочтительным для лечения и/или предотвращения гиперпролиферативных и инфекционных заболеваний и нарушений, как раскрыто выше. Таким образом, соединения настоящего изобретения неожиданно способны предотвращать иммуносупрессию и, следовательно, способны поддерживать индуцированное противораковыми Т-клетками ингибирование роста опухоли, уменьшение или разрушение метастазов и предотвращение неоваскуляризации.
Изобретение относится к бензимидазольным производным общей формулы I
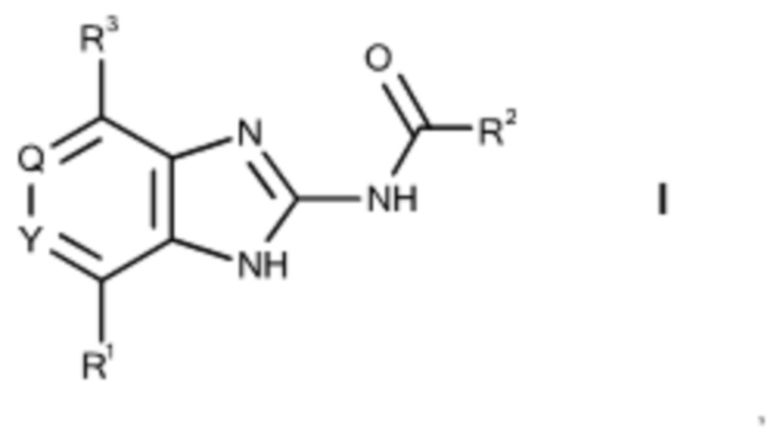
где
Q, Y независимо друг от друга означают CH или N,
R1 означает Hal или линейный или разветвленный алкил, содержащий 1-10 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством R4 и в котором 1-4 атома углерода могут быть заменены, независимо друг от друга, на группы О, S, SO, SO2, NH, NCH3, -ОСО-, -NHCONH-, -NHCO-, -NR5SO2R6-, -СОО-, -CONH-, -NCH3CO-, -CONCH3-, -С≡С- и/или группы -СН=СН-, и/или, кроме того, 1-10 атомов водорода могут быть заменены на F и/или Cl, или моно- или бициклический алкил, содержащий 3-7 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством R4 и в котором 1-4 атома углерода могут быть заменены, независимо друг от друга, на группы О, S, SO, SO2, NH, NCH3, -ОСО-, -NHCONH-, -NHCO-, -NR5SO2R6-, -СОО-, -CONH-, -NCH3CO-, -CONCH3-, -С≡С- и/или группы -СН=СН-, и/или, кроме того, 1-10 атомов водорода могут быть заменены на F и/или Cl, или моно- или бициклический гетероарил, гетероциклил, арил или циклический алкиларил, содержащий 3-14 атомов углерода и 0-4 гетероатома, независимо выбранных из N, О и S, который не замещен или моно-, ди- или тризамещен посредством R4,
R2 означает линейный или разветвленный алкил, содержащий 1-10 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством R4 и в котором 1-4 атома углерода могут быть заменены, независимо друг от друга, на группы О, S, SO, SO2, NH, NCH3, -OCO-, -NHCONH-, -NHCO-, -NR5SO2R6-, -COO-, -CONH-, -NCH3CO-, -CONCH3-, -C≡C- и/или группы -CH=CH- и/или, кроме того, 1-10 атомов водорода могут быть заменены на F и/или Cl, или циклический алкил, содержащий 3-7 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством R4 и в котором 1-4 атома углерода могут быть заменены, независимо друг от друга, на группы О, S, SO, SO2, NH, NCH3, -ОСО-, -NHCONH-, -NHCO-, -NR5SO2R6-, -СОО-, -CONH-, -NCH3CO-, -CONCH3-, -С≡С- и/или группы -СН=СН- и/или, кроме того, 1-11 атомов водорода могут быть заменены на F и/или Cl, или моно- или бициклический гетероарил, гетероциклил, арил или циклический алкиларил, содержащий 3-14 атомов углерода и 0-4 гетероатома, независимо выбранных из N, О и S, который не замещен или моно-, ди- или тризамещен посредством R4,
R3 означает линейный или разветвленный алкил или О-алкил, содержащий 1-6 атомов углерода, или циклический алкил, содержащий 3-6 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством Н, =S, =NH, =O, ОН, циклического алкила, содержащего 3-6 атомов углерода, СООН, Hal, NH2, SO2CH3, SO2NH2, CN, CONH2, NHCOCH3, NHCONH2 или NO2,
R4 означает H, R5, =S, =NR5, =O, OH, COOH, Hal, NH2, SO2CH3, SO2NH2, CN, CONH2, NHCOCH3, NHCONH2, NO2, или линейный или разветвленный алкил, содержащий 1-10 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством R5 и в котором 1-4 атома углерода могут быть заменены, независимо друг от друга, на группы О, S, SO, SO2, NH, NCH3, -ОСО-, -NHCONH-, -NHCO-, -NR5SO2R6-, -СОО-, -CONH-, -NCH3CO-, -CONCH3-, -С≡С- и/или группы -СН=СН-, и/или, кроме того, 1-10 атомов водорода могут быть заменены на F и/или Cl, или моно- или бициклический алкил, содержащий 3-7 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством R5 и в котором 1-4 атома углерода могут быть заменены, независимо друг от друга, на группы О, S, SO, SO2, NH, NCH3, -ОСО-, -NHCONH-, -NHCO-, -NRSO2R4-, -СОО-, -CONH-, -NCH3CO-, -CONCH3-, -С≡С- и/или группы -СН=СН-, и/или, кроме того, 1-10 атомов водорода могут быть заменены на F и/или Cl, или моно- или бициклический гетероарил, гетероциклил, арил или циклический алкиларил, содержащий 3-14 атомов углерода и 0-4 гетероатома, независимо выбранных из N, О и S, который не замещен или моно-, ди- или тризамещен посредством R5,
R5, R6 независимо друг от друга выбирают из группы, состоящей из Н, =S, =NH, =O, ОН, СООН, Hal, NH2, SO2CH3, SO2NH2, CN, CONH2, NHCOCH3, NHCONH2, NO2 и линейного или разветвленного алкила, содержащего 1-10 атомов углерода, в котором 1-4 атома углерода могут быть заменены, независимо друг от друга, на группы О, S, SO, SO2, NH, NCH3, -ОСО-, -NHCONH-, -NHCO-, -COO-, -CONH-, -NCH3CO-, -CONCH3-, -С≡С- и/или группы -СН=СН-, и/или, кроме того, 1-10 атомов водорода могут быть заменены на F и/или Cl,
Hal означает F, Cl, Br или I,
и его физиологически приемлемым солям, производным, сольватам, пролекарствам и стереоизомерам, включая их смеси во всех соотношениях.
Изобретение предпочтительно относится к соединению формулы I,
где R1 означает Hal или линейный или разветвленный алкил, содержащий 1-10 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством R4 и в котором 1-4 атома углерода могут быть заменены, независимо друг от друга, на группы О, S, SO, SO2, NH, NCH3, -OCO-, -NHCONH-, -NHCO-, -NR5SO2R6-, -COO-, -CONH-, -NCH3CO-, -CONCH3-, -C≡C- и/или группы -CH=CH-, и/или, кроме того, 1-10 атомов водорода могут быть заменены на F и/или Cl, или одну из следующих структур:
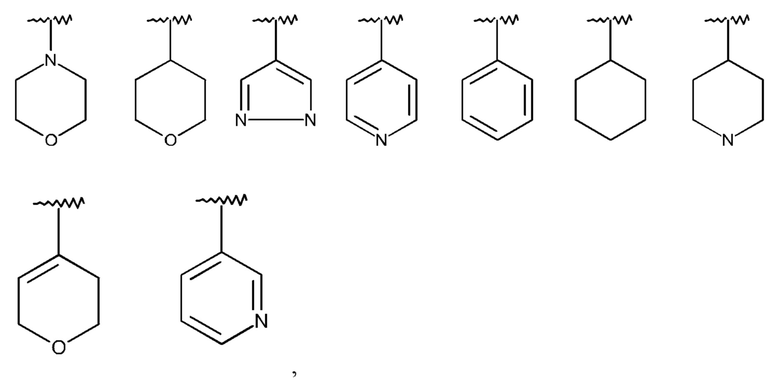
которая не замещена или моно-, ди- или тризамещена посредством R4,
и где Q, Y, R2, R3, R4, R5 и R6 имеют значения, как раскрыто выше.
Изобретение особенно предпочтительно относится к соединению формулы I, где
R1 означает Br или одну из следующих структур:
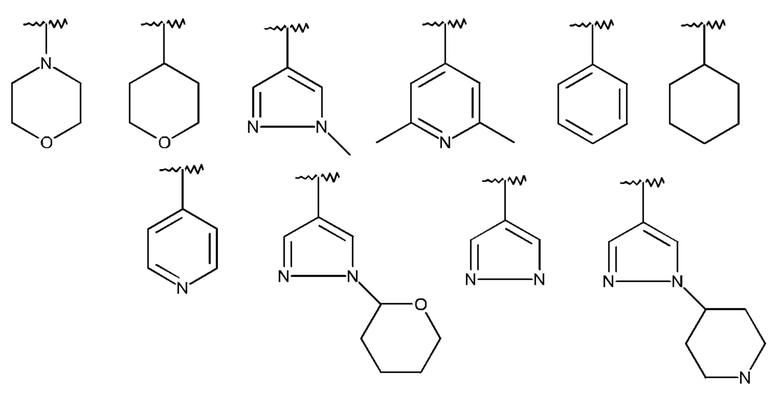
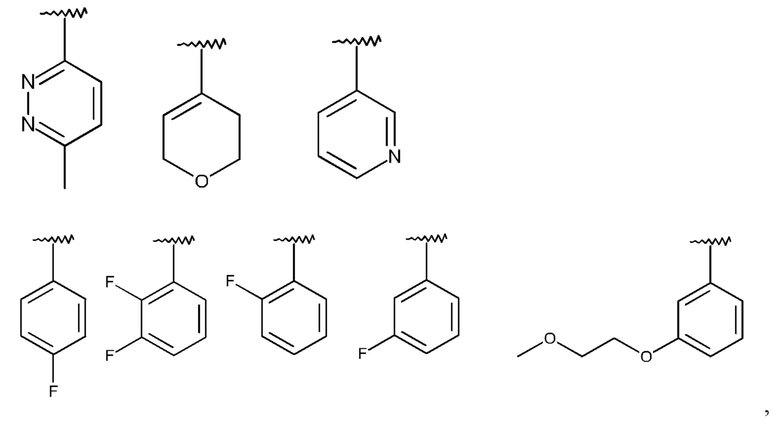
которая не замещена или моно-, ди- или тризамещена посредством R5,
и где Q, Y, R2, R3, R4, R5 и R6 имеют значения, как раскрыто выше.
Изобретение особенно предпочтительно относится к соединению формулы I, где R2 означает одну из следующих структур:
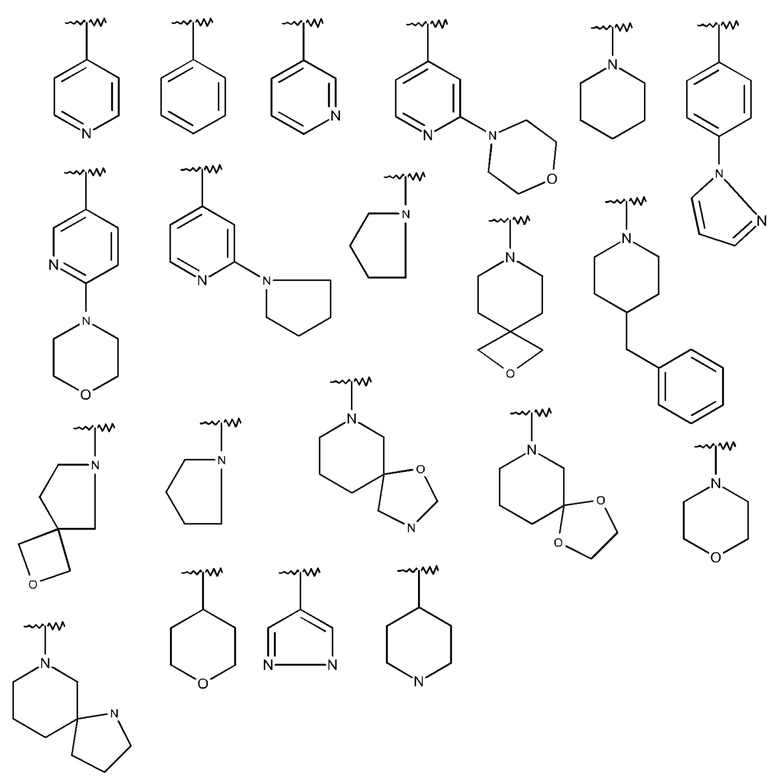
которая не замещена или моно-, ди- или тризамещена посредством R5,
и где Q, Y, R1, R3, R4, R5 и R6 имеют значения, как раскрыто выше.
Изобретение предпочтительно относится к соединению формулы I, где
R3 означает одну из следующих структур:
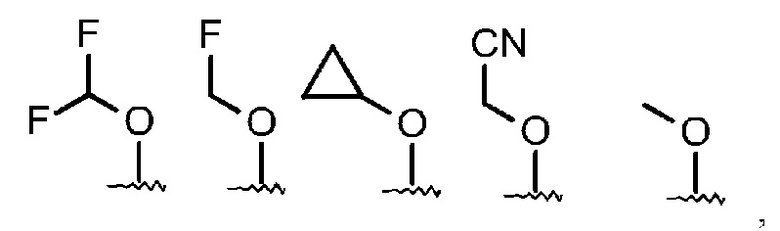
и Q, Y, R1, R2, R4, R5 и R6 имеют значения, как раскрыто выше.
Изобретение предпочтительно относится к соединению формулы I, где
R3 означает О-алкил, содержащий 1-6 атомов углерода, который не замещен или моно-, ди- или тризамещен посредством F,
и Q, Y, R1, R2, R4, R5 и R6 имеют значения, как раскрыто выше.
Изобретение предпочтительно относится к соединению формулы I, где
R3 означает ОМе,
и Q, Y, R1, R2, R4, R5 и R6 имеют значения, как раскрыто выше.
Изобретение особенно предпочтительно относится к соединению, выбранному из группы, состоящей из:
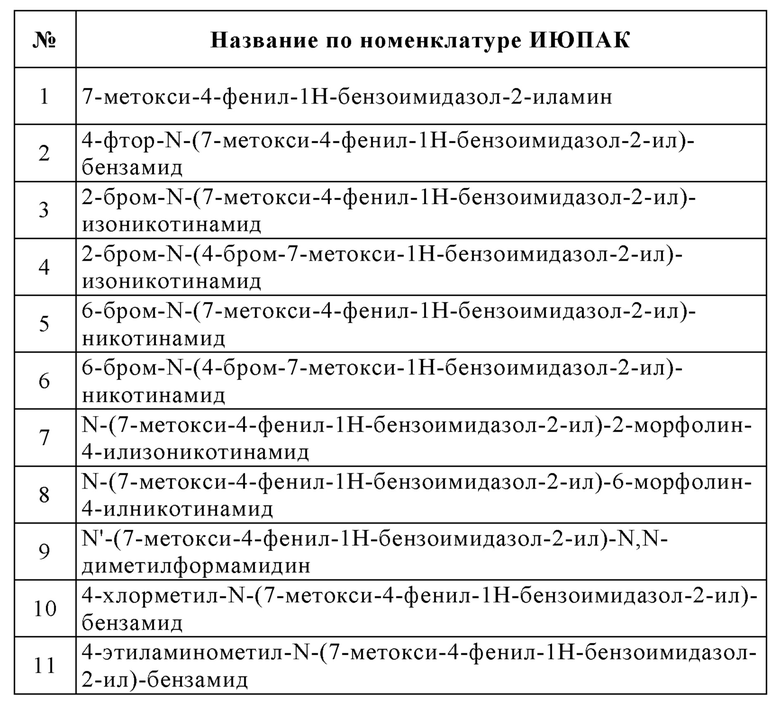

















и его физиологически приемлемым солям, производным, сольватам, пролекарствам и стереоизомерам, включая их смеси во всех соотношениях.
Все вышеупомянутые предпочтительные, особенно предпочтительные и наиболее предпочтительные значения вышеуказанных радикалов соединений формулы I следует понимать таким образом, что эти предпочтительные, особенно предпочтительные и наиболее предпочтительные значения или варианты осуществления можно комбинировать друг с другом в любой возможной комбинации с получением соединений формулы I, и что предпочтительные, особенно предпочтительные и наиболее предпочтительные соединения формулы I данного типа тем самым раскрыты явным образом.
Hal означает фтор, хлор, бром или йод, в частности, фтор, бром или хлор.
-(С=O)- или =O означает присутствие карбонильного кислорода и
обозначает 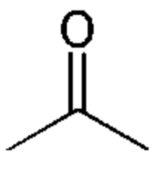 или атом кислорода, присоединенный к атому углерода двойной связью.
или атом кислорода, присоединенный к атому углерода двойной связью.
Алкил означает насыщенную неразветвленную (линейную) или разветвленную углеводородную цепь и содержит 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода. Алкил предпочтительно означает метил, кроме того этил, пропил, изопропил, бутил, изобутил, втор-бутил или трет-бутил, кроме того также пентил, 1-, 2- или 3-метилбутил, 1,1-, 1,2- или 2,2-диметилпропил, 1-этилпропил, гексил, 1-, 2-, 3- или 4-метилпентил, 1,1-, 1,2-, 1,3-, 2,2-, 2,3- или 3,3-диметилбутил, 1- или 2-этилбутил, 1-этил-1-метилпропил, 1-этил-2-метилпропил, 1,1,2- или 1,2,2-триметилпропил, линейный или разветвленный гептил, октил, нонил или децил, еще более предпочтительно, например, трифторметил.
Циклический алкил или циклоалкил означает насыщенную циклическую углеводородную цепь и содержит 3-10, предпочтительно 3-7 атомов углерода и предпочтительно означает циклопропил, циклобутил, циклопентил, циклогексил или циклогептил. Циклоалкил также означает частично ненасыщенный циклический акил, такой как, например, циклогексенил или циклогексинил.
Алкенил означает ненасыщенную неразветвленную (линейную) или разветвленную углеводородную цепь и содержит 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода.
О-алкил или OA означает линейный или разветвленный алкоксил, содержащий 1-6 атомов углерода, и предпочтительно означает метоксил, кроме того также, например, этоксил, н-пропоксил, изопропоксил, н-бутоксил, изобутоксил, втор-бутоксил или трет-бутоксил.
Алкилоксикарбонил относится к имеющим прямую или разветвленную цепь сложным эфирам карбоновых кислот - производных настоящего изобретения, т.е. метилоксикарбонилу (МеОСО-), этилоксикарбонилу или бутилоксикарбонилу.
Алкилкарбонил относится к имеющей прямую или разветвленную цепь алкильной и карбоксикислотной группе.
Арил, Ar или ароматическое кольцо означает моно- или полициклическую ароматическую или полностью ненасыщенную циклическую углеводородную цепь, например, незамещенный фенил, нафтил или бифенил, кроме того, предпочтительно фенил, нафтил или бифенил, каждый из которых является моно-, ди- или тризамещенным, например, посредством А, фтора, хлора, брома, йода, гидроксила, метокси, этокси, пропокси, бутокси, пентилокси, гексилокси, нитро, циано, формила, ацетила, пропионила, трифторметила, амино, метиламино, этиламино, диметиламино, диэтиламино, бензилокси, сульфонамидо, метилсульфонамидо, этилсульфонамидо, пропилсульфонамидо, бутилсульфонамидо, диметилсульфонамидо, фенилсульфонамидо, карбоксила, метоксикарбонила, этоксикарбонила, аминокарбонила.
Гетероцикл и гетероциклил относятся к насыщенным или ненасыщенным неароматическим кольцам или кольцевым системам, содержащим по меньшей мере один гетероатом, выбранный из О, S и N, включая, кроме того, окисленные формы серы, а именно SO и SO2. Примеры гетероциклов включают тетрагидрофуран (ТГФ), дигидрофуран, 1,4-диоксан, морфолин, 1,4-дитиан, пиперазин, пиперидин, 1,3-диоксолан, имидазолидин, имидазолин, пирролин, пирролидин, тетрагидропиран, дигидропиран, оксатиолан, дитиолан, 1,3-диоксан, 1,3-дитиан, оксатиан, тиоморфолин и т.п.
Гетероарил означает ароматический или частично ароматический гетероцикл, который содержит по меньшей мере один кольцевой гетероатом, выбранный из О, S и N. Гетероарилы таким образом включают гетероарилы, конденсированные с другими типами колец, такими как арилы, циклоалкилы и гетероциклы, которые не являются ароматическими. Примерами гетероарильных групп включают: пирролил, изоксазолил, изотиазолил, пиразолил, пиридил, оксазолил, оксадиазолил, тиадиазолил, тиазолил, имидазолил, триазолил, тетразолил, фуранил, триазинил, тиенил, пиримидил, бензизоксазолил, бензоксазолил, бензотиазолил, бензотиадиазолил, дигидробензофуранил, индолинил, пиридазинил, индазолил, изоксазолил, изоиндолил, дигидробензотиенил, индолизинил, циннолинил, фталазинил, хиназолинил, нафтиридинил, карбазолил, бензодиоксинил, бензодиоксолил, хиноксалинил, пуринил, фуразанил, тиофенил, изобензилфуранил, бензимидазолил, бензофуранил, бензотиенил, хинолил, индолил, изохинолил, дибензофуранил и т.п. В случае гетероциклильных и гетероарильных групп, включены кольца и кольцевые системы, содержащие от 3 до 15 атомов, которые образуют 1-3 кольца.
Моно- или бициклический насыщенный, ненасыщенный или ароматический гетероцикл предпочтительно означает незамещенный или моно-, ди- или тризамещенный 2- или 3-фурил, 2- или 3-тиенил, 1-, 2- или 3-пирролил, 1-, 2-, 4-или 5-имидазолил, 1-, 3-, 4- или 5-пиразолил, 2-, 4- или 5-оксазолил, 3-, 4- или 5-изоксазолил, 2-, 4- или 5-тиазолил, 3-, 4- или 5-изотиазолил, 2-, 3- или 4-пиридил, 2-, 4-, 5- или 6-пиримидинил, кроме того, предпочтительно 1,2,3-триазол-1-, -4- или -5-ил, 1,2,4-триазол-1-, -3- или 5-ил, 1- или 5-тетразолил, 1,2,3-оксадиазол-4- или -5-ил, 1,2,4-оксадиазол-3- или -5-ил, 1,3,4-тиадиазол-2-или -5-ил, 1,2,4-тиадиазол-3- или -5-ил, 1,2,3-тиадиазол-4- или -5-ил, 3- или 4-пиридазинил, пиразинил, 1-, 2-, 3-, 4-, 5-, 6- или 7-индолил, 4- или 5-изоиндолил, 1-, 2-, 4- или 5-бензимидазолил, 1-, 3-, 4-, 5-, 6- или 7-бензопиразолил, 2-, 4-, 5-, 6- или 7-бензоксазолил, 3-, 4-, 5-, 6- или 7- бензизоксазолил, 2-, 4-, 5-, 6- или 7-бензотиазолил, 2-, 4-, 5-, 6- или 7-бензизотиазолил, 4-, 5-, 6- или 7-бенз-2,1,3-оксадиазолил, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолил, 3-, 4-, 5-, 6-, 7- или 8-циннолинил, 2-, 4-, 5-, 6-, 7- или 8-хиназолинил, 5- или 6-хиноксалинил, 2-, 3-, 5-, 6-, 7- или 8-2Н-бензо-1,4-оксазинил, еще более предпочтительно 1,3-бензодиоксол-5-ил, 1,4-бензодиоксан-6-ил, 2,1,3-бензотиадиазол-4- или -5-ил или 2,1,3-бензоксадиазол-5-ил.
Гетероциклические радикалы также могут быть частично или полностью гидрированными и означать, например, 2,3-дигидро-2-, -3-, -4- или -5-фурил, 2,5-дигидро-2-, -3-, -4- или 5-фурил, тетрагидро-2- или -3-фурил, 1,3-диоксолан-4-ил, тетрагидро-2- или -3-тиенил, 2,3-дигидро-1-, -2-, -3-, -4- или -5-пирролил, 2,5-дигидро-1-, -2-, -3-, -4- или -5-пирролил, 1-, 2- или 3-пирролидинил, тетрагидро-1-, -2- или -4-имидазолил, 2,3-дигидро-1-, -2-, -3-, -4- или -5-пиразолил, тетрагидро-1-, -3- или -4-пиразолил, 1,4-дигидро-1-, -2-, -3- или -4-пиридил, 1,2,3,4-тетрагидро-1-, -2-, -3-, -4-, -5- или -6-пиридил, 1-, 2-, 3- или 4-пиперидинил, 2-, 3- или 4-морфолинил, тетрагидро-2-, -3- или -4-пиранил, 1,4-диоксанил, 1,3-диоксан-2-, -4- или -5-ил, гексагидро-1-, -3- или -4-пиридазинил, гексагидро-1-, -2-, -4- или -5-пиримидинил, 1-, 2- или 3-пиперазинил, 1,2,3,4-тетрагидро-1-, -2-, -3-, -4-, -5-, -6-, -7- или -8-хинолил, 1,2,3,4-тетрагидро-1-, -2-, -3-, -4-, -5-, -6-, -7- или -8-изохинолил, 2-, 3-, 5-, 6-, 7- или 8- 3,4-дигидро-2Н-бензо-1,4-оксазинил, еще более предпочтительно 2,3-метилендиоксифенил, 3,4-метилендиоксифенил, 2,3-этилендиоксифенил, 3,4-этилендиоксифенил, 3,4-(дифторметилендиокси)фенил, 2,3-дигидробензофуран-5- или 6-ил, 2,3-(2-оксометилендиокси)фенил или также 3,4-дигидро-2Н-1,5-бензодиоксепин-6- или -7-ил, кроме того, предпочтительно 2,3-дигидробензофуранил или 2,3-дигидро-2-оксофуранил.
Гетероцикл, кроме того, означает, например, 2-оксопиперидин-1-ил, 2-оксопирролидин-1-ил, 2-оксо-1H-пиридин-1-ил, 3-оксоморфолин-4-ил, 4-оксо-1H-пиридин-1-ил, 2,6-диоксопиперидин1-ил, 2-оксопиперазин-1-ил, 2,6-диоксопиперазин-1-ил, 2,5-диоксопирролидин-1-ил, 2-оксо-1,3-оксазолидин-3-ил, 3-оксо-2H-пиридазин-2-ил, 2-капролактам-1-ил (=2-оксоазепан-1-ил), 2-гидрокси-6-оксопиперазин-1-ил, 2-метокси-6-оксопиперазин-1-ил или 2-азабицикло[2.2.2]октан-3-он-2-ил.
Гетероциклоалкил в данном случае означает полностью гидрированный или насыщенный гетероцикл, гетероциклоалкенил (одна или несколько двойных связей) или гетероциклоалкинил (одна или несколько тройных связей) означает частично или не полностью гидрированный или ненасыщенный гетероцикл, гетероарил означает ароматический или полностью ненасыщенный гетероцикл.
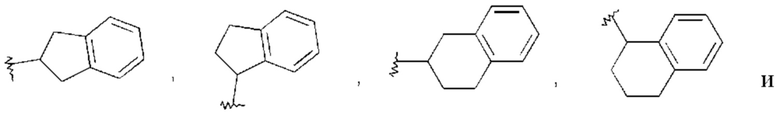
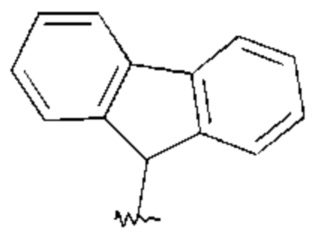
Циклическая алкиларильная группа в связи с настоящим изобретением означает, что одно или два ароматических кольца Ar конденсированы на незамещенном или моно- или дизамещенном циклическом алкиле, в котором одна или две группы СНг и/или, кроме того, 1-11 атомов водорода могут быть заменены, как, например, имеет место в радикалах, изображенных ниже:
Кроме того, нижеприведенные сокращения имеют следующие значения:
Boc трет-бутоксикарбонил
CBZ бензилоксикарбонил
DNP 2,4-динитрофенил
FMOC 9-флуоренилметоксикарбонил
ими-DNP 2,4-динитрофенил в 1-м положении имидазольного кольцо
ОМе сложный метиловый эфир
РОА феноксиацетил
DCCI дициклогексилкарбодиимид
HOBt 1-гидроксибензотриазол
Таким образом, изобретение относится к фармацевтическому препарату, содержащему соединение в соответствии с настоящим изобретением и/или одну из его физиологически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях.
Изобретение также относится к фармацевтическому препарату в соответствии с изобретением данного типа, дополнительно содержащему наполнители и/или вспомогательные вещества.
Кроме того, изобретение относится к вышеупомянутому фармацевтическому препарату в соответствии с изобретением, содержащему по меньшей мере одно дополнительное активное соединение лекарственного средства.
Под фармацевтически или физиологически приемлемыми производными подразумевают, например, соли соединения настоящего изобретения, а также так называемые пролекарственные соединения. Под пролекарственными соединениями подразумевают производные соединения настоящего изобретения, которые были модифицированы с помощью, например, алкильной или ацильной групп (см. также амино- и гидроксилзащитные ниже), Сахаров или олигопептидов и которые быстро расщепляются или высвобождаются в организме с образованием эффективных молекул. Они также включают биоразлагаемые полимерные производные соединения настоящего изобретения, как описано, например, в Int. J. Pharm. 115 (1995), 61-67.
Соединение настоящего изобретения можно применять в его заключительной несолевой форме. С другой стороны, настоящее изобретение также охватывает применение пепстатина в форме его фармацевтически приемлемых солей, которые можно получить из различных органических и неорганических оснований с помощью методик, хорошо известных в данной области техники. Фармацевтически приемлемые солевые формы пепстатина получают, главным образом, путем использования традиционных методов. В случае, если соединение настоящего изобретения содержит карбоксильную группу, одна из его пригодных солей может быть образована по реакции соединения настоящего изобретения с пригодным основанием с получением соответствующей соли присоединения основания. Такими основаниями являются, например, гидроксиды щелочных металлов, включая гидроксид калия, гидроксид натрия и гидроксид лития; гидроксиды щелочноземельных металлов, такие как гидроксид бария и гидроксид кальция; алкоголяты щелочных металлов, например, этилат калия и пропилат натрия; и различные органические основания, такие как пиперидин, диэтаноламин и N-метилглутамин. Также включены соли алюминия с пепстатином.
Кроме того, основные соли соединения настоящего изобретения включают, но не ограничиваясь только ими, соли алюминия, аммония, кальция, меди, железа(III), железа(II), лития, магния, марганца(III), марганца(II), калия, натрия и цинка.
Из вышеупомянутых солей, предпочтение отдают аммониевым солям; солям щелочных металлов - натрия и калия, и солям щелочноземельных металлов - кальция и магния. Соли соединений настоящего изобретения, которые имеют происхождение из фармацевтически приемлемых органических нетоксичных оснований, включают, но не ограничиваясь только ими, соли первичных, вторичных и третичных аминов, замещенных аминов, также включая природные замещенные амины, циклические амины, и основные ионообменные смолы, например, аргинин, бетаин, кофеин, хлорпрокаин, холин, N,N'-дибензилэтилендиамин (бензатин), дициклогексиламин, диэтаноламин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лидокаин, лизин, меглумин, N-метил-D-глюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтаноламин, триэтиламин, триметиламин, трипропиламин и трис(гидроксиметил)метиламин (трометамин).
Как уже упоминалось, фармацевтически приемлемые соли присоединения основания пепстатина образуют с металлами или аминами, такими как щелочные металлы и щелочноземельные металлы или органические амины. Предпочтительными металлами являются натрий, калий, магний и кальций. Предпочтительными органическими аминами являются N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, N-метил-D-глюкамин и прокаин.
Соли присоединения основания к соединениям настоящего изобретения получают путем приведения в контакт формы свободной кислоты с достаточным количеством желаемого основания, вызывая образование соли обычным образом. Свободную кислоту можно регенерировать путем приведения в контакт солевой формы с кислотой и выделения свободной кислоты обычным образом. Формы свободных кислот в некоторой степени отличаются от своих соответствующих солевых форм в части определенных физических свойств, таких как растворимость в полярных растворителях; для целей настоящего изобретения, однако, в остальном соли соответствуют их соответствующим формам свободных кислот.
В свете описанного выше можно увидеть, что под выражением "фармацевтически приемлемая соль" в контексте данной заявки подразумевают активное соединение, которое представляет собой соединение настоящего изобретения в форме одной из его солей, в частности, если такая солевая форма придает данному активному соединению улучшенные фармакокинетические свойства по сравнению со свободной формой данного активного соединения или любой другой солевой формой данного активного соединения, применяемой ранее. Фармацевтически приемлемая солевая форма активного соединения также может впервые обеспечить данному активному соединению желаемое фармакокинетическое свойство, которым он ранее не обладал, и может даже положительно влиять на фармакодинамику данного активного соединения в отношении его терапевтической эффективности в организме.
Сольваты соединения настоящего изобретения подразумевают аддукции молекул инертного растворителя и пепстатина, которые образуются благодаря их силе взаимного притяжения. Сольваты представляют собой, например, гидраты, такие как моногидраты или дигидраты, или алкоголяты, т.е. продукты присоединения спиртов, таких как, например, метанол или этанол, к соединениям.
Все физиологически приемлемые соли, производные, сольваты и стереоизомеры данных соединений, включая их смеси во всех соотношениях, также находятся в соответствии с настоящим изобретением.
Соединения общей формулы I могут содержать один или несколько центров хиральности, вследствие чего все стереоизомеры, энантиомеры, диастереомеры, и т.д., соединений общей формулы I также охватываются настоящим изобретением.
Изобретение также относится к оптически активным формам (стереоизомерам), энантиомерам, рацематам, диастереомерам и гидратам и сольватам этих соединений.
Соединения формулы I в соответствии с изобретением могут быть хиральными вследствие их молекулярного строения и, соответственно, могут встречаться в различных энантиомерных формах. Таким образом, они могут находиться в рацемической или оптически активной форме. Поскольку фармацевтическая эффективность рацематов или стереоизомеров соединений в соответствии с изобретением может отличаться, может оказаться желательным применение энантиомеров. В этих случаях, как конечный продукт, так даже и промежуточные соединения, могут быть разделены на энантиомерные соединения с помощью химических или физических методов, известных специалисту в данной области, или уже использоваться как таковые в синтезе.
Фармацевтически или физиологически приемлемые производные подразумевают, например, соли соединений в соответствии с изобретением, а также так называемые пролекарственные соединения. Пролекарственные соединения подразумевают соединения формулы I, которые были модифицированы, например, алкильными или ацильными группами (см. также амино- и гидроксилзащитные группы ниже), сахарами или олигопептидами и которые быстро расщепляются или высвобождаются в организме с образованием эффективных соединений в соответствии с изобретением. Таковые также включают биоразлагаемые полимерные производные соединений в соответствии с изобретением, как описано, например, в Int. J. Pharm. 115 (1995), 61-67.
Пригодными солями присоединения кислоты являются неорганические и органические соли всех физиологически или фармакологически приемлемых кислот, например, галогениды, в частности, гидрохлориды или гидробромиды, лактаты, сульфаты, цитраты, тартраты, малеаты, фумараты, оксалаты, ацетаты, фосфаты, метилсульфонаты или n-толуолсульфонаты.
Наибольшее предпочтение отдают гидрохлоридам, трифторацетатам или бистрифторацетатам соединений в соответствии с изобретением.
Сольваты соединений формулы I подразумевают аддукции молекул инертного растворителя на соединениях формулы I, которые образуются благодаря их силе взаимного притяжения. Сольваты представляют собой, например, гидраты, такие как моногидраты или дигидраты, или алкоголяты, т.е. продукты присоединения спиртов, таких как, например, метанол или этанол, к соединениям.
Кроме того, предполагается, что соединение формулы I включает его изотопно-меченные формы. Изотопно-меченная форма соединения формулы I является идентичной указанному соединению, за исключением того факта, что один или несколько атомов указанного соединения были заменены на атом или атомы, которые имеют атомную массу или массовое число, отличное от атомной массы или массового числа упомянутого атома, который обычно встречается в природе. Примеры изотопов, которые являются легко доступными коммерчески и которые могут быть введены в соединение формулы I с помощью хорошо известных методов, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, например, 2Н, 3Н, 13С, 14С, 15N, 18O, 17O, 31Р, 32Р, 35S, 18F и 36CI, соответственно. Соединение формулы I, его пролекарство или фармацевтически приемлемая соль, которые содержат один или несколько указанных выше изотопов и/или других изотопов других атомов, также составляют объем настоящего изобретения. Изотопно-меченное соединение формулы I может использоваться в ряде полезных способов. Например, меченное изотопами соединение формулы I, например, в которое введен радиоактивный изотоп, такой, как 3Н или 14С, будет полезным в анализах исследования распределения лекарственного средства и/или субстрата в ткани. Такие радиоактивные изотопы, т.е. тритий (3Н) и углерод-14 (14С), являются особенно предпочтительными вследствие простоты получения и высокой способности к выявлению. Введение более тяжелых изотопов, например, дейтерия (2Н), в соединение формулы I, будет обеспечивать терапевтические преимущества, основывающиеся на большей метаболической стабильности указанного соединения, меченного изотопами. Большая метаболическая стабильность проявляется непосредственно в увеличении времени полураспада in vivo или снижении требуемой дозы, что при большинстве условий будет представлять предпочтительный вариант осуществления указанного изобретения. Меченное изотопом соединение формулы I обычно можно получить путем осуществления методик, раскрытых в схемах синтеза и в описании, относящемся к ним, в разделах, касающихся примеров и способов получения, описанных в данной заявке, путем замены немеченого изотопами реагента его соответствующим легко доступным реагентом, меченным изотопом.
С целью изменения окислительного метаболизма соединения посредством первичного кинетического изотопного эффекта, в соединение формулы I также может быть введен дейтерий (2Н). Первичный кинетический изотопный эффект представляет собой изменение скорости химической реакции, которое происходит по причине обмена изотопных ядер, что, в свою очередь, вызывается изменением энергий основного состояния, что необходимо для образования ковалентной связи после указанного изотопного обмена. Обмен на более тяжелый изотоп обычно будет приводить к снижению энергии основного состояния для химической связи, вызывая, таким образом, уменьшение скорости скорость-лимитирующего этапа разрушения связи. Когда происходит разрушение связи в или поблизости участка седлообразной конфигурации вдоль координаты реакции образования нескольких продуктов, коэффициент распределения продуктов может существенно изменяться. Например, в случае если дейтерий связывается с атомом углерода в положении, в котором обмен не происходит, различия скорости kM/kD=2-7 являются типичными. Если такая разница в скорости успешно применяется к соединению формулы I, которое чувствительно к окислению, профиль данного соединения in vivo может таким образом в значительной степени изменяться, что и приводит к улучшению фармакокинетических свойств.
В процессе обнаружения и совершенствования терапевтических агентов средний специалист в данной области ищет пути оптимизации фармакокинетических параметров до тех пор, пока не получит желательные in vitro свойства. Рационально предположить, что многие соединения со слабыми фармакокинетическими профилями восприимчивы к окислительному метаболизму. Анализы in vitro с микросомами печени, которые сейчас являются доступными, обеспечивают ценную информацию о процессе окислительного метаболизма этого типа, что, в свою очередь, позволяет получить рациональную модель меченных дейтерием соединений формулы I с улучшенной стабильностью вплоть до резистентности к такому окислительному метаболизму. Таким образом, получают значительное улучшение фармакокинетических профилей соединений формулы I, что может быть количественно выражено в величинах увеличения времени полураспада in vivo (Т/2), в концентрации при максимальном терапевтическом эффекте (Cmax), площадью под кривой ответа на определенную дозу (AUC) и F; в величинах уменьшения клиренса, дозы и материальных затрат.
Приведенное далее предназначено для иллюстрации сказанного выше: соединение формулы I, которое имеет многочисленные потенциальные сайты для окислительного метаболизма, например, атомы водорода бензила и атомы водорода, соединенные с атомом азота, получают в виде серии аналогов, в которых различные комбинации атомов водорода заменяют на атомы дейтерия таким образом, что в результате некоторые, большинство или все эти атомы водорода заменены на атомы дейтерия. Определение времени полураспада обеспечивает подходящее и точное определение степени улучшения резистентности к окислительному метаболизму. Таким образом определяют, что время полураспада исходного соединения может быть продлено вплоть до 100% как результат такого замещения водорода дейтерием.
Замещение водорода дейтерием в соединении формулы I также можно использовать для достижения благоприятного изменения спектра метаболитов исходного соединения в целях уменьшения или устранения нежелательных токсичных метаболитов. Например, если токсичный метаболит возникает при окислительном расщеплении углерод-водородной связи С-Н, то можно рационально предположить, что меченый дейтерием аналог значительно уменьшит или устранит продуцирование нежелательного метаболита, даже в случае, когда отдельное окисление не является скорость-лимитирующим этапом. Дополнительная информация, относящаяся к уровню техники в отношении обмена водорода на дейтерий, приведена, например, в Hanzlik и др., J. Org. Chem. 55, 3992-3997, 1990; Reider и др., J. Org. Chem. 52, 3326-3334, 1987; Foster, Adv. Drug Res. 14, 1-40, 1985; Gillette и др., Biochemistry 33 (10) 2927-2937, 1994; и Jarman и др. Carcinogenesis 16 (4), 683-688, 1993.
Изобретение также относится к смесям соединений формулы I в соответствии с изобретением, например, смесям двух диастереомеров, например, в соотношении 1:1, 1:2, 1:3, 1:4, 1:5, 1:10, 1:100 или 1:1000. Эти смеси особенно предпочтительно представляют собой смеси двух стереоизомерных соединений. Однако предпочтение также отдают смесям двух или более соединений формулы I.
Кроме того, изобретение относится к способу получения соединений формулы I, отличающемуся тем, что
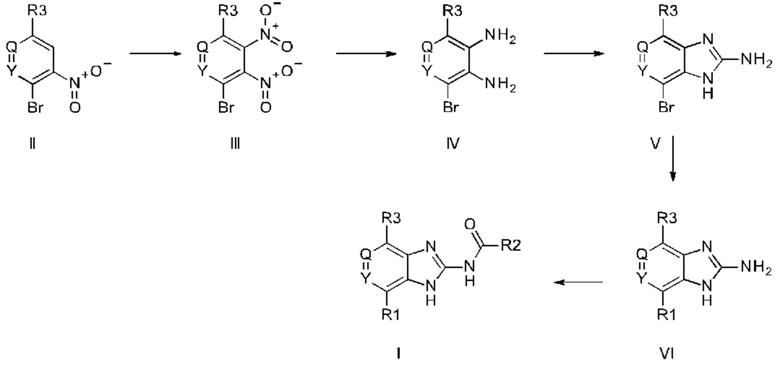
a) соединение формулы II подвергают реакции нитрования с последующим восстановлением с получением соединения формулы IV, соединение формулы IV циклизуют с получением соединения формулы V, соединение формулы V подвергают превращению в реакции типа Сузуки в соединение формулы VI с применением катализатора и основания, соединение формулы VI подвергают превращению в стандартных условиях амидирования или образования карбамида с получением соединения формулы I, где Q, Y, R1, R2, и R3 имеют значения, как раскрыто выше
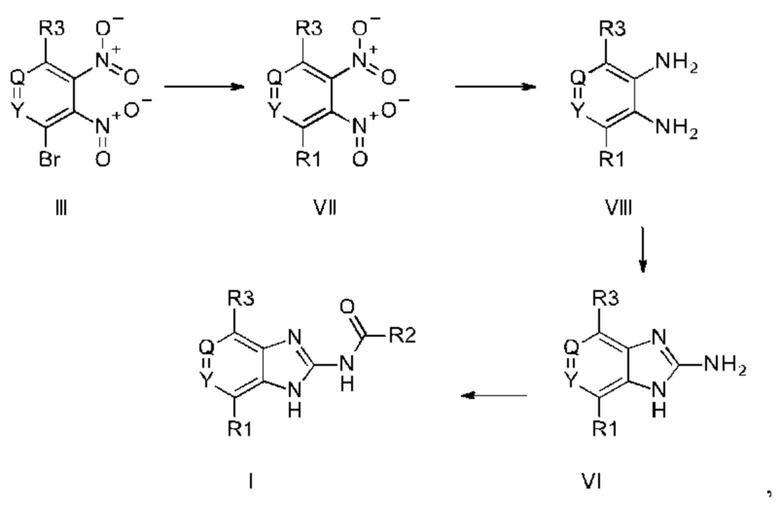
b) соединение формулы III подвергают реакции со сложным эфиром бороновой кислоты или бороновой кислотой в условиях реакции типа Сузуки с получением соединения формулы VII или подвергают реакции с амином в реакции нуклеофильного замещения при повышенной температуре с получением соединения формулы VII, соединение формулы VII восстанавливают до соединения формулы VIII и циклизуют в соединение формулы VI и в заключение подвергают превращению с получением соединения формулы I в стандартных условиях амидирования или образования карбамида, где Q, Y, R1, R2 и R3 имеют значения, как раскрыто выше,
c) основание соединения формулы I превращают в одну из его солей путем обработки кислотой, или
d) кислоту соединения формулы I превращают в одну из его солей путем обработки основанием.
Кроме того, в каждом случае реакции можно проводить постадийно, а также можно модифицировать последовательность реакций сочетания структурных элементов с адаптацией концепции использования защитных групп.
Исходные вещества или исходные соединения являются общеизвестными. Если они являются новыми, их можно получить с помощью хорошо известных способов.
При необходимости, исходные вещества также можно образовать in situ, не выделяя их из реакционной смеси, а вместо этого немедленно превращая их далее в соединения формулы I.
Соединения формулы I предпочтительно получают путем их высвобождения из функциональных производных с помощью сольволиза, в частности, гидролиза, или с помощью гидрогенолиза. Предпочтительные исходные вещества для сольволиза или гидрогенолиза представляют собой вещества, которые содержат соответствующие защищенные амино, карбоксильные и/или гидроксильные группы взамен одной или нескольких свободных амино, карбоксильных и/или гидроксильных групп, предпочтительно вещества, которые несут аминозащитную группу взамен атома Н, который присоединен к атому N. Кроме того, предпочтение отдают исходным веществам, которые несут гидроксилзащитную группу взамен атома Н гидроксильной группы. Предпочтение также отдают исходным веществам, которые несут защищенную карбоксильную группу взамен свободной карбоксильной группы.
Также в молекуле исходного вещества может присутствовать множество одинаковых или разных защищенных амино, карбоксильных и/или гидроксильных групп. Если присутствующие защитные группы отличаются друг от друга, они во многих случаях могут быть отщеплены селективно.
Термин "аминозащитная группа" является общеизвестным и относится к группам, которые пригодны для защиты (блокирования) аминогруппы от химических реакций, но которые могут быть легко удалены после проведения желаемой химической реакции в другой части молекулы. Типичными такими группами являются, в частности, незамещенные или замещенные ацильные группы, кроме того, незамещенные или замещенные арильные (например, 2,4-динитрофенил) или аралкильные группы (например, бензил, 4-нитробензил, трифенилметил). Кроме того, поскольку аминозащитные группы удаляют после желаемой реакции или последовательности реакций, их тип и размер не являются решающими, однако предпочтение отдают группам, содержащим 1-20, в частности, 1-8 атомов углерода. Термин "ацильная группа" следует понимать в самом широком смысле в связи с настоящим способом. Он охватывает ацильные группы, полученные из алифатических, аралифатических, ароматических или гетероциклических карбоновых кислот или сульфоновых кислот и, в частности, алкоксикарбонильные, арилоксикарбонильные и, в особенности, аралкоксикарбонильные группы. Примерами таких ацильных групп являются алканоил, такой как ацетил, пропионил, бутирил, аралканоил, такой как фенилацетил, ароил, такой как бензоил или толуил, арилоксиалканоил, такой как феноксиацетил, алкоксикарбонил, такой как метоксикарбонил, этоксикарбонил, 2,2,2-трихлорэтоксикарбонил, ВОС, 2-йодэтоксикарбонил, аралкоксикарбонил, такой как CBZ, 4-метоксибензилоксикарбонил или FMOC. Предпочтительными ацильными группами являются CBZ, FMOC, бензил и ацетил.
Термин "кислотозащитная группа" или "карбоксилзащитная группа" также является общеизвестным и относится к группам, которые пригодны для защиты группы -СООН от химических реакций, но которые могут быть легко удалены после проведения желаемой химической реакции в другой части молекулы. Использование сложных эфиров взамен свободных кислот, например, замещенных и незамещенных алкиловых эфиров (таких как метиловые, этиловые, трет-бутиловые и их замещенные производные), замещенных и незамещенных бензиловых эфиров или сложных силиловых эфиров, является типичным. Тип и размер кислотозащитной группы не являются решающими, однако предпочтение отдают группам, содержащим 1-20, в частности, 1-10 атомов углерода.
Термин "гидроксилзащитная группа" также является общеизвестным и относится к группам, которые пригодны для защиты гидроксильной группы от химических реакций, но которые могут быть легко удалены после проведения желаемой химической реакции в другой части молекулы. Типичными такими группами являются вышеупомянутые незамещенные или замещенные арильные, аралкильные или ацильные группы, кроме того, также алкильные группы. Тип и размер гидроксилзащитных групп не являются решающими, однако предпочтение отдают группам, содержащим 1-20, в частности, 1-10 атомов углерода. Примерами гидроксилзащитных групп являются, среди прочего, бензил, n-нитробензоил, n-толуолсульфонил и ацетил, причем бензил и ацетил являются предпочтительными.
Дополнительные типичные примеры амино-, кислото- и гидроксилзащитных групп могут быть найдены, например, в "Greene's Protective Groups in Organic Synthesis", четвертое издание, Wiley-Interscience, 2007.
Функциональные производные соединений формулы I для использования в качестве исходных веществ можно получить известными методами синтеза аминокислот и пептидов, как описано, например, в указанных стандартных работах и заявках на патенты.
Соединения формулы I высвобождают из их функциональных производных, в зависимости от используемой защитной группы, например, с помощью сильных кислот, предпочтительно, при применении трифторуксусной кислоты или перхлорной кислоты, а также при применении других сильных неорганических кислот, таких как хлористоводородная кислота или серная кислота, сильных органических кислот, таких как трихлоруксусная кислота, или сульфоновые кислоты, такие как бензоил- или n-толуолсульфоновая кислота. Присутствие дополнительного инертного растворителя и/или катализатора возможно, но не является всегда необходимым.
В зависимости от соответствующего пути синтеза, исходные вещества необязательно можно подвергать реакции в присутствии инертного растворителя.
Пригодными инертными растворителями являются, например, гептан, гексан, петролейный эфир, ДМСО, бензол, толуол, ксилол, трихлорэтилен, 1,2-дихлорэтан, тетрахлорметан, хлороформ или дихлорметан; спирты, такие как метанол, этанол, изопропанол, н-пропанол, н-бутанол или трет-бутанол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир (предпочтительно для замещения на азоте индола), тетрагидрофуран (ТГФ) или диоксан; простые эфиры гликоля, такие как монометиловый эфир или моноэтиловый эфир этиленгликоля, диметиловый эфир диэтиленгликоля (диглим); кетоны, такие как ацетон или бутанон; амиды, такие как ацетамид, диметилацетамид, N-метилпирролидон (NMP) или диметилформамид (ДМФА); нитрилы, такие как ацетонитрил; сложные эфиры, такие как этилацетат, карбоновые кислоты или ангидриды кислот, такие как, например, уксусная кислота или ангидрид уксусной кислоты, нитросоединения, такие как нитрометан или нитробензол, необязательно также смеси указанных растворителей друг с другом или смеси с водой.
Количество растворителя не является решающим; предпочтительно можно добавлять от 10 г до 500 г растворителя на г соединения формулы I, подвергаемого реакции.
Может быть предпочтительным добавление связывающего кислоту средства, например, гидроксида, карбоната или бикарбоната щелочного металла или щелочноземельного металла, или других солей щелочного или щелочноземельного металла и слабых кислот, предпочтительно солей калия, натрия или кальция, или добавление органического основания, такого как, например, триэтиламин, диметиламин, пиридин или хинолин, или избытка аминного компонента.
Полученные соединения в соответствии с изобретением можно отделить от соответствующего раствора, в котором их получили (например, с помощью центрифугирования и промывки) и можно хранить в другой композиции после отделения, или их можно оставить непосредственно в растворе, в котором их получили. Полученные соединения в соответствии с изобретением также можно внести в желаемые растворители для конкретного применения.
Продолжительность реакций зависит от выбранных условий реакций. В общем, продолжительность реакций составляет от 0.5 часа до 10 дней, предпочтительно от 1 до 24 часов. При использовании микроволновой печи, время реакций можно сократить до значений от 1 до 60 минут.
Соединения формулы I, а также исходные вещества для их получения, кроме того, получают с помощью известных методов, как описано в литературе (например, в стандартных работах, таких как Houben-Weyl, Methoden der organischen Chemie [Методы органической химии], Georg-Thieme-Verlag, Штутгарт), например, в условиях реакций, которые известны и пригодны для указанных реакций. В данном случае также можно использовать хорошо известные варианты, которые здесь не описаны более подробно.
Стадии обычной обработки, такие как, например, добавление воды к реакционной смеси и экстрагирование, позволяют получить соединения после удаления растворителя. Может оказаться предпочтительным, если для дополнительной очистки продукта затем осуществлять перегонку или кристаллизацию, или проводить хроматографическую очистку.
Кислоту формулы I можно превратить в соответствующую соль присоединения, используя основание, например, по реакции эквивалентных количеств кислоты и основания в инертном растворителе, таком как этанол, с последующим упариванием. Пригодными основаниями для этой реакции являются, в частности, те, которые дают физиологически приемлемые соли. Таким образом, кислоту формулы I можно превратить в соответствующую соль металла, в частности, соль щелочного или щелочноземельного металла, используя основание (например, гидроксид натрия, гидроксид калия, карбонат натрия или карбонат калия) или в соответствующую аммониевую соль. Органические основания, которые дают физиологически приемлемые соли, такие как, например, этаноламин, также пригодны для этой реакции.
С другой стороны, основание формулы I можно превратить в соответствующую соль присоединения кислоты, используя кислоту, например, по реакции эквивалентных количеств основания и кислоты в инертном растворителе, таком как этанол, с последующим упариванием. Пригодными кислотами для этой реакции являются, в частности, те, которые дают физиологически приемлемые соли. Таким образом, можно использовать неорганические кислоты, например, серную кислоту, азотную кислоту, галогенводородные кислоты, такие как хлористоводородная кислота или бромистоводородная кислота, фосфорную кислоту, такую как ортофосфорная кислота, сульфаминовую кислоту и, кроме того, органические кислоты, в частности, алифатические, алициклические, аралифатические, ароматические или гетероциклические, моно- или многоосновные карбоновые, сульфоновые или серные кислоты, например, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, пивалевая кислота, диэтилуксусная кислота, малоновая кислота, янтарная кислота, пимелиновая кислота, фумаровая кислота, малеиновая кислота, молочная кислота, винная кислота, яблочная кислота, лимонная кислота, глюконовая кислота, аскорбиновая кислота, никотиновая кислота, изоникотиновая кислота, метан- или этансульфоновая кислота, этандисульфоновая кислота, 2-гидроксисульфоновая кислота, бензолсульфоновая кислота, n-толуолсульфоновая кислота, нафталинмоно- и дисульфоновая кислоты или лаурилсерная кислота. Соли с физиологически неприемлемыми кислотами, например, пикраты, можно использовать для выделения и/или очистки соединений формулы I.
Было обнаружено, что соединения формулы I хорошо переносятся и обладают ценными фармакологическими свойствами.
Поскольку показано, что аденозиновые рецепторы, такие как A2A и A2B, оказывают понижающую регуляцию иммунного ответа во время воспаления и защищают ткани от иммунного повреждения, ингибирование передачи сигналов через аденозиновые рецепторы можно использовать для того, чтобы усилить и продлить иммунный ответ.
В данном документе обеспечены способы усиления иммунного ответа. В одном примере, способ увеличивает желаемое и целевое повреждение ткани, такое как повреждение опухоли, например, злокачественного новообразования. В данном документе раскрыты способы ингибирования одного или нескольких процессов, способствующих продуцированию внеклеточного аденозина и запускаемой аденозином передачи сигналов через аденозиновые рецепторы. Например, усиление иммунного ответа, местного воспаления тканей и направленного разрушения тканей достигается: путем ингибирования или снижения местной гипоксии ткани, продуцирующей аденозин; путем разложения (или превращения в неактивный) накопленного внеклеточного аденозина; путем предотвращения или снижения экспрессии аденозиновых рецепторов на иммунных клетках; и/или путем ингибирования/антагонизирования передачи сигналов аденозиновыми лигандами через аденозиновые рецепторы. Результаты, раскрытые в данном документе, демонстрируют, что in vivo введение средств, которые нарушают путь "гипоксия -> накопление аденозина -> иммуносупрессивная передачи сигналов посредством аденозиновых рецепторов иммунным клеткам" у субъектов, страдающих различными заболеваниями (например, злокачественным новообразованием и сепсисом) может in vivo привести к обеспечению лечения опухолей или улучшенной иммунизации.
В одном примере, способ включает введение одного или нескольких ингибиторов внеклеточного аденозина и/или ингибиторов аденозиновых рецепторов, таких как антагонист аденозиновых рецепторов. Для повышения эффективности вакцины, один или несколько ингибиторов аденозиновых рецепторов и/или ингибиторов внеклеточного аденозина можно вводить в сочетании с вакциной. В одном примере, один или несколько ингибиторов аденозиновых рецепторов или ингибиторов внеклеточного аденозина вводят для усиления иммунного ответа/воспаления. В другом примере, способ обеспечивает достижение целевого повреждения ткани, такого как разрушение опухоли.
Следовательно, изобретение также относится к применению соединений в соответствии с изобретением для приготовления лекарственного средства для лечения и/или профилактики заболеваний, которые вызваны, спровоцированы и/или усилены агонистами аденозиновых или других рецепторов A2A и/или A2B.
Таким образом, изобретение также относится, в частности, к лекарственному средству, содержащему по меньшей мере одно соединение в соответствии с изобретением и/или одну из его физиологически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях, для применения для лечения и/или профилактики физиологических и/или патофизиологических состояний.
Особое предпочтение отдают, в частности, физиологическим и/или патофизиологическим состояниям, которые связаны с аденозиновыми рецепторами A2A и/или A2B.
Физиологические и/или патофизиологические состояния подразумевают физиологические и/или патофизиологические состояния, которые имеют медицинское значение, такие как, например, заболевания или расстройства и медицинские нарушения, жалобы, симптомы или осложнения и т.п., в частности, заболевания.
Изобретение кроме того относится к лекарственному средству, содержащему по меньшей мере одно соединение в соответствии с изобретением и/или одну из его физиологически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях, для применения для лечения и/или профилактики физиологических и/или патофизиологических состояний, выбранных из группы, состоящей из гиперпролиферативных и инфекционных заболеваний и нарушений.
Изобретение дополнительно относится к лекарственному средству, содержащему по меньшей мере одно соединение в соответствии с изобретением и/или одну из его физиологически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях, для применения для лечения и/или профилактики физиологических и/или патофизиологических состояний, выбранных из группы, состоящей из гиперпролиферативных и инфекционных заболеваний и нарушений, где гиперпролиферативное заболевание или нарушение представляет собой злокачественное новообразование.
Таким образом, изобретение, в особенности, предпочтительно относится к лекарственному средству, содержащему по меньшей мере одно соединение в соответствии с изобретением и/или одну из его физиологически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях, как указано выше, где злокачественное новообразование выбирают из группы, состоящей из острой и хронической лимфоцитарной лейкемии, острой гранулоцитарной лейкемии, рака коры надпочечников, рака мочевого пузыря, рака головного мозга, рака молочной железы, рака шейки матки, гиперплазии шейки матки, рака шейки матки, хориокарциномы, хронической гранулоцитарной лейкемии, хронической лимфоцитарной лейкемии, рака ободочной кишки, рака эндометрия, рака пищевода, эссенциального тромбоцитоза, урогенитальной карциномы, глиомы, глиобластомы, волосатоклеточной лейкемии, карциномы головы и шеи, болезни Ходжкина, саркомы Капоши, карциномы легких, лимфомы, злокачественной карциноидной опухоли, злокачественной гиперкальциемии, злокачественной меланомы, злокачественной инсулиномы поджелудочной железы, медуллярной карциномы щитовидной железы, меланомы, множественной миеломы, фунгоидного микоза, миелоидной и лимфоцитарной лейкемии, нейробластомы, неходжкинской лимфомы, немелкоклеточного рака легкого, остеогенной саркомы, карциномы яичника, карциномы поджелудочной железы, истинной полицитемии, первичной карциномы головного мозга, первичной макроглобулинемии, рака предстательной железы, почечно-клеточного рака, рабдомиосаркомы, рака кожи, мелкоклеточного рака легкого, саркомы мягких тканей, плоскоклеточного рака, рака желудка, рака яичка, рака щитовидной железы и опухоли Вильмса.
Изобретение еще более предпочтительно относится к лекарственному средству, содержащему по меньшей мере одно соединение в соответствии с изобретением и/или одну из его физиологически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях, для применения для лечения и/или профилактики физиологических и/или патофизиологических состояний, выбранных из группы, состоящей из гиперпролиферативных и инфекционных заболеваний и нарушений, где гиперпролиферативное заболевание или нарушение выбирают из группы, состоящей из возрастной макулярной дегенерации, болезни Крона, цирроза, хронических нарушений, связанных с воспалением, пролиферативной диабетической ретинопатии, пролиферативной витреоретинопатии, ретролентальной фиброплазии, гранулематоза, иммунной гиперпролиферации, связанной с трансплантацией органа или ткани и иммунопролиферативного заболевания или нарушения, выбранного из группы, состоящей из воспалительного заболевания кишечника, псориаза, ревматоидного артрита, системной красной волчанки (SLE), гиперпролиферации сосудов на фоне гипоксии сетчатки и васкулита.
Изобретение еще более предпочтительно относится к лекарственному средству, содержащему по меньшей мере одно соединение в соответствии с изобретением и/или одну из его физиологически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях, для применения для лечения и/или профилактики физиологических и/или патофизиологических состояний, выбранных из группы, состоящей из гиперпролиферативных и инфекционных заболеваний и нарушений, где инфекционное заболевание или нарушение выбирают из группы, состоящей из
а) вирусиндуцированных инфекционных заболеваний, которые вызваны ретровирусами, гепаднавирусами, герпесвирусами, флавивирусами и/или аденовирусами, где ретровирусы выбирают из лентивирусов или онкоретровирусов, где лентивирусы выбирают из группы, состоящей из HIV-1, HIV-2, FIV, BIV, вирусов SIV, SHIV, CAEV, VMV и EIAV, и онкоретровирусы выбирают из группы, состоящей из HTLV-I, HTLV-II и BLV, гепаднавирусы выбирают из группы, состоящей из HBV, GSHV и WHV, герпесвирусы выбирают из группы, состоящей из HSV I, HSV II, EBV, VZV, HCMV или HHV 8, и флавивирусы выбирают из группы, состоящей из HCV, вируса Западного Нила и вируса желтой лихорадки,
b) бактериальных инфекционных заболеваний, которые вызваны грамположительными бактериями, где грамположительные бактерии выбирают из группы, состоящей из метициллин-чувствительных и метициллин-резистентных стафилококков (включая Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus haemolyticus, Staphylococcus hominis, Staphylococcus saprophyticus и коагулазоотрицательные стафилококки), Staphylococcus aureus с промежуточной чувствительностью к гликопептидам (GISA), пенициллин-чувствительных и пенициллин-резистентных стрептококков (включая Streptococcus pneumoniae, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus avium, Streptococcus bovis, Streptococcus lactis, Streptococcus sanguis и Streptococci группы С (GCS), Streptococci группы G (GGS) и стрептококки группы «viridans»), энтерококков (включая ванкомицин-чувствительные и ванкомицин-резистентные штаммы, такие как Enterococcus faecalis и Enterococcus faecium), Clostridium difficile, Iisteria monocytogenes, Corynebacterium jeikeium, Chlamydia spp (включая С. pneumoniae) и Mycobacterium tuberculosis,
c) бактериальных инфекционных заболеваний, которые вызваны грамотрицательными бактериями, где грамотрицательные бактерии выбирают из группы, состоящей из рода Enterobacteriacae, включая Escherichia spp. (включая Escherichia coli), Klebsiella spp., Enterobacter spp., Citrobacter spp., Serratia spp., Proteus spp., Providencia spp., Salmonella spp., Shigella spp., рода Pseudomonas (включая P. aeruginosa), Moraxella spp.(включая M. catarrhalis), Haemophilus spp. и Neisseria spp.,
d) инфекционных заболеваний, индуцированных внутриклеточными активными паразитами, выбранными из группы, состоящей из филума Apicomplexa или Sarcomastigophora (включая Trypanosoma, Plasmodia, Leishmania, Babesia или Theileria), Cryptosporidia, Sacrocystida, Amoebia, Coccidia и Trichomonadia.
Предполагается, что лекарственные средства, раскрытые выше, включают соответствующее применение соединений в соответствии с изобретением для приготовления лекарственного средства для лечения и/или профилактики вышеуказанных физиологических и/или патофизиологических состояний.
Дополнительно предполагается, что лекарственные средства, раскрытые выше, включают соответствующий способ лечения и/или профилактики вышеуказанных физиологических и/или патофизиологических состояний, в котором по меньшей мере одно соединение в соответствии с изобретением вводят пациенту, нуждающемуся в таком лечении.
Соединения в соответствии с изобретением предпочтительно проявляют выгодную биологическую активность, которая может быть легко продемонстрирована в ферментных анализах и экспериментах на животных, как описано в примерах. В таких анализах активности ферментов, соединения в соответствии с изобретением предпочтительно проявляют и взывают эффект ингибирования, который обычно подтверждается значениями IC50 в пригодном диапазоне, предпочтительно в микромолярном диапазоне и более предпочтительно в наномолярном диапазоне.
Соединения в соответствии с изобретением можно вводить людям или животным, в частности, млекопитающим, таким как обезьяны, собаки, кошки, крысы или мыши, и можно применять для терапевтического лечения человека или животных и для борьбы с вышеупомянутыми заболеваниями. Кроме того, они могут применяться в качестве диагностических средств или в качестве реагентов.
Кроме того, соединения в соответствии с изобретением можно применять для выделения и исследования активности или экспрессии аденозиновых рецепторов A2A и/или A2B. Кроме того, они особенно пригодны для применения в методах диагностики заболеваний на фоне нарушения активности аденозиновых рецепторов A2A и/или A2B. Следовательно, изобретение также относится к применению соединений в соответствии с изобретением для выделения и исследования активности или экспрессии аденозиновых рецепторов A2A и/или A2B, или в качестве связующих и ингибиторов аденозиновых рецепторов A2A и/или A2B.
Для диагностических целей, соединения в соответствии с изобретением, например, могут быть радиоактивно мечеными. Примерами радиоактивных меток являются 3Н, 14С, 231I и 125I. Предпочтительным методом мечения является способ с использованием йодогена (Fraker и др., 1978). Кроме того, соединения в соответствии с изобретением могут быть мечены с помощью ферментов, флуорофоров и хемофоров. Примерами ферментов являются щелочная фосфатаза, β-галактозидаза и глюкозооксидаза, примером флуорофора является флуоресцеин, примером хемофора является люминол, а автоматизированные системы детектирования, например, флуоресцентного окрашивания, описаны, например, в US 4,125,828 и US 4,207,554.
Более того, настоящее изобретение относится к фармацевтическим композициям, содержащим соединения настоящего изобретения, и их применению для лечения и/или профилактики заболеваний и нарушений, при которых может быть полезной частичная или полная инактивация аденозиновых рецепторов A2A и/или A2B.
Соединения формулы I можно применять для приготовления фармацевтических препаратов, в частности, с помощью нехимических способов. В этом случае, их вводят в пригодную дозированную форму вместе с по меньшей мере одним твердым, жидким и/или полужидким наполнителем или вспомогательным веществом и, необязательно, в комбинации с одним или несколькими дополнительным(-и) активным(-ыми) соединением(-ями).
Следовательно, изобретение также относится к фармацевтическим препаратам, содержащим по меньшей мере одно соединение формулы I и/или одну из его физиологически приемлемых солей, производных, сольватов и стереоизомеров, включая их смеси во всех соотношениях. В частности, изобретение также относится к фармацевтическим препаратам, которые дополнительно содержат наполнители и/или вспомогательные вещества, а также к фармацевтическим препаратам, которые содержат по меньшей мере одно дополнительное активное соединение лекарственного средства.
В частности, изобретение также относится к способу приготовления фармацевтического препарата, отличающемуся тем, что соединение формулы I и/или одну из его физиологически приемлемых солей, производных, сольватов и стереоизомеров, включая их смеси во всех соотношениях, вводят в пригодную дозированную форму вместе с твердым, жидким или полужидким наполнителем или вспомогательным веществом и необязательно с дополнительным активным соединением лекарственного средства.
Фармацевтические препараты в соответствии с изобретением можно применять в качестве лекарственных средств в медицине человека и ветеринарии. Пациент или хозяин может принадлежать к любому виду млекопитающих, например, такому как, приматы, особенно люди; грызуны, включая мышей, крыс и хомячков; кролики; лошади, крупный рогатый скот, собаки, кошки, и т.д. Животные модели представляют интерес для экспериментальных исследований, где они обеспечивают модель для лечения заболевания человека.
Пригодными веществами-носителями являются органические или неорганические вещества, которые пригодны для энтерального (например, перорального), парентерального или местного введения и не реагируют с новыми соединениями, например, вода, растительные масла (такие как подсолнечное масло или рыбий жир), бензиловые спирты, полиэтиленгликоли, желатин, углеводы, такие как лактоза или крахмал, стеарат магния, тальк, ланолин или вазелин. Благодаря своим экспертным знаниям, специалист в данной области осведомлен, какие вспомогательные вещества пригодны для желаемого состава лекарственного средства. Помимо растворителей, например, воды, физиологического солевого раствора или спиртов, таких как, например, этанол, пропанол или глицерин, растворы сахаров, такие как растворы глюкозы или маннита, или смесь указанных растворителей, также можно использовать гелеобразователи, вспомогательные средства для таблеток и другие носители активного ингредиента, например, скользящие вещества, стабилизаторы и/или смачивающие средства, эмульгаторы, соли для воздействия на осмотическое давление, антиоксиданты, диспергаторы, антивспениватели, буферные вещества, вкусовые и/или ароматические вещества или корректоры вкуса, консерванты, солюбилизаторы или красители. При необходимости, препараты или лекарственные средства в соответствии с изобретением могут содержать одно или несколько дополнительных активных соединений, например, один или несколько витаминов.
При необходимости, препараты или лекарственные средства в соответствии с изобретением могут содержать одно или несколько дополнительных активных соединений и/или один или несколько усилителей действия (вспомогательных веществ).
Термины "фармацевтический состав" и "фармацевтический препарат" в целях настоящего изобретения используются в качестве синонимов.
В данном контексте, "фармацевтически переносимый" относится к лекарственным средствам, преципитирующим реагентам, наполнителям, вспомогательным веществам, стабилизаторам, растворителям и другим средствам, которые облегчают введение фармацевтических препаратов, полученных из них, млекопитающему без нежелательных физиологических побочных действий, таких как, например, тошнота, головокружение, проблемы с пищеварением или т.п.
В случае фармацевтических препаратов для парентерального введения, существует потребность в изотоничности, нормальной гидратации и переносимости и безопасности состава (низкая токсичность), используемых вспомогательных веществ и первичной упаковки. Неожиданно, соединения в соответствии с изобретением предпочтительно обладают тем преимуществом, что возможно прямое применение и дополнительные стадии очистки для удаления токсикологически неприемлемых веществ, таких как, например, органические растворители в высоких концентрациях или другие токсикологически неприемлемые вспомогательные вещества, таким образом, не являются необходимыми перед применением соединений в соответствии с изобретением в фармацевтических составах.
Изобретение особенно предпочтительно также относится к фармацевтическим препаратам, содержащим по меньшей мере одно соединение в соответствии с изобретением в осажденной некристаллической, осажденной кристаллической или в растворенной или суспендированной форме, и необязательно наполнители и/или вспомогательные вещества и/или дополнительные фармацевтические активные соединения.
Предпочтительно, соединения в соответствии с изобретением позволяют приготовлять высококонцентрированные составы без вредной, нежелательной агрегации соединений в соответствии с изобретением. Таким образом, с помощью соединений в соответствии с изобретением можно получить готовые к применению растворы, имеющие высокое содержание активных компонентов, с водными растворителями или в водной среде.
Соединения и/или их физиологически приемлемые соли и сольваты также можно лиофилизировать и полученные в результате лиофилизаты применять, например, для приготовления препаратов для инъекций.
Водные препараты можно получить путем растворения или суспендирования соединений в соответствии с изобретением в водном растворе и необязательно добавления вспомогательных веществ. С этой целью, определенные объемы исходных растворов, включающих указанные дополнительные вспомогательные вещества в определенной концентрации, предпочтительно добавляют к раствору или суспензии, имеющему(-ей) определенную концентрацию соединений в соответствии с изобретением, и смесь необязательно разбавляют водой до предварительно рассчитанной концентрации. Альтернативно, вспомогательные вещества можно добавлять в твердой форме. Количества исходных растворов и/или воды, которые необходимы в каждом случае, вслед за этим можно добавлять к полученному(-ой) водному раствору или суспензии. Соединения в соответствии с изобретением предпочтительно также можно растворять или суспендировать непосредственно в растворе, включающем все дополнительные вспомогательные вещества.
Предпочтительно можно приготовить растворы или суспензии, которые включают соединения в соответствии с изобретением и имеют рН в диапазоне от 4 до 10, предпочтительно от 5 до 9, и осмоляльность в диапазоне от 250 до 350 мОсмол/кг. Фармацевтический препарат может таким образом быть введен непосредственно, по существу без боли внутривенно, внутриартериально, внутрисуставно, подкожно или чрескожно. Кроме того, препарат также можно добавлять к растворам для инфузий, таким как, например, раствор глюкозы, изотонический солевой раствор или раствор Рингера, который может также содержать дополнительные активные соединения, что также делает возможным введение относительно больших количеств активного соединения.
Фармацевтические препараты в соответствии с изобретением также могут содержать смеси нескольких соединений в соответствии с изобретением.
Препараты в соответствии с изобретением хорошо переносятся физиологически, их легко приготовить, их можно четко распределить и предпочтительно они являются стабильными касательно условий анализа, продуктов разложения и агрегатов во время хранения и транспортировки, и при многократном осуществлении замораживания и процедур оттаивания. Они предпочтительно могут храниться в стабильном состоянии в течение, по меньшей мере, от трех месяцев до двух лет при температуре холодильника (2-8°С) и при комнатной температуре (23-27°С) и относительной влажности воздуха (О.В.) 60%.
Например, соединения в соответствии с изобретением можно хранить в стабильном состоянии с помощью сушки и при необходимости превращать в готовый к применению фармацевтический препарат путем растворения или суспендирования. Возможными методами сушки являются, например, не ограничиваясь этими примерами, сушка газообразным азотом, сушка в вакуумной печи, лиофилизация, промывка органическими растворителями и последующая сушка воздухом, сушка в жидком слое, сушка в псевдоожижженом слое, сушка распылением, вальцовая сушка, послойная сушка, сушка воздухом при комнатной температуре и дальнейшие методы.
Термин "эффективное количество" означает количество лекарственного средства или фармацевтического активного компонента, которое вызывает в ткани, системе, животном или человеке биологическую или медицинскую реакцию, которая является искомой или желаемой, например, исследователем или врачом.
Кроме того, термин "терапевтически эффективное количество" означает количество, которое, по сравнению с соответствующим субъектом, который не получил это количество, имеет следующие последствия: улучшение лечения, излечивание, предотвращение или устранение заболевания, синдрома, болезненного состояния, жалоб, нарушения или предотвращение побочных действий или также замедление прогрессирования заболевания, ослабление жалоб или нарушения. Термин "терапевтически эффективное количество" также охватывает количества, которые являются эффективными для усиления нормальной физиологической функции.
При применении препаратов или лекарственных средств в соответствии с изобретением, соединения в соответствии с изобретением и/или их физиологически приемлемые соли и сольваты обычно применяют по аналогии с известными, коммерчески доступными препаратами или препаративными формами, предпочтительно в дозировках между 0.1 и 500 мг, в частности, 5 и 300 мг, на применяемую единицу. Суточная доза составляет предпочтительно между 0.001 и 250 мг/кг, в частности, 0.01 и 100 мг/кг, массы тела. Препарат можно вводить один или несколько раз в день, например, два, три или четыре раза в день. Однако, индивидуальная доза для пациента зависит от большого числа индивидуальных факторов, таких как, например, эффективность конкретного применяемого соединения, возраст, масса тела, общее состояние здоровья, пол, питание, время и способ введения, скорость экскреции, сочетание с другими лекарственными средствами и тяжесть и продолжительность конкретного заболевания.
Мерой поглощения активного соединения лекарственного средства в организме является его биологическая доступность. Если активное соединение лекарственного средства доставляется в организм внутривенно в виде раствора для инъекции, его абсолютная биологическая доступность, т.е. доля фармацевтического средства, которая достигает системной крови, т.е. основной системы кровообращения, в неизмененной форме, составляет 100%. В случае перорального введения терапевтически активного соединения, активное соединение обычно находится в виде твердого вещества в составе и поэтому должно сначала быть растворено для того, чтобы быть способным преодолеть барьеры, например, желудочно-кишечный тракт, слизистую оболочку полости рта, слизистую оболочку носа или кожу, в частности, роговой слой эпидермиса, или быть усвоенным организмом. Фармакокинетические данные, т.е. данные о биологической доступности, можно получить аналогично методу, описанному в J. Shaffer и др., J. Pharm. Sciences, 88 (1999), 313-318.
Кроме того, лекарственные средства этого типа можно получить с помощью одного из способов, которые являются общеизвестным в фармацевтическом уровне техники.
Лекарственные средства могут быть адаптированы для введения при помощи любого желаемого пригодного пути, например, перорального (включая буккальное или сублингвальное), ректального, легочного, назального, местного (включая буккальное, сублингвальное или трансдермальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное, внутрикожное и, в частности, внутрисуставное) пути введения. Лекарственные средства этого типа можно получить с помощью любого способа, известного в области фармацевтики, например, путем объединения активного соединения с наполнителем(-ями) или вспомогательным(-и) веществом(-ами).
Предпочтительно, для введения лекарственных средств в соответствии с изобретением пригодным является парентеральное введение. В случае парентерального введения, внутрисуставное введение является особенно предпочтительным.
Таким образом, изобретение предпочтительно также относится к применению фармацевтического препарата в соответствии с изобретением для внутрисуставного введения для лечения и/или профилактики физиологических и/или патофизиологических состояний, выбранных из группы, состоящей из остеоартрита, травматических повреждений хряща, артрита, боли, аллодинии или гипералгезии.
Преимущество внутрисуставного введения заключается в том, что соединение в соответствии с изобретением может быть введено непосредственно в синовиальную жидкость в непосредственной близости от суставного хряща и также способно распространяться оттуда в хрящевую ткань. Фармацевтические препараты в соответствии с изобретением таким образом также могут быть введены непосредственно в суставную щель и таким образом осуществлять свое действие непосредственно на месте действия, как и предполагалось. Соединения в соответствии с изобретением также пригодны для приготовления лекарственных средств для парентерального введения, имеющих медленное, устойчивое и/или контролируемое высвобождения активного соединения. Таким образом, они также пригодны для приготовления составов замедленного высвобождения, которые являются предпочтительными для пациента, поскольку введение необходимо повторять только через относительно большие промежутки времени.
Лекарственные средства, адаптированные для парентерального введения включают водные и неводные стерильные растворы для инъекции, содержащие антиоксиданты, буферы, бактериостатические и растворенные вещества, с помощью которых состав является изотоническим по отношению к крови или синовиальной жидкости реципиента, которого подвергают лечению; а также водные и неводные стерильные суспензии, которые могут содержать суспензионную среду и загустители. Составы могут поставляться в однодозовых или многодозных емкостях, например, запаянных ампулах и флаконах, и храниться в высушенном сублимацией (лиофилизированном) состоянии, так что необходимо только добавление стерильной жидкости-носителя, например, воды для инъекции, непосредственно перед применением. Растворы и суспензии для инъекции, приготовленные в соответствии с раскрытой рецептурой, можно получить из стерильных порошков, гранул и таблеток.
Соединения в соответствии с изобретением также можно вводить в виде липосомных систем доставки, таких как, например, небольшие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как, например, холестерин, стеариламин или фосфатидилхолины.
Соединения в соответствии с изобретением также можно сочетать с растворимыми полимерами в качестве нацеливающих носителей лекарственных средств. Такие полимеры могут охватывать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидофенол или полиэтиленоксид-полилизин, замещенный пальмитоиловыми радикалами. Кроме того, соединения в соответствии с изобретением можно сочетать с биоразлагаемыми полимерами, которые пригодны для обеспечения замедленного высвобождения лекарственного средства, например, такими как полимолочная кислота, поли-эпсилон-капролактон, полигидроксимасляная кислота, сложные полиортоэфиры, полиацетали, полидигидроксипираны, полицианоакрилаты, сополимер молочной кислоты с гликолевой кислотой, полимеры, такие как конъюгаты декстран -метакрилат, сложные полифосфоэфиры, различные полисахариды и полиамины, и поли-е-капролактон, альбумин, хитозан, коллаген или модифицированный желатин, и перекрестно-сшитые или амфипатические блок-сополимеры гидрогелей.
Пригодными для энтерального введения (перорального или ректального) являются, в частности, таблетки, драже, капсулы, сиропы, соки, капли или суппозитории, и пригодными для местного применения являются мази, кремы, пасты, лосьоны, гели, спреи, пены, аэрозоли, растворы (например, растворы в спиртах, таких как этанол или изопропанол, ацетонитрил, ДМФА, диметилацетамид, 1,2-пропандиол или их смеси друг с другом и/или с водой) или порошки. Также особенно пригодными для местного применения являются липосомальные препараты.
В случае состава в виде мази, активное соединение можно использовать либо с парафиновой, либо со смешивающейся с водой основой для крема. Альтернативно, активное соединение может быть введено в состав в виде крема с основой для крема типа масло-в-воде или вода-в-масле.
Лекарственные средства, адаптированные для трансдермального введения, могут доставляться в виде независимых пластырей для пролонгированного, тесного контакта с эпидермисом реципиента. Таким образом, например, активное соединение может доставляться из пластыря путем ионофореза, как описано в общих чертах в Pharmaceutical Research, 3 (6), 318 (1986).
Само собой разумеется, что кроме компонентов, особым образом упомянутых выше, лекарственные средства в соответствии с изобретением также могут содержать другие, обычные в данной области средства для конкретного типа фармацевтического состава.
Изобретение также относится к комплекту (набору), состоящему из отдельных упаковок
a) эффективного количества соединения формулы I и/или его физиологически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях, и
b) эффективного количества дополнительного активного соединения лекарственного средства.
Комплект включает пригодные емкости, такие как коробки или пачки, индивидуальные бутылки, пакеты или ампулы. Комплект может включать, например, отдельные ампулы, каждая из которых содержит эффективное количество соединения формулы I и/или его фармацевтически приемлемых солей, производных, сольватов, пролекарств и стереоизомеров, включая их смеси во всех соотношениях, и эффективное количество дополнительного активного соединения лекарственного средства в растворенной или лиофилизированной форме.
Кроме того, лекарственные средства в соответствии с изобретением можно применять с целью обеспечения аддитивных или синергетических эффектов в случае некоторых известных методов терапии и/или можно применять с целью восстановления эффективности определенных существующих методов терапии.
Помимо соединений в соответствии с изобретением, фармацевтические препараты в соответствии с изобретением также могут содержать дополнительные активные соединения лекарственного средства, например, для применения для лечения злокачественного новообразования, другие противоопухолевые лекарственные средства. Для лечения других упомянутых заболеваний, фармацевтические препараты в соответствии с изобретением также, помимо соединений в соответствии с изобретением, могут содержать дополнительные активные соединения лекарственного средства, которые, как известно специалисту в данной области, применяются для их лечения.
В одном основном варианте осуществления, обеспечиваются способы усиления иммунного ответа у хозяина, нуждающемуся в этом. Иммунный ответ может быть повышен путем уменьшения Т-клеточной переносимости, в том числе посредством усиления высвобождения IFN-γ, снижения продуцирования или активации регуляторных Т-клеток, или усиления продуцирования антиген-специфических Т-клеток памяти в хозяине. В одном варианте осуществления, способ включает введение соединения настоящего изобретения хозяину в комбинации или поочередно с антителом. В конкретных подвариантах осуществления, антитело представляет собой терапевтическое антитело. В одном отдельном варианте, обеспечивается способ повышения эффективности терапии пассивными антителами, включающий введение соединения настоящего изобретения в комбинации или поочередно с одним или нескольким пассивными антителами. Этот способ может повышать эффективность терапии антителами для лечения пролиферативных нарушений, таких как злокачественное новообразование, или может повышать эффективность терапии при лечении или предотвращении инфекционных заболеваний. Соединение настоящего изобретения можно вводить в комбинации или поочередно с антителами такими как, например, ритуксимаб, герцептин или эрбитукс.
В другом основном варианте, обеспечивается способ лечения или предотвращения аномальной клеточной пролиферации, включающий введение соединения настоящего изобретения хозяину, нуждающемуся в этом, по существу в отсутствие другого противоопухолевого средства.
В другом основном варианте, обеспечивается способ лечения или предотвращения аномальной клеточной пролиферации в хозяине, нуждающемся в этом, включающий введение хозяину первого соединения настоящего изобретения по существу в комбинации с первым противоопухолевым средством и вслед за этим введение второго антагониста рецепторов A2A и/или A2B. В одном подварианте осуществления, второй антагонист вводят по существу в отсутствие другого противоопухолевого средства. В другом основном варианте, обеспечивается способ лечения или предотвращения аномальной клеточной пролиферации в хозяине, нуждающемся в этом, включающий введение хозяину соединения настоящего изобретения по существу в комбинации с первым противоопухолевым средством и вслед за этим введение второго противоопухолевого средства в отсутствие антагониста.
Таким образом, лечение злокачественных заболеваний, раскрытое в данном случае, можно осуществлять либо в форме терапии соединением настоящего изобретения, либо в комбинации с оперативным вмешательством, облучением или химиотерапией. Химиотерапия этого типа может включать применение одного или нескольких активных соединений из следующих классов противоопухолевых активных соединений:
(i) антипролиферативные/противоопухолевые/повреждающие ДНК активные соединения и их комбинации, которые применяются в медицинской онкологии, такие как алкилирующие активные соединения (например, цисплатин, карбоплатин, циклофосфамид, азотный иприт, мельфалан, хлорамбуцил, бусульфан и нитрозомочевины); антиметаболиты (например, антифолаты, такие как фторпиримидины, такие как 5-фторурацил и тегафур, ралтитрексед, метотрексат, арабинозид цитозина, гидроксимочевина и гемцитабин); противоопухолевые антибиотики (например, антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин и митрамицин); антимитотические активные соединения (например, алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин, и таксоиды, такие как таксол и таксотер); ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан, иринотекан и камптотецин) и влияющие на дифференциацию клеток активные соединения (например, ретиноевая кислота, полностью находящаяся в транс-конфигурации, 13-цис-ретиноевая кислота и фенретинид);
(ii) цитостатические активные соединения, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен и йодоксифен), регуляторы рецепторов эстрогена (например, фульвестрант), антиандрогены (например, бикалутамид, флутамид, нилутамид и ципротерон ацетат), антагонисты LHRH или агонисты LHRH (например, гозерелин, лейпрорелин и бузерелин), прогестероны (например, мегестрол ацетат), ингибиторы ароматазы (например, анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5α-редуктазы, такие как финастерид;
(iii) активные соединения, которые ингибируют инвазию злокачественных клеток, включая, например, ингибиторы металлопротеиназы, такие как маримастат, и ингибиторы функции рецептора урокиназного активатора плазминогена;
(iv) ингибиторы функции фактора роста, например, антитела к фактору роста, антитела к рецептору фактора роста, например, анти-erbb2 антитело трастузумаб [Герцептин™] и анти-erbb1 антитело цетуксимаб [С225]), ингибиторы фарнезилтрансферазы, ингибиторы тирозинкиназы и ингибиторы серин/треонинкиназы, например, ингибиторы семейства фактора роста эпидермиса (например, ингибиторы тирозинкиназ EGFR семейства, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, AZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)хиназолин-4-амин (CI 1033), например, ингибиторы семейства фактора роста тромбоцитов и, например, ингибиторы семейства фактора роста гепатоцитов;
(v) антиангиогенные активные соединения, такие как бевацизумаб, ангиостатин, эндостатин, линомид, батимастат, каптоприл, ингибитор из хрящевой ткани, генистеин, интерлейкин 12, лавендустин, ацетат медроксипрогестерона, рекомбинантный человеческий тромбоцитарный фактор 4, текогалан, тромбоспондин, TNP-470, моноклональное антитело анти-VEGF, растворимый химерный белок VEGF-рецептора, антитела к рецептору VEGF, антитела к рецептору PDGF, ингибиторы интегринов, ингибиторы тирозинкиназы, ингибиторы серин/треонинкиназы, антисмысловые олигонуклеотиды, антисмысловые олигодезоксинуклеотиды, миРНК, анти-VEGF аптамеры, фактор пигментного эпителия и соединения, которые были опубликованы в международных заявках на патенты WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354);
(vi) средства, разрушающие сосуды, такие как комбретастатин А4 и соединения, которые были опубликованы в международных заявках на патенты WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 и WO 02/08213;
(vii) методы антисмысловой терапии, например, такая, которая направлена на упомянутые выше мишени, такие как ISIS 2503, антисмысловая терапия, направленная против гена Ras;
(viii) методы генной терапии, включая, например, методы замены атипичных, модифицированных генов, таких как атипичный р53 или атипичный BRCA1 или BRCA2, методы GDEPT (ген-направленная ферментная пролекарственная терапия), такие как методы, в которых применяют цитозиндезаминазу, тимидинкиназу или бактериальный нитроредуктазный фермент, и методы, которые повышают переносимость пациента к химиотерапии или радиотерапии, такие как терапия множественной лекарственной устойчивости; и
(ix) методы иммунотерапии, включая, например, методы повышения иммуногенности опухолевых клеток пациента в условиях ex vivo и in vivo, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, методы снижения анергии Т-клеток, методы с применением трансфектированных иммунных клеток, таких как цитокин-трансфектированные дендритные клетки, методы с применением цитокин-трансфектированных опухолевых клеток и методы с применением анти-идиотипичных антител;
(x) химиотерапевтические средства, включая, например, абареликс, альдеслейкин, алемтузумаб, алитретиноин, аллопуринол, алтретамин, амифостин, анастрозол, триоксид мышьяка, аспарагиназу, живую БЦЖ, бевацизумаб, бексаротен, блеомицин, бортезомиб, бусульфан, калустерон, камптотецин, капецитабин, карбоплатин, кармустин, целекоксиб, цетуксимаб, хлорамбуцил, цинакальцет, цисплатин, кладрибин, циклофосфамид, цитарабин, дакарбазин, дактиномицин, дарбэпоэтин альфа, дауномицин, денилейкин-дифтитокс, дексразоксан, доцетаксел, доксорубицин, дромостанолон, эпирубицин, эпоэтин альфа, эстрамустин, этопозид, эксеместан, филграстим, флоксуридин, флударабин, фторурацил, фульвестрант и гемцитабин.
Лекарственные средства из таблицы 1 можно предпочтительно, но не исключительно, комбинировать с соединениями формулы I.
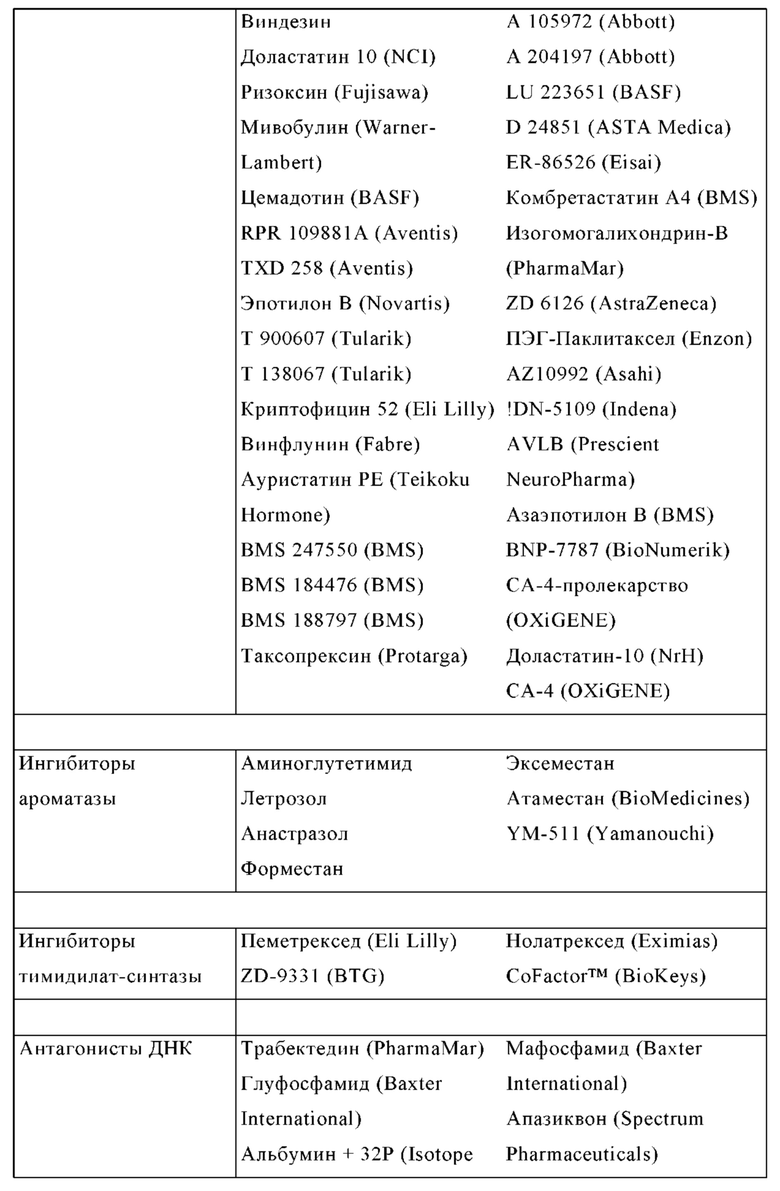
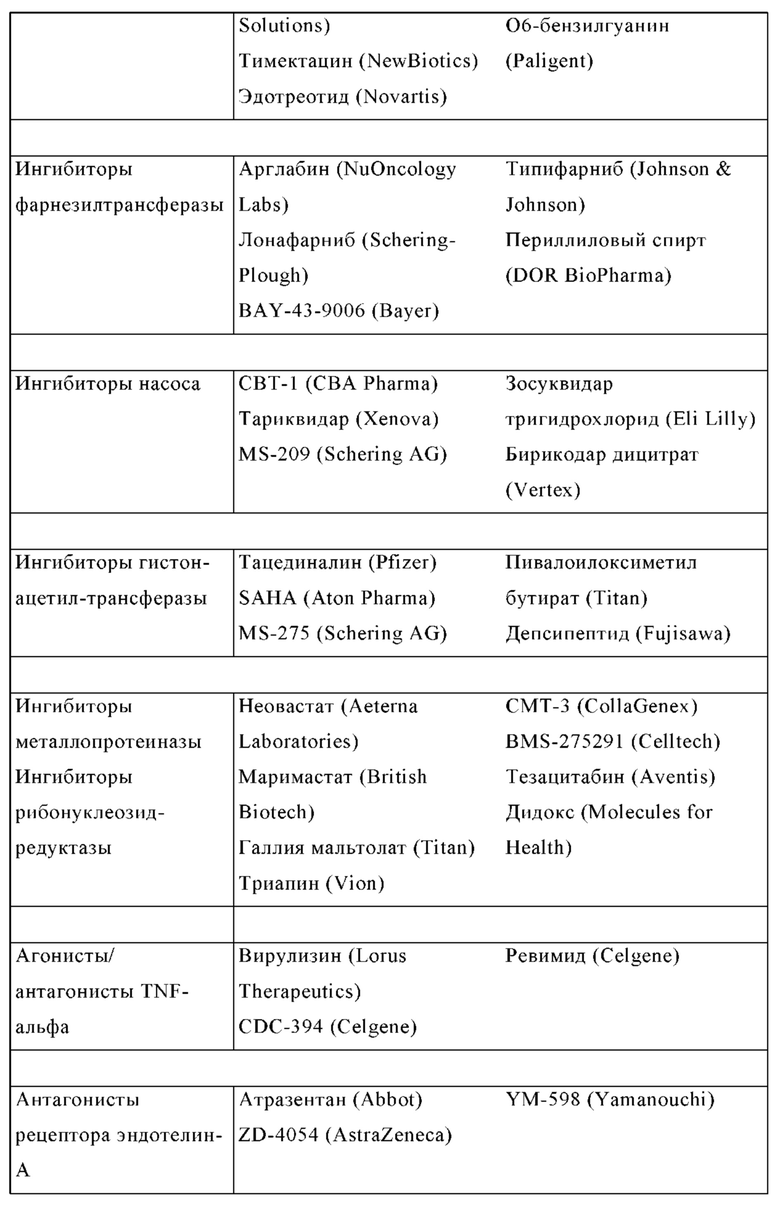




Даже без дополнительных вариантов осуществления предполагается, что специалист в данной области сможет использовать приведенное выше описание в самом широком объеме. Предпочтительные варианты осуществления, следовательно, следует рассматривать просто как описательное раскрытие, которое никоим образом не ограничивает настоящее изобретение.
Таким образом, следующие примеры предназначены для пояснения изобретения и не ограничивают его. Если не указано иное, процентные значения означают процентные значения по массе. Все температуры указаны в градусах Цельсия. "Обычная обработка": при необходимости добавляют воду, рН доводят, при необходимости, до значений между 2 и 10, в зависимости от строения конечного продукта, смесь экстрагируют этилацетатом или дихлорметаном, фазы разделяют, органическую фазу сушат над сульфатом натрия, фильтруют и упаривают, и продукт очищают с помощью хроматографии на силикагеле и/или путем кристаллизации.
Значения Rf приведены для силикагеля; масс-спектрометрия: EI (ионизация электронным ударом): М+, FAB (бомбардировка быстрыми атомами): (М+Н)+, ТГФ (тетрагидрофуран), NMP (N-метилпирролидон), ДМСО (диметилсульфоксид), ЭА (этилацетат), МеОН (метанол), ТСХ (тонкослойная хроматография).
Перечень сокращений
AUC - Площадь под кривой зависимости концентрации лекарственного вещества в плазме от времени
Cmax - Максимальная концентрация в плазме
CL - Клиренс
CV - Коэффициент вариации
CYP - Цитохром Р450
ДМСО - Диметилсульфоксид
F - Биологическая доступность
fa - Поглощенная фракция
в/в - Внутривенное
ЖХ-МС/МС - Жидкостная хроматография с тандемной масс-спектрометрией
LLOQ - Нижний предел количественного определения
NC - Не рассчитано
ND - Не определено
ПЭГ - Полиэтиленгликоль
Pgp - Гликопротеин проницаемости
PK - Фармакокинетический (фармакокинетика)
п/о - Перорально (пероральный препарат)
t1/2 - Время полураспада
tmax - Время, при котором в плазме достигается максимальная концентрация лекарственного вещества
СВЭЖХ - Сверхвысокоэффективная жидкостная хроматография
Vss - Объем распределения (в равновесном состоянии)
об./об. - Объем к объему
Пример 1: Примеры соединений настоящего изобретения
В особенности, изобретение относится к соединениям таблицы 2 и их физиологически приемлемым солям, производным, сольватам, пролекарствам и стереоизомерам, включая их смеси во всех соотношениях.
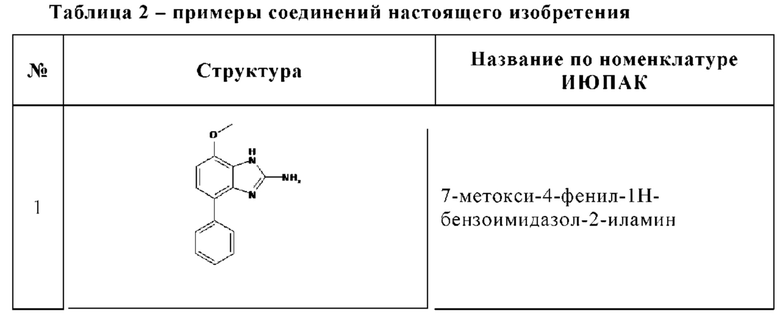
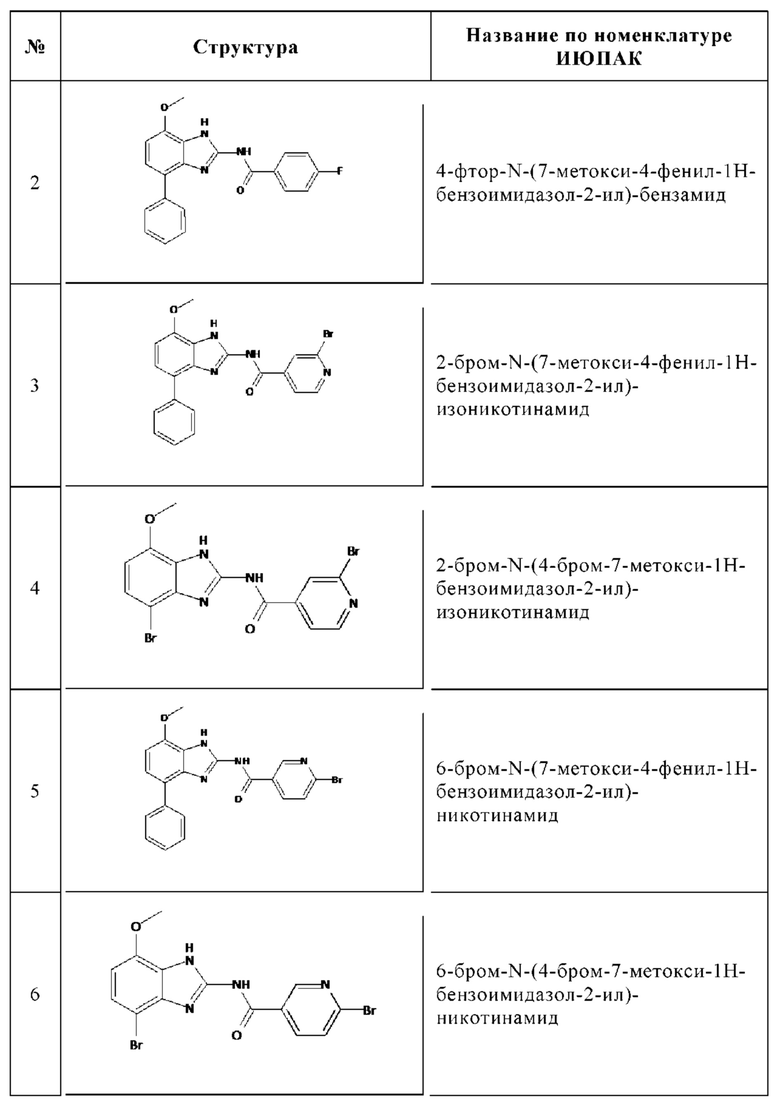
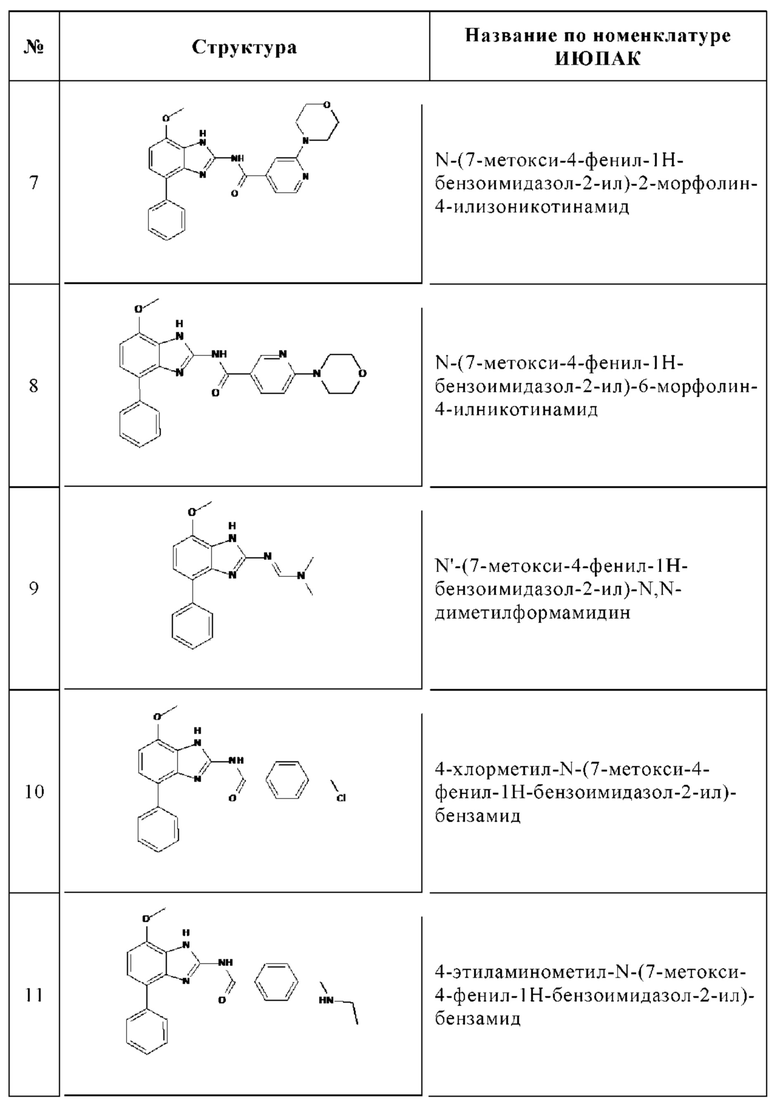
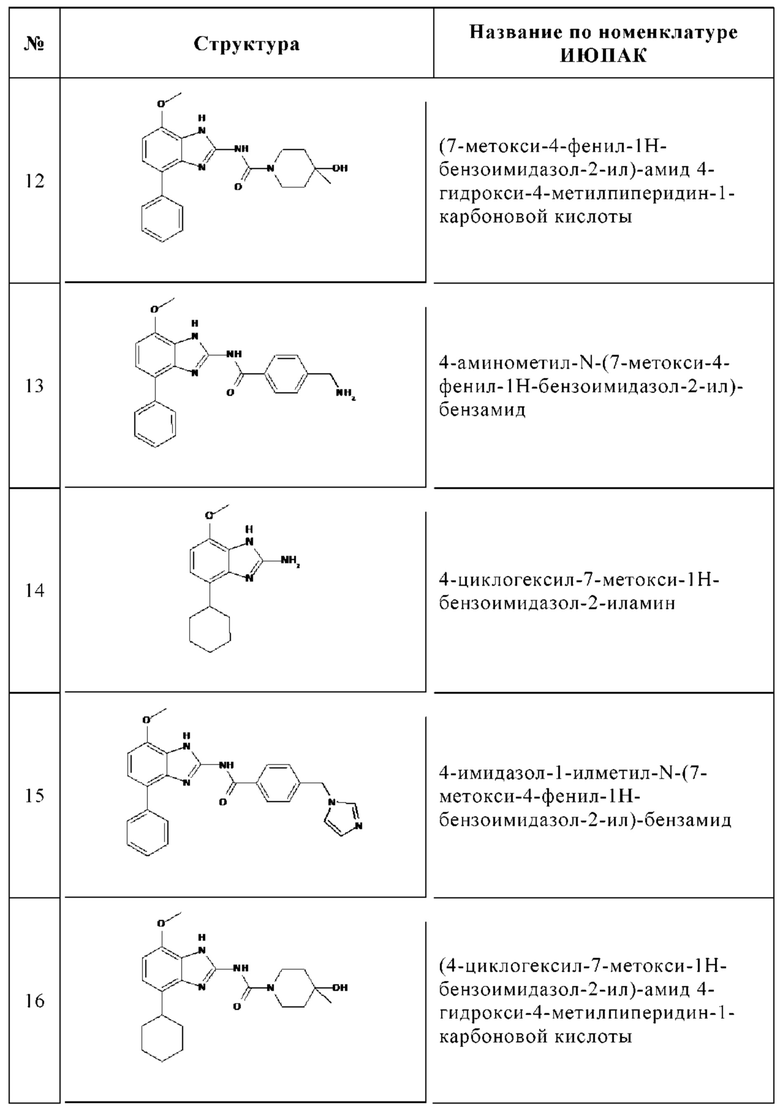
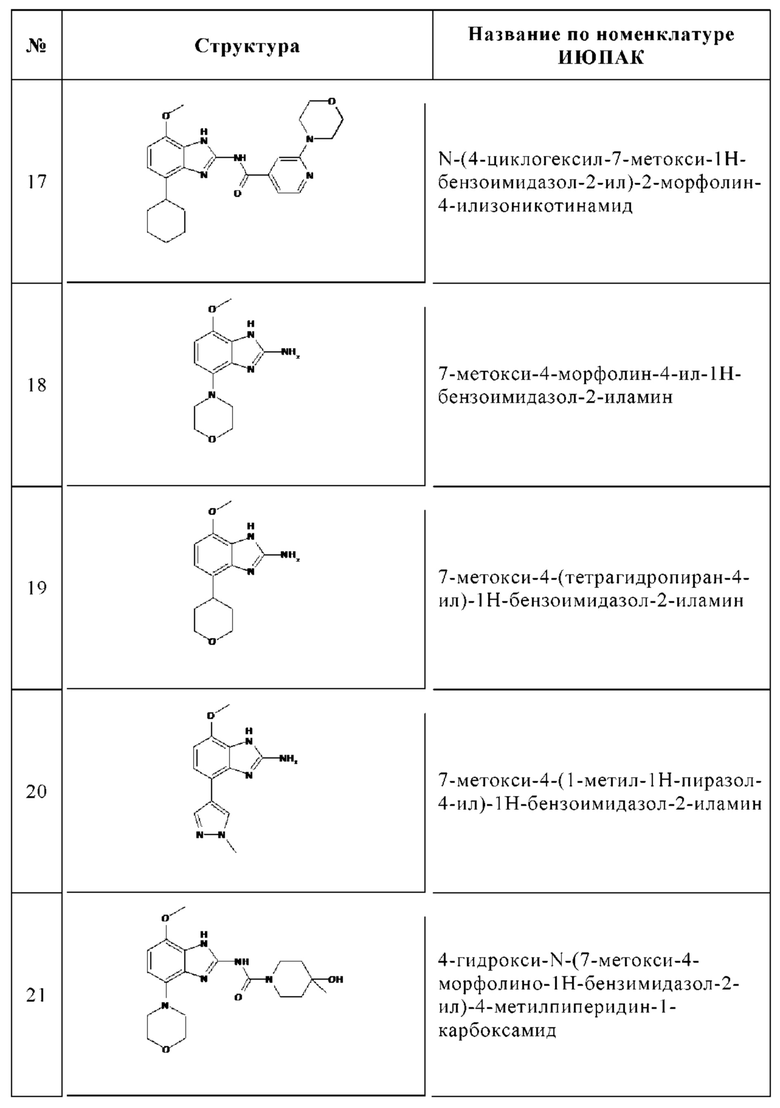
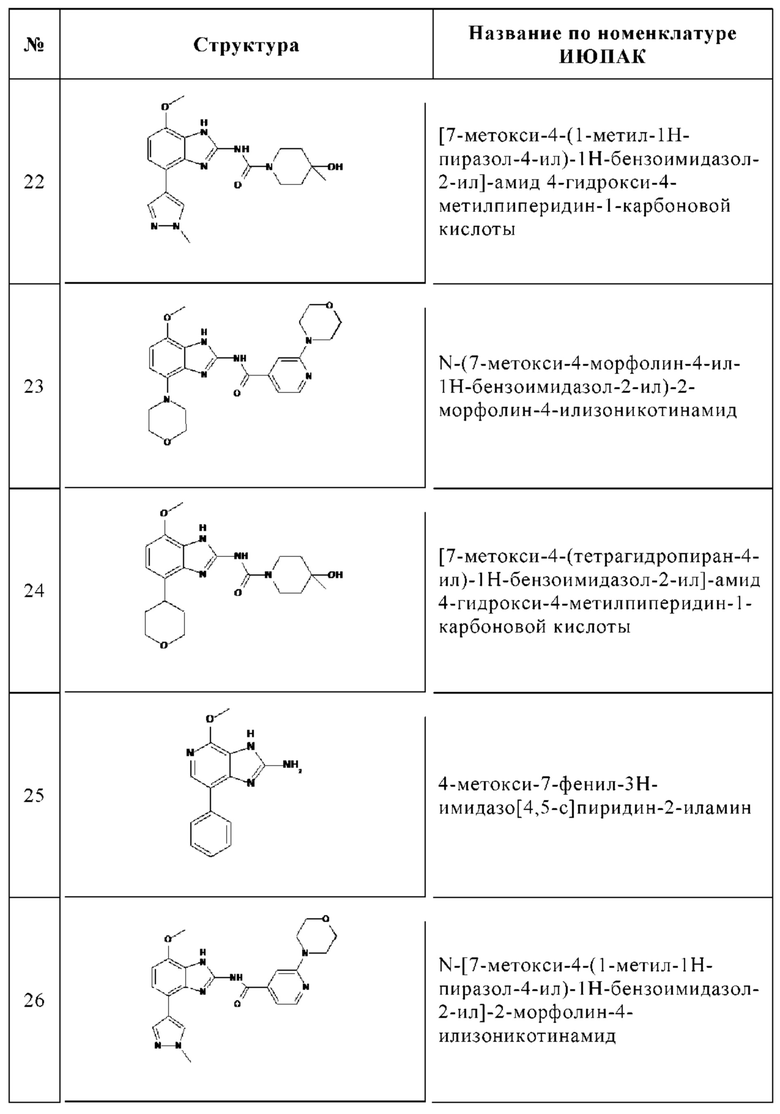
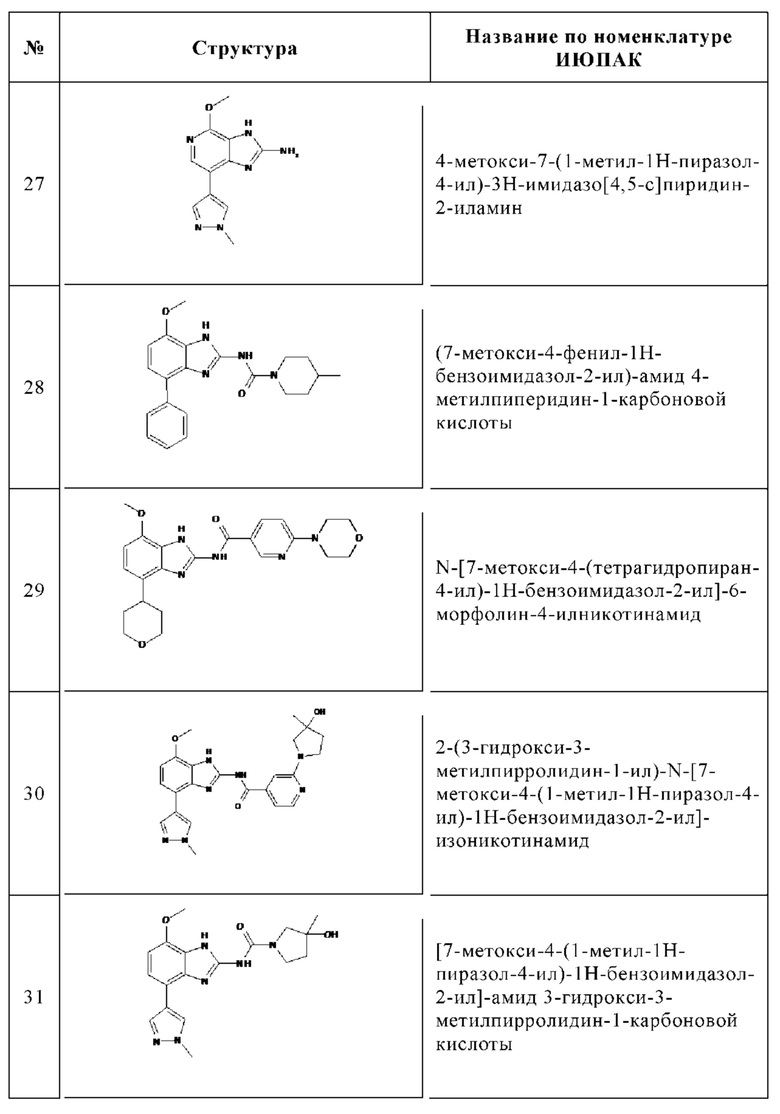
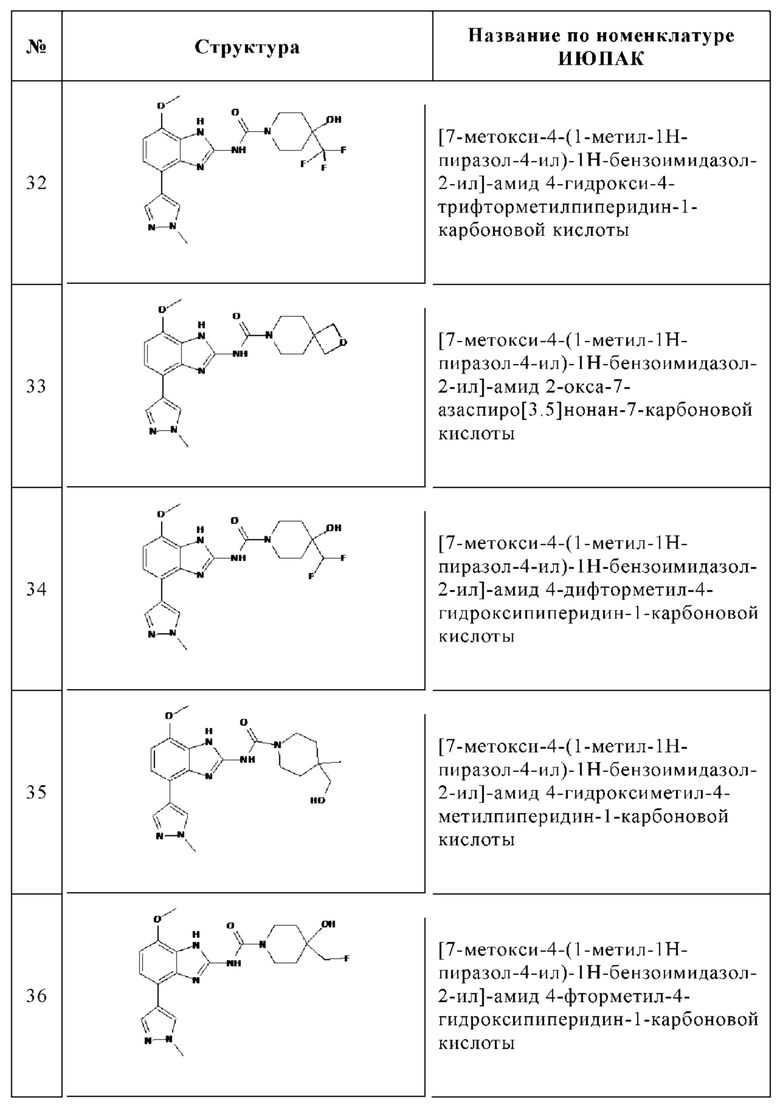
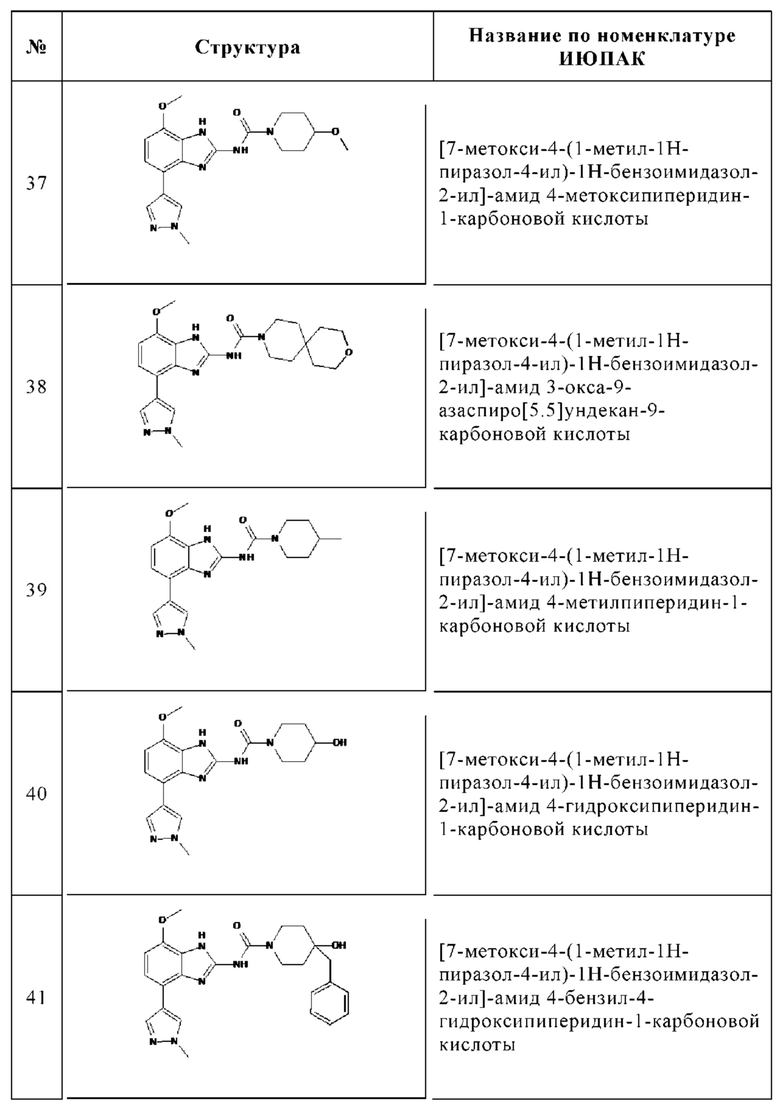
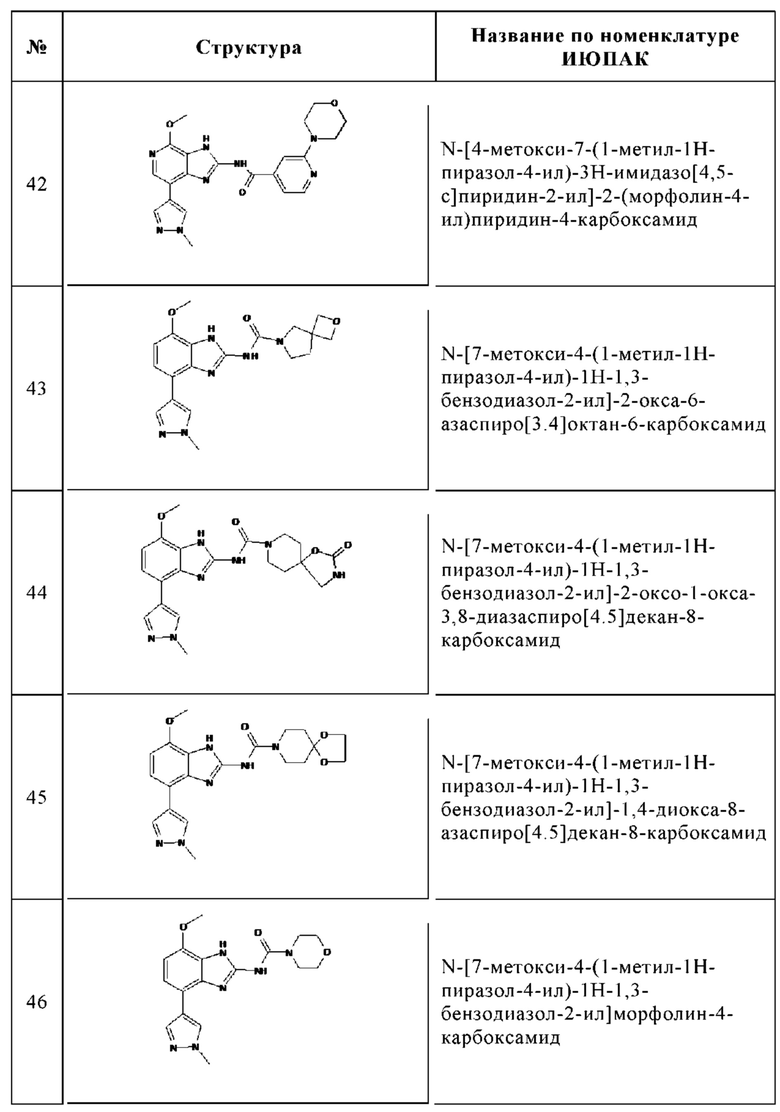
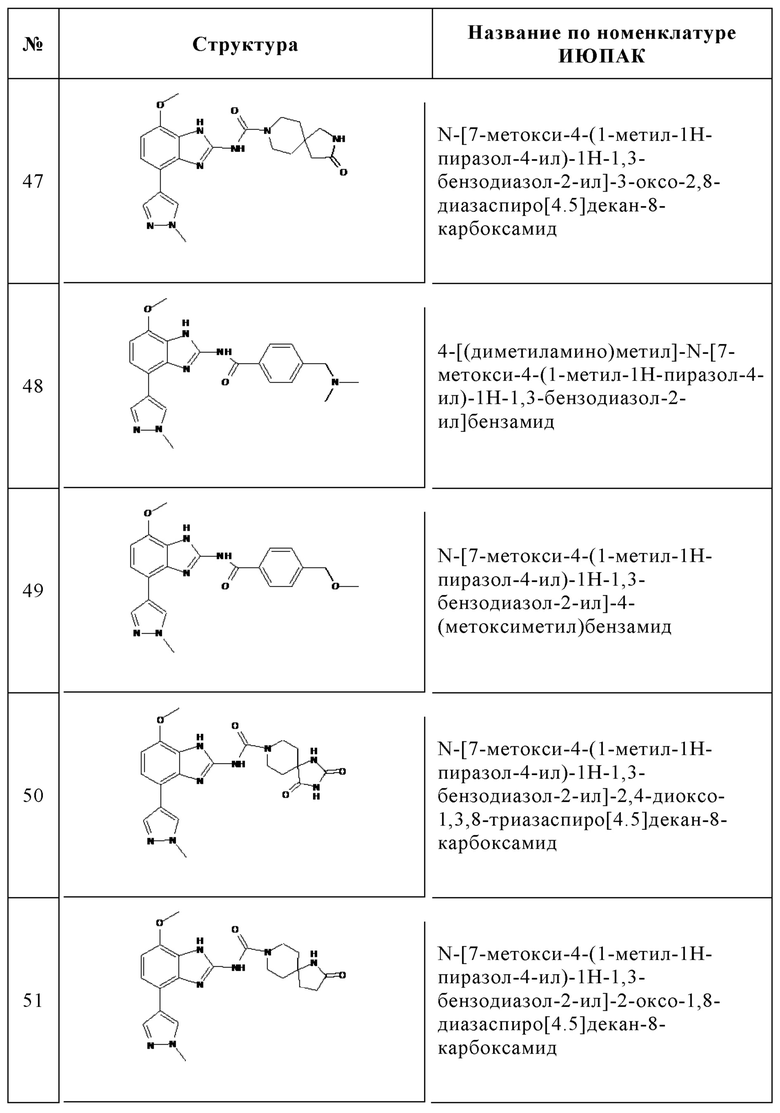
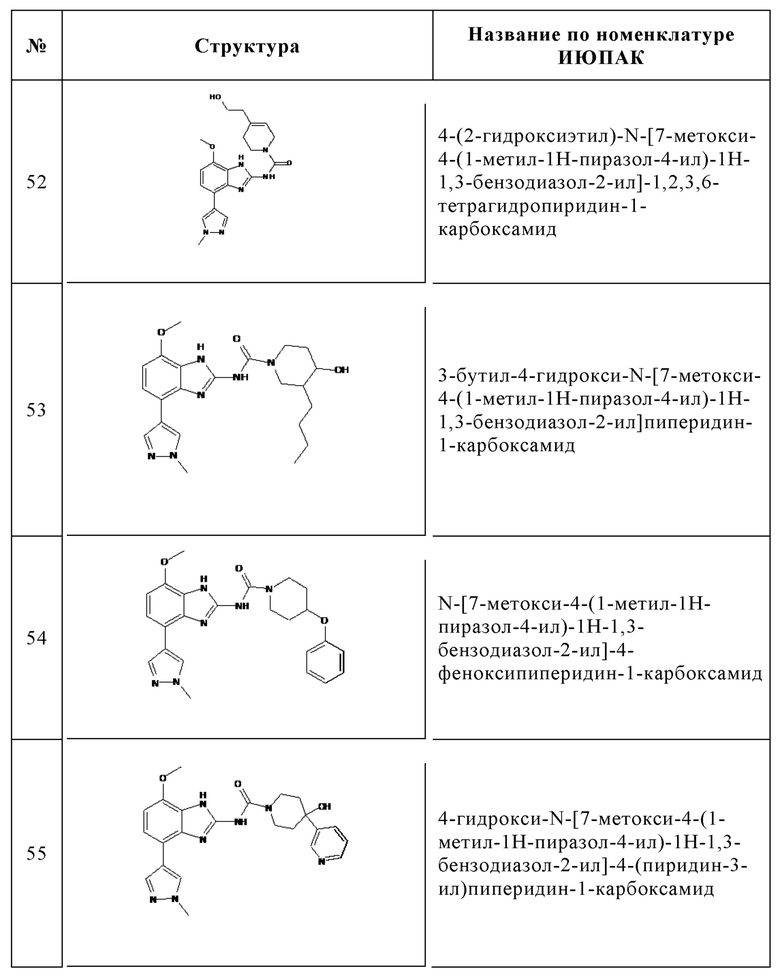
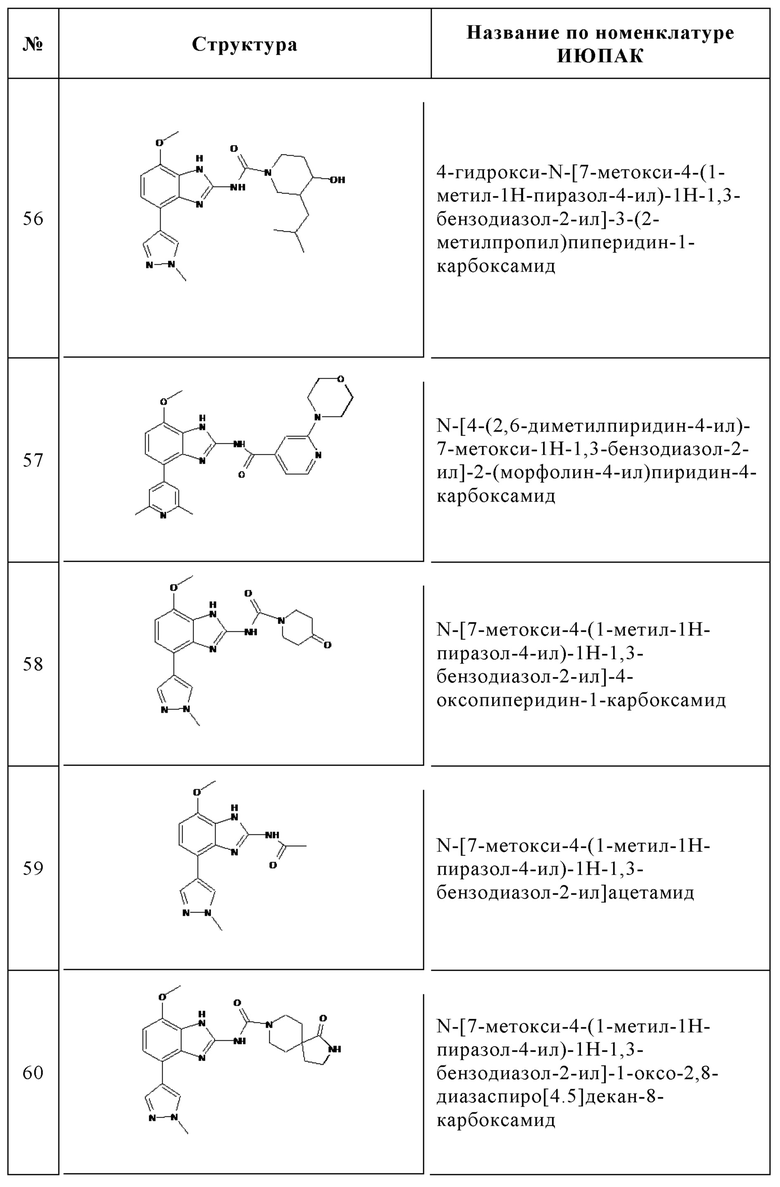
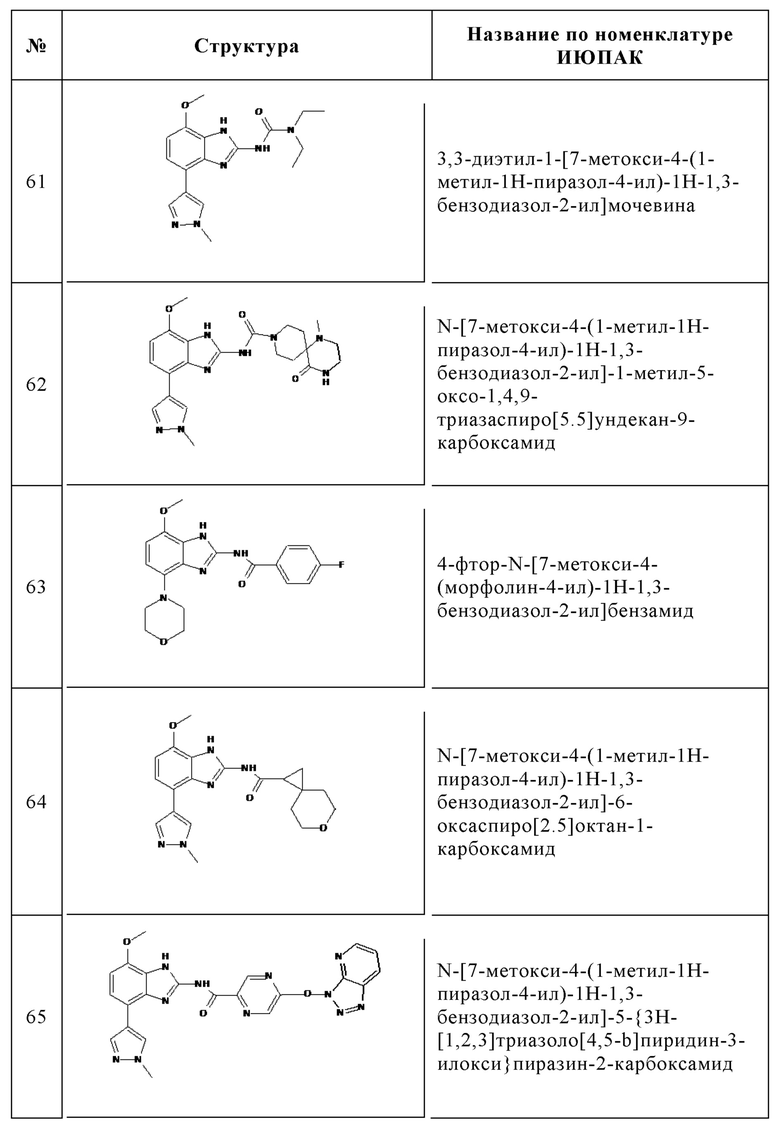
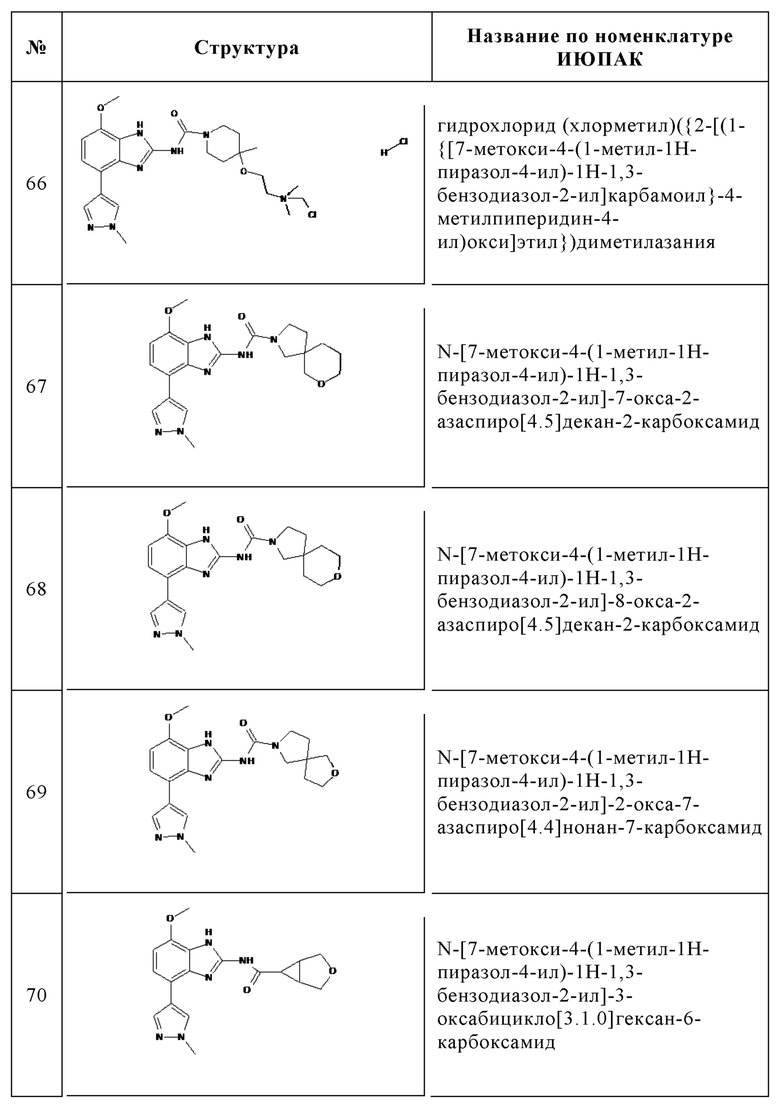
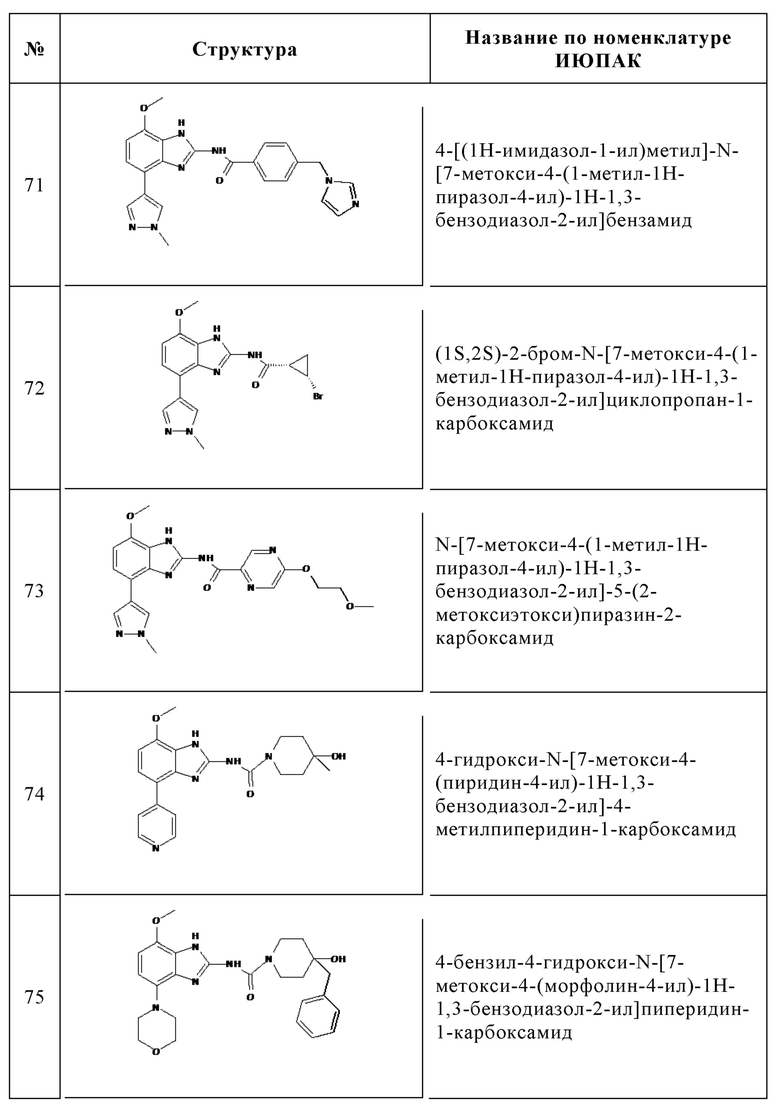
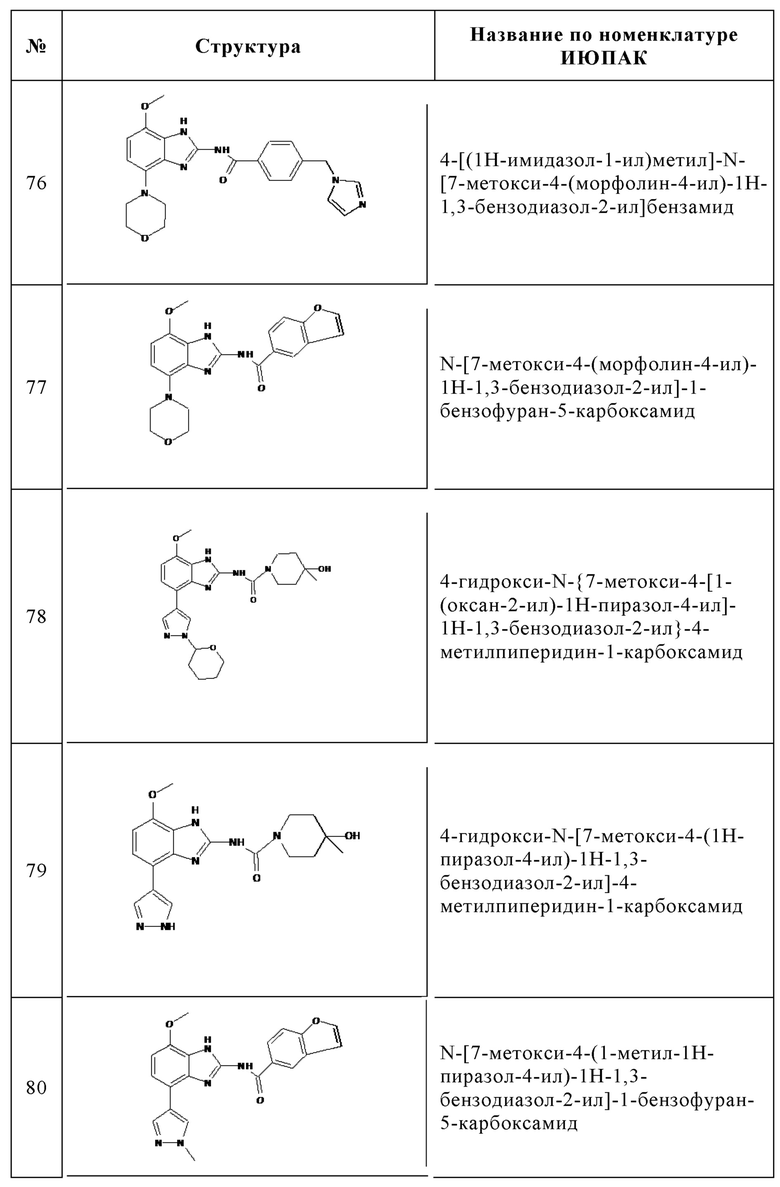
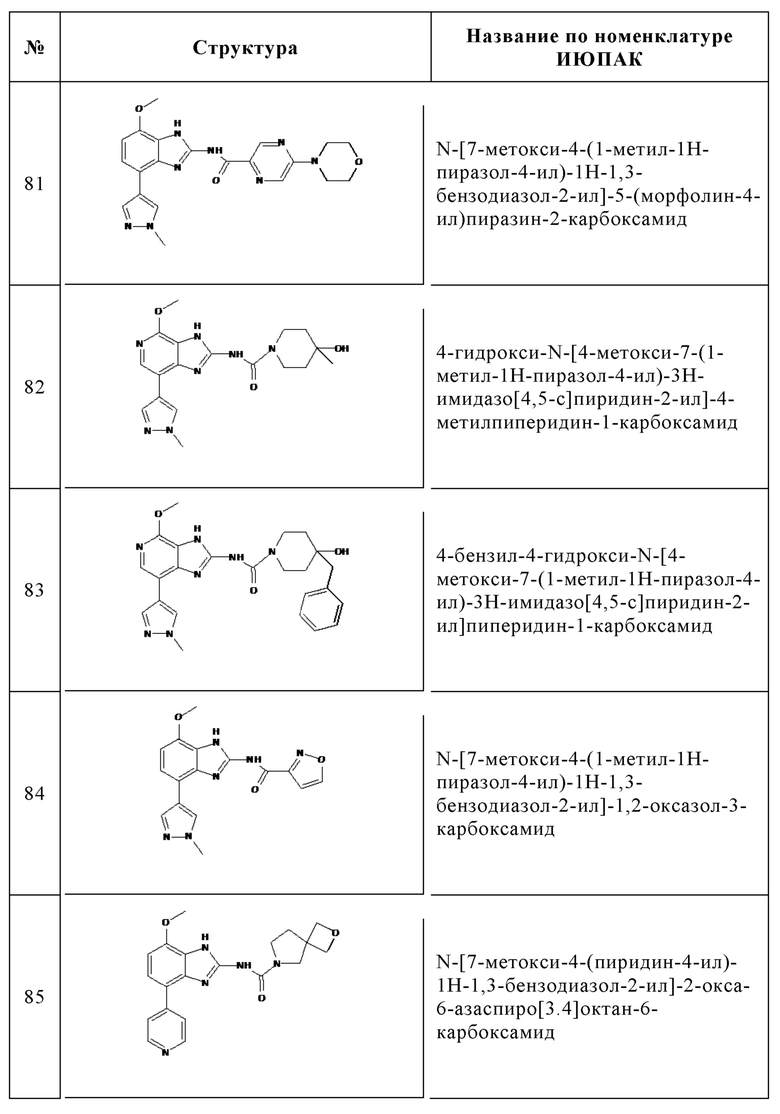
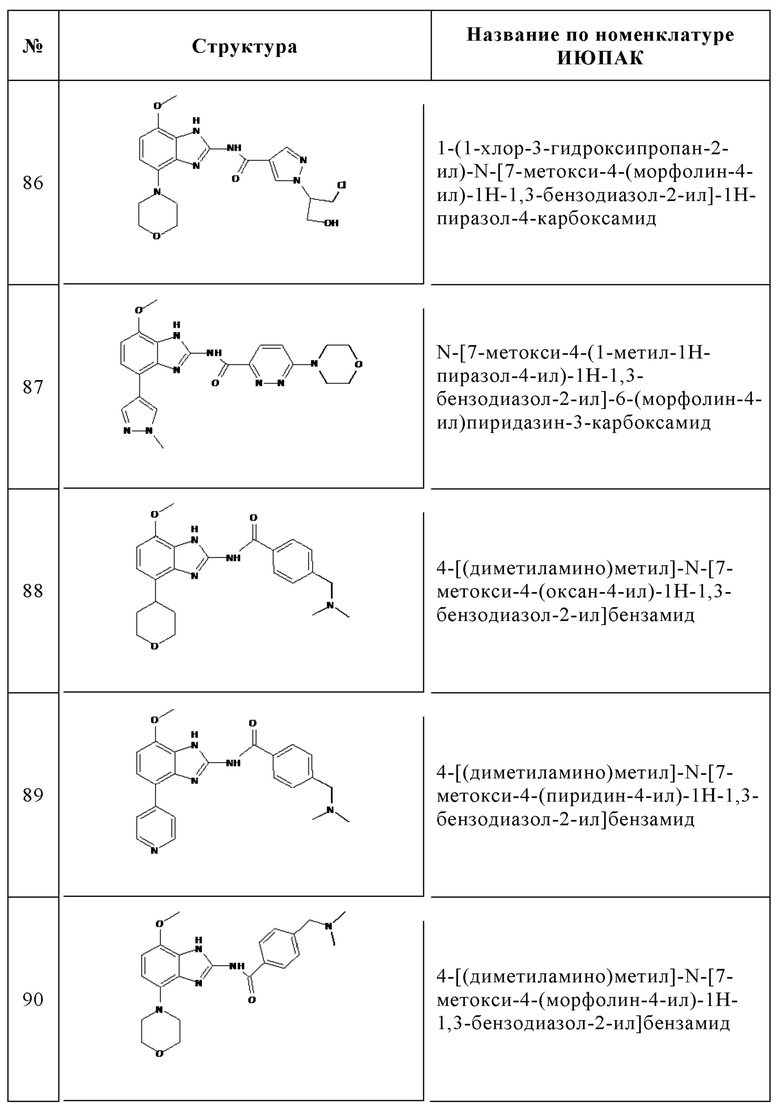
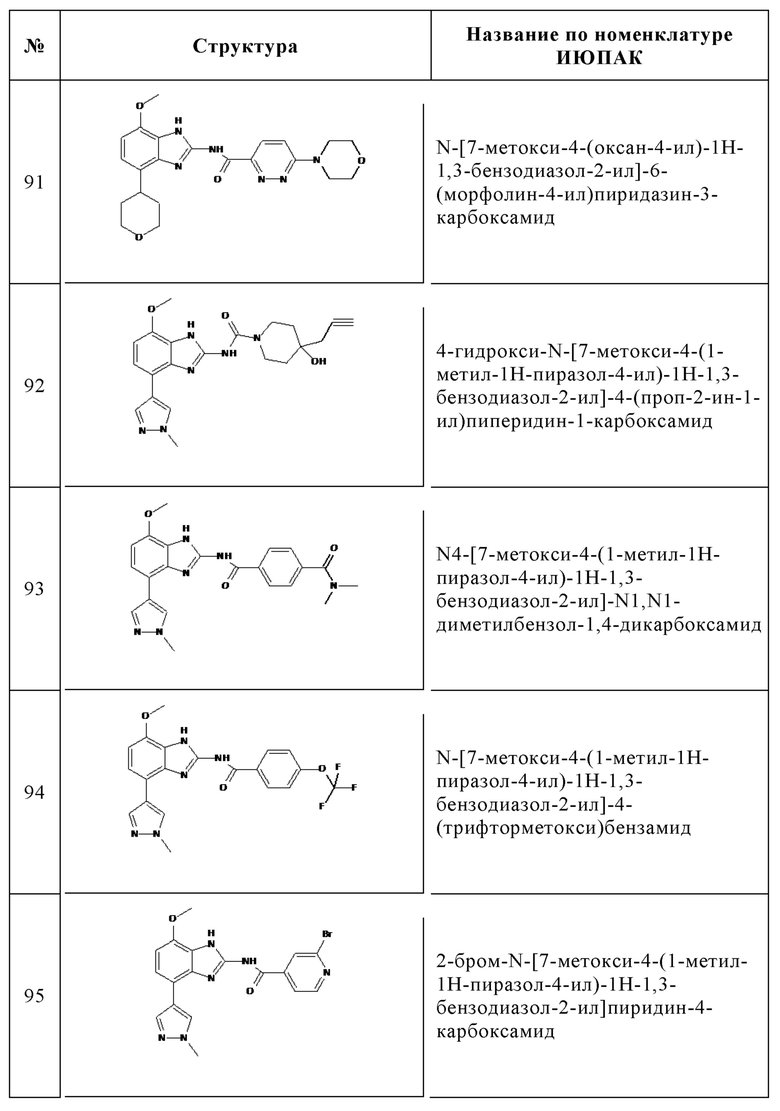
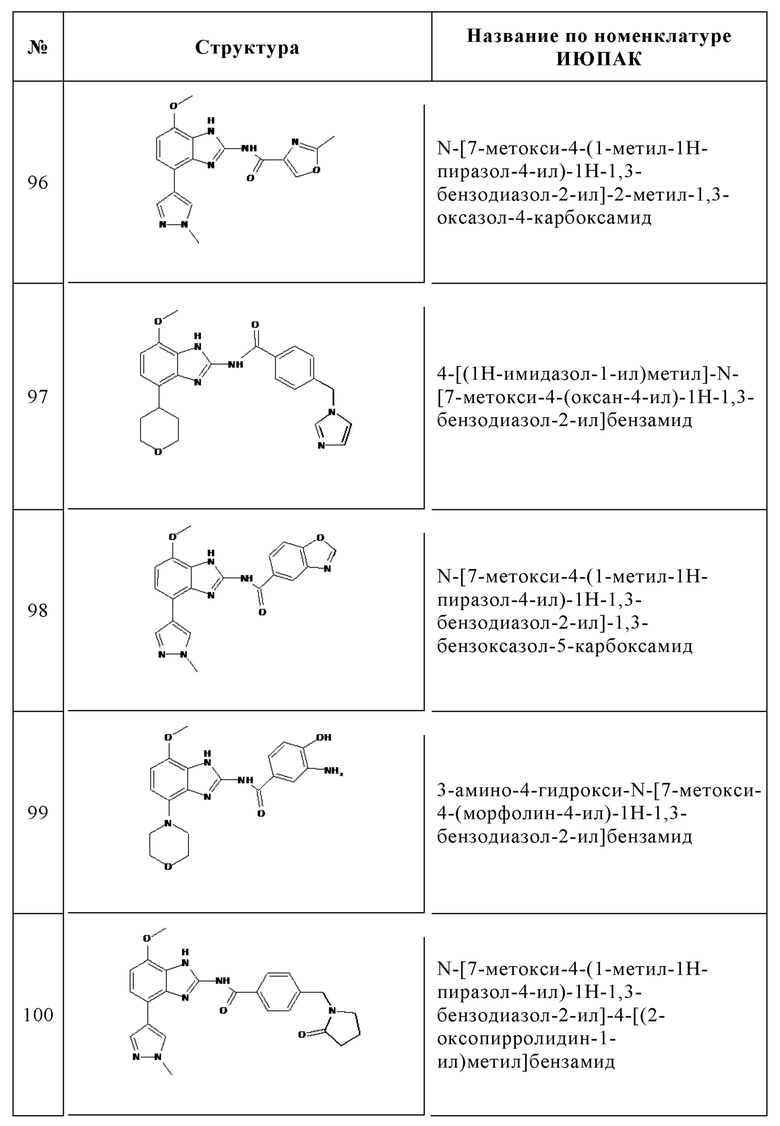
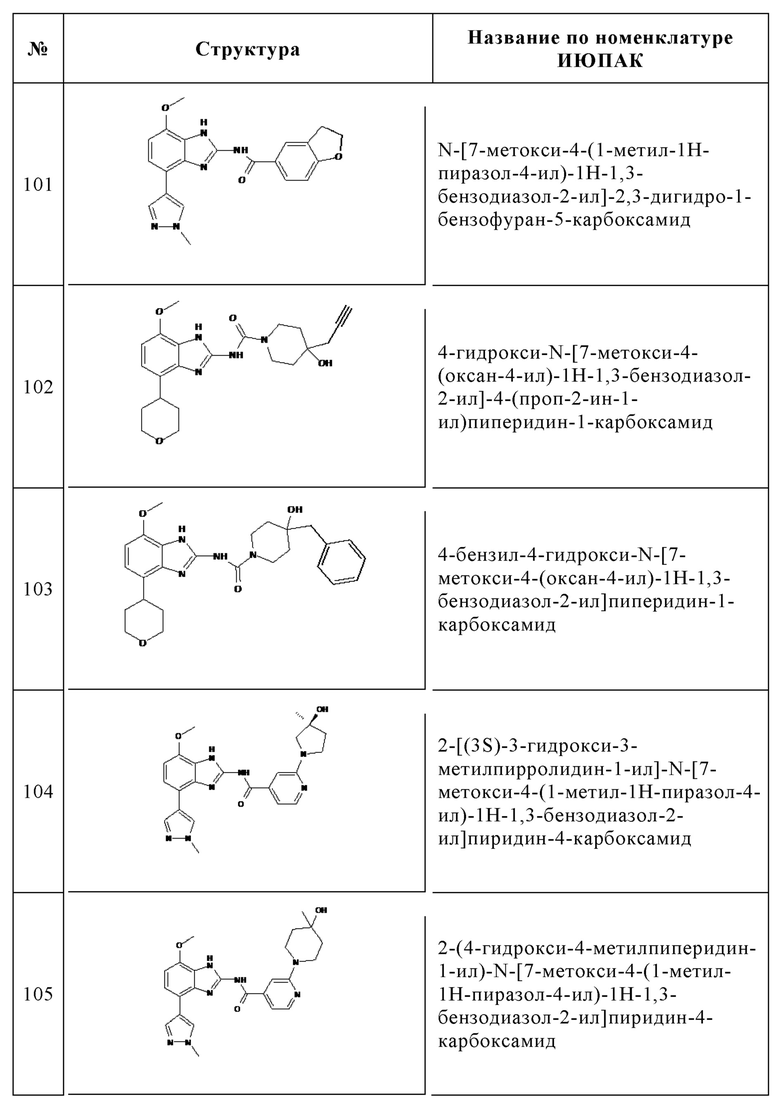
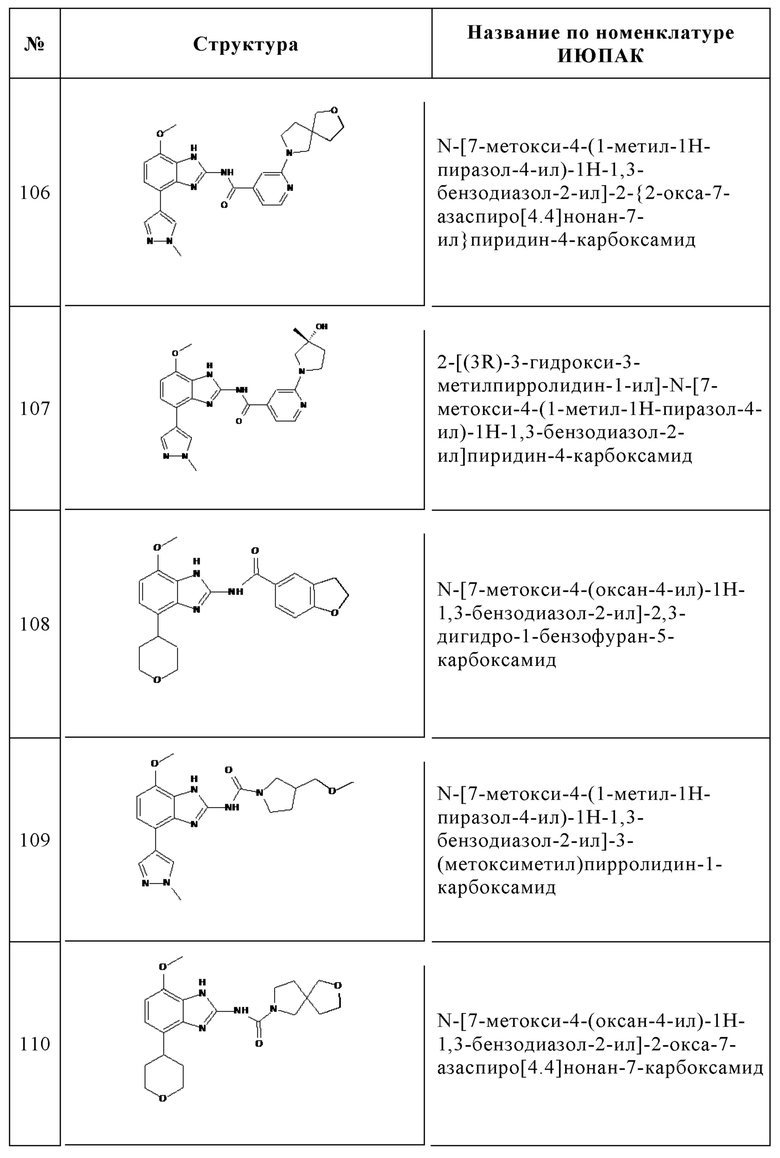
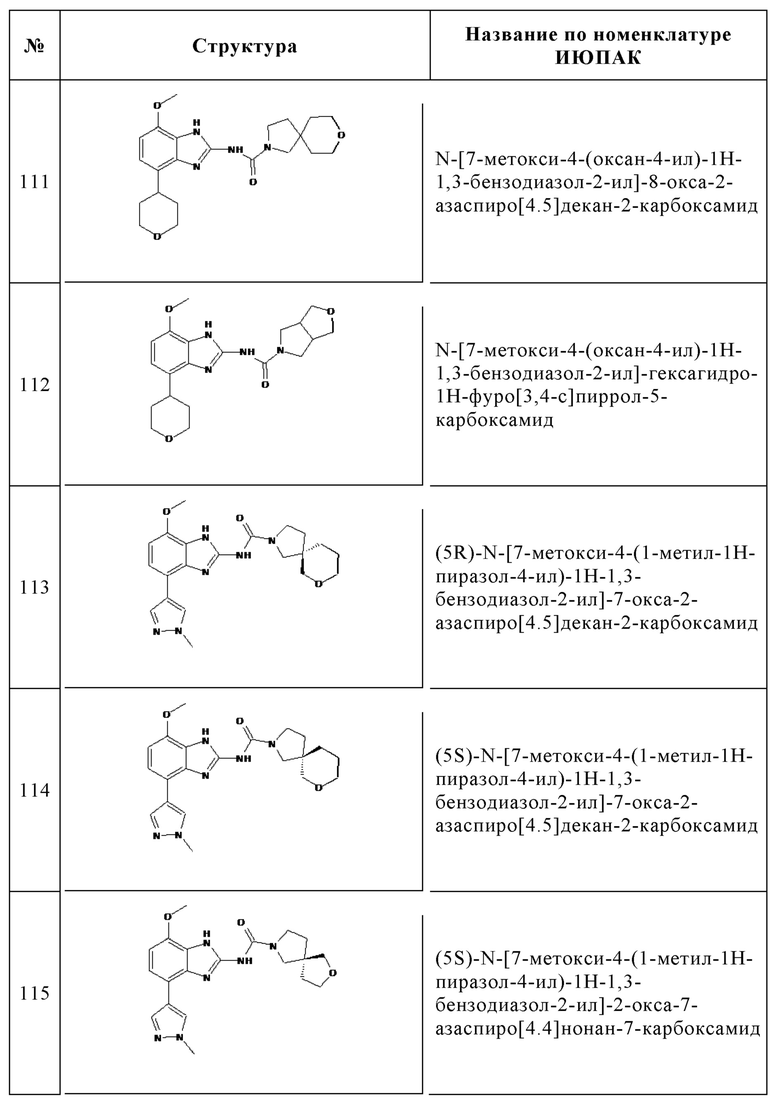
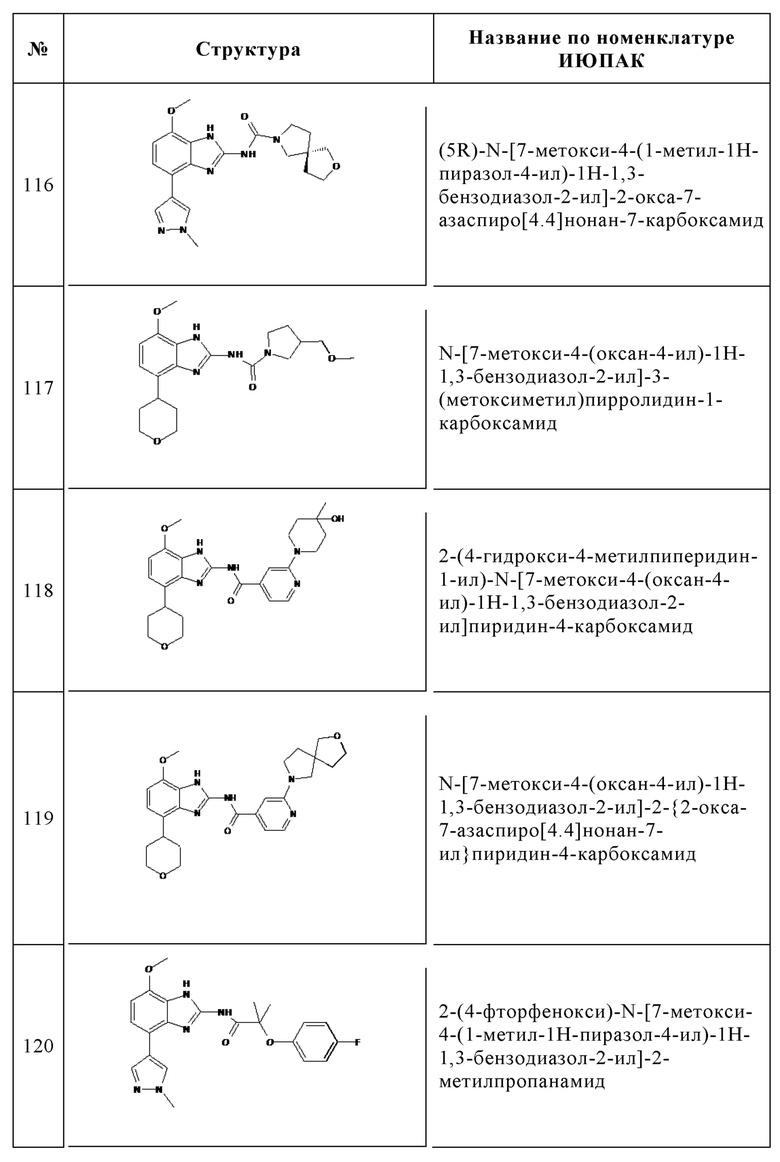
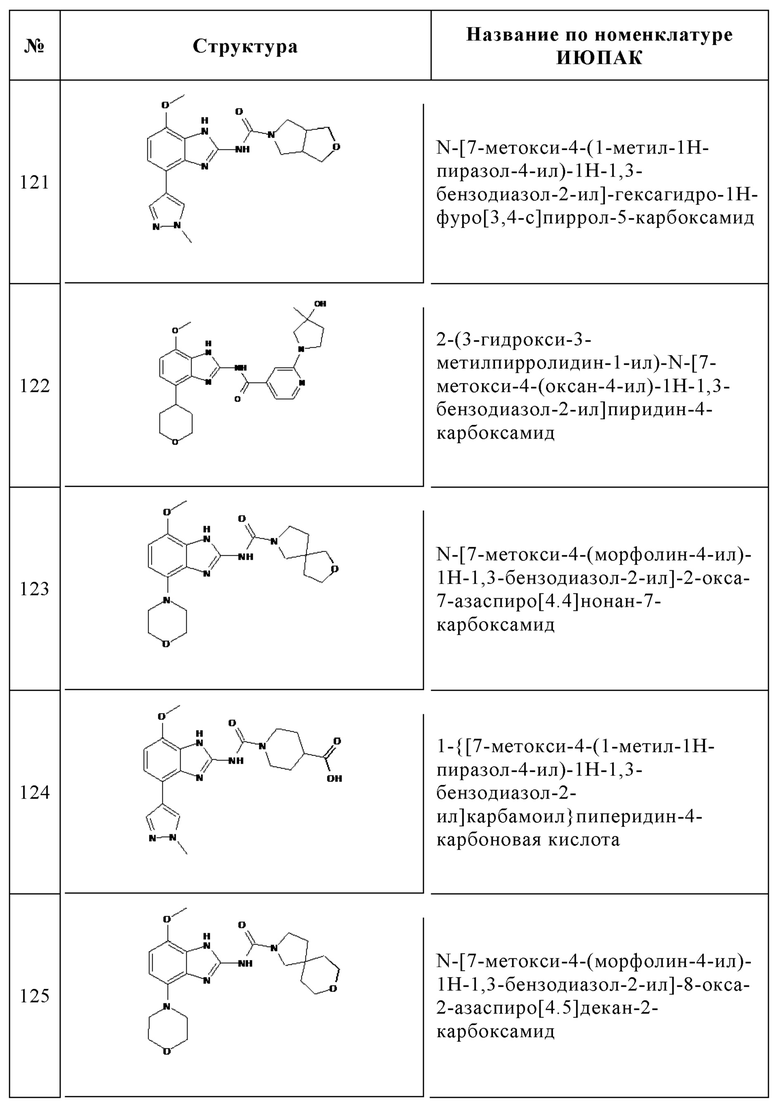
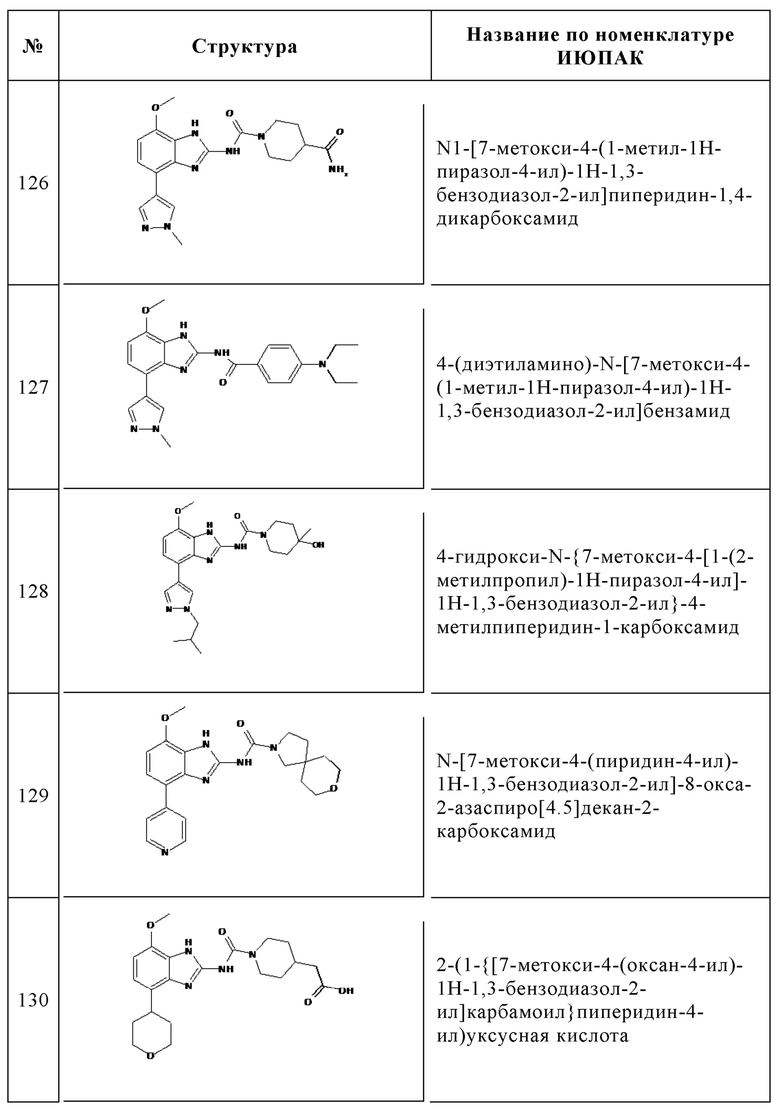
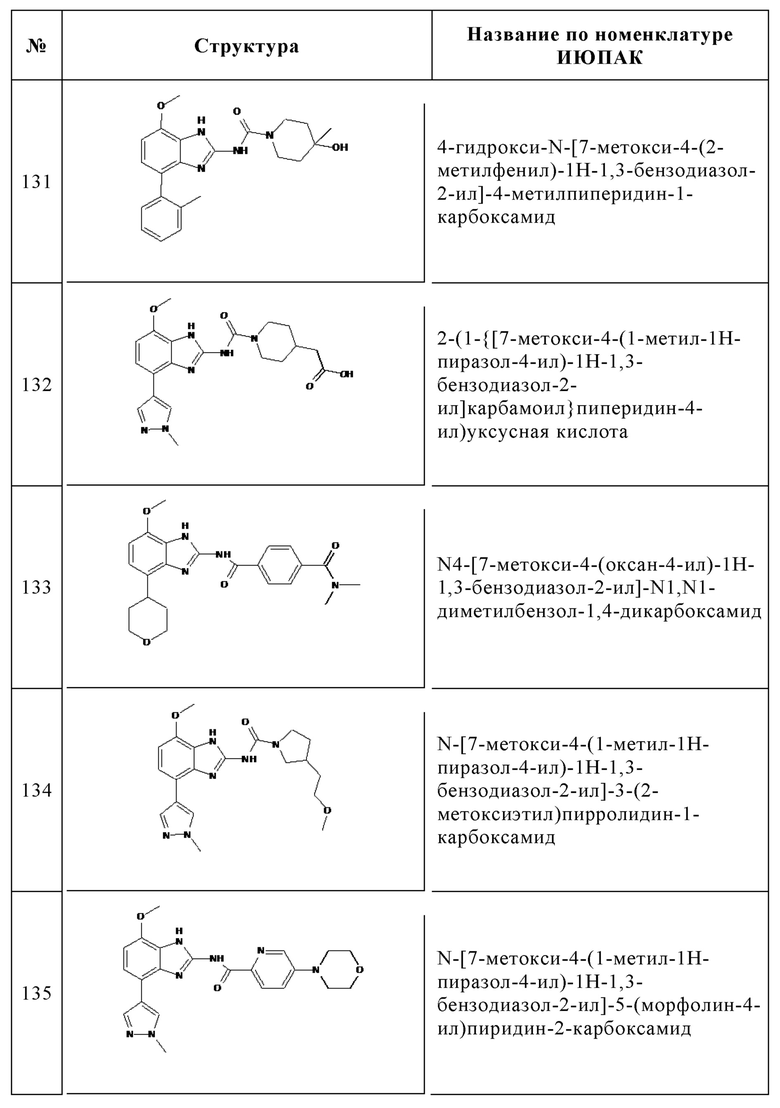
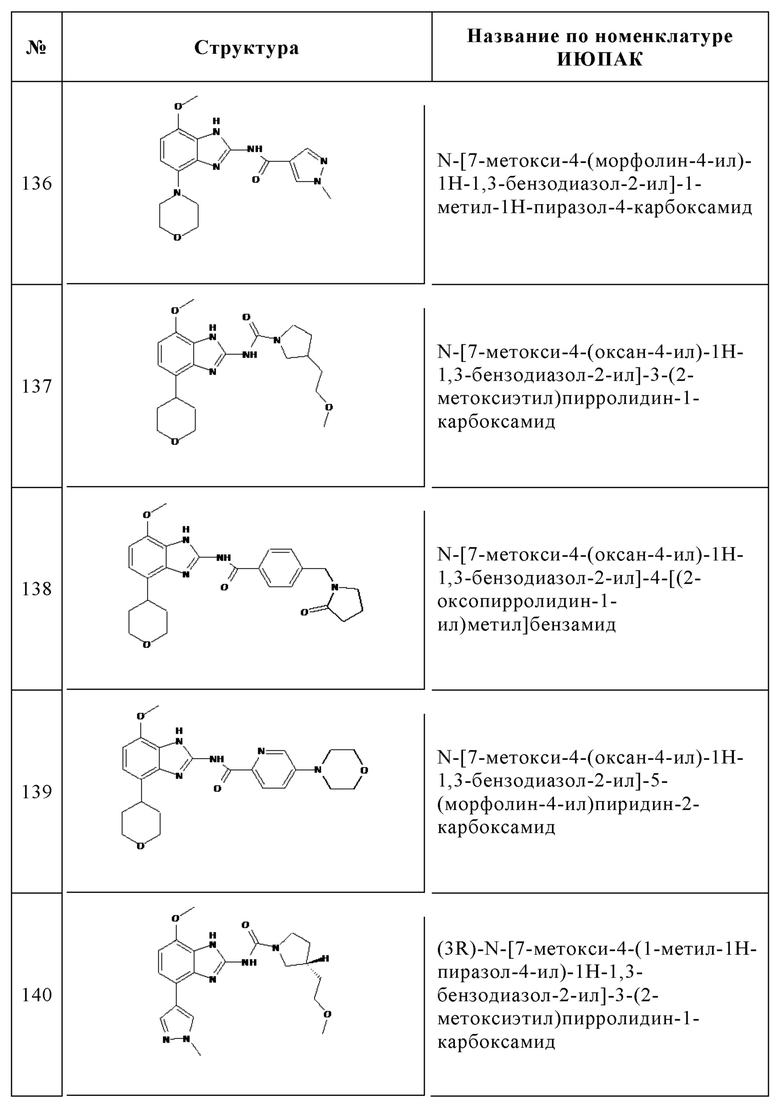
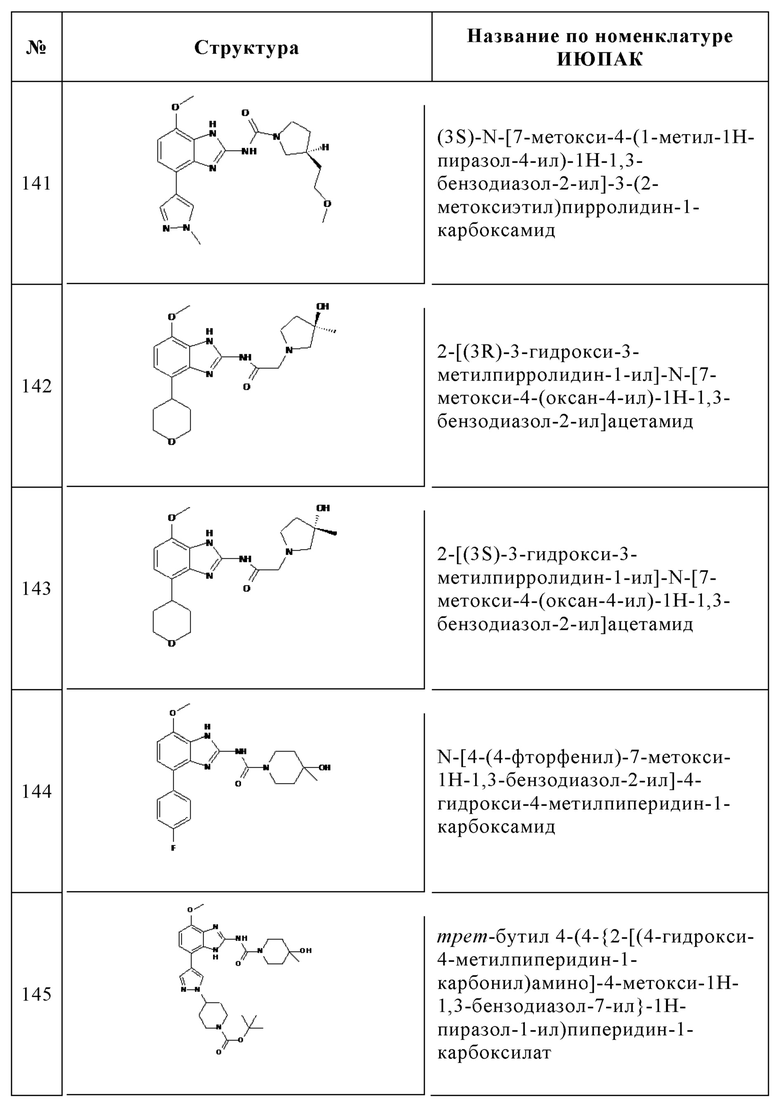
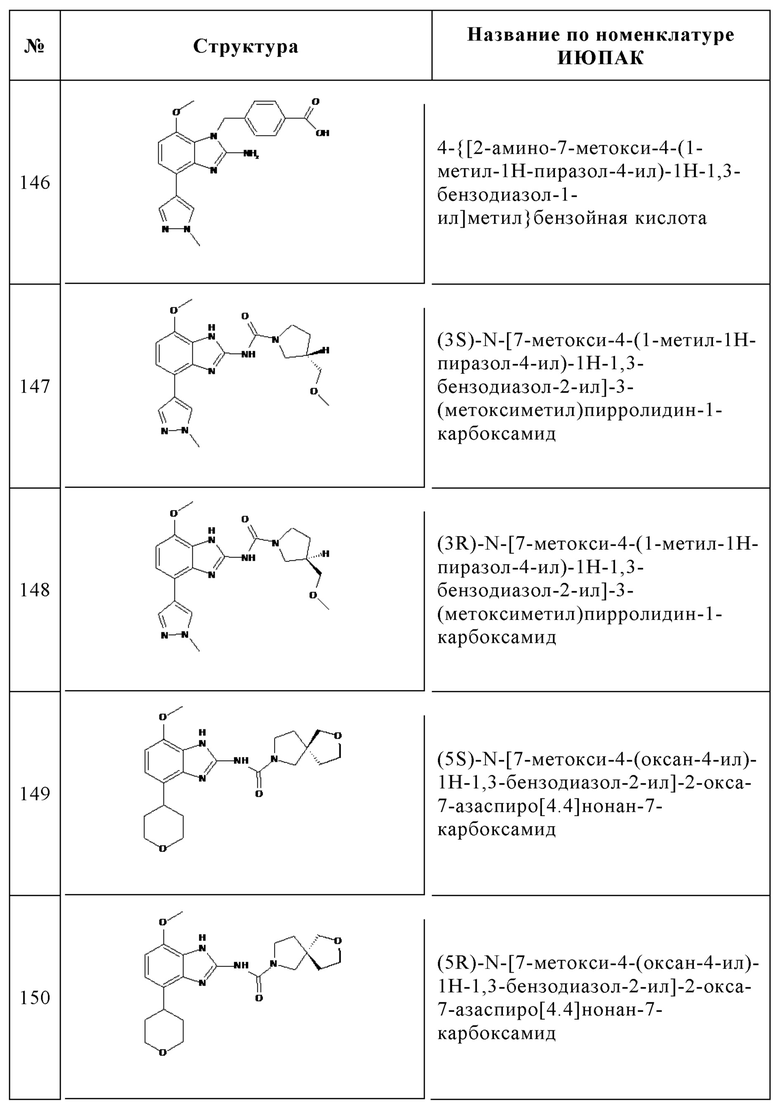
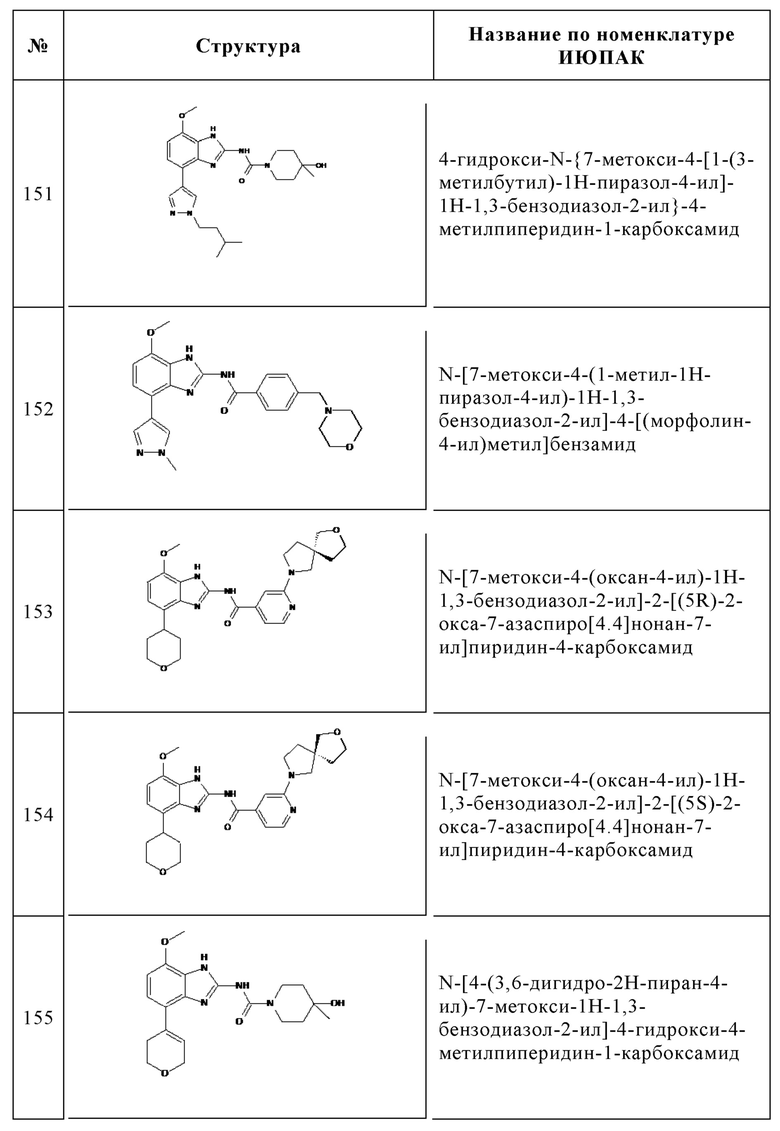
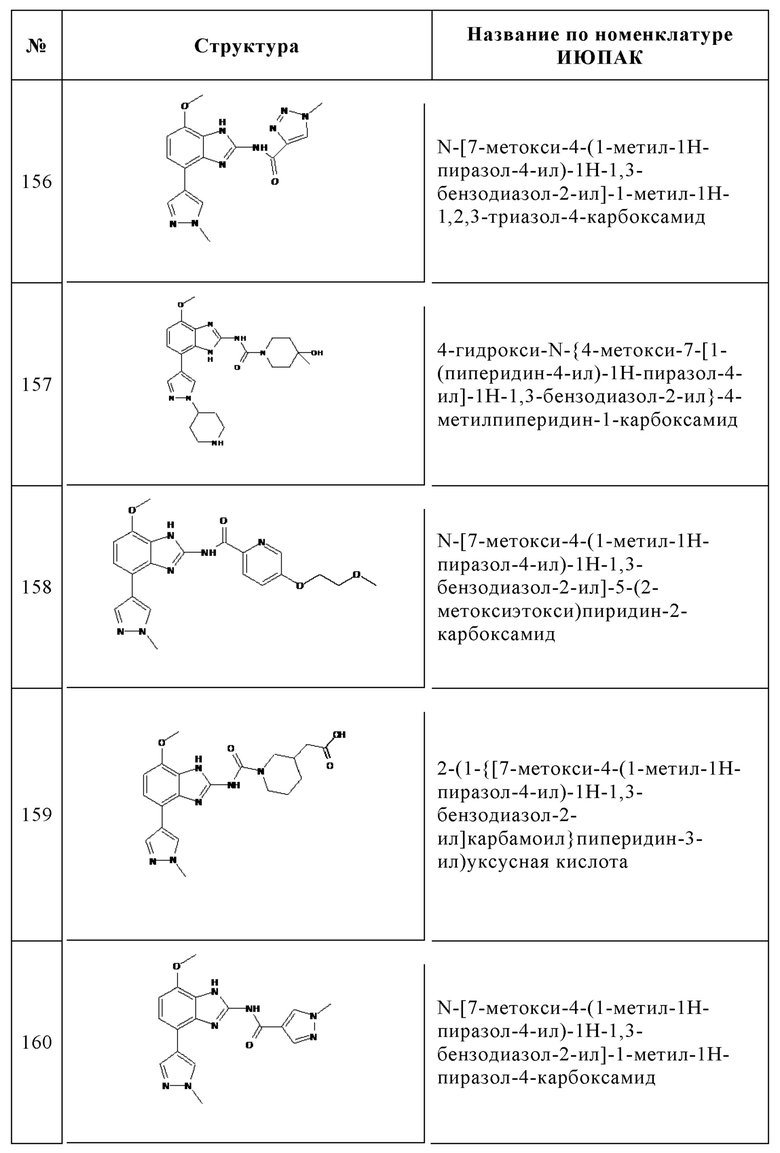
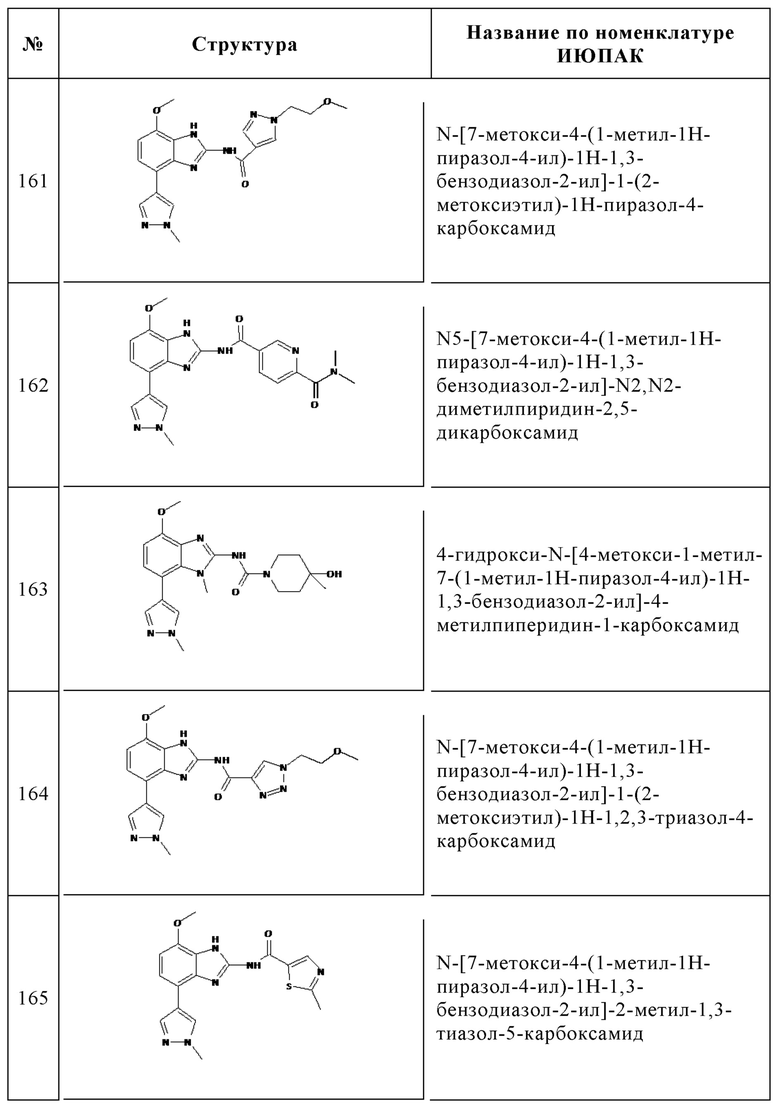
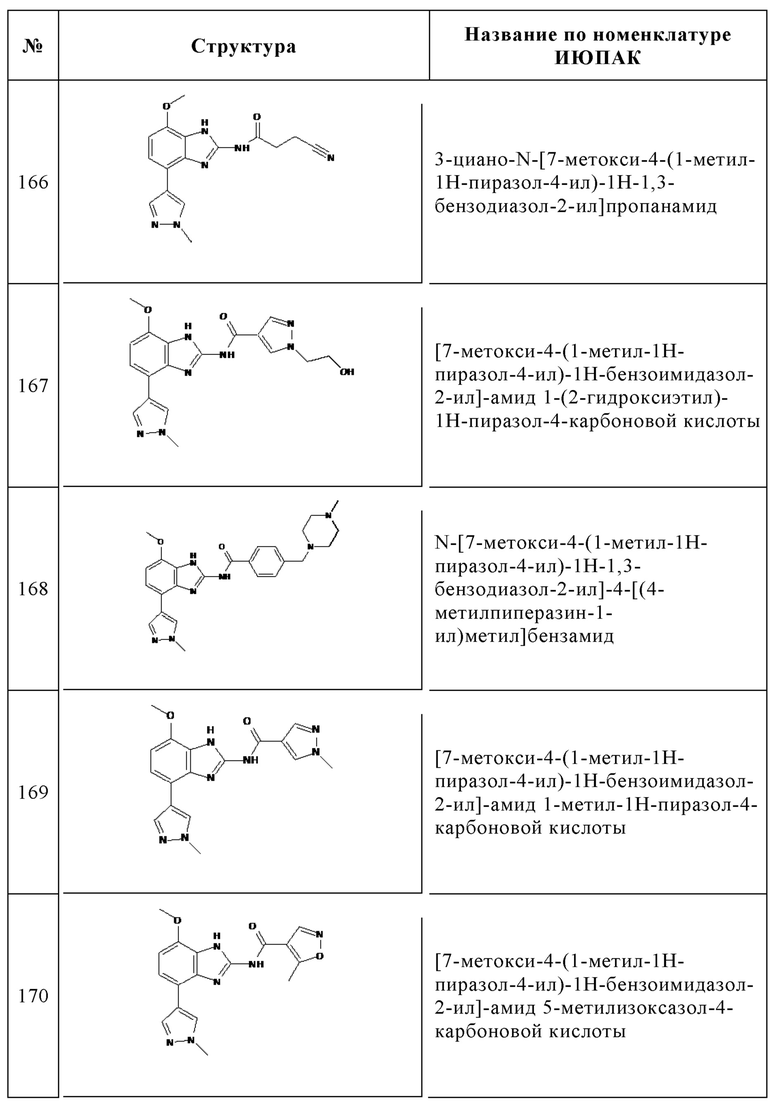
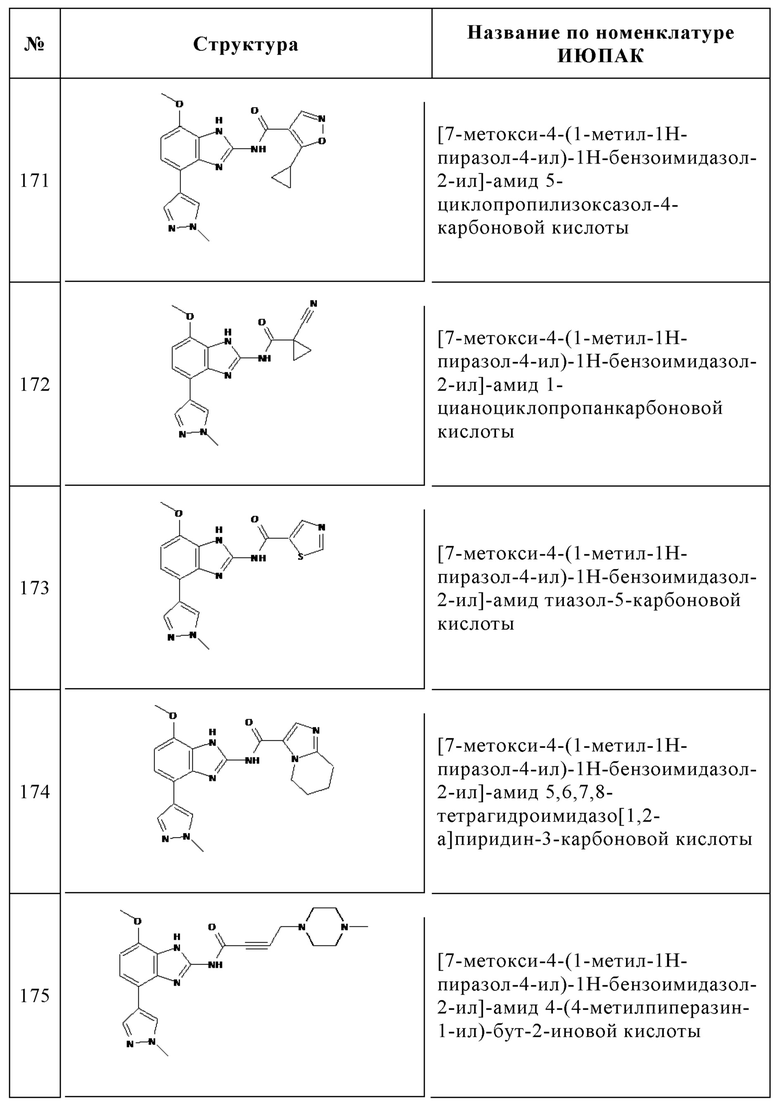
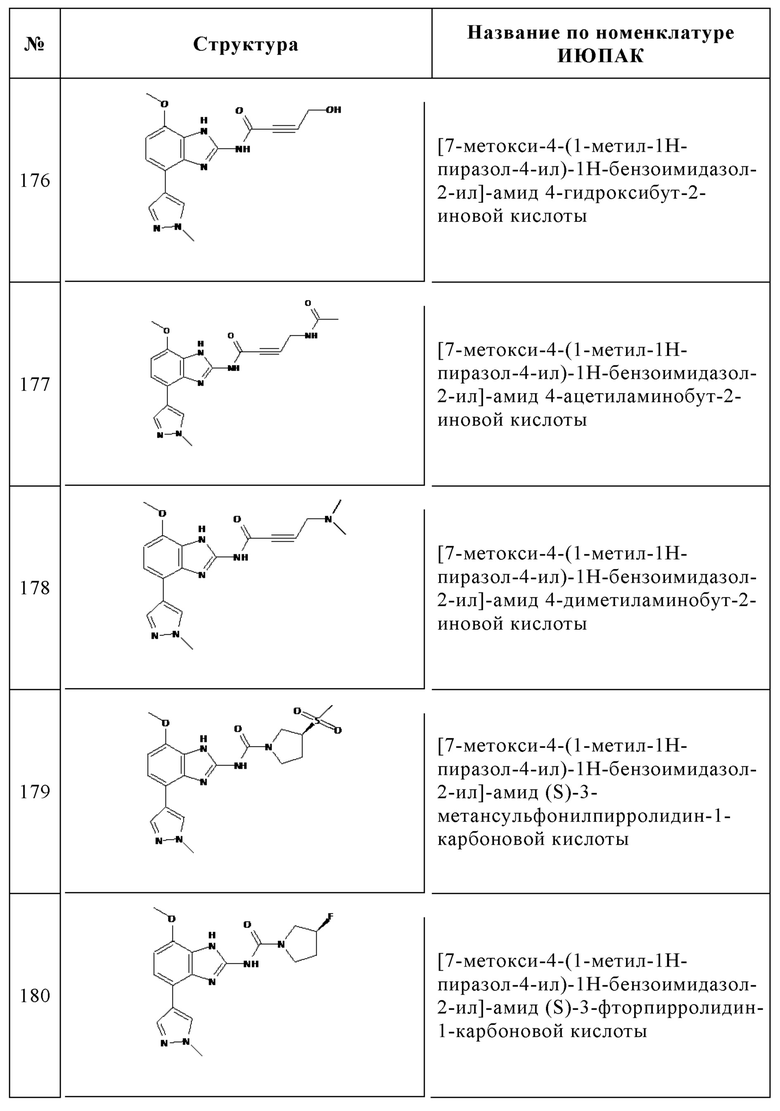
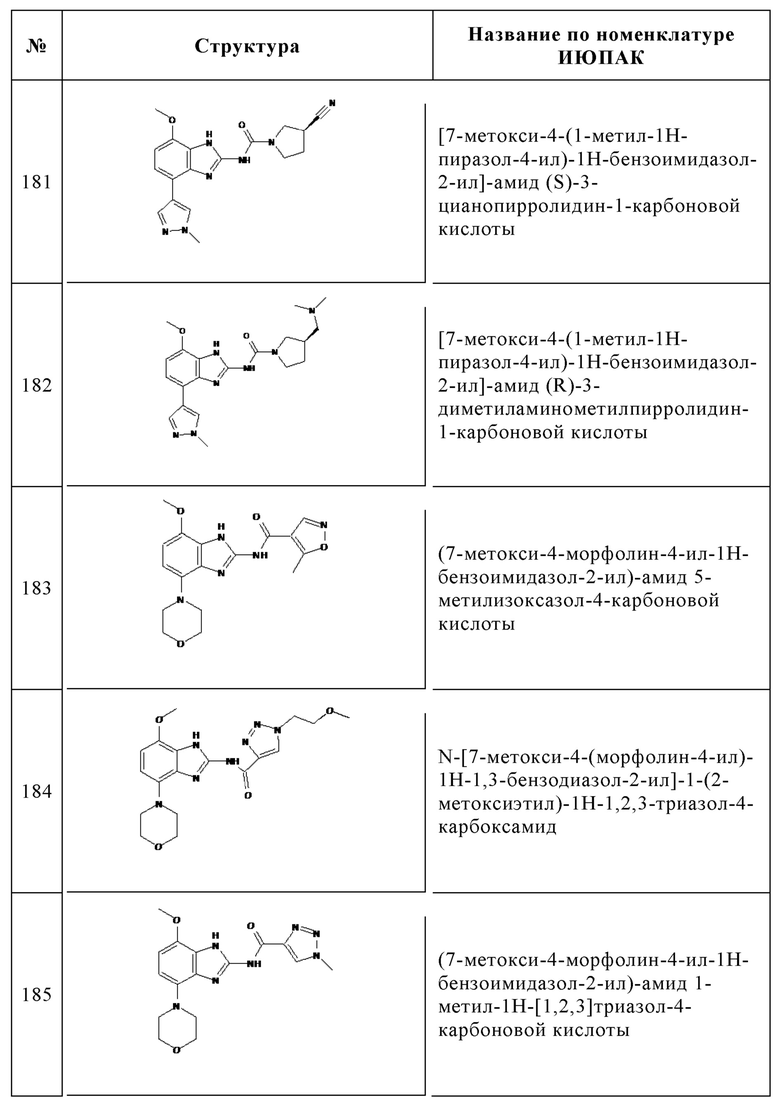
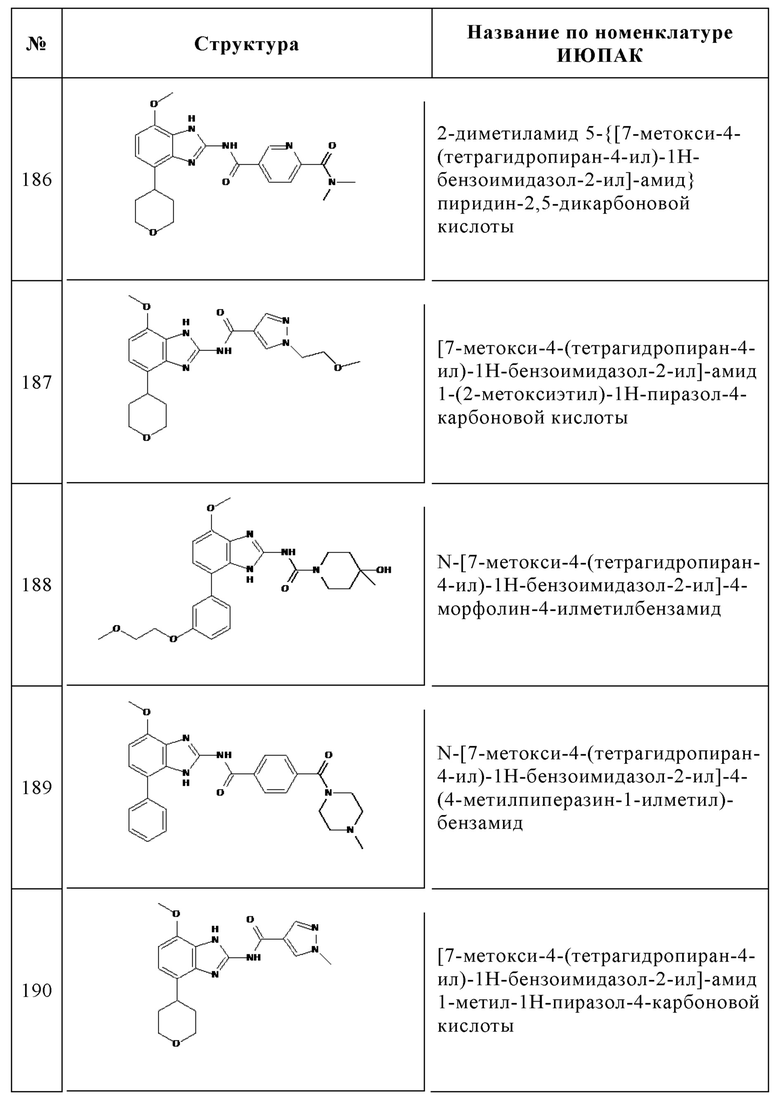
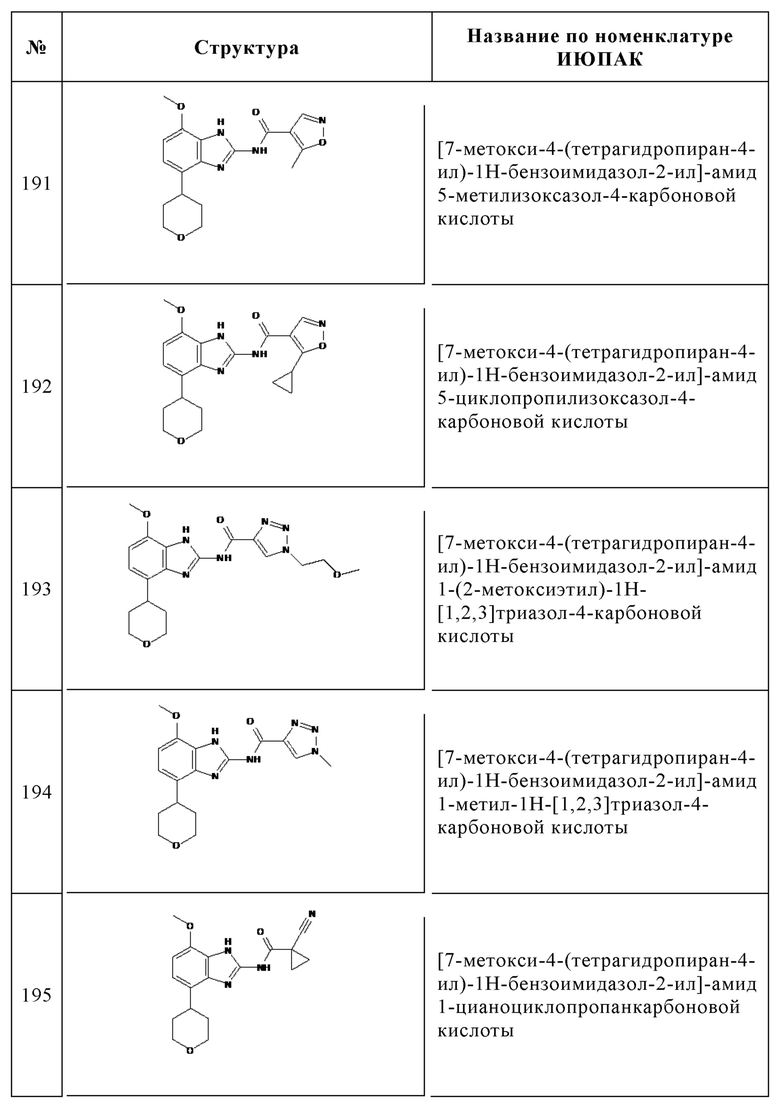
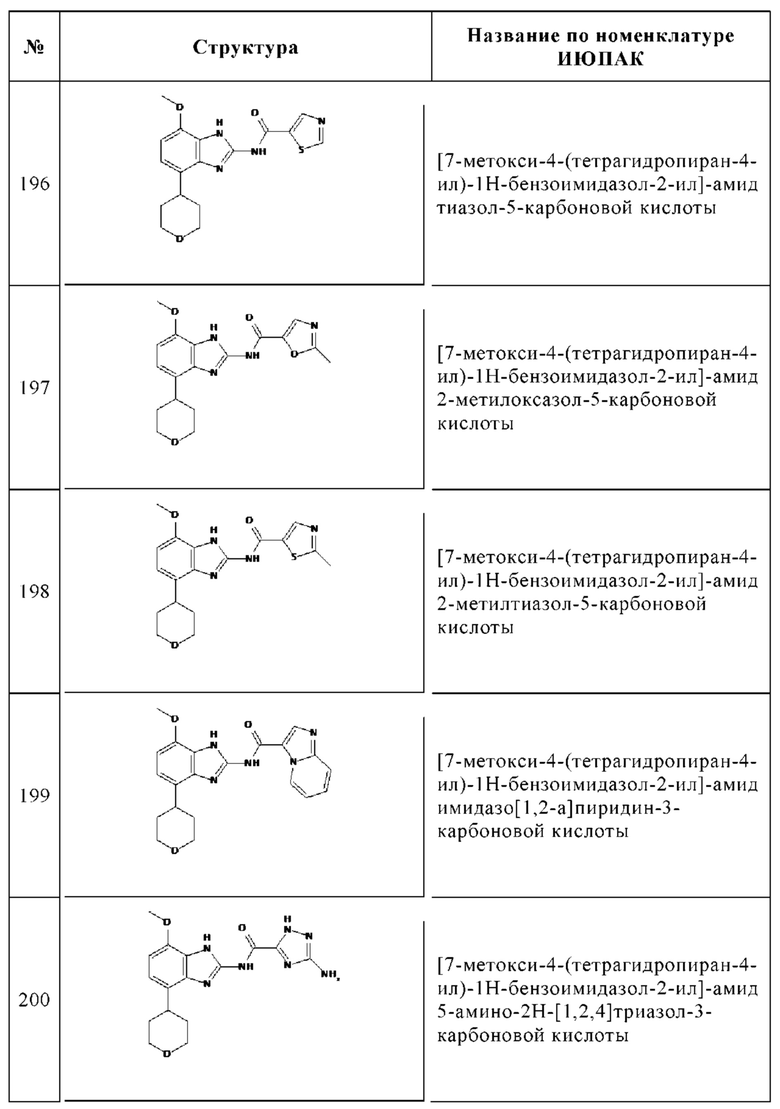
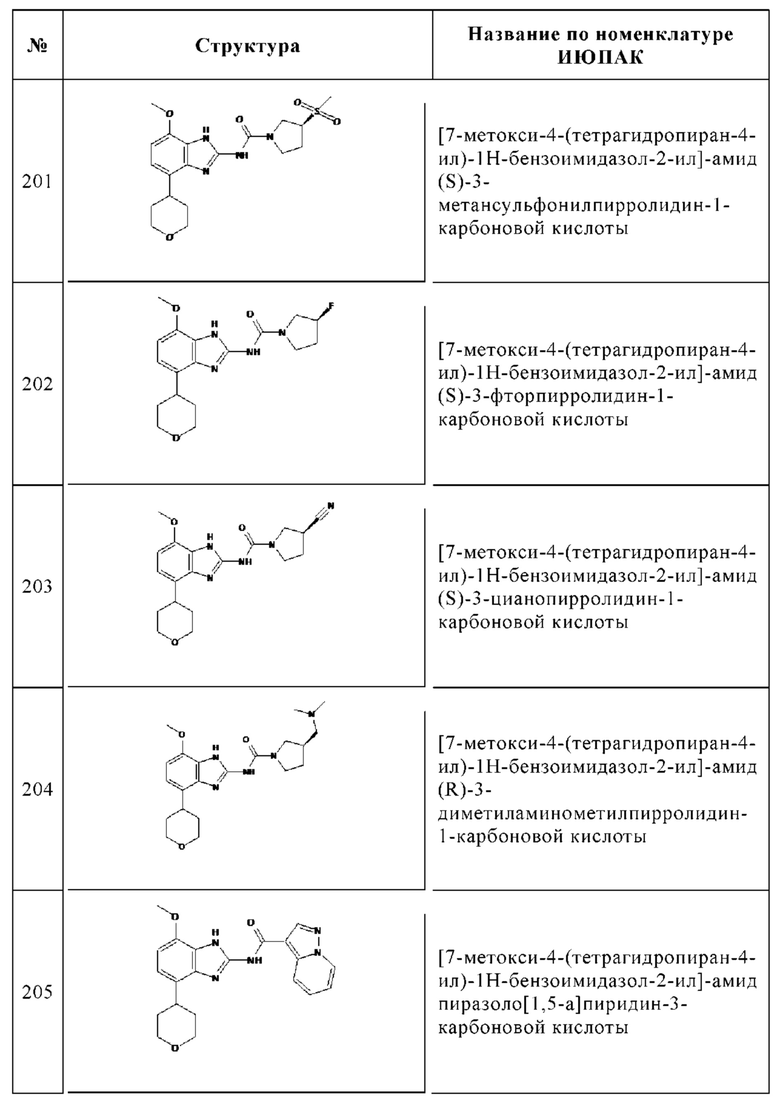
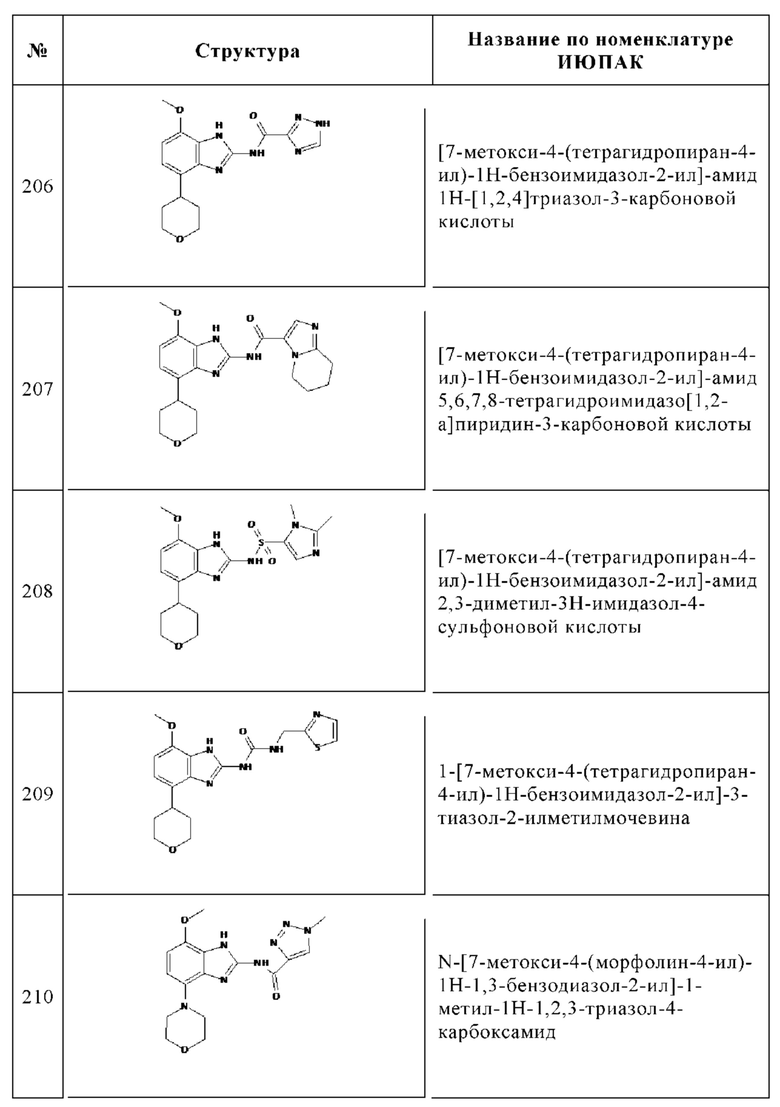
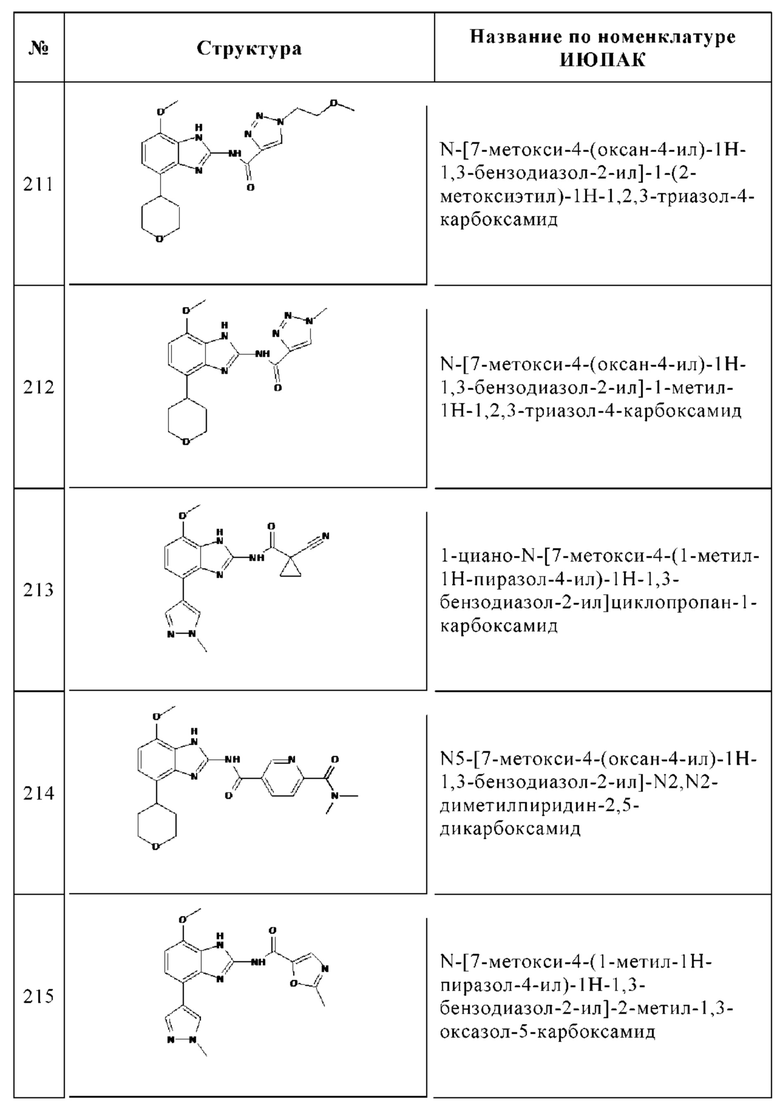
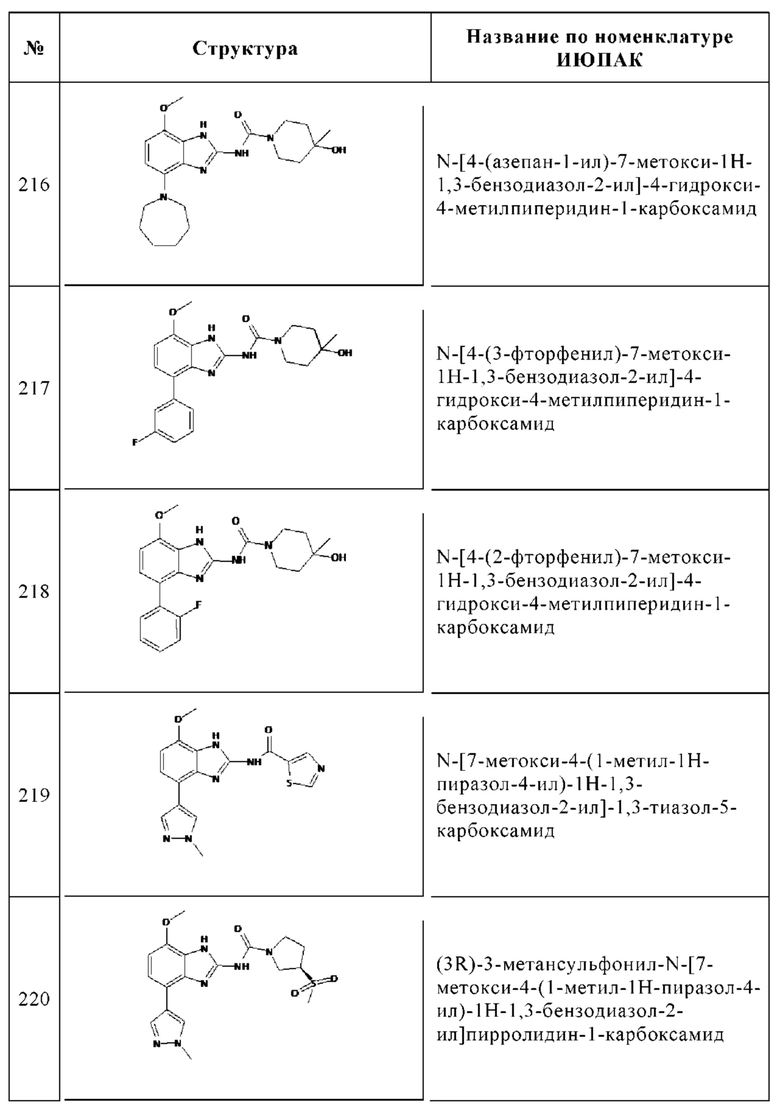
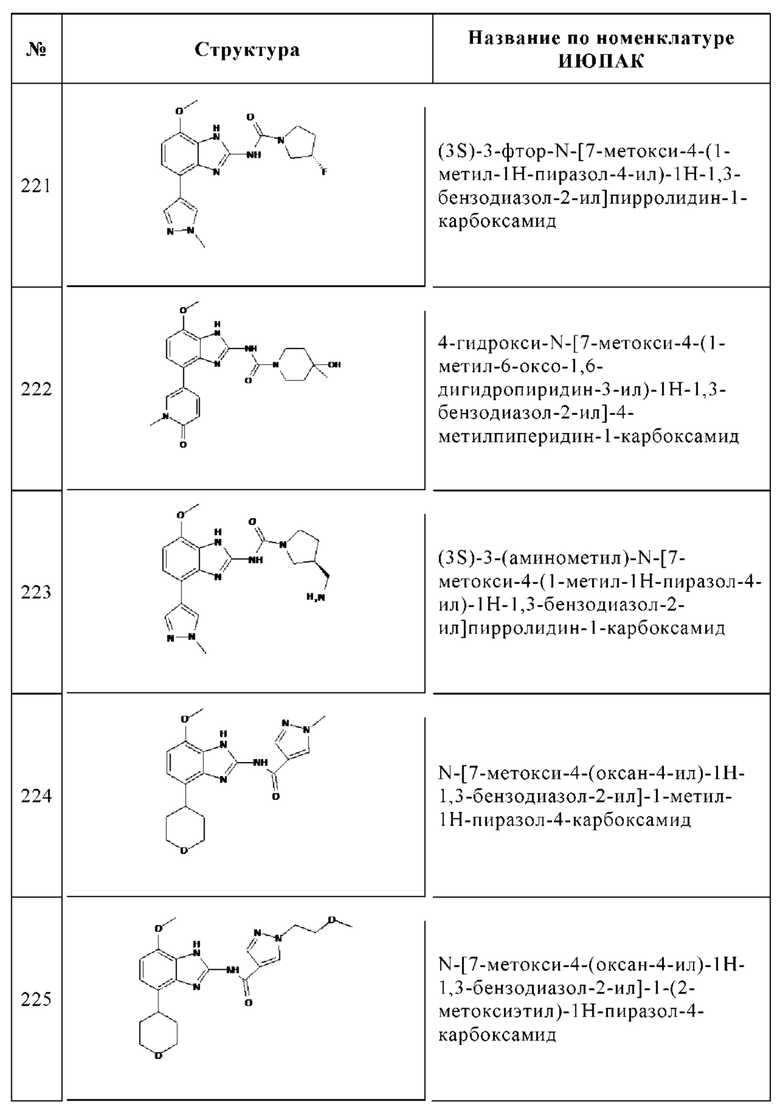
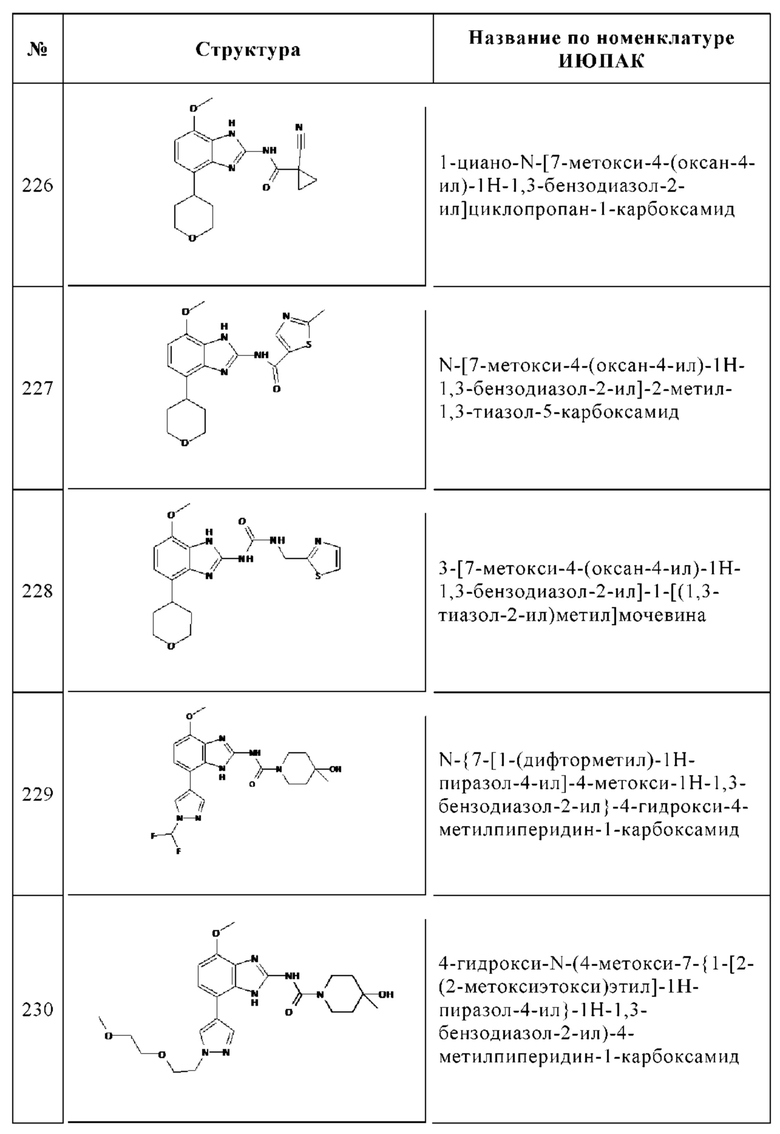
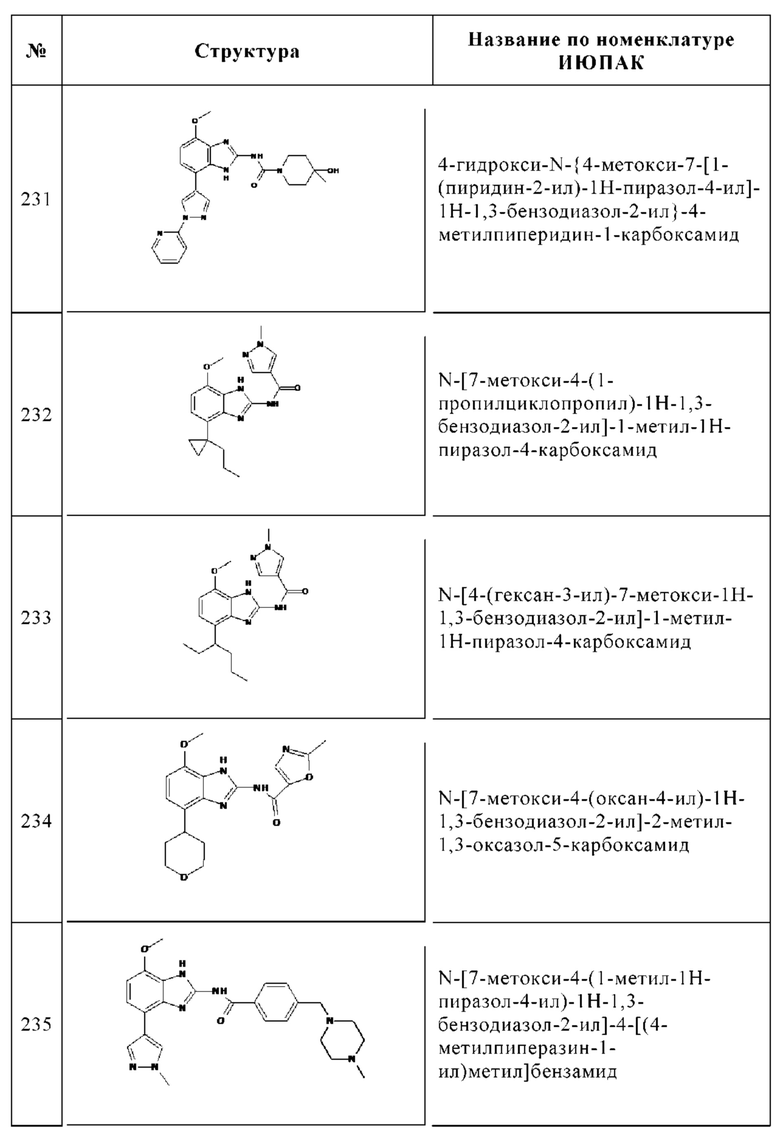
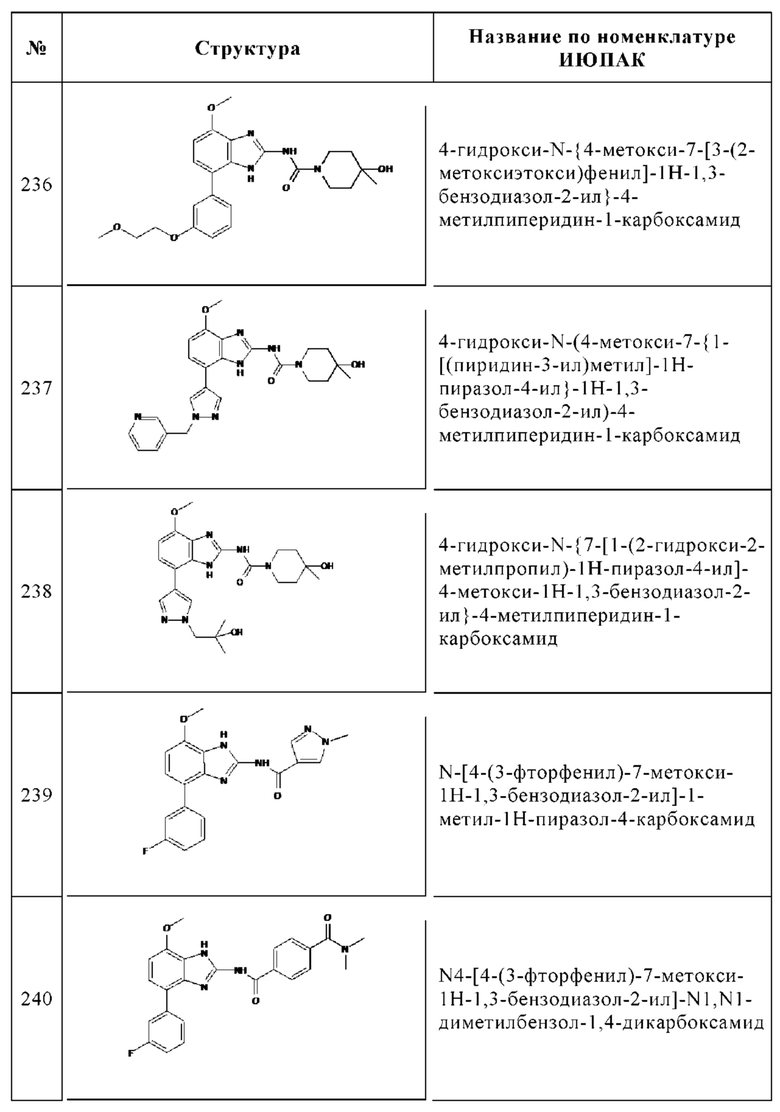
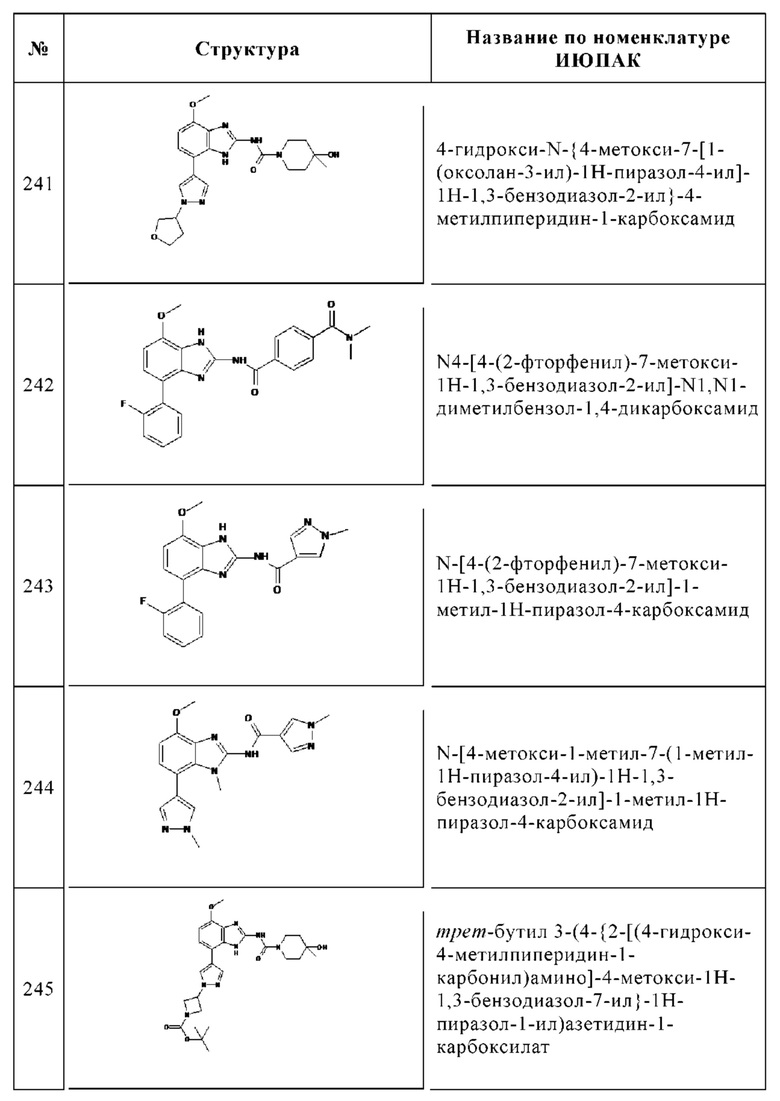
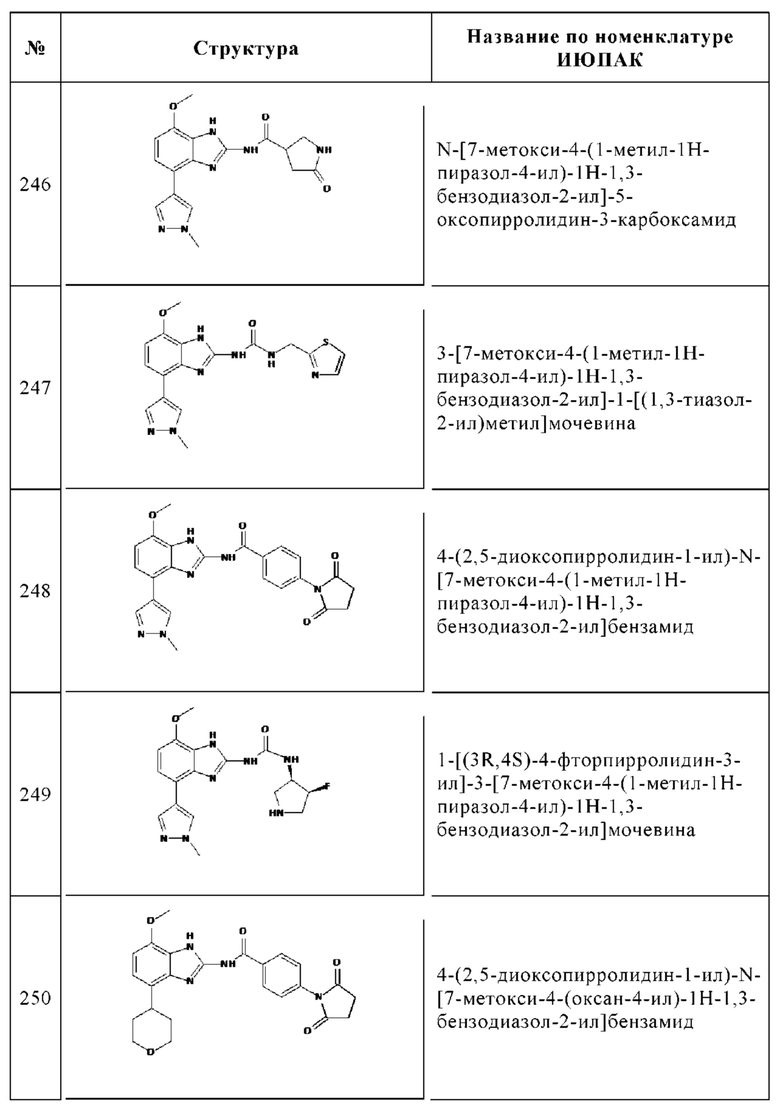
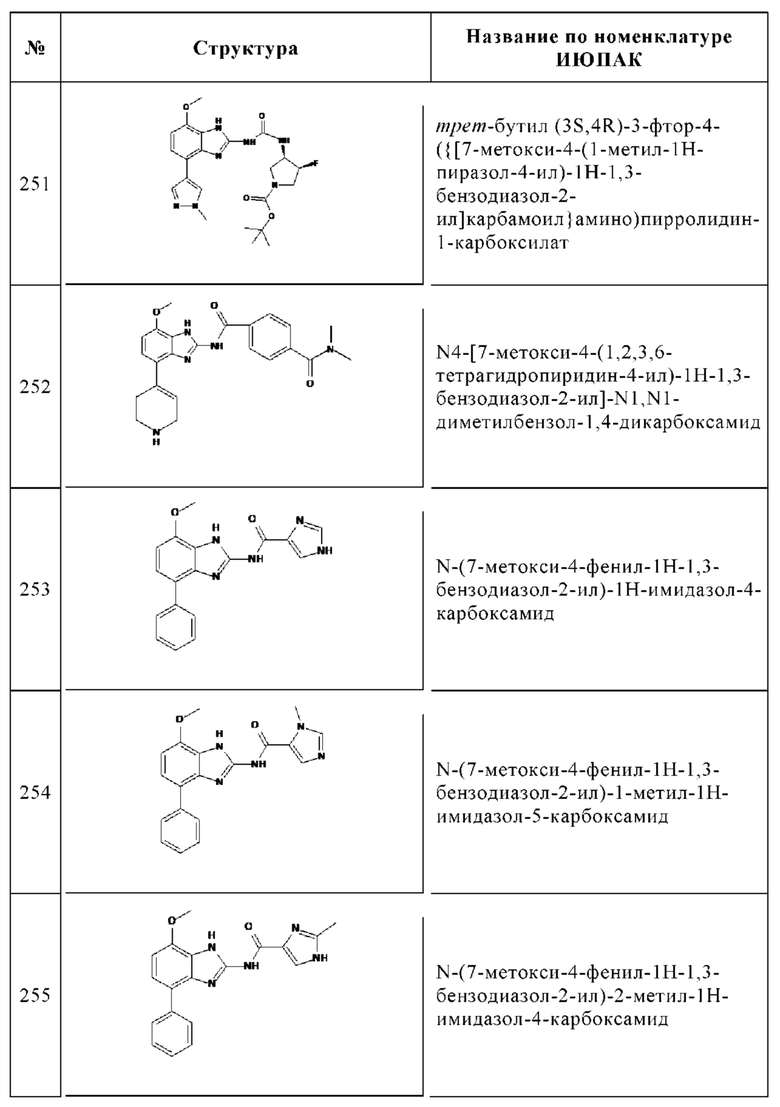
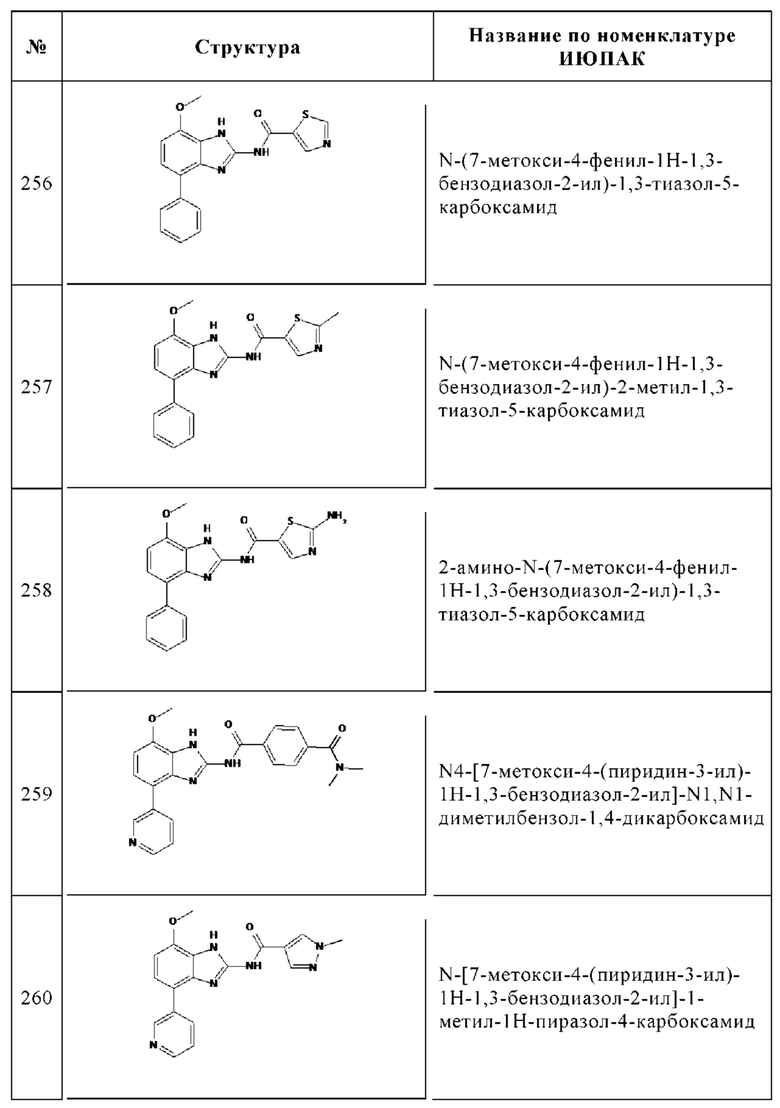
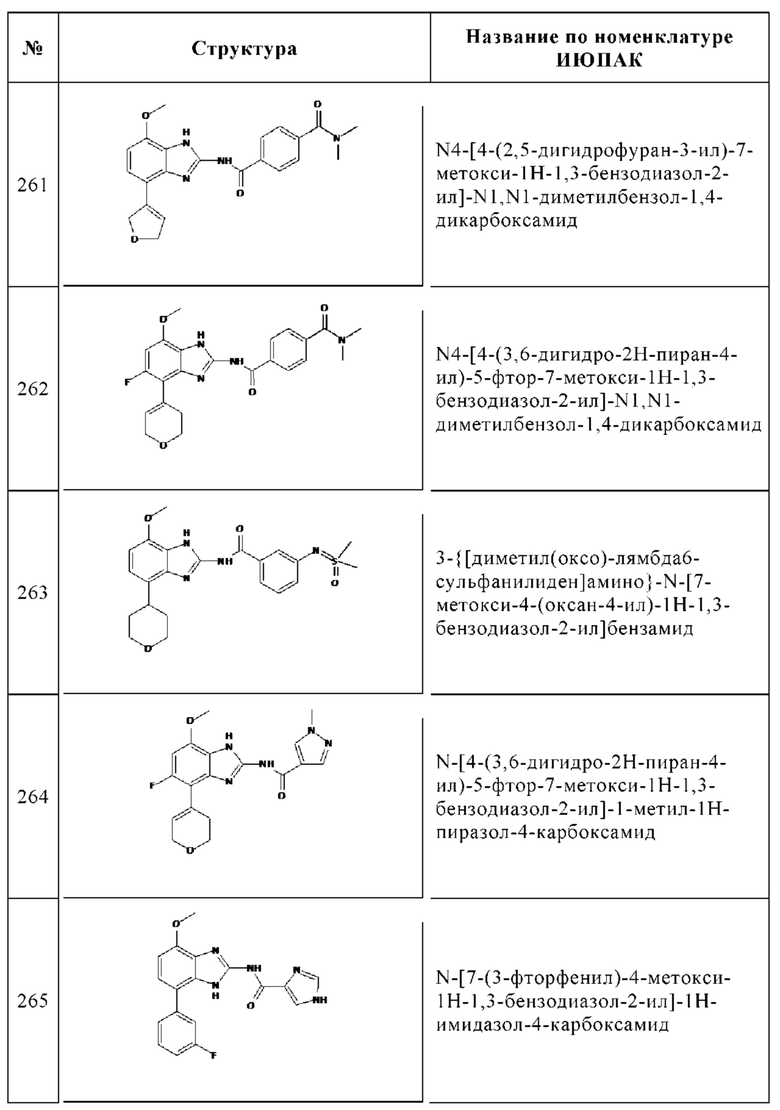
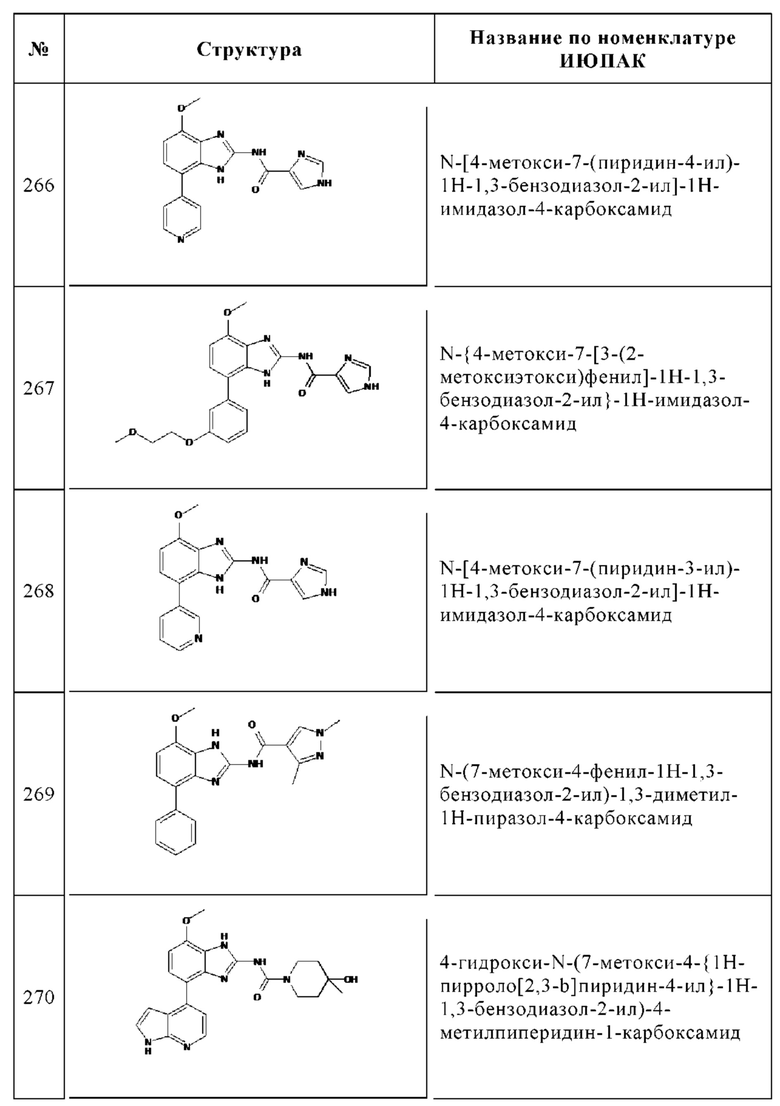
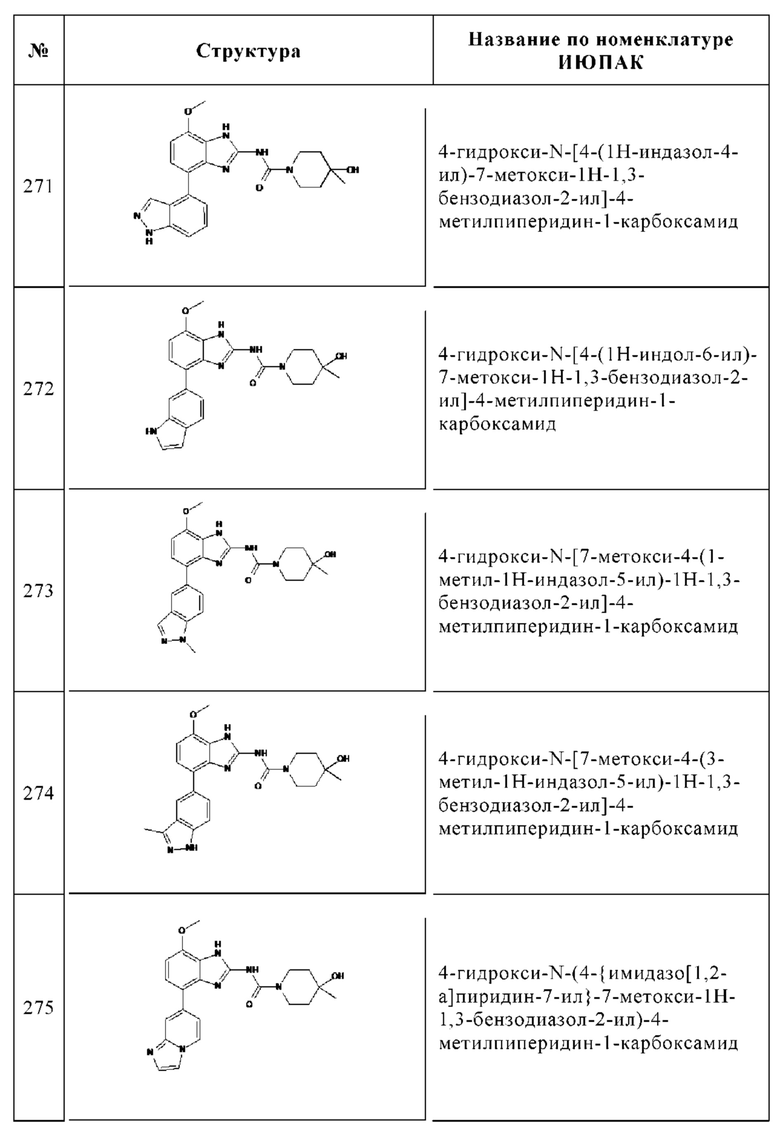
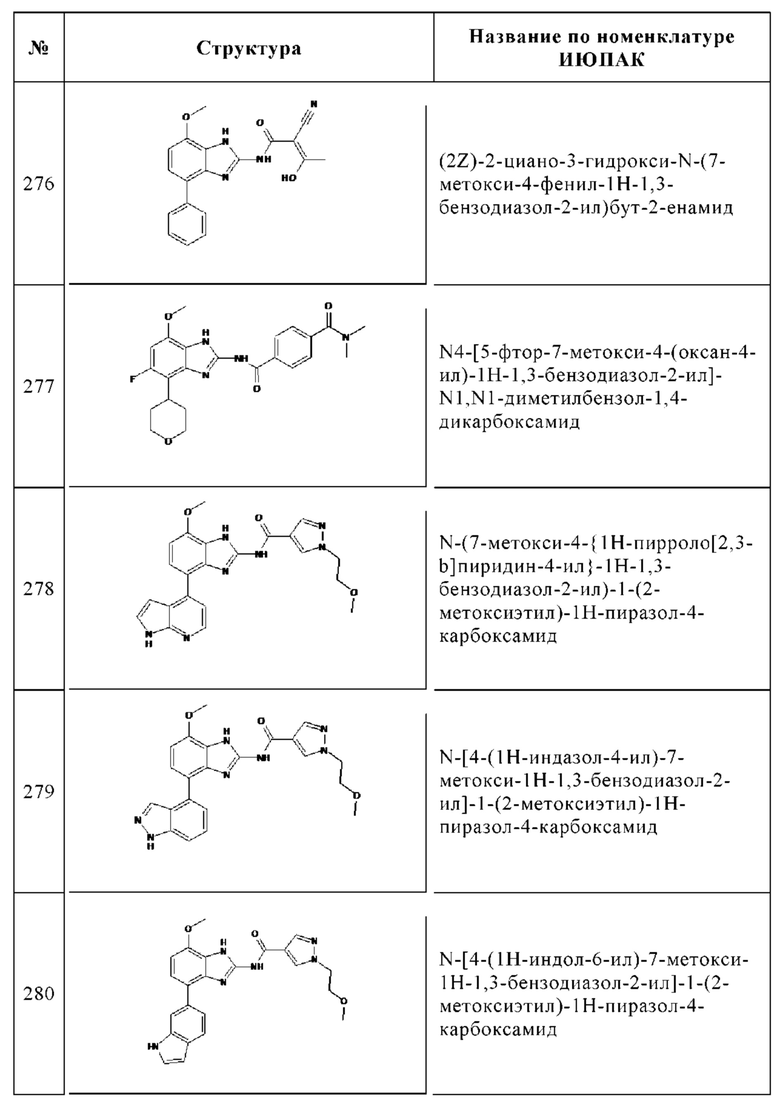
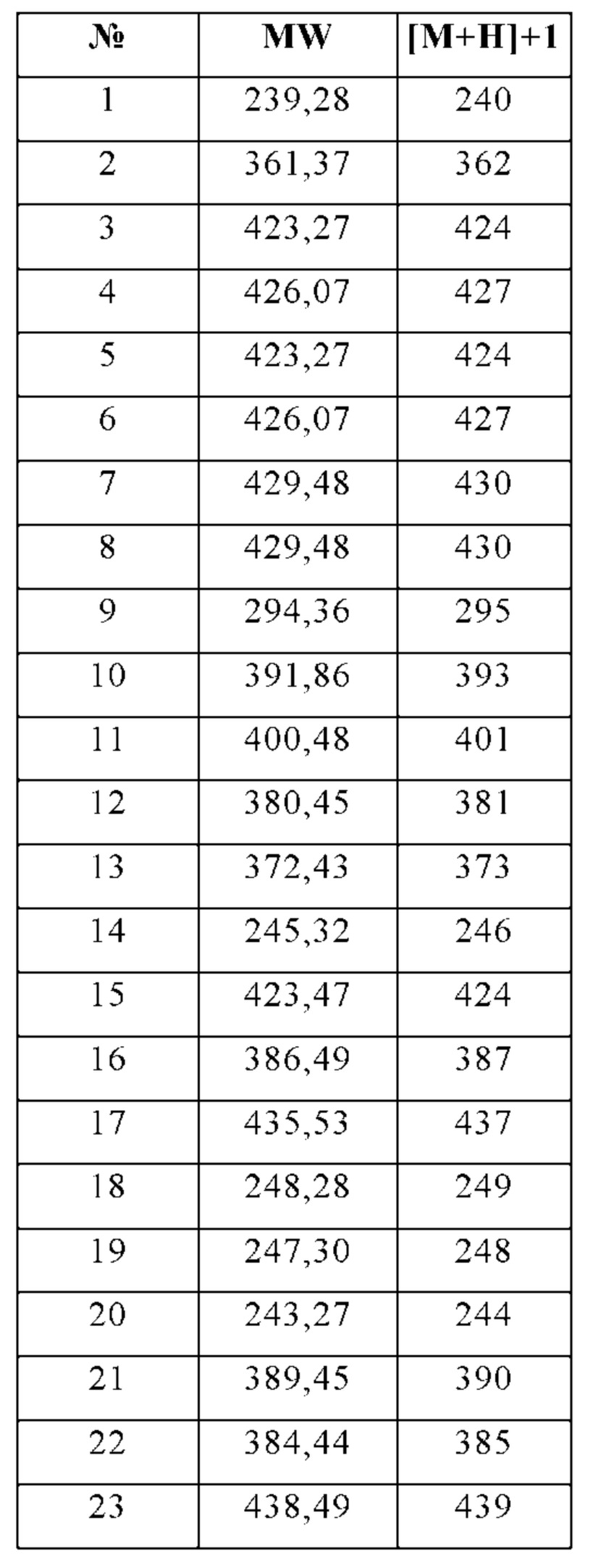
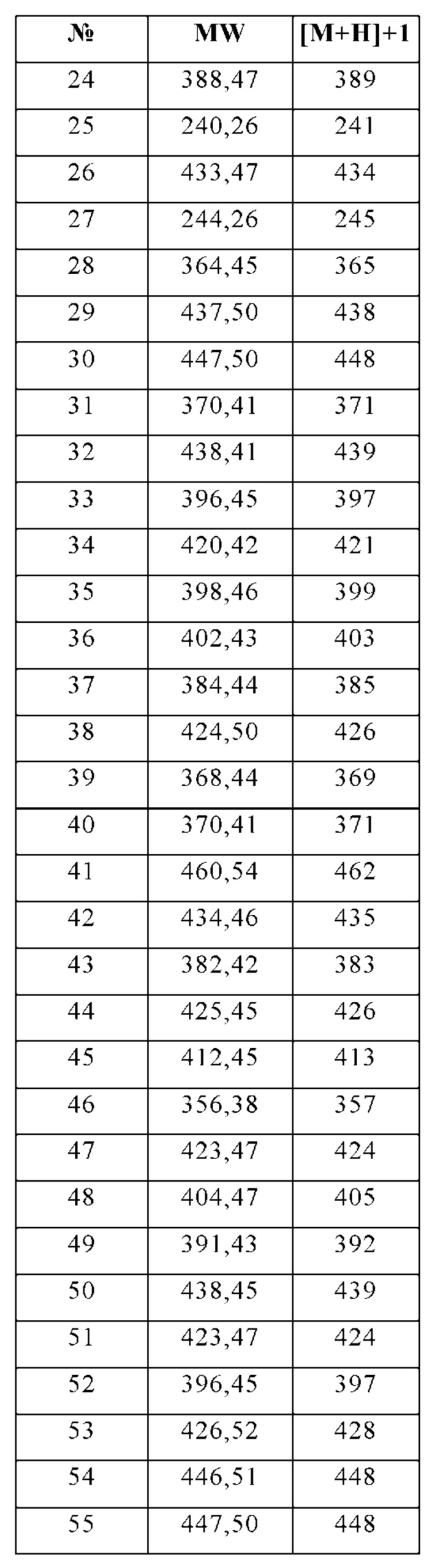
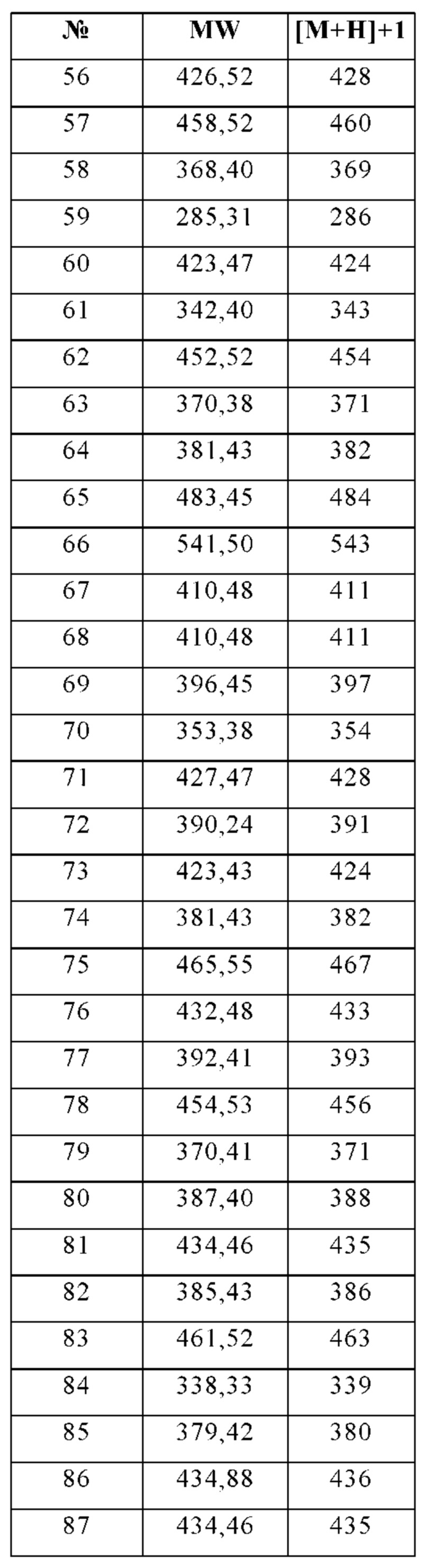
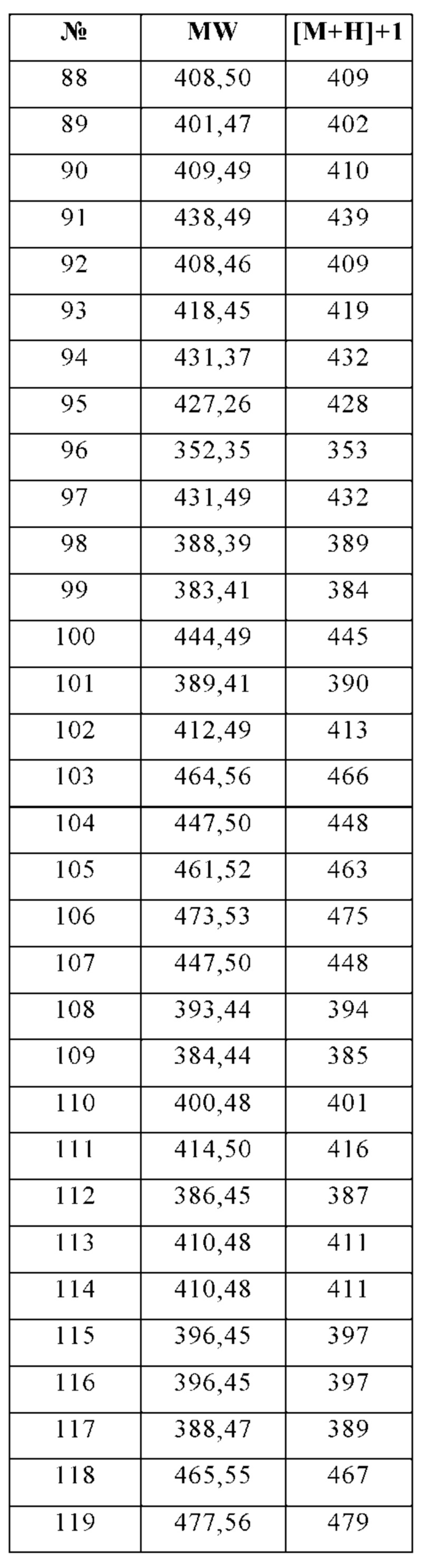
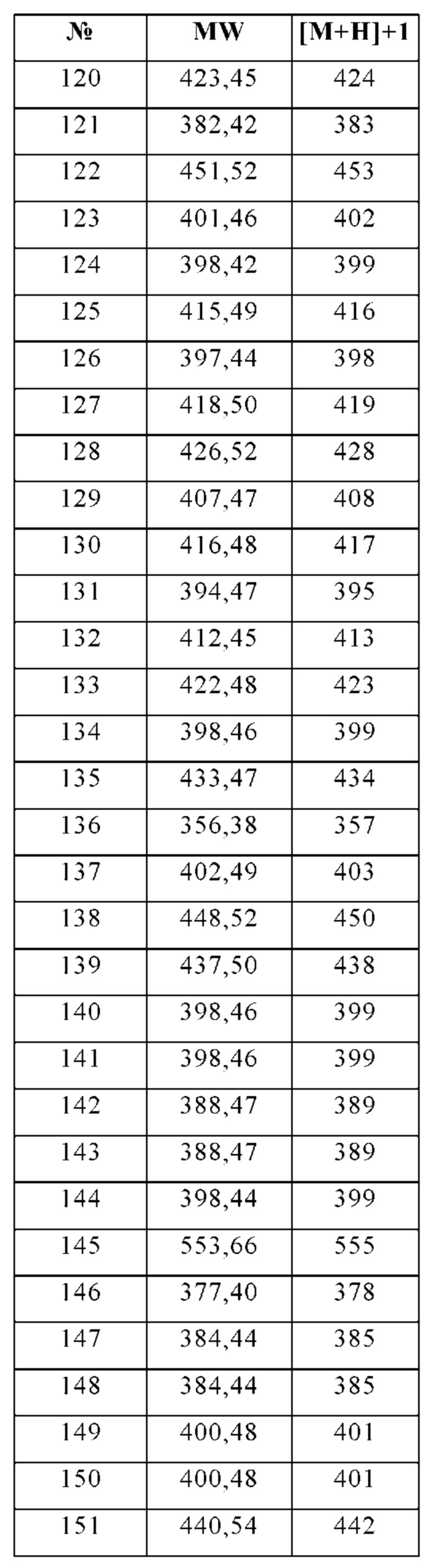
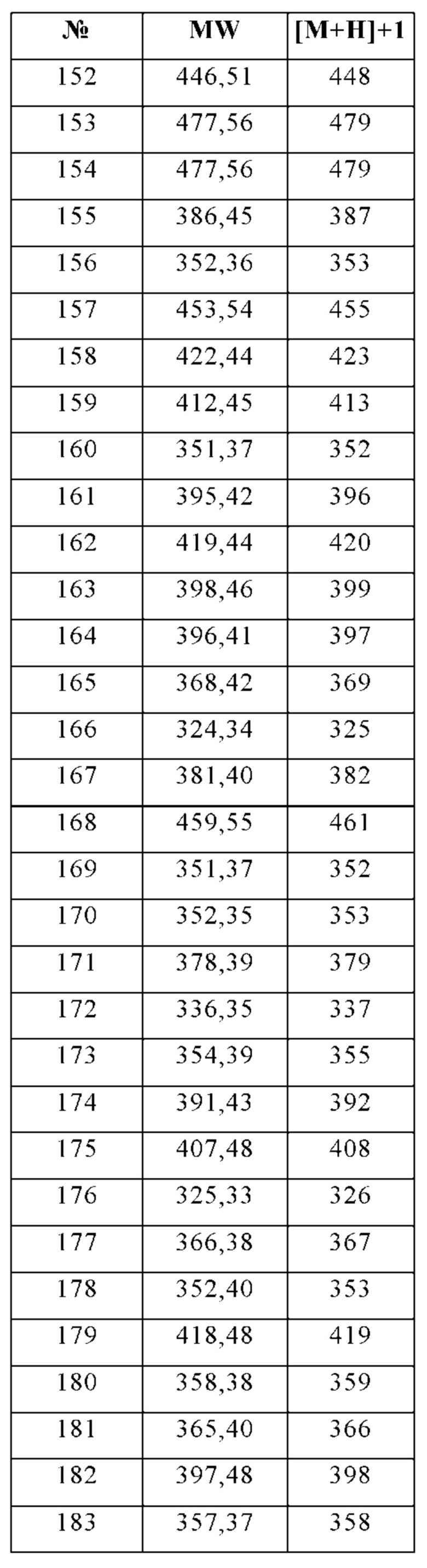
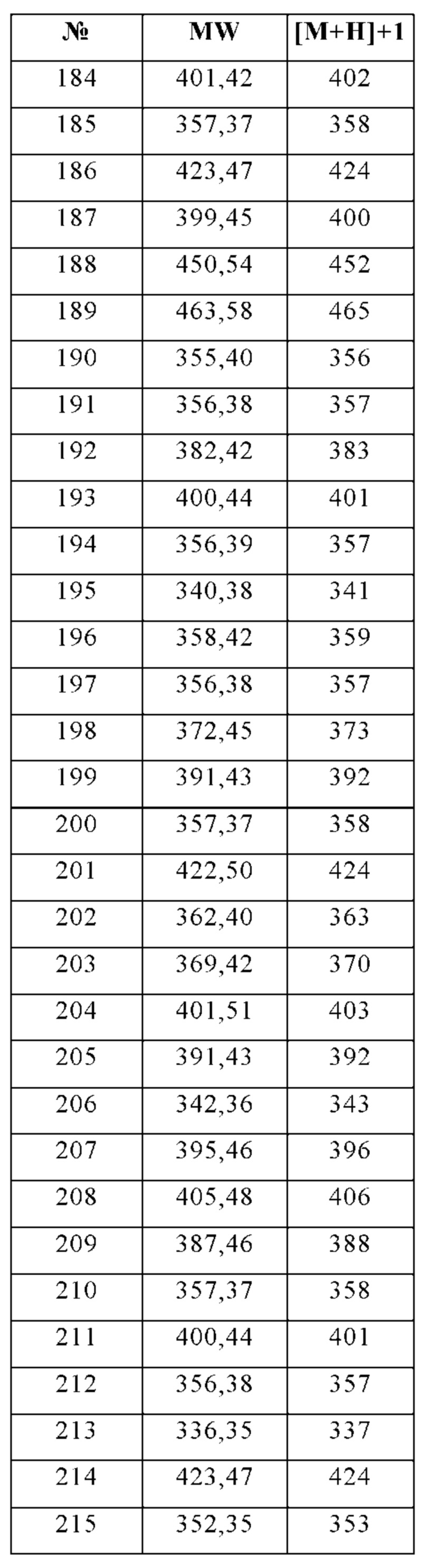
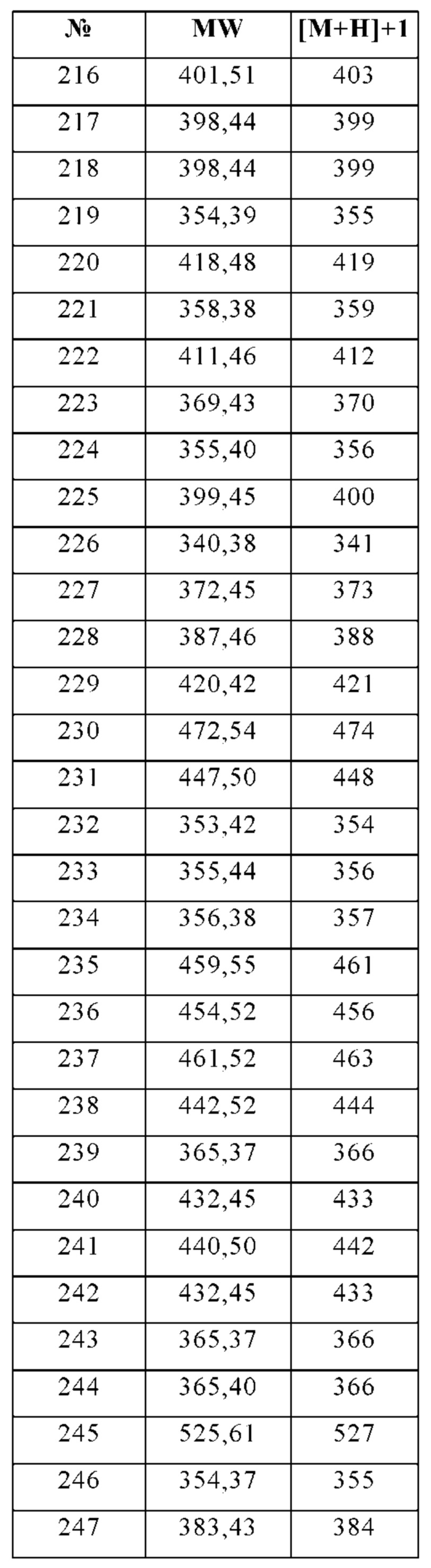
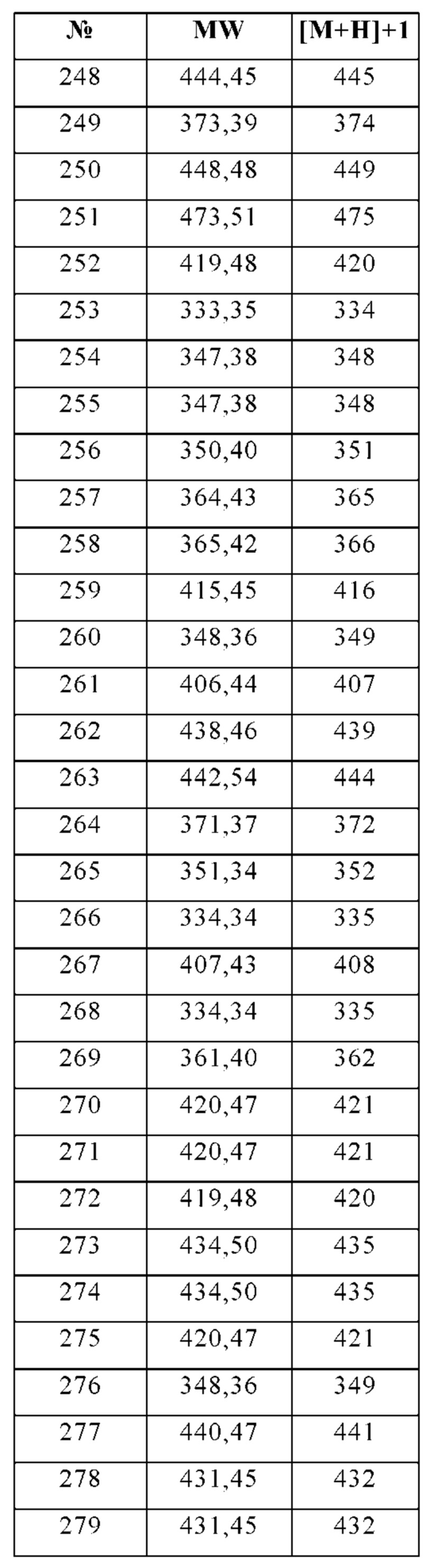
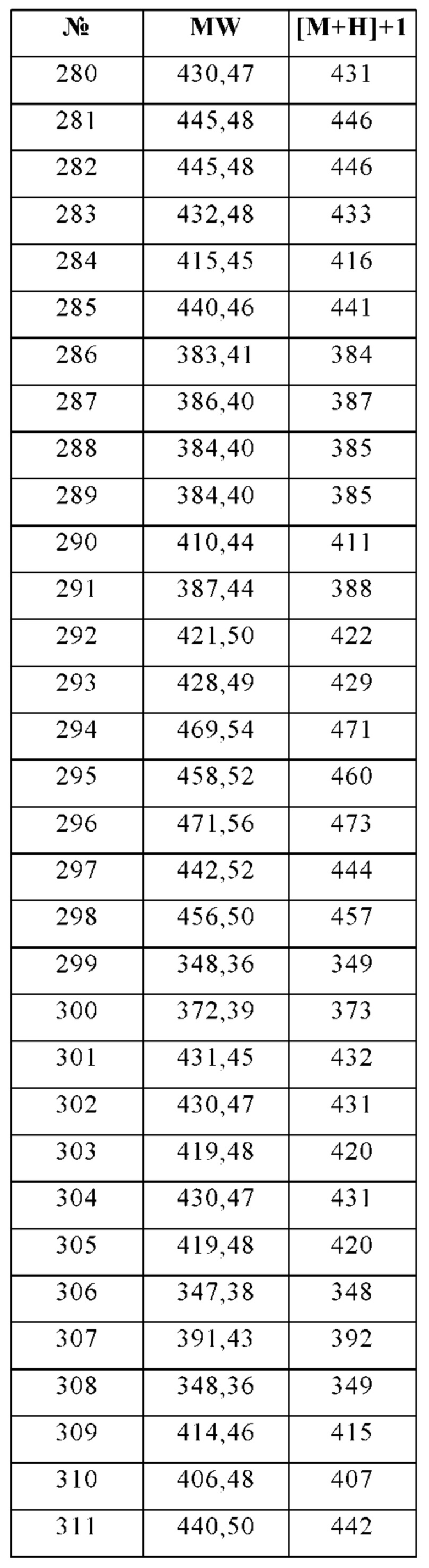
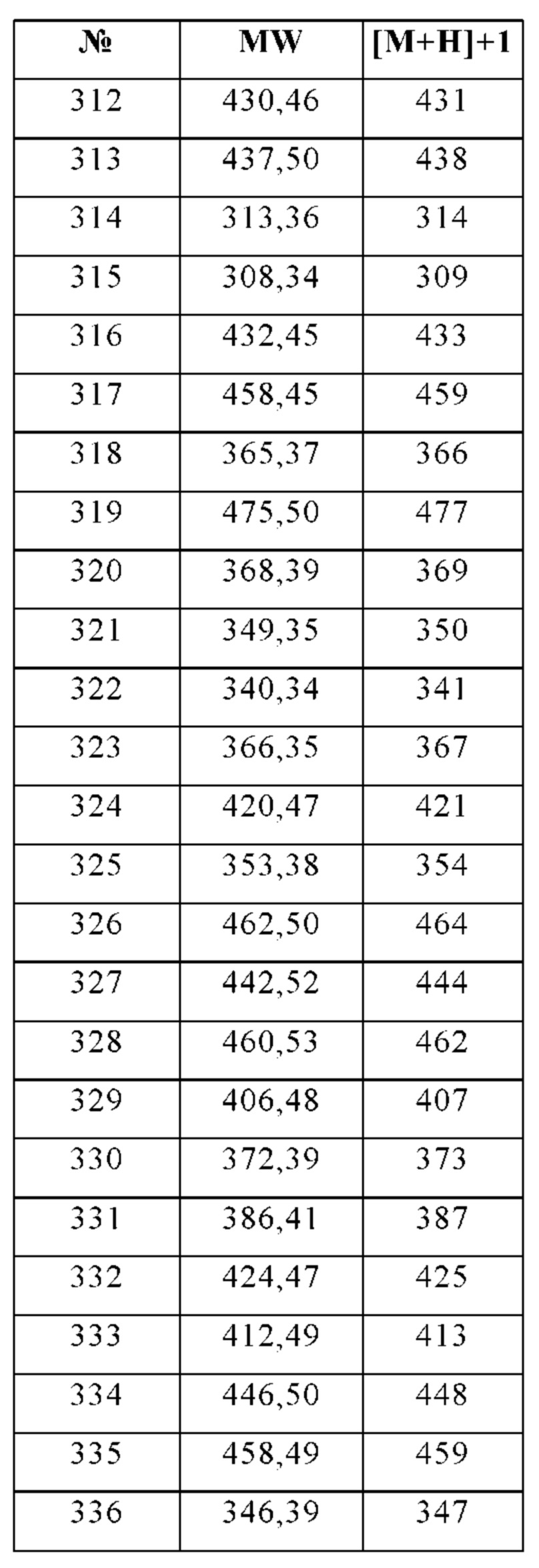



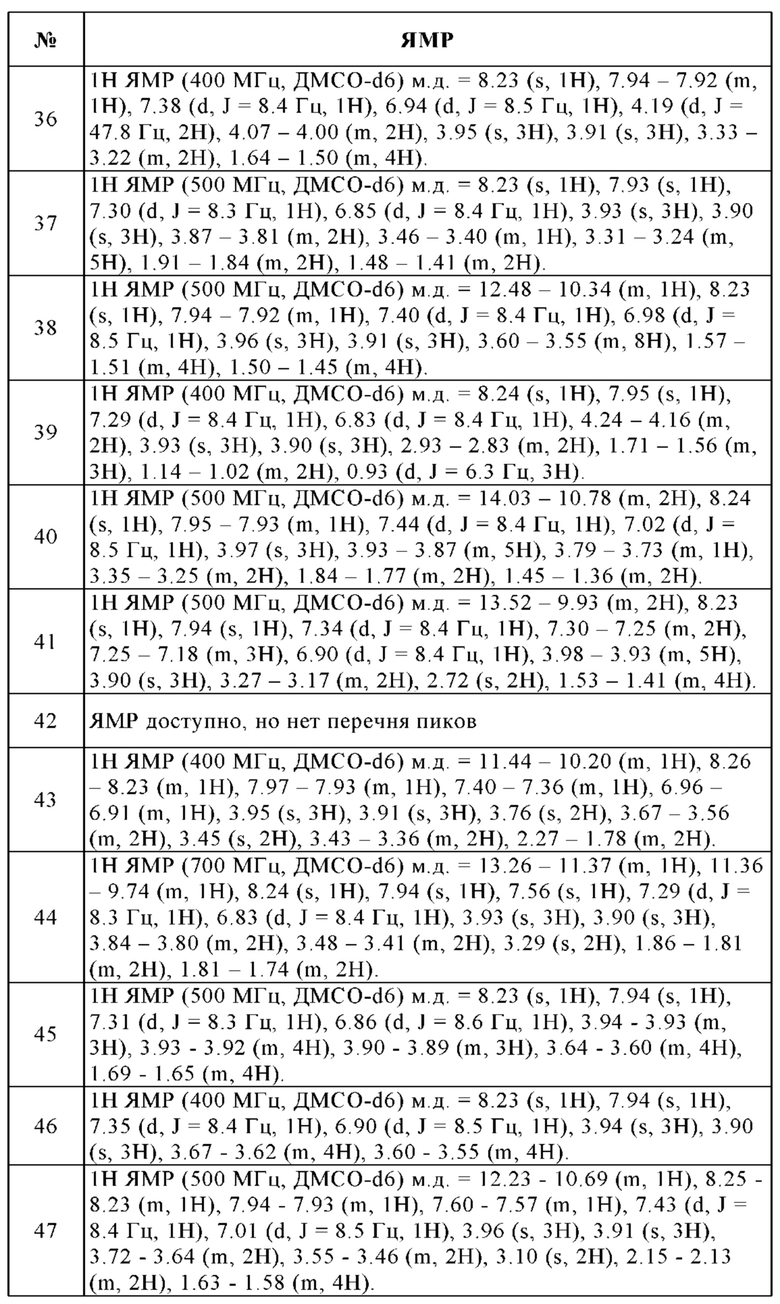
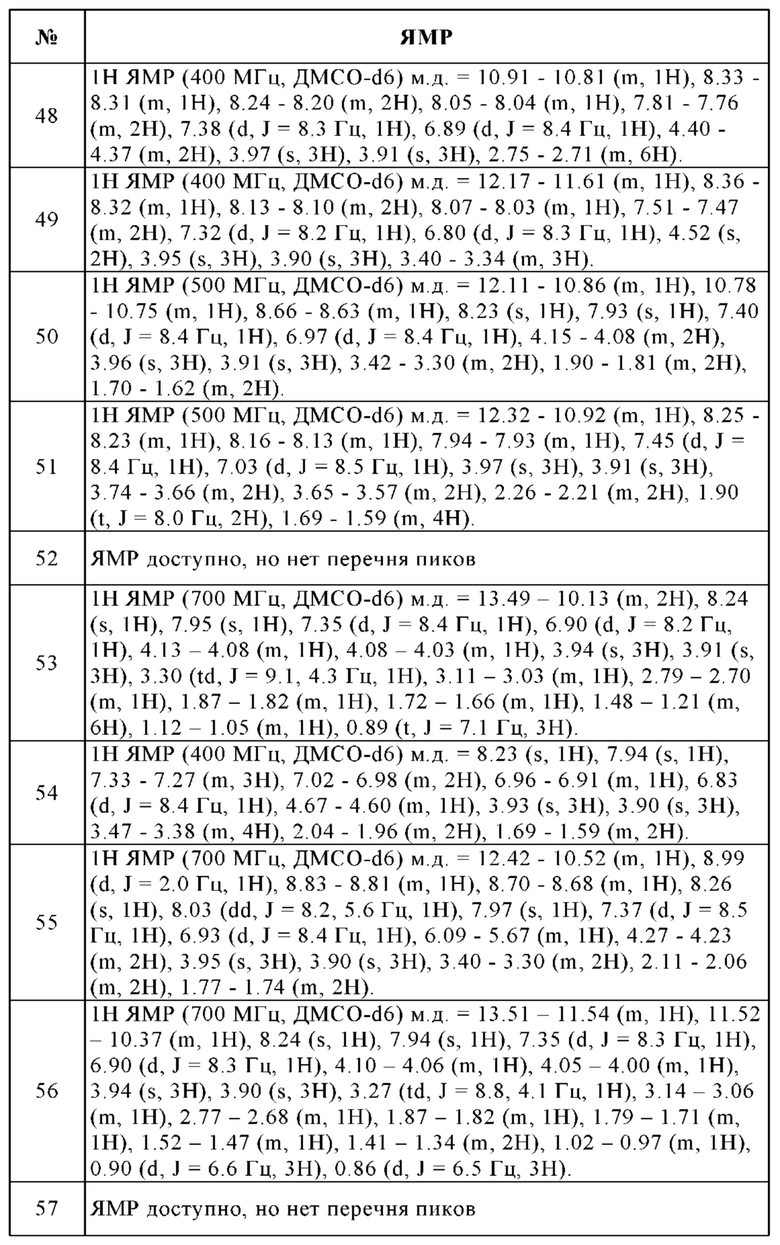



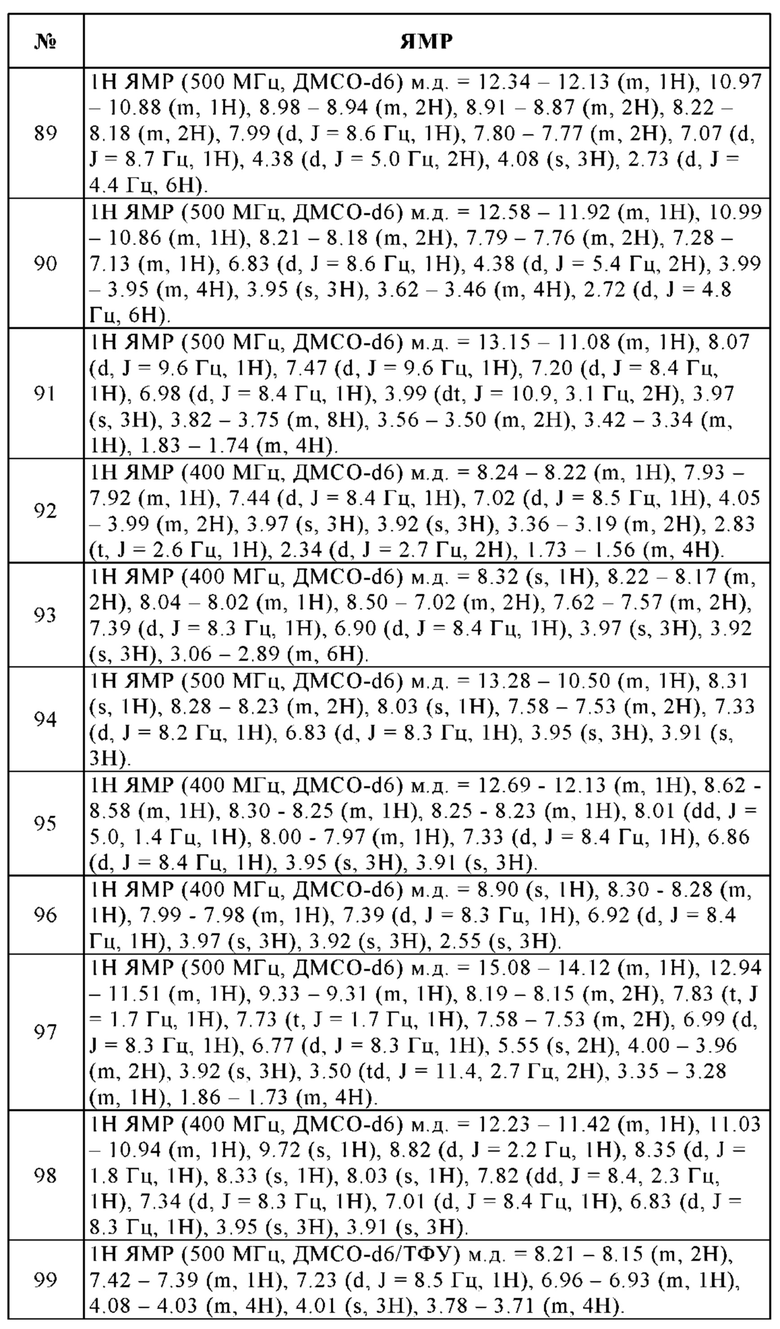
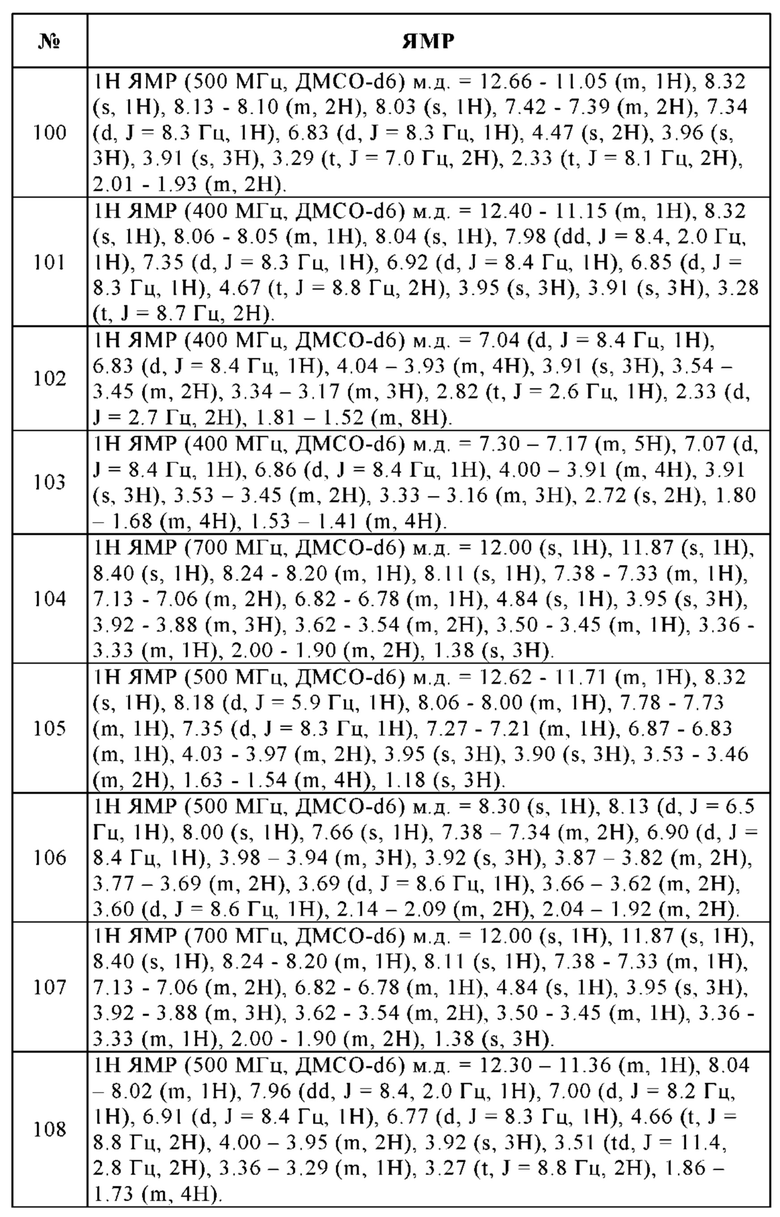
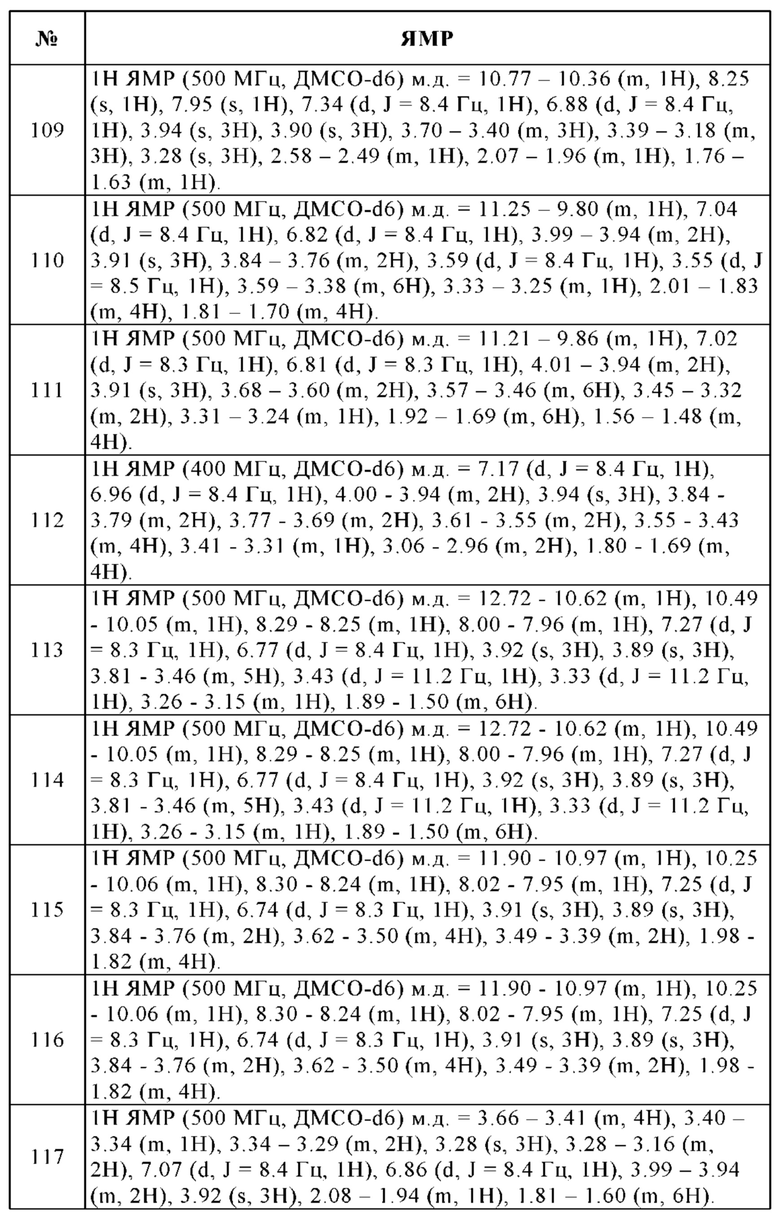

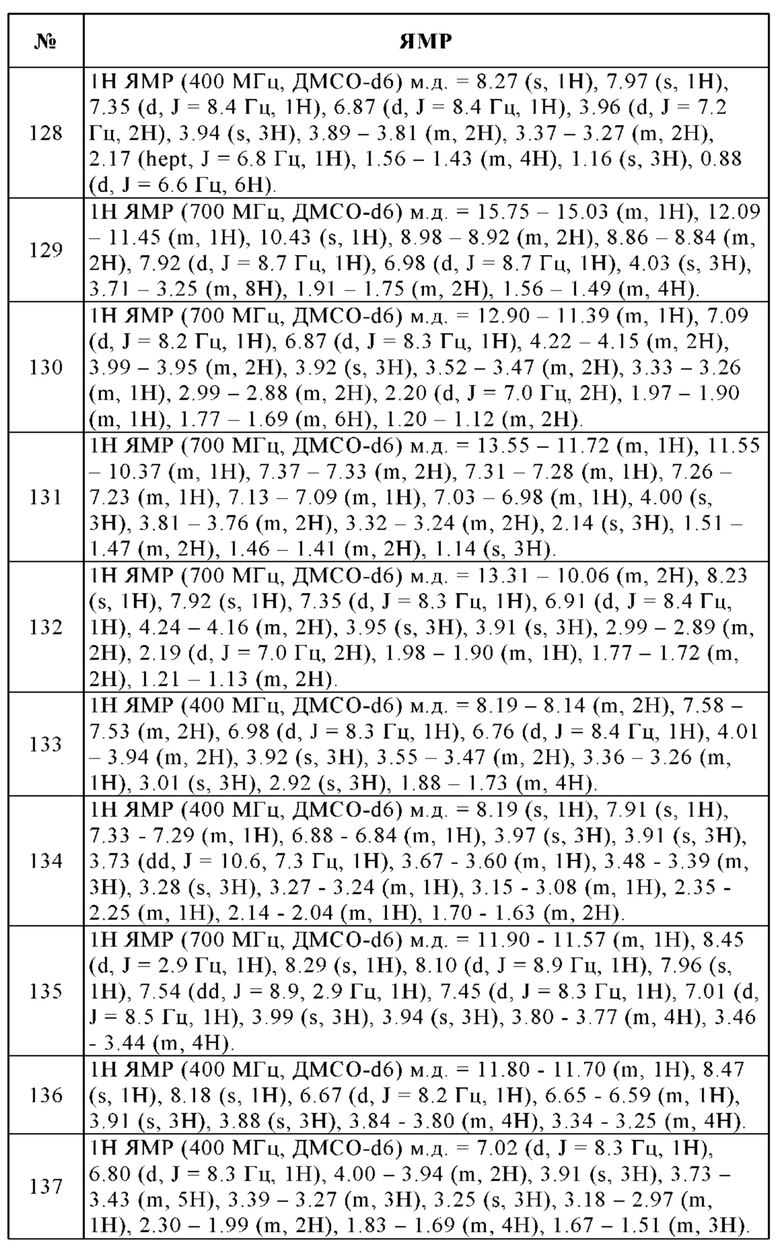
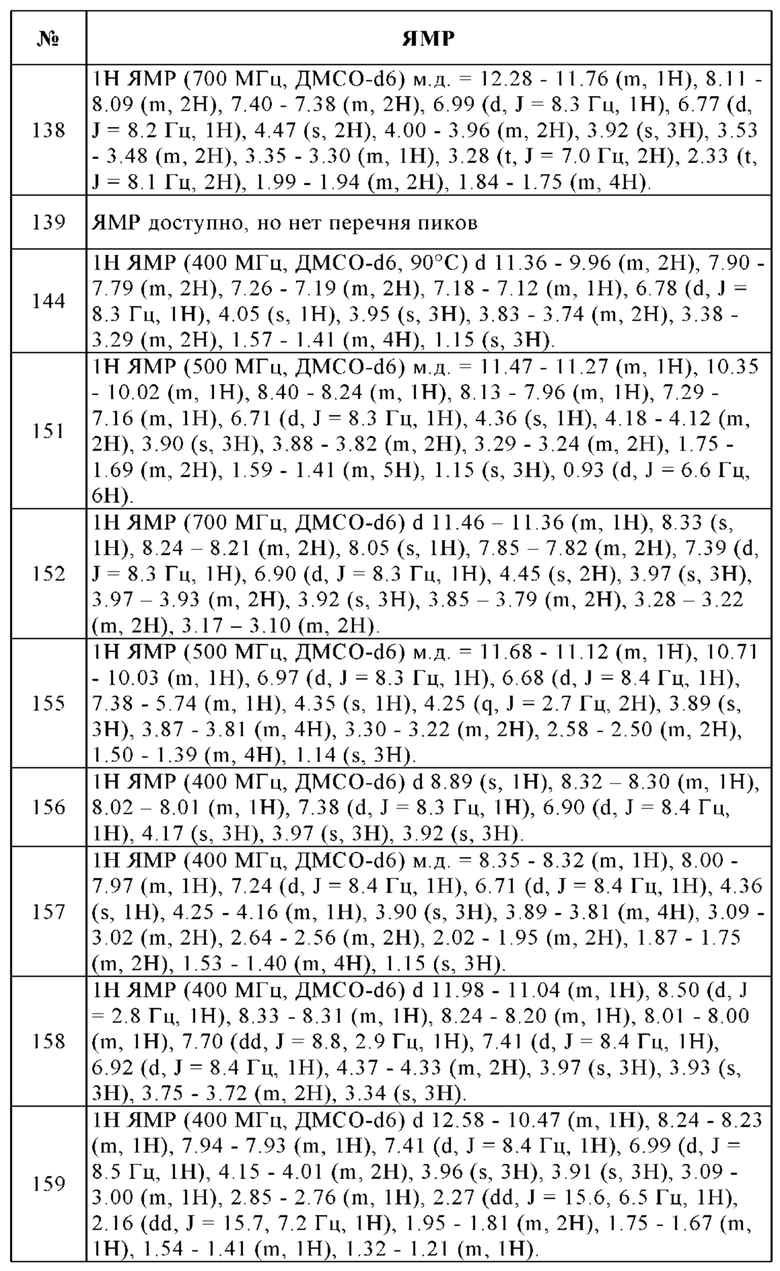
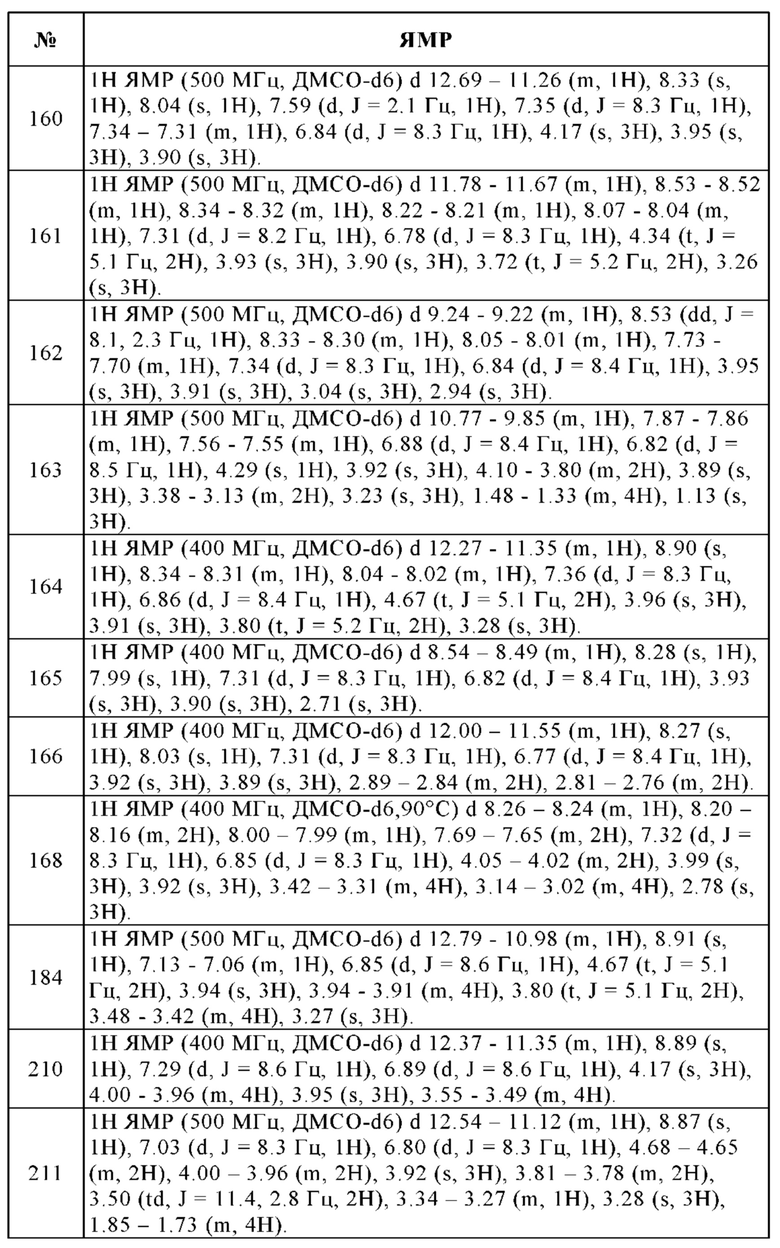
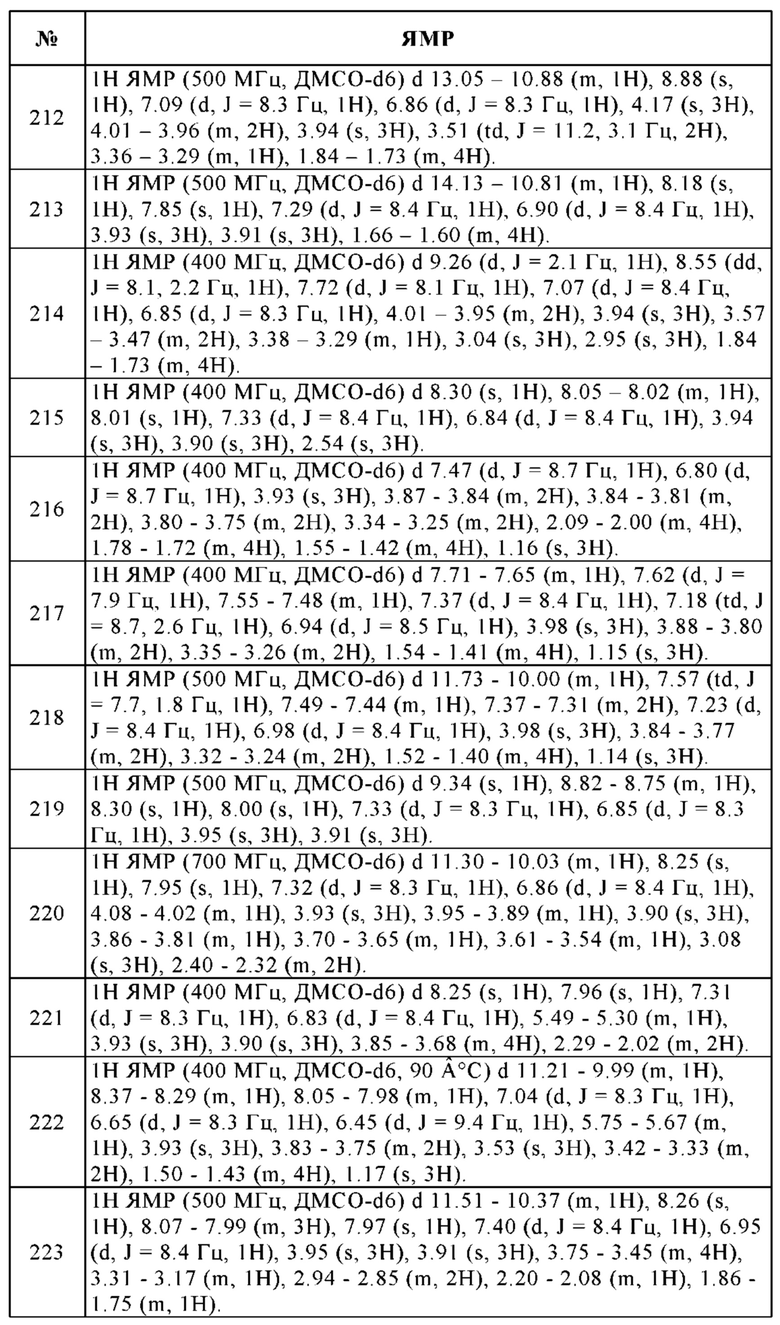
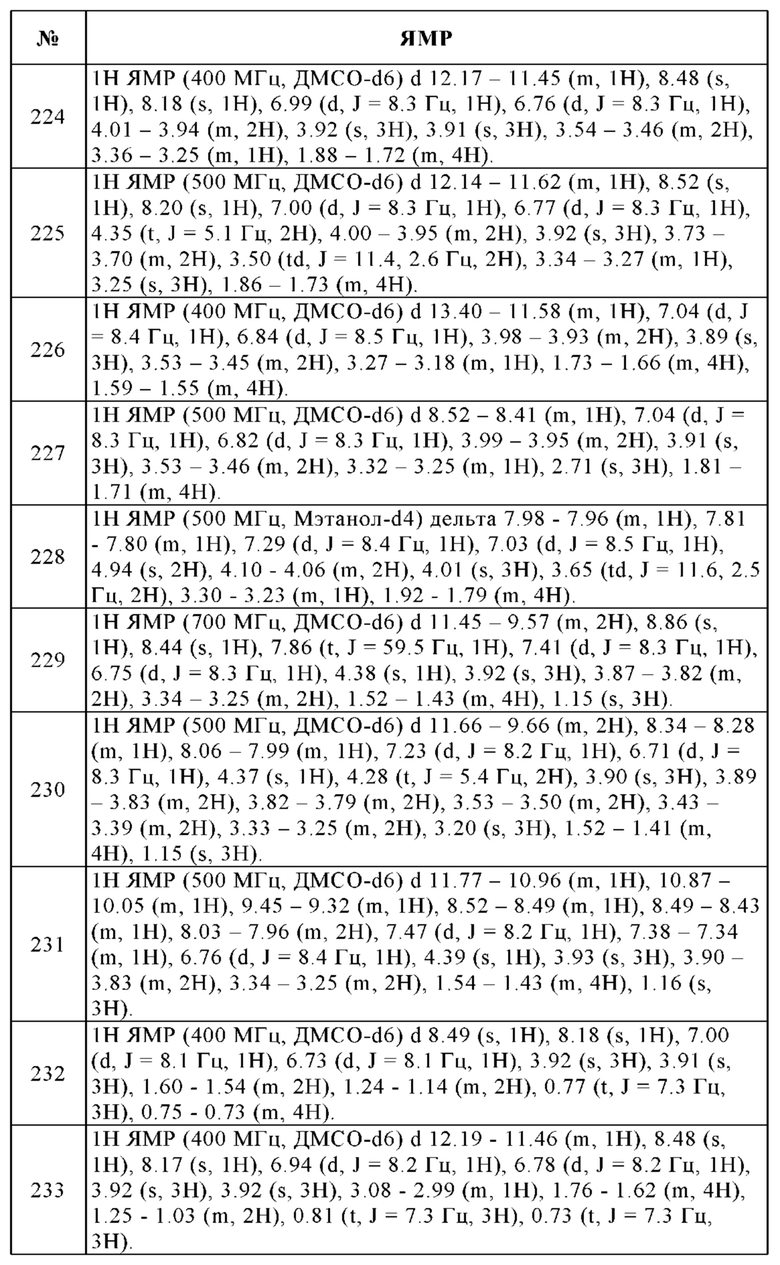
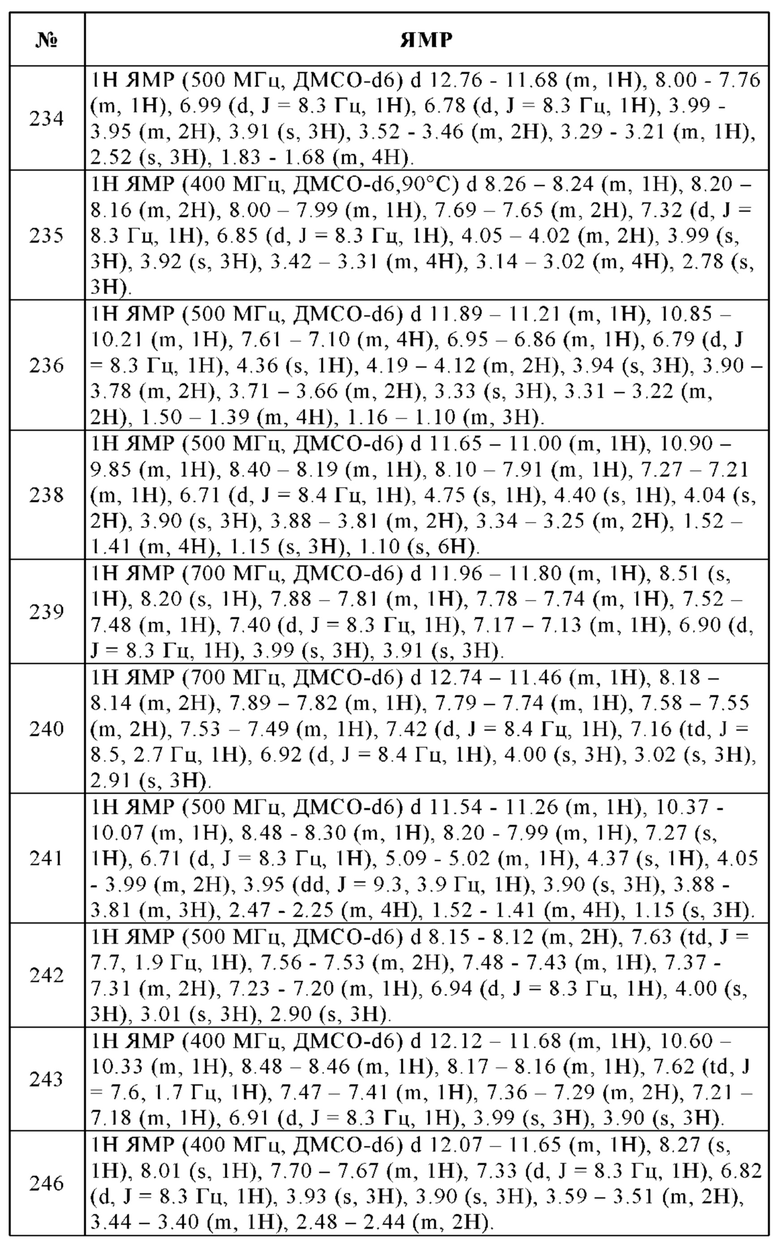
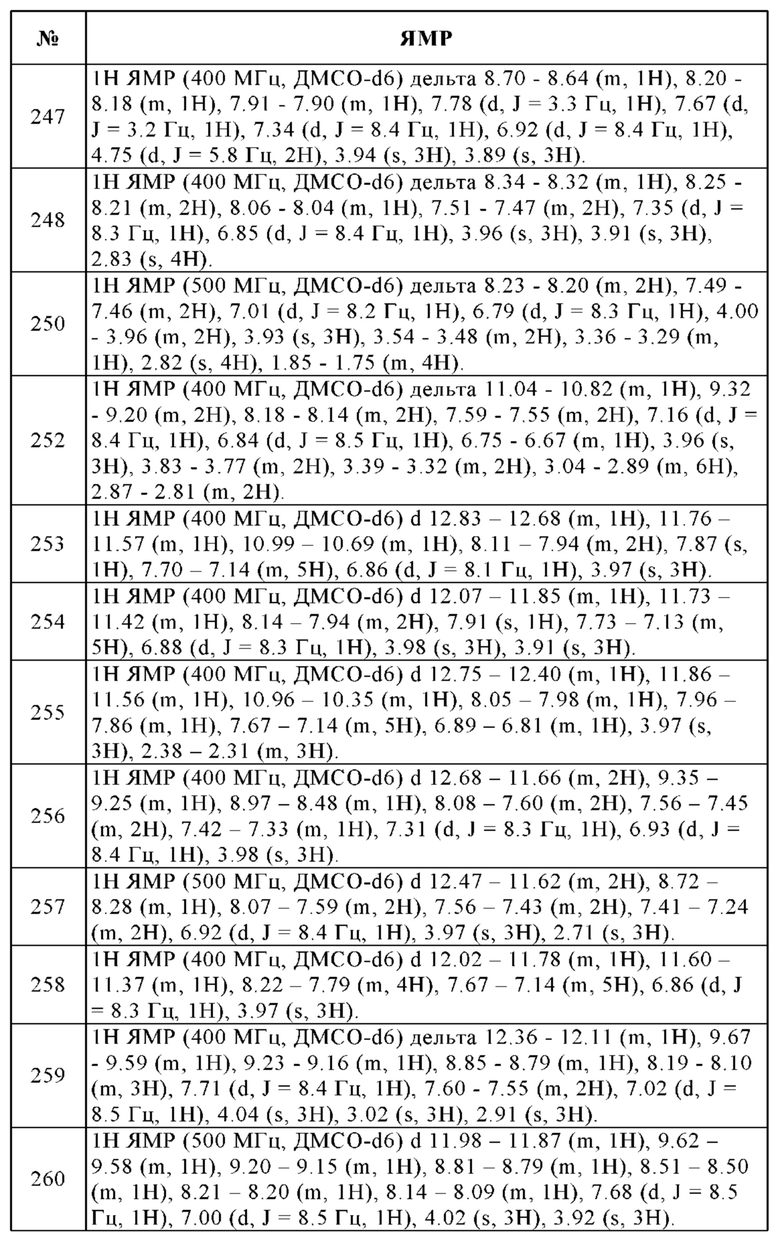
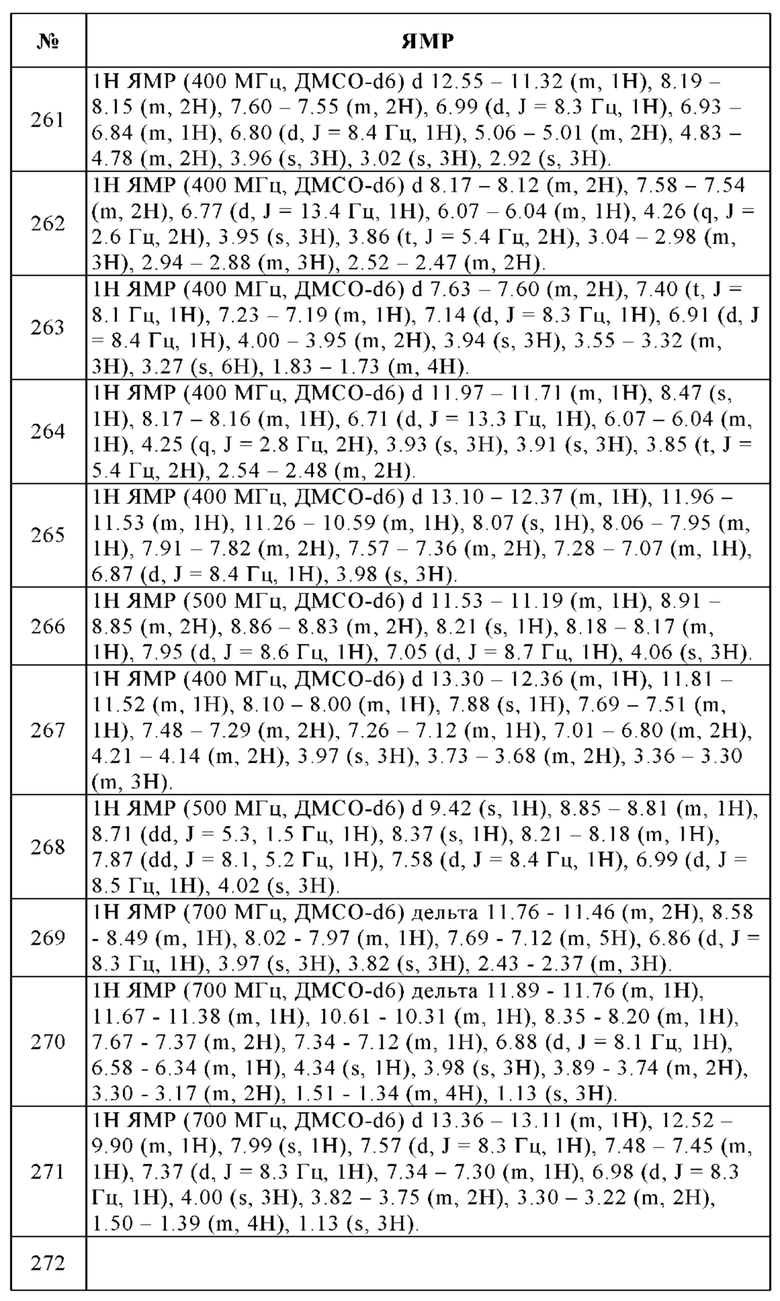
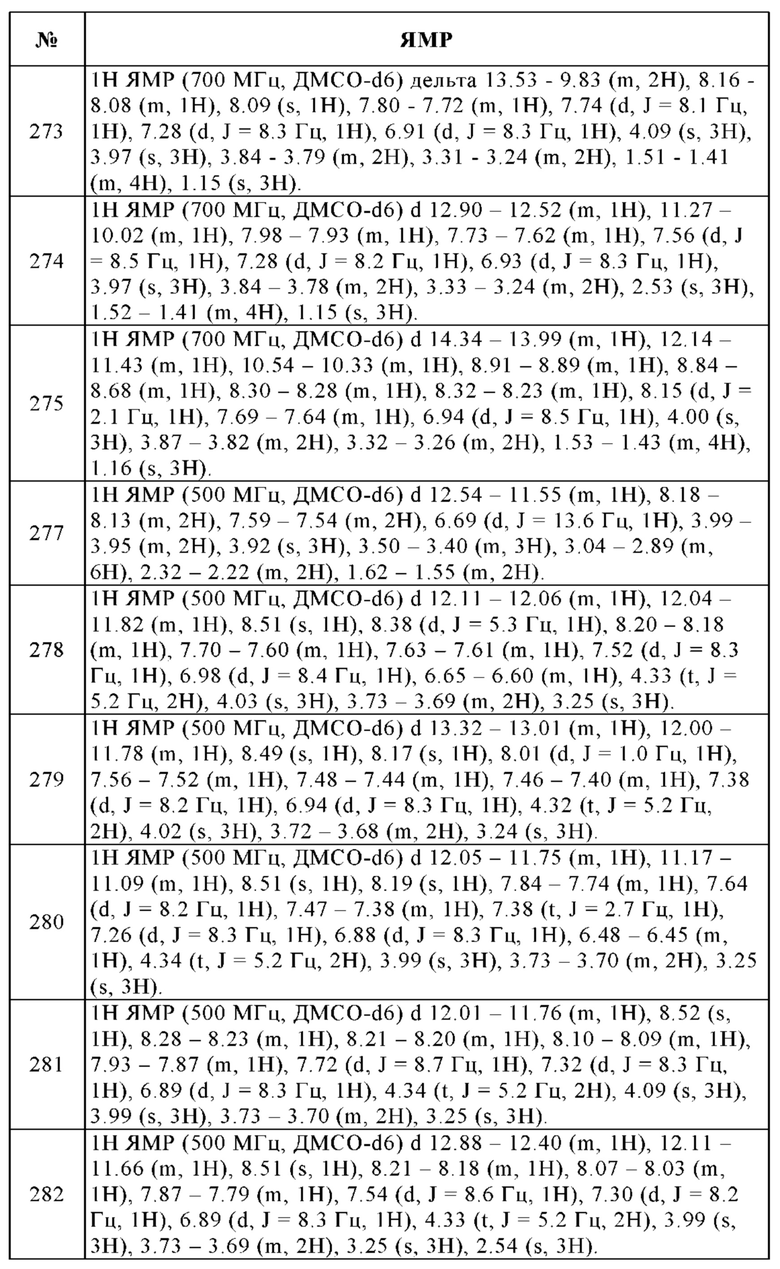
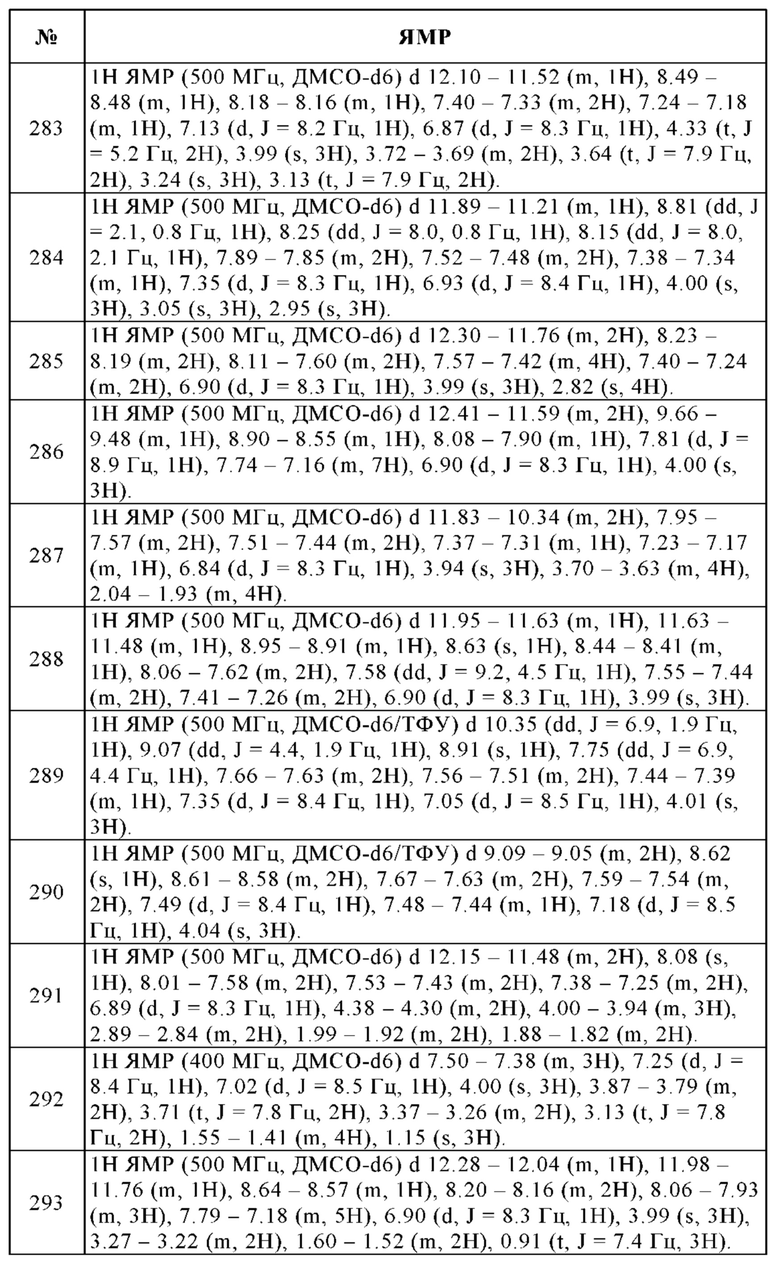
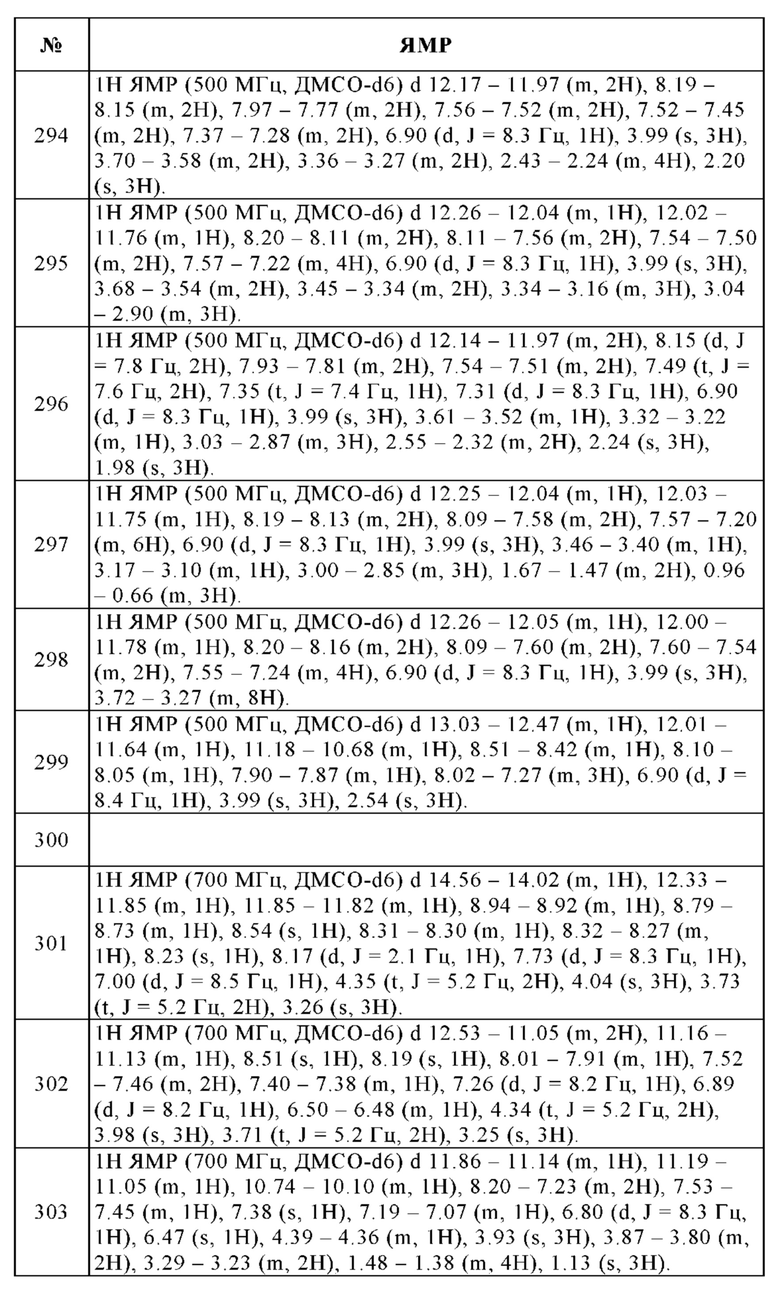
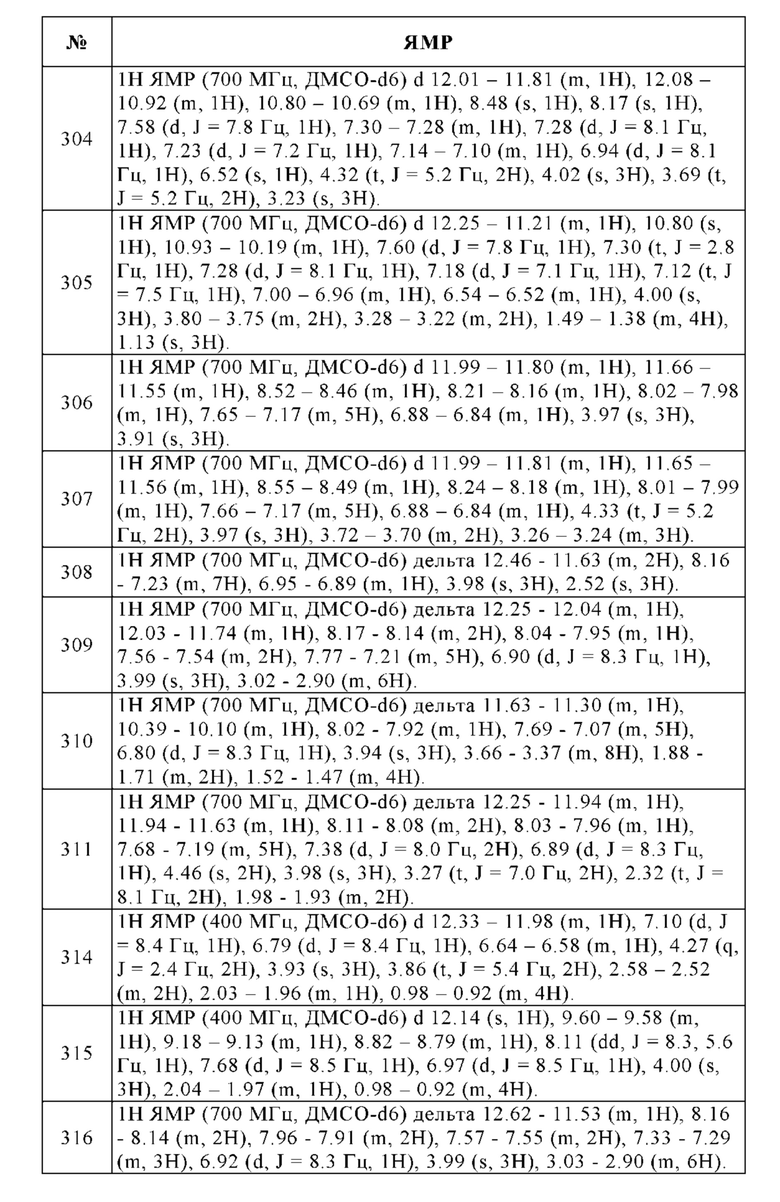

Пример 2: Получение соединений настоящего изобретения и аналитические методы
Все используемые растворители были коммерчески доступными, причем их использовали без дополнительной очистки. Реакции типично проводили с использованием безводных растворителей в инертной атмосфере азота. Колоночную флэш-хроматографию обычно проводили с использованием силикагеля Silica gel 60 (размер частиц 0.035-0.070 мм).
Данные всех ЯМР-экспериментов записывали либо на ЯМР-спектрометре Bruker Mercury Plus 400, оснащенном датчиком Bruker 400 BBFO, на 400 МГц для протонного ЯМР, либо на ЯМР-спектрометре Bruker Mercury Plus 300, оснащенном датчиком Bruker 300 BBFO, на 300 МГц для протонного ЯМР. Все дейтерированные растворители типично содержали от 0.03% до 0.05% об./об. тетраметилсилана, который использовали в качестве стандартного сигнала (установлен на м.д. = 0.00 для обоих, 1Н и 13С).
Анализы ЖХ-МС выполняли на приборе Agilent Technologies LC-MS 1200 серии, включающем МС-детектор LCMS 6110 Quadrupole. Используемая колонка и условия описаны в методах ВЭЖХ. Температура колонки составляла 40°С при заданной скорости потока. С использованием детектора на диодной матрице выполняли сканирование в диапазоне 200-400 нм. Масс-спектрометр был оснащен электрораспылительным источником ионов (ES), работающим в режиме положительных или отрицательных ионов. Сканирование на масс-спектрометре выполняли в диапазоне m/z 90-900 со временем сканирования 0.6 с.
1. 4-Этиламинометил-N-(7-метокси-4-фенил-1Н-бензоимидазол-2-ил)-бензамид, 11
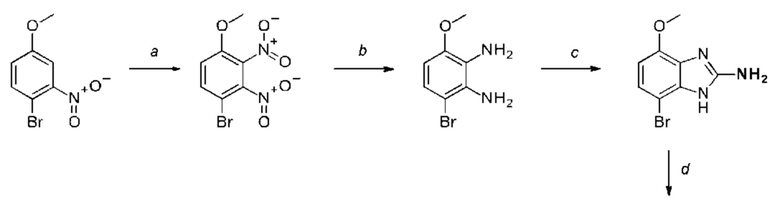
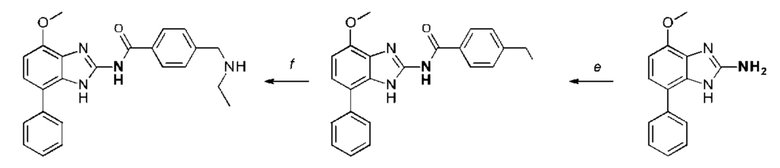
Общая методика нитрования ароматического кольца
а. 1-Бром-4-метокси-2,3-динитробензол
97% 4-Бром-3-нитроанизол (10.0 г, 43.1 ммоль) нитровали путем добавления по каплям 10 мл смеси азотной кислоты, дымящей 100% (40 мл) и 95-98% (6 мл) серной кислоты. Смесь перемешивали в течение 1 ч при КТ. Реакционную смесь выливали на ледяную воду и три раза экстрагировали этилацетатом. Объединенные органические слои промывали водой и солевым раствором, сушили над Na2SO4, фильтровали и концентрировали досуха. Сырое вещество очищали с помощью флэш-хроматографии (этилацетат/циклогексан) с получением 5.30 г (44%) указанного в заголовке соединения в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 100%. Rt 2.65 мин (метод А). [М+Н]+ 276.8, 278.9.
Общая методика восстановления нитрогруппы
b. 3-Бром-6-метоксибензол-1,2-диамин
В 250 мл круглодонную колбу помещали губчатый никелевый катализатор (2.00 г), увлажненный ТГФ, ТГФ (60 мл) и 1-бром-4-метокси-2,3-динитробензол (5.30 г, 19.1 ммоль). Смесь перемешивали в течение 6 ч при КТ в атмосфере водорода. Твердые вещества отфильтровывали и отбрасывали. Фильтрат упаривали досуха с получением 3.90 г (94%) 3-бром-6-метоксибензол-1,2-диамина в виде желтого твердого вещества, которое использовали без дополнительной очистки. ВЭЖХ/МС (чистота) 100%. Rt 1.42 мин (метод А). [М+Н]+ 217.0, 218.9.
Общая методика образования бензимидазольного кольца
c. 4-Бром-7-метокси-1Н-бензоимидазол-2-иламин
К 3-бром-6-метоксибензол-1,2-диамину (3.90 г, 18.0 ммоль), растворенному в метаноле (50 мл) и воде (25 мл), добавляли бромциан (2.86 г, 27.0 ммоль) при КТ и полученную в результате смесь перемешивали при КТ в течение 20 ч. Реакционную смесь упаривали до удаления метанола. При охлаждении водный раствор подщелачивали аммиаком. Осадок отфильтровывали и кристаллизовали из дихлорметана с получением 3.90 г (89%) указанного в заголовке соединения в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 99%. Rt 1.72 мин (метод А). [М+Н]+ 242.0, 243.9.
Общая методика реакций Сузуки:
d. 7-Метокси-4-фенил-1Н-бензоимидазол-2-иламин
В реактор высокого давления, продутый и выдерживаемый с инертной атмосферой аргона, помещали 99% 4-бром-7-метокси-1Н-бензоимидазол-2-иламин (1.68 г, 7.02 ммоль), 98% бензолбороновую кислоту (1.05 г, 8.43 ммоль), 2М карбонат калия (5 мл, 49,1 ммоль), 95% комплекс Pd(dppf)Cl2-дихлорметан (449 мг, 0.562 ммоль), этанол (2.5 мл) и толуол (25 мл). Смесь перемешивали в течение 20 ч при 90°С, охлаждали до комнатной температуры и концентрировали досуха в вакууме. Остаток очищали с помощью колоночной хроматографии (дихлорметан/этанол, градиент) с получением 1.22 г (70%) указанного в заголовке соединения в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 97%. Rt 2.09 мин (метод А). [М+Н]+ 240.1.
Общая методика образования амидной связи
e. 4-Хлорметил-N-(7-метокси-4-фенил-1Н-бензоимидазол-2-ил)-бензамид
К перемешиваемому раствору 7-метокси-4-фенил-1Н-бензоимидазол-2-иламина (300 мг, 1.22 ммоль) и N-этилдиизопропиламина (1.24 мл, 7.30 ммоль) в тетрагидрофуране (6 мл) при КТ по каплям добавляли раствор 97% 4-(хлорметил)бензоилхлорида (276 мг, 1.46 ммоль) в дихлорметане (3 мл) и перемешивали в течение 60 ч при КТ. Остаток очищали с помощью колоночной хроматографии (этилацетат/циклогексан, градиент). К растворенной чистой фракции добавляли три капли 1 н. раствора HCl, и упаривали досуха с получением 50.0 мг (10%) HCl соли указанного в заголовке соединения в виде бесцветного твердого вещества. 1Н ЯМР (500 МГц, ДМСО-d6) м.д. = 12.82-11.31 (m, 1Н), 8.148.11 (m, 2Н), 7.87-7.82 (m, 2Н), 7.64-7.60 (m, 2Н), 7.52-7.47 (m, 2Н), 7.38-7.34 (m, 1H), 7.33 (d, J = 8.3 Гц, 1H), 6.94 (d, J = 8.4 Гц, 1H), 4.86 (s, 2Н), 4.00 (s, 3Н). ВЭЖХ/МС (чистота) 100%. Rt 2.92 мин (метод А). [М+Н]+ 392.0.
f. 4-Этиламинометил-N-(7-метокси-4-фенил-1Н-бензоимидазол-2-ил)-бензамид
К перемешиваемому раствору гидрохлорида 4-хлорметил-N-(7-метокси-4-фенил-1Н-бензоимидазол-2-ил)-бензамида (44.0 мг, 0.103 ммоль) в тетрагидрофуране (2 мл), добавляли 2М этиламин в ТГФ (1 мл), и перемешивали в течение 20 ч при КТ и затем в течение дополнительных 20 ч при 50°С. Смесь упаривали досуха и остаток очищали с помощью препаративной ВЭЖХ (ацетонитрил/вода, градиент). К растворенной чистой фракции добавляли пять капель 1 н. раствора HCl и упаривали досуха с получением 10.0 мг (21%) дигидрохлорида указанного в заголовке соединения в виде бесцветного твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) м.д. = 8.91-8.82 (m, 2Н), 8.16-8.12 (m, 2Н), 7.86-7.81 (m, 2Н), 7.66-7.62 (m, 2Н), 7.51-7.45 (m, 2Н), 7.37-7.32 (m, 1Н), 7.30 (d, J = 8.3 Гц, 1H), 6.91 (d, J=8.4 Гц, 1H), 4.24-4.19 (m, 2Н), 3.97 (s, 3Н), 3.06-2.96 (m, 2Н), 1.22 (t, J=7.3 Гц, 3Н). ВЭЖХ/МС (чистота) 100%. Rt 2.42 мин (метод А). [М+Н]+ 401.1.
2. 4-Гидрокси-N-[7-метокси-4-(1-метил-1Н-пиразол-4-ил)-1Н-1,3-бензодиазол-2-ил]-4-(проп-2-ин-1-ил)пиперидин-1-карбоксамид
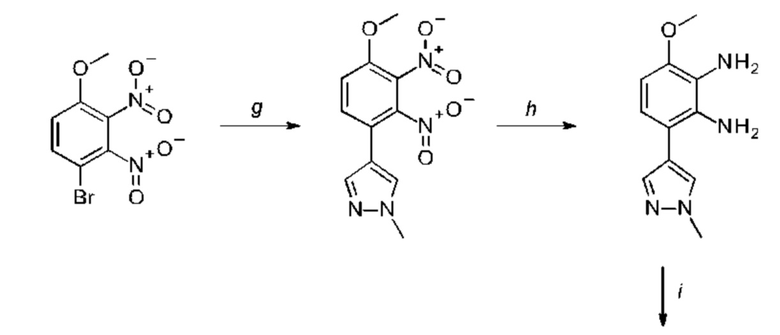
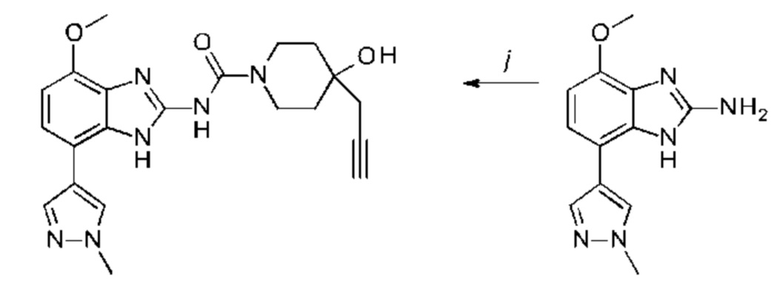
g. 4-(4-Метокси-2,3-динитрофенил)-1-метил-1Н-пиразол
В реактор высокого давления, продутый и выдерживаемый с инертной атмосферой аргона, помещали 88% 1-бром-4-метокси-2,3-динитробензол (4.00 г, 12.7 ммоль), сложный пинаколовый эфир 1-метил-1Н-пиразол-4-бороновой кислоты (3.17 г, 15.2 ммоль), 2М карбонат калия (16 мл, 157 ммоль), комплекс Pd(dppf)Cl2 - дихлорметан, (1.01 г, 1.27 ммоль), этанол (8 мл) и толуол (80 мл) Смесь перемешивали в течение 2 ч при 90°С, охлаждали до комнатной температуры и концентрировали досуха в вакууме. Остаток очищали с помощью колоночной хроматографии (этилацетат/циклогексан, градиент) с получением 2.70 г (76%) указанного в заголовке соединения в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 100%. Rt 2.38 мин (метод А). [М+Н]+ 279.0.
h. 3-Метокси-6-(1-метил-1Н-пиразол-4-ил)-бензол-1,2-диамин
В колбу помещали 5% палладий на угле Е101 R Noblyst (1.50 г, 14.1 ммоль), тетрагидрофуран (30 мл) и 4-(4-метокси-2,3-динитрофенил)-1-метил-1Н-пиразол (2.70 г, 9.71 ммоль). Смесь перемешивали в течение 18 ч при КТ в атмосфере водорода. Твердые вещества отфильтровывали и отбрасывали. Фильтрат упаривали досуха и остаток использовали без дополнительной очистки с получением 2.10 г (91%) указанного в заголовке соединения в виде коричневатого твердого вещества. ВЭЖХ/МС (чистота) 92%. Rt 1.44 мин (метод А). [М+Н]+ 219.1.
i. 7-Метокси-4-(1-метил-1Н-пиразол-4-ил)-1Н-бензоимидазол-2-иламин 92% 3-Метокси-6-(1-метил-1Н-пиразол-4-ил)-бензол-1,2-диамин (2.10 г, 8.85 ммоль) растворяли в метаноле (100 мл) и воде (20 мл). Добавляли бромциан (1.44 г, 13.3 ммоль) и реакционную смесь перемешивали при КТ в течение 2 ч. Смесь упаривали досуха и очищали с помощью колоночной хроматографии (дихлорметан/этанол, градиент) с получением 2.20 г (100%) указанного в заголовке соединения в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 98%. Rt 1.74 мин (метод А). [М+Н]+ 244.1.
Общая методика образования мочевин
j. 4-Гидрокси-N-[7-метокси-4-(1-метил-1Н-пиразол-4-ил)-1Н-1,3-бензодиазол-2-ил]-4-(проп-2-ин-1-ил)пиперидин-1-карбоксамид
К перемешиваемому раствору 1,1'-карбонилдиимидазола (84.9 мг, 0.524 ммоль) в дихлорметане (5 мл) медленно добавляли 98% 7-метокси-4-(1-метил-1Н-пиразол-4-ил)-1Н-бензоимидазол-2-иламин (100 мг, 0.403 ммоль), суспендированный в дихлорметане (1 мл) при 60°С. После 20 ч выдерживания при 70°С, добавляли гидрохлорид 4-проп-2-инилпиперидин-4-ола (92.0 мг, 0.524 ммоль) и триэтиламин (0.168 мл, 1.21 ммоль) и смесь перемешивали в течение дополнительных 2 ч при 60°С. Смесь упаривали досуха и остаток очищали с помощью препаративной ВЭЖХ (ацетонитрил/вода, градиент). К растворенной чистой фракции добавляли пять капель 1 н. раствора HCl и упаривали досуха с получением 30.0 мг (17%) гидрохлорида указанного в заголовке соединения в виде светло-бежевого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) d 8.24-8.22 (m, 1H), 7.93-7.92 (m, 1H), 7.44 (d, J=8.4 Гц, 1H), 7.02 (d, J=8.5 Гц, 1H), 4.05-3.99 (m, 2H), 3.97 (s, 3Н), 3.92 (s, 3Н), 3.36-3.19 (m, 2H), 2.83 (t, J=2.6 Гц, 1H), 2.34 (d, J=2.7 Гц, 2H), 1.73-1.56 (m, 4H). ВЭЖХ/МС (чистота) 100%. Rt 2.00 мин (метод A). [M+H]+ 409.2.
3. 2-(3-Гидрокси-3-метилпирролидин-1-ил)-N-[7-метокси-4-(оксан-4-ил)-1Н-1,3-бензодиазол-2-ил]пиридин-4-карбоксамид
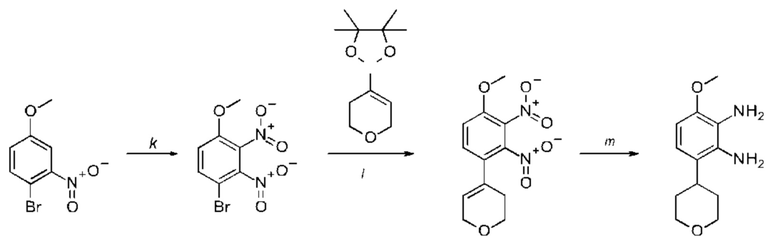
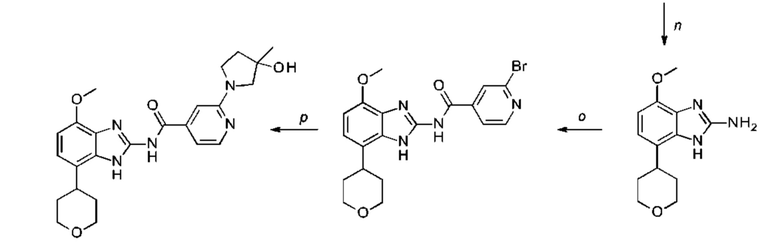
k. 1-Бром-4-метокси-2,3-динитробензол
В 3-горлую круглодонную колбу помещали 1-бром-4-метокси-2-нитробензол (50.0 г, 205 ммоль) в серной кислоте (100 мл). При перемешивании при 0°С по каплям добавляли азотную кислоту (24 мл, 530 ммоль). Раствор перемешивали в течение 1 ч при комнатной температуре и гасили 1000 мл ледяной воды. Раствор два раза экстрагировали 1000 мл этилацетата, объединенные органические слои сушили над безводным сульфатом натрия и концентрировали досуха. Сырое вещество перекристаллизовывали из смеси этилацетат/гексан (2:3) с получением 20.0 г (32%) 1-бром-4-метокси-2,3-динитробензола в виде желтого твердого вещества. Температура плавления: 150-153°С. 1H ЯМР (400 МГц, ДМСО-d6) м.д. = 8.19 (d, J=9.3 Гц, 1H), 7.70 (d, J=9.3 Гц, 1Н), 4.02 (s, 3Н). ВЭЖХ/МС (чистота) 91%. [М+Н]+ 276.8, 278.9.
l. 4-(4-Метокси-2,3-динитрофенил)-3,6-дигидро-2Н-пиран
В реактор высокого давления, продутый и выдерживаемый с инертной атмосферой аргона, помещали 91% 1-бром-4-метокси-2,3-динитробензол (15.7 г, 51.4 ммоль), 95% 2-(3,6-дигидро-2Н-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (13.7 г, 61.7 ммоль), 95% комплекс Pd(dppf)Cl2 - дихлорметан (4.42 г, 5.14 ммоль) и карбонат калия (8.53 г, 61.7 ммоль), растворенные в воде (12 мл), этаноле (31.6 мл) и толуоле (316 мл). Смесь перемешивали в течение 1 ч при 100°С, охлаждали до комнатной температуры и концентрировали досуха в вакууме. Остаток очищали с помощью колоночной хроматографии (этилацетат/петролейный эфир: 1/1) с получением 13.0 г (86%) 4-(4-метокси-2,3-динитрофенил)-3,6-дигидро-2Н-пирана в виде оранжевого твердого вещества. ВЭЖХ/МС (чистота) 95%. [М+Н]+ 281.2.
m. 3-Метокси-6-(тетрагидропиран-4-ил)-бензол-1,2-диамин
В 250 мл круглодонную колбу помещали 10% палладий на угле (4.00 г, 3.76 ммоль), метанол (100 мл) и 95% 4-(4-метокси-2,3-динитрофенил)-3,6-дигидро-2Н-пиран (10.5 г, 33.9 ммоль). Смесь перемешивали в течение 15 ч при 35°С в атмосфере водорода. Твердые вещества отфильтровывали и отбрасывали. Фильтрат упаривали досуха и остаток очищали с помощью колоночной хроматографии (этилацетат/гексан, 70/30) с получением 4.51 г (58%) 3-метокси-6-(оксан-4-ил)бензол-1,2-диамина в виде желтого твердого вещества. Температура плавления: 116-117°С. 1Н ЯМР (400 МГц, хлороформ-d) 6.67 (d, J=8.5 Гц, 1H), 6.45 (d, J=8.5 Гц, 1H), 4.18-4.09 (m, 2Н), 3.86 (s, 3Н), 3.65 -3.53 (m, 2Н), 3.46(s, 4Н), 2.82-2.64 (m, 1Н), 1.93-1.73 (m, 4Н). ВЭЖХ/МС (чистота) 97%. [М+Н]+ 223.1.
n. 7-Метокси-4-(тетрагидропиран-4-ил)-1Н-бензоимидазол-2-иламин
К 97% 3-бром-6-метоксибензол-1,2-диамину (1.86 г, 8.10 ммоль), растворенному в метаноле (40 мл) и воде (10 мл), добавляли 98% бромциан (1.31 г, 12.2 ммоль) при КТ и полученную в результате смесь перемешивали при КТ в течение 20 ч. Реакционную смесь упаривали до удаления метанола. При охлаждении водный раствор подщелачивали аммиаком и упаривали досуха. Остаток вносили в воду и 3 раза экстрагировали дихлорметаном. Объединенный органический слой сушили над Na2SO4, фильтровали и упаривали досуха. Остаток очищали с помощью колоночной хроматографии (дихлорметан/этанол, градиент) с получением 2.11 г (100%) указанного в заголовке соединения в виде бежевого твердого вещества. ВЭЖХ/МС (чистота) 95%. Rt 1.74 мин (метод А). [М+Н]+ 248.1.
о. 2-Бром-N-[7-метокси-4-(тетрагидропиран-4-ил)-1Н-бензоимидазол-2-ил]-изоникотинамид
95% 7-Метокси-4-(тетрагидропиран-4-ил)-1Н-бензоимидазол-2-иламин (1.00 г, 3.84 ммоль), 97% 2-бромпиридин-4-карбоновую кислоту (1.01 г, 4.99 ммоль), гидрат 1-гидроксибензотриазола (156 мг, 1.15 ммоль) и гексафторфосфат [диметиламино-([1,2,3]триазоло[4,5-b]пиридин-3-илокси)-метилен]-диметиламмония (HATU, 1.90 г, 4.99 ммоль) растворяли в N,N-диметилформамиде (30 мл). Затем при КТ добавляли 4-метилморфолин (1.27 мл, 11.5 ммоль) и смесь перемешивали при КТ в течение 3 дней. Реакционную смесь упаривали досуха, вносили в дихлорметан и перемешивали в течение 1 ч. Образовавшийся осадок отфильтровывали и отбрасывали. Фильтрат упаривали досуха и остаток очищали с помощью колоночной хроматографии (дихлорметан/этанол, градиент) с получением 2.67 г (100%) указанного в заголовке соединения в виде светло-желтого тонкодисперсного порошка. ВЭЖХ/МС (чистота) 62%. Rt 2.34 мин (метод А). [М+Н]+ 201.9, 203.9.
р. 2-(3-Гидрокси-3-метилпирролидин-1-ил)-N-[7-метокси-4-(оксан-4-ил)-1Н-1,3-бензодиазол-2-ил]пиридин-4-карбоксамид
62% 2-Бром-N-[7-метокси-4-(тетрагидропиран-4-ил)-1Н-бензоимидазол-2-ил]-изоникотинамид (300 мг, 0.431 ммоль), 3-метилпирролидин-3-ол (77.2 мг, 0.561 ммоль), карбонат цезия (281 мг, 0.863 ммоль) и 2,6-ди-трет-бутил-4-метилфенол (0.009 мл, 0.043 ммоль) растворяли в 1-метил-2-пирролидоне для синтеза (10 мл) и смесь перемешивали при 140°С в течение 3 дней. Реакционную смесь упаривали досуха и остаток очищали с помощью препаративной ВЭЖХ (ацетонитрил/вода, градиент). К растворенной чистой фракции добавляли три капли 1 н. раствор HCl и упаривали досуха с получением 13.0 мг (6%) гидрохлорида указанного в заголовке соединения в виде светло-бежевого твердого вещества. 1Н ЯМР (700 МГц, ДМСО-d6) м.д. = 14.22-12.03 (m, 2Н), 8.09 (d, J=6.4 Гц, 1H), 7.60 (s, 1H), 7.40-7.35 (m, 1H), 7.07 (d, J=8.3 Гц, 1H), 6.85 (d, J=8.4 Гц, 1H), 4.00-3.96 (m, 2H), 3.93 (s, 3H), 3.80-3.70 (m, 2H), 3.61-3.48 (m, 4H), 3.33-3.26 (m, 1H), 2.09-1.99 (m, 2H), 1.80-1.71 (m, 4H), 1.42 (s, ЗН). ВЭЖХ/МС (чистота) 100%. Rt 2.02 мин (метод A). [M+H]+ 452.2.
4. 4-[(Диметиламино)метил]-N-(4-метокси-7-морфолино-1Н-бензимидазол-2-ил)бензамид
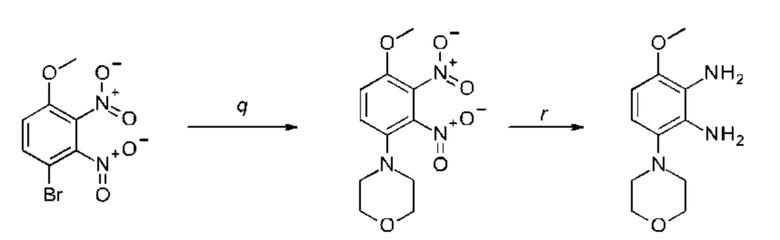
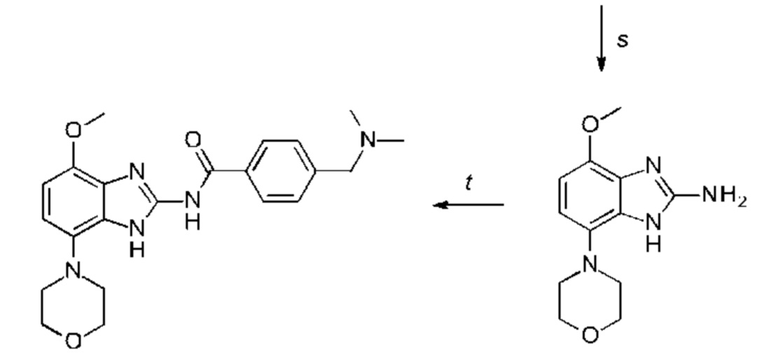
q. 4-(4-Метокси-2,3-динитрофенил)морфолин
В реактор высокого давления помещали 90% 1-бром-4-метокси-2,3-динитробензол (25.0 г, 81.2 ммоль), диоксан (300 мл) и 95% морфолин (29.8 г, 325 ммоль). Смесь перемешивали в течение 15 ч при 100°С. Твердые вещества отфильтровывали и отбрасывали. Фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии (этилацетат/гексан, 60/40) с получением 13.0 г (51%) 4-(4-метокси-2,3-динитрофенил)морфолина в виде темно-красного твердого вещества. ВЭЖХ/МС (чистота) 90%. [М+Н]+ 284.0.
r. 3-Метокси-6-морфолинобензол-1,2-диамин
В 250-мл круглодонную колбу помещали 10% палладий на угле (3.00 г, 2.82 ммоль), метанол (100 мл) и 90% 4-(4-метокси-2,3-динитрофенил)морфолин (12.7 г, 40.3 ммоль). Смесь перемешивали в течение 4 ч при КТ в атмосфере водорода. Твердые вещества отфильтровывали и отбрасывали. Фильтрат упаривали досуха и остаток очищали с помощью колоночной хроматографии (этилацетат/гексан/NEt3, 69.5/29.5/1%) с получением 7.30 г (77%) 3-метокси-6-(морфолин-4-ил)бензол-1,2-диамина в виде розового твердого вещества. 1Н ЯМР (500 МГц, ДМСО-d6) м.д. = 1Н ЯМР (400 МГц, ДМСО-d6) 6.34 (d, J=8.6 Гц, 1H), 6.22 (d, J=8.6 Гц, 1H), 4.22 (s, 4Н), 3.75-3.71 (m, 4Н), 3.70 (s, 3Н), 2.73 -2.68 (m, 4Н). Температура плавления: 113-115°С, ВЭЖХ/МС (чистота) 95%. [М+Н]+ 224.1
s. 4-Метокси-7-морфолино-1Н-бензимидазол-2-амин
К 95% 3-метокси-6-морфолин-4-илбензол-1,2-диамину (4.90 г, 20.8 ммоль), растворенному в метаноле (40 мл) и воде (10 мл) добавляли 98% бромциан (3.38 г, 31.3 ммоль) при КТ и полученную в результате смесь перемешивали при КТ в течение 20 ч. При охлаждении водный раствор подщелачивали аммиаком и упаривали досуха. Остаток непосредственно очищали с помощью колоночной хроматографии (дихлорметан/этанол, градиент) с получением 5.28 г (100%) указанного в заголовке соединения в виде желтого твердого вещества. ВЭЖХ/МС (чистота) 98%. Rt 1.64 мин (метод А). [М+Н]+ 249.1.
t. 4-[(Диметиламино)метил]-N-(4-метокси-7-морфолино-1Н-бензимидазол-2-ил)бензамид
98% 7-Метокси-4-морфолин-4-ил-1Н-бензоимидазол-2-иламин (100 мг, 0.395 ммоль), гидрохлорид 4-[(диметиламино)метил]бензойной кислоты (111 мг, 0.513 ммоль), гексафторфосфат [диметиламино-([1,2,3]триазоло[4,5-b]пиридин-3-илокси)-метилен]-диметиламмония (HATU, 195 мг, 0.513 ммоль), 4-(диметиламино)пиридин (48.2 мг, 0.395 ммоль) и гидрат 1-гидроксибензотриазола (16.0 мг, 0.118 ммоль) растворяли в N,N-диметилформамиде (5 мл). К этой смеси добавляли 4-метилморфолин (0.13 мл, 1.18 ммоль) и смесь перемешивали при КТ в течение 3 дней. Реакционную смесь упаривали досуха и остаток очищали с помощью препаративной ВЭЖХ (ацетонитрил/вода, градиент). К растворенной чистой фракции добавляли три капли 1 н. раствора HCl и упаривали досуха с получением 90.0 мг (51%) гидрохлорида указанного в заголовке соединения в виде бесцветного твердого вещества. 1Н ЯМР (500 МГц, ДМСО-d6) м.д. = 12.58-11.92 (m, 1Н), 10.99-10.86 (m, 1H), 8.21-8.18 (m, 2Н), 7.79-7.76 (m, 2Н), 7.28-7.13 (m, 1H), 6.83 (d, J=8.6 Гц, 1H), 4.38 (d, J=5.4 Гц, 2Н), 3.99-3.95 (m, 4Н), 3.95 (s, 3Н), 3.62-3.46 (m, 4H), 2.72 (d, J=4.8 Гц, 6H). ВЭЖХ/МС (чистота) 100%. Rt 1.74 мин (метод А). [М+Н]+ 410.1.
Метод А
Прибор Agilent Technologies 1200 серии; колонка: Chromolith Performance RP18e; 100×3 мм; подвижная фаза А: вода/0.1% ТФУ, подвижная фаза В: ацетонитрил/0.1% ТФУ; градиент: 1%В в течение 0.2 мин, от 1%В до 100%В за 3.8 мин, удерживание 0.4 мин; скорость потока: 2 мл/мин, длина волны: 220 нм.
Pd(dppf)Cl2 = дигидрохлорид 1,1'-бис(дифенилфосфино)ферроцен]палладия(II)
Пример 3: Исследование соединений настоящего изобретения на ингибирующую активность против человеческих аденозиновых рецепторов в рекомбинантных клетках.
Функциональную активность человеческих рецепторов А2А, А2В, A1 и А3 определяли путем определения количества цАМФ, являющегося вторичным мессенджером для аденозиновых рецепторов.
Для этой цели рекомбинантные клетки HEK293, экспрессирующие человеческие рецепторы А2А или А2В (оба сопряженные с Gs-белками), высевали в 394-луночные микротитрационные планшеты, добавляли тестируемые соединения и агонист (NECA). После 15-минутной инкубации добавляли реагенты HTRF (cAMP dynamic 2, Cis Bio) и уровни цАМФ в клетках определяли с использованием планшет-ридера ENVISION (Perkin Elmer).
Для человеческих рецепторов A1 и А3 использовали рекомбинантные клетки СНО, экспрессирующие либо A1, либо А3-рецептор. Поскольку оба рецептора сопряжены с Gi-белками, протокол анализа был адаптирован:
Клетки высевали в 384-луночные планшеты, добавляли форсколин, тестируемые соединения и агонисты (CPA для A1- и IB-MECA для А3-рецептора). После 30-минутной инкубации добавляли реагенты HTRF (сАМР dynamic 2, Cis Bio) и определяли уровни цАМФ в клетках с использованием планшет-ридера ENVISION (Perkin Elmer).
Полученные исходные данные были нормализованы по отношению к ингибиторному контролю и нейтральному контролю (ДМСО), а нормализованные данные были выравнены с использованием программного обеспечения GeneData.
Соединения настоящего изобретения демонстрируют высокую селективность в отношении аденозиновых рецепторов А2А и А2В по сравнению с аденозиновыми рецепторами A1 и А3 (см., например, данные некоторых примеров соединений настоящего изобретения в таблице 4).
В частности, в отличие от известного антагониста аденозинового рецептора А2А тозаденанта и подобных бензотиазольных производных, соединения настоящего изобретения неожиданно проявляют двойную активность А2А/А2В (см. таблицу 4), которая является предпочтительной для лечения и/или предотвращения гиперпролиферативных и инфекционных заболеваний и нарушений, как раскрыто выше, или соединения настоящего изобретения демонстрируют, по меньшей мере, высокую ингибирующую активность на А2А вместе с другими раскрытыми в данном документе неожиданными преимуществами, которые приводят к высокой эффективности в лечении и/или предотвращении гиперпролиферативных и инфекционных заболеваний и нарушений.
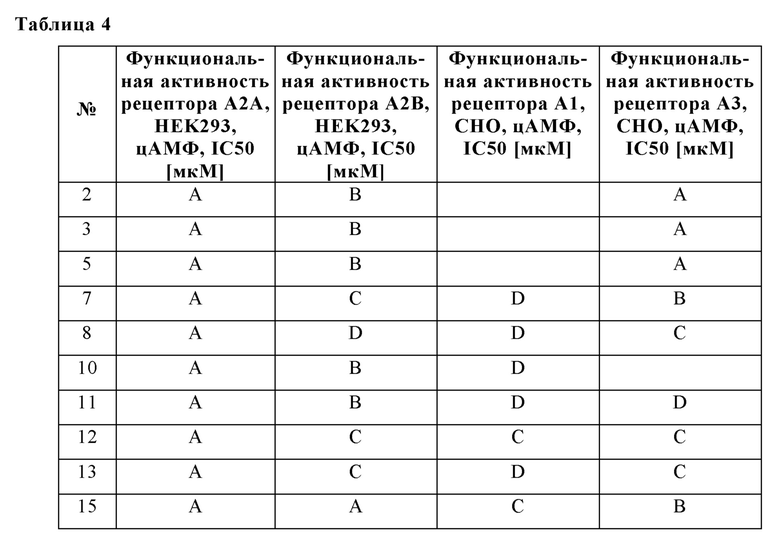
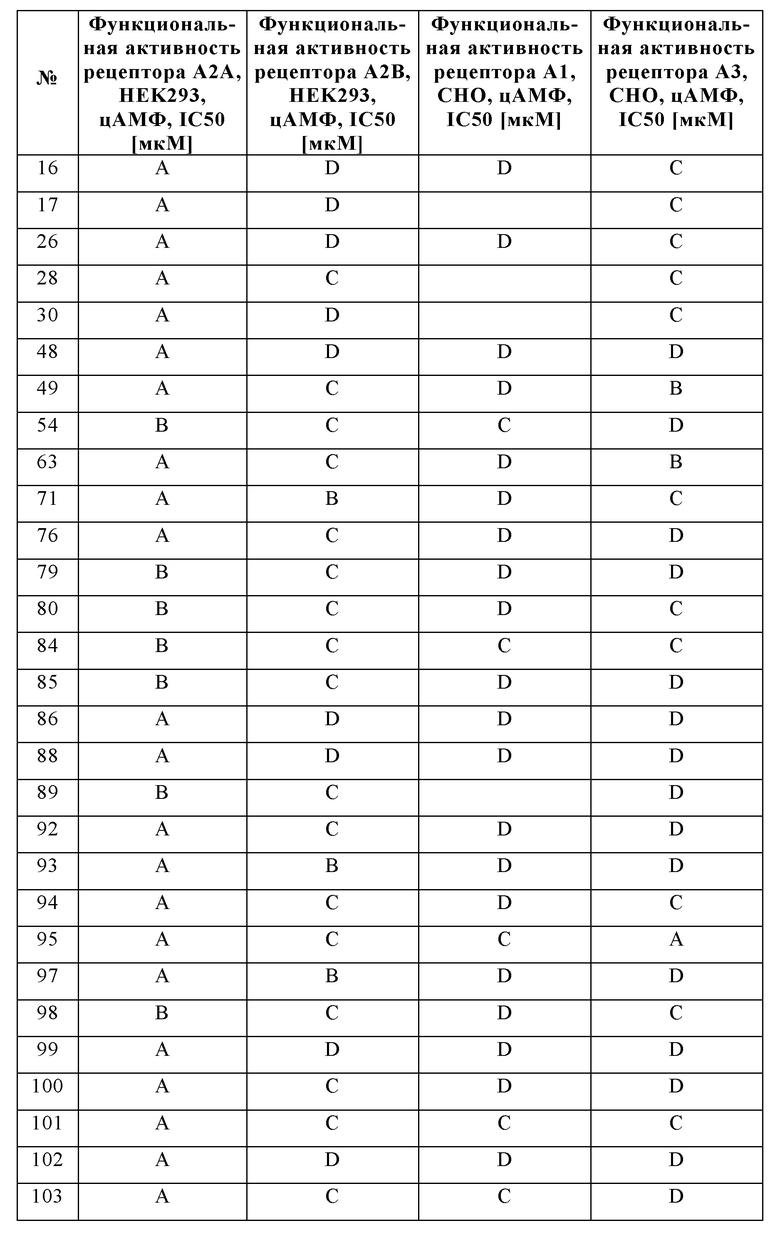
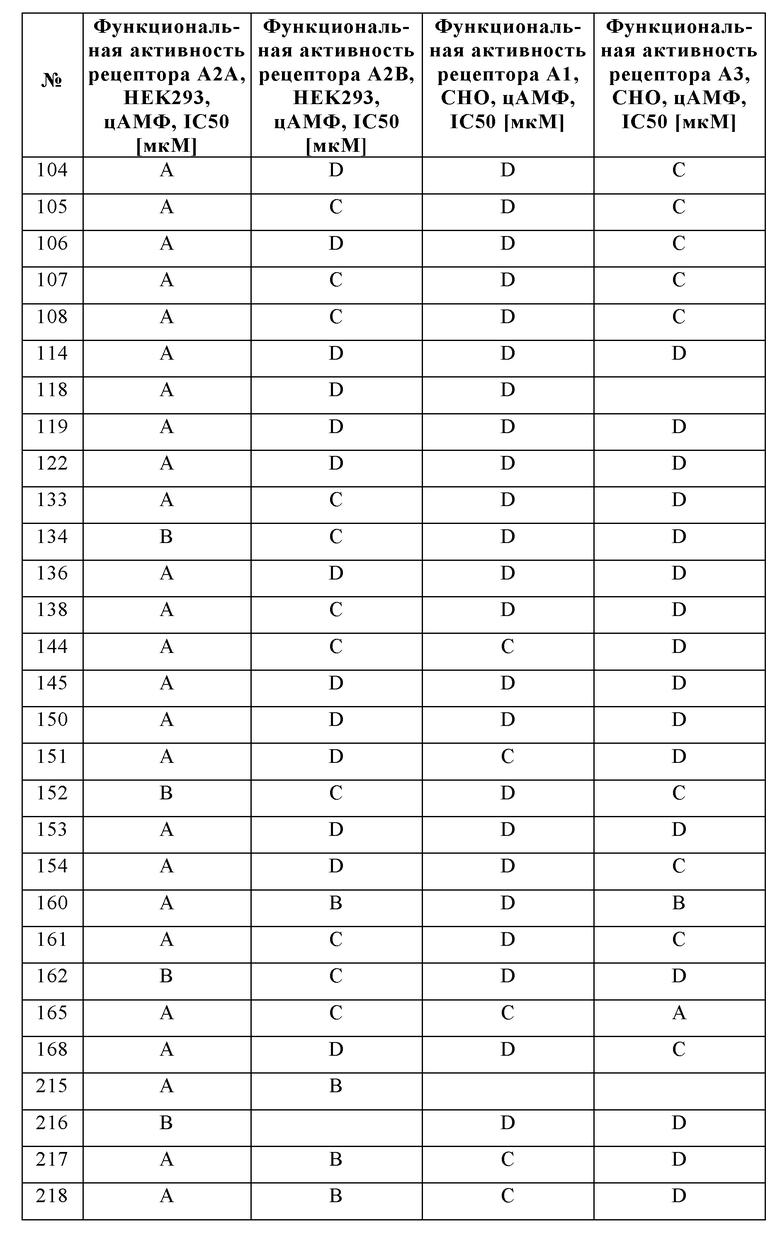
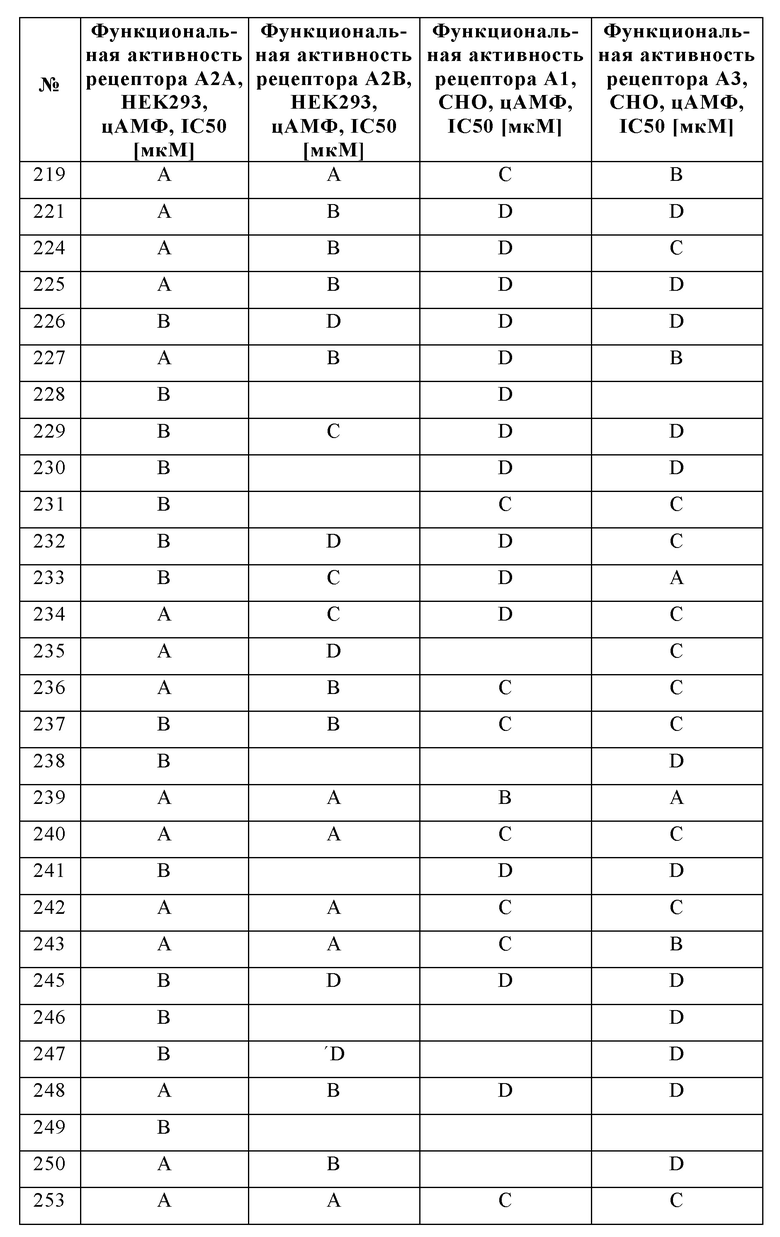
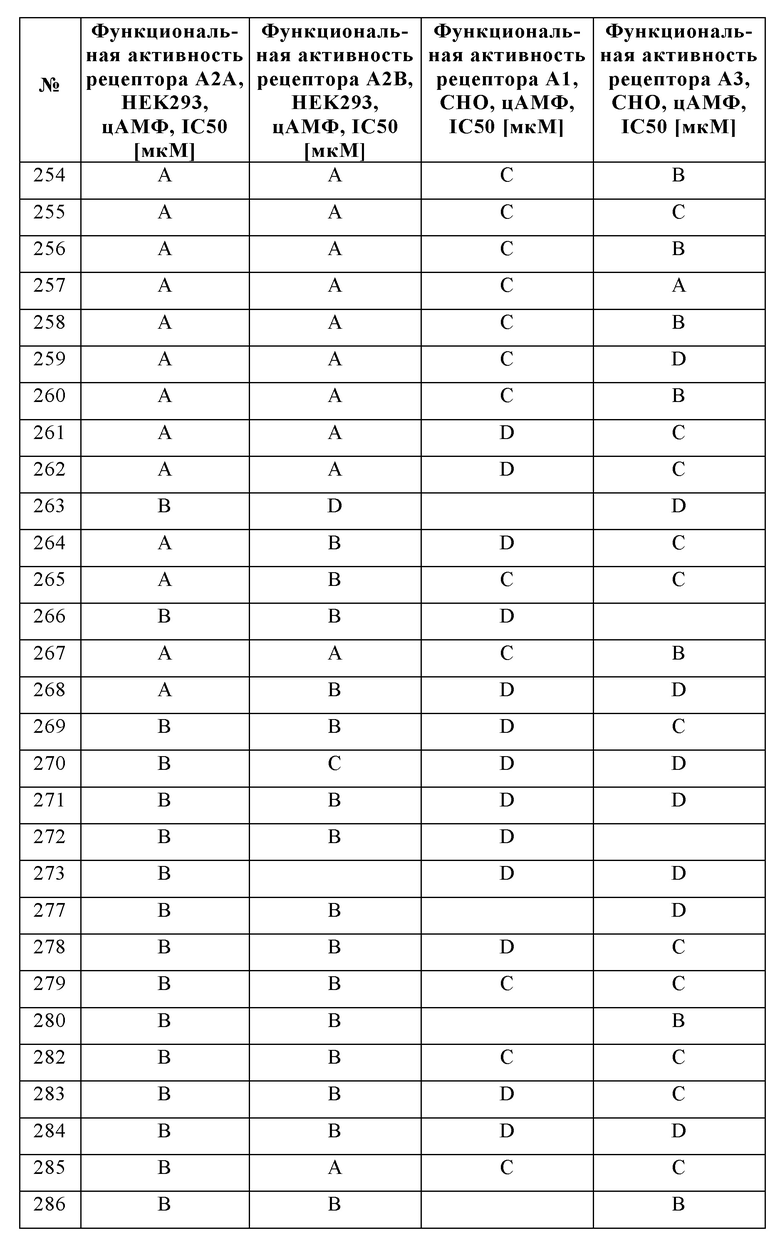
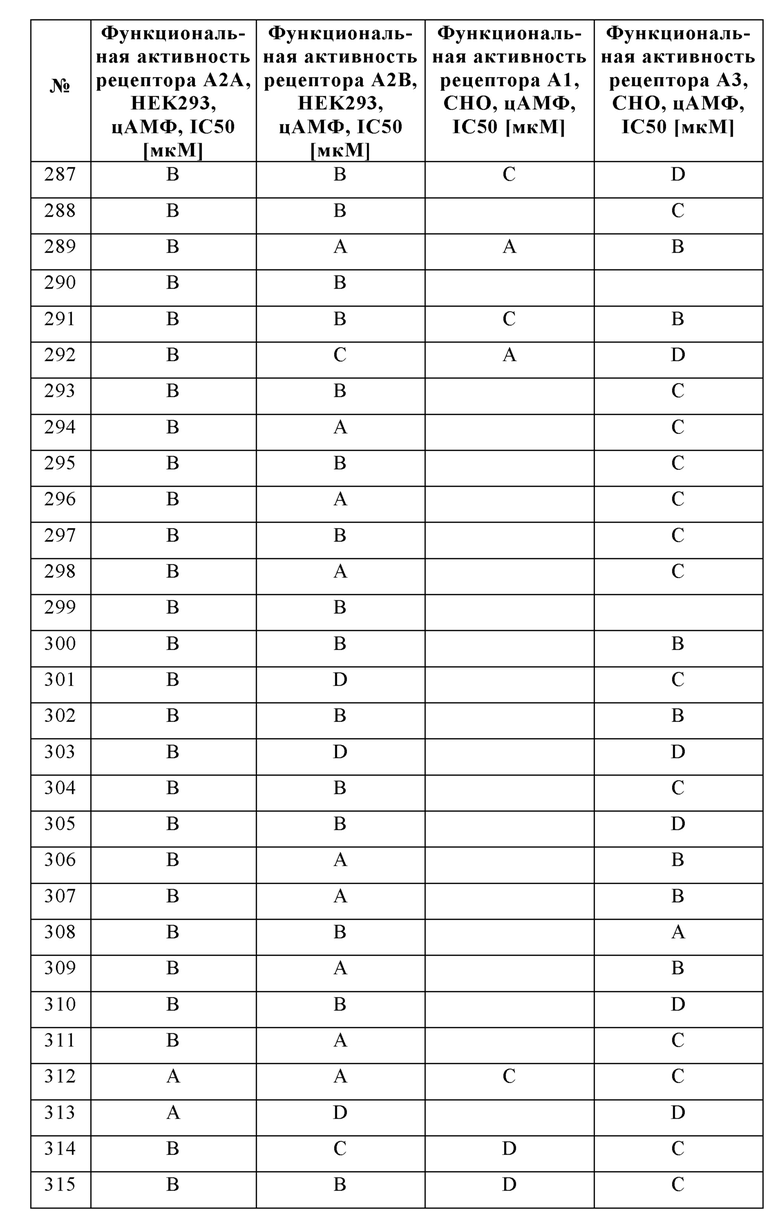
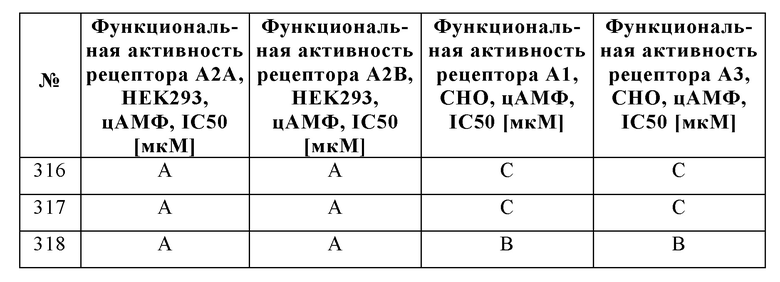
А означает, что значение IC50 составляет <10 нМ, В означает, что значение IC50 составляет <100 нМ, С означает, что значение IC50 составляет <1 мкМ, D означает, что значение IC50 составляет >1 мкМ.
Пример 4: Исследование действия соединений настоящего изобретения против эндогенного человеческого рецептора А2А
Эндогенную функциональную активность Gs-связанного человеческого рецептора А2А измеряли в Т-клетках, где этот рецептор высоко экспрессируется. Определение активности рецептора осуществляли путем определения количества цАМФ, который является вторичным мессенджером для аденозиновых рецепторов.
В общем, человеческие пан-Т-клетки выделяли из человеческих МКПК (набор для выделения пан-Т-клеток MACS, Miltenyi Biotec), которые были получены из свежей цельной крови. Т-клетки высевали в 384-луночные микротитрационные планшеты и обрабатывали тестируемыми соединениями. После 10-минутной инкубации при комнатной температуре добавляли агонист аденозинового рецептора А2А CGS-21680 и планшеты инкубировали еще в течение 45 минут. В заключение, реагенты HTRF (cAMP Femto Kit, CisBio) добавляли в лунки, и через 1 ч определяли уровни цАМФ в клетках с использованием планшет-ридера ENVISION (Perkin Elmer).
Полученные исходные данные были нормализованы по отношению к ингибиторному контролю и нейтральному контролю (ДМСО), а нормализованные данные были выравнены с использованием программного обеспечения GeneData Screener.
Соединения настоящего изобретения демонстрируют способность ингибировать рецептор А2А, экспрессируемый в человеческих Т-клетках, которые инкубируют с агонистом аденозинового рецептора А2А CGS-21680 (по данным количественного определения цАМФ), что является предпочтительным для лечения и/или предотвращения гиперпролиферативных и инфекционных заболеваний и нарушений, как раскрыто выше. Таким образом, соединения настоящего изобретения неожиданно способны предотвращать иммуносупрессию и, следовательно, способны поддерживать индуцированное противораковыми Т-клетками ингибирование роста опухоли, уменьшение или разрушение метастазов и предотвращение неоваскуляризации.
Пример 5: Исследование фармакокинетических свойств соединений настоящего изобретения у крыс и мышей
Целью исследования было получение информации о фармакокинетических свойствах соединений настоящего изобретения у самок крыс Вистар/мышей после однократного внутривенного и перорального введения.
Средства и методы:
Эксперименты на животных (прижизненная фаза)
Самкам крыс Вистар/мышей (n=6) вводили либо одну внутривенную (болюсную) инъекцию, либо выполняли пероральное введение (через желудочный зонд) тестируемого соединения. Дозы 0.2 и 10 мг/кг (в перерасчете на соединение) вводили внутривенно и перорально, соответственно, в виде раствора в ДМСО (0.2%)/ПЭГ 200 (40%)/вода для в/в введения и в виде суспензии в Methocel (0.5%)/Tween 20 (0.25%) в воде для перорального дозирования. Последующие образцы крови брали сублингвально при ингаляции изофлурана у 3 животных на один путь введения через 0.1 (только в/в), 0.25 (только перорально), 0.5, 1, 2, 4, 6 и 24 ч и дополнительно обрабатывали для получения плазмы. Также, образцы мочи и кала у 3 крыс на один путь введения собирали в течение промежутка времени от 0 до 24 ч и объединяли для анализа.
Биоаналитика:
Концентрации соединений в плазме, фекалиях определяли количественно с использованием СВЭЖХ метода вместе с масс-спектрометрическим детектированием (ЖХ-МС/МС), ранее разработанным в «Институте метаболизма лекарств и фармакокинетики». Система ЖХ-МС/МС состояла из устройства Waters Acquity UPLC в сочетании с АВ Sciex масс-спектрометром API 5500 Q-trap. Разделение посредством СВЭЖХ проводили на колонке с обращенной фазой (HSS Т3, 1.8 мкМ, 2.1×50 мм) с использованием градиента подвижной фазы с 0.1% муравьиной кислоты и ацетонитрилом в качестве элюентов. Детектирование соединений осуществляли с использованием множественного мониторинга реакций в режиме положительной ионизации. В образцы плазмы добавляли внутренний стандарт (20 мкл) и аналит экстрагировали из матрицы с использованием трет-бутилметилового эфира (tBME). Органическую фазу упаривали досуха в потоке азота. Остаток растворяли в смеси ацетонитрил/0.1% муравьиной кислоты для анализа ЖХ-МС/МС. Образцы фекалий гомогенизировали с 4-кратным их объемом смеси этанол/вода (4:1, об./об.). В аликвоты водно-этанольных экстрактов добавляли внутренний стандарт, разбавляли смесью ацетонитрил/вода (1:1, об./об.) и непосредственно вводили в систему ЖХ-МС/МС.
Фармакокинетическое оценивание:
Фармакокинетические параметры Cmax и tmax были взяты из наблюдаемых данных. Площадь под кривой (AUC), клиренс (CL), объем (V), период полураспада (t1/2), F и все нормализированные по дозе значения были рассчитаны с использованием специального программного обеспечения "DDS-ТОХ". Значения "DDS-TOX" были оценены для нескольких соединений и показаны сопоставимыми со значениями, предоставленными проверенным программным обеспечением WinNonLin. Значения AUC рассчитывали с помощью некомпартментального анализа с использованием линейно-логарифмического метода трапеций. Числовые данные для средних концентраций в плазме и полученные фармакокинетические параметры были округлены до 3 значащих цифр для представления. Данные о пероральной биодоступности и экскреции - выраженные в % дозы - отображены с использованием 2 значащих цифр.
В отличие от известного антагониста аденозинового рецептора А2А тозаденанта и подобных бензотиазольных производных, соединения настоящего изобретения неожиданно демонстрируют лучшие фармакокинетические свойства у мыши как у животной модели, актуальной для злокачественного новообразования (см. таблицу 6), что является предпочтительным для лечения и/или предотвращения гиперпролиферативных и инфекционных заболеваний и нарушений, как раскрыто выше.
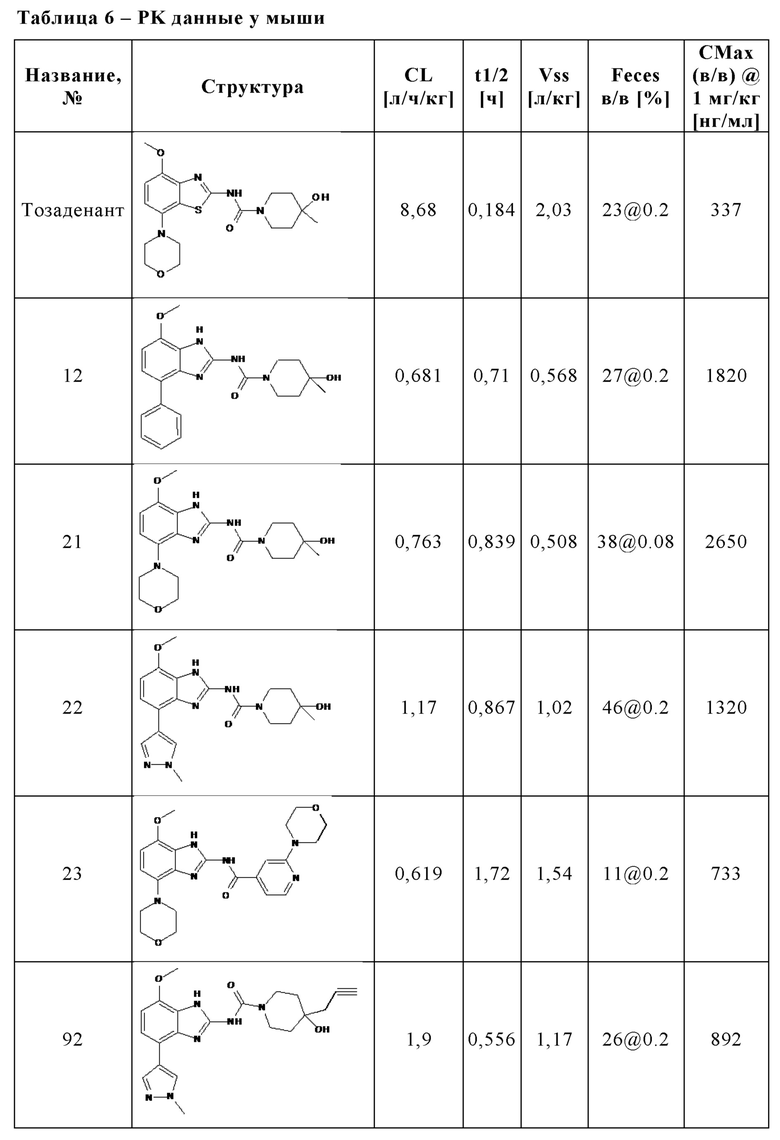
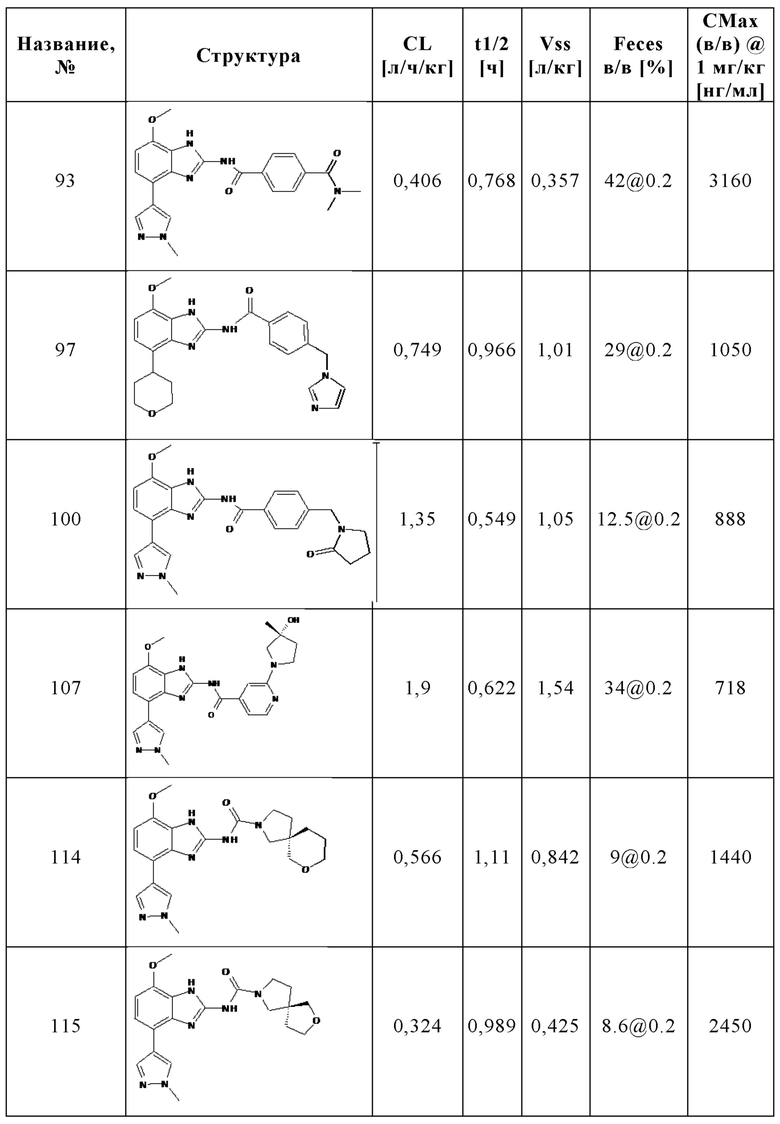
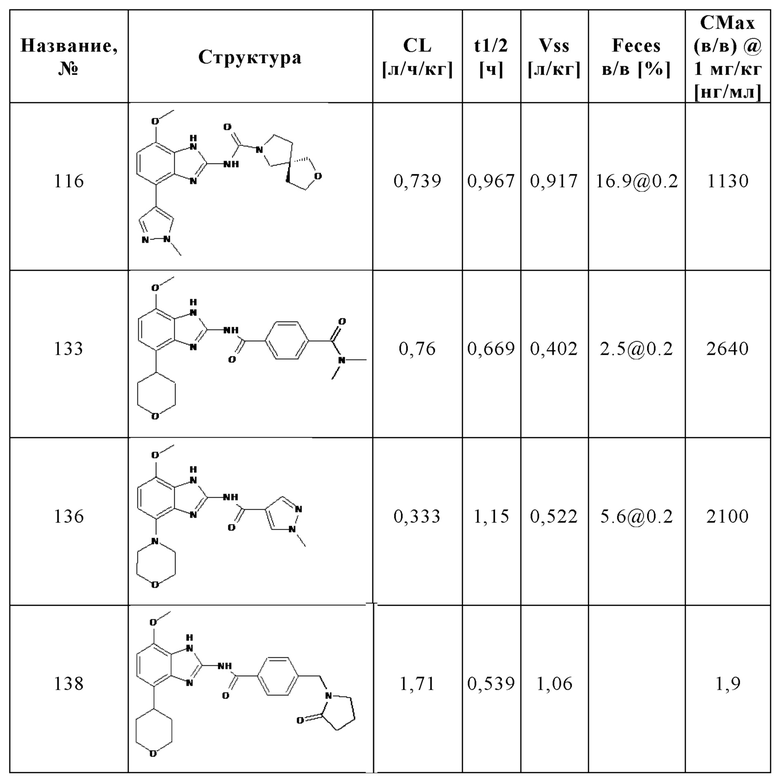
Пример 6: Исследование действия соединений настоящего изобретения на мышиных Т-клетках
Подготовка:
Аденозин (Ado) в микроокружении опухоли может ингибировать активность Т-клеток путем передачи сигналов через рецепторы А2А и подавлять секрецию цитокинов Т-клетками. А2А-специфические агонисты, такие как CGS-21680, выполняют похожую работу по ингибированию секреции Т-клеток цитокинов in vitro и in vivo. Потенциальные антагонисты А2А или двойные антагонисты А2А/А2В могут спасти Т-клетки от этого ингибирования. В данном случае мы опишем систему in vitro, которую мы создали, используя пан-Т-клетки из селезенки мыши для скрининга потенциальных антагонистов А2А или двойных антагонистов А2А/А2В на их активность. Описанный метод включает использование предварительно покрытых CD3/CD28 гранул для стимуляции пан-Т-клеток, очищенных от спленоцитов мыши, в комбинации с добавлением агониста А2А вместе с потенциальными антагонистами А2А или двойными антагонистами А2А/А2В для оценки потенцирования продукции цитокинов Т-клетками.
Описание анализа:
Вкратце, мышиные пан-Т-клетки очищают от тканей селезенки мышей BALB/c с использованием набора для выделения пан-Т-клеток Pan Т cell isolation kit Mouse II (MACS Miltenyi biotech Cat# Order №130-095-130) в соответствии с протоколом производителя. Очищенные Т-клетки высевают в 96-луночные полистирольные круглодонные микропланшеты фирмы Nunc™ в среде RPMI с 10% инактивированной нагреванием фетальной бычьей сыворотки. Клетки выдерживают при 37°С в течение 1 ч, а затем активируют предварительно покрытыми CD3/CD28 гранулами (Dynabeads™ Mouse Т-Activator CD3/CD28; Cat# 11456D). Через 30 мин клетки обрабатывают различными дозами тестируемого(-ых) антагониста(-ов). Клетки инкубируют в течение дополнительных 30 мин при 37°С перед обработкой агонистом А2А CGS-21680 (1 мкМ) или нейтральным контролем (ДМСО). После 24-часовой инкубации уровни IL-2 в супернатантах и после 48-часовой инкубации уровни IFN-γ в супернатантах измеряют с помощью ELISA в соответствии с протоколом изготовителя (R&D systems Cat# DY402 (IL-2); DY485 (IFN-γ)). Как только концентрации рассчитаны, вычисляют разницу концентрации цитокинов в контроле ДМСО и в контроле одного агониста (называется Δ), и с использованием Microsoft Excel вычисляют процент спасения при каждой концентрации антагониста. Эти проценты спасения цитокинов зависимым от дозы антагониста образом наносят на график с помощью программного обеспечения GraphPad Prism и рассчитывают значение IC50.
В отличие от известного антагониста аденозинового рецептора А2А тозаденанта, соединения настоящего изобретения демонстрируют, что они способны спасать Т-клетки от ингибирования и способны предотвращать подавление секреции цитокинов, индуцированной аденозином или А2А-специфическими агонистами, такими как CGS-2168 (см. таблицу 7),что является предпочтительным для лечения и/или предотвращения гиперпролиферативных и инфекционных заболеваний и нарушений, как раскрыто выше.
Таким образом, соединения настоящего изобретения неожиданно способны предотвращать иммуносупрессию и, следовательно, способны поддерживать индуцированное противораковыми Т-клетками ингибирование роста опухоли, уменьшение или разрушение метастазов и предотвращение неоваскуляризации.
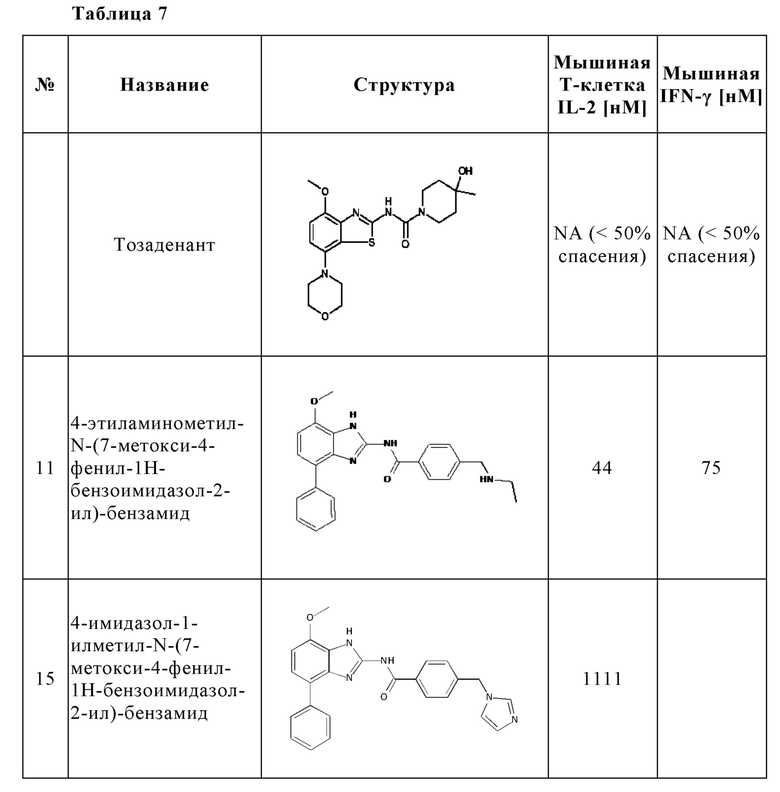
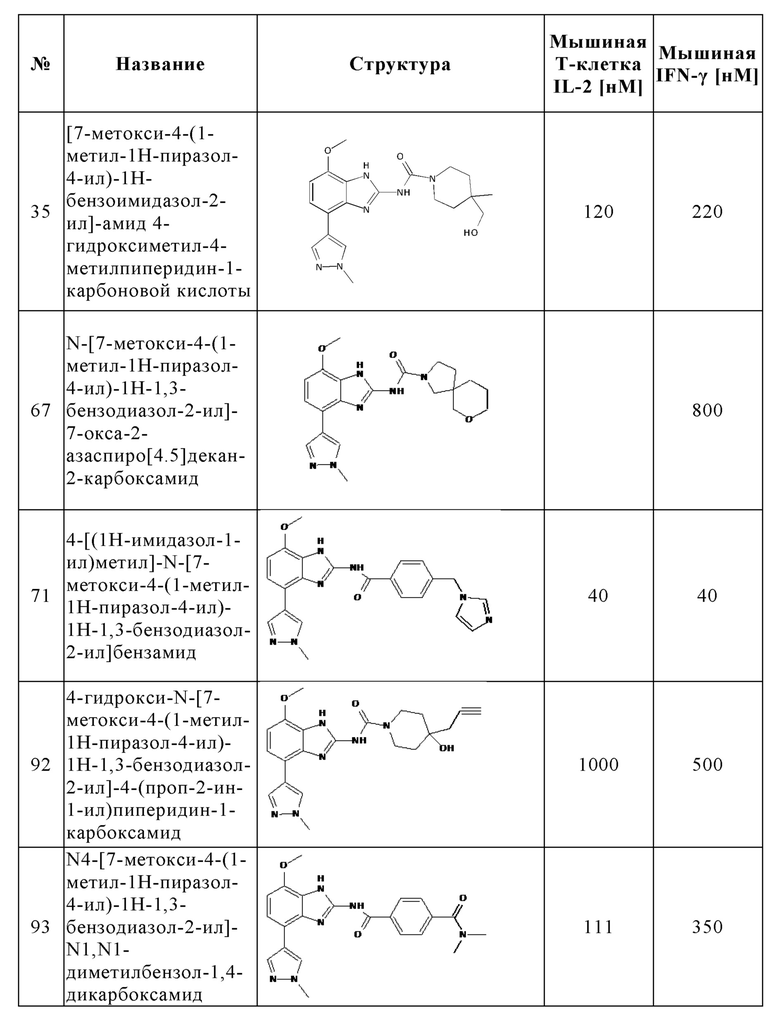
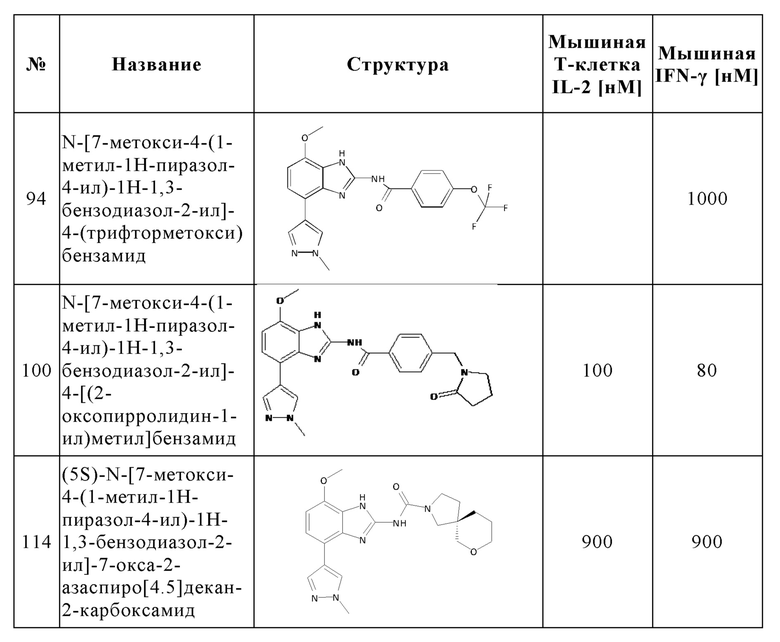
Пример 7: Инъекционные флаконы
Раствор 100 г соединения настоящего изобретения и 5 г динатрийгидрофосфата в 3 л бидистиллированной воды доводят до рН 6.5 с помощью 2 н. соляной кислоты, фильтруют в стерильных условиях, переносят в инъекционные флаконы, лиофилизируют в стерильных условиях и герметизируют в стерильных условиях. Каждый инъекционный флакон содержит 5 мг соединения настоящего изобретения.
Пример 8: Раствор
Раствор готовят из 1 г соединения настоящего изобретения, 9.38 г NaH2PO4 2 H2O, 28.48 г Na2HPO4⋅12 H2O и 0.1 г хлорида бензалкония в 940 мл бидистиллированной воды. Значение рН устанавливают на 6.8, и раствор доводят до 1 л и стерилизуют с помощью облучения.
Пример 9: Ампулы
Раствор 1 кг соединения настоящего изобретения в 60 л бидистиллированной воды фильтруют в стерильных условиях, переносят в ампулы, лиофилизируют в стерильных условиях и герметизируют в стерильных условиях. Каждая ампула содержит 10 мг соединения настоящего изобретения.
Изобретение относится к бензимидазольным производным, фармацевтическому препарату, содержащему указанные производные, который предназначен для лечения и/или профилактики физиологических и/или патофизиологических состояний, опосредованных активностью аденозинового рецептора A2A или аденозиновых рецепторов A2A и A2B, например, таких как гиперпролиферативные или инфекционные заболевания и нарушения. 5 н. и 6 з.п. ф-лы, 7 табл., 9 пр.
1. Соединение, выбранное из группы, состоящей из:

















и его физиологически приемлемые соли и стереоизомеры, включая их смеси во всех соотношениях.
2. Соединение по п. 1 и его физиологически приемлемые соли и стереоизомеры, включая их смеси во всех соотношениях, в качестве ингибитора аденозиновых рецепторов A2A и/или A2B.
3. Фармацевтический препарат для ингибирования активности аденозинового рецептора A2A или аденозиновых рецепторов A2A и A2B, содержащий эффективное количество по меньшей мере одного соединения по п. 1 и/или его физиологически приемлемые соли и стереоизомеры, включая их смеси во всех соотношениях.
4. Фармацевтический препарат по п. 3, дополнительно содержащий наполнители и/или вспомогательные вещества.
5. Способ приготовления фармацевтического препарата, отличающийся тем, что соединение по п. 1 и/или одну из его физиологически приемлемых солей и стереоизомеров, включая их смеси во всех соотношениях, вводят в пригодную дозированную форму вместе с твердым, жидким или полужидким наполнителем или вспомогательным веществом.
6. Лекарственное средство, содержащее эффективное количество по меньшей мере одного соединения по п. 1 и/или одну из его физиологически приемлемых солей и стереоизомеров, включая их смеси во всех соотношениях, для применения для лечения и/или профилактики физиологических и/или патофизиологических состояний, опосредованных активностью аденозинового рецептора А2А или аденозиновых рецепторов А2А и А2В.
7. Лекарственное средство, содержащее эффективное количество по меньшей мере одного соединения по п. 1 и/или одну из его физиологически приемлемых солей и стереоизомеров, включая их смеси во всех соотношениях, для применения для лечения и/или профилактики физиологических и/или патофизиологических состояний, выбранных из группы, состоящей из гиперпролиферативных и инфекционных заболеваний и нарушений, опосредованных активностью аденозинового рецептора А2А или аденозиновых рецепторов А2А и А2В.
8. Лекарственное средство по п. 7, где гиперпролиферативное заболевание или нарушение представляет собой злокачественное новообразование.
9. Лекарственное средство по п. 8, где злокачественное новообразование выбирают из группы, состоящей из острой и хронической лимфоцитарной лейкемии, острой гранулоцитарной лейкемии, рака коры надпочечников, рака мочевого пузыря, рака головного мозга, рака молочной железы, рака шейки матки, гиперплазии шейки матки, рака шейки матки, хориокарциномы, хронической гранулоцитарной лейкемии, хронической лимфоцитарной лейкемии, рака ободочной кишки, рака эндометрия, рака пищевода, эссенциального тромбоцитоза, урогенитальной карциномы, глиомы, глиобластомы, волосатоклеточной лейкемии, карциномы головы и шеи, болезни Ходжкина, саркомы Капоши, карциномы легких, лимфомы, злокачественной карциноидной опухоли, злокачественной гиперкальциемии, злокачественной меланомы, злокачественной инсулиномы поджелудочной железы, медуллярной карциномы щитовидной железы, меланомы, множественной миеломы, фунгоидного микоза, миелоидной и лимфоцитарной лейкемии, нейробластомы, неходжкинской лимфомы, немелкоклеточного рака легкого, остеогенной саркомы, карциномы яичника, карциномы поджелудочной железы, истинной полицитемии, первичной карциномы головного мозга, первичной макроглобулинемии, рака предстательной железы, почечно-клеточного рака, рабдомиосаркомы, рака кожи, мелкоклеточного рака легкого, саркомы мягких тканей, плоскоклеточного рака, рака желудка, рака яичка, рака щитовидной железы и опухоли Вильмса.
10. Лекарственное средство по п. 7, где гиперпролиферативное заболевание или нарушение выбирают из группы, состоящей из возрастной макулярной дегенерации, болезни Крона, цирроза, хронических нарушений, связанных с воспалением, пролиферативной диабетической ретинопатии, пролиферативной витреоретинопатии, ретролентальной фиброплазии, гранулематоза, иммунной гиперпролиферации, связанной с трансплантацией органа или ткани и иммунопролиферативного заболевания или нарушения, выбранного из группы, состоящей из воспалительного заболевания кишечника, псориаза, ревматоидного артрита, системной красной волчанки (SLE), гиперпролиферации сосудов на фоне гипоксии сетчатки и васкулита.
11. Лекарственное средство по п. 7, где инфекционное заболевание или нарушение выбирают из группы, состоящей из
a) вирусиндуцированных инфекционных заболеваний, которые вызваны ретровирусами, гепаднавирусами, герпесвирусами, флавивирусами и/или аденовирусами, где ретровирусы выбирают из лентивирусов или онкоретровирусов, где лентивирусы выбирают из группы, состоящей из HIV-1, HIV-2, FIV, BIV, вирусов SIV, SHIV, CAEV, VMV и EIAV, и онкоретровирусы выбирают из группы, состоящей из HTLV-I, HTLV-II и BLV, гепаднавирусы выбирают из группы, состоящей из HBV, GSHV и WHV, герпесвирусы выбирают из группы, состоящей из HSV I, HSV II, EBV, VZV, HCMV или HHV 8, и флавивирусы выбирают из группы, состоящей из HCV, вируса Западного Нила и вируса желтой лихорадки,
b) бактериальных инфекционных заболеваний, которые вызваны грамположительными бактериями, где грамположительные бактерии выбирают из группы, состоящей из метициллин-чувствительных и метициллин-резистентных стафилококков (включая Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus haemolyticus, Staphylococcus hominis, Staphylococcus saprophyticus и коагулазоотрицательные стафилококки), Staphylococcus aureus с промежуточной чувствительностью к гликопептидам (GISA), пенициллин-чувствительных и пенициллин-резистентных стрептококков (включая Streptococcus pneumoniae, Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus avium, Streptococcus bovis, Streptococcus lactis, Streptococcus sanguis и Streptococci группы С (GCS), Streptococci группы G (GGS) и стрептококки группы «viridans»), энтерококков (включая ванкомицин-чувствительные и ванкомицин-резистентные штаммы, такие как Enterococcus faecalis и Enterococcus faecium), Clostridium difficile, listeria monocytogenes, Corynebacterium jeikeium, Chlamydia spp (включая С. pneumoniae) и Mycobacterium tuberculosis,
c) бактериальных инфекционных заболеваний, которые вызваны грамотрицательными бактериями, где грамотрицательные бактерии выбирают из группы, состоящей из рода Enterobacteriacae, включая Escherichia spp. (включая Escherichia coli), Klebsiella spp., Enterobacter spp., Citrobacter spp., Serratia spp., Proteus spp., Providencia spp., Salmonella spp., Shigella spp., рода Pseudomonas (включая P. aeruginosa), Moraxella spp. (включая M. catarrhalis), Haemophilus spp. и Neisseria spp.,
d) инфекционных заболеваний, индуцированных внутриклеточными активными паразитами, выбранными из группы, состоящей из филума Apicomplexa или Sarcomastigophora (включая Trypanosoma, Plasmodia, Leishmania, Babesia или Theileria), Cryptosporidia, Sacrocystida, Amoebia, Coccidia и Trichomonadia.
| WO 2006099379 A2, 21.09.2006 | |||
| CN 102731488 A, 17.10.2012 | |||
| 2-ИМИДАЗОБЕНЗОТИАЗОЛЫ КАК ЛИГАНДЫ АДЕНОЗИНОВОГО РЕЦЕПТОРА | 2004 |
|
RU2340612C2 |
| ПРОИЗВОДНЫЕ БЕНЗОТИАЗОЛА | 2005 |
|
RU2382782C2 |
| ПРОИЗВОДНЫЕ БЕНЗОТИАЗОЛА И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С АДЕНОЗИНОВЫМИ А-РЕЦЕПТОРАМИ | 2004 |
|
RU2348622C2 |
| WO 2005000842 A1, 06.01.2005 | |||
| WO 2005116026 A1, 08.12.2005 | |||
| Wan, Zhao-Kui et al "Dienophilicity of imidazole in inverse electron demand Diels-Alder reactions: Cycloadditions with 1,2,4,5-tetrazines and the | |||

