ОБЛАСТЬ ТЕХНИКИ
Данное изобретение относится к способу окисления этилена до этиленоксида и гидрирования этиленоксида в этиленгликоль.
УРОВЕНЬ ТЕХНИКИ
Роль этиленгликоля в обществе значительно возросла с тех пор, как вскоре после его первого синтеза свойства молекулы стали предметом ожесточенного спора между двумя гигантами ранней органической химии, Адольфом Вюрцем и Германом Кольбе. В частности, Вюрц и Кольбе оспаривали функциональность и химическую формулу этиленгликоля среди все еще возникающих представлений о гомологах спирта, которые были ключом к великим теориям Кольбе о химической структуре. Лаборатория вскоре превратилась в опосредованную войну растущей промышленной и технической мощи растущего соперничества между Германией и Францией, когда обе страны выделяли значительные ресурсы на научные исследования, а волшебство Вюрца в области химического синтеза давало Франции значительное преимущество. Спор был завершен только применением оружия, когда Германская конфедерация Бисмарка аннексировала эльзасскую родину Вюрца в результате франко-прусской войны и, таким образом, фактически превратила международный спор во внутренний.
Сегодня интерес к этиленгликолю более мирный, но тем более конкурентный, потому что этиленгликоль является одним из наиболее широко производимых органических химикатов. С тех пор как накануне Первой мировой войны началось крупномасштабное промышленное производство этиленгликоля, резкое увеличение использования двигателей внутреннего сгорания для моторных автомобилей и других транспортных средств стимулировало спрос на этиленгликоль в качестве охлаждающей жидкости и антифриза. С тех пор рост производства этиленгликоля только ускорился, так что к 2017 году оценочное мировое производство этиленгликоля превысило 25 миллиардов тонн.
Этиленгликоль обычно получают как одно из многих производных этиленоксида, и, хотя доступны другие способы производства, большая часть производится из этиленоксида жидкофазным не каталитическим способом термической гидратации. Поскольку этиленоксид взаимодействует с этиленгликолями легче, чем с водой, неизбежно, что будут появляться смеси моноэтиленгликоля (МЭГ), а также сопутствующих продуктов с более высоким содержанием гликоля, таких как диэтиленгликоль (ДЭГ), триэтиленгликоль (ТЭГ) и даже высших этиленгликолей. Хотя эти высшие гликоли имеют значительную экономическую ценность, многие производители и заводские операторы хотели бы избежать их образования, потому что рынок конечных потребителей этих продуктов не так хорошо развит, и может быть трудно найти промышленных потребителей, которым они нужны, и продать им эти высшие гликоли.
Для подавления реакции между продуктом гликоля и этиленоксидом, и тем самым уменьшить образование этих высших гликолей, проводят обычную не каталитическую гидратацию с количеством воды, которое намного превышает стехиометрическое количество воды для гидратации этиленоксида до этиленгликоля, например, 15-40 моль воды на моль этиленоксида. Такое добавление избытка воды эффективно для уравновешивания кинетически благоприятной параллельной реакции между продуктом гликоля и этиленоксидом, которая, как упомянуто выше, конкурирует с гидратацией этиленоксида до моноэтиленгликоля. Однако, будучи эффективным для подавления образования высших этиленгликолей, использование большого избытка воды по сравнению с этиленоксидом представляет проблему для оператора установки при удалении этих больших избытков непрореагировавшей воды, поскольку такое удаление является энергоемким и требует производственных мощностей для выпаривания/дистилляции. Соответственно, проводились интенсивные исследования альтернатив стандартной термической гидратации этиленоксида для производства этиленгликоля.
Одним из первых примеров альтернативы традиционной термической гидратации является гомогенная каталитическая гидратация этиленоксида до моноэтиленгликоля. Самые ранние примеры этого подхода включали гомогенный катализ серной кислоты и связанных с ней солей (см. Othmer, D.F. and Thakar, M.S., Glycol Production - Hydration of Ethylene Oxide. Ind. Eng. Chem. 1958, 50, 1235). В европейском патенте № 0,123,700 описана модификация таких более ранних поколений кислотных катализаторов путем их обработки, например, этиламинами, чтобы частично нейтрализовать их, в надежде улучшить селективность реакции гидратации по моноэтиленгликолю. С тех пор, для гомогенных систем были предложены другие соли, такие как соли четвертичного фосфония, как описано в патенте США № 4,160,116, и соли металлатов и бикарбонатов, как описано в патенте США № 7,683,221. Все более креативные сочетания органических веществ, таких как ЭДТК и соединения Салена, также были предложены в качестве гомогенных катализаторов (см. Hal, J.W., Ledford, J. S., and Zhang, X., Catalysis Today 123 (2007), 310-315).
Гомогенные системы катализаторов часто применяют в двухстадийном способе производства этиленгликоля, см., например, патент США № 4,519,875, в котором этиленоксид сначала взаимодействует с диоксидом углерода для производства этиленкарбоната, который затем гидролизуют до этиленгликоля, где на обеих стадиях обычно применяют один и тот же катализатор. Вслед за этим новаторским патентом, постоянные исследования привели к постепенным усовершенствованиям двухстадийного способа. Например, в патенте США № 5,763,691 реакцию карбонизации катализируют в абсорбате этиленоксида в присутствии катализатора карбонизации на основе соли галогенированного органического фосфония. Дополнительные исследования значительно расширили круг известных катализаторов; см., например, макроциклические хелатирующие соединения («краун-эфиры»), описанные в патенте США № 7,453,015.
Хотя гомогенные катализаторы улучшили селективность реакции гидратации по этиленгликолю по сравнению с не каталитической гидратацией, гомогенные катализаторы имеют недостатки, которые более чем перекрывают эти улучшения. В частности, раствор продукта гликоля содержит растворенный или суспендированный гомогенный катализатор - по существу вызывая загрязнение продукта катализатором, что значительно ухудшает качество продукта, а также является очень агрессивным, как, например, в случае кислотных катализаторов. Таким образом, необходима дополнительная стадия отделения гомогенного катализатора от раствора продукта гликоля, увеличивающая стоимость и усложняющая способ. Например, хотя в патенте США № 5763691, упомянутом выше, представлены попытки упростить двухстадийное производство этиленгликоля через взаимодействие этиленоксида и диоксида углерода в абсорбате, патент все еще требует отдельной стадии дистилляции для удаления катализатора карбонизации.
Такая сложность способа приносит с собой дополнительный недостаток, заключающийся в том, что он не очень универсален и не гибок. Например, способу не хватает гибкости для того, чтобы работать частично каталитически, то есть в смешанном каталитическом и не каталитическом режимах, что позволило бы производить больше высших гликолей, если того пожелает оператор, а также адаптировать завод в случае некоторых сбоев в логистике и эксплуатации, когда он не может работать в каталитическом режиме, например, когда достаточное количество катализатора недоступно. Двухстадийному способу гидратации также не хватает универсальности для использования как на новых заводах, так и на модернизированных. Двухстадийный способ гидратации можно использовать только на новых заводах и нельзя использовать на модернизированных, потому что в случае модернизации необходимо будет удалить и заменить все «внутренние интерфейсы», что сделает такую модернизацию слишком дорогостоящей. Недостатком двухстадийного способа гидратации является необходимость инвентаризации концентрированного этиленоксида (концентрация более 70%) для производства промежуточного этиленкарбоната на первой стадии способа. Реакция карбонизации на первой стадии проводится барботированием диоксида углерода в такой концентрированный жидкий раствор этиленоксида при высокой температуре и давлении (например, более 1,6 МПа). Большой объем концентрированного этиленоксида требует значительных дополнительных мер (например, хранения в жидком хладагенте) для предотвращения реакций или событий, которые могут привести к возгоранию, горению, дефлаграции, детонации или взрыву любого газового потока, но особенно тех, которые содержат типовые концентрации этиленоксида.
Неудивительно, что с учетом вышеупомянутых недостатков как не каталитических, так и гомогенно-каталитических систем гидратации, также исследовали гетерогенный катализ с использованием твердых катализаторов или катализаторов на подложке. В поисках преимуществ более высокой селективности каталитической гидратации без вышеупомянутых недостатков, исследователи обратили свое внимание на гетерогенные системы катализаторов. Поскольку гетерогенные катализаторы не абсорбируются в жидкофазные реакционные смеси или растворы продуктов, не загрязняют растворы продуктов, не требуют стадии разделения и способствуют непрерывной работе реактора гидратации, они вызывают значительный интерес.
Множество твердых материалов было предложено в качестве гетерогенных катализаторов для гидратации. Этот список включает цеолиты, такие как описаны в патенте США № 3,028,434, а также аморфные алюминосиликаты, твердые основания и кислотные катализаторы на подложке, такие как описаны в патенте США № 4,165,440. Также были предложены ионообменные смолы, частично нейтрализованная амином стирол-дивинилбензольная смола на основе сульфоновой кислоты описана в EP 0 123 709; катализаторы на основе четвертичной фосфониевой ионообменной смолы, см. публикацию PCT WO 2000/35840; и ионообменные смолы на основе сильного основания, суммированные в патенте США № 6,211,419, были среди первых ионообменных смол, предложенных для катализа реакции воды и этиленоксида. Более поздние публикации расширили область до широкого спектра анионообменных смол, см., например, патент Китая № 1208135.
Хотя прекрасные результаты были достигнуты с использованием гетерогенных катализаторов на основе ионообменных смол, их использование также имеет определенные недостатки. В частности, ионообменная смола имеет ограниченную устойчивость к нагреванию, так что в типовых условиях реакции в течение длительного времени службы смолы набухают, что приводит к дезактивации. Набухание, которое возникает в результате внутренней полимеризации в слое смолы и общего накопления высокомолекулярных побочных продуктов, является серьезной проблемой, которая является существенным ограничивающим фактором срока службы ионообменной смолы. Смолы обычно претерпевают некоторое обратимое набухание во время работы из-за воздействия растворителей; однако при осторожном обращении это набухание можно уменьшить, а срок службы ионообменной смолы продлить. Однако воздействие этиленоксида на ионообменную смолу может вызвать более проблематичное и необратимое набухание смолы, особенно когда такое воздействие происходит при более высоких температурах реактора гидратации, которые, как упоминалось выше, могут выходить за пределы интервала температур, который смола может удовлетворительно переносить.
Набухание не только является проблемой само по себе, но также может снизить эффективность и усложнить процесс каталитической гидратации; например, когда избыточное набухание требует замены смолы и, таким образом, отключения установки каталитической гидратации, в которой она содержится. Были предприняты различные попытки решения этих проблем, включая попытки найти смолы, менее склонные к набуханию при обычных условиях эксплуатации, а также разработать модификации процесса в попытке улучшить производительность в дополнение к уменьшению набухания. Например, в патенте ЕР1828086 описано применение “защитных шариков”, созданных для удаления примесей, которые предположительно могут вызвать набухание смолы, но такие попытки были неэффективными.
Вместо того чтобы защищать смолы защитным слоем в попытке минимизировать набухание, другой более успешный подход, описанный в публикации РСТ WO2008/150338, направлен не на защиту смолы, а на облегчение процесса удаления ионообменной смолы, срок службы которой истек, за счет ранее не предлагавшейся работы каталитического реактора в режиме восходящего потока, который расширяет слой катализатора, что позволяет разделить частицы катализатора по размеру и тем самым селективно удалить набухшие частицы. Еще один подход показан в патенте США № 6,160,187, который утверждает, что уменьшение набухания может быть достигнуто в условиях адиабатической реакции и при использовании адиабатического реактора. Однако примеры, содержащиеся в патенте «187», на самом деле не демонстрируют какого-либо улучшения характеристик набухания смол, равно как и не было последующих разработок адиабатического режима, разработкой которого пренебрегли.
Соответственно, несмотря на то, что было несколько многообещающих разработок в отдельных аспектах гетерогенной каталитической гидратации, остается значительная потребность в дальнейших улучшениях каталитической гидратации в адиабатических условиях процесса.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к жидкофазному способу производства этиленгликоля, включающему: получение потока воды и сырьевого потока водного этиленоксида; разделение сырьевого потока водного этиленоксида на, по меньшей мере, первую равную часть и вторую равную часть; объединение первой части и потока воды с получением первого сырьевого потока; подачу первого сырьевого потока на вход первого адиабатического реактора, где вход первого адиабатического реактора имеет температуру на входе; взаимодействие этиленоксида и воды в присутствии первого катализатора на основе ионообменной смолы в первом адиабатическом реакторе с получением исходящего потока первого реактора, содержащего воду, этиленгликоль и непрореагировавший этиленоксид; извлечение исходящего потока первого реактора из выхода первого адиабатического реактора; охлаждение исходящего потока первого реактора через теплообменник, расположенный после первого адиабатического реактора до температуры гидратации; объединение второй части и охлажденного потока продукта с получением второго сырьевого потока; перенос второго сырьевого потока на вход второго адиабатического реактора, где вход второго адиабатического реактора имеет температуру гидролиза; взаимодействие этиленоксида и воды, содержащейся во втором сырьевом потоке, в присутствии второго катализатора на основе ионообменной смолы во втором адиабатическом реакторе с получением исходящего потока второго реактора, содержащего воду, этиленгликоль, и непрореагировавший этиленоксид; где каждый из первого адиабатического реактора и второго адиабатического реактора содержат катализатор на основе ионообменной смолы в неподвижном слое катализатора, и температура на выходе выше, чем температура на входе.
КРАТКОЕ ОПИСАНИЕ НЕСКОЛЬКИХ ВИДОВ ЧЕРТЕЖЕЙ
Вышеизложенное краткое описание, а также следующее подробное описание предпочтительных вариантов изобретения будет лучше понято при чтении вместе с приложенными чертежами. Для иллюстрации изобретения на чертежах показаны предпочтительные в настоящее время варианты. Однако следует понимать, что изобретение не ограничивается показанными точными устройствами и инструментами. На рисунках:
На фигуре 1 показана схема технологического процесса получения потока концентрированного этиленоксида в соответствии с данным изобретением;
На фигурах 2-3 показаны схемы технологического процесса вариантов получения этиленгликоля в соответствии с данным изобретением; и
На фигуре 4 показана схема технологического процесса имитированного типового способа в соответствии с данным изобретением для получения этиленгликоля.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Все части, проценты и соотношения, используемые здесь, выражены по объему, если не указано иное. Все давления являются абсолютными. Все документы, цитируемые здесь, включены в качестве ссылки.
Под «водой» подразумевается любой вид воды, подходящей для использования в химической и нефтехимической переработке, включая деионизированную, деминерализованную, промышленную, питьевую и дистиллированную воду. Также может применяться конденсат пара и конденсат, полученный в части дегидратации и выпаривания способа производства гликоля.
В данном изобретении разработан улучшенный способ адиабатической каталитической гидратации, в котором снижено набухание катализатора, значительно улучшена гибкость способа, и способ сконфигурирован так, чтобы его можно было преимущественно использовать для модернизации не каталитического способа до каталитического. Конкретным преимуществом данного изобретения является то, что применяют ряд адиабатических реакторов с впрыском свежего водного раствора этиленоксида в каждый адиабатический реактор. Обычно, реакцию не каталитической гидратации проводят в одном адиабатическом реакторе безотносительно к выходу иди максимальной температуре реактора. Такая работа может причинить конкретный вред ионообменным смолам в соответствии с данным изобретением, так как температуры вне интервала температур, обозначенного для таких смол, могут увеличивать разбухание и снижать период службы катализатора, как описано выше.
Вместо использования таких одностадийных адиабатических реакторов и сопутствующих им недостатков (таких как высокая температура в присутствии катализатора), в данном изобретении используют многостадийные адиабатические реакторы, расположенные в ряд, с внешним охлаждением между стадиями и впрыском свежего водного раствор этиленоксида на каждой стадии. Так как тепло в адиабатическом реакторе не удаляется, температура исходящего потока превышает температуру сырья, и тепло реакции необходимо удалять из исходящего потока охладителем на каждой стадии. Это особенно важно для предотвращения вышеупомянутого разбухания катализатора и для предотвращения избыточной не каталитической реакции продуктов гликоля с этиленоксидом до высших гликолей. Кроме того, обычный не каталитический адиабатический реактор не может быть преимущественно сконфигурирован так, чтобы позволять серию из множества стадий реактора с впрыском свежего водного раствора этиленоксида. В такой конфигурации, введение свежего этиленоксида в выходящий поток реактора, содержащий значительную концентрацию продуктов гликоля, вызовет избыточную реакцию продуктов гликоля до нежелательных высших гликолей. В данном изобретении, эти проблемы решаются реакторным блоком каталитической гидратации с двумя или более адиабатическими реакторами, расположенными в ряд, промежуточным охлаждением между реакторами и впрыском свежего водного раствора этиленоксида в каждый реактор. Промежуточное охлаждение удаляет тепло реакции из исходящего потока, снижая его температуру так, чтобы его можно было объединить со свежим водным раствором этиленоксида для следующей стадии реактора. В данном изобретении используется как температура охлажденного исходящего потока из каждого промежуточного охладителя, так и пропорция впрыска свежего водного этиленоксида в каждый реактор для контроля максимальной температуры в каждом реакторе в интервале, желательном для срока службы и производительности катализатора. Более того, с точки зрения процесса, охлаждение с помощью внешних межступенчатых теплообменников легче, эффективнее и проще, чем настройка соответствующего режима для внутреннего охлаждения реактора, и его можно регулировать для поглощения нормальных колебаний в процессе при сохранении производительности каталитических реакторов. Таким образом, применение ряда адиабатических реакторов в соответствии с данным изобретением улучшает универсальность процесса и позволяет модернизировать или переоборудовать обычный процесс не каталитической гидратации в каталитической, как более подробно описано ниже.
В данном изобретении эти преимущества достигаются при одновременном сохранении высокой селективности предшествующих систем каталитической гидратации к моноэтиленгликолю. В каждом слое адиабатического реактора этиленоксид и вода взаимодействуют в присутствии гетерогенного катализатора (такого как ионообменная смола) с образованием моноэтиленгликоля, при этом не менее 98% этиленоксида превращается в моноэтиленгликоль, при этом лишь незначительная доля превращается в высшие гликоли, такие как диэтиленгликоль или триэтиленгликоль.
Таким образом, в настоящем изобретении разработана улучшенная схема процесса, которая включает реакторный блок каталитической гидратации, содержащий последовательно соединенные адиабатические реакторы, который сохраняет низкое набухание смолы и превосходную селективность, а также уменьшает сложность процесса и увеличивает универсальность.
Этот блок реакторов каталитической гидратации (также “блок реакторов”), полученный в соответствии с данным изобретением, как описано выше, показан как 25 на фигуре 2 и более подробно описан ниже. Блок реакторов 25 загружают, среди прочих сырьевых потоков, сырьевым потоком воды 36, жидким холодным сырьевым потоком через трубопровод 40, который высоко обогащен этиленоксидом, и который подают под давлением, достаточно высоким для того, чтобы удерживать этиленоксид в жидкости. Блок реакторов 25 включает конечный адиабатический реактор для не катализированного термического гидролиза ЭО 26, который обеспечивает полное превращение оставшегося не превращенного ЭО (присутствующего в исходящем потоке последнего адиабатического реактора из ряда реакторов 22) до уровней ч./млн., значительно снижая общий требуемый объем каталитического реактора ряда реакторов 22.
Любые подходящие средства получения такого жидкого сырьевого потока, который высоко обогащен этиленоксидом, применимы к данному изобретательскому способу каталитического производства этиленгликоля. Одним из подходящим средств для этого является прямой двухстадийный блок испарительный абсорбер-десорбер, показанный на фигуре 1. На фигуре 2 показаны коммуникации между блоком абсорбер-десорбер 1 и блоком реакторов 25, которые получены в соответствии с данным изобретением. Способ получения этиленоксида со ссылкой на этот прямой двухстадийный блок испарительный абсорбер-десорбер далее описан более подробно.
Этиленоксид получают при непрерывном контакте кислородсодержащего газа с олефином, предпочтительно, этиленом, в присутствии катализатора этиленоксида (“эпоксидирования”) (описанного более подробно ниже). Кислород может подаваться в реакцию в по существу чистой молекулярной форме или в смеси, такой как воздух. В качестве примера, обычные сырьевые смеси реагентов в рабочих условиях могут содержать от около 0,5% до около 45%, предпочтительно, от около 5% до около 30% этилена и от около 3% до около 15% кислорода, где остальное составляют сравнительно инертные материалы, включая такие вещества, как диоксид углерода, вода, инертные газы, другие углеводороды и модераторы реакции, описанные здесь. Неограничивающие примеры инертных газов включают азот, аргон, гелий и их смеси. Неограничивающие примеры других углеводородов включают метан, этан, пропан и их смеси. Диоксид углерода и вода являются побочными продуктами процесса эпоксидирования, а также обычными примесями в сырьевых газах. Оба оказывают неблагоприятное воздействие на катализатор, поэтому концентрации этих компонентов обычно выдерживают на минимуме.
Как отмечено ранее, в реакции также присутствуют один или более модераторов реакции, неограничивающие примеры которых включают органические галогенсодержащие соединения, такие как C1-C8 галогенуглеводороды; особенно предпочтительными являются хлоридсодержащие модераторы, такие как метилхлорид, этилхлорид, этилендихлорид, винилхлорид или их смеси. Контроль уровня концентрации хлорида особенно важен для рутенийсодержащих катализаторов.
Как указано выше, обычный способ эпоксидирования этилена включает парофазное окисление этилена молекулярным кислородом в присутствии катализатора эпоксидирования, в трубчатом реакторе с неподвижным слоем. Обычно коммерческие этиленоксидные реакторы с неподвижным слоем имеют форму множества параллельно расположенных трубок (в подходящей оболочке) внешним диаметром приблизительно 0,7-2,7 дюймов и внутренним диаметром 0,5-2,5 дюймов и длиной 15-53 футов, каждая из которых заполнена и упакована катализатором. Исходную реакционную смесь (описанную выше) вводят в эти трубки, и полученный исходящий из реактора газ содержит этиленоксид, неиспользованные реагенты и побочные продукты.
Только для целей иллюстрации, далее представлены условия, которые часто применяют в современных коммерческих этиленоксидных реакторах: часовая объемная скорость газа (ЧОСГ) 1500-10000 ч-1, давление на входе реактора 150-400 ф./кв. дюйм изб., температура охладителя 180-315°C, уровень превращения кислорода 10-60% и производительность ЭО (объем выпуска) 7-20 ф. ЭО/куб.ф. катализатора/ч. Потребление сырья на входе реактора после завершения запуска и во время обычной работы обычно составляет (в % объемных) 1-40% этилена, 3-12% O2; 0,3%-20%, предпочтительно, 0,3-5%, более предпочтительно, 0,3-1% CO2; 0-3% этана, некоторое количество одного или более хлоридных модераторов, которые описаны здесь; и остаток сырья составляет аргон, метан, азот или их смеси.
Типовые условия работы для способа эпоксидирования этилена включают температуры в интервале от около 180°C до около 330°C и, предпочтительно, от около 200°C до около 325°C и более предпочтительно, от около 225°C до около 280°C. Рабочее давление может варьироваться от около атмосферного давления до около 30 атмосфер, в зависимости от желаемого массового расхода и производительности. Более высокое давление может применяться в объеме данного изобретения. Время нахождения в коммерческих реакторах обычно составляет порядка от около 2 до около 20 секунд.
На фигуре 1 жидкость из промывной колонны (далее обозначенные как поток “воды из цикла обогащения”) течет через трубопровод 1 и подается в испарительный барабан 5. Потоком воды из цикла обогащения является водный поток, содержащий от 1,5% моль до 4% моль этиленоксида с некоторым количеством растворенных газов, таких как метан, этилен и другие. Этот поток воды из цикла обогащения подают под давлением, предпочтительно, от около 0,4 МПа до около 0,6 МПа в испарительный барабан 5. Абсорбирующее устройство 9 постоянно и непосредственно закреплено в верхней части испарительного барабана 5, и в испарительном барабане имеется отверстие для соединения с абсорбером 9. В данном изобретении давление и в испарительном барабане 5 и в абсорбере 9 поддерживается на уровне, достаточном для того, чтобы при поступлении воды из цикла обогащения в испарительный барабан 5, “легкие” газы, которые растворены в воде из цикла обогащения, которые имеют летучесть более, чем у этиленоксида, вымывались из воды из обогащенного цикла при поступлении в испарительный барабан 5, и пар легких газов направляется разностью давления между испарительным барабаном 5 и абсорбером 9 вверх через отверстие в абсорбер 9 с образованием верхних слоев абсорбера 9, а растворенный этиленоксид остается в основном солюбилизированным в потоке воды из цикла обогащения в кубовом остатке испарительного барабана 5. Эти более летучие “легкие” газы включают указанные выше инертные, непрореагировавшие или побочные газы, растворенные в воде из цикла обогащения. Кроме того, следы этиленоксида также могут испаряться с более летучими легкими газами в потоке воды из цикла обогащения. Небольшие количества этиленоксида, которые испаряются из воды из цикла обогащения и поднимаются в абсорбер 9 с испарениями растворенного легкого газа, могут быть восстановлены нисходящим промывочным потоком (не показан) для абсорбции и восстановления двигающегося вверх этиленоксида.
Как указано выше, абсорбер 9 прямо и постоянно прикреплен к верхней поверхности испарительного барабана 5, например, сваркой. Абсорбер 9 прикреплен к испарительному барабану 5 так же, как в конфигурации испарительной колонны и ребойлера, хорошо известной специалисту в данной области техники. Более конкретно, абсорбер 9 прикреплен к испарительному барабану 5 вдоль продольной оси испарительного барабана 5, но предпочтительно, прикреплен в середине продольной оси. Поперечное сечение абсорбера 9 совпадает с формой отверстия в испарительном барабане 5, над которым прикреплен абсорбер 9, и что позволяет сообщение между испарительным барабаном 5 и абсорбером 9. Поперечное сечение абсорбера и форма отверстия в испарительном барабане 5 имеют одинаковую совпадающую форму, и этой формой может быть любая форма между кругом и эллипсом, между эксцентричностью 0 и эксцентричностью 1. Отверстие в испарительном барабане 5 и абсорбере 9 расположено так, что центр отверстия является коллинеарным с центром абсорбера 9. Конфигурация отверстия в испарительном барабане 5 и форма абсорбера 9 и их соответствующее расположение обеспечивает должную жидкостное сообщение между этими двумя элементами. Абсорбер 9 предпочтительно приварен плоским сварным швом к внутреннему диаметру отверстия, сделанного в испарительном барабане 5 так, что внешний диаметр абсорбера 9 находится заподлицо с диаметром отверстия в испарительном барабане 5.
Чтобы обеспечить баланс между вымыванием более летучих «легких» растворенных веществ при сохранении высокой растворимости этиленоксида в воде из цикла обогащения, давление в испарительном барабане 5 поддерживается на уровне от около 0,1 МПа до 0,3 МПа, и давление в абсорбере 9, с которым имеется прямое сообщение, лишь немного ниже давления в испарительном барабане 5. Температура газов в верхней части абсорбера 9 составляет от около 30°C до около 45°C, а температура в испарительном барабане 5 составляет от около 50°C до около 60°C.
Верхние компоненты абсорбера 9 возвращают через трубопровод 12 в реакторный блок до установки (не показан) как возможный рецикл в реактор. Вода из цикла обогащения вытекает в виде кубового остатка из испарительного барабана 5 через насос 14 и через необязательный теплообменник 20 (который предварительно нагревает поток) и в десорбер 15. Давление в десорбере 15 поддерживают через контроль обратного давления, создаваемого отводом паров из конденсатора выпара 19. Температура в верхней части десорбера 15 должна быть от около 25°C до около 40°C, и в нижней части десорбера 15 составляет от около 120°C до около 160°C. Поток воды из цикла обогащения может быть предварительно нагрет в теплообменнике 20 для снижения нагрузки, необходимой для работы десорбера 15. Давление в десорбере 15 поддерживают в интервале от около 0,2 МПа до около 0,6 МПа. В десорбере 15 также может применяться система пароструйных насосов, хорошо известной специалистам в данной области техники, где пар подают из: (1) пара, образующегося везде в системе получения этиленоксида, или (2) пара под средним или высоким давлением, подаваемого из внешних источников/ЗГУ; или сочетания этих двух источников.
Находящийся под давлением поток из верхней части десорбера вытекает через трубопровод 30 в блок реакторов 25, как показано на фигуре 2. Поток течет в конденсатор 16 и затем в испарительный барабан 17, который отделяет промытые легкие газы от концентрированного жидкого потока водного этиленоксида, который подают в насос 18. Исходящее из насоса 18 делят на два потока: один поток в виде обратного потока обратно в десорбер 15, и один жидкий водный поток чистого продукта, концнетрированного в этиленоксиде. Промытые легкие газы из барабана 17 текут в конденсатор выпара 19 для дальнейшего восстановления этиленоксида из потока промытого пара и контроля количества растворенных легких газов в концентрированном жидком растворе этиленоксида 40. По сравнению с сильно разбавленным водным раствором, найденным в сравнимом месте в блоке восстановления этиленоксида обычного способа, находящийся под давлением поток из верхней части десорбера в значительно большей степени обогащен этиленоксидом, содержа, по меньшей мере, около 40% моль, предпочтительно, по меньшей мере, около 50% моль этиленоксида, где остальным содержимым в основном является вода. Таким образом, находящийся под давлением поток из верхней части десорбера дает и высокие концентрации этиленоксида, и находится под высоким давлением. Это делает поток крайне полезным для дальнейшей обработки до этиленгликолей через каталитический гидролиз на очень экономичной основе, так как нет необходимости дополнительно обогащать поток или создавать давление на следующих стадиях обработки.
Другой предпочтительной опцией способа в соответствии с данным изобретением является то, что кубовый остаток десорбера 15, который представляет собой обеднённый водный раствор цикла, содержащий немного этиленоксида (от около 1 молярных ч./млн. до около 50 молярных ч./млн.), и который также не содержит легкие компоненты, а также примеси, может в предпочтительных вариантах применяться во множестве других схемах рационального использования тепловой энергии по всему заводу, в зависимости от нужд и условий.
В этих условиях потоков с концентрацией этиленоксида выше, чем обычно, операторы будут продолжать соблюдать соответствующие стандарты безопасности, которые всегда соблюдаются при производстве, обработке или хранении этиленоксида. Как всегда, должны быть приняты меры для предотвращения реакций или событий, которые могут привести к возгоранию, горению, дефлаграции, детонации или взрыву любого газового потока, особенно тех, которые содержат этиленоксид в концентрациях выше типовых. Соответственно, для предотвращения таких событий могут использоваться предохранительные устройства для сброса или уменьшения нежелательного повышения давления в способе, реакции или системах разделения или где-либо еще на заводе по производству этиленоксида как до, так и после того, что показано. Разделение в десорбере 15 настолько эффективно, что почти весь этиленоксид в воде из цикла обогащения, который поступает в десорбер 15, успешно отделяется и восстанавливается из воды из цикла обогащения, и покидает десорбер 15 в виде чистого жидкого верхнего потока, в то время как только очень небольшая часть ЭО покидает десорбер 15 в других потоках.
Верхний поток десорбера под давлением, полученный в десорбере 15, течет под повышенным давлением через трубопровод 30 из блока десорбера 1 в блок реакторов 25, как показано на фигуре 2. В соответствии с данным изобретением, блок реакторов содержит два или более реакторов. На фигуре 2, показан блок реакторов 25 с рядом из четырех каталитических реакторов, расположенных последовательно, каждый из которых отделен теплообменником. Более конкретно, как показано на фигуре 2, и более подробно на фигуре 3, блок реакторов 25 показывает ряд реакторов 22 из четырех расположенных в ряд адиабатических реакторов с неподвижным слоем катализатора с нисходящим потоком сырья 38, 40, 46 и 52 с тремя промежуточными охладителями, встроенными теплообменниками 39, 43 и 49. Реактор обозначен как первый адиабатический реактор 38, второй адиабатический реактор 40, третий адиабатический реактор 46 и четвертый адиабатический реактор 52, но в данном описании эти адиабатические реакторы 38, 40, 46 и 52 также могут быть обозначены как “реакторы”.
Как описано выше, такое промежуточное охлаждение необходимо, так как реакция воды с этиленоксидом является высоко экзотермической, поэтому необходимо удалять тепло реакции из продукта или исходящих потоков, если реакция происходит в адиабатическом реакторе, как в данном изобретении, чтобы избежать избыточной температуры в слое реактора, которая может снизить срок службы чувствительного к температуре катализатора, такого как ионообменные смолы. Это отличается от изотермических условий, где тепло реакции удаляют непосредственно из реактора. Сохраняя катализатор на основе ионообменной смолы при температурах, которые может выдержать смола, конечно является критическим для снижения набухания, предотвращения разложения и максимизации срока службы катализатора. При должно работе, повышение адиабатической температуры можно контролировать на уровне, который сохраняет хорошую эффективность и срок службы.
Наконец, после ряда реакторов 22 расположен обычный не каталитический адиабатический трубчатый реактор 26 (этот реактор, конечно, не содержит катализатор).
Так как верхний поток десорбера под давлением течет через трубопровод 30 в блок реактора, его делят на равные части между четырьмя реакторами в ряду; затем первую равную часть высококонцентрированного потока этиленоксида смешивают с источником воды 36 (предварительно нагретым в теплообменнике 35), в смешивающем устройстве, например, статическом смесителе, с получением разбавленного водного сырьевого потока этиленоксида 37. Поток 36 нагревают в теплообменнике 35 для придания потоку 37 температуры от 50°C до 90°C.
Также присутствует поток воды 36. Поток воды 36 может быть взят из любого источника воды, включая ЗГУ, но, предпочтительно, его берут из другого блока завода по производству этиленоксида/этиленгликоля. Например, вода может поступать из восстановленной конденсированной воды из блока выпаривания этиленгликоля завода, более конкретно, вода может быть конденсирована из верхнего пара одного или более испарителей в блоке выпаривания.
При объединении, поток воды 36 и верхний поток десорбера под давлением, текущий через трубопровод 30, образуют объединенный разбавленный сырьевой поток в ряд реакторов 22. Этот объединенный сырьевой поток будет иметь стехиометрический избыток воды к этиленоксиду, по сравнению с необходимым для гидратации этиленоксида до этиленгликоля, однако, количество этого избытка значительно меньше, чем избыточное количество, применяемое в известном уровне техники. Следовательно, в соответствии с данным изобретением, объединенные потоки 40 и 36 имеют молярное отношение воды:этиленоксида от около 5:1 до около 15:1, предпочтительно, от около 7:1 до около 12:1. И таким образом, так как объединенный общий сырьевой поток содержит меньше воды, поток продукта из последнего реактора ряда 88 также имеет соответствующие пониженные количества воды, которую необходимо удалить из продукта, как описано ниже.
Водный этиленоксид, содержащийся в жидком потоке продукта десорбера 40, делят на четыре приблизительно равных потока 57, 61,64 и 67, каждый из этих потоков содержит приблизительно равные молярные количества воды и этиленоксида. Из них, поток 57 объединяют с потоком воды 36 (после нагревания в теплообменнике 35) с получением сырьевого потока 37 в реактор 38 так, что молярное отношение воды:этиленоксида в сырьевом потоке 37 составляет от около 40:1 до около 10:1, предпочтительно, от около 30:1 до около 20:1. Это является значительным улучшением по сравнению с обычным проведением каталитической и не каталитической гидратации. При совместной подаче потока 57 вместе с потоком воды 36 в первый реактор 38 и последующей совместной подаче других равных частей (61, 64, 67) водного сырьевого потока этиленоксида 40 с исходящим потоком реактора (70, 73, 76) может быть получено высокое соотношение разбавленной воды:этиленоксида в сырьевом потоке в каждый реактор, одновременно сохраняя низкое общее соотношение воды:этиленоксида в потоке продукта 88 в конце блока реакторов, который подают в испарители для удаления воды. Таким образом, реакцию этиленоксида и воды катализируют в присутствии ионообменной смолы и при значительном избытке воды к этиленоксиду. Это способствует селективности к производству моноэтиленгликоля и подавлению образования высшего гомолога гликоля, таким образом оба эффекта дают преимущество. И еще это сохраняется при получении конечного продукта, который намного более концентрирован в этиленгликоле и, следовательно, требует намного меньше энергии в блоке выпаривания. Например, в соответствии с данным изобретением, конечный продукт блока реакторов, объединенный продукт из реактора 88, может иметь соотношение воды:этиленгликоля 10:1 или даже ниже, в то время как наоборот, на обычном заводе исходящий поток реактора, поданный в блок выпаривания, имеет молярное соотношение воды:этиленоксида около 25:1, тем самым снижая количество тепловой нагрузки для выпаривания избытка воды на коэффициент, по меньшей мере, 2,5. Значительным преимуществом данного изобретения является то, что молярное соотношение воды:этиленоксида может быть скорректировано и оптимизировано, при необходимости, и желательно для катализатора и энергозатрат, доступности этиленоксида и требований к выходу продукта.
Проиллюстрировав общую схему работы блока реакторов и преимущества настоящего изобретения, далее способ в соответствии с данным изобретением будет описан более подробно со ссылкой на Фиг.3.
Как отмечено выше, потоки 57 и 36 объединяют с получением сырьевого потока 37 в первый реактор и, таким образом, конечная температура слоя катализатора в реакторе 38 регулируется через контроль нагревания потока воды 36 в теплообменнике 35, что, в свою очередь, контролирует входящую температуру потока 37. Конечная температура слоя катализатора в каждом из ряда расположенных последовательно реакторов 40, 46 и 52 регулируется применением теплообменников 39, 43 и 49, соответственно, применяемых для охлаждения сырьевого потока в каждый реактор.
Регулирование температуры и давления являются важными частями способа в соответствии с данным изобретением. Реакцию гидратации в настоящем изобретении проводят как жидкофазный процесс. В настоящем изобретении под «жидкофазным процессом» подразумевается, что сырьевые потоки, подаваемые в реактор, продукт реактора или исходящие потоки реактора, а также реагенты внутри реактора поддерживаются в жидкой фазе. Соответственно, температура и давление в каждом реакторе таковы, что по мере того, как потоки подаются и входят в каждый реактор, они поддерживаются в жидкой фазе. Таким образом, температура и давление в каждом реакторе регулируются для поддержания содержимого реактора в жидкой фазе. Выбор температуры во многом определяется соображениями вышеупомянутых параграфов. Таким образом, при указанных ниже интервалах температур в настоящем изобретении, когда содержимое реактора находится в жидкой форме, давление будет составлять от примерно 0,5 МПа до примерно 1,5 МПа, предпочтительно от 0,8 МПа до 1,2 МПа.
При выборе температуры необходимо уравновесить два конкурирующих требования. Во-первых, температура на входе в соответствии с данным изобретением должна быть достаточно высокой для запуска реакции гидратации этиленоксида, чтобы превращение этиленоксида в моноэтиленгликоль было полным или почти полным. В частности, под «полным», «почти полным» или «завершением» подразумевается, что процент превращения этиленоксида в моноэтиленгликоль должен составлять, по меньшей мере, 97%, предпочтительно, по меньшей мере, около 99%. Таким образом, температура на входе в реакторы 38, 40, 46 и 52 должна быть в интервале от около 50°C до около 90°C, предпочтительно, в интервале от около 70°C до около 85°C. Однако температура в реакторе (и, следовательно, на выходе из реактора) не должна быть слишком высокой, потому что, как упомянуто выше, значительное повышение температуры внутри реактора вызывает набухание и разрушение ионообменной смолы. Высокие температуры в реакторе являются результатом экзотермической реакции гидратации, поэтому для сдерживания любого повышения температуры необходимо удалить это тепло реакции. В таких областях применения, адиабатические реакторы имеют значительно меньше технологических осложнений, но имеют тот недостаток, что они не отводят тепло реакции. Соответственно, в настоящем изобретении было обнаружено, что повышение температуры, тем не менее, может, по меньшей мере, частично сдерживаться избытком воды в каждом реакторе, которая действует как поглотитель тепла. Таким образом, хотя одной из целей настоящего изобретения является уменьшение количества воды, используемой при производстве этиленгликоля, тем не менее, по меньшей мере, минимальное количество воды необходимо для сдерживания повышения температуры в реакторах, возникающего в результате экзотермической реакции. Как описано выше, обе эти цели выполняются в данном изобретении разделением водного сырьевого потока этиленоксида 40 на четыре равных части 57, 61, 64 и 67 и совместной подачей (также может применяться термин “постепенная подача”) их с получением сырьевых потоков в реакторе 38, 40, 46, 52. При такой изобретательской конфигурации, высокое соотношение воды:этиленоксида получают в каждом реакторе, сдерживая экзотерму так, что на выходе из каждого реактора температура составляет от около 85°C до около 120°C, предпочтительно, от около 100°C до около 110°C.
Несмотря на успех настоящего изобретения в сдерживании повышения температуры в реакторе, тем не менее, может возникнуть необходимость в дальнейшем снижении температуры выходящего потока реактора до достижения приемлемого интервала температур на входе, упомянутого выше. Для этого потоки 70, 73 и 76 пропускают через теплообменники 39, 43 и 49, соответственно. Это дает возможность для интеграции тепла, поскольку поток (не показан) извне блока реакторов может быть нагрет в любом из теплообменников 39, 43 и 49 за счет косвенного теплообмена с исходящим потоком реактора из одного или более расположенных последовательно реакторов.
Как отмечено выше, при более высоких температурах происходит почти полное превращение этиленоксида в моноэтиленгликоль, при более низких температурах на входе не весь этиленгликоль будет реагировать, и обнаруживаемые количества этиленоксида появятся и останутся непрореагировавшими («прорвавшимися») в исходящем потоке реактора. В определенных ситуациях может потребоваться снижение температуры на входе для того, чтобы уменьшить превращение этиленоксида, чтобы обеспечить больший прорыв этиленоксида, когда, например, желательно продлить срок службы катализатора на основе ионообменной смолы или в других ситуациях, как описано ниже.
В дополнение к вышеупомянутым параметрам и рабочим условиям, другие факторы, которые могут быть учтены при эксплуатации процесса, включают, но не ограничены ими: конфигурацию реактора и процесса, скорость превращения и селективность катализатора на основе ионообменной смолы, и скорость потока реагентов (ЧОСЖ). Эта уникальная конфигурация реактора обеспечивает гибкость в выборе рабочих параметров реактора, которые наилучшим образом соответствуют целям эксплуатации установки с точки зрения использования катализатора и энергии, доступности этиленоксида и требований к выходу продукта. Кроме того, получение высококонцентрированного жидкого сырьевого потока этиленоксида 40, который составляет настоящее изобретение, значительно сокращает количество стадий, требуемых в блоке процесса эпоксидирования, по сравнению с обычными коммерческими установками, которые требуют большего количества стадий из-за необходимости в более разбавленном водном этиленоксиде и высоких температур для обычного не каталитического термического гидролиза.
Принимая во внимание вышеупомянутые соображения, сырьевой поток 37 при подходящей температуре подают в реактор 38, где этиленоксид взаимодействует с водой в присутствии катализатора на основе ионообменной смолы или слоя катализатора с получением моноэтиленгликоля. В адиабатических реакторах в соответствии с данным изобретением, например, реакторе 38, смешивание этиленоксида с горячей разбавляющей водой для получения подходящей температуры в присутствии катализатора вызывает гидратацию этиленоксида, которая высокоселективна по моноэтиленгликолю так, что, по меньшей мере, около 95%, предпочтительно, по меньшей мере, около 98% этиленоксида превращается в моноэтиленгликоль, и только незначительная доля превращается в высшие гликоли, такие как диэтиленгликоль или триэтиленгликоль. Так, например, в отношении реактора 38 на молярной основе количество молей моноэтиленгликоля в исходящем потоке реактора 70 будет приблизительно равно количеству молей этиленоксида в сырьевом потоке 38. В дополнение к продукту моноэтиленгликоля, каждый исходящий поток реакторов в соответствии с данным изобретением содержит воду (избыток непрореагировавшей воды для применения в следующем реакторе), моноэтиленгликоль и следовые количества других компонентов, включая этиленоксид и высшие гомологи гликоля.
Из-за падения давления по длине реактора 38, существует значительные перепад давления между реактором 38 и 40 для исходящего потока первого реактора 70 чтобы пройти через теплообменник 39, где он охлаждается, и затем исходящий поток первого реактора объединяют с потоком 61 с получением второго сырьевого потока 81, который подают в реактор 40, как в реакторе 38, где этиленоксид взаимодействует с водой с высокой селективностью и степенью превращения так, что по существу весь этиленоксид взаимодействует со стехиометрическим количеством воды с получением моноэтиленгликоля при вышеуказанных процентах превращения. Исходящий поток второго реактора, который как и исходящий поток первого реактора содержит непрореагировавшую воду, моноэтиленгликоль и следовые количества вышеупомянутых компонентов, проходит с перепадом давления через теплообменник 43. По сути, единственной разницей между исходящим потоком первого реактора 70 и исходящим потоком второго реактора 73 является то, что последний содержит больше моноэтиленгликоля, потому что он содержит количество моноэтиленгликоля, полученное в обоих реакторах 38 и 40. Реакторы 46 и 52 и связанные с ними потоки работают так же, как и 38 и 40, где исходящий поток второго реактора 73, охлаждают в теплообменнике 43 и объединяют с равной частью 64 водного концентрированного сырьевого потока этиленоксида 40 с получением третьего сырьевого потока 75, который взаимодействует в присутствии слоя третьего катализатора на основе ионообменной смолы в третьем реакторе 46 с получением исходящего потока третьего реактора 76. Исходящий поток третьего реактора 76 охлаждают в теплообменнике 49 и объединяют с равной частью 67 с получением четвертого сырьевого потока 77, который взаимодействует в присутствии слоя четвертого катализатора на основе ионообменной смолы в четвертом реакторе 52 с получением исходящего потока четвертого реактора 79, где каждый исходящий поток последовательно более обогащен моноэтиленгликолем, как описано выше.
В дополнение к вышеописанному ряду реакторов 22, включающему четыре последовательных реактора с неподвижным слоем катализатора с нисходящим потоком сырья, блок реакторов 25 также включает обычный трубчатый реактор 26 в который может подаваться исходящий поток четвертого реактора 79 после нагревания в теплообменнике 55. В данном изобретении этот трубчатый реактор 26 сконфигурирован и работает так, чтобы придавать ранее неизвестные уровни универсальности и управляемости схеме процесса каталитической гидратации, которая далее описана более подробно.
Во-первых, в дополнение к повышению превращения и селективности реакции этиленоксида и воды по МЭГ, данное изобретение также может использоваться и конфигурироваться для повышения универсальности и адаптируемости процесса гидратации, позволяя оператору процесса производить ди- и триэтиленгликоль. Это может быть выполнено путем физического обхода одного или нескольких каталитических реакторов 38, 40, 46, 52 (сам этот байпас не показан) и, таким образом, удаления одного или нескольких каталитических реакторов из циркуляции сырьевого потока. Этиленоксид, который «прорывается» и остается непрореагировавшим после обхода одного или более вышеупомянутых каталитических реакторов, направляют в не каталитический трубчатый реактор 26 для термического превращения этиленоксида и воды в этиленгликоль. Поскольку трубчатый реактор 26 является не каталитическим и полностью зависит от термической конверсии, исходящий поток четвертого реактора 79 предварительно нагревают в теплообменнике 55, чтобы попасть в диапазон эффективных температур для некаталитической гидратации этиленоксида в моноэтиленгликоль, предпочтительно, температура на входе в трубчатый реактор 26 составляет от около 130°C до около 160°C.
Конечно, такое не каталитическое термическое превращение является значительно менее селективным по МЭГ, с получением значительных количеств гликолей с более высоким содержанием по сравнению с МЭГ, таких как ДЭГ и ТЭГ, так, что за счет увеличения количества этиленоксида, который превращается в этиленгликоль в трубчатом реакторе, продукт объединенного реактора 88 содержит меньше МЭГ и больше ДЭГ и ТЭГ, чем было бы произведено если бы использовались только каталитические реакторы в ряду реакторов 22. Это дает оператору универсальный и гибкий процесс производства этиленгликоля, так как выпуск моно-, ди- и триэтиленгликоля может регулироваться оператором для удовлетворения спроса. В крайнем случае, все четыре последовательных реактора обходятся, так что остается только последний некаталитический трубчатый реактор 26, и вся гидратация будет происходить в этом реакторе, тем самым превращая каталитический процесс в некаталитический. Альтернативно, каталитический ряд 22 добавляет гибкость для снижения энергопотребления на заводе при сохранении фиксированного производства моноэтиленгликоля по сравнению с таким же производством на заводе, использующем некаталитический гидролиз EO. Если около половины производства этиленгликоля производится в каталитических реакторах 38 и 40 ряда реакторов 22 (с обходом реакторов 46 и 42), а остальное производится в трубчатом термическом реакторе 26, полученное распределение продукта будет таким же, как при обычном не каталитическом процессе термического гидролиза, но с преимуществом работы установки при примерно на 20% более низком молярном соотношении вода:этиленоксид, что приводит к значительной экономии за счет выпаривания гликоля.
Альтернативно, вместо того, чтобы физически обходить реакторы, может быть осуществлен «температурный байпас» путем снижения температуры на входе в один или несколько последовательных реакторов до необходимого низкого уровня, чтобы уменьшить реакцию между этиленоксидом и водой и уменьшить количество моноэтиленгликоля, образующегося в реакторах, что означает больший «прорыв» непрореагировавшего этиленоксида, наблюдаемый на выходах каталитических реакторов. Этот этиленоксид затем превращается в этиленгликоль в трубчатом реакторе, как указано выше.
Трубчатый реактор может также выполнять «завершающую» роль. Даже когда ряд реакторов 22 работает с нормальной каталитической эффективностью, разработанной для максимального превращения этиленоксида в этиленгликоль, тем не менее, небольшое количество непрореагировавшего этиленоксида останется непрореагировавшим после всех четырех реакторов в ряду 22, и этот этиленоксид будет «прорывом» в исходящем потоке четвертого реактора 79. В данном изобретении ряд 26 работает нормально, так что концентрация этиленоксида в выходящем потоке четвертого реактора 79 не будет превышать примерно 1% моль. Этот оставшийся этиленоксид затем можно превратить в моноэтиленгликоль в трубчатом реакторе 26.
Наконец, трубчатый реактор 26 также может использоваться в качестве резервного реактора в ситуациях, когда один или несколько каталитических реакторов неисправны или должны быть по иным причинам выведены из эксплуатации из-за истощения, закупорки слоя смолы или сопла или других трудностей, обычно наблюдаемых при эксплуатации слоев ионообменной смолы. Когда это случается, неисправный каталитический реактор необходимо вывести из эксплуатации. Это означает, что в ряду реакторов 22 будет избыток непрореагировавшего этиленоксида, что нежелательно. Этот избыточный прорыв этиленоксида может быть превращен в этиленгликоль в трубчатом реакторе 26. Таким образом, присутствие трубчатого реактора 26 в настоящем изобретении обеспечивает значительную гибкость и работоспособность для оператора процесса. В отличие от предшествующего уровня техники, где невозможность замены или ремонта катализатора на основе ионообменной смолы или реактора является серьезным недостатком твердых/гетерогенных каталитических систем, в данном изобретении реакторы могут быть выведены на автономный режим для такого ремонта без прерывания работы.
Хотя четыре реактора и три теплообменника показаны в блоке реакторов на фигурах 2 и 3, это сделано только для целей иллюстрации. В зависимости от конкретных потребностей и требований может использоваться больше или меньше реакторов. Увеличение количества реакторов имеет преимущество уменьшения экзотермы в каждом реакторе, тем самым уменьшая количество необратимого набухания, вызванного температурой, которое катализатор на основе ионообменной смолы испытывает внутри каждого реактора. Кроме того, увеличение количества реакторов также увеличивает разбавление этиленоксида, и поскольку этиленоксид разрушает большинство ионообменных смол, ожидается, что повышенное разбавление продлит срок службы смолы. Кроме того, снижается общий объем катализатора.
Первичными недостатками большего числа реакторов являются стоимость и сложность, включая дополнительные капитальные затраты на реакторы, насосы, теплообменники и другое оборудование, а также эксплуатационные расходы на большие объемы катализатора на основе ионообменной смолы, а также дополнительные трубопроводы, приборы и сложность эксплуатации.
Алкиленгликоль в объединенном продукте реактора 88 затем может быть восстановлен с помощью выпарной батареи или вакуумной дистилляции, или любым другим подходящим методом, известным специалистам в данной области. Методы восстановления могут быть объединены для разных стадий восстановления. Например, выпарную батарею можно использовать для удаления воды из исходящего потока реактора, в то время как вакуумная дистилляция может использоваться для сушки гликоля. Действительно, одним из преимуществ данного изобретения является то, что в постгликолевом блоке реакторов на заводе можно уменьшить сложность, поскольку количество тепловой нагрузки, необходимое для выпаривания избыточной воды, было уменьшено, как указано выше и как подробно описано в примере ниже. Таким образом, хотя заводы известного уровня техники часто включают системы выпаривания, имеющие несколько эффектов или стадий (см., например, ЕР № 2121646 В1), данное изобретение требует меньшего количества стадий.
Как обсуждалось выше, данное изобретение может включать один или несколько катализаторов на основе ионообменной смолы. Ионообменные смолы имеют полимерную матрицу, которая содержит на поверхности центры ионного обмена, заполненные ионными функциональными группами. Ионообменные смолы обычно различают на катионные и анионообменные смолы, хотя доступны и другие типы ионообменных смол. Для более полного описания катионо- и анионообменных смол см. de Dardel, F. and Arden, T. V. Ion Exchangers in Ullman’s Encyclopedia of Industrial Chemistry (2005).
Подходящие полимерные матрицы для катализатора на основе ионообменной смолы включают полистирольную матрицу, полиакриловую матрицу, полиалкиламиновую смолу, а также другие полимерные материалы. Предпочтительно, полимерная матрица поперечно сшита с дивенилбензолом в достаточной степени для увеличения рабочей мощности, но при этом, не увеличивая плотность ионообменного материала до такой степени, что ионообменный материал становится слишком физически твердым и слишком химически стойким к химической обработке. Предпочтительно, матрица представляет собой сополимер стирола и дивинилбензола.
К участкам полимерных матриц, описанных выше, прикреплены ионные стационарные группы, которые определяют, функционирует ли смола как катионная или анионная ионообменная смола. В растворах положительный или отрицательный заряд стационарных групп компенсируется ионами с противоположным зарядом, которые названы здесь функциональными группами.
Сильнокислотные катионные ионообменные смолы обычно включают сульфоновые группы в качестве стационарных групп, в свою очередь присоединенные к полимерной матрице стирол-дивенилбензол. Примеры сильнокислотных сульфокислотных катионообменных смол включают Amberlite IR 120, Dowex HCR, Lewatit S 100 и Amberlyst 15, среди прочих.
Катионные ионообменные смолы могут также включать полимерный материал, который является сополимером, полученным аддитивной полимеризацией акриловой или метакриловой кислоты и дивинилбензола, таким как описан в патенте США № 3,957,698. Другие подходящие полимерные материалы для катионообменных смол включают полимерную матрицу, полученную поперечным сшиванием поливинилпиридиновой смолы с дивенилбензолом. Такие материалы доступны под торговой маркой Reillex HPQ. Сульфированные фенольные полимерные смолы также являются подходящими катионными ионообменными смолами.
Слабокислотные катионные ионообменные смолы обычно включают карбоксильные группы в качестве стационарных групп. Примеры слабокислотных катионообменных смол включают, Amberlite IRC 86, Dowex Mac-3, Lewatit CNP, среди прочих.
В данном изобретении, сильноосновные анионообменные смолы содержат четвертичные аммониевые стационарные группы. Они далее подразделяются на I тип, полученный реакцией триметиламина с сополимером стирола и дивенилбензола после хлорметилирования, и II тип, полученный реакцией сополимера стирола и дивенилбензола с диметилэтаноламином. Подходящие примеры таких смол I типа включают Lewatit MP 500, доступный от Lanxess, и Amberlyst 26 и Amberlite IRA 402 и IRA 410, доступные от Dow. Подходящие примеры сильноосновных смол II типа включают, например, Purolite A510S (Purolite Corporation).
Слабоосновные анионообменные смолы обычно включают полиакрильные смолы, получившие стационарные группы в результате реакции с полифункциональным амином с получением анионообменных смол, таких как слабоосновные смолы на основе третичного аммония Amberlite IRA 67 и Amberlyst 21 (доступные от Dow). Следует особо отметить, что эту ионообменную смолу затем можно дополнительно обработать хлорметаном или диметилсульфатом с получением сильноосновной смолы I типа на основе четвертичного амина Amberlite IRA 458 (Dow). Слабоосновные анионообменные смолы могут также включать группу свободного основания в качестве стационарной группы, такие как смола Amberlite IRA-67 (Dow).
В конкретном варианте данного изобретения, сильноосновная анионообменная смола содержит «связывающую» группу из 3-7 связывающих атомов, в частности 3-5 атомов углерода между четвертичной аммониевой стационарной группой и бензольной группой полимерной матрицы/материала. Пример такой связывающей цепи в сильноосновной смоле проиллюстрирован в патенте США № 5,945,568 и был произведен под названием Diaion TSA1200 (Mitsubishi Chemical). Как указано в патенте '568, связывающая группа, которая связывает стационарную группу четвертичного аммония с бензольной группой полимерной матрицы, особо не ограничена, если она является достаточно длинной. Подходящими примерами связывающей группы являются алкиленовая группа или алкиленоксиалкиленовая группа. Предпочтительным примером для использования в данном изобретении является анионообменная смола, которая содержит, в качестве субстрата, полимер винилароматического соединения, и которая имеет такую структуру, что четвертичная аммониевая группа связана с ароматической группой через связывающую группу, имеющую длину цепи, по меньшей мере, 3.
Не ограничиваясь теорией, полагают, что в данном изобретении эта связывающая группа предотвращает нуклеофильную атаку на аммониевую стационарную группу из бензольного кольца, которая, в противном случае, привела бы к ускоренной дезактивации и разложению катализатора на основе ионообменной смолы и снижению стабильности и срока службы катализатора.
Множество ионов подходят для использования в качестве функциональной группы в анионных смолах в соответствии с данным изобретением, и могут быть выбраны из группы, включающей анионы бикарбоната, карбоната, гидроксида, бисульфита, карбоксилата, цитрата и металата, и молибдата. Эти функциональные группы могут использоваться с любыми стационарными группами и полимерным материалом, указанными выше.
В данном изобретении предпочтительно, чтобы катализатор на основе ионообменной смолы принадлежал к группе сильноосновных анионообменных смол I типа, более предпочтительно, сильноосновных анионообменных смол I типа с бикарбонатной или моноцитратной функциональной группой, и наиболее предпочтительно, сильноосновных анионообменных смол I типа с бикарбонатной или моноцитратной функциональной группой со связующей группой.
В дополнение к ионообменным материалам, указанным выше, может быть использована любая другая подходящая катионная или анионообменная смола, такая как катионо- и анионообменные смолы, указанные выше в de Dardel, F. and Arden, T. V. 2008, Ion Exchanger, in Ullman’s Encyclopedia of Industrial Chemistry.
В дополнение к вышеупомянутому ионообменному материалу также можно использовать сочетание гомогенного и гетерогенного катализатора, например, твердого катализатора, на котором адсорбирован растворимый металлический катализатор. В этом случае сначала получают твердый катализатор с адсорбированным на нем металлическим катализатором. Затем во время жидкофазной реакции гидратации, описанной выше, металлический катализатор десорбируется с твердого катализатора и может эффективно катализировать реакцию. Затем металлический катализатор должен быть отделен от продуктов реакции и может быть повторно использован для повторной адсорбции на твердом катализаторе. Твердым катализатором в этом случае может быть ионообменная смола.
Вышеупомянутые параграфы касаются выбора подходящего катализатора на основе ионообменной смолы для использования в каталитических реакторах в соответствии с данным изобретением. Что касается работы каталитических реакторов, содержащих ионообменную смолу, на фигурах 2 и 3 сырьевые потоки проходят в направлении от верха реактора вниз через каждый из реакторов. Это называется режимом «нисходящего потока», но в данном изобретении реакторы могут работать в режимах «нисходящего потока» или «восходящего потока». Процессы с нисходящим потоком имеют преимущество увеличения плотности или объема катализатора внутри реактора и, таким образом, уменьшения размера и стоимости самих реакторов. Работа с нисходящим потоком также минимизирует не каталитическую реакцию воды и этиленоксида до этиленгликоля. Однако во время работы с нисходящим потоком селективность катализатора может быть ухудшена как из-за локальных неоднородностей и примесей, которые могут образовываться и захватываться в слой катализатора, так и из-за «канализации», которая развивается в слое катализатора.
Как можно предположить, при работе с восходящим потоком исходный поток движется в направлении снизу реактора наверх. В публикации РСТ № WO2008/150338A1 сообщается об увеличении стабильности и срока службы ионообменной смолы в рабочем режиме с восходящим потоком. В данном изобретении с жидкофазными реагентами и твердым катализатором работа с восходящим потоком может давать умеренную степень псевдоожижения слоя во время работы с восходящим потоком, что увеличивает долю пустот между соседними частицами смолы, позволяя уменьшить неоднородности и примеси, которые в противном случае внедряются и захватываются слоем катализатора во время работы с нисходящим потоком и ухудшают эффективность катализатора. В патенте США № 6,160,187 ранее недооценивалась работа каталитических реакторов с восходящим потоком, поскольку возникающее псевдоожижение слоя якобы вызывает большее истирание катализатора и снижение селективности по моноэтиленгликолю из-за продольного перемешивания. Однако в данном изобретении было обнаружено, что в жидкофазном процессе данного изобретения эти критические замечания нереалистичны, потому что они более вероятны в системах газ/твердый катализатор, где есть гораздо более высокие скорости, большая разница в плотностях (между газообразным реагентом и твердым катализатором, по сравнению с жидким реагентом и твердым катализатором) и, таким образом, гораздо большее расширение слоя, чем в данной системе жидкость/твердый катализатор в соответствии с данным изобретением. Таким образом, работа с восходящим потоком остается подходящим режимом работы в соответствии с данным изобретением.
Катализатор эпоксидирования на основе серебра.
Катализатор эпоксидирования на основе серебра включает подложку и, по меньшей мере, каталитически эффективное количество серебра или серебросодержащего соединения; также необязательно присутствует активирующее количество рения или ренийсодержащего соединения; также необязательно присутствует активирующее количество одного или нескольких щелочных металлов или соединений, содержащих щелочные металлы. Подложка, применяемая в соответствии с данным изобретением, может быть выбрана из большого числа твердых огнеупорных подложек, которые могут быть пористыми и могут обеспечивать предпочтительную пористую структуру. Хорошо известно, что оксид алюминия полезен в качестве подложки катализатора для эпоксидирования олефина, и он является предпочтительной подложкой.
Независимо от характера используемой подложки, она обычно имеет форму частиц, больших кусков, мелких кусков, гранул, колец, сфер, вагонного колеса, полых цилиндров с поперечными перегородками и подобных, размер которых подходит для использования в реакторе эпоксидирования с неподвижным слоем. Частицы подложки, предпочтительно, будут иметь эквивалентные диаметры в интервале от около 3 мм до около 12 мм и, более предпочтительно, в интервале от около 5 мм до около 10 мм. Эквивалентным диаметром является диаметр сферы, имеющей такое же отношение внешней поверхности (т. е. без учета поверхности в порах частицы) к объему, что и у частиц используемой подложки. Подходящие подложки доступны от Saint-Gobain Norpro Co., Sud Chemie AG, Noritake Co., CeramTec AG и Industrie Bitossi S.p.A. Не ограничиваясь конкретными композициями и составами, содержащимися здесь, дополнительная информация о композициях подложки и способах получения подложек можно найти в публикации патента США № 2007/0037991.
Для получения катализатора для окисления олефина до оксида олефина, на поверхность подложки, имеющей вышеуказанные характеристики, затем наносят каталитически эффективное количество серебра. В одном варианте, каталитически эффективное количество серебра составляет от 10% массовых до 45% массовых. Катализатор получают пропиткой подложки соединением, комплексом или солью серебра, растворенными в подходящем растворителе, достаточном для того, чтобы вызвать осаждение соединения-предшественника серебра на подложку. Предпочтительно, используют водный раствор серебра.
Активирующее количество рениевого компонента, которым может быть ренийсодержащее соединение или ренийсодержащий комплекс, также может быть нанесено на подложку до, одновременно или после осаждения серебра. Рениевый промотор может присутствовать в количестве от около 0,001% масс. до около 1% масс., предпочтительно, от около 0,005% масс. до около 0,5% масс. и более предпочтительно, около 0,01% масс. до примерно 0,1% масс. в расчете на массу всего катализатора, включая подложку, в пересчете на металлический рений.
Другие компоненты, которые также могут быть нанесены на подложку до, одновременно или после осаждения серебра и рения, представляют собой активирующие количества щелочного металла или смеси двух или более щелочных металлов, а также необязательные активирующие количества компонента щелочноземельного металла группы IIA или смеси двух или более компонентов щелочноземельного металла группы IIA, и/или компонент переходного металла, или смеси двух или более компонентов переходных металлов, все которые могут быть в форме ионов металлов, соединений металлов, комплексов металлов и/или солей металлов, растворенных в подходящем растворителе. Подложка может быть пропитана одновременно или на отдельных стадиях различными промоторами катализатора. Конкретное сочетание подложки, серебра, промотора(ов) щелочного металла, рениевого компонента и необязательного дополнительного промотора(ов) в соответствии с данным изобретением обеспечивает улучшение одного или нескольких каталитических свойств по сравнению с таким же сочетанием серебра и подложки при отсутствии или применении только одного из промоторов.
Используемый здесь термин «активирующее количество» определенного компонента катализатора относится к количеству этого компонента, которое эффективно работает для улучшения каталитических характеристик катализатора по сравнению с катализатором, который не содержит этот компонент. Точные применяемые концентрации, конечно, будут зависеть, среди прочих факторов, от желаемого содержания серебра, природы подложки, вязкости жидкости и растворимости конкретного соединения, используемого для доставки промотора в пропитывающий раствор. Примеры каталитических свойств включают, среди прочего, функциональность (устойчивость к снижению качества), селективность, активность, конверсию, стабильность и выход. Специалисту в данной области понятно, что одно или несколько отдельных каталитических свойств могут быть улучшены «активирующим количеством», в то время как другие каталитические свойства могут быть улучшены, или не улучшены, или даже могут быть уменьшены.
Подходящие промоторы щелочных металлов могут быть выбраны из лития, натрия, калия, рубидия, цезия или их сочетаний, при этом цезий является предпочтительным, и сочетания цезия с другими щелочными металлами являются особенно предпочтительными. Количество щелочного металла, нанесенного или присутствующего на подложке, должно быть активирующим количеством. Предпочтительно, количество варьируется от около 10 ч./млн. до около 3000 ч./млн., более предпочтительно, от около 15 ч./млн. до около 2000 ч./млн. и даже более предпочтительно, от около 20 ч./млн. до около 1500 ч./млн. и особенно предпочтительно, от около 50 ч./млн. до около 1000 ч./млн. массовых от всего катализатора, в пересчете на металл.
Подходящие промоторы щелочноземельных металлов включают элементы из группы IIA Периодической таблицы элементов, которыми могут быть бериллий, магний, кальций, стронций и барий или их сочетания. Подходящие промоторы переходных металлов могут включать элементы из групп IVA, VA, VIA, VIIA и VIIIA Периодической таблицы элементов и их сочетания.
Количество промотора(ов) щелочноземельного металла и/или промотора(ов) переходного металла, нанесенных на подложку, является активирующим количеством. Промотор переходного металла обычно может присутствовать в количестве от около 0,1 микромоль на грамм до около 10 микромоль на грамм, предпочтительно от около 0,2 микромоль на грамм до около 5 микромоль на грамм.
Раствор серебра, используемый для пропитки подложки, может также содержать необязательный растворитель или комплексообразующий/солюбилизирующий агент, известный в данной области техники. Множество растворителей или комплексообразующих/солюбилизирующих агентов может применяться для солюбилизации серебра до желаемой концентрации в пропитывающей среде. Подходящие комплексообразующие/солюбилизирующие агенты включают амины, аммиак, щавелевую кислоту, молочную кислоту и их сочетания. Амины включают алкилендиамин, содержащий от 1 до 5 атомов углерода. В одном предпочтительном варианте, раствор содержит водный раствор оксалата серебра и этилендиамина. Комплексообразующий/солюбилизирующий агент может присутствовать в пропитывающем растворе в количестве от около 0,1 до около 5,0 моль на моль серебра, предпочтительно от около 0,2 до около 4,0 моль и более предпочтительно, от около 0,3 до около 3,0 моль на каждый моль серебра.
При применении растворителя, он может быть органическим растворителем или водой и может быть полярным или по существу или полностью неполярным. В общем, растворитель должен обладать достаточной сольватирующей способностью для растворения компонентов раствора. В то же время предпочтительно, чтобы растворитель был выбран так, чтобы избежать чрезмерного влияния на сольватированные промоторы или взаимодействия с ними. Предпочтительны растворители на органической основе, содержащие от 1 до около 8 атомов углерода на молекулу. Могут использоваться смеси нескольких органических растворителей или смеси органических растворителей с водой при условии, что такие смешанные растворители функционируют так, как желательно здесь.
Концентрация серебра в пропитывающем растворе обычно составляет от около 0,1% массовых вплоть до максимальной растворимости, предусмотренной конкретным используемым сочетанием растворитель/солюбилизирующий агент. Обычно очень удобно использовать растворы, содержащие от 0,5% до около 45% массовых серебра, при этом предпочтительны концентрации от 5 до 35% массовых серебра.
Пропитка выбранной подложки проводится любым из общепринятых способов; например, пропитка избытком раствора, пропитка по влагоемкости, нанесение распылением и т.д. Обычно материал подложки помещают в контакт с серебросодержащим раствором до тех пор, пока достаточное количество раствора не будет абсорбировано подложкой. Предпочтительно, количество серебросодержащего раствора, используемого для пропитки пористой подложки, не больше, чем необходимо для заполнения пор подложки. Можно использовать однократную пропитку или ряд пропиток с промежуточной сушкой или без нее, что частично зависит от концентрации компонента серебра в растворе. Методы пропитки описаны, например, в патентах США №№ 4,761,394, 4,766,105, 4,908,343, 5,057,481, 5,187,140, 5,102,848, 5,011,807, 5,099,041 и 5,407,888. Могут применяться известные в данной области техники методы предварительного осаждения, совместного осаждения и последующего осаждения разных промоторов.
После пропитки подложки серебросодержащим соединением, т.е. предшественником серебра, компонентом рения, компонентом щелочного металла и необязательными другими промоторами, пропитанную подложку обжигают в течение времени, достаточного для превращения серебросодержащего соединения в активные виды серебра и для удаления летучих компонентов из пропитанной подложки с получением предшественника катализатора. Обжиг может быть выполнен нагреванием пропитанной подложки, предпочтительно, постепенно, до температуры в интервале от около 200°C до около 600°C под давлением в интервале от 0,05 до около 3,5 МПа. В общем, чем выше температура, тем короче требуемый период нагревания. Широкий спектр периодов нагревания был предложен в данной области техники; например, в патенте США № 3,563,914 раскрыто нагревание в течение менее 300 секунд, и в патенте США № 3,702,259 раскрыто нагревание в течение от 2 до 8 часов при температуре от 100°C до 375°C, обычно в течение от около 0,5 до около 8 часов. Однако, важно только то, чтобы время нагревание коррелировало с температурой так, чтобы все содержащееся серебро превратилось в активные виды серебра. Для этой цели может применяться непрерывное или ступенчатое нагревание.
Во время обжига пропитанная подложка может подвергаться воздействию газовой атмосферы, содержащей инертный газ или смесь инертного газа с от около 10 ч./млн. до 21% объемных кислородсодержащего окисляющего компонента. Для целей данного изобретения инертный газ определен как газ, который по существу не реагирует с катализатором или предшественником катализатора в условиях, выбранных для обжига. Дополнительную информацию о производстве катализатора можно найти в вышеупомянутой публикации патента США № 2007/0037991.
Только для целей иллюстрации, ниже представлены условия, которые часто применяют в современных коммерческих реакторных установках для производства этиленоксида: часовая объемная скорость газа (ЧОСГ) 1500-10000 ч-1, давление на входе в реактор от 1 МПа до 3 МПа, температура охладителя 180-315°C, уровень превращения кислорода 10-60% и скорость получения ЭО (рабочая скорость) 100-350 кг ЭО/м3 катализатора/ч и изменение концентрации этиленоксида, ΔЭО, от около 1,5% до около 4,5%. Композиция сырья на входе в реактор после завершения запуска и во время обычной работы обычно содержит (в % объемных) 1-40% этилена, 3-12% O2; 0,2%-10%, предпочтительно, 0,2%-6%, более предпочтительно, 0,2%-5% CO2; 0-5% этана, некоторое количество одного или более хлоридных модераторов, которые описаны здесь; и оставшуюся часть сырья составляют аргон, метан, азот или их смеси.
ПРИМЕРЫ
Далее изобретение описано более подробно с помощью следующих не ограничивающих примеров.
Пример 1 (Известный уровень техники)
Способ получения этиленгликоля согласно известному уровню техники имитируют с применением одного не каталитического трубчатого/трубчатого реактора. В способе сырьевой поток подают в реактор, из которого получают исходящий поток реактора, и в таблице ниже даны значения в килограммах в час:
Выше «СВЭО» означает молярное отношение воды к этиленоксиду. Этот способ известного уровня техники дает этиленгликоль с хорошими степенями превращения, но использует большой избыток воды, которая затем должна быть удалена из продукта этиленгликоля, что делает способ значительно более энергоемким, чем способ в соответствии с данным изобретением, как описано ниже.
Пример 2 (данное изобретение)
Способ получения этиленгликоля в соответствии с данным изобретением имитируют согласно фигуре 4. Этот вариант сконфигурирован так же, как блок реакторов 25 на фигуре 2, описанной выше, где обычный трубчатый термический реактор 26 эквивалентен трубчатому реактору “DD”.
Как показано на фигуре 4, в имитации представлены четыре расположенных последовательно каталитических реактора с фиксированным слоем с нисходящим потоком с тремя промежуточными встроенными теплообменниками в качестве средств нагревания. В этой имитации реакторы заполняют одной или более подходящими ионообменными смолами, как описано выше. Способ работает в соответствии с данным изобретением так, что температуры и композиции потоков, точек входа и выхода и реакторов такие, как указаны ниже в таблице I, где содержимое потоков в таблице дано в единицах килограммы в час.
Как можно увидеть из таблицы выше, воду и этиленоксид подают в процесс в молярном отношении 10:1 (сочетание потоков A и B), которое намного меньше, чем в процессе известного уровня техники, например, который показан в примере 1 выше. Однако, при делении входящих потоков на множество потоков, подаваемых во множество ректоров, как указано в данном изобретении, намного более высокие соотношения воды к этиленоксиду получают в каждом реакторе, как показано выше молярное отношение воды к этиленоксиду в потоках, входящих в реактор, приблизительно составляет 32:1 в каждом случае. Такое высокое СВЭО не только улучшает селективность и превращение этиленоксида до МЭГ (как показано выше, превращение в МЭГ в этой имитации составляет 99%), но также помогает снижать рост температуры в каждом реакторе, снижая количество охладителя, необходимое в промежуточных теплообменниках, и снижая высокие температуры, которые потенциально могут повредить катализатор на основе ионообменной смолы.
Более того, как отмечено выше, применение более низкого СВЭО в способе в соответствии с данным изобретением позволяет значительное энергосбережение по сравнению с известным уровнем техники. В этой имитации способа в соответствии с данным изобретением (пример 2) и имитации сравнительного процесса известного уровня техники (пример 1) конечная испарительная нагрузка составляет:
Таким образом, способ известного уровня техники имеет испарительную нагрузку в 2,5 раза выше, чем способ в соответствии с данным изобретением.
Специалистам в данной области техники будет понятно, что изменения могут быть внесены в варианты жидкофазного процесса, описанного выше, не выходя за пределы широкой концепции изобретения. Таким образом понятно, что это изобретение не ограничено конкретными раскрытыми вариантами, конфигурациями или композициями, но оно предназначено для охвата модификаций в пределах сущности и объема данного изобретения, как определено в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕЦИРКУЛЯЦИОННЫЙ СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНГЛИКОЛЯ | 2019 |
|
RU2796340C2 |
| СПОСОБ УЛУЧШЕНИЯ ПРОИЗВОДСТВА ЭТИЛЕНГЛИКОЛЯ | 2019 |
|
RU2796341C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЕНГЛИКОЛЯ | 2008 |
|
RU2477718C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ИЗВЛЕЧЕНИЯ ЭТИЛЕНОКСИДА | 2009 |
|
RU2525237C2 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ С КОНЦЕНТРИРОВАННЫМИ РАСТВОРАМИ ЭТИЛЕНОКСИДА | 2019 |
|
RU2792388C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНГЛИКОЛЯ | 2012 |
|
RU2599828C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ЭТИЛЕНОКСИДА И ЭТИЛЕНГЛИКОЛЯ | 2008 |
|
RU2462461C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЕНГЛИКОЛЯ | 2008 |
|
RU2466123C2 |
| СПОСОБЫ И СИСТЕМЫ РЕЦИРКУЛЯЦИИ ТЕХНОЛОГИЧЕСКОЙ ВОДЫ В ПРОИЗВОДСТВЕ ЭТИЛЕНГЛИКОЛЯ | 2017 |
|
RU2737471C2 |
| ТВЕРДЫЙ КАТАЛИЗАТОР, ИСПОЛЬЗУЕМЫЙ ДЛЯ ПРЕВРАЩЕНИЯ АЛКИЛЕНОКСИДА В АЛКИЛЕНГЛИКОЛЬ | 2008 |
|
RU2470706C2 |


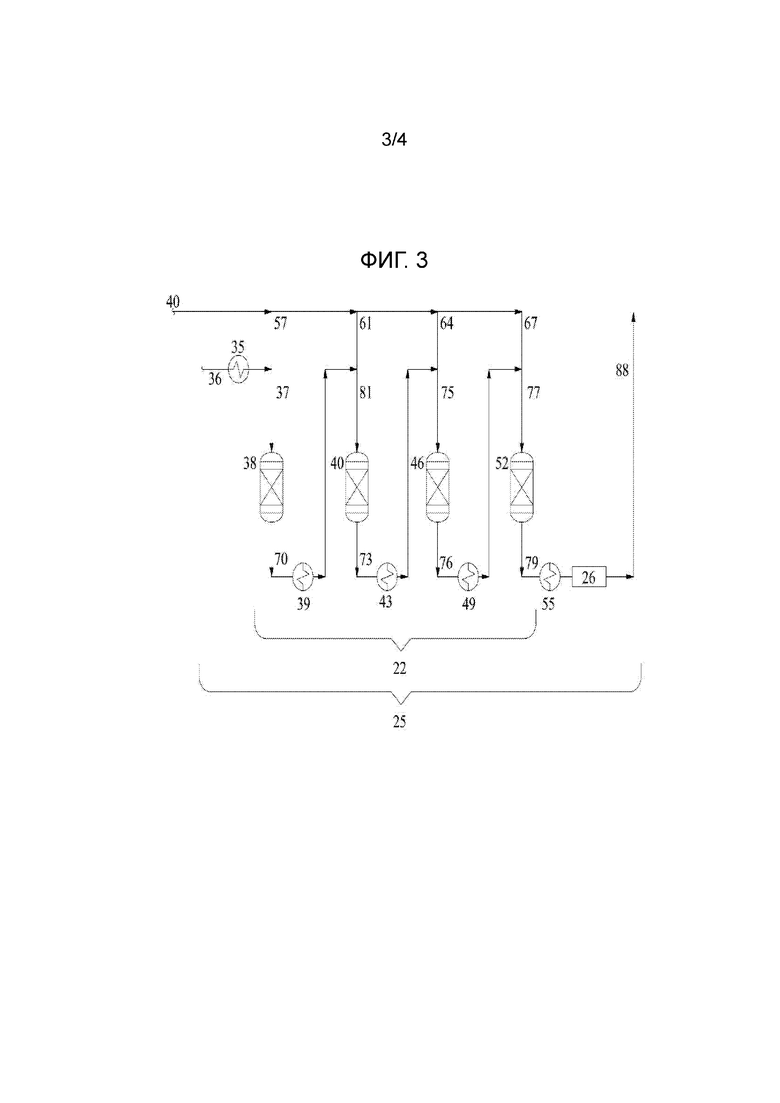
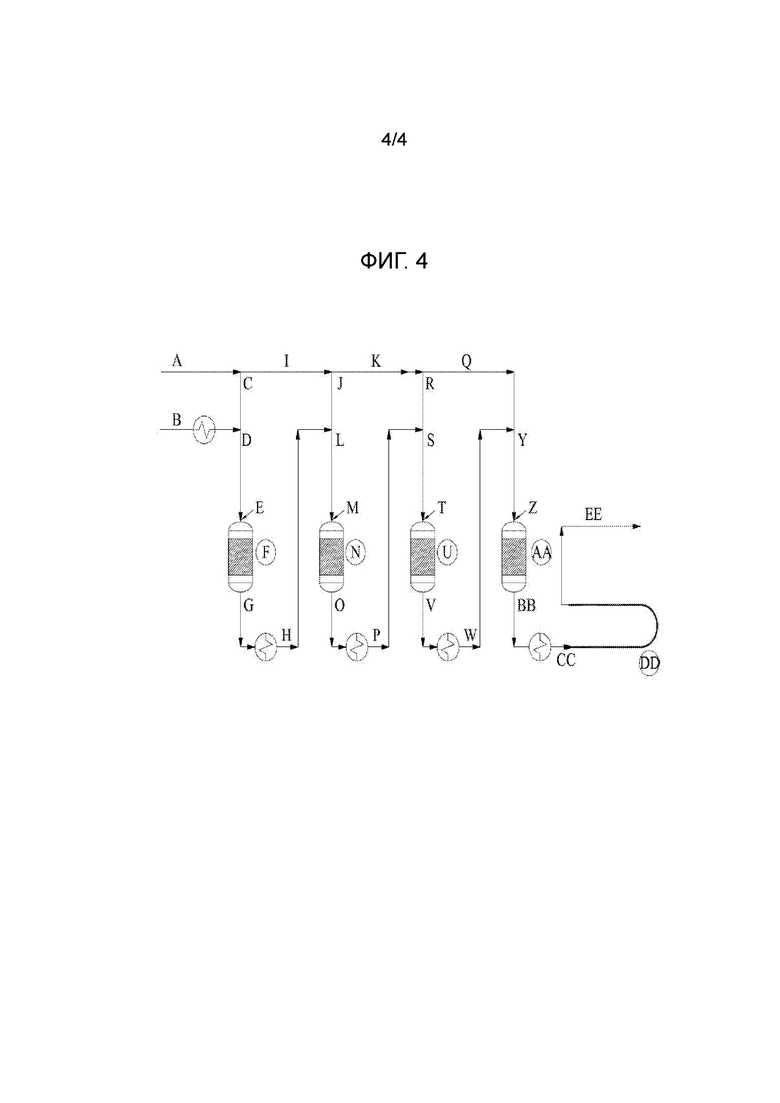
Настоящее изобретение относится к жидкофазному способу получения этиленгликоля, включающему следующие стадии: получение потока воды и сырьевого потока водного этиленоксида; разделение сырьевого потока водного этиленоксида на по меньшей мере первую равную часть и вторую равную часть; объединение первой части и потока воды с получением первого сырьевого потока; подачу первого сырьевого потока на вход первого адиабатического реактора, где вход первого адиабатического реактора имеет температуру на входе; взаимодействие этиленоксида и воды в присутствии первого катализатора на основе ионообменной смолы в первом адиабатическом реакторе с получением исходящего потока первого реактора, содержащего воду, этиленгликоль и непрореагировавший этиленоксид; извлечение исходящего потока первого реактора из выхода первого адиабатического реактора; охлаждение исходящего потока первого реактора через теплообменник, расположенный после первого адиабатического реактора до температуры гидратации; и объединение второй части и охлажденного потока продукта с получением второго сырьевого потока; перенос второго сырьевого потока на вход второго адиабатического реактора, где вход второго адиабатического реактора имеет температуру гидролиза и взаимодействие этиленоксида и воды, содержащейся во втором сырьевом потоке, в присутствии второго катализатора на основе ионообменной смолы во втором адиабатическом реакторе с получением исходящего потока второго реактора, содержащего воду, этиленгликоль и непрореагировавший этиленоксид. При этом каждый из первого адиабатического реактора и второго адиабатического реактора содержит катализатор на основе ионообменной смолы в неподвижном слое катализатора, а температура на выходе из адиабатических реакторов выше, чем температура на входе. Общее молярное отношение вода : этиленоксид в потоке воды и добавленном сырьевом потоке водного этиленоксида вместе составляет от 5:1 до 15:1. Технический результат – разработка улучшенного способа адиабатической каталитической гидратации, в котором снижено набухание катализатора, значительно улучшена гибкость способа. 12 з.п. ф-лы, 4 ил., 2 пр.
1. Жидкофазный способ получения этиленгликоля, включающий:
получение потока воды и сырьевого потока водного этиленоксида;
разделение сырьевого потока водного этиленоксида на по меньшей мере первую равную часть и вторую равную часть;
объединение первой части и потока воды с получением первого сырьевого потока;
подачу первого сырьевого потока на вход первого адиабатического реактора, где вход первого адиабатического реактора имеет температуру на входе;
взаимодействие этиленоксида и воды в присутствии первого катализатора на основе ионообменной смолы в первом адиабатическом реакторе с получением исходящего потока первого реактора, содержащего воду, этиленгликоль и непрореагировавший этиленоксид;
извлечение исходящего потока первого реактора из выхода первого адиабатического реактора;
охлаждение исходящего потока первого реактора через теплообменник, расположенный после первого адиабатического реактора до температуры гидратации; и
объединение второй части и охлажденного потока продукта с получением второго сырьевого потока;
перенос второго сырьевого потока на вход второго адиабатического реактора, где вход второго адиабатического реактора имеет температуру гидролиза;
взаимодействие этиленоксида и воды, содержащейся во втором сырьевом потоке, в присутствии второго катализатора на основе ионообменной смолы во втором адиабатическом реакторе с получением исходящего потока второго реактора, содержащего воду, этиленгликоль и непрореагировавший этиленоксид;
где каждый из первого адиабатического реактора и второго адиабатического реактора содержат катализатор на основе ионообменной смолы в неподвижном слое катализатора, а температура на выходе из адиабатических реакторов выше, чем температура на входе, и
где общее молярное отношение вода : этиленоксид в потоке воды и добавленном сырьевом потоке водного этиленоксида вместе составляет от 5:1 до 15:1, предпочтительно от 7:1 до 12:1.
2. Жидкофазный способ по п. 1, где температура на входе составляет от 50 до 90°C.
3. Жидкофазный способ по пп. 1, 2, где температура на выходе составляет от 85 до 120°C.
4. Жидкофазный способ по пп. 1-3, где молярное отношение вода : этиленоксид в первом сырьевом потоке составляет от 40:1 до 10:1, предпочтительно от 30:1 до 20:1.
5. Жидкофазный способ по пп. 1-4, проводимый непрерывно.
6. Жидкофазный способ по пп. 1-5, где сырьевой поток водного этиленоксида дополнительно делят на третью равную часть и четвертую равную часть и который включает следующие стадии:
извлечение исходящего потока второго реактора из выхода второго адиабатического реактора;
охлаждение исходящего потока второго реактора через теплообменник, расположенный после второго адиабатического реактора;
объединение третьей части и охлажденного исходящего потока второго реактора с получением третьего сырьевого потока;
перенос третьего сырьевого потока на вход третьего адиабатического реактора;
взаимодействие этиленоксида и воды, содержащейся в третьем сырьевом потоке, в присутствии третьего слоя катализатора на основе ионообменной смолы в третьем адиабатическом реакторе с получением исходящего потока третьего реактора, содержащего воду, этиленгликоль и непрореагировавший этиленоксид;
извлечение исходящего потока третьего реактора из выхода третьего адиабатического реактора;
охлаждение исходящего потока третьего реактора через теплообменник, расположенный после третьего адиабатического реактора;
объединение четвертой части и охлажденного исходящего потока третьего реактора с получением четвертого сырьевого потока;
перенос третьего сырьевого потока на вход четвертого адиабатического реактора; и
взаимодействие этиленоксида и воды, содержащейся в четвертом сырьевом потоке, в присутствии четвертого слоя катализатора на основе ионообменной смолы в четвертом адиабатическом реакторе с получением исходящего потока четвертого реактора, содержащего воду, этиленгликоль и непрореагировавший этиленоксид.
7. Жидкофазный способ по п. 6, дополнительно включающий подачу исходящего потока четвертого реактора в некаталитический реактор.
8. Жидкофазный способ по пп. 1-7, где сырьевой поток водного этиленоксида получают согласно следующим стадиям:
получение потока воды из цикла обогащения, содержащего этиленоксид, метан, этилен и другие растворенные легкие газы;
отделение, в испарительном барабане, пара растворенного легкого газа от потока воды из цикла обогащения;
направление вверх пара растворенного легкого газа через отверстие в испарительном барабане, позволяющее жидкостное сообщение с абсорбером, закрепленным на верхней поверхности испарительного барабана с образованием парового верхнего потока абсорбера;
нагревание воды из цикла обогащения, полученной из кубового остатка испарительного барабана, и закачивание ее в десорбер; и
разделение на: (1) обогащенный жидкий поток верхней части десорбера, содержащий по меньшей мере 40 мол.%, предпочтительно по меньшей мере 50 мол.% этиленоксида; и (2) обеднённый водный раствор в кубовом остатке десорбера, содержащий от 5 до 50 молярных ч./млн этиленоксида.
9. Жидкофазный способ по пп. 1-8, где катализатором на основе ионообменной смолы является сильноосновная анионообменная смола I типа.
10. Жидкофазный способ по пп. 1-9, где катализатор на основе ионообменной смолы имеет бикарбонатную или моноцитратную функциональную группу.
11. Жидкофазный способ по пп. 1-10, где катализатор на основе ионообменной смолы включает связывающую группу.
12. Жидкофазный способ по пп. 1-11, где первый и второй сырьевые потоки по существу не содержат диоксид углерода.
13. Жидкофазный способ по п. 9, где сильноосновная анионообменная смола I типа включает стационарную группу четвертичного аммония.
| US 6160187 А1, 12.12.2000 | |||
| WO 2006086642 A2, 17.08.2006 | |||
| CN 103709001 B, 11.05.2016 | |||
| US 20050119510 A1, 02.06.2005 | |||
| US 20060161026 A1, 20.07.2006 | |||
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЕНГЛИКОЛЯ | 2008 |
|
RU2477718C2 |

