Область техники, к которой относится изобретение
Настоящее изобретение относится к азабициклическим и диазепиновым производным, применяемым в качестве модуляторов мускариновых рецепторов, и к способам лечения заболеваний с их применением.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Мускариновый рецептор представляет собой мишень для возбуждающего нейротрансмиттера ацетилхолина, и был назван вследствие селективной активации рецептора мускарином. Мускариновый рецептор широко распространен в тканях человека и дополнительно классифицируется на подтипы от M1 до M5. Модуляция мускариновых рецепторов считалась терапевтической мишенью при нарушениях, начиная от гиперактивного мочевого пузыря и заканчивая когнитивными расстройствами (Abrams et al., Br. J Pharmacol 2006 July; 148(5): 565-578).
Миопия является рефракционной ошибкой глаза, вызванной чрезмерным ростом глаза в продольном направлении. Это удлинение глаза приводит к тому, что визуальное изображение фокусируется перед сетчаткой, и, как правило, обуславливает нечеткое видение отдаленных объектов. Сообщалось, что неселективный антагонист мускаринового рецептора атропин эффективен в виде 1% капель для местного применения при лечении миопии. (Chua et al., Ophthalmology 2006 Dec; 113(12):2285-91). Однако сообщались многочисленные побочные эффекты, в том числе мидриаз (расширение зрачка) и нечеткость зрения на близком расстоянии вследствие циклоплегии (неспособности к аккомодации). В настоящее время корректирующие линзы представляют собой основное средство для уменьшения интенсивности проявления нарушений, связанных с длинной глаза, таких как миопия. Однако линзы оптически исправляют рефракционные ошибки без устранения причины, которая представляет собой чрезмерный рост глаза. Таким образом, остается необходимость в способах лечения нарушений, связанных с чрезмерным ростом глаза.
Краткое описание изобретения
Остается необходимость в новых видах лечения и видах терапии в случае чрезмерного роста глаза. В настоящем изобретении предусмотрены соединения, их фармацевтически приемлемые соли, их фармацевтические композиции и их комбинации, которые являются модуляторами мускаринового рецептора. В настоящем изобретении дополнительно предусмотрены способы лечения, предупреждения или уменьшения интенсивности проявления нарушений, связанных с чрезмерным ростом глаза, предусматривающие введение субъекту, нуждающемуся в этом, эффективного количества модулятора мускаринового рецептора. В данном документе описаны различные варианты осуществления настоящего изобретения.
В некоторых аспектах в данном документе представлено соединение формулы (I) или (II) или его фармацевтически приемлемая соль:
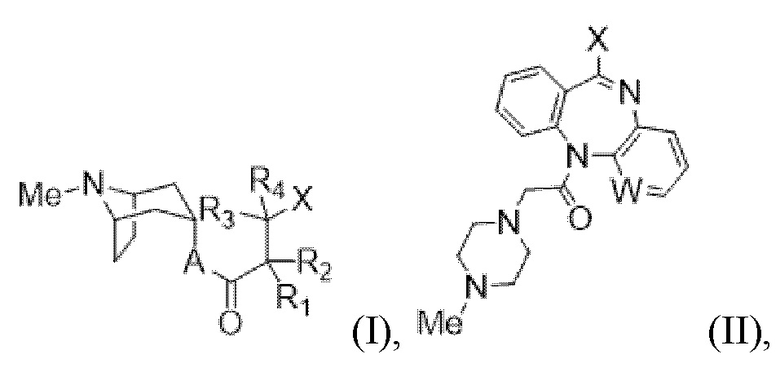
где
Me=CH3;
A=O или NR5;
W=N или CH;
X = -OH, -O-Y-Z, -S-Y-Z или -NR5-Y-Z;
R1 и R2 независимо представляют собой заместители в виде H, D, гидроксила, алкокси, нитрила, атомов галогена, прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных групп, необязательно замещенных атомами галогена; или
R1 и R2 независимо представляют собой заместители в виде фенильных или бензильных групп, необязательно замещенных одним или несколькими заместителями, выбранными из прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных групп, -галогеналкильных групп, гидроксила, алкокси, нитрила, нитро, амино, амида, сложного эфира, сульфона, сульфоксида, сульфонамида и атомов галогена; или
R1 и R2 независимо замещены гетероциклическим насыщенным, ненасыщенным или ароматическим 5- или 6-членным кольцом, содержащим один или несколько гетероатомов, выбранных из азота, кислорода и серы, и необязательно замещенных одним или несколькими заместителями, выбранными из прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных, -галогеналкильных групп, гидроксила, алкокси, нитрила, нитро, амино, амида, сложного эфира, сульфона, сульфоксида или атомов галогена;
R3 и R4 независимо замещены водородом, прямыми, или разветвленными, или циклическими C1-C10-алкильными или -галогеналкильными группами, или
R3 и R4 могут быть объединены с образованием 3-6-членных колец;
R5=H или прямые или разветвленные C1-C20-алкильные группы, предпочтительно C1-C10, прямые или разветвленные C1-C10-галогеналкильные группы;
Y представляет собой двухвалентный радикал, имеющий следующее значение:
прямой или разветвленный C1-C20 (предпочтительно C1-C10)-алкил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из атомов галогена и гидроксила;
-C(O)(C1-C10алкил)- или -C(O)(CH2)nC(O)O-(C1-C10алкил)- или -(C1-C10алкил)-;
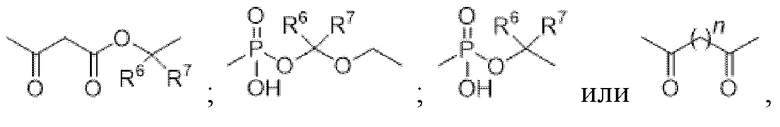
где n представляет собой целое число от 0 до 20;
R6 и R7 независимо представляют собой H или прямые или разветвленные C1-C10-алкильные группы, прямые или разветвленные C1-C10-галогеналкильные группы; или
R6 и R7 могут быть объединены с образованием 3-6-членных колец; и
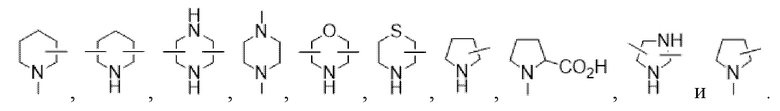
Z представляет собой H, -OH, C1-6алкокси, -COOH, -NR8R9;
R8 и R9 независимо представляют собой заместители в виде C1-C20-алкила, предпочтительно C1-C10, необязательно замещенного одним или несколькими заместителями, выбранными из гидроксила, амино, сложного эфира, карбоновой кислоты и атомов галогена; или
R8 и R9 могут быть объединены с образованием 3-6-членных колец, содержащих один или несколько гетероатомов, которые выбраны из группы, состоящей из:
В другом аспекте в настоящем изобретении предусмотрена фармацевтическая композиция, содержащая: (1) терапевтически эффективное количество (предпочтительно от приблизительно 0,01 до приблизительно 10,0 процентов по весу, более предпочтительно от приблизительно 0,01 до приблизительно 5 процентов по вес/объем или от приблизительно 0,1 до 5,0 процентов по весу) (a) соединения по настоящему изобретению и/или (b) его фармацевтически приемлемой соли и (2) один или несколько фармацевтически приемлемых носителей. В другом аспекте в настоящем изобретении предусмотрена фармацевтическая композиция, содержащая: (1) соединение по настоящему изобретению и/или его фармацевтически приемлемую соль и (2) один или несколько фармацевтически приемлемых носителей.
В другом аспекте в настоящем изобретении предусмотрена комбинация, в частности фармацевтическая комбинация, содержащая: (1) терапевтически эффективное количество (предпочтительно от приблизительно 0,01 до приблизительно 10,0 процентов по весу, более предпочтительно от приблизительно 0,01 до приблизительно 5 процентов по вес/объем или от приблизительно 0,1 до 5,0 процентов по весу) (a) соединения по настоящему изобретению и/или (b) его фармацевтически приемлемой соли и (2) одно или несколько терапевтически активных средств. В другом аспекте в настоящем изобретении предусмотрена комбинация, в частности фармацевтическая комбинация, содержащая: (1) соединение по настоящему изобретению и/или его фармацевтически приемлемую соль и (2) одно или несколько терапевтически активных средств.
Конкретные предпочтительные варианты осуществления настоящего изобретения станут понятными из следующего более подробного описания некоторых предпочтительных вариантов осуществления и формулы изобретения.
Краткое описание ЧЕРТЕЖЕЙ
На фигуре 1 представлен 1H ЯМР-спектр этил-2-фтор-2-фенилацетата;
на фигуре 2 представлен 1H ЯМР-спектр 2-фтор-2-фенилуксусной кислоты;
на фигуре 3 представлен 1H ЯМР-спектр 2-фтор-2-фенилацетилхлорида;
на фигуре 4 представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фтор-2-фенилацетата;
на фигуре 5 представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фтор-3-гидрокси-2-фенилпропаноата;
на фигуре 6 представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фтор-3-гидрокси-2-фенилпропаноата;
на фигуре 7 представлен 1H ЯМР-спектр 2-метил-2-фенилацетилхлорида;
на фигуре 8 представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-метил-2-фенилацетата;
на фигуре 9 представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-метил-3-гидрокси-2-фенилпропаноата;
на фигуре 10 представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-метил-3-гидрокси-2-фенилпропаноата;
на фигуре 11 представлен 1H ЯМР-спектр 2,3-дифенилпропаноилхлорида;
на фигуре 12 представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дифенилпропаноата;
на фигуре 13 представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-бензил-3-гидрокси-2-фенилпропаноата; и
на фигуре 14 представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-бензил-3-гидрокси-2-фенилпропаноата.
На фигуре 15A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-метил-2-(тиофен-2-ил)пропаноата. На фигуре 15B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-метил-2-(тиофен-2-ил)пропаноата.
На фигуре 16A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(гидроксиметил)-2-фенилпропаноата. На фигуре 16B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(гидроксиметил)-2-фенилпропаноата.
На фигуре 17A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(гидроксиметил)-2-фенилбутаноата. На фигуре 17B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(гидроксиметил)-2-фенилбутаноата.
На фигуре 18A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-циклопропил-2-(гидроксиметил)-2-фенилпропаноата. На фигуре 18B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-циклопропил-2-(гидроксиметил)-2-фенилпропаноата.
На фигуре 19A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-фтор-2-метил-2-фенилпропаноата. На фигуре 19B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-фтор-2-метил-2-фенилпропаноата.
На фигуре 20A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-4,4,4-трифтор-2-(гидроксиметил)-2-фенилбутаноата. На фигуре 20B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-4,4,4-трифтор-2-(гидроксиметил)-2-фенилбутаноата.
На фигуре 21A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дигидрокси-2-фенилпропаноата. На фигуре 21B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дигидрокси-2-фенилпропаноата.
На фигуре 22A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-метокси-2-метил-2-фенилпропаноата. На фигуре 22B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-метокси-2-метил-2-фенилпропаноата.
На фигуре 23A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропаноата. На фигуре 23B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропаноата.
На фигуре 24A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-хлорбензил)-2-фенилпропаноата. На фигуре 24B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-хлорбензил)-2-фенилпропаноата.
На фигуре 25A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(2-хлорбензил)-2-фенилпропаноата. На фигуре 25B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(2-хлорбензил)-2-фенилпропаноата.
На фигуре 26A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-гидроксибензил)-2-фенилпропаноатной соли муравьиной кислоты. На фигуре 26B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-гидроксибензил)-2-фенилпропаноатной соли муравьиной кислоты.
На фигуре 27A представлен 1H ЯМР-спектр 2-фтор-3-гидрокси-N-((1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил)-2-фенилпропанамида. На фигуре 27B представлен 13C ЯМР-спектр 2-фтор-3-гидрокси-N-((1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил)-2-фенилпропанамида.
На фигуре 28A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-фторбензил)-3-гидрокси-2-фенилпропаноата. На фигуре 28B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-фторбензил)-3-гидрокси-2-фенилпропаноата.
На фигуре 29A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метилбензил)-2-фенилпропаноата. На фигуре 29B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метилбензил)-2-фенилпропаноата.
На фигуре 30A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорбензил)-3-гидрокси-2-фенилпропаноата. На фигуре 30B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорбензил)-3-гидрокси-2-фенилпропаноата.
На фигуре 31A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорфенил)-3-гидрокси-2-метилпропаноата. На фигуре 31B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорфенил)-3-гидрокси-2-метилпропаноата.
На фигуре 32A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-хлорфенил)-3-гидрокси-2-метилпропаноата. На фигуре 32B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-хлорфенил)-3-гидрокси-2-метилпропаноата.
На фигуре 33A представлен 1H ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-гидроксифенил)-2-метилпропаноата. На фигуре 33B представлен 13C ЯМР-спектр (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-гидроксифенил)-2-метилпропаноата.
Подробное описание изобретения
Настоящее изобретение относится к классам соединений, каждое из которых имеет атропиновый или пирензепиновый остаток, и к их фармацевтически приемлемым солям. В предпочтительных вариантах осуществления в настоящем изобретении предусмотрено соединение формулы (I) или (II) или его фармацевтически приемлемая соль:
где
Me=CH3;
A=O или NR5;
W=N или CH;
X = -OH, -O-Y-Z, -S-Y-Z или -NR5-Y-Z;
R1 и R2 независимо представляют собой заместители в виде H, D, гидроксила, алкокси, нитрила, атомов галогена, прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных групп, необязательно замещенных атомами галогена; или
R1 и R2 независимо представляют собой заместители в виде фенильных или бензильных групп, необязательно замещенных одним или несколькими заместителями, выбранными из прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных групп, -галогеналкильных групп, гидроксила, алкокси, нитрила, нитро, амино, амида, сложного эфира, сульфона, сульфоксида, сульфонамида и атомов галогена; или
R1 и R2 независимо замещены гетероциклическим насыщенным, ненасыщенным или ароматическим 5- или 6-членным кольцом, содержащим один или несколько гетероатомов, выбранных из азота, кислорода и серы, и необязательно замещенных одним или несколькими заместителями, выбранными из прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных, -галогеналкильных групп, гидроксила, алкокси, нитрила, нитро, амино, амида, сложного эфира, сульфона, сульфоксида или атомов галогена;
R3 и R4 независимо замещены водородом, прямыми, или разветвленными, или циклическими C1-C10-алкильными или -галогеналкильными группами, или
R3 и R4 могут быть объединены с образованием 3-6-членных колец;
R5=H или прямые или разветвленные C1-C20-алкильные группы, предпочтительно C1-C10, прямые или разветвленные C1-C10-галогеналкильные группы;
Y представляет собой двухвалентный радикал, имеющий следующее значение:
прямой или разветвленный C1-C20 (предпочтительно C1-C10)-алкил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из атомов галогена и гидроксила;
-C(O)(C1-C10алкил)- или -C(O)(CH2)nC(O)O-(C1-C10алкил)- или -(C1-C10алкил)-;
где n представляет собой целое число от 0 до 20;
R6 и R7 независимо представляют собой H или прямые или разветвленные C1-C10-алкильные группы, прямые или разветвленные C1-C10-галогеналкильные группы; или
R6 и R7 могут быть объединены с образованием 3-6-членных колец; и
Z представляет собой H, -OH, C1-6алкокси, -COOH, -NR8R9;
R8 и R9 независимо представляют собой заместители в виде C1-C20-алкила, предпочтительно C1-C10, необязательно замещенного одним или несколькими заместителями, выбранными из гидроксила, амино, сложного эфира, карбоновой кислоты и атомов галогена; или
R8 и R9 могут быть объединены с образованием 3-6-членных колец, содержащих один или несколько гетероатомов, которые выбраны из группы, состоящей из:
В некоторых вариантах осуществления соединение формулы (I) представляет собой атропин формулы (IA),
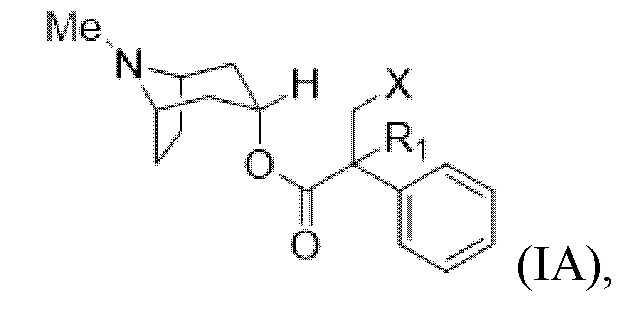
где Me, R1 и X описаны выше.
ОПРЕДЕЛЕНИЯ
Если не указано иное, термин "соединения по настоящему изобретению" или "соединение по настоящему изобретению" относится к соединениям формулы (I) или ее подформулам и иллюстративным соединениям и к их солям, а также ко всем стереоизомерам (в том числе диастереоизомерам и энантиомерам), ротамерам, таутомерам и меченным изотопом соединениям (включая замещения дейтерием), а также изначально образованным фрагментам.
Выражение "эффективное количество" соединений по настоящему изобретению, описанных ниже, относится к такому количеству терапевтического соединения, которое необходимо или является достаточным для осуществления его предполагаемой функции у млекопитающего, например, лечение у млекопитающего состояния, ассоциированного с мускариновым рецептором, или состояния болезни. Эффективное количество терапевтического соединения может варьироваться в соответствии с факторами, такими как количество этиологического фактора, уже присутствующего у млекопитающего, возраст, пол и вес млекопитающего, и способность терапевтических соединений по настоящему изобретению влиять на ассоциированное с мускариновым рецептором нарушение у млекопитающего. Специалист в данной области сможет изучить вышеупомянутые факторы и принять решение относительно эффективного количества терапевтического соединения без лишних экспериментов. Анализ in vitro или in vivo также может быть использован для определения "эффективного количества" терапевтических соединений, описанных ниже. Специалист в данной области сможет выбрать соответствующее количество терапевтического соединения для применения в вышеупомянутом анализе или в качестве терапии.
Фраза "офтальмологически совместимый" является признанной в уровне техники и относится к составам, полимерам и другим материалам и/или лекарственным формам, которые подходят для применения в контакте с тканями глаза людей и животных без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерных с обоснованным соотношением польза/риск, определяемым специалистом в данной области.
Используемая в данном документе фармацевтическая композиция представляет собой композицию, подходящую для применения в фармации. Композиция, подходящая для применения в фармации, может быть стерильной, гомогенной и/или изотонической. Фармацевтические композиции могут быть получены в определенных вариантах осуществления в водной форме, например, в предварительно заполненном шприце или в другом контейнере с одной или несколькими дозами. В некоторых вариантах осуществления фармацевтические композиции по настоящему изобретению являются офтальмологически совместимыми и подходящими для офтальмологического введения субъекту-человеку, например, посредством местного или других известных способов доставки. В другом варианте осуществления фармацевтические композиции по настоящему изобретению являются подходящими для интравитреального введения. В еще одном варианте осуществления фармацевтические композиции по настоящему изобретению являются подходящими для введения посредством интравитреальной инфузии. В еще одном варианте осуществления фармацевтические композиции вводят перорально.
Подразумевается, что применяемый в данном документе термин "алкил" включает разветвленные, прямоцепочечные и циклические замещенные или незамещенные насыщенные алифатические углеводородные группы. Алкильные группы могут содержать от приблизительно 1 до приблизительно 24 атомов углерода ("C1-C24"), от приблизительно 7 до приблизительно 24 атомов углерода ("C7-C24"), от приблизительно 8 до приблизительно 24 атомов углерода ("C8-C24") или от приблизительно 9 до приблизительно 24 атомов углерода ("C9-C24"). Алкильные группы также могут содержать от приблизительно 1 до приблизительно 8 атомов углерода ("C1-C8"), от приблизительно 1 до приблизительно 6 атомов углерода ("C1-C6") или от приблизительно 1 до приблизительно 3 атомов углерода ("C1-C3"). Примеры C1-C6алкильных групп включают без ограничения метильные, этильные, пропильные, изопропильные, н-бутильные, изобутильные, трет-бутильные, н-пентильные, неопентильные и н-гексильные радикалы.
Используемый в данном документе термин "C2-6алкенил" относится к группе, представляющей собой радикал, с прямой или разветвленной углеводородной цепью, состоящей только из атомов углерода и водорода, содержащей по меньшей мере одну двойную связь, имеющей от двух до шести атомов углерода, и которая присоединена к остальной части молекулы посредством одинарной связи. Термины "C2-C20алкенил" и "C2-C10алкенил" следует понимать соответственно. Примеры C2-6алкенила включают без ограничения, этенил, проп-1-енил, бут-1-енил, пент-1-енил, пент-4-енил и пента-1,4-диeнил.
Используемый в данном документе термин "C2-6алкинил" относится к группе, представляющей собой радикал, с прямой или разветвленной углеводородной цепью, состоящей только из атомов углерода и водорода, содержащей по меньшей мере одну тройную связь, имеющей от двух до шести атомов углерода, и которая присоединена к остальной части молекулы посредством одинарной связи. Термин "C2-4алкинил" следует понимать соответственно. Примеры C2-6алкинила включают без ограничения, этинил, проп-1-инил, бут-1-инил, пент-1-инил, пент-4-инил и пента-1,4-диинил.
Используемый в данном документе термин "C1-6алкокси" относится к радикалу формулы -ORa, где Ra представляет собой C1-6алкильный радикал, в целом определенный выше. Примеры C1-6алкокси включают без ограничения метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, пентокси и гексокси.
"Галоген" относится к брому, хлору, фтору или йоду.
Применяемый в данном документе термин "гетероциклил" или "гетероциклический" относится к устойчивому радикалу, представляющему собой 5- или 6-членное неароматическое моноциклическое кольцо, которое содержит 1, 2 или 3 гетероатома, независимо выбранных из азота, кислорода и серы. Гетероциклильный радикал может быть связан посредством атома углерода или гетероатома. Примеры гетероциклила включают без ограничения азетидинил, оксетанил, пирролинил, пирролидил, тетрагидрофурил, тетрагидротиенил, пиперидил, пиперазинил, тетрагидропиранил, морфолинил или пергидроазепинил.
Применяемый в данном документе термин "гетероарил" относится к радикалу, представляющему собой 5- или 6-членное ароматическое моноциклическое кольцо, которое содержит 1, 2, 3 или 4 гетероатома, независимо выбранных из азота, кислорода и серы. Гетероарильный радикал может быть связан посредством атома углерода или гетероатома. Примеры гетероарила включают без ограничения фурил, пирролил, тиенил, пиразолил, имидазолил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, тетразолил, пиразинил, пиридазинил, пиримидил или пиридил.
В некоторых вариантах осуществления настоящее изобретение предусматривает новые фармацевтические составы, в частности, новые фармацевтические составы, в которых активный ингредиент содержит модулятор мускаринового рецептора общей формулы (I) или (II) и/или его фармацевтически приемлемые соли:
где
Me=CH3;
A=O или NR5;
W=N или CH;
X = -OH, -O-Y-Z, -S-Y-Z или -NR5-Y-Z;
R1 и R2 независимо представляют собой заместители в виде H, D, гидроксила, алкокси, нитрила, атомов галогена, прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных групп, необязательно замещенных атомами галогена; или
R1 и R2 независимо представляют собой заместители в виде фенильных или бензильных групп, необязательно замещенных одним или несколькими заместителями, выбранными из прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных групп, -галогеналкильных групп, гидроксила, алкокси, нитрила, нитро, амино, амида, сложного эфира, сульфона, сульфоксида, сульфонамида и атомов галогена; или
R1 и R2 независимо замещены гетероциклическим насыщенным, ненасыщенным или ароматическим 5- или 6-членным кольцом, содержащим один или несколько гетероатомов, выбранных из азота, кислорода и серы, и необязательно замещенных одним или несколькими заместителями, выбранными из прямых, разветвленных или циклических C1-C20 (предпочтительно C1-C10)-алкильных, -галогеналкильных групп, гидроксила, алкокси, нитрила, нитро, амино, амида, сложного эфира, сульфона, сульфоксида или атомов галогена;
R3 и R4 независимо замещены водородом, прямыми, или разветвленными, или циклическими C1-C10-алкильными или -галогеналкильными группами, или
R3 и R4 могут быть объединены с образованием 3-6-членных колец;
R5=H или прямые или разветвленные C1-C20-алкильные группы, предпочтительно C1-C10, прямые или разветвленные C1-C10-галогеналкильные группы;
Y представляет собой двухвалентный радикал, имеющий следующее значение:
прямой или разветвленный C1-C20 (предпочтительно C1-C10)-алкил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из атомов галогена и гидроксила;
-C(O)(C1-C10алкил)- или -C(O)(CH2)nC(O)O-(C1-C10алкил)- или -(C1-C10алкил)-;
где n представляет собой целое число от 0 до 20;
R6 и R7 независимо представляют собой H или прямые или разветвленные C1-C10-алкильные группы, прямые или разветвленные C1-C10-галогеналкильные группы; или
R6 и R7 могут быть объединены с образованием 3-6-членных колец; и
Z представляет собой H, -OH, C1-6алкокси, -COOH, -NR8R9;
R8 и R9 независимо представляют собой заместители в виде C1-C20-алкила, предпочтительно C1-C10, необязательно замещенного одним или несколькими заместителями, выбранными из гидроксила, амино, сложного эфира, карбоновой кислоты и атомов галогена; или
R8 и R9 могут быть объединены с образованием 3-6-членных колец, содержащих один или несколько гетероатомов, которые выбраны из группы, состоящей из:
В некоторых вариантах осуществления соединение формулы (I) представляет собой атропин формулы (IA),
где Me, R1 и X описаны выше.
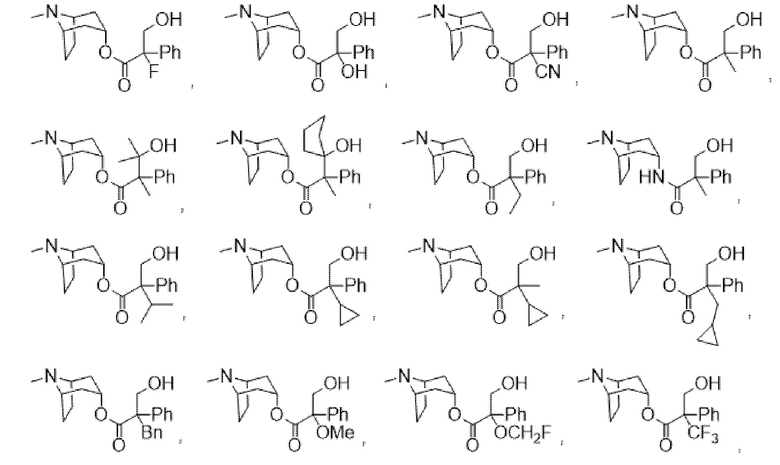
В некоторых вариантах осуществления соединения формулы (I) и формулы (II) выбраны из группы, состоящей из:
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фтор-3-гидрокси-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-метил-3-гидрокси-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-бензил-3-гидрокси-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-метил-2-(тиофен-2-ил)пропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(гидроксиметил)-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(гидроксиметил)-2-фенилбутаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-циклопропил-2-(гидроксиметил)-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-фтор-2-метил-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-4,4,4-трифтор-2-(гидроксиметил)-2-фенилбутаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дигидрокси-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-метокси-2-метил-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-хлорбензил)-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(2-хлорбензил)-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-гидроксибензил)-2-фенилпропаноатной соли муравьиной кислоты,
2-фтор-3-гидрокси-N-((1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил)-2-фенилпропанамида,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-фторбензил)-3-гидрокси-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метилбензил)-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорбензил)-3-гидрокси-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропаноата,
(1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-(бензилокси)фенил)пропаноата и
6-((11-(2-(4-метилпиперазин-1-ил)ацетил)-11H-бензо[e]пиридо[3,2-b][1,4]диазепин-6-ил)окси)гексилнитрата.
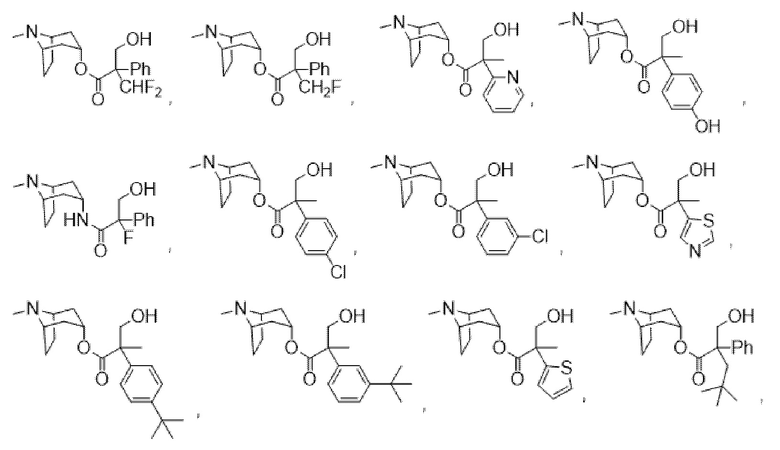
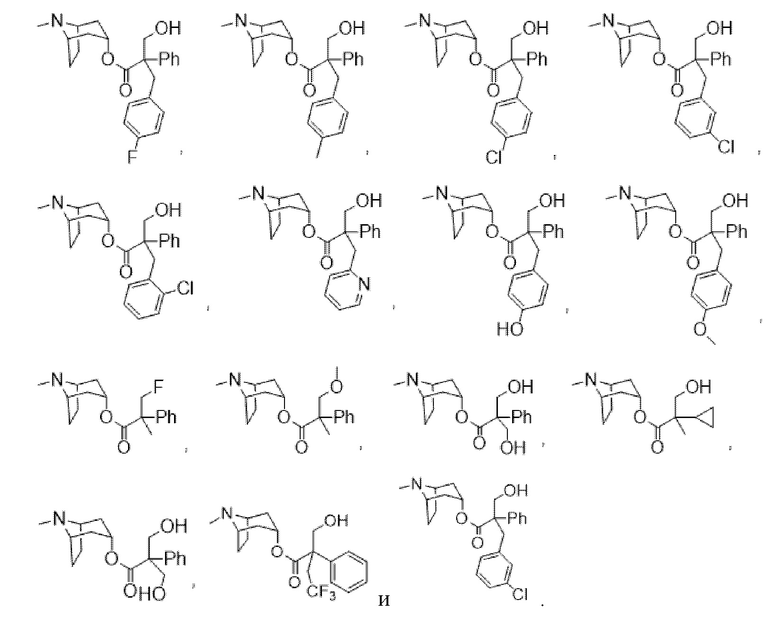
Дополнительные соединения по настоящему изобретению включают следующие:
В следующих параграфах представлены примеры соединений в соответствии с настоящим изобретением.
Примеры
Примеры 1-3

ЯМР-спектры были получены на спектрометре Bruker 400 МГц или спектрометре Bruker 300 МГц.
Способы LCMS подробно описаны ниже (если не указано иное):
Стандартный способ LCMS
Способ QC LCMS
Сокращения
DAST Трифторид диэтиламиносеры
DCM Дихлорметан
DMF Диметилформамид
к.т. Комнатная температура
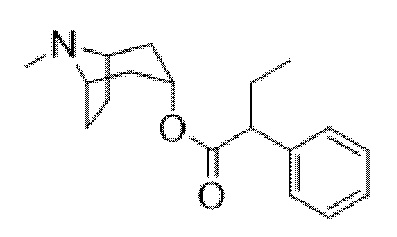
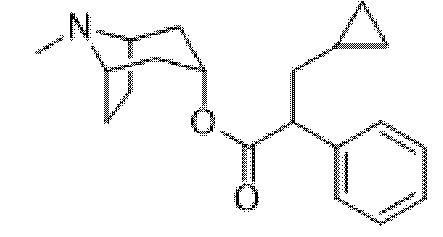
Схема синтеза согласно примеру 1: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фтор-3-гидрокси-2-фенилпропанoат
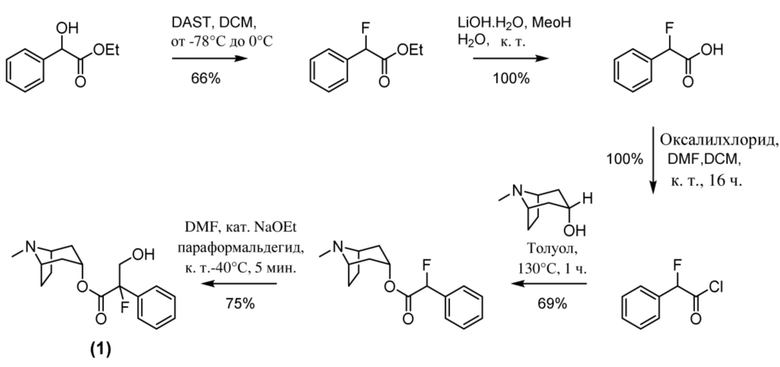
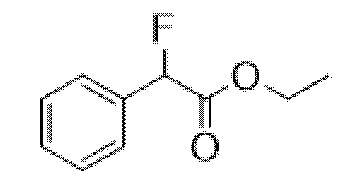
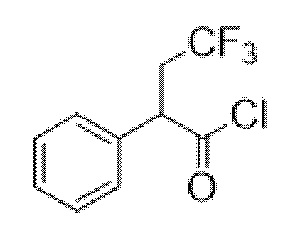
Этил-2-фтор-2-фенилацетат
В раствор этилманделата (93 г, 0,52 моль) в DCM (1,5 л) при -78°C добавляли DAST (81,8 мл, 0,62 моль) с такой скоростью, что T≤-60°C. Реакционную смесь перемешивали при обеспечении нагревания до 0°C. Через 1 ч медленно повышали основность реакционной смеси с помощью 1 н. NaOH до pH~7. Смесь экстрагировали с помощью DCM. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде масла соломенного цвета (62,4 г, 66%). Материал применяли без очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,54-7,36 (5H, m), 5,77 (1H, d, J=47,3 Гц), 4,32-4,16 (2H, m), 1,26 (3H, t, J=7,2 Гц). 1H ЯМР-спектр показан на фигуре 1.
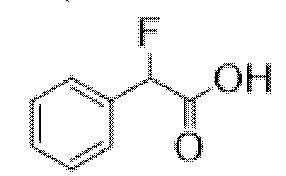
2-Фтор-2-фенилуксусная кислота
В раствор этил-2-фтор-2-фенилацетата (32,4 г, 0,18 моль) в MeOH (100 мл) добавляли LiOH.H2O (11,2 г, 0,27 моль) в воде (15 мл) и реакционную смесь перемешивали при к. т. в течение 1 ч. (был отмечен небольшой экзотермический эффект). Реакционную смесь разбавляли этилацетатом и подкисляли до ~ pH 3 с помощью 1 н. HCl. Продукт экстрагировали этилацетатом и объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде белого твердого вещества (27,4 г, 100%). Материал применяли без очистки.
1H ЯМР (400 МГц, d6-DMSO) δ 13,47 (1H, br s), 7,48-7,37 (5H, m), 5,95 (1H, d, J=47,6 Гц). 1H ЯМР-спектр показан на фигуре 2.
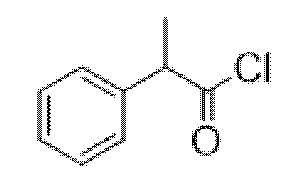
2-Фтор-2-фенилацетилхлорид
В раствор 2-фтор-2-фенилуксусной кислоты (27,4 г, 0,18 моль) в DCM (250 мл) при к. т. добавляли 1 каплю DMF. Добавляли оксалилхлорид (23,3 мл, 0,27 моль), что приводило к выделению пузырьков газа. Реакционную смесь перемешивали при к. т. в течение 16 ч. Реакционную смесь концентрировали in vacuo с получением указанного в заголовке соединения в виде масла соломенного цвета (30,1 г, 100%). Материал применяли без очистки.
1H ЯМР (400 МГц, CDCl3) δ 7,54-7,43 (5H, m), 5,90 (1H, d, 47,6 Гц). 1H ЯМР-спектр показан на фигуре 3.

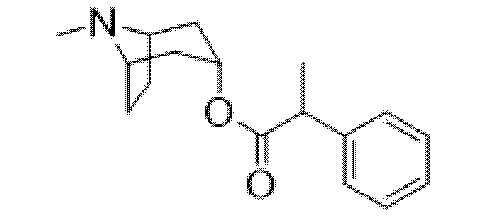
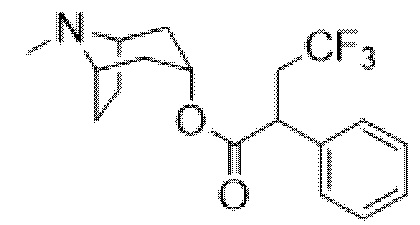
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-фтор-2-фенилацетат
В раствор тропина (27,1 г, 0,19 моль) в толуоле (450 мл) добавляли 2-фтор-2-фенилацетилхлорид (30,1 г, 0,17 моль), что приводило к образованию осадка. Реакционную смесь перемешивали с обратным холодильником в течение 1,5 ч. Реакционную смесь разбавляли с помощью этилацетата и экстрагировали с помощью 1 н. HCl. Повышали основность объединенных водных фракций до ~ pH 13 с помощью 1 н. NaOH и экстрагировали с помощью этилацетата. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде масла соломенного цвета, которое отвердевало при отстаивании (33,5 г, 69%).
1H ЯМР (400 МГц, CDCl3) δ 7,51-7,37 (5H, m), 5,73 (1H, d, J=47,9 Гц), 5,08 (1H, t, J=5,3 Гц), 3,07-2,93 (2H, m), 2,22 (3H, s), 2,16-2,01 (2H, m), 1,97-1,85 (1H, m), 1,84-1,64 (3H, m), 1,56-1,48 (1H, m), 1,37-1,29 (1H, m). 1H ЯМР-спектр показан на фигуре 4.
LCMS (ESI) [M+H]+ 278, Rt=0,70 мин.
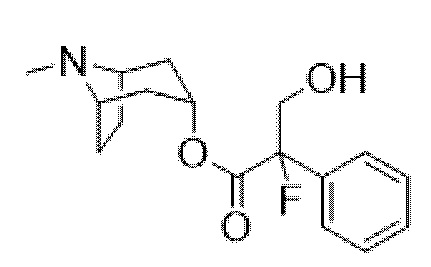
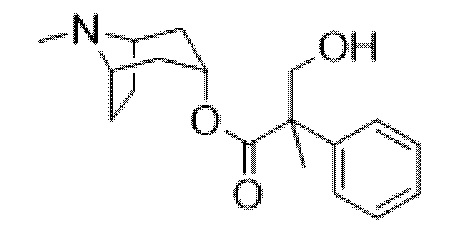
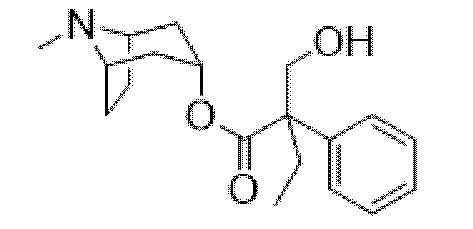
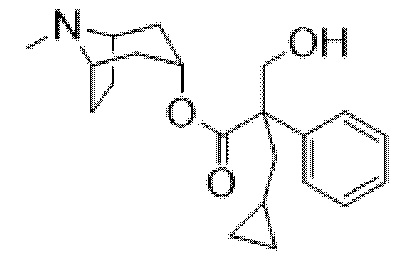
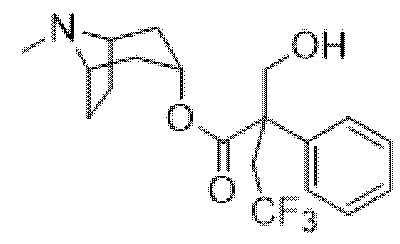
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-фтор-3-гидрокси-2-фенилпропанoат
В раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фтор-2-фенилацетата (33,5 г, 0,12 моль) в DMF (120 мл) добавляли параформальдегид (5,45 г, 0,18 моль). К данной суспензии добавляли свежеприготовленный этоксид натрия (140 мг натрия в 3,6 мл этанола), что приводило к растворению твердых веществ. Реакционную смесь перемешивали при 40°C в течение 5 мин. Реакционную смесь разбавляли с помощью этилацетата и экстрагировали с помощью 1 н. HCl и повышали основность объединенных водных фракций до ~ pH 13 с помощью 1 н. NaOH и экстрагировали с помощью этилацетата. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo до ~ 1/5 объема, при котором достигалась точка кристаллизации продукта. Твердое вещество собирали с помощью фильтрации и высушивали in vacuo с получением указанного в заголовке соединения в виде белого твердого вещества (25,0 г, 67%). Исходные растворы концентрировали in vacuo и растирали с этилацетатом с получением второй порции, по чистоте сходной с первой (2,92 г, 8%).
1H ЯМР (400 МГц, CDCl3) δ 7,54-7,48 (2H, m), 7,45-7,35 (3H, m), 5,09 (1H, t, J=5,2 Гц), 4,36 (1H, dd, J=29,2, 13,2 Гц), 4,04 (1H, dd, J=15,3, 12,9 Гц), 3,10-2,99 (2H, m), 2,46 (1H, br s), 2,24 (3H, s), 2,18-2,06 (2H, m), 2,01-1,58 (6H, m). 1H ЯМР-спектр показан на фигуре 5.
13C ЯМР (400 МГц, CDCl3) δ 168,4, 134,8, 129,0, 128,7, 124,7, 97,5 (d, J=189 Гц), 69,8, 67,1, 59,6, 40,4, 36,3, 25,3. 13C ЯМР-спектр показан на фигуре 6.
LCMS (ESI) [M+H]+ 308, Rt=0,66 мин.
QC LCMS (ESI) [M+H]+ 308,2, Rt=2,28 мин. (94,2%), чистота > 99% согласно ЯМР.
Схема синтеза согласно примеру 2: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-метил-3-гидрокси-2-фенилпропанoат
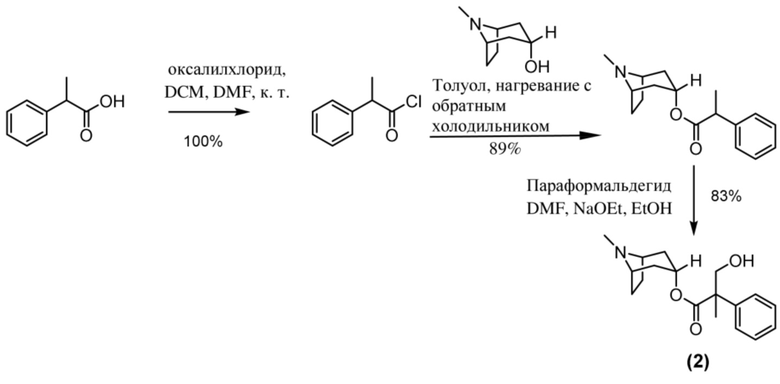
2-Метил-2-фенилацетилхлорид
В раствор 2-метил-2-фенилуксусной кислоты (7,65 г, 50,94 ммоль) в DCM (60 мл) при к. т. добавляли 1 каплю DMF. Добавляли оксалилхлорид (8,9 мл, 0,102 моль), что приводило к выделению пузырьков газа. Реакционную смесь перемешивали при к. т. в течение 18 ч., затем концентрировали in vacuo с получением указанного в заголовке соединения в виде масла соломенного цвета (8,6 г, 100%). Материал применяли без очистки.
1H ЯМР (300 МГц, CDCl3) δ 7,43-7,26 (5H, m), 4,12 (1H, квартет, J=7,1 Гц), 1,60 (3H, d, J=7,1 Гц). 1H ЯМР-спектр показан на фигуре 7.
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-метил-2-фенилацетат
В раствор тропина (6,5 г, 45,9 ммоль) в толуоле (40 мл) добавляли 2-метил-2-фенилацетилхлорид (8,6 г, 51,0 ммоль), что приводило к образованию осадка. Реакционную смесь перемешивали с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали in vacuo и остаток растирали с диэтиловым эфиром, затем твердые вещества отфильтровывали с получением белого твердого вещества. Данные твердые вещества разделяли между H2O и DCM, затем повышали основность до pH >10 с использованием 1 н. NaOH и продукт экстрагировали в DCM. Объединенные экстракты DCM промывали с помощью H2O и солевого раствора, затем пропускали через картридж для разделения фаз и концентрировали in vacuo с получением указанного в заголовке соединения в виде масла соломенного цвета (11,43 г, 81%).
1H ЯМР (300 МГц, CDCl3) δ 7,36-7,21 (5H, m), 4,96 (1H, t, J=5,4 Гц), 3,67 (1H, квартет, J=7,2 Гц), 3,06-2,90 (2H, m), 2,21 (3H, s), 2,13-1,96 (2H, m), 1,95-1,57 (4H, m), 1,54-1,44 (1H, m), 1,51 (3H, d, J=7,2 Гц), 1,40-1,28 (1H, m). 1H ЯМР-спектр показан на фигуре 8.
LCMS (ESI) [M+H]+ 274, Rt=0,79 мин.
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-метил-3-гидрокси-2-фенилпропанoат
В раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-метил-2-фенилацетата (12,4 г, 45,36 ммоль) в DMF (15 мл) добавляли параформальдегид (2,04 г, 68,04 ммоль). К данной суспензии добавляли свежеприготовленный этоксид натрия (52 мг натрия в 1,0 мл этанола), что приводило к растворению твердых веществ. Реакционную смесь перемешивали при к.т. в течение 4 ч. и затем разделяли между H2O и DCM. Объединенные экстракты DCM промывали с помощью H2O и солевого раствора, затем пропускали через картридж для разделения фаз и концентрировали in vacuo. Полученное в результате масло растирали с диэтиловым эфиром, в результате чего продукт кристаллизовался. Твердое вещество собирали с помощью фильтрации и высушивали in vacuo с получением указанного в заголовке соединения в виде белого твердого вещества (10,5 г, 76%).
1H ЯМР (400 МГц, CDCl3) δ 7,38-7,25 (5H, m), 5,06 (1H, t, J=4,1 Гц), 4,12 (1H, dd, J=8,5, 4,2 Гц), 3,62 (1H, dd, J=8,7, 6,0 Гц), 3,04-2,89 (2H, m), 2,55 (1H, t, J=5,2 Гц), 2,19 (3H, s), 2,15-2,00 (2H, m), 1,90-1,78 (1H, m), 1,74-1,57 (6H, m), 1,50-1,43 (1H, m), 1,23-1,11 (1H, m). 1H ЯМР-спектр показан на фигуре 9.
13C ЯМР (400 МГц, CDCl3) δ 175,2, 140,2, 128,6, 127,3, 126,2, 69,7, 68,0, 59,6, 59,5, 52,2, 40,4, 36,6, 36,3, 25,4, 24,9, 19,5 (наличие хирального центра приводит к неэквивалентности атомов углерода тропанового кольца, следовательно, наблюдается 16 сигналов, а не 13). 13C ЯМР-спектр показан на фигуре 10.
LCMS (ESI) [M+H]+ 304, Rt=0,69 мин.
QC LCMS (ESI [M+H]+ 304,2, Rt=2,38 мин. (98,9%)
Схема синтеза согласно примеру 3: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-бензил-3-гидрокси-2-фенилпропанoат


2,3-Дифенилпропаноилхлорид
В раствор 2,3-дифенилпропановой кислоты (16,0 г, 70,71 ммоль) в DCM (80 мл) при к. т. добавляли DMF (0,10 мл) и смесь охлаждали на ледяной бане. Добавляли оксалилхлорид (12,3 мл, 141,42 ммоль), что приводило к выделению пузырьков газа и охлаждающую баню удаляли. Реакционную смесь перемешивали при к. т. в течение 18 ч., затем концентрировали in vacuo с получением указанного в заголовке соединения в виде масла соломенного цвета (17,3 г, 100%). Материал применяли без очистки.
1H ЯМР (300 МГц, CDCl3) δ 7,33-7,07 (10H, m), 3,86 (1H, dd, J=7,0, 8,4 Гц), 3,41 (1H, dd, J=13,8, 8,4 Гц), 3,03 (1H, dd, J=13,8, 7,0 Гц). 1H ЯМР-спектр показан на фигуре 11.
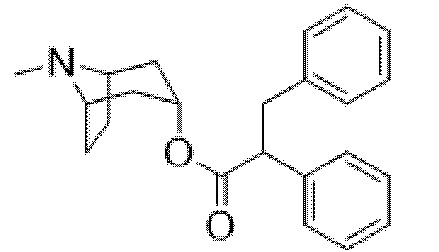
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дифенилпропанoат
В раствор тропина (9,49 г, 70,71 ммоль) в толуоле (70 мл) добавляли 2,3-дифенилпропаноилхлорид (17,30 г, 70,71 ммоль), что приводило к образованию осадка. Реакционную смесь перемешивали с обратным холодильником в течение 2 ч., затем концентрировали in vacuo. Остаток растирали с диэтиловым эфиром и твердые вещества отфильтровывали с получением белого твердого вещества. Это твердое вещество разделяли между H2O и DCM, повышали основность до pH >10 с использованием 1 н. раствора NaOH и продукт экстрагировали в DCM. Экстракты DCM промывали с помощью H2O и солевого раствора, затем пропускали через картридж для разделения фаз и концентрировали in vacuo с получением указанного в заголовке соединения в виде масла соломенного цвета (22,4 г, 90%).
1H ЯМР (300 МГц, CDCl3) δ 7,35-7,11 (10H, m), 4,90 (1H, t, J=5,4 Гц), 3,79 (1H, dd, J=6,5, 9,0 Гц), 3,43 (1H, dd, J=13,5, 9,0 Гц), 3,03 (1H, dd, J=13,5, 6,5 Гц), 2,97-2,87 (2H, m), 2,18 (3H, s), 2,05-1,92 (2H, m), 1,85-1,65 (2H, m), 1,56-1,30 (4H, m). 1H ЯМР-спектр показан на фигуре 12.
LCMS (ESI) [M+H]+ 350, Rt=0,98 мин.
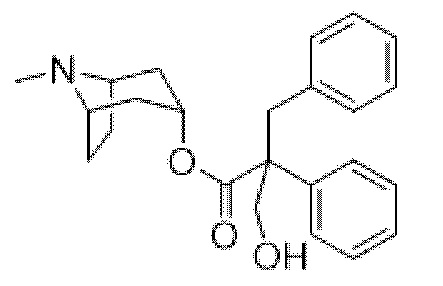
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-бензил-3-гидрокси-2-фенилпропанoат
В раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дифенилпропаноата (22,40 г, 64,10 ммоль) в DMF (30 мл) добавляли параформальдегид (3,27 г, 108,96 ммоль). К данной суспензии добавляли свежеприготовленный этоксид натрия (74 мг натрия в 1,3 мл этанола), что приводило к растворению твердых веществ. Реакционную смесь перемешивали при к. т. в течение 3 ч. перед тем, как добавляли H2O. Осадок отфильтровывали, промывали с помощью H2O и высушивали in vacuo с удалением большей части H2O. Полученное в результате "влажное" твердое вещество разделяли между MeOH и DCM и фазы разделяли. Экстракт DCM высушивали (MgSO4), фильтровали и концентрировали in vacuo, что приводило к кристаллизации продукта. Осадок отфильтровывали и высушивали in vacuo с получением указанного в заголовке соединения в виде белого твердого вещества (18,18 г, 75%).
1H ЯМР (400 МГц, CDCl3) δ 7,39-7,16 (8H, m), 7,05-6,97 (2H, m), 5,09 (1H, t, J=5,3 Гц), 4,12-3,95 (2H, m), 3,54-3,42 (2H, m), 3,02-2,90 (2H, m), 2,19 (3H, s), 2,15-2,00 (2H, m), 1,84-1,58 (4H, m), 1,54-1,41 (2H, m), 1,28-1,19 (1H, m). 1H ЯМР-спектр показан на фигуре 13.
13C ЯМР (400 МГц, CDCl3) δ 173,3, 139,9, 136,7, 130,5, 128,7, 128,1, 127,4, 126,8, 126,7, 68,1, 63,9, 59,5 (x2 сигнала), 56,5, 40,4, 38,9, 36,5, 36,4, 25,2, 25,0 (наличие хирального центра приводит к неэквивалентности атомов углерода тропанового кольца, следовательно, наблюдается 20 сигналов, а не 17). 13C ЯМР-спектр показан на фигуре 14.
LCMS (ESI) [M+H]+ 380, Rt=0,90 мин.
QC LCMS (ESI) [M+H]+ 380,3, Rt=3,19 мин. (99,2%)
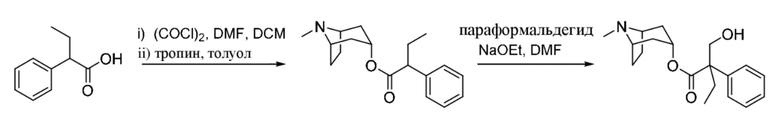
Схема синтеза согласно примеру 4: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-метил-2-(тиофен-2-ил)пропаноат
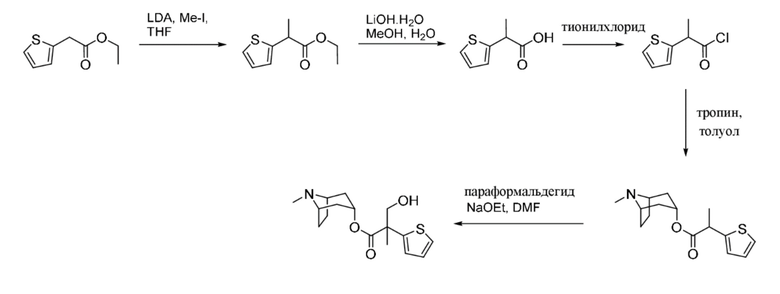
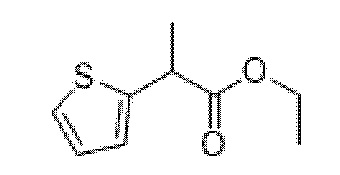
Этил-2-(тиофен-2-ил)пропаноат
В раствор диизопропиламина (4,5 мл, 32,3 ммоль) в THF (100 мл) при -40°C добавляли по каплям n-BuLi (12,9 мл, 2,5 М, 32,3 ммоль). Реакционную смесь перемешивали при -40°C в течение 20 мин., затем охлаждали до -78°C. Добавляли по каплям этил-2-(тиофен-2-ил)ацетат (5 г, 29,4 ммоль) с такой скоростью, что T < -60°C. Реакционную смесь перемешивали при -78°C в течение 30 мин., затем нагревали до 0°C. Добавляли метилиодид (2,2 мл, 35,3 ммоль) и реакционную смесь перемешивали при 0°C в течение 2 ч. Реакционную смесь разбавляли с помощью воды и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Полученный остаток очищали на диоксиде кремния (80 г, 0-50% этилацетат в циклогексане) с получением указанного в заголовке соединения в виде бледного коричневого масла (3,3 г, 62%). 1H ЯМР (400 МГц, CDCl3) δ 7,20 (1H, t, J=3,4 Гц), 6,95 (2H, d, J=3,4 Гц), 4,22-4,12 (2H, m), 3,99 (1H, q, J=7,2 Гц), 1,58 (3H, d, J=7,1 Гц), 1,30-1,19 (3H, m).
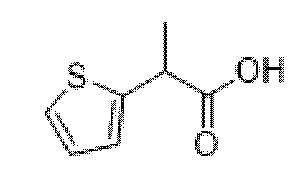
2-(Тиофен-2-ил)пропановая кислота
В раствор этил-2-(тиофен-2-ил)пропаноата (5,9 г, 32,39 ммоль) в метаноле (40 мл) и воде (15 мл) добавляли моногидрат гидроксида лития (2,0 г, 48,4 ммоль) и реакционную смесь перемешивали при к.т. в течение 3 ч. Реакционную смесь концентрировали in vacuo до примерно ¼ объема и остаток экстрагировали с помощью EtOAc. Органические фракции отбрасывали и водный слой подкисляли до ~ pH 3 с помощью 1 н. HCl и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде желтого масла (5,0 г, 100%). 1H ЯМР (400 МГц, d6-DMSO) δ 7,26 (1H, dd, J=5,2, 1,6 Гц), 6,89 (1H, dd, J=5,1, 3,5 Гц), 6,87-6,80 (1H, m), 3,74 (1H, q, J=7,0 Гц), 1,35 (3H, d, J=7,0 Гц).
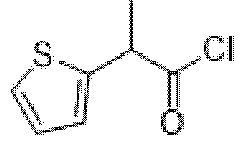
2-(Тиофен-2-ил)пропаноилхлорид
2-(Тиофен-2-ил)пропановую кислоту (2,0 г, 12,8 ммоль) растворяли в тионилхлориде (10,0 мл, 137,1 ммоль) и реакционную смесь нагревали с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали in vacuo с получением указанного в заголовке соединения в виде коричневого масла (2,24 г, 100%). 1H ЯМР (400 МГц, CDCl3) δ 7,30 (1H, dd, J=5,1, 1,2 Гц), 7,06-6,99 (2H, m), 4,39 (1H, q, J=7,1 Гц), 1,70 (3H, d, J=7,1 Гц).
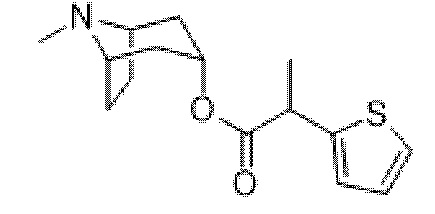
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(тиофен-2-ил)пропаноат
В раствор 2-(тиофен-2-ил)пропаноилхлорида (2,2 г, 12,6 ммоль) в толуоле (20 мл) добавляли тропин (1,62 г, 11,5 ммоль) и реакционную смесь нагревали при 100°C в течение 2 ч. Реакционную смесь разбавляли с помощью EtOAc и экстрагировали с помощью 1 н. HCl и органическую фракцию отбрасывали. Повышали основность водной фазы до ~ pH 12 с помощью 6 н. NaOH и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде коричневого масла, которое применяли неочищенным в следующей реакции (2,29 г, 71%). LCMS (ESI) [M+H]+ 280, Rt=0,72 мин.
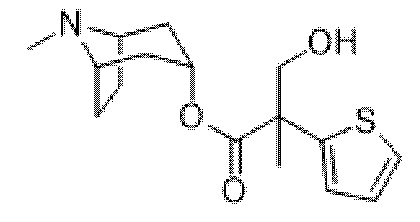
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-метил-2-(тиофен-2-ил)пропаноат
В суспензию (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(тиофен-2-ил)пропаноата (1,3 г, 4,7 ммоль) и параформальдегида (0,21 г, 6,9 ммоль) в DMF (10 мл) добавляли раствор этоксида натрия в этаноле (0,11 мл, 21%, 0,2 ммоль) и реакционную смесь перемешивали при к. т. в течение 10 мин. Реакционную смесь разбавляли с помощью EtOAc и экстрагировали с помощью 1 н. HCl. Органические фракции отбрасывали. Повышали основность водной фракции до ~ pH 12 с помощью 1 н. NaOH и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (760 мг, 52%). 1H ЯМР (400 МГц, CDCl3) δ 7,24 (1H, dd, J=4,6, 1,4 Гц), 7,02-6,98 (2H, m), 5,03 (1H, t, J=5,54 Гц), 4,13 (1H, d, J=11,3 Гц), 3,74 (1H, d, J=11,3 Гц), 3,08-2,94 (2H, m), 2,58 (1H, br s), 2,22 (3H, s), 2,16-2,01 (1H, m), 1,98-1,87 (1H, m), 1,86-1,70 (5H, m), 1,69-1,62 (1H, m), 1,57-1,50 (1H, m), 1,41-1,31 (1H, m). Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигуре 15A и фигуре 15B соответственно.
LCMS (ESI) [M+H]+ 310, Rt=2,40 мин.
Схема синтеза согласно примеру 5: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(гидроксиметил)-2-фенилпропанoат

В раствор aтропина (3,1 г, 10,8 ммоль) и параформальдегида (1,6 г, 54,1 ммоль) в DMF (5 мл) при 0°C добавляли этоксид натрия в этаноле (0,08 мл, 21%, 0,22 ммоль) и реакционную смесь перемешивали при 0°C в течение 5 мин., затем при к. т. в течение 1 ч. Реакционную смесь фильтровали через целит и фильтрат очищали на диоксиде кремния (24 г, 0-40% (2 н. NH3 в MeOH) в DCM). Половину полученного материала повторно очищали на C18 с диоксидом кремния (50 г, 0-50% 0,02 М NH3 в MeCN) с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (1,19 г, 67%). 1H ЯМР (400 МГц, d6-DMSO) δ 7,35-7,27 (2H, m), 7,26-7,14 (3H, m), 4,88 (1H, t, J=5,1 Гц), 4,74 (1H, t, J=4,62 Гц), 4,06 (2H, dd, J=10,2, 4,9 Гц), 3,97 (2H, dd, J=10,2, 4,7 Гц), 3,35 (1H, s), 2,92-2,83 (2H, m), 2,07 (3H, s), 1,92 (2H, dt, J=14,4, 4,1 Гц), 1,75-1,60 (2H, m), 1,50-1,36 (4H, m). LCMS (ESI) [M+H]+ 320, Rt=1,96 мин. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 16A и 16B соответственно.
Схема синтеза согласно примеру 6: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(гидроксиметил)-2-фенилбутаноат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-фенилбутаноат
В раствор 2-фенилмасляной кислоты (2,0 г, 12,2 ммоль) в DCM (20 мл) при 0°C добавляли оксалилхлорид (2,1 мл, 24,4 ммоль) и DMF (0,06 мл, 0,7 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 мин., затем при к. т. в течение 16 ч. Реакционную смесь концентрировали in vacuo и остаток растворяли в толуоле (10 мл). Данный раствор добавляли в раствор тропина (1,7 г, 12,18 ммоль) в толуоле (20 мл) и реакционную смесь перемешивали при к. т. в течение 10 мин., затем с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали in vacuo и остаток собирали с помощью фильтрации и промывали с помощью диэтилового эфира. Твердое вещество растворяли в 1 н. NaOH и экстрагировали с помощью DCM. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде бесцветного масла (1,76 г, 50%). 1H ЯМР (400 МГц, CDCl3) δ 7,35-7,21 (5H, m), 4,96 (1H, t, J=5,1 Гц), 3,41, (1H, t, J=7,8 Гц), 3,06-2,91 (2H, m), 2,21 (3H, s), 2,18-1,97 (3H, m), 1,94-1,70 (4H, m), 1,67-1,59 (1H, m), 1,52-1,36 (2H, m), 0,91 (3H, t, J=7,4 Гц). LCMS (ESI) [M+H]+ 288, Rt=0,84 мин.
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(гидроксиметил)-2-фенилбутаноат
В суспензию (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фенилбутаноата (1,8 г, 6,1 ммоль) и параформальдегида (0,28 г, 9,2 ммоль) в DMF (6 мл) добавляли раствор этоксида натрия в этаноле (0,11 мл, 21%, 0,2 ммоль) и реакционную смесь перемешивали при к. т. в течение 4 ч. Реакционную смесь разбавляли с помощью EtOAc, экстрагировали с помощью 1 н. HCl и органические фракции отбрасывали. Повышали основность водной фракции до ~ pH 12 с помощью 1 н. NaOH и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Осадок очищали на диоксиде кремния (50 г, 0-18% (2 н. NH3 MeOH в DCM) с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (1,56 мг, 80%). 1H ЯМР (400 МГц, CDCl3) δ 7,38-7,21 (5H, m), 5,07 (1H, t, J=5,32 Гц), 4,12 (1H, d, J=11,1 Гц), 3,99 (1H, d, J=11,1 Гц), 3,02-2,88 (2H, m), 2,57 (1H, br s), 2,28-1,99 (7H, m), 1,86-1,64 (2H, m), 1,61-1,43 (3H, m), 1,29-1,20 (1H, m), 0,94 (3H, t, J=7,6 Гц). LCMS (ESI) [M+H]+ 318, Rt=2,71 мин. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 17A и 17B соответственно.
Схема синтеза согласно примеру 7: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-циклопропил-2-(гидроксиметил)-2-фенилпропанoат
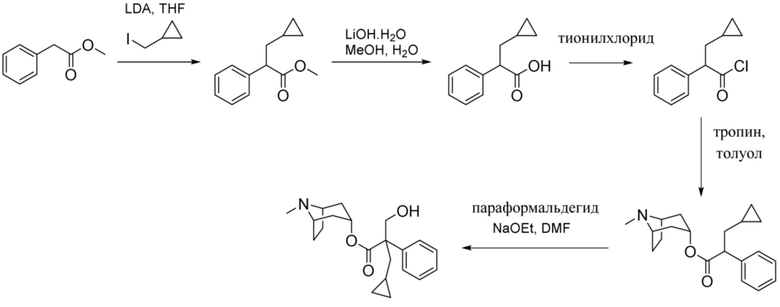

Метил-3-циклопропил-2-фенилпропанoат
В раствор метил-2-фенилацетата (2 г, 13,3 ммоль) в THF (50 мл) при -78°C добавляли LDA (7,3 мл, 2 М, 14,6 ммоль) с такой скоростью, что T < -60°C. Реакционную смесь перемешивали при -78°C в течение 1 ч. Добавляли по каплям (йодметил)циклопропан (5 г, 27,5 ммоль) в THF (10 мл) и реакционную смесь перемешивали при -78°C в течение 30 мин., затем нагревали до к. т. и перемешивали в течение 3 ч. Реакционную смесь разбавляли водой, экстрагировали с помощью EtOAc и объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Остаток очищали на диоксиде кремния (40 г, 0-100% DCM в циклогексане) с получением указанного в заголовке соединения в виде желтого масла (2,6 г, 95%). 1H ЯМР (400 МГц, CDCl3) δ 7,35-7,20 (5H, m), 3,71-3,64 (4H, m), 1,94-1,84 (1H, m), 1,78-1,69 (1H, m), 0,67-0,56 (1H, m), 0,46-0,34 (2H, m), 0,13-0,00 (2H, m).
3-Циклопропил-2-фенилпропановая кислота
В раствор метил-3-циклопропил-2-фенилпропаноата (2,59 г, 12,7 ммоль) в метаноле (100 мл) и воде (15 мл) добавляли моногидрат гидроксида лития (1,33 г, 31,7 ммоль) и реакционную смесь перемешивали при к. т. в течение 16 ч. Реакционную смесь концентрировали in vacuo до ~¼ объема, разбавляли с помощью 1 н. HCl до ~pH 1 и смесь экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде желтого масла (2,44 г, 100%). 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,21 (5H, m), 3,67 (1H, t, J=8,0 Гц), 1,97-1,87 (1H, m), 1,79-1,69 (1H, m), 0,69-0,58 (1H, m), 0,47-0,38 (2H, m), 0,15-0,00 (2H, m).
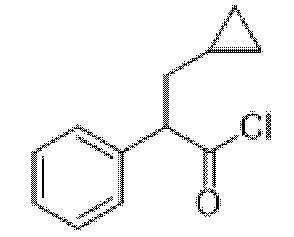
3-Циклопропил-2-фенилпропаноилхлорид
В раствор 3-циклопропил-2-фенилпропановой кислоты (2,44 г, 12,8 ммоль) в DCM (100 мл) добавляли DMF (0,01 мл) и оксалилхлорид (1,7 мл, 19,2 ммоль) и реакционную смесь перемешивали при к. т. в течение 3 ч. Реакционную смесь концентрировали in vacuo с получением указанного в заголовке соединения в виде бледно-желтого масла (2,68 г, 100%), применяемого без очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,41-7,27 (5H, m), 4,10 (1H, t, J=7,3 Гц), 2,06-1,95 (1H, m), 1,86-1,77 (1H, m), 0,70-0,58 (1H, m), 0,52-0,38 (2H, m), 0,18-0,02 (2H, m).
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-циклопропил-2-фенилпропанoат
В раствор тропина (2,68 г, 18,9 ммоль) в толуоле (20 мл) добавляли 3-циклопропил-2-фенилпропаноилхлорид (4,36 г, 20,8 ммоль) и реакционную смесь нагревали с обратным холодильником в течение 1,5 ч. Реакционную смесь экстрагировали с помощью 1 н. HCl и органическую фракцию отбрасывали. Повышали основность водной фракции до ~ pH 12 с помощью 6 н. NaOH и экстрагировали с помощью этилацетата. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде желтого масла (1,76 г, 50%). 1H ЯМР (400 МГц, CDCl3) δ 7,35-7,20 (5H, m), 4,96 (1H, t, J=5,2 Гц), 3,63 (1H, t, J=7,4 Гц), 3,06-2,91 (2H, m), 2,22 (3H, s), 2,14-1,83 (4H, m), 1,82-1,70 (3H, m), 1,68-1,60 (1H, m), 1,52-1,38 (2H, m), 0,68-0,58 (1H, m), 0,47-0,35 (2H, m), 0,14-0,01 (2H, m).
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-циклопропил-2-(гидроксиметил)-2-фенилпропанoат
В суспензию (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-циклопропил-2-фенилпропаноата (1,76 г, 5,62 ммоль) и параформальдегида (0,25 г, 8,42 ммоль) в DMF добавляли этоксид натрия в этаноле (0,07 мл, 4 М, 1,12 ммоль) и реакционную смесь перемешивали при 40°C в течение 3 ч. Добавляли дополнительную порцию этоксида натрия (0,07 мл, 4 М, 1,12 ммоль) и реакционную смесь перемешивали при 40°C в течение 16 ч. Реакционную смесь разбавляли с помощью EtOAc и экстрагировали с помощью 1 н. HCl. Органическую фракцию отбрасывали и повышали основность водной фракции до ~ pH 12 с помощью 6 н. NaOH. Смесь экстрагировали с помощью EtOAc и объединенные органические фракции промывали с помощью солевого раствора, высушивали (MgSO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (40 г, 0-10% (7 н. NH3 в MeOH) в DCM) с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (0,23 г, 12%). 1H ЯМР (400 МГц, CDCl3) δ 7,39-7,22 (5H, m), 5,08 (1H, t, J=5,48 Гц), 4,31-4,19 (2H, m), 3,04-2,92 (2H, m), 2,20 (3H, s), 2,15-2,03 (3H, m), 1,87-1,65 (3H, m), 1,64-1,47 (4H, m), 1,31-1,20 (1H, m), 0,69-0,57 (1H, m), 0,52-0,39 (2H, m), 0,21-0,07 (2H, m). LCMS (ESI) [M+H]+ 344, Rt=2,82 мин. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 18A и 18B соответственно.
Схема синтеза согласно примеру 8: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-фтор-2-метил-2-фенилпропанoат
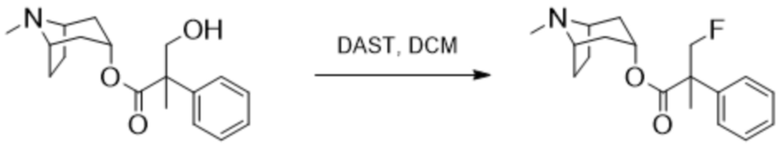
В раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-метил-2-фенилпропаноата (392 мг, 1,29 ммоль) в DCM при 0°C добавляли DAST (0,51 мл, 3,9 ммоль) и реакционную смесь перемешивали при к. т. в течение 48 ч. Реакционную смесь разбавляли с помощью насыщ. водн. раствора Na2CO3 и экстрагировали с помощью DCM. Объединенные органические фракции высушивали (Na2SO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (4 г, 0-12% (2 н. NH3 в MeOH) в DCM). Полученный остаток затем очищали с помощью SFC (YMC Amylose-C, 15% MeOH+0,1% Et2NH) с получением указанного в заголовке соединения в виде бледно-желтого масла (26 мг, 6%). 1H ЯМР (400 МГц, CDCl3) δ 7,39-7,27 (5H, m), 5,06 (1H, t, J=5,4 Гц), 4,97 (1H, dd, J=47,0, 8,7 Гц), 4,55 (1H, dd, J=47,0, 8,7 Гц), 3,05-2,94 (2H, m), 2,21 (3H, s), 2,14-2,01 (2H, m), 1,91-1,73 (2H, m), 1,72 (3H, d, J=2,1 Гц), 1,66-1,49 (4H, m). LCMS (ESI) [M+H]+ 306, Rt=3,10 мин. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 19A и 19B соответственно.
Схема синтеза согласно примеру 9: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-4,4,4-трифтор-2-(гидроксиметил)-2-фенилбутаноат
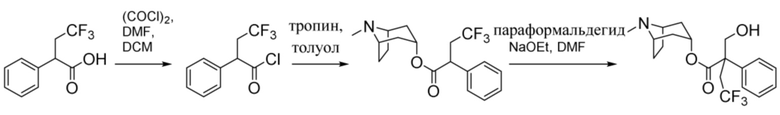
4,4,4-Трифтор-2-фенилбутаноилхлорид
В раствор 4,4,4-трифтор-2-фенилбутановой кислоты (410 мг, 1,9 ммоль) и DMF (0,1 мл) в DCM (25 мл) при 0°C добавляли оксалилхлорид (0,33 мл, 3,8 ммоль) и реакционную смесь перемешивали при 0°C в течение 1 ч. перед перемешиванием при к. т. в течение 16 ч. Реакционную смесь концентрировали in vacuo с получением продукта в виде желтого масла (445 мг, 100%). 1H ЯМР (400 МГц, CDCl3) δ 7,47-7,36 (3H, m), 7,33-7,27 (2H, m), 4,29 (1H, dd, J=10,2, 7,3 Гц), 3,24-3,05 (1H, m), 2,65-2,46 (1H, m).
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-4,4,4-трифтор-2-фенилбутаноат
В раствор тропина (236 мг, 1,7 ммоль) в толуоле (7 мл) добавляли 4,4,4-трифтор-2-фенилбутаноилхлорид (445 мг, 1,9 ммоль) в толуоле (3 мл) и реакционную смесь нагревали с обратным холодильником в течение 3 ч. Реакционную смесь концентрировали in vacuo и остаток растирали с диэтиловым эфиром. Твердое вещество растворяли в 1 н. NaOH и экстрагировали с помощью DCM. Объединенные органические фракции промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде желтого твердого вещества (390 мг, 61%). 1H ЯМР (400 МГц, CDCl3) δ 7,40-7,24 (5H, m), 4,97 (1H, t, J=5,5 Гц), 3,83 (1H, dd, J=8,7, 5,4 Гц), 3,21-3,07 (1H, m), 3,07-3,02 (1H, m), 2,95-2,89 (1H, m), 2,57-2,42 (1H, m), 2,21 (3H, s), 2,14-2,05 (1H, m), 2,05-1,85 (2H, m), 1,81-1,57 (3H, m), 1,46-1,39 (1H, m), 1,30-1,20 (1H, m). LCMS (ESI) [M+H]+ 342, Rt=0,88 мин.
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-4,4,4-трифтор-2-(гидроксиметил)-2-фенилбутаноат
В суспензию (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-4,4,4-трифтор-2-фенилбутаноата (390 мг, 1,1 ммоль) и параформальдегида (51 мг, 1,7 ммоль) в DMF (3 мл) добавляли этоксид натрия в этаноле (0,3 мл, 0,2 М, 0,06 ммоль) и реакционную смесь перемешивали при к. т. в течение 2 ч. Добавляли большее количество параформальдегида (40 мг, 1,3 ммоль) и этоксида натрия в этаноле (0,15 мл, 1,5 М, 0,2 ммоль) и реакционную смесь перемешивали при к. т. в течение 16 ч. Реакционную смесь разбавляли водой, экстрагировали с помощью DCM и объединенные органические фракции промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (12 г, 0-10% (2 н. NH3 в MeOH) в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (105 мг, 24%). 1H ЯМР (400 МГц, CDCl3) δ 7,44-7,23 (5H, m), 5,09 (1H, t, J=5,4 Гц), 4,31 (2H, br s), 3,17-3,03 (2H, m), 3,02-2,93 (2H, m), 2,19 (3H, s), 2,12-2,03 (2H, m), 1,88-1,69 (3H, m), 1,64-1,48 (2H, m), 1,42-1,23 (2H, m). LCMS (ESI) [M+H]+ 372, Rt=2,95 мин. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 20A и 20B соответственно.
Схема синтеза согласно примеру 10: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дигидрокси-2-фенилпропанoат
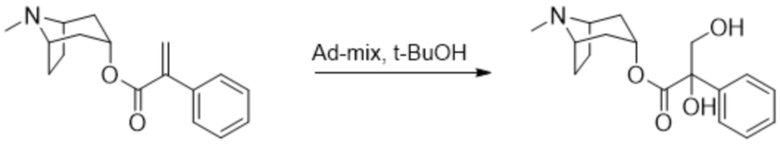
Раствор AD-mix-alpha (1,3 г) в смеси воды (5,0 мл) и трет-бутанола (5 мл) перемешивали при комнатной температуре в течение 15 мин. с получением прозрачного желтого раствора, который охлаждали с помощью ледяной бани до примерно 0°C. Добавляли раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фенилакрилата (250 мг, 0,921 ммоль) в трет-бутаноле (1,0 мл) и полученный раствор перемешивали при 0°C в течение 3 ч., затем обеспечивали его нагревание до к. т. в течение ночи. Реакционную смесь обрабатывали с помощью избытка Na2SO3, затем разделяли между водой и DCM. Водный слой отделяли и дополнительно экстрагировали с помощью DCM и объединенные органические экстракты промывали с помощью солевого раствора, высушивали (Na2SO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (12 г, 0-10% (2 н. NH3 в MeOH) в DCM) с получением энантиомерно обогащенного (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дигидрокси-2-фенилпропанoата в виде смолы (110 мг, 39%).
В отдельной процедуре раствор AD-mix-beta (1,3 г) в смеси воды (5,0 мл) и трет-бутанола (5 мл) перемешивали при комнатной температуре в течение 15 мин. с получением прозрачного желтого раствора, который охлаждали с помощью ледяной бани до примерно 0°C. Добавляли раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-фенилакрилата (250 мг, 0,921 ммоль) в трет-бутаноле (1,0 мл) и полученный раствор перемешивали при 0°C в течение 3 ч., затем обеспечивали его нагревание до к. т. в течение ночи. Реакционную смесь обрабатывали с помощью избытка Na2SO3, затем разделяли между водой и DCM. Водный слой отделяли и дополнительно экстрагировали с помощью DCM и объединенные органические экстракты промывали с помощью солевого раствора, высушивали (Na2SO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (12 г, 0-10% (2 н. NH3 в MeOH) в DCM) с получением энантиомерно обогащенного (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2,3-дигидрокси-2-фенилпропанoата с обогащением противоположными энантиомерами в виде смолы (126 мг, 45%).
Часть продукта реакции AD-mix-alpha (95 мг, 0,311 ммоль) растворяли в ацетонитриле (1 мл) с получением прозрачного раствора. Подобным образом часть продукта реакции AD-mix-beta (95 мг, 0,311 ммоль) отдельно растворяли в ацетонитриле (1,0 мл) также с получением прозрачного раствора. Затем объединяли два раствора, обогащенных противоположными энантиомерами, разбавляли водой (2,0 мл) и подвергали сублимационному высушиванию с получением рацемического указанного в заголовке соединения в виде белого твердого вещества (190 мг). 1H ЯМР (400 МГц, CDCl3) δ 7,60-7,53 (2H, m), 7,42-7,30 (3H, m), 5,06 (1H, t, J=5,5 Гц), 4,32 (1H, d, J=11,5 Гц), 4,16 (1H, br s), 3,82 (1H, d, J=11,5 Гц), 3,13-3,03 (2H, m), 2,41 (1H, br s), 2,26 (3H, s), 2,20-2,07 (2H, m), 2,06-1,88 (3H, m), 1,84-1,76 (1H, m), 1,71-1,1,53 (2H, m). LCMS (ESI) [M+H]+ 306,2, Rt=1,97 мин. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 21A и 21B соответственно.
Схема синтеза согласно примеру 11: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-метокси-2-метил-2-фенилпропанoат
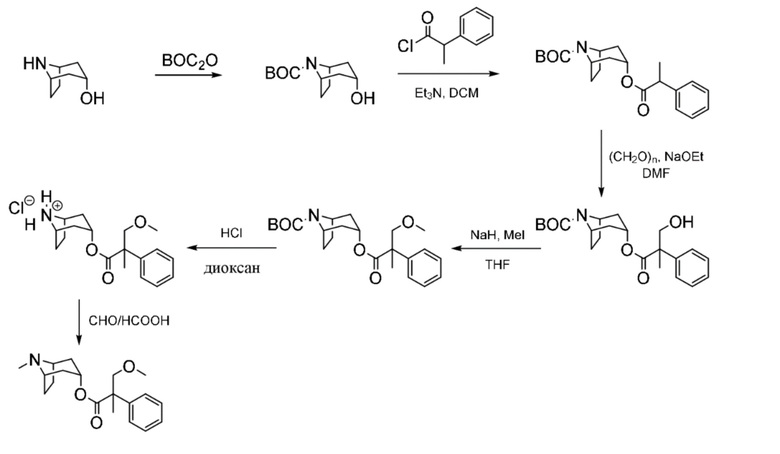
трет-Бутил-(1R,3r,5S)-3-гидрокси-8-азабицикло[3.2.1]октан-8-карбоксилат
Перемешиваемый раствор нортропина (12,72 г, 100 ммоль) в DCM (150 мл) обрабатывали с помощью триэтиламина (27,9 мл, 20,24 г, 200 ммоль) и охлаждали на льду. Затем добавляли твердый ди-трет-бутилдикарбонат (32,74 г, 150,0 ммоль) в течение 10 мин. и сосуд для добавления промывали DCM (50 мл). Наблюдалось интенсивное выделение газа, и полученную смесь перемешивали в течение 2 ч. при нагревании до к. т. и затем при к. т. в течение 16 ч. Полученную смесь разбавляли с помощью DCM (100 мл), промывали 10% раствором лимонной кислоты (водн.), солевым раствором (50 мл) и слои разделены. Объединенные органические фракции высушивали (Na2SO4), фильтровали и концентрировали in vacuo с получением твердого вещества кремового цвета. Твердый остаток растирали с диэтиловым эфиром (30 мл) и подвергали воздействию ультразвука с получением сыпучего твердого вещества, которое собирали с помощью фильтрации, 19,67 г (86%). 1H ЯМР (400 МГц, CDCl3) δ 4,29-4,06 (3H, m), 2,26-1,87 (6H, m), 1,75-1,65 (2H, m), 1,55 (1H, d, J=1,5 Гц), 1,45 (9H, s). LCMS (ESI) [M+H]+ не наблюдается, Rt=1,21 мин.
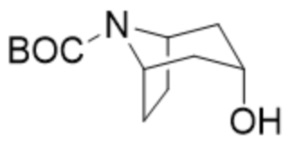
трет-Бутил-(1R,3r,5S)-3-((2-фенилпропаноил)окси)-8-азабицикло[3.2.1]октан-8-карбоксилат
Раствор 2-фенилпропионилхлорида (8,20 г, 48,63 ммоль) в толуоле (50 мл) добавляли к перемешиваемой смеси трет-бутил-(1R,3r,5S)-3-гидрокси-8-азабицикло[3.2.1]октан-8-карбоксилата (10,05 г, 44,21 ммоль) и триэтиламина (12,3 мл, 8,95 г, 88,42 ммоль) в толуоле (50 мл). Полученную суспензию нагревали с обратным холодильником при интенсивном перемешивании в течение 24 ч. с получением продукта в виде грязно-белой суспензии. Реакционную смесь охлаждали до к. т. и разбавляли с помощью EtOAc (100 мл) и H2O (100 мл). Водный слой отделяли и дополнительно экстрагировали с помощью EtOAc (2×100 мл). Объединенные экстракты промывали с помощью 5% раствора лимонной кислоты (водн.), насыщ. NaHCO3 (водн.) и солевого раствора, высушивали (Na2SO4), фильтровали и концентрировали in vacuo. Обеспечивалось получение указанного в заголовке соединения (16,21 г, 102%). 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,22 (5H, m), 5,06 (1H, t, J=5 Гц), 4,24-3,90 (2H, m), 3,68 (1H, q, J=7 Гц), 2,23-1,90 (2H, m), 1,86-1,57 (5H, m), 1,54-1,46 (1H, m), 1,52 (3H, d, J=7 Гц), 1,43 (9H, s), 1,38-1,27 (1H, m). LCMS (ESI) [M+H]+ не наблюдается, Rt=1,98 мин.
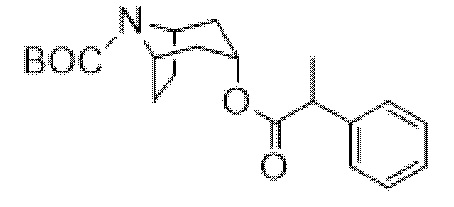
трет-Бутил-(1R,3r,5S)-3-((3-гидрокси-2-метил-2-фенилпропаноил)окси)-8-азабицикло[3.2.1]октан-8-карбоксилат
Перемешиваемую взвесь трет-бутил-(1R,3r,5S)-3-((2-фенилпропаноил)окси)-8-азабицикло[3.2.1]октан-8-карбоксилата (7,19 г, 20,0 ммоль) в DMF (15 мл) обрабатывали параформальдегидом (0,90 г, 30 ммоль), затем 21% раствором этоксида натрия (0,37 мл, 1,00 ммоль) и перемешивали при к. т. в течение 18 ч. Добавляли вторую часть 21% раствора этоксида натрия (0,37 мл, 1,00 ммоль) и продолжали перемешивание в течение дополнительных 2 ч. LCMS показала сдвинутый пик, но не массу иона. Реакционную смесь разбавляли DCM (200 мл), промывали с помощью воды и солевого раствора. Органический слой разделяли, высушивали (Na2SO4), фильтровали и концентрировали in vacuo с получением неочищенного продукта в виде желтого масла. Остаток очищали на диоксиде кремния (120 г, 0-50% EtOAc в циклогексане) с получением указанного в заголовке соединения в виде белого твердого вещества (5,29 г, 68%). 1H ЯМР (400 МГц, CDCl3) δ 7,39-7,24 (5H, m), 5,16 (1H, t, J=5 Гц), 4,14 (1H, dd, J=6, 11 Гц), 4,19-3,89 (2H, m), 3,62 (1H, dd, J=8, 11 Гц), 2,43 (1H, dd, J=6, 8 Гц), 2,29-1,91 (2H, m), 1,84-1,70 (1H, m), 1,70 (3H, s), 1,70-1,46 (4H, m), 1,43 (9H, s), 1,25-1,08 (1H, m). LCMS (ESI) [M+H-Boc]+ 290, Rt=1,60 мин.
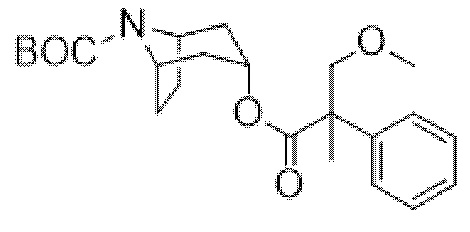
трет-Бутил-(1R,3r,5S)-3-((3-метокси-2-метил-2-фенилпропаноил)окси)-8-азабицикло[3.2.1]октан-8-карбоксилат
Перемешиваемую суспензию гидроксида натрия (0,24 г, 60%, 6,01 ммоль) в сухом THF (10 мл) охлаждали до 0°C и добавляли по каплям раствор трет-бутил-(1R,3r,5S)-3-((3-гидрокси-2-метил-2-фенилпропаноил)окси)-8-азабицикло[3.2.1]-октан-8-карбоксилата (1,95 г, 5,01 ммоль) в сухом THF (10 мл). Полученную смесь перемешивали в течение 1 ч. при 0°C и затем реакционную смесь охлаждали до -6°C и добавляли метилйодид (0,34 мл, 5,5 ммоль). Обеспечивали медленное нагревание смеси до к. т. в течение 3 ч. Реакционную смесь гасили путем добавления водн. NH4Cl (20 мл) и экстрагировали с помощью EtOAc. Объединенные органические экстракты промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo. Остаток очищали на диоксиде кремния (80 г, 0-25% EtOAc в циклогексане) с получением указанного в заголовке соединения в виде бесцветного сиропа, (1,54 г, 76%). 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,21 (5H, m), 5,12 (1H, t, J=5,3 Гц), 4,19-3,94 (2H, m), 3,99 (1H, d, J=8,7 Гц), 3,62 (1H, d, J=8,7 Гц), 3,387 (3H, s), 2,23-1,93 (2H, m), 1,80-1,66 (2H, m), 1,65 (3H, s), 1,62-1,45 (4H, m), 1,43 (9H, s). LCMS (ESI) [M+H-Boc]+ 304, Rt=1,81 мин.
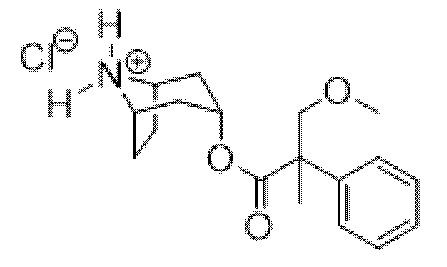
(1R,3r,5S)-8-Азабицикло[3.2.1]октан-3-ил-3-метокси-2-метил-2-фенилпропанoата гидрохлорид
К перемешиваемому раствору трет-бутил-(1R,3r,5S)-3-((3-метокси-2-метил-2-фенилпропаноил)окси)-8-азабицикло[3.2.1]октан-8-карбоксилата (1,10 г, 2,73 ммоль) в диоксане (2,0 мл) добавляли 4 н. раствор HCl в диоксане (2,0 мл) и реакционную смесь перемешивали при к. т. в течение 20 ч. Реакционную смесь концентрировали in vacuo с получением неочищенного продукта в виде бесцветного сиропа (1,18 г, 100%), который применяли без очистки. 1H ЯМР (400 МГц, CDCl3) δ 9,47 (2H, br s), 7,38-7,21 (5H, m), 5,10 (1H, t, J=4,5 Гц), 3,99 (1H, d, J=8,7 Гц), 3,93-3,83 (2H, m), 3,60 (1H, d, J=8,7 Гц), 3,37 (3H, s), 2,59-2,45 (2H, m), 2,04-1,54 (6H, m), 1,64 (3H, s). LCMS (ESI) [M+H]+ 304, Rt=1,52 мин.
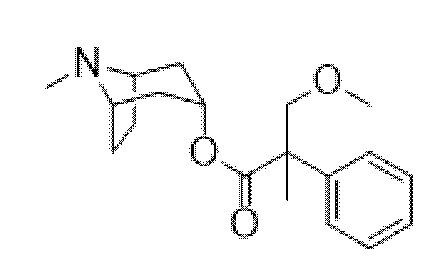
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-метокси-2-метил-2-фенилпропанoат
Неочищенный (1R,3r,5S)-8-азабицикло[3.2.1]октан-3-ил-3-метокси-2-метил-2-фенилпропанoата гидрохлорид (1,18 г, 2,73 ммоль) растворяли в муравьиной кислоте (4,0 мл) и обрабатывали с помощью 37% раствора формальдегида (0,81 мл, 10,9 ммоль) и реакционную смесь нагревали с обратным холодильником в течение 17 ч. Реакционную смесь охлаждали до к. т. и загружали в картридж SCX-2 (50 г), предварительно увлажненный с помощью DCM. Картридж промывали с помощью DCM (400 мл), MeOH (200 мл) и продукт элюировали с помощью 2 М NH3 в MeOH (200 мл). Основной элюент концентрировали in vacuo. И остаток очищали с помощью HPLC на колонке Kinetix Axia C18 RP (длинная) с применением 5-50% CH3CN в H2O (0,1% HCOOH) при 18 мл/мин. с линейным изменением температуры в течение 10 мин., УФ-излучение при 194 нм. Соответствующие фракции объединяли и подвергали сублимационному высушиванию с получением продукта в виде бесцветного сиропа (415 мг, 48%), содержащего 0,75 экв. формиата, 0,25 экв. DCM. 1H ЯМР (400 МГц, CDCl3) δ 7,37-7,24 (5H, m), 5,09 (1H, t, J=5,1 Гц), 3,99 (1H, d, J=8,7 Гц), 3,62 (1H, d, J=8,7 Гц), 3,49-3,42 (2H, m ), 3,38 (3H, s), 2,65-2,50 (2H, m), 2,50 (3H, s), 1,933-1,84 (2H, m), 1,74-1,60 (4H, m), 1,64 (3H, s). LCMS (ESI) [M+H]+ 318,3 Rt=3,05 мин. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 22A и 22B соответственно.
Схема синтеза согласно примеру 12: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропанoат

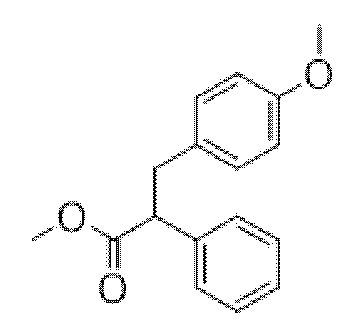
Метил-3-(4-метоксифенил)-2-фенилпропанoат
Добавляли трет-бутоксид калия (2,15 г, 19,16 ммоль) к перемешиваемому раствору метилфенилацетата (2,00 г, 13,32 ммоль) в сухом THF (20 мл) при 0°C. После перемешивания в течение 15 мин. добавляли по каплям раствор 4-метоксибензилбромида в сухом THF (10 мл) с поддержанием T ≤ 5°C. Полученную смесь перемешивали в течение 10 мин. перед тем, как обеспечивали ее нагревание до к. т. в течение ночи. Полученную смесь разбавляли с помощью EtOAc (150 мл), промывали с помощью H2O (50 мл), затем с помощью солевого раствора (50 мл) и слои разделяли. Органический слой высушивали (Na2SO4) и концентрировали in vacuo. Остаток очищали на диоксиде кремния (25 г 0-50% EtOAc в циклогексане с получением указанного в заголовке соединения в виде неочищенного бледно-желтого масла (1,0 г, 27%), применяемого при этом без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,30 (4H, d, J=4,2 Гц), 7,26 (1H, m), 7,03 (2H, m), 6,77 (2H, m), 4,06 (1H, m), 3,76 (6H, s), 3,34 (1H, m), 2,96 (1H, m).
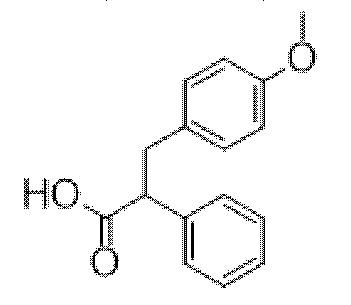
3-(4-Метоксифенил)-2-фенилпропановая кислота
Метил-3-(4-метоксифенил)-2-фенилпропанoат (1,1 г, 4,07 ммоль) в THF (20 мл) и MeOH (5 мл) при к. т. обрабатывали с помощью 2 н. NaOH (водн.) (4 мл, 8,0 ммоль) и перемешивали в течение 18 ч. Реакционную смесь концентрировали in vacuo и остаток разбавляли с помощью H2O (60 мл) и промывали с помощью EtOAc. Водную фракцию подкисляли с помощью 1 н. HCl до pH 1,0 и экстрагировали с помощью EtOAc. Объединенные органические фракции высушивали (MgSO4) и концентрировали in vacuo с получением неочищенного продукта в виде бесцветного масла, которое кристаллизовалось при отстаивании (881 мг, 84%). LCMS Rt=1,28 мин., 255,1 (M-H)-. Применяли без очистки. 1H ЯМР (400 МГц, CDCl3) δ 7,32-7,26 (5H, d, m), 7,03 (2H, m), 6,77 (2H, m), 3,82 (1H, dd, J=6,9, 8,5 Гц), 3,76 (3H, s), 3,35 (1H, dd, J=6,96, 14,7 Гц).
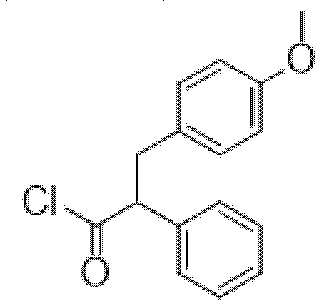
3-(4-Метоксифенил)-2-фенилпропаноилхлорид
Перемешиваемый раствор 3-(4-метоксифенил)-2-фенилпропановой кислоты (450 мг, 1,76 ммоль) в DCM (5,0 мл), содержащий DMF (10 мкл), обрабатывали по каплям при 0°C с помощью оксалилхлорида (200 мкл, 2,29 ммоль). Полученную смесь перемешивали при 0°C в течение 10 мин. и затем обеспечивали ее нагревание до к. т. и перемешивали в течение 18 ч. Реакционную смесь концентрировали in vacuo и неочищенный продукт подвергали азеотропной перегонке с толуолом (2×10 мл) с получением указанного в заголовке неочищенного соединения (483 мг, 100%). Применяли непосредственно без очистки.
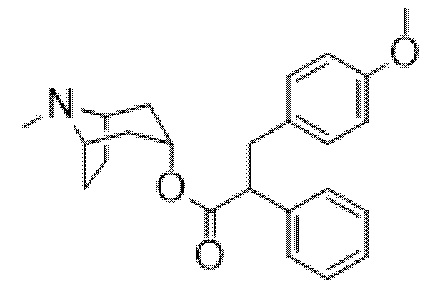
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(4-метоксифенил)-2-фенилпропанoат
Неочищенный 3-(4-метоксифенил)-2-фенилпропаноилхлорид (483 мг, 1,76 ммоль) перемешивали в сухом толуоле (5 мл) с тропином (248 мг, 1,76 ммоль) при 100°C в течение 3 ч. Реакционную смесь концентрировали in vacuo. Остаток обрабатывали с помощью DCM (30 мл) и H2O (20 мл) и повышали основность до pH 10 с помощью 1 н. NaOH (водн.). Органический слой разделяли, промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (12 г 0-10% MeOH в DCM) до получения указанного в заголовке соединения в виде светло-коричневого масла (277 мг, 41%). ¹H ЯМР (400 МГц, CDCl3) d 7,31-7,26 (m, 5H), 7,07-7,04 (m, 2H), 6,80-6,75 (m, 2H), 4,91 (dd, J=5,2, 5,2 Гц, 1H), 3,77-3,76 (m, 3H), 3,37 (dd, J=9,0, 13,8 Гц, 1H), 3,06-2,94 (m, 3H), 2,25 (s, 3H), 2,16-2,10 (m, 2H), 1,84-1,72 (m, 2H), 1,60-1,48 (m, 4H), 1,42-1,35 (m, 1H). LCMS 0,99 мин., 380,1 (M+H)+.
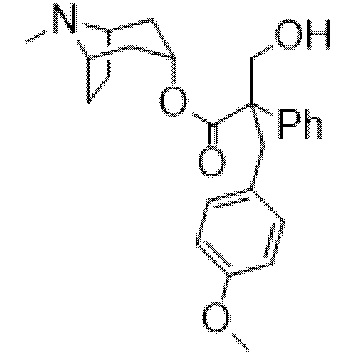
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропанoат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(4-метоксифенил)-2-фенилпропанoат (200 мг, 0,53 ммоль) в DMF (3 мл) обрабатывали параформальдегидом (160 мг, 5,33 ммоль), затем 21% раствором этоксида натрия (4 мкл, 0,05 ммоль) и реакционную смесь перемешивали при 50°C в течение 24 ч. Реакционную смесь разбавляли с помощью DCM (15 мл) и фильтровали через целит. Фильтрат концентрировали in vacuo для уменьшения объема и очищали на диоксиде кремния (12 г, 0-10% MeOH в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (187 мг, 86%) . ¹H ЯМР (400 МГц, CDCl3) 7,37-7,33 (2H, m), 7,26 (3H, s), 6,96-6,90 (2H, m), 6,76-6,71 (2H, m), 5,09 (1H, dd, J=5,3, 5,3 Гц), 4,08-3,96 (2H, m), 3,76-3,76 (3H, m), 3,49-3,34 (2H, m), 3,02-2,92 (2H, m), 2,20 (6H, s), 1,76-1,58 (3H, m), 1,56-1,44 (2H, m), 1,28-1,20 (1H, m). LCMS 3,30 мин., 410,0 (M+H)+. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 23A и 23B соответственно.
Схема синтеза согласно примеру 13: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-хлорбензил)-2-фенилпропаноат
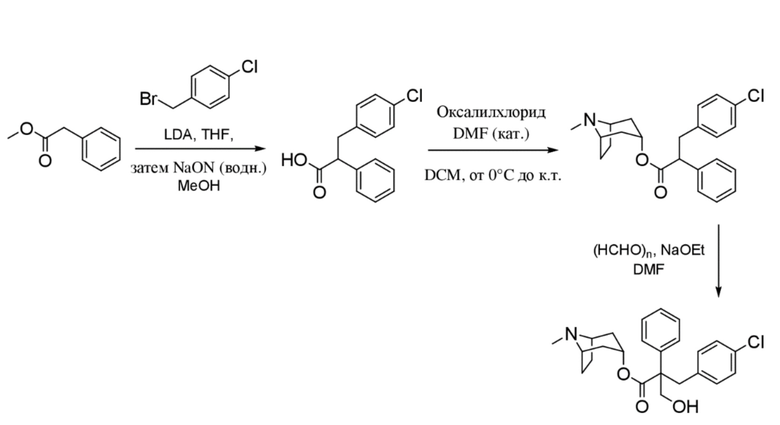

3-(4-Хлорфенил)-2-фенилпропановая кислота
Добавляли по каплям н-бутиллитий (2,5 М раствор в гексанах, 15,3 мл, 38,3 ммоль) в атмосфере азота к охлажденному раствору N, N-диизопропиламина (5,6 мл, 40,0 ммоль) в сухом THF (20 мл) с поддержанием T ≤ 0°C. Полученный раствор LDA перемешивали в течение 20 мин., затем охлаждали с помощью бани с сухим льдом/ацетоном до -78°C. Добавляли по каплям с помощью шприца раствор метилфенилацетата (5,0 г, 33,3 ммоль) в сухом THF (20 мл) с поддержанием T ≤ -55°C. После перемешивания в течение 2 ч. добавляли раствор 4-хлорбензилбромида (4,8 мл, 36,6 ммоль) в сухом THF (10 мл) и полученную смесь перемешивали в течение ночи с нагреванием до к. т. Смесь разбавляли с помощью EtOAc (100 мл) и воды (50 мл) и водный слой разделяли и дополнительно экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo, и остаток растворяли в смеси метанола (20 мл) и THF (100 мл), и добавляли водный раствор гидроксида натрия (2 М, 35 мл, 70 ммоль). Полученный непрозрачный раствор перемешивали в течение 22 ч., затем концентрировали in vacuo. Остаток разделяли между хлористоводородной кислотой (1 н., 50 мл) и EtOAc (100 мл). Водный слой отделяли и дополнительно экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (8,62 г, 99%). 1H ЯМР (400 МГц, CDCl3): δ 7,35-7,24 (5H, m), 7,21-7,15 (2H, m), 7,04-6,98 (2H, m), 3,80 (1H, dd, J=7,5, 8 Гц), 3,36 (1H, dd, J=8, 13,9 Гц), 3,0 (1H, dd, J=7, 13,9 Гц).

(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(4-хлорфенил)-2-фенилпропанoат
Перемешиваемый раствор 3-(4-хлорфенил)-2-фенилпропановой кислоты (3,30 г, 12,7 ммоль) в DCM (30 мл), содержащий DMF (45 мкл), обрабатывали по каплям при 0°C с помощью оксалилхлорида (2,2 мл, 25,3 ммоль) и полученную смесь перемешивали при 0°C в течение 10 мин., затем обеспечивали ее нагревание до к. т. и перемешивали в течение 18 ч. Реакционную смесь концентрировали in vacuo и неочищенный продукт подвергали азеотропной перегонке с толуолом (20 мл) с получением неочищенного 3-(4-хлорфенил)-2-фенилпропаноилхлорида, который растворяли в сухом толуоле (50 мл). Добавляли тропин (1,62 г, 11,51 ммоль) и триэтиламин (4,8 мл, 34,52 ммоль) и смесь перемешивали при 100°C в течение 4 ч, охлаждали до к. т. и перемешивали в течение ночи. Реакционную смесь разбавляли с помощью EtOAc и промывали с помощью насыщенного NaHCO3, воды, солевого раствора, высушивали (Na2SO4) и концентрировали in vacuo. Остаток очищали с помощью хроматографии на диоксиде кремния (80 г, 0-10% (2 М NH3 в MeOH) в градиенте DCM) с получением чистого указанного в заголовке соединения в виде вязкого оранжевого масла (2,24 г, 50%). Вторую порцию неочищенного указанного в заголовке соединения (0,54 г, 12%) также выделяли в виде коричневого масла. ¹H ЯМР (400 МГц, CDCl3): δ 7,35-7,23 (5H, m), 7,22-7,16 (2H, m), 7,09-7,02 (2H, m), 4,90 (1H, t, J=5,4 Гц), 3,73 (1H, dd, J=6,9, 8,7 Гц), 3,39 (1H, dd, J=8,7, 13,8 Гц), 3,00 (1H, dd, J=6,9, 13,8 Гц), 2,99-2,94 (1H, m), 2,96-2,88 (1H, m), 2,18 (3H, s), 2,07-1,94 (2H, m), 1,86-1,64 (2H, m), 1,56-1,41 (3H, m), 1,34-1,25 (1H, m); LCMS (ESI) [M+H]+ 384,1.
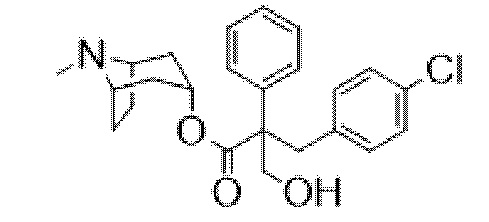
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-хлорбензил)-2-фенилпропаноат
Раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-(4-хлорфенил)-2-фенилпропанoата (2,22 г, 5,78 ммоль) в DMF (10 мл) обрабатывали с помощью параформальдегида (0,26 г, 8,7 ммоль). Добавляли 21% раствор этоксида натрия в этаноле (0,29 мл, 0,6 ммоль) и полученную смесь перемешивали при к. т. в течение 14 ч. Реакционную смесь разбавляли водой (50 мл) и EtOAc (100 мл) и водный слой отделяли и дополнительно экстрагировали с помощью EtOAc. Объединенные органические слои промывали с помощью 5% вес/вес водного раствора хлорида лития (2×25 мл), солевого раствора, высушивали (Na2SO4) и фильтровали через слой целита. Раствор концентрировали in vacuo и остаток очищали на диоксиде кремния (40 г, 0,5-10% (2 М NH3 в MeOH) в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (0,86 г, 36%). ¹H ЯМР (400 МГц, d6-DMSO): δ 7,35-7,27 (2H, m), 7,27-7,21 (1H, m), 7,20-7,14 (2H, m), 7,09-7,01 (2H, m), 6,88-6,79 (2H, m), 5,14 (1H, широкий t, J=4 Гц), 4,90 (1H, t, J=4,8 Гц), 3,98 (1H, dd, J=4,2, 10 Гц), 3,74 (1H, dd, J=3,8, 10 Гц), 3,36 (1H, d, J=13 Гц), 3,23 (1H, d, J=13 Гц), 2,90-2,84 (1H, m), 3,84-2,78 (1H, m), 2,06 (3H, s), 1,99-1,87 (2H, m), 1,72-1,27 (5H, m), 1,13-1,03 (1H, m); LCMS (ESI) [M+H]+ 414,2. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 24A и 24B соответственно.
Схема синтеза согласно примеру 14: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(2-хлорбензил)-2-фенилпропаноат
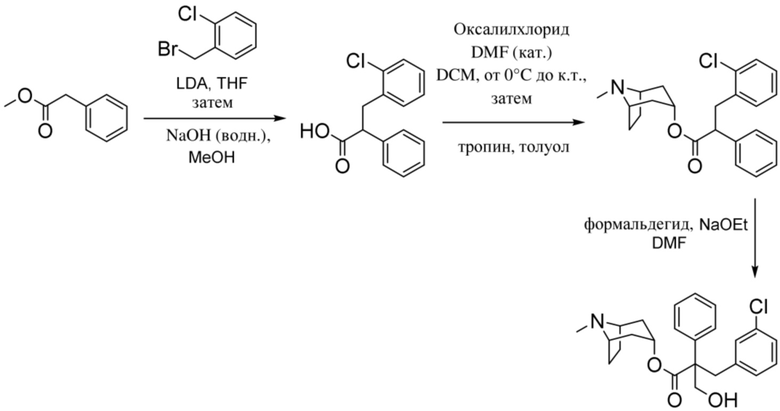
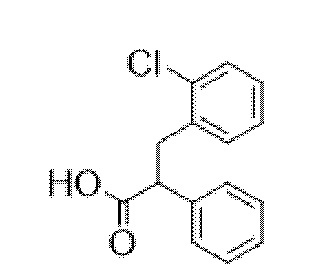
3-(2-Хлорфенил)-2-фенилпропановая кислота
Добавляли по каплям н-бутиллитий (2,5 М раствор в гексанах, 15,3 мл, 38,3 ммоль) в атмосфере азота к охлажденному раствору N, N-диизопропиламина (5,6 мл, 39,95 ммоль) в сухом THF (20 мл) с поддержанием T ≤ 0°C. Полученный раствор LDA перемешивали в течение 20 мин., затем охлаждали с помощью бани с сухим льдом/ацетоном до -74°C. Добавляли по каплям с помощью шприца раствор метилфенилацетата (5,0 г, 33,3 ммоль) в сухом THF (20 мл) с поддержанием T ≤ -55°C. После перемешивания в течение 1 ч. добавляли раствор 2-хлорбензилбромида (4,8 мл, 36,6 ммоль) в сухом THF (10 мл) и полученную смесь перемешивали в течение ночи с нагреванием до к. т. Смесь разбавляли с помощью EtOAc (50 мл) и насыщенного водного раствора хлорида аммония (10 мл). Органический слой отделяли и промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo с получением неочищенного метил-3-(2-хлорфенил)-2-фенилпропанoата в виде оранжевого сиропа. Данное вещество растворяли в смеси метанола (60 мл) и воды (20 мл) и добавляли гидроксид лития (0,80 г, 33,29 ммоль). Полученный непрозрачный раствор перемешивали в течение 40 ч., затем концентрировали in vacuo с получением оранжевого водного раствора, который разбавляли с помощью хлористоводородной кислоты (1 М, 20 мл), воды (30 мл) и EtOAc (100 мл). Водный слой отделяли и дополнительно экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде смеси с непрореагировавшим метил-3-(2-хлорфенил)-2-фенилпропанoатом. Смесь повторно растворяли в THF (50 мл), метаноле (10 мл) и водном растворе гидроксида натрия (2 М, 17 мл, 34 ммоль) и полученный непрозрачный раствор перемешивали при к. т. в течение ночи. Растворители концентрировали in vacuo и остаток разделяли между водой (50 мл), хлористоводородной кислотой (1 М, 50 мл) и EtOAc (100 мл). Водный слой отделяли и дополнительно экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (4,10 г, 94%). 1H ЯМР (400 МГц, CDCl3): δ 7,35-7,24 (6H, m), 7,16-7,08 (1H, m), 7,07-7,03 (2H, m), 4,00 (1H, dd, J=6,7, 8,5 Гц), 3,49 (1H, dd, J=8,5, 13,8 Гц), 3,15 (1H, dd, J=6,7, 13,8 Гц).
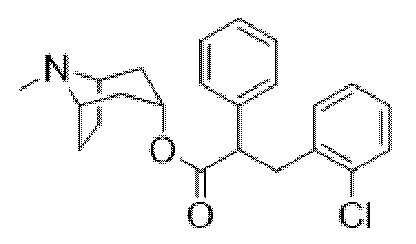
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(2-хлорфенил)-2-фенилпропанoат
Перемешиваемый раствор 3-(2-хлорфенил)-2-фенилпропановой кислоты (3,30 г, 12,7 ммоль) в DCM (30 мл), содержащий DMF (45 мкл), обрабатывали с помощью оксалилхлорида (2,2 мл, 25,3 ммоль) и полученную смесь перемешивали при к. т. в течение ночи. Реакционную смесь концентрировали in vacuo и неочищенный продукт подвергали азеотропной перегонке с помощью толуола (20 мл). Остаток растворяли в сухом толуоле (50 мл) и добавляли тропин (1,62 г, 11,5 ммоль) и триэтиламин (4,8 мл, 34,5 ммоль) и смесь перемешивали при 110°C в течение 4 ч., охлаждали до к. т. и перемешивали в течение ночи. Реакционную смесь разбавляли с помощью EtOAc (150 мл) и промывали с помощью насыщенного NaHCO3, воды, солевого раствора, высушивали (Na2SO4) и концентрировали in vacuo. Остаток очищали на диоксиде кремния (120 г, 0-10% (2 М NH3 в MeOH) в DCM) с получением чистого указанного в заголовке соединения в виде вязкого оранжевого масла (3,26 г, 73%). ¹H ЯМР (400 МГц, CDCl3) δ 7,37-7,22 (6H, m), 7,16-7,04 (3H, m), 4,91 (1H, t, J=5,4 Гц), 3,95 (1H, dd, J=6,5, 9 Гц), 3,51 (1H, dd, J=9, 13,5 Гц), 3,16 (1H, dd, J=6,5, 13,5 Гц), 2,97-2,88 (2H, m), 2,18 (3H, s), 2,05-1,94 (2H, m), 1,86-1,63 (2H, m), 1,55-1,40 (3H, m), 1,35-1,26 (1H, m); LCMS (ESI) [M+H]+ 384,1.
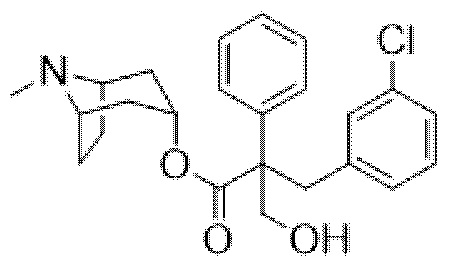
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(2-хлорбензил)-2-фенилпропаноат
Раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-(2-хлорфенил)-2-фенилпропанoата (1,92 г, 5,0 ммоль) в DMF (10 мл) обрабатывали с помощью параформальдегида (0,23 г, 7,5 ммоль). Добавляли 21% раствор этоксида натрия в этаноле (0,25 мл, 0,5 ммоль) и полученную смесь перемешивали при к. т. в течение 3 ч., затем нагревали до 45°C в течение ночи. Добавляли вторые части параформальдегида (0,23 г, 7,5 ммоль) и 21% этоксида натрия в этаноле (0,25 мл, 0,5 ммоль) и смесь перемешивали при 45°C в течение 4,5 ч., затем при 80°C в течение 1,5 ч. Нагревание останавливали и реакционную смесь перемешивали при к. т. в течение ночи. Смесь разбавляли с водой (50 мл) и EtOAc (100 мл), водный слой отделяли и дополнительно экстрагировали с помощью EtOAc. Объединенные органические слои промывали с помощью 5% вес/вес водного раствора хлорида лития (2×25 мл), солевого раствора, высушивали (Na2SO4), фильтровали через целит и концентрировали in vacuo. Остаток очищали на диоксиде кремния [40 г, 0,5-10% (2 М NH3 в MeOH) в DCM] с получением указанного в заголовке соединения в виде белого твердого вещества (1,0 г, 48%). ¹H ЯМР (400 МГц, d6-DMSO): δ 7,26-7,18 (4H, m), 7,17-7,10 (3H, m), 6,99-6,91 (2H, m), 5,21 (1H, широкий t, J=4 Гц), 4,99 (1H, t, J=5,2 Гц), 4,06-3,94 (2H, m), 3,51 (1H, d, J=14 Гц), 3,46 (1H, d, J=14 Гц), 2,93-2,87 (1H, m), 2,86-2,79 (1H, m), 2,07 (3H, s), 2,03-1,89 (2H, m), 1,76-1,62 (1H, m), 1,62-1,49 (3H, m), 1,42-1,33 (1H, m), 1,22-1,10 (1H, m); LCMS (ESI) [M+H]+ 414,2. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 25A и 25B соответственно.
Схема синтеза согласно примеру 15: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-гидроксибензил)-2-фенилпропаноатная соль муравьиной кислоты

3-(4-((трет-Бутилдиметилсилил)окси)фенил)-2-фенилпропановая кислота
трет-Бутилдиметилхлорсилан (3,16 г, 21,0 ммоль) добавляли к перемешиваемому раствору 3-(4-гидроксифенил)-2-фенилпропановой кислоты (2,42 г, 10,0 ммоль) и имидазола (2,04 г, 29,97 ммоль) в сухом DMF (40 мл) и полученную смесь перемешивали при к. т. в течение ночи. Добавляли дополнительную часть трет-бутилдиметилхлорсилана (0,75 г, 5,0 ммоль) и перемешивание продолжали в течение 2 ч. Реакционную смесь разбавляли с помощью EtOAc и промывали водой, насыщенным водным раствором хлоридом лития, солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo с получением масла соломенного цвета. Масло растворяли в метаноле (40 мл), добавляли карбонат калия (1,52 г, 11,0 ммоль) и полученную суспензию перемешивали при к. т. в течение 1,5 ч. Реакционную смесь концентрировали in vacuo и остаток разделяли между водой и DCM и водную фазу подкисляли до pH 3 посредством осторожного добавления 1 М хлористоводородной кислоты. Водный слой отделяли и дополнительно экстрагировали с помощью DCM. Объединенный органический слой промывали солевым раствором, высушивали (Na2SO4) и концентрировали in vacuo с получением неочищенного указанного в заголовке соединения в виде масла соломенного цвета. Данное вещество очищали с помощью хроматографии на диоксиде кремния (80 г, 0-20% EtOAc в градиенте DCM) с получением указанного в заголовке соединения в виде масла соломенного цвета (1,58 г, 44%). 1H ЯМР (400 МГц, CDCl3): δ 7,32-7,22 (5H, m), 6,95-6,89 (2H, m), 6,71-6,64 (2H, m), 3,79 (1H, видимый t, J=8 Гц), 3,32 (1H, dd, J=8,2, 13,9 Гц), 2,95 (1H, dd, J=7,2, 13,9 Гц), 0,95 (9H, s), 0,15 (6H, s).
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(4-((трет-бутилдиметилсилил)окси)фенил)-2-фенилпропанoат
Перемешиваемый раствор 3-(4-((трет-бутилдиметилсилил)окси)фенил)-2-фенилпропановой кислоты (1,30 г, 3,65 ммоль) в DCM (20 мл), содержащий одну каплю DMF (45 мкл), обрабатывали по каплям при 0°C с помощью оксалилхлорида (0,64 мл, 7,3 ммоль) и полученную смесь перемешивали при 0°C в течение 10 мин., затем убирали охлаждающую баню и реакционную смесь перемешивали в течение ночи с нагреванием до к. т. Смесь концентрировали in vacuo и неочищенное промежуточное соединение в виде хлорангидрида подвергали азеотропной перегонке с толуолом (20 мл) с получением 3-(4-((трет-бутилдиметилсилил)окси)фенил)-2-фенилпропаноилхлорида в виде масла соломенного цвета. Данное вещество растворяли в сухом толуоле (20 мл) и тропине (0,51 г, 3,7 ммоль) и добавляли триэтиламин (1,5 мл, 11,0 ммоль). Полученную смесь перемешивали при 110°C в течение 3 ч. Смесь охлаждали до к. т., разбавляли с помощью EtOAc (50 мл) и промывали с помощью воды, насыщенного NaHCO3, солевого раствора, высушивали (Na2SO4) и концентрировали in vacuo. Продукт очищали на диоксиде кремния (25 г, 0-10% (2 М NH3 в MeOH) в DCM) с получением чистого указанного в заголовке соединения в виде бесцветного сиропа (0,778 г, 44%). 1H ЯМР (400 МГц, CDCl3): δ 7,34-7,21 (5H, m), 7,02-6,94 (2H, m), 6,73-6,66 (2H, m), 4,90 (1H, t, J=5,3 Гц), 3,73 (1H, dd, J=6,6, 9 Гц), 3,35 (1H, dd, J=9, 13,8 Гц), 2,95 (1H, dd, J=6,6, 13,8 Гц), 2,98-2,89 (2H, m), 2,19 (3H, s), 2,05-1,94 (2H, m), 1,87-1,68 (2H, m), 1,60-1,51 (2H, m), 1,50-1,35 (3H, m), 0,96 (9H, s), 0,16 (6H, s).
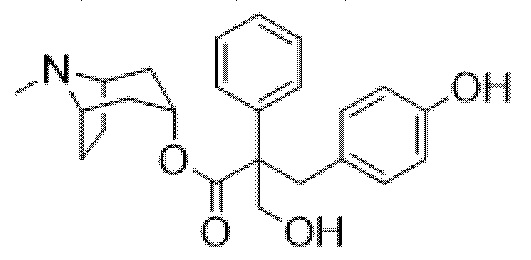
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-гидроксибензил)-2-фенилпропаноатная соль муравьиной кислоты
Раствор (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-(4-((трет-бутилдиметилсилил)окси)-фенил)-2-фенилпропанoата (0,77 г, 1,61 ммоль) в DMF (10 мл) обрабатывали с помощью параформальдегида (0,072 г, 2,4 ммоль) и добавляли 21% раствор этоксида натрия в этаноле (0,06 мл, 0,16 ммоль) при к. т. Полученную смесь перемешивали при к. т. в течение 6,5 ч., затем добавляли вторые части параформальдегида (0,072 г, 2,4 ммоль) и 21% раствор этоксида натрия в этаноле (0,06 мл, 0,15 ммоль) и смесь перемешивали в течение ночи. Добавляли третью часть параформальдегида (0,072 г, 2,4 ммоль) и 21% раствор этоксида натрия в этаноле (0,06 мл, 0,15 ммоль) и продолжали перемешивание при к. т. в течение 3 дней. Реакционную смесь разбавляли с помощью EtOAc (100 мл) и промывали с помощью воды, 5% водного раствора хлорида лития, солевого раствора, высушивали (Na2SO4) и концентрировали in vacuo. Продукт частично очищали на 15 мкм диоксиде кремния (4 г, 0-20% (2 М NH3 в MeOH) в DCM) с получением неочищенного указанного в заголовке соединения в виде белого твердого вещества, которое дополнительно очищали с помощью препаративной HPLC с обращенной фазой с применением колонки Kinetix Axia® C18 21,2×250 мм, 18 мл/мин. с градиентным элюентом 10-60% MeCN в воде с 0,1% HCOOH с монохроматическим УФ-обнаружением при 220 нм. Обеспечивалось получение указанного в заголовке соединения в виде белого твердого вещества (0,273 г, 38%). ¹H ЯМР (400 МГц, d6-DMSO): δ 8,31 (1H, s), 7,34-7,26 (2H, m), 7,26-7,19 (1H, m), 7,10-7,02 (2H, m), 6,63 (2H, dm, J=8,5 Гц), 6,49 (2H, dm, J=8,5 Гц), 4,91 (1H, t, J=4,9 Гц), 4,48-3,60 (3H, bs), 3,95 (1H, d, J=10 Гц), 3,77 (1H, d, J=10 Гц), 3,26 (1H, d, J=13,3 Гц), 3,25-3,16 (2H, m), 3,13 (1H, d, J=13,3 Гц), 2,28 (3H, s), 2,19-2,09 (2H, m), 1,80-1,41 (5H, m), 1,26-1,17 (1H, m); LCMS (ESI) [MH]+ 396,4. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 26A и 26B соответственно.
Схема синтеза согласно примеру 16: 2-фтор-3-гидрокси-N-((1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил)-2-фенилпропанамид
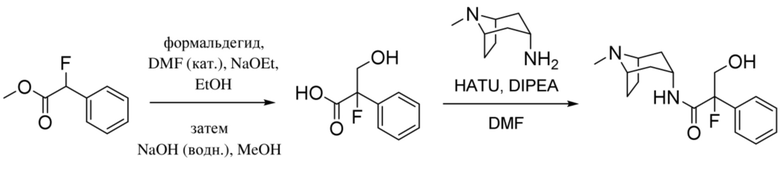
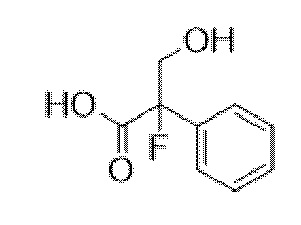
2-Фтор-3-гидрокси-2-фенилпропановая кислота
Перемешиваемую смесь метил-2-фтор-2-фенилпропанoата (1,49 г, 7,62 ммоль) и параформальдегида (0,55 г, 18,3 ммоль) в DMF (5 мл) обрабатывали с помощью 21% раствора этоксида натрия в этаноле (0,43 мл, 1,14 ммоль) и полученную суспензию оранжевого цвета перемешивали при к. т. в течение 3,5 ч. Смесь разбавляли с помощью EtOAc и промывали с помощью хлористоводородной кислоты, 5% водного раствора хлорида лития, солевого раствора, высушивали (Na2SO4) и концентрировали in vacuo с получением неочищенного метил-2-фтор-3-гидрокси-2-фенилпропанoата в виде оранжевого масла (1,82 г). Данное вещество растворяли в смеси воды (9 мл) и метанола (20 мл), затем добавляли гидроксид лития (0,44 г, 18,3 ммоль) и полученную смесь перемешивали при к. т. в течение 4 ч. Добавляли раствор хлороводорода в 1,4-диоксане (4 н., 5 мл, 20 ммоль) и смесь концентрировали in vacuo с получением указанного в заголовке неочищенного соединения в виде темно-кремового твердого вещества (2,21 г, количественно). Данный материал применяли без очистки. 1H ЯМР (400 МГц, d6-DMSO): δ 7,58-7,29 (5H, m), 4,11 (1H, dd, J=12,3, 30,7 Гц), 3,79 (1H, dd, J =12,3, 17,6 Гц), 3,10-3,09 (2H+вода, широкий s).
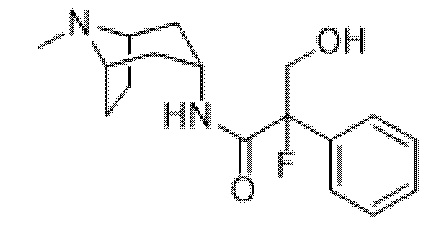
2-Фтор-3-гидрокси-N-((1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил)-2-фенилпропанамид
Тремя частями добавляли HATU (3,77 г, 9,9 ммоль) к перемешиваемому ледяному раствору неочищенной 2-фтор-3-гидрокси-2-фенилпропановой кислоты (2,21 г, 7,6 ммоль), 8-метил-8-азабицикло[3.2.1]октан-3-амина (1,18 г, 8,4 ммоль) и DIPEA (4,0 мл, 22,9 ммоль) в DMF (20 мл). Полученную оранжевую смесь нагревали до к. т. и перемешивали в течение 40 ч. Реакционную смесь концентрировали in vacuo и остаток разделяли между водой и EtOAc. Водный слой отделяли и дополнительно экстрагировали с помощью EtOAc. Объединенные органические слои промывали с помощью 5% водного раствора хлорида лития, солевого раствора, высушивали (Na2SO4) и концентрировали in vacuo с получением оранжевой смолы. Продукт очищали на диоксиде кремния (40 г, диоксид кремния с размерами пор 15 мкм, 5-20% (2 М NH3 в MeOH) в DCM) с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (0,631 г, 27%). 1H ЯМР (400 МГц, d6-DMSO): δ 7,84 (1H, bs), 7,63-7,47 (2H, m), 7,44-7,33 (3H, m), 5,45 (1H, t, J=5,8 Гц), 4,12 (1H, ddd, J=6,3, 12,3, 33 Гц), 3,80-3,67 (2H, m), 3,61-3,47 (2H, m), 2,49 (3H, bs), 2,25-1,86 (8H, m); LCMS (ESI) [M+H]+ 307,2. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 37A и 37B соответственно.
Схема синтеза согласно примеру 17: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-фторбензил)-3-гидрокси-2-фенилпропаноат
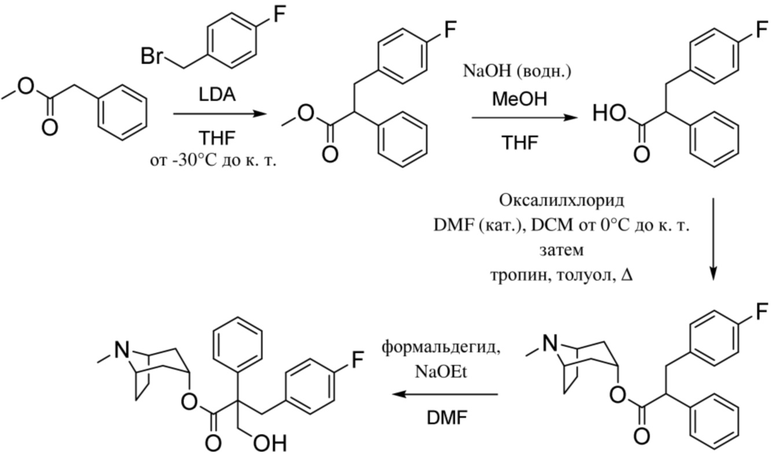

Метил-3-(4-фторфенил)-2-фенилпропанoат
2 М диизопропиламин лития в THF/гептан/этилбензол (10 мл, 20 ммоль) добавляли к перемешиваемому раствору метилфенилацетата (3,00 г, 20,0 ммоль) в сухом THF (40 мл) в атмосфере аргона при -30°C. После перемешивания в течение 15 мин. обеспечивали нагревание реакционной смеси до 0°C и перемешивали в течение 30 мин. Реакционную смесь охлаждали до -30°C и добавляли по каплям раствор 4-фторбензилбромида (4,10 г, 21,7 ммоль) в сухом THF (10 мл) с поддержанием T ≤ -25°C. Полученную смесь перемешивали в течение 10 мин. перед тем, как обеспечивали ее нагревание до к. т. в течение 16 ч. Растворитель удаляли in vacuo и остаток разбавляли с помощью EtOAc, промывали с помощью воды и солевого раствора и слои разделяли. Органическую фракцию высушивали (Na2SO4) и концентрировали in vacuo. Остаток очищали на диоксиде кремния (40 г, 0-10% MTBE в циклогексане) с получением указанного в заголовке соединения в виде неочищенного бледно-желтого масла (1,51 г, 29%), применяемого при этом без дополнительной очистки. ¹H ЯМР (400 МГц, CDCl3): δ 7,33-7,26 (5H, m), 7,08-7,03 (2H, m), 6,93-6,88 (2H, m), 3,81-3,77 (1H, m), 3,37 (1H, dd, J=8,7, 13,8 Гц), 2,99 (1H, ddd, J=4,1, 6,7, 13,5 Гц).
3-(4-Фторфенил)-2-фенилпропановая кислота
Метил-3-(4-фторфенил)-2-фенилпропанoат (1,51 г, 5,9 ммоль) перемешивали в THF (30 мл) и MeOH (3 мл) и обрабатывали с помощью 2 М NaOH (водн.) (12 мл, 24,0 ммоль). Реакционную смесь перемешивали при к. т. в течение 18 ч. Растворитель удаляли in vacuo, остаток разбавляли с помощью H2O и промывали с помощью EtOAc. Водную фракцию подкисляли с помощью 1 М HCl до pH 1,0 и экстрагировали с помощью EtOAc. Объединенные органические экстракты высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде светло-коричневого масла, которое кристаллизовалось при отстаивании (1,13 г, 79%). Материал применяли без очистки. ¹H ЯМР (400 МГц, CDCl3): δ 7,34-7,27 (5H, m), 7,06 (2H, ddd, J=3,1, 5,3, 11.8 Гц), 6,93-6,88 (2H, m), 3,81 (1H, dd, J=7,1, 8,4 Гц), 3,37 (1H, dd, J=8,4, 13,9 Гц), 3,01 (1H, dd, J=7,1, 13,9 Гц); LCMS (ESI) [M-H]- 243.
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(4-фторфенил)-2-фенилпропанoат
Перемешиваемый раствор 3-(4-фторфенил)-2-фенилпропановой кислоты (1,13 г, 4,6 ммоль) в DCM (10 мл), содержащий DMF (200 мкл), в атмосфере аргона обрабатывали по каплям при 0°C с помощью оксалилхлорида (573 мкл, 6,6 ммоль). Полученную смесь перемешивали при 0°C в течение 10 мин., затем обеспечивали ее нагревание до к. т. и перемешивали в течение 18 ч. Растворители удаляли in vacuo и остаток подвергали азеотропной перегонке с толуолом (2×15 мл) с получением неочищенного хлорангидрида, который перемешивали в сухом толуоле (10 мл) с (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-олом (790 мг, 5,6 ммоль) в атмосфере аргона при 100°C в течение 4 ч. Реакционную смесь концентрировали in vacuo и остаток обрабатывали с помощью DCM (60 мл) и H2O (40 мл) и повышали основность до pH 10 с помощью 1 М NaOH (водн.). Органические фракции отделяли, промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением неочищенного продукта в виде бледно-желтого масла (1,27 г). Данное вещество применяли неочищенным без очистки. ¹H ЯМР (400 МГц, CDCl3): δ 7,26 (4H, s), 7,19-7,07 (3H, m), 6,94-6,89 (2H, m), 4,99-4,89 (1H, m), 3,73 (1H, dd, J=6,8, 8,8 Гц), 3,40 (1H, dd, J=8,8, 13,8 Гц), 3,03-2,88 (3H, m), 2,36-2,32 (2H, m), 2,25-2,20 (3H, m), 2,09-1,43 (5H, m), 1,36-1,25 (1H, m); LCMS (ESI) [M+H]+ 368,1.
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-фторбензил)-3-гидрокси-2-фенилпропаноат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(4-фторфенил)-2-фенилпропанoат (154 мг, 0,4 ммоль) в DMF (3 мл) обрабатывали параформальдегидом (160 мг, 5,3 ммоль), затем 21% раствором этоксида натрия (4 мкл, 0,05 ммоль) и перемешивали при 50 ± 5°C в течение 24 ч. Реакционную смесь охлаждали, разбавляли с помощью DCM (15 мл) и фильтровали через целит. Фильтрат концентрировали in vacuo с уменьшением объема и очищали на диоксиде кремния (4 г, 0-10% 2 М NH3/MeOH в DCM) с получением указанного в заголовке соединения в виде 108 мг белого твердого вещества. Данное вещество дополнительно очищали с помощью HPLC на колонке Kinetix Axia C18 RP (длинная) с применением 10-60% CH3CN в H2O [0,1% HCOOH] с линейным изменением температуры в течение 10 мин. при 18 мл/мин. (УФ-излучение при 200 нм) с получением указанного в заголовке соединения в виде белого твердого вещества (64 мг, 38%). ¹H ЯМР (400 МГц, CDCl3): δ 7,38-7,27 (3H, m), 7,16-7,12 (2H, m), 6,90-6,85 (4H, m), 5,15 (1H, t, J=5,1 Гц), 4,12-4,08 (1H, m), 3,98 (1H, d, J=10,6 Гц,), 3,50-3,34 (4H, m), 2,61-2,53 (3H, m), 2,52 (4H, s), 1,91-1,80 (2H, m), 1,75 (2H, d, J=16,4 Гц), 1,62-1,48 (2H, m); LCMS (ESI) [M+H]+ 398,3. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 28A и 28B соответственно.
Схема синтеза согласно примеру 18: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метилбензил)-2-фенилпропаноат
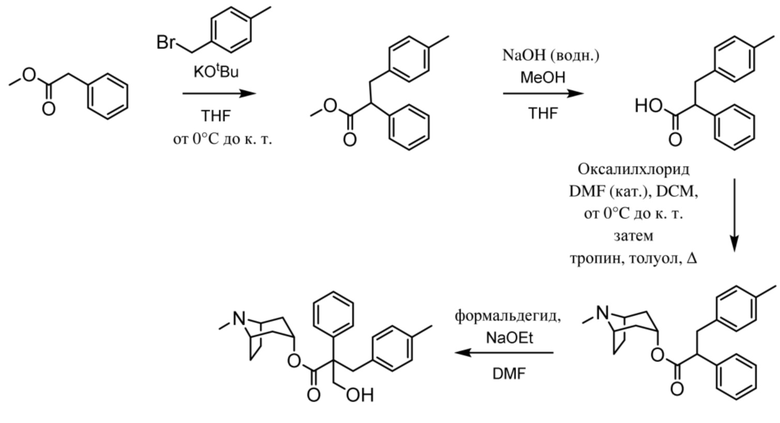

Метил-2-фенил-3-(п-толил)пропаноат
трет-Бутоксид калия (4,08 г, 22,1 ммоль) добавляли к перемешиваемому раствору метилфенилацетата (3,00 г, 20 ммоль) в сухом THF (30 мл) в атмосфере аргона при 0°C. После перемешивания в течение 15 мин. добавляли по каплям раствор 4-метилбензилбромида в сухом THF (10 мл) с поддержанием T ≤ 5°C. Полученную смесь перемешивали в течение 10 мин. перед тем, как обеспечивали ее нагревание до к. т. в течение 16 ч. Полученную смесь разбавляли с помощью EtOAc, промывали с помощью воды, затем солевым раствором. Органическую фракцию высушивали (Na2SO4) и концентрировали in vacuo с получением неочищенного продукта в виде желтого масла. Данное вещество очищали на диоксиде кремния (40 г, 0-25% EtOAc в циклогексане) с получением указанного в заголовке соединения в виде неочищенного бледно-желтого масла (3,60 г, 70%), применяемого при этом без дополнительной очистки.

2-Фенил-3-(п-толил)пропановая кислота
Метил-2-фенил-3-(п-толил)пропаноат (3,60 г, 14,2 ммоль) в THF (60 мл) и MeOH (6 мл) обрабатывали с помощью 2 М NaOH (водн.) (20 мл, 40 ммоль) и реакционную смесь перемешивали при к. т. в течение 18 ч. Реакционную смесь концентрировали in vacuo и остаток разбавляли с помощью воды (100 мл) и промывали с помощью EtOAc. Водную фракцию подкисляли с помощью 1 М HCl до pH 1,0 и экстрагировали с помощью EtOAc. Объединенные органические фракции высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке неочищенного соединения в виде светло-коричневого масла, которое кристаллизовалось при отстаивании (1,67 г, 49%). Применяли без очистки. ¹H ЯМР (400 МГц, CDCl3): δ 7,31 (3H, d, J=4,3 Гц), 7,29-7,27 (2H, m), 7,02 (4H, dd, J=8,2, 12,7 Гц), 3,84 (1H, dd, J=6,8, 8,6 Гц), 3,37 (1H, dd, J=8,6, 13,9 Гц), 3,00 (1H, dd, J=6,8, 13,9 Гц), 2,29 (3H, s).
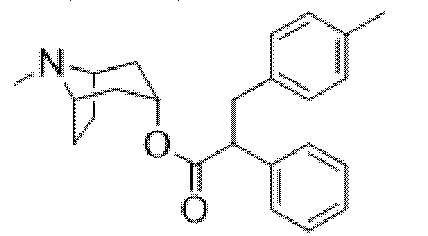
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-фенил-3-(п-толил)пропаноат
Перемешиваемый раствор 2-фенил-3-(п-толил)пропановой кислоты (1,0 г, 4,2 ммоль) в DCM (10 мл), содержащий DMF (200 мкл), в атмосфере аргона обрабатывали по каплям при 0°C с помощью оксалилхлорида (518 мкл, 5,9 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 мин., затем обеспечивали нагревание до к. т. и перемешивали в течение 18 ч. Реакционную смесь концентрировали in vacuo и остаток подвергали азеотропной перегонке с толуолом (2×15 мл) с получением неочищенного хлорангидрида. Данное вещество перемешивали в сухом толуоле (10 мл) с (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-олом (710 мл, 5,1 ммоль) в атмосфере аргона при 100°C в течение 4 ч. Реакционную смесь концентрировали in vacuo. Полученный остаток обрабатывали с помощью DCM (60 мл) и воды (40 мл) и повышали основность до pH 10 с помощью 1 М NaOH (водн.). Органическую фракцию отделяли, промывали солевым раствором, высушивали (MgSO4), концентрировали in vacuo. Остаток очищали на диоксиде кремния (12 г, 0-10% 2 М NH3/MeOH в DCM) с получением указанного в заголовке соединения в виде светло-коричневого масла (688 мг, 45%). ¹H ЯМР (400 МГц, CDCl3): δ 7,33-7,25 (5H, m), 7,03 (4H, m), 4,88 (1H, m), 3,78-3,72 (1H, m), 3,38 (1H, m), 3,18 (1H, m) 3,02-2,88 (3H, m), 2,89, (3H, s), 2,17 (3H, s), 2,07-1,91 (4H, m), 1,63-1,44 (3H, m); LCMS (ESI) [M+H]+ 364,2.
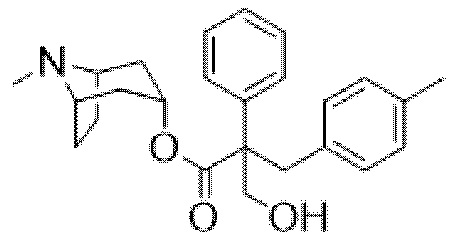
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метилбензил)-2-фенилпропаноат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-фенил-3-(п-толил)пропаноат (688 мг, 1,9 ммоль) в DMF (10 мл) обрабатывали параформальдегидом (722 мг, 24,0 ммоль), затем 21% раствором этоксида натрия (20 мкл, 0,054 ммоль) и перемешивали при 50 ± 5°C в течение 24 ч. Реакционную смесь охлаждали, разбавляли с помощью DCM (50 мл) и фильтровали через целит. Фильтрат концентрировали in vacuo и остаток очищали на диоксиде кремния (12 г, 0-10% 2 М NH3/MeOH в DCM), полученный остаток дополнительно очищали с помощью HPLC на колонке Kinetix Axia C18 RP (длинная) с применением 10-60% CH3CN в H2O [0,1% HCOOH] с линейным изменением температуры в течение 10 мин. при 18 мл/мин. (УФ-излучение=200 нм) с получением указанного в заголовке соединения в виде белого твердого вещества (260 мг, 35%). ¹H ЯМР (400 МГц, CDCl3): δ 7,38-7,33 (2H, m), 7,31-7,27 (1H, m), 7,20 (2H, d, J=7,3 Гц,), 7,01-6,97 (2H, m), 6,87-6,83 (2H, m), 5,14 (1H, t, J=5,0 Гц), 4,04 (2H, q, J=11,0 Гц), 3,49-3,34 (5H, m), 2,62-2,55 (2H, m), 2,52 (3H, s), 2,29-2,27 (3H, m), 1,87-1,69 (4H, m), 1,63-1,46 (2H, m); LCMS (ESI) [M+H]+ 394,3. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 29A и 29B соответственно.
Схема синтеза согласно примеру 19: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорбензил)-3-гидрокси-2-фенилпропаноат
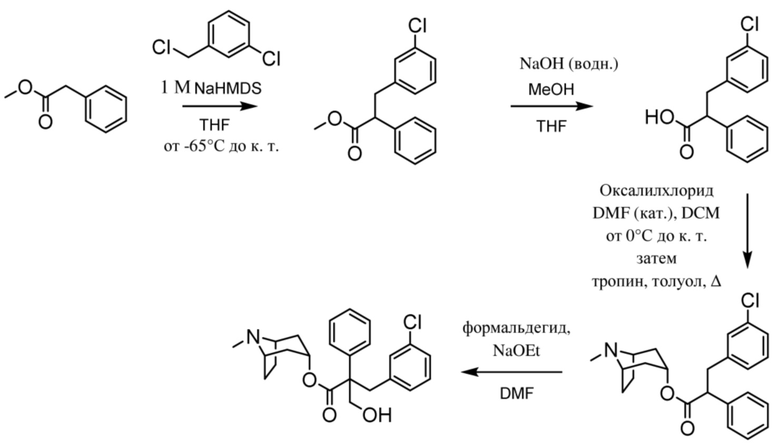

Метил-3-(3-хлорфенил)-2-фенилпропанoат
1 М бис(триметилсилил)амид натрия в THF (20 мл, 20 ммоль) добавляли по каплям к перемешиваемому раствору метилфенилацетата (3,00 г, 20,0 ммоль) и 3-хлорбензилхлорида (3,16 г, 19,6 ммоль) в сухом THF (40 мл) в атмосфере аргона при -78°C с поддержанием T ≤ -65°C. Реакционную смесь перемешивали в течение 1 ч., затем обеспечивали нагревание до к. т. в течение 16 ч. Реакционную смесь концентрировали in vacuo и остаток разбавляли с помощью EtOAc, промывали водой, затем солевым раствором. Органическую фракцию высушивали (Na2SO4) и концентрировали in vacuo с получением неочищенного продукта в виде желтого масла (5,48 г, 100%). Данное вещество применяли неочищенным без очистки. ¹H ЯМР (400 МГц, CDCl3): δ 7,32-7,26 (6H, m), 7,16-7,14 (2H, m), 7,13-7,10 (1H, m), 3,82 (1H, dd, J=6,7, 9,2 Гц), 3,61 (3H, s), 3,39 (1H, dd, J=13,6, 9,6 Гц), 2,99 (1H, dd, J=13,6, 6,8 Гц).
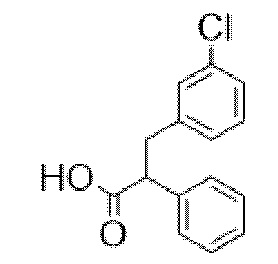
3-(3-Хлорфенил)-2-фенилпропановая кислота
Метил-3-(3-хлорфенил)-2-фенилпропанoат (5,48 г, 20 ммоль) в смеси THF (70 мл) и MeOH (7 мл) обрабатывали с помощью 2 М NaOH (водн.) (28,5 мл, 57 ммоль) и реакционную смесь перемешивали при к. т. в течение 18 ч. Реакционную смесь концентрировали in vacuo и остаток разбавляли водой и промывали с помощью EtOAc. Водную фракцию подкисляли с помощью 1 М HCl до pH 1,0 и экстрагировали с помощью EtOAc. Объединенные органические фракции высушивали (MgSO4) и концентрировали in vacuo с получением неочищенного продукта в виде желтого масла, которое кристаллизовалось при отстаивании (3,82 г, 73%). Применяли без очистки. ¹H ЯМР (400 МГц, CDCl3): δ 7,34-7,27 (5H, m), 7,16-7,11 (3H, m), 7,00-6,96 (1H, m), 3,84 (1H, dd, J=7,0, 8,5 Гц), 3,38 (1H, dd, J=8,5, 13,9 Гц), 3,01 (1H, dd, J=7,0, 13,9 Гц).
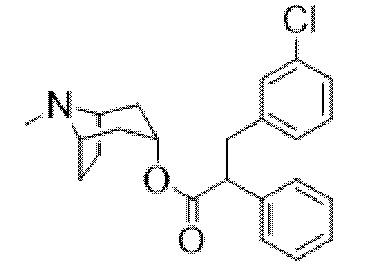
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(3-хлорфенил)-2-фенилпропанoат
Перемешиваемый раствор 3-(3-хлорфенил)-2-фенилпропановой кислоты (2,0 г, 7,7 ммоль) в DCM (20 мл), содержащий DMF (300 мкл), в атмосфере аргона обрабатывали по каплям при 0°C с помощью оксалилхлорида (955 мкл, 5,9 ммоль). Реакционную смесь перемешивали при 0°C в течение 10 мин., затем обеспечивали ее нагревание до к. т. и перемешивали в течение 18 ч. Реакционную смесь концентрировали in vacuo и остаток подвергали азеотропной перегонке с толуолом (2×15 мл) с получением неочищенного хлорангидрида. Полученный остаток перемешивали в сухом толуоле (10 мл) с (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-олом (1,31 г, 9,3 ммоль) в атмосфере аргона при 100°C в течение 3 ч. Реакционную смесь концентрировали in vacuo. Полученный остаток обрабатывали с помощью DCM (60 мл) и воды (40 мл) и повышали основность до pH 10 с помощью 1 М NaOH (водн.). Органическую фракцию отделяли, промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Остаток очищали на диоксиде кремния (40 г, 0-10% 2 М NH3/MeOH в DCM) с получением указанного в заголовке соединения в виде светло-коричневого масла (1,06 г, 36%). ¹H ЯМР (400 МГц, CDCl3): δ 7,33-7,26 (5H, m), 7,16-7,15 (3H, m), 7,03-7,00 (1H, m), 4,91 (1H, t, J=5,4 Гц), 3,75 (1H, dd, J=6,5, 9,0 Гц), 3,48 (2H, s), 3,03-2,91 (2H, m), 2,19 (3H, s), 2,07-1,94 (3H, m), 1,83-1,42 (4H, m), 1,36-1,28 (1H, m); LCMS (ESI) [M+H]+ 384,1/386,1.
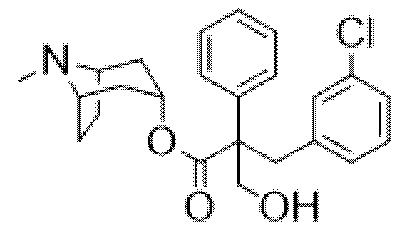
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорбензил)-3-гидрокси-2-фенилпропаноат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-(3-хлорфенил)-2-фенилпропанoат (1,10 г, 2,9 ммоль) в DMF (15 мл) обрабатывали параформальдегидом (1,10 г, 36,0 ммоль), затем 21% раствором этоксида натрия (30 мкл, 0,006 ммоль) и перемешивали при 100 ± 5°C в течение 24 ч. Реакционную смесь разбавляли с помощью DCM (50 мл) и фильтровали через целит. Фильтрат концентрировали in vacuo и остаток очищали на диоксиде кремния (24 г, 0-10% 2 М NH3/MeOH в DCM). Продукт дополнительно очищали с помощью HPLC на колонке Kinetix Axia C18 RP (длинная) с применением 10-60% CH3CN в H2O [0,1% HCOOH] с линейным изменением температуры в течение 10 мин. при 18 мл/мин. (УФ-излучение=200 нм) с получением продукта в виде белого твердого вещества (554 мг, 50%). ¹H ЯМР (400 МГц, CDCl3): δ 7,38-7,27 (3H, m), 7,16-7,06 (4H, m), 6,91 (1H, t, J=1,7 Гц), 6,79 (1H, d, J=7,6 Гц), 5,16 (1H, t, J=5,0 Гц), 4,10 (1H, d, J=10,6 Гц), 3,98 (1H, d, J=10,6 Гц), 3,52-3,45 (3H, m), 3,35 (1H, d, J=13,3 Гц), 2,64-2,55 (1H, m), 2,54 (3H, s), 1,91-1,72 (4H, m), 1,68-1,48 (2H, m); LCMS (ESI) [M+H]+ 414,2. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 30A и 30B соответственно.
Схема синтеза согласно примеру 20: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропаноат

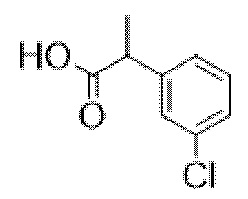
2-(3-Хлорфенил)пропановая кислота
Метил-2-(3-хлорфенил)пропаноат (590 мг, 2,97 ммоль) в THF (12 мл) и MeOH (1 мл) при к. т. обрабатывали с помощью 2 н. NaOH (водн.) (4,2 мл, 8,41 ммоль) и перемешивали в течение ночи. Реакционную смесь концентрировали in vacuo и полученный водный слой подкисляли с помощью 1 н. HCl до pH 1 и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Остаток очищали на диоксиде кремния (40 г, 0-30% EtOAc в циклогексане) с получением указанного в заголовке соединения в виде бледно-желтого масла (229 мг, 42%). 1H ЯМР (400 МГц, CDCl3): δ 7,34-7,15 (4H, m), 3,72 (1H, q, J=7,3 Гц), 1,51 (3H, d, J=7,3 Гц).
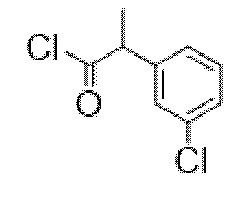
2-(3-Хлорфенил)пропаноилхлорид
Перемешиваемый раствор 2-(3-хлорфенил)пропановой кислоты (229 мг, 1,24 ммоль) в DCM (3 мл), содержащий DMF (55 мкл), обрабатывали по каплям с помощью оксалилхлорида (150 мкл, 1,77 ммоль) при 0°C в атмосфере аргона. Полученную смесь перемешивали при 0°C в течение 30 мин. перед тем, как обеспечивали ее нагревание до к. т. в течение ночи. Реакционную смесь концентрировали in vacuo с получением указанного в заголовке неочищенного соединения, которое применяли непосредственно без очистки.
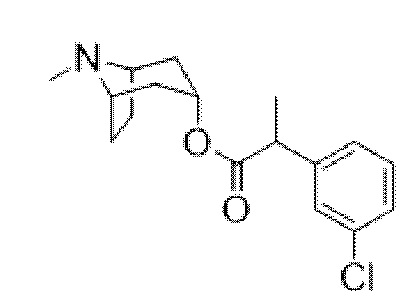
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорфенил)пропаноат
Неочищенный 2-(3-хлорфенил)пропаноилхлорид перемешивали в сухом толуоле (3 мл) с тропином (214 мг, 1,51 ммоль) при 100°C в атмосфере аргона в течение 3 ч. Реакционную смесь концентрировали in vacuo. Остаток разбавляли с помощью водного насыщенного NaHCO3 и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (40 г, 0-10% 2 н. NH3 MeOH в DCM) с получением указанного в заголовке соединения в виде желтого масла (272 мг, 71%). LCMS (ESI) [M+H]+ 308,2.
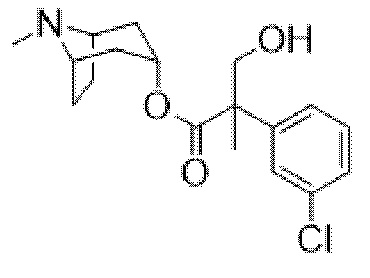
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорфенил)-3-гидрокси-2-метилпропаноат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(3-хлорфенил)пропаноат (337 мг, 1,09 ммоль) в DMF (2,5 мл) обрабатывали с помощью параформальдегида (49 мг, 1,64 ммоль), затем 21% раствором этоксида натрия (4,3 мкл, 0,055 ммоль) и реакционную смесь перемешивали при к. т. в течение 1 ч. Реакционную смесь разбавляли с помощью 1 н. HCl и экстрагировали с помощью EtOAc. Затем повышали основность объединенных водных фракций с помощью 1 н. NaOH до pH 13 и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (40 г, 0-10% 2 н. NH3 MeOH в DCM) с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (193 мг, 52%). 1H ЯМР (400 МГц, CDCl3): δ 7,34-7,26 (3H, m), 7,23-7,17 (1H, m), 5,06 (1H, t, J=5,4 Гц), 4,07 (1H, d, J=11,3 Гц), 4,64 (1H, d, J=11,3 Гц), 3,06-3,00 (1H, m), 2,99-2,93 (1H, m), 2,57 (1H, br s), 2,21 (3H, m), 2,16-2,03 (2H, m), 1,93-1,82 (1H, m), 1,80-1,69 (1H, m), 1,67 (3H, s), 1,66-1,56 (2H, m), 1,51-1,44 (1H, m), 1,27-1,17 (1H, m); LCMS (ESI) [M+H]+ 338,12. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 31A и 31B соответственно.
Схема синтеза согласно примеру 21: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-метоксибензил)-2-фенилпропаноат
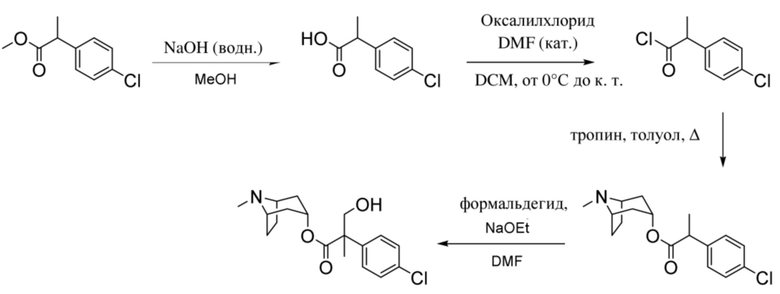
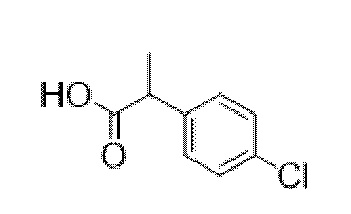
2-(4-Хлорфенил)пропановая кислота
Метил-2-(4-хлорфенил)пропаноат (547 мг, 2,75 ммоль) в THF (12 мл) и MeOH (1 мл) при к. т. обрабатывали с помощью 2 н. NaOH (водн.) (3,9 мл, 7,79 ммоль) и перемешивали в течение 8 ч. Реакционную смесь концентрировали in vacuo и полученный водный слой подкисляли с помощью 1 н. HCl до pH 1 и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Остаток очищали на диоксиде кремния (40 г, 0-30% EtOAc в циклогексане) с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (137 мг, 27%). 1H ЯМР (400 МГц, CDCl3): δ 7,35-7,16 (4H, m), 3.71 (1H, q, J=9,4 Гц), 1,49 (3H, d, J=9,6 Гц).
2-(4-Хлорфенил)пропаноилхлорид
Перемешиваемый раствор 2-(4-хлорфенил)пропановой кислоты (265 мг, 1,44 ммоль) в DCM (4 мл), содержащий DMF (70 мкл), обрабатывали по каплям с помощью оксалилхлорида (180 мкл, 2,05 ммоль) при 0°C в атмосфере аргона. Полученную смесь перемешивали при 0°C в течение 30 мин. Реакционную смесь концентрировали in vacuo с получением указанного в заголовке неочищенного соединения и применяли непосредственно без очистки.

(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-хлорфенил)пропаноат
Неочищенный 2-(4-хлорфенил)пропаноилхлорид перемешивали в сухом толуоле (4 мл) с тропином (247 мг, 1,75 ммоль) при 100°C в атмосфере аргона в течение ночи. Реакционную смесь концентрировали in vacuo. Остаток разбавляли с помощью EtOAc и экстрагировали с помощью 1 н. HCl. Затем повышали основность объединенных водных фракций с помощью 1 н. NaOH до pH 13 и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде коричневого масла, которое применяли неочищенным в следующей реакции (230 мг, 52%). LCMS (ESI) [M+H]+ 308,1.
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-хлорфенил)-3-гидрокси-2-метилпропаноат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-хлорфенил)пропаноат (230 мл, 0,75 ммоль) в DMF (2 мл) обрабатывали с помощью параформальдегида (34 мг, 1,12 ммоль), затем 21% раствором этоксида натрия, (3 мкл, 0,037 ммоль) и реакционную смесь перемешивали при к. т. в течение 1 ч. Добавляли дополнительное количество параформальдегида (34 мг, 1,12 ммоль), затем 21% раствор этоксида натрия (3 мкл, 0,037 ммоль) и продолжали перемешивание при к. т. в течение 1 ч. Реакционную смесь разбавляли с помощью 1 н. HCl и экстрагировали с помощью EtOAc. Затем повышали основность объединенных водных фракций с помощью 1 н. NaOH до pH 13 и экстрагировали с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (40 г, 0-10% 2 н. NH3 MeOH в DCM) с получением указанного в заголовке соединения в виде грязно-белого твердого вещества (118 мг, 47%). 1H ЯМР (400 МГц, CDCl3): δ 7,36-7,31 (2H, m), 7,28-7,22 (2H, m), 5,06 (1H, t, J=5,5 Гц), 4,05 (1H, d, J=11,1 Гц), 3,62 (1H, d, J=11,1 Гц), 3,06-3,00 (1H, m), 2,99-2,92 (1H, m), 2,52 (1H, br s), 2,21 (3H, s), 2,16-2,01 (2H, m), 1,93-1,82 (1H, m), 1,80 (1H, m), 1,67 (3H, s), 1,65-1,56 (2H, m), 1,49-1,42 (1H, m), 1,25-1,15 (1H, m); LCMS (ESI). [M+H]+ 338,1. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 32A и 32B соответственно.
Схема синтеза согласно примеру 22: (1R,3r,5S)-8-метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-(бензилокси)фенил)пропаноат
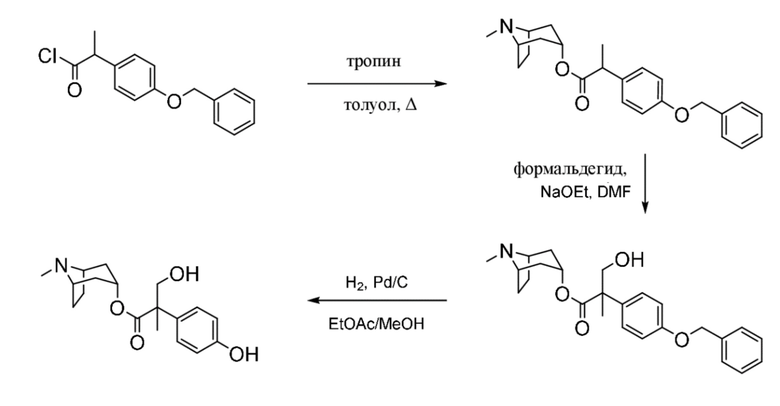
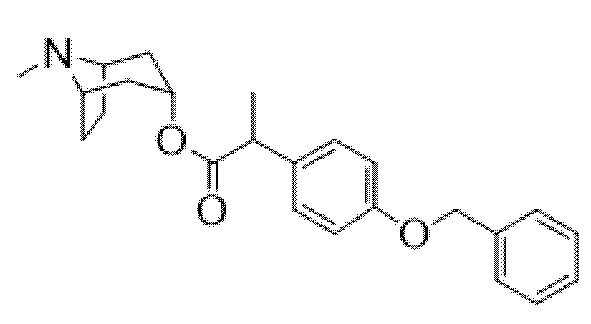
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-(бензилокси)фенил)пропаноат
В раствор 2-(4-(бензилокси)фенил)пропаноилхлорида (1,61 г, 5,86 ммоль) в толуоле (20 мл) добавляли тропин (750 мг, 5,33 ммоль) и реакционную смесь перемешивали при 90°C в течение 3 ч., затем при 50°C в течение 16 ч. Реакционную смесь разбавляли с помощью 1 н. HCl и экстрагировали с помощью EtOAc и органические фракции отбрасывали. Повышали основность объединенных водных фракций с помощью 1 н. NaOH и экстрагировали их с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения (750 мг, 37%). 1H ЯМР (400 МГц, CDCl3) δ 7,47-7,29 (5H, m), 7,23-7,19 (2H, m), 6,97-6,90 (2H, m), 5,06 (2H, s), 4,94 (1H, t, J=5,3 Гц), 3,62 (1H, q, 7,1 Гц), 3,06-3,00 (1H, m), 2,98-2,93 (1H, m), 2,22 (3H, s), 2,12-1,98 (3H, m), 1,92-1,57 (4H, m), 1,48 (3H, d, J=7,2 Гц), 1,43-1,35 (1H, m). LCMS (ESI) [M+H]+ 380,2, Rt=0,97 мин. (способ 2).
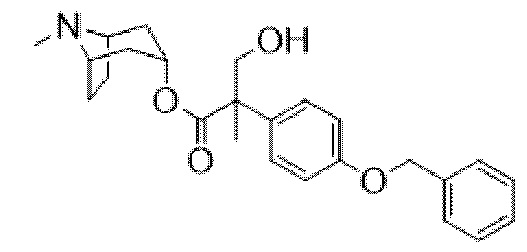
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-(бензилокси)фенил)-3-гидрокси-2-метилпропаноат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-(бензилокси)фенил)пропаноат (750 мг, 1,98 ммоль) в DMF (8 мл) обрабатывали с помощью параформальдегида (89 мг, 2,96 ммоль), затем 21% раствором этоксида натрия (37 мкл, 0,10 ммоль) и реакционную смесь перемешивали при к. т. в течение 1 ч. Реакционную смесь разбавляли с помощью EtOAc и экстрагировали с помощью 1 н. HCl. Повышали основность объединенных водных фракций с помощью 1 н. NaOH и экстрагировали их с помощью EtOAc. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo. Полученный осадок очищали на диоксиде кремния (12 г, 0-10% MeOH в DCM) с получением указанного в заголовке соединения (685 мг, 85%). 1H ЯМР (400 МГц, CDCl3) δ 7,45-7,29 (5H, m), 7,25-7,20 (2H, m), 6,99-6,92 (2H, m), 5,06 (2H, s), 5,04 (1H, t, J=5,5 Гц), 4,09 (1H, d, J=11,1 Гц), 3,59 (1H, d, J=11,2 Гц), 3,04-2,99 (1H, m), 2,94-2,89 (1H, m), 2,50 (1H, br s), 2,20 (3H, s), 2,14-1,99 (2H, m), 1,90-1,78 (1H, m), 1,74-1,58 (6H, m), 1,49-1,41 (1H, m), 1,25-1,14 (1H, m). LCMS (ESI) [M+H]+ 410,2 Rt=0,87 мин. (способ 2).
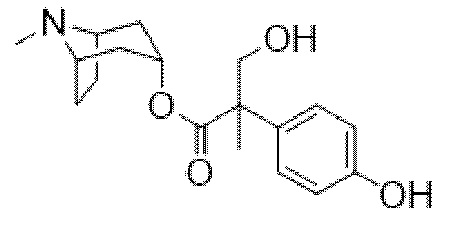
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-3-гидрокси-2-(4-гидроксифенил)-2-метилпропаноат
(1R,3r,5S)-8-Метил-8-азабицикло[3.2.1]октан-3-ил-2-(4-(бензилокси)фенил)-3-гидрокси-2-метилпропаноат (685 мг, 1,67 ммоль) в EtOAc (10 мл) и MeOH (3 мл) обрабатывали с помощью палладия на угле (10 вес. %, 150 мг) и перемешивали при к. т. в атмосфере водорода в течение 16 ч. Реакционную смесь фильтровали и фильтрат концентрировали in vacuo. Остаток очищали на диоксиде кремния (12 г, 0-10% MeOH в DCM) с получением указанного в заголовке соединения в виде белого твердого вещества (278 мг, 52%). 1H ЯМР (400 МГц, CDCl3): δ 7,18-7,12 (2H, m), 6,77-6,71 (2H, m), 5,03 (1H, t, J=5,1 Гц), 4,10 (1H, d, J=11,4 Гц), 3,57 (1H, d, J=11,4 Гц), 3,11-3,05 (1H, m), 3,01-2,92 (1H, m), 2,27 (3H, s), 2,17 (1H, dt, J=15,2, 4,1 Гц), 2,08 (1H, dt, J=15,3, 4,3 Гц), 1,92-1,78 (1H, m), 1,67-1,54 (6H, m), 1,41 (1H, d, J=15,3 Гц), 1,18-1,07 (1H, m); LCMS (ESI) [M+H]+ 320,3. Спектры 1H ЯМР и 13C ЯМР для указанного в заголовке соединения показаны на фигурах 33A и 33B соответственно.
Схема синтеза согласно примеру 23: 6-хлор-11H-бензо[e]пиридо[3,2-b][1,4]диазепин
2-Нитробензоилхлорид
В раствор 2-нитробензойной кислоты (5 г, 29,9 ммоль) в DCM (140 мл) добавляли 1 каплю DMF с последующим добавлением оксалилхлорида (3,7 мл, 41,9 ммоль), что приводило к выделению пузырьков газа. Реакционную смесь перемешивали при к. т. в течение 20 мин. Реакционную смесь концентрировали in vacuo с получением указанного в заголовке соединения в виде масла соломенного цвета (5,55 г, колич.). Материал применяли без очистки. 1H ЯМР (400 МГц, CDCl3): δ 8,13-8,07 (1H, m), 7,84-7,68 (3H, m).
N-(2-Хлорпиридин-3-ил)-2-нитробензамид
В раствор 2-нитробензоилхлорида (5,55 г, 29,9 ммоль) в THF (120 мл) добавляли пиридин (14,5 мл, 0,18 моль) и 3-амино-2-хлорпиридин (4,23 г, 32,9 ммоль), что приводило к образованию осадка. Реакционную смесь перемешивали при к. т. в течение 2 ч. перед тем, как разбавляли с помощью 10% водного раствора лимонной кислоты и экстрагировали с помощью этилацетата. Объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением продукта в виде твердого вещества светло-коричневого цвета (8,6 г, колич.). Материал применяли без очистки. 1H ЯМР (400 МГц, d6-DMSO) δ 10,61 (1H, s), 8,31 (1H, dd, J=4,3, 1,5 Гц), 8,25-8,16 (2H, m), 7,94-7,87 (1H, m), 7,84-7,75 (2H, m), 7,54 (1H, dd, J=8,3, 4,3 Гц).
2-Амино-N-(2-хлорпиридин-3-ил)бензамид
В раствор N-(2-хлорпиридин-3-ил)-2-нитробензамида (8,3 г, 29,9 ммоль) в этаноле (100 мл) добавляли воду (20 мл), железо (2,67 г, 47,8 ммоль) и хлорид аммония (16 г, 0,3 моль). Смесь нагревали с обратным холодильником в течение 40 мин., затем разбавляли водой и фильтровали. Фильтрат экстрагировали с помощью этилацетата и объединенные органические фракции промывали солевым раствором, высушивали (MgSO4) и концентрировали in vacuo с получением указанного в заголовке соединения в виде бежевого твердого вещества (7,4 г, колич.) Материал применяли без очистки. 1H ЯМР (400 МГц, d6-DMSO) δ 9,93 (1H, s), 8,29 (1H, dd, J=4,7, 1,8 Гц), 8,05 (1H, dd, J=7,9, 1,8 Гц), 7,73 (1H, dd, J=8,0, 1,5 Гц), 7,48 (1H, dd, J=7,8, 4,7 Гц), 7,24 (1H, ddd, J=8,4, 7,1, 1,5 Гц), 6,78 (1H, dd, J=8,3, 0,9 Гц), 6,61 ( 1H, ddd, J=8,1, 7,2, 1,1 Гц), 6,48 (2H, br s).
5,11-Дигидро-6H-бензо[e]пиридо[3,2-b][1,4]диазепин-6-он
2-Амино-N-(2-хлорпиридин-3-ил)бензамид (3,5 г, 14,1 ммоль) нагревали при 210°C в течение 5 мин. После достижения 200°C реакционная смесь окрашивалась в черный цвет и наблюдалось выделение пузырьков газа. Реакционную смесь охлаждали до комнатной температуры и твердый остаток промывали с помощью насыщ. водн. раствора NaHCO3, затем DCM с получением указанного в заголовке продукта в виде серого твердого вещества (2,89 г, 96%). Материал применяли без очистки. 1H ЯМР (400 МГц, d6-DMSO): δ 9,94 (1H, s), 8,63 (1H, s), 7,90 (1H, dd, J=4,84, 1,6 Гц), 7,72 (1H, dd, J=7,9, 1,6 Гц), 7,41-7,29 (2H, m), 7,13 (1H, dd, J=8,3, 0,8 Гц), 6,97 (1H, dd, J=7,8, 4,8 Гц), 6,95-6,89 (1H, m).
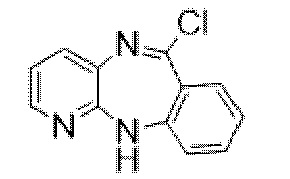
6-Хлор-11H-бензо[e]пиридо[3,2-b][1,4]диазепин
Смесь 5,11-дигидро-6H-бензо[e]пиридо[3,2-b][1,4]диазепин-6-она (500 мг, 2,37 ммоль) и пентахлорида фосфора в хлорбензоле (12 мл) нагревали при 100°C в течение 4 ч. Реакционную смесь охлаждали до ~4°C, добавляли изогексан (10 мл) и твердые вещества отфильтровывали. Твердое вещество промывали с помощью дополнительного количества изогексана (10 мл) и высушивали in vacuo с получением продукта в виде желтого твердого вещества (520 мг, 95%). Материал применяли без очистки. 1H ЯМР (400 МГц, CDCl3): δ 9,96 (1H, s), 7,72-7,59 (3H, m), 7,41 (1H, td, J=8,0, 1,3 Гц), 7,16-7,04 (3H, m).
6-Хлор-11H-бензо[e]пиридо[3,2-b][1,4]диазепин, полученный в примере 23, можно использовать для получения пирензепина формулы (II) по настоящему изобретению в соответствии со следующей схемой реакции:
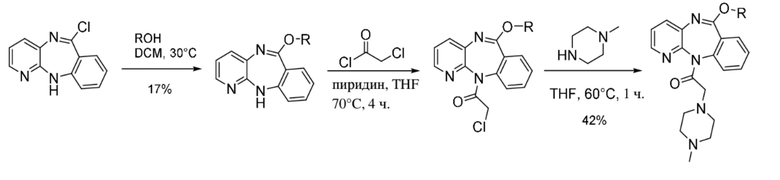 ,
,
в которой R может представлять собой гексил.
Пример 24
Анализ в отношении мускаринового рецептора человека
Протокол анализа в отношении M1-M5
M1
Гомогенаты клеточных мембран (45 мкг белка) инкубировали в течение 60 мин. при 22°С с 2 нМ [3Н]пирензепином в отсутствие или в присутствии тестируемого соединения в буфере, содержащем 50 мМ Tris-HCl (рН 7,4), 120 мМ NaCl, 5 мМ KCl, 5 мМ MgCl2 и 1 мМ EDTA. Неспецифическое связывание определяли в присутствии 1 мкМ атропина. После инкубации образцы быстро фильтровали в вакууме через фильтры из стекловолокна (GF/B, Packard), предварительно пропитанные 0,3% PEI, и несколько раз промывали ледяным 50 мМ Tris-HCl с применением сборщика клеток на 96 образцов (Unifilter, Packard). Фильтры высушивали, затем определяли радиоактивность в сцинтилляционном счетчике (Topcount, Packard) с использованием сцинтилляционного коктейля (Microscint 0, Packard). Результаты выражены в процентах ингибирования специфического связывания контрольного радиолиганда. Стандартным эталонным соединением являлся пирензепин, который тестировали в каждом эксперименте при нескольких концентрациях с получением кривой успешности конкурирования, по которой рассчитывали его Ki и IC50.
M2
Гомогенаты клеточных мембран (60 мкг белка) инкубировали в течение 60 мин. при 22°С с 2 нМ [3H]AF-DX 384 в отсутствие или в присутствии тестируемого соединения в буфере, содержащем 50 мМ Tris-HCl (рН 7,4), 120 мМ NaCl, 5 мМ KCl, 5 мМ MgCl2 и 1 мМ EDTA. Неспецифическое связывание определяли в присутствии 1 мкМ атропина. После инкубации образцы быстро фильтровали в вакууме через фильтры из стекловолокна (GF/B, Packard), предварительно пропитанные 0,3% PEI, и несколько раз промывали ледяным 50 мМ Tris-HCl с применением сборщика клеток на 96 образцов (Unifilter, Packard). Фильтры высушивали, затем определяли радиоактивность в сцинтилляционном счетчике (Topcount, Packard) с использованием сцинтилляционного коктейля (Microscint 0, Packard). Результаты выражены в процентах ингибирования специфического связывания контрольного радиолиганда. Стандартным эталонным соединением являлся метоктрамин, который тестировали в каждом эксперименте при нескольких концентрациях для получения кривой успешности конкурирования, по которой рассчитывали его Ki и IC50.
M3
Гомогенаты клеточной мембраны (8 мкг белка) инкубировали в течение 60 мин. при 22°С с 0,2 нМ [3H]4-DAMP в отсутствие или в присутствии тестируемого соединения в буфере, содержащем 10 мМ Tris-HCl (рН 7,4) и 2 мМ EDTA. Неспецифическое связывание определяли в присутствии 1 мкМ атропина. После инкубации образцы быстро фильтровали в вакууме через фильтры из стекловолокна (GF/B, Packard), предварительно пропитанные 0,3% PEI, и несколько раз промывали ледяным 50 мМ Tris-HCl с применением сборщика клеток на 96 образцов (Unifilter, Packard). Фильтры высушивали, затем определяли радиоактивность в сцинтилляционном счетчике (Topcount, Packard) с использованием сцинтилляционного коктейля (Microscint 0, Packard). Результаты выражены в процентах ингибирования специфического связывания контрольного радиолиганда. Стандартным эталонным соединением являлся 4-DAMP, который тестировали в каждом эксперименте при нескольких концентрациях для получения кривой успешности конкурирования, по которой рассчитывали его Ki и IC50.
M4
Гомогенаты клеточных мембран (16 мкг белка) инкубировали в течение 60 мин. при 22°С с 0,2 нМ [3H]4-DAMP в отсутствие или в присутствии тестируемого соединения в буфере, содержащем 10 мМ Tris-HCl (рН 7,4) и 2 мМ EDTA. Неспецифическое связывание определяли в присутствии 1 мкМ атропина. После инкубации образцы быстро фильтровали в вакууме через фильтры из стекловолокна (GF/B, Packard), предварительно пропитанные 0,3% PEI, и несколько раз промывали ледяным 50 мМ Tris-HCl с применением сборщика клеток на 96 образцов (Unifilter, Packard). Фильтры высушивали, затем определяли радиоактивность в сцинтилляционном счетчике (Topcount, Packard) с использованием сцинтилляционного коктейля (Microscint 0, Packard). Результаты выражены в процентах ингибирования специфического связывания контрольного радиолиганда. Стандартным эталонным соединением являлся 4-DAMP, который тестировали в каждом эксперименте при нескольких концентрациях для получения кривой успешности конкурирования, по которой рассчитывали его Ki и IC50.
M5
Гомогенаты клеточных мембран (15 мкг белка) инкубировали в течение 60 мин. при 22°С с 0,3 нМ [3H]4-DAMP в отсутствие или в присутствии тестируемого соединения в буфере, содержащем 10 мМ Tris-HCl (рН 7,4) и 2 мМ EDTA. Неспецифическое связывание определяли в присутствии 1 мкМ атропина. После инкубации образцы быстро фильтровали в вакууме через фильтры из стекловолокна (GF/B, Packard), предварительно пропитанные 0,3% PEI, и несколько раз промывали ледяным 50 мМ Tris-HCl с применением сборщика клеток на 96 образцов (Unifilter, Packard). Фильтры высушивали, затем определяли радиоактивность в сцинтилляционном счетчике (Topcount, Packard) с использованием сцинтилляционного коктейля (Microscint 0, Packard). Результаты выражены в процентах ингибирования специфического связывания контрольного радиолиганда. Стандартным эталонным соединением являлся 4-DAMP, который тестировали в каждом эксперименте при нескольких концентрациях для получения кривой успешности конкурирования, по которой рассчитывали его Ki и IC50.
Аффинность связывания M1-M5
Результаты анализа в отношении мускариновых рецепторов человека некоторых соединений по настоящему изобретению показаны в таблице ниже.
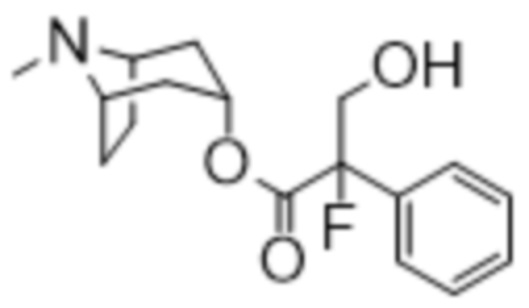
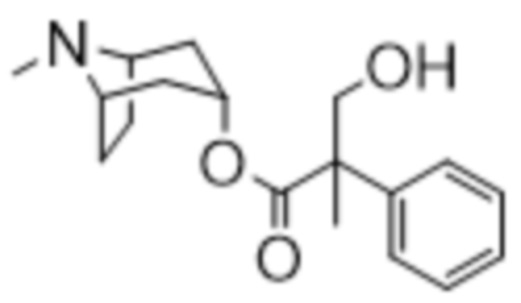
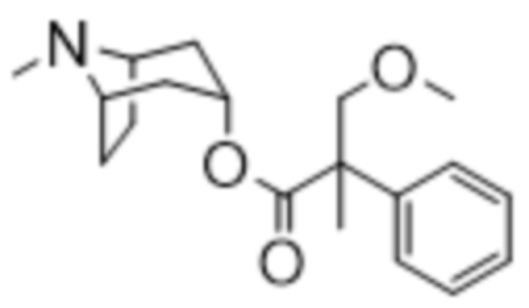
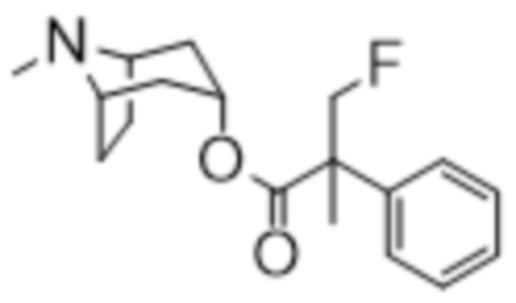
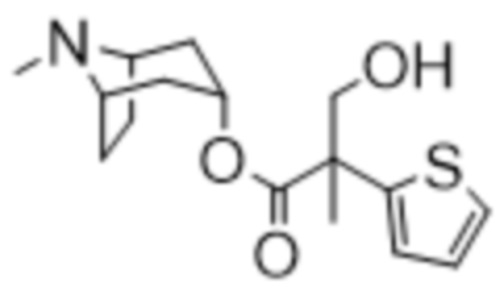
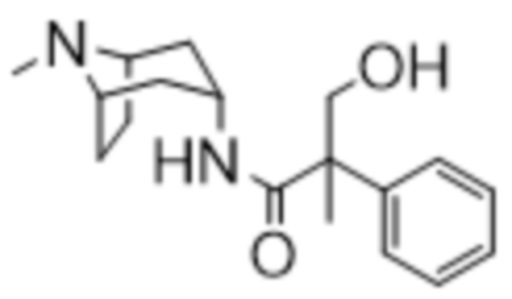
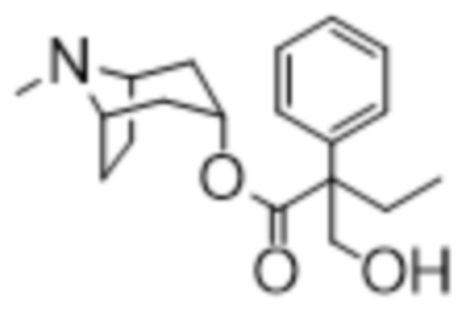
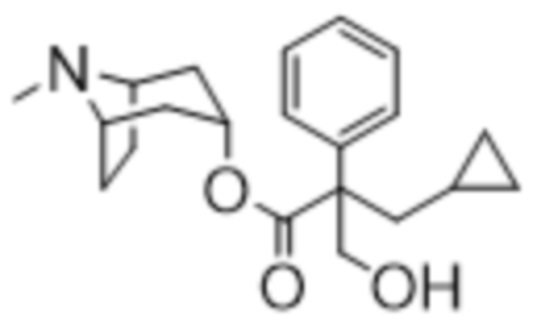
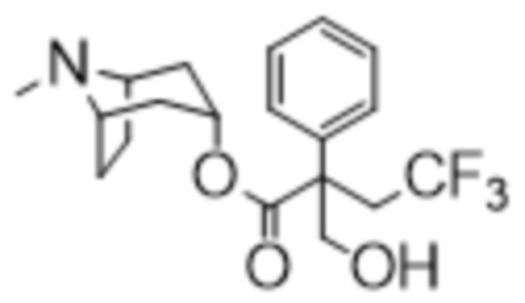
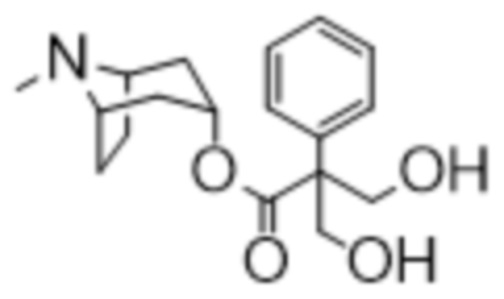
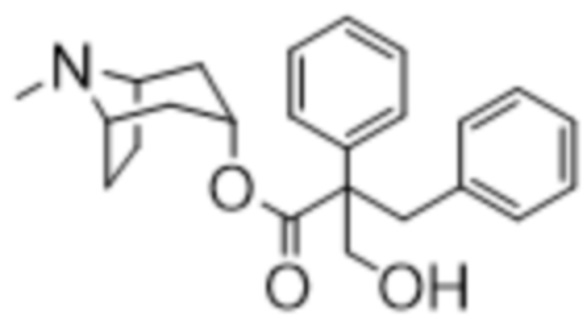
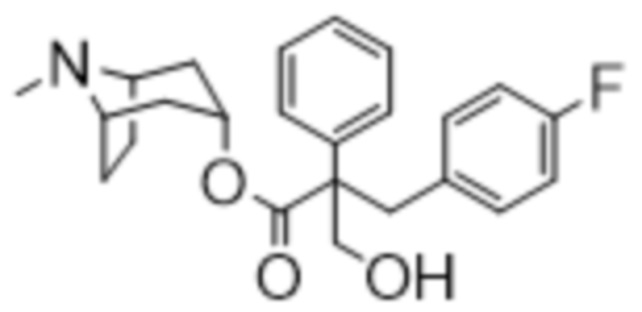
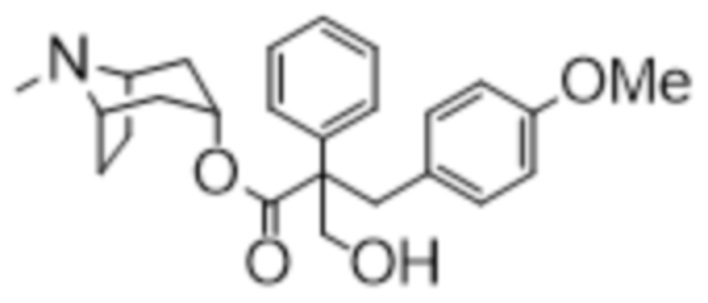
Считается, что результаты, демонстрирующие ингибирование или стимуляцию выше 50%, отображают значительные эффекты тестируемых соединений.
Соединения по настоящему изобретению и их фармацевтически приемлемые соли могут присутствовать в комбинации с одним или несколькими фармацевтическими носителями с образованием различных типов составов для доставки. Например, могут быть использованы составы для местного применения и они могут включать офтальмологически приемлемые консерванты, поверхностно-активные средства, усилители вязкости, буферы, хлорид натрия и воду для образования водных офтальмологически совместимых растворов и суспензий. Подходящие составы для местного применения описаны в обзорных статьях, например, "Recent Advances in Topical Ocular Drug Delivery", by V.K. Yellepeddi and S. Palakurthi (J. Ocul. Pharmacol. Ther. 2016, 32(2):67-82), включенной в данный документ посредством ссылки во всей своей полноте. Также предусматриваются составы для системного введения (например, перорально проглатываемые таблетки) и составы для внутриглазной инъекции. (фармацевтически приемлемые носители) Примеры подходящих фармацевтически приемлемых носителей описаны в Remington’s Pharmaceutical Sciences, Mack Publishing Company (включено в данный документ посредством ссылки во всей своей полноте), стандартном ссылочном тексте в области фармацевтики.
Конкретный тип выбранного состава будет зависеть от различных факторов, таких как подлежащие использованию соединение или его соль, частота введения дозы, место локализации подлежащего лечению заболевания, выбранный путь введения и стандартная фармацевтическая практика. Офтальмологически совместимые водные растворы, суспензии, мази и гели для местного применения являются предпочтительными лекарственными формами для лечения офтальмологических заболеваний в передней части глаза (роговица, радужная оболочка, трабекулярная сеть) или офтальмологических заболеваний задней части глаза, если соединение может быть составлено так, что оно может быть доставлено путем местного применения и способно проникать в ткани передней части глаза. Соединение в соответствии с формулой (I) обычно будет содержаться в таких составах в количестве от приблизительно 0,01 до приблизительно 10,0 вес/процент. Предпочтительные значения концентрации для местного введения варьируются от приблизительно 0,1 до приблизительно 5,0 вес/процент. Таким образом, для местного введения эти составы доставляют на поверхность глаза от одного до шести раз в сутки, что зависит от решения врача соответствующей квалификации. Системное введение, например, в форме таблеток, применимо для лечения офтальмологического заболевания, особенно задней части глаза, например, сетчатки.
Соединения по настоящему изобретению предпочтительно включены в офтальмологически совместимые составы для доставки в глаз. Соединения могут быть объединены с офтальмологически приемлемыми консервантами, поверхностно-активными средствами, усилителями вязкости, усилителями проницаемости, буферами, хлоридом натрия и водой для образования водных, стерильных офтальмологические суспензии или раствора. Составы на основе офтальмологического раствора могут быть получены путем растворения соединения в физиологически приемлемом изотоническом водном буфере. Кроме того, офтальмологический раствор может включать офтальмологически приемлемое поверхностно-активное средство для обеспечения растворения соединения. Более того, офтальмологический раствор может содержать средство для повышения вязкости, такое как гидроксиметилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилметилцеллюлоза, метилцеллюлоза, поливинилпирролидон или им подобные, для улучшения удерживания состава в конъюнктивальном мешке. Также могут быть использованы гелеобразующие средства, в том числе без ограничения геллан и ксантановая камедь. Для получения стерильных офтальмологических составов в виде мази активный ингредиент объединяют с консервантом в соответствующей среде-носителе, такой как минеральное масло, жидкий ланолин или белый вазелин. Стерильные офтальмологические составы в виде геля могут быть получены путем суспендирования соединения в гидрофильной основе, полученной из комбинации, например, Carbopol-974 или ему подобного, в соответствии с опубликованными составами для аналогичных офтальмологических составов; при этом могут быть включены консерванты и средства, регулирующие тоничность.
Фармацевтические композиции могут включать одно или несколько буферных средств или регулирующих pH средств для обеспечения улучшенного контроля pH. В некоторых вариантах осуществления для местного применения фармацевтическая композиция по настоящему изобретению имеет показатель pH от 5,0 до 8,0, от 5,0 до 7,0, от 6,0 до 8,0 или от 6,0 до 7,0. В одном варианте осуществления pH фармацевтической композиции по настоящему изобретению составляет от приблизительно 6,3 до приблизительно 7,3. В определенном варианте осуществления водная фармацевтическая композиция по настоящему изобретению имеет практически нейтральный показатель pH, составляющий приблизительно 6,8.
Другие предусматриваемые вспомогательные средства, которые могут быть использованы в фармацевтических композициях по настоящему изобретению, включают, например, противомикробные средства, антиоксиданты, антистатические средства, липиды, такие как фосфолипиды или жирные кислоты, стероиды, такие как холестерин, белковые вспомогательные средства, такие как сывороточный альбумин (человеческий сывороточный альбумин), рекомбинантный человеческий альбумин, желатин, казеин, солеобразующие противоионы, например, натрий и им подобные. Эти и дополнительные известные фармацевтические вспомогательные средства и/или добавки, подходящие для применения в составах по настоящему изобретению, известные из уровня техники, например, приведены в "The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); и Remington: the Science and Practice of Pharmacy, 21st edition, Gennaro, Ed., Lippincott Williams & Wilkins (2005).
В другом аспекте в настоящем изобретении предусмотрена фармацевтическая композиция, содержащая: (1) соединение по настоящему изобретению, его фармацевтически приемлемую соль, фармацевтически приемлемый и фармацевтически приемлемый носитель. В одном варианте осуществления композиция содержит терапевтически эффективное количество, предпочтительно от приблизительно 0,01 до приблизительно 10,0 процентов по весу, более предпочтительно от приблизительно 0,01 до приблизительно 5 процентов по вес/объем или от приблизительно от 0,1 до 5,0 процентов по весу (a) указанного соединения и/или (b) указанной фармацевтически приемлемой соли. В дополнительном варианте осуществления композиция содержит по меньшей мере два таких фармацевтически приемлемых носителя, которые описаны в данном документе. Для целей настоящего изобретения, если не обозначено иным образом, сольваты и гидраты обычно относятся к рассматриваемым композициям. Предпочтительно фармацевтически приемлемые носители являются стерильными. Фармацевтическая композиция может быть составлена для конкретных путей введения, таких как пероральное введение, парентеральное введение и ректальное введение и т. д. Кроме того, фармацевтические композиции по настоящему изобретению могут быть изготовлены в твердой форме (в том числе без ограничения в капсулах, таблетках, пилюлях, гранулах, порошках или суппозиториях) или в жидкой форме (в том числе без ограничения в растворах, суспензиях или эмульсиях). Фармацевтические композиции можно подвергать традиционным фармацевтическим технологическим операциям, таким как стерилизация, и/или они могут содержать традиционные инертные разбавители, смазывающие средства или буферные средства, а также вспомогательные средства, такие как консерванты, стабилизаторы, смачивающие средства, эмульгаторы и буферы и т. д.
В другом варианте осуществления настоящего изобретения фармацевтические композиции представляют собой таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с одним или несколькими из:
a) разбавителей, например, лактозы, декстрозы, сахарозы, маннита, сорбита, целлюлозы и/или глицина;
b) смазывающих веществ, например, диоксида кремния, талька, стеариновой кислоты, ее магниевой или кальциевой соли и/или полиэтиленгликоля; также в случае таблеток
c) связующих средств, например, алюмосиликата магния, крахмальной пасты, желатина, трагаканта, метилцеллюлозы, натрий-карбоксиметилцеллюлозы и/или поливинилпирролидона; при необходимости
d) разрыхлителей, например, видов крахмала, агара, альгиновой кислоты или ее натриевой соли или шипучих смесей; и
e) абсорбентов, красителей, ароматизаторов и подсластителей.
Таблетки могут быть либо покрыты пленочной оболочкой, либо покрыты кишечнорастворимой оболочкой в соответствии со способами, известными из уровня техники.
Фармацевтические композиции по настоящему изобретению могут включать дополнительное терапевтическое средство в дополнение к соединениям по настоящему изобретению. Дополнительные терапевтические средства могут включать, например, другие соединения и антитела, применимые для лечения офтальмологических заболеваний.
Фармацевтические композиции по настоящему изобретению можно вводить пациенту. Используемый в данном документе термин "субъект" или "пациент" относится к человеку и к отличным от человека млекопитающим, в том числе без ограничения к приматам, кроликам, свиньям, лошадям, собакам, кошкам, овцам и коровам. Предпочтительно субъектом или пациентом является человек.
Предполагаются различные способы доставки для введения фармацевтических композиций, и они могут предусматривать, например, местный, интравитреальный, пероральный, IV, в камеру глаза, и другие способы, известные специалистам в данной области техники.
В одном варианте осуществления введение, как правило, будет осуществляться с помощью шприца. Таким образом в настоящем изобретении предусматривается устройство для доставки (например, шприц), содержащее фармацевтическую композицию по настоящему изобретению (например, предварительно заполненный шприц). Пациенты будут получать эффективное количество соединения в соответствии с формулой (I) в качестве основного активного ингредиента.
В еще одном варианте осуществления применяют глазные вставки или пленки для доставки соединения по настоящему изобретению. В одном таком варианте осуществления соединение формулы (I) составлено в полимерную глазную вставку, содержащую один или несколько мукоадгезивных полимеров, которые являются биологически совместимыми с поверхностью глаза и слезной пленкой глаза. В некоторых вариантах осуществления после введения полимерной глазной вставки в слепой мешок глаза толщина слезной пленки может увеличиваться в течение по меньшей мере 30 минут после введения. Один или несколько мукоадгезивных полимеров могут быть выбраны из группы, содержащей: гиалуроновую кислоту (в форме кислоты или соли), гидроксипропилметилцеллюлозу (HPMC), метилцеллюлозу, полисахарид из семян тамаринда (TSP), гуар, гидроксипропилгуар (HP-гуар), склероглюкан, полоксамер, поли(галактуроновую)кислоту, альгинат натрия, пектин, ксантановую камедь, ксилоглюкановую камедь, хитозан, натрия карбоксиметилцеллюлозу, поливиниловый спирт, поливинилпирролидин, карбомер, полиакриловую кислоту и их комбинации. В одном варианте осуществления настоящего описания один или несколько мукоадгезивных полимеров могут представлять собой HP-гуар, гиалуроновую кислоту и гиалуронат натрия.
В настоящем изобретении дополнительно предусмотрен способ доставки соединения в соответствии с формулой (I) пациенту, предусматривающий стадию введения пациенту фармацевтической композиции по настоящему изобретению один или несколько раз в сутки.
Как используется в данном документе, все процентные содержания являются процентными содержаниями по весу, если не указано иное. Если не указано иное, термины в единственном числе означают "один", "по меньшей мере один" или "один или несколько". Если контекст не требует иного, термины в единственном числе, используемые в данном документе, должны включать множественное число, а термины во множественном числе должны включать единственное число. С целью ясности содержание любых патентов, заявок на выдачу патентов и ссылок, цитируемых во всем данном описании, тем самым включены посредством ссылки во всей своей полноте.
ПРИМЕРЫ СОСТАВА
Следующие примеры приводятся для демонстрации вариантов осуществления настоящего изобретения. Специалистам в данной области будет понятно, что можно выполнить изменения в конкретных вариантах осуществления, раскрываемых в данном документе, и все же получить подобный результат без отступления от идеи и объема настоящего изобретения.
ПРИМЕР СОСТАВА 1. Офтальмологический препарат для местного применения
Были подробно описаны настоящее изобретение и его варианты осуществления. Однако конкретные варианты осуществления любых процесса, варианта изготовления, композиции на основе соединения, соединений, средств, способов и/или стадий, описываемых в настоящем описании, не предназначены для ограничения объема настоящего изобретения. Различные модификации, замены и изменения могут быть сделаны в раскрываемом веществе без отклонения от сущности и/или существенных признаков настоящего изобретения. Соответственно, специалист в данной области техники легко поймет при ознакомлении с настоящим изобретением, что дальнейшие модификации, замены и/или варианты, выполняющие фактически ту же функцию или достигающие фактически того же результата, что и варианты осуществления, раскрываемые в данном документе, могут быть использованы в соответствии с такими родственными вариантами осуществления настоящего изобретения. Таким образом, следующая формула изобретения предназначена для охвата модификаций, замен и вариантов процессов, вариантов изготовления, композиций на основе соединения, соединений, средств, способов и/или стадий, раскрываемых в данном документе. Формула изобретения не должна рассматриваться как ограниченная описанным порядком или элементами, если это конкретно не указано. Следует учитывать, что различные изменения по форме и содержанию могут быть выполнены без отклонения от объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2840772C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2022 |
|
RU2803084C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2777979C2 |
| СПИРО-КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИПЕРИДИНА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛИЕВОГО КАНАЛА НАРУЖНОГО МЕДУЛЛЯРНОГО СЛОЯ | 2013 |
|
RU2642066C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2747318C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2023 |
|
RU2826177C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2715058C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ STING И СПОСОБЫ С ИХ ИСПОЛЬЗОВАНИЕМ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2825271C2 |
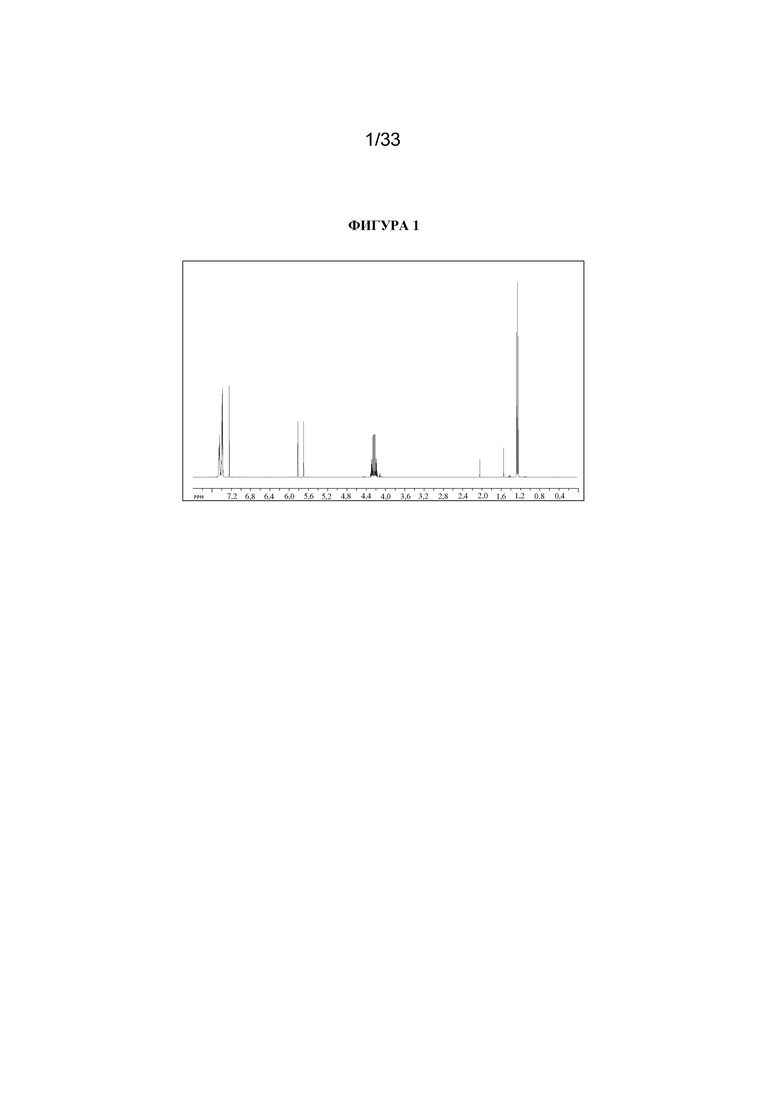
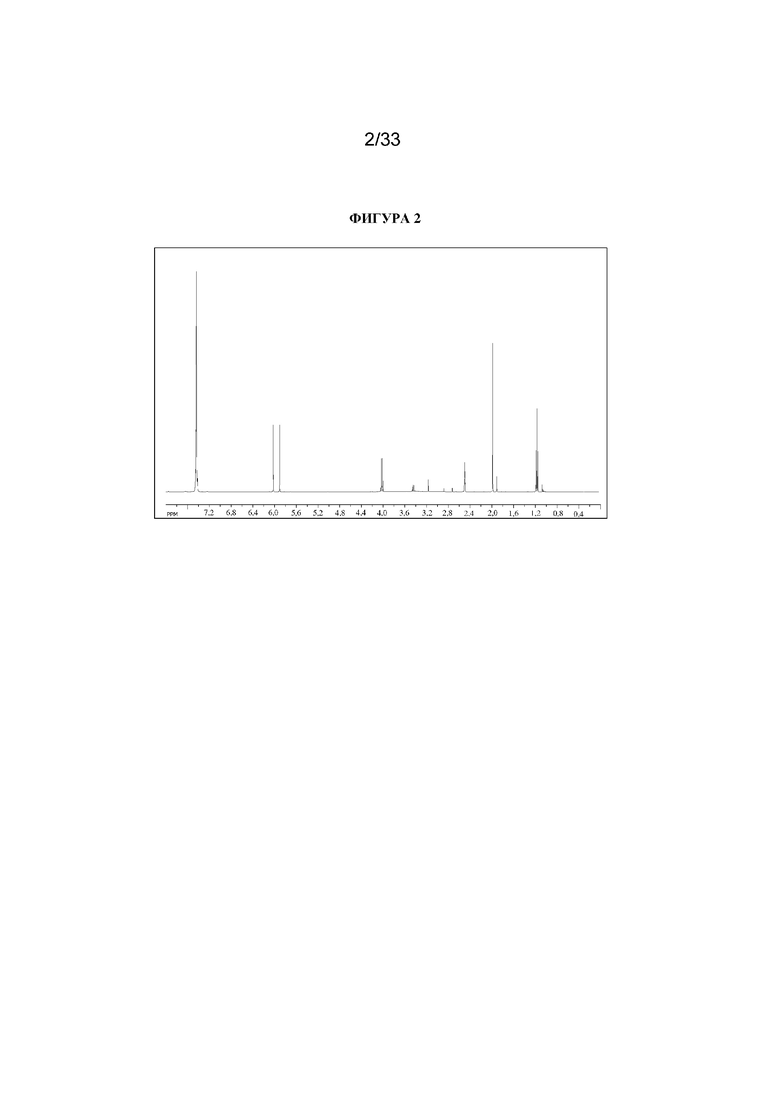
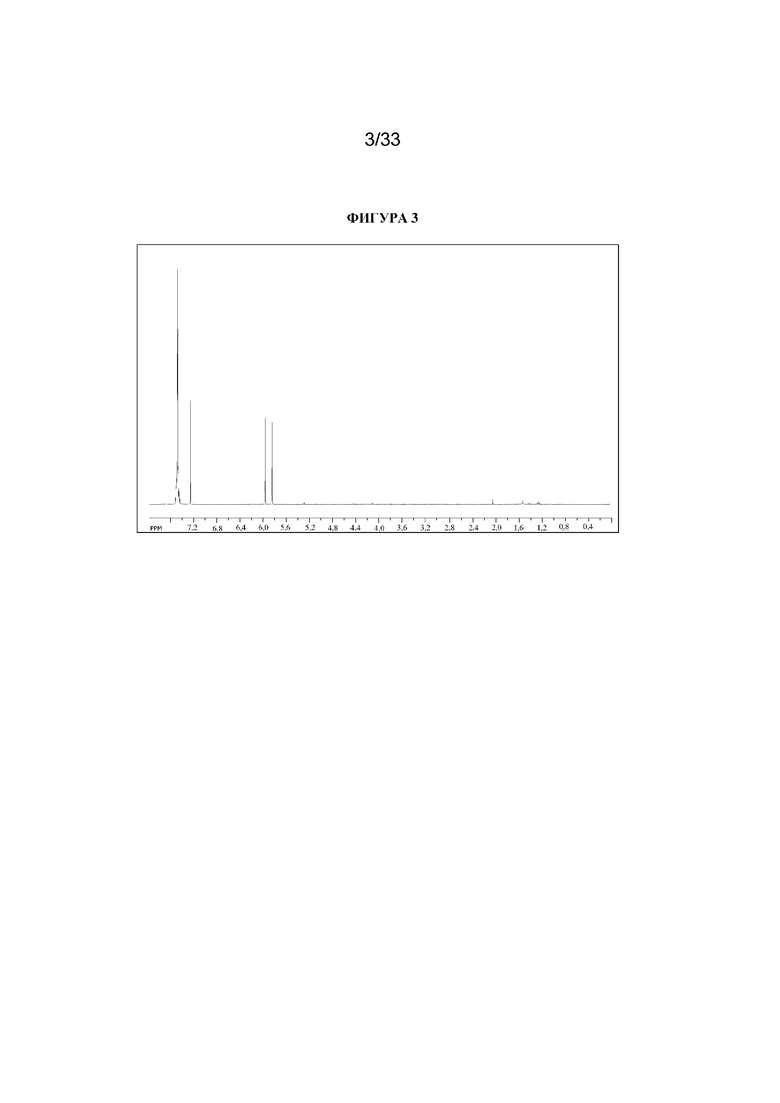
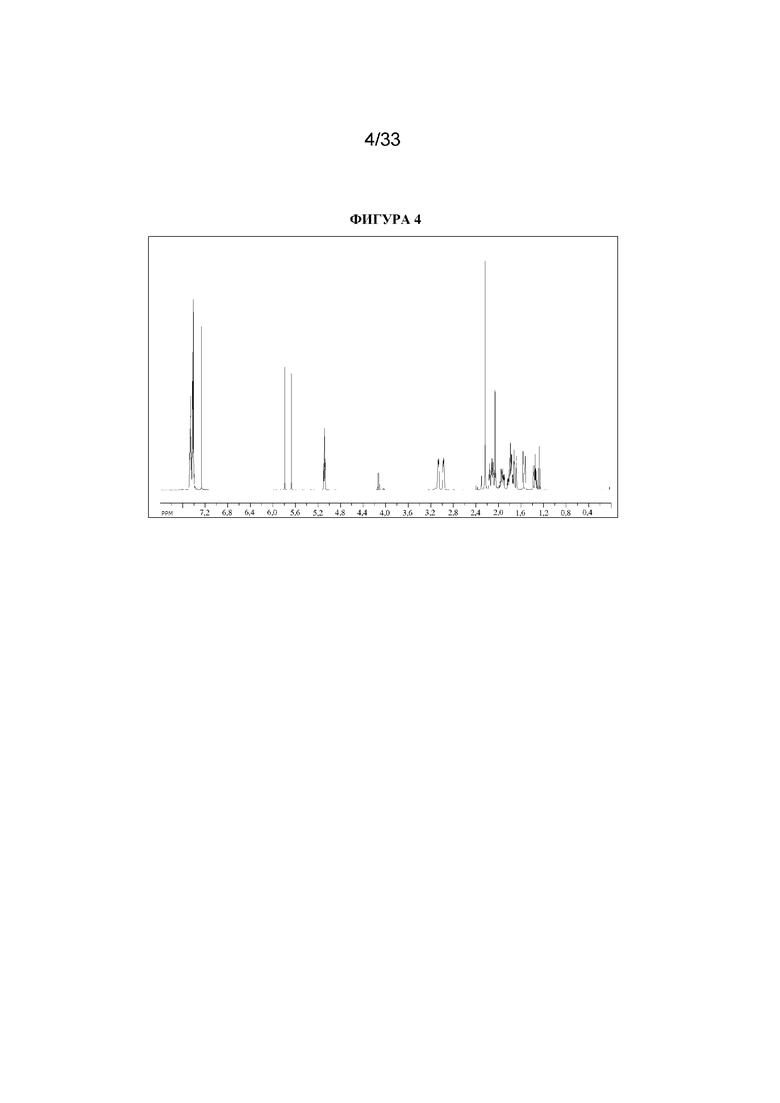
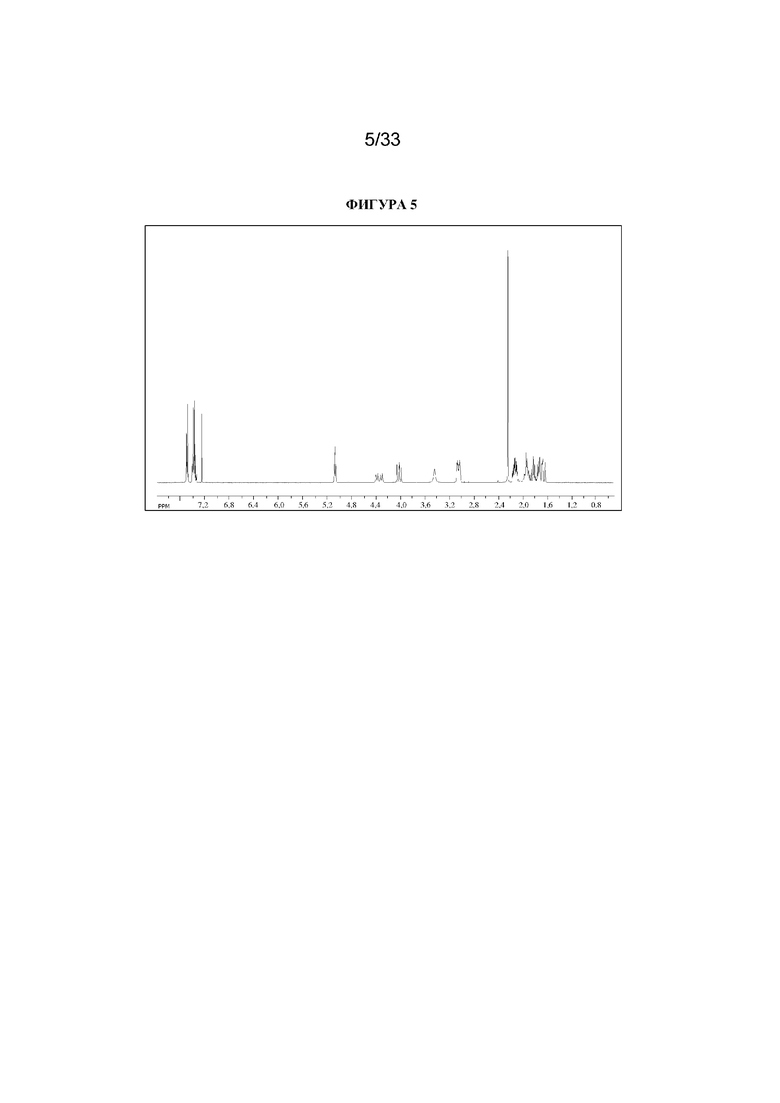

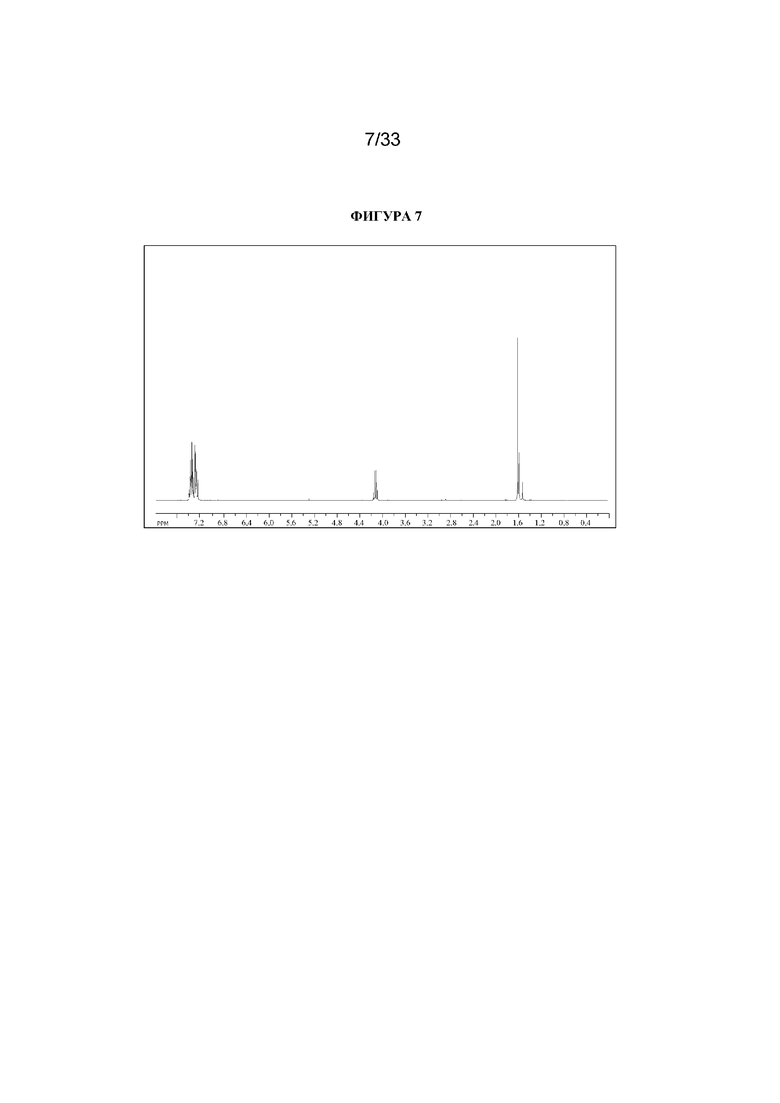
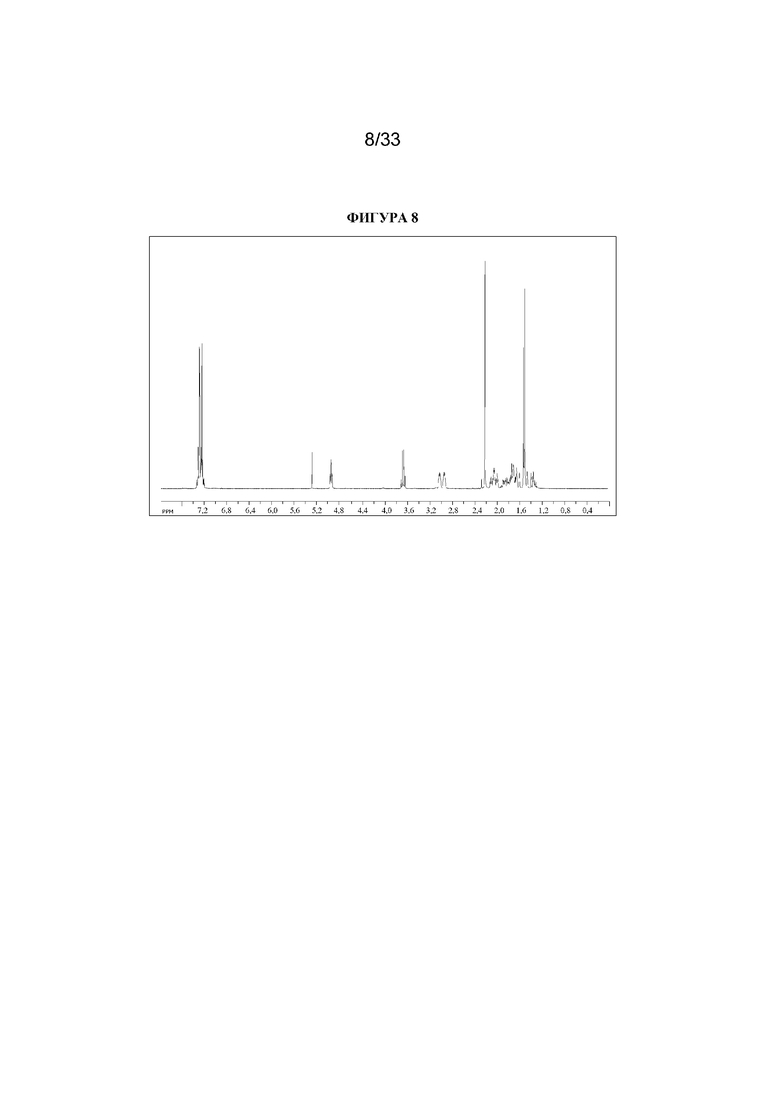
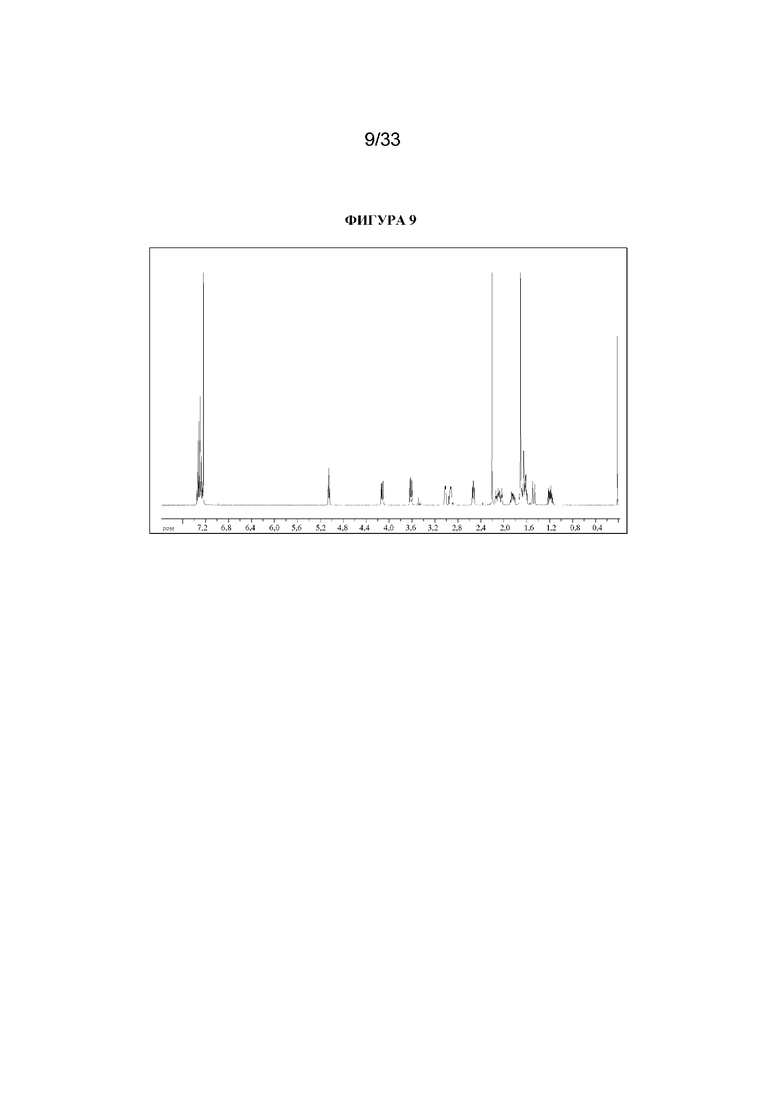
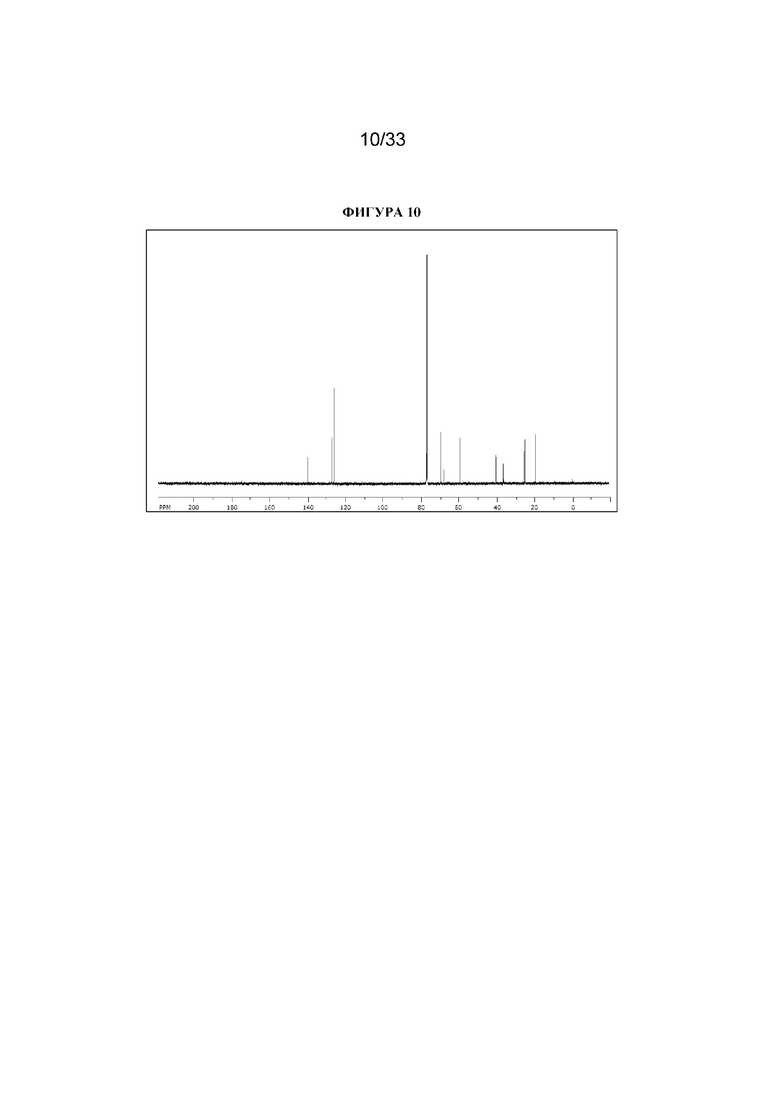
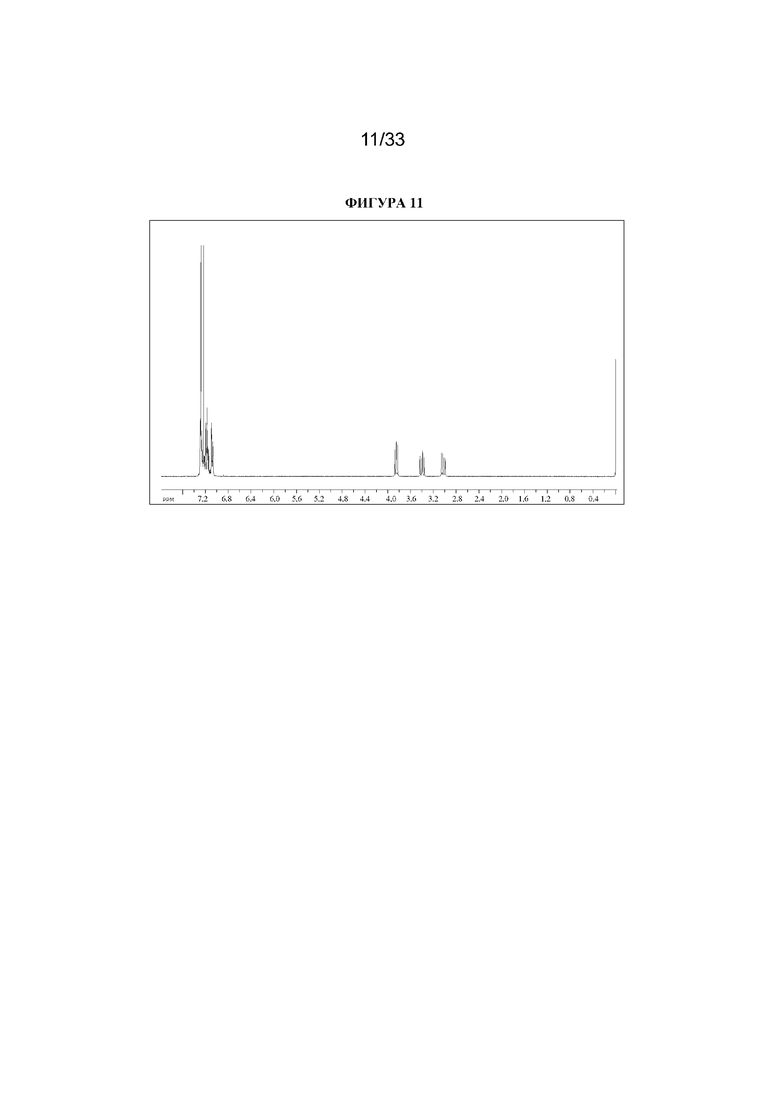
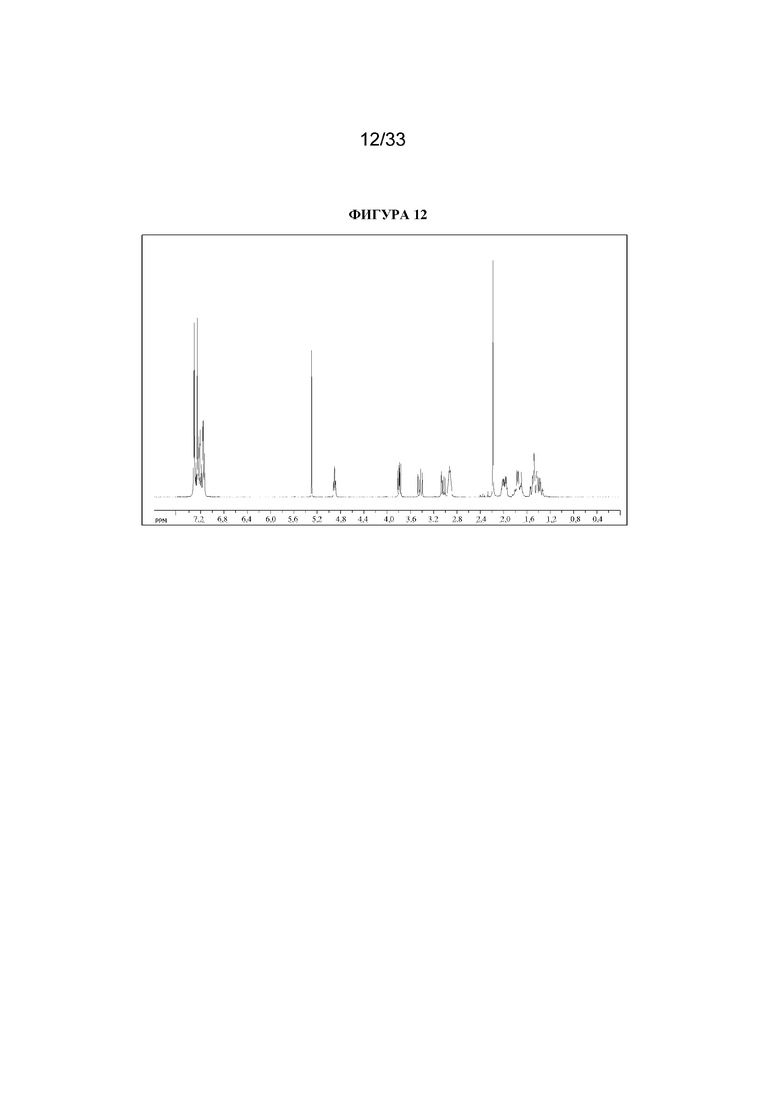
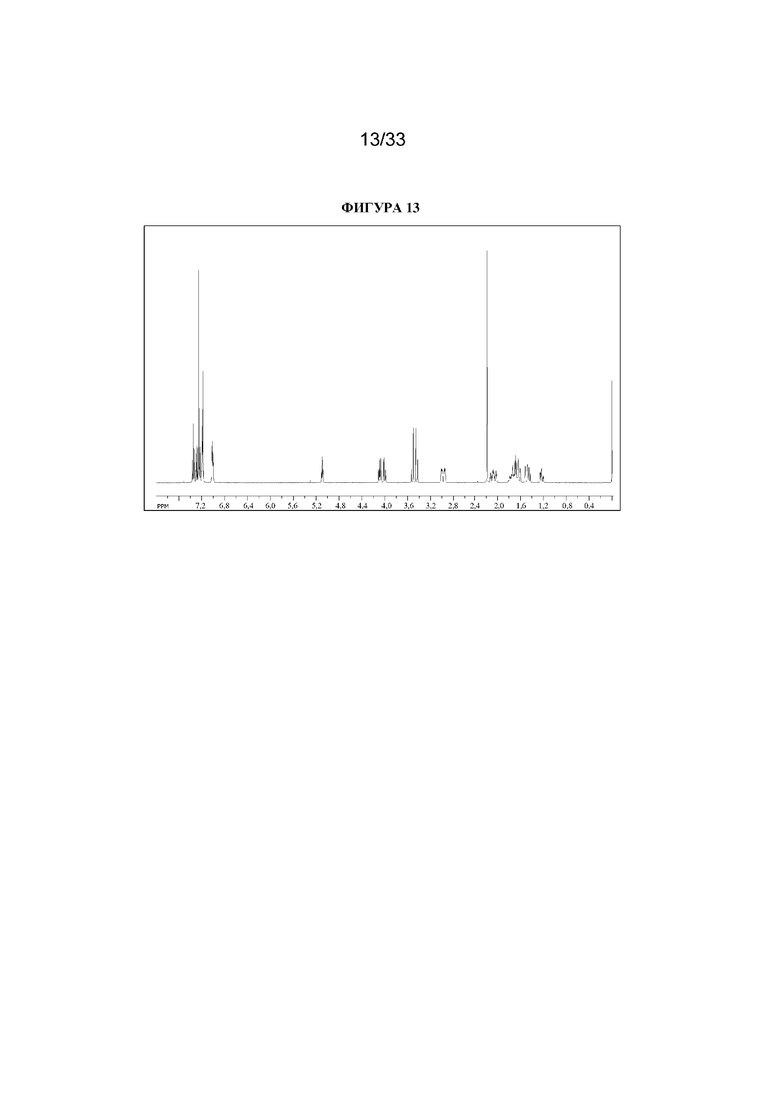

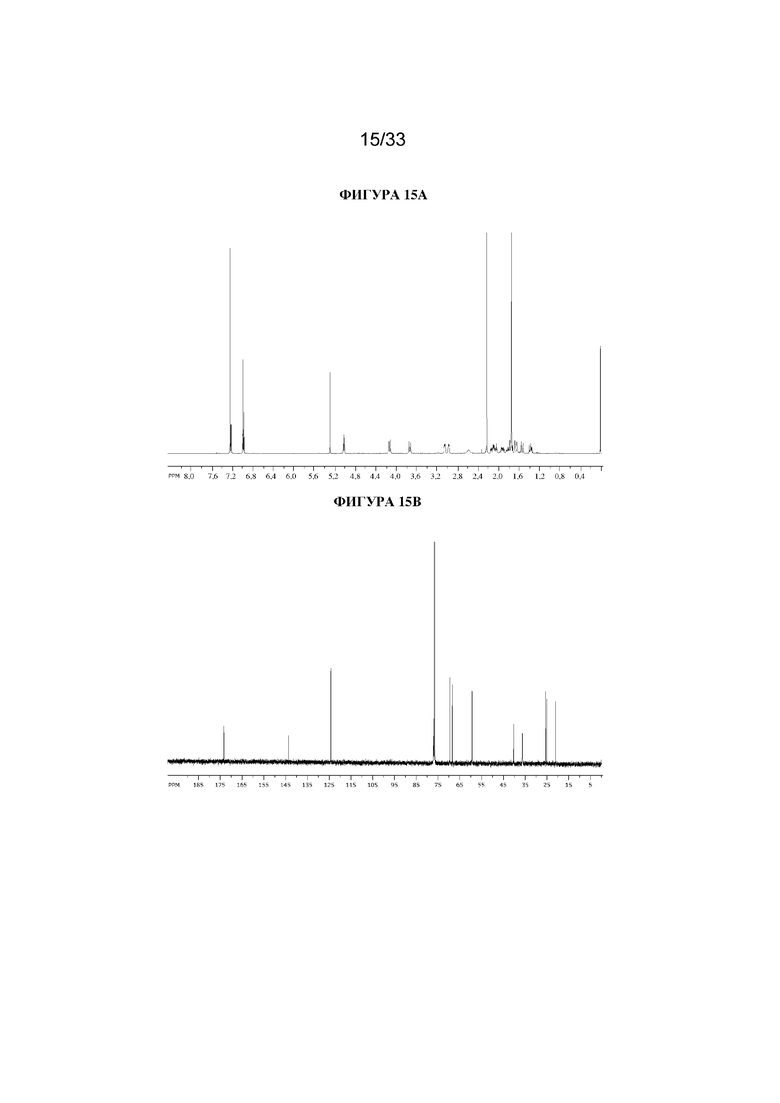

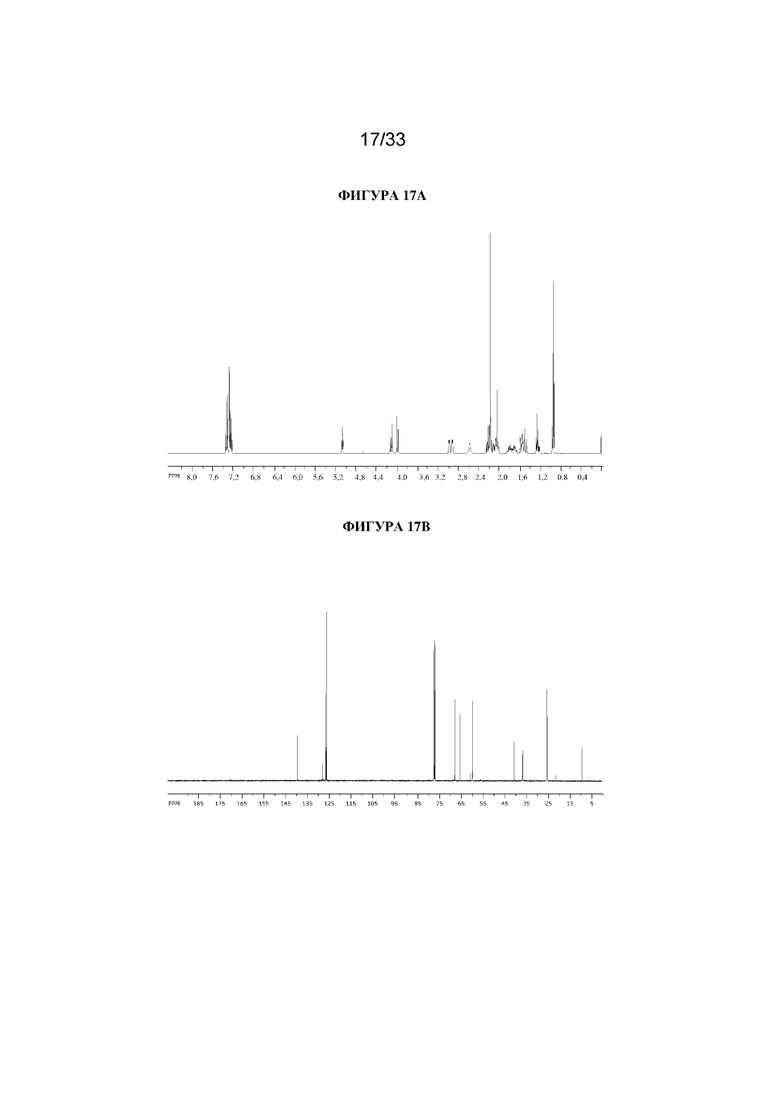

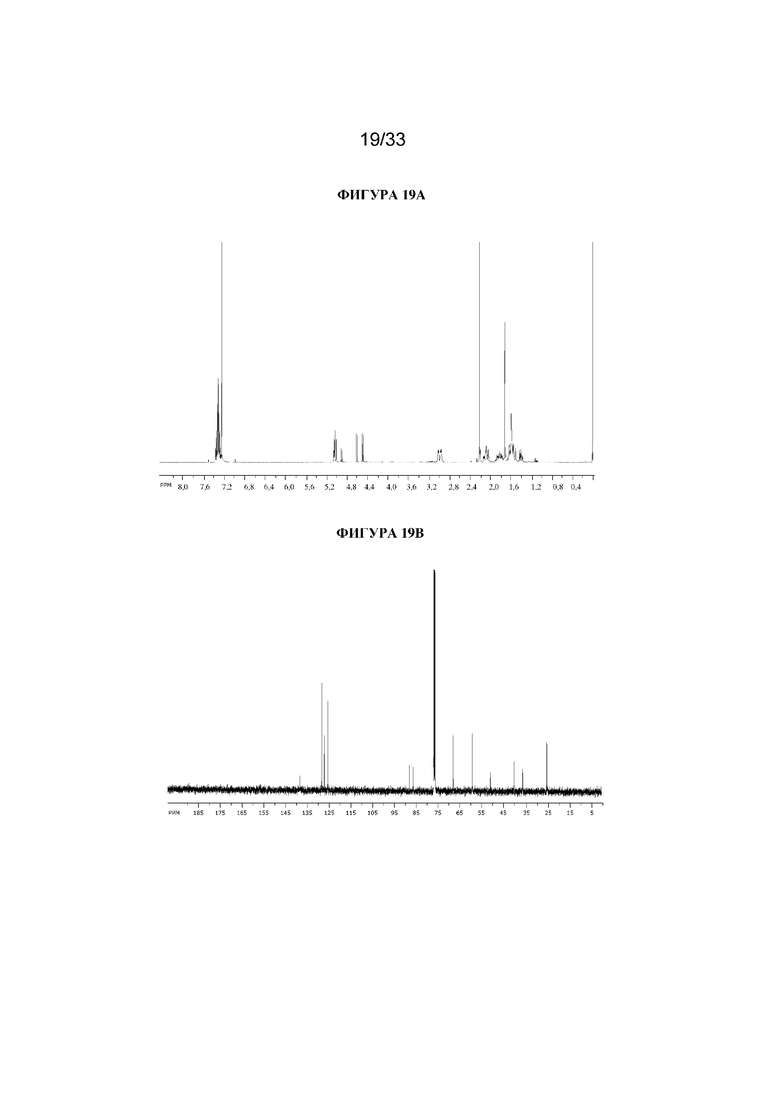
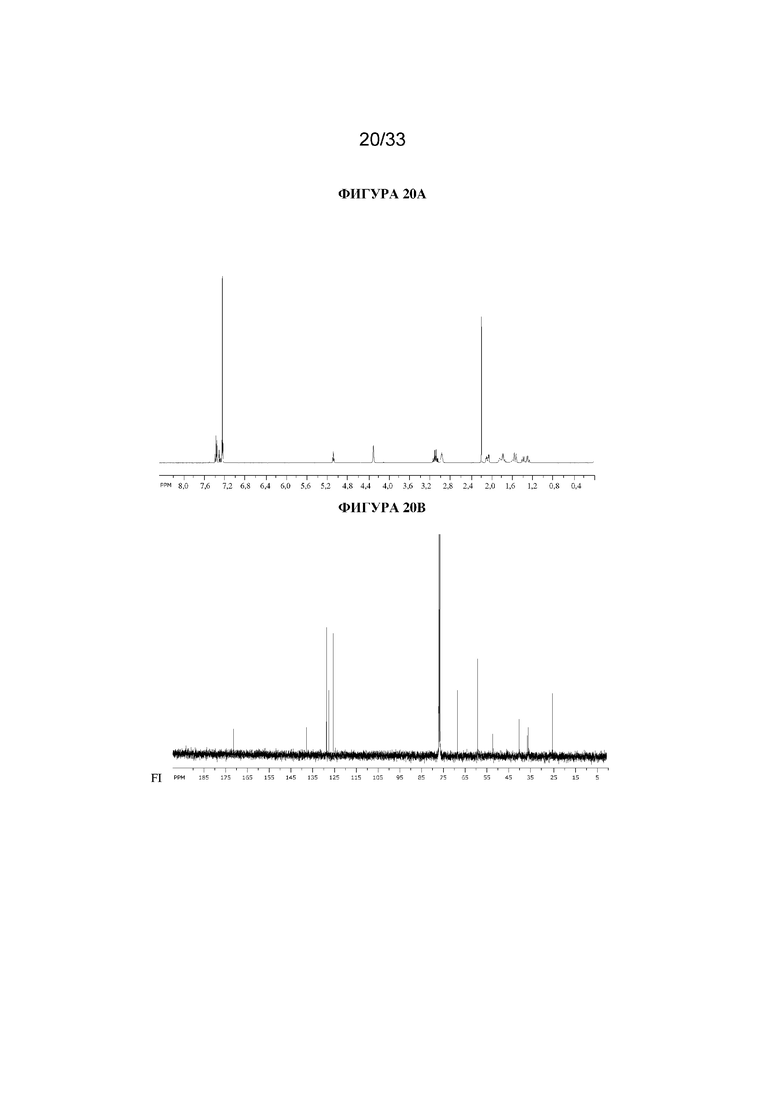
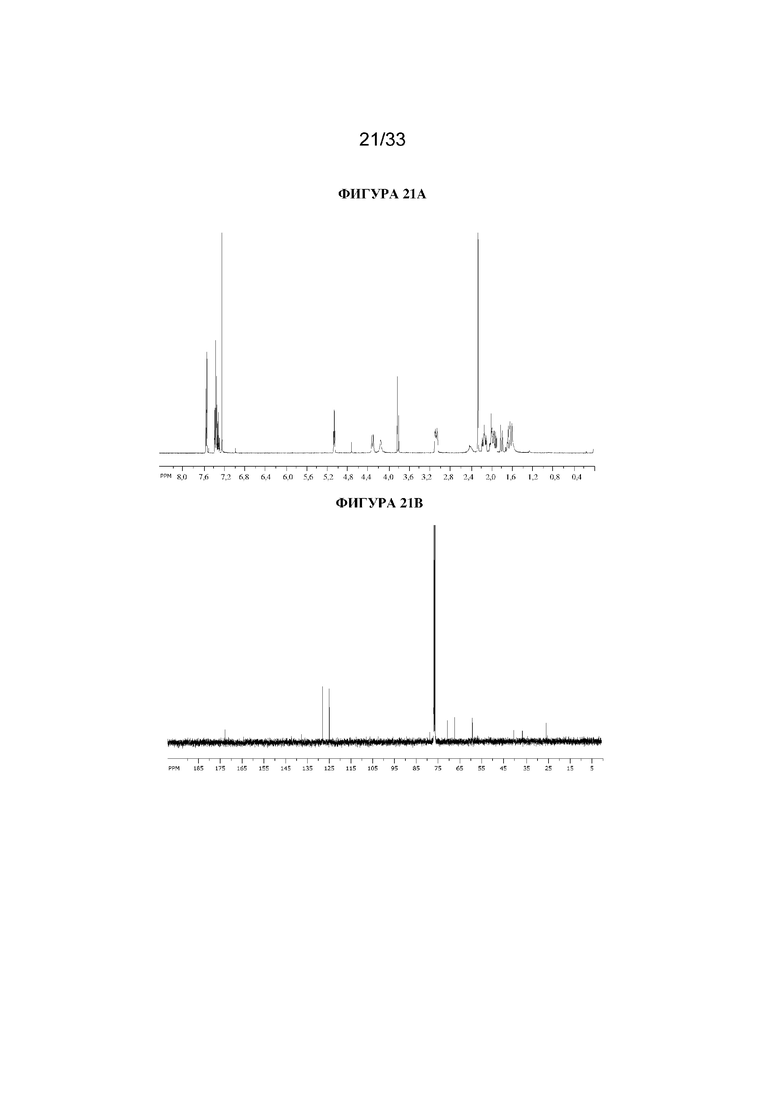

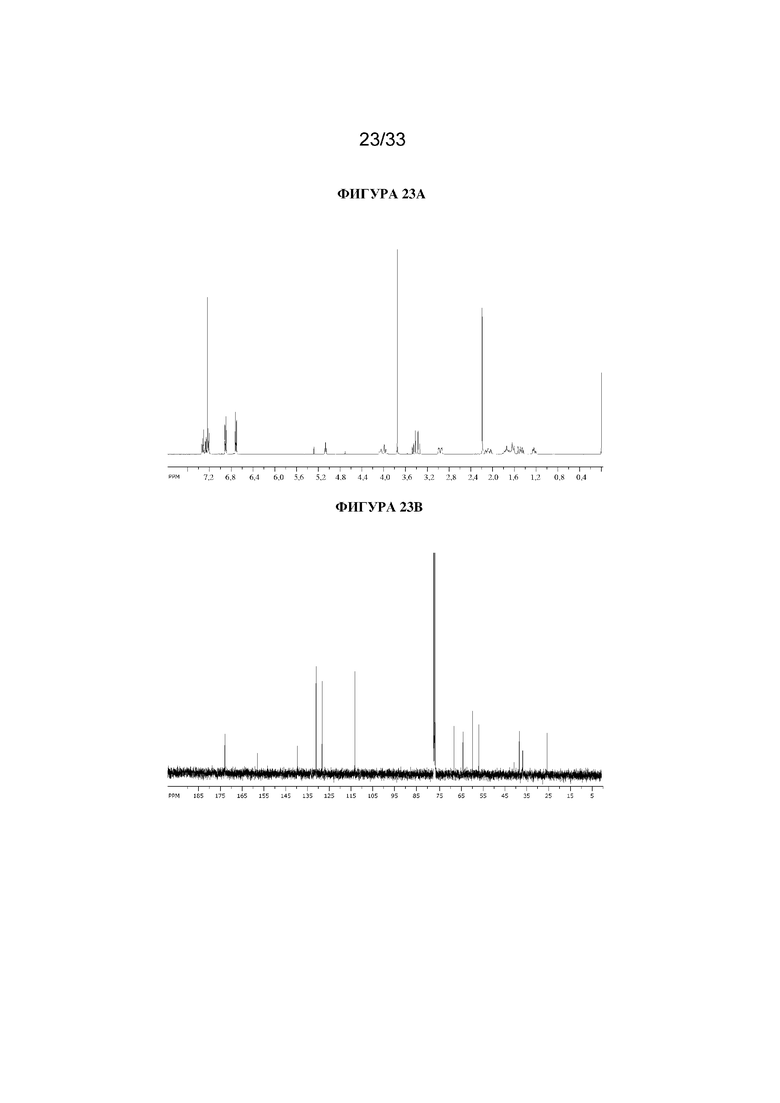
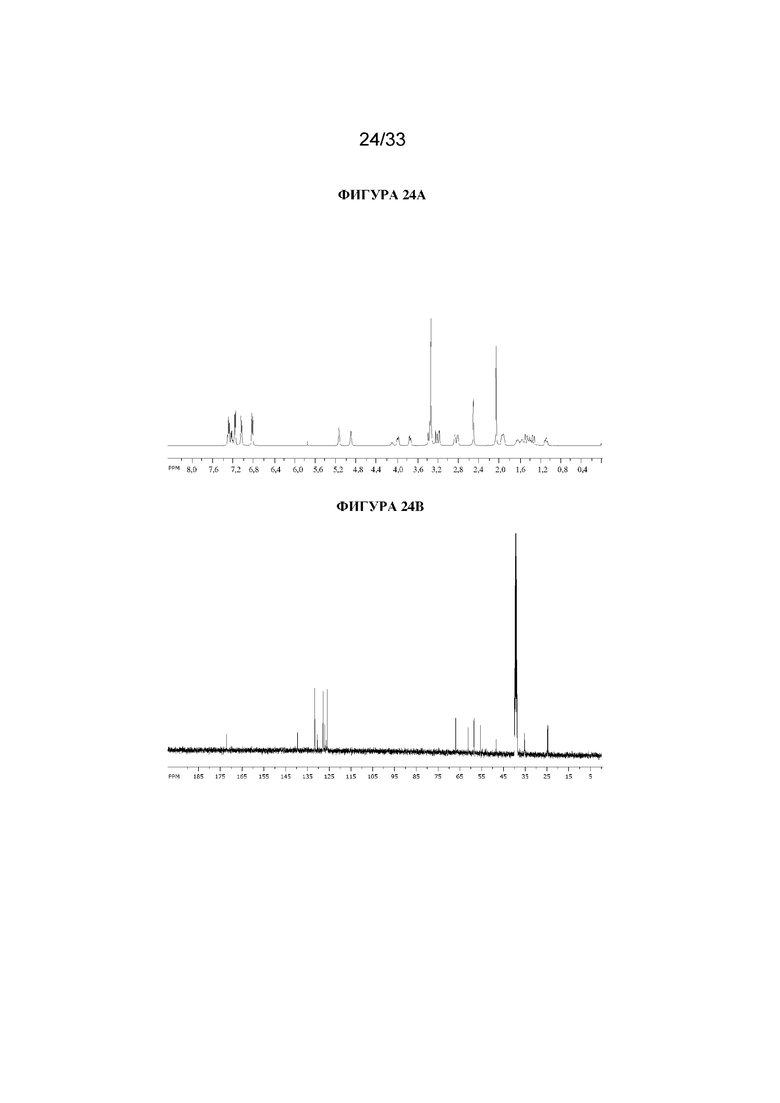
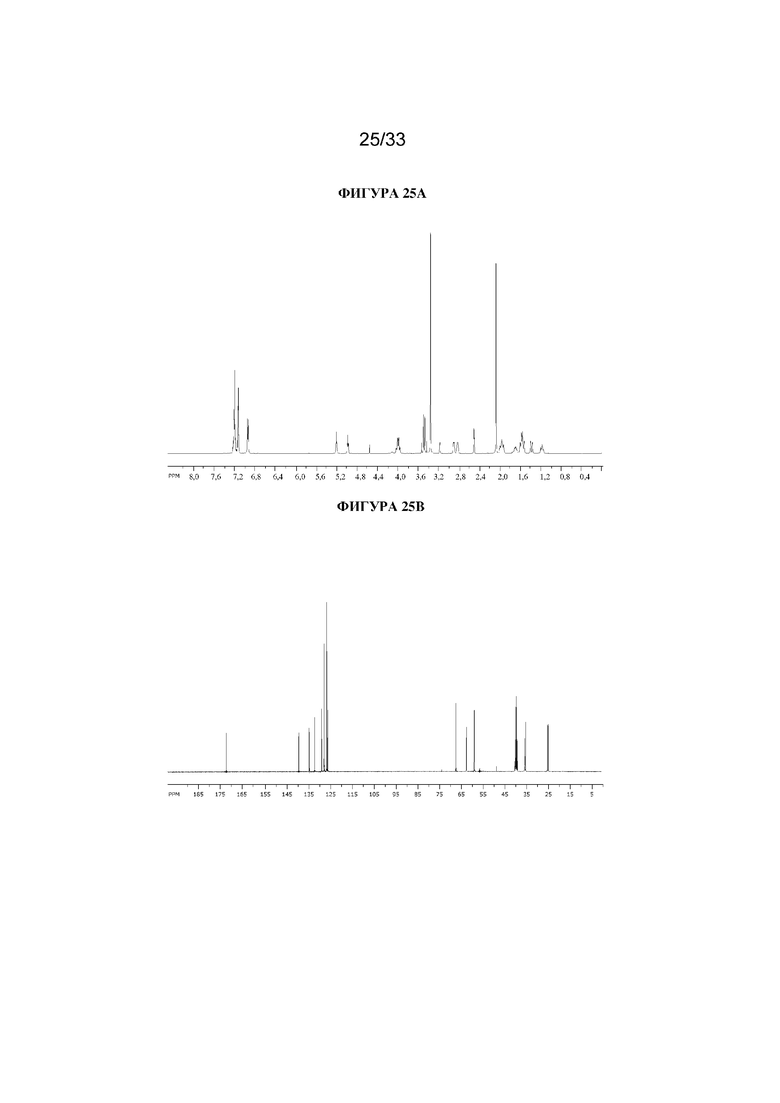
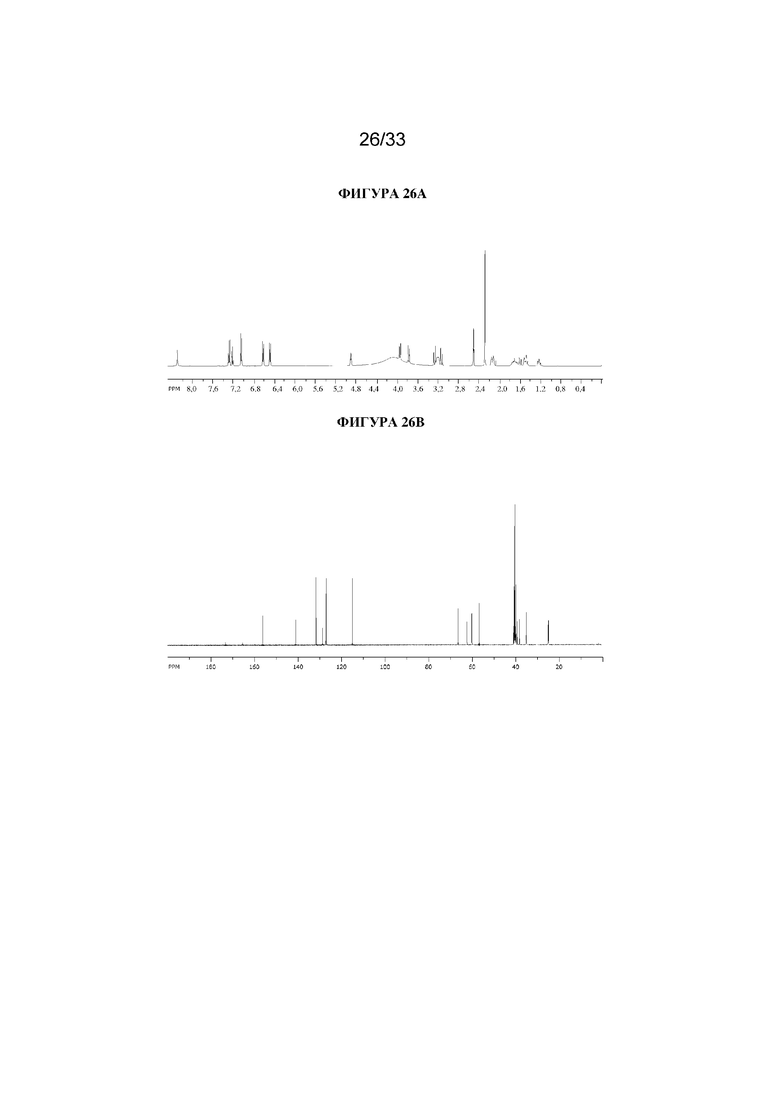
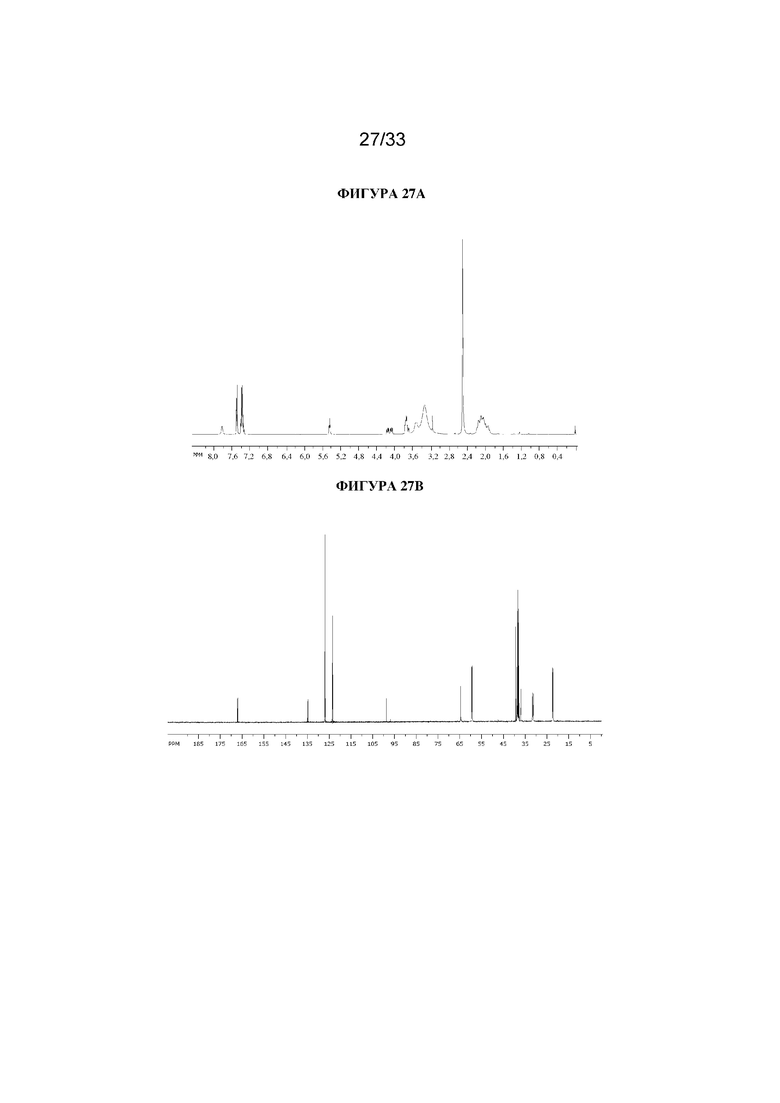
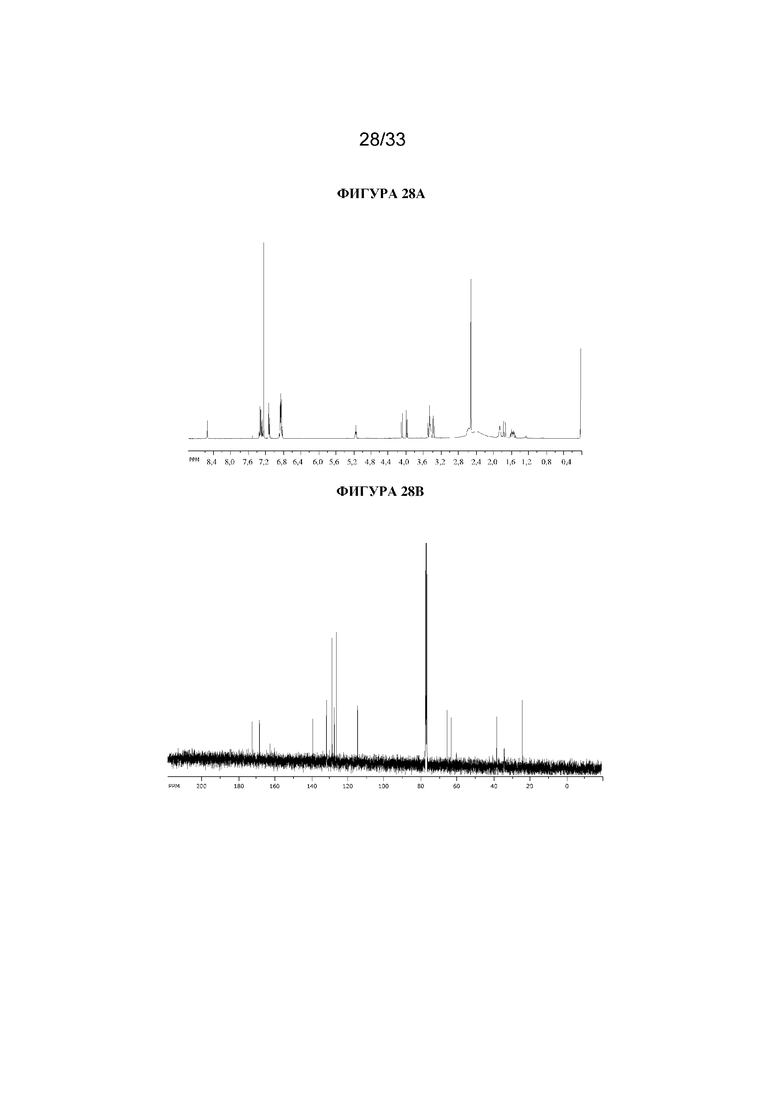
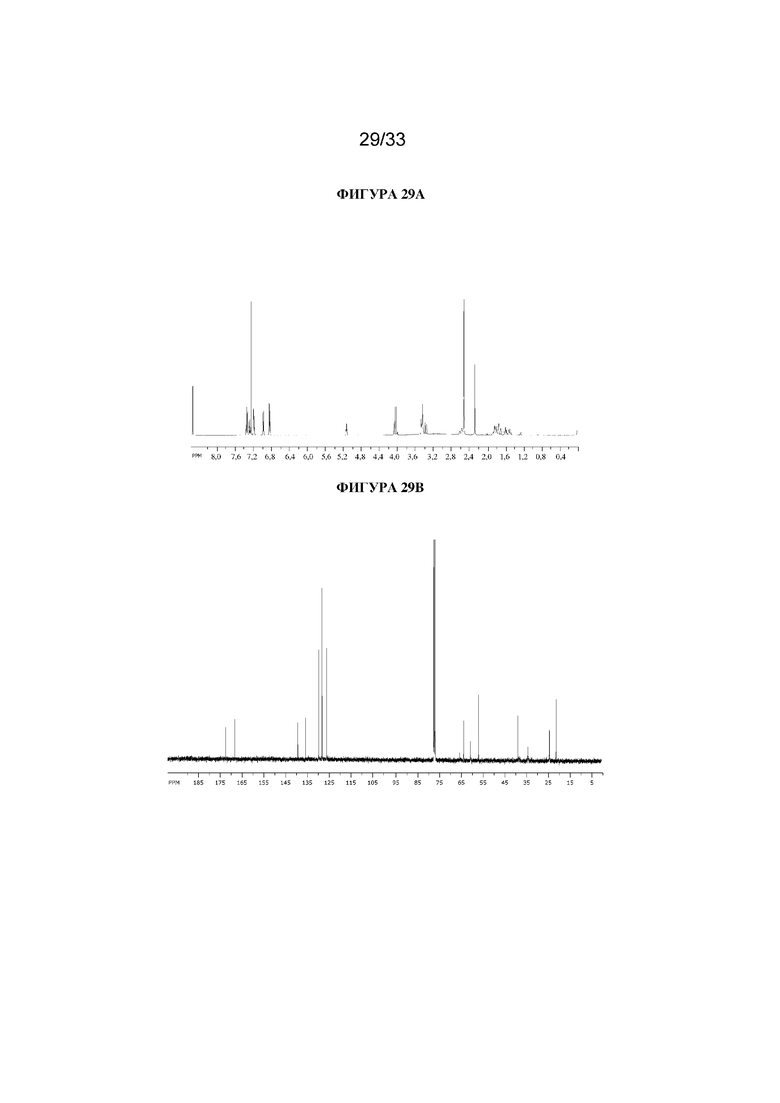
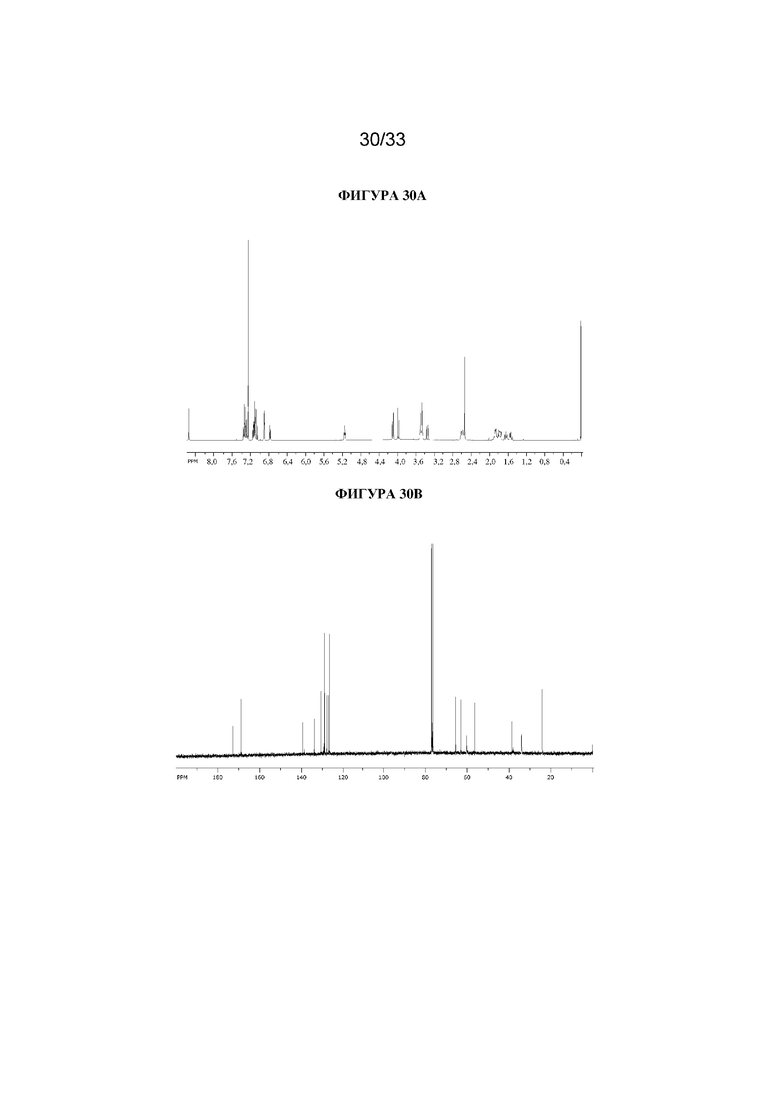
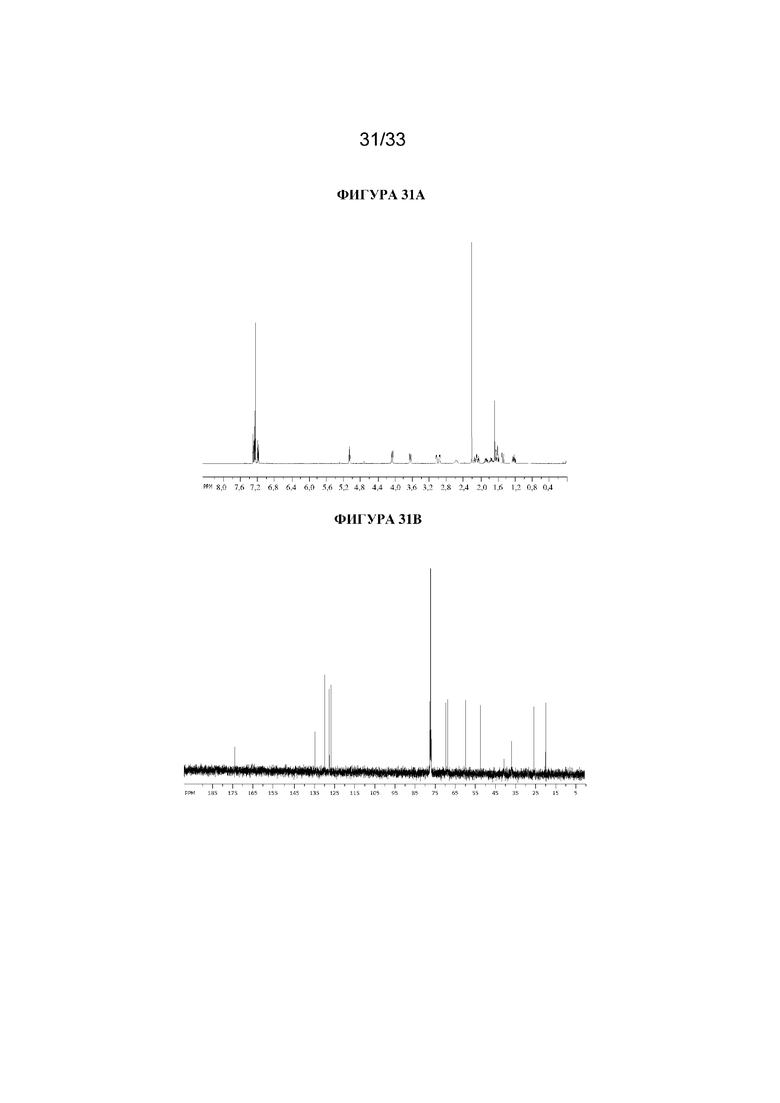
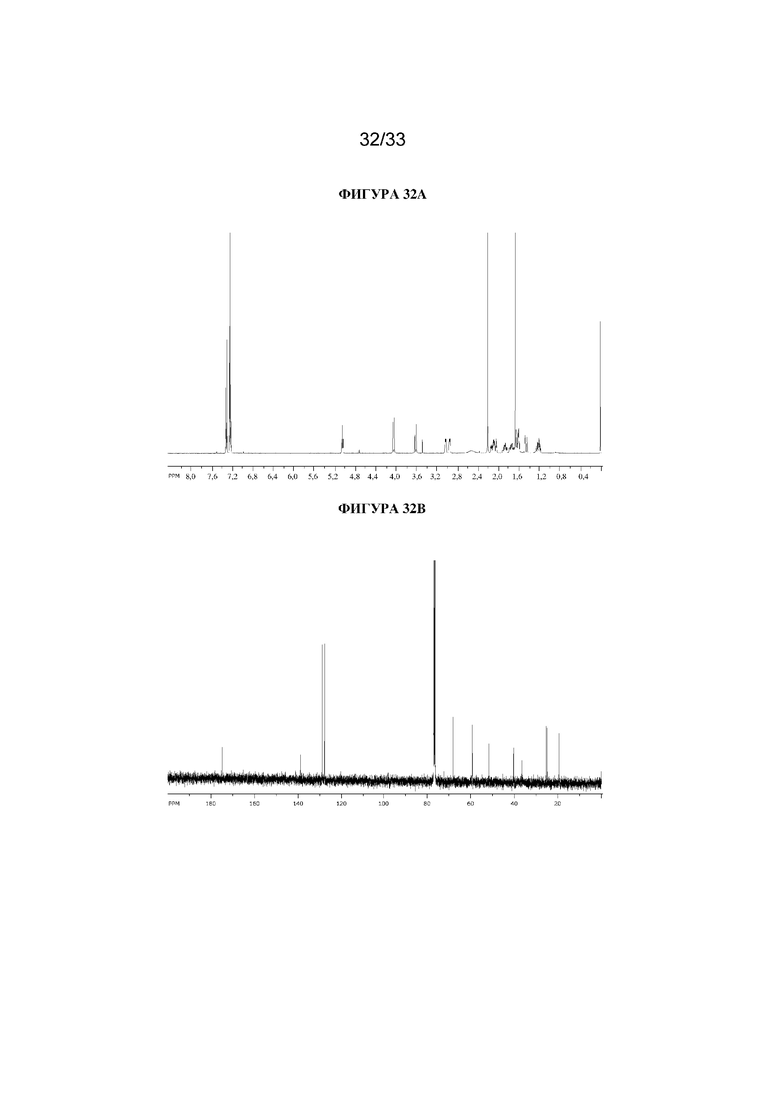
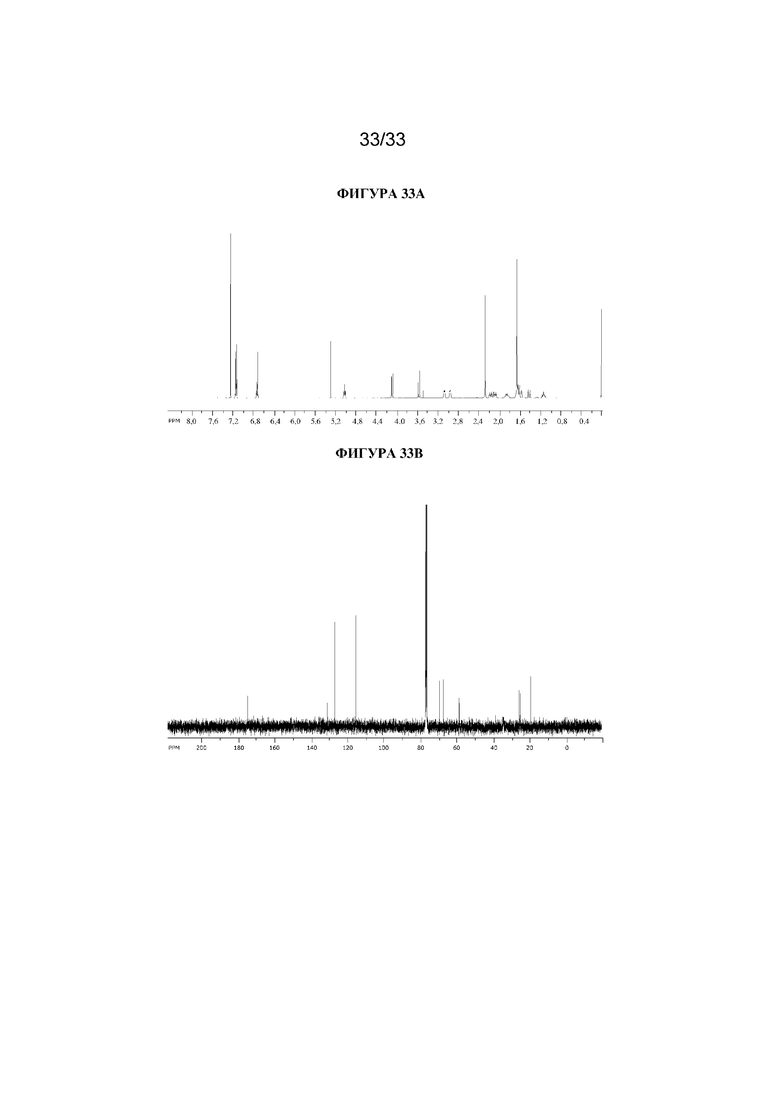
Изобретение относится к области органической химии, а именно к гетероциклическому соединению  или
или 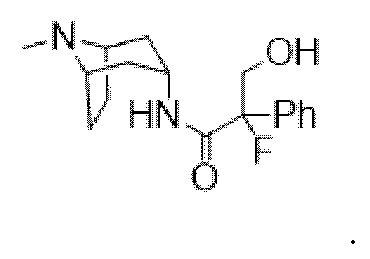 Также изобретение относится к фармацевтической композиции на основе указанного соединения. Технический результат: эффективное модулирование мускариновых рецепторов указанным соединением, полезное для лечения офтальмологических заболеваний. 2 н. и 5 з.п. ф-лы, 52 ил., 1 табл., 25 пр.
Также изобретение относится к фармацевтической композиции на основе указанного соединения. Технический результат: эффективное модулирование мускариновых рецепторов указанным соединением, полезное для лечения офтальмологических заболеваний. 2 н. и 5 з.п. ф-лы, 52 ил., 1 табл., 25 пр.
1. Соединение
 или
или 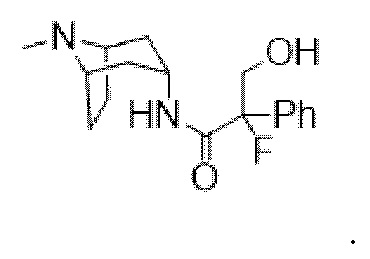
2. Фармацевтическая композиция, применяемая в качестве модулятора мускариновых рецепторов, содержащая: (1) соединение по п. 1 и/или его фармацевтически приемлемую соль; и (2) один или несколько фармацевтически приемлемых носителей.
3. Композиция по п. 2, где указанная композиция представляет собой офтальмологически совместимую композицию.
4. Композиция по п. 2 или 3, где указанная композиция содержит терапевтически эффективное количество указанного соединения и/или указанной его фармацевтически приемлемой соли.
5. Композиция по п. 2 или 3, где указанная композиция содержит от 0,01 до 10,0 процентов по весу указанного соединения и/или указанной фармацевтически приемлемой соли.
6. Композиция по любому из пп. 2-5, где указанная композиция содержит: от 0,01 процента по вес/объем до 5 процентов по вес/объем указанного соединения и/или указанной фармацевтически приемлемой соли; или от 0,1 до 5,0 процентов по весу указанного соединения и/или указанной фармацевтически приемлемой соли.
7. Композиция по любому из пп. 2-6, где указанная композиция представляет собой композицию для местного применения.
| Обратный клапан | 1977 |
|
SU979680A1 |
| Способ усиления бетонных элементов,поврежденных трещинами | 1983 |
|
SU1176049A2 |
| US 5223528 A, 29.06.1993 | |||
| TANG и др., "Human erythrocyte as a model for investigating muscarinic agonists and antagonists", 1991, 22(3), с.485-490 | |||
| DEI S | |||
| и др., "Differential analgesic activity of the enantiomers of atropine derivatives does not correlate with their muscarinic subtype | |||

