Предпосылки изобретения
ALK–2, также известный как рецептор активина A I типа (ACVR1) или как рецептор R1 (SKR1) серин–треониновых протеинкиназ, представляет собой протеинкиназу, которая у людей кодируется геном ACVR1.
ALK–2 представляет собой рецептор BMP I типа, который широко экспрессируется. Он содержит внеклеточный домен, связывающий лиганд, и регулируемый внутриклеточный серин/треонинкиназный домен, оба из которых необходимы для передачи сигнала.
Костные морфогенетические белки (BMP) представляют собой многофункциональные факторы роста, которые являются представителями суперсемейства трансформирующих факторов роста β (TGFβ). Передача сигнала BMP играет важную роль в развитии сердца, нервной системы и хрящей, а также при формировании костей в постнатальный период. BMP эктопически индуцируют формирование эндохондральных костей и играют крайне важную роль в морфогенезе скелета и суставов (Urist, Science 110:893–899 (1965); Olsen et al, Annu. Rev. Cell Dev. Biol. 16:191–220 (2000); Kronenberg, Nature 423:332–336 (2003); Thomas et al, Nat. Genet. 12:315–317 (1996); Thomas et al, Nat. Genet. 17:58–64 (1997); Polinkowsky et al, Nat. Genet. 17:18–19 (1997); Storm et al., Nature 368:639–643 (1994); и Wozney, Prog. Growth Factor Res. 1:267–280 (1989)).
Передача сигнала BMP регулируется на множестве уровней, в том числе посредством внеклеточных антагонистов, таких как ноггин (Massague, Nat. Rev. MoI. Cell. Biol. 1:169–178 (2000)). Было выдвинуто предположение, что несвоевременная или нежелательная активация путей передачи сигнала, существенно важных для нормального развития, может стимулировать патологические процессы, такие как спондилоартропатии. Также описаны эффекты передачи сигнала BMP в отношении инициирования и прогрессирования артрита посредством генетического переноса ноггина (Lories et al, J. Clin. Invest., 115, 1571 –1579 (2005)). Физиологические роли передачи сигнала BMP и рецептором BMP при нормальном формировании костей, в том числе развитии скелета и конечностей, были изучены и рассмотрены в Zhao, Genetics 35:43–56 (2003).
Эксперименты с антагонистами BMP демонстрируют, что регуляция сигнальных белков BMP является основным фактором при формировании in vivo костей (Devlin et al., Endocrinology 144:1972–1978 (2003) и Wu et al., J. Clin. Invest., 112: 924 (2003)).
Прогрессирующая оссифицирующая фибродисплазия (FOP) представляет собой редко встречающееся и обусловливающее потерю трудоспособности генетическое нарушение, характеризующееся анатомическими закономерностями врожденных пороков развития больших пальцев стоп и прогрессирующей гетеротопической эндохондральной оссификации. Эктопическая экспрессия BMP4 была выявлена у пациентов, имеющих FOP (Gannon et al., Hum. Pathol. 28:339–343 (1997) и Xu et al, Clin. Genet. 58:291–298 (2000)). Было показано, что пациенты, имеющие FOP, характеризовались активирующими мутациями в ALK–2 (Shore et al., Nat. Genet., 38(5):525–7 (2006)).
Было установлено, что избыточная передача сигнала BMP приводит к ряду состояний, описанных выше. В WO2008033408 и WO2009114180 описаны ингибиторы пути передачи сигнала BMP. Тем не менее, все еще существует постоянная потребность выявления альтернативных путей, с помощью которых передачу сигнала BMP можно регулировать.
Такая потребность может быть удовлетворена путем разработки селективных ингибиторов ALK–2.
Специфические антитела к ALK–2 описаны, например, в WO1994011502 и WO2008030611. Остеогенные белки, которые связываются с ALK–2, описаны в WO2012023113 и WO2012077031.
В WO2007123896 описан способ лечения патологии, ассоциированной с гетеротопической оссификацией, посредством введения siRNA, специфичной в отношении нуклеиновой кислоты, кодирующей мутантный ALK–2.
В WO2014160203 и WO2014138088 описаны ингибиторы пути BMP. В WO 2015152183 описаны ингибиторы ALK–2 для лечения FOP. Ингибиторы ALK–2 также описаны в WO2014151871.
Краткое описание изобретения
Существует постоянная потребность в разработке новых ингибиторов ALK–2, которые являются хорошими кандидатными лекарственными средствами. Для таких кандидатов могут быть найдены варианты применения inter alia в лечении прогрессирующей оссифицирующей фибродисплазии (FOP) или ненаследственной гетеротопической оссификации (HO).
В настоящем изобретении предусмотрены соединения, их фармацевтически приемлемые соли, фармацевтические композиции, содержащие их, при этом соединения представляют собой ингибиторы ALK–2. В настоящем изобретении дополнительно предусмотрены способы лечения, предупреждения или снижения тяжести прогрессирующей оссифицирующей фибродисплазии (FOP) или ненаследственной гетеротопической оссификации (HO), включающие введение нуждающемуся в этом субъекту эффективного количества ингибитора ALK–2.
В данном документе описаны различные варианты осуществления настоящего изобретения.
В определенных аспектах в данном документе предусмотрено соединение формулы (I) в свободной форме или в форме фармацевтически приемлемой соли
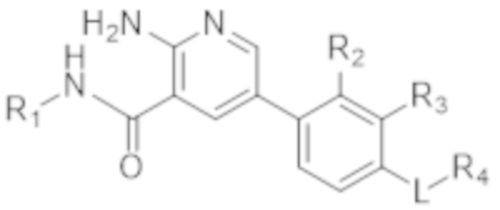
(I).
В другом варианте осуществления в настоящем изобретении в настоящем изобретении предусмотрена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I), определенного в данном документе, в свободной форме или в форме фармацевтически приемлемой соли или ее подформул (Ia), (II), (IIa), (IIb) и один или несколько фармацевтически приемлемых носителей.
В дополнительном аспекте настоящее изобретение относится к способу подавления активности рецептора ALK–2 у субъекта, где способ включает введение субъекту терапевтически эффективного количества соединения формулы (I), определенного в данном документе, в свободной форме или в форме фармацевтически приемлемой соли или ее подформул (Ia), (II), (IIa), (IIb).
В еще одном аспекте настоящее изобретение относится к способу лечения нарушения или заболевания, выбранного из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии, включающему введение субъекту терапевтически эффективного количества соединения формулы (I), определенного в данном документе, в свободной форме или в форме фармацевтически приемлемой соли или ее подформул (Ia), (II), (IIa), (IIb).
Соединения согласно настоящему изобретению демонстрируют приемлемые фармакокинетические свойства, являются нетоксичными и демонстрируют незначительное количество побочных эффектов. В частности, соединения согласно настоящему изобретению являются селективными ингибиторами ALK–2 по сравнению с другими рецепторами. Кроме того, идеальное кандидатное лекарственное средство будет находиться в форме, которая является стабильной, негигроскопичной и легкой для составления.
Фигуры
На фигуре 1 показан количественный анализ с применением μCT гетеротопической кости, сформированной в правой задней конечности мышей через 6 недель после индукции обострения FOP с помощью аденовируса/CTX, и показан эффект лечения с применением соединения A в течение 6 недель в отношении образования HO. Среднее значение +/– SEM. *: p < 0,05, **: p < 0,01, ***: p < 0,01 в сравнении с обработкой средой–носителем; односторонний ANOVA с использованием апостериорного критерия Даннетта.
На фигуре 2 показан количественный анализ с использованием μCT гетеротопической кости, сформированной в правой задней конечности мышей через 8 недель после индукции обострения FOP с помощью аденовируса/CTX, и показан эффект лечения с применением соединения A в течение 6 недель в отношении образования HO. Среднее значение +/– SEM. *: p < 0,05, **: p < 0,01, ***: p < 0,01 в сравнении с обработкой средой–носителем; t–критерий Стьюдента.
На фигуре 3 показана порошковая рентгеновская дифрактограмма кристаллической свободной формы кристаллов модификации HA соединения A.
На фигуре 4 показана диаграмма дифференциальной сканирующей калориметрии кристаллов кристаллической свободной формы модификации HA соединения A.
На фигуре 5 показана диаграмма термогравиметрического анализа кристаллической свободной формы модификации HA соединения A.
На фигуре 6 показана диаграмма Фурье–спектроскопии в инфракрасной области кристаллической свободной формы модификации HA соединения A.
На фигуре 7 показана порошковая рентгеновская дифрактограмма кристаллической свободной формы модификации A соединения A.
На фигуре 8 показана диаграмма дифференциальной сканирующей калориметрии кристаллической свободной формы модификации A соединения A.
На фигуре 9 показана диаграмма термогравиметрического анализа кристаллической свободной формы модификации A соединения A.
На фигуре 10 показана диаграмма Фурье–спектроскопии в инфракрасной области кристаллической свободной формы модификации A соединения A.
На фигуре 11 показана порошковая рентгеновская дифрактограмма кристаллической соли фумаровой кислоты модификации HA соединения A.
На фигуре 12 показана диаграмма дифференциальной сканирующей калориметрии кристаллической соли фумаровой кислоты модификации HA соединения A.
На фигуре 13 показана диаграмма термогравиметрического анализа кристаллической соли фумаровой кислоты модификации HA соединения A.
На фигуре 14 показана диаграмма Фурье–спектроскопии в инфракрасной области кристаллической соли фумаровой кислоты модификации HA соединения A.
На фигуре 15 показана диаграмма динамической сорбции паров кристаллической соли фумаровой кислоты модификации HA соединения A при 25 градусах C, способ при относительной влажности (RH) 50%–0%–90%.
На фигуре 16 показана диаграмма динамической сорбции паров кристаллической соли фумаровой кислоты модификации HA соединения A при 40 градусах C, способ при относительной влажности (RH) 50%–0%–90%.
На фигуре 17 показана порошковая рентгеновская дифрактограмма кристаллической соли фосфорной кислоты соединения A.
На фигуре 18 показана порошковая рентгеновская дифрактограмма ангидрида кристаллической свободной формы (пример 89 C).
На фигуре 19 показана порошковая рентгеновская дифрактограмма тригидрата кристаллической свободной формы (пример 89 D)).
Подробное описание изобретения
В частности, настоящее изобретение относится к соединению формулы (I) в свободной форме или в форме приемлемой соли,
(I),
где
L представляет собой связь, (CH2)n, –CH(CH3)–, –O–(CH2)n–, –C(O)–, –C(O)–NH–(CH2)n–;
n равняется 1, 2, 3;
R1 выбран из C3–C7циклоалкила, необязательно замещенного один раз или более чем один раз заместителем, независимо выбранным из гидроксила, галогена, C1–C3алкила; мостикового C5–C10циклоалкила, необязательно замещенного один раз или более чем один раз заместителем, независимо выбранным из гидроксила, гидроксиC1–C3алкила;
R2 и R3 независимо выбраны из H, галогена, C1–C3алкила;
R4 представляет собой N–содержащее гетероциклическое неароматическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7;
R7 независимо выбран из C1–C3алкила, гидроксиC1–C3алкила, галогенC1–C3алкила, C1–C3алкоксиC1–C3алкила, галогенC1–C3алкоксиC1–C3алкила, C2–C4алкинила, цианоC1–C3алкила, (CH2)m–R8;
m равняется 0, 1, 2 или 3;
R8 выбран из:
– 4–, 5– или 6–членного насыщенного или ненасыщенного неароматического гетероциклического кольца, содержащего один или несколько гетероатомов, выбранных из N, O или S, при этом указанное кольцо необязательно замещено один раз или более чем один раз заместителем, независимо выбранным из оксо, SO2C1–C3алкила, галогенC1–C3алкила; или
– C3–C6циклоалкила, необязательно замещенного один раз или более чем один раз галогеном.
Если не указано иное, термин "соединения согласно настоящему изобретению" или "соединения согласно данному изобретению" относится к соединениям формулы (I), (Ia), (II), (IIa), (IIb) и их солям, а также всем стереоизомерам (включая диастереоизомеры и энантиомеры), ротамерам, таутомерам и меченным изотопом соединениям (включая замещения дейтерием), а также изначально образованным фрагментам.
Используемый в данном документе термин "C1–C3алкил" относится к радикалу с линейной или разветвленной углеводородной цепью, состоящему только из атомов углерода и водорода, не содержащему ненасыщенных связей, имеющему от одного до трех атомов углерода, и который присоединен к остальной части молекулы посредством одинарной связи. Примеры C1–C3алкила включают метил, этил, н–пропил, 1–метилэтил (изопропил).
Используемый в данном документе термин "гидроксиC1–C3алкил" относится к радикалу формулы –Ra–OH, где Ra представляет собой C1–C3алкил, определенный выше.
Используемый в данном документе термин "C3–C7циклоалкил" относится к насыщенным моноциклическим углеводородным группам, состоящим из 3–7 атомов углерода. Примеры C3–C7циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил.
Используемый в данном документе термин "C1–C3алкокси" относится к радикалу формулы –ORa, где Ra представляет собой C1–C3алкильный радикал, в целом определенный выше. Примеры C1–C3алкокси включают без ограничения метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, пентокси и гексокси.
Используемый в данном документе термин "C1–C3алкоксиC1–C3алкил" относится к радикалу формулы –Rb–O–Ra, где Ra представляет собой C1–C3алкильный радикал, и Rb представляет собой C1–C3алкильный радикал, определенный выше. Атом кислорода может быть связан с любым атомом углерода в любом алкильном радикале. Примеры C1–C3алкоксиC1–C3алкила включают, например, метоксиметил, метоксиэтил, этоксиэтил.
"Галоген" или "галогено" относится к брому, хлору, фтору или йоду.
Используемый в данном документе термин "галогенC1–C3алкил" или "галогеноC1–C3алкил" относится к C1–C3алкильному радикалу, определенному выше, замещенному одним или несколькими галогеновыми радикалами, определенными выше. Примеры галогенC1–C3алкила включают, например, трифторметил, дифторметил, фторметил, трихлорметил, 2,2,2–трифторэтил, 1–фторметил–2–фторэтил, 3–бром–2–фторпропил и 1–бромметил–2–бромэтил.
Используемый в данном документе термин "галогенC1–C3алкоксиC1–C3алкил" относится к радикалу формулы –Rb–O–Ra, где Ra представляет собой C1–C3алкильный радикал, и Rb представляет собой галогенC1–C3алкильный радикал, определенный выше.
Используемый в данном документе термин "N–содержащее гетероциклическое неароматическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S" по отношению к R4 относится к насыщенному или ненасыщенному неароматическому кольцу или кольцевой системе, которая представляет собой 4–, 5–, 6– или 7–членное моноциклическое кольцо, содержащее 1, 2 или 3 гетероатома, выбранных из O, S и N, где по меньшей мере один гетероатом представляет собой N, 6–, 7–, 8–, 9–, 10–, 11– или 12–членную бициклическую кольцевую систему, содержащую 1, 2, 3, 4 или 5 гетероатомов, выбранных из O, S и N, где по меньшей мере один гетероатом представляет собой N, или 10–, 11–, 12–, 13–, 14– или 15–членную трициклическую кольцевую систему, содержащую 1, 2, 3, 4, 5, 6 или 7 гетероатомов, выбранных из O, S и N, где по меньшей мере один гетероатом представляет собой N, где N и S также могут быть необязательно окислены до различных степеней окисления. N–содержащее гетероциклическое неароматическое кольцо может быть присоединено посредством гетероатома или атома углерода. N–содержащее гетероциклическое неароматическое кольцо может включать конденсированные или мостиковые кольца, а также спироциклические кольца. В предпочтительном варианте осуществления "N–содержащее гетероциклическое неароматическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S" представляет собой N–содержащее 5–членное насыщенное моноциклическое кольцо, N–содержащее 6–членное насыщенное моноциклическое или бициклическое кольцо, N–содержащее 7–членное насыщенное спироциклическое кольцо. Примеры N–содержащего гетероциклического неароматического кольца включают морфолин, пиперазин, пиперидин, имидазолидин, имидазолин, пирролин, пирролидин, тиоморфолин, 3–азабицикло[3.1.0]гексан.
Используемый в данном документе термин "N–содержащее 5–членное гетероциклическое неароматическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S" по отношению к R4 включает, в качестве примеров, пирролидин, 2,3–дигидропиррол, оксазолидин, имидазолидин. В предпочтительном варианте осуществления он относится к пирролидину. Кольцо присоединено к остальной части молекулы посредством атома углерода в кольце.
Используемый в данном документе термин "N–содержащее 6–членное гетероциклическое неароматическое моноциклическое или бициклическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S" по отношению к R4 включает, в качестве примеров, морфолин, тиоморфолин, пиперидин, пиперазин, 3–азабицикло[3.1.0]гексан. В предпочтительном варианте осуществления он относится к 3–азабицикло[3.1.0]гексану. Кольцо присоединено к остальной части молекулы посредством атома углерода в кольце.
Используемый в данном документе термин "5–членное кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из N, O или S" по отношению к вариантам осуществления, где R5 и R6 вместе с атомом N, к которому они присоединены, образуют указанное кольцо, включает, в качестве примеров, пирролидин, 2,3–дигидропиррол, оксазолидин, имидазолидин. В предпочтительном варианте осуществления он относится к пирролидину.
Используемый в данном документе термин "6–членное кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из N, O или S" по отношению к вариантам осуществления, где R5 и R6 вместе с атомом N, к которому они присоединены, образуют указанное кольцо, включает, в качестве примеров, пиперидинил, морфолинил, пиперазинил.
Используемый в данном документе термин "7–членный спироцикл, необязательно содержащий один дополнительный гетероатом, выбранный из N, O или S" по отношению к вариантам осуществления, где R5 и R6 вместе с атомом N, к которому они присоединены, образуют указанное кольцо, включает, в качестве примеров, 2–азаспиро[3.3]гептан, 2–окса–6–азаспиро[3.3]гептан.
Используемый в данном документе термин "мостиковый C5–C10циклоалкил" относится к насыщенной бициклической или трициклической кольцевой системе, содержащей по меньшей мере один мостик. Примеры мостикового C5–C10циклоалкила включают, например, бицикло[1.1.1]пентан, бицикло[2.2.2]октан, бицикло[2.2.1]гептан, трицикло–[3.3.1.13,7]декан.
Используемый в данном документе термин "C2–C4алкинил" относится к группе, представляющей собой радикал с линейной цепью, состоящей только из атомов углерода и водорода, содержащей по меньшей мере одну тройную связь, имеющей от двух до четырех атомов углерода, и которая присоединена к остальной части молекулы посредством одинарной связи. Примеры C2–C4алкинила включают, например, этинил, проп–1–инил, бут–1–инил.
Используемый в данном документе термин "цианоC1–C3алкил" относится к радикалу формулы –Ra–CN, где Ra представляет собой C1–C3алкил, определенный выше.
Используемый в данном документе термин "4–, 5– или 6–членное насыщенное или ненасыщенное неароматическое гетероциклическое кольцо, содержащее один или несколько гетероатомов, выбранных из N, O или S", включает, в качестве примеров, тетрагидропиран, морфолин, пиперидин, оксетан. В предпочтительном варианте осуществления он представляет собой тетрагидропиран.
Используемый в данном документе термин "ALK–2" относится к рецептору активина A I типа (ACVRI), также известному как ACVRLK2; SKR1; ACVR1A; рецептор активина I типа; активиновая рецепторподобная киназа 2; рецептор R1 серин/треонинпротеинкиназ; рецептор I типа суперсемейства TGF–B; ACTRI; TSRI; киназа 2, подобная рецептору активина A II типа; рецептор активина 1 типа; гидроксиалкилпротеинкиназа; ACTR–I; TSR–I.
В данном документе описаны различные варианты осуществления настоящего изобретения. Необходимо признать, что характеристики, описанные в каждом варианте осуществления, можно объединять с другими описанными характеристиками с получением дополнительных вариантов осуществления настоящего изобретения.
Вариант осуществления 1. Соединение формулы (I) в свободной форме или в форме фармацевтически приемлемой соли,
(I),
где
L представляет собой связь, (CH2)n, –CH(CH3)–, –O–(CH2)n–, –C(O)–, –C(O)–NH–(CH2)n–;
n равняется 1, 2, 3;
R1 выбран из C3–C7циклоалкила, необязательно замещенного один раз или более чем один раз заместителем, независимо выбранным из гидроксила, галогена, C1–C3алкила; мостикового C5–C10циклоалкила, необязательно замещенного один раз или более чем один раз заместителем, независимо выбранным из гидроксила, гидроксиC1–C3алкила;
R2 и R3 независимо выбраны из H, галогена, C1–C3алкила;
R4 представляет собой N–содержащее гетероциклическое неароматическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7;
R7 независимо выбран из C1–C3алкила, гидроксиC1–C3алкила, галогенC1–C3алкила, C1–C3алкоксиC1–C3алкила, галогенC1–C3алкоксиC1–C3алкила, C2–C4алкинила, цианоC1–C3алкила, (CH2)m–R8;
m равняется 0, 1, 2 или 3;
R8 выбран из:
– 4–, 5– или 6–членного насыщенного или ненасыщенного неароматического гетероциклического кольца, содержащего один или несколько гетероатомов, выбранных из N, O или S, при этом указанное кольцо необязательно замещено один раз или более чем один раз заместителем, независимо выбранным из оксо, SO2C1–C3алкила, галогенC1–C3алкила; или
– C3–C6циклоалкила, необязательно замещенного один раз или более чем один раз галогеном.
Вариант осуществления 2. Соединение согласно варианту осуществления 1 формулы (Ia) в свободной форме или в форме фармацевтически приемлемой соли,
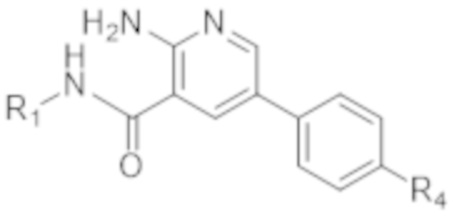
(Ia).
Вариант осуществления 3. Соединение согласно любому из вариантов осуществления 1 или 2 в свободной форме или в форме фармацевтически приемлемой соли, где R4 представляет собой NR5R6, где R5 и R6 вместе с атомом N, к которому они присоединены, образуют 5–членное кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из N, O или S, необязательно замещенный один раз или более чем один раз R7.
Вариант осуществления 4. Соединение согласно любому из вариантов осуществления 1 или 2 в свободной форме или в форме фармацевтически приемлемой соли, где R4 представляет собой NR5R6, где R5 и R6 вместе с атомом N, к которому они присоединены, образуют 6–членное кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из N, O или S, необязательно замещенный один раз или более чем один раз R7.
Вариант осуществления 5. Соединение согласно любому из вариантов осуществления 1 или 2 в свободной форме или в форме фармацевтически приемлемой соли, где R4 представляет собой NR5R6, где R5 и R6 вместе с атомом N, к которому они присоединены, образуют 7–членный спироцикл, необязательно содержащий один дополнительный гетероатом, выбранный из N, O или S, необязательно замещенный один раз или более чем один раз R7.
Вариант осуществления 6. Соединение согласно любому из вариантов осуществления 1 или 2 в свободной форме или в форме фармацевтически приемлемой соли, где R4 представляет собой N–содержащее 5–членное гетероциклическое неароматическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7 и где указанное кольцо присоединено к остальной части молекулы посредством атома углерода в кольце.
Вариант осуществления 7. Соединение согласно любому из вариантов осуществления 1 или 2 в свободной форме или в форме фармацевтически приемлемой соли, где R4 представляет собой N–содержащее 6–членное гетероциклическое неароматическое моноциклическое или бициклическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7 и где указанное кольцо присоединено к остальной части молекулы посредством атома углерода в кольце.
Вариант осуществления 8. Соединение согласно любому из вариантов осуществления 1, 2 или 7 формулы (II) в свободной форме или в форме фармацевтически приемлемой соли,
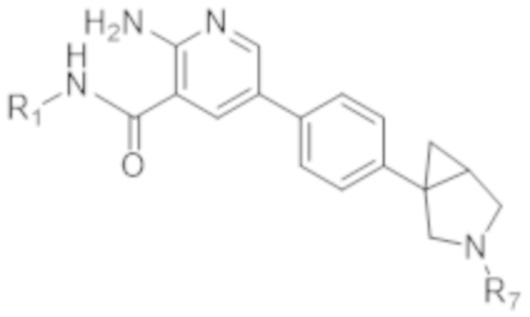
(II),
где R7 независимо выбран из C1–C3алкила, гидроксиC1–C3алкила, галогенC1–C3алкила, C1–C3алкоксиC1–C3алкила, галогенC1–C3алкоксиC1–C3алкила, C2–C4алкинила, цианоC1–C3алкила, (CH2)m–R8;
m равняется 0, 1, 2 или 3.
Вариант осуществления 9. Соединение согласно варианту осуществления 8 формулы (IIa) в свободной форме или в форме фармацевтически приемлемой соли,
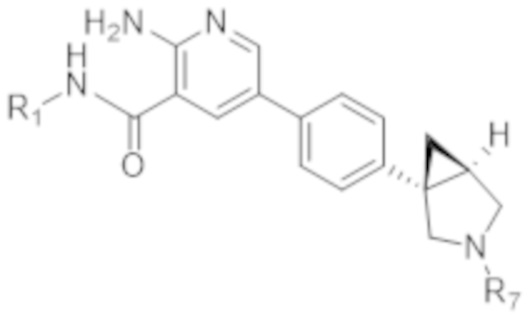
(IIa).
Вариант осуществления 10. Соединение согласно варианту осуществления 8 формулы (IIb) в свободной форме или в форме фармацевтически приемлемой соли,
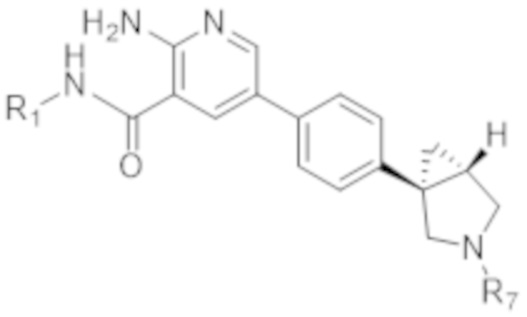
(IIb).
Вариант осуществления 11. Соединение согласно любому из предыдущих вариантов осуществления в свободной форме или в форме фармацевтически приемлемой соли, где
R7 представляет собой (CH2)m–R8;
R8 представляет собой 6–членное гетероциклическое кольцо, содержащее один или несколько гетероатомов, выбранных из N, O или S, при этом указанное кольцо необязательно замещено один раз или более чем один раз заместителем, независимо выбранным из оксо, SO2C1–C3алкила, галогенC1–C3алкила;
m равняется 0, 1, 2 или 3.
Вариант осуществления 12. Соединение согласно варианту осуществления 11 в свободной форме или в форме фармацевтически приемлемой соли, где R8 представляет собой незамещенный тетрагидропиран, и m равняется 0.
Вариант осуществления 13. Соединение согласно варианту осуществления 11 в свободной форме или в форме фармацевтически приемлемой соли, где R8 представляет собой незамещенный морфолин, и m равняется 2 или 3.
Вариант осуществления 14. Соединение в соответствии с любым из предыдущих вариантов осуществления в свободной форме или в форме фармацевтически приемлемой соли, где R1 представляет собой циклогексил, необязательно замещенный один раз или более чем один раз заместителем, независимо выбранным из гидроксила, галогена, C1–C3алкила.
Вариант осуществления 15. Соединение согласно любому из предыдущих вариантов осуществления в свободной форме или в форме фармацевтически приемлемой соли, где R1 представляет собой
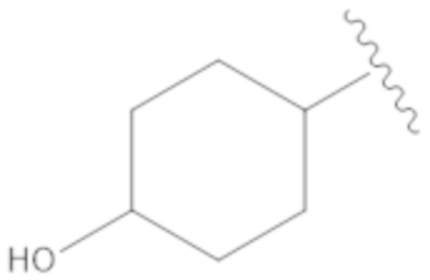 .
.
Вариант осуществления 16. Соединение согласно любому из предыдущих вариантов осуществления в свободной форме или в форме фармацевтически приемлемой соли, где R1 представляет собой
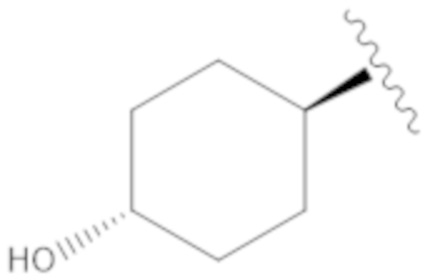 .
.
Вариант осуществления 17. Соединение согласно любому из вариантов осуществления 1–13 в свободной форме или в форме фармацевтически приемлемой соли, где R1 представляет собой бицикло[2.2.2]октанил, замещенный в одном случае гидроксилом.
Вариант осуществления 18. Соединение согласно варианту осуществления 17 в свободной форме или в форме фармацевтически приемлемой соли, где R1 представляет собой 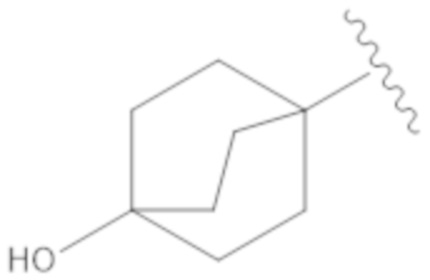 .
.
Вариант осуществления 19. Соединение согласно варианту осуществления, любому из вариантов осуществления 1–13 в свободной форме или в форме фармацевтически приемлемой соли, где R1 представляет собой бицикло[1.1.1]пентанил, замещенный гидроксиметилом.
Вариант осуществления 20. Соединение согласно варианту осуществления 19 в свободной форме или в форме фармацевтически приемлемой соли, где R1 представляет собой
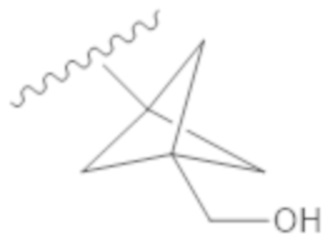 .
.
Вариант осуществления 21. Соединение согласно варианту осуществления 1 в свободной форме или в форме фармацевтически приемлемой соли, которое выбрано из
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(3–морфолинoпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–(3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–(3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(1–изопропилпирролидин–3–ил)фенил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–(3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(3,3,3–трифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(пирролидин–3–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(3–фтор–4–гидроксициклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(2–азаспиро[3.3]гептан–2–илметил)фенил)–2–амино–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(4–(3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–(3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(2–фтор–4–(пиперазин–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–((2–метилпирролидин–1–ил)метил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–(3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(4–(3–(бут–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(оксетан–3–илметил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(1–(2,2,2–трифторэтил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(2–(2,2,2–трифторэтокси)этил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида;
2–амино–5–(4–(3–(1,1–диоксидотетрагидро–2H–тиопиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–циклогексил–5–(4–(4–метилпиперазин–1–карбонил)фенил)никотинамида;
2–амино–5–(3–фтор–4–(2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–циклогексил–5–(4–морфолинoфенил)никотинамида;
2–амино–5–(4–(3–(2,2–дифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–5–(4–(3–(2–цианоэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–циклогексил–5–(4–(3–морфолинoпропокси)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(проп–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)–2–хлорфенил)–2–амино–N–(4–гидроксициклогексил)никотинамида;
2–амино–5–(2–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–5–(4–(3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(4–(3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
5–(4–(1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–циклогексил–5–(4–(пиперидин–1–ил)фенил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–циклогексил–5–(4–((2–(4–метилпиперазин–1–ил)этил)карбамоил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(2,2,2–трифторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–(3–гидроксиадамантан–1–ил)никотинамида;
2–амино–N–циклогексил–5–(4–(морфолин–4–карбонил)фенил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(4–((2–метилпирролидин–1–ил)метил)фенил)никотинамида;
2–амино–5–(2,3–дифтор–4–((2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил–4–d)–5–(4–(3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(4–метилпиперазин–1–карбонил)фенил)никотинамида;
2–амино–5–(2–хлор–4–((2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–(3–(2–гидроксипропан–2–ил)бицикло[1.1.1]пентан–1–ил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–циклогексил–5–(3–(морфолин–4–карбонил)фенил)никотинамида;
2–амино–5–(3–хлор–4–((2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
5–(4–(1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–(4–гидрокси–1–метилциклогексил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(2–метил–4–((2–метилпирролидин–1–ил)метил)фенил)никотинамида и
2–амино–N–циклогексил–5–(3–(4–(2–гидроксиэтил)пиперазин–1–карбонил)фенил)никотинамида.
Вариант осуществления 22. Соединение согласно варианту осуществления 21 в свободной форме или в форме фармацевтически приемлемой соли, которое выбрано из
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
5–(4–((S)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(3–морфолинoпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4r)–4–гидроксициклогексил)–5–(4–(1–изопропилпирролидин–3–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(3,3,3–трифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4r)–4–гидроксициклогексил)–5–(4–(пирролидин–3–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1R,3S,4R)–3–фтор–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1s,4S)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(2–азаспиро[3.3]гептан–2–илметил)фенил)–2–амино–N–((1r,4r)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(2–фтор–4–(пиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидрокси–4–метилциклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(бут–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(оксетан–3–илметил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(2,2,2–трифторэтил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–(2,2,2–трифторэтокси)этил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(1,1–диоксидотетрагидро–2H–тиопиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–5–(3–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(3,3,3–трифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(2,2–дифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(2–цианоэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(проп–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(проп–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)–2–хлорфенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–5–(2–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидрокси–4–метилциклогексил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(бут–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидрокси–4–метилциклогексил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–циклогексил–5–(4–((2–(4–метилпиперазин–1–ил)этил)карбамоил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2,2,2–трифторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1R,3R)–3–гидроксиадамантан–1–ил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(оксетан–3–илметил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1s,4R)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(2,3–дифтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(2,2–дифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2,2,2–трифторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил–4–d)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1s,4s)–4–гидроксициклогексил)–5–(4–(4–метилпиперазин–1–карбонил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(2–хлор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–(3–(2–гидроксипропан–2–ил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(3–хлор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидрокси–1–метилциклогексил)никотинамида и
2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(2–метил–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамида.
Вариант осуществления 23. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения согласно любому из предыдущих вариантов осуществления в свободной форме или в форме фармацевтически приемлемой соли и один или несколько фармацевтически приемлемых носителей.
Вариант осуществления 24. Комбинация, содержащая терапевтически эффективное количество соединения согласно любому из вариантов осуществления 1–22 в свободной форме или в форме фармацевтически приемлемой соли и один или несколько терапевтически активных средств.
Вариант осуществления 25. Соединение согласно любому из вариантов осуществления 1–22 в свободной форме или в форме фармацевтически приемлемой соли для применения в качестве лекарственного препарата.
Вариант осуществления 26. Соединение согласно любому из вариантов осуществления 1–22 в свободной форме или в форме фармацевтически приемлемой соли для применения в лечении нарушения или заболевания, выбранного из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
Вариант осуществления 27. 2–Амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в свободной форме, в частности, в свободной форме модификации HA, в частности, (i) характеризующийся значениями 2–тета в отражении XRPD первых 2, первых 3, первых 4, первых 5, первых 6, первых 8, в частности, первых десяти, или, в частности, всех значений 2–тета, приведенных в таблице A ниже; или (ii) демонстрирующий диаграмму DSC, показанную на фиг. 4; или (iii) характеризующийся диаграммой TGA, показанной на фиг. 5; или (iv) характеризующийся диаграммой FT–IR, показанной на фиг. 6; или характеризующийся двумя, или тремя, или, в частности, всеми свойствами (i)–(iv), только что упомянутыми; или, в частности, характеризующийся диаграммой XRPD, показанной на фиг. 3.
Вариант осуществления 28. 2–Амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в свободной форме, в частности, в форме модификации A, в частности, (i) характеризующийся значениями 2–тета в отражении XRPD первых 2, первых 3, первых 4, первых 5, первых 6, первых 8, в частности, первых десяти, или всех значений 2–тета, приведенных в таблице B ниже; или (ii) демонстрирующий диаграмму DSC, показанную на фиг. 8; или (iii) характеризующийся диаграммой TGA, показанной на фиг. 9; или (iv) характеризующийся диаграммой FT–IR, показанной на фиг. 10; или характеризующийся двумя, или тремя, или, в частности, всеми свойствами (i)–(iv), только что упомянутыми; или, в частности, характеризующийся диаграммой XRPD, показанной на фиг. 7.
Вариант осуществления 29. 2–Амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в виде ангидрида свободной формы, в частности, характеризующийся диаграммой XRPD, показанной на фиг. 18.
Вариант осуществления 30. 2–Амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в виде тригидрата свободной формы, в частности, характеризующийся диаграммой XRPD, показанной на фигуре 19.
Вариант осуществления 31. 2–Амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в виде соли фумаровой кислоты, в частности, характеризующейся молярным соотношением, составляющим 1: 1, соединения A и фумаровой кислоты, в частности, в форме соли фумаровой кислоты модификации HA, в частности, (i) характеризующийся значениями 2–тета в отражении XRPD первых 2, первых 3, первых 4, первых 5, первых 6, первых 8, в частности, первых десяти, или, в частности, всех значений 2–тета, приведенных в таблице C ниже; или (ii) демонстрирующий диаграмму DSC, показанную на фиг. 12; или (iii) характеризующийся диаграммой TGA, показанной на фиг. 13; или (iv) характеризующийся диаграммой FT–IR, показанной на фиг. 14; или (v) характеризующийся диаграммой DVS при 25°C, показанной на фиг. 15; или (vi) характеризующийся диаграммой DVS при 40°C, показанной на фиг. 16; или характеризующийся двумя, или тремя, или четырьмя, или пятью, или, в частности, всеми свойствами (i)–(iv), только что упомянутыми; или, в частности, характеризующийся диаграммой XRPD, показанной на фиг. 11.
Вариант осуществления 32. 2–Амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в виде варианта 1 аморфной формы соли фумаровой кислоты, в частности, характеризующийся молярным соотношением, составляющим 1: 1, соединения A и фумаровой кислоты, в частности, демонстрирующий переход в стеклообразное состояние при приблизительно 143°C в случае проведения анализа посредством модулированной DSC при скорости нагревания 1 K/мин., амплитудном значении температуры 1 K, периоде 60 секунд.
Вариант осуществления 33. 2–Амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в виде варианта 2 аморфной формы соли фумаровой кислоты, в частности, характеризующийся молярным соотношением, составляющим 1: 1, соединения A и фумаровой кислоты, в частности, демонстрирующий переход в стеклообразное состояние при приблизительно 78°C, в случае проведения анализа посредством DSC при скорости нагревания, составляющей 10 K/мин.
Вариант осуществления 34. 2–Амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в форме соли фосфорной кислоты, в частности, характеризующийся молярным соотношением, составляющим 1: 1,5, соединения A и ортофосфорной кислоты, в частности, (i) характеризующийся значениями 2–тета в отражении XRPD первых 2, первых 3, первых 4, первых 5, первых 6, первых 8, в частности, первых десяти, или, в частности, всех значений 2–тета, приведенных в таблице D ниже; или, в частности, характеризующийся диаграммой XRPD, показанной на фиг. 17.
Вариант осуществления 35. Соединение в форме согласно любому из вариантов осуществления 27–34 для применения в качестве лекарственного препарата.
Вариант осуществления 36. Соединение в форме согласно любому из вариантов осуществления 27–34 для применения в лечении нарушения или заболевания, выбранного из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
В случае если значение 2–тета (или в таблицах ниже "угол") приведено в данном изобретении, это означает относительное значение ± 0,2, или, в частности, значение как таковое.
В случае если используется термин "модификация", это относится к аморфным формам или, в частности, к кристаллическим полиморфам (т. е. формам кристаллических структур), также включающим псевдополиморфы, такие как сольваты или, в частности, гидраты свободных форм или солей соответственно.
В зависимости от выбора исходных материалов и процедур, соединения могут быть представлены в форме чистых оптических изомеров или в виде смесей изомеров, таких как рацематы и смеси диастереоизомеров, в зависимости от количества асимметрических атомов углерода. Настоящее изобретение подразумевает включение всех таких возможных изомеров, в том числе рацемических смесей, смесей диастереоизомеров и оптически чистых форм. Оптически активные (R)– и (S)–изомеры могут быть получены с использованием хиральных синтонов или хиральных реагентов или выделены с применением общепринятых методик. В случае если соединение содержит двойную связь, заместитель может иметь E– или Z–конфигурацию. В случае если соединение содержит дизамещенный циклоалкил, циклоалкильный заместитель может иметь цис– или транс–конфигурацию. Также подразумевается включение всех таутомерных форм.
Используемые в данном документе термины "соль" или "соли" относятся к соли присоединения кислоты или присоединения основания соединения согласно настоящему изобретению. "Соли" включают, в частности, "фармацевтически приемлемые соли". Термин "фармацевтически приемлемые соли" относится к солям, которые сохраняют биологическую эффективность и свойства соединений согласно данному изобретению и которые, как правило, не являются биологически или иным образом нежелательными. Во многих случаях соединения согласно настоящему изобретению способны к образованию кислых солей ввиду присутствия основного фрагмента аминопиридина. В случае если указана "свободная форма", это относится к форме без дополнительных кислот и/или оснований, т. е. соединению как таковому (которое, однако, может образовывать внутренние соли, если соединение содержит основные и кислотные группы).
Фармацевтически приемлемые соли присоединения кислоты могут быть образованы с помощью неорганических кислот и органических кислот.
Неорганические кислоты, из которых могут быть получены соли, включают, например, хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т. п.
Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту, сульфосалициловую кислоту и т. п.
В другом аспекте настоящее изобретение предусматривает соединения формулы (I) в форме соли, представляющей собой ацетат, аскорбат, адипат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, соль камфорсульфоновой кислоты, соль каприновой кислоты, хлорид/гидрохлорид, хлортеофиллонат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, глутамат, глутарат, гликолят, гиппурат, гидройодид/йодид, изетионат, лактат, лактобионат, лаурилсульфат, соль яблочной кислоты, соль малеиновой кислоты, малонат, соль миндальной кислоты, мезилат, метилсульфат, соль муциновой кислоты, соль нафтойной кислоты, напсилат, никотинaт, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионaт, себацинат, стеарат, сукцинат, сульфосалицилат, сульфат, тартрат, тозилат трифенатат, трифторацетат или ксинафоат.
Любая формула, приведенная в данном документе, также подразумевает присутствие немеченных форм, а также меченных изотопом форм соединений. Меченные изотопом соединения имеют структуры, изображенные посредством формул, приведенных в данном документе, за исключением того, что один или несколько атомов заменены атомом, характеризующимся выбранной атомной массой или массовым числом. Примеры изотопов, которые могут быть включены в соединения согласно настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 11C, 13C, 14C, 15N, 18F 31P, 32P, 35S, 36Cl, 123I, 124I, 125I соответственно. Настоящее изобретение включает различные меченные изотопом соединения, определенные в данном документе, например, соединения, в которых присутствуют радиоактивные изотопы, такие как 3H и 14C, или соединения, в которых присутствуют изотопы, не являющиеся радиоактивными, такие как 2H и 13C. Такие меченные изотопом соединения применимы в метаболических исследованиях (с использованием 14C), исследованиях кинетики реакций (с использованием, например 2H или 3H), методиках обнаружения или визуализации, таких как позитронно–эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), в том числе анализах распределения лекарственного средства или субстрата в тканях, или в лучевой терапии пациентов. В частности, меченное 18F соединение может быть, в частности, необходимо для исследований PET или SPECT. Меченные изотопом соединения формулы (I) в общих случаях могут быть получены с помощью общепринятых методик, известных специалистам в данной области техники, или посредством способов, аналогичных описанным в сопутствующих примерах, с использованием подходящих меченных изотопом реагентов вместо немеченных реагентов, используемых ранее.
Кроме того, замещение более тяжелыми изотопами, в частности, дейтерием (т. е. 2H или D), может давать определенные терапевтические преимущества, что обусловлено более высокой устойчивостью к инактивации в процессе метаболизма, например, увеличение периода полувыведения in vivo, или снижение требований в отношении дозировки, или улучшение в отношении терапевтического индекса. Следует понимать, что дейтерий в данном контексте рассматривается в качестве заместителя соединения формулы (I). Концентрация такого более тяжелого изотопа, конкретно дейтерия, может быть определена посредством коэффициента изотопного обогащения. Используемый в данном документе термин "коэффициент изотопного обогащения" означает соотношение между распространенностью изотопа и распространенностью в природе указанного изотопа. В случае если заместитель в соединении согласно данному изобретению представляет собой указанный дейтерий, такое соединение характеризуется коэффициентом изотопного обогащения для каждого обозначенного атома дейтерия, составляющим по меньшей мере 3500 (введение дейтерия 52,5% при каждом обозначенном атоме дейтерия), по меньшей мере 4000 (введение дейтерия 60%), по меньшей мере 4500 (введение дейтерия 67,5%), по меньшей мере 5000 (введение дейтерия 75%), по меньшей мере 5500 (введение дейтерия 82,5%), по меньшей мере 6000 (введение дейтерия 90%), по меньшей мере 6333,3 (введение дейтерия 95%), по меньшей мере 6466,7 (введение дейтерия 97%), по меньшей мере 6600 (введение дейтерия 99%) или по меньшей мере 6633,3 (введение дейтерия 99,5%).
Фармацевтически приемлемые сольваты в соответствии с настоящим изобретением включают таковые, где растворитель для кристаллизации может быть замещен изотопом, например, D2O, d6–ацетон, d6–DMSO.
Соединения согласно настоящему изобретению, т. е. соединения формулы (I), которые содержат группы, способные действовать в качестве доноров и/или акцепторов в отношении водородных связей, могут быть способны к образованию сокристаллов с подходящими средствами для образования сокристаллов. Такие сокристаллы могут быть получены из соединений формулы (I) посредством известных процедур образования сокристаллов. Такие процедуры включают измельчение, нагревание, совместную сублимацию, совместное плавление или приведение в контакт в растворе соединений формулы (I) со средством для образования сокристаллов в условиях кристаллизации и выделение сокристаллов, образованных таким образом. Подходящие средства для образования сокристаллов включают описанные в WO 2004/078163. Следовательно, в настоящем изобретении дополнительно предусмотрены сокристаллы, содержащие соединение формулы (I).
Используемый в данном документе термин "фармацевтически приемлемый носитель" включает любые и все растворители, дисперсионные среды, покрытия, поверхностно–активные вещества, антиоксиданты, консерванты (например, антибактериальные средства, противогрибковые средства), средства для обеспечения изотоничности, замедляющие абсорбцию средства, соли, консерванты, стабилизаторы лекарственных средств, связующие средства, вспомогательные вещества, разрыхляющие средства, смазывающие вещества, подслащивающие средства, ароматизирующие средства, красители и т. п. и их комбинации, которые будут известны специалистам в данной области техники (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289– 1329). За исключением случаев, когда любой традиционный носитель является несовместимым с активным ингредиентом, предполагается его применение в терапевтических или фармацевтических композициях.
Термин "терапевтически эффективное количество" соединения согласно настоящему изобретению относится к количеству соединения согласно настоящему изобретению, которое будет вызывать биологический или медицинский ответ у субъекта, например, снижение или подавление активности фермента или белка, или уменьшать тяжесть симптомов, облегчать состояние, замедлять или сдерживать прогрессирование заболевания, или предупреждать заболевание и т. д. В одном неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к количеству соединения согласно настоящему изобретению, которое при введении субъекту является эффективным в отношении (1) по меньшей мере частичного облегчения, подавления, предупреждения и/или снижения тяжести состояния, или нарушения, или заболевания, (i) опосредованного ALK–2, или (ii) ассоциированного с активностью ALK–2, или (iii) характеризующегося активностью (нормальной или аномальной) ALK–2; или (2) уменьшения или подавления активности ALK–2; или (3) снижения или подавления экспрессии ALK–2. В другом неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к количеству соединения согласно настоящему изобретению, которое при введении в клетку, или ткань, или неклеточный биологический материал, или среду является эффективным в отношении по меньшей мере частичного снижения или подавления активности ALK–2; или по меньшей мере частичного снижения или подавления экспрессии ALK–2.
Используемый в данном документе термин "субъект" относится к человеку.
Используемый в данном документе термин "подавлять", "подавление" или "подавляющий" относится к снижению или ослаблению данного состояния, симптома, или нарушения, или заболевания или значительному снижению исходной активности биологической активности или процесса.
Используемый в данном документе термин "лечить", "осуществлять лечение" или "лечение" любого заболевания или нарушения относится в одном варианте осуществления к снижению тяжести заболевания или нарушения (т. е. замедлению, или остановке, или снижению развития заболевания или по меньшей мере одного из его клинических симптомов). В другом варианте осуществления "лечить", "осуществление лечения" или "лечение" относится к облегчению или снижению тяжести по меньшей мере одного физического параметра, включая такие, которые могут быть неявными для пациента. В еще одном варианте осуществления "лечить", "осуществление лечения" или "лечение" относится к модулированию заболевания или нарушения либо физически, (например, стабилизация явного симптома), либо физиологически, (например, стабилизация физического параметра), либо с помощью того и другого. В еще одном варианте осуществления "лечить", "осуществление лечения" или "лечение" относится к предупреждению или задержке возникновения, или развития, или прогрессирования заболевания или нарушения.
Используемый в данном документе субъект "нуждается в" лечении, если для такого субъекта будет биологическая, медицинская польза или польза в отношении качества жизни в результате такого лечения.
Используемый в данном документе термин в формах единственного числа и подобные термины, применяемые в контексте настоящего изобретения (в частности, в контексте формулы изобретения), следует истолковывать с охватом как форм единственного числа, так и множественного числа, если в данном документе не указано иное или нет явного противоречия по контексту.
Все способы, описанные в данном документе, можно осуществлять в любом подходящем порядке, если в данном документе не указано иное или нет иного явного противоречия по контексту. Применение любых и всех примеров или примерных формулировок (например, "такой как") в данном документе предназначено только для лучшего объяснения настоящего изобретения и не предполагает ограничения объема настоящего изобретения, заявленного иным образом.
Любой асимметрический атом (например, углерод или подобный) соединения(соединений) согласно настоящему изобретению может находиться в рацемической или энантиомерно обогащенной форме, например, в (R)–, (S)– или (R,S)–конфигурации. В определенных вариантах осуществления каждый асимметрический атом предусматривает энантиомерный избыток, составляющий по меньшей мере 50%, энантиомерный избыток, составляющий по меньшей мере 60%, энантиомерный избыток, составляющий по меньшей мере 70%, энантиомерный избыток, составляющий по меньшей мере 80%, энантиомерный избыток, составляющий по меньшей мере 90%, энантиомерный избыток, составляющий по меньшей мере 95%, или энантиомерный избыток, составляющий по меньшей мере 99%, в (R)– или (S)–конфигурации. Заместители при атомах с ненасыщенными двойными связями могут, если это возможно, находиться в цис– (Z)– или транс– (E)–форме.
Соответственно, применяемое в данном документе соединение согласно настоящему изобретению может находиться в форме одного из возможных изомеров, ротамеров, атропоизомеров, таутомеров или их смесей, например, в виде практически чистых геометрических (цис– или транс–) изомеров, диастереомеров, оптических изомеров (антиподов), рацематов или их смесей.
Любые полученные в результате смеси изомеров могут быть разделены на основании физико–химических отличий составляющих на чистые или практически чистые геометрические или оптические изомеры, диастереомеры, рацематы, например, посредством хроматографии и/или фракционной кристаллизации.
Любые полученные в результате рацематы конечных продуктов или промежуточных соединений могут быть разделены на оптические антиподы посредством известных способов, например, посредством разделения их диастереомерных солей, полученных с помощью оптически активной кислоты или основания, и выделения оптически активного кислотного или основного соединения. В частности, основный фрагмент таким образом может быть использован для разделения соединений согласно настоящему изобретению на их оптические антиподы, например, посредством фракционной кристаллизации соли, образованной с помощью оптически активной кислоты, например, винной кислоты, дибензоилвинной кислоты, диацетилвинной кислоты, ди–O,O'–п–толуолвинной кислоты, миндальной кислоты, яблочной кислоты или камфор–10–сульфоновой кислоты. Рацемические продукты также могут быть разделены посредством хиральной хроматографии, например, жидкостной хроматографии высокого давления (HPLC) с применением хирального адсорбента.
Кроме того, соединения согласно настоящему изобретению, в том числе их соли, также могут быть получены в форме их гидратов или включают другие растворители, применяемые для их кристаллизации. Соединения согласно настоящему изобретению могут по своей природе или по технологическому замыслу образовывать сольваты с помощью фармацевтически приемлемых растворителей (в том числе воды); следовательно, предполагается, что настоящее изобретение охватывает как сольватированные и несольватированные формы. Термин "сольват" относится к молекулярному комплексу соединения согласно настоящему изобретению (в том числе его фармацевтически приемлемых солей) с одной или несколькими молекулами растворителя. Такие молекулы растворителя представляют собой широко используемые в фармацевтической области, которые известны как нетоксичные для реципиента, например, вода, этанол и т. п. Термин "гидрат" относится к комплексу, в котором молекула растворителя представляет собой воду.
Соединения согласно настоящему изобретению, в том числе их соли, гидраты и сольваты, могут по своей природе или по технологическому замыслу образовывать полиморфы.
Как правило, соединения формулы (I) могут быть получены в соответствии со схемами, представленными ниже.
Схема 1
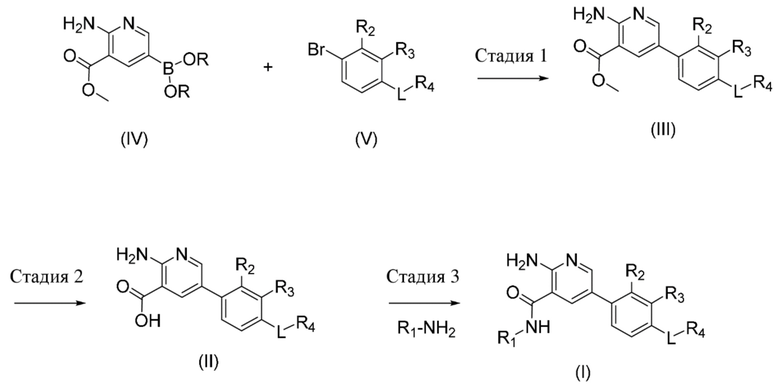
Стадия 1. Соединение формулы (III), где R2, R3, R4 и L определены в данном документе по отношению к соединению формулы (I), может быть получено посредством осуществления реакции сочетания соединения формулы (IV), где фрагмент B(OR)2 образует производное бороновой кислоты (например, где R представляет собой водород или пинакол), с соединением формулы (V), где R2, R3, R4 и L определены в данном документе по отношению к соединению формулы (I), в присутствии подходящего растворителя, такого как, например, диоксан или 2–метил–2–бутанол, основания, такого как карбонат калия, и подходящего катализатора, предпочтительно катализатора на основе палладия, такого как, например, [1,1'–бис(дифенилфосфино)ферроцен]дихлорпалладий(II). Соединения формул IV, V, III и II могут применяться в свободной форме или в форме их солей.
Стадия 2. Соединение формулы (II), где R2, R3, R4 и L определены в данном документе по отношению к соединению формулы (I), может быть получено посредством обработки соединения формулы (III), где R2, R3, R4 и L определены в данном документе по отношению к соединению формулы (I), с использованием подходящего основания, такого как, например, гидроксид лития или гидроксид натрия, в присутствии подходящего растворителя, такого как, например, тетрагидрофуран или алканол, такой как метанол, Добавление кислоты, такой как HCl, приводит к образованию соответствующей соли, например, соли, представляющей собой гидрохлорид, соединения формулы II.
Стадия 3. Соединение формулы (I), где R1, R2, R3, R4 и L определены в данном документе, может быть получено посредством осуществления реакции сочетания соединения формулы (II), где R2, R3, R4 и L определены в данном документе по отношению к соединению формулы (I), с подходящим амином, имеющим формулу R1–NH2, где R1 определен в данном документе по отношению к соединению формулы (I), таким как, например, транс–4–аминоциклогексанол, подходящим амидным реагентом для реакции сочетания, таким как, например, O–(7–азабензотриазол–1–ил)–N,N,N´,N´–тетраметилуроний–гексафторфосфат, и основанием, таким как, например, N–метилморфолин или триэтиламин, в присутствии подходящего растворителя, такого как, например, N,N–диметилформамид или ацетонитрил.
Соединения формул (IV) и (V) могут быть получены, как описано в примерах, представленных дополнительно ниже.
Схема 2
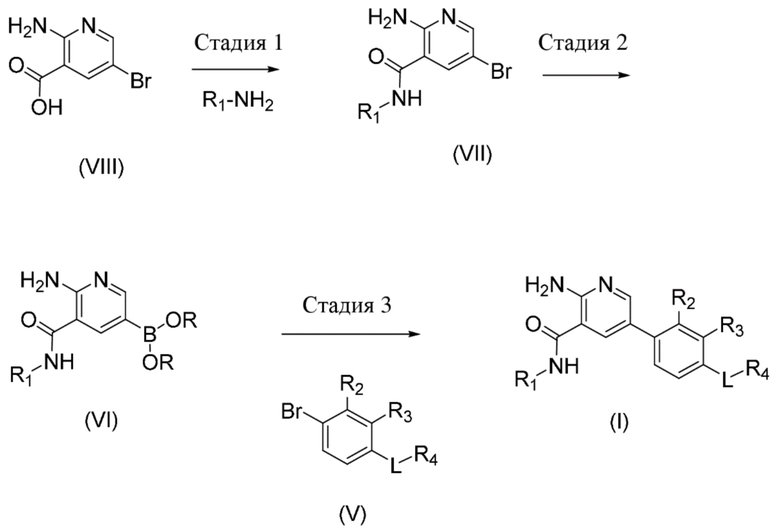
Стадия 1. Соединение формулы (VII), где R1 определен в данном документе по отношению к соединению формулы (I), может быть получено посредством обработки соединения формулы (VIII) с использованием подходящего амина формулы R1–NH2, такого как, например, транс–4–аминоциклогексанол, подходящего амидного реагента для реакции сочетания, такого как, например, O–(7–азабензотриазол–1–ил)–N,N,N´,N´–тетраметилуроний–гексафторфосфат, и основания, такого как, например, N–метилморфолин, в присутствии подходящего растворителя, такого как, например, N,N–диметилформамид.
Стадия 2. Соединение формулы (VI), где R1 определен в данном документе по отношению к соединению формулы (I), и фрагмент B(OR)2 образует производное бороновой кислоты (например, где R представляет собой водород или пинакол), может быть получено посредством осуществления реакции сочетания соединения формулы (VII), где R1 определен в данном документе по отношению к соединению формулы (I), с соединением бора, таким как, например, бис(пинаколато)дибор, в присутствии подходящего растворителя, такого как, например, диоксан, соли, такой как ацетат калия, и подходящего катализатора, предпочтительно катализатора на основе палладия, такого как, например, [1,1'–бис(дифенилфосфино)ферроцен]дихлорпалладий(II).
Стадия 3. Соединение формулы (I), где R1, R2, R3, R4 и L определены в данном документе, может быть получено посредством осуществления реакции сочетания соединения формулы (VI), где R1 определен в данном документе по отношению к соединению формулы (I), и фрагмент B(OR)2 образует производное бороновой кислоты (например, где R представляет собой водород или пинакол), с соединением формулы (V), где R2, R3, R4 и L определены в данном документе по отношению к соединению формулы (I), в присутствии подходящего растворителя, такого как, например, диоксан, основания, такого как гидроксид натрия, и подходящего катализатора, предпочтительно катализатора на основе палладия, такого как, например, [1,1'–бис(дифенилфосфино)ферроцен]дихлорпалладий(II).
Схема 3
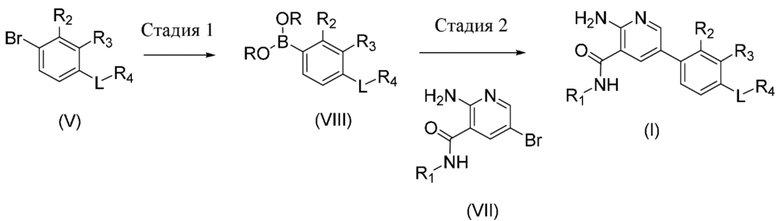
Стадия 1. Соединение формулы (VIII), где R2, R3, L, R4 определены в данном документе по отношению к соединению формулы (I), и фрагмент B(OR)2 образует производное бороновой кислоты (например, где R представляет собой водород или пинакол), может быть получено посредством осуществления реакции сочетания соединения формулы (V), где R2, R3, L, R4 определены в данном документе по отношению к соединению формулы (I), с соединением бора, таким как, например, бис(пинаколато)дибор, в присутствии подходящего растворителя, такого как, например, диоксан, соли, такой как ацетат калия, и подходящего катализатора, предпочтительно катализатора на основе палладия, такого как, например, [1,1'–бис(дифенилфосфино)ферроцен]дихлорпалладий(II), или в качестве альтернативы в подходящем растворителе, таком как тетрагидрофуран, и сильном основании, таком как н–бутиллитий или хлорид изопропилмагния, или смеси двух из них с соединением бора, таким как триметилборонат.
Стадия 2. Соединение формулы (I), где R1, R2, R3, R4 и L определены в данном документе, может быть получено посредством осуществления реакции сочетания соединения формулы (VIII), где R2, R3, L, R4 определены в данном документе по отношению к соединению формулы (I), и фрагмент B(OR)2 образует производное бороновой кислоты (например, где R представляет собой водород или пинакол), с соединением формулы (VII), где R1 определен в данном документе по отношению к соединению формулы (I), в присутствии подходящего растворителя, такого как, например, диоксан, основания, такого как карбонат калия, и подходящего катализатора, предпочтительно катализатора на основе палладия, такого как, например, [1,1'–бис(дифенилфосфино)ферроцен]дихлорпалладий(II).
Схема 4
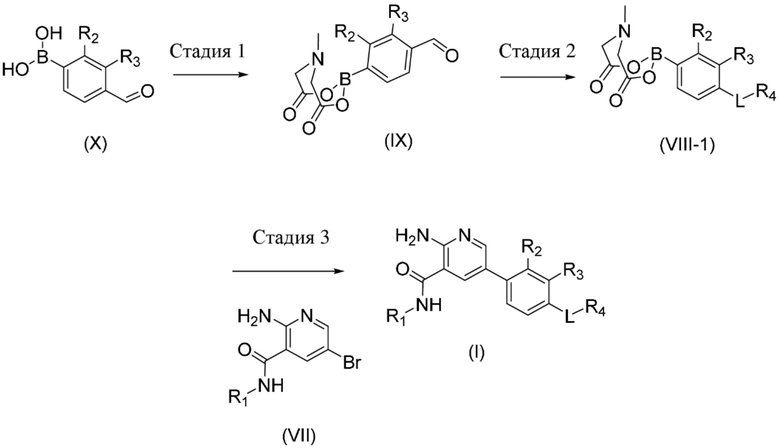
Стадия 1. Соединение формулы (IX), где R2 и R3 определены в данном документе по отношению к соединению формулы (I), может быть получено посредством обработки соединения формулы (X), где R2 и R3 определены в данном документе по отношению к соединению формулы (I), с использованием N–метилиминодиуксусной кислоты в присутствии подходящего растворителя, такого как, например, DMF.
Стадия 2. Соединение формулы (VIII–1), где R1, R2, R3, R4 и L определены в данном документе, может быть получено посредством обработки соединения формулы (IX), где R2 и R3 определены в данном документе по отношению к соединению формулы (I), с помощью подходящего амина с подходящим восстановителем, таким как например, триацетоксиборгидрид натрия, и кислотой, такой как уксусная кислота, в присутствии подходящего растворителя, такого как, например, THF.
Стадия 3. Соединение формулы (I), где R1, R2, R3, R4 и L определены в данном документе, может быть получено посредством осуществления реакции сочетания соединения формулы (VIII–1), где R2, R3, L, R4 определены в данном документе по отношению к соединению формулы (I), с соединением формулы (VII), где R1 определен в данном документе по отношению к соединению формулы (I), в присутствии подходящего растворителя, такого как, например, диоксан, основания, такого как фосфат калия, и подходящего катализатора, предпочтительно катализатора на основе палладия, такого как, например, хлорид (2–дициклогексилфосфино–2′,4′,6′–триизопропил–1,1′–бифенил)[2–(2–аминоэтил)фенил)]палладия(II).
В дополнительном аспекте настоящее изобретение относится к способу получения соединения формулы (I) в свободной форме или в форме фармацевтически приемлемой соли, включающему стадии:
a) осуществления реакции сочетания соединения формулы (II), определенного в данном документе, с соединением формулы R1–NH2, определенным в данном документе, с получением соединения формулы (I);
b) выделения получаемого таким образом соединения формулы (I) в свободной форме или в форме фармацевтически приемлемой соли.
В дополнительном аспекте настоящее изобретение относится к способу получения соединения формулы (I) в свободной форме или в форме фармацевтически приемлемой соли, включающему стадии:
a) осуществления реакции сочетания соединения формулы (VI), определенного в данном документе, с соединением формулы (V), определенным в данном документе, с получением соединения формулы (I);
b) выделения получаемого таким образом соединения формулы (I) в свободной форме или в форме фармацевтически приемлемой соли.
В дополнительном аспекте настоящее изобретение относится к способу получения соединения формулы (I) в свободной форме или в форме фармацевтически приемлемой соли, включающему стадии:
a) осуществления реакции сочетания соединения формулы (VIII) или соединения формулы (VIII–1), определенных в данном документе, с соединением формулы (VII), определенным в данном документе, с получением соединения формулы (I);
b) выделения получаемого таким образом соединения формулы (I) в свободной форме или в форме фармацевтически приемлемой соли.
Настоящее изобретение дополнительно включает любой вариант способов согласно настоящему изобретению, в котором промежуточный продукт, получаемый на любой его стадии, применяют в качестве исходного материала и проводят остальные стадии, или в котором исходные материалы получают in situ при условиях реакции, или в котором компоненты реакционной смеси применяют в форме их солей или оптически чистого материала.
Соединения согласно настоящему изобретению и промежуточные соединения также могут быть превращены друг в друга в соответствии со способами, общеизвестными специалистам в данной области техники.
В другом аспекте в настоящем изобретении предусмотрена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В дополнительном варианте осуществления композиция содержит по меньшей мере два фармацевтически приемлемых носителя, таких как описанные в данном документе. Для целей настоящего изобретения, если не обозначено иным образом, сольваты и гидраты обычно подразумевают композиции. Предпочтительно фармацевтически приемлемые носители являются стерильными. Фармацевтическая композиция может быть составлена для конкретных путей введения, таких как пероральное введение, парентеральное введение, ректальное введение, трансдермальное введение и т. д. Кроме того, фармацевтические композиции согласно настоящему изобретению могут быть изготовлены в твердой форме (в том числе без ограничения капсулы, таблетки, пилюли, гранулы, порошки или суппозитории) или в жидкой форме (в том числе без ограничения растворы, суспензии или эмульсии). Фармацевтические композиции могут подвергаться стандартным фармацевтическим операциям, таким как стерилизация, и/или могут содержать стандартные инертные разбавители, смазывающие средства или буферные средства, а также адъюванты, такие как консерванты, стабилизаторы, смачивающие средства, эмульгаторы и буферы, и т. д.
Как правило, фармацевтические композиции представляют собой таблетки или желатиновые капсулы, содержащие активный ингредиент вместе с одним или несколькими из:
a) разбавителей, например, лактозы, декстрозы, сахарозы, маннита, сорбита, целлюлозы и/или глицина;
b) смазывающих средств, например, диоксида кремния, талька, стеариновой кислоты, ее магниевой или кальциевой соли и/или полиэтиленгликоля; для таблеток также
c) связующих средств, например, алюмосиликата магния, крахмальной пасты, желатина, трагаканта, метилцеллюлозы, карбоксиметилцеллюлозы натрия и/или поливинилпирролидона; при необходимости
d) разрыхлителей, например, видов крахмала, агара, альгиновой кислоты или ее натриевой соли или шипучих смесей; и
e) абсорбентов, красителей, ароматизаторов и подсластителей.
Таблетки могут быть либо покрытыми оболочкой, либо покрытыми энтеросолюбильным покрытием в соответствии со способами, известными из уровня техники.
Подходящие композиции для перорального введения включают эффективное количество соединения согласно настоящему изобретению в форме таблеток, пастилок для рассасывания, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких капсул, или сиропов, или настоек. Композиции, предназначенные для перорального применения, получают в соответствии с любым способом, известным из уровня техники, для изготовления фармацевтических композиций, и такие композиции могут содержать одно или несколько средств, выбранных из группы, состоящей из подслащивающих веществ, ароматизирующих средств, красящих средств и консервирующих средств, с целью получения препаратов, которые являются фармацевтически приемлемыми и имеют привлекательный вкус. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, которые являются подходящими для изготовления таблеток. Такие вспомогательные вещества представляют собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие средства, например, кукурузный крахмал или альгиновая кислота; связующие средства, например, крахмал, желатин или аравийская камедь; и смазывающие средства, например, стеарат магния, стеариновая кислота или тальк. Таблетки являются непокрытыми или покрытыми посредством известных методик для отсрочки распада и всасывания в желудочно–кишечном тракте и тем самым обеспечивают устойчивое действие в течение более длительного периода. Например, можно использовать материал замедленного действия, такой как глицерилмоностеарат или глицерилдистеарат. Составы для перорального применения могут быть представлены в виде твердых желатиновых капсул, где активный ингредиент смешивают с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешивают с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Соединения формулы (I) в свободной форме или в форме фармацевтически приемлемой соли проявляют ценные фармакологические свойства, например, ALK–2–модулирующие свойства, например, показанные с помощью анализов in vitro и in vivo, представленных в следующих разделах, и, следовательно, предназначены для терапии или для применения в качестве химических веществ для исследований, например, в качестве фармакологически активных соединений.
Соединения согласно настоящему изобретению могут быть применимы в лечении показания, выбранного из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
Без ограничения теорией полагают, что соединения согласно настоящему изобретению, которые являются селективными ингибиторами ALK–2, уменьшают/подавляют передачу сигнала BMP и восстановление аномальной ткани, ассоциированной с ним.
Таким образом, в качестве дополнительного варианта осуществления в настоящем изобретении предусмотрено применение соединения формулы (I) или ее подформул (Ia), (II), (IIa), (IIb) в свободной форме или в форме фармацевтически приемлемой соли в терапии. В дополнительном варианте осуществления терапия выбрана из заболевания, лечение которого можно осуществлять посредством подавления рецептора ALK–2. В другом варианте осуществления заболевание выбрано из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
Таким образом, в качестве дополнительного варианта осуществления в настоящем изобретении предусмотрено соединение формулы (I) или ее подформул (Ia), (II), (IIa), (IIb) в свободной форме или в форме фармацевтически приемлемой соли для применения в терапии. В дополнительном варианте осуществления терапия выбрана из заболевания, лечение которого можно осуществлять посредством подавления рецептора ALK–2. В другом варианте осуществления заболевание выбрано из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
В другом варианте осуществления в настоящем изобретении предусмотрен способ лечения заболевания, лечение которого осуществляют посредством подавления рецептора ALK–2, включающий введение терапевтически приемлемого количества соединения формулы (I) или ее подформул (Ia), (II), (IIa), (IIb) в свободной форме или в форме фармацевтически приемлемой соли. В дополнительном варианте осуществления заболевание выбрано из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
Таким образом, в качестве дополнительного варианта осуществления в настоящем изобретении предусмотрено применение соединения формулы (I) или ее подформул (Ia), (II), (IIa), (IIb) в свободной форме или в форме фармацевтически приемлемой соли для изготовления лекарственного препарата. В дополнительном варианте осуществления лекарственный препарат предназначен для лечения заболевания, лечение которого можно осуществлять посредством подавления рецептора ALK–2. В другом варианте осуществления заболевание выбрано из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
В одном варианте осуществления настоящего изобретения предусмотрен 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в свободной форме или его фармацевтически приемлемая соль, в частности, в одной из модификаций, указанных в данном документе, для применения в лечении гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
В одном варианте осуществления предусмотрен 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в свободной форме или его фармацевтически приемлемая соль, в частности, в одной из модификаций, указанных в данном документе, для применения в лечении гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
В одном варианте осуществления предусмотрен 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в свободной форме или его фармацевтически приемлемая соль, в частности, в одной из модификаций, указанных в данном документе, для применения в лечении гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии
Следующие примеры предназначены для иллюстрации настоящего изобретения, и их не следует истолковывать как ограничивающие его. Значения температуры приведены в градусах Цельсия. Если не указано иное, все операции выпаривания осуществляют при пониженном давлении, как правило, от приблизительно 15 мм рт. ст. до 100 мм рт. ст. (= 20–133 мбар). Структура конечных продуктов, промежуточных соединений и исходных материалов подтверждена посредством стандартных аналитических способов, например, микроанализа и спектроскопических характеристик, например, MS, IR, NMR. Используемые аббревиатуры представляют собой стандартные аббревиатуры из уровня техники.
Все исходные материалы, структурные элементы, реагенты, кислоты, основания, дегидрирующие средства, растворители и катализаторы, используемые для синтеза соединений согласно настоящему изобретению, являются либо коммерчески доступными, либо могут быть получены посредством способов органического синтеза, известных специалисту в данной области техники, или посредством способов, описанных или аналогичных, соответственно, способам, представленным в разделе "Примеры". Также соединения согласно настоящему изобретению могут быть получены посредством способов органического синтеза, известных специалисту в данной области техники, как показано в следующих примерах.
Примеры



АНАЛИТИЧЕСКИЕ СПОСОБЫ
1H–ЯМР
Измерения проводили на спектрометре Bruker UltrashieldTM 400 (400 МГц), Bruker UltrashieldTM 600 (600 МГц), 400 MHz DRX Bruker CryoProbe (400 МГц) или 500 MHz DRX Bruker CryoProbe (500 МГц) с применением триметилсилана в качестве внутреннего стандарта или без него. Химические сдвиги (δ–значения) приведены в ppm, отсчитывая от тетраметилсилана, спектры на схеме расщепления обозначены как синглет (s), дуплет (d), триплет (t), квартет (q), мультиплет, неразделенные или более перекрывающиеся сигналы (m), широкий сигнал (bs). Растворители приведены в скобках.
UPLC–MS
Колонка: Waters Acquity HSS T3, C18, 1,8 мкм, 2,1×50 мм, термостат при 60°C. Скорость потока: 1,0 мл/мин. Градиент: от 5% до 98% B за 1,40 мин., затем 98% B в течение 0,40 мин., от 98% до 5% B за 0,10 мин., 5% B в течение 0,10 мин.; A=вода+0,05% муравьиной кислоты+3,75 мМ NH4OAc, B=ацетонитрил+0,04% муравьиной кислоты. Обнаружение UV/VIS (DAD), ESI (+/−). Диапазон масс–спектрометра: 100–1200 Да.
В случае примеров 46, 56, 74, 81, 85 и 87:
колонка: Waters Acquity BEH, C18, 1,7 мкм, 2,1×50 мм, термостат при 50°C. Расход: 1,0 мл/мин. Градиент: от 2% до 98% B за 4,40 мин., затем 98% B в течение 0,75 мин., от 98% до 2% B за 0,04 мин.; A=вода+0,1% муравьиной кислоты, B=ацетонитрил+0,1% муравьиной кислоты. Обнаружение UV/VIS (DAD), ESI (+/−). Диапазон масс–спектрометра: 100–1200 Да.
UPLC–HRMS:
Waters Acquity SDS, C18, 1,7 мкм, 2,1×50 мм, термостат при 50°C. Градиент: от 5% до 98% B за 7,50 мин., затем 98% B в течение 0,40 мин., от 98% до 5% B за 0,15 мин.; A=вода+5 мМ NH4OH, B=ацетонитрил+5 мМ NH4OH. Обнаружение UV/VIS (DAD), ESI (+/−). Диапазон масс–спектрометра: 100–1200 Да.
HPLC–MS
Колонка: Waters Symmetry C8, 3,5 мкм, 2,1×50 мм, термостат при 50°C. Скорость потока: 1,0 мл/мин.
Градиент: от 10% до 95% B за 2 мин., затем 95% B в течение 1 мин., от 95% до 10% B за 0,5 мин. 10% B в течение 0,50 мин.; A=вода+0,1% TFA, B=ацетонитрил+0,1% TFA. Обнаружение UV/VIS (DAD), APCI (+). Диапазон масс–спектрометра: 100–1200 Да.
Измерение величины вращения плоскости поляризации света
Величину вращения плоскости поляризации света измеряли с применением поляриметра Perkin Elmer серии PE241 № 5325, работающего при 589 нм с применением хлороформа в качестве растворителя.
СПОСОБЫ ОЧИСТКИ
prepHPLC
Gilson GX–281, насосы 331/332.
Колонка: Waters Sunfire C18, 30×100 мм, 5 мкм. Скорость потока: 30 мл/мин.
Подвижная фаза: вода (содержащая 0,1% TFA) и ацетонитрил (способ 1a).
Или:
Колонка: X–Bridge C18, 30×50 мм, 5 мкм. Скорость потока: 75 мл/мин.
Подвижная фаза: вода (содержащая 5 мМ NH4OH) и ацетонитрил (способ 1b).
Флэш–хроматография с нормальной фазой
Teledyne ISCO CombiFlash
Колонка: Redisep Rf для флэш–хроматографии с силикагелем.
Подвижная фаза: циклогексан/EtOAc (способ 2a) или DCM/MeOH (способ 2b).
Biotage Flash–Master II
Колонка: предварительно заполненная силикагелем 60 (40–63 мкм), от Merck.
Подвижная фаза: DCM и MeOH (содержащий 7,3 мМ NH4OH) (способ 2c).
Флэш–хроматография с обращенной фазой
Teledyne ISCO CombiFlash
Колонка: Redisep Rf Gold C18 для высокожидкостной хроматографии, предварительно заполненные колонки на 15,5 г, 50 г или 240 г, 20–40 мкм, 100A.
Подвижная фаза: вода и ацетонитрил (содержащий 7,3 мМ NH4OH) (способ 3a) или вода (содержащая 0,1% TFA) и ацетонитрил (способ 3b).
Пример 1: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
В раствор соли TFA и 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 1a, 80 мг, 0,158 ммоль) в DCM (4 мл) добавляли 1–(метилсульфонил)пиперидин–4–он (30,8 мг, 0,174 ммоль) и AcOH (0,018 мл, 0,316 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали при 45°C в течение 30 мин. Добавляли триацетоксиборогидрид натрия (84 мг, 0,395 ммоль) при к. т. и реакционную смесь перемешивали при 45°C в течение 1 ч. Затем реакционную смесь разбавляли насыщ. водн. раствором NaHCO3 и смешивали с EtOAc. После разделения фаз водн. слой снова экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,31 (d, 1H), 8,11 (d, 1H), 7,59 (d, 2H), 7,24 (d, 2H), 7,12 (s, 2H), 4,58 (d, 1H), 3,80–3,63 (m, 1H), 3,45–3,36 (m, 4H), 3,15 (dd, 1H), 2,89–2,83 (m, 5H), 2,65 (dd, 1H), 2,35–2,29 (m, 1H), 1,98–1,81 (m, 7H), 1,57–1,18 (m, 8H), 0,83–0,70 (m, 1H). (UPLC–MS) tR 0,49 мин; ESI–MS 554 [M+H]+.
Промежуточное соединение 1a: 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамид
В раствор (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b, 940 мг, 1,55 ммоль) в диоксане (10 мл) добавляли 4 М раствор HCl в диоксане (1,55 мл, 6,20 ммоль) при к. т. Реакционную смесь перемешивали при 65°C в течение 5 ч. и затем концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде гидрохлоридной соли. Необязательно указанное в заголовке соединение дополнительно очищали с помощью prepHPLC (способ 1a) с получением соли TFA после выпаривания растворителей. (UPLC–MS) tR 0,45 мин; ESI–MS 393 [M+H]+.
Промежуточное соединение 1b: (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат
В раствор 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c, 630 мг, 1,59 ммоль) в DMF (10 мл) добавляли транс–4–аминоциклогексанол (290 мг, 1,91 ммоль), HATU (909 мг, 2,39 ммоль) и N–метилморфолин (0,53 мл, 4,78 ммоль) при к. т. Реакционную смесь перемешивали в течение 2 ч., разбавляли насыщ. водн. раствором NaHCO3 и три раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. (UPLC–MS) tR 1,00 мин; ESI–MS 493 [M+H]+.
Промежуточное соединение 1c: 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота
В раствор (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d, 1,00 г, 2,44 ммоль) в THF (15 мл) добавляли 2 М водн. раствор LiOH (3,66 мл, 7,33 ммоль) при к. т. Реакционную смесь перемешивали при 65°C в течение 2 ч. и затем концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения. (UPLC–MS) tR 0,92 мин; ESI–MS 396 [M+H]+.
Промежуточное соединение 1d: (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат
Разделяли рацемический трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат (промежуточное соединение 1e, 13,5 г, 33,0 ммоль) с помощью хиральной препаративной SFC (Waters SFC 200, Waters SFC 200, CHIRALPAK AD–H 5 мкм, 30 * 250 мм, подвижная фаза CO2/i–PrOH, 70:30, скорость потока 120 г/мин.; УФ–детекция при 278 нм). Указанное в заголовке соединение получали в виде грязно–белого твердого вещества после концентрирования при пониженном давлении. Хиральная аналитическая SFC (CHIRALPAK AD–3 3 мкм, 2 * 100 мм, подвижная фаза CO2/MeOH+0,1% DEA, 60:40, скорость потока 1 мл/мин.; УФ–детекция при 274 нм): tR 2,30 мин., ee=98,6%, [α]D20 = +90°.
Промежуточное соединение 1e: трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат
В раствор трет–бутил–1–(4–бромфенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (полученного аналогично описанию в WO 2007/022935) (3,34 г, 9,38 ммоль) в диоксане (75 мл) добавляли метил–2–амино–5–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)никотинат (полученный аналогично описанию в WO 2012/087938) (3,91 г, 14,1 ммоль). Добавляли 2 М водн. раствор K2CO3 (9,38 мл, 18,8 ммоль) и аддукт Pd(dppf)Cl2 и DCM (0,766 г, 0,938 ммоль) в атмосфере азота и смесь нагревали до 80°C в атмосфере азота в течение 18 ч. Реакционную смесь разбавляли с помощью 150 мл воды и три раза экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения в виде грязно–белого твердого вещества. (UPLC–MS) tR 1,29 мин; ESI–MS 410 [M+H]+.
Пример 2: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
В раствор соли TFA и 2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2a, 117 мг, 0,259 ммоль) в DMF (3 мл) добавляли транс–4–аминоциклогексанола гидрохлорид (39,3 мг, 0,259 ммоль), HATU (148 мг, 0,389 ммоль) и N–метилморфолин (0,085 мл, 0,778 ммоль) при к. т. Реакционную смесь перемешивали при к. т. в течение 2 ч. и затем разбавляли насыщ. водн. раствором NaHCO3 и смешивали с EtOAc. После разделения фаз водн. слой снова экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d,1H), 8,31 (d, 1H), 8,11 (d, 1H), 7,58 (d, 2H), 7,23 (d, 2H), 7,12 (s, 2H), 4,58 (d, 1H), 3,82–3,66 (m, 1H), 3,48–3,34 (m, 3H), 3,07 (d, 1H), 2,64–2,55 (m, 2H), 1,96–1,77 (m, 5H), 1,50–1,19 (m, 5H), 1,04 (dd, 6H), 0,75 (dd, 1H). (UPLC–MS) tR 0,50 мин; ESI–MS 435 [M+H]+.
Промежуточное соединение 2a: соль TFA и 2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты
В раствор метил–2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 2b, 660 мг, 1,22 ммоль) в THF (10 мл) добавляли 2 М водн. раствор LiOH (1,22 мл, 2,44 ммоль) при к. т. Реакционную смесь перемешивали при 65°C в течение 2 ч. и затем концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде соли TFA. (UPLC–MS) tR 0,37 мин; ESI–MS 338 [M+H]+.
Промежуточное соединение 2b: метил–2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
В раствор метил–2–амино–5–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)никотината (полученный аналогично описанию в WO 2012/087938) (462 мг, 1,663 ммоль) в ацетонитриле (7 мл) добавляли (1R,5S)–1–(4–бромфенил)–3–изопропил–3–азабицикло[3.1.0]гексан (промежуточное соединение 2c, 466 мг, 1,66 ммоль), 2 М водн. K2CO3 (1,66 мл, 3,33 ммоль) и аддукт PdCl2(dppf) и DCM (67,9 мг, 0,083 ммоль) при к. т. Реакционную смесь закрывали и подвергали облучению в микроволновом реакторе при 120°C в течение 10 мин., затем охлаждали, фильтровали через целит, разбавляли насыщ. водн. раствором NaHCO3 и смешивали с EtOAc. После разделения фаз водн. слой снова экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт применяли без дополнительной очистки. (UPLC–MS) tR 0,62 мин; ESI–MS 352 [M+H]+.
Промежуточное соединение 2c: (1R,5S)–1–(4–бромфенил)–3–изопропил–3–азабицикло[3.1.0]гексан
В раствор (1R,5S)–1–(4–бромфенил)–3–азабицикло[3.1.0]гексана (получен аналогично описанию в WO 2007/022935) (525 мг, 2,21 ммоль) в ацетонитриле (10 мл) добавляли 2–йодпропан (0,420 мл, 2,65 ммоль) и K2CO3 (609 мг, 4,41 ммоль) при к. т. в атмосфере азота. Реакционную смесь перемешивали при 65°C в течение 3 ч., затем разбавляли с помощью EtOAc и смешивали с насыщ. водн. раствором NaHCO3. После разделения фаз водн. фазу экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде грязно–белого твердого вещества. (UPLC–MS) tR 0,72 мин; ESI–MS 280/282 [M+H]+.
Пример 3: 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамид
В раствор (6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)бороновой кислоты (промежуточного соединения 3a, 300 мг, 0,645 ммоль) в диоксане (5 мл) добавляли (R)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептан (промежуточное соединение 3b, 420 мг, 0,774 ммоль), PdCl2(dppf) (23,6 мг, 0,032 ммоль) и 2 н. водн. NaOH (0,645 мл, 1,29 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали при 80°C в течение 2 ч., затем разбавляли с помощью EtOAc и воды. После двух стадий экстракции с помощью EtOAc органические слои промывали насыщ. водн. раствором NaHCO3 и солевым раствором соответственно, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт растворяли в MeOH и пропускали через картридж с силикагелем и тиолом (загрузка 500 мг; предварительно заполненный с помощью MeOH) для удаления палладия. Фильтрат концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения в виде коричневатого твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,37 (d, 1H), 8,27 (d, 1H), 8,09–8,14 (m, 1H), 7,57 (bs, 2H), 7,33 (bs, 2H), 7,12 (bs, 2H), 4,54–4,58 (m, 1H), 4,51–4,66 (m, 3H), 3,55–3,81 (m, 1H), 3,35–3,54 (m, 2H), 3,20–3,24 (m, 1H), 3,20–3,27 (m, 2H), 3,15 (bs, 1H), 1,78–1,91 (m, 4H), 1,14–1,41 (m, 5H), 1,11 (bs, 3H). (UPLC–MS) tR 0,45 мин; ESI–MS 437 [M+H]+.
Промежуточное соединение 3a: (6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)бороновая кислота.
В раствор 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамида (промежуточного соединения 3c, 1,00 г, 2,67 ммоль) в диоксане (25 мл) добавляли бис(пинаколато)дибор (0,815 г, 3,21 ммоль), PdCl2(dppf) (0,098 г, 0,134 ммоль) и KOAc (0,525 г, 5,35 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали при 80°C в течение 18 ч., охлаждали, фильтровали через слой целита и концентрировали при пониженном давлении с получением указанного в заголовке соединения (пинаколатовый сложный эфир гидролизовался при условиях реакции) в виде неочищенного коричневатого твердого вещества, которое применяли без дополнительной очистки. (UPLC–MS) tR 0,32 мин; ESI–MS 280 [M+H]+.
Промежуточное соединение 3b: (R)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептан.
В раствор (R)–1–(4–бромфенил)этанамина (0,216 мл, 1,499 ммоль) в DMF (7 мл) добавляли DIPEA (0,524 мл, 3,00 ммоль) и 3,3–бис(бромметил)оксетан (439 мг, 1,80 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали при 100°C в течение 5 ч., охлаждали и разбавляли водой и EtOAc. После разделения фаз водный слой дважды экстрагировали с помощью EtOAc и объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде желтого масла, которое применяли без дополнительной очистки. (UPLC–MS) tR 0,52 мин; ESI–MS 282/284 [M+H]+.
Промежуточное соединение 3c: 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамид.
В раствор 2–амино–5–бромникотиновой кислоты (2,00 г, 9,22 ммоль) в DMF (30 мл) добавляли транс–4–аминоциклогексанол (1,68 г, 11,1 ммоль), HATU (7,01 г, 18,4 ммоль) и N–метилморфолин (4,05 мл, 36,9 ммоль) при к. т. После перемешивания в течение 2 ч. реакционную смесь разбавляли с помощью EtOAc и насыщ. водн. раствора NaHCO3. Водную фазу два раза экстрагировали с помощью EtOAc. Объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии с нормальной фазой (способ 2a) с получением указанного в заголовке соединения в виде желтого твердого вещества. (UPLC–MS) tR 0,62 мин; ESI–MS 314/316 [M+H]+.
Пример 4: 5–(4–((S)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамид
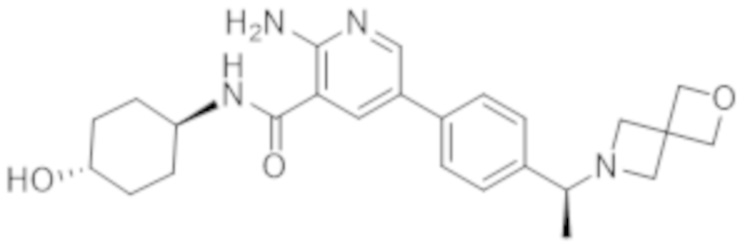
Указанное в заголовке соединение получали способом, аналогичным для 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 3), за исключением того, что применяли (S)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептан (промежуточное соединение 4a) вместо (R)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептана (промежуточного соединения 3b). 1H ЯМР (400 МГц, DMSO–d6) δ 8,37 (d, 1H), 8,27 (d, 1H), 8,09–8,14 (m, 1H), 7,57 (bs, 2H), 7,33 (bs, 2H), 7,12 (bs, 2H), 4,54–4,58 (m, 1H), 4,51–4,66 (m, 3H), 3,55–3,81 (m, 1H), 3,35–3,54 (m, 2H), 3,20–3,24 (m, 1H), 3,20–3,27 (m, 2H), 3,15 (bs, 1H), 1,78–1,91 (m, 4H), 1,14–1,41 (m, 5H), 1,11 (bs, 3H). (UPLC–MS) tR 0,45 мин; ESI–MS 437 [M+H]+.
Промежуточное соединение 4a: (S)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептан
Указанное в заголовке соединение получали способом, аналогичным для (R)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептана (промежуточного соединения 3b), за исключением того, что применяли (S)–1–(4–бромфенил)этанамин вместо (R)–1–(4–бромфенил)этанамина. (UPLC–MS) tR 0,55 мин; ESI–MS 282/284 [M+H]+.
Пример 5: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(3–морфолинопропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
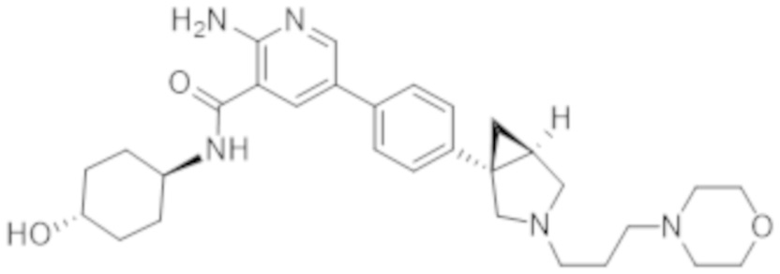
В раствор соли TFA и 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4S)–4–гидроксициклогексил)никотинамида (промежуточного соединения 5a, 70 мг, 0,138 ммоль) в ацетонитриле (3 мл) добавляли K2CO3 (47,7 мг, 0,345 ммоль) и 4–(3–бромпропил)морфолин (47,9 мг, 0,166 ммоль) при к. т. Реакционную смесь перемешивали при 65°C в течение 2 ч. Затем реакционную смесь разбавляли насыщ. водн. раствором NaHCO3 и смешивали с EtOAc. После разделения фаз водн. слой снова экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,31 (d, 1H), 8,11 (d, 1H), 7,58 (d, 2H), 7,22 (d, 2H), 7,12 (s, 2H), 4,58 (d, 1H), 3,82–3,67 (m, 1H), 3,61–3,50 (m, 4H), 3,41 (s, 2H), 3,05 (d, 1H), 2,48–2,24 (m, 9H), 1,93–1,76 (m, 5H), 1,61 (m, 2H), 1,46–1,17 (m, 6H), 0,76 (dd, 1H). (UPLC–MS) tR 0,40 мин; ESI–MS 520 [M+H]+.
Промежуточное соединение 5a: 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамид
В раствор (1S,5R)–трет–бутил–1–(4–(6–амино–5–(((1r,4S)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 5b, 1,18 г, 1,95 ммоль) в диоксане (10 мл) добавляли 4 М раствор HCl в диоксане (1,95 мл, 7,78 ммоль) при к. т. Реакционную смесь перемешивали при 65°C в течение 5 ч. и затем концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде гидрохлоридной соли. Необязательно продукт дополнительно очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде соли TFA после выпаривания растворителей. (UPLC–MS) tR 0,45 мин; ESI–MS 393 [M+H]+.
Промежуточное соединение 5b: (1S,5R)–трет–бутил–1–(4–(6–амино–5–(((1r,4S)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 5c) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c). (UPLC–MS) tR 1,00 мин; ESI–MS 493 [M+H]+.
Промежуточное соединение 5c: 2–амино–5–(4–((1S,5R)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c), за исключением того, что применяли (1S,5R)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат (промежуточное соединение 5d) вместо (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d). (UPLC–MS) tR 0,92 мин; ESI–MS 396 [M+H]+.
Промежуточное соединение 5d: (1S,5R)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат
Разделяли рацемический трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат (промежуточное соединение 1e, 13,5 г, 33,0 ммоль) с помощью хиральной препаративной SFC (CHIRALPAK AD–H 5 мкм 30 * 250 мм, подвижная фаза CO2/i–PrOH, 70:30, скорость потока 120 г/мин.; УФ–детекция при 278 нм). Указанное в заголовке соединение получали в виде грязно–белого твердого вещества после концентрирования при пониженном давлении. Хиральная аналитическая HPLC (CHIRALPAK AD–3 3 мкм, 2 * 100 мм, подвижная фаза CO2/MeOH+0,1% DEA, 60:40, скорость потока 1 мл/мин.; УФ–детекция при 274 нм): tR 2,90 мин., ee=98,7%, [α]D20 = −87°.
Пример 6: 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамид
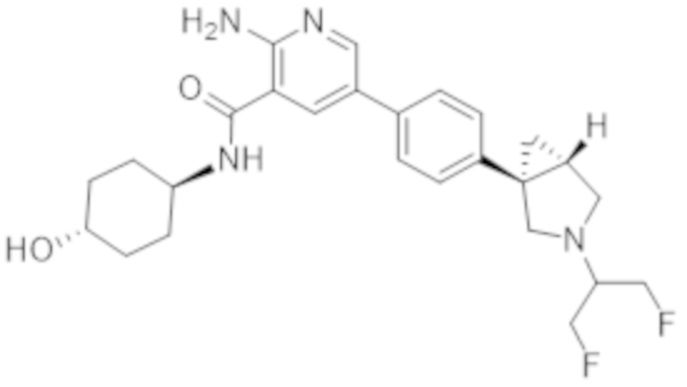
В раствор соли TFA и 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 1a, 70 мг, 0,138 ммоль) в ацетонитриле (3 мл) добавляли K2CO3 (38,2 мг, 0,276 ммоль) и 1,3–дифторпропан–2–илтрифторметансульфонат (промежуточное соединение 6a) (37,8 мг, 0,166 ммоль) при к. т. Реакционную смесь перемешивали при к. т. в течение 1 ч., затем разбавляли насыщ. водн. раствором NaHCO3 и смешивали с EtOAc. После разделения фаз водн. слой снова экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, DMSO–d6) δ 8,41 (d, 1H), 8,37 (d, 1H), 8,18 (s, 1H), 7,62 (s, 2H), 7,35 (d, 1H), 7,25 (m, 3H), 4,64 (d, 3H), 3,74 (m, 4H), 2,89 (d, 4H), 1,87 (t, 5H), 1,34 (m, 6H), 0,82 (s, 1H). (UPLC–MS) tR 0,68 мин; ESI–MS 471 [M+H]+.
Промежуточное соединение 6a: 1,3–дифторпропан–2–илтрифторметансульфонат
В раствор 1,3–дифторпропан–2–ола (300 мг, 3,12 ммоль) в DCM (8 мл) добавляли DMAP (26,7 мг, 0,219 ммоль) и TEA (0,522 мл, 3,75 ммоль) в атмосфере азота при к. т. Реакционную смесь охлаждали до 0°C и добавляли трифторметансульфоновый ангидрид (0,630 мл, 3,75 ммоль). После перемешивания при 0°C в течение 60 мин. и при к. т. в течение 3 ч. реакционную смесь разбавляли с помощью DCM. Органический слой промывали водой, дважды водн. раствором лимонной кислоты и дважды насыщ. водн. раствором NaHCO3, высушивали над MgSO4 и концентрировали при 500 мбар. Указанное в заголовке соединение получали в виде неочищенного масла, которое применяли без дополнительной очистки.
Пример 7: 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
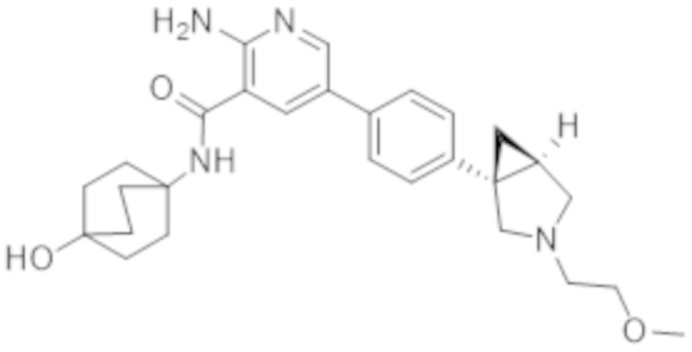
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 7a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли 4–аминобицикло[2.2.2]октан–1–ола гидрохлорид вместо транс–4–аминоциклогексанола. Неочищенный продукт сначала очищали с помощью prepHPLC (способ 1a), затем с помощью флэш–хроматографии с обращенной фазой (способ 3a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,34 (d, 1H), 7,98 (d, 1H), 7,79 (s, 1H), 7,57 (d, 2H), 7,21 (d, 2H), 6,92 (s, 2H), 4,32 (s, 1H), 3,45 (t, 2H), 3,41–3,29 (m, 2H), 3,27 (s, 3H), 3,08 (d, 1H), 2,68–2,51 (m, 3H), 2,07–2,03 (m, 6H), 1,83–1,79 (m, 1H), 1,65–1,61 (m, 6H), 1,33 (t, 1H), 0,77 (bs, 1H). (UPLC–MS) tR 0,52 мин; ESI–MS 477 [M+H]+.
Промежуточное соединение 7a: 2–амино–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2c), за исключением того, что применяли метил–2–амино–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат (промежуточное соединение 7b) вместо (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d). (UPLC–MS) tR 0,32 мин; ESI–MS 354 [M+H]+.
Промежуточное соединение 7b: метил–2–амино–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
В раствор метил–5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорида (промежуточного соединения 7c, 210 мг, 0,607 ммоль) в ацетонитриле (6 мл) добавляли K2CO3 (168 мг, 0,729 ммоль) и 1–бром–2–метоксиэтан (0,068 мл, 0,729 ммоль) при к. т. Реакционную смесь перемешивали при 65°C в течение 3 ч. Затем реакционную смесь разбавляли насыщ. водн. раствором NaHCO3 и смешивали с EtOAc. После разделения фаз водн. слой снова экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения, которое применяли без дополнительной очистки. (UPLC–MS) tR 0,62 мин; ESI–MS 368 [M+H]+.
Промежуточное соединение 7c: метил–5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорид
В раствор (1S,5R)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 5d, 500 мг, 1,22 ммоль) в диоксане (7 мл) добавляли 4 М раствор HCl в диоксане (1,22 мл, 4,88 ммоль) при к. т. Реакционную смесь перемешивали при 60°C в течение 3 ч. и затем концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде гидрохлоридной соли. (UPLC–MS) tR 0,58 мин; ESI–MS 310 [M+H]+.
Пример 8: 2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамид
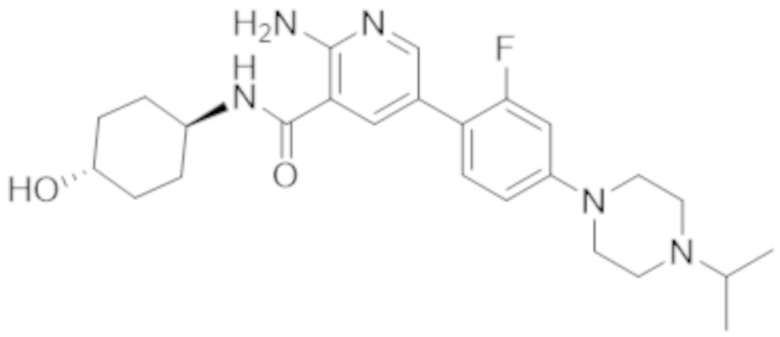
В раствор 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамида (промежуточного соединения 3c, 230 мг, 0,731 ммоль) в диоксане (4 мл) добавляли 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазин (промежуточное соединение 8a, 463 мг, 0,731 ммоль), 2 н. водн. K2CO3 (0,73 мл, 1,46 ммоль) и аддукт PdCl2(dppf) и DCM (29,9 мг, 0,037 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали при 80°C в течение 60 мин., затем разбавляли с помощью EtOAc и водн. NaHCO3. После разделения фаз водн. слой экстрагировали с помощью EtOAc. Органические слои промывали с помощью насыщ. водн. раствора NaHCO3 и солевого раствора соответственно, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,24–8,16 (m, 2H), 7,95 (d, 1H), 7,39 (t, 1H), 7,06 (s, 2H), 6,86 (d, 2H), 4,54 (d,1H), 3,69 (m, 1H), 3,41–3,33 (m, 2H), 3,15 (s, 3H), 2,66 (q, 3H), 1,82 (m, 4H), 1,42–0,88 (m, 10H). (UPLC–MS) tR 0,48 мин; ESI–MS 456 [M+H]+.
Промежуточное соединение 8a: 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазин
В раствор 1–(4–бром–3–фторфенил)–4–изопропилпиперазина (промежуточного соединения 8b, 212 мг, 0,662 ммоль) в диоксане (8 мл) добавляли бис(пинаколато)дибор (202 мг, 0,794 ммоль), KOAc (130 мг, 1,323 ммоль) и аддукт PdCl2(dppf) и DCM (27 мг, 0,033 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали при 90°C в течение 2 ч., затем фильтровали через слой целита и концентрировали при пониженном давлении с получением указанного в заголовке соединения, которое применяли без какой–либо дополнительной очистки. (UPLC–MS) tR 0,79 мин; ESI–MS 349 [M+H]+.
Промежуточное соединение 8b: 1–(4–бром–3–фторфенил)–4–изопропилпиперазин
В раствор 1–(4–бром–3–фторфенил)пиперазина (промежуточного соединения 8c, 215 мг, 0,797 ммоль) в ацетонитриле (7 мл) добавляли 2–йодпропан (0,095 мл, 0,956 ммоль) и K2CO3 (220 мг, 1,593 ммоль) при к. т. Реакционную смесь перемешивали при 65°C в течение 4 ч., затем разбавляли с помощью EtOAc и водн. NaHCO3. После разделения фаз водн. слой экстрагировали с помощью EtOAc. Органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла, которое применяли без какой–либо дополнительной очистки. (UPLC–MS) tR 0,66 мин; ESI–MS 302 [M+H]+.
Промежуточное соединение 8c: 1–(4–бром–3–фторфенил)пиперазин
В раствор трет–бутил–4–(4–бром–3–фторфенил)пиперазин–1–карбоксилата (300 мг, 0,835 ммоль) в диоксане (8 мл) добавляли 4 н. HCl в диоксане (0,84 мл, 3,34 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали в течение 2 ч., затем разбавляли с помощью EtOAc и водн. NaHCO3. После разделения фаз водн. слой экстрагировали с помощью EtOAc. Органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения, которое применяли без какой–либо дополнительной очистки. (UPLC–MS) tR 0,60 мин; ESI–MS 260 [M+H]+.
Пример 9: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
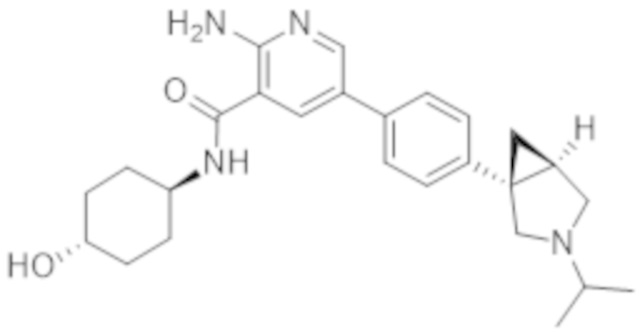
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 2), за исключением того, что применяли соль TFA и 2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 9a) вместо соли TFA и 2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d,1H), 8,31 (d, 1H), 8,11 (d, 1H), 7,58 (d, 2H), 7,23 (d, 2H), 7,12 (s, 2H), 4,58 (d, 1H), 3,82–3,66 (m, 1H), 3,48–3,34 (m, 3H), 3,07 (d, 1H), 2,64–2,55 (m, 2H), 1,96–1,77 (m, 5H), 1,50–1,19 (m, 5H), 1,04 (dd, 6H), 0,75 (dd, 1H). (UPLC–MS) tR 0,50 мин; ESI–MS 435 [M+H]+.
Промежуточное соединение 9a: соль TFA и 2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты
Указанное в заголовке соединение получали способом, аналогичным для соли TFA и 2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2a), за исключением того, что применяли метил–2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат (промежуточное соединение 9b) вместо метил–2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 2b). (UPLC–MS) tR 0,37 мин; ESI–MS 338 [M+H]+.
Промежуточное соединение 9b: метил–2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
Указанное в заголовке соединение получали способом, аналогичным для метил–2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 2b), за исключением того, что применяли (1S,5R)–1–(4–бромфенил)–3–изопропил–3–азабицикло[3.1.0]гексан (промежуточное соединение 9c) вместо (1R,5S)–1–(4–бромфенил)–3–изопропил–3–азабицикло[3.1.0]гексана (промежуточного соединения 2c). (UPLC–MS) tR 0,62 мин; ESI–MS 352 [M+H]+.
Промежуточное соединение 9c: (1S,5R)–1–(4–бромфенил)–3–изопропил–3–азабицикло[3.1.0]гексан
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–1–(4–бромфенил)–3–изопропил–3–азабицикло[3.1.0]гексана (промежуточного соединения 2c), за исключением того, что применяли (1S,5R)–1–(4–бромфенил)–3–азабицикло[3.1.0]гексан (получен аналогично описанию в WO 2007/022935) вместо (1R,5S)–1–(4–бромфенил)–3–азабицикло[3.1.0]гексана. (UPLC–MS) tR 0,70 мин; ESI–MS 280/282 [M+H]+.
Пример 10: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–морфолиноэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
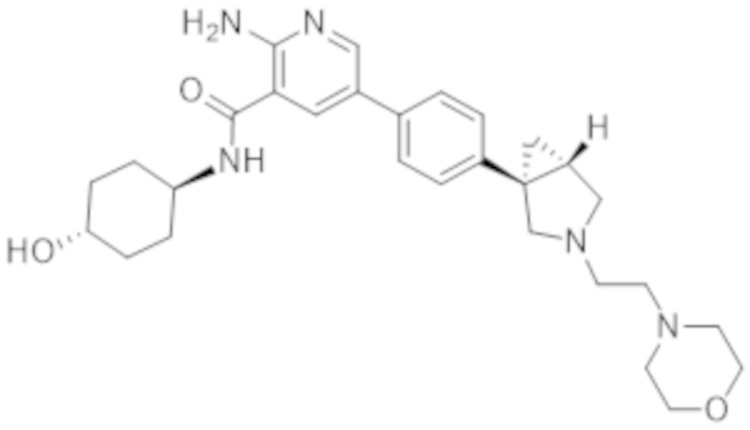
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 4–(2–бромэтил)морфолин вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a) и реакционную смесь перемешивали при 65°C в течение 1 ч. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,31 (d, 1H), 8,11 (d, 1H), 7,59 (d, 2H), 7,22 (d, 2H), 7,12 (s, 2H), 4,59 (d, 1H), 3,84–3,67 (m, 1H), 3,63–3,51 (m, 4H), 3,46–3,39 (m, 2H), 3,09 (d, 1H), 2,71–2,57 (m, 3H), 2,42 (s, 6H), 1,95–1,75 (m, 5H), 1,53–1,15 (m, 5H), 0,77 (d, 1H). (UPLC–MS) tR 0,49 мин; ESI–MS 506 [M+H]+.
Пример 11: 2–амино–5–(4–((1S,5R)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамид
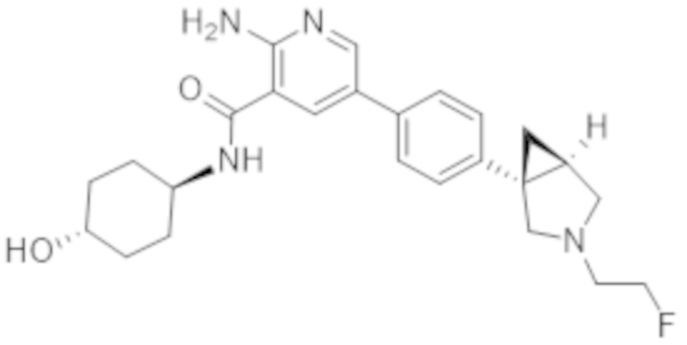
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 1–бром–2–фторэтан вместо 4–(3–бромпропил)морфолина и реакционную смесь перемешивали при 60°C в течение 5 ч. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,31 (d, 1H), 8,11 (d, 1H), 7,59 (d, 2H), 7,22 (d, 2H), 7,13 (s, 2H), 4,59 (d, 2H), 4,50 (s,1H), 3,73 (d, 1H), 3,41 (d, 3H), 2,83–2,78 (m, 3H), 1,85 (t, 5H), 1,47–1,16 (m, 6H), 0,78 (s, 1H). (UPLC–MS) tR 0,47 мин; ESI–MS 439 [M+H]+.
Пример 12: 2–амино–N–((1r,4r)–4–гидроксициклогексил)–5–(4–(1–изопропилпирролидин–3–ил)фенил)никотинамид
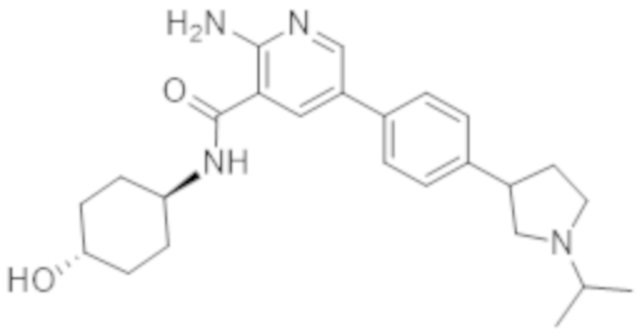
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамида (примера 8), за исключением того, что применяли 1–изопропил–3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)пирролидин (промежуточное соединение 12a) вместо 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (промежуточного соединения 8a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,36 (d,1H), 8,26 (d, 1H), 8,09 (d, 1H), 7,62–7,51 (m, 2H), 7,37–7,28 (m, 2H), 7,09 (s, 2H), 4,55 (d, 1H), 3,70 (dq, 1H), 3,44–3,34 (m, 2H), 3,03 (d, 1H), 2,75 (s, 2H), 2,29–2,06 (m, 2H), 1,87–1,68 (m, 5H), 1,42–1,17 (m, 5H), 1,06 (t, 6H). (UPLC–MS) tR 0,48 мин; ESI–MS 423 [M+H]+.
Промежуточное соединение 12a: 1–изопропил–3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)пирролидин
Указанное в заголовке соединение получали способом, аналогичным для 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (промежуточного соединения 8a), за исключением того, что применяли 3–(4–бромфенил)–1–изопропилпирролидин (промежуточное соединение 12b) вместо 1–(4–бром–3–фторфенил)–4–изопропилпиперазина (промежуточного соединения 8b). (UPLC–MS) tR 0,79 мин; ESI–MS 316 [M+H]+.
Промежуточное соединение 12b: 3–(4–бромфенил)–1–изопропилпирролидин
Указанное в заголовке соединение получали способом, аналогичным для 1–(4–бром–3–фторфенил)–4–изопропилпиперазина (промежуточного соединения 8b), за исключением того, что применяли 3–(4–бромфенил)пирролидина гидрохлорид вместо 1–(4–бром–3–фторфенил)пиперазина (промежуточного соединения 8c). (UPLC–MS) tR 0,62 мин; ESI–MS 269 [M+H]+.
Пример 13: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2–морфолиноэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
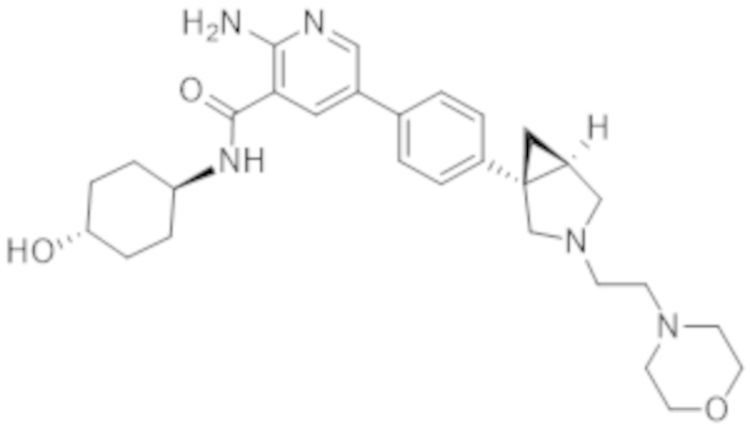
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 4–(2–бромэтил)морфолин вместо 4–(3–бромпропил)морфолина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,31 (d, 1H), 8,11 (d, 1H), 7,59 (d, 2H), 7,22 (d, 2H), 7,12 (s, 2H), 4,59 (d, 1H), 3,84–3,67 (m, 1H), 3,63–3,51 (m, 4H), 3,46–3,39 (m, 2H), 3,09 (d, 1H), 2,71–2,57 (m, 3H), 2,42 (s, 6H), 1,95–1,75 (m, 5H), 1,53–1,15 (m, 5H), 0,77 (d, 1H). (UPLC–MS) tR 0,49 мин; ESI–MS 506 [M+H]+.
Пример 14: 2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
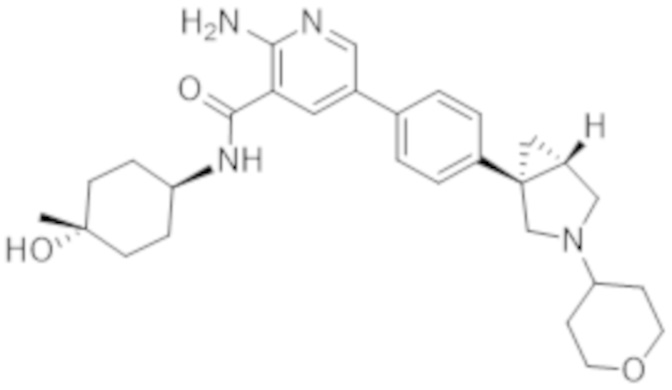
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 14a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли транс–4–амино–1–метилциклогексанол вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 8,53 (d, 1H), 8,46 (d, 1H), 8,35 (s, 1H), 7,72 (d, 2H), 7,71 (bs, 2H), 7,41 (d, 2H), 4,06 (dd, 1H), 3,98 (dd, 2H), 3,85–3,43 (m, 5H), 3,27 (t, 2H), 2,26 (dd, 1H), 1,99 (d, 2H), 1,83–1,76 (m, 4H), 1,68–1,56 (m, 3H), 1,51–1,45 (m, 5H), 1,21–1,13 (m, 5H). (UPLC–MS) tR 0,51 мин; ESI–MS 491 [M+H]+.
Промежуточное соединение 14a: 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота (соль TFA)
В раствор метил–2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 14b, 1,30 г, 2,97 ммоль) в безводном THF (16 мл) добавляли 2 М водн. раствор LiOH (4,46 мл, 8,92 ммоль) при к. т. Реакционную смесь перемешивали при 65°C в течение 140 мин. и затем концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии с обращенной фазой (способ 3b). Чистые фракции концентрировали и лиофилизировали с получением указанного в заголовке соединения в виде соли TFA грязно–белого цвета. (UPLC–MS) tR 0,40 мин; ESI–MS 380 [M+H]+.
Промежуточное соединение 14b: метил–2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
В раствор метил–5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорида (промежуточного соединения 14c, 1,18 г, 3,28 ммоль) в DCM (16 мл) добавляли дигидро–2H–пиран–4(3H)–он (0,33 мл, 3,60 ммоль) и AcOH (0,38 мл, 6,55 ммоль) при к. т. Реакционную смесь перемешивали при 45°C в течение 30 мин. Добавляли триацетоксиборогидрид натрия (1,74 г, 8,19 ммоль) при к. т. и реакционную смесь перемешивали при 45°C в течение 1 ч. Затем реакционную смесь разбавляли насыщ. раствором NaHCO3 и смешивали с EtOAc. После разделения фаз водн. слой снова экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения, которое применяли без дополнительной очистки. (UPLC–MS) tR 0,67 мин; ESI–MS 394 [M+H]+.
Промежуточное соединение 14c: метил–5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорид
В раствор (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d, 1,50 г, 3,66 ммоль) в диоксане (25 мл) добавляли 4 М раствор HCl в диоксане (3,66 мл, 14,7 ммоль) при к. т. Реакционную смесь перемешивали при 60°C в течение 3 ч. и затем концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде гидрохлоридной соли. (UPLC–MS) tR 0,58 мин; ESI–MS 310 [M+H]+.
Пример 15: 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(2–морфолиноэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
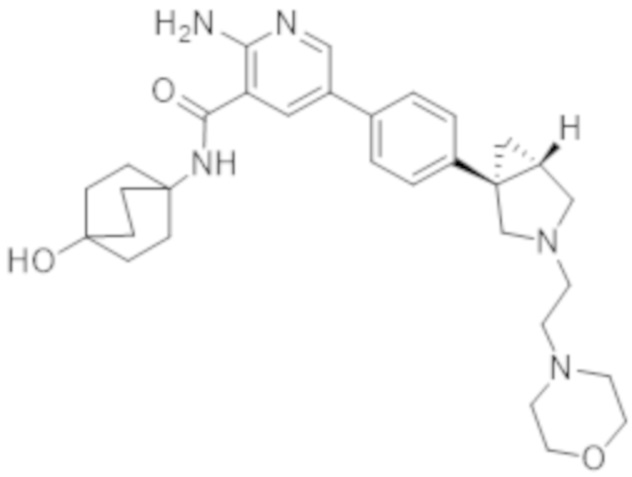
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1R,5S)–3–(2–морфолиноэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 15a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли 4–аминобицикло[2.2.2]октан–1–ола гидрохлорид вместо транс–4–аминоциклогексанола. Неочищенный продукт сначала очищали с помощью prepHPLC (способ 1a), затем с помощью флэш–хроматографии с обращенной фазой (способ 3a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,34 (d, 1H), 7,98 (d, 1H), 7,79 (s, 1H), 7,56 (d, 2H), 7,21 (d, 2H), 6,92 (s, 2H), 4,31 (s, 1H), 3,58–3,56 (m, 4H), 3,09 (d, 1H), 2,64–2,62 (m, 2H), 2,59–2,41 (m, 9H), 2,07–2,04 (m, 6H), 1,82–1,79 (m, 1H), 1,65–1,61 (m, 6H), 1,31 (t, 1H), 0,75 (dd, 1H). (UPLC–MS) tR 0,53 мин; ESI–MS 532 [M+H]+.
Промежуточное соединение 15a: 2–амино–5–(4–((1R,5S)–3–(2–морфолиноэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2c), за исключением того, что применяли метил–2–амино–5–(4–((1R,5S)–3–(2–морфолиноэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат (промежуточное соединение 15b) вместо (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d). (UPLC–MS) tR 0,39 мин; ESI–MS 409 [M+H]+.
Промежуточное соединение 15b: метил–2–амино–5–(4–((1R,5S)–3–(2–морфолиноэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
Указанное в заголовке соединение получали способом, аналогичным для метил–2–амино–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 7b), за исключением того, что применяли метил–5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорид (промежуточное соединение 14c) вместо метил–5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорида (промежуточного соединения 7c) и применяли 4–(2–бромэтил)морфолин вместо 1–бром–2–метоксиэтана. (UPLC–MS) tR 0,62 мин; ESI–MS 423 [M+H]+.
Пример 16: 2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
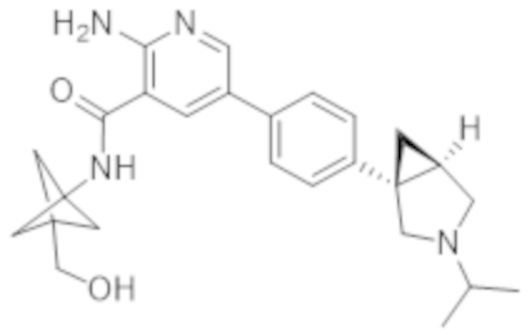
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 2), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 9a) вместо соли TFA и 2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2a) и применяли (3–аминобицикло[1.1.1]пентан–1–ил)метанол (промежуточное соединение 16a) вместо транс–4–аминоциклогексанола гидрохлорида. 1H ЯМР (400 МГц, DMSO–d6) δ 9,05 (s, 1H), 8,41 (d,1H), 8,18 (d,1H), 7,61 (d, 2H), 7,48 (dd, 1H), 7,33–7,17 (m, 4H), 4,55 (t, 1H), 3,51 (d, 3H), 3,31 (s, 2H), 3,08 (s, 2H), 2,07–1,99 (m, 1H), 1,41–1,22 (m, 3H), 1,17–0,98 (m, 9H), 0,79 (d, 1H). (UPLC–MS) tR 0,54 мин; ESI–MS 433 [M+H]+.
Промежуточное соединение 16a: (3–аминобицикло[1.1.1]пентан–1–ил)метанол
В раствор соли HCl и метил–3–аминобицикло[1.1.1]пентан–1–карбоксилата (218 мг, 1,23 ммоль) в THF (7 мл) добавляли небольшими порциями LiAlH4 (140 мг, 3,68 ммоль) при 0°C. Реакционную смесь перемешивали при к. т. в течение 2 ч. и гасили при 0°C смесью THF и воды, затем фильтровали через слой целита и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде желтого масла, которое применяли без дополнительной очистки. FIA–MS 114 [M+H]+.
Пример 17: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(3,3,3–трифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
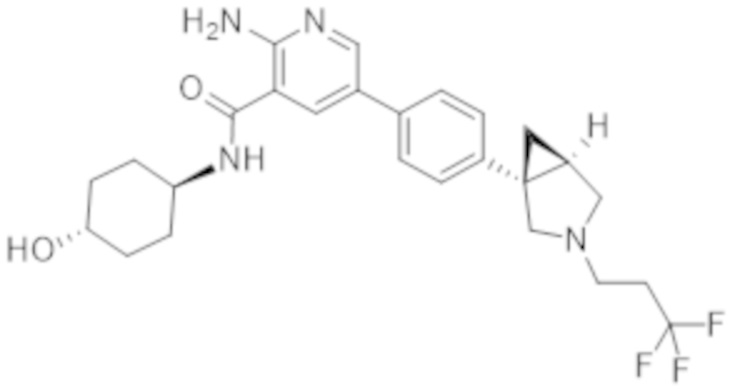
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 3,3,3–трифторпропилтрифторметансульфонат вместо 4–(3–бромпропил)морфолина и реакционную смесь перемешивали при к. т. в течение 60 мин. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,60 (d, 2H), 7,23 (d, 2H), 7,12 (s, 2H), 4,59 (d, 1H), 3,79–3,66 (m, 1H), 3,39 (m, 3H), 3,09 (s, 1H), 2,72–2,59 (m, 4H), 1,86 (m, 5H), 1,52–1,18 (m, 6H), 0,79 (s, 1H). (UPLC–MS) tR 0,59 мин; ESI–MS 489 [M+H]+.
Пример 18: 2–амино–N–((1r,4r)–4–гидроксициклогексил)–5–(4–(пирролидин–3–ил)фенил)никотинамид
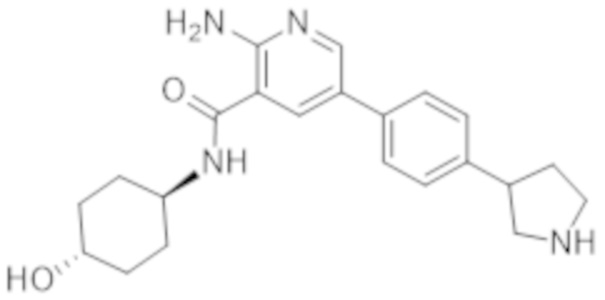
В раствор трет–бутил–3–(4–(6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)пирролидин–1–карбоксилата (промежуточного соединения 18a, 120 мг, 0,140 ммоль) в диоксане (3 мл) добавляли 4 н. HCl в диоксане (0,210 мл, 0,839 ммоль) в атмосфере азота при к. т. и полученную смесь перемешивали в течение 4 ч. После концентрирования при пониженном давлении неочищенный продукт очищали с помощью хроматографии с обращенной фазой (способ 3a). Чистые фракции разделяли между насыщ. водн. раствором NaHCO3 и EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде смеси диастереомеров. 1H ЯМР (400 МГц, DMSO–d6) δ 8,26 (d,1H), 8,09 (d, 1H), 7,57 (d, 2H), 7,35–7,29 (m, 2H), 7,09 (s, 2H), 4,55 (d, 1H), 3,77–3,69 (m, 2H), 3,22–3,15 (m, 2H), 3,12–2,91 (m, 2H), 2,74–2,62 (m, 2H), 2,24–2,07 (m, 1H), 1,83 (dd, 4H), 1,42–1,16 (m, 5H). (UPLC–MS) tR 0,42 мин; ESI–MS 381 [M+H]+.
Промежуточное соединение 18a: трет–бутил–3–(4–(6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)пирролидин–1–карбоксилат
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамида (примера 8), за исключением того, что применяли трет–бутил–3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)пирролидин–1–карбоксилат (промежуточное соединение 18b) вместо 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (промежуточного соединения 8a). (UPLC–MS) tR 0,95 мин; ESI–MS 481 [M+H]+.
Промежуточное соединение 18b: трет–бутил–3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)пирролидин–1–карбоксилат
Указанное в заголовке соединение получали способом, аналогичным для 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (промежуточного соединения 8a), за исключением того, что применяли трет–бутил–3–(4–бромфенил)пирролидин–1–карбоксилат (промежуточное соединение 18c) вместо 1–(4–бром–3–фторфенил)–4–изопропилпиперазина (промежуточного соединения 8b). (UPLC–MS) tR 1,43 мин; ESI–MS 374 [M+H]+.
Промежуточное соединение 18c: трет–бутил–3–(4–бромфенил)пирролидин–1–карбоксилат
В раствор 3–(4–бромфенил)пирролидина (200 мг, 0,885 ммоль) в DCM (7 мл) добавляли ди–трет–бутилдикарбонат (0,308 мл, 1,327 ммоль) и TEA (0,247 мл, 1,769 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали в течение 2 ч. Затем реакционную смесь разбавляли насыщ. водн. раствором NaHCO3 и смешивали с DCM. После разделения фаз водн. слой снова экстрагировали с помощью DCM. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла, которое применяли без дополнительной очистки. (UPLC–MS) tR 1,33 мин; ESI–MS 270 [M+H]+.
Пример 19: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
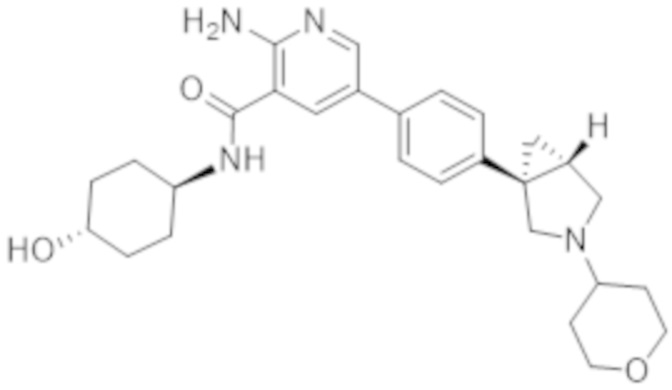
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 1), за исключением того, что применяли дигидро–2H–пиран–4(3H)–он вместо 1–(метилсульфонил)пиперидин–4–она. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (s, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,62 (dd, 2H), 7,25 (s, 2H), 7,12 (s, 2H), 4,59 (d, 1H), 3,97–3,63 (m, 4H), 3,40 (m, 2H), 3,31 (m, 2H), 3,19–3,03 (m, 2H), 1,99–1,65 (m, 8H), 1,51–1,16 (m, 8H). (UPLC–MS) tR 0,48 мин; ESI–MS 477 [M+H]+.
Пример 20: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
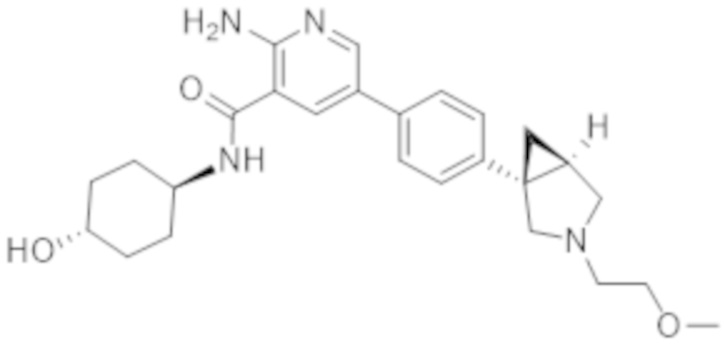
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 1–бром–2–метоксиэтан вместо 4–(3–бромпропил)морфолина и реакционную смесь перемешивали при 60°C в течение 4 ч. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (s, 1H), 8,11 (s, 1H), 7,59 (d, 2H), 7,22 (d, 2H), 7,13 (s, 2H), 4,60 (d, 1H), 3,73 (d, 1H), 3,54–3,36 (m, 4H), 3,27 (s, 3H), 2,64 (m, 4H), 1,87 (m, 6H), 1,40–1,27 (m, 6H), 0,76 (s, 1H). (UPLC–MS) tR 0,49 мин; ESI–MS 451 [M+H]+.
Пример 21: 2–амино–N–((1R,3S,4R)–3–фтор–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
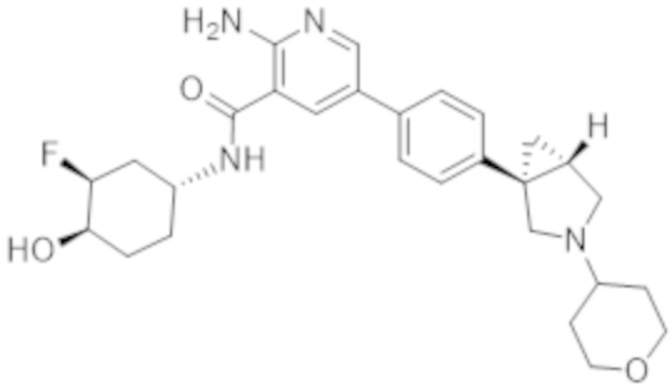
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 14a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли (1R,2S,4R)–4–амино–2–фторциклогексанола гидрохлорид (промежуточное соединение 21a) вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 9,60 (bs, 1H), 8,42 (s, 1H), 8,35 (d, 1H), 8,12 (s, 1H), 7,64 (bs, 2H), 7,36 (bs, 2H), 7,14 (s, 2H), 4,94 (t, 1H), 4,80 (d, 1H), 4,16–3,82 (m, 4H), 3,81–3,45 (m, 4H), 3,30–3,25 (m, 4H), 2,15 (bs, 1H), 1,88–1,85 (m, 2H), 1,65–1,59 (m, 5H), 1,76–1,27 (m, 3H), 1,18 (bs, 1H). (UPLC–MS) tR 0,48 мин; ESI–MS 495 [M+H]+.
Промежуточное соединение 21a: (1R,2S,4R)–4–амино–2–фторциклогексанола гидрохлорид
В раствор бензил–((1R,3S,4R)–3–фтор–4–гидроксициклогексил)карбамата (промежуточного соединения 21b, 376 мг, 1,41 ммоль) в EtOH (20 мл) добавляли палладий на угле (10%) (80 мг, 0,075 ммоль) в атмосфере азота. Реакционный сосуд три раза продували азотом, затем три раза продували водородом. После перемешивания при к. т. в течение 17 ч. реакционную смесь пропускали через слой целита и осадок на фильтре промывали с помощью EtOH с получением бесцветного раствора. Медленно добавляли хлористоводородную кислоту (1,25 М в EtOH, 14 мл, 17,5 ммоль) и смесь перемешивали при к. т. в течение 5 ч. После концентрирования при пониженном давлении получали указанное в заголовке соединение в виде грязно–белого гигроскопичного твердого вещества. FIA ESI–MS 134 [M+H]+.
Промежуточное соединение 21b: бензил–((1R,3S,4R)–3–фтор–4–гидроксициклогексил)карбамат
В раствор бензил–((1R,3S)–3–фтор–4–оксоциклогексил)карбамата (промежуточного соединения 21c, 768 мг, 2,90 ммоль) в MeOH (15 мл) добавляли порциями NaBH4 (274 мг, 7,24 ммоль) при 0°C. После перемешивания в течение 30 мин. при 0°C реакционную смесь разбавляли насыщ. водн. раствором NH4Cl и обеспечивали его нагревание до к. т. Растворитель удаляли при пониженном давлении и добавляли DCM и воду. После разделения фаз водн. слой три раза экстрагировали с помощью DCM. Объединенные органические слои промывали солевым раствором, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении с получением белого твердого вещества. Неочищенный продукт очищали с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. (UPLC–MS) tR 0,76 мин; ESI–MS 268 [M+H]+. Изображенную абсолютную конфигурацию подтверждали с помощью рентгеноструктурной кристаллографии.
Промежуточное соединение 21c: бензил–((1R,3S)–3–фтор–4–оксоциклогексил)карбамат
В раствор бензил–((1R,3S,4S)–3–фтор–4–гидроксициклогексил)карбамата (промежуточного соединения 21d, 710 мг, 2,66 ммоль) в DCM (26 мл) добавляли хлорхромат пиридиния (859 мг, 3,98 ммоль). Реакционную смесь перемешивали в течение 6 ч. при к. т. Добавляли другую порцию хлорхромата пиридиния (573 мг, 2,66 ммоль) и реакционную смесь перемешивали в течение еще 18 ч. После добавления DCM и насыщ. водн. раствора NaHCO3 обе фазы разделяли, водный слой дважды экстрагировали с помощью DCM и объединенные органические слои промывали солевым раствором, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения в виде бесцветного масла. (UPLC–MS) tR 0,82 мин; ESI–MS 266 [M+H]+.
Промежуточное соединение 21d: бензил–((1R,3S,4S)–3–фтор–4–гидроксициклогексил)карбамат
В сосуд загружали бензил–(1R,3R,6S)–7–оксабицикло[4.1.0]гептан–3–илкарбамат (промежуточное соединение 21e, 1,50 г, 6,07 ммоль) и добавляли тригидрофторид TEA (4,94 мл, 30,3 ммоль). Сосуд закрывали и его содержимое перемешивали при 100°C в течение 2 ч. После охлаждения реакционную смесь медленно выливали в перемешиваемый раствор K2CO3 (5,87 г, 42,5 ммоль) в воде (400 мл) и три раза экстрагировали с помощью DCM. Объединенные органические слои промывали водой и солевым раствором, высушивали над безводным MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный материал очищали с помощью хроматографии с нормальной фазой (способ 2b) с получением смеси региоизомеров, которую разделяли с помощью препаративной хиральной HPLC (колонка: ChiralPak AD, 20 мкм, 50×5 см, скорость потока: 90 мл/мин., длина волны для выявления: 220 нм, подвижная фаза: н–гептан:EtOH, 90:10 до t=52 мин., 85:15 до t=69 мин., затем 80:20). Указанное в заголовке соединение выделяли в виде бесцветного масла и определяли его характеристики с помощью UPLC–MS (SQ13, колонка: Acquity HSS T3 1,8 мкм 2,1×50 мм при 60°C, элюент A: вода+0,05% муравьиной кислоты+3,75 мМ NH4OAc, элюент B: ацетонитрил+0,04% муравьиной кислоты, градиент: от 5 до 98% B за 1,4 мин., поток 1,0 мл/мин., tR 0,81 мин.; ESI–MS 268 [M+H]+).
Промежуточное соединение 21e: (1R,3R,6S)–7–оксабицикло[4.1.0]гептан–3–илкарбамат
Разделяли син–бензил–(7–оксабицикло[4.1.0]гептан–3–ил)карбамат (полученный, как описано у Gómez–Sánchez et al, Tetrahedron 2005, 61(5), 1207–1219) с помощью препаративной хиральной HPLC (колонка: ChiralPak AY, 10 мкм, 25×5 см, скорость потока: 30 мл/мин., длина волны для выявления: 214 нм, подвижная фаза: н–гептан:изопропанол, 80:20). Указанное в заголовке соединение выделяли в виде бесцветного масла и определяли его характеристики с помощью хиральной HPLC (ChiralPak AY–H, 5 мкм, 15×0,46 см, скорость потока: 1 мл/мин., длина волны для выявления: 214 нм, подвижная фаза: гексан:изопропанол, 70:30, tR 2,24 мин., ee 99,2%). (UPLC–MS) tR 0,90 мин; ESI–MS 248 [M+H]+.
Пример 22: 2–амино–N–((1s,4S)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
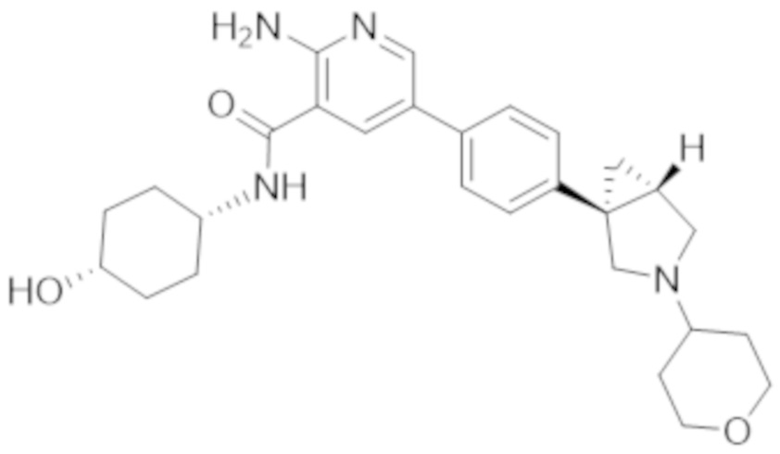
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 14a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли цис–4–аминоциклогексанол вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 9,50 (bs, 1H), 8,41–8,33 (m, 2H), 8,17 (d, 1H), 7,62 (bs, 2H), 7,26 (bs, 2H), 7,14 (s, 1H), 4,42 (d, 1H), 4,05–3,79 (m, 5H), 3,34–3,24 (m, 4H), 3,11 (bs, 1H), 2,70–2,50 (m, 1H), 1,83–1,71 (m, 7H), 1,62–1,47 (m, 7H), 1,35–1,29 (m, 1H). (UPLC–MS) tR 0,52 мин; ESI–MS 477 [M+H]+.
Пример 23: 5–(4–(2–азаспиро[3.3]гептан–2–илметил)фенил)–2–амино–N–((1r,4r)–4–гидроксициклогексил)никотинамид
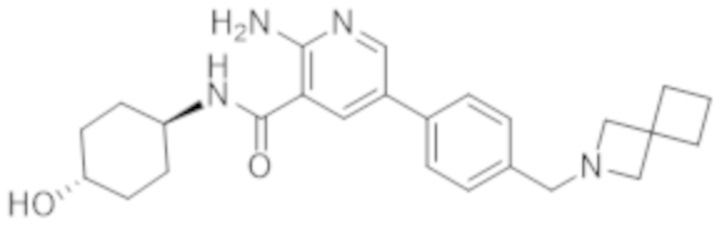
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамида (примера 8), за исключением того, что применяли 2–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)бензил)–2–азаспиро[3.3]гептан (промежуточное соединение 23a) вместо 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (промежуточного соединения 8a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,40 (d, 1H), 8,30 (d, 1H), 8,14 (d, 1H), 7,61 (d, 2H), 7,33 (d, 2H), 7,14 (s, 2H), 4,58 (d, 1H), 3,79–3,66 (m, 1H), 3,55 (s, 2H), 3,41 (dt, 1H), 3,14 (s, 3H), 2,07 (t, 4H), 1,94–1,72 (m, 6H), 1,47–1,20 (m, 5H) (UPLC–MS) tR 0,51 мин; ESI–MS 421 [M+H]+.
Промежуточное соединение 23a: 2–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)бензил)–2–азаспиро[3.3]гептан
В раствор 2–(4–(бромметил)фенил)–4,4,5,5–тетраметил–1,3,2–диоксаборолана (300 мг, 1,01 ммоль) в ацетонитриле (8 мл) добавляли 2–азаспиро[3.3]гептан (148 мг, 1,11 ммоль) и карбонат цезия (428 мг, 1,31 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали в течение 60 мин., затем разбавляли насыщ. водн. раствором NaHCO3 и EtOAc. После разделения фаз водн. слой экстрагировали с помощью EtOAc и объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения, которое применяли без дополнительной очистки. (UPLC–MS) tR 0,82 мин; ESI–MS 314 [M+H]+.
Пример 24: 2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
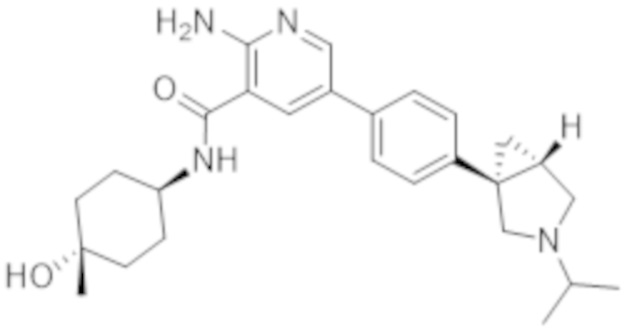
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 2), за исключением того, что применяли транс–4–амино–1–метилциклогексанол вместо транс–4–аминоциклогексанола гидрохлорида. 1H ЯМР (400 МГц, DMSO–d6) δ 8,43 (d, 1H), 8,34 (d, 1H), 8,16 (d, 1H), 7,68 (d, 2H), 7,39 (d, 2H), 4,06 (dd, 1H), 3,86–3,41 (m, 9H), 2,26 (dt, 1H), 1,78 (d, 2H), 1,68–1,55 (m, 2H), 1,47 (t, 4H), 1,33 (dd, 6H), 1,18 (s, 4H). (UPLC–MS) tR 0,53 мин; ESI–MS 449 [M+H]+.
Пример 25: 2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамид
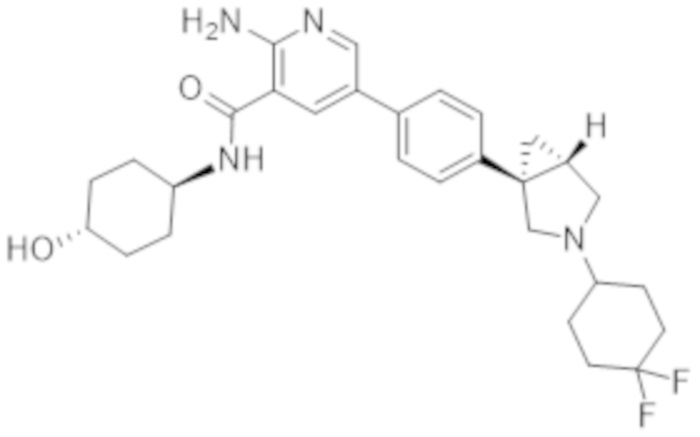
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 1), за исключением того, что применяли 4,4–дифторциклогексанон вместо 1–(метилсульфонил)пиперидин–4–она. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,32 (d, 1H), 8,11 (d, 1H), 7,59 (d, 2H), 7,23 (d, 2H), 7,13 (s, 2H), 4,59 (d, 1H), 3,73 (d, 1H), 3,39 (d, 2H), 3,09 (d, 1H), 2,68–2,55 (m, 2H), 2,37 (d, 2H), 2,00 (s, 2H), 1,84 (d, 8H), 1,64 (d, 2H), 1,46–1,16 (m, 5H), 0,78 (s, 1H). (UPLC–MS) tR 0,56 мин; ESI–MS 511 [M+H]+.
Пример 26: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
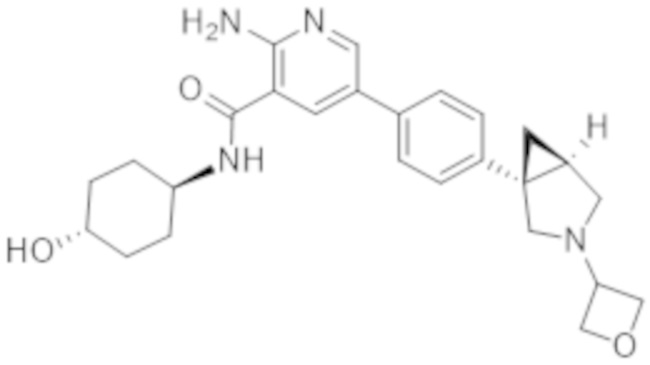
В раствор соли TFA и 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4S)–4–гидроксициклогексил)никотинамида (промежуточного соединения 5a, 70 мг, 0,138 ммоль) в DCM (4 мл) добавляли оксетан–3–он (12,0 мг, 0,166 ммоль) и AcOH (0,012 мл, 0,207 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали при 45°C в течение 30 мин. Добавляли триацетоксиборогидрид натрия (58,6 мг, 0,276 ммоль) при к. т. и реакционную смесь перемешивали при 45°C в течение 3 ч. Затем реакционную смесь разбавляли насыщ. водн. раствором NaHCO3 и смешивали с EtOAc. После разделения фаз водн. слой снова экстрагировали с помощью EtOAc. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт сначала очищали с помощью prepHPLC (способ 1a), затем с помощью флэш–хроматографии с обращенной фазой (способ 3a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. (UPLC–MS) tR 0,48 мин; ESI–MS 449 [M+H]+.
Пример 27: 2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамид
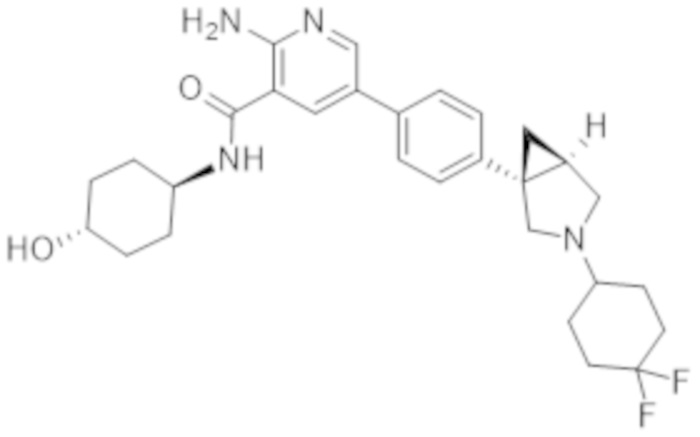
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 26), за исключением того, что применяли 4,4–дифторциклогексанон вместо оксетан–3–она. После очистки указанное в заголовке соединение получали в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 9,50 (bs, 1H), 8,38 (d, 1H), 8,32 (d, 1H), 8,11 (d, 1H), 7,59 (d, 2H), 7,23 (d, 2H), 7,13 (s, 2H), 4,59 (d, 1H), 4,12 (bs, 1H), 3,73 (d, 1H), 3,39 (d, 2H), 3,09 (d, 1H), 2,68–2,55 (m, 2H), 2,37 (d, 2H), 2,00 (s, 2H), 1,84 (d, 8H), 1,64 (d, 2H), 1,46–1,16 (m, 5H). (UPLC–MS) tR 0,56 мин; ESI–MS 511 [M+H]+.
Пример 28: 2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
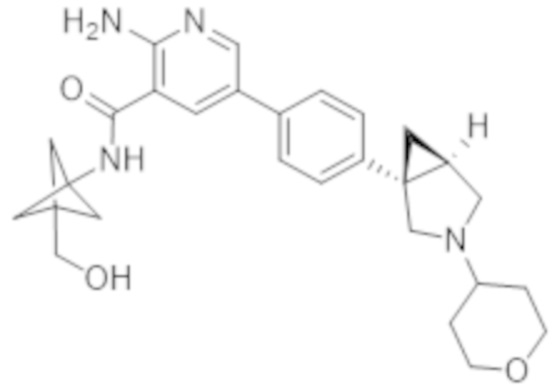
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 28a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли (3–аминобицикло[1.1.1]пентан–1–ил)метанол (промежуточное соединение 16a) вместо транс–4–аминоциклогексанола. Неочищенный продукт сначала очищали с помощью prepHPLC (способ 1a), затем с помощью флэш–хроматографии с обращенной фазой (способ 3a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 9,35 (bs, 1H), 9,06 (s, 1H), 8,44 (s, 1H), 8,19 (s,1H), 7,68 (d, 2H), 7,37 (d, 2H), 7,26 (bs, 2H), 4,57 (t, 1H), 4,12 (d, 1H), 3,97 (bs, 1H), 3,80–3,56 (m, 2H), 3,51 (d, 2H), 3,35–3,20 (m, 6H), 2,29–2,23 (m, 1H), 2,11–2,05 (m, 1H), 1,99 (bs, 1H), 1,97 (s, 6H), 1,67–1,61 (m, 1H), 1,19–1,14 (bs, 1H). (UPLC–MS) tR 0,50 мин; ESI–MS 475 [M+H]+.
Промежуточное соединение 28a: 2–амино–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2c), за исключением того, что применяли метил–2–амино–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат (промежуточное соединение 28b) вместо (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d). (UPLC–MS) tR 0,37 мин; ESI–MS 380 [M+H]+.
Промежуточное соединение 28b: метил–2–амино–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
Указанное в заголовке соединение получали способом, аналогичным для метил–2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 14b), за исключением того, что применяли метил–5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорид (промежуточное соединение 7c) вместо метил–5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорида (промежуточного соединения 14c). (UPLC–MS) tR 0,62 мин; ESI–MS 394 [M+H]+.
Пример 29: 2–амино–5–(2–фтор–4–(пиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4r)–4–гидроксициклогексил)–5–(4–(пирролидин–3–ил)фенил)никотинамида (примера 18), за исключением того, что применяли трет–бутил–4–(4–(6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)–3–фторфенил)пиперазин–1–карбоксилат (промежуточное соединение 29a) вместо 3–(4–(6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)пирролидин–1–карбоксилата (промежуточного соединения 18a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,29–8,16 (m, 2H), 7,95 (d, 1H), 7,38 (t, 1H), 7,06 (s, 2H), 6,88–6,75 (m, 2H), 4,54 (d, 1H), 3,69 (m,1H), 3,42–3,32 (m, 2H), 3,17 (dd, 4H), 2,92 (dt, 4H), 1,88–1,73 (m, 4H), 1,43–1,15 (m, 4H). (UPLC–MS) tR 0,42 мин; ESI–MS 414 [M+H]+.
Промежуточное соединение 29a: трет–бутил–4–(4–(6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)–3–фторфенил)пиперазин–1–карбоксилат
Указанное в заголовке соединение получали способом, аналогичным для 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (примера 8), за исключением того, что применяли трет–бутил–4–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)пиперазин–1–карбоксилат (промежуточное соединение 29b) вместо 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (промежуточного соединения 8a). Реакционную смесь перемешивали при 80°C в течение 60 мин. и проводили очистку с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения в виде коричневатого твердого вещества. (UPLC–MS) tR 0,96 мин; ESI–MS 514 [M+H]+.
Промежуточное соединение 29b: трет–бутил–4–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)пиперазин–1–карбоксилат
Указанное в заголовке соединение получали способом, аналогичным для трет–бутил–4–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)пиперазин–1–карбоксилата (промежуточного соединения 8a), за исключением того, что применяли трет–бутил–4–(4–бром–3–фторфенил)пиперазин–1–карбоксилат вместо 1–(4–бром–3–фторфенил)пиперазина (промежуточного соединения 8b). (UPLC–MS) tR 1,38 мин; ESI–MS 407 [M+H]+.
Пример 30: 2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 14a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли (3–аминобицикло[1.1.1]пентан–1–ил)метанол (промежуточное соединение 16a) вместо транс–4–аминоциклогексанола. Неочищенный продукт сначала очищали с помощью prepHPLC (способ 1a), затем с помощью флэш–хроматографии с обращенной фазой (способ 3a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 9,05 (s, 1H), 8,41 (d,1H), 8,18 (d,1H), 7,61 (d, 2H), 7,24–7,22 (m, 4H), 4,55 (t, 1H), 3,87–3,84 (m, 2H), 3,51 (d, 2H), 3,48–3,25 (m, 2H), 3,10 (bs, 1H), 2,56–2,47 (m, 4H), 1,97 (s, 6H), 1,96–1,74 (m, 3H), 1,39–0,30 (m, 3H), 0,77 (bs, 1H). (UPLC–MS) tR 0,51 мин; ESI–MS 475 [M+H]+.
Пример 31: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамида (примера 8), за исключением того, что применяли (R)–(4–((2–метилпирролидин–1–ил)метил)фенил)бороновую кислоту (промежуточное соединение 31a) вместо 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (промежуточного соединения 8a). Очистку проводили с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,75 (d, 1H), 8,65 (d, 1H), 8,48 (d, 1H), 7,81 (dd, 2H), 7,63 (d, 2H), 4,52 (dd, 1H), 4,18 (dd, 1H), 3,69 (ddt, 1H), 3,51–3,31 (m, 2H), 3,25–3,04 (m, 3H), 2,19 (ddt, 1H), 1,82 (td, 6H), 1,64–1,52 (m, 1H), 1,39–1,15 (m, 7H). (UPLC–MS) tR 0,44 мин; ESI–MS 409 [M+H]+.
Промежуточное соединение 31a: (R)–(4–((2–метилпирролидин–1–ил)метил)фенил)бороновая кислота
В раствор (4–(бромметил)фенил)бороновой кислоты (125 мг, 0,582 ммоль) в ацетонитриле (4 мл) добавляли K2CO3 (161 мг, 1,164 ммоль) и (R)–2–метилпирролидин (54,5 мг, 0,640 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали в течение 60 мин., затем фильтровали через слой целита. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде грязно–белого твердого вещества, которое применяли без дополнительной очистки. (UPLC–MS) tR 0,33 мин; ESI–MS 220 [M+H]+.
Пример 32: 2–амино–N–((1r,4S)–4–гидрокси–4–метилциклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
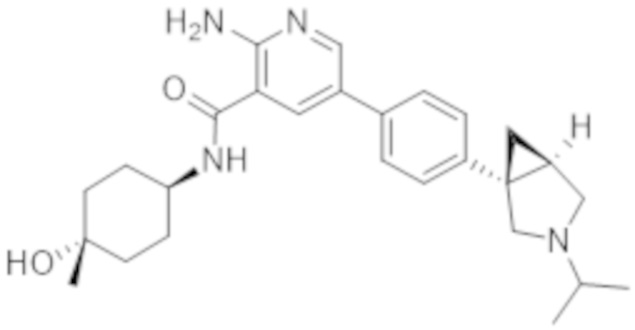
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 2), за исключением того, что применяли соль TFA и 2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 9a) вместо соли TFA и 2–амино–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2a) и применяли транс–4–амино–1–метилциклогексанол вместо транс–4–аминоциклогексанола гидрохлорида. 1H ЯМР (400 МГц, DMSO–d6) δ 8,43 (d, 1H), 8,34 (d, 1H), 8,16 (d, 1H), 7,68 (d, 2H), 7,39 (d, 2H), 4,06 (dd, 1H), 3,86–3,41 (m, 9H), 2,26 (dt, 1H), 1,78 (d, 2H), 1,68–1,55 (m, 2H), 1,47 (t, 4H), 1,33 (dd, 6H), 1,18 (s, 4H). (UPLC–MS) tR 0,50 мин; ESI–MS 449 [M+H]+.
Пример 33: 2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамид
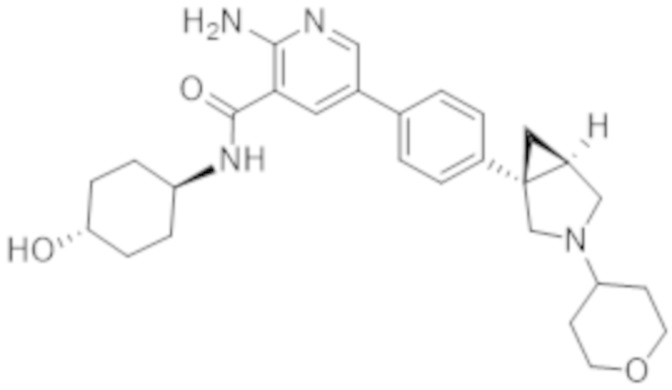
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 26), за исключением того, что применяли дигидро–2H–пиран–4(3H)–он вместо оксетан–3–она. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (s, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,62 (dd, 2H), 7,25 (s, 2H), 7,12 (s, 2H), 4,59 (d, 1H), 3,97–3,63 (m, 4H), 3,40 (m, 2H), 3,31 (m, 2H), 3,19–3,03 (m, 2H), 1,99–1,65 (m, 8H), 1,51–1,16 (m, 8H). (UPLC–MS) tR 0,47 мин; ESI–MS 477 [M+H]+.
Пример 34: 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид (также называемый соединением A в данном документе)
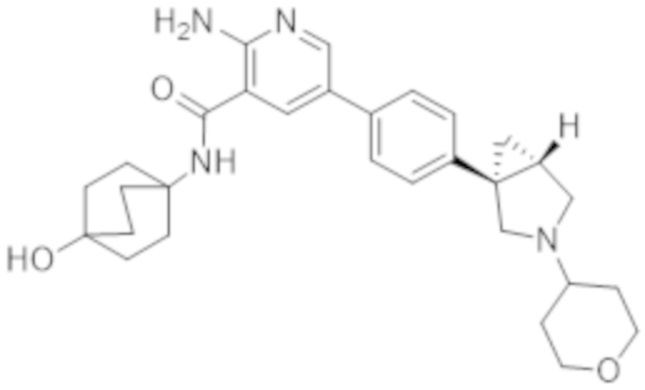
В раствор соли TFA и 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 14a, 4,10 г, 8,14 ммоль) и 4–аминобицикло[2.2.2]октан–1–ола гидрохлорида (2,17 г, 12,2 ммоль) в безводном DMF (60 мл) добавляли N–метилморфолин (2,24 мл, 20,4 ммоль) и HATU (4,64 г, 12,2 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали в течение 2 ч. и затем разбавляли насыщ. водн. раствором NaHCO3 и три раза экстрагировали с помощью EtOAc. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью хроматографии с обращенной фазой (способ 3b). Чистые фракции обрабатывали насыщ. водн. раствором NaHCO3 и три раза экстрагировали с помощью EtOAc. Объединенные органические экстракты промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде грязно–белого твердого вещества. Изображенную абсолютную конфигурацию подтверждали с помощью рентгеноструктурной кристаллографии указанного в заголовке соединения в комплексе с доменом ALK–2–киназы. 1H ЯМР (400 МГц, DMSO–d6) δ 8,34 (d, 1H), 7,98 (d, 1H), 7,79 (s, 1H), 7,57 (d, 2H), 7,23 (d, 2H), 6,92 (s, 2H), 4,31 (s, 1H), 3,91–3,78 (m, 2H), 3,40 (bs, 1H), 3,33–3,24 (m, 2H), 3,11 (d, 1H), 2,57 (bs, 1H), 2,50–2,34 (m, 1H), 2,34 (bs, 1H), 2,12–1,94 (m, 6H), 1,90–1,72 (m, 3H), 1,71–1,51 (m, 6H), 1,51–1,34 (m, 2H), 1,31 (t, 1H), 0,82–0,68 (m, 1H). (UPLC–MS) tR 0,54 мин; ESI–MS 503 [M+H]+. Хиральная HPLC (ChiralPak Id, 5 мкм, скорость потока: 1 мл/мин., длина волны для выявления: 270 нм, подвижная фаза: гептан:изопропанол, 60:40 (+ 0,1% диэтиламина)): tR 18,7 мин., ee 92,3%.
Альтернативный пример 34A. В раствор 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–
азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты гидрохлорида (1 кг, 1,683 моль) и 4–аминобицикло[2.2.2]октан–1–ола гидрохлорида (343,9 г, 1,935 моль) в DMF (3500 мл) добавляли Et3N (681,2 г, 6,732 моль) и HATU (767,9 г, 2,019 моль) при к. т. Реакционную смесь перемешивали при к. т. в течение 1 ч. Смесь нагревали до IT=45°C, добавляли 5% раствор NH3.H2O (5200 г). Перемешивали в течение приблизительно 30 мин., добавляли дополнительное количество 5% раствора NH3.H2O (1800 г). Смесь нагревали до IT=45°C в течение 2 ч. Смесь охлаждали до IT=22°C., фильтровали, влажный осадок на фильтре промывали с помощью H2O (1500 мл x 3). Влажный осадок на фильтре высушивали под вакуумом при 45°C в течение 24 ч. Неочищенный продукт растворяли в ацетоне (3000 мл), затем фильтровали с удалением некоторого количества нерастворенного твердого вещества. Фильтрат нагревали до IT=50°C. Добавляли H2O (2000 мл). Смесь перемешивали при IT=50°C в течение 30 мин., пока не образовывался белый осадок. Медленно добавляли H2O (4000 мл). Смесь перемешивали при IT=50°C в течение 2 ч. Смесь охлаждали до IT=22°C за 2 ч., фильтровали, влажный осадок на фильтре промывали ацетоном:H2O=1:2 (об./об., 1000 мл x 2). Влажный осадок на фильтре высушивали под вакуумом при 45°C в течение 24 ч. Всего получали 760 г белого твердого вещества (выход 89%, ee 99,4%).
1H ЯМР (DMSO–d6) δ: 8,32 (d, J=2,3 Гц, 1H), 7,97 (d, J=2,3 Гц, 1H), 7,77 (s, 1H), 7,54 (d, J=8,3 Гц, 2H), 7,20 (d, J=8,4 Гц, 2H), 6,90 (s, 2H), 4,31 (s, 1H), 3,82 (m, 2H), 3,29 (m, 2H), 3,07 (d, J=8,5 Гц, 1H), 2,54 (d, J=8,3 Гц, 1H), 2,44 (dd, J=8,5, 3,5 Гц, 1H), 2,37 (m, 1H), 2,31 (td, J=10,2, 5,0 Гц, 1H), 2,04 (m, 6H), 1,80 (dt, J=7,9, 3,8 Гц, 1H), 1,71 (d, J=12,3 Гц, 1H), 1,65 (d, J=11,5 Гц, 1H), 1,62 (m, 6H), 1,38 (m, 1H), 1,34 (m, 1H), 1,29 (t, J=3,9 Гц, 1H), 0,73 (dd, J=7,9, 3,6 Гц, 1H).
13C ЯМР (DMSO–d6) δ: 167,80, 157,69, 148,28, 141,27, 134,91, 134,79, 126,40, 125,66, 123,53, 111,01, 66,22, 65,59, 59,10, 55,46, 52,04, 33,72, 31,92, 31,77, 30,59, 29,61, 24,14, 17,20.
MS(ESI–TOF): 503,3018 [M+H]+.
Исходный материал (гидрохлоридную соль) получали следующим образом.
Суспендировали метил–2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината дигидрохлорид (10 г, 19,5 ммоль, 1,0 экв.) в MeOH (31,7 г). Затем добавляли раствор NaOH (2,9 г, 72,2 ммоль, 3,7 экв.) в H2O (10 г). Реакционную смесь нагревали до 45 ± 5°C и перемешивали в течение более 3 ч. с получением суспензии.
В другой флакон, содержащий ацетон (200 г), добавляли 5–6 н. HCl в i–PrOH (14,8 г, 97,6 ммоль, 5 экв.). Раствор нагревали до 47 ± 3°C. Затем в смесь добавляли по каплям вышеуказанную суспензию MeOH и перемешивали при 47 ± 3°C в течение 3 ч. Смесь охлаждали до 23 ± 3°C и перемешивали в течение 3 ч. После фильтрации влажный осадок на фильтре промывали ацетоном (40 г). Влажный осадок на фильтре высушивали под вакуумом при 55°C в течение 8 ч. Получали 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты гидрохлорид (12,3 г, чистота 99,3% в соответствии с HPLC, выход при анализе 62,1%) в виде грязно–белого твердого вещества.
1H ЯМР (DMSO–d6) δ: 11,53 (br s, 1H), 8,64 (br s, 1H), 8,54 (br s, 1H), 7,72–8,42 (m, 2H), 7,64 (br d, J=7,9 Гц, 2H), 7,38 (br d, J=7,8 Гц, 2H), 3,85–4,04 (m, 3H), 3,40–3,73 (m, 4H), 3,15–3,33 (m, 2H), 2,18 (br d, J=3,9 Гц, 1H), 1,95–2,12 (m, 4H), 1,88 (br d, J=10,0 Гц, 1H), 1,05 (br t, J=6,4 Гц, 1H).
13C ЯМР (DMSO–d6) δ: 167,2, 155,8, 144,3, 142,0, 139,6, 133,8, 127,7, 126,3, 124,1, 110,0, 65,8, 62,5, 55,7, 53,2, 29,9, 28,8, 28,7, 23,5, 16,6.
MS(ESI–TOF): 380,1974 [M+H]+.
Исходный материал, 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината дигидрохлорид, получали следующим образом.
В круглодонную колбу объемом 500 мл загружали 1R,5S)–1–(4–бромфенил)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–3–ия хлорид (20 г, 1 экв.), EA (200 мл) и 25% K2CO3 (62 г). Смесь перемешивали в течение 30 мин., пока все твердые вещества не растворялись. После разделения фаз органический слой концентрировали. Добавляли 2–метил–2–бутанол (48 г, 60 мл). Органический слой концентрировали. Добавляли 2–метил–2–бутанол (144 г, 180 мл).
Смесь переносили в Redlay объемом 500 мл. Добавляли K2CO3 (18,8 г, 2,5 экв.) и метил–2–амино–5–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)никотинат (16,8 г, 1,04 экв.; см. промежуточное соединение 1e). Смесь три раза дегазировали с помощью N2. Смесь нагревали до IT=50 ± 5°C за 1 ч. Добавляли Pd(dppf)Cl2 (1,2 г, 0,03 экв.). Смесь нагревали до IT=70 ± 3°C и перемешивали в течение 2 ч. После охлаждения до 22°C добавляли H2O (120 г) и EA (180 г) и перемешивали в течение 30 мин. Добавляли MCC (6 г) и смесь фильтровали через MCC. Осадок на фильтре промывали с помощью EA (54 г). После разделения фаз органический слой промывали с помощью 5% NaCl (124 г). Затем в органический слой добавляли Quadrasil MP (поглотитель тяжелых металлов от Johnson Matthey, 6 г). Смесь нагревали до IT=55°C в течение 8 ч., фильтровали через MCC и промывали с помощью EA (54 г). В органический слой добавляли Quadrasil MP (2 г). Смесь нагревали до IT=55°C в течение 6 ч., фильтровали через CMC и промывали с помощью EA (54 г). Органический слой концентрировали. Добавляли ацетон (158 г, 200 мл). После перемешивания при IT=22 ± 3°C в течение 30 мин. смесь нагревали до IT=40 ± 3°C. Добавляли по каплям 15,5% HCl (38,4 г) при IT <50°C. Смесь перемешивали при IT=45 ± 3°C в течение 1 ч. Смесь охлаждали до 22 ± 3°C. Смесь перемешивали при 22 ± 3°C в течение 1 ч. и фильтровали. Осадок на фильтре промывали ацетоном (32 г x 2). Влажный осадок на фильтре высушивали под вакуумом при 50°C в течение по меньшей мере 8 ч. Получали исходный материал, 22,5 г белого твердого вещества (чистота 97,1% в соответствии с HPLC, содержание воды 5,2%, выход при анализе 87%).
1H ЯМР (DMSO–d6) δ: 11,43 (br d, J=5,7 Гц, 1H), 8,62–8,80 (m, 2H), 7,85–8,58 (m, 2H), 7,67 (d, J=8,3 Гц, 2H), 7,39 (d, J=8,4 Гц, 2H), 3,85–4,06 (m, 6H), 3,60–3,69 (m, 2H), 3,50–3,59 (m, 1H), 3,44 (br d, J=7,7 Гц, 1H), 3,14–3,31 (m, 2H), 2,21 (dt, J=8,4, 4,2 Гц, 1H), 1,94–2,12 (m, 4H), 1,76–1,93 (m, 1H), 1,07 (br t, J=7,1 Гц, 1H).
13C ЯМР (DMSO–d6) δ: 165,0, 154,0, 143,5, 142,0, 140,1, 132,8, 127,7, 126,5, 124,2, 110,7, 65,8, 62,5, 55,6, 53,3, 53,3, 29,9, 28,9, 28,8, 23,6, 16,8.
MS(ESI–TOF): 394,2071 [M+H]+.
Исходный материал, 1R,5S)–1–(4–бромфенил)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–3–ия хлорид, получали следующим образом.
В Redlay объемом 1 л загружали (1R,5S)–1–(4–бромфенил)–3–азабицикло[3.1.0]гексан–3–ия хлорид (30 г, 1 экв.), дигидро–2H–пиран–4(3H)–он (13,13 г, 1,2 экв.) и THF (300 мл). Смесь перемешивали при IT=22 ± 5°C в течение 1 ч. Добавляли порциями NaBH(OAc)3 (30,1 г, 1,3 экв.) с поддержанием IT < 30°C. Смесь перемешивали при IT=22 ± 5°C в течение 2 ч. Добавляли 6,2% HCl (93 г, 90 мл, 1,5 экв.) с поддержанием IT < 30°C и pH < 2. Смесь перемешивали в течение 10 мин. Добавляли 25% K2CO3 (259 г, 210 мл) с регулированием pH=8–9. Добавляли IPAc (300 мл). Смесь перемешивали в течение 10 мин. После разделения фаз в органический слой добавляли H2O (150 г). Смесь перемешивали в течение 10 мин. После разделения фаз органический слой концентрировали под вакуумом (50–100 мбар, водяная баня 50°C). Добавляли IPA (120 г, 150 мл). Органический слой концентрировали под вакуумом (50–100 мбар, водяная баня 50°C). Добавляли IPA (144 г, 180 мл). Смесь фильтровали через CMC. Осадок на фильтре промывали с помощью IPA (24 г x 2). В органический слой добавляли H2O (5 г). Добавляли по каплям 31% HCl (19,3 г) при IT < 35°C. Смесь перемешивали при IT=22 ± 5°C в течение 2 ч. и фильтровали. Осадок на фильтре промывали с помощью IPA (48 г x 2). Влажный осадок на фильтре высушивали под вакуумом при 50°C в течение по меньшей мере 6 ч. Получали необходимый продукт (31,4 г, чистота 98% в соответствии с HPLC, выход 78%) в виде белого твердого вещества.
1H ЯМР (DMSO–d6 и D2O) δ: 7,46 (br d, J=8,4 Гц, 2H), 7,15 (br d, J=8,4 Гц, 2H), 3,90 (br d, J=7,8 Гц, 3H), 3,62 (br s, 1H), 3,51 (br s, 2H), 3,16–3,38 (m, 3H), 2,05–2,21 (m, 1H), 1,93 (br s, 2H), 1,49–1,71 (m, 2H), 1,05–1,30 (m, 1H).
13C ЯМР (DMSO–d6) δ: 138,3, 131,9, 129,2, 120,4, 65,5, 62,3, 56,2, 53,9, 29,1, 28,9, 24,8, 23,0.
MS(ESI–TOF): 322,0761 [M+H]+.
Полный способ изготовления альтернативного примера 34 A изображен на следующей реакционной схеме 34A:
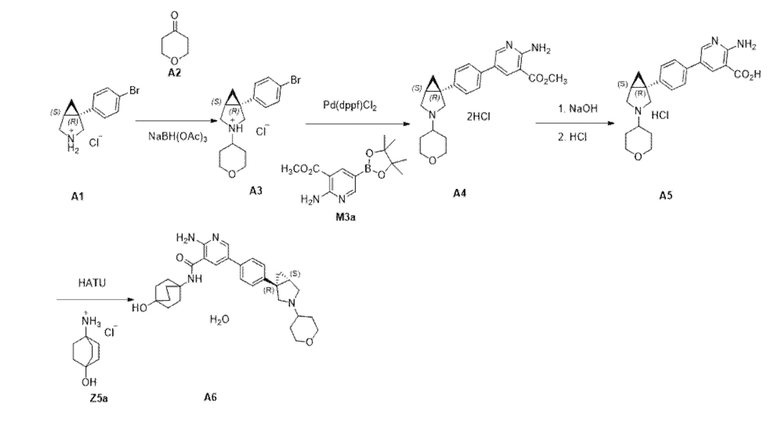
Первое соединение на данной схеме, A1, может быть получено следующим образом.
Стадия 1–2. Синтез 1c и 1d
Схема 34A1
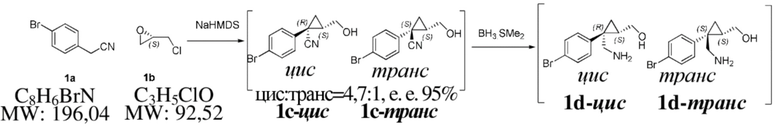
Процедура реакции (см. также Xu, Feng et al., Org. Lett. Vol 8, No.17, 2006, pages 3885–3888):
Добавляли по каплям NaHMDS (2,0 М в THF, 19,2 л, 38,4 моль, 2,5 экв.) в раствор 1a (3,00 кг, 15,3 моль, 1,0 экв. получаемый, например, от Sigma–Aldrich) и 1b (1,80 кг, 19,47 ммоль, 1,27 экв.; получаемый, например, от Sigma–Aldrich) в THF (30 л) при –15–20°C в атмосфере N2 в течение 5 ч. Реакционную смесь перемешивали в течение дополнительных 3 ч. при –15°C, затем постепенно нагревали до к. т. и перемешивали в течение 16 ч. С помощью HPLC показали, что 4–бромфенилацетонитрил полностью израсходовался.
Медленно добавляли BF3.Et2O (4,74 кг, 15,3 моль, 1,0 экв.) при 20–40°C. После добавления медленно добавляли BH3.DMS (19,2 л, 38,4 моль, 2,5 экв.) при 20–40°C. После добавления смесь перемешивали при 50–55°C в течение 16 ч. С помощью HPLC показали, что промежуточное соединение полностью израсходовалось.
Смесь охлаждали до –5–0°C, медленно добавляли AcOH (4,5 кг, 74,4 моль, 4,86 экв.). После добавления медленно добавляли 3 н. HCl (36 кг). Смесь перемешивали в течение 1 часа и большую часть THF удаляли под вакуумом при 40–45°C. Затем остаток экстрагировали с помощью MTBE (3×10 л). Фазу на основе MTBE промывали с помощью 3 н. HCl (4×5 л). Объединенный водный слой охлаждали до 0°C. Затем водный слой нейтрализовали с помощью 30% NaOH до pH >10, экстрагировали с помощью IPAc (3×15 кг), промывали солевым раствором (5 л), выпаривали под вакуумом с получением неочищенного соединения 1d (3,71 кг), которое применяли непосредственно на следующей стадии.
Стадия 3–4. Синтез 1e и 1f
Схема 34A2
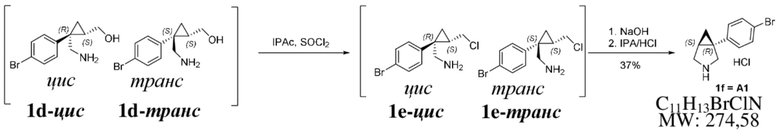
Процедура реакции
Охлаждали IPAc (12 л) до 0°C и добавляли SOCl2(5,46 кг, 45,9 моль, 1,5 экв.) с поддержанием температуры при 0–4°C. После добавления медленно добавляли раствор неочищенного соединения 1d (7,46 кг, две партии, объединенные из 6 кг 1a) в IPAc (24 л). После добавления смесь перемешивали в течение 1 ч. при 0°C, затем нагревали до к. т. и перемешивали в течение 4 ч. С помощью HPLC показали, что промежуточное соединение полностью израсходовалось.
Смесь охлаждали до –5–0°C и гасили водой (30 л), затем медленно добавляли 30% водный раствор NaOH с регулированием pH до 8,5–9 и перемешивали в течение ночи при 0–5°C. С помощью HPLC показали, что промежуточное соединение полностью израсходовалось.
Медленно добавляли 30% водный раствор NaOH с регулированием pH до 11–12 при 0–10°C и перемешивали при к. т. в течение 30 мин. Фазы разделяли, водный слой экстрагировали с помощью IPAc (10 кг), объединенную органическую фазу промывали солевым раствором (5 л), высушивали над Na2SO4 и фильтровали. Затем фильтрат охлаждали до 0–5°C. Медленно добавляли 4 М HCl\IPA (8,0 кг, 151 моль) при 0–5°C. После добавления реакционную смесь перемешивали в течение 4 ч. при 0–5°C. Смесь фильтровали, собирали твердое вещество, промывали с помощью MTBE (6 л) и высушивали под вакуумом с получением соединения 1f (3,10 кг, анализ 98%, выход 37%)
Альтернативный пример 34B.
Исходный материал A5 на схеме 34A в качестве альтернативы также можно получать следующим образом (реакционная схема 34B) и затем применять, как показано в альтернативном примере 34A выше.
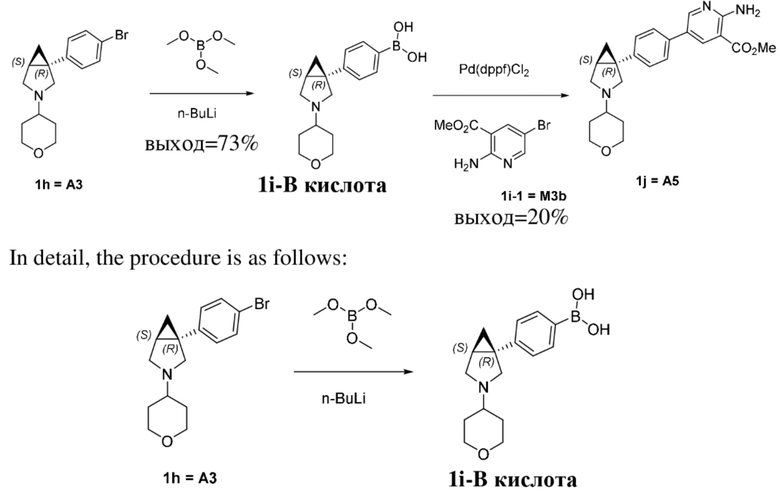
Подробно процедура состоит в следующем.
В трехгорлой колбе объемом 100 мл охлаждали раствор (1R,5S)–1–(4–бромфенил)–3–тетрагидропиран–4–ил–3–азабицикло[3.1.0]гексана (5,00 г, 15,52 ммоль, 1,00 экв.) в безводном THF (50,00 мл) до –78°C в атмосфере N2. Добавляли по каплям н–BuLi (2,5 М в гексане, 7,45 мл, 1,20 экв.) при –78°C и перемешивали в течение 1 ч. при –78°C. Добавляли по каплям раствор триметилбората (4,84 г, 46,56 ммоль, 5,26 мл, 3,00 экв.) в безводном THF (10,00 мл) при –78°C. После добавления обеспечивали нагревание реакционной смеси до 25°C и ее перемешивали при 25°C в течение 16 ч. После того, как исходный материал полностью расходовался, проводили контроль с помощью LC–MS и обнаруживали 74,23% целевого соединения. Реакционную смесь охлаждали до 0°C на ледяной бане и гасили насыщенным раствором NH4Cl (50 мл). Регулировали pH смеси до 12–13 с помощью раствора NaOH (1 М) и экстрагировали ее с помощью EtOAc (30 мл x 3). Водный слой концентрировали при пониженном давлении с получением остатка (18 г). Остаток очищали с помощью prep–HPLC (условия с TFA) с получением [4–[(1R,5S)–3–тетрагидропиран–4–ил–3–азабицикло[3.1.0]гексан–1–ил]фенил]бороновой кислоты (4,60 г, 11,47 ммоль, выход 73,88%, соль TFA) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ: 7,72 (d, 2H), 7,24 (d, 2H), 4,13 (d, 1H), 4,06–4,03 (m, 2H), 3,86 (d, 1H), 3,70–3,67 (m, 2H), 3,45–3,32 (m, 3H), 2,26–2,23 (m, 1H), 2,11–2,05 (m, 2H), 1,81–1,77 (m, 2H), 1,34–1,20 (m, 2H). ESI–MS 288 [M+H]+.
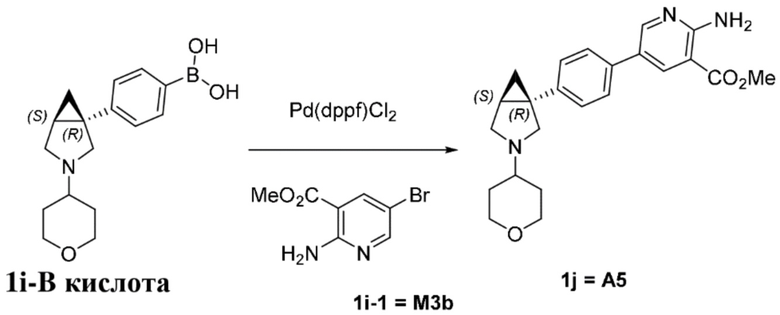
Нагревали смесь [4–[(1R,5S)–3–тетрагидропиран–4–ил–3–азабицикло[3.1.0]гексан–1–ил]фенил]бороновой кислоты (500,00 мг, 1,25 ммоль, 1,00 экв. соль TFA), метил–2–амино–5–бромпиридин–3–карбоксилата (433,22 мг, 1,88 ммоль, 1,50 экв.), Pd(dppf)Cl2 (91,46 мг, 0,125 ммоль, 0,10 экв.) и K2CO3 (518,29 мг, 3,75 ммоль, 3,00 экв.) в диоксане (5,00 мл) и H2O (1,00 мл) до 100°C и перемешивали в течение 16 ч. при 100°C в атмосфере N2. Реакционную смесь охлаждали до 30°C и концентрировали при пониженном давлении. Остаток очищали с помощью prep–HPLC (нейтральные условия) с получением метил–2–амино–5–[4–[(1R,5S)–3–тетрагидропиран–4–ил–3–азабицикло[3.1.0]гексан–1–ил]фенил]пиридин–3–карбоксилата (100,00 мг, выход 20,02%, чистота 98,47% в соответствии с HPLC) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, DMSO–d6) δ: 8,49 (d, 1H), 8,35 (d, 1H), 7,46 (d, 2H), 7,24 (d, 2H), 4,01–3,98 (m, 2H), 3,94 (s, 3H), 3,47–3,41 (m, 3H), 3,19 (d, 1H), 2,65 (d, 1H), 2,50–2,55 (m, 1H), 2,45–2,35 (m, 1H), 1,81–1,76 (m, 3H), 1,64–1,46 (m, 2H), 1,45–1,40 (m, 1H), 0,86–0,83 (m, 1H). ESI–MS 394 [M+H]+.
Пример 35: 2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 35a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли 4–аминобицикло[2.2.2]октан–1–ола гидрохлорид вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 9,05 (bs, 1H), 8,42 (d, 1H), 8,22 (s, 1H), 8,02 (s, 1H), 7,69 (d, 2H), 7,50 (bs, 2H), 7,39 (d, 2H), 4,83 (bs, 1H), 4,08 (dd, 1H), 3,80–3,25 (m, 6H), 2,31–2,09 (m, 4H), 2,09–1,99 (m, 6H), 1,98–1,71 (m, 4H), 1,70–1,56 (m, 6H), 1,42–1,40 (m, 1H), 1,10 (t, 1H). (UPLC–MS) tR 0,62 мин; ESI–MS 537 [M+H]+.
Промежуточное соединение 35a: 2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2c), за исключением того, что применяли метил–2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат (промежуточное соединение 35b) вместо (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d). (UPLC–MS) tR 0,46 мин; ESI–MS 414 [M+H]+.
Промежуточное соединение 35b: метил–2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
Указанное в заголовке соединение получали способом, аналогичным для метил–2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 14b), за исключением того, что применяли метил–5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорид (промежуточное соединение 7c) вместо метил–5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–аминоникотината гидрохлорид (промежуточного соединения 14c) и применяли 4,4–дифторциклогексанон вместо дигидро–2H–пиран–4(3H)–она. (UPLC–MS) tR 0,73 мин; ESI–MS 428 [M+H]+.
Пример 36: 2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 2), за исключением того, что применяли (3–аминобицикло[1.1.1]пентан–1–ил)метанол (промежуточное соединение 16a) вместо транс–4–аминоциклогексанола гидрохлорида. 1H ЯМР (400 МГц, DMSO–d6) δ 9,05 (s, 1H), 8,41 (d,1H), 8,18 (d,1H), 7,61 (d, 2H), 7,48 (dd, 1H), 7,33–7,17 (m, 4H), 4,55 (t, 1H), 3,51 (d, 3H), 3,31 (s, 2H), 3,08 (s, 2H), 2,07–1,99 (m, 1H), 1,41–1,22 (m, 3H), 1,17–0,98 (m, 9H), 0,79 (d, 1H). (UPLC–MS) tR 0,54 мин; ESI–MS 433 [M+H]+.
Пример 37: 2–амино–5–(4–((1S,5R)–3–(бут–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 1–бромбут–2–ин вместо 4–(3–бромпропил)морфолина и реакционную смесь перемешивали при к. т. в течение 30 мин. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (s, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,59 (d, 2H), 7,21 (s, 2H), 7,13 (s, 2H), 4,60 (d1H), 3,72 (s, 1H), 3,39 (s, 3H), 3,21 (s,1H), 2,98–2,58 (m, 3H), 1,84 (dd, 8H), 1,49–1,17 (m, 5H), 0,78 (s, 1H). (UPLC–MS) tR 0,54 мин; ESI–MS 445 [M+H]+.
Пример 38: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(оксетан–3–илметил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 3–(йодметил)оксетан вместо 4–(3–бромпропил)морфолина и реакционную смесь перемешивали при 60°C в течение 60 мин. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,30 (d, 1H), 8,11 (d, 1H), 7,59 (d, 2H), 7,22 (d, 2H), 7,12 (s, 2H), 4,73–4,61 (m, 2H), 4,58 (d, 1H), 4,29 (s, 2H), 3,84–3,67 (m, 1H), 3,52–3,38 (m, 2H), 3,27–3,12 (m, 2H), 2,94 (d, 1H), 2,80 (s, 2H), 1,95–1,76 (m, 5H), 1,54–1,14 (m, 6H), 0,76 (s, 1H). (UPLC–MS) tR 0,49 мин; ESI–MS 463 [M+H]+.
Пример 39: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(2,2,2–трифторэтил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
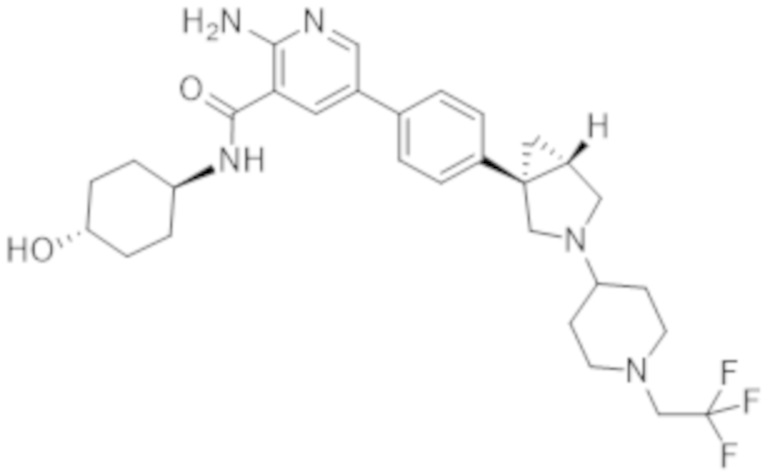
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 1), за исключением того, что применяли 1–(2,2,2–трифторэтил)пиперидин–4–он вместо 1–(метилсульфонил)пиперидин–4–она. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,31 (d,1H), 8,11 (d, 1H), 7,58 (d, 2H), 7,23 (d, 2H), 7,12 (s, 2H), 4,55 (bs, 1H), 3,80–3,62 (m, 1H), 3,20–3,04 (m, 3H), 2,88 (d, 2H), 2,57 (d, 1H), 2,46 (dd, 1H), 2,41–2,29 (m, 2H), 2,15 (t, 1H), 1,99–1,69 (m, 8H), 1,49–1,17 (m, 7H), 1,15 (t, 1H), 0,75 (dd, 1H). (UPLC–MS) tR 0,62 мин; ESI–MS 558 [M+H]+.
Пример 40: 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(2–морфолиноэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
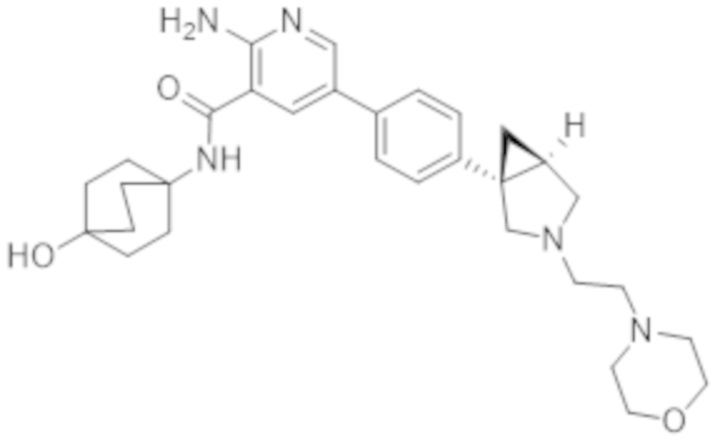
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли соль HCl и 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида (промежуточного соединения 40a) вместо 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 5a) и применяли 4–(2–бромэтил)морфолин вместо 4–(3–бромпропил)морфолина, и реакционную смесь перемешивали при 60°C в течение 3 ч. 1H ЯМР (400 МГц, DMSO–d6) δ 8,34 (d, 1H), 7,98 (d, 1H), 7,79 (s, 1H), 7,57 (d, 2H), 7,21 (d, 2H), 6,92 (s, 2H), 4,32 (s, 1H), 3,65–3,49 (m, 4H), 3,08 (d, 1H), 2,61 (t, 2H), 2,59–2,39 (m, 9H), 2,15–1,98 (m, 6H), 1,85–1,73 (m, 1H), 1,70–1,54 (m, 6H), 1,31 (t, 1H), 0,75 (dd, 1H). (UPLC–MS) tR 0,52 мин; ESI–MS 532 [M+H]+.
Промежуточное соединение 40a: 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 5a), за исключением того, что применяли (1S,5R)–трет–бутил–1–(4–(6–амино–5–((4–гидроксибицикло[2.2.2]октан–1–ил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат (промежуточное соединение 40b) вместо (1S,5R)–трет–бутил–1–(4–(6–амино–5–(((1r,4S)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 5b). Одну фракцию указанного в заголовке соединения получали в виде гидрохлоридной соли после выпаривания диоксана; другую фракцию получали в виде соли TFA после дополнительной очистки с помощью prepHPLC (способ 1a). (UPLC–MS) tR 0,48 мин; ESI–MS 419 [M+H]+.
Промежуточное соединение 40b: (1S,5R)–трет–бутил–1–(4–(6–амино–5–((4–гидроксибицикло[2.2.2]октан–1–ил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилат
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 5c) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли 4–аминобицикло[2.2.2]октан–1–ола гидрохлорид вместо транс–4–аминоциклогексанола. (UPLC–MS) tR 1,05 мин; ESI–MS 519 [M+H]+.
Пример 41: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–(2,2,2–трифторэтокси)этил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
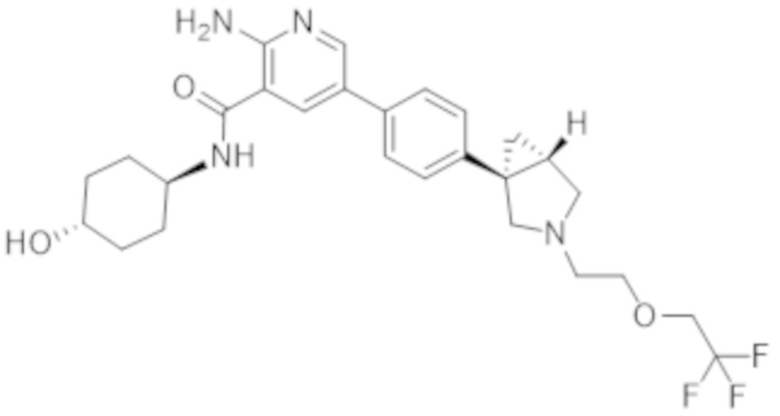
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 2–(2,2,2–трифторэтокси)этилтрифторметансульфонат (промежуточное соединение 41a) вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a). После очистки указанное в заголовке соединение получали в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 9,15 (bs, 1H), 8,40 (s, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,63 (d, 2H), 7,23 (s, 2H), 7,13 (s, 2H), 4,59 (d, 1H), 4,11 (s, 2H), 3,80–3,63 (m, 3H), 3,40 (m, 4H), 2,75–2,60 (m, 3H), 1,86 (m, 5H), 1,51–1,15 (m, 6H). (UPLC–MS) tR 0,60 мин; ESI–MS 519 [M+H]+.
Промежуточное соединение 41a: 2–(2,2,2–трифторэтокси)этилтрифторметансульфонат
Указанное в заголовке соединение получали способом, аналогичным для 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a), за исключением того, что применяли 2–(2,2,2–трифторэтокси)этанол вместо 1,3–дифторпропан–2–ола, и его получали в виде неочищенного масла, которое применяли без дополнительной очистки.
Пример 42: 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
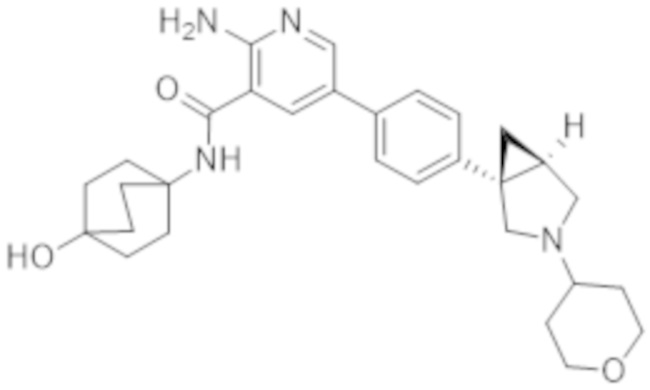
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 1), за исключением того, что применяли соль HCl и 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида (промежуточного соединения 40a) вместо 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 1a) и применяли дигидро–2H–пиран–4(3H)–он вместо 1–(метилсульфонил)пиперидин–4–она. После очистки указанное в заголовке соединение получали в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 9,62 (bs, 1H), 8,38 (d, 1H), 7,98 (d, 1H), 7,81 (s, 1H), 7,65 (d, 2H), 7,37 (d, 2H), 6,95 (s, 2H), 4,33 (s, 1H), 4,06–3,97 (m, 3H), 3,75–3,43 (m, 4H), 3,29–3,26 (m, 2H), 2,28–2,19 (m, 1H), 2,07–2,03 (m, 6H), 2,03–1,96 (m, 2H), 1,75–1,63 (m, 1H), 1,63–1,61 (m, 6H), 1,63–1,61 (m, 1H), 1,25–1,17 (m, 1H). (UPLC–MS) tR 0,52 мин; ESI–MS 503 [M+H]+.
Пример 43: 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамид
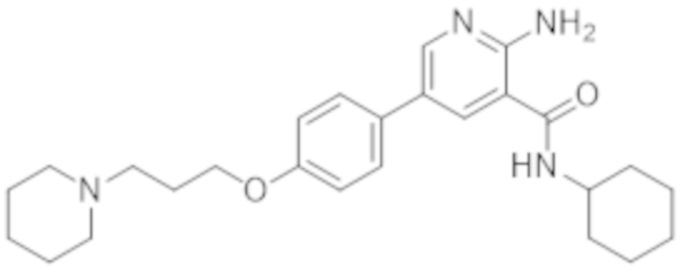
В раствор 2–амино–5–бром–N–циклогексилникотинамида (промежуточного соединения 43a, 50 мг, 0,168 ммоль) в 2 мл DMF/EtOH/воды (2:1:1) добавляли 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидин (60,9 мг, 0,168 ммоль), K2CO3 (69,5 мг, 0,503 ммоль) и PdCl2(PPh3)2 (11,8 мг, 0,017 ммоль). Реакционную смесь перемешивали в течение 15 мин. при 80°C, затем охлаждали и разбавляли с помощью 5 мл EtOAc и фильтровали через слой Na2SO4. После концентрирования при пониженном давлении неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,34 (d, 1H), 8,33 (d, 1H), 8,09 (d, 1H), 7,59 (d, 2H), 7,06 (s, 2H), 7,01 (d, 2H), 4,03 (t, 2H), 3,75 (s, 1H), 2,50–2,22 (m, 6H), 1,95–1,65 (m, 6H), 1,62 (d, 1H), 1,52–1,48 (m, 4H), 1,46–1,34 (m, 6H), 1,32–1,15 (m, 1H). (HPLC–MS) tR 1,46 мин; APCI–MS 437,3 [M+H]+.
Промежуточное соединение 43a: 2–амино–5–бром–N–циклогексилникотинамид
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–бромникотиновую кислоту вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли циклогексанамин вместо транс–4–аминоциклогексанола. Неочищенный продукт применяли без дополнительной очистки. (HPLC–MS) tR 0,98 мин.; ESI–MS 208/300 [M+H]+.
Пример 44: 2–амино–5–(4–((1R,5S)–3–(1,1–диоксидотетрагидро–2H–тиопиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамид
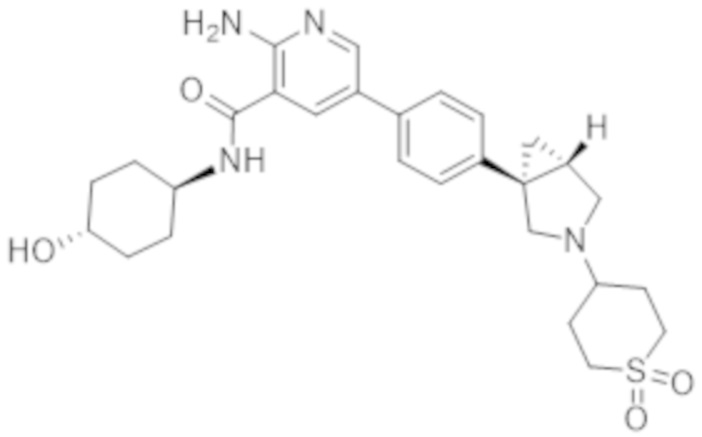
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (1), за исключением того, что применяли дигидро–2H–тиопиран–4(3H)–он–1,1–диоксид вместо 1–(метилсульфонил)пиперидин–4–она. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,59 (d, 2H), 7,24 (d, 2H), 7,12 (s, 2H), 4,58 (d, 1H), 3,82–3,65 (m, 1H), 3,48–3,35 (m, 2H), 3,19–2,91 (m, 5H), 2,62–2,51 (m, 2H), 2,17–1,97 (m, 4H), 1,89–1,83 (m, 5H), 1,45–1,19 (m, 6H), 0,81 (dd, 1H). (UPLC–MS) tR 0,48 мин; ESI–MS 525 [M+H]+.
Пример 45: 2–амино–N–циклогексил–5–(4–(4–метилпиперазин–1–карбонил)фенил)никотинамид
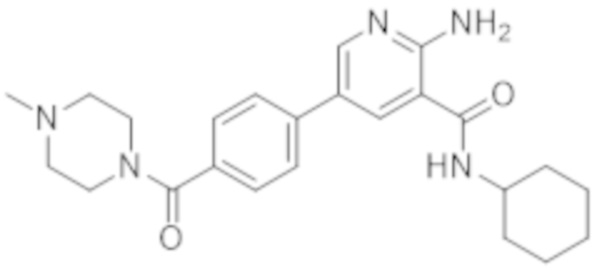
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли (4–метилпиперазин–1–ил)–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)метанон вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,46 (d, 1H), 8,37 (s, 1H), 8,21 (d, 1H), 7,76 (d, 2H), 7,47 (d, 2H), 7,22 (s, 2H), 3,82–3,55 (m, 1H), 3,35 (t, 4H), 2,31–2,22 (t, 4H), 2,21 (s, 3H), 1,90–1,80 (m, 2H), 1,80–1,70 (m, 2H), 1,70–1,55 (m, 1H), 1,35–1,29 (m, 4H), 1,28–1,05 (m, 1H). (HPLC–MS) tR 1,29 мин; APCI–MS 422 [M+H]+.
Пример 46: 2–амино–5–(3–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамид
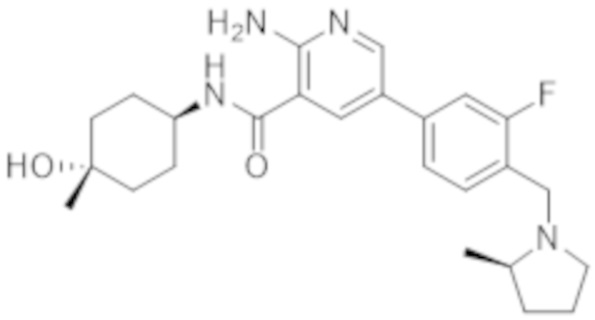
В сосуд для микроволновой обработки загружали 2–амино–5–бром–N–((1r,4r)–4–гидрокси–4–метилциклогексил)никотинамид (промежуточное соединение 59b, 33 мг, 0,10 ммоль), (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион (промежуточное соединение 46a, 38,3 мг, 0,11 ммоль) и XPhos Pd (7,39 мг, 0,01 ммоль). Добавляли диоксан (1 мл) с последующим добавлением 3 н. водн. раствора K3PO4 (0,10 мл, 0,30 ммоль). Реакционную смесь продували потоком азота в течение 5 мин. и затем подвергали облучению в микроволновом реакторе при 120°C в течение 30 мин. После охлаждения реакционную смесь концентрировали при пониженном давлении и очищали с помощью prepHPLC (способ 1b) с получением указанного в заголовке соединения. UPLC–HRMS tR 3,21 мин.; ESI 441,26 [M+H]+.
Промежуточное соединение 46a: (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион
Встряхивали раствор 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b, 112 мг, 0,40 ммоль), (R)–(2)–метилпирролидина (37,5 мг, 0,44 ммоль) и уксусной кислоты (2,4 мг, 0,040 ммоль) в THF (2 мл) в течение 2 ч. при к. т. и добавляли триацетоксиборогидрид натрия (110 мг, 0,52 ммоль). Полученную реакционную смесь встряхивали в течение 18 ч. и затем фильтровали через карбонатный картридж SPE с последующим промыванием с помощью THF. Фильтрат и промывные растворы концентрировали при пониженном давлении с получением указанного в заголовке соединения, которое применяли без дополнительной очистки. ESI–MS 349 [M+H]+.
Промежуточное соединение 46b: 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид
Перемешивали раствор (3–фтор–4–формилфенил)бороновой кислоты (67 мг, 0,40 ммоль) и N–метилиминодиуксусной кислоты (65 мг, 0,44 ммоль) в DMF (2 мл) при 90°C в течение 18 ч. Реакционную смесь охлаждали и фильтровали через карбонатный картридж SPE с последующим промыванием ацетонитрилом три раза. Фильтрат и промывные растворы концентрировали при пониженном давлении с получением указанного в заголовке соединения, которое применяли без дополнительной очистки. ESI–MS 558 [2M+H]+.
Пример 47: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(3,3,3–трифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 3,3,3–трифторпропилтрифторметансульфонат вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (s, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,60 (d, 2H), 7,23 (d, 2H), 7,13 (s, 2H), 4,59 (d, 1H), 3,74 (m, 1H), 3,40 (m, 3H), 3,09 (m, 1H), 2,72–2,64 (m, 4H), 1,86 (m, 5H), 1,49–1,14 (m, 6H), 0,78 (s, 1H). (UPLC–MS) tR 0,60 мин; ESI–MS 489 [M+H]+.
Пример 48: 2–амино–5–(4–((1R,5S)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 1–бром–2–фторэтан вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a) и реакционную смесь перемешивали при 60°C в течение 3 ч. После очистки получали указанное в заголовке соединение в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 9,95 (bs, 1H), 8,41 (s, 1H), 8,32 (d, 1H), 8,13 (d, 1H), 7,70–7,57 (m, 2H), 7,36 (bs, 2H), 7,16 (s, 2H), 4,80–4,62 (m, 2H), 4,59 (d, 1H), 4,08 (bs, 1H), 3,77–3,52 (m, 4H), 3,51–3,35 (m, 3H), 2,24 (bs, 1H), 1,87 (t, 4H), 1,59–1,13 (m, 6H). (UPLC–MS) tR 0,46 мин; ESI–MS 439 [M+H]+.
Пример 49: 2–амино–N–циклогексил–5–(4–морфолинофенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли 4–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)морфолин вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,35 (d, 1H), 8,34 (s, 1H), 8,08 (d, 1H), 7,55 (d, 2H), 7,03 (d, 2H), 7,03 (s, 2H), 3,85–3,65 (m, 1H), 3,77 (t, 4H), 3,14 (t, 4H), 1,90–1,80 (m, 2H), 1,80–1,70 (m, 2H), 1,80 (dd, 1H), 1,38–1,20 (m, 4H), 1,19–1,05 (m, 1H). (HPLC–MS) tR 1,60 мин; APCI–MS 381 [M+H]+.
Пример 50: 2–амино–5–(4–((1R,5S)–3–(2,2–дифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 2,2–дифторпропилтрифторметансульфонат вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a) и реакционную смесь перемешивали при к. т. в течение 60 мин. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (s, 1H), 8,33 (s, 1H), 8,14 (s, 1H), 7,59 (d, 2H), 7,32–7,11 (m, 4H), 4,58 (bs, 1H), 3,74 (bs, 1H), 3,11 (bs, 1H), 3,00–2,66 (m, 5H), 1,86 (bs, 5H), 1,63 (t, 3H), 1,48–1,18 (m, 6H), 0,81 (bs, 1H). (UPLC–MS) tR 0,81 мин; ESI–MS 471 [M+H]+.
Пример 51: 2–амино–5–(4–((1S,5R)–3–(2–цианоэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамид
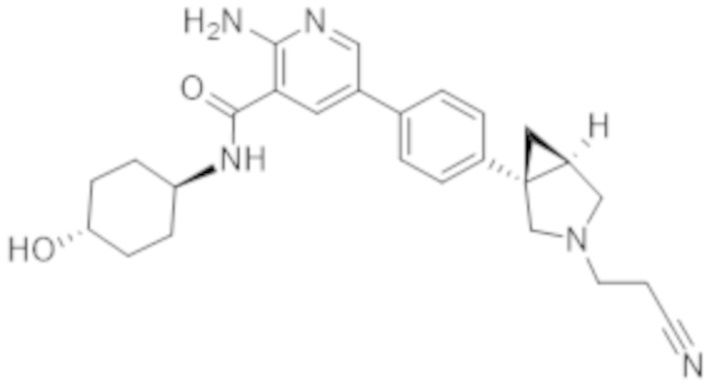
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 3–бромпропаннитрил вместо 4–(3–бромпропил)морфолина и реакционную смесь перемешивали при 60°C в течение 24 ч. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,30 (d, 1H), 8,12 (d, 1H), 7,60 (d, 2H), 7,23 (d, 2H), 7,12 (s, 2H), 4,59 (d, 1H), 3,73 (d, 1H), 3,18 (d, 1H), 3,09 (d, 1H), 2,81–2,62 (m, 6H), 1,99–1,78 (m, 5H), 1,48–1,18 (m, 6H), 0,80 (dd, 1H). (UPLC–MS) tR 0,50 мин; ESI–MS 446 [M+H]+.
Пример 52: 2–амино–N–циклогексил–5–(4–(3–морфолинопропокси)фенил)никотинамид
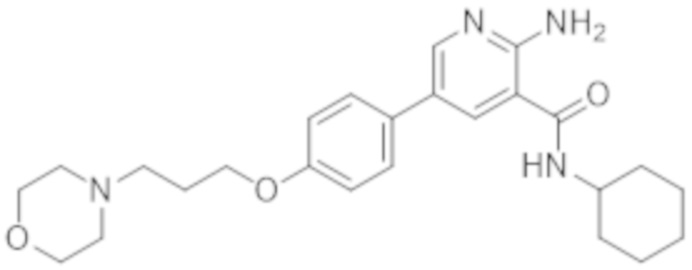
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли 4–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)морфолин вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,35 (d, 1H), 8,34 (s, 1H), 8,10 (d, 1H), 7,59 (d, 2H), 7,06 (s, 2H), 7,01 (d, 2H), 4,04 (t, 2H), 3,82–3,68 (m, 1H), 3,58 (t, 4H), 2,50–2,22 (m, 6H), 1,90–1,78 (m, 4H), 1,78–1,69 (m, 2H), 1,63 (dd, 1H), 1,38–1,20 (m, 4H), 1,19–1,05 (m, 1H). (HPLC–MS) tR 1,40 мин; APCI–MS 439 [M+H]+.
Пример 53: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(проп–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
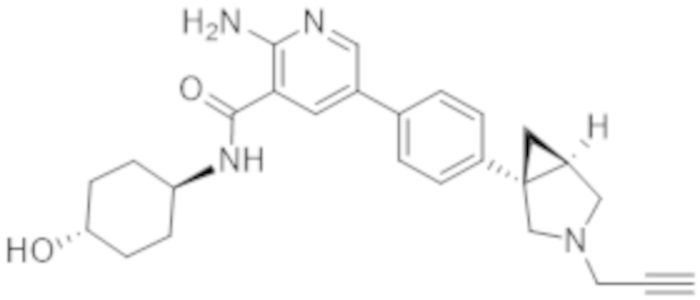
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(3–морфолинопропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 5), за исключением того, что применяли 3–бромпроп–1–ин вместо 4–(3–бромпропил)морфолина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,59 (d, 2H), 7,21 (d, 2H), 7,13 (s, 2H), 4,59 (d, 1H), 3,73 (d, 1H), 3,52–3,38 (m, 2H), 3,21 (d, 1H), 2,93 (d, 1H), 2,83 (d, 1H), 2,72 (d, 1H), 2,40 (s, 3H), 1,95–1,75 (m, 4H), 1,44–1,19 (m, 5H), 0,80 (dd, 1H). (UPLC–MS) tR 0,49 мин; ESI–MS 431 [M+H]+.
Пример 54: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(проп–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
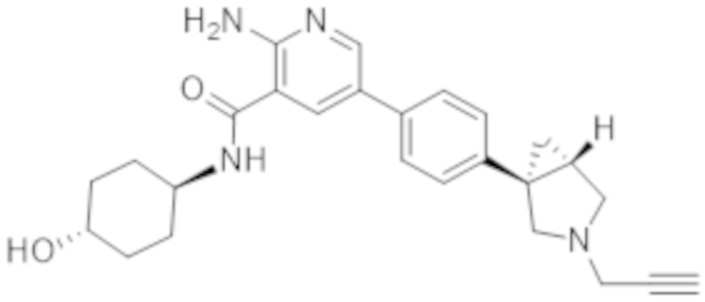
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 3–бромпроп–1–ин вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,59 (d, 2H), 7,21 (d, 2H), 7,12 (s, 2H), 4,59 (d, 1H), 3,76–3,70 (m, 1H), 3,46 (s, 2H), 3,45–3,37 (m, 1H), 3,22 (d, 1H), 2,83 (d, 1H), 2,72 (d, 1H), 2,71–2,69 (m, 2H), 1,92–1,77 (m, 5H), 1,51–1,17 (m, 5H), 0,80 (dd, 1H). (UPLC–MS) tR 0,52 мин; ESI–MS 431 [M+H]+.
Пример 55: 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)–2–хлорфенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамид
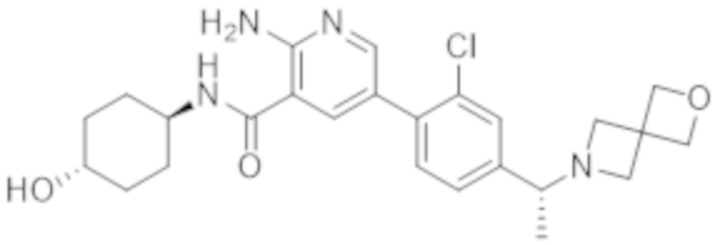
Указанное в заголовке соединение получали способом, аналогичным для 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 3), за исключением того, что применяли (R)–6–(1–(4–бром–3–хлорфенил)этил)–2–окса–6–азаспиро[3.3]гептан (промежуточное соединение 55a) вместо (R)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептана (промежуточного соединения 3b). 1H ЯМР (400 МГц, DMSO–d6) δ 8,17 (m, 2H), 8,00 (s, 1H), 7,42–7,44 (m, 2H), 7,40 (m, 1H), 7,34 (bs, 2H), 4,62 (s, 3H), 4,55 –4,57 (m, 1H), 3,72 (m, 1H), 3,35–3,54 (m, 4H), 3,20–3,24 (m, 2H), 3,15 (bs, 1H), 1,78–1,88 (m, 4H), 1,20–1,41 (m, 4H), 1,11 (bs, 3H). (UPLC–MS) tR 0,47 мин; ESI–MS 471/473 [M+H]+.
Промежуточное соединение 55a: (R)–6–(1–(4–бром–3–хлорфенил)этил)–2–окса–6–азаспиро[3.3]гептан
Указанное в заголовке соединение получали способом, аналогичным для (R)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептана (промежуточного соединения 3b), за исключением того, что применяли (R)–1–(4–бром–3–хлорфенил)этанамин вместо (R)–1–(4–бромфенил)этанамина. (UPLC–MS) tR 0,63 мин; ESI–MS 316/318 [M+H]+.
Пример 56: 2–амино–5–(2–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамид
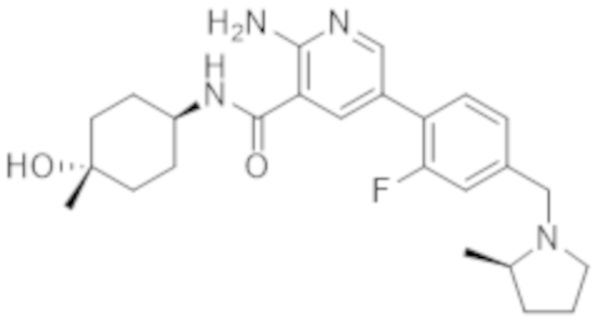
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(3–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида (примера 46), за исключением того, что применяли (R)–2–(2–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион (промежуточное соединение 56a) вместо (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a). UPLC–HRMS tR 3,22 мин.; ESI 441,26 [M+H]+.
Промежуточное соединение 56a: (R)–2–(2–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион
Указанное в заголовке соединение получали способом, аналогичным для (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a), за исключением того, что применяли 3–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид (промежуточное соединение 56b) вместо 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b). ESI–MS 349 [M+H]+.
Промежуточное соединение 56b: 3–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид
Указанное в заголовке соединение получали способом, аналогичным для 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b), за исключением того, что применяли (2–фтор–4–формилфенил)бороновую кислоту вместо (3–фтор–4–формилфенил)бороновой кислоты. ESI–MS 576 [2M+H2O+H]+.
Пример 57: 2–амино–5–(4–((1R,5S)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамид
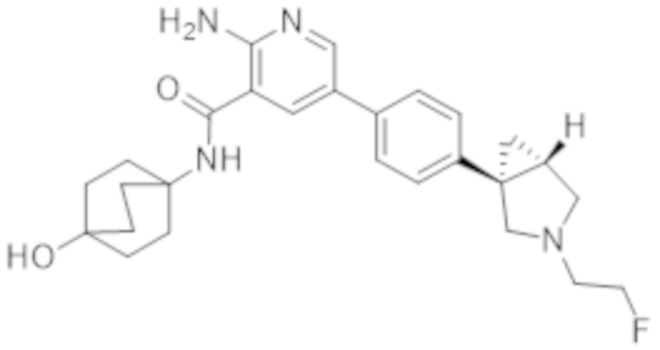
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли соль HCl и 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида (промежуточного соединения 34a) вместо 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 1a) и применяли 1–бром–2–фторэтан вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a). После очистки указанное в заголовке соединение получали в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 10,00 (bs, 1H), 8,38 (d, 1H), 8,04 (d, 1H), 7,85 (s, 1H), 7,77 (d, 2H), 7,35 (d, 2H), 7,08 (s, 2H), 4,83 (td, 2H), 4,74 (bs, 1H), 4,08 (bs, 1H), 3,89–3,57 (m, 4H), 3,18 (d, 1H), 2,24 (bs, 1H), 2,13–1,97 (m, 6H), 1,74–1,53 (m, 6H), 1,48 (bs, 1H), 1,17 (bs, 1H). (UPLC–MS) tR 0,53 мин; ESI–MS 465 [M+H]+.
Пример 58: 2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидрокси–4–метилциклогексил)никотинамид
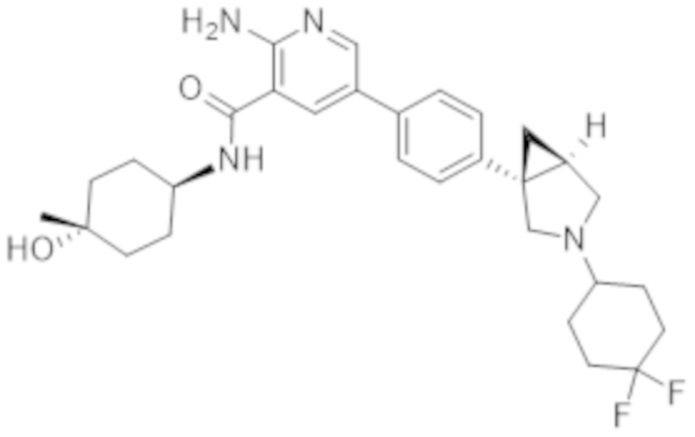
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 35a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли транс–4–амино–1–метилциклогексанол вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) и пропускали через ионообменный картридж (PL–HCO3) для обессоливания с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,29 (d, 1H), 8,10 (d,1H), 7,59 (d, 2H), 7,25 (d, 2H), 7,10 (s, 2H), 4,33 (s, 1H), 3,81 (bs, 1H), 3,39 (d, 2H), 2,62–2,56 (m, 1H), 2,00 (bs, 2H), 1,88–1,76 (m, 8H), 1,67–1,53 (m, 4H), 1,50–1,43 (m, 4H), 1,30 (t, 1H), 1,17 (s, 3H), 0,86–0,68 (m, 1H). (UPLC–MS) tR 0,61 мин; ESI–MS 525 [M+H]+.
Пример 59: 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамид
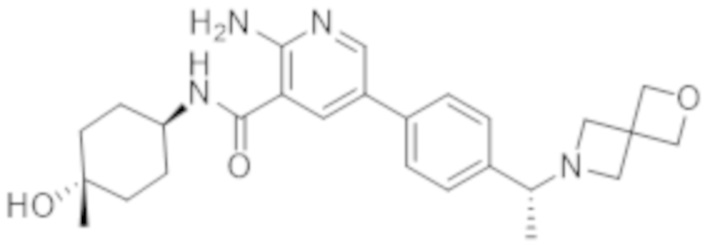
Указанное в заголовке соединение получали способом, аналогичным для 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 3), за исключением того, что применяли (6–амино–5–(((1r,4r)–4–гидрокси–4–метилциклогексил)карбамоил)пиридин–3–ил)бороновую кислоту (промежуточное соединение 59a) вместо (6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)бороновой кислоты (промежуточного соединения 3a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (s, 1H), 8,25 (bs, 1H), 8,11 (d, 1H) 7,71–7,54 (m, 2H), 7,49–7,28 (m, 2H), 7,12 (bs, 2H), 4,61 (bs, 4H), 4,31 (s, 1H), 3,89–3,68 (m, 2H), 3,31 (s, 15H), 3,27–2,99(m, 2H), 1,86–1,69 (m, 2H), 1,66–1,52 (m, 2H), 1,51–1,35 (m, 5H), 1,16 (s, 1H), 1,30–0,95 (m, 2H). (UPLC–MS) tR 0,47 мин; ESI–MS 451 [M+H]+.
Промежуточное соединение 59a: (6–амино–5–(((1r,4r)–4–гидрокси–4–метилциклогексил)карбамоил)пиридин–3–ил)бороновая кислота
Указанное в заголовке соединение получали способом, аналогичным для (6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)бороновой кислоты (промежуточного соединения 3a), за исключением того, что применяли 2–амино–5–бром–N–((1r,4r)–4–гидрокси–4–метилциклогексил)никотинамид (промежуточное соединение 59b) вместо 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамида (промежуточного соединения 3c). (UPLC–MS) tR 0,33 мин; ESI–MS 394 [M+H]+.
Промежуточное соединение 59b: 2–амино–5–бром–N–((1r,4r)–4–гидрокси–4–метилциклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамида (промежуточного соединения 3c), за исключением того, что применяли транс–4–амино–1–метилциклогексанол вместо транс–4–аминоциклогексанола. (UPLC–MS) tR 0,68 мин; ESI–MS 328/330 [M+H]+.
Пример 60: 2–амино–5–(4–((1R,5S)–3–(бут–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамид
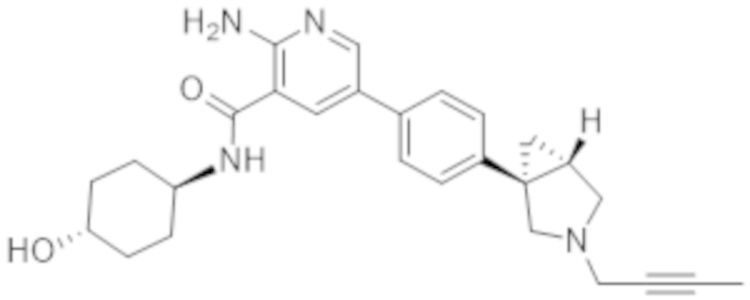
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 1–бромбут–2–ин вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,59 (d, 2H), 7,21 (d, 2H), 7,12 (s, 2H), 4,58 (d, 1H), 3,77–3,71 (m, 1H), 3,43–3,37 (m, 3H), 3,22 (d, 1H), 2,92 (d, 1H), 2,80 (d, 1H), 1,93–1,78 (m, 8H), 1,44–1,19 (m, 6H), 0,79 (dd, 1H). (UPLC–MS) tR 0,54 мин; ESI–MS 445 [M+H]+.
Пример 61: 2–амино–N–циклогексил–5–(4–(пиперидин–1–ил)фенил)никотинамид
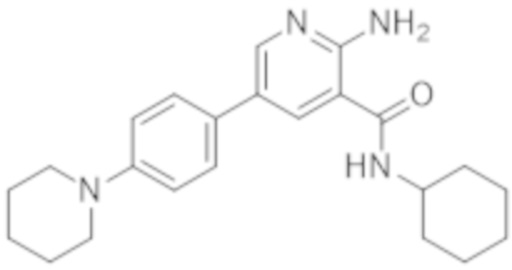
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли 4–(пиперидин–1–ил)фенилбороновую кислоту вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,33 (d, 1H), 8,33 (s, 1H), 8,08 (d, 1H), 7,51 (d, 2H), 7,01 (s, 2H), 7,00 (d, 2H), 3,82–3,68 (m, 1H), 3,17 (t, 4H), 1,90–1,78 (m, 2H), 1,78–1,69 (m, 2H), 1,68–1,49 (m, 7H), 1,38–1,20 (m, 4H), 1,19–1,05 (m, 1H). (HPLC–MS) tR 1,40 мин; APCI–MS 379 [M+H]+.
Пример 62: 2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
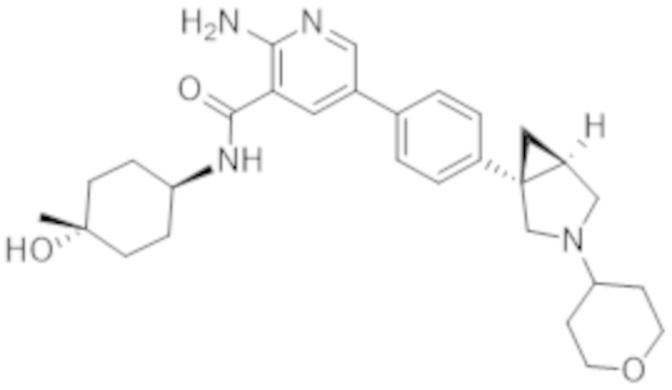
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 28a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли транс–4–амино–1–метилциклогексанол вместо транс–4–аминоциклогексанола. Неочищенный продукт сначала очищали с помощью prepHPLC (способ 1a), затем флэш–хроматографии с обращенной фазой (способ 3a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,92 (d, 1H), 8,46 (s, 1H), 8,22 (s,1H), 7,61 (d, 2H), 7,25 (d, 2H), 7,18 (s, 2H), 4,15–4,09 (m, 1H), 3,88–3,82 (m, 2H), 3,42–3,36 (m, 3H), 3,10 (bs, 1H), 2,65–2,34 (m, 12H), 1,85–1,75 (m, 3H), 1,45–1,31 (m, 6H), 0,77 (bs, 1H). (UPLC–MS) tR 0,49 мин; ESI–MS 491 [M+H]+.
Пример 63: 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
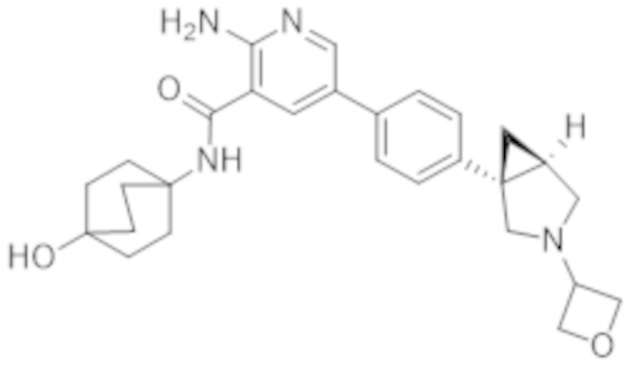
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 1), за исключением того, что применяли соль HCl и 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида (промежуточного соединения 40a) вместо 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 1a) и применяли оксетан–3–он вместо 1–(метилсульфонил)пиперидин–4–она. 1H ЯМР (400 МГц, DMSO–d6) δ 8,34 (d, 1H), 7,98 (d, 1H), 7,80 (s, 1H), 7,57 (d, 2H), 7,22 (d, 2H), 6,92 (s, 2H), 4,61–4,56 (m, 2H), 4,52–4,48 (m, 2H), 4,32 (s, 1H), 3,80–3,74 (m, 1H), 3,03 (d, 1H), 2,57–2,51 (m, 3H), 2,14–1,98 (m, 6H), 1,88–1,85 (m, 1H), 1,71–1,55 (m, 6H), 1,45–1,35 (m, 1H), 0,83–0,80 (m, 1H). (UPLC–MS) tR 0,51 мин; ESI–MS 475 [M+H]+.
Пример 64: 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
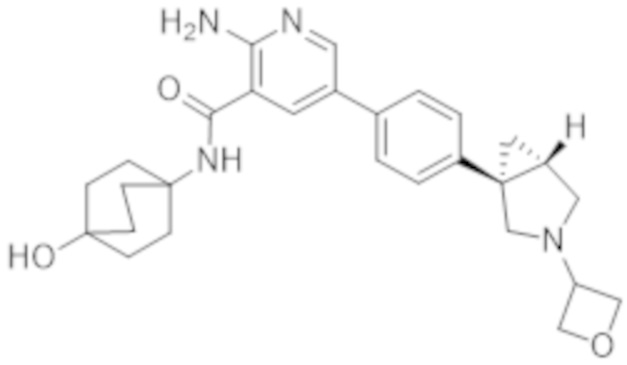
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 1), за исключением того, что применяли соль HCl и 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида (промежуточного соединения 34a) вместо 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 1a) и применяли оксетан–3–он вместо 1–(метилсульфонил)пиперидин–4–она. 1H ЯМР (400 МГц, DMSO–d6) δ 8,34 (d, 1H), 7,98 (d, 1H), 7,80 (s, 1H), 7,57 (d, 2H), 7,22 (d, 2H), 6,92 (s, 2H), 4,61–4,56 (m, 2H), 4,52–4,48 (m, 2H), 4,31 (s, 1H), 3,81–3,76 (m, 1H), 3,03 (d, 1H), 2,57–2,51 (m, 3H), 2,07–2,03 (m, 6H), 1,88–1,85 (m, 1H), 1,65–1,61 (m, 6H), 1,45–1,35 (m, 1H), 0,83–0,80 (m, 1H). (UPLC–MS) tR 0,55 мин; ESI–MS 475 [M+H]+.
Пример 65: 2–амино–N–циклогексил–5–(4–((2–(4–метилпиперазин–1–ил)этил)карбамоил)фенил)никотинамид
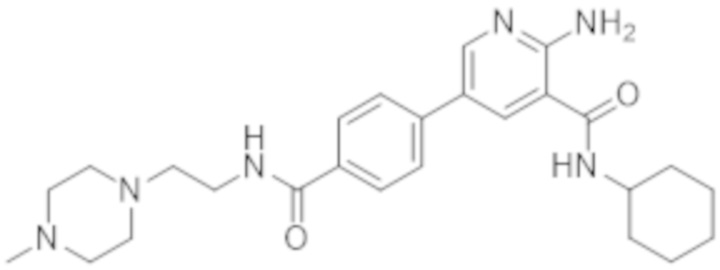
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 4–(6–амино–5–(циклогексилкарбамоил)пиридин–3–ил)бензойную кислоту (промежуточное соединение 65a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли 2–(4–метилпиперазин–1–ил)этанамин вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью хроматографии с нормальной фазой (способ 2c) с получением указанного в заголовке соединения в виде грязно–белого твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,50 (d, 1H), 8,42 (t, 1H), 8,37 (d, 1H), 8,23 (d, 1H), 7,91 (d, 2H), 7,80 (d, 2H), 7,25 (s, 2H), 3,82–3,65 (m, 1H), 3,40 (q, 2H), 3,32–3,13 (m, 3H), 2,99–2,83 (m, 1H), 2,43–2,31 (m, 6H), 2,17 (s, 3H), 1,90–1,80 (m, 2H), 1,80–1,70 (m, 2H), 1,70–1,55 (m, 1H), 1,40–1,25 (m, 4H), 1,20–1,10 (m, 1H). (HPLC–MS) tR 1,28 мин; APCI–MS 465 [M+H]+.
Промежуточное соединение 65a: 4–(6–амино–5–(циклогексилкарбамоил)пиридин–3–ил)бензойная кислота
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли 4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)бензойную кислоту вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. Неочищенный продукт растирали с 2–пропанолом с получением коричневатого твердого вещества, которое очищали с помощью prepHPLC (способ 1a). Чистые фракции собирали и концентрировали при пониженном давлении, затем повышали основность с помощью Na2CO3, после чего указанное в заголовке соединение осаждалось в виде бесцветного твердого вещества, которое отфильтровывали и высушивали при пониженном давлении. (HPLC–MS) tR 1,52 мин.; ESI–MS 340 [M+H]+.
Пример 66: 2–амино–5–(4–((1S,5R)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамид
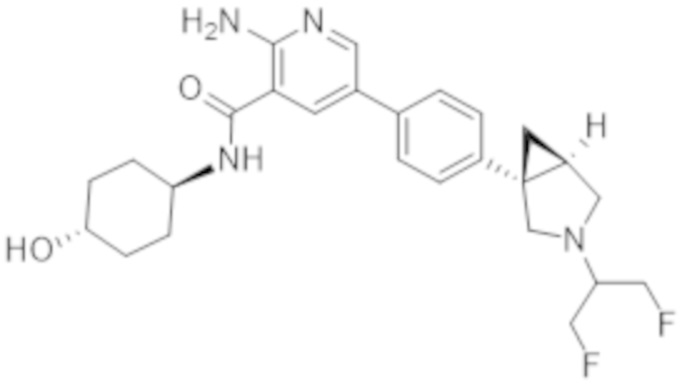
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 1,3–дифторпропан–2–илтрифторметансульфонат (промежуточное соединение 6a) вместо 4–(3–бромпропил)морфолина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,59 (d, 2H), 7,24 (d, 2H), 7,12 (s, 2H), 4,68 (t, 2H), 4,57 (dd, 3H), 3,81–3,65 (m, 1H), 3,46–3,35 (m, 2H), 3,08 (d, 1H), 2,93 (d, 1H), 2,83 (dd, 1H), 2,05–1,76 (m, 5H), 1,53–1,16 (m, 6H), 0,78 (dd, 1H). (UPLC–MS) tR 0,68 мин; ESI–MS 471 [M+H]+.
Пример 67: 2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2,2,2–трифторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
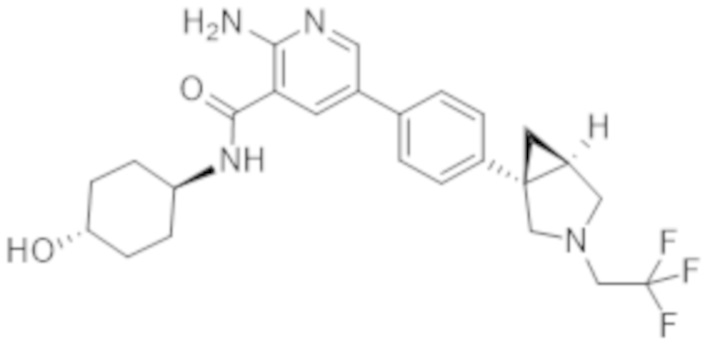
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 2,2,2–трифторэтилтрифторметансульфонат вместо 4–(3–бромпропил)морфолина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,46–8,31 (m, 2H), 8,18 (bs, 1H), 7,59 (d, 2H), 7,29 (bs, 1H), 7,23 (d, 2H), 3,83–3,54 (m, 1H), 3,51–3,24 (m, 5H), 3,11 (d, 1H), 2,92 (d, 1H), 2,86 (m, 1H), 1,92–1,77 (m, 5H), 1,29–1,21 (m, 1H), 1,42–1,18 (m, 4H), 1,14 (bs, 1H), 0,85–0,79 (m, 1H). (UPLC–MS) tR 1,04 мин; ESI–MS 475 [M+H]+.
Пример 68: 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,3R)–3–гидроксиадамантан–1–ил)никотинамид
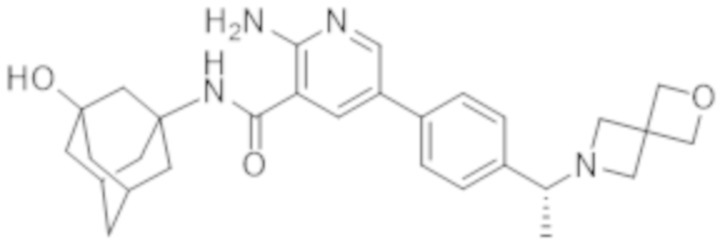
Указанное в заголовке соединение получали способом, аналогичным для 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 3), за исключением того, что применяли (6–амино–5–(((1r,3r)–3–гидроксиадамантан–1–ил)карбамоил)пиридин–3–ил)бороновую кислоту (промежуточное соединение 68a) вместо (6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)бороновой кислоты (промежуточного соединения 3a). 1H ЯМР (400 МГц, DMSO–d6) δ 8,65 (bs, 1H), 8,49 (s, 1H), 8,35 (s, 1H), 7,87 (d, 2H), 7,59 (d, 2H), 4,73 (dd, 2H), 4,65–4,54 (m, 4H), 4,45–4,39 (m, 1H), 4,16–4,10 (m, 1H), 3,98 (s, 1H), 3,90 (bs, 1H), 2,21 (s, 2H), 2,04–1,94 (m, 6H), 1,60 (bs, 2H), 1,49–1,43 (m, 3H). (UPLC–MS) tR 0,56 мин; ESI–MS 489 [M+H]+.
Промежуточное соединение 68a: 6–амино–5–(((1r,3r)–3–гидроксиадамантан–1–ил)карбамоил)пиридин–3–ил)бороновая кислота
Указанное в заголовке соединение получали способом, аналогичным для (6–амино–5–(((1r,4r)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)бороновой кислоты (промежуточного соединения 3a), за исключением того, что применяли 2–амино–5–бром–N–((1r,3r)–3–гидроксиадамантан–1–ил)никотинамид (промежуточное соединение 68b) вместо 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамида (промежуточного соединения 3c). (UPLC–MS) tR 0,47 мин; ESI–MS 332 [M+H]+.
Промежуточное соединение 68b: 2–амино–5–бром–N–((1r,3r)–3–гидроксиадамантан–1–ил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамида (промежуточного соединения 3c), за исключением того, что применяли 3–амино–1–адамантанол вместо транс–4–аминоциклогексанола. (UPLC–MS) tR 0,82 мин; ESI–MS 366/368 [M+H]+.
Пример 69: 2–амино–N–циклогексил–5–(4–(морфолин–4–карбонил)фенил)никотинамид
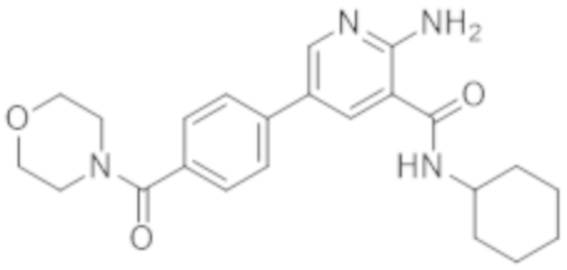
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли 4–(морфолин–4–карбонил)фенилбороновую кислоту вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,47 (d, 1H), 8,42 (s, 1H), 8,30 (d, 1H), 7,78 (d, 2H), 7,51 (d, 2H), 7,49 (s, 2H), 3,85–3,70 (m, 1H), 3,70 –3,50 (m, 8H), 1,90–1,80 (m, 2H), 1,80–1,70 (m, 2H), 1,70–1,55 (m, 1H), 1,35–1,29 (m, 4H), 1,28–1,05 (m, 1H). (HPLC–MS) tR 1,51 мин; APCI–MS 409 [M+H]+.
Пример 70: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(оксетан–3–илметил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
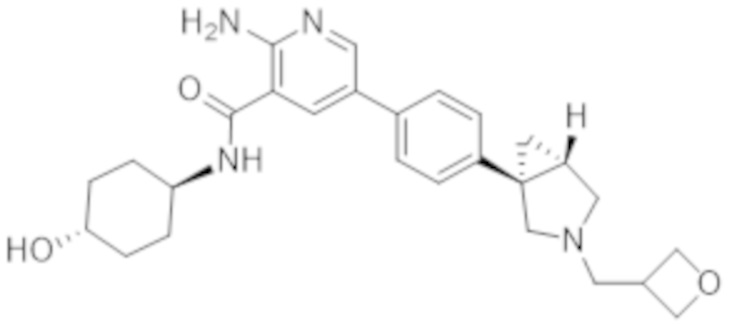
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли оксетан–3–илметилтрифторметансульфонат (промежуточное соединение 70a) вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a) и реакционную смесь перемешивали при 60°C в течение 3 ч. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,30 (d, 1H), 8,11 (d, 1H), 7,58 (d, 2H), 7,21 (d, 2H), 7,12 (s, 2H), 4,66 (dt, 2H), 4,58 (d, 1H), 4,28 (t, 2H), 3,90–3,66 (m, 1H), 3,43–3,35 (m, 1H), 3,25 (dd, 1H), 3,19–3,09 (m, 1H), 2,97 (d, 1H), 2,79 (d, 2H), 2,62–2,51 (m, 2H), 1,98–1,75 (m, 5H), 1,48–1,13 (m, 5H), 0,75 (dd, 1H). (UPLC–MS) tR 0,46 мин; ESI–MS 463 [M+H]+.
Промежуточное соединение 70a: оксетан–3–илметилтрифторметансульфонат
Указанное в заголовке соединение получали способом, аналогичным для 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a), за исключением того, что применяли оксетан–3–илметанол вместо 1,3–дифторпропан–2–ола, и его получали в виде неочищенного масла, которое применяли без дополнительной очистки.
Пример 71: 2–амино–N–((1s,4R)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
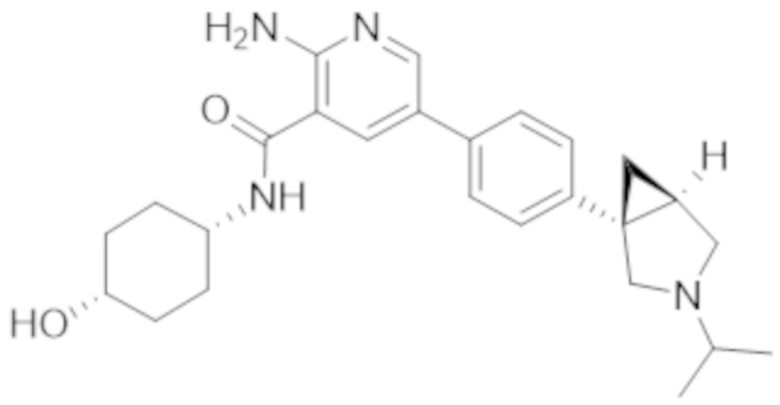
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 71a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли цис–4–аминоциклогексанола гидрохлорид вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,49–8,34 (m, 2H), 8,17 (s, 1H), 7,67 (s, 2H), 7,36 (s, 2H), 7,16 (s, 2H), 4,43 (d, 1H), 3,81 (s, 2H), 3,81–3,59 (m, 4H), 1,88–1,62 (m, 5H), 1,61–1,44 (m, 5H), 1,35 (s, 6H), 1,16 (s, 2H). (UPLC–MS) tR 0,52 мин; ESI–MS 435 [M+H]+.
Промежуточное соединение 71a: 2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c), за исключением того, что применяли метил–2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат (промежуточное соединение 71b) вместо (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d). (UPLC–MS) tR 0,40 мин; ESI–MS 338 [M+H]+.
Промежуточное соединение 71b: метил–2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
Указанное в заголовке соединение получали способом, аналогичным для метил–2–амино–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 7b), за исключением того, что применяли 2–йодпропан вместо 1–бром–2–метоксиэтана. (UPLC–MS) tR 0,65 мин; ESI–MS 352 [M+H]+.
Пример 72: 2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамид
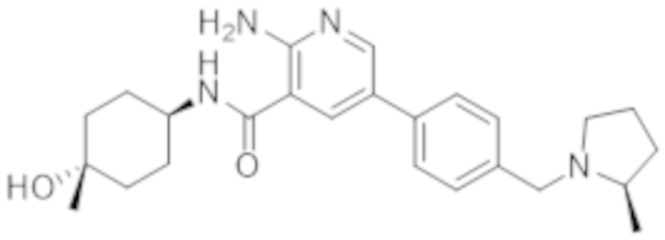
В раствор (4–(бромметил)фенил)бороновой кислоты (73,1 мг, 0,340 ммоль) в ацетонитриле (2 мл) добавляли (R)–2–метилпирролидин (29,0 мг, 0,340 ммоль) и K2CO3 (78 мг, 0,567 ммоль) при к. т. в атмосфере азота и полученную суспензию белого цвета перемешивали при 60°C в течение 2 ч. Добавляли воду (1 мл) при к. т. с последующим добавлением 2–амино–5–бром–N–((1r,4r)–4–гидрокси–4–метилциклогексил)никотинамида (промежуточного соединения 59b, 150 мг, 0,283 ммоль) в ацетонитриле (2 мл). Затем добавляли PdCl2(dppf) (10,37 мг, 0,014 ммоль) и реакционную смесь перемешивали при 90°C в течение 60 мин. После фильтрации через слой целита и концентрирования при пониженном давлении неочищенный продукт разбавляли насыщ. водн. раствором NaHCO3 и EtOAc. После разделения фаз водный слой экстрагировали с помощью EtOAc и объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт сначала очищали с помощью prepHPLC (способ 1a), затем с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 9,75 (d, 1H), 8,52 (t, 1H), 8,41 (d, 1H), 8,27 (d, 1H), 7,85 (d, 2H), 7,66 (d, 2H), 7,38 (bs, 2H), 4,67–4,53 (m, 1H), 4,25 (dd, 1H), 3,85 (s, 1H), 3,58–3,49 (m, 1H), 3,37–3,18 (m, 3H), 2,29 (m, 1H), 2,06–1,88 (m, 2H), 1,83 (s, 2H), 1,65 (dd, 2H), 1,51 (d, 4H), 1,39 (dd, 3H), 1,22 (d, 3H). (UPLC–MS) tR 0,47 мин; ESI–MS 423 [M+H]+.
Пример 73: 2–амино–5–(4–((1S,5R)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамид
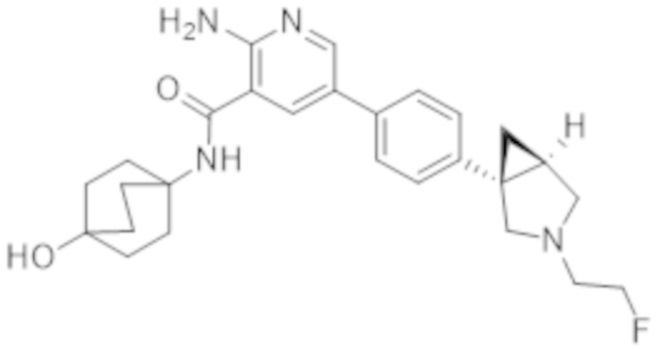
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли соль HCl и 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида (промежуточного соединения 40a) вместо 5–(4–((1S,5R)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 5a) и применяли 1–бром–2–фторэтан вместо 4–(3–бромпропил)морфолина, и реакционную смесь перемешивали при 60°C в течение 3 ч. 1H ЯМР (400 МГц, DMSO–d6) δ 8,35 (d, 1H), 7,98 (d, 1H), 7,79 (s, 1H), 7,57 (d, 2H), 7,22 (d, 2H), 6,92 (s, 2H), 4,55 (td, 2H), 4,31 (s, 1H), 3,38 (d, 1H), 3,10 (d, 1H), 2,84 (t, 1H), 2,76 (t, 1H), 2,64 (d, 1H), 2,54–2,51 (m, 1H), 2,13–1,96 (m, 6H), 1,83 (dd, 1H), 1,73–1,54 (m, 6H), 1,34 (t, 1H), 0,78 (dd, 1H). (UPLC–MS) tR 0,51 мин; ESI–MS 465 [M+H]+.
Пример 74: 2–амино–5–(2,3–дифтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамид
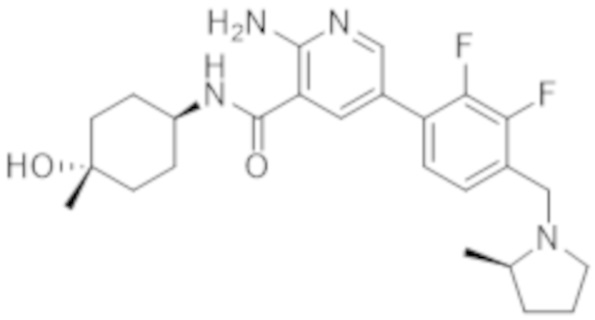
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(3–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида (примера 46), за исключением того, что применяли (R)–2–(2,3–дифтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион (промежуточное соединение 74a) вместо (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a). (UPLC–MS) tR 0,76 мин; ESI–MS 459 [M+H]+.
Промежуточное соединение 74a: (R)–2–(2,3–дифтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион
Указанное в заголовке соединение получали способом, аналогичным для (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a), за исключением того, что применяли 2,3–дифтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид (промежуточное соединение 74b) вместо 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b). ESI–MS 367 [M+H]+.
Промежуточное соединение 74b: 2,3–дифтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид
Указанное в заголовке соединение получали способом, аналогичным для 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b), за исключением того, что применяли (2,3–дифтор–4–формилфенил)бороновую кислоту вместо (3–фтор–4–формилфенил)бороновой кислоты. ESI–MS 595 [2M+H]+.
Пример 75: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
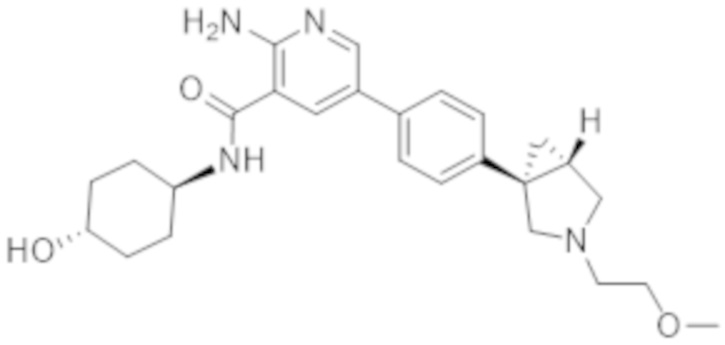
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 1–бром–2–метоксиэтан вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a) и реакционную смесь перемешивали при 60°C в течение 3 ч. После очистки получали указанное в заголовке соединение в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 9,70 (bs, 1H), 8,41 (s, 1H), 8,31 (d, 1H), 8,12 (d, 1H), 7,66–7,60 (m, 2H), 7,30 (bs, 2H), 7,14 (s, 2H), 4,59 (d, 1H), 3,81–3,38 (m, 8H), 3,31 (s, 3H), 2,75–2,25 (m, 4H),1,86 (t, 4H), 1,50–1,21 (m, 6H). (UPLC–MS) tR 0,48 мин; ESI–MS 451 [M+H]+.
Пример 76: 2–амино–5–(4–((1S,5R)–3–(2,2–дифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамид
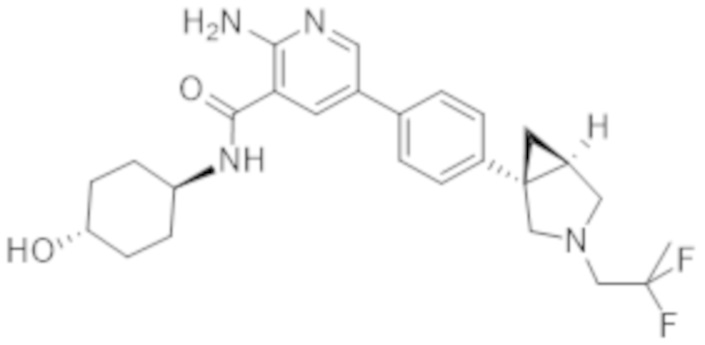
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 5), за исключением того, что применяли 2,2–дифторпропилтрифторметансульфонат вместо 4–(3–бромпропил)морфолина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,33 (d, 1H), 8,14 (d, 1H), 7,60 (d, 2H), 7,32–7,09 (m, 4H), 4,59 (bs, 1H), 3,74 (bs, 1H), 3,11 (d, 1H), 2,98–2,63 (m, 5H), 1,89–1,83 (m, 5H), 1,63 (t, 3H), 1,42–1,25 (m, 6H), 0,81 (bs, 1H). (UPLC–MS) tR 0,82 мин; ESI–MS 471 [M+H]+.
Пример 77: 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2,2,2–трифторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
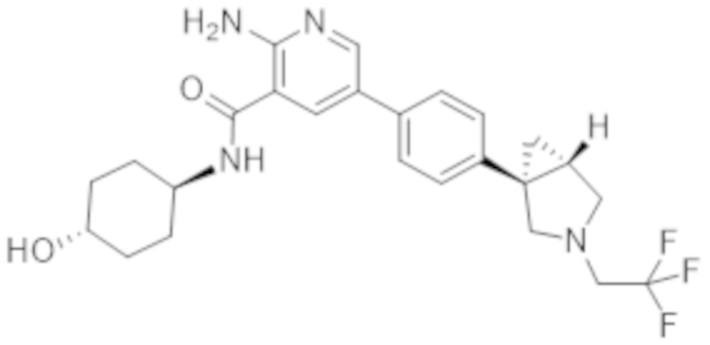
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида (примера 6), за исключением того, что применяли 2,2,2–трифторэтилтрифторметансульфонат вместо 1,3–дифторпропан–2–илтрифторметансульфоната (промежуточного соединения 6a) и реакционную смесь перемешивали при 60°C в течение 1 ч. 1H ЯМР (400 МГц, DMSO–d6) δ 8,39 (d, 1H), 8,32 (d, 1H), 8,13 (d, 1H), 7,60 (d, 2H), 7,24 (d, 2H), 7,16 (s, 2H), 4,58 (s, 1H), 3,73 (dt, 1H), 3,41 (m, 4H), 3,13 (d, 1H), 2,93 (d, 1H), 2,84 (dd, 1H), 1,92–1,77 (m, 5H), 1,51–1,15 (m, 5H), 0,84 (dd, 1H). (UPLC–MS) tR 1,03 мин; ESI–MS 475 [M+H]+.
Пример 78: 2–амино–N–((1r,4S)–4–гидроксициклогексил–4–d)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
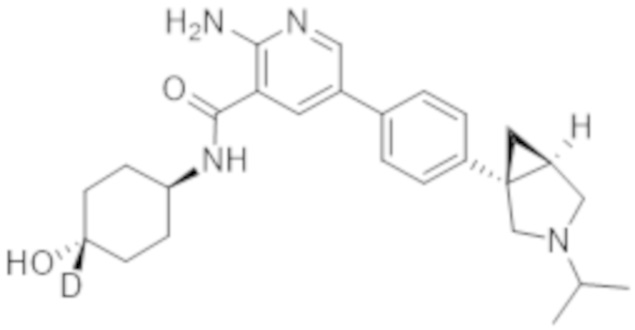
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 71a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли транс–4–аминоциклогексан–1–d–1–ола гидрохлорид (промежуточное соединение 78a) вместо транс–4–аминоциклогексанола. Неочищенный продукт сначала очищали с помощью prepHPLC (способ 1a), затем с помощью флэш–хроматографии с обращенной фазой (способ 3a) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. 1H ЯМР (400 МГц, DMSO–d6) δ 8,38 (d, 1H), 8,31 (d, 1H), 8,11 (d, 1H), 7,58 (d, 2H), 7,22 (d, 2H), 7,12 (s, 2H), 4,56 (s, 1H), 3,73 (dt, 1H), 3,37 (s, 1H), 3,06 (d, 1H), 2,57 (d, 1H), 2,49–2,41 (m, 2H), 1,93–1,73 (m, 5H), 1,31 (m, 5H), 1,04 (dd, 6H), 0,74 (dd, 1H). (UPLC–MS) tR 0,50 мин; ESI–MS 436 [M+H]+.
Промежуточное соединение 78a: транс–4–аминоциклогексан–1–d–1–ола гидрохлорид
Дейтерированные исходные материалы на основе аминоциклогексила можно получать с применением способов, подобных известным в уровне техники (например, Quirante, J. et al, J. Org. Chem. 67(7): 2323–2328 (2002)). Например, трет–бутилбензил(4–оксоциклогексил)карбамат можно восстанавливать с помощью NaBD4 с получением дейтерированного трет–бутилбензил((1r,4r)–4–гидроксициклогексил)карбамата, который после удаления защитных групп трет–бутилоксикарбонила и бензила может быть задействован в способах синтеза, описанных в данном документе.
Пример 79: соль TFA и 2–амино–N–((1s,4s)–4–гидроксициклогексил)–5–(4–(4–метилпиперазин–1–карбонил)фенил)никотинамида
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли 2–амино–5–бром–N–((1s,4s)–4–гидроксициклогексил)никотинамид (промежуточное соединение 79a) вместо 2–амино–5–бром–N–циклогексилникотинамида (промежуточного соединения 43a) и применяли (4–метилпиперазин–1–ил)(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)метанон вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. Реакцию проводили в микроволновом реакторе (время облучения 15 мин. при 80°C). Указанное в заголовке соединение получали в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 10,01 (s, 2H), 8,56 (d, 1H), 8,51 (d, 1H), 8,43 (d, 1H), 7,85 (d, 2H), 7,57 (d, 2H), 3,86–3,55 (m, 3H), 3,55–3,20 (m, 4H), 3,40–3,15 (m, 3H), 2,85 (s, 3H), 1,85–1,70 (m, 2H), 1,70–1,65 (m, 2H), 1,65–1,40 (m, 4H). (HPLC–MS) tR 0,35 мин; ESI–MS 438 [M+H]+.
Промежуточное соединение 79a: 2–амино–5–бром–N–((1s,4s)–4–гидроксициклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамида (промежуточного соединения 3c), за исключением того, что применяли цис–4–аминоциклогексанол вместо транс–4–аминоциклогексанола. (UPLC–MS) tR 0,53 мин; ESI–MS 314/316 [M+H]+.
Пример 80: 2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида (примера 1), за исключением того, что применяли соль HCl и 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида (промежуточного соединения 34a) вместо 5–(4–((1R,5S)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида (промежуточного соединения 1a) и применяли 4,4–дифторциклогексанон вместо 1–(метилсульфонил)пиперидин–4–она. 1H ЯМР (400 МГц, DMSO–d6) δ 8,32 (d, 1H), 7,95 (d, 1H), 7,76 (s, 1H), 7,55 (d, 2H), 7,21 (d, 2H), 6,88 (s, 2H), 4,28 (s, 1H), 3,36 (s, 1H), 3,07 (s, 1H), 2,55 (s, 1H), 2,45 (s, 1H), 2,32 (d, 1H), 2,30–1,95 (m, 8H), 1,77 (d, 5H), 1,70–1,32 (m, 8H), 1,27 (t, 1H), 0,75 (s, 1H). (UPLC–MS) tR 0,63 мин; ESI–MS 537 [M+H]+.
Пример 81: соль TFA и 2–амино–5–(2–хлор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(3–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида (примера 46), за исключением того, что применяли (R)–2–(2–хлор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион (промежуточное соединение 81a) вместо (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a). UPLC–HRMS tR 3,45 мин., 457,24/459,24 [M+H]+.
Промежуточное соединение 81a: (R)–2–(2–хлор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион
Указанное в заголовке соединение получали способом, аналогичным для (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a), за исключением того, что применяли 3–хлор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид (промежуточное соединение 81b) вместо 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b). ESI–MS 365/367 [M+H]+.
Промежуточное соединение 81b: 3–хлор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид
Указанное в заголовке соединение получали способом, аналогичным для 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b), за исключением того, что применяли (2–хлор–4–формилфенил)бороновую кислоту вместо (3–фтор–4–формилфенил)бороновой кислоты. ESI–MS 313/315 [M+H2O+H]+.
Пример 82: 2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 82a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли транс–4–амино–1–метилциклогексанол вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения, которое получали в виде соли TFA. 1H ЯМР (400 МГц, DMSO–d6) δ 8,37 (s, 1H), 8,26 (d, 1H), 8,09 (d,1H), 7,57 (d, 2H), 7,23 (d, 2H), 7,08 (s, 2H), 4,33 (s, 1H), 3,81 (bs, 1H), 3,39 (d, 2H), 2,62–2,56 (m, 2H), 2,00 (bs, 2H), 1,88–1,76 (m, 8H), 1,67–1,53 (m, 4H), 1,50–1,43 (m, 4H), 1,30 (t, 1H), 1,17 (s, 3H), 0,76 (bs, 1H). (UPLC–MS) tR 0,61 мин; ESI–MS 525 [M+H]+.
Промежуточное соединение 82a: 2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновая кислота
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 2c), за исключением того, что применяли метил–2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат (промежуточное соединение 82b) вместо (1R,5S)–трет–бутил–1–(4–(6–амино–5–(метоксикарбонил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1d). (UPLC–MS) tR 0,46 мин; ESI–MS 414 [M+H]+.
Промежуточное соединение 82b: метил–2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинат
Указанное в заголовке соединение получали способом, аналогичным для метил–2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотината (промежуточного соединения 14b), за исключением того, что применяли 4,4–дифторциклогексанон вместо дигидро–2H–пиран–4(3H)–она. (UPLC–MS) tR 0,72 мин; ESI–MS 428 [M+H]+.
Пример 83: 2–амино–N–(3–(2–гидроксипропан–2–ил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для (1R,5S)–трет–бутил–1–(4–(6–амино–5–(((1r,4R)–4–гидроксициклогексил)карбамоил)пиридин–3–ил)фенил)–3–азабицикло[3.1.0]гексан–3–карбоксилата (промежуточного соединения 1b), за исключением того, что применяли 2–амино–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновую кислоту (промежуточное соединение 14a) вместо 2–амино–5–(4–((1R,5S)–3–(трет–бутоксикарбонил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотиновой кислоты (промежуточного соединения 1c) и применяли 2–(3–аминобицикло[1.1.1]пентан–1–ил)пропан–2–ола гидрохлорид (промежуточное соединение 83a) вместо транс–4–аминоциклогексанола. Неочищенный продукт очищали с помощью prepHPLC (способ 1a) с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, DMSO–d6) δ 8,98 (s, 1H), 8,37 (s, 1H), 8,15 (s, 1H), 7,57 (d, 2H), 7,20 (d, 2H), 7,18 (s, 2H), 4,15 (s, 1H), 3,85–3,79 (m, 2H), 3,38 (bs, 1H), 3,08 (bs, 1H), 2,55–2,25 (m, 5H), 1,90 (s, 6H), 1,89–1,71 (m, 3H), 1,39–1,22 (m, 3H), 1,06 (s, 6H), 0,75 (bs, 1H). (UPLC–MS) tR 0,59 мин; ESI–MS 503 [M+H]+.
Промежуточное соединение 83a: 2–(3–аминобицикло[1.1.1]пентан–1–ил)пропан–2–ола гидрохлорид
Перемешивали трет–бутил(3–(2–гидроксипропан–2–ил)бицикло[1.1.1]пентан–1–ил)карбамат (промежуточное соединение 83b, 298 мг, 1,235 ммоль) в насыщенном растворе HCl в этаноле (3,8 мл) в течение 29 ч. при к. т. Реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде неочищенной гидрохлоридной соли, которую применяли без дополнительной очистки. 1H–ЯМР (400МГц, DMSO–d6) δ 8,51 (s, 3H), 1,78 (s, 6H), 1,03 (s, 6H).
Промежуточное соединение 83b: трет–бутил(3–(2–гидроксипропан–2–ил)бицикло[1.1.1]пентан–1–ил)карбамат
В раствор метил–3–((трет–бутоксикарбонил)амино)бицикло[1.1.1]пентан–1–карбоксилата (679 мг, 2,81 ммоль) в безводном THF добавляли по каплям 3 М бромметилмагний в THF (4,13 мл, 12,4 ммоль) при –78°C в атмосфере аргона. Обеспечивали нагревание реакционной смеси до к. т. и ее перемешивали в течение 4 ч., затем медленно разбавляли с помощью 2 мл MeOH при регулировании температуры. Смесь концентрировали при пониженном давлении и неочищенный продукт очищали с помощью хроматографии с нормальной фазой (способ 2a) с получением указанного в заголовке соединения. (UPLC–MS) tR 0,22 мин; ESI–MS 142 [M+H]+ указывала на потерю BOC–группы при условиях ионизации.
Пример 84: 2–амино–N–циклогексил–5–(3–(морфолин–4–карбонил)фенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли 3–(морфолин–4–карбонил)фенилбороновую кислоту вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,45 (s, 1H), 8,38 (d, 1H), 8,18 (s, 1H), 7,78 (d, 1H), 7,71 (s, 1H), 7,52 (t, 1H), 7,34 (d, 1H), 7,20 (s, 2H), 3,85–3,70 (m, 1H), 3,70 –3,50 (m, 8H), 1,90–1,80 (m, 2H), 1,80–1,70 (m, 2H), 1,70–1,55 (m, 1H), 1,35–1,29 (m, 4H), 1,28–1,05 (m, 1H). (HPLC–MS) tR 1,52 мин; APCI–MS 409 [M+H]+.
Пример 85: 2–амино–5–(3–хлор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(3–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида (примера 46), за исключением того, что применяли (R)–2–(3–хлор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион (промежуточное соединение 85a) вместо (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a). (UPLC–MS) tR 0,79 мин; ESI–MS 457/459 [M+H]+.
Промежуточное соединение 85a: (R)–2–(3–хлор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–дион
Указанное в заголовке соединение получали способом, аналогичным для (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a), за исключением того, что применяли 2–хлор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид (промежуточное соединение 85b) вместо 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b). ESI–MS 365/367 [M+H]+.
Промежуточное соединение 85b: 2–хлор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид
Указанное в заголовке соединение получали способом, аналогичным для 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b), за исключением того, что применяли (3–хлор–4–формилфенил)бороновую кислоту вместо (3–фтор–4–формилфенил)бороновой кислоты. ESI–MS 591 [2M+H]+.
Пример 86: 5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидрокси–1–метилциклогексил)никотинамид
В раствор 2–амино–5–бром–N–((1r,4r)–4–гидрокси–1–метилциклогексил)никотинамида (промежуточного соединения 86a, 100 мг, 0,152 ммоль) в диоксане (3 мл) добавляли (R)–6–(1–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)этил)–2–окса–6–азаспиро[3.3]гептан (промежуточное соединение 86b, 100 мг, 0,152 ммоль), PdCl2(dppf) (5,57 мг, 7,62 мкмоль) и 2 н. водн. NaOH (0,152 мл, 0,305 ммоль) в атмосфере азота при к. т. Реакционную смесь перемешивали при 80°C в течение 2 ч., затем разбавляли с помощью EtOAc и воды. После двух стадий экстракции с помощью EtOAc объединенные органические слои промывали насыщ. водн. раствором NaHCO3 и солевым раствором соответственно. После высушивания над MgSO4, фильтрации и концентрирования при пониженном давлении неочищенный продукт очищали с помощью хроматографии с нормальной фазой (способ 2b) с получением указанного в заголовке соединения. 1H ЯМР (400 МГц, DMSO–d6) δ 8,37 (s, 1H), 8,05 (bs, 1H), 7,70 (s, 1H), 7,62 (bs, 2H), 7,38 (bs, 2H), 6,92 (bs, 2H), 4,62 (s, 4H), 4,47 (s, 1H), 3,48–3,42 (m, 1H), 3,40–3,35 (m, 4H), 3,27–3,20 (m, 2H), 3,15 (bs, 1H), 1,60 (m, 2H), 1,65–1,16 (m, 13H). (UPLC–MS) tR 0,52 мин; ESI–MS 451 [M+H]+.
Промежуточное соединение 86a: 2–амино–5–бром–N–((1r,4r)–4–гидрокси–1–метилциклогексил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–бром–N–((1r,4r)–4–гидроксициклогексил)никотинамида (промежуточного соединения 3c), за исключением того, что применяли транс–4–амино–4–метилциклогексанол вместо транс–4–аминоциклогексанола. (UPLC–MS) tR 0,70 мин; ESI–MS 328/330 [M+H]+.
Промежуточное соединение 86b: (R)–6–(1–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)этил)–2–окса–6–азаспиро[3.3]гептан
Указанное в заголовке соединение получали способом, аналогичным для 1–(3–фтор–4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенил)–4–изопропилпиперазина (промежуточного соединения 8a), за исключением того, что применяли (R)–6–(1–(4–бромфенил)этил)–2–окса–6–азаспиро[3.3]гептан (промежуточное соединение 3b) вместо 1–(4–бром–3–фторфенил)–4–изопропилпиперазина (промежуточного соединения 8b). (UPLC–MS) tR 0,70 мин; ESI–MS 330 [M+H]+.
Пример 87: 2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(2–метил–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамид
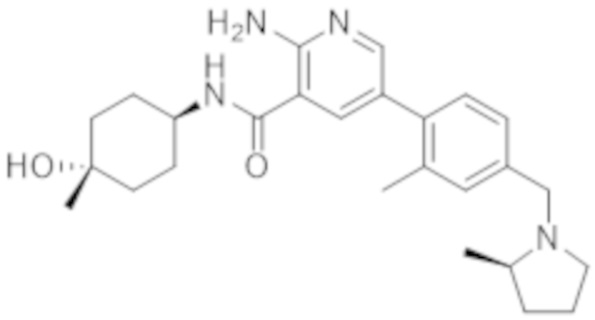
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–5–(3–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида (примера 46), за исключением того, что применяли (R)–6–метил–2–(2–метил–4–((2–метилпирролидин–1–ил)метил)фенил)–1,3,6,2–диоксазаборокан–4,8–дион (промежуточное соединение 87a) вместо (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a). (UPLC–MS) tR 0,65 мин; ESI–MS 437 [M+H]+.
Промежуточное соединение 87a: (R)–6–метил–2–(2–метил–4–((2–метилпирролидин–1–ил)метил)фенил)–1,3,6,2–диоксазаборокан–4,8–дион
Указанное в заголовке соединение получали способом, аналогичным для (R)–2–(3–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–6–метил–1,3,6,2–диоксазаборокан–4,8–диона (промежуточного соединения 46a), за исключением того, что применяли 3–метил–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид (промежуточное соединение 87b) вместо 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b). ESI–MS 345 [M+H]+.
Промежуточное соединение 87b: 3–метил–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегид
Указанное в заголовке соединение получали способом, аналогичным для 2–фтор–4–(6–метил–4,8–диоксо–1,3,6,2–диоксазаборокан–2–ил)бензальдегида (промежуточного соединения 46b), за исключением того, что применяли (4–формил–2–метилфенил)бороновую кислоту вместо (3–фтор–4–формилфенил)бороновой кислоты. ESI–MS 568 [2M+H2O+H]+.
Пример 88: 2–амино–N–циклогексил–5–(3–(4–(2–гидроксиэтил)пиперазин–1–карбонил)фенил)никотинамид
Указанное в заголовке соединение получали способом, аналогичным для 2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида (примера 43), за исключением того, что применяли 3–(4–(2–гидроксиэтил)пиперазин–1–карбонил)фенилбороновую кислоту вместо 1–(3–(4–(4,4,5,5–тетраметил–1,3,2–диоксаборолан–2–ил)фенокси)пропил)пиперидина. 1H ЯМР (400 МГц, DMSO–d6) δ 8,44 (s, 1H), 8,38 (d, 1H), 8,17 (s, 1H), 7,77 (d, 1H), 7,68 (s, 1H), 7,51 (t, 1H), 7,31 (d, 1H), 7,19 (s, 2H), 4,44 (t, 1H), 3,85–3,70 (m, 1H), 3,70 –3,58 (m, 2H), 3,58–3,48 (m, 2H), 3,48–3,35 (m, 2H), 2,50–2,32 (m, 6H), 1,90–1,80 (m, 2H), 1,80–1,70 (m, 2H), 1,70–1,55 (m, 1H), 1,35–1,29 (m, 4H), 1,28–1,05 (m, 1H). (HPLC–MS) tR 1,31 мин; APCI–MS 452 [M+H]+.
Пример 89: соли, аморфные формы и кристаллические полиморфы и псевдополиморфы соединения из примера 34.
A) Свободная форма модификации HA. Растворяли 800 г свободной формы 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида в 3 литрах ацетона. Полученный раствор перемешивали при 50°C и постепенно добавляли 2 литра воды. Смесь перемешивали при 50°C в течение 30 мин., затем постепенно добавляли 4 литра воды. Смесь перемешивали при 50°C в течение 2 часов и охлаждали до 22°C в течение периода, составляющего 2 часа. Твердое вещество разделяли с помощью фильтрации с отсасыванием, дважды промывали 1 литром ацетона:воды 1:2 и высушивали при 45°C в течение 24 часов без применения вакуума. Получали приблизительно 660 г свободной формы модификации HA 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида в виде белого твердого вещества. Свободная форма модификации HA представляет собой моногидрат.
На фиг. 3 показана диаграмма XRPD свободной формы модификации HA.
В таблице A ниже подробно приведены соответствующие пики и их значения 2–тета ("угол"), а также их относительные значения интенсивности (все значения 2–тета составляют +/− 0,2).
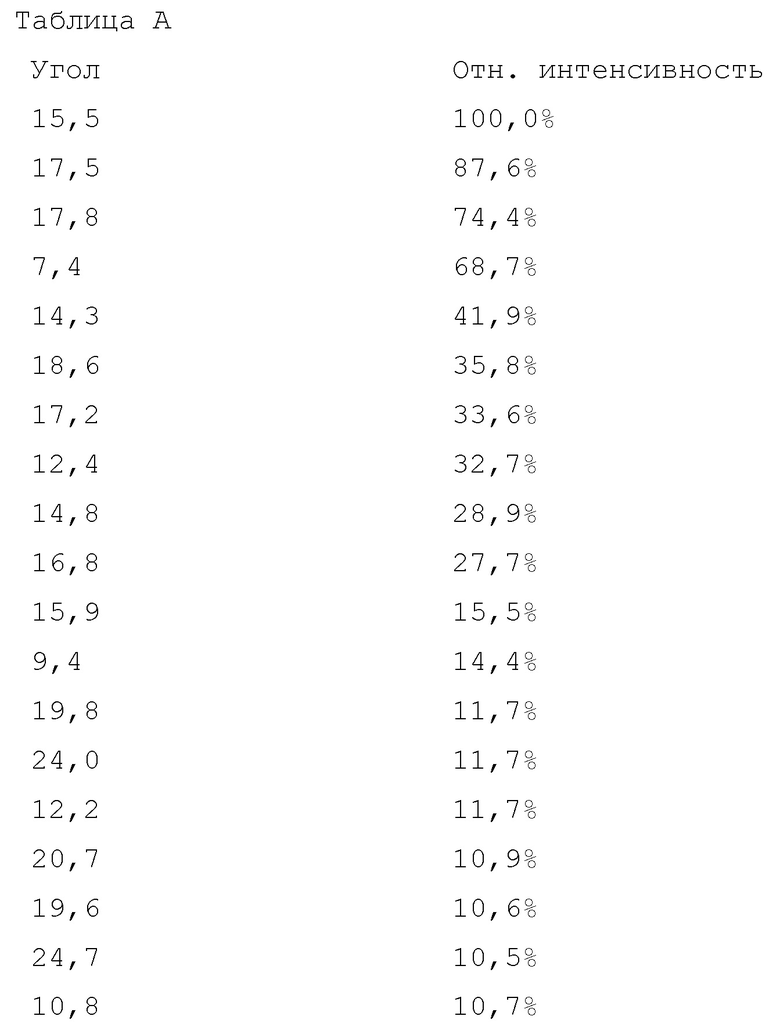
На фиг. 4 показана диаграмма открытой дифференциальной сканирующей калориметрии (DSC) свободной формы модификации HA. Первый эндотермический пик на диаграмме DSC соответствует дегидратации свободной формы модификации HA. Второй эндотермический пик соответствует плавлению безводной формы, полученной в результате дегидратации.
На фиг. 5 показана диаграмма термогравиметрического анализа (TGA) свободной формы модификации HA.
Результаты TGA демонстрируют, что свободная форма модификации HA содержит приблизительно 3,5% воды по весу, что соответствует приблизительно 1 молекуле воды.
На фиг. 6 представлена диаграмма FT–IR свободной формы модификации HA. Обнаруженные пики:
волновое число (в см−1): 3481(w), 3328(m), 2931(m), 2886(w), 2863(w), 2787(w), 1632(s), 1617(m), 1584(w), 1524(s), 1459 (s), 1242(m), 1090(m), 885(m), 769(m). (s=сильный, m=средний, w=слабый).
B) Свободная форма модификации A. Добавляли 0,5 г свободной формы модификации HA 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида в 3,5 мл ацетона. Смесь перемешивали при 40°C в течение 1 часа и охлаждали до 22°C. Твердое вещество фильтровали и высушивали при 40°C в течение 12 часов. Получали приблизительно 0,4 г свободной формы модификации A в виде белого твердого вещества.
На фиг. 7 показана диаграмма XRPD свободной формы модификации A.
В таблице B ниже подробно определены соответствующие пики и их значения 2–тета ("угол"), а также их относительные значения интенсивности (все значения 2–тета составляют +/− 0,2).
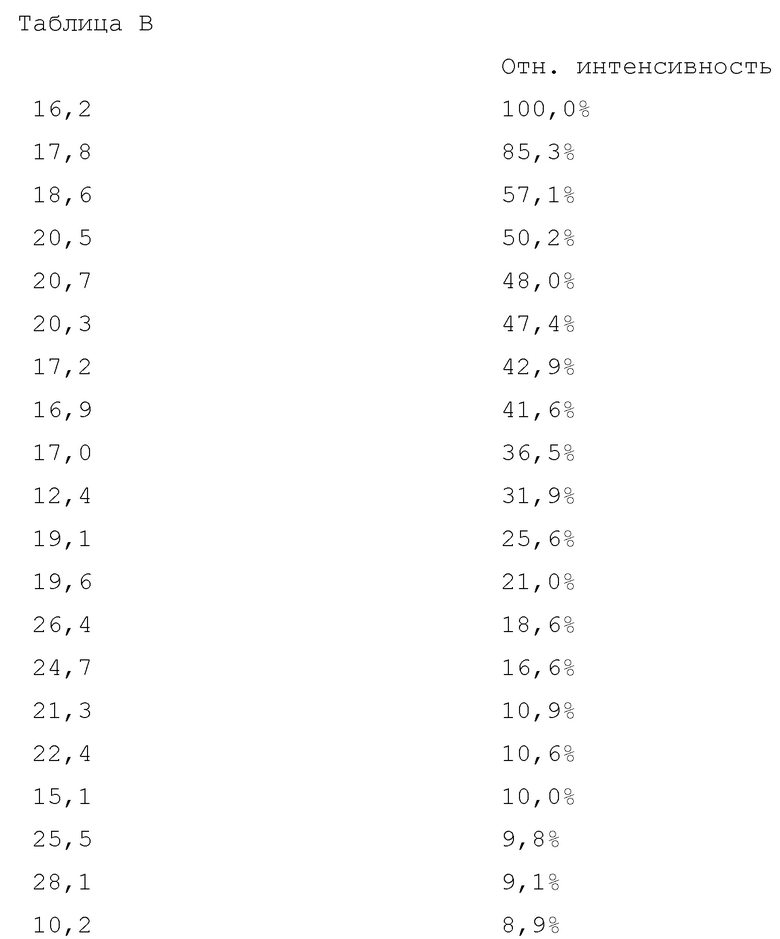
На фиг. 8 показана диаграмма открытой дифференциальной сканирующей калориметрии (DSC) свободной формы модификации A.
На фиг. 9 показана диаграмма термогравиметрического анализа (TGA) свободной формы модификации A. Результаты TGA указывают на то, что свободная форма модификации A представляет собой ангидрид.
На фиг. 10 представлена диаграмма FT–IR свободной формы модификации A. Обнаруженные пики:
волновое число (в см−1): 3480(w), 3436(w), 3401(w), 3301(m), 2944(w), 2864(w), 2805(w), 1644(s), 1615(m), 1583(w), 1520(s), 1459(m), 1247(m), 1098(m), 883(m), 801(m). (s=сильный, m=средний, w=слабый).
C) Свободная форма ангидрида. Подвергали 100 мг свободной формы модификации HA 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида воздействию атмосферы N2 при 25°C в течение 6 часов. Определяли характеристики полученного в результате вещества с помощью XRPD в защитной атмосфере N2. Твердое вещество представляет собой свободную форму ангидрида 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида.
На фигуре 18 показана диаграмма XRPD данной формы ангидрида.
D) Свободная форма тригидрата. Подвергали 100 мг свободной формы модификации HA 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида воздействию 80% относительной влажности при 25°C в течение ночи. Определяли характеристики твердого вещества с помощью XRPD при 80% относительной влажности при 25°C. Твердое вещество представляет собой свободную форму тригидрата 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида.
На фигуре 19 показана диаграмма XRPD данной формы тригидрата.
E) Фумаратная соль модификации HA. К 3,03 г свободной формы 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида добавляли 30 мл ацетона:воды (80:20, об./об.). Смесь перемешивали при 50°C в течение 0,5 часа и получали прозрачный раствор. В раствор медленно добавляли раствор 708,4 мг фумаровой кислоты в 15 мл ацетона:воды (80:20, об./об.). Полученную суспензию постепенно охлаждали до 25°C и продолжали перемешивание в течение 12 часов. Твердое вещество разделяли с помощью фильтрации с отсасыванием, один раз промывали с помощью 10 мл ацетона и подвергали воздействию условий окружающей среды (приблизительно 25°C, RH 50%) в течение ночи. Получали 3,3 г фумаратной соли модификации HA 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида в виде белого твердого вещества. Фумаратная соль модификации HA представляет собой дигидрат.
На фиг. 11 показана диаграмма XRPD фумаратной соли модификации HA.
В таблице C ниже подробно определены соответствующие пики и их значения 2–тета ("угол"), а также их относительные значения интенсивности (все значения 2–тета составляют +/− 0,2).
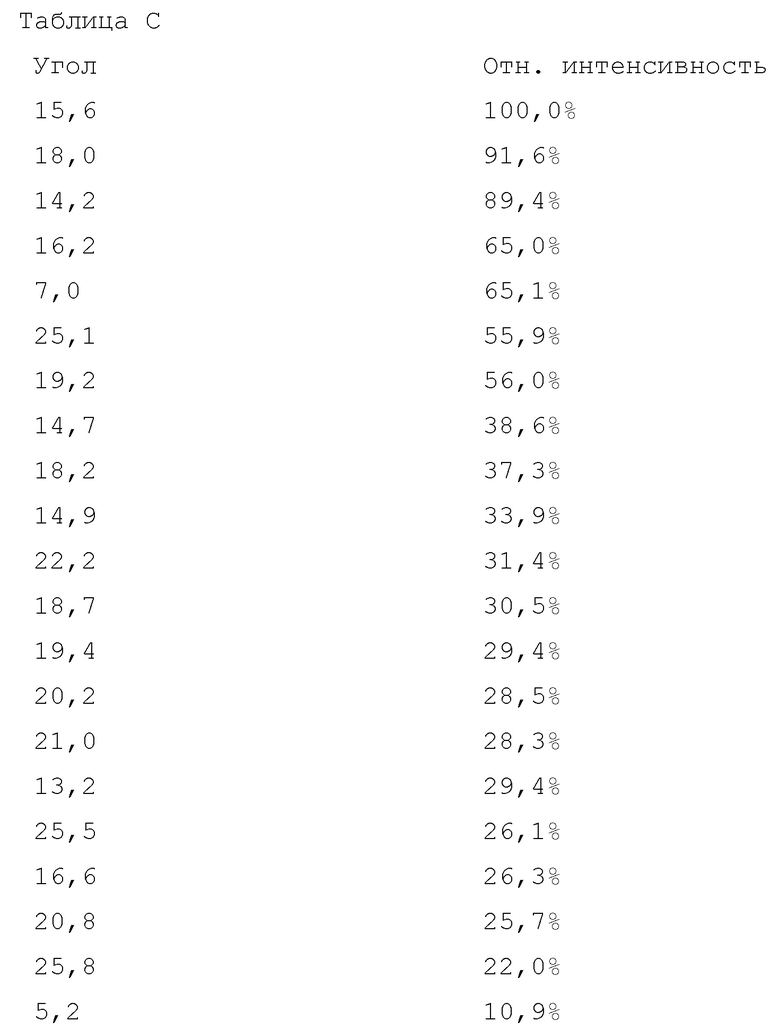
На фиг. 12 показана диаграмма DSC фумаратной соли модификации HA. Широкий эндотермический пик до 150°C соответствует дегидратации фумаратной соли модификации HA.
На фиг. 13 показана диаграмма термогравиметрического анализа (TGA) фумаратной соли модификации HA. Результаты TGA показывают, что фумаратная соль модификации HA содержит приблизительно 5,9% воды по весу, что соответствует приблизительно 2 молекулам воды.
На фиг. 14 представлена диаграмма FT–IR фумаратной соли модификации HA. Обнаруженные пики:
волновое число (в см−1): 3241(m), 2951(w), 2867(w), 1669(m), 1538(m), 1456(w), 1355(m), 1249(m), 1087(w), 979(w), 885(w), 797(w). (s=сильный, m=средний, w=слабый).
На фиг. 15 показана диаграмма DVS фумаратной соли модификации HA при 25°C. На ее основе можно сделать вывод, что фумаратная соль модификации HA является физически стабильной при RH от по меньшей мере 0% до 90% при 25°C.
На фиг. 16 показана диаграмма DVS фумаратной соли модификации HA при 40°C. На ее основе можно сделать вывод, что фумаратная соль модификации HA является физически стабильной при RH от по меньшей мере 10% до 90% при 40°C.
Следует отметить, что фумаратная соль модификации HA является стабильной при более широком диапазоне влажности, чем свободная форма полиморфов (данные не показаны).
F) Аморфная форма фумаратной соли (вариант 1). Добавляли 500 мг фумаратной соли модификации HA 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида в 10 мл этанола при 25°C с образованием прозрачного раствора. Затем в раствор добавляли 3 мл гептана. Белое твердое вещество выпадало в осадок. Твердое вещество разделяли с помощью фильтрации с отсасыванием и высушивали при 40°C под вакуумом в течение ночи. Получали 380 мг аморфной формы фумаратной соли 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида в виде белого твердого вещества. Аморфная форма демонстрирует переход в стекловидное состояние при приблизительно 143°C в случае проведения анализа с помощью модулированной DSC при скорости нагревания 2 K/мин., амплитудном значении температуры 1 K, периоде 60 секунд.
G) Аморфная форма фумаратной соли (вариант 2). Нагревали фумаратную соль модификации HA 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида с помощью DSC до 150°C при 10 K/мин., затем охлаждали до –20°C при 20 K/мин. Полученный материал представлял собой аморфную форму фумаратной соли 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида, который являлся белым твердым веществом, и демонстрирует переход в стеклообразное состояние при приблизительно 78°C в случае проведения анализа с помощью DSC при скорости нагревания 10 K/мин.
H) Фосфатная соль. В 3,01 г свободной формы 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида добавляли 30 мл ацетона:воды (75:25, об./об.). Полученную смесь перемешивали при 50°C в течение 0,5 часа и получали прозрачный раствор. В раствор медленно добавляли раствор 695,2 мг в 10 мл ацетона:воды (95:5, об./об.). Полученную суспензию постепенно охлаждали до 25°C и продолжали перемешивание в течение 24 часов. В суспензию добавляли 1,5 мл воды. Продолжали перемешивание смеси при 50°C в течение 12 часов и ее медленно охлаждали до 25°C. Твердое вещество разделяли с помощью фильтрации с отсасыванием, один раз промывали с помощью 10 мл ацетона:воды (90:10, об./об.) и подвергали воздействию условий окружающей среды (приблизительно 25°C, RH 50%) в течение ночи. Получали 2,7 г фосфатной соли 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида в виде белого твердого вещества.
На фиг. 17 показана диаграмма XRPD фосфатной соли.
В таблице D ниже подробно определены соответствующие пики и их значения 2–тета ("угол"), а также их относительные значения интенсивности (все значения 2–тета составляют +/− 0,2).
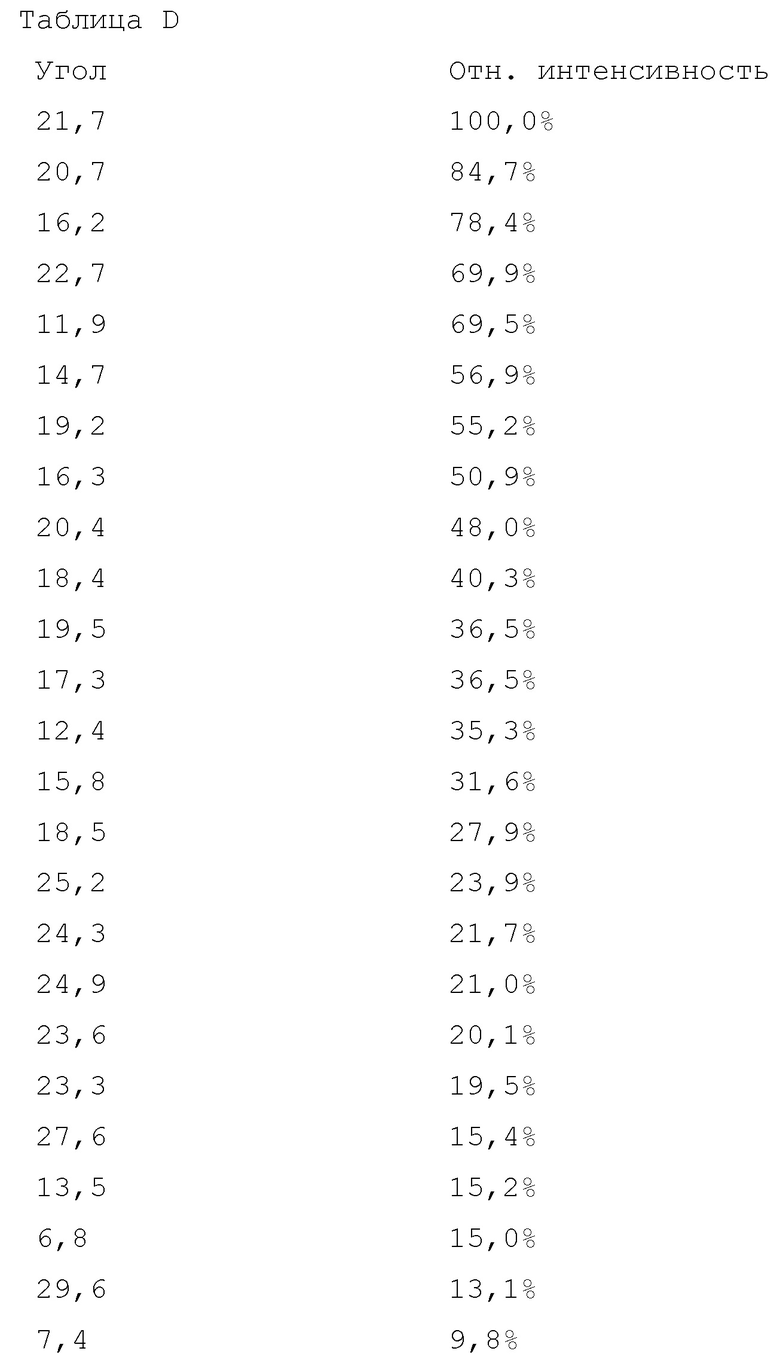
I) Фумаратная соль модификации HA (F) выше) демонстрирует более высокую растворимость, чем свободная форма модификации HA в некоторых водных буферах.
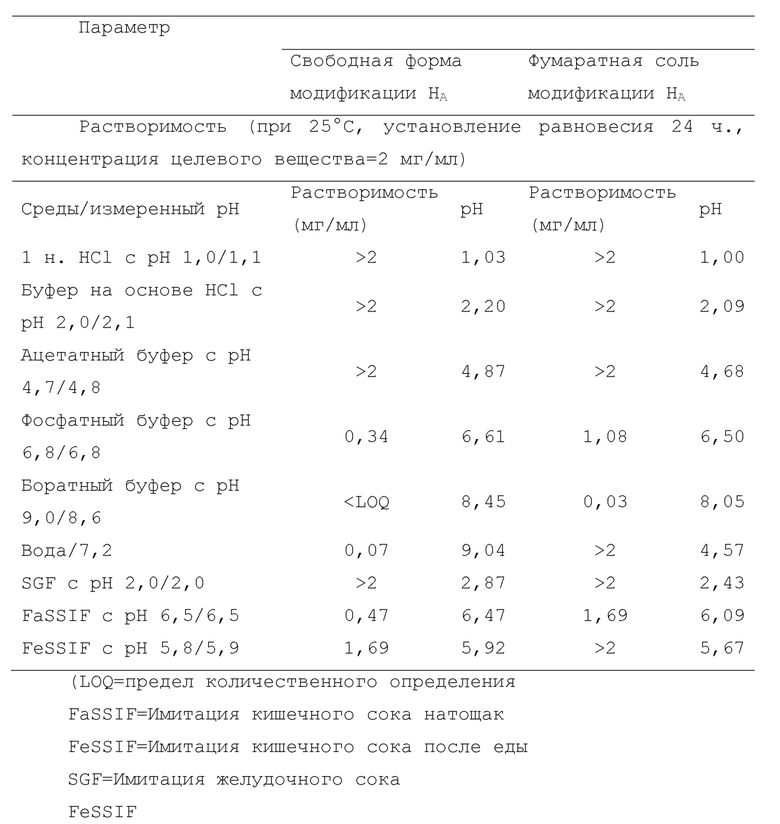
Rylo MG 19 PHARMA [мг]
FaSSIF
Получение буфера FaSSIF
SGF
J) Соль молочной кислоты, соль винной кислоты и соль яблочной кислоты соединения A можно получать способами, аналогичными приведенным выше.
K) Различные сольваты можно получать из многих солей и солевых форм соединения A, указанных выше (подробно не показаны).
Упоминаемые в примерах выше данные XRPD, TGA, DSC, FT–IR и DVS получены с применением следующих способов.
a) Способ TGA
Устройство: TA Discovery, TA Discovery, Нью–Касл, Делавэр, США
Диапазон значений температуры: от 30 до 300°C
Скорость сканирования: 10°C/мин.
Поток азота: 20 мл/мин.
Масса образца: ~2–10 мг
b) Способ DSC
Устройство: TA Discovery
Диапазон значений температуры: от 30 до 250 или 300°C
Скорость сканирования: 10°C/мин.
Поток азота: 50 мл/мин.
Масса образца: ~2 мг
c) Способ XRPD. Все диаграммы XRPD получали в режиме работы в отраженном свете.
(i) Способ XRPD 1 (фиг. 3, 11, 17)
Устройство: Bruker D8 Advance (Bruker, Брухзаль, Германия))
Детектор: LYNXEYE (режим 1D), открытый угол: 1,996°
Излучение: CuKα (0,15406 нм)
Монохроматор: никелевый фильтр
Мощность генератора рентгеновского излучения: 40 кВ, 40 мА
Размер шага, разделение: 0,041 градуса
Диапазон сканирования: от 2° до 45° (значение 2–тета)
Длительность сканирования: 36 мин.
Щели: первичная: размер закрепленного подсвеченного образца: 5 мм, вторичная щель: 5 мм, угловая щель Соллера: 2,5°
(ii) Способ XRPD 2 (фиг. 7)
Устройство: Bruker D8 Advance
Детектор: LYNXEYE (режим 1D), открытый угол: 1,996°
Излучение: CuKα (0,15406 нм)
Монохроматор: никелевый фильтр
Мощность генератора рентгеновского излучения: 40 кВ, 40 мА
Размер шага, разделение: 0,041 градуса
Диапазон сканирования: от 2° до 45° (значение 2–тета)
Длительность сканирования: 330 секунд
Щели: первичная: размер закрепленного подсвеченного образца: 5 мм, вторичная щель: 5 мм, угловая щель Соллера: 2,5
Способ XRPD 3 (фиг. 18, 19)
Устройство: Bruker D8 Advance XRPD с камерой Cryo–RH (Bruker, Брухзаль, Германия)
Детектор: VANTEC–1 (детектор 1D)
Излучение: CuKα (0,15406 нм)
Монохроматор: никелевый фильтр
Мощность генератора рентгеновского излучения: 40 кВ, 40 мА
Размер шага, разделение: 0,0165 градуса
Диапазон сканирования: от 2° до 45° (значение 2–тета)
Длительность сканирования: 17 мин.
Щели: рассеивающая щель: V12, антирассеивающая щель: 10,0 мм, детекторная щель: 17,43 мм,
первичная щель Соллера: 2,5°, вторичная щель Соллера: 2,5°
d) DVS
Устройство: Advantage (Surface Measurement Systems, Лондон, Великобритания)
Масса образца: ~10 мг
Температура: 25°C
dm/dt: 0,002%/мин.
e) FT–IR
Thermo Fisher Nicolet 6700 с ослабленной полной отражательной способностью (ATR), Thermo Fisher Scientific, Уолтем, Массачусетс, США.
Биохимические анализы (примеры 90 и 91)
В случае всех биохимических анализов рекомбинантные белки человека экспрессировали в клетках насекомых, трансфицированных бакуловирусом, и очищали из них. Конструкции содержали GS–домен и киназный домен ALK2 дикого типа (aa172–499), FOP–мутантного ALK2 (aa172–499 R206H), ALK3 (aa198–525), ALK5 (aa162–503) и ALK6 (aa168–495).
Пример 90. Анализ ингибирования фермента in vitro с применением анализа биохимического аутофосфорилирования (анализ киназной активности ADPGlo на основе люминесценции) – "анализ ADPGlo"
Панель для определения селективности в отношении киназ, в которой измерение аутофосфорилирования осуществляется с применением тест–набора для определения активности киназ ADP–Glo™ (Promega, V9101), настраивали для ALK2 дикого типа (aa172–499) и ALK3 (aa198–525).
Анализы проводили в 384–луночных малообъемных аналитических микротитрационных планшетах с конечным объемом реакционной смеси 6 мкл. Кривые зависимости доза–эффект получали путем инкубирования 10 нМ каждой киназы в 50 мМ Hepes с pH 7,5, 0,02% Tween 20, 0,02% BSA, 1 мМ DTT, 10мкм Na3VO4, 10 мМ β–глицеринфосфата, 1 мМ MgCl2, 12 мМ MnCl2 и 15 мкм ATP в течение 60 мин. при 32°C в присутствии или отсутствие соединения, разбавленного в DMSO. Количество образующегося ADP представляет собой меру киназной активности, и его количественное определение осуществляли с применением тест–набора для определения активности киназ ADP–Glo™ (Promega) в соответствии с инструкциями изготовителя. ADP превращается в ATP при добавлении 3 мкл реактива ADP–Glo™ и инкубации при 32°C в течении 60 мин. Впоследствии ATP превращается в биолюминесцентный сигнал при добавлении 6 мкл реактивов тест–набора для определения люциферазы (буфер для обнаружения киназы+субстрат для обнаружения киназы (Promega)) и дополнительной инкубации при 32°C в течении 60 мин. Для измерения люминесценции применяли многоканальный ридер PHERAstar™ при времени интервала измерений 0,1 секунд (оптический модуль для измерений люминесценции в диапазоне длин волн от 230 нм до 750 нм). Люминесцентный сигнал положительно коррелировал с киназной активностью.
Значения специфической активности показаны в таблице ниже.
л–1]
л–1]
Пример 91. Анализ ингибирования фермента in vitro с применением биохимического анализа фосфорилирования пептидов – "Caliper–анализ»
Панель для определения селективности в отношении киназ, в которой осуществляется измерение уровня фосфорилирования субстрата, представляющего собой пептид, настраивали для ALK2 дикого типа (aa172–499), FOP–мутантов ALK2 (aa172–499 R206H), ALK1 (aa166–493), ALK5 (aa162–503) и ALK6 (aa168–495). Технология, которую применяли для описанного анализа, основывается на разделении и количественном определении субстрата и продукта в электрическом поле. В ходе осуществляемой киназой реакции субстрат, представляющий собой пептид, фосфорилируется киназой. Перенос фосфатного остатка также приводит к введению двух дополнительных отрицательных зарядов и, следовательно, к изменению общего заряда фосфопептида по сравнению с нефосфорилированным пептидом. Благодаря этой разнице заряда фосфорилированный и нефосфорилированный пептиды мигрируют в электрическом поле с разными скоростями.
В применяемом способе это разделение происходит внутри чипа, который содержит сложную капиллярную систему для одновременного анализа 12 образцов ("12–канальный чип", Caliper Technologies Corp., Маунтин–Вью, США). Для того, чтобы обеспечить обнаружение и количественное определение пептидов в капиллярной системе, пептиды снабжали флуоресцентной меткой (флуоресцеином). При наличии этой метки количество пептидов можно определять по интенсивности флуоресценции с помощью инструментов лазерной системы и системы обнаружения (LC3000, Caliper Life Sciences).
Анализы проводили в 384–луночных малообъемных аналитических микротитрационных планшетах с конечным объемом реакционной смеси 9 мкл. Кривые зависимости доза–эффект получали путем инкубирования 10 нМ каждой киназы вместе с 2 мкмоль флуоресцентно меченного субстрата, представляющего собой пептид 5–Fluo–Ahx–KKYQAEEN–T–YDEYENKK–амид (10 мМ исходный раствор в DMSO), в 50 мМ Hepes с pH 7,5, 0,02% Tween 20, 0,02% BSA, 1 мМ DTT, 10мкМ Na3VO4, 10 мМ ß–глицеринфосфата, 1 мМ MgCl2, 12 мМ MnCl2 (для 7 мМ ALK1 и ALK6) и 15мкМ ATP в течение 60 мин. при 30°C в присутствии или отсутствие соединения, разбавленного в DMSO.
Осуществляемую киназой реакцию останавливали путем добавления 15 мкл STOP–буфера (100 мМ HEPES с pH 7,5, 5% DMSO, 0,1% покрывающего реагента Caliper, 10 мМ EDTA и 0,015% Brij35.
Планшеты, в которых осуществляемые киназой реакции были остановлены, перемещали в рабочую станцию Caliper LC3000 (Caliper Technologies Corp., Маунтин–Вью, США) для считывания. Относительное количество фосфорилированного пептида r рассчитывали с применением значений высоты пика субстрата, s, и высоты пика продукта, p: r=p/(p+s).
Значения специфической активности показаны в таблице ниже.
л–1])
л–1])
л–1])
л–1])
л–1])
Пример 92. Анализ передачи сигнала BMP с применением репортерного гена
Клеточную линию гепатоцеллюлярной карциномы человека (HuH7), стабильно трансфицированную репортерной плазмидой, состоящей из элемента отклика на BMP (BRE) из промотора Id1, слитого с репортерным геном, представляющим собой ген люциферазы, получали посредством лентивирусной трансдукции.
Клетки содержали в DMEM (GIBCO № 41965 с высоким содержанием глюкозы плюс L–глутамин), 10% FCS (Amimed № 2–01F10–I), 1% Pen/Strp (Amimed № 4–01F00) и 5 мкг/мл бластидицина (InvivoGen № ant–bl–1) при 37°C, 5% CO2. Анализы проводили в 384–луночных плоскодонных полистироловых микротитрационных планшетах (обработанных культурой клеток) со стерильной крышкой. За 16 ч. до начала анализа клетки подвергали голоданию путем замены среды на среду без бластицидина и FCS. Перед анализом клетки отделяли от флакона для исходной культуры с применением трипсина/EDTA и подсчитывали. Клеточную суспензию получали в той же среде без бластицидина и FCS. В каждую лунку планшета, уже содержащего последовательные разведения каждого соединения в DMSO (конечная концентрация DMSO 0,5%), добавляли по 2×104 клеток в общем объеме 40 мкл. Клетки и соединение инкубировали в течение 1 ч. при 37°C, 5% CO2, после чего осуществляли стимуляцию с помощью рекомбинантного BMP6 (R&D Systems № 507–BP/CF) в количестве 5 мкл на лунку при конечной концентрации 100 нг/мл. Аналитические планшеты инкубировали еще 5 часов при 37°C, 5% CO2, после чего проводили измерение уровней люциферазы.
Количественное определение экспрессированной люциферазы осуществляли с применением системы для анализа люциферазы Steady–Glo® (Promega № E2520). В каждую лунку добавляли по 5 мкл реагента Steady–Glo®, образцы перемешивали путем интенсивного встряхивания планшета, после чего проводили измерение люминесценции на многоканальном ридере PHERAstar™ в режиме 1 секунда/лунка (оптический модуль для измерений люминесценции в диапазоне длин волн от 230 нм до 750 нм).
Значения специфической активности соединений по настоящему изобретению дополнительно описаны в таблице ниже.
[мкмоль л–1]
Пример 93. Эффективность in vivo в мышиной модели FOP (прогрессирующей оссифицирующей фибродисплазии) у детей
Универсальная гетерозиготная экспрессия мутантного рецептора BMP типа I Alk2 R206H, обуславливающего FOP, у мышей приводит к перинатальной смертности (Chakkalakal et al., 2012).
Для исследования роли Alk2(R206H) в постнатальный период в организме молодой особи в качестве доклинической мышиной модели FOP необходима мутантная мышь с индуцируемым Alk2(R206H) (Prof. D. Goldhamer, UConn, ASBMR реферат, 2013) со встроенной stop–кассетой, фланкированной loxP–сайтами, расположенной выше мутантного экзона, что делает экспрессию мутантного аллеля зависимой от рекомбиназы Cre. Их скрещивали с мышами Rosa26–CreERt2, что позволяло осуществлять временное и пространственное регулирование гетерозиготной экспрессии Alk2(R206H).
В данном случае применяли мышей, гетерозиготных по универсально индуцируемому Alk2(R206H), в комбинации с технологией на основе CreER–loxP, индуцируемого тамоксифеном (tam). Успешная индукция экспрессии Alk2(R206H) была достигнута в результате введения тамоксифена 1–недельным самцам и самкам мышей Alk2(R206H); Rosa26–CreERt2.
Обострения FOP индуцировали посредством глубокой травмы мышц, осуществляемой путем местной внутримышечной (im.) инъекции кардиотоксина (CTX) и сопутствующей внутримышечной инъекции аденовируса, с получением Alk2(R206H)–зависимого местного обострения FOP. CTX вызывает повреждение скелетных мышц путем ингибирования Ca/Mg–ATPase в плазматической мембране и индуцирования высвобождения кальция из саркоплазматического ретикулума.
Массу тела 1–недельных самцов и самок Alk2(R206H); Rosa26–CreERt2 измеряли в начале эксперимента и далее ежедневно. Всех мышат обрабатывали один раз в день путем подкожного (sc.) введения 20 мг/кг тамоксифена (тамоксифен в виде свободного основания (Sigma T5648), растворенный в 10% абсолютного спирта, 90% подсолнечного масла (Sigma S5007), при 8 мг/мл), начиная с возраста 1 неделя в течение 10 дней.
На 3–й день инъекции тамоксифена всех мышат подвергали местному повреждению мышц путем однократной внутримышечной инъекции 100 мкл аденовируса+кардиотоксина (CTX) в икроножную мышцу правой задней конечности, принимая меры, чтобы не осуществить инъекцию в кровеносные сосуды и не касаться иглой костей. Для этого 90 мкл аденовируса (Ad–GFP–2A–iCre, № в кат. 1772, Vector Biolabs; титр: 1×1010 БОЕ (бляшкообразующих единиц)/мл) смешивали с 10 мкл 100 мкМ исходного раствора кардиотоксина (получали из кардиотоксина, № в кат. L8102, Latoxan, или кардиотоксина, № в кат. C9759, Sigma, с применением стерильного физиологического раствора). Животных анестезировали с помощью ингаляции низкой дозы изофлурана, с правой задней конечности сбривали шерсть и кожу дезинфицировали с помощью бетасептика, после чего осуществляли внутримышечную инъекцию, которую проводили медленно и осторожно с применением инсулинового шприца.
Терапевтическую пероральную обработку мышей, гетерозиготных по Alk2(R206H), с частотой b.i.d. начинали через 3 дня после повреждения мышц и осуществляли с помощью 2, 5, 10 и 25 мг/кг соединения A (соединение из примера 34=2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабициклилo[3.1.0]гексан–1–ил)фенил)никотинамид) или соответствующей среды–носителя в течение 6 недель. У всех мышей гетеротопическую оссификацию оценивали раз в две недели с помощью рентгенографии всего тела и задней конечности (устройство Faxitron) и визуализации in vivo с помощью компьютерной микротомографии (µCT), начиная с 2 недели после нанесения Ad/CTX.
С целью определения полного масштаба HO при вскрытии собирали правые задние конечности для визуализации с помощью микро–CT ex vivo. Целую правую заднюю конечность с прикрепленной мышцей как можно быстрее разрезали и переносили в 70% этанол при 4°C. Через 24 ч. образцы переносили в свежий 70% этанол для осуществления визуализации с помощью μCT с высоким разрешением с применением устройства μCT40 от Scanco Medical (размер вокселя: 16 мкм, время интеграции: 250 мс, среднее разрешение, 55 E(кВп), 145 мкA, режим: непрерывное вращение конусообразного луча, порог 275, фильтр Гаусса: среднее квадратическое отклонение 0,7, окрестность 1). Определяли общий объем костной ткани (мм3) эктопической кости в задней конечности. Результаты показаны на фигуре 1. Из фигуры видно, что применение соединения A (соединение из примера 34) приводило к уменьшению объема костной ткани правой задней конечности ex vivo.
Для исследования эффекта соединения A (соединение из примера 34) в отношении прогрессирования гетеротопической оссификации после запуска процесса мышам, гетерозиготным по Alk2(R206H), подобным образом вводили 100 мкл аденовируса+кардиотоксина (CTX), но позволяли гетеротопической кости развиваться в течении 2,5 недели. После подтверждения образования HO через 2 недели мышат рандомизировали на основе наличия HO, генетического фона и пола. Обработку начинали через 2,5 недели после повреждения и осуществляли с применением среды–носителя или 10 мг/кг соединения A b.i.d. и продолжали в течение 6 недель. У всех мышей гетеротопическую оссификацию оценивали раз в две недели с помощью рентгенографии всего тела и задней конечности (устройство Faxitron) и визуализации in vivo с помощью компьютерной микротомографии (μCT), начиная с 2 недели после применения Ad/CTX.
С целью определения полного масштаба HO при вскрытии собирали правые задние конечности для визуализации с помощью микро–CT ex vivo. Целую правую заднюю конечность с прикрепленной мышцей как можно быстрее разрезали и переносили в 70% этанол при 4°C. Через 24 ч. образцы переносили в свежий 70% этанол для осуществления визуализации с помощью μCT с высоким разрешением с применением устройства μCT40 от Scanco Medical (размер вокселя: 16 мкм, время интеграции: 250 мс, среднее разрешение, 55 E(кВп), 145 мкA, режим: непрерывное вращение конусообразного луча, порог 275, фильтр Гаусса: среднее квадратическое отклонение 0,7, окрестность 1). Определяли общий объем костной ткани (мм3) эктопической кости в задней конечности. Результаты показаны на фигуре 2. Из результатов видно, что применение соединения A (соединение из примера 34) предотвращало увеличение объема костной ткани правой задней конечности ex vivo.
Из результатов, показанных на фигурах 1 и 2, видно, что соединение по настоящему изобретению (соединение A) подавляло гетеротопическую оссификацию, если введение дозы осуществляли вскоре после повреждения мышцы, и предотвращало дополнительное прогрессирование гетеротопической оссификации у приплода мышей ALK2(R206H), если введение дозы осуществляли после того, как гетеротопическая оссификация уже началась.
Пример 94. Гетеротопическая оссификация у крыс, вызванная тенотомией ахиллова сухожилия в срединной точке
Чтобы проверить, способны ли соединения по настоящему изобретению предотвращать гетеротопическую оссификацию (НО) мягких тканей, вызванную травмой, можно применять терапевтическую эффективность в крысиной модели односторонней тенотомии ахиллова сухожилия в срединной точке (Rooney et al., Matrix 12: 274–281, 1992). Для этого левое ахиллово сухожилие 8–недельных самок крыс линии Wistar (масса тела в диапазоне 190–265 г) полностью рассекали с применением стерильного скальпеля (номер лезвия 11) под действием ингаляционного наркоза с помощью изофлурана с сопутствующей обезболивающей обработкой с применением 0,03 мг/кг бупренорфина в течение 48 часов через каждые 10–12 ч. подкожно. Профилактическую пероральную обработку с помощью соединения по настоящему изобретению (10 мг/кг q.d.) или среды–носителя (натрия карбоксиметилцеллюлоза:вода:Tween 80, 0,5:99:0,5) проводили в течение 10 недель, начиная со дня оперативного вмешательства (n=11–12 крыс на группу). После оперативного вмешательства крыс содержали по отдельности в течение 3–4 дней, а после этого содержали группами по два животных на клетку при 25°C с циклом чередования света и темноты 12:12 ч. и кормили стандартным рационом для грызунов, содержащим 18,2% белка и 3,0% жира, с калорийностью 15,8 МДж/кг (3890, Provimi Kliba SA), при этом еду и воду предоставляли ad libitum. Эффективность обработки оценивали в продольном направлении путем получения рентгенограмм оперированной дистальной конечности (система Faxitron LX–60) через 4 и 10 недель после тенотомии. Объем гетеротопической костной ткани количественно определяли in vivo с помощью компьютерной микротомографии (микро–CT) под действием ингаляционного наркоза с помощью изофлурана (прибор vivaCT40, Scanco Medical AG; разрешение 17,5 мкм) через 6 и 9 недель после оперативного вмешательства.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2022 |
|
RU2803084C1 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2017 |
|
RU2777979C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ ALK-2 | 2023 |
|
RU2826177C1 |
| АЗАИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ПОВРЕЖДЕНИЯХ СУХОЖИЛИЙ И/ИЛИ СВЯЗОК | 2017 |
|
RU2758256C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ KDM5, И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2840256C1 |
| ПРОИЗВОДНЫЕ ПИРРОЛОПИРИМИДИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2012 |
|
RU2618673C2 |
| ПРОИЗВОДНЫЕ АЗАБИЦИКЛО [3.1.0]ГЕКСАНА, ПРИМЕНИМЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ДОПАМИНОВЫХ РЕЦЕПТОРОВ D3 | 2005 |
|
RU2434011C2 |
| МАКРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ КИНАЗ TRK | 2019 |
|
RU2778294C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| ЗАМЕЩЕННЫЕ ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2840772C1 |
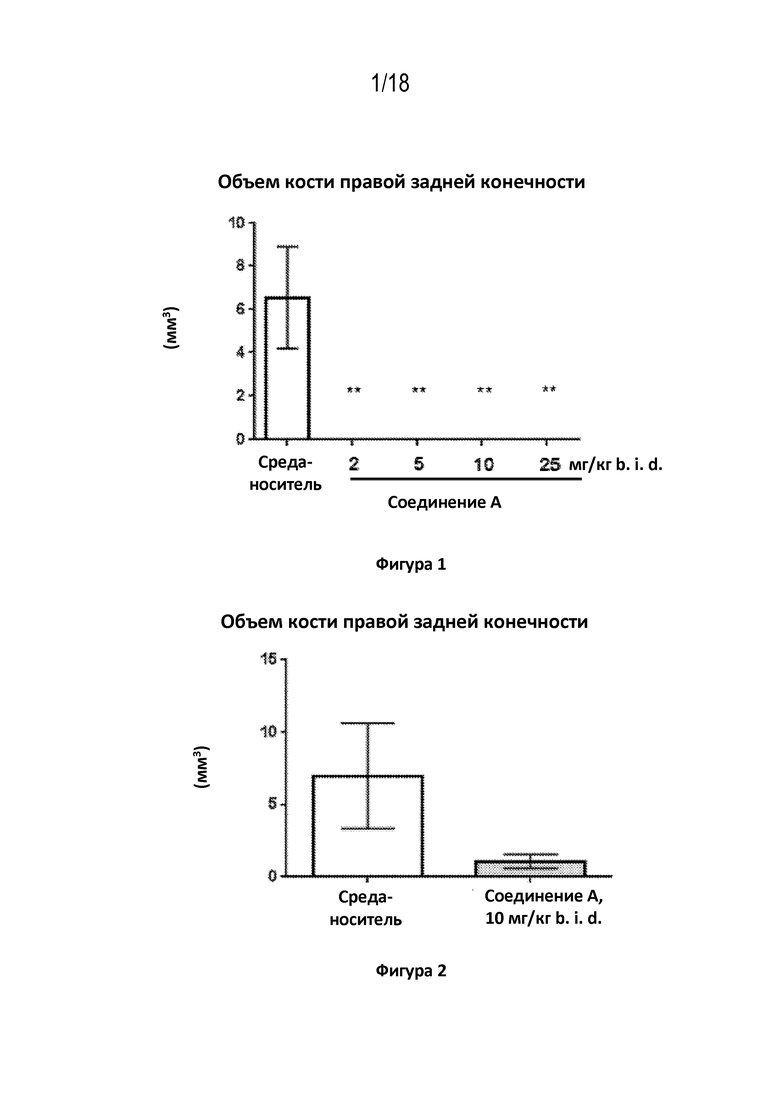
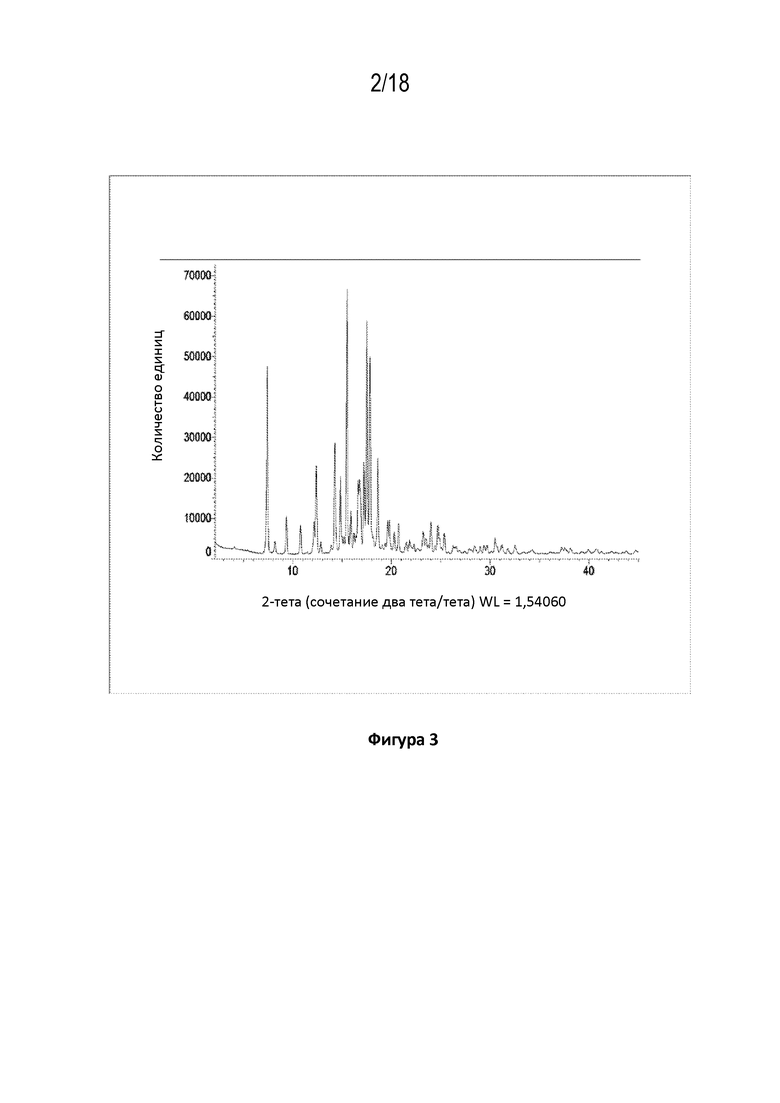
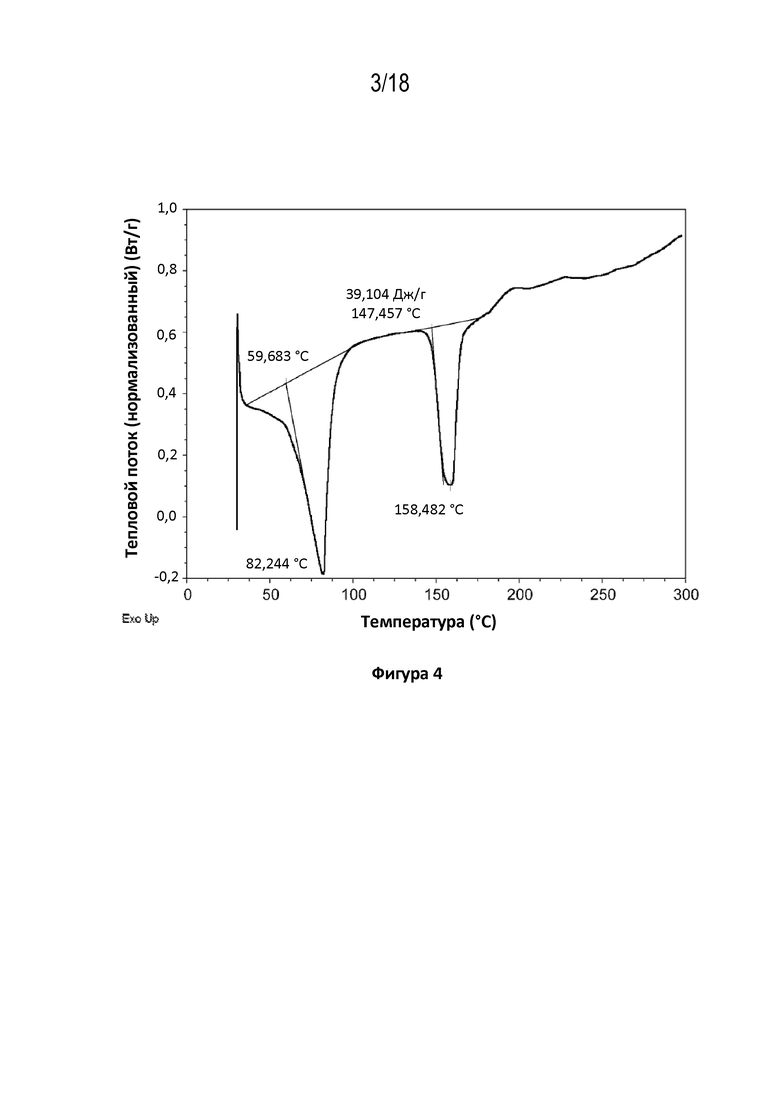
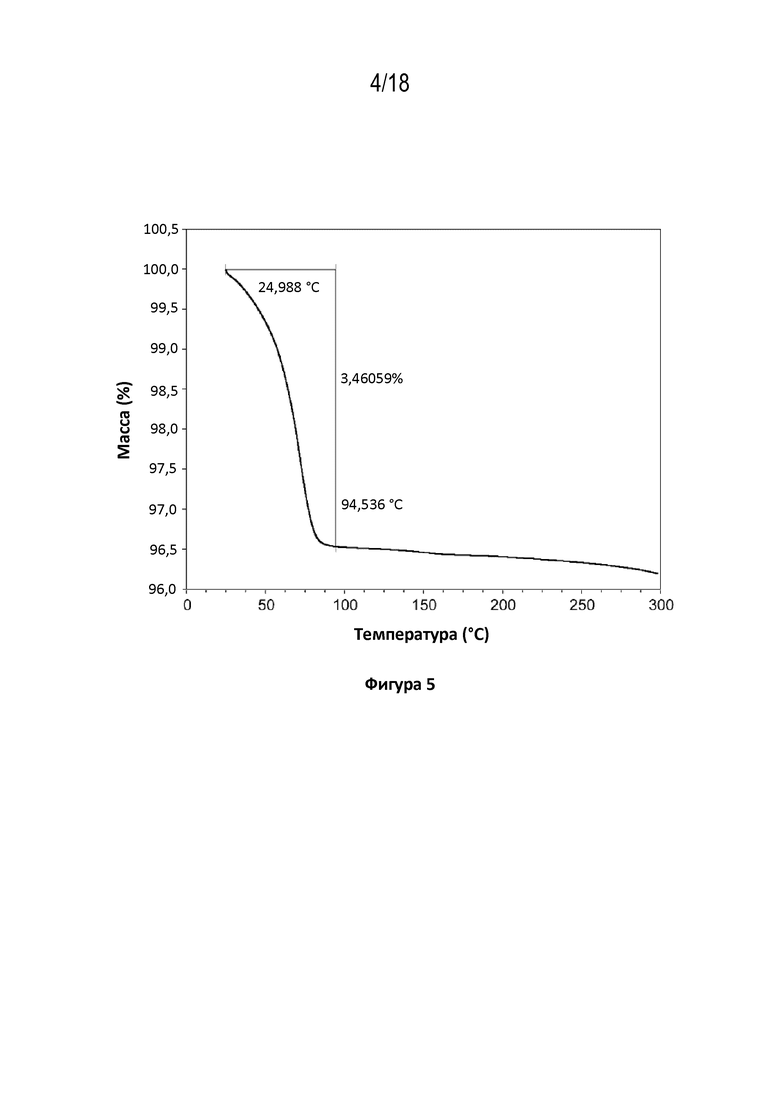
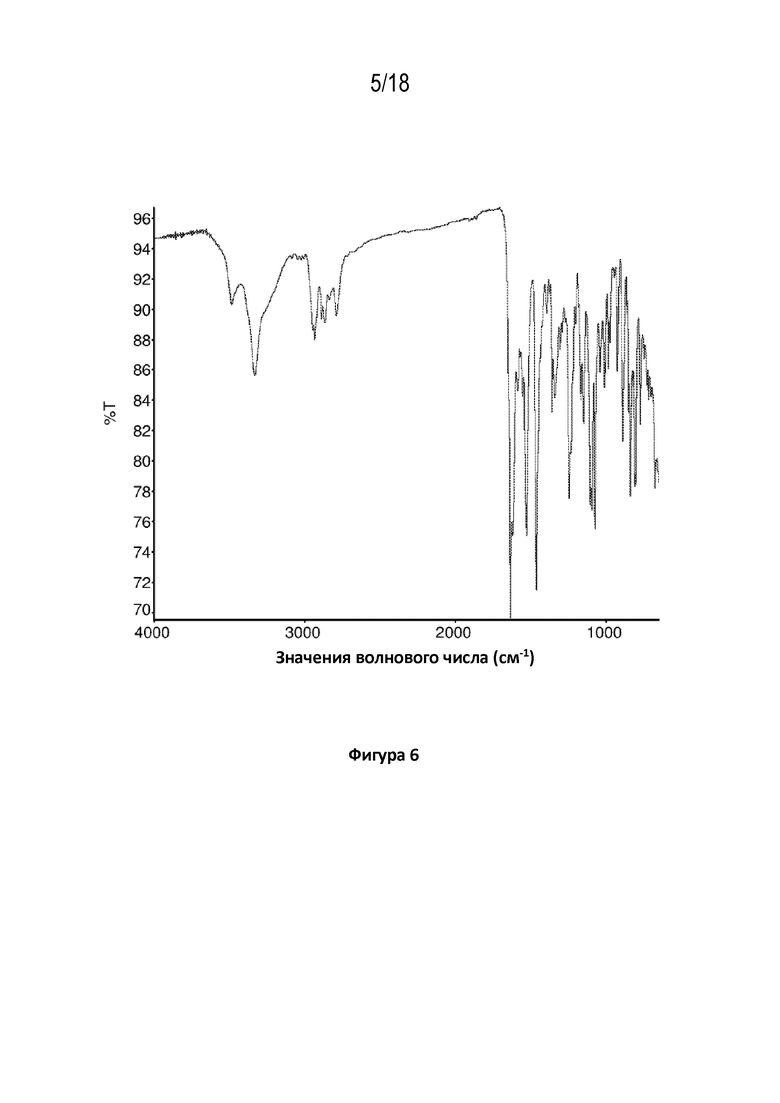
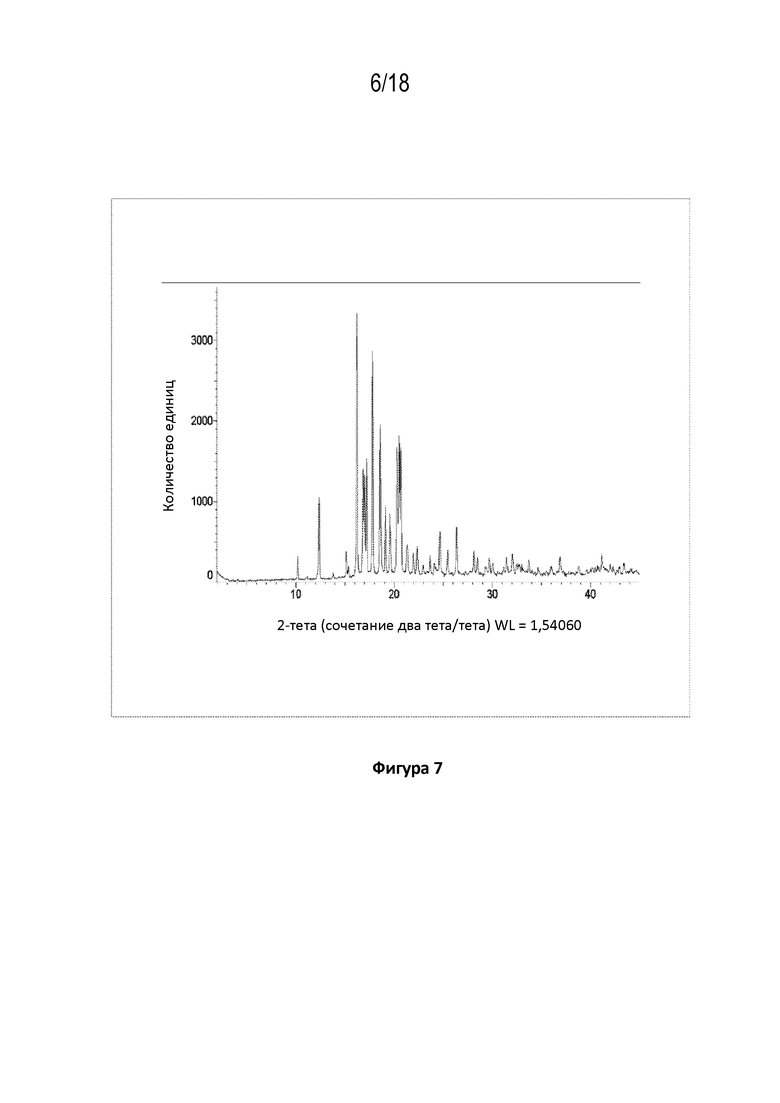
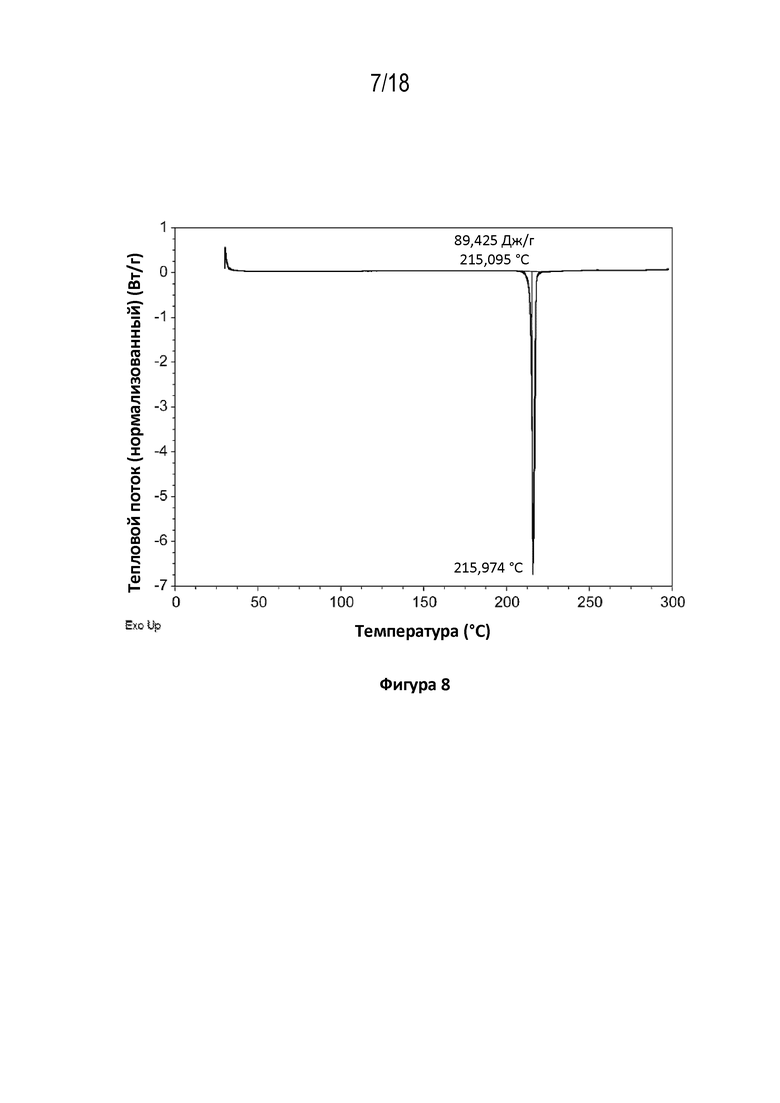
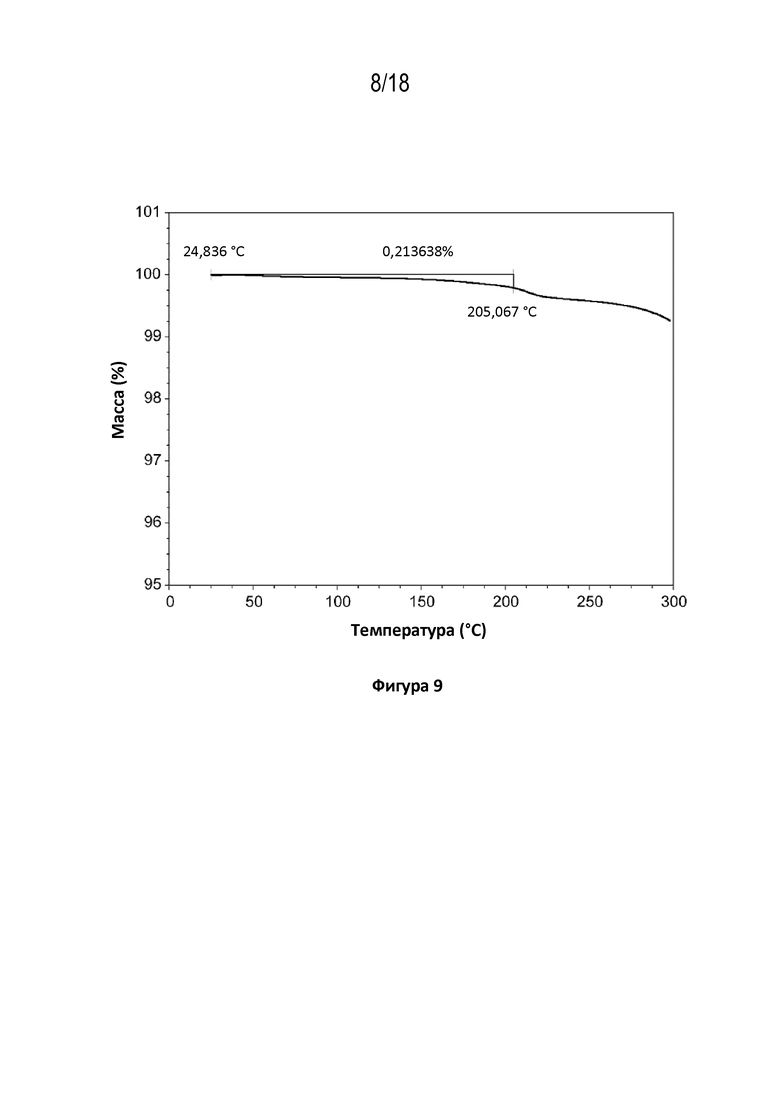
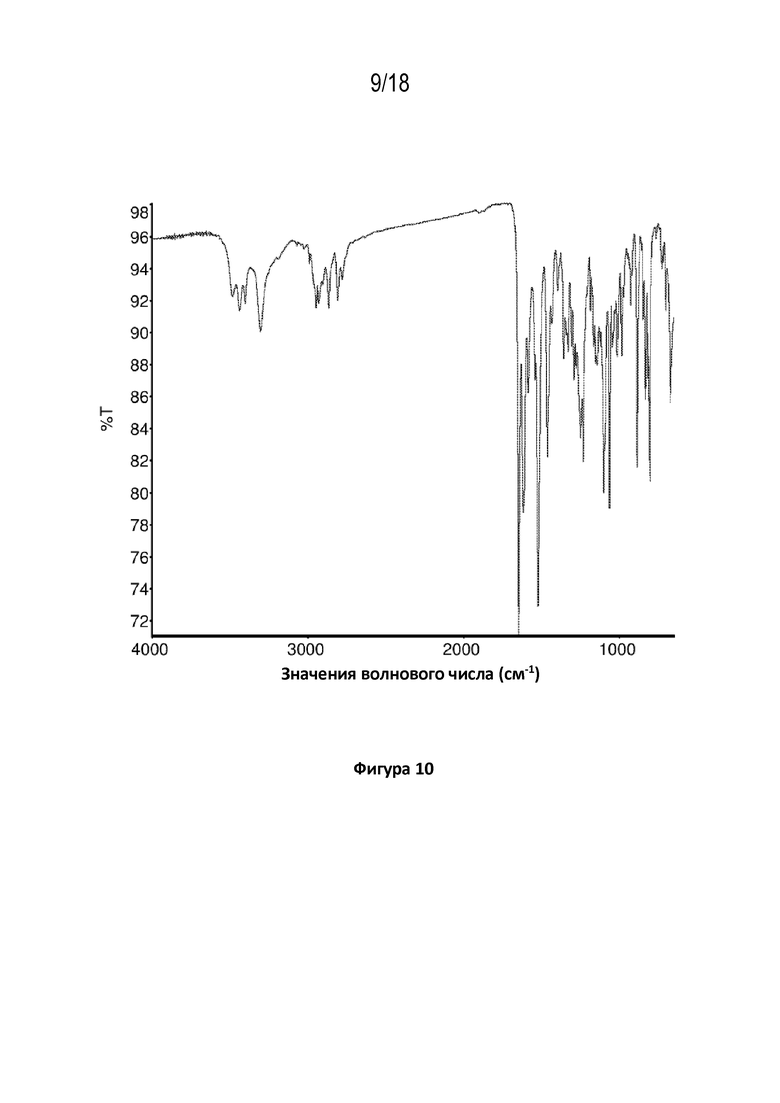
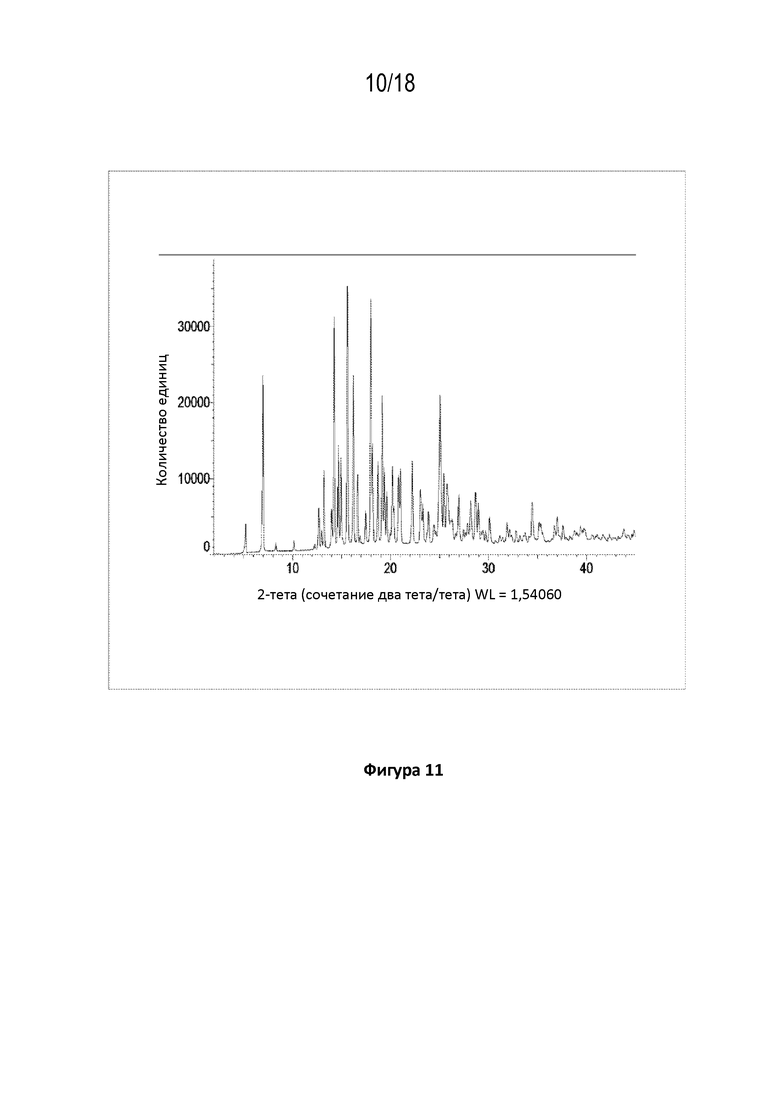
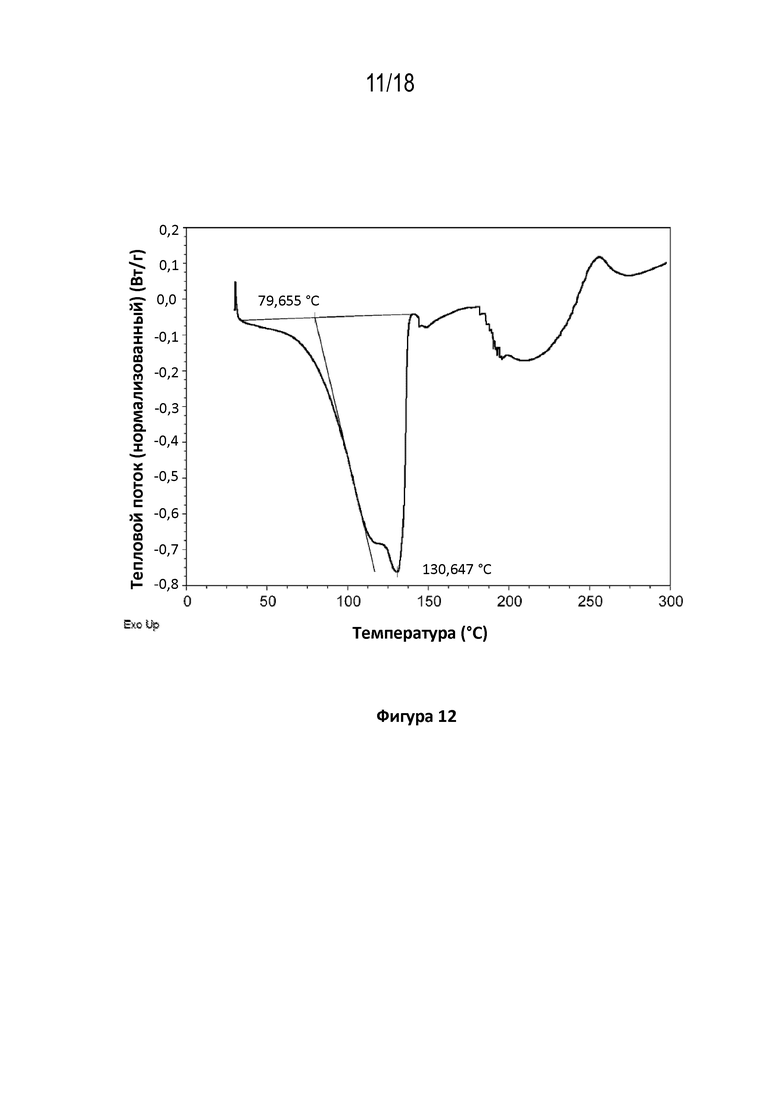
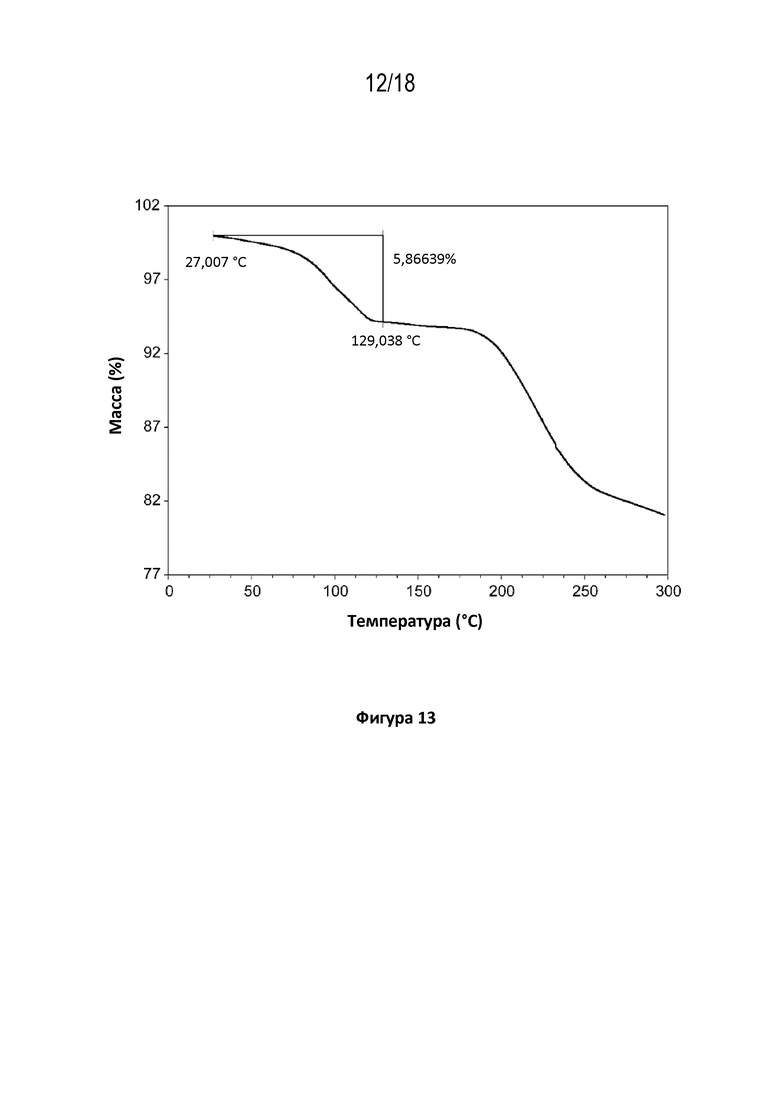
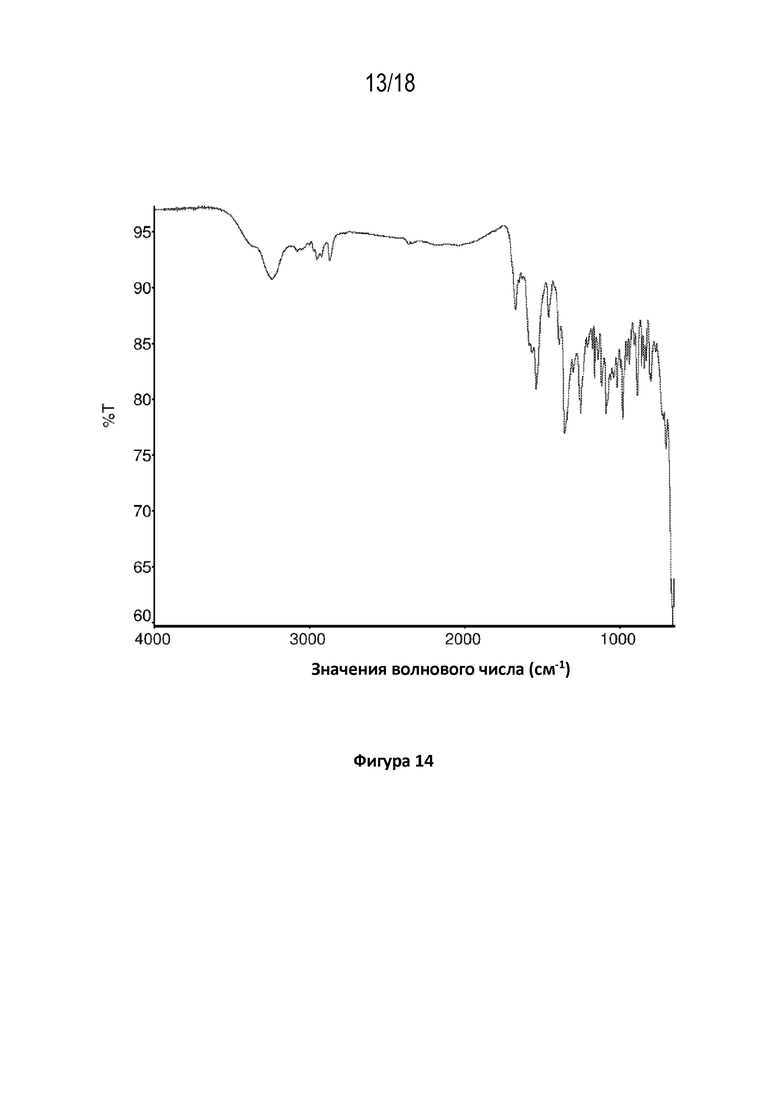
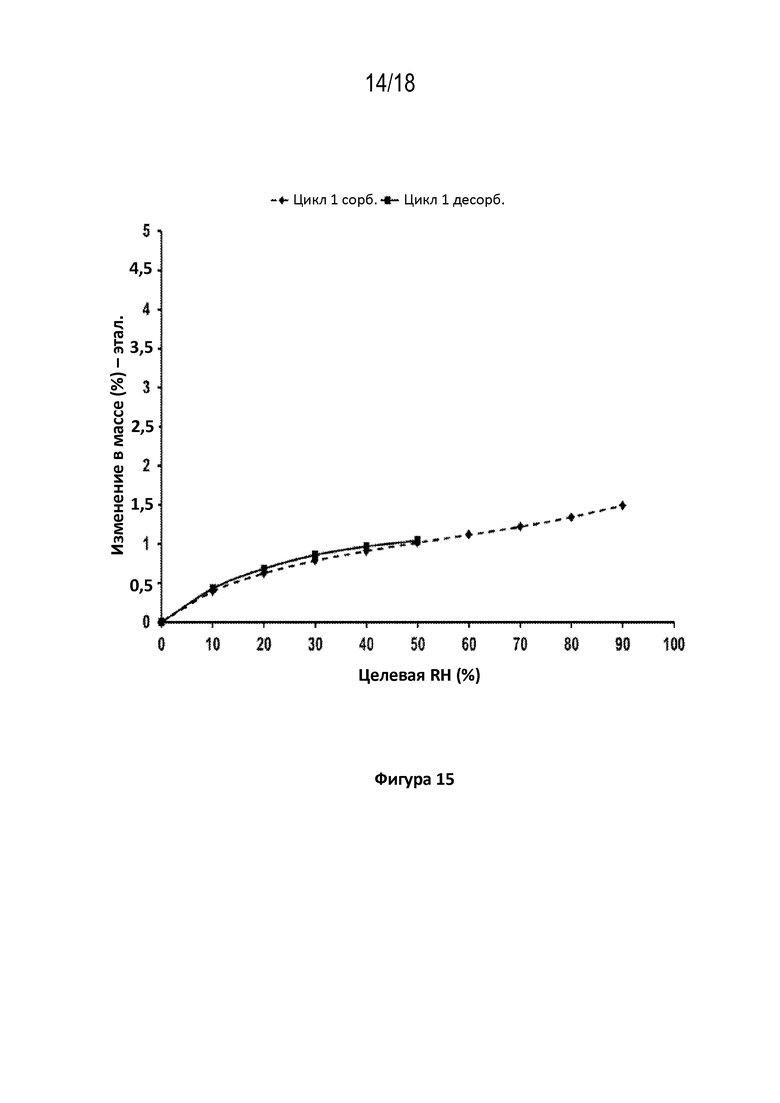
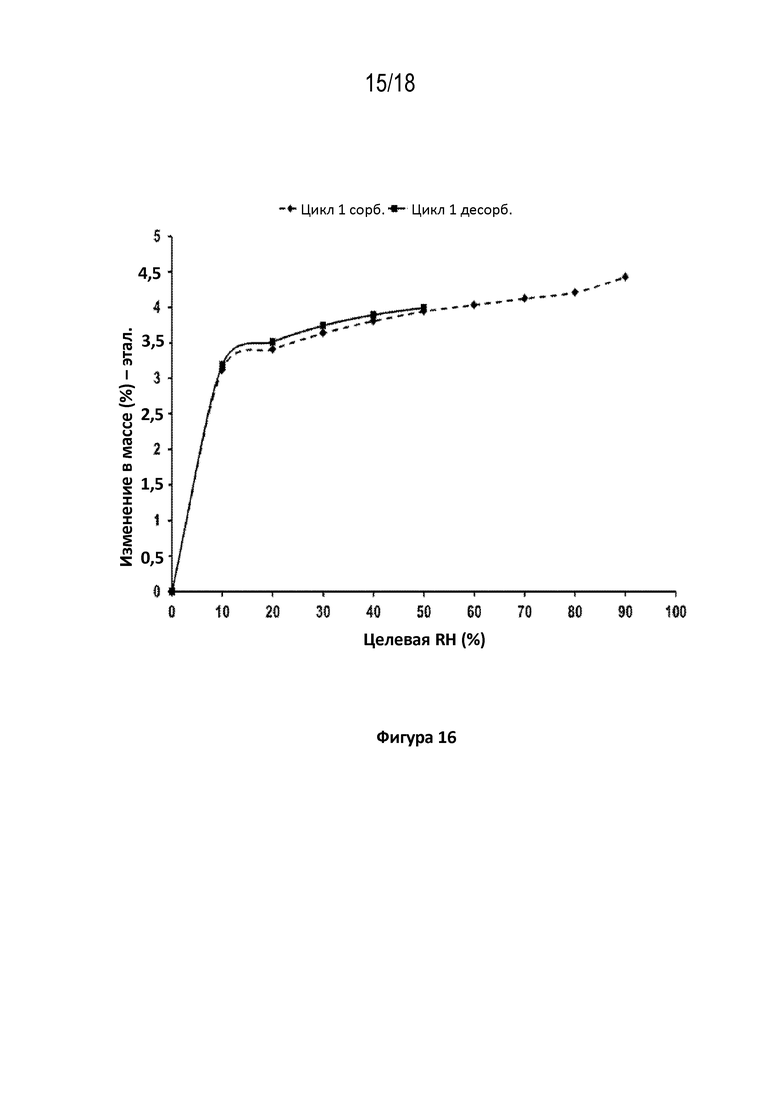
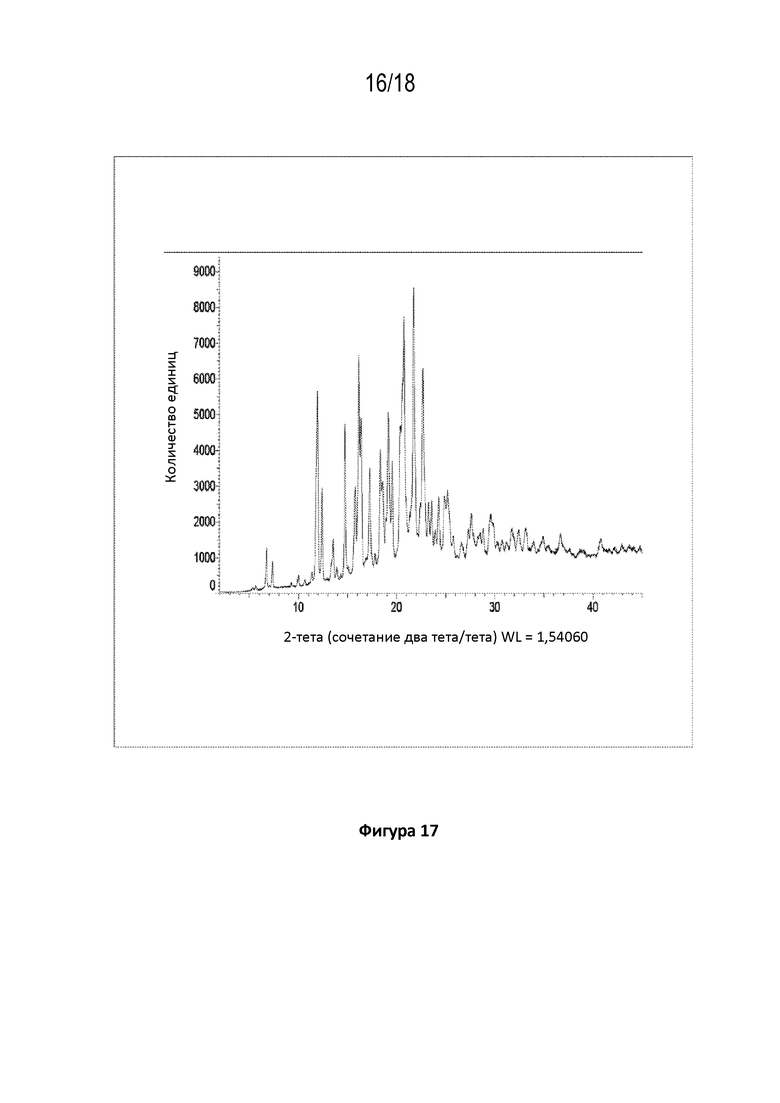
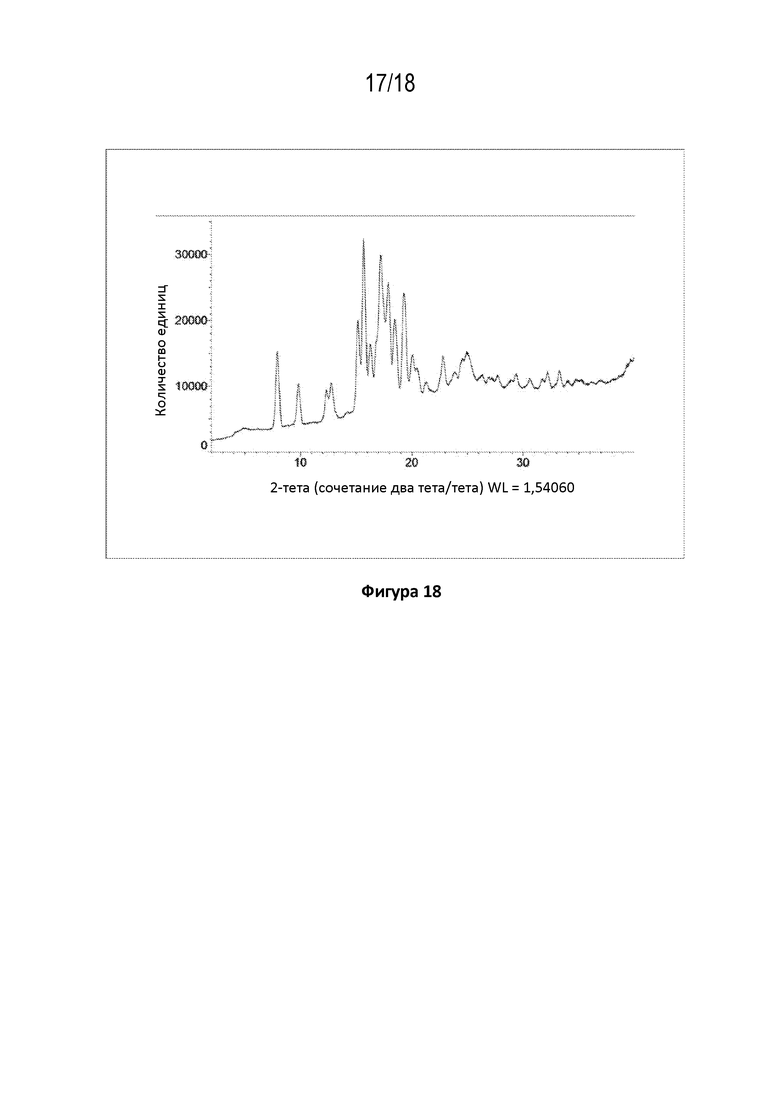
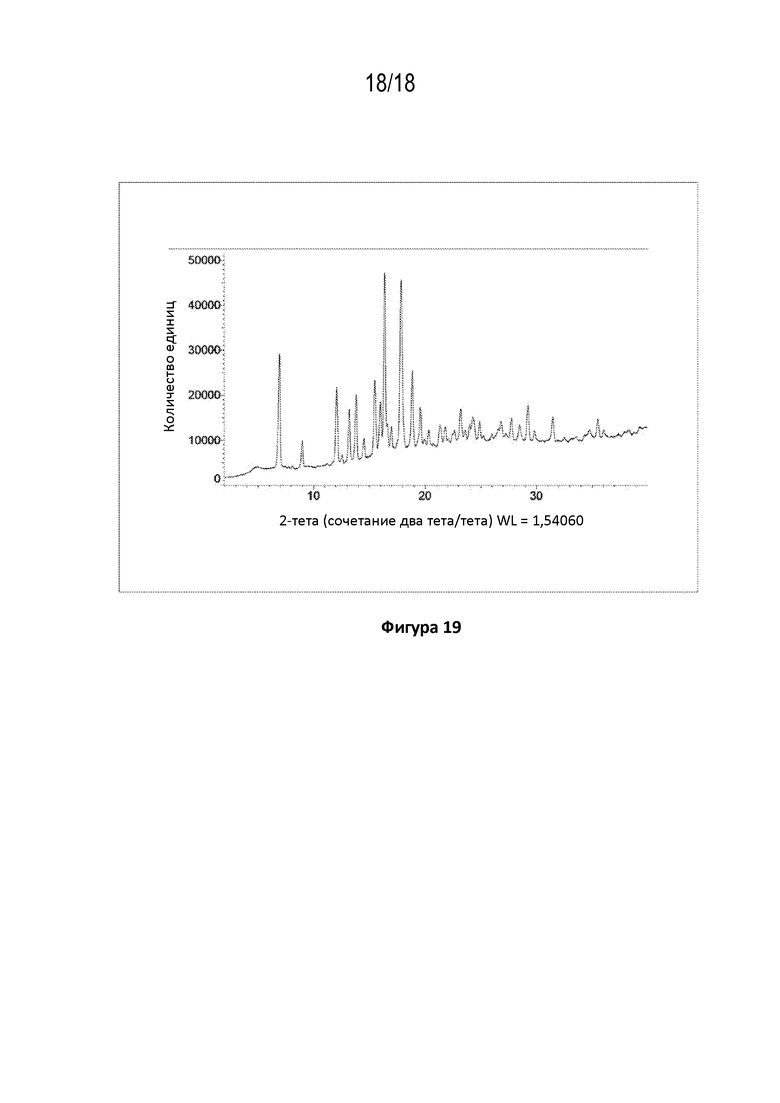
Изобретение относится к соединению формулы (I) в свободной форме или в форме фармацевтически приемлемой соли
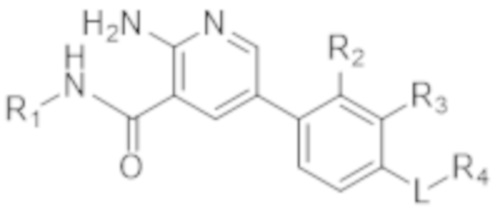
(I),
где L представляет собой связь, (CH2)n, –CH(CH3)–, –O–(CH2)n–, –C(O)–, –C(O)–NH–(CH2)n–; n равняется 1, 2 или 3; R1 представляет собой C3–C7циклоалкил, необязательно замещенный один раз или более чем один раз заместителем, независимо выбранным из гидроксила, галогена, C1–C3алкила; или мостиковый C5–C10циклоалкил, необязательно замещенный один раз или более чем один раз заместителем, независимо выбранным из гидроксила и гидроксиC1–C3алкила; R2 и R3 независимо выбраны из H, галогена и C1–C3алкила; R4 представляет собой N–содержащее гетероциклическое неароматическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7; каждый R7 независимо выбран из C1–C3алкила, гидроксиC1–C3алкила, галогенC1–C3алкила, C1–C3алкоксиC1–C3алкила, галогенC1–C3алкоксиC1–C3алкила, C2–C4алкинила, цианоC1–C3алкила и (CH2)m–R8; m равняется 0, 1, 2 или 3; R8 выбран из: 4–, 5– или 6–членного насыщенного или ненасыщенного неароматического гетероциклического кольца, содержащего один или несколько гетероатомов, выбранных из N, O или S, при этом указанное кольцо необязательно замещено один раз или более чем один раз заместителем, независимо выбранным из оксо, SO2C1–C3алкила и галогенC1–C3алкила; или C3–C6циклоалкила, необязательно замещенного один раз или более чем один раз галогеном, которое применяют для подавления активности рецептора ALK–2; а также к фармацевтическим композициям, содержащим указанное соединение, и применению указанного соединения в лечении гетеротопической оссификации и прогрессирующей оссифицирующей фибродисплазии. 5 н. и 13 з.п. ф-лы, 19 ил., 12 табл., 93 пр.
1. Соединение формулы (I) в свободной форме или в форме фармацевтически приемлемой соли
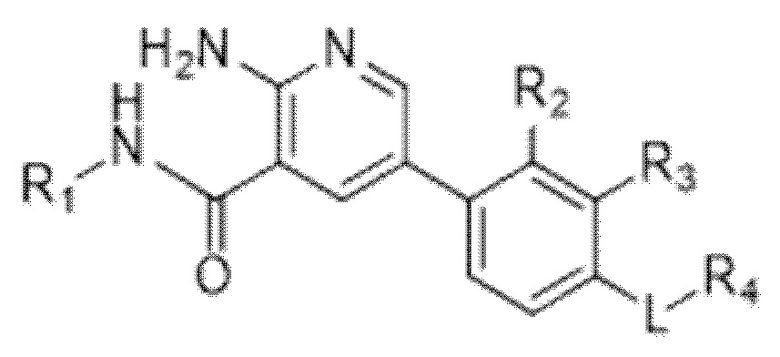
(I),
где
L представляет собой связь, (CH2)n, –CH(CH3)–, –O–(CH2)n–, –C(O)–, –C(O)–NH–(CH2)n–;
n равняется 1, 2 или 3;
R1 представляет собой C3–C7циклоалкил, необязательно замещенный один раз или более чем один раз заместителем, независимо выбранным из гидроксила, галогена, C1–C3алкила; или мостиковый C5–C10циклоалкил, необязательно замещенный один раз или более чем один раз заместителем, независимо выбранным из гидроксила и гидроксиC1–C3алкила;
R2 и R3 независимо выбраны из H, галогена и C1–C3алкила;
R4 представляет собой N–содержащее гетероциклическое неароматическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7;
каждый R7 независимо выбран из C1–C3алкила, гидроксиC1–C3алкила, галогенC1–C3алкила, C1–C3алкоксиC1–C3алкила, галогенC1–C3алкоксиC1–C3алкила, C2–C4алкинила, цианоC1–C3алкила и (CH2)m–R8;
m равняется 0, 1, 2 или 3;
R8 выбран из:
– 4–, 5– или 6–членного насыщенного или ненасыщенного неароматического гетероциклического кольца, содержащего один или несколько гетероатомов, выбранных из N, O или S, при этом указанное кольцо необязательно замещено один раз или более чем один раз заместителем, независимо выбранным из оксо, SO2C1–C3алкила и галогенC1–C3алкила; или
– C3–C6циклоалкила, необязательно замещенного один раз или более чем один раз галогеном.
2. Соединение формулы (I) в свободной форме или в форме фармацевтически приемлемой соли по п. 1,
где R4 выбран из NR5R6; N–содержащего 5–членного гетероциклического неароматического кольца, необязательно содержащего один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7 и где указанное кольцо присоединено к остальной части молекулы посредством атома углерода в кольце; и N–содержащего 6–членного гетероциклического неароматического моноциклического или бициклического кольца, необязательно содержащего один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7 и где указанное кольцо присоединено к остальной части молекулы посредством атома углерода в кольце;
R5 и R6 вместе с атомом N, к которому они присоединены, образуют
5–членное кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из N, O или S, необязательно замещенный один раз или более чем один раз R7;
6–членное кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из N, O или S, необязательно замещенный один раз или более чем один раз R7; или
7–членный спироцикл, необязательно содержащий один дополнительный гетероатом, выбранный из N, O или S, необязательно замещенный один раз или более чем один раз R7.
3. Соединение формулы (I) в свободной форме или в форме фармацевтически приемлемой соли по п. 1 или 2,
где
R2 и R3 представляют собой водород;
L представляет собой связь;
R4 представляет собой N–содержащее 6–членное гетероциклическое неароматическое бициклическое кольцо, необязательно содержащее один или несколько дополнительных гетероатомов, выбранных из N, O или S, где указанное кольцо необязательно замещено один раз или более чем один раз R7 и где указанное кольцо присоединено к остальной части молекулы посредством атома углерода в кольце,
R7 представляет собой (CH2)m–R8;
m равняется 0, 1, 2 или 3;
R8 представляет собой 6–членное насыщенное или ненасыщенное неароматическое гетероциклическое кольцо, содержащее один или несколько гетероатомов, выбранных из N, O или S, при этом указанное кольцо необязательно замещено один раз или более чем один раз заместителем, независимо выбранным из оксо, SO2C1–C3алкила и галогенC1–C3алкила.
4. Соединение формулы (I) в свободной форме или в форме фармацевтически приемлемой соли по любому из пп. 1–3, где
R1 представляет собой циклогексил, замещенный один раз гидроксилом.
5. Соединение формулы (I) в свободной форме или в форме фармацевтически приемлемой соли по любому из пп. 1–3, где
R1 представляет собой бицикло[2.2.2]октанил, замещенный гидроксилом.
6. Соединение формулы (I) в свободной форме или в форме фармацевтически приемлемой соли по любому из пп. 1–3, где
R1 представляет собой бицикло[1.1.1]пентанил, замещенный гидроксиметилом.
7. Соединение по п. 1 в свободной форме или в форме фармацевтически приемлемой соли, которое выбрано из
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(3–морфолинoпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–(3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–(3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(1–изопропилпирролидин–3–ил)фенил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–(3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(3,3,3–трифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(пирролидин–3–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(3–фтор–4–гидроксициклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(2–азаспиро[3.3]гептан–2–илметил)фенил)–2–амино–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(4–(3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–(3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(2–фтор–4–(пиперазин–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–((2–метилпирролидин–1–ил)метил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–(3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(4–(3–(бут–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(оксетан–3–илметил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(1–(2,2,2–трифторэтил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(2–(2,2,2–трифторэтокси)этил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–циклогексил–5–(4–(3–(пиперидин–1–ил)пропокси)фенил)никотинамида;
2–амино–5–(4–(3–(1,1–диоксидотетрагидро–2H–тиопиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–циклогексил–5–(4–(4–метилпиперазин–1–карбонил)фенил)никотинамида;
2–амино–5–(3–фтор–4–(2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–циклогексил–5–(4–морфолинoфенил)никотинамида;
2–амино–5–(4–(3–(2,2–дифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–5–(4–(3–(2–цианоэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксициклогексил)никотинамида;
2–амино–N–циклогексил–5–(4–(3–морфолинoпропокси)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(проп–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)–2–хлорфенил)–2–амино–N–(4–гидроксициклогексил)никотинамида;
2–амино–5–(2–фтор–4–((2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–5–(4–(3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(4–(3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
5–(4–(1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–циклогексил–5–(4–(пиперидин–1–ил)фенил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–(3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–циклогексил–5–(4–((2–(4–метилпиперазин–1–ил)этил)карбамоил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(3–(2,2,2–трифторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–(3–гидроксиадамантан–1–ил)никотинамида;
2–амино–N–циклогексил–5–(4–(морфолин–4–карбонил)фенил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(4–((2–метилпирролидин–1–ил)метил)фенил)никотинамида;
2–амино–5–(2,3–дифтор–4–((2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–(4–гидроксициклогексил–4–d)–5–(4–(3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксициклогексил)–5–(4–(4–метилпиперазин–1–карбонил)фенил)никотинамида;
2–амино–5–(2–хлор–4–((2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–(3–(2–гидроксипропан–2–ил)бицикло[1.1.1]пентан–1–ил)–5–(4–(3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–циклогексил–5–(3–(морфолин–4–карбонил)фенил)никотинамида;
2–амино–5–(3–хлор–4–((2–метилпирролидин–1–ил)метил)фенил)–N–(4–гидрокси–4–метилциклогексил)никотинамида;
5–(4–(1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–(4–гидрокси–1–метилциклогексил)никотинамида;
2–амино–N–(4–гидрокси–4–метилциклогексил)–5–(2–метил–4–((2–метилпирролидин–1–ил)метил)фенил)никотинамида и
2–амино–N–циклогексил–5–(3–(4–(2–гидроксиэтил)пиперазин–1–карбонил)фенил)никотинамида.
8. Соединение по п. 1 в свободной форме или в форме фармацевтически приемлемой соли, которое выбрано из
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(метилсульфонил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
5–(4–((S)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(3–морфолинoпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(2–фтор–4–(4–изопропилпиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4r)–4–гидроксициклогексил)–5–(4–(1–изопропилпирролидин–3–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(3,3,3–трифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4r)–4–гидроксициклогексил)–5–(4–(пирролидин–3–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1R,3S,4R)–3–фтор–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1s,4S)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–(2–азаспиро[3.3]гептан–2–илметил)фенил)–2–амино–N–((1r,4r)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(2–фтор–4–(пиперазин–1–ил)фенил)–N–((1r,4r)–4–гидроксициклогексил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидрокси–4–метилциклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–N–(3–(гидроксиметил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(бут–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(оксетан–3–илметил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(1–(2,2,2–трифторэтил)пиперидин–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(2–морфолинoэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–(2,2,2–трифторэтокси)этил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(1,1–диоксидотетрагидро–2H–тиопиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–5–(3–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(3,3,3–трифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(2,2–дифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(2–цианоэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(проп–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(проп–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)–2–хлорфенил)–2–амино–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–5–(2–фтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидрокси–4–метилциклогексил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(бут–2–ин–1–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидрокси–4–метилциклогексил)–5–(4–((1S,5R)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1S,5R)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(оксетан–3–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–циклогексил–5–(4–((2–(4–метилпиперазин–1–ил)этил)карбамоил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(1,3–дифторпропан–2–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–(2,2,2–трифторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1R,3R)–3–гидроксиадамантан–1–ил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(оксетан–3–илметил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1s,4R)–4–гидроксициклогексил)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(2–фторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(2,3–дифтор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2–метоксиэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(4–((1S,5R)–3–(2,2–дифторпропил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4S)–4–гидроксициклогексил)никотинамида;
2–амино–N–((1r,4R)–4–гидроксициклогексил)–5–(4–((1R,5S)–3–(2,2,2–трифторэтил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1r,4S)–4–гидроксициклогексил–4–d)–5–(4–((1S,5R)–3–изопропил–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–N–((1s,4s)–4–гидроксициклогексил)–5–(4–(4–метилпиперазин–1–карбонил)фенил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–(4–гидроксибицикло[2.2.2]октан–1–ил)никотинамида;
2–амино–5–(2–хлор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–5–(4–((1R,5S)–3–(4,4–дифторциклогексил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
2–амино–N–(3–(2–гидроксипропан–2–ил)бицикло[1.1.1]пентан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамида;
2–амино–5–(3–хлор–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)–N–((1r,4R)–4–гидрокси–4–метилциклогексил)никотинамида;
5–(4–((R)–1–(2–окса–6–азаспиро[3.3]гептан–6–ил)этил)фенил)–2–амино–N–((1r,4R)–4–гидрокси–1–метилциклогексил)никотинамида и
2–амино–N–((1r,4R)–4–гидрокси–4–метилциклогексил)–5–(2–метил–4–(((R)–2–метилпирролидин–1–ил)метил)фенил)никотинамида.
9. Соединение по п. 1, где соединение представляет собой 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в свободной форме, в форме соли фумаровой кислоты или в форме соли фосфорной кислоты.
10. Соединение по п. 1, где соединение представляет собой 2–амино–N–(4–гидроксибицикло[2.2.2]октан–1–ил)–5–(4–((1R,5S)–3–(тетрагидро–2H–пиран–4–ил)–3–азабицикло[3.1.0]гексан–1–ил)фенил)никотинамид в форме соли фумаровой кислоты или в форме соли фосфорной кислоты.
11. Фармацевтическая композиция для подавления активности рецептора ALK–2, содержащая терапевтически эффективное количество соединения по любому из пп. 1–10 или его фармацевтически приемлемой соли и один или несколько фармацевтически приемлемых носителей.
12. Способ подавления активности рецептора ALK–2 у субъекта, где способ включает введение субъекту терапевтически эффективного количества соединения по любому из пп. 1–10 или его фармацевтически приемлемой соли.
13. Способ лечения нарушения или заболевания, выбранного из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии, включающий введение субъекту терапевтически эффективного количества соединения по любому из пп. 1–10 или его фармацевтически приемлемой соли.
14. Соединение по любому из пп. 1–10 или его фармацевтически приемлемая соль для применения в качестве лекарственного средства для лечения нарушения или заболевания, выбранного из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
15. Соединение по любому из пп. 1–10 или его фармацевтически приемлемая соль для применения в лечении нарушения или заболевания, выбранного из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
16. Применение соединения по любому из пп. 1–10 или его фармацевтически приемлемой соли в получении лекарственного средства для лечения нарушения или заболевания, выбранного из гетеротопической оссификации или прогрессирующей оссифицирующей фибродисплазии.
17. Соединение по п. 9, где соединение находится в модификации HA или А свободной формы.
18. Соединение по п. 10, где соединение находится в модификации HA фумаратной соли.
| WO 2014151871 A9, 31.12.2014 | |||
| WO 2014138088 A1, 12.09.2014 | |||
| НОВЫЕ ПРОИЗВОДНЫЕ ЦИКЛОМОЧЕВИНЫ, ИХ ПОЛУЧЕНИЕ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2004 |
|
RU2341523C2 |

