Область техники, к которой относится изобретение
Настоящее изобретение касается в целом соединений, которые могут применяться в терапии, в частности в лечении состояний, вызванных определенными вирусами, таких как обычная простуда, энцефалит, менингит, миокардит, конъюнктивит, панкреатит, а также диабет, рак и нейродегенеративные заболевания, такие как болезнь Альцгеймера и боковой амиотрофический склероз. Более предпочтительно, настоящее изобретение касается определенных аминозамещенных гетероароматических соединений и их применения в терапии.
Предшествующий уровень техники
Пиразоло[1,5-a]пиримидин представляет собой широко применяющийся в медицинской химии фрагмент, и его производные известны своей потенциальной применимостью в качестве анальгетиков, антагонистов рецепторов бензодиазепина, антагонистов рецепторов ангиотензина II, ингибиторов ангиогенеза, противовоспалительных средств, антагонистов рецептора нейропептида Y, COX2-ингибитора, антагонистов рецептора кортикотропин-рилизинг гормона II типа и ингибиторов CHK1 (например, Mayo et al (Adv. Synth. Catal. 2003, 345, 620-624; Tellew et al (Bioorg. Med. Chem. Lett. 2010, 20, 7259-7264); Chen et al (Bioorg. Med. Chem. Lett. 2004, 14, 3669-3673); Labroli et al (Bioorg. Med. Chem. Lett. 2011, 21, 471-474); Griffith et al (Bioorg. Med. Chem. Lett. 2011, 21, 2641-2645); Gilligan et al, (J. Med. Chem. 2009, 52, 3073-3083); He et al. (US Patent No. 6,313,124 B1); и Wren et al. (WO 2010/086040).
Этот фрагмент был также описан в ингибиторах фосфатидилинозитол-4-киназы (PI4K).
В работах Bianco et al (PLoS Pathogens, 2012, 8(3), 1-17) и LaMarche et al (Antimicr. Agents and Chemother. 2012, 56(10), 5149-5156) было показано, что PI4K инграют важную роли в репликации вируса гепатита С (HCV), а работе Yang et al (J. Biol. Chem. 2012, 287(11), 8547-8467) было показано то же самое для коронавируса. В работах McLeod et al (ACS Med. Chem. Lett. 2013, 4(7), 585-589) и van der Schaar et al (Antimicrobial Agents Chemother. 2013, 57(10), 4971-4981) были показаны некоторые имидазопиразиновые производные, ингибирующие PI4K, которые являются сильными противовирусными средствами против пикорнавируса.
В работе Gudmundsson et al (Bioorg. Med. Chem. Lett. 2009, 19, 5689-5692) были раскрыты некоторые 3-арилпиразоло[1,5-a]пиримидины с потенциальной активностью против вирусов герпеса.
В работе Hwang et al (Bioorg. Med. Chem. Lett. 2012, 22, 7297-7301) описаны 3-арилпиразоло[1,5-a]пиримидины в качестве PI4K ингибиторов, оказывающих анти-HCV действие.
В работе Décor et al (Bioorg Med Chem Lett. 2013, 23, 3841-7) было также показано, что PI4K играет важную роль в репликации энтеровируса. Однак в ней было также показано, что PI4K ингибиторы (не 3-арилпиразоло[1,5-a]пиримидины и не 3-арилпиразоло[1,5-a]пиримидин 3-(3,4-диметоксифенил)-2,5-диметил-N-(2-морфолиноэтил)пиразоло[1,5-a]пиримидин-7-амин (называемый T-00127-HEV1)) в тестах in-vivo вызывали гибель мышей, что вызвало сомнение в безопасности ингибирования PI4K.
В WO 2015/110491 некоторые 3-арилпиразоло[1,5-a]пиримидины описаны в качестве PI4K ингибиторов для лечения заболеваний, вызываемых вирусами.
Производные имидазо[1,2-b]пиридазина были описаны в качестве ингибиторов Mps1 киназы (Kusakabe, J. Med. Chem. 2015, 58, 1760-1775). Похожие структурные фрагменты также были описаны как присутствующие в ингибиторах фосфатидилинозитол 4-киназы (PI4K) (McLeod et al (ACS Med. Chem. Lett. 2013, 4(7), 585-589) и van der Schaar et al (Antimicrobial Agents Chemother. 2013, 57(10), 4971-4981), и было показано, что ингибиторы PI4K являются потенциальными противовирусными средствами (Bianco et al, PLoS Pathogens, 2012, 8(3), 1-17; LaMarche et al, Antimicr. Agents and Chemother. 2012, 56(10), 5149-5156; Décor et al, Bioorg. Med. Chem. Lett. 2013, 23, 3841-7).
Аутофагия представляет собой процесс гомеостатического разложения в клетках, который используется для генерации питательных веществ во время стресса и как механизм для рециклизации поврежденных органелл или микробов в цитозоле (Karanasios et al, 2016, Autophagy at the cell, tissue and organismal level (Springer)). Многие патогены взаимодействуют с аутофагическими путями передачи сигналов у хозяина и могут нарушать нормальную аутофагию. В работе Lai et al (Viruses, 2016, 8(32), 1-13) описано, что вирусы разрушают механизм аутофагии, чтобы способствовать репликации вирусов и выходу из хозяина, и что ингибирование PI4KIIIβ влияет на процессы аутофагии и тем самым ингибирует репликацию вируса. В работе Sridhar et al (EMBO J. 2013,32, 324-339) описана роль PI4KIIIβ как ключевого фактора в аутофагии, и считается, что многие заболевания вызваны или связаны с нарушенной или патологической аутофагией, например, нейродегенеративные и нервно-психиатрические заболевания, рак, сердечные заболевания, воспалительные заболевания и диабет (Polajnar et al J. Cell. Mol. Med. 2014, 9(18). 1705-1711; Levine et al, Cell, 2008, 132(1), 27-42; Barlow, et al, DNA Cell. Biol, 2015, 34(4), 252-260).
Цитохром P450 3A4 (сокращенно CYP3A4) является одним из наиболее важных цитохром P450 ферментов и участвует в окислительной трансформации многих применяющихся в клинической практике терапевтических средств. Поэтому ингибиторы CYP3A4 могут влиять на метаболизм ряда лекарственных средств, повышая их биодоступность и тем самым вызывая побочные эффекты из-за несоразмерно высокой экспозиции лекарственного средства. У пациентов, получающих параллельно несколько разных лекарственных средств, необходимо избегать таких межлекарственных взаимодействий, и это иногда является препятствием для применения лекарственных средств, которые по всем остальным показателям пригодны для использования.
Соответственно, имеется постоянная потребность в новых лекарственных средствах, которые не только обладают искомой терапевтической активностью, но и не влияют или слабо влияют на активность ферментов, участвующих в метаболизме лекарственных средств, в частности CYP3A4.
Краткое описание изобретения
Первым аспектом настоящего изобретения является соединение, имеющее формулу (I)

или его фармацевтически приемлемая соль, где
один из X и Y представляет собой C, а другой представляет собой N;
R1 выбран из галогена, CN, NH(CH3), N(CH3)2, C1-C4 алкокси-группы, необязательно замещенной 1-3 галогенами, и C1-C4 алкила, необязательно замещенного 1-3 галогенами, CN, NH(CH3) или N(CH3)2; и
R2 представляет собой 3,4-диметоксифенил или 1,3-диметил-1H-индазол-5-ил.
Соединение, имеющее формулу (I), объединяет высокую противовирусную активность с низкой, предпочтительно – практически пренебрежимо малой – CYP3A4-ингибирующей активностью. Соответственно, в настоящем изобретении описано лекарственное средство со значительно уменьшенным риском межлекарственных взаимодействий.
Другим аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль для применения в терапии.
Другим аспектом настоящего изобретения является фармацевтическая композиция, содержащая соединение, имеющее формулу (I), или его фармацевтически приемлемую соль и, необязательно, фармацевтически приемлемое вспомогательное вещество.
Другим аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль для применения в лечении вирусной инфекции, например, РНК-вирусной инфекции.
Другим аспектом является применение соединения, имеющего формулу (I), или его фармацевтически приемлемой соли в производстве лекарственного средства для лечения вирусной инфекции, например, РНК-вирусной инфекции.
Другим аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль, способные улучшать нарушенную или модулировать патологическую аутофагию в клетке, для применения в лечении указанных в настоящем тексте заболеваний.
Другим аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль, для применения в лечении заболевания, связанного с нарушенной или патологической аутофагией.
Другим аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль, для применения в лечении заболевания связанного с нарушенной аутофагией.
Другим аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль, для применения в лечении заболевания, связанного с патологической аутофагией.
Другим аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль, для применения в лечении инфекции, вызванной безоболочечным одноцепочечным (+) РНК-вирусом.
Другим аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль, для применения в лечении энтеровирусной инфекции, например, пикорнавирусной инфекции.
Еще одним аспектом настоящего изобретения является соединение, имеющее формулу (I), или его фармацевтически приемлемая соль, для применения в лечении заболевания, выбранного из панкреатита, полиомиелита, энцефалита, менингита, сепсиса, рака, такого как рак груди, простаты, яичника или кишечника, паралича, сердечных заболеваний, таких как миокардит, диабета, обычной простуды, энтеровирусного везикулярного стоматита, герпетической ангины, плевродинии, диареи, слизисто-кожных поражений, респираторного заболевания, конъюнктивита, миозита, синдрома хронической усталости, нервно-психиатрических заболеваний и нейродегенеративных заболеваний, таких как рассеянный склероз, болезнь Паркинсона, амиотрофический боковой склероз, болезнь Альцгеймера и болезнь Хантингтона, или воспалительных состояний.
Другим аспектом настоящего изобретения является способ лечения вирусной инфекции, например, РНК-вирусной инфекции, посредством введения терапевтически эффективного количества соединения, имеющего формулу (I), или его фармацевтически приемлемой соли, млекопитающему, нуждающемуся в таком лечении.
Подробное описание изобретения
Если иное не указано или не следует явным образом из контекста, все технические и научные термины и сокращения, использующиеся в настоящем тексте, имеют свои обычные значения, известные квалифицированным специалистам в данной области. Тем не менее, определения некоторых использующихся в настоящем тексте терминов приведены ниже.
“Фармацевтически приемлемый” означает, что может применяться для приготовления фармацевтической композиции, которая будет безопасной, нетоксичной и ни биологически, ни каким-либо иным образом нежелательной, и включает допустимость применения как для ветеринарного, так и для фармацевтического применения в отношении людей.
Термин “лечение" (или "лечить") заболевание или нарушение может означать ослабление заболевания или нарушения (т.е. остановку или замедление прогресса заболевания или по меньшей мере одного из его клинических симптомов) и/или облегчение по меньшей мере одного физического параметра, который может не ощущаться пациентом. Данный термин может также относиться к ингибированию заболевания или нарушения физически (например, стабилизация ощущаемого симптома), физиологически (например, стабилизация физического параметра), или оба указанных случая сразу.
"Терапевтически эффективное количество" означает количество соединения, которое при введении пациенту для лечения заболевания является достаточным для обеспечения лечения целевого заболевания. "Терапевтически эффективное количество" варьируется в зависимости от соединения, заболевания и степени его тяжести, а также от возраста, массы тела и других параметров пациента, подвергающегося лечению.
Термин “вирусная инфекция” означает инфекцию, вызванную вирусом, у млекопитающего.
Термин “РНК-вирусная инфекция” означает вирусную инфекцию, при которой вирус в качестве генетического материала имеет РНК (рибонуклеиновую кислоту).
Термин “инфекция, вызванная безоболочечным одноцепочечным (+) РНК-вирусом” означает инфекцию, которую вызывает безоболочечный одноцепочечный (+) РНК-вирус.
“Безоболочечный вирус” представляет собой вирус, не имеющий оболочки.
“Безоболочечный одноцепочечный (+) РНК-вирус” представляет собой вирус, который в качестве генетического материала имеет одноцепочечную РНК, и эта РНК может быть немедленно транслирована в вирусный белок клеткой, инфицированной этим вирусом.
Под “патологической аутофагией" понимается, например, аутофагия, которая благоприятствует репликации и высвобождению вируса.
Под “нарушенной аутофагией” понимается ненормально работающая аутофагия в клетке.
Заболевание, связанное с нарушенной или патологической аутофагией, которое можно лечить по настоящему изобретению, например, может быть выбрано из нейродегенеративных и нервно-психиатрических заболеваний, рака, сердечных заболеваний, воспалительных заболеваний и диабета, например, из указанных в настоящем тексте заболеваний.
Термин “млекопитающее” означает человека или любое млекопитающее животное, например, примата, сельскохозяйственное животное, домашнее животное или лабораторное животное. Примерами таких животных являются обезьяны, коровы, овцы, козы, лошади, свиньи, собаки, кошки, кролики, мыши, крысы и т.д. Предпочтительно, млекопитающим является человек. В некоторых вариантах осуществления, однако, млекопитающим является другое животное, например, сельскохозяйственное животное, такое как корова, овца, коза, лошадь или свинья. В некоторых других вариантах осуществления, млекопитающее представляет собой домашнее животное, например, собаку, кошку или кролика.
Термин “вспомогательное вещество” означает фармацевтически приемлемые химические вещества, такие как вещества, известные квалицированным специалистам в области фармацевтики как помогающие вводить лекарственные средства. Оно представляет собой вещество, которое может применяться для приготовления фармацевтической композиции, является безопасным, нетоксичным и не является биологически или каким-либо иным образом нежелательным, и включает вспомогательные вещества, приемлемые для ветеринарного применения, а также для фармацевтического применения для человека. Примеры вспомогательных веществ включают связующие вещества, поверхностно-активные вещества, разбавители, разрыхлители, антиадгезивы и лубриканты.
Если иное не указано или не следует явным образом из контекста, термин “галоген” означает F (фтор), Cl (хлор), Br (бром) или I (иод).
Если иное не указано или не следует явным образом из контекста, термин “Cm-Cn алкил” означает линейную или разветвленную алкильную группу, содержащую от m до n атомов углерода. Например, термин “C1-C4 алкил” означает линейную или разветвленную алкильную группу, содержащую от 1 до 4 атомов углерода. Такие C1-C4 алкилы включают метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил и трет-бутил.
Термин “Cm-Cn алкокси-группа” означает фрагмент, имеющий формулу
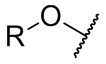 ,
,
где R представляет собой Cm-Cn алкил.
Термин NH(CH3) означает фрагмент, имеющий формулу
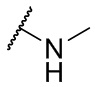 .
.
Термин N(CH3)2 означает фрагмент, имеющий формулу
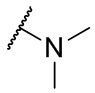 .
.
Термин CN означает фрагмент, имеющий формулу
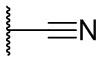 .
.
В соединении, имеющем формулу (I), один из X и Y представляет собой C, а другой представляет собой N. В некоторых вариантах осуществления, X представляет собой C, а Y представляет собой N, т.е. соединение представлено формулой (Ia)
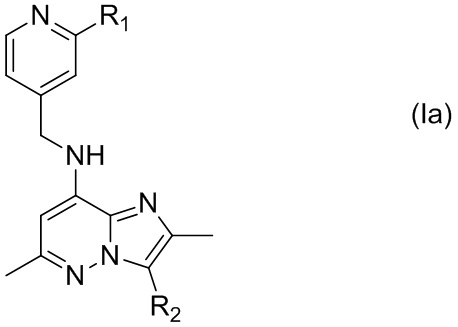
где R1 и R2 имеют указанные в настоящем тексте значения.
В некоторых вариантах осуществления, X представляет собой N, а Y представляет собой C, т.е. соединение представлено формулой (Ib)
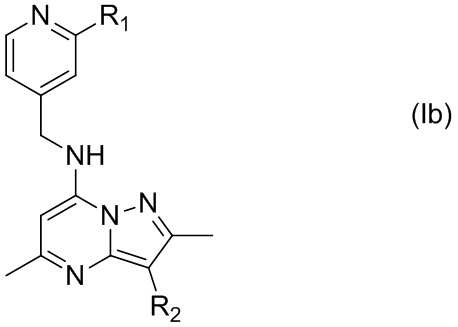
где R1 и R2 имеют указанные в настоящем тексте значения.
В соединении, имеющем формулу (I), R1 выбран из галогена, CN, NH(CH3), N(CH3)2, C1-C4 алкокси-группы, необязательно замещенной 1-3 галогенами, C1-C4 алкила, необязательно замещенного 1-3 галогенами, CN, NH(CH3) или N(CH3)2.
В некоторых вариантах осуществления, R1 выбран из галогена, CN, C1-C4 алкокси-группы, необязательно замещенной 1-3 галогенами, и C1-C4 алкила, необязательно замещенного 1-3 галогенами.
В некоторых вариантах осуществления, когда R1 представляет собой галоген, этот галоген выбран из F, Cl и Br. В некоторых вариантах осуществления, галоген представляет собой F или Cl. В некоторых вариантах осуществления, галоген представляет собой F.
В некоторых вариантах осуществления, когда R1 представляет собой необязательно замещенный C1-C4 алкил, этот алкил более предпочтительно представляет собой C1-C3 алкил. В некоторых вариантах осуществления, алкил представляет собой метил или этил. В некоторых вариантах осуществления, алкил представляет собой метил. В некоторых вариантах осуществления, алкил представляет собой этил.
В некоторых вариантах осуществления, когда R1 представляет собой необязательно замещенный C1-C4 алкил, этот алкил является незамещенным. В некоторых других вариантах осуществления, алкил замещен 1-3 галогенами, CN, NH(CH3) или N(CH3)2. В некоторых вариантах осуществления, когда алкил замещен 1-3 галогенами, эти галогены выбраны из F и Cl, в частности F. В некоторых вариантах осуществления, алкил представляет собой алкил, замещенный 1-3 галогенами и/или одним заместителем, выбранным из CN, NH(CH3) и N(CH3)2.
В некоторых вариантах осуществления, когда R1 представляет собой замещенный алкил, любой заместитель в этом алкиле выбран из галогена и CN. В некоторых других вариантах осуществления, когда R1 представляет собой замещенный алкил, любой заместитель в этом алкиле выбран из галогена, NH(CH3) и N(CH3)2. В некоторых других вариантах осуществления, когда R1 представляет собой замещенный алкил, любой заместитель в этом алкиле выбран из NH(CH3) и N(CH3)2. В некоторых других вариантах осуществления, когда R1 представляет собой замещенный алкил, любой заместитель в этом алкиле выбран из галогена, например, F и Cl, в частности F.
В некоторых вариантах осуществления, когда R1 представляет собой C1-C4 алкокси-группу, эта алкокси-группа более предпочтительно представляет собой C1-C3 алкокси-группу. В некоторых вариантах осуществления, алкокси-группа представляет собой C1-C2 алкокси-группу. В некоторых вариантах осуществления, алкокси-группа представляет собой метокси-группу. В некоторых вариантах осуществления, когда R1 представляет собой необязательно замещенную C1-C4 алкокси-группу, эта алкокси-группа является незамещенной. В некоторых других вариантах осуществления, алкокси-группа замещен 1-3 галогенами. В некоторых вариантах осуществления, когда алкокси-группа замещен 1-3 галогенами, любой такой галоген выбран из F и Cl, в частности F.
В некоторых вариантах осуществления, R1 выбран из галогена, CN, необязательно замещенной C1-C4 алкокси-группы и необязательно замещенного C1-C4 алкила. В некоторых из этих вариантов осуществления, R1 выбран из галогена, необязательно замещенной C1-C3 алкокси-группа и необязательно замещенного C1-C3 алкила, например, из F, Cl, CN, необязательно замещенной C1-C3 алкокси-группы и необязательно замещенного C1-C3 алкила; или из F, Cl, CN, необязательно замещенной метокси-группы и необязательно замещенного метила и этила; или из F и необязательно замещенного метила. В некоторых из этих вариантов осуществления, любой заместитель в любом алкиле или алкокси-группе представляет собой F или Cl, в частности F. В некоторых других из этих вариантов осуществления, R1 выбран из галогена, незамещенной C1-C4 алкокси-группы и незамещенного C1-C4 алкила, например, из F, Cl, CN, C1-C3 алкокси-группы и C1-C3 алкила; или из F, Cl, CN, метокси-группы, метила и этила; или из F и метила.
В некоторых вариантах осуществления, R1 выбран из галогена и необязательно замещенного C1-C4 алкила. В некоторых из этих вариантов осуществления, R1 выбран из галогена и необязательно замещенного C1-C3 алкила, например, из F, Cl и необязательно замещенного C1-C3 алкила; или из F, Cl и необязательно замещенного метила или этила; или из F и необязательно замещенного метила или этила; или из F и необязательно замещенного метила. В некоторых из этих вариантов осуществления, любой заместитель в алкиле представляет собой F или Cl, в частности F. В некоторых других из этих вариантов осуществления, R1 выбран из галогена и незамещенного C1-C4 алкила, например, из F, Cl и C1-C3 алкила; или из F, Cl, метила и этила; или из F, метила и этила; или из F и метила.
В некоторых других вариантах осуществления, R1 выбран из галогена, незамещенного C1-C4 алкила и незамещенной C1-C4 алкокси-группы, например, из F, Cl, C1-C3 алкила и C1-C3 алкокси-группы; или из F, Cl, метила, этила и метокси-группы; или из F, метила, этила и метокси-группы; или из F, метила и метокси-группы; или из метила и метокси-группы.
В некоторых вариантах осуществления, R1 выбран из необязательно замещенного C1-C4 алкила и необязательно замещенной C1-C4 алкокси-группы. В некоторых вариантах осуществления, R1 выбран из незамещенного C1-C4 алкила и незамещенной C1-C4 алкокси-группы. В других вариантах осуществления, R1 выбран из необязательно замещенного C1-C4 алкила и незамещенной C1-C4 алкокси-группы. В других вариантах осуществления, R1 выбран из C1-C4 алкила и C1-C4 алкокси-группы, где эта алкокси-группа и алкил необязательно замещены 1-3 галогенами, например, 1-3 галогенами, выбранными из F и Cl, в частности 1-3 атомами F. В некоторых вариантах осуществления, R1 выбран из метила, этила, трифторметила и метокси-группы.
В других вариантах осуществления, R1 выбран из галогена, например, R1 представляет собой F или Cl, в частности F.
В некоторых других вариантах осуществления, R1 выбран из F, CN, метокси-группы, метила, трифторметила и этила; например, из F, метила, трифторметила и этила.
В соединении, имеющем формулу (I), R2 представляет собой 3,4-диметоксифенил или 1,3-диметил-1H-индазол-5-ил, т.е. R2 представляет собой фрагмент, имеющий формулу (II) или (III)
 .
.
В некоторых вариантах осуществления, R2 представляет собой 3,4-диметоксифенил, т.е. фрагмент, имеющий формулу (II), и соединение по настоящему изобретению может быть представлено формулой (Ic)

где X, Y и R1 имеют указанные в настоящем тексте значения.
В некоторых других вариантах осуществления, R2 представляет собой 1,3-диметил-1H-индазол-5-ил, т.е. фрагмент, имеющий формулу (III), и соединение по настоящему изобретению может быть представлено формулой (Id)
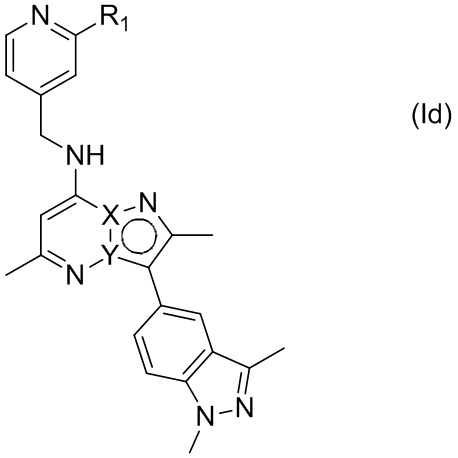
где X, Y и R1 имеют указанные в настоящем тексте значения.
В некоторых вариантах соединения, имеющего формулу (Ic), X представляет собой C, а Y представляет собой N, т.е. соединение по настоящему изобретению может быть представлено формулой (Ie)
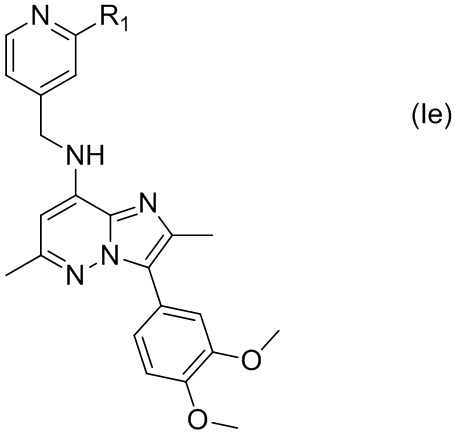
где R1 имеет указанное в настоящем тексте значение.
В некоторых других вариантах соединения, имеющего формулу (Ic), X представляет собой N, а Y представляет собой C, т.е. соединение по настоящему изобретению может быть представлено формулой (If)
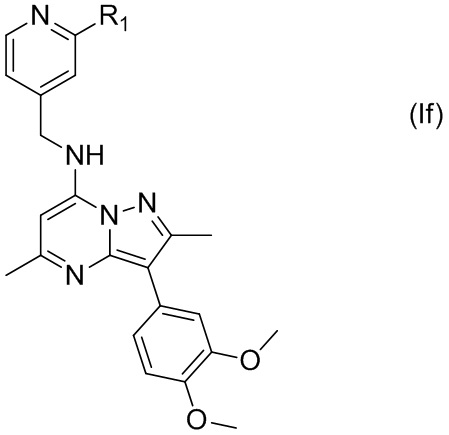
где R1 имеет указанное в настоящем тексте значение.
В некоторых вариантах соединения, имеющего формулу (Id), X представляет собой C, а Y представляет собой N, т.е. соединение по настоящему изобретению может быть представлено формулой (Ig)
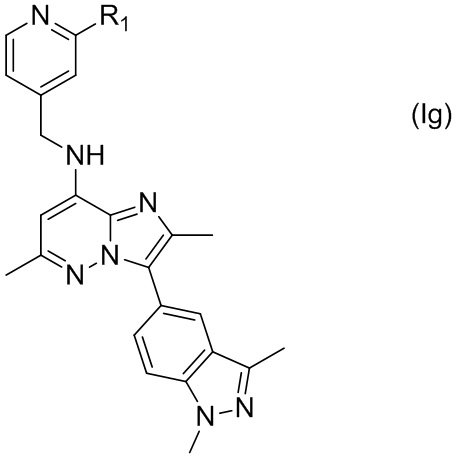
где R1 имеет указанное в настоящем тексте значение.
В некоторых других вариантах соединения, имеющего формулу (Id), X представляет собой N, а Y представляет собой C, т.е. соединение по настоящему изобретению может быть представлено формулой (Ih)
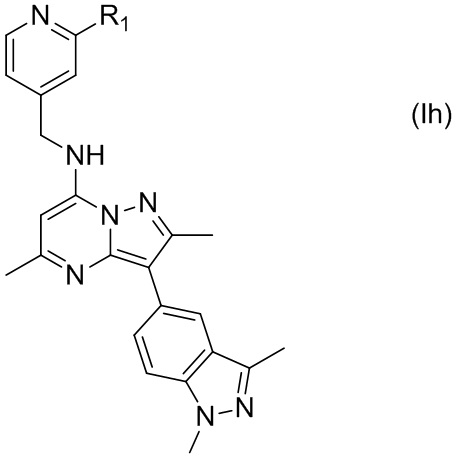
где R1 имеет указанное в настоящем тексте значение.
Если иное не указано или не следует явным образом из контекста, любое упоминание соединения, имеющего формулу (I), должно также пониматься как указание соединения, представленного любой из формул (Ia), (Ib), (Ic), (Id), (Ie), (If), (Ig) и (Ih).
В некоторых вариантах осуществления, соединение, имеющее формулу (I), более предпочтительно представляет собой соединение, имеющее формулу (Ie) или (Ih), т.е. соединение, имеющее формулу (I)

или его фармацевтически приемлемая соль, где
R1 имеет указанное в настоящем тексте значение; и
X представляет собой C, Y представляет собой N, и R2 представляет собой 3,4-диметоксифенил; или
X представляет собой N, Y представляет собой C, и R2 представляет собой 1,3-диметил-1H-индазол-5-ил.
Соединения, имеющие формулу (I) могут быть также переведены в их фармацевтически приемлемые соли. Термин “фармацевтически приемлемая соль” соединения означает соль, которая является фармацевтически приемлемой согласно определению в настоящем тексте, и которая имеет целевую фармакологическую активность материнского соединения. Фармацевтически приемлемые соли включают кислотно-аддитивные соли, сформированные с неорганическими кислотами, например, с хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, азотной кислотой, фосфорной кислотой; или сформированные с органическими кислотами, например, с уксусной кислотой, бензолсульфокислотой, бензойной кислотой, камфорсульфокислотой, лимонной кислотой, этансульфокислотой, фумаровой кислотой, глюкогептоновой кислотой, глюконовой кислотой, глютаминовой кислотой, гликолевой кислотой, гидроксинафтойной кислотой, 2-гидроксиэтансульфокислотой, молочной кислотой, малеиновой кислотой, яблочной кислотой, малоновой кислотой, миндальной кислотой, метансульфокислотой, муконовой кислотой, 2-нафталинсульфокислотой, пропионовой кислотой, салициловой кислотой, янтарной кислотой, винной кислотой, п-толуолсульфокислотой, триметилуксусной кислотой и т.д.
При получении кислотно-аддитивных солей предпочтительно применяют такие кислоты, которые дают терапевтически приемлемые соли. Примерами таких кислот являются галогеноводородные кислоты, серная кислота, фосфорная кислота, азотная кислота, алифатическая, алициклическая, ароматическая или гетероциклическая карбоксильная или сульфокислота, такая как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, п-гидроксибензойная кислота, эмбоновая кислота, метансульфокислота, этансульфокислота, гидроксиэтансульфокислота, галогенбензолсульфокислота, толуолсульфокислота или нафталинсульфокислота.
Когда в химической структуре присутствует хиральный атом, все стереоизомеры, возникающие вследствие присутствия хирального атома, охватываются этой структурой, если иное не указано особо. При использовании RS-системы Кана-Ингольда-Прелога, любой асимметрический атом может быть представлен в (R)- или (S)-конфигурации, и соединение может быть представлено в виде смеси его стереоизомеров, например, рацемической смеси, или в виде только одного стереоизомера, и оба эти случая входят в объем настоящего изобретения.
Настоящее изобретение включает фармацевтические композиции, содержащие соединение, имеющее формулу (I), или его индивидуальный изомер, рацемическую или нерацемическую смесь изомеров, или его фармацевтически приемлемую соль, вместе с по меньшей мере одним фармацевтически приемлемым вспомогательным веществом, например, носителем, и, необязательно, другими терапевтическими и/или профилактическими ингредиентами.
Настоящее изобретение включает фармацевтические композиции, содержащие по меньшей мере одно соединение, имеющее формулу (I), или его индивидуальный изомер, рацемическую или нерацемическую смесь изомеров, или его фармацевтически приемлемую соль, вместе с по меньшей мере одним фармацевтически приемлемым вспомогательным веществом, например, носителем, и, необязательно, другими терапевтическими и/или профилактическими ингредиентами.
Фармацевтическая композиция по настоящему изобретению может быть для наружного (местного) или системного применения, например, для энтерального введения, например, для ректального или перорального введения, или для парентерального введения млекопитающему (в особенности, человеку), и cодержит терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли в качестве действующего вещества, в комбинации с фармацевтически приемлемым вспомогательным веществом, например, фармацевтически приемлемым носителем. Терапевтически эффективное количество действующего вещества имеет указанное выше в тексте определение и зависит, например, от вида млекопитающего, веса тела, возраста, индивидуального состояния, индивидуальных фармакокинетических параметров, заболевания, которое подвергается лечению, и способа введения.
Для целей энтерального, например, перорального, введения соединения по настоящему изобретению могут вводиться в широкий ряд дозированных готовых форм. Фармацевтические композиции и дозированные формы могут содержать соединение или соединения по настоящему изобретению или их фармацевтически приемлемую соль(соли) в качестве действующего вещества. Фармацевтически приемлемые носители могут быть твердыми или жидкими. Твердые формы препаратов включают порошки, таблетки, пилюли, литые таблетки, саше, суппозитории и диспергируемые гранулы. Твердым носителем может являться одно или больше веществ, которые могут служить также разбавителями, ароматизаторами, солюбилизаторами, лубрикантами, суспендирующими агентами, связующими веществами, консервантами, разрыхлителями для таблеток или инкапсулирующим материалом. В порошках носитель обычно представляет собой тонкоизмельченное твердое вещество, которое представляет собой смесь с тонкоизмельченным действующим веществом. В таблетках действующее вещество обычно смешивают в подходящих пропорциях с носителем, имеющим необходимую связующую способность, и затем прессуют в таблетки целевой формы и размера. Подходящие носители включают (но не ограничиваются только ими) карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакантовую камедь, метилцеллюлозу, натрия карбоксиметилцеллюлозу, низкоплавкий воск, масло какао и т.п. Препарат действующего вещества может содержать инкапсулирующий материал в качестве носителя, дающий капсулу, в которой действующее вещество, с носителями или без носителей, окружено носителем и находится с ним в ассоциации.
Другие формы, подходящие для перорального введения, включают жидкие препараты, включая эмульсии, сиропы, эликсиры, водные растворы, водные суспензии или твердые препараты, которые предназначены для превращения в жидкие готовые формы незадолго до применения. Эмульсии можно готовить в виде растворов, например, в водном растворе пропиленгликоля, или они могут содержать эмульгаторы, например, такие как лецитин, сорбитан моноолеат или смола акации. Водные растворы можно приготовить растворением действующего вещества в воде и добавлением подходящих красителей, ароматизаторов, стабилизаторов и загустителей. Водные суспензии можно получить посредством диспергирования тонкоизмельченного активного компонента в воде с вязким материалом, таким как природные или синтетические смолы, полимеры, метилцеллюлоза, натрия карбоксиметилцеллюлоза и другие хорошо известные суспендирующие агенты. Жидкие препараты включают растворы, суспензии и эмульсии, и они помимо действующего вещества могут содержать красители, ароматизаторы, стабилизаторы, буферные добавки, искусственные и натуральные подсластители, диспергирующие добавки, загустители, солюбилизаторы и т.п.
Примеры композиций для ректального введения включают суппозитории, которые могут содержать, например, подходящее нераздражающее вспомогательное вещество, такое как масло какао, синтетические эфиры глицеридов или полиэтиленгликоли, которые являются твердыми при комнатной температуре, но плавятся и/или растворяются в ректальной полости, высвобождая лекарственное средство.
Соединения по настоящему изобретению можно также вводить парентерально, например, посредством ингаляции, инъекции или инфузии, например, внутривенной, внутриартериальной, внутрикостной, внутримышечной, интрацеребральной, интрацеребровентрикулярной, внутрисуставной, внутригрудинной, внутриоболочечной, внутриочаговой, внутричерепной, внутрикожной и подкожной инъекцией или инфузией.
Так, для целей парентерального введения, фармацевтические композиции по настоящему изобретению могут быть в форме стерильного инъекционного или инфузионного препарата, например, в форме водной или масляной суспензии. Такую суспензию можно приготовить по методикам, известным в данной области, с использованием подходящих диспергирующих или смачивающих агентов (например, Tween 80), и суспендирующих агентов. Стерильный инъекционный или инфузионный препарат может также представлять собой стерильный инъекционный или инфузионный раствор, или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе. Например, фармацевтическая композиция может представлять собой раствор в 1,3-бутандиоле. Другие примеры подходящих носителей и растворителей, которые можно применять в композициях по настоящему изобретению, включают (но не ограничиваются только ими) маннит, воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспензионной среды широко применяются стерильные жирные масла. Для этой цели можно применять любые марки жирных масел, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, могут применяться для приготовления инъекционных составов, так же как природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масла, особенно в их полиэтоксилированных версиях. Эти масляные растворы или суспензии могут также содержать длинноцепочечный спирт в качестве разбавителя или диспергирующего агента.
Растворы для парентерального применения могут также содержать подходящие стабилизаторы и, при необходимости, буферные вещества. Подходящие стабилизаторы включают антиоксиданты, такие как бисульфат натрия, сульфит натрия или аскорбиновая кислота, по отдельности или в комбинации, лимонную кислоту и ее соли, и натриевую соль ЭДТА. Парентеральные растворы могут также содержать консерванты, такие как бензалконий хлорид, метил- или пропил-парабен и хлорбутанол.
Для введения ингаляционно или назально, подходящие фармацевтические препараты представляют собой частицы, аэрозоли, порошки, туманы или капельки, например, имеющие средний размер около 10 мкм в диаметре или меньше. Например, композиции для ингаляций можно готовить в виде растворов в физрастворе, с применением бензилового спирта или других подходящих консервантов, промоторов абсорбции для улучшения биодоступности, фторуглеродов и/или других солюбилизирующих или диспергирующих агентов, известных в данной области.
В некоторых вариантах осуществления, препарат по настоящему изобретению представляет собой липосомальный препарат. Липосомальные препараты хорошо известны в области фармацевтики и описаны, например, в Remington, Essentials of Pharmaceutics, Ed. Linda Felton (Pharmaceutical Press 2012), p. 456-7, и во многих других публикациях. Например, информация по выбору подходящих липосомных формул, подходящих липидов, способам получения и т.п. легко доступна квалифицированному специалисту. Примерами липидов для липосомных препаратов являются фосфолипиды, сфинголипиды, стерольные липиды и жирные кислоты. Липиды, подходящие для липосомных препаратов можно приобрести, например, у компании Avanti® Polar Lipids, Inc.
Фармацевтические композиции по настоящему изобретению можно также наносить наружно, на кожу или на слизистые оболочки. Для целей наружного применения фармацевтическая композиция может представлять собой, например, лосьон, гель, пасту, настой, чрезкожный пластырь или гель для трансмукозальной доставки.
Композицию можно вводить в состав подходящей пасты, содержащей действующие вещества, суспендированные или растворенные в носителе. Носители для наружного применения соединений по настоящему изобретению включают (но не ограничиваются только ими) минеральное масло, жидкий парафин, белый петролатум, пропиленгликоль, полиоксиэтилен/полиоксипропиленовые соединения, эмульсионный воск и воду.
Альтернативно, фармацевтическую композицию можно вводить в состав подходящего лосьона или крема, содержащего действующее вещество, суспендированное или растворенное в носителе. Подходящие носители включают (но не ограничиваются только ими) минеральное масло, сорбитан моностеарат, полисорбат 60, воск цетиловых эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
Фармацевтические композиции по настоящему изобретению можно также наносить наружно в области нижнего отдела пищеварительного тракта в составе ректальных суппозиториев или в виде препаратов для введения посредством клизмы.
Подходящие вспомогательные фармацевтические вещества, например, носители, и способы приготовления дозированных фармацевтических форм описаны в Remington’s Pharmaceutical Sciences, Mack Publishing Company, который является стандартным справочником в области приготовления лекарственных препаратов.
Фармацевтические композиции могут содержать от примерно 1% до примерно 95%, предпочтительно от примерно 20% до примерно 90% соединения, имеющего формулу (I), вместе с по меньшей мере одним фармацевтически приемлемым вспомогательным веществом. В целом, соединения по настоящему изобретению вводят в терапевтически эффективном количестве одним из общепринятых способов введения лекарственных средств, имеющих похожее применение. Подходящие суточные дозировки находятся в диапазоне от 1 до 1000 мг, например, 1-500 мг в сутки или 1-50 мг в сутки, в зависимости от многих факторов, таких как степень тяжести заболевания, подвергающегося лечению, возраст и состояние здоровья пациента, эффективность применяемого соединения, способ введения и готовая форма, а также от показаний к применению, и т.д. Квалифицированный специалист в области лечения таких заболеваний может без излишних экспериментов и на основе профессиональных знаний и информации, раскрытой в настоящей заявке, рассчитать терапевтически эффективное количество соединений по настоящему изобретению в каждом конкретном случае. Соединения по настоящему изобретению можно вводить в виде фармацевтических препаратов, включая препараты, подходящие для энтерального или парентерального введения. Предпочтительным путем введения является пероральный с применением удобного режима ежедневных дозировок, который можно подбирать в зависимости от степени тяжести заболевания.
Соединение по настоящему изобретению может применяться для лечения заболеваний, вызванных РНК-вирусной инфекцией у млекопитающего, например, инфекцией безоболочечного одноцепочечного (+) РНК-вируса, в частности – заболеваний, вызванных пикорнавирусами.
Пикорнавирус может представлять собой, например, парэховирус (например, Ljungan или Parecho), кардиовирус (например, вирус ЭМК или вирус Тэйлера), энтеровирус (например, энтеровирус, вирус коксаки, полиомиелит, риновирус) или гепатовирус. В случае ветеринарного применения, пикорнавирус может представлять собой, например, афтовирус или тешовирус.
Заболеваниями, которые считаются связанными или вызванными или каким-либо иным образом ассоциированными с вирусной инфекцией, например, пикорнавирусами, являются, например, нейродегенеративные заболевания, такие как рассеянный склероз, болезнь Паркинсона, амиотрофический боковой склероз, болезнь Альцгеймера, болезнь Хантингтона, полиомиелит, энцефалит, менингит, сепсис, рак, паралич, миокардит, диабет, обычная простуда, энтеровирусный везикулярный стоматит, герпетическая ангина, плевродиния, диарея, слизисто-кожное поражение, респираторное заболевание, конъюнктивит, миозит и синдром хронической усталости.
В рамки знаний и навыков квалифицированного специалиста в данной области входит синтез и идентификация соединений, имеющих формулу (I) по настоящему изобретению, согласно способам, описанным ниже в рамках неограничивающих Примеров, а также общим способам, описанным в литературе, например, в PCT/EP2018/058522 (опубликован как WO 2018/185120 A1) и PCT/EP2015/051177 (опубликован как WO 2015/110491 A2), содержание которых включено в настоящий текст посредством ссылки.
Примеры
Общие методы
Реакции проводили в высушенных в пламени горелки герметично закрывающихся пробирках или в высушенной в термошкафу стеклянной посуде под избыточным давлением аргона или азота, если иное не указано специально. Чувствительные к воздуху или влажности жидкости и растворы переносили шприцом. Тетрагидрофуран (ТГФ) перегоняли над системой натрий/бензофенон-кетил. Дихлорметан (CH2Cl2) перегоняли над гидридом кальция. Все остальные химические реактивы получали от коммерческих поставщиков и использовали без дополнительной очистки, если иное не указано особо. Молекулярные сита активировали при 350°C и размалывали непосредственно перед использованием, затем сушили в вакууме в пламени горелки. Прохождение реакций отслеживали методом тонкослойной хроматографии (ТСХ), используя пластинки с нанесенным слоем силикагеля толщиной 0.25 мм (E. Merck). Растворы в органических растворителях упаривали на роторном испарителе при температуре ниже 50°C. Колоночную флэш-хроматографию проводили с силикагелем 60-120, 230-400 меш или с нейтральным оксидом алюминия. Выходы приведены для хроматографически и спектрально чистых соединений, если не указано иное.
Приборы
1H и 13C спектры записывали на спектрометре Bruker AVANCE III HD с рабочей частотой 400 МГц или Bruker AVANCE II с рабочей частотой 300 МГц. Значения хим.сдвигов приведены в миллионных долях (м.д, шкала δ) в слабое поле относительно тетраметилсилана, в качестве референсов использовали остаточные сигналы растворителя в ЯМР-спектре (CHCl3: δ 7.26 для 1H-ЯМР, δ 77.16 для 13C-ЯМР). Анализы методом LC-MS проводили на приборе Agilent XCT Ion Trap, оснащенном программами Chemstation и Bruker daltonics.
Синтез 3-(3,4-диметоксифенил)-8-иод-2,6-диметилимидазо[1,2-b]пиридазина
Промежуточный продукт 3-(3,4-диметоксифенил)-8-иод-2,6-диметилимидазо[1,2-b] пиридазин получали многостадийным синтезом согласно описанной ниже методике:
Стадия 1: 2,6-диметилимидазо[1,2-b]пиридазин
В перемешиваемый раствор 6-метилпиридазин-3-иламина (50 г, 455.3 ммоль) в этаноле (500 мл) добавляли хлорацетон (58 мл, 683 ммоль), и раствор нагревали при 85°C в течение 10 часов. После завершения реакции отгоняли этанол из реакционной смеси. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение 2,6-диметилимидазо[1,2-b] пиридазин (36 г, 53.4%) 10%-ным этилацетатом в гексане и получая его в виде темно-коричневого твердого вещества.
Стадия 2: 3-бром-2,6-диметилимидазо[1,2-b]пиридазин
В перемешиваемый раствор 2,6-диметилимидазо[1,2-b]пиридазина (36 г, 244.5 ммоль) в ацетонитриле (360 мл) добавляли N-бромсукцинимид (NBS) (52.2 г, 293.4 ммоль) и перемешивали при комнатной температуре в течение 1 часа. После завершения реакции отгоняли ацетонитрил из реакционной смеси. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение 3-бром-2,6-диметилимидазо[1,2-b]пиридазин (18 г, 32.5%) 15%-ным этилацетатом в гексане и получая его в виде твердого вещества.
Стадия 3: 3-(3,4-диметоксифенил)-2,6-диметилимидазо[1,2-b]пиридазин
В 100-миллилитровую круглодонную колбу загружали 3-бром-2,6-диметилимидазо [1,2-b]пиридазин (5 г, 17.6 ммоль), эфир бороновой кислоты (4.4 г, 19.4 ммоль), карбонат калия (7.5 г, 54.3 ммоль) и смесь диоксан : вода (45 мл : 5 мл). Полученный раствор дегазировали пропусканием азота в течение 10 минут и затем добавляли комплекс [1,1′-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) с дихлорметаном (Pd(dppf)Cl2.ДХМ) (1.8 г, 2.2 ммоль). Реакционную смесь нагревали при 100oC в течение 16 часов. После завершения реакции смесь разбавляли этилацетатом и фильтровали через слой целита. Фильтрат разделяли между этилацетатом и водой. Этилацетатный слой сушили над сульфатом натрия и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение 3-(3,4-диметоксифенил)-2,6-диметилимидазо[1,2-b] пиридазин (4.2 г, 67.7%) 30%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества.
Стадия 4: 3-(3,4-диметоксифенил)-8-иод-2,6-диметилимидазо[1,2-b]пиридазин
В перемешиваемый 2М раствор диизопропиламида лития (LDA) (0.7 мл, 1.4 ммоль) в ТГФ при -78oC по каплям добавляли раствор 3-(3,4-диметоксифенил)-2,6-диметилимидазо[1,2-b]пиридазина (0.2 г, 0.705 ммоль) в ТГФ (5 мл). Через 10 минут добавляли иод (0.178 г, 0.705 ммоль), растворенный в ТГФ (3 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После этого реакцию гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом. Этилацетатные слои сушили над Na2SO4 и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение 3-(3,4-диметоксифенил)-8-иод-2,6-диметилимидазо[1,2-b] пиридазин (30 мг, 10.7%) 30%-ным этилацетатом в гексане и получая его в виде бледно-желтого твердого вещества.
Пример 1
3-(3,4-диметоксифенил)-2,6-диметил-N-((2-метилпиридин-4-ил)метил)имидазо[1,2-b] пиридазин-8-амин
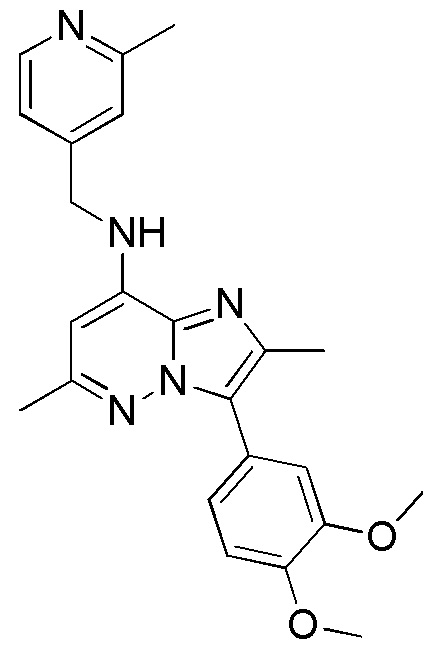
В перемешиваемый раствор 3-(3,4-диметоксифенил)-8-иод-2,6-диметилимидазо[1,2-b]пиридазина (0.1 г, 0.240 ммоль) и амина (0.06 г, 0.312 ммоль) в толуоле (2 мл) добавляли карбонат цезия (0.156 г, 0.48 ммоль), 2,2′-бис(дифенилфосфино)-1,1′-бинафтил (BINAP (7 мг, 0.012 ммоль) и Pd(OAc)2 (2 мг, 0.012 ммоль). Реакционную смесь перемешивали при 105°C в течение 16 часов. После завершения реакции смесь разбавляли 10%-ным MeOH-CH2Cl2 и фильтровали через слой целита. Фильтрат упаривали и полученный твердый остаток промывали ацетонитрилом, получая Пр. 2 (0.09 г, 91.83%) в виде светло-коричневого твердого вещества.
Пример 2
3-(3,4-диметоксифенил)-N-((2-фторпиридин-4-ил)метил)-2,6-диметилимидазо[1,2-b] пиридазин-8-амин
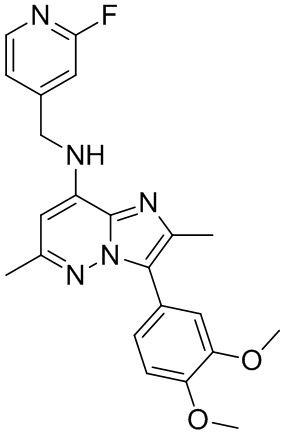
В перемешиваемый раствор 3-(3,4-диметоксифенил)-8-иод-2,6-диметилимидазо[1,2-b]пиридазина (0.15 г, 0.360 ммоль) и амина (0.093 г, 0.46 ммоль) в толуоле (3 мл) добавляли карбонат цезия (0.23 г, 0.72 ммоль), 2,2′-бис(дифенилфосфино)-1,1′-бинафтил (BINAP) (11 мг, 0.018 ммоль) и Pd(OAc)2 (4 мг, 0.018 ммоль). Реакционную смесь перемешивали при 105°C в течение 16 часов. После завершения реакции смесь разбавляли 10%-ным MeOH-CH2Cl2 и фильтровали через слой целита. Фильтрат упаривали, и полученный твердый остаток промывали ацетонитрилом, получая Пр. 1 (0.120 г, 80%) в виде светло-коричневого твердого вещества.
Синтез 7-хлор-3-иод-2,5-диметилпиразоло[1,5-a]пиримидина
Промежуточный продукт 7-хлор-3-иод-2,5-диметилпиразоло[1,5-a]пиримидин получали многостадийным синтезом согласно описанной ниже методике:
Стадия 1: 2,5-диметилпиразоло[1,5-a]пиримидин-7-ол
В круглодонную колбу загружали 3-амино-5-метилпиразол (100 г, 1.02 моль), этил ацетоацетат (161 мл, 1.23 моль), уксусную кислоту (300 мл) и 1,4-диоксан (1000 мл). Реакционную смесь кипятили в течение 16 часов при 105°C. После завершения реакции получали не совсем белое твердое вещество, которое отфильтровывали при отсасывании. Осадок промывали холодным гексаном и сушили в вакууме, получая 2,5-диметилпиразоло [1,5-a]пиримидин-7-ол (85 г, 49%) в виде не совсем белого твердого вещества.
Стадия 2: 7-хлор-2,5-диметилпиразоло[1,5-a]пиримидин
В раствор 2,5-диметилпиразоло[1,5-a]пиримидин-7-ола (120 г, 0.73 моль) в ацетонитриле (1200 мл) по каплям добавляли POCl3 (103 мл, 1.1 моль). После окончания добавления реакционную смесь нагревали при 80°C в течение 12 часов. После завершения реакции отгоняли POCl3 из реакционной смеси. Полученный сырой продукт разбавляли водой, нейтрализовывали насыщенным раствором NaHCO3 и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной флэш-хроматографии, элюируя 7-хлор-2,5-диметилпиразоло[1,5-a]пиримидин 20%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества (108 г, 80.8%).
Стадия 3: 7-хлор-3-иод-2,5-диметилпиразоло[1,5-a]пиримидин
В ледяной раствор 7-хлор-2,5-диметилпиразоло[1,5-a]пиримидина (120 г, 0.66 моль) в ацетонитриле (1200 мл) при -10°C порциями добавляли N-иодсукцинимид (163.5 г, 0.726 моль). Реакционную смесь перемешивали при той же температуре в течение 1 часа. После завершения реакции наблюдали осадок. Реакционную смесь гасили ледяной водой и фильтровали при отсасывании. Полученное твердое вещество промывали гексаном и сушили в вакууме, получая 7-хлор-3-иод-2,5-диметилпиразоло[1,5-a]пиримидин в виде белого твердого вещества (182.8 г, 89.9%).
Синтез 1,3-диметил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-индазола
Эфир бороновой кислоты 1,3-диметил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-индазол получали многостадийным синтезом согласно описанной ниже методике:
Стадия 1: 5-бром-1,3-диметил-1H-индазол
Перемешиваемый раствор 1-(5-бром-2-фторфенил)этанона (50 г, 230 ммоль) и N-метил-гидразина (42.4 мл, 805 ммоль) в пиридине (500 мл) нагревали при 90°C в течение 10 часов. После завершения реакции отгоняли пиридин из реакционной смеси. Полученный сырой продукт разделяли между водой и этилацетатом. Этилацетатные слои сушили над сульфатом натрия и упаривали. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение 5-бром-1,3-диметил-1H-индазол (20.23 г, 41.8%) 2%-ным этилацетатом в гексане и получая его в виде светло-коричневого вязкого вещества.
Стадия 2: 1,3-диметил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-индазол
В раствор 5-бром-1,3-диметил-1H-индазола (26 г, 115 ммоль) и биспинаколато-диборона (32.3 г, 127 ммоль) в 1,4-диоксане (260 мл) добавляли KOAc (34 г, 345 ммоль). Реакционную смесь дегазировали азотом 10 минут, затем добавляли Pd(PPh3)4 (6.6 г, 5.57 ммоль) и нагревали при 95°C в течение 16 часов. После окончания добавления реакционную смесь нагревали при 80°C в течение 12 часов. После завершения реакции смесь фильтровали через целит, фильтрат упаривали. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение 1,3-диметил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-индазол (18 г, 52.1%) 5%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества.
Пример 3
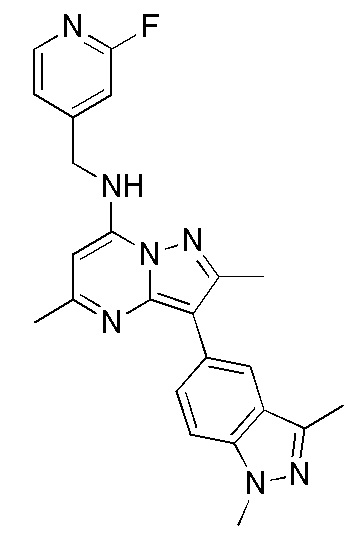
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-фторпиридин-4-ил)метил] пиразоло[1,5-a]пиримидин-7-амин
Стадия 1: N-[(2-фторпиридин-4-ил)метил]-3-иод-2,5-диметилпиразоло[1,5-a]пиримидин-7-амин
В перемешиваемый раствор 7-хлор-3-иод-2,5-диметилпиразоло[1,5-a]пиримидина (1.1 г, 3.57 ммоль) и 1-(2-фторпиридин-4-ил)метанамина (0.586 г, 4.6 ммоль) в этаноле (5.5 мл) добавляли диизопропилэтиламин (5.5 мл, 5 объемов) и перемешивали при 80°C в течение 6 часов. После завершения реакции отгоняли этанол из реакционной смеси. Полученный сырой продукт разделяли между водой и этилацетат. Этилацетатные слои сушили над сульфатом натрия и упаривали при пониженном давлении. Целевое соединение выделяли методом колоночной флэш-хроматографии, элюируя 35%-ным этилацетатом в гексане. После упаривания получали N-[(2-фторпиридин-4-ил)метил]-3-иод-2,5-диметилпиразоло [1,5-a]пиримидин-7-амин (0.9 г, 63.82%) в виде светло-желтого вещества.
Стадия 2: трет-бутил [(2-фторпиридин-4-ил)метил](3-иод-2,5-диметилпиразоло[1,5-a] пиримидин-7-ил)карбамат
В перемешиваемый раствор N-[(2-фторпиридин-4-ил)метил]-3-иод-2,5-диметилпиразоло[1,5-a]пиримидин-7-амина (0.5 г, 1.26 ммоль) в дихлорметане (ДХМ) (5 мл) добавляли 4-диметиламинопиридин (DMAP) (0.153 г, 1.26 ммоль) при 0°C. Затем добавляли по каплям Boc-ангидрид (ди-трет-бутилдикарбонат) (0.33 мл, 1.51 ммоль) при той же температуре и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции смесь промывали водой. Органический слой сушили над сульфатом натрия и упаривали. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение трет-бутил [(2-фторпиридин-4-ил)метил](3-иод-2,5-диметилпиразоло[1,5-a]пиримидин-7-ил)карбамат (0.51 г, 82.2% ) 10%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества.
Стадия 3: трет-бутиловый эфир [3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-пиразоло[1,5-a]пиримидин-7-ил]-(2-фтор-пиридин-4-илметил)-карбаминовой кислоты
В 25-миллилитровую круглодонную колбу загружали трет-бутил [(2-фторпиридин-4-ил)метил](3-иод-2,5-диметилпиразоло[1,5-a]пиримидин-7-ил)карбамат (0.45 г, 0.904 ммоль), 1,3-диметил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-индазол (0.322 г, 1.17 ммоль), карбонат калия (0.311 г, 2.26 ммоль) и смесь диоксан:вода (4.5 мл:0.5 мл). Полученный раствор дегазировали пропусканием азота 10 минут и затем добавляли комплекс Pd(dppf)Cl2.ДХМ (0.110 г, 0.135 ммоль). Реакционную смесь нагревали при 100°C в течение 16 часов. После завершения реакции смесь разбавляли этилацетатом и фильтровали через слой целита. Фильтрат разделяли между этилацетатом и водой. Этилацетатный слой сушили над сульфатом натрия и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение трет-бутиловый эфир [3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-пиразоло[1,5-a] пиримидин-7-ил]-(2-фтор-пиридин-4-илметил)-карбаминовой кислоты (0.2 г, 42.9%) 30%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества.
Стадия 4: 3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-фторпиридин-4-ил)метил] пиразоло[1,5-a]пиримидин-7-амин
В ледяной раствор трет-бутилового эфира [3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-пиразоло[1,5-a] пиримидин-7-ил]-(2-фтор-пиридин-4-илметил)-карбаминовой кислоты (0.2 г, 0.388 ммоль) в дихлорметане (2 мл) по каплям добавляли ТФУК (2 мл). После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение 6 часов. После окончания реакции отгоняли растворитель и избыток ТФУК. Полученный сырой продукт подщелачивали 1н. раствором NaOH и экстрагировали 10%-ным MeOH-ДХМ. Органические слои сушили над сульфатом натрия и упаривали. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение Пр. 3 (0.06 г, 37.5%) 70%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества.
Пример 4
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-метилпиридин-4-ил)метил] пиразоло[1,5-a]пиримидин-7-амин

Стадия 1: 3-иод-2,5-диметил-N-[(2-метилпиридин-4-ил)метил]пиразоло[1,5-a] пиримидин-7-амин
В перемешиваемый раствор 7-хлор-3-иод-2,5-диметилпиразоло[1,5-a]пиримидина (1.1 г, 3.57 ммоль) и 1-(2-метилпиридин-4-ил)метанамина (0.586 г, 4.6 ммоль) в этаноле (5.5 мл) добавляли диизопропилэтиламин (5.5 мл, 5 объемов) и перемешивали при 80°C в течение 6 часов. После завершения реакции отгоняли этанол из реакционной смеси. Полученный сырой продукт разделяли между водой и этилацетат. Этилацетатные слои сушили над сульфатом натрия и упаривали при пониженном давлении. Целевое соединение выделяли методом колоночной флэш-хроматографии, элюируя 35%-ным этилацетатом в гексане. После упаривания получали 3-иод-2,5-диметил-N-[(2-метилпиридин-4-ил)метил]пиразоло [1,5-a]пиримидин-7-амин (0.9 г, 40.1%) в виде светло-желтого вещества.
Стадия 2: трет-бутиловый эфир (3-иод-2,5-диметил-пиразоло[1,5-a]пиримидин-7-ил)-(2-метил-пиридин-4-илметил)-карбаминовой кислоты
В перемешиваемый раствор 3-иод-2,5-диметил-N-[(2-метилпиридин-4-ил)метил] пиразоло[1,5-a]пиримидин-7-амина (0.5 г, 1.26 ммоль) в дихлорметане (5 мл) добавляли DMAP (0.153 г, 1.26 ммоль) при 0°C. Затем добавляли по каплям Boc-ангидрид (0.33 мл, 1.51 ммоль) при той же температуре и перемешивали при комнатной температуре в течение 2 часов. После завершения реакции смесь промывали водой. Органический слой сушили над сульфатом натрия и упаривали. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение трет-бутиловый эфир (3-иод-2,5-диметил-пиразоло[1,5-a]пиримидин-7-ил)-(2-метил-пиридин-4-илметил)-карбаминовой кислоты (0.51 г, 82.2 %) 10%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества.
Стадия 3: трет-бутиловый эфир [3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-пиразоло[1,5-a]пиримидин-7-ил]-(2-метил-пиридин-4-илметил)-карбаминовой кислоты
В 25-миллилитровую круглодонную колбу загружали трет-бутиловый эфир (3-иод-2,5-диметил-пиразоло[1,5-a]пиримидин-7-ил)-(2-метил-пиридин-4-илметил)-карбаминовой кислоты (0.45 г, 0.904 ммоль), 1,3-диметил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-индазол (0.322 г, 1.17 ммоль), карбонат калия (0.311g , 2.26 ммоль) и смесь диоксан:вода (4.5 мл : 0.5 мл). Полученный раствор дегазировали пропусканием азота 10 минут и затем добавляли комплекс Pd(dppf)Cl2.ДХМ (0.110 г, 0.135 ммоль). Реакционную смесь нагревали при 100°C в течение 16 часов. После завершения реакции смесь разбавляли этилацетатом и фильтровали через слой целита. Фильтрат разделяли между этилацетатом и водой. Этилацетатный слой сушили над сульфатом натрия и упаривали при пониженном давлении. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение трет-бутиловый эфир [3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-пиразоло[1,5-a] пиримидин-7-ил]-(2-метил-пиридин-4-илметил)-карбаминовой кислоты (0.2 г, 43.4%) 30%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества.
Стадия 4: 3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-метилпиридин-4-ил) метил]пиразоло[1,5-a]пиримидин-7-амин
В ледяной раствор трет-бутилового эфира [3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-пиразоло[1,5-a]пиримидин-7-ил]-(2-метил-пиридин-4-илметил)-карбаминовой кислоты (0.2 г, 0.388 ммоль) в дихлорметане (2 мл) по каплям добавляли трифторуксусную кислоту (ТФУК) (2 мл). После окончания добавления реакционную смесь перемешивали при комнатной температуре в течение 6 часов. После окончания реакции отгоняли растворитель и избыток ТФУК. Полученный сырой продукт подщелачивали 1н. раствором NaOH и экстрагировали 10%-ным MeOH-ДХМ. Органические слои сушили над сульфатом натрия и упаривали. Полученный сырой продукт очищали методом колоночной флэш-хроматографии (нейтральный оксид алюминия), элюируя целевое соединение Пр. 4 (0.06 г, 37.7%) 70%-ным этилацетатом в гексане и получая его в виде не совсем белого твердого вещества.
Примеры 5-8 синтезировали по тем же общим методикам, которые описаны для Примеров 3 и 4.
Пример 5
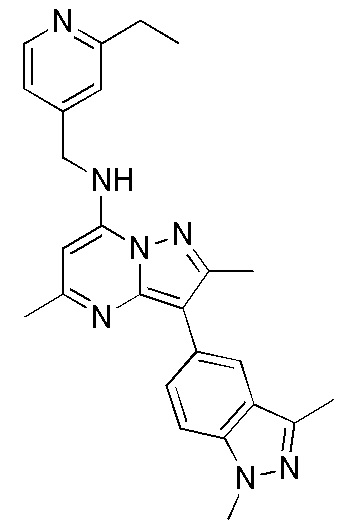
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-этилпиридин-4-ил)метил] пиразоло[1,5-a]пиримидин-7-амин
Пример 6
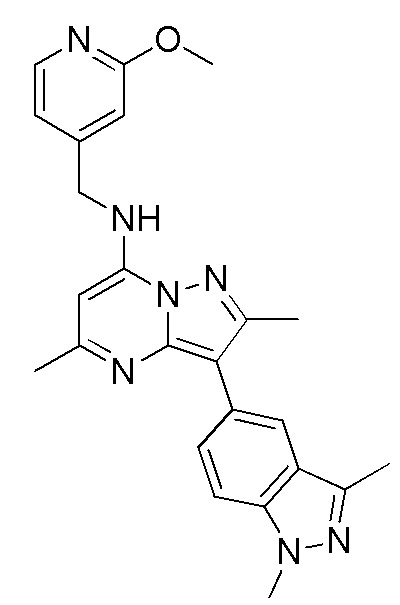
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-метоксипиридин-4-ил)метил] пиразоло[1,5-a]пиримидин-7-амин
Пример 7
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-цианопиридин-4-ил)метил] пиразоло[1,5-a]пиримидин-7-амин
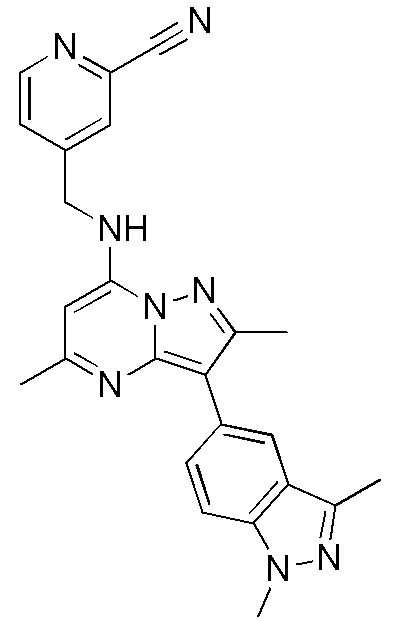
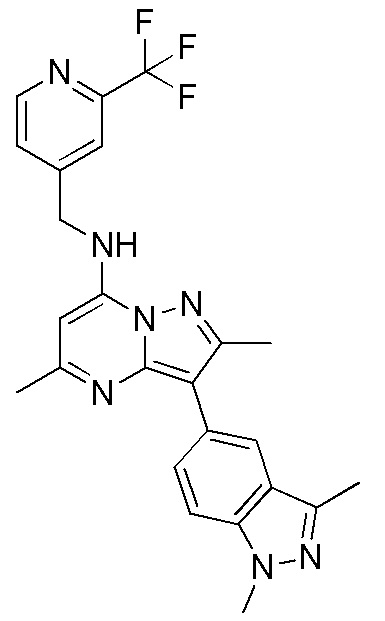
Пример 8
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-трифторметилпиридин-4-ил) метил]пиразоло[1,5-a]пиримидин-7-амин
Данные анализа соединений из Примеров 1-8 приведены в таблице 1.
Таблица 1
Пример 9
Липосомный состав
50 мг 3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-фторпиридин-4-ил)метил] пиразоло[1,5-a]пиримидин-7-амина (Пример 3) и 500 мг соевого лецитина переносили в стакан. Добавляли хлороформ (около 10 мл) и перемешивали смесь до полного растворения компонентов. Затем растворитель удаляли на роторном испарителе, получая тонкую липидную пленку. Добавляли воду (10 мл) к полученной липидной пленке и оставляли для регидратации пленки при комнатной температуре. Состав тщательно перемешивали и обрабатывали ультразвуком до образования гомогенной дисперсии. Состав имел значение pH = 6.42.
Биологические тесты
In vitro анализ в культуре клеток млекопитающих
Противовирусную активность соединений по настоящему изобретению оценивали по их способности предотвращать вызывание вирусом вирусных цитопатических эффектов (ЦПЭ) в культуре клеток млекопитающих. Время инкубирования, линия клеток, плотность клеток и титр вируса отличались от анализа к анализу, но общая методика была следующая: Клетки выращивали на 96-луночных плоскодонных микропланшетах до конфлюэнтности примерно 90% (20000-90000 клеток на лунку) в подходящей среде. Титр вируса определяли на клетках стандартным методом дозы заражения 50% культуры ткани (TCID50). Вкратце, клетки инфицировали добавлением 50 мкл суспензии вируса и разбавляли в 10 раз добавлением среды. Планшеты инкубировали при 37°C с 5% CO2 в течение 3-7 дней, и клетки ежедневно осматривали на предмет ЦПЭ. После детектирования ЦПЭ планшеты окрашивали раствором кристаллического фиолетового по Граму и считывали оптическую плотность при 540 нм. В тестах использовали наивысшее разбавление вируса, которое давало > 95% ЦПЭ. Вещества в конечной концентрации 2.5-20 мкM и вирус добавляли к клеткам и инкубировали 3-7 дней в зависимости от используемого вируса и линии клеток. В качестве контрольных образцов в каждый планшет включали неинфицированные клетки и клетки, инфицированные вирусом (без добавления соединения). Клетки окрашивали кристаллическим фиолетовым после детектирования ЦПЭ на инфицированных контролях, и считывали оптическую плотность при 540 нм. Потенциал ингибирования вычисляли как % в сравнении с неинфицированным и инфицированным контролем.
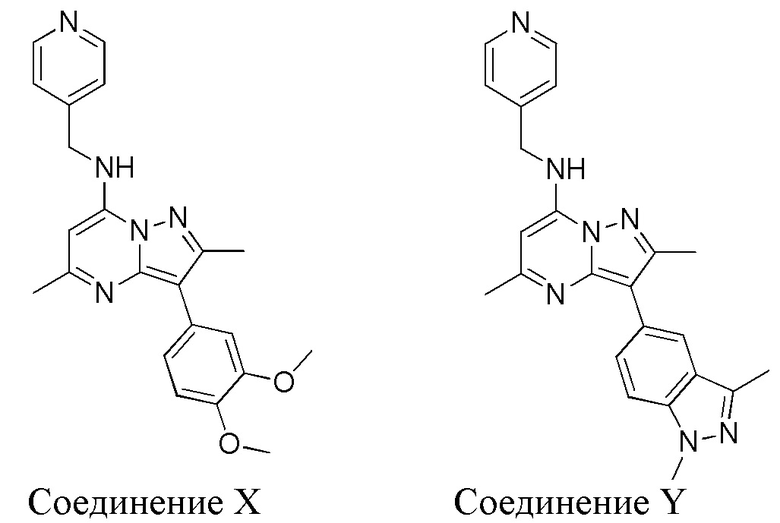
Соединения по настоящему изобретению из Примеров 1-4 тестировали, используя описанную выше методику. Кроме того, такой же тест проводили с соединениями, не имеющими заместителя в положении 2 в пиридиновом кольце, а именно: 3-(3,4-диметоксифенил)-2,5-диметил-N-(пиридин-4-илметил)пиразоло[1,5-a]пиримидин-7-амин (Соединение “X”) и 3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(пиридин-4-ил) метил]пиразоло[1,5-a]пиримидин-7-амин (Соединение “Y”):
В таблицах 2 и 3 показан потенциал ингибирования для протестированных соединений на разных пикорнавирусах в концентрации 10 нM и 100 нM, соответственно. EV6: энтеровирус линия 6; EV30: энтеровирус линия 30; EV-D68: энтеровирус D68; EV71: энтеровирус линия 71; Cox B1: вирус коксаки B линия 1; Cox B2: вирус коксаки B линия 2; Cox B3: вирус коксаки B линия 3; Cox B5: вирус коксаки B линия 5; Polio 1: вирус полиомиелита линия 1.
Таблица 2
Таблица 3
In Vitro тест ингибирования фермента CYP 3A4 в микросомах печени человека методом маркерного субстрата
In vitro тест ингибирования фермента CYP3A4 проводили с использованием микросом печени человека в концентрации 0.5 мг/мл. Кетоконазол использовали в качестве ингибитора сравнения, и мидазолам использовали как селективный маркерный субстрат для фермента CYP 3A4. Серийные разведения тестируемых/контрольных образцов делали в калий-фосфатном буфере (50 мM, pH 7.40), получая восемь концентраций, получаемых разбавлением в соотношении 1:4. Процент ацетонитрил/ ДМСО сохраняли равным 2.5%/0.25% в 2.5Х серийно разводимых растворах контроль/тестовый образец, соответственно. Финальная концентрация ацетонитрил/ ДМСО составляла 1%/0.1%, соответственно. Промежуточные растворы, такие как 10X рабочий раствор (5 мг/мл) микросом печени человека, 10X концентрации маркерных субстратов в буфере, 5X концентрации кофакторов (5.0 мM NADP+, 25.0 мM G-6-P, 3.0 МЕ/мл G-6-PDH и 10.0 мM MgCl2), готовили в калий-фосфатном буфере. Финальные концентрации микросом печени в реакционной смеси составляли 0.5 мг/мл, кофакторами служили 1.0 мM NADP+, 5.0 мM G-6-P, 0.6 МЕ/мл G-6-PDH и 2.0 мM MgCl2. Мидазолам тестировали в тестовой концентрации 5 мкМ. 40 мкл 2.5X серийно разведенных растворов тестируемого соединения/контроля и 10 мкл 5X раствора микросом печени добавляли в 96-луночный планшет и инкубировали 10 минут при 37°C при встряхивании (400 об/мин) на аппарате Thermomixer. После предварительного инкубирования добавляли 20 мкл калий-фосфатного буфера, 10 мкл рабочего раствора соответствующего селективного субстрата и 20 мкл смеси кофакторов. Результаты тестов, выраженные как IC50 в мкM, приведены в таблице 4.
Таблица 4
Фармакокинетические характеристики
Приготовление составов
Отвешивали 10 мг соединения и переносили в мерную пробирку. Затем добавляли 500 мкл ДМА (5% об/об), тщательно перемешивали на вихревой мешалке и обрабатывали ультразвуком до полного растворения тестируемых образцов. Затем добавляли 5 мл ПЭГ 200 (50% об/об) и тщательно перемешивали на вихревой мешалке. Затем добавляли стерильную воду для контроля (SWFI) небольшими порциями и тщательно перемешивали на вихревой мешалке. Наконец доводили объем до 10 мл добавлением SWFI (45% об/об), получая прозрачный раствор с финальной концентрацией 1 мг/мл. Значение pH состава составляло 6.51. Состав был приготовлен незадолго до введения животным.
Введение дозы
Взрослые здоровые самцы крыс Sprague Dawley возрастом 8-10 недель использовали для тестов после минимум 3 дней акклиматизации. Животным натощак вводили тестируемое соединение в рекомендованном носителе (5% ДМА + 50% ПЭГ 200 + 45% SWFI) перорально в дозировке 10 мг/кг веса тела и в объемной дозировке 10 мл/кг веса тела.
Под мягкой изофлурановой анестезией брали образцы крови в промаркированные пробирки, содержащие антикоагулянт (K2ЭДТА - 2 мг/мл крови), в определенные моменты времени после введения дозы. Взятые образцы крови центрифугировали при 4000 об/мин, 4°C в течение 10 минут, плазму отделяли и хранили при -80°C до проведения анализа. Полученные результаты приведены в таблице 5. AUC = площадь под кривой; MRT = среднее время удержания состава.
Таблица 5
* Липосомный состав
Метаболическая стабильность
Анализ метаболической стабильности проводили с использованием микросом человека/крысы. Финальный состав для анализа содержал 5 мкM тестируемого соединения и контрольного соединения (диклофенак или имипрамин), полученных из стоковых растворов в ДМСО, так что финальная концентрация ДМСО составляла 0.1%, 0.25 мг/мл микросомного белка и кофакторы (5.0 мM G-6-P, 0.06 Ед/мл G-6-PDH, 2.0 мM MgCl2, 1.0 мM NADP+). Тестируемое соединение/контрольные соединения инкубировали с микросомами печени человека/крысы с кофакторами и без кофакторов. Реакционную смесь (100 мкл) удаляли через определенный период времени, и реакцию останавливали добавлением стоп-реагента. Образцы экстрагировали в присутствии внутреннего стандарта и анализировали с применением метода LC-MS/MS. Процент тестируемого/контрольного соединения, оставшегося после указанного периода инкубирования, вычисляли относительно площади пика в момент времени 0 минут.
Краткая методика
4X рабочую концентрацию тестируемого соединения (20 мкM) / контрольного образца (20 мкM) готовили в 50 мM калий-фосфатного буфера (pH 7.4), используя 5 мM стоковые растворы в ДМСО.
10X рабочую концентрацию раствора микросом печени человека/крысы (2.5 мг/мл) готовили в 50 мM калий-фосфатном буфере (pH 7.40), используя стоковый раствор микросом печени человека/крысы (концентрация белка 20 мг/мл). Реакционную смесь в количестве 100 мкл (для каждой отсечки времени) инкубировали при добавлении 45 мкл калий-фосфатного буфера, 10 мкл разведенного раствора микросом печени человека/крысы (2.5 мг/мл), 25 мкл тестируемых/контрольных соединений (20 мкM) и 20 мкл смеси кофактор/буфер. Реакционную смесь инкубировали далее при 37°C в течение указанного времени (с кофакторами: 0, 15, 30, 60 и 120 мин; без кофакторов: 0 и 120 мин). 100 мкл инкубированных образцов тестируемых соединений и контрольных соединений в указанные моменты времени инкубирования переносили в соответствующие пробирки для экстрагирования образца.
Метод экстракции белка осаждением
Тестируемые и контрольные образцы экстрагировали методом осаждения белка. 100 мкл инкубированного образца в каждый момент времени добавляли в пробирки, содержащие 200 мкл ледяного ацетонитрила и 50 мкл раствора галоперидола (0.5 мкг/мл). Затем пробирки тщательно перемешивали на вихревой мешалке и центрифугировали при 10000 об/мин в течение 10 минут при 4°C. Прозрачный надосадочный раствор (200 мкл) из образцов анализировали методом LC-MS/MS. Полученные результаты, выраженные в процентах сохранения после 60 минут инкубирования, приведены в таблице 6.
Таблица 6
Связывание с белками плазмы человека и крысы
Методология
Анализ связывания с белками плазмы проводили на приборе для анализа быстрого равновесного диализа (Rapid Equilibrium Dialysis (RED)), имеющем диализную мембрану с отсечкой по молекулярному весу 8000 дальтон.
Каждая диализная вставка имела две камеры. Красная камера – для плазмы, а белая камера – для буфера. Тестируемые соединения и контрольные соединения (варфарин и пропранолол) готовили в необходимых для теста концентрациях 10 мкM в плазме крови человека/крысы (pH доводили до 7.40), используя 10 мM стоковые растворы в ДМСО (финальная концентрация ДМСО составляла 0.1%). 300 мкл образца плазмы помещали в камеру для образца. 500 мкл буфера помещали в камеру для буфера. После герметизации RED-прибора липкой пленкой, инкубировали при 37°C со встряхиванием (300 об/мин) в течение 4 часов. После инкубирования отбирали аликвоту 50 мкл из каждой лунки (со стороны плазмы и со стороны буфера) и разводили равным объемом противоположного матрикса для нейтрализации эффекта матрикса, после чего проводили экстракцию образца.
Метод экстракции белка осаждением
Все образцы экстрагировали методом осаждения белка посредством добавления 200 мкл ледяного ацетонитрила и 50 мкл внутреннего стандарта (раствор галоперидола 0.1 мкг/мл). Пробирки тщательно перемешивали на вихревой мешалке и центрифугировали при 10000 об/мин в течение при 4°C в течение 10 минут. Прозрачный надосадочный раствор (200 мкл) из образцов анализировали методом LC-MS/MS. Полученные результаты, выраженные в % связывания с белками плазмы, приведены в таблице 7.
Таблица 7
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[1,5-A]ТРИАЗИН-4-АМИНА, ПРИМЕНИМЫЕ В ТЕРАПИИ | 2016 |
|
RU2712196C2 |
| ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ, ПРИГОДНЫЕ В ТЕРАПИИ | 2018 |
|
RU2770363C2 |
| N-ПИРИДИН-3-ИЛ ИЛИ N-ПИРАЗИН-2-ИЛ КАРБОКСАМИДЫ В КАЧЕСТВЕ АГЕНТОВ, ПОВЫШАЮЩИХ УРОВЕНЬ ХОЛЕСТЕРИНА ЛПВП | 2011 |
|
RU2540069C2 |
| 1-N-АЛКИЛ-N-АРИЛПИРИМИДИНАМИНЫ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2153494C2 |
| СОЕДИНЕНИЯ ЗАМЕЩЕННОГО N-(1Н-ИНДАЗОЛ-4-ИЛ)ИМИДАЗОЛ[1,2-a]ПИРИДИН-3-КАРБОКСАМИДА КАК ИНГИБИТОРЫ cFMS | 2010 |
|
RU2562977C2 |
| N1-(4-(5-(ЦИКЛОПРОПИЛМЕТИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-ИЛ)ПИРИДИН-2-ИЛ)ЦИКЛОГЕКСАН-1,4-ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ СK1 И/ИЛИ IRAK1 ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2761457C2 |
| N-(4-(АЗАИНДАЗОЛ-6-ИЛ)-ФЕНИЛ)-СУЛЬФОНАМИДЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2014 |
|
RU2671864C2 |
| ЗАМЕЩЕННЫЕ N-(1H-ИНДАЗОЛ-4-ИЛ) ИМИДАЗОЛ [1,2-А]ПИРИДИН-3- КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ III ТИПА | 2011 |
|
RU2591195C2 |
| ПИРАЗОЛО[1,5-а]ПИРИМИДИНЫ В КАЧЕСТВЕ ПРОТИВОВИРУСНЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2689788C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
Изобретение относится к соединению, имеющему формулу (I), или к его фармацевтически приемлемой соли, где один из X и Y представляет собой C, а другой представляет собой N; R1 выбран из галогена, CN, C1-C4 алкокси-группы, и C1-C4 алкила, необязательно замещенного 1-3 галогенами; и R2 представляет собой 3,4-диметоксифенил или 1,3-диметил-1H-индазол-5-ил. Изобретение также относится к фармацевтической композиции для ингибирования энтеровирусов на основе соединения формулы (I). Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине в качестве эффективных лекарственных средств для лечения энтеровирусной инфекции, где энтеровирусы выбраны из EV, вируса Коксаки, полиомиелита, риновируса. 4 н. и 14 з.п. ф-лы, 7 табл., 9 пр.
 .
.
1. Соединение, имеющее формулу (I)
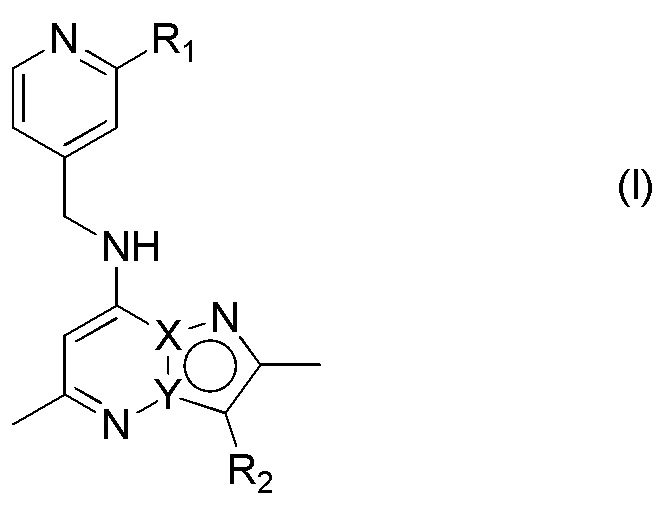 ,
,
или его фармацевтически приемлемая соль, где
один из X и Y представляет собой C, а другой представляет собой N;
R1 выбран из галогена, CN, C1-C4 алкокси-группы, и C1-C4 алкила, необязательно замещенного 1-3 галогенами; и
R2 представляет собой 3,4-диметоксифенил или 1,3-диметил-1H-индазол-5-ил.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R2 представляет собой 3,4-диметоксифенил.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где R2 представляет собой 1,3-диметил-1H-индазол-5-ил.
4. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R1 выбран из F, CN, метокси-группы, метила, трифторметила и этила.
5. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R1 выбран из галогена и C1-C4 алкила, необязательно замещенного 1-3 галогенами.
6. Соединение по п. 5 или его фармацевтически приемлемая соль, где R1 выбран из F, метила, трифторметила и этила.
7. Соединение по п. 6 или его фармацевтически приемлемая соль, где R1 представляет собой метил.
8. Соединение по любому из пп. 1-7, где X представляет собой N, а Y представляет собой C.
9. Соединение по любому из пп. 1-7, где X представляет собой C, а Y представляет собой N.
10. Соединение по п. 1, выбранное из следующих:
3-(3,4-диметоксифенил)-2,6-диметил-N-((2-метилпиридин-4-ил)метил)имидазо[1,2-b]пиридазин-8-амин,
3-(3,4-диметоксифенил)-N-((2-фторпиридин-4-ил)метил)-2,6-диметилимидазо[1,2-b]пиридазин-8-амин,
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-фторпиридин-4-ил)метил]пиразоло [1,5-a]пиримидин-7-амин,
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-метилпиридин-4-ил)метил]пиразоло[1,5-a]пиримидин-7-амин,
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-этилпиридин-4-ил)метил]пиразоло[1,5-a]пиримидин-7-амин,
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-метоксипиридин-4-ил)метил]пиразоло[1,5-a]пиримидин-7-амин,
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-цианопиридин-4-ил)метил]пиразоло [1,5-a]пиримидин-7-амин, и
3-(1,3-диметил-1H-индазол-5-ил)-2,5-диметил-N-[(2-трифторметилпиридин-4-ил)метил]пиразоло[1,5-a]пиримидин-7-амин,
или его фармацевтически приемлемая соль.
11. Фармацевтическая композиция для ингибирования энтеровирусов, содержащая эффективное количество соединения по любому из пп. 1-10 или его фармацевтически приемлемую соль и фармацевтически приемлемое вспомогательное вещество.
12. Фармацевтическая композиция по п. 11, в которой энтеровирусы выбраны из EV, вируса Коксаки, полиомиелита, риновируса.
13. Соединение по любому из пп. 1-10 или его фармацевтически приемлемая соль для применения в лечении энтеровирусной инфекции.
14. Соединение или его фармацевтически приемлемая соль для применения по п. 13, где энтеровирусы выбраны из EV, вируса Коксаки, полиомиелита, риновируса.
15. Применение соединения по любому из пп. 1-10 или его фармацевтически приемлемой соли в производстве лекарственного средства для лечения энтеровирусной инфекции.
16. Применение по п. 17, где энтеровирусы выбраны из EV, вируса Коксаки, полиомиелита, риновируса.
17. Способ лечения энтеровирусной инфекции у млекопитающего, который включает введение млекопитающему эффективного количества соединения по любому из пп. 1-10 или его фармацевтически приемлемой соли.
18. Способ по п. 17, где энтеровирусы выбраны из EV, вируса Коксаки, полиомиелита, риновируса.
| WO 2010086040 A1, 05.08.2010 | |||
| US 6313124 B1, 06.11.2001 | |||
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИИ | 2005 |
|
RU2351592C2 |
| Электромагнитное приспособление к весам для автоматического отвешивания сыпучих тел | 1929 |
|
SU28041A1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
