Область техники
[0001] Настоящее изобретение относится к способу получения кристаллических форм производного диазабициклооктана, представленного формулой (VII), в частности формулой (VII-1).
Уровень техники
[0002] В патенте Японии 4515704 (патентный документ 1) описано новое гетероциклическое соединение, способы его получения и его применение в качестве фармацевтического средства, и в качестве его примера раскрыто конкретное соединение транс-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид натрия (NXL104). Способы получения конкретного производного пиперидина в качестве промежуточного соединения также описаны в публикации не прошедшей экспертизы патентной заявке Японии № 2010-138206 (патентный документ 2) и публикации не прошедшей экспертизы патентной заявке Японии (перевод заявки PCT) № 2010-539147 (патентный документ 3), в то время как способы получения NXL104 и его кристаллических форм описаны в международной публикации № WO 2011/042560 (патентный документ 4).
[0003] Кроме того, в патенте Японии 5038509 (патентный документ 5) описан (2S,5R)-7-оксо-N-(пиперидин-4-ил)-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (MK7655), в то время как способ получения конкретного производного пиперидина и MK7655 описаны в публикации не прошедшей экспертизы патентной заявке Японии № 2011-207900 (патентный документ 6) и Международной публикации WO 2010/126820 (патентный документ 7).
[0004] Авторы настоящего изобретения также раскрыли новое производное диазабициклооктана, представленное следующей формулой (VII), в патентной заявке Японии № 2012-122603 (патентный документ 8):
[0005] [Химическая формула 1]
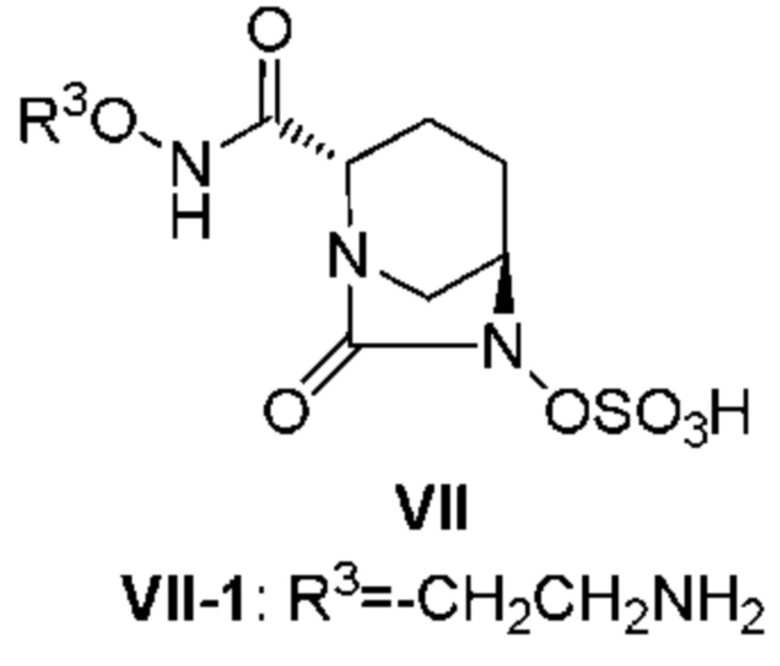
(где R3 является таким, как будет описано далее).
Документы известного уровня техники
Патентные документы
[0006] Патентный документ 1: описание патента Японии 4515704
Патентный документ 2: описание не прошедшей экспертизы патентной заявки Японии № 2010-138206
Патентный документ 3: публикация не прошедшей экспертизы патентной заявки Японии (перевод заявки PCT) № 2010-539147
Патентный документ 4: Международная публикация WO 2011/042560
Патентный документ 5: описание патента Японии 5038509
Патентный документ 6: описание не прошедшей экспертизы патентной заявки Японии № 2011-207900
Патентный документ 7: Международная публикация WO 2010/126820
Патентный документ 8: описание патентной заявки Японии № 2012-122603
Сущность изобретения
Проблемы, решаемые настоящим изобретением
[0007] [Химическая формула 2]
Схема 1
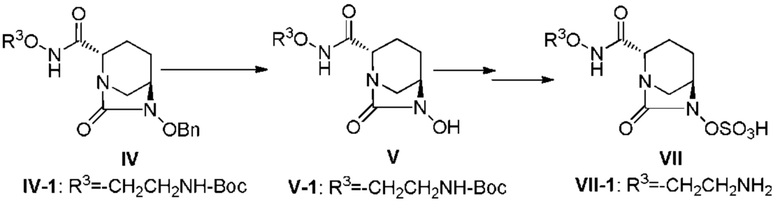
(в приведенных выше формулах R3 является таким, как будет описано ниже, OBn представляет собой бензилокси, и Boc представляет собой трет-бутоксикарбонил.)
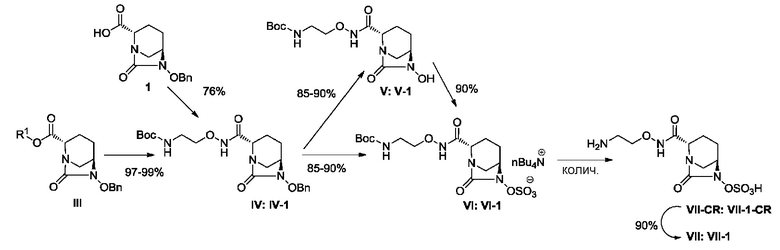
[0008] В ходе изучения промышленного производства (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида, представленного приведенной ниже формулой (VII), в частности формулой (VII-1), возникают различные проблемы, относящиеся к производству, такие как 1) необходимость предоставления соединения в стабильной кристаллической форме из-за сложности обработки в ходе производства аморфного активного фармацевтического ингредиента (API), в частности лиофилизованного продукта, и трудности в обеспечении стабильности; 2) необходимость отделения неочищенного активного фармацевтического ингредиента, содержащего кислоту после удаления защитной группы в боковой цепи группы R3ONHC(=O)-, и проведения процедуры доведения pH до стабильного уровня; 3) необходимость улучшения выхода и избегания загрязнения побочными продуктами путем контролирования чрезмерной реакции, которая проистекает в боковой цепи группы R3ONHC(=O)-, которая также сульфатируется в ходе сульфатирования соединения, представленного формулой (V); 4) невозможность не принимать во внимание низкое выделение в добавление к нестабильности в растворенном состоянии, особенно нестабильности во время выпаривания растворителя реакции для концентрирования соединения, представленного приведенной ниже формулой (V); и 5) неудовлетворительный выход соединения, представленного приведенной ниже формулой (IV). В частности, стадия выделения и регулирования pH неочищенного соединения, представленного формулой (VII-1-CR), содержащего кислоту, для кристаллизации соединения, представленного формулой (VII-1), весьма затруднительна из-за комплекса факторов, вызывающих нестабильность и повышенную растворимость соединения, разложение продуктов и загрязнение.
Средства для решения указанных проблем
[0009] Авторы настоящего изобретения провели подробное изучение способов получения соединения, представленного приведенной ниже формулой (VII), и создали серию способов получения сверхчистого раствора соединения, представленного формулой (VII), который не влияет на кристаллизацию соединения, представленного формулой (VII), а также способ получения высокостабильных кристаллических форм.
[0010] А именно: (1) настоящее изобретение относится к способу получения соединения, представленного следующей формулой (VII):
[Химическая формула 3]
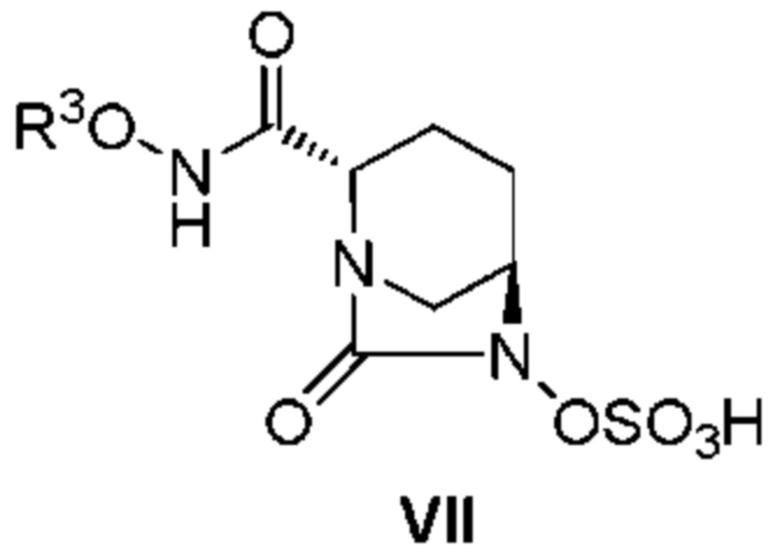 ,
,
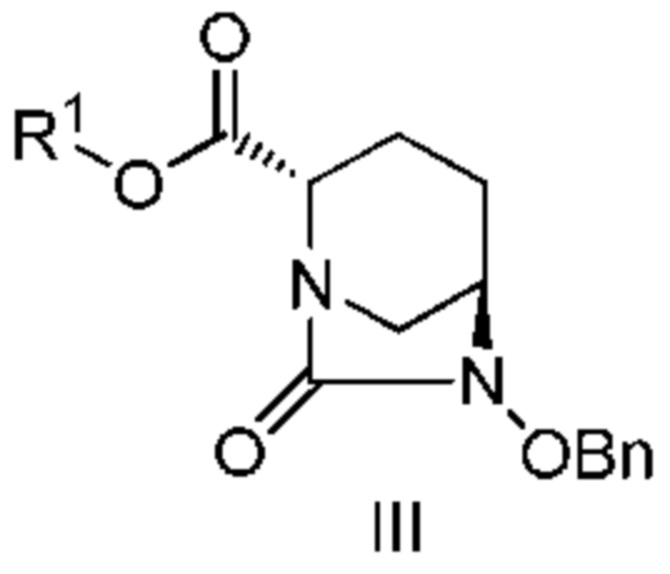
включающему взаимодействие соединения, представленного следующей формулой (III):
[Химическая формула 4]
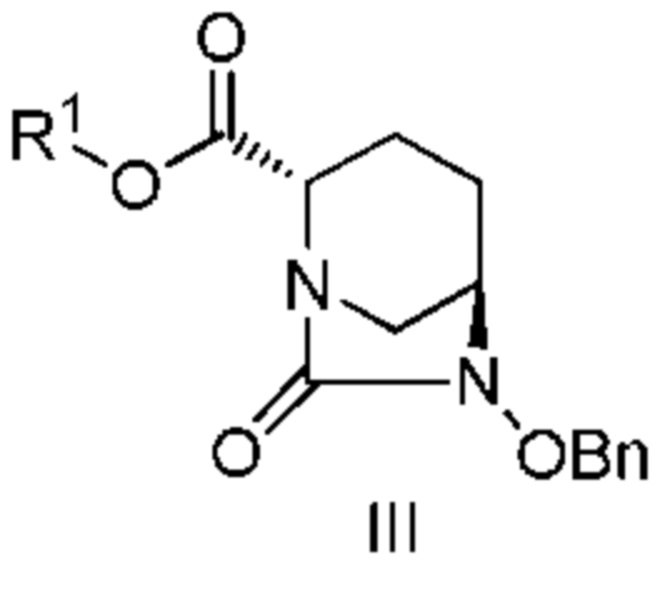
с соединением: R3ONH2, с получением соединения, представленного следующей формулой (IV):
[Химическая формула 5]
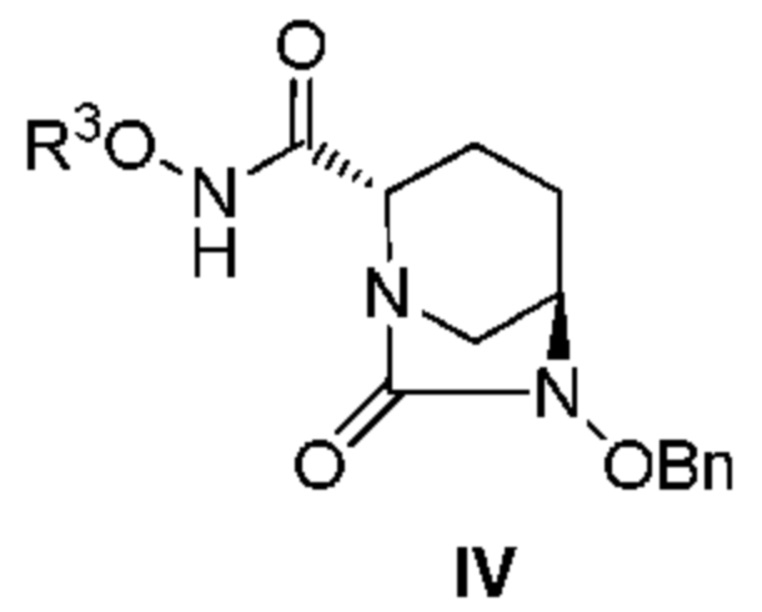 ,
,
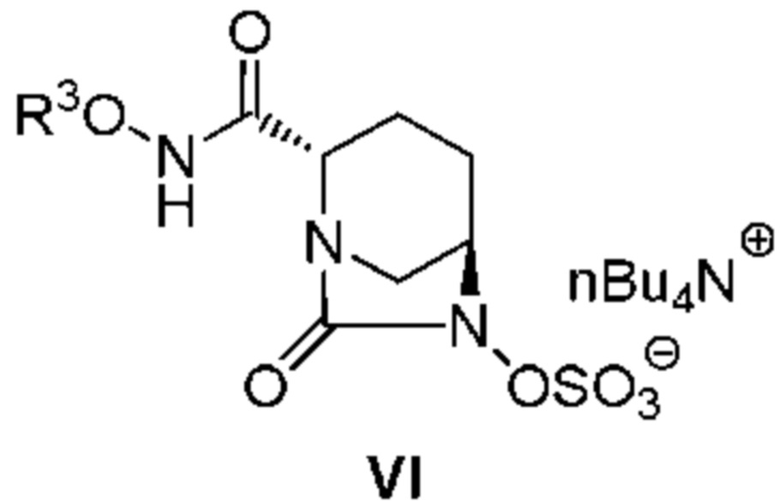
обработку катализатором палладий на угле в атмосфере водорода, при одновременном или последовательном проведении реакции сульфатирования с использованием комплекса триоксид серы-триметиламин в присутствии каталитического количества основания в водном растворителе, и обработку гидросульфатом тетрабутиламмония, с получением соединения, представленного следующей формулой (VI):
[Химическая формула 6]
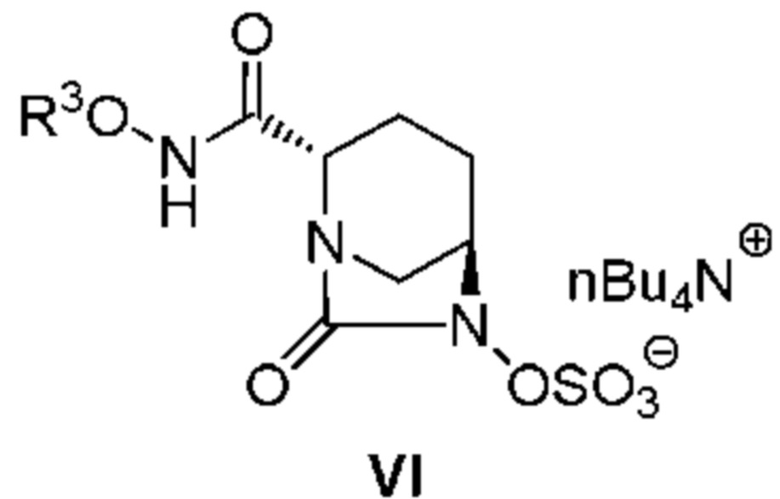 ,
,
с последующим, в случае, когда боковая цепь R3ONHC(=O)- имеет защитную группу, удалением защитной группы кислоты, и осаждением неочищенного продукта добавлением слабого растворителя к реакционному раствору, с получением неочищенного соединения, представленного следующей формулой (VII-CR):
[Химическая формула 7]
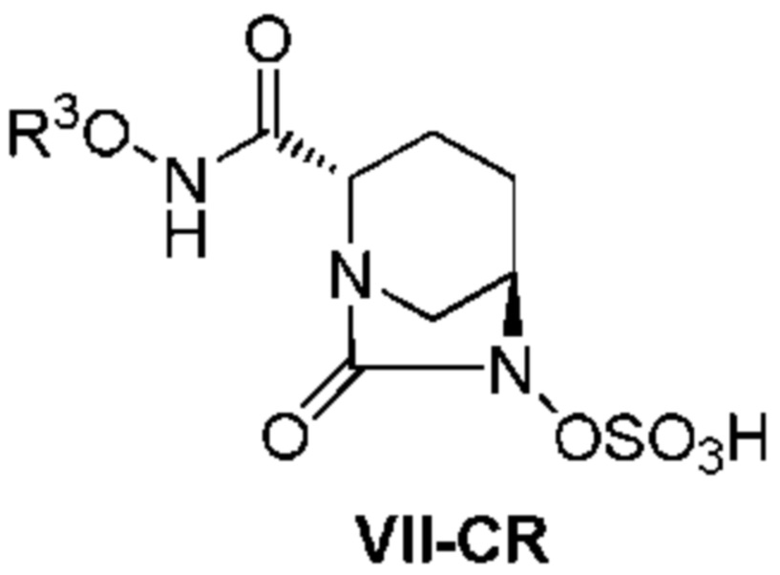
(в каждой из указанных выше формулах OBn представляет собой бензилокси, R1 представляет собой 2,5-диоксопирролидин-1-ил, 1,3-диоксо-3a,4,7,7a-тетрагидро-1H-изоиндол-2(3H)-ил, 1,3-диоксогексагидро-1H-изоиндол-2(3H)-ил или 3,5-диоксо-4-азатрицикло[5.2.1.02,6]дек-8-ен-4-ил, R3 представляет собой C1-6алкил или гетероциклил, R3 может быть модифицирован 0-5 R4; R4 может быть последовательно замещенным, при этом R4 представляет собой C1-6алкил, гетероциклил, R5(R6)N- или защитную группу; R5 и R6, каждый независимо, представляет собой водород или C1-6алкил или вместе образуют гетероциклил; дополнительно, R3, R5 и R6 могут подвергаться закрытию кольца в произвольном положении),
с последующим поочередным добавлением неочищенного соединения, представленного формулой (VII-CR), и охлажденного льдом буфера для получения раствора, имеющего pH от 4 до 5,5, концентрированием после обессоливания синтетическим адсорбентом, при необходимости, регулированием температуры, внесением затравки, при необходимости, и кристаллизацией добавлением слабого растворителя.
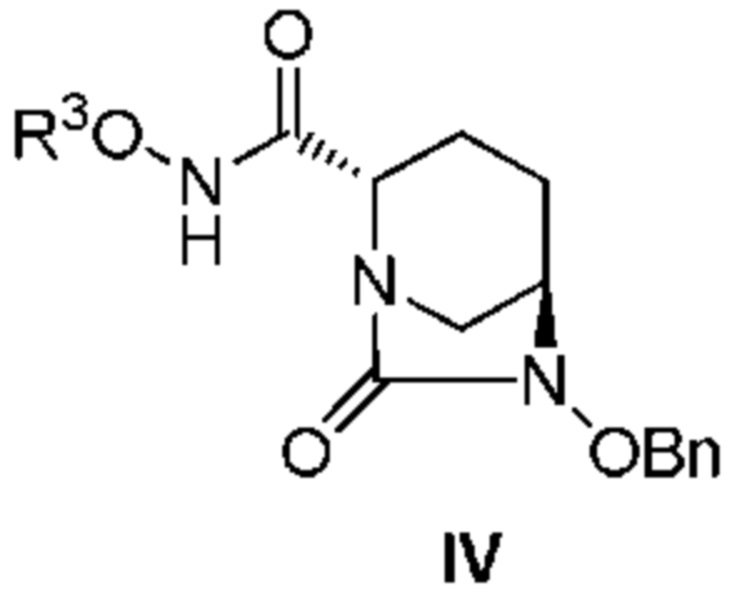
[0011] Кроме того, (2) другой аспект настоящего изобретения относится к способу получения соединения, представленного следующей формулой (IV):
[Химическая формула 8]
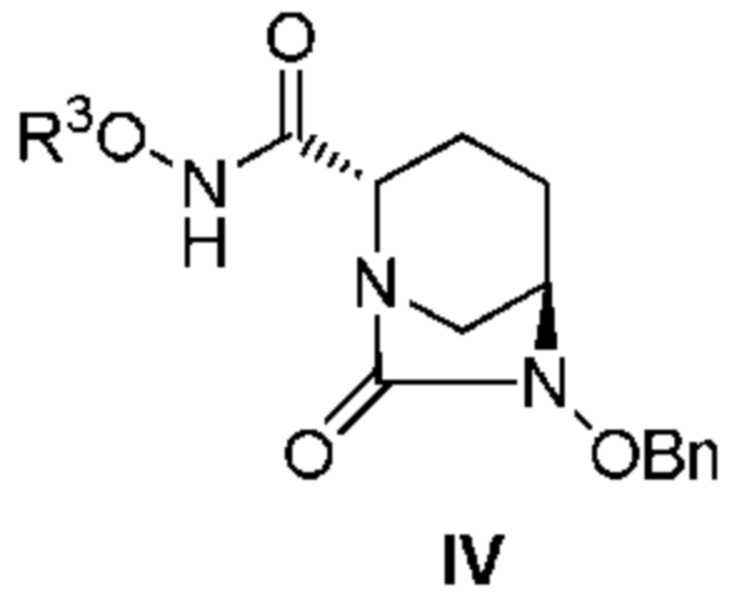 ,
,
включающему взаимодействие соединения, представленного следующей формулой (III):
[Химическая формула 9]
(в каждой из указанных выше формулах R1, R3 и OBn являются такими, как описано выше) с соединением: R3ONH2.
[0012] Кроме того, (3) другой аспект настоящего изобретения относится к способу получения соединения, представленного следующей формулой (VI):
[Химическая формула 10]
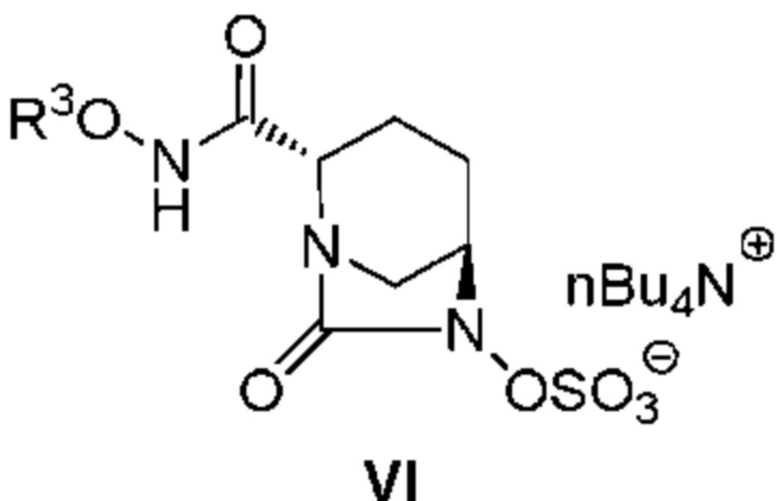 ,
,
включающему обработку соединения, представленного следующей формулой (IV):
[Химическая формула 11]
(в каждой из указанных выше формулах R3 и OBn являются такими, как описано выше)
катализатором палладий на угле в атмосфере водорода, при одновременном или последовательном проведении реакции сульфатирования с использованием комплекса триоксид серы-триметиламин в присутствии каталитического количества основания в водном растворителе, и обработку гидросульфатом тетрабутиламмония.
[0013] Кроме того, (4) другой аспект настоящего изобретения относится к способу получения неочищенного соединения, представленного следующей формулой (VII-CR):
[Химическая формула 12]
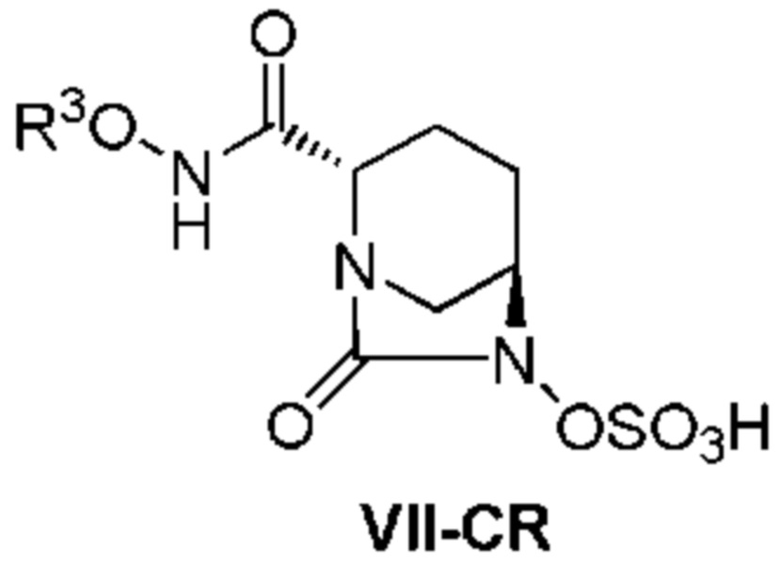 ,
,
включающему, в случае, когда боковая цепь R3ONHC(=O)- имеет защитную группу, удаление защитной группы кислоты из соединения, представленного следующей формулой (VI):
[Химическая формула 13]
(в каждой из указанных выше формулах R3 является таким, как описано выше),
с последующим добавлением слабого растворителя на основе сложного эфира к реакционному раствору для осаждения неочищенного продукта.
[0014] Кроме того, (5) другой аспект настоящего изобретения относится к способу получения соединения, представленного следующей формулой (VII):
[Химическая формула 14]
 ,
,
включающему поочередное добавление неочищенного соединения, представленного следующей формулой (VII-CR):
[Химическая формула 15]
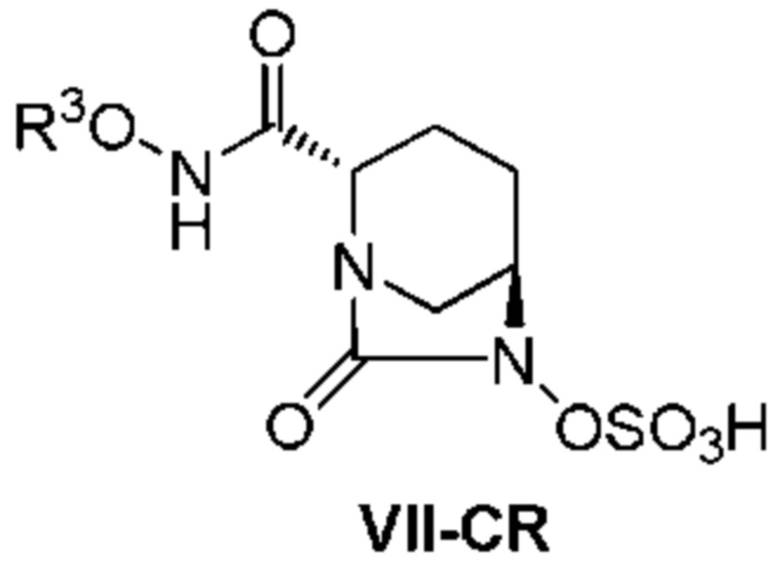
(в каждой из указанных выше формулах R3 является таким, как описано выше)
и охлажденного льдом буфера для получения раствора, имеющего pH от 4 до 5,5, концентрирование после обессоливания синтетическим адсорбентом, при необходимости, регулирование температуры, внесение затравки, при необходимости, и кристаллизацию добавлением слабого растворителя на основе спирта.
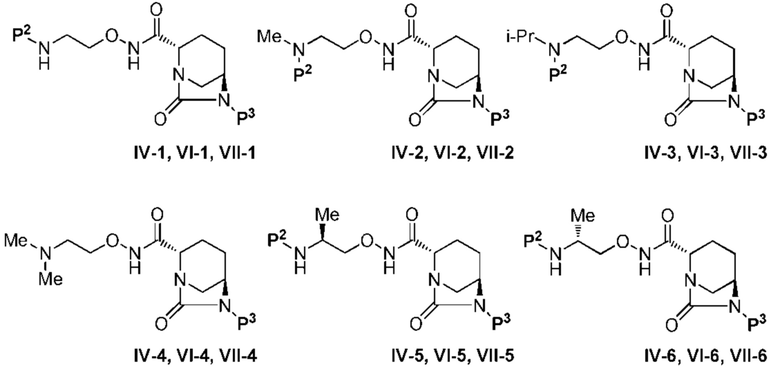
[0015] Кроме того, (6) другой аспект настоящего изобретения относится к способу получения, описанному в любом из пунктов (1)-(5) выше, где R3 в формулах (IV), (VI), (VII-CR) и (VII) выбран из
2-(трет-бутоксикарбониламино)этила;
2-аминоэтила;
2-((трет-бутоксикарбонил)(метил)амино)этила;
2-(метиламино)этила;
2-((трет-бутоксикарбонил)(изопропил)амино)этила;
2-(изопропиламино)этила;
2-(диметиламино)этила;
(2S)-2-((трет-бутоксикарбонил)амино)пропила;
(2S)-2-(амино)пропила;
(2R)-2-((трет-бутоксикарбонил)амино)пропила;
(2R)-2-(амино)пропила;
3-((трет-бутоксикарбонил)амино)пропила;
3-(амино)пропила;
(2S)-трет-бутоксикарбонилазитидин-2-илметила;
(2S)-азитидин-2-илметила;
(2R)-трет-бутоксикарбонилпирролидин-2-илметила;
(2R)-пирролидин-2-илметила;
(3R)-трет-бутоксикарбонилпиперидин-3-илметила;
(3R)-пиперидин-3-илметила;
(3S)-трет-бутоксикарбонилпирролидин-3-ила;
(3S)-пирролидин-3-ила;
1-(трет-бутоксикарбонил)азитидин-3-ила и
азитидин-3-ила.
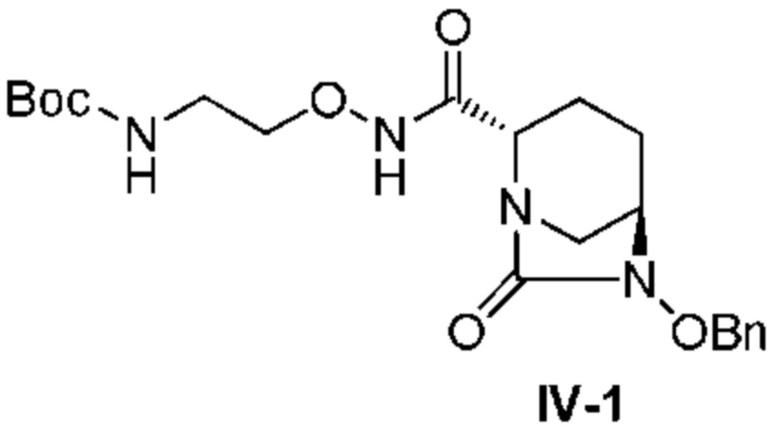
[0016] Кроме того, (7) другой аспект настоящего изобретения относится к способу получения соединения, представленного следующей формулой (VII-1):
[Химическая формула 16]
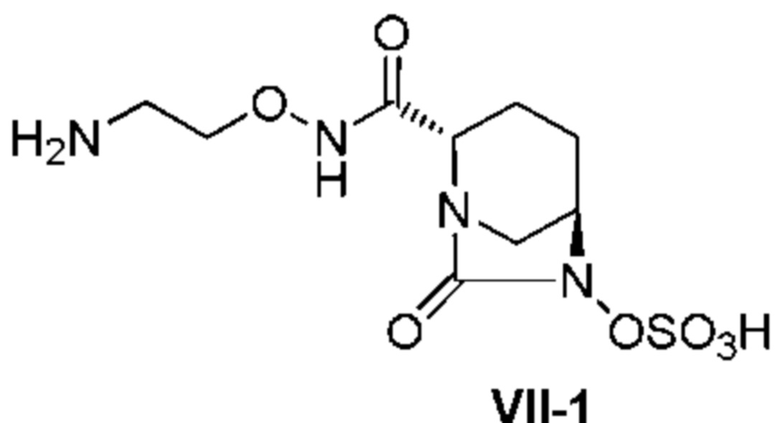 ,
,
включающему взаимодействие соединения, представленного следующей формулой (III):
[Химическая формула 17]
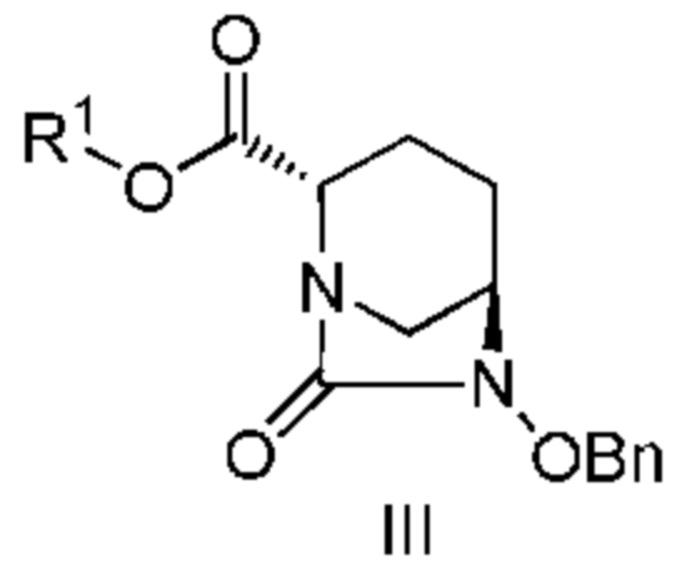
с трет-бутил 2-(аминоокси)этилкарбаматом в присутствии основания, с получением соединения, представленного следующей формулой (IV-1):
[Химическая формула 18]
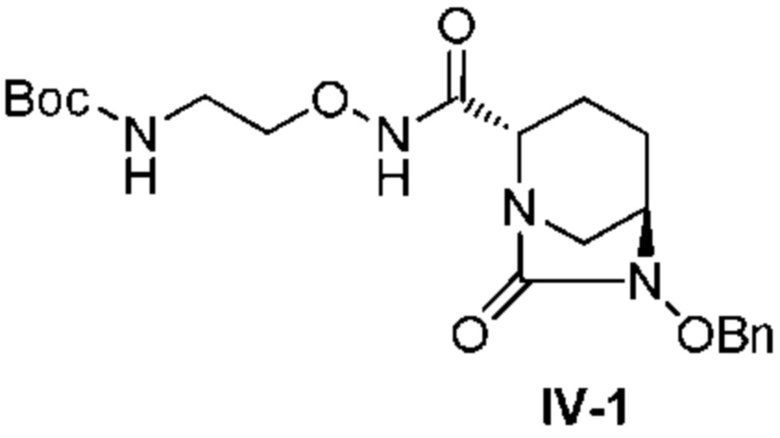 ,
,
с последующей обработкой катализатором палладий на угле в атмосфере водорода, при одновременном или последовательном проведении реакции сульфатирования с использованием катализатора триоксид серы-триметиламин в присутствии каталитического количества основания в водном растворителе, и обработку гидросульфатом тетрабутиламмония, с получением соединения, представленного формулой (VI-1):
[Химическая формула 19]
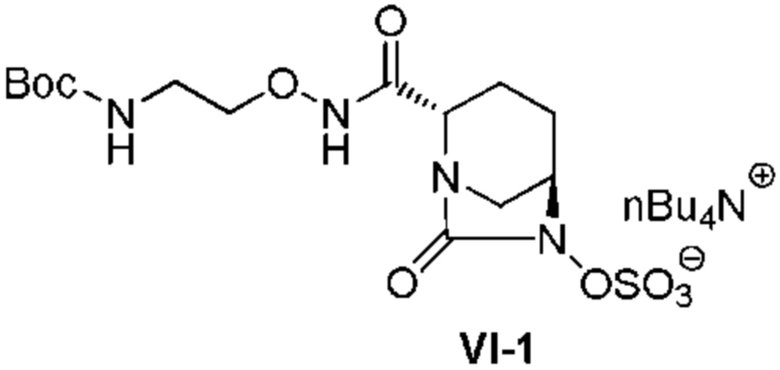 ,
,
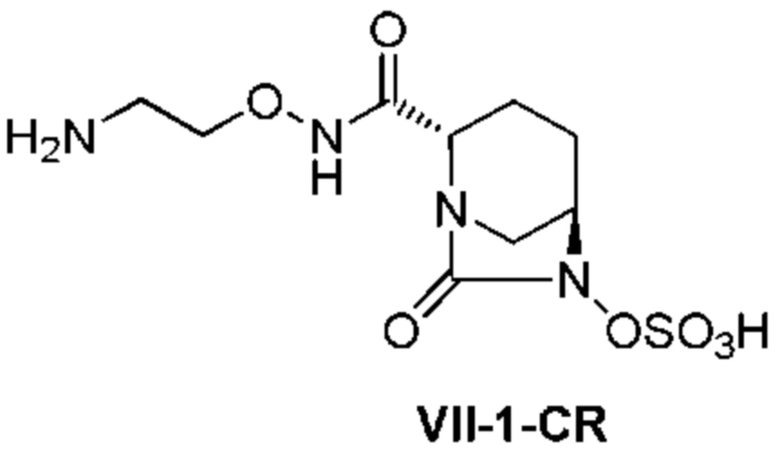
с последующим удалением трет-бутоксикарбонильной (Boc) группы с помощью трифторуксусной кислоты и добавлением по каплям этилацетата в реакционный раствор для осаждения неочищенного продукта и получения неочищенного соединения, представленного следующей формулой (VII-1-CR):
[Химическая формула 20]
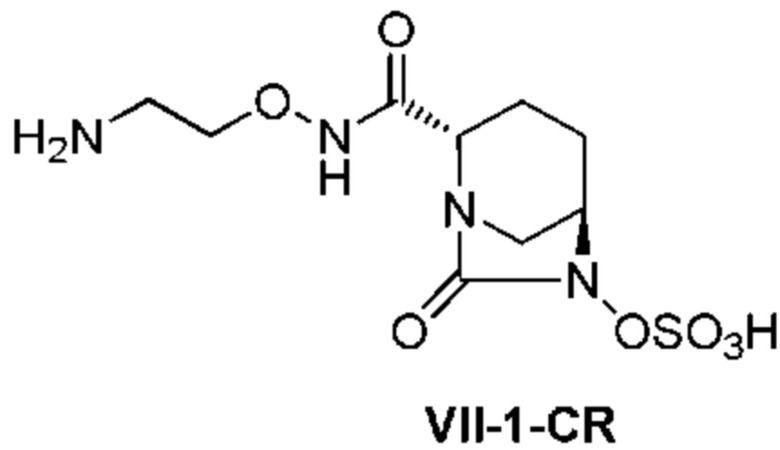
(в каждой из указанных выше формулах R1 и OBn являются такими, как описано выше),
с последующим поочередным добавлением неочищенного соединения, представленного формулой (VII-1-CR), и охлажденного льдом фосфатного буфера для получения раствора, имеющего pH от 4 до 5,5, концентрированием после обессоливания синтетическим адсорбентом, при необходимости, регулированием температуры, внесением затравки, при необходимости, и добавлением изопропанола для кристаллизации.
[0017] Кроме того, (8) другой аспект настоящего изобретения относится к способу получения соединения, представленного следующей формулой (IV-1):
[Химическая формула 21]
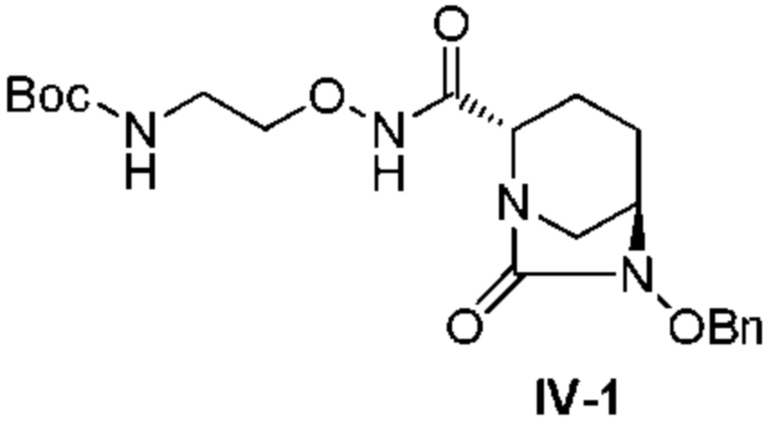 ,
,
включающему взаимодействие соединения, представленного следующей формулой (III):
[Химическая формула 22]
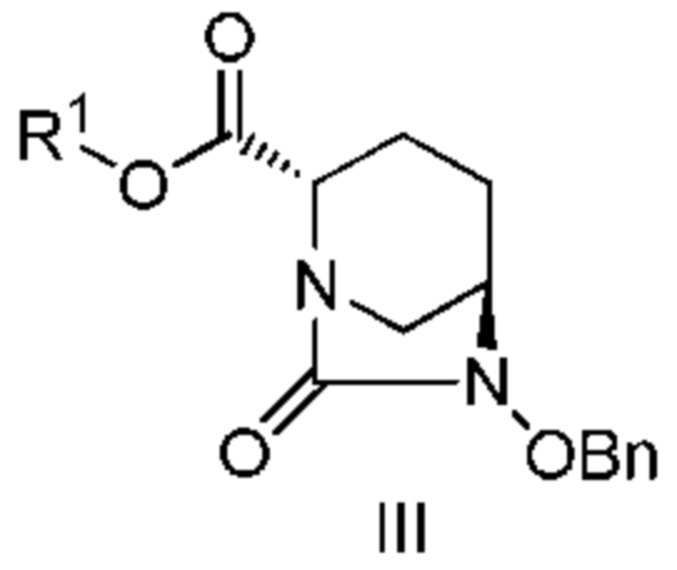
(в каждой из указанных выше формулах R1 и OBn являются такими, как описано выше)
с трет-бутил 2-(аминоокси)этилкарбаматом в присутствии основания.
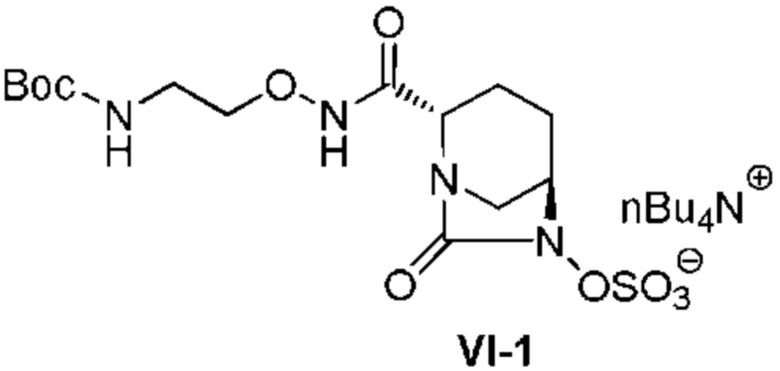
[0018] Кроме того, (9) другой аспект настоящего изобретения относится к способу получения соединения, представленного следующей формулой (VI-1):
[Химическая формула 23]
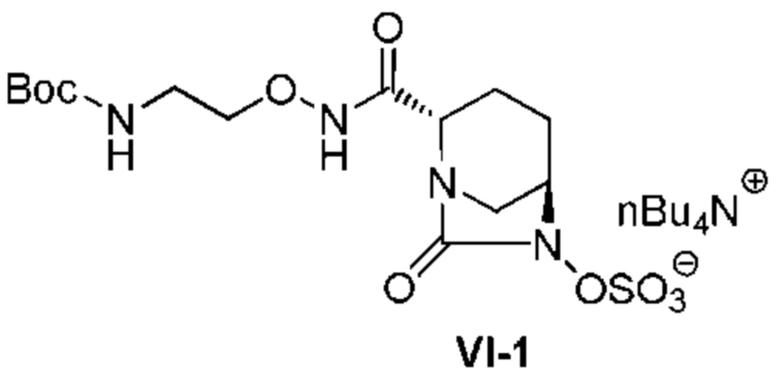 ,
,
включающему обработку соединения, представленного следующей формулой (IV-1):
[Химическая формула 24]
(в каждой из указанных выше формулах OBn является таким, как описано выше), катализатором палладий на угле в атмосфере водорода, при одновременном или последовательном проведении реакции сульфатирования с использованием комплекса триоксид серы-триметиламин в присутствии каталитического количества основания в водном растворителе, и обработку гидросульфатом тетрабутиламмония.
[0019] Кроме того, (10) другой аспект настоящего изобретения относится к способу получения неочищенного соединения, представленного следующей формулой (VII-1-CR):
[Химическая формула 25]
 ,
,
включающему удаление трет-бутоксикарбонильной (Boc) группы с помощью трифторуксусной кислоты из соединения, представленного следующей формулой (VI-1):
[Химическая формула 26]
 ,
,
с последующим добавлением по каплям этилацетата в реакционный раствор для осаждения неочищенного продукта.
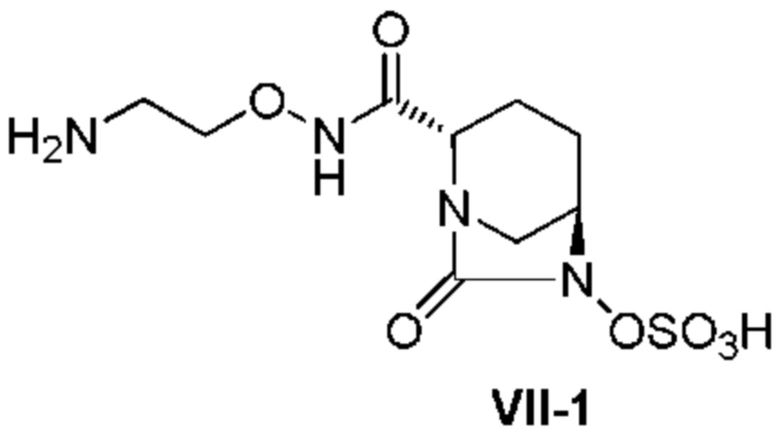
[0020] Кроме того, (11) другой аспект настоящего изобретения относится к способу получения соединения, представленного следующей формулой (VII-1):
[Химическая формула 27]
 ,
,
включающему поочередное добавление неочищенного соединения, представленного следующей формулой (VII-1-CR):
[Химическая формула 28]
и охлажденного льдом фосфатного буфера для получения раствора, имеющего pH от 4 до 5,5, концентрирование после обессоливания синтетическим адсорбентом, при необходимости, и регулирование температуры, с последующим внесением затравки, при необходимости, и добавлением изопропанола для кристаллизации.
[0021] Кроме того, (12) другой аспект настоящего изобретения относится к кристаллической форме I соединения, представленного формулой (VII-1):
[Химическая формула 29]
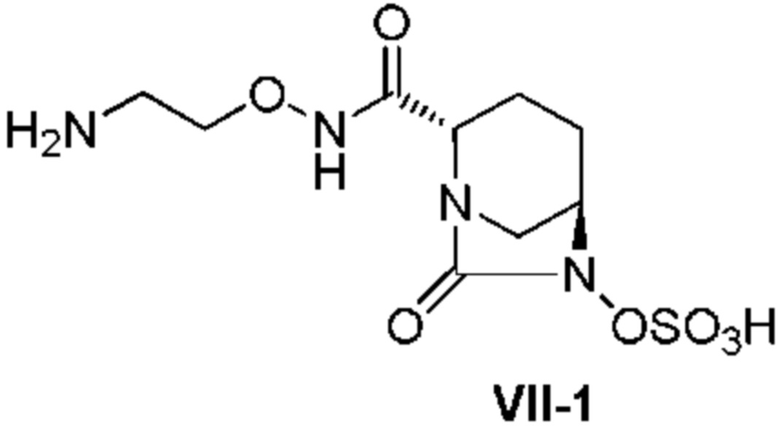 ,
,
имеющей характеристические пики, наблюдаемые при периоде кристаллической решетки (d) 7,34, 5,66, 5,53, 5,30, 5,02, 4,66, 4,37, 4,28, 4,06, 3,68, 3,62, 3,47, 3,36, 3,30, 3,16, 3,11, 3,03, 2,99 и 2,50 Å на диаграмме порошковой дифракции рентгеновских лучей.
[0022] Кроме того, (13) другой аспект настоящего изобретения относится к способу, описанному в любом из пунктов (1)-(11), получения кристаллической формы I, описанной в пункте (12).
[0023] Кроме того, (14) другой аспект настоящего изобретения относится к способу получения кристаллической формы I, описанной в пункте (12), включающему доведение температуры раствора соединения, представленного формулой (VII-1), до 20-25°C, внесение затравки кристаллической формы I и перемешивание, с последующим дополнительным добавлением изопропанола.
[0024] Кроме того, (15) другой аспект настоящего изобретения относится к применению кристаллической формы I, описанной в пункте (12), для получения фармацевтической композиции, необязательно содержащей фармацевтически приемлемый носитель.
[0025] Кроме того, (16) другой аспект настоящего изобретения относится к применению кристаллической формы I, описанной в пункте (12), для получения фармацевтической композиции, содержащей β-лактамовый антибиотик, выбранный из группы, состоящей из ампициллина, амоксициллина, пиперациллина, тикарциллина, фломоксефа, цефотаксима, цефтриаксона, цефтазидима, цефепима, цефтаролина, цефтолозана, имипенема, меропенема, биапенема, дорипенема, эртапенема и азтреонама, и необязательно фармацевтически приемлемый носитель.
[0026] Кроме того, (17) другой аспект настоящего изобретения относится к кристаллической форме II соединения, представленного формулой (VII-1):
[Химическая формула 30]
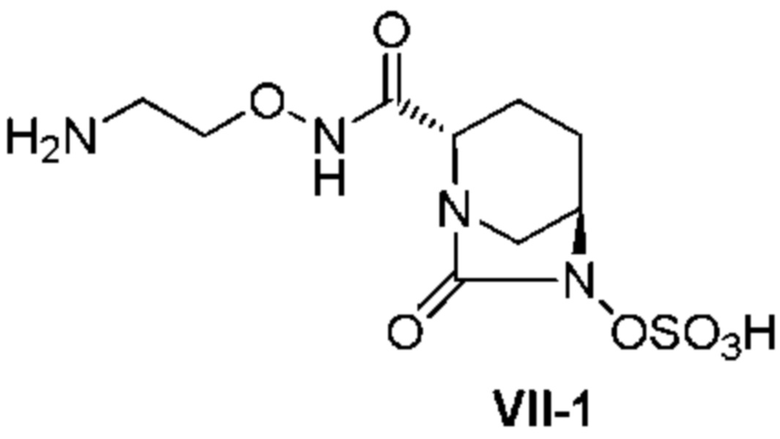 ,
,
имеющей характеристические пики, наблюдаемые при периоде кристаллической решетки (d) 9,46, 5,62, 5,23, 5,10, 5,00, 4,91, 4,67, 4,45, 4,29, 3,96, 3,78, 3,71, 3,52, 3,24, 3,18, 3,10, 3,02, 2,88, 2,81, 2,77, 2,67, 2,50 и 2,45 Å на диаграмме порошковой дифракции рентгеновских лучей.
[0027] Кроме того, (18) другой аспект настоящего изобретения относится к способу, описанному в любом из пунктов (1)-(11), получения кристаллической формы II, описанной в пункте (17).
[0028] Кроме того, (19) другой аспект настоящего изобретения относится к способу получения кристаллической формы II, описанной в пункте (17), включающему регулирование температуры раствора соединения, представленного формулой (VII-1), от 10 до 15°C, добавление изопропанола и перемешивание.
[0029] Кроме того, (20) другой аспект настоящего изобретения относится к применению кристаллической формы II, описанной в пункте (17), для получения фармацевтической композиции, необязательно содержащей фармацевтически приемлемый носитель.
[0030] Кроме того, (21) другой аспект настоящего изобретения относится к применению кристаллической формы II, описанной в пункте (17), для получения фармацевтической композиции, содержащей β-лактамовый антибиотик, выбранный из группы, состоящей из ампициллина, амоксициллина, пиперациллина, тикарциллина, фломоксефа, цефотаксима, цефтриаксона, цефтазидима, цефепима, цефтаролина, цефтолозана, имипенема, меропенема, биапенема, дорипенема, эртапенема и азтреонама, и необязательно фармацевтически приемлемый носитель.
[0031] Кроме того, (22) другой аспект настоящего изобретения относится к кристаллической форме III соединения, представленного формулой (VII-1):
[Химическая формула 31]
,
имеющей характеристические пики, наблюдаемые при периоде кристаллической решетки (d) 8,32, 6,10, 5,98, 5,51, 5,16, 5,07, 4,85, 4,70, 4,61, 4,35, 4,20, 4,06, 4,00, 3,95, 3,77, 3,73, 3,65, 3,42, 3,39, 3,36, 3,26, 3,23, 3,13, 3,09, 2,99, 2,81 и 2,52 Å на диаграмме порошковой дифракции рентгеновских лучей.
[0032] Кроме того, (23) другой аспект настоящего изобретения относится к способу, описанному в любом из пунктов (1)-(11), получения кристаллической формы III, описанной в пункте (22).
[0033] Кроме того, (24) другой аспект настоящего изобретения относится к способу получения кристаллической формы III, описанной в пункте (22), включающему доведение
температуры раствора соединения, представленного формулой (VII-1), до 20-25°C, внесение затравки кристаллической формы III, добавление изопропанола и перемешивание.
[0034] Кроме того, (25) другой аспект настоящего изобретения относится к применению кристаллической формы III, описанной в пункте (22), для получения фармацевтической композиции, необязательно содержащей фармацевтически приемлемый носитель.
[0035] Кроме того, (26) другой аспект настоящего изобретения относится к применению кристаллической формы III, описанной в пункте (22), для получения фармацевтической композиции, содержащей β-лактамовый антибиотик, выбранный из группы, состоящей из ампициллина, амоксициллина, пиперациллина, тикарциллина, фломоксефа, цефотаксима, цефтриаксона, цефтазидима, цефепима, цефтаролина, цефтолозана, имипенема, меропенема, биапенема, дорипенема, эртапенема и азтреонама, и необязательно содержащей фармацевтически приемлемый носитель.
[0036] Кроме того, (27) другой аспект настоящего изобретения относится к кристаллической форме IV соединения, представленного формулой (VII-1):
[Химическая формула 32]
,
имеющей характеристические пики, наблюдаемые при периоде кристаллической решетки (d) 7,88, 6,41, 5,20, 4,67, 4,50, 4,02, 3,81, 3,75, 3,70, 3,62, 3,38, 3,23, 3,20 и 2,74 Å на диаграмме порошковой дифракции рентгеновских лучей.
[0037] Кроме того, (28) другой аспект настоящего изобретения относится к способу, описанному в любом из пунктов (1)-(11), получения кристаллической формы IV, описанной в пункте (27).
[0038] Кроме того, (29) другой аспект настоящего изобретения относится к способу получения кристаллической формы IV, описанной в пункте (27), включающему доведение температуры раствора соединения, представленного формулой (VII-1), до 20-25°C, добавление метанола и перемешивание.
[0039] Кроме того, (30) другой аспект настоящего изобретения относится к способу получения кристаллической формы IV, описанной в пункте (27), включающему перемешивание кристаллической формы I, II или III, описанной в пункте (12), (17) или (22), в метаноле, этаноле или изопропаноле.
[0040] Кроме того, (31) другой аспект настоящего изобретения относится к применению кристаллической формы IV, описанной в пункте (27), для получения фармацевтической композиции, необязательно содержащей фармацевтически приемлемый носитель.
[0041] Кроме того, (32) другой аспект настоящего изобретения относится к применению кристаллической формы IV, описанной в пункте (27), для получения фармацевтической композиции, содержащей β-лактамовый антибиотик, выбранный из группы, состоящей из ампициллина, амоксициллина, пиперациллина, тикарциллина, фломоксефа, цефотаксима, цефтриаксона, цефтазидима, цефепима, цефтаролина, цефтолозана, имипенема, меропенема, биапенема, дорипенема, эртапенема и азтреонама, и необязательно содержащей фармацевтически приемлемый носитель.
[0042] Кроме того, (33) другой аспект настоящего изобретения относится к применению смеси кристаллических форм I, II, III или IV, описанных в пунктах (12), (17), (22) или (27), для получения фармацевтической композиции, необязательно содержащей фармацевтически приемлемый носитель.
[0043] Кроме того, (34) другой аспект настоящего изобретения относится к применению смеси кристаллических форм I, II, III или IV, описанных в пунктах (12), (17), (22) или (27), для получения фармацевтической композиции, содержащей β-лактамовый антибиотик, выбранный из группы, состоящей из ампициллина, амоксициллина, пиперациллина, тикарциллина, фломоксефа, цефотаксима, цефтриаксона, цефтазидима, цефепима, цефтаролина, цефтолозана, имипенема, меропенема, биапенема, дорипенема, эртапенема и азтреонама, и необязательно фармацевтически приемлемый носитель.
Эффекты настоящего изобретения
[0044] Согласно серии способов получения по настоящему изобретению, кристаллические формы соединения, представленного приведенной ниже формулой (VII), в частности соединение, представленное формулой (VII-1), и его кристаллические формы, обладающие благоприятной стабильностью, могут быть получены с хорошей воспроизводимостью и высоким выходом.
[0045] [Химическая формула 33]
Схема 2
[0046] Хотя существуют факторы, которые непосредственно влияют на кристаллизацию соединения, представленного формулой (VII), и вызывают разложение соединения, представленного формулой (VII), в ходе установления pH и перенос продуктов разложения с различных стадий на следующую стадию, разложение соединения, представленного формулой (VII), полностью контролируется, и раствор с высокой чистотой соединения, представленного формулой (VII), которое должно быть кристаллизовано, может быть получен с высоким выходом путем применения способов получения соединения, представленного формулой (VI), и соединения, представленного формулой (VII-CR), разработанного в настоящем изобретении, и дополнительное применение серии стадий для выделения, нейтрализации и обессоливания соединения, представленного формулой (VII-CR), содержащего кислоту.
[0047] Соединение, представленное приведенной ниже формулой (III), в частности соединение, в котором R1 представляет собой 2,5-диоксопирролидин-1-ил, 1,3-диоксо-3a,4,7,7a-тетрагидро-1H-изоиндол-2(3H)-ил, 1,3-диоксогексагидро-1H-изоиндол-2(3H)-ил или 3,5-диоксо-4-азатрицикло[5.2.1.02.6]дек-8-ен-4-ил, приводит к получению соединения, представленного формулой (IV), с высокой чистотой и высоким выходом, по сравнению с синтезом из приведенного выше соединения формулы (1).
[0048] Хотя выход соединения, представленного приведенной выше формулой (V), которое, как правило, является нестабильным в растворе, снижается при увеличении масштаба производства, формирование разложившихся продуктов во время концентрирования соединения, представленного формулой (V), уменьшение выхода, вызванное потерей при выделении, и загрязнение излишком реакционных продуктов на стадии сульфатирования предотвращается путем образования соединения, представленного формулой (VI), из соединения, представленного формулой (IV), при проведении реакции в одном сосуде без выделения или последовательных реакций. Хотя растворитель на основе спирта является наиболее предпочтительным для реакции дебензилирования соединения, представленного формулой (IV), комплекс триоксида серы-пиридина, который обычно используют на последующей стадии сульфатирования, дезактивируется в растворителях на основе спирта, и поэтому не может быть использован. Комплекс триоксида серы-триметиламина, обнаруженный для настоящего изобретения, проявляет великолепную стабильность в растворителях на основе спирта, и дает возможность осуществления реакции сульфатирования в одном сосуде без выделения или последовательных реакций.
[0049] На стадии получения соединения, представленного формулой (VII-CR), соединение представленное формулой (VII-CR), может быть получено с благоприятной воспроизводимостью в виде легко обрабатываемых твердых веществ, обладающих низкой гигроскопичностью и небольшим количеством продуктов разложения, путем осаждения слабым растворителем, таким как растворитель на основе сложного эфира или растворитель на основе простого эфира, имеющим низкую гигроскопичность, в частности универсальным этилацетатом из данного реакционного раствора. Соотношением загрязнения кислотными компонентами, оказывающих влияние на стадии нейтрализации, можно управлять в пределах допустимого диапазона для последующих стадий от 10 до 30% моль промывкой влажных твердых веществ, тем самым делая возможным поддерживать высокий уровень чистоты и демонстрировать посредством ВЭЖХ соотношение 99% или более.
[0050] Преобразование кристаллов в полиморфную форму у соединения, представленного формулой (VII-1), полученного в настоящем изобретении, не наблюдалось в тесте на стабильность, осуществленном при 40°C в твердом состоянии или в перемешиваемой суспензии в водном растворителе в течение длительного времени. Когда кристаллическую форму III соединения, представленного формулой (VII-1), подвергали эксперименту XRD-ДСК, нагревали до 160°C при 60% RH и затем давали охладиться при выстаивании до 63°C, кристаллическая форма изменялась до безводной кристаллической формы примерно при 145°C, и затем возвращалась к кристаллической форме III примерно при температуре ниже 90°C после охлаждения. На основе этих выводов, кристаллическая форма соединения, представленного формулой (VII-1), в частности кристаллическая форма III, представляет стабильную форму в обычных условиях.
[0051] Когда изменения с течением времени содержания воды, общего количества связанных веществ и содержания аморфной формы и кристаллических форм I, II, III и IV соединения, представленного формулой (VII-1), одновременно сопоставляли в условиях 40°C (75% RH), как показано в следующей таблице 1, в отличие от общего количества связанных веществ аморфной формы, составлявшего 0,5% в начале эксперимента, впоследствии количество значительно увеличилось до 6,6% после 1 месяца и до 12,3% после 3 месяцев, а также в отличие от содержания, составлявшего 99,4% в начале эксперимента, оно снизилось до 93,3% после 1 месяца и 87,5% после 3 месяцев. С другой стороны, в отличие от общего количества связанных веществ кристаллических форм I, II, III и IV, составлявшего 0,0-0,1% в начале эксперимента, количества были неизменными, демонстрируя 0,0% после 1 месяца и 0,0-0,5% после 3 месяцев, а также в отличие от содержания, составлявшего 99,8-99,9% в начале эксперимента, оно оставалось неизменным и стабильным, демонстрируя 99,8-100,0% после 1 месяца и 99,3-99,9% после 3 месяцев, а также в отличие от содержания воды в кристаллических формах I, II, III и IV, составлявшего 5,3-5,7% и 0,1% в начале эксперимента, оно оставалось неизменным и стабильным, демонстрируя 5,5-5,9% и 0,1% после 3 месяцев и 1 месяца.
[0052]
Условия хранения: 40°C/75% RH, герметичный контейнер
[0053] Кроме того, когда за прочным контактом кристаллической формы III в упаковочном контейнере для большого количества лекарственного средства наблюдали в течение длительного времени, как приведено в таблице 2 и таблице 3, в отличие от содержания, общего количества связанных веществ и содержания воды в начале эксперимента, составлявших 99,9%, 0,09% и 5,20%, соответственно, спустя 3 месяца при 40°C (75% RH) значения составляли 99,9%, 0,06% и 5,29%, соответственно, и спустя 1 месяц при 60°C значения составляли 99,9%, 0,04% и 5,08%, соответственно, указывая на то, что кристаллы были неизменными и сохраняли стабильность.
[0054]
Внутренняя упаковка: полиэтиленовый пакет низкой плотности, нейлоновая связывающая лента
Наружная упаковка: алюминиевый ламинированный пакет, запаянный
Условия хранения: 40°C/75% RH
[0055]
Внутренний пакет: полиэтиленовый пакет низкой плотности, нейлоновая связывающая лента
Наружная упаковка: алюминиевый ламинированный пакет, запаянный
Условия хранения: 60°C
Краткое описание фигур
[0056] На фиг.1 показана диаграмма порошковой рентгеновской дифракции кристаллической формы I.
На фиг.2 показана диаграмма порошковой рентгеновской дифракции кристаллической формы II.
На фиг.3 показана диаграмма порошковой рентгеновской дифракции кристаллической формы III.
На фиг.4 показана диаграмма порошковой рентгеновской дифракции кристаллической формы IV.
Способы осуществления настоящего изобретения
[0057] Как описано выше, настоящее изобретение обеспечивает высокую стабильность кристаллических форм соединения, представленного приведенной ниже формулой (VII), в частности соединения, представленного формулой (VII-1), и способы его получения.
[0058] [Химическая формула 34]

(в формуле (VII) выше, R3 представляет собой C1-6алкил или гетероциклил; R3 может быть модифицирован 0-5 R4, R4 может быть последовательно замещенным; при этом R4 представляет собой C1-6лкил, гетероциклил, R5(R6)N- или защитную группу; R5 и R6, каждый независимо, представляет собой водород или C1-6алкил или вместе образуют гетероциклил; кроме того, R3, R5 и R6 могут подвергаться закрытию кольца в произвольном положении.)
[0059] Ниже приводится подробное объяснение способа получения кристаллической формы соединения, представленного формулой (VII), по настоящему изобретению, но настоящее изобретение не ограничивается объемом приведенных конкретных их примеров.
[0060] "C1-6алкил" обозначает алкильную группу, имеющую 1-6 атомов углерода, которая может быть линейной, разветвленной или циклической.
[0061] "Гетероциклил" означает 3-7-членное моноциклическое гетероциклическое насыщенное кольцо или неароматическое кольцо, имеющее в сумме 1-3 гетероатома, выбранных из атома азота, атома кислорода и атома серы, в качестве составляющих кольца.
[0062] "R5(R6)N-" обозначает амино, а именно амино, моно-C1-6алкиламино или ди-C1-6алкиламино, замещенные R5 и R6, или гетероциклил, образованный R5 и R6 вместе с атомом азота.
[0063] "Модифицированный" означает водород в R3, замененный или присоединенный к R4.
[0064] "R3 может быть модифицирован 0-5 R4, R4 может быть последовательно замещенным", означает, что R4, который модифицирует R3, может быть дополнительно модифицирован R4, и его примеры включают R3-(R4)0-5, R3-(R4-R40-4), R3-(R4-R40-3)2, R3-(R4-R40-2)3 и R3-(R4-R40-1)4.
[0065] Конкретные примеры "защитной группы" включают защитные группы карбаматного типа и триалкилсилильные группы, которые являются аминозащитными группами и гидроксилзащитными группами, как описано в Protective Groups in Organic Synthesis (T. W. Greene et al., Wiley, New York (1999)), и их предпочтительные примеры включают триизопропилсилил (TIPS), трет-бутилдиметилсилил (TBDMS или TBS), трет-бутоксикарбонил (Boc), триметилсилилэтоксикарбонил (Teoc), 4-метоксибензилоксикарбонил (PMZ, Moz) и дифенилметоксикарбонил.
[0066] Конкретные примеры "C1-6алкила" включают линейные или разветвленные C1-6алкильные группы, такие как метильная, этильная, пропильная, изопропильная, бутильная, трет-бутильная, втор-бутильная, изобутильная, пентильная, 1,1-диметилпропильная, 1,2-диметилпропильная, неопентильная, 1-метилбутильная, 2-метилбутильная, изопентильная или гексильная группа; C3-6циклоалкильные группы, такие как циклопропильная, циклобутильная, циклопентильная или циклогексильная группа; и метильные группы, замещенные C3-5циклоалкильной группой, такой как циклопропилметильная, циклобутилметильная или циклопентилметильная группа, и предпочтительно включают метильную, этильную, пропильную, изопропильную, бутильную, трет-бутильную, циклопропильную, циклобутильную, циклопропилметильную и циклобутилметильную группы.
[0067] Конкретные примеры "гетероциклильных" групп включают азиридин, оксиран, трииран, азитидин, оксетан, тиэтан, пирролидин, тетрагидрофуран, тетрагидротиофен, имидазолидин, оксазолидин, тиазолидин, пиразолидин, пиперидин, тетрагидро-2H-пиран, тетрагидро-2H-тиопиран, гексагидропиридазин, пиперазин, морфорин, тиоморфорин, 1,2-оксазолидин, 1,3-оксазолидин, 1,2-оксазинан, 1,3-оксазинан, 1,4-диоксан, 1,2-тиазолидин, 1,3-тиазолидин, 1,2-тиазинан, 1,3-тиазинан, азепан, оксепан, тиепан, 1,4-диазепан, 1,4-оксазепан, 1,4-тиазепан, 1,2,5-триазепан, 1,4,5-оксадиазепан, 1,2,5-оксадиазепан, 1,4,5-тиадиазепан, 1,5,2-диоксазепан, 1,5,2-оксатиазепан, 3,4-дигидро-2H-пиррол, 4,5-дигидро-1H-пиразол, 4,5-дигидро-1H-имидазол, 4,5-дигидро-1,2-оксазол, 4,5-дигидро-1,3-оксазол, 4,5-дигидро-1,3-тиазол, 2,3,4,5-тетрагидропиридин, 1,2,3,6-тетрагидропиразин, 5,6-дигидро-4H-1,2-оксазин и 3,6-дигидро-2-H-1,4-оксазин, и предпочтительно включают азитидин, пирролидин, тетрагидрофуран, пиперидин, тетрагидро-2H-пиран, имидазолидин, 1,3-оксазолидин, 1,3-тиазолидин, гексагидропиридазин, пиперазин, морфорин, 1,2-оксазинан, азепан, 1,4-диазепан и 1,2-оксазепан. При этом само собой разумеется, что приведенные выше конкретные примеры включают такие группы, которые связываются с защитной группой, такой как трет-бутоксикарбонильная (Boc) группа.
[0068] Конкретные примеры "R5(R6)N-" включают амино, метиламино, этиламино, пропиламино, изопропиламино, бутиламино, трет-бутиламино, втор-бутиламино, изобутиламино, пентиламино, 1,1-диметилпропиламино, 1,2-диметилпропиламино, неопентиламино, 1-метилбутиламино, 2-метилбутиламино, изопентиламино, гексиламино, N,N-диметиламино, N,N-диэтиламино, N,N-дипропиламино, N,N-ди(изопропил)амино, N,N-дибутиламино, N,N-ди(трет-бутил)амино, N,N-ди(втор-бутил)амино, N,N-ди(изобутил)амино, N,N-дипентиламино, N,N-ди(1,1-диметилпропил)амино, N,N-ди(1,2-диметилпропил)амино, N,N-ди(неопентил)амино, N,N-ди(1-метилбутил)амино, N,N-ди(2-метилбутил)амино, N,N-ди(изопентил)амино и N,N-ди(гексил)амино, и предпочтительно включают амино, метиламино, этиламино, пропиламино, изопропиламино, N,N-диметиламино и N,N-диэтиламино. При этом само собой разумеется, что приведенные выше конкретные примеры включают такие группы, которые связываются с защитной группой, такой как трет-бутоксикарбонильная (Boc) группа.
[0069] Конкретные примеры групп, образующихся в случае, когда R5 и R6 группы R5(R6)N- соединяются с образованием гетероциклильной группы, включают азитидин-1-ильную, пирролидин-1-ильную, пиперидин-1-ильную и азепан-1-ильную группы. При этом само собой разумеется, что приведенные выше конкретные примеры включают такие группы, которые связываются с защитной группой, такой как трет-бутоксикарбонильная (Boc) группа.
"R3, R5 и R6 могут подвергаться закрытию кольца в произвольном положении" означает, что, в случае, когда R3 представляет собой C1-6алкил, и R4, который модифицирует R3, а именно R5 или R6, содержащиеся в R5(R6)N-, представляют собой C1-6алкил, R3 и R5 или R6 могут вместе образовывать 3-7-членное насыщенное кольцо.
[0070] В продолжение, хотя конкретные примеры соединений, образующихся в случае заместителя, определенного как R4, модифицирующего C1-6алкил или гетероциклил, который образует R3O-, разъясняются перечислением их даже более конкретных примеров, само собой разумеется, что такие соединения не ограничиваются объемом указанных конкретных примеров.
[0071] Конкретные примеры "C1-6алкила", модифицированного аминогруппой (H2N-), конкретного примера группы R5(R6)N-, включают 2-аминоэтил, 2-аминопропил, 3-аминопропил, 2-амино-1-метилэтил, 2-аминобутил, 3-аминобутил, 4-аминобутил, 2-амино-1,1-диметилэтил, 2-амино-1-метилпропил или 3-амино-2-метилпропил. При этом само собой разумеется, что приведенные выше конкретные примеры включают такие группы, которые связываются с защитной группой, такой как трет-бутоксикарбонильная (Boc) группа, содержащаяся в R5OCO-.
[0072] Конкретные примеры гетероциклила, модифицированного метильной группой, конкретного примера C1-6алкила, включают 1-метилазитидин, 3-метилазитидин, 1-метилпирролидин, 3-метилпирролидин, 1-метилимидазолидин, 3-метилоксазолидин, 1-метилпиразолидин, 1-метилпиперидин, 4-метилпиперидин, 2-метилтетрагидро-2H-пиран, 4-метилтетрагидро-2H-пиран, 1-метилпиперазин, 1,4-диметилпиперазин, 4-метилморфолин, 4-метилтиоморфолин, 1-метилазепан, 1-метил-1,4-диазепан и 1,4-диметил-1,4-диазепан. При этом само собой разумеется, что приведенные выше конкретные примеры включают такие группы, которые связываются с защитной группой, такой как трет-бутоксикарбонильная (Boc) группа.
[0073] Конкретные примеры гетероциклила, модифицированного аминогруппой (H2N-), конкретного примера R5(R6)N-, включают 3-аминоазитидин, 3-аминопирролидин, 3-аминотетрагидрофуран, 3-аминотетрагидротиофен, 4-аминопиразолидин, 4-аминопиперидин, 4-аминотетрагидро-2H-пиран, 4-аминотетрагидро-2H-тиопиран, 4-аминогексагидропиридазин, 4-амино-1,2-оксазолидин, 4-амино-1,2-оксазинан, 4-аминоазепан, 4-аминооксепан и 6-амино-1,4-диазепан. При этом само собой разумеется, что приведенные выше конкретные примеры включают такие группы, которые связываются с защитной группой, такой как трет-бутоксикарбонильная (Boc) группа.
[0074] Конкретные примеры гетероциклила, модифицированного метилом или этилом, конкретными примерами C1-6алкила, включают азитидин-2-илметил, азитидин-3-илметил, пирролидин-2-илметил, пирролидин-3-илметил, тетрагидрофуран-3-илметил, тетрагидротиофен-3-илметил, пиразолидин-4-илметил, 1,2-оксазолидин-3-илметил, пиперидин-2-илметил, пиперидин-3-илметил, пиперидин-4-илметил, тетрагидро-2H-пиран-4-илметил, тетрагидро-2H-тиопиран-4-илметил, гексагидропиридазин-4-илметил, пиперазин-2-илметил, 1,2-оксазинан-3-илметил, морфорин-2-илметил, морфорин-3-илметил, тиоморфорин-2-илметил, тиоморфорин-3-илметил, азепан-2-илметил, азепан-4-илметил, оксепан-2-илметил, оксепан-4-илметил, 1,4-диазепан-2-илметил, 1,4-диазепан-6-илметил, 2-(азитидин-1-ил)этил, 2-(пирролидин-1-ил)этил, 2-(пиразолидин-1-ил)этил, 2-(пиперидин-1-ил)этил, 2-(гексагидропиридазин-1-ил)этил, 2-(пиперазин-1-ил)этил, 2-(морфолин-4-ил)этил, 2-(тиоморфолин-4-ил)этил, 2-(1,2-оксазолидин-2-ил)этил, 2-(1,2-оксазинан-2-ил)этил, 2-(азепан-1-ил)этил или 2-(1,4-диазепан-1-ил)этил. При этом само собой разумеется, что приведенные выше конкретные примеры включают такие группы, которые связываются с защитной группой, такой как трет-бутоксикарбонильная (Boc) группа.
[0075] Конкретными примерами соединений, представленных химическими формулами (IV), (VI), (VII-CR) и (VII), по настоящему изобретению, являются соединения, выбранные из
трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата;
тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата;
(2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(метил)карбамата;
тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(метил)карбамата;
(2S,5R)-N-[2-(метиламино)этокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(пропан-2-ил)карбамата;
тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(пропан-2-ил)карбамата;
(2S,5R)-7-оксо-N-[2-(пропан-2-иламино)этокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
(2S,5R)-6-бензилокси-N-[2-(диметиламино)этокси]-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
тетрабутиламмоний (2S,5R)-N-[2-(диметиламино)этокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
(2S,5R)-N-[2-(диметиламино)этокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил{(2S)-1-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамата;
тетрабутиламмоний трет-бутил{(2S)-1-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамата;
(2S,5R)-N-{[(2S)-2-аминопропил]окси}-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил{(2R)-1-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамата;
тетрабутиламмоний трет-бутил{(2R)-1-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамата;
(2S,5R)-N-{[(2R)-2-аминопропил]окси}-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил{3-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропил}карбамата;
тетрабутиламмоний трет-бутил{3-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропил}карбамата;
(2S,5R)-N-(3-аминопропокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил (2S)-2-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилата;
тетрабутиламмоний трет-бутил (2S)-2-{[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилата;
(2S,5R)-N-[(2S)-азитидин-2-илметокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил (2R)-2-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пирролидин-1-карбоксилата;
тетрабутиламмоний трет-бутил (2R)-2-{[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пирролидин-1-карбоксилата;
(2S,5R)-7-оксо-N-[(2R)-пирролидин-2-илметокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил (3R)-3-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пиперидин-1-карбоксилата;
тетрабутиламмоний трет-бутил (3R)-3-{[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пиперидин-1-карбоксилата;
(2S,5R)-7-оксо-N-[(3R)-пиперидин-3-илметокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил (3S)-3-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пирролидин-1-карбоксилата;
тетрабутиламмоний трет-бутил (3S)-3-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пирролидин-1-карбоксилата;
(2S,5R)-7-оксо-N-[(3S)-пирролидин-3-илокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида;
трет-бутил 3-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилата;
тетрабутиламмоний трет-бутил 3-{[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилата и
(2S,5R)-N-(азитидин-3-илметокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида,
и включают следующую группу соединений:
[0076] [Химическая формула 35]
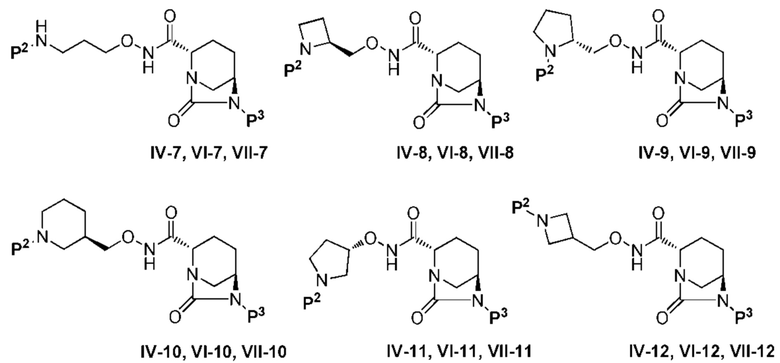
(где P2 является защитной группой, такой как трет-бутоксикарбонил (Boc), или водородом, и P3 представляет собой бензилокси (OBn), тетрабутиламмонийсульфокси или сульфокси).
[0077] Более предпочтительно, конкретные примеры включают
трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат;
тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат;
(2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид;
и они представлены приведенными выше формулами (IV-1), (VI-1), (VII-1-CR) и (VII-1).
[0078] Соединение по настоящему изобретению является ингибитором β-лактамазы и проявляет активность, которая ингибирует разложение β-лактамовых противомикробных средств этим ферментом. Таким образом, фармацевтический препарат, предоставленный настоящим изобретением, основан на сочетанном применении β-лактамового противомикробного средства.
Фармацевтический препарат, предоставленный настоящим изобретением, характеризуется содержанием веществ активных ингредиентов, выбранных из группы, состоящей из соединения, представленного формулой (VII), его фармацевтически приемлемой соли и их гидрата или сольвата, и хотя он может быть введен перорально или парентерально, предпочтительно его вводить парентерально. Соединение по настоящему изобретению и β-лактамовый антибиотик могут вводиться путем получения фармацевтической композиции способом, состоящим в сочетанном применении каждого фармацевтического средства, индивидуально полученного для применения, и введении одновременно или по отдельности, или путем предварительного смешивания обоих фармацевтических средств и обычно с использованием одной или двух или более препаративных добавок (носителей).
[0079] Конкретные примеры фармацевтических композиций для перорального введения включают таблетки, капсулы, гранулы, порошки, пилюли, водные и неводные пероральные растворы и суспензии.
[0080] Примеры путей введения для парентерального введения включают интраназальное введение, глазные капли, ушные капли, чрескожное введение, интратрахейное введение, ректальное введение, урологическое введение, подкожное введение, внутримышечное введение и внутривенное введение.
[0081] Конкретные примеры фармацевтических композиций для парентерального введения включают растворы для внутривенной инъекции при использовании фармацевтической композиции в порошковой форме с приемлемым растворителем для внутривенного введения. Примеры приемлемых растворителей включают стерильную воду для инъекций, физиологический солевой раствор, раствор глюкозы, раствор Рингера, стерильную воду для инъекций, содержащую метилпарагидроксибензоат или пропилпарагидроксибензоат, и стерильную воду для инъекций, содержащую бензиловый спирт.
[0082] Порошкообразную форму фармацевтической композиции для внутривенного введения получают путем диспергирования активного фармацевтического ингредиента в виде соединения по настоящему изобретению и β-лактамового антибиотика в запечатанном флаконе после прохождения стадии стерилизации.
[0083] При этом примеры β-лактамовых антибиотиков, которые могут сочетанно применяться с соединением по настоящему изобретению, включают пенициллиновые, цефемовые и карбапенемовые антибиотики.
[0084] Конкретные примеры пенициллина включают бензилпенициллин, фенетициллин, клоксациллин, диклоксациллин, ампициллин, циклациллин, амоксициллин, талампициллин, бекампициллин, ленампициллин, аспоксициллин, пиперациллин, сульбенициллин, пивмециллинам, сультамициллин, феноксиметилпенициллин, карбенициллин, азидоциллин, пропициллин, эпициллин, тикарциллин, пирбенициллин, азлоциллин, мезлоциллин и другие известные пенициллины.
[0085] Конкретные примеры цефема включают цефаклор, цефазолин, цефатризин, цефадроксил, цефапирин, цефамандол нафат, цефрадин, цефалексин, цефалотин, цефепим, цефокситин, цефиксим, цефзидим, цефдиторен, цефдинир, цефсулодин, цефселис, цефзопран, цефтаксим, цефтазидим, цефтаролин, цефтиам, цефтизоксим, цефтибутен, цефтезол, цефтетам, цефтриаксон, цефницид, цефпирамид, цефпиром, цефбуперазон, цефпрозил, цефперазон, цефподоксим, цефминокс, цефметазол, цефменоксим, цефрадин, цефроксадин, цефроксадин, цефтолозан (CXA101; гидросульфатная соль (6R,7R)-3-[5-амино-4-[3-(2-аминоэтил)уреид]-1-метил-1H-пиразол-2-ий-2-илметил]-7-[2-(5-амино-1,2,4-тиадиазол-3-ил)-2-[(Z)-1-карбокси-1-метилэтоксиимино]ацетамид]-3-цефем-4-карбоновой кислоты) и другие известные цефемы.
[0086] Примеры карбапенемовых антибиотиков включают имипенем, панипенем, меропенем, биапенем, дорипенем, эртапенем и тебипенем, и они могут быть сочетанно использованы с ингибиторами DHP-1, такими как циластанин натрия, при необходимости.
[0087] Примеры β-лактамовых антибиотиков, отличных от карбепенема, пенициллина и цефема, включают азтреонам, карумонам, латамоксеф, лоракарбеф, фаропенем и ритипенем.
[0088] Примеры таких пенициллинов, в частности, являющихся предпочтительными для сочетанного введения с соединением согласно настоящему изобретению, включают ампициллин, амоксициллин, карбенициллин, пиперациллин, азлоциллин, мезлоциллин и тикарциллин. Такие пенициллины могут быть использованы в форме фармацевтически приемлемой соли, например в виде натриевой соли. С другой стороны, ампициллин или амоксициллин может быть использован сочетанно с соединением, представленным формулой (VII), в форме суспензии для инъекций или цвиттерионных (ампициллина тригидрат или амоксициллина тригидрат) тонких гранул для использования в суспензии для инъекций. Примеры антибиотиков на основе цефема, в частности, предпочтительных для сочетанного введения с соединением согласно настоящему изобретению, включают цефотаксим, цефтриаксон, цефтазидим и цефепим, и они могут быть использованы в виде фармацевтически приемлемой соли, например в виде натриевой соли. Примеры карбапенемовых антибиотиков, в частности предпочтительных для сочетанного введения с соединением согласно настоящему изобретению, включают имипенем, меропенем, биапенем, дорипенем и эртапенем.
[0089] Примером β-лактамового антибиотика, отличного от карбапенемовых, пенициллиновых и цефемовых антибиотиков, которые в частности являются предпочтительными для сочетанного введения с соединением согласно настоящему изобретению, является азтреонам.
[0090] Ниже приводится последовательное объяснение способов получения соединения, представленного следующей формулой (VII), и его кристаллических форм, предоставленных настоящим изобретением.
[0091] [Химическая формула 36]
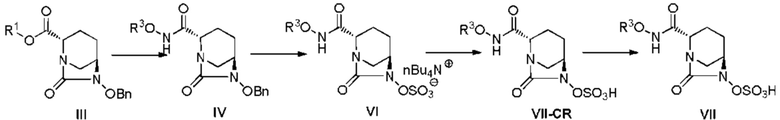
(в приведенных выше формулах (III), (IV), (VI), (VII-CR) и (VII), OBn, R1 и R3 являются такими, как описано выше.)
[0092] Стадию получения соединения, представленного формулой (IV), из соединения, представленного следующей формулой (III):
[Химическая формула 37]
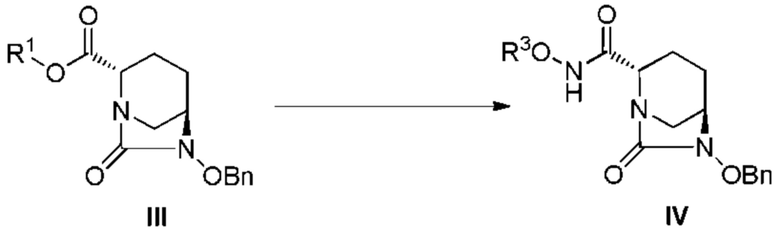
(где OBn, R1 и R3 являются такими, как описано выше) осуществляют по методике, описанной ниже.
[0093] Примеры используемого растворителя включают воду, метанол, этанол, изопропанол, этилацетат, тетрагидрофуран, диоксан, дихлорметан, хлороформ, 1,2-дихлорэтан и 2,2,2-трифторэтанол, предпочтительные примеры включают этилацетат, диоксан, дихлорметан, хлороформ и дихлорэтан, и такие растворители могут использоваться по отдельности или в виде смеси.
[0094] Соединение: R3ONH2, используемое в данной реакции, выбрано из таких соединений, у которых перечислены конкретные примеры R3, и используется в количестве в интервале 1-2 эквивалента, и предпочтительно 1-1,3 эквивалента, из расчета на соединение, представленное формулой (III).
[0095] Примеры основания, используемого в данной реакции, включают гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия, гидроксид калия, триэтиламин, диизопропилэтиламин, диметилбутиламин, трибутиламин, N-метилморфолин, пиридин, N-метилимидазол, 4-диметиламинопиридин, и использование триэтиламина является предпочтительным, и данное основание может быть использовано в виде водного раствора, в случае использования неорганического основания. Основание используют в количественном интервале от 0 до 2 эквивалентов, и предпочтительно от 0 до 1,5 эквивалентов, из расчета на соединение, представленное формулой (III). Температура реакции составляет интервал от -25 до 50°C и предпочтительно интервал от -10 до 10°C. Время реакции составляет интервал 1-24 часа и предпочтительно интервал 1-16 часов.
[0096] Соединение, представленное формулой (IV), может быть выделено после завершения реакции разбавлением реакционного раствора подходящим растворителем и последовательным промыванием водой, разбавленной кислотой и водным раствором основания (такими как разбавленная хлористоводородная кислота, гидросульфат калия, лимонная кислота и водный бикарбонат натрия или насыщенный раствор соли), с последующим концентрированием путем выпаривания растворителя. Примеры органических растворителей, используемых для разбавления, включают диэтиловый эфир, этилацетат, бутилацетат, толуол, дихлорметан и хлороформ, и этилацетат является предпочтительным. Хотя продукт выделяют обычно используемыми методами выделения и очистки, он может быть использован на следующей стадии только после соответствующей обработки.
[0097] Стадия преобразования соединения, представленного следующей формулой (IV):
[Химическая формула 38]
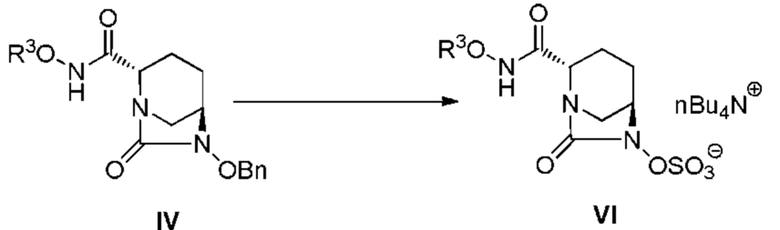
в соединение, представленное формулой (VI) выше, осуществляют по методике, описанной ниже.
[0098] Примеры растворителей, используемых в данной реакции, включают воду, метанол, этанол, изопропанол и ацетонитрил, и такие растворители могут использоваться по отдельности или в виде смеси. В случае осуществления реакции бензилирования и реакции сульфатирования одновременно, водный растворитель является предпочтительным, и в случае непрерывного проведения, воду предпочтительно добавляют в ходе сульфатирования. Количество добавляемой воды составляет интервал от 50 до 200% по объему, и предпочтительно 75-125% по объему растворителя.
[0099] Количество используемого палладия на угле составляет интервал от 5 до 100% по массе, и предпочтительно 5-30% по массе, из расчета на соединение, представленное формулой (IV).
Источником подаваемого водорода, используемого при гидрогенолизе, является газообразный водород, и давление водорода выбирают в интервале от атмосферного до 1 МПа и предпочтительно от атмосферного давления до 0,5 МПа. Количество подаваемого водорода составляет по меньшей мере суммарное количество, равное или большее, чем стехиометрическое.
[0100] Температура реакции гидрогенолиза составляет интервал от 10 до 50°C и предпочтительно интервал от 20 до 30°C. Время реакции составляет интервал от 0,5 до 3 часов и предпочтительно интервал от 0,5 до 2 часов.
[0101] Комплекс триоксида серы-триметиламина, используемый в качестве реагента сульфатирования, используют в количественном интервале 1-2 эквивалента, и предпочтительно 1-1,3 эквивалента, из расчета на соединение, представленное формулой (IV).
[0102] Примеры основания, используемого при сульфатировании, включают триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин и динатрийгидрофосфат, и предпочтительно используют триэтиламин, и данное основание может быть использовано в количественном интервале 0,1-1 эквивалент, и предпочтительно 0,1-0,3 эквивалента, из расчета на соединение, представленное формулой (IV).
[0103] Температура реакции сульфатирования составляет интервал от 0 до 50°C и предпочтительно интервал от 15 до 30°C. Время реакции составляет интервал от 12 до 48 часов и предпочтительно интервал от 12 до 24 часов.
[0104] После завершения реакции, соединение, представленное формулой (VI), может быть получено отфильтровыванием примесей, таких как кристаллические вещества, и добавлением концентрата, полученного при выпаривании растворителя, к водному раствору дигидрофосфата натрия, с последующим добавлением 1-3 молярных эквивалентов гидрофосфата тетрабутиламмония, экстракцией органическим растворителем, таким как этилацетат, и концентрированием путем выпаривания растворителя. Полученное в результате соединение, представленное формулой (VI), может быть использовано в виде концентрированного раствора на следующей стадия без выделения и очистки.
[0105] Стадия преобразования соединения, представленного следующей формулой (VI):
[Химическая формула 39]
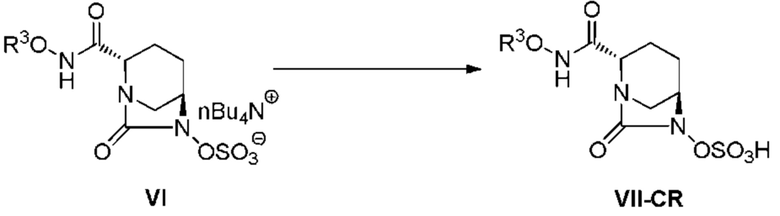
в соединение, представленное формулой (VII-CR) выше, осуществляют по методике, описанной ниже.
[0106] Примеры растворителей, используемых на данной стадии, для удаления защитной группы, необязательно содержащейся в R3O-NHC(=O)-, в частности трет-бутоксикарбонильной (Boc) группы, включают этилацетат, тетрагидрофуран, диоксан, дихлорметан, хлороформ, 1,2-дихлорэтан и 2,2,2-трифторэтанол, и предпочтительно используют дихлорметан или этилацетат, и данный растворитель используют по отдельности или в виде смеси. Количество используемого растворителя составляет интервал от 2 до 10 об./масс., и предпочтительно 2-6 об./масс., из расчета на чистый вес соединения, представленного формулой (VI).
[0107] Примеры кислот, используемых при снятии защиты, включают хлористоводородную кислоту, серную кислоту, фосфорную кислоту, муравьиную кислоту, трифторуксусную кислоту, метансульфоновую кислоту, трифторметансульфоновую кислоту, хлорметансульфоновую кислоту и тетрафторборную кислоту, и трифторуксусная кислота является предпочтительной. Данную кислоту используют в количественном интервале от 2 до 10 об./масс., и предпочтительно 2-6 об./масс., из расчета на чистый вес соединения, представленного формулой (VI). Данную кислоту добавляют в температурном интервале от -50 до 0°C и предпочтительно в интервале от -20 до 0°C. Температура реакции составляет интервал от -5 до 20°C и предпочтительно интервал от -5 до 5°C. Время реакции составляет интервал от 0,5 до 5 часов и предпочтительно интервал от 0,5 до 3 часов.
[0108] После завершения удаления защиты, стадию получения соединения, представленного формулой (VII-CR), путем добавления реакционного раствора и добавления слабого растворителя, осуществляют по методике, описанной ниже. Примеры используемого слабого растворителя включают слабые растворители на основе простых эфиров и слабые растворители на основе сложных эфиров, предпочтительные примеры включают слабые растворители на основе сложных эфиров, такие как этилацетат, изопропилацетат или бутилацетат, и этилацетат является даже более предпочтительным. Количество используемого слабого растворителя составляет 1-3-кратный и предпочтительно 1,5-2-кратный объем реакционного раствора. Слабый растворитель, предварительно охлажденный до -5 до 5°C, добавляют к полученному реакционному раствору отдельными порциями или по каплям. После завершения добавления слабого растворителя, реакционный раствор дополнительно перемешивают. Осажденные вещества отфильтровывают и промывают несколько раз, при необходимости, полученное в результате влажное твердое вещество подвергают вакуумной сушке и сушат в течение 10 часов или более, до тех пор, пока температура данного вещества не достигнет комнатной температуры, с получением соединения, представленного формулой (VII-CR).
[0109] Стадия преобразования соединения, представленного следующей формулой (VII-CR):
[Химическая формула 40]
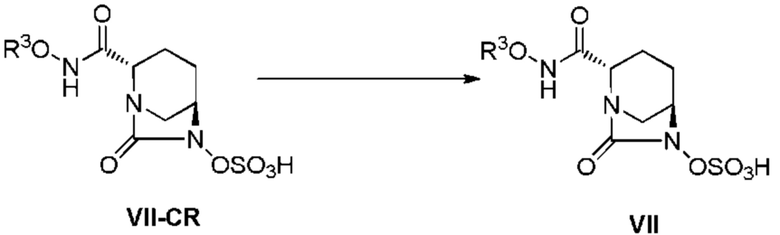
в соединение, представленное формулой (VII) выше, осуществляют по методике, описанной ниже.
[0110] Фосфатный буфер, имеющий pH 6,5, является предпочтительным буфером для использования при регулировании pH соединения, представленного приведенной выше формулой (VII-CR). Концентрацию буфера выбирают в интервале 0,1-0,5M, и предпочтительно она составляет 0,2M, хотя варьирует в зависимости от оставшейся трифторуксусной кислоты в соединении, представленном приведенной выше формулой (VII-CR). Общее количество используемого буфера составляет интервал от 10 до 50 об./масс., из расчета на чистый вес соединения, представленного формулой (VII-CR).
[0111] После предварительного охлаждения 5-7 об./масс. фосфатного буфера до 0-10°C, pH регулируют поочередным добавлением и растворением небольшой части соединения, представленного формулой (VII-CR), и так, чтобы pH охлажденного фосфатного буфера составлял 4-5,5 и предпочтительно 4,2-4,8, и в конечном итоге, был доведен до pH 4,6, с последующим разбавлением путем добавления воды до 25 об./масс., в случае общего количества, составляющего менее чем 25 об./масс., из расчета на чистый вес соединения, представленного формулой (VII-CR), и концентрируют при пониженном давлении до 20 об./масс. при температуре раствора 15°C или ниже. Кроме того, pH водного раствора доводят до 5,4 фосфатным буфером, с последующим разбавлением водой, с получением водного раствора, имеющего концентрацию 36 об./масс.
[0112] Обессоливание водного раствора после приведенного выше регулирования pH осуществляют, при необходимости, очисткой на колонке с использованием синтетического адсорбента. Примеры используемых синтетических адсорбентов включают Diaion HP-20 и SepaBeads SP-207, Mitsubishi Chemical, и предпочтительно используют SepaBeads SP-207. Синтетический адсорбент используют в интервале от 55 до 65 об./масс., из расчета на чистый вес соединения, представленного формулой (VII-CR). Обессоливание осуществляют путем абсорбции приведенного выше водного раствора на синтетический адсорбент, обессоливанием 65-70 об./масс. воды и элюированием 150-200 об./масс. 10% изопропанолом. Активная фракция содержится в интервале от 20 до 25 об./масс. Кристаллизацию осуществляют при использовании концентрата, полученного концентрированием полученной активной фракции, при пониженном давлении до 5-7 об./масс. и при температуре раствора 20°C или ниже.
[0113] Кристаллические полиморфы могут быть сольватированными или гидратированными (такими как ангидрид, моногидрат или дигидрат). Кристаллические формы I, II, III и IV существуют в виде кристаллических полиморфов соединения, представленного формулой (VII-1). Кристаллические формы I и III кристаллизуются при 0-35°C и предпочтительно при комнатной температуре от 20 до 25°C, более предпочтительно в результате внесения затравки затравочного кристалла, в то время как кристаллическая форма II может кристаллизоваться без присутствия затравочного кристалла в сверх насыщенных условиях путем охлаждения при 15°C или ниже и добавления слабого растворителя. Кристаллическая форма IV может кристаллизоваться при комнатной температуре с использованием растворителей на основе спирта, таких как метанол, который растворяет соединение, представленное формулой (VII-1), а также слабого растворителя, или может быть получена перекристаллизацией кристаллической формы I до формы III из спиртового растворителя в дегидратирующих условиях.
Кристаллические формы I, II и III отличаются по ДСК, растворимости в водном изопропаноле и размерам кристаллической решетки на диаграммах порошковой рентгеновской дифракции. Кристаллическая форма I обладает пониженной растворимостью в водном изопропаноле, в то время как кристаллические формы II и III имеют одинаковую растворимость.
[0114] Кристаллизацию соединения, представленного приведенной выше формулой (VII), осуществляют по методике, описанной ниже. Первоначальное количество химического раствора доводят до концентрации, которая дает возможность кристаллической форме, имеющей пониженную растворимость, адекватно раствориться. В случае соединения, представленного формулой (VII-1), первоначальное количество составляет интервал от 10% до 30% и предпочтительно от 10% до 20%. В случае кристаллической формы, для которой внесение затравки является предпочтительным, затравочные кристаллы получают заранее. Затравочные кристаллы кристаллической формы I получают при комнатной температуре, в то время как затравочные кристаллы кристаллической формы III могут быть получены без загрязнения путем последовательной кристаллизации в интервале от 15% до 25%. Количество используемых затравочных кристаллов составляет 0,01-20% и предпочтительно 0,01%-2%.
[0115] Примеры используемых слабых растворителей включают метанол, этанол, изопропанол, ацетон, ацетонитрил и тетрагидрофуран, и предпочтительно включают слабые растворители на основе спирта, такие как метанол, этанол или изопропанол. Количество слабого растворителя регулируется на основании растворимости, так что выделение составляет 1% или меньше. В случае соединения, представленного формулой (VII-1), слабый растворитель используют в 1-10-кратном и предпочтительно в 6-7-кратном объеме от первоначального объема данного раствора. Время добавления слабого растворителя является таким, что слабый растворитель по каплям вводят в полученную смесь, сформированную в суспензию, с последующим внесением затравки в случае кристаллической формы I, сразу после внесения затравки в случае кристаллических форм II и III, и без внесения затравки в случае кристаллической формы IV.
Кристаллическая форма IV также может быть получена суспендированием и перемешиванием кристаллической формы I, II или III в метаноле, этаноле или изопропаноле вместо их водного раствора.
[0116] Регулирование температуры раствора является важным фактором в условиях управления требуемым кристаллическим полимофизмом, и оно обусловлено скоростью осаждения полиморфных кристаллов при заданной температуре. В случае соединения, представленного формулой (VII-1), кристаллические формы I, III и IV поддерживают в температурном интервале от 20 до 25°C, в то время как кристаллическую форму II поддерживают при температуре 15°C или ниже. Кроме того, в случае кристаллической формы IV, полученной суспендированием и перемешиванием кристаллической формы I, II или III в растворителе, используемом вместо их водного раствора, в зависимости от растворимости кристаллической формы I, II или III в данном растворителе, ее регулируют при температуре в диапазоне от 20 до 100°C и предпочтительно в интервале от 20 до 65°C.
Время перемешивания зависит от скорости осаждения, и перемешивание осуществляют в течение 1-24 часов и предпочтительно в течение 1-15 часов.
Кристаллические формы соединения, представленного формулой (VII), могут быть получены путем обычного фильтрования, промывания и вакуумной сушки или проточной сушки осажденных кристаллов. В случае сольватированных кристаллов, пересушивания можно избежать при использовании средств, регулирующих температуру материала, потери при высушивании, увлажняющих и ограничивающих вакуумную сушку средств, или увлажненной проточной сушки.
ПРИМЕРЫ
[0117] Настоящее изобретение будет описано более подробно посредством примеров, но настоящее изобретение не предназначено для ограничения этими примерами, и могут быть произведены различные модификации.
[0118] Ссылочный пример 1
Дигидрохлорид метил (2S,5R)-5-(бензилоксиамино)пиперидин-2-карбоксилата
Стадия 1
Метил (2S,5S)-5-гидроксипиперидин-2-карбоксилат
К 2M раствору хлористого водорода в метаноле (12,8 л) добавляли коммерчески доступную (2S,5S)-5-гидроксипиперидин-2-карбоновую кислоту (по данным ВЭЖХ содержание 84%, чистый выход 912,22 г, промытая 3,1 л 2M хлористого водорода в метаноле), с последующим кипячением с обратным холодильником в течение 3 часов (внутренние температуры 63-67°C). Затем реакционный раствор охлаждали, добавляли 1,4-диоксан (12,8 л), и растворитель отгоняли при пониженном давлении. К полученному остатку (4,1 кг) добавляли этилацетат (18,3 л) и охлажденный льдом 44% водный раствор карбоната калия (23,7 л) до отделения органического слоя, и водный слой дополнительно экстрагировали этилацетатом (3×18,3 л). Каждый из органических слоев промывали 50% водным раствором карбоната калия (7,3 л). Органические слои объединяли, сушили над безводным карбонатом калия (2,37 кг) и фильтровали, и растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в толуоле (9,1 л), и добавляли 9,2 г активированного угля, с последующим перемешиванием в течение 30 минут и фильтрованием. Растворитель затем отгоняли при пониженном давлении. Растворитель полученного остатка заменяли этилацетатом (9,1 л), с получением 1130 г указанного в заголовке соединения в виде бледно-желтого масла (по данным ВЭЖХ содержание 78,9%, чистый выход 891,57 г, выход 89%).
[0119] Стадия 2
Метил (2S,5S)-5-гидрокси-1-(2,2,2-трифторацетил)пиперидин-2-карбоксилат
Дегидратированный этилацетатный раствор (7,4 л) метил (2S,5S)-5-гидроксипиперидин-2-карбоксилата (по данным ВЭЖХ содержание 78,8%, чистый выход 459,48 г) охлаждали до -40°C, с последующим добавлением триэтиламина (1300 г), и затем по каплям добавляли трифторуксусный ангидрид (1349 г, промытый 100 мл дегидратированного этилацетата) при (-40)-(-12)°C в течение 30 минут. После завершения добавления по каплям, температуру повышали до -2°C за 15 минут, и полученную смесь перемешивали в течение 75 минут, и к полученной смеси дополнительно добавляли воду (1277 мл), с последующим перемешиванием при 25°C в течение 1 часа. Полученную смесь выливали в воду (8,4 л) (промывали 4,5 л этилацетата) и затем экстрагировали этилацетатом (2×9,8 л), и объединенный органический слой последовательно промывали 1M хлористоводородной кислотой (8,5 л), насыщенным бикарбонатом натрия (8,5 л) и насыщенным раствором соли (8,5 л), сушили над безводным сульфатом натрия (1,8 кг) и фильтровали. Растворитель органического слоя отгоняли при пониженном давлении, и к полученному остатку добавляли этилацетат (3,6 л), с последующим замещающим концентрированием. Полученный остаток затем сушили в вакууме, с получением 793,4 г указанного в заголовке соединения (ВЭЖХ содержание 81,5%, чистый выход 648,66 г, выход 88%).
[0120] Стадия 3
Метил (2S,5R)-5-(бензилоксиамино)-1-(2,2,2-трифторацетил)пиперидин-2-карбоксилат
4,0 л раствора метил (2S,5S)-5-гидрокси-1-(2,2,2-трифторацетил)пиперидин-2-карбоксилата (ВЭЖХ содержание 81,5%, чистый выход 556,23 г) в дегидратированном ацетонитриле охлаждали до -40°C и добавляли 2,6-лютидин (259,24 г) (промытый 100 мл ацетонитрила), с последующим добавлением по каплям трифторметансульфонового ангидрида (645,72 г) при (-43)-(-37)°C в течение 1 часа и 10 минут (промытый 100 мл ацетонитрила). Реакционный раствор перемешивали при -35°C в течение 50 минут, и затем по каплям добавляли бензилоксиамин (550,27 г) при -35°C или ниже, с последующим промыванием ацетонитрилом (500 мл). После градиентного повышения температуры реакционного раствора до -5°C, добавляли 2,6-лютидин (259,24 г), с последующим перемешиванием при 5°C в течение 40 часов. После концентрирования до 1,8 л, полученную смесь разбавляли этилацетатом (12,4 л) и последовательно промывали водой (12,4 л), 10% лимонной кислотой (4×8 л+4,7 л), насыщенным бикарбонатом натрия (6,3 л) насыщенным раствором соли (7,2 л). Органический слой сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. Полученный остаток сушили в вакууме с получением 867,73 г указанного в заголовке соединения (ВЭЖХ содержание 71,56%, выход 79%).
[0121] Стадия 4
Гидрохлорид метил (2S,5R)-5-(бензилоксиамино)-1-(2,2,2-трифторацетил)пиперидин-2-карбоксилата
Метил (2S,5R)-5-(бензилоксиамино)-1-(2,2,2-трифторацетил)пиперидин-2-карбоксилат (ВЭЖХ содержание 70,13%, чистый выход 673,20 г) разбавляли этилацетатом (4,8 л) и добавляли 48 г активированного угля, с последующим перемешиванием в течение 1 часа. Полученную смесь фильтровали и промывали 2 л этилацетата. Фильтрат разбавляли 4,7 л этилацетата, и добавляли 1М раствор хлористого водорода в этилацетате (2,7 л) при комнатной температуре, с последующим перемешиванием в течение 15 минут, и затем добавляли 28,6 л гексана, с последующим охлаждением до 0°C. После перемешивания в течение 3 часов, кристаллическое твердое вещество отфильтровывали, промывали смесью гексан/этилацетат=4/1 (3 л) и сушили в вакууме, с получением 724,0 г указанного в заголовке соединения (ВЭЖХ содержание 91,72%, выход 90%).
[0122] Стадия 5
Дигидрохлорид метил (2S,5R)-5-(бензилоксиамино)пиперидин-2-карбоксилата
Гидрохлорид метил (2S,5R)-5-(бензилоксиамино)-1-(2,2,2-трифторацетил)пиперидин-2-карбоксилата (ВЭЖХ содержание 92,01%, чистый выход 732,25 г) растворяли в 2М метанольном растворе хлористого водорода (15 л), с последующим нагреванием при кипячении с обратным холодильником в течение 27 часов. Полученную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении до 3 л. Полученную смесь разбавляли 2,7 л метанола, и затем добавляли 16,3 л этилацетата, с последующим перемешиванием в течение 1 часа. Выпавшее в осадок твердое кристаллическое вещество отфильтровывали, промывали этилацетатом (3×1,1 л) и сушили в вакууме, с получением 572,0 г указанного в заголовке соединения (ВЭЖХ содержание 98,06%, выход 92%).
1H ЯМР (400 МГц, D2O) δ 1,40-1,51 (м, 1H), 1,61-1,72 (м, 1H), 1,90-1,94 (м, 1H), 2,25-2,30 (м, 1H), 2,80 (т, J=11,2 Гц, 1H), 3,19-3,27 (м, 1H), 3,51-3,55 (м, 1H), 3,66 (с, 3H), 3,87-3,91 (м, 1H), 4,68 (с, 2H), 7,27 (с, 5H); МС m/z 265 [M-2HCl+H]+.
[0123] Ссылочный пример 2
(2S,5R)-6-(Бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота
Стадия 1
Метил (2S,5R)-5-(бензилоксиамино)пиперидин-2-карбоксилат
К дигидрохлориду метил (2S,5R)-5-(бензилоксиамино)пиперидин-2-карбоксилата (ссылочный пример 1, 1,319 г) добавляли этилацетат (20 мл) и 50% карбонат калия (20 мл) для разделения слоев. Водный слой экстрагировали три раза этилацетатом (15 мл). Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали, концентрировали при пониженном давлении и затем сушили в вакууме в течение ночи, с получением 975 мг указанного в заголовке соединения (выход 94%).
1H ЯМР (400 МГц, CDCl3) δ 1,25-1,35 (м, 1H), 1,49-1,59 (м, 1H), 1,89-2,11 (м, 2H), 2,45 (т, J=11,7 Гц, 1H), 2,96-3,03 (м, 1H), 3,28-3,39 (м, 2H), 3,72 (с, 3H), 4,68 (с, 2H), 7,26-7,35 (м, 5H); МС m/z 265 [M+H]+.
[0124] Стадия 2
Метил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
К метил (2S,5R)-5-(бензилоксиамино)пиперидин-2-карбоксилату (1,154 г, 4,37 ммоль) добавляли дегидратированный ацетонитрил (198 мл), с последующим охлаждением льдом. При 5°C или ниже триэтиламин (1,60 мл) и дифосген (0,389 мл) последовательно добавляли по каплям, с последующим перемешиванием при 2°C в течение 20 минут. К полученному реакционному раствору затем добавляли 4-диметиламинопиридин (70,0 мг), с последующим перемешиванием при комнатной температуре в течение 10 часов. Реакционный раствор концентрировали при пониженном давлении и заменой растворителя три раза на этилацетат, и полученный раствор концентрировали до 30 мл. К полученному добавляли этилацетат (20 мл) и воду (40 мл) для разделения слоев. Отделенный водный слой экстрагировали дважды этилацетатом (30 мл). Объединенный органический слой последовательно промывали 5% лимонной кислотой (40 мл), 6,5% бикарбонатом натрия (30 мл) и 5% насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. 1,16 г Полученного в результате остатка разбавляли этилацетатом (5,5 мл), добавляли н-гексан (11 мл) и вносили затравочные кристаллы для затравки и кристаллизовали. Дополнительно добавляли н-гексан (49 мл) и перемешивали при 0°C в течение 1 часа, и затем твердое кристаллическое вещество отфильтровывали, промывали н-гексаном (60 мл) и сушили в вакууме, с получением 882,3 мг указанного в заголовке соединения в виде бесцветного кристаллического порошка (выход 71%).
1H ЯМР (400 МГц, CDCl3) δ 1,65-1,70 (м, 1H), 2,03-2,12 (м, 3H), 2,90 (д, J=12,0 Гц, 1H), 3,07 (м, 1H), 3,32 (м, 1H), 4,12 (дд, J=4,6 и 4,4 Гц, 1H), 4,91 (д, J=11,2 Гц, 1H), 5,06 (д, J=11,2 Гц, 1H), 7,35-7,44 (м, 5H); МС m/z 291 [M+H]+.
[0125] Стадия 3
(2S,5R)-6-(Бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота
К метил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилату (809,0 мг, 2,79 ммоль) добавляли тетрагидрофуран (8 мл) и воду (3,6 мл), с последующим добавлением по каплям 0,5M гидроксида лития (6,41 мл) при 4,9°C или ниже в течение 10 минут. После перемешивания реакционного раствора при 2°C в течение 2 часов, добавляли воду (30 мл), с последующим промыванием этилацетатом (25 мл). В отделенный водный слой добавляли этилацетат (15 мл), и pH доводили до 4,0 1М хлористоводородной кислотой, с последующей экстракцией дважды этилацетатом (этилацетат: 65 мл в общей сложности). Отделенный водный слой доводили до pH 3,4 1М хлористоводородной кислотой, один раз экстрагировали этилацетатом, и затем водный слой доводили до pH 2,4 и дважды экстрагировали этилацетатом. Этилацетатный экстракт, проэкстрагированный в общей сложности пять раз (175 мл), промывали насыщенным раствором соли (40 мл), сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. 759,1 мг полученного в результате остатка разбавляли этилацетатом (5 мл), добавляли н-гексан (3 мл), вносили затравочные кристаллы для затравки и кристаллизовали. Дополнительно добавляли раствор этилацетат/н-гексан (5/3) (8 мл) и перемешивали, и затем добавляли н-гексан (20 мл), с последующим перемешиванием при 4°C в течение 14 часов. Твердое кристаллическое вещество отфильтровывали, промывали н-гексаном (55 мл) и затем сушили в вакууме, с получением 633,6 мг указанного в заголовке соединения в виде бесцветного кристаллического порошка (выход 82%).
1H ЯМР (400 МГц, CDCl3) δ 1,67 (м, 1H), 2,04-2,26 (м, 3H), 2,85 (д, J=12,0 Гц, 1H), 3,13 (м, 1H), 3,35 (м, 1H), 4,12 (м, 1H), 4,91 (д, J=11,3 Гц, 1H), 5,06 (д, J=11,3 Гц, 1H), 7,37-7,44 (м, 5H); МС m/z 277 [M+H]+.
[0126] Ссылочный пример 3
2,5-Диоксопирролидин-1-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
[Химическая формула 41]
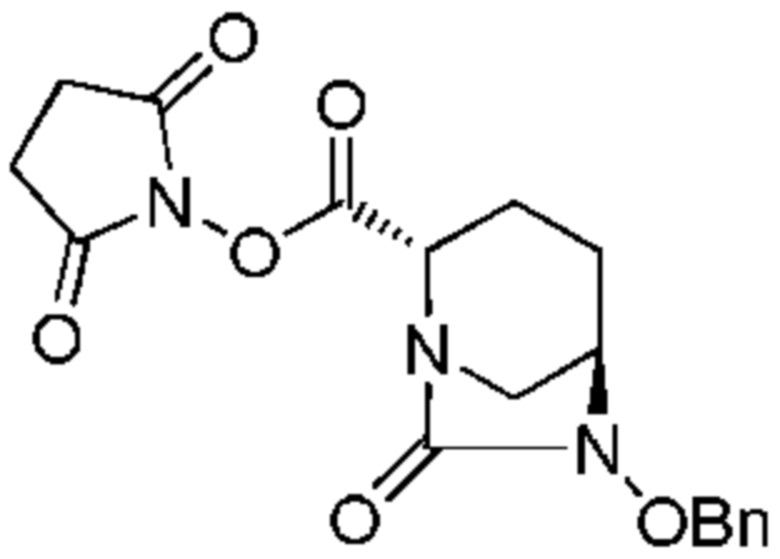
Стадия 1
(2S,5R)-5-((Бензилокси)амино)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновая кислота
Дигидрохлорид метил (2S,5R)-5-(бензилоксиамино)пиперидин-2-карбоксилата (ссылочный пример 1, 65,4 г, 200 ммоль) растворяли в воде (400 мл) и 1,4-диоксане (270 мл), и полученный раствор охлаждали льдом. Затем добавляли 5M гидроксид натрия (132 мл), с последующим перемешиванием в течение 1 часа. К полученному реакционному раствору добавляли 5M хлористоводородную кислоту (12 мл), карбонат калия (27,6 г) и ди-трет-бутилдикарбонат (48 г), и температуру полученной смеси поднимали до комнатной температуры, с последующим перемешиванием в течение ночи. Концентрированный раствор реакционной смеси промывали этилацетатом, доводили до pH 3,3 моногидратом лимонной кислоты, дважды экстрагировали этилацетатом (500 мл), промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и фильтровали, и растворитель концентрировали при пониженном давлении и замене растворителя этилацетатом, с получением 68,7 г указанного в заголовке соединения (количественный). Соединение использовали на следующей стадии без очистки. Часть полученного соединения кристаллизовали из смеси этилацетат/гексан для подтверждения его структуры.
1H ЯМР (400 МГц, CDCl3) δ 1,46 (с, 9H), 1,50-1,72 (м, 2H), 1,98-2,10 (м, 2H), 3,12-3,19 (м, 2H), 4,13-4,20 (м, 1H), 4,76 (д, J=11,5 Hz), 4,70 (д, J=11,5 Hz), 4,85-4,92 (м, 1H), 7,26-7,35 (м, 5H); МС m/z 351 [M+H]+.
[0127] Стадия 2
2,5-Диоксопирролидин-1-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
(2S,5R)-5-((Бензилокси)амино)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновую кислоту (ссылочный пример 3, стадия 1, 700 мг, 2 ммоль) растворяли в дегидратированном тетрагидрофуране (10 мл) и охлаждали до -20°C. К полученной смеси последовательно по каплям добавляли изобутилхлорформиат (300 мг) и триэтиламин (444 мг), с последующим перемешиванием в течение 15 минут. К полученному реакционному раствору добавляли 1-гидроксипирролидин-2,5-дион (253 мг), с последующим перемешиванием в течение 30 минут и дополнительном перемешивании при комнатной температуре в течение 30 минут. Реакционный раствор разбавляли этилацетатом (35 мл), последовательно промывали 10% лимонной кислотой (10 мл), насыщенным бикарбонатом натрия (10 мл) и насыщенным раствором соли (10 мл), сушили над безводным сульфатом магния и фильтровали, и растворитель отгоняли при пониженном давлении, с получением 985 мг остатка. Общее количество полученного остатка растворяли в дегидратированном хлороформе (10 мл), и в полученный раствор добавляли триэтиламин (303 мг), с последующим охлаждением льдом. К полученной смеси добавляли трифосген (237 мг), с последующим перемешиванием в течение 30 минут. К полученному затем добавляли метанол (0,1 мл), с последующим перемешиванием в течение 30 минут. Затем по каплям добавляли раствор метансульфоновой кислоты (1,3 мл) в дихлорметане (4,0 мл) и дополнительно перемешивали в течение 30 минут. Полученную смесь по каплям добавляли к охлажденному льдом 1М гидрокарбонату калия (2,4 г/20 мл), с последующим перемешиванием в течение 30 минут. Затем добавляли хлороформ (10 мл) для разделения слоев. Органический слой последовательно промывали 1М хлористоводородной кислотой (10 мл), насыщенным бикарбонатом натрия (10 мл) и насыщенным раствором соли (10 мл). Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток затравляли кристаллами, и к полученному твердому веществу добавляли смесь гексан/этилацетат (1/2, 3 мл). Полученную смесь затем перемешивали, фильтровали и последовательно промывали смесью гексан/этилацетат (1/1, 3 мл) и гексаном (3 мл), с получением 556 мг указанного в заголовке соединения в виде кристаллов (выход 75%).
1H ЯМР (400 МГц, CDCl3) δ 1,70-1,77 (м, 1H), 2,04-2,27 (м, 3H), 2,80-2,90 (м, 4H), 3,09-3,19 (м, 2H), 3,35 (ушир.с, 1H), 4,48 (д, J=6,9 Гц, 1H), 4,92 (д, J=11,3 Гц, 1H), 5,07 (д, J=11,3 Гц, 1H), 7,35-7,45 (м, 5H); МС m/z 374 [M+H]+.
[0128] Ссылочный пример 4
(1R,2S,6R,7S)-3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дек-8-ен-4-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
[Химическая формула 42]
(2S,5R)-5-((Бензилокси)амино)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновую кислоту (ссылочный пример 3, стадия 1, 14,0 г, 41,09 ммоль) растворяли в дегидратированном тетрагидрофуране (200 мл), с последующим охлаждением до около -20°C. К полученной смеси по каплям добавляли изобутилхлорформиат (6,11 г) и затем триэтиламин (8,86 г), с последующим перемешиванием при этой же температуре в течение 15 минут. К полученному реакционному раствору затем добавляли (1R,2S,6R,7S)-4-гидрокси-4-азатрицикло[5.2.1.02,6]дек-8-ен-3,5-дион (7,87 г), с последующим перемешиванием при этой же температуре в течение 30 минут и дополнительном перемешивании при комнатной температуре в течение 30 минут. Реакционный раствор разбавляли этилацетатом (700 мл), последовательно промывали охлажденной льдом 10% лимонной кислотой (200 мл), насыщенным бикарбонатом натрия (200 мл) и насыщенным раствором соли (200 мл), сушили над безводным сульфатом магния и фильтровали. Растворитель отгоняли при пониженном давлении, и снова концентрировали с заменой этилацетатом. Общее количество полученного в результате остатка (25,1 г) (чистый выход 92%) растворяли в дегидратированном хлороформе (180 мл), и в полученный раствор добавляли триэтиламин (5,5 г), с последующим охлаждением льдом. К полученной смеси добавляли трифосген (4,29 г), с последующим перемешиванием в течение 30 минут. К полученному затем добавляли метанол (1 мл), с последующим перемешиванием в течение 30 минут. К полученному реакционному раствору по каплям добавляли раствор метансульфоновой кислоты (23,5 мл) в дихлорметане (30 мл), с последующим дополнительным перемешиванием в течение 30 минут. Полученную смесь по каплям добавляли к охлажденному льдом 1М гидрокарбонату калия (43,5 г/200 мл), с последующим перемешиванием в течение 30 минут. Затем добавляли хлороформ (100 мл) для разделения слоев. Органический слой последовательно промывали 1M хлористоводородной кислотой (200 мл), насыщенным бикарбонатом натрия (200 мл) и насыщенным раствором соли (200 мл). Каждый из водных слоев последовательно обратно экстрагировали хлороформом (100 мл). Органические слои объединяли, сушили над безводным сульфатом магния и фильтровали. Растворитель концентрировали при пониженном давлении, и полученный в результате остаток растворяли в хлороформе (70 мл). К полученному раствору добавляли гексан (100 мл), с последующим перемешиванием в течение 30 минут для кристаллизации. К полученному дополнительно добавляли гексан (100 мл), с последующим перемешиванием в течение 1 часа. Кристаллические вещества отфильтровывали и сушили, с получением 15,4 г указанного в заголовке соединения (содержание 100%, выход 88%).
ВЭЖХ: COSMOSIL 5C18 MS-II 4,6×150 мм, 35°C, 0,02M TFA/CH3CN=50/50, 1,0 мл/мин, UV 210 нм, RT 7,1 мин; энантиомерный избыток 99,9% эи или более: CHIRALPAK AD-H, 4,6×150 мм, 40°C, гексан/EtOH=1/1, UV 210 нм, 1 мл/мин, RT 37,3 мин (цф. энантиомер 16,5 мин); т.пл. 196°C; [α]26D+12,686° (c 0,885, CHCl3);
1H ЯМР (400 МГц, CDCl3) δ 1,52 (д, J=9,1 Гц, 1H), 1,70 (м, 1H), 1,78 (д, J=9,1 Гц, 1H), 2,01-2,26 (м, 3H), 3,04-3,17 (м, 2H), 3,32 (м, 3H), 3,45 (ушир.с, 2H), 4,41 (д, J=6,7 Гц, 1H), 4,91 (д, J=11,4 Гц, 1H), 5,06 (д, J=11,4 Гц, 1H), 6,19 (ушир.с, 2H), 7,33-7,46 (м, 5H); МС m/z 438 [M+H]+.
[0129] Ссылочный пример 5
(2S,5R)-N-(2-Аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1)
[Химическая формула 43]
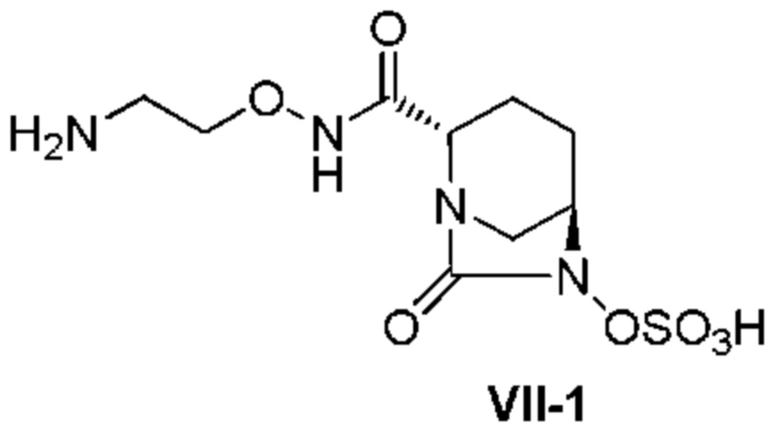
Стадия 1
трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (IV-1)
Раствор (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (4,30 г, 15,56 ммоль) в дегидратированном этилацетате (47 мл) охлаждали до -30°C, и последовательно по каплям добавляли изобутилхлорформиат (2,17 г, промытый дегидратированным этилацетатом 1 мл) и триэтиламин (1,61 г, промытый 1 мл дегидратированного этилацетата) при -30°C, с последующим перемешиванием в течение 1 часа. К полученному реакционному раствору добавляли раствор трет-бутил 2-(аминоокси)этилкарбамата (3,21 г) в дегидратированном этилацетате (4 мл) (промытый 1 мл дегидратированного этилацетата), и температуру полученной смеси поднимали до 0°C в течение 1,5 часов, с последующим перемешиванием в течение ночи. Полученную смесь последовательно промывали 8% водной лимонной кислотой (56 мл), насыщенным бикарбонатом натрия (40 мл) и насыщенным раствором соли (40 мл) и сушили над безводным сульфатом магния. Полученную смесь затем фильтровали, концентрировали до 5 мл и дополнительно концентрировали при замене этанолом (10 мл) до 6 мл. К полученному в результате раствору добавляли этанол (3 мл) и гексан (8 мл), и полученную смесь охлаждали льдом и затравливали кристаллами, с последующим перемешиванием в течение 15 минут. К полученной смеси по каплям добавляли гексан (75 мл) около 2 часов, с последующим перемешиванием в течение ночи. Кристаллические вещества отфильтровывали, промывали гексаном и сушили в вакууме, с получением 5,49 г указанного в заголовке соединения (чистый выход 4,98 г, выход 74%).
ВЭЖХ: COSMOSIL 5C18 MS-II 4,6×150 мм, 33,3 мМ фосфатный буфер/MeCN=50/50, 1,0 мл/мин, UV 210 нм, RT 4,4 мин;
1H ЯМР (400 МГц, CDCl3) δ 1,44 (с, 9H), 1,56-1,70 (м, 1H), 1,90-2,09 (м, 2H), 2,25-2,38 (м, 1H), 2,76 (д, J=11,6 Гц, 1H), 3,03 (ушир.д, J=11,6 Гц, 1H), 3,24-3,47 (м, 3H), 3,84-4,01 (м, 3H), 4,90 (д, J=11,6 Гц, 1H), 5,05 (д, J=11,6 Гц, 1H), 5,44 (ушир.с, 1H), 7,34-7,48 (м, 5H), 9,37 (ушир.с, 1H); МС m/z 435 [M+H]+.
[0130] Стадия 2
трет-бутил{2-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (V-1)
К раствору трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (3,91 г, 9,01 ммоль) в метаноле (80 мл) добавляли 10% катализатор палладий на угле (50% масс., 803 мг), с последующим перемешиванием в течение 45 минут в атмосфере водорода. Реакционный раствор фильтровали через рыхлый слой целита и концентрировали при пониженном давлении, с получением 3,11 г указанного в заголовке соединения (количественный).
ВЭЖХ: COSMOSIL 5C18 MS-II 4,6×150 мм, 33,3 мМ фосфатный буфер/MeCN=75/25, 1,0 мл/мин, UV 210 нм, RT 3,9 мин;
1H ЯМР (400 МГц, CD3OD) δ 1,44 (с, 9H), 1,73-1,83 (м, 1H), 1,86-1,99 (м, 1H), 2,01-2,12 (м, 1H), 2,22 (ушир.дд, J=15,0, 7,0 Гц, 1H), 3,03 (д, J=12,0 Гц, 1H), 3,12 (ушир.д, J=12,0 Гц, 1H), 3,25-3,35 (м, 2H), 3,68-3,71 (м, 1H), 3,82-3,91 (м, 3H); МС m/z 345 [M+H]+.
[0131] Стадия 3
Тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (VI-1)
К раствору трет-бутил{2-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (3,09 г, 8,97 ммоль) в дихлорметане (80 мл) добавляли 2,6-лютидин (3,20 мл) и комплекс триоксид серы-пиридин (3,58 г), с последующим перемешиванием при комнатной температуре в течение ночи. Реакционный раствор выливали в полунасыщенный бикарбонат натрия, и водный слой промывали хлороформом. К водному слою добавляли тетрабутиламмонийгидросульфат (3,47 г) и хлороформ (30 мл), с последующим перемешиванием в течение 10 минут. Затем водный слой экстрагировали хлороформом, полученный в результате органический слой сушили над безводным сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении, с получением 5,46 г указанного в заголовке соединения (выход 91%).
ВЭЖХ: COSMOSIL 5C18 MS-II 4,6×150 мм, 33,3 мМ фосфатный буфер/MeCN=80/20, 1,0 мл/мин, UV 210 нм, RT 2,0 мин;
1H ЯМР (400 МГц, CDCl3) δ 1,01 (т, J=7,4 Гц, 12H), 1,37-1,54 (м, 8H), 1,45 (с, 9H), 1,57-1,80 (м, 9H), 1,85-1,98 (м, 1H), 2,14-2,24 (м, 1H), 2,30-2,39 (м, 1H), 2,83 (д, J=11,6 Гц, 1H), 3,20-3,50 (м, 11H), 3,85-3,99 (м, 3H), 4,33-4,38 (м, 1H), 5,51 (ушир.с, 1H), 9,44 (ушир.с, 1H); МС m/z 425 [M-Bu4N+2H]+.
[0132] Стадия 4
(2S,5R)-N-(2-Аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1)
К раствору тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (5,20 г, 7,82 ммоль) в дихлорметане (25 мл) добавляли трифторуксусную кислоту (25 мл) при охлаждении льдом, с последующим перемешиванием при 0°C в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении. Полученный в результате остаток промывали диэтиловым эфиром, доводили до pH 7 водным раствором бикарбоната натрия, очищали колоночной хроматографией на октадецилсиликагеле (вода) и лиофилизировали, с получением 1,44 г указанного в заголовке соединения (выход 57%).
ВЭЖХ: COSMOSIL 5C18 MS-II 4,6×150 мм, 33,3 мМ фосфатный буфер/MeCN=99/1, 1,0 мл/мин, UV 210 нм, RT 3,1 мин;
1H ЯМР (400 МГц, D2O) δ 1,66-1,76 (м, 1H), 1,76-1,88 (м, 1H), 1,91-2,00 (м, 1H), 2,00-2,08 (м, 1H), 3,02 (д, J=12,0 Гц, 1H), 3,15 (т, J=5,0 Гц, 2H), 3,18 (ушир.д, J=12,0 Гц, 1H), 3,95 (дд, J=7,8, 2,2 Гц, 1H), 4,04 (т, J=5,0 Гц, 2H), 4,07 (дд, J=6,4 и 3,2 Гц, 1H); МС m/z 325 [M+H]+.
[0133] Ссылочный пример 6
(2S,5R)-N-[2-(Метиламино)этокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-2)
[Химическая формула 44]
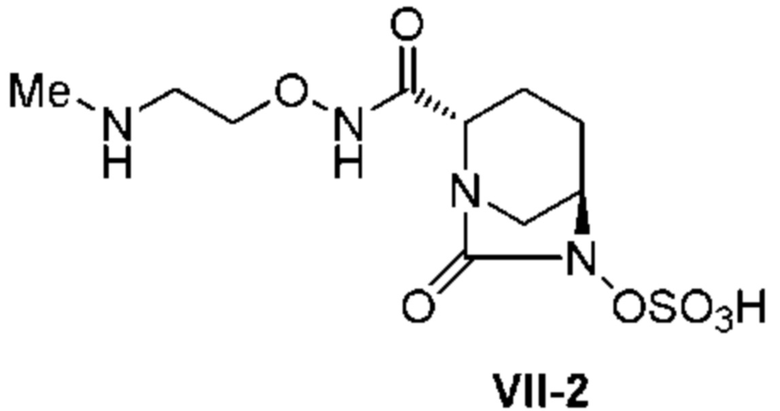
Стадия 1
трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(метил)карбамат (IV-2)
По аналогичной методике ссылочного примера 5, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (390 мг, 1,41 ммоль) и трет-бутил(2-(аминоокси)этил)(метил)карбамата (436 мг) и затем подвергали очистке колоночной хроматографией на силикагеле, с получением 347,8 мг указанного в заголовке соединения (выход 55%).
1H ЯМР (400 МГц, CDCl3) δ 1,46 (с, 9H), 1,58-1,70 (м, 1H), 1,88-2,07 (м, 2H), 2,25-2,36 (м, 1H), 2,70-3,08 (м, 2H), 2,88 (с, 3H), 3,23-3,41 (м, 2H), 3,51-3,68 (м, 1H), 3,83-4,10 (м, 3H), 4,90 (д, J=11,4 Гц, 1H), 5,06 (д, J=11,4 Гц, 1H), 7,32-7,47 (м, 5H), 10,11 (ушир.с, 1H); МС m/z 449 [M+H]+.
[0134] Стадия 2
трет-бутил{2-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(метил)карбамат (V-2)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из общего количества соединения стадии 1 (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,46 (с, 9H), 1,73-1,83 (м, 1H), 1,86-2,00 (м, 1H), 2,01-2,13 (м, 1H), 2,14-2,28 (м, 1H), 2,93 (с, 3H), 3,04 (д, J=10,8 Гц, 1H), 3,08-3,18 (м, 1H), 3,43-3,55 (м, 2H), 3,65-3,72 (м, 1H), 3,79-3,88 (м, 1H), 3,92-4,05 (м, 2H); МС m/z 359 [M+H]+.
[0135] Стадия 3
(2S,5R)-N-[2-(Метиламино)этокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-2)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(метил)карбамат получали из общего количества соединения стадии 2 (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,01 (т, J=7,2 Гц, 12H), 1,36-1,53 (м, 8H), 1,47 (с, 9H), 1,57-1,77 (м, 9H), 1,83-1,98 (м, 1H), 2,13-2,25 (м, 1H), 2,28-2,40 (м, 1H), 2,82-2,96 (м, 4H), 3,22-3,42 (м, 11H), 3,60-4,08 (м, 3H), 4,34 (ушир.с, 1H), 10,15 (ушир.с, 1H); МС m/z 437 [M-Bu4N]-.
[0136] Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 149,4 мг указанного в заголовке соединения (3 стадия выход 57%).
1H ЯМР (500 МГц, D2O) δ 1,73-1,97 (м, 2H), 1,98-2,07 (м, 1H), 2,08-2,18 (м, 1H), 2,74 (с, 3H), 3,09 (д, J=12,0 Гц, 1H), 3,21-3,32 (м, 3H), 4,04 (дд, J=7,5, 2,0 Гц, 1H), 4,10-4,23 (м, 3H); МС m/z 337 [M-H]-.
[0137] Ссылочный пример 7
(2S,5R)-7-Оксо-N-[2-(пропан-2-иламино)этокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-3)
[Химическая формула 45]
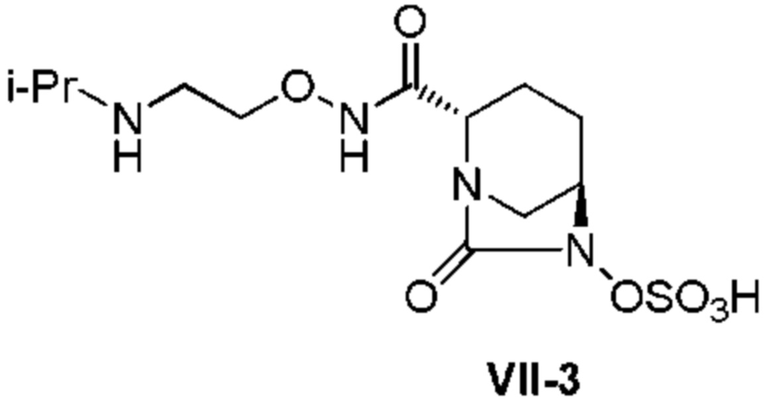
Стадия 1
трет-Бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(пропан-2-ил)карбамат (IV-3)
Раствор (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (414 мг, 1,50 ммоль) в дегидратированном дихлорметане (14,1 мл) охлаждали в атмосфере аргона до 0°C, и к полученному добавляли последовательно изобутилхлорформиат (245,9 мг) и затем триэтиламин (197 мг), с последующим перемешиванием в течение 30 минут. К полученной реакционной смеси по каплям добавляли трет-бутил(2-(аминоокси)этил)(изопропил)карбамат (596 мг). После завершения добавления, температуру повышали до комнатной температуры, и полученную смесь перемешивали в течение 1 часа. Эту реакционную смесь последовательно промывали 0,5M хлористоводородной кислотой и насыщенным раствором соли, и органический слой сушили над сульфатом магния и концентрировали при пониженном давлении. Полученный в результате остаток подвергали колоночной хроматографии на силикагеле, с получением 578,4 мг указанного в заголовке соединения (выход 81%).
1H ЯМР (400 МГц, CDCl3) δ 1,15 (д, J=6,8 Гц, 6H), 1,46 (с, 9H), 1,55-1,70 (м, 1H), 1,89-2,07 (м, 2H), 2,25-2,37 (м, 1H), 2,73-2,90 (м, 1H), 2,98-3,08 (м, 1H), 3,22-3,38 (м, 2H), 3,40-3,60 (м, 1H), 3,83-4,06 (м, 4H), 4,90 (д, J=11,2 Гц, 1H), 5,06 (д, J=11,2 Гц, 1H), 7,35-7,46 (м, 5H), 10,29 (ушир.с, 1H); МС m/z 477 [M+H]+.
[0138] Стадия 2
трет-бутил{2-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(пропан-2-ил)карбамат (V-3)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из общего количества соединения стадии 1 (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,09-1,23 (м, 6H), 1,46 (с, 9H), 1,73-2,27 (м, 4H), 3,06 (д, J=11,6 Гц, 1H), 3,08-3,50 (м, 4H), 3,64-3,73 (м, 1H), 3,79-3,98 (м, 3H); МС m/z 387 [M+H]+.
[0139] Стадия 3
(2S,5R)-7-Оксо-N-[2-(пропан-2-иламино)этокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-3)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(пропан-2-ил)карбамат получали из общего количества соединения стадии 2 (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,01 (д, J=7,4 Гц, 12H), 1,10-1,20 (м, 6H), 1,33-1,77 (м, 17H), 1,46 (с, 9H), 1,84-1,97 (м, 1H), 2,12-2,25 (м, 1H), 2,28-2,40 (м, 1H), 2,79-2,95 (м, 1H), 3,17-3,45 (м, 9H), 3,50-3,67 (м, 1H), 3,80-4,07 (м, 5H), 4,34 (ушир.с, 1H), 10,36 (ушир.с, 1H); МС m/z 465 [M-Bu4N]-.
[0140] Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 252,1 мг указанного в заголовке соединения (3 стадия выход 57%).
1H ЯМР (500 МГц, D2O) δ 1,28 (д, J=6,5 Гц, 6H),1,74-1,83 (м, 1H), 1,85-1,96 (м, 1H), 1,98-2,14 (м, 2H), 3,11 (д, J=12,5 Гц, 1H), 3,22-3,30 (м, 3H), 3,40 (квинт, J=6,5 Гц, 1H), 4,01 (ушир.д, J=5,5 Гц, 1H), 4,09-4,18 (м, 3H); МС m/z 367 [M+H]+.
[0141] Ссылочный пример 8
(2S,5R)-N-[2-(Диметиламино)этокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-4)
[Химическая формула 46]

Стадия 1
(2S,5R)-6-Бензилокси-N-[2-(диметиламино)этокси]-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (IV-4)
Раствор (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (553 мг, 2,00 ммоль) в дегидратированном дихлорметане (10 мл) охлаждали до 0°C в атмосфере аргона, и по каплям добавляли изобутилхлорформиат (289 мкл, 2,20 ммоль). Затем добавляли триэтиламин (293 мкл), и полученную смесь перемешивали в течение 30 минут, с получением смешанного ангидрида кислоты в реакционной системе. К полученной реакционной смеси медленно добавляли дигидрохлорид 2-(аминоокси)-N,N-диметилэтанамина (591 мг) и триэтиламин (930 мкл), одновременно промывая дегидратированным дихлорметаном (7,0 мл), с последующим перемешиванием в течение 1 часа при этой же температуре. После фильтрования реакционной смеси, полученный остаток промывали метанолом, и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток растворяли в дихлорметане и воде, и органический слой, экстрагированный дихлорметаном, сушили над сульфатом магния и затем концентрировали при пониженном давлении. Полученный в результате остаток подвергали колоночной хроматографии на силикагеле (амино-модифицированное соединение кремния, хлороформ/метанол=10/1), с получением 291,1 мг указанного в заголовке соединения в виде бесцветного масла (выход 40%).
1H ЯМР (400 МГц, CDCl3) δ 1,45-1,85 (м, 4H), 2,29 (с, 6H), 2,60 (т, J=5,2 Гц, 2H), 2,81 (д, J=11,6 Гц, 1H), 2,97 (ушир.д, J=11,6 Гц, 1H), 3,28-3,34 (м, 1H), 3,92-4,07 (м, 3H), 4,90 (д, J=11,6 Гц, 1H), 5,05 (д, J=11,6 Гц, 1H), 7,35-7,48 (м, 5H); МС m/z 363 [M+H]+.
[0142] Стадия 2
(2S,5R)-N-[2-(Диметиламино)этокси]-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (V-4)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из общего количества соединения стадии 1 (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,74-1,84 (м, 1H), 1,87-1,98 (м, 1H), 2,03-2,12 (м, 1H), 2,15-2,24 (м, 1H), 2,36 (с, 6H), 2,67-2,74 (м, 2H), 3,07 (ушир.д, J=11,6 Гц, 1H), 3,12 (ушир.д, J=11,6 Гц, 1H), 3,67-3,72 (м, 1H), 3,83 (ушир.д, J=6,4 Гц, 1H), 3,96-4,06 (м, 2H); МС m/z 273 [M+H]+.
[0143] Стадия 3
(2S,5R)-N-[2-(Диметиламино)этокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-4)
Реакционную смесь, полученную по аналогичной методике ссылочного примера 5, разбавляли хлороформом и промывали водой, с получением пиридиний (2S,5R)-N-[2-(диметиламино)этокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида. Его нейтрализовали насыщенным бикарбонатом натрия и затем очищали колоночной хроматографией на октадецилсиликагеле, с получением 130,7 мг указанного в заголовке соединения (2 стадия выход 43%).
1H ЯМР (400 МГц, D2O) δ 1,68-1,84 (м, 2H), 1,86-2,04 (м, 2H), 2,80 (с, 6H), 3,09-3,17 (м, 2H), 3,17-3,29 (м, 2H), 3,80-3,90 (м, 1H), 4,02-4,13 (м, 3H); МС m/z 353 [M+H]+.
[0144] Ссылочный пример 9
(2S,5R)-N-{[(2S)-2-Аминопропил]окси}-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-5)
[Химическая формула 47]
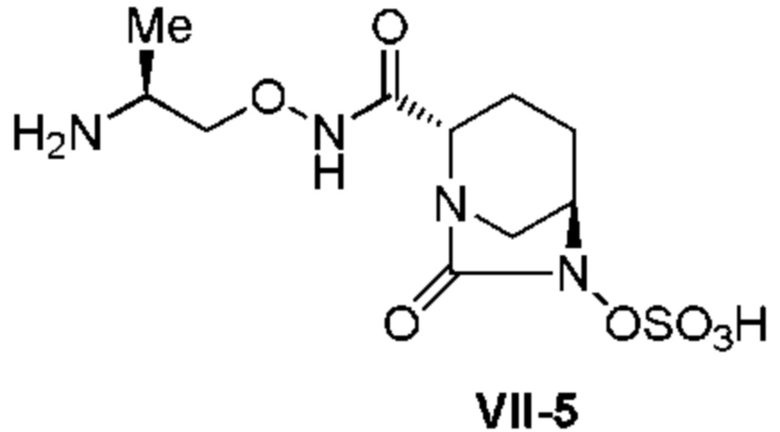
Стадия 1
трет-бутил{(2S)-1-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамат (IV-5)
По аналогичной методике ссылочного примера 7, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (414 мг, 1,50 ммоль) и (S)-трет-бутил(1-(аминоокси)пропан-2-ил)карбамата (550 мг) и затем подвергали очистке колоночной хроматографией на силикагеле, с получением 585,6 мг указанного в заголовке соединения (выход 87%).
1H ЯМР (400 МГц, CDCl3) δ 1,17 (д, J=6,4 Гц, 3H), 1,44 (с, 9H), 1,55-1,70 (м, 1H), 1,90-2,10 (м, 2H), 2,26-2,34 (м, 1H), 2,80 (д, J=12,0 Гц, 1H), 3,06 (ушир.д, J=12,0 Гц, 1H), 3,27-3,34 (м, 1H), 3,64-3,74 (м, 1H), 3,86-3,98 (м, 3H), 4,81 (ушир.д, J=7,6 Гц, 1H), 4,90 (д, J=11,6 Гц, 1H), 5,05 (д, J=11,6 Гц, 1H), 7,34-7,45 (м, 5H), 9,68 (ушир.с, 1H); МС m/z 449 [M+H]+.
[0145] Стадия 2
трет-бутил{(2S)-1-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамат (V-5)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из общего количества соединения стадии 1 (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,16 (д, J=6,4 Гц, 3H), 1,44 (с, 9H), 1,74-1,84 (м, 1H), 1,86-1,98 (м, 1H), 2,03-2,12 (м, 1H), 2,21 (ушир.дд, J=15,2, 6,8 Гц, 1H), 3,06 (д, J=12,0 Гц, 1H), 3,14 (ушир.д, J=12,0 Гц, 1H), 3,68-3,72 (м, 1H), 3,74-3,87 (м, 4H); МС m/z 359 [M+H]+.
[0146] Стадия 3
(2S,5R)-N-{[(2S)-2-аминопропил]окси}-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-5)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил{(2S)-1-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамат получали из общего количества соединения стадии 2 (количественный). МС m/z 437[M-Bu4N]-.
Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 117,1 мг указанного в заголовке соединения (3 стадия выход 26%).
1H ЯМР (400 МГц, D2O) δ 1,17 (д, J=6,8 Гц, 3H), 1,66-1,89 (м, 2H), 1,91-2,08 (м, 2H), 3,02 (д, J=12,0 Гц, 1H), 3,18 (ушир.д, J=12,0 Гц, 1H), 3,47-3,58 (м, 1H), 3,82 (дд, J=11,8, 9,4 Гц, 1H), 3,92-4,02 (м, 2H), 4,05-4,10 (м, 1H); МС m/z 339 [M+H]+.
[0147] Ссылочный пример 10
(2S,5R)-N-{[(2R)-2-Аминопропил]окси}-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-6)
[Химическая формула 48]
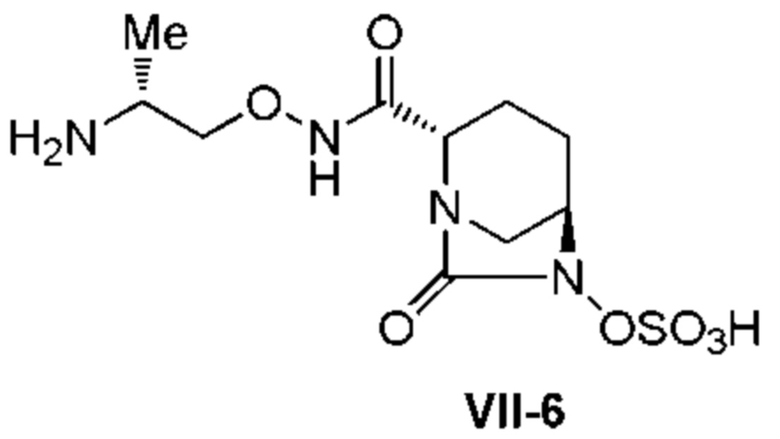
Стадия 1
трет-Бутил{(2R)-1-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамат (IV-6)
По аналогичной методике ссылочного примера 7, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (414 мг, 1,50 ммоль) и (R)-трет-бутил(1-(аминоокси)пропан-2-ил)карбамата (569 мг) и затем подвергали очистке колоночной хроматографией на силикагеле, с получением 625 мг указанного в заголовке соединения (выход 93%).
1H ЯМР (400 МГц, CDCl3) δ 1,14 (д, J=6,4 Гц, 3H), 1,43 (с, 9H), 1,53-1,70 (м, 1H), 1,90-2,06 (м, 2H), 2,28-2,36 (м, 1H), 2,79 (д, J=12,0 Гц, 1H), 3,02 (ушир.д, J=12,0 Гц, 1H), 3,28-3,33 (м, 1H), 3,56-3,68 (м, 1H), 3,84 (дд, J=11,2, 3,6 Гц, 1H), 3,92-4,04 (м, 2H), 4,66 (ушир.д, J=8,0 Гц, 1H), 4,91 (д, J=11,2 Гц, 1H), 5,06 (д, J=11,2 Гц, 1H), 7,35-7,45 (м, 5H), 9,94 (ушир.с, 1H); МС m/z 449 [M+H]+.
[0148] Стадия 2
трет-бутил{(2R)-1-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамат (V-6)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из общего количества соединения стадии 1 (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,15 (д, J=6,4 Гц, 3H), 1,44 (с, 9H), 1,73-1,84 (м, 1H), 1,86-2,00 (м, 1H), 2,01-2,12 (м, 1H), 2,19-2,29 (м, 1H), 3,06 (д, J=11,6 Гц, 1H), 3,10-3,20 (м, 1H), 3,67-3,72 (м, 1H), 3,73-3,92 (м, 4H); МС m/z 359 [M+H]+.
[0149] Стадия 3
(2S,5R)-N-{[(2R)-2-Аминопропил]окси}-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-6)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил{(2R)-1-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропан-2-ил}карбамат получали из общего количества соединения стадии 2 (количественный). МС m/z 437 [M-Bu4N]-.
[0150] Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 212,6 мг указанного в заголовке соединения (3 стадия выход 45%).
1H ЯМР (400 МГц, D2O) δ 1,17 (д, J=6,8 Гц, 3H), 1,66-1,78 (м, 1H), 1,78-1,88 (м, 1H), 1,90-2,06 (м, 2H), 3,02 (д, J=12,0 Гц, 1H), 3,18 (ушир.д, J=12,0 Гц, 1H), 3,48-3,58 (м, 1H), 3,83 (дд, J=11,8, 9,0 Гц, 1H), 3,94 (ушир.д, J=7,2 Гц, 1H), 3,98 (дд, J=11,8, 3,4 Гц, 1H), 4,06-4,10 (м, 1H); МС m/z 339 [M+H]+.
[0151] Ссылочный пример 11
(2S,5R)-N-(3-Аминопропокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-7)
[Химическая формула 49]

Стадия 1
трет-Бутил{3-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропил}карбамат (IV-7)
По аналогичной методике ссылочного примера 5, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (390 мг, 1,41 ммоль) и трет-бутил(3-(аминоокси)пропил)карбамата (730 мг) и затем подвергали очистке колоночной хроматографией на силикагеле, с получением 398,1 мг указанного в заголовке соединения (выход 63%).
1H ЯМР (400 МГц, CDCl3) δ 1,44 (с, 9H), 1,50-1,67 (м, 1H), 1,75-1,86 (м, 2H), 1,88-2,07 (м, 2H), 2,28-2,37 (м, 2H), 2,77 (д, J=11,0 Гц, 1H), 3,01 (ушир.д, J=11,0 Гц, 1H), 3,20-3,38 (м, 3H), 3,89-4,04 (м, 3H), 4,90 (д, J=11,4 Гц, 1H), 5,05 (д, J=11,4 Гц, 1H), 5,17 (ушир.с, 1H), 7,36-7,45 (м, 5H), 9,21 (ушир.с, 1H); МС m/z 449 [M+H]+.
[0152] Стадия 2
трет-бутил{3-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропил}карбамат (V-7)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из соединения приведенной выше стадии 1 (392,8 мг, 876 мкмоль) (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,43 (с, 9H), 1,73-1,99 (м, 4H), 2,01-2,12 (м, 1H), 2,13-2,24 (м, 1H), 3,07 (д, J=11,6 Гц, 1H), 3,09-3,21 (м, 3H), 3,69 (ушир.с, 1H), 3,80-3,96 (м, 3H); МС m/z 359 [M+H]+.
[0153] Стадия 3
(2S,5R)-N-(3-Аминопропокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-7)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил{3-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропил}карбамат получали из общего количества соединения стадии 2 (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,01 (т, J=7,4 Гц, 12H), 1,33-1,53 (м, 8H), 1,47 (с, 9H), 1,55-1,96 (м, 12H), 2,14-2,23 (м, 1H), 2,31-2,41 (м, 1H), 2,85 (ушир.д, J= 11,2 Гц, 1H), 3,15-3,42 (м, 11H), 3,88-4,07 (м, 3H), 4,35 (ушир.с, 1H), 5,27 (ушир.с, 1H), 9,26 (ушир.с, 1H); МС m/z 437 [M-Bu4N]-.
[0154] Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 138,4 мг указанного в заголовке соединения (3 стадия выход 47%).
1H ЯМР (400 МГц, D2O) δ 1,67-2,05 (м, 6H), 3,00-3,19 (м, 4H), 3,82-3,94 (м, 3H), 4,05-4,10 (м, 1H); МС m/z 337 [M-H]-.
[0155] Ссылочный пример 12
(2S,5R)-N-[(2S)-Азитидин-2-илметокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-8)
[Химическая формула 50]
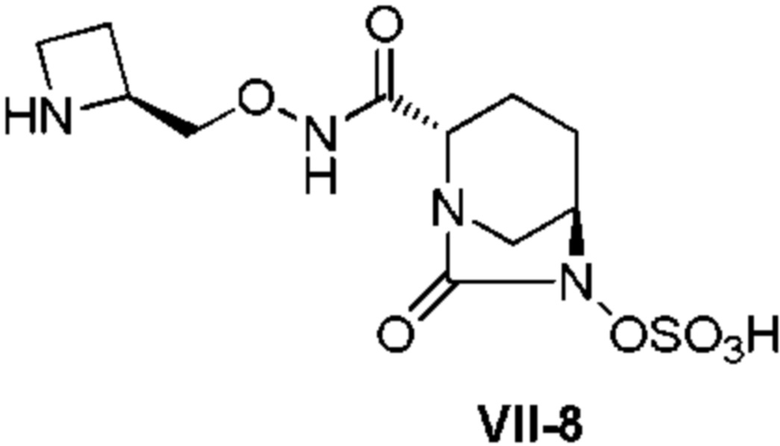
Стадия 1
трет-бутил-(2S)-2-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилат (IV-8)
По аналогичной методике ссылочного примера 8, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (553 мг, 2,00 ммоль) и (S)-трет-бутил 2-((аминоокси)метил)азитидин-1-карбоксилата (578 мг) и затем подвергали очистке колоночной хроматографией на силикагеле, с получением 760,1 мг указанного в заголовке соединения (выход 83%).
1H ЯМР (400 МГц, CDCl3) δ 1,46 (с, 9H), 1,56-1,70 (м, 1H), 1,88-2,07 (м, 3H), 2,23-2,34 (м, 2H), 2,84 (д, J=11,6 Гц, 1H), 3,02 (д, J=11,6 Гц, 1H), 3,28 (ушир.с, 1H), 3,77-4,03 (м, 4H), 4,06-4,15 (м, 1H), 4,37-4,48 (м, 1H), 4,89 (д, J =11,6 Гц, 1H), 5,04 (д, J=11,6 Гц, 1H), 7,34-7,44 (м, 5H), 10,63 (ушир.с, 1H); МС m/z 461 [M+H]+.
[0156] Стадия 2
трет-бутил-(2S)-2-{[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилат (V-8)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из соединения приведенной выше стадии 1 (699 мг, 1,52 ммоль) (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,44 (с, 9H), 1,74-1,85 (м, 1H), 1,86-1,99 (м, 1H), 2,02-2,14 (м, 1H), 2,16-2,40 (м, 3H), 3,06 (д, J=11,6 Гц, 1H), 3,10-3,17 (м, 1H), 3,67-3,74 (м, 1H), 3,75-3,93 (м, 3H), 4,01 (дд, J=10,6, 10,6 Гц, 1H), 4,14 (дд, J=10,6, 10,6 Гц, 1H), 4,37-4,47 (м, 1H); МС m/z 371 [M+H]+.
[0157] Стадия 3
(2S,5R)-N-[(2S)-Азитидин-2-илметокси]-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-8)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил (2S)-2-{[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилат получали из общего количества соединения стадии 2 (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,01 (т, J=7,2 Гц, 12H), 1,30-2,10 (м, 19H), 1,46 (с, 9H), 2,12-2,39 (м, 3H), 2,89 (ушир.д, J=12,0 Гц, 1H), 3,23-3,39 (м, 9H), 3,76-3,93 (м, 3H), 3,95-4,06 (м, 1H), 4,08-4,18 (м, 1H), 4,33 (ушир.с, 1H), 4,37-4,50 (м, 1H); МС m/z 449 [M-Bu4N]-.
[0158] Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 172,3 мг указанного в заголовке соединения (3 стадия выход 32%).
1H ЯМР (500 МГц, D2O) δ 1,71-1,83 (м, 1H), 1,84-1,97 (м, 1H), 1,98-2,16 (м, 2H), 2,36-2,49 (м, 1H), 2,50-2,61 (м, 1H), 3,10 (д, J=12,0 Гц, 1H), 3,22-3,30 (м, 1H), 3,92-4,12 (м, 5H), 4,25-4,36 (м, 1H), 4,68-4,77 (м, 1H); МС m/z 351 [M+H]+.
[0159] Ссылочный пример 13
(2S,5R)-7-Оксо-N-[(2R)-пирролидин-2-илметокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-9)
[Химическая формула 51]
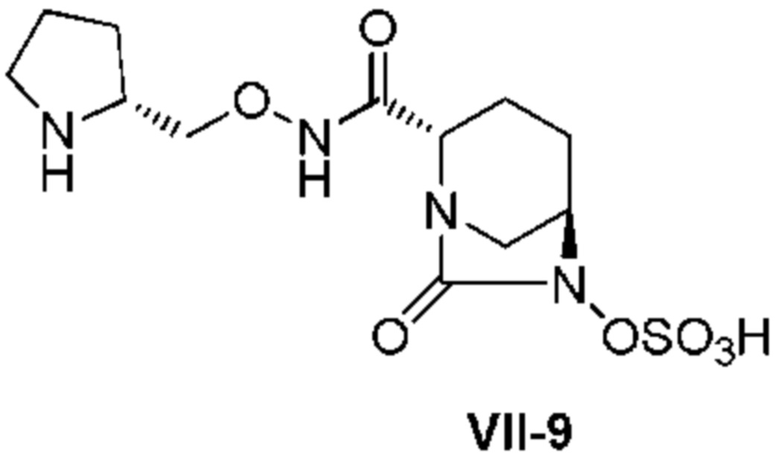
Стадия 1
трет-бутил-(2R)-2-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пирролидин-1-карбоксилат (IV-9)
По аналогичной методике ссылочного примера 5, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (390 мг, 1,41 ммоль) и (R)-трет-бутил 2-((аминоокси)метил)пирролидин-1-карбоксилата (796 мг) и затем подвергали очистке колоночной хроматографией на силикагеле, с получением 336 мг указанного в заголовке соединения (выход 50%).
1H ЯМР (400 МГц, CDCl3) δ 1,45 (с, 9H), 1,52-1,72 (м, 1H), 1,80-2,09 (м, 6H), 2,27-2,39 (м, 1H), 2,84 (ушир.д, J=12,4 Гц, 1H), 2,96-3,08 (м, 1H), 3,28-3,44 (м, 3H), 3,60-3,86 (м, 2H), 3,89-4,06 (м, 1H), 4,14-4,29 (м, 1H), 4,90 (д, J=11,2 Гц, 1H), 5,06 (д, J=11,2 Гц, 1H), 7,32-7,47 (м, 5H), 10,56 (с, 1H); МС m/z 475 [M+H]+.
[0160] Стадия 2
трет-бутил-(2R)-2-{[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пирролидин-1-карбоксилат (V-9)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из общего количества соединения стадии 1 (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,46 (с, 9H), 1,73-2,27 (м, 8H), 3,06 (д, J=11,6 Гц, 1H), 3,09-3,18 (м, 1H), 3,24-3,40 (м, 2H), 3,67-3,71 (м, 1H), 3,73-4,12 (м, 4H); МС m/z 385 [M+H]+.
[0161] Стадия 3
(2S,5R)-7-Оксо-N-[(2R)-пирролидин-2-илметокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-9)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил (2R)-2-{[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пирролидин-1-карбоксилат получали из общего количества соединения стадии 2 (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,01 (т, J=7,4 Гц, 12H), 1,34-1,51 (м, 8H), 1,46 (с, 9H), 1,55-1,78 (м, 10H), 1,80-2,01 (м, 4H), 2,11-2,23 (м, 1H), 2,29-2,42 (м, 1H), 2,88 (ушир.д, J=11,2 Гц, 1H), 3,21-3,43 (м, 10H), 3,60-3,86 (м, 2H), 3,88-4,07 (м, 2H), 4,16-4,28 (м, 1H), 4,34 (ушир.с, 1H), 10,62 (ушир.с, 1H); МС m/z 463 [M-Bu4N+2H]+.
[0162] Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 77,4 мг указанного в заголовке соединения (3 стадия выход 30%).
1H ЯМР (500 МГц, D2O) δ 1,66-2,18 (м, 8H), 3,14 (д, J=12,8 Гц, 1H), 3,23 (ушир.д, J=12,8 Гц, 1H), 3,30 (т, J=7,3 Гц, 2H), 3,89 (ддд, J=8,2, 8,2, 3,4 Гц, 1H), 3,92-4,01 (м, 2H), 4,09-4,18 (м, 2H); МС m/z 365 [M+H]+.
[0163] Ссылочный пример 14
(2S,5R)-7-Оксо-N-[(3R)-пиперидин-3-илметокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-10)
[Химическая формула 52]
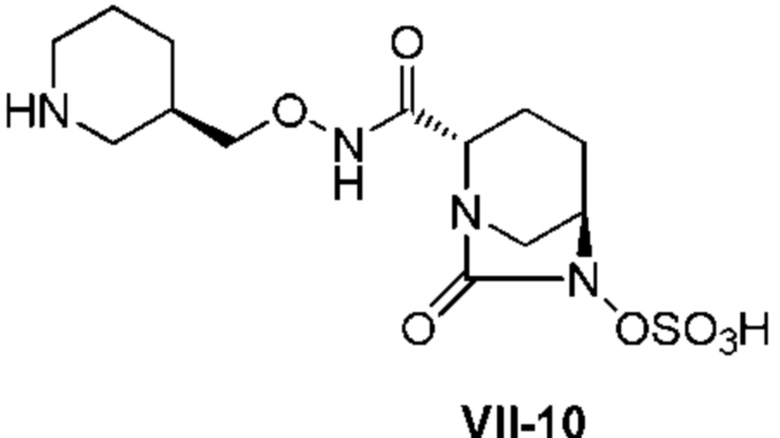
Стадия 1
трет-Бутил (3R)-3-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пиперидин-1-карбоксилат (IV-10)
По аналогичной методике ссылочного примера 8, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (390 мг, 1,41 ммоль) и (R)-трет-бутил 3-((аминоокси)метил)пиперидин-1-карбоксилата (527 мг) и затем подвергали очистке колоночной хроматографией на силикагеле, с получением 333 мг указанного в заголовке соединения (выход 48%).
1H ЯМР (400 МГц, CDCl3) δ 1,15-2,10 (м, 8H), 1,45 (с, 9H), 2,25-2,40 (м, 1H), 2,70-3,08 (м, 4H), 3,27-3,37 (м, 1H), 3,65-4,00 (м, 5H), 4,90 (д, J=11,2 Гц, 1H), 5,05 (д, J=11,2 Гц, 1H), 7,34-7,46 (м, 5H), 9,22 (ушир.с, 1H); МС m/z 489 [M+H]+.
[0164] Стадия 2
трет-Бутил (3R)-3-{[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пиперидин-1-карбоксилат (V-10)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из общего количества соединения стадии 1 (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,24-1,37 (м, 1H), 1,40-1,56 (м, 1H), 1,45 (с, 9H), 1,64-1,73 (м, 1H), 1,75-2,00 (м, 4H), 2,03-2,13 (м, 1H), 2,15-2,26 (м, 1H), 2,65-2,95 (м, 2H), 3,06 (д, J=12,0 Гц, 1H), 3,13 (ушир.д, J=12,0 Гц, 1H), 3,67-3,91 (м, 5H), 4,01-4,08 (м, 1H); МС m/z 399 [M+H]+.
[0165] Стадия 3
(2S,5R)-7-Оксо-N-[(3R)-пиперидин-3-илметокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-10)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил (3R)-2-{[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}пиперидин-1-карбоксилат получали из общего количества соединения стадии 2 (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,01 (дд, J=7,6 и 6,8 Гц, 12H), 1,11-1,99 (м, 23H), 1,46 (с, 9H), 2,12-2,24 (м, 1H), 2,30-2,42 (м, 1H), 2,67-2,96 (м, 3H), 3,19-3,38 (м, 9H), 3,70-3,99 (м, 5H), 4,35 (ушир.с, 1H), 9,16 (ушир.с, 1H); МС m/z 477 [M-Bu4N]-.
[0166] Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 106 мг указанного в заголовке соединения (3 стадия выход 41%).
1H ЯМР (400 МГц, D2O) δ 1,16-1,28 (м, 1H), 1,54-1,88 (м, 5H), 1,92-2,16 (м, 3H), 2,72 (т, J=12,2 Гц, 1H), 2,81 (ддд, J=12,8 и 12,8 и 3,5 Гц, 1H), 3,02 (д, J=12,0 Гц, 1H), 3,15-3,28 (м, 2H), 3,37-3,44 (м, 1H), 3,70 (дд, J=10,3 и 7,6 Гц, 1H), 3,79 (дд, J=10,3 и 5,0 Гц, 1H), 3,88-3,94 (м, 1H), 4,06-4,10 (м, 1H); МС m/z 377 [M-H]-.
[0167] Ссылочный пример 15
(2S,5R)-7-Оксо-N-[(3S)-пирролидин-3-илокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-11)
[Химическая формула 53]
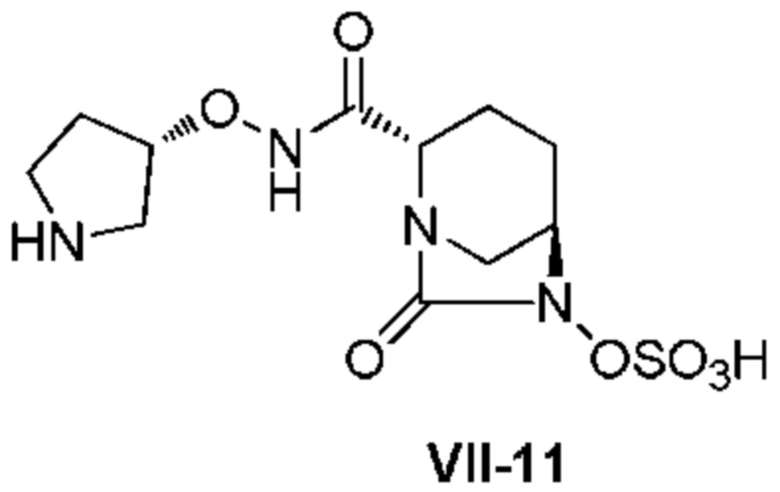
Стадия 1
трет-Бутил (3S)-3-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пирролидин-1-карбоксилат (IV-11)
По аналогичной методике ссылочного примера 5, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (553 мг, 2,00 ммоль) и (S)-трет-бутил 3-(аминоокси)пирролидин-1-карбоксилата (606 мг) и подвергали колоночной хроматографии на силикагеле, с получением 920,4 мг указанного в заголовке соединения (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,46 (с, 9H), 1,61-1,68 (м, 1H), 1,89-2,09 (м, 3H), 2,15-2,19 (м, 1H), 2,28-2,34 (м, 1H), 2,75 (д, J=11,6 Гц, 1H), 2,95-3,06 (м, 1H), 3,31 (ушир.с, 1H), 3,35-3,68 (м, 4H), 3,97 (д, J=7,6 Гц, 1H), 4,60 (ушир.д, J= 23,2 Гц, 1H), 4,90 (д, J=11,6 Гц, 1H), 5,05 (д, J=11,6 Гц, 1H), 7,26-7,43 (м, 5H), 9,08 (ушир.д, J=23,2 Гц, 1H); МС m/z 461 [M+H]+.
[0168] Стадия 2
трет-Бутил (3S)-3-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пирролидин-1-карбоксилат (V-11)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из соединения приведенной выше стадии 1 (869 мг, 1,89 ммоль) (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,47 (с, 9H), 1,75-2,12 (м, 4H), 2,13-2,25 (м, 2H), 3,05 (д, J=12,0 Гц, 1H), 3,13 (ушир.д, J=12,0 Гц, 1H), 3,25-3,50 (м, 2H), 3,61 (ушир.д, J=13,2 Гц, 1H), 3,70 (ушир.с, 1H), 3,86 (ушир.д, J=7,2 Гц, 1H), 4,32-4,38 (м, 1H), 4,54-4,62 (м, 1H); МС m/z 371 [M+H]+.
[0169] Стадия 3
(2S,5R)-7-Оксо-N-[(3S)-пирролидин-3-илокси]-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-11)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил (3S)-3-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пирролидин-1-карбоксилат (количественный) получали из общего количества соединения стадии 2. МС m/z 449 [M-Bu4N]-.
Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 170,7 мг указанного в заголовке соединения (3 стадия выход 26%).
1H ЯМР (400 МГц, D2O) δ 1,71-1,92 (м, 2H), 1,95-2,18 (м, 3H), 2,21-2,30 (м, 1H), 3,07 (д, J=12,2 Гц, 1H), 3,24 (ушир.д, J=12,2 Гц, 1H), 3,31-3,45 (м, 3H), 3,51 (д, J=13,6 Гц, 1H), 3,99 (ушир.д, J=6,0 Гц, 1H), 4,10-4,14 (м, 1H), 4,72-4,77 (м, 1H); МС m/z 349 [M-H]-.
[0170] Ссылочный пример 16
(2S,5R)-N-(Азитидин-3-илметокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-12)
[Химическая формула 54]
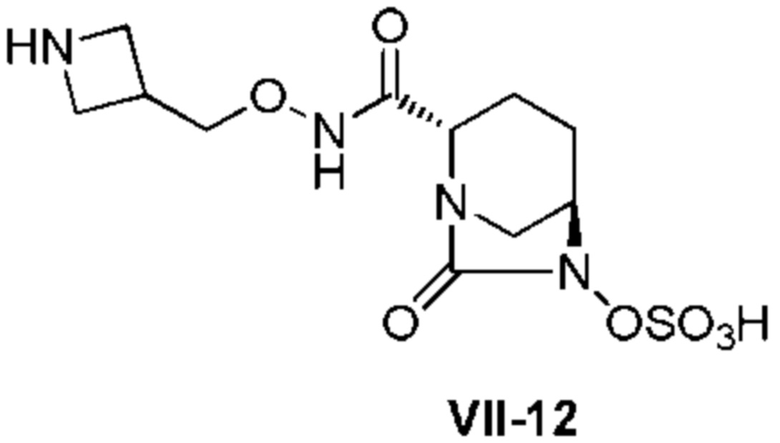
Стадия 1
трет-бутил 3-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилат (IV-12)
По аналогичной методике ссылочного примера 8, неочищенный продукт получали из (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (553 мг, 2,00 ммоль) и трет-бутил 3-((аминоокси)метил)азитидин-1-карбоксилата (564 мг) и подвергали колоночной хроматографии на силикагеле, с получением 699,7 мг указанного в заголовке соединения (выход 76%).
1H ЯМР (400 МГц, CDCl3) δ 1,43 (с, 9H), 1,54-1,70 (м, 1H), 1,87-2,06 (м, 2H), 2,27-2,35 (м, 1H), 2,75 (д, J=11,6 Гц, 1H), 2,80-2,90 (м, 1H), 3,01 (ушир.д, J=11,6 Гц, 1H), 3,32 (ушир.с, 1H), 3,68-3,76 (м, 2H), 3,94 (ушир.д, J=7,6 Гц, 1H), 4,00-4,15 (м, 4H), 4,90 (д, J=11,8 Гц, 1H), 5,05 (д, J=11,8 Гц, 1H), 7,35-7,44 (м, 5H), 9,08 (ушир.с, 1H); МС m/z 461 [M+H]+.
[0171] Стадия 2
трет-бутил 3-{[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилат (V-12)
По аналогичной методике ссылочного примера 5, указанное в заголовке соединение получали из соединения приведенной выше стадии 1 (642 мг, 1,39 ммоль) (количественный).
1H ЯМР (400 МГц, CD3OD) δ 1,43 (с, 9H), 1,74-1,85 (м, 1H), 1,86-1,97 (м, 1H), 2,04-2,13 (м, 1H), 2,16-2,24 (м, 1H), 2,84-2,94 (м, 1H), 3,05 (д, J=11,6 Гц, 1H), 3,13 (ушир.д, J=11,6 Гц, 1H), 3,68-3,82 (м, 3H), 3,83 (ушир.д, J=6,8 Гц, 1H), 3,97-4,06 (м, 4H); МС m/z 371 [M+H]+.
[0172] Стадия 3
(2S,5R)-N-(Азитидин-3-илметокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-12)
По аналогичной методике ссылочного примера 5, тетрабутиламмоний трет-бутил 3-{[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилат получали из общего количества соединения стадии 2 (количественный).
1H ЯМР (400 МГц, CDCl3) δ 1,01 (т, J=7,2 Гц, 12H), 1,37-1,51 (м, 8H), 1,46 (с, 9H), 1,54-1,75 (м, 9H), 1,82-1,97 (м, 1H), 2,13-2,25 (м, 1H), 2,29-2,40 (м, 1H), 2,77-2,95 (м, 2H), 3,24-3,40 (м, 9H), 3,64-4,16 (м, 7H), 4,36 (ушир.с, 1H), 9,16 (ушир.с, 1H); МС m/z 449 [M-Bu4N]-.
[0173] Общее количество приведенной выше тетрабутиламмониевой соли подвергали удалению защиты с помощью трифторуксусной кислоты, и после очистки колоночной хроматографией на октадецилсиликагеле, получали 164,7 мг указанного в заголовке соединения (3 стадия выход 34%).
1H ЯМР (400 МГц, D2O) δ 1,65-1,89 (м, 2H), 1,92-2,06 (м, 2H), 3,06 (д, J=12,4 Гц, 1H), 3,10-3,22 (м, 2H), 3,90-4,00 (м, 5H), 4,07-4,14 (м, 3H); МС m/z 351 [M+H]+.
[0174] Ссылочный пример 17
(3aR,7aS)-2-Гидрокси-3a,4,7,7a-тетрагидро-1H-изоиндол-1,3(2H)-дион
[Химическая формула 55]
[0175] Гидроксиламинсульфат (24,975 г, 0,152 моль) растворяли в воде (100 мл) и добавляли (3aR,7aS)-3a,4,7,7a-тетрагидроизобензофуран-1,3-дион (45,228 г). К полученной смеси добавляли 25% водный раствор гидроксида натрия (50 г) небольшими порциями в течение 15 минут, с последующим перемешиванием при 90°C в течение 2 часов. Полученную смесь охлаждали до комнатной температуры. Выпавшее в осадок твердое кристаллическое вещество подвергали фильтрованию при отсасывании, с последующим подсушиванием в течение 30 минут. Влажные кристаллы сушили в вакууме при 50°C в течение 2 дней, с получением 42,87 г указанного в заголовке соединения (выход 87%).
1H ЯМР (400 МГц, CDCl3) δ 2,20-2,31 (м, 2H), 2,56-2,65 (м, 2H), 3,08-3,14 (м, 2H), 5,91 (дт, J=0,9, 2,7 Гц, 2H); МС m/z 166 [M-H]-.
[0176] Ссылочный пример 18
(3aR,7aS)-1,3-Диоксо-3a,4,7,7a-тетрагидро-1H-изоиндол-2(3H)-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
[Химическая формула 56]
[0177] Стадия 1
1-трет-бутил 2-((3aR,7aS)-1,3-диоксо-3a,4,7,7a-тетрагидро-1H-изоиндол-2(3H)-ил) (2S,5R)-5-((бензилокси)амино)пиперидин-1,2-дикарбоксилат
[Химическая формула 57]
[0178] (2S,5R)-5-((Бензилокси)амино)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновую кислоту (ссылочный пример 3, стадия 1, 3,504 г, 10 ммоль) растворяли в дегидратированном тетрагидрофуране (50 мл), с последующим охлаждением до около -20°C. К полученной смеси по каплям добавляли изобутилхлорформиат (1,51 г) и затем триэтиламин (2,17 г), с последующим перемешиванием при этой же температуре в течение 15 минут. Затем к полученному реакционному раствору добавляли (3aR,7aS)-2-гидрокси-3a,4,7,7a-тетрагидро-1H-изоиндол-1,3(2H)-дион (ссылочный пример 17, 1,84 г), с последующим перемешиванием при этой же температуре в течение 30 минут и затем при комнатной температуре в течение 30 минут. Реакционный раствор разбавляли этилацетатом (200 мл), последовательно промывали охлажденной льдом 10% лимонной кислотой (60 мл), насыщенным бикарбонатом натрия (60 мл) и насыщенным раствором соли (60 мл), сушили над безводным сульфатом магния и фильтровали. Остаток, полученный при концентрировании фильтрата при пониженном давлении, подвергали колоночной хроматографии на силикагеле (гексан/этилацетат=2/1), с получением 4,689 г указанного в заголовке соединения в виде бесцветного пенообразного твердого вещества (выход 94%).
1H ЯМР (400 МГц, CDCl3) δ 1,47 (ушир.с, 9H), 1,59-1,75 (м, 2H), 2,04-2,32 (м, 2H), 2,16-2,35 (м, 2H), 2,61 (д, J=15,2 Гц, 2H), 3,14-3,24 (м, 4H), 4,15-4,22 (м, 1H), 4,71 (q, J=11,6 Гц, 2H), 5,03 (ушир.с, 1H), 5,97 (ушир.с, 2H), 7,26-7,38 (м, 5H); МС m/z 500 [M+H]+.
[0179] Стадия 2
Соединение приведенной выше стадии 1 (4,689 г, 9,386 ммоль) растворяли в дегидратированном хлороформе (50 мл) и добавляли триэтиламин (1,40 г), с последующим охлаждением льдом. К полученной смеси добавляли трифосген (1,09 г), с последующим перемешиванием в течение 0,5 часа, и завершение реакции получения указанного в заголовке соединения подтверждали ТСХ. К полученной смеси добавляли метанол (0,255 мл) при охлаждении льдом, с последующим перемешиванием в течение 30 минут. Последовательно добавляли метансульфоновую кислоту (8,89 г), с последующим перемешиванием в течение 30 минут. Завершение реакции получения указанного в заголовке соединения подтверждали ТСХ. Полученную смесь по каплям добавляли к охлажденному льдом 1М водному раствору гидрокарбоната калия (11,1 г/100 мл), с последующим перемешиванием в течение 0,5 часа. Затем добавляли хлороформ (30 мл) для разделения слоев. Органический слой последовательно промывали 1M хлористоводородной кислотой (70 мл), насыщенным бикарбонатом натрия (70 мл) и насыщенным раствором соли (70 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток растворяли в хлороформе (16 мл) и добавляли гексан (24 мл), с последующим перемешиванием в течение 15 минут. Дополнительно добавляли гексан (8 мл), с последующим перемешиванием еще в течение 15 минут. Выпавшее в осадок твердое вещество отфильтровывали, промывали смесью хлороформ/гексан (2/3) и сушили при пониженном давлении, с получением 3,51 г указанного в заголовке соединения в виде бесцветного кристаллического порошка (выход 88%).
1H ЯМР (400 МГц, CDCl3) δ 1,67-1,77 (м, 1H), 2,08 (д, J=14,2 Гц, 1H), 2,14-2,26 (м, 2H), 2,30 (д, J=13,8 Гц, 2H), 2,55-2,66 (м, 2H), 3,10-3,24 (м, 4H), 3,34 (ушир.с, 1H), 4,45 (д, J=6,4 Гц, 1H), 4,91 (д, J=11,2 Гц, 1H), 5,06 (д, J=11,4 Гц, 1H), 5,97 (ушир.с, 2H), 7,34-7,45 (м, 5H); МС m/z 426 [M+H]+.
[0180] Ссылочный пример 19
(3aR,7aS)-2-Гидроксигексагидро-1H-изоиндол-1,3(2H)-дион
[Химическая формула 58]
[0181] Гидроксиламинсульфат (24,975 г, 0,152 моль) растворяли в воде (75 мл) и добавляли (3aR,7aS)-гексагидроизобензофуран-1,3-дион (48,000 г). К полученной смеси добавляли 25% водный раствор гидроксида натрия (50 г) небольшими порциями в течение 15 минут, с последующим перемешиванием при 90°C в течение 1 часа. Полученную смесь охлаждали до комнатной температуры, дважды экстрагировали хлороформом 50 мл, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, полученный остаток растворяли в хлороформе, примеси отфильтровывали, и растворитель концентрировали при пониженном давлении. Полученный остаток растворяли добавлением этилацетата, полученный раствор концентрировали при пониженном давлении и сушили в вакууме в течение следующих 2 дней, с получением 49,35 г указанного в заголовке соединения в виде бесцветного твердого вещества (выход 94%).
1H ЯМР (400 МГц, CDCl3) δ 1,45 (дт, J=3,0, 5,9 Гц, 4H), 1,71-1,90 (м, 4H), 2,84-2,92 (м, 2H), 6,01 (ушир.с, 1H); МС m/z 168 [M-H]-.
[0182] Ссылочный пример 20
(3aR,7aS)-1,3-Диоксогексагидро-1H-изоиндол-2(3H)-ил (2S,5R)-6-(бензилокси)- 7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат
[Химическая формула 59]
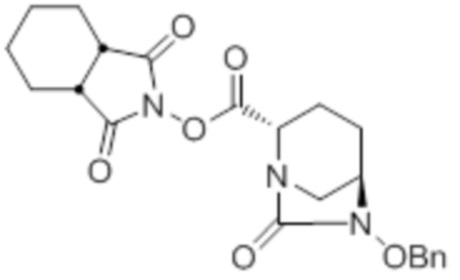
[0183] Стадия 1
1-трет-бутил 2-((3aR,7aS)-1,3-диоксогексагидро-1H-изоиндол-2(3H)-ил) (2S,5R)-5-((бензилокси)амино)пиперидин-1,2-дикарбоксилат
[Химическая формула 60]
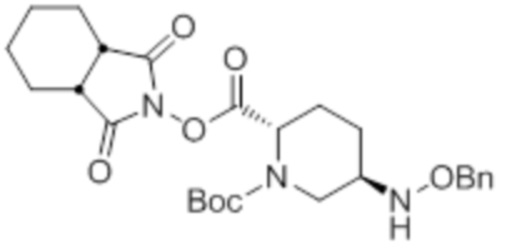
[0184] (2S,5R)-5-((Бензилокси)амино)-1-(трет-бутоксикарбонил)пиперидин-2-карбоновую кислоту (ссылочный пример 3, стадия 1, 3,504 г, 10 ммоль) растворяли в дегидратированном тетрагидрофуране (50 мл), с последующим охлаждением до около -20°C. К полученной смеси по каплям добавляли изобутилхлорформиат (1,51 г) и затем триэтиламин (2,17 г), с последующим перемешиванием при этой же температуре в течение 15 минут. Затем к полученному реакционному раствору добавляли (3aR,7aS)-2-гидроксигексагидро-1H-изоиндол-1,3(2H)-дион (ссылочный пример 19, 1,86 г), с последующим перемешиванием при этой же температуре в течение 30 минут и затем при комнатной температуре в течение 30 минут. Реакционный раствор разбавляли этилацетатом (200 мл), последовательно промывали охлажденной льдом 10% лимонной кислотой (60 мл), насыщенным бикарбонатом натрия (60 мл) и насыщенным раствором соли (60 мл), сушили над безводным сульфатом магния и фильтровали. Остаток, полученный при концентрировании фильтрата при пониженном давлении, подвергали колоночной хроматографии на силикагеле (гексан/этилацетат=2/1), с получением 4,521 г указанного в заголовке соединения в виде бесцветного пенообразного твердого вещества (выход 90%).
1H ЯМР (400 МГц, CDCl3) δ 1,35-1,58 (м, 13H), 1,62 (ушир.с, 1H), 1,76 (ушир.с, 2H), 1,90 (ушир.с, 4H), 1,95-2,15 (м, 2H), 3,00 (ушир.с, 2H), 3,15-3,30 (м, 2H), 4,16-4,25 (м, 1H), 4,72 (q, J=11,6 Гц, 2H), 5,30-5,53 (м, 1H), 7,26-7,38 (м, 5H); МС m/z 502 [M+H]+.
[0185] Стадия 2
Соединение приведенной выше стадии 1 (4,521 г, 9,01 ммоль) растворяли в дегидратированном хлороформе (50 мл) и добавляли триэтиламин (1,350 г), с последующим охлаждением льдом. К полученной смеси добавляли трифосген (1,043 г), с последующим перемешиванием в течение 0,5 часов, и завершение реакции получения указанного в заголовке соединения подтверждали ТСХ. К полученной смеси добавляли метанол (0,245 мл) при охлаждении льдом, с последующим перемешиванием в течение 30 минут. Впоследствии добавляли метансульфоновую кислоту (8,53 г), с последующим перемешиванием в течение 30 минут. Завершение реакции получения указанного в заголовке соединения подтверждали ТСХ. Полученную смесь по каплям добавляли к охлажденному льдом 1M водному раствору гидрокарбоната калия (10,668 г/90 мл), с последующим перемешиванием в течение 0,5 часа. Затем добавляли хлороформ (33 мл) для разделения слоев. Органический слой последовательно промывали 1M хлористоводородной кислотой (70 мл), насыщенным бикарбонатом натрия (70 мл) и насыщенным раствором соли (70 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии на силикагеле (хлороформ/этилацетат=6/1), с получением 3,106 г указанного в заголовке соединения в виде бесцветного твердого вещества (выход 81%).
1H ЯМР (400 МГц, CDCl3) δ 1,50 (ушир.с, 4H), 1,62 (ушир.с, 1H), 1,68-1,84 (м, 1H), 1,91 (ушир.с, 4H), 2,04-2,27 (м, 2H), 3,02 (ушир.с, 2H), 3,15 (с, 2H), 3,35 (ушир.с, 1H), 4,47 (д, J=6,6 Гц, 1H), 4,92 (д, J=11,2 Гц, 1H), 5,07 (д, J=11,4 Гц, 1H), 7,34-7,45 (м, 5H); МС m/z 428 [M+H]+.
[0186] Пример 1
трет-Бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (IV-1)
Пример 1a
[Химическая формула 61]
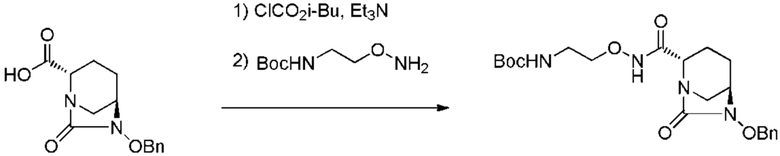
Раствор (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (4,80 кг, 17,373 моль) в дегидратированном этилацетате (62 л) охлаждали до -30°C, последовательно по каплям добавляли изобутилхлорформиат (2,52 кг) и триэтиламин (1,85 кг), с последующим перемешиванием при -30°C в течение 15 минут. К полученному реакционному раствору добавляли раствор трет-бутил 2-(аминоокси)этилкарбамата в дегидратированном этилацетате (15% масс., 23,45 кг) в течение 30 минут (промытый 2 л дегидратированного этилацетата), и температуру повышали до 0°C в течение 1 часа. Полученную смесь последовательно промывали 8% лимонной кислотой (65 л), 5% бикарбонатом натрия (60 л) и водой (60 л), и концентрировали до 24 л. К концентрату добавляли этилацетат (24 л), и полученную смесь концентрировали с заменой растворителя дважды до 24 л. К полученному в результате концентрату добавляли этилацетат (29 л) и гексан (72 л), с последующим перемешиванием в течение ночи. К полученной смеси по каплям добавляли гексан (82 л), с последующим перемешиванием в течение 2 часов. Выпавшие в осадок кристаллы отфильтровывали, промывали гексаном и сушили в вакууме, с получением 5,51 кг указанного в заголовке соединения (выход 76%). Инструментальные данные согласуются с данными ссылочного примера 5, стадия 1.
[0187] Пример 1b
[Химическая формула 62]
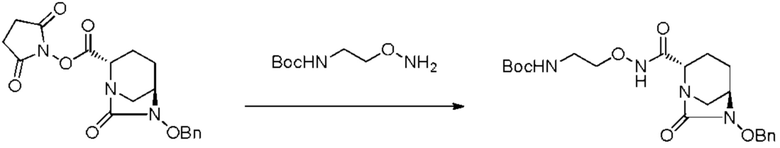
2,5-Диоксопирролидин-1-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (ссылочный пример 3, 373 мг, 1 ммоль) растворяли в дегидратированном дихлорметане (5 мл), и к полученному добавляли раствор трет-бутил 2-(аминоокси)этилкарбамата (194 мг) в дегидратированном дихлорметане (2 мл, промытый 1 мл) при охлаждении льдом, с последующим перемешиванием в течение 1 часа. Реакционный раствор разбавляли этилацетатом (65 мл), последовательно промывали 10% лимонной кислотой (20 мл), насыщенным бикарбонатом натрия (20 мл) и насыщенным раствором соли (20 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, с получением 362 мг указанного в заголовке соединения (выход 83%). Инструментальные данные согласуются с данными для соединения ссылочного примера 5, стадия 1.
[0188] Пример 1c
[Химическая формула 63]
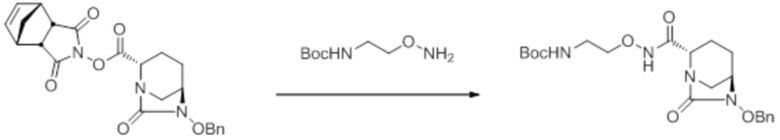
(1R,2S,6R,7S)-3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дек-8-ен-4-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (ссылочный пример 4, 49,7 г, 113,6 ммоль) суспендировали в дегидратированном этилацетате (650 мл). К полученной суспензии добавляли раствор трет-бутил 2-(аминоокси)этилкарбамата (24,2 г) в дегидратированном этилацетате (134 мл) и триэтиламин (13,8 г) при комнатной температуре, с последующим перемешиванием в течение 2,5 часов. Реакционный раствор разбавляли этилацетатом (0,8 л), последовательно промывали охлажденной 0,25M хлористоводородной кислотой (1 л), насыщенным бикарбонатом натрия (1 л) и водой (1 л), и концентрировали при пониженном давлении, с получением 48,08 г указанного в заголовке соединения (выход 98%, ВЭЖХ соотношение площади 99% или более). Инструментальные данные согласуются с данными для соединения ссылочного примера 5, стадия 1.
[0189] Пример 1d
[Химическая формула 64]
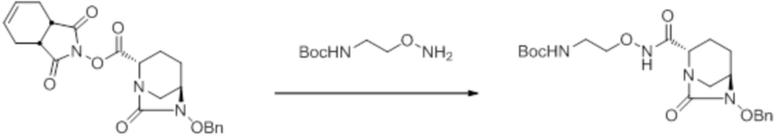
[0190] (3aR,7aS)-1,3-Диоксо-3a,4,7,7a-тетрагидро-1H-изоиндол-2(3H)-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (ссылочный пример 18, 425 мг, 1 ммоль) растворяли в дегидратированном хлороформе (5 мл), и добавляли раствор трет-бутил 2-(аминоокси)этилкарбамата (211 мг) в дегидратированном этилацетате (1,41g) и триэтиламин (121 мг) при охлаждении льдом, с последующим перемешиванием в течение 30 минут. Реакционный раствор разбавляли этилацетатом (75 мл), последовательно промывали 10% лимонной кислотой (35 мл), насыщенным бикарбонатом натрия (35 мл) и насыщенным раствором соли (35 мл), и сушили над безводным сульфатом магния, и фильтрат концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии на силикагеле (гексан/этилацетат=1/2), с получением 481 мг указанного в заголовке соединения (количественный). Инструментальные данные согласуются с данными для соединения ссылочного примера 5, стадия 1.
[0191] Пример 1e
[Химическая формула 65]
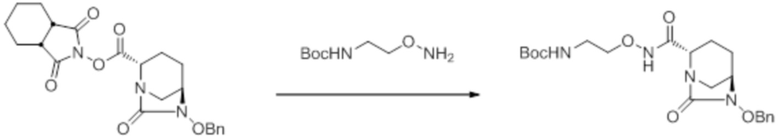
[0192] (3aR,7aS)-1,3-Диоксогексагидро-1H-изоиндол-2(3H)-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (ссылочный пример 20, 427 мг, 1 ммоль) растворяли в дегидратированном хлороформе (5 мл), и добавляли раствор трет-бутил 2-(аминоокси)этилкарбамата (211 мг) в дегидратированном этилацетате (1,41 г) и триэтиламин (121 мг) при охлаждении льдом, с последующим перемешиванием в течение 30 минут. Реакционный раствор разбавляли этилацетатом (75 мл), последовательно промывали 10% лимонной кислотой (35 мл), насыщенным бикарбонатом натрия (35 мл) и насыщенным раствором соли (35 мл), и сушили над безводным сульфатом магния, и фильтрат концентрировали при пониженном давлении. Полученный остаток подвергали колоночной хроматографии на силикагеле (гексан/этилацетат=1/2), с получением 418 мг указанного в заголовке соединения (выход 96%). Инструментальные данные согласуются с данными для соединения ссылочного примера 5, стадия 1.
[0193] Пример 2
трет-Бутил{2-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (V-1)
[Химическая формула 66]
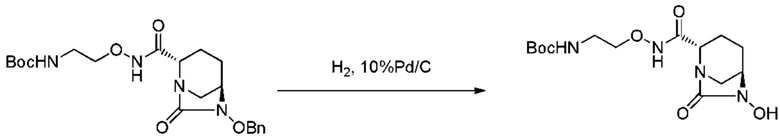
К раствору трет-Бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (IV-1, 5,52 кг, 12,705 моль) в метаноле (85 л) добавляли 10% катализатор палладий на угле (50% масс., 0,55 кг), с последующим перемешиванием в течение 1 часа в атмосфере водорода под давлением (0,1 МПа). Катализатор отфильтровывали, и твердое вещество промывали метанолом (25 л). Фильтраты объединяли и концентрировали при пониженном давлении до 39 л при температуре жидкости 10°C или ниже. К концентрату добавляли ацетонитрил (44 л), и полученную смесь концентрировали с заменой растворителя до 39 л при температуре жидкости 10°C или ниже. Данную операцию проводили дважды. Полученную смесь охлаждали до 0°C, с последующим перемешиванием в течение ночи. Выпавшие в осадок кристаллы отфильтровывали, промывали ацетонитрилом (24 л) и сушили в вакууме, с получением 3,63 кг указанного в заголовке соединения (выход 83%). Инструментальные данные согласуются с данными для соединения ссылочного примера 5, стадия 2.
[0194] Пример 3
Тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (VI-1)
Пример 3a
[Химическая формула 67]
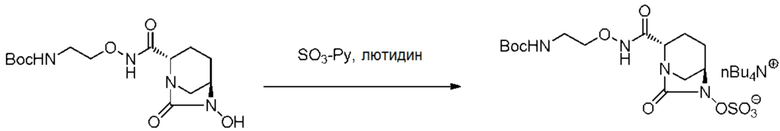
К ацетонитрилу (51 л) последовательно добавляли воду (51 мл), трет-бутил{2-[({[(2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (V-1, 3,53 кг, 10,251 моль), комплекс триоксида серы-пиридина (3,95 кг) и 2,6-лютидин (2,21 кг), с последующим перемешиванием при 35-45°C в течение ночи. Полученную смесь фильтровали для удаления не растворившихся веществ, и твердое вещество промывали ацетонитрилом (11 л). Фильтраты объединяли и концентрировали до 17 л. Концентрат охлаждали до 10°C или ниже, и слои разделяли добавлением 9% дигидрофосфата натрия (60 л) и этилацетата (113 л). Органический слой снова экстрагировали 9% дигидрофосфатом натрия (11 л). К полученному в результате водному слою добавляли этилацетат (113 л), водный раствор 30% тетрабутиламмонийгидросульфата (12,87 кг) и 37% дигидрофосфат натрия (56,5 кг), с последующим перемешиванием в течение 15 минут. Органический слой отделяли, промывали 20% дигидрофосфатом натрия (60 л), сушили над безводным сульфатом магния (2,5 кг), фильтровали и затем концентрировали при пониженном давлении. Кристаллы указанного в заголовке соединения осаждали из концентрированных растворов и растворяли в этилацетате, и общий объем жидкости доводили до 20 л, с получением 32,55 кг раствора указанного в заголовке соединения в этилацетате (чистый выход 6,25 кг, выход 92%). Полученный раствор подвергали следующей стадии без дополнительной очистки.
[0195] Пример 3b: Синтез в одном реакторе из трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (IV-1)
[Химическая формула 68]
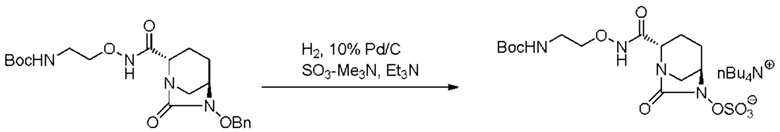
К раствору трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (IV-1, 515 мг, 1,16 ммоль) в изопропаноле (7 мл) добавляли воду (5 мл), комплекс триоксид серы-триметиламин (196 мг), триэтиламин (0,0407 мл) и 10% палладиевый катализатор на углероде (53,3% масс., 95,0 мг) в атмосфере водорода, с последующим перемешиванием при комнатной температуре в течение 2 часов. Дополнительно добавляли 10% палладиевый катализатор на углероде (53,3% масс., 63,5 мг), и полученную смесь перемешивали при комнатной температуре в течение 3 часов в атмосфере водорода и затем заменяли газообразным аргоном, с последующим перемешиванием при комнатной температуре в течение 1 часа. Катализатор из реакционного раствора фильтровали через рыхлый слой целита и промывали смесью изопропанол/вода (1/1, 40 мл), и затем фильтровали через MF (Millipore) и промывали смесью изопропанол/вода (1/1, 15 мл). Изопропанол затем отгоняли при пониженном давлении. К полученному в результате водному раствору добавляли дигидрофосфат натрия (5,29 г), этилацетат (20 мл) и тетрабутиламмонийгидросульфат (476 мг), с последующим перемешиванием при комнатной температуре в течение 10 минут. Полученную смесь затем дважды экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, с получением 702 мг указанного в заголовке соединения (выход 91%).
[0196] Пример 3c: Последовательный синтез из трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (IV-1)
[Химическая формула 69]
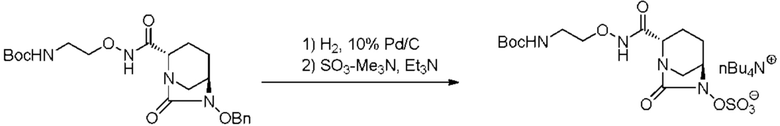
К раствору трет-бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (IV-1, 5,0 г, 11,51 ммоль) в изопропаноле (80 мл) добавляли 10% катализатор палладий на угле (50% масс., 0,5 г), с последующим перемешиванием в течение 2 часов в атмосфере водорода. Катализатор из реакционного раствора отфильтровывали через рыхлый слой целита, и твердое вещество промывали изопропанолом (15 мл). Фильтраты затем объединяли, и к полученному добавляли воду (47,5 мл), комплекс триоксида серы-триметиламина (1,8 г) и триэтиламин (0,237 г), с последующим перемешиванием при 25-30°C в течение 24 часа. Полученную смесь концентрировали до 47 мл при пониженном давлении. К полученному добавляли дигидрофосфат натрия (11,87 г), этилацетат (200 мл) и тетрабутиламмонийгидросульфат (4,688 г), с последующим перемешиванием в течение 10 минут. Органический слой отделяли, и водный слой дополнительно экстрагировали этилацетатом (2×100 мл). Объединенные органические фазы сушили над сульфатом магния и фильтровали. Органический растворитель фильтрата концентрировали при пониженном давлении, с получением раствора указанного в заголовке соединения в этилацетате (чистый выход 6,522 г, выход 85%)
[0197] Пример 4
(2S,5R)-N-(2-Аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1-CR)
[Химическая формула 70]
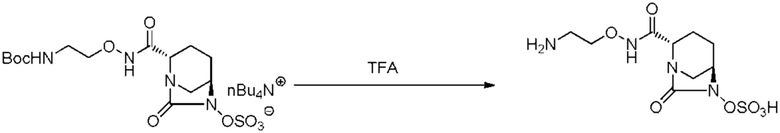
Пример 4a
Раствор тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамата (VI-1, 788 г, чистый выход 467,1 г, 0,701 моль) в дихлорметане (934 мл) охлаждали до -20°C в струе азота, по каплям добавляли трифторуксусную кислоту (934 мл) в течение 15 минут, и температуру повышали до 0°C, с последующим перемешиванием в течение 1 часа. Реакционный раствор охлаждали до -20°C, к полученному по каплям добавляли диизопропиловый эфир (4,17 л), температуру полученной смеси поднимали до -6°C, и полученную смесь перемешивали в течение 1 часа. Выпавшие в осадок вещества отфильтровывали и промывали диизопропиловым эфиром (2×1 л), и влажное твердое вещество сушили в вакууме, с получением 342,08 г указанного в заголовке соединения (чистый выход 222,35 г, выход 98%, ВЭЖХ соотношение площади 96,1%, CE/TFA 27% моль).
[0198] Пример 4b
Тетрабутиламмоний трет-бутил{2-[({[(2S,5R)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}карбамат (VI-1, этилацетатный раствор 15,60 кг, чистый выход 2,98 кг) охлаждали до температуры жидкости 0°C или ниже и концентрировали до 9 л. К концентрату добавляли дихлорметан (9 л), и полученную смесь охлаждали до -20°C в струе азота. К полученной смеси по каплям добавляли трифторуксусную кислоту (16,5 л) при -5°C или ниже в течение около 1 часа, с последующим перемешиванием при (-5)-(-0)°C в течение 1 часа. К полученному реакционному раствору порциями добавляли этилацетат, охлажденный до 0°C, при 7°C или ниже (4×8,3 л, 24,6 л, 57,8 л в общей сложности), с последующим перемешиванием при 0°C в течение ночи. Выпавшие в осадок вещества отфильтровывали, промывали этилацетатом (13,5 л, 9 л) и сушили в вакууме, с получением 1,74 кг указанного в заголовке соединения (чистый выход 1,43 кг, выход 99%, ВЭЖХ соотношение площади 99,1%, CE/TFA 10% моль, GC/EtOAc 14%).
[0199] Пример 5
Кристаллическая форма I (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Пример 5a
0,5M Буфер уксусной кислоты (pH 5,5, 35 мл) охлаждали льдом, и к полученному поочередно добавляли (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1-CR, 36 г) и охлажденный 5M гидроксид натрия с доведением pH до 5,5. Полученную смесь подвергали колоночной хроматографии на октадецилсиликагеле (3,6 л) и элюировали водой. Активные фракции собирали и концентрировали при пониженном давлении на водяной бане при 35°C. Выпавшие в осадок кристаллы сушили в вакууме в течение ночи. 2,10 г Полученных в результате кристаллов растирали в порошок, и затем добавляли смесь изопропанол/вода (19/1, 13 мл) при охлаждении льдом, с последующим растиранием при 0°C в течение 1 часа. Полученную суспензию фильтровали, с последующим промыванием охлажденной смесью изопропанол/вода (19/1, 80 мл). Полученные в результате кристаллы сушили в вакууме, с получением 1,68 г указанного в заголовке соединения (выход 80%). ДСК эндотермический пик: 111°C. Растворимость в водном 60% изопропанольном растворе: 0,44% (10°C), 0,48% (20°C). Указанное в заголовке соединение показало характеристические пики на диаграмме порошковой дифракции рентгеновских лучей, как приведено в таблице 4 и на фиг.1 ниже. Инструменты и параметры анализа были следующими: рентгеновский порошковый дифрактометр: RINT2100 от Rigaku Corporation; источник рентгеновского излучения: CuKα1, напряжение трубки: 40 кВ, сила тока трубки: 40 мА, скорость сканирования 4°/мин, диапазон сканирования: 2θ=3-40°.
[0200]
Порошковая рентгеновская дифракция кристаллической формы I
[0201] Пример 5b
(2S,5R)-N-(2-Аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1-CR, чистый выход 4,253 г) растворяли в 0,2M фосфатном буфере (pH 6,5, 73 мл) и pH доводили до 5,5, с последующим разбавлением водой (20 мл). Полученную смесь концентрировали до 130 мл, подвергали очистке на смоле (SP207, 260 мл) и элюировали водой (238 мл) и 10% водным раствором изопропанола (780 мл). Активные фракции собирали и концентрировали до 30 мл при пониженном давлении. К полученному добавляли активированный уголь (Seisei Shirasagi, 87 мг), с последующим перемешиванием при комнатной температуре в течение 30 минут. Активированный уголь отфильтровывали через мембранный фильтр, и фильтрат подвергали лиофилизации, с получением 4,07 г (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1) в аморфной форме (выход 95,7%). Полученное аморфное соединение (0,2 г) растворяли в воде (0,8 мл), и в полученный раствор добавляли изопропанол (1,2 мл) и затравляли кристаллической формой I (пример 5a, 1 мг) при комнатной температуре, с последующим перемешиванием на магнитной мешалке в течение 3 часов. Выпавшие в осадок кристаллы отфильтровывали и сушили, с получением 0,1 г указанного в заголовке соединения (выход 50%). Данные кристаллы показали такую же картину пиков, как у кристаллов примера 5a на диаграмме порошковой рентгеновской дифракции.
[0202] Пример 5c
(2S,5R)-N-(2-Аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1-CR, чистый выход 2,113 г) и 0,2M фосфатный буфер (pH 6,5, 73 мл) добавляли поочередно, и pH доводили до 4,6, с последующим разбавлением водой (27 мл). Полученную смесь концентрировали до 80 мл при пониженном давлении, и затем pH доводили до 5,4 добавлением 0,2M фосфатного буфера (pH 6,5, 16 мл), с последующим разбавлением водой (48 мл). Полученную смесь подвергали очистке на смоле (SP207, 240 мл) и элюировали водой (276 мл) и 10% водным раствором изопропанола (720 мл). Активные фракции собирали и концентрировали при пониженном давлении до 12 мл. К полученному добавляли активированный уголь (Seisei Shirasagi, 40 мг), с последующим перемешиванием при комнатной температуре в течение 30 минут. Активированный уголь отфильтровывали через мембранный фильтр, с последующим разбавлением водой до 14 мл. Водный раствор затравливали кристаллической формой I (пример 5b, 6 мг), перемешивали на магнитной мешалке при комнатной температуре. К полученной в результате суспензии по каплям добавляли изопропанол (84 мл) в течение 1 часа. После завершения добавления по каплям, полученную смесь перемешивали в течение 3 часов. Выпавшие в осадок кристаллы отфильтровывали и сушили, с получением 1,834 г указанного в заголовке соединения (выход 86,8%). Содержание воды: 5,37%, содержание водного продукта: 95,3%, ВЭЖХ соотношение площади 99,3%. Данные кристаллы показали такую же картину пиков, как у кристаллов примера 5a на диаграмме порошковой рентгеновской дифракции.
[0203] Пример 6
Кристаллическая форма II (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Пример 6a
0,2M фосфатный буфер (pH 6,5, 0,8 л) охлаждали до 10°C или ниже. К полученному добавляли при одновременном перемешивании (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1-CR, чистый выход 49,96 г) и охлажденный 0,2M фосфатный буфер (pH 6,5) поочередно небольшими порциями для доведения pH от 4,2 до 5,5 и, наконец, до pH 5,5. Полученную смесь разбавляли водой (общее количество 1,8 л) и концентрировали при пониженном давлении при температуре жидкости 18°C или ниже до 1,6 л. Концентрат разбавляли водой до 1,8 л (ВЭЖХ соотношение площади 96,7%), подвергали очистке на смоле (Sepabeads SP207, 3 л), и элюировали водой (0,83 л) и 10% водным раствором изопропанола, собирая активные фракции. Активные фракции объединяли (1,5 л) и концентрировали при пониженном давлении при температуре жидкости 15°C или ниже до 0,5 л. К полученному добавляли активированный уголь (0,88 г), с последующим перемешиванием в течение 30 минут. Активированный уголь отфильтровывали через мембранный фильтр и промывали водой (0,05 л×2). Фильтраты объединяли и концентрировали при пониженном давлении при температуре жидкости 15°C или ниже до 0,2 л, и температуру жидкости доводили до 10-15°C. К полученной смеси по каплям добавляли изопропанол (0,25 л) в течение 10 минут. После перемешивания в течение 1 часа, по каплям дополнительно добавляли изопропанол (0,6 л) в течение 15 минут. Полученную смесь перемешивали в течение 1 часа, и выпавшие в осадок кристаллы отфильтровывали, промывали изопропанолом (0,2 л) и сушили в вакууме до тех пор, пока температура вещества не устанавливалась в 20°C, с получением 44,69 г указанного в заголовке соединения (выход 85%, содержание воды 5,9%, ВЭЖХ соотношение площади 100%). ДСК эндотермический пик: 92°C. Растворимость в водном 60% изопропанольном растворе: 0,67% (10°C), 0,74% (20°C).
[0204] Указанное в заголовке соединение также показало картину характеристических пиков на диаграмме порошковой рентгеновской дифракции, как приведено в таблице 5 и на фиг.2 ниже. Инструменты и параметры анализа были следующими: рентгеновский порошковый дифрактометр: RINT2100 от Rigaku Corporation; источник рентгеновского излучения: CuKα1, напряжение трубки: 40 кВ, сила тока трубки: 40 мА, скорость сканирования 4°/мин, диапазон сканирования: 2θ=3-40°.
[0205]
Данные порошкового рентгеноструктурного анализа
Порошковая рентгеновская дифракция кристаллической формы II
[0206] Пример 6b
Эксперимент на преобразование кристаллической формы II (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Отбирали небольшое количество суспендированных кристаллов примера 6a и перемешивали при комнатной температуре в течение одного дня. Выпавшие в осадок кристаллы собирали и подвергали порошковому рентгеноструктурному анализу. Не наблюдалось преобразования кристаллов в различные кристаллические формы.
[0207] Пример 7
Кристаллическая форма III (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Пример 7a
0,2M фосфатный буфер (pH 6,5, 3,0 л) охлаждали до 10°C или ниже, и к полученному добавляли при одновременном перемешивании (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1-CR, чистый выход 133,334 г) и охлажденный 0,2M фосфатный буфер (pH 6,5, 1,8 л) поочередно небольшими порциями для доведения pH от 5,1 до 5,5, и, наконец, до pH 5,3. Полученную смесь концентрировали при пониженном давлении при температуре жидкости 18°C или ниже до 3,6 л. pH Концентрата доводили до pH 5,5 добавлением 0,2M фосфатного буфера (pH 6,5). Концентрат разбавляли водой до 4,8 л, подвергали очистке на смоле (Sepabeads SP207, 7L) и элюировали водой (7,2 л) и 10% водным раствором изопропанола, собирая активные фракции. Активные фракции объединяли (3,1 л) и концентрировали при пониженном давлении при температуре жидкости 15°C или ниже до 1,8 л. К полученному добавляли активированный уголь (2,66 г), с последующим перемешиванием в течение 30 минут. Активированный уголь отфильтровывали через мембранный фильтр и промывали водой (0,39 л). Фильтраты объединяли и концентрировали при пониженном давлении при температуре жидкости 18°C или ниже до 0,6 л. Температуру жидкости концентрата доводили до 20-25°C, и к полученному по каплям добавляли изопропанол (0,77 л). Кристаллическую форму II (пример 6a, 0,63 г) затравляли кристаллами, с последующим перемешиванием в течение 1 часа. К полученной смеси дополнительно по каплям добавляли изопропанол (1,93 л) около 1,5 часов, с последующим перемешиванием в течение 30 минут. Выпавшие в осадок кристаллы отфильтровывали, промывали изопропанолом (1 л) и сушили в вакууме до тех пор, пока температура вещества не устанавливалась в 20°C. Получали 127,3 г указанного в заголовке соединения, содержащего небольшое количество кристаллической формы II (выход 90%, содержание воды 5,3%, ВЭЖХ соотношение площади 99,9%). Кристаллы, полученные на данной стадии, использовали в качестве затравочных кристаллов, и эту же стадию повторяли. Полученные в результате кристаллы дополнительно использовали в качестве затравочных кристаллов на следующей стадии, с получением указанного в заголовке соединения только в виде кристаллической формы III по данным порошкового рентгеноструктурного анализа кристаллов. ДСК эндотермический пик: 102°C. Растворимость в водном 60% изопропанольном растворе: 0,76% (10°C), 0,80% (20°C). Указанное в заголовке соединение показало картину характеристических пиков на диаграмме порошковой рентгеновской дифракции, как приведено в таблице 6 и на фиг.3 ниже. Инструменты и параметры анализа были следующими: рентгеновский порошковый дифрактометр: RINT2100 от Rigaku Corporation; источник рентгеновского излучения: CuKα1, напряжение трубки: 40 кВ, сила тока трубки: 40 мА, скорость сканирования 4°/мин, диапазон сканирования: 2θ=3-40°.
[0208]
Данные порошкового рентгеноструктурного анализа
Порошковая рентгеновская дифракция кристаллической формы III
[0209] Пример 7b
0,2M фосфатный буфер (pH 6,5, 7,2 л) охлаждали до 10°C или ниже, и к полученному добавляли при одновременном перемешивании (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (VII-1-CR, чистый выход 1,2 кг) и охлажденный льдом 0,2M фосфатный буфер (pH 6,5, 3,5 л) поочередно небольшими порциями для доведения pH от 4,2 до 4,8, и, наконец, до pH 4,6. Полученную смесь разбавляли водой (19,3 л) (общее количество 30 л) и концентрировали при пониженном давлении при температуре жидкости 18°C или ниже до 24 л. pH концентрата доводили до pH 5,4 добавлением 0,2M фосфатного буфера (pH 6,5, 2,4 л) (ВЭЖХ соотношение площади 98,5%). Концентрат разбавляли водой до 43,2 л, подвергали очистке на смоле (Sepabeads SP207, 75 л) и элюировали водой (83 л) и 10% водным раствором изопропанола, собирая активные фракции. Активные фракции объединяли (33 л) и концентрировали при температуре жидкости 15°C или ниже до 7,2 л. К полученному добавляли активированный уголь (24 г), с последующим перемешиванием в течение 30 минут. Активированный уголь отфильтровывали через мембранный фильтр и промывали водой (0,4 л×2). Фильтраты объединяли, и температуру жидкости доводили до 20-25°C. Их затравливали кристаллами кристаллической формы III (пример 7a, 3,6 г). К полученной смеси по каплям добавляли изопропанол (50,4 л) в течение 1 часа, с последующим перемешиванием в течение ночи. Выпавшие в осадок кристаллы отфильтровывали, промывали изопропанолом (4,8 л) и сушили в вакууме до тех пор, пока температура вещества не устанавливалась в 20°C, с получением 1,17 кг указанного в заголовке соединения (выход 90%, содержание воды 5,3%, ВЭЖХ соотношение площади 100%). Данные кристаллы показали такую же картину пиков, как у кристаллов примера 7a на диаграмме порошковой рентгеновской дифракции.
[0210] Пример 7c
Эксперимент на преобразование кристаллов кристаллической формы III (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Отбирали небольшое количество суспензии соединения со стадии примера 7a сразу после внесения затравки и суспензии после добавления по каплям изопропанола, и каждую из взятых порций перемешивали на магнитной мешалке при комнатной температуре в течение одного дня и четырех дней. Выпавшие в осадок кристаллы собирали и подвергали порошковому рентгеноструктурному анализу. Не наблюдалось преобразования кристаллов в различные кристаллические формы.
[0211] Пример 7d
Эксперимент дифракции рентгеновских лучей-дифференциальной сканирующей калометрии (XRD-ДСК) кристаллической формы III (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Преобразованием кристаллов при нагревании в полиморф кристаллической формы III примера 7a исследовали при использовании XRD-ДСК. Условия нагревания и охлаждения были следующими: температуру повышали со скоростью 2°C/мин от комнатной температуры до 160°C и затем охлаждали до 63°C при постоянной относительной влажности (RH) 60%. ДСК и XRD образцов последовательно измеряли. Инструменты и параметры анализа были следующими: рентгеновский порошковый дифрактометр: SmartLab и XRD-ДСК от Rigaku Corporation; источник рентгеновского излучения: CuKα1, напряжение трубки: 45 кВ, сила тока трубки: 200 мА, скорость сканирования 80°/мин, диапазон сканирования: 2θ=5-35°.
[0212] Пример 8
Кристаллическая форма IV (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Пример 8a
0,20 г кристаллической формы III примера 7a растворяли в 2 мл воды. К полученному по каплям добавляли при одновременном перемешивании метанол (30 мл), с последующим выстаиванием при 20-25°C в течение ночи. Полученную смесь фильтровали, промывали метанолом (2×2 мл) и сушили в вакууме при комнатной температуре в течение ночи, с получением 0,13 г указанного в заголовке соединения (выход 68%).
Указанное в заголовке соединение показало картину характеристических пиков на диаграмме порошковой рентгеновской дифракции, как приведено в таблице 7 и на фиг.4 ниже. Инструменты и параметры анализа были следующими: рентгеновский порошковый дифрактометр: RINT2100 от Rigaku Corporation; источник рентгеновского излучения: CuKα1, напряжение трубки: 40 кВ, сила тока трубки: 40 мА, скорость сканирования 4°/мин, диапазон сканирования: 2θ=3-40°.
[0213]
Данные порошкового рентгеноструктурного анализа
Порошковая рентгеновская дифракция кристаллической формы IV
[0214] Пример 8b
К 25 г кристаллической формы III примера 7a добавляли метанол (200 мл), с последующим перемешиванием при 20-25°C в течение 3,5 часов. Полученную смесь фильтровали, промывали метанолом (2×20 мл) и сушили в вакууме при комнатной температуре в течение ночи, с получением 23 г указанного в заголовке соединения (выход 99%). Данные кристаллы показали такую же картину пиков, как у кристаллов примера 8a на диаграмме порошковой рентгеновской дифракции.
[0215] Пример 8c
К 25 г кристаллической формы III примера 7a добавляли этанол (200 мл), с последующим перемешиванием при 20-25°C в течение 3,5 часов. Полученную смесь фильтровали, промывали этанолом (2×20 мл) и сушили в вакууме при комнатной температуре в течение ночи, с получением 23 г указанного в заголовке соединения (выход 99%). Данные кристаллы показали такую же картину пиков, как у кристаллов примера 8a на диаграмме порошковой рентгеновской дифракции.
[0216] Пример 8d
Эксперимент на преобразование кристаллов кристаллической формы IV (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Отбирали кристаллическую форму IV примеров 8a-c и добавляли туда смесь изопропанол/вода (6/1), и суспендировали при перемешивании при 25°C или 40°C в течение недели. Образцы отбирали спустя 12 часов, 24 часа (один день), 48 часов (два дня), 72 часов (три дня), 96 часов (четыре дня) и 168 часов (одна неделя) и подвергали порошковому рентгеноструктурному анализу после проточной сушки. Не наблюдалось преобразования кристаллов в различные кристаллические формы за все время перемешивания.
[0217] Пример 9a
Повышение стабильности кристаллических форм I-IV (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1)
Кристаллическую форму III примера 7a растворяли в воде и подвергали лиофилизации, с получением (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1) в аморфной форме. Каждое соединение в аморфной форме и кристаллические формы I-IV примеров 5-8 взвешивали во флаконе с закручивающейся крышкой, и тесты на стабильность проводили в каждом из температурных и влажных условий. Способы измерения связанных веществ, измерения содержания и измерения содержания воды были следующими. Результаты показаны в таблице 9.
[0218] Измерение связанных веществ и содержания
Образцы растворяли в воде, и полученные растворы использовали в качестве растворов образцов. Растворы образцов, каждый в количестве 5 мкл, тестировали в следующих условиях с помощью JP16, жидкостной хроматографии <2,01> с получением количества каждого связанного вещества (%) и общего количества связанных веществ (%), и содержания.
[0219] Условия тестирования:
Колонка: водная Atlantis dc18, 5 мкм, 4,6×250 мм
Температура колонки: постоянная температура около 35°C
Вводимое количество: 5 мкл
Детектор: поглощающий ультрафиолет фотометр (длина волны: 210 нм)
Подвижная фаза A: 1,32 г гидрофосфата диаммония растворяли в 900 мл воды и добавляли фосфорную кислоту для доведения pH до 3,0. К полученному затем добавляли воду до объема 1000 мл.
Подвижная фаза B: Ацетонитрил для жидкостной хроматографии
Градиентная программа: соотношение смешивания подвижной фазы A с подвижной фазой B регулировали по следующей временной программе.
Скорость потока: 1,0 мл/мин
Время удерживания: около 6,5 минут
Время измерения: 30 минут
[0220] Измерение содержания воды
Около 20 мг продукта точно отмеряли и тестировали с помощью JP16, определения воды <2,48> кулонометрическим титрованием.
[0221]
Условия хранения: 40°C/75% RH, герметичный контейнер
[0222] Пример 9b
Повышение стабильности кристаллической формы III (2S,5R)-N-(2-аминоэтокси)-7-оксо-6-(сульфокси)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида (VII-1) в контейнере упаковки
Кристаллическую форму III упаковывали в следующих условиях, и тестирование на стабильность проводили в условиях каждой из температур и влажности согласно условиям анализа примера 9a. Результаты показаны в таблицах 10-12.
Контейнер для упаковки
Внутренний пакет: полиэтилен низкой плотности нейлоновая связывающая лента
Наружный пакет: алюминиевый ламинированный пакет запаянный
[0223]
Стабильность кристаллической формы III
Внутренний пакет: полиэтиленовый пакет низкой плотности нейлоновая связывающая лента
Наружный пакет: алюминиевый ламинированный пакет запаянный
Условия хранения: 25°C/60% RH
[0224]
Стабильность кристаллической формы III
Внутренний пакет: полиэтиленовый пакет низкой плотности нейлоновая связывающая лента
Наружный пакет: алюминиевый ламинированный пакет·запаянный
Условия хранения: 40°C/75% RH
[0225]
Стабильность кристаллической формы III
Внутренний пакет: полиэтиленовый пакет низкой плотности·нейлоновая связывающая лента
Наружный пакет: алюминиевый ламинированный пакет·запаянный
Условия хранения: 60°C
[0226] Пример 10
трет-Бутил{2-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]этил}(метил)карбамат (IV-2)
[Химическая формула 71]
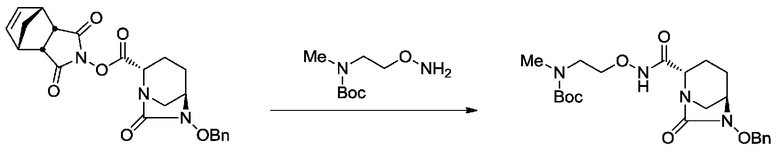
(1R,2S,6R,7S)-3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дек-8-ен-4-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (ссылочный пример 4, 144 мг, 0,329 ммоль) растворяли в дегидратированном дихлорметане (2,5 мл), и к полученному добавляли раствор трет-бутил(2-(аминоокси)этила)(метил)карбамата (88,8 мг) в дегидратированном дихлорметане (0,5 мл), с последующим перемешиванием при комнатной температуре в течение 18 часов. Реакционный раствор разбавляли этилацетатом (10 мл) и последовательно промывали 0,25M хлористоводородной кислотой, насыщенным бикарбонатом натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, с получением 132 мг указанного в заголовке соединения (выход 89%). Инструментальные данные согласуются с данными соединения ссылочного примера 6, стадия 1.
[0227] Пример 11
трет-Бутил{3-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пропил}карбамат (IV-7)
[Химическая формула 72]
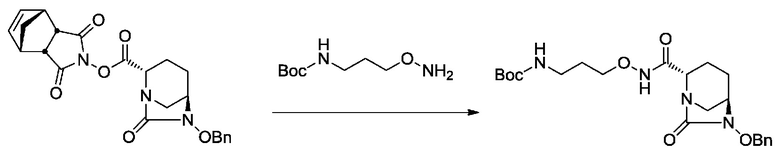
(1R,2S,6R,7S)-3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дек-8-ен-4-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (ссылочный пример 4, 148 мг, 0,339 ммоль) растворяли в дегидратированном дихлорметане (2,5 мл). К полученному добавляли раствор трет-бутил 3-(аминоокси)пропилкарбамата (90,9 мг) в дегидратированном дихлорметане (0,5 мл), с последующим перемешиванием в течение 18 часов при комнатной температуре. Реакционный раствор разбавляли этилацетатом (10 мл), последовательно промывали 0,25M хлористоводородной кислотой, насыщенным бикарбонатом натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, с получением 134 мг указанного в заголовке соединения (выход 88%). Инструментальные данные согласуются с данными соединения ссылочного примера 11, стадия 1.
[0228] Пример 12
трет-Бутил (2S)-2-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилат (IV-8)
[Химическая формула 73]
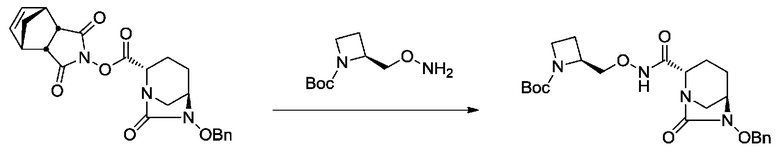
(1R,2S,6R,7S)-3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дек-8-ен-4-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (ссылочный пример 4, 145 мг, 0,331 ммоль) растворяли в дегидратированном дихлорметане (2,5 мл). К полученному добавляли раствор (S)-трет-бутил 2-((аминоокси)метил)азитидин-1-карбоксилата (93,2 мг) в дегидратированном дихлорметане (0,5 мл), с последующим перемешиванием при комнатной температуре в течение 21 часов. Реакционный раствор разбавляли этилацетатом (10 мл), последовательно промывали 0,25M хлористоводородной кислотой, насыщенным водным бикарбонатом натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, с получением 127 мг указанного в заголовке соединения (выход 83%). Инструментальные данные согласуются с данными соединения ссылочного примера 12, стадия 1.
[0229] Пример 13
трет-Бутил (3S)-3-[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]пирролидин-1-карбоксилат (IV-11)
[Химическая формула 74]
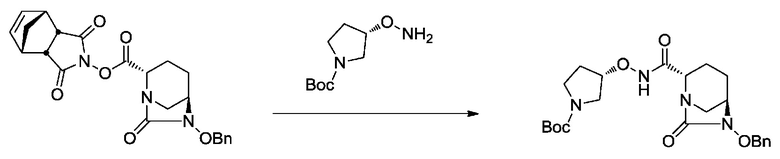
(1R,2S,6R,7S)-3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дек-8-ен-4-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (ссылочный пример 4, 145 мг, 0,332 ммоль) растворяли в дегидратированном дихлорметане (2,5 мл). К полученному добавляли раствор (S)-трет-бутил 3-(аминоокси)пирролидин-1-карбоксилата (91,6 мг) в дегидратированном дихлорметане (0,5 мл), с последующим перемешиванием при комнатной температуре в течение 19 часов. Реакционный раствор разбавляли этилацетатом (10 мл), последовательно промывали 0,25M хлористоводородной кислотой, насыщенным бикарбонатом натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, с получением 145 мг указанного в заголовке соединения (выход 95%). Инструментальные данные согласуются с данными соединения ссылочного примера 15, стадия 1.
[0230] Пример 14
трет-бутил 3-{[({[(2S,5R)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]окт-2-ил]карбонил}амино)окси]метил}азитидин-1-карбоксилат (IV-12)
[Химическая формула 75]
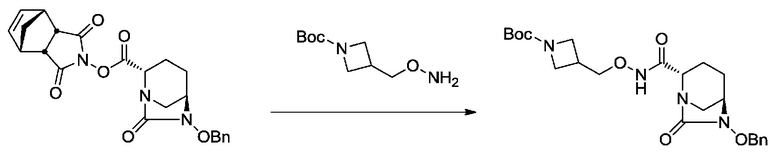
(1R,2S,6R,7S)-3,5-Диоксо-4-азатрицикло[5.2.1.02,6]дек-8-ен-4-ил (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (пример 4, 140 мг, 0,320 ммоль) растворяли в дегидратированном дихлорметане (2,5 мл). К полученному добавляли раствор трет-бутил 3-((аминоокси)метил)азитидин-1-карбоксилата (91,5 мг) в дегидратированном дихлорметане (0,5 мл), с последующим перемешиванием при комнатной температуре в течение 20 часов. Реакционный раствор разбавляли этилацетатом (10 мл), последовательно промывали 0,25M хлористоводородной кислотой, насыщенным бикарбонатом натрия и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, с получением 132 мг указанного в заголовке соединения (выход 90%). Инструментальные данные согласуются с данными соединения ссылочного примера 16, стадия 1.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2838718C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2840014C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2014 |
|
RU2719480C2 |
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| ОПТИЧЕСКИ АКТИВНОЕ ПРОИЗВОДНОЕ ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2591701C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
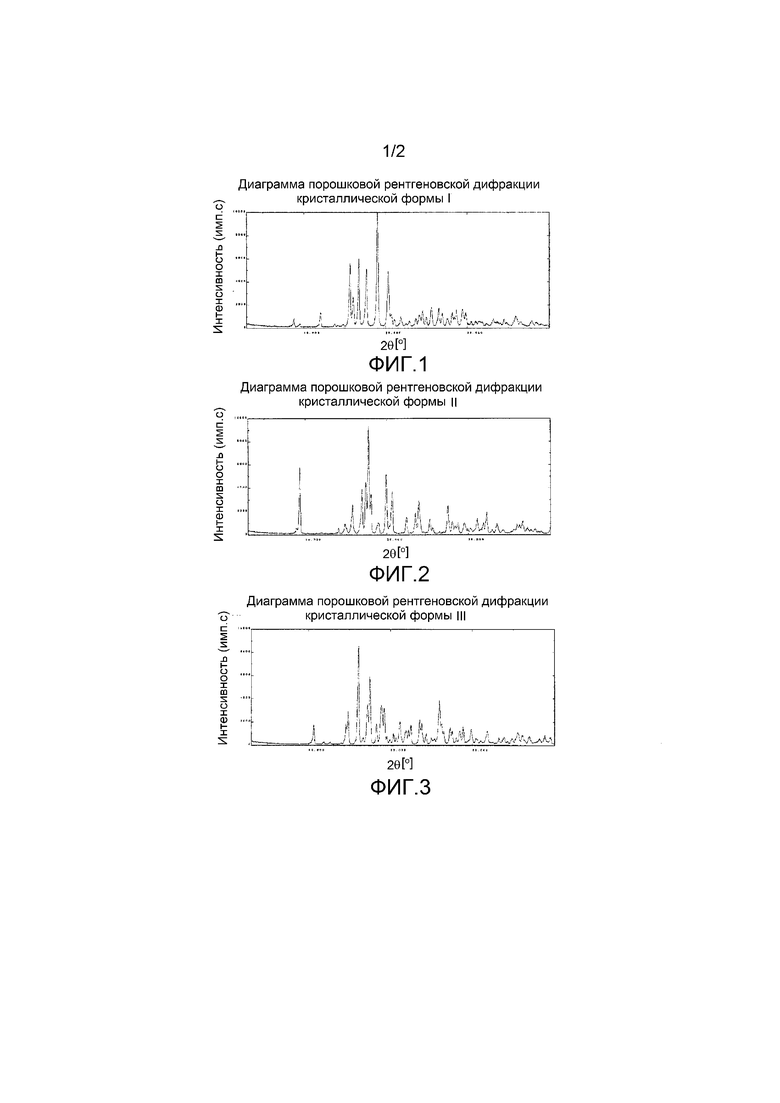
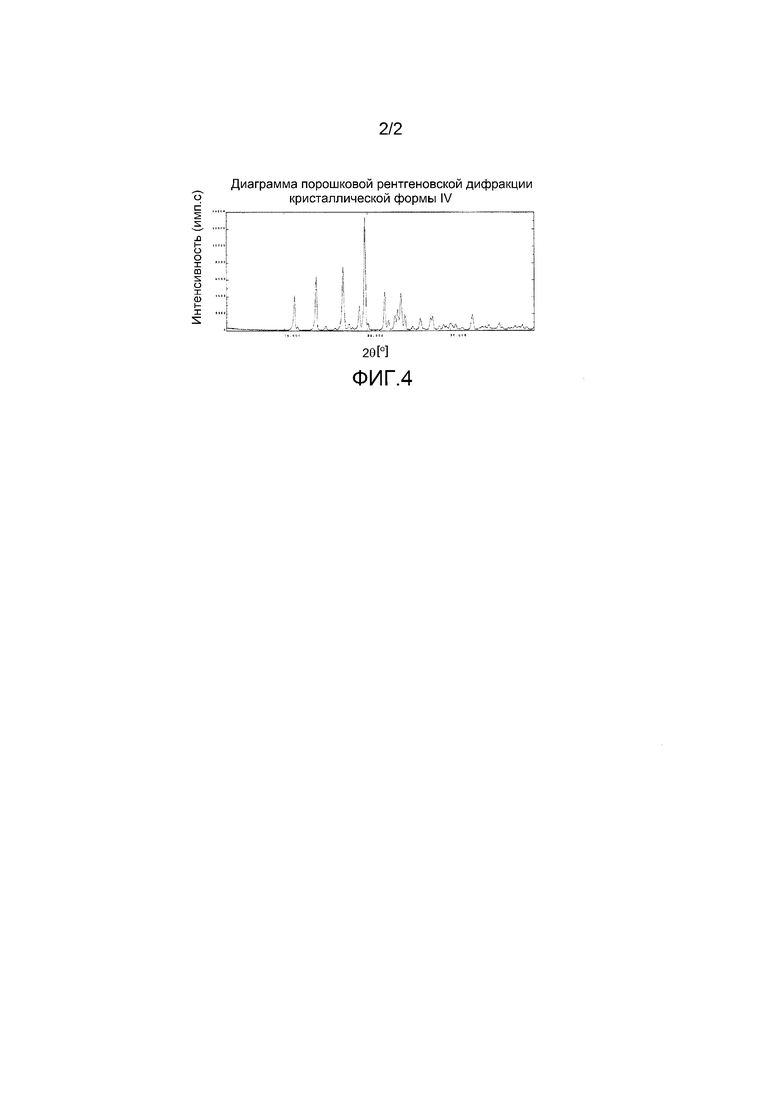
Изобретение относится к области органической химии, а именно к способу получения кристаллической формы IV соединения формулы (VII-1), имеющей характеристические пики, наблюдаемые при следующих значениях периода кристаллической решетки (d) 7,88, 6,41, 5,20, 4,67, 4,50, 4,02, 3,81, 3,75, 3,70, 3,62, 3,38, 3,23, 3,20 и 2,74 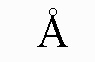 на диаграмме порошковой дифракции рентгеновских лучей, где способ включает поочередное добавление неочищенного соединения, представленного формулой (VII-1-CR), и охлажденного льдом фосфатного буфера для получения раствора, имеющего pH от 4 до 5,5; доведение температуры полученного раствора соединения до 20-25°C, добавление метанола и перемешивание. Изобретение обеспечивает получение стабильной кристаллической формы соединения формулы (VII-1). 2 з.п. ф-лы, 4 ил., 12 табл., 14 пр.
на диаграмме порошковой дифракции рентгеновских лучей, где способ включает поочередное добавление неочищенного соединения, представленного формулой (VII-1-CR), и охлажденного льдом фосфатного буфера для получения раствора, имеющего pH от 4 до 5,5; доведение температуры полученного раствора соединения до 20-25°C, добавление метанола и перемешивание. Изобретение обеспечивает получение стабильной кристаллической формы соединения формулы (VII-1). 2 з.п. ф-лы, 4 ил., 12 табл., 14 пр.
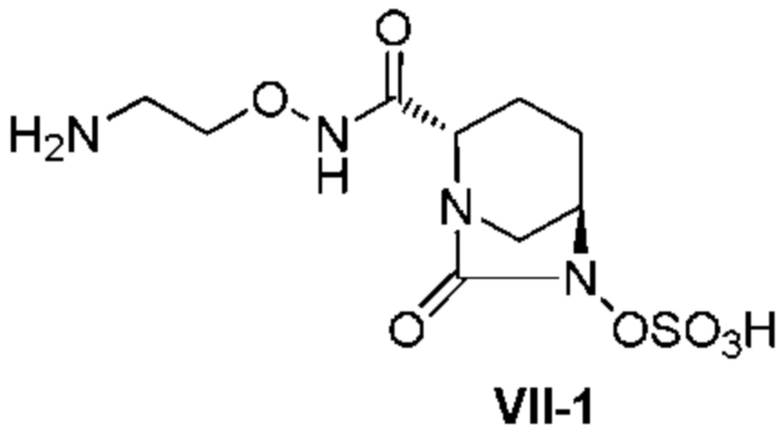
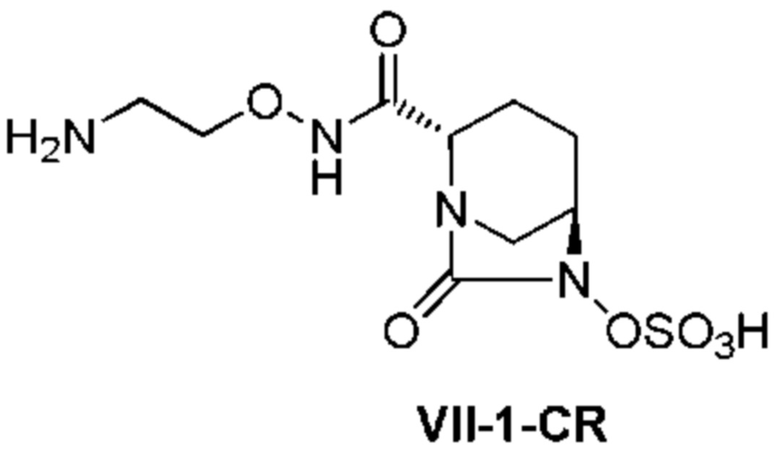
1. Способ получения кристаллической формы IV соединения, представленного формулой (VII-1):
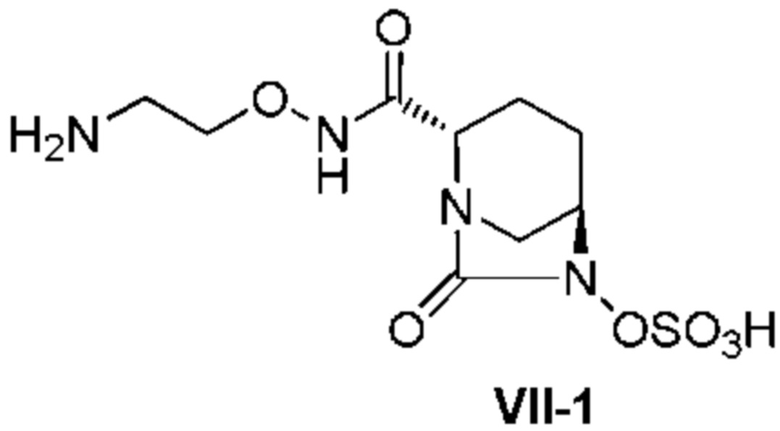 ,
,
имеющей характеристические пики, наблюдаемые при следующих значениях периода кристаллической решетки (d) 7,88, 6,41, 5,20, 4,67, 4,50, 4,02, 3,81, 3,75, 3,70, 3,62, 3,38, 3,23, 3,20 и 2,74  на диаграмме порошковой дифракции рентгеновских лучей, где способ включает :
на диаграмме порошковой дифракции рентгеновских лучей, где способ включает :
поочередное добавление неочищенного соединения, представленного формулой (VII-1-CR):
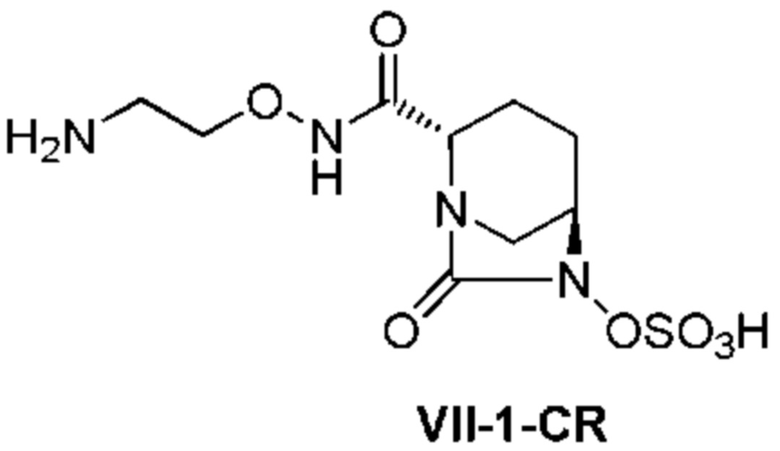 ,
,
и охлажденного льдом фосфатного буфера для получения раствора, имеющего pH от 4 до 5,5; и
доведение температуры полученного раствора соединения до 20-25°C, добавление метанола и перемешивание.
2. Способ получения кристаллической формы IV по п.1, где неочищенное соединение, представленное формулой (VII-1-CR), загрязнено от 10 до 30 мольных % кислотными компонентами и демонстрирует соотношение площадей ВЭЖХ, равное 99 % или больше.
3. Способ получения кристаллической формы IV по п.1 или 2, где раствор, имеющий рН от 4 до 5,5, концентрируют после обессоливания синтетическим адсорбентом.
| US 2013225554 A1, 29.08.2013 | |||
| ARMANDO J | |||
| AGUIAR et al | |||
| "Effect of Polymorphism on the Absorption of Chloramphenicol from Chloramphenicol Palmitate", Journal of Pharmaceutical Sciences, 56(7), 1967, с.847-853 | |||
| M.R.CAIRA | |||
| Crystalline polymorphism of organic compounds, TOPICS IN CURRENT CHEMISTRY, Springer Verlag Berlin Heidelberg, 1998, V.198, |
