ТЕХНИЧЕСКАЯ ОБЛАСТЬ
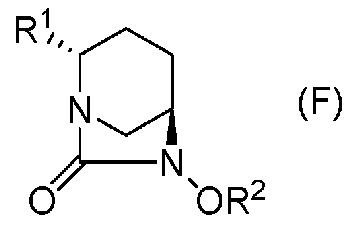
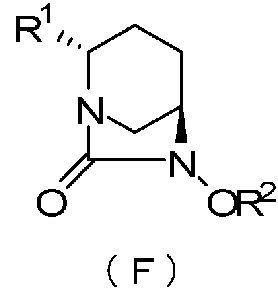
Настоящее изобретение относится к оптически активному производному диазабициклооктана, определенному приведенной ниже формулой (F), которое может быть использовано как фармацевтическое промежуточное соединение для получения ингибитора β-лактамазы, и к способу его получения.
Химическая формула 1
 .
.
В формуле (F), выше, R1 обозначает CO2R, CO2M или CONH2, где R обозначает метильную группу, трет-бутильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу, и М обозначает атом водорода, неорганический катион или органический катион; и R2 обозначат бензильную группу или аллильную группу.
УРОВЕНЬ ТЕХНИКИ
Пенициллины и цефалоспорины представляют собой β-лактамовые антибиотики, которые наиболее широко и часто используются в клинике. Однако развитие у различных патогенов резистентности к β-лактамовым антибиотикам оказало строго разрушительный эффект на поддержание эффективного лечения бактериальных инфекций. Самым значимым известным механизмом, связанным с развитием резистентности у бактерий, является продукция β-лактамаз класса A, C и D, имеющих сериновый остаток в активном центре. Эти ферменты расщепляют β-лактамовый антибиотик, приводя к потере противомикробных активностей. Класс А β-лактамаз избирательно гидролизуют пенициллины, в то время как класс C β-лактамаз имеет субстратный профиль, благоприятный для цефалоспоринов. Как коммерчески доступные ингибиторы β-лактамаз известны клавулановая кислота, сулбактам и тазобактам, и эти ингибиторы эффективны главным образом против бактерий, продуцирующих класс А β-лактамаз, и используемы в форме смеси с пенициллиновым антибиотиком. Однако до настоящего времени сообщалось о приблизительно 250 типах или более β-лактамаз, и среди них, в дополнение к экспансии класса C β-лактамаз, а также β-лактамаз расширенного спектра (ESBL), принадлежащих классам A и D β-лактамаз, другие резистентные бактерии, которые продуцируют класс А KPC-2 β-лактамаз, расщепляющих даже карбапенем, как последнее прибежище для β-лактамового антибиотика, рассматривают как проблему. Хотя разработка нового ингибитора сильно востребована, поскольку коммерчески доступные ингибиторы неэффективны против этих β-лактамаз и раскрыты потенциальные ингибиторы, в разработке имеются только несколько кандидатов.
В последние годы в US 7112592 (патентный документ 1) и US 7612087 (патентный документ 2) было раскрыто, что рацемическое производное диазабициклооктана является многообещающим соединением для лечения инфекционных заболеваний в качестве не-β-лактамового противомикробного средства или ингибитора β-лактамазы, и был приведен рабочий Пример рацемических диазабициклооктановых производных из рацемического производного цис-5-гидроксипиперидин-2-карбоновой кислоты и их биологической активности.
Относительно оптически активного диазабициклооктанового производного, в рабочем Примере 1 WO2009/091856 A2 (патентный документ 3) и WO2010/126820 A2 (патентный документ 4) описан способ получения производного, имеющего особую боковую амидную цепь. Дополнительно, рабочий Пример 1 патентного документа 3 содержит просто описание химического названия (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты как промежуточного соединения для исследования, и точно также в WO2009/133442 A1 (патентный документ 5) описано химическое название (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, и в ЕР 2135959 A1 (патентный документ 6) описано химическое название (2S,5R)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, 7-оксо-6-(сульфокси)-мононатриевая соль.
С другой стороны, относительно (2S,5S)-5-гидроксипиперидин-2-карбоновой кислоты и (2S,5R)-5-(бензилоксиамино)пиперидин-2-карбоновой кислоты, которые рассматриваются как важный исходный материал диазабициклооктанового производного и его производных, об одном соединении, имеющем сложноэфирную боковую цепь, сообщалось в Tetrahedron Asymmetry 2006, 17(17), 2479-2486 (непатентный документ 2) и J. Chem. Soc., Chem. Commun., 1993, 1434 (непатентный документ 3), и о соединении, имеющем боковую амидную цепи сообщалось в рабочем Примере 1C патентного документа 3, Org. Lett., 2009, 11(16), 3566-3569 (непатентный документ 3) и патентном документе 4. Далее, в качестве способа получения производного не через производное (2S,5S)-5-гидроксипиперидин-2-карбоновой кислоты, в US 2010/197928 (патентный документ 7) раскрыт способ получения бензил (2S)-5-(бензилоксиимино)пиперидин-2-бензилкарбоксилата или (2S,5R/S)-5-(бензилоксиамино)пиперидин-2-бензилкарбоксилата.
ЛИТЕРАТУРА ИЗ УРОВНЯ ТЕХНИКИ
[Патентный документ 1] патент США 7112592.
[Патентный документ 2] патент США 7612087.
[Патентный документ 3] Международная Публикация 2009/091856 A2.
[Патентный документ 4] Международная Публикация 2010/126820 A2.
[Патентный документ 5] Международная Публикация 2009/133442 A1.
[Патентный документ 6] Публикация европейской заявки на патент 2135959 A1.
[Патентный документ 7] Публикация заявки на патент США 2010/197928 A1.
[Непатентный документ 1] Jung, JC.; Avery, MA. "Diastereoselective synthesis of (2S,5S)- and (2S,5R)-N-benzyloxycarbonyl-5-hydroxypipecolic acids from trans-4-hydroxy-L-proline" Tetrahedron Asymmetry 2006, 17(17), 2479-2486.
[Непатентный документ 2] Baldwin, JE.; Adlington, RM.; Godfrey, CRA.; Gollins, DW.; Vaughan, JG. "A Novel Entry to Carbenoid Species via β-Ketosulfoxonium Ylides" Journal of the Chemical Society Chemical Communications 1993, 1434-1435.
[Непатентный документ 3] Mangion, IK.; Nwamba, IK.; Shevlin, M.; Huffman MA. "Iridium-Catalyzed X-H Insertions of sulfoxonium Ylides" Organic Letters 2009, 11(16), 3566-3569.
[Непатентный документ 4] Dolence, EK.; Lin, CE.; Miller, MJ.; Payne, SM. "Synthesis and siderophore activity of albomycin-like peptides derived from N5-acetyl-N5-hydroxy-L-ornithine" Journal of Medicinal Chemistry 1991, 34(3), 956-968.
[Непатентный документ 5] King, FE.; King, TJ.; Warwick, AJ. "The Chemistry of Extractives from Hardwoods. Part III. Baikiain, an Amino-acid Present in Baikiaea plurijuga" Journal of the Chemical Society 1950, 3590-3597.
[Непатентный документ 6] Witkop, B.; Folts, CM. "The Configuration of 5-Hydroxypipecolic Acid from Dates" Journal of the American Chemical Society 1957, 79(1), 192-197.
[Непатентный документ 7] Freed, ME.; Day AR. "Synthesis of 5-Ketopipecolic Acid from Glutamic Acid" The Journal of Organic Chemistry 1960, 25(12), 2105-2107.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ЗАДАЧИ, РЕШАЕМЫЕ В ИЗОБРЕТЕНИИ
Однако уровень техники в отношении ингибитора β-лактамазы, имеющего диазабициклооктановый скелет, особенно в отношении диазабициклооктанового производного, как общего промежуточного соединения, используемого для получения ингибитора β-лактамазы, имеет множество технических проблем, которые решаются, как показано ниже.
В патентных документах 1 и 2 показан рабочий пример рацемического диазабициклооктанового производного, но не раскрыт способ получения оптически активного диазабициклооктанового производного и способ оптического разделения этого производного и данные инструментальных исследований для оптически активного соединения, особенно данные, демонстрирующие получение оптически активного соединения, такие как угол вращения, и там не продемонстрировано, что оптически активное соединение действительно получено в независимой форме.
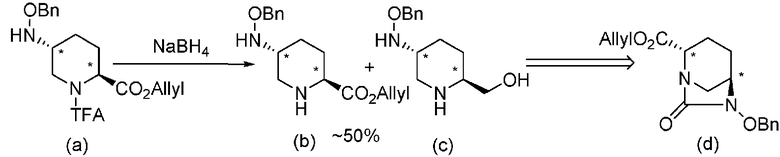
В способах, описанных в патентных документах 1 и 2, выбор защитной группы для сложного эфира карбоксилата в положении 2 является несоответствующим, и поэтому транс-5-(бензилоксиамино)пиперидин-2-аллилкарбоксилат, представленный формулой (b) в реакционной схеме, приведенной ниже, как предшественник для промежуточного соединения, и транс-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилат, представленный формулой (d), приведенной ниже, в качестве желаемого важного промежуточного соединения не могут быть эффективно получены. В области производства лекарственных средств, когда соединение имеет асимметрический атом углерода, желательно, чтобы только единственный энантиомер был селективно получен в зависимости от цели, но непросто применить непосредственно способы согласно патентным документам 1 и 2 к отдельно полученному оптически активному производному (2S,5S)-5-гидроксипиперидин-2-карбоновой кислоты или для оптического разделения массового рацемического диазабициклооктанового производного и внедрения полученного оптически активного соединения в областях исследования и производства лекарственных средств.
Химическая формула 2
 .
.
В приведенной выше реакционной схеме TFA обозначает 2,2,2-трифторацетильную группу, NaBH4 обозначает боргидрид натрия, и BnO обозначает бензилоксигруппу.
В патентных документах 3, 5 и 6 описаны химические названия: (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота, (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, (1R,2S,5R)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид и 7-оксо-6-(сульфокси)-мононатриевая соль, как оптически активные соединения, но, относительно способа их получения, просто даны ссылки на патентные документы 1 и 2, в которых раскрыт способ получения рацемической модификации.
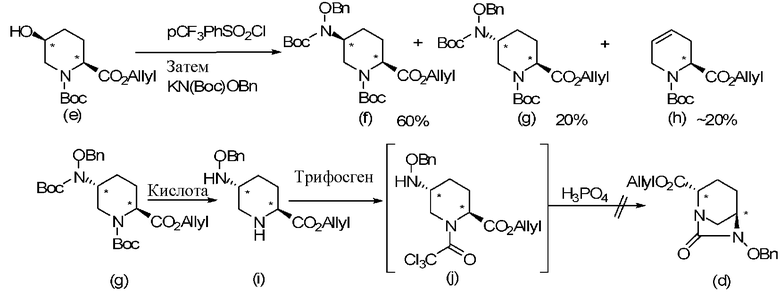
Только патентные документы 3 и 4 демонстрируют способ получения оптически активного диазабициклооктанового производного, но этот способ является специфическим только для соединения, имеющего особую боковую амидную цепь в положении 2, и таким образом, не предлагается применение способа к общему промежуточному соединению. Далее, была сделана попытка реакцию введения бензилоксиаминогруппы через п-трифторметилбензолсульфонилоксигруппу или реакцию образования внутримолекулярного карбамида с использованием трифосгена, раскрытую в патентных документах 3 и 4, применить к соединению, имеющему сложноэфирную боковую цепь в положении 2, но стереоселективность бензилоксиаминогруппы не наблюдалась, и реальная реакция формирования внутримолекулярного карбамида не происходила, что указывает, что вышеупомянутые реакции не могут быть непосредственно применены к соединению, имеющему сложноэфирную боковую цепь в положении 2.
Химическая формула 3
 .
.
В вышеописанной реакционной схеме Вос обозначает трет-бутоксикарбонильную группу, pCF3PhSO2Cl обозначает п-трифторметилбензолсульфонилхлорид, KN(Вос)OBn обозначает N-трет-бутоксикарбонилбензилоксиамид калия, и BnO обозначает бензилоксигруппу; соединения формул (f) и (g), показанные в вышеописанной реакционной схеме, не могут быть непосредственно разделены, и поэтому их структуры были определены ЯМР после удаления защитной группы Вос, формирования внутримолекулярного карбамида с использованием дифосгена и выделения продукта.
Далее, патентный документ 4 также демонстрирует способ получения (2S,5S)-ди-трет-бутил-5-гидроксипиперидин-1,2-дикарбоксилата, но селективное удаление защитной трет-бутоксикарбонильной группы и сложного трет-бутилового эфира на кольце пиперидина затруднительно, и дальнейшая селективная этерификация трет-бутила только карбоксильной группы отдельно от гидроксильной группы после удаления всех защитных групп нелегка. Поэтому трудно в промышленном отношении использовать раскрытое соединение как исходный материал непосредственно для общего промежуточного соединения, к которому относится настоящее изобретение.
В патентном документе 7 не раскрыто количество триметилсульфоксония йодида, используемого в получении важного исходного материала, и неясно, является ли способ пригодным для осуществления на практике процессом без побочной реакции, такой как разложение сложного эфира или возможности рацемизации вследствие избытка реагента. В действительности, описаны данные инструментального анализа, показывающие плоскую конфигурацию сформированного кетосульфоксонийилидного соединения, но относительно соединений, включая продукты, формирующиеся на последующих стадиях, данные инструментального анализа, показывающие оптическую чистоту, особенно такую как угол вращения, не демонстрируется. Далее, стереоселективность бензилоксиаминогруппы в положении 5 является низкой, поскольку цис-транс=1:1, и таким образом процесс не является эффективным. Сформированный изомер цис-транс присутствует в форме смеси, которую трудно разделить, и нет никакого описания, показывающего, что диазабициклооктановое производное может быть действительно получено из готовой смеси.
Как описано выше, способ получения оптически активного диазабициклооктанового производного, в частности, 2-карбоновой кислоты или сложноэфирного производного, которое может быть использовано в качестве общего промежуточного соединения, не был раскрыт до настоящего времени. Поэтому разработка легкоосуществимого способа получения оптически активного диазабициклооктанового производного, имеющего боковую цепь карбоновой кислоты и сложноэфирную боковую цепь, которое может использоваться как общее промежуточное соединение, было желательно для исследования более эффективного нового соединения и фармацевтических разработок.
В этой ситуации, авторы настоящего изобретения провели обширные и интенсивные исследования с целью разработки оптически активного диазабициклооктанового производного, в частности, 2-карбоновой кислоты или сложноэфирного производного, которое может быть использовано как фармацевтическое промежуточное соединение для ингибитора β-лактамазы, и легкоосуществимого способа его получения. В результате было обнаружено, что, при использовании в качестве исходного материала производного (2S,5S)-5-гидроксипиперидин-2-карбоновой кислоты, которое является известным соединением, оптически активное диазабициклооктановое производное может быть промышленно получено с превосходной воспроизводимостью с высоким выходом с помощью относительно короткого процесса, без снижения оптической чистоты производного, и далее, что оптически активное диазабициклооктановое производное, полученное таким способом, может использоваться как фармацевтическое промежуточное соединение для ингибитора β-лактамазы, и было осуществлено настоящее изобретение.
СРЕДСТВА РЕШЕНИЯ ЗАДАЧИ
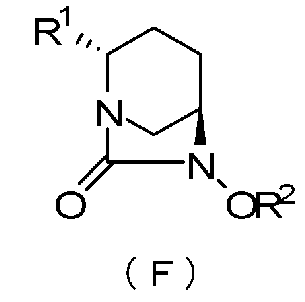
В частности, настоящее изобретение относится к оптически активному производному (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, определяемому следующей формулой (F):
Химическая формула 4
 ,
,
в которой:
R1 обозначает CO2R, CO2M или CONH2,
причем R обозначает метильную группу, трет-бутильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу, и
М обозначает атом водорода, неорганический катион или органический катион; и
R2 обозначает бензильную группу или аллильную группу.
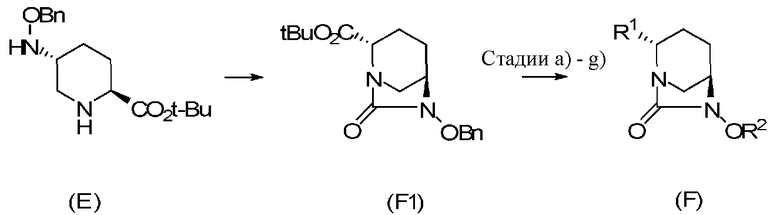
Настоящее изобретение также относится к способу получения соединения, определяемого формулой (F), приведенной выше, причем способ включает подвергание соединения, представленного формулой (E), приведенной ниже, формированию внутримолекулярного карбамида и затем подвергание полученного соединения, представленного ниже формулой (F1), по меньшей мере одной из стадий, описанных ниже:
Химическая формула 5
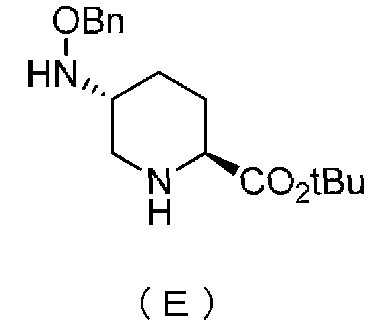 ,
,
в которой Bn обозначает бензильную группу, и tBu обозначает трет-бутильную группу,
Химическая формула 6
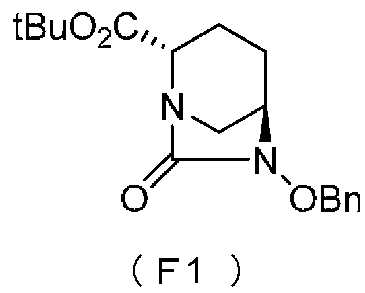 ,
,
в которой Bn обозначает бензильную группу, и tBu обозначает трет-бутильную группу,
стадии а для расщепления сложного эфира,
стадии b для преобразования соединения в форму соли неорганического катионного или органического катиона,
стадии c для обработки соединения кислотой, чтобы преобразовать соединение в свободную кислоту,
стадии d для осуществления карбамоилирования для карбоновой кислоты,
стадии e для преобразования карбоновой кислоты в сложный эфир,
стадии f для удаления бензильной группы бензилоксигруппы в положении 6, и
стадии g для преобразования группы в положении 6 в аллилокси.
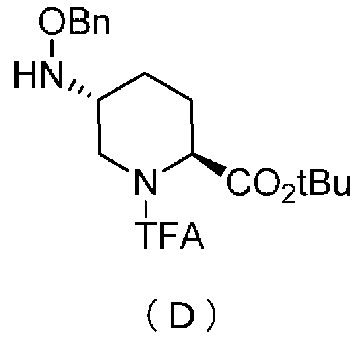
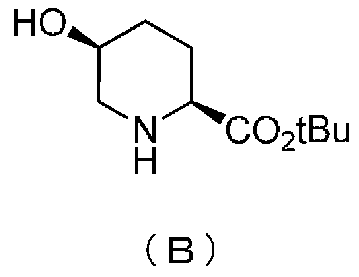
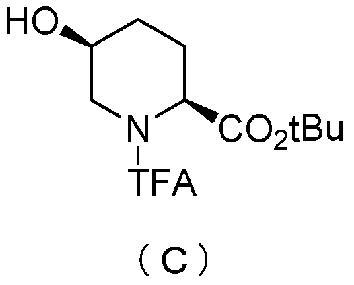
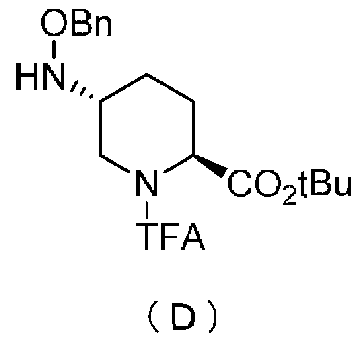
Кроме того, настоящее изобретение также относится к способу получения соединения, представленного формулой (E), приведенной выше, причем способ включает подвергание соединения, представленного формулой (B), приведенной ниже, трифторацетилированию и введение полученного соединения, представленного формулой (C), приведенной ниже, в реакцию с бензилоксиамином в присутствии активатора гидроксильной группы и подвергание полученного соединения, представленного формулой (D), приведенной ниже, детрифторацетилированию:
Химическая формула 7
 ,
,
в которой tBu обозначает трет-бутильную группу,
Химическая формула 8
 ,
,
в которой tBu обозначает трет-бутильную группу, и TFA обозначает трифторацетильную группу,
Химическая формула 9
 ,
,
в которой Bn обозначает бензильную группу, tBu обозначает трет-бутильную группу, и TFA обозначает трифторацетильную группу.
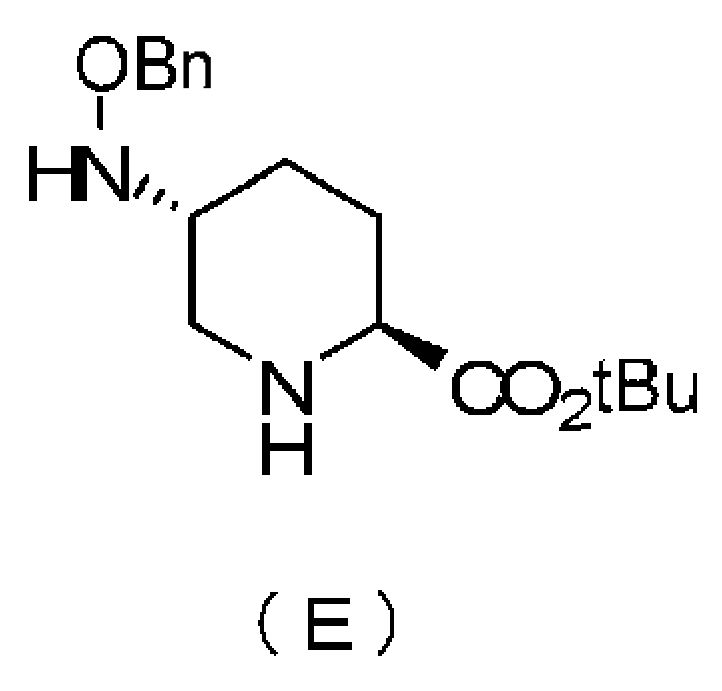
Кроме того, настоящее изобретение также относится к промежуточному соединению для применения в получении соединения, представленного формулой (F), приведенной выше, то есть, соединению, представленному следующими формулами (B), (C), (D) и (E):
Химическая формула 10
 ,
,
в которой tBu обозначает трет-бутильную группу,
Химическая формула 11
 ,
,
в которой tBu обозначает трет-бутильную группу, и TFA обозначает трифторацетильную группу,
Химическая формула 12
 ,
,
в которой Bn обозначает бензильную группу, tBu обозначает трет-бутильную группу, и TFA обозначает трифторацетильную группу,
Химическая формула 13
 ,
,
в которой Bn обозначает бензильную группу, и tBu обозначает трет-бутильную группу.
ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
С помощью способа получения оптически активного диазабициклооктанового производного согласно настоящему изобретению, оптически активное производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты может быть промышленно получено с превосходной воспроизводимостью с высоким выходом с помощью относительно короткого процесса, без снижения оптической чистоты производного. Кроме того, таким образом полученное оптически активное производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты согласно настоящему изобретению легко кристаллизуется, и следовательно, легко в обращении и может использоваться в массовом производстве оптически активного соединения для ингибитора β-лактамазы, имеющего диазабициклооктановый скелет, или может использоваться как важное промежуточное соединение в исследовании и массовом производстве более эффективного нового ингибитора β-лактамазы, и поэтому особенно превосходно как промежуточное соединение для промышленной продукции.
ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Как упомянуто выше, настоящее изобретение относится к оптически активному производному (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, определяемому следующей формулой (F):
Химическая формула 14
 ,
,
в которой: R1 обозначает CO2R, CO2M или CONH2,
причем R обозначает метильную группу, трет-бутильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу, и М обозначает атом водорода, неорганический катион или органический катион; и R2 обозначает бензильную группу или аллильную группу.
Неорганический катион представляет собой, например, натрий, калий, литий или кальций и предпочтительно натрий, калий или кальций. Органический катион представляет собой соль аммония, образованную из амина, такого как триметиламин, триэтиламин, циклогексиламин или дициклогексиламин; или соль четвертичного аммониевого основания, такого как тетраметиламмоний, тетраэтиламмоний, тетрабутиламмоний или триэтилбензиламмоний, и предпочтительно соль циклогексиламмония.
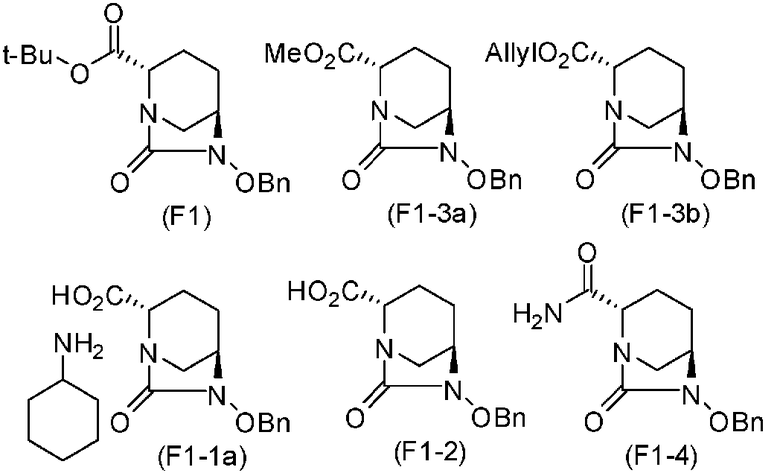
Предпочтительные примеры соединений, определяемых формулой (F), включают следующие соединения:
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота соль циклогексиламина,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилат,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилат,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилат,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-2,5-диоксопирролидин-1-ил-карбоксилат,
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат,
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота соль циклогексиламина,
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота и
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилат.
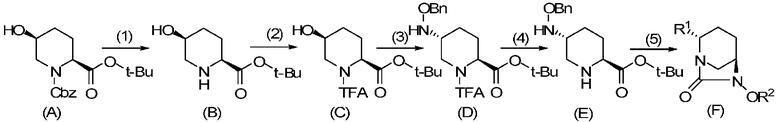
Оптически активное производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, определяемое общей формулой (F), получаемое в соответствии с настоящим изобретением, может быть получено из соединения формулы (E), и соединение формулы (E) может быть получено из соединения формулы (A), которое является известным соединением, в качестве исходного материала в основном в соответствии с процессом, показанным в соответствии со схемой химической реакции, приведенной ниже.
Химическая формула 15
 .
.
В приведенной выше схеме химической реакции, Cbz обозначает бензилоксикарбонильную группу, t-Bu обозначает трет-бутильную группу, TFA обозначает 2,2,2-трифторацетильную группу, OBn обозначает бензилоксигруппу, R2 обозначает бензильную группу или аллильную группу, предпочтительно бензильную группу, и числа в круглых скобках указывают число соответствующих стадий.
В частности, соединение, определяемое формулой (F) согласно настоящему изобретению, может быть получено через первую стадию удаления бензилоксикарбонильной группы соединения, представленного формулой (A), с получением соединения, представленного формулой (B), вторую стадию трифторацетилирования атома азота пиперидина с получением соединения, представленного формулой (C), третью стадию замещения гидроксильной группы в положении 5 бензилоксиамином в присутствии активатора гидроксильной группы с получением соединения, представленного формулой (D), четвертую стадию удаления трифторацетильной группы с получением соединения, представленного формулой (E), и пятую стадию осуществления формирования внутримолекулярного карбамида и затем преобразования боковых цепей R1, R2 с получением оптически активного производного (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, определяемого формулой (F).
Выбор сложного трет-бутилового эфира соединения, представленного формулой (A), который может использоваться как исходный материал в настоящем изобретении, имеет очень важную роль в селективном удалении трифторацетильной группы соединения, представленного формулой (D). Кроме того, бензилоксикарбонильная группа, которая является защитной группой для NH на кольце пиперидина, может быть легко разблокирована отдельно от сложного трет-бутилового эфира, и следовательно, соединение формулы (A) представляет собой оптимальный исходный материал как предшественник для соединения, представленного формулой (B). Далее, способ согласно настоящему изобретению представляет собой чрезвычайно полезный процесс, такой, что транс-оксиамино соединение может быть селективно получено без образования ненужного цис-бензилоксиамино соединения. Таким образом, вышеупомянутый способ с использованием соединения, представленного формулой (A), в качестве исходного материала чрезвычайно полезен так же, как способ эффективного получения оптически активного (2S,5R)-5-(бензилоксиамино)пиперидин-2-трет-бутилкарбоксилата, представленного формулой (E).
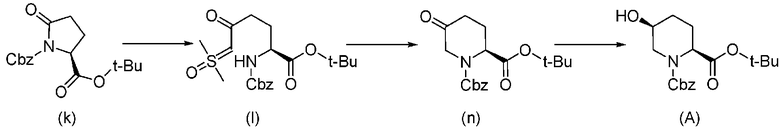
Соединение, представленное формулой (A), которое используется как исходный материал в способе согласно настоящему изобретению, может быть получено способом, описанным в непатентном документе 1, но может также быть получено более эффективным способом, показанным на реакционной схеме, приведенной ниже, из известного соединения, представленного формулой (k), приведенной ниже, описанного в непатентном документе 4.
Химическая формула 16
 .
.
В приведенной выше схеме химической реакции, Cbz обозначает бензилоксикарбонильную группу, и t-Bu обозначает трет-бутильную группу.
Оптически активное производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, определяемое формулой (F), получаемое в соответствии с настоящим изобретением, может быть получено, например, способом, который включает подвергание соединения, представленного формулой (E), формированию внутримолекулярного карбамида и затем подвергание полученного соединения, представленного формулой (F1), по меньшей мере одной из стадий: стадии (стадия a) расщепления сложного эфира, стадии (стадия b) преобразования соединения в форму соли неорганического катиона или органического катиона, стадии (стадия c) обработки соединения кислотой с преобразованием соединения в свободную кислоту, стадии (стадия d) осуществления карбамоилирования карбоновой кислоты, стадии (стадия e) преобразования карбоновой кислоты в сложный эфир, стадии (стадия f) удаления бензильной группы бензилоксигруппы в положении 6 и стадии (стадия g) преобразования группы в положении 6 в аллилокси.
Химическая формула 17
 .
.
Символы, показанные в формулах, имеют значения, определенные выше.
В некоторых вариантах осуществления вышеописанного способа получения соединения, определяемого формулой (F), из соединения, определяемого формулой (E), как показано ниже, соединение, представленное формулой (F), в каждом варианте осуществления может быть получено через стадию (5-1) для формирования внутримолекулярного карбамида и затем по меньшей мере одну из стадий с (5-2) по (5-8). В частности, эти стадии могут быть проведены в соответствии со способом, показанным в соответствии со схемой химической реакции, приведенной ниже.
Химическая формула 18
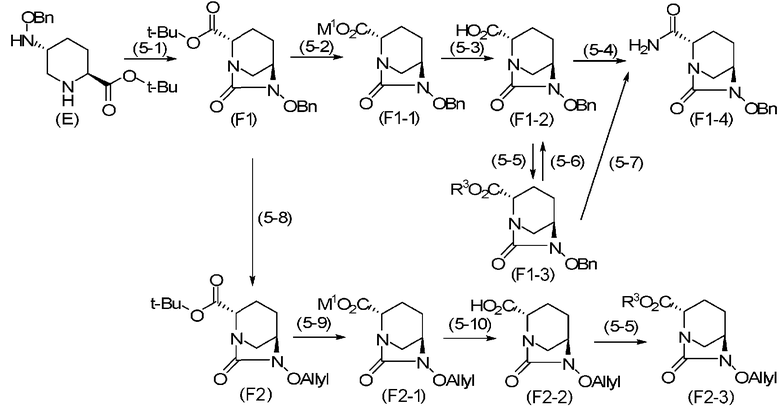 .
.
В приведенной выше реакционной схеме OBn обозначает бензилоксигруппу, t-Bu обозначает трет-бутильную группу, M1 обозначает циклогексил аммоний, R3 обозначает метильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу, OAllyl обозначает аллилоксигруппу, и числа в круглых скобках указывают число соответствующих стадий.
В частности, процесс согласно показанному выше варианту осуществления настоящего изобретения включает стадию 5-1 подвергания соединения, представленного формулой (E), формированию внутримолекулярного карбамида с получением соединения, представленного формулой (F1), стадию 5-2 расщепления сложного трет-бутилового эфира с получением соли циклогексил аммония, представленной формулой (F1-1), стадию 5-3 обработки соли циклогексиламмония кислотой с получением свободной кислоты, представленной формулой (F1-2), стадии 5-4 или 5-5 с получением формулы (F1-4) или формулы (F1-3) из карбоновой кислоты, или стадию 5-8 удаления бензильной группы из формулы (F1) и преобразования ее в аллильную группу с получением формулы (F2), стадии 5-9 и 5-10 расщепления сложного трет-бутилового эфира с получением формул (F2-1) и (F2-2), и стадию 5-5 с получением формулы (F2-3) из карбоновой кислоты.
Среди соединения, определяемого формулой (F) согласно настоящему изобретению, полученного вышеупомянутым способом согласно настоящему изобретению, (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат, (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилат, (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилат, (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота соль циклогексиламмония, (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3,2,1]октан-2-карбоновая кислота и (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, которые, соответственно, представлены ниже формулами (F1), (F1-3a), (F1-3b), (F1-1a), (F1-2) и (F1-4), могут быть индивидуально получены в форме кристалла оптически активного диазабициклооктанового производного и поэтому имеют то преимущество, что они являются легкими в выделении, очистке, хранении и транспортировке. Это указывает, что настоящее изобретение является полезным в промышленном отношении изобретением.
Химическая формула 19
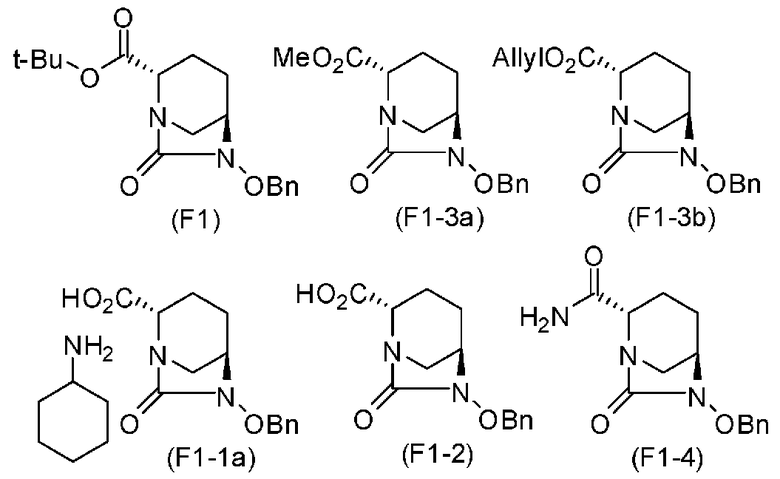 .
.
В приведенных выше формулах t-Bu обозначает трет-бутильную группу, OBn обозначает бензилоксигруппу, и Ме обозначает метильную группу.
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат, представленный формулой (F1), присутствует в форме кристалла, имеющего характеристические пики, появляющиеся в интервалах решетки (d) 11,56, 10,96, 6,55, 6,00, 5,79, 5,56, 5,47, 5,25, 4,90, 4,35, 4,23 и 3,86 Å, и особенно предпочтительно, чтобы соединение было получено как кристалл с высокой чистотой, который является легким в работе, выделении или очистке, особенно в промышленном масштабе.
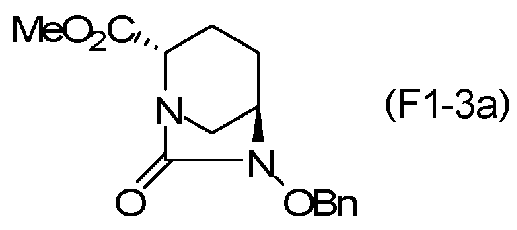
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилат, представленный формулой (F1-3a), присутствует в форме кристалла, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 10,39, 5,86, 5,69, 5,34, 4,81, 4,44, 3,98, 3,78, 3,11, 3,03, 2,93 и 2,77 Å, и особенно предпочтительно, чтобы соединение было получено как кристалл с высокой чистотой, который является легким в работе, выделении или очистке, особенно в промышленном масштабе.
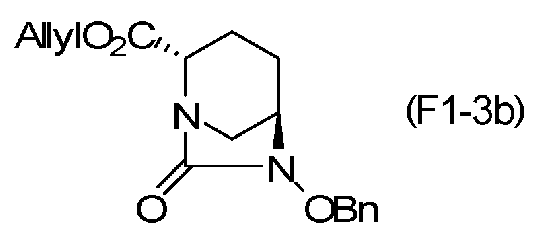
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилат, представленный формулой (F1-3b), присутствует в форме кристалла, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки 14,72, 4,91, 4,46, 4,24 и 3,67 Å, и особенно предпочтительно, чтобы соединение было получено как кристалл с высокой чистотой, который является легким в работе, выделении или очистке, особенно в промышленном масштабе.
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота соль циклогексиламмония, представленная формулой (F1-1a), присутствует в форме кристалла, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 9,95, 8,45, 6,26, 5,87, 5,52, 5,22, 5,10, 4,96, 4,73, 4,54, 4,16, 3,93 и 3,55 Å, и особенно предпочтительно, чтобы соединение было получено как кристалл с высокой чистотой, который является легким в работе, выделении или очистке, особенно в промышленном масштабе.
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота, представленная формулой (F1-2), присутствует в форме кристалла, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 8,19, 7,14, 6,64, 6,29, 5,60, 5,21, 4,91, 4,60, 4,21, 3,69, 3,45 и 3,13 Å, и особенно предпочтительно, чтобы соединение было получено как кристалл с высокой чистотой, который является легким в работе, выделении или очистке, особенно в промышленном масштабе.
Далее, (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, представленный формулой (F1-4), присутствует в форме кристалла, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 13,06, 6,52, 5,14, 4,74, 4,63, 4,34, 3,85 и 3,72 Å, и особенно предпочтительно, чтобы соединение было получено как кристалл с высокой чистотой, который является легким в работе, выделении или очистке, особенно в промышленном масштабе.
Ниже будет описан более подробно способ в соответствии с настоящим изобретением, который включает ряд стадий для получения оптически активного диазабициклооктанового производного, определяемого формулой (F), из соединения, определяемого формулой (A), в качестве исходного материала.
Синтез соединения формулы (B) из соединения формулы (A)
Бензилоксикарбонильную группу (2S,5S)-1-бензил-2-трет-бутил-5-гидроксипиперидин-1,2-дикарбоксилата, который используется как исходный материал в настоящем изобретении и который представлен формулой (A):
Химическая формула 20
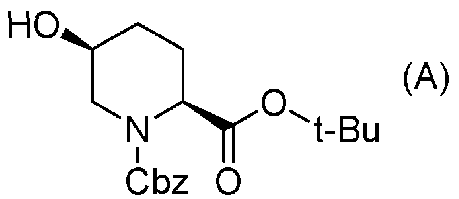 ,
,
причем в формуле (A), приведенной выше, Cbz обозначает бензилоксикарбонильную группу, и t-Bu обозначает трет-бутильную группу,
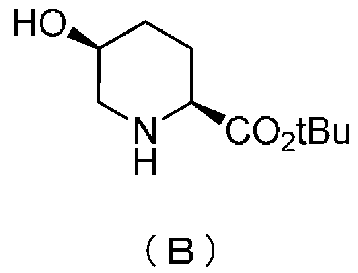
удаляют реакцией каталитического гидрирования в атмосфере газообразного водорода в присутствии катализатора, получая (2S,5S)-5-гидроксипиперидин-2-трет-бутилкарбоксилат, представленный формулой (B):
Химическая формула 21
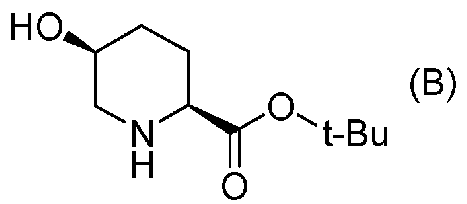 ,
,
причем в формуле (B), приведенной выше, t-Bu обозначает трет-бутильную группу.
Относительно катализатора, используемого в реакции, может использоваться произвольный гидрирующий катализатор, но предпочтительно может использоваться, например, оксид платины, оксид палладия, палладиевая чернь или палладий на углероде. Катализатор может использоваться в диапазоне от 0,05 до 1 вес./вес. в терминах весового отношения катализатора к соединению формулы (A). Давление водорода может быть от атмосферного давления до 0,5 МПа.
Растворитель, используемый в реакции, может быть выбран из воды, метанола, этанола, пропанола, изопропанола, бутанола, простого эфира, простого диизопропилового эфира, этилацетата, бутилацетата, толуола, тетрагидрофурана и 1,4-диоксана, и эти растворители могут использоваться индивидуально или в комбинации.
Предпочтительно, катализатор, выбранный из оксида платины, оксида палладия, палладиевой черни и палладия на углероде, может использоваться в весовом отношении 0,05 к 0,5 вес./вес. в метаноле или этаноле.
Более предпочтительно, палладий на углероде в весовом отношении 0,05 к 0,25 вес./вес. может использоваться как катализатор в этаноле.
Соединение, представленное формулой (B), полученное на первой стадии, может быть выделено, например, в форме свободного основания с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как фильтрация для катализатора, концентрация растворителя, обмен растворителя, формирование соли и кристаллизация, и использовано на следующей стадии, или оно может использоваться на следующей стадии без очистки после обработки.
Синтез соединения формулы (C) из соединения формулы (B)
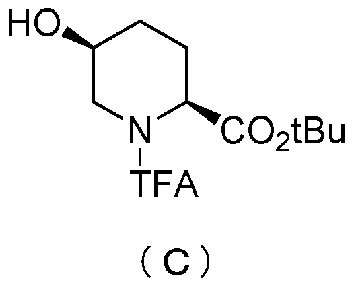
Полученное выше соединение формулы (B) обрабатывают трифторацетилирующим средством в присутствии основания, получая (2S,5S)-5-гидрокси-1-(2,2,2-трифторацетил)пиперидин-2-трет-бутилкарбоксилат, представленный формулой (C):
Химическая формула 22
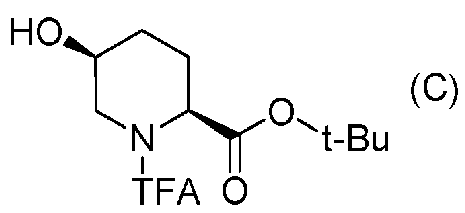 ,
,
причем в формуле (C), приведенной выше, TFA обозначает 2,2,2-трифторацетильную группу, и t-Bu обозначает трет-бутильную группу.
В частности, трифторацетилирование соединения, представленного формулой (B), осуществляют, растворяя соединение формулы (B) в подходящем растворителе и вводя его в реакцию с избытком трифторацетилирующего средства в присутствии избытка основания, чтобы сформировать 1,5-дитрифторацетильное соединение, и затем отщепляя только трифторацетильную группу в положении 5.
Основание, используемое в реакции, может быть выбрано из неорганических оснований, таких как гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, гидроксид натрия и гидроксид калия, и органических оснований, таких как триэтиламин, диизопропилэтиламин, трибутиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен, пиридин, 2-пиколин и 2,6-диметилпиридин, и используется в количестве в диапазоне от 2 до 6 молярных эквивалентов относительно соединения формулы (B).
Трифторацетилирующее средство может быть выбрано из трифторуксусной кислоты, трифторэтилацетата, ангидрида трифторуксусной кислоты, хлорангидрида трифторуксусной кислоты, сложного эфира трифторацетилсукцинимида, сложного эфира трифторацетилбензотриазола, сложного эфира трифторацетилпентафторфенила, 2-трифторацетоксипиридина и додецил трифтортиоацетата и может использоваться в количестве в диапазоне от 1,5 до 3 молярных эквивалентов относительно соединения формулы (B). Реакцию трифторацетилирования проводят при температуре в диапазоне от -30 до +50°C. Отщепление трифторацетоксигруппы в положении 5 можно осуществлять, после постобработки для трифторацетилирования или немедленно после трифторацетилирования, перемешивая смесь в воде или спиртовом растворителе, таком как метанол или этанол, в присутствии указанного основания при комнатной температуре или при нагревании.
Растворитель, используемый в реакции, может быть выбран из воды, метанола, этанола, пропанола, изопропанола, бутанола, дихлорметана, 1,2-дихлорэтана, хлороформа, простого эфира, простого диизопропилового эфира, этилацетата, бутилацетата, тетрагидрофурана, 1,4-диоксан, N,N-диметилформамида и N,N-диметилацетамида, и эти растворители могут использоваться индивидуально или в комбинации.
Предпочтительно, реакцию проводят, добавляя по каплям от 2 до 2,5 молярных эквивалентов трифторуксусного ангидрида к соединению в дегидрированном дихлорметане или тетрагидрофуране в присутствии 4-5 молярных эквивалентов третичного амина, выбранного из триэтиламина, диизопропилэтиламина и трибутиламина при температуре от -20 до +10°C и обрабатывая смесь водой при комнатной температуре.
Более предпочтительно, реакцию проводят, добавляя по каплям 2 молярных эквивалента трифторуксусного ангидрида к соединению в дегидрированном тетрагидрофуране в присутствии 4 молярных эквивалентов триэтиламина при температуре от -10 до 0°C и затем обрабатывая смесь водой при комнатной температуре.
Соединение, представленное формулой (C), полученное на второй стадии, может быть легко выделено с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как экстракция, промывка, высушивание, концентрация растворителя и обмен растворителя, и использовано на следующей стадии, или оно может использоваться на следующей стадии без очистки после постобрабоки.
Синтез соединения формулы (D) из соединения формулы (C)
Полученное выше соединение формулы (C) вводят в реакцию с активатором гидроксильной группы и затем с бензилоксиамином в присутствии основания, чтобы получить (2S,5R)-5-(бензилоксиамино)-1-(2,2,2-трифторацетил)пиперидин-2-трет-бутилкарбоксилат, представленный формулой (D):
Химическая формула 23
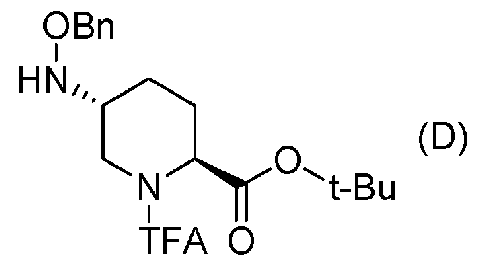 ,
,
причем в формуле (D), приведенной выше, TFA обозначает 2,2,2-трифторацетильную группу, t-Bu обозначает трет-бутильную группу, и OBn обозначает бензилоксигруппу.
В частности, реакцию можно осуществлять, растворяя соединение формулы (C) в подходящем растворителе и охлаждая полученный раствор, и добавляя к раствору, например, по каплям активатор гидроксильной группы в присутствии основания и затем добавляя бензилоксиамин и основание, чтобы осуществить реакцию.
Основание, которое может присутствовать в реакционном растворе, может быть выбрано из органических оснований, таких как триэтиламин, диизопропилэтиламин, трибутиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен, пиридин, 2-пиколин и 2,6-диметилпиридин, и используется в количестве в диапазоне от 2 до 3 молярных эквивалентов относительно соединения, представленного формулой (C).
Активатор гидроксильной группы может быть выбран из трифторметансульфонилхлорида и трифторметансульфонового ангидрида и используется в количестве в диапазоне от 1 до 1,5 молярных эквивалентов относительно соединения, представленного формулой (C). Реакцию проводят при температуре в диапазоне от -50 до +30°C.
Бензилоксиамин используется в количестве в диапазоне от 2 до 3 молярных эквивалентов относительно соединения, представленного формулой (C).
Растворитель, используемый в реакции, может быть выбран из дихлорметана, 1,2-дихлорэтана, толуола, этилацетата, бутилацетата, тетрагидрофурана, 1,4-диоксана, ацетонитрила, N,N-диметилформамида и N,N-диметилацетамида.
Предпочтительно, реакцию проводят, добавляя по каплям от 1,0 до 1,2 молярных эквивалентов трифторметансульфонового ангидрида к соединению в дегидратированном ацетонитриле или тетрагидрофуране в присутствии от 1,0 до 1,5 молярных эквивалентов ароматического амина, выбранного из пиридина, 2-пиколина и 2,6-диметилпиридина, при температуре от -40 до -20°C и перемешивая полученную смесь при той же самой температуре, пока соединение, представленное формулой (C), не исчезнет, и затем добавляя от 2 до 3 молярных эквивалентов бензилоксиамина и от 1,0 до 1,5 молярных эквивалентов 2,6-диметилпиридина, чтобы осуществить реакцию при температуре от -5 до +15°C в течение от 2 до 3 дней.
Более предпочтительно, реакцию проводят, добавляя по каплям 1,05 молярных эквивалентов трифторметансульфонового ангидрида к соединению в дегидратированном ацетонитриле в присутствии 1,1 молярных эквивалентов 2,6-диметилпиридина при температуре от -35 до -25°C и перемешивая полученную смесь при той же самой температуре, пока соединение, представленное формулой (C), не исчезнет, и затем добавляя 2 молярных эквивалента бензилоксиамина и 1,1 молярных эквивалентов 2,6-диметилпиридина, чтобы осуществить реакцию при температуре от 0 до 10°C в течение от 2 до 3 дней.
Соединение, представленное формулой (D), полученное на третьей стадии, может быть легко выделено с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как экстракция, промывка, высушивание, концентрация растворителя и обмен растворителя, и использовано на следующей стадии, или может использоваться на следующей стадии без очиски после постобрабоки.
Синтез соединения формулы (E) из соединения формулы (D)
Полученное выше соединение формулы (D) подвергают удалению трифторацетильной группы в присутствии неорганического основания, чтобы получить оптически активный (2S,5R)-5-(бензилоксиамино)пиперидин-2-трет-бутилкарбоксилат, представленный формулой (E):
Химическая формула 24
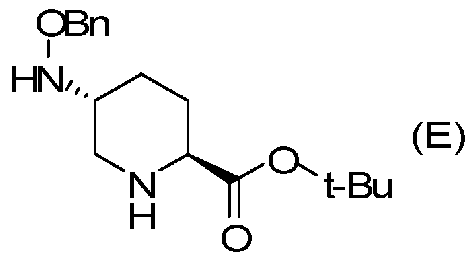 ,
,
причем в формуле (E), приведенной выше, t-Bu обозначает трет-бутильную группу, и OBn обозначает бензилоксигруппу.
В частности, удаление трифторацетильной группы из соединения, представленного формулой (D), можно осуществить, растворяя соединение формулы (D) в подходящем растворителе и подвергая его сольволизу в присутствии неорганического основания.
Неорганическое основание может быть выбрано из неорганических оснований, таких как гидроксид натрия, гидроксид калия, гидроксид лития, гидроксид цезия, карбонат натрия, карбонат калия и карбонат цезия, и используется в количестве в диапазоне от 1 до 3 молярных эквивалентов относительно соединения, представленного формулой (D).
Растворитель, используемый в реакции, может быть выбран из воды, метанола, этанола, тетрагидрофурана и 1,4-диоксана, и эти растворители могут использоваться индивидуально или в комбинации. Температура реакции предпочтительно составляет 30°C или ниже.
Предпочтительно, гидролиз осуществляют, используя от 1,5 до 2,5 молярных эквивалентов неорганического основания, выбранного из гидроксида натрия, гидроксида калия, гидроксида лития и гидроксида цезия в содержащем воду диоксане или тетрагидрофуране при температуре от 0°C до комнатной температуры.
Более предпочтительно, гидролиз осуществляют, используя 2 молярных эквивалента гидроксида натрия в содержащем воду диоксане при температуре от 0 до 30°C.
Соединение, представленное формулой (E), полученное на четвертой стадии, может быть легко выделено, например, в форме свободного основания с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как нейтрализация избытка основания, экстракция, промывка, высушивание, концентрация растворителя, обмен растворителя, формирование соли и кристаллизация, и использован на следующей стадии, или может использоваться на следующей стадии без очистки после постобрабоки.
Синтез соединения формулы (F1) из соединения формулы (E)
(5-1) Синтез соединения формулы (F1) из соединения формулы (E)
Соединение, представленное формулой (E), вводят в реакцию с эквивалентом фосгена в присутствии основания, чтобы осуществить формирование внутримолекулярного карбамида, таким образом получая (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат, представленный формулой (F1):
Химическая формула 25
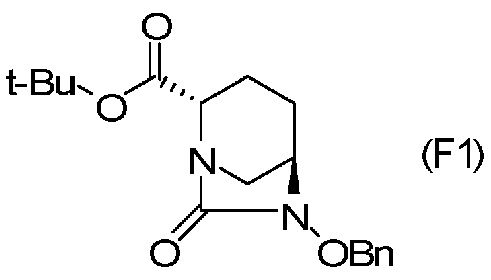 ,
,
причем в формуле (F1), приведенной выше, t-Bu обозначает трет-бутильную группу, и OBn обозначает бензилоксигруппу.
Основание, используемое в реакции, может быть выбрано из триэтиламина, диизопропилэтиламина, трибутиламина, 1,8-диазабицикло[5.4.0]ундец-7-ена, пиридина, 2-пиколина, 2,6-диметилпиридина и 4-диметиламинопиридина и может быть предпочтительно выбрано из третичного амина, выбранного из триэтиламина, диизопропилэтиламина и трибутиламина, и органического основания, например, ароматического амина, такого как 4-диметиламинопиридин, и используется в количестве в диапазоне от 2 до 4 молярных эквивалентов относительно соединения, представленного формулой (E). Когда 4-диметиламинопиридин используется в качестве основания, он используется в количестве в диапазоне от 0,01 до 2 молярных эквивалентов относительно соединения, представленного формулой (E).
Эквивалентный фосген может быть выбран из фосгена, дифосгена и трифосгена, предпочтительно из фосгена и дифосгена, и используется в количестве в диапазоне от 0,5 до 2 молярных эквивалентов относительно соединения, представленного формулой (E).
Растворитель, используемый в реакции, может быть выбран из, например, дихлорметана, 1,2-дихлорэтана, толуола, этилацетата, бутилацетата, тетрагидрофурана, 1,4-диоксана, ацетонитрила, N,N-диметилформамида и N,N-диметилацетамида.
Реакцию проводят при реакционной концентрации в диапазоне от 0,01 до 0,1 М. Реакцию проводят при температуре реакции в диапазоне от -20 до +30°C.
Предпочтительно, реакцию проводят, добавляя к соединению в дегидратированном ацетонитриле или тетрагидрофуране в концентрации от 0,01 до 0,1 М при температуре от -5 до 30°C от 2 до 3 молярных эквивалентов третичного амина, выбранного из триэтиламина, диизопропилэтиламина и трибутиламина, или от 0,05 до 1,5 молярных эквивалентов 4-диметиламинопиридина и от 0,5 до 1,0 молярных эквивалентов дифосгена или от 1,0 до 2,0 молярных эквивалентов фосгена, и перемешивая полученную смесь при комнатной температуре.
Более предпочтительно, реакцию проводят, добавляя к соединению в дегидратированном ацетонитриле в концентрации от 0,025 до 0,05 М при температуре от -5 до +25°C от 2,6 до 2,8 молярных эквивалентов триэтиламина или от 0,1 до 1,0 молярных эквивалентов 4-диметиламинопиридина и от 0,6 до 0,7 молярных эквивалентов дифосгена или от 1,2 до 1,4 молярных эквивалентов фосгена и перемешивая полученную смесь при комнатной температуре.
Соединение, представленное формулой (F1), полученное на стадии 5-1, может быть легко выделено с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как нейтрализация избытка основания, концентрация растворителя, экстракция, промывка, высушивание, концентрация растворителя, обмен растворителя и кристаллизация.
(5-2) Синтез соединения формулы (F1-1a) из соединения формулы (F1)
Сложный трет-бутиловый эфир полученного выше соединения формулы (F1) в положении 2 расщепляют, используя кислоту или соль металла, и затем добавляют циклогексиламин, чтобы получить (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты соль циклогексиламина, представленную формулой (F1-1a):
Химическая формула 26
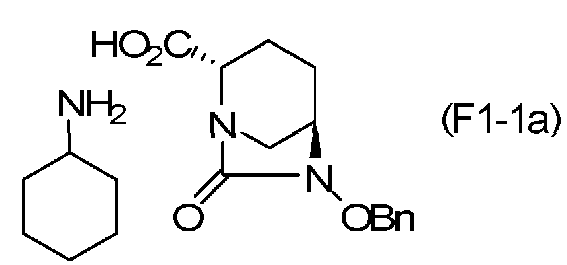 ,
,
причем в формуле (F1-1a), приведенной выше, OBn обозначает бензилоксигруппу.
Расщепление сложного трет-бутилового эфира соединения, представленного формулой (F1), с использованием кислоты или соли металла осуществляют, растворяя соединение формулы (F1) в подходящем растворителе и обрабатывая полученный раствор кислотой или солью металла.
Кислота, используемая в реакции, может быть выбрана из неорганических кислот, таких как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота и азотная кислота, и органических кислот, таких как муравьиная кислота, уксусная кислота, трифторуксусная кислота, тетрафторборная кислота, метансульфоновая кислота, п-толуолсульфоновая кислота и трифторметансульфоновая кислота. Кислота может быть предпочтительно выбрана из трифторуксусной кислоты, муравьиной кислоты, метансульфоновой кислоты, п-толуолсульфоновой кислоты, соляной кислоты и серной кислоты и используется в количестве в диапазоне от 1 молярного эквивалента относительно соединения, представленного формулой (F1), до количества растворителя.
Соль металла, используемая в реакции, может быть выбрана из йодида лития, йодида магния, бромида цинка, хлорида церия, тетрахлорида титана, трифторида бора, хлорида алюминия и бромида алюминия и используется в количестве в диапазоне от 1 до 6 молярных эквивалентов относительно соединения, представленного формулой (F1).
Растворитель, используемый в реакции, может быть выбран из воды, метанола, этанола, изопропанола, этилацетата, бутилацетата, 1,4-диоксана, дихлорметана, 1,2-дихлорэтана и толуола, и эти растворители могут использоваться индивидуально или в комбинации.
Реакцию проводят при температуре в диапазоне от -25 до +25°C.
Предпочтительно, соединение перемешивают в муравьиной кислоте или в дихлорметане с 2-3 молярными эквивалентами серной кислоты, или в смеси трифторуксусная кислота/дихлорметан (1/1) при температуре от 0 до +25°C.
Более предпочтительно, соединение перемешивают в смеси трифторуксусная кислота/дихлорметан (1/1) при температуре от 0 до +25°C.
Затем формирование соли с циклогексиламином можно осуществить, проводя, после завершения вышеописанной реакции, в случае необходимости, концентрацию растворителя, экстракцию, промывку, высушивание, концентрацию растворителя и обмен растворителя и затем добавляя к полученному продукту в подходящем растворителе циклогексиламин.
Эквивалент добавленного циклогексиламина выбирают из 1-4 молярных эквивалентов относительно соединения формулы (F1).
Эта стадия представляет собой стадию формирования соли для синтеза соединения формулы (F1-1), в которой М обозначает циклогексиламмоний, но с получением соединения формулы (F), в которой М обозначает неорганический катион или органический катион, отличный от циклогексиламмония, основание, используемое в формировании соли, может быть выбрано из аминов, таких как триметиламин, триэтиламин, циклогексиламин и дициклогексиламин; органических солей аммония, таких как тетраметиламмоний гидроксид, тетраэтиламмония гидроксид, тетрабутиламмония гидроксид и триэтилбензиламмония гидроксид; и солей 2-этилгексановой кислоты с щелочным или щелочноземельным металлом, таким как натрий, калий, литий или кальций. Эквивалент добавленного основания выбирают из 1-5 молярных эквивалентов относительно соединения формулы (F1).
В любом из случаев, когда М обозначает циклогексиламмоний, и случаев, когда М обозначает неорганический катион или органический катион, отличный от циклогексиламмония, растворитель, используемый в формировании соли, может быть выбран из метанола, этанола, изопропанола, ацетона, метилэтил кетона, этилацетата, бутилацетата, простого диэтилового эфира, простого диизопропилового эфира, тетрагидрофурана, 1,4-диоксана, дихлорметана, 1,2-дихлорэтана, толуола и гексана, и эти растворители могут использоваться индивидуально или в комбинации.
Предпочтительно, от 1 до 4 молярных эквивалентов циклогексиламина относительно соединения формулы (F1) добавляют к соединению в этилацетате, чтобы сформировать соль, с последующей кристаллизацией.
Более предпочтительно, от 1 до 3 молярных эквивалентов циклогексиламина добавляют к соединению в этилацетате, чтобы сформировать соль, с последующей кристаллизацией.
Соль, представленная формулой (F1-1), полученная на стадии 5-2, может быть легко выделена и сохранена с использованием обычных средств процедуры обработки, обычно используемых в органической химии, таких как фильтрация, промывка и высушивание, после формирования соли и кристаллизации, и, следовательно, особенно превосходна, также как промежуточное соединение в промышленном производстве.
(5-3) Синтез соединения формулы (F1-2) из соединения формулы (F1-1)
Полученное выше соединение формулы (F1-1) обрабатывают кислотой, чтобы получить свободную карбоновую кислоту, получая (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновую кислоту, представленную формулой (F1-2):
Химическая формула 27
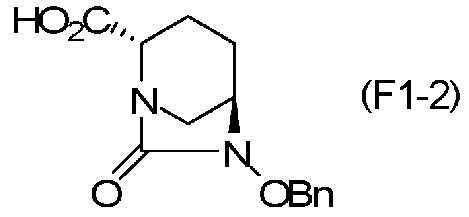 ,
,
причем в формуле (F1-2), приведенной выше, OBn обозначает бензилоксигруппу.
Для обработки соли соединения, представленного формулой (F1-1), кислотой, чтобы получить свободную карбоновую кислоту, соединение формулы (F1-1) растворяют в водном растворе подходящей кислоты и экстрагируют органическим растворителем.
Кислота, используемая в реакции, может быть выбрана из неорганических кислот, таких как соляная кислота, серная кислота, гидросульфат калия, фосфорная кислота, азотная кислота и дигидрофосфат натрия.
Органический растворитель, используемый в экстракции, может быть выбран из таких органических растворителей, как дихлорметан и этилацетат.
Предпочтительно, соединение формулы (F1-1) растворяют в водном растворе неорганической кислоты, выбранной из соляной кислоты, серной кислоты, гидросульфата калия и дигидрофосфата натрия, и экстрагируют с органическим растворителем, таким как этилацетатом.
Более предпочтительно, соединение формулы (F1-1) растворяют в насыщенном водном растворе дигидрофосфата натрия или разбавленной соляной кислоты и экстрагируют органическим растворителем, таким как этилацетат.
Карбоновая кислота, представленная формулой (F1-2), полученная на стадии 5-3, может быть выделена с использованием обычных средств процедуры обработки, обычно используемых в органической химии, таких как экстракция растворителем, концентрация, обмен растворителя и кристаллизация, или может использоваться на следующей стадии без выделения.
(5-4) Синтез соединения формулы (F1-4) из соединения формулы (F1-2)
Полученное выше соединение формулы (F1-2) вводят в реакцию с концентрированным водным раствором аммиака в присутствии основания и активатора карбоновой кислоты, чтобы получить оптически активный (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, представленный формулой (F1-4):
Химическая формула 28
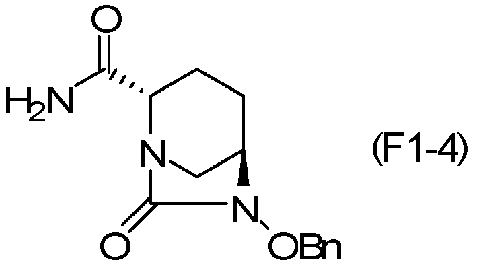 ,
,
причем в формуле (F1-4), приведенной выше, OBn обозначает бензилоксигруппу.
В частности, соединение, представленное формулой (F1-2), вводят в реакцию с активатором карбоновой кислоты и концентрированным водным раствором аммиака в подходящем растворителе в присутствии основания, или активный сложный эфир выделяют и затем вводят в реакцию с концентрированным водным раствором аммиака, чтобы получить соединение карбоксамида.
Основание, используемое в реакции, может быть выбрано из неорганических оснований, таких как гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, гидроксид натрия и гидроксид калия, и органических оснований, таких как триэтиламин, диизопропилэтиламин, трибутиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен, пиридин, 2-пиколин, 2,6-диметилпиридин и 4-диметиламинопиридин, и может быть предпочтительно выбрано из триэтиламина, диизопропилэтиламина и трибутиламина и может использоваться в количестве в диапазоне от 0,8 до 1,5 молярных эквивалентов относительно соединения, представленного формулой (F1-2).
Активатор карбоновой кислоты, используемый в реакции, может быть выбран из хлорангидридов кислоты, таких как этил хлорформиат, изобутил хлорформиат, пивалоил хлорид и 2,4,6-трихлорбензоил хлорид, и ангидридов кислоты, таких как изовалериановый ангидрид и триметилуксусный ангидрид, предпочтительно из этила хлорформиата, изобутила хлорформиата и пивалоил хлорида, и используется в количестве в диапазоне от 0,8 до 1,5 молярных эквивалентов относительно соединения, представленного формулой (F1-2).
Растворитель, используемый в реакции, может быть выбран из воды, дихлорметана, 1,2-дихлорэтана, толуола, этилацетата, бутилацетата, тетрагидрофурана, 1,4-диоксана, ацетонитрила, N,N-диметилформамида, N,N-диметилацетамида и пиридина, и эти растворители могут использоваться индивидуально или в комбинации.
Эта стадия может протекать в присутствии конденсирующего средства. Конденсирующее средство может быть выбрано из единственного карбодиимида, такого как N,N'-дициклогексилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид; и комбинации катализаторов, таких как 1-гидроксибензотриазол, N-гидроксисукцинимид или 2-гидроксипиридин-N-оксид и бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат, 2-хлор-1-метилпиридиния йодид или (4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид. Конденсирующее средство может использоваться в количестве в диапазоне от 0,8 до 1,5 молярных эквивалентов относительно соединения, представленного формулой (F1-2).
Концентрированный водный раствор аммиака используется в количестве в диапазоне от 5 до 100 молярных эквивалентов относительно соединения, представленного формулой (F1-2).
Реакцию проводят при температуре реакции в диапазоне от -20 до +25°C.
Предпочтительно, на этой стадии соединение вводят в реакцию с 1,1 молярными эквивалентами реагента на основе смешанного ангидрида кислоты, выбранного из этил хлорформиата, изобутил хлорформиата и пивалоил хлорида, в дегидратированном дихлорметане в присутствии 1,2 молярных эквивалентов третичного амина, выбранного из триэтиламина, диизопропилэтиламина и трибутиламина, при температуре от -5 до +5°C и затем вводят в реакцию с 5-50 молярными эквивалентами концентрированного водного раствора аммиака.
Более предпочтительно, соединение вводят в реакцию с 1,1 молярными эквивалентами изобутил хлорформиата в дихлорметане в присутствии 1,2 молярных эквивалентов триэтиламина при температуре от -5 до +5°C и затем вводят в реакцию с 5-20 молярными эквивалентами концентрированного водного раствора аммиака.
Соединение карбоксамида, представленное формулой (F1-4), полученное на стадии 5-4, может быть выделено с использованием обычных средств процедуры обработки, обычно используемых в органической химии, таких как экстракция растворителем, промывка, высушивание, концентрация растворителя, обмен растворителя и кристаллизация.
(5-5) Синтез соединения формулы (F1-3a), формулы (F1-3b), формулы (F1-3c) или формулы (F1-3d) из соединения формулы (F1-2); и синтез соединения формулы (F2-3) из соединения формулы (F2-2)
Полученное выше соединение, представленное формулой (F1-2), или нижеупомянутое соединение формулы (F2-2) подвергают этерификации карбоновой кислоты в положении 2, чтобы получить (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилат, представленный формулой (F1-3a):
Химическая формула 29
 ,
,
причем в формуле (F1-3a) Ме обозначает метильную группу, и OBn обозначает бензилоксигруппу;
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилат, представленный формулой (F1-3b):
Химическая формула 30
 ,
,
причем в формуле (F1-3b) OBn обозначает бензилоксигруппу;
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилат, представленный формулой (F1-3c):
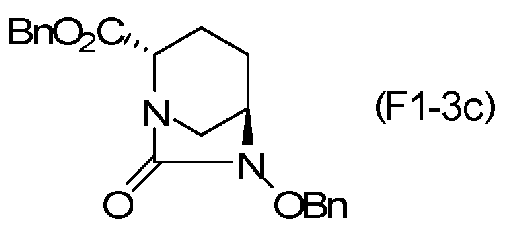
Химическая формула 31
 ,
,
причем в формуле (F1-3c) Bn обозначает бензильную группу, и OBn обозначает бензилоксигруппу;
(2S,5R)-2,5-диоксопирролидин-1-ил-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат, представленный формулой (F1-3d):
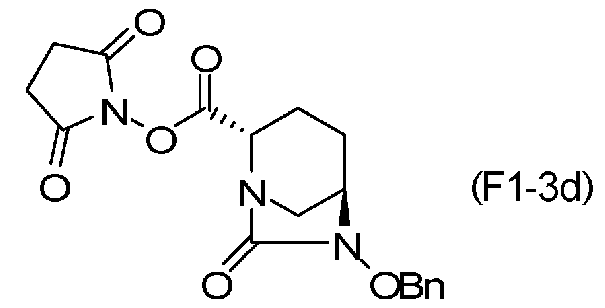
Химическая формула 32
 ,
,
причем в формуле (F1-3d) OBn обозначает бензилоксигруппу; или
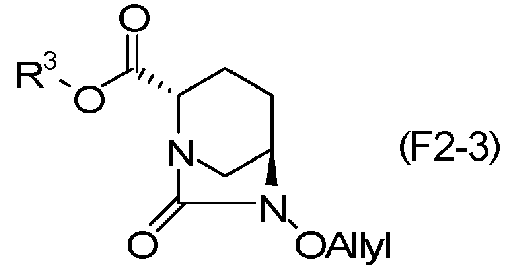
соединение, определенное формулой (F2-3):
Химическая формула 33
 ,
,
причем в формуле (F2-3) R3 обозначает метильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу.
В частности, этерификацию соединения, представленного формулой (F1-2), и соединения, представленного формулой (F2-2), можно осуществлять, вводя соединение в реакцию с галогеналкилом, аллилгалогенидом или бензилгалогенидом в подходящем растворителе в присутствии алкилирующего агента и основания; или вводя соединение в реакцию с активатором карбоновой кислоты или дегидратирующим конденсирующим средством и спиртом в присутствии основания.
Алкилирующий агент, используемый в реакции, может быть выбран из диазоалкилов, таких как диазометан, триметилсилилдиазометан и дифенилдиазометан, и соединений галогенов, таких как метилйодид, этилйодид, аллилхлорид, аллилбромид, бензилхлорид, бензилбромид, п-нитробензилбромид и п-метоксибензилбромид.
Основание, используемое в реакции, может быть выбрано из неорганических оснований, таких как гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидроксид калия, и органических оснований, таких как триэтиламин, диизопропилэтиламин, трибутиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен, пиридин, 2-пиколин, 2,6-диметилпиридин и 4-диметиламинопиридин.
Активатор карбоновой кислоты или конденсирующее средство, используемое в реакции, могут быть выбраны из единственного карбодиимида, такого как N,N'-дициклогексилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид; комбинации катализаторов, таких как 1-гидроксибензотриазол или 2-гидроксипиридин-N-оксид, и активатора карбоновой кислоты, такого как бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат, 2-хлор-1-метилпиридиния йодид или (4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид; и реагентов на основе смешанного ангидрида кислоты, включающих хлорангидрид кислоты, такой как этил хлорформиат, изобутил хлорформиат, пивалоил хлорид или 2,4,6-трихлорбензоил хлорид, и ангидрид кислоты, такой как изовалериановый ангидрид или триметилуксусный ангидрид.
Растворитель, используемый в реакции этерификации, может быть выбран из воды, дихлорметана, 1,2-дихлорэтана, толуола, этилацетата, бутилацетата, тетрагидрофурана, 1,4-диоксана, ацетонитрила, N,N-диметилформамида, N,N-диметилацетамида и пиридина, и эти растворители могут использоваться индивидуально или в комбинации.
Спирт, используемый в реакции, может быть выбран из метанола, аллилового спирта, бензилового спирта и 2,5-диоксопирролидин-1-ола.
На этой стадии, когда проводится этерификация метила, предпочтительно вводить соединение в реакцию с 1-1,5 молярными эквивалентами триметилсилилдиазометана в смеси растворителей толуол-метанол при охлаждении льдом.
Когда проводится этерификация аллила, предпочтительно вводить соединение в реакцию с 1-3 молярными эквивалентами аллилбромида в N,N-диметилформамиде в присутствии от 1 до 3 молярных эквивалентов гидрокарбоната натрия при комнатной температуре.
Когда проводится этерификация бензила, предпочтительно вводить соединение в реакцию с 1,5-2,5 молярными эквивалентами бензилового спирта в дихлорметане в присутствии от 1,3 до 1,7 молярных эквивалентов 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида при комнатной температуре.
Когда проводится этерификация 2,5-диоксопирролидин-1-ила, предпочтительно вводить соединение в реакцию с изобутил хлорформиатом в дихлорметане в присутствии третичного амина и затем с N-гидроксисукцинимидом при охлаждении льдом.
Соединения, представленные формулами (F1-3a), (F1-3b), (F1-3c) и (F1-3d), полученные на стадии 5-5, и соединение, представленное формулой (F2-3c), которое является частным случаем соединения, представленного формулой (F2-3), могут быть выделены с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как экстракция растворителем, разделение и промывка, высушивание, концентрация растворителя и кристаллизация.
(5-6) Синтез соединения формулы (F1-2) из соединения формулы (F1-3a) (стадии 5-6.1)
Сложный метиловый эфир полученного выше соединения формулы (F1-3a) гидролизуют, используя неорганическое основание, чтобы получить соединение формулы (F1-2).
В частности, расщепление сложного метилового эфира соединения, представленного формулой (F1-3a), полученного вышеописанным способом, можно осуществить, растворяя соединение формулы (F1-3a) в подходящем растворителе с последующим сольволизом в присутствии подходящего основания.
Неорганическое основание, используемое в реакции, может быть выбрано из таких неорганических оснований, как карбонат натрия, карбонат калия, карбонат цезия, гидроксид лития, гидроксид натрия, гидроксид калия и гидроксид цезия, и используется в количестве в диапазоне от 1,0 до 1,5 молярных эквивалентов относительно соединения, представленного формулой (F1-3a).
Растворитель, используемый в реакции, может быть выбран из воды, метанола, этанола, пропанола, изопропанола, бутанола, простого эфира, простого диизопропилового эфира, толуола, тетрагидрофурана и 1,4-диоксана, и эти растворители могут использоваться индивидуально или в комбинации.
Реакцию проводят при температуре реакции в диапазоне от -20 до +25°C.
Предпочтительно, соединение формулы (F1-3a) перемешивают в водном растворе тетрагидрофурана при температуре от -10 до +10°C, вместе с 1,0-1,2 эквивалентами гидроксида лития.
Более предпочтительно, соединение формулы (F1-3a) перемешивают в водном растворе тетрагидрофурана при температуре от -5 до +5°C, вместе с 1,0-1,1 эквивалентами гидроксида лития.
Карбоновая кислота, представленная (F1-2), полученная на стадии 5-6.1, может быть выделена с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как концентрация растворителя, подкисление, экстракция растворителем, разделение и промывка, высушивание, концентрация растворителя и формировании соли, и использована на следующей стадии, или она может использоваться на следующей стадии без выделения.
(5-6) Синтез соединения формулы (F1-2) из соединения формулы (F1-3b) через соединение формулы (F1-1a) (стадии 5-6.2)
Полученное выше соединение, представленное формулой (F1-3b), вводят в реакцию с нуклеофилом в присутствии катализатора для расщепления сложного эфира аллилового спирта и затем добавляют циклогексиламин, чтобы получить соединение формулы (F1-1a), с последующей обработкой неорганической кислотой с получением свободной карбоновой кислоты с получением соединения формулы (F1-2).
В частности, расщепление сложного эфира аллилового спирта соединения, представленного формулой (F1-3b), можно осуществлять, растворяя соединение формулы (F1-3b) в подходящем растворителе и обрабатывая полученный раствор подходящим нуклеофилом в присутствии катализатора.
Катализатор, используемый в реакции, может быть выбран из ацетата палладия, тетракис(трифенилфосфин)палладия, дихлорбис(трифенилфосфин)палладия, хлортрис(трифенилфосфин)родия и литий диметилмеди, и может использоваться в количестве в диапазоне от 0,01 до 0,1 молярных эквивалентов относительно соединения, представленного формулой (F1-3b).
Нуклеофил, используемый в реакции, может быть выбран из 2-этилгексаноата натрия, 2-метилгексаноата натрия, пирролидина, димедона, бензилоксиамина и бензолсульфената натрия, и может использоваться в количестве в диапазоне от 1 до 2 молярных эквивалентов относительно соединения, представленного формулой (F1-3b).
Растворитель, используемый в реакции, может быть выбран из воды, метанола, этанола, пропанола, изопропанола, бутанола, простого эфира, простого диизопропилового эфира, этилацетата, бутилацетата, дихлорметана, дихлорэтана, толуола, тетрагидрофурана, 1,4-диоксана и ацетонитрила, и эти растворители могут использоваться индивидуально или в комбинации.
Температуру реакции выбирают в диапазоне от -20 до +25°C.
На этой стадии, предпочтительно, соединение, представленное формулой (F1-3b), перемешивают в тетрагидрофуране, ацетонитриле или дихлорметане при комнатной температуре вместе с 1-2 молярными эквивалентами 2-этилгексаноата натрия, пирролидина или димедона, в присутствии от 0,01 до 0,05 молярных эквивалентов ацетата палладия, дихлорбис(трифенилфосфин)палладия или тетракис(трифенилфосфин)палладия.
Более предпочтительно, соединение, представленное формулой (F1-3b), перемешивают в дихлорметане при 20°C вместе с 1-1,5 молярными эквивалентами 2-этилгексаноата натрия в присутствии от 0,01 до 0,03 молярных эквивалентов тетракис(трифенилфосфин)палладия.
Таким образом полученное соединение с расщепленным сложным эфиром аллилового спирта обрабатывают циклогексиламином, чтобы получить соединение формулы (F1-1a), и затем соединение обрабатывают неорганической кислотой, выбранной из соляной кислоты, серной кислоты, гидросульфата калия и дигидрофосфата натрия, чтобы получить свободную карбоновую кислоту, получая соединение формулы (F1-2).
Карбоновая кислота, представленная формулой (F1-2), полученная на стадии 5-6.2, может быть выделена с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как концентрация растворителя, подкисление, экстракция растворителем, разделение и промывка, высушивание, концентрация растворителя и формирование соли, и использована на следующей стадии, или она может использоваться на следующей стадии без выделения.
(5-7) Синтез соединения формулы (F1-4) из соединения формулы (F1-3d)
Полученное выше соединение, представленное формулой (F1-3d), вводят в реакцию с водным раствором аммиака, чтобы получить соединение, представленное формулой (F1-4).
В частности, реакцию можно осуществить, растворяя соединение, представленное формулой (F1-3d), в подходящем растворителе и обрабатывая полученный раствор концентрированным водным раствором аммиака.
Концентрированный водный раствор аммиака используется в количестве в диапазоне от 5 до 100 молярных эквивалентов относительно соединения, представленного формулой (F1-3d).
Растворитель, используемый в реакции, может быть выбран из воды, дихлорметана, 1,2-дихлорэтана, толуола, этилацетата, бутилацетата, тетрагидрофурана, 1,4-диоксана, ацетонитрила, N,N-диметилформамида и N,N-диметилацетамида, и эти растворители могут использоваться индивидуально или в комбинации.
Реакцию проводят при температуре реакции в диапазоне от -20 до +25°C.
На этой стадии, предпочтительно, соединение, представленное формулой (F1-3d), вводят в реакцию с 5-50 молярными эквивалентами концентрированного водного раствора аммиака в дегидратированном дихлорметане при температуре от -5 до +5°C.
Более предпочтительно, соединение, представленное формулой (F1-3d), вводят в реакцию с 5-20 молярными эквивалентами концентрированного водного раствора аммиака в дихлорметане при температуре от -5 до +5°C.
Соединение карбоксамида, представленное формулой (F1-4), полученное на стадии 5-7, может быть выделено с использованием обычных средств процедуры обработки, обычно используемых в органической химии, таких как экстракция растворителем, промывка, высушивание, концентрация растворителя, обмен растворителя и кристаллизация.
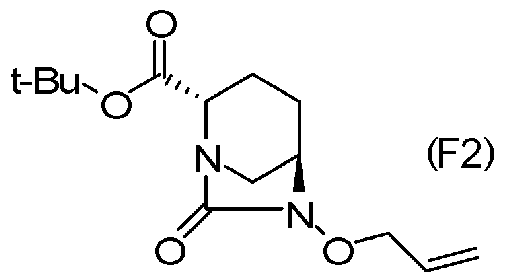
(5-8) Синтез соединения формулы (F2) из соединения формулы (F1)
Бензильную группу полученного выше соединение, представленного формулой (F1), удаляют реакцией каталитического гидрирования, и затем полученное соединение вводят в реакцию с аллилирующим агентом в присутствии основания, чтобы получить (2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат, представленный формулой (F2):
Химическая формула 34
 ,
,
причем в формуле (F2), приведенной выше, t-Bu обозначает трет-бутильную группу.
В частности, преобразование бензильной группы соединения, представленного формулой (F1), в аллильную группу можно осуществить, растворяя соединение формулы (F1) в подходящем растворителе и подвергая реакции гидрирования с использованием катализатора и затем реакции аллилирования в присутствии основания.
Катализатор, используемый в реакции, может быть выбран из произвольных гидрирующих катализаторов, и предпочтительно может использоваться никель Ренея, оксид платины, оксид палладия, палладиевая чернь или палладий на углероде.
Давление водорода может быть от атмосферного давления до 0,5 МПа.
Растворитель, используемый в реакции гидрирования, может быть выбран из воды, метанола, этанола, пропанола, изопропанола, бутанола, простого эфира, простого диизопропилового эфира, этилацетата, бутилацетата, толуола, тетрагидрофурана и 1,4-диоксана, и эти растворители могут использоваться индивидуально или в комбинации.
Стадию гидрирования предпочтительно проводят в метаноле или этаноле, используя катализатор, выбранный из оксида платины, оксида палладия, палладиевой черни и палладия на углероде.
Более предпочтительно, эту стадию проводят в этаноле, используя палладий на углероде в качестве катализатора.
6-гидрокси соединение с удаленной бензильной группой, полученное на вышеописанной стадии гидрирования, может использоваться на следующей стадии без выделения с использованием, после завершения реакции, обычных средств процедуры обработки, обычно используемых в органической химии, таких как фильтрация для катализатора, концентрация растворителя и обмен растворителя.
Основание, используемое в реакции аллилирования, может быть выбрано из неорганических оснований, таких как гидрокарбонат натрия, гидрокарбонат калия, карбонат натрия, карбонат калия, карбонат цезия, гидроксид натрия и гидроксид калия, и органических оснований, таких как триэтиламин, диизопропилэтиламин, трибутиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен, пиридин, 2-пиколин, 2,6-диметилпиридин и 4-диметиламинопиридин, и может использоваться в количестве в диапазоне от 1,0 до 3 молярных эквивалентов относительно соединения, представленного формулой (F1).
Аллилирующий агент, используемый в реакции аллилирования, может быть выбран из аллилхлорида и аллилбромида и может использоваться в количестве в диапазоне от 1,0 до 3 молярных эквивалентов относительно соединения, представленного формулой (F1).
Растворитель, используемый в реакции аллилирования, может быть выбран из дихлорметана, 1,2-дихлорэтана, толуола, этилацетата, бутилацетата, тетрагидрофурана, 1,4-диоксана, ацетонитрила, N,N-диметилформамида и N,N-диметилацетамида.
Реакцию проводят при температуре реакции от 0 до +25°C.
На стадии реакции аллилирования, предпочтительно, соединение перемешивают в дегидратированном ацетонитриле, N,N-диметилформамиде или N,N-диметилацетамиде при комнатной температуре, вместе с 1-2 молярными эквивалентами аллилбромида, в присутствии 1-2 молярных эквивалентов неорганического основания, выбранного из безводного карбоната натрия, карбоната калия и карбоната цезия.
Более предпочтительно, соединение перемешивают в дегидратированном ацетонитриле при комнатной температуре вместе с 1-2 молярными эквивалентами аллилбромида в присутствии 1 молярного эквивалента безводного карбоната калия.
Соединение, представленное формулой (F2), полученное на стадии 5-8, может быть выделено с использованием, после завершения реакции, обычных средств обработки, обычно используемых в органической химии, таких как концентрация растворителя, обмен растворителя, разделение и промывка, высушивание и концентрация растворителя.
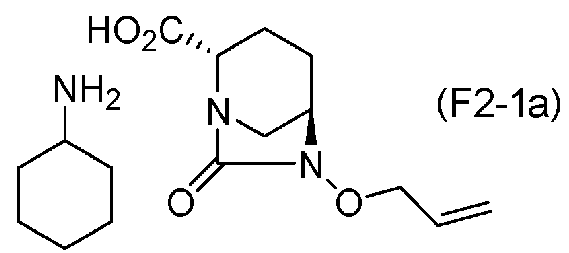
(5-9) Синтез соединения формулы (F2-1a) из соединения формулы (F2)
Сложный трет-бутиловый эфир полученного выше соединения формулы (F2) в положении 2 расщепляют, используя кислоту, и затем добавляют циклогексиламин, чтобы получить соль циклогексиламина (2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, представленную формулой (F2-1a):
Химическая формула 35
 .
.
Расщепление сложного трет-бутилового эфира соединения, представленного формулой (F2), с использованием кислоты проводят, растворяя соединение формулы (F2) в подходящем растворителе и выполняя тот же самый процесс как на стадии 5-2. Относительно кислоты, предпочтительно может использоваться трифторуксусная кислота, метансульфоновая кислота, п-толуолсульфоновая кислота, соляная кислота или серная кислота, и после расщепления сложного трет-бутилового эфира с использованием кислоты, обработка циклогексиламином приводит к циклогексиламин соли.
Соль, представленная формулой (F2-1a), полученная на стадии 5-9, может быть легко выделена и сохранена с использованием обычных средств процедуры обработки, обычно используемых в органической химии, таких как фильтрация, промывка и высушивание, после формирования соли и кристаллизации, и следовательно, особенно превосходна также в качестве промежуточного соединения в промышленном производстве.
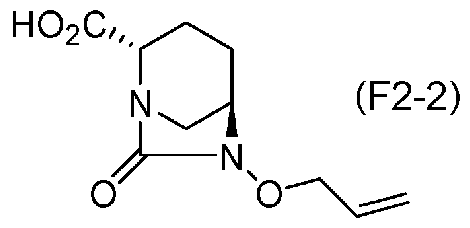
(5-10) Синтез соединения формулы (F2-2) из соединения формулы (F2-1a)
Полученное выше соединение формулы (F2-1a) обрабатывают кислотой, чтобы получить свободную карбоновую кислоту, получая (2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновую кислоту, представленную формулой (F2-2):
Химическая формула 36
 .
.
Для обработки соли, представленной формулой (F2-1a), кислотой с получением свободной карбоновой кислоты, соединение формулы (F2-1a) растворяют в водном растворе подходящей кислоты и подвергают тому же самому процессу, как на стадии 5-3. Относительно кислоты, может использоваться неорганическая кислота, такая как соляная кислота, серная кислота, гидросульфат калия или дигидрофосфат натрия.
Карбоновая кислота, представленная формулой (F2-2), полученная на стадии 5-10, может быть выделена с использованием обычных средств процедуры обработки, обычно используемых в органической химии, таких как экстракция растворителем, концентрация и обмен растворителя, и использована на следующей стадии, или она может использоваться на следующей стадии без выделения.
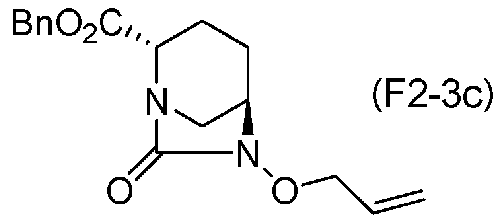
(5-5) Синтез соединения формулы (F2-3c) из соединения формулы (F2-2)
Карбоновую кислоту полученного выше соединения, представленного формулой (F2-2), в положении 2 вводят в реакцию с бензиловым спиртом в присутствии дегидрирующего конденсирующего средства, чтобы получить (2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилат, который является частным случаем соединения, представленного формулой (F2-3), и который представлен формулой (F2-3c):
Химическая формула 37
 ,
,
причем в формуле (F2-3c), приведенной выше, Bn обозначает бензильную группу.
Соединения, представленные формулами (F1), (F1-3a) и (F1-3b), приведенными ниже, полученные на вышеописанных стадиях, могут быть кристаллизованы из, например, раствора этилацетата и гексана. Соединение, представленное формулой (F1-1a), может быть кристаллизовано из, например, смеси этилацетат-простой эфир. Далее, соединение, представленное формулой (F1-2), может быть кристаллизовано из, например, смеси этилацетат-гексан. Кроме того, соединение, представленное формулой (F1-4), может быть кристаллизовано из, например, раствора хлороформа и гексана.
Химическая формула 38
 .
.
В приведенных формулах t-Bu обозначает трет-бутильную группу, OBn обозначает бензилоксигруппу, и Ме обозначает метильную группу.
Относительно соединений, представленных формулами (F1), (F1-3a), (F1-3b), (F1-1a), (F1-2) и (F1-4), которые могут быть получены, как описано выше, наблюдение под поляризационным микроскопом и порошковая рентгеновская дифрактометрия подтвердили, что каждое из соединений может быть получено в форме кристалла, и особенно в порошковой рентгеновской дифрактометрии каждое соединение идентифицировано характеристическими пиками. Пиковые структуры соединений показаны ниже в таблицах 1-6.
Порошковая дифракция рентгеновских лучей соединения (F1)
Порошковая дифракция рентгеновских лучей соединения (F1)
Порошковая дифракция рентгеновских лучей соединения (F1)
Порошковая дифракция рентгеновских лучей соединения (F1)
Порошковая дифракция рентгеновских лучей соединения (F1)
Порошковая дифракция рентгеновских лучей соединения (F1)
Соединение, определенное формулой (F), согласно настоящему изобретению может использоваться как промежуточное соединение для получения соединения, представленного формулой (H), приведенной ниже. Соединение, представленное формулой (H), приведенной ниже, и его антипод получали из соединения, представленного формулой (F), согласно настоящему изобретению в качестве исходного материала, и сравнивали относительно биологической активности.
Химическая формула 39
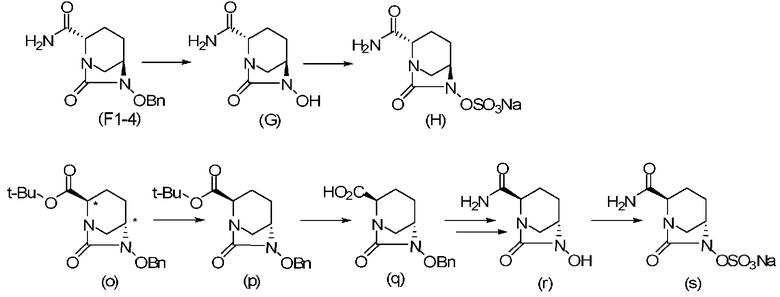
В приведенной выше схеме химической реакции, t-Bu обозначает трет-бутильную группу, и OBn обозначает бензилоксигруппу.
Соединение, представленное формулой (H), было получено из соединения, представленного формулой (F1-4), среди соединений, представленных формулой (F), полученных способом согласно настоящему изобретению. Кроме того, рацемический (2R/S,5S/R)-6-(бензилокси)-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат, представленный формулой (o), был получен и подвергнут оптическому разделению с использованием хиральной колонки с получением антипода, представленного формулой (p), с получением, таким образом, (2R,5S)-1,6-диазабицикло[3.2.1]октан-2-карбоксамида, 7-оксо-6-(сульфокси)-мононатриевой соли, представленная формулой (s) через антипод.
Относительно каждого из полученных соединений формул (H) и (s), оценивали ингибирующую активность в отношении β-лактамазного фермента и эффект каждого соединения в комбинации с антибиотиком. В результате было обнаружено, что соединение, представленное формулой (H), продемонстрировало активность, но соединение, представленное формулой (s), не показало никакой активности. Результаты подтвердили, что соединение формулы (F), которое может быть получено способом согласно настоящему изобретению, представляет собой энантиомер, особенно пригодный в качестве сырья для получения лекарственного средства и промежуточного соединения для его получения.
В таблице 7, приведенной выше, TAZ обозначает Tazobactam, IC50 указывает ингибирующую активность против фермента AmpC, и MIC указывает противомикробную активность пиперациллина (PIPC) при использовании в комбинации с 4 мкг/мл соединения.
В этом случае, антипод, представленный формулой (p), получают из рацемического соединения оптическим разделением, но антиподы, представленные формулами (r) и (s), не могут быть отделены от соответствующих рацемических соединений с использованием хиральной колонки с нормальной фазой или с обратной фазой. Также было подтверждено, что рацемические соединения, представленные формулой (o), имеющие сложный трет-бутиловый эфир, демонстрируют превосходные свойства в качестве промежуточного соединения, так, что специальный растворитель не является необходимым в качестве мобильной фазы, что отделение от его антипода является легким, и что его разложение в ходе концентрации активной фракции не является вероятным.
Далее, соединения, представленные формулами (F1-2) и (F1-4), могут использоваться также в качестве важного промежуточного соединения для получения оптически активного соединения для ингибитора β-лактамазы, имеющего диазабициклооктановый скелет, показанный в патентных документах 1-6, или для исследования более эффективного нового ингибитора β-лактамазы и для фармацевтических разработок.
Химическая формула 40
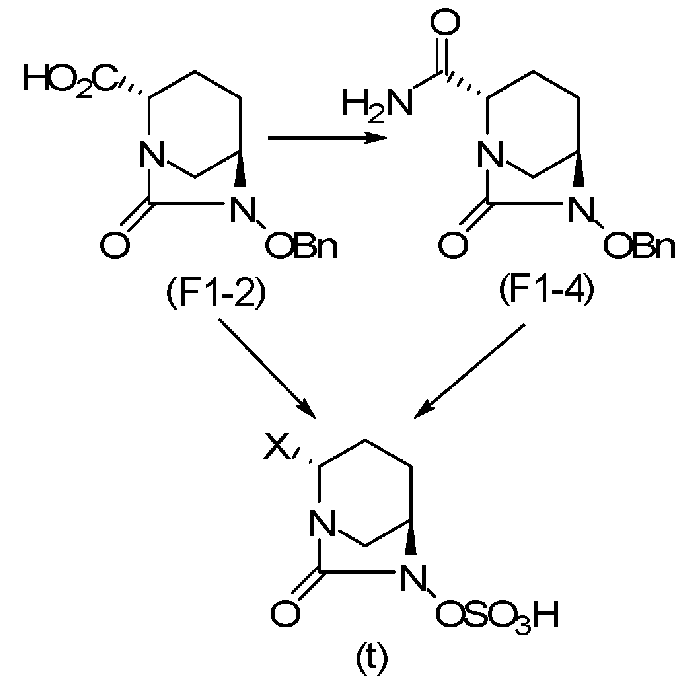
В описанной выше схеме химической реакции OBn обозначает бензилоксигруппу, и X обозначает активный заместитель.
Биологическая активность вышеописанных соединений, представленных формулами (H) и (s), может быть измерена следующим образом. В частности, ингибирующую активность (значение IC50) против фермента AmpC, который является β-лактамазой класса C, определяли, используя нитроцефин в качестве субстрата, чтобы проверить, действительно ли соединения имели ингибирующую активность, и сравнить их активности. Далее, используя конститутивно продуцирующую AmpC Pseudomonas aeruginosa, объединенную противомикробную активность (MIC) измеряли, используя пиперациллин (PIPC) в качестве антибиотика и соединение формулы (H) или (s) в комбинации, чтобы проверить, действительно ли противомикробная активность PIPC могла быть восстановлена.
Таким образом, настоящее изобретение также относится к применению следующих конкретных соединений, представленных формулой (F), для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты:
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат;
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилат;
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилат;
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилат;
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота соль циклогексиламмония;
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота;
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид; и
(2S,5R)-5-(бензилоксиамино)пиперидин-2-трет-бутилкарбоксилат.
ПРИМЕРЫ
Далее настоящее изобретение будет проиллюстрировано более подробно примерами, но настоящее изобретение не ограничено примерами, поскольку возможны различные модификации.
Справочный пример 1
(2S,5S)-1-бензил-2-трет-бутил-5-гидроксипиперидин-1,2-дикарбоксилат (A)
Стадия 1: (S)-1-бензил-2-трет-бутил-5-оксопирролидин-1,2-дикарбоксилат
Химическая формула 41
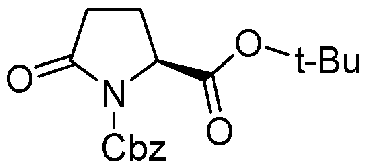 .
.
100 г (S)-1-(бензилоксикарбонил)-5-оксопирролидин-2-карбоновой кислоты растворяли в дегидратированном дихлорметане (2 л) и при охлаждении льдом добавляли концентрированную серную кислоту (10 мл) и 213 г изобутена, с последующим перемешиванием в течение ночи при +20°C или меньше. Реакционную смесь добавляли к холодному водному раствору карбоната натрия, отмечая вскипание, с последующим жидкостным разделением органической фазы, промывали насыщенным солевым раствором и высушивали над безводным сульфатом магния, и растворитель концентрировали при пониженном давлении. Остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=7/3) и кристаллизовали с гексаном/этилацетатом, получая 80 г целевого соединения в форме бесцветного кристаллического порошка (выход 67%). Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, УФ 210 нм, гексан/этанол=2/1, объемная скорость потока 1 мл/мин, время удерживания 4,2 мин).
[α]20 D-43,3° (c 0,52 в CHCl3), согласно непатентному документу 4 -41,8° (c 6,71, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,39 (с, 9H), 2,04 (м, 1H), 2,32 (м, 1H), 2,51 (ддд, J=17,6, 9,5, 3,2 Гц, 1H), 2,62 (ддд, J=17,6, 10,5, 9,5 Гц, 1H), 4,55 (дд, J=9,5, 2,7 Гц, 1H), 5,25 (д, J=12,2 Гц, 1H), 5,30 (д, J=12,2 Гц, 1H), 7,26-7,41 (м, 5H); MS m/z: 320 (M+1).
Стадия 2: (S)-2-(бензилоксикарбониламино)-5-оксо-6-диметил-трет-бутилсульфоксонийгексаноат
Химическая формула 42
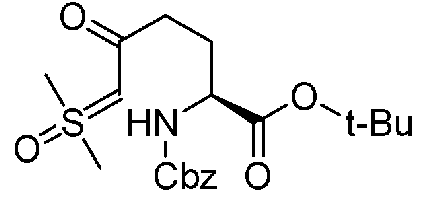 .
.
К раствору 70,2 г (313 ммоль) триметилсульфоксония йодида в дегидратированном N,N-диметилформамиде (585 мл) под атмосферой аргона добавляли 36,8 г (279 ммоль) трет-бутоксида калия с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем при 5°C или меньше в течение 20 минут добавляли 87,0 г (272 ммоль) (S)-1-бензил-2-трет-бутил-5-оксопирролидин-1,2-дикарбоксилата (промытого дегидратированным N,N-диметилформамидом (87 мл)), с последующим введением в реакцию при той же самой температуре в течение 1 часа. Реакционную смесь добавляли к ледяной воде (2,6 л), насыщали хлоридом натрия, экстрагировали этилацетатом (2,6 л × однократно, 1,3 л × дважды, 650 мл × 4 раза), и растворитель органического слоя отгоняли при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гептан/этилацетат=1/2→этилацетат/метанол=19/1→9/1), получая 112,3 г целевого соединения в форме светло-желтого масла (количественный выход). 1H ЯМР (400 МГц, CDCl3, δ): 1,46 (с, 9H), 1,95 (м, 1H), 2,09 (м, 1Н), 2,23-2,32 (м, 2H), 3,32 (с, 3H), 3,33 (с, 3Н), 4,22 (м, 1H), 4,37 (с, 1H), 5,07 (д, J=12,0 Гц, 1H), 5,13 (д, J=12 Гц, 1H), 5,75 (шир. д, J=8,0 Гц, 1H), 7,30-7,36 (м, 5H); MS m/z: 412 (M+1).
Стадия 3: (S)-1-бензил-2-трет-бутил-5-оксопиперидин-1,2-дикарбоксилат
Химическая формула 43
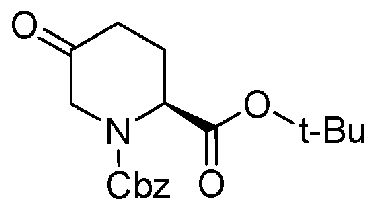 .
.
24,8 г (57,84 ммоль) (S)-2-(бензилоксикарбониламино)-5-оксо-6-диметил-трет-бутилсульфоксония гексаноата растворяли в 1,2-дихлорэтане (774 мл) и, после деаэрации, 388,5 мг (0,58 ммоль) ди-μ-хлорбис-[(η-циклоокт-1,5-диен)]дииридия(I) добавляли под атмосферой аргона с последующим повышением температуры и введением в реакцию при +70°C в течение 2 часов. Растворитель реакционной смеси отгоняли при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=2/1), получая 14,55 г целевого соединения в форме красного масла (выход 76%). 1H ЯМР (400 МГц, CDCl3, δ): 1,38 (с, 4,5H), 1,47 (с, 4,5H), 2,12-2,48 (м, 4H), 3,93 (д, J=19,0 Гц, 0,5H), 4,00 (д, J=18,8 Гц, 0,5H), 4,37 (д, J=18,8 Гц, 0,5H), 4,46 (д, J=19,0 Гц, 0,5H), 4,62 (дд, J=7,3, 6,6 Гц, 0,5H), 4,77 (дд, J=6,6, 5,9 Гц, 0,5H), 5,10-5,23 (м, 2H), 7,34-7,35 (м, 5H); MS m/z: 334 (M+1).
Стадия 4: (2S,5S)-1-бензил-2-трет-бутил-5-гидроксипиперидин-1,2-дикарбоксилат (A)
Химическая формула 44
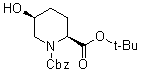 .
.
Раствор 14,55 г (43,66 ммоль) (S)-1-бензил-2-трет-бутил-5-оксопиперидин-1,2-дикарбоксилата в этаноле (437 мл) охлаждали льдом и добавляли 1,65 г (43,62 ммоль) боргидрида натрия с последующим введением в реакцию при охлаждении льдом в течение 20 минут. Насыщенный водный раствор хлорида аммония добавляли по каплям к реакционной смеси, пока вскипание не прекращалось, и полученную соль растворяли добавлением воды. Органический растворитель смеси отгоняли при пониженном давлении, и водный слой остатка экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором с последующим высушиванием над безводным сульфатом магния, и растворитель отгоняли при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=3/1→2/1), получая 13,35 г целевого соединения в форме бесцветного масла (выход 91%). Энантиомерный избыток: 98,8% энантиомерного избытка (CHIRALPAK AD-H, 4,6×150 мм, УФ 210 нм, гексан/этанол=4/1, объемная скорость потока 1 мл/мин, время удерживания 9,1 мин).
[α]20 D-29,7° (c 1,3, CHCl3), согласно непатентному документу 1 -27,9° (c 2,0, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,42 (с, 4,5H), 1,46 (с, 4,5H), 1,66-1,75 (м, 2H), 1,96-2,00 (м, 2H), 2,24-2,30 (м, 1H), 2,74-2,80 (м, 0,5H), 2,84-2,90 (м, 0,5H), 3,64 (шир.с, 1H), 4,15-4,20 (м, 0,5H), 4,23-4,27 (м, 0,5H), 4,65 (д, J=5,4 Гц, 0,5H), 4,78 (д, J=4,6 Гц, 0,5H), 5,07 (д, J=12,5 Гц, 1H), 5,21 (д, J=12,5 Гц, 1H), 7,26-7,37 (м, 5H); MS m/z: 334 (M+1).
Последовательный синтез (2S,5S)-1-бензил-2-трет-бутил-5-гидроксипиперидин-1,2-дикарбоксилата (A)
Химическая формула 45
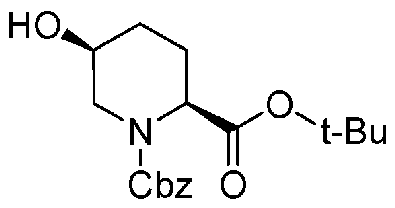 .
.
112,3 г (272 ммоль) (S)-трет-бутил-2-(бензилоксикарбониламино)-5-оксо-6-диметилсульфоксония гексаноата растворяли в 1,2-дихлорэтане (3,4 л) и, после деаэрации, 1,83 г (2,72 ммоль) ди-μ-хлорбис-[(η-циклоокт-1,5-диен)]дииридия(I) добавляли под атмосферой аргона с последующим повышением температуры до +70°C в течение 1,75 часов и реакцией в течение 1 часа. После охлаждения до комнатной температуры, растворитель реакционной смеси отгоняли при пониженном давлении, и полученный остаток растворяли в этаноле (1,1 л). Смесь охлаждали льдом, и 5,14 г (136 ммоль) боргидрида натрия добавляли в течение 10 минут, с последующим введением в реакцию при охлаждении льдом в течение 20 минут. Насыщенный водный раствор хлорида аммония (265 мл) добавляли по каплям к реакционной смеси, пока вскипание не прекращалось, и полученную соль растворяли добавлением воды (250 мл). Органический растворитель смеси отгоняли при пониженном давлении, и водный слой остатка экстрагировали этилацетатом (0,9 л × 3 раза). Растворитель отгоняли при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (гептан/этилацетат=3/1→2/1), получая 66,82 г целевого соединения в форме бесцветного масла (выход 73%). Инструментальные данные были совместимы с таковыми со стадией 4 из Справочного примера 1.
Пример 1
(2S,5S)-5-гидроксипиперидин-2-трет-бутилкарбоксилат (B)
Химическая формула 46
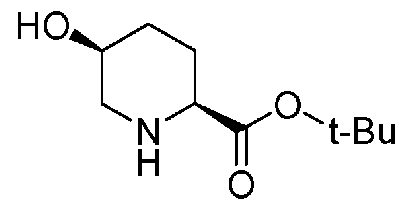 .
.
К раствору 67,2 г (200,4 ммоль) (2S,5S)-1-бензил-2-трет-бутил-5-гидроксипиперидин-1,2-дикарбоксилата в этаноле (900 мл) добавляли 10,1 г 10%-го палладия на углероде (содержание воды приблизительно 50%), с последующим энергичным перемешиванием в течение ночи при комнатной температуре в атмосфере водорода. Катализатор смеси фильтровали через слой целита, чтобы сконцентрировать фильтрат, посредством чего получали 39,3 г целевого соединения в форме бесцветного твердого вещества (выход 97%). Энантиомерный избыток: 99% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, УФ 210 нм, диэтиламин/гексан/этанол=0,1/80/20, объемная скорость потока 1 мл/мин, время удерживания 6,3 мин).
[α]20 D-28,7°(c 1,01, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,47 (с, 9H), 1,63 (м, 1H), 1,79-1,84 (м, 3H), 2,82 (дд, J=12,2, 2,2 Гц, 1H), 3,02 (ддд, J=12,2, 3,7, 1,7 Гц, 1H), 3,21 (м, 1H), 3,80 (м, 1H); MS m/z: 202 (M+1).
Пример 2
(2S,5S)-5-гидрокси-1-(2,2,2-трифторацетил)пиперидин-2-трет-бутилкарбоксилат (C)
Химическая формула 47
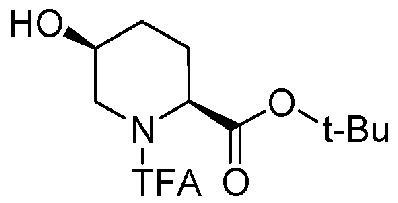 .
.
Раствор 39,14 г (194 ммоль) (2S,5S)-5-гидроксипиперидин-2-трет-бутилкарбоксилата в дегидратированном тетрагидрофуране (450 мл) охлаждали до температуры от -3 до -5°C под атмосферой аргона и добавляли 78,7 г (776 ммоль) триэтиламина с последующим добавлением по каплям 81,5 г (388 ммоль) ангидрида трифторуксусной кислоты в течение 30 минут. Реакционной смеси давали реагировать при температуре от -3 до -5°C в течение 1 часа и добавляли воду (90 мл) с последующим повышением температуры до комнатной температуры и перемешиванием в течение 1 часа. К реакционной смеси добавляли воду (740 мл) с последующей экстракцией этилацетатом (450 мл × 3 раза), и объединенный органический слой промывали последовательно 5%-ым водным раствором лимонной кислоты (450 мл), водным 6,5% раствором гидрокарбоната натрия (450 мл) и водой (450 мл). Растворитель отгоняли при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=2/1), получая 50,06 г целевого соединения в форме твердого вещества светло-желтого цвета (выход 87%). Энантиомерный избыток: 99% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, УФ 210 нм, гексан/этанол=4/1, объемная скорость потока 1 мл/мин, время удерживания 4,2 мин).
[α]20 D-54,1°(c 0,73, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): представлено как смесь двух ротамеров (7:3), 1,26-1,43 (м, 1H), 1,46 (с, 2,7H), 1,47 (с, 6,3H), 1,68-1,77 (м, 1H), 1,81 (д, J=4,8 Гц, 0,3H), 1,89 (д, J=5,2 Гц, 0,7H), 2,05-2,08 (м, 1H), 2,36-2,42 (м, 1H), 2,77 (дд, J=12,2, 12,0 Гц, 0,3H), 3,12 (дд, J=13,2, 10,7 Гц, 0,7H), 3,68-3,77 (м, 1H), 4,00 (м, 1H), 4,52-4,60 (м, 0,6H), 5,07 (д, J=5,9 Гц, 0,7H); MS m/z: 298 (M+1).
Пример 3
(2S,5R)-5-(бензилоксиамино)-1-(2,2,2-трифторацетил)пиперидин-2-трет-бутилкарбоксилат (D)
Химическая формула 48
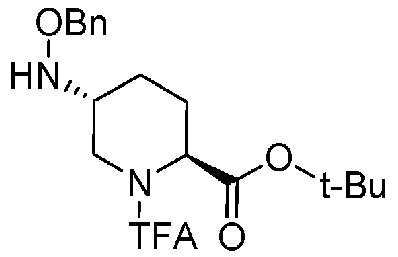 .
.
После охлаждения раствора 10,22 г (34,38 ммоль) (2S,5S)-5-гидрокси-1-(2,2,2-трифторацетил)пиперидин-2-трет-бутилкарбоксилата в дегидратированном ацетонитриле (113 мл) до температуры от -30 до -40°C под атмосферой аргона добавляли 4,4 мл (37,78 ммоль) 2,6-диметилпиридина и затем 5,92 мл (36,09 ммоль) ангидрида трифторметансульфоновой кислоты добавляли по каплям за 10 минут с последующей реакцией при -30°C в течение 15 минут. К этой реакционной смеси добавляли 8,46 г (68,73 ммоль) бензилоксиамина (промытого ацетонитрилом (5 мл)), с последующим повышением температуры до 0°C в течение 30 минут, и добавляли еще 4,4 мл (37,78 ммоль) 2,6-диметилпиридина с последующей реакцией при температуре от 0 до 5°C в течение 3,5 дней. Эту реакционную смесь концентрировали при пониженном давлении, и полученный остаток разбавляли этилацетатом (200 мл) и последовательно промывали водой (200 мл), 10%-ым водным раствором лимонной кислоты (200 мл × 3 раза), водным 6,5% раствором гидрокарбоната натрия (100 мл) и насыщенным солевым раствором (100 мл). Каждый водный слой снова экстрагировали этилацетатом (100 мл), органические слои объединяли и высушивали над безводным сульфатом магния, и растворитель отгоняли при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=4/1), получая 11,69 г целевого соединения в форме бесцветного масла (выход 85%). Энантиомерный избыток: 99,0% энантиомерного избытка (CHIRALPAK AD-H, 4,6×150 мм, УФ 210 нм, гексан/этанол=9/1, объемная скорость потока 1 мл/мин, время удерживания 4,5 мин).
[α]20 D-45,6°(c 0,73, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): представлено как смесь двух ротамеров (7-3), 1,46 (с, 2,7H), 1,48 (с, 6,3H), 1,62-1,65 (м, 2H), 1,93-2,05 (м, 2H), 3,13 (м, 0,3H), 3,24-3,29 (м, 1H), 3,46 (м, 0,7H), 4,12 (м, 0,3H), 4,58-4,77 (м, 2,7H), 5,06 (м, 0,7H), 5,38 (м, 1H), 7,30-7,36 (м, 5H); MS m/z: 403 (M+1).
Пример 4
(2S,5R)-5-(бензилоксиамино)пиперидин-2-трет-бутилкарбоксилат (E)
Химическая формула 49
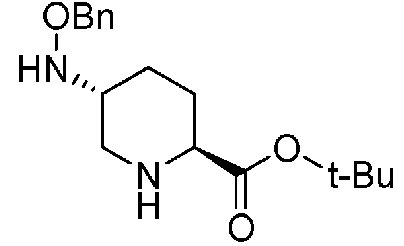 .
.
Воду (9,2 мл) добавляли к раствору 6,91 г (17,17 ммоль) (2S,5R)-5-(бензилоксиамино)-1-(2,2,2-трифторацетил)пиперидин-2-трет-бутилкарбоксилата в 1,4-диоксане (34 мл), и, при охлаждении льдом, 2,5M NaOH (13,7 мл) добавляли по каплям с последующей реакцией при той же самой температуре в течение 0,5 часов. К реакционной смеси добавляли уксусную кислоту (приблизительно 1 мл), с последующей концентрацией при пониженном давлении, и затем полученный сконцентрированный остаток экстрагировали этилацетатом (58 мл, 29 мл). После того, как органические слои были вымыты соответственно 50%-ым водным раствором карбоната калия, их объединяли, затем высушивали безводным сульфатом натрия, и растворитель отгоняли при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=4/1→0/1→этилацетат/метанол=19/1), получая 4,74 г целевого соединения в форме бесцветного масла (выход 90%). Энантиомерный избыток: 98,9% энантиомерного избытка (CHIRALPAK AD-H, 4,6×150 мм, УФ 210 нм, диэтиламин/гексан/этанол=0,1/80/20, объемная скорость потока 1 мл/мин, время удерживания 5,5 мин).
[α]20 D-2,8°(c 0,73, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,28 (м, 1H), 1,42-1,46 (м, 10H), 1,92 (м, 1H), 2,04 (ддд, J=12,9, 7,3, 4,0 Гц, 1H), 2,43 (дд, J=12,0, 9,8 Гц, 1H), 2,98 (м, 1H), 3,16 (дд, J=11,0, 3,2 Гц, 1H), 3,57 (ддд, J=12,0, 4,2, 2,0 Гц, 1H), 4,68 (с, 2H), 7,29-7,35 (м, 5H); MS m/z: 307 (M+1).
Пример 5
Последовательный синтез (2S,5R)-5-(бензилоксиамино)пиперидин-2-трет-бутилкарбоксилата (E)
Химическая формула 50
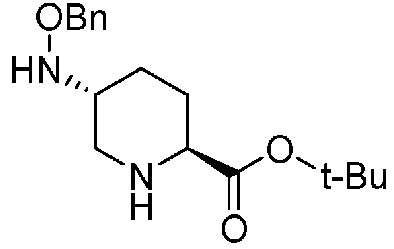 .
.
Раствор 47,9 г (161 ммоль) (2S,5S)-5-гидрокси-1-(2,2,2-трифторацетил)пиперидин-2-трет-бутилкарбоксилата в дегидратированном ацетонитриле (318 мл) охлаждали до температуры от -30 до -40°C под атмосферой аргона и добавляли 20,5 мл (177 ммоль) 2,6-диметилпиридина, и затем 28,4 мл (169 ммоль) ангидрида трифторметансульфоновой кислоты добавляли по каплям за 40 минут с последующей дальнейшей реакцией при -30°C в течение 15 минут. К этой реакционной смеси добавляли 39,7 г (322 ммоль) бензилоксиамина (промытого ацетонитрилом (11 мл)) в течение 8 минут, с последующим повышением температуры до 0°C в течение 30 минут, и добавляли еще 20,5 мл (177 ммоль) 2,6-диметилпиридина с последующей реакцией при температуре от 0 до 5°C в течение 2 дней. Эту реакционную смесь концентрировали при пониженном давлении, и полученный остаток разбавляли этилацетатом (960 мл) и промывали последовательно водой (960 мл), 10%-ым водным раствором лимонной кислоты (960 мл × 3 раза), водным 6,5% раствором гидрокарбоната натрия (480 мл) и насыщенным солевым раствором (480 мл). Каждый водный слой снова экстрагировали этилацетатом (960 мл), органические слои объединяли, и растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в растворе 1,4-диоксана (320 мл) и воды (86 мл), и, при охлаждении льдом, 2,5M, NaOH (128 мл) добавляли по каплям с последующей реакцией при той же самой температуре в течение 0,5 часов. К реакционной смеси добавляли уксусную кислоту (приблизительно 9,3 мл), с последующей концентрацией при пониженном давлении, и затем полученный сконцентрированный остаток экстрагировали этилацетатом (580 мл, 290 мл). После того, как органические слои были вымыты соответственно 50%-ым водным раствором карбоната калия (580 мл), их объединяли, и растворитель отгоняли при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=4/1→0/1→этилацетат/метанол=100/1→19/1), получая 36,58 г целевого соединения в форме бесцветного масла (выход 74%). Инструментальные данные были совместимы с таковыми из Примера 4.
Пример 6
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат (F1)
Химическая формула 51
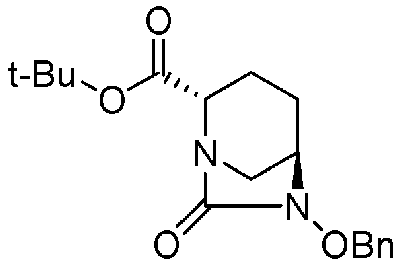 .
.
К раствору 4,14 г (13,51 ммоль) (2S,5R)-5-(бензилоксиамино)пиперидин-2-трет-бутилкарбоксилата в дегидратированном ацетонитриле (615 мл) под атмосферой аргона при 0°C добавляли триэтиламин 4,9 мл (35,16 ммоль), и затем 1,18 мл (9,78 ммоль) дифосгена добавляли по каплям в течение 5 минут с последующим перемешиванием при той же самой температуре в течение 10 минут. К этому раствору добавляли 182 мг (1,623 ммоль) 4-диметиламинопиридина, и температуру повышали до комнатной температуры с последующей реакцией в течение 3 часов. После концентрации реакционной смеси при пониженном давлении до объема одной десятой, полученный концентрированный раствор разбавляли этилацетатом, промывали последовательно водой, 5%-ым водным раствором лимонной кислоты, водным 6,5% раствором гидрокарбоната натрия и насыщенным солевым раствором и высушивали над безводным сульфатом магния, и растворитель отгоняли при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=2/1), получая 3,09 г целевого соединения (выход 69%). Полученное твердое вещество перекристаллизовывали из смеси этилацетат-гексан, и полученный осадок отфильтровывали. Влажный кристалл промывали гексаном и затем высушивали при пониженном давлении при комнатной температуре, получая целевое соединение в форме бесцветного кристаллического порошка. Энантиомерный избыток: 99,4% энантиомерного избытка (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=2/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 8,0 мин).
Т. пл. 83°C; [α]20 D+5,9°(c 0,61, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,48 (с, 9H), 1,62 (м, 1H), 2,00-2,10 (м, 3H), 2,98 (д, J=11,7 Гц, 1H), 3,03 (м, 1H), 3,30 (м, 1H), 4,01 (м, 1H), 4,90 (д, J=11,5 Гц, 1H), 5,06 (д, J=11,5 Гц, 1H), 7,35-7,42 (м, 5H); MS m/z: 333 (M+1).
На диаграмме порошковой дифракции рентгеновских лучей,кристалл целевого соединения демонстрировал характерные пиковые структуры, как показано в следующей таблице 8. Для измерения RINT 2100 от Rigaku Corporation использовался как устройство для порошковой дифракции рентгеновских лучей, в котором измерение проводили с CuKα1 в качестве рентгеновского источника, напряжение трубки 40 кВ, ток электронной лампы 40 мА, скорость сканирования 4°/мин и диапазон сканирования 2θ = от 3 до 40°.
Пример 7
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат (F1)
Реакция с газообразным фосгеном
К раствору 3,0 г (9,791 ммоль) (2S,5R)-5-(бензилоксиамино)пиперидин-2-трет-бутилкарбоксилата в дегидратированном ацетонитриле (150 мл) под атмосферой аргона при комнатной температуре добавляли 3,82 мл (27,4 ммоль) триэтиламина и 120 мг (0,979 ммоль) 4-диметиламинопиридина, и газообразный фосген (полученный добавлением 1,548 г (7,83 ммоль) дифосгена по каплям на активированном угле (1 г) при 60°C в течение 1,5 часов) вводили посредством потока аргона с последующим перемешиванием в течение ночи. Избыток фосгена расщепляли концентрированным водным раствором аммиака (0,6 мл), и растворитель реакционной смеси концентрировали при пониженном давлении. Остаток разбавляли этилацетатом (50 мл), промывали последовательно водой (50 мл), 5%-ым водным раствором лимонной кислоты (50 мл), водным 6,5% раствором гидрокарбоната натрия (25 мл) и насыщенным солевым раствором (25 мл) и высушивали над безводным сульфатом магния, и растворитель отгоняли при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=2/1), получая 2,25 г целевого соединения (выход 69%). Полученное твердое вещество перекристаллизовывали из смеси этилацетат-гексан, и полученный осадок отфильтровывали. Влажный кристалл промывали гексаном и затем высушивали при пониженном давлении при комнатной температуре, получая целевое соединение в форме бесцветного кристаллического порошка. Инструментальные данные были совместимы с таковыми для целевого соединения Примера 6.
Пример 8
Соль циклогексиламина (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (F1-1a)
Химическая формула 52
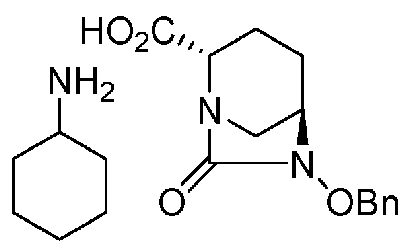 .
.
К раствору 270 мг (0,842 ммоль) (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилата в дихлорметане (2 мл) под атмосферой аргона при 0°C, добавляли трифторуксусную кислоту (2 мл), и температуру повышали до комнатной температуры с последующей реакцией в течение 4 часов. Реакционную смесь концентрировали, и полученный остаток разбавляли этилацетатом, промывали последовательно водой и насыщенным солевым раствором и высушивали над безводным сульфатом магния, и растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в этилацетате (2,5 мл), и раствор 149 мг циклогексиламина в простом диэтиловом эфире добавляли при комнатной температуре с последующим перемешиванием при 0°C в течение 1 часа. Полученный осадок отфильтровывали, и осадок промывали простым диэтиловым эфиром и затем высушивали при пониженном давлении при комнатной температуре, получая 270 мг целевого соединения в форме бесцветного кристаллического порошка (выход 86%).
Т. пл. 175°C; [α]20 D-36,8°(c 0,50, H2O); 1H ЯМР (400 МГц, DMSO-d6, δ): 1,00-1,30 (м, 5H), 1,53-1,95 (м, 8H), 2,04-2,09 (м, 1H), 2,76 (д, J=11,6 Гц, 1H), 2,80-2,93 (м, 1H), 3,19 (д, J=11,2 Гц, 1H), 3,33 (шир.с, 2H), 3,40 (д, J=7,2 Гц, 1H), 3,51 (шир.с, 1H), 4,87 (д, J=11,6 Гц, 1H), 4,93 (д, J=11,6 Гц, 1H), 7,30-7,45 (м, 5H), 8,04 (шир.с, 1H); MS m/z: 100, 277 (M+1).
На диаграмме порошковой дифракции рентгеновских лучей кристалл целевого соединения демонстрировал характерные пиковые структуры, как показано в следующей таблице 9. Для измерения RINT 2100 от Rigaku Corporation использовался как устройство для порошковой дифракции рентгеновских лучей, в котором измерение проводили с CuKα1 в качестве рентгеновского источника, напряжение трубки 40 кВ, ток электронной лампы 40 мА, скорость сканирования 4°/мин и диапазон сканирования 2θ = от 3 до 40°.
Пример 9
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота (F1-2)
Химическая формула 53
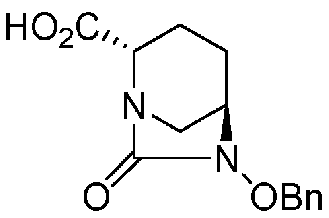 .
.
230 мг соли циклогексиламина (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в насыщенном водном растворе дигидрофосфата натрия с последующей экстракцией 4 раза этилацетатом, и объединенный органический слой промывали насыщенным солевым раствором и затем высушивали над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении и высушивали под вакуумом, получая 161 мг целевого соединения в форме бесцветного пенистого твердого вещества (выход 87%). Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, трифторуксусная кислота/гексан/этанол=0,1/80/20, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 10,5 мин).
[α]20 D+11,5°(c 0,56, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,67 (м, 1H), 2,04-2,26 (м, 3H), 2,85 (д, J=12,0 Гц, 1H), 3,13 (м, 1H), 3,35 (м, 1H), 4,12 (м, 1H), 4,91 (д, J=11,3 Гц, 1H), 5,06 (д, J=11,3 Гц, 1H), 7,37-7,44 (м, 5H); MS m/z: 277 (M+1).
Пример 10
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота (F1-2), обработка разбавленной соляной кислотой с последующей кристаллизацией
3,75 г (10,0 ммоль) соли циклогексиламина (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в 50 мл воды и добавляли 100 мл этилацетата и 20 мл 1н. соляной кислоты. Смесь перемешивали с последующей экстракцией этилацетатом (100 мл каждый раз) 3 раза. Органический слой высушивали безводным сульфатом магния, и растворитель концентрировали до 10 мл при пониженном давлении. 120 мл гексана постепенно добавляли при перемешивании при охлаждении льдом, и полученный осадок отфильтровывали. Сырой кристалл промывали гексаном и высушивали при комнатной температуре при пониженном давлении, получая 2,44 г (8,83 ммоль) целевого соединения в форме бесцветного кристаллического порошка.
Т. пл. 116°C; другие инструментальные данные были совместимы с таковыми для целевого соединения Примера 9.
На диаграмме порошковой дифракции рентгеновских лучей кристалл целевого соединения демонстрировал характерные пиковые структуры, как показано в следующей таблице 10. Для измерения RINT 2100 от Rigaku Corporation использовался как устройство для порошковой дифракции рентгеновских лучей, в котором измерение проводили с CuKα1 в качестве рентгеновского источника, напряжение трубки 40 кВ, ток электронной лампы 40 мА, скорость сканирования 4°/мин и диапазон сканирования 2θ = от 3 до 40°.
Пример 11
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота (F1-2), синтез из (F1-3a)
Химическая формула 54]
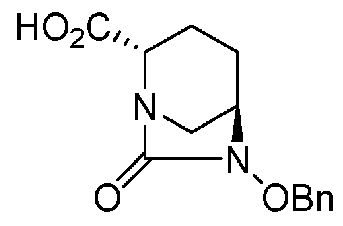 .
.
К раствору 100 мг (0,345 ммоль) (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилата в тетрагидрофуране (3 мл) добавляли воду (3 мл) с последующим охлаждением до 0°C и добавляли 15,2 мг (0,362 ммоль) моногидрата гидроксида лития с последующим перемешиванием при той же самой температуре в течение 15 минут. Реакционную смесь промывали этилацетатом, и водный слой подкисляли насыщенным водным раствором дигидрофосфата натрия с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором, высушивали над безводным сульфатом магния, и затем растворитель концентрировали при пониженном давлении, получая 93,1 мг целевого соединения в форме бесцветного пенистого твердого вещества (выход 98%). Инструментальные данные были совместимы с таковыми из Примера 9.
Пример 12
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота (F1-2), синтез из (F1-3b)
Химическая формула 55
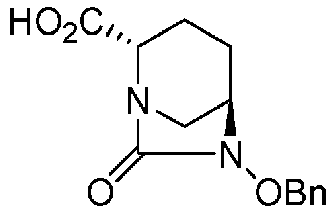 .
.
К раствору 100 мг (0,316 ммоль) (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилата в дихлорметане (2 мл) добавляли раствор 0,5M 2-этилгексаноата натрия в этилацетате (1 мл) и 12 мг тетракис(трифенилфосфин)палладия(0) с последующим перемешиванием при комнатной температуре в течение 1 часа. Реакционную смесь разбавляли этилацетатом с последующим жидкостным разделением насыщенным водным раствором дигидрофосфата натрия, водный слой экстрагировали дважды этилацетатом, и объединенный органический слой высушивали над безводным сульфатом натрия. Остаток, полученный после концентрации растворителя при пониженном давлении, растворяли в этилацетате с последующим добавлением циклогексиламина (33 мг), и осажденное твердое вещество отфильтровывали и промывали простым эфиром. Полученное твердое вещество растворяли в насыщенном водном растворе дигидрофосфата натрия с последующей экстракцией этилацетатом, и органический слой промывали насыщенным солевым раствором и высушивали над безводным сульфатом магния. Затем растворитель концентрировали при пониженном давлении, получая 68 мг целевого соединения в форме бесцветного пенистого твердого вещества (выход 75%). Инструментальные данные были совместимы с таковыми для соединения из Примера 9.
Пример 13
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилат (F1-3a)
Химическая формула 56
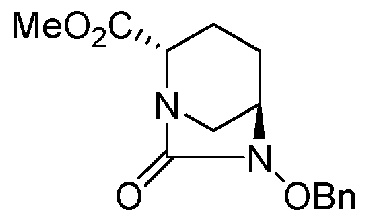 .
.
66 мг (0,239 ммоль) (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в толуоле (0,6 мл) и метаноле (0,6 мл) и, при охлаждении льдом, добавляли 0,54 мл (0,324 ммоль) 0,6 М раствора триметилсилилдиазометан-гексана с последующим перемешиванием в течение 20 минут. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=2/1), получая 21,5 мг целевого соединения в форме бесцветного твердого вещества (выход 31%). Полученное твердое вещество перекристаллизовывали из смеси этилацетат-гексан, полученный осадок отфильтровывали, влажный кристалл промывали гексаном и затем высушивали при пониженном давлении при комнатной температуре, получая целевое соединение в форме бесцветного кристаллического порошка. Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=2/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 12,8 мин).
Т. пл. 86°C; [α]20 D+5,3°(c 1,10, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,65-1,70 (м, 1H), 2,03-2,12 (м, 3H), 2,90 (д, J=12,0 Гц, 1H), 3,07 (м, 1H), 3,79 (с, 3H), 4,12 (дд, J=4,6, 4,4 Гц, 1H), 4,91 (д, J=11,2 Гц, 1H), 5,06 (д, J=11,2 Гц, 1H), 7,35-7,44 (м, 5H); MS m/z: 291 (M+1).
На диаграмме порошковой дифракции рентгеновских лучей кристалл целевого соединения демонстрировал характерные пиковые структуры, как показано в следующей таблице 11. Для измерения RINT 2100 от Rigaku Corporation использовался как устройство для порошковой дифракции рентгеновских лучей, в котором измерение проводили с CuKα1 в качестве рентгеновского источника, напряжение трубки 40 кВ, ток электронной лампы 40 мА, скорость сканирования 4°/мин и диапазон сканирования 2θ = от 3 до 40°.
Пример 14
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилат (F1-3b)
Химическая формула 57
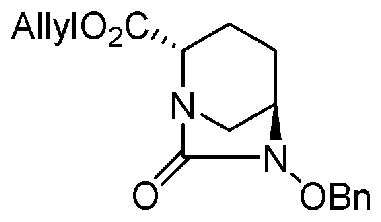 .
.
46 мг (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в N,N-диметилформамиде (0,5 мл) и добавляли 21 мг гидрокарбоната натрия и 30 мкл аллилбромида с последующим перемешиванием в течение 6,5 часов. К реакционному раствору добавляли этилацетат с последующими последовательными промывками водой и насыщенным солевым раствором и высушиванием над безводным сульфатом натрия, и растворитель концентрировали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=3/1), получая 7,5 мг целевого соединения в форме бесцветного твердого вещества (выход 14%). Полученное твердое вещество перекристаллизовывали из смеси этилацетат-гексан, полученный осадок отфильтровывали, влажный кристалл промывали гексаном и затем высушивали при пониженном давлении при комнатной температуре, получая целевое соединение в форме бесцветного кристаллического порошка. Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=2/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 8,0 минут).
Т. пл. 60-62°C; [α]20 D+4,0°(c 1,05, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,69 (м, 1H), 2,02-2,15 (м, 3H), 2,93 (д, J=12,0 Гц, 1H), 3,07 (м, 1H), 3,31 (м, 1H), 4,14 (дд, J=6,5, 2,6 Гц, 1H), 4,67 (ддд, J=5,9, 1,5, 1,2 Гц, 1H), 4,91 (д, J=11,5 Гц, 1H), 5,06 (д, J=11,5 Гц, 1H), 5,26 (м, 1H), 5,34 (м, 1H), 5,92 (м, 1H), 7,36-7,42 (м, 5H); MS m/z: 317 (M+1).
На диаграмме порошковой дифракции рентгеновских лучей кристалл целевого соединения демонстрировал характерные пиковые структуры, как показано в следующей таблице 12. Для измерения RINT 2100 от Rigaku Corporation использовался как устройство для порошковой дифракции рентгеновских лучей, в котором измерение проводили с CuKα1 в качестве рентгеновского источника, напряжение трубки 40 кВ, ток электронной лампы 40 мА, скорость сканирования 4°/мин и диапазон сканирования 2θ = от 3 до 40°.
Пример 15
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилат (F1-3c)
Химическая формула 58
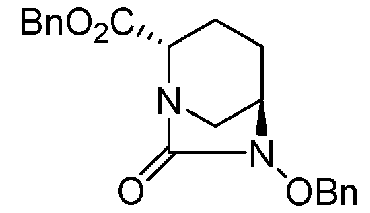 .
.
94 мг (0,346 ммоль) (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в дихлорметане (3,4 мл) и добавляли 70 мкл (0,676 ммоль) бензилового спирта и 98 мг (0,511 ммоль) 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида с последующим перемешиванием при комнатной температуре в течение 24 часов. Реакционную смесь концентрировали при пониженном давлении, и затем остаток разбавляли этилацетатом и промывали водой и затем насыщенным солевым раствором. Органический слой высушивали над безводным сульфатом магния, и растворитель концентрировали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=3/1), получая 41,2 мг целевого соединения (выход 33%). Энантиомерный избыток: 99,8% энантиомерного избытка (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=2/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 33,2 мин).
[α]20 D+3,3°(c 0,82, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,58-1,65 (м, 1H), 2,01-2,12 (м, 3H), 2,86 (д, J=12,0 Гц, 1H), 3,03 (м, 1H), 3,28 (м, 1H), 4,15 (м, 1H), 4,89 (д, J=11,5 Гц, 1H), 5,05 (д, J=11,5 Гц, 1H), 5,22 (с, 2H), 7,26-7,43 (м, 10H); MS m/z: 367 (M+1).
Пример 16
(2S,5R)-2,5-диоксопирролидин-1-ил-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат (F1-3d)
Химическая формула 59
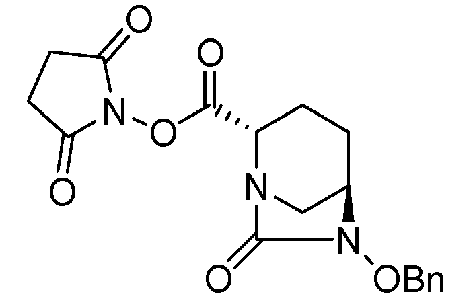 .
.
201 мг (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в дегидратированном дихлорметане (3,6 мл) и добавляли 162 мг N-метилморфолина с последующим охлаждением до 0°C. К смеси добавляли 198,8 мг изобутилхлорформиата с последующим перемешиванием в течение 10 минут и затем добавляли 167 мг N-гидроксисукцинимида с последующим дальнейшим перемешиванием в течение 0,5 часов. Реакционную смесь промывали водой и высушивали над безводным сульфатом магния, и растворитель концентрировали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=1/2), получая 161 мг целевого соединения в форме бесцветного твердого вещества (выход 59%).
[α]20 D+4,76°(c 0,88, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,74 (м, 1H), 2,08 (м, 1H), 2,16-2,29 (м, 2H), 2,85 (м, 4H), 3,11-3,18 (м, 2H), 3,34 (с, 1H), 4,48 (д, J=6,4 Гц, 1H), 4,92 (д, J=11,2 Гц, 1H), 5,06 (д, J=11,2 Гц, 1H), 7,35-7,45 (м, 5H); MS m/z: 274 (M+1).
Пример 17
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат (F2)
Химическая формула 60
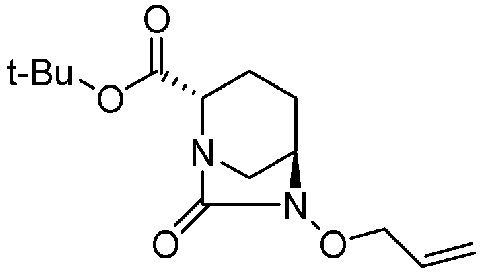 .
.
140 мг (0,421 ммоль) (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилата растворяли в этаноле (3,1 мл) и добавляли 14 мг 10%-го палладия на углероде (50%-ое содержание воды) с последующим перемешиванием при комнатной температуре в течение 1 часа в атмосфере водорода. Катализатор реакционной смеси фильтровали через целит, и остаток, полученный в результате концентрации растворителя при пониженном давлении, растворяли в ацетонитриле (4,1 мл) и добавляли 62 мг (0,449 ммоль) безводного карбоната калия и 70 мкл (0,809 ммоль) аллилбромида, с последующим перемешиванием при комнатной температуре в течение 3 часов. После концентрации реакционной смеси при пониженном давлении, остаток разбавляли этилацетатом и промывали последовательно водой, насыщенным водным раствором хлорида аммония и насыщенным солевым раствором, затем органический слой высушивали над безводным сульфатом магния, и растворитель концентрировали при пониженном давлении. Полученный остаток подвергали хроматографии на колонках с силикагелем (н-гексан/этилацетат=5/2), получая 60,8 мг целевого соединения (выход 54%). Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=2/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 4,8 мин).
[α]20 D-39,3°(c 1,11, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,50 (с, 9H), 1,70-1,80 (м, 1H), 2,04-2,12 (м, 3H), 3,08 (д, J=12,0 Гц, 1H), 3,14 (м, 1H), 3,74 (м, 1H), 4,01 (м, 1H), 4,45 (м, 2H), 5,29-5,39 (м, 2H), 5,98-6,08 (м, 1H); MS m/z: 283 (M+1).
Пример 18
Соль циклогексиламина (2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (F2-1a)
Химическая формула 61]
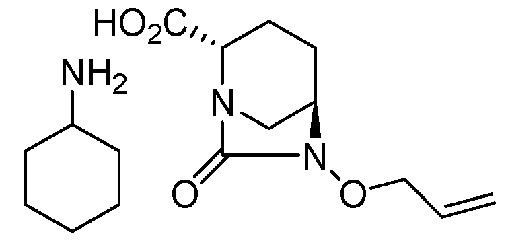 .
.
Из (2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилата согласно способу из Примера 8 целевое соединение получали в форме бесцветного твердого вещества.
[α]20 D-44,4°(c 0,25, H2O); 1H ЯМР (400 МГц, D2O, δ): 0,95-1,24 (м, 5H), 1,48-1,81 (м, 8H), 2,02 (дд, J=14,6, 7,1 Гц, 1H), 2,92 (д, J=11,7 Гц, 1H), 3,00 (м, 1H), 3,62 (д, J=7,6 Гц, 1H), 3,88 (с, 1H), 4,33-4,36 (м, 2H), 5,23-5,33 (м, 2H), 5,85-5,95 (м, 1H); MS m/z: 100, 227 (M+1).
Пример 19
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота (F2-2)
Химическая формула 62
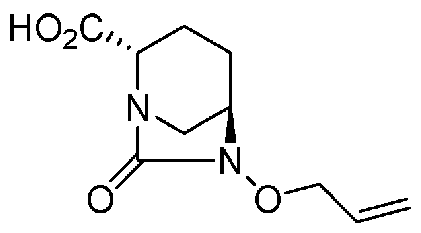 .
.
Из соли циклогексиламина (2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты согласно способу из Примера 9 получали целевое соединение. Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, трифторуксусная кислота/гексан/этанол=0,1/80/20, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 5,5 мин).
[α]20 D-32,3°(c 1,59, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,60-1,81 (м, 1H), 2,01-2,13 (м, 2H), 2,25-2,31 (м, 1H), 3,07 (д, J=11,7 Гц, 1H), 3,33 (шир. д, J=11,2 Гц, 1H), 3,86 (с, 1H), 4,19 (д, J=7,3 Гц, 1H), 4,42-4,52 (м, 2H), 5,33-5,42 (м, 2H), 5,96-6,06 (м, 1H); MS m/z: 227 (M+1).
Пример 20
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилат (F2-3c)
Химическая формула 63
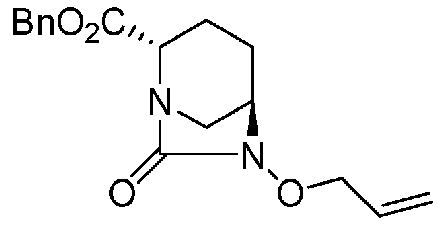 .
.
Из (2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты согласно способу из Примера 14 получали целевое соединение. Энантиомерный избыток: 98,5% энантиомерного избытка (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=2/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 15,5 мин).
[α]20 D-42,5°(c 0,252, CHCl3); 1H ЯМР (400 МГц, CDCl3, δ): 1,67-1,77 (м, 1H), 2,08-2,15 (м, 3H), 2,97 (д, J=12,0 Гц, 1H), 3,14 (м, 1H), 3,73 (м, 1H), 4,16 (м, 1H), 4,39-4,51 (м, 2H), 5,23 (м, 2H), 5,29-5,38 (м, 2H), 5,96-6,05 (м, 1H), 7,33-7,38 (м, 5H); MS m/z: 317 (M+1).
Пример 21
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (F1-4), синтез из (F1-3d)
Химическая формула 64
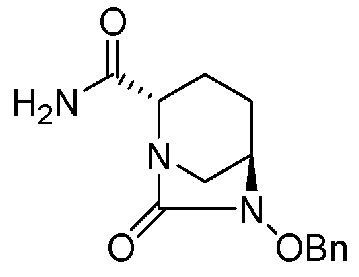 .
.
60 мг (2S,5R)-2,5-диоксопирролидин-1-ил 6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилата растворяли в дегидратированном дихлорметане (0,8 мл) с последующим охлаждением до 0°C. К реакционному раствору добавляли 0,12 мл концентрированного водного раствора аммиака с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем добавляли воду (10 мл), и органический слой разделяли на фракции с последующими последовательными промывками водой и насыщенным солевым раствором и высушиванием над безводным сульфатом магния. Остаток, полученный в результате концентрации растворителя при пониженном давлении, подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=1/3) и затем кристаллизовали смесью хлороформ/гексан=1:3, получая 30,4 мг целевого соединения в форме бесцветного кристаллического порошка.
[α]20 D-26,1° (c 0,498, MeOH); 1H ЯМР (400 МГц, CDCl3, δ): 1,60 (м, 1H), 1,90-2,03 (м, 2H), 2,36 (м, 1H), 2,76 (д, J=11,6 Гц, 1H), 3,03 (д, J=11,6 Гц, 1H), 3,31 (с, 1H), 3,95 (д, J=7,6 Гц, 1H), 4,91 (с, J=11,2 Гц, 1H), 5,06 (д, J=11,6 Гц, 1H), 5,45 (с, 1H), 6,56 (с, 1H), 7,26-7,44 (м, 5H); MS m/z: 276 (M+1).
Пример 22
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (F1-4)
Химическая формула 65
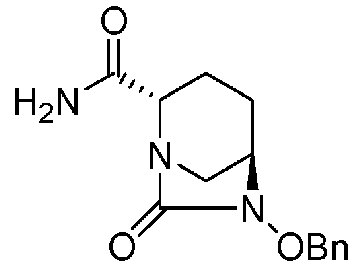 .
.
400 мг (1,44 ммоль) (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в дегидратированном дихлорметане (14,4 мл) и добавляли 176 мг триэтиламина с последующим охлаждением до 0°C. К смеси добавляли 237 мг изобутилхлорформиата с последующим перемешиванием при той же самой температуре в течение 20 минут. К реакционной смеси добавляли 1,0 мл концентрированного водного раствора аммиака с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем добавляли воду (10 мл), и органический слой разделяли на фракции с последующими последовательными промывками водой и насыщенным солевым раствором и высушиванием над безводным сульфатом магния. Остаток, полученный в результате концентрации растворителя при пониженном давлении, подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=1/3) и затем кристаллизовали смесью хлороформ/гексан=1:3, получая 315 мг целевого соединения в форме бесцветного кристаллического порошка (выход 79%). Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=4/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 16,2 мин).
Т. пл. 169°C; [α]20 D-22,0° (c 1,26, MeOH); 1H ЯМР и MS были эквивалентны таковым для целевого соединения из Примера 21.
На диаграмме порошковой дифракции рентгеновских лучей кристалл целевого соединения демонстрировал характерные пиковые структуры, как показано в следующей таблице 13. Для измерения RINT 2100 от Rigaku Corporation использовался как устройство для порошковой дифракции рентгеновских лучей, в котором измерение проводили с CuKα1 в качестве рентгеновского источника, напряжение трубки 40 кВ, ток электронной лампы 40 мА, скорость сканирования 4°/мин и диапазон сканирования 2θ = от 3 до 40°.
Пример 23
(2S,5R)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, 7-оксо-6-(сульфокси)-мононатриевая соль (H)
Стадия 1: (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (G)
Химическая формула 66
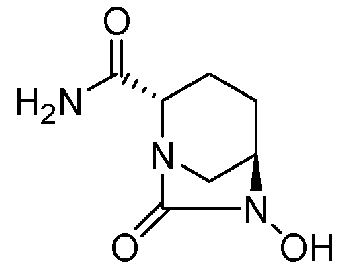 .
.
445 мг (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида растворяли в метаноле (16 мл) и добавляли 80 мг 10%-го палладия на углероде (50%-ое содержание воды) с последующим перемешиванием в течение 0,75 часов в атмосфере водорода. Катализатор реакционной смеси фильтровали через целит, и растворитель концентрировали при пониженном давлении и высушивали под вакуумом, получая 357 мг целевого соединения в форме бесцветного твердого вещества (количественный выход).
[α]20 D-66,7°(c 1,22, MeOH); 1H ЯМР (400 МГц, CD3OD, δ): 1,74 (м, 1H), 1,89 (м, 1H), 2,04 (м, 1H), 2,26 (м, 1H), 2,96 (д, J=11,6 Гц, 1H), 3,15 (м, 1H), 3,69 (с, 1H), 3,84 (д, J=8,0 Гц, 1H); MS m/z: 186 (M+1).
Стадия 2: (2S,5R)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, 7-оксо-6-(сульфокси)-мононатриевая соль (H)
Химическая формула 67
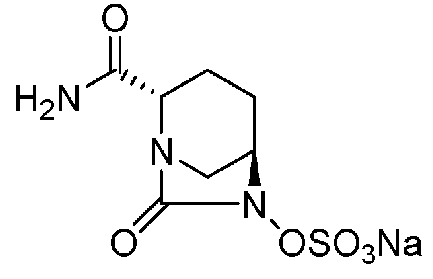 .
.
317 мг (2S,5R)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида растворяли в дегидратированном пиридине (17 мл) и добавляли 1360 мг комплекса триоксид серы·пиридин с последующим перемешиванием при комнатной температуре в течение 20 часов. Твердое вещество в реакционном растворе отфильтровывали, растворитель фильтрата концентрировали при пониженном давлении, и остаток растворяли в насыщенном водном растворе дигидрофосфата натрия (30 мл) с последующей промывкой этилацетатом (50 мл). 609 мг тетрабутиламмония гидросульфата растворяли в водной фазе с последующей экстракцией этилацетатом (100 мл × 4 раза) и высушиванием над безводным сульфатом натрия, и растворитель концентрировали при пониженном давлении. Соль тетрабутиламмония (сырой выход 86%), полученную в результате хроматографии остатка на колонках с силикагелем (дихлорметан/ацетон=50/50), растворяли в 50%-ом водном растворе ацетона, загружали на DOWEX 50W×8 (тип Na, 150 мл) и элюировали водой, и активную фракцию лиофилизировали, получая 338 мг целевого соединения в форме бесцветного твердого вещества (выход 80%). Чистота LC-MS 100%.
[α]20 D-37,1°(c 0,496, H2O); 1H ЯМР (400 МГц, D2O, δ): 1,68 (м, 1H), 1,81 (м, 1H), 1,95 (м, 1H), 2,07 (м, 1H), 3,00 (д, J=12,4 Гц, 1H), 3,22 (д, J=12,0 Гц, 1H), 3,94 (д, J=7,6 Гц, 1H), 4,08 (с, 1H); MS m/z: 264 (M-1).
Пример 24
(2R,5S)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, 7-оксо-6-(сульфокси)-мононатриевая соль (r)
Стадия 1: (2R/S,5S/R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат
Химическая формула 68
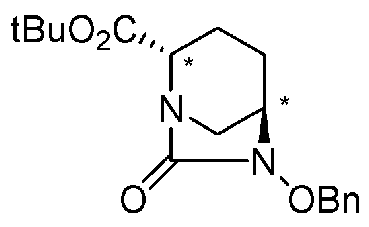 .
.
Водный раствор соляной кислоты рацемической 5-кетопиперидин-2-карбоновой кислоты, полученной способами, описанными в непатентном документе 5, непатентном документе 6 и непатентном документе 7, бензилоксикарбонилировали бензилхлорформиатом, поддерживая рН 10,5 с помощью гидроксида натрия, затем сырой продукт обрабатывали в дегидратированном дихлорметане трет-бутиловым спиртом, 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлоридом и 4-диметиламинопиридином, и сырой продукт далее восстанавливали в метаноле боргидридом натрия и очищали на колонке, получая (2S/R, 5S/R)-1-бензил-2-трет-бутил-5-гидроксипиперидин-1,2-дикарбоксилат, который использовали, получая целевое соединение в форме бесцветного твердого вещества согласно Примерам 1-6. Энантиомерный избыток: 3% энантиомерного избытка (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=2/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 4,2 мин (2R,5S), 7,9 мин (2S,5R)).
Т. пл. 100°C; 1H ЯМР и MS были эквивалентны таковым для целевого соединения Примера 6.
Стадия 2: (2R,5S)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат (p)
Химическая формула 69
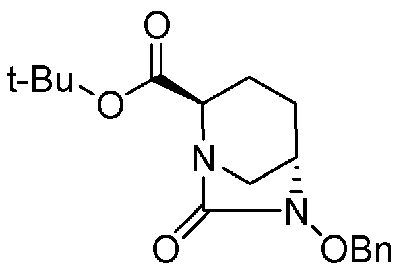 .
.
30,3 г (2R/S,5S/R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилата подвергали хиральной хроматографии на колонках (CHRAL PAK IA, метанол/ацетонитрил=95/5), и активную фракцию, соответствующую первому пику, собирали, получая 13,9 г целевого соединения в форме бесцветного твердого вещества (выход 46%). Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=2/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 4,2 мин).
Т. пл. 84°C; [α]20 D-6,1° (c 0,83, CHCl3); 1H ЯМР и MS были эквивалентны таковым для целевого соединения Примера 6.
Стадия 3: соль циклогексиламина (2R,5S)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (F1-1a)
Химическая формула 70
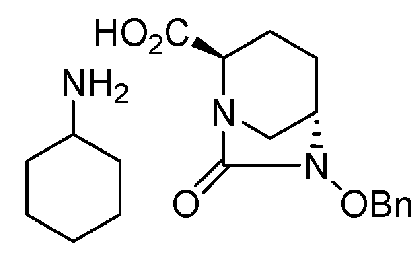 .
.
К раствору 3,34 г (10,0 ммоль) (2R,5S)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилата в дихлорметане (25 мл) в атмосфере аргона при 0°C добавляли трифторуксусную кислоту (25 мл) с последующим повышением температуры до комнатной температуры и реакцией в течение 4 часов. После концентрации реакционной смеси, полученный остаток разбавляли этилацетатом, затем промывали последовательно водой и насыщенным солевым раствором и высушивали над безводным сульфатом магния, и растворитель отгоняли при пониженном давлении. Полученный остаток растворяли в этилацетате (10 мл), и раствор 256 мг циклогексиламина в простом диэтиловом эфире добавляли при комнатной температуре с последующим выдерживанием при 0°C в течение 1 часа. Полученный осадок отфильтровывали, и осадок промывали простым диэтиловым эфиром, затем высушивали при пониженном давлении, получая 3,36 г целевого соединения в форме бесцветного кристаллического порошка (выход 89%).
[α]20 D+35,7° (c 0,51, H2O); 1H ЯМР и MS были эквивалентны таковым для целевого соединения из Примера 8.
Стадия 4: (2R,5S)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота (q)
Химическая формула 71
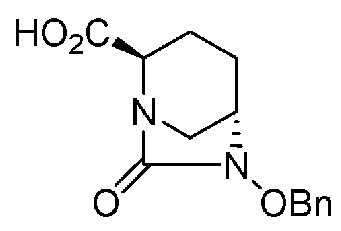 .
.
Соль циклогексиламина 750 мг (2R,5S)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в насыщенном водном растворе дигидрофосфата натрия с последующей экстракцией три раза этилацетатом, и объединенный органический слой промывали насыщенным солевым раствором и затем высушивали над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении, получая 507 мг целевого соединения в форме бесцветного масла (выход 91,5%). Энантиомерный избыток: 98,6% энантиомерного избытка (CHIRALPAK AD-H, 4,6×150 мм, трифторуксусная кислота/гексан/этанол=0,1/80/20, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 6,2 мин).
[α]20 D-11,1° (c 0,90, CHCl3); 1H ЯМР и MS были эквивалентны таковым для целевого соединения из Примера 9.
Стадия 5: (2R,5S)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид
Химическая формула 72
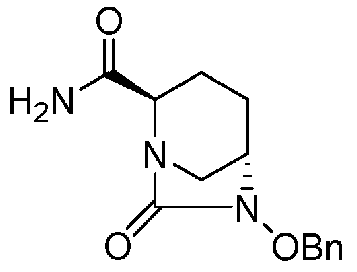 .
.
230 мг (0,84 ммоль) (2R,5S)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты растворяли в дегидратированном дихлорметане (4,2 мл) и добавляли 110 мг триэтиламина с последующим охлаждением до 0°C. К смеси добавляли 137 мг изобутилхлорформиата с последующим перемешиванием при той же самой температуре в течение 20 минут. К реакционной смеси добавляли 0,6 мл водного раствора аммиака с последующим перемешиванием при комнатной температуре в течение 1 часа. Затем добавляли воду (10 мл), и органический слой разделяли на аликвоты с последующими последовательными промывками водой и насыщенным солевым раствором и высушиванием над безводным сульфатом магния. Остаток, полученный в результате концентрации растворителя при пониженном давлении, подвергали хроматографии на колонках с силикагелем (гексан/этилацетат=1/3) и затем кристаллизовали смесью хлороформ/гексан=1:3, получая 202 мг целевого соединения в форме бесцветного кристаллического порошка (выход 87%). Энантиомерный избыток: 99,9% энантиомерного избытка или больше (CHIRALPAK AD-H, 4,6×150 мм, гексан/этанол=4/1, УФ 210 нм, объемная скорость потока 1 мл/мин, время удерживания 10,3 мин).
[α]20 D+24,5° (c 0,61, MeOH); 1H ЯМР и MS были эквивалентны таковым для целевого соединения из Примера 22.
Стадия 6: (2R,5S)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид (r)
Химическая формула 73
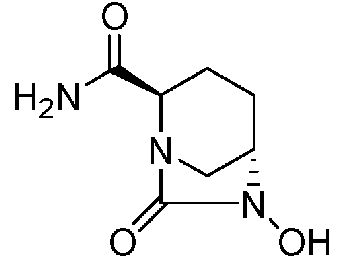 .
.
190 мг (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида растворяли в метаноле (6,9 мл) и добавляли 40 мг 10%-го палладия на углероде (50%-ое содержание воды) с последующим перемешиванием в течение 1,5 часов в атмосфере водорода. Катализатор реакционной смеси фильтровали через целит, и растворитель концентрировали при пониженном давлении и высушивали под вакуумом, получая 126 мг целевого соединения в форме бесцветного твердого вещества (количественный выход).
[α]20 D-55,7° (c 0,52, MeOH); 1H ЯМР и MS были эквивалентны таковым для целевого соединения из Примера 23, Стадия 1.
Стадия 7: (2R,5S)-1,6-диазабицикло[3.2.1]октан-2-карбоксамид, 7-оксо-6-(сульфокси)-мононатриевая соль (s)
Химическая формула 74
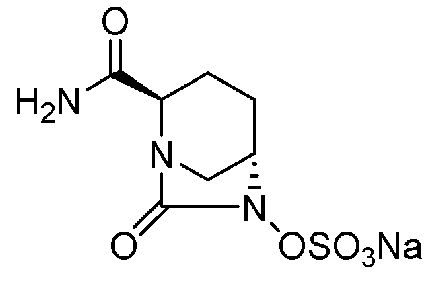 .
.
112 мг (2R,5S)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида растворяли в дегидратированном пиридине (6 мл) и добавляли 481 мг комплекса триоксид серы·пиридин с последующим перемешиванием при комнатной температуре в течение 20 часов. Твердое вещество реакционного раствора отфильтровывали, растворитель фильтрата концентрировали при пониженном давлении, и остаток растворяли в насыщенном водном растворе дигидрофосфата натрия (30 мл) и промывали этилацетатом (50 мл). 190 мг тетрабутиламмония гидросульфата растворяли в водной фазе с последующим перемешиванием в течение 10 минут. Реакционный раствор экстрагировали этилацетатом (100 мл × 5 раз) и высушивали над безводным сульфатом натрия, и затем растворитель концентрировали при пониженном давлении. Соль тетрабутиламмония (сырой выход 85%), полученную хроматографией остатка на колонках с силикагелем (дихлорметан/ацетон=50/50), растворяли в 50%-ом водном растворе ацетона, загружали на DOWEX5WX8 (тип Na, 61 мл) и элюировали водой, и активную фракцию лиофилизировали, получая 109 мг целевого соединения в форме бесцветного твердого вещества (выход 63%). Чистота LC-MS 100%.
[α]20 D+38,1° (c 0,496, H2O); 1H ЯМР и MS были эквивалентны таковым для целевого соединения из Примера 22, Стадия 2.
Пример 25
Определяли ингибирующую активность соединений, полученных в Примерах 23 и 24, в отношении фермента β-лактамазы и антибактериальную активность PIPC в комбинации с этими соединениями. Структурные формулы тестируемого соединения показаны в следующей таблице 14.
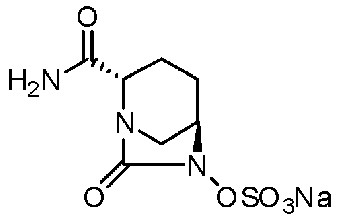
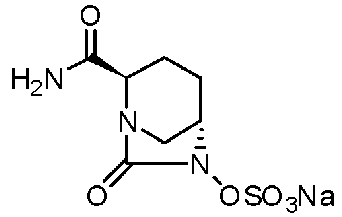
Ингибирующая активность фермента β-лактамазы
Используя геном P. aeruginosa PAO1 в качестве шаблона, ДНК для кодирования домена AmpC β-лактамазы, исключая сигнальный пептид, амплифицировали с помощью ПЦР. Это продукт ПЦР встраивали в вектор рЕТ-28b(+) (Merck), вводили в E. coli BL21 (Merck) и, под индукцией 1 мМ изопропил-β-D-(-)-тиогалактопиранозида (Nacalai Tesque), культивировали в течение ночи при 20°C, чтобы экспрессировать AmpC. После сбора бактериальных клеток, AmpC очищали от клеточного экстракта, полученного ультразвуковой обработкой с использованием CM Sepharose Fast Flow (GE Healthcare) и HiTrap Heparin HP (GE Healthcare) при 4°C.
Для измерения ингибирующей активности в отношении β-лактамазы 100 мМ (конечная концентрация) нитроцефина (Oxoid) использовали как субстрат, и 2,5% ДМСО, 10 мкг/мл бычьего сывороточного альбумина (Sigma-Aldrich) и 50 мМ фосфатного буфера при pH 7,0 использовали как реакционный раствор. В каждую лунку планшета с 96 лунками добавляли тестируемые соединения (соединения, показанные в таблице 14) и AmpC (конечная концентрация 0,5 нМ) с последующей преинкубацией при 30°C в течение 10 минут. К каждой лунке добавляли нитроцефин для смешивания с последующей инкубацией при 30°C в течение 20 минут, и Multiskan Ascent (Thermo Fisher Scientific) использовали для измерения при длины волны 492 нм, таким образом измеряя гидролитическую активность AmpC в отношении нитроцефина, чтобы определить ингибирующую активность в отношении фермента. Как контроль, был получен реакционный раствор, не включающий AmpC, и концентрацию тестируемого соединения, показывающую 50%-ое ингибирование, принимали в качестве значения IC50. Результаты показаны в таблице 15.
Комбинаторный эффект
Комбинаторный эффект тестируемого соединения β-лактамным агентом против бактерий оценивали, используя штамм, конститутивно экспрессирующий AmpC, выбранный из P. aeruginosa PAO1 через экспонирование к этому агенту. Используя пиперациллин (PIPC, Sigma-Aldrich) в качестве β-лактамного агента, измерение проводили процессом разбавления агаровой пластинки, в котором минимальная ингибирующая концентрация (MIC) PIPC основана на Clinical and Laboratory Standards Institute (способ CLSI). Таким образом, получали агаровый планшет, содержащий 4 мкг/мл (конечная концентрация) тестируемого соединения и PIPC в каждой концентрации в агар-агаре Mueller-Hinton (Becton, Dickinson and Company), и бактерии, культивируемые в течение ночи в отрегулированном в отношении катионов бульоне Muller-Hinton (Becton, Dickinson and Company), разбавляли в той же самой среде до концентрации 104 КОЕ/пятно и инокулировали на планшет, содержащий агент. Этот планшет, содержащий агент, культивировали в течение ночи при 35°C, и минимальную концентрацию агента, при которой не наблюдается рост бактерий, принимали за MIC. Результаты показаны в таблице 16.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ ИНГИБИТОР бета-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2013 |
|
RU2693898C2 |
| НОВЫЙ ИНГИБИТОР β-ЛАКТАМАЗЫ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2800050C2 |
| ПРОСТОЙ СПОСОБ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2711358C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2014 |
|
RU2719480C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2838718C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2023 |
|
RU2840014C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722932C1 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |
Изобретение относится к оптически активному диазабициклооктановому производному формулы (F), где R1 обозначает CO2R, CO2M или CONH2, причем R обозначает метильную группу, трет-бутильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу, и М обозначает атом водорода, неорганический катион, выбранный из лития, калия, натрия, или органический катион, причем органическим катионом является соль аммония, образованная из амина, выбранного из циклогексиламина; и R2 обозначает бензильную группу или аллильную группу. Также изобретение относится к способу получения соединения формулы (F) и промежуточных соединений, к промежуточным соединениям, кристаллическим формам соединения формулы (F), а также применению соединения формулы (F) в качестве фармацевтического промежуточного соединения для получения ингибитора β-лактамазы, и к способу его получения. Технический результат: получены новые производные (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, а также их кристаллические формы, полезные в синтезе ингибитора β-лактамазы. 26 н. и 1 з.п. ф-лы, 16 табл., 25 пр.
1. Оптически активное производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, определяемое следующей формулой (F):
Химическая формула 75
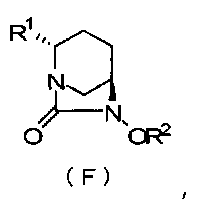
в которой:
R1 обозначает CO2R, CO2M или CONH2,
причем R обозначает метильную группу, трет-бутильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу, и
М обозначает атом водорода, неорганический катион, выбранный из лития, калия, натрия, или органический катион, причем органическим катионом является соль аммония, образованная из амина, выбранного из циклогексиламина; и
R2 обозначает бензильную группу или аллильную группу.
2. Способ получения соединения, представленного формулой (F1), приведенной ниже, который включает
подвергание соединения, представленного формулой (Е), приведенной ниже, формированию внутримолекулярного карбамида с эквивалентом фосгена в присутствии основания с получением соединения, представленного ниже формулой (F1):
Химическая формула 76
,
в которой Вn обозначает бензильную группу и tBu обозначает трет-бутильную группу,
Химическая формула 77
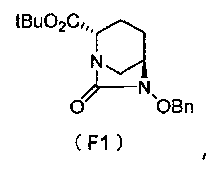
в которой Bn обозначает бензильную группу и tBu обозначает трет-бутильную группу.
3. Способ получения соединения, представленного формулой (F1-1a) или (F2-1a) приведенной ниже, который включает
расщепление трет-бутилового эфира соединения, представленного формулой (F1) или (F2), приведенной ниже, в положении 2 с использованием кислоты или соли металла; и последующее
добавление циклогексиламино с получением соединения, представленного формулой (F1-1a) или (F2-1a), приведенной ниже:
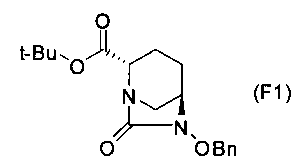
в которой t-Bu обозначает трет-бутильную группу и OBn обозначает бензилоксигруппу,
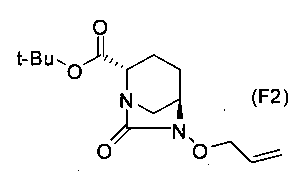
в которой t-Bu обозначает трет-бутильную группу,
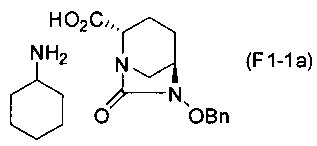
в которой OBn обозначает бензилоксигруппу,
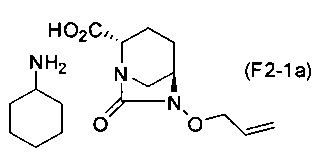 .
.
4. Способ получения соединения, представленного формулой (F1-2) или (F2-2), приведенной ниже, который включает
обработку соединения, представленного формулой (F1-1a) или (F2-1a), приведенной ниже, кислотой с получением соединения, представленного формулой (F1-2) или (F2-2), приведенной ниже:
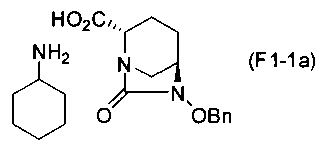
в которой OBn обозначает бензилоксигруппу,
в которой OBn обозначает бензилоксигруппу,
5. Способ получения соединения, представленного формулой (F1-4), приведенной ниже, который включает
реакцию соединения, представленного формулой (F1-2), приведенной ниже, с концентрированным водным раствором гидроксида аммония в присутствии основания и активирующего агента для карбоновой кислоты с получением соединения, представленного формулой (F1-4), приведенной ниже:
в которой OBn обозначает бензилоксигруппу,
в которой OBn обозначает бензилоксигруппу.
6. Способ получения соединения, представленного формулой (F1-3) или (F2-3), приведенной ниже, который включает
подвергание соединения, представленного формулой (F1-2) или (F2-2), приведенной ниже, этерификации карбоновой кислоты в положении 2 путем реакции этого соединения с метилгалогенидом, аллилгалогенидом или бензилгалогенидом в подходящем растворителе в присутствии алкилирующего агента и основания; или путем реакции этого соединения с активирующим агентом для карбоновой кислоты или дегидрирующим конденсирующим агентом и спиртом, выбранным из метанола, аллилового спирта, бензилового спирта и 2,5-диоксопирролидин-1-ола в присутствии основания с получением соединения, представленного формулой (F1-3) или (F2-3), приведенной ниже:
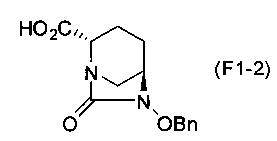
в которой OBn обозначает бензилоксигруппу,
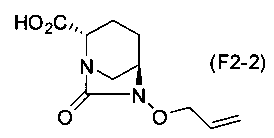
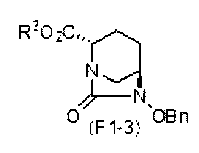
в которой R3 обозначает метильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу и OBn обозначает бензилоксигруппу,
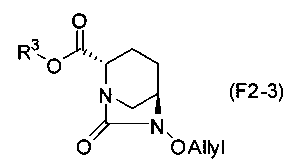
в которой R3 обозначает метильную группу, аллильную группу, бензильную группу или 2,5-диоксопирролидин-1-ильную группу и OAllyl обозначает аллилоксигруппу.
7. Способ получения соединения, представленного формулой (F2), приведенной ниже, который включает
удаление бензильной группы из бензилоксигруппы в положении 6 соединения, представленного формулой (F1), приведенной ниже, путем реакции каталитического гидрирования, с последующим
превращением группы в положении 6 полученного соединения с использованием аллилирующего агента в присутствии основания в аллилокси с получением, таким образом, соединения, представленного формулой (F2), приведенной ниже:
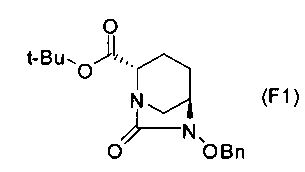
в которой t-Bu обозначает трет-бутильную группу и OBn обозначает бензилоксигруппу,
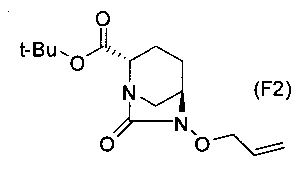
в которой t-Bu обозначает трет-бутильную группу.
8. Способ получения соединения, представленного формулой (Е), приведенной ниже
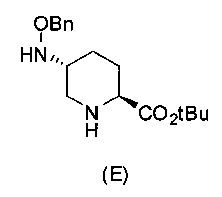
в которой Bn обозначает бензильную группу и t-Bu обозначает трет-бутильную группу,
который включает подвергание соединения, представленного формулой (В), приведенной ниже, трифторацетилированию и введение полученного соединения, представленного формулой (С), приведенной ниже, в реакцию с бензилоксиамином в присутствии активатора гидроксильной группы и подвергание полученного соединения, представленного формулой (D), приведенной ниже, детрифторацетилированию:
Химическая формула 78
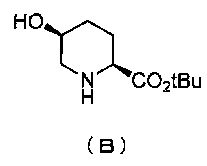 ,
,
в которой tBu обозначает трет-бутильную группу,
Химическая формула 79
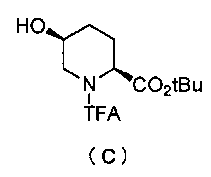 ,
,
в которой tBu обозначает трет-бутильную группу и TFA обозначает трифторацетильную группу,
Химическая формула 80
,
в которой Вn обозначает бензильную группу, tBu обозначает трет-бутильную группу и TFA обозначает трифторацетильную группу.
9. Соединение, представленное следующей формулой (В):
Химическая формула 81
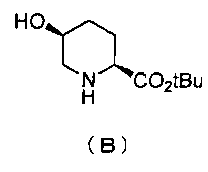 ,
,
в которой tBu обозначает трет-бутильную группу.
10. Соединение, представленное следующей формулой (С):
Химическая формула 82
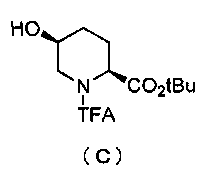 ,
,
в которой tBu обозначает трет-бутильную группу и TFA обозначает трифторацетильную группу.
11. Соединение, представленное следующей формулой (D):
Химическая формула 83
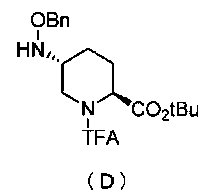 ,
,
в которой Bn обозначает бензильную группу, tBu обозначает трет-бутильную группу и TFA обозначает трифторацетильную группу.
12. Соединение, представленное формулой (Е):
Химическая формула 84
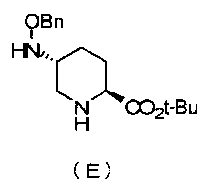 ,
,
в которой Bn обозначает бензильную группу и t-Bu обозначает трет-бутильную группу.
13. Соединение по п.1, которое является любым из следующих соединений:
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота соль циклогексиламина,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамид,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилат,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилат,
(2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилат,
(2S,5R)-2,5-диоксопирролидин-1-ил-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксилат,
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилат,
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота соль циклогексиламина,
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновая кислота и
(2S,5R)-6-(аллилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-
2-бензилкарбоксилат.
14. Кристалл (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилата, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 11,56, 10,96, 6,55, 6,00, 5,79, 5,56, 5,47, 5,25, 4,90, 4,35, 4,23 и 3,86 Å.
15. Кристалл (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилата, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 10,39, 5,86, 5,69, 5,34, 4,81, 4,44, 3,98, 3,78, 3,11, 3,03, 2,93 и 2,77 Å.
16. Кристалл (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилата, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 14,72, 4,91, 4,46, 4,24 и 3,67 Å.
17. Кристалл (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты циклогексиламмониевой соли, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 9,95, 8,45, 6,26, 5,87, 5,52, 5,22, 5,10, 4,96, 4,73, 4,54, 4,16, 3,93 и 3,55 Å.
18. Кристалл (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 8,19, 7,14, 6,64, 6,29, 5,60, 5,21, 4,91, 4,60, 4,21, 3,69, 3,45 и 3,13 Å.
19. Кристалл (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида, который показывает порошковую структуру дифракции рентгеновских лучей, имеющую характеристические пики, появляющиеся в интервалах решетки (d) 13,06, 6,52, 5,14, 4,74, 4,63, 4,34, 3,85 и 3,72 Å.
20. Применение (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-трет-бутилкарбоксилата для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты по п.1.
21. Применение (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-метилкарбоксилата для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты по п.1.
22. Применение (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-аллилкарбоксилата для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты по п.1.
23. Применение (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-бензилкарбоксилата для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты по п.1.
24. Применение циклогексиламмониевой соли (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты по п.1.
25. Применение (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты по п.1.
26. Применение (2S,5R)-6-(бензилокси)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоксамида для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты по п.1.
27. Применение (2S,5R)-5-(бензилоксиамино)пиперидин-2-трет-бутилкарбоксилата для получения лекарственного средства для лечения инфекционного заболевания, причем лекарственное средство включает ингибитор β-лактамазы, содержащий производное (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты по п.1.
| US 7638529 B2, 29.12.2009 | |||
| US 0007612087 B2, 03.11.2009 | |||
| ДИАЗАБИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2124014C1 |

