Настоящее изобретение относится к дозировке и схеме введения антител против С5, в частности, антитела против С5 кровалимаба, для применения в способе лечения или предупреждения у субъекта связанного с С5 заболевания, включая пароксизмальную ночную гемоглобинурию (PNH). Дозировка и схема введения по настоящему изобретению включает введение антитела против С5, предпочтительно антитела против С5 кровалимаба, с ударными дозами, с последующим введением субъекту (а) поддерживающей(щих) дозы(доз) антитела против С5, где исходно вводимая ударная доза дается субъекту внутривенно, а остальные ударные и поддерживающие дозы вводятся подкожно в меньшей дозировке, чем вводимая внутривенно ударная доза.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Система комплемента играет центральную роль в клиренсе иммунных комплексов и в иммунных ответах на инфекционные агенты, чужеродные антигены, инфицированные вирусом клетки и опухолевые клетки. Имеются примерно 25-30 белков комплемента, которые находятся в виде набора комплексов белков плазмы и мембранных кофакторов. Компоненты комплемента достигают их иммунозащитных функций посредством взаимодействия в ряде сложных событий ферментативных расщеплений и связывания с мембраной. Образующиеся каскады комплемента приводят к образованию продуктов с опсоническими, иммунорегулирующим и и литическими функциями.
Система комплемента может активироваться посредством трех отличных путей: классического пути, лектинового пути и альтернативного пути. Данные пути имеют многие общие компоненты и, в то время как они отличаются по их исходным стадиям, они сходятся и имеют те же самые общие конечные компоненты комплемента (от С5 до С9), ответственные за активацию и разрушение клеток-мишеней.
Классический путь обычно активируется посредством образования комплексов антиген-антитело. Независимо первой стадией в активации лектинового пути является связывание специфичных лектинов, таких как маннансвязывающий лектин (MBL), Н-фиколин, М-фиколин, L-фиколин и лектин С-типа CL-11. В отличие от этого, при альтернативном пути спонтанно осуществляется низкий уровень активации обмена, который может легко усиливаться на чужеродных или других ненормальных поверхностях (бактерии, дрожжи, инфицированные вирусом клетки или поврежденная ткань). Данные пути сходятся в точке, где компонент комплемента С3 расщепляется активной протеазой с образованием С3а и C3b.
С3а представляет собой анафилотоксин. C3b связывается с бактериальными и другими клетками, а также с определенными вирусами и иммунными комплексами, и метит их для удаления из системы кровообращения (роль, известная как опсонин). C3b также образует комплекс с другими компонентами с образованием конвертазы С5, которая расщепляет С5 до С5а и C5b.
С5 представляет собой 190 кДа белок, находящийся в нормальной сыворотке в концентрации приблизительно 80 мкг/мл (0,4 мкМ). С5 является гликозилированным, причем примерно 1,5-3,0% его массы приписывается углеводу. Зрелый С5 представляет собой гетеродимер из 115 кДа альфа-цепи, которая связывается дисульфидом с 75 кДа бета-цепью. С5 синтезируется в виде одноцепочечного белка-предшественника (про-С5 предшественника) из 1676 аминокислот (см., например, US-B1 6355245 и US-B1 7432356). Данный про-С5 предшественник расщепляется с образованием бета-цепи в виде аминоконцевого фрагмента и альфа-цепи в виде карбоксиконцевого фрагмента. Полипептидные фрагменты - альфа-цепь и бета-цепь - соединяются друг с другом посредством дисульфидной связи и составляют зрелый белок С5.
Терминальный путь системы комплемента начинается с захвата и расщепления С5. Зрелый С5 расщепляется на фрагменты С5а и C5b во время активации путей комплемента. С5а отщепляется от альфа-цепи С5 конвертазой С5 в виде аминоконцевого фрагмента, содержащего первые 74 аминокислоты альфа-цепи. Остальная часть зрелого С5 представляет собой фрагмент C5b, который содержит остальную альфа-цепь, связанную дисульфидной связью с бета-цепью. Приблизительно 20% от 11 кДа массы С5а приписываеся углеводу.
С5а представляет собой другой анафилатоксин. C5b объединяется с С6, С7, С8 и С9 с образованием комплекса атаки мембраны (MAC, C5b-9, терминального комплекса комплемента (ТСС)) на поверхности клетки-мишени. При вставке в мембраны клеток-мишеней достаточного числа MAC образуются поры MAC для опосредования быстрого осмотического лизиса клеток-мишеней.
Как упомянуто выше, С3а и С5а представляют собой анафилатоксины. Они могут запускать дегранулирование тучных клеток, что высвобождает гистамин и другие медиаторы воспаления, приводя к сокращению гладких мышц, повышенной проницаемости сосудов, активации лейкоцитов и другим воспалительным явлениям, включающим пролиферацию клеток, приводящую к гиперцеллюлярности. С5а также функционирует в качестве хемотаксического пептида, который служит для привлечения гранулоцитов, таких как нейтрофилы, эозинофилы, базофилы и моноциты, к сайту активации комплемента.
Активность С5а регулируется плазматическим ферментом карбоксипептидазой N, который удаляет карбоксиконцевой аргинин от С5а, образуя С5а-дез-Arg производное. С5а-дез-Arg демонстрирует только 1% анафилактической активности и полиморфоядерной хемотаксической активности немодифицированного С5а.
В то время как правильно функционирующая система комплемента обеспечивает надежную защиту против инфицирующих микробов, неподходящая регуляция или активация комплемента вовлечена в патогенез целого ряда расстройств, включающих, например, пароксизмальную ночную гемоглобинурию (PNH); ревматоидный артрит (RA); волчаночный нефрит; ишемическое реперфузионное повреждение; атипический гемолитический уремический синдром (aHUS); болезнь плотного осадка (DDD); макулодистрофию (например, возрастную макулодистрофию (AMD)); синдром гемолиза, повышенного уровня ферментов печени и уменьшенного числа тромбоцитов (HELLP); тромботическую тромбоцитопеническую пурпуру (ТТР); спонтанную потерю плода; слабоиммунный васкулит; буллезный эпидермолиз; рецидивирующую потерю плода; рассеянный склероз (MS); травматическое повреждение мозга и повреждение, возникающее из-за инфаркта миокарда, сердечно-легочного шунтирования и гемодиализа (см., например, Holers et al., Immunol. Rev. (2008), Vol. 223, pp. 300-316). Следовательно, ингибирование избыточных или неконтролируемых активаций каскада комплемента может давать клиническую пользу пациентам с такими расстройствами.
Пароксизмальная ночная гемоглобинурия (PNH) представляет собой необычное расстройство крови, при котором красные кровяные клетки (эритроциты) подвергаются риску и, таким образом, разрушаются быстрее, чем нормальные эритроциты. PNH возникает из-за клонального размножения гематопоэтических стволовых клеток с соматическими мутациями в гене PIG-A (фосфатидилинозитолгликан класса А), который расположен на X хромосоме.
Мутации в PIG-A приводят к ранней блокировке в синтезе гликозилфосфатидилинозитола (GPI) - молекулы, которая требуется для заякоривания многих белков на поверхностях клеток. Следовательно, кровяные клетки PNH являются дефицитными по GPI-заякоренным белкам, которые включают комплемент-регулирующие белки CD55 и CD59. При нормальных обстоятельствах данные комплемент-регулирующие белки блокируют образование MAC на поверхностях клеток, предотвращая, посредством этого, лизис эритроцитов. Отсутствие GPI-заякоренных белков вызывает опосредованный комплементом гемолиз при PNH.
PNH отличается гемолитической анемией (пониженное число эритроцитов), гемоглобинурией (присутствие гемоглобина в моче, особенно очевидное после сна) и гемоглобинемией (присутствие гемоглобина в кровотоке). Известно, что субъекты, пораженные PNH, имеют приступы, которые здесь определяются как возникновения случаев темноокрашенной мочи. Гемолитическая анемия обусловлена внутрисосудистым разрушением эритроцитов компонентами комплемента. Другие известные симптомы включают дисплазию, слабость, эректильную дисфункцию, тромбоз и рецидивирующую абдоминальную боль.
Экулизумаб представляет собой гуманизированное моноклональное антитело, направленное против белка комплемента С5, и первую терапию, одобренную для лечения пароксизмальной ночной гемоглобинурии (PNH) и атипического гемолитического уремического синдрома (aHUS) (см., например, Dmytrijuk et al., The Oncologist (2008), 13(9), pp. 993-1000). Экулизумаб ингибирует расщепление C5 на C5a и C5b посредством C5 конвертазы, что предотвращает образование терминального комплекса комплемента C5b-9. И С5а, и C5b-9 вызывают события, опосредованные терминальным комплементом, которые характерны для PNH и aHUS (см., например, WO-A2 2005/074607, WO-А1 2007/106585, WO-A2 2008/069889 и WO-A2 2010/054403). Для лечения PNH антитела против С5 экулизумаб или равулизумаб представляют собой обычную терапию. Однако вплоть до 3,5% индивидов азиатского происхождения несут полиморфизмы в С5, влияющие на Arg885, который соответствует сайту связывания экулизумаба или равулизумаба (Nishimura et al., N Engl J Med, Vol.370, pp.632-639 (2014); DOI: 10.1056/NEJMoa 1311084). Пациенты с PNH с данными полиморфизмами испытывают плохой контроль внутрисосудистого гемолиза экулизумабом или равулизумабом, таким образом, составляя группу с высокой неудовлетворенной медицинской потребностью.
В нескольких сообщениях были описаны антитела против С5. Например, в WO 95/29697 было описано антитело против С5, которое связывается с альфа-цепью С5, но не связывается с С5а и блокирует активацию С5. В WO-A2 2002/30985 описано моноклональное антитело против С5, которое ингибирует образование С5а. С другой стороны, в WO-A1 2004/007553 описано антитело против С5, которое распознает протеолитический сайт для С5 конвертазы на альфа-цепи С5 и ингибирует превращение С5 до С5а и C5b. В WO-A1 2010/015608 описано антитело против С5, которое имеет константу аффинности по меньшей мере 1×107 М-1. Кроме того, в WO-A1 2017/123636 и WO-A1 2017/132259 описаны антитела против С5. Кроме того, в WO-A 2016/098356 раскрыто получение антитела против С5, отличающегося связыванием с эпитопом в пределах бета-цепи С5 с более высокой аффинностью при нейтральном рН, чем при кислотном рН. Одно из антител против С5, раскрытое в WO-A1 2016/098356, относится к антителу против С5 кровалимабу (см. Пример 1 ниже относительно подробностей). Кровалимаб представляет собой антитело против С5, которое связывается с отличным эпитопом на бета-субъединице С5, который является отличным от эпитопа связывания экулизумаба/равулизумаба. Исследования in vitro продемонстрировали то, что антитело против С5 кровалимаб одинаково связывается с и ингибирует активность С5 дикого типа и мутанта по Arg885 (Fukuzawa et al., Sci Rep, 7(1): 1080. doi: 10.1038/s41598-017-01087-7 (2017)). В отличие от этого в WO-A1 2017/104779 на Фиг. 21 приводится то, что антитело против С5 экулизумаб не ингибирует мутант С5 по Arg855. Кроме того, WO-A1 2018/143266 относится к фармацевтическим композициям для применения в лечении или предупреждении заболевания, связанного с С5. Кроме того, в WO-A1 2018/143266 раскрыты дозировки и схемы введения антитела против С5 кровалимаба в том виде, в котором они используются в исследовании COMPOSER (ВР39144). Исследование COMPOSER относится к глобальному, многоцентровому открытому исследованию фазы I/II для оценки безопасности и эффективности, фармакокинетики (PK) и фармакодинамики (PD) антитела против С5 кровалимаба у здоровых субъектов и субъектов с PNH. Исследование COMPOSER содержало три части: часть 1 у здоровых участников, часть 2 и часть 3 у пациентов с пароксизмальной ночной гемоглобинурией (PNH). Дополнительно пациенты, охватываемые в части 3 данного исследования, были пациентами, которых лечили антителом против С5 экулизумабом в течение по меньшей мере 3 месяцев. Было задумано так, что участники части 1 исследования COMPOSER включали три группы здоровых пациентов: согласно исходному замыслу протокола первая группа представляет собой группу пациентов, которым антитело против С5 кровалимаб вводится внутривенно (в.в.) один раз в дозе 75 мг/организм; вторая группа пациентов представляет собой группу участников, которым антитело против С5 кровалимаб вводится внутривенно (в.в.) один раз в дозе 150 мг/организм, и третья группа представляет собой группу субъектов, которым антитело против С5 кровалимаб вводится подкожно (п.к.) один раз в дозе 170 мг/организм. Поскольку часть 1 исследования COMPOSER является адаптивной по природе (на основе продолжающейся оценки данных безопасности, переносимости, фармакокинетики (PK) и фармакодинамики (PD)), фактические дозы, которые давали для части 1, составляли: 75 мг в.в. для первой группы пациентов, 125 мг в.в. для второй группы пациентов, и 100 мг п. к. для третьей группы пациентов, зарегистрированных в части 1 исследования COMPOSER.
Часть 2 исследования COMPOSER была разработана для включения группы субъектов, которым антитело против С5 кровалимаб вводится внутривенно три раза: согласно исходному замыслу протокола антитело против С5 кровалимаб исходно вводилось в дозе 300 мг/организм (в.в.), затем - 500 мг/организм (в.в.) через неделю после исходного введения и наконец - 1000 мг/организм (в.в.) через две недели после второго введения. Начиная с двух недель после последнего внутривенного введения, антитело против С5 кровалимаб вводится подкожно один раз в неделю в дозе 170 мг/организм. На основе возникающих клинических данных из части 1 и симуляции PK исходная доза для пациентов в части 2 исследования COMPOSER была изменена от 300 мг до 375 мг в.в. Таким образом, фактические дозы, которые дают в части 2 исследования COMPOSER являются следующими: антитело против С5 кровалимаб исходно вводится внутривенно (в.в.) в дозе 375 мг/организм, с последующей дозой 500 мг/организм (в.в.) через неделю после исходного введения и, наконец, 1000 мг/организм (в.в.) через две недели после второго введения. Начиная с двух недель после конечного внутривенного введения, антитело против С5 кровалимаб вводится подкожно (п.к.) один раз в неделю в дозе 170 мг/организм.
Часть 3 данного исследования включала пациентов, которых лечили антителом против С5 экулизумабом в течение трех месяцев до регистрации в данном испытании, и данные пациенты должны были получать регулярные инфузии экулизумаба. Часть 3 данного исследования была спланирована для включения трех групп субъектов. Антитело против С5 кровалимаб исходно вводится субъектам всех групп внутривенно один раз в дозе 1000 мг/организм. Начиная с одной недели после исходного внутривенного введения (сутки 8 после исходного в.в. введения), антитело против С5 кровалимаб подкожно вводится субъектам первой группы один раз каждую неделю в дозе 170 мг/организм, субъектам второй группы один раз каждые две недели в дозе 340 мг/организм и субъектам третьей группы один раз каждые четыре недели в дозе 680 мг/организм. В части 3 COMPOSER у всех пациентов с PNH, которые переключились с антитела против С5 экулизумаба на кровалимаб, были выявлены комплексы лекарственное средство-мишень-лекарственное средство (DTDC) между кровалимабом, человеческим С5 и антителом экулизумабом. DTDC запускают временное увеличение клиренса кровалимаба, что может потенциально увеличивать риск временной потери полного ингибирования терминального пути комплемента (см. Röth et al., Blood (2020), Vol. 135, pp. 912-920; doi: 10.1182/blood.2019003399 и Sostelly et al., Blood (2019), Vol. 134, p. 3745).
Кроме того, в WO-A1 2018/143266 описано то, что иммунокомплексы (лекарственное средство-мишень-лекарственное средство) между кровалимабом, человеческим С5 и антителом экулизумабом могли образоваться у субъектов, которых лечили экулизумабом. При переключении субъектов, особенно субъектов, для которых необходимо сохраняющееся полное ингибирование С5, таких как пациенты с PNH или aHUS, с антитела против С5 экулизумаба на кровалимаб, оба антитела против С5 присутствуют в системе кровообращения и образуют комплексы лекарственное средство-мишень-лекарственное средство (DTDC), так как они связываются с разными эпитопами человеческого С5. Данные DTDC строятся из повторения цепи молекул экулизумаб-С5-кровалимаб-С5 и могут расти при сборке двух DTDC с образованием большего DTDC. Целью лечения пациентов, охватываемых в части 3 исследования COMPOSER, кровалимабом является обеспечение быстрого и сохраняющегося полного ингибирования терминального пути комплемента. Однако комплексы лекарственное средство-мишень-лекарственное средство (DTDC), состоящие из кровалимаба, человеческого С5 и экулизумаба, были выявлены у всех пациентов, переключающихся с экулизумаба в части 3 COMPOSER. DTDC и особенно большие DTDC более медленно подвергаются клиренсу и с большей вероятностью вызывают токсичность. Поскольку образование таких DTDC может вызывать потенциальные риски, такие как ухудшение кровообращения, риск васкулита из-за размеров комплексов, реакции гиперчувствительности типа III или ненормальную активацию системы комплемента, следует избегать образования таких DTDC (см. также Röth et al., Blood (2020), Vol., 135, pp. 912-920; doi: 10.1182/blood.2019003399).
Кроме того, антитело против С5 кровалимаб на основе его механизма действия ингибирует опосредованный комплементом лизис красных кровяных клеток (эритроцитов), не имеющих регуляторных белков комплемента. Если терминальный путь комплемента временно не блокируется во время интервала лечения, данные красные кровяные клетки (эритроциты) будут лизированы, и это может приводить к прорывному гемолизу, который является тяжелым клиническим осложнением у пациентов с PNH. Биологический стресс (инфекция, хирургическое вмешательство, беременность) приводит к физиологической активации пути комплемента с повышающей регуляцией С5 (Schutte et al., Int Arch Allergy Appl Immunol. (1975), Vol. 48(5), pp. 706-720). У пациентов с PNH, следовательно, важно не только поддерживать полную блокаду активности терминального комплемента во всем интервале дозирования, но также сохранять резерв свободных сайтов связывания кровалимаба для минимизации появления прорывного гемолиза.
Соответственно, существует потребность в идентификации дозировки и схемы введения, которая (1) минимизирует образование DTDC у пациентов, страдающих от заболеваний, связанных с С5, и, в частности, у пациентов, переключающихся с антитела против С5 экулизумаба на кровалимаб, (2) максимизирует уровень свободных сайтов связывания кровалимаба и (3) обеспечивает то, что у пациентов сохраняется превышение целевой пороговой концентрации антитела против С5, требующейся для ингибирования терминального комплемента, несмотря на вариабельность между индивидами.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данная потребность решается настоящим изобретением посредством предоставления воплощений, как определено в формуле изобретения.
Настоящее изобретение относится к антителу против С5 для применения в способе лечения или предупреждения связанного с С5 заболевания у субъекта, где данный способ включает следующие последовательные стадии:
(а) внутривенное введение субъекту ударной дозы 1000 мг антитела против С5 один раз, с последующим подкожным введением данному субъекту по меньшей мере одной ударной дозы 340 мг антитела против С5; и
(б) подкожное введение данному субъекту по меньшей мере одной поддерживающей дозы 680 мг антитела против С5.
В контексте настоящего изобретения субъект, подлежащий лечению, предпочтительно представляет собой пациента с массой тела от 40 кг до 100 кг. В контексте настоящего изобретения субъект, подлежащий лечению, представляет(ют) собой субъекта(тов), который(рые) страдает(ют) от связанного с С5 заболевания, которое требует ингибирования активности комплемента (например, PNH и aHUS). Кроме того, данное изобретение направлено на применение антитела против С5 для лечения или предупреждения связанного с С5 заболевания, в частности, PNH. В контексте настоящего изобретения настоящее изобретение направлено на лечение или предупреждение связанного с С5 заболевания, в частности, PNH, у пациентов, которых лечили одним пригодным фармацевтическим продуктом для лечения или предупреждения связанного с С5 заболевания, в частности, PNH, и где вводимая внутривенно ударная доза антитела против С5 вводится субъекту после последней дозы фармакологического продукта. Соответственно, описанная в данном документе дозировка и схема введения антитела против С5, в частности, антитела против С5 кровалимаба, дается пациентам, которых лечили одним полезным фармацевтическим продуктом для лечения или предупреждения связанного с С5 заболевания, в частности, PNH. Как более подробно объясняется ниже, полезный фармацевтический продукт для лечения связанного с С5 заболевания, который давали субъектам до начала заявленной дозировки и схемы лечения, относится к антителу против С5 экулизумабу или равулизумабу, предпочтительно - к антителу против С5 экулизумабу.
Как показано в приложенных Примерах, доза и схема лечения, как определено в формуле изобретения, обеспечивают длительную и согласованную блокаду активности терминального комплемента (с приблизительно больше, чем 95% субъектов, поддерживаемых выше целевого порога 100 мкг/мл); см. Фиг. 4 и 7. Кроме того, ингибирование терминального комплемента достигалось немедленно после исходной дозы и обычно поддерживалось на протяжении всего интервала дозирования; см. Фиг. 8. Кроме того, дозировка и схема лечения по настоящему изобретению также обеспечивают достаточный резерв свободных сайтов связывания для большей части интервала дозирования как у не получавших лечения, так и у подвергавшихся предварительному лечению экулизумабом пациентов; см. Фиг. 2. Кровалимаб и экулизумаб связываются с разными эпитопами С5, и, таким образом, ожидается то, что образуются DTDC. Ожидается, что DTDC развиваются, если пациенты одновременно подвергаются воздействию кровалимаба и экулизумаба (см. Фиг. 5), во время периода переключения от экулизумаба на антитело против С5 кровалимаб. Образование DTDC может способствовать увеличению клиренса кровалимаба и может вызывать потенциальные риски, такие как реакции гиперчувствительности типа III, как объясняется выше. У пациентов, переключающихся с экулизумаба на кровалимаб, доза и схема лечения, как определено в формуле изобретения, уменьшают образование DTDC; см. Фиг. 3 и 12. Соответственно, описанные в данном документе дозировка и схема лечения описывают новую и улучшенную схему дозировки антител против С5, предпочтительно антитела против С5 кровалимаба, для лечения или предупреждения связанного с С5 заболевания, предпочтительно PNH. Безопасность и терапевтическая эффективность заявленной дозировки и схемы лечения дополнительно описываются на Фиг. 9-11.
Соответственно, настоящее изобретение относится к антителу против С5, предпочтительно антителу против С5 кровалимабу, для применения в способе лечения или предупреждения связанного с С5 заболевания у субъекта, предпочтительно у субъекта с массой тела от 40 до 100 кг, где данный способ включает следующие последовательные стадии:
(а) внутривенное введение субъекту ударной дозы 1000 мг антитела против С5 один раз, с последующим подкожным введеннием данному субъекту по меньшей мере одной ударной дозой 340 мг антитела против С5; и
(б) подкожное введение данному субъекту по меньшей мере одной поддерживающей дозы 680 мг антитела против С5.
Термин «ударная доза» относится к дозе антитела против С5, введенной субъекту, страдающему от связанного с С5 заболевания, предпочтительно PNH, в начале лечения, т.е. в начале схемы лечения. В фармакокинетике (PK) «ударная доза» представляет собой исходную более высокую дозу лекарственного средства, которую можно давать пациенту в начале курса лечения перед снижением до меньшей дозы. В контексте настоящего изобретения ударную дозу сперва дают субъектам, подлежащим лечению посредством внутривенного введения, с последующим подкожным введением. В контексте настоящего изобретения ударную дозу дают один раз в дозе 1000 мг. Соответственно, в контексте настоящего изобретения ударную дозу композиции, приготовленной для внутривенного введения, дают субъекту внутривенно один раз перед тем, как подкожно дается одна ударная доза или более чем одна ударная доза фармацевтической композиции, приготовленной для подкожного введения.
В контексте настоящего изобретения одна ударная доза или более чем одна ударная доза антитела против С5 вводится пациентам подкожно после внутривенного введения ударной дозы 1000 мг антитела против С5. Подкожно вводимая(ые) ударная(ные) доза(зы) вводится(дятся) подкожно в дозе 340 мг антитела против С5 по меньшей мере один раз данному субъекту от 1 суток до 3 недель (21 суток) после начала внутривенного введения антитела против С5. Соответственно, в контексте настоящего изобретения ударная доза 340 мг антитела против С5 вводится субъекту подкожно по меньшей мере один раз через 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 или 21 сутки после начала внутривенного введения антитела против С5. Предпочтительно ударная доза 340 мг антитела против С5 вводится субъекту через 1 сутки после начала внутривенного введения антитела против С5. Более предпочтительно одна ударная доза 340 мг антитела против С5 вводится подкожно через 1 сутки после начала внутривенного введения. В контексте настоящего изобретения по меньшей мере одна дополнительная ударная доза 340 мг антитела против С5 вводится субъекту подкожно через 1 неделю (7 суток), 2 недели (14 суток) или 3 недели (21 сутки) после начала внутривенного введения антитела против С5. Наиболее предпочтительно дополнительные ударные дозы 340 мг антитела против С5 вводятся подкожно через 1 неделю (7 суток), 2 недели (14 суток) и 3 недели (21 сутки) после начала внутривенного введения антитела против С5. Соответственно, в пределах контекста настоящего изобретения 1, 2, 3, 4 и/или 5 ударных доз дается(ются) субъекту, где одна ударная доза, предпочтительно исходная ударная доза вводится субъекту внутривенно в дозе 1000 мг, и где 1, 2, 3 или 4 ударные дозы дается(ются) пациенту подкожно в дозе 340 мг. В контексте настоящего изобретения предпочтительным является подкожное введение 4 ударных доз, причем каждая имеет дозировку 340 мг антитела против С5, где дополнительные ударные дозы вводятся подкожно через 1 сутки после начала внутривенного введения антитела против С5, с последующим подкожным введением ударных доз один раз в неделю через 1 неделю, 2 недели и 3 недели после начала внутривенного введения антитела против С5. Соответственно, пациенту с использованием ударных доз может вводиться общее количество 2360 мг антитела против С5. Данное общее количество относится к общим дозам антитела против С5, введенного после 22 суток лечения, т.е. к дозе, достигаемой в конце суток 22 лечения, которая рассчитывается посредством сложения ударных доз в сутки 1 (ударная доза 1000 мг, исходно введенная внутривенно), 2 (первая подкожно вводимая ударная доза 340 мг, которая дается пациенту через 1 сутки после начала внутривенного введения антитела против С5), 8 (вторая подкожно вводимая ударная доза 340 мг, которая дается через 1 неделю после начала внутривенного введения), 15 (третья подкожно вводимая ударная доза 340 мг, которая дается через 2 недели после начала внутривенного введения) и 22 (четвертая подкожно вводимая ударная доза 340 мг, которая дается через 3 недели после начала внутривенного введения). Например, общее количество антитела против С5, которое дается через (а) ударную(ные) дозу(зы), соответствующую(щие) внутривенному введению 1000 мг (сутки 1), с последующим подкожным введением 340 мг (сутки 2), 340 мг (сутки 8), 340 мг (сутки 15) и 340 мг (сутки 22) составляет 2360 мг.
Согласно настоящему изобретению после исходной дозы или доз следует(ют) последующие дозы равных или меньших количеств антитела против С5 с достаточно близкими интервалами для поддержания концентрации антитела против С5 на уровне или выше эффективного целевого уровня. Соответственно, в контексте настоящего изобретения (а) поддерживающая(щие) доза(зы) вводится(дятся) пациентам после ударной(ных) дозы(доз). «Поддерживающая доза» относится к дозе антитела против С5, которую дают субъекту, страдающему от связанного с С5 заболевания, для поддержания концентрации антитела против С5 выше определенного эффективного порога концентрации антитела против С5. В контексте настоящего изобретения целевой уровень антитела против С5 составляет приблизительно 100 мкг/мл или больше. Целевой уровень концентрации антитела против С5 в пределах настоящего изобретения может определяться в биологическом образце субъекта, подлежащего лечению. Средства и способы для определения концентрации антитела против С5 в биологическом образце находятся в пределах обычного знания специалиста, и она, например, может определяться посредством иммуноанализа. Предпочтительно в контексте настоящего изобретения иммуноанализ представляет собой ELISA (твердофазный иммуноферментный анализ). Подобным образом, гемолитическую активность можно использовать в качестве параметра для эффективного лечения пациентов, страдающих от связанного с С5 заболевания, посредством заявленной дозировки и схемы лечения. В контексте настоящего изобретения полное ингибирование терминального комплемента (полное ингибирование терминального пути системы комплемента) может определяться по гемолитической активности, которая составляет меньше, чем 10 U/мл. В данном контексте гемолитическая активность может быть определена в биологическом образце пациента, подлежащего лечению. Предпочтительным является то, что гемолитическая активность составляет меньше, чем 10 U/мл, т.е. 10, 9, 8, 7, 6, 5, 4, 3, 2, 1 или 0 U/мл. Средства и способ определения гемолитической активности в биологическом образце пациентов, подлежащих лечению, посредством дозировки и схемы введения по изобретению известны специалисту. В качестве примера, гемолитическую активность можно определять иммуноанализом. Предпочтительно в контексте настоящего изобретения иммуноанализ представляет собой липосомный иммуноанализ ex vivo (LIA). В контексте настоящего изобретения биологический образец представляет собой образец крови. Предпочтительно образец крови представляет собой образец красной крови (эритроцитов). Предпочтительно поддерживающую(щие) дозу(зы) вводят пациентам подкожно в дозе или дозах, составляющих 680 мг антитела против С5. Соответственно, в пределах контекста настоящего изобретения субъекту дают по меньшей мере одну поддерживающую или более чем одну поддерживающую дозу, где данную(ные) поддерживающую(щие) дозу(зы) вводят подкожно в дозе 680 мг. В контексте настоящего изобретения по меньшей мере одну поддерживающую дозу 680 мг антитела против С5 вводят субъекту подкожно через 4 недели (28 суток) после начала внутривенного введения антитела против С5. Предпочтительно поддерживающая доза 680 мг вводится субъектам подкожно один раз через 4 недели после начала внутривенного введения антитела против С5. Соответственно, в контексте настоящего изобретения по меньшей мере одна поддерживающая доза 680 мг вводится пациенту подкожно через 4 недели (28 суток) после начала внутривенного введения антитела против С5, т.е. на сутки 29 схемы лечения. Соответственно, в контексте настоящего изобретения поддерживающая доза 680 мг вводится подкожно, предпочтительно один раз в 4 недели (28 суток) после начала внутривенного введения антитела против С5. В контексте настоящего изобретения пациенту может вводиться общее количество 3040 мг антитела против С5 с ударными дозами и поддерживающей дозой согласно настоящему изобретению. Данное общее количество относится к общим дозам антитела против С5, введенным после 29 суток лечения, т.е. дозе, достигаемой в конце суток 29 лечения, которая рассчитывается путем сложения ударных доз в сутки 1 (ударная доза 1000 мг, исходно вводимая внутривенно), 2 (первая вводимая подкожно ударная доза 340 мг, которую дают пациенту через 1 сутки после начала внутривенного введения антитела против С5), 8 (вторая вводимая подкожно ударная доза 340 мг, которую дают через 1 неделю после начала внутривенного введения), 15 (третья вводимая подкожно ударная доза 340 мг, которую дают через 2 недели после начала внутривенного введения), 22 (четвертая вводимая подкожно ударная доза 340 мг, которую дают через 3 недели после начала внутривенного введения) и подкожно вводимой поддерживающей дозы 680 мг (сутки 29). Например, общее количество антитела против С5, которое дают посредством ударной дозы и поддерживающей дозы, соответствующей внутривенному введению 1000 мг (сутки 1), с последующим подкожным введением 340 мг (сутки 2), 340 мг (сутки 8), 340 мг (сутки 15), 340 мг (сутки 22) и 680 мг (сутки 29) составляет 3040 мг.
Подкожное введение поддерживающей дозы 680 мг можно повторять несколько раз с интервалами времени 4 недели (Q4W). В контексте настоящего изобретения предпочтительно то, что поддерживающую дозу 680 мг повторяют по меньшей мере 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 24, 36, 48 месяцев. Предпочтительным в контексте настоящего изобретения является повторение поддерживающей дозы 680 мг с интервалами времени 4 недели и продолжение в течение всей жизни пациента.
В частности, настоящее изобретение относится к антителу против С5 для применения в способе лечения или предупреждения связанного с С5 заболевания у субъекта, предпочтительно у субъекта с массой тела от 40 кг до 100 кг, где данный способ включает следующие последовательные стадии:
(i) внутривенное введение ударной дозы 1000 мг антитела против С5 субъекту один раз;
(ii) подкожное введение ударной дозы 340 мг антитела против С5 субъекту через 1 сутки после начала внутривенного введения антитела против С5;
(iii) подкожное введение ударной дозы 340 мг антитела против С5 субъекту через 1 неделю (7 суток), 2 недели (14 суток) и 3 недели (21 сутки) после начала внутривенного введения антитела против С5 один раз в неделю;
(iv) подкожное введение поддерживающей дозы 680 мг антитела против С5 субъекту через 4 недели (28 суток) после начала внутривенного введения антитела против С5; и
(v) повторение стадии (iv) несколько раз с интервалами времени 4 недели (28 суток).
Термины «внутривенное введение»/«осуществление внутривенного введения» относятся в контексте настоящего изобретения к введению антитела против С5 в вену субъекта таким образом, что организм пациента, подлежащего лечению, получает антитело против С5 за приблизительно 15 минут или меньше, предпочтительно за 5 минут или меньше. Для внутривенного введения антитело против С5 должно быть приготовлено таким образом, что его следует вводить посредством подходящего устройства, такого как шприц (но не ограничиваясь им). В контексте настоящего изобретения препарат для внутривенного введения содержит от 50 до 350 мг антитела против С5, от 1 до 100 мМ буферизующего агента, такого как гистидин/аспарагиновая кислота, имеющего рН 5,5 плюс/минус 1,0, от 1 до 100 мМ аминокислоты, такой как аргинин, и от 0,01 до 0,1% неионного поверхностно-активного вещества, такого как полоксамер. Предпочтительно в контексте настоящего изобретения предложен препарат для внутривенного введении в 2 мл стеклянном флаконе, содержащий следующие компоненты: 170 мг/мл кровалимаба, 30 мМ гистидин/аспарагиновую кислоту (рН 5,8), 100 мМ аргинина гидрохлорид и 0,05% Poloxamer 188™. Данный препарат затем вводится пациенту в пределах переносимого периода времени, такого как 5 минут, 15 минут, 30 минут, 90 минут или меньше. Кроме того, препарат для внутривенного введения дается пациентам, подлежащим лечению, с объемом инъекции от 1 мл до 15 мл, предпочтительно примерно 6 мл.
Термины «подкожное введение»/«осуществление подкожного введения» относятся в контексте настоящего изобретения к введению антитела против С5 под кожу животного или человеческого пациента, предпочтительно в пределы кармана между кожей и подлежащей тканью, посредством относительно медленной, замедленной доставки из вместилища лекарственного средства. Данный карман может быть создан посредством сжатия или оттягивания кожи вверх и от подлежащей ткани. Для подкожного введения антитело против С5 должно быть приготовлено таким образом, что его можно вводить посредством подходящего устройства, такого как (но не ограничиваясь) шприцем, предварительно заполненным шприцем, инъецирующим устройством, инфузионным насосом, инъекционным шприцем-ручкой, безыгольным устройством или посредством системы доставки на основе трансдермального пластыря. В контексте настоящего изобретения препарат для подкожного введения содержит от 50 до 350 мг антитела против С5, от 1 до 100 мМ буферизующего агента, такого как гистидин/аспарагиновая кислота, имеющего рН 5,5 плюс/минус 1,0, от 1 до 100 мМ аминокислоты, такой как аргинин, и от 0,01 до 0,1% неионного поверхностно-активного вещества, такого как полоксамер. Предпочтительно в контексте настоящего изобретения предложен препарат для внутривенного введения в 2,25 мл предварительно заполненном шприце, содержащем следующие компоненты: 170 мг/мл кровалимаба, 30 мМ гистидин/аспарагиновая кислота (рН 5,8), 100 мМ аргинина гидрохлорид и 0,05% Poloxamer 188™. В контексте настоящего изобретения препарат для подкожного введения предложен в виде предварительно заполненного шприца с приспособлением для безопасности иглы. Инъекционные устройства для подкожного введения содержат примерно от 1 до 15 мл или более, предпочтительно 2,25 мл препарата для подкожного введения, содержащего антитело против С5. При нормальных условиях инъекционный объем, подлежащий подкожному введению, составляет от 1 до 15 мл, предпочтительно либо 2 мл (340 мг кровалимаба), либо 6 мл (680 мг кровалимаба). В контексте настоящего изобретения подкожное введение относится к введению антитела против С5 под кожу пациента, подлежащего лечению, посредством относительно медленной, замедленной доставки из вместилища лекарственного средства в течение периода времени, включающего 30 минут или меньше, 90 минут или меньше, но не ограничиваясь ими. Возможно введение может осуществляться посредством подкожной имплантации насоса для доставки лекарственного средства, имплантированного под кожу пациента, подлежащего лечению, где данный насос доставляет заданное количество антитела против С5 в течение заданного периода времени, такого как 30 минут, 90 минут, или периода времени, охватывающего продолжительность схемы лечения.
В контексте настоящего изобретения приведенные выше дозировки и схемы лечения могут быть полезными для лечения или предупреждения связанного с С5 заболевания у субъекта, которого лечили по меньшей мере одним фармакологическим продуктом для применения в лечении или предупреждении заболевания один или более чем один раз. Например, схема лечения по настоящему изобретению может быть полезна для лечения пациента, имеющего связанное с С5 заболевание, который получал предшествующее лечение по меньшей мере одним фармакологическим продуктом для применения в способе лечения или предупреждения заболевания, но ожидается, что он будет лучше отвечать на схему лечения согласно настоящему изобретению. В таких случаях может производиться переключение лекарственного средства от данного фармакологического продукта на антитела против С5 для применения в лечении или предупреждении связанного с С5 заболевания согласно настоящему изобретению. Предпочтительно вводимая внутривенно ударная доза антитела против С5 дается субъекту, подлежащему лечению, после конечной дозы данного фармацевтического продукта. Данная вводимая внутривенно ударная доза антитела против С5 предпочтительно имеет дозу 1000 мг.
В контексте настоящего изобретения фармакологический продукт содержит активное вещество, которое отличается от антитела против С5, которое дают согласно настоящему изобретению либо внутривенно, либо подкожно. Данное активное вещество фармакологического продукта в контексте настоящего изобретения может представлять собой миРНК (малая интерферирующая РНК), нацеленную на мРНК С5, или антитело против С5, которое отличается от антитела против С5, вводимого подкожно или внутривенно субъекту, подлежащему лечению согласно настоящему изобретению. Данный фармакологический продукт может содержать антитело против С5, которое представляет собой отличное антитело от антитела против С5, которое дают пациентам в контексте настоящего изобретения. Антитело, содержащееся в данном фармацевтическом продукте, которое использовали в предшествующем лечении, может представлять собой равулизумаб или экулизумаб, или их варианты. Предпочтительно антитело, содержащееся в данном фармакологическом продукте, которое использовали в предшествующем лечении, представляет собой экулизумаб или его варианты. Типичные варианты последовательности антитела против С5 экулизумаба показаны в SEQ ID NO: 11 и 12.
Вариантами антитела в контексте настоящего изобретения могут быть антитела против С5, которые содержат вариант области Fc, в который была введена одна или более чем одна модификация аминокислоты в область Fc антитела с природной последовательностью. Данный вариант области Fc может содержать последовательность человеческой области Fc (например, области Fc человеческого IgG1, IgG2, IgG3 или IgG4), содержащую аминокислотную модификацию (например, замену) в одном или более чем одном положении аминокислоты. В контексте настоящего изобретения вариант антитела обладает некоторыми, но не всеми эффекторными функциями, что делает его желательным кандидатом для применений, при которых период полувыведения антитела in vivo является важным, тем не менее определенные эффекторные функции (такие как комплемент и ADCC (антителозависимая клеточная цитотоксичность)) являются необязательными или вредными. Можно проводить анализы цитотоксичности in vitro и/или in vivo для подтверждения уменьшения/истощения активностей CDC (комплементзависимая цитотоксичность) и/или ADCC. Например, можно проводить анализы связывания с рецептором Fc (FcR) для обеспечения того, что данное антитело не имеет связывания с R (рецептор) Fc гамма (следовательно, вероятно не имеет активности ADCC), но сохраняет способность к связыванию с FcRn. Первичные клетки для опосредования ADCC - клетки NK (природные киллеры) - экспрессируют только RIII Fc гамма, тогда как моноциты экспрессируют RI Fc гамма, RII Fc гамма и RIII Fc гамма. Экспрессия FcR на гематопоэтических клетках обобщается в Таблице 3 на странице 464 Ravetch and Kinet, Annu. Rev. Immunol. 9:457-492 (1991). Неограничивающие примеры анализов in vitro для оценки активности ADCC интересующей молекулы описываются в US-B1 5500362 (см., например, Hellstrom et al., Proc. Nat'l Acad. Sci. USA (1983), Vol.83, pp.7059-7063) и Hellstrom et al., Proc. Nat'l Acad. Sci. USA (1985), Vol. 82, pp. 1499-1502; US-B1 5821337 (см. Bruggemann et al., J. Exp.Med. (1987), Vol.166, pp.1351-1361). В качестве альтернативы, можно использовать нерадиоактивные способы анализа (см., например, нерадиоактивный анализ цитотоксичности ACTI™ для проточной цитометрии (CellTechnology, Inc. Mountain View, CA) и нерадиоактивный анализ цитотоксичности CytoTox 96 (зарегистрированный товарный знак) (Promega, Madison, WI)). Полезные эффекторные клетки для таких анализов включают одноядерные клетки периферической крови (РВМС) и клетки-природные киллеры (NK). Альтернативно или дополнительно, активность ADCC интересующей молекулы может оцениваться in vivo, например, в животной модели, такой как модель, раскрытая в dynes et al., Proc. Nat'l Acad. Sci. USA (1998), Vol.95, pp.652-656. Анализы связывания C1q также могут проводиться для подтверждения того, что антитело не способно связываться с C1q и, следовательно, не имеет активности CDC. См., например, ELISA (твердофазный иммуноферментный анализ) связывания с C1q и С3 с в WO-A2 2006/029879 и WO-A1 2005/100402. Для оценки активации комплемента можно проводить анализ CDC (см., например, Gazzano-Santoro et al., J. Immunol. Methods (1996), Vol. 202, pp. 163; Cragg et al., Blood (2003), Vol. 101, pp. 1045-1052 и Cragg et al., Blood (2004), Vol. 103, pp. 2738-2743). Определения связывания с FcRn и клиренса/периода полувыведения in vivo также можно осуществлять с использованием способов, известных в данной области (см., например, Petkova et al., Int'l. Immunol. (2006), Vol. 18(12), pp. 1759-1769).
Антитела с пониженной эффекторной функцией включают антитела с заменой одного или более чем одного остатка области Fc 238, 265, 269, 270, 297, 327 и 329 (US-B1 6737056). Такие мутанты Fc включают мутантов Fc с заменами по двум или более чем двум положениям аминокислот 265, 269, 270, 297 и 327, включая так называемого мутанта Fc "DANA" с заменой остатков 265 и 297 на аланин (US-B1 7332581).
Описываются определенные варианты антител с улучшенным или уменьшенным связыванием с FcRn. (См., например, US-B1 6737056; WO-A2 2004/056312 и Shields et al., J. Biol. Chem. (2001), Vol. 9(2), pp. 6591-6604).
В некоторых воплощениях вариант антитела содержит область Fc с одной или более чем одной аминокислотной заменой, которая улучшает ADCC, например, с заменами в положениях 298, 333 и/или 334 области Fc (нумерация остатков EU).
В некоторых воплощениях в области Fc делаются изменения, которые приводят к измененному (т.е. либо улучшенному, либо уменьшенному) связыванию с C1q и/или комплементзависимой цитотоксичности (CDC), например, как описано в US-B1 6194551, WO 1999/51642 и Idusogie et al., J. Immunol. (2000), Vol. 164, pp. 4178-4184.
Антитела с увеличенными периодами полувыведения и улучшенным связыванием с неонатальным рецептором Fc (FcRn), который отвечает за перенос материнских IgG в плод (Guyer et al., J. Immunol. (1976), Vol. 117, pp. 587 и Kim et al., J. Immunol. (1994), Vol. 24, pp. 249), описываются в US 2005/0014934. Данные антитела содержат область Fc с одной или более чем одной заменой в ней, которая улучшает связывание области Fc с FcRn. Такие варианты Fc включают варианты с заменами в одном или более чем одном остатке области Fc: 238, 256, 265, 272, 286, 303, 305, 307, 311, 312, 317, 340, 356, 360, 362, 376, 378, 380, 382, 413, 424 или 434, например, замену остатка 434 области Fc (US-B1 7371826). См. также Duncan, Nature (1988), Vol. 322, pp. 738-740, US-B1 5648260; US-B15624821 и WO 1994/29351 относительно дургих примеров вариантов области Fc.
В контексте настоящего изобретения исходная доза композиции для внутривенной инъекции в настоящем изобретении вводится в те же самые сутки или через 1 сутки, 2 суток, 3 суток, 4 суток, 5 суток, 6 суток, 7 суток (1 неделя), 8 суток, 9 суток, 10 суток, 11 суток, 12 суток, 13 суток, 14 суток (две недели), 15 суток, 16 суток, 17 суток, 18 суток, 19 суток, 20 суток, 21 сутки (3 недели) или больше суток после того, как пациенту, подлежащему лечению, вводится последняя доза фармакологического продукта. Предпочтительно в контексте настоящего изобретения вводимая внутривенно ударная доза антитела против С5 вводится в сутки 3 или через 3 суток, 4 суток, 5 суток, 6 суток, 7 суток (1 неделя), 8 суток, 9 суток, 10 суток, 11 суток, 12 суток, 13 суток, 14 суток (две недели), 15 суток, 16 суток, 17 суток, 18 суток, 19 суток, 20 суток, 21 сутки (3 недели) или больше суток после конечной дозы фармакологического продукта. Предпочтительно вводимая внутривенно ударная доза антитела против С5 дается пациенту через 7 суток (1 неделя) или больше суток после последней дозы фармакологического продукта. Также предпочтительным в контексте настоящего изобретения является внутривенное введение ударной дозы через 14 суток (2 недели) или больше суток после конечной дозы фармакологического продукта. Наиболее предпочтительным в контексте настоящего изобретения является внутривенное введение антитела против С5 через 21 сутки (3 недели) после последней дозы фармакологического продукта.
В контексте настоящего изобретения термин «неделя» относится к периоду времени 7 суток.
В контексте настоящего изобретения термин «месяц» относится к периоду времени 4 недели.
Термин «лечение» в контексте настоящего изобретения включает последовательный ряд «индуцирующего лечения» и по меньшей мере одного «поддерживающего лечения». Типично лечение согласно данному изобретению включает «индуцирующее лечение» и по меньшей мере одно «поддерживающее лечение». Типично лечение согласно данному изобретению может продолжаться 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год (12 месяцев), 2 года (24 месяца), 3 года (36 месяцев) или 4 года (48 месяцев). Предпочтительным в контексте настоящего изобретения является лечение, которое продолжается в течение всей жизни пациента.
«Индуцирующее лечение» состоит из последовательного ряда (i) внутривенного введения субъекту ударной дозы, предпочтительно дозы 1000 мг, антитела против С5 и (ii) подкожного введения субъекту по меньшей мере одной ударной дозы, предпочтительно дозы 340 мг, антитела против С5. Как объясняется в данном документе выше, в пределах контекста настоящего изобретения предпочтительным является то, что ударная доза 340 мг антитела против С5 дается субъекту через 1 сутки, 1 неделю (7 суток), 2 недели (14 суток) и 3 недели (21 сутки) после вводимой внутривенно ударной дозы. Предпочтительно ударная доза, подлежащая введению внутривенно, имеет дозу 1000 мг. Ударная доза, которая дается субъекту, подлежащему лечению, подкожно, имеет дозу 1360 мг. Таким образом, в контексте настоящего изобретения во время индуцирующего лечения субъекту, подлежащему лечению, вводится ударная доза 2360 мг либо внутривенно, либо подкожно. «Поддерживающее лечение» состоит из последовательного ряда (i) поддерживающего периода, где субъектам подкожно дается одна или более чем одна поддерживающая доза. В контексте настоящего изобретения предпочтительным является то, что субъекту дается поддерживающая доза 680 мг антитела против С5 предпочтительно один раз через 4 недели (1 месяц) после начала внутривенного введения ударной дозы антитела против С5. Как объясняется выше, подкожное введение поддерживающей дозы 680 мг можно повторять несколько раз с интервалами времени 4 недели (Q4W). Предпочтительным в контексте настоящего изобретения является повторение поддерживающей дозы 680 мг с интервалами времени 4 недели и продолжающееся в течение всей жизни пациента.
В контексте настоящего изобретения связанное с С5 заболевание представляет собой опосредованное комплементом заболевание или состояние, которое включает избыточную или неконтролируемую активацию С5. В некоторых воплощениях связанное с С5 заболевание представляет собой по меньшей мере одно, выбранное из группы, состоящей из пароксизмальной ночной гемоглобинурии (PNH), ревматоидного артрита (RA), волчаночного нефрита, ишемического реперфузионного повреждения, атипического гемолитического уремического синдрома (aHUS), болезни плотного осадка (DDD), макулодистрофии, синдрома гемолиза, повышенного уровня ферментов печени и уменьшенного числа тромбоцитов (HELLP), тромботической тромбоцитопенической пурпуры (ТТР), спонтанной потери плода, слабоиммунного васкулита, буллезного эпидермолиза, рецидивирующей потери плода, рассеянного склероза (MS), травматического повреждения мозга, повреждения, возникающего из-за инфаркта миокарда, сердечно-легочного шунтирования и гемодиализа, рефрактерной генерализованной тяжелой миастении (gMG) и оптиконевромиелита (NMO). Предпочтительно в контексте настоящего изобретения связанное с С5 заболевание представляет собой по меньшей мере одно, выбранное из группы, состоящей из PNH, aHUS, gMG и NMO. Наиболее предпочтительно связанное с С5 заболевание представляет собой PNH. Кроме того, в контексте настоящего изобретения субъект, страдающий от связанного с С5 заболевания PNH, может быть подвергнут анализу на присутствие мутации Arg885 С5. Соответственно, раскрытую в данном документе схему дозировки также можно использовать для лечения и/или предупреждения у субъектов, страдающих от PNH, отличающихся тем, что данные субъекты имеют мутацию Arg885 С5. В данном контексте мутация Arg885 означает генетическое изменение С5, где Arg в положении 885 заменяется His. В данном контексте термин «С5» относится к белку, имеющему аминокислотную последовательность, как показано в SEQ ID NO: 13.
В контексте настоящего изобретения антитело против С5 предпочтительно представляет собой кровалимаб. Подробности о последовательности антитела против С5 кровалимаба (номер CAS: 1917321-26-6) раскрываются в списке №119 предложенных международных непатентованных названий для фармацевтических веществ (INN), как опубликовано на страницах 302 и 303 WHO Drug Information (2018), Vol. 32, №2. Последовательности антитела против С5 кровалимаба также демонстрируются в SEQ ID NO: 3 (тяжелая цепь) и SEQ ID NO: 4 (легкая цепь). Получение антитела против С5 кровалимаба, используемого в настоящем изобретении, описывается в WO 2016/098356 (см. Пример 1 относительно подробностей). Кроме того, в контексте настоящего изобретения антитело против С5 кровалимаб вводится пациентам посредством препарата либо для внутривенного, либо для подкожного введения. Предпочтительным в контексте настоящего изобретения является внутривенное или подкожное введение предложенных в данном документе дозировок в виде (а) фиксированной(ных) дозы(доз).
Препарат для внутривенного введения содержит от 50 до 350 мг антитела против С5 кровалимаба, от 1 до 100 мМ буферизующего агента, такого как гистидин/аспарагиновая кислота, имеющего рН 5,5 плюс/минус 1,0, от 1 до 100 мМ аминокислоты, такой как аргинин, и от 0,01 до 0,1% неионного поверхностно-активного вещества, такого как полоксамер. Предпочтительный в контексте настоящего изобретения препарат для внутривенного введения предложен в 2 мл стеклянном флаконе, содержащем следующие компоненты: 170 мг/мл кровалимаба, 30 мМ гистидин/аспарагиновая кислота (рН 5,8), 100 мМ аргинина гидрохлорид и 0,05% Poloxamer 188™.
Препарат для подкожного введения содержит от 50 до 350 мг антитела против С5 кровалимаба, от 1 до 100 мМ буферизующего агента, такого как гистидин/аспарагиновая кислота, имеющего рН 5,5 плюс/минус 1,0, от 1 до 100 мМ аминокислоты, такой как аргинин, и от 0,01 до 0,1% неионного поверхностно-активного вещества, такого как полоксамер. Предпочтительный в контексте настоящего изобретения препарат для внутривенного введения предложен в 2,25 мл предварительно заполненном шприце, содержащем следующие компоненты: 170 мг/мл кровалимаба, 30 мМ гистидин/аспарагиновая кислота (рН 5,8), 100 мМ аргинина гидрохлорид и 0,05% Poloxamer 188™.
Антитело против С5 экулизумаб продается под торговым наименованием Солирис® компанией Alexion Pharmaceuticals, Inc. Последовательности антитела против С5 экулизумаба показаны в SEQ ID NO: 1 (тяжелая цепь) и SEQ ID NO: 2 (легкая цепь). Кроме того, варианты последовательности антитела против С5 экулизумаба показаны в SEQ ID NO: 11 и 12.
Последовательности антитела против С5 равулизумаба продаются под торговым наименованием Ультомирис® компанией Alexion Pharmaceuticals, Inc. Последовательности антитела против С5 равулизумаба (номер CAS: 1803171-55-2) раскрываются в списке №117 предложенных международных непатентованных названий фармацевтических веществ (INN), как опубликовано на страницах 319 и 320 WHO Drug Information (2017), Vol. 31, №2. Последовательности антитела против С5 равулизумаба также показаны в SEQ ID NO: 5 (тяжелая цепь) и SEQ ID NO: 6 (легкая цепь).
Пациенты, описанные в контексте настоящего изобретения, представляют собой пациентов, страдающих от связанного с С5 заболевания. Предпочтительными пациентами в контексте настоящего изобретения являются пациенты с массой тела от 40 кг до 100 кг. В контексте настоящего изобретения связанное с С5 заболевание представляет собой опосредованное комплементом заболевание или состояние, которое включает избыточную или неконтролируемую активацию С5. В некоторых воплощениях связанное с С5 заболевание представляет собой по меньшей мере одно, выбранное из группы, состоящей из пароксизмальной ночной гемоглобинурии (PNH), ревматоидного артрита (RA), волчаночного нефрита, ишемического реперфузионного повреждения, атипического гемолитического уремического синдрома (aHUS), болезни плотного осадка (DDD), макулодистрофии, синдрома гемолиза, повышенного уровня ферментов печени и уменьшенного числа тромбоцитов (HELLP), тромботической тромбоцитопенической пурпуры (ТТР), спонтанной потери плода, слабоиммунного васкулита, буллезного эпидермолиза, рецидивирующей потери плода, рассеянного склероза (MS), травматического повреждения мозга, повреждения, возникающего из-за инфаркта миокарда, сердечно-легочного шунтирования или гемодиализа, рефрактерной генерализованной тяжелой миастении (gMG) и оптиконевромиелита (NMO). Предпочтительно в контексте настоящего изобретения связанное с С5 заболевание представляет собой по меньшей мере одно, выбранное из группы, состоящей PNH, aHUS, gMG и NMO. Наиболее предпочтительно связанное с С5 заболевание представляет собой PNH.
Кроме того, настоящее изобретение относится к способу лечения или предупреждения связанного с С5 заболевания у субъекта, где данный способ включает следующие последовательные стадии:
(а) осуществление внутривенного введения ударной дозы 1000 мг антитела против С5 субъекту один раз, с последующим подкожным введением по меньшей мере одной ударной дозы 340 мг антитела против С5 субъекту; и
(б) подкожное введение по меньшей мере одной поддерживающей дозы 680 мг антитела против С5 субъекту.
В контексте настоящего изобретения является предпочтительным то, что способ лечения или предупреждения связанного с С5 заболевания у субъекта проводится посредством следующих стадий введения:
(i) внутривенное введение ударной дозы 1000 мг антитела против С5 субъекту один раз;
(ii) подкожное введение ударной дозы 340 мг антитела против С5 субъекту через 1 сутки после начала внутривенного введения антитела против С5;
(iii) подкожное введение ударной дозы 340 мг антитела против С5 субъекту через 1 неделю, 2 недели и 3 недели после начала внутривенного введения антитела против С5 один раз в неделю;
(iv) подкожное введение поддерживающей дозы 680 мг антитела против С5 субъекту через 4 недели после начала внутривенного введения антитела против С5; и
(v) повторение стадии (iv) несколько раз с интервалами времени 4 недели.
Как объясняется выше, в контексте настоящего изобретения является предпочтительным то, что антитело против С5, используемое в контексте дозировки и схемы введения, представляет собой кровалимаб. Кроме того, приведенное выше определение подобным образом применимо к приведенным выше способам лечения или предупреждения связанного с С5 заболевания. В контексте настоящего изобретения также предпочтительно, что субъект, подлежащий лечению, имеет массу тела от 40 кг до 100 кг. На Фиг. показано:
Фиг. 1: связь между антителом против С5 кровалимабом и гемолитической активностью, измеренная липосомным иммуноанализом (LIA), у здоровых субъектов и субъектов со связанным с С5 заболеванием - пароксизмальной ночной гемоглобинурией (PNH)
Оценка связи воздействие-ответ демонстрирует то, что для достижения полного ингибирования терминального комплемента требуется приблизительно 100 мкг/мл кровалимаба. Полное ингибирование терминального комплемента (полное ингибирование терминального пути системы комплемента) определяется как гемолитическая активность меньше 10 U/мл. Вертикальная пунктирная линия отмечает порог фармакодинамики (PD) 100 мкг/мл кровалимаба.
Фиг. 2: доступные свободные сайты связывания антитела против С5 кровалимаба
Серые линии соответствуют симуляции 15 индивидов на основе параметров, оцениваемых из данных COMPOSER (ВР39144). Для симуляций использовали данные исследования COMPOSER. По ординате показана концентрация антитела против С5 кровалимаба (RO7112689; SKY59). По абсциссе показано время в сутках. Темно-серые линии соответствуют значениям медианы для данных 15 пациентов: S0: схема части 3 COMPOSER; S5: предложенная схема в части 4 исследования COMPOSER и фазе III.
Фиг. 3: профиль во времени комплекса лекарственное средство-мишень-лекарственное средство (DTDC)
Серые линии соответствуют симуляции 15 индивидов на основе параметров, оцениваемых из данных COMPOSER (ВР39144). Для симуляций использовали данные исследования COMPOSER. Темно-серые линии соответствуют значениям медианы для данных 15 пациентов: S0: схема части 3 COMPOSER; S5: предложенная схема в части 4 исследования COMPOSER и фазе III; RO7112689: кровалимаб (SKY59).
Фиг. 4: симулированные профили концентрация-время кровалимаба при лечении не получавших лечения пациентов (верхняя панель) и пациентов с PNH, переключающихся с лечения экулизумабом на кровалимаб (нижняя панель)
Серый интервал соответствует интервалу 90%-ного прогнозирования, а серая линия - прогнозируемой медиане. Черная пунктирная линия соответствует целевому уровню концентрации 100 мкг/мл антитела против С5 кровалимаба.
Фиг. 5: модель, описывающая как комплексы лекарственное средство-мишень-лекарственное средство (DTDC) между кровалимабом, человеческим С5 и антителом экулизумабом подвергаются клиренсу, рециклируют и последовательно строятся из меньших DTDC
При переключении пациентов от антитела против С5 экулизумаба на кровалимаб в системе кровообращения присутствуют оба антитела против С5 и образуют DTDC, так как они связываются с разными эпитопами человеческого С5. Данные DTDC строятся из повторения цепи молекул экулизумаб-С5-кровалимаб-С5 и растут с течением времени при сборке двух DTDC с образованием большего DTDC. Данная модель (Фиг. 5) описывает как DTDC подвергаются клиренсу и рециклируют посредством рецепторов FcRn антитела против С5 кровалимаба. (1) DTDC развиваются, если пациенты одновременно подвергаются воздействию кровалимаба и экулизумаба во время периода переключения с 1 лекарственного средства на другое из-за дифференциального распознавания эпитопа С5 данными антителами. Данные DTDC поглощаются в эндосомы посредством фагоцитоза. (2) Антитело кровалимаб, которое рН-зависимо связывается с человеческим С5, диссоциирует от растворимого человеческого С5, который был связан с антителом против С5 кровалимабом, при кислотных условиях (рН 6,0) в эндосоме, в то время как антитело против С5 экулизумаб все еще связано с растворимым человеческим С5 при кислотных условиях в эндосоме. (3) Антитела против С5 (антитело против С5 кровалимаб и комплекс С5-экулизумаб) поглощаются клетками посредством связывания с FcRn, экспрессируемым на клеточной мембране. Комплекс С5-экулизумаб транслоцируется в лизосому для деградации или рециклирования с белком С5, все еще связанным с антителом. В отличие от этого, антитело против С5 кровалимаб имеет улучшенную функциональность/эффективность, так как оно диссоциирует от FcRn в эндосоме при кислотных условиях для высвобождения обратно в плазму без белка С5. (4), (5) Высвобожденное антитело против С5 кровалимаб вновь доступно для связывания с человеческим С5 и для создания других, меньших DTDC. Это имеет эффект «рециклирования» антитела против С5 кровалимаба. DTDC и, в частности, комплексы С5-экулизумаб затем вновь деградируют посредством эндосом, тогда как антитело против С5 кровалимаб вновь рециклирует с построением меньших DTDC.
Фиг. 6: часть 4 COMPOSER включала пациентов с PNH
В части 4 COMPOSER оценивали эффекты на безопасность, фармакокинетику (PK) и фармакодинамику (PD) оптимизированной схемы введения кровалимаба у пациентов с PNH, которые не получали терапии против С5, предпочтительно терапии кровалимабом, или которые переключались с экулизумаба, с первичной оценкой через 20 недель. Из 15 зарегистрированных пациентов 8 (53%) ранее не получали терапию ингибитором С5, а 7 (47%) переключались с экулизумаба на кровалимаб.
Фиг. 7: воздействие кровалимаба у пациентов, зарегистрированных в части 4 исследования COMPOSER
У всех пациентов поддерживались уровни кровалимаба больше значения Ctrough (минимальная концентрация) приблизительно 100 мкг/мл, что ассоциировано с ингибированием активности терминального комплемента. Линии представляют среднее значение, и затемненная область показывает доверительный интервал 95%.
Фиг. 8: ход липосомного иммуноанализа (LIA), показывающего медианную активность комплемента у пациентов, зарегистрированных в части 4 исследования COMPOSER
Ингибирование терминального комплемента достигалось сразу после исходной дозы и обычно сохранялось на протяжении всего периода исследования. Линии представляют значение медианы, а усы показывают 95%-ный доверительный интервал. Нижняя граница количественного измерения анализа LIA составляет 10 U/мл. LIA - липосомный иммуноанализ.
Фиг. 9: измерение общего уровня и уровня свободного С5 у пациентов, зарегистрированных в части 4 исследования COMPOSER
(А) У не получавших лечения пациентов наблюдали ограниченное общее накопление С5, а у пациентов с переключением наблюдали снижение. (В) Уровни свободного С5 быстро снижались после исходной дозы и оставались низкими на протяжении всего периода последующего наблюдения.
Фиг. 10: измерение нормированного уровня лактатдегидрогеназы (LDH) у пациентов, зарегистрированных в части 4 исследования COMPOSER
У наивных пациентов медианные уровни лактатдегидрогеназы (LDH) снижались до уровней, меньших или равных 1,5× верхней границы нормы (ULN) к суткам 15 и оставались ниже этого уровня на протяжении всего периода наблюдения. У пациентов, которые переключались с экулизумаба на кровалимаб, медианный исходный уровень LDH был меньше или равен 1,5× ULN и оставался таким на протяжении всего периода наблюдения. LDH - лактатдегидрогеназа; ULN - верхняя граница нормы.
Фиг. 11: краткое изложение неблагоприятных событий (АЕ), связанных с лечением кровалимабом
Кровалимаб хорошо переносился, и не наблюдали серьезных неблагоприятных событий (АЕ), связанных с лечением.
Фиг. 12: наблюдаемые профили DTDC с течением времени с частью 3 и частью 4 схем с кровалимабом исследования COMPOSER
Сплошные линии представляют собой сумму медианных процентных содержаний кровалимаба, элюированного во фракциях 1-4 (левые панели) и фракциях 5-6 (правые панели) гель-фильтрации (SEC). Схема дозировки части 3 исследования COMPOSER показана светло-серым, и схема дозировки части 4 показана темно-серым.
Фиг. 13: нормированные уровни LDH пациентов с PNH, несущих мутацию С5 Arg885His, которых лечили кровалимабом
Кровалимаб достигал поддерживающегося ингибирования терминального комплемента у пациентов с PNH с полиморфизмом Arg885. У всех пациентов достигалось полное ингибирование терминального комплемента при измерении посредством липосомного иммуноанализа (LIA). Уровни LIA варьировали от 32-42 U/мл в начале исследования, снижались до меньше, чем 10 U/мл к суткам 2 и поддерживались далее. Нижняя граница количественного измерения для анализа LIA составляет 10 U/мл. LIA - липосомный иммуноанализ.
Следующие примеры иллюстрируют данное изобретение
Пример 1: антитела против С5
Последовательности антитела против С5 кровалимаба демонстрируются в SEQ ID NO: 3 (тяжелая цепь) и SEQ ID NO: 4 (легкая цепь). Кроме того, получение антитела против С5 кровалимаба, используемого в настоящем изобретении, описывается в WO 2016/098356. Вкратце, гены, кодирующие вариабельный домен тяжелой цепи (VH) 305LO15 (SEQ ID NO: 7)), объединяли с генами, кодирующими модифицированный вариант SG115 константного домена (СН) тяжелой цепи человеческого IgG1 (SEQ ID NO: 8). Гены, кодирующие вариабельный домен легкой цепи (VL) 305LO15 (SEQ ID NO: 9)), объединяли с генами, кодирующими константный домен челевеческой легкой цепи (CL) (SK1, SEQ ID NO: 10). Антитела экспрессировались в клетках HEK293, сотрансфицированных комбинацией экспрессионных векторов тяжелой и легкой цепи, и очищали посредством белка.
Пример 2: дозировки и схемы введения, использованные в исследовании COMPOSER (ВР39144; идентификатор ClinicalTrials.gov: NCT03157635)
Для определения подходящих дозировок и схемы введения инициировали исследование фазы I/II COMPOSER (ВР39144). Данное исследование исходно состояло из трех частей: часть 1 - у здоровых участников, часть 2 и часть 3 - у пациентов с пароксизмальной ночной гемоглобинурией (PNH). Кроме того, пациенты, охватываемые в части 3 данного исследования, представляли собой пациентов, которых лечили антителом против С5 экулизумабом в течение по меньшей мере 3 месяцев.
Часть 1 данного исследования была разработана для включения трех групп здоровых пациентов. Первая группа представляет собой группу пациентов, кому антитело против С5 кровалимаб вводится внутривенно (в.в.) один раз в дозе 75 мг/организм. Вторая группа пациентов представляет собой группу участников, кому антитело против С5 кровалимаб вводится внутривенно (в.в.) один раз в дозе 150 мг/организм. Третья группа представляет собой группу субъектов, кому антитело против С5 кровалимаб вводится подкожно (п.к.) один раз в дозе 170 мг/организм. Поскольку часть 1 исследования COMPOSER является адаптивной по природе (на основе продолжающейся оценки данных по безопасности, переносимости, фармакокинетике (PK) и фармакодинамике (PD)), фактические дозы, приведенные для части 1, составляли: 75 мг в.в. для первой группы пациентов, 125 мг в.в. для второй группы пациентов и 100 мг п. к. для третьей группы пациентов, зарегистрированных в части 1 исследования COMPOSER.
Часть 2 данного исследования была разработана для включения группы субъектов, кому антитело против С5 кровалимаб вводится внутривенно три раза: согласно исходной схеме протокола антитело против С5 кровалимаб исходно вводилось в дозе 300 мг/организм (в.в.), затем 500 мг/организм (в.в.) через неделю после исходного введения и, наконец, 1000 мг/организм (в.в.) через две недели после второго введения. Начиная с двух недель после последнего внутривенного введения, антитело против С5 кровалимаб вводится подкожно (п.к.) один раз в неделю в дозе 170 мг/организм. На основе появляющихся клинических данных из части 1 и симуляции PK исходная доза для пациентов в части 2 исследования COMPOSER была заменена от 300 мг до 375 мг в.в. Таким образом, фактические дозы, приведенные в части 2 исследования COMPOSER, являются следующими: антитело против С5 кровалимаб исходно вводится внутривенно (в.в.) в дозе 375 мг/организм, с последующей дозой 500 мг/организм (в.в.) через неделю после исходного введения и, наконец - 1000 мг/организм (в.в.) через две недели после второго введения. Начиная с двух недель после последнего внутривенного введения, антитело против С5 кровалимаб вводится подкожно (п.к.) один раз в неделю в дозе 170 мг/организм.
Часть 3 данного исследования включала пациентов, которых лечили антителом против С5 экулизумабом в течение по меньшей мере трех месяцев до регистрации в данном клиническом испытании, и данные пациенты должны были получать регулярные инфузии экулизумаба. Часть 3 данного исследования была разработана для включения трех групп субъектов. Антитело против С5 кровалимаб исходно вводится субъектам во всех группах внутривенно один раз в дозе 1000 мг/организм. Начиная с одной недели после исходного внутривенного введения (сутки 8 после в.в. введения), антитело против С5 кровалимаб подкожно (п.к.) вводится субъектам первой группы один раз каждую неделю в дозе 170 мг/организм, субъектам второй группы один раз каждые две недели в дозе 340 мг/организм и субъектам третьей группы один раз каждые четыре недели в дозе 680 мг/организм. 15 здоровых пациентов регистрировали в части 1 исследования COMPOSER. Часть 1 была рандомизированной таким образом, что только 9 из исходных 15 пациентов получали кровалимаб. 19 пациентов регистрировали в части 3 исследования COMPOSER, но три пациента прервали участие.
Подробности относительно пациентов, включенных: в исследование COMPOSER (часть 1, часть 2 и часть 3) могут быть обобщены следующим образом:
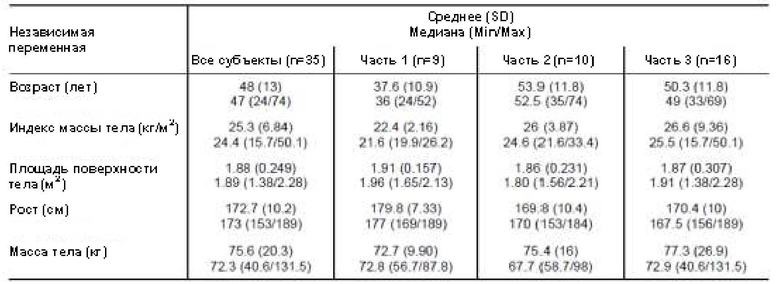
После получения приведенных выше подробностей о пациентах, включенных в части 1-3 исследования COMPOSER, один дополнительный пациент части 3 исследования COMPOSER прервал участие в исследовании.
Пример 3: определение схемы дозирования для достижения полного и поддерживающегося ингибирования терминального комплемента на всем протяжении лечения антителом против С5 кровалимабом
Целью лечения для кровалимаба при связанных с С5 заболеваниях, таких как предпочтительно пароксизмальная ночная гемоглобинурия (PNH), является обеспечения быстрого и поддерживающегося полного ингибирования терминального пути комплемента. У пациентов, переключающихся от экулизумаба на кровалимаб, период отмывки является клинически неподходящим. Следовательно, по замыслу, остаточные концентрации экулизумаба присутствуют при инициации дозирования кровалимаба. Комплексы лекарственное средство-мишень-лекарственное средство (DTDC), состоящие из кровалимаба, человеческого С5 и экулизумаба, были выявлены у всех пациентов, переключающихся с экулизумаба в части 3 COMPOSER с использованием мультиплексного анализа, объединяющего гель-фильтрацию (SEC) с твердофазным иммуноферментным анализом (ELISA). SEC представляет собой методику разделения на основе различия радиуса Стокса и геометрии белков: SEC разделяет молекулы согласно различиям в размере по мере того, как они проходят через среду гель-фильтрации, упакованную в колонку, с образованием упакованного слоя. В отличие от ионообменной или аффинной хроматографии молекулы не связываются с хроматографической средой таким образом, что состав буферизованной среды прямо не влияет на разрешение (степень разделения между пиками). Среда представляет собой пористую матрицу сферических частиц с химической и физической стабильностью и инертностью (отсутствием реакционной способности и адсорбционных свойств). SEC использовали в режиме фракционирования для разделения многочисленных компонентов в образце на основе различий в их размере. Для состава сложного образца с разными белками, подобного сыворотке, комбинация SEC с ELISA, специфичной в отношении аналита (кровалимаба), обеспечивала желательную специфичность и чувствительность для выявления концентраций кровалимаба в каждой из разделенных фракций. Для обеспечения выявления концентраций кровалимаба с использованием ELISA разделение SEC фракционируется на восемь фракций. Для каждого индивида с использованием данного подхода описывали профиль DTDC с течением времени. Для определения схемы дозирования, для которой ожидается достижение полного и поддерживаемого ингибирования терминального комплемента на протяжении всего интервала дозирования, разработали два дополняющих информированных о модели подхода разработки лекарственного средства (MIDD) для рекомендации дозы, подлежащей применению в данном клиническом испытании (доза фазы III):
• Эмпирическая модель популяционной фармакокинетики, использованная для рекомедации подкожной (п.к.) дозы и схемы поддержания концентраций кровалимаба выше целевой пороговой концентрации 100 мкг/мл на протяжении всего интервала дозирования у пациентов.
• Биохимическая модель, одновременно описывающая кинетику общего и свободного С5, фармакокинетику кровалимаба и экулизумаба, и кинетику DTDC, использованную для рекомендации дозы и схемы, минимизирующей образование больших DTDC у пациентов, переключающихся с экулизумаба на кровалимаб, и максимизирующей уровень свободных сайтов связывания кровалимаба у всех пациентов.
3.1 Модель популяционной фармакокинетики
Профили концентрация-время антитела против С5 кровалимаба лучше всего описывались с использованием двухкамерной открытой модели с устранением первого порядка и адсорбцией первого порядка для описания подкожного (п.к.) введения (см. Betts A. et al., mAbs (2018), Vol. 10, No. 5, pp. 751-764). Фармакокинетические (PK) профили у пациентов, переключающихся с лечения экулизумабом в части 3 COMPOSER, демонстрируют кратковременное более быстрое выведение, не наблюдаемое у здоровых добровольцев и не получавших лечение пациентов с PNH. Для описания фармакокинетики (PK) для пациентов, переключающихся с лечения экулизумабом на антитело против С5 кровалимаб, выведение кровалимаба моделировали как комбинацию выведения первого порядка, использованного для не получавших лечение пациентов, и более быстрого клиренса, который экспоненциально снижается во времени. Массу тела (медиана: 72,3 (40,6-131,5) [кг]) анализировали в качестве независимой переменной для клиренсов и объемов, и обнаружили то, что она значимо влияет на данные параметры при включении с использованием аллометрического масштабирования с коэффициентом, зафиксированным на уровне 0,75 для клиренсов и 1 для объемов. Параметр «клиренс» является мерой способности организма выводить лекарственное средство. Клиренс выражается как объем в единицу времени. Параметр «объемы» обозначает объем распределения - меру кажущегося пространства в организме, доступного для содержания антитела против С5 кровалимаба. Возраст также был обнаружен в качестве независимой переменной, влияющей на скорость поглощения, и был введен в модель в качестве категориальной независимой переменной. Пациенты в возрасте, большем или равном 50 лет, по-видимому, имеют меньшую скорость поглощения, чем более молодые пациенты. Биодоступность после подкожного (п.к.) введения оценивается как составляющая приблизительно 100%.
Данная модель могла точно оценивать РК параметры и имеет хорошую прогностическую эффективность, которая определяет ее применение в целях симуляции.
3.2 Биохимическая модель комплексов лекарственное средство-мишень-лекарственное средство (DTDC)
Разработали биохимическую математическую модель для исследования кинетики образования и выведения DTDC при предположении того, что комплексы увеличенного размера образуются посредством обратимого связывания меньших комплексов (см. Фиг. 5). Данная модель учитывает все комплексы, образованные повторением звена Ab1-Ag-Ab2 (антитело 1 (Ab1), антитело 2 (Ab2) и антиген (Ag) представляют собой кровализумаб, экулизумаб и С5 соответственно), начиная с наименьших комплексов (Ab1-Ag-Ab2) вплоть до наибольших комплексов, содержащих 4 Ab1, 4 Ab2 и 8 Ag (например, комплекс Ab1-Ag-Ab2-Ag-Ab1-Ag-Ab2-Ag-Ab1-Ag-Ab2-Ag-Ab1-Ag-Ab2-Ag) в том виде, в котором он наблюдается в анализах SEC in vitro. Каждую возможную биохимическую реакцию, описывающую образование комплекса через связывание 2 меньших комплексов, описывали с использованием модели связывания лиганда. В каждой реакции связывания также учитывали клиренс комплексов и рециклирование свободного кровалимаба из DTDC (из-за SMART-Ig Recycling®, высвобождающего С5 от кровалимаба в кислотных условиях лизосомы). Подробности системы SMART-Ig Recycling® были описаны Fukuzawa et al., Sci Rep.(2017), Vol.7(1): 1080; doi: 10.1038/s41598-017-01087-7. Параметры модели оценивали с использованием нелинейного подхода со смешанными эффектами с использованием данных, собранных в исследовании COMPOSER. Для разработки модели использовали общий уровень кровалимаба, общий уровень С5 и 8 фракций SEC, где DTDC выявляются согласно их молекулярной массе. Оценка адекватности модели была удовлетворительной для целей симуляции. Данную модель калибровали с использованием концентраций экулизумаба во время переключения, профилей во времени общего уровня кровалиаба, общих концентраций С5 и основанных на хроматографии измерений распределения размера DTDC, полученных из исследования COMPOSER фазы I/II (см. Röth et al., Blood (2020), Vol., 135, pp. 912-920; doi: 10.1182/blood.2019003399).
3.3 Определение дозы для фазы III
Параллельное применение обеих моделей - модели популяционной фармакокинетики и биохимической модели DTDC - обеспечивало идентификацию фиксированной дозы и схемы дозирования, которая (1) минимизирует образование больших DTDC у пациентов, переключающихся с экулизумаба на кровалимаб, (2) максимизирует уровень свободных сайтов связывания кровалимаба и (3) обеспечивает то, что пациенты остаются выше целевой пороговой концентрации, требующейся для ингибирования терминального комплемента (целевая Cthrough выше приблизительно 100 мкг/мл кровалимаба), несмотря на присущую вариабельность между индивидами.
На основе его механизма действия кровалимаб ингибирует опосредованный комплементом лизис эритроцитов, не имеющих регуляторных белков комплемента. Если терминальный путь комплемента временно не блокируется во время интервала обработки, данные эритроциты будут лизироваться, и это может приводить к прорывному гемолизу, который является тяжелым клиническим осложнением у пациентов с PNH. Биологический стресс (инфекция, хирургия, беременность) приводит к физиологической активации пути комплемента с повышающей регуляцией С5 (Schutte et al., Int Arch Allergy Appl Immunol (1975), Vol. 48(5), pp. 706-720.). У пациентов с PNH, следовательно, важно не только поддерживать полную блокаду активности терминального комплемента посредством интервала дозирования, но также поддерживать резерв свободных сайтов связывания кровалимаба для минимизации появления прорывного гемолиза.
Доступные данные по фармакокинетике (PK) и фармакодинамике (PD) из частей 1, 2 и 3 из исследования COMPOSER интегрировали для обеспечения характеристики связи PK/PD кровалимаба после в.в. и п. к. введения и для идентификации уровней воздействия, требующихся для полного ингибирования активности системы терминального комплемента. Посредством объединения данных РК и PD от 9 здоровых волонтеров в части 1, 10 пациентов с PNH в части 2 и 16 пациентов с PNH в части 3 было показано, что кровалимаб индуцирует зависимое от концентрации ингибирование сывороточной гемолитической активности при измерении посредством липосомного иммуноанализа ex vivo (LIA). Оценка связи воздействие-ответ демонстрирует, что требуется приблизительно 100 мкг/мл кровалимаба для достижения полного ингибирования терминального комплемента, определяемого как гемолитическая активность меньше 10 U/мл (см. Фиг. 1).
В модели популяционной PK массу тела анализировали в качестве независимой переменной для клиренса кровалимаба и объема распределения, и обнаружили, что она статистически влияет на данные параметры при включении с использованием алло метрического масштабирования. Как следствие, для данной дозы большие пациенты имеют тенденцию к тому, чтобы иметь меньшее воздействие для того, чтобы быть подвергнутыми недостаточному воздействию по сравнению с меньшими пациентами. Для учета компенсации на эффект массы тела предлагается ярусный подход дозирования на основе массы для обеспечения того, что у всех получивших пациентов достигается сравнимое воздействие кровалимаба по всему интервалу дозирования.
Были определены следующие две схемы дозирования:
• Для пациентов с массой тела больше 40 кг, но меньше 100 кг
Ударные дозы: 1000 мг кровалимаба, введенного внутривенно (в.в.) в сутки 1, с последующими 340 мг кровалимаба, введенными подкожно (п.к.) в сутки 2, 8, 15 и 22.
Поддерживающие дозы: 680 мг кровалимаба п. к. в сутки 29, с последующим подкожным введением 680 мг кровалимаба п. к. один раз каждые 4 недели (Q4W) далее.
• Для пациентов с массой тела, большей или равной 100 кг
Ударные дозы: 1500 мг кровалимаба в.в. в сутки 1, с последующими 340 мг кровалимаба п. к. в сутки 2, 8, 15 и 22.
Поддерживающие дозы: 1020 мг кровалимаба п. к. в сутки 29, с последующим подкожным введением 1020 мг кровалимаба п. к. один раз каждые 4 недели (Q4W) далее.
Пример 4: результаты симуляций модели DTDC
Симуляции, проведенные от данной модели были нацелены на идентификацию дозы и схемы дозирования, минимизирующих образование больших DTDC у пациентов, переключающихся с экулизумаба на кровалимаб, и обеспечивающих достаточные резервы свободных сайтов связывания кровалимаба у пациентов, переключающихся с экулизумаба, или не получавших лечения пациентов с PNH. Последний критерий обеспечивает объективную оценку границы контроля гемолиза, которую обеспечивает схема дозирования для защиты от прорывного гемолиза. Симуляции проводили только с использованием оценок параметров от пациентов в части 3 COMPOSER, которые переключались с экулизумаба на кровалимаб. Схема дозирования, обеспечивающая достаточный резерв свободных эпитопов кровалимаба у пациентов, которых предварительно лечили экулизумабом, также подходит для лечения пациентов, не получавших лечения. Как показано на Фиг. 2 и Фиг. 3, ожидается то, что вышеупомянутые схемы дозирования максимизируют доступность свободных эпитопов при минимизации образования наибольших DTDC.
Пример 5: результаты симуляций модели популяционной фармакокинетики
Проводили симуляции из модели популяционной РК для рекомендации дозы и схемы дозирования для обеспечения быстрого установления стационарных концентраций, а также поддержки наименьших концентраций выше 100 мкг/мл у большинства пациентов во всем интервале дозирования как у наивных в отношении лечения, так и у подвергавшихся предварительному лечению экулизумабом пациентов с PNH.
Профили концентрации кровалимаба относительно времени симулировали для 20000 не получавших лечения пациентов с PNH и 20000 пациентов с PNH, которые переключали лечение с экулизумаба на кровалимаб, с медианной массой тела 75,6 кг (стандартное отклонение плюс/минус 20,3; с 42,2 кг и 109,0 кг 5-ым и 95-ым процентилями соответственно). Симуляции объясняли эффект возраста с 50% симулированной популяции, находящейся в возрасте младше 50 лет, и с 50% симулированной популяции, находящейся в возрасте старше 50 лет. Выбор распределения массы тела основывается на наблюдаемом распределении в исследовании COMPOSER.
На основе результатов симуляции (Фиг. 4) прогнозируется, что вышеупомянутые дозировки и схема лечения приведут к быстрому установлению стационарных концентраций и поддерживаемым значениям Ctrough больше, чем 100 мкг/мл у приблизительно 95% индивидов во всем интервале дозирования, независимо от массы тела. Прогнозируется то, что данная схема дозирования будет поддерживать концентрации выше 100 мкг/мл как у не получавших лечения пациентов, так и у пациентов, переключающихся с экулизумаба, несмотря на наблюдаемое кратковременное увеличение клиренса кровалимаба и являющееся следствием этого более длительное время для достижения стационарных концентраций у последних.
Ожидается, что предложенные выше доза и схема дозирования обеспечивают полную и устойчивую блокаду активности терминального комплемента (причем поддерживаемую выше целевого порога у приблизительно 95% пациентов) и также обеспечивают достаточный резерв свободных сайтов связывания для большей части интервала дозирования как у не получавших лечения, так и у подвергавшихся предварительному лечению экулизумабом пациентов. Также ожидается, что у пациентов, переключающихся с экулизумаба, будет снижаться образование больших DTDC. Приведенные выше дозировки были подтверждены в части 4 исследования COMPOSER у семи пациентов, переключающихся с экулизумаба на кровалимаб. Часть 4 оценивала эффекты на безопаснось, фармакокинетику (PK) и фармакодинамику (PD) приведенной выше оптимизированной схемы с кровалимабом у 15 пациентов (дата отсечения - 29 января 2020 г. ) с PNH, которые не получали терапию антителом против С5 (8 пациентов (53%)), или которых ранее лечили антителом против С5 экулизумабом (7 пациентов (47%)). Исходные характеристики пациентов, зарегистрированных в части 4 исследования COMPOSER, показаны на Фиг. 6. Самая подходящая дозировка для уменьшения сохранения DTDC, в частности, больших DTDC, состояла из серии ударных доз (1000 мг кровалимаба, введенного внутривенно (в.в.) в сутки 1, с последующими 340 мг кровалимаба, введенного подкожно (п.к.) в сутки 2, 8, 15 и 22), с последующим поддерживающим дозированием (680 мг кровалимаба п. к. в сутки 29, с последующим подкожным введением 680 мг кровалимаба п.к. один раз каждые 4 недели (Q4W) далее). Данные части 4 COMPOSER подтвердили то, что распределение размера DTDC сдвигалось к меньшим комплексам с использованием заявленной оптимизированной схемы дозирования. Дальнейшие результаты в отношении описанных выше дозы и схемы введения кровалимаба (1000 мг кровалимаба, введенного внутривенно (в.в.) в сутки 1, с последующими 340 мг кровалимаба, введенного подкожно (п.к.) в сутки 2, 8, 15 и 22), с последующим поддерживающим дозированием (680 мг кровалимаба п. к. в сутки 29, с последующим подкожным введением 680 мг кровалимаба п.к. один раз каждые 4 недели (Q4W) далее) приводятся на Фиг. 7-11.
Как показано на Фиг. 7, с данной оптимизированной схемой дозирования воздействие кровалимаба устойчиво поддерживалось выше значения Cthrough приблизительно 100 мкг/мл (уровень, ассоциированный с ингибированием комплемента) на протяжении всего периода последующего наблюдения 20 недель (140 суток).
Кроме того, ингибирование терминального комплемента достигалось сразу после исходной дозы и сохранялось на протяжении всего периода исследования (см. Фиг. 8).
Кроме того, ограниченное общее накопление С5 наблюдали у пациентов с PNH, которые не получали терапии антителом против С5 (8 пациентов; Фиг. 9(A)), а снижение уровней С5 наблюдали у переключившихся пациентов (пациенты с PNH, которых ранее лечили антителом против С5 экулизумабом (7 пациентов; Фиг. 9(B)). Кроме того, на Фиг. 10 описано то, что внутрисосудистый гемолиз контролировался, большинство пациентов имели стабилизацию уровня гемоглобина и избегали переливания крови: всего у 10 (67%) пациентов, включая 5 из 8 пациентов, не получавших лечения, и 5 из 7 переключившихся пациентов, достигалась стабилизация гемоглобина (избежание большего или равного 2 г/дл снижения уровня гемоглобина от исходного в отсутствие переливания крови) в неделю 20. От исходного момента времени до недели 20 11 (73%) пациентов, включая 5 из 8 пациентов, не получавших лечения, и 6 из 7 переключившихся пациентов, освобождались от переливания крови. Свыше 7,2 общих лет подвергания пациентов риску ни один из пациентов не испытывал событий прорывного гемолиза (ВТН), как определяется в Kulasekararaj et al., Blood (2019), Vol. 33, pp. 540-549.
Кроме того, выявили то, что приведенная выше доза и схема лечения антитела против С5 кровалимаба хорошо переносилась, и не наблюдали серьезных связанных с лечением нежелательных явлений (АЕ) (см. Фиг. 11).
Таким образом, описанный в данном документе подход моделирования доказывает то, что заявленная схема дозировки является превосходной для лечения или предупреждения связанного с С5 заболевания, такого как PNH, как у не получавших лечения, так и, в частности, у получавших предварительное лечение экулизумабом субъектов.
Пример 6: результаты сравнения распределения размера DTDC между частью 3 и частью 4 исследования COMPOSER
В части 3 COMPOSER комплексы лекарственное средство-мишень-лекарственное средство (DTDC) между кровалимабом, человеческим С5 и антителом экулизумабом были выявлены у всех пациентов с PNH, которые переключились с антитела против С5 экулизумаба на кровалимаб. Целью настоящего примера является описание результатов сравнения распределения размера DTDC между схемой дозировки части 3 и части 4 исследования COMPOSER. В части 3 исследования COMPOSER антитело против С5 кровалимаб исходно вводится субъектам внутривенно один раз в дозе 1000 мг/организм. Начиная с одной недели после исходного внутривенного введения (сутки 8 после в.в. введения) антитело против С5 кровалимаб вводится подкожно (п.к.) один раз в каждую неделю в дозе 170 мг/организм, один раз каждые две недели в дозе 340 мг/организм или один раз каждые четыре недели в дозе 680 мг/организм. В части 4 исследования COMPOSER кровалимаб вводили согласно приведенной выше схеме дозирования и лечения: данными оптимизированной дозой и схемой были серия ударных доз 1000 мг в сутки 1 и 340 мг п. к. в сутки 2, 8, 15 и 22, с последующим поддерживающим дозированием 680 мг п. к. каждые 4 недели, начиная с суток 29 (неделя 5). Серия ударных доз увеличивала общую дозу кровалимаба, полученную во время первого месяца лечения, с уменьшением образования больших DTDC, в соответствии с теорией «решетки» образования комплексов. Эту оптимизированную стратегию дозирования исследовали в части 4 на пациентах, которые переключали лечения, и сравнивали с 19 пациентами с PNH, которые были зарегистрированы в части 3 и переключались с экулизумаба на кровалимаб. Распределения размера DTDC измеряли с использованием гель-фильтрации (SEC) в сочетании с ELISA. SEC разделяла DTDC на фракции согласно их размеру: большие DTDC находятся во фракциях 1-4, а меньшие комплексы, такие как одиночные мотивы и не DTDC, находятся во фракциях 5-6. DTDC наблюдали у всех пациентов из части 3 (Фиг. 12; большие DTDC находятся во фракциях 1-4, а меньшие комплексы, такие как одиночные мотивы и не DTDC находятся во фракциях 5-6). Два пациента из части 3 испытывали клинические проявления, сравнимые с реакциями гиперчувствительности типа III, которые были приписаны DTDC. Распределение размера DTDC у пациентов в части 4, которые получали оптимизированную стратегию дозирования, изменялось по-другому, чем у пациентов в части 3, согласно предсказаниям модели. У переключившихся пациентов из части 4 (n равно 7; дата отсечения 29 января 2020 г. ) сумма DTDC во фракции 1-4 начинала снижаться в сутки 8 и продолжала снижаться, в отличие от части 3. В сутки 22 средняя процентная доля наибольших DTDC снижалась на 56% у пациентов в части 4 относительно пациентов в части 3. Кроме того, сывороточные концентрации кровалимаба оставались выше 100 мкг/мл для пациентов части 4, на уровне, ассоциированном с ингибированием комплемента. Несмотря на то, что DTDC наблюдаются у всех пациентов части 4, которые переключились с экулизумаба, не происходило нежелательных явлений, свидетельствующих о реакции гиперчувствительности типа III. В заключение, оптимизированная схема с кровалимабом приводила к меньшей концентрации больших DTDC, чем у пациентов, которые получали схему части 3.
Пример 7: результаты ответа на кровалимаб пациентов с PNH с полиморфизмом С5
Пароксизмальная ночная гемоглобинурия (PNH) отличается потерей эндогенных регуляторов комплемента CD59 и CD55 на гематопоэтических клетках. Периферические элементы крови являются чувствительными к разрушению комплементом, приводя к внутрисосудистому гемолизу и тромбозу. Стандартная терапия представляет собой ингибирование терминального комплемента экулизумабом - моноклональным антителом против С5 (mAb). Однако вплоть до 3,5% индивидов азиатского происхождения несут полиморфизмы в С5, влияющие на Arg885, который соответствует сайту связывания экулизумаба и равулизумаба (см. Nishimura et al., N Engl J Med, Vol. 370, pp. 632-639 (2014); DOI: 10.1056/NEJMoa1311084). Пациенты с PNH с данными полиморфизмами испытывают плохой контроль над внутрисосудистым гемолизом с использованием экулизумаба, таким образом, составляя группу с большой неудовлетворенной медицинской потребностью. Кровалимаб представляет собой новое mAb (моноклональное антитело) против С5, которое связывается с отличным эпитопом на бета-субъединице С5. Исследования in vitro продемонстрировали то, что кровалимаб одинаково связывается и ингибирует активность С5 дикого типа и мутанта по Arg885 (Fukuzawa et al., Sci Rep, 7(1): 1080. doi: 10.1038/s41598-017-01087-7 (2017)).
Цели: целью настоящего примера является описание ответа на кровалимаб пациентов с PNH с полиморфизмом С5.
Способы: приведенную выше дозу и схему введения кровалимаба (1000 мг кровалимаба, введенного внутривенно (в.в.) в сутки 1, с последующими 340 мг введенного подкожно (п.к.) в сутки 2, 8, 15 и 22), с последующим поддерживающим дозированием (680 мг кровалимаба п. к. в сутки 29, с последующим подкожным введением 680 мг кровалимаба п. к. один раз каждые 4 недели (Q4W) далее) вводили пациентам с PNH с полиморфизмом С5 (мутация Arg885 С5 (SEQ ID NO: 13)). При каждом посещении определяли плазматическую концентрацию кровалимаба, лактатдегидрогеназы (LDH), свободного и общего С5, и активность комплемента. Пациентов отслеживали на наличие переливаний крови, событий прорывного гемолиза (ВТН) и на безопасность.
Результаты: из 44 пациентов, зарегистрированных в части 2 (n равно 10), части 3 (n равно 19) и части 4 (n равно 15) исследования COMPOSER (идентификатор ClinicalTrials.gov: NCT03157635), четыре имели полиморфизм нуклеотидов c.2654G->A, предсказывающий замену Arg885His. В дату отсечения -в сентябре 2019 г. период последующего наблюдения варьировал от 12,4 до 98,3 недель. Все четыре пациента были мужчинами, продиагностированными за 44-734 недели до регистрации с размером клона гранулоцитов PNH, варьирующим от 89 до 95%. При регистрации один пациент переключался с продолжающейся терапии экулизумабом, тогда как три ранее прервали лечение экулизумабом. Все пациенты имели уровень LDH, в 3 раза превышающий верхнюю границу нормы (ULN) при регистрации, который быстро падал и поддерживался на уровне меньше, чем 1,5× ULN на протяжении всего последующего периода наблюдения (Фиг. 13). Для одного пациента требовались переливания после регистрации (12 единиц эритроцитов (RBC) на протяжении 6 месяцев); данный пациент имел лежащий в основе диагноз апластической анемии, и ему требовалось 198 единиц RBC за 12 месяцев до регистрации. Ни один из четырех пациентов на испытывал событий прорывного гемолиза (ВТН). У всех четырех пациентов достигалось полное ингибирование терминального комплемента при измерении посредством липосомного иммуноанализа (LIA). Уровни LIA варьировали от 32 до 42 U/мл на входе в исследование, падали до меньше 10 U/мл (нижний уровень количественного измерения) к суткам 2 и затем поддерживались. Аналогичным образом, свободный С5 поддерживался на уровне меньше 0,5 мкг/мл после недели 6 (сутки 43). Профиль безопасности данных пациентов был аналогичным для остальных участников. Было описано три серьезных нежелательных явления (SAE), ни одно из которых не было связано с исследуемым лечением. Один пациент имел два SAE: камни желчных протоков и желчекаменную болезнь. Второй пациент имел SAE в виде инфекции верхних дыхательных путей с госпитализацией, которая происходила через 20 месяцев и завершалась во время нахождения на лечении.
Заключения: кровалимаб достигал полного и устойчивого ингибирования терминального комплемента у пациентов с PNH с полиморфизмом Arg885. Таким образом, кровалимаб представляет собой многообещающее антитело против С5 для лечения и/или предупреждения у пациентов, страдающих от PNH, где данные пациенты отличаются наличием мутации Arg885His С5.
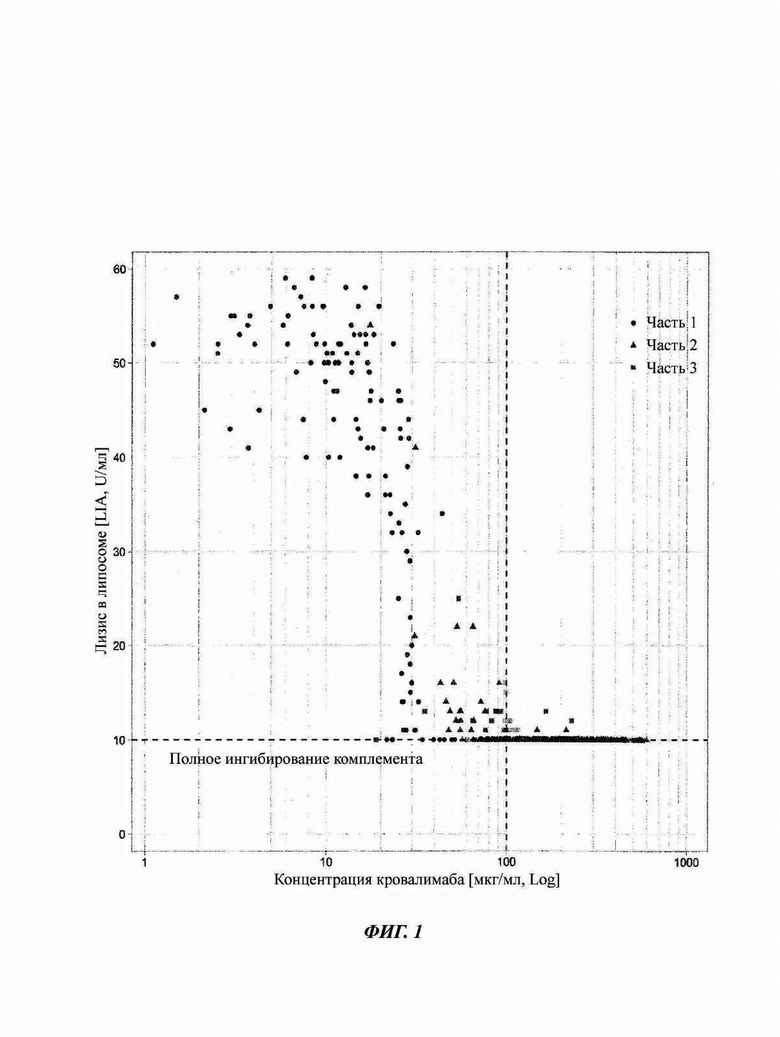
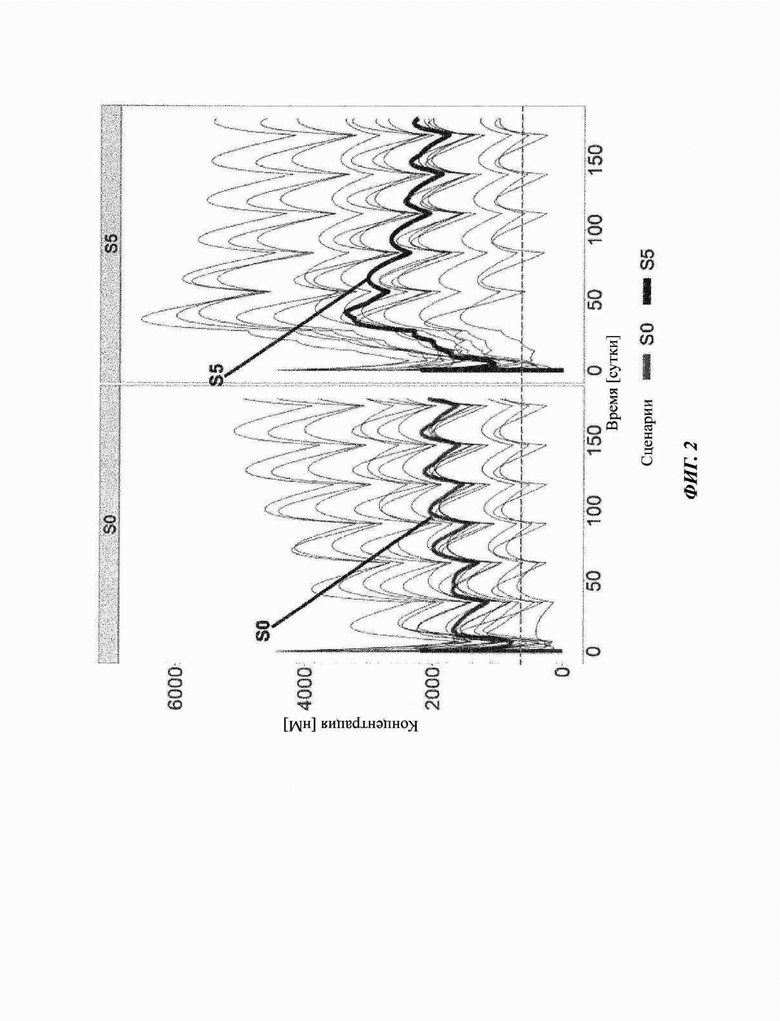
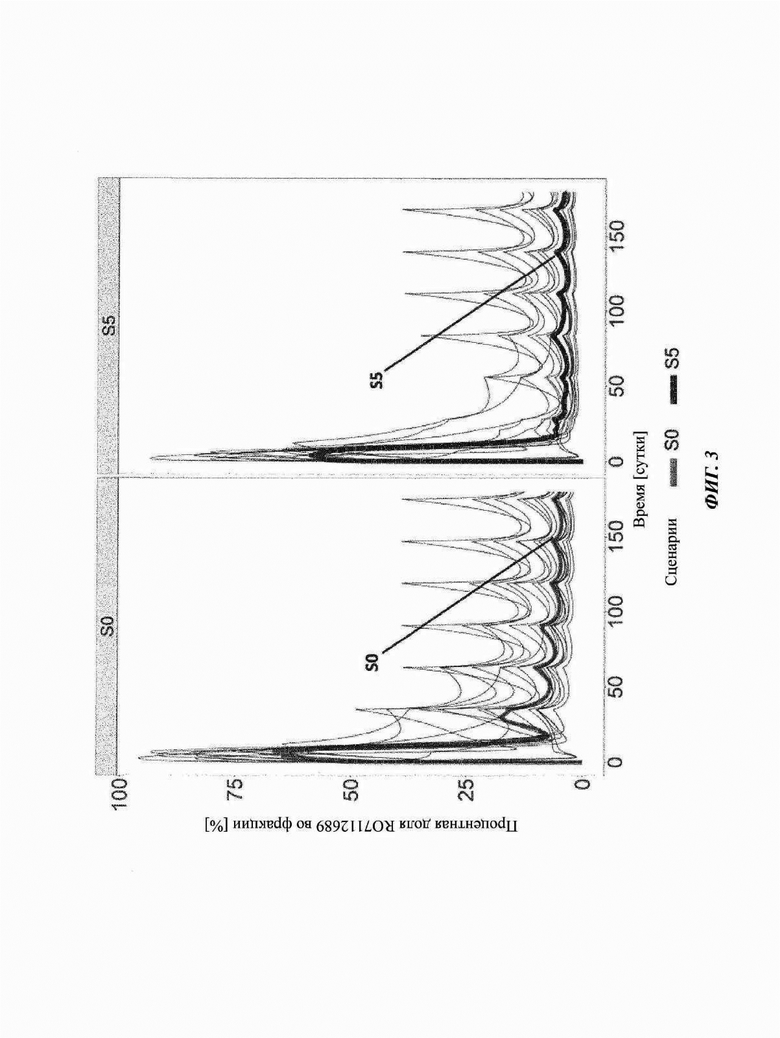
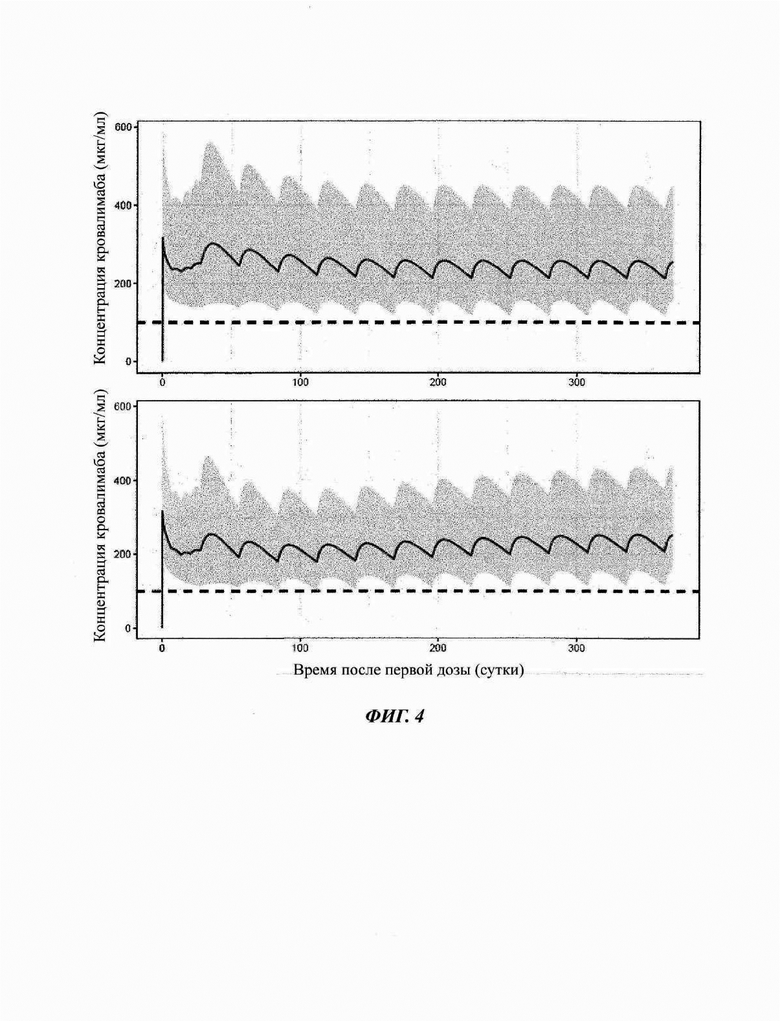
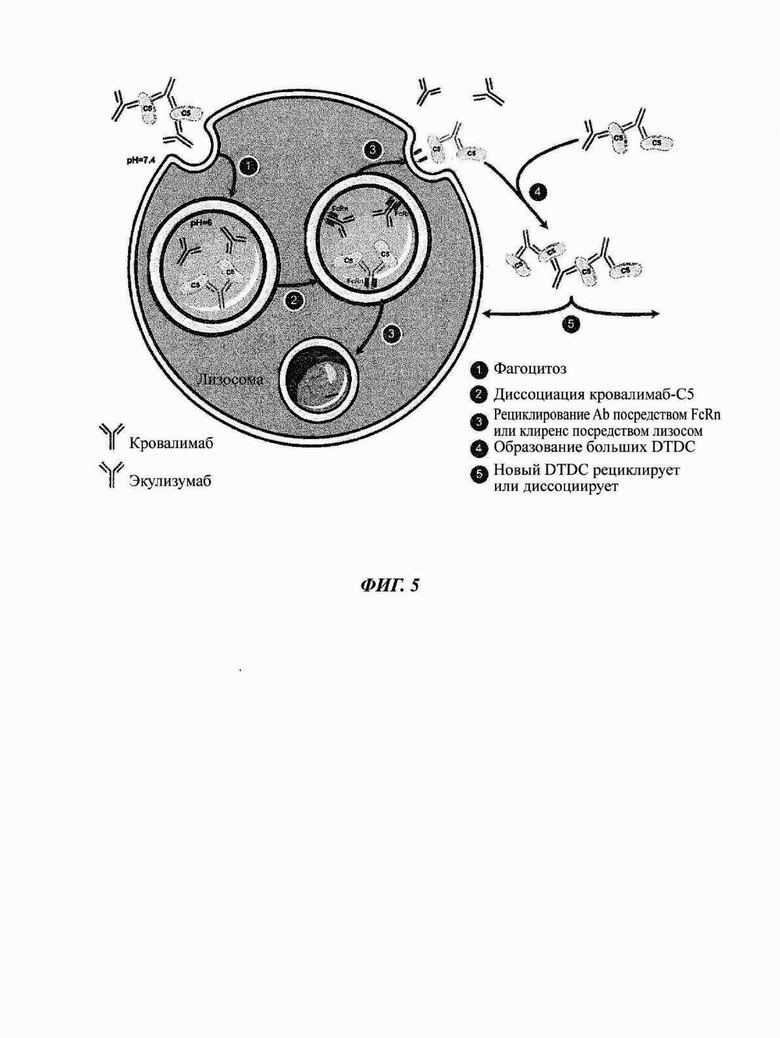
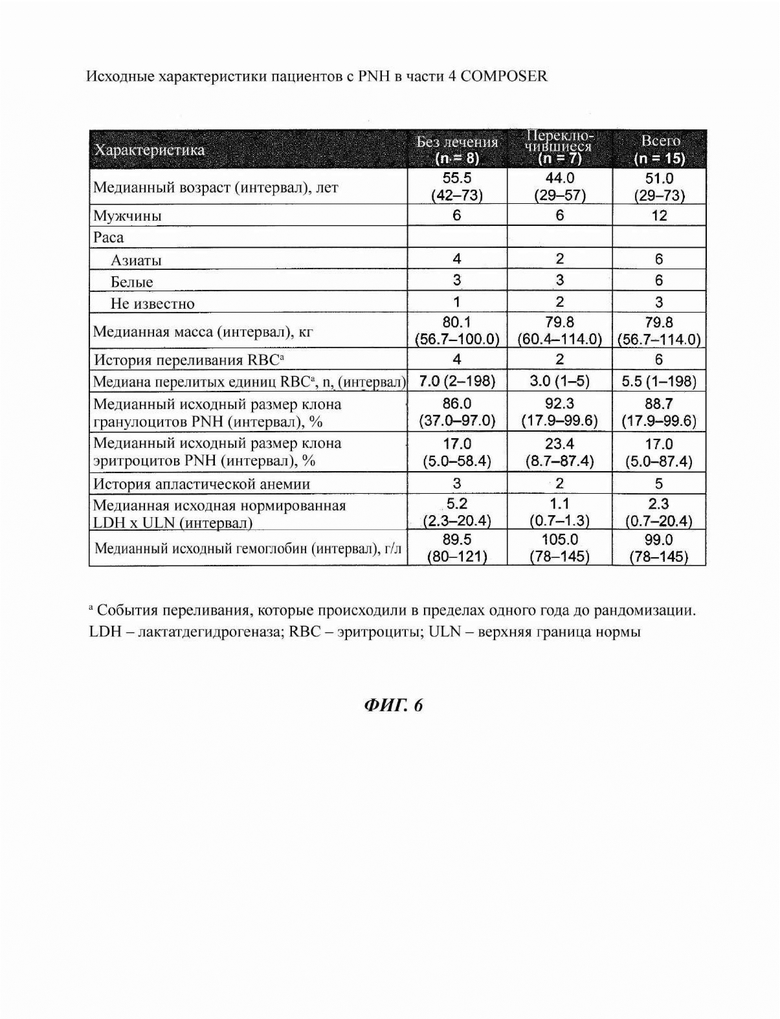
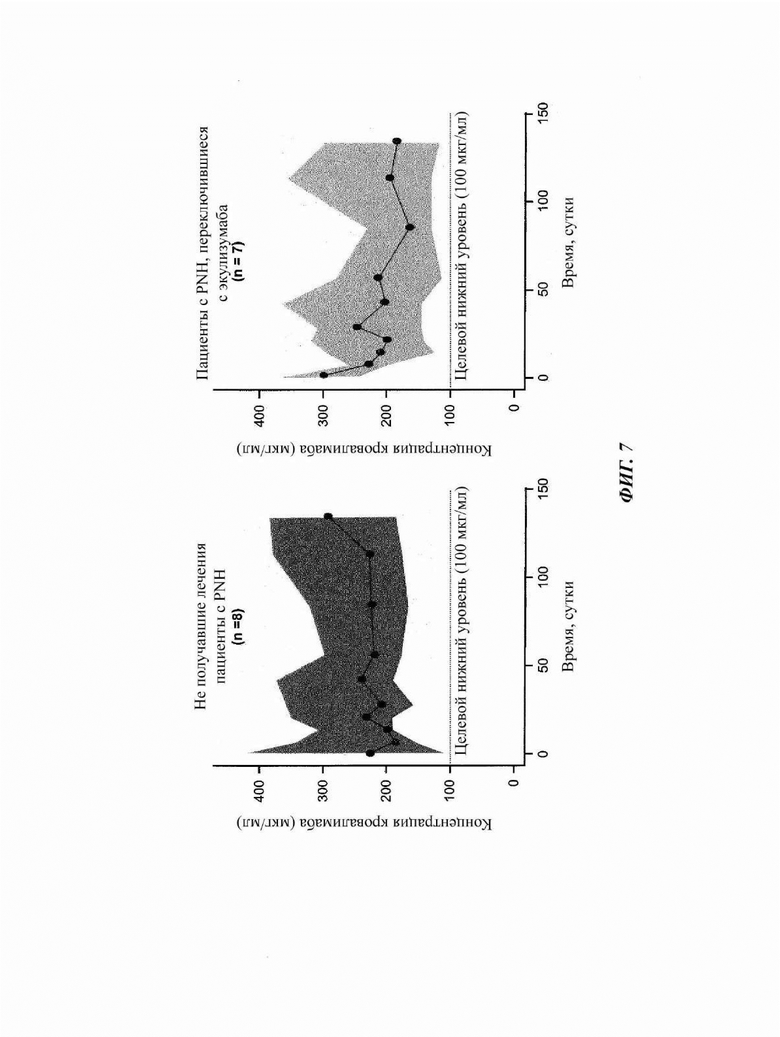
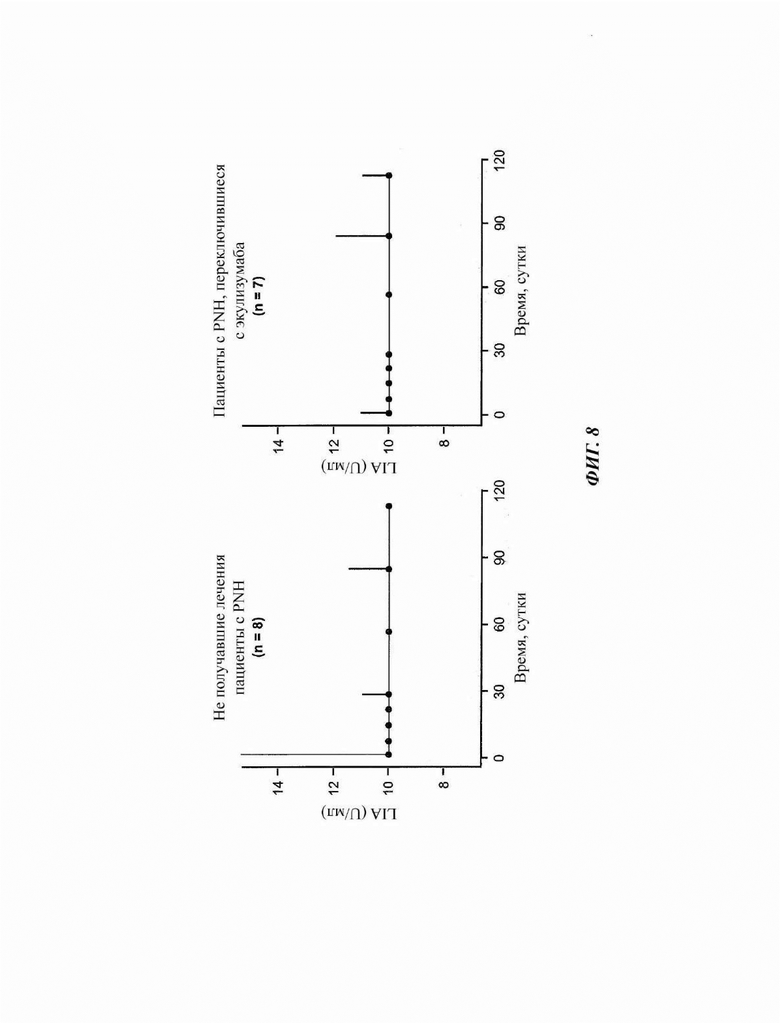
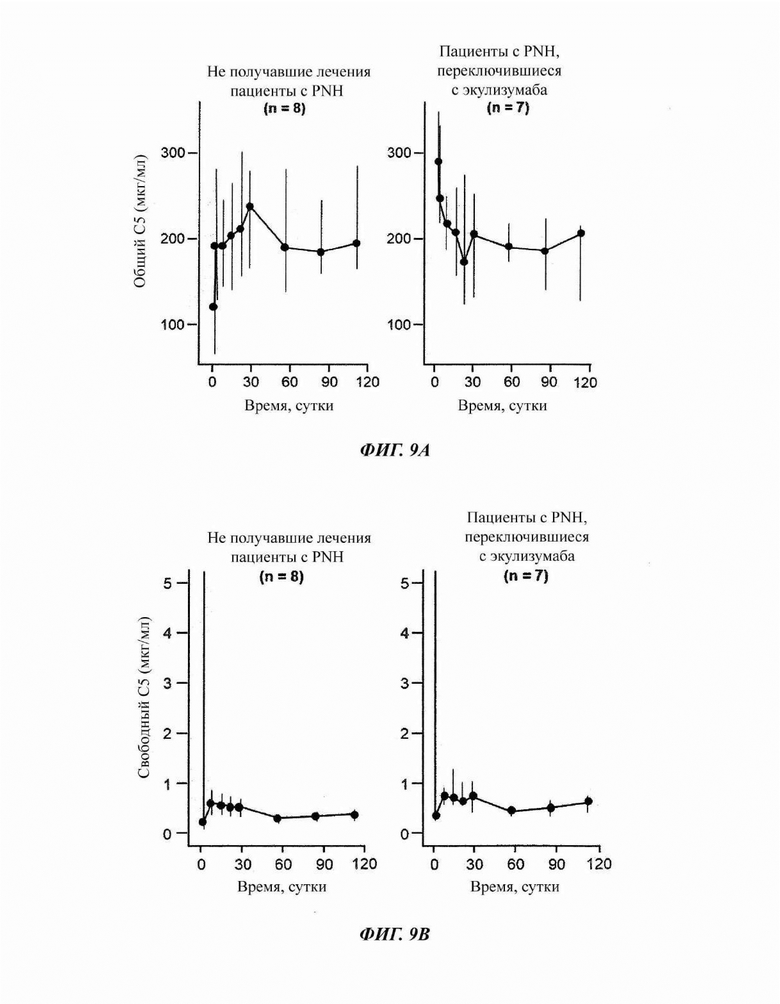
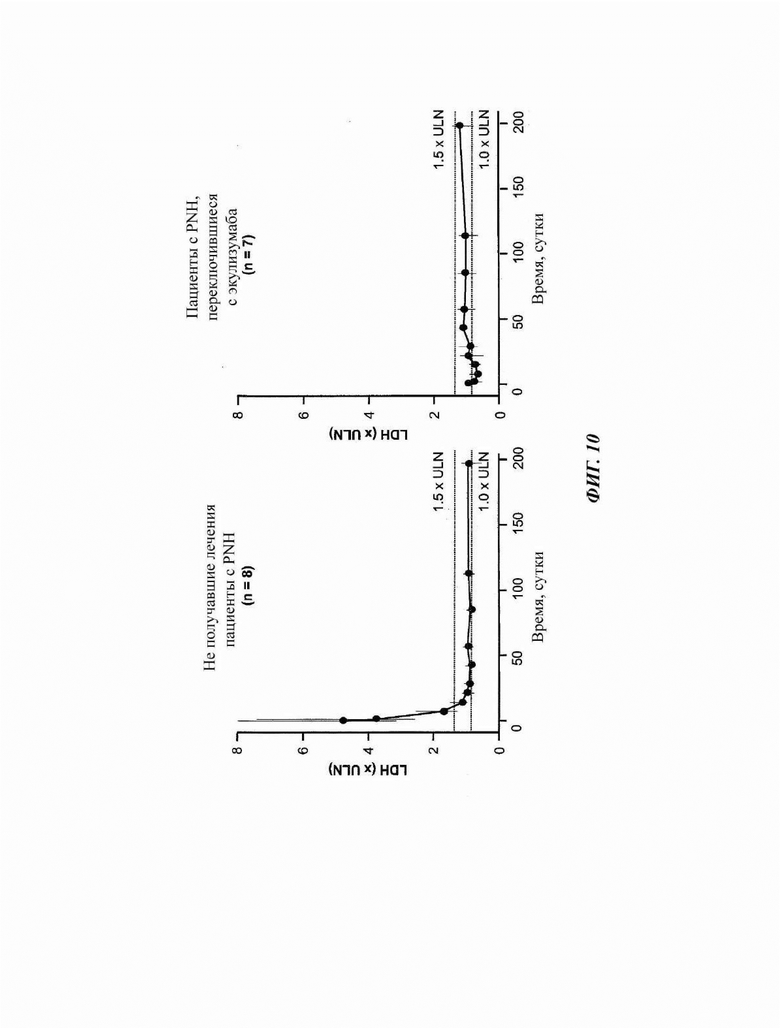

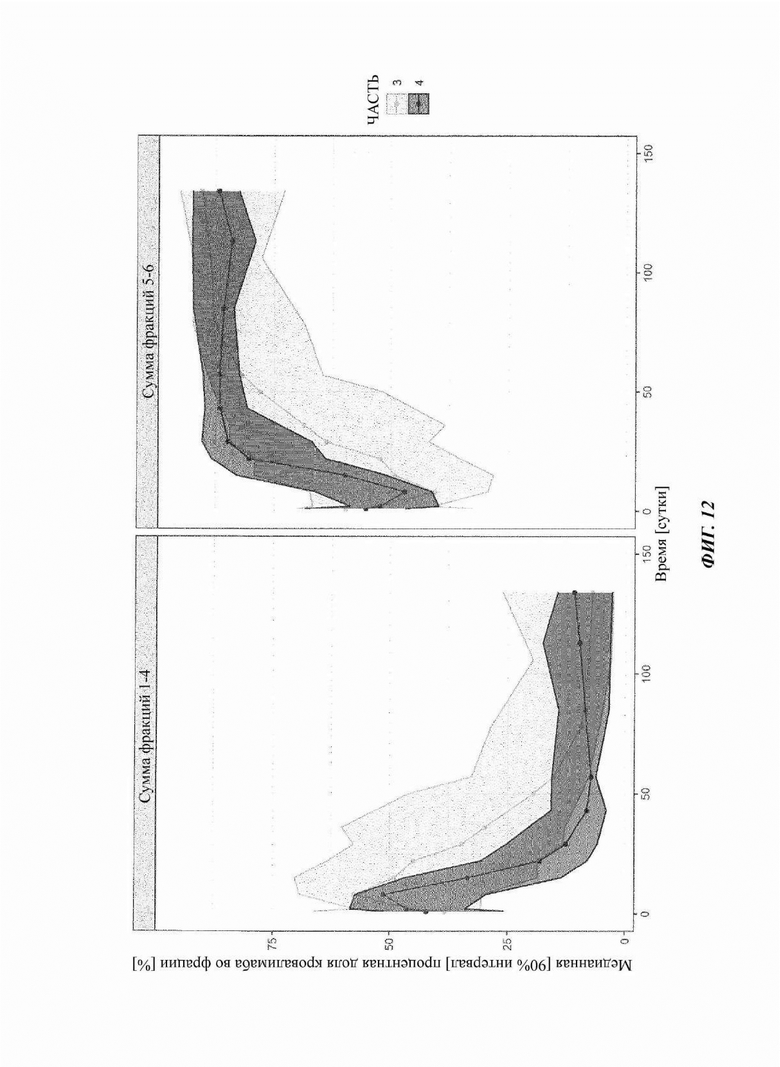
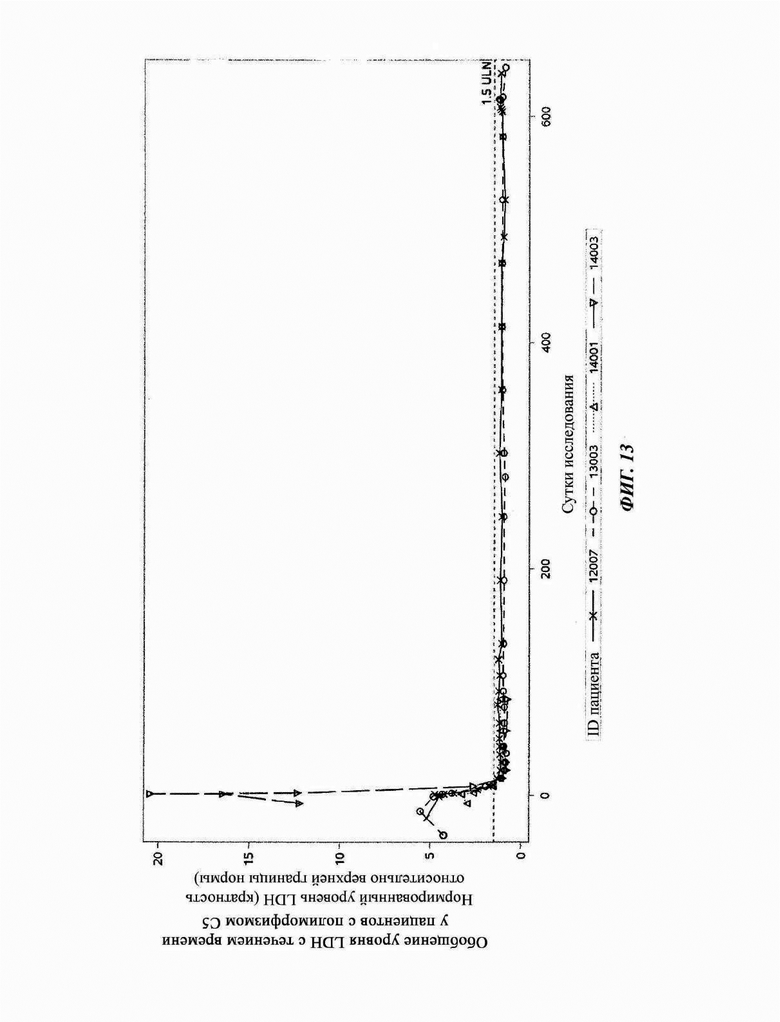
Изобретение относится к медицине. Раскрыт способ лечения или предупреждения связанного с С5 заболевания у субъекта, причем указанный способ проводят посредством следующих стадий введения: (i) внутривенное введение ударной дозы 1000 мг антитела против С5 субъекту один раз; (ii) подкожное введение ударной дозы 340 мг антитела против С5 субъекту через 1 сутки после начала внутривенного введения антитела против С5; (iii) подкожное введение ударной дозы 340 мг антитела против С5 субъекту через 1 неделю, 2 недели и 3 недели после начала внутривенного введения антитела против С5 один раз в неделю; (iv) подкожное введение поддерживающей дозы 680 мг антитела против С5 субъекту через 4 недели после начала внутривенного введения антитела против С5; и (v) повторение стадии (iv) несколько раз с интервалами времени 4 недели, где указанное антитело против С5 представляет собой кровалимаб, а указанное С5 заболевание представляет собой пароксизмальную ночную гемоглобинурию (PNH). Изобретение обеспечивает длительную и согласованную блокаду активности терминального комплемента, достаточный резерв свободных сайтов связывания для большей части интервала дозирования, безопасность и терапевтическую эффективность. 8 з.п. ф-лы, 13 ил., 1 табл., 7 пр.
1. Способ лечения или предупреждения связанного с С5 заболевания у субъекта, причем указанный способ проводят посредством следующих стадий введения:
(i) внутривенное введение ударной дозы 1000 мг антитела против С5 субъекту один раз;
(ii) подкожное введение ударной дозы 340 мг антитела против С5 субъекту через 1 сутки после начала внутривенного введения антитела против С5;
(iii) подкожное введение ударной дозы 340 мг антитела против С5 субъекту через 1 неделю, 2 недели и 3 недели после начала внутривенного введения антитела против С5 один раз в неделю;
(iv) подкожное введение поддерживающей дозы 680 мг антитела против С5 субъекту через 4 недели после начала внутривенного введения антитела против С5; и
(v) повторение стадии (iv) несколько раз с интервалами времени 4 недели,
где указанное антитело против С5 представляет собой кровалимаб, а указанное С5 заболевание представляет собой пароксизмальную ночную гемоглобинурию (PNH).
2. Способ по п. 1, где указанный субъект получал предшествующее лечение по меньшей мере одним фармакологическим продуктом, пригодным для лечения или предупреждения связанного с С5 заболевания, где вводимую внутривенно ударную дозу 1000 мг антитела против С5 вводят субъекту после последней дозы фармакологического продукта.
3. Способ по п. 2, где внутривенно вводимую ударную дозу 1000 мг антитела против С5 вводят субъекту на третьи сутки или через 3 суток после введения последней дозы фармакологического продукта.
4. Способ по п. 2 или 3, где указанный фармакологический продукт содержит миРНК, нацеленную на мРНК С5, или антитело против С5, которое отличается от антитела против С5, содержащегося в композиции для подкожной или внутривенной инъекции.
5. Способ по любому из пп. 2-4, где указанный фармакологический продукт содержит экулизумаб, равулизумаб или их варианты.
6. Способ по любому из пп. 1-5, где указанный субъект имеет массу тела от 40 до 100 кг.
7. Способ по любому из пп. 1-6, где концентрация антитела против С5, определенная в биологическом образце указанного субъекта, составляет 100 мкг/мл или более.
8. Способ по любому из пп. 1-6, где гемолитическая активность, определенная в биологическом образце указанного субъекта, составляет менее 10 U/мл.
9. Способ по п. 7 или 8, где указанный биологический образец представляет собой образец крови, предпочтительно образец красной крови.
| RISITANO A.M | |||
| et al | |||
| Anti-complement Treatment for Paroxysmal Nocturnal Hemoglobinuria: Time for Proximal Complement Inhibition? A Position Paper From the SAAWP of the EBMT // Front Immunol, 14.06.2019, V | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 2016098356 A1, 23.06.2016 | |||
| International Nonproprietary Names for Pharmaceutical Substances // WHO Drug Information, | |||

