УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Система комплемента действует вместе с другими иммунологическими системами организма, защищая от вторжения клеточных и вирусных патогенов. Существует по меньшей мере 25 белков комплемента, которые представляют собой сложную совокупность белков плазмы крови и мембранных кофакторов. Белки плазмы крови составляют около 10% глобулинов в сыворотке крови позвоночных. Компоненты комплемента выполняют свои иммунные защитные функции путем взаимодействия в серии сложных, но точных процессов ферментативного расщепления и связывания с мембраной. Возникающий в результате каскад комплемента приводит к продукции продуктов с опсонической, иммунорегуляторной и литической функциями. Краткое изложение биологической активности, связанной с активацией комплемента, представлено, например, в The Merck Manual, 16th Edition.
В то время как правильно функционирующая система комплемента обеспечивает надежную защиту от инфицирующих микробов, было выявлено, что ненадлежащая регуляция или активация путей комплемента вовлечены в патогенез различных нарушений, включая атипичный гемолитико-уремический синдром (аГУС). аГУС представляет собой крайне редкое нарушение, обусловленное хронической неконтролируемой активацией комплемента. Возникающее в результате воспаление и повреждение клеток приводят к разрушительным клиническим проявлениям данного заболевания.
Гемолитико-уремический синдром (ГУС) характеризуется тромбоцитопенией, микроангиопатической гемолитической анемией и острой почечной недостаточностью. Классифицируют два типа ГУС: связанный с диареей (Д+ГУС; также называемый ГУС с продукцией шигатоксина Е. coli (STEC)-ГУС или типичный ГУС) и бездиарейный или атипичный ГУС (аГУС). Д+ГУС является наиболее распространенной формой, на которую приходится более 90% случаев, и вызывается он предшествующим заболеванием бактерией, продуцирующей шигаподобный токсин, например, Е coli О157:Н7.
аГУС может быть наследственным, приобретенным или идиопатическим. Наследуемые формы аГУС могут быть связаны с мутациями в ряде компонентов комплемента человека, включая, например, фактор Н комплемента (CFH), мембранный кофакторный белок (МСР), фактор I комплемента (CFI), C4b-связывающий белок (С4ВР), фактор В комплемента (CFB) и компонент 3 комплемента (С3). См., например, Caprioli et al. (2006) Blood 108:1267-1279. Определенные мутации в гене, кодирующем CD55, хотя они еще не были охарактеризованы как связанные с аГУС, связаны с тяжестью аГУС. См., например, Esparza-Gordillo et al. (2005) Hum Mot Genet 14:703-712.
аГУС встречается редко и имеет смертность до 25%. У многих пациентов с данным заболеванием сохраняется постоянная неврологическая или почечная недостаточность, например, у по меньшей мере 50% пациентов с аГУС почечная недостаточность прогрессирует до терминальной стадии хронической почечной недостаточности (ТХПН). См., например, Kavanagh et al. (2006) British Medical Bulletin 77 and 78:5-22. До недавнего времени возможности лечения пациентов с аГУС были ограничены и часто включали в себя инфузию плазмы или плазмообмен. В некоторых случаях пациенты с аГУС подвергаются одно- или двусторонней нефрэктомии или трансплантации почки (см. Artz et al. (2003) Transplantation 76:821-826). Все же рецидивы данного заболевания у пролеченных пациентов встречаются часто.
Пациенты с аГУС подвержены значительному риску морбидности и смертности. Соответственно, целью данного изобретения является обеспечение улучшенных способов лечения пациентов с аГУС.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В данном документе представлены композиции и способы лечения аГУС у пациента-человека (например, взрослого пациента в возрасте 18 лет и старше), включающие в себя введение указанному пациенту антитела против С5 или его антигенсвязывающего фрагмента, при этом указанное антитело против С5 или его антигенсвязывающий фрагмент вводят (или предназначены для введения) в соответствии с конкретной клинической схемой применения (т.е.. в конкретном количестве дозы и в соответствии с конкретным графиком введения доз).
В способах, описанных в данном документе, могут применяться любое подходящее антитело против С5 или его антигенсвязывающий фрагмент. Иллюстративное антитело против С5 представляет собой равулизумаб (также известный как ULTOMIRIS®, ALXN1210 и антитело BNJ441), содержащий тяжелую и легкую цепи, имеющие последовательности, приведенные в SEQ ID NO: 14 и 11, соответственно, или их антигенсвязывающие фрагменты и варианты. В других вариантах осуществления антитело содержит определяющие комплементарность области тяжелой и легкой цепей (CDR) или вариабельные области (VR) равулизумаба. Соответственно, в одном варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 вариабельной области тяжелой цепи (VH) равулизумаба, имеющей последовательность, приведенную в SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 вариабельной области легкой цепи (VL) равулизумаба, имеющей последовательность, приведенную в SEQ ID NO: 8. В другом варианте осуществления антитело антитело содержит последовательности CDR1, CDR2 и CDR3 тяжелой цепи, указанные SEQ ID NO: 19, 18 и 3, соответственно, и последовательности CDR1, CDR2 и CDR3 легкой цепи, указанные SEQ ID NO: 4, 5 и 6, соответственно. В другом варианте осуществления антитело содержит области VH и VL, имеющие аминокислотные последовательности, указанные в SEQ ID NO: 12 и SEQ ID NO: 8, соответственно. В другом варианте осуществления антитело содержит константную область тяжелой цепи, указанную в SEQ ID NO: 13.
В другом варианте осуществления антитело содержит вариантную константную область Fc человека, которая связывается с неонатальным Fc-рецептором человека (FcRn), при этом вариантная константная область СН3 Fc человека содержит замены Met-429-Leu и Asn-435-Ser в остатках, соответствующих метионину 428 и аспарагину 434 константной области Fc нативного IgG человека, каждый остаток пронумерован согласно нумерации ЕС.
В другом варианте осуществления антитело содержит последовательности CDR1, CDR2 и CDR3 тяжелой цепи, указанные SEQ ID NO: 19, 18 и 3, соответственно, и последовательности CDR1, CDR2 и CDR3 легкой цепи, указанные SEQ ID NO: 4, 5 и 6, соответственно, и вариантную константная область Fc человека, которая связывается с неонатальным Fc-рецептором человека (FcRn), при этом вариантная константная область СН3 Fc человека содержит замены Met-429-Leu и Asn-435-Ser в остатках, соответствующих метионину 428 и аспарагину 434 константной области Fc нативного IgG человека, каждый остаток пронумерован согласно нумерации ЕС.
В другом варианте осуществления антитело связывается с С5 человека при рН 7,4 и 25°С с аффинной константой диссоциации (KD), которая находится в диапазоне от 0,1 нМ до 1 нМ. В другом варианте осуществления антитело связывается с С5 человека при рН 6,0 и 25°С с KD ≥ 10 нМ. В еще одном варианте осуществления показатель антитела [(KD антитела или его антигенсвязывающего фрагмента для С5 человека при рН 6,0 и при 25°C)/(KD антитела или его антигенсвязывающего фрагмента для С5 человека при рН 7,4 и при 25°С)] составляет больше чем 25.
Другое иллюстративное антитело против С5 представляет собой антитело 7086, описанное в патентах США №8241628 и №8883158. В одном варианте осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области антитела 7086 (см. патенты США №8241628 и №8883158). В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 21, 22 и 23, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 24, 25 и 26, соответственно. В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат область VH антитела 7086, имеющую последовательность, указанную в SEQ ID NO: 27, и область VL антитела 7086, имеющую последовательность, указанную в SEQ ID NO: 28.
Другое иллюстративное антитело против С5 представляет собой антитело 8110, также описанное в патентах США №8241628 и №8883158. В одном варианте осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области антитела 8110. В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 29, 30 и 31, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 32, 33 и 34, соответственно. В другом варианте осуществления антитело содержит область VH антитела 8110, имеющую последовательность, указанную в SEQ ID NO: 35, и область VL антитела 8110, имеющую последовательность, указанную в SEQ ID NO: 36.
Другое иллюстративное антитело против С5 представляет собой антитело 305LO5, описанное в US 2016/0176954 A1. В одном варианте осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области антитела 305LO5. В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 37, 38 и 39, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 40, 41 и 42, соответственно. В другом варианте осуществления антитело содержит область VH антитела 305LO5, имеющую последовательность, указанную в SEQ ID NO: 43, и область VL антитела 305LO5, имеющую последовательность, указанную в SEQ ID NO: 44.
Другое иллюстративное антитело против С5 представляет собой антитело SKY59, описанное в Fukuzawa Т., et at, Rep.2017 Apr 24;7(1): 1080). В одном варианте осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области антитела SKY59. В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат тяжелую цепь, содержащую SEQ ID NO: 45, и легкую цепь, содержащую SEQ ID NO: 46.
Другое иллюстративное антитело против С5 представляет собой антитело REGN3918 (также известное как Н4Н12166РР), описанное в US 20170355757. В одном варианте осуществления антитело содержит вариабельную область тяжелой цепи, содержащую SEQ ID NO: 47, и вариабельную область легкой цепи, содержащую SEQ ID NO: 48. В другом варианте осуществления антитело содержит тяжелую цепь, содержащую SEQ ID NO: 49, и легкую цепь, содержащую SEQ ID NO: 50.
В другом варианте осуществления антитело конкурирует за связывание и/или связывается с одним и тем же эпитопом на С5, что и указанные выше антитела (например, экулизумаб, равулизумаб, антитело 7086, антитело 8110, антитело 305LO5, антитело SKY59 или антитело REGN3918). В другом варианте осуществления антитело имеет по меньшей мере около 90% идентичности аминокислотной последовательности вариабельной области с вышеупомянутыми антителами (например, по меньшей мере около 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% идентичности вариабельной области).
В одном варианте осуществления доза антитела против С5 или его антигенсвязывающего фрагмента основана на массе тела пациента. Например, в одном варианте осуществления, 2400 мг или 3000 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела от ≥ 40 до < 60 кг. В другом варианте осуществления 2700 мг или 3300 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела от ≥ 60 до < 100 кг. В другом варианте осуществления 3000 мг или 3600 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела ≥ 100 кг. В определенных вариантах осуществления схемы применения подбираются так, чтобы обеспечить оптимальный желаемый ответ (например, эффективный ответ).
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят в одном или большем числе циклов введения. В одном варианте осуществления цикл введения составляет 26 недель. В одном варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят один раз на 1-е сутки цикла введения, один раз на 15-е сутки цикла введения и после этого - каждые восемь недель. В одном варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят каждые восемь недель после цикла введения во время периода продления, составляющего до двух лет включительно (например, в дозе 3000 мг, 3300 мг или 3600 мг).
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят в одном или большем числе циклов введения. В одном варианте осуществления цикл введения составляет 26 недель. В другом варианте осуществления лечение состоит из по меньшей мере 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11 циклов. В другом варианте осуществления лечение продолжается в течение всей жизни пациента- че л ов ека.
В другом варианте осуществления представлен способ лечения пациента-человека с аГУС, при этом указанный способ включает в себя введение пациенту (например, во время цикла введения) эффективного количества антитела против С5 или его антигенсвязывающего фрагмента, содержащих последовательности CDR1, CDR2 и CDR3 тяжелой цепи, указанные SEQ ID NO: 19, 18 и 3, соответственно, и последовательности CDR1, CDR2 и CDR3 легкой цепи, указанные SEQ ID NO: 4, 5 и 6, соответственно, при этом указанные антитело против С5 или его антигенсвязывающий фрагмент вводят:
(a) один раз на 1-е сутки в дозе, составляющей: 2400 мг - пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг - пациенту с массой тела ≥ 100 кг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей: 3000 мг - пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг - пациенту с массой тела ≥ 100 кг.
В другом варианте осуществления представлен способ лечения пациента-человека с аГУС, при этом указанный способ включает в себя введение пациенту (например, во время цикла введения) эффективного количества антитела против С5 или его антигенсвязывающего фрагмента, содержащих последовательности CDR1, CDR2 и CDR3 тяжелой цепи, указанные SEQ ID NO: 19, 18 и 3, соответственно, последовательности CDR1, CDR2 и CDR3 легкой цепи, указанные SEQ ID NO: 4, 5 и 6, соответственно, и вариантную константную область Fc человека, которая связывается с неонатальным Fc-рецептором человека (FcRn), при этом вариантная константная область СН3 Fc человека содержит замены Met-429-Leu и Asn-435-Ser в остатках, соответствующих метионину 428 и аспарагину 434 нативной константной области Fc IgG человека, каждый остаток пронумерован согласно нумерации ЕС, при этом указанные антитело против С5 или его антигенсвязывающий фрагмент вводят:
(a) один раз на 1-е сутки в дозе, составляющей: 2400 мг пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг - пациенту с массой тела ≥ 100 кг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей:
3000 мг - пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг - пациенту с массой тела ≥ 100 кг.
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела от ≥ 40 до < 60 кг:
(a) один раз на 1-е сутки в дозе, составляющей: 2400 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей:
3000 мг.
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела от ≥ 60 до < 100 кг:
(a) один раз на 1-е сутки в дозе, составляющей: 2700 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей:
3300 мг.
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела ≥ 100 кг:
(a) один раз на 1-е сутки в дозе, составляющей: 3000 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей:
3600 мг.
В некоторых вариантах осуществления пациент ранее не получал лечения ингибитором комплемента (например, пациент является таким, который ранее не проходил лечение ингибитором комплемента).
В других вариантах осуществления пациент ранее получал лечение одним антителом против С5 или его антигенсвязывающим фрагментом, а затем был переведен на другое антитело против С5 или его антигенсвязывающий фрагмент во время курса лечения. Например, в определенных вариантах осуществления вводят различные антитела против С5 во время курса лечения. В одном варианте осуществления различные антитела против С5 вводят во время отдельных периодов лечения и продления. Например, в одном варианте осуществления проводят лечение пациента экулизумабом во время периода лечения (например, в течение 26 недель), после чего проводят лечение другим антителом против С5 (например, равулизумабом, антителом 7086, антителом 8110, антителом 305LO5, антителом SKY59 или антителом REGN3918), например, во время периода продления. В другом варианте осуществления экулизумаб вводят пациенту в дозе 600 мг на 1-е, 8-е, 15-е и 22-е сутки цикла введения во время фазы индукции, после чего вводят поддерживающую дозу экулизумаба, составляющую 900 мг, в 19-е сутки цикла введения и каждые две недели после этого (например, в общей сложности на протяжении 26 недель), после чего проводят лечение равулизумабом в течение периода продления, составляющего до двух лет.В другом варианте осуществления проводят лечение пациента равулизумабом (например, в течение 26 недель), после чего проводят лечение другим антителом против С5 (например, экулизумабом, антителом 7086, антителом 8110, антителом 305LO5, антителом SKY59 или антителом REGN3918) в течение, например, периода продления.
Иллюстративные альтернативные антитела против С5 включают в себя, но не ограничиваются ими: (i) равулизумаб, (ii) антитело или его антигенсвязывающий фрагмент, содержащие домены CDR1, CDR2 и CDR3 тяжелой цепи, содержащие SEQ ID NO: 21, 22 и 23, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, содержащие SEQ ID NO: 24, 25 и 26, соответственно, (iii) антитело или его антигенсвязывающий фрагмент, содержащие вариабельную область тяжелой цепи, содержащую SEQ ID NO: 27, и вариабельную область легкой цепи, содержащую SEQ ID NO: 28, (iv) антитело или его антигенсвязывающий фрагмент, содержащие домены CDR1, CDR2 и CDR3 тяжелой цепи, содержащие SEQ ID NO: 29, 30 и 31, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, содержащие SEQ ID NO: 32, 33 и 34, соответственно, (v) антитело или его антигенсвязывающий фрагмент, содержащие вариабельную область тяжелой цепи, содержащую SEQ ID NO: 35, и вариабельную область легкой цепи, содержащую SEQ ID NO: 36, (vi) антитело или его антигенсвязывающий фрагмент, содержащие домены CDR1, CDR2 и CDR3 тяжелой цепи, содержащие SEQ ID NO: 37, 38 и 39, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, содержащие SEQ ID NO: 40, 41 и 42, соответственно, (vii) антитело или его антигенсвязывающий фрагмент, содержащие вариабельную область тяжелой цепи, содержащую SEQ ID NO: 43, и вариабельную область легкой цепи, содержащую SEQ ID NO: 44, (viii) антитело или его антигенсвязывающий фрагмент, содержащие тяжелую цепь, содержащую SEQ ID NO: 45, и легкую цепь, содержащую SEQ ID NO: 46, соответственно, (vii) антитело или его антигенсвязывающий фрагмент, содержащие вариабельную область тяжелой цепи, содержащую SEQ ID NO: 47, и вариабельную область легкой цепи, содержащую SEQ ID NO: 48, (х) антитело или его антигенсвязывающий фрагмент, содержащие тяжелую цепь, содержащую SEQ ID NO: 49, и легкую цепь, содержащую SEQ ID NO: 50.
В некоторых вариантах осуществления пациент ранее получал лечение в течение по меньшей мере 1 месяца, по меньшей мере 2 месяцев, по меньшей мере 3 месяцев, по меньшей мере 4 месяцев, по меньшей мере 5 месяцев, по меньшей мере 6 месяцев, по меньшей мере 7 месяцев, по меньшей мере 8 месяцев, по меньшей мере 9 месяцев, по меньшей мере 10 месяцев, по меньшей мере 11 месяцев, по меньшей мере 12 месяцев, по меньшей мере 18 месяцев или по меньшей мере 24 месяцев антителом против С5 или его антигенсвязывающим фрагментом (например, экулизумабом) до перехода на другое антитело против С5 или его антигенсвязывающий фрагмент (например, равулизумаб). В конкретном варианте осуществления пациент ранее получал лечение экулизумабом в течение по меньшей мере 6 месяцев.
В другом варианте осуществления, когда пациент (например, пациент с аГУС) получает лечение первым антителом против С5, а затем переводится на лечение вторым другим антителом против С5, особенно, когда указанное второе другое антитело против С5 связывается с эпитопом на С5, отличным от эпитопа, с которым связывается указанное первое антитело против С5, графики введения учитывают период полужизни указанного первого антитела против С5. Например, для обеспечения того, чтобы первое антитело против С5 было выведено (например, «вымыто») из пациента перед введением второго (другого) антитела против С5 (например, чтобы избежать проблем, связанных с агрегацией, образованием иммунных комплексов и т.д.), учитывают период полужизни указанного первого антитела против С5. В одном варианте осуществления второе (другое) антитело против С5 не вводят до тех пор, пока не пройдет период времени, соответствующий 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7 или 7,5 периодам полужизни первого антитела против С5 после заключительного введения указанного первого антитела против С5.
В другом варианте осуществления пациент ранее получал лечение экулизумабом, а затем был переведен на лечение вторым (другим) антителом против С5 (например, равулизумабом, антителом 7086, антителом 8110, антителом 305LO5, антителом SKY59 или антителом REGN3918). В одном варианте осуществления, когда первое вводимое антитело представляет собой экулизумаб, второе (другое) антитело против С5 не вводят, например, до тех пор, пока не пройдет по меньшей мере 36, 45, 54, 63, 72, 81, 90, 99, 108, 117 или 126 суток после заключительного введения экулизумаба.
В другом варианте осуществления пациент ранее получал лечение равулизумабом, а затем был переведен на лечение другим антителом против С5 (например, экулизумабом, антителом 7086, антителом 8110, антителом 305LO5, антителом SKY59 или антителом REGN3918). В одном варианте осуществления, когда первое вводимое антитело представляет собой равулизумаб, второе (другое) антитело против С5 не вводят, например, до тех пор, пока не пройдет по меньшей мере 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 375 или 400 суток после заключительного введения равулизумаба.
В качестве дополнения или альтернативы, используются методики для выведения или усиления клиренса первого антитела против С5 до перехода на лечение вторым (другим) антителом против С5. Иллюстративные методики включают в себя, но не ограничиваются ими, плазмаферез или гемотрансфузии. В другом варианте осуществления антитело против первого антитела против С5 (например, антитело против экулизумаба, антитело против равулизумаба, антитело против 7086, антитело против 8110, антитело против 305LO5, антитело против SKY59 или антитело против REGN3918) вводят для выведения или усиления клиренса указанного первого антитела против С5 до введения второго (другого) антитела против С5.
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент (например, равулизумаб) вводят пациенту, при этом лечение (например, цикл введения) начинается через по меньшей мере около две недели, по меньшей мере около три недели, по меньшей мере около четыре недели, по меньшей мере около шесть недель, по меньшей мере около семь недель или по меньшей мере около восемь недель после введения указанному пациенту заключительной дозы экулизумаба. В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент (например, равулизумаб) вводят пациенту, при этом лечение (например, цикл введения) начинается через по меньшей мере две недели после введения указанному пациенту заключительной дозы экулизумаба.
В некоторых вариантах осуществления пациенты, получавшие лечение в соответствии с описанными в данном документе способами, были вакцинированы против менингококковых инфекций в течение 3 лет до или во время начала лечения. В одном варианте осуществления пациенты, которые получали лечение меньше чем через 2 недели после введения менингококковой вакцины, также получают соответствующие профилактические антибиотики в течение 2 недель после вакцинации. В еще одном варианте осуществления пациенты, получающие лечение в соответствии с описанными в данном документе способами, вакцинированы против серотипов менингококка А, С, Y, W135 и/или В.
В другом аспекте схемы лечения, описанные в данном документе, достаточны для поддержания определенных остаточных концентраций антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови. Например, в одном варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 200, 205, 210, 215, 220, 225, 230, 240, 245, 250, 255, 260, 265, 270, 280, 290, 300, 305, 310, 315, 320, 325, 330, 335, 340, 345, 350, 355, 360, 365, 370, 375, 380, 385, 390, 395 или 400 мкг/мл или больше во время лечения. В одном варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 100 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 150 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 200 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 250 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 300 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне от 100 мкг/мл до 200 мкг/мл. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне около 175 мкг/мл.
В другом варианте осуществления для получения эффективного ответа антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание по меньшей мере 50 мкг, 55 мкг, 60 мкг, 65 мкг, 70 мкг, 75 мкг, 80 мкг, 85 мкг, 90 мкг, 95 мкг, 100 мкг, 105 мкг, 110 мкг, 115 мкг, 120 мкг, 125 мкг, 130 мкг, 135 мкг, 140 мкг, 145 мкг, 150 мкг, 155 мкг, 160 мкг, 165 мкг, 170 мкг, 175 мкг, 180 мкг, 185 мкг, 190 мкг 195 мкг, 200 мкг, 205 мкг, 210 мкг, 215 мкг, 220 мкг, 225 мкг, 230 мкг, 235 мкг, 240 мкг, 245 мкг, 250 мкг, 255 мкг или 260 мкг антитела на миллилитр крови пациента. В другом варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание от 50 мкг до 250 мкг антитела на миллилитр крови пациента. В другом варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание от 100 мкг до 200 мкг антитела на миллилитр крови пациента. В другом варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание около 175 мкг антитела на миллилитр крови пациента.
В другом варианте осуществления для получения эффективного ответа пациенту вводят антитело против С5 в количестве и с частотой, обеспечивающими минимальную концентрацию свободного С5. Например, в одном варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание концентрации свободного С5 на уровне 0,2 мкг/мл, 0,3 мкг/мл, 0,4 мкг/мл, 0,5 мкг/мл или ниже. В другом варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание концентрации свободного С5 на уровне от 0,309 мкг/мл до 0,5 мкг/мл или ниже. В другом варианте осуществления описанное в данном документе лечение снижает концентрацию свободного С5 больше чем на 99% в течение периода лечения. В другом варианте осуществления указанное лечение снижает концентрацию свободного С5 больше чем на 99,5% в течение периода лечения.
Антитела против С5 или их антигенсвязывающие фрагменты могут вводиться пациенту любыми подходящими средствами. В одном варианте осуществления антитела сформулированы для внутривенного введения.
Эффективность способов лечения, представленных в данном документе, можно оценить с помощью любых подходящих средств. В одном варианте осуществления для пациента с аГУС лечение вызывает по меньшей мере один терапевтический эффект, выбранный из группы, состоящей из снижения или прекращения тяжелой гипертензии, протеинурии, уремии, вялости/усталости, раздражительности, тромбоцитопении, микроангиопатической гемолитической анемии и нарушения функции почек (например, острой почечной недостаточности).
В других вариантах осуществления лечение приводит к ингибированию терминального комплемента.
В других вариантах осуществления лечение вызывает сдвиг в сторону нормальных уровней связанного с гемолизом гематологического биомаркера, выбранного из группы, состоящей из свободного гемоглобина, гаптоглобина, количества ретикулоцитов, ПНГ-клона эритроцитов и D-димера.
В другом варианте осуществления лечение вызывает повышение стабилизации гемоглобина по сравнению с исходным уровнем у пациента до лечения. В другом варианте осуществления лечение приводит к повышению уровня гемоглобина на ≥ 20 г/л. В другом варианте осуществления лечение приводит к предотвращению снижения уровня гемоглобина на ≥ 2 г/дл по сравнению с исходным уровнем при отсутствии трансфузии с момента определения исходного уровня к 183-м суткам.
В других вариантах осуществления лечение приводит к нормализации уровня тромбоцитов (≥ 150 × 109/л). В других вариантах осуществления лечение приводит к нормализации уровня тромбоцитов (≥ 150 × 109/л) на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение приводит к нормализации уровня ЛДГ (≤246 Ед/л). В других вариантах осуществления лечение приводит к нормализации уровня ЛДГ (≤246 Ед/л) на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение приводит к улучшению уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем. В других вариантах осуществления лечение приводит к улучшению уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение приводит к полному ответу ТМА (т.е. нормализации уровня тромбоцитов (≥ 150 × 109/л), нормализации ЛДГ (≤246 Ед/л) и улучшению уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем). В других вариантах осуществления лечение приводит к полному ответу ТМА на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение приводит к модифицированному полному ответу ТМА (т.е. нормализации уровня тромбоцитов (≥ 150 × 109/л), нормализации уровня ЛДГ (≤246 Ед/л), и пациент прекращает диализ, если он находился на диализе в исходном состоянии, или улучшению уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем для пациента, который не находился на диализе в исходном состоянии). В других вариантах осуществления лечение приводит к модифицированному полному ответу ТМА на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение снижает потребность в гемотрансфузиях. В другом варианте осуществления лечение приводит к повышению отмены трансфузий больше чем на 70%. В другом варианте осуществления лечение приводит к отмене трансфузии в период с момента определения исходного уровня до 183-х суток.
В других вариантах осуществления лечение приводит к устранению прорывного гемолиза во время периода лечения. В другом варианте осуществления лечение приводит к снижению прорывного гемолиза по сравнению с исходным уровнем прорывного гемолиза до лечения.
В других вариантах осуществления лечение приводит к снижению значительных неблагоприятных сосудистых событий (MAVE).
В других вариантах осуществления лечение вызывает изменение качества жизни по сравнению с исходным уровнем, оцениваемое с помощью шкалы функциональной оценки терапии хронических заболеваний (FACIT) для показателя утомляемости версии 4 и опросника Европейской организации по изучению и лечению злокачественных опухолей для оценки качества жизни из 30 вопросов. В одном варианте осуществления лечение вызывает изменение качества жизни по сравнению с исходным уровнем, оцениваемое по шкале FACIT для показателя утомляемости, на один или большее число (например, 1, 2 или 3) баллов. В другом варианте осуществления лечение вызывает изменение качества жизни по сравнению с исходным уровнем, оцениваемое по шкале FACIT для показателя утомляемости, на 3 балла через 150 суток или через большее число суток (, например, 150 суток, 151 сутки, 152 суток, 153 суток, 154 суток, 155 суток, 156 суток, 157 суток, 158 суток, 159 суток, 160 суток, 161 сутки, 162 суток, 163 суток, 164 суток, 165 суток, 166 суток, 167 суток, 168 суток, 169 суток, 170 суток, 171 сутки, 172 суток, 173 суток, 174 суток, 175 суток, 176 суток, 177 суток, 178 суток, 179 суток, 180 суток, 181 сутки, 182 суток 183 суток, 184 суток, 185 суток, 186 суток, 187 суток, 188 суток, 189 суток, 190 суток, 191 сутки, 192 суток, 193 суток, 194 суток, 195 суток, 196 суток, 197 суток, 198 суток, 199 суток, 200 суток, 205 суток, 210 суток, 215 суток, 220 суток или 225 суток) после начала лечения.
Стадия хронической болезни почек (ХБП) классифицируется на основе стадий хронической болезни почек, определенных Национальным фондом почки США. Стадии ХБП и соответствующие значения расчетной скорости клубочковой фильтрации (рСКФ) следующие: Стадия 1: рСКФ >= 90 (норма), Стадия 2: рСКФ = 60-89, Стадия 3А: рСКФ = 45-59, Стадия 3В: рСКФ = 30-44, Стадия 4: рСКФ = 15-29 и стадия 5: рСКФ < 15 (включая диализ: Конечная стадия). Стадия 1 считается наилучшей категорией. Стадия 5 считается наихудшей категорией. Улучшение рСКФ (например, > 15) соответствует улучшению стадии ХБП (например, более низкой стадии ХБП). Соответственно, в других вариантах осуществления хроническая болезнь почек (ХБП) пациента улучшается на одну или большее число стадий после начала лечения. Например, ХБП пациента улучшается на одну, две, три, четыре или пять стадий). В другом варианте осуществления ХБП пациента улучшается на одну или большее число стадий через 150 суток или через большее число суток (например, 150 суток, 151 сутки, 152 суток, 153 суток, 154 суток, 155 суток, 156 суток, 157 суток, 158 суток, 159 суток, 160 суток, 161 сутки, 162 суток, 163 суток, 164 суток, 165 суток, 166 суток, 167 суток, 168 суток, 169 суток, 170 суток, 171 сутки, 172 суток, 173 суток, 174 суток, 175 суток, 176 суток, 177 суток, 178 суток, 179 суток, 180 суток, 181 сутки, 182 суток 183 суток, 184 суток, 185 суток, 186 суток, 187 суток, 188 суток, 189 суток, 190 суток, 191 сутки, 192 суток, 193 суток, 194 суток, 195 суток, 196 суток, 197 суток, 198 суток, 199 суток, 200 суток, 205 суток, 210 суток, 215 суток, 220 суток или 225 суток) после начала лечения.
В других вариантах осуществления лечение приводит к повышению рСКФ по сравнению с исходным уровнем. В других вариантах осуществления лечение приводит к сдвигу в сторону нормальных уровней рСКФ (например, ≥ 90). В других вариантах осуществления лечение приводит к повышению рСКФ по сравнению с исходным уровнем, и ХБП пациента улучшается на одну или большее число стадий. В других вариантах осуществления лечение приводит к сдвигу в сторону нормальных уровней рСКФ (например, ≥ 90) по сравнению с исходным уровнем, и ХБП пациента улучшается на одну или большее число стадий.
В других вариантах осуществления лечение приводит к значению временного компромисса по опроснику EQ-5D-3L для США (US ТТО), которое составляет > 0,94.
В другом аспекте представлено антитело против С5 или его антигенсвязывающий фрагмент, содержащие домены CDR1, CDR2 и CDR3 вариабельной области тяжелой цепи, имеющей последовательность, указанную в SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 вариабельной области легкой цепи, имеющей последовательность, указанную в SEQ ID NO: 8, для введения пациенту, имеющему аГУС:
(a) один раз на 1-е сутки в дозе, составляющей: 2400 мг пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг - пациенту с массой тела ≥ 100 кг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей: 3000 мг - пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг - пациенту с массой тела ≥ 100 кг.
В одном варианте осуществления антитело определено как безопасное, переносимое и достаточно неиммуногенное после многократного введения внутривенных доз для применения у пациентов с аГУС.
Также представлены наборы, которые включают в себя фармацевтическую композицию, содержащую антитело против С5 или его антигенсвязывающий фрагмент, такое как равулизумаб, и фармацевтически приемлемый носитель, в терапевтически эффективном количестве, адаптированном для применения в описанных в данном документе способах. В одном варианте осуществления набор содержит:
(a) дозу антитела против С5 или его антигенсвязывающего фрагмента, содержащих домены CDR1, CDR2 и CDR3 вариабельной области тяжелой цепи, имеющей последовательность, указанную в SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 вариабельной области легкой цепи, имеющей последовательность, указанную в SEQ ID NO: 8; и
(b) инструкции по применению антитела против С5 или его антигенсвязывающего фрагмента в способах, описанных в данном документе.
В одном варианте осуществления 2400 мг или 3000 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела от ≥ 40 до < 60 кг. В другом варианте осуществления 2700 мг или 3300 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела от ≥ 60 до < 100 кг. В другом варианте осуществления 3000 мг или 3600 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела ≥ 100 кг.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг.1 представлен план исследования для ALXN1210-aHUS-311.
На фиг.2 обобщены первичные, вторичные критерии оценки, и критерии оценки безопасности для ALXN1210-aHUS-311.
На фиг.3 обобщены критерии включения и исключения для ALXN1210-aHUS-311.
На фиг.4 показано распределение пациентов для ALXN1210-aHUS-311.
На фиг.5 указаны данные для деривационного примера с подтвержденным полным ответом ТМА.
Фиг. 6 представляет собой диаграмму Венна, показывающую ключевые данные об эффективности, относящиеся к первичному полному ответу ТМА в течение периода первичной оценки.
Фиг. 7 представляет собой график, отображающий время до полного ответа ТМА.
Фиг. 8 представляет собой график, отображающий средние концентрации в плазме крови (мкг/мл) с течением времени (линейная шкала). Основанное на массе тела введение доз приводило к максимальной, равновесной и остаточной концентрациям препарата, как и было спрогнозировано, без неожиданных фармакокинетических результатов.
Фиг. 9 представляет собой серию гистограмм, отображающих ключевые данные об эффективности, относящиеся к первичному полному ответу ТМА в течение периода первичной оценки и до конца сбора данных. 95%-е доверительные интервалы представлены линиями в верхней части каждого столбца.
На фиг.10 показан полный ответ ТМА в общем и по подгруппам в течение 26-недельного периода первичной оценки.
На фиг.11 показаны ключевые результаты эффективности для статуса полного ответа ТМА с течением времени (незакрашенный круг), включая нормализацию количества тромбоцитов (незакрашенный треугольник), нормализацию гематологических показателей (+), повышение уровня креатинина в сыворотке крови на 25% по сравнению с исходным уровнем (незакрашенный квадрат) и нормализацию ЛДГ (X).
На фиг.12 показаны среднее изменение рСКФ (мл/мин/1,73 м2) по сравнению с исходным уровнем и 95%-й доверительный интервал с течением времени.
На фиг.13 показан сдвиг стадий хронической болезни почек (ХБП) в 183-е сутки по сравнению с исходным уровнем.
На фиг.14 показаны наблюдаемое и основанное на модели средние изменения уровня тромбоцитов (109/л) по сравнению с исходным уровнем и 95%-й доверительный интервал с течением времени.
На фиг.15 показаны наблюдаемый средний уровень тромбоцитов (109/л) и 95%-й доверительный интервал с течением времени.
На фиг.16 показаны наблюдаемое и основанное на модели средние изменения уровня ЛДГ (Ед/л) по сравнению с исходным уровнем и 95%-й доверительный интервал с течением времени.
На фиг.17 показаны наблюдаемый средний уровень ЛДГ (Ед/л) и 95%-й доверительный интервал с течением времени.
На фиг.18 показаны наблюдаемое и основанное на модели средние изменения уровня гемоглобина (НЬ) (г/л) по сравнению с исходным уровнем и 95%-й доверительный интервал с течением времени.
На фиг.19 показаны наблюдаемый средний уровень Hb (г/л) и 95%-й доверительный интервал с течением времени.
На фиг.20 показаны среднее изменение показателя утомляемости по шкале FACIT по сравнению с исходным уровнем и 95%-й доверительный интервал с течением времени. Оценка по FACIT варьирует от 0 до 52, при этом более высокая оценка указывает на меньшую утомляемость
На фиг.21 показаны среднее изменение EQ-5D-3L по сравнению с исходным уровнем и 95%-й доверительный интервал с течением времени.
На фиг.22 представлены средние концентрации (мкг/л) свободного компонента С5 комплемента в сыворотке крови с течением времени с момента определения исходного уровня до 183-х суток.
На фиг.23 приведено сравнение исследуемой популяции и результатов между исследованием ALXN1210-aHUS-311 и исследованием экулизумаба у взрослых (С10-004).
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
I. Антитела против С5
Описанные в данном документе антитела против С5 связываются с компонентом С5 комплемента (например, с С5 человека) и ингибируют расщепление С5 на фрагменты С5а и C5b. Как описано выше, такие антитела также обладают, например, улучшенными фармакокинетическими свойствами по сравнению с другими антителами против С5 (например, экулизумабом), применяемыми в терапевтических целях.
Термин «антитело» описывает полипептиды, содержащие по меньшей мере один антигенсвязывающий сайт, производный от антитела (например, область VH/VL или Fv, или CDR). Антитела включают в себя известные формы антител. Например, антитело может быть человеческим антителом, гуманизированным антителом, биспецифическим антителом или химерным антителом. Антитело также может представлять собой Fab, Fab'2, ScFv, SMIP, аффитело®, наноантитело или доменное антитело. Антитело также может представлять собой антитело любого из следующих изотипов: IgG1, IgG2, IgG3, IgG4, IgM, IgAl, IgA2, IgAsec, IgD и IgE. Антитело может представлять собой встречающееся в природе антитело или может представлять собой антитело, которое было изменено с помощью методики белковой инженерии (например, путем мутации, делеции, замены, конъюгации с отличным от антитела фрагментом). Например, антитело может содержать одну или большее число вариантных аминокислот (по сравнению со встречающимся в природе антителом), которые изменяют свойство (например, функциональное свойство) данного антитела. Например, в данной области техники известны многочисленные такие изменения, которые влияют, например, на период полужизни, эффекторную функцию и/или иммунные ответы на антитело у пациента. Термин антитело также включает в себя искусственные или сконструированные полипептидные конструкции, которые содержат по меньшей мере один антигенсвязывающий сайт, производный от антитела.
Антитела против С5 (или производные от них домены VH/VL), подходящие для применения в данном изобретении, могут быть получены с использованием способов, хорошо известных в данной области техники. В качестве альтернативы, могут применяться признанные в данной области техники антитела против С5. Также можно применять антитела, которые конкурируют с любыми из указанных признанных в данной области техники антител за связывание с С5.
Экулизумаб (также известный как SOLIRIS®) представляет собой антитело против С5, содержащее домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 1, 2 и 3, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 4, 5 и 6, соответственно. Экулизумаб содержит вариабельную область тяжелой цепи, имеющую аминокислотную последовательность, указанную в SEQ ID NO: 7, и вариабельную область легкой цепи, имеющую аминокислотную последовательность, указанную в SEQ ID NO: 8. Вариабельные области экулизумаба описаны в PCT/US1995/005688 и патенте США №6355245, содержание которых включено в данный документ посредством ссылки. Экулизумаб содержит тяжелую цепь, содержащую аминокислотную последовательность, указанную в SEQ ID NO: 10, и легкую цепь, имеющую аминокислотную последовательность, указанную в SEQ ID NO: 11. Полные тяжелая и легкая цепи экулизумаба описаны в PCT/US2007/006606, содержание которой включено в данный документ посредством ссылки.
Иллюстративное антитело против С5 представляет собой равулизумаб, содержащий тяжелую и легкую цепи, имеющие последовательности, приведенные в SEQ ID NO: 14 и 11, соответственно, или их антигенсвязывающие фрагменты и варианты. Равулизумаб (также известный как ULTOMIRIS®, BNJ441 и ALXN1210) описан в PCT/US2015/019225 и патенте США №9079949, содержание которых включено в данный документ посредством ссылки. Термины равулизумаб, BNJ441 и ALXN1210 могут использоваться взаимозаменяемо в данном документе, но все они относятся к одному и тому же антителу. Равулизумаб избирательно связывается с белком С5 комплемента человека, ингибируя его расщепление до С5а и C5b во время активации комплемента. Это ингибирование предотвращает высвобождение провоспалительного медиатора С5а и образование цитолитического порообразующего мембрано атакующе го комплекса (MAC) C5b-9, сохраняя при этом проксимальные или ранние компоненты активации комплемента (например, С3 и C3b), необходимые для опсонизации микроорганизмов и клиренса иммунных комплексов.
В других вариантах осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области равулизумаба. Например, в одном варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 области VH равулизумаба, имеющей последовательность, указанную в SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 области VL равулизумаба, имеющей последовательность, указанную в SEQ ID NO: 8. В другом варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 19, 18 и 3, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 4, 5 и 6, соответственно. В другом варианте осуществления антитело содержит области VH и VL, имеющие аминокислотные последовательности, указанные в SEQ ID NO: 12 и SEQ ID NO: 8, соответственно.
Другое иллюстративное антитело против С5 представляет собой антитело BNJ421, содержащее тяжелую и легкую цепи, имеющие последовательности, приведенные в SEQ ID NO: 20 и 11, соответственно, или их антигенсвязывающие фрагменты и варианты. BNJ421 (также известный как ALXN1211) описан в PCT/US 2015/019225 и патенте США №9079949, содержание которых включено в данный документ посредством ссылки.
В других вариантах осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области BNJ421. Соответственно, в одном варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 области VH BNJ421, имеющей последовательность, указанную в SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 области VL BNJ421, имеющей последовательность, указанную в SEQ ID NO: 8. В другом варианте осуществления антитело содержит домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 19, 18 и 3, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 4, 5 и 6, соответственно.
Точные границы CDR определяли по-разному в зависимости от разных способов. В некоторых вариантах осуществления положения CDR или каркасных областей в вариабельном домене легкой или тяжелой цепи могут быть такими, как определено по Кабату - Kabat et at. [(1991) "Sequences of Proteins of Immunological Interest." NIH Publication No. 91-3242, U.S. Department of Health and Human Services, Bethesda, MD]. В таких случаях CDR могут называться «CDR по Кабату» (например, «LCDR2 по Кабату» или «HCDR1 по Кабату»). В некоторых вариантах осуществления положения CDR вариабельных областей легкой и тяжелой цепи могут быть такими, как определено по Чотиа - Chothia et al. (1989) Nature 342:877-883. Соответственно, эти области могут называться «CDR по Чотиа» (например, «LCDR2 по Чотиа» или «HCDR3 по Чотиа»). В некоторых вариантах осуществления положения CDR вариабельных областей легкой и тяжелой цепи могут быть такими, как определено по комбинированному определению Кабата-Чотиа. В таких вариантах осуществления эти области могут называться «CDR по комбинированному определению Кабата-Чотиа». Thomas et al. [(1996) Mot Immunol 33(17/18): 1389-1401] иллюстрирует идентификацию границ CDR в соответствии с определениями по Кабату и по Чотиа.
В другом варианте осуществления антитело содержит области VH и VL, имеющие аминокислотные последовательности, указанные в SEQ ID NO: 12 и SEQ ID NO: 8, соответственно. В другом варианте осуществления антитело содержит константную область тяжелой цепи, указанную в SEQ ID NO: 13. В другом варианте осуществления антитело содержит полипептид тяжелой цепи, указанный в SEQ ID NO: 14, и полипептид легкой цепи, указанный в SEQ ID NO: 11. В другом варианте осуществления антитело содержит вариантную константную область Fc человека, которая связывается с неонатальным Fc-рецептором человека (FcRn), при этом вариантная константная область СН3 Fc человека содержит замены Met-429-Leu и Asn-435-Ser в остатках, соответствующих метионину 428 и аспарагину 434 константной области Fc нативного IgG человека, каждый остаток пронумерован согласно нумерации ЕС.
В другом варианте осуществления антитело содержит последовательности CDR1, CDR2 и CDR3 тяжелой цепи, указанные SEQ ID NO: 19, 18 и 3, соответственно, и последовательности CDR1, CDR2 и CDR3 легкой цепи, указанные SEQ ID NO: 4, 5 и 6, соответственно, и вариантную константная область Fc человека, которая связывается с неонатальным Fc-рецептором человека (FcRn), при этом вариантная константная область СНЗ Fc человека содержит замены Met-429-Leu и Asn-435-Ser в остатках, соответствующих метионину 428 и аспарагину 434 константной области Fc нативного IgG человека, каждый остаток пронумерован согласно нумерации ЕС.
В других вариантах осуществления описанное в данном документе антитело против С5 содержит CDR1 тяжелой цепи, содержащую или состоящую из следующей аминокислотной последовательности: GHIFSNYWIQ (SEQ ID NO: 19). В другом варианте осуществления описанное в данном документе антитело против С5 содержит CDR2 тяжелой цепи, содержащую или состоящую из следующей аминокислотной последовательности: EILPGSGHTEYTENFKD (SEQ ID NO: 18).
В другом варианте осуществления антитело связывается с С5 человека при рН 7,4 и 25°С с аффинной константой диссоциации (KD), которая находится в диапазоне от 0,1 нМ до 1 нМ. В другом варианте осуществления антитело связывается с С5 человека при рН 6,0 и 25°С с KD ≥ 10 нМ. В еще одном варианте осуществления показатель антитела [(KD антитела или его антигенсвязывающего фрагмента для С5 человека при рН 6,0 и при 25°C)/(KD антитела или его антигенсвязывающего фрагмента для С5 человека при рН 7,4 и при 25°С)] составляет больше чем 25.
Другое иллюстративное антитело против С5 представляет собой антитело 7086, описанное в патентах США №8241628 и №8883158. В одном варианте осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области антитела 7086 (см. патенты США №8241628 и №8883158). В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 21, 22 и 23, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 24, 25 и 26, соответственно. В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат область VH антитела 7086, имеющую последовательность, указанную в SEQ ID NO: 27, и область VL антитела 7086, имеющую последовательность, указанную в SEQ ID NO: 28.
Другое иллюстративное антитело против С5 представляет собой антитело 8110, также описанное в патентах США №8241628 и №8883158. В одном варианте осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области антитела 8110. В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 29, 30 и 31, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 32, 33 и 34, соответственно. В другом варианте осуществления антитело содержит область VH антитела 8110, имеющую последовательность, указанную в SEQ ID NO: 35, и область VL антитела 8110, имеющую последовательность, указанную в SEQ ID NO: 36.
Другое иллюстративное антитело против С5 представляет собой антитело 305LO5, описанное в US 2016/0176954 A1. В одном варианте осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области антитела 305LO5. В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат домены CDR1, CDR2 и CDR3 тяжелой цепи, имеющие последовательности, указанные в SEQ ID NO: 37, 38 и 39, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, имеющие последовательности, указанные в SEQ ID NO: 40, 41 и 42, соответственно. В другом варианте осуществления антитело содержит область VH антитела 305LO5, имеющую последовательность, указанную в SEQ ID NO: 43, и область VL антитела 305LO5, имеющую последовательность, указанную в SEQ ID NO: 44.
Другое иллюстративное антитело против С5 представляет собой антитело SKY59, описанное в Fukuzawa Т., et at, Rep.2017 Apr 24;7(1): 1080). В одном варианте осуществления антитело содержит CDR тяжелой и легкой цепей или вариабельные области антитела SKY59. В другом варианте осуществления антитело или его антигенсвязывающий фрагмент содержат тяжелую цепь, содержащую SEQ ID NO: 45, и легкую цепь, содержащую SEQ ID NO: 46.
Другое иллюстративное антитело против С5 представляет собой антитело REGN3918 (также известное как Н4Н12166РР), описанное в US 20170355757. В одном варианте осуществления антитело содержит вариабельную область тяжелой цепи, содержащую SEQ ID NO: 47, и вариабельную область легкой цепи, содержащую SEQ ID NO: 48. В другом варианте осуществления антитело содержит тяжелую цепь, содержащую SEQ ID NO: 49, и легкую цепь, содержащую SEQ ID NO: 50.
В другом варианте осуществления антитело конкурирует за связывание и/или связывается с одним и тем же эпитопом на С5, что и указанные выше антитела (например, экулизумаб, равулизумаб, антитело 7086, антитело 8110, антитело 305LO5, антитело SKY59 или антитело REGN3918). В другом варианте осуществления антитело имеет по меньшей мере около 90% идентичности аминокислотной последовательности вариабельной области с вышеупомянутыми антителами (например, по меньшей мере около 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98% или 99% идентичности вариабельной области).
Описанное в данном документе антитело против С5 может, в некоторых вариантах осуществления, содержать вариантную константную область Fc человека, которая связывается с неонатальным Fc-рецептором человека (FcRn) с большей аффинностью, чем аффинность нативной константной области Fc человека, из которой получена указанная вариантная константная область Fc человека. Например, константная область Fc может содержать одну или большее число (например, две, три, четыре, пять, шесть, семь или восемь или больше) аминокислотных замен относительно нативной константной области Fc человека, из которой была получена вариантная константная область Fc. Замены могут повышать аффинность связывания антитела IgG, содержащего вариантную константную область Fc, с FcRn при рН 6,0, сохраняя при этом зависимость взаимодействия от рН. Способы тестирования того, повышают ли одна или большее число замен в константной области Fc антитела аффинность константной области Fc к FcRn при рН 6,0 (при сохранении зависимости взаимодействия от рН), известны в данной области техники и представлены в примерах, касающихся экспериментальной части. См., например, PCT/US2015/019225 и патент США №9079949, описания каждого из которых включены в данный документ посредством ссылки в полном объеме.
Замены, которые повышают аффинность связывания константной области Fc антитела с FcRn, известны в данной области техники и включают в себя, например: (1) тройную замену M252Y/S254T/T256E, описанную в работе Dall' Acqua et al. (2006) J Biol Chem 281: 23514-23524; (2) замены M428L или T250Q/M428L, описанные в работе Hinton et al. (2004) J Biol Chem 279:6213-6216 и Hinton et al. (2006) J Immunol 176:346-356; и (3) замены N434A или T307/E380A/N434A, описанные в работе Petkova et al. (2006) Int Immunol 18f 12): 1759-69. Дополнительные пары замен - P257I/Q311I, P257I/N434H и D376V/N434H - описаны например, в работе Datta-Mannan et al. (2007) J Biol Chem 282(3): 1709-1717, содержимое которой включено в данный документ посредством ссылки в полном объеме.
В некоторых вариантах осуществления вариантная константная область имеет замену в аминокислотном остатке 255, пронумерованном согласно нумерации ЕС, на валин. В некоторых вариантах осуществления вариантная константная область имеет замену в аминокислотном остатке 309, пронумерованном согласно нумерации ЕС, на аспарагин. В некоторых вариантах осуществления вариантная константная область имеет замену в аминокислотном остатке 312, пронумерованном согласно нумерации ЕС, на изолейцин. В некоторых вариантах осуществления вариантная константная область имеет замену в аминокислотном остатке 386, пронумерованном согласно нумерации ЕС.
В некоторых вариантах осуществления вариантная константная область Fc содержит не больше чем 30 (например, не больше чем 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, девять, восемь, семь, шесть, пять, четыре, три или две) аминокислотных замен, вставок или делеций относительно нативной константной области, из которой она была получена. В некоторых вариантах осуществления вариантная константная область Fc содержит одну или большее число аминокислотных замен, выбранных из группы, состоящей из: M252Y, S254T, Т256Е, N434S, M428L, V259I, T250I и V308F. В некоторых вариантах осуществления вариантная константная область Fc человека содержит метионин в положении 428 и аспарагин в положении 434, каждый остаток пронумерован согласно нумерации ЕС. В некоторых вариантах осуществления вариантная константная область Fc содержит двойную замену 428L/434S, как описано, например, в патенте США №8088376.
В некоторых вариантах осуществления точное местоположение этих мутаций может быть смещено от положения константной области нативного Fc человека из-за инженерии антител. Например, двойная замена 428L/434S при использовании в химерном Fc IgG2/4 может соответствовать 429L и 435S, как в вариантах M429L и N435S, обнаруживаемых в BNJ441 (равулизумаб) и описанных в патенте США №9079949, описание которого включено в данный документ посредством ссылки в полном объеме.
В некоторых вариантах осуществления вариантная константная область содержит замену в аминокислотном положении 237, 238, 239, 248, 250, 252, 254, 255, 256, 257, 258, 265, 270, 286, 289, 297, 298, 303, 305, 307, 308, 309, 311, 312, 314, 315, 317, 325, 332, 334, 360, 376, 380, 382, 384, 385, 386, 387, 389, 424, 428, 433, 434 или 436 (нумерация ЕС) относительно нативной константной области Fc человека. В некоторых вариантах осуществления замена выбрана из группы, состоящей из следующего: метионин на глицин в положении 237; аланин на пролин в положении 238; лизин на серии в положении 239; изолейцин на лизин в положении 248; аланин, фенилаланин, изолейцин, метионин, глутамин, серии, валин, триптофан, или тирозин для треонина в положении 250; фенилаланин, триптофан или тирозин на метионин в положении 252; треонин на серии в положении 254; глутаминовая кислота на аргинин в положении 255; аспарагиновая кислота, глутаминовая кислота или глутамин на треонин в положении 256; аланин, глицин, изолейцин, лейцин, метионин, аспарагин, серии, треонин или валин на пролин в положении 257; гистидин на глутаминовую кислоту в положении 258; аланин на аспарагиновую кислоту в положении 265; фенилаланин на аспарагиновую кислоту в положении 270; аланин или глутаминовая кислота на аспарагин в положении 286; гистидин на треонин в положении 289; аланин на аспарагин в положении 297; глицин на серии в положении 298; аланин на валин в положении 303; аланин на валин в положении 305; аланин, аспарагиновая кислота, фенилаланин, глицин, гистидин, изолейцин, лизин, лейцин, метионин, аспарагин, пролин, глутамин, аргинин, серии, валин, триптофан или тирозин на треонин в положении 307; аланин, фенилаланин, изолейцин, лейцин, метионин, пролин, глутамин или треонин на валин в положении 308; аланин, аспарагиновая кислота, глутаминовая кислота, пролин или аргинин на лейцин или валина в положении 309; аланин, гистидин или изолейцин на глутамин в положении 311; аланин или гистидин на аспарагиновую кислоту в положении 312, лизин или аргинин на лейцин в положении 314; аланин или гистидин на аспарагин в положении 315; аланин на лизин в положении 317; глицин на аспарагин в положении 325; валин на изолейцин в положении 332; лейцин на лизин в положении 334; гистидин на лизин в положении 360; аланин на аспарагиновую кислоту в положении 376; аланин на глутаминовую кислоту в положении 380; аланин на глутаминовую кислоту в положении 382; аланин на аспарагин или серии в положении 384; аспарагиновая кислота или гистидин на глицин в положении 385; пролин на глутамин в положении 386; глутаминовая кислота на пролин в положении 387; аланин или серии на аспарагин в положении 389; аланин на серии в положении 424; аланин, аспарагиновая кислота, фенилаланин, глицин, гистидин, изолейцин, лизин, лейцин, аспарагин, пролин, глутамин, серии, треонин, валин, триптофан или тирозин на метионин в положении 428; лизин на гистидин в положении 433; аланин, фенилаланин, гистидин, серии, триптофан или тирозин на аспарагин в положении 434; и гистидин на тирозин или фенилаланин в положении 436, каждый остаток пронумерован согласно нумерации ЕС.
Подходящие антитела против С5 для применения в способах, описанных в данном документе, в некоторых вариантах осуществления содержат полипептид тяжелой цепи, содержащий аминокислотную последовательность, представленную в SEQ ID NO: 14, и/или полипептид легкой цепи, содержащий аминокислотную последовательность, представленную в SEQ ID NO: 11. В качестве альтернативы, антитела против С5 для применения в способах, описанных в данном документе, в некоторых вариантах осуществления содержат полипептид тяжелой цепи, содержащий аминокислотную последовательность, представленную в SEQ ID NO: 20, и/или полипептид легкой цепи, содержащий аминокислотную последовательность, представленную в SEQ ID NO: 11.
В одном варианте осуществления антитело связывается с С5 при рН 7,4 и 25°С (и, в иных случаях, в физиологических условиях) с аффинной константой диссоциации (KD), которая составляет по меньшей мере 0,1 (например, по меньшей мере 0,15, 0,175, 0,2, 0,25, 0,275, 0,3, 0,325, 0,35, 0,375, 0,4, 0,425, 0,45, 0,475, 0,5, 0,525, 0,55, 0,575, 0,6, 0,625, 0,65, 0,675, 0,7, 0,725, 0,75, 0,775, 0,8, 0,825, 0,85, 0,875, 0,9, 0,925, 0,95 или 0,975) нМ. В некоторых вариантах осуществления KD антитела против С5 или его антигенсвязывающего фрагмента составляет не больше чем 1 (например, не больше чем 0,9, 0,8, 0,7, 0,6, 0,5, 0,4, 0,3 или 0,2) нМ.
В других вариантах осуществления показатель [(KD антитела для С5 при рН 6,0 при C)/(KD антитела для С5 при рН 7,4 при 25°С)] составляет больше чем 21 (например, больше чем 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, ПО, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 350, 400, 450, 500, 600, 700, 800, 900, 1000, 1500, 2000, 2500, 3000, 3500, 4000, 4500, 5000, 5500, 6000, 6500, 7000, 7500 или 8000).
Способы определения того, связывается ли антитело с белковым антигеном и/или определения аффинности антитела к белковому антигену, известны в данной области техники. Например, связывание антитела с белковым антигеном может быть обнаружено и/или количественно определено с использованием различных методик, таких как следующие, но не ограничиваясь ими: вестерн-блоттинг, дот-блоттинг, метод поверхностного плазмонного резонанса (ППР) (например, система BIAcore; Pharmacia Biosensor АВ, Упсала, Швеция и Пискатауэй, Нью-Джерси, США), или твердофазный иммуноферментный анализ (твердофазный ИФА). См., например, Benny К. С.Lo (2004) "Antibody Engineering: Methods and Protocols," Humana Press (ISBN: 1588290921); Johne et al. (1993) J Immunol Meth 160:191-198; Jonsson et al. (1993) Ann Biol Clin 51:19-26; и Jonsson et al. (1991) Biotechniques 11.:620-627. Кроме того, в примерах, касающихся экспериментальной части, представлены способы измерения аффинности (например, констант диссоциации и ассоциации).
В контексте данного документа термин «ка» относится к константе скорости ассоциации антитела с антигеном. Термин «kd» относится к константе скорости диссоциации антитела из комплекса антитело/антиген. А термин «KD» относится к равновесной константе диссоциации взаимодействия антитело-антиген. Равновесная константа диссоциации выводится из соотношения кинетических констант скорости: KD = ka/kd. Такие определения предпочтительно проводить при 25°С или 37°С (см. примеры, касающихся экспериментальной части). Например, кинетика связывания антитела с С5 человека может быть определена при рН 8,0, 7,4, 7,0, 6,5 и 6,0 с помощью поверхностного плазмонного резонанса (ППР) на приборе BIAcore 3000 с использованием метода анти-Fc захвата для иммобилизации антитела.
В одном варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент блокируют образование или активность активных фрагментов С5а и/или C5b белка С5 (например, белка С5 человека). Благодаря этому блокирующему эффекту антитела подавляют, например, провоспалительные эффекты С5а и образование мембраноатакующего комплекса (MAC) C5b-9 на поверхности клетки.
Способы определения того, ингибирует ли конкретное антитело, описанное в данном документе, расщепление С5, известны в данной области техники. Ингибирование компонента С5 комплемента человека может снизить способность комплемента лизировать клетки в биологических жидкостях организма субъекта. Такое снижение способности комплемента, присутствующего в жидкости (-ях) организма, лизировать клетки можно измерить способами, хорошо известными в данной области техники, такими как, например, обычный тест на гемолиз, такой как анализ гемолиза, описанный в Kabat and Mayer (eds.), "Experimental Immunochemistry, 2nd Edition," 135-240, Springfield, IL, CC Thomas (1961), страницы 135-139, или обычный вариант данного анализа, такой как анализ гемолиза эритроцитов курицы, как описано, например, в e.g., Hillmen et al. (2004) N Engl J Med 350(6): 552. Способы определения того, ингибирует ли соединение-кандидат расщепление С5 человека на формы С5а и C5b, известны в данной области техники и описаны в Evans et al. (1995) Mol Immunol 32(16): 1183-95. Например, концентрацию и/или физиологическую активность С5а и C5b в жидкости организма можно измерить способами, хорошо известными в данной области техники. Для C5b можно использовать анализы на гемолиз или анализы растворимого C5b-9, как обсуждается в данном документе. Также можно использовать другие анализы, известные в данной области техники. Используя анализы этих или других подходящих типов, можно проводить скрининг агентов-кандидатов, способных ингибировать компонент С5 комплемента человека.
Иммунологические методики, такие как твердофазный ИФА, но не ограничиваясь им, могут использоваться для измерения концентрации белка С5 и/или его продуктов расщепления для определения способности антитела против С5 или его антигенсвязывающего фрагмента ингибировать превращение С5 в биологически активные продукты. В некоторых вариантах осуществления измеряют образование С5а. В некоторых вариантах осуществления используются C5b-9 неоэпитоп-специфические антитела для обнаружения образования терминального комплемента.
Анализы на гемолиз можно использовать для определения ингибирующей активности антитела против С5 или его антигенсвязывающего фрагмента при активации комплемента. Для определения эффекта антитела против С5 или его антигенсвязывающего фрагмента на гемолиз, опосредованный классическим путем комплемента, в растворе сыворотки крови для тестирования in vitro, в качестве клеток-мишеней используют, например, эритроциты овцы, покрытые гемолизином, или эритроциты курицы, сенсибилизированные антителом против эритроцитов курицы. Процент лизиса нормализуют, считая 100% лизис равным лизису, происходящему в отсутствие ингибитора. В некоторых вариантах осуществления классический путь комплемента активируется антителом IgM человека, например, как используется в наборе Classical Pathway Complement Kit от Wieslab® (Wieslab® COMPL CP310, Euro-Diagnostica, Швеция). Кратко, исследуемую сыворотку крови инкубируют с антителом против С5 или его антигенсвязывающим фрагментом в присутствии антитела IgM человека. Количество образующегося C5b-9 измеряют путем приведения указанной смеси в контакт с антителом против C5b-9, конъюгированным с ферментом, и флуорогенным субстратом, и измерения оптической плотности при соответствующей длине волны. В качестве контроля тестируемую сыворотку крови инкубируют в отсутствие антитела против С5 или его антигенсвязывающего фрагмента. В некоторых вариантах осуществления тестируемая сыворотка крови представляет собой сыворотку крови с дефицитом С5, восстановленную полипептидом С5.
Для определения эффекта антитела против С5 или его антигенсвязывающего фрагмента на гемолиз, опосредованный альтернативным путем комплемента, в качестве клеток-мишеней можно использовать несенсибилизированные эритроциты кролика или морской свинки. В некоторых вариантах осуществления исследуемый раствор сыворотки крови представляет собой сыворотку крови с дефицитом С5, восстановленную полипептидом С5. Процент лизиса нормализуют, считая 100% лизис равным лизису, происходящему в отсутствие ингибитора. В некоторых вариантах осуществления альтернативный путь комплемента активируется молекулами липополисахаридов, например, как используется в наборе Alternative Pathway Complement Kit от Wieslab® (Wieslab® COMPL AP330, Euro-Diagnostica, Швеция). Кратко, исследуемую сыворотку крови инкубируют с антителом против С5 или его антигенсвязывающим фрагментом в присутствии липополисахарида. Количество образующегося C5b-9 измеряется путем приведения указанной смеси в контакт с антителом против C5b-9, конъюгированным с ферментом, и флуорогенным субстратом, и измерения флуоресценции при соответствующей длине волны. В качестве контроля тестируемую сыворотку крови инкубируют в отсутствие антитела против С5 или его антигенсвязывающего фрагмента.
В некоторых вариантах осуществления активность С5 или ее ингибирование количественно определяют с помощью анализа CH50eq. Анализ CH50eq представляет собой способ измерения общей активности классического пути комплемента в сыворотке крови. Данный литический тест представляет собой анализ, в котором используются сенсибилизированные антителами эритроциты в качестве активатора классического пути комплемента и различные разведения исследуемой сыворотки крови для определения количества, необходимого для 50% лизиса (СН50). Процент гемолиза можно определить, например, с помощью спектрофотометра. Анализ CH50eq обеспечивает косвенную оценку образования терминальных комплексов комплемента (ТСС), поскольку сами ТСС непосредственно отвечает за измеряемый гемолиз.
Данный анализ хорошо известен и широко практикуется специалистами в данной области техники. Кратко, для активации классического пути комплемента неразбавленные образцы сыворотки крови (например, восстановленные образцы сыворотки крови человека) добавляют в лунки для микроанализа, содержащие сенсибилизированные антителами эритроциты, чтобы таким образом генерировать ТСС. Затем активированные сыворотки крови разводят в лунках для микроанализа, которые покрыты реагентом захвата (например, антителом, которое связывается с одним или большим числом компонентов ТСС). ТСС, присутствующий в активированных образцах, связывается с моноклональными антителами, покрывающими поверхность лунок для микроанализа. Лунки промывают и в каждую лунку добавляют реагент обнаружения, меченный обнаруживаемым образом, который распознает связанный ТСС.Обнаруживаемая метка может быть, например, флуоресцентной меткой или ферментной меткой. Результаты данного анализа выражаются в единицах эквивалента СН50 на миллилитр (СН50, Ед. экв./мл).
Ингибирование, например, в том, что касается активности терминального комплемента, включает в себя по меньшей мере 5 (например, по меньшей мере 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55 или 60) % снижения активности терминального комплемента, например, в анализе на гемолиз или анализе CH50eq, по сравнению с эффектом контрольного антитела (или его антигенсвязывающего фрагмента) в аналогичных условиях и в эквимолярной концентрации. Существенное ингибирование, как этот термин используется в данном документе, относится к ингибированию данной активности (например, активности терминального комплемента) по меньшей мере на 40 (например, по меньшей мере на 45, 50, 55, 60, 65, 70, 75, 80, 85, 90 или на 95 или больше) %. В некоторых вариантах осуществления антитело против С5, описанное в данном документе, содержит одну или большее число аминокислотных замен относительно CDR экулизумаба (т.е. SEQ ID NO: 1-6), но сохраняет по меньшей мере 30 (например, по меньшей мере 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90 или 95) % ингибирующей активности комплемента экулизумаба в анализе на гемолиз или анализе CH50eq.
Описанное в данном документе антитело против С5 имеет период полужизни в сыворотке крови человека, который составляет по меньшей мере 20 (например, по меньшей мере 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54 или 55) суток. В другом варианте осуществления описанное в данном документе антитело против С5 имеет период полужизни в сыворотке крови человека, который составляет по меньшей мере 40 суток. В другом варианте осуществления описанное в данном документе антитело против С5 имеет период полужизни в сыворотке крови человека, который составляет около 43 суток. В другом варианте осуществления описанное в данном документе антитело против С5 имеет период полужизни в сыворотке крови человека, который составляет 39-48 суток. Способы измерения периода полужизни в сыворотке кров известны в данной области техники. В некоторых вариантах осуществления антитело против С5 или его антигенсвязывающий фрагмент, описанные в данном документе, имеют период полужизни в сыворотке крови, который по меньшей мере на 20 (например, по меньшей мере на 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 125, 150, 175, 200, 250, 300, 400, 500) % больше чем период полужизни экулизумаба в сыворотке крови, например, как измерено в одной из мышиных модельных систем, описанных в примерах, касающихся экспериментальной части (например, С5-дефицитная/NOD/scid мышь или модельная система hFcRn трансгенной мыши).
В одном варианте осуществления антитело конкурирует за связывание и/или связывается с одним и тем же эпитопом на С5, что и антитела, описанные в данном документе. Термин «связывается с одним и тем же эпитопом» применительно к двум или более антителам означает, что данные антитела связываются с одним и тем же сегментом аминокислотных остатков, как определено при помощи данного способа. Методики определения того, связываются ли антитела с «одним и тем же эпитопом на С5», для описанных в данном документе антител включают в себя, например, методы картирования эпитопа, такие как рентгеноструктурный анализ кристаллов комплексов антиген:антитело, который обеспечивает атомарное разрешение эпитопа, и масс-спектрометрия водородно-дейтериевого обмена (HDX-MS). В других методах исследуется связывание антитела с фрагментами пептидного антигена или мутированными вариациями антигена, где потеря связывания из-за модификации аминокислотного остатка в последовательности антигена часто считается признаком компонента эпитопа. Кроме того, также могут использоваться вычислительные комбинаторные методы для картирования эпитопов. Эти методы основаны на способности представляющего интерес антитела к аффинному выделению специфических коротких пептидов из фагового дисплея пептидных комбинаторных библиотек. Ожидается, что антитела, имеющие одинаковые последовательности VH и VL или одинаковые последовательности CDR1, 2 и 3, будут связываться с одним и тем же эпитопом.
Антитела, которые «конкурируют с другим антителом за связывание с мишенью», относятся к антителам, которые ингибируют (частично или полностью) связывание другого антитела с мишенью. Конкурируют ли два антитела друг с другом за связывание с мишенью, т.е. ингибирует ли и в какой степени одно антитело связывание другого антитела с мишенью, можно определить с помощью известных экспериментов по конкурентному связыванию. В определенных вариантах осуществления антитело конкурирует с другим антителом и ингибирует его связывание с мишенью по меньшей мере на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90% или 100%. Уровень ингибирования или конкурирования может быть различным в зависимости от того, какое антитело является «блокирующим антителом» (т.е. немеченое антитело, которое сначала инкубируют с мишенью). Конкурирующие антитела связываются с одним и тем же эпитопом, перекрывающимся эпитопом или с соседними эпитопами (например, что подтверждается стерическими затруднениями).
Антитела против С5 или их антигенсвязывающие фрагменты, описанные в данном документе, используемые в описанных в данном документе способах, могут быть получены с использованием множества методик, признанных в данной области техники. Моноклональные антитела могут быть получены с помощью различных методик, знакомых специалистам в данной области техники. Кратко, клетки селезенки животного, иммунизированного желаемым антигеном, иммортализуют, обычно путем слияния с клеткой миеломы (см. Kohler & Milstein, Eur. J. Immunol. 6: 511-519 (1976)). Альтернативные способы иммортализации включают в себя трансформацию вирусом Эпштейна-Барр, онкогенами или ретровирусами, или другие способы, хорошо известные в данной области техники. Колонии, возникающие из единичных иммортализованных клеток, подвергаются скринингу на продукцию антител с желаемой специфичностью и аффинностью к антигену, и выход моноклональных антител, продуцируемых такими клетками, может быть увеличен с помощью различных методик, включая инъекцию в перитонеальную полость позвоночного-хозяина. В качестве альтернативы, можно выделить последовательности ДНК, которые кодируют моноклональное антитело или его связывающий фрагмент, путем скрининга библиотеки ДНК из В-клеток человека в соответствии с общим протоколом, изложенным в Huse, et al., Science 246: 1275-1281 (1989).
II. Композиции
Также в данном документе представлены композиции, содержащие антитело против С5 или его антигенсвязывающий фрагмент.В одном варианте осуществления композиция содержит антитело против С5, содержащее домены CDR1, CDR2 и CDR3 в вариабельной области тяжелой цепи, имеющей последовательность, указанную в SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 в вариабельной области легкой цепи, имеющей последовательность, указанную в SEQ ID NO: 8. В другом варианте осуществления антитело против С5 содержит тяжелую и легкую цепи, имеющие последовательности, приведенные в SEQ ID NO: 14 и 11, соответственно. В другом варианте осуществления антитело против С5 содержит тяжелую и легкую цепи, имеющие последовательности, приведенные в SEQ ID NO: 20 и 11, соответственно.
Композиции могут быть составлены в виде фармацевтического раствора, например, для введения субъекту для лечения или профилактики нарушения, связанного с комплементом, такого как аГУС.Фармацевтические композиции, как правило, содержат фармацевтически приемлемый носитель. В контексте данного документа термин «фармацевтически приемлемый носитель» относится и включает в себя любые и все растворители, диспергирующие среды, покрытия, антибактериальные и противогрибковые агенты, изотонические и замедляющие всасывание агенты и тому подобное, которые являются физиологически совместимыми. Композиции могут содержать фармацевтически приемлемую соль, например, соль присоединения кислоты или соль присоединения основания, сахара, углеводы, полиолы и/или модификаторы тоничности.
Композиции могут быть составлены стандартными способами. Составление фармацевтических композиций является хорошо налаженным искусством и дополнительно описано, например, в Gennaro (2000) "Remington: The Science and Practice of Pharmacy," 20th Edition, Lippincott, Williams & Wilkins (ISBN: 0683306472); Ansel et al. (1999) "Pharmaceutical Dosage Forms and Drug Delivery Systems," 7th Edition, Lippincott Williams & Wilkins Publishers (ISBN: 0683305727); и в Kibbe (2000) "Handbook of Pharmaceutical Excipients American Pharmaceutical Association," 3rd Edition (ISBN: 091733096X). В некоторых вариантах осуществления композиция может быть составлена, например, в виде забуференного раствора подходящей концентрации и подходящего для хранения при 2-8°С (например, 4°С). В некоторых вариантах осуществления композиция может быть составлена для хранения при температуре ниже 0°С (например, -20°С или -80°С). В некоторых вариантах осуществления композиция может быть составлена для хранения в течение периода времени до 2 лет (например, в течение одного месяца, двух месяцев, трех месяцев, четырех месяцев, пяти месяцев, шести месяцев, семи месяцев, восьми месяцев, девяти месяцев, 10 месяцев, 11 месяцев, 1 года, 1½ лет или 2 лет) при 2-8°С (например, 4°С). Таким образом, в некоторых вариантах осуществления описанные в данном документе композиции стабильны при хранении в течение по меньшей мере 1 года при 2-8°С (например, 4°С).
Фармацевтические композиции могут быть в различных формах. Эти формы включают в себя, например, жидкие, полутвердые и твердые дозированные формы, такие как жидкие растворы (например, растворы для инъекций и инфузий), дисперсии или суспензии, таблетки, пилюли, порошки, липосомы и суппозитории. Предпочтительная форма частично зависит от предназначенного способа введения и терапевтического применения. Например, композиции, содержащие композицию, предназначенную для системной или местной доставки, могут быть в форме растворов для инъекций или инфузий. Соответственно, композиции могут быть составлены для введения парентеральным способом (например, внутривенной, подкожной, внутрибрюшинной или внутримышечной инъекцией). «Парентеральное введение», «вводить парентерально» и другие грамматически эквивалентные фразы, используемые в данном документе, относятся к способам введения, отличным от энтерального и местного введения, как правило, путем инъекции, и включают в себя следующие, но не ограничиваются ими: внутривенную, интраназальную, интраокулярную, легочную, внутримышечную, внутриартериальную, интратекальную, интракапсулярную, интраорбитальную, интракардиальную, интрадермальную, внутрилегочную, внутрибрюшинную, транстрахеальную, подкожную, субкутикулярную, внутрисуставную, субкапсулярную, субарахноидальную, внутриспинальную, эпидуральную, интрацеребральную, интракраниальную, интракаротидную и интрастернальную инъекции и инфузии.
В одном варианте осуществления композиция содержит равулизумаб (также известный как ULTOMIRIS®, антитело BNJ441 или ALXN1210) для инъекций. В одном варианте осуществления инъекция представляет собой стерильный, прозрачный до полупрозрачного, слегка беловатого цвета раствор без консервантов для внутривенного применения. В другом варианте осуществления каждый флакон с однократной дозой содержит 300 мг равулизумаба для инъекций в концентрации 10 мг/мл с рН 7,0. В другом варианте осуществления равулизумаб для инъекций требует разведения до конечной концентрации 5 мг/мл. В другом варианте осуществления каждый мл дополнительно содержит полисорбат 80 (0,2 мг) (растительного происхождения), хлорид натрия (8,77 мг), двухосновный фосфат натрия (1,78 мг), одноосновный фосфат натрия (0,46 мг) и воду для инъекций.
III. Способы лечения
В данном документе представлены способы лечения аГУС у пациента-человека, включающие в себя введение указанному пациенту антитела против С5 или его антигенсвязывающего фрагмента, при этом указанные антитело против С5 или его антигенсвязывающий фрагмент вводят (или предназначены для введения) в соответствии с конкретной клинической схемой применения (т.е. в конкретном количестве дозы и в соответствии с конкретным графиком введения доз).
В контексте данного документа термины «индукция» и «фаза индукции» используются взаимозаменяемо и относятся к первой фазе лечения в клиническом испытании.
В контексте данного документа термины «поддержание» и «фаза поддержания» используются взаимозаменяемо и относятся ко второй фазе лечения в клиническом испытании. В определенных вариантах осуществления лечение продолжают до тех пор, пока наблюдается клиническое улучшение или пока не возникнут неконтролируемая токсичность или прогрессирование заболевания.
В контексте данного документа термин «субъект» или «пациент» означает пациента-человека (например, пациента, имеющего нарушение, связанное с комплементом). В одном варианте осуществления нарушение, связанное с комплементом, представляет собой атипичный гемолитико-уремический синдром (аГУС). Патология и клинические проявления у пациентов с аГУС также обусловлены активацией терминального комплемента. Более конкретно, активация С5 и нарушение регуляции активации комплемента приводят к повреждению эндотелия, потреблению тромбоцитов и тромботическим микроангиопатическим (ТМА) явлениям, характеризующимся тромбоцитопенией, механическим внутрисосудистым гемолизом и повреждением почек. Важно отметить, что у около 20% пациентов наблюдаются также внепочечные проявления заболевания, включающие в себя проявления в центральной нервной системе, сердце, желудочно-кишечном тракте, дистальных отделах конечностей, и тяжелое системное поражение органов (Loirat, et al., Orphanet. J. Rare Dis. 2011; 6:60). Симптомы аГУС хорошо известны специалистам в области медицины редких заболеваний или заболеваний почек и включают в себя, например, тяжелую гипертензию, протеинурию, уремию, вялость/усталость, раздражительность, тромбоцитопению, микроангиопатическую гемолитическую анемию и нарушение функции почек (например, острую почечную недостаточность).
аГУС может быть наследственным, приобретенным или идиопатическим. аГУС можно считать наследственным, когда два или большее число (например, три, четыре, пять или шесть, или больше) членов одной семьи страдают данным заболеванием с разницей во времени, составляющей по меньшей мере шесть месяцев, и исключено воздействие общего провоцирующего агента, или когда у субъекта идентифицирована одна или большее число мутаций гена, ассоциированного с аГУС (например, одна или большее число мутаций в CFH, MCP/CD46, CFB или CFI). Например, субъект может иметь ассоциированный с CFH аГУС, ассоциированный с CFB аГУС, ассоциированный с CFI аГУС или ассоциированный с МСР аГУС.До 30% наследственных аГУС связано с мутациями в CFH, 12% с мутациями в МСР, 5-10% с мутациями в CFI и меньше чем 2% - с мутациями в CFB. Наследственные аГУС могут быть мультиплексными (т.е. семейными; когда данное заболевание имеют два или большее число членов семьи) или симплексными (т.е. однократное появление в семье). аГУС можно считать приобретенным, когда можно определить вызывающий его фактор окружающей среды (например, лекарственное средство, системное заболевание или вирусные агенты, или бактериальные агенты, которые не приводят к появлению шигаподобных экзотоксинов) или триггер. аГУС можно считать идиопатическим, когда нет очевидного триггера (генетического или экологического).
Могут быть выполнены лабораторные тесты для определения того, есть ли у субъекта-человека тромбоцитопения, микроангиопатическая гемолитическая анемия или острая почечная недостаточность. Тромбоцитопения может быть диагностирована медицинским работником как одно или большее число из следующего: (i) количество тромбоцитов, которое составляет меньше чем 150000/мм3 (например, меньше чем 60000/3); (ii) снижение времени выживания тромбоцитов, отражающее усиленное разрушение тромбоцитов в кровотоке; и (iii) гигантские тромбоциты, наблюдаемые в мазке периферической крови, что согласуется с вторичной активацией тромбоцитопоэза. Микроангиопатическая гемолитическая анемия может быть диагностирована медицинским работником как одно или большее число из следующего: (i) концентрации гемоглобина, которые составляют меньше чем 10 мг/дл (например, меньше чем 6,5 мг/дл); (ii) повышенные концентрации лактатдегидрогеназы (ЛДГ) в сыворотке крови (>460 Ед/л); (iii) гипербилирубинемия, ретикулоцитоз, циркулирующий свободный гемоглобин и низкие либо необнаруживаемые концентрации гаптоглобина; и (iv) обнаружение фрагментированных эритроцитов (шистоцитов) с типичными признаками пойкилоцитов с шиловидными отростками или шлемовидных эритроцитов в мазке периферической крови в комбинации с отрицательным тестом Кумбса. См., например, Kaplan et al. (1992) "Hemolytic Uremic Syndrome and Thrombotic Thrombocytopenic Purpura," Informa Health Care (ISBN 0824786637), и Zipfel (2005) "Complement and Kidney Disease," Springer (ISBN 3764371668). Концентрации С3 и C4 в крови также могут быть использованы в качестве меры активации или нарушения регуляции комплемента. Кроме того, состояние субъекта может быть дополнительно охарактеризовано путем идентификации субъекта как носителя одной или большего числа мутаций в гене, ассоциированном с аГУС, таком как CFI, CFB, CFH или МСР (см. выше). Подходящие способы обнаружения мутации в гене включают в себя, например, методики секвенирования ДНК и мультиплексные методики анализа нуклеиновых кислот.См., например, Breslin et al. (2006) Clin Am Soc Nephrol 1:88-99, и Goicoechea de Jorge et al. (2007) P'roc Natl Acad Sci USA 104:240-245.
В контексте данного документа термин «эффективное лечение» относится к лечению, которое приводит к положительному эффекту, например, облегчению по меньшей мере одного симптома заболевания или нарушения. Положительный эффект может иметь форму улучшения по сравнению с исходным уровнем, т.е. улучшения по сравнению с измерением или наблюдением, сделанным до начала терапии в соответствии с указанным способом. В контексте аГУС, например, эффективное лечение может относиться к облегчению одного или большего числа симптомов, выбранных из группы, состоящей из тяжелой гипертензии, протеинурии, уремии, вялости/усталости, раздражительности, тромбоцитопении, микроангиопатической гемолитической анемии и/или нарушения функции почек (например, острой почечной недостаточности)).
Термин «эффективное количество» относится к количеству агента, которое обеспечивает желаемый биологический, терапевтический и/или профилактический результат.Этим результатом может быть снижение, облегчение, временное ослабление, уменьшение, отсрочка и/или смягчение одного или большего числа проявлений, симптомов или причин заболевания, или любое другое желаемое изменение биологической системы. В одном примере «эффективное количество» представляет собой такое количество антитела против С5 или его антигенсвязывающего фрагмента, которое с клинически доказанной эффективностью облегчает по меньшей мере один симптом аГУС (например, тяжелую гипертензию, протеинурию, уремию, вялость/усталость, раздражительность, тромбоцитопению, микроангиопатическую гемолитическую анемию и нарушение функции почек (например, острую почечную недостаточность)). Эффективное количество можно вводить посредством одного или большего числа введений.
В одном варианте осуществления доза антитела против С5 или его антигенсвязывающего фрагмента основана на массе тела пациента. Например, в одном варианте осуществления, 2400 мг или 3000 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела от ≥ 40 до < 60 кг. В другом варианте осуществления 2700 мг или 3300 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела от ≥ 60 до < 100 кг. В другом варианте осуществления 3000 мг или 3600 мг антитела против С5 или его антигенсвязывающего фрагмента вводят пациенту с массой тела ≥ 100 кг. В определенных вариантах осуществления схемы применения подбираются так, чтобы обеспечить оптимальный желаемый ответ (например, эффективный ответ).
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят в одном или большем числе циклов введения. В одном варианте осуществления цикл введения составляет 26 недель. В одном варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят один раз на 1-е сутки цикла введения, один раз на 15-е сутки цикла введения и после этого - каждые восемь недель. В одном варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят каждые восемь недель после цикла введения во время периода продления, составляющего до двух лет включительно (например, в дозе 3000 мг, 3300 мг или 3600 мг).
В другом варианте осуществления представлен способ лечения пациента-человека с аГУС, при этом указанный способ включает в себя введение указанному пациенту эффективного количества антитела против С5 или его антигенсвязывающего фрагмента, содержащих последовательности CDR1, CDR2 и CDR3 тяжелой цепи, указанные SEQ ID NO: 19, 18 и 3, соответственно, и последовательности CDR1, CDR2 и CDR3 легкой цепи, указанные SEQ ID NO: 4, 5 и 6, соответственно, при этом указанные антитело против С5 или его антигенсвязывающий фрагмент вводят:
(a) один раз на 1-е сутки в дозе, составляющей: 2400 мг пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг - пациенту с массой тела ≥ 100 кг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей:
3000 мг - пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг - пациенту с массой тела ≥ 100 кг.
В другом варианте осуществления представлен способ лечения пациента-человека с аГУС, при этом указанный способ включает в себя введение пациенту (например, во время цикла введения) эффективного количества антитела против С5 или его антигенсвязывающего фрагмента, содержащих последовательности CDR1, CDR2 и CDR3 тяжелой цепи, указанные SEQ ID NO: 19, 18 и 3, соответственно, последовательности CDR1, CDR2 и CDR3 легкой цепи, указанные SEQ ID NO: 4, 5 и 6, соответственно, и вариантную константную область Fc человека, которая связывается с неонатальным Fc-рецептором человека (FcRn), при этом вариантная константная область СНЗ Fc человека содержит замены Met-429-Leu и Asn-435-Ser в остатках, соответствующих метионину 428 и аспарагину 434 нативной константной области Fc IgG человека, каждый остаток пронумерован согласно нумерации ЕС, при этом указанные антитело против С5 или его антигенсвязывающий фрагмент вводят:
(a) один раз на 1-е сутки в дозе, составляющей: 2400 мг - пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг -пациенту с массой тела ≥ 100 кг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей:
3000 мг - пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг - пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг - пациенту с массой тела ≥ 100 кг.
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела от ≥ 40 до < 60 кг:
(a) один раз на 1-е сутки в дозе, составляющей: 2400 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей: 3000 мг.
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела от ≥ 60 до < 100 кг:
(a) один раз на 1-е сутки в дозе, составляющей: 2700 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей: 3300 мг.
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела ≥ 100 кг:
(a) один раз на 1-е сутки в дозе, составляющей: 3000 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей: 3600 мг.
В некоторых вариантах осуществления пациент ранее не получал лечения ингибитором комплемента (например, пациент является таким, который ранее не проходил лечение ингибитором комплемента).
В других вариантах осуществления пациент ранее получал лечение одним антителом против С5 или его антигенсвязывающим фрагментом, а затем был переведен на другое антитело против С5 или его антигенсвязывающий фрагмент во время курса лечения. Например, в определенных вариантах осуществления вводят различные антитела против С5 во время курса лечения. В одном варианте осуществления различные антитела против С5 вводят во время отдельных периодов лечения и продления. Например, в одном варианте осуществления проводят лечение пациента экулизумабом во время периода лечения (например, в течение 26 недель), после чего проводят лечение другим антителом против С5 (например, равулизумабом, антителом 7086, антителом 8110, антителом 305LO5, антителом SKY59 или антителом REGN3918), например, во время периода продления. В другом варианте осуществления экулизумаб вводят пациенту в дозе 600 мг на 1-е, 8-е, 15-е и 22-е сутки цикла введения во время фазы индукции, после чего вводят поддерживающую дозу экулизумаба, составляющую 900 мг, в 19-е сутки цикла введения и каждые две недели после этого (например, в общей сложности на протяжении 26 недель), после чего проводят лечение равулизумабом в течение периода продления, составляющего до двух лет.В другом варианте осуществления проводят лечение пациента равулизумабом (например, в течение 26 недель), после чего проводят лечение другим антителом против С5 (например, экулизумабом, антителом 7086, антителом 8110, антителом 305LO5, антителом SKY59 или антителом REGN3918) в течение, например, периода продления.
Иллюстративные альтернативные антитела против С5 включают в себя, но не ограничиваются ими: (i) равулизумаб, (ii) антитело или его антигенсвязывающий фрагмент, содержащие домены CDR1, CDR2 и CDR3 тяжелой цепи, содержащие SEQ ID NO: 21, 22 и 23, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, содержащие SEQ ID NO: 24, 25 и 26, соответственно, (iii) антитело или его антигенсвязывающий фрагмент, содержащие вариабельную область тяжелой цепи, содержащую SEQ ID NO: 27, и вариабельную область легкой цепи, содержащую SEQ ID NO: 28, (iv) антитело или его антигенсвязывающий фрагмент, содержащие домены CDR1, CDR2 и CDR3 тяжелой цепи, содержащие SEQ ID NO: 29, 30 и 31, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, содержащие SEQ ID NO: 32, 33 и 34, соответственно, (v) антитело или его антигенсвязывающий фрагмент, содержащие вариабельную область тяжелой цепи, содержащую SEQ ID NO: 35, и вариабельную область легкой цепи, содержащую SEQ ID NO: 36, (vi) антитело или его антигенсвязывающий фрагмент, содержащие домены CDR1, CDR2 и CDR3 тяжелой цепи, содержащие SEQ ID NO: 37, 38 и 39, соответственно, и домены CDR1, CDR2 и CDR3 легкой цепи, содержащие SEQ ID NO: 40, 41 и 42, соответственно, (vii) антитело или его антигенсвязывающий фрагмент, содержащие вариабельную область тяжелой цепи, содержащую SEQ ID NO: 43, и вариабельную область легкой цепи, содержащую SEQ ID NO: 44, (viii) антитело или его антигенсвязывающий фрагмент, содержащие тяжелую цепь, содержащую SEQ ID NO: 45, и легкую цепь, содержащую SEQ ID NO: 46, соответственно, (vii) антитело или его антигенсвязывающий фрагмент, содержащие вариабельную область тяжелой цепи, содержащую SEQ ID NO: 47, и вариабельную область легкой цепи, содержащую SEQ ID NO: 48, (х) антитело или его антигенсвязывающий фрагмент, содержащие тяжелую цепь, содержащую SEQ ID NO: 49, и легкую цепь, содержащую SEQ ID NO: 50.
В некоторых вариантах осуществления пациент ранее получал лечение в течение по меньшей мере 1 месяца, по меньшей мере 2 месяцев, по меньшей мере 3 месяцев, по меньшей мере 4 месяцев, по меньшей мере 5 месяцев, по меньшей мере 6 месяцев, по меньшей мере 7 месяцев, по меньшей мере 8 месяцев, по меньшей мере 9 месяцев, по меньшей мере 10 месяцев, по меньшей мере 11 месяцев, по меньшей мере 12 месяцев, по меньшей мере 18 месяцев или по меньшей мере 24 месяцев антителом против С5 или его антигенсвязывающий фрагментом (например, экулизумабом) до перехода на другое антитело против С5 или его антигенсвязывающий фрагмент (например, равулизумаб). В конкретном варианте осуществления пациент ранее получал лечение экулизумабом в течение по меньшей мере 6 месяцев.
В другом варианте осуществления, когда пациент (например, пациент с аГУС) получает лечение первым антителом против С5, а затем переводится на лечение вторым другим антителом против С5, особенно, когда указанное второе другое антитело против С5 связывается с эпитопом на С5, отличным от эпитопа, с которым связывается указанное первое антитело против С5, графики введения учитывают период полужизни указанного первого антитела против С5. Например, для обеспечения того, чтобы первое антитело против С5 было выведено (например, «вымыто») из пациента перед введением второго (другого) антитела против С5 (например, чтобы избежать проблем, связанных с агрегацией, образованием иммунных комплексов и т.д.), учитывают период полужизни указанного первого антитела против С5. В одном варианте осуществления второе (другое) антитело против С5 не вводят до тех пор, пока не пройдет период времени, соответствующий 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7 или 7,5 периодам полужизни первого антитела против С5 после заключительного введения указанного первого антитела против С5.
В другом варианте осуществления пациент ранее получал лечение экулизумабом, а затем был переведен на лечение вторым (другим) антителом против С5 (например, равулизумабом, антителом 7086, антителом 8110, антителом 305LO5, антителом SKY59 или антителом REGN3918). В одном варианте осуществления, когда первое вводимое антитело представляет собой экулизумаб, второе (другое) антитело против С5 не вводят, например, до тех пор, пока не пройдет по меньшей мере 36, 45, 54, 63, 72, 81, 90, 99, 108, 117 или 126 суток после заключительного введения экулизумаба.
В другом варианте осуществления пациент ранее получал лечение равулизумабом, а затем был переведен на лечение другим антителом против С5 (например, экулизумабом, антителом 7086, антителом 8110, антителом 305LO5, антителом SKY59 или антителом REGN3918). В одном варианте осуществления, когда первое вводимое антитело представляет собой равулизумаб, второе (другое) антитело против С5 не вводят, например, до тех пор, пока не пройдет по меньшей мере 100, 125, 150, 175, 200, 225, 250, 275, 300, 325, 375 или 400 суток после заключительного введения равулизумаба.
В качестве дополнения или альтернативы, используются методики для выведения или усиления клиренса первого антитела против С5 до перехода на лечение вторым (другим) антителом против С5. Иллюстративные методики включают в себя, но не ограничиваются ими, плазмаферез или гемотрансфузии. В другом варианте осуществления антитело против первого антитела против С5 вводят для выведения или усиления клиренса указанного первого антитела против С5 (например, антитела против экулизумаба, антитела против равулизумаба, антитела против 7086, антитела против 8110, антитела против 305LO5, антитела против SKY59 или антитела против REGN3918) до введения второго (другого) антитела против С5.
В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент (например, равулизумаб) вводят пациенту, при этом цикл введения начинается через по меньшей мере около две недели, по меньшей мере около три недели, по меньшей мере около четыре недели, по меньшей мере около шесть недель, по меньшей мере около семь недель или по меньшей мере около восемь недель после введения указанному пациенту заключительной дозы экулизумаба. В другом варианте осуществления антитело против С5 или его антигенсвязывающий фрагмент (например, равулизумаб) вводят пациенту, при этом лечение (например, цикл введения) начинается через по меньшей мере две недели после введения указанному пациенту заключительной дозы экулизумаба.
В некоторых вариантах осуществления пациенты, получавшие лечение в соответствии с описанными в данном документе способами, были вакцинированы против менингококковых инфекций в течение 3 лет до или во время начала лечения. В одном варианте осуществления пациенты, которые получали лечение меньше чем через 2 недели после введения менингококковой вакцины, также получают соответствующие профилактические антибиотики в течение 2 недель после вакцинации. В еще одном варианте осуществления пациенты, получающие лечение в соответствии с описанными в данном документе способами, вакцинированы против серотипов менингококка А, С, Y, W135 и/или В.
В контексте данного документа термин «остаточный уровень в сыворотке крови» относится к самому низкому уровню, при котором данный агент (например, антитело против С5 или его антигенсвязывающий фрагмент) или медицинский препарат присутствуют в сыворотке крови. Напротив, «пиковый уровень в сыворотке крови» относится к наивысшему уровню данного агента в сыворотке крови. «Средний уровень в сыворотке крови» относится к среднему уровню данного агента в сыворотке крови с течением времени.
В одном варианте осуществления описанные схемы лечения достаточны для поддержания определенных остаточных концентраций в сыворотке крови антитела против С5 или его антигенсвязывающего фрагмента. Например, в одном варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 200, 205, 210, 215, 220, 225, 230, 240, 245, 250, 255, 260, 265, 270, 280, 290, 300, 305, 310, 315, 320, 325, 330, 335, 340, 345, 350, 355, 360, 365, 370, 375, 380, 385, 390, 395 или 400 мкг/мл или больше. В одном варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 100 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 150 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 200 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 250 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне 300 мкг/мл или больше. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне от 100 мкг/мл до 200 мкг/мл. В другом варианте осуществления лечение поддерживает остаточную концентрацию антитела против С5 или его антигенсвязывающего фрагмента в сыворотке крови на уровне около 175 мкг/мл.
В другом варианте осуществления для получения эффективного ответа антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание по меньшей мере 50 мкг, 55 мкг, 60 мкг, 65 мкг, 70 мкг, 75 мкг, 80 мкг, 85 мкг, 90 мкг, 95 мкг, 100 мкг, 105 мкг, 110 мкг, 115 мкг, 120 мкг, 125 мкг, 130 мкг, 135 мкг, 140 мкг, 145 мкг, 150 мкг, 155 мкг, 160 мкг, 165 мкг, 170 мкг, 175 мкг, 180 мкг, 185 мкг, 190 мкг 195 мкг, 200 мкг, 205 мкг, 210 мкг, 215 мкг, 220 мкг, 225 мкг, 230 мкг, 235 мкг, 240 мкг, 245 мкг, 250 мкг, 255 мкг или 260 мкг антитела на миллилитр крови пациента. В другом варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание от 50 мкг до 250 мкг антитела на миллилитр крови пациента. В другом варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание от 100 мкг до 200 мкг антитела на миллилитр крови пациента. В другом варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание около 175 мкг антитела на миллилитр крови пациента.
В другом варианте осуществления для получения эффективного ответа пациенту вводят антитело против С5 в количестве и с частотой, обеспечивающими минимальную концентрацию свободного С5. Например, в одном варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание концентрации свободного С5 на уровне 0,2 мкг/мл, 0,3 мкг/мл, 0,4 мкг/мл, 0,5 мкг/мл или ниже. В другом варианте осуществления антитело против С5 вводят пациенту в количестве и с частотой, обеспечивающими поддержание концентрации свободного С5 на уровне от 0,309 мкг/мл до 0,5 мкг/мл или ниже. В другом варианте осуществления описанное в данном документе лечение снижает концентрацию свободного С5 больше чем на 99% в течение периода лечения. В другом варианте осуществления указанное лечение снижает концентрацию свободного С5 больше чем на 99,5% в течение периода лечения.
IV. Результаты
В данном документе представлены способы лечения аГУС у пациента, включающие в себя введение указанному пациенту антитела против С5 или его антигенсвязывающего фрагмента.
Симптомы аГУС включают в себя, но не ограничиваются ими, тяжелую гипертензию, протеинурию, уремию, вялость/усталость, раздражительность, тромбоцитопению, микроангиопатическую гемолитическую анемию и нарушение функции почек (например, острую почечную недостаточность). Пациенты, получающие лечение в соответствии с описанными в данном документе способами, предпочтительно испытывают улучшение по меньшей мере одного из проявлений аГУС.
В других вариантах осуществления лечение приводит к ингибированию терминального комплемента.
В других вариантах осуществления лечение вызывает сдвиг в сторону нормальных уровней связанного с гемолизом гематологического биомаркера, выбранного из группы, состоящей из свободного гемоглобина, гаптоглобина, количества ретикулоцитов, ПНГ-клона эритроцитов и D-димера.
В другом варианте осуществления лечение вызывает повышение стабилизации гемоглобина по сравнению с исходным уровнем у пациента до лечения. В другом варианте осуществления лечение приводит к повышению уровня гемоглобина на ≥ 20 г/л. В другом варианте осуществления лечение приводит к предотвращению снижения уровня гемоглобина на ≥ 2 г/дл по сравнению с исходным уровнем при отсутствии трансфузии с момента определения исходного уровня к 183-м суткам.
В других вариантах осуществления лечение приводит к нормализации уровня тромбоцитов (≥ 150 × 109/л). В других вариантах осуществления лечение приводит к нормализации уровня тромбоцитов (≥ 150 × 109/л) на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение приводит к нормализации уровня ЛДГ (≤246 Ед/л). В других вариантах осуществления лечение приводит к нормализации уровня ЛДГ (≤246 Ед/л) на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение приводит к улучшению уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем. В других вариантах осуществления лечение приводит к улучшению уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение приводит к полному ответу ТМА (т.е. нормализации уровня тромбоцитов ( ≥ 150 × 10/л), нормализации ЛДГ (≤246 Ед/л) и улучшению уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем). В других вариантах осуществления лечение приводит к полному ответу ТМА на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение приводит к модифицированному полному ответу ТМА (т.е. нормализации уровня тромбоцитов (≥ 150 × 109/л), нормализации уровня ЛДГ (≤246 Ед/л), и пациент прекращает диализ, если он находился на диализе в исходном состоянии, или улучшению уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем для пациента, который не находился на диализе в исходном состоянии). В других вариантах осуществления лечение приводит к модифицированному полному ответу ТМА на период, составляющий по меньшей мере 28 суток (например, по меньшей мере 28 суток, 1 месяц, 2 месяца, 3 месяца, 4 месяца, 5 месяцев, 6 месяцев, 7 месяцев, 8 месяцев, 9 месяцев, 10 месяцев, 11 месяцев, 1 год или два года).
В других вариантах осуществления лечение снижает потребность в гемотрансфузиях. В другом варианте осуществления лечение приводит к повышению отмены трансфузий больше чем на 70%. В другом варианте осуществления лечение приводит к отмене трансфузии в период с момента определения исходного уровня до 183-х суток.
В других вариантах осуществления лечение приводит к устранению прорывного гемолиза во время периода лечения. В другом варианте осуществления лечение приводит к снижению прорывного гемолиза по сравнению с исходным уровнем прорывного гемолиза до лечения.
В других вариантах осуществления лечение приводит к снижению значительных неблагоприятных сосудистых событий (MAVE).
В других вариантах осуществления лечение вызывает изменение качества жизни по сравнению с исходным уровнем, оцениваемое с помощью шкалы функциональной оценки терапии хронических заболеваний (FACIT) для показателя утомляемости версии 4 и опросника Европейской организации по изучению и лечению злокачественных опухолей для оценки качества жизни из 30 вопросов. В одном варианте осуществления лечение вызывает изменение качества жизни по сравнению с исходным уровнем, оцениваемое по шкале FACIT для показателя утомляемости, на один или большее число (например, 1, 2 или 3) баллов. В другом варианте осуществления лечение вызывает изменение качества жизни по сравнению с исходным уровнем, оцениваемое по шкале FACIT для показателя утомляемости, на 3 балла через 150 суток или через большее число суток (, например, 150 суток, 151 сутки, 152 суток, 153 суток, 154 суток, 155 суток, 156 суток, 157 суток, 158 суток, 159 суток, 160 суток, 161 сутки, 162 суток, 163 суток, 164 суток, 165 суток, 166 суток, 167 суток, 168 суток, 169 суток, 170 суток, 171 сутки, 172 суток, 173 суток, 174 суток, 175 суток, 176 суток, 177 суток, 178 суток, 179 суток, 180 суток, 181 сутки, 182 суток 183 суток, 184 суток, 185 суток, 186 суток, 187 суток, 188 суток, 189 суток, 190 суток, 191 сутки, 192 суток, 193 суток, 194 суток, 195 суток, 196 суток, 197 суток, 198 суток, 199 суток, 200 суток, 205 суток, 210 суток, 215 суток, 220 суток или 225 суток) после начала лечения.
Стадия хронической болезни почек (ХБП) классифицируется на основе стадий хронической болезни почек, определенных Национальным фондом почки США. Стадии ХБП и соответствующие значения расчетной скорости клубочковой фильтрации (рСКФ) следующие: Стадия 1: рСКФ >= 90 (норма), Стадия 2: рСКФ = 60-89, Стадия 3А: рСКФ = 45-59, Стадия 3В: рСКФ = 30-44, Стадия 4: рСКФ = 15-29 и стадия 5: рСКФ < 15 (включая диализ: Конечная стадия). Стадия 1 считается наилучшей категорией. Стадия 5 считается наихудшей категорией. Улучшение рСКФ (например, > 15) соответствует улучшению стадии ХБП (например, более низкой стадии ХБП). Соответственно, в других вариантах осуществления хроническая болезнь почек (ХБП) пациента улучшается на одну или большее число стадий после начала лечения. Например, ХБП пациента улучшается на одну, две, три, четыре или пять стадий). В другом варианте осуществления ХБП пациента улучшается на одну или большее число стадий через 150 суток или через большее число суток (например, 150 суток, 151 сутки, 152 суток, 153 суток, 154 суток, 155 суток, 156 суток, 157 суток, 158 суток, 159 суток, 160 суток, 161 сутки, 162 суток, 163 суток, 164 суток, 165 суток, 166 суток, 167 суток, 168 суток, 169 суток, 170 суток, 171 сутки, 172 суток, 173 суток, 174 суток, 175 суток, 176 суток, 177 суток, 178 суток, 179 суток, 180 суток, 181 сутки, 182 суток 183 суток, 184 суток, 185 суток, 186 суток, 187 суток, 188 суток, 189 суток, 190 суток, 191 сутки, 192 суток, 193 суток, 194 суток, 195 суток, 196 суток, 197 суток, 198 суток, 199 суток, 200 суток, 205 суток, 210 суток, 215 суток, 220 суток или 225 суток) после начала лечения.
В других вариантах осуществления лечение приводит к повышению рСКФ по сравнению с исходным уровнем. В других вариантах осуществления лечение приводит к сдвигу в сторону нормальных уровней рСКФ (например, ≥ 90). В других вариантах осуществления лечение приводит к повышению рСКФ по сравнению с исходным уровнем, и ХБП пациента улучшается на одну или большее число стадий. В других вариантах осуществления лечение приводит к сдвигу в сторону нормальных уровней рСКФ (например, ≥ 90) по сравнению с исходным уровнем, и ХБП пациента улучшается на одну или большее число стадий.
В других вариантах осуществления лечение приводит к значению временного компромисса по опроснику EQ-5D-3L для США (US ТТО), которое составляет > 0,94.
V. Наборы и стандартные лекарственные формы
Также в данном документе представлены наборы, которые включают в себя фармацевтическую композицию, содержащую антитело против С5 или его антигенсвязывающий фрагмент, такое как равулизумаб, и фармацевтически приемлемый носитель, в терапевтически эффективном количестве, адаптированном для применения в описанных в данном документе способах. Наборы, необязательно, также могут включать в себя инструкции, содержащие, например, графики введения, позволяющие практикующему человеку (например, врачу, медсестре или пациенту) вводить содержащуюся в них композицию для введения композиции пациенту, имеющему аГУС. Набор также может включать в себя шприц.
Необязательно, наборы включают в себя несколько упаковок фармацевтических композиций с однократной дозой, каждая из которых содержит эффективное количество антитела против С5 или его антигенсвязывающего фрагмента для однократного введения в соответствии с представленными выше способами. Инструменты или устройства, необходимые для введения фармацевтической (-их) композиции (-й), также могут быть включены в указанные наборы. Например, набор может содержать один или большее число предварительно заполненных шприцев, содержащих количество антитела против С5 или его антигенсвязывающего фрагмента.
В одном варианте осуществления, в данном изобретении представлен набор для лечения аГУС у пациента-человека, содержащий:
(a) дозу антитела против С5 или его антигенсвязывающего фрагмента, содержащих домены CDR1, CDR2 и CDR3 вариабельной области тяжелой цепи, имеющей последовательность, указанную в SEQ ID NO: 12, и домены CDR1, CDR2 и CDR3 вариабельной области легкой цепи, имеющей последовательность, указанную в SEQ ID NO: 8; и
(b) инструкции по применению антитела против С5 или его антигенсвязывающего фрагмента в соответствии с любым из способов, описанных в данном документе.
В одном варианте осуществления набор содержит дозу антитела против С5 или его антигенсвязывающего фрагмента, при этом указанные антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела от ≥ 40 до < 60 кг:
(a) один раз на 1-е сутки в дозе, составляющей: 2400 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей: 3000 мг.
В другом варианте осуществления набор содержит дозу антитела против С5 или его антигенсвязывающего фрагмента, при этом указанные антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела от ≥ 60 до < 100 кг:
(a) один раз на 1-е сутки в дозе, составляющей: 2700 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей: 3300 мг.
В другом варианте осуществления набор содержит дозу антитела против С5 или его антигенсвязывающего фрагмента, при этом указанные антитело против С5 или его антигенсвязывающий фрагмент вводят пациенту с массой тела ≥ 100 кг:
(а) один раз на 1-е сутки в дозе, составляющей: 3000 мг; и
(b) на 15-е сутки и после этого - каждые восемь недель в дозе, составляющей: 3600 мг.
Следующие ниже примеры являются просто иллюстративными и не должны рассматриваться как ограничивающие объем этого описания каким-либо образом, поскольку многие вариации и эквиваленты станут очевидными для специалистов в данной области техники после прочтения настоящего описания.
Содержание всех ссылок, записей Genbank, патентов и опубликованных патентных заявок, цитируемых в этой заявке, прямо включено в нее посредством ссылки.
ПРИМЕРЫ
ПРИМЕР 1: Несравнительное многоцентровое исследование 3-й фазы равулизумаба (ALXN1210) у взрослых пациентов с атипичным гемолитико-уремическим синдромом (аГУС), ранее не получавших лечение ингибиторами комплемента
Несравнительное исследование равулизумаба (ALXN1210-aHUS-311) проводится у взрослых пациентов и пациентов-подростков с атипичным гемолитико-уремическим синдромом (аГУС), ранее не получавших лечение ингибиторами комплемента. План данного исследования представлен на фиг.1.
аГУС представляет собой тромботическую микроангиопатию (ТМА), чаще всего вызванную мутациями в генах, кодирующих белки, участвующие в альтернативном пути комплемента (АПК), или аутоантителами против регуляторных белков АПК (Noris, et al., Clin. J. Am. Soc. Nephrol 2010; 5:1844-59). Пациенты с аГУС подвержены риску опасных для жизни проявлений заболевания, возникающих в результате повреждения эндотелия, включая тромбоцитопению, внутрисосудистый гемолиз, острую почечную недостаточность и внепочечное повреждение тканей. Важно отметить, что у около 20% пациентов наблюдаются внепочечные проявления заболевания, включающие в себя проявления в центральной нервной системе, сердце, ЖКТ, дистальной конечности, и тяжелое системное поражение органов (Loirat, et al, Orphanet J. Rare Dis. 2011; 6:60 и Brodsky, Blood. 2015; 126:2459-65). До появления экулизумаба показатели смертности среди пациентов с аГУС достигали 15% во время острой прогрессирующей фазы данного заболевания (Noris, et al., Clin. J. Am. Soc. Nephrol 2010; 5:1844-59) и Sellier-Leclerc, J. Am. Soc. Nephrol 2007; 18:2392-2400). У no меньшей мере 50% пациентов почечная недостаточность прогрессировала до терминальной стадии хронической почечной недостаточности (ТХПН), часто в течение года после начала заболевания, и для поддержания жизни им требовался диализ или пересадка почки. Хроническая неконтролируемая терминальная активация комплемента, в частности, активация компонента 5 комплемента (С5) и нарушение регуляции активности комплемента занимают центральное место в патогенезе аГУС и разрушительных проявлениях данного заболевания. Вследствие этого, целенаправленная блокада С5 с селективным ингибированием генерации С5а и C5b-9 представляет собой важный терапевтический механизм лечения.
1. Цели
Первичной целью данного исследования является оценка эффективности равулизумаба у пациентов-подростков и взрослых пациентов с аГУС, ранее не получавших лечение ингибиторами комплемента, в ингибировании опосредованной комплементом ТМА, характеризующейся тромбоцитопенией, гемолизом и нарушением функции почек.
Вторичными целями данного исследования являются: (1) характеризация безопасности и переносимости равулизумаба в данной популяции пациентов, (2) оценка эффективности равулизумаба с помощью дополнительных показателей (например, статуса потребности в диализе, времени до полного ответа ТМА, статуса полного ответа ТМА с течением времени, наблюдаемого значения и изменения от исходного уровня расчетной скорости клубочковой фильтрации (рСКФ), стадии хронической болезни почек (ХБП) (на основании оценки в отдельные целевые дни и классифицированной как улучшенная, стабильная (без изменений) или ухудшенная по сравнению с исходным уровнем), наблюдаемого значения и изменения от исходного уровня гематологических параметров (тромбоцитов, ЛДГ, гемоглобина), повышения уровня гемоглобина ≥ 20 г/л от исходного уровня (сохраняемого в течение по меньшей мере 2 последовательных измерений, произведенных с интервалом по меньшей мере в 4 недели), изменения от исходного уровня качества жизни (КЖ) (на основании оценки с помощью Европейского опросника для оценки качества жизни в 5 категориях по 3 уровням (EQ-5D-3L; все пациенты), опросника функциональной оценки терапии хронических заболеваний (FACIT) для показателя утомляемости версии 4 (пациенты возрастом < 18 лет) и педиатрических опросников FACIT для показателя утомляемости (пациенты возрастом < 18 лет)), (3) характеризация фармакокинетики/фармакодинамики (ФК/ФД) равулизумаба по изменениям концентрации равулизумаба в сыворотке крови с течением времени и изменениям концентрации свободного С5 с течением времени, и (4) оценка безопасности и эффективности равулизумаба при длительном применении. 2. Критерии оценки
Первичные, вторичные критерии оценки и критерии оценки безопасности для данного исследования обобщены на фиг.2. Первичным критерием оценки эффективности является полный ответ ТМА в течение 26-недельного периода первичной оценки, о котором свидетельствует нормализация гематологических параметров (количество тромбоцитов и уровень ЛДГ) и повышение уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем, что подтверждается 2 последовательными измерениями, произведенными с интервалом не меньше чем в 4 недели.
Вторичными критериями оценки эффективности в данном исследовании являются следующие:
A. статус потребности в диализе;
B. время до полного ответа ТМА;
C. статус полного ответа ТМА с течением времени;
D. наблюдаемое значение и изменение по сравнению с исходным уровнем рСКФ;
E. стадия ХБП на основании оценки исследователя в определенные целевые дни и классифицированная как улучшенная, стабильная (без изменений) или ухудшенная по сравнению с исходным уровнем;
F. наблюдаемое значение и изменение по сравнению с исходным уровнем гематологических параметров (тромбоциты, ЛДГ, гемоглобин);
G. повышение уровня гемоглобина на ≥ 20 г/л по сравнению с исходным уровнем, сохраняющееся по меньшей мере в течение 2 последовательных измерений, произведенных с интервалом не меньше чем в 4 недели;
H. изменение КЖ по сравнению с исходным уровнем, измеренное с помощью опросников EQ-5D-3L (все пациенты), FACIT версии 4 для показателя утомляемости (пациенты возрастом ≥ 18 лет) и педиатрического FACIT для показателя утомляемости (пациенты возрастом < 18 лет).
Фармакокинетическими (ФК) и фармакодинамическими (ФД) критериями оценки в данном исследовании являются изменения концентрации равулизумаба в сыворотке крови с течением времени и изменения концентрации свободного С5 с течением времени.
Безопасность и переносимость равулизумаба оцениваются с помощью физических обследований, показателей жизнедеятельности, электрокардиограмм (ЭКГ), лабораторных исследований и частоты нежелательных явлений (НЯ) и серьезных нежелательных явлений (СНЯ). Также оценивается относительное число пациентов, у которых развиваются антитела против лекарственного средства (АЛС).
Исследуемые биомаркеры ФД-эффекта включают в себя следующие, но не ограничиваются ими: изменение по сравнению с исходным уровнем уровней маркеров нарушенной регуляции комплемента (например, фактора Ва), воспаления сосудов (например, растворимого рецептора фактора некроза опухоли 1 [sTNFR1]), активации/повреждения эндотелия (например, растворимой молекулы адгезии сосудов 1 [sVCAM1], тромбомодулина), коагуляции (например, D-димера) и повреждения почек (например, цистатина С). Дополнительные оценки могут включать в себя измерения экскреции равулизумаба с мочой, гемолиза эритроцитов курицы, общего С5, аутоантител против белков комплемента (например, антитела против фактора Н) и активности АПК (например, модифицированный тест Хэма, анализ отложения комплемента).
Экспериментальные генетические исследования могут быть выполнена для изучения генетических вариантов в генах, которые, как известно, ассоциированы с аГУС, а также для выявления новых генетических вариантов, ассоциированных с аГУС, нарушением регуляции комплемента, метаболизмом или эффективностью равулизумаба. Пациенты могут отказаться от предоставления образца для экспериментального генетического исследования и по-прежнему участвовать в данном клиническом исследовании.
3. Краткое изложение плана исследования
Исследование ALXN1210-aHUS-311 представляет собой нез ас лепленное несравнительное многоцентровое исследование 3-й фазы для оценки безопасности и эффективности равулизумаба, вводимого путем внутривенной (в/в) инфузии пациентам-подросткам (возрастом от 12 до < 18 лет) и взрослым пациентам (возрастом ≥ 18 лет) с аГУС. В данном исследовании принимают участие около 55 пациентов, получающих равулизумаб. План данного исследования представлен на фиг.1. Все пациенты никогда ранее не получали лечения ингибиторами комплемента и включают в себя по меньшей мере от 6 до 10 подростков (возрастом от 12 до < 18 лет во время скрининга) и по меньшей мере от 10 до 25 пациентов с предшествующими трансплантациями почек.
Данное исследование состоит из 7-дневного периода скрининга, 26-недельного периода первичной оценки и периода продления продолжительностью до 2 лет.Дозы препарата основаны на последней зарегистрированной массе тела пациента во время визита исследования (таблица 5). Пациенты получают насыщающую дозу равулизумаба в/в (2400 мг - для пациентов с массой тела от ≥ 40 до < 60 кг, 2700 мг - для пациентов с массой тела от ≥ 60 до < 100 кг, 3000 мг - для пациентов с массой тела ≥ 100 кг) на 1-е сутки, а затем -поддерживающие дозы равулизумаба в/в (3000 мг - для пациентов с массой тела от ≥ 40 до < 60 кг, 3300 мг - для пациентов с массой тела от ≥ 60 до < 100 кг, 3600 мг - для пациентов с массой тела ≥ 100 кг) на 15-е сутки и один раз в 8 недель (1 р./8 нед.) после этого в общей сложности в течение 26 недель лечения. После периода первичной оценки пациенты начинают период продления и получают равулизумаб до тех пор, пока препарат не будет зарегистрирован или одобрен (в соответствии с нормативно-правовыми актами конкретной страны), или в течение периода до 2 лет, в зависимости от того, что произойдет раньше. Завершение данного клинического испытания определяется как последний визит последнего пациента.
В данном незаслепленном несравнительном исследовании 3-й фазы оцениваются безопасность и эффективность лечения равулизумабом. Хотя для данного исследования не планируется проведение формального сравнительного анализа, данные от пациентов, получавших равулизумаб, оцениваются в контексте данных, наблюдавшихся в ретроспективной контрольной группе пациентов, получавших лечение экулизумабом. Ретроспективная контрольная группа состоит из пациентов с аГУС, которые получали лечение экулизумабом в проспективных регистрационных исследованиях С08-002А/В, С10-003 и С10-004, для которых представляющие интерес план исследования и особенности проведения, которые могут повлиять на величину эффекта, были аналогичны данному исследованию. Кроме того, данная контрольная группа ограничена пациентами в возрасте ≥ 12 лет, проходившими плазмообмен/плазмотрансфузию (ПО/ПТ) за 4 или меньше недели до начала лечения экулизумабом для дальнейшего соответствия критериям включения в данное исследование.
График проведения оценок для периода скрининга и периода первичной оценки представлены в таблице 1. График проведения оценок для периода продления представлен в таблице 2. Дополнительные (внеплановые) визиты за пределами указанных визитов допускаются по усмотрению исследователя. Процедуры, анализы и оценки проводятся по усмотрению исследователя. Любые анализы, процедуры или оценки, проведенные во время внеплановых визитов, регистрируются в электронных индивидуальных регистрационных картах (эИРК). Для анализов во время внеплановых визитов используются анализы лабораторные анализы, выполняемые местной лабораторией или центральной лабораторией. Однако, если необходимо использовать результаты анализов, выполняемых местной лабораторией, во время внеплановых визитов собираются дублирующие образцы для выполнения анализов в центральной лаборатории.
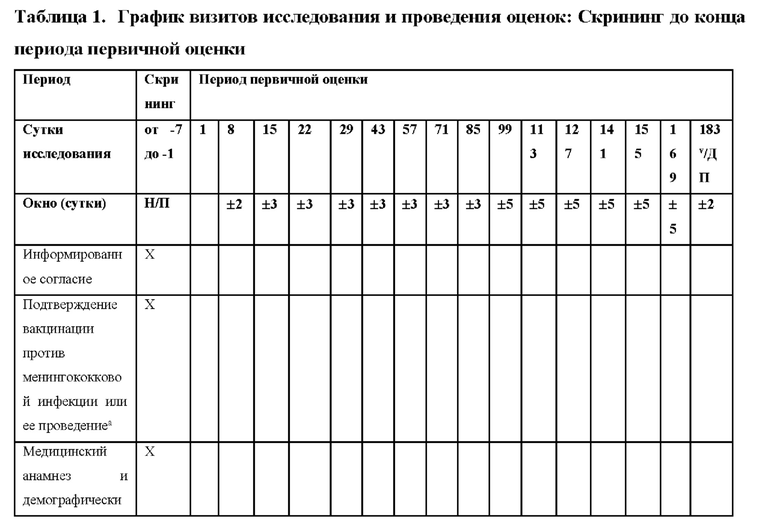
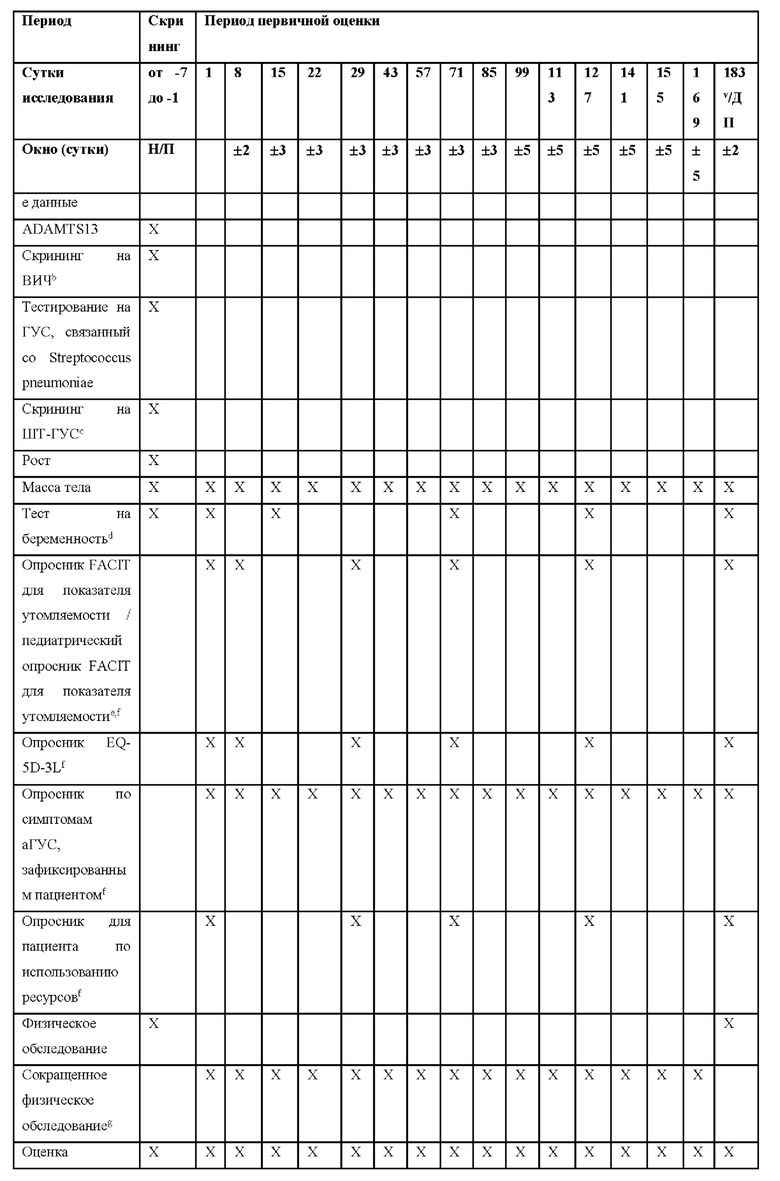
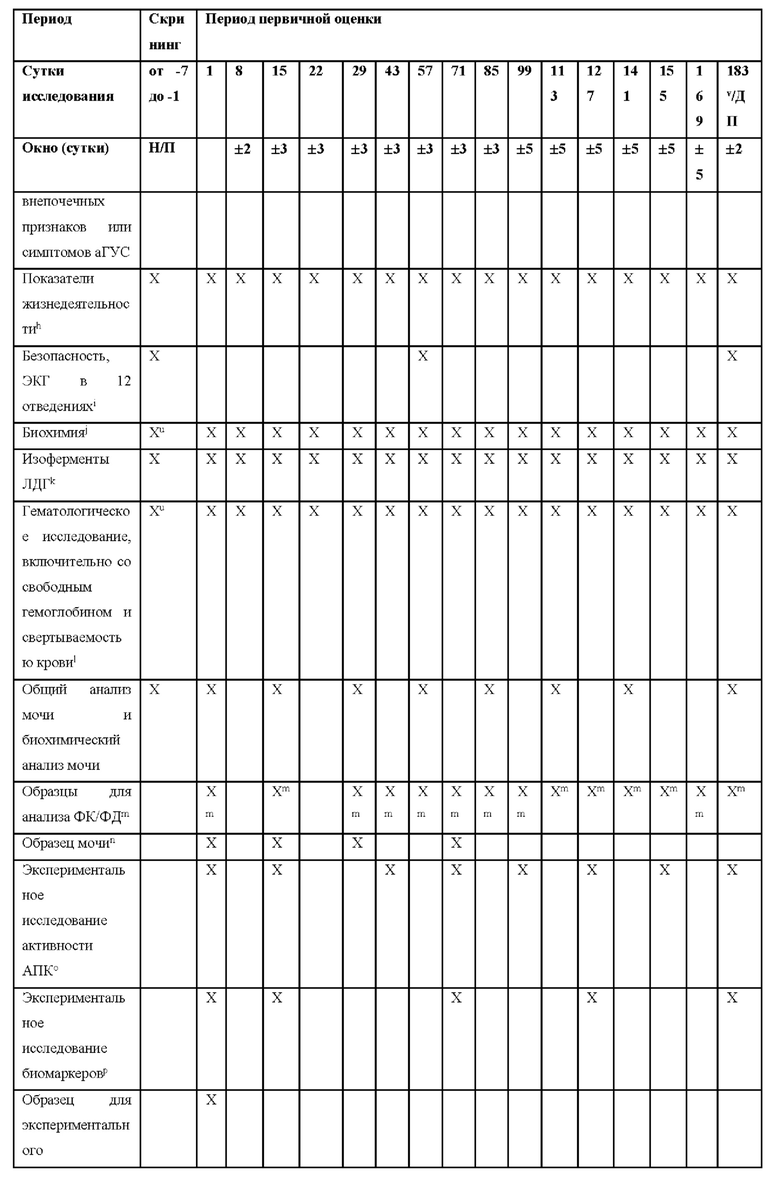
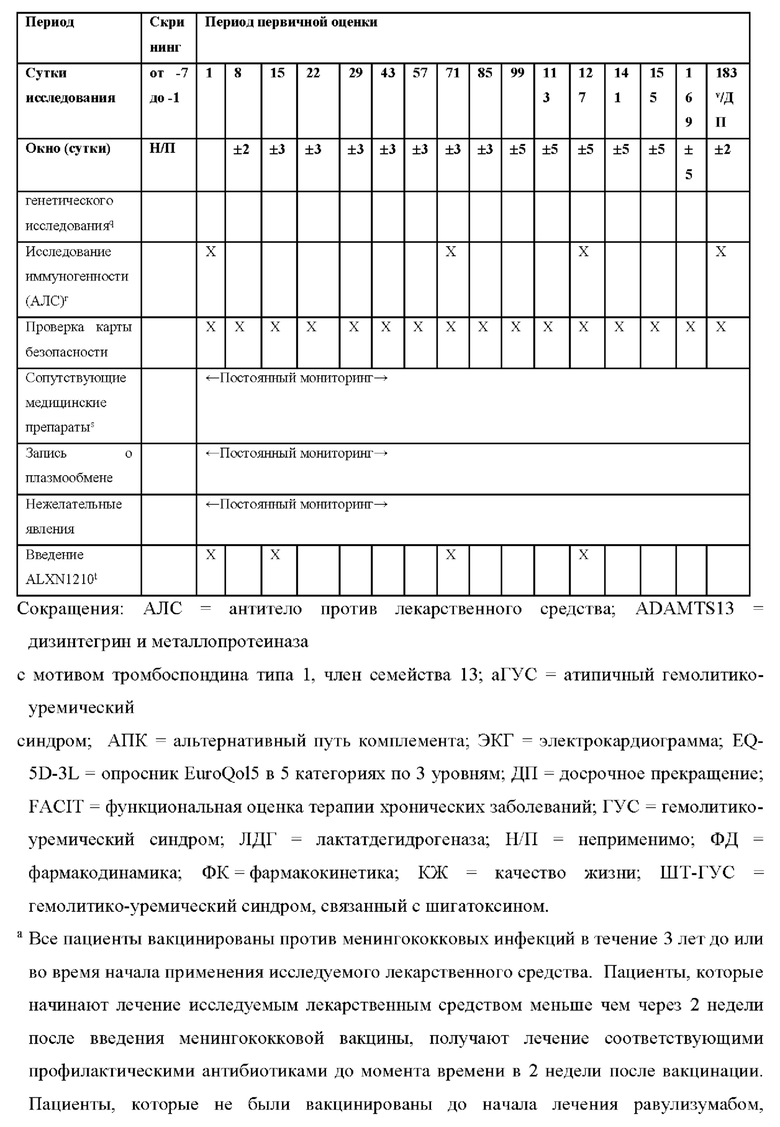



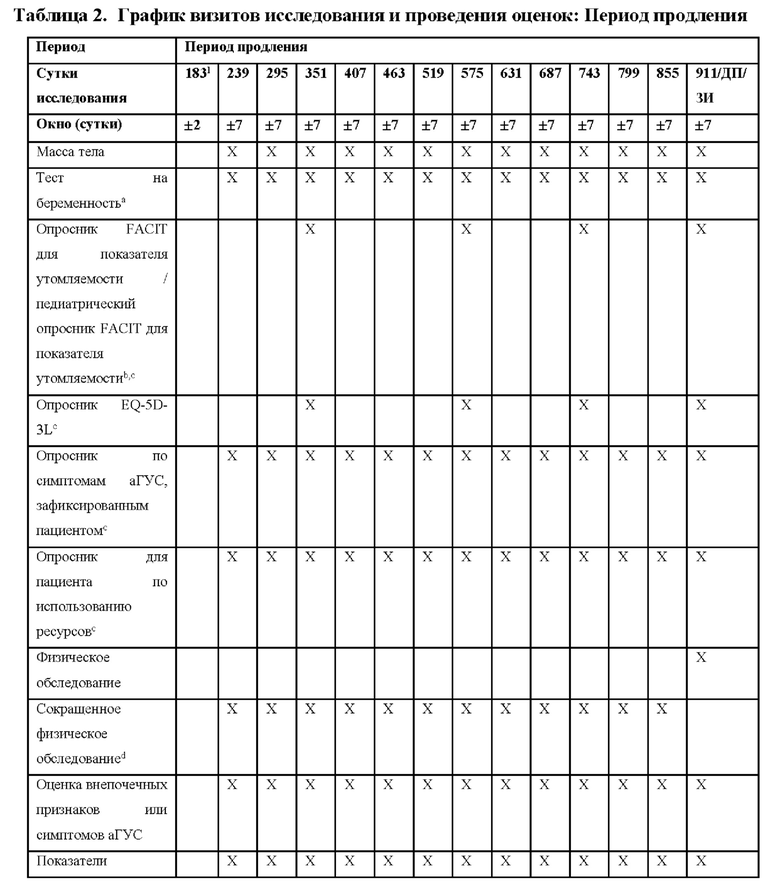
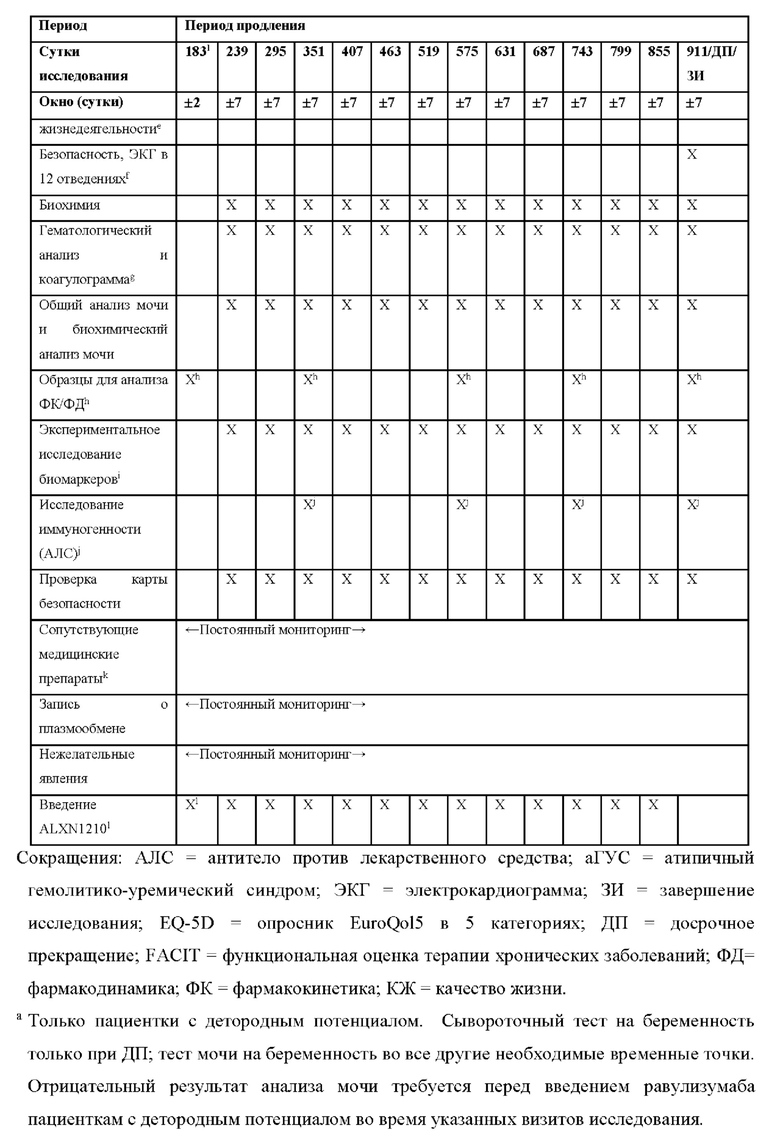


4. Популяция для исследования
В данном исследовании принимают участие в общей сложности около 55 пациентов с задокументированным аГУС, которые распределены для лечения равулизумабом в около 200 исследовательских центрах по всему миру. В данном исследовании принимают участие по меньшей мере от 6 до 10 пациентов-подростков (возрастом от 12 до < 18 лет во время скрининга) и по меньшей мере от 10 до 25 пациентов с предшествующими трансплантациями почек.
Субъекты, которые не соответствуют критериям для участия в данном исследовании (отсеивание по результатам скрининга), могут пройти скрининг повторно. Пациенты могут пройти скрининг повторно не больше чем 2 раза. Проспективное утверждение отклонений от протокола касательно критериев набора и зачисления, также известных как отступления от или исключения из протокола, не допускается.
Краткое изложение критериев включения и исключения представлено на фиг.3. Пациенты могут принимать участие в данном исследовании, если они соответствуют всем нижеследующим критериям и ни одному из критериев исключения:
Пациенты-мужчины или пациенты-женщины возрастом ≥ 12 лет с массой тела ≥ 40 кг на момент получения согласия.
• Признаки ТМА, включающие в себя тромбоцитопению, признаки гемолиза и дисфункцию почек, основанные на следующих результатах лабораторных исследований скринингового визита: Количество тромбоцитов < 150000 на микролитр (мкл) и ЛДГ ≥ 1,5 × верхняя граница нормы (ВГН), и гемоглобин < нижняя граница нормы (НГН) для соответствующих возраста и пола, и уровень креатинина в сыворотке крови ≥ ВГН у взрослых (возраст ≥ 18 лет) или ≥ 97,5-й процентиль для соответствующего возраста при скрининге у подростков (возраст от 12 до < 18 лет) (пациенты, которым требуется диализ вследствие острого повреждения почек, также могут принимать участие).
• Среди пациентов с трансплантацией почки: аГУС в анамнезе до текущей трансплантации почки или отсутствие аГУС в анамнезе, и стойкие признаки ТМА после приостановки приема ингибитора кальцинейрина ([CNI]; например, циклоспорина, такролимуса) или ингибитора мишени рапамицина млекопитающих ([mTORi]; например, сиролимуса, эверолимуса) в течение по меньшей мере 4 суток и по большей мере 7 суток.
• Среди пациенток с началом ТМА в послеродовом периоде - стойкие признаки ТМА в течение ≥ 3 суток после дня родов.
• Чтобы снизить риск менингококковой инфекции (Neisseria meningitidis), все пациенты должны быть вакцинированы против менингококковых инфекций в течение 3 лет до или во время начала применения исследуемого лекарственного средства. Пациенты, которые вакцинируются против менингококка меньше чем за 2 недели до начала лечения равулизумабом, получают лечение соответствующими профилактическими антибиотиками до момента времени в 2 недели после вакцинации. Пациенты, которые не были вакцинированы до начала лечения равулизумабом, получают профилактические антибиотики до и в течение по меньшей мере 2 недель после вакцинации против менингококка.
• Пациенты возрастом < 18 лет должны быть вакцинированы против Haemophilus influenzae типа b (Hib) и Streptococcus pneumoniae в соответствии с национальными и местными рекомендациями касательно графика вакцинаций.
• Пациентки с детородным потенциалом и пациенты с женщинами-партнершами с детородным потенциалом должны соблюдать указанные в протоколе рекомендации по контрацепции во время лечения и в течение по меньшей мере 8 месяцев после последней дозы исследуемого лекарственного средства.
• Готовы и способны дать письменное информированное согласие и соблюдать график визитов исследования. Для пациентов возрастом < 18 лет: законный опекун данного пациента должен быть готов и способен дать письменное информированное согласие, а данный пациент должен быть готов дать письменное информированное согласие несовершеннолетнего.
Образцы, собранные при скрининге, могут быть исследованы как в местной лаборатории, так и в центральной лаборатории. Если используются результаты анализов, выполняемых местной лабораторией, на ЛДГ, количество тромбоцитов, гемоглобин и креатинин сыворотки крови, собираются дублирующие образцы для выполнения анализов в центральной лаборатории, чтобы гарантировать, что исходные измерения и измерения после определения исходных уровней для данных анализов будут получены из центральной лаборатории. Несмотря на то, что результаты анализов, выполняемых местной лабораторией, могут быть использованы для ускорения оценки возможности участия в данном исследовании, окончательное определение данных критериев включения должно основываться на результатах, полученных из центральной лаборатории.
Пациенты исключаются из участия в данном исследовании, если они соответствуют любому из следующих критериев:
A. Известный дефицит (активность < 5%) «дизинтегрина и металлопротеиназы с мотивом тромбоспондина типа 1, члена семейства 13».
B. Гемолитико-уремический синдром, связанный с шигатоксином (ШТ-ГУС).
C. Гемолитико-уремический синдром (ГУС), связанный со Streptococcus pneumoniae, о чем свидетельствует положительный прямой тест Кумбса и инфицирование Streptococcus pneumoniae (например, культура, тест на антиген).
D. Известное инфицирование вирусом иммунодефицита человека (ВИЧ).
E. Неизлеченный системный менингококковый менингит.
F. Пациенты с подтвержденным диагнозом продолжающегося сепсиса, определенным на основании положительных культур крови в пределах 7 суток до начала скрининга и не получавшие лечения антибиотиками.
G. Наличие или подозрение на активную и нелеченую системную бактериальную инфекцию, которая, по мнению исследователя, затрудняет точный диагноз аГУС или препятствует способности контролировать болезнь аГУС.
H. Беременность или период лактации.
I. Пересадка сердца, легкого, тонкого кишечника или печени.
J. Среди пациентов с трансплантацией почки любое из следующих:
a. острая почечная дисфункция в пределах 4 недель после трансплантации, согласующаяся с диагнозом острого антитело-опосредованного отторжения (АОО), согласно критериям Banff 2013, или
b. острая почечная дисфункция в пределах 4 недель после трансплантации и повышение уровня донор-специфических антител (DSA), согласующиеся с клиническим диагнозом острого АОО.
c. Поликистозная болезнь почек в анамнезе.
K. Среди пациентов возрастом ≥ 18 лет с систолическим артериальным давлением
(САД) ≥ 170 мм рт.ст. или пациентов возрастом от 12 до < 18 лет с клиническим диагнозом артериальной гипертензии - любое из следующих:
а. Стойкие признаки ТМА (критерий включения №2) после меньше чем 4 суток снижения артериального давления (АД) до ≤ 140 мм рт.ст.
b. Известная гипертрофия левого желудочка.
с. Известные мелкие и гиперэхогенные почки в УЗИ.
L. Выявленный ГУС, связанный с воздействием лекарственного средства.
М. Получение ПО/ПТ в течение 28 суток или дольше до начала скрининга на текущую ТМА.
N. Злокачественное новообразование в анамнезе в пределах 5 лет по отношению к моменту скрининга, за исключением немеланомного рака кожи или карциномы шейки матки in situ, которые были вылечены без признаков рецидива.
О. Трансплантация костного мозга (ТКМ) / трансплантация гемопоэтических стволовых клеток (ТГСК) в пределах последних 90 суток до начала скрининга.
Р. ГУС, связанный с дефицитом витамина В12.
Q. Известный системный склероз (склеродермия), системная красная волчанка (СКВ) или положительная реакция на антифосфолипидные антитела, или антифосфолипидный синдром.
R. Хронический диализ (определяется как регулярный диализ в качестве заместительной почечной терапии при ТХПН).
S. Пациенты, хронически получающие внутривенный иммуноглобулин (ВВИГ) в пределах 8 недель до начала скрининга, за исключением случаев, не связанных с исследуемым заболеванием (например, гипогаммаглобулинемия); или хроническую терапию ритуксимабом в пределах 12 недель до начала скрининга.
Т. Пациенты, получающие другие иммуносупрессивные виды терапии, такие как стероиды, mTORi (например, сиролимус, эверолимус), CNI (например, циклоспорин или такролимус), исключаются, если: а) получают лечение по установленной схеме против отторжения после трансплантации, или Ь) у пациента подтверждены антитела к фактору комплемента, требующие иммуносупрессивной терапии, или с) стероиды применяются в связи с состоянием, отличным от аГУС (например, астмой).
U. Участие в другом исследовании с интервенционным лечением или получение любого экспериментального вида терапии в пределах 30 суток до начала получения исследуемого лекарственного средства на 1-е сутки в данном исследовании или в пределах 5 периодов полужизни данного исследуемого препарата, в зависимости от того, какое из значений больше.
V. Предшествующее применение экулизумаба или других ингибиторов комплемента.
W. Повышенная чувствительность к мышиным белкам или к одному из эксципиентов.
X. Любое медицинское или психологическое патологическое состояние, которое, по мнению исследователя, может повысить риск для пациента, если данный пациент будет участвовать в данном исследовании, или исказить результаты данного исследования.
Y. Известное или предполагаемое злоупотребление наркотиками или алкоголем, или зависимости от них в анамнезе в пределах 1 года до начала скрининга. Лабораторные результаты по критерию исключения номер 1 могут быть недоступны до введения первой дозы. Более поздние результаты по критерию исключения номер А могут привести к прекращению участия и замене данного пациента.
Пациент имеет право прекратить свое участие в данном исследовании в любое время. Если пациент отзывает свое согласие, проводятся оценки, указанные для посещения с целью досрочного прекращения (ДП). Пациенты, которые прекращают участие в данном исследовании, не заменяются. Прием пациентом исследуемого препарата может быть прекращен, если у исследователя или спонсора есть основания полагать, что в наилучших интересах пациента прекратить лечение.
Пациенты с предшествующей трансплантацией почки, у которых развивается АОО (C4d-положительная биопсия почки) и для которых ритуксимаб считается подходящим видом терапии, должны прекратить участие в данном исследовании и получать стандартный вид терапии. Основная причина и любая (-ые) другая (-ие) причина (-ы) прекращения регистрируются в эИРК.
Если пациент прекращает участие в данном исследовании и имеет текущее НЯ или не вернувшийся к норме лабораторный показатель, который значительно выходит за пределы референсного диапазона и клинически значим, исследователь пытается обеспечить последующее наблюдение до тех пор, пока не будет достигнуто удовлетворительное клиническое значение данного лабораторного показателя или нежелательного явления.
Спонсор или компетентный орган может прекратить данное исследование по разумной причине. Условия, которые служат основанием для прекращения данного исследования, включают в себя следующие, но не ограничиваются ими: (1) обнаружение неожиданного, серьезного или неприемлемого риска для пациентов, включенных в данное исследование, (2) решение спонсора приостановить или прекратить тестирование, оценку или разработку исследуемого препарата, (3) несоблюдение исследователем утвержденного протокола, применимых руководящих принципов и/или правил, и (4) предоставление заведомо ложной информации от исследователя спонсору и/или регулятивным органам.
Если в какой-либо момент будет установлено, что данные скрининга пациента не удовлетворяют одному или большему числу из следующих критериев включения/исключения (критерий включения номер 2 или критерий исключения номер 1), после получения по меньшей мере 1 дозы исследуемого препарата (например, данные анализа пациента, выполненного местной лабораторий, используемые для подтверждения критериев включения, впоследствии определяются центральной лабораторией, как больше не соответствующие критериям включения), участие данного пациента в данном исследовании прекращают, и данный пациент может быть заменен. Процедуры досрочного прекращения выполняются применимо к пациентам, участие которых было прекращено досрочно, и все НЯ собираются до 60 суток после введения указанному пациенту последней дозы исследуемого препарата.
Завершение исследования определяется как дата последнего визита последнего пациента в период продления.
5. Исследуемое лечение
Равулизумаб - гуманизированное моноклональное антитело против С5, состоящее из двух тяжелых цепей из 448 аминокислот и двух легких цепей из 214 аминокислот, представляет собой иммуноглобулин IgG2/4-каппа, состоящий из константных областей человека и областей, определяющих комплементарность, мыши, пересаженных на каркас из вариабельных областей легких и тяжелых цепей человека. Равулизумаби экулизумаб имеют более чем 99% идентичности первичной аминокислотной последовательности и очень схожую фармакологию.
Лекарственный продукт равулизумаб поставляется для клинических исследований в виде стерильного, не содержащего консервантов раствора концентрацией 10 мг/мл в одноразовых флаконах, и предназначен для инфузии путем разбавления в коммерчески доступном физиологическом растворе (0,9% раствор хлорида натрия для инъекций; фармакопея для конкретной страны) для введения путем внутривенной (в/в) инфузии. Таблица 3 и текущая брошюра исследователя (БИ) предоставляют дополнительную информацию
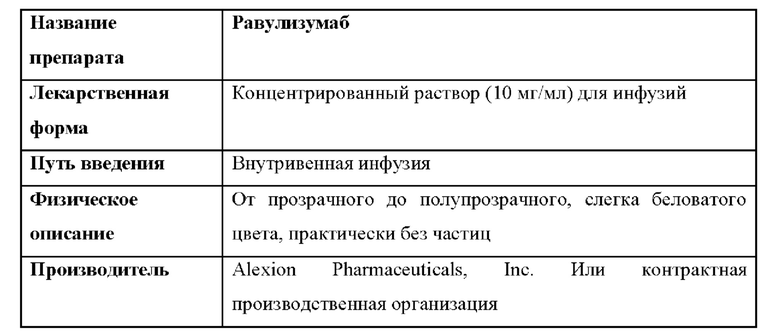
Равулизумаб упакован во флаконы из боросиликатного стекла 1-го типа согласно Фармакопеи США (USP) / Фармакопеи Европейского союза (ЕР) и закупорен бутилкаучуковой пробкой с алюминиевым колпачком и комбинированным колпачком «флип-офф». Исследуемое лекарственное средство поставляется в наборах. Равулизумаб поставляется в каждый исследовательский центр после получения всех необходимых обязательных документов в соответствии с применимыми нормативно-правовыми актами.
По прибытии наборов данного исследуемого лекарственного средства в исследовательский центр фармацевт (или назначенное квалифицированное лицо) незамедлительно извлекает наборы данного исследуемого лекарственного средства из транспортировочного холодильника и хранит их в оригинальных картонных упаковках в условиях охлаждения при температуре от 2°С до 8°С (от 35°F до 47°F) и в защищенном от света месте. Равулизумаб не замораживается. Исследуемое лекарственное средство хранится в защищенном помещении с ограниченным доступом, и температура контролируется ежедневно.
Лекарственное средство перед введением находится при комнатной температуре. Материал не нагревается никакими средствами (например, с помощью микроволновой печи или другого источника тепла), кроме температуры окружающего воздуха.
Равулизумаб не вводят путем в/в струйного введения или болюсной инъекции. Инфузионные растворы исследуемого лекарственного средства готовят в асептических условиях. Требуемую для пациента дозу равулизумаба дополнительно разбавляют в коммерчески доступном физиологическом растворе (0,9% хлорид натрия; фармакопея для конкретной страны) в объеме, указанном в таблице 4. Раствор равулизумаба в разбавителе вводят пациенту посредством системы для внутривенного введения с помощью инфузионной помпы. Для инфузии необходимо использовать встроенный в систему фильтр.
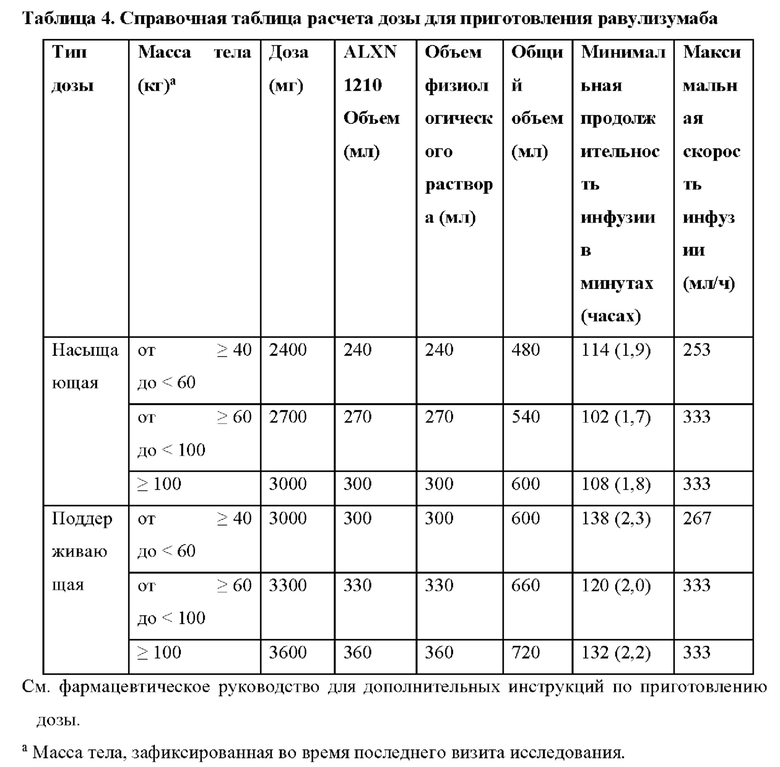
Дозы исследуемого лекарственного средства готовятся и отпускаются только фармацевтом или квалифицированным медицинским работником. Исследуемое лекарственное средство отпускаются только зарегистрированным пациентам с подтвержденным правом на участие в данном исследовании. Как только исследуемое лекарственное средство приготовлено для пациента, оно вводится только этому пациенту. Флаконы с исследуемым лекарственным средством предназначены только для одноразового использования, и любой лекарственный продукт, оставшийся во флаконе, не используется для другого пациента. Любое лекарственное средство, оставшееся в системе для инфузий или инфузионном мешке, не используется для другого пациента.
Все материалы клинического исследования хранятся в надежном месте, распределяются и отпускаются надлежащим образом обученными лицами. Ведется подробный учет полученных, отпущенных и уничтоженных количеств исследуемого препарата. Если не указано иное, пустые флаконы и флаконы с остаточными материалами хранятся для проверки и отчетности наблюдателем за исследованием до их уничтожения или обрабатываются в соответствии с местными фармацевтическими стандартными операционными процедурами (СОП) для лекарственных средств в клинических исследованиях. Чтобы выполнить требования регуляторных органов, касающиеся учета лекарственного средства, в конце исследования все оставшиеся запасы равулизумаба сверяются и уничтожаются или возвращаются в Alexion в соответствии с применимыми нормативно-правовыми актами.
Пациенты получают равулизумаб в течение 26 недель. Равулизумаб вводят путем медленной в/в инфузии в течение около 2 часов. Равулизумаб не вводят путем в/в струйного введения или болюсной инъекции.
Клиническая схема применения равулизумаба в течение периода первичной оценки основана на последней зарегистрированной массе тела пациента во время визита исследования (таблица 5). Пациенты получают насыщающую дозу равулизумаба в/в на 1-е сутки, а затем поддерживающую дозу равулизумаба в/в на 15-е сутки и 1 р./8 нед (каждые восемь недель) после этого.
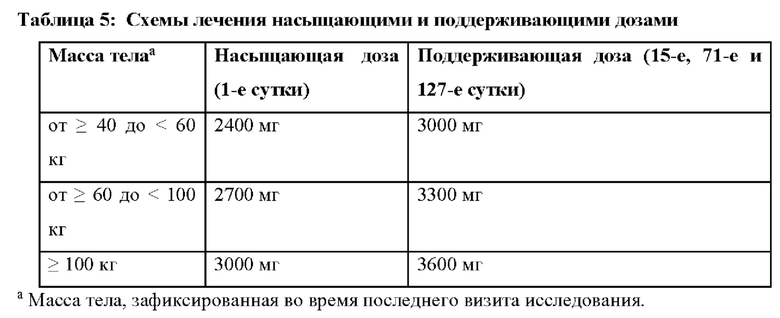
После периода первичной оценки все пациенты переходят в период продления продолжительностью до 2 лет, в течение которого все пациенты получают равулизумаб 1 р./8 нед. (каждые восемь недель). Фактическое время введения всех доз регистрируется в эИРК пациента.
Данное исследование является незас лепленным. Пациентам, которые соответствуют всем критериям для участия в данном исследовании, назначается исследуемое лечение равулизумабом во время первоначального визита (1-е сутки). Интерактивная система голосового ответа или веб-ответа (IxRS) используется для назначения флаконов, содержащих равулизумаб, каждому пациенту.
Инфузия других моноклональных антител была связана с инфузионными реакциями, которые обычно начинаются во время инфузий или вскоре после завершения инфузий.
Ранее назначенные медицинские препараты (включая витамины и растительные препараты), в том числе те, которые обсуждаются в критериях исключения, и процедуры (любое терапевтическое вмешательство, такое как хирургическое вмешательство / биопсия или физиотерапия), которые пациент принимает или которым подвергается в пределах 28 суток (или 3 лет - для документирования вакцинации против менингококка) до начала скрининга до введения первой дозы равулизумаба, регистрируются в эИРК пациента.
Для аналитических целей любой диализ в течение 14-дневного периода, непосредственно следующего за введением первой дозы равулизумаба, не считается «новым диализом».
Все приемы медицинских препаратов и процедуры, предпринятые во время исследования, регистрируются в исходном документе / медицинской карте и в эИРК пациента. Регистрация данной информации включает в себя все отпускаемые по рецепту лекарственные средства, растительные препараты, витамины, минералы, отпускаемые без рецепта лекарственные средства и принимаемые на данный момент медицинские препараты. Сопутствующие медицинские препараты регистрируются с момента первой инфузий исследуемого лекарственного средства до 56 суток после введения пациенту последней дозы исследуемого лекарственного средства. Любые изменения в сопутствующих медицинских препаратах также регистрируются в исходном документе / медицинской карте и в эИРК пациента. Любой сопутствующий медицинский препарат, который считается необходимым для стандартного лечения пациента во время данного исследования или для лечения любого НЯ, наряду с разрешенными медицинскими препаратами, описанными ниже, может быть назначен по усмотрению исследователя. Тем не менее, исследователь несет ответственность за то, чтобы сведения обо всех медицинских препаратах были полностью зарегистрированы в исходном документе / медицинской карте и в эИРК пациента.
Пациентам запрещается принимать любые из следующих медицинских препаратов и процедур в любое время после введения первой дозы исследуемого лекарственного средства: экулизумаб или другие ингибиторы комплемента, использование любого другого исследуемого лекарственного средства или устройства в рамках клинического испытания, ВВИГ (за исключением медицинской необходимости, не связанной с исследуемым заболеванием, например, гипогаммаглобулинемия), ритуксимаб, ПО/ПТ после введения первой дозы и новый диализ в течение первых 48 часов после введения первой дозы равулизумаба, если нет настоятельной медицинской необходимости, оцениваемой как: (1) гиперволемия, не отвечающая на диуретики, (2) рефракторный дисбаланс электролитов, или (3) впервые выявленная уремическая энцефалопатия. Исключения должны быть одобрены спонсором до проведения диализа в зависимости от конкретного случая.
Следующие сопутствующие медицинские препараты и процедуры разрешены при определенных обстоятельствах и со следующими ограничениями: использование других иммуносупрессивных видов терапии (таких как стероиды, mTORi [например, сиролимус, эверолимус], CNI [например, циклоспорин или такролимус]) до скрининга или во время данного исследования не допускается, если: а) является частью лечения по установленной схеме против отторжения после трансплантации, или Ь) у пациента подтверждены антитела против факторов комплемента, требующие иммуносупрессивной терапии, или с) стероиды используются для лечения состояния, отличного от аГУС (например, астмы).
Любые пациенты, получающие другие ингибиторы комплемента (включая экулизумаб) или проходящие ПО/ПТ после введения первой дозы исследуемого лекарственного средства, прекращают участие в данном исследовании.
Из-за его механизма действия, применение равулизумаба повышает восприимчивость пациента к инфекции. Чтобы снизить риск инфекции, все пациенты вакцинируются против N. meningitidis, Hib и Streptococcus pneumoniae.
Пациенты вакцинируются против N. meningitidis в пределах 3 лет до или к моменту приема первой дозы равулизумаба. Пациенты, которые получают лечение исследуемым лекарственным средством меньше чем через 2 недели после введения менингококковой вакцины, получают лечение соответствующими профилактическими антибиотиками до момента времени в 2 недели после вакцинации. Вакцины против серотипов А, С, Y, W135 и В, где они доступны, рекомендуются для предотвращения распространенных патогенных серотипов менингококка. Пациенты вакцинируются или ревакцинируются в соответствии с действующими национальными руководящими принципами вакцинации или местной практикой для использования вакцинации с ингибиторами комплемента (например, экулизумабом).
Следует признать, что некоторые пациенты, которые не были вакцинированы против N. meningiditis в пределах 3 лет до получения первой дозы равулизумаба, могут не иметь возможности пройти вакцинацию к моменту приема первой дозы. Пациенты, которые не были вакцинированы до начала лечения равулизумабом, получают профилактические антибиотики до и в течение по меньшей мере 2 недель после вакцинации против менингококка.
Вакцинации может быть недостаточно для предотвращения менингококковой инфекции. Следует принимать во внимание официальное руководство и местную практику по надлежащему использованию антибактериальных агентов. Все пациенты находятся под наблюдением на предмет ранних проявлений менингококковой инфекции, немедленно оцениваются при подозрении на инфекцию и при необходимости лечатся соответствующими антибиотиками.
Чтобы повысить осведомленность о рисках и способствовать быстрому выявлению любых потенциальных проявлений или симптомов инфекции, с которыми пациенты сталкиваются в ходе данного исследования, пациентам предоставляется карта безопасности для постоянного ношения с собой. Дополнительное обсуждение и объяснение потенциальных рисков, признаков и симптомов происходят в определенные моменты времени в рамках проверки карты безопасности пациента и на протяжении всего исследования, как описано в графике проведения оценок (таблица 1 и таблица 2).
Пациенты вакцинируются против Haemophilus influenzae типа b (Hib) и Streptococcus pneumoniae в соответствии с национальными и местными рекомендациями по графику вакцинации до или во время приема первой дозы равулизумаба. Статус вакцинации против N. meningitidis, Hib и S. pneumoniae регистрируется в эИРК пациента.
Пациентам вводят исследуемое лекарственное средство в контролируемых условиях под наблюдением исследователя или назначенного им лица, тем самым обеспечивая соблюдение режима введения исследуемого лекарственного средства. Исследователь или назначенное лицо обеспечивают, чтобы все пациенты были надлежащим образом проинформированы о конкретной схеме применения, необходимой для соблюдения протокола исследования, гарантируют, что пациент получает соответствующую дозу в назначенные моменты времени во время исследования и что во время инфузий осуществляется надлежащий мониторинг безопасности.
Перед приемом исследуемого лекарственного средства пациентки, которые считают себя в постменопаузе, должны предоставить доказательства менопаузы, основанные на сочетании аменореи в течение по меньшей мере 1 года и повышенного уровня фолликулостимулирующего гормона (ФСГ) в сыворотке крови (> 30 МЕ/л) (например, при отсутствии заместительной гормональной терапии, пищевых фитоэстрогенов).
Пациентки с детородным потенциалом используют высокоэффективный способ контрацепции (как определено ниже), начиная с момента скрининга и продолжая в течение по меньшей мере 8 месяцев после введения последней дозы исследуемого лекарственного средства. Высокоэффективные способы контрацепции* включают: гормональную контрацепцию, связанную с ингибированием овуляции, внутриматочное устройство, внутриматочную систему высвобождения гормонов, двустороннюю окклюзию маточных труб, вазэктомию у партнера (при условии, что партнер является единственным сексуальным партнером пациента), половое воздержание (определяется как воздержание от гетеросексуальных половых контактов в течение всего периода риска, связанного с лечением исследуемым лекарственным средством; надежность полового воздержания необходимо оценивать относительно продолжительности клинического исследования и предпочтительного и обычного образа жизни пациента), комбинации мужского презерватива с колпачком, диафрагмой или губкой со спермицидом (способы двойной барьерной контрацепции). Пациенты мужского пола, у которых есть супруга / партнер женского пола, потенциально способная к деторождению, или беременная либо кормящая грудью супруга либо партнер, соглашаются использовать средства двойной барьерной контрацепции (мужской презерватив плюс соответствующий барьерный способ для партнера женского пола) во время лечения и в течение по меньшей мере 8 месяцев после введения последней дозы исследуемого лекарственного средства. Двойная барьерная контрацепция необходима даже при документально подтвержденной медицинской оценке хирургически успешной вазэктомии.
Пациенты мужского пола не сдают сперму во время лечения и в течение по меньшей мере 8 месяцев после введения последней дозы исследуемого лекарственного средства.
6. Оценки эффективности
Первичной оценкой эффективности является полный ответ ТМА в течение 26-недельного периода первичной оценки. Критериями полного ответа ТМА являются: (1) нормализация количества тромбоцитов, (2) нормализация уровня ЛДГ и (3) повышение уровня креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем.
Пациенты, которые отвечают всем критериям полного ответа ТМА, подтвержденным 2 последовательными измерениями, полученными с интервалом по меньшей мере 4 недель, классифицируются как соответствующие первичному критерию оценки эффективности.
В ходе данного исследования измеряются следующие вторичные оценки эффективности:
A. статус потребности в диализе;
B. время до полного ответа ТМА;
C. статус полного ответа ТМА с течением времени;
D. наблюдаемое значение и изменение по сравнению с исходным уровнем рСКФ;
E. стадия ХБП на основании оценки исследователя в определенные целевые дни и классифицированная как улучшенная, стабильная (без изменений) или ухудшенная по сравнению с исходным уровнем;
F. наблюдаемое значение и изменение по сравнению с исходным уровнем гематологических параметров (тромбоциты, ЛДГ, гемоглобин);
G. повышение уровня гемоглобина на ≥ 20 г/л по сравнению с исходным уровнем, сохраняющееся по меньшей мере в течение 2 последовательных измерений, произведенных с интервалом по меньшей мере в 4 недели;
H. изменение КЖ по сравнению с исходным уровнем, измеренное с помощью опросников EQ-5D-3L (все пациенты), FACIT версии 4 для показателя утомляемости (пациенты возрастом ≥ 18 лет) и педиатрического FACIT для показателя утомляемости (пациенты возрастом < 18 лет).
7. Оценки безопасности
Исследователь или назначенное лицо встречаются с пациентами, чтобы обсудить потенциальные риски касательно безопасности равулизумаба и предоставить исследователю возможность рассмотреть любые опасения пациента касательно безопасности, связанные с данным исследованием.
Мониторинг сбора НЯ проводится с момента получения информированного согласия до завершения исследования. Исследователи следят за любыми НЯ до их завершения (устранения или стабилизации). В случае выхода пациента из данного исследования мониторинг НЯ продолжается до последнего визита последнего пациента, если это возможно. Сроки проведения клинических и лабораторных оценок определяются в соответствии с графиком проведения оценок (таблицы 1 и 2). За любыми клинически значимыми отклонениями от нормы следят до их устранения или стабилизации.
Проводят обзор демографических параметров, включая возраст, пол, расу и этническую принадлежность. Собирают и документируют полный медицинский анамнез. Регистрируют массу тела и рост. Рост измеряется только во время скрининга.
Медицинский анамнез пациента с аГУС, включая появление первого симптома аГУС и дату постановки диагноза, документируют при скрининговом визите.
Медицинский анамнез пациента, включая предшествующие и сопутствующие патологические состояния / расстройства, регистрируют при скрининговом визите. В дополнение к информации о вакцинации против менингококка также регистрируют использование медицинских препаратов (отпускаемых по рецепту или без рецепта, включая витамины и/или растительные добавки) в течение 28 дней (или 3 лет - для документирования вакцинации против менингококка) до начала скрининга.
Физическое обследование включает в себя следующие оценки: внешний вид пациента; кожа; голова, ухо, глаз, нос и горло; шея; лимфатические узлы; грудь; сердце; брюшная полость; конечности; центральная нервная система; и опорно-двигательный аппарат. Сокращенное физическое обследование состоит из обследования систем организма, основанного на оценке исследователя и симптомах пациента. Измерения показателей жизнедеятельности производят после того, как пациент отдыхал по меньшей мере 5 минут, и включают в себя систолическое и диастолическое АД (в миллиметрах ртутного столба [мм рт.ст.]), пульсоксиметрию, частоту сердечных сокращений (ударов в минуту), частоту дыхания (вдохов в минуту) и пероральное или тимпаническое измерение температуры (в градусах Цельсия [°С] или градусах Фаренгейта [°F]).
Сбор образцов для теста на беременность по сыворотке крови, гематологического исследования, биохимического исследования, коагулограммы и анализа мочи проводят в сроки, указанные в графике проведения оценок (таблицы 1 и 2). Образцы для лабораторных исследований собирают перед каждым введением исследуемого лекарственного средства.
Образцы, собранные при скрининге, могут быть исследованы как в местной лаборатории, так и в центральной лаборатории. Если используются результаты анализов, выполняемых местной лабораторией, на ЛДГ, количество тромбоцитов, гемоглобин и креатинин сыворотки крови, собираются дублирующие образцы для выполнения анализов в центральной лаборатории, чтобы гарантировать, что исходные измерения и измерения после определения исходных уровней для данных анализов будут получены из центральной лаборатории. В случае дублирования образцов из местных и центральных лабораторий для анализа используются результаты центральной лаборатории.
Ожидается, что некоторые лабораторные показатели могут находиться за пределами нормального диапазона значений из-за основного заболевания. Исследователи должны использовать медицинское суждение при оценке клинической значимости этих показателей.
Клиническая значимость определяется как любая вариация лабораторных измерений, имеющая медицинское значение и приводящая к изменению медицинской помощи. Если отмечаются клинически значимые лабораторные изменения по сравнению с исходным значением, такие изменения документируются как НЯ в эИРК для НЯ. Исследователь оценивает взаимосвязь с исследуемым лечением для всех клинически значимых параметров, находящихся вне нормального диапазона. Исследователь продолжает наблюдать за пациентом посредством дополнительных лабораторных исследований до тех пор, пока: (1) параметры вернутся в нормальный диапазон или к исходному уровню, или (2) по мнению исследователя, параметры, которые выходят за пределы нормального диапазона, не связаны с введением исследуемого лекарственного средства или другими процедурами, определяемыми протоколом.
Для женщин с детородным потенциалом тест на беременность по сыворотке крови или моче (т.е. бета-хорионический гонадотропин человека [β-ХГЧ]) проводится в соответствии с графиком проведения оценок (таблицы 1 и 2). Образцы крови анализируют относительно гематологических параметров.
Образцы крови анализируют относительно биохимических параметров сыворотки крови. Непрямой билирубин рассчитывают на основании значений общего билирубина и прямого билирубина; следовательно, результаты определения непрямого билирубина недоступны, если прямой билирубин ниже предела количественной оценки. Уровень ФСГ в сыворотке крови измеряют во время скрининга пациенток в постменопаузе для подтверждения их постменопаузального статуса.
Биохимические исследования проводят в моменты времени, указанные в графике проведения оценок (таблицы 1 и 2). рСКФ рассчитывают для всех визитов, во время которых собирают показатели биохимии сыворотки крови, с использованием формулы модификации диеты при заболевании почек для пациентов возрастом ≥ 18 лет и формулы Шварца для модификации диеты при заболевании почек для пациентов возрастом < 18 лет.
Образцы крови анализируют относительно показателей свертывания крови.
Анализируют образцы мочи. Проводят микроскопию образцов мочи, если результаты макроскопического анализа не являются нормальными. Образцы мочи также анализируют для измерения уровней белков и креатинина, чтобы рассчитать соотношение общего белка и креатинина в моче.
Для каждого пациента снимают однократные цифровые ЭКГ в 12 отведениях в соответствии с графиком проведения оценок (таблицы 1 и 2). Пациенты должны лежать на спине в течение около 5-10 минут перед снятием ЭКГ и продолжать лежать на спине, но не в состоянии сна, во время снятия ЭКГ. Исследователь или назначенное лицо несут ответственность за анализ ЭКГ, чтобы оценить, находится ли ЭКГ в пределах нормы, и определить клиническую значимость результатов. Эти оценки указывают в ИРК.
Собирают образцы крови для тестирования на наличие и титр АЛС к равулизумабу в сыворотке крови до введения исследуемого лекарственного средства, как указано в графике проведения оценок (см. таблицы 1 и 2). Если результаты данного анализа положительные, то данный анализ могут повторять каждые 3 месяца до тех пор, пока результаты станут отрицательными или стабилизируются на основании измеренного титра и оценок безопасности. При необходимости можно провести дополнительную характеризацию образования антител, включая связывание и нейтрализацию антител, ФК/ФД, безопасность и активность равулизумаба.
НЯ представляет собой любое нежелательное медицинское происшествие у пациента, которому вводят фармацевтический препарат, и которое не обязательно имеет причинно-следственную связь с данным лечением. Следовательно, НЯ может быть любым неблагоприятным или непредусмотренным признаком (например, отклонением от нормы в лабораторном исследовании), симптомом или заболеванием, временно связанными с применением медицинского продукта, независимо от того, считается или не считается оно связанным с данным медицинским продуктом.
Ситуации, в которых неблагоприятного медицинского события не произошло (например, госпитализация для плановой операции, если она запланирована до начала данного исследования, госпитализация по социальным причинам или для удобства), и ожидаемые ежедневные колебания предшествующего (-их) заболевания (-й) или патологического (-их) состояния (-й), которые присутствуют или обнаружены в начале данного исследования, которые не ухудшаются, не являются НЯ.
Отсутствие эффекта от лекарственного средства не является НЯ в клинических исследованиях, поскольку целью клинического исследования является установление эффекта от лекарственного средства.
Ошибка в приеме медицинского препарата (включая преднамеренное неправильное использование, злоупотребление и передозировку медицинским продуктом) или применение, отличное от того, что определено в протоколе, не считается НЯ, если только в результате ошибки в приеме медицинского препарата не произошло неблагоприятное медицинское событие.
О случаях беременности, которые происходят на протяжении периода, когда мать или отец подвергаются воздействию исследуемого препарата, следует сообщать в пределах 24 часов с момента информирования исследователя / исследовательского участка. Данные о влиянии на плод и грудном вскармливании собираются для нормативной отчетности и оценки безопасности.
Нежелательные явления регистрируются с момента подписания согласия. НЯ, о котором сообщается после получения информированного согласия, но до введения исследуемого лекарственного средства, считается предшествующим лечению НЯ.
Следующие явления являются важными выявленными рисками в данном исследовании: менингококковые инфекции.
Степень тяжести НЯ оценивается с использованием Общих терминологических критериев нежелательных явлений (СТСАЕ) версии 4.03 или выше. Для каждого термина НЯ предусмотрена шкала оценки (тяжести). Каждый термин СТСАЕ является термином низкого уровня (LLT) в Медицинском словаре для регуляторной деятельности (MedDRA®). Каждый LLT кодируется в соответствии с предпочтительным термином MedDRA. Оценка относится к степени тяжести НЯ. СТСАЕ присваивает оценку от 1 до 5, с однозначными клиническими описаниями степени тяжести для каждого НЯ (таблица 6).

Любое изменение тяжести НЯ документируется на основе конкретных рекомендаций, содержащихся в Руководстве по заполнению эИРК. Степень тяжести и серьезность различаются: степень тяжести описывает интенсивность НЯ, в то время как термин «серьезность» относится к НЯ, которое соответствует определенным критериям серьезного нежелательного явления (СНЯ).
Исследователь должен предоставить оценку достоверности причинно-следственной связи (не связано, связь маловероятна, связь возможна, связь вероятна или определенно связано) для всех НЯ (как серьезных, так и несерьезных) на основе медицинского суждения исследователя и наблюдаемых симптомов, связанных с данным явлением (таблица 7). Данная оценка регистрируется в эИРК и любых дополнительных формах по мере необходимости.
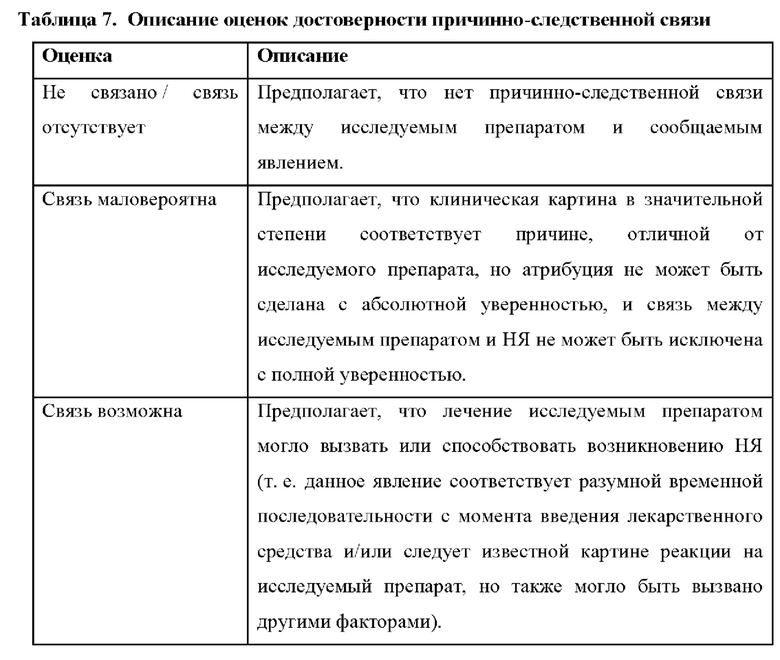
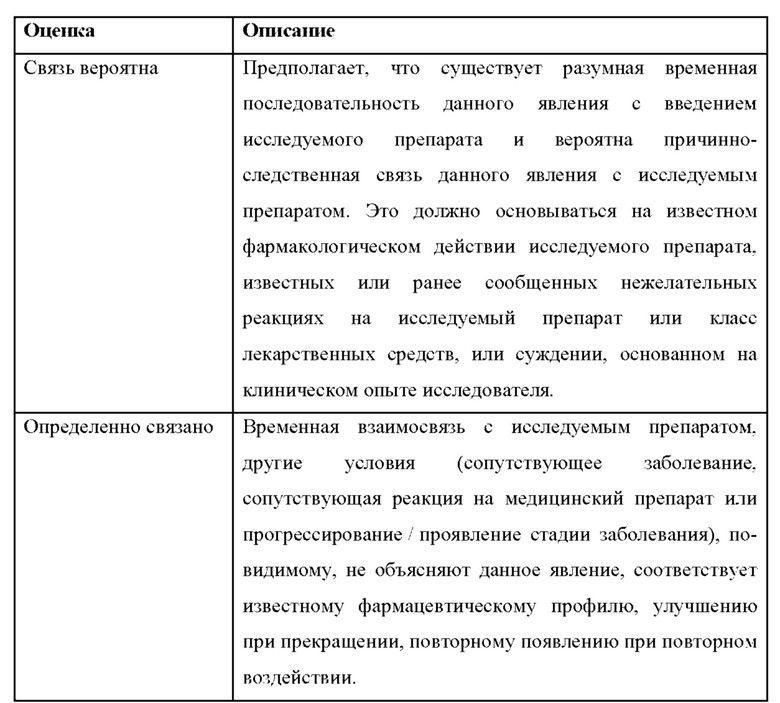
Серьезным нежелательным явлением (СНЯ) является любое неблагоприятное медицинское событие, которое:
• Приводит к смерти
• Является опасным для жизни (т.е. пациент находился под угрозой смерти во время протекания данного явления)
• Требует госпитализации в стационар или продления существующей госпитализации.
• Приводит к стойкой или значительной инвалидизации / нетрудоспособности
• Является врожденной аномалией / пороком развития
Важные медицинские явления, которые могут не привести к смерти, могут не быть непосредственно опасными для жизни или не требовать госпитализации, могут рассматриваться как серьезные нежелательные явления в тех случаях, когда, на основании соответствующего медицинского суждения, они могут поставить пациента под угрозу или могут потребовать вмешательства для предотвращения одного из последствий, перечисленных выше.
Предполагаемые непредвиденные серьезные нежелательные явления (ПНСНЯ, англ. «SUSAR») представляют собой серьезные явления, которые не указаны в БИ и которые исследователь определяет как связанные с исследуемыми препаратом или процедурой. Раздел 21 свода федеральных нормативных актов США (CFR) 312.32 и Директива Европейского союза по клиническим испытаниям 2001/20/ЕС, и связанные с ними подробные руководства или национальные нормативно-правовые требования в странах-участницах требуют отчетности о ПНСНЯ.
Все НЯ (серьезные и несерьезные) собираются с момента подписания формы информированного согласия (ФИС) до 60 суток после введения последней дозы исследуемого лекарственного средства для пациентов, досрочно прекратившим участие в исследовании, или до 56 суток после введения последней дозы исследуемого лекарственного средства для пациентов, которые завершили исследование. Все НЯ регистрируются в эИРК после того, как исследователь или его/ее сотрудники узнают об их возникновении.
Все СНЯ регистрируются независимо от оценки причинно-следственной связи исследователем. Не существует ограничений по времени для сообщения о СНЯ, которые, как считается, причинно связаны с исследуемым лекарственным средством. Исследователи могут в любое время сообщать о СНЯ, независимо от причинно-следственной связи.
Для всех СНЯ исследователь должен предоставить следующее: соответствующую и запрошенную дополнительную информацию, причинно-следственную связь СНЯ, лечение / вмешательство, проведенные относительно СНЯ, результат СНЯ и подтверждающую медицинскую документацию, и лабораторную / диагностическую информацию.
Данные о беременности собираются в ходе данного исследования для всех пациенток и женщин-супругов/партнеров пациентов мужского пола. Воздействие во время беременности (также называемое воздействием in utero) может быть результатом либо воздействия на организм матери, либо передачи лекарственного препарата через сперму после воздействия на организм отца. Беременность сама по себе не рассматривается как НЯ, если только нет подозрения, что исследуемый препарат мог повлиять на эффективность противозачаточных препаратов. Однако осложнения беременности и отклоняющиеся от нормы исходы беременности являются НЯ и могут соответствовать критериям для СНЯ (например, эктопическая беременность, самопроизвольный аборт, внутриутробная гибель плода, смерть новорожденного или врожденный порок развития). Плановые аборты без осложнений не должны регистрироваться как НЯ.
8. Оценки фармакокинетики и фармакодинамики
Образцы крови для определения концентрации лекарственного средства в сыворотке крови и оценки ФД собирают до и после введения исследуемого лекарственного средства в моменты времени, указанные в графике проведения оценок (см. таблицы 1 и 2). Регистрируют фактические дату и время (в 24-часовом формате) сбора каждого образца. Число моментов времени для ФК-выборки для любого конкретного пациента не превышает ныне запланированное число моментов времени.
Образцы крови для оценки ФК и ФД берут из руки, противоположной руке, используемой для введения лекарственного средства. Оценки для ФК/ФД следующие: (1) изменения концентрации равулизумаба в сыворотке крови с течением времени и (2) изменение концентраций свободного С5.
9. Экспериментальные оценки
Для экспериментальных исследований биомаркеров приводятся сводные статистические данные о значении параметра, изменениях и процентных изменениях по сравнению с исходным уровнем.
Взаимосвязь между концентрацией равулизумаба и экспериментально исследуемыми биомаркерами или корреляция между клинической пользой и ключевыми экспериментально исследуемыми биомаркерами могут быть оценены с помощью графического представления. Также могут быть проведены экспериментальный анализ и анализ потенциальных взаимосвязей между клиническими исходами, ФК/ФД, генетическим профилем и уровнями биомаркеров. Кратко излагаются данные об активности АПК и результаты анализа аутоантител, если проводилась их оценка.
Экспериментальные генетические исследования могут быть выполнена для изучения генетических вариантов в генах, которые, как известно, ассоциированы с аГУС, а также для выявления новых генетических вариантов, ассоциированных с аГУС, нарушением регуляции комплемента, метаболизмом или эффективностью равулизумаба.
Исследователь сообщает пациенту или опекуну пациента о генетических мутациях, имеющих известное клиническое значение при аГУС, и проводит соответствующую генетическую консультацию. Генетические варианты неизвестного клинического значения не сообщают пациентам или их исследователю.
Дополнительные признаки или симптомы аГУС оцениваются с помощью Опросника для пациента по использованию ресурсов и Опросника о сообщаемых пациентом симптомах аГУС.
Компоненты внепочечных признаков или симптомов аГУС, включая показатели жизнедеятельности и результаты клинических лабораторных исследований, могут быть описательно обобщены для исходных моментов времени, моментов времени после определения исходных уровней и относительно изменений по сравнению с исходным уровнем. Могут быть предоставлены списки пациентов.
Анализ признаков, симптоматики и использования ресурсов может включать в себя стандартные подходы к категориальным результатам с повторными измерениями или без повторных измерений.
Если оценка для 1-х суток отсутствует, в качестве исходной оценки используется скрининговая оценка.
Для оценки полного ответа ТМА в течение 26-недельного периода первичной оценки (первичный критерий оценки) для пациентов, у которых отсутствует оценка эффективности, которая является частью определения полного ответа ТМА во время участия в исследовании, используют последнее задокументированное значение (методика LOCF). Для пациентов, которые досрочно прекратили участие в данном исследовании до 26-й недели, для оценки полного ответа ТМА используются их данные до момента прекращения участия в данном исследовании.
Недостающие данные для систем определения КЖ обрабатываются в соответствии с инструкциями для каждой системы определения.
Промежуточный анализ для данного исследования запланирован на конец 26-недельного периода первичной оценки после того, как все пациенты завершат 26-недельный период первичной оценки или досрочно выйдут из указанного периода. В дополнение к этому, в конце 2-летнего периода продления проводится второй анализ для обобщения долгосрочных показателей эффективности, безопасности и ФК.
ПРИМЕР 2: Данные для несравнительного многоцентрового исследования 3-й фазы равулизумаба (ALXN1210) у взрослых пациентов с атипичным гемолитико-уремическим синдромом (аГУС), ранее не получавших лечение ингибиторами комплемента
Ниже приводится краткое изложение данных из одного исследования равулизумаба (ALXN1210-aHUS-311) у пациентов с атипичным гемолитико-уремическим синдромом (аГУС), ранее не получавших лечение ингибиторами комплемента, которое было проведено по существу в соответствии с протоколом, описанным выше в примере 1. Период первичной оценки составлял 26 недель, а далее следовал период продления продолжительностью до 2 лет.План данного исследования представлен на фиг.1.
Целью данного исследования была оценка эффективности равулизумаба у взрослых пациентов с аГУС, ранее не получавших лечение ингибиторами комплемента, в ингибировании опосредованной комплементом тромботической микроангиопатии (ТМА), характеризующейся тромбоцитопенией, гемолизом и нарушением функции почек. Первичным критерием оценки был полный ответ ТМА в пределах 26-недельного периода первичной оценки. Первичные, вторичные критерии оценки и критерии оценки безопасности для данного исследования обобщены на фиг.2. Краткое изложение критериев включения и исключения представлено на фиг.3.
Требования к зачислению заключались в следующем: (1) по меньшей мере 6 подростков (отложено до ALXN1210-aHUS-312), (2) по меньшей мере 10 пациентов с предшествующей трансплантацией почки (зачислено 8) и (3) по меньшей мере 30 пациентов, отвечающих критериям лабораторной оценки ТМА на 1-е сутки на основе результатов центральной лаборатории (зачислено 32).
Объем собираемых данных включает в себя данные за период первичной оценки для всех пациентов, а также любые доступные данные за период продления до октября 2019 года включительно.
Пятьдесят восемь (58) субъектов были зачислены и получили по меньшей мере одну дозу (включены в выборку для анализа безопасности), два субъекта были зачислены и заменены (признаны неприемлемыми после введения первой дозы и исключены, как предопределено в протоколе), пятьдесят шесть (56)субъектов входили в выборку для полного анализа, одиннадцать (11) субъектов прекратили лечение и девять (9) субъектов прекратили участие в данном исследовании. Распределение пациентов схематически указано на фиг.4. Комплаентность лечению составляла 100%. Исходные демографические показатели указаны в таблице 8, исходные показатели заболевания указаны в таблице 9, а исходные лабораторные показатели указаны в таблице 10.
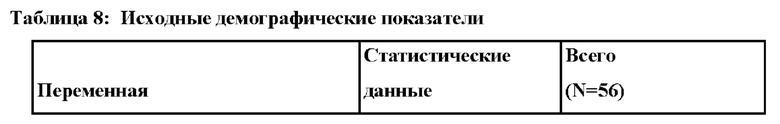
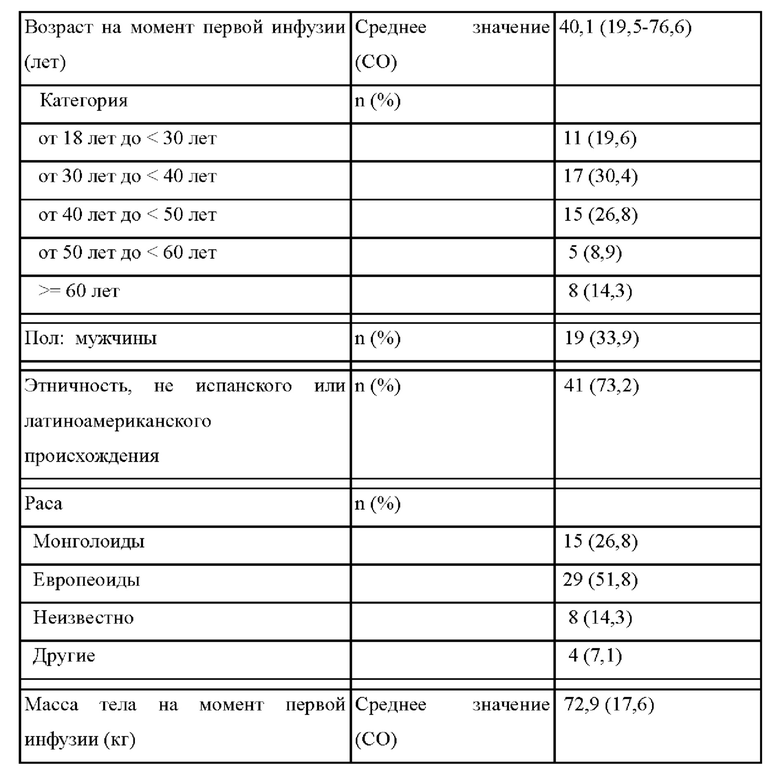
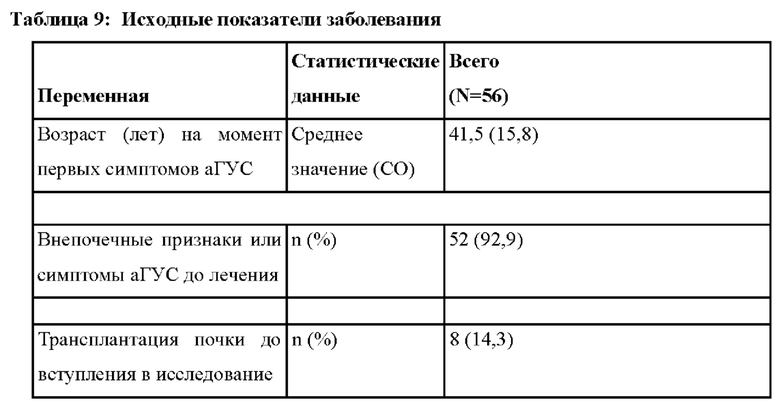
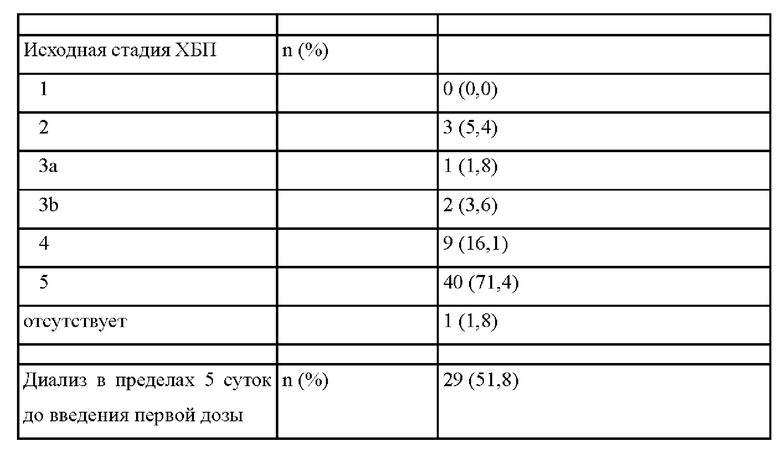
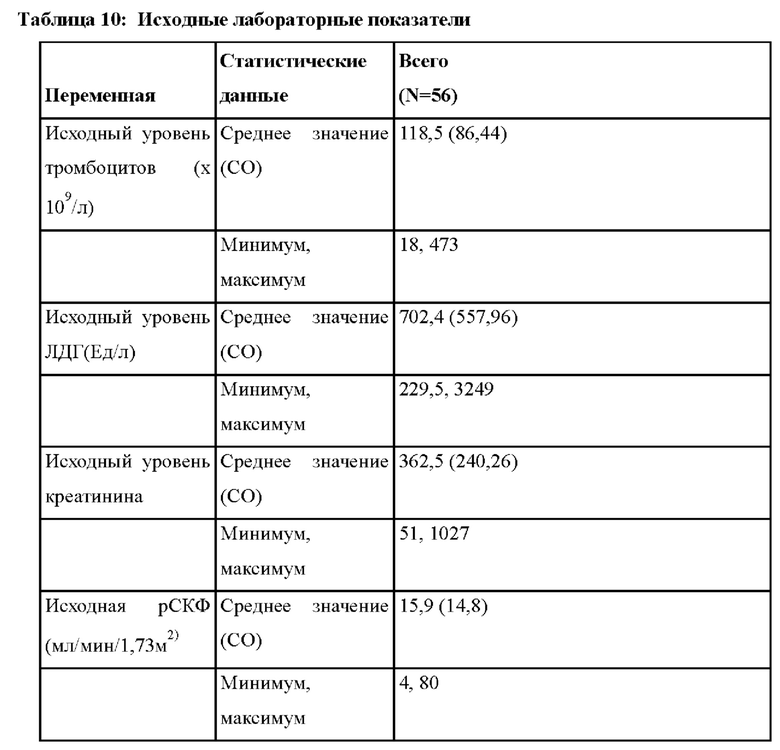
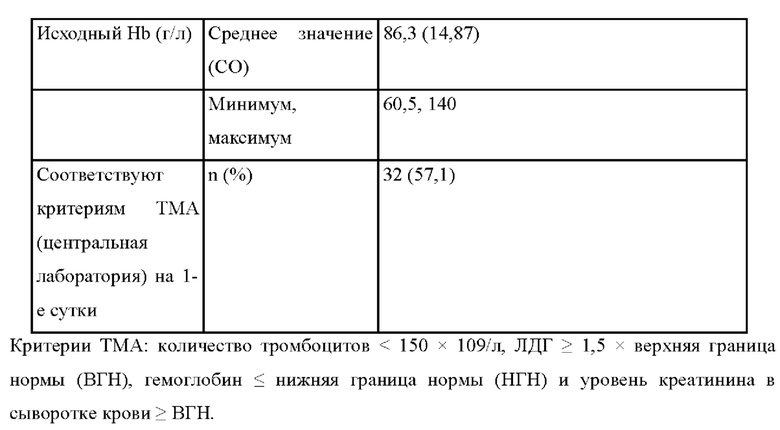
Определения ответа ТМА указаны в таблице 11. Ответ достигался, когда было соответствие всем критериям одновременно и соответствие каждому критерию длилось по меньшей мере 28 суток. Значения уровней тромбоцитов, полученные со дня трансфузии тромбоцитов крови до 3-х суток после трансфузии, исключались из всех анализов. Все значения уровней креатинина в сыворотке крови, полученные в период, когда пациент находился на диализе, исключались из всех анализов. Если пациент находился на диализе в исходном состоянии, то первым допустимым значением уровня креатинина, используемым в качестве исходного значения, была первая оценка через ≥ 6 суток после диализа. Если пациент находился на диализе в течение всего 26-недельного периода первичной оценки, то исходный уровень креатинина не рассчитывался.


Ключевые результаты анализа эффективности указаны в таблицах 12-14. На фиг.5 представлен деривационный пример, показывающий результаты подтвержденного полного ответа ТМА в 57-е сутки, включающий нормализацию тромбоцитов на 112 суток, нормализацию ЛДГ на 35 суток и улучшение креатинина на ≥ 25% на 70 суток.
Критерии полного ответа ТМА достигались, когда было соответствие всем критериям одновременно и соответствие каждому критерию длилось по меньшей мере 28 суток. Компоненты полного ответа ТМА, а также другие вторичные критерии оценки эффективности указывают на устойчивый ответ на лечение. Как показано на фиг.9, 53,6% (30/56) пациентов достигли полного ответа ТМА в течение периода первичной оценки. У 33,9% (19/56) пациентов наблюдался частичный ответ.
Нормализация гематологических показателей включает в себя одновременную нормализацию количества тромбоцитов и нормализацию уровня ЛДГ. Соответствие каждому критерию продолжалось по меньшей мере 28 суток. 73,2% (41/56) субъектов достигли нормализации гематологических показателей в течение периода первичной оценки (см. таблицу 12). 83,9% (47/56) субъектов достигли нормализации количества тромбоцитов в течение периода первичной оценки (см. таблицу 12, фиг.10, фиг.14 и фиг.15).
76,8% (43/56) субъектов достигли нормализации ЛДГ в течение периода первичной оценки (см. таблицу 12, фиг.10, фиг.16 и фиг.17).
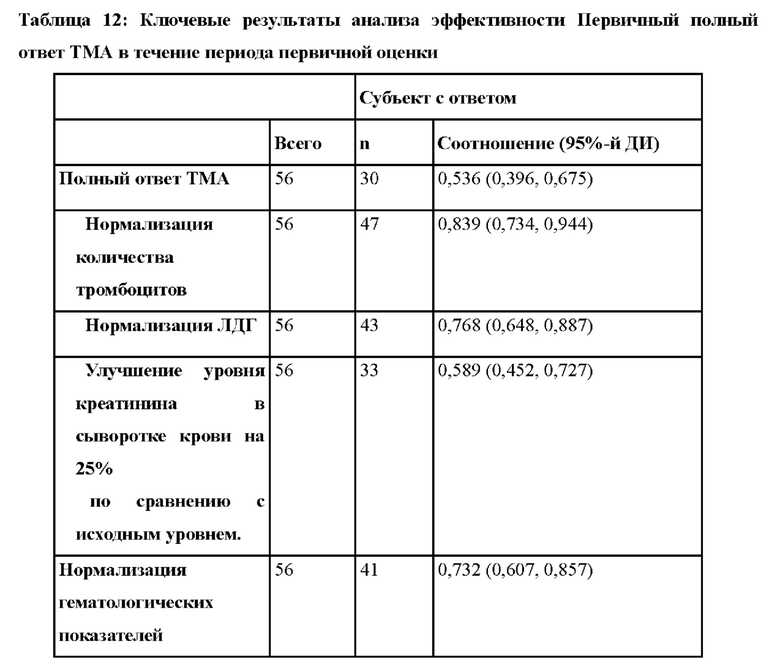
Как показано на фиг.6, было тридцать (30) субъектов с полным ответом ТМА. Сорок (40) из пятидесяти шести (56)субъектов (71,4%) достигли ответа гемоглобина (Hb) (повышение на ≥ 20 г/л). Из тридцати (30) полных респондентов ТМА у четырех (4) не было ответа на гемоглобин (Hb). На фиг.10 показан полный ответ ТМА в общем и по подгруппам в течение 26-недельного периода первичной оценки.
На Фиг. 7 показано время до полного ответа ТМА. Среднее время до полного ответа ТМА составило 86 суток. Пациенты, у которых не было ответа, были цензированы в день последнего визита или прекращения исследования.
На фиг.11 показан статус полного ответа ТМА с течением времени (незакрашенный круг), включая нормализацию количества тромбоцитов (незакрашенный треугольник), нормализацию гематологических показателей (+), 25% улучшение уровня креатинина в сыворотке крови по сравнению с исходным уровнем (незакрашенный квадрат) и нормализацию ЛДГ (X).
У семи (7) субъектов ответа не было. Результаты для субъектов, у которых не было ответа, указаны в таблице 13.
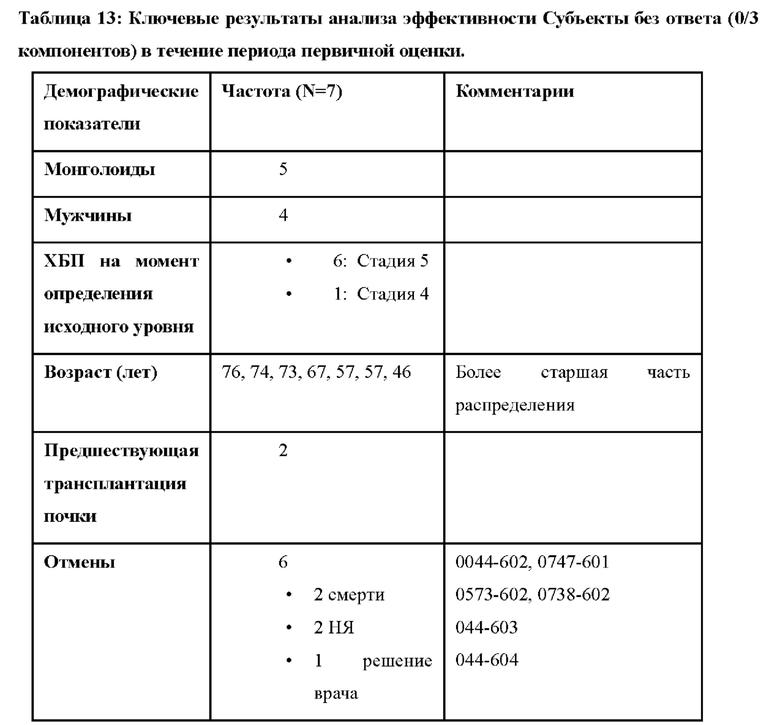
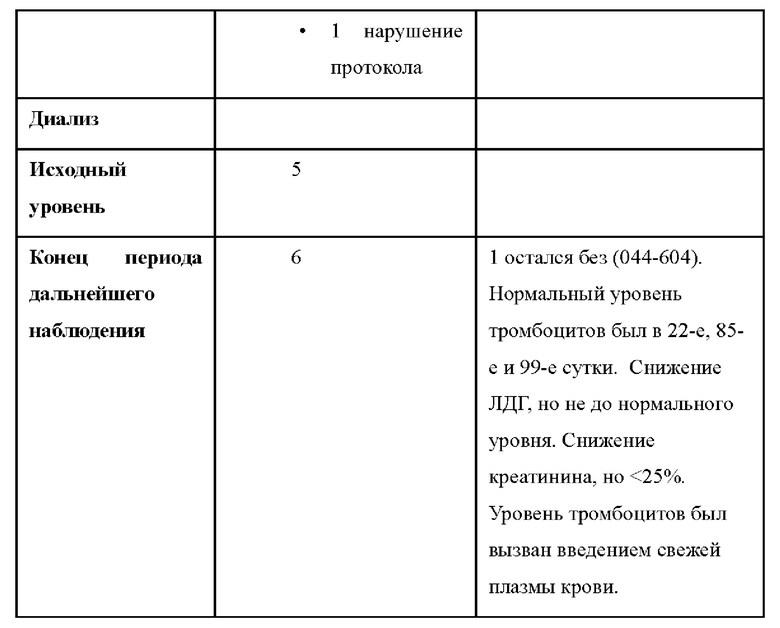
Как показано в таблице 14, у 58,6% (17/29) пациентов, которые были на диализе на момент определения исходного уровня, диализ был отменен на момент последнего доступного наблюдения. Из 27 пациентов, которые не были на диализе на момент определения исходного уровня, 21 (78%) пациент был по-прежнему без диализа на момент последнего наблюдения.
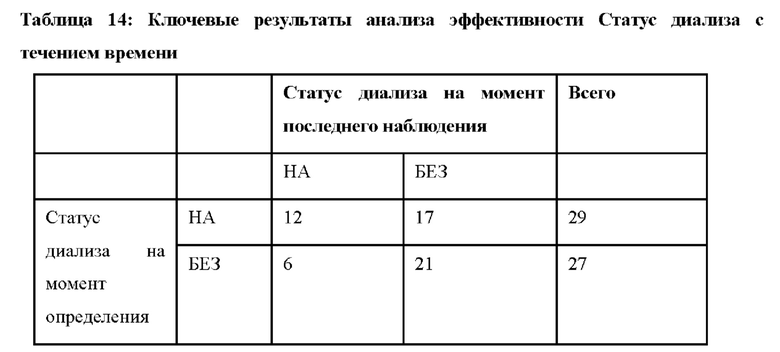
Что касается фармакокинетики / фармакодинамики, 99,53% всех результатов определения концентрации свободного С5, полученных после введения первой дозы в течение периода первичной оценки, составляли ≤ 0,5 мг/мл - установленный порог для ингибирования терминального комплемента (см. фиг.22). Основанное на массе тела введение доз приводило к максимальной, равновесной и остаточной концентрациям препарата, как и было спрогнозировано, без неожиданных фармакокинетических результатов (см. фиг.8).
Ключевые результаты анализа эффективности указаны в таблице 15. Группа пациентов, оцениваемая на предмет безопасности, включает в себя всех пациентов, получивших ≥ 1 дозы исследуемого лекарственного средства (N=58). Двое из этих пациентов были исключены из анализа эффективности по протоколу в связи с непригодностью (идентификация STEC-ГУС).
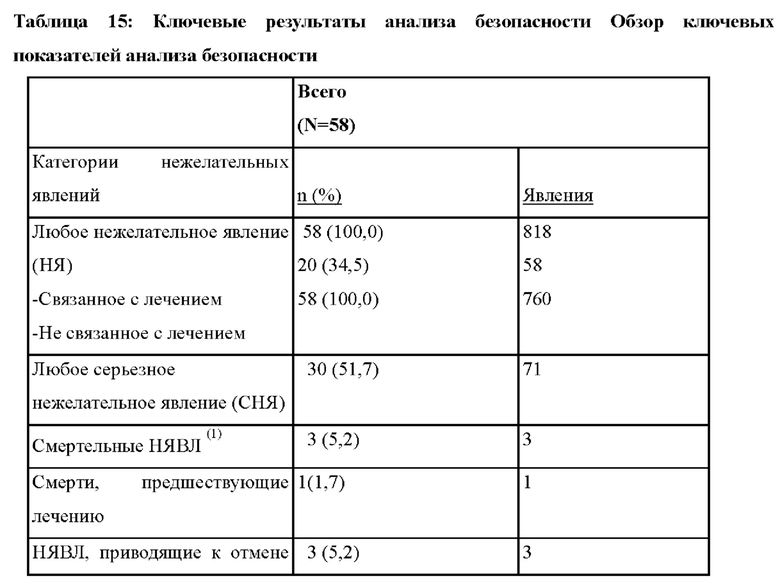

Произошло четыре смерти: 1 - вследствие нежелательного явления, предшествующего лечению (тромбоз церебральных артерий) и 3 - вследствие нежелательных явлений, не связанных с лечением (2 септических шока, 1 внутричерепное кровоизлияние). Сводная информация о смертности пациентов, получивших по меньшей мере одну дозу равулизумаба, указана в таблице 16.
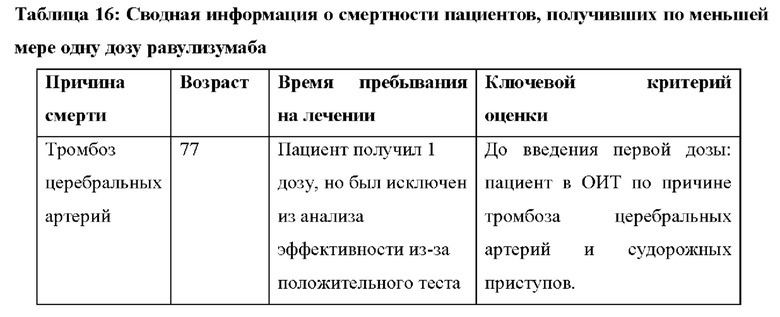
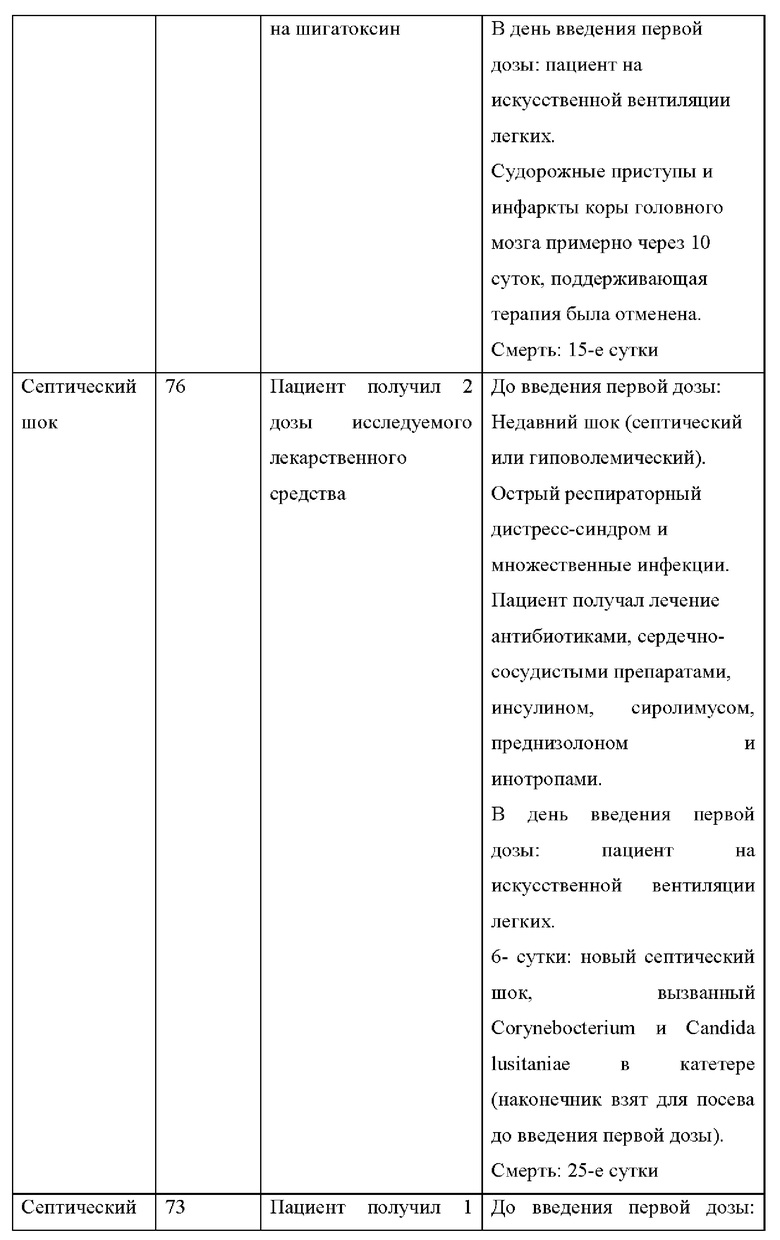
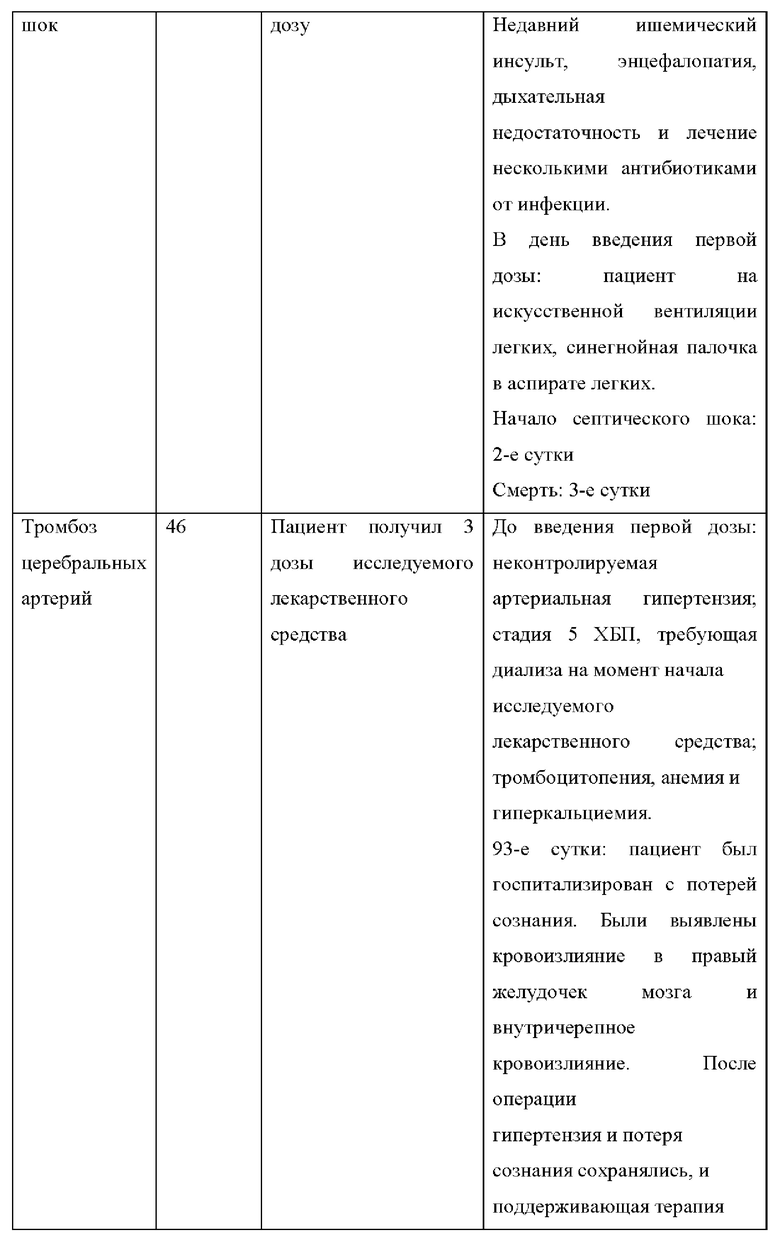

Как показано в таблице 17, наиболее частыми нежелательными явлениями были головная боль (N=21), диарея (N=18), рвота (N=15), тошнота (N=13) и гипертензия (N=13). Как показано в таблице 18, наиболее распространенным серьезным нежелательным явлением была пневмония (N=3). Три субъекта прекратили участие в исследовании вследствие нежелательного явления. Случаев заболевания менингококком не было.

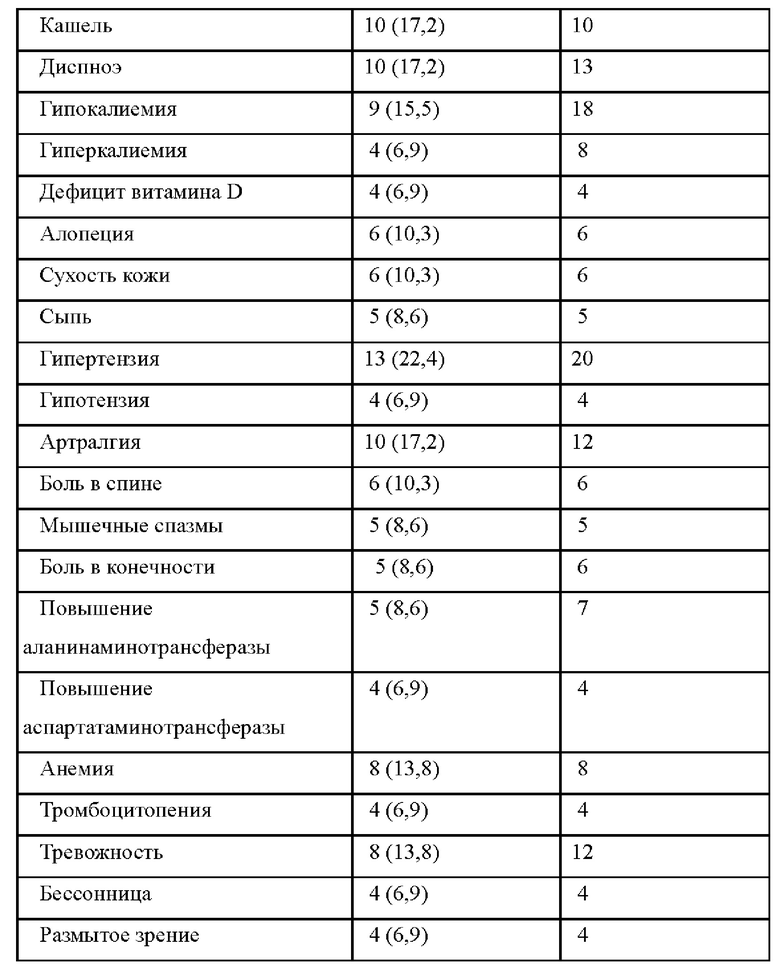
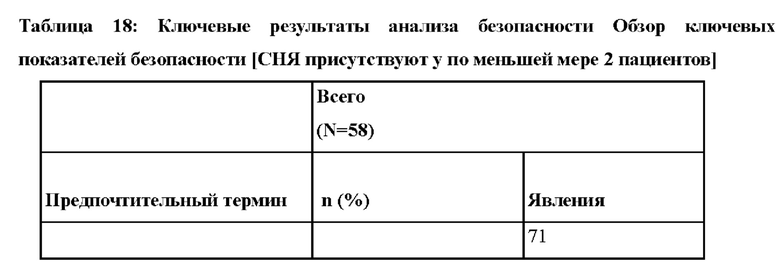
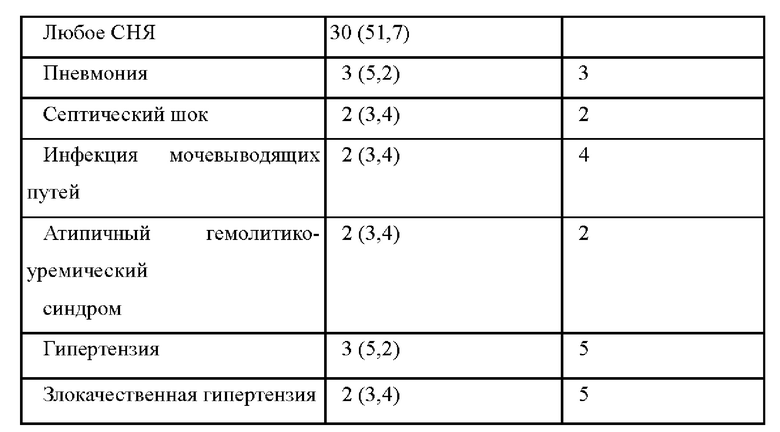
71,4% (40/56) субъектов достигли ответа гемоглобина (Hb) в течение периода первичной оценки (см. таблицу 19 и фиг.6). На фиг.18 показаны наблюдаемое и основанное на модели средние изменения уровня Hb по сравнению с исходным уровнем и 95%-й доверительный интервал с течением времени. На фиг.19 показаны наблюдаемый средний уровень Hb и 95%-й доверительный интервал с течением времени.
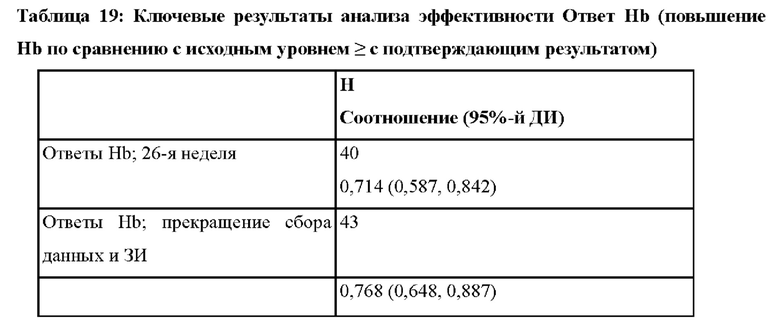
Стадия хронической болезни почек (ХБП) классифицируется на основе стадий хронической болезни почек, определенных Национальным фондом почки США. Стадии ХБП и соответствующие значения расчетной скорости клубочковой фильтрации (рСКФ) следующие: Стадия 1: рСКФ >= 90 (норма), Стадия 2: рСКФ = 60-89, Стадия 3А: рСКФ = 45-59, Стадия 3В: рСКФ = 30-44, Стадия 4: рСКФ = 15-29 и стадия 5: рСКФ < 15 (включая диализ: Конечная стадия). Стадия 1 считается наилучшей категорией. Стадия 5 считается наихудшей категорией. Улучшение рСКФ (например, > 15) соответствует улучшению стадии ХБП (например, более низкой стадии ХБП).
На фиг.12 показаны средние рСКФ по сравнению с исходным уровнем и 95%-й доверительный интервал. На фиг.13 показан сдвиг в категориях ХБП / рСКФ по сравнению с исходным уровнем к 183-м суткам. Данные представлены в виде n (%). Категории рСКФ показаны в мл/мин/1,73 м2. Исходные значения рассчитаны на основе последнего доступного значения рСКФ до началом лечения. Нижний треугольник (обозначенный наклонными штрихами) отображает улучшение по сравнению с исходным уровнем к 183-м суткам, верхний треугольник (обозначенный точками) отображает ухудшение, а белые клетки отображают отсутствие изменений.
Кроме того, как показано в таблице 20 и на фиг.13, у тридцати двух (32) из сорока семи (47) субъектов стадия ХБП улучшилась к 183-м суткам по сравнению с исходным уровнем (у шести - на 5 стадий, у семи - на 4 стадии, у пяти - на 3 стадии,у четырех - на 2 стадии и у десятерых - на 1 стадию). У тринадцати (13) из сорока семи (47) субъектов изменений не было. Состояние двух (2) из тринадцати (13) ухудшилось.

В таблице 21 и на фиг.20 показано изменение показателя утомляемости с течением времени. Данные на фиг.20 представлены как среднее значение (столбики ошибок 95%-й ДИ). Наблюдалось быстрое улучшение показателя утомляемости, т.е. среднее улучшение на 9 пунктов к 8-м суткам. Клинически значимое улучшение показателя утомляемости ( ≥ 3 баллов) наблюдалось у 84,1% (37/44) пациентов в 183-е сутки. Среднее увеличение составило 20 пунктов по сравнению с исходным уровнем к 183-м суткам.


EQ-5D-3L оценивается с использованием соотношения, полученного в соответствии со значением временного компромисса по опроснику для США (US ТТО), а также ответом по визуальной аналоговой шкале (VAS). US ТТО > 0,94 указывает на полное здоровье. Исходный уровень представляет собой значение для 1-х суток. На фиг.21 показаны наблюдаемый средняя оценка EQ-5D-3L и 95%-й доверительный интервал. Что касается иммуногенности, у одного пациента наблюдался связанный с лечением положительный результат анализа на антитела против лекарственного средства (АЛС) без нейтрализующих антител и без видимого влияния на фармакокинетику / фармакодинамику (см. таблицу 22).
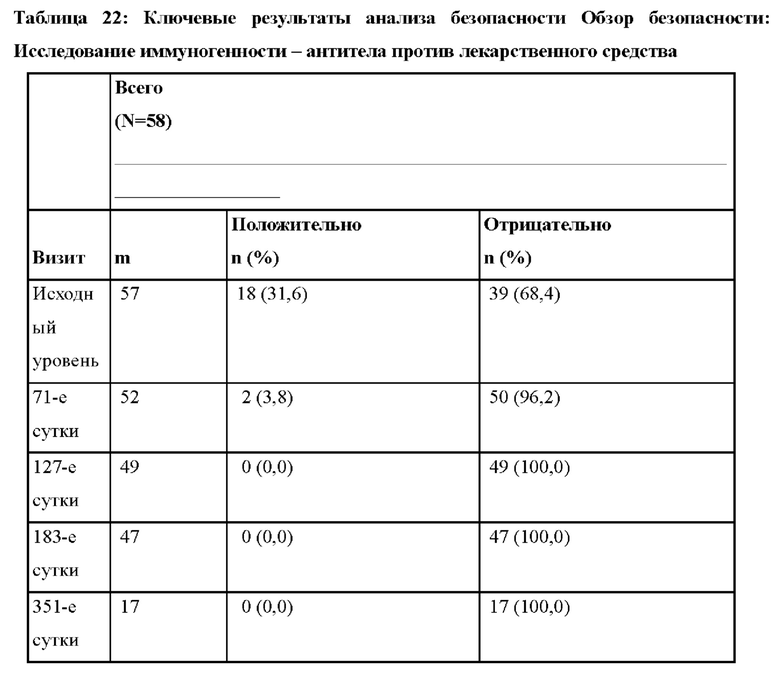

На фиг.22 показаны концентрации свободного комплемента С5 в сыворотке крови с течением времени (полулогарифмическая шкала). Пунктирная горизонтальная линия отображает концентрацию свободного С5 в сыворотке крови, равную 0,5 мкг/мл, при этом полное ингибирование терминального комплемента определяется как концентрация свободного С5 в сыворотке крови ≤ 0,5 мкг/мл. Последующие образцы свободного С5 для 1-х суток были исключены, поскольку они расценивались как биологически неправдоподобные. Исключения были подтверждены парными данными по ФК, т.к. образцы для анализа ФК и свободного С5 были взяты из одного и того же забора крови. [N=2, образцы для 1-х суток перед введением дозы / N=1, образец для 1-х суток по окончании инфузии]. Как следует из фиг.22, равулизумаб продемонстрировал немедленное, полное и устойчивое ингибирование терминального комплемента в течение 8-недельного интервала между введением доз.
Наконец, исследуемая популяция в данном исследовании равулизумаба (ALXN1210-aHUS-311) и в исследовании экулизумаба у взрослых (С10-004) была аналогичной в 183-е сутки. Однако, как показано на фиг.23, равулизумаб привел к улучшению показателей по сравнению с экулизумабом в отношении следующих вторичных клинических параметров: (а) категория рСКФ / стадия хронической болезни почек (ХБП) и (b) повышение расчетной скорости клубочковой фильтрации (рСКФ). В частности, из пациентов, получавших лечение равулизумабом в данном исследовании, 68% достигли улучшения в категории рСКФ / стадия ХБП по меньшей мере на одну стадию, а среднее увеличение рСКФ составило 35±35. В отличие от этого, из пациентов, получавших лечение экулизумабом в исследовании С10-004, только 63% достигли улучшения в категории рСКФ / стадия ХБП по меньшей мере на одну стадию, а среднее увеличение рСКФ составило 29±24. «Полный ответ ТМА» в данном исследовании эквивалентен «модифицированному полному ответу ТМА» в С10-004.
Таким образом, равулизумаб обеспечивал немедленное и полное ингибирование С 5, которое сохранялось в течение 8-недельного интервала между введением доз. Полный ответ ТМА был достигнут у 54% пациентов, что аналогично данным по экулизумабу (56% в исследовании С10-004). Наблюдалось быстрое улучшение количества тромбоцитов. Кроме того, существенно улучшилась функция почек. В частности, 58,6% пациентов, находившихся на диализе в исходном состоянии, не нуждались в диализе в конце данного исследования. Более того, никаких неожиданных проблем с безопасностью выявлено не было. Таким образом, результаты данного исследования свидетельствуют в пользу применения равулизумаба с 8-недельными интервалами введения доз у взрослых пациентов с опосредованной комплементом ТМА.








--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> АЛЕКСИОН ФАРМАСЬЮТИКАЛС, ИНК.
<120> ДОЗЫ И ВВЕДЕНИЕ АНТИТЕЛ ПРОТИВ C5 ДЛЯ ЛЕЧЕНИЯ АТИПИЧНОГО ГЕМОЛИТИКО-УРЕМИЧЕСКОГО СИНДРОМА (АГУС)
<130> AXJ-257PC
<140> PCT/US2020/014998
<141> 2020-01-24
<160> 50
<170> PatentIn version 3.5
<210> 1
<211> 10
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 1
Gly Tyr Ile Phe Ser Asn Tyr Trp Ile Gln
1 5 10
<210> 2
<211> 17
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 2
Glu Ile Leu Pro Gly Ser Gly Ser Thr Glu Tyr Thr Glu Asn Phe Lys
1 5 10 15
Asp
<210> 3
<211> 13
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 3
Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val
1 5 10
<210> 4
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 4
Gly Ala Ser Glu Asn Ile Tyr Gly Ala Leu Asn
1 5 10
<210> 5
<211> 7
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 5
Gly Ala Thr Asn Leu Ala Asp
1 5
<210> 6
<211> 9
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 6
Gln Asn Val Leu Asn Thr Pro Leu Thr
1 5
<210> 7
<211> 122
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 7
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly Ser Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser
115 120
<210> 8
<211> 107
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 8
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Gly Ala Ser Glu Asn Ile Tyr Gly Ala
20 25 30
Leu Asn Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Gly Ala Thr Asn Leu Ala Asp Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Asn Val Leu Asn Thr Pro Leu
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys
100 105
<210> 9
<211> 326
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 9
Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Cys Ser Arg
1 5 10 15
Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr
20 25 30
Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser
35 40 45
Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser
50 55 60
Leu Ser Ser Val Val Thr Val Pro Ser Ser Asn Phe Gly Thr Gln Thr
65 70 75 80
Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn Thr Lys Val Asp Lys
85 90 95
Thr Val Glu Arg Lys Cys Cys Val Glu Cys Pro Pro Cys Pro Ala Pro
100 105 110
Pro Val Ala Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp
115 120 125
Thr Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp
130 135 140
Val Ser Gln Glu Asp Pro Glu Val Gln Phe Asn Trp Tyr Val Asp Gly
145 150 155 160
Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn
165 170 175
Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp
180 185 190
Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Gly Leu Pro
195 200 205
Ser Ser Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu
210 215 220
Pro Gln Val Tyr Thr Leu Pro Pro Ser Gln Glu Glu Met Thr Lys Asn
225 230 235 240
Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile
245 250 255
Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr
260 265 270
Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg
275 280 285
Leu Thr Val Asp Lys Ser Arg Trp Gln Glu Gly Asn Val Phe Ser Cys
290 295 300
Ser Val Met His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu
305 310 315 320
Ser Leu Ser Leu Gly Lys
325
<210> 10
<211> 448
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 10
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly Ser Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Pro Ser Ser Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Thr Val Glu Arg Lys Cys
210 215 220
Cys Val Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Pro Ser
225 230 235 240
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg
245 250 255
Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser Gln Glu Asp Pro
260 265 270
Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala
275 280 285
Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Tyr Arg Val Val
290 295 300
Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr
305 310 315 320
Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ser Ser Ile Glu Lys Thr
325 330 335
Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu
340 345 350
Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys
355 360 365
Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser
370 375 380
Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp
385 390 395 400
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg Leu Thr Val Asp Lys Ser
405 410 415
Arg Trp Gln Glu Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala
420 425 430
Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Leu Gly Lys
435 440 445
<210> 11
<211> 214
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 11
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Gly Ala Ser Glu Asn Ile Tyr Gly Ala
20 25 30
Leu Asn Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Gly Ala Thr Asn Leu Ala Asp Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Asn Val Leu Asn Thr Pro Leu
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 12
<211> 122
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 12
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly His Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly His Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser
115 120
<210> 13
<211> 326
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 13
Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Cys Ser Arg
1 5 10 15
Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr
20 25 30
Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser
35 40 45
Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser
50 55 60
Leu Ser Ser Val Val Thr Val Pro Ser Ser Asn Phe Gly Thr Gln Thr
65 70 75 80
Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn Thr Lys Val Asp Lys
85 90 95
Thr Val Glu Arg Lys Cys Cys Val Glu Cys Pro Pro Cys Pro Ala Pro
100 105 110
Pro Val Ala Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp
115 120 125
Thr Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp
130 135 140
Val Ser Gln Glu Asp Pro Glu Val Gln Phe Asn Trp Tyr Val Asp Gly
145 150 155 160
Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn
165 170 175
Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp
180 185 190
Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Gly Leu Pro
195 200 205
Ser Ser Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu
210 215 220
Pro Gln Val Tyr Thr Leu Pro Pro Ser Gln Glu Glu Met Thr Lys Asn
225 230 235 240
Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile
245 250 255
Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr
260 265 270
Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg
275 280 285
Leu Thr Val Asp Lys Ser Arg Trp Gln Glu Gly Asn Val Phe Ser Cys
290 295 300
Ser Val Leu His Glu Ala Leu His Ser His Tyr Thr Gln Lys Ser Leu
305 310 315 320
Ser Leu Ser Leu Gly Lys
325
<210> 14
<211> 448
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 14
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly His Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly His Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Pro Ser Ser Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Thr Val Glu Arg Lys Cys
210 215 220
Cys Val Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Pro Ser
225 230 235 240
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg
245 250 255
Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser Gln Glu Asp Pro
260 265 270
Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala
275 280 285
Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Tyr Arg Val Val
290 295 300
Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr
305 310 315 320
Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ser Ser Ile Glu Lys Thr
325 330 335
Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu
340 345 350
Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys
355 360 365
Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser
370 375 380
Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp
385 390 395 400
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg Leu Thr Val Asp Lys Ser
405 410 415
Arg Trp Gln Glu Gly Asn Val Phe Ser Cys Ser Val Leu His Glu Ala
420 425 430
Leu His Ser His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Leu Gly Lys
435 440 445
<210> 15
<211> 326
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 15
Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Cys Ser Arg
1 5 10 15
Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr
20 25 30
Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser
35 40 45
Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser
50 55 60
Leu Ser Ser Val Val Thr Val Thr Ser Ser Asn Phe Gly Thr Gln Thr
65 70 75 80
Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn Thr Lys Val Asp Lys
85 90 95
Thr Val Glu Arg Lys Cys Cys Val Glu Cys Pro Pro Cys Pro Ala Pro
100 105 110
Pro Val Ala Gly Pro Ser Val Phe Leu Phe Pro Pro Lys Pro Lys Asp
115 120 125
Thr Leu Tyr Ile Thr Arg Glu Pro Glu Val Thr Cys Val Val Val Asp
130 135 140
Val Ser His Glu Asp Pro Glu Val Gln Phe Asn Trp Tyr Val Asp Gly
145 150 155 160
Met Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn
165 170 175
Ser Thr Phe Arg Val Val Ser Val Leu Thr Val Val His Gln Asp Trp
180 185 190
Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Gly Leu Pro
195 200 205
Ala Pro Ile Glu Lys Thr Ile Ser Lys Thr Lys Gly Gln Pro Arg Glu
210 215 220
Pro Gln Val Tyr Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn
225 230 235 240
Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile
245 250 255
Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr
260 265 270
Thr Pro Pro Met Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys
275 280 285
Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys
290 295 300
Ser Val Met His Glu Ala Leu His Asn His Tyr Thr Gln Lys Ser Leu
305 310 315 320
Ser Leu Ser Pro Gly Lys
325
<210> 16
<211> 448
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 16
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Tyr Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly Ser Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Thr Ser Ser Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Thr Val Glu Arg Lys Cys
210 215 220
Cys Val Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Pro Ser
225 230 235 240
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Tyr Ile Thr Arg
245 250 255
Glu Pro Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp Pro
260 265 270
Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Met Glu Val His Asn Ala
275 280 285
Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Phe Arg Val Val
290 295 300
Ser Val Leu Thr Val Val His Gln Asp Trp Leu Asn Gly Lys Glu Tyr
305 310 315 320
Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ala Pro Ile Glu Lys Thr
325 330 335
Ile Ser Lys Thr Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu
340 345 350
Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys
355 360 365
Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser
370 375 380
Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Met Leu Asp
385 390 395 400
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser
405 410 415
Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala
420 425 430
Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys
435 440 445
<210> 17
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 17
Gly Ala Ser Glu Asn Ile Tyr His Ala Leu Asn
1 5 10
<210> 18
<211> 17
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 18
Glu Ile Leu Pro Gly Ser Gly His Thr Glu Tyr Thr Glu Asn Phe Lys
1 5 10 15
Asp
<210> 19
<211> 10
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 19
Gly His Ile Phe Ser Asn Tyr Trp Ile Gln
1 5 10
<210> 20
<211> 448
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 20
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ala
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly His Ile Phe Ser Asn Tyr
20 25 30
Trp Ile Gln Trp Val Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Glu Ile Leu Pro Gly Ser Gly His Thr Glu Tyr Thr Glu Asn Phe
50 55 60
Lys Asp Arg Val Thr Met Thr Arg Asp Thr Ser Thr Ser Thr Val Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Tyr Phe Phe Gly Ser Ser Pro Asn Trp Tyr Phe Asp Val Trp
100 105 110
Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro
115 120 125
Ser Val Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr
130 135 140
Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr
145 150 155 160
Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro
165 170 175
Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr
180 185 190
Val Pro Ser Ser Asn Phe Gly Thr Gln Thr Tyr Thr Cys Asn Val Asp
195 200 205
His Lys Pro Ser Asn Thr Lys Val Asp Lys Thr Val Glu Arg Lys Cys
210 215 220
Cys Val Glu Cys Pro Pro Cys Pro Ala Pro Pro Val Ala Gly Pro Ser
225 230 235 240
Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg
245 250 255
Thr Pro Glu Val Thr Cys Val Val Val Asp Val Ser Gln Glu Asp Pro
260 265 270
Glu Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala
275 280 285
Lys Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Tyr Arg Val Val
290 295 300
Ser Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr
305 310 315 320
Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ser Ser Ile Glu Lys Thr
325 330 335
Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu
340 345 350
Pro Pro Ser Gln Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys
355 360 365
Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser
370 375 380
Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp
385 390 395 400
Ser Asp Gly Ser Phe Phe Leu Tyr Ser Arg Leu Thr Val Asp Lys Ser
405 410 415
Arg Trp Gln Glu Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala
420 425 430
Leu His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Leu Gly Lys
435 440 445
<210> 21
<211> 5
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 21
Ser Tyr Ala Ile Ser
1 5
<210> 22
<211> 17
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 22
Gly Ile Gly Pro Phe Phe Gly Thr Ala Asn Tyr Ala Gln Lys Phe Gln
1 5 10 15
Gly
<210> 23
<211> 7
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 23
Asp Thr Pro Tyr Phe Asp Tyr
1 5
<210> 24
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 24
Ser Gly Asp Ser Ile Pro Asn Tyr Tyr Val Tyr
1 5 10
<210> 25
<211> 7
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 25
Asp Asp Ser Asn Arg Pro Ser
1 5
<210> 26
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 26
Gln Ser Phe Asp Ser Ser Leu Asn Ala Glu Val
1 5 10
<210> 27
<211> 116
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 27
Gln Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Ser
1 5 10 15
Ser Val Lys Val Ser Cys Lys Ala Ser Gly Gly Thr Phe Ser Ser Tyr
20 25 30
Ala Ile Ser Val Trp Arg Gln Ala Pro Gly Gln Gly Leu Glu Trp Met
35 40 45
Gly Gly Ile Gly Pro Phe Phe Gly Thr Ala Asn Tyr Ala Gln Lys Phe
50 55 60
Gln Gly Arg Val Thr Ile Thr Ala Asp Glu Ser Thr Ser Thr Ala Tyr
65 70 75 80
Met Glu Leu Ser Ser Leu Arg Ser Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Arg Asp Thr Pro Tyr Phe Asp Tyr Trp Gly Gln Gly Thr Leu Val
100 105 110
Thr Val Ser Ser
115
<210> 28
<211> 108
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 28
Asp Ile Glu Leu Thr Gln Pro Pro Ser Val Ser Val Ala Pro Gly Gln
1 5 10 15
Thr Ala Arg Ile Ser Cys Ser Gly Asp Ser Ile Pro Asn Tyr Tyr Val
20 25 30
Tyr Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Val Leu Val Ile Tyr
35 40 45
Asp Asp Ser Asn Arg Pro Ser Gly Ile Pro Glu Arg Phe Ser Gly Ser
50 55 60
Asn Ser Gly Asn Thr Ala Thr Leu Thr Ile Ser Gly Thr Gln Ala Glu
65 70 75 80
Asp Glu Ala Asp Tyr Tyr Cys Gln Ser Phe Asp Ser Ser Leu Asn Ala
85 90 95
Glu Val Phe Gly Gly Gly Thr Lys Leu Thr Val Leu
100 105
<210> 29
<211> 4
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 29
Asn Tyr Ile Ser
1
<210> 30
<211> 17
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 30
Ile Ile Asp Pro Asp Asp Ser Tyr Thr Glu Tyr Ser Pro Ser Phe Gln
1 5 10 15
Gly
<210> 31
<211> 8
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 31
Tyr Glu Tyr Gly Gly Phe Asp Ile
1 5
<210> 32
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 32
Ser Gly Asp Asn Ile Gly Asn Ser Tyr Val His
1 5 10
<210> 33
<211> 7
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 33
Lys Asp Asn Asp Arg Pro Ser
1 5
<210> 34
<211> 9
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 34
Gly Thr Tyr Asp Ile Glu Ser Tyr Val
1 5
<210> 35
<211> 116
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 35
Glu Val Gln Leu Val Gln Ser Gly Ala Glu Val Lys Lys Pro Gly Glu
1 5 10 15
Ser Leu Lys Ile Ser Cys Lys Gly Ser Gly Tyr Ser Phe Thr Asn Tyr
20 25 30
Ile Ser Trp Val Arg Gln Met Pro Gly Lys Gly Leu Glu Trp Met Gly
35 40 45
Ile Ile Asp Pro Asp Asp Ser Tyr Thr Glu Tyr Ser Pro Ser Phe Gln
50 55 60
Gly Gln Val Thr Ile Ser Ala Asp Lys Ser Ile Ser Thr Ala Tyr Leu
65 70 75 80
Gln Trp Ser Ser Leu Lys Ala Ser Asp Thr Ala Met Tyr Tyr Cys Ala
85 90 95
Arg Tyr Glu Tyr Gly Gly Phe Asp Ile Trp Gly Gln Gly Thr Leu Val
100 105 110
Thr Val Ser Ser
115
<210> 36
<211> 106
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 36
Ser Tyr Glu Leu Thr Gln Pro Pro Ser Val Ser Val Ala Pro Gly Gln
1 5 10 15
Thr Ala Arg Ile Ser Cys Ser Gly Asp Asn Ile Gly Asn Ser Tyr Val
20 25 30
His Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Val Leu Val Ile Tyr
35 40 45
Lys Asp Asn Asp Arg Pro Ser Gly Ile Pro Glu Arg Phe Ser Gly Ser
50 55 60
Asn Ser Gly Asn Thr Ala Thr Leu Thr Ile Ser Gly Thr Gln Ala Glu
65 70 75 80
Asp Glu Ala Asp Tyr Tyr Cys Gly Thr Tyr Asp Ile Glu Ser Tyr Val
85 90 95
Phe Gly Gly Gly Thr Lys Leu Thr Val Leu
100 105
<210> 37
<211> 6
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 37
Ser Ser Tyr Tyr Val Ala
1 5
<210> 38
<211> 17
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 38
Ala Ile Tyr Thr Gly Ser Gly Ala Thr Tyr Lys Ala Ser Trp Ala Lys
1 5 10 15
Gly
<210> 39
<211> 13
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 39
Asp Gly Gly Tyr Asp Tyr Pro Thr His Ala Met His Tyr
1 5 10
<210> 40
<211> 11
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 40
Gln Ala Ser Gln Asn Ile Gly Ser Ser Leu Ala
1 5 10
<210> 41
<211> 7
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 41
Gly Ala Ser Lys Thr His Ser
1 5
<210> 42
<211> 12
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 42
Gln Ser Thr Lys Val Gly Ser Ser Tyr Gly Asn His
1 5 10
<210> 43
<211> 123
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 43
Gln Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Gly
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Ser His Ser Ser
20 25 30
Tyr Tyr Val Ala Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp
35 40 45
Val Gly Ala Ile Tyr Thr Gly Ser Gly Ala Thr Tyr Lys Ala Ser Trp
50 55 60
Ala Lys Gly Arg Phe Thr Ile Ser Lys Asp Thr Ser Lys Asn Gln Val
65 70 75 80
Val Leu Thr Met Thr Asn Met Asp Pro Val Asp Thr Ala Thr Tyr Tyr
85 90 95
Cys Ala Ser Asp Gly Gly Tyr Asp Tyr Pro Thr His Ala Met His Tyr
100 105 110
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser
115 120
<210> 44
<211> 110
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 44
Asp Val Val Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Gln Ala Ser Gln Asn Ile Gly Ser Ser
20 25 30
Leu Ala Trp Tyr Gln Gln Lys Pro Gly Gln Ala Pro Arg Leu Leu Ile
35 40 45
Tyr Gly Ala Ser Lys Thr His Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Val Ala Thr Tyr Tyr Cys Gln Ser Thr Lys Val Gly Ser Ser
85 90 95
Tyr Gly Asn His Phe Gly Gly Gly Thr Lys Val Glu Ile Lys
100 105 110
<210> 45
<211> 451
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 45
Gln Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Arg
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Val His Ser Ser
20 25 30
Tyr Tyr Met Ala Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp
35 40 45
Val Gly Ala Ile Phe Thr Gly Ser Gly Ala Glu Tyr Lys Ala Glu Trp
50 55 60
Ala Lys Gly Arg Val Thr Ile Ser Lys Asp Thr Ser Lys Asn Gln Val
65 70 75 80
Val Leu Thr Met Thr Asn Met Asp Pro Val Asp Thr Ala Thr Tyr Tyr
85 90 95
Cys Ala Ser Asp Ala Gly Tyr Asp Tyr Pro Thr His Ala Met His Tyr
100 105 110
Trp Gly Gln Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly
115 120 125
Pro Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser Thr Ser Gly Gly
130 135 140
Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val
145 150 155 160
Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe
165 170 175
Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val
180 185 190
Thr Val Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr Ile Cys Asn Val
195 200 205
Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys Lys Val Glu Pro Lys
210 215 220
Ser Cys Asp Lys Thr His Thr Cys Pro Pro Cys Pro Ala Pro Glu Leu
225 230 235 240
Arg Arg Gly Pro Lys Val Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr
245 250 255
Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys Val Val Val Asp Val
260 265 270
Ser His Glu Asp Pro Glu Val Lys Phe Asn Trp Tyr Val Asp Gly Val
275 280 285
Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu Gln Tyr Asn Ser
290 295 300
Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu His Gln Asp Trp Leu
305 310 315 320
Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys Gly Leu Pro Ser
325 330 335
Ser Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro
340 345 350
Gln Val Tyr Thr Leu Pro Pro Ser Arg Glu Glu Met Thr Lys Asn Gln
355 360 365
Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala
370 375 380
Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr
385 390 395 400
Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu Tyr Ser Lys Leu
405 410 415
Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn Val Phe Ser Cys Ser
420 425 430
Val Leu His Glu Ala Leu His Ala His Tyr Thr Arg Lys Glu Leu Ser
435 440 445
Leu Ser Pro
450
<210> 46
<211> 217
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 46
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Gly Ile Ser Ser Ser
20 25 30
Leu Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Gly Ala Ser Glu Thr Glu Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Gln Asn Thr Lys Val Gly Ser Ser
85 90 95
Tyr Gly Asn Thr Phe Gly Gly Gly Thr Lys Val Glu Ile Lys Arg Thr
100 105 110
Val Ala Ala Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu
115 120 125
Lys Ser Gly Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro
130 135 140
Arg Glu Ala Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly
145 150 155 160
Asn Ser Gln Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr
165 170 175
Ser Leu Ser Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His
180 185 190
Lys Val Tyr Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val
195 200 205
Thr Lys Ser Phe Asn Arg Gly Glu Cys
210 215
<210> 47
<211> 120
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 47
Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu
1 5 10 15
Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Asp Ser Val Ser Ser Ser
20 25 30
Tyr Trp Thr Trp Ile Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile
35 40 45
Gly Tyr Ile Tyr Tyr Ser Gly Ser Ser Asn Tyr Asn Pro Ser Leu Lys
50 55 60
Ser Arg Ala Thr Ile Ser Val Asp Thr Ser Lys Asn Gln Phe Ser Leu
65 70 75 80
Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala
85 90 95
Arg Glu Gly Asn Val Asp Thr Thr Met Ile Phe Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser
115 120
<210> 48
<211> 107
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 48
Ala Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Gly Ile Arg Asn Asp
20 25 30
Leu Gly Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ala Ala Ser Ser Leu Gln Ser Gly Val Pro Ser Arg Phe Ala Gly
50 55 60
Arg Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Leu Gln Asp Phe Asn Tyr Pro Trp
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys
100 105
<210> 49
<211> 447
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 49
Gln Val Gln Leu Gln Glu Ser Gly Pro Gly Leu Val Lys Pro Ser Glu
1 5 10 15
Thr Leu Ser Leu Thr Cys Thr Val Ser Gly Asp Ser Val Ser Ser Ser
20 25 30
Tyr Trp Thr Trp Ile Arg Gln Pro Pro Gly Lys Gly Leu Glu Trp Ile
35 40 45
Gly Tyr Ile Tyr Tyr Ser Gly Ser Ser Asn Tyr Asn Pro Ser Leu Lys
50 55 60
Ser Arg Ala Thr Ile Ser Val Asp Thr Ser Lys Asn Gln Phe Ser Leu
65 70 75 80
Lys Leu Ser Ser Val Thr Ala Ala Asp Thr Ala Val Tyr Tyr Cys Ala
85 90 95
Arg Glu Gly Asn Val Asp Thr Thr Met Ile Phe Asp Tyr Trp Gly Gln
100 105 110
Gly Thr Leu Val Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val
115 120 125
Phe Pro Leu Ala Pro Cys Ser Arg Ser Thr Ser Glu Ser Thr Ala Ala
130 135 140
Leu Gly Cys Leu Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser
145 150 155 160
Trp Asn Ser Gly Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val
165 170 175
Leu Gln Ser Ser Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro
180 185 190
Ser Ser Ser Leu Gly Thr Lys Thr Tyr Thr Cys Asn Val Asp His Lys
195 200 205
Pro Ser Asn Thr Lys Val Asp Lys Arg Val Glu Ser Lys Tyr Gly Pro
210 215 220
Pro Cys Pro Pro Cys Pro Ala Pro Glu Phe Leu Gly Gly Pro Ser Val
225 230 235 240
Phe Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg Thr
245 250 255
Pro Glu Val Thr Cys Val Val Val Asp Val Ser Gln Glu Asp Pro Glu
260 265 270
Val Gln Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala Lys
275 280 285
Thr Lys Pro Arg Glu Glu Gln Phe Asn Ser Thr Tyr Arg Val Val Ser
290 295 300
Val Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys
305 310 315 320
Cys Lys Val Ser Asn Lys Gly Leu Pro Ser Ser Ile Glu Lys Thr Ile
325 330 335
Ser Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu Pro
340 345 350
Pro Ser Gln Glu Glu Met Thr Lys Asn Gln Val Ser Leu Thr Cys Leu
355 360 365
Val Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser Asn
370 375 380
Gly Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser
385 390 395 400
Asp Gly Ser Phe Phe Leu Tyr Ser Arg Leu Thr Val Asp Lys Ser Arg
405 410 415
Trp Gln Glu Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala Leu
420 425 430
His Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Leu Gly Lys
435 440 445
<210> 50
<211> 214
<212> PRT
<213> Искусственная последовательность
<220>
<223> синтетическая
<400> 50
Ala Ile Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Gly Ile Arg Asn Asp
20 25 30
Leu Gly Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Ala Ala Ser Ser Leu Gln Ser Gly Val Pro Ser Arg Phe Ala Gly
50 55 60
Arg Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Thr Tyr Tyr Cys Leu Gln Asp Phe Asn Tyr Pro Trp
85 90 95
Thr Phe Gly Gln Gly Thr Lys Val Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| ДОЗИРОВКА И СХЕМА ВВЕДЕНИЯ ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ СВЯЗАННЫХ С С5 ЗАБОЛЕВАНИЙ ПОСРЕДСТВОМ ПРИМЕНЕНИЯ АНТИТЕЛА ПРОТИВ С5 КРОВАЛИМАБА | 2020 |
|
RU2832188C2 |
| АНТИТЕЛА ЧЕЛОВЕКА К PCSK9 ДЛЯ ПРИМЕНЕНИЯ В СПОСОБАХ ЛЕЧЕНИЯ КОНКРЕТНЫХ ГРУПП ИНДИВИДУУМОВ | 2012 |
|
RU2721279C2 |
| СТАБИЛЬНЫЕ СОСТАВЫ АНТИТЕЛ ПРОТИВ TIGIT, ОТДЕЛЬНО И В КОМБИНАЦИИ С АНТИТЕЛАМИ ПРОТИВ РЕЦЕПТОРА 1 ПРОГРАММИРУЕМОЙ СМЕРТИ (PD-1), И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2820576C2 |
| АНТИТЕЛО ПРОТИВ В7-Н3, ЕГО АНТИГЕНСВЯЗЫВАЮЩИЙ ФРАГМЕНТ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2765306C2 |
| АНТИТЕЛО ПРОТИВ СТОЛБНЯЧНОГО ТОКСИНА И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2815280C1 |
| Антитела против ингибитора пути тканевого фактора и их применения | 2016 |
|
RU2815681C1 |
| АНТИТЕЛО ПРОТИВ БЕТА-АМИЛОИДА, ЕГО АНТИГЕНСВЯЗЫВАЮЩИЙ ФРАГМЕНТ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2777844C1 |
| Прогнозирование риска развития нежелательной реакции, связанной с введением антитела к ALK2, и способности отвечать на лечение антителом к ALK2 | 2019 |
|
RU2783762C2 |
| БИСПЕЦИФИЧЕСКИЕ АНТИГЕНСВЯЗЫВАЮЩИЕ МОЛЕКУЛЫ, СОДЕРЖАЩИЕ АНТИ-FAP КЛОН 212 | 2019 |
|
RU2799429C2 |
| АНТИТЕЛО ПРОТИВ IL-5, ЕГО АНТИГЕНСВЯЗЫВАЮЩИЙ ФРАГМЕНТ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2772716C2 |
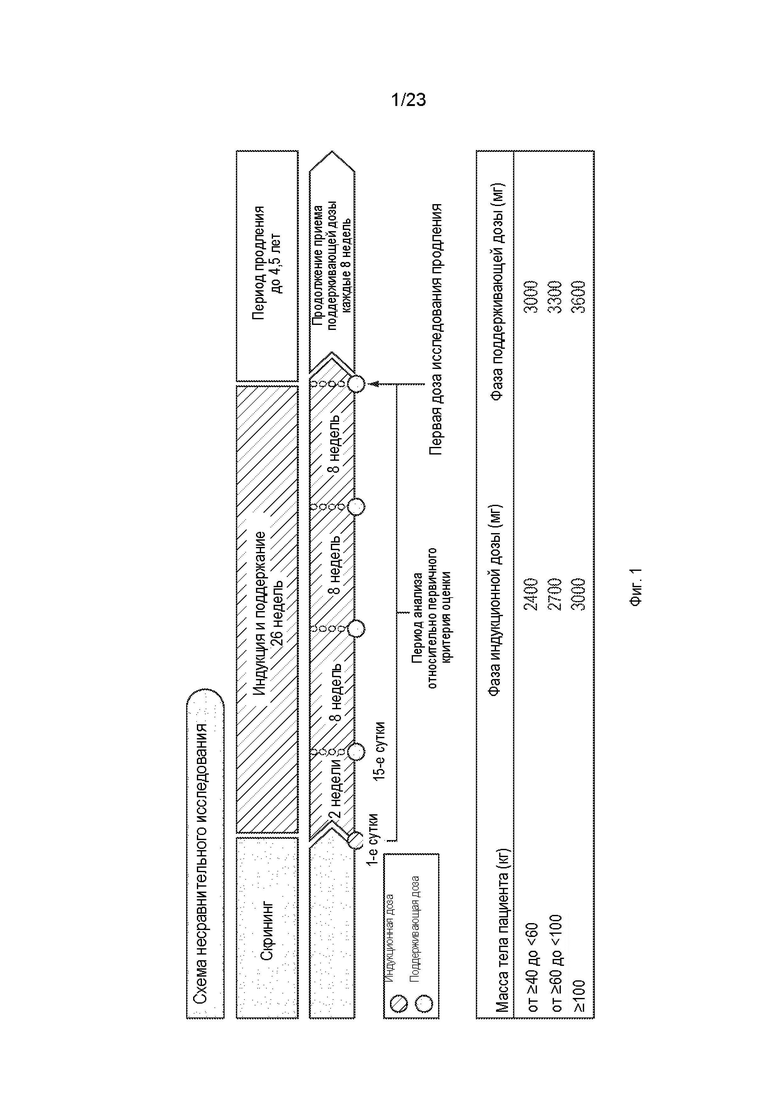

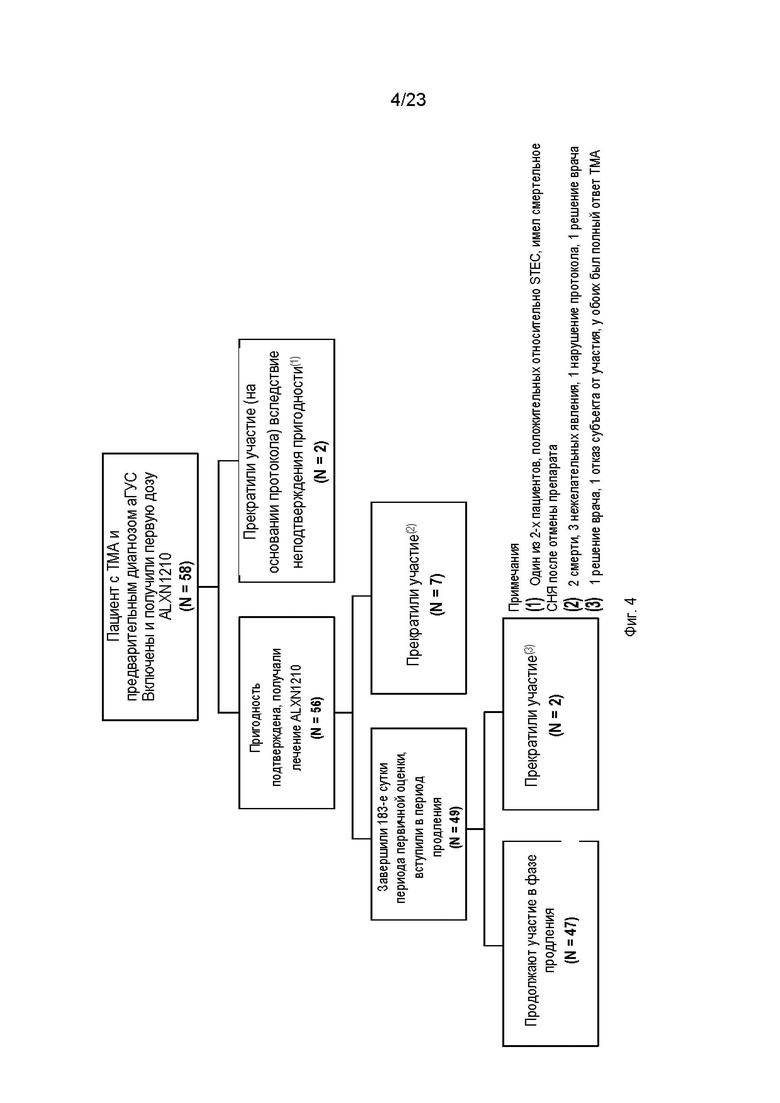

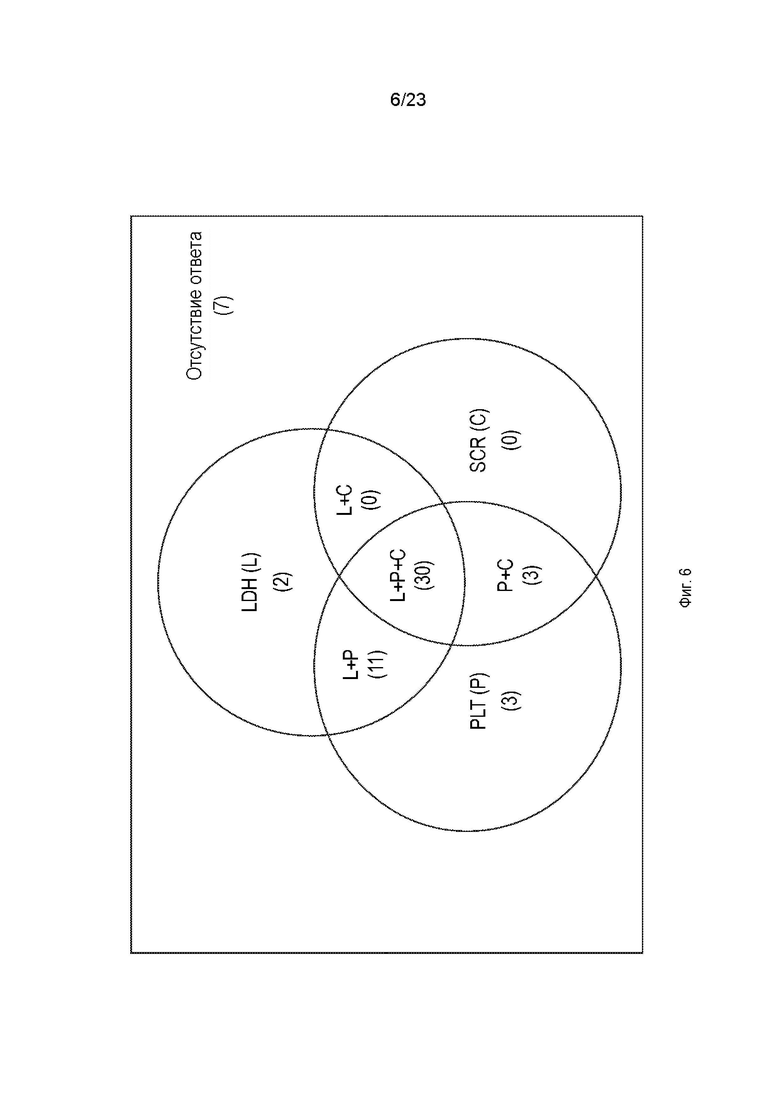
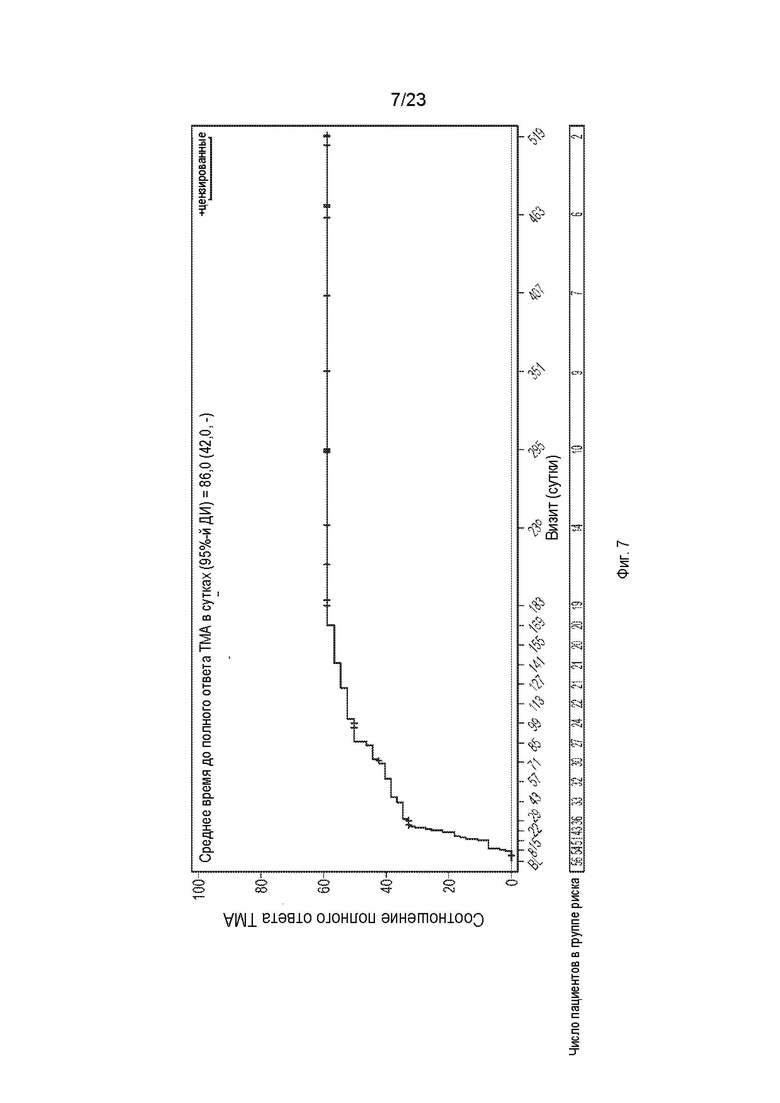
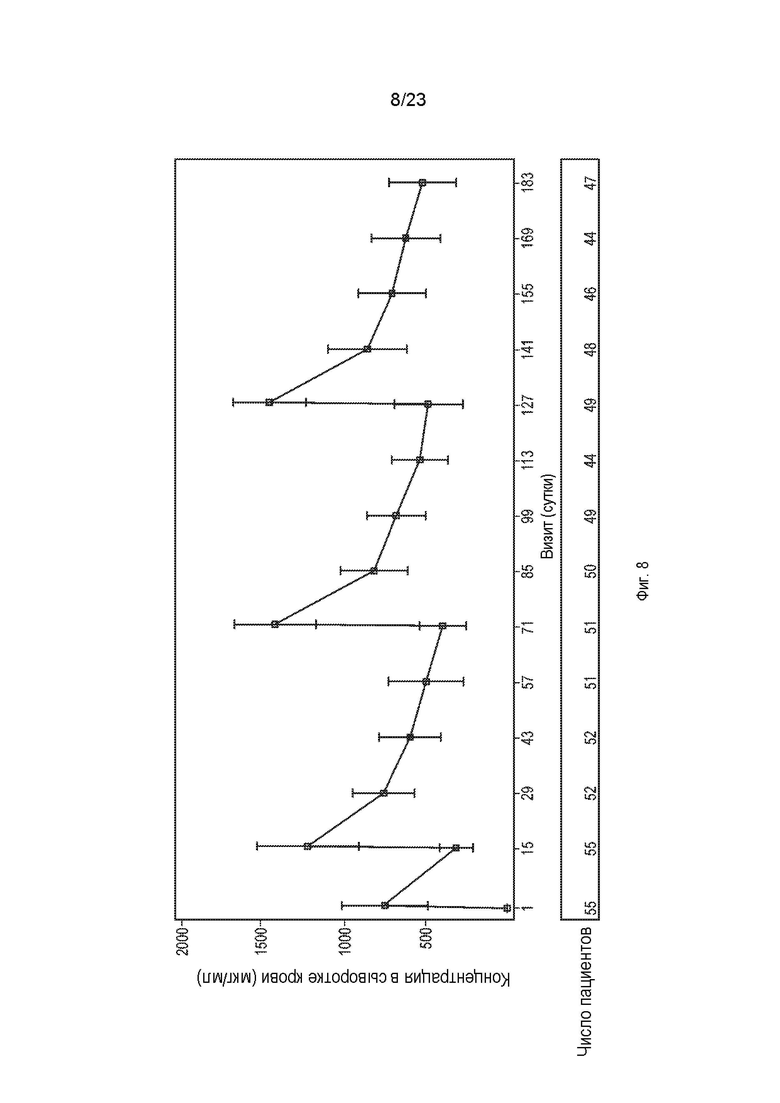
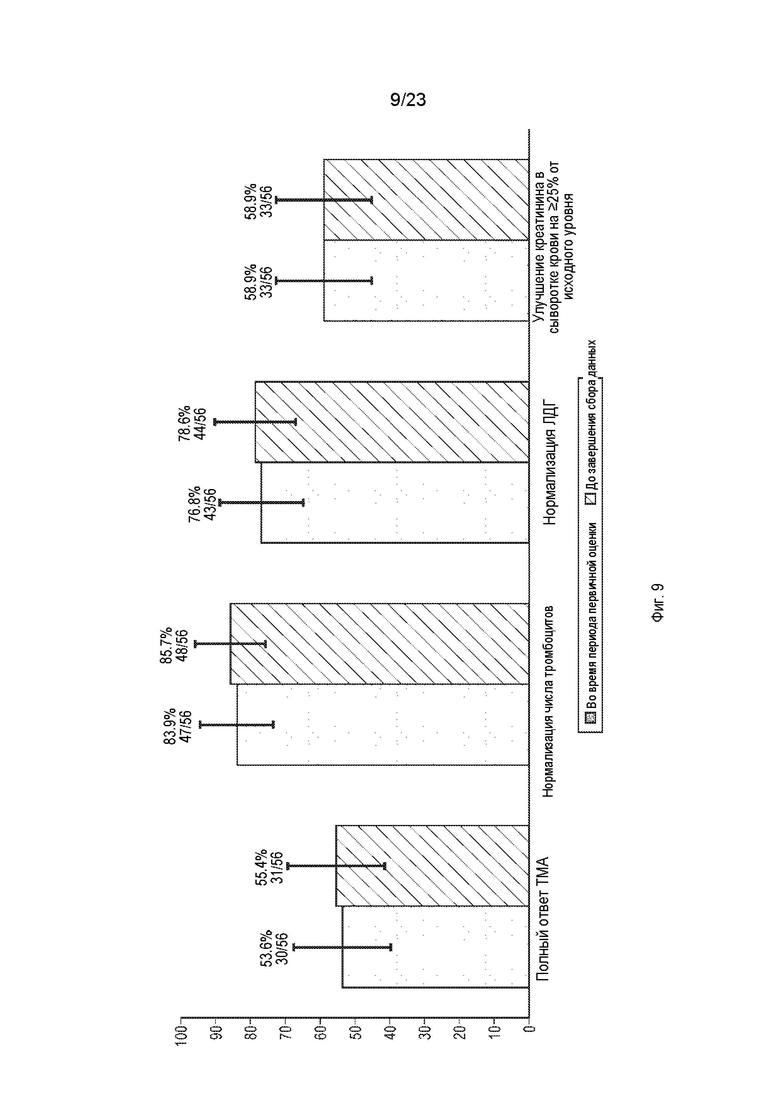

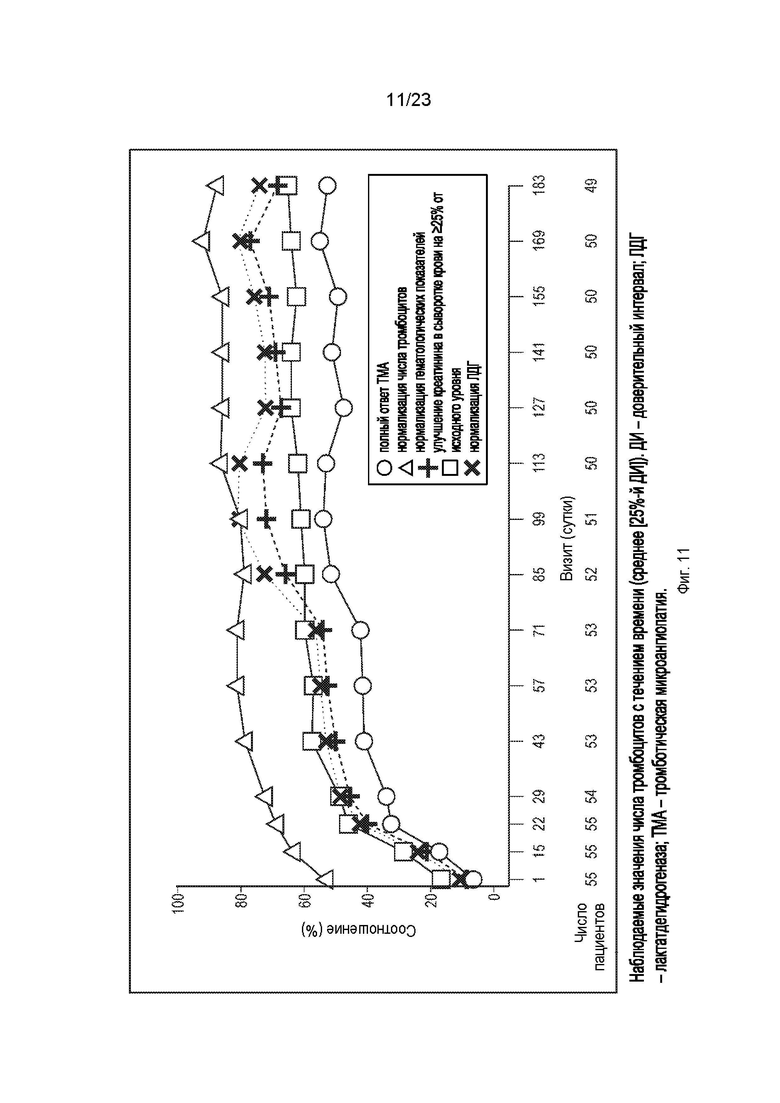
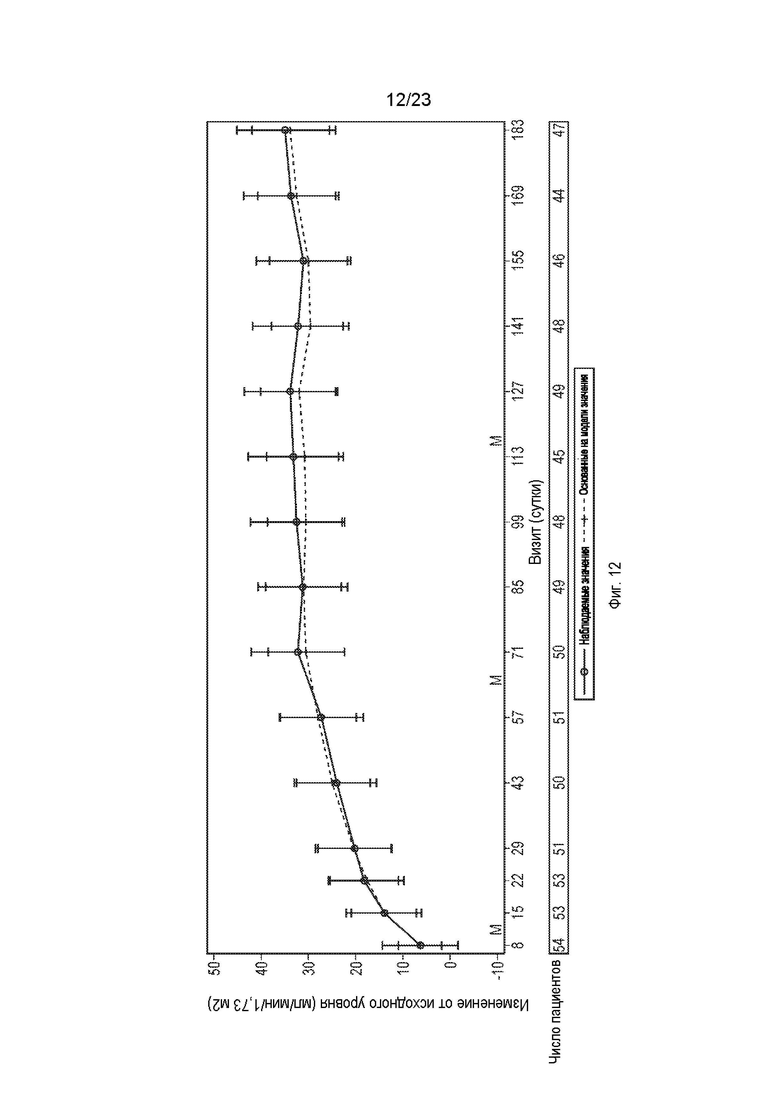
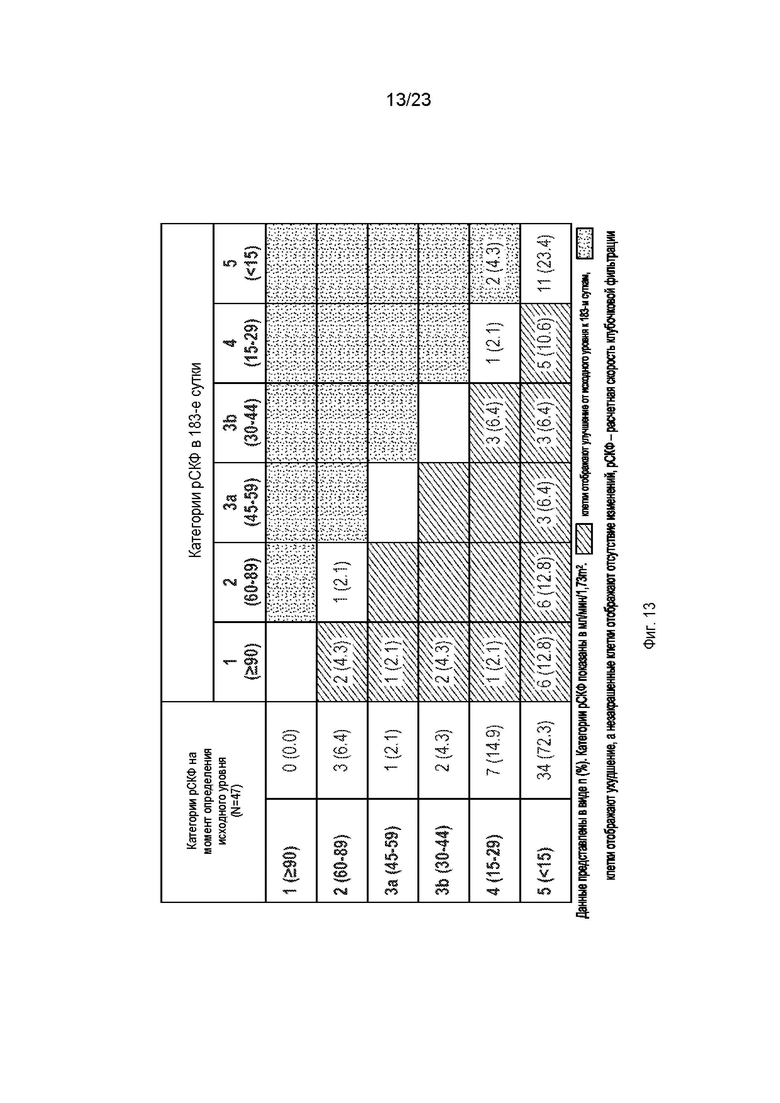
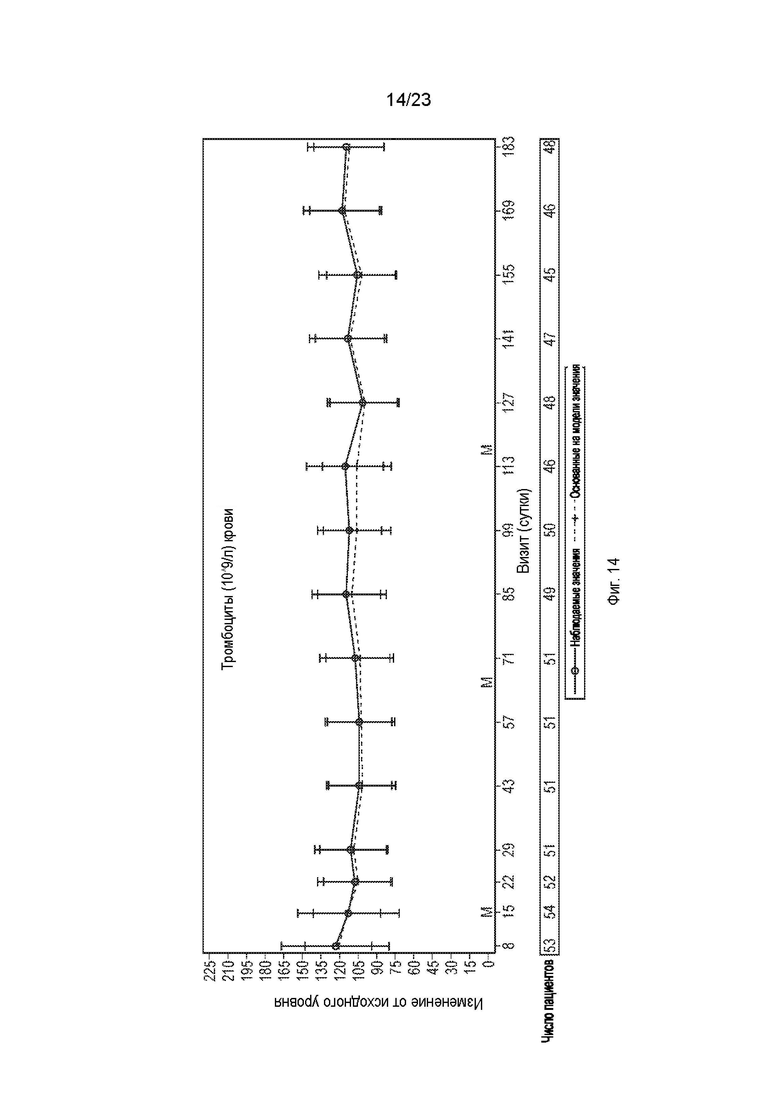
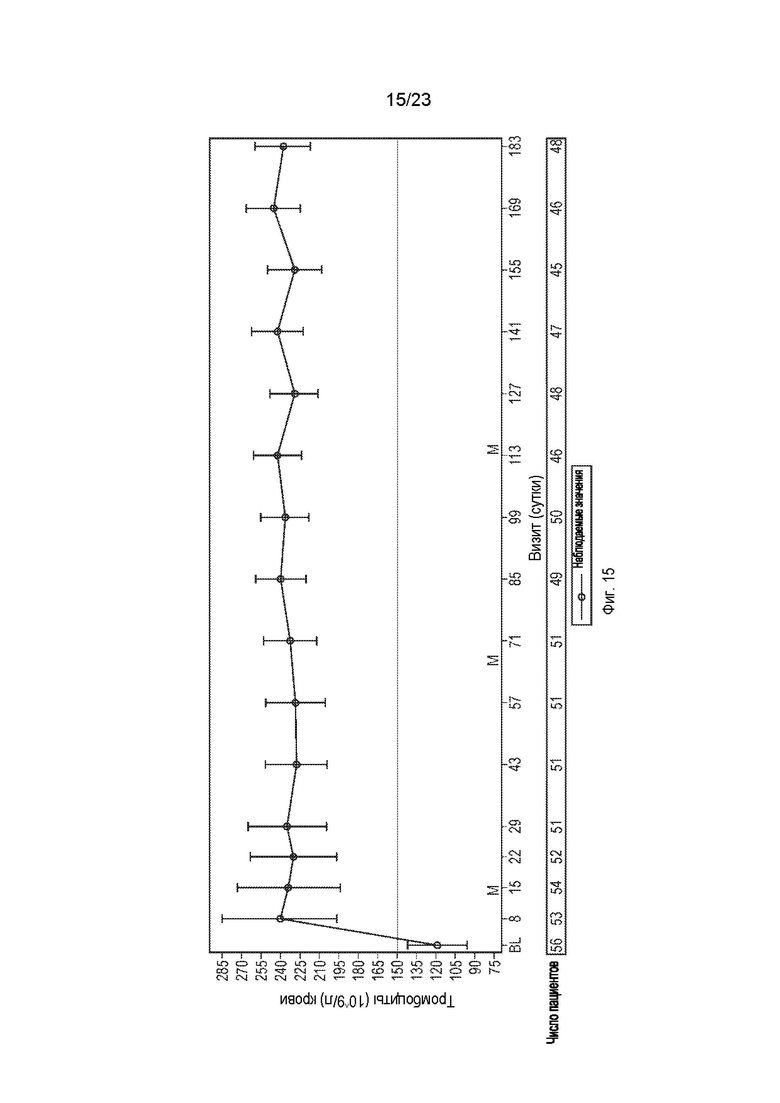
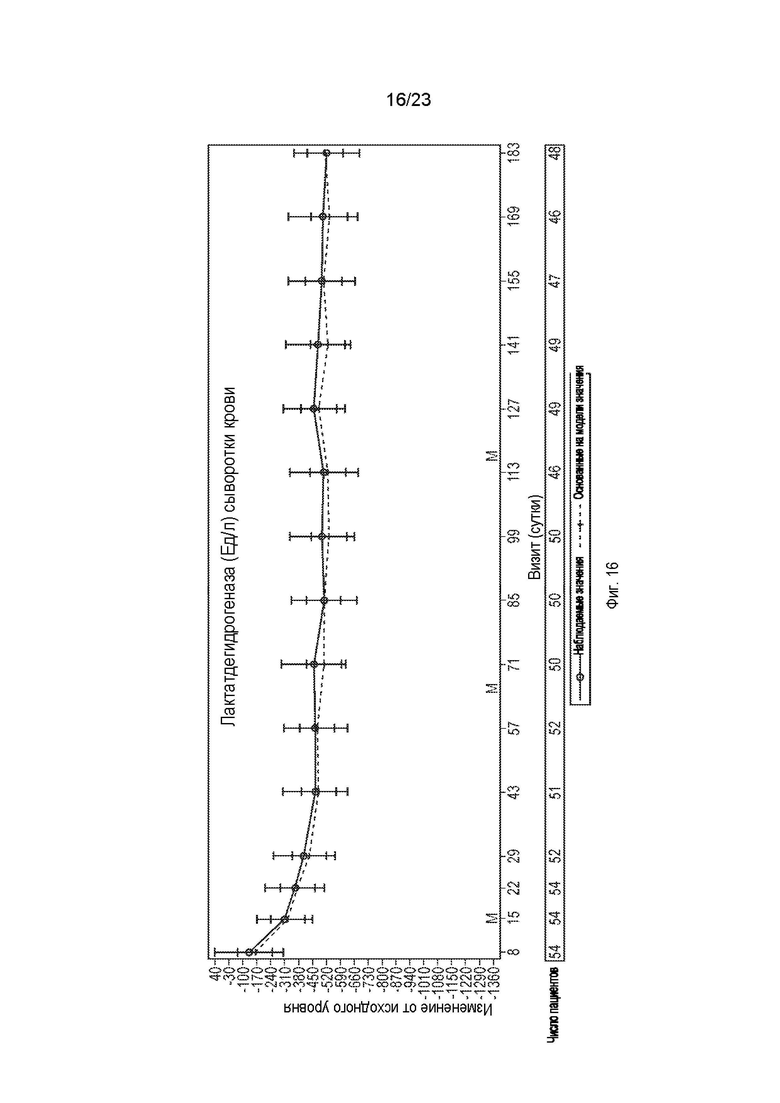
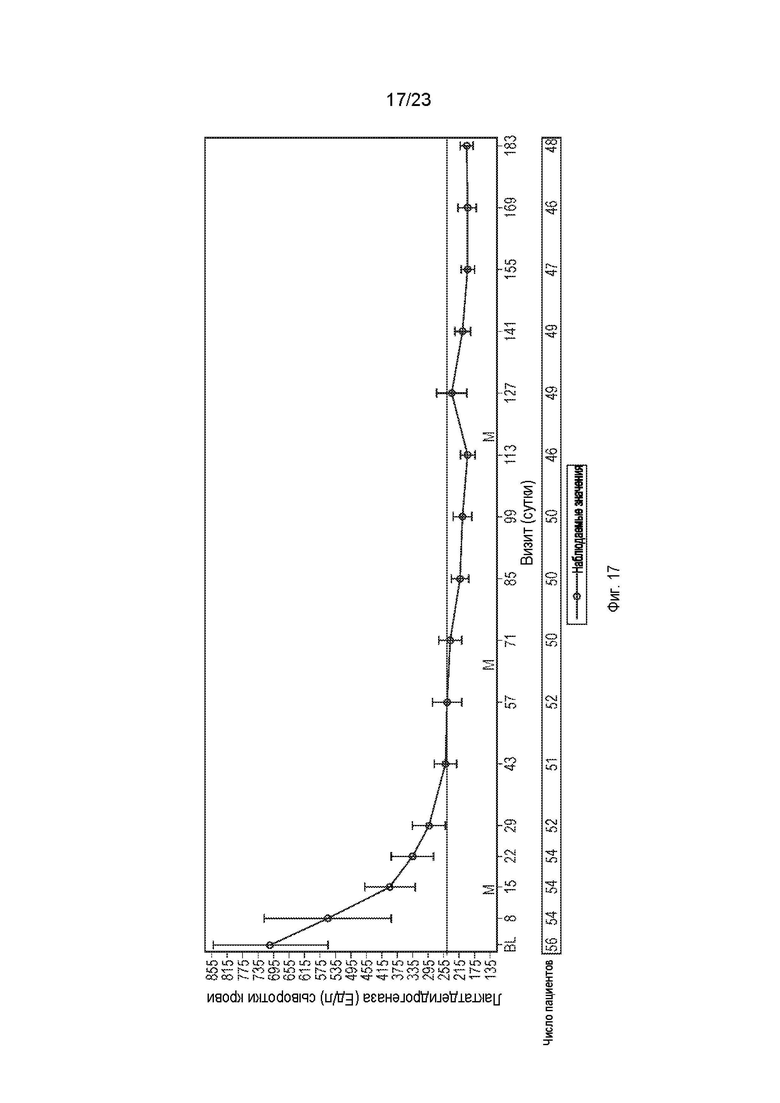
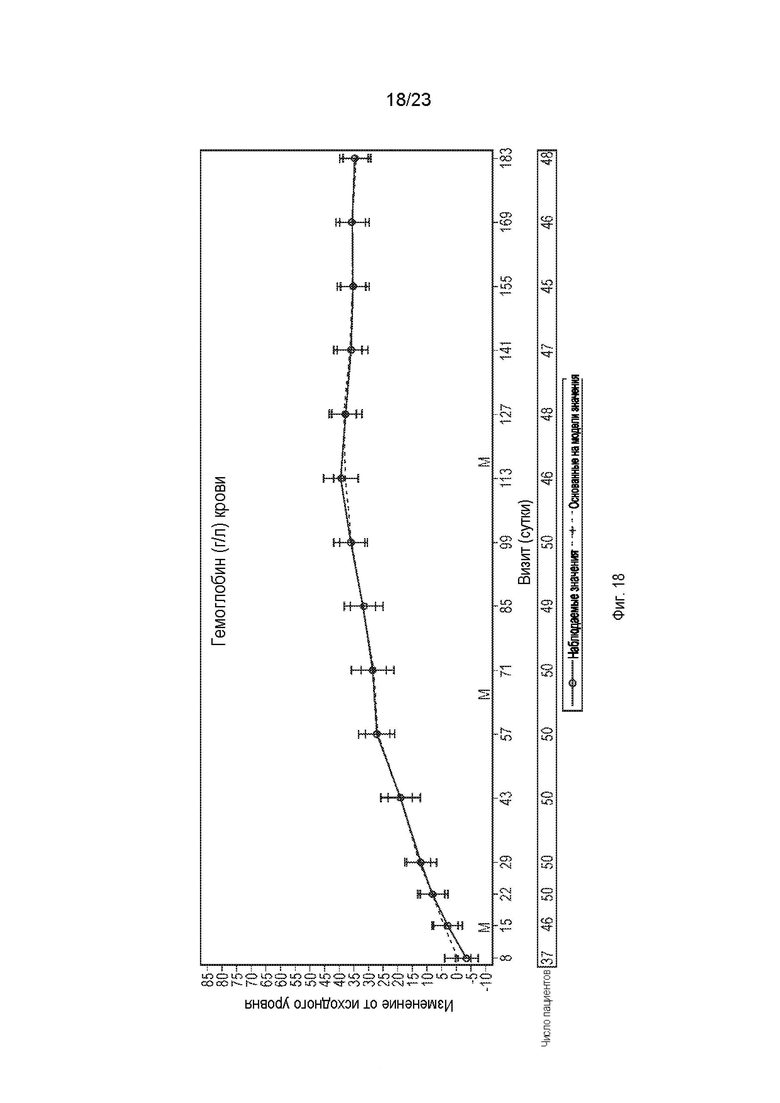
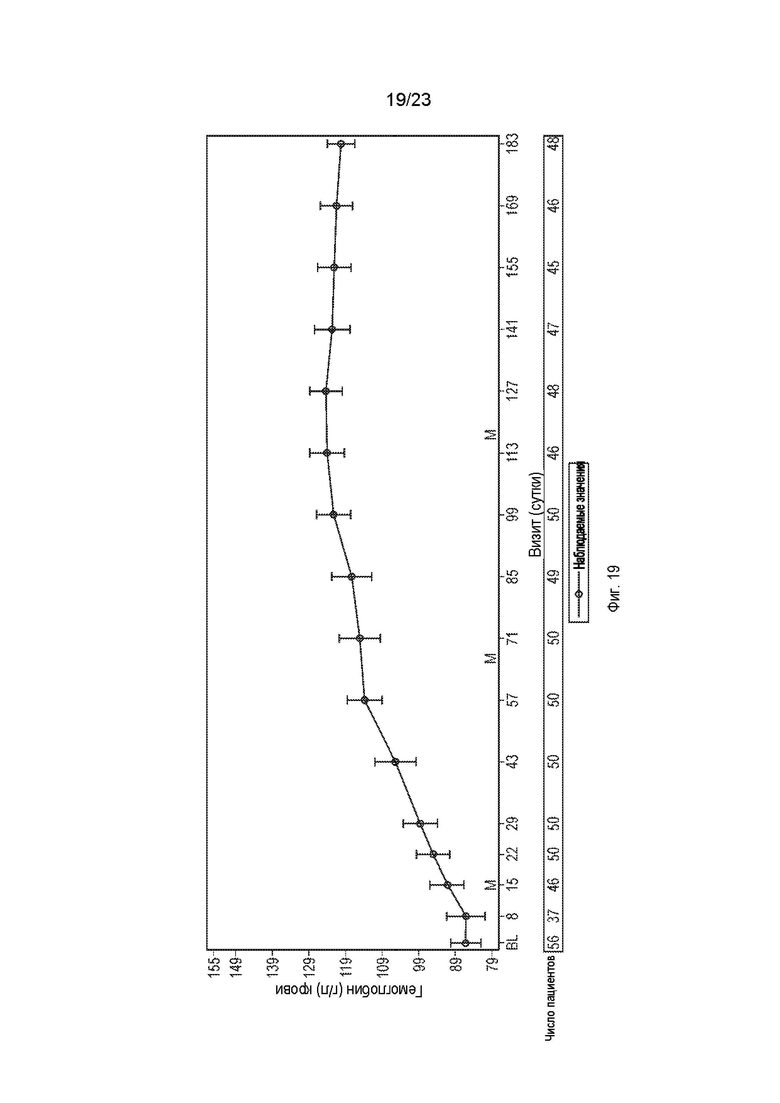
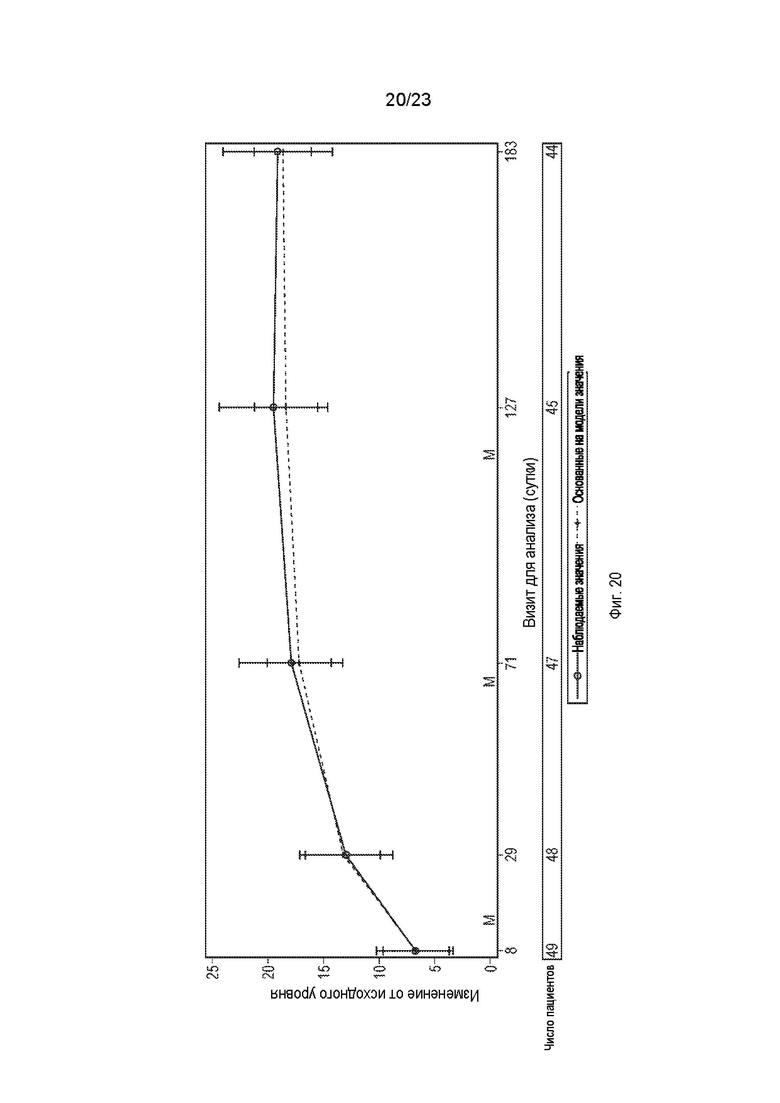
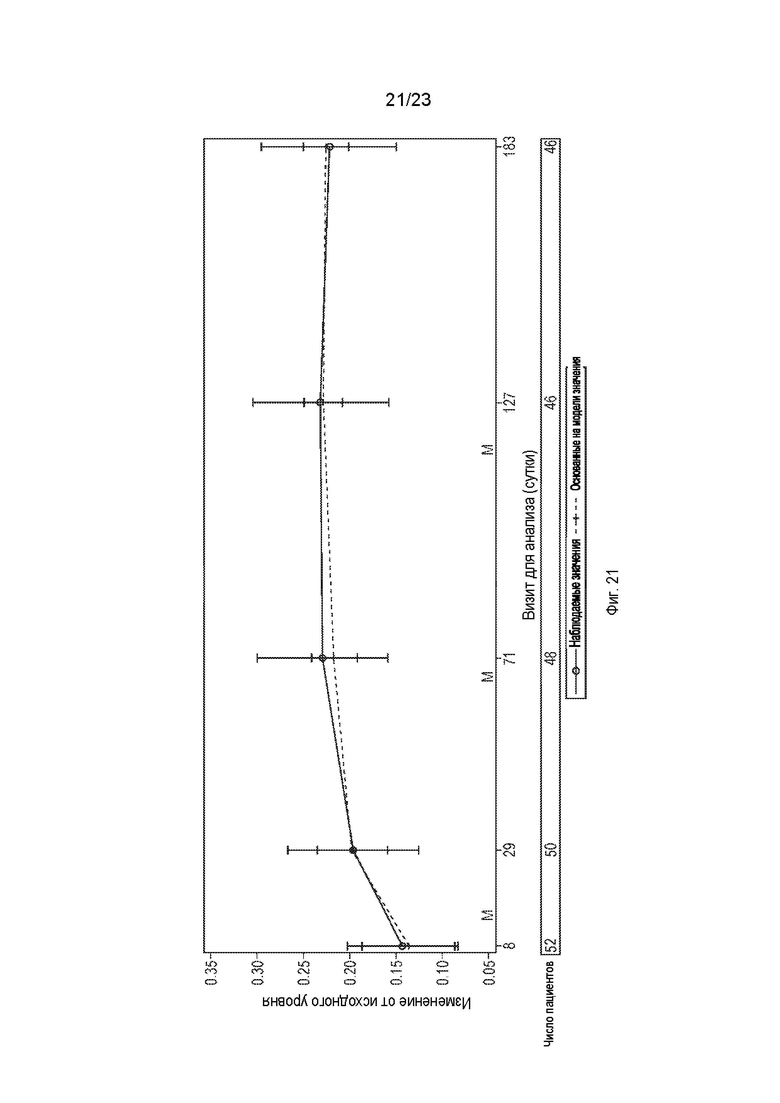
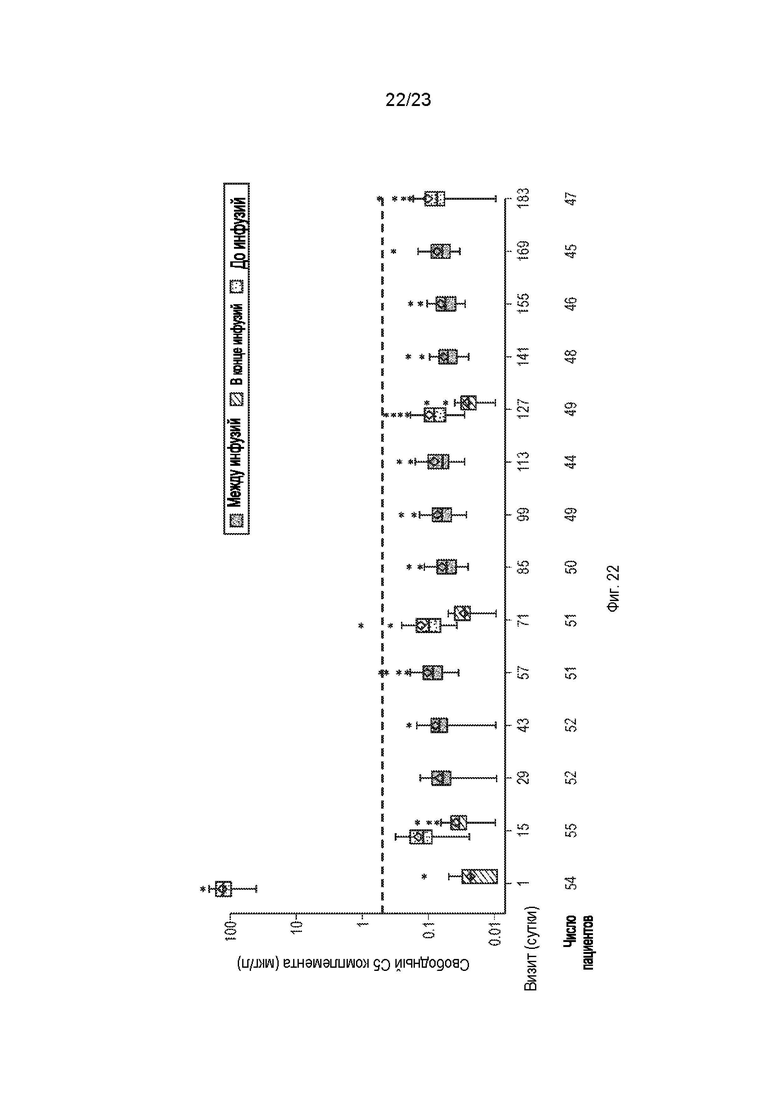
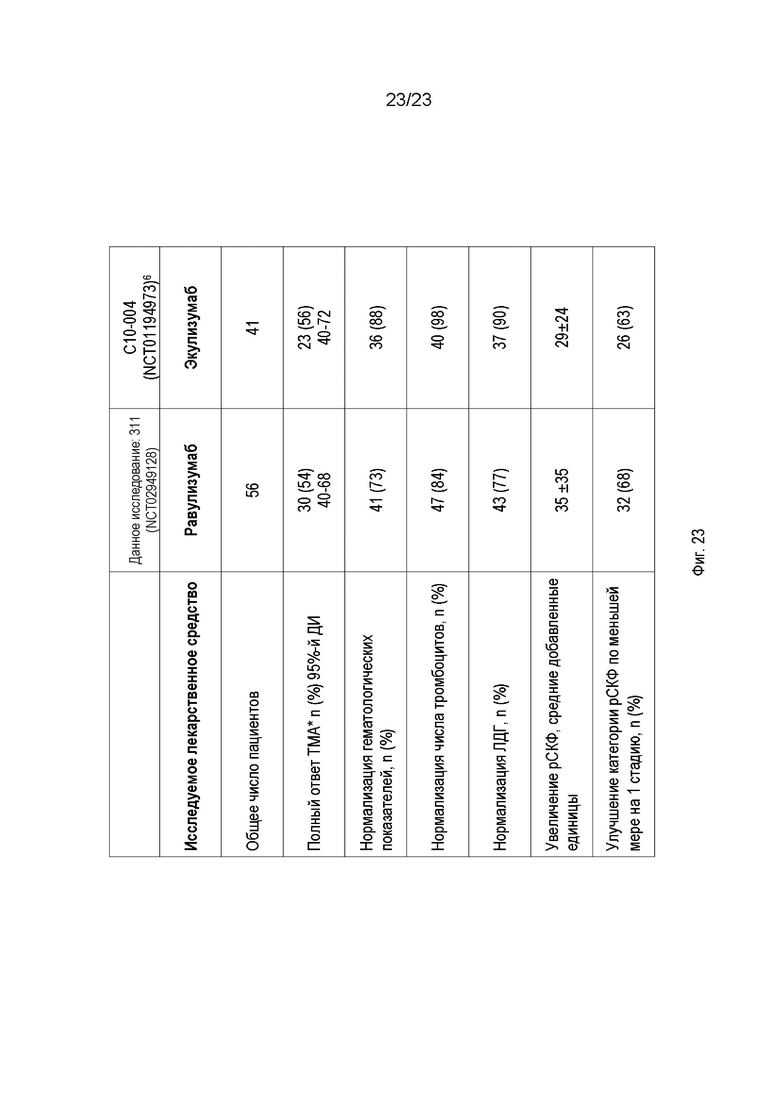
Группа изобретений относится к способам лечения аГУС у взрослого пациента-человека в возрасте 18 лет или старше с применением антитела против С5. Антитело против C5 представляет собой антитело, содержащее полипептид тяжелой цепи с SEQ ID NO: 14, полипептид легкой цепи с SEQ ID NO: 11, или представляет собой равулизумаб. Антитело против C5 вводят в конкретном количестве дозы, основанной на массе тела пациента, и в соответствии с конкретным графиком введения доз. Изобретения обеспечивают улучшенные способы лечения пациентов с аГУС. 4 н. и 38 з.п. ф-лы, 23 ил., 22 табл., 2 пр.
1. Способ лечения атипичного гемолитико-уремического синдрома (аГУС) у взрослого пациента-человека в возрасте 18 лет или старше, включающий введение указанному пациенту эффективного количества антитела против C5, где антитело против C5 представляет собой антитело, содержащее полипептид тяжелой цепи, содержащий аминокислотную последовательность, представленную в SEQ ID NO: 14, полипептид легкой цепи, содержащий аминокислотную последовательность, представленную в SEQ ID NO: 11, при этом указанное антитело против C5 вводят:
(а) один раз на 1-е сутки в дозе, составляющей: 2400 мг – пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг – пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг – пациенту с массой тела ≥ 100 кг; и
(b) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей: 3000 мг – пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг – пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг – пациенту с массой тела ≥ 100 кг.
2. Способ лечения атипичного гемолитико-уремического синдрома (аГУС) у взрослого пациента-человека в возрасте 18 лет или старше, включающий введение указанному пациенту эффективного количества антитела против C5, где антитело против C5 представляет собой равулизумаб, при этом указанное антитело против C5 вводят:
(а) один раз на 1-е сутки в дозе, составляющей: 2400 мг – пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг – пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг – пациенту с массой тела ≥ 100 кг; и
(b) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей: 3000 мг – пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг – пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг – пациенту с массой тела ≥ 100 кг.
3. Способ по п. 1 или 2, отличающийся тем, что указанный пациент ранее получал лечение экулизумабом.
4. Способ по любому из предшествующих пунктов, отличающийся тем, что лечение начинают по меньшей мере через две недели после введения указанному пациенту последней дозы экулизумаба.
5. Способ по любому из предшествующих пунктов, отличающийся тем, что указанный пациент получал лечение экулизумабом в течение по меньшей мере 6 месяцев до 1-х суток лечения.
6. Способ по любому из предшествующих пунктов, отличающийся тем, что указанный пациент ранее получал лечение экулизумабом в дозе 900 мг каждые 2 недели.
7. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное антитело против С5 связывается с C5 человека при pH 7,4 и 25°C с аффинной константой диссоциации (KD), которая находится в диапазоне от 0,1 нМ до 1 нМ.
8. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное антитело против С5 связывается с C5 человека при pH 6,0 и 25°C с KD ≥ 10 нМ.
9. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное антитело против С5 вводят пациенту с массой тела от ≥ 40 до < 60 кг:
(а) один раз на 1-е сутки в дозе, составляющей: 2400 мг; и
(b) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей 3000 мг.
10. Способ по любому из пп. 1-8, отличающийся тем, что указанное антитело против С5 вводят пациенту с массой тела от ≥ 60 до < 100 кг:
(а) один раз на 1-е сутки в дозе, составляющей: 2700 мг; и
(b) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей 3300 мг.
11. Способ по любому из пп. 1-8, отличающийся тем, что указанное антитело против С5 вводят пациенту с массой тела ≥ 100 кг:
(а) один раз на 1-е сутки в дозе, составляющей: 3000 мг; и
(b) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей 3600 мг.
12. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение поддерживает остаточную концентрацию указанного антитела против C5 в сыворотке крови на уровне 100 мкг/мл или больше во время лечения.
13. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение поддерживает остаточную концентрацию указанного антитела против C5 в сыворотке крови на уровне 200 мкг/мл или больше во время лечения.
14. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение поддерживает концентрацию свободного C5 на уровне от 0,309 до 0,5 мкг/мл или ниже.
15. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение снижает концентрацию свободного C5 больше чем на 99% в течение периода лечения.
16. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение снижает концентрацию свободного C5 больше чем на 99,5% в течение периода лечения.
17. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное антитело против C5 вводят в дозе 3000 мг, 3300 мг или 3600 мг каждые восемь недель в течение периода времени до двух лет.
18. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное антитело против C5 составлено для внутривенного введения.
19. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение в общей сложности составляет 26 недель лечения.
20. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к ингибированию терминального комплемента.
21. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к снижению гемолиза по сравнению с исходным уровнем на основании оценки уровней лактатдегидрогеназы (ЛДГ).
22. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к нормализации уровней ЛДГ.
23. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к устранению прорывного гемолиза во время периода лечения.
24. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение вызывает сдвиг в сторону нормальных уровней связанного с гемолизом гематологического биомаркера, выбранного из группы, состоящей из свободного гемоглобина, гаптоглобина, количества ретикулоцитов, ПНГ-клона эритроцитов и D-димера.
25. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение оказывает по меньшей мере один терапевтический эффект, выбранный из группы, состоящей из снижения или прекращения тяжелой гипертензии, протеинурии, уремии, вялости, усталости, раздражительности, тромбоцитопении, микроангиопатической гемолитической анемии и нарушения функции почек по сравнению с исходным уровнем.
26. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение вызывает сдвиг в сторону нормальных уровней фактора Ва, растворимого рецептора фактора некроза опухоли 1 [sTNFR1], растворимой молекулы адгезии сосудов 1 [sVCAM1], тромбомодулина, D-димера и цистатина С.
27. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение вызывает повышение стабилизации гемоглобина по сравнению с исходным уровнем.
28. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение вызывает снижение потребности в гемотрансфузиях по сравнению с исходным уровнем.
29. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение вызывает снижение количества значительных неблагоприятных сосудистых событий (MAVE - англ.: major adverse vascular events).
30. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение вызывает изменение качества жизни по сравнению с исходным уровнем, оцениваемое с помощью шкалы функциональной оценки терапии хронических заболеваний (FACIT) для показателя утомляемости версии 4 и опросника Европейской организации по изучению и лечению злокачественных опухолей для оценки качества жизни из 30 вопросов.
31. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к нормализации уровня тромбоцитов.
32. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к улучшению уровней креатинина в сыворотке крови на ≥ 25% по сравнению с исходным уровнем.
33. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к полному ответу ТМА.
34. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к модифицированному полному ответу тромботической микроангиопатии (ТМА).
35. Способ по любому из предшествующих пунктов, отличающийся тем, что хроническая болезнь почек (ХБП) пациента улучшается на одну или большее число стадий после начала лечения.
36. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к сдвигу в сторону нормальных уровней рСКФ.
37. Способ по любому из предшествующих пунктов, отличающийся тем, что указанное лечение приводит к значению временного компромисса по опроснику EQ-5D-3L для США (US TTO), которое составляет > 0,94.
38. Применение набора для лечения атипичного гемолитико-уремического синдрома (аГУС) у взрослого пациента-человека в возрасте 18 лет или старше, содержащий:
(a) дозу антитела против C5, где антитело против C5 представляет собой равулизумаб; и
(b) инструкции по применению равулизумаба согласно способу по п. 2,
при этом равулизумаб вводят 1) один раз на 1-е сутки в дозе, составляющей: 2400 мг – пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг – пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг – пациенту с массой тела ≥ 100 кг; и
2) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей: 3000 мг – пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг – пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг – пациенту с массой тела ≥ 100 кг.
39. Применение по п. 38, отличающийся тем, что указанное антитело против С5 вводят пациенту с массой тела от ≥ 40 до < 60 кг:
(а) один раз на 1-е сутки в дозе, составляющей 2400 мг; и
(b) на 15-е сутки.
40. Применение по п. 38, отличающийся тем, что указанное антитело против С5 вводят пациенту с массой тела от ≥ 60 до < 100 кг:
(а) один раз на 1-е сутки в дозе, составляющей 2700 мг; и
(b) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей 3300 мг.
41. Применение по п. 40, отличающийся тем, что указанное антитело против С5 вводят пациенту с массой тела ≥ 100 кг:
(а) один раз на 1-е сутки в дозе, составляющей 3000 мг; и
(b) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей 3600 мг.
42. Применение антитела против C5 для лечения атипичного гемолитико-уремического синдрома (аГУС) у взрослого пациента-человека в возрасте 18 лет или старше, где антитело против C5 представляет собой равулизумаб, при этом указанное антитело против C5 вводят:
(а) один раз на 1-е сутки в дозе, составляющей: 2400 мг – пациенту с массой тела от ≥ 40 до < 60 кг, 2700 мг – пациенту с массой тела от ≥ 60 до < 100 кг, или 3000 мг – пациенту с массой тела ≥ 100 кг; и
(b) на 15-е сутки и после этого – каждые восемь недель в дозе, составляющей: 3000 мг – пациенту с массой тела от ≥ 40 до < 60 кг, 3300 мг – пациенту с массой тела от ≥ 60 до < 100 кг, или 3600 мг – пациенту с массой тела ≥ 100 кг.
| Автомобиль-сани, движущиеся на полозьях посредством устанавливающихся по высоте колес с шинами | 1924 |
|
SU2017A1 |
| EA 201691764 A1, 30.12.2016 | |||
| Ravulizumab for atypical haemolytic uraemic syndrome in adults and children - first line, EVIDENCE BRIEFING, 01.08.2018, pp | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Alexion | |||

