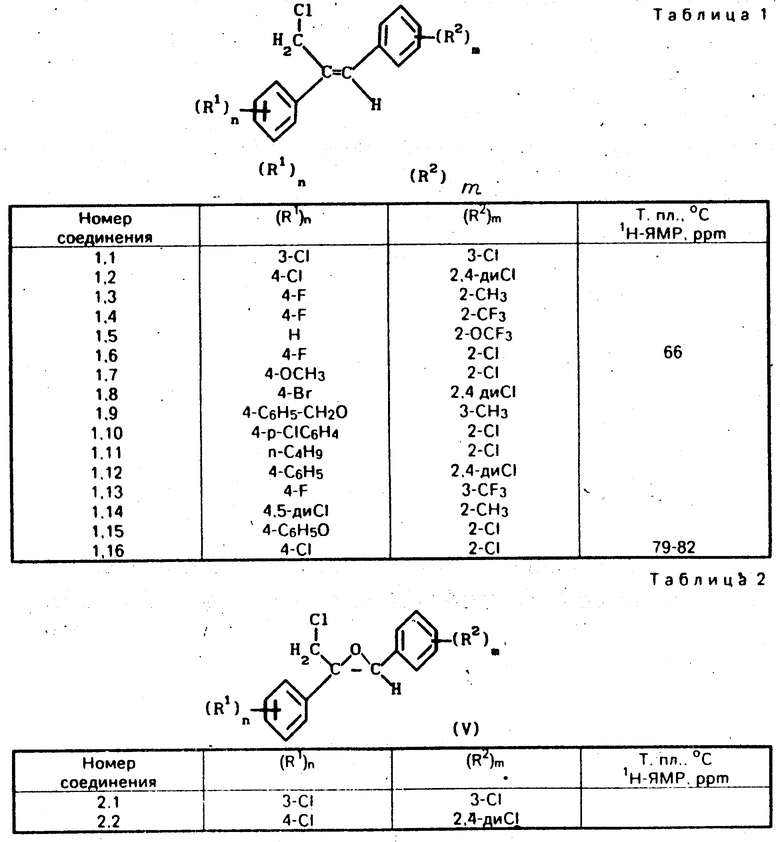
Изобретение относится к стереоизбирательному получению 1/2-диарилаллилхлоридов общей формулы
(R1)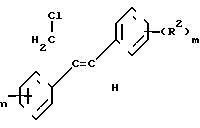 (I) где остатки R1 и R2 независимо друг от друга означают водород/ галоген/ алкил/ галогеналкил/ алкокси/ галогеналкокси или незамещенный или замещенный ароматический остаток/ а n и m означают 1/2 или 3.
(I) где остатки R1 и R2 независимо друг от друга означают водород/ галоген/ алкил/ галогеналкил/ алкокси/ галогеналкокси или незамещенный или замещенный ароматический остаток/ а n и m означают 1/2 или 3.
Согласно выкладкам ФРГ NN 3218129 и 3218130/ а также патенту Европы 196038 и патенту США 3422153/ соединения с типом структуры l представляют собой ценные промежуточные продукты в получении фармакологических/ фунгицидных и противогрибковых действующих начал. До сих пор их получали путем радикального галогенирования соответствующих диарилпропеновых соединений или путем окисления с последующей реакцией замещения. Недостатком известных из данного уровня техники способов является применение дорогостоящих реагентов/ например/ таких дорогих реагентов галогенирования/ как N-бромсукцинимид в реакции радикального бромирования/ число ступеней реакции синтеза и/ в частности/ низкая стереоизбирательность реакции.
Общеизвестно/ что молекулы/ проявляющие специфическое биологическое или фармакологическое действие/ во многих случаях требуют определенного геометрического расположения определенных функциональных групп. Например/ среди фунгицидных действующих начал особенно высокой эффективностью в качестве средств защиты растений обладают в первую очередь соединения с Z-конфигурацией (см.правило последовательности заместителей по Кану/ Ингольду и Прелогу)/ т. е. соединения/ в которых незамещенные или замещенные фенилы один относительно другого находятся в транс-положении.
Поэтому задача настоящего изобретения заключалась в разработке способа/ согласно которому удается получить с высоким выходом промежуточные продукты 1 с максимальной чистотой изомеров/ т.е. с высокой избирательностью относительно Е- и транс-конфигурации фенильных остатков/ присоединенных к двойной связи соединения. Другая задача заключалась в том/ чтобы с применением выгодных промежуточных продуктов разработать способ получения фунгицидных азолилметилоксиранов IV/ отличающийся высокими общими выходами и меньшим числом стадий реакций/ чем у известных способов.
Согласно уровню техники арилзамещенные спирты в кислых условиях реакции/ например/ с применением серной кислоты в органической фазе можно перевести в соответствующие арилзамещенные олефины или стиролы (см./ например/ Houben-Weyl, Methoden der organischen Chemie, 4-e изд./ т. 5/1 b Alkene, Cycloalkene, Arylalkene изд-во Georg Thieme Verlag г. Штуттгарт/ 1972 г./ стр. 62 и сл./ в частности стр. 70 и 71; Tetrahedron т.26. стр. 4277 и сл. (1790 г)).
Известно также/ что подобного рода реакции можно осуществить с использованием акцепторов воды/ например ацетангидрида. Однако для осуществления этих реакций отщепления обычно требуются высокие температуры. В таких условиях реакции получают только недостаточное соотношение Е- и Z-изомеров относительно расположения арила по отношению к арилу.
Теперь найден способ стереоизбирательного получения Z-1/2-диарил-аллилхлоридов общей формулы I
(R1)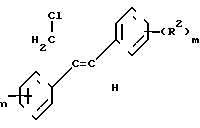 где остатки R1 и R2 независимо друг от друга означают водород/ галоген/ алкил/ галогеналкил/ алкокси/ галогеналкокси или незамещенный или замещенный ароматический остаток/ а n и m означают 1/2 или 3/ отличающийся тем/ что хлоргидрины формулы II
где остатки R1 и R2 независимо друг от друга означают водород/ галоген/ алкил/ галогеналкил/ алкокси/ галогеналкокси или незамещенный или замещенный ароматический остаток/ а n и m означают 1/2 или 3/ отличающийся тем/ что хлоргидрины формулы II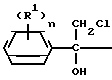 CH
CH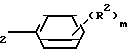 где остатки имеют вышеуказанное значение/ дегидратируют в инертном растворителе/ представляющем собой простой эфир или сложный эфир карбоновой кислоты/ в присутствии ангидрида карбоновой кислоты или органической кислоты или неорганической кислоты или олеума при температурах до 50°С.
где остатки имеют вышеуказанное значение/ дегидратируют в инертном растворителе/ представляющем собой простой эфир или сложный эфир карбоновой кислоты/ в присутствии ангидрида карбоновой кислоты или органической кислоты или неорганической кислоты или олеума при температурах до 50°С.
Согласно предлагаемому способу/ обеспечена высокая стереоизбирательность получения 1/2 -диарилаллилхлоридов с Z-конфигурацией. Как правило/ в частности при осуществлении предпочтительных вариантов выполнения предлагаемого способа соотношение Z:E составляет 8:1 - 15:1. Неожиданно высока и региоселективность/ с которой протекает удаление воды/ потому что можно было ожидать/ что в качестве побочной реакции в усиленной мере будет протекать реакция отщепления воды в сторону хлорметильной боковой цепи с образованием хлорвинилдиарильных соединений. Далее/ удается подавить такие ожидаемые конкурирующие реакции/ как реакция замещения воды вместо ее удаления. Ожидаемое ацилирование спиртовой группы тоже фактически отсутствует.
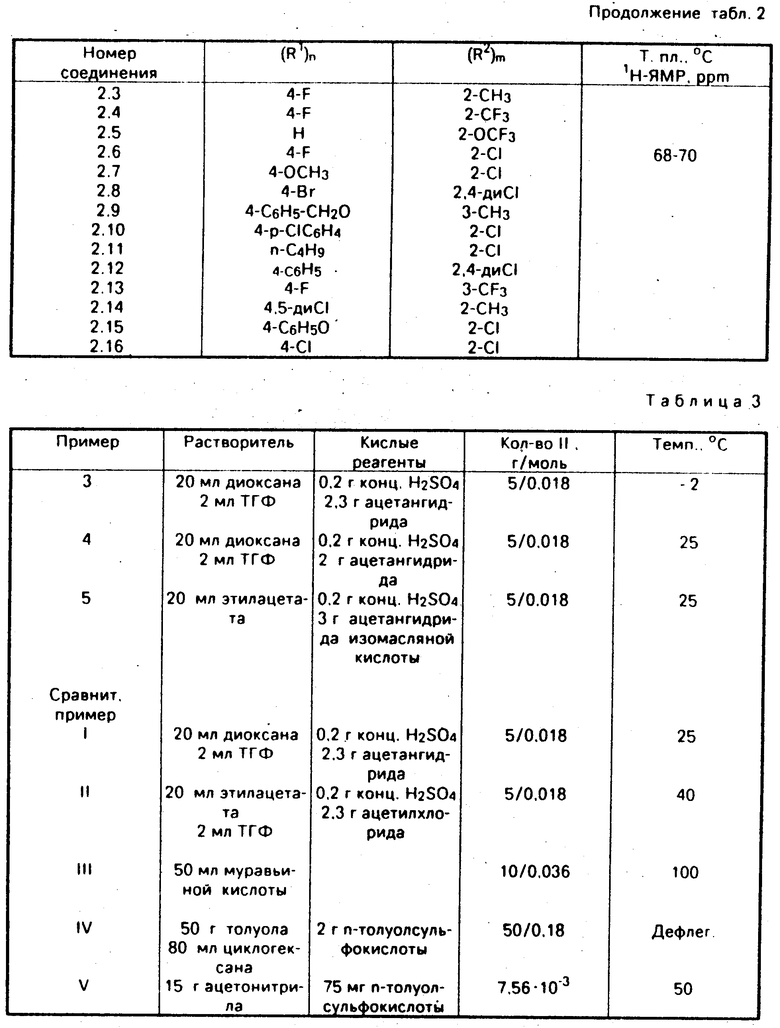
Хлоргидрины общей формулы II общеизвестны и могут быть получены с хорошими входами/ например/ реакцией присоединения бензильных соединений Гриньяра IV к ω-хлорацетофенонам VII согласно следующей схеме реакции: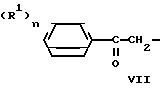 Cl+XMg
Cl+XMg CH
CH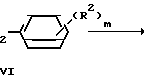 II
II
Что касается способа получения Z-аллилхлоридов целесообразно провести реакцию дегидратации отдноступенчатым способом/ т. е. получить сначала хлоргидрин в простом диэтиловом эфире и добавить к полученному диэтилэфирному раствору неорганическую кислоту/ например/ конц.серную/ и ангидрид карбоновой кислоты при температуре ок. 10-0°С.
Кроме того/ хлоргидрин можно получить не только в виде водного раствора в результате вышеописанного синтеза/ но и путем его выделения из предшествующей его стадии/ т.е. из алкоксилата магния в результате добавления эквимолярных количеств кислоты/ например серной/ с последующей дегидратацией.
Согласно изобертению/ целесообразно постепенное добавление ангидрида карбоновой кислоты/ причем процесс О-ацилирования хлоргидрина в значительной мере удается подавить в пользу процесса дегидратации.
Предлагаемую реакцию дегидратации хлоригидринов II осуществляют в простом или сложном эфире в качестве растворителя. В случае использования нециклических простых эфиров предпочтение следует отдать эфирам с числом атомов кислорода не менее 2/ например эфирам гликолей и низкомолекулярных алифатических спиртов/ например этиленгликольдиметиловому или -диэтиловому эфиру. Особенно выгодным оказались циклические простые эфиры/ например ТГФ и в частности диоксан. Для улучшения сольволиза при низких температурах/ например/ ниже  10°C можно добавить незначительные количества апротонных растворителей/ например этилацетата/ галогенуглеводородов/ таких как метиленхлорид или ТГФ/ например к диоксану в качестве растворителя.
10°C можно добавить незначительные количества апротонных растворителей/ например этилацетата/ галогенуглеводородов/ таких как метиленхлорид или ТГФ/ например к диоксану в качестве растворителя.
Для предлагаемого способа особенно выгодными сложными эфирами оказались эфиры низкомолекулярных алифатических карбоновых кислот/ в частности монокарбоновых/ с низкомолекулярными алифатическими спиртами/ причем понятие низкомолекулярных спиртов включает спирты с числом атомов С от одного до шести. В качестве примеров эфиров можно назвать этиловый эфир уксусной кислоты/ этиловый эфир муравьиной кислоты/ метиловый эфир пропионовой кислоты/ метиловый эфир масляной кислоты/ метиловый или этиловый эфир изомасляной кислоты/ причем предпочтение отдается этилацетату. Количества растворителей не играют решающей роли и колеблются в широких пределах. они обычно составляют 1 - 50 мас.%/ в частности 2/5-10 мас.%/ считая на хлоргидрин II. Более высокие количества растворителей вполне возможны. Для дегидратации можно также использовать смеси растворителей/ упомянутых/ например/ в п. 1-5 формулы изобретения/ причем соотношение компонентов может колебаться в широких пределах  от 10:1 до 1:10. В целях достижения высоких выходов за единицу времени и высоких долей Z-изомеров рекомендуется прибавить добавки в количествах 5-20 мас.% считая на диоксан.
от 10:1 до 1:10. В целях достижения высоких выходов за единицу времени и высоких долей Z-изомеров рекомендуется прибавить добавки в количествах 5-20 мас.% считая на диоксан.
В качестве акцептора кислоты к реакционной массе добавляют ангидрид карбоновой кислоты. В частности применяют такие ангидриды алифатических низкомолекулярных монокарбоновых кислот/ как ацетангидрид/ ангидриды пропионовой/ масляной и изомасляной кислот. Но можно также использовать и ангидриды алифатических или ароматических дикарбоновых кислот/ например малоновой/ малеиновой/ янтарной или фталевой кислот.
В реакции дегидратации/ как правило/ используются 0/5-3/ в частности 1-2 мольных эквивалента ангидрида/ считая на хлоргидрин II. Возможно также применение больших количеств/ но они не дают каких либо дополнительных выгод.
Особенно выгодные результаты достигаются комбинацией диоксана и/или ТГФ в качестве растворителя и ацетангидрида и серной кислоты или в случае применения этилового эфира уксусной кислоты в качестве растворителя в комбинации с ангидридом изомасляной кислоты и серной кислотой.
Дегидратация осуществляется в кислых реакционных условиях/ для создания которых применяют обычные кислоты/ например такие органические сульфокислоты/ как трифторметансульфоновая/ метансульфоновая/ п-толуолсульфоновая или нафталинсульфоновая/ и в частности такие концентрированные минеральные кислоты/ как хлорная/ фосфорная и в частности серная с концентрацией 30-99/9 % / предпочтительно 50-99 %/ или олеум. В случае применения кислот с большим содержанием воды обычно применяют большее количество ангидрида карбоновой кислоты.
Кислоту применяют в каталитическом/ стехиометрическом или избыточном количестве/ считая на соединение II. Предпочитаются количества около 0/01-4 мольн. эквивалента/ считая на соединение II.В случае применения олеума целесообразно использовать меньшие количества - 0/05-1 мольн.эквивалент/ считая на соединение II.
Выгодный вариант выполнения предлагаемого изобретения заключается в том/ что в качестве акцептора воды вместо ангидрида карбоновой кислоты применяют кетен как таковой или в сочетании/ считая на соединение II/ со стехиометрическими или каталитическими количествами алифатической карбоновой кислоты. В этом случае целесообразно поместить в реактор карбоновую кислоту/ например одну из вышеприведенных низкомлекулярных алифатических/ и добавить в реакционную массу газообразный кетен или же в растворенный в растворителе хлоргидрин II добавляют кетен в газообразном виде без добавки карбоновой кислоты. Количество добавляемого кетена соответствует вышеуказанным количествам ангидрида карбоновой кислоты.
Для достижения высоких долей Z-изомеров рекомендуется провести дегидратацию при минимальных температурах/ т.е. при температурах до 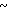 50°С/ предпочтительно от -25 до +40°С/ в частности от - 25 до +30°С.
50°С/ предпочтительно от -25 до +40°С/ в частности от - 25 до +30°С.
Как правило/ дегидратацию осуществляют под обычным давлением. Осуществление реакции под повышенным или пониженным давлением тоже возможно и повышение давления в некоторых случаях может привести к увеличению выхода за единицу времени.
Таким образом/ согласно предлагаемому способу/ получают Z-1/2-диарилаллилхлориды формулы I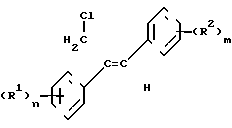 где остатки R1 и R2 независимо друг от друга означают водород/ галоген/ алкил С1-С7/ галогеналкил С1-С5 алкокси С1-С5/ галогеналкокси С1-С5 или незамещенный или замещенный одним- тремя остатками/ указанными для радикалов R1 и R2/ ароматический остаток;
где остатки R1 и R2 независимо друг от друга означают водород/ галоген/ алкил С1-С7/ галогеналкил С1-С5 алкокси С1-С5/ галогеналкокси С1-С5 или незамещенный или замещенный одним- тремя остатками/ указанными для радикалов R1 и R2/ ароматический остаток;
n-1 или 2/ или 3.
В формуле I показатели n и m предпочтительно означают 1/ а заместители R1 и R2 в этой формуле независимо друг от друга водород/ галоген/ например фтор/ хлор/ бром/ йод/ предпочтительно хлор и фтор; разветвленный или неразветвленный алкил C1-C7/ например метил/ этил/ пропил/ 1-метилэтил/ бутил/ 1-метилпропил/ 2-метилпропил/ 1/1-диметилэтил/ пентил/ 1-метилбутил/ 2-метилбутил/ 3-метилбутил/ 1/1-диметилпропил/ 1/2-диметилпропил/ 2/2-диметилпропил/ 1-этилпропил/ гексил/ 1-метилпентил/ 2-метилпентил/ 3-метилпентил/ 4-метилпентил/ 1/1-диметилбутил/ 1/2-диметилбутил/ 1/3-диметилбутил/ 2/2-диметилбутил/ 2/3-диметилбутил/ 3/3-диметилбутил/ 1-этилбутил/ 2-этилбутил/ 1/1/2-триметилпропил/ 1/2/2/-триметилпропил/ 1-этил-1-метилпропил/ 1-этил-2-метилпропил;
галогеналкил C1-C6/например фторметил/ дифторметил/ трифторметил/ хлордифторметил/ дихлорфторметил/ трихлорметил/ 1-фторэтил/ 2-фторэтил/ 2/2-дифторэтил/ 2/2/2-трифторэтил/ 2-хлор-2/2-дифторэтил/ 2/2-дихлор-2-фторэтил/ 2/2/2-трихлорэтил/ пентафторэтил/ предпочтительно трифторметил;
алкокси C1-C5/ например/ метокси/ этокси/ пропокси/ 1-метилэтокси/ бутокси/ 1-метилпропокси/ 2-метилпропокси/ 1/1-диметилэтокси/ предпочтительно метокси/ этокси и пропокси:
галогеналкокси C1-C5/ например дифторметокси/ трифторметокси/ хлордифторметокси/ дихлорфторметокси/ 1-фторэтокси/ 2-фторэтокси/ 2/2-дифторэтокси/ 1/1/2/2-тетрафторэтокси/ 2/2/2-трифторэтокси/ 2-хлор-1/1/2-трифторэтокси/ пентафторэтокси/ предпочтительно трифторметокси;
ароматический остаток/ например фенил/ который незамещен или одно-/ двух-или трехкратно замещен остатком R3/ имеющий предпочтительное значение/ указанное для R1 или R2/ т.е. который означает водород/ галоген/ разветвленный или неразветвленный алкил C1-C7/ галогеналкил C1-C6/ алкокси C1-C5/ или галогеналкокси C1-C5.
Предпочитаются остатки R1-2-F и R2-2-Ce.
По сравнению с известными из патента ДЕ-А 3218 129 Z-1/2- диарилаллилбромидами/ Z-1/2-диарилаллилхлориды общей формулы I обладают неожиданными преимуществами. Помимо очень простого их эпоксидирования до диарилоксиранов следует назвать еще то преимущество/ что благодаря стереоизбирательности эпоксидирования получают не смеси изомеров оксиранов/ что имеет место/ когда исходным материалом являются известные Z-1/2-диарилаллилбромиды/ а оксираны/ у которых арилы имеют трансоидное расположение.
Могут присутствовать/ например/ указанные в табл.1 типы замещения:
Определение соотношения изомеров Z: Е в диарилаллилхлоридах I проводится известным образом/ например ВСЖХ (высокоскоростной жидкостной хроматографией)/ газовой хгроматографией или методами 1H-ЯМР-спектрометрии с применением чистых Z- и E-изомеров для сравнения и стандартизацией соответствующих соотношений изомерных компонентов смеси.
Получение фунгицидных действующих начал III и IV на основе исходных материалов - диарилаллилхлоридов I и хлоргидринов II изображено в нижеследующей схеме реакций.
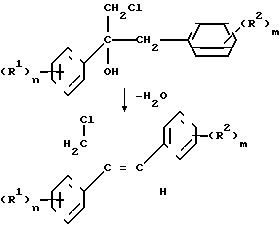
 I
I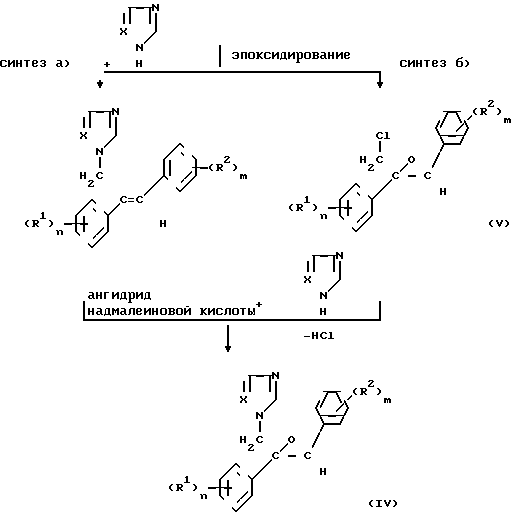
Последовательность реакций согласно синтезу б) можно осуществить известным образом/ например по методу/ принцип которого описан в патенте ДЕ-А 3218129. Реакция замещения хлорного атома азольной или имидазольной группой в соединении V обычно осуществляется в инертном растворителе/ например диметилформамиде или N-метилпирролидоне/ в присутствии неорганического или органического основания/ такого как гидроксид натрия или калия/ карбоната натрия или калия/ дициклогексиламина/ диметилциклогексиламина.
Промежуточные продукты V являются новыми соединениями. Предпочтительные остатки R1 и R2/ а также показатели n и m имеют значение/ аналогичное определениям/ изложенным в связи с описанием соединения I (см.табл.2).
В случае синтеза а) первая стадия/ т.е. стадия замещения соответствует последней стадии синтеза б). Целесообразно осуществить процесс дегидратации и последующей процесс замещения одностадийным способом без выделения и очистки промежуточного продукта II. В целях эпоксидирования соединений III целесообразно использовать высокий избыток надмалеиновой кислоты/ которую получают "ин ситу" путем взаимодействия 5-30 мольн.экв./ в частности 5-10 мольн. экв. ангидрида малеиновой кислоты/ считая на соединение III/ с раствором перекиси водорода в количествах ниже стехиометрических/ считая на ангидрид малеиновой кислоты. Обычно используют мольное соотношение ангидрида с пероксидом водорода 1/5-10/ в частности 2-4. Предпочтительно используют 30-50 %-ный водный раствор пероксида водорода.
Температура реакции для процесса эпоксидирования 0-100°С/ в частности 20-80°С.
Эпоксидирование осуществляют в присутствии апротонного полярного растворителя. В качестве таких растворителей можно использовать/ например/ такие галогенуглеводороды/ как дихлорметан/ дихлорэтан/ хлорбензол или хлортолуол/ или такие ароматические углеводороды/ как бензол/ толуол или ксилол. Количество растворителя не имеет решающего значения. Оно обычно составляет 5-50/ в частности 10-20 мас.%/ считая на олефин.
Согласно этому методу эпоксидирования/ можно получить намного более высокие выходы азолилметилоксиранов IV/ чем по способу/ описанному в патенте ДЕ-А 3218129.
Отдельные стадии синтеза описаны в последующих примерах.
Пример 1. Получение исходных веществ II.
1-Хлор-2-(4-хлорфенил)-3-(2-хлорфенил)-пропан-2-ол.
К 9/7 г (0/404 моль) магниевой стружки в 20 мл абс. диэтилового эфира при 24-36°С в течение 5 мин добавляют 5/0 г (0/031 моль) 2-хлорбензилхлорида. После того/ как начнется реакция/ по каплям добавляют раствор 200 мл. абс диэтилового эфира и 50/2 г (0/31 моль) 2-хлорбензилхлорида. Затем продолжают нагревать массу при температуре дефлегмации в течение еще 10 мин. В атмосфере азота декантируют избыточный магний. Полученный в результате раствор Гриньяра помещают в реактор при 0°С. Затем к нему по каплям добавляют 55/7 г (0/3 моль) n-хлор- ω-хлорацетофенона/ растворенного в 350 мл толуола/ и продолжают перемешивать массу при 0°С в течение еще 1/5 ч. При температуре 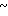 2-6°С реакционную массу по каплям добавляют в 1/5 л конц. раствора хлористого аммония. После экстрагирования метил-трет-бутиловым эфиром с последующей обычной переработкой получают 92/9 г (выход 99%/ степень чистоты по данным ВСЖХ - 68/2 %) 1-хлор-2-(4-хлорфенил)-3-(2-хлорфенил)пропан-2-ол в виде сырого масла/ которое подвергается последующей реакции. Для определения продукта проводили перекристаллизацию из н-гексана; т.пл. 64-69°С.
2-6°С реакционную массу по каплям добавляют в 1/5 л конц. раствора хлористого аммония. После экстрагирования метил-трет-бутиловым эфиром с последующей обычной переработкой получают 92/9 г (выход 99%/ степень чистоты по данным ВСЖХ - 68/2 %) 1-хлор-2-(4-хлорфенил)-3-(2-хлорфенил)пропан-2-ол в виде сырого масла/ которое подвергается последующей реакции. Для определения продукта проводили перекристаллизацию из н-гексана; т.пл. 64-69°С.
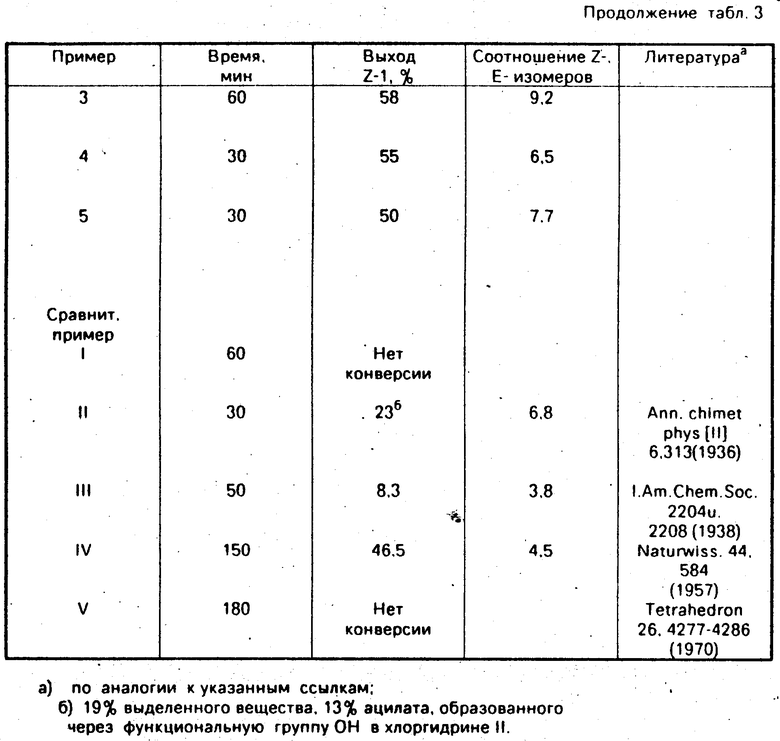
Примеры 2-5 и сравнительные примеры I-IV.
Дегидратация хлоргидринов II.
Z-3-хлор-2-(4-хлорфенил)-1-(2-хлорфенил)пропен.
(Соединение N 1.16 в табл.1).
При -2°C к 60 г (0/2 моль) хлорного спирта/ описанного в примере 1/ в 230 мл диоксана и 23 мл ТГФ добавляют 24/5 г (0/24 моль) ацетангидрида. Затем к массе по каплям добавляют 2/36 г (0/024 моль) конц. серной кислоты. После перемешивания в течение 3 ч при 0° по данным ВСЖХ фактически весь исходный материал прореагировал.
Затем при 0°С в течение 30 мин добавляют смесь из полунасыщенного раствора хлористого натрия и 50 %-ного раствора едкого натра для того/ чтобы установилось значение рН 8-9.
Наконец/ высушивают органическую фазу и концентрируют ее в вакууме/ после чего она может применяться для последующих реакций без какой-либо дополнительной очистки.
Выход 55/7 (ZE= 9,1/1) сырого масла/ которое после перекристаллизации из н-гексана дает чистый Z-изомер с т.пл. 79-82°С.
Аналогично можно получить Z-1/2-диарилаллилхлориды (См.табл.1).
Z-3-хлор-2-(4-фторфенил)-1-(2-хлорфенил) пропен.
(примеры N 1/6 в табл.1)
1-хлор-2-(4-фторфенил)-3-(2-хлофенил)-пропен-2-ол/ полученный реакцией присоединения по Гриньяру хлористого 2-хлорбензилмагния к п-фтор- ω-хлорацетофенону и применяемого в качестве сырого материала со степенью чистоты по данным ВСЖХ 78-87%/ в условиях реакции/ описанных в табл.2/ подвергали реакции/ как описано в примере 2. Долю Z- и Е-изомеров определяли методом высокоскоростной жидкостной хроматографии (нескорректированный относительный процент площади) (см.табл.3).
Дегидратация 1-хлор-2-(4-фторфенил)пропан-2-ола.
F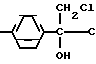 H
H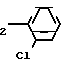 ____→
____→ 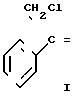
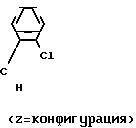
Пример 6. Получение хлоргидрина и процесс дегидратации "ин ситу" 1-хлор-2-(4-хлорфенил)-3-(2-хлорфенил)пропан-2-ол.
К 36/0 (1/5 моль) магниевой стружки в 200 мл простого диэтилового эфира по каплям добавляют раствор 170 г (1/0 моль) 2-хлорбензилхлорида в 400 мл диэтилового эфира. Затем при -10°С по каплям добавляют раствор 155 г (0/9 моль) п-фтор-ω-хлоранцетофенона в 450 мл диэтилового эфира. Затем еще 2 ч перемешивают при 25°С.
Потом при -10°С по каплям добавляют 49/0 г (0/5 моль) конц.серной кислоты в 300 мл диэтилового эфира. Потом нагревают до 25°С и отсасывают от выпавшей соли. Полученный сырой эфирный раствор хлоргидрина используют для нижеуказанной реакции.
Z-3-хлор-2-(4-фторфенил)-1-(2-хлорфенил)пропен.
К 525 мл вышеописанного сырого раствора/ содержащего ок. 134/5 г хлоргидрина (0/45 моль)/ при -10°С добавляют 8/0 г (0/08 моль) конц.среной кислоты/ после чего в течение 2 ч добавляют 57/1 г (0/58 моль) ацетангидрида. Затем выпавшую соль вновь отделяют фильтрацией. Полученный в результате выпаривания растворителя из фильтрата сырой аллилхлорид используют для тризолового замещения или для эпоксидирования.
Пример 7. Кетеновый вариант.
Z-3-хлор-2-(4-фторфенил)-1-(2-хлорфенил)пропен.
При 0°С через исходную смесь 250 мл диоксана/ 25 мл ТГФ/ 12/4 г уксусной кислоты (0/2 моль) и 69 г (0/23 моль) сырого 1-хлор-2-(4-фторфенил)-3-(2-хлорфенил)пропан-2-ола/ полученного в результате реакции Гриньяра по примеру 1/ в течение  1 ч пропускают 43 г (1/02 моль) кетена. После обычной переработки продукта на основе данных ВСЖХ получают выход/ который практически равняется выходу/ получаемому в случае применения ацетангидрида в соответствии с примером 2. Соотношение Z- и Е-изомеров для данного типа ведения реакции составляет
1 ч пропускают 43 г (1/02 моль) кетена. После обычной переработки продукта на основе данных ВСЖХ получают выход/ который практически равняется выходу/ получаемому в случае применения ацетангидрида в соответствии с примером 2. Соотношение Z- и Е-изомеров для данного типа ведения реакции составляет  11:1.
11:1.
Примеры 8 и 9. Получение азолилметилоксиранов IV по синтезу а)
Z-3-(1/2/4-триазол-1-ил)-2-(4-хлорфенил)-1-(2-хлорфенил)пропен.
К раствору 11/5 г (0/17 моль) триазола в 150 мл диметилформамида добавляют 6/6 г гидроксида натрия и с перемешиванием нагревают до температуры  70° до тех пор/ пока не образуется прозрачный раствор. Затем охлаждают до 10°С/ после чего в течение 1 ч по каплям добавляют 49/5 г полученного по примеру 2 Z-3-хлор-2-(4-хлорфенил)-1-(2-хлорфенил)пропен в виде сырого продукта в 50 мл диметилформамида. Затем массу перемешивают еще 4 ч при комнатной температуре.
70° до тех пор/ пока не образуется прозрачный раствор. Затем охлаждают до 10°С/ после чего в течение 1 ч по каплям добавляют 49/5 г полученного по примеру 2 Z-3-хлор-2-(4-хлорфенил)-1-(2-хлорфенил)пропен в виде сырого продукта в 50 мл диметилформамида. Затем массу перемешивают еще 4 ч при комнатной температуре.
Затем добавляют 200 мл воды и неоднократно экстрагируют простым метил-трет-бутиловым эфиром. Собранные органические фазы промывают/ высушивают и концентрируют в вакууме. После перекристаллизации из метил-трет-бутилового эфира и н-гексана получают
24/4 г Z-3-(1/2/4-триазол-1-ил)-2-(4-хлорфенил)-1-(2-хлорфенил)пропена с т.пл. 106-110°С.
Цис -2-(1/2/4-триазол-1-илметил)-2-(4-фторфенил)-3-(2-хлорфенил)-оксиран.
84 г (0/9 моль) ангидрида малеиновой кислоты и 6 капель конц. серной кислоты в 90 мл дихлорэтана вместе с 22 г 50%-ной перекиси водорода нагревают до 50°С. Затем по каплям добавляют 28 г (0/089 моль) Z-3-(1/2/4-триазол-1-ил)-2-(4-фторфенил)-1- (2-хлорфенил)пропена в 75 мл дихлорэтана. Массу перемешивают 3 ч при указанной температуре и затем еще 2/5 ч при 70°С.
После охлаждения реакционной массы отсасывают продукт от выпавшей малеиновой кислоты и встряхивают его с раствором тиосульфата и разбавленным раствором едкого натра. Высушенная и максимально выпаренная в вакууме при температуре около 50°С органическая фаза после охлаждения и повторного выпаривания маточного раствора дает 14 г целевого продукта (что равняется выходу в 50 %).
Примеры 10 и 11.
Получение азолилметилоксиранов IV согласно синтезу б).
Цис-1-хлорметил-2-(2-хлорфенил)-1-(4-фторфенил)оксиран (соединение N 2.6 по табл.2).
Исходят из 56/2 г (0/2 моль) Z-3-хлор-2-(4-фторфенил)-1-(2-хлорфенил)пропена в 530 млм ледяной уксусной кислоты и добавляют 196 г (2 моль) ангидрида малеиновой кислоты. Затем в течение 1 ч при 25°С добавляют 68 г (1 моль) 50%-ного раствора перекиси водорода. Массу перемешивают в течение еще 3-4 ч при 40°С/ затем в течение 10 ч при 25°С.
Наконец/ реакционную массу с перемешиванием вводят в 3 л воды и 50 мл 10%-ного раствора тиосульфата натрия и потом/ если необходимо/ вновь добавляют раствор тиосульфата натрия до исчезновения перекиси. Получаемый в результате бесцветный осадок отсасывают и высушивают. Сырое вещество/ полученное в результате перекристаллизации из п-гексана можно использовать без очистки (т.пл.68-70°С).
Цис-2-(1/2/4-триазол-1-илметил)-2-(4-фторфенил)-3-(2-хлорфенил) оксиран.
1/5 г (5 ммоль) цис-1-хлорметил-2-(2-хлорфенил)-1-(4-фторфенил)оксирана и 0/69 г (75 ммоль) 1/2/4-триазолида натрия перемешивают в течение 5 ч при 75°С в 7 мл ДМФ. После охлаждения реакционную массу нейтрализуют путем добавления незначительного количества уксусной кислоты/ после чего добавляют около 10 мл воды/ в результате чего выпадает кристаллический продукт (выход 1/4 г). Полученный продукт отсасывают/ промывают водой и высушивают в вакууме.
| название | год | авторы | номер документа |
|---|---|---|---|
| Z-1,2,-ДИАРИЛАЛЛИЛХЛОРИДЫ | 1992 |
|
RU2096401C1 |
| ХЛОРМЕТИЛДИАРИЛОКСИРАНЫ | 1991 |
|
RU2125997C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЦИС-2-(1Н-1,2,4-ТРИАЗОЛ-1-ИЛ-МЕТИЛ)-2-(ГАЛОГЕНФЕНИЛ)-3-(ГАЛОГЕНФЕНИЛ)-ОКСИРАНА | 1990 |
|
RU2071473C1 |
| ПРОИЗВОДНЫЕ N-ЗАМЕЩЕННОГО 3-АЗАБИЦИКЛО[3.2.0]ГЕПТАНА | 1994 |
|
RU2136678C1 |
| Акарицидное средство | 1981 |
|
SU1152508A3 |
| СПОСОБ ПОЛУЧЕНИЯ КВАДРАТНОЙ КИСЛОТЫ | 1991 |
|
RU2017719C1 |
| ЦИКЛОГЕКСЕНОНОКСИМОВЫЙ ЭФИР, ГЕРБИЦИДНОЕ СРЕДСТВО И СПОСОБ БОРЬБЫ | 1991 |
|
RU2074617C1 |
| СПОСОБ ПОЛУЧЕНИЯ DL-АЛЬФА-ТОКОФЕРОЛА ИЛИ ЕГО АЦЕТАТА | 1994 |
|
RU2098416C1 |
| 3-ОКСИ-2-ЦИКЛОБУТЕН-1-ОН-ОСНОВНЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2078757C1 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2047604C1 |
Использование: Z-1,2-диарилаллилхлоридов ф-лы I, где остатки R1и R2 независимо друг от друга означают водород, галоген, алкил, галогеналкил, алкокси, галогеналкоксиокси или незамещенный или замещенный ароматический остаток, а n и m означают 1, 2 или 3, могут быть промежуточными продуктами в получении фармакологических и противогрибковых действующих начал. Получение их ведут путем дегидратации хлоргидринов ф-лы II, где остатки имеют вышеуказанные значения, в инертном простом эфире или сложном эфире карбоновой кислоты в качестве растворителя, в присутствии ангидрида карбоновой кислоты и органической или неорганической кислоты при температуре до 50°С. 8 з.п. ф-лы, 3 табл. Структура соединений ф-л I и II: 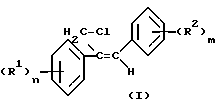
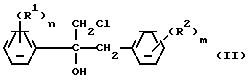
(R2)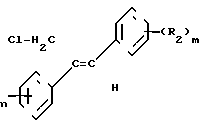
где R1 и R2 независимо друг от друга - водород, галоген, алкил, галогеналкил, алкокси, галогеналкокси или незамещенный или замещенный ароматический остаток;
n и m=1,2 или 3,
отличающийся тем, что хлоргидрины общей формулы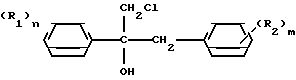
где R1 и R2 имеют указанное значение,
дегидратируют в инертном простом эфире или сложном эфире карбоновой кислоты в качестве растворителя в присутствии ангидрида карбоновой кислоты или соответствующего кетена и органической или неорганической кислоты или олеума при температурах до 50oС.
