Изобретение относится к области органической химии, а также к сельскому хозяйству, конкретно к синтезу органических соединений, которые могут найти использование в композициях для предотвращения или ингибирования роста растений.
Известно использование акриламидов в качестве бактерицидов. Однако известные соединения проявляют недостаточное биологическое действие.
Целью изобретения является синтез соединений, а также повышение бактерицидного действия.
Указанная цель достигается соединением формулы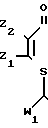
 (I) где R1 представляет С5-С10-алкил, С5-С7-циклоалкил, фенил, фенэтил или бензил, где арил возможно замещен Сl; Z1 и Z2 каждый независимо представляет водород, хлор или С1-С4-алкил;
(I) где R1 представляет С5-С10-алкил, С5-С7-циклоалкил, фенил, фенэтил или бензил, где арил возможно замещен Сl; Z1 и Z2 каждый независимо представляет водород, хлор или С1-С4-алкил;
W1 и W2 каждый независимо представляет соответственно СОR2 и СОR3,
где R2 - С1-С8-алкоксигруппа или С1-С4-алкиламиногруппа,
R3 - С1-С4-алкил, С1-С4-алкоксигруппа, С3-С13-алкиламиногруппа или циклопропиламиногруппа, каждый возможно замещен одним или более хлором, метилом, метоксигруппой, нитрогруппой; или бензиламиногруппа, цианопентиламиногруппа, аминоаллил, аминотиенил, аминотиазолил, аминопиридил, аминоэтилпиридил, аминопиридинил, N-метилпиразолил, аминоморфолил или аминопропилморфолил,
а также композицией на основе соединений формулы (I) в количестве 1-90 мас.%, сельскохозяйственно приемлемый носитель - остальное.
Данные соединения обычно используют в комбинации с носителем или приготавливают в таком виде, чтобы в дальнейшем они могли быть пригодны для рассеивания фунгицидов. Так, например, эти β-тиоакриламиды могут быть приготовлены в форме смачиваемых порошков, эмульсионных концентратов, пылевидных препаратов, гранулированных рецептур, аэрозолей или текучих эмульсионных концентратов. В таких рецептурах β-тиоакриламиды разбавляют жидким или твердым носителем, и, при желании, в них вводят подходящие поверхностно-активные вещества.
Обычно, желательно, особенно в случае рецептур для опрыскивания листвы, включать вспомогательные вещества, такие как смачивающие агенты, распределяющие вещества, диспергирующие вещества, загустители, адгезивы, в соответствии с существующей в сельском хозяйстве практикой.
Обычно β-тиоакриламиды, отвечающие настоящему изобретению, могут растворяться в подходящих растворителях, таких как ацетон, метанол, этанол, диметилформамид или диметилсульфоксид, а такие растворы разбавляются водой. Концентрации данного раствора могут изменяться от 1 до 90 мас.%, предпочтительно от 5 до 50 мас. % . Для получения эмульсионных концентратов β-тиоакриламиды могут растворяться в подходящих органических растворителях или смеси растворителей, вместе с эмульгирующим агентом, который обеспечивает диспергирование фунгицида в воде. Концентрация активного ингредиента в эмульсионных концентратах обычно составляет от 10-90 мас.% и в текучих эмульсионных концентратах она может составлять до 75 мас.%. Могут быть получены водные текучие препараты β-тиоакриламидов с концентрацией активных ингредиентов 5-70 мас.%, предпочтительно 20-50 мас.%.
Типичная текучая рецептура приготавливается путем мокрого измельчения смеси 35 ч. β-тиоакриламидов, 10 ч. глины Бардена, 4 ч. натрийлигносульфоната, 1 ч. анионного смачивающего агента и 50 ч. воды. Смачиваемые порошки, пригодные для опрыскивания, приготавливаются путем перемешивания β-тиоакриламидного соедине- ния с тонкоизмельченным твердым веществом, таким как глина, неорганические силикаты и карбонаты, а также кремнеземы, и путем ввода в эти смеси смачивающих веществ, загустителей и/или диспергирующих агентов. Концентрация активных ингредиентов в таких композициях обычно находится в пределах 5-98 мас.%, предпочтительно 40-75 мас.%, и это достигается путем перемешивания 50 ч. активного ингредиента, выбранного из замещенных S-β-дикарбо- нилом β-тиоакриламидов примеров 1-69, 45 ч. синтетической осажденной гидратированной двуокиси кремния, 1 ч. анионного нафталиносульфоната как смачивающего агента и 4 ч. лигносульфоната натрия. Согласно другому способу приготов- ления, вместо двуокиси кремния в указанном выше смачиваемом порошке, используют глину каолинового типа, и в другом таком препарате 25% двуокиси кремния заменяется синтетическим натрийсиликоалюминатом. Пылевидные препараты приготав- ливают путем перемешивания амидов и солей и их комплексных соединений с тонкоизмельченными инертными твердыми веществами, которые по своей природе могут быть органическими или неорганическими. Материалы, используемые для этой цели, включают ботанические цветы, кремнеземы, силикаты, карбонаты, тальк и глины. Желаемый способ приготовления пылевидного препарата заключается в разбавлении смачиваемого порошка тонкоизмельченным носителем. Обычно приготав- ливают пылевидные препараты с концентрацией активного ингредиента 20-80 мас.%, и затем их разбавляют до используемой концентрации 1-10%.
β-Тиоакриламиды, отвечающие настоящему изобретению, особенно полезны для обработки локуcа, подвергнутого загрязнению бактериями или грибами. Типичные локуcы, подвергаемые загрязнению бактериями, находятся в водных системах, таких как системы водного охлаждения, промывочные воды в прачечных, масляно-водные системы, такие как смазочно-охлаждающие эмульсии, зоны нефтяных промыслов и т. д., где микроорганизмы должны быть уничтожены, или где необходимо предотвращение их роста.
Изобретение иллюстрируется следующими примерами.
П р и м е р 1А. Получение N-н-октил-цис-3[3-(2,4-пентандионил)тио]акриламида.
В перемешиваемый раствор 2,41 мл (0,0235 моль) 2,4-пентадиона в 20 мл абсолютного этанола в атмосфере азота вводят 8,78 (0,0235 моль) 21 мас.% раствора этилата натрия в абсолютном этаноле. Через 20 мин образуется светло-желтая суспензия. Эта смесь охлаждается до 0оС и в нее вводят по каплям раствор 5 г (0,0235 моль) 2-н-октил-4-изотиазолин-3-она в 5 мл абсолютного этанола. После этого добавления смесь нагревается до комнатной температуры и перемешивается в течение 5 ч. Затем эта смесь вливается в 200 мл 10%-ной водной соляной кислоты и трехкратно экстрагируется этилацетатом. Объединенные органические слои промываются один раз водой и несколько раз насыщенным водным раствором бикарбоната натрия до тех пор, пока промывочные растворы не будут иметь нейтральное значение рН. Затем органический слой промывается солевым раствором, высушивается над сульфатом магния и фильтруется. После удаления растворителей получается бледно-желтый твердый продукт, который перекристаллизовывается из смеси гексан-этилацетат, и в результате получается 4,84 г (66% выход) N-н-октил-цис[3-(2,4-пентандионил)тио]акриламида в виде белого твердого кристаллического вещества, т. пл. 87-89оС.
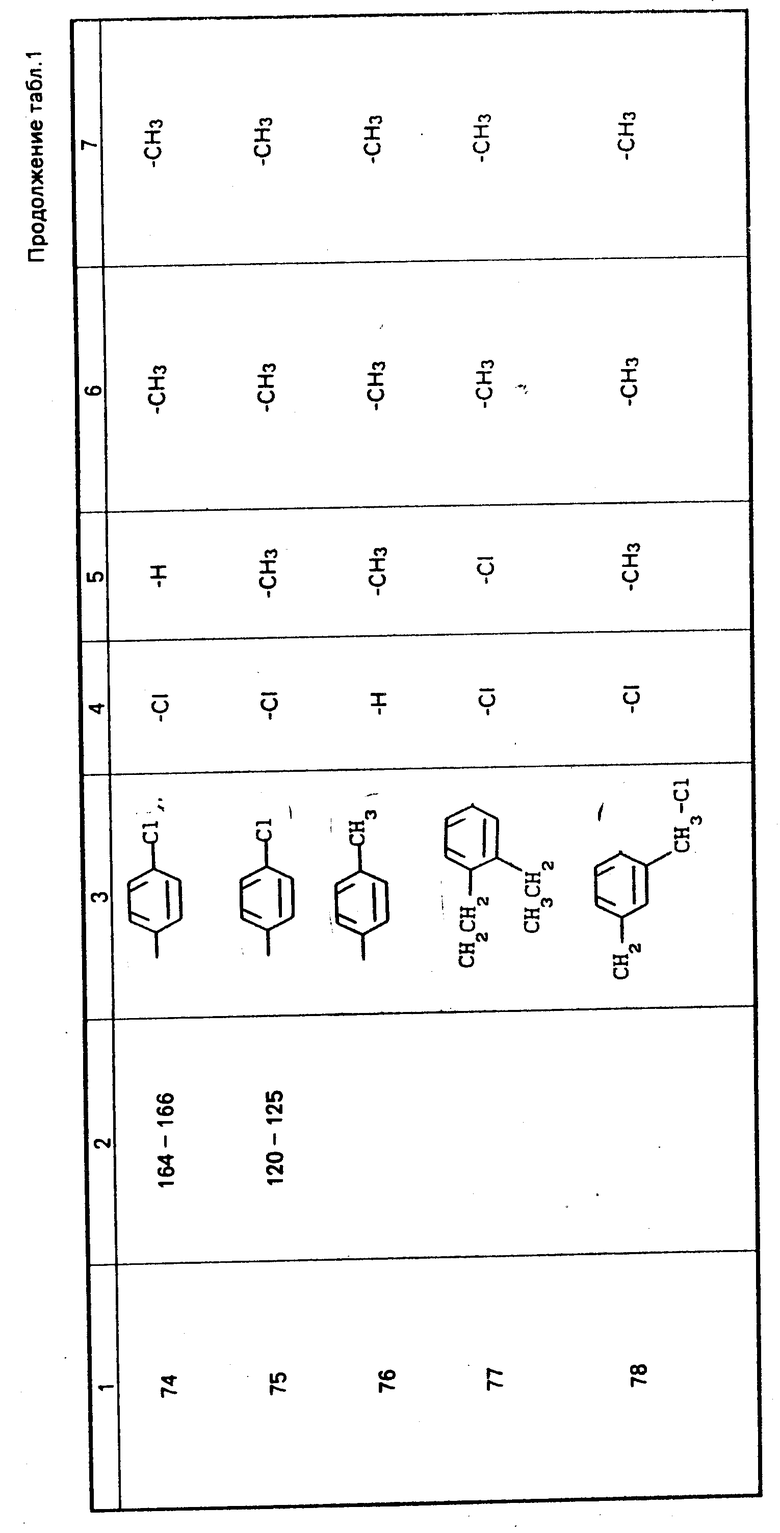
П р и м е р 1В. Синтез N-(п-хлорфенил)-2-метил-3-хлор-цис-3-тио(1-ацетилпропан-2-он-1-ил)акриламида .
А. В четырехгорлую колбу, вместимостью 500 мл, снабженную механической мешалкой, воронкой для ввода и термометром, вводят 80 мл этилацетата, который охлаждают до 0оС. Затем вводят одновременно 12,0 г (0,026 моль) N, N'-бис-(п-хлорфенил)-3,3'-дитиодиизобутирамида и 17,7 г (0,131 моль) сульфурилхлорида, каждый в виде 24 равных порций, с перемешиванием, в течение 1 ч. После нагрева образующейся смеси до комнатной температуры, желтоватый раствор концентрируется при пониженном давлении. Остаточный твердый продукт перекристаллизовывается из 2-пропанола, и в результате получается 6,1 г (выход 89%) 2-(п-хлорфенил)-4-метил-5-хлор-изотиазо-лин-3-онового промежуточного продукта в виде игольчатых кристаллов; т. пл. 104,5-106оС.
В. В сухую, продутую азотом трехгорлую колбу вместимостью 500 мл, снабженную обратным холодильником, магнитной стержневой мешалкой и термометром, вводят 75 мл абсолютного этанола. В виде отдельных свеженарезанных кусочков добавляют металлический натрий (0,53 г, 0,023 моль) с одновременным перемешиванием с такой скоростью, чтобы температура смеси осталась ниже 35оС. После того, как весь натрий растворяется, добавляют по каплям 2,31 г (0,023 моль) 2,4-пентандиона в течение 10 мин с одновременным перемешиванием. После нагрева раствора в течение 15 мин вводят в течение 20 мин 6,0 г (0,023 моль) 2-(п-хлорфенил)-4-метил-5-хлор-4-изотиазолин-3-она в 60 мл нагретого абсолютного этанола, поддерживая при этом температуру реакционной смеси ниже 30оС. Смесь дополнительно перемешивается в течение 1 ч и затем вливается в охлажденный льдом 2 н. раствор НСl с одновременным перемешиванием. Твердый осадок удаляется путем фильтрации и трехкратно промывается свежей водой. Твердый продукт высушивается в вакууме при 40оС, и в результате получается 7,8 г (93,5%-ный выход) продукта, который перекристаллизовывается из абсолют- ного этанола в виде белых хлопкообразных игл; т. пл. 164-166оС;
ИК-спектр (КВr): 3300 см-1 (NН), 1660 см-1 (C=О);
Спектр ЯМР (СDCl3): 2,2 м. д. (с., СН3); 2,4 м. д. (с., 2СН3); 7,2-7,6 м. д. (м., 5Н), ароматика и NН; 17,3 м. д. (с., СН). Т. пл. 164-166оС.
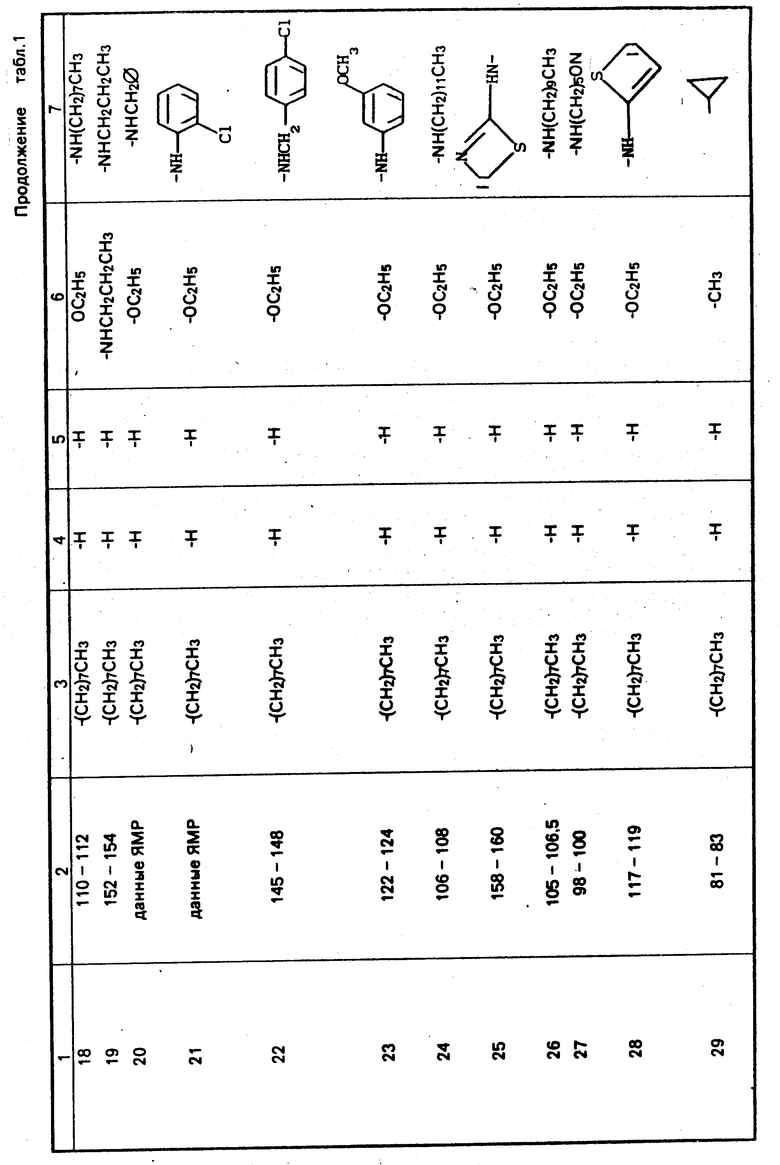
П р и м е р ы 2-7. Получение других соединений, отвечающих данному изобретению.
Осуществляется процедура таким же образом, как в примере 2, с той разницей, что вместо 2-н-октил-4-изотиазолин-3-она используются 2-н-октил-4,5-дихлор-4-изотиазолин-3-он, 2-н-пентил-4-изотиазолин-3-он, 2-н-гептил-4-изотиазолин-3-он, 2-н-гек- сил-4-изотиазолин-3-он, 2-циклогексил-4,5-дихлор-4-изотиазолин-3-он и 2-н-гексил-4,5-дихлор-4-изотиазолин-3-он, и в результате получаются продукты примеров 2, 3, 4, 5, 6 и 7 соответственно, как показано в табл. 1. Эти изотиазолин-3-оновые промежуточные соединения получаются как описано ниже:
А. Получение промежуточного соединения 2-н-октил-4,5-дихлор-4-изотиазолин-3-она, для примера 2.
В четырехгорлой колбе, вместимостью 3 л, снабженной металлической мешалкой, термометром, впускным отводом для хлора и обратным холодильником, сообщающимся с кислотным промывателем, суспендируют 432,0 г (1,0 моль) N, N'-ди-н-октил-3,3'- дитиодипропионамида в 930 мл этилацетата. Суспензия нагревается до 60оС и все количество хлора 490 г (6,9 моль) вводится ниже уровня поверхности с постоянной скоростью потока в течение 3 ч. В течение первого часа ввода хлора реакция является слабо экзотермической, и температура поддерживается равной 60оС за счет наружного охлаждения. После начального периода слабо экзотермической реакции температура начинает снижаться, и требуется наружный нагрев для поддержания ее на уровне 60оС. После ввода хлора растворитель удаляется в вакууме, и в результате получается 590 г вязкой жидкости янтарного цвета. Этот сырой продукт растворяется в 500 мл гексана с одновременным перемешиванием и охлаждается до 0оС. Отделяемый твердый продукт удаляют путем фильтрации и высушивают, и в результате получается примерно 310 г (55%-ный) выход промежуточного продукта 2-н-октил-4,5-дихлор-4-изотиазолин-3-она, т. пл. 44-47оС.
В. Получение промежуточного продукта 2-н-пентил-4-изотиазолин-3-она, для примера 3
В суспензию 209 г (0,6 моль) N, N'-бис-(н-пентил)-3,3'-дитиодипропионамида в 600 мл этилацетата при 40оС вводят в течение 1 ч 134,0 г (1,9 моль) хлора. После этого ввода полученная смесь нагревается до комнатной температуры, дегазируется и выпаривается в вакууме, и в результате получается хлоргидратная соль 2-н-пентил-4-изотиазолин-3-она в виде твердого вещества. Это твердое вещество вводится в смесь 300 мл воды и 200 мл хлороформа. Вводится в виде отдельных порций твердый бикарбонат натрия с одновременным перемешиванием до тех пор, пока величина рН водной фазы не становится равной 7-8. Происходит разделение слоев и водная фаза экстрагируется дополнительной порцией хлороформа. Соединенные хлороформные экстракты высушиваются над сульфатом магния и выпаривается, и в результате получается коричневое масло. Отгонка в вакууме этого масла приводит к образованию 161 г (82%-ный выход) промежуточного продукта 2-н-пентил-4-изотиазолин-3-она; т. кип. 118оС/0,05 мм.
С. Получение промежуточного продукта 2-н-гептил-4-изотиазолин-3-она, для примера 4
В суспензию 75 г (0,185 моль) N, N'-бис-(н-гептил)-3,3'-дитиодипропионамида в 600 мл толуола вводят 39,4 г (0,555 моль) хлора в течение 1 ч при 25-40оС. Смесь перемешивается в течение еще 1 ч и охлаждается до комнатной температуры. Полученный желтый раствор промывается водой, и толуольная фаза высушивается над сульфатом магния и концентрируется. Остаточное масло растворяется в простом эфире и затем насыщается газообразным НСl. Полученный твердый осадок удаляется путем фильтрации и растворяется в воде.
Водный раствор несколько раз экстрагируется простым эфиром. Объединенные эфирные экстракты высушиваются над сульфатом магния и концентрируется в вакууме, и в результате получается 30,7 г (83%-ный выход) 2-н-гептил-4-изотиазолин-3-она (промежуточный продукт) в виде тяжелого бесцветного масла, которое не отгоняется.
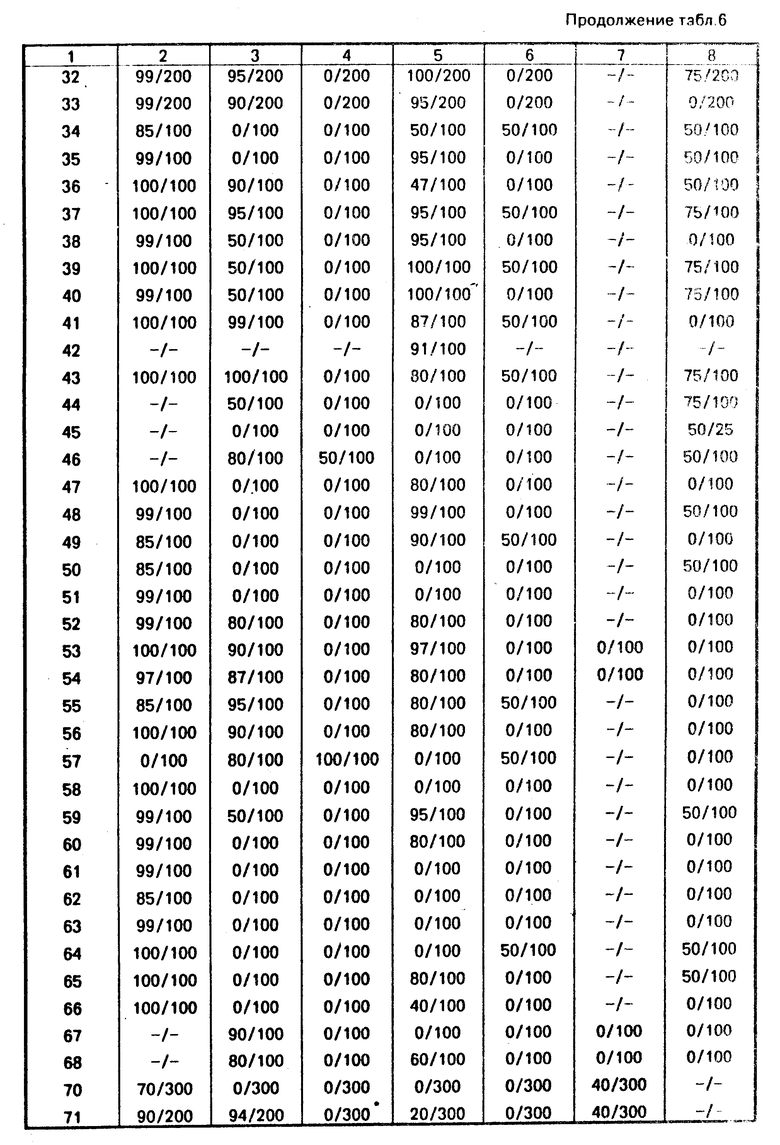
Д. Получение промежуточного продукта, 2-н-гексил-4-изотиазолин-3-она, для примера 5
В смесь 37,6 г (0,1 моль) N, N'-бис-(н-гексил)3,3'-дитиодипропионамида и 400 мл дихлорида этилена при комнатной температуре вводят 42,2 г (0,315 моль) сульфурилхлорида в течение 1 ч. Этот прозрачный желтый раствор перемешивается при комнатной температуре в течение ночи и затем концентрируется. Остаток разбавляется простым эфиром, который промывается раствором бикарбоната натрия, высушивается над сульфатом магния, и затем концентрируется, и в результате получается коричневое масло. Это масло разгоняется при 105-115оС/0,04 мм; и в результате получается 35 г сырого продукта. Дистиллят растворяется в простом эфире и обрабатывается газообразным НСl, и в результате осаждается хлористоводородная соль 2-н-гексил-4-изотиазолин-3-она. Эта соль удаляется путем фильтрации, перемешивается с водой и затем экстрагируется несколько раз простым эфиром. Соединенный эфирный экстракт высушивается над сульфатом магния и концентрируется. Остаточное масло отгоняется в вакууме, и в результате получается 28,8 г (87%-ный выход) чистого промежуточного продукта, 2-н-гексил-4-изотиазолин-3-она, в виде желтого масла, т. кип. 102оС/0,03 мм.
Е. Получение 2-циклогексил-4,5-дихлор-4-изотиазолин-3-она, промежуточного продукта для примера 6.
В 400 мл этилацетата при 0оС вводят одновременно в течение 1 ч 148 г (0,4 моль) N, N'-бис-(циклогексил)-3,3'-дитиодипроциоамида в виде 40 порций по 3,7 г с интервалами 15 мин, и 116,5 г (1,64 моль) хлора. Температура поддерживается в пределах 0-5оС во время введения и затем смесь нагревается до 15оС. Полученный твердый продукт, представляющий собой хлоргидрат циклогексиламина, извлекается. После концентрирования фильтрата получается дополнительная порция хлоргидрата циклогексиламина, смешанного с маслом. Масло растворяется путем растирания его с ацетоном и затем фильтруется. Фильтрат обесцвечивается посредством активированного угля и выпаривается до кашицы. После перекристаллизации этой кашицы из 300 мл метанола получается 21,6 г (11% -ный выход) промежуточного 2-циклогексил-4,5-дихлор-4-изотиазолин-3-она; т. пл. 113,5-115,5оС.
Г. Получение промежуточного продукта - 2-н-гексил-4,5-дихлор-4-изотиазолин-3-она - для примера 7
В раствор 9,2 г (0,05 моль) N, N'-бис-(н-гексил)-3,3'-дитиодипропинамида в 100 мл этилацетата при комнатной температуре вводят 10,65 г (0,15 моль) хлора в течение 30 мин. Во время этого добавления температура смеси повышается до 53оС, и в конце медленно снижается. После охлаждения до комнатной температуры смесь концентрируется в вакууме. Полученное масло янтарного цвета, (17,8 г) подвергается хроматографическому разделению в колонке с силикагелем, с использованием толуола в качестве элюента. Сначала элюиpуется побочный продукт - 2-н-гексил-4,4,5,5-тетрахлор-4-изотиазолин-3-он - в виде масла (48 г), которое отгоняется при 128-133оС/0,3 мм.
Затем из колонки элюируется целевой 2-н-гексил-4,5-дихлор-4-изотиазолин-3-он (промежуточный продукт); 8,2 г (выход 60%), в виде масла, которое отгоняется при 130-136оС/0,35 мм.
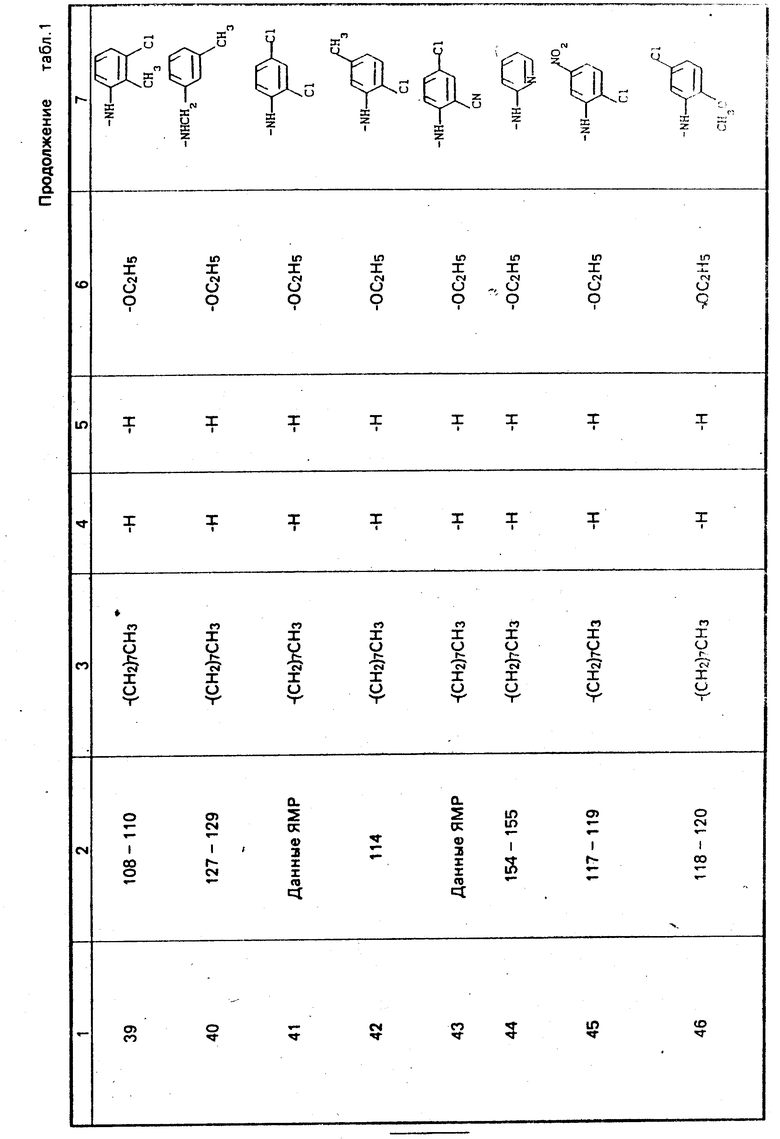
П р и м е р ы 8, 9, 10 и 69. Получение других соединений, отвечающих настоящему изобретению
Осуществляются процедуры так же, как и в примере 1, но с использованием 3,5-гептандиона, 5,5-диметил-1,3-циклогександиона, 2,4-гександиона и этилбензоилацетона соответственно, вместо 2,4-пентадиона, и в результате получаются продукты примеров 8, 9, 10 и 69, как показано в табл. 1.
П р и м е р 11. Получение N-н-октил-цис-3[2-(этокси-3-оксобутаноил)тио] акриламида
В сухую колбу вместимостью 250 мл, снабженную механической мешалкой, термометром и отводом для впуска азота, вводят 75 мл абсолютного этанола. Вводят металлический натрий в виде кусочков (0,36 г, 0,016 моль) с одновременным перемешиванием в атмосфере азота. После растворения всего натрия вводится раствор 2,23 г (0,017 моль) этилацетоацетата в 10 мл хлористого метилена в течение 5 мин. По истечении 15 мин вводятся раствор 2-н-октил-4-изотиазолин-3-она (3,0 г, 0,014 моль) в 10 мл хлористого метилена в течение 10 мин. Смесь перемешивается еще в течение 20 мин и затем охлаждается до 5оС. Эта смесь вливается в 30 мл охлажденного льдом 2 н. раствора H Сl, с одновременным перемешиванием. Смесь разбавляется водой и экстрагируется хлористым метиленом. Экстракт хлористого метилена высушивается над сульфатом магния и концентрируется. Полученное в остатке густое желтое масло отверждается при стоянии. Эта твердая масса растирается с небольшим количеством простого эфира, который удаляется путем фильтрации, и в результате получается 4,1 г (85%-ный выход) N-н-октил-цис-3-[2-(этокси-3-оксобутаноил)тио]акриламид с т. пл. 84-86оС.
П р и м е р 12. Получение N-н-октил-цис-3[2-(N-бензоилацетамидо)тио] акриламида
Осуществляется процедура таким же образом, как в примере 11, с той разницей, что вместо этилацетоацетата используется 3,27 г (0,017 моль) N-бензилацетоацетамида, и в результате получается 5,2 г (62%-ный выход) N-н-октил-цис-3-[2-(N-бензоилацетоацетамидо)тио]акриламида с т. пл. 91-94оС.
П р и м е р 18. Получение N-н-октил-цис-3-[2-(этил-N-н-октилмалонамидо)тио]акри-ламида
В перемешанный охлажденный (0оС) раствор 21,5 г (0,143 моль) этилмалонилхло рида в 250 мл метиленхлорида в атмосфере азота вводят по каплям раствор 18,47 г (0,143 моль) н-октиламина в 100 мл метиленхлорида в течение 0,5 ч. После введения реакционная смесь нагревается до комнатной температуры и перемешивается в течение 2 ч. Реакционная смесь охлаждается до 0оС и вводится по каплям в течение 2 ч. 100 мл метиленхлорида. Вводится дополнительная порция метиленхлорида для эффективного перемешивания суспензии солей. После этого ввода реакционная смесь помещается в холодильник. После 12 ч реакции смесь нагревается до комнатной температуры и тотчас промывается трехкратно водой с целью удаления солей. Органический слой высушивается над сульфатом магния и фильтруется. После удаления растворителя получается 29,5 г (84%-ный выход) этил N-н-октилмалонамида в виде бледно-желтого твердого вещества.
В высушенную в сушильной печи вместимостью 1 л колбу помещают 2,9 г (0,12 моль) промытого гексаном гидрида натрия в 250 мл татрагидрофурана (свежеотогнанного из натрийбензофенонкетона). К этой суспензии добавляют по каплям раствор 30 г (0,12 моль) N-н-октилмалонамида в 100 мл свежеотогнанного тетрагидрофурана в течение 0,5 ч с одновременным перемешиванием. Затем смесь дополнительно перемешивается в течение 1,5 ч и в течение этого времени она становится гомогенной. Смесь охлаждается до 0оС, и в нее вводится по каплям с одновременным перемешиванием раствор 25,5 г (0,12 моль) 2-н-октил-4-изотиазолин-3-она в 150 мл свежеотогнан- ного тетрагидрофурана. После этого ввода смесь нагревается до комнатной температуры и перемешивается в течение 2 ч. Затем эта смесь вливается в 10%-ную водную соляную кислоту и трехкратно экстрагируется хлористым метиленом. Соединенные органические слои промываются водой до тех пор, пока промывание воды не становится нейтральным (с нейтральным рН). Органический раствор промывается солевым раствором, высушивается над сульфатом магния и фильтруется. После удаления растворителей получается 54 г сырого твердого продукта. После перекристаллизации из смеси гексан/этилацетат получается 32,8 г (выход 60%) N-н-октил-цис-3-[2-(этил-N-н-октилмалонамидо)тио] акриламида, в виде белого кристаллического твердого вещества, с т. пл. 110-112оС, как показано в табл. 1.
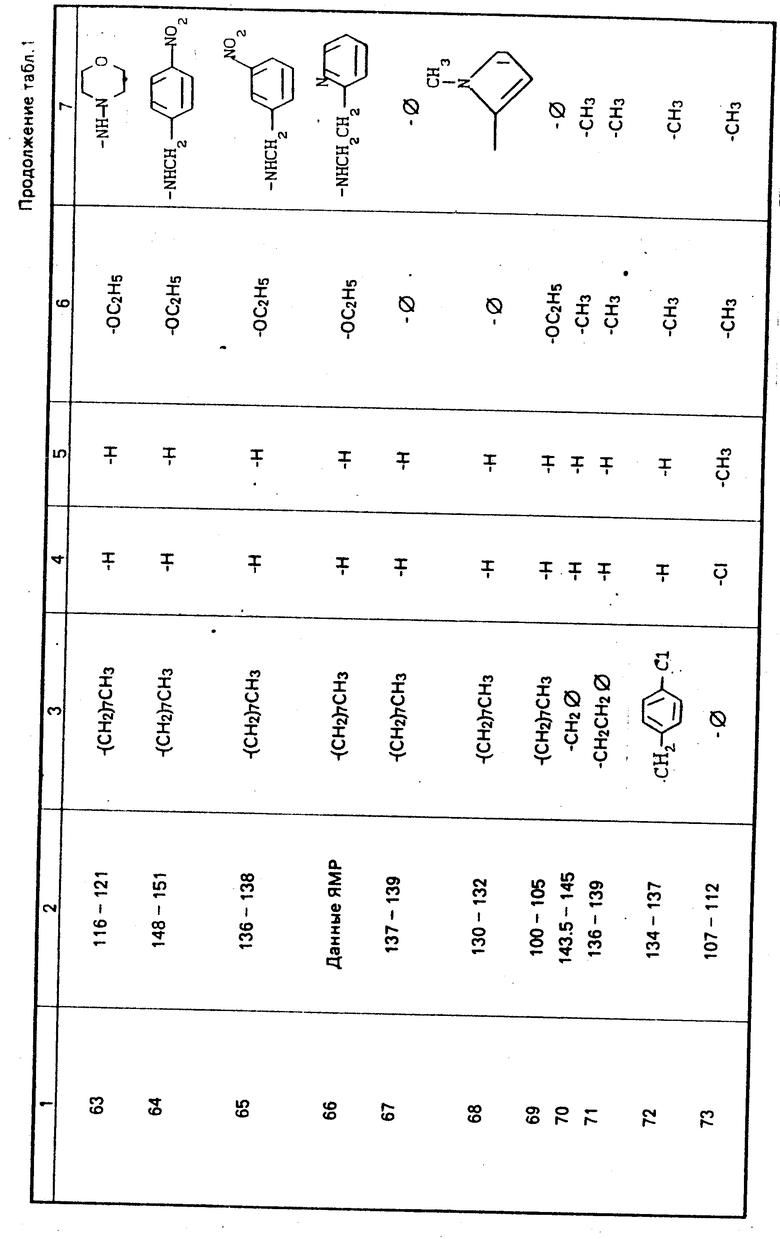
П р и м е р ы 13-17, 36, 37, 43, 49-51, 60 и 62-66.
Процедура осуществляется таким же образом, как описано в примере 18, но вместо н-октиламина в первом этапе процесса используются следующие коммерчески доступные амины: н-бутиламин, изобутиламин, н-пропиламин, фтор-бутиламин, н-гексиламин, циклопропиламин, 2-пропениламин, 2-циано-4-хлоранилин, 3-(аминометил) пиридин, 3-хлор-4-метоксианилин, 3-морфолинопропиламин, 4-метоксибензиламин, 2-аминопиримидин, 4-аминоморфолин, 4-нитробензиламин, 3-нитробензил- амин и 2-(2-аминоэтил)пиридин с получением соответственно продуктов примеров 13, 14, 15, 16, 17, 36, 37, 43, 50, 51, 60, 62, 63, 64, 65 и 66, как показано в табл. 1.
П р и м е р 19. Получение N-н-октил-цис-3-[2(N, N'-ди-н-пропилмалонодиамидо)тио]акриламида
В 2,28 мл (0,015 моль) диэтилмалоната в атмосфере азота вводят 2,5 мл (0,03 моль) н-пропиламина с одновременным перемешиванием. Смесь перемешивается в течение ночи, и в течение этого времени продукт кристаллизуется из раствора. Кристаллы фильтруются и промываютcя этилацетатом, и в результате получается 1,2 г (43%-ный выход) белых кристаллов N, N'-ди-н-пропилмалонамида.
В раствор 0,055 г (2,5 ммоль) промытого гексаном гидрида натрия в 15 мл тетрагидрофурана в атмосфере азота вводят 0,465 г (2,5 ммоль) N, N'-ди-н-пропилмалонамида в 15 мл N, N'-диметилформамида и 15 мл тетрагидрофурана. Данная смесь перемешивается при комнатной температуре в течение 1 ч и затем охлаждается до 0оС. Раствор 0,5 г (2,3 ммоль) 2-н-октил-4-изотиазолин-3-она в 5 мл тетрагидрофурана вводится в эту смесь по каплям. Смесь нагревается до комнатной температуры и перемешивается в течение 2 ч. Эту смесь вливают в 2 н. раствор соляной кислоты, разбавляют водой и экстрагируют этилацетатом. Соединенные органические соли высушивают над сульфатом магния, фильтруют и отделяют от растворителя, и в результате получается желтый твердый продукт. После перекристаллизации из 75% этилацетата - гексана получается 0,27 г (30% -ный выход) N-н-октил-цис-3-[2-(N, N'-ди-н-пропилмалонодиамидо)тио]акрилами- да в виде белого твердого вещества с т. пл. 152-154оС, как показано в табл. 1.
П р и м е р 23. Получение N-н-октил-цис-3-{2-[этил-N-(3-метоксифенил)малоноимидо] тио}акриламида
В перемешанный, охлажденный (0оС) раствор 2,13 мл (0,017 моль) этилмалонилхлорида в 10 мл хлористого метилена в атмосфере азота вводят по каплям раствор 2,03 мл (0,017 моль) м-анизидина в 10 мл хлористого метилена в течение 0,5 ч. После этого добавления реакционная смесь нагревается до комнатной температуры и перемешивается в течение 2 ч. Реакционная смесь охлаждается до 0оС, и в нее вводят по каплям в течение 2 ч раствор 2,36 мл (0,017 моль) триэтиламина в 10 мл хлористого метилена. Вводят дополнительную порцию хлористого метилена для поддержания эффективного помешивания суспензии солей. После этого ввода реакционная смесь поддерживается в холодном состоянии путем помещения ее в холодильник. По истечении 12 ч реакционная смесь нагревается до комнатной температуры и тотчас трехкратно промывается водой для удаления солей. Органический слой высушивается над сульфатом магния и фильтруется. После удаления растворителя получается сырое масло. После очистки методом мгновенной хроматографии (на силикагеле, с использованием в качестве элюента 25% этилацетата/гексана) получается 2,5 г (64%-ный выход) этил-N-3-метоксифенилмалонамида в виде красного масла.
В 10 мл абсолютного этанола вводят (0,1 г (0,005 моль) металлического натрия с одновременным перемешиванием. После полной реакции натрия вводят раствор 1,09 г (0,005 моль) этил-N-3-метоксифенилмалонамида в 5 мл абсолютного этанола. По прошествии 15 мин раствор охлаждается до 0оС, и вводится по каплям раствор 1,0 г (0,005 моль) 2-н-октил-4-изотиазолин-3-она в 10 мл абсолютного этанола. По прошествии 30 мин смесь вливают в 2 н. водный раствор соляной кислоты, разбавляют водой и экстрагируют трехкратно простым этиловым эфиром. Соединенные органические слои промывают солевым раствором, высушивают над сульфатом магния и фильтруют. После удаления растворителей получается сырой твердый продукт, который перемешивается со смесью 25% этилацетата/гексана и фильтруется, и в результате получается 1,01 г (52%-ный выход) N-н-октил-цис-3-{ 2-[этил-N-(3-метоксифенил)мало- намидо]тио} акриламида в виде белого твердого кристаллического вещества; т. пл. 122-124оС, как показано в табл. 1.
П р и м е р ы 20-22.
Процедура осуществляется таким же образом, как и в примере 23, с использованием вместо 3-анизидина одного из следующих промышленно доступных аминов: бензиламин, 2-хлоранилин и 4-хлорбензиламин, и в результате получают продукты соответственно примеров 20, 21 и 22, как показано в табл. 1.
П р и м е р 29. Получение N-н-октил-цис-3-[2-(1-циклопропил-1,3-бутандионил)тио] акриламида
В перемешанный раствор 8,4 г (0,1 моль) циклопропилметилкетона в 100 мл этилацетата в атмосфере азота вводят по каплям 39 мл (0,1 моль) 21%-ного раствора этилата натрия в абсолютном этаноле. Осуществляют нагрев колбы, снабженной конденсатором и ловушкой Дина-Старка. Этанол удаляется путем азеотропной отгонки. В колбу при необходимости вводят дополнительную порцию этилацетата. По прошествии 3 ч температура дистиллята достигает 75оС, и реакционная смесь охлаждается и отстаивается в течение ночи. Осаждаемое более твердое вещество извлекается путем фильтрации, и фильтрат удаляется. Этот твердый продукт растворяется в воде, и данный раствор подкисляется при 0оС 10%-ным водным раствором соляной кислоты. Этот раствор трехкратно экстрагируется простым этиловым эфиром. Этилацетатный фильтрат трехкратно промывается водой, подкисляется 10%-ный водной соляной кислотой и экстрагируется простым этиловым эфиром. Эфирные экстракты от обеих экстракций объединяются, высушиваются над сульфатом магния и фильтруются. После удаления растворителей получается 3,5 г 1-циклопропил-1,3-бутандиона в виде масла. Анализ методом газовой хроматографии показывает, что данный продукт имеет степень чистоты 75%. Этот промежуточный продукт используется без его дополнительной очистки.
В 25 мл абсолютного этанола в атмосфере азота вводят 3,0 мл (0,009 моль) 21%-ного этилата натрия в абсолютном этаноле. В этот перемешанный раствор вводят раствор 1,63 г (0,009 моль) 1-циклопропил-1,3-бутадиона (промежуточного продукта) в 15 мл абсолютного этанола. По прошествии 15 мин этот раствор охлаждают до 0оС, и в него вводят по каплям при перемешивании раствор 2,0 г (0,009 моль) 2-н-октил-4-изотиазолин-3-она в 10 мл абсолютного этанола. После прекращения добавления смесь нагревается до комнатной температуры и перемешивается в течение 12 ч. Эта смесь вливается в охлажденный льдом 10% -ный водный раствор соляной кислоты, отгоняется с водой и трехкратно экстрагируется этиловым эфиром. Соединенные органические слои промываются солевым раствором, высушиваются над сульфатом магния и фильтруются. После удаления растворителей получается твердое вещество, которое перекристаллизовывается из смеси этилацетата/гексана, и в результате получается 1,2 г (выход 40%) N-н-октил-цис-3-[2-(1-циклопропил-1,3-бутандиолил)тио]акриламида в виде белого твердого вещества, т. пл. 81-83оС, как показано в табл. 1.
П р и м е р 31. Получение N-н-октил-цис-3-[2-(N-фенилацетоацетамидо)тио]акрил- амида
В перемешанный, охлажденный (0оС) раствор 1,26 мл (0,01 моль) диизопропиламина в 25 мл свежеотогнанного тетрагидрофурана в атмосфере азота вводят 6,43 мл (0,01 моль) 1,4М раствора н-бутиллития в гексане. По прошествии 10 мин вводится по каплям раствор 1,66 г (0,01 моль) ацетоацетанилида в 10 мл тетрагидрофуране. Через 15 мин вводится по каплям раствор 2,0 г (0,01 моль) 2-н-октил-4-изотиазолин-3-она в 15 мл тетрагидрофурана. После добавления реакционная смесь нагревается до комнатной температуры и перемешивается в течение 2 дней. Смесь вливается в охлажденный льдом 10%-ный водный раствор соляной кислоты, разбавляется 25 мл воды и экстрагируется трехкратно этилацетатом. Соединенные органические слои промываются водой, солевым раствором и высушиваются над сульфатом магния. После фильтрации и удаления растворителей получается коричневое масло, которое подвергается хроматографическому разделению (силикагель. этилацетат - гексан в качестве элюента), и в результате получается коричневое твердое вещество. Этот твердый продукт перекристаллизовывается из этилацетата-гексана, и в результате получается 0,78 г (20%-ный выход) N-н-октил-цис-3-[2-(N-фенилацетоацетамидо)тио]акриламида в виде рыжевато-коричневого твердого вещества; т. пл. 130-133оС, как показано в табл. 1.
П р и м е р ы 30, 47 и 57.
Осуществляется процедура таким же образом, как в примере 31, с использованием вместо ацетоацетанилида одного из нижеследующих промышленно допустимых аминов: 2-ацетоацетанизидин, 4-ацетоацетанизидин в 2-ацетоацеттолуидин, в результате получаются соединения примеров 30, 47 и 57 соответственно, как показано в табл. 1.
П р и м е р 39. Получение N-н-октил-цис-3-{2-[этил-N-(2-метил-3-хлорфенил)малонамидо]тио}акриламида
В перемешанный, охлажденный (0оС) раствор 2,0 г (0,013 моль) этилмалонилхлорида в 15 мл метиленхлорида в азоте вводят по каплям раствор 1,88 г (0,013 моль) 2-метил-3-хлоранилина в 10 мл метиленхлорида в течение 0,5 ч. После этого ввода реакционная смесь нагревается до комнатной температуры и перемешивается в течение 2 ч. Реакционная смесь охлаждается до 0оС, и вводится по каплям в течение 2 ч раствор 1,87 мл (0,013 моль) триэтиламина в 10 мл метиленхлорида. Вводится дополнительная порция метиленхлорида для эффективного перемешивания суспензии солей. После этого ввода реакционная смесь охлаждается путем ввода ее в холодильник. Спустя 12 ч реакционная смесь нагревается до комнатной температуры и тотчас промывается трехкратно водой для удаления солей. Органический слой высушивается над сульфатом магния и фильтруется. После удаления растворителя получается 3,28 г (96%-ный выход) этил-N-2-метил-3-хлорфенилмалонимида в виде бледно-желтого твердого продукта.
К перемешанному охлажденному (0оС) раствору 0,75 г (0,0075 моль) диизопропиламина в 50 мл свежеотогнанного тетрагидрофурана в атмосфере азота добавляется 5,35 мл (0,0075 моль) 1,4М раствора н-бутиллития в гексане. Спустя 10 мин вводится по каплям 1,91 г (0,0075 моль) этил-N-2-метил-3-хлорфенилмалонамида в 25 мл тетрагидрофурана. Спустя 15 мин вводится 1,6 г (0,0075 моль) 2-н-октил-4-изотиазолин-3-она в 25 мл тетрагидрофурана. После этого реакционная смесь нагревается до комнатной температуры и перемешивается в течение 2 дн. Эту смесь вливают в охлажденный льдом 10%-ный водный раствор соляной кислоты, разбавляют 25 мл воды и трехкратно экстрагируют хлороформом. Соединенные органические слои двухкратно промывают водой, солевым раствором и высушивают над сульфатом магния. После фильтрации и удаления растворителей получается коричневый твердый продукт. После переме- шивания со смесью 25% этилацетата/гексана и фильтрации получается 1,82 г (52% -ный выход) N-н-октил-цис-3-{2[этил-N-(2-метил-3-хлорфенил)малонамидо]тио} акриламида в виде белого твердого вещества; т. пл. 108-110оС, как показано в табл. 1.
П р и м е р ы 24-28, 32-35, 38, 40, 41, 44-46, 48, 52-56, 58 и 59.
Процедура осуществляется таким же образом, как и в примере 39, с той разницей, что вместо 3-анизидина используется один из следующих промышленно доступных аминов: н-додециламин, 2-аминотиазол, н-дециламин, 5-цианопентиламин, 2-тиофенметиламин, 3-метоксибензиламин, 4-метил- бензиламин, 3-хлоранилин, 2,3-дихлор-3,4-анилин, 2-хлорбензиламин, 3-метилбензиламин, 2,3-дихлоранилин 2-аминопиридин, 2-хлор-5-нитроанилин, 2-метокси-5-хлоранилин, 3-нитро-4-хлоранилин, 2,4-дихлоранилин, 2-анизидин, 3-толуидин, 3,5-дихлор- анилин, 2-метоксибензиламин, 4-анизидин и 2-хлор-6-метиланилин, и в результате получаются продукты примеров 24, 25, 26, 27, 28, 32, 33, 34, 35, 38, 40, 41, 44, 45, 46, 48, 52, 53, 54, 55, 56, 58 и 59 соответственно, как указано в табл. 1.
П р и м е р 42. Получение N-н-октил-цис-3-{2-[этил-N-(2-хлор-5-метилфенил)малон- амидо]тио}акриламида
В перемешанный охлажденный раствор (0оС) 2,0 г (0,013 моль) этилмалонилхлорида в 10 мл метиленхлорида в атмосфере азота вводят по каплям раствор 1,88 г (0,013 моль) 2-хлор-5-метиланилина в 10 мл метиленхлорида в течение 0,5 ч. После этого реакционная смесь нагревается до комнатной температуры и перемешивается в течение 2 ч. Реакционная смесь охлаждается до 0оС, и вводится по каплям в течение 2 ч раствор 1,87 г (0,013 моль) триэтиламина в 10 мл хлористого метилена. После этого реакционная смесь охлаждается при вводе ее в холодильник. Спустя 12 ч реакционная смесь нагревается до комнатной температуры и тотчас промывается трехкратно водой для удаления солей. Органический слой высушивается над сульфатом магния и фильтруется. После удаления растворителя получается 2,8 г (84%-ный выход) этил-N-2-хлор-5-метиленфенилмалонамида в виде рыжевато-коричневого твердого вещества. В высушенную в сушильной печи вместимостью 1 л колбу в атмосфере азота вводят 0,12 г (0,005 моль) промытого гексаном гидрида натрия в 15 мл тетрагидрофурана (свежеотогнанный из найтрийбензофенонкетила). В эту суспензию вводят по каплям раствор 1,28 г (0,005 моль) этил-N-2-хлор-5-метилфенилмалонамида в 10 мл свежеотогнанного тетрагидрофурана при одновременном перемешивании в течение 0,5 ч. Смесь перемешивается дополнительно в течение 1,5 ч, и за это время она становится однородной. Эта смесь охлаждается до 0оС, и в ее вводится по каплям с одновременным перемешиванием раствор 1,06 г (0,005 моль) 2-н-октил-4-изотиазолин-3-она в 10 мл свежеотогнанного тетрагидрофурана. После этого ввода реакционная смесь нагревается до комнатной температуры и перемешивается в течение 2,5 ч. Затем эту смесь вливают в 10% -ный водный раствор соляной кислоты и трехкратно экстрагируются хлористым метиленом. Соединенные органические слои промывают водой до тех пор, пока промывки не становятся нейтральными (с нейтральной величиной рН). Органический раствор промывают солевым раствором, высушивают над сульфатом магния и фильтруют. После удаления растворителя получается сырое масло. После очистки путем хроматографического разделения (силикагель, этиленацетат/гексан в качестве элюента) получается 0,3 г (13%-ный выход) N-н-октил-цис-3-{2-[этил-N-(2-хлор-5-метил- фенил)малонамидо] тио} акриламида в виде бледного желтого твердого вещества, т. пл. 114оС. Структура этого продукта показана в табл. 1.
П р и м е р 61.
Получение N-н-октил-цис-3-[2-(N-бензил-N'-н-октилмалондиамидо)тио]-акрил- амида
В перемешанный охлажденный (0оС) раствор 14 г (0,093 моль) этилмалонилхлорида в 50 мл метиленхлорида в атмосфере азота вводят по каплям раствор 10 г (0,093 моль) бензиламина в 50 мл хлористого метилена в течение 0,5 ч. После этого ввода реакционная смесь нагревается до комнатной температуры в течение 2 ч. Реакционная смесь охлаждается до 0оС, и вводится по каплям в течение 2 ч 13 мл (0,093 моль) триэтиламина в 50 мл метиленхлорида. Вводится дополнительная порция метиленхлорида для эффективного перемешивания суспензии солей. После этого реакционная смесь охлаждается путем помещения в холодильник. По прошествии 12 ч реакционная смесь нагревается до комнатной температуры и тотчас промывается трехкратно водой для удаления солей. Органический слой высушивается над сульфатом магния и фильтруется. После удаления растворителя получается сырой продукт, который очищается путем испарительной хроматографии (на силикагеле, 50% этилацетат/гексан в качестве элюента) и в результате получается 12,5 г (выход 61%) этил-N-бензилмалонамида.
Раствор 1,1 г (5,0 моль) этил-N-бензилмалонамида в 30 мл толуола нагревается с обратным холодильником в атмосфере азота, и вводится 1,29 г (10,0 моль) н-октиламина. Реакционная смесь нагревается с обратным холодильником в течение 5 ч и вводится 5 мл N, N'-диметилформамида. Смесь нагревается с обратным холодильником в течение 12 ч и вливается в 5%-ный водный раствор соляной кислоты. Органический слой промывают насыщенным водным раствором бикарбоната натрия, водой, солевым раствором и высушивают над сульфатом магния. После фильтрации и удаления растворителей получается сырое твердое вещество, которое перекристаллизовывается из 50% этилацетата/гексана, и в результате получается 0,8 г (выход 53%) N-н-октил-N'-бензилмалонамида.
В высушенную в сушильной печи колбу вместимостью 1 л вводится 0,071 г (2,8 моль) промытого гексаном гидрида натрия в 10 мл тетрагидрофурана (свежеотогнанного из натрийбензофенонкетила). В эту суспензию вводится по каплям с одновременным перемешиванием в течение 0,5 ч раствор 0,8 г (2,6 моль) N-н-октил-N'-бензилмалонамида в 10 мл свежеотогнанного тетрагидрофурана. Затем смесь перемешивается еще в течение 1,5 ч при 0оС с последующим осторожным нагреванием в течение 1 ч. Смесь охлаждается до 0оС, и к ней по каплям добавляется раствор 0,55 г (2,6 моль) 2-н-октил-4-изотиазолин-3-она в 10 мл свежеотогнанного тетрагидрофурана с одновременным перемешиванием. После этого ввода смесь нагревается до комнатной температуры и перемешивается в течение 2 ч. Затем эту смесь вливают в 10%-ный раствор соляной кислоты (водный) и трехкратно экстрагируют метиленхлоридом. Соединенные органические слои промывают водой до нейтрального значения рН. Органический раствор промывают солевым раствором, высушивают над сульфатом магния и фильтруют. После удаления растворителей получается сырое твердое вещество. После перекристаллизации из этилацетата/гексана получается 0,36 г (выход 27% ) N-н-октил-цис-3-[2-(N-бензил-N'-н-октил-малоноди- амидо)тио]акриламида в виде белого твердого кристаллического вещества; т. пл. 131-135оС. Данный продукт приведен в табл. 1.
П р и м е р 67. Получение N-н-октил-цис-3-[2-(1,3-дифенил-1,3-пропандионил)тио]ак- риламида
В перемешанный раствор 2,25 г (0,01 моль) 1,3-дифенил-1,3-пропандиона в 25 мл N, N'-диметилформамида в атмосфере азота вводят по каплям 10 мл (0,01 моль) 1М раствора тетрабутиламмонийфторида в тетрагидрофуране. По прошествии 15 мин вводят раствор 2,13 г (0,01 моль) 2-н-октил-4-изотиазолин-3-она в 5 мл N, N'-диметилформамида. Реакционную смесь перемешивают в течение одной недели и вливают в 10%-ный водный раствор соляной кислоты. Эту смесь трехкратно экстрагируют этилацетатом. Объединенные органические слои перемешивают солевым раствором, высушивают над сульфатом магния и фильтруют. После удаления растворителей получают сырое твердое вещество, которое очищается путем испарительной хроматографии (силикагель, этилацетат/гексан в качестве элюента) и в результате получается 0,1 г N-н-октил-цис-3-[2-(1,3-дифенил-1,3-пропандионил)тио]акриламида в виде белого твердого вещества; т, пл. 107-139оС. (См. табл. 1).
П р и м е р 68. Получение N-н-октил-цис-3-{2-[1-фенил-3-(1-метилпиррол-2-ил)-1,3- пропандионил]тио}акриламида
В перемешанную охлажденную (5оС) суспензию 0,96 г (0,04 моль) промытого гексаном натрия в 2,72 г (0,02 моль) метилбензоата в атмосфере азота вводят раствор 5,0 г (0,04 моль) 2-ацетил-1-метилпиррола в 30 мл простого этилового эфира. Вводят несколько капель этанола. Реакционная смесь нагревается при 5оС в течение 12 ч. Эту смесь вливают в воду и экстрагируют простым этиловым эфиром. Объединенные органические слои промывают солевым раствором, высушивают над сульфатом магния и фильтруют. После удаления растворителей получается воскообразное твердое вещество, которое очищается методом испарительной хроматографии (силикагель, этилацетат/гексан в качестве элюента) и в результате получается 0,8 г 1-(1-метилпиррол-2-ил)-3-фенил-1,3-пропандиона.
В перемешанный раствор 0,78 г (0,0034 моль) 1-(1-метилпиррол-2-ил)-3-фенил-1,3-пропандиона в 5 мл N, N'-диметилформамида в атмосфере азота вводят 3,4 мл (0,0034 моль) 1М раствора тетрабутиламмонийфторида в тетрагидрофуране. По прошествии 10 мин вводят раствор 0,73 г (0,0034 моль) 2-н-октил-4-изотиазолин-3-она в 5 мл N, N'-диметилформамида. По прошествии 12 ч вводят еще 3,4 мл 1М раствора тетрабутиламмонийфторида. Через 12 ч реакционную смесь разбавляют 10%-ным водным раствором соляной кислоты и трехкратно экстрагируют этилацетатом. Соединенные органи- ческие соли несколько раз промывают водой, один раз солевым раствором и высушивают над сульфатом магния. После удаления растворителей и перекристаллизации сырого продукта из этилацетата/гексана получается 0,28 г (выход 19%), N-н-октил-цис-3- { [1-фенил-3-(1-метилпиррол-2-мл)-1,3-про-пандионил] тио} акриламида в виде белого твердого вещества; т. пл. 131-132оС. (См. табл. 1).
Промежуточные продукты примеров 70-75, получаемые таким же образом, как описано в примере 1В, приготавливают следующим образом.
А. Получение 2-бензил-4-изотиазолин-3-онового промежуточного продукта для примера 70.
В суспензию 58,2 (0,15 моль) N, N'-бис-бензил-3,3'-дитиодипропионамида в 500 мл этилендихлорида при 10-15оС вводят по каплям 63,6 г (0,473 моль) сульфурилхлорида. В результате получается прозрачный раствор светло-янтарного цвета. После перемешивания в течение ночи раствор кон центрируется примерно до половины его объема. Твердый продукт кремового цвета отделяется и извлекается путем фильтрации, и получается 36,1 г хлоргидратной соли 2-бензил-4-изотиазолин-3-она, в фильтрат выпадает в осадок дополнительно 1,5 г твердого вещества, но было определено, что это исходный продукт. Фильтрат дополнительно выпаривается до получения коричневого масла. Это масло снова растворяется в бензоле, обрабатывается обесцвечивающим углем и выпаривается еще раз. Полученное масло светло-янтарного цвета отверждается при отстаивании. Этот светлый рыжевато-коричневый твердый продукт перекристаллизовывается из гептана, и результате получается 8,0 г (12%-ный выход) белых кристаллов хлоргидратной соли 2-бензил-5-хлор-4-изотиазолин-3-она, с т. пл. 58-59оС. Эта хлоргидратная соль нехлорированного бензилового соединения превращается в свободное основание путем перемешивания с водой и сушки в вакууме, и в результате получается 27,0 г (выход 47%) 2-бензил-4-изотиазолин-3-она (промежуточного продукта) с т. пл. 78-80оС.
В. Получение 2-(β-фенетил)-4-изотиазолин-3-онового промежуточного продукта для примера 71
В суспензию 103,9 г (0,25 моль) N, N'-бис-(β-фенетил)-3,3-дитиодипропионамида в 1000 мл этилендихлорида при 10-15оС вводят 101,3 г (0,75 моль) сульфурилхлорида в течение 1 ч. После этого смесь нагревается до комнатной температуры. Эта желтоватая суспензия концентрируется с удалением примерно 2/3 этилендихлорида, и отделенную твердую массу удаляют путем фильтрации и промывают простым эфиром. Этот твердый продукт вводят в смесь 350 мл воды и 200 мл хлороформа. Вводят твердый бикарбонат натрия в виде отдельных порций с одновременным перемешиванием до тех пор, пока не достигается значение рН водной фазы равное 7-8. Происходит разделение слоев, и водную фазу экстрагируют дополнительно вводимым хлороформом. Соединенные хлороформные слои высушивают над сульфатом магния и концентрируют, и в результате получается белый твердый осадок, который перекристаллизовывается из смеси бензол/гексан, и в результате получается 44,8 г (выход 44%) 2-(β-фенетил)-4-изотиазолин-3-онового промежуточного продукта с т. пл. 76-78оС.
С. Получение 2-(п-хлорбензил)-4-изотиазолин-3-онового промежуточного продукта для примера 72
В суспензию 292,0 г (0,639 моль) N, N'-бис-(4-хлорбензил)-3,3'-дитиодипропион- амида в 2 л этилацетата вводят 136,0 г (1,9717 моль) хлора в течение 1 ч, и в течение этого времени температура реакции повышается до 47оС. Смесь охлаждается до комнатной температуры, дегазируется и затем охлаждается до 10оС. Твердый продукт удаляется путем фильтрации, перемешивается с 200 мл воды, фильтруется и высушивается. Высушенное твердое вещество растворяется в 500 мл кипящего этилацетата и фильтруется с удалением нежелательного амидного исходного материала. Этилацетатный раствор при его охлаждении осаждает кристаллическое твердое вещество, которое фильтруют и высушивают, и в результате получается 132,2 г (46%-ный выход) 2-(п-хлорбензил)-4-изотиазолин-3-онового промежуточного продукта с т. пл. 88-90оС.
D. Получение 2-фенил-4-метил-5-хлор-4-изотиазолин-3-онового промежуточного продукта для примера 73
В 400 мл этилацетата при 0оС вводят в течение одного часа 155,2 г (0,4 моль) N, N'-бис-фенил-3,3'-дитиодиизобутирамида в виде 40 равных порций и 116,5 г (1,64 моль) хлора. В течение этих вводов температура поддерживается равной 0,5оС. Затем смесь нагревается до 15оС. После этого раствор выпаривается в вакууме, оставляя коричневое масло, которое при отстаивании частично отверждается. Этот материал раство- ряется в теплом этаноле. При охлаждении отделяется твердый продукт розового цвета, который удаляется путем фильтрации. Фильтрат выпаривается до получения темного масла; это масло экстрагируется простым эфиром. После выпаривания простого эфира получается 112,7 г желтой кашицы. 3 г этой кашицы подвергается хроматографическому разделению в колонке сухим с кремнеземом с проявлением толуолом.
Основная фракция (1,92 г) с величиной R примерно 0,7 экстрагируется из кремнезема простым эфиром, который затем испаряется. При стоянии остаточное масло отверждается. Этот материал перекристаллизовывается из этанола, и в результате получается промежуточный продукт 2-фенил-4-метил-5-хлор-4-изотиазолин-3-он кремового цвета с т. пл. 60-68оС.
Е. Получение 2-(п-хлорфенил)-5-хлор-4-изотиазолин-3- основного промежуточного продукта для примера 74
В суспензию 21 г (0,05 моль) N, N'-бис-(п-хлорфенил)-3,3'-дитиодипропионамида в 75 мл этилацетата при 15оС вводят 18,6 г (0,26 моль) хлора в течение 30 мин. После прекращения этого ввода смесь отстаивает ся в течение нескольких часов и затем фильтруется. Извлеченный твердый продукт перемешивается в метаноле и в результате получается белое твердое вещество. Этот материал кристаллизуется из 140 мл толуола с обесцвечиванием, с использованием активированного угля, и в результате получается 5,82 г (выход 19,5% ) 2-(хлорфенил)-5-хлор-4-изотиазолин-3-онового промежуточного продукта с т. пл. 117-119оС.
F. Получение 2-(п-хлорфенил)-4-метил-4-изотиазолин-3-онового промежуточного продукта для примера 75
В колбу, вместимостью 2 л, снабженную механической мешалкой, двумя воронками для ввода и термометром, вводят 1 л этилацетата, который охлаждают до температуры от -5 до 0оС. Одновременно вводят N, N'-бис(п-хлорфенил)-3,3'-дитиодиизобутир- амид (116,3 г, 0,254 моль), сульфурилхлорид (141,7 г 1,05 моль) и пиридин (40,0 г, 0,05 моль) в течение 75 мин при поддержании температуры 0оС. Смесь нагревают до комнатной температуры и затем снова охлаждают до 2-40оС посредством бани с сухим льдом. Твердый осадок удаляют путем фильтрации, промывают холодным этилацетатом, перемешивают с 1 л воды, повторно фильтруют и высушивают, и в результате получают 46 г 2-(п-хлорфенил)-4-метил-4-изотиазолин-3-она и 2-(п-хлорфенил)-4-метил-5-хлор-4-изотиазолин-3-она в соотношении 2:1. Эту высушенную твердую смесь (45,5 г) и сульфурилхлорид (19,0 г, 0,135 моль) вводят одновременно в 300 мл этилацетата при 0оС в течение 1 ч. После этого смесь нагревают до комнатной температуры и перемешивают в течение ночи. Эту твердую суспензию удаляют путем фильтрации, высушивают и перекристаллизовывают из 2-пропанола, и в результате получается 9,5 г 2-(п-хлорфенил)-4-метил-4-изотиазолин-3-онового промежуточного продукта с т. пл. 157-159оС.
Нижеследующая таблица 1 охватывает все классы соединений, имеющих общую формулу

 к которой W1 и W2 представляют собой соответственно СОR2 и СОR3.
к которой W1 и W2 представляют собой соответственно СОR2 и СОR3.
Данные ЯМР для некоторых примеров приведены в табл.2.
Названия соединений, представленных в табл. 1, даны в табл.3.
П р и м е р 79.
Бактерицидная активность
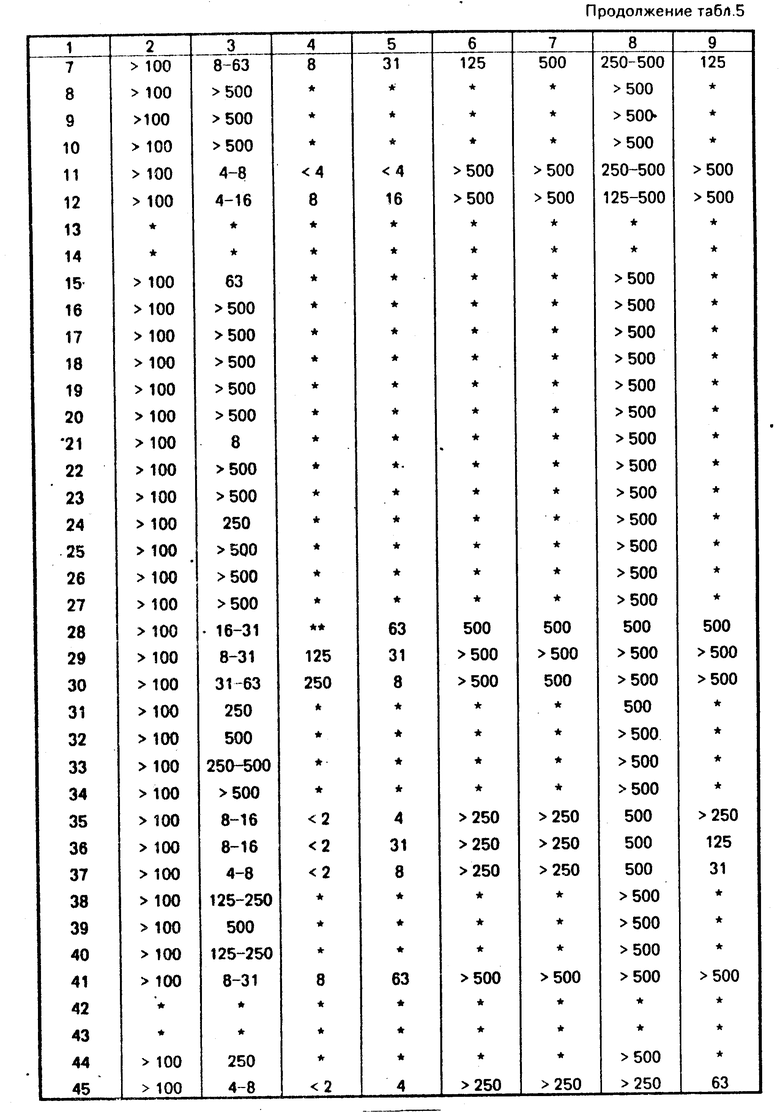
Соединения из примеров 1А, 1В, 2, 6, 18, 42, 49, 69 и 73 подвергались испытанию посредством описанных ниже процедур, результаты этих испытаний приводятся в табл. 2.
Скорость умерщвления (SOK). В данном испытании определяется потеря клеточной жизнеспособности в водной суспензии бактериальных клеток в течение 4 ч, когда эти клетки контактируют с испытываемыми соединением определенной концентрации в приготовленной искусственно жесткой воде (SНW). Основной раствор или дисперсия испытываемого соединения, обычно концентрацией 1% , приготавливается в растворе, состоящем из смеси растворителей ацетона, метанола и воды, взятых в соотношении 5:3:2. SHW приготавливается из 1 л стерилизованной деионизириванной воды, в которую вводится нижеследующие растворы:
1. 2 мл. раствора 31,74 г МgCl2 и 73,99 г CaCl2 в 1000 мл стерилизованной нагреванием дистиллированной воды.
2. 4 мл. раствора 56,03 г NaHCO3 в 1000 мл воды, которая была стерилизована путем пропускания через фильтр.
Этот объединенный раствор затем стерилизуется пропусканием через фильтр, и в результате получается SHW. Объем основного раствора вводится в SHW и получается исходное испытываемое соединение с начальной концентрацией 500 ч/млн.
Когда это испытываемое соединение готово к проведению испытания, каждый сосуд последовательных серий разбавлений, за исключением первого сосуда, содержит равный объем смеси SHW с испытываемым соединением. Первый сосуд содержит двойной объем SHW с исходной концентрацией испытываемого соединения. Половина SHW из первого сосуда вводится во второй сосуд. После перемешивания, половина полученного объема удаляется из второго сосуда и подается в третий сосуд. Полный цикл повторяется в такой степени, чтобы получилась последовательная серия концент- раций, составляющих 500, 250, 125, 63, 31, 16, 8 и 4 ч/млн.
Затем каждый сосуд инокулируется клеточной суспензией бактерий рs. fl. Эти бактерии выращиваются в пробирке в 1%-ным сланцевым агаром и инкубируются в течение 18-24 ч при 30оС. После этого пробирка промывается 4 мл стерилизованной воды. Она разбавляется до плотности 60-80 ед. мутности по Hефелометру (NТU), в 100 мл SHW, содержащей различные концентрации испытываемого соединения, вводится 0,75 мл инокулума от промывки 60-80 NТU с тщательным перемешиванием. По прошествии 4 ч от времени введения при 30оС, 5 мкл раствора переносится в 100 мкл раствора питательного бульона с целью извлечения любых живых клеток. Смесь инкубируется в течение 24 ч при 30оС до определения концентрации в ч/млн, при которой каждое соединение умерщвляет < 99,999% клеток в SHW растворе.
Минимальная ингибирующая концентрация (МIC) получается с использованием питательного бульона в испытании с двухкратным последовательным разбавлением, осуществляемом следующим образом. Основной раствор или дисперсия испытываемого соединения, обычно концентраций 1%, приготавливается в растворе, состоящем из смеси растворителей ацетон, метанол и вода в соотношении 5: 3:2. Объем основного раствора вводится в питательную среду с культурой и получается исходная концентрация соединения 500 ч/млн.
Когда все подготовлено для проведения испытания, каждый сосуд последовательной серии разбавления, за исключением первого сосуда, содержит одинаковый объем питательного бульона, свободного от соединения. Первый сосуд содержит двойной объем питательного бульона с начальной концентрацией испытываемого соединения. Половина питательного бульона из первого сосуда переносится во второй сосуд. После перемешивания половина полученного объема удаляется из второго сосуда и переносится в третий сосуд. Весь цикл повторяется, чтобы получилась последовательная серия концентраций, составля- ющих соответственно 500, 250, 125, 63, 31, 16, 8 и 4 ч/млн. Затем каждый сосуд инокулируется клеточной суспензией соответствующего испытываемого организма. Бактерии выращиваются в питательном бульоне, выращиваются на сланцевом агаре в течение времени и при температуре, которые достаточны для подвергаемых испытанию видов организмов. В случае грибков споры собираются путем пипеточного ввода воды на сланцевый агар и удаления спор стерильной проволочной петлей. Клеточно-споровая суспензия стандартизируется по времени регулирующего инкубирования, температуре и объему разбавителя. Затем данная суспензия используется для инокулирования сосудов, содержащих соединение питательного бульона. Затем эти сосуды инкубируются при подходящей температуре. После инкубирования сосуды исследуются на определение роста/отсутствия роста. Минимальная ингибирующая концентрация (МIC) определяется, как самая низкая концентрация соединения, которая не приводит в результате к полному ингибированию роста испытываемого организма.
Испытываемые организмы, которые демонстрируют активность предлагаемого изобретения, включают следующие бактерии: Рseudomonas fluorescenc (Рs. fl) грамм-отрицательные Рseudomonas aerugenosa (Рs. ае) грамм-отрицательные Еscherichia coli (Е. с.) грамм-отрицательные Staрhylocоcus aureus (S. а) грамм-положительные Грибы: Aspergillus niger (A. n) Aureobasidium pullulans (A. p.)
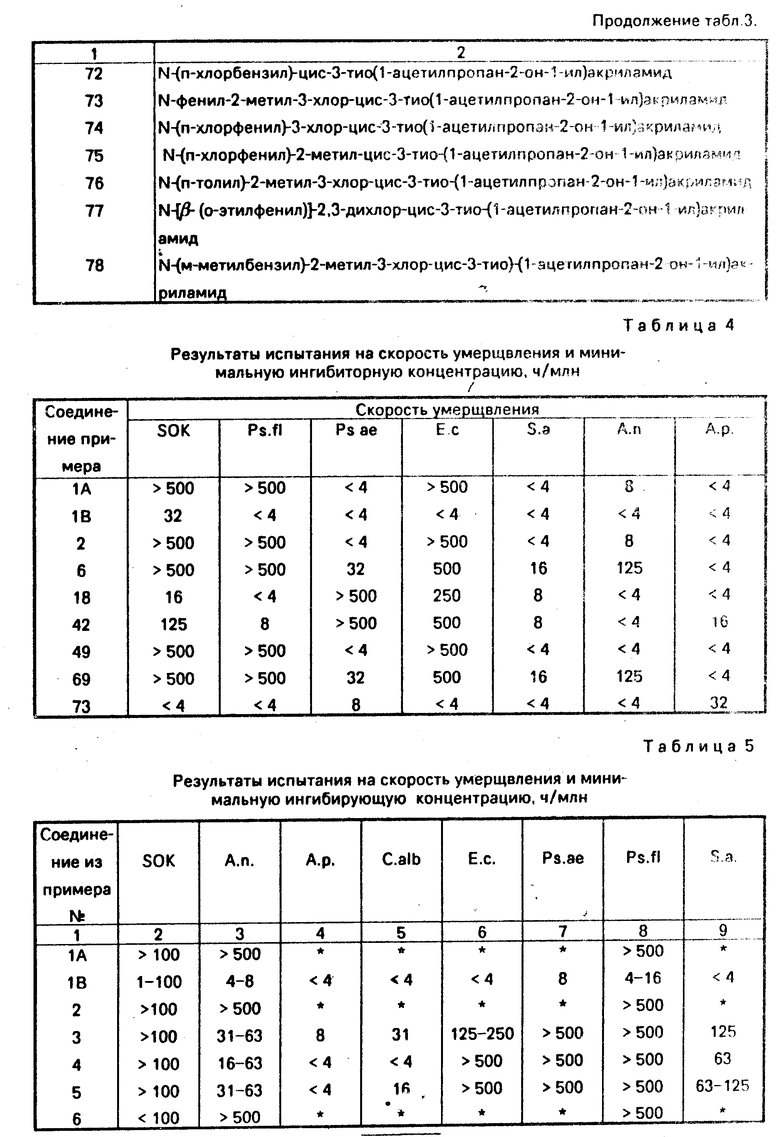
Соединения примеров 1-68 и 70-75 подвергались также другой испытательной процедуре на бактерицидную активность, полученные результаты представлены в табл. 3.
Испытание на скорость умерщвления
При испытании на SOK определяется жизнеспособность инокулума Pseudomonas fluorescens в искусственно полученной жесткой воде (SHW) при экспонировании в течение 24 ч в ацетоновом растворе концентрацией данного соединения 100 ч/млн.
Приготавливается ацетоновый раствор данного соединения концентрацией 10000 ч/млн, (и 0.1 мл этого раствора вводится в 9,8 млн SHW 0,1 мл инокулума Ps. fl с концентрацией 10000000 клеток на 1 мл вводится в SHW, и получается 10 мл раствора, который инкубируется в течение 24 ч для извлечения клеток в триптиновом осевом питательном бульоне (ТSВ). Для извлечения и замера количества живых клеток 2,5 мл смеси SHW переносится в сосуд, из которого затем 2,5 мкл подается 8-кратно в ячейки микротитрования, содержащие 225 мкл ТSВ. Затем каждая из 8 переносимых порций последовательно разбавляется семикратно, с получением восьми репликатов восьми разбавлений. Концентрация, при которой не извлекается ни одной живой клетки, используется для обратного расчета логарифма снижения. Эти данные включаются в базу данных, и автоматически рассчитываются логарифмические снижения.
Испытание на минимальную ингибирующую концентрацию
Испытание на минимальную ингибирующую концентрацию (МIC) определяет жизнеспособность инокулума Pseudomonas fluorescens в ТSВ при экспонировании в течение 72 ч с получением различных концентраций испытываемого соединения.
Аликвота 125 мкл испытываемого соединения концентрацией 1000 ч/млн в ацетоне вводится в 4,88 мл ТSВ с получением 250 ч/млн раствора. 100 мкл от этого раствора переносится в первый ряд двух пластинчатых колонок микротитрования. Оба репликата и пять дополнительных соединений последовательно разбавляются 1:1 до конечной концентрации 0,8 ч/млн в ТSВ.
Осуществляется инокуляция путем разбавления в течение 24 ч культуры Рs. fl, 4 мл на 36 мл буферного фосфатного раствора. Для переноса 1,5 мкл этой клеточной суспензии на пластинки микротитрования используется автоинокулятор Dynaterh. Пластинки инкубируются при 30оС в течение 3 дн, после чего определяется минимальная концентрация, при которой не происходит никакого роста. Данные SOK и МIC испытаний на бактериях Ps. fl, Ps. аe, Е. с. и Sа. и на грибах Саndida albicans (с. аlb), А. п. и А. р. приведены в табл. 3.
П р и м е р 80.
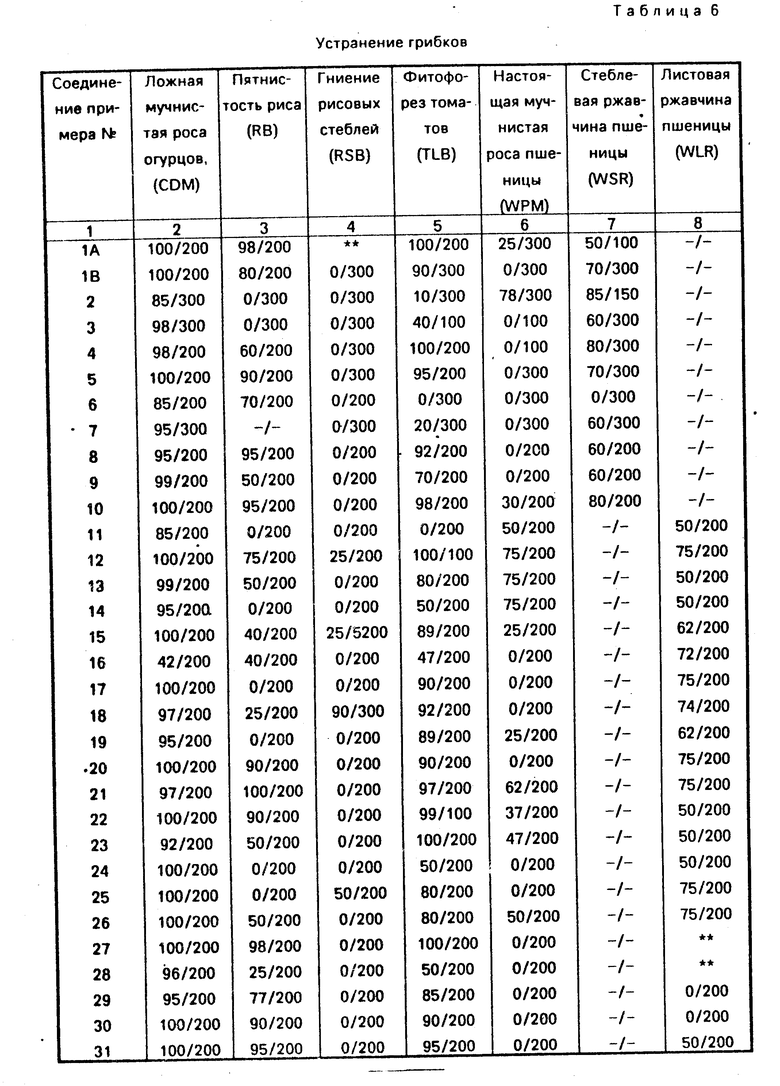
Соединения из примеров 1-68 и 70-75 испытывались на фунгицидную активность согласно следующим процедурам; результаты испытаний даны в табл. 4.
Соединения, отвечающие данному изобретению, испытывались на фунгицидную активность в условиях "ин виво" против увядания огуречных растений, вызванного ложной мучнистой росой (СДМ), пятнистости рисовых растений (RВ), гниения рисовых стеблей (RSВ), фитофореза томатов (ТLВ), настоящей мучнистой росы пшеницы (WPM), стеблевой ржавчины пшеницы (WPM) и листовой ржавчины пшеницы (WLR).
При проведении испытания на злаках (за исключением рисовых растений, испытываемых на пятнистость риса), растения подрезались примерно за 24 ч до внесения фунгицидного соединения для получения одинаковой высоты растений с тем, чтобы облегчить равномерное нанесение данного соединения и инокуляцию грибами. Данные соединения разбавлялись в смеси вода:ацетон:метанол, взятых в соотношении 2:1:1, распылялись на растения, высушивались (в течение 4-6 ч) и затем растения инокулировались грибами. Каждое испытание использовалось для контроля растений, которые подвергались обработке распылением на них смеси воды, ацетона и метанола и инокулировались грибами. Остальная процедура каждого испытания описана ниже, и полученные результаты даются как процент подавления заболевания (процент растений, обработанных соединениями, отвечающими данному изобретению, которые не обнаруживают признаков или симптомов заболевания, но сравнению с необработанными контрольными растениями).
Ложная мучнистая роса огурцов (СДМ) Pseudoperonospora cubensis оставалось на листьях огуречных растений при постоянной комнатной температуре от 65 до 75оF (от 18 до 24оС) в условиях влажного воздуха с умеренной интенсивностью света в течение 7-8 дн. Получали водную суспензию спор от зараженных листьев, и доводили концентрацию этих спор примерно до 100000 на 1 мл воды.
Продажная огуречная рассада инокулировалась путем опрыскивания нижней части листьев посредством амортизатора Deviliss до тех пор, пока на листьях не наблюдалось наличие маленьких капелек. Инокулированные растения инкубировались во влажной камере в течение 24 ч при температуре примерно 70оF (21оС) и затем инкубировались в течение 6-7 дн в условиях регулируемой комнатной температуры во влажных условиях при 65-75оF (18-24оС). Через 7 дней после инокулирования определяли процент подавления заболевания.
Пятнистость рисового растения (RВ)
Рисовые растения Nato инокулировали Pivicularia ovyzal (примерно 20000 конидий на 1 мл) путем опрыскивания листьев и стеблей щелочным воздушным распылителем до тех пор, пока на листьях не наблюдалось наличие равномерной пленки инокулума. Инокулированные растения инкубировались во влажной среде (75-85оF или 24-29оС) в течение примерно 24 ч, затем они помещались в условиях теплицы (70-75оF или 21-24оС). Через 7-8 дней после инокулирования определяли процент подавления заболевания.
Гниение рисовых стеблей (RSВ) Pellicularia bilamentosce (P. sp. sasiki) культивировались на автоклавной смеси рисовых семян и питательного бульона картофельной декстрозы (100 г рисовых семян на 30 мл питательного бульона картофельной декстрозы) в колбе Эрленмейера вместимостью 500 мл. Спустя 10 дней данная культура перемешивалась в смесителе с образованием однородного инокулума. Примерно одну чайную ложку инокулума распределяли между рисовыми саженцами Lebonnet на поверхности почвы каждого горшка (диаметром 76,2 мм). Инокулированные саженцы инкубировались в течение 5 дн во влажной камере (85-90оF или 29-32оС). После удаления саженцев из камеры тотчас же определяли процент подавления заболевания.
Фитофорез томатов (ТLВ)
Phytophtora inbentoens культивировались на четырехнедельных томатных растениях Diхie в регулируемых комнатных условиях (65-70оF или 18-21оС, относительная влажность 100%). После выдержки споры вымывались из листьев водой и диспергировались посредством атомизатора De vilbiss на трехнедельные томатные растения Piхie, которые предварительно опрыскивались экспериментальными фунгицидами. Инокулированные растения помещались во влажную камеру при 70оF (21оС) с постоянной влажностью сроком на 24 ч для их инфекционного заражения. Затем эти растения переносились в условия регулируемой комнатной температуры, как указано выше, и оценка результатов осуществлялась после трехдневного инокулирования. Степень подавления заболевания определялась, как процент подавления, через 4 дня после инокуляции и через 5 дней после опрыскивания соединений.
Настоящая мучнистая роса пшеницы (WPM)
Еrysiphe graminis (f. sp. tritici) культивировались на саженцах пшеницы в условиях регулируемой температуры от 65 до 75оF (18-24оС). Споры мучнистой росы встряхивались с культивируемых растений на саженцы пшеницы Pennol, которые предвари- тельно опрыскивались данным фунгицидным соединением. Инокулированные саженцы выдерживались в условиях регулируемой комнатной температуры 65-75оF (18-24оС), и осуществлялось подпочвенное орошение. Процент подавления заболевания определяли через 8-10 дней после инокулирования.
Стеблевая ржавчина пшеницы (WSP)
Puccinia graminis (f. sp. tritici разновидности 15В-2) культивировали на саженцах пшеницы Wanzer в течение 14 дн в теплице. Получали водную суспензию спор от зараженных растений, и концентрацию этих спор доводили примерно от 200000 спор на 1 мл деионизированной воды. Растения пшеницы Wanzer, которые предварительно обрабатывали фунгицидными соединениями, инокулировали путем нанесения на них споровой суспензии стеблевой ржавчины до момента стока, используя для этого распылитель De vilbiss, при давлении 15 фунт/дюйм2 (1,05 атм). После инокуляции растения помещали во влажные условия с температурой примерно 75оF (24оС), где они экспонировались 12 ч в постоянной темноте и затем минимум в течение 3-4 ч при небольшом освещении с интенсивностью света примерно 500 фут-свечей (5350 лк). Температура в камере не превышала 85оF (29оС). В конце периода светового воздействия на растения помещали в теплицу, где они выращивались в течение 2 недель, после которых определяли процент подавления заболевания.
Листовая ржавчина пшеницы (WLR)
Puccinia recondita разновидности РКВ и РLD) культивировали на семидневной пшенице (сultivar Fielder) в течение 14 дн в теплице. Споры собирались с листьев путем вакуумирования в циклоне или путем их оcедания на алюминиевой фольге. Эти споры очищали путем просеивания через сито размером отверстий 250 мкм и хранили или использовались сразу после их получения. Хранение осуществляли в закрытых мешках, помещенных холодильник сверхнизкого замораживания. После хранения споры должны быстро нагреваться, в течение 2 мин, до их непосредственного использования. Споровая суспензия приготавливалась из уреида путем добавления 20 мг (9,3 млн) на 1 мл масла soltrol. Эту суспензию вводили в желатиновые капсулы (объемом 0,7 мл), которые подсоединяли к масляным распыли- телям. Одна капсула использовалась на площадь, включающую 20 горшков площадью поверхности по 2 дюйм2, в которых произрастала семидневная пшеница Fielder. По прошествии примерно 15 мин, в течение которых происходило испарение масла из листьев пшеницы, растения помещали в темную влажную камеру (18-20оС), относительная влажность 100%) сроком на 24 ч. Затем эти растения помещали в теплицу на латентный период и через 10 дн осуществлялась оценка степени заболевания. Испытания на защитное и лечебное действие проводили с инокулированием растений за 1 и за 2 дня соответственно до опрыскивания растений испытываемыми химическими соединениями.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОБРАБОТКИ ДУБЛЕНОЙ КОЖИ | 1989 |
|
RU2025490C1 |
| СРЕДСТВО ДЛЯ СНИЖЕНИЯ ЛЕТУЧЕСТИ ИНГРЕДИЕНТОВ КОЖИ | 1992 |
|
RU2078829C1 |
| ФУНГИЦИДНОЕ СРЕДСТВО | 1991 |
|
RU2041627C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРА В ВОДНОЙ СИСТЕМЕ, СОПОЛИМЕР, КОМПЛЕКС И РЕАКЦИОННАЯ СМЕСЬ | 1995 |
|
RU2177953C2 |
| МИКРОБИЦИДНАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), А ТАКЖЕ СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2010 |
|
RU2552815C2 |
| СШИВАЮЩИЙ АГЕНТ И СПОСОБ СШИВАНИЯ СВЯЗУЮЩЕГО ПОЛИМЕРА ПОКРЫТИЯ | 1994 |
|
RU2135525C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРА | 1991 |
|
RU2091404C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРОВ | 1995 |
|
RU2228338C2 |
| Способ получения дихлортиолфосфатов | 1976 |
|
SU718012A3 |
| ПРИМЕНЕНИЕ МЕТИЛ-β-ЦИКЛОДЕКСТРИНА, СПОСОБ ОБРАТИМОГО СНИЖЕНИЯ ВЯЗКОСТИ ВОДНОГО РАСТВОРА И ЗАГУЩЕННАЯ ВОДНАЯ СИСТЕМА | 1994 |
|
RU2132344C1 |









Использование: продукт, химическое средство защиты растений. Сущность: производные S -b тиоакриламидов ф-лы I, в которой R1- C5-C10 - алкил, C5-C7 - циклоалкил, фенил, фенэтил, бензил, где арил возможно замещен хлором; Z1 и Z2 каждый независимо представляет водород, хлор, C1-C4- алкил, W1и W2 каждый COR2 и COR3, где R2-C1-C8- алкил, C1-C4 - алкоксигруппа, C1-C4-алкиламиногруппа; R3-C1-C6-алкил, C1-C4-алкоксигруппа, C3-C13-алкиламиногруппа, циклопропиламиногруппа; каждый возможно замещен одним или более хлором, метилом, метоксигруппой, нитрогруппой, цианогруппой; фениламиногруппа, возможно замещенная одним или более хлором, метилом, метоксигруппой или нитрогруппой; бензиламиногруппа, цианопентиламиногруппа, аминоаллил, аминотиенил, аминотиазонил, аминопиридил, аминоэтилпиридил, аминопиридинил, N-метилпиразолил, аминоморфолил, аминопропилморфолил, и композиция для предотвращения или ингибирования роста бактерий с использованием соединения ф-лы I в количестве 1-90 мас.%, сельскохозяйственно применимый носитель - остальное. 2 с. и 5 з.п. ф-лы, 4 табл. Структура соединения I - см. рис.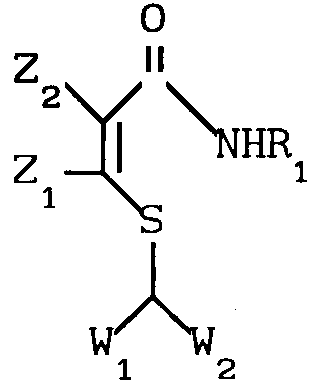
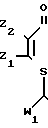

где R1 - C5 - C10-алкил, C5 - C7-циклоалкил, фенил, фенэтил или бензил, где арил возможно замещен хлором;
Z1 и Z2 - каждый независимо водород, хлор, C1 - C4-алкил;
W1 и W2 - каждый независимо соответственно COR2 и COR3, где R2-C1-C8-алкил, C1-C4-алкокси- или C1-C4-алкиламиногруппа R3-C1-C6-алкил,
C1-C4-алкоксигруппа, C3-C13-алкиламино- или циклопропиламиногруппа, причем каждый возможно замещен одним или более атомом хлора, метила, метоксигруппой, нитро- или цианогруппой, фениламиногруппа, возможно замещенная одним или более хлором, метилом, метокси- или нитрогруппой, или бензиламиногруппа, цианопентиламиногруппа, аминоаллил, аминотиенил, аминотиазолил, аминопиридил, аминоэтилпиридил, аминопиридинил, N-метилпиризолил, аминоморфолил, аминопропилморфолил.
-NH-  или NHC8H17.
или NHC8H17.
7. Композиция для предотвращения или ингибирования роста бактерий, включающая производное акриламида и сельскохозяйственный приемлемый носитель, отличающаяся тем, что в качестве производного акриламида используют S- β -тиоакриламид общей формулы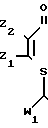

где R1 - C5-C10-алкил, C5-C7-циклоалкил, фенил, фенэтил или бензил, где арил возможно замещен хлором;
Z1 и Z2 - каждый независимо водород, хлор или C1-C4-алкил;
W1 и W2 - каждый независимо соответственно COR2 и COR3, где R2 - C1-C8-алкил, C1-C4-алкокси- или C1-C4-алкиламиногруппа, R3 - C1-C6-алкил,
C1-C4-алкоксигруппа, C3-C13-алкиламино- или циклопропиламиногруппа, причем каждый возможно замещен одним или более атомом хлора, метила, метоксигруппой, нитрогруппой или цианогруппой, фениламиногруппа, возможно замещенная одним или более хлором, метилом, метоксигруппой или нитрогруппой, бензиламиногруппа, цианопентиламиногруппа, аминоаллил, аминотиенил, аминотиазолил, аминопиридил, аминоэтилпиридил, аминопиридинил, N-метилпиразолил, аминоморфолил, аминопропилморфолил,
в количестве 1 - 90 мас.%, сельскохозяйственно приемлемый носитель - остальное.
| Патент США N 3914301, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Сплав для отливки колец для сальниковых набивок | 1922 |
|
SU1975A1 |
