Изобретение относится к органическому синтезу, в частности к способам получения хлористого винила, одного из важнейших мономеров современной химии.
В промышленной практике винилхлорид получают в основном из 1,2-дихлорэтана отщеплением хлористого водорода. В целом схема этого производства выглядит следующим образом.
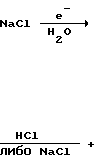 2
2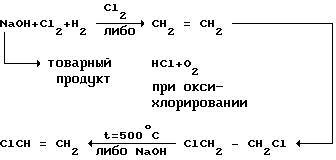 Такая схема отражает лишь принципиальную эквивалентность получения и расхода химических продуктов. В действительности товарный NaOH при пиролизе дихлорэтана частично используется в собственном производстве винилхлорида и, естественно, нарушается стехиометричность всей технологической цепочки.
Такая схема отражает лишь принципиальную эквивалентность получения и расхода химических продуктов. В действительности товарный NaOH при пиролизе дихлорэтана частично используется в собственном производстве винилхлорида и, естественно, нарушается стехиометричность всей технологической цепочки.
В виду достигнутого уровня химической науки первая и вторая стадия технологической схемы не определяют в целом экономичность и экологичность процесса, в особенности, если вторая стадия осуществляется аддитивным хлорированием этилена (более сложная ситуация складывается при получении дихлорэтана окислительным хлорированием этилена). Наиболее уязвимым узлом производства винилхлорида является непосредственно синтез винилхлорида, осуществляемый пиролизом дихлорэтана (500-550оС, давление 20-30 атм.), который служит основным источником загрязнения окружающей среды, хотя в настоящее время этот метод наиболее широко применяется в промышленности.
Термолиз дихлорэтана сопровождается большим количеством трудно утилизируемых отходов (7-10%), и для уменьшения их образования степень превращения дихлорэтана должна составлять не более 50-60%. Селективность образования винилхлорида 95-98% [1].
Однако, в условиях эксплуатации степень конверсии нередко уменьшается до 40 и даже 30%. Причиной снижения степени конверсии является состав рециркулируемого дихлорэтана, содержащего ряд примесей, получаемых при пиролизе [2]. Выделение винилхлорида ректификацией из продуктов пиролиза дихлорэтана довольно трудоемкий процесс [3-4] . После ректификации проводят дополнительную очистку винилхлорида. Например, с целью удаления ненасыщенных органических соединений с превращением их в высококипящие продукты, винилхлорид обрабатывают при 40-45оС в присутствии хлористого алюминия, оксидов алюминия, ванадия и др. соединений.
Следы хлористого водорода удаляют, смещением винилхлорида с одним из алифатических спиртов фракций С4-С11 с последующим разделением. К возвращаемому в процессе дихлорэтану предъявляются жесткие требования.
Присутствие FeCl3 (более 30 млн-1) или свободного хлора (20 млн-1) способствует закоксование труб печи. Поэтому для снижения содержания вредных примесей проводят обработку дихлорэтана в специальных реакторах очистки, где находится катализатор или адсорбент. Поступающий на стадию пиролиза дихлорэтан должен быть сухим с чистотой не менее 99,5%.
Для утилизации образующегося хлористого водорода в специальных реакторах в присутствии различных катализаторов проводят процесс оксихлорирования этилена, которому присущи также многие недостатки.
Таким образом к наиболее существенным недостаткам пиролиза дихлорэтана следует отнести:
низкая степень превращения дихлорэтана и необходимость специальной его очистки при возвращении в рецикл;
высокая температура и давление процесса;
жесткие требования к чистоте исходного дихлорэтана;
образование значительных количеств хлорорганических отходов;
сложное аппаратурное оформление и применение специальных материалов для его изготовления;
сложное регулирование и управление процессом;
невысокая чистота получаемого винилхлорида.
Жидкофазный метод дегидрохлорирования дихлорэтана, осуществляемый действием водных либо спиртово-водных растворов гидроксидов щелочных металлов практически лишен недостатков характерных для пиролизного метода.
Однако периодичность жидкофазного дегидрохлорирования, применение избытка NaOH и в особенности утилизация образующегося NaCl являются причинами препятствующими широкому промышленному применению этого метода получения винилхлорида.
Например, в известном способе [5], получение хлористого винила осуществляется щелочным дегидрохлорированием 1,2-дихлорэтана. Исходными продуктами по этому методу служит чистый перегнанный дихлорэтан, техническая 42%-ная каустическая сода и технический метанол.
Реакция между дихлорэтаном и едким натром проводится периодически в реакторе, представляющим собой вертикальный цилиндрический сосуд, снабженный рубашкой для обогрева паром и мешалкой. Для достижения полного превращения дихлорэтана количество едкого натра должно на 15-20% превышать теоретическое. Метанол для лучшего использования объема реакционного сосуда, берут в минимальном количестве, обеспечивающим достаточную скорость реакции. Практически это составляет примерно 500 л 100%-ного метанола на 1 т. едкого натра.
Процесс проводят при температуре 85-90оС, под избыточным давлением до 3 атм. , при перемешивании. Образующийся хлористый винил отгоняется от реакционной массы через холодильник и поступает на ректификацию.
Как видно, проведение процесса требует аппаратурного оформления под давлением, хотя оно и не велико. Кроме того, в процессе производства расходуется токсичный метанол, который в реакции образования хлористого винила не участвует. Большим недостатком этого метода является периодичность процесса; что резко снижает производительность.
Целью изобретения является создание производства винилхлорида из этилена и хлорида натрия по малоотходной, экологически чистой, высокопроизводительной циклической схеме, при стехиометрическом расходе гидроксида натрия к дихлорэтану с получением винилхлорида и каустической соды в эквимолярных количествах.
CH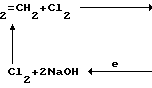
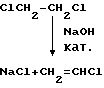
Как видно с учетом стехиометрического расхода реагентов, такая схема производства позволяет на каждый эквивалент винилхлорида получать дополнительно эквивалент товарной гидроокиси натрия, что в принципе стимулирует наращивание мощности по этим важным химическим продуктам.
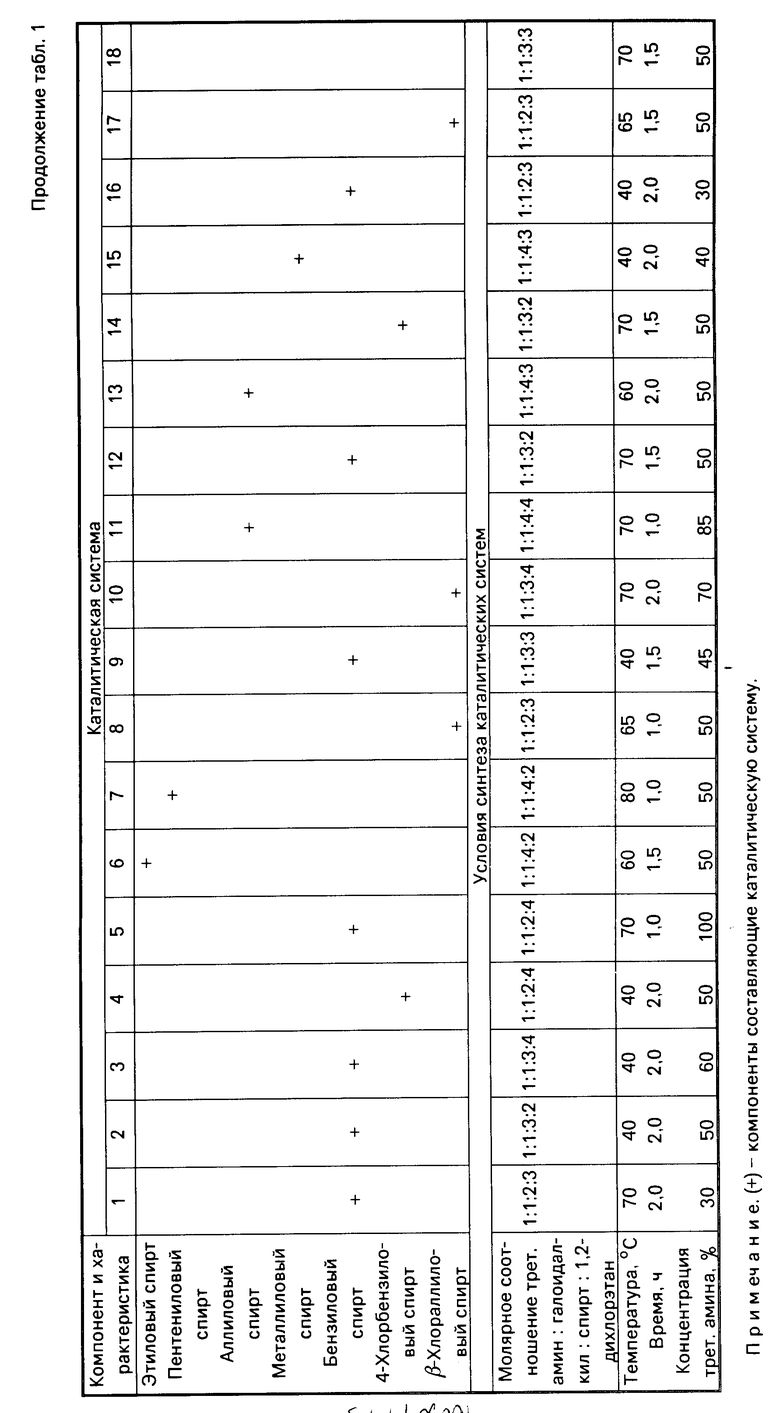
Поставленная цель достигается тем, что дегидрохлорирование 1,2-дихлорэтана проводят водным гидроксидом натрия непрерывно, в каскаде реакторов при температуре 20-70оС, при конверсии субстрата 80-95%, с применением 0,09-0,3 мас. % каталитической системы, получаемой взаимодействием водного или безводного амила (формулы - R1R2R3N, где R1R2R3 - алкил С1-С5, аллил, γ-хлораллил, бензил, непредельные С4-С5 - углеводороды), галоидалкила (преимущественно аллильного или бензильного типа), спирта (преимущественно содержащего непредельную или ароматическую группу в β-положении к атому кислорода) и 1,2-дихлорэтан при их молярном соотношении 1:1:(2-4):(2-4) соответственно.
Получаемая каталитическая система обеспечивает высокую скорость дегидрохлорирования, аналогично применяемым ранее алкоксидам, причем метод получения системы значительно проще чем синтез алкоксидов, а каталитическая активность сохраняется при длительном хранении и повышенной температуре. С учетом достаточно хорошо освоенными в промышленности хлорирования этилена и электролиза NaCl, разработанный каталитический метод позволяет на практике реализовать сбалансированное по хлору производство винилхлорида.
Сущность изобретения заключается в следующем.
Получаемый после узла хлорирования дихлорэтан (99,5-99,8%) без дополнительной очистки и осушки подается в первый реактор, каскада из двух реакторов. В реактор подается 10-50%-ный раствор водного гидроксида натрия при молярном соотношении к дихлорэтану 1:(2,2-3), либо 1:(0,25-0,4) и 0,09-0,4 мас. % каталитической системы. Расход каталитической системы ведется в расчете на третичный амин, взятый для получения этой системы, к дихлорэтану. Образующийся в реакторах хлористый винил через обратные холодильники поступает в ректификационную колонну для отделения увлеченных дихлорэтана и воды. Хлористый винил высокой чистоты с верха колонны выводится с установки. Реакционная масса из первого реактора подается во второй реактор, в котором достигается заданная конверсия дихлорэтана. Далее реакционная масса поступает в разбавитель, куда подается расчетное количество воды. После чего, поток поступает в фазоразделитель, где разделяется на органический слой и щелока. Органический слой, содержащий дихлорэтан и 85-95% поданного катализатора, возвращается в первый реактор, щелока поступают в выпарной аппарат, в котором каустик укрепляется до 35-50%-ной концентрации, для возврата в процесс. Часть отгонной воды из выпарного аппарата используется для растворения соли в разбавителе, другая часть для растворения соли после центрифуги, выделяющаяся при выпарке щелоков соль отфильтровывается, разбавляется водой и направляется на электролиз.
Особо следует отметить, что получаемый по предлагаемой технологии хлорид натрия не требует дополнительной очистки.
П р и м е р 1. Синтез каталитической системы.
В термостатируемый реактор, снабженный обратным холодильником и перемешивающим устройством, помещают 25,30 г (250 ммоль) триэтиламина. При перемешивании и комнатной температуре прибавляют 19,30 г (250 ммоль) бензилхлорида, 81,08 г (750 ммоль) бензилового спирта и 99 г (1 моль) 1,2-дихлорэтана. Смесь при перемешивании и 80оС выдерживают 1 ч. После охлаждения получают гомогенную, прозрачную жидкость.
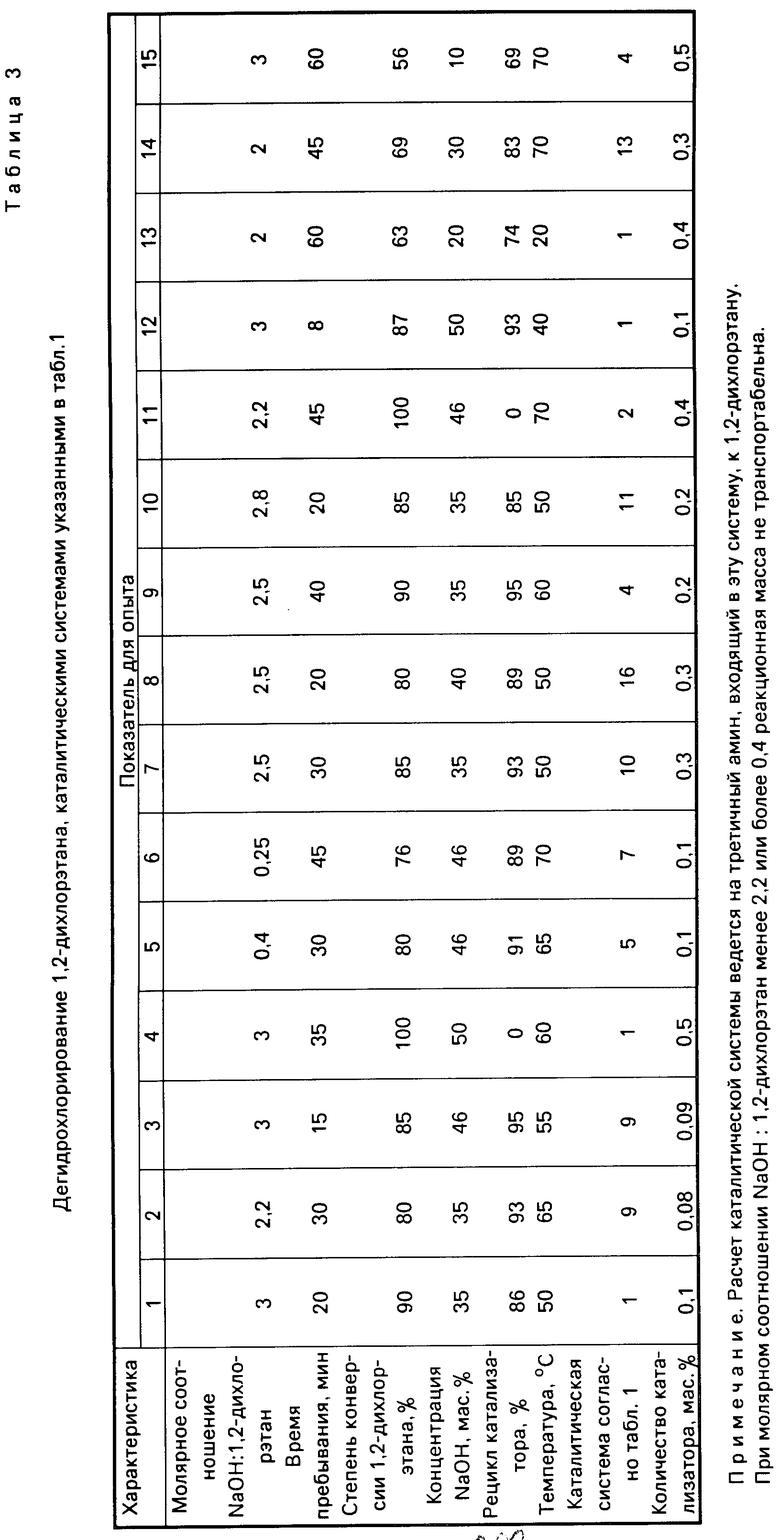
Другие примеры и составы катализаторов щелочного дегидрохлорирования 1,2-дихлорэтана приведены в табл. 1.
Кроме указанных в примере 1 и табл. 1 катализаторов дегидрохлорирования, в предлагаемом способе получения хлористого винила, возможно применение катализаторов известных способов.
П р и м е р 2. Синтез винилхлорида.
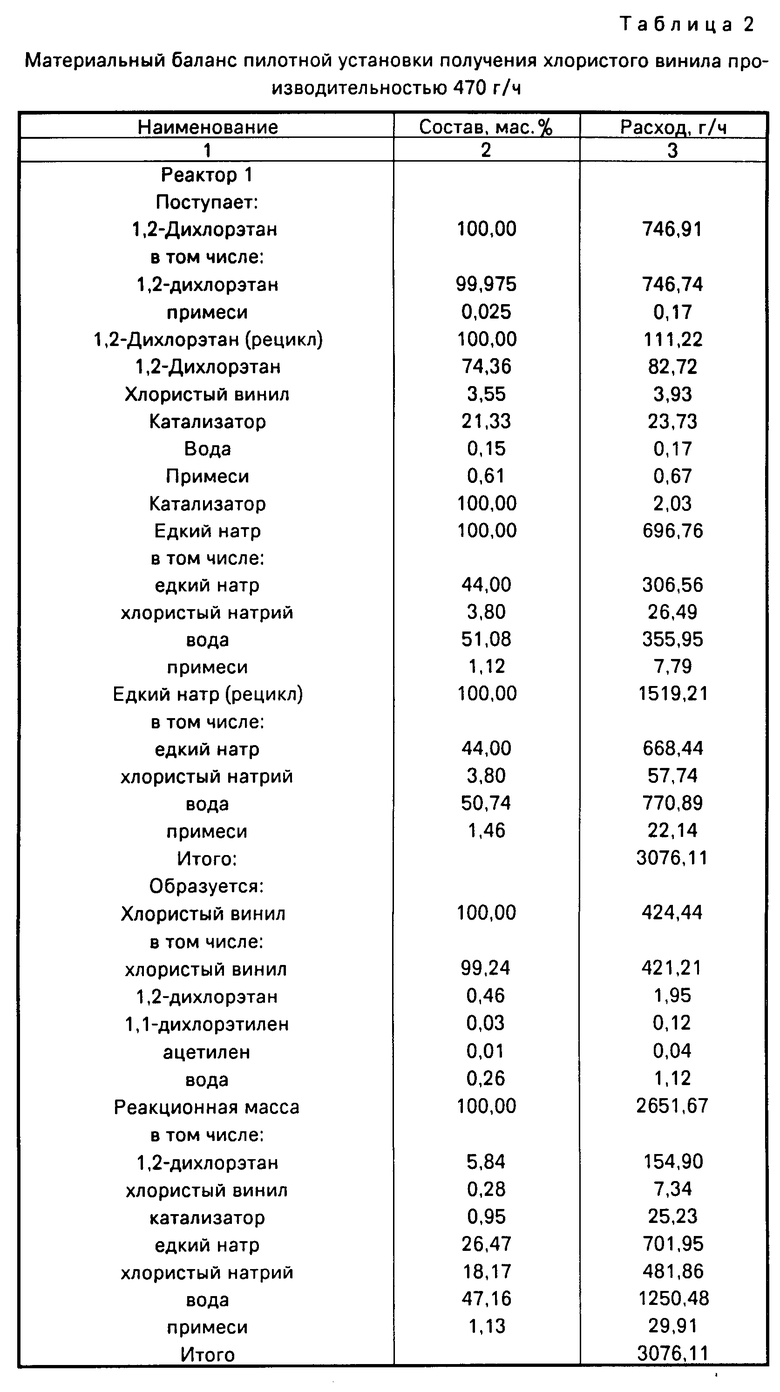
Дегидрохлорирование 1,2-дихлорэтана проводили в каскаде из двух реакторов емкостью 1 л каждый. На установке использовался 1,2-дихлорэтан чистотой 99,975% ГОСТ 1942-74, едкий натр технический 44% ГОСТ 2263-79 и катализатор, приготовленный в условиях примера 1, представляющий смесь 50,60 г триэтиламина, 36,60 г бензилхлорида, 162,16 г бензилового спирта и 198 г 1,2-дихлорэтана.
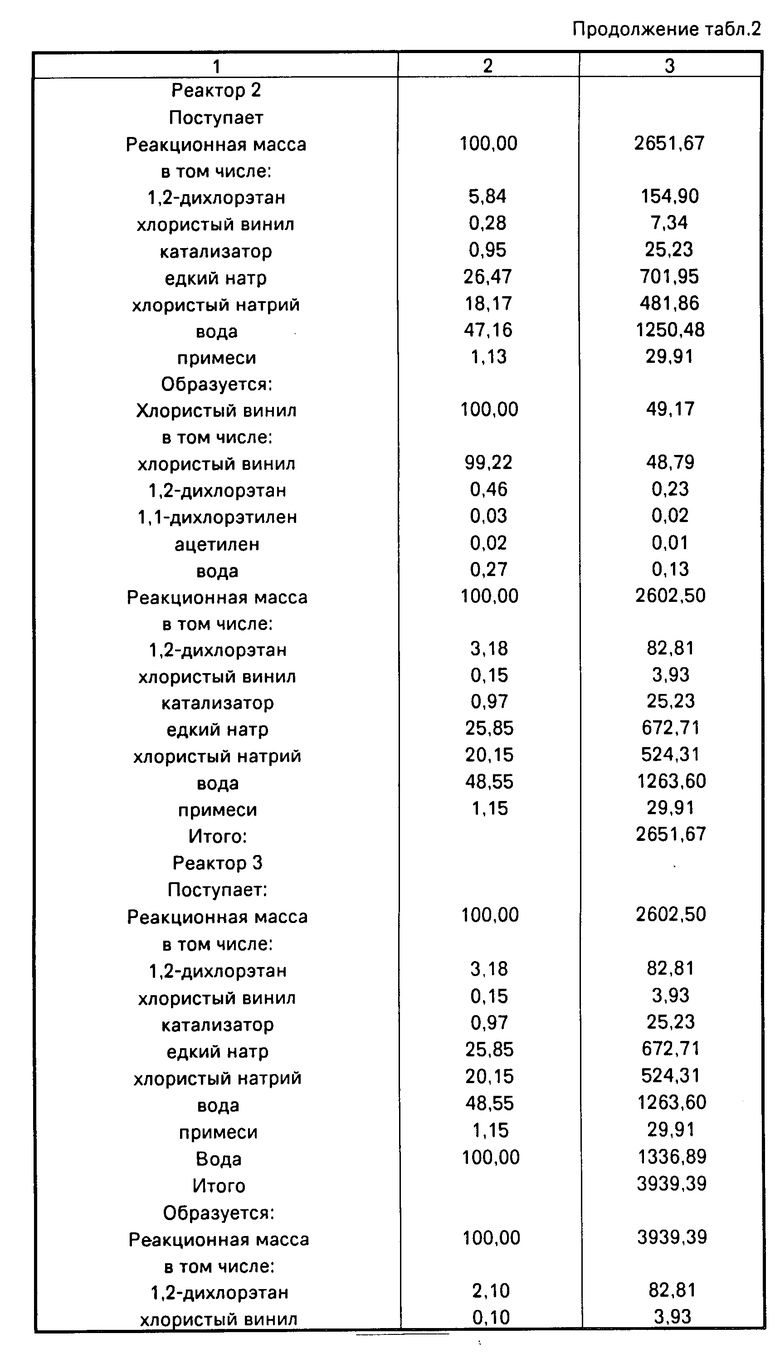
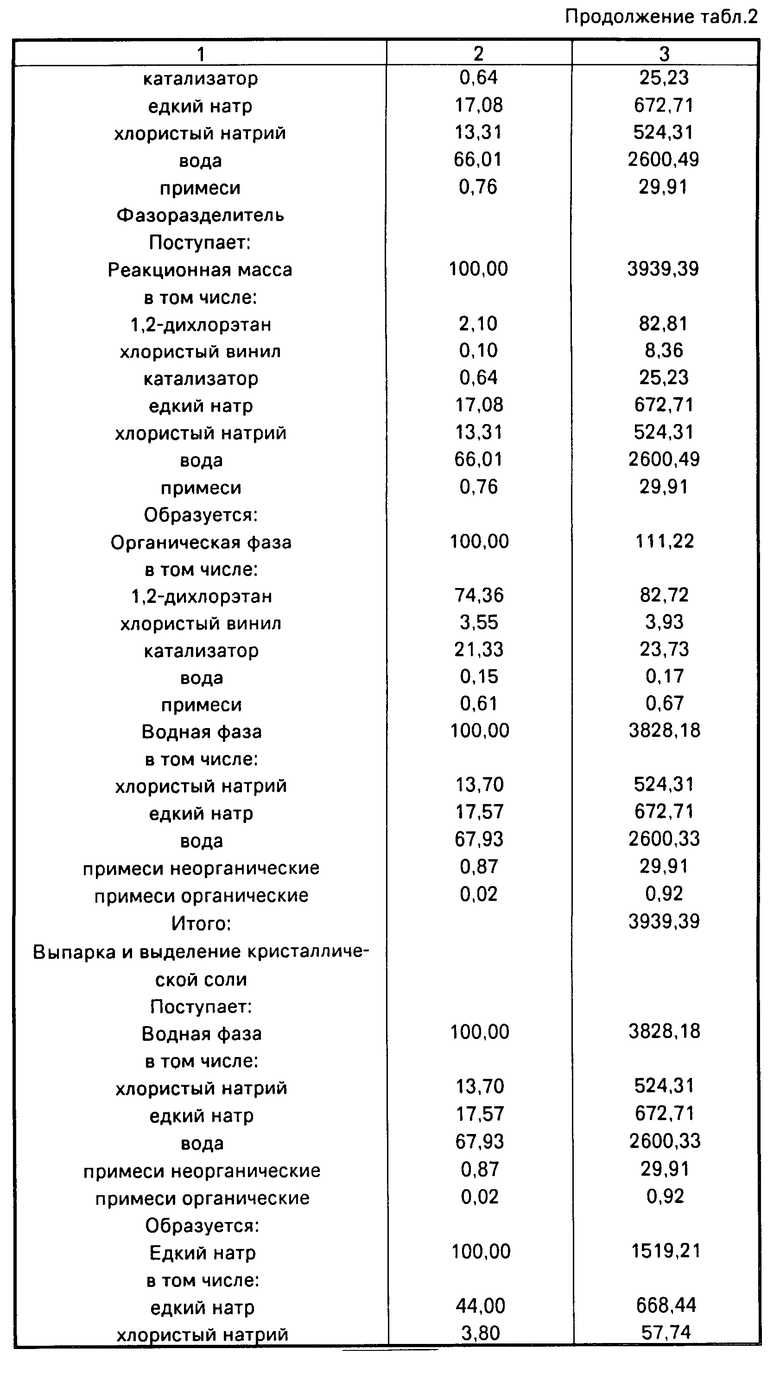
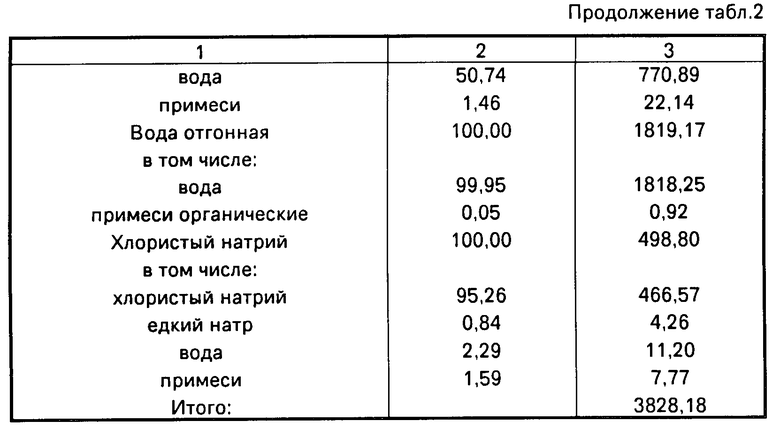
В первый реактор дозирующими насосами непрерывно подавали 764,91 г/ч свежего и 111,22 г/ч возвратного дихлорэтана, 221,91 г/ч 44,00%-ного едкого натра и 2,03 г/ч катализатора. Температура в первом и втором реакторе поддерживалась 45оС, в смесителе 18-20оС. Время пребывания в каждом реакторе составляло 15 мин, при конверсии дихлорэтана 90%. Образующийся хлористый винил 473,61 г/ч через обратный холодильник и газовый счетчик выводился с установки. В смеситель в количестве 1336,89 г/ч подавалась вода. Реакционная масса из смесителя поступала в фазоразделитель, где органическая фаза отделялась от водной. Органическая фаза, содержащая 91% катализатора возвращалась в первый реактор. Водная фаза 3828,18 г/ч подавалась в выпарной аппарат. Едкий натр укреплялся до 44%, фильтровался от хлористого натрия и возвращался в первый реактор. Хлористый натрий 498,80 г/ч и отгонная вода 1819,17 г/ч выводилась с установки. Материальный баланс представлен в табл. 2.
Другие примеры и основные показатели процесса представлены в табл. 3.
Таким образом изобретение имеет следующее преимущество: проведение процесса дегидрохлорирования дихлорэтана в непрерывном режиме при стехиометрическом соотношении реагентов; высокая чистота получаемого винилхлорида; простой и удобный метод получения высокоактивной каталитической системы; низкий расход катализатора; применение технологического оборудования, не требующего для его изготовления специальных материалов; реализуется возможность создания производства винилхлорида из этилена и хлорида натрия по малоотходной экологически чистой технологической цепочке предусматривающей электролиз NaCl, хлорирование этилена и дегидрохлорирование дихлорэтана; стимулируется увеличение мощности по винилхлориду за счет повышения выпуска товарного NaOH, при полном потреблении хлора на месте его производства.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРОВ ВОДНО-ЩЕЛОЧНОГО ДЕГИДРОХЛОРИРОВАНИЯ | 2003 |
|
RU2247601C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРИРОВАННЫХ ПРОИЗВОДНЫХ ЭТИЛЕНА | 2005 |
|
RU2288909C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛХЛОРИДА И КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2338736C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛХЛОРИДА | 1993 |
|
RU2072976C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВИНИЛХЛОРИДА | 1997 |
|
RU2129115C1 |
| КАТАЛИТИЧЕСКИЙ СПОСОБ ПЕРЕРАБОТКИ МЕТАНА | 2008 |
|
RU2394805C2 |
| ИНТЕГРИРОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ ВИНИЛХЛОРИДА | 2001 |
|
RU2184721C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ХЛОРА ИЗ ОТХОДОВ В ПРОИЗВОДСТВЕ ХЛОРА И ВИНИЛХЛОРИДА | 2012 |
|
RU2498937C1 |
| СПОСОБ КАТАЛИТИЧЕСКОГО ОКСИХЛОРИРОВАНИЯ ЭТАНА ДО ВИНИЛХЛОРИДА | 1994 |
|
RU2133729C1 |
| СПОСОБ ПРОИЗВОДСТВА ВИНИЛХЛОРИДА ИЗ ЭТАНА И ЭТИЛЕНА (ВАРИАНТЫ) | 2000 |
|
RU2259989C2 |

Использование: в производстве галоидуглеводородов, в частности в способе получения хлористого винила -мономера для синтеза полимерных материалов. Сущность изобретения: способ предусматривает реакцию 1,2-дихлорэтана с водным раствором гидроксида натрия в присутствии каталитической системы. Последнюю получают из третичных аминов, галоидалкилов и спиртов при 40-80°С с конверсией 80-90%. 2 з.п. ф-лы, 3 табл.
| Хрулев М.В | |||
| Поливинилхлорид, М.: Химия, 1964, с.264. |

