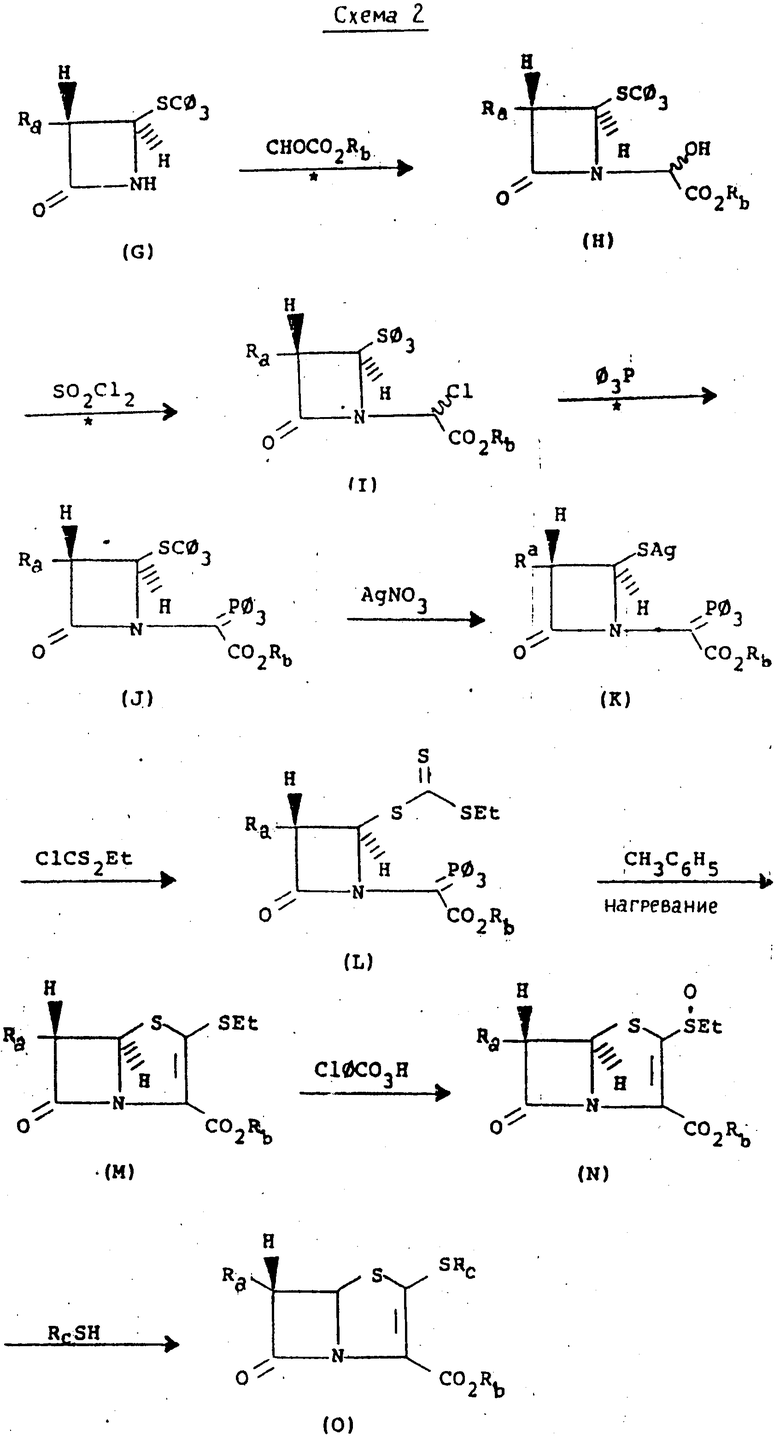
Изобретение относится к эффективным многостадийным способам получения соединений формулы (6), как показано на схеме 3 ниже. Соединения формулы (6) полезны как предшественники различных пенемных антибиотиков.
Ранее был описан ряд способов получения пенемных антибиотиков, замещенных в положение 2 алкильной группой или тиоэфирной группой, - SR2, как видно из формулы (6), представленной ниже. Два или более общих способов их получения представлены на схемах 1 и 2. На схеме 1 альтернативным интермедиатом серебряной соли меркаптана является сам меркаптан, который получают Zn/H+ восстановлением тритилированного тиола (Girijavallabhan и др., J. Antibiotics 39, 1182 (1986), патент США 4584133).
Ссылки: Girijavallabhan et al. - J.Antibiotics 39, 1182 (1986) патент США 4584133, где
Rd-CH Re - -CH2CH=CH2,
Re - -CH2CH=CH2,
Rg =C2H5, CH2N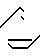 N Rf = бета-нафтил, Ха - отщепляемая группа.
N Rf = бета-нафтил, Ха - отщепляемая группа.
DiNinno et al., патент США 4610823 (1986), Leanza et al., Tetrahedron 39, 2505 (1983) где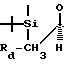 Re - -CH2CH=CH2, или -СH2 ⊘ NO2, Rf = C6H5 Rg - алкил, аралкил и т.п.,
Re - -CH2CH=CH2, или -СH2 ⊘ NO2, Rf = C6H5 Rg - алкил, аралкил и т.п.,
Ха - отщепляемая группа.
См. также Girijavallabhan et al., патенты США 4 443 373 и 4 530 793, в которых представлен альтернативный синтез соединений (Е), где Rd представляет собой CH3CHOH- и Re представляет собой CH2CH=CH2 или CH2CH2OSi(CH3)3, из соединения (А).
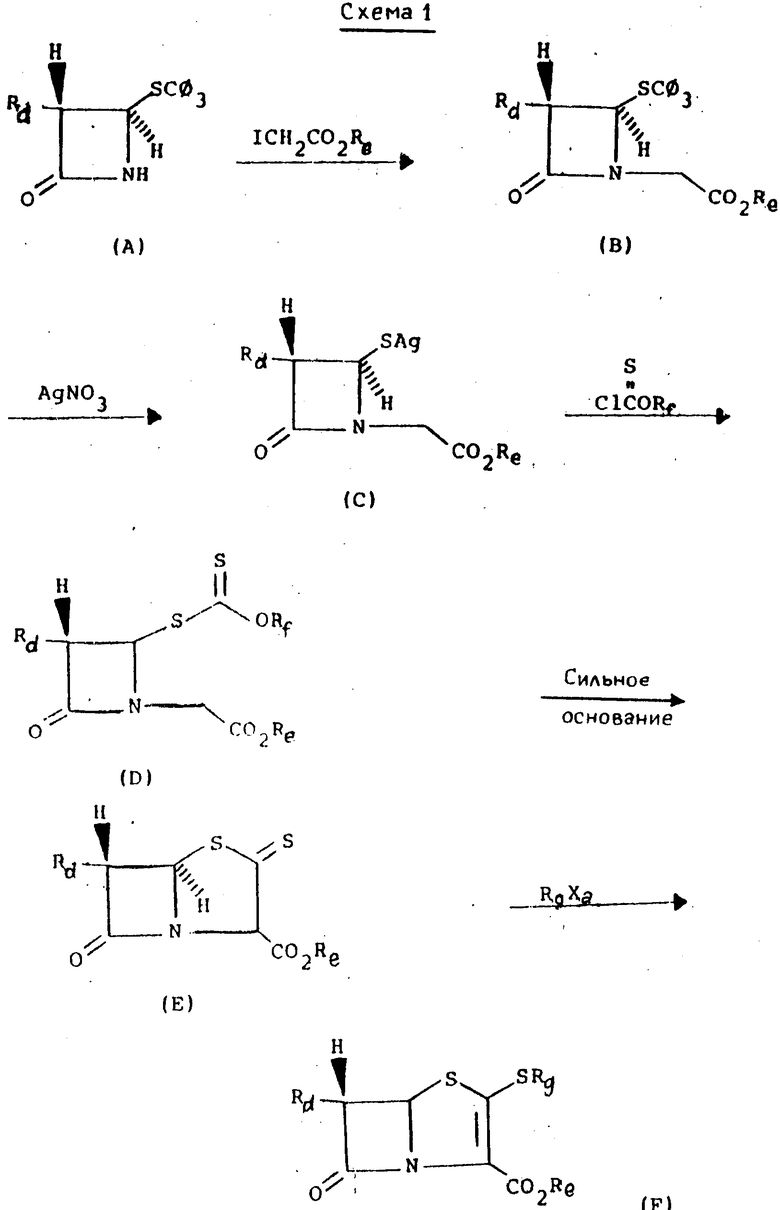
Ссылка: DiNinno et al., Tetrahedron Letters 23, 3535 (1982) где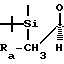 , Rb=-CH2CH=CH2,
, Rb=-CH2CH=CH2,
Rc=-CH(CH3)2, - СН2СН2OH, и т.д.
Эти стадии приведены на основании сноски 16 в ссылке на патент Великобритании 2 042 514.
См. также Ganguly et al., - J. Antimicrob. Chemo. 9, Suppl. Cl, (1982), где используются некоторые аналогичные стадии в другой последовательности.
Ghoser и др., Tetrahedron Letters 39, 2493 (1983) описали синтез 2-оксопенамов из пенициллины G и превращение в 2-алкоксипенемные производные пенициллина G. Японец Kokai 84-115, 788 (Chem. Abst 96; 34979y, Derwent аbst. 78700D) также описал превращение гидрокси и карбокси защищенных 6-(1-гидроксиэтил)-2-оксо-пенамов в соответствующие алкоксианалоги.
Кроме того, альтернативные способы синтеза пенемов включают способы, описанные Dextraz et al. в патенте США 4 769 451, Piriе et al. в патенте США 4 751 297, Volkmann et al., в патенте США 4 739 047, Brighty в патенте США 4 695 626, Brighty et al., в патенте США 4 782 145, Perrone et al. - J. Org. Chem. , 51, 3413 (1986); Batastini et al. в патенте США 4 631 150, заявке на патент Великобритании 2 187 448, Alpe giani et al, в патенте США 4 577 016 и Franceschi et al. - J. Antibiotics 36, 938 (1983).
В литературе имеются многочисленные сообщения, относящиеся к превращению 2-оксокарбапенамов и 3-оксоцефамов в 2-(алкилтио)-2-карбапенемы и 3-алкилтио- 3-цефемы через енольные эфиры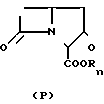 ___→
___→
 Rj где Rh представляет собой обычную карбоксизащитную группу;
Rj где Rh представляет собой обычную карбоксизащитную группу;
Ri представляет собой, например, дефинил- или диэтилфосфорил, тозил, мезил или трифторметансульфонил.
См. например Sletzinger et al., Tetrahedron Letters 21, 4221 (1980); Andrus et al. - J. Am. Chem. Soc. 106, 18080 (1984); Evans et al., Tetrahedron Letters 26, 3787 (1985) и 27, 3119 (1986) и патент США 4 673 737, Ratcliff et al., 21, 1193 (1980); Melillo et al., ibid. 21, 2783, (1980); Iimori et al. и J. Am. Chem. Soc. 105, 1659 (1983).
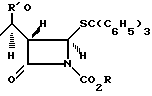
Однако химия этих карбапенемовых кетонных групп несовместима с тиолактонкарбонильной группой 2-оксопенемов. Например, в результате реакции мезилхлорида или мезил ангидрида с соединением типа (4) получают соединение типа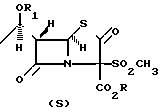 в то время, как тозилхлорид или трифлилхлорид и соединение типа (4) дают соединение типа
в то время, как тозилхлорид или трифлилхлорид и соединение типа (4) дают соединение типа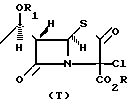 Недавно в опубликованной заявке на Европейский патент 257 419 было сообщено, что соединение типа (4), представленное ниже, ввели во взаимодействие с дифенилфосфорилхлоридом и получили дифенилфосфориловый эфир на месте, который затем ввели во взаимодействие с фенолом и получили соединение типа
Недавно в опубликованной заявке на Европейский патент 257 419 было сообщено, что соединение типа (4), представленное ниже, ввели во взаимодействие с дифенилфосфорилхлоридом и получили дифенилфосфориловый эфир на месте, который затем ввели во взаимодействие с фенолом и получили соединение типа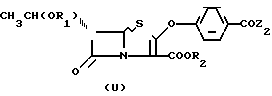 с очень малым выходом. В этой заявке не предложено подтверждение более широкого использования других сильных энольный эфир образующих реагентов, таких как трифлил хлорид, который является известным хлорирующим реагентом, а не трифлат эфир образующим реагентом (vide supra; и Hakimelahi et al., Tetrahedron Letters 1979, pp. 3643-3644).
с очень малым выходом. В этой заявке не предложено подтверждение более широкого использования других сильных энольный эфир образующих реагентов, таких как трифлил хлорид, который является известным хлорирующим реагентом, а не трифлат эфир образующим реагентом (vide supra; и Hakimelahi et al., Tetrahedron Letters 1979, pp. 3643-3644).
Мы открыли эффективный многостадийный способ синтеза пенемовых соединений, который представлен на схеме 3.
На схеме 3 различные обозначения имеют следующие значения:
R представляет собой -CH2CX=CH2, или -CH2CH2Si(CH3)3;
X - представляет собой Н;
R1 представляет собой силилзащитную группу,
R2 представляет собой С1-С4 алкил, 2-гидроксиэтил, 2-(n-нитробензилкарбониламино)-этил, 1-(n-нитробензилкарбонил) -3S-пирролидинил, 1R-оксо-3S-тиоланил или 1,1-диоксо-3R-и-3S-тиоланил.
Среди обычных силилзащитных групп находятся триметилсилил и диметил-трет-бутилсилил. Последний наиболее предпочтительный в связи с легкостью введения и удаления, одновременно обладая превосходной стабильностью как защитная группа в течение различных других стадий настоящего изобретения.
Изобретение представляет эффективный способ получения.
На первой стадии этого способа трифенилметилтио - соединение формулы (1) в присутствии одного или более молярных эквивалентов слабого основного амина, такого как пиридин, в темноте вводят во взаимодействие с нитратом серебра (по меньшей мере один молярный эквивалент, обычно в избытке, например 1,5-2-молярных эквивалента) и получают серебряную соль соответствующего меркаптана. Эту реакцию обычно проводят в реакционно инертном растворителе, таком как метанол. Температура не критична, но более низкие температуры, например - 25 до 25оС обычно являются предпочтительными, причем температуры в диапазоне 0-5оС особенно удобны и удовлетворительны. Обычно без выделения из реакционной среды промежуточную соль серебра превращают непосредственно при избытке газообразного сульфида водорода в меркаптан. Серебро выводится в виде сульфида с помощью фильтрования и меркаптан (2) выводится из меточной жидкости обычными способами, такими как экстракция и выпаривание растворителя.
Как используется в данном описании, термин "реакционно-инертный растворитель" относится к растворителю, который не взаимодействует с исходными материалами, реагентами, интермедиатами или продуктами таким образом, который мог бы оказать неблагоприятное воздействие на выход целевого продукта.
На второй стадии меркаптан (2) вводят во взаимодействие с одним молярным эквивалентом 4-нитрофенилхлорформата и получают промежуточное соединение формулы (3). Эту стадию проводят в присутствии одного молярного эквивалента третичного амина, предпочтительно диизопропил - этиламина и/или диметиламинопиридина, обычно в реакционно инертном растворителе, таком как тетрагидрофуран, и обычно реакцию проводят при пониженных температурах, например (- 25)-(+ 25)оС, удобнее при температурах в диапазоне 0-5оС. Если желательно, интермедиат (3) выделяют и определяют обычными способами. Однако предпочтительно просто использовать первоначально полученный раствор соединения формулы (3) непосредственно на следующей стадии.
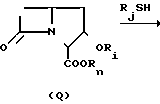
На третьей стадии интермедиат (3) подвергают циклизации в присутствии сильного основания и получают желаемый 2-оксопенем формулы (4), известное соединение, например, когда R представляет собой аллил. Предпочтительно эту стадию проводить раствором соединения формулы (3) в реакционно инертном растворителе, таком как тетрагидрофуран. Предпочтительным сильным основанием является литий гексаметилдисилиламид в том же реакционно инертном растворителе, обычно используемом в большом избытке (например 3-5 молярных эквивалента). Это основание, обычно имеющееся на рынке в виде 1 М раствора в тетрагидрофуране, разбавляют (например до 0,1-0,2 М) тетрагидрофураном и охлаждают до более низкой температуры (например, - 50 до - 100оС, обычно до 78оС, температура бани ацетон-сухой лед). Раствор соединения формулы (3) в том же растворителе добавляют по частям, поддерживая ту же низкую температуру. Реакцию, которая по существу завершается после завершения добавления, обычно гасят избыточным количеством уксусной кислоты и 2-оксопенем (4) выделяют обычными способами концентрации и экстрагирования.
На следующей стадии 2-оксопенем (4) вводят во взаимодействие со свежедистиллированным трифликовым ангидридом, обычно в молярном избытке, при пониженной температуре (от 0 до - 90оС, обычно - 78оС) в реакционно-инертном растворителе, таком как метилен хлорид, в присутствии молярного избытка (обычно 4-6 молярных эквивалентов) третичного амина, предпочтительно диизопропилэтиламин. Если желательно, полученный энольный трифлатовый эфир формулы (5) выделяют с помощью хроматографии реакционной смеси на силикагеле и идентифицируют. Однако в этом нет необходимости, так как реакционный раствор хорошо подходит для непосредственной реакции с соответствующим реагентом на следующей стадии.
На пятой стадии предлагаемого способа в одном из его предпочтительных воплощений раствор соответствующего маркаптина, R2SН, обычно растворенного в том же реакционно инертном растворителе, таком как метилен хлорид, добавили по частям в холодный раствор трифлатного эфира (5), позволяя температуре подняться не выше чем на 10-40оС от ее первоначального значения от 0 до - 90оС. После завершения реакции желаемый пенемовый интермедиат формулы (6) выделяют обычными способами, как показано ниже.
Меркаптаны, необходимые для данной последовательности реакций, известны или их можно получить обычными способами. Предпочтительные способы синтеза 3S-меркаптотиолана 1R-оксида описаны ниже.
П р и м е р 1. Аллил-2-(4R-меркапто-3S-(1R-(диметил-трет-бутилсилилокси)-этил)- 2-азетидинон-1-ил)ацетат.
Раствор 20 г (33,2 ммоль) аллил 2-(4R-(трифенилметилтио)-3S-(1R-(диметил-трет-бутилсилилокси)этил)- 2-азетидион-1-ил)ацетата (Jeff et al., Tetrahedron, vol. 39, 2505-2513, 1983, патент США 4 610 823) в 600 мл метанола охладили до 0оС, и обработали 5,94 мл (73 ммоль) пиридина. Следующую реакцию провели в колбе, защищенной от света. В указанный раствор добавили твердый нитрат серебра (10,2 г, 60 ммоль) и реакционную смесь оставили перемешиваться в течение 1,5 ч, поддерживая температуру 0оС. Как только реакция закончилась, при постоянном перемешивании медленно ввели газообразный сероводород. Темную смесь затем профильтровали через целит, сульфид серебра удалили, а фильтрат сконцентрировали. Органический остаток разделили между этилацетатом и рассолом. Слои разделили и водную фазу реэкстрагировали свежим этилацетатом. Объединенные органические слои обезводили над сульфатом натрия и затем выпаривали и получили указанное в названии соединение, которое непосредственно используют на следующей стадии.
П р и м е р 2. Аллил 2-(4R-(4-нитрофенилоксикарбонилтио)-3-(1R-диметилтрет- бутилсилилокси)этил)-2 азетидинон-1-ил) ацетат.
Раствор 4,06 г (33,2 ммоль) диметиламинопиридина и 6,69 г (33,2 ммоль) 4-нитрофенилхлороформата приготовили в 700 мл ТГФ. Раствор охладили до 0оС и обработали последовательно раствором полной загрузки указанного в названии предыдущего примера продукта в 60 мл ТГФ и отдельным раствором 5,78 мл (33,2 ммоль) диизопропилэтиламина в 60 мл ТГФ. Это добавление заняло 0,5 ч и в результате получили белый осадок. После перемешивания смеси в течение 5 мин ее профильтровали в изоляции от атмосферной влаги и профильтрованный раствор указанного в названии продукта поместили в капельную воронку и немедленно использовали на следующей стадии.
Часть этого раствора после фильтрования через малую порцию силикагеля, используя CDCl3 в качестве элюента, подвергли 1Н-ЯМР (300 МГц) и получили следующие значения дельты: 8,22 (2Н, д, J = 8 Гц), 7,29 (2Н, д, J = 8 Гц), 5,74-5,89 (1Н, ддд, J = 18 Гц, 12 Гц, J = 6 Гц), 5,46 (1Н, д, J = 2 Гц), 5,25 (1Н, д, J = 18 Гц), 5,17 (1Н, д, J = 12 Гц), 4,57 (2Н, д, J = 6 Гц), 4,25 (1Н, дв. кв. J = 5 Гц, J = 6 Гц), 4,10 (1Н, д, J = 10 Гц), 3,90 (1Н, д, J = 19 Гц), 3,27 (1Н, дв. д, J = 5 Гц, J = 2 Гц), 1,26 (3Н, д, J = 6 Гц), 0,84 (9Н, с), 0,06 (3Н, с), 0,04 (3Н, с).
П р и м е р 3. Аллил 5R, 6S-2-оксо-6-(1R-(диметил-трет-бутилсилилокси) этил)пенам-3-карбоксилат.
Весь раствор продукта из предшествующего примера добавили в 133 мл (133 ммоль), 1,01 М гексаметилдисилиламида лития (в ТГФ), который предварительно разбавили 1000 мл ТГФ и охладили до - 78оС. Добавление требовало 0,5 ч и раствор стал светло-желтым. Добавили уксусную кислоту (38 мл, 664 ммоль) и реакционную смесь перемешивали 10 мин. Примерно половину растворителя удаляли концентрацией и остаток разбавили диэтиловым эфиром до объема 2,7 л. Эфировый раствор промыли насыщенным раствором бикарбоната, насыщенным раствором рассола, обезводили над сульфатом натрия. Органическую фазу сконцентрировали и остаток профильтровали через слой силикагеля, элюируя 15% этилацетатом в гексане. Получили 6,89 г (56%) целевого продукта в виде воскового твердого вещества; т.пл. 45-48оС.
1Н-ЯМР (CDCl3, 300 МГц) дельта): 5,78-5,94 (1Н, дв, дв. д J = 18 Гц, J = 11 Гц, J = 7 Гц), 5,51 (1Н, д, J = 2 Гц), 5,32 (1Н, с), 5,25 (1Н, д, J = 11 Гц), 5,25 (1Н, д, J = 11 Гц), 5,00 (1Н, с), 4,65 (2Н, д, J = 7 Гц), 4,32 (1Н, дв. т. J = 7 Гц, J = 4 Гц), 3,54 (1Н, дв. д) J = 4 Гц, J = 2 Гц), 1,28 (3Н, д, J = 7 Гц), 0,86 (9Н, с), 0,07 (3Н, с), 0,05 (3Н, с);
С13-ЯМР (CDCl3, 75, 43 МГц) дельта: 199,0, 169,0; 163,4; 130,4; 119,6; 71,7; 67,1; 66,1; 64,6; 62,3; 25,6; 22,5; 17,9; - 4,2; - 5,1; м/е, вычислено для C13H18NO5SSi (P - tBu) 328, 0675; найдено 323, 0615.
П р и м е р 4. Аллил 5R, 6S-6 (1R-(диметил-трет-бутилсилилокси) этил)-2-(трифторметансульфонилокси)пенем-3-карбокси- лат.
Раствор 100 г (0,260 ммоль) указанного в названии предыдущего примера продукта в 5 мл метиленхлорида обработали 0,180 мл (1,03 ммоль) диизопропилэтиламина. Этот прозрачный раствор затем охладили до - 78оС в бане из ацетона-сухого льда. Добавили свежедистиллированный трифликовый ангидрид (0,045 мл, 0,270 ммоль) и прозрачный раствор перемешивали в течение 1 ч при - 78оС и получили холодный раствор указанного в названии продукта, который непосредственно использовали на следующей стадии. Малую порцию этого раствора очистили с помощью хроматографии на силикагеле, после чего провели низкотемпературную (- 78оС) кристаллизацию из патента; т.п. 40оС;
1Н-ЯМР (CDCl, 300 МГц) дельта: 5,84-5,98 (1Н, ддд, J = 18 Гц), J = 12 Гц, J = 6 Гц), 5,73 (1Н, д, J = 2 Гц), 5,37 (1Н, дд, J = 18 Гц, J = 1 Гц), 5,25 (1Н, дд, J = 12 Гц, J = 1 Гц), 4,73 (2Н, дд, J = 6 Гц, J = 1 Гц), 4,25 (1Н, дв. кв., J= = 6 Гц, J = 4 Гц), 3,86 (1Н, дд, J = 4 Гц, J = 2 Гц), 1,24 (3Н, д, J = 6 Гц), 0,87 (9Н, с), 0,08 (6Н, с); m/e вычислено для C14H17NO7S2SiF3/P-t Bu/: 460, 0168, найдено: 460, 0246.
П р и м е р 5. Аллил 5R, 6S-6-(1R-(диметил-трет-бутилсилилокси)этил)-2-((1R-ок- со-3S- тиоланил)тио)пенем-3-карбоксилат
Раствор 69 г (0,388 ммоль) 3S-(ацетилтио)тиолан-1R-оксила в 5 мл метилен хлорида обрабатывали 5 мл воды и охладили до 0оС. В перемешиваемую смесь добавили 0,78 мл (1,56 ммоль) 2,0 М гидроокиси натрия и дали выстоять в течение 0,5 ч. Реакционную смесь погасили 0,089 мл (1,56 ммоль) уксусной кислоты и экстрагировали 5 х 10 мл метиленхлорида. Органическую фазу обезводили с помощью сульфата натрия, профильтровали и обработали 0,135 мл (0,780 ммоль) диизопропилэтиламина. Этот раствор 3S-меркапттиолан-1R-оксида оставили выстояться, пока не завершили процедуру предыдущего примера. Затем этот раствор добавили в холодный предыдущего примера в течение 0,5 ч, поддерживая температуру ниже - 65оС в течение всего времени. Через 18 ч при - 78оС реакционную смесь обработали 10 мл воды и оставили нагреваться до комнатной температуры. Продукт экстрагировали метиленхлоридом и органическую фазу промыли рассолом, затем обезводили и выпарили. После фильтрования на силикагеле, получили 129 мг (98%) указанного в названии продукта, т.е. 131-134оС;
1Н-ЯМР (CDCl3, 300 МГц) дельта: 5,80-5,96 (1Н, ддд, J = 18 Гц, J = 12 Гц, J = 6 Гц), 5,62 (1Н, д. J = 2 Гц), 5,35 (1Н, дв. кв., J = 18 Гц, J = 2 Гц), 5,19 (1Н, дв. кв., J = 12 Гц, J = 2 Гц), 4,66 (2Н, м), 4,21 (1Н, дв. кв. , J = 7 Гц, J= = 3 Гц), 3,93 (1Н, дд, J = 14 Гц, J = 7 Гц), 3,67 (1Н, дд, J = 3 Гц, J = 2 Гц), 3,56-3,72 (1Н, м), 3,09 (1Н, м), 2,54-2,84 (4Н, м), 1,23 (3Н, д, J = Гц), 0,85 (9Н, с), 0,05 (6Н, с);
С13-ЯМР (CDCl3, 75,43 МГц) дельта: 171,9, 159,4, 150,8, 131,7, 118,7, 118,5, 71,8, 65,7, 65,2, 64,1, 61,7, 52,7, 46,7, 33,2, 25,7, 22,5, 17,9;
m/e вычислено для C17H24NO5S3Si/P- 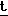 Bu/: 446, 0587, найдено: 446, 0597.
Bu/: 446, 0587, найдено: 446, 0597.
П р и м е р 6. Аллил 5R, 6S-6-(1R-(диметил-трет-бутилсилилокси)этил)-2-((1,1-дио- ксо-3R- и 3S-тиоланил)тио)пенем-3-карбоксилат.
Раствор 50 мг (0,129 ммоль) указанного в названии примера 3 продукта при 0оС в 4 мл метиленхлорида обработали 0,089 мл (0,51 ммоль) диизопропилэтил амина. Этот раствор затем охладили до - 78оС в боне сухой лед-ацетон. Добавили свеже дистиллированный трифторметансульфоангидрид (0,024 мл, 0,142 ммоль) и полученный раствор перемешивали в течение 1 ч при - 78оС. Полученный холодный раствор указанного в названии примера 4 продукта обработали раствором 19,6 мг (0,129 ммоль) рацемического 3-меркаптотиолан-1,1-диоксида (Bezmenova et al. , Khim. Geterotsikl. Soedin. 1975, 188, 2; Chem. Abstr. 1975, 170558) и 0,022 мл (0,129 ммоль) диизопропилэтиламина в 1 мл метиленхлорида. Добавление потребовало 0,5 мин и все это время температуру поддерживали ниже - 70оС. После выдержки в течение 2 ч при - 78оС реакционной смеси дали нагреваться до комнатной температуры и ее перемешивали в течение ночи. Затем раствор обработали 10 мл воды и экстрагировали этилацетатом. Органическую фазу промыли рассолом, обезводили и выпарили. После фильтрования через силикагель (3: 2 гексан:этилацетат) получили 66,7 мг (100%) указанного в названии продукта в виде смеси диастереомеров. Эти диастереомеры разделили с помощью хроматографии на силикагеле, используя в качестве элюента раствор 6:3:1 гексан:этилацетат:бензол. Более полярный диастереомер имел следующие свойства: т.п. 180-181оС (альфа)D = + 57,14о (с = 0,49 г) 100 мл)
Н-ЯМР вычислено для C17H24NO6S3Si: 462, 0536 (Р-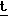 Bu), найдено: 462,0473. Менее полярный диастереомер имел следующие характеристики: т.п. 169-170оС (альфа)D = =+ 111,78о (с = 0,73 г) 100 мл);
Bu), найдено: 462,0473. Менее полярный диастереомер имел следующие характеристики: т.п. 169-170оС (альфа)D = =+ 111,78о (с = 0,73 г) 100 мл);
Н-ЯМР вычислено для C17H24NO6S3Si: 462, 0536 (P-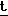 Bu), найдено: 462,0506.
Bu), найдено: 462,0506.
П р и м е р 7. Аллил 5R, 6S-6-(1R-(диметил-трет-бутилсилилокси) этил)-2-(этилтио)пенем-3-карбоксилат.
Указанный в названии примера 3 продукт (100 мг, 0,262 ммоль) превратили в холодный раствор указанного в названии примера 4 продукта по способу примера 4. Этот раствор при - 78оС обработали раствором 0,096 мл (1,3 ммоль) этантиола и 0,226 мл (1,3 ммоль) диизопропилэтиламина и 1 мл ацетонитрила. Добавление потребовало 0,5 мин и температуру раствора поддерживали ниже - 70оС в течение этого времени. Через 5 мин выдержки при - 78оС реакционной смеси дали нагреться до 0оС и ее перемешивали в течение 2 ч. Затем раствор обработали 10 мл воды и экстрагировали этилацетатом. Органическую фазу промыли рассолом и затем обезводили и выпарили. После фильтрации через силикагель (4:1 гексан: этилацетат) получили 110 мг указанного в названии продукта, т.п. 83-84оС;
Н-ЯМР вычислено для C13H31NO4S2Si: 429,1464, найдено: 429,1036; соединение, ранее описанное в родемической форме Leanza et al., Tetrahedron, vol. 39, 2505-2513 (1983).
П р и м е р 8. Аллил 5R, 6S-6-(1R-(диметил-трет-бутилсилилокси)этил)-2-(изо- пропилтио) пенем-3-карбоксилат.
По методике предыдущего примера указанное в названии примера 3 соединение (105,3 мг, 0,274 ммоль) и изопропил меркаптан (0,239 мл 1,37 ммоль) превратили в указанное в названии соединение, очистили с помощью хроматографии на силикагеле, используя 19:1 гексан:этилацетат в качестве элюента, 60 мг, т.п. 104-106оС, ранее известное в рацемической форме, Leanza и др. loc.cit.
П р и м е р 9. Аллил 5R, 6S-6-(1R-(диметил-трет-бутилсилилокси)этил-2-(гидро- ксиэтил) тио)пенем-3-карбоксилат.
По способу примера 6 указанный в названии примера 3 продукт (61 мг, 0,158 ммоль) и 2-меркаптоэтанол (0,012 мл, 0,174 ммоль) превратили в указанный в названии продукт, очистили с помощью хроматографии на силикагеле, используя 3: 2 гексан: этилацетат в качестве элюента, 60 мг, т.п. 80оС; (альфа)D = + 160,4о (с = 2,22 г/100 мл);
Н-ЯМР вычислено: для C19H31NO5S2Si: 445,1412, найдено: 445,1420.
П р и м е р 10. Аллил 5R, 6S-6-(1R-(диметил-трет-бутилсилилокси)этил)-2-(2-(4- нитробензилоксикарбониламино)этил)пен- ем-3-карбоксилат
По способу предыдущего примера указанный в названии примера 3 продукт (49,5 мг, 0,129 ммоль) и 2-((4-нитробензилоксикарбонил))-амино)этил меркаптан (33 мг, 0,129 ммоль, Shinkai et al., Synthesis 1980, 924) превратили в указанный в названии хроматографированный продукт, 71 мг, т.п. 103-105оС, (альфа)D = + 88,34о (с = 3,26 г/100 мл),
Н-ЯМР вычислено для C23H28N3O8S2Si:
566,1088 (P-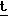 Bu), найдено: 566,1119.
Bu), найдено: 566,1119.
П р и м е р 11. Аллил 5R, 6S-6-(1R-(диметил-трет-бутилсилилокси)этил)-2-(1-(4- нитробензилоксикарбонил)-3S-пирроли-динил-тио)пенем-3-карбоксилат.
По способу примера 6 указанный в названии примера 3 продукт (101,7 мг, 0,264 ммоль) и 3S-меркапто-1-(пара-нитробензилоксикарбонил)пирролидин (0,050 мл, 0,289 ммоль Sigimura и др. Heterocycles 24, 1331, 1986) превратили в указанное в названии соединение, которое после экстрагирования в этилацетат очистили с помощью хроматографии на силикагеле, используя 2:1 гексан-:этилацетат как элюат, 147 мг, т.п. 105-106оС, (альфа)D = + 260о(с = 0,84 CHCl3)
П р и м е р 12. 2-(Триметилсилил)этил 5R, 6S-2-оксо-6-(1R-(диметил-трет-бутилсилилокси)этил)пенем-3- карбоксилат.
По способам примеров 1-3, описанным выше, 2-(триметилсилил-этил-2-(4R-(трифенилметилтио)-3S-(1S- (диметилтрет-бутилсилилокси)этил)-2-азетидион-1-ил)аце-тат превратили в указанный в названии продукт.
1Н-ЯМР (CDCl3, 300 МГц) дельта: 5,52 (1Н, д, J = Гц), 4,96 (1Н, с), 4,35 (1Н, кв., J = 8 Гц, J = 5 Гц), 4,26 (2Н, дв. т., J = 12 Гц), 3,56 (1Н, дв. д., J = 5 Гц, J = 3 Гц), 1,30 (3Н, д, J = =8 Гц), 1,06 (2Н, дв. т. J = 12 Гц), 0,89 (9Н, с), 0,1 (3Н, с), 0,08 (3Н, с), 0,05 (9Н, с),
13С-ЯМР (CDCl3, 62,89 МГц) дельта: 199,3, 169,2, 163,9, 71,8, 66,4, 65,5, 64,7, 62,5, 25,7, 22,5, 17,9, 17,4, - 1,5, - 4,2, - 5,1,
m/e вычислено для C15H26NO5SSi2 (P-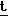 -Bu): 388,1179, найдено: 388,1125.
-Bu): 388,1179, найдено: 388,1125.
В соответствии с последовательностью стадий и способов примеров 4-5, полученный продукт далее превратили через интермедиат 2-(триметилсилил)этил 5R, 6S-6-(R-(ди-метил-трет-бутилсилил- окси)этил)-2-(трифторметансульфонилкси)пе- нем-3-карбоксилат в 2-(триметилсилил)этил 5R, 6S-6-(1R-(диметил-трет-бутилсилилокси)этил)-2-(1R-оксо-3S- тиоланил)тио)пенем-3-карбоксилат.
Приготовление 1.
(R)-3-Tиоланил п-толуолсульфонат.
(R)-4-(Метилтио) 1,2-бутандиол (1,0 г, 7,35 ммоль) и п-толуолсульфонил хлорид (3,0 г, 15,8 ммоль) соединили в 10 мл пиридина при 0-5оС, затем перемешали при комнатной температуре и в это время ТЖХ (3:1 гексан:этил ацетат) показала отсутствие диола (Rf 0,1), существенное количество диола дитосилата (Rf 0,53), некоторое количество интермедиата тиолановой соли (Rf 0,03) и следы указанного в названии продукта (Rf 0,72). Реакционную смесь затем нагревали до 60оС в течение 8 ч, после чего ТЖХ (5:1 гексан: этилацетат) показала значительное количество указанного в названии продукта (Rf 0,45), только следы дитосилата (Rf 0,22), некоторое количество тиолановой соли (Rf 0,01) и другие в основном малополярные примеси. Охлажденную реакционную смесь разбавили эквивалентным количеством воды и двумя объемами этилацетата. Органический слой отделили, промыли насыщенным NaCl, обезводили (MgSO4), десорбировали и остаток хроматографировали на силикагеле, используя 10:1 гексан:этилацетат в качестве элюента и получили 0,1 г менее полярных примесей (зловоние!) и 0,25 настоящего очищенного указанного в названии продукта, ТЖХ Rf 0,55 (4:1 гексан:этилацетат); (альфа)D = =+15,87 (с = 0,6), СH3OH).
Приготовление 2.
3R-(п-Толуолсульфонилокси)тиолан 1R-оксид
Раствор 46,30 г (0,179 моль) указанного в названии предыдущего приготовления продукта в 600 мл ацетона в потоке азота охладили до 0оС. В отдельной колбе 61,73 г (0,100 моль) пероксимоносульфат калия перемешивали в 500 мл дистиллированной воды до получения прозрачного раствора. Этот раствор добавили в раствор ацетона при 0оС и дали смеси нагреться до комнатной температуры. Через 25 мин 75 мл 10 мас.% водного раствора сульфита натрия добавили, ацетон выпарили, 300 мл этилацетата добавили и водный слой экстрагировали этилацетатом (3 х 100 мл). Экстракты объединили, обезводили (MgSO4) и сконцентрировали досуха, получив при этом 48,57 г сырого продукта. Последний очистили на силикагеле с помощью хроматографии, используя 10: 10: 1 этилацетат: CH2Cl2:CH3OH в качестве элюента и получили очищенный указанный в названии продукт, 34,67 г 71% (альфа)D = +4,26о (с = 3,0 CHCl3).
Приготовление 3.
3S-(Ацетилтио)тиолан 1R-оксид.
В высушенной над огнем колбе в атмосфере азота 31,67 г (0,1156 моль) указанного в названии предыдущего приготовления продукта растворили в 300 мл ацетона и 19,81 г (0,1734 моль) тиоацетат калия добавили.
Смесь нагревали при температуре дефлегмации в течение 3,5 ч и оставили перемешиваться при комнатной температуре в течение ночи. Смесь профильтровали, промыли 500 мл ацетона и фильтрат и смывки выпарили в вакууме и получили 23,96 г целевого продукта в виде масла. Это масло подвергли очистке с помощью испарительной хроматографии на 120 ммх25 см силикагелевой колонке, используя в качестве элюента 19: 1 этилацетат:метанол, собирая 125 мл фракции. Фракция 42-64 собрали, отогнали легкие фракции и получили очищенный указанный в названии продукт в виде масла, которое кристаллизуется при выдержке, 16,46 г (80%), т.п. 51-52оС, (альфа)D) = -83,41о (с = 0,86, CHCl3).
Вычислено,%: С 40,4; H 5,6.
C6H10S2O2
Найдено,%: C 40,15; Н 5,53.
Сущность изобретения: продукт-стереоспецифический пенем ф-лы 1, где R-алкил, /триметилсилил/-этил; R1 -силильная защита, R2 -C1-C4 -алкил; 2-гидроксиэтил; 2-/п-нитробензилкарбониламино/-этил, 1-/п-нитробензилоксикарбонил/-3S-пирролидинил, 1R-оксо-3S-тиоланил или 1,1-диоксо-3R- и 3S-тиоланин. Реагент 1: соединение ф-лы 2. Реагент 2: Ag(NO2)2 . Условия реакций: в присутствии пиридина с последовательной обработкой продукта H2S , п-нитрофенилхлормиатом, гексаметилдисилиламидомлития, ангидридом трифторметансульфокислоты, HSR2 , где R, R1 , R2 -указано выше. Структура ф-л 1, 2 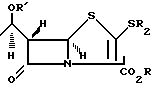
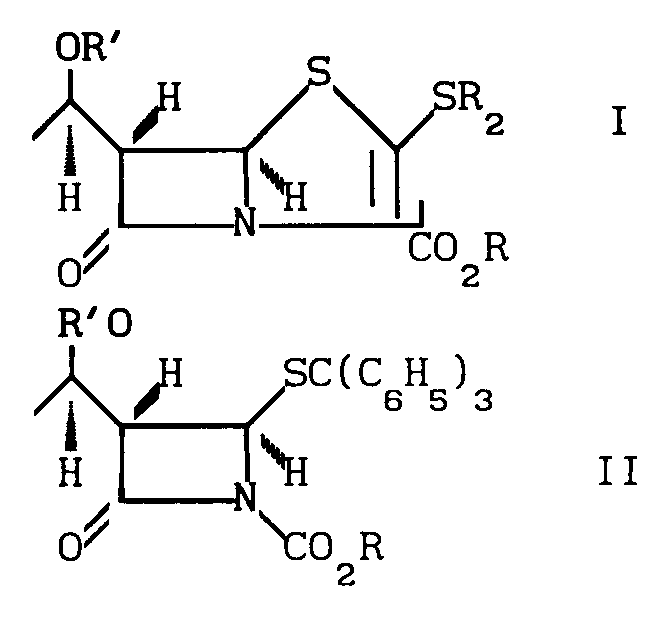
СПОСОБ ПОЛУЧЕНИЯ СТЕРЕОСПЕЦИФИЧЕСКОГО СОЕДИНЕНИЯ общей формулы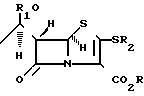
где R - аллил, (триметилсилил)-этил;
R1 - силильная защитная группа;
R2 - C1 - C4-алкил, 2-гидроксиэтил, 2-(п-нитробензилкарбониламино)-этил, 1-(п-нитробензилоксикарбонил)-3S=пирролидинил, 1R-оксо-3S-тиоланил или 1,1-диоксо-3R- и 3S-тиоланин,
включающий взаимодействие трифенилметилтиопроизводного ацетидинола с молярным избытком нитрата серебра в присутствии молярного избытка пиридина в реакционно-инертном растворителе при температуре -25...+25oС, тиоацилирование и циклизацию полученного продукта с образованием пенема, взаимодействие пенема с меркаптаном, отличающийся тем, что в качестве трифенилметилтиопроизводного азетидинола используют соединение общей формулы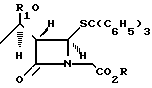
где R и R1 имеют указанные значения,
которое подвергают взаимодействию с молярным избытком нитрата серебрав присутствии молярного избытка пиридина в реакционно-инертном растворителе при температуре -25. . . +25oС с последующей обработкой реакционной смеси сероводородом в присутствии молярного избытка пиридина в реакционно-инертном растворителе при температуре -25...+25oС с получением соединения общей формулы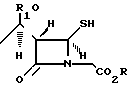
где R и R1 имеют указанные значения,
ацилированием его одним молярным эквивалентом п-нитрофенилхлорформиата в присутствии одного молярного эквивалента 4-(диметиламино) пиридина в реакционно-инертном растворителе при температуре -25...+25oС с последующей циклизацией полученного соединения общей формулы
где R и R1 имеют указанные значения,
молярным избытком гексаметилдисилиламида лития при температуре -50... -100oС с образованием пенема общей формулы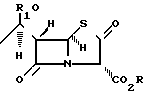
где R и R1 имеют указанные значения,
с последующим его взаимодействием с молярным эквивалентом ангидрида трифторметансульфокислоты в присутствии молярного избытка диизопропилэтиламина в реакционно-инертном растворителе при температуре -40...-78oС с образованием соединения общей формулы
где R и R1 имеют указанные значения,
и последующей обработкой его по крайней мере одним молярным эквивалентом общей формулы
HS - R2,
где R2 имеет указанные значения,
при температуре 0...-78oС.
| Di nippon и др | |||
| "Tetrahedron Hetter", 1982, 23, N 35, р.3535-3538. |
