тетрабутиламмонийфторида вСН СТСООН с выделением соединения, где R-аллилок- сикарбонильная группа. Аллальные защитные группы удаляют с помощью три- фенилфосфина и тетракис(трифенилфосс-
15794614
фин)палладия, Целевой продукт выделяют в виде кислоты или ее соли. Новые соединения малотоксичны и активны против кишечных грамположительных и грамотри- цательных бактерий,- 1 табл.









| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения пенемовых соединений или их фармацевтически приемлемых солей щелочных металлов | 1986 |
|
SU1586516A3 |
| Способ получения 5(R) пенемовых производных | 1983 |
|
SU1375139A3 |
| Способ получения оптически чистых (5R, 6S)-6-[1(R)-гидроксиэтил]-2-метоксиметилпенем-3-карбоновой кислоты или ее сложных эфиров, или ее солей с щелочными металлами | 1988 |
|
SU1586517A3 |
| Способ получения замещенных производных пенем-3-карбоновой кислоты или их сложных эфиров или их солей с щелочными металлами | 1983 |
|
SU1299512A3 |
| СОЕДИНЕНИЯ ПЕНЕМА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2079498C1 |
| Способ получения 2-оксиметилпенемов | 1987 |
|
SU1625333A3 |
| СПОСОБ ПОЛУЧЕНИЯ 2-МЕТОКСИМЕТИЛПЕНЕМОВ | 1990 |
|
RU2049786C1 |
| Способ получения соединений @ -лактама | 1980 |
|
SU1186086A3 |
| Способ получения оптически активных пенемов или их солей с щелочными металлами | 1982 |
|
SU1389680A3 |
| Способ получения 6-дезоксиантрациклинов | 1984 |
|
SU1561821A3 |
Изобретение касается производных пенема, в частности получения соединений общей ф-лы I: CH 3-C(←OH)-CH-C(O)-N-C←S-CK=CR, где K - -CH 2-X-C 6H 4-N-CH 2Q
R - C(O)OH, аллилоксикарбонил, карбоксилат-анион
X - сера, кислород, оксикарбонил, оксикарбониламино-, оксикарбонилокси-или сульфонильная группа
Q - OH, карбамоилокси, 1-метилтетразол-5-илтио-, пиридинио, N-метилпирролидинио, 3,5-диметилпиридинио, 3-гидроксипиридинио, N-метилпиперазинио, 4-сульфонатоэтанпиридинио, 3-карбоксилатэтенилпиридинио, 4-сульфоэтилпиридинио, 4-карбоксипиридинио, 4-диметиламинопиридинио, 3-карбоксилатметилпиридинио, 4-аминометилпиридинио или 4-аллилоксикарбониламинометилпиридинио, или их фармацевтически приемлемых солей, обладающих антибактериальными свойствами, что может быть использовано в медицине и ветеринарии. Цель - создание новых более активных веществ указанного класса. Синтез ведут реакцией соединений общих ф-л II и III: CH 3-C(←OY)-CH 2-C(O)-N-C←S-C(CH 2L)=C-C(O)-OCH 2-CH=CH 2 (II), A-C 6H 4-N-CH 2Q 1 (III), где L - OH, CL
Y - SI(CH 3) 2C 4H 9-T
Q 1 - галоген, O-SI(C 6H 5) 2C 4H 9-T
A - OH, SH, C(O)OH, хлоркарбонил, изоцианато, хлоркарбонилокси- или сульфиногруппа, при (-10)-(+65)°С. Далее при наличии трет-бутилфенольной группы проводят ее удаление с помощью тетрабутиламмонийхлорида в CH 3C(O)OH. Полученное при этом соединение, содержащее вместо Q 1 заместитель Q 2 - галоген, OH, в среде диметилформамида, CH 2CL 2 или тетрагидрофурана при (-70)-(+25)°С обрабатывают одним из следующих соединений: трихлорацетилизоцианатом, 1-метил-5-меркапто-1,2,3,4-тетразолом, пиридином, N-метилпирролидином, 3,5-диметилпиридином, 3-гидроксипиридином, натрий пиридин-4-этансульфонатом, 4-аллилоксикарбонилпиридином, 4-диметиламинопиридином, 4-аллилоксикарбонил-1-метилпиперазином, пиридин-4-этансульфокислотой, 3-аллилоксикарбонилэтенилпиридином, 3-аллилоксикарбонилметилпиридином, 4-аллилоксикарбониламинометилпиридином. Когда Q 2 - OH, удаляют трет-бутилдиметилсилильную группу с помощью тетрабутиламмонийфторида в CH 3C(O)OH с выделением соединения, где R - аллилоксикарбонильная группа. Аллальные защитные группы удаляют с помощью трифенилфосфина и тетракис(трифенилфосфин)палладия. Целевой продукт выделяют в виде кислоты или ее соли. Новые соединения малотоксичны и активны против кишечных грамположительных и грамотрицательных бактерий. 1 табл.
1C Изобретение относится к способу
получения новых антибиотиков ряда пе- нема или их фармацевтически приемлемых солей, которые обладают противо- бактериальными свойствами, и могут най- ти применение в медицине или в ветеринарии.
Целью изобретения является создание новых пенемовых антибиотиков, обладающих улучшенными антибактериальны- д ми свойствами.
Пример 1, Получение (5R, 6S)- -6- Ј( 1R) -оксиэтилП-2- JJ4- (1 -пиридинио) метилфеншП-оксиметилпенем-З-карбокси- лата (соединение 4).25
Стадия А.
НО-(о)-СН7ОН - НО-(о)-рНг08 РЬ гВа-1
В перемешиваемый раствор 4-гидрок- симетилфенола (6,2 г) в безводном ди- метилформамиде (100 мл) добавляют последовательно имидазол (9,5 г) и трет бутилдифенилсилилхлорид (12,8 мл) при комнатной температуре. Полученный раствор перемешивают в течение 2 ч при комнатной температуре, затем вливают в простой диизопропиловый эфир и дважды промывают водой. Органический слой обезвоживают над сульфатом натрия и растворитель отгоняют в вакууме После очистки на колонке с силикаге- лем (230-400 меш, элюент-смесь н-гек- сана с этилацетатом) получают 4-трет- бутилдифенилсилилоксиметилфенол в виде масла (12,0 г),
ЯМР (60 МГц, CDC1,)) , МЛНАД.: 1,13 (9Н, с); 4,72 (2Н, с); 5,53 (1Н, шир,с, ); 6,73 (2Н, д, ,0 Гц); 7,17 (2Н, д, J 9,0 Гц); 7,3-7,7 (ЮН, м).
Стадия В.
OSiMe Bu-t „S, ОН
OSiMeBu-t
:ОгСНгСН-СН НО -@-CH7OSsPh 78u -t
CO CH-jCV CH
д
5
0
5
0
5
0
5
Раствор диэтилазодикарбоксилата (4 мМ) и трифенилфосфина (4 мМ) в тет- рагидрофуране (10 мл) перемешивают при температуре 0°С в течение 1 ч.Эту смесь добавляют в раствор аллил (5R, 6S)-6- Ј( 1R)-трет-бутилдиметилсилилок- сиэтшГ -2-оксиметиленом-3-карбоксила- та (1,0 г) и 4-трет-бутилдифенилсили- локсиметилфенола (1,0 г) в безводном тетрагидрофуране (20 мл) при 0°С.Полученный раствор перемешивают в течение 30 мин при комнатной температуре, затем концентрируют в вакууме и очищают с помощью испарительной хроматографии на силикагеле, в результате чего получают аллил (5R, 6S)-6- (1 0-трет-бу- тилдиметилсилилокси-этшГ -2-(4-трет- -бутилдифенилсилилоксиметилфенил) ок- симетилпенем-3-карбоксилат в виде светло-желтого сиропа (600 мг),
Раствор этого продукта в тетрагидрофуране (20 мл) перемешивают в течение 8 ч при комнатной температуре в присутствии уксусной кислоты (0,40 мл) и тригидрата тетрабутиламмонийфторида (0,40 г). Растворитель отгоняют в вакууме и остаток фракционируют на колонке силикагеля (мерк 60 HP 230-400 Меш, элюент - смесь н-гексана с этилацетатом), в результате чего получают аллил (5R, 6S)- -6-р1R)-трет-бутилдиметилсилилокси- (4-оксиметилфенил)оксиметил- пенем-3-карбоксилат (370 мг).
ИК (СНСЦ) )fM01KC: 1787, 1702, 1605, 1580 .
ЯМР (200 МГц, CDC13)Ј , млн.д.: 0,06 (6Н, с); 0,86 (9Н, с); 1,21 (ЗН, д, J 6,4 Гц); 3,70 (Ш, дд, J 1,7 и 4,6 Гц); 4,22 (1Н, дк, J 4,6 и 6,4 Гц); 4,62 (2Н, с); 4,7 (2Н, м); 5,26 (1Н, д, J 10,3 Гц); 5,13 и 5,39 (2Н, АВк, J 15,6 Гц); 5,41 (1Н, д, J 17 Гц); 5,58 (1Н, Д, J 1,7 Гц); 5,9t (1H, м); 6,91 и 7,29 (каждый 2Н, д, J 8,7 Гц).
Стадия С.
OSiMejBu-l
i
:N-x Ч со7сн7сн сн7
Соответствующие фракции объединяют, очищают замораживанием и получают 30 мг указанного в названии белого порошка. .УФ (H20) AMak,c: 258 и 307 нм. (
ИК (KBr)3Maxc: 1765, 1600 см. Раствор аллил (5R, 6S)-6-plR)- ЖР (200-МГц, ) & , млн.д.: 1,22
-трет-бутилдиметилсилилоксиметил -2-уН д J 63 Гц . 372 (-1Н дд J
-(4-оксиметилфенил) оксиметилпенем-3- 10 Ь5 и r«J5 (1Н дк J
-карбоксилата(200 мг) в безводном и 63 502 и 2Н каж
Дьй д, J 14.4 Гц); 5,43 (1Н, д, J
свободном от этанола дихлорметане , ,,„. , q /ic л ., 1«5 Гц): 5j7U t/H, Ь.УЬ и b,jy
(15 мл) охлаждают в атмосфере азота,„ /}„ t Q П1 /ou
до - 70°С и затем последовательно до-С-2Н каадыи, Д, J - 8,6 Гц); 8,01 (2Н,
бавляют при перемешивании пиридин t5ВД1 ,6 7,0 Гц); 8,50 (1Н, т J-
,(0,23 мл) и трифторметансульЛангнд- 7}° Гц); ( Д) J , 6 Гц) РИД (0,22 мл). Температуре реакцион- П р и м е р 2. Таким же образом,
ной смеси дают подняться до -5°С и за-как °Писан° В 1 но заменив
тем реакцию гасят с помощью 0,1 М вод-.,ПИрИДИН «а N-метилпирролидин получаного раствора НС1. Органический слойют (5R б8)-6-(1Ю-оксиэтил -2- 4г(1-.
отделяют, промывают рассолом, обезвожи--метил-Ьпирролидинио) метилфенил оквают, выпаривают и получают смолопо- симетилпенем-3-карбоксилат (соединедобнь й остаток.ние °л
ЯМР (200МГц, ВгО)Д млн.д: 1,27
25(ЗН, д, J - 6,3 Гц); 2,22 (4Н, м);
.Продукт растворяют в тетрагидрофу-2 93 (ЗН, с); 3,30-3,65 (4Н, м): 3,84
ране (8 мл) и обрабатывают последова-(1Н, д, J 6,0 Гц); 4,23 (1Н, м);
тельно уксусной кислотой (0,55 мл) (2Н с). 5J9 и (2Н два
тетрабутиламмонийфторидом ; ЗН40j 14 Гц). (1Н с); 6,95-7,55
(0,5 г). Прозрачный раствор оставляют 39(4н м)
при комнатной температуре на 20 ч, уф (н20)макс: 308 нм. после чего его концентрируют и пропус- ик (КВг) кс . 1760, 1605,
кают через колонку с силикагелем (230-1580
400 меш, диаметр 2 см, высота 8 см).
П р и м е, р 3. Натрий (5R, 6S)-6П р и м е, р 3. Натрий (5R, 6S)-6.Элюирование проводят сначала чистым -(1К)-0ксиэтил -2-(4-оксиметилфенокPU .ГЧоптлил PU PT I CV РМ W4-J
СН2С12, затем смесью CH2C1Z/CH3CN (70:30, затем 50:50), затем чистым ацетонитрилом, после чего проводят окончательную промывку смесью (1:2). В последние фракции добавляют ,л NaCl, экстрагируют , затем концентрируют в вакууме и получают сырой сложный аллиловый эфир указанного в названии продукта в виде-пены.
Это- соединение помещают в смесь «j 5 мл СН2С12 и 0,1 мл уксусной кислоты и перемешивают с трифенилфосфином (0,025 г) и тетракис(трифенилфосфин) Pd(0) (0,025 г) в течение 30 мин, посей) метилпенем-3-карбоксилат .(соединение 2)
OSiMe,Bu-t
ОН
, я-
С07СН1СН СН7C07NQ
ш
Раствор аллил (5R, 6S)-6-Ј-(1R)- -трет-бутилдиметилсилилоксиэтил -2-(4- -оксиметилфенокси)метилпенем-3-карбок- силата (0., 5 г) в тетрагидрофуране (7 мл) последовательно обрабатывают
ле чего для завершения реакции (еще .50 уксусной кислотой (0,25 мл) и трет- 15 мин) добавляют РРЬЭ(0,02 г) и Ph бутиламмонийфторидом тригидратом (PPh) 4(0,02 -г).(0,44 г). Эту смесь перемешивают в теРеакционную смесь концентрируют и чение 15 мин, затем выдерживают при остаток смешивают с этилацетатом. По- комнатной температуре в течение 6 ч. лученное вещество растворяют в демине- -с Растворитель удаляют в вакууме, оста- рализованной воде и хроматографируют ток очищают при -помощи хроматографии на колонке Lichroprep RP-18, элюируя на силикагеле и получают аллиловый сначала водой, а затем 10%-ным раство- сложный эфир указанного в названии ром CH-jCN в воде.соединения (0,35 г). Это вещество
.157946 Т6
Соответствующие фракции объединяют, очищают замораживанием и получают 30 мг указанного в названии белого порошка. .УФ (H20) AMak,c: 258 и 307 нм. (
ИК (KBr)3Maxc: 1765, 1600 см. ЖР (200-МГц, ) & , млн.д.: 1,22
П р и м е, р 3. Натрий (5R, 6S)-61К)-0ксиэтил -2-(4-оксиметилфенок-(1К)-0ксиэтил -2-(4-оксиметилфенокW4-J
ей) метилпенем-3-карбоксилат .(соединение 2)
OSiMe,Bu-t
ОН
, я-
С07СН1СН СН7C07NQ
Раствор аллил (5R, 6S)-6-Ј-(1R)- -трет-бутилдиметилсилилоксиэтил -2-(4- -оксиметилфенокси)метилпенем-3-карбок- силата (0., 5 г) в тетрагидрофуране (7 мл) последовательно обрабатывают
(0,28 г) в смеси тетрагидрофурана (5 мл) и дихлорметана (4 мт) обрабатывают при перемешивании тоифенилфос- фином (25 мл) и тетракис (грифенилфос фин)палладием (0,25 мг), а затем натрий 2-этилгексаноатом (0,11 г). После перемешивания в течение 30 мин добавляют простой этиловый эфир и осадок разделяют центрифугированием. Полу- ченное твердое вещество растворяют в небольшом количестве воды и пропускают через колонну с обращенной фазой (Merck Lichroprep C-18), гшюируя дистиллированной водой. Сушкой выморажи- ванием получают указанной в названии соединение в виде белого порошка (0,15 г).
ИК (КВгЯ)шхс: 1770, 1600 см .
П р и м е р 4-. Натрий (5R, 6S)-6- -(1R)-оксиэтил -2-(4-карбамоилокси- метилфенокси) метилпенем-3-карбокси- лат (соединение 1). Стадия А„
ОН
OCONH7
AtA
г
0С01СН7СН СН2
Трихлорацетилизоцианат (0,15 мл) по каплям добавляют в раствор аллил (5R, б8)-6-Ј(1К)-трет-бутилдиметил- силилоксиметшГ -2-(4-оксиметилфенок- си) метилпенем-3-карбоксилата, по- лученного, как описано в примере 1, стадия В, (0,3 г) в холодном (-40°С) дихлорметане. Смеси дают нагреться до комнатной температуры, а затем концентрируют в вакууме. Оотаток растворяют в ТГФ (8 мл) и обрабатывают уксусной кислотой (0,6 мл) и тетрабутлл- аммонийфторидтригидратом (0,75 г). Через 20 ч при комнатной температуре.растворитель удаляют, остаток очищают при помощи испарительной хроматографии на силикагеле и получают аллил (5R, 6S)-6-(1R)-оксиэтил -2-(4-кар- бамоилоксиметилфенокси)метилпенем-3- карбоксилат (0,25 г) в виде аморфного твердого вещества.
Стадия В.
Указанное выше вещество СО,2 г) деаллилируют трифенилфосфином и тетракис (трифенилфосфин)палладием (0) в присутствии натрий этилгексаноата, как описано в примере 3.
При помощи хроматографии с обращенной фазой получают указанное в названии соединение (0,13 г).
ИК (КВг)а)с : 1760, 1715, 1605 см4.
ЯМР (200 МГц, DZ0): 1,26 (ЗН, д, J 6,3 Гц); 3,79 (1Н, дв, д, J 1,6, 5,8 Гц); 4,19 (1Н, дв, кв. J 5,8, 6,3 Гц); 4,98 (2Н, с); 5,44 и 5,12 (2Н, АВ кв., J 14-,6 Гц); 5,52 (Ш, д, J 1,6 Гц)-; 6,97 (2Н, д, J 8,5 Гц); 7,31 (2Н, д, J 8,5 Гц).
Пример 5 о Натрий (5R, 6S)-6-, - Ј( 1R) -оксиэтют -2- ft- (1 -метил-1,2, 3,4-тетразол-5-ил) тиометилфенокси ме- тилпенем-3-карБоксилат (соединение 3).
Стадия А.
OSiMeBu-t 1н s
С02СН7СН СНг
gsiMegu-tN N
СН3 СОгСН7СН- СН7
0
to
5
5
50
Смесь трифенилфосфина (700 мг) и диэтилазодикарбоксилата (0,42 г) в сухом ТГФ (15 мл) предварительно перемешивают при 0°С в течение 30 мин, а затем добавляют в раствор аллил (5R, 6S)-6- Ј(Ш)-трет-бутилдиметилси- лилоксиэтил -2-(4-оксиметШ1фенокси)ме- тилпенем-3-карбоксилата (400 мг) и 1-метил-5-меркапто-1,2,3,4-тетразола (100 мг) в том же растворителе (30мл), сохраняя температуру ниже 10°С. Через 15 мин растворитель удаляют в вакууме, остаток очищают при помощи хроматографии на силикагеле (циклогек- сан-этилацетат) и получают аллил (5R, 6S)-6- (Ш)-трет-бутилдиметилсилилок- .сиэтил (1-метил-1,2,3,4- -тетразол-5-ил) тиометилфенокси ме- тилпенем-3-карбоксилат в виде пены (405чмг).
ИК ( 1790, 1705 .
Стадия В.
Из указанного выше вещества (0,4 г) /путем последовательного десилилирова- ния и деаллилирования по методике, описанной в примере 3, получают указанное в названии соединение,(0,12 г) в виде аморфного твердого вещества после сушки вымораживанием.
ИК (КВг)Змсп(С: 1765, 1605 . П р и м е р 6. (5R, 6S)-6-(1R)- -Оксиэтил - 4-(1-пиридиниометил)-бензоилокси метилпенем-3-карбоксилат (соединение 6) .
OSiMeiBu-t
OSiMe-jBa-t
re.ЈU |
ИЗ ОН -Чт-У
... .
OSiPhjBu-t
C07CH2CH CHjСОгСНгСН СН7
ИК (СНС1г),3МакС : 1790, 1720 см4 ЯМР (60 МГц, CDCl), млн.д. 0,05 (6Н, с); 0,9 (9Н, с); 1,2 (ЗН, д, J 6 Гц);2,35 (1Н, шир. с);3,65 (1Н, дв.д. J 2 и 5 Гц); 4,2 (1Н, м); 4,65 (4Н, м); 5,1-5,5 (2Н, м); 5,45 ,(2Н, АВ, кв., J 15 Гц); 5,55 (1Н, И, J 2 Гц); 5,9 (1Н, м); 7,2-8,1 (4Н, м).
i Стадия С.
OSiMeiBu-t lH х
ОН
©
°Y@«L JbSWoY© n®
согснгсн сн7
Диизопропилазодикарбоксилат (0,2 мл) добавляют в холодный (0°С) раствор аллил (5R, 6S)-6- Ј(1R)-TpeT- -бутилдиметилсилилоксиэтилJ-2-окси- , 5 метилпенем-3-карбоксилата (0,21 г), трифенилфосфина (0,25 г) и 4-(трет- -бутилДиметилсилилоксиметил)бензойной кислоты (0,2 г) в сухом тетрагид- рофуране (10 мл), .После перемешивания в .течение 15 мин реакционную смесь разводят этилацетатом, промытым раст- варом уксусной кислоты (1 мл) в воде .(50 мл) . Органический слой дополнительно промывают рассолом, обезвожи- 25 -трет-бутилдиметилсилилоксиэтшЛ-2- вают MgS04 и растворитель выпаривают. (оксиметил)бензоилокси метилпе- При помощи хроматографии на силикаге- нем-3-карбоксилата (250 мг) в сухом ле получают аллил (5R, 6S)-6-(1R)- без этанола дихлорметане (10 мл) пос- -трет-бутш1Диметилсилилоксиэтил -2- ледовательно обрабатывают при -40°С 4-(трет-бутилдифенилсилилоксиметил) зо под азотом пиридином (0,25 мл) и трн- -бензоилокси -метилпенем-3-карбокси- фторметансульфангидридом (0,125 мл).
со®
Раствор аллил (5R, 6S)-6-(1R)лат (примерно 400 мг).
ИК (СНС13Ямст: 1790, 1720 см.
ЯМР (60 МГц, СОС1г)Ј , млн.д.: 0,1 (6Н, с); 0,9 (9Н, с); 1,12 (9Н, с); 1,20 (ЗН, д, J Гц); 3,70 (1Н, дв.д. J 2 и 4,5 Гц); 4,2 (1Н, м); 4,6-4,8 (АН, м); 5,1-5,6 (2Н, м); 5,55 (2Н, АВ кв.); 4,65 (1Н, д, J 2 Гц); 5,9 (1Н, м), 7,8-8,2 (14Н, м) .
Стадия В.
35
Обработку реакционной смеси и последующие десилилирование и деаллилиро- вание ведут, как описано в примере 1, и получают указанное в названии соединение (60 мг),
40
236 и 307 нм. 1765, 1720,
УФ (
ИК (КВг)макс 1600 .
ЯМР (200 МГц,020)Ј , млн.д.: 1,27 (ЗН, д, J 6,3 Гц); 3,79 (Ш, дв, д, J 1,5 и 5,9 Гц); 4,21 (1Н, м); 5,07 и 5,74 (2Н, каждый, д, J 14,8); 5,51 (1Н, д, J 1,5 Гц); 5,92 (2Н, с); 7,50 и 7,94 (4Н, каждый д, J 8,4 Гц); 8,14 (1Н, дв, д. J 6,5 и и 7,7 Гц); 8,63 (1Н, т, J 7,7 Гц); 8,99 (1Н, д, J 6,5 Гц).
SiMejBu-l U OSitle2Bu-t
1Н S cKJOJOSiPh u-l H S n. lOJOH
f °
сОгСНгСН СН2 С07СНгСН СНг
Раствор вещества из стадии А (400 мг) в тетрагидрофуране (10 мл) обрабатывают уксусной кислотой (0,1 мл) и тетрабутиламмонийфторид- тригидратом (225 мг). После перемешивания в течение 2 ч растворитель удаляют в вакууме, остаток очищают при помощи испарительной хроматографии и получают аллил (5R, 6S)-6- (1К)-трет- -бутилдиметилсилилоксиэтил -2- 4-(ок
симетил) бензоилокси метилпенем-З-кар- боксилат в виде светло-желтого масла (250 мг).
ИК (СНС1г),3МакС : 1790, 1720 см4. ЯМР (60 МГц, CDCl), млн.д. 0,05 (6Н, с); 0,9 (9Н, с); 1,2 (ЗН, д, J 6 Гц);2,35 (1Н, шир. с);3,65 (1Н, дв.д. J 2 и 5 Гц); 4,2 (1Н, м); 4,65 (4Н, м); 5,1-5,5 (2Н, м); 5,45 ,(2Н, АВ, кв., J 15 Гц); 5,55 (1Н, И, J 2 Гц); 5,9 (1Н, м); 7,2-8,1 (4Н, м).
i Стадия С.
-трет-бутилдиметилсилилоксиэтшЛ-2- (оксиметил)бензоилокси метилпе- нем-3-карбоксилата (250 мг) в сухом без этанола дихлорметане (10 мл) пос- ледовательно обрабатывают при -40°С под азотом пиридином (0,25 мл) и трн- фторметансульфангидридом (0,125 мл).
OSiMeiBu-t lH х
ОН
©
°Y@«L JbSWoY© n®
согснгсн сн7
ет-бутилдиметилсилилоксиэтшЛ-2 -(оксиметил)бензоилокси метилпе -3-карбоксилата (250 мг) в сухо этанола дихлорметане (10 мл) п овательно обрабатывают при -40° азотом пиридином (0,25 мл) и т рметансульфангидридом (0,125 мл
со®
ет-бутилдиметилсилилоксиэтшЛ-2- -(оксиметил)бензоилокси метилпе- -3-карбоксилата (250 мг) в сухом этанола дихлорметане (10 мл) по овательно обрабатывают при -40°С азотом пиридином (0,25 мл) и тр рметансульфангидридом (0,125 мл)
Раствор аллил (5R, 6S)-6-(1R)
Обработку реакционной смеси и последующие десилилирование и деаллилиро- вание ведут, как описано в примере 1, и получают указанное в названии соединение (60 мг),
0
236 и 307 нм. 1765, 1720,
5
0
УФ (
ИК (КВг)макс 1600 .
ЯМР (200 МГц,020)Ј , млн.д.: 1,27 (ЗН, д, J 6,3 Гц); 3,79 (Ш, дв, д, J 1,5 и 5,9 Гц); 4,21 (1Н, м); 5,07 и 5,74 (2Н, каждый, д, J 14,8); 5,51 (1Н, д, J 1,5 Гц); 5,92 (2Н, с); 7,50 и 7,94 (4Н, каждый д, J 8,4 Гц); 8,14 (1Н, дв, д. J 6,5 и и 7,7 Гц); 8,63 (1Н, т, J 7,7 Гц); 8,99 (1Н, д, J 6,5 Гц).
Пример. (5R, 6S)-6-(1R)- -оксиэтил -2-Гк-4 ((1 -пиридинио)ме- тил фенилТ карбамоилоксиметилпенем-3- -карбоксилат (соединение 7).
Стадия А.
ноос
;- oXcH2os1Ph2Ba-t- NV(SrOSiPh2Bu 1
В раствор 4(трет-бутилдифенилсили- локси)-метилбензойной кислоты (2 г) в безводном дихпорметане (25 мл) добавляют тионилхлорид (1j1 мл) и безводный диметилформамяд (3 капли). После перемешивания в течение 2 ч при комнатной температуре в вакууме удаляют оставшиеся летучие компоненты. Полученный таким образом сырой хлор- ангидрид растворяют в ацетоне (25 мл) и при перемешивании при 0°С смешивают с раствором азида натрия (0,97 г) в воде (4 мл). Через несколько минут большую часть ацетона удаляют в ваку- уме, а реакционную смесь экстрагируют бензолом. Органические экстракты промывают рассолом, обезвоживают и выпаривают до малого объема, оставляя концентрированный раствор 4-(трет-бутил- дифенилсилилокси) метил-бензоилазида, который в таком виде используют на следующей стадии.
ИК (пленка): 2155, 1705 см.
Стадия В.
iiPhjBu-t - )-CH7.0SiPh2Bu-t
Раствор в бензоле ацилазида, полу- ченного на стадии А, нагревают при температуре дефлегмации в течение 4 ч. Растворитель отгоняют в вакууме, получая таким образом сырой 4-(трет- -бутилдифенилсилилоксиметил)фенилизо- цианат.(
ИК (пленка) м«кс : 2190 см .
Стадия С.
«не.-. о ЧоУ051 8 1-4 А ОН+ OSiMeBu-t JtSTbsiPthBu-t
Ж-AjKONH
согснгсн снг &Ј&-%
СОгСНгСН СНг
Раствор сырого 4-(трет-бутилдифе- нилсилилоксиметил) фенилизоцианата, полученного на стадии С (2 г), в ево- бодном от этанола хлороформе (80 мл) последовательно обрабатывают аллил- (5R, 6S)-6-Ј(1R)-трет-бутилдиметил- силилоксиэт илЗ-2-оксиметилпенем-3 кар боксилатом (2 г) и 4-диметиламинопи- ,ридином (0,06 г). Раствор нагревают при температуре дефлегмации в течение 2 ч, затем промывают разбавленной соляной кислотой и водой. После удаления растворителя и хроматографии получают аллил (5R, 6S)-b- (1Ю-трет-бу- тилдиметилсилилоксиэтил -2-|11-(трет- бутилдифенилсилилоксиметил) бамоилоксиметилпенем-3-карбоксилат
(1,9 г).
Стадия Д.
/-OOSiPh-jBu-t O
J-iL N-OCONHs
(
СОгСН7СН СН7
Раствор бис -силилированного продукта со стадии С (1,6 г) в тетрагидро- фуране (50 мл) перемешивают с тетра- бутиламмонийфторидтригидратом (0,95 г) и уксусной кислотой (0,35 мл), пока тонкослойная хроматография (Si02 этилацетат - циклогексан 1:2) не покажет полное исчезновение исходного соединения. По данным испарительной хроматографии получают аллил (5R, 6S)- -6- Ј(1К.)-трет-бутилдиметилсилилоксиме- тшГ|-2-Н-Г4-(оксиметил)феншГ{карба- моилоксиметилпенем-3-карбоксилат (0,6 г).
Стадия Е.
он/§По)
ДУ.,5 OCONH
САллил (5R, б5)-6-С()-трет-бутил- диметилсилилоксиэтил)2-М-Ј4(оксиме- тил)феншт карбамоилоксиметилпенем-3- -карбоксилат (0,1 г) в безводном (10 мл) последовательно обрабатывают пиридином (0,1 мл) и трифтор- метансульфоангидридом (0,05 мл) при - 40 С в атмосфере аргона. Реакционную смесь последовательно промывают разбавленной соляной кислотой и рассолом и растворитель отгоняют в вакууме. Остаток разводят простым ди- этиловым эфиром и полученную в результате промежуточную соль пиридина (70 мг) растворяют в тетрагидрофура- не (5 мл) и уксусной кислоте (0,06 мл) и перемешивают в течение 30 ч с тет- рабутиламмонийфторид тригидратом (95 мг). После удаления растворителя родукт очищают на SiOa-колонке, снаала используя в качестве элюента
1315
смесь СКаС1г и MeCN, затем чистый MeCN, затем смесь MeCN и Н20. Десили- лированный промежуточный продукт экстрагируют из последних фракций, промывают рассолом и выпаривают органический слой. Это соединение (50 мг) растворенное в смеси ТГФ и (каждого по 1 мл), обрабатывают уксусной кислотой (0,1 мл), трифенилфосфи- ном (50 мг) и тетракис(трифенилфос- фин) Pd (50 мг). Через 15 мин добавляют простой этиловый эфир, собирают осадок, растворяют в воде и пропускают через колонку Lichroprep RP-18, используя в качестве элюента воду и затем смесь MeCN с водой. После сушки с помощью вымораживания соответствующих фракций получают указанное в названии соединение (25 мг).
ЯМР (200МГц, )Ј , млн.д.: 1,24 (ЗН, д, J 6,4 Гц); 3,82 (1Н, дд, J 1,6 и 5,8 Гц); 4,18 (1Н, дк, J 5,8 и 6,4 Гц); 5,05 и 5,42 (2Н АВк, -J 14,6 Гц); 5,55 (1Н, fl,J 1,6 Гц); 5,75 (2Н, м); 7,43 (4Н, м); 8,05 (2Н, м); 8,53 (1Н, м); 8,89 (2Н, м).
ПримерЗ. (5R, 6S)-6-(1R)-OK сиэтил -2- 4-(3,5-диметил-1-пиридинио) метилфеншт оксиметилпенем-3-кар- боксилат (соединение 11).
Раствор аллил (5R, 6S)-6-(1К)-три метилсилилоксиэтшГ -2 4-(бромметил)фенил оксиметилпенем-3-карбоксилата (О,1 г) в безводном диметилформамиде (2 мл) перемешивают в течение ночи с 3,5-диметилпиридином (0,15 мл). Раствор удаляют в вакууме и остаток раз- водяют простым диэтиловым эфиром, получая желтоватое твердое вещество (0,1 г), которое перемешивают в течение 30 мин в ТГФ - вода - уксусная кислота (6:2:1, 9 мл). Смесь выпаривают и получают сырой сложный аллило- вый эфир указанного в названии продук та, который затем получают путем деал лилирования в присутствии Pd катализатора, как описано в примере 1.
ИК (КВг)ЗмаКС: 1765, 1605 см.
УФ (КгО)ма(с : 308 нм.
ПримерЭ. По методике, описанной в примере 8, заменив 3,5-диметил- пиридин на 3-оксипиридин и натрий-4- -пиридинэтансульфонат, получают следующие продукты.
(5R, 6S)-6- (1R)-Оксиэтил 2- b-(3- 1-гидрокси-1-пиридинио)-метилфенил ок- симетшшенем-3-карбоксилат (соединение 12).
1
14
ИК (KBr)}Walcc. : 1763, 1600 . УФ (H80) s 256 и 307 нм. (5R, б8)-6-ГОЮ-оксиэтил -2- 4-(4- -сул ьфоэтил-1 -пиридинио) -метилфенил оксиметилпенем-3-карбоксилат (соединение 13).
ИК (KBr))MOIKC : 1765 см . УФ (HjO) макс : 308 нм. Пример 10. (5R, б5)-6-(1Ю- -Оксиэтил -2-Ј4-(1-пиридинио)метилфе- нокси карбонилоксиметилпенем-3-карбок- силат (соединение 8).
В раствор аллил (5R, 6S)-5-(1-R)- -трет-бутилдиметилсилилоксиэтилЗ-2-ок- симетилпенем-3-карбоксилата (400 мг) в безводном дихлорметане (20 мл) добавляют при -10°С в атмосфере азота 4-(трет-бутилдифенилсилилоксиметил)- фенилхлоркарбонат (500 мг) и триэтил- амин (0,2 мл). Реакционную смесь нагревают до комнатной температуры, промывают водным раствором NaHCOj, обезвоживают и выпаривают. После хроматографии на силикагеле получают аллил (5R, б5)-5-Ц(1)трет-бутилдиметилси- лилоксиэтилД-2- 4-(трет-бутилднфенил- силилокси)метилфеноксиЗкарбонилоксиме- тилпенем-3-карбоксилат (0,6 г). Раствор этого продукта в ТГФ (20 мл) перемешивают в течение 8 ч в присутствии уксусной кислоты (0,4 мл) и тетрабу- тиламмонийфторид тригидрата (0,4 г). Растворитель удаляют в вакууме, остаток фракционируют на колонке с сили- кагелем (н-гексан - этилацетат) и получают аллил (5R, 6S)-6- (j(tR)-TpeT- бутилдиметилсилилоксиэтил2 2-(4-окси- метилфенокси)карбонилоксиметилпенем- 3карбоксилат (0,35 г). Это соединение последовательно вводят во взаимодействие с пиридин/трифторметансуль- фоангидридом, десилилируют и деалли- лируют по методике, описанной в примере 1, стадия С, и получают указанное в названии соединение в виде аморфного твердого вещества (75 мг).
ИК (KBr)3MC,KC : 1765, 1750, 1600 .
УФ aizO)MOMЈ0: 307 нм. Пример 11. (5R, 6S)-6-j(1R)- -Оксиэтшу-2- 4-( 1-пиридинио)метил- фенил тиометилпенем-3-карбоксилат (соединение 9).
В раствор 4-метилтиобензальдегида (15,2 г) в метаноле (200 мл) добавляют при 0°С боргидрид натрия (3,8 г). После нейтрализации НС1 реакционную смесь концентрируют и разделяют меж10
ду EtOAc и рассолом. После удаления растворителя получают 4-ме тилтиобен- зиловый спирт. Это соединение (13,3 г) обрабатывают ХПВК (15,5 г) в СНС1Э 300 мл) при 0°С. Через 30 мин раствор промывают водным раствором NaHCO $ и выпаривают, в результате чего получают 4-метилсульфонилбеняиловый спирт. Это соединение (7.9 г) в безводном дихлорметане (100 мл) обрабатывают трифгорметансульфоангидридом (19,5 мл) и нагревают при температуре дефлегмации 30 мин. Реакционную смесь вливают в раствор NaOH в этаноле и получают сырой 4-меркаптобензиловый спирт, раствор которого в хлороформе сразу же окисляют водным раствором KI (добавляют по каплям до появления устойчивой окраски). После промывки NaHCO и рассолом и удаления растворителя получают 4-оксиметилфенилдисульфид. Это соединение (3 г) обрабатывают трет-бутилдифенилсилилхлоридом (6,5 мл) и имидазолом (5 г) в безводном диме- 25 тилформамиде. Через 2 ч перемешивания при комнатной температуре и хроматографии получают 4-трет-бутилдифенилси- лилоксиметилфенилдисульфид (5,5 г) в
лилоксиэтил -2-хлорметилпенем-З-кар- боксилата. Реакционную смесь разделяют между водой и этилацетатом, остаток из сухого органического слоя очищают при помощи хроматографии на сили- кагеле и получают аллил (5R, 6S)-6- Ј( 1R) -трет-бутилдиметилсилилоксиэтшГ -2-(4-оксиметилфенил) сульфонилметил- пенем-3-карбоксилат. Это вещество обрабатывают пиридином/трифторметансуль- фоангидридом, затем десилилируют и де- аллилиру ют в соответствии с методикой, описанной в примере 1, и получают указанное в названии соединение.
ИКумакс: 1765, 1605 ,
Пример 13. (5R, 6S)-6-(lR)-ОксиэтшГ|-2-Ј4-(4-карбокси-1-пириди- нио)-метилфенил оксимгтилпенем-3-кар- Ьоксилат (соединение 14).
Стадия А. Защита карбоксильной Аункции, представленной в этой реакции.
В суспензию изоникотиновой кислоты (6,15 г) в сухом ДМФ (100 мл) добавляют триэтиламин (8,4 мл) и полученный в результате раствор перемешивают всю ночь в присутствии аллилбромида
5
35
виде сиропа. Непосредственно перед ис-зд (5,1 мл). Реакционную смесь разделяют
между водой и этилацетатом и органический слой разделяют рассолом, обезвоживают на ,)., выпаривают в вакууме и получают 4-аллилоксикарЬонил- пиридин (3,1 г) в виде бесцветного масла.
ЯМР (90 МГц, СОСЦ) Ј, млн.д.: 4,77 (2Н, м); 5,24 (1Н, д, J 10 Гц); 5,37 (1Н, д, J 12 Гц); 5,94 (1Н, м); 7,80 (2Н, м); 8,/О (2Н, м).
Стадия В. Конденсация с аллил (5R, 6S) -6- Ч 1R)-оксиэтил1-2- (4-бромметил- фенил)оксиметилпенем-3-карбоксилатом.
Указанный в названии промежуточный ., продукт, содержа щий заместитель бром- метилфенил (ЬОО мг), перемешивают в течение ночи с 4-аллилоксикарбонилпи- ридином (800 мг) в сухом ДМФ. После удаления растворителя под высоким вакуумом, остаток поглощают дихлормета- ном и по каплям вливают в этиловый простой эсЬир, осадок собирают (500 мг) и используют для следующей стадии.
Стадия С. Удаление защитных групп.
Сырой аллил (5R, 6S)-6- (Ш)-Окси- 3THLnJ-2- j4- (4-аллилкарбонил-1-пириди- нио)метилфеншт оксиметштенем-3-кар- боксилат (бромид), полученный на предыдущей стадии, перемешивают в течепользованием вышеуказанный дисульфид (1 г) восстанавливают до 4-(трет-бу- тилдифенилсилилокси -етил) тиофенола путем кратковременной обработки порошком цинка (1 г) в смеси уксусной кислоты и дихлорметана (по 20 мл). Полученный таким образом сырой меркаптан соединяют с аллил-(5R, 6S)-5-(1R) -трёт-бутилдиметилсилилоксиэтил -2-ок- симетилпенем-3-карбоксилатом при усло- Q виях, описанных в примере 1, стадия Б. Полученный аллил (5R, 6S)-6-f(1R) -трет-бутилдиметилсилилоксиэтшт -2-(4- -оксиметилфенил)тиометилпенем-3-кар- боксилат обрабатывают смесью пиридина и трифторметансульфоангидрида, десилилируют и деаллилируют в соответствии с методикой примера 1, в результате чего получают указанное в названии соединение.п
ИК (KBr)3Mcm: 3300, 1765,1600 гмЛ50 Пример 12. (5R, 6S)-6-((1R)- -Юксиэтшт -2-Ј4-(1-пиридинио) -метил- фенил сульфонилметилпенем-З-карбокси- лат (соединение 10).
Раствор натрий 4-(оксиметил) фенил- сульфината в сухом ТГФ перемешивают всю ночь с 1 М эквивалентом аллил (, 6S)-b ( Ш)-трет-бутилдиметилси
5
лилоксиэтил -2-хлорметилпенем-З-кар- боксилата. Реакционную смесь разделяют между водой и этилацетатом, остаток из сухого органического слоя очищают при помощи хроматографии на сили- кагеле и получают аллил (5R, 6S)-6- Ј( 1R) -трет-бутилдиметилсилилоксиэтшГ -2-(4-оксиметилфенил) сульфонилметил- пенем-3-карбоксилат. Это вещество обрабатывают пиридином/трифторметансуль- фоангидридом, затем десилилируют и де- аллилиру ют в соответствии с методикой, описанной в примере 1, и получают указанное в названии соединение.
ИКумакс: 1765, 1605 ,
Пример 13. (5R, 6S)-6-(lR)-ОксиэтшГ|-2-Ј4-(4-карбокси-1-пириди- нио)-метилфенил оксимгтилпенем-3-кар- Ьоксилат (соединение 14).
Стадия А. Защита карбоксильной Аункции, представленной в этой реакции.
В суспензию изоникотиновой кислоты (6,15 г) в сухом ДМФ (100 мл) добавляют триэтиламин (8,4 мл) и полученный в результате раствор перемешивают всю ночь в присутствии аллилбромида
5
ние 30 мин с трифенилфосфином (250 мг и Pd (PhjP)(250 мг), ацетонитрилом (50 мг) и уксусной кислотой (5 мл). Затем добавляют дополнительное количество PPh и Pd ()Ч (каждого по 250 мг) и смесь перемешивают в течение 30 мин, после чего разбавляют простым этиловым эфиром (190 мл). Осажденное твердое вещество собирают фильтрацией, промывают диэтиловым эфиром, растворяют в водном NaHCO и подвергают хроматографии на Lichrop- rep RP-18, выполнив элюирование сначала водой, а затем смесью ацетон - вода.
Соответствующие части объединяют, сушат замораживанием и получают 240 мг указанного в названии соединения в виде белого порошка.
ЯМР (200МГц, UZ0) $ , млн.д.: 1,31 (ЗН, д, J 6,6 Гц); 3,98, д, J 1,6 и 6,0 Гц); 4,26 (1Н, дв. д, J 6,0 и 6,6 Гц); 5,26 (2Н, АВ кв); 5,77 (1Н, д, J 1,6 Гц); 5,84 (2Н с); 7,46 (4Н, м); 8,25 (2Н, м); 8,95 (2Н, м).
У ,
ИК (КВг)у МО(КС
Пример 14.
-ОксиэтшГ|-2-П4-(4-диметиламино-1-пи- ридинио)-метилфеншГ оксиметилпенем-3- -карбоксилат (соединение 15).
Раствор 4-диметиламинопиридина (1 г) и аллил (5R, 6S)-6- (1К)-окси- (4-бромметилфенил) оксиметил- г енем-3-карбоксилата (500 мг) в сухом ДМФ (5 мл) перемешивают при комнатной температуре 12 ч. Растворитель отгоняют под высоким вакуумом и остаток растирают в порошок с простым этиловым эфиром и получают смолистый осадок аллилового сложного эфира (бромид) , указанного в названии соединения. Без дополнительной очистки это вещество растворяют в 50 мл дихлорме- тана и добавляют уксусную кислоту (1 мл), трифенилфосфин (0,25 г) и тетракис(трифенилфосфин) Pd (0) (0,25 г) в указанном порядке. После перемешивания в течение 30 мин реакционную смесь концентрируют и остаток растирают в порошок с этилацетатом и подвергают хроматографии на Lichrop- rep RP-18, элюируя сначала водой, а затем 10%-ным CHjCN в воде. Сушка вымораживанием соответствующих фракций дает указанное в названии соединение (270 мг) в виде белого порошка.
308 нм.
1765, 1630 . (5R, 6S)-6-(1R)
0
Q
УФ (НгОПма)м;: 307 нм.
ИК (КВг)макс : 1760, 1600 см .
Пример 15. (5R, 6S)-5-(1R)- -оксиэтил -2- 4-(1-метилпиперазинио)- -метилфеншт оксиметилпенем-3-карбок- силат (соединение 16).
Стадия А. Защита N-функциональной группы, присутствующей в реагенте.
Раствор аллилхлорформата (15 мл) в сухом дихлорметане добавляют при -20°С при перемешивании в раствор N- метил-пиперазина (10 г) в том же растворителе. Через 30 мин температуру повышают до 15°С и реакционную смесь выливают в воду. Органический слой сливают, добавляют до рН 7 1н,КаОН и смесь экстрагируют дихлорметаном. Экстракты СНгС12 обезвоживают (MgSOjj.) , выпаривают, полученное масло| перегоняют при 118°(при 18 мм рт.ст.) и получают 4-аллилоксикарбонил-1-метил- -пиперазин (15,3 г).
Стадия В. Конденсация с аллил (5R, 5 6S)-6- Г( Ш)-оксиэтил-2-(бромметилфе- нил) оксиметилпенем-3-карбоксилатом.
Конденсацию указанного в названии соединения с 4-аллилоксикарбонкп-1-ме- тилпиперазином проводят по методике, описанной в примере 13, стадия В,
Стадия С. Удаление защитных групп.
Полученный на предыдущей стадии сырой аллил (5R, б5)-(т)оксиэтшт 2- -р4-аллилоксикарбонил-2-метилпипера- зинио)-метилфеншГ|оксиметшшенем-3- -карбоксилат обрабатывают PPhj Pd(PPh)jHOAc при условиях, описанных в принрре 13, стадия С. Проводя очистку при помощи хроматографии с обращенной фазой (Lichroprep RP-fB) и сушку вымораживанием, получают указанное в названии соединение.
№Br)JMaVC
%опма(сс
1765, 1600 308 нм.
см
Пример 16. По методике примера 13 (стадии В и С), заменив 4-ал- лилоксикарбонилпиридин 4-сульфоэтан- пиридином, 3-аллилоксикарбонил-эте- ншширидином, 3-аллилоксикарбонилме- тилпиридином и 4-аллилоксикарбонил- аминометилпиридином, получают соответственно следующие соединения:
a) (5R, 65)-6-Ј(1Я)-гидроксиэтил - -2- Ч-(4-сульфонатоэтанпиридинио)ме- тилфеноксиметилЛ-пенем-З-карбоновая кислота
19
он
ж
COiH
УФ (H50)iMOUO: 258, 308 нм; ИК (КВг) акс : 1760, 1570 - 1610
V.
в) (5R, 6S)-6-Ј(1К)-гидроксиэтил - -2-Ј4-(3-карбоксилатэтенил)-пипириди- ниометилфенил оксиметилпенем-3-кар- боновая кислота
Т S СНг (Й1
-Vf Т о-1
uCOjH
.СН7ч®
4J/-V
Кф0
сн сн-сог
УФ (H10)MO|VC: 306 нм;
с) (5R, б8)-6-(1К)-гидроксиэтил - -2-Ј4-(3-карбоксилатметил)-пиридинио- метилфенил1оксометилпенем-3-карбоновая кислота
2Q
CH7cof
УФ (Н СМиакс: 303 нм 4225;
ИК -(KBr) 3 1610 см 1;
макс
1665, 1580 d) (5R, 6S)-6-Г(Ш-гидроксиметил -2- 4- (4-аллилоксикарбониламиноме- тил) пиpидиниoмeтилфeнилJоксиметилпе- нем-3-карбоксилат
©
снг-о- -снг-т« -сн7 тсо2 снг сн-сн i
со
УФ (НгО)1мв1се: 307 нм; ИК (KBr) 1765, 1705, 1750 - 1610 см- ;
При испытаниях in vivo после внут рибркпщнного введения соединения формулы I показали очень высокую терапев ическую активность при лечении инфек |ций, вызванных как грамположительными, так и грамотрицательными бактерия ми, причем их токсичность незначитель на.
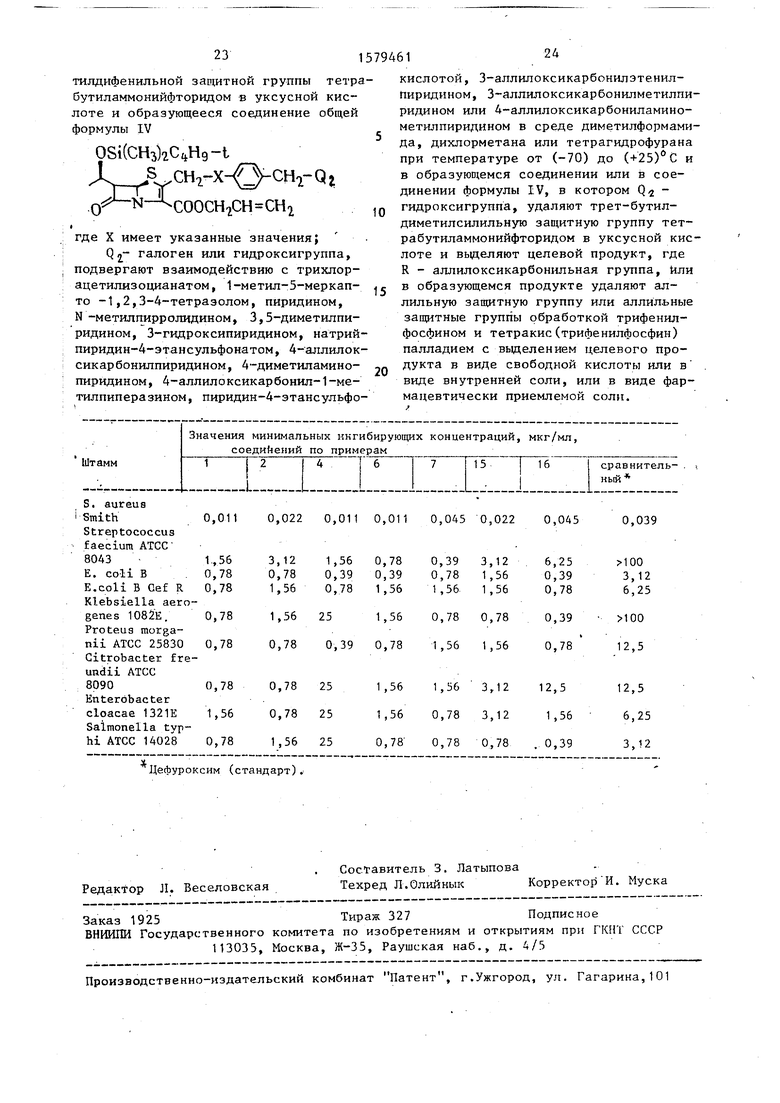
В таблице приведены данные по изучению минимальных ингибирующих кон45
е) (5R, 6S)-6-(1К)-гидроксиметил центраций пенемовых соединений. -2-{Ч-(4-аминометил)-пиридиниометил-Соединения, полученные по предлафенил } оксиметилпенем-3-карбоксилат, ,Q гидрохлорид
гаемому способу, обладают противобак- териальной активностью и поэтому могут быть использованы при лечении инфекций дыхательных путей, например бронхитов, бронхопневмонии, ллеврита гепатобилиарных и абдоминальных инфекций, например септицемии, инфекций мочепроводящих путей, например пиелонефритов, циститов, акушерских и гине кологических инфекций,например цер-
ь
.1Х,сн7-о-(о -сн1-н@)-сн%3а г
со:
УФ (): 257 нм, 308 нм; ИК (КВг)мокс :1/65, 1560 - 1U10 см- .
10
я
579461 2°
Соединения формулы I, получаемые по предлагаемому способу, представляют собой сильные противобактериальные препараты широкого спектра действия.
При испытаниях in vivo они показали очень интересные фармакокинетичес- кие свойства и высокую терапевтическую эффективность при лечении инфекций, вызванных грамположительными и грамотрицательными бактериями. Кроме того, соединения формулы I проявили высокую эффективность при оральном введении.
Соединения формулы I эффективны и in vitro при следующих дозировках: против Coeci (включая пенициллиназооб- разующие) 0,001-1 мкг/мл, против кишечных бактерий (включая |3 -лактама- 2Q зообразующие) 0,5-50 мкг/мл. Данные по испытаниям (5R,6S)-6- (Ш)-гидрок- сиэтил1-2- 4-(1-пиридинио)метилфе- нил оксиметилненем-3-карбоксилата (пример 1, соединение 4) in vivo:
t мкг/мл.мин
15
1/2 р.
Абсорбция,
% 100
-
35
Мыши 3361 27 Крысы 6150 28 3Q Влияние на вызванную в эксперименте у мышей( внутрибрюшинно ввели 3 х х ДЦгЈ- для контрольного заражения, лечение через 30,90 и 360 мин после заражения, 3JL мг/кг, кумуляторная доза :
Staphilococcus aureus Smith 4.0,032 Escherichia coli Gil1,55
При испытаниях in vivo после внут- рибркпщнного введения соединения формулы I показали очень высокую терапев- ическую активность при лечении инфек- |ций, вызванных как грамположительными, так и грамотрицательными бактериями, причем их токсичность незначительна.
В таблице приведены данные по изучению минимальных ингибирующих кон40
45
центраций пенемовых соединений. Соединения, полученные по предла
гаемому способу, обладают противобак- териальной активностью и поэтому могут быть использованы при лечении инфекций дыхательных путей, например бронхитов, бронхопневмонии, ллеврита, гепатобилиарных и абдоминальных инфекций, например септицемии, инфекций мочепроводящих путей, например пиелонефритов, циститов, акушерских и гинекологических инфекций,например цер-
вицитов, эндометритов, инфекций уха, горлд, носа, например отитов, синуситов, паротитов.
Эти соединения можно вводить как животным, так и людям в различных формах, например орально в форме табле- ток, капсул, капель или сиропов, рек- тально в форме свечей, парентерально, например внутривенно или внутримышеч- I но (как растворы или суспензии), причем в экстренных ситуациях предпочтительны растворы, путем ингаляции в форме аэрозолей или растворов для рас- внутривагинально в форме пес- / сариев, или топически в форме ма зей, кремов. Фармацевтические или ветеринарные композиции, содержащие соединения формулы I, можно получить, используя обычные носители или разбави- 2 тели, например используемые для цефа- лоспоринов.
Обычные растворители или разбавители - вода, желатин, лактоза,, крахмалы, стеарат магния, тальк, расти- 2 тельные млела, целлюлоза и т-.п. Суточная доза составляет 0,5-100 мг/кг веса тела, причем для различных пациентов конкретная доза зависит от возраста, веса и состояния и от способа 3 введения препарата.
Предпочтительным для введения сое-, динений формулы I является паренте- ральный способ. В этих случаях соединения взрослым пациентам можно в-водитьз в дозе 200-500 мг на прием, предпочтительно 400 мг на прием, 1-4 раза в день, растворенными в подходящем растворителе, таком как стерильная. вода или раствор лидокаина гидрохлорида 4 для внутримышечных инъекций, стерильная вода, физиологический раствор, раствор декстрозы или обычные жидкости для внутривенного вливания или электролиты для внутримышечных инъекций. 4 Кроме того, соединения формулы I можно использовать как противобакт ериаль- ные препараты в профилактических целях, например, в чистящих или моющих или дезинфицирующих поверхность ком- позициях, например, в концентрации 0,2-1 вес.% в смеси с суспендированными или растворенными инертными сухими или водными носителями для мытья или опрыскивания.
i
Эти соединения можно также использовать как полезные пищевые добавки в корм животным.
Формула изобретения
Способ получения производных пе- нема общей формулы I
9Н ; /-ч
,А- гснг-х- -сн2а
где R - свободная карбоксигруппа, ал лилоксикарбонильная. группа и карбоксилатанион;
X - кислород или сера, оксикарбо нильная группа, оксикарбонил аминогруппа, оксикарбонилокс группа или сульфонильная група;
Q - гидрокси, карбамоилокси, 1-мтил тетразол-5-илтио, пириди- нио, N - метилпирролидинио, 3,5-диметилпиридинИо, 3- гидроксипиридинио, 4-сульфо- - этилпиридинио, 4-карбок- , сипиридинио, 4-ди- метиламинопиридинио, N-ме- тилпицеразинио, 4-сульфона- тоэтанпиридинио, 3-карбок- силатэтенилпиридинно, 3-карбоксилатметилпири- динио, 4-аминометилпири- динио или 4-аллилоксикар- бониламинометилпиридинио, или их фармацевтически приемлемых солей, отличающийся тем, что соединение общей формулы II
OSi(CH3)
VCHjL
COOCH2CH CH2
где L - гидроксигруппа или хлор, подвергают взаимодействию с соединением общей формулы III
A- -CH2Qi
где QJ- галоген или группа формулы
-OSi(CgH5-)2C(}H9-t;
А - гидроксильная, меркапто, карбоксильная, хлоркарбонильная изоцианато, хлоркарбонилокси- группа или сульфиногруппа, в среде тетрагидрофурана, хлороформа или дихлорметана при температуре от (-10) до (+65)°С с .последующим, в случае ее наличия, удалением трет-бу23
тилдифенильной защитной группы тетра- бутиламмонийфторидом в уксусной кислоте и образующееся соединение общей формулы IV
osKow Hg-t
VCH x 0-CHrQ
.-Т-Х
о
СООСН2СН СН2
15
где X имеет указанные значения;
Qj- галоген или гидроксигруппа, подвергают взаимодействию с трихлор- ацетилизоцианатом, 1-метил-5-меркап- то -1,2,3-4-тетраэолом, пиридином, N -метилпирролидином, 3,5-диметилпи- ридином, 3-гидроксипиридином, натрий- пиридин-4-этансульфонатом, 4-аллилок- сикарбонилпиридином, 4-диметиламино- пиридином, 4-аллилоксикарбонил-1-ме- тилпиперазином, пиридин-4-этансульфо-
10
5794612
кислотой, 3-аллилоксикарбонилэтенил- Пиридином, 3-аллилокгикарбонилметилпи- ридином или 4-аллилоксикарбониламино- метилпиридином в среде диметилформами- да, дихлорметана или тетрагидрофурана при температуре от (-70) до (+25)°С и в образующемся соединении или в соединении формулы IV, в котором Q.2 - гидроксигруппа, удаляют трет-бутил- диметилсшшльную защитную группу тет- рабутиламмонийфторидом в уксусной кислоте и выделяют целевой продукт, где R - аллилоксикарбонильная группа, или в образующемся продукте удаляют ал- лильную защитную группу или аллильные защитные группы обработкой трифенил- фосфином и тетракис(трифенилфосфин) палладием с выделением целевого продукта в виде свободной кислоты или в виде внутренней соли, или в виде фарсоли.
15
20
мацевтически приемлемой
г
