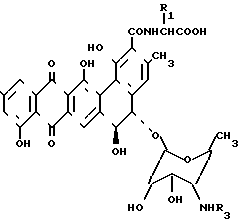
Изобретение относится к способам получения N-алкилпроизводных антибиотиков общей формулы I
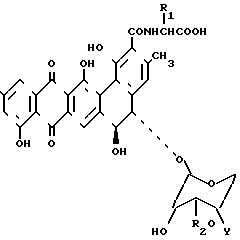 или их фармацевтически приемлемых солей, где R1 - водород или метил, причем в этом случае получается D-аланил,
или их фармацевтически приемлемых солей, где R1 - водород или метил, причем в этом случае получается D-аланил,
R2 - водород или β-D-ксилозил
Y -  R3R4R5X-, где R3 R4 и R5 - одинаковый или различный - С1-С5алкил, а Х - анион, активных в качестве противогрибковых средств.
R3R4R5X-, где R3 R4 и R5 - одинаковый или различный - С1-С5алкил, а Х - анион, активных в качестве противогрибковых средств.
Цель изобретения - получение новых водорастворимых производных антибиотиков производных 1.
Соединения формулы I получают взаимодействием соответствующего производного 1, где Y замещено группой NR7 R8, где
R7 и R8 независимо - Н или С1-С5 алкил с С1-С5 алкилгалогенидом до получения соответствующего аммонийного продукта.
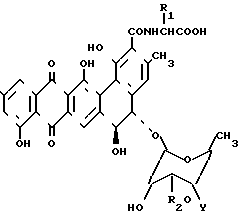 где 1a : BU - 3508; R1 = CH3; R2 - 0 - ксилозил; R3 = CH3;
где 1a : BU - 3508; R1 = CH3; R2 - 0 - ксилозил; R3 = CH3;
1b : BU - 3608 B; R1 = CH3; R2 = H; R3 = CH3;
1c : BU - 3608 C; R1 = CH3; R2 = 0-ксилозил; R3 = H;
1d : BU - 3608 D; R1 = H; R2 = 0-ксилозил; R3 = CH3;
1e : BU - 3608 b; R1 = H; R2 = 0-ксилозил; R3 = H.
BU - 3608, однако имеет очень ограниченную растворимость в воде. Таким образом, целью данного изобретения является получение водорастворимых производных различных компонентов антибиотического комплекса BU - 3608.
Изобретение рассматривает соединения, имеющие формулу II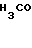
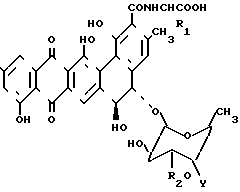 где R1 - Н либо метил, в этом случае полученный аланил - D-аланил; R2 - Н или β-D-ксилозил: Y -
где R1 - Н либо метил, в этом случае полученный аланил - D-аланил; R2 - Н или β-D-ксилозил: Y - 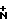 R3R4R5X, где R3, R4 и R5 - одинаковые или разные С1-С5 алкилы, а Х - это какой-либо анион, или их фармакологически приемлемые соли.
R3R4R5X, где R3, R4 и R5 - одинаковые или разные С1-С5 алкилы, а Х - это какой-либо анион, или их фармакологически приемлемые соли.
β-D-ксилозил представляет собой фрагмент
HO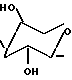
Другим аспектом изобретения являются бесксилозильные производные BU - 3608 C (IIIa), В - 3608 Д (IIIб) и BU - 3608 Е (III в), либо их фармакологически приемлемые соли.
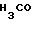
 где IIIa : R1 = CH3, R3 = H,
где IIIa : R1 = CH3, R3 = H,
IIIb : R1 = H; R3 = CH3,
IIIc : R1 = H; R3 = H.
Антибиотики BU - 3608, BU - 3608, BU - 3608, C, BU - 3608 и BU - 3608 Е использованы в качестве исходных материалов для соединений данного изобретения и могут быть получены при культивировании производящего антибиотик вида Actinomadura hibina N Р157-2 и 0278-4, которые хранятся в Американском собрании культур видов (American Type Culture Collection) (Рокквилл, Мериленд) и имеют вспомогательные номера АТСС 53557 и АТСС 53646 соответственно. Мутантная линия, полученная из линии Р157-2 обработкой N-метил-N-нитро-N-нитрозогуанидина (НТГ) производит Д и К компоненты в больших количествах чем и Р157-2, и 0278-4. Эта мутантная линия, обозначенная как А2660 также хранится в АТСС и имеет вспомогательный номер АТСС 53762.
BU-3608 В является бесксилозильным производным BU-3608, BU-3608 B, бесксилозильные BU-3608 С, Д и Е могут быть получены при нагревании BU-3608, BU-3608 С, Д и Е соответственно с соляной кислотой столько времени, сколько достаточно для отщепления ксилозной группы и установлении рН раствора для осаждения желаемого продукта.
Аминогруппы BU-3608, BU-3608 С, Д, Е или их соответствующие бесксилозильные производные могут быть алкилированы реакцией с галоидалкилами и четвертичные аммониевые соли могут быть получены при исчерпывающем алкилировании соединений формулы I, III или II, где Y это NR3 R4.
Растворимость различных антибиотиков была определена в фосфатном буферном солевом растворе (ФБР) и полученные данные приведены ниже. ФБР (-) означает раствор, в каждом литре которого содержится 0,2 г KCl, 0,2 г КН2PO4, 8 г NaCl и 1:15 г Na2HPO4; ФБР (+) дополнительно содержит 100 мг MgCl2 - 6H2O и 100 мг CaCl2. Данные приведены в табл.1.
Для сравнения, растворимость BU-3608 составляет 16-18 мг/мл и 9-23 мг/мл в растворах ФБР (-) и ФБР (+) соответственно. Таким образом, соединения формулы II демонстрируют повышенную растворимость по сравнению с BU-3608.
Биологические свойства.
Противогрибковое действие представленных соединений данного изобретения было определено как in vitro, так и in vivo. Минимальная ингибирующая концентрация (МИК) против различных грибков, была определена методом последовательных разбавлений с использованием агара декстрозы Laboranel. Примерно 0,003 мл грибковой суспензии, содержащей 106 клеток на мл было нанесено на поверхность агарового слоя, содержащего испытуемый антибиотик. Значения МИК, записанные после инкубации культур в течение 44 ч при 28оС представлены в табл.2.
Действие соединений данного изобретения in vivo было испытано против Сandida albicans А9540 на мышах. Исследуемые организмы содержались 18 ч при 28оС в среде ДГП (дрожжевой экстракт, глюкоза, пепион, Н2НРО4, MgSO4) затем суспендировались солевом растворе. Самцы мышей 10R, весом 20-24 г инфицировались внутривенно примерно 10-кратной летальной дозой испытуемого препарата. Антибиотик в различных дозах вводили внутривенно по группам из 5 мышей в каждой сразу же после инфицирования. Доза, которая защищает 50% животных от инфекции (PD50мг/кг), была вычисленa из доли выживших животных на 20 день после грибковой инфекции. Все контрольные животные умерли с 7 по 15 день после инфекции. PD50 для соединений примеров 5, 7 и 8 составляет 14 мг/кг (токсическая), 11 мг/кг и 3,5 мг/кг соответственно.
П р и м е р 1. Ферментация Actinomadura hibina штамма 0157-2.
а. Агаровый носитель Actinomadura hibina штамма 0157-2 проращивают на агаровом носителе, состоящем из 0,5%-ного растворимого крахмала (Ничиден Кагаку Ко), 0,5% глюкозы, 0,1% экстракта из мяса рыбы (Микуни Кагаку), 0,1% дрожжевого экстракта (Ориентл Уэст Ко), 0,2% N /фактора (Шеффил), 0,1% CaCO3, 0,2% NaCl, 1,6% агара. Культуру ингибируют при 28оС в течение 7 дней.
б. Рассада культуры. Порцию выросшей культуры переносят в колбу Эрленмейера на 500 мл, содержащую питательную среду следующего состава: 1% глюкозы, 2% растворимого крахмала (Ничиден Кагаку Ко), 0,5% N N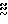 амина А (Шеффилд), 0,5% дрожжевого экстракта (Ориентл Уэст), 0,1% СаСО3. рН среды устанавливают 7,2 до стерилизации. Рассаду культуры инкубируют при 28оС 4 дня на роторном смесителе при 200 об/мин.
амина А (Шеффилд), 0,5% дрожжевого экстракта (Ориентл Уэст), 0,1% СаСО3. рН среды устанавливают 7,2 до стерилизации. Рассаду культуры инкубируют при 28оС 4 дня на роторном смесителе при 200 об/мин.
в. Ферментация в колбе. 5 мл микрофлоры переносят из рассады культуры в колбу Эрленмейера на 500 мл, которая содержит 100 мл среды следующего состава: 3% глюкозы, 3% соевой муки (Никко Сейю Ко), 0,5 "Фармамедиа" (Трейдерс Протеин) 0,1% дрожжевого экстракта (Ориентл Уэст), 0,3% СаСО3. Ферментацию проводят при 28оС в течение 5-6 дней на роторном смесителе. Производство антибиотика при ферментации контролируют методом разбавления с использованием Candida albicаns А9540 как индикатор в бульоне декстрозы. Также используется наблюдение в УФ при 500 нм в 0,01 н NaOH-MeOH растворе. Производство антибиотика достигает максимума 650 мкг/мл на пятый день.
г. Ферментация в резервуаре. 3 л рассады культуры прививают на 120 л стерильной среды, содержащейся в 200 л ферментаторе. Состав среды такой же, как использовался при ферментации в колбе. Ферментатор выдерживают при 28оС при перемешивании со скоростью 250 об/мин и аэрации со скоростью 120 л/мин. После 96 ч ферментации достигается содержание антибиотика 500 мкг/мл и рН бульона 7,9.
П р и м е р 2. Выделение и очистка антибиотика.
Созревший бульон (рН 7,8) центрифугируют, всплывающий слой подкисляют до рН 2,0 6 н. HCl для осаждения био-негативных соединений. После того, как осадок удален, рН фильтрата устанавливают 5,0 при помощи 6 н. NaOH и раствор перемешивают 30 мин при комнатной температуре. Полученный темно-красный осадок отфильтровывают и сушат в вакууме. Этот осадок затем растворяют в смеси 3: 1: 4 н-бутанол-метанол-1% NaCl и перемешивают смесь 30 мин. Нижний водный слой отделяют, промывают еще раз свежим верхним слоем, подкисляют до рН, 2,0 6 н. HCl и затем экстрагируют н-бутанолом. Экстракт промывают водой, упаривают в вакууме и лиофилизируют, получая полуочищенный гидрохлорид В-3608. Раствор твердого продукта в н-бутаноле встряхивают со щелочной водой (рН 9,0). Водный слой подкисляют до рН 2,0 и промывают этилацетатом. Экстракция н-бутанолом с последующим упариванием растворителя дает более чистый образец BV-3608 HCl. Этот продукт затем подвергают хроматографии c обращенной фазой на силикагеле (ODS-60, 350/250 меш, Ямамура Кемикл Лэб. колонка 4,5 х 90 см). Образец растворяют в воде и наносят на колонку, которая находится в равновесии при помощи смеси ацетонитрил-0,15% КН2РО4(рН 3,5) = 17: 83 (об.). Эту колонку последовательно промывают 5 л каждой из смесей ацетонитрил-0,15% КН2РО4 с соотношением компонентов 17:83, 18:82, 19:81, 20:80 и затем той же смесью растворителей с соотношением 22:78. Элюат собирают во фракции по 100 мл, за которыми следят при помощи микротекста с C. Albicans А9540 и ТСХ (силикагель, метилацетат-н-пропанол-28% водный аммиак 45:105:60). Фракции, содержащие в основном одно соединение объединяют и очищают далее, получая BV-3608.
В хроматографии с обращенной фазой на силикагеле, описанной выше, фракции, элюирующие до и после гомогенного BV-3608, объединяют. Объединенный элюат раскисляют, используя смолу НР-20, чтобы получить продукты, содержащие BV-3608 В и С. Продукт, содержащий BV-3608 В растворяют в воде и наносят на колонку ODS-60 (8,0х90 см) и промывают смесью ацетонитрил - 0,15% КН2РО4 22: 78 (рН 3,5). Фракции собирают и анализируют при помощи HPLC. Фракции, содержащие BV-3608 В, объединяют, упаривают в вакууме, и раскисляют при помощи хроматографии на смоле НР-20, получая загрязненный BV-3608, чистый BV-3608 и BV-3608 В. BV-3608 В затем очищают при помощи препаративной HPLC на колонке Microsorb Short One C18 (4,6 мм IDх100 мм, 3 мм, Рейнин Инструмент Ко), элюируя смесью ацетонитрил-0,15% КН2РО4 21:79 (рН 3,5). Для текущего анализа элюата используется НР-С, фракции, содержащие BV-3608 С, собирают и упаривают в вакууме. Полученный водный раствор раскисляют с помощью хроматографии на смоле НР-20, получая почти чистый BV-3608 С и почти чистый BV-3608.
В процессе хроматографии с обращением фаз на силикагеле, описанном выше, фракции, элюирующиеся ранее BV-3608 С, отдельно собирают и объединяют. Объединенные бледно-оранжевые фракции раскисляют с использованием хроматографии на Diaion НР-20. Полученный продукт относительно обогащен Д и Е компонентами, однако еще содержит большое количество С компонентов. Объединенные продукты наносят на колонку с обращенной фазой (ODS-60, силикагель, 8,0х90 см) и элюируют смесью ацетонитрил - 0,15% КН2РО4 21:79 (рН 3,5). Элюат анализируют при помощи HPIC с использованием вышеописанной колонки и смеси тех же растворителей 7:17 (рН 3,5) в качестве подвижной фазы при скорости течения 1,2 мл/мин и УФ-детектирования (254 нм). BV-3608 Е элюируется первым, за ним следует BV-3608 Д. Фракции, содержащие BV-3608 Е объединяют, концентрируют в вакууме и раскисляют при помощи НР-20, получая почти однородный BV-3608 Е HCl. При установлении рН водного раствора BV-3608 Е HCl равным 5,0 0,1 н. NaOH чистый BV-3608 Е осаждается в виде цвиттер-иона. Аналогично в виде цвиттер-иона получается и BV-3608 Д.
П р и м е р 3. Получение бесксилозильного BV-3608 E (III с).
Раствор гидрохлорида BV-3608 Е (97 мг) в 2 н. HCl (12 мл) нагревают при 115оС 70 мин в запаянной ампуле. рН полученного раствора устанавливают равным 5,5 прибавлением 1 н. NaOH и затем центрифугируют. Полученный таким образом твердый продукт промывают изопропанолом и ацетоном, получая 87 мл бесксилозильного BV-3608 Е. т.пл. 205-209оС (с разл.).
УФ 0,01 н. NaOH λмакс нм (ε): 235,2 (23600), 319,2 (11100), 498,4 (10800).
П р и м е р 4. Получение бесксилозильного BV-3608 С (III a).
Используя методику примера 3, из гидрохлорида BV-3608 С получают названное соединение.
т.пл. 208-215оС (разл.).
П р и м е р 5. Получение N-метил BV-3608 B (II, R2 = H R1 = R3 = R4 = CH3),
Смесь BV-3608 (540 мл) в 2 н. HCl (60 мл) нагревают при 115оС 70 мин в запаянной ампуле и затем охлаждают. Полученный твердый продукт отделяют центрифугированием (3000 об/мин), суспендируют в воде и устанавливают рН 11,7 6 н. NaOH. Раствор (60 мл) прибавляют к ацетону (300 мл) и отделяют полученный твердый BV-3608 В. Продукт растворяют в воде (20 мл) и добавляют 1 н. HCl до установления рН 8,3 и затем разбавляют 20 мл ацетонитрила. Водный НСНО (37%, 0,8 мл) и NaBH3CN (120 мг) последовательно добавляют к раствору при комнатной температуре и раствор перемешивают 15 ч. Растворитель удаляют в вакууме и водный остаток по каплям при перемешивании прибавляют к ацетону. Полученный осадок промывают ацетоном и сушат, получая 440 мг N-метил BV-3608 В в виде натриевой соли. Часть этой соли растворяют в воде и подкисляют 1 н. HCl до рН 6,0. Полученный осадок промывают водой и лиофилизируют, получая 33 мг цвиттер-ионной формы.
т.пл. 211-215оС (разл.).
УФ 0,01 н. NaOH-MeOH λмакс нм (ε): 240,8 (29700), 319,2 (13400), 499,2 (13100).
П р и м е р 6. Получение N,N-диметил бесксилозильного BV-3608 Е (II, R1 = R2 = H, R3 = R4 = CH3).
К раствору бесксилозильного BV-3608 Е (52,9 мг) в воде (5 мл) приливают MeCN (5 мл). Водный НСНО (0,2 мл) и NaBH3CN (30 мг) последовательно прибавляют к раствору при комнатной температуре и полученный раствор перемешивают 14 ч. Растворитель удаляют в вакууме и водный остаток (рН 11,3) при перемешивании прибавляют к ацетону. Полученный осадок растворяют в воде и устанавливают рН 5,5, получая твердый продукт, который последовательно промывают водой, изопропанолом, ацетоном и сушат, получая 28,7 мг N,N-диметил бесксилозильного BV-3608 Е, т.пл. 205-208оС.
УФ 0,01 н. NaOH λмакс нм (ε): 232,8 (34000), 319,2 (15700), 497,6 (14300).
П р и м е р 7. Получение N,N-диметил BV-3608 Е (II, R1 = H; R2 = ксилозил; R3 = =R4 = CH3).
К раствору BV-3608 Е (485 мг) в смеси воды (40 мл) и MeCN (40 мл) при рН 8,0 при комнатной температуре последовательно прибавляют водный НСНО (37% 1,6 мл) и NaBH3CN (240 мг). Раствор перемешивают при комнатной температуре 15 ч и растворитель удаляют в вакууме. Остаток растворяют в воде и устанавливают рН 11,0 и по каплям прибавляют к перемешиваемому ацетону (300 мл). Полученный осадок выделяют, растворяют в воде и подкисляют до рН 2,0 6 н. HCl. Раствор раскисляют при пропускании через НР-20. рН раствора, содержащего продукт, устанавливают 5,5 для его осаждения, осадок отделяют, промывают водой и ацетоном и сушат, получая 364 мг N,N-диметил BV-3608 Е.
Т.пл. 214-218 (разл.).
УФ 0,01 н. NaOH λмакс нм (ε): 233,6 (32900), 319,2 (15500), 497,6 (15100).
Вычислено, %: C 55,36; H 5,46; N 3,23.
C40H44N2O18˙1,5H2O.
Найдено, %: C 55,26; H 6,45; N 3,19.
П р и м е р 8. Получение N-метил BV-3608 (II, R1 = R3 = R4 = CH3, R2 = D-ксилозил).
К перемешиваемому раствору BV-3608 натриевой соли (550 мг) 50% водном MeCN (55 мл) прибавляют НСНО (37%, 0,75 мл) и NaBH3CN(150 мг) и смесь перемешивают 18 ч при комнатной температуре. После концентрирования водный концентрат разбавляют (200 мл), подкисляют до рН 3,0 и подвергают колоночной хроматографии на HР-20 (300 мл). После промывания водой и последующего элюирования 60% водным ацетоном элюат красного цвета концентрируют в вакууме и устанавливают рН 5,5 для осаждения N-метил BV-3608, который отфильтровывают (425 мг).
T.пл. 190-195оС.
ИК (KBr) см: 3400, 1605, 1450, 1295.
УФ 50% MeOH λмакс нм (ε): 220 (33700), 278 (26800), 488 (11400).
Масс-спектр: 855 (М + Н)+.
П р и м е р 9. Получение четвертичного аммониевого производного BV-3608 (I. Y = =N(CH3)3Cl).
Натриевую соль ВV-3608 обрабатывают метилтиодилом (1,5 мл) и бикарбонатом калия (200 мг) в ДМСО (5 мл) и метаноле (20 мл) при комнатной температуре 43 ч. Смесь концентрируют, разбавляют 0,5 н. NaOH (20 мл) и выдерживают 30 мин при 70оС. Устанавливают рН раствора 3,0 и подвергают хроматографии на НР-20 (150 мл). Элюат, содержащий названной соединение, упаривают и получают грязный продукт - четвертичное аммониевое производное (144 мг). Этот продукт подвергают хроматографии на силикагеле с обращенной фазой (2,0х45 см) с элюентом МеС - 0,15% КН2РО4 20:80 (рН 3,0). Элюат анализируют при помощи НР-С и фракции, содержащие чистое четвертичное основание, объединяют и раскисляют при помощи хроматографии на НР-20 (150 мл), получая чистое названное вещество (71 мг).
Т.пл. 205-210оС.
ИК (KBr) см-1: 3400, 1620, 1600, 1440, 1255.
УФ 50 0 МеОН λмакс нм (ε): 276 (22300), 498 (9800).
Масс-спектр: 869 (М+) Молекулярная формула C42H49N2O18Cl.
П р и м е р 10. Получение бесксилозильного BV-3608 Д (II в).
Методику, описанную в примере 3, повторяют, используя BV-3608 Д HCl, и получают названное соединение, т.пл. 205-211оС.
Использование в качестве противогрибкового препарата. Сущность: продукт N-алкилпроизводные антибиотиков или их фармацевтические соли ф-лы I, где R1 - H, CH3 (при этом получается D-аланил); R2 - H, β - D-кислозил; Y- 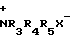 , где R3, R4, R5 - C1-C5 - алкил, X - анион. Выход 80%. БФ C42H42N2O18Cl, Y-N(CH3)3Cl . Реагент 1: соответствующее замещенное 1, где Y- NR7R8 , R7 и R8 - H или C1-C5 -алкил. Реагент 2: C1-C5 -алкилгалогенид. Условия реакции: при комнатной температуре в присутствии бикарбоната натрия. 2 табл.
, где R3, R4, R5 - C1-C5 - алкил, X - анион. Выход 80%. БФ C42H42N2O18Cl, Y-N(CH3)3Cl . Реагент 1: соответствующее замещенное 1, где Y- NR7R8 , R7 и R8 - H или C1-C5 -алкил. Реагент 2: C1-C5 -алкилгалогенид. Условия реакции: при комнатной температуре в присутствии бикарбоната натрия. 2 табл.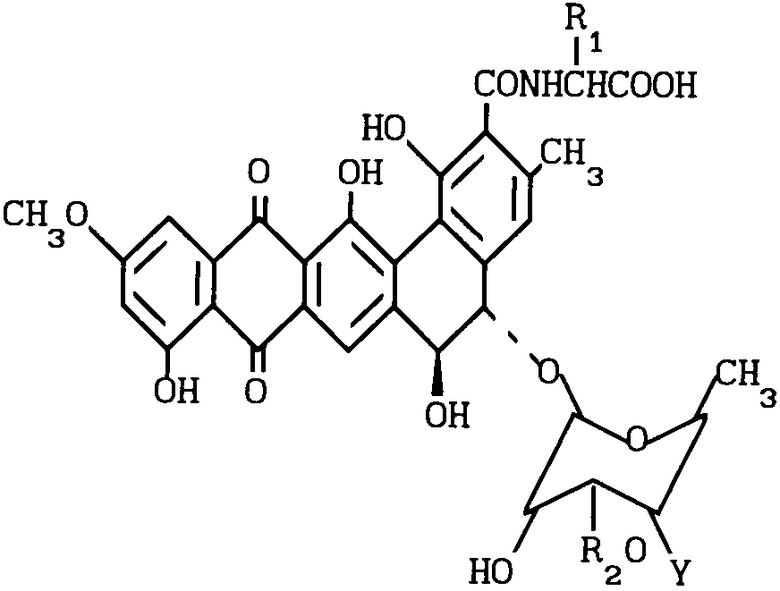
СПОСОБ ПОЛУЧЕНИЯ N-АЛКИЛПРОИЗВОДНЫХ АНТИБИОТИКОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ.
Способ получения N-алкилпроизводных антибиотиков общей формулы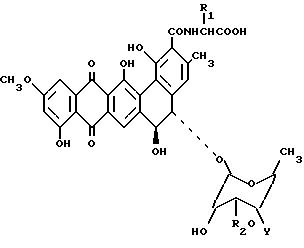
где R1 - водород или метил, причем в этом случае получается D-аланил;
R2 - водород или β = D - ксилозил;
Y - N+ R3 R4 R5 X-, где R3 R4 и R5, одинаковые или различные, - C1 - C5-алкил, а X-анион,
или их фармацевтически приемлемых солей, отличающийся тем, что проводят взаимодействие соединения общей формулы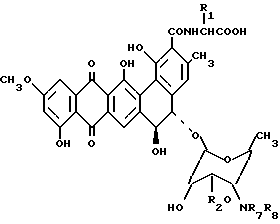
где R1 и R2 имеют указанные значения;
а R7 и R8 независимо - водород, C1 - C5-алкил,
или его соли с алкил-C1 - C5-галогенидом в количестве, достаточном для получения четвертичной аммониевой соли.
| Патент США N 4301277, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
