Заявка является частичным продолжением заявки на патент США рег. N 08/052434, поданной 23 апреля 1993 г.
Настоящее изобретение относится к способам получения N-незамещенных 2-азетидинонов, а также к новым соединениям, используемым в указанных способах.
Недавно было обнаружено, что таксол (соединение, выделяемое из коры тихоокеанского тиса) обладает многообещающим противораковым действием, особенно при лечении рака яичника. Поскольку таксол присутствует лишь в небольших количествах в коре медленно растущего тихоокеанского тиса, то для удовлетворения все возрастающего спроса на это лекарственное средство, особенно актуальной представляется разработка синтетического или полусинтетического метода получения таксола, тем более что этот метод позволил бы избежать нежелательной порчи тисовых деревьев.
В патенте США 5175315, выданном R.A.Holton 29 декабря 1992 г., раскрывается реакция взаимодействия защищенного баккатина III с защищеным N-бензол-3-гидрокси-4-фенил-2-азетидиноном (A) и получение в результате этой реакции таксола.

В патенте США 5175315 раскрывается получение 3-ацетокси-4-фенил-2-азетидинона (B), предшественника соединения (A), с помощью реакции ацетоксиацетилхлорида с N-бензилиден-4-метоксианилином, в результате которой получали N-(4-метоксифенил)-3-ацетокси-4-фенил-2-азетидинон, а после удаления 4-метоксифенильной группы осуществляли реакцию с нитратом цезия и аммония (CAN).
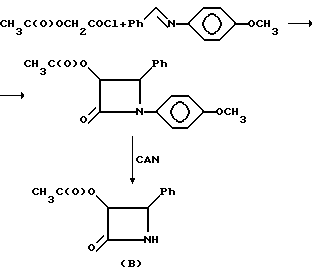
Описанный выше способ получения β- лактама (B) требует использования больших количеств CAN, что делает его непрактичным с точки зрения промышленного производства. Поэтому для крупномашстабного производства необходимо разработать новый улучшенный способ получения 3,4-замещенных-2-азетидинонов.
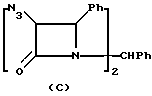
Manhas и др. ("Cyanuric Chloride: A Mild Reagent for β- Lactam Synthesis" Synthesis, 1981, 209-211) описывают синтез 3-азидо-4-фенил-2-азетидинона из азидоацетата калия, гидробензамида и циануровой кислоты в присутствии триэтиламина с последующей обработкой 10% HCl. Well и Lee ("The Synthesis of 2-Azeti dinones" J.Org.Chem., 1969, 34:1477-1479) описывают синтез 3-азидо-4-фенил (и замещенный фенил)-2-азетидинона из азидоацетилхлорида и гидробензамида в присутствии триэтиламина с последующей обработкой 10% HCl. Однако ни Manhal, ни Wells не раскрывают выделения продукта циклоприсоединения; и лишь позже сообщалось (см. Synthesis, Sept. 1975, стр. 557), что таким продуктом циклоприсоединения является димерный азетидинон (С).
Настоящее изобретение относится к новым цис-N-иминометил-3,4-дизамещенным -β- лактамом, имеющим формулы (I):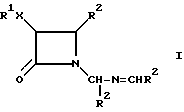
где R1 выбирают из группы, включающей в себя алкил, галогенозамещенный алкил, арил, циклоалкил и углеводное производное; X выбирают из O, N, S, C(O)O и прямой связи; R2 выбирают из группы, состоящей из арила, замещенного арила и гетероарила.
В другом своем варианте настоящее изобретения относится к способу получения цис-3,4-дизамещенных -β- лактамов, имеющих формулу (II):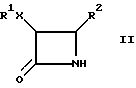
где R1, X и R2 определены выше, который заключается в том, что либо (1) соединение формулы (I) подвергают каталитическому гидрогенолизу; либо (2) соединение формулы (I) обрабатывают водным раствором кислоты, где указанную кислоту выбирают из группы, состоящей из бисульфита натрия, уксусной кислоты и муравьиной кислоты.
В еще одном своем варианте настоящее изобретение относится к способу получения β- лактама формулы (II), который заключается в том, что соединение формулы R1-X-CH2C(O)-L, а предпочтительно формулы R1C(O)OCH2C(O)-L, подвергают реакции с соединением формулы R2-CH-(N=CHR2)2 (L является уходящей группой; R1, X и R2 определены выше) в присутствии основания и при температуре около 5oC или ниже; а затем полученное в результате указанной реакции соединение либо (1) подвергают каталитическому гидрогенолизу; либо (2) обрабатывают водным раствором кислоты, где указанную кислоту выбирают из группы, состоящей из бисульфита натрия, уксусной кислоты и муравьиной кислоты.
Подробное описание изобретения
В настоящей заявке, если это не оговорено особо, используются следующие термины и определения. Термин "алкил" означает насыщенную прямую пли разветвленную углеродную цепь, имеющую от 1 до 6 атомов углерода; при этом в качестве примеров указанного алкила могут служить метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, нео-пентил и н-гексил. Термин "арил" означает моно- или бициклическую ароматическую карбоциклическую группу, примерами которой могут служить фенил и нафтил. Термин "галогено-замещенный алкил" означает алкильную группу, содержащую, по крайней мере, один атом галогена, выбранный из фтора, хлора, брома и иода; при этом в качестве примеров указанного галогено-замещенного алкила могут служить хлорметил, бромометил, трифторометил, трихлороэтил и иодоэтил. Термин "циклоалкил" означает насыщенную карбоциклическую группу, имеющую от 3 до 6 атомов углерода; при этом, в качестве примеров циклоалкила могут служить циклопропил, циклобутил, циклопентил и циклогексил. Термин "замещенный арил" означает арильную группу, имеющую от 1 до 3 одинаковых или различных заместителей, выбранных из C1-3-алкила, C1-3-алкокси, гидрокси, трифторометила и галогена. Термин "гетероарил" означает моно- или бициклическую ароматическую группу, имеющую в каждом кольце 5 или 6 атомов и имеющую, по крайней мере, один кольцевой гетероатом, выбранный из атомов азота, серы и кислорода; в частности, примерами указанного гетероарила могут служить фурил и тиенил. Термин "аралкил", означает группы, соответствующие структуре: арил CH(алкил)-. Термин "углеводное производное" означает группы, происходящие из углеводов, содержащих пиранозильное или фуранозильное кольцо; в частности, примерами углеводных производных могут служить 4,6-ди-O-ацетокси-2,3-дидезокси -α- D-глюкопиранозильная группа и т.п. Используемые в настоящем описании термины " β- лактам" и "2-азетидинон" являются взаимозаменяемыми.
Соединения формулы (I) могут быть использованы в качестве промежуточных соединений для получения цис-3-ацилокси-4-замещенные-2-азетидинонов, которые, в свою очередь, могут быть превращены в цис-1-ацил-3-защищенный гидрокси-4-замещенные-2-азетидиноны, которые затем используются для ацилирования баккатина III в целях получения таксола или его производных.
Цис-N-иминометил-2-азетидиноны формулы (I) могут быть получены путем реакции циклоприсоединения карбоксилата (а) с бис-имином (b) в присутствии основания, как указано на схеме 1.
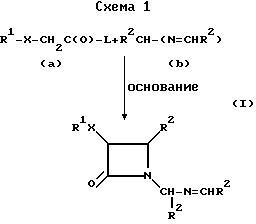
В схеме I R1, X и R2 являются такими, как они были определены выше. L является соответствующей уходящей группой, такой, как ацетат, например, трифторацетат; алкоксидная или алкилтио-группа, например, метоксид, этоксид или метилтио; игалид; или галид, а предпочтительно, если L является хлоридом. Предпочтительно, если в соединениях формулы (I), R1 представляет собой алкил, галогено-замещенный алкил, арил, арилалкил и группы углеводного производного. Более предпочтительно, если R1 представляет собой алкил, особенно метил и изопропил; хлороалкил, особенно, хлорометил; арил, особенно фенил; аралкил, особенно фенилэтил; и углеводное производное, особенно 4,6-ди-O-ацетокси-2', 3' - дидезокси -α- D-глюкопиранозил. Наиболее предпочтительно, если R1 является метилом. X выбирают из O, N, S, C(O)O и прямой связи. Предпочтительно, если X является C(O)O, а наиболее предпочтительно C(O)O. Предпочтительно, если R2 является арилом, замещенным арилом, фурилом и тиенилом; наиболее предпочтительно, фенилом, 4-метилфенилом, 4-метоксифенилом, 2-фурилом и 2-тиенилом.
Реакцию циклоприсоединения осуществляют в инертном органическом растворителе. Выбор конкретного растворителя не имеет решающего значения, если только этот растворитель не оказывает нежелательного воздействия на реакцию и не вступает в реакцию с исходными материалами и образующимися продуктами. Так, например, подходящими растворителями являются углеводороды, галогенированные углеводороды, сложные эфиры, простые эфиры, нитрилы, тиоэфиры и т.п. В качестве конкретных примеров могут быть упомянуты, например, бензол, толуол, ксилол, метиленхлорид, хлороформ, 1,2-дихлороэтан, этилацетат, н-пропилацетат, изобутиацетат, н-бутилацетат, тетрагидрофуран, н-бутилметиловый эфир и ацетонитрил. Предпочтительными растворителями являются метиленхлорид и этилацетат.
Основанием, используемым в данной реакции, может быть третичный органический амин, такой, как триметиламин, триэтилаин, диизопропилэтиламин, пиридин и диметиламинопиридин; либо более сильное металл-содержащее основание, такое как диизопропиламид лития, C1-6-алкиллитий, бис(триметилсилил)амидлития и фениллитий. Если L является галидом, то в данной реакции, предпочтительно, использовать третичный амин, такой как триэтиламин и диизопропилэтиламин, а если L не является галидом, то предпочтительно использовать более сильное основание, например, такое как диизопропиламид лития.
При осуществлении реакции циклоприсоединения имеется тенденция к образованию нежелательного димерного 2-азетидиона (с). Неожиданно было обнаружено, что димер (с) является непригодным для превращения в N-незамещенный 2-азетидинон формулы (II).
Таким образом, для максимизации выхода целевого соединения формулы (I), необходимо регулировать температуру реакции и относительные количества используемых реагентов. Эту реакцию проводят при пониженной температуре, предпочтительно при температуре около 5oC или ниже. А более предпочтительно, если температура реакции составляет от около -20oC до около 5oC.
В основном, для каждого эквивалента бис-имина (реагента (b)) используют от около 0,9 до около 1,5 эквивалента карбоксилата (реагента (a)) и от около 1 до около 1,5 эквивалента основания. Обычно на один эквивалент реагента (b) используют от около 1 до около 1,2 эквивалентов реагента (a), и от около 1 до около 1,2 эквивалентов основания. Реакцию осуществляют предпочтительно в инертной атмосфере, например в атмосфере аргона или азота, и в течение 24 часов. За ходом реакции следили с использованием стандартной хроматографической техники, например, такой как жидкости хроматография высокого давления с применением УФ-детектора.
Продукт, полученный в результате реакции циклоприсоединения, представляет собой смесь двух пар диастереомеров, где 3- и 4-заместители на азетидиновом кольце находятся по отношению друг к другу в цис-конфигурации. Хотя эти пары диастереомеров могут быть разделены, однако в этом нет необходимости. Отдельные стереоизомеры формулы (I) и их смеси также входят в объем настоящего изобретения.
Исходные материалы (a) и (b) либо являются коммерческими готовыми продуктами, либо они могут быть легко получены способами, хорошо известными специалистам. Так, например, одна группа соединений, используемых в качестве реагентов (a), может быть получена с помощью реакции соответствующего хлорангидрида карбоновой кислоты, R1C(O)Cl, с гликолевой кислотой, с получением ацилоксиуксусной кислоты, которая в свою очередь может быть дироватизирована, например, путем обработки тионилхлоридом с получением соответствующего ацилоксиацетилхлорида.
Другая группа используемых карбоксил-содержащих реагентов (a) может быть получена с помощью реакции этилбромоацетата с соответствующим алкоксидом, например, с алкоксидом, полученным из α- метилбензилового спирта. В результате гидролиза этилового спирта получают алкилоксиуксусную кислоту, которую, в свою очередь, подвергают дериватизации путем обработки тионилхлоридом с получением соответствующего алкоксиацетилхлорида.
Другие группы карбоксил-содержащих реагентов (a) могут быть получены с использованием процедуры, описанной Borer и Balogn в "An Asymmetric Synthesis of 3-Hydroxy -β- Lactam by Ketenelmi ne CycLoadditi on: UtiLicati on of Chirol Ketenes from Carbohydrates", Tetrahedron Lett., 1991, 32: 1039-1040.
Бис-иминовый реагент (b) может быть легко получен из соответствующего альдегида R2-C(O)H, и концентрированного гидроксида аммония в изопропиловом спирте.
В другом своем варианте, настоящее изобретение относится к удалению N-заместителя β- лактама формулы (I) с полученным соответствующего N-незамещенного β- лактама формулы (II), как показано в схеме II (R1 и R2 определены выше). В одном способе, N-заместитель удаляют путем каталитического гидрогенолиза, а в другом способе - это удаление осуществляют с использованием водного раствора кислоты, где указанной кислотой может быть уксусная кислота, бисульфит натрия, или муравьиная кислота.
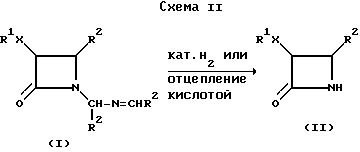
Каталитический гидрогенолиз β- лактама формулы (I) осуществляют в инертном органическом растворителе, который не вступает в реакцию с реагентами или образующимися продуктами, и не подвержен восстановлению в условиях гидрогенизации. Примерами подходящих растворителей являются сложные эфиры, такие как этилацетат; простые эфиры, такие как т-бутилметиловый эфир; спирт, такой как этанол; кетоны, такие как ацетон; углеводороды, такие как циклогексан; или амиды, такие как диметилформамид.
Катализатором может быть любой из обычно используемых катализаторов гидрирования, например, такой как платиновый, палладиевый, никелевый, родиевый и рутениевый катализатор. Предпочтительным является палладиевый катализатор. Этот катализатор может быть использован в виде сухого или водосодержащего палладированного угля, либо в виде гидроксида палладия, нанесенного на уголь. Гидрогенизации может быть проведена при давлении от около 2 фунт/кв. дюйм (0,14 кг/см2) до около 4 атм, и при температуре от около 20oC до около 30oC, а предпочтительно, при температуре окружающей среды.
В одном из предпочтительных вариантов осуществления настоящего изобретения, каталитической гидрогенизации подвергают β- лактамы формулы (I), где R1 является алкилом, а X является C(O)O. В другом предпочтительном варианте, каталитический гидрогенизации подвергают β- лактамы формулы (I), где R2 является арилом, замещенным арилом, или фурилом. В еще одном предпочтительном варианте осуществления настоящего изобретения, каталитической гидрогенизации подвергают β- лактамы формулы (I), где R1 является алкилом; R2 является арилом, замещенным арилом, или фурилом; X является C(O)O, а катализатор выбирают из палладированного угля и угля, покрытого гидроксидом палладия, а более предпочтительно, если R2 является фенилом, 4-метилфенилом, или фурилом; и наиболее предпочтительно, если R1 является метолом, R2 является фенилом, а X является C(O)O.
N-заместительт β- лактама формулы (I) может быть также отщеплен при соответствующим образом выбранных кислотных условиях. Так, например, в одном варианте настоящего изобретения β- лактам формулы (I) обрабатывают водной уксусной кислотой. Реакцию осуществляют в инертном органическом растворителе, таком как метиленхлорид, этилацетат, толуол, и т-бутилметиловый эфир. Уксусная кислота может быть использована в виде 60% - 80% (по объему), а предпочтительно от около 70% до около 75%-ного водного раствора, и в количестве от около 2,5 до около 65 эквивалентов по отношению к соединению формулы (I), а предпочтительно, от около 5 до около 8 эквивалентов. Эту реакцию проводят при повышенной температуре, например, при температуре перегонки реакционного раствора и завершают, в основном, примерно через 24 часа.
В другом способе, для удаления N-заместителя соединения формулы (I) используют водный бисульфит натрия. В этих целях может быть непосредственно использован коммерческий бисульфит натрия, смесь бисульфита натрия и метансульфита натрия. Бисульфит натрия используют в количестве от около 300 до около 600 г на один моль субстрата формулы (I). Данную реакцию осуществляют в инертном органическом растворителе при комнатной или повышенной температуре, например, при температуре от около 20 до около 60oC, а предпочтительно при около 50oC.
В еще одном варианте осуществления настоящего изобретения используют муравьиную кислоту в виде 70 - 98% - ного водного раствора (по массе), а предпочтительно в виде 90 - 95% - ного раствора (по массе), а предпочтительно в виде 90 - 95% - ного водного раствора. В основном, количество муравьиной кислоты составляет от около 4 до около 10 эквивалентов по отношению к соединению формулы (I), а предпочтительно от около 5 до около 7 эквивалентов. Обсуждаемые выше растворители обычно используются в сочетании с муравьиной кислотой.
В одном из предпочтительных вариантов осуществления настоящего изобретения β- лактам формулы (I), где R2 является арилом, замещенным арилом или тиенилом, превращают в соответствующий N-незамещенный β- лактам формулы (II) с использованием от около 70% до около 75%-ной водной уксусной кислоты. В другом предпочтительном варианте реакцию осуществляют в органическом растворителе, выбранном из галогенированного углеводорода, алкилацетата, углеводорода и простого эфира. В более предпочтительном варианте, R1 является метилом, R2 является фенилом, X является C(O)O, а реакцию осуществляют в метиленхлориде, с использованием 75% (по объему) водной уксусной кислоты.
В другом предпочтительном варианте β- лактам формулы (I), где R2 является арилом или замещенным арилом, превращают в соответствующий N-незамещенный β- лактам формулы (II) с использованием водного бисульфита натрия. В еще одном предпочтительном варианте реакцию осуществляют в органическом растворителе, выбранном из галогенированного углеводорода, алкилацетата, углеводорода и простого эфира. В более предпочтительном варианте R1 является метилом, R2 является фенилом, X является C(O)O, а реакцию осуществляют в метиленхлориде или этилацетате.
Хотя β- лактам формулы (I) может быть выделен, однако часто бывает предпочтительней проводить реакцию циклоприсоединения с последующим отщеплением N-заместителя без выделения промежуточного β- лактама формулы (I). Поэтому, в соответствии с другим вариантом своего осуществления, настоящее изобретение относится к способу получения N-незамещенного β- лактама формулы (II), заключающемуся в том, что соединение формулы R1X-CH2C(O)-L, где L является уходящей группой, подвергают реакции взаимодействия с соединением формулы R2-CH-(N= CHR2)2 в присутствии основания и при температуре около 5oC или ниже, без выделения продукта циклоприсоединения; причем полученное таким образом соединение либо (I) подвергают каталитическому гидрогенолизу, либо (2) обрабатывают водным раствором кислоты, где указанную кислоту выбирают из группы, включающей в беся бисульфит натрия, уксусную кислоту и муравьиную кислоту, а A1, R2, X и L являются такими, как они были определены выше.
В одном из предпочтительных вариантов настоящего изобретения, R1 является алкилом, арилалкилом или углеводным производным, R2 является арилом, замещенным арилом или фуранилом, X является C(O)O или O, а продукт циклоприсоединения подвергают каталитическому гидрогенолизу в присутствии палладиевого катализатора. Более предпочтительно, если реакционным растворителем является алкилацетат, такой как этилацетат, а основанием является триэтиламин. В наиболее предпочтительном варианте R2 является метилом, R2 является фенилом, а X является C(O)O.
В другом предпочтительном варианте настоящего изобретения R2 является арилом, замещенным арилом или тиенилом, а продукт циклоприсоединения обрабатывают водной уксусной кислотой. Более предпочтительно, если реакционным растворителем является галогенированный углеводород, такой как метиленхлорид, основанием является диизопропилэтиламин, а кислотой является 75% - ный водный раствор уксусной кислоты. Особенно предпочтительно, если R1 является метилом, R2 является фенилом, а X является C(O)O.
N-незамещенный β- лактам формулы (II), полученный способом настоящего изобретения, представляет собой рацемическую смесь двух энантиомеров, а именно цис-(3R)-3-ацилокси-4-замещенного-2-азетидинона (IIa) и цис-(3S)-3-ацилокси-4-замещенного-2-азетидинона (IIb).
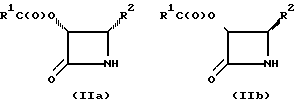
Указанная рацемическая смесь может быть разделена стандартными способами, например, такими, как превращение в диастереомеры; дифференциальная абсорбция на колонке, упакованной хиральными адсорбентами; или ферментативное разделение. Например, рацемическая смесь может быть подвергнута взаимодействию с ферментом, который катализирует гидролиз сложного эфира, такого, как эстераза или липаза, для селективного отщепления 3-ацильной группы одного энантиомера, не затрагивая при этом другой энантиомер. Альтернативно, рацемическая смесь может быть сначала подвергнута основному каталитическому гидролизу для удаления 3-ацильной группы и получения рацемической смеси, соответствующей 3-гидрокси- -β- лактаму; а затем эта рацемическая смесь 3-гидрокси -β- лактама может быть подвергнута взаимодействию с ферментом, катализирующим ацилирование гидроксигруппы, для селективного ацилирования гидрокси-группы одного энантимера, не затрагивания при этом другой энантиомер. Либо рацемическая смесь 3-гидрокси -β- лактама может быть ацилирована с использованием хиральной карбоновой кислоты, а полученная в результате диастереомерная смесь может быть затем разделена стандартными методами, с последующим удалением вспомогательного хирального соединения и получением нужного энантиомера. Энантиометрически чистый N-незамещенный 2-азетидинон может быть затем диреватизирован с получением N-ацил-3-защищенного гидроксизамещенного-2-азетидинона, который затем используют для ацилирования производного баккатина III для получения производного таксола.
Альтернативно, рацемическая смесь (IIa) и (IIb) может быть превращена в рацемическую смесь N-ацил-3-защищенный гидрокси-4-замещенный 2-азетидинона в соответствии с процедурой, описанной в патенте США 5175315; и эта рацемическая смесь может быть непосредственно использована для реакции с 13-металл-алкоксидом (например, литийалкоксидом) баккатина III в соответствии с высокой степени дистереоселективным методом, описанным в опубликованной европейской заявке 534708 (опубл. 31 марта 1993 г.), с получением таксола или его производных (III, где R может быть, например, фенилом или т-бутокси, а R2 является таким, как он было определен выше).
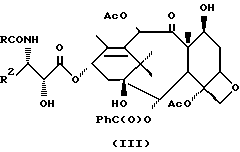
Использование таксола (R = R2 = фенил) и таксотера (R = т-бутилокси, R2 = фенил) в качестве противоопухолевых средств хорошо известно специалистам. Использование других производных таксола формулы (III) в качестве противоопухолевых средств описывалось в европейской заявке 534708 (опубликованной 31 марта 1993 г.) и в заявке PCT 92/09589 (опубликованной 11 июня 1992 г.).
Приведенные ниже примеры представлены в целях иллюстрации настоящего изобретения и не должны рассматриваться как некое ограничение объема изобретения.
Получение гидроксибензамида
В трехлитровую и трехгорловую колбу, снабженную механической мешалкой и термометром, добавляли 1 литр концентрированного гидроксида аммония, NH4OH (около 30%, 14,8 моль). Затем сразу, одной порцией, добавляли раствор бензальдегида (265 г, 2,50 М) с 500 мл 2-пропанола. Полученную смесь энергично размешивали в течение 43 часов при температуре около 22oC. Образовавшуюся в результате суспензию фильтровали, а остаток на фильтре промывали водой (1 литр). После осушки в вакууме получали 242,4 г гидроксибензамида в виде белого твердого вещества (т.пл. 100-102oC). Выход - 97,4%
В соответствии с процедурой, описанной выше, были получены следующие бис-имины формулы R2CH-(N = CHR2)2:
гидротолуамид (R2 = 4-метилфенил);
гидроанизамид (R2 = 4-метоксифенил);
гидрофуран (R2 = 2-фурил); и
гидротиенамид (R2 = 2-тиенил).
Пример 1.
(±)-цис-3-Бензиоилокси-1-[фенил(бензилидениминометил)]-4-фенил- азетидин-2-он (1)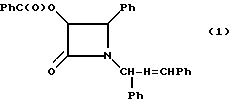
В 100-миллилитровую трехгорловую колбу, снабженную термометром, капельной воронкой и механической мешалкой, добавляли метиленхлорид (23 мл) и гидробензамид (7,04 г, 0,024 М). Полученный раствор охлаждали (сухим льдом/ацетоном) до - 20oC, в атмосфере сухого аргона. После этого сразу добавляли диизопропилэтиламин (3,52 г, 0,027 М). Затем добавляли по капле раствор (бензоилокси)ацетилхлорида (полученного в соответствии с процедурой, описанной S.J. Danishnefsky и др., Am. Chem. Soc. 1985, 107, 1280) (5,16 г, 0,026 М) в метиленхлориде (13 мл), поддерживая при этом температуру от - 20 до -15oC (прибл. 1,5 часа). Полученную смесь перемешивали еще 1 час при -20oC, а затем разводили деионизованной водой (13 мл) (экзотермическая реакция протекала до 0oC). Органическую фазу отделяли, промывали водой (2 • 10 мл), осушали безводным раствором сульфата магния и выпаривали досуха в вакууме, в результате чего получали целевое соединение в виде вязкого маслообразного вещества (11,97 г), состоящего из двух диастереомеров (прибл. 1: 1).
Образец полученного материала (2,97 г) очищали с помощью колоночной хроматографии с использованием неподвижной фазы окиси магния-двуокиси кремния (Florosil®, Fkuka Chemie AG) и элюировали 20% EtOAC/н-гексаном. Затем фракции центрального продукта объединяли и выпаривали досуха, в результате чего получали белое твердое вещество (0,803 г). Полученный материал перекристаллизовывали из смеси EtOAc (5 мл) и n-гексана (10 мл), в результате чего получали целевой продукт в виде белого твердого вещества (0,330 г), состоящий из двух диастереомеров (96:4).
Чистота (ВЖХР - площадь): 98,3%;
ЯМР (200 МГц, CACl3): δ 8,49 (с. 1H, NCH); 7,68-7,17 (м, 20H, Ar); 6,34 (с, 1H, NCH); 6,06 (д, 1H, J = 4,9 Гц, H-3); 4,85 (д, 1H, J = 4,9 Гц, H-4);
ИК (KBr) : ν (см-1): 1760 (C=O β- лактам), 1730 (C=O, сложный эфир); 1460 (C=N).
Элементный анализ для C30H24N2O3 (460, 53):
Вычислено: C 78,24; H 5,25; N 6,08;
Найдено: C 77,96; H 5,24; N 6,12.
Пример 2: (±)-цис-3-Бензоилокси-4-фенилазетидин-2-он (2)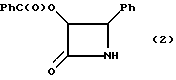
В соответствии с процедурой, описанной в примере 1, неочищенное вещество (9,00 г) растворяли в этилацетате (90 мл). К полученному раствору добавляли раствор бисульфата натрия (9,00 г, 58,5% мин. SO2 в виде смеси бисульфита и метансульфита) в воде (45 мл) и полученную двухфазную смесь нагревали до 50oC. После этого полученную смесь энергично перемешивали при 50oC до тех пор, пока тонкослойная хроматография (ТСХ) (SG 60 F254; 50% EtOAc-n-гексан; УФ254) не указывала на завершение реакции (4 ч.). Затем двухфазовый раствор разделяли в разделительной воронке. Органическую фазу отделяли, промывали водой (40 мл), осушали безводным сульфатом магния и выпаривали досуха в вакууме. Полученный твердый осадок (2,25 г) перекристаллизовывали из EtOAc (8 мл) и получали соединение в виде белого твердого вещества (3,55 г, полный выход исходя из гидроксибензамида - 74,9%) с т.пл. 118-119oC. Чистота (ВЭЖХ-площадь): 99,4%.
ЯМР (200 МГц, CDCl3): δ 7,68-7,21 (м, 10H, Ar); 6,49 (шир.с., 1H, NH); 6,18 (дд, 1H, J = 4,8; 2,7 Гц, H-3); 5,15 (д, 1H, J = 4,8 Гц, H-4).
ИК(KBr): ν (см-1) = 3260 (NH); 1755 (C=O β- лактам), 1730 (C=O, сложный эфир).
Элементный анализ для C16H13NO3(267, 28):
Вычислено: C 71,90; H 4,90; N 5,24
Найдено: C 71,76; H 4,93; N 5,31.
Пример 3: (±)-цис-3-Хлороацетокси-1-[(фенил)-(бензилидениминометил)]-4- фенилазетидин-2-он (3)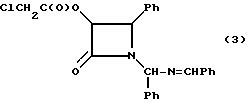
В 250-миллилитровую трехгорлую колбу, снабженную термометром, капельной воронкой и механической мешалкой, добавляли метиленхлорид (90 мл) и гидробензамид (22,38 г, 0,082 М). Полученный раствор охлаждали смесью сухого льда и ацетона до -20oC, в атмосфере сухого аргона. После этого сразу добавляли диизопропилэтиламин (11,19 г, 0,087 М). Затем к полученной смеси по капле добавляли раствор хлороацетоксиацетилхлорида (полученного в соответствии с процедурой, описанной R. Lattre-II и G. Lohaus, Liebigs Ann Chem, 1974, 870-900) (14,10 г, 0,082 М) в метиленхлориде (50 мл), поддерживая при этом температуру от -20oC до -15oC (прибл. 2 ч). Полученную смесь перемешивали еще 1 час при -15oC, разводили деионизированной водой (100 мл), экзотермическая реакция протекала до 0oC). Органическую фазу отделяли, промывали водой (2 • 25 мл), осушали безводным сульфатом магния и выпаривали в вакууме досуха, в результате чего получали неочищенный целевой продукт в виде вязкого маслообразного вещества (38,00 г), состоящий из двух диастереомеров (прибл. 1:1).
Образец полученного материала (3,22 г) очищали с помощью колоночной хроматографии с использованием неподвижной фазы окиси магния двуокиси кремния (Florosil®) и элюировали 20% EtOAc/n-гексаном. Фракции центрального продукта объединяли и выпаривали досуха, в результате чего получали желтое маслообразное вещество (0,480 г). Полученное вещество кристаллизовали из EtOAc (3 мл)/n-гексана (10 мл) и получали целевой продукт в виде белого твердого вещества (0,310 г), состоящего из двух диастереомеров (70:30). Чистота (ВЭЖХ-площадь): 98,6%.
ЯМР (200 МГц, CDCl3): δ 8,45 (с, 1H,N=CH); 7,92-6,99 (м, 15H, Ar); 6,28, 6,23 (два диастереомера) 2с, 1H, NCH); 5,88, 5,82 (два диастереомера) (2д, 1H, J = 4,8 Гц, H-3); 5,35 (4,80 (два диастереомера) (2д, 1H, J = 4,8 Гц, H-4); 3,57 (кв., 2H, J = 15,4, CH2).
ИК(KBr): ν (см-1) = 1760 (C=O β- лактам и сложный эфир); 1650 (C=N).
Элементный анализ для C25H21ClN2O3 (432, 90):
Вычислено: C 69,42; H 4,90; Cl 8,09; N 6,48
Найдено: C 69,27; H 4,89; Cl 8,40; N 6,39.
Пример 4 (±)-цис-3-Хлороацетокси-4-фенилазетидин-2-он (4)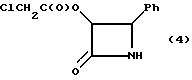
Часть неочищенного продукта 3 (4,26 г) растворяли в метиленхлориде (22 мл). Затем к этому раствору добавляли ледяную уксусную кислоту (4,60 мл) и деионизованную воду (1,46 мл), и полученный раствор нагревали с обратным холодильником. После этого смесь энергично перемешивали, продолжая нагревание до тех пор, пока тонкослойная хроматография не указывала (SG 60F254; 50% этилацетат/н-генсан; УФ254) на завершение реакции (4 часа). Полученный раствор нейтрализовали (pH 7,2) путем добавления (17 мл) по капле водного (3,5 н.) раствора NaOH при 5-10oC. Затем фазы разделяли, а водную фазу отбрасывали. Органическую фазу промывали водным раствором бисульфата натрия (25 мл, 10 мас.%), осушали безводным сульфатом магния и выпаривали досуха в вакууме. Полученный твердый остаток (2,30 г) перекристаллизовывали из толуола (10 мл) и получали целевое соединение в виде коричневого твердого вещества (0,727 г, полный выход исходя их гидробензамида - 36,1%). Чистота (ВЭЖХ - площадь): 91,4%.
ЯМР (200 МГц, CDCl3): δ = 7,41 - 7,27 (м, 5H, Ar); 6,43 (шир.с, 1H, NH); 5,94 (дд, 1H, J = 2,7, 4,7 Гц, H-3); 5,09 (д, 1H, J = 4,7 Гц; NH-4); 3,62 (кв., 2H, J = 15,3, CH2).
Пример 5. (±)-цис-3-Ацетилокси-1-[(фенил)(бензилденимино)- метил]-4-фенилазетидин-2-он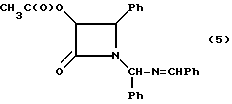
В литровую трехгорлую круглодонную колбу, снабженную термометром, магнитной мешалкой и капельной воронкой, добавляли гидробензамид (30,00 г; 100,5 мМ) и этилацетат (150 мл). При перемешивании, в атмосфере аргона, реакционную смесь охлаждали до 5oC и добавляли триэтиламин (16,8 мл, 121 мМ). После этого к смеси, по капле, добавляли в течение 90 минут раствор ацетоксиацетилхлорида (12,4 мл, 116 мМ) в этилацетате (30 мл). После выдерживания 16 часов, при той же температуре, реакционную смесь нагревали до 20oC (1,5 ч.) и переносили в разделительную воронку. Полученный органический слой последовательно промывали насыщенным водным раствором NH4Cl (150 мл, 100 мл), насыщенным водным раствором NaHCO3 (120 мл) и солевым раствором (120 мл). На этой стадии, в целях характеризации, целевое соединение может быть выделено путем осушки органической фазы сульфатом магния, фильтрацией и удалением растворителя в вакууме. В результате этой процедуры получали неочищенный наружный продукт с количественным выходом в виде красного стеклообразного вещества. Чистота (ВЭЖХ-площадь): 87,9% (1:1, смесь диастереомеров);
1H-ЯМР (CDCl3, 200 МГц): δ = 8,45 (с, 1H, N=CH); 7,80-7,85 (м, 1H, Ph); 7,60-7,65 (м, 1H, Ph); 7,26-7,50 (м, 9H, Ph); 7,00-7,10 (м, 4H, Ph); 6,28 (с, 0,5H, NCHN); 6,23 (с, 0,5H, NCHN); 6,23 (с, 0,5H, NCHN); 5,81 (д, J = 4,8 Гц, 0,5H, H-3): 5,76 (д, J = 4,8 Гц, 0,5 H, H-3); 5,30 (д, J = 4,8 Гц, 0,5H, H-4), 4,75 (д, J = 4,8 Гц, 0,5H, H-4), 1,63 (с, 3H, CH3CO).
ИК(KBr): ν (см-1)= 1763 (C= O), 1641 (C=N); УФ (метанол): λмакс (нм)= 216,252.
Пример 6. (±)-цис-3-Ацетолокси-4-фенилазетидин-2-он (6)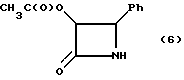
Раствор соединения примера 5 в этилацетате (500 мл) (в потоке аргона) осторожно переносили в 2,0-литровую колбу Парра, содержащую 10% палладия на активированном угле (6,00 г). Полученную смесь обрабатывали водородом (4 атм) в течение 20 часов, после чего катализатор удаляли путем фильтрации через слой целита® (диатомовая земля, Johns Man Ville). Затем осадок на фильтре суспендировали в этилацетате (200 мл), перемешивали в течение 10 минут и фильтровали. После этого осадок на фильтре промывали этилацетатом (100 мл), а фильтраты объединили. Органический слой промывали 10% HCl (300 мл) и оба слоя фильтровали через воронку из спеченного стекла для удаления белого осадка (дибензиламин, HCl), который промывали этилацетатом (100 мл). Затем фазы отделяли, а органический слой промывали еще один раз порцией раствора 10% HCl (200 мл). После этого 10% раствор HCl промывали и снова экстрагировали этилацетатом (200 мл), а объединенные органические слой промывали водным раствором NaHCO3 (нас.) (300 мл) и солевым раствором (250 мл). Органический слой осушали сульфатом магния, фильтровали и концентрировали в вакууме до объема 75 мл. Полученную смесь охлаждали до 4oC и осажденный продукт выделяли путем фильтрации. Осадок на фильтре промывали гексаном (200 мл) и получали 16,12 г (полный выход, исходя из гидробензамида 78,1%) целевого соединения в виде белых игольчатых кристаллов.
(т.пл. 150-151oC); Чистота (ВЭЖХ - площадь): 99,8%.
1H-ЯМР (CDCl3, 200 МГц): δ 7,30-7,38 (м, 5H, Ph); 6,54 (шир.с. изменяющийся, 1H, NH), 5,87 (дд, J = 2,7, 4,7 Гц, 1H, H-3), 5,04 (д, J = 4,7 Гц, 1H, H-4), 1,67 (с, 3H, CH3CO);
ИК(KBr): ν (см-1)=3210 (N-H), 1755, 1720 (C=O); KF: 0,17%;
Элементный анализ для C11H11NO3:
Вычислено: C 64,38; H 5,40; N 6,83
Найдено: C 64,07; H 5,34; N 6,77
Пример 7: (±)-цис-3-изо-Бутирилокси-1-[(фенил)бензилидениминометил]-4- фенилазетидин-2-он (7)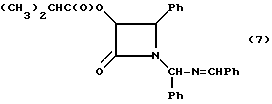
Целевое соединение получали в соответствии с процедурой описанной в примере 5, за исключением того, что изо-бутирилоксиацетилхлорид использовали вместо ацетоксиацетилхлорида. Таким образом, в результате реакции гидробензамида (30,00 г) (100,5 мМ), триэтиламина (16,8 мл, 121 мМ) и изо-бутирилоксиацетилхлорида (18,9 г, 115 мМ), проведенной в соответствии с процедурой, описанной Benington and Corin, J., Org. Chem., 26, 194 (1961), получали 50,65 г (118,8%) целевого соединения в виде темного оранжевого сиропа. В целях характеризации, порцию полученного раствора (4,65 г) очищали с помощью хроматографии Florosil® (элюент:этилацетат/гексан, 25:75) и получали целевое соединение в виде желтого сиропа.
1H-ЯМР(CDCl3, 200 МГц) (смесь диастереомеров, 1:1): δ = 8,47 (с, 0,5H, N= CH), 8,46 (с, 0,5H, N=CH), 7,80-7,91 (м, 2H, Ph), 7,29-7,68 (м, 9H, Ph), 6,94-7,11 (м, 4H, Ph); 6,29 (с, 0,5H, NCHN); 6,25 (с, 0,5H, NCHN); 5,81 (д, J = 4,9 Гц, 0,5H, H-3); 5,75 (д, J = 4,8 Гц, 0,5H, H-3); 5,32 (д, J = 4,9 Гц, 0,5H, H-4); 4,76 (д, J = 4,8 Гц, 0,5H, H-4); 2,20 (р, J = 7,0 Гц, 1H, CH(CH3)2), 0,80 (д, J= 7,0 Гц, 3H, CH(CH3)2), 0,56 (д, J = 7,0 Гц, 1,5H, CH(CH3)2), 0,54 (д, J = 7,0 Гц, 1,5 H, CH(CH3)2);
ИК (пленка): ν (см-1(=1771, 1748 (C=O), 1646 (C=N); УФ (метанол) λмакс (нм)-220,254
Пример 8: (±)-цис-3-(изо-бутирилокси)-4-фенилазетидин- 2-он (8)
Целевые соединения получали в соответствии с процедурой, описанной в примере 6, за исключением того, что был использован увлажненный катализатор, а реакцию осуществляли с масштабе 90,8 мМ, исходя из первоначального количества гидробензамида. Полученный таким образом неочищенный продукт, описанный в примере 7 (46,0 г) снова растворяли в этилацетате (460 мл) и добавляли к увлажненному 10% палладию на активированном угле (6,00 г катализатора и 6 мл воды), в результате чего получали 10,35 г целевого соединения (полный выход исходя из гидробензамида - 48,9%) в виде белых кристаллов (т. пл. 121-122oC).
Чистота (ВЭЖХ - площадь): 99,6%.
1H-ЯМР (CDCl3, 200 МГц): δ = 7,27-7,39 (м, 5H, Ph), 6,33 (шир.с, обмениваемый, 1H, NH), 5,87 (дд, J = 2,6, 4,7 Гц, 1H, H-3); 5,05 (д, J = 4,7 Гц, 1H, H-4), 2,24 (р, J = 7,0 Гц, 1H, CH(CH3)2), 0,38 (д, J = 7, OH, 3H, CH(CH3)), 0,58 (д, J = 7, OH, 3H, CH(CH3)); ИК(KBr): ν (см-1)=3203 (N-H), 1778, 1739 (C=O).
Пример 9. (±)-дис-3-Ацетилокси-1-[(4-метилфенил)(4- метилбензилиденимино]-4-(4-метилфенил)азетидин-2-он (9)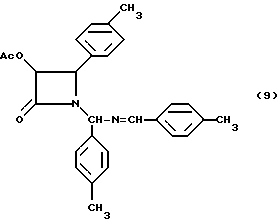
Целевое соединение получали в соответствии с процедурой, описанной в примере 5, за исключением того, что вместо гидробензамина использовали гидротолуамид. Таким образом, гидротолуамид (34,05 г, 100,0 мМ) триэтиламин (20,9 мл, 150 мМ) и ацетоксиацетилхлорид (18,9 г, 135 мМ) подвергали реакции и получали 52,0 г (118%) целевого соединения в виде коричневого сиропа. В целях характеризации, часть (5,2 г) полученного соединения очищали с помощью хроматографии Florosil® (элюент: этилацетат/гексан, 25:75) и получали целевое соединение в виде белого твердого вещества (т.пл. 110-128oC).
ВЭЖХ - чистота (площадь): 97,8% (1:1, смесь диастереомеров);
1H-ЯМР (CDCl3, 200 МГц):δ= 8,36 (с, 1H, N=CH), 7,75-7,80 (м, 1H, Ar); 7,53-7,56 (м, 1H, Ar); 7,06-7,35 (м, 7H, Ar); 6,81-6,90 (м, 3H, Ar); 6,19 (с, 0,5H, NCHN); 6,15 (с, 0,5H, NCHN); 5,79 (д, J = 4,8 Гц, 0,5H, H-3); 5,73 (д, J = 4,8 Гц, 0,5H, H-3); 5,27 (шир.с., 0,5H, H-4); 4,77 (шир.с., 0,5H, H-4); 2,40 (с, 1,5H, PhCH3); 2,39 (с, 1,5H, PhCH3); 2,35 (с, 3H, PhCH3); 2,24 (с, 1,5H, PhCH3), 2,20 (с, 1,5H, PhCH3), 1,661 (с, 1,5H, CH3CO), 1,658 (с, 1,5H, CH3CO);
ИК(KBr):ν (см-1)=1763, 1751 (C=O), 1635(C=N);
УФ(метанол): λмакс (нм) = 214, 260.
Пример 10: (±)-цис-3-(Ацетилокси)-4-(4-метилфенил)азетидин-2-он(10)
Метод 1: Гидрогенизация
Целевое соединение получали в соответствии с процедурой, описанной в примере 6, за исключением того, что был использован увлажненный катализатор, а реакцию осуществляли в масштабе 45 мМ, исходя из первоначального количества гидротолуамида. Полученный таким образом неочищенный продукт примера 9 (23,4 г) в этилацетате (315 мл) добавляли к увлажненному 10% палладию на активированном угле (3,00 г катализатора и 3 мл воды), в результате чего получали 3,50 г (35,5% - полный выход исходя из гидротолуамида) целевого соединения в виде белого твердого рыхлого вещества.
Метод 2: Бисульфит
К неочищенному продукту примера 9 (23,4 г) в этилацетате (315 мл) добавляли воду (75 мл) и бисульфит натрия (35 г). Затем эту двухфазовую смесь энергично перемешивали в течение 23 часов при 50oC, после чего органический и водный слой отделяли. Органический слой промывали водой (50 мл) и солевым раствором (150 мл), а затем осушали сульфатом магния и концентрировали в вакууме до объема 30 мл. Полученную смесь охлаждали до 5oC, а затем осажденное целевое соединение выделяли путем фильтрации и промывали холодным раствором этилацетата (10 мл), в результате чего получали 4,27 г (полный выход исходя из гидротолуамида - 43,3%) целевого соединения в виде белого твердого вещества (т. пл. 130-131oC) ВЭЖХ - чистота (площадь) : 98,6%.
1H-ЯМР (CDCl3; 200 МГц): δ = 7,13 - 7,22 (м, 4H, Ar): 6,29 (шир. с., обмененный, 1H, NH); 5,85 (дд, J= 2,6; 4,7 Гц, 1H, H-3); 5,00 (д, J= 4,7 Гц, 1H, H -4); 2,35 (с, 3H, PhCH-3); 1,70 (с, 3H, CH3CO); ИК (КВг): ν (см-1) = 3192 (N-H), 1778, 1752 (C=O); УФ (метанол) : λмакс (нм) = 222, 266.
ПримерII.(±)-цис-3-Ацетилокси-1-[(4'-метоксифенил)(4-метоксибензилиденимино)метил] -4-(4'-метоксифенил)азетидин-2-он(II)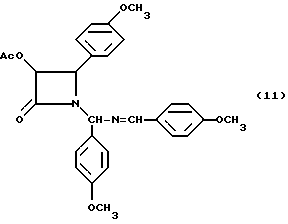
Целевое соединение получали в соответствии с процедурой, описанной в примере 5, за исключением того, что вместо гидробензамида был использован гидроанизамид, а реакцию осуществляли в масштабе 12,9 мМ (объем 100 мМ). Гидроанизамид (5,00 г, 12,9 мМ), триэтиламин (2,15 мл, 15,4 мМ) и ацетиоксиацетилхлорид (1,59 мл, 14,8 мМ) подвергали реакции и получали 6,38 г (101,2%) целевого соединения в виде бледного красного сиропа (смесь диастереомеров, 1:1);
1H-ЯМР (CDCl3; 200 МГц) : δ = 8,34 (с, 0,5H, N=CH), 8,33 (с, 0,5H, N= CH), 7,75 (д, J= 8,8 Гц, 0,5H, A); 7,58 (д, J=8,7 Гц, 0,5H) 7,14 - 7,27 (м, 3H, Ar), 6,78 - 7,03 (м, 6H, Ar); 6,63 (д, J=2,6 Гц, 1H, Ar); 6,58 (д, J= 2,5 Гц, 1H, Ar); 6,15 (с, 0,5H, NCHN), 6,11 (с, 0,5H, NCHN), 5,75 (д, J= 4,8 Гц, 0,5H, H-3), 5,69 (д, J= 4,7 Гц, 0,5H, H-3); 5,21 (д, J= 4,8 Гц, 0,5H H-4), 4,69 (д, J= 4,7 Гц, 0,5H, H-4), 3,89 (с. 3H, PhOCH3), 3,85 (с, 3H, PhOCH3), 3,81 (с, 1,5H, PhOCH3), 3,79 (с, 1,5H, PhOCH3), 1,68 (с, 1,5,), 1,67 (C, 1,5H, CH3CO);
УФ(метанол): λмакс (нм) = 216, 252.
Пример 12. (±-цис-3-(Ацетилокси)-4-(4'-метоксифенил)азетидин-2-он(12)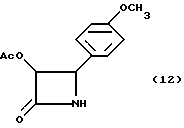
Целевое соединение получали в соответствии с процедурой, описанной в примере 6, за исключением того, что продукт был выделен с помощью препаративной ТСХ, а реакцию осуществляли в масштабе 12,9 мМ, исходя из первоначального количества гидроанизамида. Полученный сырой продукт примера 11 (6,38 снова растворяли в этилацетате (80 мл) и добавляли к 10% палладию на активированном угле (1,00 г), в результате чего получали 4,277 г (141%) сырого твердого вещества. Порцию (200 мг) полученного твердого вещества очищали с помощью препаративной ТСХ (2 мм; силикагель, этилацетат/гексан, 1:1) и получали 160 мг (полный выход из гидроанизамида - 75,5%) целевого соединения в виде светло-желтого порошка, который затем перекристаллизовывали из метиленхлорида/гексана (т. пл. 110- 111oC); Чистота (ВЭЖХ-площадь): 99,7%;
1H-ЯМР(CDCl3, 200 МГц): δ = 7,24 (д, 9,0 Гц, 2H, Ar); 6,89 (д, J=8,7 Гц, 2H, Ar); 6,23 (шир. с, обмененный, 1H, NH); 5,83 (дд, J= 2,7, 4,6 Гц, 1H, H-3), 4,99 (д, J= 4,6 Гц, 1H, H-4), 3,81 (с, 3H, PhOCH3) 1,73 (с, 3H, CH3CO);
ИК(KBr) : ν (cм-1)=3218 (N-H), 1751, 1728 (C=O);
УФ(метанол) : λмакс (нм) = 208, 230, 276.
Пример13.(±)-цис-3-Ацетилокси-1-[(2'-фурил)(2'-ферилметиленимино)метил]-4- (2'-фуранил)азетидин-2-он (13)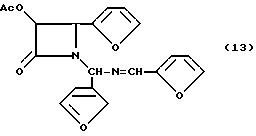
Целевое соединение получали в соответствии с процедурой, описанной в примере 5, за исключением того, что вместо гидробензамида был использован гидрофурамид, а реакцию осуществляли в масштабе 18,6 мМ (объем 100 мМ). Гидрофурамид (5,00 г, 18,6 мМ), триэтиламин (3,11 мл, 22, 3 мМ) и ацетоксиацетилхлорид (2,30 мл, 21,4 мМ) подвергали реакции и получали 6,192 г (90,4%) целевого соединения в виде бледного красного сиропа ( в виде смеси диастереомеров, 1:1).
1H-ЯМР(CDCl3, 200 МГц): δ = 8,211 (с, 0,5H, N=CH); 8,208 (с, 0,5H, N= CH), 7,14-7,59 (м, 3H, фурил), 6,90 (д, J= 3,5 Гц, 0,5H, фурил), 6,83 (д, J= 3,5 Гц, 0,5H, фурил), 6,10 - 6,53 (м, 6H, фурил, NCHN), 5,90 (д, J= 4,9 Гц, 0,5H, H-3), 5,86 (д, J= 4,8 Гц, 0,5H, H-3), 5,35 (д, J= 4,8 Гц, 0,5H, H-4), 4,90 (д, J= 4,9 Гц, 0,5H, H-4), 1,91) с, 1,5H, CH3CO), 1,88)с, 1,5H, CH3CO);
ИК(пленка): ν (см-1=1778, 1753(C=O), 1642(C=N);
УФ(метанол): λмакс = (нм) = 20, 278.
Пример 14. (±)-цис-3-(Ацетилокси)-4-(2'-фуранил)азетидин-2-он(14)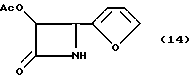
Целевое соединение получали в соответствии с процедурой, описанной в примере 6, за исключением того, что продукт выделяли с помощью препаративной ТСХ, а реакцию осуществляли в масштабе 2,7 мМ, исходя из первоначального количества гидрофурамида. Полученный неочищенный продукт Примера 13 (1,00 г ) снова растворяли в этилацетате (50 мл) и добавляли к 10% палладию на активированном угле (150 мг). После очистки неочищенного твердого вещества с помощью препаративной ТСХ (2 мм, силикагель, этилацетат/гексан, 1:1) получали 386 мг (полный выход, исходя из гидрофурамида - 65,8%) целевого соединения в виде желтого твердого вещества, которое затем перекристаллизовывали из этилацетата/гексана (т. пл 118-119oC; Чистота (ВЭЖХ - площадь) : 99,4%;
1H-ЯМР(CDCl3, 200 МГц): δ = 7,44 (т, J=1,3 Гц, 2H, фурил), 6,39 (д, J= 1,3 Гц, 1H, фурил), 6,21 (шир. с, обмененный, 1H, NH); 5,88 (дд, J=2,2, 4,6 Гц 1H, H-3); 5,05 (д, J=4,6 Гц, 1H, H-4), 2,92 (с, 3H, CH3CO);
ИК(KBr) : ν = (см-1 (=3203 (N-H), 1756, 1726, (C=O);
УФ(метанол) : λмакс (нм) = 222.
Пример15(±)-цис-3-Ацетилокси-1-[(2'-тиенил)(2'-тиенилметиленимино)метил] -4-(2'-тиенил)азетидин-2-он(15)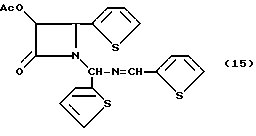
Целевое соединение получали в соответствии с процедурой, описанной в примере 5, за исключением того, что вместо гидробензамида был использован гидротиенамид. Гидротиенамид (30 г, 94,7 мМ), триэтиламин (15,84 мл, 114 мМ) м ацетоксиацетилхлорид (11,6 мл, 108 мМ) подвергали реакции и получали целевое соединение в виде вязкого маслообразного вещества. Полученный продукт содержит также смесь диастереомеров.
1H-ЯМР (CDCl3): δ 8,52 (с, 1H); 8,502 (с, 1H); 7,51 (д, J = 4,9 Гц, 1H); 7,45 (д, J= 4,4 Гц, 1H); 7,41 (д, J= 3,1 Гц, 1H): 7,37 (д, 1H); 7,30 (м, 3H); 7,16 (м, 1H); 7,16 (м, 3); 7,09 (м, 2H); 6,94 (м, 1H); 6,89 (м, 1H); 6,81-6,74 (м, 4H); 6,48 (с, 1H); 6,43 (с,1H); 5,85 (м, 2H); 5,59 (д, J= 4,8 Нц, 1H); 5,17 (д,J =4,8 Гц, 1H); 1,87 (с, 3H); 1,86 (с, 3H).
Пример 16.(±)-цис-3-(Ацетилокси)-4-(2'-тиенил)азетидин-2-он (16)
К перемешанному раствору примера 15 (0,431 г, 1,03 мМ), и дихлорметана (2,93 мл) при 25oC, добавляли одну порцию 70% водного раствора уксусной кислоты (0,35 мл ледяной уксусной кислоты и 0,15 мл воды). Затем полученную смесь нагревали с обратным холодильником и перемешивали в течение 2,5 часов. После того реакционную смесь разбавляли 50 мл дихлорметана и затем промывали двумя порциями 75 мл насыщенного водного раствора бикарбоната натрия и одной порцией 50-миллилитрового насыщенного солевого раствора. Полученный органический экстракт концентрировали в вакууме до получения коричневого маслообразного вещества, растворяли в минимальном количестве дихлорметана и затем помещали в колонку с силикагелем (101,6х12,7 мм). После элюирования градиентом смеси 10 - 60% EtOAc/гексана побочные продукты удаляли и получали целевое соединение (0,154 г, 75%) в виде белого твердого вещества.
1H-ЯМР (CDCl3): δ 7,32 (дд, J=47,, 1,5 Гц 1H), 7,03 (м, 2H), 6,75 (шир. с., 1H), 5,86 (дд, J=4,6, 2,7 Гц, 1H), 5,27 (д, J=5,3 Гц, 1H), 1,83 (с, 3H).
13C-ЯМР (CDCl3): δ 169,3, 165,5, 138,4, 127,1, 127,07, 126,2, 78,3, 54,0, 20,0.
Пример 17 (±)-цис-2-Ацетилокси-4-фенилазетидин-2-он (6).
Гидрогенолиз
В 2,0-литровую трехгорлую колбу, снабженную термометром, капельной воронкой и механической мешалкой, добавляли метиленхлорид (500 мл) гидробензамид (100,00 г, 0,335 м) и триэтиламин (56,0 мл, 0,402 М). Полученную смесь охлаждали в потоке аргона до 40oC с использованием ледяной бани, а затем по капле в течение 1 часа добавляли раствор ацетоксиацетилхлорида (41,35 мл, 0,385 мМ) в метиленхлориде (1000 мл). Через 1 час перемешивание прекращали, а реакционную колбу охлаждали до комнатной температуры в течение 15 часов, до тех пор, пока ТСХ не свидетельствовала о завершении реакции. После этого реакционную смесь переносили в 4-литровую делительную воронку и дважды промывали водным раствором NH4Cl (нас.) (500 мл, 250 мл). Органическую фазу осторожно промывали водным раствором NaHCO3) (нас.) (400 мл) и водным раствором NaCl (нас.) (250 мл), а затем осушали сульфатом магния (прибл. 100 г), фильтровали и концентрировали с помощью роторного испарителя (т. бани -40oC). Образовавшееся вязкое маслообразное вещество, кроме того, осушали путем откачки насосом (2 Торр.) в течение 1 дня, а затем отверждали и получали красное стеклообразное вещество (136,88 г, 102,5%), которое использовали без дополнительной очистки.
Неочищенное твердое вещество, описанное выше (136,88 г), растворяли в 800 мл этилацетата и переносили (в потоке аргона) в 2-литровую колбу Парра, содержащую 20,0 г 10% палладия на активированном угле (Aldri ch). После этого полученную смесь обрабатывали водородом (4 атм) в течение 1 дня при 23oC, а катализатор удаляли путем фильтрации через фильтровальную бумагу Whatman с использованием воронки Бэхнера. Полученный осадок на фильтре снова суспендировали в этилацетате (500 мл), перемешивали (10 минут) и снова фильтровали. Затем процедуру повторяли. Осадок на фильтре промывали 100 миллилитрами этилацетата, а фильтраты соединяли. Полученный органический слой промывали 1 н. раствором HCl (500 мл), а затем оба слоя фильтровали через фильтрованную бумагу Whatman для удаления белого осадка, который промывали этилацетатом (100 мл). Органический и водный слой разделяли, и органический слой промывали одной порцией 1н. раствора HCl (250 мл). После этого объединение 1 н. HCl - промывки снова экстрагировали этилацетатом (500 мл), а затем органические слои объединяли и последовательно промывали водным раствором NaHCO3 (нас.) (500 мл) и водным раствором NaCl (нас.) (300 мл). После этого органический слой осушали сульфатом магния (приблизительно 100 граммов), фильтровали и концентрировали в вакууме до объема 250 мл. Затем смесь охлаждали до 4oC (в течение ночи в холодной комнате), а осажденное твердое вещество выделяли путем фильтрации через фильтровальную бумагу Whatman. Осадок на фильтре промывали гексаном (200 мл) и осушали в вакууме, в результате чего получали чистый (±)-цис-3-ацетилокси-4-фенилазетидин-2-он (49,97 г, 72,7%) в виде белых игольчатых кристаллов (т.пл. 150 - 151oC).
Пример 18. (±)-цис-3-Ацетилокси-4-фенилазетидин-2-он (6) - Гидролиз с использованием 70% уксусной кислоты
1-литровую сухую трехгорлую круглодонную колбу (в атмосфере N2) загружали гидробензамидом (100,0 г, 335 мМ, 1 экв) и метиленхлоридом (333 мл). Полученный раствор охлаждали до 5oC и добавляли одну порцию диихопропилэтиламина (61,3 мл, 352 мМ, 1,05 экв). Реакционную смесь, кроме того, охлаждали до -20oC и температуру поддерживали постоянной. Ацетоксиацетилхлорид (167 мл) и метиленхлорид (36,0 мл, 335 мМ, 12 экв) смешивали и переносили в капельную воронку (полный объем: 203 мл). К полученному раствору в течение 2 часов и 10 минут добавляли предварительно полученный раствор гидробензамида. Скорость перемешивания корректировали так, чтобы температура не поднималась выше -16oC. Полученную светло-коричневую реакционную смесь перемешивали в течение 1 часа при -20oC. Аликвоты реакционной смеси удаляли (100 мл) через 5 минут и 30 минут после добавления ацетоксиацетилхлорида. Если реакционная смесь содержала > 5% гидробензамида, то к этой смеси через 30 минут добавляли диизопропилэтиламин (6,13 мл, 33,5 мМ, 0,10 экв), а затем раствор ацетоксиацетилхорида (3,60 мл), 33,5 мМ, 0,10 экв) в метиленхлориде (15 мл). После окончания реакции к смеси добавляли деионизованную воду (333 мл), а смесь перемешивали в течение 5 минут. Фазы отделяли, а реакционный сосуд промывали метиленхлоридом (110 мл). Полученный экстракт промывали метиленхлоридом, органические фазы объединяли (полный объем: 750 мл), а водную фазу отбрасывали.
Обогащенный органический экстракт переносили в 2-литровую круглодонную колбу, снабженную конденсатором и мешалкой, закрепленной в верхней части. К перемешанному раствору добавляли 70% водную уксусную кислоту (156 мл). Полный объем реакционной смеси составлял приблизительно 906 мл. Полученный раствор нагревали до температуры перегонки. Температуру резервуара поддерживали при 42 - 45oC. Хотя реакция завершилась через 8 часов, нагревание продолжали еще в течение 16 часов. Затем реакционную смесь охлаждали до 10oC. С помощью добавления водного раствора NaOH (3,75 н, 440 мл), pH реакционной смеси доводили до 5,10 - 7,10, а температуру поддерживали не ниже 20oC. После этого реакционную смесь переносили в 4-литровую колбу Эрленмейера и добавляли одну порцию водного раствора метабисульфита натрия (1110 мл, 10% об/об). Реакционную смесь перемешивали 10 минут при 30oC. Затем фазы отделяли, а обогащенный метиленхлоридный поток концентрировали в вакууме (25 мм рт.ст., т.бани 30oC) до объема 200 мл. После этого добавляли этилацетат, и концентрирование повторяли. Полученную суспензию перемешивали в течение 2 - 3 часов при -5oC. Образовавшееся твердое вещество собирали с помощью вакуумной фильтрации (диаметр фильтра - 15 см). Плотность продукта составляла приблизительно 3 мл/г. Полученный продукт промывали холодным (5oC) этилацетатом (100 мл). Затем этот продукт осушали до постоянной массы (1,0 мм рт. ст. , 25oC, 5 ч.) и (49,3 г, 240 мМ, полный выход - 717,%) и получали вещество в виде беловатых игольчатых кристаллов. Чистота (ВЭЖХ - площадь): 100,8%.
Пример 18. (±)-цис-3-Ацетилокси-4-фенилазетидин-2-он (6) - гидролиз с использованием водного бисульфита натрия А.1. Реакция циклоприсоединения в этилацетате
В 2-литровую трехгорлую колбу, снабженную термометром, капельной воронкой и механической мешалкой, добавляли этилацетат (500 мл) и гидробензамид (50,00 г, 0,167 М). Полученный раствор охлаждали в ледяной бане до 0oC, в атмосфере сухого аргона. После этого сразу добавляли триэтиламин (19,50 г, 0,193 М). Затем к раствору по капле приблизительно в течение 1,5 часа, при температуре 0 - 5oC, добавляли раствор ацетоксиацетилхлорида (24,02 г, 0,176 М) в этилацетате (20 мл). Полученную смесь перемешивали еще 2 часа при 0oC, а затем оставляли на 17 часов в холодной комнате (прибл. 5oC). После этого смесь разводили деионизованной водой (250 мл) и охлаждали (экзотермическая реакция протекала до 10oC). После энергичного перемешивания водную фазу отделяли и экстрагировали этилацетатом (100 мл). Органические фазы объединяли, а полученный раствор неочищенного 3-ацетокси-1-[(фенил)(бензилиденимино)метил] -4- фенилазетидин-2-она обрабатывали водным бисульфитом (как описано ниже в части B).
А.2. Реакция циклоприсоединения в метиленхлориде
В 1-литровую трехгорлую колбу, снабженную термометром, капельной воронкой и механической мешалкой, добавляли метиленхлорид (166 мл) и гидробензамид (50,00 г, 0,167 М). Полученный раствор охлаждали в бане из сухого льда/ацетона до -20oC (в атмосфере сухого аргона). Затем сразу добавляли диизопропилэтиламин (25,01 г, 0,193 М). После этого к раствору, по капле, при температуре от -20oC до -15oC (прибл. в течение 1,5 часа) добавляли раствор ацетоксиацетилхлорида )25,14 г, 0,184 М) в метиленхлориде (91 мл). Смесь перемешивали еще 1 час при -20oC, а затем разводили деионизованной водой (166 мл) (экзотермич. до 0oC). После тщательного перемешивания, водную фазу отделяли и экстрагировали еще 55 миллилитрами метиленхлорида. Объединенные органические фазы выпаривали вместе с этилацетатом (400 мл) для удаления метиленхлорида. Полученный этилацетатный раствор (приблизительно 450 мл) неочищенного 3-ацетокси-1-[(фенил)(бензилиденимино)-метил] -4-фенилазетидин-2-она обрабатывали водным бисульфитом (как описано ниже в части B).
В. Удаление бисульфита
К этилацетатному раствору неочищенного 3-ацетокси-1-[(фенил)(бензилиденимино)метил]-4-фенилазетидин-2-она, описанного в части А. 1 или А.2, добавляли деионизованную воду (250 мл) и бисульфит натрия (75,00 г), а затем двухфазовый раствор энергично перемешивали при 50±2oC до тех пор, пока ТСХ не указывала на завершение реакции (3 - 4 часа). После этого полученный двухфазовый раствор отделяли. Водную фазу (pH 6,0) отбрасывали, а органическую фазу промывали водой (150 мл), осушали сульфатом магния и концентрировали на роторном испарителе (35oC) до объема 100 мл. Полученную густую суспензию охлаждали и перемешивали при 0 - 5oC в течение 2 часов. Полученное таким образом твердое вещество (белые игольчатые кристаллы) фильтровали на воронке Бюхнера, промывали холодным этилацетатом (25 мл) и осушали в вакууме до постоянного объема.
В результате процедуры, описанной в части А.1., получали 20,63 г (полный выход -60oC) (±)-цис-3-ацетилокси-4-фенил-азетидин- 2-она. В результате процедуры, описанной в части А.2., получали 22,94 г (полный выход - 66,7%) (±)-цис-3-ацетилокси-4-фенилазетидин-2-она;
Соответственно, чистота (ВЭЖХ-площадь): 99,3% и 99,4%
Пример 20. (±)-цис-3-ацетокси-4-фенилазетидин-2-он(6)- гидролиз с использованием 75% уксусной кислоты
Сухую литровую круглодонную трехгорлую колбу, снабженную механической мешалкой, капельной воронкой, мембраной и отверстием для впуска азота, загружали гидробензамидом (100 г, 335 мМ, 1,00 экв) и метиленхлоридом (333 мл). Полученный раствор энергично перемешивали при приблизительно 160 об/мин, а затем охлаждали до 5oC. Затем сразу (одной порцией), в атмосфере азота, добавляли диизопропилэтиламин (67,1 мл, 385,2 мМ, 1,15 экв). Полученную реакционную смесь охлаждали до -20oC, поддерживая температуру бани при -30oC. Отдельно, ацетоксиацетилхлорид (39,6 мл, 368,4 мМ, 1,10 экв) и метиленхлорид (184 мл) смешивали при комнатной температуре и переносили, в атмосфере азота, в капельную воронку, после чего раствор добавляли в течение 5 часов. Первоначальную скорость регулировали таким образом, чтобы температура оставалась ниже -16oC. Протекание реакции прослеживали с помощью ВЭЖХ. Аликвоту реакционной смеси (100 мкл) удаляли через 10 минут после добавления раствора ацетоксиацетилхлорида. Затем полученную светло-коричневую реакционную смесь перемешивали при -20oC, до тех пор, пока ВЭЖХ-анализ не указывал на содержание >5% гидробензамида в растворе (% анализ по площади). Если реакционная смесь содержала >5% гидробензамида по отношению к площади, то реакционную смесь загружали дополнительным количеством диизопропиламина и ацетоксиацетилхлорида. Например, если указанная смесь содержала 10% гидробензамида, то добавляли еще диизопропилэтиламин (6,13 мл, 35,2 мМ, 0,105 экв), а затем раствор ацетоксиацетилхлорида (3,60 мл, 33,5 мМ, 0,10 экв) в метиленхлориде (16,7 мл). После этого добавляли раствор ацетоксиацетилхлорида при скорости, описанной выше. Затем реакционную смесь перемешивали 10 минут при -20oC, после чего брали образец для ВЭЖХ-анализа. После завершения реакции, к смеси добавляли воду (333 мл) (одной порцией) в течение приблизительно 10 секунд. Температуру смеси повышали до 5oC, а затем смесь перемешивали в течение 5-10 минут. После этого фазы отделяли при 5oC (время отстаивания <1 мин), а реакционный сосуд промывали метиленхлоридом (100 мл). Метиленхлоридные промывки использовали для экстракции водной фазы. Экстрагирование осуществляли при комнатной температуре, а органические фазы объединяли.
Обогащенный органический поток переносили в 2-литровую трехгорлую круглодонную колбу, снабженную обратным холодильником, верхней мешалкой и пробкой. К перемешанному раствору, при комнатной температуре, добавляли уксусную кислоту (150 мл, 7,9 экв), а затем воду (50 мл, 8,28 экв.). Добавление осуществляли приблизительно в течение 20 секунд, после чего полный объем смеси составлял приблизительно 953 мл. Полученный раствор нагревали с обратным холодильником (температура резервуара -42 -45oC). Аликвоты (100 мкл) брали каждый час и анализировали с помощью ВЭЖХ. Реакцию считали завершенной, если процент площади (±)-цис-3-ацетилокси-1-[(фенил)(бензилиденимино) метил] -4-фенилазетидин-2-она составлял менее 2%. Гидролиз, описанный в этой процедуре, завершали через 4 часа. Реакционную смесь охлаждали до 10-15oC, а pH реакционной смеси доводили до 4,64-6,92 путем добавления водного раствора NaOH (3,75 н, 7705 мл), поддерживая при этом температуру между 10oC и 20oC. После этого к реакционной смеси в течение 2 часов добавляли раствор NaOH. Во время нейтрализации указанной смеси, целевое соединение частично осаждалось, и затем его снова растворяли путем нагревания раствора до 25oC. Фазы разделяли при 25oC (время отстаивания < 1 мин), а реакционный сосуд промывали метиленхлоридом (100 мл). Метиленхлоридные промывки использовали для экстракции водной фазы. Объединенный обогащенный метиленхлоридный поток (850 мл) переносили в 2-литровую трехгорлую круглодонную колбу, снабженную верхней мешалкой, и охлаждали до -5oC -0oC, перемешивая при этом. Полученный продукт частично осаждался, затем в течение 1 часа добавляли гептан (850 мл, равный объему обогащенного метиленхлоридного потока) и полученную суспензию перемешивали еще 1 час при -5oC -0oC. Полученное целевое соединение в виде твердого вещества собирали путем вакуумной фильтрации (диаметр фильтра - 9 см), промывали холодным раствором (-5oC - 0oC) 10% метиленхлорида и гептане (200 мл) и осушали до постоянного объема (24-25 мм. рт. ст., 35-38oC, 12-15 часов), в результате чего получали целевое соединение (54,42 г, полный выход - 77,7%) в виде беловатых игольчатых кристаллов.
Чистота (ВЭЖХ - площадь): 100%.
Пример 21
Альтернативный метод получения (±)-цис-3-ацетилокси-1-[(2'- фуранил)(2'-фуранилметиленимино)метил] -4-(2'-фуранил)азетидин-2-она (13) и (±)-цис-3-(ацетилокси)-4-(2'-фуранил)азетидин-2-она (14)
В 2-литровую трехгорлую круглодонную колбу, снабженную термометром, магнитной мешалкой и капельной воронкой, добавляли гидрофурамид (80,48 г, 300 мМ) и этилацетат (1,0 л). При перемешивании (в потоке аргона), реакционную смесь охлаждали до 5oC и добавляли триэтиламин (50,2 мл, 360 мМ). После этого по капле в течение часа добавляли раствор ацетоксиацетилхлорида (37,0 мл, 344 мМ) в EtOAc (500 мл). После выдерживания 16 часов при 5oC, реакционную смесь нагревали до 20oC (1,5 ч.) и переносили в делительную воронку. Органический слоя промывали водным насыщенным раствором NH4Cl (500 мл). Оба слоя фильтровали через мелкозернистую фильтровальную бумагу из стекловолокна (Whatman), а осадок на фильтре промывали этилацетатом (50 мл). Полученный фильтрат снова переносили в делительную воронку, а водный слой удаляли. Затем органический слой последовательно промывали водным раствором NH4Cl (нас.) (250 мл), водным раствором NaHCO3 (нас.) (400 мл) и солевым раствором (400 мл). Органический слой, содержащий целевое соединение примера 13, фильтровали через мелкозернистую фильтровальную бумагу из стекловолокна (Whatman).
Раствор, описанный выше, разделяли на две равные части (приблизительно 750 мл каждая) и осторожно переносили (в потоке аргона) в две 2,0-литровые колбы Парра, каждая из которых содержала 10% палладий на активированном угле (6 г). Полученную смесь обрабатывали водородом (4 атм) в течение 1 часа, а катализатор удаляли путем фильтрации через слой целита™. Осадок на фильтре промывали этилацетатом (100 мл), а фильтраты объединяли. Органический слой дважды промывали 10% раствором соляной кислоты (500 мл, 250 мл) и снова экстрагировали этилацетатом (500 мл). Затем объединенные органические слои промывали водным раствором NaHCO3 (нас.) (400 мл) и солевым раствором (400 мл). Органический слой осушали сульфатом магния и обрабатывали 30 граммами обесцвеченного активированного угля. Через 15 минут, указанную смесь фильтровали через слой Целита™ и концентрировали в вакууме до конечного объема 160 мл. Полученную смесь охлаждали до 4oC, а осажденный продукт выделяли путем фильтрации. Осадок на фильтре промывали диэтиловым эфиром и гексаном (каждый по 100 мл), в результате чего получали 35,98 г (полный выход исходя из гидрофурамида - 61,4%) целевого соединения (14) в виде белых игольчатых кристаллов (т.пл. 118-119oC).
Пример 22
Альтернативный способ получения (±)-цис-3-ацетилокси-1- [(фенил)(бензилиденимино)метил] -4-фенилазетидин-2-она (5) и (±)-цис-3-ацетилокси-4-фенилазетидин-2-она (6).
В литровую трехгорлую круглодонную колбу, снабженную термометром, магнитной мешалкой и капельной воронкой, добавляли гидробензамид (30,00 г, 100,5 мМ) и этилацетат (150 мл). При перемешивании, в атмосфере аргона, реакционную смесь охлаждали до 5oC и добавляли триэтиламин (16,8 мл, 121 мМ). Затем к полученной смеси по капле в течение 90 минут, добавляли раствор ацетилоксиацетилхлорида (12,4 мл, 115 мМ) в этилацетате (300 мл). После выдерживания в течение 16 часов при 5oC, реакционную смесь нагревали до 20oC (1,5 ч. ) и переносили в делительную воронку. Органический слой последовательно промывали водным насыщенным раствором NH4Cl (150 мл, 75 мл), водным насыщенным раствором NaHCO3 (100 мл) и солевым раствором (75 мл).
К вышеуказанному органическому слою добавляли 90% водный раствор муравьиной кислоты (22,0 мл, 0,57 М) и смесь перемешивали при комнатной температуре в течение 2 дней. Затем смесь переносили в делительную воронку и промывали водой (200 мл), а затем насыщенным водным раствором NaHCO3 (200 мл) (при медленном добавлении). К водным NaHCO3 (нас.)-промывкам осторожно добавляли твердый NaHCO3 до тех пор, пока pH не достигала значения 7,5. После этого органический слой промывали насыщенным NaCl, осушали сульфатом магния, фильтровали и концентрировали в вакууме до объема 75 мл. Полученную смесь охлаждали до 4oC, а осажденный продукт выделяли путем фильтрации. Осадок на фильтре промывали гексаном (200 мл) и получали 14,26 г (полный выход исходя из гидробензамида - 69,1%) целевого соединения в виде белых игольчатых кристаллов (т.пл. 150-151oC). ВЭЖХ - чистота (площадь): 96,5%.
Пример 23. (±)-Цис-3-(1-фенетилокси)-1-(фенил)- (бензилиденимино)метил-4-фенилазетидин-2-он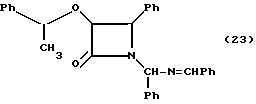
В 100-миллитровую трехгорлую круглодонную колбу, снабженную термометром, магнитной мешалкой и капельной воронкой, добавляли гидробензамид (2,98 г, 10 мМ) и дихлорметан (15 мл). При перемешивании (в потоке аргона), реакционную смесь охлаждали до 5oC и добавляли триэтиламин 91,67 мл, 12,0 мМ). Затем к раствору по капле в течение 1 часа добавляли раствор (±)-фенэтилоксиацетилхлорида (2,58 г, 13,0 мМ) в CH2Cl2 (30 мл). После выдерживания 16 часов при 5oC, реакционную смесь нагревали до 20oC и переносили в делительную воронку. Затем органический слой последовательно промывали водным раствором NH4Cl (нас.) (30 мл, 15 мл), водным раствором NaHCO3 (нас) (25 мл) и солевым раствором (25 мл). Органический слой осушали сульфатом магния, фильтровали, а растворитель удаляли в вакууме, в результате чего получали целевое соединение в виде красного маслообразного вязкого вещества (4,57 г, 99,2%) (смесь диастереомеров).
1H-ЯМР (CDCl3, 200 МГц): δ = 8,443 (с, 0,25H, N=CH), 8,414 (с, 0,25H, N= CH), 8,406 (с, 0,5H, N=CH), 7,77-7,91 (м, 2H, Ar), 6,82-7,69 (м, 18H, Ar), 6,28 (с, 0,25H, NCHN), 6,22 (с, 0,5H, NCHN), 6,17 (с, 0,25H, NCHN), 5,02 (д, J = 4,4 Гц, 0,25H, H-3), 4,89 (д, J = 5,0 Гц, 0,25H, H-3), 4,77 (д, J = 5,0 Гц, 0,25H, H-3), 4,70 (д, J = 4,9 Гц, 0,25H, H-3), 4,44-4,67 (м, 0,75H, H-4), 4,35 (д, J = 5,0 Гц, 0,25H, H-4), 3,73-3,94 (м, 1H, MeCHPh), 1,51 (д, J = 6,4 Гц, 0,75H, CH3, CH), 1,33 (д, J = 6,4 Гц, 0,75H, CH3CH), 1,32 (д, J = 6,5 Гц, 0,75H, CH3CH), 0,89 (д, J = 6,5 Гц, 0,75H, CH3CH).
Пример 24.
(±)-цис-3-(1'-фенэтилокси)-4-фенилазетидин-2-он (24)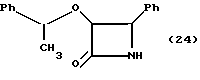
Раствор соединения примера 23 в этилацетате (50 мл) осторожно переносили (в атмосфере аргона) в 200-миллилитровую колбу Парра, содержащую 10% палладий на активированном угле (0,60 г). Полученную смесь обрабатывали водородом (4 атм) в течение 16 часов, а затем катализатор удаляли путем фильтрации через слой Целита™. Затем осадок на фильтре суспендировали в этилацетате (50 мл), перемешивали (10 минут) и фильтровали. После этого указанный осадок на фильтре промывали 10 миллилитрами этилацетата, а фильтраты объединяли. Органический слой промывали 10% раствором HCl (50 мл), а затем оба слоя фильтровали через воронку из спеченного стекла, до тех пор, пока не будет удален белый осадок (дибензиламин HCl). Затем фазы отделяли, а органический слой промывали еще одной порцией 10% раствора HCl (30 мл). Объединенные слои промывали 10% раствором HCl и снова экстрагировали этилацетатом (100 мл). Объединенные органические слои промывали водным раствором. NaHCO3 (нас. ) (50 мл) и солевым раствором (50 мл). Органический слой осушали сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали желтое маслообразное вещество (2,967 г), которое затем очищали с помощью препаративной ТСХ (2 мм; силикагель, Этил/ацетат/гексан, 3:7) и получали 2,41 г (91%) целевого соединения (24) в виде маслообразного вещества и смеси диастереомеров.
1H-ЯМР (CDCl3, 200 МГц): δ = 7,18 - 7,50 (м, 9H, Ar), 6,92-6,97 (м, 1H, Ar), 6,33 (шир. с, 0,5H, обмененный, NH), 6,28 (шир. с, 0,5H, обмениваемый, NH), 4,60-4,82 (м, 2H, H-3, H-4), 3,94 (кв., J = 6,5 Гц, 1H, CHCH3), 1,34 (д, J = 6,5 Гц, 1,5H, CHCH3), 0,91 (д, J = 6,5 Гц, 1,5H, CHCH3).
Пример 25.
(±)-цис-3-(4,6'-ди-O-ацетокси-2', 3'-дидезокси -α- D-глюкопиранозилокси)-4-фенилазетидин-2-он (25)
В 50-миллилитровую трехгорлую круглодонную колбу, снабженную термометром, магнитной мешалкой и капельной воронкой, добавляли (4,6-ди-O-ацетокси-2,3-дидезокси -α- D-глюкорапиранозилокси)ацетилхлорид (774 мг, 2,51 мМ) и дихлорметан (20 мл), после чего полученный раствор охлаждали до -78oC и добавляли триэтиламин (0,55 мл, 4,02 мМ). После перемешивании в течение 15 минут, к смеси добавляли раствор гидробензамида (824 мг, 2,76 мМ) в толуоле (5 мл). Полученную реакционную смесь нагревали до 5oC. После выдерживания 16 часов при той же температуре, реакционную смесь разбавляли дихлорметаном (50 мл) и переносили в делительную воронку. Органический слой промывали последовательно водным раствором NH4Cl (нас.) (30 мл, 15 мл), водным раствором NaHCO3 (нас. ) (25 мл) и солевым раствором (25 мл). Затем органический слой осушали сульфатом магния, фильтровали и концентрировали в вакууме. В результате этой процедуры получали 1,435 г (100,2% (±)-цис-3-(4',6'-ди-O-ацетокси-2',3'- дидезокси -α- D-глюкопиранозилокси)-1-[(фенил)бензилиденимино)метил]-4-фенилазетидин-2-она в виде вязкого маслообразного вещества и смеси диастереомеров.
Раствор вышеописанного соединения (1,435 г) в этилацетате (50 мл) осторожно переносили (в потоке аргона) в 200-миллитровую колбу Парра, содержащую 10% палладий на активированном угле (315 мг). Полученную смесь обрабатывали водородом (4 атм.) в течение 16 часов, после чего катализатор удаляли путем фильтрации через слой целита™. Полученный осадок на фильтре суспендировали в этилацетате (50 мл), перемешивали (10 мин) и фильтровали. Затем осадок на фильтре промывали этилацетатом (10 мл), а фильтраты объединяли. Органический слой промывали 10% раствором соляной кислоты (25 мл), а оба слоя фильтровали через воронку из спеченного стекла до тех пор, пока не был удален белый осадок (дибензиламин HCl). Затем фазы разделяли, а органический слой промывали еще одной порцией 10% раствора соляной кислоты (25 мл), водным раствором NaHCO3 (нас. ) (25 мл) и солевым раствором (25 мл). Органический слой осушали сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали вязкое маслообразное вещество (0,50 г). Полученное вещество очищали с помощью препаративной ТСХ (2 мм-силикагель, этилацетат/гексан, 6,5:3,5) и получали 320 мг (полный выход исходя из гидробензамида 33,8%) целевого соединения (25) в виде смеси диастереомеров (2,7:1), [(3R,4S)/3S,4R) и маслообразного вещества.
1H-ЯМР [(3R, 4S)-диастереомер, CDCl3, 200 МГц]: δ = 7,27-7,44 (м, 5H, Ph), 6,54 (шир. с, обмениваемый, 1H, NH), 5,07 (дд, J = 2,7, 4,5 Гц, 1H, H-3), 5,00 (шир. с, 1H), 4,89 (д, J = 4,5 Гц, H-4), 4,49 (тд, J = 4,0, 10,5 Гц, 1H), 4,01-4,14 (м, 1H), 3,93 (дд, J = 4,5, 12,5 Гц, 1H), 3,70 (дд, J = 1,5, 12,5 Гц, 1H), 2,49-2,59 (м, 1H), 2,06 (с, 3H, OAc), 1,94 (с, 3H, OAc), 1,57-1,93 (м, 3H).
1H-ЯМР [(3S, 4R)-диастереомер, CDCl3, 200 МГц]: δ = 7,28-7,39 (м, 5H, Ph), 6,30 (шир. с, обмениваемый, 1H, NH), 5,17 (дд, J = 2,7, 4,4 Гц, 1H, H-3), 4,88 (д, J = 4,4 Гц, H-4), 4,64 (тд, J = 5,3, 10,1 Гц, 1H), 4,13-4,35 (м, 2H), 4,04-4,12 (м, 2H), 2,04 (с, 3H, OAc), 2,01 (с, 3H, OAc), 1,61-1,86 (м, 2H), 1,34-1,54 (м, 1H), 1,04-1,19 (м, 1H).
Каждому специалисту очевидно, что настоящее изобретение может быть осуществлено и в других вариантах, не выходящих за рамки нижеследующей формулы изобретения.
Изобретение относится к новым цис-N-иминометил-3,4-дизамещенным-2-азетидинонам и к их использованию для получения N-ацил-2-азетидинонов, которые являются промежуточными соединениями в полусинтезе таксола и его производных, обладающих противораковым действием. Описываются цис-N-иминометил-3,4-дизамещенные β-лактамы формулы I, где R1 выбирают из группы, включающей С1-C6-алкил, С1-C6-алкил, замещенный галогеном, фенил, нафтил, алкилфенил или углеводное производное, содержащее пиранозильную группу; Х выбирают из группы, включающей фенил, нафтил, их замещенные 1-3 одинаковыми или различными заместителями из группы С1-C3-алкил, С1-C3-алкокси, гидрокси, трифторметил и галоген; фурил, тиенил. 5 с. и 27 з.п. ф-лы.
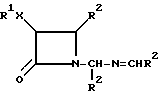
где R1 выбирают из группы, включающей С1 - С6-алкил, С1 - С6-алкил, замещенный галогеном, фенил, нафтил, алкилфенил или углеводное производное, содержащее пиранозильную группу;
Х выбирают из О и С(О)О;
R2 выбирают из группы, включающей фенил, нафтил, их замещенные 1 - 3 одинаковыми или различными заместителями из группы С1 - С3-алкил, С1 - С3-алкокси, гидрокси, трифторметил и галоген; фурил, тиенил.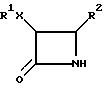
где R1, Х и R2 определены в п.1,
заключающийся в том, что соединение по п.1 подвергают каталитическому гидрогенолизу.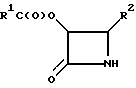
где R1 и R2 определены в п.1,
заключающийся в том, что соединение по п.1 обрабатывают водным раствором кислоты, где указанную кислоту выбирают из группы, включающей в себя бисульфит натрия, уксусную кислоту и муравьиную кислоту.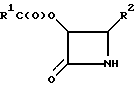
где R1 и R2 определены в п.1,
заключающийся в том, что соединение формулы
R1C(O)OCH2C(O) - L,
где L является уходящей группой,
подвергают реакции взаимодействия с соединением формулы
R2-СН (N=СНR2)2
в присутствии основания, поддерживая при этом реакционную температуру приблизительно при 5oC или ниже, и полученное таким образом соединение подвергают каталитическому гидрогенолизу.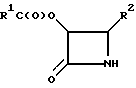
где R1 и R2 определены в п.1,
который заключается в том, что соединение формулы
R1C(O)OCH2C(O) - L,
где L является уходящей группой,
подвергают реакции взаимодействия с соединением формулы
R2-СН (N=СНR2)2
в присутствии основания, поддерживая при этом температуру реакционной смеси приблизительно 5oC или ниже, и полученное таким образом соединение подвергают обработке водным раствором кислоты, где указанную кислоту выбирают из группы, включающей в себя бисульфит натрия, уксусную кислоту и муравьиную кислоту.
Приоритет по пунктам:
23.04.93 по пп.1, 15 и 26;
13.12.93 по пп.2 - 14, 16 - 25, 27 - 32.
| ПРИВОД КРИВОШИПНОГО ПРЕССА | 0 |
|
SU388909A1 |
| Способ получения 1,3-бис(1,1,3,3,3-пентаметилдисилазанил)-2,2,4,4-тетраметилциклодисилазана | 1983 |
|
SU1087523A1 |
| US 5175315A, 29.12.92 | |||
| Journal organic chemistry, 1969, v.34, p.1477 - 1479. | |||
