Изобретение относится к способам получения производных тиозолидинона формулы II: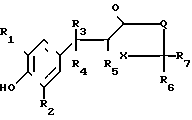 в которой R1 представляет собой С2-С6 алкенил, С2-С6 алкинил или группу (CH2)n-S
в которой R1 представляет собой С2-С6 алкенил, С2-С6 алкинил или группу (CH2)n-S , в которой n представляет собой целое число в интервале 0-3, включая крайние значения интервала;
, в которой n представляет собой целое число в интервале 0-3, включая крайние значения интервала;
R2 представляет собой водород, С1-С6 алкил, С1-С6 алкокси,
С2-С6 алкенил, С2-С6 алкинил, С1-С4-алкил -O-
-C C1-C
C1-C алкил или -
алкил или -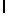 CH
CH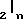 -S
-S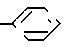 где n представляет собой целое число в интервале 0-3, включая оба крайних значения;
где n представляет собой целое число в интервале 0-3, включая оба крайних значения;
R3 представляет собой водород или С1-С6 алкил;
R4 и R5 представляет собой водород или совместно образуют связь;
R6 и R7 каждый представляет собой водород или совместно образуют группу S, или один из радикалов;
R6 и R7 представляет собой водород, а другой группы -OH или SCH3;
Х представляет собой группу 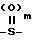 где m равно 0,1 или 2, а
где m равно 0,1 или 2, а
Q представляет собой NR8, где R8 представляет собой водород, С1-С6 алкил, С2-С6 алкенил, С3-С8 циклоалкил, SO2CH3 или -(СН2)n-Y; где n представляет собой целое число в интервале 0-3, включая оба крайних значения, а
Y представляет собой циано, OR9, -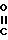 R10
R10
-NR11К12, S-(С1 С4) алкил или группу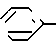 O-C1-C4 алкил где R9 представляет собой водород, С1-С4 алкил или группу -
O-C1-C4 алкил где R9 представляет собой водород, С1-С4 алкил или группу - -C1-C4-алкил
-C1-C4-алкил
R10 представляет собой С1-С4 алкил, С1-С4 алкокси, или -NН2
R11 и R12 независимо друг от друга представляют собой водород С2-С6- алкенил, С2-С6 алкинил, С1-С6 алкил, (СН2)qOH, -(СН2)q-N(С1-С4алкил)2,
-(СН2)q-S(C1-С4 алкил) или
-(CH2)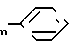 в которой n имеет указанные значения; q представляет собой целое число в интервале 1-6, включая оба крайних значения; либо R11 и R12 совместно образуют морфолиновой, пиперидинильное, пиперазинильное или N-метил-пиперазинильное кольцо, или его фармацевтически приемлемой соли, отличающейся тем, что осуществляют: реакцию соединения формулы
в которой n имеет указанные значения; q представляет собой целое число в интервале 1-6, включая оба крайних значения; либо R11 и R12 совместно образуют морфолиновой, пиперидинильное, пиперазинильное или N-метил-пиперазинильное кольцо, или его фармацевтически приемлемой соли, отличающейся тем, что осуществляют: реакцию соединения формулы
HO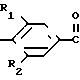 -R3 с соединением формулы
-R3 с соединением формулы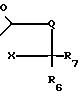 в которой R1, R2, R3 и Х имеют указанные значения, Q представляет собой N-R8 (где R8 имеет указанные значения), а R6 и R7 совместно образуют группу S, с целью получения соединения формулы
в которой R1, R2, R3 и Х имеют указанные значения, Q представляет собой N-R8 (где R8 имеет указанные значения), а R6 и R7 совместно образуют группу S, с целью получения соединения формулы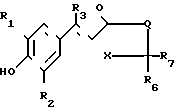 в которой R1, R2, R3, R6, R7, Х и Q имеют указанные значения;
в которой R1, R2, R3, R6, R7, Х и Q имеют указанные значения;
(а) с последующим необязательным восстановлением соединения формулы II, в которой R6 и R7 совместно образуют группу S с тем, чтобы получить соединение формулы II, в которой R6 и R7 представляет собой водород;
(в) восстановлением соединения формулы II, в которой R4 и R5совместно образуют связь с целью получения соединения формулы II, в которой R4 и R5 представляют собой водород;
(с) восстановлением соединения формулы II, в которой R4 и R5совместно образуют связь, а R6 и R7 совместно образуют группу=S с целью получения соединения формулы II, в которой R4, R5, R6 и R7представляют собой водород;
(d) алкилированием соединения формулы II, в которой R3 представляет собой водород с получением соединений формулы II, в которой R8представляет собой С1-С6 алкил, С2-С6 алкенил, С3-С6 циклоалкил или -(СН2)n-Y (где n представляет собой целое число в интервале 0-3, включая крайние значения, а Y представляет собой циано, OR9, -SH, -S(C1-С4 алкил)-NR11R12 или O-C1-C4 алкил, где R9, R11 и R12 имеют указанные значения: (е) ацелированием соединения формулы II, в которой R8 представляет собой водород с целью получения соединения формулы II, в которой R8представляет собой -(СН2)n-Y, где n представляет собой целое число в интервале 0-3, включая оба крайних значения интервала, а Y представляет собой -
O-C1-C4 алкил, где R9, R11 и R12 имеют указанные значения: (е) ацелированием соединения формулы II, в которой R8 представляет собой водород с целью получения соединения формулы II, в которой R8представляет собой -(СН2)n-Y, где n представляет собой целое число в интервале 0-3, включая оба крайних значения интервала, а Y представляет собой - R10 где R10 имеет указанные значения;
R10 где R10 имеет указанные значения;
(f) окислением соединений формулы II, в которой Х представляет собой группу -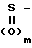 в которой m равно 0, с целью получения соединения формулы II, в которой Х представляет собой группу -
в которой m равно 0, с целью получения соединения формулы II, в которой Х представляет собой группу -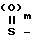 , m равно 1;
, m равно 1;
(к) восстановлением соединения формулы II, в которой R3представляет собой группу -(СН2)n-Y- в которой n имеет значения в интервале 0-3, включая крайние значения интервала, Y представляет собой OR9, где R9 представляет собой группу -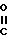 -C1-C4 алкил с целью получения соединения формулы II, в которой R8представляет собой группу -(СН2)n-Y, в которой n равно 0-3, включая оба крайних значения, а Y представляет собой OR9, где R9 представляет собой водород;
-C1-C4 алкил с целью получения соединения формулы II, в которой R8представляет собой группу -(СН2)n-Y, в которой n равно 0-3, включая оба крайних значения, а Y представляет собой OR9, где R9 представляет собой водород;
(l) осуществлением реакции соединения формулы II, в которой R8представляет собой группу -(СН2)n-Y, где n равно 0-3, включая оба крайних значения интервала, Y представляет собой -OR9, где R9представляет собой водород, с тозилгалогенидом, с целью получения соединения формулы II, в которой R8 представляет собой -(СН2)n-Y, где n равно 0-3, включая оба крайних значения интервала, а Y представляет собой OR9, где R9 представляет собой тозил;
(м) осуществлением реакции соединения формулы II, в которой R8представляет собой -(СН2)n-Y-, в которой n равно 0-3, включая оба крайних значения, а Y представляет собой -OR9, где R9 тозил, с амином формулы HNR11R12 (где R11 и R12 имеют указанные значения), с целью получения соединения формулы II, в которой R8 представляет собой группу -(СН2)n-Y, где n равно 0-3, включая оба крайних значения, а Y представляет собой NR11, R12;
(n) нагреванием соединения формулы II, в которой R8 представляет собой -(СН2)n-Y, а Y представляет собой NR11R12(R11, R12 не водород) в смеси этанол/вода в присутствии катализатора с целью получения соединения формулы II, в которой R8 представляет собой -(СН2)n-Y, Y означает NR11R12, где R11, R12 один водород, а другой не является водородом.
(о) реакцией соединения формулы II, в которой R6 и R7 представляет собой водород, с трифторуксусным ангидридом с получением соединения формулы II, в которой один из радикалов R6 и R7 представляет собой водород, а другой группу -OH;
(р) образованием соли соединения формулы II по реакции несолевой в нормы соединения с сильной кислотой или сильным основанием.
Соединения по изобретению, а также те соединения, используемые в способе по изобретению, в которых R4 и R5 представляют собой водород содержат асимметричный центр на атоме углерода в положении 5 роданина или его производного. В связи с этим такие соединения могут существовать в виде рацемической смеси или как индивидуальные стереоизомеры. Способ и соединения по изобретению охватывают как рацемат, так и его индивидуальные стереоизомеры. Способ по изобретению обеспечивает метод получения стереоизомеров некоторых соединений изобретения, а также некоторых соединений, используемых в методе изобретения.
Соединения и способ изобретения охватывают также фармацевтически применимые соли. Такие соли могут быть получены по реакции соединения формулы I с таким сильным основанием, как гидроксид натрия или такой сильной кислотой, как хлористоводородная кислота.
Соединения по изобретению включает следующие вещества: 5-[[3,5-бис(4-пентил)-4-гидроксифенил] -метил] -3-этиламино-4-тиазолидинон; 5-[[3-этилтиофенил-4-гидрокси-5-метилфенил] метилен] -2-тиоксо-4-тиазолидинон; 5-[[3-(2-бутен)-4-гидрокси-5-изопропоксифенил] -метил]-3-(2-припенил)-4-гидро кси-5-[[3,5-(метилтиофенил))-4-гидроксифенил] метилен] -3-пропил-2-тиоксо-4-ти азолидин5-[[3,5-диацетилен-4-гидроксифенил] метил] -4-тиазолидинон; 5-[[3-(3-метил-1-бутен)-4-гидрокси-5-пропилфенил] -метилен] -3-этилциано-4-тиа золи5-[[3-(2-пропенил)-4-гидрокси-5-метоксифенил] метил] -3-этокси-4-тиазолидо н; 5-[[3,5-ди-2-пропенил)-4-гидроксифенил]метилен]-3-(метиламино-метил)-2-тиокс о-4-
Следующие соединения помимо указанных являются примерами веществ, подходящих для использования в способе изобретения. 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-3-(3-метоксипропил)-2- тио5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] -метилен] -2-тиоксо-4-тиазоли динон; 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] метилен] -4-тиазолидинон; 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] -метил] -4-тиазолидинон; 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] -метил] -2-тиоксо-4-тиазолидинон 3-ацетил-5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] -метилен] -4-тиазолидин он; 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] метил-]3-метил](1-метилэтил)ами но] -5-[4-гидроксибензаль] роданин; 5-(4-гидрокси-3-метоксибензилиден)роданин,5-[(4-гидрокси-3,5-дипропилфенил)м етилен]-3-[2-(диметиламино)этил] -4-тиазолидинон; 5-[[3,5-бис(1-метилпропил)-4-гидроксифенил]метил]-3-метил-4-тиазолидон; 5-[[3,5-диметил-4-гидроксифенил]-метилен] -3-метил-4-диазолидинон; 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]-метил] -3-(метилсульфонил)-4-ти азолидинон; 5-[[4-гидрокси-3,5-бис(1,1-диметилэтил)фенил] метил] -3-(пропиламино)-4-тиазол идинон; 3-амино-5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] -метилен] -2-тиоксо-4-ти азол5-[[3,5-бис(1-метилэтил)-4-гидроксифенил] -метил] -3-метил-4-тиазолидинон; 5-[(4-гидрокси-3,5-диметоксифенил)метил] -3-метил-2-тиоксо-4-тиазолидинон; 5-[(4-гидрокси-3,5-диметоксифенил)метилен] -3-метил-2-тиоксо-4-тиазолидинон. 5-[[3,5-бис(1,1-Диметилэтил)-4-гидроксифенил]метилен]-2-тиоксо-4-тиазолидино н (на которое в последующем обслуживании ссылаются, как на соединение А). Такое соединение получают по реакции 3,5-ди-трет-бутил-4-гидроксибензальдегида с роданином при температуре дефлегмации в ледяной к уксусной кислоте с использованием в качестве катализатора плавленого ацетата натрия. 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-4-тиазолидинон-соедине ние В), 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метил]-4-тиазолидинон (соединение С) и 5-[[-3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метил]-2-тиоксо-4-тиазолидинон (соединение D) могут быть получены из соединения А.
Так, например, подвергая соединение А каталитическому гидрированию можно получать как соединение В, так и соединение С. Получаемые соотношения зависят от температуры, давления и длительности гидрирования, типа используемого растворителя и природы применяемого катализатора. Так, например, в том случае, когда соединение А обрабатывают 5% палладия на угле в среде этанола при 100оС в течение 18 ч, соотношение между соединениями В и С составляет 60:40. С другой стороны, такие трансформации могут реализоваться путем нагревания соединения А в смеси соляной кислоты и такого спирта, как этанол в присутствии цинка. Восстановление тиона без затрагивания бензильной двойной связи может осуществляться путем нагревания тиона в присутствии такого восстанавливающего агента, как гидрид три-н-бутил олова в среде такого же нереакционноспособного растворителя, как толуол и, предпочтительно, в присутствии свободно-радикального инициатора такого как азобисизобутиронитрил. Однако для осуществления такого восстановления следует использовать N-замещенный роданиновый субстрат (т. е. Q не может быть -NH).
Трансформация соединения А в Д может осуществляться различными способами, известными из литературы. В этой реакции соединение А обрабатывают таким дигидропиридином, как диэтил 2,6-диметил-1,4-дигидро-3,5-пиридинкарбоксилат в присутствии силикагеля. Реакцию удобно проводить в присутствии такого нереакционноспособного растворителя, как бензол или толуол, предпочтительно, в инертной атмосфере. Реакцию можно осуществлять при температуре в интервале от 25оС до температуры дефлегмации смеси. При предпочтительной температуре порядка 80оС реакция завершается через 12-18 ч.
Аналогичным способом, в зависимости от назначения различных заместителей, могут быть получены другие тиазолидиноны. Так, например, соединения формулы II, в которой Q представляет собой NR8, а R8представляет собой водород, С1-С6 алкил, С3-С8 циклоалкил или -(СН2)n-Y, где n представляет собой целое число а в интервале 0-3, включая крайние значения, а Y представляет собой циано или NR11 и R12, где R11 и R12 независимо друг от друга, представляют собой водород или С1-С6 алкил, могут быть получены по методу Тьюбера с сотр. описанному выше, с использованием соответствующего N-замещенного роданина и R1, R2-замещенного-4-гидроксибензальдегида. С другой стороны роданин может использоваться для конденсации с альдегидом с образованием таких соединений, в которых Q представляет собой NR8, а R8 представляет собой водород, с последующим алкилированием соответствующим R8 содержащим галогенидом, таким как иодин или бромид, с получением соответствующего N-замещенных производных, например, таких соединений формулы в II, в которых R8 представляет собой С1-С6 алкил, С2-С6 алкенил, С3-С8 циклоалкил, или -(СН2)n-Y, где Y представляет собой циано, OR9, S-)С1-С4алкил), -NR11R12 или группу 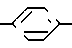 O-C1-C4- алкил, а n, R9, R11 и R12 имеют значения, указанные для формулы II. Алкилирование обычно осуществляют в среде такого инертного растворителя, как тетрагидрофуран (ТГФ) или диметилформамид (ДМФ) в присутствии такого сильного основания, как гидрид натрия. Аналогичным образом роданин может использоваться для конденсации с альдегидом с образованием соединений, в которых Q представляет собой NR8, а R8 представляет собой водород с последующим ацилированием соответствующим R8 содержащим галогенидом с получением N-замещенных производных формулы II, в которых R8 представляет (СН2)n-Y, а Y представляет собой -
O-C1-C4- алкил, а n, R9, R11 и R12 имеют значения, указанные для формулы II. Алкилирование обычно осуществляют в среде такого инертного растворителя, как тетрагидрофуран (ТГФ) или диметилформамид (ДМФ) в присутствии такого сильного основания, как гидрид натрия. Аналогичным образом роданин может использоваться для конденсации с альдегидом с образованием соединений, в которых Q представляет собой NR8, а R8 представляет собой водород с последующим ацилированием соответствующим R8 содержащим галогенидом с получением N-замещенных производных формулы II, в которых R8 представляет (СН2)n-Y, а Y представляет собой -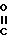 R10, где n и R10 имеют значения, указанные для формулы I.
R10, где n и R10 имеют значения, указанные для формулы I.
Соединения формулы II, в которых Q представляет собой NR8, R8-(СН2)n-Y (Y представляет собой OR9 или NR11R12, где R9 водород, ацетил или тозил; R11 и R12 имеют значения, указанные для формулы I) такие могут быть получены, согласно следующей реакционной схеме
Гидроксиалкил роданин III получают конденсацией сероуглерода, хлоруксусной кислоты и соответствующего гидроксиалкиламина стандартными способами. При конденсации с соответствующим R1, R2-замещенным 4-гидроксибензальдегидом, в соответствии с описанным выше, полученный в результате продукт представляет собой конденсированный 2-тиоксо-4-тиазолидинон IV, который трансформировали в ацетильное производное. Тиоксо-соединение может быть необязательно превращено в метиленовое соединение формулы Y в соответствии с описанным выше. Ацетильная группа интермедиата Y может быть удалена в результате обработки водным раствором аммиака в таком растворителе, как ацетонитрил, с получением соединения VI (т. е. соединения формулы II, в которых представляет собой NR8, а R8 представляет собой -(СН2)n-Y, где Y представляет собой OR9, а R9 водород). Затем гидрокси соединение VI превращают в производное тозила (VII) в ее результате обработки п-толуолсульфонил хлоридом в среде пиридина, предпочтительно при температуре около 0оС. Затем тозильное производное VII может быть трансформировано в дополнительные соединения формулы II в ходе обработки соответствующим HNR11R12 амином, в котором R11 и R12 имеют значения, указанные в предыдущем параграфе. Последнюю трансформацию удобно проводить путем реакции соединения VII в присутствии молярного избытка амина. И в этом случае для реализации указанной трансформации используют такой растворитель, как ацетонитрил.
Соединения формулы II, в которых Q представляет собой NR8, R8представляет собой -(СН2)n-Y (n=0) и Y представляет собой NR11R12, где R11 и R12 имеют значения, указанные в формуле II могут быть получены согласно следующей реакционной схеме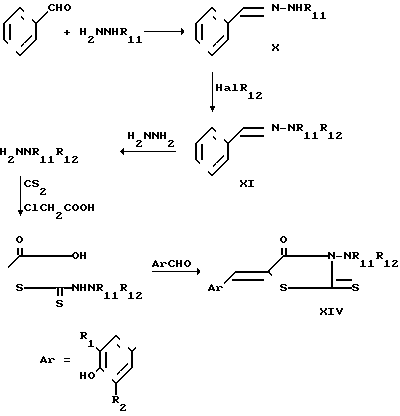
R11 замещенный гидразин обрабатывают бензальдегидом в спиртовом (предпочтительно метанольном) растворителе с образованием промежуточного соединения Х, которое, в свою очередь, реагирует с соответствующим R12 галогенидом в присутствии триэтиламина и ацетонитрила с образованием промежуточного соединения ХI. Затем соединение ХI обрабатывают гидразином с получением R11, R12 гидразина ХII. С другой стороны ХII может быть получен путем восстановления нитрозо-R11R12-амина с использованием цинковой пыли и уксусной кислоты либо алюминия и сильного основания. Сам нитрозо-R11R12-амин может быть получен из R11, R12-амина, в результате обработки нитритом натрия в среде НСl. Затем соединение ХII обрабатывают сероуглеродом, хлоруксусной кислотой и триэтиламином с получением промежуточного соединения ХIII. Конденсацией соединения ХIII с соответствующим R1, R2 замещенным 4-гидроксибензальдегидом (т. е. АrСНO) получают соединение ХIV. Как отмечалось ранее тион может быть восстановлен путем обработки таким восстанавливающим агентом, как гидрид три-н-бутил олова в таком нереакционноспособном растворителе, как толуол, предпочтительно, в присутствии такого свободно-радикального инициатора, как азобис-изобутиронитрил. Получение соединений, в которых из радикалов R11 и R12 представляет собой водород, может быть осуществлено до или после восстановления тиона в результате нагревания дизамещенного соединения в смеси этанол вода в присутствии такого катализатора, как родий.
Соединение формулы II, в которых Х представляет собой 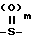 , равно I или 2 могут быть легко получены из сульфида (т. е. m 0) путем обработки таким окисляющим агентом, как м-пербензойная кислота в среде соответствующего органического растворителя, такого как хлороформ, в течение времени, достаточного для осуществления желаемого окисления.
, равно I или 2 могут быть легко получены из сульфида (т. е. m 0) путем обработки таким окисляющим агентом, как м-пербензойная кислота в среде соответствующего органического растворителя, такого как хлороформ, в течение времени, достаточного для осуществления желаемого окисления.
Соединения формулы II, в которых R3 представляет собой С1-С6 алкил, получают традиционным алкилированием по Фриделю-Крафтсу соответствующего R1, R2-замещенного фенола с последующей конденсацией с роданином или желаемым N-запмещенным роданином, в соответствии с описанным выше, либо используют в соответствии с указанным в других реакционных схемах, приведенных в тексте.
Специалисту в данной области должно быть понятно, что арильный фрагмент соединений формулы II является доступным веществом или может быть легко получен известными методами из доступных исходных материалов. Так например, п-гидроксибензальдегид может быть проалкилирован в условиях Фриделя-Крафтса с получением алкилбензальдегида, который в свою очередь сам может быть проалкилирован. Аналогичным образом, исходный роданин или N-замещенный роданин представляет собой доступное соединение или может быть получен хорошо известными способами из выпускаемых промышленностью исходных материалов.
Соединения формулы II, в которых один из радикалов R6 или R7представляет собой водород, а другой -OH (а Х представляет собой группу 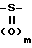 , где m равно 0, обычно получают из их предшественников формулы II, в которых R6 и R7представляют собой водород (а Х представляет собой группу
, где m равно 0, обычно получают из их предшественников формулы II, в которых R6 и R7представляют собой водород (а Х представляет собой группу  в которой m равно 1, путем обработки предшественника, например, трифторуксусным ангидридом в среде инертного растворителя (предпочтительно хлористого метилена) при пониженной температуре. Аналогичным образом, соединения формулы II, в которых в определении Q Y представляет собой циано, получают обработкой нецианированного аналога желаемым галоген-замещенным алифатическим нитрилом. Из цианопроизводного получают тетразолил путем обработки азидом три-н.бутил олова в среде, например, диметилового эфира этилен гликоля. Другие соединения формулы II могут быть получены, в соответствии с более полным объяснением, приведенным ниже, из соединений, синтез которых описан выше.
в которой m равно 1, путем обработки предшественника, например, трифторуксусным ангидридом в среде инертного растворителя (предпочтительно хлористого метилена) при пониженной температуре. Аналогичным образом, соединения формулы II, в которых в определении Q Y представляет собой циано, получают обработкой нецианированного аналога желаемым галоген-замещенным алифатическим нитрилом. Из цианопроизводного получают тетразолил путем обработки азидом три-н.бутил олова в среде, например, диметилового эфира этилен гликоля. Другие соединения формулы II могут быть получены, в соответствии с более полным объяснением, приведенным ниже, из соединений, синтез которых описан выше.
Способ и соединения по изобретению охватывают как рацемат, так и его индивидуальные стереоизомеры. Как правило, стереоизомеры могут быть получены в соответствии с методиками, хорошо известными из литературы. Однако для соединений формулы II, в которых Х представляется собой -S-; R4 и R5 представляет собой водород; а R1, R2, R3, R6, R7 и Q имеют значения, указанные для таких формул, индивидуальные стереоизомеры могут быть выделены в практически чистой изомерной форме в соответствии с новым способом, описанным ниже. В следующем способе предпочтительными соединениями, изомеры которых могут быть выделены, являются те соединения формулы II, в которых Х представляет собой группу -S-; R4 и R5 представляют собой водород; а R1, R2, R3, R6 R7 и Q имеют значения, указанные для предпочтительных, в некоторой степени более предпочтительных, более предпочтительных, особенно предпочтительных и наиболее предпочтительных соединений изобретения.Рацемическое сульфидное соединение формулы 11 реагирует с реагентом, полученным объединением тартратного лиганда, алкоксида титана, гидропероксида и, необязательно, воды. Алкоксидами титана, проходящими для использования в настоящем способе, могут служить алкоксиды титана формулы Тi (С1-С4 алкокси)4. Особенно предпочтительным алкоксидом титана является алкоксид, в котором С1-С4 алкокси группа представляет собой изопропокси. Аналогичным образом, подходящие тартратные лиганды для использования в настоящем способе включают ди(С1-С4 алкил) тартраты, причем особенно предпочтительными являются диэтилтартрат или диизопропилтартрат. Наконец, подходящие гидропероксиды, которые могут использоваться в настоящем способе, включают гидропероксид кумола, трет-бутилгидропероксид и т. п. Особенно предпочтительным гидропероксидом является трет-бутилгидропероксид.
Такую реакцию проводят путем смешивания указанных реагентов в среде инертного растворителя. Подходящие инертные растворители включают такие ароматические растворители, как толуол и т. п. такие галогенированные алканы, как хлористый метилен, 1,2-дихлорэтан, хлороформ и т. п. такие эфиры, как тетрагидрофуран, диэтиловый эфир и т. п. такие кетоны, как ацетон и т. п. Особенно предпочтительным инертным растворителем является хлористый метилен. Как правило, используемое количество растворителя должно быть достаточным для того, чтобы все соединения оставались в растворе в ходе реакции. Однако следует избегать использования избыточных количеств растворителя, поскольку в ходе выделения продукта могут иметь место нежелательные его потери.
Количество алкоксида титана, используемое в такой реакции не имеет решающего значения. Алкоксид титана может использоваться в количествах 0,4-2,0 эквивалентов относительно исходного рацемического сульфида. По причинам, подробно освещенным ниже, алкоксид титана предпочтительно используют в количествах, достаточных для достижения соотношения алкоксид титана (сульфидный субстрат, лежащих в интервале 0,5/1,0-0,75/1,0. Если алкоксид титана используют в количествах ниже эквимолярных относительно сульфидного исходного материала, то, если желательно, в систему можно добавлять молекулярные сита 3 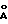 или 4
или 4 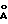 с тем, чтобы избежать дезактивации титанового комплекса водой. Количество используемого тартратного лиганда, гидропероксида и воды связано с количеством алкоксида титана и также не имеют решающего значения. Как правило тартратный лиганд используется в количествах, достаточных для достижения соотношения тартратных лиганд/алкоксид титана, лежащего в интервале 1/1-5/1, причем предпочтительное соотношение составляет 2/1. Аналогичным образом используемое количество гидропероксида может составлять от одного до двух эквимолярных количеств относительно алкоксида титана. Используемое количество воды может изменяться в интервале от безводных реакционных условий (т. е. отсутствие эквтивалентов воды) до 5 эквивалентов воды относительно количества присутствующего алкоксида титана. При использовании безводных реакционных условий, тартратный лиганд должен использоваться в количестве, достаточном для достижения соотношения тартратный лиганд/алкоксид титана, соответствующего верхнему значению указанного выше интервала соотношений тартратный лиганд/алкоксид титана. Стереохимия тартратного лиганда определяет природу стереоизомера, который будет получен из рацемического сульфидного субстрата. Так например, если в настоящей реакции используют (+) диизопропилтартрат, то будет выделен в практически чистой изомерной форме (-) энантиомер исходного сульфидного материала. Соответственно, если используют (-) диэтилтартрат, то получают практический чистый (+) энантиомер сульфидного субстрата. В соответствии с этим, тартратный лиганд должен выбираться таким образом, чтобы его стереохимия была обратной стереохимии желаемой изомерной формы.
с тем, чтобы избежать дезактивации титанового комплекса водой. Количество используемого тартратного лиганда, гидропероксида и воды связано с количеством алкоксида титана и также не имеют решающего значения. Как правило тартратный лиганд используется в количествах, достаточных для достижения соотношения тартратных лиганд/алкоксид титана, лежащего в интервале 1/1-5/1, причем предпочтительное соотношение составляет 2/1. Аналогичным образом используемое количество гидропероксида может составлять от одного до двух эквимолярных количеств относительно алкоксида титана. Используемое количество воды может изменяться в интервале от безводных реакционных условий (т. е. отсутствие эквтивалентов воды) до 5 эквивалентов воды относительно количества присутствующего алкоксида титана. При использовании безводных реакционных условий, тартратный лиганд должен использоваться в количестве, достаточном для достижения соотношения тартратный лиганд/алкоксид титана, соответствующего верхнему значению указанного выше интервала соотношений тартратный лиганд/алкоксид титана. Стереохимия тартратного лиганда определяет природу стереоизомера, который будет получен из рацемического сульфидного субстрата. Так например, если в настоящей реакции используют (+) диизопропилтартрат, то будет выделен в практически чистой изомерной форме (-) энантиомер исходного сульфидного материала. Соответственно, если используют (-) диэтилтартрат, то получают практический чистый (+) энантиомер сульфидного субстрата. В соответствии с этим, тартратный лиганд должен выбираться таким образом, чтобы его стереохимия была обратной стереохимии желаемой изомерной формы.
Рацемический сульфидный субстрат настоящего способа реагирует с реагентом, полученным из алкоксида титана, тартратного лиганда, гидроперексида и, необязательно, воды до тех пор пока практически весь нежелаемый энантиомер сульфидного исходного материала не превратиться в его сульфоксидный аналог. Конверсия в сульфоксид осуществляется при температуре, лежащей в интервале (-50)-(+50)оС, причем предпочтительная температура составляет -20оС. После того, как практически весь нежелательный энантиомер превратился в его сульфоксидный аналог, реакцию обрывают быстрым охлаждением вмеси в соответствии с хорошо известными методами.
Для обеспечения превращения практически всего нежелаемого энантиомера в сульфоксид, при минимизации превращения желаемого энантиомера, лишь 50-70% рацемического субстрата должно вступать в реакцию с реагентом, содержащим алкоксид титана. Ограничение реакции 50-70% может осуществляться по крайней мере двумя путями. Во-первых, гидропероксид может использоваться в количествах, которые обеспечивают соотношение гидропероксид/сульфидный субстрат 0,5/1-0,75/1,0. С другой стороны гидропероксид может использоваться в количествах выше 0,75 эквивалентов относительно сульфидного субстрата при условии слежения за ходом реакции с помощью стандартной аналитической техники, например, тонкослойной хроматографии (ТСХ) или жидкостной хроматографии высокого разрешения (НРIС).
Если с помощью такой техники установлено, что превратилось 50-70% сульфидного исходного материала, то реакцию прекращают с целью предотвращения дальнейшей конверсии.
После прекращения реакции непрореагировавшую часть сульфидного субстрата можно выделить из охлажденной реакционной смеси с использованием методов, хорошо известных специалистам. Такая непрореагировавшая часть будет состоять из желаемого энантиомера в практически чистой энантиомерной форме.
Следующие примеры дополнительно иллюстрируют получение соединений по изобретению, а также соединений, используемых в способе изобретения. Эти примеры также иллюстрируют способ селективного выделения энантиомеров, предусматриваемый изобретением. Эти примеры носят лишь иллюстративный характер и не ограничивают каким-либо образом сферу изобретения.
П р и м е р 1. 5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-2-тиоксо-4-тиазолидино н (соединение А).
В атмосфере азота 117-2 г 3,5-ди-трет.-бутил-4-гидроксибензальдегида, 66,6 г роданина и 143,5 г сплавленного ацетата натрия нагревали с обратным холодильником в 2500 мл ледяной уксусной кислоты. После нагревания в течение 23 ч реакционную смесь охлаждали в переливали в смесь 1 л этанола и 1 л в льда при перемешивании. Добавляли воду (500 мл) и после перемешивания в течение 30 мин полученный в результате осадок выделяли фильтрацией. Твердое вещество суспендировали с 500 мл этилацетата и фильтровали. Осадок растворяли в 3 л этанола, нагревали до кипения и добавляли воду до сохранения мутности раствора (примерно 450 мл воды). В ходе охлаждения до комнатной температуры 99,6 г целевого продукта выделяли фильтрацией, т. пл. примерно 260оС.
Вычислено, С 61,86; Н 6,83; N 4,01.
С18Н23NO2S2:
Найдено, С 62,13; Н 6,55; N 4,15.
П р и м е р ы 2-3. 5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]метилен] -4-тиазолидинон (соединение В) и 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метил]-4-тиазолидинон (соединение С).
Раствор 69,90 г 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]-метилен] -2-тиоксо-4-тиазолидин она в 4 л этанола гидрировали при давлении 500 фунт. /дюйм2) в присутствии 200 г 5% палладия на угле в течение ночи при 100оС. Реакционную смесь фильтровали выпаривали досуха. Полученный материал последовательно растворяли в 1 объеме горячего этилацетата, разбавляли 2 объемами гексана, фильтровали и загружали в хроматографическую колонку с силикагелем. Путем элюирования 35% этилацетатом в гексане получали различные фракции, которые объединяли в соответствии с чистотой соответствующих соединений. Методом хроматографии выделяли 4,6 г соединения В. Фракции, содержащие преимущественно соединение В, перекристаллизовывали из системы этилацетат/гексан, получая соединение В с общим выходом 13,79 г. В результате повторной хроматографии фракции, содержащих загрязненное соединение С на оксиде кремния, при элюировании 25%-ным раствором этилацетата в гексане, получали 9,82 г соединения С. 2,5-[[3,5-бис(1,1-Диметилэтил)-4-гидроксифенил]-метилен]-4-тиазолидинон, т. пл. 209-213оС.
Вычислено, С 67,67; Н 7,89; N 4,38.
С18Н25NO2S.
Найдено, С 67,44; Н 8,11; N 4,65. 3,5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]метил]-4-тиазолидинон, т. пл. 149-152оС.
Вычислено, С 67,25; Н 8,47; N 4,36.
С18Н27NO2S.
Найдено, C 67,43; Н 8,44; N 4,21.
П р и м е р 4. 5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]-метил]-2-тиоксо-4-тиазолидинон (соединение Д).
В атмосфере азота 13,98 г 5-[[3,5-бис(1,1-диметил-этил-4-гидроксифенил] -метилен] -2-тиоксо-4-тиазолидин она, 13,17 г диэтил-2,6-диметил-1,4-дигидро-3,5-пиридинкарбоксилата и 600 мл толуола перемешивали до получения раствора. В реакционную смесь добавляли 40 г силикагеля 60 (размер частиц менее 230 меш) предварительно высушенного в вакууме в течение 7 ч при 50оС. Реакционную смесь нагревали в течение 18 ч с обратным холодильником и отфильтровывали в горячем состоянии. Фильтрат выпаривали досуха. Остаток растворяли в 500 мл этилацетата, 5 раз промывали 400 мл порциями 1 н. хлористоводородной кислоты, сушили над сульфатом натрия, фильтровали и выпаривали в вакууме с получением желтого твердого вещества. В результате хроматографической очистки на силикагеле при элюировании 2,5% этилацетата в толуоле получали 8,0 г целевого продукта, т. пл. 178-179оС.
Вычислено, С 61,50; Н 7,17; N 3,98.
С18Е25NO2S2.
Найдено, С 61,28; Н 7,19; N 3,94.
П р и м е р 5. 5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-3-метил-2-тиоксо-4-тиа золи
Целевое соединение получали с выходом 76% из 3,5-ди-трет-бутил-4-гидроксибензальдегида и N-метилроданина согласно методике примера 1, т. пл. > 230оС.
Вычислено, С 62,77; Н 6,93; N 3,85; S 17,64.
С19Н25NO2S2.
Найдено, С 62,54; Н 7,05; N 3, 66; S 17,82.
П р и м е р 6. 5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-3-метил-4-тиазолидинон
Целевое соединение получали с выходом 71% из 10,31 г тиона из примера 5 в ходе нагревания с 38,51 мл гидрида три-н.-бутилолова и 1,16 г азобисизобутиронитрила (АИБН) в 142 мл толуола при температуре дефлегмации в течение 1 ч. Продукт реакции выделяли путем добавления воды к охлажденной реакционной смеси, разделения слоев, промывания органического слоя 1 н. хлористоводородной кислотой и насыщенным раствором хлористого натрия, сушки над сульфатом магния, концентрации в вакууме и очистки остатка хроматографированием на силикагеле, проводя элюирование 10-50% гексана в этилацетатном градиенте. Очищенный продукт имел точку плавления 142-144оС.
Вычислено, С 68,43; Н 8,16; N 4,20.
С19Н27NO2S.
Найдено, С 68,68; Н 8,00; N 3,97.
П р и м е р 7. 5-[[3,5-Бис(1,1-Диметилэтил)-4-гидроксифенил]-метил]-3-метил-4-тиазолидинон.
К 100 мл ТГФ добавляли 6,43 г соединения примера 3. Добавляли гидрид натрия (0,9 г), в результате чего выделялся газ. Добавляли иодометан (1,25 мл, 1,0 экв) и полученную в результате смесь перемешивали при комнатной температуре в течение 23 ч после чего его разбавляли объемом диэтилового эфира 1 н. HCl. Органический слой отделяли и сушили над сульфатом натрия, фильтровали и выпаривали. Полученное в результате твердое вещество обрабатывали хлороформом с образованием оранжевой пены. Образец полученного материала в количестве 5,93 г растворяли в 14 мл горячей смеси этил ацетата, разбавленного 225 мл гексана, и полученной смеси давали остывать в течение ночи до комнатной температуры. Растворитель выпаривали и полученное в результате твердое вещество растворяли в 40 мл горячей смеси диэтилового эфира, разбавленного 400 мл гексана. Такой смеси давали охлаждаться до комнатной температуры в течение ночи и образовавшийся осадок собирали фильтрацией, промывали гексаном и сушили в вакууме с образованием 3,98 г целевого соединения, т. пл. 102-105оС.
Вычислено, С 68,02; Н 8,71; N 4,17.
С19Н29NO2S.
Найдено, С 68,22; Н 8,80; N 4,21.
П р и м е р 8. 5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-3-диметил- амина-2-тиоксо-4-тиазолидинон.
Целевое соединение получали с выходом 65% из 3,5-ди-трет.-бутил-4-гидроксибензальдегида и N-диметил-аминороданина, следуя методике примера 1.
П р и м е р 9. 5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-3-диметиламино- -4-г тиазолидинон.
Соединение примера 8 восстанавливали с использованием методики примера 6 с получением целевого соединения с выходом 41% т. пл. 138-141оС.
Вычислено, С 66,26; Н 8,34; N 7,73.
С20Н30N2O2S.
Найдено, С 66,56; Н 8,59; N 7,47.
П р и м е р 10. 5-[[3,5-Бис-(1,1-диметилэтил)-4-гидроксифенил]метилен] -3-(метиламино)- -4-тиазолидинон.
А. Получение бензальдегидметилгидразона.
Бензальдегид (50,8 мл, 500 ммоля) и 26,5 мл (500 ммоля) метилгидразина растворяли в 1 л метанола. Полученную смесь перемешивали в течение 75 мин при комнатной температуре и затем отпаривали от растворителя с получением 67,8 г замещенного интермидиата.
В. Получение бензальдегид N-метил, N-2-пропенилгидразона.
Указанное соединение (67,8 г), 60,5 г бромистого алкила 50,5 г триэтиламина растворяли в 1л ацетонитрила. Полученную смесь нагревали в течение 16 ч с обратным холодильником и затем охлаждали. Добавляли еще 45 г бромистого аллила и 38 г триэтиламина и смесь снова нагревали в течение 7 ч с обратным холодильником охлаждали и затем отпаривали от растворителя с получением 268 г остатка. К этому остатку добавляли 500 мл ТГФ. Фильтрат отпаривали от растворителя с получением 67 г указанного выше промежуточного соединения.
C. Получение N-метил, N-2-пропилгидразина.
Указанное соединение (59,9 г), 44 г гидразина и 137 мл этанола нагревали с обратным холодильником в течение 21,5 и смеси давали охлаждаться. Обратный холодильник заменяли на дистилляционную насадку и смесь перегоняли при атмосферном давлении. Три первых дистиллата собирали, объединяли и добавляли 100 мл 1 н. НСl. Добавляли еще 100 мл концентрированной 1 н. НСl при охлаждении системы льдом и полученную в результате смесь отделяли и промывали небольшим количеством этил ацетата. Полученные в результате слои разделяли и отгоняли воду до начала осаждения твердых веществ на лопасти мешалки. Твердые вещества отфильтровывали и фильтрат отпаривали и добавляли к 125 мл охлажденного 50% -ного раствора NaOH. Полученное в результате твердое вещество отфильтровывали и отбрасывали. Отделяли фильтрат, содержащий два слоя. Верхний слой содержал замещенный интермедиат, а нижний, водный слой, экстрагировали диэтиловым эфиром, который после отпаривания давал дополнительное количество продукта.
Д. Получение N-метил, N-3-пропенил-5-карбокси-метил-дитиокарбамата.
К 12,67 г указанного соединения в 23 мл этанола, охлажденного до 0оС, добавляли раствор 11,18 г сероуглерода в 26 мл диэтилового эфира. Полученную в результате смесь удаляли из бани со льдом и выстаивали при комнатной температуре в течение 15,5 ч, после чего растворитель удаляли с образованием примерно 36,5 г остатка. К такому остатку добавляли 13,9 г хлоруксусной кислоты, растворенной в 29,5 мл 5 n. NaOH (охлажденной на бане со льдом). Полученный в результате раствор выстаивали в течение 3 ч при комнатной температуре. рН раствора понижали до 3 путем добавления 8 мл концентрированной хлористоводородной кислоты. Затем добавляли диэтиловый эфир (50 мл) в результате чего происходило разделение на ри фазы. Водные фазы сливали и экстрагировали 50 мл хлороформа, затем отпаривали от растворителя с получением, примерно, 40,4 г целевого соединения.
Е. Получение 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]-метилен]-2-тиоксо-3- -[метил-2-пропениламино]-4-тиазолидона.
3,5-ди-трет-бутил-4-гидроксибензальдегид (29,3 г), 38,8 г указанного соединения и 40,34 г ацетата натрия смешивали в 810 мл уксусной кислоты и полученный в результате раствор нагревали с обратным холодильником в течение 24 ч. Раствору давали охлаждаться и его отпаривали в течение 50 ч при комнатной температуре. Затем раствор переливали в 2 л ледяной воды, разделяли и промывали дополнительным объемом воды с образованием 44 г указанного выше промежуточного соединения.
F. Получение 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-3-[метил-2- -пропениламино]-4-тиазолидинона.
С использованием методики, описанной в примере 6, 42,8 г указанного тиона восстанавливали в названный выше интермедиат (8,34 г).
G. Получение 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]-метилен]-3-(метиламино)- -4-тиазолидинон.
Указанное соединение (6,11) растворителя в смеси 135 мл этанола и 15,3 мл воды и полученную смесь нагревали до 70оС. Добавляли трис-трифенилфосфин-родий (I) хлорид (50 мг) и полученную смесь нагревали с обратным холодильником в течение 50 мин после чего добавляли еще 550 мг катализатора с последующим нагреванием при температуре дефлегмации в течение 2,5 ч. Смесь охлаждали и перемешивали при комнатной температуре в течение ночи, после чего отпаривали растворитель с получением 2,05 г целевого продукта после дополнительной обработки, т. пл. 151-153; 5оС.
Вычислено, С 65,86; Н 7,56; N 8,09.
С19Н28N2O2.
Найдено, С 65,67; Н 7,81; N 8,34.
П р и м е р ы 11 и 12
5-[[3,5-Ди-2-пропенил-4-гидроксифенил] -метилен] -4-тиазолидинон и 5-[[3,5-дис-2-пропенил-4-гидроксифенил]метил]-4-тиазолидинон.
А. Получение 3,5-бис-[2-пропенил]-4-гидроксибензальдегида.
В атмосфере и с использованием механической мешалки, 250 парагидроксибензальдегида, 247,6 г бромистого аллила, 311,7 г бикарбоната калия и 650 мл ацетона нагревали с обратным холодильником в течение 18 ч. Смеси давали охлаждаться, после чего добавляли 1 л воды с последующей экстракцией двумя порциями по 800 мл диэтилового эфира. В результате последующей дистилляции органической фазы получали 299 г 4-[2-пропенил]-оксибензальдегида, который затем нагревали с 300 мл диэтиланилина и в течение 5,5 ч при 195-205оС. Смесь охлаждали и добавляли 750 мл этил ацетата. Смесь трижды промывали и добавляли 750 мл этил ацетата. Смесь трижды промывали тремя а порциями по 500 мл 1N. НСl и после последующей обработки получали около 138 г 3-(2-пропенил)-4-гидроксибензальдегида. Монозамещенный альдегид (159 г) снова нагревали с обратным холодильником в присутствии 152 г карбоната калия и 465 мл ацетона в течение 3 ч и затем давали остывать. Полученную смесь переливали в 900 мл ледяной воды и после этого экстрагировали двумя порциями по 430 мл диэтилового эфира с получением 170 г 3-(2-пропенил)-4-(2-пропенилокси)бензальдегида. Затем дизамещенный альдегид нагревали в 500 мл диэтиланилина в атмосфере азота до 195-205оС в течение 6,5 ч. Полученную смесь охлаждали и растворяли в 800 мл этил ацетата, промывали тремя 1 л порциями 1N. НСl и, после последующей обработки, получали 121,9 г замещенного промежуточного соединения.
В. Получение 5-[[3,5-ди-2-пропенил-4-гидроксифенил]-метилен]-2-тиоксо-4-тиазолидинона.
Указанное соединение (50,5 г), 36,6 г роданина и 164 г ацетата натрия при температуре дефлегмации в 1,25 л уксусной кислоты в течение 14,5 ч. Полученный в результате раствор охлаждали, переливали в 2 л ледяной воды с образованием, после разделения, около 75 г замещенного интермедиата, т. пл. 157-160оС.
Получение 5-[[3,5-ди-2-пропенил-4-гидроксифенил]метилен]-4-тиазолидинона и 5-[[3,5-ди-2-пропенил-4-гидроксифенил]-метил]-4-тиазолидинона.
Указанное соединение (74,8 г) подвергали восстановительной обработке цинковой пылью (62 г) и концентрированной соляной кислотой (950 мл) в 2,1 л горячего (примерно 82оС) этанола. После смешивания реагентов раствору давали охлаждаться до комнатной температуры, перемешивали в течение 1 ч и затем добавляли к 3,75 л ледяной воды. Полученный в результате раствор осаждался в течение ночи с образованием смолы. Жидкий слой декантировали и экстрагировали 750 мл хлороформа, а смолу растворяли в 560 мл хлороформа и полученный раствор последовательно промывали 75 мл насыщенного раствора карбоната натрия, 75 мл воды и 75 мл насыщенного раствора рассола. Полученные хлороформные растворы объединяли и затем обрабатывали 100 мл хлористого метилена. Целевые продукты получали с использованием хроматографирования на силикагеле. Элюированием с помощью градиентов 25-60% этилацетата в гексане получали различные фракции, которые обрабатывали следующим образом.
Фракции 13-15 концентрировали и затем промывали этилацетатом с получением 2,91 г 5-[[3,5-ди-2-пропенил-4-гидроксифенил] -метил]-4-тиазолидинона. Фракции 16-18 концентрировали с образованием остатка, который обрабатывали 30 мл хлористого метилена. Фракции 19-23 концентрировали с образованием остатка, который обрабатывали 35 мл хлористого метилена. После обработки оставшиеся нерастворенные вещества выделяли фильтрацией и обрабатывали 40 мл этилацетата с получением 3,85 г 5-[[3,5-ди-2-пропенил-4-гидроксифенил]-4-тиазолидинона.
Этилацетатную промывную жидкость из фракции 13-15, метиленхлоридный раствор, соединяющий фракции 16-18, и метиленхлоридный и этилацетатный растворы, полученные из фракции 19-23, объединяли и загружали в колонку с силикагелем для хроматографического разделения. В результате элюирования смесью этилацетат-гексан в соотношении 1:1 получили различные фракции, которые объединяли в соответствии с чистотой соответствующих соединений. Фракции, содержащие преимущественно 5-[[3,5-ди-2-пропенил-4-гидроксифенил]метил]-4-тиазолидинон, перекристаллизовывали из горячего этилацетата с получением 1,24 г указанного соединения (общий выход 5-[[3,5-ди-2-пропенил-4-гидроксифенил]-метил] -4-тиазолидинона 4,14 г). Фракции, содержащие преимущественно 5-[[3,5-ди-2-пропенил-4-гидроксифенил]-метилен]-4-диазолидинон обрабатывали 30 мл горячего этилацетата с получением 1,73 г указанного вещества (общий выход 5-[[3,5-ди-2-пропенил-4-гидроксифенил]-метилен]-4-диазолидинона 5,85 г).
11. 5-[[3,5-ди-2-Пропенил-4-гидроксифенил] метилен] -4-тиазолидинон, т. пл. 184-188оС.
Вычислено, С 66,87; Н 5,96; N 4,87;
С16Н17NO2S.
Найдено, С 66,62; Н 5,92; N 4,89.
12. 5-[[3,5-ди-2-Пропенил-4-гидроксифенил] метил] -4-тиазолидинон т. п. 142-144оС.
Вычислено, С 66,41; Н 6,62; N 4,84.
С16Н19NO2S
Найдено, С 66,18; Н 6,69; N 4,60.
В соответствии с методиками, указанными в примерах 11 и 12 и в других частях описания, получали следующие соединения.
П р и м е р 13. 5-[[3,5-ди-2-Пропенил-4-гидроксивенил]-метилен]-3-метил-4-тиазолидинон, т. пл. 155-159оС.
Вычислено, С 67,74; Н 6,35; N 4,65.
С17Н19NO2S.
Найдено, С 67,53; Н 6,09; N 4,45.
П р и м е р 14. 5-[[3,5-Дипропил-4-гидроксифенил]-метилен]-3-метил-4-тиазолидинон, т. пл. 162-165оС.
Вычислено, С 66,85; Н 7,59; N 4,59.
С17Н23NO2S
Найдено, С 67,12; Н 7,37; N 4,52.
П р и м е р 15. 5-[[3,5-Дипропил-4-гидроксифенил]-метилен]-4-ти-азолидинон, т. пл. 202-205оС.
Вычислено, С 65,95; Н 7,26; N 4,81.
С16Н21NO2S.
Найдено, С 66,16; Н 7,49; N 4,79.
П р и м е р 16. 5-[[3,5-Дипропил-4-гидроксифенил]метил]-4-тиазолидинон, т. пл. 155-157оС.
Вычислено, С 65,49; Н 7,90; N 4,77.
С16Н23NO2S
Найдено, С 65,71; Н 7,73; N 4,99.
П р и м е р 17. 5-[[3-(1,1-Диметилэтил)-4-гидрокси-5-метилфенил]метилен] -4-тиазолидинон.
А. Получение 4-гидрокси-3-метил-5-(1,1-диметилэтил)бензальдегида.
В атмосфере азота 76,65 г 2-(1,1-диметилэтил)-6-метилфенола(Алдрич), 65,42 г гексаметилентетрамина и 700 мл трифторуксной кислоты перемешивали при температуре дефлегмации в течение 24 ч. Реакционному раствору давали охлаждаться и жидкость удаляли выпариванием. Полученный в результате осадок переносили в 1500 мл воды и 1000 мл хлороформа и затем нейтрализовали до рН 7 с помощью твердого карбоната натрия. Полученные в результате слои разделяли и водный слой промывали хлороформом. Органический слой объединяли с хлороформной промывной жидкостью и полученный в результате раствор промывали водой, затем сушили в течение ночи над сульфатом натрия. После удаления сульфата натрия хлороформ выпаривали. Полученный в результате остаток переносили в 375 мл толуола, нагревали на паровой бане и затем давали охлаждаться до комнатной температуры в течение ночи.
В результате последующей обработки получали 28,3 г замещенного промежуточного соединения.
В. Получение 5-[[3-(1,1-диметилэтил)-4-гидрокси-5-метилфенил]-метилен] -2-тиоксо- -4-тиазолидинона. Полученное промежуточное соединение (28,3 г), 24 г N-аминороданина, 48,3 г ацетата натрия и 735 мл уксусной кислоты нагревали до температуры дефлегмации в течение 7 ч и затем давали охлаждаться до комнатной температуры при непрерывном перемешивании в течение ночи. Полученную в результате смесь переливали в 1500 мл ледяной воды при перемешивании и затем фильтровали. Влажный осадок на фильтре переносили в стакан и растворяли в смеси этилацетата и воды. Полученные в результате органические и водные слои разделяли. Органический слой сушили над сульфатом натрия и затем фильтровали сцелью удаления указанного вещества. В результате последующей обработки, с последующим распределением в горячем хлороформе и последующей сушкой в вакууме получали около 18 г целевого промежуточного вещества, т. пл. 210-216оС.
С. Получение 5-[[3-(1,1-диметилэтил)-4-гидрокси-5-метилфенил]-метилен] -4-тиазолидинон.
Восстановление полученного тиона осуществляли описанными методами и после соответствующей обработки получали 1,56 г целевого продукта, т. пл. 162-165оС.
Вычислено, С 64,95; Н 6,90; N 5,05.
С15Н19NO2S.
Найдено, С 65,12; Н 7,05; N 4,99.
Используя методики, описанные в примере 17 и других частях описания, получали следующие дополнительные соединения.
П р и м е р 18. 5-[[3,5-Бис(1-метилэтил)-4-гидроксифенил]-метилен]-3-метил-4-тиазолидинон, т. пл. 200-210оС.
Вычислено, С 66,85; Н 7,59; N 4,59.
С17Н23NO2S
Найдено, С 67,03; Н 7,55; N 4,37.
П р и м е р 19. 5-[[3,5-Бис(1-метилэтил)-4-гидроксифенил]-метил]-2-тиоксо-4-тиазолидинон.
П р и м е р 20. 5-[[3-(1,1-Диметилэтил)-4-гидрокси-5%метилфенил]метил] -4-тиазолидинон.
Раствор 0,28 г соединения примера 17 в 30 мл тетрагидрофурана гидрировали при давлении 60 фунтов/дюйм2 в присутствии 1,12 г 5%-ного палладия на угле в течение ночи при 60оС. Реакционную смесь отфильтровывали и выпаривали досуха. Полученный в результате осадок растворяли в 3,5 мл смеси этилацетат-гексан в соотношении 1:1,5 и загружали в хроматографическую колонку с силикагелем. Проводя элюирование 40%-ным этилацетатом в гексане получали фракции, которые после выпаривания досуха давали 0,05 г целевого соединения, т. пл. 64-68оС.
Вычислено, С 64,48; Н 7,58; N 5,01.
С15Н21NO2S.
Найдено, С 64,32; Н 7,66; N 4,79.
П р и м е р 21. 5-[[3,5-Бис(1-метилэтил)-4-гидроксифенил]-метил]-4-тиазолидинон.
С использованием способа, описанного в примере 20, 4,73 г соединения примера 19 превращали в 1,88 целевого соединения, т. пл. 136-139оС.
Вычислено, С 65,49; Н 7,90; N 4,77.
С16Н23NO2S.
Найдено, С 65,79; Н 7,90; N 4,81.
П р и м е р 22. 5-[[3-(1,1-Диметилэтил)-4-гидрокси-5-пропилфенил]метил] -4-тиазолидинон.
А. Получение 3-2-(1,1-диметилэтил)-феноксипропена.
Бромистый аллил (69,2 мл), 2-трет-бутилфенол (122,9 мл) и карбонат калия (121,6 г) перемешивали в 265 мл ацетона при температуре дефлегмации в течение 50 ч и затем охлаждали до 35оС. Добавляли воду (600 мл) и полученные в результате слои разделяли. Водный слой экстрагировали 600 мл диэтилового эфира. Органический слой объединяли эфирным экстрактом водных слоев и полученный в результате раствор сушили в течение ночи над сульфатом натрия. После удаления сульфата натрия выпаривали растворитель и получали, после дополнительной обработки, 147 г целевого промежуточного соединения.
В. Получение 2-(1,1-диметилэтил)-6-(2-пропенил)фенола.
Все 147 г полученного соединения подвергали перегруппировке в соответствии с описанным в примерах 11А и 12А с получением 100,8 г целевого промежуточного соединения.
С. Получение 2-(1,1-диметилэтил)-6-пропилфенола.
Раствор 54,9 г указанного соединения в 575 мл толуола гидрировали при давлении 60 фунтов/дюйм2, в присутствии 55 г никеля Рэнея, в течение 3 ч при комнатной температуре. Реакционную смесь фильтровали и выпаривали досуха с получением 59,2 г целевого интермедиата.
Д. Получение 3-(1,1-диметилэтил)-4-гидрокси-5-пропилбензальдегида.
Указанное соединение (55,48 г) превращали в 23,33 г целевого промежуточного соединения с использованием способа, описанного а в примере 17А.
Е. Получение 5-[[3-(1,1-диметилэтил)-4-гидрокси-5-пропилфенил]-метилен] -2-тиоксо-4-тиазол идин
С использованием способа, описанного в примере 17В, 5,51 г указанного соединения превращали в 6,26 г целевого промежуточного соединения, т. пл. 190,5-192оС.
Получение 5-[[3-(1,1-диметилэтил)-4-гидрокси-5-пропилфенил] метил]-2-тиоксо-4-тиазолиди нона
С использованием способа, описанного в примере 4, 4,73 г указанного соединения превращали в 2,1 г целевого промежуточного соединения.
С. Получение 5-[[3-(1,1-диметилэтил)-4-гидрокси-5-пропилфенил]-метил]-4-тиазолидинона.
Раствор 2,1 г указанного соединения в 185 мл этанола гидрировали при давлении 500 фунтов/дюйм2 в присутствии 8,4 г 5% палладия на угле в течение 20 ч при 100оС. Реакционную смесь фильтровали и выпаривали досуха. Полученный остаток растворяли в 25 мл хлористого метилена и загружали в хроматографическую колонку с силикагелем. Проводя элюирование 2000 мл 10-50% этилацетата в гексановом градиенте, с последующим элюированием 2000 мл смеси ацетат-гексан в соотношении 1:1, получали фракции, которые после выпаривания досуха давали 0,75 г целевого продукта, т. пл. 50-55оС.
Вычислено, С 66,41; Н 8,20; N 4,56.
С17Н25NO2S
Найдено, С 66,61; Н 8,22; N 4,55.
П р и м е р 23. 5-[[3-Метилтиофенил-4-гидрокси-5-этоксифенил]-метилен] -3-диметиламино-4-тиаз олид
А. Получение 5-[[3-этокси-4-гидроксифенил]метилен]-3-диметиламино-2-тиоксо-4-тиазолидинон а.
3-Этокси-4-гидроксибензальдегид (45,7 г), N-диметиламинороданин (53,35 г) и плавленый ацетат натрия (92,4) реагировали друг с другом в соответствии со способом, описанным в примере 1, с получением 52,92 г целевого промежуточного соединения, т. пл. 194-198оС.
В. Получение 5-[[3-этокси-4-гидроксифенил]метилен]-3-диметиламино-4-тиазолидинона.
С использованием способа, описанного в примере 6, 47,66 г указанного соединения превращали в 14,02 г целевого промежуточного соединения.
С. Получение 5-[[3-этокси-4-гидрокси-5- (метилтиофенилфенил]-метилен]-3-диметиламино-4-тиазолидинола.
Гидроксид натрия (0,95 г) и 17,3 мл 40 мас. раствора формальдегида растворяли в 50 мл 2-этоксиэтанола, добавляли фенилтиол (2,62 г) и 7,0 г указанного соединения и полученный в результате раствор нагревали с обратным холодильником в течение 4 ч, затем охлаждали. К охлажденной реакционной смеси добавляли этил ацетат (50 мл) и воду (25 мл) и рН полученного в результате двухфазного раствора понижали до значения 5 с использованием концентрированной хлористоводородной кислоты. Органическую фазу отделяли от двойной фазы, промывали насыщенным раствором рассола и затем загружали в хроматографическую колонку с силикагелем. В результате элюирования 4 л хлористого метилена и 4 л смеси на 3% метанола и 97% хлористого метилена получали фракции, содержащие целевой продукт. Эти фракции объединяли и снова загружали в хроматографическую колонку с силикагелем. В результате элюирования 4 л хлористого метилена с последующим элюированием 1 л 22,5% ацетонитрила в хлористом метилене получали фракции, которые после выпаривания растворителя давали целевой продукт. Этот продукт дополнительно очищали путем обработки горячим раствором 50 мл гексана и 30 мл этилацетата с получением 6,20 г 5-[[3-метилтиофенил-4-гидрокси-5-этоксифенил] -метилен]-3-диметиламино-4-тиазолилинола. Т. пл. 118-120оС.
Вычислено, С 60,55; Н 5,81; N 6,73; S 15,39.
С21Н24N2O3S2.
Найдено, С 60,75; Н 5,76; N 6,76; S 15,58.
П р и м е р 24. (+)-5-[[3,5-(1,1-диметилэтил)-4-гидроксифенил]метил]-4-тиазолидинон.
В трехгорлую круглодонную колбу емкостью 50 мл, содержащую 25 мл хлористого метилена добавляли 1,31 г 4 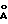 -молекулярных сит, 0,56 мл (1,88 ммоля) изопропилата титана, 0,79 мл (3,75 ммоля) /+/-диизопропил тартрата и 34 мкл (1,88 ммоля) деионизированной воды. Полученный в результате раствор перемешивали в течение 20 мин и затем добавляли 0,8 г (2,5 ммоля) рацемической смеси 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] -метил] -4-тиазолидинона. Полученный в результате раствор охлаждали до -20оС и добавляли 0,73 мл (1,88 ммоля) 2,57 М раствора трет-бутилгидропероксида в изооктане. Затем реакционный раствор перемешивали в течение 6 ч при -20оС. Через 6 ч реакционный раствор быстро охлаждали, переливая его в 50 мл раствора, полученного из 9,9 г гептагидрата сульфата железа (II), 3,3 г лимонной кислоты и воды. Полученный в результате раствор перемешивали в течение 30 мин и затем перемешивание прекращали, так чтобы можно было разделить органический и водный слои. Водный слой декантировали и промывали хлористым метиленом. Промывную метиленхлоридную жидкость объединяли с указанным органическим слоем и полученный в результате раствор промывали насыщенным раствором рассола и затем сушили над сульфатом натрия. Сульфат натрия удаляли фильтрацией и оставшуюся жидкость выпаривали с получением 1,81 г остатка.
-молекулярных сит, 0,56 мл (1,88 ммоля) изопропилата титана, 0,79 мл (3,75 ммоля) /+/-диизопропил тартрата и 34 мкл (1,88 ммоля) деионизированной воды. Полученный в результате раствор перемешивали в течение 20 мин и затем добавляли 0,8 г (2,5 ммоля) рацемической смеси 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] -метил] -4-тиазолидинона. Полученный в результате раствор охлаждали до -20оС и добавляли 0,73 мл (1,88 ммоля) 2,57 М раствора трет-бутилгидропероксида в изооктане. Затем реакционный раствор перемешивали в течение 6 ч при -20оС. Через 6 ч реакционный раствор быстро охлаждали, переливая его в 50 мл раствора, полученного из 9,9 г гептагидрата сульфата железа (II), 3,3 г лимонной кислоты и воды. Полученный в результате раствор перемешивали в течение 30 мин и затем перемешивание прекращали, так чтобы можно было разделить органический и водный слои. Водный слой декантировали и промывали хлористым метиленом. Промывную метиленхлоридную жидкость объединяли с указанным органическим слоем и полученный в результате раствор промывали насыщенным раствором рассола и затем сушили над сульфатом натрия. Сульфат натрия удаляли фильтрацией и оставшуюся жидкость выпаривали с получением 1,81 г остатка.
Этот остаток растворяли в 25 мл хлористого метилена и полученный в результате раствор хроматографировали на колонке с силикагелем. В результате элюирования 6000 мл 10-51% этилацетата в гексановом градиенте получали различные фракции, содержащие указанное целевое соединение. Эти фракции объединяли и жидкость выпаривали с получением 0,19 г целевого соединения. ( α )25 -73,6о (С 1,0 МеOH).
Вычислено, С 67,25; Н 8,47; N 4,36.
С18Н27NO2S.
Найдено, С 67,50; Н, 8,53; N 4,48.
П р и м е р ы 25, 26 и 27. (+)-5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил] метил] -4-тиазолидинон, (-)-5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] метил]-4-тиазолидинон-1-окс ид и (+)-5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]-метил]-4-тиазолидинон-1-ок сид.
Согласно способу, аналогичному описанному в примере 24, проводили реакцию между 0,89 мл (3,0 ммоля) изопропилата титана, 1,27 мл (6,0 ммоля) (-)-диизопропил тартрата, 54 мкл (3,0 ммоля) деионизированной воды, 1,61 г (5,0 ммоля) рацемического 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]-метил] -4-тиазолидинона и 2,4 мл (6,5 ммоля) 2,57М раствора трет-бутилгидропероксида в изооктане с получением остатка. Остаток растворяли в 75 мл хлористого метилена и полученный в результате раствор хроматографировали на хроматографической колонке с силикагелем. Проводя элюирование с помощью 6000 мл 10-50% этилацетата в гексановом градиенте получали различные фракции, содержащие (+)-5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метил]-4-тиазолидинона. Такие фракции объединяли и выпаривали жидкую фазу с получением 0,43 г продукта. В результате дальнейшего элюирования с помощью 4000 мл 50%-ного раствора изопропанола в гексане получали различные фракции. Фракции, которые содержали (-)-5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метил]-4-тиазолидинон-1-окс ид, объединяли и выпаривали жидкость с получением 0,87 г продукта. Фракции, содержащие (+)-5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] метил]-4-тиазолидинон-1-окс ид объединяли и выпаривали жидкость с получением 0,27 г продукта.
25. (+)-5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метил]-4-тиазолидинон.
[ α25 +70,41о (C 1,0, МеOH).
Вычислено, С 67,25; Н 8,47; N 4,36.
С18Н27NO2S.
Найдено, С 66,95; Р 8,22; N 4,26.
26. (-)-5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]метил]-4-тиазолидинон-1-окс ид, т. пл. 182-184оС.
[ α25 -21,84о (C 1,0, МеOH).
Вычислено, С 64,06; Н 8,06; N 4,15.
С18Н27NO3S.
Найдено, С 63,84; Н 8,09; N 4,12.
27. (+)-5-[3,5-[бис(1,1-диметилэтил)-4-гидроксифенил]-метил]-4-тиазолидинон-1-ок сид,т, пл. 177-181оС.
[ α25 +163,05о (С 1,0, МеOH).
Вычислено, С 64,06; Н 8,06; N 4,15.
С18Н27NO3S.
Найдено, С 63,88; Н 8,12; N 4,29.
П р и м е р 28. (-)-5-[[3,5-Бис(1,1-диметилэтил)-4-гидроксифенил]-метил] -3-метил-4-тиазолиди нон.
Согласно способу, аналогичному описанному в примере 24, проводили реакцию между 0,45 мл (1,5 ммоля) изопропилата титана, 0,63 мл (3,0 ммоля) (+)-диизопропилтартрата, 27 мкл (1,5 ммоля) воды, 0,84 г (2,5 ммоля) рацемического 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил] -метил]-3-метил-4-тиазолидинона и 0,58 мл (1,5 ммоля) 2,75М раствора трет-бутилгидропероксида в изооктане с получением остатка. Этот остаток растворяли в 25 мл хлористого метилена и полученный в результате раствор хроматографировали на колонке с силикагелем. Элюированием с помощью 1000 мл хлористого метилена и затем с помощью 6000 мл 0-10%-ного этилацетата с метиленхлоридном градиенте, затем с помощью 4000 мл 20-50%-ного изопропилового спирта с гексановом градиенте и далее с помощью 2000 мл 50%-ного раствора изопропилового спирта в гексане, получали различные фракции, содержащие целевое соединение. Эти фракции объединяли и выпаривали жидкость с получением 0,35 г целевого соединения.
Вычислено, С 68,02; Н 8,71; N 4,17.
C19H29NO2S.
Найдено, С 67,95; Н 8,55; N 4,18.
ЯМР (300 Мгц; СДCl3) σ 1,4(синглет, 18Н,2,9(синглет,3,H), 3,0(двойной дублет, 1Н), 3,8(двойной дублет, 1Н), 4,0(дублет, 1Н), 4,2(дублет, 1Н), 5,1(синглет,1Н), 7,1(синглет,2Н).
П р и м е р 29. 5-[[3-(1,1-Диметилэтил)-4-гидроксифенил]-метил]-4-тиезолидинон.
А. Получение 3-(1,1-диметилэтил)-4-гидроксибензальдегида.
В 184,4 мл (1,494 ммоля) N-метилформанилида при охлаждении прикапывали 130,9 мл (1,404 моля) хлористого фосфорида в течение 20 мин. Смеси давали нагреваться до комнатной температуры и перемешивали в течение 1 ч. Затем к реакционной смеси прикапывали в течение 25 мин орто-трет-бутилфенол (138,2 мл, 0,9 моля). После завершения добавления фенола полученную в результате реакционную смесь перемешивали в течение еще 30 мин при комнатной температуре и затем нагревали до 60оС и перемешивали в течение 5 ч при этой температуре. Реакционную смесь переливали в объем измельченного льда и экстрагировали. Водный слой отделяли и снова промывали хлороформом. Хлороформовые слои объединяли и экстрагировали 2000 мл 5%-ного раствора гидроксида калия. Затем водный экстракт гидроксида калия добавляли к 1000 мл хлороформа. рН полученной в результате двухфазной смеси устанавливали на значении 2,0 с помощью концентрированной хлористоводородной кислоты. Слои полученной смеси разделяли и водный слой снова экстрагировали хлороформом. Органический слой двухфазной смеси и хлороформный экстракт объединяли, промывали водой и затем сушили над сульфатом натрия. Летучие компоненты раствора удаляли при пониженном давлении с образованием остатка. Этот остаток растворяли в 100 мл горячего толуола и полученный в результате раствор разбавляли 100 мл гексана. Этот раствор медленно охлаждали до комнатной температуры, что сопровождалось образованием осадка. Этот осадок выделяли фильтрацией, промывали гексаном и затем сушили в вакууме с получением 20,0 г желаемого замещенного промежуточного соединения.
В. Получение 5-[[3-(1,1-диметилэтил)-4-гидроксифенил] метилен] -3-амино-2-тиоксо-4-тиазолидинона.
Бензальдегидное промежуточное вещество из примера 29А (20,0 г; 112,2 ммоля), N-аминороданин (18,29 г; 123,4 ммоля) и ацетат натрия (36,8 г; 338,8 ммоля) суспендировали в 560 мл уксусной кислоты. Суспензию нагревали до температуры дефлегмации, перемешивали при этой температуре в течение 7 ч (при этом наблюдалось образование осадка) и затем охлаждали до комнатной температуры при перемешивании. Осадок выделяли фильтрацией и затем последовательно промывали раствором этил ацетата и диэтилового эфира в соотношении 1:1, затем диэтиловым эфиром. Выделенный осадок сушили в вакууме при 60оС в течение 2 ч с получением 14,5 г желаемого целевого интермедиата, т. пл. 225оС.
С. Получение 5-[[3-(1,1-диметилэтил)-4-гидроксифенил]-метилен]-4-тиазолидинона.
Промежуточное соединение примера 29В (14,3 г; 46,4 ммоля) суспендировали в 230 мл горячего (60оС) толуола. К такой суспензии добавляли три-н.-бутилового гидрид (62,4; 232 ммоля) и АИБН (1,14 г; 6,96 ммоля). Полученную в результате суспензию нагревали до температуры дефлегмации и при этом суспендированные твердые вещества медленно растворялись. Через 30 и 55 мин после начала нагревания добавляли дополнительное количество АИБН (две порции по 1,14 г). Через 80 мин после начала нагрева горячий реакционный раствор переносили в делительную воронку и добавляли 1N, раствор хлористоводородной кислоты. Полученную в результате двухфазную смесь разбавляли этилацетатом и разделяли слои. Водный слой промывали этил ацетатом и полученную промывную жидкость объединяли с органическим слоем из двухфазной смеси. Объединенный раствор промывали насыщенным раствором хлористого натрия и затем сушили над сульфатом натрия. Летучие компоненты раствора удаляли при пониженном давлении при получении 87,7 г желтого твердого вещества. Это твердое вещество суспендировали в 1000 мл гексана и полученную суспензию перемешивали в течение 15 мин. Через 15 мин суспензию отфильтровывали и выделенное твердое вещество растворяли в 500 мл диэтилового эфира. Эфирный раствор хроматографировали на колонке с силикагелем с использованием 8000 мл градиента 5-20% изопропилового спирта в гексане, затем 2000 мл градиента 20-30% изопропилового спирта в гексане и далее 2000 ил градиента 30-30% изопропилового спирта в гексане. Фракции, в которых было установлено наличие продукта, выпаривали и обрабатывали хлористым метиленом. Полученный в результате осадок растворяли в этил ацетате, высушивали досуха при пониженном давлении и затем обрабатывали этанолом с получением 4,31 г желаемого целевого промежуточного соединения, т. пл. 110оС (разложение).
Вычислено, С 63,85; Н 7,51; N 5,32.
С14Н17NO2S.
Найдено, С 64,15; Н 6,73; N 5,60.
Д. Получение 5-[[3-(1,1диметилэтил)-4-гидроксифенил]-метил]-4-тиазолидинона. Часть промежуточного вещества из примера 29С (395,1 мг; 1,5 ммоля) растворяли в 9 мл метанола. Затем к раствору добавляли магний (72,9 мг; 3,0 ммоля) и полученную в результате реакционную смесь в течение 3 ч перемешивали при комнатной температуре. Через 3 ч было установлено, что большая часть добавленного магния вошла в реакцию и добавляли еще 182,3 мг (7,5 ммоля) магния. Перемешивание реакционной смеси при комнатной температуре продолжали в течение ночи. На следующее утро наблюдалось образование желтого осадка. Этот осадок растворяли путем добавления метанольного реакционного раствора в смеси этил ацетата с 1N. хлористоводородной кислотой. Органический слой из полученной в результате двухфазной смеси выделяли и затем сушили над сульфатом натрия. Летучие компоненты органического слоя удаляли и полученный в результате остаток обрабатывали хлористым метиленом. Затем остаток растворяли в 25 мл хлористого метилена и полученный в результате раствор хроматографировали на колонке с силикагелем с использованием 5-20% градиента изопропилового спирта в гексане. Фракции, содержащие практический чистый продукт, выпаривали с использованием 0,29 г целевого соединения, т. пл. 65-70оС.
Вычислено, С 63,37; Н 7,22; N 5,28.
С14Н19NO2S.
Найдено, С 63,08; Н 7,30; N 4,39.
Изобретение обеспечивает способ лечения воспалительных заболеваний кишечника у млекопитающих. Такая активность продемонстрирована на следующих тестовых системах.
Крысам Sрrague-Dawley из лаборатории Чарльз Ривер, Портэжд, МI (группа из шести животных массой примерно 250 г) дважды в день орально вводили испытуемое соединение (10 мг/кг) или носитель (контроль) в течение трех дней. На третий день животным ставили внутрикишечную клизму с 2%-ным раствором уксусной кислоты, причем наконечник клизмы помещали на 8 см выше края анального отверстия. Такая концентрация с уксусной кислоты обеспечивает тяжелую воспалительную реакцию толстой кишки, характеризующуюся ректальным кровотечением, поносом, эрозией эпителиальной ткани и разрушением крипт и клеток желез. Через 24 ч испытуемых и контрольных животных умертвляли и дисталь толстой кишки длиной в 10 см удаляли и разрезали вдоль продольной оси. Нарушения тканей внутри удаленного открытого участка толстой кишки оценивалась тремя независимыми наблюдателями в масштабе оценок 0-4 (0 нормальное состояние; 4 наиболее сильное воспаление). В каждой группе использовали 5-7 крыс. Результаты таких испытаний представлены ниже.
Ингибирование колитов, вызванных действием уксусной кислоты.
Соединение примера Оценка поражений Контроль 3,4 ± 0,3 1 2,2 ± 0,5 2 1,1 ± 0,5 3 0,4 ± 0,1 4 1,5 ± 0,3 6 2,4 ± 0,5
7 2,1 ± 0,1 8 2,2 ± 0,5 10 1,2 ± 0,3
11 2,0 ± 0,6 12 2,0 ± 0,6 16 1,2 ± 0,5 18 2,8 ± 0,6 21 1,5 ± 0,5 22 0,8 ± 0,2 23 2,7 ± 0,6 24 1,0 ± 0,2 25 2,5 ± 0,7 26 2,4 ± 0,6 27 2,2± 0,5 29 2,2 ± 0,5
Крысам, разновидности Sрrague-Dawley из Чарльз Ривер Лабораториз Портайдж, МI (самцы весом примерно 300 г), не давали пищи в течение 24 ч. Через 24 ч животным орально вводили по 3 мл/кг массы крысы растворителя (контрольное вещество) или испытуемого соединения, растворенного в таком растворителе. Через 30 мин каждому животному давали раствор, состоящий из 100% этанола. Через 60 мин после применения всех животных умертвляли и их желудки удаляли и промывали. Повреждение тканей внутри удаленного, открытого желудка, оценивались тремя независимыми, действующими вслепую экспериментами в шкале оценок 0-5 (где 0 нормальное состояние; 5 серьезное повреждение тканей). В каждой испытательной группе использовали по шесть крыс. Результаты испытания на животных, принявших испытуемое соединение, растворенное в растворителе, сравнивали с результатами, полученными на животных, принявших только растворитель с целью определения ингибирования повреждения тканей в процентах, которое может быть приписано действию испытуемого соединения. Результаты каждого испытания представлены в табл.
Приведенные данные показывают, что соединения, используемые в способе по изобретению, способы лечить воспалительные заболевания пищеварительного тракта. Термин "воспалительное заболевание желудка", используемый в тексте изобретения, относится к любому расстройству пищеварительной системы, которое характеризуется воспалением. Примеры таких расстройств включают заболевание Крона, слизистые колиты, псевдомембранные энтероколиты, неспецифические язвы толстой кишки, коллагенные колиты, расслабление толстой кишки, язвенный простит, эволюционный энтерит и колит, идиофатический диффузионный язвенный негрануломатозный энтерит, вызванные нестероидным противовоспалительным лекарством воспаления, клеточная спру и т. п.
Соединения формулы II также применяются при лечении воспалительных заболеваний кишечника. В связи с этим настоящее изобретение также относится к фармацевтическим композициям, которые включают по крайней мере одно соединение формулы II совместно с одним из более фармацевтически применимыми разбавителями, эксипиентами или носителями.
При приготовлении фармацевтических композиций изобретения одно или более соединений формулы II смешивают с носителем или разбавляют носителем либо капсулируют в носитель, который может иметь форму капсулы, саше, бумаги или другого резервуара. В том случае, когда носитель выполняет функции разбавителя, он может представлять собой твердый, полутвердый или жидкий материал, который выполняет функции связующего вещества, эксипиента или среды для активного ингредиента. Так например, композиции могут иметь форму таблеток, пилюль, порошков, лепешек, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как в твердой, так и в жидкой среде), мазей содержащих например, до 10 мас. активного соединения, мягких и твердых желатиновых капсул, свечей, стерильных растворов для инъекций и стерильно упакованных порошков.
Примерами подходящих носителей, эксипиентов и разбавителей могут служить лактоза, декстроза, сахароза, сорбит, маннит, крахмал, аравийская камедь, фосфат кальция, альгинаты, трагакант, желатина, силикат кальция, микрокристаллическая целлюлоза, поливинил-пирролидон целлюлоза, вода, сироп, метилцеллюлоза, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. Такие рецептуры могут дополнительно включать смазочные агенты, смачивающие агенты, эмульгирующие и суспендирующие агенты, предохраняющие агенты, подслащивающие агенты или отдушки. Композиции по изобретению могут формироваться таким образом, чтобы обеспечивалось быстрое, длительное или замедленное выделение активного ингредиента после применения на пациенте с использованием методик, хорошо известных в данной области.
Композиции формируют, предпочтительно, в единичной дозировочной форме, таким образом, чтобы каждая дозировка содержала 5-500 мг, обычно 25-300 мг активного ингредиента. Термин "единичная дозировочная форма" относится к физически дискретным единицам, предназначенным для единичной дозировки при применении на людях и других млекопитающих, причем каждая единица содержит определенное количество активного материала, рассчитанное таким образом, чтобы обеспечить желаемый терапевтический эффект, совместно с одним или более подходящими фармацевтическими разбавителями, эксипиентами или носителями.
В следующих рецептурных примерах в качестве активных ингредиентов могут использоваться любые соединения формулы II.
П р и м е р 30. Жесткие желатиновые капсулы готовили с использованием следующих ингредиентов, мг/капсулу: Соединение примера 11 250 Сухой крахмал 200 Стеарат магния 10
Указанные ингредиенты смешивали друг с другом и заполняли ими жесткие желатиновые капсулы в количестве 460 мг на капсулу.
П р и м е р 31. Таблетированную форму готовили с использованием следующих ингредиентов, мг/таблетку: Соединение примера 11 250 Микрокристаллическая целлю- лоза 400 Плавленная двуокись кремния 10 Стеариновая кислота 5
Указанные компоненты смешивали друг с другом и прессовали в таблетки весом 665 мг каждая.
П р и м е р 32. Аэрозольный раствор готовили из следующих компонентов, мас. Соединение примера 12 0,25 Этанол 29,75 Пропеллант 22 (хлордифтор- метан) 70,00
Активное соединение смешивали с этанолом и смесь добавляли к части пропелланта 22, охлажденного до -30оС, и переносили в заполняющее устройство. Затем требуемое количество подавали в контейнер из нержавеющей стали и разбавляли оставшимся количеством пропелланта. Затем на контейнер устанавливали клапанное устройство.
П р и м е р 33. Таблетки, каждая из которых содержала 60 мг активного ингредиента, готовили следующим образом, мг: Соединение примера 12 60 Крахмал 45 Микрокристаллическая целлюлоза 35 Поливинилпирролидон (в виде 10% -ного раствора в воде) 4 Натрий карбоксиметил крахмал 4,5 Стеарат магния 0,5 Тальк 1 Всего 150 мг
Активный ингредиент, крахмал и целлюлозу пропускали через сито N 45 меш США и тщательно перемешивали. Раствор поливинилпирролидона смешивали с полученным в результате порошком, который затем пропускали через сито N 14 меш США. Полученные таким образом гранулы сушили при 50-60оС и пропускали через сито N 18 меш США. Натрийкарбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш США, затем добавляли к гранулам, которые после смешивания прессовали с помощью таблетирующей машины с получением таблеток, каждая из которых весила 150 мг.
П р и м е р 34. Капсулы, содержащие по 80 мг медикамента, получали следующим образом, мг: Соединение примера 12 80 Крахмал 59 Микрокристаллическая целлю- лоза 59 Стеарат магния 2 Всего 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивали друг с другом, пропуская через сито N 45 меш США и заполняли смесью желатиновые капсулы в количестве 200 мг на капсулу.
П р и м е р 35. Свечи, содержащие по 225 мг активного ингредиента готовили следующим образом, мг: Соединение примера 13 225 Глицериды насыщенных жирных кислот До 2000
Активный ингредиент пропускали через сито N 60 меш США и суспендировали в глицеридах насыщенных кислот, которые предварительно расплавляли с подводом минимального количества тепла. Затем смесь переливали в свечевую форму с номинальным объемом 2 г и давали содержимому охлаждаться.
П р и м е р 36. Суспензии, содержащие по 50 мг медикамента в 5 мл дозировке, получали следующим образом, мг: Соединение примера 11 50 Натрий карбоксиметилцеллюлоза 50 Сироп 1,25 мл Раствор бензойной кислоты 0,10 мл Отдушка q.Y. Цветовой агент q.Y. Очищенная вода До 5 мл
Медикамент пропускали через сито N 45 меш США и смешивали с натрийкарбоксиметилцеллюлозой и сиропом с получением однородной пасты. Раствор бензойной кислоты, отдушку и цветовой агент разбавляли некоторым количеством воды и при перемешивании добавляли в систему. Затем добавляли воду в количестве, достаточном для достижения требуемого объема.
П р и м е р 37. Капсулы, содержащие по 150 мг медикамента, получали следующим образом, мг: Соединение примера 13 150 Крахмал 164 Микрокристаллическая целлю лоза 164 Стеарат магния 22 Всего 500 мг
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивали друг с другом, пропускали через сито N 45 меш США и заполняли полученной смесью твердые желатиновые капсулы по 500 мг на капсулу.
П р и м е р 38. Жесткие желатиновые капсулы готовили с использованием следующих ингредиентов, мг/капсулу: Соединение примера 3 250 Сухой крахмал 200 Стеарат магния 10
Указанные ингредиенты смешивали друг с другом и заполняли смесью твердые желатиновые капсулы в количестве 460 мг на капсулу.
П р и м е р 39. Таблетки, содержащие по 60 мг активного ингредиента готовили следующим способом, мг: Соединение примера 3 60 Крахмал 45 Микрокристаллическая целлюлоза 35 Поливинилпирролидон (в виде 10%-ного раствора в воде) 4 Натрий карбоксиметил крахмал 4,5 Стеарат магния 0,5 Тальк 1 Всего 150
Активный ингредиент, крахмал и целлюлозу пропускали через сито N 45 меш США и тщательно перемешивали. Раствор поливинилпиpролидона смешивали с полученными в результате порошками, которые далее пропускали через сито N 14 меш США. Полученные таким образом гранулы сушили при 50-60оС и пропускали через сито N 18 меш США. Натрийкарбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш США, затем добавляли к гранулам, которые после перемешивания прессовали в таблетирующей машине с получением таблеток, каждая из которых весила 150 мг.
П р и м е р 40. Свечи, содержащие по 225 мг активного ингредиента, готовили следующим образом, мг: Соединение примера 3 225 Глицериды насыщенных жирных кислот 2000
Активный ингредиент пропускали через сито N 60 меш США и суспендировали в глицеридах насыщенных жирных кислот, предварительно расплавленных с подводом минимального количества тепла. Затем смесь переливали в свечную форму с номинальным количеством 2 г и давали в смеси охлаждаться.
Ниже сравнивается процент ингибирования колита, вызванного уксусной кислотой, в отношении контрольного образца, для соединения 13 по изобретению (5-[[3,5-ди-2-пропенил-4-гидроксифенил] метилен] -3-метил-4-тиазолиденом с 5-[[3,5-бис(1,1-диметилэтил)-4-гидроксифенил]метилен]-3-метил-4-тиазолидинон ом. (Известное соединение).
Сравнение ингибирования колита, вызванного уксусной кислотой
Соединение по примеру 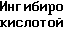 в
в и
и
 ы
ы у
у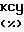 снойной 13 37,4
снойной 13 37,4
Известное 22
Как ясно видно соединения по изобретению значительно более активны, чем соединения,описанные в ЕP 211670.
Использование: в медицине в качестве противовоспалительных средств. Сущность изобретения: продукт ф-лы 1, где R′- C2-C6- алкенил, C2-C6- алкинил или группу ф-лы 2, где n - целое число 0 - 3, включая оба крайних значения; R2- Н, C1-C6- алкил, C1-C6- алкокси, C2-C6- алкенил, C2-C6 -алкил - группа ф-лы 3 или 4, где n указано выше. R3- Н или C1-C6- алкил; R4 и R5- Н или совместно образуют связь; R6 и R7 каждый -Н или совместно образуют группу S, или один из радикалов: R6 и R7- Н, а другой - группы ОН или -SCH; Х - группа ф-лы 5, m - 0,1 или 2; а Q NR8, где R8 -Н, C1-C6 -алкил, C2-C6 алкенил, C3-C8 -циклоалкил, -SO2CH3 или -(CH2)n-Y, где n указано выше, а Y - циано, OR9 -C(O)R10 -NR11R12 C1-C4 -алкил или группу ф-лы 6, где R9 Н или C1-C4- алкил, или группу -С(О) C1-C4- алкил R10- C1-C4- алкил, C1-C4- алкокси, или -NH2 R11 и R12 независимо друг от друга - Н, C2-C6- алкенил; C2-C6- алкинил ,C1-C6- алкил, -(CH2)qOH -(CH2)q-N(C1-C4алкил)2/ -(CH2)q-S(C1-C4) или группа ф-лы 7, где указано выше, а целое число 1 - 6, включая оба крайних значения: либо R11 и R12 совместно образуют морфолиновое, пиперидильное, пиперазинильное или N-метил-пиперазинильное кольцо; или его фармацевтически приемлемой соли. Реагент 1: соединение ф-лы 8. Реагент 2: соединение ф-лы 9. Соединения ф-лы 1 - 9 соответственно. Ф-ла 1 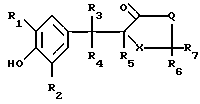 ф-ла 2:
ф-ла 2: 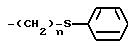 ф-ла 3:
ф-ла 3: 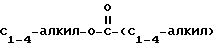 ф-ла 4:
ф-ла 4: 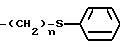 ф-ла 5:
ф-ла 5: 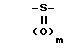 ф-ла 6:
ф-ла 6: 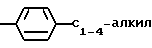 ф-ла 7:
ф-ла 7: 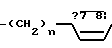 ф-ла 8:
ф-ла 8: 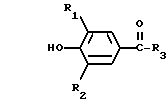 ф-ла 9:
ф-ла 9:  1 табл.
1 табл.
Способ получения производных тиазолидинона общей формулы I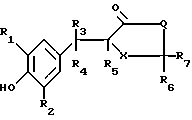
где R1 С2-С6-алкенил, С2-С6-алкинил или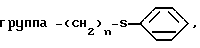
где n 0 3;
R2 водород, С1-С6-алкил, С1-С6-алкокси, С2-С6-алкенил, С2-С6-алкинил, С1-С4-алкил О -,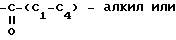
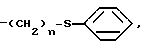
где n 0 3,
R3 водород или С1-С6-алкил;
R4 и R5 водород или совместно образуют связь;
R6 и R7 каждый водород или совместно образуют группуS, или один из радикалов;
R6 и R7 водород, а другой группа ОН или S CH3;
где m 0 или 1;
Q NR8,
где R8 водород, С1-С6-алкил, С2-С6-алкенил, С3-С8-циклоалкил, SO2CH3 или -(CH2)n Y,
где n 0 3,
Y- циано, OR9, 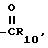 -NR11R12, -S(С1-С4)-алкил или группа
-NR11R12, -S(С1-С4)-алкил или группа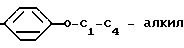
где R9 водород, С1-С4-алкил или группа 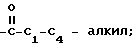
R10 С1-С4-алкил, С1-С4-алкокси или -NH2;
R11 и R12 независимо друг от друга водород, С1-С6-алкил, С2-С6-алкенил, С2-С6-алкинил, (CH2)qOH,
-(CH2)q N(С1-С4-алкил)2 - (CH2)q S-(С1-С4-алкил)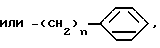
где n имеет указанные значения;
q 1 6,
либо его фармацевтически приемлемой соли, отличающийся тем, что осуществляют реакцию соединения общей формулы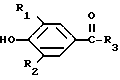
с соединением общей формулы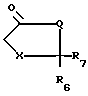
где R1, R2, R3 и Х имеют указанные значения;
Q N R8, где R8 имеет указанные значения;
R6 и R7 совместно образуют группуS,
с целью получения соединения общей формулы II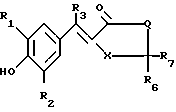
где R1, R2, R3, R6, R7, Х и Q имеют указанные значения;
(а) с последующим необязательным восстановлением соединения формулы II, где R6 и R7 совместно образуют группу S с тем, чтобы получить соединение формулы II, где R6 и R7 водород,
(b) восстановлением соединения формулы II, где R4 и R5 совместно образуют связь, с целью получения соединения формулы II, где R4 и R5 водород,
(c) восстановлением соединения формулы II, где R4 и R5 совместно образуют связь, а R6 и R7 совместно образуют группуS, с целью получения соединения формулы II, где R4, R5, R6 и R7 водород;
(d) алкилированием соединения формулы II, где R8 водород, с получением соединений формулы II, где R8 С1-С6-алкил, С2-С6-алкенил, С3-С8-циклоалкил или -(CH2)n Y, где n 0 3, а Y циано, OR9, - S-(С1-С4)-алкил, -NR11R12 или 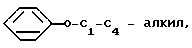 где R9, R11 и R12 имеют указанные значения,
где R9, R11 и R12 имеют указанные значения,
(e) ацилированием соединения формулы II, где R8 водород, с целью получения соединения формулы II, где R8 (CH2)n Y, где n 0 3, 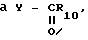 где R10 имеет указанные значения,
где R10 имеет указанные значения,
(f) окислением соединения формулы II, где Х группа 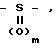 где m 0, с целью получения соединения формулы II, где Х группа
где m 0, с целью получения соединения формулы II, где Х группа 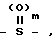 а m 1,
а m 1,
(k) восстановлением соединения формулы II, где R8- группа - (CH2)n Y, где n 0 3, а Y OR9, где R9- группа 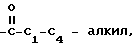
с целью получения соединения формулы II, где R8- группа - (CH2)n Y, где n 0 3, а Y OR9, где R9 - водород;
(l) осуществлением реакции соединения формулы II, где R8 группа -(CH2)n Y, где n 0 3, а Y -OR9, где R9 - водород, с тозилгалогенидом с целью получения соединения формулы II, где R8 -(CH2)n Y, где n 0 3, а Y OR9, где R9 тозил;
(m) осуществлением реакции соединения формулы II, где R8 - (CH2)n Y, где n 0 3, а Y -OR9, где R9 тозил, с амином формулы -NR11R12, где R11 и R12 имеют указанные значения, с целью получения соединения формулы II, где R8 группа -(CH2)n Y, где n 0 3, а Y группа N R11 R12;
(n) нагреванием соединения формулы II, где R8 -(CH2)n Y, а Y -N R11 R12, причем R11 и R12 не являются водородом, в смеси этанол:вода в присутствии катализатора с целью получения соединения формулы II, где R8 - -(CH2)n Y, а Y -N R11 R12, где один из радикалов R11 или R12 водород, а другой не водород;
(o) реакцией соединения формулы II, где R6 и R7 водород, с трифторуксусным ангидридом с получением соединения формулы II, где один из радикалов R6 и R7 водород, а другой группу -ОН;
(p) образованием соли соединения формулы II по реакции несолевой нормы соединения с сильной кислотой или сильным основанием.
| УСТРОЙСТВО для НАМОТКИ СЕКЦИЙ КОНДЕНСАТОРОВ | 0 |
|
SU211670A1 |
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
