Изобретение касается цефалоспориновых антибиотиков, фармацевтических композиций, содержащих эти антибиотики, и способа лечения инфекционных заболеваний у человека и животных.
Цефалоспориновые антибиотики имеют бициклическую кольцевую систему, представленную следующей формулой, в которой применена нумерационная система, обычно применяемая в условной терминологической системе цефама: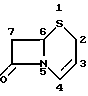
В области антимикробной терапии всегда необходимы новые химиотерапевтические средства. Мутантные штаммы, устойчивые к существующим противомикробным средствам, часто появляются неожиданно. В частности, многие штаммы Staph. aureus и Staph. epi (так называемые устойчивые против метициллина штаммы Staph. ) становятся значительно устойчивее к доступным противомикробным средствам (см. , например, Phillips, J. and Cookson, B. J. Appl. Bacteriology. 61 (6) 1989).
Чтобы удовлетворить эту потребность, продолжаются сследования, направленные на разработку таких новых средств. Изобретение предусматривает новые антимикробные средства против широкого спектра грамположительных и грамотрицательных микроорганизмов. Соединения, согласно изобретению, особенно полезны против этих устойчивых к метициллину штаммов стафилококков.
Изобретение предусматривает различные 3-тиазоло-тиоцефалоспорины, полезные в качестве антибактериальных средств. В частности, изобретение предусматривает 7β-(2-аминотиазол-4-ил)-оксимино-(или алкоксимино)-ацетиламино-3-необязательно замещенные триазолотио-3-цефем-4-карбоновые кислоты, полезные в качестве антибактериальных средств.
Изобретение предусматривает также фармацевтические композиции и терапевтические методы, полезные при лечении противомикробных инфекций у человека и животных.
Изобретение предусматривает соединения формулы (1):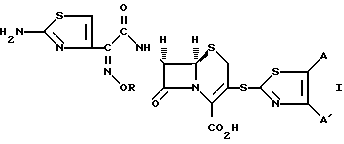
в которых R представляет собой атом водорода, C1-C6- алкил, C2-C6-алкенил, C2-C6-алкинил, C3-C6-циклоалкил или C1-C6-галоалкил и A и A' независимо друг от друга являются атомами водорода, C1-C6-алкилом, нитро, амино, C1-C6-алкокси, 5- или 6-членным гетероциклом, содержащим азот или серу, или фенилом; или A и A', вместе взятые, образуют группу формул: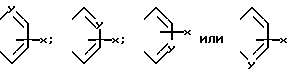 ,
,
в которых X представляет собой атом водорода, галоид, C1- C6-алкил, C1-C6-алкокси, C1-C6- алкоксикарбонил, амино, нитро или карбокси и Y является атомом азота или углерода, или их фармацевтически приемлемые соли.
Термин "фармацевтически приемлемая соль" включает те соли, которые образованы с карбоксилатными анионами, и включает соли, образованные органическими и неорганическими катионами, такими, как противоионы, выбранные из щелочных и щелочноземельных металлов (таких, как литий, натрий, калий, барий и кальций), соли аммония, и образованные органическими катионами (такими, как дибензил-аммоний, бензиламмоний, 2-гидроксиэтиламмоний, бис-(2-гидрокси-этил)-аммоний, фенилэтилбензиламмоний, дибензилэтилендиаммоний и подобными катионами). Другие катионы, подпадающие под определение названного выше термина, включают протонированные формы прокаина, хинина и N-метилглюкозамина и протонированные формы основных аминокислот, таких, как глицин, орнитин, гистидин, фенилглицин, лизин и аргинин. Кроме того, любая цветтерионная форма соединений, представленных формулой (1), образованных карбоновой кислотой и аминогруппой, относится к этому термину. Предпочтительным катионом для карбоксилатного аниона является катион натрия. Далее термин включает соли, которые образованы в стандартных кислотно-основных реакциях с основными группами (такими, как аминогруппы) и органическими или неорганическими кислотами. Такие кислоты включают хлористоводородную, серную, фосфорную, уксусную, янтарную, лимонную, молочную, малеиновую, фумаровую, пальмитиновую, холевую, памоиновую, муциновую, D-глутаминовую, d-камфорную, глутаровую, фталевую, винную, лауриновую, стеариновую, салициловую, метансульфоновую, бензолсульфоновую, сорбиновую, пикриновую, бензойную, коричную и тому подобные кислоты.
В формуле (1) термин "C1-C6-алкил" означает такие радикалы, как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, амил, трет-амил, гексил и тому подобные. Предпочтительной C1-C6-алкильной группой является метил.
Термин "C2-C6-алкенил" означает низший алкенил с прямой или разветвленной цепью, например винил, аллил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил, металлил или 1,1-диметилаллил.
Термин "C2-C6-алкинил" представляет низшую алкинильную группу с прямой или разветвленной цепью, например, этинил, 1-пропинил или пропаргил.
Термин "C3-C10-циклоалкил" включает, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил или адамантаил.
Термин "C1-C6-галоалкил" означает упомянутые выше C1-C6-алкильные группы, которые замещены одним галогеном, при этом "галоид" или "галоген" означает атом хлора, брома, иода или фтора. Предпочтителен фторзамещенный C1-C6-галоидалкил.
Термин "C1-C6-алкокси" относится к таким группам, как метокси, этокси, 3-пропокси, бутокси и тому подобным.
Термин "галоид" включает атомы фтора, брома, хлора и иода.
Термин "C1-C6-алкоксикарбонил" относится к таким группам, как метоксикарбонил, этоксикарбонил, 3-пропоксикарбонил, 3-этоксикарбонил, 4-трет-бутилоксикарбонил, 3-метоксикарбонил, 6-метоксикарбонил и тому подобным.
Термин 5- или 6-членный гетероцикл, содержащий азот или серу", относится к пиридину и тиофену и может включать более, чем азот или серу и их сочетания. Другие примеры включают группы, описанные Fletcher, Dermer and Otis, Nomenclature of Org. Compounds стр. 49-64 (1974).
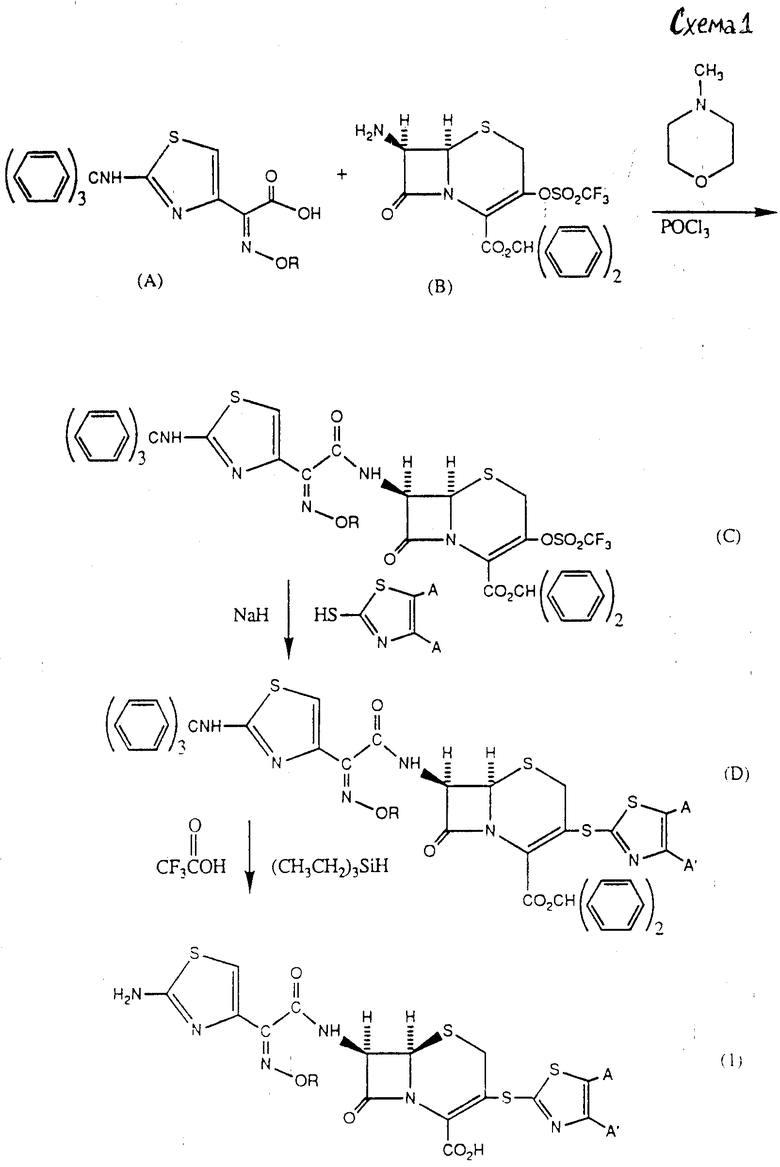
Соединения формулы (I) могут быть получены согласно схеме реакций 1.
Исходное соединение (A), (в котором R представляет собой метил), 2-(тритил)-амино-α-(метокcимино)-4-триазолуксусная кислота, может быть получено из соответствующего свободного амина (поставляемого Aldrich Chemical Co. , Jnc., 940 West Saint Paul Avenue. Milwaukee, Wisconsin 53233), используя методы, широко известные в области получения β-лактама. Исходное соединение (B), или бензгидриловый эфир 7-амино-3-трифтор-метансульфонокси-3-цефем-4-карбоновой кислоты, может быть получено с использованием известной методологии из соответствующего 3-енол-3-цефема и трифторметансульфоноксиуксусного ангидрида (Syn. Commun., 20 (14), 2185-2189 (1990).
На схеме 1 хлорангидрид соединения (A) может быть получен известным методом, например взаимодействием с хлористым фосфорилом, и может реагировать со свободным амином (B) с образованием 7-ацил-3-трифлата (C). Тиазолотиогруппа может быть затем введена взаимодействием трифлата (C) с соединением формулы: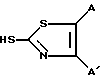
в присутствии основания, такого, как гидрид натрия. Конечный продукт формулы (1) может быть получен затем удалением амино- и карбоксизащитных групп. В схеме I трифторуксусная кислота и (CH3CH2)3SiH использованы для удаления тритильной и бензгидрильной групп. Специалисту в области химии β-лактамов будет понятно, что эффективными могут быть и другие защитные группы. Далее, можно также ввести тиазолотиофункцию в 3-е положение ядра цефема (B) до введения 7-ацильных функций, чтобы получить полезные промежуточные соединения, представленные в формуле (2).
Альтернативно соединения могут быть получены по схеме реакций 2.
На схеме 2 уксусная кислота (A), растворенная в диметилформамиде, обработана N-метилморфолином и хлористым оксалилом. Смесь 7β-амино-3-хлорцефема, растворенную в диметилформамиде и обработанную бис-(диметилсилил)-мочевиной (BSU) и пиридином, смешивали с уксусной кислотой, чтобы получить соединение (C). Затем соединение (C) обрабатывали дифенилдиазометаном и тиазолотиогруппу вводили в присутствии основания, такого, как гидрид натрия, с образованием соединения (D). Удаление бензгидрильных и тритильных групп может быть осуществлено, как описано на схеме 1.
Соединения формулы: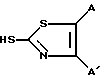 ,
,
где A и A', вместе взятые, образуют группу формул: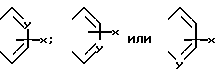
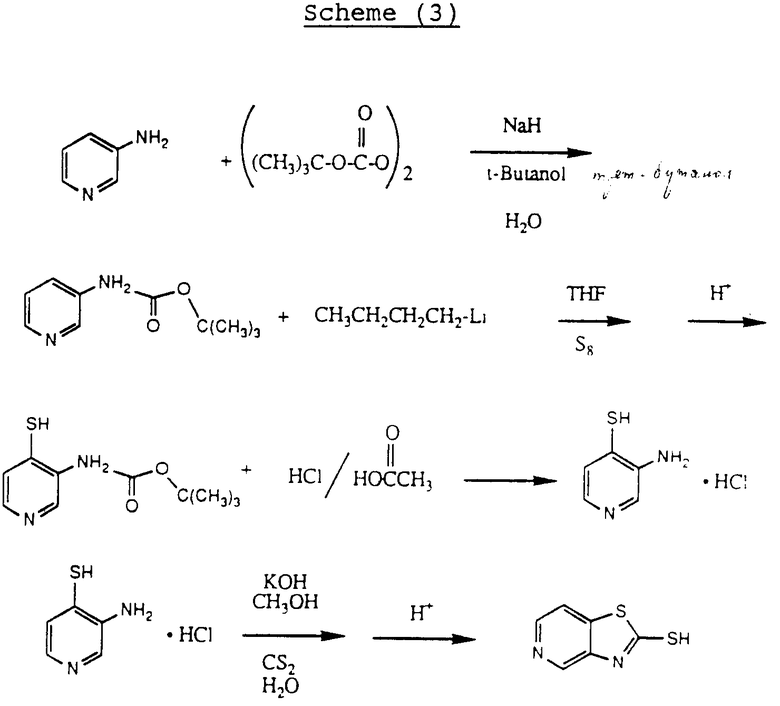
и Y является атомом азота, могут быть получены по схеме реакций 3.
На схеме 3 3-аминопиридин ацилируют ди-трет-бутилдикарбонатом, чтобы ввести трет-бутоксикарбонильную защитную группу. (Следует иметь в виду, что два других пиридинотиазолотиомеркаптана могут быть получены известными методами, используя изомеры аминопиридина. ) Трет-бутоксикарбонильный защищенный 3-аминопиридин обрабатывают затем н-бутиллитием в тетрагидрофуране, затем элементарной серой (S8), с последующей обработкой насыщенным хлористым аммонием. Полученный 3-трет-бутоксикарбониламино-4-тиапиридин обрабатывали смесью уксусной кислоты и хлористоводородной кислоты с получением гидрохлорида 3-амино-4-меркаптопиридина. Желаемый 5-пиридино-тиазолотиомеркаптен затем может быть получен обработкой этого соединения сероуглеродом в щелочной среде.
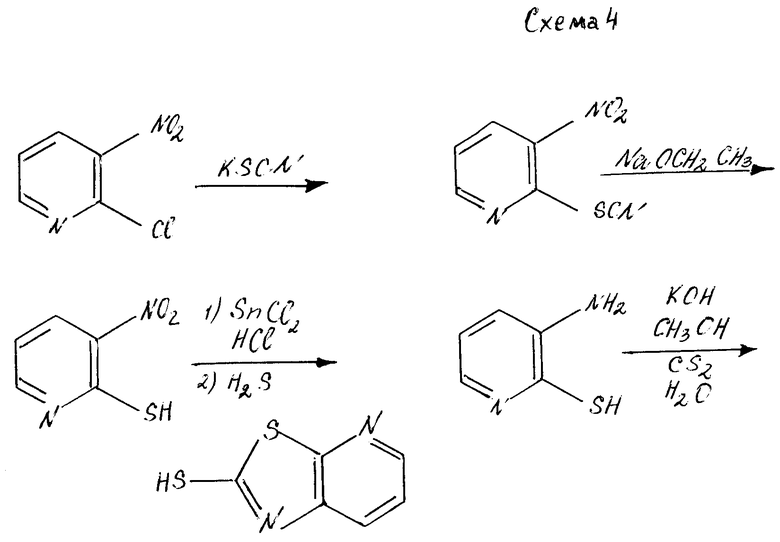
Если A и A', вместе взятые, образуют группу формулы: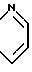 ,
,
то желаемый тиол формулы: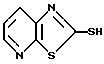
может быть получен, как показано на схеме 4.
На схеме 4 2-хлор-3-нитропиридин обрабатывают изотиоцианатом калия, чтобы получить 2-изоцианато-3-нитропиридин, который в свою очередь гидролизуют до получения 2-меркапто-3-нитро-пиридина. Промежуточное 3-нитросоединение затем восстанавливают обработкой смесью хлористого олова и хлористоводородной кислоты с получением 2-меркапто-3-аминопиридина. Желаемый пиридинотиазоло- тиомеркаптан затем получают щелочной каталитической конденсацией с сероуглеродом (KOH-CH3OH-CS2-H2O).
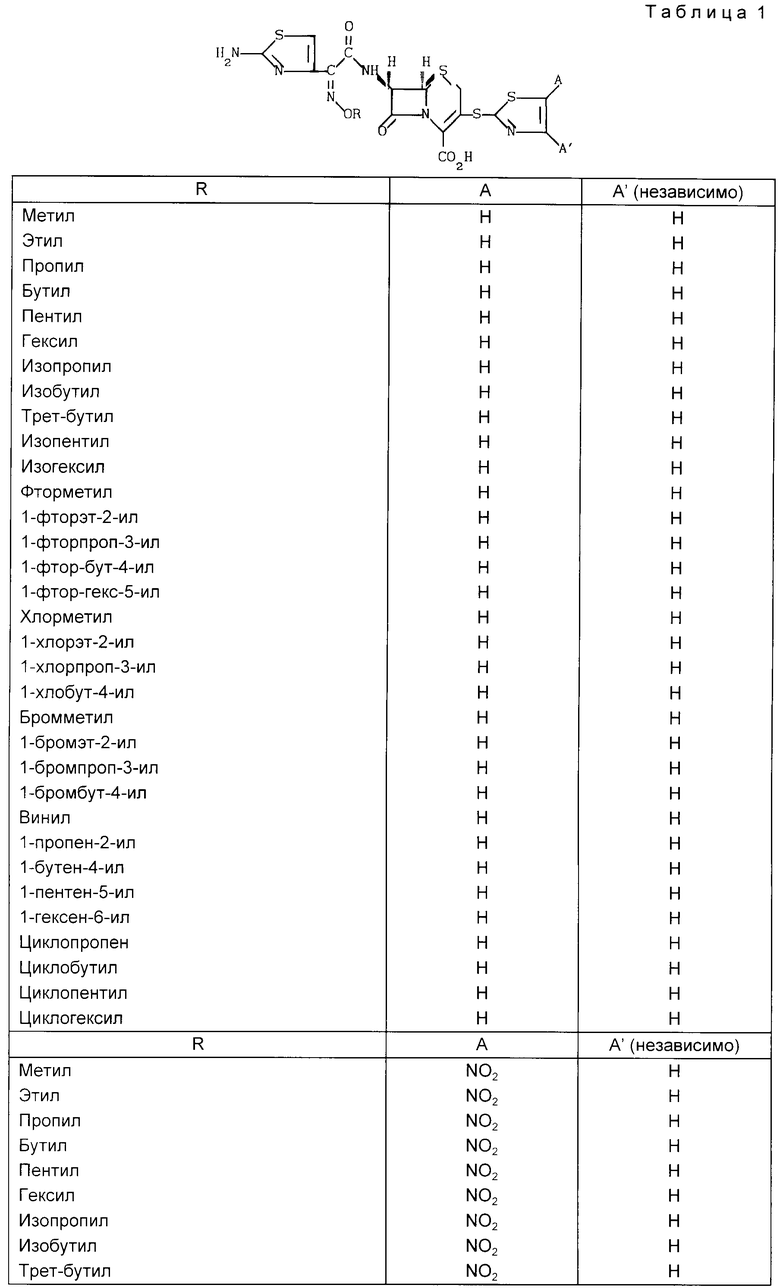
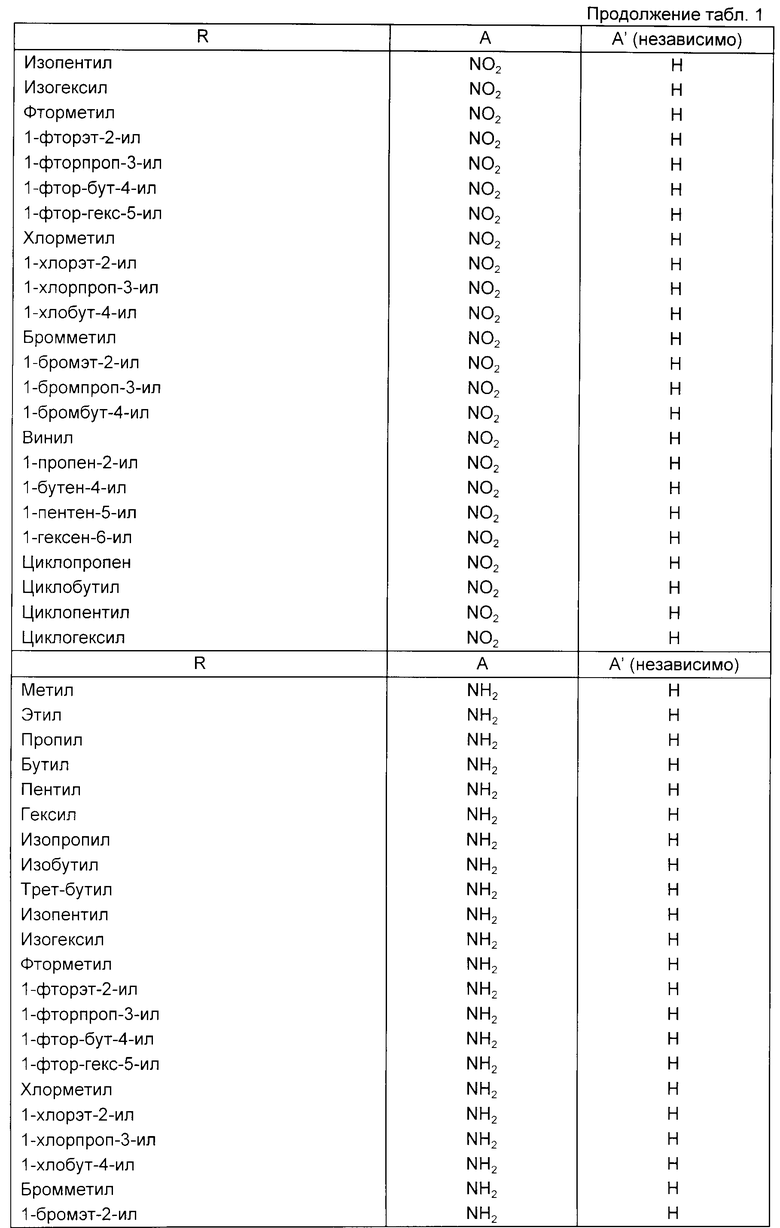
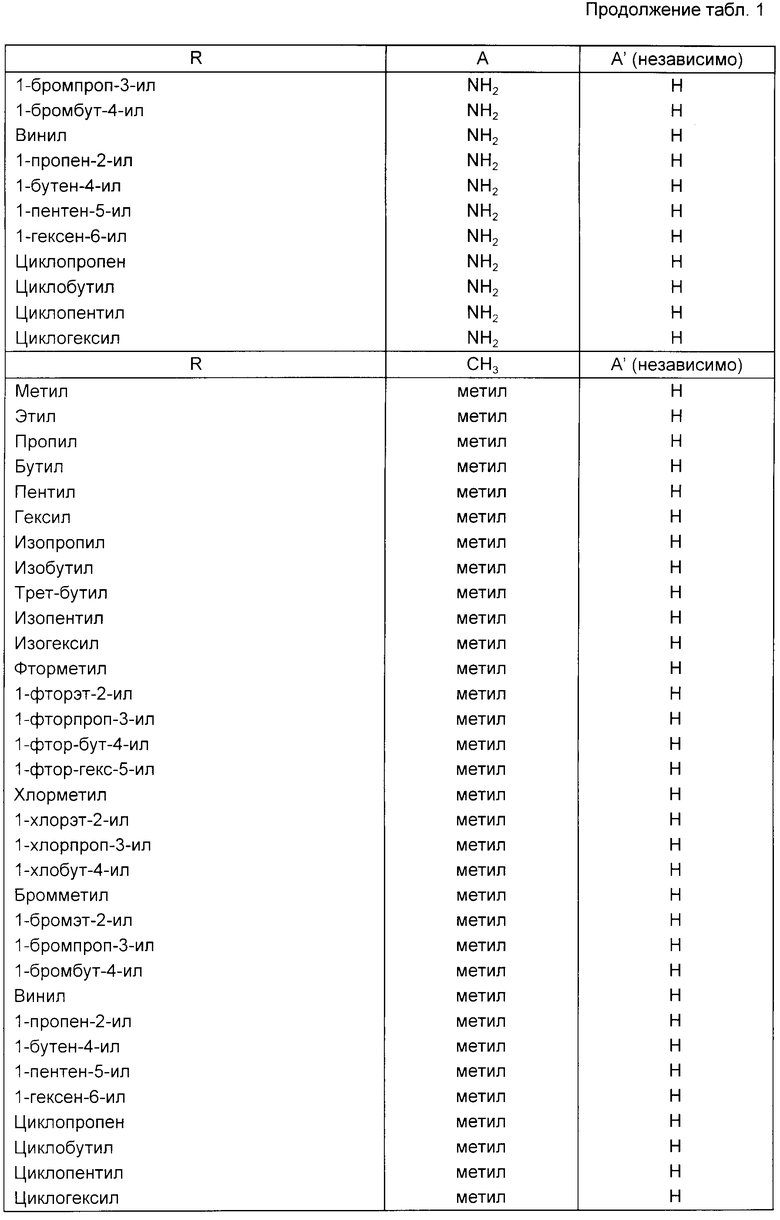
Примеры соединений, подпадающих под действие формулы (1) представлены в таблице.
В формуле (1) R предпочтительно представляет собой C1-C6-алкил или C1-C6-галоидный алкил. Предпочтительно C1-C6-алкильной группой является метил. Предпочтительной C1-C6-галоалкильной группой является фтор-C1-C6-алкил. Дальнейшей предпочтительной C1-C6-фторалкильной группой является 2-фторэт-1-ильная группа.
В формуле (1) предпочтительно что A и A', вместе взятые, образуют группу формул: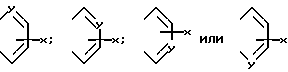
Далее предпочтительно, что Y представляет собой атом азота и A и A', взятые вместе, образуют группу формулы: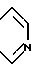
например, образуя соединение формулы: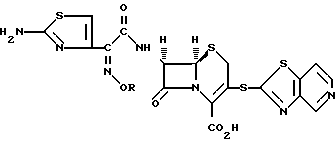
или его фармацевтически приемлемую соль. Два других предпочтительных соединения приведенной выше формулы являются соединениями, в которых R является метилом или 2-фторэт-1-илом.
Изобретение предусматривает также способ лечения инфекционных заболеваний у человека и животных и фармацевтические композиции, пригодные для введения этим методом лечения. Терапевтический метод согласно изобретению включает введение человеку или животному антибиотически эффективную нетоксичную дозу соединения, представленного формулой (1) или его фармацевтически приемлемой соли.
Антибиотически эффективным количеством является количество в пределах от 25 мг до 2 г. Соединение, соль или сложный эфир могут быть введены в однократной дозе или множественными дробными дозами в течение дня. Лечение может продолжаться от недели до 10 дней или больше в зависимости от тяжести инфекции. Конкретная доза и схема приема лекарственного средства могут зависеть от таких факторов, как вес и возраст больного, конкретный возбудитель инфекции, тяжесть инфекции, общее состояние здоровья пациента и индивидуальная терпимость к антибиотику.
Цефалоспорин может быть введен парентерально, подкожно или ректально. Как и с другими β-лактамовыми антибиотиками, способ лечения согласно изобретению может быть использован профилактически для предотвращения инфекции после контактирования или до возможного заражения, например предоперационно. Антибиотик может быть введен традиционными методами, например шприцем или капельницей для внутривенного вливания.
Фармацевтически приемлемые соли, как отмечалось ранее, могут быть полезными формами антибиотиков для получения препаратов антибиотиков.
Фармацевтические композиции согласно изобретению содержат антибиотически эффективное нетоксичное количество соединения, представленного формулой (1) или его фармацевтически приемлемой нетоксичной соли и фармацевтически приемлемого носителя.
Парентеральные композиции антибактериального средства для инъекции готовят с водой для инъекции, раствором Рингера, физиологическим раствором или раствором глюкозы. Антибиотик может также вводиться во внутривенную жидкость капельным методом.
Для парентерального применения противомикробного средства формулы (1) или его фармацевтически приемлемой соли оно может быть предпочтительно приготовлено в форме сухого кристаллического порошка или в виде лиофилизированного порошка и помещено в ампулы. Такие ампулы могут содержать от 100 мг до 2 г антибиотика на ампулу.
В качестве дальнейшего аспекта изобретение предусматривает новые промежуточные соединения формулы (2)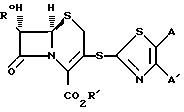 ,
,
в которой R0 является амино- или замещенной аминогруппой, R1 является водородом или карбоксизащитной группой и A и A' независимо друг от друга представляют собой водород, C1-C6-алкил, фенил, нитро, амино, 5- или 6-членный гетероцикл, содержащий азот или серу; или C1-C6-алкокси, или A и A', вместе взятые, образуют группу формул: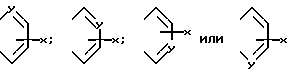
в которых X представляет собой водород, C1-C6-алкил, C1-C6-алкокси, C1-C6-алкоксикарбонил амино, нитро, или карбокси и Y является азотом или углеродом.
В формуле (2) термин "карбоксизащитная группа" относится к группе производных сложного эфира группы карбоновой кислоты, обычно применяемой для блокирования или защиты группы карбоновой кислоты в то время, когда реакции проводят на других функциональных группах соединения. Примеры таких защитных групп карбоновой кислоты включают 4-нитробензил, 4-метоксибензил, 3,4-диметоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, 3,4-метилендиоксибензил, бензгидрил, 4,4'-диметокcибензгидрил, 2,2', 4,4'-тетраметоксибензгидрил, трет-бутил, трет-амил, тритил, 4-метокситритил, 4,4'-диметокситритил, 4,4',4"-триметокситритил, 2-фенилпроп-2-ил, триметилсилил, трет-бутилдиметилсилил, фенацил, 2,2,2-трихлорэтил, β-(триметилсилил)-этил, β-/ди-(н-бутил)-метилдилил/-этил, пара-толуолсульфонилэтил, 4-нитробензилсульфонил-этил, аллил, циннамил, 1-(триметилсилилметил)-проп-1-ен-3-ил и тому подобные радикалы. Вид применяемой карбоксизащитной группы не является критическим постольку, поскольку производная карбоновая кислота остается стабильной в условиях последующей(их) реакции(ий) в других положениях молекулы и защитная группа может быть удалена в подходящий момент без разрушения остатка молекулы. В частности, важно не подвергать карбоксизащитную молекулу воздействию сильными нуклеофильными основаниями или восстановительными условиями, применяя высокоактивные металлические катализаторы, такие, как никель Ренея. (Таких жестких условий отщепления следует также избегать при удалении упоминавшихся здесь аминозащитных групп.) Предпочтительными защитными группами карбоновой кислоты являются аллил, бензгидрил и пара-нитробензильная группа. Аналогичные карбоксизащитные группы, используемые в технологии получения цефалоспорина, пенициллина и пептидов, могут быть также использованы для защиты карбокси-группы. Другие примеры этих групп можно найти у E. Haslam, "Protective Groups in Organic Chemistry", J.G.W. McOmie, Ed., Plenum Press, New York, N.Y., 1973, Chapter 5, and T.W.Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N.Y., 1981, Chapter 5.
Термин "защищенная аминогруппа", использованный в связи с формулой (2), касается аминогруппы, замещенной группой, обычно используемой для блокирования или защиты аминофункциональности в процессе взаимодействия других функциональных групп соединения. Примеры таких аминозащитных групп включают формильную группу, тритильную группу, трет-бутоксикарбонильную группу, фталимидогруппу, хлорацетильную, бромацетильную и иодацетильную группы, трихлорацетильную группу, блокирующие группы типа уретана, такие, как бензилоксикарбонил, 4-фенилбензилоксикарбонил, 2-метилбензилоксикарбонил, 4-метоксибензилоксикарбонил, 3-хлорбензилокcикарбонил, 2-хлорбензилокcикарбонил, 2,4-дихлорбензилоксикарбонил, 4-бромбензилоксикарбонил, 3-бромбензилоксикарбонил, 4-нитробензилоксикарбонил, 4-цианобензилоксикарбонил, 2-(4-бифенил)-изопропоксикарбонил, 1,1-дифенилэт-1-илоксикарбонил, 1,1-дифенилпроп-1-илоксикарбонил, 2-фенилпроп-2-ил-оксикарбонил, 2-(пара-толуил)-проп-2-илоксикарбонил, циклопентанилоксикарбонил, 1-метилциклопентанилоксикарбонил, циклогексанилоксикарбонил, 1-метилциклогексанилоксикарбонил, 2-метилциклогексанилоксикарбонил, 2-(4-толуилсульфонил)-этоксикарбонил, 2-(метилсульфонил)-этоксикарбонил, 2-(трифенилфосфино)-этоксикарбонил, 9-флуоренилметоксикарбонил ("FMOC") 2-(триметилсилил)-этоксикарбонил, аллилоксикарбонил, 1-(триметилсилилметил)-проп-1-енилоксикарбонил, 5-бензизоксалилметокcикарбонил, 4-ацетоксибензилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, 2-этинил-2-пропоксикарбонил, циклопропилметоксикарбонил, 4-(децилокси)-бензилоксикарбонил, изоборнилоксикарбонил, 1-пиперидинилоксикарбонил и тому подобные; бензоилметилсульфонильная группа, 2-(нитро)-фенилсульфенильная группа, группа дифенилфосфиноксида и подобные аминозащитные группы. Вид применяемой аминозащитной группы не является критическим, поскольку производная аминогруппа стабильна в условиях последующей(их) реакции(ий) в других положениях молекулы и может быть удалена в нужный момент без разрушения остатка молекулы. Предпочтительными аминозащитными группами являются аллилоксикарбонил, феноксиацетил, трет-бутоксикарбонил и тритил. Аналогичные аминозащитные группы, используемые в области химии цефалоспорина, пенициллина и пептидов, также входят в объем понятия упомянутых выше терминов. Другие примеры групп, относящихся к этим определениям, описаны J.W. Bouton, "Protective Groups in Organic Chemistry", J.G.W. McOmie, Ed., Plemum Press, New York, N.Y., 1973, Chapter 2 and T.W.Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N.Y., 1981, Chapter 7.
В формуле (2) предпочтительно, когда A и A', взятые вместе, для образования группы формул: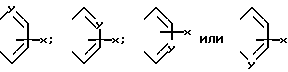
Особенно предпочтительно, когда A и A', взятые вместе, образуют группу формулы: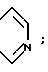
давая таким образом соединение формулы: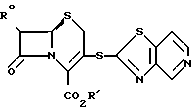
Соединения формулы (2) являются полезными промежуточными продуктами при получении противомикробных средств формулы (1). Соединения формулы (2) могут быть получены, как показано на схеме реакций 1, заменяя 3-трифлатную часть молекулы на желаемый тиол формулы: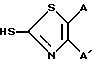 ,
,
используя 7-защищенное амино-ядро.
Конечные продукты формулы (1) могут быть получены затем из промежуточных соединений формулы (2) снятием защиты 7-аминофункции, если необходимо, с последующим ацилированием желаемой ацильной группой и дальнейшим удалением амино- и карбоксизащитных групп.
Экспериментальная часть
Получение 1. 7 бета-амино-3-хлор-3-цефем-4-карбоновая кислота
Указанное в названии примера соединение может быть получено методом Chavvette, описанным в патенте США N 4064343.
Получение 2. 3-(трет-бутилоксикарбонил)-аминопиридин
76,13 г (0,81 ммоля) 3-аминопиридина растворяли в 500 мл воды вместе со 150 мл трет-бутанола и 34 г (0,85 ммоля) гидроокиси натрия, охлаждали в ледяной бане и обрабатывали 200 г (0,92 ммоля) ди-трет-бутилбикарбоната. Через 2,5 дня добавляли еще 100 г ди-трет-бутилового эфира угольной кислоты. Затем реакционную смесь выливали в смесь этилацетата и воды. Органическую фазу отделяли, а оставшуюся водную фазу экстрагировали этилацетатом. Объединенные органические порции сушили над безводным сульфатом натрия, концентрировали в вакууме, очищали экспресс-хроматографией и получали 97 г (80%) целевого соединения, указанного в названии примера.
Спектр ЯМР (на ядрах водорода): (300 МГц, CDCl3), δ: 8,34 (д., J = 1,5 Гц, 1Н), 8,26 (д. , J = 3 Гц, 1Н), 7,97 (широкий дублет, J = 6 Гц, 1Н), 7,24-7,70 (м., 1Н), 6,81 (широкий синглет, 1Н), 1,51 (с., 9Н).
ИК-спектр (KBr, см-1): 3167, 2986, 1716, 1598, 1545, 1407, 1566, 1288, 1233, 1154, 1017.
Масс-спектр: FDMS м/е 195 (М+).
УФ-спектр (этанол) λ = 281 нм (ε = 3350)
λ = 235 нм (ε = 15200).
Получение 3. 3-(трет-бутилоксикарбонил)-амино-4-меркаптопиридин
10 г (51,5 ммоля) 3-(трет-бутилокcикарбонил)-аминопиридина растворяли в 110 мл тетрагидрофурана и охлаждали до -78oC под азотом. Затем добавляли двумя порциями 80 мл (120 ммолей), 1,6 М в гексане н-бутиллития. Затем реакционную смесь охлаждали в ледяной бане с ацетатом и позволили раствориться полученному твердому веществу. Примерно через два часа реакционную смесь охлаждали до -78oC и обрабатывали 2 г (7,8 ммоля) элементарной серы. Примерно через полчаса реакционную смесь нагревали до комнатной температуры и реакцию прекращали насыщенным раствором хлористого аммония. Обработкой и экспресс-хроматографией (50% смесь гексана и этилацетата) получали 5,24 (45%) целевого соединения, указанного в названии примера.
Точка плавления: 170-171oС (разлагается).
Спектр ЯМР на 1H (300 МГц, ДМСО-d6), δ: 12,88 (широкий синглет, 1Н), 8,95 (с., 1Н), 8,45 (шир.с., 1Н), 7,62 (шир. дублет, J = 3 Гц, 1Н), 7,44 (д. , = 3 Гц, 1Н), 1,49 (с., 9Н).
ИК-спектр (KBr, см-1): 3239, 2978, 2885, 2741, 1721, 1608, 1530, 1492, 1436, 1384, 1213, 1161, 1085.
Масс-спектр: FDMS м/е 227 (М+).
УФ-спектр (этанол): λ = 345 нм (ε = 19600)
λ = 259 нм (ε = 10200)
λ = 224 нм (ε = 17200).
Получение 4. Гидрохлорид 3-амино-4-меркаптопиридина
13,78 г (0,06 ммоля) 3-(трет-бутилоксикарбонил)-амино-4- меркаптопиридина растворяли уксусной кислотой (250 мл) и добавляли к охлажденному льдом раствору 3 н. хлористоводородной кислоты в уксусной кислоте, который получали барботированием газообразного хлористого водорода через ледяную уксусную кислоту (100 мл). Примерно через 4 ч полученное твердое вещество отфильтровывали, промывали диэтиловым эфиром, сушили в вакууме и получали 10,4 г (примерно 100% выход) целевого соединения.
Точка плавления: выше 200oC.
Спектр ЯМР на 1H (300 МГц, ДМСО-d6, δ: 8,17 (с., 1Н), 7,99 (д., J = 3 Гц, 1Н), 7,81 (д., J = 3 Гц, 1Н), 5,60-4,00 (широкий, 4Н).
ИК-спектр (KBr, см-1): 3184, 3054, 2848, 1639, 1586, 1482, 1442, 1134, 1123.
Масс-спектр: FDMS м/е 126 (М-36).
УФ-спектр (этанол): λ = 355 нм (ε = 13900)
λ = 264 нм (ε = 6830)
λ = 223 нм (ε = 13100)
Получение 5. 2-меркапто-5-пиридинотиазол
13 г (0,198 ммоля) гидроокиси калия растворяли в 32 мл воды и 154 мл метанола. Этот раствор обрабатывали затем 3,8 мл (0,063 ммоля) сероуглерода с последующей обработкой 10,4 г (0,06 ммоля) гидрохлорида 3-амино-4-меркаптопиридина. После нагревания с обратным холодильником и перемешиванием при оставлении на ночь реакционную смесь обрабатывали активированным углем и фильтровали через Hyflo Super Celтм. Фильтрат подкисляли уксусной кислотой, вызывающей образование твердого вещества. Полученное твердое вещество сушили в вакууме при 50oC в течение 3 ч и при комнатной температуре 2,5 дня, и получали 8,19 г (81%) целевого соединения, указанного в названии примера.
Точка плавления: выше 310oC (разлагается).
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 14.03 (широкий синглет, 1Н), 8,46 (с., 1Н), 8,33 (д., J = 6 Гц, 1Н), 7,75 (д., J = 6 Гц 1Н).
ИК-спектр (KBr, см-6): 3440 (широкая полоса), 2650 (шир.), 2510 (шир.), 1528, 1457, 1305, 1294, 1265, 1256, 1039, 1024, 815.
Масс-спектр: Е1 MS м/е 168 (М+).
Получение 6. 2-изотиоцианато-3-нитропиридин
10 г 2-хлор-3-нитропиридина, 8 г изотиоцианата калия и 75 г уксусной кислоты смешивали и нагревали с обратным холодильником 2 ч. Затем реакционную смесь охлаждали и выливали в 400 мл смеси воды со льдом. Полученное твердое вещество промывали водой, повторно растворяли в этилацетате и 4 раза промывали водой. Этилацетатный раствор затем обрабатывали активированным углем, сушили над безводным сульфатом натрия, фильтровали, выпаривали досуха и получали 3,72 г целевого соединения.
Точка плавления: 115-116oC.
Спектр ЯМР на 1H (300 МГц, CDCl3), δ : 8,62 (м., 1Н), 8,22 (д., J = 6 Гц, 1Н), 7,46 (м., 1Н).
Получение 7. 2-меркапто-3-нитропиридин
50 мл этанола обрабатывали 612 мг натрия при пониженной температуре (в ледяной бане) в безводных условиях. Затем реакционную смесь обрабатывали 3,6 г (0,02 ммоля) порциями соединения, полученного по примеру получения 6. Реакционную смесь перемешивали 2 ч, разбавляли 250 мл воды и выпаривали в вакууме. Затем раствор подкисляли уксусной кислотой до pH 4,5, и образовывались желтовато-красные кристаллы. Целевое соединение отфильтровывали, промывали, водой, сушили в вакуумном эксикаторе и получали 1,1 г.
Точка плавления: 185-187oC (разлагается).
Спектр ЯМР на 1H (300 МГц, CDCl3), δ : 8,09 (д., J = 7 Гц, 1Н), 7,89 (д. , J = 7 Гц, 1Н), 6,84 (дд., J = 6 и 3 Гц, 1Н).
ИК-спектр (KBr, см-1): 3119, 2872, 1611, 1577, 1527, 1349, 1330, 1240, 1141.
Масс-спектр: EIMS м/е 126 (М+).
Получение 8. 2-меркапто-3-аминопиридин
100 мл концентрированной водной хлористоводородной кислоты охлаждали в ледяной бане и обрабатывали 100 г (0,53 ммоля) хлористого олова (SnCl2). Затем реакционную смесь обрабатывали 14 г (0,11 ммоля) соединения, полученного по примеру получения 7, порционно и перемешивали 3 ч.
Затем реакционную смесь выпаривали до появления твердого остатка, растворяли в 1 л воды и обрабатывали газообразным сероводородом в течение 30 мин при нагревании на паровой бане. Полученное твердое вещество отфильтровывали, промывали теплой водой и сливали. Объединенные водные части выпаривали для получения твердого вещества. Полученное твердое вещество два раза варили с горячей концентрированной гидроокисью аммония. Полученное твердое вещество отфильтровывали и выгружали, а раствор гидроокиси аммония выпаривали для получения влажного твердого вещества, в которое в свою очередь переводили в воду. Полученное желто-зеленое целевое соединение фильтровали, промывали водой, сушили в вакууме над осушителем при 40oC и получали 4,20 г (выход 30%).
Спектр ЯМР на 1H (300 МГц, CDCl3 и ДМСО-d6), δ : 6,91 (м., 1Н), 6,65 (д. , J = 5 Гц, 1Н), 6,46 (м., 1Н), 5,03 (с., 2Н).
Получение 9. 2-меркапто-7-пиридинотиазол
2,8 г (85%) гидроокись калия растворяли в 16 мл воды и 50 мл метанола. Затем добавляли 2,6 г сероуглерода и промывали 30 мл метанола. Добавляли 4 г (23,8 ммоля) 2-меркапто-3-аминопиридина и реакционную смесь оставляли на ночь при нагревании с обратным холодильником. После охлаждения реакционную смесь обрабатывали активированным углем и фильтровали через super Celтм, промывая остаток на фильтре небольшим количеством метанола. Затем раствор подкисляли до pH 5,5 уксусной кислотой. Целевое соединение выпадало в осадок из этого раствора в виде желтоватого твердого вещества, которое сушили при 60oC над сиккативом. Выход составил 3,29 г.
Точка плавления: 285-287oC (разлагается).
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 8,38 (дд., J = 3 и 1,5 Гц, 1Н), 7,61 (дд. , J = 4 и 1,5 Гц, 1Н), 7,43 (дд., J = 5 и 3 Гц, 1Н), 3,33 (широкий синглет, 1Н).
ИК-спектр (KBr, см-1): 3040, 270oC 2540, 1597, 1523, 1399, 1311, 1302, 1274, 1132, 876.
Масс-спектр: Е1 М м/е 169 (М+1).
Получение 10. Этиловый эфир /2-(трифенилметил)-аминотиазол-4-ил/-2-бромэт-1-илоксиминоуксусной кислоты
9,88 г (0,02 ммоля) этил-/2-(трифенилметил)-аминотиазол-4-ил/-оксиминоацетата растворяли в 20 мл N,N'-диметилформамида и обрабатывали 8,28 г (0,06 ммоля) порошкообразного карбоната калия. После перемешивания в течение полчаса добавляли 17,3 мл 1,2-дибромэтана и реакционную смесь оставляли на ночь при перемешивании в атмосфере аргона.
Затем реакционную смесь переливали в 100 мл смеси дихлорметана на 200 мл воды. Водный слой снова экстрагировали дихлорметаном. Объединенные фазы дихлорметана промывали водой и рассолом, сушили над безводным сульфатом магния, фильтровали, выпаривали в вакууме и получали масло. Жидкостная хроматография (25% смесь гексана и дихлорметана давала 7,16 г (63,4%) целевого соединения.
Точка плавления: 55oC.
Спектр ЯМР на 1H (300 МГц, СДС13), δ : 7,32 (с., 15Н), 6,52 (с., 1Н), 4,55-4,46 (м., 2Н), 4,38 (кв., J = 4 Гц, 2Н), 3,63-3,53 (м., 2Н), 1,37 (т., J = 4 Гц, 3Н).
Элементарный анализ:
вычислено: C 59, 58; H 4,64; N 7,44
найдено: C 59,36; H 4,61; N 7,18.
Получение 11. Этил-/2-(трифенилметил)-аминотиазол-4-ил/- 2-фторэт-1-ил-оксиминоацетат
Указанное в названии примера целевое соединение получали способом, аналогичным описанному в примере получения 10, заменяя 1-бром-2-фторэтаном алкилирующий агент. Выход составил 3,3 г.
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 8,77 (с., 1Н), 7,39-7,12 (м., 15Н), 6,92 (с., 1Н), 4,60 (т., J = 3 Гц, 1Н), 4,44 (т., J = 3 Гц, 1Н), 4,26 (т. , J = 3 Гц, 1Н), 4,16 (т., J = 3 Гц 1Н), 3,90 (кв., J = 4 Гц, 2Н), 1,06 (т., J = 4 Гц, 3Н).
Получение 12. /2-(трифенилметил)-аминотиазол-4-ил/-Z- фторэт-1-ил-оксиминоуксусная кислота
2,5 г (5 ммолей) соединения, полученного по примеру получения 11, растворяли в 20 мл этанола и 5 мл (10 ммолей) 2 н. гидроокиси натрия.
После перемешивания в течение 2 ч при 50oC кристаллизовалась натриевая соль кислоты. Это твердое вещество диспергировали в смеси вода-хлороформ и подкисляли 1 н. хлористоводородной кислотой. Водный слой снова экстрагировали хлороформом и объединенная хлороформная фаза промывалась водой, рассолом и сушилась над безводным сульфатом магния. Затем хлороформную фазу испаряли в вакууме и получали 1,52 г (63,9%) целевого соединения в виде пены.
Точка плавления: 125, 33oC (разлагается).
Спектр ЯМР на 1Н (300 МГц, CDCl3), δ : 9,70 (шир.с., 1Н), 7,30-7,22 (м., 15Н), 6,52 (с., 1Н), 4,65 (т., J = 3 Гц, 1Н), 4,49 (т., J = 3 Гц, 1Н), 4,37 (т., J = 3 Гц, 1Н), 4,27 (т., J = 3 Гц, 1Н).
ИК-спектр (CDCl3, см-1): 3000, 1735, 1592, 1529, 1449, 1186, 1070, 1035.
Пример 1. 7 β-/(2-аминотиазол-4-ил)-(Z)-метоксиминоацетил/-амино-3-/2-(5- пиридинотиазолотио)/-3-цефем-4-карбоновая кислота
A. 7 бета-/(2-трифенилметил)-аминотиазол-4-ил-(Z)-метоксиминоацетил/-амино-3-хлор-3-цефем-4-карбоновая кислота
39,8 г (0,17 ммоля) 7 β-амино-3-хлор-3-цефем-4-карбоновой кислоты суспендировали в 800 мл N,N'-диметилформамида и обрабатывали 100 г (0,49 моля) бис-(диметилсилил)-мочевины, затем нагревали при 50-65oC в течение одного часа.
В другом реакционном сосуде 100 г (0,21 ммоля) 2-(трифенил-метил)-аминотиазол-4-ил-(Z)-метокcиминоукcусной кислоты растворяли в 800 мл N, N'-диметилформамида и охлаждали в ледяной бане с ацетоном. Затем реакционную смесь обрабатывали 23 мл (0,21 ммоля) N-метилморфолина, затем 25 г хлористого оксалина.
В первом реакционном сосуде реакционную смесь обрабатывали 32 мл (0,40 ммоля) пиридина и переносили с помощью канюли во второй реакционный сосуд в течение 50 мин.
Затем реакционную смесь выливали в 2 л смеси воды со льдом, полученное твердое вещество сушили на воздухе и получали 116 г соединения, указанного в названии примера A 1 (смесь 3:1, Δ3:Δ2).
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 9,61 (д., J = 9 Гц, 1Н), 8,83 (с. , 1Н х 1/4), 8,80 (с., 1Н х 3/4), 7,46-7,10 (широкий мультиплет, 15Н), 6,83 (с., 1Н), 6,68 (с., 1Н х 1/4), 5,72- 5,66 (м., 1Н х 3/4), 5,60-5,54 (м. , 1Н х 1/4), 5,23-5,17 (м., 1Н х 1/4), 5,20 (д., J = 5 Гц, 1Н х 3/4), 4,83 (с., 1Н х 1/4), 3,80 (с., 3Н), 3,79 (АВ кв., J = 20 Гц, 2Н х 3/4).
B. Бензгидриловый эфир 7 β-/2-(трифенилметил)-аминотиазол-4-ил-(Z)-метоксиминоацетил/-амино-3-хлор-3-цефем-4-карбоновой кислоты
Соединение, полученное в части А примера 1, растворяли в 500 мл цианистого метила (CH3CN) и обрабатывали 10 г особо чистого дифенилдиазометана и перемешивали при комнатной температуре примерно 2,5 дня. Затем реакционную смесь резко охлаждали уксусной кислотой и для превращения реакции концентрировали в вакууме, используя толуол для азеотропного избытка уксусной кислоты. Очистка экспресс-хроматографией (25% и 50% смесь этилацетат-гексан) давала 15,46 г смеси целевого соединения в отношении 2:1 Δ2-Δ3).
Спектр ЯМР на 1H (300 МГц, ДМСО-d6), δ : 9,60 (д., J = 6 Гц, 1Н 8,80 (с. , 1Н), 7,46-7,02 (широкий м. , 25Н), 6,92 (с., 1Н х 1/3), 6,88 (с., 1Н х 2/3), 6,84 (с., 1Н х 2/3), 6,78 (с., 1Н х 2/3), 6,67 (с., 1Н х 1/3), 5,76-5,70 (м. , 1Н х 1/3), 5,51-5,45 (м., 1H х 2/3), 5,76-5,70 (м., 1Н х 1/3), 5,51-5,45 (м. , 1Н х 2/3), 5,12 (д., J = 4 Гц, 1Н х 2/3), 3,79 (AB кв., J = 19 Гц, 2Н х 1/3), 3,77 (с., 3Н).
C. Бензгидриловый эфир 7 β-/2-(трифенилметил)-аминотиазол-4-ил-(Z)-метоксимино/-ацетил-3-/2-(5-пиридинотиазолотио)/-3-цефем-4-карбоновой кислоты
92 мг (2,3 ммоля, 60% в масле) гидрида натрия промывали гексаном и суспендировали в 50 мл тетрагидрофурана и обрабатывали 390,9 мг (2,3 ммоля) 2-меркапто-5-пиридинотиазола и нагревали. Раствор переносили посредством канюли к 5,7 г (2,3 ммоля) соединения, полученного в части В примера 1, растворенного в 50 мл тетрагидрофурана. Затем реакционную смесь обрабатывали 15 мл 1 н. хлористоводородной кислоты и выливали в смесь этилацетата и воды. Органическую фазу промывали рассолом, сушили над безводным сульфатом натрия. Фильтровали и концентрировали в вакууме, после колоночной хроматографии (75-90% смесь этилацетат-гексан) кристаллизовался чистый Δ3-изомер (0,31 г, 34%).
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 9,70 (д., J= 9 Гц, 1Н), 9,10 (с. , 1Н), 8,79 (с., 1Н), 8,47 (д., J = 7 Гц, 1Н), 8,08 (д., J = 7 Гц, 1Н), 7,38-7,03 (шир. мультиплет, 25Н), 6,91 (с., 1Н), 6,68 (с., 1Н), 5,90-5,82 (м. , 1Н), 5,37 (д., J = 8 Гц, 1Н), 3,83 (AB.кв., J = 20 Гц, 2Н), 3,78 (с., 3Н).
ИК-спектр (KBr, см-1): 3402 (широкий), 3030, 2938, 1786, 1738, 1695, 1522, 1496, 1371, 1278, 1223, 1044, 700.
Масс-спектр: FABMS м/е 958 (М+).
Оптическое вращение: (α)D = -133,33o, 589 нм, 5 мг диметилсульфоксида.
Элементарный анализ:
вычислено: C 63,93; H 4,10; N 10,23
найдено: C 64,19; H 4,06; N 10,43.
D. Снятие защиты для получения целевого соединения, указанного в названии примера 1.
0,42 г (438 ммолей) соединения, полученного в части C, суспендировали с 7 мл триэтилсилана и 10 мл дихлорметана и обрабатывали 5 мл трифторуксусной кислоты и перемешивали при комнатной температуре. Затем реакционную смесь концентрировали в вакууме, используя толуол, чтобы привести к азеотропной смеси избыток трифторуксусной кислоты. Полученный остаток очищали обращенно-фазовой хроматографией (10-20% смесь цианистого метила c водой).
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 9,75 (д., J = 9 Гц, 1Н), 9,18 (с. , 1Н), 8,49 (д., J = 6 Гц, 1Н), 8,19 (д., J = 6 Гц, 1Н), 7,21 (широкий синглет, 2Н), 6,71 (с., 1Н), 5,94 (дд., J = 6 Гц, и 10 Гц, 1Н), 5,35 (д., J = 6 Гц, 1Н), 3,88 (AB кв., J = 15 Гц, 2Н), 3,85 (с., 3Н).
ИК-спектр (KBr, см-1): 3395, 1782, 1621, 1532, 1381, 1037.
Масс-спектр: FABMS м/е 550 (М+).
Уф-спектр (этанол): λ = 286 нм (ε = 22700)
λ = 231 нм (ε = 34200).
Оптическое вращение (α)D = -123,26o, 589 нм.
Примеры 2-5. Соединения по примерам 2-5 были получены в основном способом, аналогичным описанному в примере 1, но используя различные меркаптаны формулы: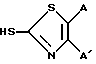
Пример 2. 7 β-/(2-аминотиазол-4-ил)-(Z)-(2-фторэт-1-ил)-оксиминоацетил/-амино-3-/2-(5-пиридиноатиазоло)/-тио-3-цефем-4-карбоновая кислота
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 9,70 (д., J = 10 Гц, 1Н), 9,03 (с. , 1Н), 8,39 (д., 1Н, J = 5 Гц), 8,03 (д., J = 5 Гц, 1Н), 7,20 (с., 2Н), 6,72 (с., 1Н), 5,73 (м., 1Н), 5,19 (д., 1Н, J = 7 Гц), 4,67 (т., J = 5 Гц, 1Н), 4,55 (т., J = 5 Гц, 1Н), 3,63 (AB кв., J = 18 Гц, 2Н).
ИК-спектр (KBr, см-1): 3420, 1774, 1668, 1663, 1653, 1617, 1534, 1388.
Масс-спектр: ("FAB") м/e 604 (М+1).
УФ-спектр (этанол): λ = 288 нм (ε = 21700)
λ = 232 нм (ε = 31400).
Оптическое вращение: (α)
Пример 3. 7 β-/(2-аминотиазол-4-ил)-(Z)-(2-фторэт- 1-ил)-оксиминоацетил/-амино-3-/2-(7-пиридинотиазолотио)/3-цефем-4-карбоновая кислота
Выход общий 13% (22,8 мг).
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 9,68 (д., 1Н, J = 10 Гц), 8,25 (д., 1Н, J = 5 Гц), 8,14 (д., 1Н, J = 10 Гц), 7,45 (м., 1Н), 7,20 (с., 2Н), 6,72 (с., 1Н), 5,70 (м., 1Н), 5,20 (д., 1Н, J = 5 Гц), 4,70 (т., 1Н, J = 5 Гц), 4,53 (т., 1Н, J = 5 Гц), 4,30 (т., 1Н, J = 5 Гц), 4,20 (т., 1Н, J = 5 Гц), 3,63 (AB кв., 2Н, J = 15 Гц).
Масс-спектр ("FAB"): м/e = 604 (М+1).
Пример 4. 7 β-/(2-аминотиазол-4-ил)-(Z)-(2-фторэт- 1-ил)-оксиминоацетил/-амино-3-(тиазол-2-ил)-тио-3-цефем-4-карбоновая кислота
Выход 63 мг (71%).
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 9,67 (д., 1Н, J = 10 Гц), 7,76 (с. , 2Н), 7,20 (с., 2Н), 6,72 (с., 1Н), 5,80-5,70 (м., 1Н), 5,20 (д., 1Н, J = 5 Гц), 4,66 (т., 1Н, J = 5 Гц), 4,50 (т., 1Н, J = 5 Гц), 4,28 (т., 1Н, J = 5 Гц), 4,19 (т., 1Н, J = 5 Гц), 3,50 (AB кв., 2Н, J = 15 Гц).
ИК-спектр (KBr, см-1): 3400, 1768, 1653, 1614, 1535, 1389, 1350, 1035.
Масс-спектр: ("FAB") м/e = 553 (М-1).
Уф-спектр (этанол): λ = 284 нм (ε = 14900)
λ = 231 нм (ε = 21800)
Пример 5. Натриевая соль 7 бета-/2-аминотиазол-4-ил- (z)-(2-фторэт-1-ил)-оксиминоацетил/-амино-3/-(бензотиазол-2-ил)-тио/-3-цефем-4-карбоновой кислоты
Спектр ЯМР на 1H (300 МГц, ДМСО - d6), δ : 9,67 (д., 1Н, J = 10 Гц), 7,92 (д., 1Н, J = 10 Гц), 7,78 (д., 1Н, J = 10 Гц), 7,43-7,26 (м., 2Н), 7,20 (с. , 2Н), 6,73 (с., 1Н), 5,66 (м., 1Н), 5,15 (д., 1Н, J = 5 Гц), 4,70 (т., 1Н, J = 5 Гц), 4,20 (т., 1Н, J = 5 Гц), 3,64 (AB кв., 2Н, J = 15 Гц).
Масс-спектр ("FAB"): м/e = 603 (М+1).
Примеры готовых форм композиций
Пример 1. Твердые желатиновые капсулы приготавливаются с использованием следующих ингредиентов. - Количество (мг/капсулу)
Активный ингредиент - 250
Крахмал, высушенный - 200
Стеарат магния - 10
Всего - 460 мг
Пример 2. Таблетки получают с использованием следующих ингредиентов - Количество (мг/таблетку)
Активный ингредиент - 250
Целлюлоза, микрокристаллическая - 400
Двуокись кремния, измельченная в тонкий порошок - 10
Стеариновая кислота - 5
Всего - 665 мг
Компоненты смешиваются и прессуются в форму таблеток с образованием таблеток, каждая весом 665 мг.
Пример 3. Приготавливается аэрозольный раствор, содержащий следующие компоненты: - Вес, %
Активный ингредиент - 0,25
Метанол - 29,75
Пропеллент 22 (хлордифторметан) - 70.00
Итого - 100,00
Активный ингредиент смешивается с этанолом, и смесь добавляется к части пропеллента 22, охлаждается до -30oC и переносится в заполняющее устройство. В контейнер из нержавеющей стали помещается требуемое количество препарата и разбавляется остатком пропеллента. Затем на контейнере устанавливается дозирующий клапан.
Пример 4. Таблетки, каждая из которых содержит 60 мг активного ингредиента, изготавливаются следующим образом, мг:
Активный ингредиент - 60
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде 10% раствора в воде) - 4
Стеарат магния - 0,5
Тальк - 1
Итого - 150
Активный ингредиент, крахмал и целлюлоза пропускаются через сито США 45 меш и тщательно смешиваются. С полученным порошком смешивается водный раствор, содержащий поливинил-пирролидон, и смесь затем пропускается через сито 14 меш США. Полученные таким образом гранулы сушатся при 50oC и пропускаются через сито 18 меш США. Затем к гранулам добавляются натриевый карбоксиметил-крахмал, стеарат магния и тальк, предварительно пропущенные через сито 60 меш, и гранулы после смешения прессуются на таблетирующем устройстве, давая таблетки, каждая весом 150 мг.
Пример 5. Капсулы, каждая из которых содержит 80 мг активного ингредиента, приготавливаются из следующих компонентов, мг
Активный ингредиент - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Итого - 200
Активный ингредиент, целлюлоза, крахмал и стеарат магния смешиваются, пропускаются через сито 45 меш (США) и заполняются в твердые желатиновые капсулы в количестве по 200 мг.
Пример 6. Медицинские свечи, каждая с содержанием активного ингредиента 225 мг, приготавливаются следующим образом, мг
Активный ингредиент - 225
Глицериды насыщенной жирной кислоты - 2000
Итого - 2225
Активный ингредиент пропускается через сито 60 меш США и суспендируется в глицеридах насыщенных жирных кислот, предварительно расплавленных с использованием минимально необходимого тепла. Смесь затем выливается в форму для медицинских свечей номинальной емкостью 2 г и оставляется охлаждаться.
Пример 7. Суспензионные препараты, каждый содержащий 50 мг активного ингредиента на 5 мл дозу, приготавливаются следующим образом:
Активный ингредиент - 50 мг
Натриевая карбоксиметилцеллюлоза - 50 мг
Сироп - 1,25 мл
Раствор бензойной кислоты - 0,10 мл
Вкусовая добавка - g•v
Красящий агент - g•v
Очищенная вода до общего объема - 5 мл
Активный ингредиент пропускается через сито 45 меш США и смешивается с натриевой карбоксиметилцеллюлозой и сиропом с образованием однородной пасты. Разбавляются частичным количеством воды и добавляются при перемешивании раствор бензойной кислоты, вкусовая добавка и красящий агент. Затем добавляется достаточное количество воды для получения требуемого объема.
Пример 8. Препаративная форма для внутривенного применения приготавливается из следующих компонентов:
Активный ингредиент - 100 мг
Изотонический физиологический раствор - 1000 мл
Раствор указанных выше ингредиентов обычно назначается для внутривенного введения пациенту со скоростью 1 мл в минуту.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-КАРБА-(1-ДЕТИА)-ЦЕФЕМА | 1993 |
|
RU2107067C1 |
| ПРОИЗВОДНЫЕ 3-ТРИФТОРМЕТИЛ-1-КАРБА-1-ДЕТИА-3-ЦЕФЕМ-4-КАРБОНОВОЙ КИСЛОТЫ И ПРОМЕЖУТОЧНЫЙ ПРОДУКТ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2089551C1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2076100C1 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНОГО ЭФИРА СУЛЬФОКСИДА 3-ЭКЗОМЕТИЛЕНЦЕФАМА | 1992 |
|
RU2010795C1 |
| Способ получения тиенопиридиний- или фуропиридиний-замещенных производных цефалоспорина | 1982 |
|
SU1169542A3 |
| ЗАМЕЩЕННЫЕ ФЕНИЛФЕНОЛЬНЫЕ СОЕДИНЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2095340C1 |
| НАФТИЛПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ ВЕЩЕСТВА | 1996 |
|
RU2167849C2 |
| Способ получения производных цефалоспорина | 1981 |
|
SU1087076A3 |
| СПОСОБ МИНИМИЗАЦИИ ГИСТЕРОТРОФНОГО ЭФФЕКТА ТАМОКСИФЕНА И АНАЛОГОВ ТАМОКСИФЕНА | 1995 |
|
RU2158589C2 |
| БЕНЗОТИОФЕНОВЫЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1996 |
|
RU2158737C2 |




Сущность изобретения: производные цефалоспорина общей формулы (I), в которой R означает атом водорода, C1-C6-алкил или C1-C6-галоидалкил; A и A', взятые вместе, означают группу формулы (II-V), в которой у означает атом азота или водорода, а X означает атом водорода, атом галогена, C1-C6-алкил, C1-C6-алкоксил, амино- или нитрогруппу, или их фармацевтически приемлемые соли и фармацевтическая композиция, содержащая в качестве активного компонента производное цефалоспорина общей формулы (I) и фармацевтически приемлемый носитель. Общая формула (I):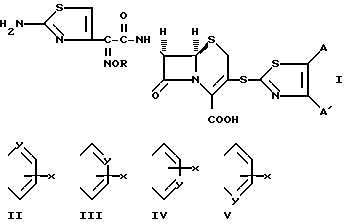
2 с. и 7 з.п. ф-лы, 1 табл.
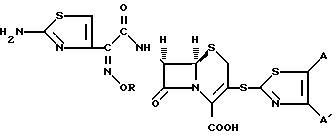
в которой R водород, С1 С6-алкил или С1 - С6-галоидалкил;
A и A1, взятые вместе, образуют группу формул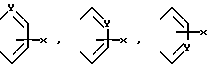
или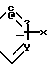
где X водород, галоген, С1 С6-алкил, С1 - С6-алкоксил, амино- или нитрогруппа;
Y азот или углерод,
и их фармацевтически приемлемые соли.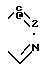
7. Соединения по п.6, в которых R метил или фторметил.
| EP, патент, 212923, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 228906, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 323177, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 329457, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 182301, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
